WO2008017827A2 - Azole and thiazole derivatives and their uses - Google Patents
Azole and thiazole derivatives and their uses Download PDFInfo
- Publication number
- WO2008017827A2 WO2008017827A2 PCT/GB2007/002992 GB2007002992W WO2008017827A2 WO 2008017827 A2 WO2008017827 A2 WO 2008017827A2 GB 2007002992 W GB2007002992 W GB 2007002992W WO 2008017827 A2 WO2008017827 A2 WO 2008017827A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- hydroxy
- alkyl
- group
- compound
- methyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *C(*)(*)C1=NC(S)=C(*N(C(I)(I)IC=O)P)*1 Chemical compound *C(*)(*)C1=NC(S)=C(*N(C(I)(I)IC=O)P)*1 0.000 description 7
- MOWQGCIZQXWZDV-QFIPXVFZSA-N CC(C)(C)[Si](C)(C)O[C@@H](CN)c(c(C=C1)c2NC1=O)ccc2OCc(cc1)ccc1OC Chemical compound CC(C)(C)[Si](C)(C)O[C@@H](CN)c(c(C=C1)c2NC1=O)ccc2OCc(cc1)ccc1OC MOWQGCIZQXWZDV-QFIPXVFZSA-N 0.000 description 1
- YKDIBVQSVMDVQN-UHFFFAOYSA-N CCC(c(cc1)c(C=CC(N2)O)c2c1O)O Chemical compound CCC(c(cc1)c(C=CC(N2)O)c2c1O)O YKDIBVQSVMDVQN-UHFFFAOYSA-N 0.000 description 1
- TXCHAJAWFWXYFJ-UHFFFAOYSA-N CN(C)Cc1cnc(C(c2ccccc2)(c2ccccc2)O)[o]1 Chemical compound CN(C)Cc1cnc(C(c2ccccc2)(c2ccccc2)O)[o]1 TXCHAJAWFWXYFJ-UHFFFAOYSA-N 0.000 description 1
- VRTKVWXXBNNXKS-OEEKEXDXSA-N CN(CCCCCCCCCCNC[C@@H](c(cc1)c(C=CC(N2)=O)c2c1O)O)Cc1cnc(C(C2CCCC2)(c2ccccc2)O)[o]1 Chemical compound CN(CCCCCCCCCCNC[C@@H](c(cc1)c(C=CC(N2)=O)c2c1O)O)Cc1cnc(C(C2CCCC2)(c2ccccc2)O)[o]1 VRTKVWXXBNNXKS-OEEKEXDXSA-N 0.000 description 1
- UGJFLUUUNWTLOG-OEEKEXDXSA-N CN(CCCCCCCCCNC[C@@H](c(c(C=C1)c2NC1=O)ccc2O)O)Cc1cnc(C(C2CCCCC2)(c2ccccc2)O)[o]1 Chemical compound CN(CCCCCCCCCNC[C@@H](c(c(C=C1)c2NC1=O)ccc2O)O)Cc1cnc(C(C2CCCCC2)(c2ccccc2)O)[o]1 UGJFLUUUNWTLOG-OEEKEXDXSA-N 0.000 description 1
- ZMEVAJOHVFUPMR-UHFFFAOYSA-N NCC(c(c(S1)c2NC1=O)ccc2O)O Chemical compound NCC(c(c(S1)c2NC1=O)ccc2O)O ZMEVAJOHVFUPMR-UHFFFAOYSA-N 0.000 description 1
- IMROOZIAGHAUBL-UHFFFAOYSA-N OC(c1ncc(CBr)[o]1)(c1ccccc1)c1ccccc1 Chemical compound OC(c1ncc(CBr)[o]1)(c1ccccc1)c1ccccc1 IMROOZIAGHAUBL-UHFFFAOYSA-N 0.000 description 1
- XTEGVFVZDVNBPF-UHFFFAOYSA-N OS(c1cccc2c1cccc2S(O)(=O)=O)(=O)=O Chemical compound OS(c1cccc2c1cccc2S(O)(=O)=O)(=O)=O XTEGVFVZDVNBPF-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/42—Oxazoles
- A61K31/421—1,3-Oxazoles, e.g. pemoline, trimethadione
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/425—Thiazoles
- A61K31/426—1,3-Thiazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- This invention relates to oxazole and thiazole derivatives, pharmaceutical compositions, methods for their preparation and use in the treatment of diseases where enhanced M3 receptor activation is implicated.
- Anti-cholinergic agents prevent the passage of, or effects resulting from the passage of, impulses through the parasympathetic nerves. This is a consequence of the ability of such compounds to inhibit the action of acetylcholine (Ach) by blocking its binding to the muscarinic cholinergic receptors.
- M1 -M5 muscarinic acetylcholine receptors
- M1 -M5 muscarinic acetylcholine receptors
- M3 mAChRs mediate contractile responses (reviewed by Caulfield, 1993, Pharmac. Ther., 58, 319 - 379).
- muscarinic receptors M1 , M2 and M3 have been demonstrated to be important and are localized to the trachea, the bronchi, submucosal glands and parasympathetic ganglia (reviewed in Fryer and Jacoby, 1998, Am J Resp Crit Care Med., 158 (5 part 3) S 154 - 160).
- M3 receptors on airway smooth muscle mediate contraction and therefore bronchoconstriction. Stimulation of M3 receptors localised to submucosal glands results in mucus secretion.
- vagal tone may either be increased (Gross et al. 1989, Chest; 96:984-987) and/or may provoke a higher degree of obstruction for geometric reasons if applied on top of oedematous or mucus-laden airway walls (Gross et al. 1984, Am Rev Respir Dis; 129:856-870).
- M3 mAChR antagonists may be useful as therapeutics in these mAChR-mediated diseases.
- Tiotropium (Spiriva TM) is a long-acting muscarinic antagonist currently marketed for the treatment of chronic obstructive pulmonary disease, administered by the inhaled route.
- ipratropium is a muscarinic antagonist marketed for the treatment of COPD.
- WO97/30994 describes oxadiazoles and thiadiazoles as muscarinic receptor antagonists.
- EP0323864 describes oxadiazoles linked to a mono- or bicyclic ring as muscarinic receptor modulators.
- ⁇ 2 adrenergic receptor agonists The class of ⁇ 2 adrenergic receptor agonists is well known. Many known ⁇ 2-agonists, in particular, long-acting ⁇ 2-agonists such as salmeterol and formoterol, have a role in the treatment of asthma and COPD. These compounds are also generally administered by inhalation. Compounds currently under evaluation as once-daily ⁇ 2 agonists are described in Expert Opin. Investig. Drugs 14 (7), 775-783 (2005). A well known ⁇ 2-agonist pharmacophore is the moiety:
- compositions that contain both a muscarinic antagonist and a ⁇ 2-agonist for use in the treatment of respiratory disorders.
- US2005/0025718 describes a ⁇ 2-agonist in combination with tiotropium, oxotropium, ipratropium and other muscarinic antagonists;
- WO02/060532 describes the combination of ipratropium with ⁇ 2-agonists and
- WO02/060533 describes the combination of oxotropium with ⁇ 2-agonists.
- Other M3 antagonist / ⁇ 2-agonist combinations are described in WO04/105759 and WO03/087097.
- Such bifunctional molecules provide bronchodilation through two separate modes of action whilst possessing single molecule pharmacokinetics.
- Such a molecule might be easier to formulate for therapeutic use as compared to two separate compounds and might be more easily co-formulated with a third active ingredient, for example a steroid.
- Such molecules are described in for example, WO04/074246, WO04/089892, WO05/111004, WO06/023457 and WO06/023460, all of which use different linker radicals for covalently linking the M3 antagonist to the ⁇ 2-agonist.
- R 1 is CrC 6 -alkyl or hydrogen; and R 2 is hydrogen or a group -R 7 , -Z-Y-R 7 , -Z-NR 9 R 10 ; -Z-CO-NR 9 R 10 , -Z-NR 9 -C(O)O-R 7 , or ; -Z-C(O)-R 7 ; and R 3 is a lone pair, or R 3 is C r C 6 -alkyl in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge; or
- R 1 and R 3 together with the nitrogen to which they are attached form a heterocycloalkyl ring
- R 2 is a lone pair or R 2 a group -R 7 , -Z-Y-R 7 , -Z-NR 9 R 10 , -Z-CO-NR 9 R 10 , -Z-NR 9 -C(O)O-R 7 ; or -Z-C(O)-R 7 , in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge; or
- R 1 and R 2 together with the nitrogen to which they are attached form a heterocycloalkyl ring, said ring being substituted by a group -Y-R 7 , -Z-Y-R 7 , -Z- NR 9 R 10 ; -Z-CO-NR 9 R 10 ; -Z-NR 9 -C(O)O-R 7 ; or ; -Z-C(O)-R 7 ; and R 3 is a lone pair, or R 3 is Ci-C 6 -alkyl in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge;
- R 4 and R 5 are independently selected from the group consisting of aryi, aryl-fused- heterocycloalkyl, heteroaryl, C r C 6 -alkyl, cycloalkyl;
- R 6 is -OH, C r C 6 -alkyl, C r C 6 -alkoxy hydroxy-C r C 6 -alkyl, nitrile, a group CONR 8 2 or a hydrogen atom;
- A is an oxygen or a sulfur atom
- X is a C r C 12 -alkylene, C 2 -C 12 -alkenylene or C 3 -C 12 -alkynylene group;
- R 7 is an C r C 6 -alkyl, aryl, aryl-fused-cycloalkyl, aryl-fused-heterocycloalkyl, heteroaryl, aryl(CrC 8 -alkyl)-, heteroary ⁇ CrCs-alkyl)-, cycloalkyl or heterocycloalkyl group;
- R 8 is C r C 6 -alkyl or a hydrogen atom
- Z is a CrC ⁇ -aikylene, C 2 -C 16 -alkenylene or C 2 -Ci 6 -alkynylene group;
- Y is a bond or oxygen atom
- R 9 and R 10 are independently a hydrogen atom, C r C 6 -alkyl, aryl, aryl-fused- heterocycloalkyl, aryl-fused-cycloalkyl, heteroaryl, aryl(CrC 6 -alkyl)-, or heteroaryl(C r C 6 -alkyl)- group; or R 9 and R 10 together with the nitrogen atom to which they are attached form a heterocyclic ring of 4-8 atoms, optionally containing a further nitrogen or oxygen atom;
- each occurrence of alkyl, heterocycloalkyl, aryl, aryl-fused-heterocycloalkyl, heteroaryl, cycloalkyl, alkoxy, alkylene, alkenylene, alkynylene or aryl-fused-cycloalkyl may be optionally substituted;
- R 1 is C r C 6 -alkyI
- R 2 is a group -Z-NR 9 R 10
- R 3 is a lone pair or R 3 is C r C 6 -alkyl in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge.
- R groups of the present invention include:
- R ,10 groups of the present invention include:
- X is an optionally substituted C 1 -C 3 alkylene group. More conveniently, X is a C 1 -C 2 alkylene group. Most conveniently, X is methylene.
- each alkyl, heterocycloalkyl, aryl, aryl- fused-heterocycloalkyl, heteroaryl, cycloalkyl, alkoxy, alkylene, alkenylene, alkynylene or aryl-fused-cycloalkyl group of the compounds of fomula (I) is unsubstituted.
- each alkenylene chain may contain 1 , 2 or 3 carbon-carbon double bonds and each alkynylene chain may contain up to 1 , 2 or 3 carbon-carbon triple bonds.
- R 1 is CrC 6 -alkyl or hydrogen; and R 2 is a group-Z-NR 9 R 10 ; and R 3 is a lone pair, or R 3 is CrC 6 -a!kyl in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge; or
- R 1 and R 3 together with the nitrogen to which they are attached form a heterocycloalkyl ring
- R 2 is a group -Z-NR 9 R 10 , in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge
- R 1 and R 2 together with the nitrogen to which they are attached form a heterocycloalkyl ring, said ring being substituted by a group , -Z-NR 9 R 10
- R 3 is a lone pair, or R 3 is C r C 6 -alkyl in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge
- R 4 and R 5 are independently selected from the group consisting of aryl, aryl-fused- heterocycloalkyl, heteroaryl, C r C 6 -alkyl, cycloalkyl;
- R 6 is -OH, C r C 6 -alkyl, C r C 6 -alkoxy hydroxy-C r C 6 -alkyl, nitrite, a group CONR 8 2 or a hydrogen atom;
- A is an oxygen or a sulfur atom
- X is a C r Ci 2 -alkylene, C 2 -C 12 -alkenylene or C 3 ⁇ C 12 -alkynylene group;
- R 8 is CrCg-alkyl or a hydrogen atom
- Z is a C 7 -C 1 i-alkylene, C 7 -C 1 r alkenylene or C 7 -C 1 r alkynylene group;
- L represents a linker comprising a hydrocarbyl chain of 7 to 11 carbon atoms, wherein the chain may additionally comprise up to three carbon-carbon double bonds, and, wherein the chain may additionally comprise up to three carbon-carbon triple bonds;
- L 1 and L 2 each independently represent hydrogen, Ci -6 alkyl or C 3-6 cycloalkyl;
- L 3 and L 4 each independently represent hydrogen, C 1 ⁇ alkyl or C 3 . 6 cycloalkyl, wherein C 1-6 alkyl and C 3-6 cycloalkyl may be optionally substituted by one or more substituents independently selected from halogen and hydroxyl; and * denotes the point of attachement of the group of formula (I) to the non- aromatic nitrogen bearing R 1 and R 3 , and * * denotes the point of attachment to the group NR 9 R 10 ;
- R 9 is a hydrogen atom or CVC 6 -alkyl
- R 10 is an aryl(C r C 6 -alkyl)-, or heteroaryl(C r C 6 -alkyl) group, in which the C r C 6 -a!kyl group is optionally substituted by hydroxy;
- alkyl, heterocycloalkyl, aryl, aryl-fused-heterocycloalkyl, heteroaryl, cycloalkyl, alkoxy, alkylene, alkenylene and alkynylene may be optionally substituted;
- the present invention provides a prodrug of a compound of formula (Ia) as herein defined, or a pharmaceutically acceptable salt thereof.
- the present invention provides an N-oxide of a compound of formula (Ia) as herein defined, or a prodrug or pharmaceutically acceptable salt thereof.
- the present invention provides a solvate (such as a hydrate) of a compound of formula (Ia) as herein defined, or an N-oxide, prodrug or pharmaceutically acceptable salt thereof.
- R 1 is Ci-C ⁇ -alkyl ;
- R 2 is a group -Z-NR 9 R 10 and R 3 is a lone pair or
- R 3 is C r C 6 -alkyl, in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge, or
- R 1 and R 2 together with the nitrogen to which they are attached represent a heterocycloalkyl ring, said ring being substituted by a group -Z-NR 9 R 10 and R 3 is a lone pair or R 3 is C r C 6 -alkyl in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge; or R 1 and R 3 together with the nitrogen to which they are attached represent a heterocycloalkyl ring, and R 2 is a group -Z-NR 9 R 10 in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge;
- R 4 is phenyl and R 5 is selected from the group consisting of aryl, heteroaryl, C 1 -C 6 - alkyl, cycloalkyl; or R 4 and R 5 are both heteroaryl;
- R 6 is -OH or, C r C 6 -alkyl
- R 8 is a hydrogen atom
- A is an oxygen or a sulfur atom
- X is a C r C 2 -alkylene group
- Z is a divalent linker radical of formula (A):
- L represents a linker comprising a hydrocarbyl chain of 7 to 11 carbon atoms, wherein the chain may additionally comprise up to three carbon-carbon double bonds, and, wherein the chain may additionally comprise up to three carbon-carbon triple bonds;
- L 1 and L 2 each independently represent hydrogen, C 1-6 alkyl or C 3 . 6 cycloalkyl
- L 3 and L 4 each independently represent hydrogen, Ci. 6 alkyl or C 3 . 6 cycloalkyl, wherein C 1-6 alkyl and C 3 . 6 cycloalkyl may be optionally substituted by one or more substituents independently selected from halogen and hydroxyl; and * denotes the point of attachement of the group of formula (I) to the non- aromatic nitrogen bearing R 1 and R 3 , and * * denotes the point of attachment to the group NR 9 R 10 ;
- R 9 is a hydrogen atom; and R 10 is selected from the group
- the present invention provides a compound of formula (Ia), or a pharmaceutically acceptable salt thereof, wherein R 1 is Ci-C 6 -alkyl; R 2 is a group -Z- NR 9 R 10 ; and R 3 is a lone pair or R 3 is a C r C 6 -alkyl in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge.
- the present invention provides compounds of formula (Ia) wherein the carbon to which R 4 , R 5 and R 6 are attached has the (R)- absolute configuration.
- the present invention provides compounds of formula (Ia) wherein the non-aromatic nitrogen shown in formula (Ia) is a tertiary nitrogen.
- the present invention provides quaternary ammonium salts of formula (Ia) wherein the non-aromatic nitrogen shown in formula (Ia) is quaternary nitrogen, carrying a positive charge.
- the present invention provides a compound of formula (Ia), or a pharmaceutically acceptable salt thereof, wherein R 4 and R 5 are independently selected from methyl, ethyl, n- or isopropyl, n-, sec- and tertbutyl; phenyl, 3,4- methylenedioxyphenyl, 3,4-ethylenedioxyphenyl, dihydrobenzofuranyl, naphthyl; pyridyl, pyrrolyl, pyrimidinyl, oxazolyl, isoxazolyl, benzisoxazolyl, benzoxazolyl, thiazolyl, benzthiazolyl, quinolyl, thienyl, benzthienyl, furyl, benzfuryl, imidazolyl, benzimidazolyl, isothiazolyl, benzisothiazolyl, pyrazolyl, isothiazolyl, triazolyl, benztriazo
- the present invention provides a compound of formula (Ia), or a pharmaceutically acceptable salt thereof, wherein (i) each of R 4 and R 5 is thienyl; or (ii) each of R 4 and R 5 is phenyl; or (iii) one of R 4 and R 5 is phenyl and the other is cyclopentyl or cyclohexyl; or (iv) one of R 4 and R 5 is thienyl, and the other is cyclopentyl or cyclohexyl.
- the present invention provides a compound of formula (Ia), or a pharmaceutically acceptable salt thereof, wherein R 6 is -OH.
- the present invention provides a compound of formula (Ia), or a pharmaceutically acceptable salt thereof, wherein R 8 is hydrogen.
- the present invention provides a compound of formula (Ia), or a pharmaceutically acceptable salt thereof, wherein R 10 is selected from:
- the present invention provides a compound of formula (Ia), or a pharmaceutically acceptable salt thereof, wherein A is an oxygen atom.
- the present invention provides a compound of formula (Ia), or a pharmaceutically acceptable salt thereof, wherein X is -CH 2 - or -CH 2 CH 2 -.
- Compounds of the invention may be useful in the treatment or prevention of diseases in which activation of muscarinic receptors are implicated, for example the present compounds are useful for treating a variety of indications, including but not limited to respiratory-tract disorders such as chronic obstructive lung disease, chronic bronchitis of all types (including dyspnoea associated therewith), asthma (allergic and non-allergic; 'whez-infant syndrome'), adult/acute respiratory distress syndrome (ARDS), chronic respiratory obstruction, bronchial hyperactivity, pulmonary fibrosis, pulmonary emphysema, and allergic rhinitis, exacerbation of airway hyperreactivity consequent to other drug therapy, particularly other inhaled drug therapy, pneumoconiosis (for example aluminosis, anthracosis, asbestosis, chalicosis, ptilosis, siderosis, silicosis, tabacosis and byssinosis); gastrointestinal-tract
- quaternary ammonium salts of the invention administered by inhalation is may be more than 12, or more than 24 hours for a typical dose.
- parenteral route usually the oral route, may be preferred.
- Another aspect of the invention is a pharmaceutical composition
- a pharmaceutical composition comprising a compound of the invention as shown in formula (Ia) and a pharmaceutically acceptable carrier or excipient.
- Another aspect of the invention is the use of a compound of the invention for the manufacture of a medicament for the treatment or prevention of a disease or condition in which muscarinic M3 receptor activity is implicated.
- Acyl means a -CO-alkyl group in which the alkyl group is as described herein.
- exemplary acyl groups include -COCH 3 and -COCH(CH 3 ) 2 .
- Acylamino means a -NR-acyl group in which R and acyl are as described herein.
- Exemplary acylamino groups include -NHCOCH 3 and -N(CH 3 )COCH 3 .
- Alkoxy and “alkyloxy” means an -O-alkyl group in which alkyl is as described below.
- Exemplary alkoxy groups include methoxy (-OCH 3 ) and ethoxy (-OC 2 H 5 ).
- Alkoxycarbonyl means a -COO-alkyl group in which alkyl is as defined below.
- Exemplary alkoxycarbonyl groups include methoxycarbonyl and ethoxycarbonyl.
- Alkyl as a group or part of a group refers to a straight or branched chain saturated hydrocarbon group having from 1 to 12, preferably 1 to 6, carbon atoms, in the chain.
- exemplary alkyl groups include methyl, ethyl, 1 -propyl and 2-propyl.
- Alkenyl as a group or part of a group refers to a straight or branched chain hydrocarbon group having from 2 to 12, preferably 2 to 6, carbon atoms and one carbon-carbon double bond in the chain.
- Exemplary alkenyl groups include ethenyl, 1-propenyl, and 2-propenyl.
- Alkylamino means a -NH-alkyl group in which alkyl is as defined above.
- exemplary alkylamino groups include methylamino and ethylamino.
- Alkylene means an -alkyl- group in which alkyl is as defined previously.
- exemplary alkylene groups include -CH 2 -, -(CH 2 ) 2 - and -C(CH 3 )HCH 2 -.
- Alkenylene means an -alkenyl- group in which alkenyl is as defined previously.
- Alkynylene means an -alkynyl- group in which -alkynyl- refers to a straight or branched chain hydrocarbon group having from 2 to 12, preferably 2 to 6, carbon atoms and one carbon-carbon triple bond in the chain.
- exemplary alkynylene groups include ethynyl and propargyl.
- Alkylsulfinyl means a -SO-alkyl group in which alkyl is as defined above.
- alkylsulfinyl groups include methylsulfinyl and ethylsulfinyl.
- Alkylsulfonyl means a -SO 2 -alkyl group in which alkyl is as defined above.
- Exemplary alkylsulfonyl groups include methylsulfonyl and ethylsulfonyl.
- Alkylthio means a -S-alkyl group in which alkyl is as defined above.
- exemplary alkylthio groups include methylthio and ethylthio.
- Aminoacyl means a -CO-NRR group in which R is as herein described.
- Exemplary aminoacyl groups include -CONH 2 and -CONHCH 3 .
- Aminoalkyl means an alkyl-NH 2 group in which alkyl is as previously described.
- exemplary aminoalkyl groups include -CH 2 NH 2 .
- aminosulfonyl means a -SO 2 -NRR group in which R is as herein described.
- exemplary aminosulfonyl groups include -SO 2 NH 2 and -SO 2 NHCH 3 .
- Aryl as a group or part of a group denotes an optionally substituted monocyclic or multicyclic aromatic carbocyclic moiety of from 6 to 14 carbon atoms, preferably from 6 to 10 carbon atoms, such as phenyl or naphthyl.
- the aryl group may be substituted by one or more substituent groups.
- Arylalkyl means an aryl-alkyl- group in which the aryl and alkyl moieties are as previously described. Preferred arylalkyl groups contain a C 1 4 alkyl moiety.
- arylalkyl groups include benzyl, phenethyl and naphthlenemethyl.
- Arylalkyloxy means an aryl-alkyloxy- group in which the aryl and alkyloxy moieties are as previously described. Preferred arylalkyloxy groups contain a C 1 4 alkyl moiety. Exemplary arylalkyl groups include benzyloxy.
- Aryl-fused-heterocycloalkyl means a monocyclic aryl ring, such as phenyl, fused to a heterocycloalkyl group, in which the aryl and heterocycloalkyl are as described herein.
- Exemplary aryl-fused-heterocycloalkyl groups include tetrahydroquinolinyl, indolinyl, benzodioxinyl, benxodioxolyl, dihydrobenzofuranyl and isoindolonyl.
- the aryl and heterocycloalkyl rings may each be substituted by one or more substituent groups.
- the aryl-fused-heterocycloalkyl group may be attached to the remainder of the compound by any available carbon or nitrogen atom.
- Aryloxy means an -O-aryl group in which aryl is described above.
- Exemplary aryloxy groups include phenoxy.
- Cyclic amine means an optionally substituted 3 to 8 membered monocyclic cycloalkyl ring system where one of the ring carbon atoms is replaced by nitrogen, and which may optionally contain an additional heteroatom selected from O, S or NR (where R is as described herein).
- Exemplary cyclic amines include pyrrolidine, piperidine, morpholine, piperazine and ⁇ /-methylpiperazine. The cyclic amine group may be substituted by one or more substituent groups.
- Cycloalkyl means an optionally substituted saturated monocyclic or bicyclic ring system of from 3 to 12 carbon atoms, preferably from 3 to 8 carbon atoms, and more preferably from 3 to 6 carbon atoms.
- Exemplary monocyclic cycloalkyl rings include cyclopropyl, cyclopentyl, cyclohexyl and cycloheptyl.
- the cycloalkyl group may be substituted by one or more substituent groups.
- Dialkylamino means a -N(alkyl)2 group in which alkyl is as defined above.
- dialkylamino groups include dimethylamino and diethylamino.
- Halo or halogen means fluoro, chloro, bromo, or iodo. Preferred are fluoro or chloro.
- Haloalkoxy means an -O-alkyl group in which the alkyl is substituted by one or more halogen atoms. Exemplary haloalkyl groups include trifluoromethoxy and difluoromethoxy.
- Haloalkyl means an alkyl group which is substituted by one or more halo atoms. Exemplary haloalkyl groups include trifiuoromethyl.
- Heteroaryl as a group or part of a group denotes an optionally substituted aromatic monocyclic or multicyclic organic moiety of from 5 to 14 ring atoms, preferably from 5 to 10 ring atoms, in which one or more of the ring atoms is/are element(s) other than carbon, for example nitrogen, oxygen or sulfur.
- Examples of such groups include benzimidazolyl, benzoxazolyl, benzothiazolyl, benzofuranyl, benzothienyl, furyl, imidazolyl, indolyl, indolizinyl, isoxazolyl, isoquinolinyl, isothiazolyl, oxazolyl, oxadiazolyl, pyrazinyl, pyridazinyl, pyrazolyl, pyridyl, pyrimidinyl, pyrrolyl, quinazolinyl, quinolinyi, tetrazolyl, 1 ,3,4-thiadiazolyl, thiazolyl, thienyl and triazolyl groups.
- the heteroaryl group may be substituted by one or more substituent groups.
- the heteroaryl group may be attached to the remainder of the compound of the invention by any available carbon or nitrogen atom.
- ⁇ eteroarylalkyl means a heteroaryl-alkyl- group in which the heteroaryl and alkyl moieties are as previously described.
- Preferred heteroarylalkyl groups contain a lower alkyl moiety.
- Exemplary heteroarylalkyl groups include pyridylmethyl.
- Heteroarylalkyloxy means a heteroaryl-alkyloxy- group in which the heteroaryl and alkyloxy moieties are as previously described.
- Preferred heteroarylalkyloxy groups contain a lower alkyl moiety.
- Exemplary heteroarylalkyloxy groups include pyridylmethyloxy.
- Heteroaryloxy means a heteroaryloxy- group in which the heteroaryl is as previously described.
- Exemplary heteroaryloxy groups include pyridyloxy.
- Heterocycloalkyl means: (i) an optionally substituted cycloalkyl group of from 4 to 8 ring members which contains one or more heteroatoms selected from O, S or NR; (ii) a cycloalkyl group of from 4 to 8 ring members which contains CONR and CONRCO (examples of such groups include succinimidyl and 2-oxopyrrolidinyl).
- the heterocycloalkyl group may be substituted by one or more substituent groups.
- the heterocycloalkyl group may be attached to the remainder of the compound by any available carbon or nitrogen atom.
- “Lower alkyl” as a group means unless otherwise specified, an aliphatic hydrocarbon group which may be straight or branched having 1 to 4 carbon atoms in the chain, i.e. methyl, ethyl, propyl (propyl or /so-propyl) or butyl (butyl, /so-butyl or tert- butyl).
- Sulfonyl means a -SO 2 -alkyl group in which alkyl is as described herein.
- exemplary sulfonyl groups include methanesulfonyl.
- “Sulfonylamino” means a -NR-sulfonyl group in which R and sulfonyl are as described herein.
- Exemplary sulfonylamino groups include -NHSO 2 CH 3 .
- R means alkyl, aryl, or heteroaryl as described herein.
- “Pharmaceutically acceptable salt” means a physiologically or toxicologically tolerable salt and includes, when appropriate, pharmaceutically acceptable base addition salts, pharmaceutically acceptable acid addition salts, and pharmaceutically acceptable quaternary ammonium salts.
- pharmaceutically acceptable base addition salts that may be formed include sodium, potassium, calcium, magnesium and ammonium salts, or salts with organic amines, such as, diethylamine, ⁇ /-methyl-glucamine, diethanolamine or amino acids (e.g.
- a compound of the invention contains a basic group, such as an amino group
- pharmaceutically acceptable acid addition salts that may be formed include hydrochlorides, hydrobromides, sulfates, phosphates, acetates, citrates, lactates, tartrates, mesylates, napadisylates (naphthalene- 1 ,5-disulfonates or naphthalene-1 -(sulfonic acid)-5-sulfonates), edisylates (ethane-1 ,2-disulfonates or ethane-1 -(sulfonic acid)-2-sulfonates), maleates, fumarates, succinates and the like; (iii) where a compound contains a quaternary ammonium group acceptable counter- ions may be, for example, chlorides, bromides, sulfates, methanesulfonates, benzenes
- Prodrug refers to a compound which is convertible in vivo by metabolic means (e.g. by hydrolysis, reduction or oxidation) to a compound of the invention. Suitable groups for forming pro-drugs are described in The Practice of Medicinal Chemistry, 2 nd Ed. pp561-585 (2003) and in F. J. Leinweber, Drug Metab. Res., , 18, 379. (1987)
- cyclic groups referred to above namely, aryl, heteroaryl, cycloalkyl, heterocycloalkyl, aryl-fused-heterocycloalkyl and cyclic amine may be substituted by one or more substituent groups.
- Suitable optional substituent groups include acyl (e.g. -COCHJ, alkoxy (e.g., -OCHJ, alkoxycarbonyl (e.g. -COOCHJ, alkylamino (e.g.
- alkylsulfinyl e.g. -SOCH 3
- alkylsulfonyl e.g. -SO 2 CH 3
- alkylthio e.g. - SCH 3
- -NH 2 aminoacyl (e.g. -CON(CHs) 2 ), aminoalkyl (e.g. -CH 2 NH 2 ), arylalkyl (e.g. -CH Ph or -CH -CH -Ph), cyano, dialkylamino (e.g. -N(CHJJ, halo, haloalkoxy (e.g.
- haloalkyl e.g. -CF 3
- alkyl e.g. -CH 3 Or -CH 2 CH 3
- -OH, -CHO, - NO 2 aryl (optionally substituted with alkoxy, haloalkoxy, halogen, alkyl or haloalkyl), heteroaryl (optionally substituted with alkoxy, haloalkoxy, halogen, alkyl or haloalkyl), heterocycloalkyl, aminoacyl (e.g. -CONH 2 , -CONHCH 3 ), aminosulfonyl (e.g.
- acylamino e.g. -NHCOCH 3
- sulfonylamino e.g. -NHSO 2 CH 3
- heteroarylalkyl cyclic amine (e.g. morpholine), aryloxy, heteroaryloxy, arylalkyloxy (e.g. benzyloxy) and heteroarylalkyloxy.
- Alkyl, alkoxy, alkylene or alkenylene groups may be optionally substituted. Suitable optional substituent groups include alkoxy (e.g., -OCH 3 ), alkylamino (e.g. -NHCH 3 ), alkylsulfinyl (e.g. -SOCHJ, alkylsulfonyl (e.g. -SO 0 CHJ, alkylthio (e.g. -SCHJ, -NH 2 , aminoalkyl (e.g. -CH 2 NH 2 ), arylalkyl (e.g. -CH 2 Ph or -CH 2 -CH 3 -Ph), cyano, dialkylamino (e.g.
- halo haloalkoxy (e.g. -OCF 3 or -OCHF 2 ), haloalkyl (e.g. -CFJ, alkyl (e.g. -CH or -CH 0 CH ), -OH, -CHO, and -NO 2 .
- Compounds of the invention may exist in one or more geometrical, optical, enantiomeric, diastereomeric and tautomeric forms, including but not limited to cis- and frans-forms, £- and Z-forms, R-, S- and meso-forms, keto-, and enol-forms. Unless otherwise stated a reference to a particular compound includes all such isomeric forms, including racemic and other mixtures thereof. Where appropriate such isomers can be separated from their mixtures by the application or adaptation of known methods (e.g. chromatographic techniques and recrystallisation techniques). Where appropriate such isomers may be prepared by the application of adaptation of known methods (e.g. asymmetric synthesis).
- Convenient compounds of the invention include:
- the present invention is also concerned with pharmaceutical formulations comprising, as an active ingredient, a compound of the invention.
- Other compounds may be combined with compounds of this invention for the prevention and treatment of inflammatory diseases of the lung.
- the present invention is also concerned with pharmaceutical compositions for preventing and treating respiratory-tract disorders such as chronic obstructive lung disease, chronic bronchitis, asthma, chronic respiratory obstruction, pulmonary fibrosis, pulmonary emphysema, and allergic rhinitis comprising a therapeutically effective amount of a compound of the invention and one or more other therapeutic agents.
- the invention includes a combination of an agent of the invention as hereinbefore described with one or more anti-inflammatory, bronchodilator, antihistamine, decongestant or anti-tussive agents, said agents of the invention hereinbefore described and said combination agents existing in the same or different pharmaceutical compositions, administered separately or simultaneously.
- Preferred combinations would have two or three different pharmaceutical compositions.
- Suitable therapeutic agents for a combination therapy with compounds of the invention include: One or more other bronchodilators such as PDE3 inhibitors; Methyl xanthines such as theophylline;
- a corticosteroid for example fluticasone propionate, ciclesonide, mometasone furoate or budesonide, or steroids described in WO02/88167, WO02/12266, WO02/100879, WO02/00679, WO03/35668, WO03/48181 , WO03/62259, WO03/64445, WO03/72592, WO04/39827 and WO04/66920;
- a non-steroidal glucocorticoid receptor agonist for example fluticasone propionate, ciclesonide, mometasone furoate or budesonide, or steroids described in WO02/88167, WO02/12266, WO02/100879, WO02/00679, WO03/35668, WO03/48181 , WO03/62259, WO03/64445, WO03/72592, WO04/39827 and WO04/66920;
- a leukotriene modulator for example montelukast, zafirlukast or pranlukast; protease inhibitors, such as inhibitors of matrix metalloprotease for example MMP12 and TACE inhibitors such as marimastat, DPC-333, GW-3333; Human neutrophil elastase inhibitors, such as sivelestat and those described in WO04/043942, WO05/021509, WO05/021512, WO05/026123, WO05/026124, WO04/024700, WO04/024701 , WO04/020410, WO04/020412, WO05/080372,
- Phosphodiesterase-4 (PDE4) inhibitors for example roflumilast, arofylline, cilomilast,
- An antitussive agent such as codeine or dextramorphan
- P2X7 anatgonists P2X7 anatgonists; iNOS inhibitors; A non-steroidal anti-inflammatory agent (NSAID), for example ibuprofen or ketoprofen;
- NSAID non-steroidal anti-inflammatory agent
- a dopamine receptor antagonist A dopamine receptor antagonist
- TNF- ⁇ inhibitors for example anti-TNF monoclonal antibodies, such as Remicade and CDP-870 and TNF receptor immunoglobulin molecules, such as Enbrel; A2a agonists such as those described in EP1052264 and EP1241176;
- A2b antagonists such as those described in WO2002/42298;
- Modulators of chemokine receptor function for example antagonists of CCR1 , CCR2,
- DP1 or CRTH2 a thromboxane A 2 antagonist eg ramatrobant
- Th1 or Th2 function Compounds which modulate Th1 or Th2 function, for example, PPAR agonists; lnterleukin 1 receptor antagonists, such as Kineret; lnterleukin 10 agonists, such as llodecakin; HMG-CoA reductase inhibitors (statins); for example rosuvastatin, mevastatin, lovastatin, simvastatin, pravastatin and fluvastatin;
- Mucus regulators such as INS-37217, diquafosol, sibenadet, CS-003, talnetant, DNK-
- Antiinfective agents antibiotic or antiviral
- antiallergic drugs including, but not limited to, anti-histamines.
- the weight ratio of the first and second active ingredients may be varied and will depend upon the effective dose of each ingredient. Generally, an effective dose of each will be used. Any suitable route of administration may be employed for providing a mammal, especially a human, with an effective dosage of a compound of the present invention. In therapeutic use, the active compound may be administered by any convenient, suitable or effective route. Suitable routes of administration are known to those skilled in the art, and include oral, intravenous, rectal, parenteral, topical, ocular, nasal, buccal and pulmonary.
- prophylactic or therapeutic dose of a compound of the invention will, of course, vary depending upon a range of factors, including the activity of the specific compound that is used, the age, body weight, diet, general health and sex of the patient, time of administration, the route of administration, the rate of excretion, the use of any other drugs, and the severity of the disease undergoing treatment.
- the daily dose range for inhalation will lie within the range of from about 0.1 ⁇ g to about 10 mg per kg body weight of a human, preferably 0.1 ⁇ g to about 0.5 mg per kg, and more preferably 0.1 ⁇ g to 50 ⁇ g per kg, in single or divided doses. On the other hand, it may be necessary to use dosages outside these limits in some cases.
- compositions suitable for administration by inhalation are known, and may include carriers and/or diluents that are known for use in such compositions.
- the composition may contain 0.01-99% by weight of active compound.
- a unit dose comprises the active compound in an amount of 1 ⁇ g to 10 mg.
- suitable doses are 10 ⁇ g per kg to 100mg per kg, preferably 40 ⁇ g per kg to 4 mg per kg.
- compositions which comprise a compound of the invention and a pharmaceutically acceptable carrier.
- composition as in pharmaceutical composition, is intended to encompass a product comprising the active ingredient(s), and the inert ingredient(s) (pharmaceutically acceptable excipients) that make up the carrier, as well as any product which results, directly or indirectly, from combination, complexation or aggregation of any two or more of the ingredients, or from dissociation of one or more of the ingredients, or from other types of reactions or interactions of one or more of the ingredients.
- pharmaceutical compositions of the present invention encompass any composition made by admixing a compound of the invention, additional active ingredient(s), and pharmaceutically acceptable excipients.
- compositions of the present invention comprise a compound of the invention as an active ingredient or a pharmaceutically acceptable salt thereof, and may also contain a pharmaceutically acceptable carrier and optionally other therapeutic ingredients.
- pharmaceutically acceptable salts refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic bases or acids and organic bases or acids, and salts of quaternary ammonium compounds with pharmaceutically acceptable counter-ions.
- the active compound is preferably in the form of microparticles. They may be prepared by a variety of techniques, including spray- drying, freeze-drying and micronisation.
- a composition of the invention may be prepared as a suspension for delivery from a nebuliser or as an aerosol in a liquid propellant, for example for use in a pressurised metered dose inhaler (PMDI).
- PMDI pressurised metered dose inhaler
- Propellants suitable for use in a PMDI are known to the skilled person, and include CFC-12, HFA-134a, HFA-227, HCFC-22 (CCI 2 F 2 ) and HFA-152 (C 2 H 4 F 2 ) and isobutane.
- a composition of the invention is in dry powder form, for delivery using a dry powder inhaler (DPI).
- DPI dry powder inhaler
- Microparticles for delivery by administration may be formulated with excipients that aid delivery and release.
- microparticles may be formulated with large carrier particles that aid flow from the DPI into the lung.
- Suitable carrier particles are known, and include lactose particles; they may have a mass median aerodynamic diameter of greater than 90 ⁇ m.
- the active compounds may be dosed as described depending on the inhaler system used.
- the administration forms may additionally contain excipients, such as, for example, propellants (e.g. Frigen in the case of metered aerosols), surface-active substances, emulsifiers, stabilizers, preservatives, flavorings, fillers (e.g. lactose in the case of powder inhalers) or, if appropriate, further active compounds.
- a large number of systems are available with which aerosols of optimum particle size can be generated and administered, using an inhalation technique which is appropriate for the patient.
- adaptors spacers, expanders
- pear-shaped containers e.g. Nebulator®, Volumatic®
- automatic devices emitting a puffer spray Autohaler®
- metered aerosols in particular in the case of powder inhalers
- a number of technical solutions are available (e.g. Diskhaler®, Rotadisk®, Turbohaler® or the inhalers for example as described EP-A-0505321 ).
- compounds of the invention may be delivered in multi-chamber devices thus allowing for delivery of combination agents.
- the compounds of the invention of the present invention can be prepared according to the procedures of the following schemes and examples, using appropriate materials, and are further exemplified by the following specific examples. Moreover, by utilising the procedures described with the disclosure contained herein, one of ordinary skill in the art can readily prepare additional compounds of the present invention claimed herein. The compounds illustrated in the examples are not, however, to be construed as forming the only genus that is considered as the invention. The examples further illustrate details for the preparation of the compounds of the present invention. Those skilled in the art will readily understand that known variations of the conditions and processes of the following preparative procedures can be used to prepare these compounds.
- the compounds of the invention may be isolated in the form of their pharmaceutically acceptable salts, such as those described previously herein above. It may be necessary to protect reactive functional groups (e.g. hydroxy, amino, thio or carboxy) in intermediates used in the preparation of compounds of the invention to avoid their unwanted participation in a reaction leading to the formation of the compounds.
- reactive functional groups e.g. hydroxy, amino, thio or carboxy
- Conventional protecting groups for example those described by T. W. Greene and P. G. M. Wuts in "Protective groups in organic chemistry” John Wiley and Sons, 1999, may be used.
- the invention further provides a process for the preparation of a compound of formula (I) or (Ia) or a pharmaceutically acceptable salt thereof as defined above which comprises:
- LG 1 represents a leaving group such as chloride, bromide, iodide, methanesulfonate or para-toluenesulfonate
- L, L 2 , L 3 , L 4 , R 4 , R 5 , R 6 , and X and R 8 are as defined in formula (Ia), with a compound of formula (III), or a suitable salt thereof such as a hydrobromide, acetate or hydrochloride salt
- compounds of formula (III) represent compounds HNR 9 R 10 wherein R 9 and R 10 are as described above for compounds of formula (I) or (Ia), and P 1 is hydrogen or a protective group such as te/t-butyldimethyl silyl in the presence of a base such as potassium carbonate, triethyiamine or diisopropylethylamine, followed by removal of the protective group (e.g. using a hydrofluoric acid-pyridine complex); or (b) when L 1 represents hydrogen and R 1 does not represent hydrogen, reacting a compound of formula (IV), or a suitable salt thereof
- L, L 2 , L 3 , L 4 , R 4 , R 5 , R 6 R 8 and X are as defined in formula (Ia), with a compound of formula (III) or a suitable salt thereof in the presence of a suitable reducing agent such as sodium cyanoborohydride, sodium triacetoxyborohydride, or hydrogen in the presence of a suitable palladium on carbon or platinum oxide catalyst; or
- LG 1 represents a leaving group such as chloride, bromide, iodide, methanesulfonate or para-toluenesulfonate
- P 2 represents a protective group (e.g. tert-butylcarbonyl)
- L, L 2 , L 3 , L 4 , R 4 , R 5 , R 6 , R 8 and X are as defined in formula (Ia), with a compound of formula (III), or a suitable salt thereof (e.g. hydrobromide, hydrochloride salt or acetate), in the presence of a base (e.g. potassium carbonate, triethylamine or diisopropylethylamine) followed by removal of the protective group (e.g. treatment with hydrochloric or trifluoroacetic acid); or
- a base e.g. potassium carbonate, triethylamine or diisopropylethylamine
- P 2 represents a protective group (e.g. tert-butylcarbonyl) with a compound of formula (III), or a suitable salt thereof (e.g. hydrobromide, hydrochloride salt or acetate), in the presence of a suitable reducing agent (e.g. sodium cyanoborohydride, sodium triacetoxyborohydride, or hydrogen in the presence of a suitable palladium on carbon or platinum oxide catalyst), followed by removal of the protective group (e.g. treatment with hydrochloric or trifluoroacetic acid); or
- R 4 when R 4 does not represent hydrogen, reacting a compound of formula (VII), or a suitable salt thereof wherein L 1 L 1 , L 2 , L 3 , L 4 , R 4 , R 5 , R 6 , R 8 and X are as defined in formula (Ia), P 3 represents hydrogen or an activating group (e.g. 3-nitrophenylsulfonyl) with a compound of formula (VIII), or a suitable salt thereof,
- an activating group e.g. 3-nitrophenylsulfonyl
- LG 2 represents a leaving group (e.g. chloride, bromide, iodide, methanesulfonate or para-toluenesulfonate) and P 1 is as defined in compound of formula (III) in the presence of a base (e.g. when P 3 is hydrogen, potassium carbonate, triethylamine, diisopropylethylamine and, when P 3 is 3-nitrophenylsulfonyl, sodium hydride or lithium di-/so-propylamide), followed by removal of the protective groups (e.g. using hydrofluoric acid-pyridine complex, thiophenol, thioacetic acid); or with a compound of formula (IX), or a suitable salt thereof,
- a base e.g. when P 3 is hydrogen, potassium carbonate, triethylamine, diisopropylethylamine and, when P 3 is 3-nitrophenylsulfonyl, sodium hydride or lithium di-/so-
- a base e.g. when P 3 is hydrogen, potassium carbonate, triethylamine, diisopropylethylamine and, when P 3 is 3-nitrophenylsulfonyl, sodium hydride or lithium di-/so-propylamide
- the protective groups e.g. trifluoroacetic acid, thiophenol, thioacetic acid
- a compound of formula (X) or a suitable salt thereof
- LG 2 represents a leaving group (e.g. chloride, bromide, iodide, methanesulfonate or para-toluenesulfonate) in the presence of a base (e.g. when P 3 is hydrogen, potassium carbonate, triethylamine, diisopropylethylamine and, when P 3 is 3- nitrophenylsulfonyl, sodium hydride or lithium di-/so-propylamide), followed by reduction of the ketone (e.g. using sodium borohydride or a borane/chiral catalyst complex), followed by removal of the protective groups (e.g. trifluoroacetic acid, thiophenol, thioacetic acid); or (f)When R 4 represents hydrogen, reacting a compound of formula (Xl)
- a base e.g. when P 3 is hydrogen, potassium carbonate, triethylamine, diisopropylethylamine and, when P 3 is 3- nitropheny
- L, L 1 , L 2 , L 3 , L 4 , R 4 , R 5 , R 6 , R 8 and X are as defined in formula (Ia),
- P 2 represents a protective group (e.g. tert-butylcarbonyl)
- P 3 represents hydrogen or an activating group (e.g. 3-nitrophenylsulfonyl)
- ith a compound of formula (VIII), (IX) or (X), or a suitable salt thereof, in the presence of a base (e.g.
- L, L 1 , and L 2 are as defined in formula (Ia)
- P 1 is as defined in compound of formula (III)
- P 3 represents a protective group (e.g. terf-butylcarbonyl or 3- nitrophenylsulfonyl) with a compound of formula (XIII), or a suitable salt thereof,
- R 4 , R 5 , R 6 , R 1 , R 8 , A and X are as defined in formula (I), in the presence of a suitable reducing agent (e.g. sodium cyanoborohydride, sodium triacetoxyborohydride, or hydrogen in the presence of a suitable palladium on carbon or platinum oxide catalyst), followed by removal of the protective groups (e.g. treatment with hydrochloric or trifluoroacetic acid thiophenol, thioacetic acid); or (h) when one or both of L 3 and L 4 represents hydrogen, reacting a compound of formula (XIV)
- a suitable reducing agent e.g. sodium cyanoborohydride, sodium triacetoxyborohydride, or hydrogen in the presence of a suitable palladium on carbon or platinum oxide catalyst
- the protective groups e.g. treatment with hydrochloric or trifluoroacetic acid thiophenol, thioacetic acid
- L, L 1 , and L 2 are as defined in formula (Ia), P 1 is as defined in compound of formula (III), P 3 represents a protective group (e.g. terf-butylcarbonyl or 3- nitrophenylsulfonoyl), LG 3 represents a leaving group (e.g. chloride, bromide, iodide, methanesulfonate or para-toluenesulfonate), with a compound of formula (XIII) or a suitable salt thereof, in the presence of a base (e.g. potassium carbonate, triethylamine, diisopropylethylamine), followed by removal of the protective groups (e.g. trifluoroacetic acid, thiophenol, thioacetic acid); or (i) when L 1 and L 2 each represents hydrogen and R 4 do not represent hydrogen, reacting a compound of formula (XV), or a suitable salt thereof,
- a base e.g. potassium carbonate, triethy
- L, L 3 , L 4 , R 1 , R 4 , R 5 R 6 , R 8 , A, and X are as defined in formula (I) and P 1 is as defined in formula (III) with a suitable reducing agent (e.g. borane tetrahydrofuran complex), followed by removal of the protective group (e.g. using hydrofluoric acid- pyridine complex); or,
- a suitable reducing agent e.g. borane tetrahydrofuran complex
- the reaction may conveniently be carried out in an organic solvent such as ⁇ /, ⁇ /-dimethylformamide, ethanol, n-butanol or dimethyl sulfoxide, at a temperature, for example, in the range from 50 to 140 0 C.
- the reaction may conveniently be carried out in an organic solvent such as methanol, ethanol, dichloromethane, acetic acid N- methylpyrolidinone, or ⁇ /, ⁇ /-dimethylformamide containing up to 10%w of water and acetic acid.
- reaction may conveniently be carried out in an organic solvent such as tetrahydrofuran, at a temperature, for example, in the range from 0 to 80 0 C.
- organic solvent such as tetrahydrofuran
- Compounds of formula (II) may be prepared by reacting a compound of formula (XVII), or a suitable salt thereof,
- L Mt (XVlK) wherein L 2 is as defined in formula (II) and Mt represents a metal such as lithium or magnesium, or aluminium or boron (e.g. methyllithium, methylmagnesium bromide, lithium aluminium hydride, sodium borohydride) in an organic solvent, for example, tetrahydrofuran or ether, at a temperature, for example in the range from 0 to 6O 0 C, followed by conversion of the resulting hydroxyl group into a suitable leaving group (e.g. chloride, bromide, iodide, methanesulfonate or para-toluenesulfonate).
- a suitable leaving group e.g. chloride, bromide, iodide, methanesulfonate or para-toluenesulfonate.
- Compounds of formula (IV) may be prepared by reacting a compound of formula (XVII) with a compound of formula (XVIII) in an organic solvent, for example, tetrahydrofuran or ether, at a temperature, for example in the range from 0 to 6O 0 C, followed by oxidation of the resulting hydroxyl group with a suitable oxidating agent (e.g. Swern reagent, Dess-Martin reagent or pyridiniumchlorochromate) in an organic solvent such as dichloromethane, ⁇ /, ⁇ /-dimethylformamide or dimethylsulfoxide at a temperature, for example in the range from -78 to 6O 0 C.
- a suitable oxidating agent e.g. Swern reagent, Dess-Martin reagent or pyridiniumchlorochromate
- an organic solvent such as dichloromethane, ⁇ /, ⁇ /-dimethylformamide or dimethylsulfoxide at a temperature
- P 2 , L, L 3 , L 4 , R 4 , R 5 , R 6 , R 8 , A, and X are as defined in formula (V), with a compound of formula (XVIII) in an organic solvent, for example, tetrahydrofuran or ether, at a temperature, for example in the range from 0 to 60 0 C, followed by conversion of the resulting hydroxyl group into a suitable leaving group (e.g. chloride, bromide, iodide, methanesulfonate or para-toluenesulfonate).
- an organic solvent for example, tetrahydrofuran or ether
- Compounds of formula (Vl) may be prepared by reacting a compound of formula (XVIII) with a compound of formula (XIX), followed by oxidation of the resulting hydroxyl group with a suitable oxidating agent (e.g. Swern reagent, Dess-Martin reagent or pyridiniumchlorochromate) in an organic solvent such as dichloromethane, ⁇ /, ⁇ /-dimethylformamide or dimethylsulfoxide at a temperature, for example in the range from -78 to 6O 0 C.
- a suitable oxidating agent e.g. Swern reagent, Dess-Martin reagent or pyridiniumchlorochromate
- an organic solvent such as dichloromethane, ⁇ /, ⁇ /-dimethylformamide or dimethylsulfoxide at a temperature, for example in the range from -78 to 6O 0 C.
- Compounds of formula (VII) in which L 1 represents hydrogen and L, L 2 , L 3 , L 4 , R 1 , R 4 , R 5 , R 6 , R 8 , A, and X are as defined in formula (VII) may be prepared by (a) reacting a compound of formula (II) with sodium azide, in an organic solvent for example, tetrahydrofuran, ⁇ /, ⁇ /-dimethylformamide or dimethylsulfoxide at a temperature, for example in the range from 25 to 85 0 C, followed by reduction of the resulting azido compound using a suitable reducing agent (e.g.
- triphenylphosphine in an organic solvent for example, tetrahydrofuran and water, and eventually followed by protection of the resulting amine (e.g. treatment with 3-nitrophenylsulfonyl chloride in the presence of a base such as pyridine); or, (b) reacting a compound of formula (IV) with an amine (e.g. benzylamine, ⁇ -methyl benzylamine, 4-methoxybenzylamine or 2,4-methoxybenzylamine) followed by reduction of the resulting imine using a suitable reducing agent (e.g.
- sodium cyanoborohydride or sodium triacetoxyborohydride in an organic solvent such as methanol, ethanol, dichloromethane, acetic acid, ⁇ /-methylpyrolidinone or N, N- dimethylformamide containing up to 10%w of water and acetic acid, followed by removal of the resulting benzyl protective group using the appropriate reagent (e.g.
- a suitable catalyst Palladium on carbon or palladium hydroxide
- DDQ 2,3- dichloro-5,6-dicyanobenzoquinone
- CAN ammonium cerium nitrate
- an organic solvent for example, ethanol, methanol, tetrahydrofuran, dichloromethane, acetonitrile, water, or a mixture thereof, at a temperature ranging from 25 to 80 0 C, and eventually followed by protection of the resulting amine (e.g. treatment with 3- nitrophenylsulfonyl chloride in the presence of a base such as pyridine);
- LG 4 is a leaving group (e.g. hydroxyl or chloride), L, L 1 , L 2 , L 3 , L 4 , R 1 , R 4 , R 5 , R 6 , R 8 , A, and X are as defined in formula (VII), with reagents such as, when LG 4 is hydroxyl, diphenylphosphonic azide, in a presence of an amine (e.g.
- triethylamine in an organic solvent, for example, fert-butanol, tetrahydrofuran, dichloromethane, water, or a mixture thereof, at a temperature ranging from 25 to 100°C, or when LG 4 is chloride, sodium azide, in an organic solvent, for example, ether, tert-butanol, tetrahydrofuran, water, or a mixture thereof, at a temperature ranging from 25 to 100 0 C (Angewandte Chemie, 2005, 54, 5188), eventually followed by protection of the resulting amine (e.g. treatment with 3-nitrophenylsuifonyl chloride in the presence of a base such as pyridine).
- a base such as pyridine
- Compounds of formula (Xl) in which L 1 represents hydrogen may be prepared by (a) reacting a compound of formula (V) with sodium azide in an organic solvent, for example, tetrahydrofuran, ⁇ /, ⁇ /-dimethylformamide or dimethylsulfoxide at a temperature, for example in the range from 25 to 85 0 C, followed by reduction of the resulting azido compound using a suitable reducing agent (e.g. triphenylphosphine or hydrogen) in an organic solvent for example, tetrahydrofuran and water, eventually followed by protection of the resulting amine (e.g.
- a suitable reducing agent e.g. triphenylphosphine or hydrogen
- a compound of formula (Vl) with an amine (e.g. benzylamine, a- methyl benzylamine, 4-methoxybenzyl amine or 2,4-methoxybenzyl amine), followed by reduction of the resulting imine using a suitable reducing agent (e.g.
- a suitable catalyst Palladium on carbon or palladium hydroxide
- an organic solvent for example, ethanol, methanol, tetrahydrofuran, dichloromethane, acetonitrile, water, or a mixture thereof, at a temperature ranging from 25 to 80 0 C, eventually followed by protection of the resulting amine (e.g. treatment with 3- nitrophenylsulfonyl chloride in the presence of a base such as pyridine).
- L 4 is a leaving group (e.g. hydroxyl or chloride)
- L, L 1 , L 2 , L 3 , L 4 , R 4 , R 5 , R 6 , R 8 , A, X and P 2 are as defined in formula (Xl), with reagents such as, when LG 4 is hydroxyl, diphenylphosphonic azide, in a presence of an amine (e.g.
- P 5 is hydrogen or a protective group (e.g. tert-butyldimethylsilyl, tetrahydropyran) and L, L 1 and L 2 are as defined in formula (XII), with a compound of formula (VIII), (IX) or (X), or a suitable salt thereof, in the presence of a base (e.g.
- potassium carbonate triethylamine or diisopropylethylamine when P 3 is hydrogen and sodium hydride or lithium di ⁇ /so-propylamide when P 3 is 3-nitrophenylsulfonyl) in an organic solvent such as ⁇ /, ⁇ /-dimethylformamide, ⁇ /-methylpyrolidinone, tetrahydrofuran, ethanol, n-butanol or dimethyl sulfoxide, at a temperature, for example, in the range from 50 to 140 0 C.
- an organic solvent such as ⁇ /, ⁇ /-dimethylformamide, ⁇ /-methylpyrolidinone, tetrahydrofuran, ethanol, n-butanol or dimethyl sulfoxide, at a temperature, for example, in the range from 50 to 140 0 C.
- P 5 is hydrogen or a protective group (e.g. tert-butyldimethylsilyl, tetrahydropyran) and, L and L 2 are as defined in formula (XII), with a compound of formula (III), or a suitable salt thereof, in the presence of a suitable reducing agent (e.g.
- P 6 and P 7 represent an acyclic or cyclic carbonyl protective group (e.g. dimethoxy or diethoxy acetal, 1 ,3-dioxolane or 1 ,3-dioxane) and, L and L 2 are as defined in formula (XII), with a compound of formula (III), or a suitable salt thereof, in the presence of a suitable reducing agent (e.g.
- an organic solvent such as methanol, ethanol, dichloromethane, acetic acid, ⁇ /-methypyrolidinone or /V, ⁇ /-dimethylformamide containing up to 10%w of water and acetic acid, followed by removal of the protective group (e.g. diluted hydrochloric acid or amberlyst-15 resin in methanol).
- Compounds of formula (XIV) can be prepared by converting compound of formula (XII), or a precursor to compound of formula (XII) as decribed above, chosing an appropriate sequence of reactions such as, for example, reduction of an aldehyde to an alcohol (e.g. sodium borohydride), appropriate selective removal of the protective group (e.g. hydrofluoric acid-pyridine complex, tetrabutylamonium fluoride, diluted hydrochloric acid or amberlyst-15 resin in methanol) and conversion of an alcohol into a suitable leaving group (e.g. halogen, mesylate, tosylate); or, Compounds of formula (XV) and (XVI) can be prepared by similar methods by reacting a compound of formula (XXVI)
- L, L 3 , L 4 , R 4 , R 5 , R 6 , R 8 , A, and X are as defined in formula (XV)
- P 8 represents either R 3 as defined in compound of formula (XV) or P 2 as defined in compound of formula (XVI) and LG 6 represent hydroxyl or a leaving group (e.g. chloride) with a compound of formula (111), or a suitable salt thereof.
- the reaction is conveniently carried out in the presence of an activating reagent, for example, carbonyldiimidazole or O-(7- azabenzotriazol-1 -yl)- ⁇ /, ⁇ /, ⁇ /', ⁇ /-tetramethyluroniumhexafluorophosphate (HATU), in an organic solvent, for example, ⁇ /, ⁇ /-dimethylformamide or dichloromethane, at a temperature, for example in the range from 0 to 6O 0 C
- an activating reagent for example, carbonyldiimidazole or O-(7- azabenzotriazol-1 -yl)- ⁇ /, ⁇ /, ⁇ /', ⁇ /-tetramethyluroniumhexafluorophosphate (HATU)
- an organic solvent for example, ⁇ /, ⁇ /-dimethylformamide or dichloromethane
- LG 6 represents chloride
- the reaction is conveniently carried out in the presence of a base, for example, trieth
- LG 7 represent a hydroxyl, an esther (e.g. methoxy, ethoxy), a leaving group (e.g.
- L, L 1 , L 2 , L 3 , L 4 and P 3 are as defined in compound of formula (VII); - for compound of formula (Xl), P 9 represents P 2 , P 10 represents
- P 9 and P 10 represents an appropriate nitrogen protecting group, such as te/f-butoxycarbonyl, followed by suitable deprotection (e.g. trifluoroacetic acid acid);
- P 9 represents R 4
- P 10 represents
- L, L 3 , and L 4 are as defined in compound of formula (XVII), wherein P 11 and P 12 represent an acyclic or cyclic carbonyl protective group (e.g. dimethoxy or diethoxy acetal, 1 ,3-dioxolane or 1 ,3-dioxane), followed by suitable deprotection (e.g. diluted hydrochloric acid or amberlyst-15 resin in methanol);
- P 11 and P 12 represent an acyclic or cyclic carbonyl protective group (e.g. dimethoxy or diethoxy acetal, 1 ,3-dioxolane or 1 ,3-dioxane), followed by suitable deprotection (e.g. diluted hydrochloric acid or amberlyst-15 resin in methanol);
- P 9 represents P 2
- P 10 represents wherein L, L 3 , and L 4 are as defined in compound of formula (XIX) 1 wherein P 11 and P 12 represent an acyclic or cyclic carbonyl protective group (e.g. dimethoxy or diethoxy acetal, 1 ,3-dioxolane or 1 ,3-dioxane), followed by suitable deprotection (e.g. diluted hydrochloric acid or amberlyst-15 resin in methanol);
- acyclic or cyclic carbonyl protective group e.g. dimethoxy or diethoxy acetal, 1 ,3-dioxolane or 1 ,3-dioxane
- suitable deprotection e.g. diluted hydrochloric acid or amberlyst-15 resin in methanol
- L, L 1 , L 2 , L 3 , and L 4 are as defined in compound of formula (XX), wherein P 14 represent an acid protective group (e.g. methyl, ethyl or tert-butyl), followed by suitable deprotection (e.g. lithium hydroxide or sodium hydroxide, trifluoroacetic acid, hydrochloric acid);
- P 14 represent an acid protective group (e.g. methyl, ethyl or tert-butyl)
- suitable deprotection e.g. lithium hydroxide or sodium hydroxide, trifluoroacetic acid, hydrochloric acid
- L, L 1 , L 2 , L 3 , and L 4 are as defined in compound of formula (XXI), wherein P 14 represent an acid protective group (e.g. methyl, ethyl or tert-butyl), followed by suitable deprotection (e.g. lithium hydroxide or sodium hydroxide, trifluoroacetic acid, hydrochloric acid);
- P 14 represent an acid protective group (e.g. methyl, ethyl or tert-butyl)
- suitable deprotection e.g. lithium hydroxide or sodium hydroxide, trifluoroacetic acid, hydrochloric acid
- L, L 3 , and L 4 are as defined in compound of formula (XXVI), wherein P 14 represent an acid protective group (e.g. methyl, ethyl or tert-butyl), followed by suitable deprotection (e.g. lithium hydroxide or sodium hydroxide, trifluoroacetic acid, hydrochloric acid);
- P 14 represent an acid protective group (e.g. methyl, ethyl or tert-butyl)
- suitable deprotection e.g. lithium hydroxide or sodium hydroxide, trifluoroacetic acid, hydrochloric acid
- R in compounds of formula (I) or (Ia) is a group of formula it will be recognised by those skilled in the art that a similar set of reactions as described above may be used, employing a compound of formula (Ilia) in place of a compound of formula (III), optionally suitably protected, or a suitable salt thereof such as a hydrobromide, hydrochloride or acetate salt
- R 5 M (XLVI) wherein R 4 and R 5 are as defined in general formula (I) and M represents a metallic counterion such as Li or MgBr.
- the reaction may take place in an aprotic organic solvent such as THF or diethyl ether at a range of temperatures, preferably between -78 0 C and the reflux temperature of the solvent.
- R 1 is as defined in general formula (I).
- the reaction is performed in a range of solvents, preferably THF/DCM at a range of temperatures, preferably between 0 and 100 0 C.
- reaction by reaction with a brominating agent such as N-bromosuccinimide in the presence of a radical initiator such as AIBN or benzoyl peroxide.
- a brominating agent such as N-bromosuccinimide
- a radical initiator such as AIBN or benzoyl peroxide.
- the reaction can be carried out in suitable solvents, such as CCI 4 , or chlorobenzene at a range of temperatures, preferably between ambient temperature and the reflux temperature of the solvent.
- compounds of formula (XXXI) can be prepared from compounds of general formula (XXX) by palladium-catalysed cyclisation using a palladium catalyst such as bis(dibenzylideneacetone)palladium in the presence of a ligand such as triphenylphosphine and a base such as sodium tert-butoxide in a solvent such as THF from room temperature to the reflux temperature of the solvent.
- a palladium catalyst such as bis(dibenzylideneacetone)palladium in the presence of a ligand such as triphenylphosphine and a base such as sodium tert-butoxide in a solvent such as THF from room temperature to the reflux temperature of the solvent.
- Compounds of general formula (XLV) can be prepared from compounds of formula (XLIII) using methods described above for the preparation of compounds of formula (XXX) from compounds of formula (XXIX).
- Compounds of formula (Xll-a) wherein -NR 1 R 3 is a secondary amine may be prepared from compounds of formula (Xll-a) wherein -NR 1 R 3 is a -NH 2 group by reductive alkylation with a suitably substituted aldehyde.
- the reaction is carried out in the presence of a reducing agent such as sodium cyanoborohydride or sodium borohydride, preferably sodium triacetoxyborohydride in a range of organic solvents, preferably dichloroethane.
- Compounds of formula (Xll-e) may be prepared from compounds of formula (Xll-d) by alkylation or reductive alkylation methods as described above and according to standard methods well-known to those skilled in the art.
- Compounds of formula (Xll-d) can be prepared from compounds of general formula (XXXV):
- a reducing agent such as lithium aluminium hydride, diisobutyl aluminium hydride, or borane in a range of aprotic solvents such as diethyl ether, or THF or preferably by hydrogenation in the presence of a catalyst such as Raney Nickel in a suitable solvent such as EtOAc or EtOH at a range of temperatures from room temperature to the reflux temperature of the solvent.
- Compounds of general formula (XXXV) can be prepared from compounds of general formula (XXVI l-a) by reaction with a source of cyanide ion such as acetone cyanohydrin or an inorganic cyanide, preferably sodium cyanide, in the presence of a non-nucleophilic base such as tetramethyl guanidine, in a range of solvents, preferably ethanol, at a range of temperatures, preferably between ambient temperature and the reflux temperature of the solvent.
- a source of cyanide ion such as acetone cyanohydrin or an inorganic cyanide, preferably sodium cyanide
- a non-nucleophilic base such as tetramethyl guanidine
- Compounds of formula (Xlll-c) can be prepared from compounds of formula (Xlll-a) by reaction with a reducing agent such as triethylsilane in the presence of an acid such as trifluoroacetic acid in a solvent such as DCM from room temperature to the reflux temperature of the solvent.
- a reducing agent such as triethylsilane
- an acid such as trifluoroacetic acid
- Compounds of formula (Xlll-b) can be prepared from compounds of formula (Xlll-a) by reaction with an alkylating agent of formula (XLVIII):
- R f is a C 1-6 alkyl group and Y is a leaving group such as halogen, tosylate, mesylate.
- the reaction is performed in the presence of a base such as sodium hydride in a solvent such as THF from O 0 C to the reflux temperature of the solvent.
- a base such as sodium hydride
- a solvent such as THF from O 0 C to the reflux temperature of the solvent.
- Compounds of general formula (Xlll-h) can be prepared from compounds of formula (Xlll-g) using methods described above for the preparation of compounds of formula (Xlll-e) from compounds of formula (Xlll-d).
- Compounds of general formula (Xlll-g) can be prepared from compounds of formula (XXXIX) using methods described above for the preparation of compounds of formula (Xlll-d) from compounds of formula (XXXV).
- Compounds of general formula (Xlll-f) can be prepared from compounds of formula (XXVII-b) using methods described above for the preparation of compounds of formula (Xlll-a) from compounds of formula (XXVII-a).
- Compounds of general formula (XXVII-b) can be prepared from compounds of formula (XXXVIII) using methods described above for the preparation of compounds of formula (XXXIl) from compounds of formula (XXXI).
- Compounds of Formula (LII) may be prepared from compounds of Formula (L) by employing a similar sequence of reactions as used to prepare compounds of Formula (Xlll-a) from compounds of Formula (XXXIV) in Scheme 1 above.
- Compounds of formula (L) wherein R 4 and R 5 are the same may be prepared from compounds of Formula (XLIX) where R is a suitable alkyl group (such as ethyl or methyl) by treatment with an appropriate organometallic reagent such as a Grignard reagent, in a suitable solvent such as THF or diethyl ether.
- Compounds of Formula (L) wherein R 4 and R 5 are dissimilar may be prepared from compounds of Formula (XLIX) by converting to an intermediate amide, preferably a Weinreb amide, and performing the introduction of R 4 and R 5 through their respective organometallic reagents in a stepwise manner.
- the present invention also comprises intermediate compounds having utility in the synthesis of the compounds of formula (I) or (Ia).
- such intermediate compounds are selected from the group including 5-[(R)-1-(tert-butyl ⁇ dimethyl-silanyloxy)-2-(9- ⁇ [2-(cyclohexyl-hydroxy-phenyl-methyl)-oxazol-5-ylmethyl]- methyl-amino ⁇ -nonylamino)-ethyl]-8-hydroxy-1 H-quinolin-2-one; 5-[(R)-1-(tert-butyl- dimethyl-silanyloxy)-2-(9- ⁇ [2-(hydroxy-diphenyl-methyl)-oxazol-5-ylmethyl]-methyl- amino ⁇ -nonylamino)-ethyl]-8-(4-methoxy-benzyloxy)-1 /-/-quinolin-2-one; 5-[(R)-1 - hydroxy-2-(9- ⁇ [2-(hydroxy-)-(
- such intermediate compounds are selected from the group including 5-[(f?)-1 -(tert-butyl-dimethyl-silanyloxy)-2-(9- ⁇ [2-(hydroxy-diphenyl- methyl)-oxazol-5-ylmethyl]-methyl-amino ⁇ -nonylamino)-ethyl]-8-(4-methoxy- benzyloxy)-1 H-quinolin-2-one; 5-[(R)-1 -hydroxy-2-(9- ⁇ [2-(hydroxy-diphenyl-methyl)- oxazol-5-ylmethyl]-methyl-amino ⁇ -nonylamino)-ethyl]-8-(4-methoxy-benzyloxy)-1 H- quinolin-2-one; and [2-(hydroxy-diphenyl-methyl)-oxazol-5-ylmethyl]-dimethyl-(9-oxo- nonyl)-ammonium bromide.
- the compounds of formula (I) and (Ia) have activity as pharmaceuticals, in particular as dual adrenergic ⁇ 2 receptor agonists and anticholinergic agents including muscarinic receptor (M1 , M2, and M3) antagonists, in particular M3 antagonists.
- Diseases and conditions which may be treated with the compounds of formula (I) or (Ia) and their pharmaceutically acceptable salts include:
- respiratory tract obstructive diseases of the airways including: asthma, including bronchial, allergic, intrinsic, extrinsic, exercise-induced, drug-induced (including aspirin and NSAID-induced) and dust-induced asthma, both intermittent and persistent and of all severities, and other causes of airway hyper-responsiveness; chronic obstructive pulmonary disease (COPD); bronchitis, including infectious and eosinophilic bronchitis; emphysema; bronchiectasis; cystic fibrosis; sarcoidosis; farmer's lung and related diseases; hypersensitivity pneumonitis; lung fibrosis, including cryptogenic fibrosing alveolitis, idiopathic interstitial pneumonias, fibrosis complicating anti-neoplastic therapy and chronic infection, including tuberculosis and aspergillosis and other fungal infections; complications of lung transplantation; vasculitic and thrombotic disorders of the lung vasculature
- osteoarthritides associated with or including osteoarthritis/osteoarthrosis both primary and secondary to, for example, congenital hip dysplasia; cervical and lumbar spondylitis, and low back and neck pain; osteoporosis; rheumatoid arthritis and Still's disease; seronegative spondyloarthropathies including ankylosing spondylitis, psoriatic arthritis, reactive arthritis and undifferentiated spondarthropathy; septic arthritis and other infection- related arthopathies and bone disorders such as tuberculosis, including Potts' disease and Poncet's syndrome; acute and chronic crystal-induced synovitis including urate gout, calcium pyrophosphate deposition disease, and calcium apatite related tendon, bursal and synovial inflammation; Behcet's disease; primary and secondary Sjogren's syndrome; systemic sclerosis and limited scleroderma; systemic lupus erythemato
- arthitides for example rheumatoid arthritis, osteoarthritis, gout or crystal arthropathy
- other joint disease such as intervertebral disc degeneration or temporomandibular joint degeneration
- bone remodelling disease such as osteoporosis, Paget's disease or osteonecrosis
- polychondritits such as osteoporosis, Paget's
- skin psoriasis, atopic dermatitis, contact dermatitis or other eczematous dermatoses, and delayed-type hypersensitivity reactions; phyto- and photodermatitis; seborrhoeic dermatitis, dermatitis herpetiformis, lichen planus, lichen sclerosus et atrophica, pyoderma gangrenosum, skin sarcoid, discoid lupus erythematosus, pemphigus, pemphigoid, epidermolysis bullosa, urticaria, angioedema, vasculitides, toxic erythemas, cutaneous eosinophilias, alopecia areata, male-pattern baldness, Sweet's syndrome, Weber-Christian syndrome, erythema multiforme; cellulitis, both infective and non-infective; panniculitis;cutaneous lymphomas, non-melanoma
- eyes blepharitis; conjunctivitis, including perennial and vernal allergic conjunctivitis; ulceris; anterior and posterior uveitis; choroiditis; autoimmune; degenerative or inflammatory disorders affecting the retina; ophthalmitis including sympathetic ophthalmitis; sarcoidosis; infections including viral , fungal, and bacterial;
- gastrointestinal tract glossitis, gingivitis, periodontitis; oesophagitis, including reflux; eosinophilic gastro-enteritis, mastocytosis, Crohn's disease, colitis including ulcerative colitis, proctitis, pruritis ani; coeliac disease, irritable bowel syndrome, and food-related allergies which may have effects remote from the gut (for example migraine, rhinitis or eczema);
- abdominal hepatitis, including autoimmune, alcoholic and viral; fibrosis and cirrhosis of the liver; cholecystitis; pancreatitis, both acute and chronic; 8. genitourinary: nephritis including interstitial and glomerulonephritis; nephrotic syndrome; cystitis including acute and chronic (interstitial) cystitis and Hunner's ulcer; acute and chronic urethritis, prostatitis, epididymitis, oophoritis and salpingitis; vulvovaginitis; Peyronie's disease; erectile dysfunction (both male and female);
- allograft rejection acute and chronic following, for example, transplantation of kidney, heart, liver, lung, bone marrow, skin or cornea or following blood transfusion; or chronic graft versus host disease;
- CNS Alzheimer's disease and other dementing disorders including CJD and nvCJD; amyloidosis; multiple sclerosis and other demyelinating syndromes; cerebral atherosclerosis and vasculitis; temporal arteritis; myasthenia gravis; acute and chronic pain (acute, intermittent or persistent, whether of central or peripheral origin) including visceral pain, headache, migraine, trigeminal neuralgia, atypical facial pain, joint and bone pain, pain arising from cancer and tumor invasion, neuropathic pain syndromes including diabetic, post-herpetic, and HIV-associated neuropathies; neurosarcoidosis; central and peripheral nervous system complications of malignant, infectious or autoimmune processes;
- cardiovascular atherosclerosis, affecting the coronary and peripheral circulation; pericarditis; myocarditis , inflammatory and auto-immune cardiomyopathies including myocardial sarcoid; ischaemic reperfusion injuries; endocarditis, valvulitis, and aortitis including infective (for example syphilitic); vasculitides; disorders of the proximal and peripheral veins including phlebitis and thrombosis, including deep vein thrombosis and complications of varicose veins;
- oncology treatment of common cancers including prostate, breast, lung, ovarian, pancreatic, bowel and colon, stomach, skin and brain tumors and malignancies affecting the bone marrow (including the leukaemias) and lymphoproliferative systems, such as Hodgkin's and non-Hodgkin's lymphoma; including the prevention and treatment of metastatic disease and tumour recurrences, and paraneoplastic syndromes; and,
- gastrointestinal tract Coeliac disease, proctitis, eosinopilic gastro-enteritis, mastocytosis, Crohn's disease, ulcerative colitis, microscopic colitis, indeterminant colitis, irritable bowel disorder, irritable bowel syndrome, non-inflammatory diarrhea, food-related allergies which have effects remote from the gut, e.g., migraine, rhinitis and eczema.
- the present invention provides a compound of formula (Ia) or a pharmaceutically-acceptable salt thereof as hereinbefore defined for use in therapy.
- the present invention provides the use of a compound of formula (Ia) or a pharmaceutically acceptable salt thereof as hereinbefore defined in the manufacture of a medicament for use in therapy.
- the term “therapy” also includes “prophylaxis” unless there are specific indications to the contrary.
- the terms “therapeutic” and “therapeutically” should be construed accordingly.
- Prophylaxis is expected to be particularly relevant to the treatment of persons who have suffered a previous episode of, or are otherwise considered to be at increased risk of, the disease or condition in question.
- Persons at risk of developing a particular disease or condition generally include those having a family history of the disease or condition, or those who have been identified by genetic testing or screening to be particularly susceptible to developing the disease or condition.
- the invention still further provides a method of treating, or reducing the risk of, an inflammatory disease or condition (including a reversible obstructive airways disease or condition) which comprises administering to a patient in need thereof a therapeutically effective amount of a compound of formula (Ia) or a pharmaceutically acceptable salt thereof as hereinbefore defined.
- the compounds of this invention may be used in the treatment of adult respiratory distress syndrome (ARDS), pulmonary emphysema, bronchitis, bronchiectasis, chronic obstructive pulmonary disease (COPD), asthma and rhinitis.
- ARDS adult respiratory distress syndrome
- COPD chronic obstructive pulmonary disease
- the daily dosage of the compound of the invention if inhaled, may be in the range from 0.05 micrograms per kilogram body weight ( ⁇ g/kg) to 100 micrograms per kilogram body weight ( ⁇ g/kg).
- the daily dosage of the compound of the invention may be in the range from 0.01 micrograms per kilogram body weight ( ⁇ g/kg) to 100 milligrams per kilogram body weight (mg/kg).
- the compounds of formula (Ia) and pharmaceutically acceptable salts thereof may be used on their own but will generally be administered in the form of a pharmaceutical composition in which the formula (Ia) compound/salt (active ingredient) is in association with a pharmaceutically acceptable adjuvant, diluent or carrier.
- a pharmaceutically acceptable adjuvant diluent or carrier.
- Conventional procedures for the selection and preparation of suitable pharmaceutical formulations are described in, for example, "Pharmaceuticals - The Science of Dosage Form Designs", M. E. Aulton, Churchill Livingstone, 1988.
- the pharmaceutical composition will preferably comprise from 0.05 to 99 %w (per cent by weight), more preferably from 0.05 to 80 %w, still more preferably from 0.10 to 70 %w, and even more preferably from 0.10 to 50 %w, of active ingredient, all percentages by weight being based on total composition.
- the present invention also provides a pharmaceutical composition comprising a compound of formula (Ia) or a pharmaceutically acceptable salt thereof as hereinbefore defined, in association with a pharmaceutically acceptable adjuvant, diluent or carrier.
- the invention further provides a process for the preparation of a pharmaceutical composition of the invention which comprises mixing a compound of formula (Ia) or a pharmaceutically acceptable salt thereof as hereinbefore defined with a pharmaceutically acceptable adjuvant, diluent or carrier.
- compositions may be administered topically (e.g. to the skin or to the lung and/or airways) in the form, e.g., of creams, solutions, suspensions, heptafluoroalkane (HFA) aerosols and dry powder formulations, for example, formulations in the inhaler device known as the Turbuhaler ® ; or systemically, e.g. by oral administration in the form of tablets, capsules, syrups, powders or granules; or by parenteral administration in the form of solutions or suspensions; or by subcutaneous administration; or by rectal administration in the form of suppositories; or transdermally.
- HFA heptafluoroalkane
- Dry powder formulations and pressurized HFA aerosols of the compounds of the invention may be administered by oral or nasal inhalation.
- the compound is desirably finely divided.
- the finely divided compound preferably has a mass median diameter of less than 10 ⁇ m, and may be suspended in a propellant mixture with the assistance of a dispersant, such as a C 8 -C 20 fatty acid or salt thereof, (for example, oleic acid), a bile salt, a phospholipid, an alkyl saccharide, a perfluorinated or polyethoxylated surfactant, or other pharmaceutically acceptable dispersant.
- a dispersant such as a C 8 -C 20 fatty acid or salt thereof, (for example, oleic acid), a bile salt, a phospholipid, an alkyl saccharide, a perfluorinated or polyethoxylated surfactant, or other pharmaceutically acceptable dispersant.
- the compounds of the invention may also be administered by means of a dry powder inhaler.
- the inhaler may be a single or a multi dose inhaler, and may be a breath actuated dry powder inhaler.
- a carrier substance for example, a mono-, di- or polysaccharide, a sugar alcohol, or another polyol.
- Suitable carriers are sugars, for example, lactose, glucose, raffinose, melezitose, lactitol, maltitol, trehalose, sucrose, mannitol; and starch.
- the finely divided compound may be coated by another substance.
- the powder mixture may also be dispensed into hard gelatine capsules, each containing the desired dose of the active compound.
- This spheronized powder may be filled into the drug reservoir of a multidose inhaler, for example, that known as the Turbuhaler ® in which a dosing unit meters the desired dose which is then inhaled by the patient.
- a multidose inhaler for example, that known as the Turbuhaler ® in which a dosing unit meters the desired dose which is then inhaled by the patient.
- the active ingredient with or without a carrier substance, is delivered to the patient.
- the compound of the invention may be admixed with an adjuvant or a carrier, for example, lactose, saccharose, sorbitol, mannitol; a starch, for example, potato starch, corn starch or amylopectin; a cellulose derivative; a binder, for example, gelatine or polyvinylpyrrolidone; and/or a lubricant, for example, magnesium stearate, calcium stearate, polyethylene glycol, a wax, paraffin, and the like, and then compressed into tablets.
- an adjuvant or a carrier for example, lactose, saccharose, sorbitol, mannitol
- a starch for example, potato starch, corn starch or amylopectin
- a cellulose derivative for example, gelatine or polyvinylpyrrolidone
- a lubricant for example, magnesium stearate, calcium stearate, polyethylene glycol, a wax
- the cores may be coated with a concentrated sugar solution which may contain, for example, gum arabic, gelatine, talcum and titanium dioxide.
- a concentrated sugar solution which may contain, for example, gum arabic, gelatine, talcum and titanium dioxide.
- the tablet may be coated with a suitable polymer dissolved in a readily volatile organic solvent.
- the compound of the invention may be admixed with, for example, a vegetable oil or polyethylene glycol.
- Hard gelatine capsules may contain granules of the compound using either the above- mentioned excipients for tablets. Also liquid or semisolid formulations of the compound of the invention may be filled into hard gelatine capsules.
- Liquid preparations for oral application may be in the form of syrups or suspensions, for example, solutions containing the compound of the invention, the balance being sugar and a mixture of ethanol, water, glycerol and propylene glycol.
- Such liquid preparations may contain colouring agents, flavouring agents, saccharine and/or carboxymethylcellulose as a thickening agent or other excipients known to those skilled in art.
- the compounds of the invention may also be administered in conjunction with other compounds used for the treatment of the above conditions.
- the invention therefore further relates to combination therapies wherein a compound of the invention, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition or formulation comprising a compound of the invention, is administered concurrently or sequentially or as a combined preparation with another therapeutic agent or agents, for the treatment of one or more of the conditions listed.
- NSAIDs nonsteroidal anti-inflammatory agents
- COX-1 / COX-2 inhibitors whether applied topically or systemically
- piroxicam diclofenac
- propionic acids such as naproxen, flurbiprofen, fenoprofen, ketoprofen and ibuprofen
- fenamates such as mefenamic acid, indomethacin, sulindac, azapropazone, pyrazolones such as phenylbutazone, salicylates such as aspirin
- selective COX-2 inhibitors such as meloxicam
- the present invention still further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, together with a cytokine or agonist or antagonist of cytokine function, (including agents which act on cytokine signalling pathways such as modulators of the SOCS system) including alpha-, beta-, and gamma-interferons; insulin-like growth factor type I (IGF-1); interleukins (IL) including IL1 to 17, and interleukin antagonists or inhibitors such as anakinra; tumour necrosis factor alpha (TNF- ⁇ ) inhibitors such as anti-TNF monoclonal antibodies (for example infliximab; adalimumab, and CDP-870) and TNF receptor antagonists including immunoglobulin molecules (such as etanercept) and low-molecular-weight agents such as pentoxyfyliine.
- a cytokine or agonist or antagonist of cytokine function including agents which act on cytokine signal
- the invention relates to a combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, with a monoclonal antibody targeting B-Lymphocytes (such as CD20 (rituximab), MRA-alLI6R) or T-Lymphocytes (CTLA4-lg, HuMax 11-15).
- B-Lymphocytes such as CD20 (rituximab), MRA-alLI6R
- T-Lymphocytes CLA4-lg, HuMax 11-15.
- the present invention still further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, with a modulator of chemokine receptor function such as an antagonist of CCR1 , CCR2, CCR2A, CCR2B, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR9, CCR10 and CCR1 1 (for the C-C family); CXCR1 , CXCR2, CXCR3, CXCR4 and CXCR5 (for the C-X-C family) and CX 3 CRI for the C-X 3 -C family.
- a modulator of chemokine receptor function such as an antagonist of CCR1 , CCR2, CCR2A, CCR2B, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR9, CCR10 and CCR1 1 (for the C-C family); CXCR1 , CXCR2, CXCR3, CXCR4 and CXCR5 (for the C-
- the present invention further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, with an inhibitor of matrix metalloprotease (MMPs), i.e., the stromelysins, the collagenases, and the gelatinases, as well as aggrecanase; especially collagenase-1 (MMP-1), collagenase-2 (MMP-8), collagenase-3 (MMP-13), stromelysin-1 (MMP-3), stromelysin-2 (MMP-10), and stromelysin-3 (MMP-11) and MMP-9 and MMP-12, including agents such as doxycycline.
- MMPs matrix metalloprotease
- the present invention still further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, and a leukotriene biosynthesis inhibitor, 5-lipoxygenase (5-LO) inhibitor or 5-lipoxygenase activating protein (FLAP) antagonist such as; zileuton; ABT-761 ; fenleuton; tepoxalin; Abbott- 79175; Abbott-85761 ; a N-(5-substituted)-thiophene-2-alkylsulfonamide; 2,6-di-tert- butylphenolhydrazones; a methoxytetrahydropyrans such as Zeneca ZD-2138; the compound SB-210661 ; a pyridinyl-substituted 2-cyanonaphthalene compound such as L- 739,010; a 2-cyanoquino!ine compound such as L-746,530; or an indole or quinoline compound such as MK-591
- the present invention further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, and a receptor antagonist for leukotrienes (LT) B4, LTC4, LTD4, and LTE4.
- a receptor antagonist for leukotrienes (LT) B4, LTC4, LTD4, and LTE4 selected from the group consisting of the phenothiazin-3-1s such as L-651 ,392; amidino compounds such as CGS-25019c; benzoxalamines such as ontazolast; benzenecarboximidamides such as BIIL 284/260; and compounds such as zafirlukast, ablukast, montelukast, pranlukast, verlukast (MK-679), RG-12525, Ro-245913, iralukast (CGP 45715A), and BAY x 7195.
- the present invention still further relates to the combination of a compound of the invention, or
- PDE PDE inhibitor
- a methylxanthanine including theophylline and aminophylline a selective PDE isoenzyme inhibitor including a PDE4 inhibitor an inhibitor of the isoform PDE4D, or an inhibitor of PDE5.
- the present invention further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, and a histamine type 1 receptor antagonist such as cetirizine, loratadine, desloratadine, fexofenadine, acrivastine, terfenadine, astemizole, azelastine, levocabastine, chlorpheniramine, promethazine, cyclizine, or mizolastine; applied orally, topically or parenterally.
- a histamine type 1 receptor antagonist such as cetirizine, loratadine, desloratadine, fexofenadine, acrivastine, terfenadine, astemizole, azelastine, levocabastine, chlorpheniramine, promethazine, cyclizine, or mizolastine
- the present invention still further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, and a proton pump inhibitor (such as omeprazole) or a gastroprotective histamine type 2 receptor antagonist.
- a proton pump inhibitor such as omeprazole
- a gastroprotective histamine type 2 receptor antagonist such as a gastroprotective histamine type 2 receptor antagonist.
- the present invention further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, and an antagonist of the histamine type 4 receptor.
- the present invention still further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, and an alpha-1/alpha-2 adrenoceptor agonist vasoconstrictor sympathomimetic agent, such as propylhexedrine, phenylephrine, phenylpropanolamine, ephedrine, pseudoephedrine, naphazoline hydrochloride, oxymetazoline hydrochloride, tetrahydrozoline hydrochloride, xylometazoline hydrochloride, tramazoline hydrochloride or ethylnorepinephrine hydrochloride.
- the present invention further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, and a chromone, such as sodium cromoglycate or nedocromil sodium.
- the present invention still further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, with a glucocorticoid, such as flunisolide, triamcinolone acetonide, beclomethasone dipropionate, budesonide, fluticasone propionate, ciclesonide or mometasone furoate.
- a glucocorticoid such as flunisolide, triamcinolone acetonide, beclomethasone dipropionate, budesonide, fluticasone propionate, ciclesonide or mometasone furoate.
- the present invention further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, with an agent that modulates a nuclear hormone receptor such as PPARs.
- the present invention still further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, together with an immunoglobulin (Ig) or Ig preparation or an antagonist or antibody modulating Ig function such as anti-lgE (for example omalizumab).
- the present invention further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, and another systemic or topically-applied anti-inflammatory agent, such as thalidomide or a derivative thereof, a retinoid, dithranol or calcipotriol.
- a compound of the invention or a pharmaceutically acceptable salt thereof
- another systemic or topically-applied anti-inflammatory agent such as thalidomide or a derivative thereof, a retinoid, dithranol or calcipotriol.
- the present invention still further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, and combinations of aminosalicylates and sulfapyridine such as sulfasalazine, mesalazine, balsalazide, and olsalazine; and immunomodulatory agents such as the thiopurines, and corticosteroids such as budesonide.
- aminosalicylates and sulfapyridine such as sulfasalazine, mesalazine, balsalazide, and olsalazine
- immunomodulatory agents such as the thiopurines, and corticosteroids such as budesonide.
- the present invention further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, together with an antibacterial agent such as a penicillin derivative, a tetracycline, a macrolide, a beta-lactam, a fluoroquinolone, metronidazole, an inhaled aminoglycoside; an antiviral agent including acyclovir, famciclovir, valaciclovir, ganciclovir, cidofovir, amantadine, rimantadine, ribavirin, zanamavir and oseltamavir; a protease inhibitor such as indinavir, nelfinavir, ritonavir, and saquinavir; a nucleoside reverse transcriptase inhibitor such as didanosine, lamivudine, stavudine, zalcitabine or zidovudine; or a non-nucleoside reverse transcripta
- the present invention still further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, and a cardiovascular agent such as a calcium channel blocker, a beta-adrenoceptor blocker, an angiotensin-converting enzyme (ACE) inhibitor, an angiotensin-2 receptor antagonist; a lipid lowering agent such as a statin or a fibrate; a modulator of blood cell morphology such as pentoxyfylline; thrombolytic, or an anticoagulant such as a platelet aggregation inhibitor.
- a cardiovascular agent such as a calcium channel blocker, a beta-adrenoceptor blocker, an angiotensin-converting enzyme (ACE) inhibitor, an angiotensin-2 receptor antagonist
- ACE angiotensin-converting enzyme
- angiotensin-2 receptor antagonist angiotensin-2 receptor antagonist
- a lipid lowering agent such as a statin or a fibrate
- a modulator of blood cell morphology such as
- the present invention further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, and a CNS agent such as an antidepressant (such as sertraline), an anti-Parkinsonian drug (such as deprenyl, L- dopa, ropinirole, pramipexole, a MAOB inhibitor such as selegine and rasagiline, a comP inhibitor such as tasmar, an A-2 inhibitor, a dopamine reuptake inhibitor, an NMDA antagonist, a nicotine agonist, a dopamine agonist or an inhibitor of neuronal nitric oxide synthase), or an anti-Alzheimer's drug such as donepezil, rivastigmine, tacrine, a COX-2 inhibitor, propentofylline or metrifonate.
- a CNS agent such as an antidepressant (such as sertraline), an anti-Parkinsonian drug (such as deprenyl, L-
- the present invention still further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, and an agent for the treatment of acute or chronic pain, such as a centrally or peripherally-acting analgesic (for example an opioid or derivative thereof), carbamazepine, phenytoin, sodium valproate, amitryptiline or other anti-depressant agent-s, paracetamol, or a nonsteroidal anti-inflammatory agent.
- analgesic for example an opioid or derivative thereof
- carbamazepine for example an opioid or derivative thereof
- phenytoin for example an opioid or derivative thereof
- sodium valproate for example an opioid or derivative thereof
- amitryptiline or other anti-depressant agent-s sodium valproate
- paracetamol paracetamol
- nonsteroidal anti-inflammatory agent for example an opioid or derivative thereof
- the present invention further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, together with a parenterally or topically-applied (including inhaled) local anaesthetic agent such as lignocaine or a derivative thereof.
- a parenterally or topically-applied (including inhaled) local anaesthetic agent such as lignocaine or a derivative thereof.
- a compound of the present invention can also be used in combination with an anti-osteoporosis agent including a hormonal agent such as raloxifene, or a biphosphonate such as alendronate.
- an anti-osteoporosis agent including a hormonal agent such as raloxifene, or a biphosphonate such as alendronate.
- the present invention still further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, together with a: (i) tryptase inhibitor; (ii) platelet activating factor (PAF) antagonist; (iii) interleukin converting enzyme (ICE) inhibitor; (iv) IMPDH inhibitor; (v) adhesion molecule inhibitors including VLA-4 antagonist; (vi) cathepsin; (vii) kinase inhibitor such as an inhibitor of tyrosine kinase (such as Btk, Itk, Jak3 or MAP, for example Gefitinib or lmatinib mesylate), a serine / threonine kinase (such as an inhibitor of a MAP kinase such as p38, JNK, protein kinase A, B or C, or IKK), or a kinase involved in cell cycle regulation (such as a cylin dependent kinase
- - or B.sub2. -receptor antagonist for example colchicine;
- xi xanthine oxidase inhibitor, for example allopurinol;
- uricosuric agent for example probenecid, sulfinpyrazone or benzbromarone;
- xiii growth hormone secretagogue;
- PDGF platelet-derived growth factor;
- PDGF platelet-derived growth factor
- fibroblast growth factor for example basic fibroblast growth factor (bFGF);
- GM-CSF granulocyte macrophage colony stimulating factor (GM-CSF);
- GM-CSF granulocyte macrophage colony stimulating factor
- capsaicin cream for example tachykinin NK.subi .
- NKP-608C SB-233412 (talnetant) or D-4418
- elastase inhibitor such as UT-77 or ZD-0892
- TACE TNF-alpha converting enzyme inhibitor
- iNOS induced nitric oxide synthase
- chemoattractant receptor-homologous molecule expressed on TH2 cells such as a CRTH2 antagonist
- inhibitor of P38 agent modulating the function of Toll-like receptors (TLR),
- agent modulating the activity of purinergic receptors such as P2X7
- inhibitor of transcription factor activation such as NFkB, API or STATS
- a glucocorticoid receptor GR-receptor
- the present invention provides a combination (for example for the treatment of COPD, asthma or allergic rhinitis) of a compound of formula (I) and one or more agents selected from the list comprising: o a non-steroidal glucocorticoid receptor (GR-receptor) agonist; o a PDE4 inhibitor including an inhibitor of the isoform PDE4D; o a modulator of chemokine receptor function (such as a CCR1 receptor antagonist); o a steroid (such as budesonide); and o an inhibitor of p38 kinase function.
- GR-receptor non-steroidal glucocorticoid receptor
- PDE4 inhibitor including an inhibitor of the isoform PDE4D
- o a modulator of chemokine receptor function such as a CCR1 receptor antagonist
- o a steroid such as budesonide
- an inhibitor of p38 kinase function for example for the treatment of COPD, asthma
- a compound of the invention, or a pharmaceutically acceptable salt thereof, can also be used in combination with an existing therapeutic agent for the treatment of cancer
- suitable agents include: (i) an antiproliferative/antineoplastic drug or a combination thereof, as used in medical oncology, such as an alkylating agent (for example cis-platin, carboplatin, cyclophosphamide, nitrogen mustard, melphalan, chlorambucil, busulphan or a nitrosourea); an antimetabolite (for example an antifolate such as a fluoropyrimidine like 5-fluorouracil or tegafur, raltitrexed, methotrexate, cytosine arabinoside, hydroxyurea, gemcitabine or paclitaxel); an antitumour antibiotic (for example an anthracycline such as adriamycin, bleomycin, doxorubicin, daunomycin, epirubicin, idarubicin,
- a cytostatic agent such as an antioestrogen (for example tamoxifen, toremifene, raloxifene, droloxifene or iodoxyfene), an oestrogen receptor down regulator (for example fulvestrant), an antiandrogen (for example bicalutamide, flutamide, nilutamide or cyproterone acetate), a LHRH antagonist or LHRH agonist (for example goserelin, leuprorelin or buserelin), a progestogen (for example megestrol acetate), an aromatase inhibitor (for example as anastrozole, letrozole, vorazole or exemestane) or an inhibitor of 5 ⁇ -reductase such as finasteride; (iii) an agent which inhibits cancer cell invasion (for example a metalloproteinase inhibitor like marimastat or an inhibitor of urokinase plasm
- an inhibitor of growth factor function for example: a growth factor antibody (for example the anti-erbb2 antibody trastuzumab, or the anti-erbb1 antibody cetuximab [C225]), a farnesyl transferase inhibitor, a tyrosine kinase inhibitor or a serine/threonine kinase inhibitor, an inhibitor of the epidermal growth factor family (for example an EGFR family tyrosine kinase inhibitor such as N.-(3-chloro-4- fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)quinazolin-4-amine (gefitinib, AZD1839), N-(3-ethynylphenyl)-6,7-bis(2-methoxyethoxy)quinazolin-4-amine (erlotinib, OSI-774) or 6-acrylamido-N.-(3-chloro-4-fluorophen
- an agent used in antisense therapy for example one directed to one of the targets listed above, such as ISIS 2503, an anti-ras antisense;
- an agent used in a gene therapy approach for example approaches to replace aberrant genes such as aberrant p53 or aberrant BRCA1 or BRCA2, GDEPT
- Silica gel used for medium pressure column chromatography is 0.035 to 0.070 mm (220 to 440 mesh) (e.g. Fluka silica gel 60), and an applied pressure up to 10 psi accelerated column elution.
- TLC thin layer chromatography
- it refers to silica gel TLC using plates, typically 3 x 6 cm silica gel on aluminium foil plates with a fluorescent indicator (254 nm) (e.g. Fluka 60778). All solvents and commercial reagents were used as received.
- Purification by prepacked SCX-2 cartridge refers to Isolute ® SCX-2, a strong cation exchange sorbent (Argonaut/IST).
- Purification over NH 2 -silica gel refers to Isolute ® flash NH 2 prepacked cartridges (Argonaut/IST).
- Phenyl hexyl column 250 x 21.20 mm i.d. Luna column with 5 ⁇ m particle size
- Micromass Platform LCT with a C18-reverse-phase column (100 x 3.0 mm i.d. Higgins Clipeus with 5 ⁇ m particle size), elution with solvent A (water with 0.1% formic acid) and solvent B (acetonitrile with 0.1% formic acid).
- MS ionisation method Electrospray (positive ion).
- MS ionisation method Electrospray (positive and negative ion).
- MS ionisation method Electrospray (positive and negative ion).
- AIBN (2,2'-azobis(2-methylproprionitrile)
- HATU 0-(7-azabenzotriazol-1 -y ⁇ )-N,N,N'N -tetramethyluroniumhexaf luoro- phosphate
- NaHCO 3 sodium hydrogen carbonate
- TFA trifluoroacetic acid
- THF tetrahydrofuran
- Example 1 5-F(/?)-2-(9-fr2-(Cvclohexyi-hvdroxy-phenyl-methyl)-oxazol-5-ylmethvil-methvl- aminol-nonylaminoV1-hvdroxy-ethyll-8-hydroxy-1 /y-quinolin-2-one naphthalene-1 ,5-disulfonate.
- Oxaly) chloride (19 g, 0.15 mol) was added over 15 min to a solution of phenylglyoxylic acid (20 g, 0.13 mol) and 5 drops of DMF in dry DCM (100 m!_).
- the reaction mixture was stirred at RT for 3 hours after which the solvent was removed in vacuo.
- the residue was taken up in dry DCM (50 ml_) and the solution was added over 30 minutes to a mixture of propargyl amine (7.3 g, 0.13 mol) and triethylamine (13.5 g, 0.13 mol) in dry DCM (100 ml_) at 0 0 C.
- the mixture was allowed to warm to RT overnight.
- Trifluoromethane sulphonic acid (42.5 mL, 0.48 mol) was added dropwise to a solution of 2-oxo-2-phenyl- ⁇ /-prop-2-ynyl-acetamide (74.88 g, 0.40 mol) in 1 ,4- dioxane (600 mL). The resulting solution was heated at 90 0 C for 20 hours. The reaction mixture was cooled to RT and concentrated in vacuo. The residue was partitioned between DCM and concentrated aqueous ammonia and adjusted to pH 10 by addition of 1 M NaOH (aq). The aqueous phase was separated and then further extracted with DCM.
- the aqueous phase was extracted with EtOAc and the combined organic layers were washed with 10% citric acid solution (aq), saturated NaHCO 3 (aq), brine, dried (Na 2 SO 4 ), and filtered.
- the solvent was evaporated in vacuo to afford a white solid. This was triturated with diethyl ether and collected by filtration, washed with diethyl ether, and dried at 45 °C in vacuo to afford the title compound as a white solid.
- the mother liquors were concentrated and the residue purified by column chromatography over silica gel using a gradient of 5-10% ETOAc/cyclohexane to afford a second batch of semi-pure product.
- Example 1 step h, diastereomer 2).
- Example 1 The title compound was prepared by methods analogous to those used in Example 1 step c and d, employing (5 ⁇ methyl-oxazol-2-yl)-phenyl ⁇ methanone (Example 1 , intermediate b) and cyclopentylmagnesium chloride.
- the reaction mixture was partitioned between 10% lithium chloride (aq) and EtOAc and the aqueous layer was extracted with EtOAc. The combined organic layers were washed with brine, dried (Na 2 SO 4 ), filtered, and concentrated in vacuo.
- the residual yellow oil was purified by column chromatography over silica gel using a gradient of 0-10% MeOH/DCM as eluent, then passed through a SCX-2cartridge, and liberated using a 2 M ammonia solution in MeOH. Evaporation of the solvent in vacuo afforded the title compound.
- the title compound was prepared from 9- ⁇ [2-(cyclopentyl-hydroxy-phenyl-methyl)- oxazol-5-ylmethyl]-methyl-amino ⁇ -nonanal and 5-[(f?)-2-annino ⁇ 1 -(terf-butyl-dimethyl- silanyloxy)-ethyl]-8-hydroxy-1 W-quinolin-2-one (prepared according to
- Example 1 The title compound was prepared by methods analogous to those used in Example 1 step c and d, employing (5-methyl-oxazol-2-yl)-phenyl-methanone (Example 1 , intermediate b) and phenylmagnesium bromide.
- Example 3 The title compound was prepared from (5-bromomethyl-oxazol-2-yl)-diphenyl- methanol (Example 3, intermediate a) and 9-methylamino-nonan-1 -ol (prepared according to JP1992-31818) by a similar method to that employed in Example 1 , steps f.
- the title compound was prepared from 5-[(F?)-1-(tert-butyl-dimethyl-silanyloxy)-2-(9- ⁇ [2-(hydroxy-diphenyl-methyl)-oxazol-5-ylmethyl]-methyl-amino ⁇ -nonylamino)-ethyl]-8- (4-methoxy-benzyloxy)-1 /-/-quinolin-2-one by a similar method to that employed in Example 1 , step i.
- the crude product was purified by HPLC (HPLC system 3), eluting with water/MeCN (+0.1% diethylamine) instead of water/MeCN (+0.1 % formic acid).
- LC-MS (method 2): Rt 2.21 min, m/z 759 [MH]+.
- step b A mixture in DMF (2 ml_) of cyclopentyl-(5-methylaminomethyl-oxazol-2-yl)-phenyl- methanol (Example 2, step b: first eluting enantiomer) (0.25 g, 0.87 mmol), 8-bromo- 1 ,1 -dimethoxy-octane (J. Med. Chem. 1989, 32(6), 1319) (0.22 g, 0.88 mmol), caesium carbonate (0.58 g, 1.78 mmol) and water (50 ⁇ l_, 2.77 mmol) was stirred at RT for 48 hours. The resulting solution was concentrated in vacuo and the residue was partitioned between water (20 m!_) and DCM (20 ml_). The aqueous layer was further extracted with DCM (20 ml_) and the combined organic layer was dried
- the title compound was prepared from cyclopentyl-(5-methylaminomethyl-oxazol-2- yl)-phenyl-methanol (Example 2, step b: first eluting enantiomer), 10-bromo-1 ,1- dimethoxy-decane and 5-[(fl)-2-amino-1 -(tert-butyl-dimethylsilanyloxy) ethyl]-8-(4- methoxybenzyloxy)-1 H-quinolin-2-one (Example 3, intermediate f) by similar methods to those employed in Example 4, steps a, b and Example 3, steps g, h and i.
- the crude product was purified by HPLC (HPLC system 3) to give the desired product as a diformate salt,
- Example 3 The title compound was prepared from (5-bromomethyl-oxazol-2-yl)-diphenyl- methanol (Example 3, intermediate a) and dimethylamine by a similar method to that employed in Example 2, step b.
- the title compound was prepared from [2-(hydroxy-diphenyl-methyl)-oxazol-5- ylmethyl]-dimethyl-(9-oxo-nonyl)-ammonium bromide and 5-[(F?)-2-amino-1-(tert- butyl-dimethylsilanyloxy) ethyl]-8-(4-methoxybenzyloxy)-1 H-quinolin-2-one (Example 3, intermediate f) by similar methods to those employed in Example 3, steps g, h and i.
- the crude product was purified by HPLC (HPLC system 3) to give the desired product as a diformate salt,
- the title compound was prepared from (5-methylaminomethyl-oxazol-2-yl)-diphenyl ⁇ methanol (Example 7, intermediate a), 10-bromo-1 ,1 -dimethoxy-decane (Example 5, intermediate a) and 5-[(R)-2-a.m ⁇ uo- ⁇ -(tert-butyl-dimethyls ⁇ anyloxy) ethyl]-8-(4- methoxy benzyloxy)-1 /-/-quinolin-2-one (Example 3, intermediate f) by similar methods to those employed in Example 4, steps a, b and Example 3, steps g, h and i.
- the crude product was purified by HPLC (HPLC system 3) to give the desired product as a formate salt.
- Example 9 was prepared using similar methods to those used for the preparation of Example 6, employing (/ I ?)-cyclohexyl-(5-dimethyIaminomethyl-oxazol-2-yI)-phenyl- methanol in place of (5-dimethylaminomethyl-oxazol-2-yl)-diphenyl-methanol.
- LC-MS (method 5): Rt 5.75 min, m/z 659 [M]+.
- Example 10 was prepared using similar methods to those used for the preparation of Example 6, employing (fl)-cyclohexyl-(5-dimethylaminomethyl-oxazol-2-yl)-phenyl- methanol in place of (5-dimethylaminomethyl-oxazol-2-yl)-diphenyl-methanol and 7- ((F?)-2-amino-1 -hydroxy-ethyl)-4-hydroxy-3H-benzothiazol-2-one (prepared according to WO 2007027134) instead of 5-[(R)-2-Amino-1 -(tert-butyl-dimethylsilanyloxy)ethyl]- 8-(4-methoxybenzyloxy)-1 H-quinolin-2-one.
- the compounds of the invention may be tested for pharmaceutical activity using assays know in the art, such as for example:
- Radioligand binding studies utilising [ 3 H]-N-methyl scopolamine ([ 3 H]-NMS) and commercially available cell membranes expressing the human muscarinic receptors (M2 and M3) were used to assess the affinity of muscarinic antagonists for M2 and M3 receptors.
- Membranes in TRIS buffer were incubated in 96-well plates with [ 3 H]- NMS and M3 antagonist at various concentrations for 3 hours. Membranes and bound radioligand were then harvested by filtration and allowed to dry overnight. Scintillation fluid was then added and the bound radioligand counted using a Canberra Packard Topcount scintillation counter
- the half-life of antagonists at each muscarinic receptor was measured using the alternative radioligand [ 3 H]-QNB and an adaptation of the above affinity assay. Antagonists were incubated for 3 hours at a concentration 10-fold higher than their Ki, as determined with the [ 3 H]-QNB ligand, with membranes expressing the human muscarinic receptors. At the end of this time, [ 3 H]-QNB was added to a concentration 25-fold higher than its Kd for the receptor being studied and the incubation continued for various time periods from 15 minutes up to 180 minutes. Membranes and bound radioligand were then harvested by filtration and allowed to dry overnight. Scintillation fluid was then added and the bound radioligand counted using a Canberra Packard Topcount scintillation counter.
- the rate at which [3H]-QNB is detected binding to the muscarinic receptors is related to the rate at which the antagonist dissociates from the receptor, ie. to the half life of the antagonists on the receptors.
- Example 2 had a binding Ki of 0.85nM at the M3 receptor, whilst Example 7 had a binding Ki of 1.1nM.
- Radioligand binding studies utilising [ 125 l]-lodocyanopindolol and commercially available cell membranes expressing the human ⁇ 2 adrenergic receptor were used to assess the affinity of antagonists for ⁇ 2 -adrenergic receptor.
- Membranes and SPA- beads were incubated with [ 125 l]-lodocyanopindolol and ⁇ 2 antagonist at various concentrations for 3 hours at room temperature in TRIS buffer. The assay was performed in 96-well plates which were read using the Wallac Microbeta counter.
- Example 5 exhibited a binding Ki of 43nM
- Example 8 a binding Ki of 69nM
- Example 2 had a binding Ki of 99nM.
- CHO cells expressing the human M3 receptor were seeded and incubated overnight in 96 well collagen coated plates (black-wall, clear bottom) at a density of 50000 / 75 ⁇ l of medium in 3% serum.
- a calcium-sensitive dye (Molecular Devices, Cat # R8041) was prepared in HBSS buffer with the addition of 5mM probenecid (pH 7.4).
- An equal volume of the dye solution (75 ⁇ l) was added to the cells and incubated for 45 minutes followed by addition of 50 ⁇ l of muscarinic antagonists or vehicle. After a further 15 minutes the plate was read on a FLEXstationTM (excitation 488nm, emission 525nm) for 15 seconds to determine baseline fluorescence.
- the muscarinic agonist Carbachol was then added at an EC 80 concentration and the fluorescence measured for a further 60 seconds.
- the signal was calculated by subtracting the peak response from the mean of the baseline fluorescence in control wells in the absence of antagonist. The percentage of the maximum response in the presence of antagonist was then calculated in order to generate IC 50 curves.
- Some compounds of the invention were tested in this assay and were found to have IC50 values of ⁇ 500nM.
- Example 2 had an 1C50 value of of 209nM, whilst Example 7 had an IC50 value of 35nM.
- Tracheae were removed from adult male Dunkin Hartley Guinea pigs and dissected free of adherent tissue before being cut open longitudinally in a line opposite the muscle. Individual strips of 2-3 cartilage rings in width were cut and suspended using cotton thread in 10ml water-jacketed organ baths and attached to a force transducer ensuring that the tissue is located between two platinum electrodes. Responses were recorded via a MPI OOW/Ackowledge data acquisition system connected to a PC. Tissues were equilibrated for one hour under a resting tone of 1g and were then subjected to electrical field stimulation at a frequency of 80Hz with a pulse width of 0.1 ms, a unipolar pulse, triggered every 2 minutes.
- a "voltage-response" curve was generated for each tissue and a submaximal voltage then applied to every piece of tissue according to its own response to voltage. Tissues were washed with Krebs solution and allowed to stabilize under stimulation prior to addition of test compound. Concentration response curves were obtained by a cumulative addition of test compound in half-log increments. Once the response to each addition had reached a plateau the next addition was made. Percentage inhibition of EFS-stimulated contraction is calculated for each concentration of each compound added and dose response curves constructed using Graphpad Prism software and the EC 50 calculated for each compound. Onset time and duration of action studies were performed by adding the previously determined EC 50 concentration of compound to EFS contracted tissues and the response allowed to plateau.
- Example 2 had an EC50 value of 153nM.
- Control 9 ⁇ M Isoproterenol (max stimulation obtained, lowest counts) Blank: 0.1 nM Isoproterenol 1 % DMSO (no stimulation, highest counts)
- Detection positive control 10 ⁇ M cAMP (1/200 dilution of 1OmM stock, then 1/5 in assay)
- Detection negative control stimulation buffer Standard Isoproterenol and Formoterol (EC50 ⁇ 7nM, 0.05nM respectively)
- Example 2 had an EC50 value of 37nM.
- mice Male Guinea pigs (Dunkin Hartley), weighing 500-60Og housed in groups of 5 were individually identified. Animals were allowed to acclimatize to their local surroundings for at least 5 days. Throughout this time and study time animals were allowed access to water and food ad libitum.
- Guinea pigs were anaesthetized with the inhaled anaesthetic Halothane (5%).
- Test compound or vehicle (0.25 - 0.50 ml/kg) was administered intranasally. Animals were placed on a heated pad and allowed to recover before being returned to their home cages.
- guinea pigs were terminally anaesthetized with Urethane (250 ⁇ g/ml, 2ml/kg).
- Urethane 250 ⁇ g/ml, 2ml/kg.
- the jugular vein was cannulated with a portex i.v. cannula filled with heparinised phosphate buffered saline (hPBS) (10U/ml) for i.v. administration of methacholine.
- hPBS heparinised phosphate buffered saline
- the trachea was exposed and cannulated with a rigid portex cannula and the oesophagus cannulated transorally with a flexible portex infant feeding tube.
- the spontaneously breathing animal was then connected to a pulmonary measurement system (EMMS, Hants, UK) consisting of a flow pneumotach and a pressure transducer.
- EMMS pulmonary measurement system
- Hants UK
- the tracheal cannula was attached to a pneumotach and the oesophageal cannula attached to a pressure transducer.
- the oesophageal cannula was positioned to give a baseline resistance of between 0.1 and 0.2cmH20/ml/s.
- a 2 minute baseline reading was recorded before i.v. administration of methacholine (up to 30 ⁇ g/kg, 0.5ml/kg).
- a 2 minute recording of the induced constriction was taken from the point of i.v. administration.
- the software calculated a peak resistance and a resistance area under the curve (AUC) during each 2 minute recording period which were used to analyse the bronchoprotective effects of test compounds
- Guinea pigs (450-55Og) supplied by Harlan UK or David Hall, Staffs UK and acclimatised to the in-house facilities for a minimum of three days before use. Guinea pigs were randomly assigned into treatment groups and weighed. Each animal was lightly anaesthetised (4% Halothane) and administered compound or vehicle intranasally (0.5ml/kg) at up to 24 hours before challenge with pilocarpine. At the test time point, guinea pigs were terminally anaesthetised with urethane (25% solution in H20, 1.5g/kg).
- Saliva production was calculated by subtracting the pre-weighed weight of the pad from each 5 minute period post weighed pad and these numbers added together to produce an accumulation of saliva over 15 minutes. Each 5 minute period could be analysed in addition to the whole 15 minute recording period. Baseline production of saliva was assumed to be constant and multiplied by three to produce a reading for baseline saliva production over 15 minutes.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
The present invention provides compounds of formula (Ia): wherein A, X, and R<SUP>1</SUP>-R<SUP>8</SUP> are as defined herein, such compounds having utility in the treatment of diseases where M3 and beta2 receptors are implicated, such as respiritory tract diseases; compositions comprising such compounds; uses of such compounds in therapy (such as asthma or COPD); and methods of treating a patient with such compounds.
Description
Chemical Compounds
Field of the Invention
This invention relates to oxazole and thiazole derivatives, pharmaceutical compositions, methods for their preparation and use in the treatment of diseases where enhanced M3 receptor activation is implicated.
Background to the invention
Anti-cholinergic agents prevent the passage of, or effects resulting from the passage of, impulses through the parasympathetic nerves. This is a consequence of the ability of such compounds to inhibit the action of acetylcholine (Ach) by blocking its binding to the muscarinic cholinergic receptors.
There are five subtypes of muscarinic acetylcholine receptors (mAChRs), termed M1 -M5, and each is the product of a distinct gene and each displays unique pharmacological properties. mAChRs are widely distributed in vertebrate organs, and these receptors can mediate both inhibitory and excitatory actions. For example, in smooth muscle found in the airways, bladder and gastrointestinal tract, M3 mAChRs mediate contractile responses (reviewed by Caulfield, 1993, Pharmac. Ther., 58, 319 - 379).
In the lungs, muscarinic receptors M1 , M2 and M3 have been demonstrated to be important and are localized to the trachea, the bronchi, submucosal glands and parasympathetic ganglia (reviewed in Fryer and Jacoby, 1998, Am J Resp Crit Care Med., 158 (5 part 3) S 154 - 160). M3 receptors on airway smooth muscle mediate contraction and therefore bronchoconstriction. Stimulation of M3 receptors localised to submucosal glands results in mucus secretion.
Increased signalling through muscarinic acetylcholine receptors has been noted in a variety of different pathophysiological states including asthma and COPD. In COPD, vagal tone may either be increased (Gross et al. 1989, Chest; 96:984-987) and/or may provoke a higher degree of obstruction for geometric reasons if applied on top of oedematous or mucus-laden airway walls (Gross et al. 1984, Am Rev Respir Dis; 129:856-870). In addition, inflammatory conditions can lead to a loss of inhibitory
M2 receptor activity which results in increased levels of acetylcholine release following vagal nerve stimulation (Fryer et al, 1999, Life Sci., 64, (6-7) 449-455). The resultant increased activation of M3 receptors leads to enhanced airway obstruction. Thus the identification of potent muscarinic receptor antagonists would be useful for the therapeutic treatment of those disease states where enhanced M3 receptor activity is implicated. Indeed, contemporary treatment strategies currently support regular use of M3 antagonist bronchodilators as first-line therapy for COPD patients (Pauwels et al. 2001 , Am Rev Respir Crit Care Med; 163:1256-1276)
Incontinence due to bladder hypercontractility has also been demonstrated to be mediated through increased stimulation of M3 mAChRs. Thus M3 mAChR antagonists may be useful as therapeutics in these mAChR-mediated diseases.
Despite the large body of evidence supporting the use of anti-muscarinic receptor therapy for treatment of airway disease states, relatively few anti-muscarinic compounds are in use in the clinic for pulmonary indications. Thus, there remains a need for novel compounds that are capable of causing blockade at M3 muscarinic receptors, especially those compounds with a long duration of action, enabling a once-daily dosing regimen. Since muscarinic receptors are widely distributed throughout the body, the ability to deliver anticholinergic drugs directly to the respiratory tract is advantageous as it allows lower doses of the drug to be administered. The design and use of topically active drugs with a long duration of action and that are retained on the receptor or in the lung would allow reduction of unwanted side effects that could be seen with systemic administration of the same drugs.
Tiotropium (Spiriva ™) is a long-acting muscarinic antagonist currently marketed for the treatment of chronic obstructive pulmonary disease, administered by the inhaled route.

Additionally ipratropium is a muscarinic antagonist marketed for the treatment of COPD.

Ipratropium
Chem. Pharm. Bull. 27 (12) 3149-3152 (1979) and J. Pharm. Sci 69 (5) 534-537
(1980) describe furyl derivatives as possessing atropine-like activities.
Med. Chem. Res 10 (9), 615-633 (2001) describes isoxazoles and Δ2-isoxazolines as muscarinic antagonists.
WO97/30994 describes oxadiazoles and thiadiazoles as muscarinic receptor antagonists.
EP0323864 describes oxadiazoles linked to a mono- or bicyclic ring as muscarinic receptor modulators.
The class of β2 adrenergic receptor agonists is well known. Many known β2-agonists, in particular, long-acting β2-agonists such as salmeterol and formoterol, have a role in the treatment of asthma and COPD. These compounds are also generally administered by inhalation. Compounds currently under evaluation as once-daily β2 agonists are described in Expert Opin. Investig. Drugs 14 (7), 775-783 (2005). A well known β2-agonist pharmacophore is the moiety:

Also known in the art are pharmaceutical compositions that contain both a muscarinic antagonist and a β2-agonist for use in the treatment of respiratory disorders. For example, US2005/0025718 describes a β2-agonist in combination with tiotropium, oxotropium, ipratropium and other muscarinic antagonists; WO02/060532 describes the combination of ipratropium with β2-agonists and WO02/060533 describes the combination of oxotropium with β2-agonists. Other M3 antagonist / β2-agonist combinations are described in WO04/105759 and WO03/087097.
Also known in the art are compounds possessing both muscarinic receptor antagonist and β2-agonist activity present in the same molecule. Such bifunctional molecules provide bronchodilation through two separate modes of action whilst possessing single molecule pharmacokinetics. Such a molecule might be easier to formulate for therapeutic use as compared to two separate compounds and might be more easily co-formulated with a third active ingredient, for example a steroid. Such molecules are described in for example, WO04/074246, WO04/089892, WO05/111004, WO06/023457 and WO06/023460, all of which use different linker radicals for covalently linking the M3 antagonist to the β2-agonist.
Summary of the Invention
According to the present invention we provide the use of a compound of formula (I):

wherein
(i) R1 is CrC6-alkyl or hydrogen; and R2 is hydrogen or a group -R7, -Z-Y-R7,
-Z-NR9R10; -Z-CO-NR9R10, -Z-NR9-C(O)O-R7, or ; -Z-C(O)-R7; and R3 is a lone pair, or R3 is CrC6-alkyl in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge; or
(ii) R1 and R3 together with the nitrogen to which they are attached form a heterocycloalkyl ring, and R2 is a lone pair or R2 a group -R7, -Z-Y-R7, -Z-NR9R10, -Z-CO-NR9R10, -Z-NR9-C(O)O-R7; or -Z-C(O)-R7, in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge; or
(iii) R1 and R2 together with the nitrogen to which they are attached form a heterocycloalkyl ring, said ring being substituted by a group -Y-R7, -Z-Y-R7, -Z- NR9R10; -Z-CO-NR9R10; -Z-NR9-C(O)O-R7; or ; -Z-C(O)-R7; and R3 is a lone pair, or R3 is Ci-C6-alkyl in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge;
R4 and R5 are independently selected from the group consisting of aryi, aryl-fused- heterocycloalkyl, heteroaryl, CrC6-alkyl, cycloalkyl;
R6 is -OH, CrC6-alkyl, CrC6-alkoxy hydroxy-CrC6-alkyl, nitrile, a group CONR8 2 or a hydrogen atom;
A is an oxygen or a sulfur atom;
X is a CrC12-alkylene, C2-C12-alkenylene or C3-C12-alkynylene group;
R7 is an CrC6-alkyl, aryl, aryl-fused-cycloalkyl, aryl-fused-heterocycloalkyl, heteroaryl, aryl(CrC8-alkyl)-, heteroary^CrCs-alkyl)-, cycloalkyl or heterocycloalkyl group;
R8 is CrC6-alkyl or a hydrogen atom;
Z is a CrC^-aikylene, C2-C16-alkenylene or C2-Ci 6-alkynylene group;
Y is a bond or oxygen atom;
R9 and R10 are independently a hydrogen atom, CrC6-alkyl, aryl, aryl-fused- heterocycloalkyl, aryl-fused-cycloalkyl, heteroaryl, aryl(CrC6-alkyl)-, or heteroaryl(Cr C6-alkyl)- group; or R9 and R10 together with the nitrogen atom to which they are attached form a heterocyclic ring of 4-8 atoms, optionally containing a further nitrogen or oxygen atom;
wherein, unless otherwise specified, each occurrence of alkyl, heterocycloalkyl, aryl, aryl-fused-heterocycloalkyl, heteroaryl, cycloalkyl, alkoxy, alkylene, alkenylene, alkynylene or aryl-fused-cycloalkyl may be optionally substituted;
or a pharmaceutically acceptable salt, solvate, N-oxide or prodrug thereof;
in the manufacture of a medicament for the treatment of prevention of a disease or condition in which M3 muscarinic receptor activity and β-adrenergic receptor activity is implicated.
Conveniently, R1 is CrC6-alkyI; R2 is a group -Z-NR9R10; and R3 is a lone pair or R3 is CrC6-alkyl in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge.
Convenient R groups of the present invention include:


More conveniently, R ,10 groups of the present invention include:

Conveniently, X is an optionally substituted C1-C3 alkylene group. More conveniently, X is a C1-C2 alkylene group. Most conveniently, X is methylene.
Conveniently, unless otherwise specified, each alkyl, heterocycloalkyl, aryl, aryl- fused-heterocycloalkyl, heteroaryl, cycloalkyl, alkoxy, alkylene, alkenylene, alkynylene or aryl-fused-cycloalkyl group of the compounds of fomula (I) is unsubstituted.
Conveniently, each alkenylene chain may contain 1 , 2 or 3 carbon-carbon double bonds and each alkynylene chain may contain up to 1 , 2 or 3 carbon-carbon triple bonds.
According to a futher aspect of the invention, there is provided a compound of formula (Ia):

wherein
(i) R1 is CrC6-alkyl or hydrogen; and R2 is a group-Z-NR9R10; and R3 is a lone pair, or R3 is CrC6-a!kyl in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge; or
(ii) R1 and R3 together with the nitrogen to which they are attached form a heterocycloalkyl ring, and R2 is a group -Z-NR9R10, in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge, or
(iii) R1 and R2 together with the nitrogen to which they are attached form a heterocycloalkyl ring, said ring being substituted by a group , -Z-NR9R10; and R3 is a lone pair, or R3 is CrC6-alkyl in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge;
R4 and R5 are independently selected from the group consisting of aryl, aryl-fused- heterocycloalkyl, heteroaryl, CrC6-alkyl, cycloalkyl;
R6 is -OH, CrC6-alkyl, CrC6-alkoxy hydroxy-CrC6-alkyl, nitrite, a group CONR8 2 or a hydrogen atom;
A is an oxygen or a sulfur atom;
X is a CrCi2-alkylene, C2-C12-alkenylene or C3~C12-alkynylene group;
R8 is CrCg-alkyl or a hydrogen atom;
Z is a C7-C1 i-alkylene, C7-C1 ralkenylene or C7-C1 ralkynylene group;
or Z is a divalent linker radical of formula (A):

wherein L represents a linker comprising a hydrocarbyl chain of 7 to 11 carbon atoms, wherein the chain may additionally comprise up to three carbon-carbon double bonds, and, wherein the chain may additionally comprise up to three carbon-carbon triple bonds;
L1 and L2 each independently represent hydrogen, Ci-6 alkyl or C3-6 cycloalkyl;
L3 and L4 each independently represent hydrogen, C1^ alkyl or C3.6 cycloalkyl, wherein C1-6 alkyl and C3-6 cycloalkyl may be optionally substituted by one or more substituents independently selected from halogen and hydroxyl;
and * denotes the point of attachement of the group of formula (I) to the non- aromatic nitrogen bearing R1 and R3, and ** denotes the point of attachment to the group NR9R10;
R9 is a hydrogen atom or CVC6-alkyl;
R10 is an aryl(CrC6-alkyl)-, or heteroaryl(CrC6-alkyl) group, in which the CrC6-a!kyl group is optionally substituted by hydroxy;
wherein, unless otherwise specified, each occurrence of alkyl, heterocycloalkyl, aryl, aryl-fused-heterocycloalkyl, heteroaryl, cycloalkyl, alkoxy, alkylene, alkenylene and alkynylene may be optionally substituted;
or a pharmaceutically acceptable salt thereof.
In another aspect the present invention provides a prodrug of a compound of formula (Ia) as herein defined, or a pharmaceutically acceptable salt thereof.
In yet another aspect the present invention provides an N-oxide of a compound of formula (Ia) as herein defined, or a prodrug or pharmaceutically acceptable salt thereof.
In a further aspect the present invention provides a solvate (such as a hydrate) of a compound of formula (Ia) as herein defined, or an N-oxide, prodrug or pharmaceutically acceptable salt thereof.
In one subset of the compounds of the invention:
R1 is Ci-Cβ-alkyl ; R2 is a group -Z-NR9R10 and R3 is a lone pair or R3 is CrC6-alkyl, in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge, or
R1 and R2 together with the nitrogen to which they are attached represent a heterocycloalkyl ring, said ring being substituted by a group -Z-NR9R10 and R3 is a lone pair or R3 is CrC6-alkyl in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge;
or R1 and R3 together with the nitrogen to which they are attached represent a heterocycloalkyl ring, and R2 is a group -Z-NR9R10 in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge;
R4 is phenyl and R5 is selected from the group consisting of aryl, heteroaryl, C1-C6- alkyl, cycloalkyl; or R4 and R5 are both heteroaryl;
R6 is -OH or, CrC6-alkyl;
R8 is a hydrogen atom;
A is an oxygen or a sulfur atom;
X is a CrC2-alkylene group;
Z is a divalent linker radical of formula (A):

wherein L represents a linker comprising a hydrocarbyl chain of 7 to 11 carbon atoms, wherein the chain may additionally comprise up to three carbon-carbon double bonds, and, wherein the chain may additionally comprise up to three carbon-carbon triple bonds;
L1 and L2 each independently represent hydrogen, C1-6 alkyl or C3.6 cycloalkyl;
L3 and L4 each independently represent hydrogen, Ci.6 alkyl or C3.6 cycloalkyl, wherein C1-6 alkyl and C3.6 cycloalkyl may be optionally substituted by one or more substituents independently selected from halogen and hydroxyl; and * denotes the point of attachement of the group of formula (I) to the non- aromatic nitrogen bearing R1 and R3, and ** denotes the point of attachment to the group NR9R10;
R9 is a hydrogen atom; and
R10 is selected from the group


In one aspect, the present invention provides a compound of formula (Ia), or a pharmaceutically acceptable salt thereof, wherein R1 is Ci-C6-alkyl; R2 is a group -Z- NR9R10; and R3 is a lone pair or R3 is a CrC6-alkyl in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge.
It will be appreciated that the carbon atom to which R4, R5 and R6 are attached can be an asymmetric centre so compounds of the invention may be in the form of single enantiomers or mixtures of enantiomers.
In one aspect, the present invention provides compounds of formula (Ia) wherein the carbon to which R4, R5 and R6 are attached has the (R)- absolute configuration.
In one aspect, the present invention provides compounds of formula (Ia) wherein the non-aromatic nitrogen shown in formula (Ia) is a tertiary nitrogen.
In another aspect, the present invention provides quaternary ammonium salts of formula (Ia) wherein the non-aromatic nitrogen shown in formula (Ia) is quaternary nitrogen, carrying a positive charge.
In one aspect, the present invention provides a compound of formula (Ia), or a pharmaceutically acceptable salt thereof, wherein R4 and R5 are independently
selected from methyl, ethyl, n- or isopropyl, n-, sec- and tertbutyl; phenyl, 3,4- methylenedioxyphenyl, 3,4-ethylenedioxyphenyl, dihydrobenzofuranyl, naphthyl; pyridyl, pyrrolyl, pyrimidinyl, oxazolyl, isoxazolyl, benzisoxazolyl, benzoxazolyl, thiazolyl, benzthiazolyl, quinolyl, thienyl, benzthienyl, furyl, benzfuryl, imidazolyl, benzimidazolyl, isothiazolyl, benzisothiazolyl, pyrazolyl, isothiazolyl, triazolyl, benztriazolyl, thiadiazolyl, oxadiazolyl, pyridazinyl, pyridazinyl, triazinyl, indolyl or indazolyl; indanyl and 1 ,2,3,4-tetrahydronaphthalenyl; cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl; and R6 is -OH, a hydrogen atom, methyl, ethyl, hydroxymethyl, nitrile, or a group CONR8 2 wherein each R8 is independently methyl, ethyl, or a hydrogen atom.
In another aspect, the present invention provides a compound of formula (Ia), or a pharmaceutically acceptable salt thereof, wherein (i) each of R4 and R5 is thienyl; or (ii) each of R4 and R5 is phenyl; or (iii) one of R4 and R5 is phenyl and the other is cyclopentyl or cyclohexyl; or (iv) one of R4 and R5 is thienyl, and the other is cyclopentyl or cyclohexyl.
In one aspect, the present invention provides a compound of formula (Ia), or a pharmaceutically acceptable salt thereof, wherein R6 is -OH.
In one aspect, the present invention provides a compound of formula (Ia), or a pharmaceutically acceptable salt thereof, wherein R8 is hydrogen.
In one aspect, the present invention provides a compound of formula (Ia), or a pharmaceutically acceptable salt thereof, wherein R10 is selected from:

In one aspect, the present invention provides a compound of formula (Ia), or a pharmaceutically acceptable salt thereof, wherein A is an oxygen atom.
In one aspect, the present invention provides a compound of formula (Ia), or a
pharmaceutically acceptable salt thereof, wherein X is -CH2- or -CH2CH2-.
Compounds of the invention may be useful in the treatment or prevention of diseases in which activation of muscarinic receptors are implicated, for example the present compounds are useful for treating a variety of indications, including but not limited to respiratory-tract disorders such as chronic obstructive lung disease, chronic bronchitis of all types (including dyspnoea associated therewith), asthma (allergic and non-allergic; 'wheezy-infant syndrome'), adult/acute respiratory distress syndrome (ARDS), chronic respiratory obstruction, bronchial hyperactivity, pulmonary fibrosis, pulmonary emphysema, and allergic rhinitis, exacerbation of airway hyperreactivity consequent to other drug therapy, particularly other inhaled drug therapy, pneumoconiosis (for example aluminosis, anthracosis, asbestosis, chalicosis, ptilosis, siderosis, silicosis, tabacosis and byssinosis); gastrointestinal-tract disorders such as irritable bowel syndrome, spasmodic colitis, gastroduodenal ulcers, gastrointestinal convulsions or hyperanakinesia, diverticulitis, pain accompanying spasms of gastrointestinal smooth musculature; urinary-tract disorders accompanying micturition disorders including neurogenic pollakisuria, neurogenic bladder, nocturnal enuresis, psychosomatic bladder, incontinence associated with bladder spasms or chronic cystitis, urinary urgency or pollakiuria; motion sickness; and cardiovascular disorders such as vagally induced sinus bradycardia.
For treatment of respiratory conditions, administration by inhalation will often be preferred, and in such cases administration of compounds (Ia) which are quaternary ammonium salts will often be preferred. In many cases, the duration of action of quaternary ammonium salts of the invention administered by inhalation is may be more than 12, or more than 24 hours for a typical dose. For treatment of gastrointestinal-tract disorders and cardiovascular disorders, administration by the parenteral route, usually the oral route, may be preferred.
Another aspect of the invention is a pharmaceutical composition comprising a compound of the invention as shown in formula (Ia) and a pharmaceutically acceptable carrier or excipient.
Another aspect of the invention is the use of a compound of the invention for the manufacture of a medicament for the treatment or prevention of a disease or condition in which muscarinic M3 receptor activity is implicated.
Description of Definitions
Unless otherwise qualified in the context in which they are used, the following terms have the following meanings when used herein:
"Acyl" means a -CO-alkyl group in which the alkyl group is as described herein. Exemplary acyl groups include -COCH3 and -COCH(CH3)2. "Acylamino" means a -NR-acyl group in which R and acyl are as described herein. Exemplary acylamino groups include -NHCOCH3 and -N(CH3)COCH3.
"Alkoxy" and "alkyloxy" means an -O-alkyl group in which alkyl is as described below. Exemplary alkoxy groups include methoxy (-OCH3) and ethoxy (-OC2H5). "Alkoxycarbonyl" means a -COO-alkyl group in which alkyl is as defined below. Exemplary alkoxycarbonyl groups include methoxycarbonyl and ethoxycarbonyl.
"Alkyl" as a group or part of a group refers to a straight or branched chain saturated hydrocarbon group having from 1 to 12, preferably 1 to 6, carbon atoms, in the chain. Exemplary alkyl groups include methyl, ethyl, 1 -propyl and 2-propyl. "Alkenyl" as a group or part of a group refers to a straight or branched chain hydrocarbon group having from 2 to 12, preferably 2 to 6, carbon atoms and one carbon-carbon double bond in the chain. Exemplary alkenyl groups include ethenyl, 1-propenyl, and 2-propenyl.
"Alkylamino" means a -NH-alkyl group in which alkyl is as defined above. Exemplary alkylamino groups include methylamino and ethylamino.
"Alkylene" means an -alkyl- group in which alkyl is as defined previously. Exemplary alkylene groups include -CH2-, -(CH2)2- and -C(CH3)HCH2-.
"Alkenylene" means an -alkenyl- group in which alkenyl is as defined previously. Exemplary alkenylene groups include -CH=CH-, -CH=CHCH2-, and - CH2CH=CH-.
"Alkynylene" means an -alkynyl- group in which -alkynyl- refers to a straight or branched chain hydrocarbon group having from 2 to 12, preferably 2 to 6, carbon atoms and one carbon-carbon triple bond in the chain. Exemplary alkynylene groups include ethynyl and propargyl. "Alkylsulfinyl" means a -SO-alkyl group in which alkyl is as defined above.
Exemplary alkylsulfinyl groups include methylsulfinyl and ethylsulfinyl.
"Alkylsulfonyl" means a -SO2-alkyl group in which alkyl is as defined above. Exemplary alkylsulfonyl groups include methylsulfonyl and ethylsulfonyl.
"Alkylthio" means a -S-alkyl group in which alkyl is as defined above. Exemplary alkylthio groups include methylthio and ethylthio. "Aminoacyl" means a -CO-NRR group in which R is as herein described.
Exemplary aminoacyl groups include -CONH2 and -CONHCH3.
"Aminoalkyl" means an alkyl-NH2 group in which alkyl is as previously described. Exemplary aminoalkyl groups include -CH2NH2.
"Aminosulfonyl" means a -SO2-NRR group in which R is as herein described. Exemplary aminosulfonyl groups include -SO2NH2 and -SO2NHCH3.
"Aryl" as a group or part of a group denotes an optionally substituted monocyclic or multicyclic aromatic carbocyclic moiety of from 6 to 14 carbon atoms, preferably from 6 to 10 carbon atoms, such as phenyl or naphthyl. The aryl group may be substituted by one or more substituent groups. "Arylalkyl" means an aryl-alkyl- group in which the aryl and alkyl moieties are as previously described. Preferred arylalkyl groups contain a C1 4 alkyl moiety.
Exemplary arylalkyl groups include benzyl, phenethyl and naphthlenemethyl.
"Arylalkyloxy" means an aryl-alkyloxy- group in which the aryl and alkyloxy moieties are as previously described. Preferred arylalkyloxy groups contain a C1 4 alkyl moiety. Exemplary arylalkyl groups include benzyloxy.
"Aryl-fused-heterocycloalkyl" means a monocyclic aryl ring, such as phenyl, fused to a heterocycloalkyl group, in which the aryl and heterocycloalkyl are as described herein. Exemplary aryl-fused-heterocycloalkyl groups include tetrahydroquinolinyl, indolinyl, benzodioxinyl, benxodioxolyl, dihydrobenzofuranyl and isoindolonyl. The aryl and heterocycloalkyl rings may each be substituted by one or more substituent groups. The aryl-fused-heterocycloalkyl group may be attached to the remainder of the compound by any available carbon or nitrogen atom.
"Aryloxy" means an -O-aryl group in which aryl is described above. Exemplary aryloxy groups include phenoxy. "Cyclic amine" means an optionally substituted 3 to 8 membered monocyclic cycloalkyl ring system where one of the ring carbon atoms is replaced by nitrogen, and which may optionally contain an additional heteroatom selected from O, S or NR (where R is as described herein). Exemplary cyclic amines include pyrrolidine, piperidine, morpholine, piperazine and Λ/-methylpiperazine. The cyclic amine group may be substituted by one or more substituent groups.
"Cycloalkyl" means an optionally substituted saturated monocyclic or bicyclic ring system of from 3 to 12 carbon atoms, preferably from 3 to 8 carbon atoms, and more preferably from 3 to 6 carbon atoms. Exemplary monocyclic cycloalkyl rings include cyclopropyl, cyclopentyl, cyclohexyl and cycloheptyl. The cycloalkyl group may be substituted by one or more substituent groups.
"Dialkylamino" means a -N(alkyl)2 group in which alkyl is as defined above.
Exemplary dialkylamino groups include dimethylamino and diethylamino.
"Halo" or "halogen" means fluoro, chloro, bromo, or iodo. Preferred are fluoro or chloro. "Haloalkoxy" means an -O-alkyl group in which the alkyl is substituted by one or more halogen atoms. Exemplary haloalkyl groups include trifluoromethoxy and difluoromethoxy.
"Haloalkyl" means an alkyl group which is substituted by one or more halo atoms. Exemplary haloalkyl groups include trifiuoromethyl. "Heteroaryl" as a group or part of a group denotes an optionally substituted aromatic monocyclic or multicyclic organic moiety of from 5 to 14 ring atoms, preferably from 5 to 10 ring atoms, in which one or more of the ring atoms is/are element(s) other than carbon, for example nitrogen, oxygen or sulfur. Examples of such groups include benzimidazolyl, benzoxazolyl, benzothiazolyl, benzofuranyl, benzothienyl, furyl, imidazolyl, indolyl, indolizinyl, isoxazolyl, isoquinolinyl, isothiazolyl, oxazolyl, oxadiazolyl, pyrazinyl, pyridazinyl, pyrazolyl, pyridyl, pyrimidinyl, pyrrolyl, quinazolinyl, quinolinyi, tetrazolyl, 1 ,3,4-thiadiazolyl, thiazolyl, thienyl and triazolyl groups. The heteroaryl group may be substituted by one or more substituent groups. The heteroaryl group may be attached to the remainder of the compound of the invention by any available carbon or nitrogen atom.
Ηeteroarylalkyl" means a heteroaryl-alkyl- group in which the heteroaryl and alkyl moieties are as previously described. Preferred heteroarylalkyl groups contain a lower alkyl moiety. Exemplary heteroarylalkyl groups include pyridylmethyl. "Heteroarylalkyloxy" means a heteroaryl-alkyloxy- group in which the heteroaryl and alkyloxy moieties are as previously described. Preferred heteroarylalkyloxy groups contain a lower alkyl moiety. Exemplary heteroarylalkyloxy groups include pyridylmethyloxy.
"Heteroaryloxy" means a heteroaryloxy- group in which the heteroaryl is as previously described. Exemplary heteroaryloxy groups include pyridyloxy.
"Heterocycloalkyl" means: (i) an optionally substituted cycloalkyl group of from 4 to 8 ring members which contains one or more heteroatoms selected from O, S or NR; (ii) a cycloalkyl group of from 4 to 8 ring members which contains CONR and CONRCO (examples of such groups include succinimidyl and 2-oxopyrrolidinyl). The heterocycloalkyl group may be substituted by one or more substituent groups. The heterocycloalkyl group may be attached to the remainder of the compound by any available carbon or nitrogen atom.
"Lower alkyl" as a group means unless otherwise specified, an aliphatic hydrocarbon group which may be straight or branched having 1 to 4 carbon atoms in the chain, i.e. methyl, ethyl, propyl (propyl or /so-propyl) or butyl (butyl, /so-butyl or tert- butyl).
"Sulfonyl" means a -SO2-alkyl group in which alkyl is as described herein. Exemplary sulfonyl groups include methanesulfonyl.
"Sulfonylamino" means a -NR-sulfonyl group in which R and sulfonyl are as described herein. Exemplary sulfonylamino groups include -NHSO2CH3. R means alkyl, aryl, or heteroaryl as described herein.
"Pharmaceutically acceptable salt" means a physiologically or toxicologically tolerable salt and includes, when appropriate, pharmaceutically acceptable base addition salts, pharmaceutically acceptable acid addition salts, and pharmaceutically acceptable quaternary ammonium salts. For example (i) where a compound of the invention contains one or more acidic groups, for example carboxy groups, pharmaceutically acceptable base addition salts that may be formed include sodium, potassium, calcium, magnesium and ammonium salts, or salts with organic amines, such as, diethylamine, Λ/-methyl-glucamine, diethanolamine or amino acids (e.g. lysine) and the like; (ii) where a compound of the invention contains a basic group, such as an amino group, pharmaceutically acceptable acid addition salts that may be formed include hydrochlorides, hydrobromides, sulfates, phosphates, acetates, citrates, lactates, tartrates, mesylates, napadisylates (naphthalene- 1 ,5-disulfonates or naphthalene-1 -(sulfonic acid)-5-sulfonates), edisylates (ethane-1 ,2-disulfonates or ethane-1 -(sulfonic acid)-2-sulfonates), maleates, fumarates, succinates and the like; (iii) where a compound contains a quaternary ammonium group acceptable counter- ions may be, for example, chlorides, bromides, sulfates, methanesulfonates, benzenesulfonates, toluenesulfonates (tosylates), napadisylates (naphthalene-1 ,5- disulfonates or naphthalene-1 -(sulfonic acid)-5-sulfonates), edisylates (ethane-1 ,2- disulfonates or ethane-1 -(sulfonic acid)-2-sulfonates), isethionates (2- hydroxyethylsulfonates), phosphates, acetates, citrates, lactates, tartrates,
mesylates, maleates, fumarates, xinafoates, p-acetamidobenzoates, succinates and the like; wherein the number of quaternary ammonium species balances the pharmaceutically acceptable counter-ion D- such that compound of formula (I) has no net charge.
It will be understood that, as used herein, references to the compounds of the invention are meant to also include the pharmaceutically acceptable salts.
"Prodrug" refers to a compound which is convertible in vivo by metabolic means (e.g. by hydrolysis, reduction or oxidation) to a compound of the invention. Suitable groups for forming pro-drugs are described in The Practice of Medicinal Chemistry, 2nd Ed. pp561-585 (2003) and in F. J. Leinweber, Drug Metab. Res., , 18, 379. (1987)
It will be understood that, as used in herein, references to the compounds of the invention are meant to also include the prodrug forms. "Saturated" pertains to compounds and/or groups which do not have any carbon-carbon double bonds or carbon-carbon triple bonds.
The cyclic groups referred to above, namely, aryl, heteroaryl, cycloalkyl, heterocycloalkyl, aryl-fused-heterocycloalkyl and cyclic amine may be substituted by one or more substituent groups. Suitable optional substituent groups include acyl (e.g. -COCHJ, alkoxy (e.g., -OCHJ, alkoxycarbonyl (e.g. -COOCHJ, alkylamino (e.g.
-NHCH3), alkylsulfinyl (e.g. -SOCH3), alkylsulfonyl (e.g. -SO2CH3), alkylthio (e.g. - SCH3), -NH2, aminoacyl (e.g. -CON(CHs)2), aminoalkyl (e.g. -CH2NH2), arylalkyl (e.g. -CH Ph or -CH -CH -Ph), cyano, dialkylamino (e.g. -N(CHJJ, halo, haloalkoxy (e.g. -OCF3 or -OCHF2), haloalkyl (e.g. -CF3), alkyl (e.g. -CH3Or -CH2CH3), -OH, -CHO, - NO2, aryl (optionally substituted with alkoxy, haloalkoxy, halogen, alkyl or haloalkyl), heteroaryl (optionally substituted with alkoxy, haloalkoxy, halogen, alkyl or haloalkyl), heterocycloalkyl, aminoacyl (e.g. -CONH2, -CONHCH3), aminosulfonyl (e.g. -SO2NH2, -SO2NHCH3), acylamino (e.g. -NHCOCH3), sulfonylamino (e.g. -NHSO2CH3), heteroarylalkyl, cyclic amine (e.g. morpholine), aryloxy, heteroaryloxy, arylalkyloxy (e.g. benzyloxy) and heteroarylalkyloxy.
Alkyl, alkoxy, alkylene or alkenylene groups may be optionally substituted. Suitable optional substituent groups include alkoxy (e.g., -OCH3), alkylamino (e.g. -NHCH3), alkylsulfinyl (e.g. -SOCHJ, alkylsulfonyl (e.g. -SO0CHJ, alkylthio (e.g. -SCHJ, -NH2, aminoalkyl (e.g. -CH2NH2), arylalkyl (e.g. -CH2Ph or -CH2-CH3-Ph), cyano,
dialkylamino (e.g. -N(CH3)2), halo, haloalkoxy (e.g. -OCF3 or -OCHF2), haloalkyl (e.g. -CFJ, alkyl (e.g. -CH or -CH0CH ), -OH, -CHO, and -NO2.
Compounds of the invention may exist in one or more geometrical, optical, enantiomeric, diastereomeric and tautomeric forms, including but not limited to cis- and frans-forms, £- and Z-forms, R-, S- and meso-forms, keto-, and enol-forms. Unless otherwise stated a reference to a particular compound includes all such isomeric forms, including racemic and other mixtures thereof. Where appropriate such isomers can be separated from their mixtures by the application or adaptation of known methods (e.g. chromatographic techniques and recrystallisation techniques). Where appropriate such isomers may be prepared by the application of adaptation of known methods (e.g. asymmetric synthesis).
Convenient compounds of the invention include:
5-[(R)-2-(9-{[2-(Cyclohexyl-hydroxy-phenyl-methyl)-oxazol-5-ylmethyl]-methyl- amino}-nonylamino)-1-hydroxy-ethyl]-8-hydroxy-1 H-quinolin-2-one;
5-[(fl)-2-(9-{[2-(Cyclopentyl-hydroxy-phenyl-methyl)-oxazol-5-ylmethyl]- methyl-amino} -nonylamino)-1 -hydroxy-ethyl]-8-hydroxy-1 H-quinolin-2-one;
8-Hydroxy-5-[(R)-1-hydroxy-2-(9-{[2-(hydroxy-diphenyl-methyl)-oxazol-5- ylmethyl] -methyl-amino}-nonylamino)-ethyl]-1 H-quinolin-2-one;
5-[(f?)-2-(8-{[2-(Cyclopentyl-hydroxy-phenyl-methyl)-oxazol-5-ylmethyl]- methyl-amino}-octylamino)-1-hydroxy-ethyl]-8-hydroxy-1 /-/-quinolin-2-one;
5-[(fl)-2-(10-{[2-(Cyclopentyl-hydroxy-phenyl-methyl)-oxazol-5-ylmethyl]- methyl-amino}-decylamino)-1-hydroxy-ethyl]-8-hydroxy-1 H-quinolin-2-one; [2-(Hydroxy-diphenyl-methyl)-oxazol-5-ylmethyl]-{9-[(R)-2-hydroxy-2-(8- hydroxy-2-oxo-1 ,2-dihydro-quinolin-5-yl)-ethylamino]-nonyl}-dimethyl-ammonium; 8-Hydroxy-5-[(fi)-1-hydroxy-2-(8-{[2-(hydroxy-diphenyl-methyl)-oxazol-5- ylmethyl]-methyl-amino}-octylamino)-ethyl]-1 H-quinolin-2-one;
8-Hydroxy-5-[(fi)-1 -hydroxy-2-(10-{[2-(hydroxy-diphenyl-methyl)-oxazol-5- ylmethyl]-methyl-amino}-decylamino)-ethyl]-1 H-quinolin-2-one;
[2-((R)-Cyclohexyl-hydroxy-phenyl-methyl)-oxazol-5-ylmethyl]-{9-[(R)-2- hydroxy-2-(8-hydroxy-2-oxo-1 ,2-dihydro-quinolin-5-yl)-ethylamino]-nonyl}-dimethyl- ammonium diformate; and
^-((^-Cyclohexyl-hydroxy-phenyl-methyO-oxazol-δ-ylmethylHS-KR)^- hydroxy-2~(4-hydroxy-2-oxo-2,3-dihydro-benzothiazol-7-yl)-ethylamino]-nonyl}- dimethyl-ammonium diformate.
The present invention is also concerned with pharmaceutical formulations comprising, as an active ingredient, a compound of the invention. Other compounds may be combined with compounds of this invention for the prevention and treatment of inflammatory diseases of the lung. Thus the present invention is also concerned with pharmaceutical compositions for preventing and treating respiratory-tract disorders such as chronic obstructive lung disease, chronic bronchitis, asthma, chronic respiratory obstruction, pulmonary fibrosis, pulmonary emphysema, and allergic rhinitis comprising a therapeutically effective amount of a compound of the invention and one or more other therapeutic agents.
Other compounds may be combined with compounds of this invention for the prevention and treatment of inflammatory diseases of the lung. Accordingly the invention includes a combination of an agent of the invention as hereinbefore described with one or more anti-inflammatory, bronchodilator, antihistamine, decongestant or anti-tussive agents, said agents of the invention hereinbefore described and said combination agents existing in the same or different pharmaceutical compositions, administered separately or simultaneously. Preferred combinations would have two or three different pharmaceutical compositions. Suitable therapeutic agents for a combination therapy with compounds of the invention include: One or more other bronchodilators such as PDE3 inhibitors; Methyl xanthines such as theophylline;
A corticosteroid, for example fluticasone propionate, ciclesonide, mometasone furoate or budesonide, or steroids described in WO02/88167, WO02/12266, WO02/100879, WO02/00679, WO03/35668, WO03/48181 , WO03/62259, WO03/64445, WO03/72592, WO04/39827 and WO04/66920; A non-steroidal glucocorticoid receptor agonist;
A leukotriene modulator, for example montelukast, zafirlukast or pranlukast; protease inhibitors, such as inhibitors of matrix metalloprotease for example MMP12 and TACE inhibitors such as marimastat, DPC-333, GW-3333; Human neutrophil elastase inhibitors, such as sivelestat and those described in WO04/043942, WO05/021509, WO05/021512, WO05/026123, WO05/026124,
WO04/024700, WO04/024701 , WO04/020410, WO04/020412, WO05/080372,
WO05/082863, WO05/082864, WO03/053930;
Phosphodiesterase-4 (PDE4) inhibitors, for example roflumilast, arofylline, cilomilast,
ONO-6126 or lC-485; Phosphodiesterase-7 inhibitors;
An antitussive agent, such as codeine or dextramorphan;
Kinase inhibitors, particularly P38 MAPKinase inhibitors;
P2X7 anatgonists; iNOS inhibitors; A non-steroidal anti-inflammatory agent (NSAID), for example ibuprofen or ketoprofen;
A dopamine receptor antagonist;
TNF-α inhibitors, for example anti-TNF monoclonal antibodies, such as Remicade and CDP-870 and TNF receptor immunoglobulin molecules, such as Enbrel; A2a agonists such as those described in EP1052264 and EP1241176;
A2b antagonists such as those described in WO2002/42298;
Modulators of chemokine receptor function, for example antagonists of CCR1 , CCR2,
CCR3, CXCR2, CXCR3, CX3CR1 and CCR8, such as SB-332235, SB-656933, SB-
265610, SB-225002, MCP-1 (9-76), RS-504393, MLN-1202, INCB-3284; Compounds which modulate the action of prostanoid receptors, for example a PGD2
(DP1 or CRTH2), or a thromboxane A2 antagonist eg ramatrobant;
Compounds which modulate Th1 or Th2 function, for example, PPAR agonists; lnterleukin 1 receptor antagonists, such as Kineret; lnterleukin 10 agonists, such as llodecakin; HMG-CoA reductase inhibitors (statins); for example rosuvastatin, mevastatin, lovastatin, simvastatin, pravastatin and fluvastatin;
Mucus regulators such as INS-37217, diquafosol, sibenadet, CS-003, talnetant, DNK-
333, MS1-1956, gefitinib;
Antiinfective agents (antibiotic or antiviral), and antiallergic drugs including, but not limited to, anti-histamines.
The weight ratio of the first and second active ingredients may be varied and will depend upon the effective dose of each ingredient. Generally, an effective dose of each will be used.
Any suitable route of administration may be employed for providing a mammal, especially a human, with an effective dosage of a compound of the present invention. In therapeutic use, the active compound may be administered by any convenient, suitable or effective route. Suitable routes of administration are known to those skilled in the art, and include oral, intravenous, rectal, parenteral, topical, ocular, nasal, buccal and pulmonary.
The magnitude of prophylactic or therapeutic dose of a compound of the invention will, of course, vary depending upon a range of factors, including the activity of the specific compound that is used, the age, body weight, diet, general health and sex of the patient, time of administration, the route of administration, the rate of excretion, the use of any other drugs, and the severity of the disease undergoing treatment. In general, the daily dose range for inhalation will lie within the range of from about 0.1 μg to about 10 mg per kg body weight of a human, preferably 0.1 μg to about 0.5 mg per kg, and more preferably 0.1 μg to 50μg per kg, in single or divided doses. On the other hand, it may be necessary to use dosages outside these limits in some cases. Compositions suitable for administration by inhalation are known, and may include carriers and/or diluents that are known for use in such compositions. The composition may contain 0.01-99% by weight of active compound. Preferably, a unit dose comprises the active compound in an amount of 1 μg to 10 mg. For oral administration suitable doses are 10μg per kg to 100mg per kg, preferably 40μg per kg to 4 mg per kg.
Another aspect of the present invention provides pharmaceutical compositions which comprise a compound of the invention and a pharmaceutically acceptable carrier.
The term "composition", as in pharmaceutical composition, is intended to encompass a product comprising the active ingredient(s), and the inert ingredient(s) (pharmaceutically acceptable excipients) that make up the carrier, as well as any product which results, directly or indirectly, from combination, complexation or aggregation of any two or more of the ingredients, or from dissociation of one or more of the ingredients, or from other types of reactions or interactions of one or more of the ingredients. Accordingly, the pharmaceutical compositions of the present invention encompass any composition made by admixing a compound of the invention, additional active ingredient(s), and pharmaceutically acceptable excipients.
The pharmaceutical compositions of the present invention comprise a compound of the invention as an active ingredient or a pharmaceutically acceptable salt thereof, and may also contain a pharmaceutically acceptable carrier and optionally other therapeutic ingredients. The term "pharmaceutically acceptable salts" refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic bases or acids and organic bases or acids, and salts of quaternary ammonium compounds with pharmaceutically acceptable counter-ions.
For delivery by inhalation, the active compound is preferably in the form of microparticles. They may be prepared by a variety of techniques, including spray- drying, freeze-drying and micronisation.
By way of example, a composition of the invention may be prepared as a suspension for delivery from a nebuliser or as an aerosol in a liquid propellant, for example for use in a pressurised metered dose inhaler (PMDI). Propellants suitable for use in a PMDI are known to the skilled person, and include CFC-12, HFA-134a, HFA-227, HCFC-22 (CCI2F2) and HFA-152 (C2H4F2) and isobutane.
In a preferred embodiment of the invention, a composition of the invention is in dry powder form, for delivery using a dry powder inhaler (DPI). Many types of DPI are known.
Microparticles for delivery by administration may be formulated with excipients that aid delivery and release. For example, in a dry powder formulation, microparticles may be formulated with large carrier particles that aid flow from the DPI into the lung. Suitable carrier particles are known, and include lactose particles; they may have a mass median aerodynamic diameter of greater than 90 μm.
In the case of an aerosol-based formulation, an example is: Compound of the invention 24 mg / canister
Lecithin, NF Liq. Cone. 1.2 mg / canister
Trichlorofluoromethane, NF 4.025 g / canister Dichlorodifluoromethane, NF 12.15 g / canister.
The active compounds may be dosed as described depending on the inhaler system used. In addition to the active compounds, the administration forms may additionally
contain excipients, such as, for example, propellants (e.g. Frigen in the case of metered aerosols), surface-active substances, emulsifiers, stabilizers, preservatives, flavorings, fillers (e.g. lactose in the case of powder inhalers) or, if appropriate, further active compounds.
For the purposes of inhalation, a large number of systems are available with which aerosols of optimum particle size can be generated and administered, using an inhalation technique which is appropriate for the patient. In addition to the use of adaptors (spacers, expanders) and pear-shaped containers (e.g. Nebulator®, Volumatic®), and automatic devices emitting a puffer spray (Autohaler®), for metered aerosols, in particular in the case of powder inhalers, a number of technical solutions are available (e.g. Diskhaler®, Rotadisk®, Turbohaler® or the inhalers for example as described EP-A-0505321 ). Additionally, compounds of the invention may be delivered in multi-chamber devices thus allowing for delivery of combination agents.
Methods of Synthesis
The compounds of the invention of the present invention can be prepared according to the procedures of the following schemes and examples, using appropriate materials, and are further exemplified by the following specific examples. Moreover, by utilising the procedures described with the disclosure contained herein, one of ordinary skill in the art can readily prepare additional compounds of the present invention claimed herein. The compounds illustrated in the examples are not, however, to be construed as forming the only genus that is considered as the invention. The examples further illustrate details for the preparation of the compounds of the present invention. Those skilled in the art will readily understand that known variations of the conditions and processes of the following preparative procedures can be used to prepare these compounds.
The compounds of the invention may be isolated in the form of their pharmaceutically acceptable salts, such as those described previously herein above. It may be necessary to protect reactive functional groups (e.g. hydroxy, amino, thio or carboxy) in intermediates used in the preparation of compounds of the invention to avoid their unwanted participation in a reaction leading to the formation of the compounds. Conventional protecting groups, for example those described by T. W. Greene and P.
G. M. Wuts in "Protective groups in organic chemistry" John Wiley and Sons, 1999, may be used.
The invention further provides a process for the preparation of a compound of formula (I) or (Ia) or a pharmaceutically acceptable salt thereof as defined above which comprises:
(a) when L1 represents hydrogen and R1 does not represent hydrogen, reacting a compound of formula (II)



(c) when L1 represents hydrogen and R1 represents hydrogen, reacting a compound of formula (V)

(d) when L1 represents hydrogen and R1 represents hydrogen, reacting a compound of formula (Vl)

(e) when R4 does not represent hydrogen, reacting a compound of formula (VII), or a suitable salt thereof
 wherein L1 L1, L2, L3, L4, R4, R5, R6, R8 and X are as defined in formula (Ia), P3 represents hydrogen or an activating group (e.g. 3-nitrophenylsulfonyl) with a compound of formula (VIII), or a suitable salt thereof,
wherein L1 L1, L2, L3, L4, R4, R5, R6, R8 and X are as defined in formula (Ia), P3 represents hydrogen or an activating group (e.g. 3-nitrophenylsulfonyl) with a compound of formula (VIII), or a suitable salt thereof,



LG2 represents a leaving group (e.g. chloride, bromide, iodide, methanesulfonate or para-toluenesulfonate) in the presence of a base (e.g. when P3 is hydrogen, potassium carbonate, triethylamine, diisopropylethylamine and, when P3 is 3- nitrophenylsulfonyl, sodium hydride or lithium di-/so-propylamide), followed by
reduction of the ketone (e.g. using sodium borohydride or a borane/chiral catalyst complex), followed by removal of the protective groups (e.g. trifluoroacetic acid, thiophenol, thioacetic acid); or (f)When R4 represents hydrogen, reacting a compound of formula (Xl)





(j)when L1 and L2 each represents hydrogen and R4 represents hydrogen, reacting a compound of formula (XVI)
 wherein L, L3, L4, R4, R5, R6, A, and X are as defined in formula (I) and P2 is as defined in compound of formula (Xl) with a suitable reducing agent (e.g. borane tetrahydrofuran complex), followed by removal of the protective group (e.g. using hydrofluoric acid-pyridine complex); and optionally after (a), (b), (c), (d), (e), (f), (g), (h), (i) or (j) carrying out one or more of the following:
wherein L, L3, L4, R4, R5, R6, A, and X are as defined in formula (I) and P2 is as defined in compound of formula (Xl) with a suitable reducing agent (e.g. borane tetrahydrofuran complex), followed by removal of the protective group (e.g. using hydrofluoric acid-pyridine complex); and optionally after (a), (b), (c), (d), (e), (f), (g), (h), (i) or (j) carrying out one or more of the following:
• converting the compound obtained to a further compound of the invention
• forming a pharmaceutically acceptable salt of the compound.
In process variants (a), (c), (e), (f) and (h), the reaction may conveniently be carried out in an organic solvent such as Λ/,Λ/-dimethylformamide, ethanol, n-butanol or dimethyl sulfoxide, at a temperature, for example, in the range from 50 to 1400C. In process variants (b), (d) and (g), the reaction may conveniently be carried out in an organic solvent such as methanol, ethanol, dichloromethane, acetic acid N- methylpyrolidinone, or Λ/,Λ/-dimethylformamide containing up to 10%w of water and acetic acid.
In process variants (i) and (j), the reaction may conveniently be carried out in an organic solvent such as tetrahydrofuran, at a temperature, for example, in the range from 0 to 800C.
Compounds of formula (II) may be prepared by reacting a compound of formula (XVII), or a suitable salt thereof,
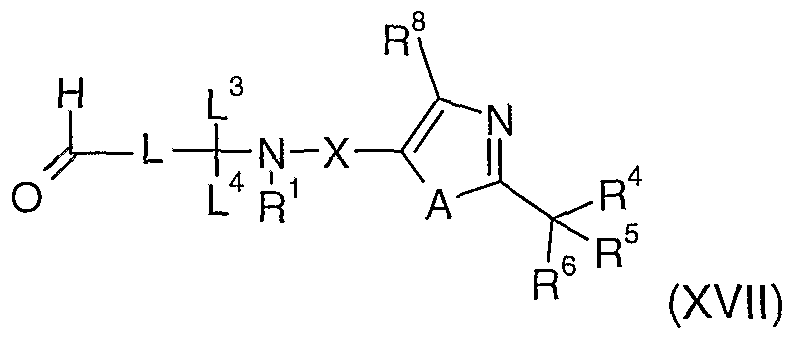
L Mt (XVlK) wherein L2 is as defined in formula (II) and Mt represents a metal such as lithium or
magnesium, or aluminium or boron (e.g. methyllithium, methylmagnesium bromide, lithium aluminium hydride, sodium borohydride) in an organic solvent, for example, tetrahydrofuran or ether, at a temperature, for example in the range from 0 to 6O0C, followed by conversion of the resulting hydroxyl group into a suitable leaving group (e.g. chloride, bromide, iodide, methanesulfonate or para-toluenesulfonate).
Compounds of formula (IV) may be prepared by reacting a compound of formula (XVII) with a compound of formula (XVIII) in an organic solvent, for example, tetrahydrofuran or ether, at a temperature, for example in the range from 0 to 6O0C, followed by oxidation of the resulting hydroxyl group with a suitable oxidating agent (e.g. Swern reagent, Dess-Martin reagent or pyridiniumchlorochromate) in an organic solvent such as dichloromethane, Λ/,Λ/-dimethylformamide or dimethylsulfoxide at a temperature, for example in the range from -78 to 6O0C. Compounds of formula (V) may be prepared by reacting a compound of formula (XIX)

Compounds of formula (VII) in which L1 represents hydrogen and L, L2, L3, L4, R1, R4, R5, R6, R8, A, and X are as defined in formula (VII) may be prepared by (a) reacting a compound of formula (II) with sodium azide, in an organic solvent for example, tetrahydrofuran, Λ/,Λ/-dimethylformamide or dimethylsulfoxide at a temperature, for example in the range from 25 to 850C, followed by reduction of the
resulting azido compound using a suitable reducing agent (e.g. triphenylphosphine) in an organic solvent for example, tetrahydrofuran and water, and eventually followed by protection of the resulting amine (e.g. treatment with 3-nitrophenylsulfonyl chloride in the presence of a base such as pyridine); or, (b) reacting a compound of formula (IV) with an amine (e.g. benzylamine, α-methyl benzylamine, 4-methoxybenzylamine or 2,4-methoxybenzylamine) followed by reduction of the resulting imine using a suitable reducing agent (e.g. sodium cyanoborohydride or sodium triacetoxyborohydride) in an organic solvent such as methanol, ethanol, dichloromethane, acetic acid, Λ/-methylpyrolidinone or N, N- dimethylformamide containing up to 10%w of water and acetic acid, followed by removal of the resulting benzyl protective group using the appropriate reagent (e.g. hydrogen and a suitable catalyst (Palladium on carbon or palladium hydroxide), 2,3- dichloro-5,6-dicyanobenzoquinone (DDQ), or ammonium cerium nitrate (CAN)) in an organic solvent, for example, ethanol, methanol, tetrahydrofuran, dichloromethane, acetonitrile, water, or a mixture thereof, at a temperature ranging from 25 to 800C, and eventually followed by protection of the resulting amine (e.g. treatment with 3- nitrophenylsulfonyl chloride in the presence of a base such as pyridine);
Compounds of formula (VII) in which L, L1, L2, L3, L4, R1, R4, R5, R6, R8, A, and X are as defined in formula (VII) may be prepared by reacting a compound of formula (XX)

Compounds of formula (Xl) in which L1 represents hydrogen may be prepared by (a) reacting a compound of formula (V) with sodium azide in an organic solvent, for example, tetrahydrofuran, Λ/,Λ/-dimethylformamide or dimethylsulfoxide at a temperature, for example in the range from 25 to 850C, followed by reduction of the resulting azido compound using a suitable reducing agent (e.g. triphenylphosphine or hydrogen) in an organic solvent for example, tetrahydrofuran and water, eventually followed by protection of the resulting amine (e.g. treatment with 3-nitrophenylsulfonyl chloride in the presence of a base such as pyridine); or (b) reacting a compound of formula (Vl) with an amine (e.g. benzylamine, a- methyl benzylamine, 4-methoxybenzyl amine or 2,4-methoxybenzyl amine), followed by reduction of the resulting imine using a suitable reducing agent (e.g. sodium cyanoborohydride, sodium triacetoxyborohydride) in an organic solvent such as methanol, ethanol, dichloromethane, acetic acid Λ/-methylpyrolidinone, or N,N- dimethylformamide containing up to 10%w of water and acetic acid, followed by removal of the resulting benzyl protective group using the appropriate reagent (e.g. hydrogen and a suitable catalyst (Palladium on carbon or palladium hydroxide), 2,3- dichloro-5,6-dicyanobenzoquinone (DDQ), or ammonium cerium nitrate (CAN)) in an organic solvent, for example, ethanol, methanol, tetrahydrofuran, dichloromethane, acetonitrile, water, or a mixture thereof, at a temperature ranging from 25 to 800C, eventually followed by protection of the resulting amine (e.g. treatment with 3- nitrophenylsulfonyl chloride in the presence of a base such as pyridine).
Compounds of formula (Xl) may be prepared by reacting a compound of formula (XXI)
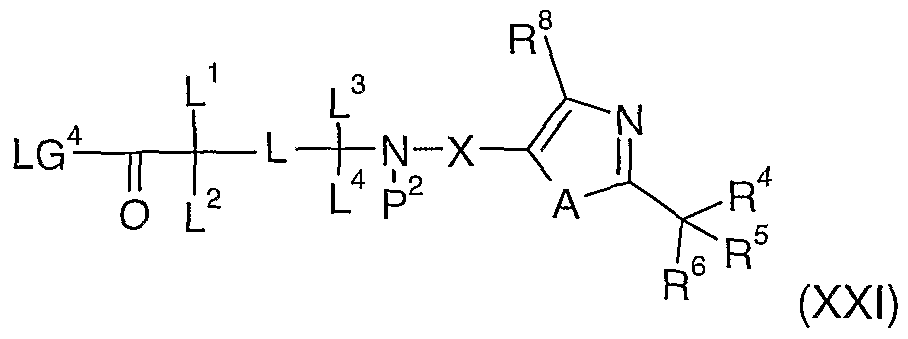
(a) reacting a compound of formula (XXII)

(b) reacting a compound of formula (XXIII)
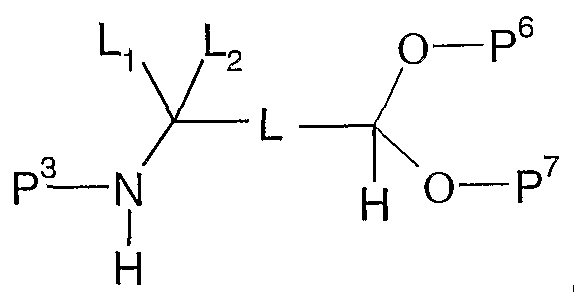



Compounds of formula (VIl), (XIX), (XX), (XXI) can be accessed through a general amination reaction of a compound of formula (XXVII)


- for compound of formula (VII), P9 represents R4, P10 represents


- for compound of formula (XIII), P9 and P10 represents represents an appropriate nitrogen protecting group, such as te/f-butoxycarbonyl, followed by suitable deprotection (e.g. trifluoroacetic acid acid); - for compound of formula (XVII), P9 represents R4, P10 represents

- for compound of formula (XIX), P9 represents P2, P10 represents
 wherein L, L3, and L4 are as defined in compound of formula (XIX)1 wherein P11 and P12 represent an acyclic or cyclic carbonyl protective group (e.g. dimethoxy or diethoxy acetal, 1 ,3-dioxolane or 1 ,3-dioxane), followed by suitable deprotection (e.g. diluted hydrochloric acid or amberlyst-15 resin in methanol);
wherein L, L3, and L4 are as defined in compound of formula (XIX)1 wherein P11 and P12 represent an acyclic or cyclic carbonyl protective group (e.g. dimethoxy or diethoxy acetal, 1 ,3-dioxolane or 1 ,3-dioxane), followed by suitable deprotection (e.g. diluted hydrochloric acid or amberlyst-15 resin in methanol);
- for compound of formula (XX), P9 represents R4, P10 represents

- for compound of formula (XXI), P9 represents P2, P10 represents

- for compound of formula (XXVI), P9 represents P8, P10 represents

When R in compounds of formula (I) or (Ia) is a group of formula
 it will be recognised by those skilled in the art that a similar set of reactions as described above may be used, employing a compound of formula (Ilia) in place of a compound of formula (III), optionally suitably protected, or a suitable salt thereof such as a hydrobromide, hydrochloride or acetate salt
it will be recognised by those skilled in the art that a similar set of reactions as described above may be used, employing a compound of formula (Ilia) in place of a compound of formula (III), optionally suitably protected, or a suitable salt thereof such as a hydrobromide, hydrochloride or acetate salt

Compounds of formula (HIa) are known in the art, for example J. Med. Chem. 1987, 30, 1166.
Compounds of formula (XIII) and (XXVII) may be prepared using methods described, in the following schemes;


Scheme 1
 (XXXIX)
(XXXIX)
 (Xlll-g)
(Xlll-g)
 (Xlll-h)
(Xlll-h)
Scheme 2

(XXIX)
(XLII)

(XLIII)


(XLV)
Scheme 3
It will be apparent that some compounds can contain a chiral centre and thus exist in enatiomeric forms which can be separated by chiral preparative HPLC techniques using conditions know to those skilled in the art and exemplified below.
Compounds of general formula (Xlll-a) may be prepared from compounds of general formula (XXXIII):

by reaction with a compound of general formula (XLVI):
R5M (XLVI)
wherein R4 and R5 are as defined in general formula (I) and M represents a metallic counterion such as Li or MgBr. The reaction may take place in an aprotic organic solvent such as THF or diethyl ether at a range of temperatures, preferably between -78 0C and the reflux temperature of the solvent.
Compounds of general formula (XLVI) are well known in the art and are readily available or can be prepared by known methods.
Compounds of general formula (XXXIII) can be prepared from compounds of general formula (XXXII):

by reaction with an amine of formula (XLVII):
R1NH2 (XLVII)
wherein R1 is as defined in general formula (I). The reaction is performed in a range of solvents, preferably THF/DCM at a range of temperatures, preferably between 0 and 1000C.
Compounds of general formula (XLVII) are well known in the art and can be prepared by known methods, or are commercially available.
Compounds of formula (XXXII) can be prepared from compounds of general formula (XXXI):

by reaction with a brominating agent such as N-bromosuccinimide in the presence of a radical initiator such as AIBN or benzoyl peroxide. The reaction can be carried out
in suitable solvents, such as CCI4, or chlorobenzene at a range of temperatures, preferably between ambient temperature and the reflux temperature of the solvent.
Compounds of formula (XXXI) can be prepared from compounds of general formula (XXX):

by reaction with an acid such as hydrochloric acid, sulphuric acid, methanesulfonic or trifluoromethansulfonic acid in a range of solvents such as THF, DCM, water, and preferably 1 ,4-dioxan at a range of temperatures, preferably between ambient temperature and the reflux temperature of the solvent.
Alternatively compounds of formula (XXXI) can be prepared from compounds of general formula (XXX) by palladium-catalysed cyclisation using a palladium catalyst such as bis(dibenzylideneacetone)palladium in the presence of a ligand such as triphenylphosphine and a base such as sodium tert-butoxide in a solvent such as THF from room temperature to the reflux temperature of the solvent.
Alternatively compounds of formula (XXXI) can be prepared from compounds of formula (XLI):

according to the method described in J. Chem. Soc. 1948, 1960. Compounds of general formula (XLI) are known in the art and can be prepared by known methods such as those described in Tetrahedron 2002, 58(14), 2813.
Alternatively compounds of formula (XXXI) can be prepared from compounds of formula (XLII):

according to the method described in J. Org. Chem., 1938, _?, 319. Compounds of general formula (XL) are well known in the art and can be prepared by known methods such as those described in GB2214180.
Compounds of general formula (XXX) can be prepared from compounds of general formula (XXIX):

by reaction with propargylamine in the presence of a suitable coupling agent, such as DCC/HOBt or many other known coupling methodologies. Alternatively compounds of formula (XXIX) may be converted to, for example, the acid chloride and amide formation effected optionally in the presence of a suitable non-nucleophilic base and compatible solvent under well known conditions. Compounds of general formula (XXIX) are readily available or can be prepared by known methods.
Compounds of general formula (XXVI l-a) can be prepared from compounds of formula (XXXIV):

according to methods similar to those used to prepare compounds of formula (XXXII) from compounds of formula (XXXI) as described above.
Compounds of general formula (XXXIV) can be prepared from compounds of formula (XXXI) using methods described above for the preparation of compounds of formula (Xlll-a) from compounds of formula (XXXIII).
Alternatively compounds of formula (XXXIV) may be prepared from compounds of formula (XLIV):

using methods described above for the preparation of compounds of formula (XXXI) from compounds of formula (XXIX). Compounds of general formula (XLIV) can be prepared by known methods such as those described in GB2214180.
Alternatively compounds of formula (XXXIV) may be prepared from compounds of formula (XLV):
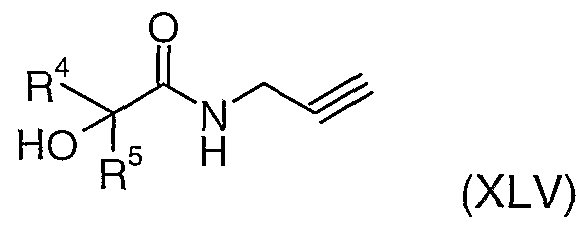
using methods described above for the preparation of compounds of formula (XXXI) from compounds of formula (XXX).
Compounds of general formula (XLV) can be prepared from compounds of formula (XLIII) using methods described above for the preparation of compounds of formula (XXX) from compounds of formula (XXIX).
Compounds of formula (Xll-a) wherein -NR1R3 is a secondary amine (i.e. one of R1 or R3 is a hydrogen atom) may be prepared from compounds of formula (Xll-a) wherein -NR1R3 is a -NH2 group by reductive alkylation with a suitably substituted aldehyde. The reaction is carried out in the presence of a reducing agent such as sodium cyanoborohydride or sodium borohydride, preferably sodium triacetoxyborohydride in a range of organic solvents, preferably dichloroethane.
Compounds of formula (Xll-e) may be prepared from compounds of formula (Xll-d) by alkylation or reductive alkylation methods as described above and according to standard methods well-known to those skilled in the art.
Compounds of formula (Xll-d) can be prepared from compounds of general formula (XXXV):

by reaction with a reducing agent such as lithium aluminium hydride, diisobutyl aluminium hydride, or borane in a range of aprotic solvents such as diethyl ether, or THF or preferably by hydrogenation in the presence of a catalyst such as Raney Nickel in a suitable solvent such as EtOAc or EtOH at a range of temperatures from room temperature to the reflux temperature of the solvent.
Compounds of general formula (XXXV) can be prepared from compounds of general formula (XXVI l-a) by reaction with a source of cyanide ion such as acetone cyanohydrin or an inorganic cyanide, preferably sodium cyanide, in the presence of a non-nucleophilic base such as tetramethyl guanidine, in a range of solvents, preferably ethanol, at a range of temperatures, preferably between ambient temperature and the reflux temperature of the solvent.
Compounds of formula (Xlll-c) can be prepared from compounds of formula (Xlll-a) by reaction with a reducing agent such as triethylsilane in the presence of an acid such as trifluoroacetic acid in a solvent such as DCM from room temperature to the reflux temperature of the solvent.
Compounds of formula (Xlll-b) can be prepared from compounds of formula (Xlll-a) by reaction with an alkylating agent of formula (XLVIII):
FtV (XLVIII)
wherein Rf is a C1-6 alkyl group and Y is a leaving group such as halogen, tosylate, mesylate. The reaction is performed in the presence of a base such as sodium hydride in a solvent such as THF from O0C to the reflux temperature of the solvent.
Compounds of general formula (Xlll-h) can be prepared from compounds of formula (Xlll-g) using methods described above for the preparation of compounds of formula (Xlll-e) from compounds of formula (Xlll-d).
Compounds of general formula (Xlll-g) can be prepared from compounds of formula (XXXIX) using methods described above for the preparation of compounds of formula (Xlll-d) from compounds of formula (XXXV).
Compounds of general formula (Xlll-f) can be prepared from compounds of formula (XXVII-b) using methods described above for the preparation of compounds of formula (Xlll-a) from compounds of formula (XXVII-a).
Compounds of general formula (XXVII-b) can be prepared from compounds of formula (XXXVIII) using methods described above for the preparation of compounds of formula (XXXIl) from compounds of formula (XXXI).
Compounds of general formula (XXXVIII) may be prepared from compounds of general formula (XXXV):
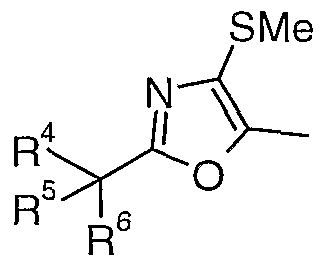
by reaction with a reducing agent such as Raney Nickel in a solvent such as ethanol at a temperature from room temperature to the reflux temperature of the solvent according to the method described in J. Org. Chem. 2006, 71 (8), 3026.
Compounds of general formula (XXXVII) may be prepared from compounds of general formula (XXXVI):

Compounds of general formula (XXXVI) are well known in the art and can be prepared by known methods, or are commercially available.


(LII) Scheme 4
Compounds of Formula (LII) may be prepared from compounds of Formula (L) by employing a similar sequence of reactions as used to prepare compounds of Formula (Xlll-a) from compounds of Formula (XXXIV) in Scheme 1 above.
Compounds of formula (L) wherein R4 and R5 are the same may be prepared from compounds of Formula (XLIX) where R is a suitable alkyl group (such as ethyl or methyl) by treatment with an appropriate organometallic reagent such as a Grignard reagent, in a suitable solvent such as THF or diethyl ether. Compounds of Formula (L) wherein R4 and R5 are dissimilar may be prepared from compounds of Formula (XLIX) by converting to an intermediate amide, preferably a Weinreb amide, and performing the introduction of R4 and R5 through their respective organometallic reagents in a stepwise manner.
Compounds of Formula (XLIX) are known in the literature - for example, HeIv. Chim. Acta 1946, 29, 1957.
Compounds of formula (I) or (Ia) wherein R3 is a lone pair may be converted into compounds of formula (I) or (Ia) wherein R3 is CrC6-alkyl by alkylation with a suitable alkylating agent, eg MeI optionally in the presence of a solvent such as dichloromethane, acetonitrile, isopropanol, THF, chloroform or mixtures thereof. It will be appreciated by those skilled in the art that the order of alkylation can be reversed, so that the group R3 is introduced first, followed by alkylation with a suitable group R2.
It will be appreciated by those skilled in the art that in the processes of the present invention certain functional groups such as hydroxyl or amino groups in the reagents may need to be protected by protecting groups. Thus, the preparation of the compounds of formula (I) or (Ia) may involve, at an appropriate stage, the removal of one or more protecting groups.
The protection and deprotection of functional groups is described in 'Protective
Groups in Organic Chemistry', edited by J.W.F. McOmie, Plenum Press (1973) and 'Protective Groups in Organic Synthesis', 3rd edition, T.W. Greene and P.G.M. Wuts,
Wiley-lnterscience (1999).
Compounds of formula (I) or (Ia) can be converted into further compounds of formula (I) or (Ia) using standard procedures.
The present invention also comprises intermediate compounds having utility in the synthesis of the compounds of formula (I) or (Ia). In a first embodiment, such intermediate compounds are selected from the group including 5-[(R)-1-(tert-butyl~ dimethyl-silanyloxy)-2-(9-{[2-(cyclohexyl-hydroxy-phenyl-methyl)-oxazol-5-ylmethyl]- methyl-amino}-nonylamino)-ethyl]-8-hydroxy-1 H-quinolin-2-one; 5-[(R)-1-(tert-butyl- dimethyl-silanyloxy)-2-(9-{[2-(hydroxy-diphenyl-methyl)-oxazol-5-ylmethyl]-methyl- amino}-nonylamino)-ethyl]-8-(4-methoxy-benzyloxy)-1 /-/-quinolin-2-one; 5-[(R)-1 - hydroxy-2-(9-{[2-(hydroxy-diphenyl-methyl)-oxazol-5-ylmethyl]-methyl-amino}- nonylamino)-ethyl]-8-(4-methoxy-benzyloxy)-1 H-quinolin-2-one; and [2-(hydroxy- diphenyl-methyl)-oxazol-5-ylmethyl]-dimethyl-(9-oxo-nonyl)-ammonium bromide.
In an alternative embodiment such intermediate compounds are selected from the group including 5-[(f?)-1 -(tert-butyl-dimethyl-silanyloxy)-2-(9-{[2-(hydroxy-diphenyl- methyl)-oxazol-5-ylmethyl]-methyl-amino}-nonylamino)-ethyl]-8-(4-methoxy- benzyloxy)-1 H-quinolin-2-one; 5-[(R)-1 -hydroxy-2-(9-{[2-(hydroxy-diphenyl-methyl)- oxazol-5-ylmethyl]-methyl-amino}-nonylamino)-ethyl]-8-(4-methoxy-benzyloxy)-1 H-
quinolin-2-one; and [2-(hydroxy-diphenyl-methyl)-oxazol-5-ylmethyl]-dimethyl-(9-oxo- nonyl)-ammonium bromide.
The compounds of formula (I) and (Ia) have activity as pharmaceuticals, in particular as dual adrenergic β2 receptor agonists and anticholinergic agents including muscarinic receptor (M1 , M2, and M3) antagonists, in particular M3 antagonists. Diseases and conditions which may be treated with the compounds of formula (I) or (Ia) and their pharmaceutically acceptable salts include:
1. respiratory tract: obstructive diseases of the airways including: asthma, including bronchial, allergic, intrinsic, extrinsic, exercise-induced, drug-induced (including aspirin and NSAID-induced) and dust-induced asthma, both intermittent and persistent and of all severities, and other causes of airway hyper-responsiveness; chronic obstructive pulmonary disease (COPD); bronchitis, including infectious and eosinophilic bronchitis; emphysema; bronchiectasis; cystic fibrosis; sarcoidosis; farmer's lung and related diseases; hypersensitivity pneumonitis; lung fibrosis, including cryptogenic fibrosing alveolitis, idiopathic interstitial pneumonias, fibrosis complicating anti-neoplastic therapy and chronic infection, including tuberculosis and aspergillosis and other fungal infections; complications of lung transplantation; vasculitic and thrombotic disorders of the lung vasculature, and pulmonary hypertension; antitussive activity including treatment of chronic cough associated with inflammatory and secretory conditions of the airways, and iatrogenic cough; acute and chronic rhinitis including rhinitis medicamentosa, and vasomotor rhinitis; perennial and seasonal allergic rhinitis including rhinitis nervosa (hay fever); nasal polyposis; acute viral infection including the common cold, and infection due to respiratory syncytial virus, influenza, coronavirus (including SARS) or adenovirus; or eosinophilic esophagitis;
2. bone and joints: arthritides associated with or including osteoarthritis/osteoarthrosis, both primary and secondary to, for example, congenital hip dysplasia; cervical and lumbar spondylitis, and low back and neck pain; osteoporosis; rheumatoid arthritis and Still's disease; seronegative spondyloarthropathies including ankylosing spondylitis, psoriatic arthritis, reactive arthritis and undifferentiated spondarthropathy; septic arthritis and other infection- related arthopathies and bone disorders such as tuberculosis, including Potts' disease and Poncet's syndrome; acute and chronic crystal-induced synovitis including urate gout, calcium pyrophosphate deposition disease, and calcium apatite related tendon, bursal and synovial inflammation; Behcet's disease; primary and
secondary Sjogren's syndrome; systemic sclerosis and limited scleroderma; systemic lupus erythematosus, mixed connective tissue disease, and undifferentiated connective tissue disease; inflammatory myopathies including dermatomyositits and polymyositis; polymalgia rheumatica; juvenile arthritis including idiopathic inflammatory arthritides of whatever joint distribution and associated syndromes, and rheumatic fever and its systemic complications; vasculitides including giant cell arteritis, Takayasu's arteritis, Churg-Strauss syndrome, polyarteritis nodosa, microscopic polyarteritis, and vasculitides associated with viral infection, hypersensitivity reactions, cryoglobulins, and paraproteins; low back pain; Familial Mediterranean fever, Muckle-Wells syndrome, and Familial Hibernian Fever, Kikuchi disease; drug-induced arthalgias, tendonititides, and myopathies;
3. pain and connective tissue remodelling of musculoskeletal disorders due to injury [for example sports injury] or disease: arthitides (for example rheumatoid arthritis, osteoarthritis, gout or crystal arthropathy), other joint disease (such as intervertebral disc degeneration or temporomandibular joint degeneration), bone remodelling disease (such as osteoporosis, Paget's disease or osteonecrosis), polychondritits, scleroderma, mixed connective tissue disorder, spondyloarthropathies or periodontal disease (such as periodontitis);
4. skin: psoriasis, atopic dermatitis, contact dermatitis or other eczematous dermatoses, and delayed-type hypersensitivity reactions; phyto- and photodermatitis; seborrhoeic dermatitis, dermatitis herpetiformis, lichen planus, lichen sclerosus et atrophica, pyoderma gangrenosum, skin sarcoid, discoid lupus erythematosus, pemphigus, pemphigoid, epidermolysis bullosa, urticaria, angioedema, vasculitides, toxic erythemas, cutaneous eosinophilias, alopecia areata, male-pattern baldness, Sweet's syndrome, Weber-Christian syndrome, erythema multiforme; cellulitis, both infective and non-infective; panniculitis;cutaneous lymphomas, non-melanoma skin cancer and other dysplastic lesions; drug-induced disorders including fixed drug eruptions;
5. eyes: blepharitis; conjunctivitis, including perennial and vernal allergic conjunctivitis; iritis; anterior and posterior uveitis; choroiditis; autoimmune; degenerative or inflammatory disorders affecting the retina; ophthalmitis including sympathetic ophthalmitis; sarcoidosis; infections including viral , fungal, and bacterial;
6. gastrointestinal tract: glossitis, gingivitis, periodontitis; oesophagitis, including reflux; eosinophilic gastro-enteritis, mastocytosis, Crohn's disease, colitis including ulcerative colitis, proctitis, pruritis ani; coeliac disease, irritable bowel syndrome, and
food-related allergies which may have effects remote from the gut (for example migraine, rhinitis or eczema);
7. abdominal: hepatitis, including autoimmune, alcoholic and viral; fibrosis and cirrhosis of the liver; cholecystitis; pancreatitis, both acute and chronic; 8. genitourinary: nephritis including interstitial and glomerulonephritis; nephrotic syndrome; cystitis including acute and chronic (interstitial) cystitis and Hunner's ulcer; acute and chronic urethritis, prostatitis, epididymitis, oophoritis and salpingitis; vulvovaginitis; Peyronie's disease; erectile dysfunction (both male and female);
9. allograft rejection: acute and chronic following, for example, transplantation of kidney, heart, liver, lung, bone marrow, skin or cornea or following blood transfusion; or chronic graft versus host disease;
10. CNS: Alzheimer's disease and other dementing disorders including CJD and nvCJD; amyloidosis; multiple sclerosis and other demyelinating syndromes; cerebral atherosclerosis and vasculitis; temporal arteritis; myasthenia gravis; acute and chronic pain (acute, intermittent or persistent, whether of central or peripheral origin) including visceral pain, headache, migraine, trigeminal neuralgia, atypical facial pain, joint and bone pain, pain arising from cancer and tumor invasion, neuropathic pain syndromes including diabetic, post-herpetic, and HIV-associated neuropathies; neurosarcoidosis; central and peripheral nervous system complications of malignant, infectious or autoimmune processes;
11. other auto-immune and allergic disorders including Hashimoto's thyroiditis, Graves' disease, Addison's disease, diabetes mellitus, idiopathic thrombocytopaenic purpura, eosinophilic fasciitis, hyper-lgE syndrome, antiphospholipid syndrome;
12. other disorders with an inflammatory or immunological component; including acquired immune deficiency syndrome (AIDS), leprosy, Sezary syndrome, and paraneoplastic syndromes;
13. cardiovascular: atherosclerosis, affecting the coronary and peripheral circulation; pericarditis; myocarditis , inflammatory and auto-immune cardiomyopathies including myocardial sarcoid; ischaemic reperfusion injuries; endocarditis, valvulitis, and aortitis including infective (for example syphilitic); vasculitides; disorders of the proximal and peripheral veins including phlebitis and thrombosis, including deep vein thrombosis and complications of varicose veins;
14. oncology: treatment of common cancers including prostate, breast, lung, ovarian, pancreatic, bowel and colon, stomach, skin and brain tumors and malignancies affecting the bone marrow (including the leukaemias) and lymphoproliferative systems, such as Hodgkin's and non-Hodgkin's lymphoma;
including the prevention and treatment of metastatic disease and tumour recurrences, and paraneoplastic syndromes; and,
15. gastrointestinal tract: Coeliac disease, proctitis, eosinopilic gastro-enteritis, mastocytosis, Crohn's disease, ulcerative colitis, microscopic colitis, indeterminant colitis, irritable bowel disorder, irritable bowel syndrome, non-inflammatory diarrhea, food-related allergies which have effects remote from the gut, e.g., migraine, rhinitis and eczema.
Thus, the present invention provides a compound of formula (Ia) or a pharmaceutically-acceptable salt thereof as hereinbefore defined for use in therapy. In a further aspect, the present invention provides the use of a compound of formula (Ia) or a pharmaceutically acceptable salt thereof as hereinbefore defined in the manufacture of a medicament for use in therapy.
In the context of the present specification, the term "therapy" also includes "prophylaxis" unless there are specific indications to the contrary. The terms "therapeutic" and "therapeutically" should be construed accordingly.
Prophylaxis is expected to be particularly relevant to the treatment of persons who have suffered a previous episode of, or are otherwise considered to be at increased risk of, the disease or condition in question. Persons at risk of developing a particular disease or condition generally include those having a family history of the disease or condition, or those who have been identified by genetic testing or screening to be particularly susceptible to developing the disease or condition.
The invention still further provides a method of treating, or reducing the risk of, an inflammatory disease or condition (including a reversible obstructive airways disease or condition) which comprises administering to a patient in need thereof a therapeutically effective amount of a compound of formula (Ia) or a pharmaceutically acceptable salt thereof as hereinbefore defined.
In particular, the compounds of this invention may be used in the treatment of adult respiratory distress syndrome (ARDS), pulmonary emphysema, bronchitis, bronchiectasis, chronic obstructive pulmonary disease (COPD), asthma and rhinitis. For the above-mentioned therapeutic uses the dosage administered will, of course, vary with the compound employed, the mode of administration, the treatment desired and the disorder indicated. For example, the daily dosage of the compound of the invention, if inhaled, may be in the range from 0.05 micrograms per kilogram body weight (μg/kg) to 100 micrograms per kilogram body weight (μg/kg). Alternatively, if the compound is administered orally, then the daily dosage of the
compound of the invention may be in the range from 0.01 micrograms per kilogram body weight (μg/kg) to 100 milligrams per kilogram body weight (mg/kg).
The compounds of formula (Ia) and pharmaceutically acceptable salts thereof may be used on their own but will generally be administered in the form of a pharmaceutical composition in which the formula (Ia) compound/salt (active ingredient) is in association with a pharmaceutically acceptable adjuvant, diluent or carrier. Conventional procedures for the selection and preparation of suitable pharmaceutical formulations are described in, for example, "Pharmaceuticals - The Science of Dosage Form Designs", M. E. Aulton, Churchill Livingstone, 1988. Depending on the mode of administration, the pharmaceutical composition will preferably comprise from 0.05 to 99 %w (per cent by weight), more preferably from 0.05 to 80 %w, still more preferably from 0.10 to 70 %w, and even more preferably from 0.10 to 50 %w, of active ingredient, all percentages by weight being based on total composition. The present invention also provides a pharmaceutical composition comprising a compound of formula (Ia) or a pharmaceutically acceptable salt thereof as hereinbefore defined, in association with a pharmaceutically acceptable adjuvant, diluent or carrier.
The invention further provides a process for the preparation of a pharmaceutical composition of the invention which comprises mixing a compound of formula (Ia) or a pharmaceutically acceptable salt thereof as hereinbefore defined with a pharmaceutically acceptable adjuvant, diluent or carrier.
The pharmaceutical compositions may be administered topically (e.g. to the skin or to the lung and/or airways) in the form, e.g., of creams, solutions, suspensions, heptafluoroalkane (HFA) aerosols and dry powder formulations, for example, formulations in the inhaler device known as the Turbuhaler®; or systemically, e.g. by oral administration in the form of tablets, capsules, syrups, powders or granules; or by parenteral administration in the form of solutions or suspensions; or by subcutaneous administration; or by rectal administration in the form of suppositories; or transdermally.
Dry powder formulations and pressurized HFA aerosols of the compounds of the invention may be administered by oral or nasal inhalation. For inhalation, the compound is desirably finely divided. The finely divided compound preferably has a mass median diameter of less than 10 μm, and may be suspended in a propellant mixture with the assistance of a dispersant, such as a C8-C20 fatty acid or salt thereof, (for example, oleic acid), a bile salt, a phospholipid, an alkyl saccharide, a
perfluorinated or polyethoxylated surfactant, or other pharmaceutically acceptable dispersant.
The compounds of the invention may also be administered by means of a dry powder inhaler. The inhaler may be a single or a multi dose inhaler, and may be a breath actuated dry powder inhaler.
One possibility is to mix the finely divided compound of the invention with a carrier substance, for example, a mono-, di- or polysaccharide, a sugar alcohol, or another polyol. Suitable carriers are sugars, for example, lactose, glucose, raffinose, melezitose, lactitol, maltitol, trehalose, sucrose, mannitol; and starch. Alternatively the finely divided compound may be coated by another substance. The powder mixture may also be dispensed into hard gelatine capsules, each containing the desired dose of the active compound.
Another possibility is to process the finely divided powder into spheres which break up during the inhalation procedure. This spheronized powder may be filled into the drug reservoir of a multidose inhaler, for example, that known as the Turbuhaler® in which a dosing unit meters the desired dose which is then inhaled by the patient. With this system the active ingredient, with or without a carrier substance, is delivered to the patient.
For oral administration the compound of the invention may be admixed with an adjuvant or a carrier, for example, lactose, saccharose, sorbitol, mannitol; a starch, for example, potato starch, corn starch or amylopectin; a cellulose derivative; a binder, for example, gelatine or polyvinylpyrrolidone; and/or a lubricant, for example, magnesium stearate, calcium stearate, polyethylene glycol, a wax, paraffin, and the like, and then compressed into tablets. If coated tablets are required, the cores, prepared as described above, may be coated with a concentrated sugar solution which may contain, for example, gum arabic, gelatine, talcum and titanium dioxide. Alternatively, the tablet may be coated with a suitable polymer dissolved in a readily volatile organic solvent.
For the preparation of soft gelatine capsules, the compound of the invention may be admixed with, for example, a vegetable oil or polyethylene glycol. Hard gelatine capsules may contain granules of the compound using either the above- mentioned excipients for tablets. Also liquid or semisolid formulations of the compound of the invention may be filled into hard gelatine capsules.
Liquid preparations for oral application may be in the form of syrups or suspensions, for example, solutions containing the compound of the invention, the balance being sugar and a mixture of ethanol, water, glycerol and propylene glycol.
Optionally such liquid preparations may contain colouring agents, flavouring agents, saccharine and/or carboxymethylcellulose as a thickening agent or other excipients known to those skilled in art.
The compounds of the invention may also be administered in conjunction with other compounds used for the treatment of the above conditions.
The invention therefore further relates to combination therapies wherein a compound of the invention, or a pharmaceutically acceptable salt thereof, or a pharmaceutical composition or formulation comprising a compound of the invention, is administered concurrently or sequentially or as a combined preparation with another therapeutic agent or agents, for the treatment of one or more of the conditions listed.
In particular, for the treatment of the inflammatory diseases such as (but not restricted to) rheumatoid arthritis, osteoarthritis, asthma, allergic rhinitis, chronic obstructive pulmonary disease (COPD), psoriasis, and inflammatory bowel disease, the compounds of the invention may be combined with the following agents: nonsteroidal anti-inflammatory agents (hereinafter NSAIDs) including non-selective cyclo- oxygenase COX-1 / COX-2 inhibitors whether applied topically or systemically (such as piroxicam, diclofenac, propionic acids such as naproxen, flurbiprofen, fenoprofen, ketoprofen and ibuprofen, fenamates such as mefenamic acid, indomethacin, sulindac, azapropazone, pyrazolones such as phenylbutazone, salicylates such as aspirin); selective COX-2 inhibitors (such as meloxicam, celecoxib, rofecoxib, valdecoxib, lumarocoxib, parecoxib and etoricoxib); cyclo-oxygenase inhibiting nitric oxide donors (CINODs); glucocorticosteroids (whether administered by topical, oral, intramuscular, intravenous, or intra-articular routes); methotrexate; leflunomide; hydroxychloroquine; d-penicillamine; auranofin or other parenteral or oral gold preparations; analgesics; diacerein; intra-articular therapies such as hyaluronic acid derivatives; and nutritional supplements such as glucosamine.
The present invention still further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, together with a cytokine or agonist or antagonist of cytokine function, (including agents which act on cytokine signalling pathways such as modulators of the SOCS system) including alpha-, beta-, and gamma-interferons; insulin-like growth factor type I (IGF-1); interleukins (IL) including IL1 to 17, and interleukin antagonists or inhibitors such as anakinra; tumour necrosis factor alpha (TNF-α) inhibitors such as anti-TNF monoclonal antibodies (for example infliximab; adalimumab, and CDP-870) and TNF receptor antagonists
including immunoglobulin molecules (such as etanercept) and low-molecular-weight agents such as pentoxyfyliine.
In addition the invention relates to a combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, with a monoclonal antibody targeting B-Lymphocytes (such as CD20 (rituximab), MRA-alLI6R) or T-Lymphocytes (CTLA4-lg, HuMax 11-15).
The present invention still further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, with a modulator of chemokine receptor function such as an antagonist of CCR1 , CCR2, CCR2A, CCR2B, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR9, CCR10 and CCR1 1 (for the C-C family); CXCR1 , CXCR2, CXCR3, CXCR4 and CXCR5 (for the C-X-C family) and CX3CRI for the C-X3-C family.
The present invention further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, with an inhibitor of matrix metalloprotease (MMPs), i.e., the stromelysins, the collagenases, and the gelatinases, as well as aggrecanase; especially collagenase-1 (MMP-1), collagenase-2 (MMP-8), collagenase-3 (MMP-13), stromelysin-1 (MMP-3), stromelysin-2 (MMP-10), and stromelysin-3 (MMP-11) and MMP-9 and MMP-12, including agents such as doxycycline. The present invention still further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, and a leukotriene biosynthesis inhibitor, 5-lipoxygenase (5-LO) inhibitor or 5-lipoxygenase activating protein (FLAP) antagonist such as; zileuton; ABT-761 ; fenleuton; tepoxalin; Abbott- 79175; Abbott-85761 ; a N-(5-substituted)-thiophene-2-alkylsulfonamide; 2,6-di-tert- butylphenolhydrazones; a methoxytetrahydropyrans such as Zeneca ZD-2138; the compound SB-210661 ; a pyridinyl-substituted 2-cyanonaphthalene compound such as L- 739,010; a 2-cyanoquino!ine compound such as L-746,530; or an indole or quinoline compound such as MK-591 , MK-886, and BAY x 1005.
The present invention further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, and a receptor antagonist for leukotrienes (LT) B4, LTC4, LTD4, and LTE4. selected from the group consisting of the phenothiazin-3-1s such as L-651 ,392; amidino compounds such as CGS-25019c; benzoxalamines such as ontazolast; benzenecarboximidamides such as BIIL 284/260; and compounds such as zafirlukast, ablukast, montelukast, pranlukast, verlukast (MK-679), RG-12525, Ro-245913, iralukast (CGP 45715A), and BAY x 7195.
The present invention still further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, and a phosphodiesterase
(PDE) inhibitor such as a methylxanthanine including theophylline and aminophylline; a selective PDE isoenzyme inhibitor including a PDE4 inhibitor an inhibitor of the isoform PDE4D, or an inhibitor of PDE5.
The present invention further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, and a histamine type 1 receptor antagonist such as cetirizine, loratadine, desloratadine, fexofenadine, acrivastine, terfenadine, astemizole, azelastine, levocabastine, chlorpheniramine, promethazine, cyclizine, or mizolastine; applied orally, topically or parenterally.
The present invention still further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, and a proton pump inhibitor (such as omeprazole) or a gastroprotective histamine type 2 receptor antagonist. The present invention further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, and an antagonist of the histamine type 4 receptor.
The present invention still further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, and an alpha-1/alpha-2 adrenoceptor agonist vasoconstrictor sympathomimetic agent, such as propylhexedrine, phenylephrine, phenylpropanolamine, ephedrine, pseudoephedrine, naphazoline hydrochloride, oxymetazoline hydrochloride, tetrahydrozoline hydrochloride, xylometazoline hydrochloride, tramazoline hydrochloride or ethylnorepinephrine hydrochloride. The present invention further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, and a chromone, such as sodium cromoglycate or nedocromil sodium.
The present invention still further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, with a glucocorticoid, such as flunisolide, triamcinolone acetonide, beclomethasone dipropionate, budesonide, fluticasone propionate, ciclesonide or mometasone furoate.
The present invention further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, with an agent that modulates a nuclear hormone receptor such as PPARs. The present invention still further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, together with an
immunoglobulin (Ig) or Ig preparation or an antagonist or antibody modulating Ig function such as anti-lgE (for example omalizumab).
The present invention further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, and another systemic or topically-applied anti-inflammatory agent, such as thalidomide or a derivative thereof, a retinoid, dithranol or calcipotriol.
The present invention still further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, and combinations of aminosalicylates and sulfapyridine such as sulfasalazine, mesalazine, balsalazide, and olsalazine; and immunomodulatory agents such as the thiopurines, and corticosteroids such as budesonide.
The present invention further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, together with an antibacterial agent such as a penicillin derivative, a tetracycline, a macrolide, a beta-lactam, a fluoroquinolone, metronidazole, an inhaled aminoglycoside; an antiviral agent including acyclovir, famciclovir, valaciclovir, ganciclovir, cidofovir, amantadine, rimantadine, ribavirin, zanamavir and oseltamavir; a protease inhibitor such as indinavir, nelfinavir, ritonavir, and saquinavir; a nucleoside reverse transcriptase inhibitor such as didanosine, lamivudine, stavudine, zalcitabine or zidovudine; or a non-nucleoside reverse transcriptase inhibitor such as nevirapine or efavirenz.
The present invention still further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, and a cardiovascular agent such as a calcium channel blocker, a beta-adrenoceptor blocker, an angiotensin-converting enzyme (ACE) inhibitor, an angiotensin-2 receptor antagonist; a lipid lowering agent such as a statin or a fibrate; a modulator of blood cell morphology such as pentoxyfylline; thrombolytic, or an anticoagulant such as a platelet aggregation inhibitor.
The present invention further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, and a CNS agent such as an antidepressant (such as sertraline), an anti-Parkinsonian drug (such as deprenyl, L- dopa, ropinirole, pramipexole, a MAOB inhibitor such as selegine and rasagiline, a comP inhibitor such as tasmar, an A-2 inhibitor, a dopamine reuptake inhibitor, an NMDA antagonist, a nicotine agonist, a dopamine agonist or an inhibitor of neuronal nitric oxide synthase), or an anti-Alzheimer's drug such as donepezil, rivastigmine, tacrine, a COX-2 inhibitor, propentofylline or metrifonate.
The present invention still further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, and an agent for the treatment of acute or chronic pain, such as a centrally or peripherally-acting analgesic (for example an opioid or derivative thereof), carbamazepine, phenytoin, sodium valproate, amitryptiline or other anti-depressant agent-s, paracetamol, or a nonsteroidal anti-inflammatory agent.
The present invention further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, together with a parenterally or topically-applied (including inhaled) local anaesthetic agent such as lignocaine or a derivative thereof.
A compound of the present invention, or a pharmaceutically acceptable salt thereof, can also be used in combination with an anti-osteoporosis agent including a hormonal agent such as raloxifene, or a biphosphonate such as alendronate.
The present invention still further relates to the combination of a compound of the invention, or a pharmaceutically acceptable salt thereof, together with a: (i) tryptase inhibitor; (ii) platelet activating factor (PAF) antagonist; (iii) interleukin converting enzyme (ICE) inhibitor; (iv) IMPDH inhibitor; (v) adhesion molecule inhibitors including VLA-4 antagonist; (vi) cathepsin; (vii) kinase inhibitor such as an inhibitor of tyrosine kinase (such as Btk, Itk, Jak3 or MAP, for example Gefitinib or lmatinib mesylate), a serine / threonine kinase (such as an inhibitor of a MAP kinase such as p38, JNK, protein kinase A, B or C, or IKK), or a kinase involved in cell cycle regulation (such as a cylin dependent kinase); (viii) glucose-6 phosphate dehydrogenase inhibitor; (ix) kinin-B.subi . - or B.sub2. -receptor antagonist; (x) anti- gout agent, for example colchicine; (xi) xanthine oxidase inhibitor, for example allopurinol; (xii) uricosuric agent, for example probenecid, sulfinpyrazone or benzbromarone; (xiii) growth hormone secretagogue; (xiv) transforming growth factor (TGFβ); (xv) platelet-derived growth factor (PDGF); (xvi) fibroblast growth factor for example basic fibroblast growth factor (bFGF); (xvii) granulocyte macrophage colony stimulating factor (GM-CSF); (xviii) capsaicin cream; (xix) tachykinin NK.subi . or NK.sub3. receptor antagonist such as NKP-608C, SB-233412 (talnetant) or D-4418; (xx) elastase inhibitor such as UT-77 or ZD-0892; (xxi) TNF-alpha converting enzyme inhibitor (TACE); (xxii) induced nitric oxide synthase (iNOS) inhibitor; (xxiii) chemoattractant receptor-homologous molecule expressed on TH2 cells, (such as a CRTH2 antagonist); (xxiv) inhibitor of P38; (xxv) agent modulating the function of Toll-like receptors (TLR), (xxvi) agent modulating the activity of purinergic receptors
such as P2X7; (xxvii) inhibitor of transcription factor activation such as NFkB, API or STATS; or (xxviii) a glucocorticoid receptor (GR-receptor) agonist.
In a further aspect the present invention provides a combination (for example for the treatment of COPD, asthma or allergic rhinitis) of a compound of formula (I) and one or more agents selected from the list comprising: o a non-steroidal glucocorticoid receptor (GR-receptor) agonist; o a PDE4 inhibitor including an inhibitor of the isoform PDE4D; o a modulator of chemokine receptor function (such as a CCR1 receptor antagonist); o a steroid (such as budesonide); and o an inhibitor of p38 kinase function.
A compound of the invention, or a pharmaceutically acceptable salt thereof, can also be used in combination with an existing therapeutic agent for the treatment of cancer, for example suitable agents include: (i) an antiproliferative/antineoplastic drug or a combination thereof, as used in medical oncology, such as an alkylating agent (for example cis-platin, carboplatin, cyclophosphamide, nitrogen mustard, melphalan, chlorambucil, busulphan or a nitrosourea); an antimetabolite (for example an antifolate such as a fluoropyrimidine like 5-fluorouracil or tegafur, raltitrexed, methotrexate, cytosine arabinoside, hydroxyurea, gemcitabine or paclitaxel); an antitumour antibiotic (for example an anthracycline such as adriamycin, bleomycin, doxorubicin, daunomycin, epirubicin, idarubicin, mitomycin-C, dactinomycin or mithramycin); an antimitotic agent (for example a vinca alkaloid such as vincristine, vinblastine, vindesine or vinorelbine, or a taxoid such as taxol or taxotere); or a topoisomerase inhibitor (for example an epipodophyllotoxin such as etoposide, teniposide, amsacrine, topotecan or a camptothecin);
(ii) a cytostatic agent such as an antioestrogen (for example tamoxifen, toremifene, raloxifene, droloxifene or iodoxyfene), an oestrogen receptor down regulator (for example fulvestrant), an antiandrogen (for example bicalutamide, flutamide, nilutamide or cyproterone acetate), a LHRH antagonist or LHRH agonist (for example goserelin, leuprorelin or buserelin), a progestogen (for example megestrol acetate), an aromatase inhibitor (for example as anastrozole, letrozole, vorazole or exemestane) or an inhibitor of 5α-reductase such as finasteride;
(iii) an agent which inhibits cancer cell invasion (for example a metalloproteinase inhibitor like marimastat or an inhibitor of urokinase plasminogen activator receptor function);
(iv) an inhibitor of growth factor function, for example: a growth factor antibody (for example the anti-erbb2 antibody trastuzumab, or the anti-erbb1 antibody cetuximab [C225]), a farnesyl transferase inhibitor, a tyrosine kinase inhibitor or a serine/threonine kinase inhibitor, an inhibitor of the epidermal growth factor family (for example an EGFR family tyrosine kinase inhibitor such as N.-(3-chloro-4- fluorophenyl)-7-methoxy-6-(3-morpholinopropoxy)quinazolin-4-amine (gefitinib, AZD1839), N-(3-ethynylphenyl)-6,7-bis(2-methoxyethoxy)quinazolin-4-amine (erlotinib, OSI-774) or 6-acrylamido-N.-(3-chloro-4-fluorophenyl)-7-(3- morpholinopropoxy)quinazolin-4-amine (Cl 1033)), an inhibitor of the platelet-derived growth factor family, or an inhibitor of the hepatocyte growth factor family; (v) an antiangiogenic agent such as one which inhibits the effects of vascular endothelial growth factor (for example the anti-vascular endothelial cell growth factor antibody bevacizumab, a compound disclosed in WO 97/22596, WO 97/30035, WO 97/32856 or WO 98/13354), or a compound that works by another mechanism (for example linomide, an inhibitor of integrin αvβ3 function or an angiostatin); (vi) a vascular damaging agent such as combretastatin A4, or a compound disclosed in WO 99/02166, WO 00/40529, WO 00/41669, WO 01/92224, WO 02/04434 or WO 02/08213;
(vii) an agent used in antisense therapy, for example one directed to one of the targets listed above, such as ISIS 2503, an anti-ras antisense; (viii) an agent used in a gene therapy approach, for example approaches to replace aberrant genes such as aberrant p53 or aberrant BRCA1 or BRCA2, GDEPT
(gene-directed enzyme pro-drug therapy) approaches such as those using cytosine deaminase, thymidine kinase or a bacterial nitroreductase enzyme and approaches to increase patient tolerance to chemotherapy or radiotherapy such as multi-drug resistance gene therapy; or (ix) an agent used in an immunotherapeutic approach, for example ex-vivo and in-vivo approaches to increase the immunogenicity of patient tumour cells, such as transfection with cytokines such as interleukin 2, interleukin 4 or granulocyte-macrophage colony stimulating factor, approaches to decrease T-cell anergy, approaches using transfected immune cells such as cytokine-transfected dendritic cells, approaches using cytokine-transfected tumour cell lines and approaches using anti-idiotypic antibodies.
The invention will now be illustrated but not limited by reference to the following Examples:
General Experimental Details:
Silica gel used for medium pressure column chromatography is 0.035 to 0.070 mm (220 to 440 mesh) (e.g. Fluka silica gel 60), and an applied pressure up to 10 psi accelerated column elution. Where thin layer chromatography (TLC) has been used, it refers to silica gel TLC using plates, typically 3 x 6 cm silica gel on aluminium foil plates with a fluorescent indicator (254 nm) (e.g. Fluka 60778). All solvents and commercial reagents were used as received. Purification by prepacked SCX-2 cartridge refers to Isolute® SCX-2, a strong cation exchange sorbent (Argonaut/IST). Purification over NH2-silica gel refers to Isolute® flash NH2 prepacked cartridges (Argonaut/IST).
All compounds containing a basic centre(s) and purified by reversed-phase HPLC were obtained as the TFA salt, unless stated otherwise.
Preparative HPLC conditions: HPLC system 1
C18-reverse-phase column (100 x 22.5 mm i.d. Genesis column with 7 μm particle size), eluting using linear gradients of mixtures of solvent A (water with 0.1 % TFA) and solvent B (acetonitrile with 0.1 % TFA) at a flow rate of 5 mL/min with UV detection set at 230 nm.
HPLC system 2
Phenyl hexyl column (250 x 21.20 mm i.d. Luna column with 5 μm particle size), eluting using linear gradients of mixtures of solvent A (water with 0.1 % TFA) and solvent B (acetonitrile with 0.1 % TFA) at a flow rate of 18 mL/min with UV detection set at 254 nm.
HPLC system 3
C18-reverse-phase column (250 x 21.20 mm Phenomenex Gemini column with 5 μm particle size), eluting using linear gradients of mixtures of solvent A (water with 0.1% formic acid) and solvent B (acetonitrile with 0.1 % formic acid) at a flow rate of 5-10 mL/min with UV detection set at 230 nm.
The Liquid Chromatography Mass Spectroscopy (LC-MS) systems used: LC-MS method 1
Micromass Platform LCT with a C18-reverse-phase column (100 x 3.0 mm i.d. Higgins Clipeus with 5 μm particle size), elution with solvent A (water with 0.1% formic acid) and solvent B (acetonitrile with 0.1% formic acid). Gradient:
Gradient - Time flow mLΛnin %A %B
0.00 1.0 95 5
1.00 1.0 95 5
15.00 1.0 5 95
20.00 1.0 5 95
22.00 1.0 95 5
25.00 1.0 95 5
Detection - MS, ELS, UV (100 μL split to MS with in-line UV detector). MS ionisation method - Electrospray (positive ion).
LC-MS method 2 Micromass Platform LCT with a C18-reverse-phase column (30 x 4.6 mm i.d. Phenomenex Luna 3 μm particle size), elution with solvent A (water with 0.1% formic acid) and solvent B (acetonitrile with 0.1 % formic acid). Gradient:
Gradient - Time flow mL/min %A %B
0.00 2.0 95 5
0.50 2.0 95 5
4.50 2.0 5 95
5.50 2.0 5 95
6.00 2.0 95 5
Detection - MS, ELS, UV (100 μL split to MS with in-line UV detector). MS ionisation method - Electrospray (positive and negative ion).
LC-MS method 3
Waters Micromass ZQ with a C18-reverse-phase column (30 x 4.6 mm i.d. Phenomenex Luna 3 μm particle size), elution with solvent A (water with 0.1 % formic acid) and solvent B (acetonitrile with 0.1 % formic acid). Gradient:
Gradient - Time flow mL/min %A %B
0.00 2.0 95 5
0.50 2.0 95 5
4.50 2.0 5 95
5.50 2.0 5 95
6.00 2.0 95 5
Detection - MS, ELS, UV (100 μL split to MS with in-line UV detector). MS ionisation method - Electrospray (positive and negative ion).
LC-MS method 4
Waters ZMD with a C18-reverse-phase column (30 x 4.6 mm i.d. Phenomenex Luna with 3 μm particle size), elution with solvent A (water with 0.1% formic acid) and solvent B (acetonitrile with 0.1% formic acid). Gradient:
Gradient - Time flow mL/min %A %B
0.00 2.0 95 5
0.50 2.0 95 5
4.50 2.0 5 95
5.50 2.0 5 95
6.00 2.0 95 5
Detection - MS, ELS, UV (200μl_/min split to MS with in-line Waters 996 DAD detection).
MS ionisation method - Electrospray (positive and negative ion).
LC-MS method 5
Waters Micromass ZQ with a C18-reverse-phase column (100 x 3.0 mm Higgins
Clipeus with 5 μm particle size), elution with A: water + 0.1% formic acid; B: acetonitrile + 0.1 % formic acid. Gradient:
Gradient - Time flow ml/min %A %B
0.00 1 .0 95 5
1.00 1.0 95 5
15.00 1.0 5 95
20.00 1.0 5 95
22.00 1.0 95 5
25.00 1 .0 95 5
Detection - MS, ELS, UV (100 μl split to MS with in-line UV detector at 254nm) MS ionisation method - Electrospray (positive ion)
Abbreviations used in the experimental section:
AIBN = (2,2'-azobis(2-methylproprionitrile)
DCE = 1 ,2-dichloroethane DCM = dichloromethane
DMF = Λ/,Λ/-dimethyl formamide
DMSO = dimethylsulfoxide
EtOAc = ethyl acetate
EtOH = ethanol HCI = hydrochloric acid
IMS = industrially methylated spirit
HATU = 0-(7-azabenzotriazol-1 -y\)-N,N,N'N -tetramethyluroniumhexaf luoro- phosphate
HPLC = high performance liquid chromatography MeOH = methanol
Min = minutes
NaHCO3 = sodium hydrogen carbonate
NaOH = Sodium hydroxide
Na2SO4 = Sodium sulphate NH4CI = Ammonium chloride
RT = room temperature
Rf = retention factor (TLC)
Rt = retention time (LCMS or HPLC)
TBDMS = te/f-butyldimethyl silyl TEMPO = 2,2,6,6-tetramethyl-1 -piperidinyloxy free radical
TFA = trifluoroacetic acid
THF = tetrahydrofuran
TLC = thin layer chromatography
Compound names were generated using the commercially available chemical naming software package MDL® AutoNom.
Example 1 5-F(/?)-2-(9-fr2-(Cvclohexyi-hvdroxy-phenyl-methyl)-oxazol-5-ylmethvil-methvl- aminol-nonylaminoV1-hvdroxy-ethyll-8-hydroxy-1 /y-quinolin-2-one naphthalene-1 ,5-disulfonate.

a) 2-Oxo-2-phenyl-/V-prop-2-ynyl-acetamide

Oxaly) chloride (19 g, 0.15 mol) was added over 15 min to a solution of phenylglyoxylic acid (20 g, 0.13 mol) and 5 drops of DMF in dry DCM (100 m!_). The reaction mixture was stirred at RT for 3 hours after which the solvent was removed in vacuo. The residue was taken up in dry DCM (50 ml_) and the solution was added over 30 minutes to a mixture of propargyl amine (7.3 g, 0.13 mol) and triethylamine (13.5 g, 0.13 mol) in dry DCM (100 ml_) at 0 0C. The mixture was allowed to warm to RT overnight. Water (10 ml_) was added and the mixture was washed twice with 1 M HCI (aq), saturated NaHCO3 (aq), and brine. The organic phase was dried (Na2SO4), filtered, and concentrated in vacuo to 100 ml_. Cyclohexane was added until the solution became slightly turbid. The suspension was left at RT for 1 hour. The solid precipitate was collected by filtration and dried at 45 0C in vacuo to afford the title
compound as a light brown crystalline solid. A second crop was collected by re- crystallisation of the mother liquors. Yield: 22 g, 88%.
LC-MS (Method 3): Rt 2.47 min, m/z 188 [MH]+.
b) (5-methyl-oxazol-2-yl)-phenyl-methanone

Trifluoromethane sulphonic acid (42.5 mL, 0.48 mol) was added dropwise to a solution of 2-oxo-2-phenyl-Λ/-prop-2-ynyl-acetamide (74.88 g, 0.40 mol) in 1 ,4- dioxane (600 mL). The resulting solution was heated at 90 0C for 20 hours. The reaction mixture was cooled to RT and concentrated in vacuo. The residue was partitioned between DCM and concentrated aqueous ammonia and adjusted to pH 10 by addition of 1 M NaOH (aq). The aqueous phase was separated and then further extracted with DCM. The organic extracts were combined, washed with brine, dried (Na2SO4), filtered and concentrated in vacuo. The crude product was purified by column chromatography (Biotage Companion automated system) over silica gel eluting with 0-60% ethyl acetate/cyclohexane to afford the title compound as an orange waxy solid.
Yield: 63.69 g, 85%.
LC-MS (Method 3): Rt 2.94 min, m/z 188 [MH]+.
c) Cyclohexyl-(5-methyl-oxa2ol-2-yl)-phenyl-methanol

To a stirred, cooled 0 0C solution of (5-methyl-oxazol-2-yl)-phenyl-methanone (5.0 g, 27 mmol) in dry THF (50 mL) under nitrogen was added dropwise a solution of
cyclohexylmagnesium chloride (2 M in diethyl ether; 16.7 ml_, 33 mmol) over 10 minutes. After 1 hour the reaction mixture was quenched with saturated NH4CI (aq) and stirred at 0 0C for a further 1 hour. Water was added and the phases were separated. The aqueous phase was extracted with EtOAc and the combined organic layers were washed with 10% citric acid solution (aq), saturated NaHCO3 (aq), brine, dried (Na2SO4), and filtered. The solvent was evaporated in vacuo to afford a white solid. This was triturated with diethyl ether and collected by filtration, washed with diethyl ether, and dried at 45 °C in vacuo to afford the title compound as a white solid. The mother liquors were concentrated and the residue purified by column chromatography over silica gel using a gradient of 5-10% ETOAc/cyclohexane to afford a second batch of semi-pure product. Trituration with diethyl ether and drying the solids at 45 °C under vacuum afforded the pure product. Yield: 4.82 g, 67%. LC-MS (Method 2): Rt 4.03 min, m/z 272 [MH]+. Rf = 0.24 (10% EtOAc/cyclohexane).
d) (S-Bromomethyl-oxazol^-ylJ-cyclohexyl-phenyl-methanol

To a solution of cyclohexyl-(5-methyl-oxazol-2-yl)-phenyl-methanol (2.80 g, 10.3 mmol) and Λ/-bromosuccinimide (2.05 g, 11.5 mmol) in dry DCE (40 mL) under nitrogen was added AIBN (0.168 g, 1.02 mmol), and the reaction mixture was heated at 80 °C for 2 hours. The reaction was cooled to RT and washed with saturated NaHCO3 (aq). The phases were separated and the aqueous layer was extracted with DCM. The combined organic layers were washed with brine, dried (Na2SO4), filtered, and concentrated in vacuo to afford the product as a light yellow solid. Yield: 3.62 g. Quant. LC-MS (Method 4): Rt 4.15 min, m/z 350, 352 [MH]+. Rf = 0.24 (10% ethyl acetate/cyclohexane).
e) (9-Hydroxy-nonyl)-methyl-ammonium bromide.

To a solution of 9-bromo-1-nonanol (10.2 g, 45.7 mmol) in IMS (50 ml_) at 0 0C was added a solution of methylamine (57 ml_, 8 M in EtOH, 0.46 mol). After 30 minutes at 0 CC the reaction mixture was allowed to warm to RT and stirred for 26 hours. The solvent was removed in vacuo to afford a white solid, which was triturated with diethyl ether to provide the title compound as a white solid.
Yield: 9.97 g, 86%. LC-MS (Method 3): Rt 1.51 min, m/z 175 [MH]+.
f) 9-{[2-(Cyclohexyl-hydroxy-phenyl-methyl)-oxazol-5-ylmethyl]-methyl-amϊno}- nonan-1-ol.

(5-Bromomethyl-oxazol-2-yl)-cyclohexyl-phenyl-methanol (1.0 g, 2.86 mmol) was added to a solution of (9-hydroxy-nonyl)-methyl-ammonium bromide (0.69 g, 2.86 mmol) and Λ/,Λ/-diisopropylethylamine (1.0 mL, 5.7 mmol) in DCM. After stirring the mixture at RT for 4 hours saturated NaHCO3 (aq) was added. The phases were separated and the aqueous layer extracted with DCM. The combined organic layers were dried (Na2SO4), filtered, and concentrated in vacuo to afford a yellow oil. Purification by column chromatography over silica gel and gradient eluting from 5- 10% MeOH/DCM provided the title compound as a colourless oil. Yield: 0.80 g, 63%.
LC-MS (Method 2): Rt 2.42 min, m/z 443 [MH]+.
g) ^{^-(Cyclohexyl-hydroxy-phenyl-methylJ-oxazol-S-ylmethyll-methyl-amino}- nonanal

A solution of DMSO (0.17 mL, 2.3 mmol) in DCM (3 ml_) was added dropwise to a solution of oxalyl chloride (94 μl_) in DCM (3 mL) at -78 °C under nitrogen. Then a solution of θ-^^cyclohexyl-hydroxy-phenyl-methyO-oxazol-S-ylmethylj-methyl- amino}-nonan-1-ol (0.49 g, 1.1 mmol) in DCM (5 mL) was added and the reaction mixture was stirred at -78 0C for 15 minutes. Triethylamine (0.62 mL, 4.4 mmol) was added and the reaction mixture was allowed to warm to RT. After 1 hour saturated NaHCO3 (aq) was added, the phases separated, and the aqueous layer extracted with DCM. The combined organic layers were washed with brine, dried (Na2SO4), filtered, and concentrated in vacuo to afford a yellow/brown viscous oil. The crude product was resubmitted to the same reaction conditions to further the conversion of starting material to afford a yellow viscous oil plus solids. This was triturated with diethyl ether and the supernatant was concentrated in vacuo to afford a foam, which was used without further purification. Yield: 0.59 g. LC-MS (method 2): Rt 2.74 min, m/z 441 [MH]+.
h) 5-[(f?)-1-(fert-Butyl-dimethyl-silanyloxy)-2-(9-{[2-(cyclohexyl-hydroxy-phenyl- methyl)-oxazol-5-ylmethyl]-methyl-amino}-nonylamino)-ethyl]-8-hydroxy-1 H- quinolin-2-one

A mixture of 9-{[2-(cyclohexyl-hydroxy-phenyl-methyl)-oxazol-5-ylmethyl]-methyl- aminoj-nonanal (0.49 g, 1.1 mmol), 5-[(F?)-2-amino-1-(terf-butyl-dimethyl-silanyloxy)-
ethyl]-8-hydroxy-1 H-quinolin-2-one (prepared according to US20040167167), (0.37 g, 1.1 mmol), and sodium triacetoxyborohydride (0.33 g, 1.6 mmol) in dry DCE (10 ml_) was stirred under nitrogen at RT for 19 hours. The solvent was evaporated in vacuo and the residue was purified by column chromatography over silica gel using a mixture of DCM/MeOH/acetic acid/water (120:15:3:2) as eluent to afford the product as a pale brown gum. The residue was taken up in MeOH, passed through a SCX-2 cartridge, and liberated using a 2 M ammonia solution in MeOH. Evaporation of the solvent in vacuo afforded the title compound as a mixture of diastereomers as a yellow/green gum. Yield: 78 mg, 9%.
LC-MS (method 2): Rt 2.64 min, m/z 760 [MH]+.
Separation of the diastereomers of 5-[(/?)-1-(te/?-butyl-dirnethyl-silanyloxy)-2-(9- {[2-(cyclohexyl-hydroxy-phenyl-methyl)-oxazol-5-ylmethyl]-methyl-amino}- nonylamino)-ethyl]-8-hydroxy-1 H-quinolin-2-one
5-[(/?)-1-(terf-Butyl-dimethyl-silanyloxy)-2-(9-{[2-(cyclohexyl-hydroxy-phenyl-methyl)- oxazol-5-ylmethyl]-methyl-amino}-nonylamino)-ethyl]-8-hydroxy-1 H-quinolin-2-one (0.27 g) could be separated into the two individual diastereomers by preparative chiral HPLC (Chiralpak IA, 250 x 20 mm i.d.) using 10% EtOH/heptane (+ 0.1 % diethylamine) at 15 mL/min. After evaporation of the volatiles the diastereomers were obtained as brown viscous oils.
Yield: 50.8 mg (diastereomer 1). Chiral HPLC: Rt 20 min. LC-MS (method 2): Rt 2.65 min, m/z 760 [MH]+.
Yield: 64.2 mg (diastereomer 2). Chiral HPLC: Rt 23 min.
LC-MS (method 2): Rt 2.55 min, m/z 760 [MH]+.
iJ S-KffJ^^Θ-f^^Cyclohexyl-hydroxy-phenyl-methylJ-oxazol-δ-ylmethyll-methyl- amino}-nonylamino)-1-hydroxy-ethyl]-8-hydroxy-1 H-quinolin-2-one.
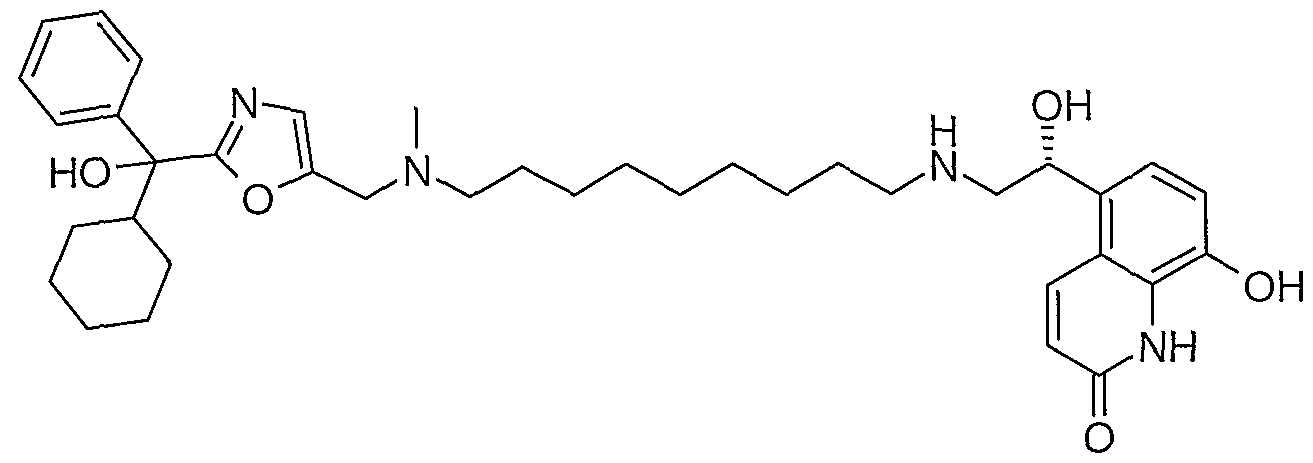
A solution of 5-[(R)-1-(tert-butyl-dimethyl-silanyloxy)-2-(9-{[2-(cyclohexyl-hydroxy- phenyl-methyl)-oxazol-5-ylmethyl]-methyl-amino}-nonylamino)-ethyl]-8-hydroxy-1 /-/- quinolin-2-one (75 mg, 0.10 mmol) in THF (1 ml_) under nitrogen was treated with triethylamine trihydrofluoride (48 μl_, 0.30 mmol). After stirring at RT overnight, the reaction mixture was neutralised with saturated NaHCO3 (aq) and extracted with DCM. The combined organic extracts were washed with brine, dried (Na2SO4), filtered, and concentrated to dryness to afford a green/brown gum. This was purified by preparative HPLC (systemi , 25% B + 1.7% B/min). The product fractions were concentrated, the residue taken up in MeOH/DCM, passed through a SCX-2 cartridge, and liberated using a 2 M ammonia solution in MeOH. Evaporation of the solvent in vacuo afforded the title compound as a yellow gum.
Yield: 21 mg, 33%.
LC-MS (method 2): Rt 2.22 min, m/z 645 [MH]+.
The separate diastereomers of 5-[(R)-1-(te/t-butyl-dimethyl-silanyloxy)-2-(9-{[2- (cyclohexyl-hydroxy-phenyl-methyl)-oxazol-5-ylmethyl]-methyl-amino}-nonylamino)- ethyl]-8-hydroxy-1 H-quinolin-2-one were deprotected analogously to the procedure described above. The analysis data were in accordance with the title compound.
j) 5-[(/?)-2-(9-{[2-(Cyclohexyl-hydroxy-phenyl-methyl)-oxazol-5-ylmethyl]-methyl- amino}-nonylamino)-1-hydroxy-ethyl]-8-hydroxy-1 H-quinolin-2-one naphthalene- 1 ,5-disulfonate.

Naphthalene-1 ,5-disuIfonic acid tetrahydrate (16 mg, 0.043 mmol) was added to a solution of 5-[(f?)-2-(9-{[2-(cyclohθxyl-hydroxy-phenyl-methyl)-oxazol-5-ylmethyl]- methyl-amino} nonylamino)-1-hydroxy-ethyl]-8-hydroxy-1 H-quinolin-2-one (28 mg, 0.043 mmol) in MeOH (1 mL). The reaction mixture was stirred at RT for 1 hour and then warmed to 35 0C for 20 minutes. The suspension was cooled to RT and the solids collected by filtration, washed with MeOH and dried at 45 0C in vacuo to afford the title compound as an off-white solid.
Yield: 35 mg, 89% (Example 1 a, diastereomer 1 , derived from the product of
Example 1 , step h, diastereomer 1).
LC-MS (method 1): Rt 6.25 min, m/z 645 [MH]+.
Yield: 33 mg, 82% (Example 1b, diastereomer 2, derived from the product of
Example 1 , step h, diastereomer 2).
LC-MS (method 1): Rt 6.25 min, m/z 645 [MH]+.
Example 2
5-r(fi1-2-(9-fr2-(Cvclopentyl-hvdroxy-phenyl-methyl)-oxazol-5-ylmethyll-methyl- amino} -nonylamino)-1-hvdroxy-ethvn-8-hvdroxy-1 H-quinolin-2-one naphthalene-1 ,5-disulfonate.


The title compound was prepared by methods analogous to those used in Example 1 step c and d, employing (5~methyl-oxazol-2-yl)-phenyl~methanone (Example 1 , intermediate b) and cyclopentylmagnesium chloride.
Rf = 0.61 (20% ethyl acetate/pentane).
b) Cyclopentyl-(5-methylaminomethyl-oxazol-2-yl)-phenyl-methanol

A solution of methylamine (18.4 ml_, 127 mmol) in THF was added to a stirred solution of (5-bromomethyl-oxazol-2-yl)-cyclopentyl-phenyl-methanol (4.66 g, 12.7 mmol) in THF (80 ml_) and the reaction mixture was stirred at RT for 1 hour. The suspension was partitioned between EtOAc and saturated NaHCO3 (aq) and the aqueous layer was extracted with EtOAc. The combined organic layers were washed with brine, dried (Na2SO4), filtered, and concentrated in vacuo to afford a light yellow/brown oil. This was passed through a SCX-2 cartridge, and liberated using a 2 M ammonia solution in MeOH. After evaporation of the solvent in vacuo the residue was purified by column chromatography over NH2-silica gel using a gradient of 0-70% ethyl acetate/DCM as eluent to afford the title compound as a yellow oil. Yield: 1.91 g, 53%.
LC-MS (method 4): Rt 2.11 min, m/z 287 [MH]+.
Separation of the enantiomers of cyclopentyl-(5-methylaminomethyl-oxazol~2- yl)-phenyl-methanol
rac-Cyclopentyl-(5-methylaminomethyl-oxazol-2-yI)-phenyl-methanol (1.91 g) could be separated into the two individual enantiomers by preparative chiral HPLC (Chiralpak IA1 250 x 20 mm i.d.) using 13% EtOH/heptane (+ 0.1 % diethylamine) at 10 ml_/min. After evaporation of the volatiles the enantiomers were obtained as colourless oils.
Yield: 0.83 g (first eluting enantiomer).
Chiral HPLC: Rt 14 min. LC-MS (method 4): Rt 2.11 min, m/z 287 [MH]+.
Yield: 0.73 g (second eluting enantiomer). Chiral HPLC: Rt 16 min.
LC-MS (method 4): Rt 2.11 min, m/z 287 [MH]+.
c) ^{^-(Cyclopentyl-hydroxy-phenyl-methylJ-oxazol-S-ylmethyll-methyl- amino}-nonan -1-ol

A mixture of cyclopentyl-(5-methylaminomethyl-oxazol-2-yl)-phenyl-methanol (first eluting enantiomer; Example 2, step b), (0.80 g, 2.8 mmol), 9-bromo-nonan-1 -ol (0.69 g, 3.1 mmol), caesium carbonate (1.82 g, 5.6 mmol) and water (151 μL, 8.4 mmol) in DMF (10 mL) was stirred at RT for 16hrs. More 9-bromo-nonan-1-ol (0.1 equiv, 0.31 mmol, 69 mg) was added and the mixture was stirred for a further 16hrs. The reaction mixture was partitioned between 10% lithium chloride (aq) and EtOAc and the aqueous layer was extracted with EtOAc. The combined organic layers were washed with brine, dried (Na2SO4), filtered, and concentrated in vacuo. The residual yellow oil was purified by column chromatography over silica gel using a gradient of 0-10%
MeOH/DCM as eluent, then passed through a SCX-2cartridge, and liberated using a 2 M ammonia solution in MeOH. Evaporation of the solvent in vacuo afforded the title compound.
Yield: 0.56 g, 47%.
LC-MS (method 4): Rt 2.47 min, m/z 429 [MH]+.
d) 9-{[2-(Cyclopentyl-hydroxy-phenyl-methyl)-oxazol-5-ylmethyl]-methyl- amino}-nonanal

A solution of ^{^-(cyclopentyl-hydroxy-phenyl-methyO-oxazol-S-ylmethylJ-methyl- amino}-nonan-1-ol, (0.52 g, 1.21 mmol) in DCM (10.5 ml_) was treated with acetic acid-1 ,1 -diacetoxy-3-oxo-1 lambda*5*-ioda-2-oxa-indan-1 -yl ester (Dess Martin reagent), (0.82 g, 1.9 mmol), (0.17 ml_, 2.3 mmol) and allowed to stir at RT for 9 hours. The reaction mixture was washed with saurated. NaHCO3 (aq) and the organic phase was separated, dried (Na2SO4), filtered, and concentrated in vacuo. The crude product was used without further purification. Yield: 0.40 g. 78% LC-MS (method 4): Rt 2.61 min, m/z 427 [MH]+.
e) 5-[(/?)-2-(9-{[2-(Cyclopentyl-hydroxy-phenyl-methyl)-oxazol-5-ylmethyl]- methyl-amino}-nonylamino)-1-hydroxy-ethyl]-8-hydroxy-1 H-quinolin-2-one naphthalene-1 ,5-disulfonate.
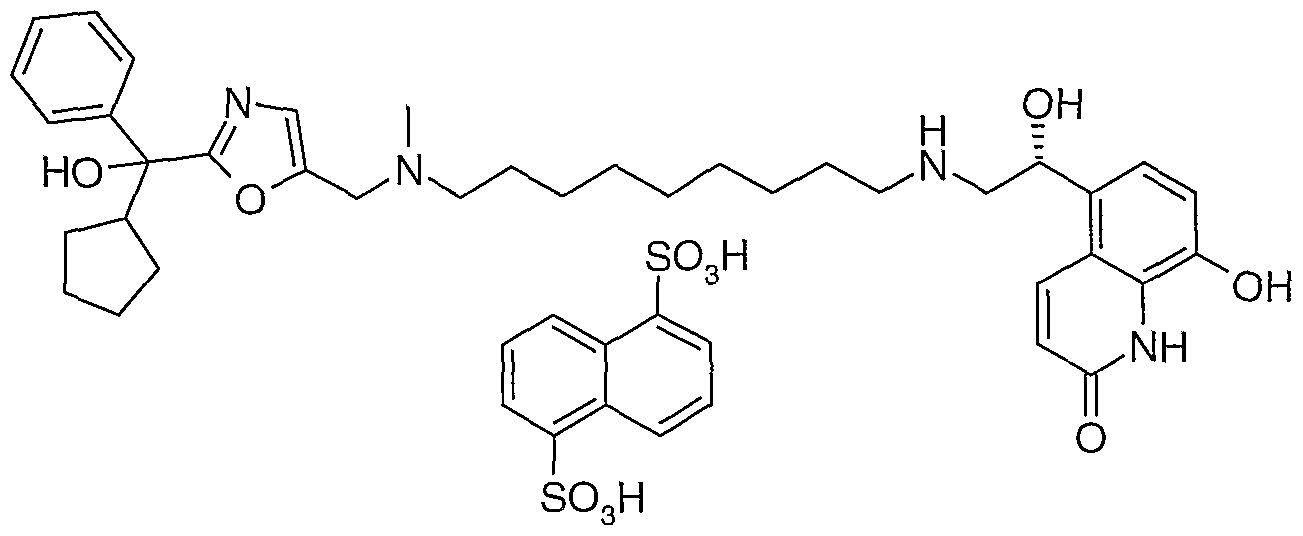
The title compound was prepared from 9-{[2-(cyclopentyl-hydroxy-phenyl-methyl)- oxazol-5-ylmethyl]-methyl-amino}-nonanal and 5-[(f?)-2-annino~1 -(terf-butyl-dimethyl- silanyloxy)-ethyl]-8-hydroxy-1 W-quinolin-2-one (prepared according to
US20040167167) by similar methods to those employed in Example 1 , steps h, i and j-
LC-MS (method 1): Rt 6.05 min, m/z 631 [MH]+.
Example 3
8-Hydroxy-5-r(R)-1-hvdroxy-2-(9-{r2-(hvdroxy-diphenyl-methyl)-oxazol-5- ylmethyli -methyl-amino}-nonylamino)-ethyll-1 H-quinolin-2-one naphthalene- 1 ,5-disulf onate

a) (5-Bromomethyl-oxazol-2-y!)-diphenyl-methanol

The title compound was prepared by methods analogous to those used in Example 1 step c and d, employing (5-methyl-oxazol-2-yl)-phenyl-methanone (Example 1 , intermediate b) and phenylmagnesium bromide.
LC-MS (method 2): Rt 3.53min, m/z 344, 346 [MH]+.
b) 9-{[2-(Hydroxy-diphenyl-methyl)-oxazol-5-ylmethyl]-methyl-amino}-nonan-1- ol

The title compound was prepared from (5-bromomethyl-oxazol-2-yl)-diphenyl- methanol (Example 3, intermediate a) and 9-methylamino-nonan-1 -ol (prepared according to JP1992-31818) by a similar method to that employed in Example 1 , steps f.
LC-MS (method 2): Rt 2.30 min, m/z 437 [MH]+.
c) 9-{[2-(Hydroxy-diphenyl-methyl)-oxazol-5-ylmethyl]-methyl-amino}-nonanal

To a cooled (0 0C) solution of 9-{[2-(hydroxy-diphenyl-methyl)-oxazol-5-ylmethyl]- methyl-amino}-nonan-1-o) (0.28 g, 0.64 mmol) in DCM (7 mL) was added DIPEA
(0.22 ml_, 1.28 ml_), then DMSO (0.17 ml_, 2.3 mmol). The reaction mixture was stirred at O 0C for 20 minutes and then treated portionwise over 10 minutes with pyridine sulphur trioxide (0.20 g, 1.28 mmol). The reaction mixture was stirred at 0 0C for a further 30 minutes and then at RT for 18 hours. Sat. NaHCO3 (aq) was added and the phases separated. The organic layer was dried (Na2SO4), filtered, and concentrated in vacuo to afford an orange oil. The residual orange oil was purified by column chromatography over silica gel eluting initially with DCM then 5% MeOH/DCM to afford the title compound.
Yield: 0.23 g. 81%
LC-MS (method 4): Rt 2.47 min, m/z 435 [MH]+.
d) [(/?)-2-(tert-Butyl-dimethylsilanyloxy)-2-(8-hydroxy-2-oxo-1,2-dihydroquinolin- 5-yl) ethyl]carbamic acid benzyl ester

A suspension of 5-[(R)-2-amino-1-(terf-butyl-dimethyl-silanyloxy)-ethyl]-8-hydroxy-1 /-/- quinolin-2-one (prepared according to US20040167167), (1.00 g, 3.0 mmol) in THF (30 ml_) was treated with N-(benzyloxycarbonyloxy)succinimide (0.75 g, 3.0 mmol) and the mixture was stirred at RT for 4 hours. After diluting with sat. ammonium chloride (aq) (100 ml_), water was added to dissolve the solid which had precipitated and the phases were separated. The aqueous was further extracted with EtOAc and the combined organics were washed with 10% citric acid (aq), satd NaHCO3 (aq) and brine, dried (Na2SO4) and cocentrated in vacuo. The resulting brown gum was purified by flash silica gel chromatography eluting with 3-5% MeOH in DCM, to give the product as a brown foam. Yield: 1.19g, 85% LC-MS (Method 2): Rt 3.79 min, m/z 469 [MH]+.
e) {(/?)-2-(tert-Butyl-dimethylsilanyloxy)-2-[8-(4-methoxybenzyloxy)-2-oxo-1 ,2- dihydroquinolin-5-yl]ethyl}carbamic acid benzyl ester

[(/?)-2-(tert-Butyl-dimethylsilanyloxy)-2-(8-hydroxy-2-oxo-1 ,2-dihydroquinolin-5- yl)ethyl] carbamic acid benzyl ester (0.50 g, 1.07 mmol) was dissolved in DMF (11 ml_) and treated with potassium carbonate (0.22 g, 1.60 mmol) and 4-methoxybenzyl chloride (0.16 ml_, 1.2 mmol). The reaction mixture was stirred at RT for 17 hours and the volatiles were evaporated. The residue was partitioned between EtOAc and water. The phases were separated and the aqueous phasewas extracted further with EtOAc. The combined organics were washed with 10% citric acid (aq), sat. NaHCO3 (aq) and brine, dried (Na2SO4) and evaporated. Purification by flash silica gel chromatography, eluting with EtOAc/cyclohexane (1 :1), afforded a pale yellow gum. Yield: 446mg, 71 % LC-MS (Method 2): Rt 4.55 min, m/z 589 [MH]+.
f) 5-[(/?)-2-Amino-1 -(tert-butyl-dimethylsilanyloxy)ethyl]-8-(4- methoxybenzyloxy)-1 H-quinolin-2-one

To a solution of {(/r?)-2-(tert-butyl-dimethylsilanyloxy)-2-[8-(4-methoxybenzyloxy)-2- oxo-1 ,2-dihydroquinolin-5-yl]ethyl}carbamic acid benzyl ester (0.41 g, 0.69 mmol) in
IMS (7 ml_) was added pyridine (28 μl, 0.35 mmol) and 5% palladium on carbon (40 mg). The flask was evacuated and back-filled with hydrogen three times. The reaction mixture was stirred under a hydrogen atmosphere for 2 days and the catalyst was removed by filtration. The volatiles were evaporated to give a bright yellow gum which was purified on an lsolute Si Il SPE cartridge (10 g) eluting with 0-10% MeOH in DCM. The desired product was obtained as a yellow solid.
Yield: 161 mg, 51 %
LC-MS (Method 3): Rt 2.72 min, m/z 455 [MH]+.
g) 5-[(R)-1-(tert-Butyl-dimethyl-silanyloxy)-2-(9-{[2-(hydroxy-diphenyl-methyl)- oxazol-5-ylmethyl]-methyl-amino}-nonylamino)-ethyl]-8-(4-methoxy-ben2yloxy)- 1 tø-quinolin-2-one

A solution in MeOH (3 mL) of 9-{[2-(hydroxy-diphenyl-methyl)-oxazol-5-ylmethyl]- methyl-amino}-nonanal (0.64 g, 1.47 mmol) was added to a solution in MeOH (12 mL) of 5-[(/:?)-2-amino-1-(tert-butyl-dimethyl-silanyloxy)-ethyl]-8-(4-methoxy- benzyloxy)-1 W-quinolin-2-one (0.67 g, 1.47 mmol) and the reaction mixture was stirred over 3A molecular sieves for 48 hours at RT. The solution was cooled to 0 0C and treated portionwise with sodium borohydride (0.11 g, 2.94 mmol) and allowed to stir at RT for 2 hours. NaHCO3 (aq) was added, the phases separated, and the organic layer was dried (Na2SO4), filtered, and concentrated in vacuo to give a pale yellow oil. The residue was re-dissolved in DCM and stirred with polystyrene-bound benzaldehyde resin to remove excess starting amine. The resin was removed by filtration and the solution was concentrated in vacuo to afford the title compound. This material was used in the next step without further purification.
Yield: 0.63 g. 49%
LC-MS (method 2): Rt 2.21 min, m/z 759 [MH]+.
h) 5-[(/iO-1-Hydroxy-2-(9-{[2-(hydroxy-diphenyl-methyl)-oxa2ol-5-ylmethyl]- methyl-amino}-nonylamino)-ethyl]-8-(4-methoxy-benzyloxy)-1 H-quinolin-2-one

The title compound was prepared from 5-[(F?)-1-(tert-butyl-dimethyl-silanyloxy)-2-(9- {[2-(hydroxy-diphenyl-methyl)-oxazol-5-ylmethyl]-methyl-amino}-nonylamino)-ethyl]-8- (4-methoxy-benzyloxy)-1 /-/-quinolin-2-one by a similar method to that employed in Example 1 , step i. The crude product was purified by HPLC (HPLC system 3), eluting with water/MeCN (+0.1% diethylamine) instead of water/MeCN (+0.1 % formic acid). LC-MS (method 2): Rt 2.21 min, m/z 759 [MH]+.
i) 8-Hydroxy-5-[(f?)-1-hydroxy-2-(9-{[2-(hydroxy-diphenyl-methyl)-oxazol-5- ylmethyl]-methyl-amino}-nonylamino)-ethyl]-1 H-quinolin-2-one

5-[(fl)-1-Hydroxy-2-(9-{[2-(hydroxy-diphenyl-methyl)-oxazol-5-ylmethyl]-methyl- amino}-nonylamino)-ethyl]-8-(4-methoxy-benzyloxy)-1 /-/-quinolin-2-one (33 mg, 43 μmol) was dissolved in a solution of 30% TFA in DCM (1.5 mL). After stirring at RT for 30 minutes the purple solution was cooled to 0 0C and treated with water (5 mL) and DCM (5 mL). To the rapidly stirring solution was cauteously added solid NaHCO3. The organic phase was separated and the aqueous phase was further extracted with DCM (5 mL). The combined organic layer was dried (Na2SO4), filtered, and concentrated in vacuo. The residue was purified by column chromatography
(Biotage Companion automated system) over silica gel eluting initially with DCM gradient eluting to 40% MeOH/DCM to afford the title compound.
Yield: 13 mg, 47%
LC-MS (Method 1 ): Rt 5.65 min, m/z 638 [MH]+
j) 8-Hydroxy-5-[(/?)-1-hydroxy-2-(9-{[2-(hydroxy-diphenyl-methyl)-oxazol-5- ylmethyl]-methyl-amino}-nonylamino)-ethyl]-1 H-quinolin-2-one naphthalene- 1,5-disulfonate

The title compound was prepared from 9-{[2-(hydroxy-diphenyl-methyl)-oxazol-5- ylmethyl]-methyl-amino}-nonanal by similar methods to those employed in Example I . step j.
LC-MS (method 1): Rt 6.05 min, m/z 630 [MH]+.
Example 4
S-rfffl-Σ-fS-ffΣ-fCvclopentyl-hvdroxy-phenyl-methvπ-oxazol-S-ylmethyll-methyl- aminoj-octylaminoVi-hydroxy-ethyll-S-hydroxy-IH-quinolin^-one diformate

a) Cyclopentyl-(5-{[(8,8-dimethoxy-octyl)-methyl-amino]-methyl}-oxazol-2-yl)- phenyl-methanol

A mixture in DMF (2 ml_) of cyclopentyl-(5-methylaminomethyl-oxazol-2-yl)-phenyl- methanol (Example 2, step b: first eluting enantiomer) (0.25 g, 0.87 mmol), 8-bromo- 1 ,1 -dimethoxy-octane (J. Med. Chem. 1989, 32(6), 1319) (0.22 g, 0.88 mmol), caesium carbonate (0.58 g, 1.78 mmol) and water (50 μl_, 2.77 mmol) was stirred at RT for 48 hours. The resulting solution was concentrated in vacuo and the residue was partitioned between water (20 m!_) and DCM (20 ml_). The aqueous layer was further extracted with DCM (20 ml_) and the combined organic layer was dried
(MgSO4), filtered, and concentrated in vacuo. The residue was purified by silica gel column chromatography (5 g lsolute cartridge), eluting with 1 % (2M NH3 in MeOH)/10% cyclohexane/89% DCM to afford the title compound. Yield: 0.32g, 80% LC-MS (Method 2): Rt 2.47 min, m/z 459 [MH]+
bJ δ-i^^Cyclopentyl-hydroxy-phenyl-methyO-oxazol-S-ylmethyll-methyl- amino}-octanal

Cyclopentyl-(5-{[(8,8-dimethoxy-octyl)-methyl-amino]-methyl}-oxazol-2-yl)-phenyl- methanol (0.32 g, 0.70 mmol) was dissolved in acetonitrile (15 ml_) and 1 M HCI (aq) (10 ml_) was added. The solution was stirred at RT for 4 hours and then the acetonitrile was removed in vacuo and the aqueous solution was basified by the addition of sat. NaHCO3 (aq). The product was extracted with DCM (3 x 50 mL) and the combined organic extracts were washed with sat. brine (20 mL), dried (MgSO4), filtered and concentrated in vacuo. The residue was purified by column
chromatography (Biotage Companion automated system) over silica gel eluting initially with (0.2% ammonia/MeOH) in DCM:cyclohexane (1 :1 ) to (2% ammonia/MeOH) in DCM:cyclohexane (1 :1) to afford the title compound as a colourless gum. Yield: 0.12 g, 40%
LC-MS (Method 2): Rt 2.32 + 2.44 min, m/z 413 [MNa]+.
c) 5-[(/?)-2-(8-{[2-(Cyclopentyl-hydroxy-phenyl-methyl)-oxa2θl-5-ylmethyl]- methyl-amino}-octylamino)-1-hydroxy-ethyl]-8-hydroxy-1 H-quinolin-2-one diformate

The title compound was prepared from 8-{[2-(cyclopentyl-hydroxy-phenyl-methyl)- oxazol-5-ylmethyl]-methyl-amino}-octanal and 5-[(F?)-2-amino-1-(tert-butyl-dimethyl silanyloxy) ethyl]-8-(4-methoxybenzyloxy)-1 H-quinolin-2-one (Example 3, intermediate f) by similar methods to those employed in Example 3, steps g, h and i.
The crude product was purified by HPLC (HPLC system 3) to give the desired product as a diformate salt, LC-MS (method 5): Rt 5.30 min, m/z 617 [MH]+.
Example 5
5-r(ft)-2-(10-{r2-(Cvclopentyl-hvdroxy-phenyl-methvπ-oxazol-5-ylmethvn-methyl- amino>-decylamino)-1-hvdroxy-ethyll-8-hvdroxy-1 /-/-quinolin-2-one diformate

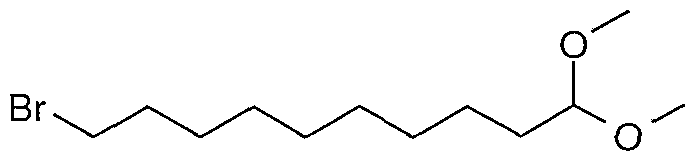
A solution of 10-bromo-decanal {Synthesis 1997, 10, 1 195) (2.16 g, 9.2 mmol) and trimethyl orthoformate (1.07 g, 10.1 mmol) in MeOH (20 ml_) was treated with 1.25 M HCI in MeOH (0.18 ml_, 0.23 mol) and heated to reflux for 4 hours. Solid NaHCO3 was added and the MeOH was removed in vacuo. The resulting solution was concentrated in vacuo and the residue was partitioned between water and DCM. The aqueous layer was further extracted with DCM and the combined organic layer was dried (Na2SO4), filtered, and concentrated in vacuo to give the title compound as a colourless oil.
Yield: 2.42 g, 93% 1H NMR (CDCI3): £ 1.22-1.37 (m, 10H), 1.42 (m, 2H), 1.58 (m, 2H), 1.85 (m,
2H), 3.31 (s, 6H), 3.40 (t, J = 6.4 Hz, 2H), 4.35 (t, J = 5.6 Hz, 1 H).
b) 5-[(f7)-2-(10-{[2-(Cyclopentyl-hydroxy-phenyl-methyl)-oxazol-5-ylmethyl]- methyl-amino}-decylamino)-1-hydroxy-ethyl]-8-hydroxy-1 H-quinolin-2-one diformate

The title compound was prepared from cyclopentyl-(5-methylaminomethyl-oxazol-2- yl)-phenyl-methanol (Example 2, step b: first eluting enantiomer), 10-bromo-1 ,1- dimethoxy-decane and 5-[(fl)-2-amino-1 -(tert-butyl-dimethylsilanyloxy) ethyl]-8-(4- methoxybenzyloxy)-1 H-quinolin-2-one (Example 3, intermediate f) by similar methods to those employed in Example 4, steps a, b and Example 3, steps g, h and i. The
crude product was purified by HPLC (HPLC system 3) to give the desired product as a diformate salt,
LC-MS (method 5): Rt 5.78 min, m/z 645 [MH]+.
Example 6 r2-(Hvdroxy-diphenyl-methyl)-oxazol-5-ylmethvn-(9-r(/?)-2-hvdroxy-2-(8- hvdroxy-2-oxo-1 ,2-dihvdro-quinolin-5-yl)-ethylamino1-nonyll-dinnethyl- ammonium diformate

a) (5-Dimethylaminomethyl-oxazol-2-yl)-diphenyl-methanol
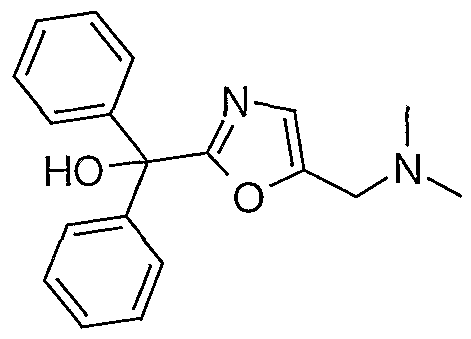
The title compound was prepared from (5-bromomethyl-oxazol-2-yl)-diphenyl- methanol (Example 3, intermediate a) and dimethylamine by a similar method to that employed in Example 2, step b.
LC-MS (method 2): Rt 0.32 & 2.07 min, m/z 309 [MH]+.
b) [2-(Hydroxy-diphenyl-methyl)-oxazol-5-ylmethyl]-dimethyl-(9-oxo-nonyl)- ammonium bromide

A solution of (5-dimethylaminomethyl-oxazol-2-yl)-diphenyl-methanol (0.30 g, 0.93 mmol) and 9-bromo-1 ,1-dimethoxy-nonane (1.04 g, 3.70 mmol) in a mixture of acetonitrile/chloroform (1 :1 ) (8 mL) was heated at 50 0C for 18 hours. The reaction mixture was diluted with DCM and the organic layer was washed with water, separated and concentrated in vacuo. The residue was purified by silica gel column chromatography (5 g lsolute cartridge), eluting with 2-5% MeOH/DCM to afford a mixture of the desired aldehyde and dimethyl acetal intermediate. Conversion to the title compound was undertaken using a similar method to that employed in Example 4, step b.
LC-MS (method 2): Rt 2.42 min, m/z 449 [MH]+.
c) [2-(Hydroxy-diphenyl-methyl)-oxazol-5-ylmethyl]-{9-[(/7)-2-hydroxy-2-(8- hydroxy-2-oxo-1 ,2-dihydro-quinolin-5-yl)-ethylamino]-nonyl}-dimethyI- ammonium formate formate

The title compound was prepared from [2-(hydroxy-diphenyl-methyl)-oxazol-5- ylmethyl]-dimethyl-(9-oxo-nonyl)-ammonium bromide and 5-[(F?)-2-amino-1-(tert- butyl-dimethylsilanyloxy) ethyl]-8-(4-methoxybenzyloxy)-1 H-quinolin-2-one (Example 3, intermediate f) by similar methods to those employed in Example 3, steps g, h and i. The crude product was purified by HPLC (HPLC system 3) to give the desired product as a diformate salt,
LC-MS (method 1): Rt 5.54 min, m/z 653 [M]+.
Example 7
8-Hvdroxy-5-r(ff)-1-hvdroxy-2-(8-{r2-(hydroxy-diphenyl-methyl)-oxazol-5- vlmethvn-methvl-aminol-octvlaminoVethyll-I H-quinolin-Σ-one formate

a) (5-Methylaminomethyl-oxazol-2-yl)-diphenyl-methanol

The title compound was prepared from (5-bromomethyl-oxazol-2-yl)-diphenyl- methano! by a similar method to that employed in Example 2, step b.
LC-MS (method 4): Rt 1.91 min, m/z 295 [MH+], 336 [MH-MeCN+].
b) 8-Hydroxy-5-[(/τf)-1-hydroxy-2-(8-{[2-(hydroxy-diphenyl-methyl)-oxazol-5- ylmethyl]-methyl-amino}-octylamino)-ethyl]-1H-quinolin-2-one formate
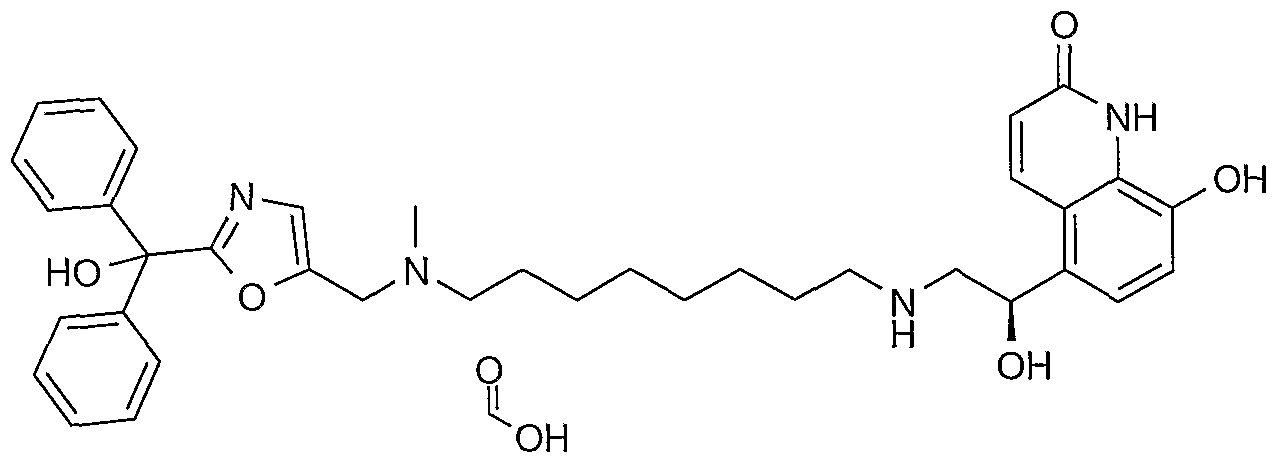
Example 8 8-Hvdroxy-5-r(ff)-1-hvdroxy-2-(10-fr2-(hvdroxy-diphenyl-methyl)-oxazol-5- ylmethyll-methyl-amino>-decylamino)-ethvn-1 H-quinolin-2-one formate

The title compound was prepared from (5-methylaminomethyl-oxazol-2-yl)-diphenyl~ methanol (Example 7, intermediate a), 10-bromo-1 ,1 -dimethoxy-decane (Example 5, intermediate a) and 5-[(R)-2-a.m\uo-λ -(tert-butyl-dimethylsϋanyloxy) ethyl]-8-(4- methoxy benzyloxy)-1 /-/-quinolin-2-one (Example 3, intermediate f) by similar methods to those employed in Example 4, steps a, b and Example 3, steps g, h and i. The crude product was purified by HPLC (HPLC system 3) to give the desired product as a formate salt.
LC-MS (method 1 ): Rt 5.94 min, m/z 653 [MH]+.
Example 9 ra-αffl-Cvclohexyl-hvdroxy-phenyl-methvπ-oxazol-δ-ylmethvn-O-rfRVa- hvdroxy-2-(8-hvdroxy-2-oxo-1,2-dihvdro-quinolin-5-vπ-ethylaminol-nonyl>- dimethyl-ammonium diformate

Example 9 was prepared using similar methods to those used for the preparation of Example 6, employing (/I?)-cyclohexyl-(5-dimethyIaminomethyl-oxazol-2-yI)-phenyl- methanol in place of (5-dimethylaminomethyl-oxazol-2-yl)-diphenyl-methanol. LC-MS (method 5): Rt 5.75 min, m/z 659 [M]+.
Example 10 r2-((/?)-Cvclohexyl-hvdroxy-phenyl-methyl)-oxazol-5-ylmethvπ-{9-r(ffl-2- hvdroxy-2-(4-hvdroxy-2-oxo-2,3-dihvdro-benzothiazol-7-yl)-ethylamino1-nonyl>- dimethyl-ammonium diformate

Example 10 was prepared using similar methods to those used for the preparation of Example 6, employing (fl)-cyclohexyl-(5-dimethylaminomethyl-oxazol-2-yl)-phenyl- methanol in place of (5-dimethylaminomethyl-oxazol-2-yl)-diphenyl-methanol and 7- ((F?)-2-amino-1 -hydroxy-ethyl)-4-hydroxy-3H-benzothiazol-2-one (prepared according to WO 2007027134) instead of 5-[(R)-2-Amino-1 -(tert-butyl-dimethylsilanyloxy)ethyl]- 8-(4-methoxybenzyloxy)-1 H-quinolin-2-one.
LC-MS (method 5): Rt 5.96 min, m/z 665 [M]+.
The compounds of the invention may be tested for pharmaceutical activity using assays know in the art, such as for example:
Biological Examples
Muscarinic Receptor Radioligand Binding Assays
Radioligand binding studies utilising [3H]-N-methyl scopolamine ([3H]-NMS) and commercially available cell membranes expressing the human muscarinic receptors (M2 and M3) were used to assess the affinity of muscarinic antagonists for M2 and M3 receptors. Membranes in TRIS buffer were incubated in 96-well plates with [3H]- NMS and M3 antagonist at various concentrations for 3 hours. Membranes and bound radioligand were then harvested by filtration and allowed to dry overnight. Scintillation fluid was then added and the bound radioligand counted using a Canberra Packard Topcount scintillation counter
The half-life of antagonists at each muscarinic receptor was measured using the alternative radioligand [3H]-QNB and an adaptation of the above affinity assay. Antagonists were incubated for 3 hours at a concentration 10-fold higher than their Ki, as determined with the [3H]-QNB ligand, with membranes expressing the human muscarinic receptors. At the end of this time, [3H]-QNB was added to a concentration 25-fold higher than its Kd for the receptor being studied and the incubation continued for various time periods from 15 minutes up to 180 minutes. Membranes and bound radioligand were then harvested by filtration and allowed to dry overnight. Scintillation fluid was then added and the bound radioligand counted using a Canberra Packard Topcount scintillation counter.
The rate at which [3H]-QNB is detected binding to the muscarinic receptors is related to the rate at which the antagonist dissociates from the receptor, ie. to the half life of the antagonists on the receptors.

M3 Binding K, < 1 nM "+++"; Ki 1 -1 OnM "++"; K, >10nM "+"; NT - Not Tested
All compounds tested in this assay exhibited binding Ki potencies greater than 2OnM. By way of further exemplification, Example 2 had a binding Ki of 0.85nM at the M3 receptor, whilst Example 7 had a binding Ki of 1.1nM.
3- Adrenergic Receptor Radioligand Binding Assays
Radioligand binding studies utilising [125l]-lodocyanopindolol and commercially available cell membranes expressing the human β2 adrenergic receptor were used to assess the affinity of antagonists for β2-adrenergic receptor. Membranes and SPA- beads were incubated with [125l]-lodocyanopindolol and β2 antagonist at various concentrations for 3 hours at room temperature in TRIS buffer. The assay was performed in 96-well plates which were read using the Wallac Microbeta counter.

β2 Binding Ki <50nM "+++"; Ki 50-10OnM "++"; Ki > 10OnM V; NT- Not Tested
All compounds tested in this assay exhibited binding Ki potencies greater than 20OnM. By way of further exemplification, Example 5 exhibited a binding Ki of 43nM, and Example 8 a binding Ki of 69nM and Example 2 had a binding Ki of 99nM..
Analysis of Inhibition of M3 Receptor Activation via Calcium Mobilization
CHO cells expressing the human M3 receptor were seeded and incubated overnight in 96 well collagen coated plates (black-wall, clear bottom) at a density of 50000 / 75μl of medium in 3% serum. The following day, a calcium-sensitive dye (Molecular Devices, Cat # R8041) was prepared in HBSS buffer with the addition of 5mM probenecid (pH 7.4). An equal volume of the dye solution (75μl) was added to the cells and incubated for 45 minutes followed by addition of 50μl of muscarinic
antagonists or vehicle. After a further 15 minutes the plate was read on a FLEXstation™ (excitation 488nm, emission 525nm) for 15 seconds to determine baseline fluorescence. The muscarinic agonist Carbachol was then added at an EC80 concentration and the fluorescence measured for a further 60 seconds. The signal was calculated by subtracting the peak response from the mean of the baseline fluorescence in control wells in the absence of antagonist. The percentage of the maximum response in the presence of antagonist was then calculated in order to generate IC50 curves. Some compounds of the invention were tested in this assay and were found to have IC50 values of <500nM. By way of further exemplification, Example 2 had an 1C50 value of of 209nM, whilst Example 7 had an IC50 value of 35nM.
Evaluation of potency and duration of action in Isolated Guinea Pig Trachea
Experiments were carried out at 370C in modified Krebs-Henseleit solution, (114mM NaCI, 15mM NaHCO3, 1mM MgSO4, 1.3mM CaCI2, 4.7mM KCI, 11.5mM glucose and 1.2mM KH2PO4 , pH 7.4) gassed with 95% O2/5% CO2. lndomethacin was added to a final concentration of 3μM
Tracheae were removed from adult male Dunkin Hartley Guinea pigs and dissected free of adherent tissue before being cut open longitudinally in a line opposite the muscle. Individual strips of 2-3 cartilage rings in width were cut and suspended using cotton thread in 10ml water-jacketed organ baths and attached to a force transducer ensuring that the tissue is located between two platinum electrodes. Responses were recorded via a MPI OOW/Ackowledge data acquisition system connected to a PC. Tissues were equilibrated for one hour under a resting tone of 1g and were then subjected to electrical field stimulation at a frequency of 80Hz with a pulse width of 0.1 ms, a unipolar pulse, triggered every 2 minutes. A "voltage-response" curve was generated for each tissue and a submaximal voltage then applied to every piece of tissue according to its own response to voltage. Tissues were washed with Krebs solution and allowed to stabilize under stimulation prior to addition of test compound. Concentration response curves were obtained by a cumulative addition of test compound in half-log increments. Once the response to each addition had reached a plateau the next addition was made. Percentage inhibition of EFS-stimulated contraction is calculated for each concentration of each compound added and dose response curves constructed using Graphpad Prism software and the EC50 calculated for each compound.
Onset time and duration of action studies were performed by adding the previously determined EC50 concentration of compound to EFS contracted tissues and the response allowed to plateau. The time taken to reach 50% of this response was determined to be the onset time. Tissues were then washed free of compound by flushing the tissue bath with fresh Krebs solution and the time taken for the contraction in response to EFS to return to 50% of the response in the presence of compound is measured. This is termed the duration of action. Some compounds of the invention were tested in this assay and were found to have EC50 values of <500nM. By way of further exemplification, Example 2 had an EC50 value of 153nM.
Alphascreen cAMP beta functional assay
Cell line
CHO-B2 (clone 11)
5000 cells/well
Stimulation Buffer pH7.4
IxHBSS
0.1 % BSA (1 mg/ml)
5mM HEPES (1/200 dilution of 1 M stock) 0.5mM IBMX (1/1000 dilution of 50OmM stock)
Lysis Buffer pH7.4 MiIIi-Q H2O 0.1% BSA (1 mg/ml) 5mM HEPES (1/200 dilution of 1 M stock) 0.3% Tween-20 (1/333 of stock)
Control: 9μM Isoproterenol (max stimulation obtained, lowest counts) Blank: 0.1 nM Isoproterenol 1 % DMSO (no stimulation, highest counts) Detection positive control: 10μM cAMP (1/200 dilution of 1OmM stock, then 1/5 in assay)
Detection negative control: stimulation buffer Standard Isoproterenol and Formoterol (EC50~7nM, 0.05nM respectively)
Compound Dilutions
Prepare compound dilutions x11 1.1 desired required final concentration in DMSO. -1/55.6 from DMSO to stimulation buffer (1.8% DMSO) -1/2 dilution in well
Streptavidin Donar bead/biotinylated cAMP detection mix (15tιl/well)
Donar bead (1/150 dilution)
Biotinylated cAMP (1/2000 dilution)
Make up in lysis buffer
Prepare at least 30 minutes before use.
Cell preparation
Remove growth medium. Add 1-2ml cell dissociation buffer for ~5mins to detach cells. Collect cells and centrifuge for 5 minutes at 1200rpm. Aspirate supernatant and resuspend pellet in stimulation buffer. Perform a cell count and adjust cell number to 2x106/ml in stimulation buffer.
Prepare immediately prior to use.
Cells/anti-cAMP Acceptor bead mix (5ul/well) Beads (1/25 dilution) Make in stimulation buffer (10U/μl stock to 0.4U/μl).
Dilute above solution 1 :1 with cells (2x106/ml) to give 0.2U/μl beads and 2000cell/μl Final well conditions: 1 U/well acceptor beads and 5000 cells/well/5μl
Addition to Plate To a 384 well Optiplate add in triplicate;
5μl cells/anti-cAMP acceptor bead mix
5μl compound or control/blank
Shake plate for 3 mins and leave in dark for 30 mins at room temperature
15μl donor beads/biotinylated cAMP mix Shake plate for 3 mins and leave in dark for 2 hours at room temperature
Read on Packard Fusion
Final Well Conditions 5μl acceptor beads/5000 cells 5μl agonist in buffer
15μl donor beads/biotinylated cAMP 25μl final volume 0.4% DMSO
Data Calculation The data is analysed using an in house Excel-Fit template.
Some of the compounds were tested in the adrenergic functional assay and were shown to have an EC50 of < 50OnM. By way of further exemplification, Example 2 had an EC50 value of 37nM.
Methacholine Induced Bronchoconstriction in vivo
Male Guinea pigs (Dunkin Hartley), weighing 500-60Og housed in groups of 5 were individually identified. Animals were allowed to acclimatize to their local surroundings for at least 5 days. Throughout this time and study time animals were allowed access to water and food ad libitum.
Guinea pigs were anaesthetized with the inhaled anaesthetic Halothane (5%). Test compound or vehicle (0.25 - 0.50 ml/kg) was administered intranasally. Animals were placed on a heated pad and allowed to recover before being returned to their home cages.
Up to 72hrs post dosing guinea pigs were terminally anaesthetized with Urethane (250μg/ml, 2ml/kg). At the point of surgical anaesthesia, the jugular vein was cannulated with a portex i.v. cannula filled with heparinised phosphate buffered saline (hPBS) (10U/ml) for i.v. administration of methacholine. The trachea was exposed and cannulated with a rigid portex cannula and the oesophagus cannulated transorally with a flexible portex infant feeding tube. The spontaneously breathing animal was then connected to a pulmonary measurement system (EMMS, Hants, UK) consisting of a flow pneumotach and a pressure transducer. The tracheal cannula was attached to a pneumotach and the oesophageal cannula attached to a pressure transducer.
The oesophageal cannula was positioned to give a baseline resistance of between 0.1 and 0.2cmH20/ml/s. A 2 minute baseline reading was recorded before i.v. administration of methacholine (up to 30μg/kg, 0.5ml/kg). A 2 minute recording of the induced constriction was taken from the point of i.v. administration.
The software calculated a peak resistance and a resistance area under the curve (AUC) during each 2 minute recording period which were used to analyse the bronchoprotective effects of test compounds
Inhibition of pilocarpine induced salivation by i.n. administered compounds.
Guinea pigs (450-55Og) supplied by Harlan UK or David Hall, Staffs UK and acclimatised to the in-house facilities for a minimum of three days before use. Guinea pigs were randomly assigned into treatment groups and weighed. Each animal was lightly anaesthetised (4% Halothane) and administered compound or vehicle intranasally (0.5ml/kg) at up to 24 hours before challenge with pilocarpine. At the test time point, guinea pigs were terminally anaesthetised with urethane (25% solution in H20, 1.5g/kg). Once sufficient anaesthesia had developed (absence of toe pinch reflex) each animal had an absorbent pad placed in the mouth for 5 minutes to dry residual saliva, this pad was removed and replaced with a new pre-weighed pad for 5 minutes to establish a reading of baseline saliva production. At the end of this 5 minute period the pad was removed and weighed. A new pre-weighed pad was inserted into the mouth before each animal received s.c. pilocarpine administered under the skin at the back of the neck (0.6mg/kg @ 2ml/kg). The pad was removed, weighed and replaced with a new pre-weighed pad every 5 minutes up to 15 minutes.
Saliva production was calculated by subtracting the pre-weighed weight of the pad from each 5 minute period post weighed pad and these numbers added together to produce an accumulation of saliva over 15 minutes. Each 5 minute period could be analysed in addition to the whole 15 minute recording period. Baseline production of saliva was assumed to be constant and multiplied by three to produce a reading for baseline saliva production over 15 minutes.
Inhibition of saliva produced by the compound could be calculated by using the following equation:
(1 -(Test-baseline)/(Veh-baseline))*100.
Claims
1. Use of a compound of formula (I):

wherein
(i) R1 is CrCe-alkyl or hydrogen; and R2 is hydrogen or a group -R7, -Z-Y-R7, -Z-NR9R10; -Z-CO-NR9R10, -Z-NR9-C(O)O-R7, or ; -Z-C(O)-R7; and R3 is a lone pair, or R3 is Ci-C6-alkyl in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge; or
(ii) R1 and R3 together with the nitrogen to which they are attached form a heterocycloalkyl ring, and R2 is a lone pair or R2 is a group -R7, -Z-Y-R7, -Z-NR9R10, -Z-CO-NR9R10, -Z-NR9-C(O)O-R7; or -Z-C(O)-R7 in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge; or
(iii) R1 and R2 together with the nitrogen to which they are attached form a heterocycloalkyl ring, said ring being substituted by a group -Y-R7, -Z-Y-R7, -Z- NR9R10; -Z-CO-NR9R10; -Z-NR9-C(O)O-R7; or ; -Z-C(O)-R7; and R3 is a lone pair, or R3 is CrC6-alkyl in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge;
R4 and R5 are independently selected from the group consisting of aryl, aryl-fused- heterocycloalkyl, heteroaryl, CrC6-alkyl, cycloalkyl;
R6 is -OH, Ci-C6-alkyl, CrC6-alkoxy hydroxy-CrC6-alkyl, nitrile, a group CONR8 2 or a hydrogen atom;
A is an oxygen or a sulfur atom;
X is a Ci-Ci2-alkylene, C2-C12-alkenylene or C3-C12-alkynylene group; R7 is an Ci-C6-alkyl, aryl, aryl-fused-cycloalkyl, aryl-fused-heterocycloalkyl, heteroaryl, aryl(CrC8-alkyl)-, heteroaryl(CrC8-alkyl)-, cycloalkyl or heterocycloalkyl group;
R8 is CrC6-alkyl or a hydrogen atom;
Z is a CrC16-alkylene, C2-Ci6-alkenylene or C2-C16-alkynylene group;
Y is a bond or oxygen atom;
R9 and R10 are independently a hydrogen atom, CrC6-alkyl, aryl, aryl-fused- heterocycloalkyl, aryl-fused-cycloalkyl, heteroaryl, aryl(CrC6-alkyl)-, or heteroaryl(Cr C6-alkyl)- group; or R9 and R10 together with the nitrogen atom to which they are attached form a heterocyclic ring of 4-8 atoms, optionally containing a further nitrogen or oxygen atom;
wherein, unless otherwise specified, each occurrence of alkyl, heterocycloalkyl, aryl, aryl-fused-heterocycloalkyl, heteroaryl, cycloalkyl, alkoxy, alkylene, alkenylene, alkynylene or aryl-fused-cycloalkyl may be optionally substituted;
or a pharmaceutically acceptable salt, solvate, N-oxide or prodrug thereof;
in the manufacture of a medicament for the treatment of prevention of a disease or condition in which M3 muscarinic receptor activity and β-adrenergic receptor activity is implicated.
2. A compound of formula (Ia):

wherein (i) R1 is CrC6-alkyl or hydrogen; and R2 is a group-Z-NR9R10; and R3 is a lone pair, or R3 is CrC6-alkyl in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge; or
(ii) R1 and R3 together with the nitrogen to which they are attached form a heterocycloalkyl ring, and R2 is a group -Z-NR9R10, in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge, or
(iii) R1 and R2 together with the nitrogen to which they are attached form a heterocycloalkyl ring, said ring being substituted by a group , -Z-NR9R10; and R3 is a lone pair, or R3 is Ci-C6-alkyl;
R4 and R5 are independently selected from the group consisting of aryl, aryl-fused- heterocycloalkyl, heteroaryl, C1-CVaIkVl, cycloalkyl;
R6 is -OH, CrC6-alkyl, CrC6-alkoxy hydroxy-CrC6-alkyl, nitrile, a group CONR8 2 or a hydrogen atom;
A is an oxygen or a sulfur atom;
X is a CrC12-alkylene, C2-C12-alkenylene or C3-C12-alkynylene group;
R8 is CrC6-alkyl or a hydrogen atom;
Z is a Cy-Cn-alkylene, C7-C1 ralkenylene or C7-C1 ralkynylene group;
or Z is a divalent linker radical of formula (A):

wherein L represents a linker comprising a hydrocarbyl chain of 7 to 11 carbon atoms, wherein the chain may additionally comprise up to three carbon-carbon double bonds, and, wherein the chain may additionally comprise up to three carbon-carbon triple bonds; L1 and L2 each independently represent hydrogen, C1-6 alky] or C3.6 cycloalkyl;
L3 and L4 each independently represent hydrogen, C1-6 alkyl or C3-6 cycloalkyl, wherein C1-6 alkyl and C3.6 cycloalkyl may be optionally substituted by one or more substituents independently selected from halogen and hydroxyl; and * denotes the point of attachement of the group of formula (I) to the non- aromatic nitrogen bearing R1 and R3, and ** denotes the point of attachment to the group NR9R10;
R9 is a hydrogen atom or CrC6-alkyl;
R10 is an aryl(Ci-C6-alkyl)-, or heteroaryl(CrC6-alkyl) group, in which the CrC6-alkyl group is optionally substituted by hydroxy;
wherein, unless otherwise specified, each occurrence of alkyl, heterocycloalkyl, aryl, aryl-fused-heterocycloalkyl, heteroaryl, cycloalkyl, alkoxy, alkylene, alkenylene and alkynylene may be optionally substituted;
or a pharmaceutically acceptable salt thereof.
3. A compound, or a pharmaceutically acceptable salt thereof, as claimed in claim 2 wherein:
R1 is d-Ce-alkyl ; R2 is a group -Z-NR9R10 and R3 is a lone pair or R3 is CrC6-alkyl, in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge, or
R1 and R2 together with the nitrogen to which they are attached represent a heterocycloalkyl ring, said ring being substituted by a group -Z-NR9R10 and R3 is a lone pair or R3 is CrC6-alkyl in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge; or R1 and R3 together with the nitrogen to which they are attached represent a heterocycloalkyl ring, and R2 is a group -Z-NR9R10 in which case the nitrogen atom to which it is attached is a quaternary nitrogen and carries a positive charge;
R4 is phenyl and R5 is selected from the group consisting of aryl, heteroaryl, C1-C6- alkyl, cycloalkyl; or
R4 and R5 are both heteroaryl; R6 is -OH or, d-Cβ-alkyl;
R8 is a hydrogen atom;
A is an oxygen or a sulfur atom;
X is a CrC2-alkylene group;
Z is a divalent linker radical of formula (A):

wherein L represents a linker comprising a hydrocarbyl chain of 7 to 11 carbon atoms, wherein the chain may additionally comprise up to three carbon-carbon double bonds, and, wherein the chain may additionally comprise up to three carbon-carbon triple bonds;
L1 and L2 each independently represent hydrogen, Ci-6 alkyl or C3.6 cycloalkyl;
L3 and L4 each independently represent hydrogen, C1-6 alkyl or C3-6 cycloalkyl, wherein Ci-6 alkyl and C3.6 cycloalkyl may be optionally substituted by one or more substituents independently selected from halogen and hydroxyl; and * denotes the point of attachement of the group of formula (I) to the non- aromatic nitrogen bearing R1 and R3, and ** denotes the point of attachment to the group NR9R10;
R9 is a hydrogen atom;
R10 is selected from the group 

4. A compound or a pharmaceutically acceptable salt thereof as claimed in claim 2 wherein R10 is a group selected from
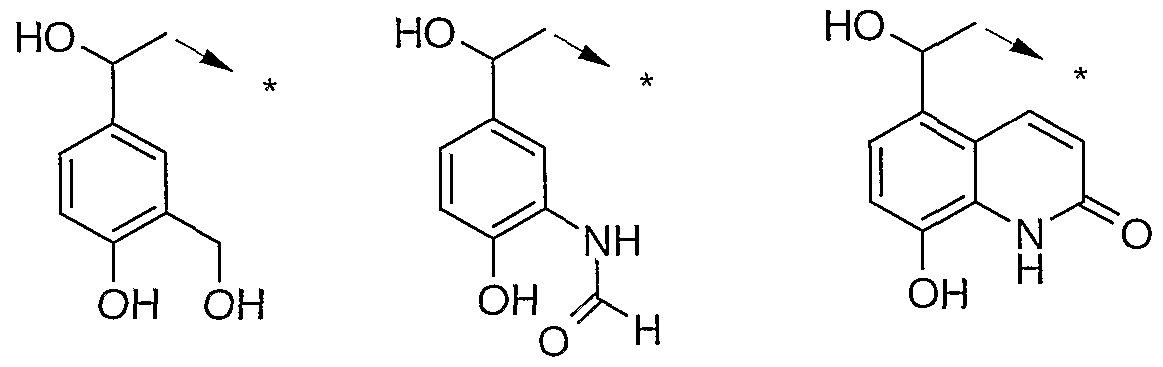

5. A compound, or a pharmaceutically acceptable salt thereof, as claimed in claim 2 or claim 4, wherein R4 and R5 are independently selected from methyl, ethyl, n- or isopropyl, n-, sec- and tertbutyl; phenyl, 3,4-methylenedioxyphenyl, 3,4- ethylenedioxyphenyl, dihydrobenzofuranyl, naphthyl; pyridyl, pyrrolyl, pyrimidinyl, oxazolyl, isoxazolyl, benzisoxazolyl, benzoxazolyl, thiazolyl, benzthiazolyl, quinolyl, thienyl, benzthienyl, furyl, benzfuryl, imidazolyl, benzimidazolyl, isothiazolyl, benzisothiazolyl, pyrazolyl, isothiazolyl, triazolyl, benztriazolyl, thiadiazolyl, oxadiazolyl, pyridazinyl, pyridazinyl, triazinyl, indolyl or indazolyl; indanyl and 1 ,2,3,4- tetrahydronaphthalenyl; cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl; and R6 is -OH, a hydrogen atom, methyl, ethyl, hydroxymethyl, nitrile, or a group CONR8 2 wherein each R8 is independently methyl, ethyl, or a hydrogen atom.
6. A compound, or a pharmaceutically acceptable salt thereof, as claimed in claim 5, wherein (i) each of R4 and R5 is thienyl; or (ii) each of R4 and R5 is phenyl; or (iii) one of R4 and R5 is phenyl and the other is cyclopentyl or cyclohexyl; or (iv) one of R4 and R5 is thienyl, and the other is cyclopentyl or cyclohexyl.
7. A compound, or a pharmaceutically acceptable salt thereof, as claimed in any one of claims 2 to 6, wherein R6 is -OH.
8. A compound, or a pharmaceutically acceptable salt thereof, as claimed in any one of claims 2 to 7, wherein R8 is hydrogen.
9. A compound, or a pharmaceutically acceptable salt thereof, as claimed in any one of claims 2 to 8, wherein A is an oxygen atom.
10. A compound, or a pharmaceutically acceptable salt thereof, as claimed in any one of claims 2 to 9, wherein X is -CH2- or -CH2CH2-.
1 1. A compound as claimed in claim 2, selected from the group consisting of 5-[(R)-2-(9-{[2-(Cyclohexyl-hydroxy-phenyl-methyl)-oxazol-5-ylmethyl]-methyl- amino}-nonylamino)-1-hydroxy-ethyl]-8-hydroxy-1 H-quinolin-2-one; 5-[(R)-2-(9-{[2-(Cyclopentyl-hydroxy-phenyl-methyl)-oxazol-5-ylmethyl]- methyl-amino} -nonylamino)-1 -hydroxy-ethyl]-8-hydroxy-1 H-quinolin-2-one;
8-Hydroxy-5-[(R)-1-hydroxy-2-(9-{[2-(hydroxy-diphenyl-methyl)-oxazol-5- ylmethyl] -methyl-amino}-nonylamino)-ethyl]-1 H-quinolin-2-one;
5-[(R)-2-(8-{[2-(Cyclopentyl-hydroxy-phenyl-methyl)-oxazol-5-ylmethyl]- methyl-amino}-octylamino)-1 -hydroxy-ethyl]-8-hydroxy-1 /-/-quinolin-2-one;
5-[(/:?)-2-(10-{[2-(CycIopentyl-hydroxy-phenyl-methyl)-oxazol-5-ylmethyl]- methyl-amino}-decylamino)-1-hydroxy-ethyl]-8-hydroxy-1 H-quinolin-2-one;
[2-(Hydroxy-diphenyl-methyl)-oxazol-5-ylmethyl]-{9-[(fl)-2-hydroxy-2-(8- hydroxy-2-oxo-1 ,2-dihydro-quinolin-5-yl)-ethylamino]-nonyl}-dimethyl-ammonium; 8-Hydroxy-5-[(f?)-1 -hydroxy-2-(8-{[2-(hydroxy-diphenyl-methyl)-oxazol-5- ylmethyl]-methyl-amino}-octylamino)-ethyl]-1 H-quinolin-2-one; 8-Hydroxy-5-[(fl)-1-hydroxy-2-(10-{[2-(hydroxy-diphenyl-methyl)-oxazol-5- ylmethyl]-methyl-amino}-decylamino)-ethyl]-1 /-/-quinolin-2-one;
[2-((R)-Cyclohexyl-hydroxy-phenyl-methyl)-oxazol-5-ylmethyl]-{9-[(R)-2- hydroxy-2-(8-hydroxy-2-oxo-1 ,2-dihydro-quinolin-5-yl)-ethylamino]-nonyl}-dimethyl- ammonium diformate; and
[2-((R)-Cyclohexyl-hydroxy-phenyl-methyl)-oxazol-5-ylmethyl]-{9-[(R)-2- hydroxy-2-(4-hydroxy-2-oxo-2,3-dihydro-benzothiazol-7-yl)-ethylamino]-nonyl}- dimethyl-ammonium diformate; or a pharmaceutically acceptable salt thereof.
12. A compound or a pharmaceutically acceptable salt thereof as claimed in any one of claims 2 to 11 , for use in therapy.
13. A pharmaceutical composition comprising a compound or a pharmaceutically acceptable salt thereof as claimed in any one of claims 2 to 12 and a pharmaceutically acceptable carrier or excipient.
14. A pharmaceutical composition as claimed in claim 13 in a form suitable for inhalation.
15. Use of a compound or a pharmaceutically acceptable salt thereof as claimed in any one of claims 2 to 11 for the manufacture of a medicament for use in the treatment of prevention of a disease or condition in which M3 muscarinic receptor activity and β-adrenergic receptor activity is implicated.
16. A method of treatment of a disease or condition in which M3 muscarinic receptor activity and β-adrenergic receptor activity is implicated comprising administration to a subject in need thereof of an effective amount of a compound or a pharmaceutically acceptable salt thereof as claimed in any one of claims 2 to 11.
17. Use as claimed in claim 15 or a method as claimed in claim 16, wherein the disease or condition is a respiratory-tract disorder.
18. Use as claimed in claim 15 or a method as claimed in claim 16, wherein the disease or condition is chronic obstructive lung disease, chronic bronchitis, asthma, chronic respiratory obstruction, bronchial hyperactivity, pulmonary fibrosis, pulmonary emphysema, or allergic rhinitis.
19. Use of a compound or a pharmaceutically acceptable salt thereof as claimed in any one of claims 2 to 1 1 in combination with (i) an inhaled corticosteroid such as fluticasone propionate, ciclesonide, mometasone furoate or budesonide and/or (ii) an inhaled PDE4 inhibitor, such as roflumilast, cilomilast or tofimilast.
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBPCT/GB2006/002956 | 2006-08-08 | ||
| PCT/GB2006/002956 WO2007017669A1 (en) | 2005-08-08 | 2006-08-08 | Azole and thiazole derivatives and their use |
| GB0702383.1 | 2007-02-07 | ||
| GB0702383A GB0702383D0 (en) | 2007-02-07 | 2007-02-07 | Chemical compound |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2008017827A2 true WO2008017827A2 (en) | 2008-02-14 |
| WO2008017827A3 WO2008017827A3 (en) | 2008-04-03 |
Family
ID=38926156
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2007/002992 Ceased WO2008017827A2 (en) | 2006-08-08 | 2007-08-07 | Azole and thiazole derivatives and their uses |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2008017827A2 (en) |
Cited By (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008096128A1 (en) * | 2007-02-07 | 2008-08-14 | Argenta Discovery Ltd | Oxazole and thiazole derivatives and their uses 2 |
| WO2008096126A1 (en) * | 2007-02-07 | 2008-08-14 | Argenta Discovery Ltd | Combination of a muscarinic receptor antagonist and a beta-2-adrenoceptor agonist |
| WO2008096143A1 (en) * | 2007-02-07 | 2008-08-14 | Argenta Discovery Ltd | Napadisylate salt of a muscarinic m3 antagonist |
| WO2008096129A1 (en) * | 2007-02-07 | 2008-08-14 | Argenta Discovery Ltd | Nitrogen containing hetrocyclic compounds useful as bifunctional modulators of m3 receptors and beta- 2 receptors |
| WO2008096136A1 (en) * | 2007-02-07 | 2008-08-14 | Argenta Discovery Ltd | Combinations with a muscarinic receptor antagonist |
| WO2008096149A3 (en) * | 2007-02-07 | 2008-10-16 | Argenta Discovery Ltd | Napadisylate salt of a muscarinic m3 antagonist |
| WO2010015792A1 (en) * | 2008-08-06 | 2010-02-11 | Argenta Discovery Limited | Nitrogen containing heterocyclic compounds useful as bifunctional modulators of m3 receptors and beta-2 receptors |
| WO2011081937A1 (en) | 2009-12-15 | 2011-07-07 | Gilead Sciences, Inc. | Corticosteroid-beta-agonist-muscarinic antagonist compounds for use in therapy |
| US8207193B2 (en) | 2006-11-14 | 2012-06-26 | Astrazeneca Ab | Quiniclidine derivatives of (hetero) arylcycloheptanecarboxylic acid as muscarinic receptor antagonists |
| US8329729B2 (en) | 2008-05-13 | 2012-12-11 | Astrazeneca Ab | Quinuclidine derivatives as muscarinic M3 receptor antagonists |
| US9233108B2 (en) | 2011-11-11 | 2016-01-12 | Almirall, S.A. | Cyclohexylamine derivatives having β2 adrenergic agonist and M3 muscarinic antagonist activities |
| US9315463B2 (en) | 2010-05-13 | 2016-04-19 | Almirall, S.A. | Cyclohexylamine derivatives having β2 adrenergic agonist and M3 muscarinic antagonist activities |
| US9518050B2 (en) | 2012-12-18 | 2016-12-13 | Almirall, S.A. | Cyclohexyl and quinuclidinyl carbamate derivatives having β2 adrenergic agonist and M3 muscarinic antagonist activity |
| US9562039B2 (en) | 2013-02-27 | 2017-02-07 | Almirall, S.A. | Salts of 2-amino-1-hydroxyethyl-8-hydroxyquinolin-2(1H)-one derivatives having both β2 adrenergic receptor agonist and M3 muscarinic receptor antagonist activities |
| US9579316B2 (en) | 2013-07-25 | 2017-02-28 | Almirall, S.A. | Salts of 2-amino-1-hydroxyethyl-8-hydroxyquinolin-2(1H)-one derivatives having both muscarinic receptor antagonist and β2 adrenergic receptor agonist activities |
| US9643961B2 (en) | 2010-05-13 | 2017-05-09 | Almirall, S.A. | Cyclohexylamine derivatives having β2 adrenergic antagonist and M3 muscarinic antagonist activities |
| WO2018108089A1 (en) | 2016-12-14 | 2018-06-21 | 北京硕佰医药科技有限责任公司 | Class of bifunctional compounds with quaternary ammonium salt structure |
| US10005771B2 (en) | 2014-09-26 | 2018-06-26 | Almirall, S.A. | Bicyclic derivatives having β2 adrenergic agonist and M3 muscarinic antagonist activities |
| US10287282B2 (en) | 2014-12-31 | 2019-05-14 | Angion Biomedica Corp. | Methods and agents for treating disease |
| US10456390B2 (en) | 2013-07-25 | 2019-10-29 | Almirall, S.A. | Combinations comprising MABA compounds and corticosteroids |
| US11459319B2 (en) | 2014-08-11 | 2022-10-04 | Angion Biomedica Corp. | Cytochrome P450 inhibitors and uses thereof |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3877470T2 (en) * | 1987-09-10 | 1993-06-24 | Merck Sharp & Dohme | OXAZOLES AND THIAZOLES FOR TREATING SENILE DEMENTIA. |
| IL88846A0 (en) * | 1988-01-08 | 1989-07-31 | Merck Sharp & Dohme | Lipophilic oxadiazoles,their preparation and pharmaceutical compositions containing them |
| AU640239B2 (en) * | 1990-06-18 | 1993-08-19 | Mitsubishi Chemical Corporation | Pyrazolecarboxamide, insecticidal and miticidal composition, and fungicidal composition for use in agriculture and horticulture |
| GB9603755D0 (en) * | 1996-02-22 | 1996-04-24 | Pfizer Ltd | Therapeutic agents |
| DE69828207T2 (en) * | 1997-10-09 | 2005-12-01 | Ortho-Mcneil Pharmaceutical, Inc. | Thiophene-substituted piperazines for the treatment of benign prostatic hyperplasia |
| US6242448B1 (en) * | 1998-12-17 | 2001-06-05 | American Home Products Corporation | Trisubstituted-oxazole derivatives as serotonin ligands |
-
2007
- 2007-08-07 WO PCT/GB2007/002992 patent/WO2008017827A2/en not_active Ceased
Cited By (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8207193B2 (en) | 2006-11-14 | 2012-06-26 | Astrazeneca Ab | Quiniclidine derivatives of (hetero) arylcycloheptanecarboxylic acid as muscarinic receptor antagonists |
| WO2008096128A1 (en) * | 2007-02-07 | 2008-08-14 | Argenta Discovery Ltd | Oxazole and thiazole derivatives and their uses 2 |
| WO2008096126A1 (en) * | 2007-02-07 | 2008-08-14 | Argenta Discovery Ltd | Combination of a muscarinic receptor antagonist and a beta-2-adrenoceptor agonist |
| WO2008096143A1 (en) * | 2007-02-07 | 2008-08-14 | Argenta Discovery Ltd | Napadisylate salt of a muscarinic m3 antagonist |
| WO2008096129A1 (en) * | 2007-02-07 | 2008-08-14 | Argenta Discovery Ltd | Nitrogen containing hetrocyclic compounds useful as bifunctional modulators of m3 receptors and beta- 2 receptors |
| WO2008096136A1 (en) * | 2007-02-07 | 2008-08-14 | Argenta Discovery Ltd | Combinations with a muscarinic receptor antagonist |
| WO2008096149A3 (en) * | 2007-02-07 | 2008-10-16 | Argenta Discovery Ltd | Napadisylate salt of a muscarinic m3 antagonist |
| AU2008212649B2 (en) * | 2007-02-07 | 2011-05-19 | Astrazeneca Ab | Combination of a muscarinic receptor antagonist and a beta-2-adrenoceptor agonist |
| US8329729B2 (en) | 2008-05-13 | 2012-12-11 | Astrazeneca Ab | Quinuclidine derivatives as muscarinic M3 receptor antagonists |
| WO2010015792A1 (en) * | 2008-08-06 | 2010-02-11 | Argenta Discovery Limited | Nitrogen containing heterocyclic compounds useful as bifunctional modulators of m3 receptors and beta-2 receptors |
| WO2011081937A1 (en) | 2009-12-15 | 2011-07-07 | Gilead Sciences, Inc. | Corticosteroid-beta-agonist-muscarinic antagonist compounds for use in therapy |
| US9315463B2 (en) | 2010-05-13 | 2016-04-19 | Almirall, S.A. | Cyclohexylamine derivatives having β2 adrenergic agonist and M3 muscarinic antagonist activities |
| US9643961B2 (en) | 2010-05-13 | 2017-05-09 | Almirall, S.A. | Cyclohexylamine derivatives having β2 adrenergic antagonist and M3 muscarinic antagonist activities |
| US9757383B2 (en) | 2011-11-11 | 2017-09-12 | Almirall, S.A. | Cyclohexylamine derivatives having β2 adrenergic agonist and M3 muscarinic antagonist activities |
| US9549934B2 (en) | 2011-11-11 | 2017-01-24 | Almirall, S.A. | Cyclohexylamine derivatives having β2 adrenergic agonist and M3 muscarinic antagonist activities |
| US9233108B2 (en) | 2011-11-11 | 2016-01-12 | Almirall, S.A. | Cyclohexylamine derivatives having β2 adrenergic agonist and M3 muscarinic antagonist activities |
| US10300072B2 (en) | 2011-11-11 | 2019-05-28 | Almirall, S.A. | Cyclohexylamine derivatives having β2 adrenergic agonist and M3 muscarinic antagonist activities |
| US9518050B2 (en) | 2012-12-18 | 2016-12-13 | Almirall, S.A. | Cyclohexyl and quinuclidinyl carbamate derivatives having β2 adrenergic agonist and M3 muscarinic antagonist activity |
| US9562039B2 (en) | 2013-02-27 | 2017-02-07 | Almirall, S.A. | Salts of 2-amino-1-hydroxyethyl-8-hydroxyquinolin-2(1H)-one derivatives having both β2 adrenergic receptor agonist and M3 muscarinic receptor antagonist activities |
| US9579316B2 (en) | 2013-07-25 | 2017-02-28 | Almirall, S.A. | Salts of 2-amino-1-hydroxyethyl-8-hydroxyquinolin-2(1H)-one derivatives having both muscarinic receptor antagonist and β2 adrenergic receptor agonist activities |
| US10456390B2 (en) | 2013-07-25 | 2019-10-29 | Almirall, S.A. | Combinations comprising MABA compounds and corticosteroids |
| US11459319B2 (en) | 2014-08-11 | 2022-10-04 | Angion Biomedica Corp. | Cytochrome P450 inhibitors and uses thereof |
| US10005771B2 (en) | 2014-09-26 | 2018-06-26 | Almirall, S.A. | Bicyclic derivatives having β2 adrenergic agonist and M3 muscarinic antagonist activities |
| US10287282B2 (en) | 2014-12-31 | 2019-05-14 | Angion Biomedica Corp. | Methods and agents for treating disease |
| US10851095B2 (en) | 2014-12-31 | 2020-12-01 | Angion Biomedica Corp. | Methods and agents for treating disease |
| US11434234B2 (en) | 2014-12-31 | 2022-09-06 | Angion Biomedica Corp. | Methods and agents for treating disease |
| WO2018108089A1 (en) | 2016-12-14 | 2018-06-21 | 北京硕佰医药科技有限责任公司 | Class of bifunctional compounds with quaternary ammonium salt structure |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2008017827A3 (en) | 2008-04-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2008017827A2 (en) | Azole and thiazole derivatives and their uses | |
| WO2010015792A1 (en) | Nitrogen containing heterocyclic compounds useful as bifunctional modulators of m3 receptors and beta-2 receptors | |
| US8148373B2 (en) | Compounds | |
| WO2008149110A1 (en) | Bicyclor [2.2.1] hept-7-ylamine derivatives and their use in the treatment of diseases and conditions in which m3 muscarinic receptor activity and beta-adrenergic activity are implicated | |
| WO2008096127A2 (en) | Bicyclo[2.2.1]hept-7-ylamine derivatives and their use as m3 muscarinic receptor modulators | |
| US7951954B2 (en) | Bezothiazol derivatives as Beta2 adrenoreceptor agonists | |
| WO2008017824A1 (en) | Bicyclo [2. 2. 1] hept-7-ylamine derivatives and their use | |
| WO2008096129A1 (en) | Nitrogen containing hetrocyclic compounds useful as bifunctional modulators of m3 receptors and beta- 2 receptors | |
| US7741360B2 (en) | Bi-aryl or aryl-heteroaryl substituted indoles | |
| AU2010202319B2 (en) | Novel benzothiazolone derivatives | |
| US7700782B2 (en) | Compounds 569 | |
| EP2225222B1 (en) | A quinoline derivative acting as a p2x7-receptor antagonist | |
| US8476265B2 (en) | Compounds-801 | |
| WO2011048409A1 (en) | Cyclic amine derivatives having beta2 adrenergic receptor agonist and muscarinic receptor antagonist activity | |
| US20090221653A1 (en) | 7-(2-amino-1-hydroxy-ethyl)-4-hydroxybenzothiazol-2(3H)-one-derivatives as beta2 adrenoreceptor agonists | |
| US20080058309A1 (en) | Novel Compounds 171 | |
| WO2010067102A1 (en) | Diazaspiro [5.5] undecane derivatives and related compounds as muscarinic-receptor antagonists and beta-adrenoreceptor agonists for the treatment of pulmonary disorders | |
| US20090203753A1 (en) | 7-(2-amino-1-hydroxy-ethyl)-4-hydroxybenzothiazol-2(3H)-one-derivatives as beta2 adrenoreceptor agonists | |
| US20130289043A1 (en) | (4-tert-butylpiperazin-2-yl)(piperazin-1-yl)methanone-n-carboxamide derivatives | |
| WO2008099165A1 (en) | Piperidine derivatives and their use for treatment of ccr8 mediated diseases | |
| US20090118288A1 (en) | N-Benzyl-Morpholine Derivatives as Modulators of the Chemokine Receptor | |
| WO2010018352A1 (en) | Heterocyclic compounds used in the treatment of diseases where enhanced m3 receptor activation is implicated | |
| WO2011154678A1 (en) | Compounds | |
| HK1148002B (en) | A quinoline derivative acting as a p2x7-receptor antagonist |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07789128 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| NENP | Non-entry into the national phase |
Ref country code: RU |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 07789128 Country of ref document: EP Kind code of ref document: A2 |