WO2008012283A1 - Preparation of (2r,3r)-3-(3-methoxyphenyl)-n,n,2-trimethylpentanamine - Google Patents
Preparation of (2r,3r)-3-(3-methoxyphenyl)-n,n,2-trimethylpentanamine Download PDFInfo
- Publication number
- WO2008012283A1 WO2008012283A1 PCT/EP2007/057559 EP2007057559W WO2008012283A1 WO 2008012283 A1 WO2008012283 A1 WO 2008012283A1 EP 2007057559 W EP2007057559 W EP 2007057559W WO 2008012283 A1 WO2008012283 A1 WO 2008012283A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- anhydride
- methoxyphenyl
- process according
- compound
- chloride
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- YHCVGGJYRMYIGG-UHFFFAOYSA-N CC(CN(C)C)C(c1cccc(OC)c1)=O Chemical compound CC(CN(C)C)C(c1cccc(OC)c1)=O YHCVGGJYRMYIGG-UHFFFAOYSA-N 0.000 description 1
- LPDJHUUWTGXTCU-UHFFFAOYSA-N CCC(c1cccc(OC)c1)=O Chemical compound CCC(c1cccc(OC)c1)=O LPDJHUUWTGXTCU-UHFFFAOYSA-N 0.000 description 1
- OHIXXXFGGLGZAB-UHFFFAOYSA-N CCO[NH+]([NH+]([O-])O)[O-] Chemical compound CCO[NH+]([NH+]([O-])O)[O-] OHIXXXFGGLGZAB-UHFFFAOYSA-N 0.000 description 1
- 0 CC[C@@]([C@@](C)CN(C)C)(c1cc(OC)ccc1)O* Chemical compound CC[C@@]([C@@](C)CN(C)C)(c1cc(OC)ccc1)O* 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C217/00—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton
- C07C217/54—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton
- C07C217/64—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains further substituted by singly-bound oxygen atoms
- C07C217/66—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains further substituted by singly-bound oxygen atoms with singly-bound oxygen atoms and six-membered aromatic rings bound to the same carbon atom of the carbon chain
- C07C217/72—Compounds containing amino and etherified hydroxy groups bound to the same carbon skeleton having etherified hydroxy groups bound to carbon atoms of at least one six-membered aromatic ring and amino groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton with amino groups linked to the six-membered aromatic ring, or to the condensed ring system containing that ring, by carbon chains further substituted by singly-bound oxygen atoms with singly-bound oxygen atoms and six-membered aromatic rings bound to the same carbon atom of the carbon chain linked by carbon chains having at least three carbon atoms between the amino groups and the six-membered aromatic ring or the condensed ring system containing that ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C213/00—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton
- C07C213/08—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton by reactions not involving the formation of amino groups, hydroxy groups or etherified or esterified hydroxy groups
Definitions
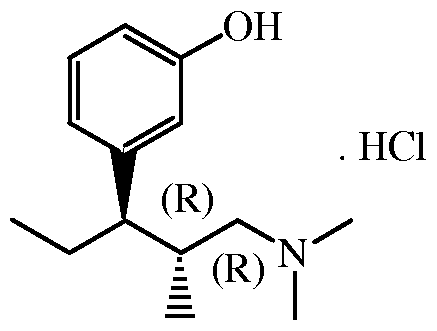
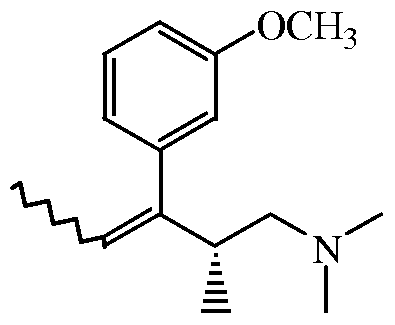
- the present invention relates to an improved process for the preparation of (2R,3R)-3- (3-methoxyphenyl)-N,N,2-trimethylpentanamine which is an intermediate for the preparation of the analgesic tapentadol.
- Tapentadol is the INN (International Non-proprietary Name) of 3-[(lR,2R)-3- (dimethylamino)-l-ethyl-2-methylpropyl]phenol monohydrochloride which compound is represented by the formula :
- the synthetic precursor of tapentadol in the above scheme is (2R,3R)-3-(3-methoxy- phenyl)-N,N,2-trimethylpentanamine (intermediate (+23) in the above scheme) which can be obtained by removing the tertiary hydroxy group of (2 l S r ,3 ⁇ )-l-(dimethylamino)- 3-(3-methoxyphenyl)-2-methyl-3-pentanol by consecutive conversion into the corresponding halogenide with thionyl chloride and subsequent removal of the Cl by treatment with zinc borohydride, zinc cyanoborohydride and/or tin cyanoborohydride.
- WO-2004/108658 discloses an alternative process for obtaining (2R,3R)-3-(3-methoxy- phenyl)-N,N,2-trimethylpentanamine by converting (2S,3S)-l-(dimethylamino)-3-(3- methoxyphenyl)-2-methyl-3-pentanol into a mixture of (2R,3R) and (2R,3S)-3-(3- methoxyphenyl)-N,N,2-trimethylpentanamine as outlined below.
- WO-2005/000788 discloses an alternative process for obtaining (2R,3R)-3-(3-methoxy- phenyl)-N,N,2-trimethylpentanamine by converting (2S,3S)-l-(dimethylamino)-3-(3- methoxyphenyl)-2-methyl-3-pentanol into a mixture of (2R,3R) and (2R,3S)-3-(3- methoxyphenyl)-N,N,2-trimethylpentanamine as outlined below.
- the object of the present invention is to provide an improved method for the synthesis of (2R,3R)-3-(3-methoxyphenyl)-N,N,2-trimethylpentanamine which is more convenient and more efficient than the previously known methods.
- - A
- the present invention achieves this object by providing an improved process for the preparation of (2R,3R)-3-(3-methoxyphenyl)-N,N,2-trimethylpentanamine, or an acid addition salt thereof, which is characterized by the steps of a) acylating (2 l S r ,3R)-l-(dimethylamino)-3-(3-methoxyphenyl)-2-methyl-3-pentanol
- the acylating agent of step a) is an organic acyl halide or organic acid anhydride selected from acetic anhydride, acetyl chloride, trifluoroacetic anhydride, chloroacetic anhydride, chloro acetylchloride, dichloroacetic anhydride, trichloroacetic anhydride, benzoic anhydride, benzoyl chloride, phthalic anhydride, phtaloyl dichloride, terephthaloyldichloride, succinic anhydride, succinyl chloride, ethyl oxalyl chloride, methyl oxalyl chloride, Meldrum's acid, ethyl chloroformate, methylchloroformate, acetylsalicyloyl chloride, or any other suitable acylating agent.
- the acylation reaction of step a) may be performed in the presence of a suitable base, such as e.g
- the catalyst of step b) is selected from a palladium catalyst, or any other suitable catalyst such as e.g. Raney nickel, platinum, platinum on carbon, ruthenium or rhodium on carbon.
- the palladium (Pd) catalyst may be a homogeneous Pd catalyst, such as for example Pd(OAc) 2 , PdCl 2 , Pd(PPh 3 ) 4 , Pd(PPh 3 ) 2 Cl 2 , Pd 2 (dba) 3 (tris(dibenzylidene acetone) dipalladium), palladium thiomethylphenylglutaramide metallacycle and the like, or a heterogeneous Pd catalyst, such as for example palladium on charcoal, palladium on metal oxides, palladium on zeolites.
- a homogeneous Pd catalyst such as for example Pd(OAc) 2 , PdCl 2 , Pd(PPh 3 ) 4 , Pd(PPh 3 ) 2 Cl 2 , Pd 2 (dba) 3 (tris(dibenzylidene acetone) dipalladium), palladium thiomethylphenylglutaramide metallacycle
- the palladium catalyst is a heterogeneous Pd catalyst, more preferably palladium on charcoal or palladium on carbon (Pd/C).
- Pd/C is a recoverable catalyst, is stable and relatively inexpensive. It can be easily separated (filtration) from the reaction mixture thereby reducing the risk of Pd traces in the final product.
- the use of Pd/C also avoids the need for ligands, such as for example phosphine ligands, which are expensive, toxic and contaminants of the synthesized products.
- the reaction-inert solvent of step b) is selected from diethyl ether, tetrahydrofuran, 2-methyltetrahydrofuran or mixtures thereof.
- steps a) and b) are executed as a "one pot synthesis" procedure.
- the present invention also relates to novel compounds of formula (III)
- the acyl group in compounds of formula (III) represents CH3-CO-, CF3-CO-, CH 2 Cl-CO-, CHCl 2 -CO-, CCl 3 -CO-, CH 3 O-CO-CO-, CH 3 O-CO-, CH 3 CH 2 O-CO-, CH 3 CH 2 O-CO-CO, phenyl-CO-, or meta-CH 3 COO-phenyl-CO- when the acylating agent used to prepared the compounds of formula (III) as set out above is selected from acetic anhydride, acetyl chloride, trifluoroacetic anhydride, chloroacetic anhydride, chloro acetylchloride, dichloroacetic anhydride, trichloroacetic anhydride, methyl oxalyl chloride, ethyl oxalyl chloride, methyl chloroformate, ethyl chloroformate, benzoic anhydride, benzoyl chloride, or
- the starting material for the process of the present invention i.e. (2S, 3R)-I- (dimethylamino)-3-(3-methoxyphenyl)-2-methyl-3-pentanol (compound 4), was prepared by reacting (2S)-3-(dimethylairrino)-l-(3-methoxyphenyl)-2-methyl-l- propanone (compound 3) with ethylmagnesium chloride in THF under Grignard reaction conditions.
- the reaction of the Grignard reagent with the ketone compound (3) introduces a second asymmetric carbon atom.
- the Grignard reaction of (2S)-3-(dimethylamino)-l-(3- methoxyphenyl)-2-methyl-l-propanone (compound 3) with an ethylmagnesium halide is highly stereospecific.
- the optical purity of the starting compound (3) was found to be 98.0%.
- Compound (4) was analysed to comprise 96.8% of the desired (2S,3R) enantiomer, less than 0.4% of the (2S,3S) enantiomer and 3.0% of the (2R,3S) enantiomer.
- Table 1 lists the stereoisomeric purity of the compound (4) when prepared as outlined above.
- Compound (4) can be converted into compound (5) by acylating compound (4) with trifluoroacetic anhydride and subsequent hydrogenolysis over a palladium catalyst, using 2-methyltetrahydrofuran as a solvent, in a "one pot synthesis" procedure.
- optical purity of starting compound (4) was 96.8% enantiomer (2S,3R). It was found that hydrogenolysis after acylation of compound (4) is highly stereospecific to give the desired enantiomer (2R,3R)-enantiomer of compound (5) with an optical purity of 96.3%.
- the reaction mixture is heated to reflux temperature and stirred and refluxed for 5 hours.
- the reaction mixture is allowed to cool to 20 0 C, and water (730 ml) and toluene (146 ml) are added.
- the upper organic layer is discarded and an aqueous NaOH solution (50% w/w, 175.2 ml) is added to the water layer while stirring for 10 minutes and keeping the temperature below 25 0 C. After 10 minutes the layers are allowed to separate, the upper organic layer is isolated and washed with water (219 ml). The organic layer is isolated and concentrated to obtain 3-(dimethylamino)-l- (3-methoxyphenyl)-2-methyl-l-propanone as on oily residue (294.9 g).
- Compound (3) prepared according to procedure of Example 2 typically has an enantiomeric purity of 97% or higher.
- Compound (4) prepared according to the procedure of Example 4 comprises 96.8% of the desired (2S,3R) enantiomer, less than 0.4% of the (2S,3S) enantiomer and 3.0% of the (2R,3S) enantiomer.
- Example 4 Synthesis of (2fl,3/O-3-(3-methoxyphenyl)-N,N,2- trimethylpentanamine (5)
- Compound (5) prepared according to the procedure of Example 5 comprises 96.3% of the desired (2R,3R) enantiomer, 2.5% of the (2S,3S) enantiomer and 1.2% of the (2R,3S) enantiomer.
- Compound (6) prepared according to the procedure of Example 6 comprises 99.7% of the desired (2R,3R) enantiomer, and 0.3% of the (2R,3S) enantiomer.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
Abstract
Description



















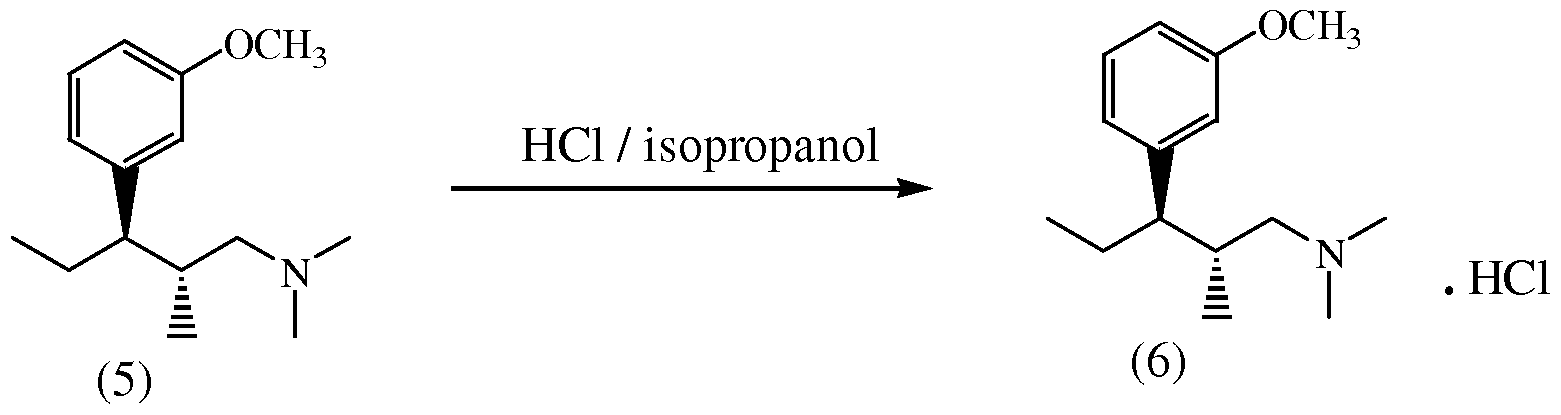
Claims




Priority Applications (18)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP07787803A EP2046726B1 (en) | 2006-07-24 | 2007-07-23 | Preparation of (2r,3r)-3-(3-methoxyphenyl)-n,n,2-trimethylpentanamine |
| DE602007004494T DE602007004494D1 (en) | 2006-07-24 | 2007-07-23 | Preparation of (2R, 3R) -3- (3-methoxy-phenyl) -N, N, 2-trimethylpentanamine |
| PL07787803T PL2046726T3 (en) | 2006-07-24 | 2007-07-23 | Preparation of (2r,3r)-3-(3-methoxyphenyl)-n,n,2-trimethylpentanamine |
| HR20100197T HRP20100197T1 (en) | 2006-07-24 | 2007-07-23 | Preparation of (2r,3r)-3-(3-methoxyphenyl)-n,n,2-trimethylpentanamine |
| HK09111685.8A HK1134280B (en) | 2006-07-24 | 2007-07-23 | Preparation of (2r,3r)-3-(3-methoxyphenyl)-n,n,2-trimethylpentanamine |
| KR1020097000899A KR101370398B1 (en) | 2006-07-24 | 2007-07-23 | Preparation of (2r,3r)-3-(3-methoxyphenyl)-n,n,2-trimethylpentanamine |
| CA2656696A CA2656696C (en) | 2006-07-24 | 2007-07-23 | Preparation of (2r,3r)-3-(3-methoxyphenyl)-n,n,2-trimethylpentanamine |
| CN2007800284726A CN101495447B (en) | 2006-07-24 | 2007-07-23 | Preparation of (2R,3R)-3-(3-methoxyphenyl)-N,N,2-trimethylpentanamine |
| JP2009521242A JP5208933B2 (en) | 2006-07-24 | 2007-07-23 | Production of (2R, 3R) -3- (3-methoxyphenyl) -N, N, 2-trimethylpentanamine |
| US12/374,894 US8138376B2 (en) | 2006-07-24 | 2007-07-23 | Preparation of (2R,3R)-3-(3-methoxyphenyl)-N,N,2-trimethylpentanamine |
| MX2009000911A MX2009000911A (en) | 2006-07-24 | 2007-07-23 | Preparation of (2r,3r)-3-(3-methoxyphenyl)-n,n,2-trimethylpentana mine. |
| AT07787803T ATE455752T1 (en) | 2006-07-24 | 2007-07-23 | PREPARATION OF (2R,3R)-3-(3-METHOXYPHENYL)-N,N,2-TRIMETHYLPENT NAMIN |
| AU2007278224A AU2007278224C1 (en) | 2006-07-24 | 2007-07-23 | Preparation of (2R,3R)-3-(3-methoxyphenyl)-N,N,2-trimethylpentanamine |
| EA200970147A EA015003B1 (en) | 2006-07-24 | 2007-07-23 | Preparation of (2r,3r)-3-(3-methoxyphenyl)-n,n,2-trimethylpentanamine |
| BRPI0714671A BRPI0714671B8 (en) | 2006-07-24 | 2007-07-23 | PREPARATION OF (2R,3R)-3-(3-METHOXYPHENYL)-N,N,2-TRIMETHYLPENTANAMINE AND INTERMEDIATE COMPOUNDS |
| DK07787803.1T DK2046726T3 (en) | 2006-07-24 | 2007-07-23 | Preparation of (2R, 3R) -3-3-Methoxyphenyl) -N, N, 2-trimethylpentanamine |
| IL196627A IL196627A (en) | 2006-07-24 | 2009-01-21 | Preparation of (2r,3r)-3-(3-methoxyphenyl)-n,n,2-trimethylpentanamine |
| US13/397,186 US8440863B2 (en) | 2006-07-24 | 2012-02-15 | Preparation of (2R,3R)-3-(3-methoxyphenyl)-N,N,2-trimethylpentanamine |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP06117708 | 2006-07-24 | ||
| EP06117708.5 | 2006-07-24 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/374,894 A-371-Of-International US8138376B2 (en) | 2006-07-24 | 2007-07-23 | Preparation of (2R,3R)-3-(3-methoxyphenyl)-N,N,2-trimethylpentanamine |
| US13/397,186 Division US8440863B2 (en) | 2006-07-24 | 2012-02-15 | Preparation of (2R,3R)-3-(3-methoxyphenyl)-N,N,2-trimethylpentanamine |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2008012283A1 true WO2008012283A1 (en) | 2008-01-31 |
Family
ID=37772663
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2007/057559 Ceased WO2008012283A1 (en) | 2006-07-24 | 2007-07-23 | Preparation of (2r,3r)-3-(3-methoxyphenyl)-n,n,2-trimethylpentanamine |
Country Status (21)
| Country | Link |
|---|---|
| US (2) | US8138376B2 (en) |
| EP (1) | EP2046726B1 (en) |
| JP (1) | JP5208933B2 (en) |
| KR (1) | KR101370398B1 (en) |
| CN (1) | CN101495447B (en) |
| AT (1) | ATE455752T1 (en) |
| AU (1) | AU2007278224C1 (en) |
| BR (1) | BRPI0714671B8 (en) |
| CA (1) | CA2656696C (en) |
| CY (1) | CY1110075T1 (en) |
| DE (1) | DE602007004494D1 (en) |
| DK (1) | DK2046726T3 (en) |
| EA (1) | EA015003B1 (en) |
| ES (1) | ES2339403T3 (en) |
| HR (1) | HRP20100197T1 (en) |
| IL (1) | IL196627A (en) |
| MX (1) | MX2009000911A (en) |
| PL (1) | PL2046726T3 (en) |
| PT (1) | PT2046726E (en) |
| SI (1) | SI2046726T1 (en) |
| WO (1) | WO2008012283A1 (en) |
Cited By (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ITMI20092110A1 (en) * | 2009-12-01 | 2011-06-02 | Archimica Srl | NEW PROCESS FOR THE PREPARATION OF TAPENTADOL AND ITS INTERMEDIATES. |
| WO2011080736A1 (en) | 2009-12-29 | 2011-07-07 | Mapi Pharma Hk Limited | Intermediate compounds and processes for the preparation of tapentadol and related compounds |
| WO2011107876A2 (en) | 2010-03-05 | 2011-09-09 | Actavis Group Ptc Ehf | Improved resolution methods for isolating desired enantiomers of tapentadol intermediates and use thereof for the preparation of tapentadol |
| WO2011092719A3 (en) * | 2010-02-01 | 2011-09-22 | Ind-Swift Laboratories Limited | Process for preparing 1 -phenyl-3 -dimethylaminopropane derivatives |
| WO2011128784A2 (en) | 2010-04-05 | 2011-10-20 | Actavis Group Ptc Ehf | Novel process for preparing highly pure tapentadol or a pharmaceutically acceptable salt thereof |
| CZ302993B6 (en) * | 2010-12-30 | 2012-02-08 | Zentiva, K.S. | Process for preparing (2R,3R)-N,N-dimethyl-3-(hydroxyphenyl)-2-methylpentylamine (tapentadol) |
| WO2012038974A1 (en) | 2010-09-20 | 2012-03-29 | Ind-Swift Laboratories Limited | Process for preparing l-phenyl-3-dimethylaminopropane derivative |
| CZ303115B6 (en) * | 2010-12-30 | 2012-04-11 | Zentiva, K.S. | Process for preparing (2R,3R)-N,N-dimethyl-3-(3-hydroxyphenyl)-2-methylpentylamine |
| WO2012089181A1 (en) * | 2010-12-30 | 2012-07-05 | Zentiva, K.S. | O-substituted (2r,3r)-3-(3-hydroxyphenyl)-2-methyl-4-pentenoic acids and a method of obtaining the same |
| WO2012089177A1 (en) * | 2010-12-30 | 2012-07-05 | Zentiva, K.S. | Method of producing (2r,3r)-na-dimethyl-3-(3-hydroxyphenyi)-2-methylpentylamine (tapentadol) |
| US8288592B2 (en) | 2009-09-22 | 2012-10-16 | Actavis Group Ptc Ehf | Solid state forms of tapentadol salts |
| WO2012146978A2 (en) | 2011-04-28 | 2012-11-01 | Actavis Group Ptc Ehf | A novel process for the preparation of tapentadol or a pharmaceutically acceptable salt thereof |
| WO2013090161A1 (en) | 2011-12-12 | 2013-06-20 | Boehringer Ingelheim International Gmbh | Stereoselective synthesis of tapentadol and its salts |
| WO2013120466A1 (en) | 2012-02-17 | 2013-08-22 | Zentiva, K.S. | A new solid form of tapentadol and a method of its preparation |
| WO2015075678A1 (en) | 2013-11-21 | 2015-05-28 | Unimark Remedies Ltd. | A novel process for the preparation of 1-phenyl-3-aminopropane derivatives |
| WO2015091068A1 (en) | 2013-12-16 | 2015-06-25 | Farma Grs, D.O.O. | Crystalline forms of tapentadol intermediate |
| WO2015117576A1 (en) | 2014-02-04 | 2015-08-13 | Zentiva, K.S. | A solid form of tapentadol maleate and a method of its preparation |
| US9446008B2 (en) | 2011-03-04 | 2016-09-20 | Gruenenthal Gmbh | Semisolid aqueous pharmaceutical composition containing tapentadol |
| US9914695B2 (en) | 2015-07-10 | 2018-03-13 | Mallinckrodt Llc | Two-step process for preparing 3-substituted phenylalkylamines |
| WO2019035775A1 (en) * | 2017-08-18 | 2019-02-21 | Saneca Pharmaceuticals A.S. | Method for the preparation of tapentadol and its intermediates |
| WO2019238267A1 (en) | 2018-06-15 | 2019-12-19 | Pharmathen S.A. | A novel process for the preparation of tapentadol |
| US10898452B2 (en) | 2016-09-23 | 2021-01-26 | Gruenenthal Gmbh | Stable formulation for parenteral administration of Tapentadol |
| US11013701B2 (en) | 2015-03-27 | 2021-05-25 | Grünenthal GmbH | Stable formulation for parenteral administration of tapentadol |
| US11547678B2 (en) | 2011-03-04 | 2023-01-10 | Gruenenthal Gmbh | Aqueous pharmaceutical formulation of tapentadol for oral administration |
| EP4116288A1 (en) | 2021-07-08 | 2023-01-11 | KRKA, d.d., Novo mesto | Racemization of (s) and/or (r)-3-(dimethylamino)-1-(3-methoxyphenyl)-2-methylpropan-1- one and its mixtures |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI401237B (en) * | 2006-07-24 | 2013-07-11 | Preparation of 3-[(1R,2R)-3-(dimethylamino)-1-ethyl-2-methylpropyl]phenol | |
| CN102002065B (en) * | 2009-09-02 | 2014-09-10 | 上海特化医药科技有限公司 | Method for preparing tapentadolhydrochloride and intermediate thereof |
| IT1401109B1 (en) * | 2010-07-02 | 2013-07-12 | Archimica Srl | NEW PROCESS FOR THE PREPARATION OF TAPENTADOL AND ITS INTERMEDIATES. |
| EP3650439B1 (en) * | 2010-07-23 | 2021-02-24 | Grünenthal GmbH | Salts or co-crystals of 3-(3-dimethylamino-1-ethyl-2-methyl-propyl)-phenol |
| CN102477016B (en) * | 2010-11-26 | 2014-12-17 | 中国医学科学院药物研究所 | Synthesis and application of intermediate of tapentadol |
| CN102617501A (en) * | 2011-01-31 | 2012-08-01 | 中国科学院上海药物研究所 | Substituted valeramide compound, preparation method and application thereof |
| CN103501775A (en) * | 2011-03-04 | 2014-01-08 | 格吕伦塔尔有限公司 | Parenteral administration of tapentadol |
| CN102206164A (en) * | 2011-04-11 | 2011-10-05 | 中国药科大学 | Method for preparing tapentadol intermediate |
| EP2674414A1 (en) | 2012-06-15 | 2013-12-18 | Siegfried AG | Method for the preparation of 1-aryl-1-alkyl-3-dialkylaminopropane compounds |
| CN103159633B (en) * | 2012-07-06 | 2015-08-12 | 江苏恩华药业股份有限公司 | The preparation method of tapentadol hydrochloride and the compound for the preparation of tapentadol hydrochloride |
| CN104725287A (en) * | 2013-12-20 | 2015-06-24 | 北京蓝贝望生物医药科技股份有限公司 | Preparation method and application of para-toluenesulfonate |
| CN106278915A (en) * | 2015-05-19 | 2017-01-04 | 重庆圣华曦药业股份有限公司 | A kind of method preparing tapentadol hydrochloride intermediate |
| AU2020245333A1 (en) * | 2019-03-28 | 2021-10-28 | Council Of Scientific And Industrial Research | Process for the preparation of tapentadol and analogs thereof |
| EP4269384A1 (en) | 2022-04-26 | 2023-11-01 | KRKA, d.d., Novo mesto | Method for crystallizing tapentadol intermediate, tapentadol intermediate of high purity, method of making tapentadol and tapentadol of high purity |
| CN119192000B (en) * | 2024-11-26 | 2025-07-15 | 苏州盛迪亚生物医药有限公司 | Method for preparing tapentadol or its pharmaceutically acceptable salt by continuous flow reaction |
| CN119176758B (en) * | 2024-11-26 | 2025-05-09 | 苏州盛迪亚生物医药有限公司 | A method for preparing tapentadol or its pharmaceutically acceptable salt by solid-support catalysis |
| CN119191999A (en) * | 2024-11-26 | 2024-12-27 | 苏州盛迪亚生物医药有限公司 | A method for preparing tapentadol or its pharmaceutically acceptable salt |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5811582A (en) * | 1996-03-13 | 1998-09-22 | Gruenenthal Gmbh | Dimethyl-(3-aryl-but-3-enyl)-amine compounds as pharmaceutical active ingredients |
| US6344558B1 (en) * | 1994-07-23 | 2002-02-05 | Gruenthal Gmbh | 1-phenyl-3-dimethylaminopropane compounds with a pharmacological effect |
| WO2004108658A1 (en) * | 2003-06-06 | 2004-12-16 | Grünenthal GmbH | Method for the production of substituted 3-aryl-butyl amine compounds |
| WO2005000788A1 (en) * | 2003-06-23 | 2005-01-06 | Grünenthal GmbH | Method for dehydrating substituted 4-dimethylamino-2-aryl-butan-2-ol compounds and method for producing substituted dimethyl-(3-aryl-butyl)-amine compounds by means of heterogeneous catalysis |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS60188333A (en) * | 1984-03-07 | 1985-09-25 | Ihara Chem Ind Co Ltd | Method for producing halogenated benzenes |
| ES2278664T3 (en) * | 2000-09-14 | 2007-08-16 | Firmenich Sa | USE OF Unsaturated ESTERS AS PERFUMING INGREDIENTS. |
| TWI401237B (en) * | 2006-07-24 | 2013-07-11 | Preparation of 3-[(1R,2R)-3-(dimethylamino)-1-ethyl-2-methylpropyl]phenol |
-
2007
- 2007-07-23 MX MX2009000911A patent/MX2009000911A/en active IP Right Grant
- 2007-07-23 BR BRPI0714671A patent/BRPI0714671B8/en active IP Right Grant
- 2007-07-23 HR HR20100197T patent/HRP20100197T1/en unknown
- 2007-07-23 CN CN2007800284726A patent/CN101495447B/en not_active Expired - Fee Related
- 2007-07-23 EP EP07787803A patent/EP2046726B1/en active Active
- 2007-07-23 JP JP2009521242A patent/JP5208933B2/en active Active
- 2007-07-23 PT PT07787803T patent/PT2046726E/en unknown
- 2007-07-23 CA CA2656696A patent/CA2656696C/en active Active
- 2007-07-23 WO PCT/EP2007/057559 patent/WO2008012283A1/en not_active Ceased
- 2007-07-23 DE DE602007004494T patent/DE602007004494D1/en active Active
- 2007-07-23 US US12/374,894 patent/US8138376B2/en active Active
- 2007-07-23 EA EA200970147A patent/EA015003B1/en not_active IP Right Cessation
- 2007-07-23 PL PL07787803T patent/PL2046726T3/en unknown
- 2007-07-23 DK DK07787803.1T patent/DK2046726T3/en active
- 2007-07-23 SI SI200730205T patent/SI2046726T1/en unknown
- 2007-07-23 KR KR1020097000899A patent/KR101370398B1/en active Active
- 2007-07-23 AT AT07787803T patent/ATE455752T1/en active
- 2007-07-23 ES ES07787803T patent/ES2339403T3/en active Active
- 2007-07-23 AU AU2007278224A patent/AU2007278224C1/en active Active
-
2009
- 2009-01-21 IL IL196627A patent/IL196627A/en active IP Right Grant
-
2010
- 2010-04-20 CY CY20101100353T patent/CY1110075T1/en unknown
-
2012
- 2012-02-15 US US13/397,186 patent/US8440863B2/en active Active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6344558B1 (en) * | 1994-07-23 | 2002-02-05 | Gruenthal Gmbh | 1-phenyl-3-dimethylaminopropane compounds with a pharmacological effect |
| US5811582A (en) * | 1996-03-13 | 1998-09-22 | Gruenenthal Gmbh | Dimethyl-(3-aryl-but-3-enyl)-amine compounds as pharmaceutical active ingredients |
| WO2004108658A1 (en) * | 2003-06-06 | 2004-12-16 | Grünenthal GmbH | Method for the production of substituted 3-aryl-butyl amine compounds |
| WO2005000788A1 (en) * | 2003-06-23 | 2005-01-06 | Grünenthal GmbH | Method for dehydrating substituted 4-dimethylamino-2-aryl-butan-2-ol compounds and method for producing substituted dimethyl-(3-aryl-butyl)-amine compounds by means of heterogeneous catalysis |
Non-Patent Citations (4)
| Title |
|---|
| DATABASE BEILSTEIN [cdrom] Beilstein Institut zur Förderung der Chemischen Wissenschaften, Frankfurt am Main, DE; 2004, XP002423030, Database accession no. RID:9554854 * |
| DATABASE BEILSTEIN [online] Beilstein Institut zur Förderung der Chemischen Wissenschaften, Farnkfurtam Main, DE; 1986, XP002423029, Database accession no. RID:2562465 * |
| DATABASE CA [online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; SPALVA, E. A.: "Relations between structure and anesthetic activity of some esters of aralkanols", XP002423028, retrieved from STN Database accession no. 1964:85583 * |
| FARMAKOLOGIYA I TOKSIKOLOGIYA (MOSCOW) , 26(5), 606-11 CODEN: FATOAO; ISSN: 0014-8318, 1963 * |
Cited By (32)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9512060B2 (en) | 2009-09-22 | 2016-12-06 | Actavis Group Ptc Ehf | Solid state forms of tapentadol salts |
| US8981154B2 (en) | 2009-09-22 | 2015-03-17 | Actavis Group Ptc Ehf | Solid state forms of tapentadol salts |
| US8288592B2 (en) | 2009-09-22 | 2012-10-16 | Actavis Group Ptc Ehf | Solid state forms of tapentadol salts |
| WO2011067714A1 (en) | 2009-12-01 | 2011-06-09 | Archimica Srl | New process for the preparation of tapentadol and intermediates thereof |
| ITMI20092110A1 (en) * | 2009-12-01 | 2011-06-02 | Archimica Srl | NEW PROCESS FOR THE PREPARATION OF TAPENTADOL AND ITS INTERMEDIATES. |
| CN102711461A (en) * | 2009-12-29 | 2012-10-03 | Mapi医药公司 | Intermediate compounds and processes for the preparation of tapentadol and related compounds |
| WO2011080736A1 (en) | 2009-12-29 | 2011-07-07 | Mapi Pharma Hk Limited | Intermediate compounds and processes for the preparation of tapentadol and related compounds |
| US8410176B2 (en) | 2009-12-29 | 2013-04-02 | Mapi Pharma Ltd. | Intermediate compounds and processes for the preparation of tapentadol and related compounds |
| WO2011092719A3 (en) * | 2010-02-01 | 2011-09-22 | Ind-Swift Laboratories Limited | Process for preparing 1 -phenyl-3 -dimethylaminopropane derivatives |
| WO2011107876A2 (en) | 2010-03-05 | 2011-09-09 | Actavis Group Ptc Ehf | Improved resolution methods for isolating desired enantiomers of tapentadol intermediates and use thereof for the preparation of tapentadol |
| WO2011128784A2 (en) | 2010-04-05 | 2011-10-20 | Actavis Group Ptc Ehf | Novel process for preparing highly pure tapentadol or a pharmaceutically acceptable salt thereof |
| WO2011128784A3 (en) * | 2010-04-05 | 2012-01-19 | Actavis Group Ptc Ehf | Novel process for preparing highly pure tapentadol or a pharmaceutically acceptable salt thereof |
| WO2012038974A1 (en) | 2010-09-20 | 2012-03-29 | Ind-Swift Laboratories Limited | Process for preparing l-phenyl-3-dimethylaminopropane derivative |
| US8552219B2 (en) | 2010-09-20 | 2013-10-08 | Ind-Swift Laboratories Limited | Process for preparing L-phenyl-3-dimethylaminopropane derivative |
| CZ302993B6 (en) * | 2010-12-30 | 2012-02-08 | Zentiva, K.S. | Process for preparing (2R,3R)-N,N-dimethyl-3-(hydroxyphenyl)-2-methylpentylamine (tapentadol) |
| WO2012089181A1 (en) * | 2010-12-30 | 2012-07-05 | Zentiva, K.S. | O-substituted (2r,3r)-3-(3-hydroxyphenyl)-2-methyl-4-pentenoic acids and a method of obtaining the same |
| CZ303115B6 (en) * | 2010-12-30 | 2012-04-11 | Zentiva, K.S. | Process for preparing (2R,3R)-N,N-dimethyl-3-(3-hydroxyphenyl)-2-methylpentylamine |
| WO2012089177A1 (en) * | 2010-12-30 | 2012-07-05 | Zentiva, K.S. | Method of producing (2r,3r)-na-dimethyl-3-(3-hydroxyphenyi)-2-methylpentylamine (tapentadol) |
| US11547678B2 (en) | 2011-03-04 | 2023-01-10 | Gruenenthal Gmbh | Aqueous pharmaceutical formulation of tapentadol for oral administration |
| US9446008B2 (en) | 2011-03-04 | 2016-09-20 | Gruenenthal Gmbh | Semisolid aqueous pharmaceutical composition containing tapentadol |
| WO2012146978A2 (en) | 2011-04-28 | 2012-11-01 | Actavis Group Ptc Ehf | A novel process for the preparation of tapentadol or a pharmaceutically acceptable salt thereof |
| WO2013090161A1 (en) | 2011-12-12 | 2013-06-20 | Boehringer Ingelheim International Gmbh | Stereoselective synthesis of tapentadol and its salts |
| WO2013120466A1 (en) | 2012-02-17 | 2013-08-22 | Zentiva, K.S. | A new solid form of tapentadol and a method of its preparation |
| WO2015075678A1 (en) | 2013-11-21 | 2015-05-28 | Unimark Remedies Ltd. | A novel process for the preparation of 1-phenyl-3-aminopropane derivatives |
| WO2015091068A1 (en) | 2013-12-16 | 2015-06-25 | Farma Grs, D.O.O. | Crystalline forms of tapentadol intermediate |
| WO2015117576A1 (en) | 2014-02-04 | 2015-08-13 | Zentiva, K.S. | A solid form of tapentadol maleate and a method of its preparation |
| US11013701B2 (en) | 2015-03-27 | 2021-05-25 | Grünenthal GmbH | Stable formulation for parenteral administration of tapentadol |
| US9914695B2 (en) | 2015-07-10 | 2018-03-13 | Mallinckrodt Llc | Two-step process for preparing 3-substituted phenylalkylamines |
| US10898452B2 (en) | 2016-09-23 | 2021-01-26 | Gruenenthal Gmbh | Stable formulation for parenteral administration of Tapentadol |
| WO2019035775A1 (en) * | 2017-08-18 | 2019-02-21 | Saneca Pharmaceuticals A.S. | Method for the preparation of tapentadol and its intermediates |
| WO2019238267A1 (en) | 2018-06-15 | 2019-12-19 | Pharmathen S.A. | A novel process for the preparation of tapentadol |
| EP4116288A1 (en) | 2021-07-08 | 2023-01-11 | KRKA, d.d., Novo mesto | Racemization of (s) and/or (r)-3-(dimethylamino)-1-(3-methoxyphenyl)-2-methylpropan-1- one and its mixtures |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2046726B1 (en) | Preparation of (2r,3r)-3-(3-methoxyphenyl)-n,n,2-trimethylpentanamine | |
| US8263809B2 (en) | Preparation of 3-[(1R,2R)-3-(dimethylamino)-1ethyl-2-methylpropyl]phenol | |
| HK1134280B (en) | Preparation of (2r,3r)-3-(3-methoxyphenyl)-n,n,2-trimethylpentanamine | |
| HK1130247B (en) | Preparation of 3-[(1r,2r)-3-(dimethylamino)-1ethyl-2-methylpropyl]phenol |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780028472.6 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07787803 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2656696 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007787803 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020097000899 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009521242 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12374894 Country of ref document: US Ref document number: MX/A/2009/000911 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007278224 Country of ref document: AU Ref document number: 719/DELNP/2009 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200970147 Country of ref document: EA |
|
| NENP | Non-entry into the national phase |
Ref country code: RU |
|
| ENP | Entry into the national phase |
Ref document number: 2007278224 Country of ref document: AU Date of ref document: 20070723 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: PI0714671 Country of ref document: BR Kind code of ref document: A2 Effective date: 20090123 |