WO2008008517A2 - Bridged diazepan orexin receptor antagonists - Google Patents
Bridged diazepan orexin receptor antagonists Download PDFInfo
- Publication number
- WO2008008517A2 WO2008008517A2 PCT/US2007/016037 US2007016037W WO2008008517A2 WO 2008008517 A2 WO2008008517 A2 WO 2008008517A2 US 2007016037 W US2007016037 W US 2007016037W WO 2008008517 A2 WO2008008517 A2 WO 2008008517A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- diazabicyclo
- methyl
- triazol
- benzoyl
- substituted
- Prior art date
Links
- 0 *C(N1CC(CC2)N(*)C2CC1)=O Chemical compound *C(N1CC(CC2)N(*)C2CC1)=O 0.000 description 2
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/08—Bridged systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/08—Bridged systems
Definitions
- the orexins (hypocretins) comprise two neuropeptides produced in the hypothalamus: the orexin A (OX-A) (a 33 amino acid peptide) and the orexin B (OX-B) (a 28 amino acid peptide) (Sakurai T. et al., Cell, 1998, 92, 573-585). Orexins are found to stimulate food consumption in rats suggesting a physiological role for these peptides as mediators in the central feedback mechanism that regulates feeding behaviour (Sakurai T. et al., Cell, 1998, 92, 573-585).
- Orexins also regulate states of sleep and wakefulness opening potentially novel therapeutic approaches for narcoleptic or insomniac patients (Chemelli R.M. et al., Cell, 1999, 98, 437-451).
- Two orexin receptors have been cloned and characterized in mammals. They belong to the super family of G-protein coupled receptors (Sakurai T. et al., Cell, 1998, 92, 573- 585): the orexin-1 receptor (OX or OXlR) is selective for OX-A and the orexin-2 receptor (OX2 or OX2R) is capable to bind OX-A as well as OX-B.
- the physiological actions in which orexins are presumed to participate are thought to be expressed via one or both of OX 1 receptor and OX 2 receptor as the two subtypes of orexin receptors.
- Orexin receptors are found in the mammalian brain and may have numerous implications in pathologies such as depression; anxiety; addictions; obsessive compulsive disorder; affective neurosis; depressive neurosis; anxiety neurosis; dysthymic disorder; behaviour disorder; mood disorder; sexual dysfunction; psychosexual dysfunction; sex disorder; schizophrenia; manic depression; delirium; dementia; severe mental retardation and dyskinesias such as Huntington's disease and Tourette syndrome; eating disorders such as anorexia, bulimia, cachexia, and obesity; addictive feeding behaviors; binge/purge feeding behaviors; cardiovascular diseases; diabetes; appetite/taste disorders; emesis, vomiting, nausea; asthma; cancer; Parkinson's disease; Cushing's syndrome/disease; basophile adenoma; prolactinoma; hyperprolactinemia; hypophysis tumour/adenoma; hypothalamic diseases; inflammatory bowel disease; gastric diskinesia; gastric ulcers; Froehlich'
- HIV post-chemotherapy pain; post-stroke pain; post-operative pain; neuralgia; emesis, nausea, vomiting; conditions associated with visceral pain such as irritable bowel syndrome, and angina; migraine; urinary bladder incontinence e.g. urge incontinence; tolerance to narcotics or withdrawal from narcotics; sleep disorders; sleep apnea; narcolepsy; insomnia; parasomnia; jet lag syndrome; and neurodegenerative disorders including nosological entities such as disinhibition-dementia-parkinsonism-amyotrophy complex; pallido-ponto-nigral degeneration; epilepsy; seizure disorders and other diseases related to general orexin system dysfunction.
- Certain orexin receptor antagonists are disclosed in PCT patent publications WO
- the present invention is directed to diazepan compounds which are antagonists of orexin receptors, and which are useful in the treatment or prevention of neurological and psychiatric disorders and diseases in which orexin receptors are involved.
- the invention is also directed to pharmaceutical compositions comprising these compounds and the use of these compounds and compositions in the prevention or treatment of such diseases in which orexin receptors are involved.
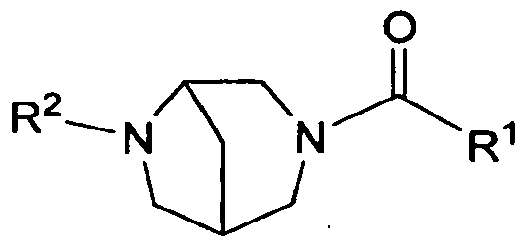
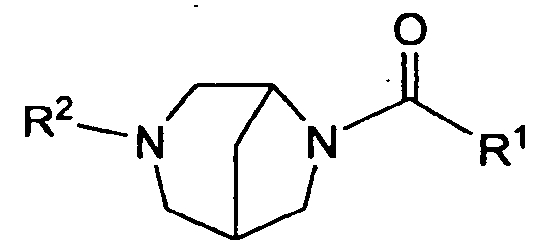
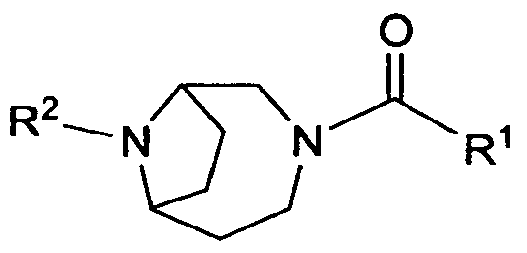
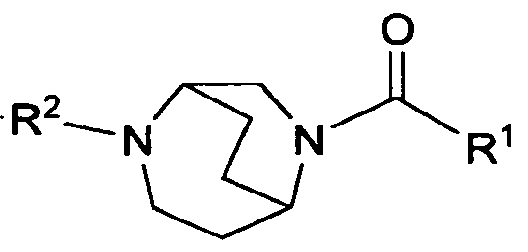
- the present invention is directed to compounds of the formula I:
- a is 0 or 1
- b is 0 or 1
- m is 0 or 1
- p is 1 or 2;
- Rl is selected from the group consisting of: ( 1 ) phenyl, where the phenyl is substituted with Rl &, Rl b and Rl c,
- heteroaryl where the heteroaryl is substituted with Rl a, Rib and Rl C;
- R2 is heteroaryl, where the heteroaryl is substituted with R2a, R2b and R2c;
- Rla, RIb 3 Rl c ; R2a, R2b and R2c are independently selected from the group consisting of:
- RlO and Rl 1 are independently selected from the group consisting of: (a) hydrogen,
- Ci-6alkyl which is unsubstituted or substituted with one or more substituents selected from Rl 3,
- R 14 is selected from the group consisting of:
- Rl and R2 are defined herein; or a pharmaceutically acceptable salt thereof.
- Rl and R2 are defined herein; or a pharmaceutically acceptable salt thereof.
- An embodiment of the present invention includes compounds wherein X is -SO2-.
- An embodiment of the present invention includes compounds wherein
- An embodiment of the present invention includes compounds wherein Rl is phenyl, which is unsubstituted or substituted with one or more of:
- Ci-6alkyl which is unsubstituted or substituted with one or more substituents selected from Rl 3,
- Rl is phenyl, which is unsubstituted or substituted with one or more methyl, -CF3, halo, -OCF3, -OCH3, -OCH2CH3, -CO2CH3, -CN, -N(CH3), -NH(CH2CH3), -NO 2 , triazolyl or phenyl.
- Rl is phenyl, which is unsubstituted or substituted with one or more methyl, -CF3, chloro, fluro, -OCF3, -OCH3, -OCH2CH3, -CO2CH3, triazolyl or phenyl.
- Rl is phenyl, which is unsubstituted or substituted with one or more methyl, -CF3, fluro, -OCF3, -OCH3, -CO2CH3 or phenyl.
- An embodiment of the present invention includes compounds wherein Rl is phenyl.
- An embodiment of the present invention includes compounds wherein Rl is 2,6-dimethoxyphenyl.
- An embodiment of the present invention includes compounds wherein R2 is heteroaryl, which is unsubstituted or substituted with one or more of: (1) halogen,
- heterocycle where the heterocycle is unsubstituted or substituted with one or more substituents selected from Rl 3,
- RlO and Rl 1 are independently selected from the group consisting of:
- An embodiment of the present invention includes compounds wherein R2 is heteroaryl, which is unsubstituted or substituted with halogen, hydroxyl, Ci-6alkyl, -O-Cl_6alkyl or phenyl.
- R2 is selected from the group consisting of:
- R2 is selected from the group consisting of:
- R2 is benzothiazolyl, which is unsubstituted or substituted with chloro.
- An embodiment of the present invention includes compounds wherein R2 is 6-chloro-benzothiazolyl.
- An embodiment of the present invention includes compounds wherein R2 is benzoxazolyl.
- An embodiment of the present invention includes compounds wherein R2 is quinazolinyl or quinolinyl.
- Specific embodiments of the present invention include a compound which is selected from the group consisting of the subject compounds of the Examples herein or a pharmaceutically acceptable salt thereof.
- the compounds of the present invention may contain one or more asymmetric centers and can thus occur as racemates and racemic mixtures, single enantiomers, diastereomeric mixtures and individual diastereomers. Additional asymmetric centers may be present depending upon the nature of the various substituents on the molecule. Each such asymmetric center will independently produce two optical isomers and it is intended that all of the possible optical isomers and diastereomers in mixtures and as pure or partially purified compounds are included within the ambit of this invention. The present invention is meant to comprehend all such isomeric forms of these compounds.
- Formula I shows the structure of the class of compounds without preferred stereochemistry.
- the coupling reaction is often the formation of salts using an enantiomerically pure acid or base.
- the diasteromeric derivatives may then be converted to the pure enantiomers by cleavage of the added chiral residue.
- the racemic mixture of the compounds can also be separated directly by chromatographic methods utilizing chiral stationary phases, which methods are well known in the art.
- any enantiomer of a compound may be obtained by stereoselective synthesis using optically pure starting materials or reagents of known configuration by methods well known in the art.
- Ci- ⁇ alkyl is defined to identify the group as having 1, 2, 3, 4, 5 or 6 carbons in a linear or branched arrangement, such that Ci_8alkyl specifically includes methyl, ethyl, n-propyl, iso-propyl, n-butyl, iso-butyl, tert- butyl, pentyl, and hexyl.
- a group which is designated as being independently substituted with substituents may be independently substituted with multiple numbers of such substituents.
- heterocycle includes both unsaturated and saturated heterocyclic moieties, wherein the unsaturated heterocyclic moieties (i.e. "heteroaryl”) include benzoimidazolyl, benzimidazolonyl, benzofuranyl, benzofurazanyl, benzopyrazolyl, benzothiazolyl, benzotriazolyl, benzothiophenyl, benzoxazepin, benzoxazolyl, carbazolyl, carbolinyl, cinnolinyl, furanyl, imidazolyl, indolinyl, indolyl, indolazinyl, indazolyl, isobenzofuranyl, isoindolyl, isoquinolyl, isothiazolyl, isoxazolyl, naphthpyridinyl, oxadiazolyl, oxazolyl, oxazoline, isoxazoline, oxazoline, oxazoline,
- salts refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic or organic bases and inorganic or organic acids.
- Salts derived from inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic salts, manganous, potassium, sodium, zinc, and the like. Particularly preferred are the ammonium, calcium, magnesium, potassium, and sodium salts. Salts in the solid form may exist in more than one crystal structure, and may also be in the form of hydrates.
- Salts derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, and basic ion exchange resins, such as arginine, betaine, caffeine, choline, N,N'-dibenzylethylene-diamine, diethylamine, 2- diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethyl- morpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine, and the like.
- basic ion exchange resins
- salts may be prepared from pharmaceutically acceptable non-toxic acids, including inorganic and organic acids.
- acids include acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethanesulfonic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, nitric, pamoic, pantothenic, phosphoric, succinic, sulfuric, tartaric, p- toluenesulfonic acid, and the like.
- Specific compounds within the present invention include a compound which selected from the group consisting of the compounds disclosed in the following Examples and pharmaceutically acceptable salts thereof and individual diastereomers thereof.
- the subject compounds are useful in a method of antagonizing orexin receptor activity in a patient such as a mammal in need of such inhibition comprising the administration of an effective amount of the compound.
- the present invention is directed to the use of the compounds disclosed herein as antagonists of orexin receptor activity. In addition to primates, especially humans, a variety of other mammals can be treated according to the method of the present invention.
- the present invention is directed to a compound of the present invention or a pharmaceutically acceptable salt thereof for use in medicine.
- the present invention is further directed to a use of a compound of the present invention or a pharmaceutically acceptable salt thereof for the manufacture of a medicament for antagonizing orexin receptor activity or treating the disorders and diseases noted herein in humans and animals.
- the subject treated in the present methods is generally a mammal, preferably a human being, male or female.
- the term "therapeutically effective amount” means the amount of the subject compound that will elicit the biological or medical response of a tissue, system, animal or human that is being sought by the researcher, veterinarian, medical doctor or other clinician. It is recognized that one skilled in the art may affect the neurological and psychiatric . disorders by treating a patient presently afflicted with the disorders or by prophylactically treating a patient afflicted with the disorders with an effective amount of the compound of the present invention.
- treatment and “treating” refer to all processes wherein there may be a slowing, interrupting, arresting, controlling, or stopping of the progression of the neurological and psychiatric disorders described herein, but does not necessarily indicate a total elimination of all disorder symptoms, as well as the prophylactic therapy of the mentioned conditions, particularly in a patient who is predisposed to such disease or disorder.
- administration of and or “administering a” compound should be understood to mean providing a compound of the invention or a prodrug of a compound of the invention to the individual in need thereof.
- composition as used herein is intended to encompass a product comprising the specified ingredients in the specified amounts, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts.
- Such term in relation to pharmaceutical composition is intended to encompass a product comprising the active ingredient(s), and the inert ingredient(s) that make up the carrier, as well as any product which results, directly or indirectly, from combination, complexation or aggregation of any two or more of the ingredients, or from dissociation of one or more of the ingredients, or from other types of reactions or interactions of one or more of the ingredients.
- the pharmaceutical compositions of the present invention encompass any composition made by admixing a compound of the present invention and a pharmaceutically acceptable carrier.
- pharmaceutically acceptable it is meant the carrier, diluent or excipient must be compatible with the other ingredients of the formulation and not deleterious to the recipient thereof.
- CHO cells expressing the rat orexin- 1 receptor or the human orexin-2 receptor, are grown in Iscove's modified DMEM containing 2 mM L-glutamine, 0.5 g/ml G418, 1% hypoxanthine-thymidine supplement, 100 U/ml penicillin, 100 ug/ml streptomycin and 10 % heat-inactivated fetal calf serum (FCS).
- FCS heat-inactivated fetal calf serum
- the cells are seeded at 20,000 cells / well into Becton-Dickinson black 384-well clear bottom sterile plates coated with poly-D- lysine. All reagents were from GIBCO-Invitrogen Corp.
- Ala 6 ' 12 human orexin-A as the agonist is prepared as a 1 mM stock solution in 1% bovine serum albumin (BSA) and diluted in assay buffer (HBSS containing 20 mM HEPES, 0.1% BSA and 2.5mM probenecid, pH7.4) for use in the assay at a final concentration of 7OpM.
- Test compounds are prepared as 10 mM stock solution in DMSO, then diluted in 384-well plates, first in DMSO, then assay buffer.
- Fluorescence is measured for each well at 1 second intervals for 5 minutes and the height of each fluorescence peak is compared to the height of the fluorescence peak induced by 70 pM Ala 6 ' 12 orexin-A with buffer in place of antagonist.
- IC50 value the concentration of compound needed to inhibit 50 % of the agonist response
- the intrinsic orexin receptor antagonist activity of a compound which may be used in the present invention may be determined by these assays.
- the compounds of the following examples had activity in antagonizing the rat orexin- 1 receptor and/or the human orexin-2 receptor in the aforementioned assays, generally with an IC50 of less than about 50 ⁇ M.
- Preferred compounds within the present invention had activity in antagonizing the rat orexin- 1 receptor and/or the human orexin- 2 receptor in the aforementioned assays with an IC50 of less than about 100 nM. Such a result is indicative of the intrinsic activity of the compounds in use as antagonists of orexin- 1 receptor and/or the orexin-2 receptor.
- the present invention also includes compounds within the generic scope of the invention which possess activity as agonists of the orexin- 1 receptor and/or the orexin-2 receptor. With respect to other diazepan compounds, the present compounds exhibit unexpected properties, such as with respect to increased oral bioavailability, metabolic stability, and/or selectivity with respect to other receptors.
- the orexin receptors have been implicated in a wide range of biological functions. This has suggested a potential role for these receptors in a variety of disease processes in humans or other species.
- the compounds of the present invention have utility in treating, preventing, ameliorating, controlling or reducing the risk of a variety of neurological and psychiatric disorders associated with orexin receptors, including one or more of the following conditions or diseases: sleep disorders, sleep disturbances, including enhancing sleep quality, improving sleep quality, increasing sleep efficiency, augmenting sleep maintenance; increasing - the value which is calculated from the time that a subject sleeps divided by the time that a subject is attempting to sleep; improving sleep initiation; decreasing sleep latency or onset (the time it takes to fall asleep); decreasing difficulties in falling asleep; increasing sleep continuity; decreasing the number of awakenings during sleep; decreasing intermittent wakings during sleep; decreasing nocturnal arousals; decreasing the time spent awake following the initial onset of sleep; increasing the total amount of sleep; reducing the fragmentation of sleep; altering the timing, frequency or duration of REM
- the present invention provides methods for: enhancing the quality of sleep; augmenting sleep maintenance; increasing REM sleep; increasing stage 2 sleep; decreasing fragmentation of sleep patterns; treating insomnia; enhancing cognition; increasing memory retention; treating or controlling obesity; treating or controlling depression; treating, controlling, ameliorating or reducing the risk of epilepsy, including absence epilepsy; treating or controlling pain, including neuropathic pain; treating or controlling Parkinson's disease; treating or controlling psychosis; or treating, controlling, ameliorating or reducing the risk of schizophrenia, in a mammalian patient in need thereof which comprises administering to the patient a therapeutically effective amount of a compound of the present invention.
- the subject compounds are further useful in a method for the prevention, treatment, control, amelioration, or reducation of risk of the diseases, disorders and conditions noted herein.
- the dosage of active ingredient in the compositions of this invention may be varied, however, it is necessary that the amount of the active ingredient be such that a suitable dosage form is obtained.
- the active ingredient may be administered to patients (animals and human) in need of such treatment in dosages that will provide optimal pharmaceutical efficacy.
- the selected dosage depends upon the desired therapeutic effect, on the route of administration, and on the duration of the treatment.
- the dose will vary from patient to patient depending upon the nature and severity of disease, the patient's weight, special diets then being followed by a patient, concurrent medication, and other factors which those skilled in the art will recognize.
- dosage levels of between 0.0001 to 10 mg/kg. of body weight daily are administered to the patient, e.g., humans and elderly humans, to obtain effective antagonism of orexin receptors.
- the dosage range will generally be about 0.5 mg to 1.0 g. per patient per day which may be administered in single or multiple doses.
- the dosage range will be about 0.5 mg to 500 mg per patient per day; more preferably about 0.5 mg to 200 mg per patient per day; and even more preferably about 5 mg to 50 mg per patient per day.
- Pharmaceutical compositions of the present invention may be provided in a solid dosage formulation preferably comprising about 0.5 mg to 500 mg active ingredient, more preferably comprising about 1 mg to 250 mg active ingredient.
- the pharmaceutical composition is preferably provided in a solid dosage formulation comprising about 1 mg, 5 mg, 10 mg, 25 mg, 50 mg, 100 mg, 200 mg or 250 mg active ingredient.
- the compositions are preferably provided in the form of tablets containing 1.0 to 1000 milligrams of the active ingredient, particularly 1, 5, 10, 15, 20, 25, 50, 75, 100, 150, 200, 250, 300, 400, 500, 600, 750, 800, 900, and 1000 milligrams of the active ingredient for the symptomatic adjustment of the dosage to the patient to be treated.
- the compounds may be administered on a regimen of 1 to 4 times per day, preferably once or twice per day.
- the compounds of the present invention may be used in combination with one or more other drugs in the treatment, prevention, control, amelioration, or reduction of risk of diseases or conditions for which compounds of the present invention or the other drugs may have utility, where the combination of the drugs together are safer or more effective than either drug alone.
- Such other drug(s) may be administered, by a route and in an amount commonly used therefor, contemporaneously or sequentially with a compound of the present invention.
- a pharmaceutical composition in unit dosage form containing such other drugs and the compound of the present invention is preferred.
- the combination therapy may also includes therapies in which the compound of the present invention and one or more other drugs are administered on different overlapping schedules.
- the compounds of the present invention and the other active ingredients may be used in lower doses than when each is used singly.
- the pharmaceutical compositions of the present invention include those that contain one or more other active ingredients, in addition to a compound of the present invention.
- the above combinations include combinations of a compound of the present invention not only with one other active compound, but also with two or more other active compounds.
- compounds of the present invention may be used in combination with other drugs that are used in the prevention, treatment, control, amelioration, or reduction of risk of the diseases or conditions for which compounds of the present invention are useful.
- Such other drugs may be administered, by a route and in an amount commonly used therefor, contemporaneously or sequentially with a compound of the present invention.
- a pharmaceutical composition containing such other drugs in addition to the compound of the present invention is preferred.
- the pharmaceutical compositions of the present invention include those that also contain one or more other active ingredients, in addition to a compound of the present invention.
- the weight ratio of the compound of the compound of the present invention to the second active ingredient may be varied and will depend upon the effective dose of each ingredient. Generally, an effective dose of each will be used. Thus, for example, when a compound of the present invention is combined with another agent, the weight ratio of the compound of the present invention to the other agent will generally range from about 1000:1 to about 1:1000, preferably about 200:1 to about 1 :200. Combinations of a compound of the ⁇ present invention and other active ingredients will generally also be within the aforementioned range, but in each case, an effective dose of each active ingredient should be used. In such combinations the compound of the present invention and other active agents may be administered separately or in conjunction. In addition, the administration of one element may be prior to, concurrent to, or subsequent to the administration of other agent(s).
- the compounds of the present invention may be administered in conbination with other compounds which are known in the art to be useful for enhancing sleep quality and preventing and treating sleep disorders and sleep disturbances, including e.g., sedatives, hypnotics, anxiolytics, antipsychotics, antianxiety agents, antihistamines, benzodiazepines, barbiturates, cyclopyrrolones, GABA agonists, 5HT-2 antagonists including 5HT-2A antagonists and 5HT-2A/2C antagonists, histamine antagonists including histamine H3 antagonists, histamine H3 inverse agonists, imidazopyridines, minor tranquilizers, melatonin agonists and antagonists, melatonergic agents, other orexin antagonists, orexin agonists, prokineticin agonists and antagonists, pyrazolopyrimidines, T-type calcium channel antagonists, triazolopyridines, and the like, such as: adinazolam, allobar
- the subject compound may be employed in combination with other compounds which are known in the art, either administered separately or in the same pharmaceutical compositions, include, but are not limited to: insulin sensitizers including (i) PPAR ⁇ antagonists such as glitazones (e.g.
- ciglitazone darglitazone; englitazone; isaglitazone (MCC-555); pioglitazone; rosiglitazone; troglitazone; tularik; BRL49653; CLX-0921 ; 5-BTZD), GW-0207, LG- 100641, and LY-300512, and the like);
- biguanides such as metformin and phenformin
- insulin or insulin mimetics such as biota, LP-100, novarapid, insulin detemir, insulin lispro, insulin glargine, insulin zinc suspension (lente and ultralente); Lys-Pro insulin, GLP-I (73-7) (insulintropin); and GLP-I (7-36)-NH2)
- sulfonylureas such as acetohexamide; chlorpropamide; diabinese; glibenclamide; glipizide; g
- CNTF Central neurotrophic factors
- GI-181771 Gaxo-SmithKline
- SR146131 Sanofi Synthelabo
- butabindide PD170,292, and PD 149164 (Pfizer)
- CNTF derivatives such as axokine (Regeneron)
- monoamine reuptake inhibitors such as sibutramine
- UCP-I uncoupling protein- 1
- activators such as phytanic acid, 4-[(E)-2-(5,6,7,8-tetrahydro-5,5,8,8-tetramethyl-2-napthalenyl)-l- propenyl]benzoic acid (TTNPB), retinoic acid
- thyroid hormone ⁇ agonists such as KB- 2611 (KaroBioBMS)
- FAS fatty acid synthase inhibitors, such as Cerulen
- dipeptidyl peptidase IV (DP-IV) inhibitors such as isoleucine thiazolidide, valine pyrrolidide, NVP-DPP728, LAF237, MK-431 , P93/01, TSL 225, TMC-2A/2B/2C, FE 999011, P9310/K364, VIP 0177, SDZ 274-444; (46) dicarboxylate transporter inhibitors; (47) glucose transporter inhibitors; (48) phosphate transporter inhibitors; (49) Metformin (Glucophage®); and (50) Topiramate (Topimax®); and (50) peptide YY, PYY 3-36, peptide YY analogs, derivatives, and fragments such as BIM- 43073D, BIM-43004C (Olitvak, D.A. et al., Dig. Dis. Sci. 44(3):643-
- Neuropeptide Y2 (NPY2) receptor agonists such NPY3-36, N acetyl [Leu(28,31)] NPY 24-36, TASP-V, and cyclo-(28/32)-Ac-[Lys28-Glu32]-(25-36)-pNPY;
- Neuropeptide Y4 (NPY4) agonists such as pancreatic peptide (PP), and other Y4 agonists such as 1229U91;
- cyclooxygenase-2 inhibitors such as etoricoxib, celecoxib, valdecoxib, parecoxib, lumiracoxib, BMS347070, tiracoxib or JTE522, ABT963, CS502 and GW406381, and pharmaceutically acceptable salts thereof;
- Neuropeptide Yl (NPYl) antagonists such as BIBP3226, J-115814, BIBO 3304, LY-357897, CP-671906, GI
- the subject compound may be employed in combination with an anti-depressant or anti-anxiety agent, including norepinephrine reuptake inhibitors (including tertiary amine tricyclics and secondary amine tricyclics), selective serotonin reuptake inhibitors (SSRIs), monoamine oxidase inhibitors (MAOIs), reversible inhibitors of monoamine oxidase (RIMAs), serotonin and noradrenaline reuptake inhibitors (SNRIs), corticotropin releasing factor (CRF) antagonists, ⁇ -adrenoreceptor antagonists, neurokinin- 1 receptor antagonists, atypical anti-depressants, benzodiazepines, 5-HTi A agonists or antagonists, especially 5-HT JA partial agonists, and corticotropin releasing factor (CRF) antagonists.
- norepinephrine reuptake inhibitors including tertiary amine tricyclics and secondary amine tricyclics
- Specific agents include: amitriptyline, clomipramine, doxepin, imipramine and trimipramine; amoxapine, desipramine, maprotiline, nortriptyline and protriptyline; fluoxetine, fluvoxamine, paroxetine and sertraline; isocarboxazid, phenelzine, tranylcypromine and selegiline; moclobemide: venlafaxine; aprepitant; bupropion, lithium, nefazodone, trazodone and viloxazine; alprazolam, chlordiazepoxide, clonazepam, chlorazepate, diazepam, halazepam, lorazepam, oxazepam and prazepam; buspirone, flesinpxan, gepirone and ipsapirone, and pharmaceutically acceptable salts thereof.
- the subject compound may be employed in combination with anti -Alzheimer's agents; beta-secretase inhibitors; gamma-secretase inhibitors; growth hormone secretogogues; recombinant growth hormone; HMG-CoA reductase inhibitors; NS AID's including ibuprofen; vitamin E; anti-amyloid antibodies; CB-I receptor antagonists or CB-I receptor inverse agonists; antibiotics such as doxycycline and rifampin; N-methyl-D- aspartate (NMDA) receptor antagonists, such as memantine; cholinesterase inhibitors such as galantamine, rivastigmine, donepezil, and tacrine; growth hormone secretagogues such as ibutamoren, ibutamoren mesylate, and capromorelin; histamine H3 antagonists; AMPA agonists; PDE IV inhibitors; GABAA inverse agonists; or neuronal nicot
- the subject compound may be employed in combination with sedatives, hypnotics, anxiolytics, antipsychotics, antianxiety agents, cyclopyrrolones, imidazopyridines, pyrazolopyrimidines, minor tranquilizers, melatonin agonists and antagonists, melatonergic agents, benzodiazepines, barbiturates, 5HT-2 antagonists, and the like, such as: adinazolam, allobarbital, alonimid, alprazolam, amitriptyline, amobarbital, amoxapine, bentazepam, benzoctamine, brotizolam, bupropion, busprione, butabarbital, butalbital, capuride, carbocloral, chloral betaine, chloral hydrate, chlordiazepoxide, clomipramine, clonazepam, cloperidone, clorazepate, clorethate
- the subject compound may be employed in combination with levodopa (with or without a selective extracerebral decarboxylase inhibitor such as carbidopa or benserazide), anticholinergics such as biperiden (optionally as its hydrochloride or lactate salt) and trihexyphenidyl (benzhexol) hydrochloride, COMT inhibitors such as entacapone, MOA-B inhibitors, antioxidants, A2a adenosine receptor antagonists, cholinergic agonists, NMDA receptor antagonists, serotonin receptor antagonists and dopamine receptor agonists such as alentemol, bromocriptine, fenoldopam, lisuride, naxagolide, pergolide and pramipexole.
- levodopa with or without a selective extracerebral decarboxylase inhibitor such as carbidopa or benserazide
- anticholinergics such as biperi
- the dopamine agonist may be in the form of a pharmaceutically acceptable salt, for example, alentemol hydrobromide, bromocriptine mesylate, fenoldopam mesylate, naxagolide hydrochloride and pergolide mesylate.
- a pharmaceutically acceptable salt for example, alentemol hydrobromide, bromocriptine mesylate, fenoldopam mesylate, naxagolide hydrochloride and pergolide mesylate.
- Lisuride and pramipexol are commonly used in a non-salt form.
- the subject compound may be employed in combination with acetophenazine, alentemol, benzhexol, bromocriptine, biperiden, chlorpromazine, chlorprothixene, clozapine, diazepam, fenoldopam, fluphenazine, haloperidol, levodopa, levodopa with benserazide, levodopa with carbidopa, lisuride, loxapine, mesoridazine, molindolone, naxagolide, olanzapine, pergolide, perphenazine, pimozide, pramipexole, risperidone, sulpiride, tetrabenazine, trihexyphenidyl, thioridazine, thiothixene or trifluoperazine.
- the subject compound may be employed in combination with a compound from the phenothiazine, thioxanthene, heterocyclic dibenzazepine, butyrophenone, diphenylbutylpiperidine and indolone classes of neuroleptic agent.
- phenothiazines include chlorpromazine, mesoridazine, thioridazine, acetophenazine, fluphenazine, perphenazine and trifluoperazine.
- Suitable examples of thioxanthenes include chlorprothixene and thiothixene.
- An example of a dibenzazepine is clozapine.
- An example of a butyrophenone is haloperidol.
- An example of a diphenylbutylpiperidine is pimozide.
- An example of an indolone is molindolone.
- Other neuroleptic agents include loxapine, sulpiride and risperidone.
- the neuroleptic agents when used in combination with thesubject compound may be in the form of a pharmaceutically acceptable salt, for example, chlorpromazine hydrochloride, mesoridazine besylate, thioridazine hydrochloride, acetophenazine maleate, fluphenazine hydrochloride, flurphenazine enathate, fluphenazine decanoate, trifluoperazine hydrochloride, thiothixene hydrochloride, haloperidol decanoate, loxapine succinate and molindone hydrochloride.
- Perphenazine, chlorprothixene, clozapine, haloperidol, pimozide and risperidone are commonly used in a non-salt form.
- the subject compound may be employed in combination with an anoretic agent such as aminorex, amphechloral, amphetamine, benzphetamine, chlorphentermine, clobenzorex, cloforex, clominorex, clortermine, cyclexedrine, dexfenfluramine, dextroamphetamine, diethylpropion, diphemethoxidine, N-ethylamphetamine, fenbutrazate, fenflviraniine, fenisorex, fenproporex, fludorex, fluminorex, furfurylmethylamphetamine, levamfetamine, levophacetoperane, mazindol, mefenorex, metamfepramone, methamphetamine, norpseudoephedrine, pentorex, phendimetrazine, phenmetrazine, phentermine, phenylpropanolamine
- the subject compound may be employed in combination with an opiate agonist, a lipoxygenase inhibitor, such as an inhibitor of 5-lipoxygenase, a cyclooxygenase inhibitor, such as a cyclooxygenase-2 inhibitor, an interleukin inhibitor, such as an interleukin- 1 inhibitor, an NMDA antagonist, an inhibitor of nitric oxide or an inhibitor of the synthesis of nitric oxide, a non-steroidal antiinflammatory agent, or a cytokine-suppressing antiinflammatory agent, for example with a compound such as acetaminophen, asprin, codiene, fentanyl, ibuprofen, indomethacin, ketorolac, morphine, naproxen, phenacetin, piroxicam, a steroidal analgesic, sufentanyl, sunlindac, tenidap, and the like.
- a lipoxygenase inhibitor such as an inhibitor of 5-lip
- the subject compound may be administered with a pain reliever; a potentiator such as caffeine, an H2-antagonist, simethicone, aluminum or magnesium hydroxide; a decongestant such as phenylephrine, phenylpropanolamine, pseudophedrine, oxymetazoline, ephinephrine, naphazoline, xylometazoline, propylhexedrine, or levo-desoxy-ephedrine; an antiitussive such as codeine, hydrocodone, caramiphen, carbetapentane, or dextramethorphan; a diuretic; and a sedating or non-sedating antihistamine.
- a pain reliever such as caffeine, an H2-antagonist, simethicone, aluminum or magnesium hydroxide
- a decongestant such as phenylephrine, phenylpropanolamine, pseudophedrine, oxymetazoline, ephinep
- the compounds of the present invention may be administered by oral, parenteral (e.g., intramuscular, intraperitoneal, intravenous, ICV, intracistemal injection or infusion, subcutaneous injection, or implant), by inhalation spray, nasal, vaginal, rectal, sublingual, or topical routes of administration and may be formulated, alone or together, in suitable dosage unit formulations containing conventional non-toxic pharmaceutically acceptable carriers, adjuvants and vehicles appropriate for each route of administration.
- parenteral e.g., intramuscular, intraperitoneal, intravenous, ICV, intracistemal injection or infusion, subcutaneous injection, or implant
- inhalation spray nasal, vaginal, rectal, sublingual, or topical routes of administration
- nasal, vaginal, rectal, sublingual, or topical routes of administration may be formulated, alone or together, in suitable dosage unit formulations containing conventional non-toxic pharmaceutically acceptable carriers, adjuvants and vehicles appropriate for each route of administration.
- the compounds of the invention are effective for
- compositions for the administration of the compounds of this invention may conveniently be presented in dosage unit form and may be prepared by any of the methods well known in the art of pharmacy. All methods include the step of bringing the active ingredient into association with the carrier which constitutes one or more accessory ingredients.
- the pharmaceutical compositions are prepared by uniformly and intimately bringing the active ingredient into association with a liquid carrier or a finely divided solid carrier or both, and then, if necessary, shaping the product into the desired formulation.
- the active object compound is included in an amount sufficient to produce the desired effect upon the process or condition of diseases.
- composition is intended to encompass a product comprising the specified ingredients in the specified amounts, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts.
- compositions intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions and such compositions may contain one or more agents selected from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents in order to provide pharmaceutically elegant and palatable preparations.
- Tablets contain the active ingredient in admixture with non-toxic pharmaceutically acceptable excipients which are suitable for the manufacture of tablets.
- excipients may be for example, inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, for example, corn starch, or alginic acid; binding agents, for example starch, gelatin or acacia, and lubricating agents, for example magnesium stearate, stearic acid or talc.
- the tablets may be uncoated or they may be coated by known techniques to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period.
- compositions for oral use may also be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, for example peanut oil, liquid paraffin, or olive oil.
- Aqueous suspensions contain the active materials in admixture with excipients suitable for the manufacture of aqueous suspensions.
- Oily suspensions may be formulated by suspending the active ingredient in a suitable oil. Oil-in-water emulsions may also be employed.
- Dispersible powders and granules suitable for preparation of an aqueous suspension by the addition of water provide the active ingredient in admixture with a dispersing or wetting agent, suspending agent and one or more preservatives.
- Pharmaceutical compositions of the present compounds may be in the form of a sterile injectable aqueous or oleagenous suspension.
- the compounds of the present invention may also be administered in the form of suppositories for rectal administration. For topical use, creams, ointments, jellies, solutions or suspensions, etc., containing ; the compounds of the present invention may be employed.
- the compounds of the present invention may also be formulated for administered by inhalation.
- the compounds of the present invention may also be administered by a transdermal patch by methods known in the art.
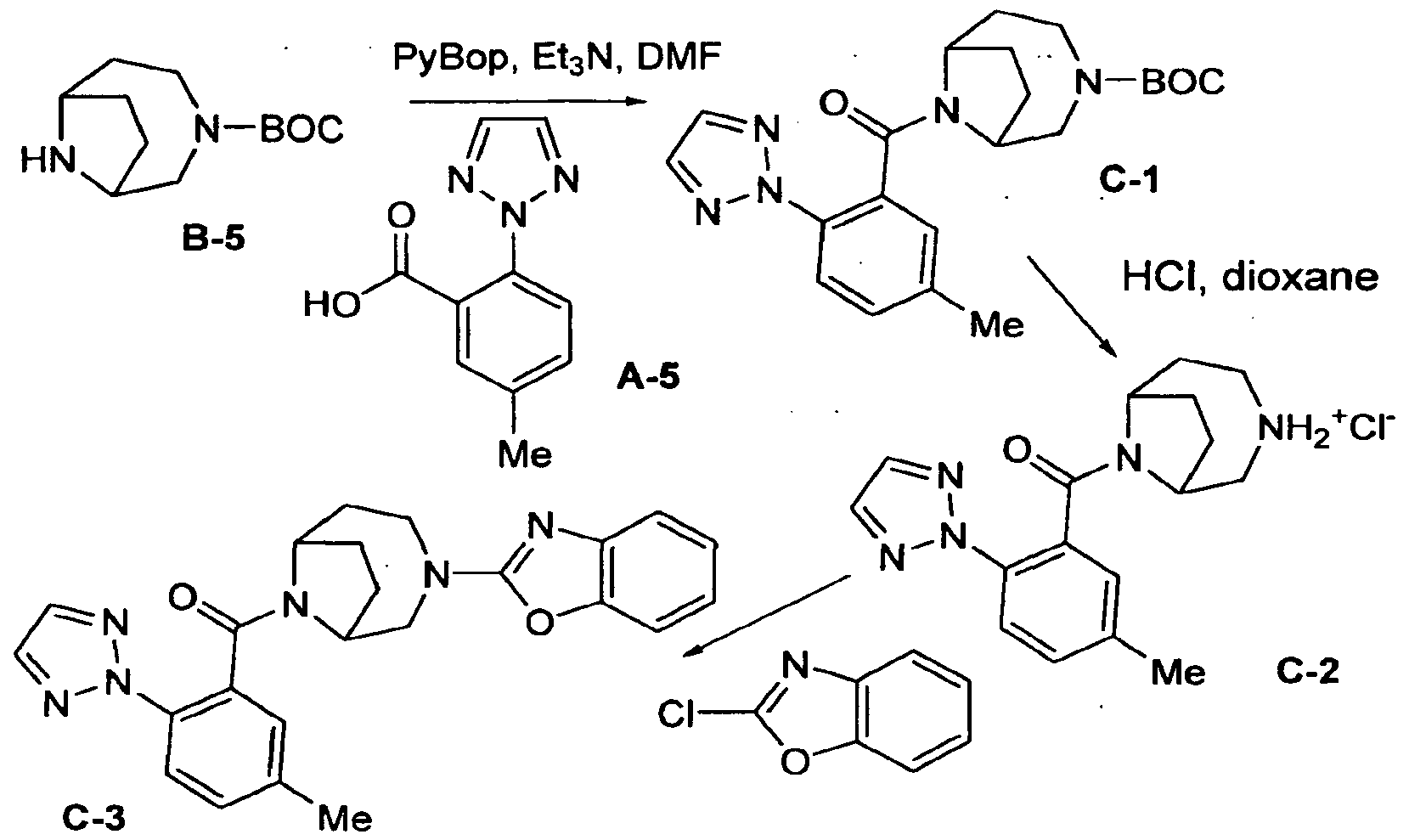
- Several methods for preparing the compounds of this invention are illustrated in the following Schemes and Examples. Starting materials are made according to procedures known in the art or as illustrated herein. The following abbreviations are used herein: Me: methyl; Et: ethyl; t-Bu: tert-butyl; Ar: aryl; Ph: phenyl; Bn: ben2yl; Ac: acetyl; THF: tetrahydrofuran; DEAD: diethylazodicarboxylate; DIPEA: N,N-diisopropylethylamine; DMSO: dimethylsulfoxide; EDC: N-(3-Dimethylaminopropyl)-N'-ethylcarbodiimide; HOBT: hydroxybenzotriazole hydrate; Boc: tert-butyloxy carbonyl; Et3N: trie
- the final product may be further modified, for example, by manipulation of substituents.
- substituents may include, but are not limited to, reduction, oxidation, alkylation, acylation, and hydrolysis reactions which are commonly known to those skilled in the art.
- the order of carrying out the foregoing reaction schemes may be varied to facilitate the reaction or to avoid unwanted reaction products.
- the following examples are provided so that the invention might be more fully understood. These examples are illustrative only and should not be construed as limiting the invention in any way.
- a solution Of LiAlH 4 (800 mL of a IM soln in THF) was diluted with THF (800 mL) and cooled to O 0 C.
- the enantiomers of A-7 were resolved by preparative chiral chromatography (ChiralPak AD 2 x 25 cm column; eluting with 40% hexanes/60% EtOAc).
- This material (128 mg, 0.423 mmol) was diluted in MeOH (5 ml) and treated with 10 wt.% Pd(OH) 2 .
- the flask was evacuated and backfilled with H2 ( g ) three times and stirred under a H 2 ( g ) atmosphere (1 atm) at RT for 1 h.
- the mixture was filtered through a syringe filter and the filtrate was concentrated to yield D-5.
- EtOAc/hexanes were isolated as the free-base; alternately, some products were purified by reverse phase HPLC (CH3CN/H2O containing 0.1% TFA as a modifier) and isolated as the TFA salt, in which case the masses reported and found are for the free-base.
- fractions containing the product could be basified with NaHCO 3 and extracted with EtOAc, dried over Na 2 SO 4 , and concentrated to provide the free-base.
- EtOAc/hexanes were isolated as the free-base; alternately, some products were purified by reverse phase HPLC (CH3CN/H2O containing 0.1% TFA as a modifier) and isolated as the TFA salt, in which case the masses reported and found are for the free-base.
- fractions containing the product could be basified with NaHC ⁇ 3 and extracted with EtOAc, dried over Na 2 SO 4 , and concentrated to provide the free-base.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Anesthesiology (AREA)
- Child & Adolescent Psychology (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
Description





















Claims






Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12/309,243 US8618102B2 (en) | 2006-07-14 | 2007-07-13 | Bridged diazepan orexin receptor antagonists |
| EP07796851.9A EP2049110B1 (en) | 2006-07-14 | 2007-07-13 | Bridged diazepan orexin receptor antagonists |
| JP2009519551A JP2009543785A (en) | 2006-07-14 | 2007-07-13 | Cross-linked diazepan orexin receptor antagonist |
| AU2007272854A AU2007272854B2 (en) | 2006-07-14 | 2007-07-13 | Bridged diazepan orexin receptor antagonists |
| CA002657623A CA2657623A1 (en) | 2006-07-14 | 2007-07-13 | Bridged diazepan orexin receptor antagonists |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US83117806P | 2006-07-14 | 2006-07-14 | |
| US60/831,178 | 2006-07-14 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2008008517A2 true WO2008008517A2 (en) | 2008-01-17 |
| WO2008008517A3 WO2008008517A3 (en) | 2008-12-04 |
Family
ID=38923948
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/016037 WO2008008517A2 (en) | 2006-07-14 | 2007-07-13 | Bridged diazepan orexin receptor antagonists |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US8618102B2 (en) |
| EP (1) | EP2049110B1 (en) |
| JP (1) | JP2009543785A (en) |
| AU (1) | AU2007272854B2 (en) |
| CA (1) | CA2657623A1 (en) |
| WO (1) | WO2008008517A2 (en) |
Cited By (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008147518A1 (en) | 2007-05-23 | 2008-12-04 | Merck & Co., Inc. | Pyridyl piperidine orexin receptor antagonists |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2010028033A1 (en) * | 2008-09-05 | 2010-03-11 | Targacept, Inc. | Amides of diazabicyclooctanes and uses thereof |
| WO2010028011A1 (en) * | 2008-09-05 | 2010-03-11 | Targacept, Inc. | Amides of diazabicyclononanes and uses thereof |
| JP2010527924A (en) * | 2007-05-18 | 2010-08-19 | メルク・シャープ・エンド・ドーム・コーポレイション | Oxo-bridged diazepan orexin receptor antagonist |
| WO2011050202A1 (en) | 2009-10-23 | 2011-04-28 | Janssen Pharmaceutica Nv | Fused heterocyclic compounds as orexin receptor modulators |
| WO2011050200A1 (en) | 2009-10-23 | 2011-04-28 | Janssen Pharmaceutica Nv | Fused heterocyclic compounds as orexin receptor modulators |
| WO2011050198A1 (en) | 2009-10-23 | 2011-04-28 | Janssen Pharmaceutica Nv | Disubstituted octahy - dropyrrolo [3,4-c] pyrroles as orexin receptor modulators |
| US7951797B2 (en) | 2006-12-01 | 2011-05-31 | Merck Sharp & Dohme Corp. | Substituted diazepan orexin receptor antagonists |
| WO2012085857A1 (en) | 2010-12-22 | 2012-06-28 | Actelion Pharmaceuticals Ltd | 3,8-diaza-bicyclo[4.2.0]oct-3-yl amides |
| WO2012145581A1 (en) | 2011-04-20 | 2012-10-26 | Janssen Pharmaceutica Nv | Disubstituted octahy-dropyrrolo [3,4-c] pyrroles as orexin receptor modulators |
| WO2013050938A1 (en) | 2011-10-04 | 2013-04-11 | Actelion Pharmaceuticals Ltd | 3,7-diazabicyclo[3.3.1]nonane and 9-oxa-3,7-diazabicyclo[3.3.1]nonane derivatives |
| WO2013068935A1 (en) | 2011-11-08 | 2013-05-16 | Actelion Pharmaceuticals Ltd | 2-(1,2,3-triazol-2-yl)benzamide and 3-(1,2,3-triazol-2-yl)picolinamide derivatives as orexin receptor antagonists |
| WO2014006402A1 (en) * | 2012-07-03 | 2014-01-09 | Heptares Therapeutics Limited | Orexin receptor antagonists |
| WO2014057435A1 (en) | 2012-10-10 | 2014-04-17 | Actelion Pharmaceuticals Ltd | Orexin receptor antagonists which are [ortho bi (hetero )aryl]-[2-(meta bi (hetero )aryl)-pyrrolidin-1-yl]-methanone derivatives |
| WO2014141065A1 (en) | 2013-03-12 | 2014-09-18 | Actelion Pharmaceuticals Ltd | Azetidine amide derivatives as orexin receptor antagonists |
| WO2015055994A1 (en) * | 2013-10-15 | 2015-04-23 | Takeda Cambridge Limited | Cyclopentylbenzamide derivatives and their use for the treatment of psychotic and cognitive disorders |
| US9156819B2 (en) | 2011-10-19 | 2015-10-13 | Merck Sharp & Dohme Corp. | 2-pyridyloxy-4-nitrile orexin receptor antagonists |
| WO2016084866A1 (en) * | 2014-11-26 | 2016-06-02 | 持田製薬株式会社 | Novel diazabicyclo derivative |
| US9440982B2 (en) | 2012-02-07 | 2016-09-13 | Eolas Therapeutics, Inc. | Substituted prolines/piperidines as orexin receptor antagonists |
| US9499517B2 (en) | 2012-02-07 | 2016-11-22 | Eolas Therapeutics, Inc. | Substituted prolines / piperidines as orexin receptor antagonists |
| WO2017194548A1 (en) | 2016-05-10 | 2017-11-16 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods and pharmaceutical compositions for the treatment of autoimmune inflammatory diseases |
| CN107573270A (en) * | 2017-09-29 | 2018-01-12 | 河南师范大学 | A kind of synthetic method of α formylpyrroles alkanes compound |
| CN107759620A (en) * | 2016-08-16 | 2018-03-06 | 广东东阳光药业有限公司 | Octahydro pyrrolo- [3,4 c] azole derivatives and its application method and purposes |
| US9914721B2 (en) | 2013-12-04 | 2018-03-13 | Idorsia Pharmaceuticals Ltd | Use of benzimidazole-proline derivatives |
| WO2018146466A1 (en) | 2017-02-09 | 2018-08-16 | Benevolentai Bio Limited | Orexin receptor antagonists |
| WO2018206959A1 (en) | 2017-05-10 | 2018-11-15 | Benevolentai Bio Limited | Orexin receptor antagonists |
| WO2018206956A1 (en) | 2017-05-10 | 2018-11-15 | Benevolentai Bio Limited | Orexin receptor antagonists |
| US10221170B2 (en) | 2014-08-13 | 2019-03-05 | Eolas Therapeutics, Inc. | Difluoropyrrolidines as orexin receptor modulators |
| WO2019073251A1 (en) * | 2017-10-13 | 2019-04-18 | The Institute Of Cancer Research: Royal Cancer Hospital | Lysyl oxidase inhibitors |
| US10329287B2 (en) | 2012-06-04 | 2019-06-25 | Idorsia Pharmaceuticals Ltd | Benzimidazole-proline derivatives |
| US10828302B2 (en) | 2016-03-10 | 2020-11-10 | Janssen Pharmaceutica Nv | Methods of treating depression using orexin-2 receptor antagonists |
| US10894789B2 (en) | 2016-02-12 | 2021-01-19 | Astrazeneca Ab | Halo-substituted piperidines as orexin receptor modulators |
Families Citing this family (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2011502146A (en) | 2007-10-29 | 2011-01-20 | メルク・シャープ・エンド・ドーム・コーポレイション | Substituted diazepanorexin receptor antagonist |
| EP2632465B1 (en) | 2010-10-27 | 2015-12-30 | Merck Sharp & Dohme Corp. | Inhibitors of the renal outer medullary potassium channel |
| WO2015048091A1 (en) * | 2013-09-24 | 2015-04-02 | The Board Of Regents Of The University Of Texas System | Orexin-control of bone formation and loss |
| WO2015095111A1 (en) | 2013-12-18 | 2015-06-25 | Merck Sharp & Dohme Corp. | Diazepane orexin receptor antagonists |
| TW201613864A (en) * | 2014-02-20 | 2016-04-16 | Takeda Pharmaceutical | Novel compounds |
| MX2017003254A (en) * | 2014-09-11 | 2017-10-12 | Janssen Pharmaceutica Nv | Substituted 2-azabicycles and their use as orexin receptor modulators. |
| WO2016065585A1 (en) | 2014-10-30 | 2016-05-06 | Merck Sharp & Dohme Corp. | Piperidine isoxazole and isothiazole orexin receptor antagonists |
| WO2016065584A1 (en) | 2014-10-30 | 2016-05-06 | Merck Sharp & Dohme Corp. | Piperidine oxadiazole and thiadiazole orexin receptor antagonists |
| WO2016065586A1 (en) | 2014-10-30 | 2016-05-06 | Merck Sharp & Dohme Corp. | Pyrazole, triazole and tetrazole orexin receptor antagonists |
| WO2016065587A1 (en) | 2014-10-30 | 2016-05-06 | Merck Sharp & Dohme Corp. | Pyrazole orexin receptor antagonists |
| WO2016065583A1 (en) | 2014-10-30 | 2016-05-06 | Merck Sharp & Dohme Corp. | Oxazole orexin receptor antagonists |
| WO2016085784A1 (en) | 2014-11-26 | 2016-06-02 | Merck Sharp & Dohme Corp. | Methyl diazepane orexin receptor antagonists |
| WO2016085783A1 (en) * | 2014-11-26 | 2016-06-02 | Merck Sharp & Dohme Corp. | Bridged diazepane orexin receptor antagonists |
| WO2016086358A1 (en) | 2014-12-02 | 2016-06-09 | Merck Sharp & Dohme Corp. | Hydroxymethyl piperidine orexin receptor antagonists |
| WO2016086357A1 (en) | 2014-12-02 | 2016-06-09 | Merck Sharp & Dohme Corp. | Methyl oxazole orexin receptor antagonists |
| JP2018016544A (en) * | 2014-12-03 | 2018-02-01 | 持田製薬株式会社 | Novel diazabicyclo [2.2.2]octane derivative |
| WO2016095205A1 (en) | 2014-12-19 | 2016-06-23 | Merck Sharp & Dohme Corp. | Heteroaryl orexin receptor antagonists |
| US9938276B2 (en) | 2014-12-19 | 2018-04-10 | Merck Sharp & Dohme Corp. | 6,5-bicyclic octahydropyrrolopyridine orexin receptor antagonists |
| US9987255B2 (en) | 2014-12-19 | 2018-06-05 | Merck Sharp & Dohme Corp. | 5,5-bicyclic oxazole orexin receptor antagonists |
| WO2016095204A1 (en) | 2014-12-19 | 2016-06-23 | Merck Sharp & Dohme Corp. | Pyrrolidine orexin receptor antagonists |
| US10011595B2 (en) | 2014-12-19 | 2018-07-03 | Merck Sharp & Dohme Corp. | Ethyldiamine orexin receptor antagonists |
| WO2016101118A1 (en) | 2014-12-23 | 2016-06-30 | Merck Sharp & Dohme Corp. | Amidoethyl azole orexin receptor antagonists |
| WO2016101119A1 (en) | 2014-12-23 | 2016-06-30 | Merck Sharp & Dohme Corp. | Fused heteroaryl derivatives as orexin receptor antagonists |
| WO2017012502A1 (en) | 2015-07-17 | 2017-01-26 | Sunshine Lake Pharma Co., Ltd. | Substituted quinazoline compounds and preparation and uses thereof |
| CN106749269B (en) | 2015-11-23 | 2019-01-04 | 广东东阳光药业有限公司 | Octahydro pyrrolo- [3,4-c] azole derivatives and application thereof |
| CN106986859B (en) * | 2016-01-20 | 2020-02-11 | 广东东阳光药业有限公司 | Indole derivatives and uses thereof |
| JOP20190284A1 (en) * | 2017-06-14 | 2019-12-11 | Bayer Pharma AG | Diazabicyclic substituted imidazopyrimidines and their use for the treatment of breathing disorders |
Citations (42)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0885888A1 (en) | 1996-02-02 | 1998-12-23 | Nippon Shinyaku Company, Limited | Isoquinoline derivatives and drugs |
| WO1999009024A1 (en) | 1997-08-14 | 1999-02-25 | Smithkline Beecham Plc | Phenyl urea and phenyl thiourea derivatives as hfgan72 antagonists |
| WO1999058533A1 (en) | 1998-05-08 | 1999-11-18 | Smithkline Beecham Plc | Phenylurea and phenylthio urea derivatives |
| WO2000044755A1 (en) | 1999-01-29 | 2000-08-03 | Abbott Laboratories | Diazabicyclic derivatives as nicotinic acetylcholine receptor ligands |
| WO2000047580A2 (en) | 1999-02-12 | 2000-08-17 | Smithkline Beecham Plc | Phenyl urea and phenyl thiourea derivatives |
| WO2000047576A1 (en) | 1999-02-12 | 2000-08-17 | Smithkline Beecham Plc | Cinnamide derivatives as orexin-1 receptors antagonists |
| WO2000047577A1 (en) | 1999-02-12 | 2000-08-17 | Smithkline Beecham Plc | Phenyl urea and phenyl thiourea derivatives as orexin receptor antagonists |
| WO2001068609A1 (en) | 2000-03-14 | 2001-09-20 | Actelion Pharmaceuticals Ltd. | 1,2,3,4-tetrahydroisoquinoline derivatives |
| WO2001085693A1 (en) | 2000-05-11 | 2001-11-15 | Banyu Pharmaceutical Co., Ltd. | N-acyltetrahydroisoquinoline derivatives |
| WO2001096302A1 (en) | 2000-06-16 | 2001-12-20 | Smithkline Beecham P.L.C. | Piperidines for use as orexin receptor antagonists |
| WO2002044172A1 (en) | 2000-11-28 | 2002-06-06 | Smithkline Beecham P.L.C. | Morpholine derivatives as antagonists of orexin receptors |
| WO2002051838A1 (en) | 2000-12-27 | 2002-07-04 | Actelion Pharmaceuticals Ltd. | Novel benzazepines and related heterocyclic derivatives which are useful as orexin receptor antagonists |
| US6440970B1 (en) | 2000-05-25 | 2002-08-27 | Targacept, Inc. | Pharmaceutical compositions and methods for use |
| WO2002089800A2 (en) | 2001-05-05 | 2002-11-14 | Smithkline Beecham P.L.C. | N-aroyl cyclic amine derivatives as orexin receptor antagonists |
| WO2002090355A1 (en) | 2001-05-05 | 2002-11-14 | Smithkline Beecham P.L.C. | N-aroyl cyclic amines |
| WO2003002561A1 (en) | 2001-06-28 | 2003-01-09 | Smithkline Beecham P.L.C. | N-aroyl cyclic amine derivatives as orexin receptor antagonists |
| WO2003002559A2 (en) | 2001-06-28 | 2003-01-09 | Smithkline Beecham P.L.C. | Piperidine compounds for use as orexin receptor antagonist |
| WO2003032991A1 (en) | 2001-10-11 | 2003-04-24 | Smithkline Beecham Plc | N-aroyl piperazine derivatives as orexin receptor antagonists |
| WO2003037847A1 (en) | 2001-11-01 | 2003-05-08 | Smithkline Beecham P.L.C. | Benzamide derivatives as antagonists of orexin receptors |
| WO2003041711A1 (en) | 2001-11-10 | 2003-05-22 | Smithkline Beecham P.L.C. | Piperazine bis-amide derivatives and their use as antagonists of the orexin receptor |
| WO2003051972A1 (en) | 2001-12-14 | 2003-06-26 | Basf Aktiengeselschaft | Stabilising composition ii |
| WO2003051873A1 (en) | 2001-12-19 | 2003-06-26 | Smithkline Beecham Plc | Piperazine compounds and their phamaceutical use |
| US20030225268A1 (en) | 1999-01-29 | 2003-12-04 | Bunnelle William H. | Diazabicyclic CNS active agents |
| WO2003105136A1 (en) | 2002-06-07 | 2003-12-18 | Lg Electronics Inc. | High-density multi-layer optical disc, method for recording data thereon on layer-by-layer basis, and method for managing spare areas thereof |
| WO2004004733A1 (en) | 2002-07-09 | 2004-01-15 | Actelion Pharmaceuticals Ltd. | 7,8,9,10-tetrahydro-6h-azepino, 6,7,8,9-tetrahydro-pyrido and 2,3-dihydro-2h-pyrrolo[2,1-b]-quinazolinone derivatives |
| WO2004026866A1 (en) | 2002-09-18 | 2004-04-01 | Glaxo Group Limited | N-aroyl cyclic amines as orexin receptor antagonists |
| WO2004033418A2 (en) | 2002-10-11 | 2004-04-22 | Actelion Pharmaceuticals Ltd. | Sulfonylamino-acetic derivatives and their use as orexin receptor antagonists |
| WO2004041807A1 (en) | 2002-11-06 | 2004-05-21 | Glaxo Group Limited | Novel compounds |
| WO2004041816A1 (en) | 2002-11-06 | 2004-05-21 | Glaxo Group Limited | Azacyclic compounds as orexin receptor antagonist |
| WO2004052876A1 (en) | 2002-12-12 | 2004-06-24 | Janssen Pharmaceutica, N.V. | Substituted 4-phenyl-[1,3]-dioxanes |
| WO2004083218A1 (en) | 2003-03-20 | 2004-09-30 | Actelion Pharmaceuticals Ltd | Guanidine derivatives and use thereof as neuropeptide ff receptor antagonists |
| WO2004085403A1 (en) | 2003-03-26 | 2004-10-07 | Actelion Pharmaceuticals Ltd | Tetrahydroisoquinolyl acetamide derivatives for use as orexin receptor antagonists |
| WO2004096780A1 (en) | 2003-04-28 | 2004-11-11 | Actelion Pharmaceuticals Ltd | Quinoxalinone-3- one derivatives as orexin receptor antagonists |
| US20050101602A1 (en) | 2003-09-19 | 2005-05-12 | Anwer Basha | Substituted diazabicycloalkane derivatives |
| WO2005060959A1 (en) | 2003-12-22 | 2005-07-07 | Sanofi-Aventis | Pyrazole derivatives and use thereof as orexin receptor antagonists |
| WO2005075458A1 (en) | 2004-02-10 | 2005-08-18 | Sanofi-Aventis | Pyrimidine derivatives as orexin receptors antagonists |
| WO2005118548A1 (en) | 2004-03-01 | 2005-12-15 | Actelion Pharmaceuticals Ltd | Substituted 1,2,3,4-tetrahydroisoquinoline derivatives |
| WO2006067224A2 (en) | 2004-12-23 | 2006-06-29 | Biovitrum Ab (Publ) | Spiro-benzodioxole and spiro-benzodioxane compounds as orexin receptor antagonists |
| WO2006110626A1 (en) | 2005-04-12 | 2006-10-19 | Merck & Co., Inc. | Amidopropoxyphenyl orexin receptor antagonists |
| WO2006127550A1 (en) | 2005-05-23 | 2006-11-30 | Merck & Co., Inc. | Proline bis-amide orexin receptor antagonists |
| WO2007019234A2 (en) | 2005-08-04 | 2007-02-15 | Merck & Co., Inc. | Aminoethane sulfonamide orexin receptor antagonists |
| WO2007025069A2 (en) | 2005-08-26 | 2007-03-01 | Merck & Co., Inc. | Diazaspirodecane orexin receptor antagonists |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2175395A1 (en) * | 1995-06-02 | 1996-12-03 | Ronald J. Mattson | Melatonergic indanyl piperazines or homopiperazines |
| ES2242749T3 (en) * | 2000-05-25 | 2005-11-16 | Targacept, Inc. | HETOROARILDIAZABICYCLOALKANOS AS LIGANDOS OF NICOTINIC COLINERGIC RECEPTORS. |
| TW200300354A (en) * | 2001-11-12 | 2003-06-01 | Jms Co Ltd | Injection needle retractable injector |
| US6676630B2 (en) | 2002-06-04 | 2004-01-13 | Bioject Medical Technologies, Inc. | Needle-free injection system |
| US20050065178A1 (en) | 2003-09-19 | 2005-03-24 | Anwer Basha | Substituted diazabicycloakane derivatives |
| US8685961B2 (en) | 2006-03-29 | 2014-04-01 | Merck Sharp & Dohme Corp. | Diazepan orexin receptor antagonists |
| DE602007008434D1 (en) | 2006-07-14 | 2010-09-23 | Merck Sharp & Dohme | RECEPTORS |
| PE20081229A1 (en) | 2006-12-01 | 2008-08-28 | Merck & Co Inc | DIAZEPAM OREXIN RECEPTOR ANTAGONISTS REPLACED |
| CA2688776A1 (en) | 2007-05-18 | 2008-11-27 | Merck & Co., Inc. | Oxo bridged diazepan orexin receptor antagonists |
-
2007
- 2007-07-13 EP EP07796851.9A patent/EP2049110B1/en not_active Not-in-force
- 2007-07-13 AU AU2007272854A patent/AU2007272854B2/en not_active Expired - Fee Related
- 2007-07-13 JP JP2009519551A patent/JP2009543785A/en not_active Ceased
- 2007-07-13 WO PCT/US2007/016037 patent/WO2008008517A2/en active Application Filing
- 2007-07-13 CA CA002657623A patent/CA2657623A1/en not_active Abandoned
- 2007-07-13 US US12/309,243 patent/US8618102B2/en active Active
Patent Citations (43)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0885888A1 (en) | 1996-02-02 | 1998-12-23 | Nippon Shinyaku Company, Limited | Isoquinoline derivatives and drugs |
| WO1999009024A1 (en) | 1997-08-14 | 1999-02-25 | Smithkline Beecham Plc | Phenyl urea and phenyl thiourea derivatives as hfgan72 antagonists |
| WO1999058533A1 (en) | 1998-05-08 | 1999-11-18 | Smithkline Beecham Plc | Phenylurea and phenylthio urea derivatives |
| WO2000044755A1 (en) | 1999-01-29 | 2000-08-03 | Abbott Laboratories | Diazabicyclic derivatives as nicotinic acetylcholine receptor ligands |
| US20030225268A1 (en) | 1999-01-29 | 2003-12-04 | Bunnelle William H. | Diazabicyclic CNS active agents |
| WO2000047580A2 (en) | 1999-02-12 | 2000-08-17 | Smithkline Beecham Plc | Phenyl urea and phenyl thiourea derivatives |
| WO2000047576A1 (en) | 1999-02-12 | 2000-08-17 | Smithkline Beecham Plc | Cinnamide derivatives as orexin-1 receptors antagonists |
| WO2000047577A1 (en) | 1999-02-12 | 2000-08-17 | Smithkline Beecham Plc | Phenyl urea and phenyl thiourea derivatives as orexin receptor antagonists |
| WO2001068609A1 (en) | 2000-03-14 | 2001-09-20 | Actelion Pharmaceuticals Ltd. | 1,2,3,4-tetrahydroisoquinoline derivatives |
| WO2001085693A1 (en) | 2000-05-11 | 2001-11-15 | Banyu Pharmaceutical Co., Ltd. | N-acyltetrahydroisoquinoline derivatives |
| US6440970B1 (en) | 2000-05-25 | 2002-08-27 | Targacept, Inc. | Pharmaceutical compositions and methods for use |
| WO2001096302A1 (en) | 2000-06-16 | 2001-12-20 | Smithkline Beecham P.L.C. | Piperidines for use as orexin receptor antagonists |
| WO2002044172A1 (en) | 2000-11-28 | 2002-06-06 | Smithkline Beecham P.L.C. | Morpholine derivatives as antagonists of orexin receptors |
| WO2002051232A2 (en) | 2000-12-27 | 2002-07-04 | Actelion Pharmaceuticals Ltd. | Novel benzazepines and related heterocyclic derivatives |
| WO2002051838A1 (en) | 2000-12-27 | 2002-07-04 | Actelion Pharmaceuticals Ltd. | Novel benzazepines and related heterocyclic derivatives which are useful as orexin receptor antagonists |
| WO2002089800A2 (en) | 2001-05-05 | 2002-11-14 | Smithkline Beecham P.L.C. | N-aroyl cyclic amine derivatives as orexin receptor antagonists |
| WO2002090355A1 (en) | 2001-05-05 | 2002-11-14 | Smithkline Beecham P.L.C. | N-aroyl cyclic amines |
| WO2003002561A1 (en) | 2001-06-28 | 2003-01-09 | Smithkline Beecham P.L.C. | N-aroyl cyclic amine derivatives as orexin receptor antagonists |
| WO2003002559A2 (en) | 2001-06-28 | 2003-01-09 | Smithkline Beecham P.L.C. | Piperidine compounds for use as orexin receptor antagonist |
| WO2003032991A1 (en) | 2001-10-11 | 2003-04-24 | Smithkline Beecham Plc | N-aroyl piperazine derivatives as orexin receptor antagonists |
| WO2003037847A1 (en) | 2001-11-01 | 2003-05-08 | Smithkline Beecham P.L.C. | Benzamide derivatives as antagonists of orexin receptors |
| WO2003041711A1 (en) | 2001-11-10 | 2003-05-22 | Smithkline Beecham P.L.C. | Piperazine bis-amide derivatives and their use as antagonists of the orexin receptor |
| WO2003051972A1 (en) | 2001-12-14 | 2003-06-26 | Basf Aktiengeselschaft | Stabilising composition ii |
| WO2003051873A1 (en) | 2001-12-19 | 2003-06-26 | Smithkline Beecham Plc | Piperazine compounds and their phamaceutical use |
| WO2003105136A1 (en) | 2002-06-07 | 2003-12-18 | Lg Electronics Inc. | High-density multi-layer optical disc, method for recording data thereon on layer-by-layer basis, and method for managing spare areas thereof |
| WO2004004733A1 (en) | 2002-07-09 | 2004-01-15 | Actelion Pharmaceuticals Ltd. | 7,8,9,10-tetrahydro-6h-azepino, 6,7,8,9-tetrahydro-pyrido and 2,3-dihydro-2h-pyrrolo[2,1-b]-quinazolinone derivatives |
| WO2004026866A1 (en) | 2002-09-18 | 2004-04-01 | Glaxo Group Limited | N-aroyl cyclic amines as orexin receptor antagonists |
| WO2004033418A2 (en) | 2002-10-11 | 2004-04-22 | Actelion Pharmaceuticals Ltd. | Sulfonylamino-acetic derivatives and their use as orexin receptor antagonists |
| WO2004041807A1 (en) | 2002-11-06 | 2004-05-21 | Glaxo Group Limited | Novel compounds |
| WO2004041816A1 (en) | 2002-11-06 | 2004-05-21 | Glaxo Group Limited | Azacyclic compounds as orexin receptor antagonist |
| WO2004052876A1 (en) | 2002-12-12 | 2004-06-24 | Janssen Pharmaceutica, N.V. | Substituted 4-phenyl-[1,3]-dioxanes |
| WO2004083218A1 (en) | 2003-03-20 | 2004-09-30 | Actelion Pharmaceuticals Ltd | Guanidine derivatives and use thereof as neuropeptide ff receptor antagonists |
| WO2004085403A1 (en) | 2003-03-26 | 2004-10-07 | Actelion Pharmaceuticals Ltd | Tetrahydroisoquinolyl acetamide derivatives for use as orexin receptor antagonists |
| WO2004096780A1 (en) | 2003-04-28 | 2004-11-11 | Actelion Pharmaceuticals Ltd | Quinoxalinone-3- one derivatives as orexin receptor antagonists |
| US20050101602A1 (en) | 2003-09-19 | 2005-05-12 | Anwer Basha | Substituted diazabicycloalkane derivatives |
| WO2005060959A1 (en) | 2003-12-22 | 2005-07-07 | Sanofi-Aventis | Pyrazole derivatives and use thereof as orexin receptor antagonists |
| WO2005075458A1 (en) | 2004-02-10 | 2005-08-18 | Sanofi-Aventis | Pyrimidine derivatives as orexin receptors antagonists |
| WO2005118548A1 (en) | 2004-03-01 | 2005-12-15 | Actelion Pharmaceuticals Ltd | Substituted 1,2,3,4-tetrahydroisoquinoline derivatives |
| WO2006067224A2 (en) | 2004-12-23 | 2006-06-29 | Biovitrum Ab (Publ) | Spiro-benzodioxole and spiro-benzodioxane compounds as orexin receptor antagonists |
| WO2006110626A1 (en) | 2005-04-12 | 2006-10-19 | Merck & Co., Inc. | Amidopropoxyphenyl orexin receptor antagonists |
| WO2006127550A1 (en) | 2005-05-23 | 2006-11-30 | Merck & Co., Inc. | Proline bis-amide orexin receptor antagonists |
| WO2007019234A2 (en) | 2005-08-04 | 2007-02-15 | Merck & Co., Inc. | Aminoethane sulfonamide orexin receptor antagonists |
| WO2007025069A2 (en) | 2005-08-26 | 2007-03-01 | Merck & Co., Inc. | Diazaspirodecane orexin receptor antagonists |
Non-Patent Citations (2)
| Title |
|---|
| OKUMURA ET AL., BIOCHEM. BIOPHYS. RES. COMM., vol. 280, 2001, pages 976 - 981 |
| See also references of EP2049110A4 |
Cited By (69)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7951797B2 (en) | 2006-12-01 | 2011-05-31 | Merck Sharp & Dohme Corp. | Substituted diazepan orexin receptor antagonists |
| JP2010527924A (en) * | 2007-05-18 | 2010-08-19 | メルク・シャープ・エンド・ドーム・コーポレイション | Oxo-bridged diazepan orexin receptor antagonist |
| US8569311B2 (en) | 2007-05-23 | 2013-10-29 | Merch Sharp & Dohme Corp. | Pyridyl piperidine orexin receptor antagonists |
| US8242121B2 (en) | 2007-05-23 | 2012-08-14 | Merck Sharp & Dohme Corp. | Pyridyl piperidine orexin receptor antagonists |
| WO2008147518A1 (en) | 2007-05-23 | 2008-12-04 | Merck & Co., Inc. | Pyridyl piperidine orexin receptor antagonists |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2010028033A1 (en) * | 2008-09-05 | 2010-03-11 | Targacept, Inc. | Amides of diazabicyclooctanes and uses thereof |
| WO2010028011A1 (en) * | 2008-09-05 | 2010-03-11 | Targacept, Inc. | Amides of diazabicyclononanes and uses thereof |
| WO2011050200A1 (en) | 2009-10-23 | 2011-04-28 | Janssen Pharmaceutica Nv | Fused heterocyclic compounds as orexin receptor modulators |
| JP2013508405A (en) * | 2009-10-23 | 2013-03-07 | ヤンセン ファーマシューティカ エヌ.ベー. | Fused heterocyclic compounds as orexin receptor modulators |
| WO2011050198A1 (en) | 2009-10-23 | 2011-04-28 | Janssen Pharmaceutica Nv | Disubstituted octahy - dropyrrolo [3,4-c] pyrroles as orexin receptor modulators |
| US9062044B2 (en) | 2009-10-23 | 2015-06-23 | Janssen Pharmaceutica Nv | Disubstituted octahydropyrrolo[3,4-c]pyrroles as orexin receptor modulators |
| JP2016102118A (en) * | 2009-10-23 | 2016-06-02 | ヤンセン ファーマシューティカ エヌ.ベー. | Fused heterocyclic compounds as orexin receptor modulators |
| JP2013508404A (en) * | 2009-10-23 | 2013-03-07 | ヤンセン ファーマシューティカ エヌ.ベー. | Fused heterocyclic compounds as orexin receptor modulators |
| JP2013508403A (en) * | 2009-10-23 | 2013-03-07 | ヤンセン ファーマシューティカ エヌ.ベー. | Disubstituted octahydropyrrolo [3,4-c] pyrrole as an orexin receptor modulator |
| EP3093291A1 (en) | 2009-10-23 | 2016-11-16 | Janssen Pharmaceutica N.V. | Disubstituted octahy - dropyrrolo [3,4-c]pyrroles as orexin receptor modulators |
| EP3581575A1 (en) | 2009-10-23 | 2019-12-18 | Janssen Pharmaceutica NV | Disubstituted octahydropyrrolo[3,4-c]pyrroles as orexin receptor modulators |
| US9079911B2 (en) | 2009-10-23 | 2015-07-14 | Janssen Pharmaceutica Nv | Disubstituted octahydropyrrolo[3,4-c]pyrroles as orexin receptor modulators |
| WO2011050202A1 (en) | 2009-10-23 | 2011-04-28 | Janssen Pharmaceutica Nv | Fused heterocyclic compounds as orexin receptor modulators |
| US11667644B2 (en) | 2009-10-23 | 2023-06-06 | Janssen Pharmaceutica Nv | Disubstituted octahydropyrrolo[3,4-c]pyrroles as orexin receptor modulators |
| US8653263B2 (en) | 2009-10-23 | 2014-02-18 | Janssen Pharmaceutica | Disubstituted octahydropyrrolo[3,4-c]pyrroles as orexin receptor modulators |
| US8680275B2 (en) | 2009-10-23 | 2014-03-25 | Janssen Pharmaceutica Nv | Fused heterocyclic compounds as orexin receptor modulators |
| USRE48841E1 (en) | 2009-10-23 | 2021-12-07 | Janssen Pharmaceutica Nv | Disubstituted octahydropyrrolo[3,4-c]pyrroles as orexin receptor modulators |
| US11059828B2 (en) | 2009-10-23 | 2021-07-13 | Janssen Pharmaceutica Nv | Disubstituted octahydropyrrolo[3,4-C]pyrroles as orexin receptor modulators |
| WO2012085857A1 (en) | 2010-12-22 | 2012-06-28 | Actelion Pharmaceuticals Ltd | 3,8-diaza-bicyclo[4.2.0]oct-3-yl amides |
| WO2012085852A1 (en) | 2010-12-22 | 2012-06-28 | Actelion Pharmaceuticals Ltd | 3,8-diaza-bicyclo[4.2.0]oct-8-yl amides |
| WO2012145581A1 (en) | 2011-04-20 | 2012-10-26 | Janssen Pharmaceutica Nv | Disubstituted octahy-dropyrrolo [3,4-c] pyrroles as orexin receptor modulators |
| US9586962B2 (en) | 2011-04-20 | 2017-03-07 | Janssen Pharmaceutica Nv | Disubstituted octahydropyrrolo [3,4-C] pyrroles as orexin receptor modulators |
| WO2013050938A1 (en) | 2011-10-04 | 2013-04-11 | Actelion Pharmaceuticals Ltd | 3,7-diazabicyclo[3.3.1]nonane and 9-oxa-3,7-diazabicyclo[3.3.1]nonane derivatives |
| US9156819B2 (en) | 2011-10-19 | 2015-10-13 | Merck Sharp & Dohme Corp. | 2-pyridyloxy-4-nitrile orexin receptor antagonists |
| WO2013068935A1 (en) | 2011-11-08 | 2013-05-16 | Actelion Pharmaceuticals Ltd | 2-(1,2,3-triazol-2-yl)benzamide and 3-(1,2,3-triazol-2-yl)picolinamide derivatives as orexin receptor antagonists |
| US9150566B2 (en) | 2011-11-08 | 2015-10-06 | Actelion Pharmaceuticals Ltd. | 2-(1,2,3-triazol-2-yl)benzamide and 3-(1,2,3-triazol-2-YL)picolinamide derivatives as orexin receptor antagonists |
| US9440982B2 (en) | 2012-02-07 | 2016-09-13 | Eolas Therapeutics, Inc. | Substituted prolines/piperidines as orexin receptor antagonists |
| US9896452B2 (en) | 2012-02-07 | 2018-02-20 | Eolas Therapeutics, Inc. | Substituted prolines/piperidines as orexin receptor antagonists |
| US9499517B2 (en) | 2012-02-07 | 2016-11-22 | Eolas Therapeutics, Inc. | Substituted prolines / piperidines as orexin receptor antagonists |
| US11040966B2 (en) | 2012-06-04 | 2021-06-22 | Idorsia Pharmaceuticals Ltd | Benzimidazole-proline derivatives |
| US10329287B2 (en) | 2012-06-04 | 2019-06-25 | Idorsia Pharmaceuticals Ltd | Benzimidazole-proline derivatives |
| US10316028B2 (en) | 2012-07-03 | 2019-06-11 | Heptares Therapeutics Limited | Orexin receptor antagonists |
| WO2014006402A1 (en) * | 2012-07-03 | 2014-01-09 | Heptares Therapeutics Limited | Orexin receptor antagonists |
| US9850237B2 (en) | 2012-07-03 | 2017-12-26 | Heptares Therapeutics Limited | Orexin receptor antagonists |
| US9555044B2 (en) | 2012-07-03 | 2017-01-31 | Heptares Therapeutics Limited | Orexin receptor antagonists |
| US9249160B2 (en) | 2012-07-03 | 2016-02-02 | Heptares Therapeutics Limited | Orexin receptor antagonists |
| US9493446B2 (en) | 2012-10-10 | 2016-11-15 | Actelion Pharmaceuticals Ltd. | Orexin receptor antagonists which are [ortho bi-(hetero-)aryl]-[2-(meta bi-(hetero-)aryl)-pyrrolidin-1-yl]-methanone derivatives |
| WO2014057435A1 (en) | 2012-10-10 | 2014-04-17 | Actelion Pharmaceuticals Ltd | Orexin receptor antagonists which are [ortho bi (hetero )aryl]-[2-(meta bi (hetero )aryl)-pyrrolidin-1-yl]-methanone derivatives |
| US9403813B2 (en) | 2013-03-12 | 2016-08-02 | Actelion Pharmaceuticals Ltd. | Azetidine amide derivatives as orexin receptor antagonists |
| WO2014141065A1 (en) | 2013-03-12 | 2014-09-18 | Actelion Pharmaceuticals Ltd | Azetidine amide derivatives as orexin receptor antagonists |
| US9493432B2 (en) | 2013-10-15 | 2016-11-15 | Takeda Pharmaceuticals Company Limited | Cyclopentylbenzamide derivatives and their use for the treatment of psychotic and cognitive disorders |
| WO2015055994A1 (en) * | 2013-10-15 | 2015-04-23 | Takeda Cambridge Limited | Cyclopentylbenzamide derivatives and their use for the treatment of psychotic and cognitive disorders |
| US9914721B2 (en) | 2013-12-04 | 2018-03-13 | Idorsia Pharmaceuticals Ltd | Use of benzimidazole-proline derivatives |
| US10221170B2 (en) | 2014-08-13 | 2019-03-05 | Eolas Therapeutics, Inc. | Difluoropyrrolidines as orexin receptor modulators |
| WO2016084866A1 (en) * | 2014-11-26 | 2016-06-02 | 持田製薬株式会社 | Novel diazabicyclo derivative |
| US10894789B2 (en) | 2016-02-12 | 2021-01-19 | Astrazeneca Ab | Halo-substituted piperidines as orexin receptor modulators |
| US12084437B2 (en) | 2016-02-12 | 2024-09-10 | Astrazeneca Ab | Halo-substituted piperidines as orexin receptor modulators |
| US11434236B2 (en) | 2016-02-12 | 2022-09-06 | Astrazeneca Ab | Halo-substituted piperidines as orexin receptor modulators |
| US11241432B2 (en) | 2016-03-10 | 2022-02-08 | Janssen Pharmaceutica Nv | Methods of treating depression using orexin-2 receptor antagonists |
| US10828302B2 (en) | 2016-03-10 | 2020-11-10 | Janssen Pharmaceutica Nv | Methods of treating depression using orexin-2 receptor antagonists |
| WO2017194548A1 (en) | 2016-05-10 | 2017-11-16 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods and pharmaceutical compositions for the treatment of autoimmune inflammatory diseases |
| CN107759620A (en) * | 2016-08-16 | 2018-03-06 | 广东东阳光药业有限公司 | Octahydro pyrrolo- [3,4 c] azole derivatives and its application method and purposes |
| CN107759620B (en) * | 2016-08-16 | 2021-11-12 | 广东东阳光药业有限公司 | Octahydropyrrolo [3,4-c ] pyrrole derivatives, methods of use, and uses thereof |
| WO2018146466A1 (en) | 2017-02-09 | 2018-08-16 | Benevolentai Bio Limited | Orexin receptor antagonists |
| WO2018206959A1 (en) | 2017-05-10 | 2018-11-15 | Benevolentai Bio Limited | Orexin receptor antagonists |
| WO2018206956A1 (en) | 2017-05-10 | 2018-11-15 | Benevolentai Bio Limited | Orexin receptor antagonists |
| CN107573270A (en) * | 2017-09-29 | 2018-01-12 | 河南师范大学 | A kind of synthetic method of α formylpyrroles alkanes compound |
| CN107573270B (en) * | 2017-09-29 | 2019-11-29 | 河南师范大学 | A kind of synthetic method of α-formylpyrrole alkanes compound |
| CN111212836A (en) * | 2017-10-13 | 2020-05-29 | 癌症研究所:皇家癌症医院 | Lysyl oxidase inhibitors |
| US11325915B2 (en) | 2017-10-13 | 2022-05-10 | The Institute Of Cancer Research: Royal Cancer Hospital | Lysyl oxidase inhibitors |
| CN111212836B (en) * | 2017-10-13 | 2023-12-15 | 癌症研究所:皇家癌症医院 | Lysyl oxidase inhibitors |
| US12060360B2 (en) | 2017-10-13 | 2024-08-13 | The Institute Of Cancer Research: Royal Cancer Hospital | Lysyl oxidase inhibitors |
| WO2019073251A1 (en) * | 2017-10-13 | 2019-04-18 | The Institute Of Cancer Research: Royal Cancer Hospital | Lysyl oxidase inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| EP2049110B1 (en) | 2014-08-20 |
| WO2008008517A3 (en) | 2008-12-04 |
| US20090239843A1 (en) | 2009-09-24 |
| US8618102B2 (en) | 2013-12-31 |
| EP2049110A2 (en) | 2009-04-22 |
| JP2009543785A (en) | 2009-12-10 |
| CA2657623A1 (en) | 2008-01-17 |
| AU2007272854B2 (en) | 2013-08-01 |
| EP2049110A4 (en) | 2011-08-10 |
| AU2007272854A1 (en) | 2008-01-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2049110B1 (en) | Bridged diazepan orexin receptor antagonists | |
| AU2008255005B2 (en) | OXO bridged diazepan orexin receptor antagonists | |
| AU2007272855B2 (en) | Substituted diazepan orexin receptor antagonists | |
| EP2001485B1 (en) | Diazepan orexin receptor antagonists | |
| EP2214676B1 (en) | Substituted diazepan orexin receptor antagonists | |
| EP2089382B1 (en) | Substituted diazepan compounds as orexin receptor antagonists | |
| EP2348846B1 (en) | Disubstituted azepan orexin receptor antagonists | |
| US20090176789A1 (en) | Diazaspirodecane orexin receptor antagonists | |
| EP2049526A2 (en) | 2-substituted proline bis-amide orexin receptor antagonists | |
| WO2010048010A1 (en) | 2,5-disubstituted piperidine orexin receptor antagonists | |
| EP2349270A1 (en) | 2,5-disubstituted morpholine orexin receptor antagonists | |
| WO2007126934A2 (en) | Amidoethylthioether orexin receptor antagonists | |
| WO2009011775A1 (en) | Amidoethyl alkylamino orexin receptor antagonists |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07796851 Country of ref document: EP Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007272854 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12309243 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007796851 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2657623 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009519551 Country of ref document: JP |
|
| ENP | Entry into the national phase |
Ref document number: 2007272854 Country of ref document: AU Date of ref document: 20070713 Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| NENP | Non-entry into the national phase |
Ref country code: RU |