WO2008005572A2 - Substituted 4-aryl-chromene as activator of caspases and inducer of apoptosis and as antivascular agent and the use thereof - Google Patents
Substituted 4-aryl-chromene as activator of caspases and inducer of apoptosis and as antivascular agent and the use thereof Download PDFInfo
- Publication number
- WO2008005572A2 WO2008005572A2 PCT/US2007/015676 US2007015676W WO2008005572A2 WO 2008005572 A2 WO2008005572 A2 WO 2008005572A2 US 2007015676 W US2007015676 W US 2007015676W WO 2008005572 A2 WO2008005572 A2 WO 2008005572A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- cancer
- stereoisomer
- carcinoma
- disorder
- Prior art date
Links
- 0 COc1c*([C@]2C(C#N)=C(N)Oc3c2ccc(NC(CN*)=O)c3N)cc(Br)c1OC Chemical compound COc1c*([C@]2C(C#N)=C(N)Oc3c2ccc(NC(CN*)=O)c3N)cc(Br)c1OC 0.000 description 3
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D311/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings
- C07D311/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only hetero atom, condensed with other rings ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D311/04—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring
- C07D311/58—Benzo[b]pyrans, not hydrogenated in the carbocyclic ring other than with oxygen or sulphur atoms in position 2 or 4
Definitions
- This invention is in the field of medicinal chemistry.
- the invention relates to the discovery that (i?)(-)-2,7,8-triamino-4-(3-bromo-4,5-dimethoxyphenyl)-3- cyano-4H-chromene (IR), substantially free from the corresponding ( ⁇ -stereoisomer, is an inducer of apoptosis and is a vascular disrupting agent.
- the invention also relates to the use of compound IR, substantially free from the corresponding ( ⁇ -stereoisomer, as a therapeutically effective anti -cancer agent, and ' combination with other anticancer agents, as well as for the treatment of diseases due to overgrowth of vasculature, such as ocular neovascularization.
- Organisms eliminate unwanted cells by a process variously known as regulated cell death, programmed cell death, or apoptosis. Such cell death occurs as a normal aspect of animal development, as well as in tissue homeostasis and aging (Glucksmann, A., Biol. Rev. Cambridge Philos. Soc. 25:59-86 (1951); Glucksmann, A., Archives de Biologie 76:419-431 (1965); Ellis, et al, Dev. /72:591-603 (1991); Vaux, et al, Cell 76:111-119 (1994)).
- Apoptosis regulates cell number, facilitates morphogenesis, removes harmful or otherwise abnormal cells and eliminates cells that have already performed their function. Additionally, apoptosis occurs in response to various physiological stresses, such as hypoxia or ischemia (WO96/20721).
- a cell activates its internally encoded suicide program as a result of either internal or external signals.
- the suicide program is executed through the activation of a carefully regulated genetic program (Wyllie, et al, Int. Rev. Cyt. (55:251 (1980); Ellis, et al., Ann. Rev. Cell Bio. 7:663 (1991)).
- Apoptotic cells and bodies are usually recognized and cleared by neighboring cells or macrophages before lysis. Because of this clearance mechanism, inflammation is not induced despite the clearance of great numbers of cells (Orrenius, S., J. Internal Medicine 237:529-536 (1995)).
- apoptotic cell death involves at least 14 genes, 2 of which are the pro-apoptotic (death-promoting) ced (for cell death abnormal) genes, ced-3 and ced-4.
- CED-3 is homologous to interleukin 1 beta-converting enzyme, a cysteine protease, which is now called caspase-1.
- caspase family of cysteine proteases comprises 14 different members, and more can be discovered in the future. All known caspases are synthesized as zymogens that require cleavage at an aspartyl residue prior to forming the active enzyme. Thus, caspases are capable of activating other caspases, in the manner of an amplifying cascade.
- BCL-like proteins include BCL-xL and BAX-alpha, which appear to function upstream of caspase activation.
- BCL-xL appears to prevent activation of the apoptotic protease cascade, whereas BAX-alpha accelerates activation of the apoptotic protease cascade.
- chemotherapeutic (anti-cancer) drugs can trigger cancer cells to undergo suicide by activating the dormant caspase cascade. This can be a crucial aspect of the mode of action of most, if not all, known anticancer drugs (Los, et al., Blood 90:3118-3129 (1997); Friesen, et al., Nat.
- antineoplastic drugs frequently involves an attack at specific phases of the cell cycle.
- the cell cycle refers to the stages through which cells normally progress during their lifetime. Normally, cells exist in a resting phase termed G 0 . During multiplication, cells progress to a stage in which DNA synthesis occurs, termed S. Later, cell division, or mitosis occurs, in a phase called M.
- Antineoplastic drugs such as cytosine arabinoside, hydroxyurea, 6-mercaptopurine, and methotrexate are S phase specific, whereas antineoplastic drugs, such as vincristine, vinblastine, and paclitaxel are M phase specific.
- tumor vasculature is essential for the growth and metastasis of solid tumors. Therefore tumor vasculature is an attractive target for therapy because damaging or blocking a single tumor vessel can kill many tumor cells.
- Antiangiogenic approaches using antiangiogenic agents such as small molecular inhibitors of VEGF receptors or monoclonal antibody targeting VEGF receptors, are designed to prevent the neovascularization processes in tumors, thus blocking the formation of new blood vessels and tumor growth.
- VDAs vascular disrupting agents
- VTAs vascular targeting agents
- VDAs Two types have been developed.
- the first types are biological or ligand-directed VDAs which use antibodies, peptides or growth factors to target toxins or pro-coagulants to the tumor endothelium.
- the second types are small molecule VDAs, and most of them are tubulin-binding agents.
- DMXAA 5,6-dimethylxanthenone-4-acetic acid
- VDAs are most effective at killing cells in the poorly perfused hypoxic core of tumors, and leaving a viable rim of well-perfused tumor tissues at the periphery, which can rapidly regrow if not treated. Therefore VDAs as single agents in general have poor anti-tumor effects.
- VDAs are well tolerated and have different side-effect profiles than other types of anticancer therapies. Since VDAs target tumor vasculature, they can kill tumor cells that are resistant to conventional chemotherapy and radiotherapy. In addition, VDAs also should be useful for the treatment of other diseases due to overgrowth of vasculature, such as ocular neovascularization (Numbu, H. et al., Invest Ophthalmol. Vis. ScL 44: 3650-5 (2003)).
- Vinca alkaloids and colchicine are known to induce haemorrhagic necrosis of solid tumors.
- these antivascular effects were only observed at doses approaching or exceeding their maximum tolerated doses, therefore they could not be used for therapeutic application.
- tubulin-binding agents interacting at the colchicine-binding site have been found to preferentially target tumor endothelial cells while sparing normal vasculature, and to induce haemorrhagic necrosis of solid tumors at doses that are well tolerated.
- VDAs vascular disrupting agents
- CA4P was found to selectively target endothelial cells, but not smooth muscle cells, and to induce regression of unstable nascent tumor neovessels by rapidly disrupting the molecular engagement of the endothelial cell-specific junctional molecule vascular endothelial- cadherin both in vitro and in vivo.
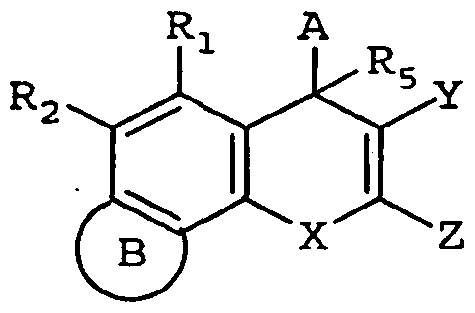
- EP537949 discloses derivatives of 4H-naphthoI[l,2-6]pyran as antiproliferatives:
- each R 1 is independently halo, trifluoromethyl, Ci -4 alkoxy, hydroxy, nitro, alkyl, C M alkylthio, hydroxy-Ci-4 alkyl, hydroxy-Ci- 4 alkoxy, trifiuoromethoxy, carboxy, -COOR 5 where R 5 is an ester group, -CONR 6 R 7 or -NR 6 R 7 where R 6 and R 7 are each hydrogen or Ci -4 alkyl;
- R 2 is phenyl, naphthyl or heteroaryl selected from thienyl, pyridyl, benzothienyl, quinolinyl, benzofiiranyl or benzimidazolyl, wherein said phenyl, naphthyl and heteroaryl groups are optionally substituted, or R 2 is furanyl optionally substituted with C1-4 alkyl;
- R 3 is nitrile, carboxy, -COOR 8 where R 8 is an ester group, -CONR 9 R 10 where R 9 and R 10 are each hydrogen or Ci -4 alkyl or R 11 SO 2 where R 11 is C M alkyl or optionally substituted phenyl;
- R 1 is C 1-4 alkoxy, OH or COOH
- R 2 is optionally substituted phenyl
- R 3 is nitrile, or R 3 is carboxy or -COOR 8 when R 2 is phenyl substituted with 3-nitro or 3- trifluoromethyl and R 8 is an ester group;
- EP599514 discloses the preparation of pyranoquinoline derivatives as inhibitors of cell proliferation:
- R 1 is optionally substituted phenyl or optionally substituted heteroaryl selected from thienyl, pyridyl, benzothienyl, quinolinyl, benzofuranyl or benzimidazolyl, or R 1 is furanyl optionally substituted with Ci -4 alkyl;
- R 2 is nitrile, carboxy, — CO 2 R 4 wherein R 4 is an ester group, -CON(R 5 )R 6 where R 5 and R 6 are independently H or Ci.4 alkyl, or R 7 SO 2 where R 7 is Q_4 alkyl or optionally substituted phenyl;
- X is C 2-4 alkylene; and the ring P represents a pyridine fused to the benzopyran nucleus.
- EP618206 discloses the preparation of naphthopyran and pyranoquinoline as immunosuppressants and cell proliferation inhibitors:
- R 2 is phenyl, naphthyl or heteroaryl selected from thienyl, pyridyl, benzothienyl, quinolinyl, benzofuranyl or benzimidazolyl, wherein said phenyl, naphthyl and heteroaryl groups are optionally substituted, or R 2 is furanyl optionally substituted with C 1.4 alkyl;
- R 3 is nitrile, carboxy, -COOR 8 where R 8 is an ester group, -CONR 9 R 10 where R 9 and R 10 are each hydrogen or C1-4 alkyl, or -SO2R 11 where R 11 is Ci ⁇ alkyl or optionally substituted phenyl-Cj.4 alkyl;
- R 4 is 1-pyrrolyl, 1-imidazolyl or 1-pyrazolyl, each of which is optionally substituted by one or two C 1-4 alkyl, carboxyl, hydroxyl-Ci-4 alkyl or -CHO groups, or R 4 is l-(l,2,4-triazolyl), l-(l,3,4-triazolyl) or 2-(l,2,3-triazolyl), each of which is optionally substituted by a Ci -4 alkyl or Ci -4 perfluoroalkyl group, or R 4 is 1-tetrazolyl optionally substituted by Ci -4 alkyl;
- EP619314 discloses the preparation of 4-phenyl-4//-naphtho[2,l-6]pyran derivatives:
- Ri and R 2 are independently halo, trifluoromethyl, C 1 -C 4 alkoxy, hydroxy, nitro, Ci-C 4 alkyl, Ci-C 4 alkylthio, hydroxy-Ci-C 4 alkyl, hydroxy-Ci-C 4 alkoxy, trifluoromethoxy, carboxy, -COORs where Rg is an ester group, -COR 9 , -CONR 9 R 10 or - NR9R10 where R 9 and Rio are each hydrogen or C 1 -C 4 alkyl;
- R 3 is nitrile, carboxy or -CO 2 Ri 1 wherein Rj 1 is an ester group;
- X is C 2 -C 4 alkylene, or R4 is optionally substituted 1-pyrrolyl; and m and n are each independently 0-2.
- the compounds are said to be useful for the treatment of restenosis, immune disease, and diabetic complications.
- X is O, S or NR. 6 , wherein R 6 is hydrogen or optionally substituted alkyl;
- Y is CN, COR 7 , CO 2 R 7 or CONR x Ry, wherein R 7 , R x and R y are independently hydrogen, C M O alkyl, haloalkyl, aryl, fused aryl, carbocyclic, a heterocyclic group, a heteroaryl group, alkenyl, alkynyl, arylalkyl, arylalkenyl, arylalkynyl, heteroarylalkyl, heteroarylalkenyl, heteroarylalkynyl, carbocycloalkyl, heterocycloalkyl, hydroxyalkyl or aminoalkyl; or R x and R y are taken together with the nitrogen to which they are attached to form a heterocycle;
- A is optionally substituted and is aryl, heteroaryl, saturated carbocyclic, partially saturated carbocylic, saturated heterocyclic, partially saturated heterocyclic, arylalkyl or heteroaryl alkyl;
- B is an optionally substituted aromatic or heteroaromatic ring.
- X is O, S or NR O , wherein R $ is hydrogen or optionally substituted alkyl or aryl;
- Y is CN, COR 7 , CO 2 R 7 or CONR x Ry, wherein R 7 , R x and R y are independently hydrogen, Ci-io alkyl, haloalkyl, aryl, fused aryl, carbocyclic, a heterocyclic group, a heteroaryl group, alkenyl, alkynyl, arylalkyl, arylalkenyl, arylalkynyl, heteroarylalkyl, heteroarylalkenyl, heteroarylalkynyl, carbocycloalkyl, heterocycloalkyl, hydroxyalkyl or aminoalkyl; or R x and R y are taken together with the nitrogen to which they are attached to form a heterocycle;
- Z is O, S, halo, NR 8 , or NCORs, wherein R 8 is independently H, Ci -4 alkyl or aryl;
- A is optionally substituted and is aryl, heteroaryl, saturated carbocyclic, partially saturated carbocyclic, saturated heterocyclic, partially saturated heterocyclic, arylalkyl or heteroarylalkyl;
- B is optionally substituted and is an aryl, heteroaryl, saturated carbocyclic, partially saturated carbocyclic, saturated heterocyclic, or partially saturated heterocyclic ring.
- X is O, S or NR O , wherein R 6 is hydrogen or optionally substituted alkyl
- Y is CN, COR 7 , CO 2 R 7 or CONR x Ry, wherein R 7 , R x and R y are independently hydrogen, C MO alkyl, haloalkyl, aryl, fused aryl, carbocyclic, a heterocyclic group, a heteroaryl group, alkenyl, alkynyl, arylalkyl, arylalkenyl, arylalkynyl, heteroarylalkyl, heteroarylalkenyl, heteroarylalkynyl, carbocycloalkyl, heterocycloalkyl, hydroxyalkyl or aminoalkyl; or R x and R y are taken together with the nitrogen to which they are attached to form a heterocycle;
- R1-R2 are independently hydrogen, halo, haloalkyl, aryl, fused aryl, carbocyclic, a heterocyclic group, a heteroaryl group, d-io alkyl, alkenyl, alkynyl, arylalkyl, arylalkenyl, arylalkynyl, heteroarylalkyl, heteroarylalkenyl, heteroarylalkynyl, carbocycloalkyl, heterocycloalkyl, hydroxyalkyl, aminoalkyl, carboxyalkyl, nitro, amino, cyano, acylamido, hydroxy, thiol, acyloxy, azido, alkoxy, carboxy, methylenedioxy, carbonylamido or alkylthiol;
- R 5 is hydrogen or C MO alkyl
- A is optionally substituted and is aryl, heteroaryl, saturated carbocyclic, partially saturated carbocylic, saturated heterocyclic, partially saturated heterocyclic, arylalkyl or heteroarylalkyl;
- B is optionally substituted and is a fused thiazole, oxazole, 2-imino-imidazole, 2,l ,3-thiadiazo-2-one, thiazol-2-one, oxazol-2-one, imidazol-2-thione, thiazol-2-thione, oxazol-2-thione, imidazoline, oxazoline, thiazoline, triazole, oxazine, oxazine-2,3-dione, or piperazine ring.
- PCT published patent application WO02/092594 disclosed substituted 4H- chromenes and analogs as activators of caspases and inducers of apoptosis:
- R 1 -R 4 are independently hydrogen, halo, haloalkyl, aryl, fused aryl, carbocyclic, a heterocyclic group, a heteroaryl group, C 1-1O alkyl, alkenyl, alkynyl, arylalkyl, arylalkenyl, arylalkynyl, heteroarylalkyl, heteroarylalkenyl, heteroarylalkynyl, carbocycloalkyl, heterocycloalkyl, hydroxyalkyl, aminoalkyl, carboxyalkyl, nitro, amino, cyano, acylamido, hydroxy, thiol, acyloxy, azido, alkoxy, carboxy, methylenedioxy, carbonylamido or alkylthiol; or Ri and R 2 , or R 2 and K 3 , or R3 and R4, taken together with the atoms to which they are attached form an aryl, heteroaryl, partially saturated carbo
- R 5 is hydrogen or Ci -io alkyl
- A is optionally substituted and is aryl, heteroaryl, saturated carbocyclic, partially saturated carbocylic, saturated heterocyclic, partially saturated heterocyclic or arylalkyl;
- Y is CN, COR 7 , CO2R7 or CONR x Ry, wherein R 7 , R x and R y are independently hydrogen, Ci-io alkyl, haloalkyl, aryl, fused aryl, carbocyclic, a heterocyclic group, a heteroaryl group, alkenyl, alkynyl, arylalkyl, arylalkenyl, arylalkynyl, heteroarylalkyl, heteroarylalkenyl, heteroarylalkynyl, carbocycloalkyl, heterocycloalkyl, hydroxyalkyl or aminoalkyl; or R x and R y are taken together with the nitrogen to which they are attached to form a heterocycle; and
- PCT published patent application WO03/097806 disclosed substituted 4-aryl-4//- pyrrolo[2,3- ⁇ ]chromenes and analogs as activators of caspases and inducers of apoptosis:
- Ri is selected from the group consisting of alkyl, cycloalkyl, cycloalkylalkyl, hydroxyalkyl, haloalkyl, alkoxyalkyl, aminoalkyl and oxiranylalkyl;
- R 3 and R 4 are independently hydrogen, halo, haloalkyl, aryl, fused aryl, carbocyclic, a heterocyclic group, a heteroaryl group, C LIO alkyl, alkenyl, alkynyl, arylalkyl, arylalkenyl, arylalkynyl, heteroarylalkyl, heteroarylalkenyl, heteroarylalkynyl, carbocycloalkyl, heterocycloalkyl, hydroxyalkyl, aminoalkyl, carboxyalkyl, nitro, amino, cyano, acylamido, hydroxy, thiol, acyloxy, azido, alkoxy, carboxy, methylenedioxy, carbonylamido or alkylthiol;
- R 5 is hydrogen or Ci-io alkyl
- A is optionally substituted and is aryl, heteroaryl, saturated carbocyclic, partially saturated carbocyclic, saturated heterocyclic, partially saturated heterocyclic or arylalkyl;
- D is optionally substituted and is a heteroaromatic, partially saturated heterocyclic or saturated heterocyclic fused ring, wherein said fused ring has 5 or 6 ring atoms, wherein one or two of said ring atoms are nitrogen atoms and the others of said ring atoms are carbon atoms;
- Y is CN, CORi 9 , CO 2 Ri 9 or CONR 20 R 2 I, wherein Ri 9 , R 20 and R 2 i are independently hydrogen, Ci-io alkyl, haloalkyl, aryl, fused aryl, carbocyclic, a heterocyclic group, a heteroaryl group, alkenyl, alkynyl, arylalkyl, arylalkenyl, arylalkynyl, heteroarylalkyl, heteroarylalkenyl, heteroarylalkynyl, carbocycloalkyl, heterocycloalkyl, hydroxyalkyl or aminoalkyl; or
- R 2O and R 2 1 are taken together with the nitrogen to form a heterocycle
- Kasibhatla, et al., (MoI. Cancer Ther. 3:1365-74 (2004)) reported a novel series of
- MX-116407 2-amino-4-(3-bromo-4,5-dimethoxy-phenyl)-3-cyano-4H-chromenes to disrupt tumor vasculature and to induce tumor necrosis in vivo.
- MX-116407 One of the compounds, named MX-116407, was found to be highly active and produced tumor regressions in all testing animals in a human lung tumor xenograft (Calu-6) model.
- MX-116407 significantly enhanced the antitumor activity of cisplatin, resulting in 40% tumor-free animals.
- 4-aryl-4H-chromenes as a new series of apoptosis inducers and the structure-activity relationships (SAR) of the 4-aryl group.
- 2-Amino-4-(3-bromo-4,5-dirnethoxyphenyi)-3- cyano-7-(dimethylamino)-4H-chromene (MX-58151) and 2-amino-3-cyano-7- (dimethylamino)-4-(5-methyl-3-pyridyl)-4H-chromene were identified as the lead compounds from these studies.
- Kemnitzer, et al., (Bioorg. Med. Chem. Lett. 15:4745-51 (2005)) reported the exploration of the SAR of 4-aryl-4//-chromenes via modifications at the 7- and 5-, 6-, 8- positions.
- Kemnitzer, et al, reported the exploration of the SAR of 4-aryl-4H-chromenes with fused rings at the 7,8-positions.
- 2-amino-4-(3-bromo-4,5-dimethoxyphenyl)-3-cyano-4,7- dihydropyrano[2,3-e]indole and 2-amino-4-(3-bromo-4,5-dimethoxyphenyl)-3-cyano-4,9- dihydropyrano[3,2-g]indole were found to be highly active both as caspase activators and inhibitors of cell proliferation.
- An embodiment of the present invention relates to the discovery that the R- stereoisomer (IR) of 2,7,8-triamino-4-(3-bromo-4,5-dimethoxyphenyl)-3-cyano-4//- chromene (1) is an activator of the caspase cascade and inducer of apoptosis, and has antivascular effects and is a vascular disrupting agent (VDA) or vascular targeting agent (VTA).
- VDA vascular disrupting agent
- VTA vascular targeting agent
- an embodiment of the present invention relates to the use of compound IR as an inducer of apoptosis and as an antivascular agent.
- the present invention relates to a compound of Formula
- the present invention relates to a prodrug of IR having
- the present invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising the compound of Formula IR, or a pharmaceutically acceptable salt or prodrug thereof, and a pharmaceutically acceptable excipient or carrier
- the present invention relates to a method of treating a disorder responsive to the induction of apoptosis in an animal suffering therefrom, comprising administering to a mammal in need of such treatment an effective amount of a compound of Formula IR, substantially free from the corresponding ( ⁇ -stereoisomer, or a pharmaceutically acceptable salt or prodrug thereof.
- the present invention relates to a method of treating, preventing or ameliorating neoplasia and cancer by administering compound IR, substantially free from the corresponding (5)-stereoisomer, to a mammal in need of such treatment.
- the present invention relates to a method of treating a disorder responsive to an antivascular agent in an animal suffering therefrom, comprising administering to a mammal in need of such treatment an effective amount of a compound of Formula IR, substantially free from the corresponding ( ⁇ -stereoisomer, or a pharmaceutically acceptable salt or prodrug thereof.
- the present invention relates to a method for inhibiting the growth of endothelial cells of an animal in need thereof, comprising delivering to the cells a compound of Formula IR, substantially free from the corresponding ( ⁇ -stereoisomer, or a pharmaceutically acceptable salt or prodrug thereof.
- the present invention relates to a method for inhibiting vascularization in a tissue of an animal in need thereof comprising delivering to the tissue a compound of Formula IR, substantially free from the corresponding ( ⁇ -stereoisomer, or a pharmaceutically acceptable salt or prodrug thereof.
- the present invention relates to a method for the separation of an stereoisomer having Formula IR from the corresponding (S)- stereoisomer, comprising contacting a mixture comprising solvent and a racemic mixture comprising IR and the corresponding ( ⁇ -stereoisomer with a chiral stationary phase, contacting the mixture and the chiral stationary phase with an eluting solvent, and isolating the stereoisomer IR from the eluting solvent, wherein the stereoisomer IR is isolated substantially free of the corresponding (. ⁇ -stereoisomer.
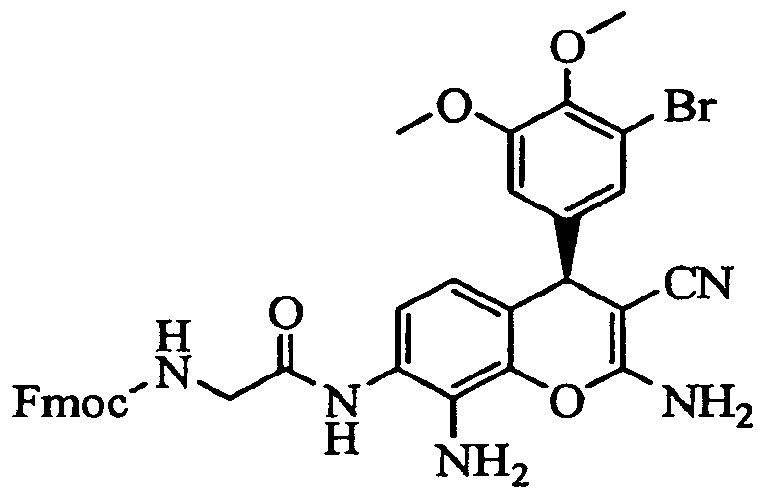
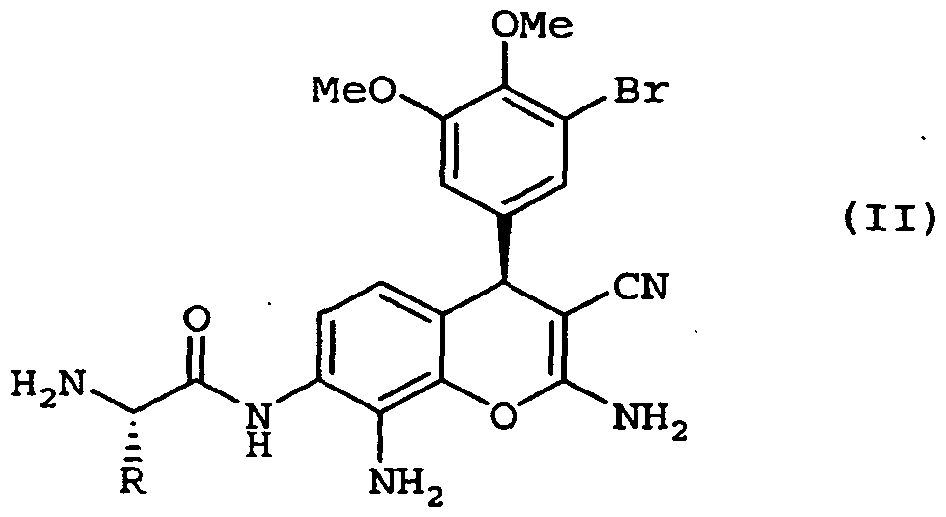
- the present invention relates to a method of preparing a prodrug of Formula II, comprising contacting a compound of Formula IR with a protected amino acid and coupling reagent to form a protected prodrug, and deprotecting the protected prodrug to form the compound of Formula II, wherein the compound of Formula IR is substantially free of the corresponding (5)-stereoisomer.
- substantially free of a given stereoisomer refers to the stereoisomeric purity of the stereoisomer, and is used herein to mean there is greater than 95% of the given stereoisomer present.
- the (i?)-stereoisomer is present in greater than 95%.
- Stereoisomers can be detected using a variety of techniques, for example, chiral HPLC is used.
- the present invention arises out of the surprising discovery that when compound 1 , 2,7,8-triamino-4-(3-bromo-4,5-dimethoxyphenyl)-3-cyano-4H-chromene, which is a potent and efficacious activator of the caspase cascade and inducer of apoptosis, and exists as a racemic mixture, was separated into the corresponding R-isomer (-) and S-isomer (+), the R-isomer IR is found to be the active isomer and the S-isomer (+) is found to be essentially inactive. In addition, the R-isomer (-) is found to be an efficacious antivascular agent.
- the present invention relates to a compound of Formula IR, substantially free from the corresponding ( ⁇ -stereoisomer:
- the compound IR is about 95%, 96%, 97%, 98%, 99% or greater free from the corresponding (S)- stereoisomer. In another example, the compound IR is about 99.9% free from the corresponding (S ⁇ -stereoisomer.
- compositions include, but are not limited to, inorganic and organic acid addition salts, such as hydrochloride, hydrobromide, phosphate, sulphate, citrate, lactate, tartrate, maleate, fumarate, mandelate and oxalate.
- Prodrugs for use in the present invention include, but are not limited to, amides
- Amino acids for use in the present invention include natural and non-natural amino acids.
- Natural amino acids for use in the present invention include alanine, arginine, asparagine, aspartic acid, cysteine, glutamine, glutamic acid, glycine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, proline, serine, threonine, tryptophan, tyrosine and valine.
- prodrugs are amides prepared using amino acid, especially natural amino acid.
- R is hydrogen, alkyl and alkyl substituted with hydroxy, carboxy, carbamoyl, mercapto, imidazolyl, methylthio, aryl, amino or guanidine, or
- R and the NH 2 group that is bonded to the carbon atom to which R is bonded are taken together to form a ring such as in proline.
- Compound 1 can be prepared using any method known in the art.
- compound 1 is prepared as illustrated in Scheme 1. Reaction of 2,3-diaminophenol with 5-bromoveratraldehyde (3-bromo-4,5-dimethoxybenzaldehyde) and malononitrile in the presence of a base such as dimethylisopropylamine (DMIPA) in a solvent such as ethanol produced the racemic mixture 1 in yield of 82-90%.
- DMIPA dimethylisopropylamine
- the present invention relates to a method for the separation of stereoisomers having Formula IR or IS from their racemic mixture having Formula 1.
- the stereoisomer IR or IS can be isolated from the eluting solvent substantially free from the other corresponding stereoisomer.
- the stereoisomer IR can be isolated from the eluting solvent about 96, 97, 98, 99 % or greater free from the other corresponding stereoisomer.
- the stereoisomer IR can be isolated from the eluting solvent about 99% free from the other corresponding stereoisomer.
- the stereoisomer IR can be isolated from the eluting solvent about 99.9% free from the other corresponding stereoisomer.
- the present invention relates to a method for the separation of a stereoisomer having Formula IR from the corresponding (. ⁇ -stereoisomer.
- the method includes contacting a mixture comprising solvent and a racemic mixture comprising IR and the corresponding ( ⁇ -stereoisomer with a chiral stationary phase, contacting the mixture and the chiral stationary phase with an eluting solvent, and isolating said stereoisomer IR from the eluting solvent, wherein the stereoisomer IR is isolated substantially free of the corresponding ( ⁇ -stereoisomer.
- the stereoisomer IR is isolated about 99% free from the corresponding (.S)-St ereo isomer.
- chiral stationary phase refers to separation media capable of separating corresponding stereoisomers (enantiomeric compounds).
- the chiral stationary phase can include chiral molecules and/or polymers bonded to solid supports, chiral phases created in situ on the surface of the solid adsorbent, or surface cavities that allow for specific interactions with one stereoisomer.
- chiral stationary phases for use in the present invention include, but are not limited to, stationary phases in which chiral proteins, small chiral molecules, polymers of cellulose or amylose, marocyclic glycopeptides or cyclodextrins are coated, bonded or otherwise adsorbed to silica or other solid matrices.
- a chiral stationary phase for use in the present invention is an Amylose tris[(-S)- ⁇ -methylbenzylcarbamate] coated on 20 ⁇ m silica gel (available from Daicel Chemical Industries, Ltd. as the CHIRALPAK ® AS-V, Tokyo, Japan).
- Example solvents for use in the chiral separation include MeOH and acetonitrile.
- solvents can be used, for example, ethyl acetate and ethanol.
- racemic mixture 1 can also be separated using other methods, including supercritical fluid conditions (SFC) or using simulated moving bed (SMB) technology.
- SFC supercritical fluid conditions
- SMB simulated moving bed
- a simulated moving bed apparatus which allows for the separation of IR from a mixture of IR and IS is commercially available from, for example, NovaSep, Inc., Boothwyn, PA, or from Knauer, ASI, Franklin, MA (CSEP ® Models). See, for example, the apparatuses disclosed in U.S. Patent Nos. 3,268,605; 4,434,051 and 5,456,825. See also U.S. Patent Nos. 5,126,055; 5,434,298 and 6,533,936 for methods of purifying stereoisomers using simulated moving bed technology.
- Another embodiment of the present invention relates to a method of producing a prodrug of Formula II.
- the method includes contacting a compound of Formula IR with a protected amino acid and coupling reagent to form a protected prodrug, and deprotecting the protected prodrug to form the compound of Formula II.
- the compound of Formula IR is substantially free of the corresponding (5)-stereoisomer.
- Protected amino acids for use in the present invention include protected natural and non-natural amino acids.
- the protected amino acid is 9- fluorenylmethyl carbamate protected L-alanine (Fmoc-L-alanine).
- Coupling reagents for use in the present invention include those reagents that efficiently, and in high yield, couple amino acids to amino groups.
- the coupling reagent is a mixture that includes dicyclohexylcarbodiimide (DCC) and hydroxybenzotriazole (HOBt), or in another example, the coupling reagent is a mixture that includes l-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDC) and hydroxybenzotriazole (HOBt). It is understood by one ordinary skill in the art that other coupling reagents can be used.
- Protected prodrugs can be deprotected using any method that efficiently, and in high yield, cleaves the protecting group from the prodrug.
- the protected prodrug is deprotected using aqueous base.
- base for use in the deprotection includes, but are not limited to, aqueous hydroxide bases, including aqueous solutions of a hydroxide salt.
- Hydroxide salts include, for example, ammonium, sodium, calcium potassium and magnesium hydroxide.
- amino acid prodrugs of compound IR can be prepared as shown in Scheme 3. Reaction of IR with a 9-fluorenylmethyl carbamate (Fmoc) protected amino acid, such as Fmoc-L-alanine, in the presence of coupling agents, such as dicyclohexylcarbodiimide (DCC) and hydroxybenzotriazole (HOBt), produced the protected prodrug, Fmoc-alanine amide, of IR.
- the Fmoc protecting group can be removed under basic conditions, such as using 2N NaOH, to produce the alanine amide of IR, which is expected to have better aqueous solubility than that of IR due to the presence of the more basic amino group.
- the amide prodrug is injected into animals, such as mice or human, it is expected that the amino acid will be removed by amino peptidase to produce the active drug IR.
- An embodiment of the present invention relates to the discovery that compound
- IR is an activator of caspases and inducer of apoptosis. Therefore, compound IR is useful in a variety of clinical conditions in which there is uncontrolled cell growth and spread of abnormal cells, such as in the case of cancer.
- Another embodiment of the present invention relates to the discovery that compound IR is a potent and highly efficacious activator of caspases and inducer of apoptosis in drug resistant cancer cells, such as breast and prostate cancer cells, which enables it to kill these drug resistant cancer cells.
- drug resistant cancer cells such as breast and prostate cancer cells
- compound IR is useful for the treatment of drug resistant cancer in animals.
- compound IR is a potent anti vascular agent. Therefore, compound IR is useful for inhibiting the growth of endothelial cells and inhibiting the vascularization of a tissue.
- compound IR is useful for the treatment of cancer in animals via targeting or disrupting vasculature in rumors, blocking blood supply to the tumors and causing tumor cell death.
- Compound IR also is expected to be useful for the treatment of other diseases due to overgrowth of vasculature, such as ocular neovascularization.
- Another embodiment of the present invention relates to a therapeutic method useful to modulate in vivo apoptosis or in vivo neoplastic disease, comprising administering to a subject in need of such treatment an effective amount of compound IR, substantially free from the corresponding ( ⁇ -stereoisomer, or a pharmaceutically acceptable salt or prodrug, which functions as a caspase cascade activator and inducer of apoptosis.
- Another embodiment of the present invention relates to a therapeutic method comprising administering to an animal an effective amount of compound IR, substantially free from the corresponding ( ⁇ -stereoisomer, or a pharmaceutically acceptable salt or prodrug of said compound, wherein said therapeutic method is useful to treat cancer, which is a group of diseases characterized by the uncontrolled growth and spread of abnormal cells.
- Such diseases include, but are not limited to, Hodgkin's disease, non-Hodgkin's lymphoma, acute lymphotic leukemia, chronic lymphocytic leukemia, multiple myeloma, neuroblastoma, breast carcinoma, ovarian carcinoma, lung carcinoma, Wilms 1 tumor, cervical carcinoma, testicular carcinoma, soft-tissue sarcoma, primary macroglobulinemia, bladder carcinoma, chronic granulocytic leukemia, primary brain carcinoma, malignant melanoma, small-cell lung carcinoma, stomach carcinoma, colon carcinoma, malignant pancreatic insulinoma, malignant carcinoid carcinoma, choriocarcinomas, mycosis fungoides, head or neck carcinoma, osteogenic sarcoma, pancreatic carcinoma, acute granulocytic leukemia, hairy cell leukemia, neuroblastoma, rhabdomyosarcoma, Kaposi's sarcoma, genitourinary carcinoma, thyroid carcinoma, esoph
- compositions containing therapeutically effective concentrations of the compound formulated for oral, intravenous, local and topical application, for the treatment of cancer, neoplastic diseases and other diseases in which caspase cascade mediated physiological responses are implicated are administered to an individual exhibiting the symptoms of one or more of these disorders.
- the amounts are effective to ameliorate or eliminate one or more symptoms of the disorders.
- An effective amount of a compound for treating a particular disease is an amount that is sufficient to ameliorate, or in some manner reduce, the symptoms associated with the disease.
- Such amount can be administered as a single dosage or can be administered according to a regimen, whereby it is effective.
- the amount can cure the disease but, typically, is administered in order to ameliorate the disease. Typically, repeated administration is required to achieve the desired amelioration of symptoms.
- Another embodiment of the present invention relates to a pharmaceutical composition comprising compound IR, or a pharmaceutically acceptable salt or prodrug of said compound, which functions as a caspase cascade activator and inducer of apoptosis in combination with a pharmaceutically acceptable vehicle is provided.
- the pharmaceutical composition comprising compound IR is substantially free of the corresponding (5)-stereoisomer.
- the pharmaceutical composition comprising compound IR is about 95, 96, 97, 98 or 99 % or greater free of the corresponding (_S)-stereoisomer.
- the pharmaceutical composition comprising compound IR is about 99.9% free from the corresponding (. ⁇ -stereoisomer.
- Another embodiment of the present invention relates to a composition effective to treat cancer comprising compound IR, or a pharmaceutically acceptable salt or prodrug of said compound, which functions as a caspase cascade activator and inducer of apoptosis, in combination with at least one known cancer chemotherapeutic agent, or a pharmaceutically acceptable salt of said agent.
- alkylating agents such as busulfan, cis-platin, mitomycin C, and carboplatin
- antimitotic agents such as colchicine, vinblastine, paclitaxel, and
- Another embodiment of the present invention relates to a composition effective to treat cancer comprising compound IR, or a pharmaceutically acceptable salt or prodrug of said compound, which functions as a caspase cascade activator, inducer of apoptosis, and as a vascular disrupting agent, in combination with at least one approved cancer therapeutic agent (approved now or in the future), or a pharmaceutically acceptable salt of said agent.
- approved cancer therapeutic agents include, as examples, combination with sunitinib, e.g for the treatment of renal cell cancer; combination with sorafenib, e.g. for the treatment of liver cancer; combination with carbotaxel or bevacuzimab, e.g.
- the approved cancer therapeutic can be used at its approved dose and schedule, such as once every 21 days.
- Compound IR can be administered, for example, starting the day before, the same day or the day after the approved therapeutic agent.
- Compound IR can be administered once a day for 3 consecutive days on a 21 days cycle.
- Alternative cycles for compound IR can be, as an example, once a week for 3 weeks on a 28 days cycle.
- the compound of the invention can be administered together with at least one known chemotherapeutic agent as part of a unitary pharmaceutical composition.
- the compound of the invention can be administered apart from the at least one known cancer chemotherapeutic agent.
- the compound of the invention and the at least one known cancer chemotherapeutic agent are administered substantially simultaneously, i.e. the compounds are administered at the same time or one after the other, so long as the compounds reach therapeutic levels in the blood at the same time.
- the compound of the invention and the at least one known cancer chemotherapeutic agent are administered according to their individual dose schedule, so long as the compounds reach therapeutic levels in the blood.
- Another embodiment of the present invention relates to a composition effective to treat cancer comprising a bioconjugate of said compound IR, which functions as a caspase cascade activator and inducer of apoptosis, in bioconjugation with at least one known therapeutically useful antibodies, such as Herceptin ® or Rituxan ® , growth factors such as DGF, NGF, cytokines, such as IL-2, EL-4, or any molecule that binds to cell surface.
- the antibodies and other molecules will deliver compound IR to its targets and make it an effective anticancer agent.
- the bioconjugates also could enhance the anticancer effect of therapeutically useful antibodies, such as Herceptin ® or Rituxan ® .
- Another embodiment of the present invention relates to a composition effective to treat cancer comprising compound IR or a pharmaceutically acceptable salt or prodrug of said compound, which functions as a caspase cascade activator and inducer of apoptosis, in combination with radiation therapy.
- the compound of the invention can be administered at the same time as the radiation therapy is administered or at a different time.
- Another embodiment of the present invention relates to a composition effective for post-surgical treatment of cancer, comprising compound IR, or a pharmaceutically acceptable salt or prodrug of said compound, which functions as a caspase cascade activator and inducer of apoptosis.
- the invention also relates to a method of treating cancer by surgically removing the cancer and then treating the animal with one of the pharmaceutical compositions described herein.
- a wide range of immune mechanisms operate rapidly following exposure to an infectious agent. Depending on the type of infection, rapid clonal expansion of the T and B lymphocytes occurs to combat the infection. The elimination of the effector cells following an infection is one of the major mechanisms maintaining immune homeostasis. This deletion of reactive cells has been shown to be regulated by a phenomenon known as apoptosis. Autoimmune diseases have been identified as a consequence of deregulated cell death.
- the immune system directs its powerful cytotoxic effector mechanisms against specialized cells, such as oligodendrocytes in multiple sclerosis, the beta cells of the pancreas in diabetes mellitus, and thyrocytes in Hashimoto's thyroiditis (Ohsako, S. & Elkon, K.B., Cell Death Differ. 5:13-21 (1999)). Mutations of the gene encoding the lymphocyte apoptosis receptor Fas/APO-l/CD95 are reported to .
- APS autoimmune lymphoproliferative syndrome
- Fas-Fas ligand (FasL) interaction is known to be required for the maintenance of immune homeostasis.
- Experimental autoimmune thyroiditis (EAT) characterized by autoreactive T and B cell responses and a marked lymphocytic infiltration of the thyroid, is a good model to study the therapeutic effects of FasL. Batteux, F., et al., (J. Immunol. 162:603-608 (1999)) reported that by direct injection of DNA expression vectors encoding FasL into the inflamed thyroid, the development of lymphocytic infiltration of the thyroid was inhibited and induction of infiltrating T cells death was observed. These results show that FasL expression on thyrocytes can have a curative effect on ongoing EAT by inducing death of pathogenic autoreactive infiltrating T lymphocytes.
- Bisindolylmaleimide VIII is known to potentiate Fas-mediated apoptosis in human astrocytoma 132 INl cells and in Molt-4T cells, both of which were resistant to apoptosis induced by anti-Fas antibody in the absence of bisindolylmaleimide VIII. Potentiation of Fas-mediated apoptosis by bisindolylmaleimide VIII was reported to be selective for activated, rather than non-activated, T cells, and was Fas-dependent. Zhou, T., et al., ⁇ Nat. Med.
- Psoriasis is a chronic skin disease that is characterized by scaly red patches.
- Psoralen plus ultraviolet A is a widely used and effective treatment for psoriasis vulgaris and Coven, et al. (Photodermatol. Photoimmunol. Photomed. 15:22-27 (1999)) reported that lymphocytes treated with psoralen 8-MOP or TMP plus UVA displayed DNA degradation patterns typical of apoptotic cell death. Ozawa, et al. (J. Exp. Med. 189:111-718 (1999)) reported that induction of T cell apoptosis could be the main mechanism by which 312-nm UVB resolves psoriasis skin lesions.
- methotrexate Low doses of methotrexate can be used to treat psoriasis to restore a clinically normal skin.
- Heenen, et al. reported that low doses of methotrexate can induce apoptosis and this mode of action could explain the reduction in epidermal hyperplasia during treatment of psoriasis with methotrexate. Therefore, an effective amount of compound IR, or a pharmaceutically acceptable salt or prodrug of compound IR, which functions as a caspase cascade activator and inducer of apoptosis, is an effective treatment for hyperproliferative diseases, such as psoriasis.
- Synovial cell hyperplasia is a characteristic of patients with rheumatoid arthritis
- RA synovial cells Excessive proliferation of RA synovial cells, as well as those defective in synovial cell death, might be responsible for the synovial cell hyperplasia.
- Wakisaka, et al. found that although RA synovial cells could die via apoptosis through Fas/FasL pathway, apoptosis of synovial cells was inhibited by proinflammatory cytokines present within the synovium, and suggested that inhibition of apoptosis by the proinflammatory cytokines can contribute to the outgrowth of synovial cells, and lead to pannus formation and the destruction of joints in patients with RA.
- an effective amount of compound IR, or a pharmaceutically acceptable salt or prodrug of compound IR, which functions as a caspase cascade activator and inducer of apoptosis is an effective treatment for rheumatoid arthritis.
- compositions within the scope of the present invention include all compositions wherein compound IR is contained in an amount that is effective to achieve its intended purpose. While individual needs vary, determination of optimal ranges of effective amounts of each component is within the skill of the art.
- the compounds can be administered to mammals, e.g., humans, orally at a dose of 0.0025 to 50 mg/kg, or an equivalent amount of the pharmaceutically acceptable salt thereof, per day, of the body weight of the mammal being treated for apoptosis-mediated disorders. In one example, about 0.01 to about 10 mg/kg is orally administered to treat or prevent such disorders. For intramuscular injection, the dose is generally about one-half of the oral dose.
- a suitable intramuscular dose would be about 0.0025 to about 25 mg/kg, and another example would be from about 0.01 to about 5 mg/kg.
- a known cancer chemotherapeutic agent is also administered, it is administered in an amount which is effective to achieve its intended purpose.
- the amounts of such known cancer chemotherapeutic agents effective for cancer are well known to those of skill in the art.
- the unit oral dose comprises about 0.01 to about 50 mg.
- the unit oral dose comprises about 0.1 to about 10 mg of the compounds of the present invention.
- the unit dose can be administered one or more times daily as one or more tablets, each containing from about 0.1 to about 10, conveniently about 0.25 to 50 mg of the compound or its solvates.
- the compound in a topical formulation, can be present at a concentration of about
- the compounds of the present invention can be administered as part of a pharmaceutical preparation.
- the pharmaceutical preparation can contain suitable pharmaceutically acceptable carriers that include, for example, excipients and auxiliaries.
- the excipients and auxiliaries facilitate processing the compounds into preparations which can be used pharmaceutically.
- the orally administered preparations comprise 0.01 to 99 percent, of active compound(s), together with the excipient. In another example, the orally administered preparations comprise about 0.25 to 75 percent of active compound(s), together with the excipient.
- tablets, dragees, or capsules can be used as the forms for orally administering the compounds of the present invention.
- the rectally administered preparations as well as oral solutions and solutions for injection, comprise about 0.01 to 99 percent of active compound(s), together with the excipient. In another example, these preparations comprise from about 0.25 to 75 percent of active compound(s), together with the excipient.
- suppositories can be used as the form for rectally administering the compounds of the present invention.
- compositions of the invention can be administered to any animal that can experience the beneficial effects of the compounds of the invention.
- animals are mammals, e.g., humans and veterinary animals, although the invention is not intended to be so limited.
- compositions of the present invention can be administered by any means that achieve their intended purpose.
- administration can be by parenteral, subcutaneous, intravenous, intramuscular, intraperitoneal, transdermal, buccal, intrathecal, intracranial, intranasal or topical routes.
- administration can be by the oral route.
- the dosage administered will be dependent upon the age, health, and weight of the recipient, kind of concurrent treatment, if any, frequency of treatment, and the nature of the effect desired.
- compositions of the present invention are manufactured in a manner which is itself known, e.g., by means of conventional mixing, granulating, dragee-making, dissolving, or lyophilizing processes.
- pharmaceutical preparations for oral use can be obtained by combining the active compounds with solid excipients, optionally grinding the resultant mixture and processing the mixture of granules, after adding suitable auxiliaries, if desired or necessary, to obtain tablets or dragee cores.
- Excipients for use in the present invention include, but are not limited to, fillers, such as saccharides, e.g., lactose or sucrose, mannitol or sorbitol; cellulose preparations and/or calcium phosphates, e.g., tricalcium phosphate or calcium hydrogen phosphate; and binders, such as starch paste, using, e.g., maize starch, wheat starch, rice starch, potato starch, gelatin, tragacanth, methyl cellulose, hydroxypropylmethylcellulose, sodium carboxymethylcellulose, and/or polyvinyl pyrrolidone.
- fillers such as saccharides, e.g., lactose or sucrose, mannitol or sorbitol
- cellulose preparations and/or calcium phosphates e.g., tricalcium phosphate or calcium hydrogen phosphate
- binders such as starch paste, using, e.g., maize starch,
- disintegrating agents can be added such as the above-mentioned starches and also carboxymethyl-starch, cross-linked polyvinyl pyrrolidone, agar, or alginic acid or a salt thereof, such as sodium alginate.
- Auxiliaries are, above all, flow-regulating agents and lubricants, e.g., silica, talc, stearic acid or salts thereof, such as magnesium stearate or calcium stearate, and/or polyethylene glycol.
- Dragee cores are provided with suitable coatings which, optionally, are resistant to gastric juices.
- concentrated saccharide solutions can be used, which can optionally contain gum arabic, talc, polyvinyl pyrrolidone, polyethylene glycol and/or titanium dioxide, lacquer solutions and suitable organic solvents or solvent mixtures.
- suitable cellulose preparations such as acetylcellulose phthalate or hydroxypropymethyl-cellulose phthalate, are used.
- Dye stuffs or pigments can be added to the tablets or dragee coatings, e.g., for identification or in order to characterize combinations of active compound doses.
- Other pharmaceutical preparations which can be used orally, include push-fit capsules made of gelatin, as well as soft, sealed capsules made of gelatin and a plasticizer, such as glycerol or sorbitol.
- the push-fit capsules can contain the active compounds in the form of granules, which can be mixed with fillers, such as lactose, binders such as starches, and/or lubricants such as talc or magnesium stearate and, optionally, stabilizers.
- the active compounds are dissolved or suspended in suitable liquids, such as fatty oils, or liquid paraffin. Stabilizers can optionally be added.
- compositions which can be used rectally include, e.g., suppositories, which consist of a combination of one or more of the active compounds with a suppository base.
- suitable suppository bases are natural or synthetic triglycerides, or paraffin hydrocarbons.
- gelatin rectal capsules which consist of a combination of the active compounds with a base.
- Possible base materials include, e.g., liquid triglycerides, polyethylene glycols, or paraffin hydrocarbons.
- Formulations for parenteral administration include aqueous solutions of the active compounds in water-soluble form, e.g., water-soluble salts and alkaline solutions.
- the active compound can be present in about 0.01 to about 50 mg/mL.
- suspensions of the active compounds as appropriate oily injection suspensions can be administered.
- Suitable lipophilic solvents, vehicles, excipients or carriers include fatty oils, e.g., sesame oil, or synthetic fatty acid esters, e.g., ethyl oleate or triglycerides; polyethylene glycols (“PEG”), e.g., PEG-200, 400, 600, 800 or 1000; cremophor; cyclodextrins; or block copolymers of poly(ethylene glycol) and poly(propylene glycol) (“poloxamers”), e.g., LUTROL ® .
- fatty oils e.g., sesame oil, or synthetic fatty acid esters, e.g., ethyl oleate or triglycerides
- PEG polyethylene glycols
- PEG-200 polyethylene glycols
- cremophor cremophor
- cyclodextrins or block copolymers of poly(ethylene glycol) and poly(propy
- the excipient or carrier is selected from the group consisting of poly(ethylene glycol), block copolymers of poly(ethylene glycol) and poly(propylene glycol), and saline.
- a pharmaceutical composition for use in the present invention includes about 10 mg/mL of the compound IR, substantially free from the corresponding (S)-stereoisomer, about 25% (v/v) poly(ethylene glycol), about 5% (v/v) block copolymers of poly(ethylene glycol) and poly(propylene glycol) and saline.
- Aqueous injection suspensions can contain substances, which increase the viscosity of the suspension, e.g., sodium carboxymethyl cellulose, sorbitol, polysorbate, e.g. polysorbate 20, 80, 81, 90 and 94 (e.g., TWEEN ® ), dextrose, e.g. 1%, 2%, 5%, 10% or 20% solutions of dextrose in water (e.g., 5% dextrose in water "D5W”), and/or dextran.
- the suspension can also contain stabilizers.
- the excipient or carrier is selected from the group consisting of poly(ethylene glycol), polysorbate, and a solution of 5% dextrose in water.
- a pharmaceutical composition for use in the present invention includes about 10 mg/mL of the compound IR, substantially free from the corresponding ( ⁇ -stereoisomer, about 7% (v/v) poly(ethylene glycol) 400, about 9% (v/v) polysorbate 80 and about 84% (v/v) of a solution of 5% dextrose in water.
- compounds of the invention are employed in topical and parenteral formulations and are used for the treatment of skin cancer.
- the topical compositions of this invention are formulated as oils, creams, lotions, ointments and the like by choice of appropriate carriers.
- suitable carriers include vegetable or mineral oils, white petrolatum (white soft paraffin), branched chain fats or oils, animal fats and high molecular weight alcohol (greater than C 12 ).
- Particular examples of carriers include those in which the active ingredient is soluble.
- Emulsifiers, stabilizers, humectants and antioxidants can also be included, as well as agents imparting color or fragrance, if desired.
- transdermal penetration enhancers can be employed in these topical formulations. Examples of such enhancers can be found in U.S. Patent Nos. 3,989,816 and 4,444,762.
- creams are formulated from a mixture of mineral oil, self- emulsifying beeswax and water, in which the active ingredient, dissolved in a small amount of an oil, such as almond oil, is admixed.
- an oil such as almond oil
- a typical example of such a cream is one which includes about 40 parts water, about 20 parts beeswax, about 40 parts mineral oil and about 1 part almond oil.
- Ointments can be formulated by mixing a solution of the active ingredient in a vegetable oil, such as almond oil with warm soft paraffin, and allowing the mixture to cool.
- a vegetable oil such as almond oil
- a typical example of such an ointment is one which includes about 30% almond oil and about 70% white soft paraffin by weight.
- DLD-I and human non-small cell lung cancer cell line H1299 were grown according to media component mixtures designated by American Type Culture Collection + 10% FCS (Invitrogen Corporation), in a 5% CO 2 -95% humidity incubator at 37 0 C.
- T-47D and ZR-75-1 cells were maintained at a cell density between 30 and 80% confluency at a cell density of 0.1 to 0.6 x 10 6 cells/mL.
- Cells were harvested at 600xg and resuspended at 0.65 x 10 6 cells/mL into appropriate media + 10% FCS.
- the samples were mixed by agitation and then incubated at 37 0 C for 24 h in a 5% CO2-95% humidity incubator. After incubation, the samples were removed from the incubator and 50 ⁇ l of a solution containing 20 ⁇ M of N-(Ac-DEVD)-W- ethoxycarbonyl-R110 (SEQ ID No:l) fluorogenic substrate (Cytovia, Inc.; US 6,335,429), 20% sucrose (Sigma), 20 mM DTT (Sigma), 200 mM NaCl (Sigma), 40 mM Na PIPES buffer p ⁇ 7.2 (Sigma), and 500 ⁇ g/ml lysolecithin (Calbiochem) was added.
- a solution containing 20 ⁇ M of N-(Ac-DEVD)-W- ethoxycarbonyl-R110 (SEQ ID No:l) fluorogenic substrate (Cytovia, Inc.; US 6,335,429)
- 20% sucrose Sigma
- R(-)-2,7,8-triamino-4-(3-bromo-4,5-dimethoxy-phenyl)-3-cyano-4//- chromene is a potent caspase cascade activator and inducer of apoptosis in solid tumor cells, and is the active isomer of the racemate 2,7,8-triamino-4-(3-bromo-4,5- dimethoxy-phenyl)-3-cyano-4//-chromene.
- IS 5'(+)-2,7,8-triamino-4-(3-bromo-4,5- dimethoxy-phenyl)-3-cyano-4//-chromene
- IS is the inactive isomer of the racemate 2,7,8-triamino-4-(3-bromo-4,5-dimethoxy-phenyl)-3-cyano-4H-chromene.
- the observed activity of IS might be due to the presence of small percent (-1%) of IR in the sample tested.
- Compound 4 the alanine amide prodrug of IR, also is a potent caspase cascade activator and inducer of apoptosis.
- SNU398, colon cancer cells HCTl 16 and HeLa cells were grown and harvested as in Example 7.
- An aliquot of 90 ⁇ L of cells (4.4 x 10 4 cells/mL) was added to a well of a 96-well microtiter plate containing 5 ⁇ L of a 10 % DMSO in RPMI-1640 media solution containing 10 nM to 100 ⁇ M of R(-)-2,7,8-triamino-4-(3-bromo-4,5-dimethoxy-phenyl)- 3-cyano-4H-chromene (1 nM to 10 ⁇ M final).
- Baseline for GI5 0 dose for 50% inhibition of cell proliferation
- GI5 0 dose for 50% inhibition of cell proliferation
- the samples were mixed by agitation and then incubated at 37 0 C for 0.5 h in a 5% CO 2 -95% humidity incubator. After incubation, the samples were removed from the incubator and 25 ⁇ L of CellTiter-Glo TM reagent (Promega) was added.
- GI 50 (nM) are summarized in Table II: Table II. GI 50 in Cancer Cells
- R(-)-2,7,8-triamino-4-(3-bromo-4,5-dimethoxy-phenyl)-3-cyano-4H- chromene is identified as antineoplastic compound that inhibits cell proliferation.
- Miroscopy images were recorded with a Zeiss LSM 510 confocal microscope (Zeiss Canada Ltd, Toronto, Ontario, Canada). The effect of compound IR on capillary tube disruption was evaluated by light microscopy (x40 magnification) and the results are summarized in Table m.
- Compound IR was prepared as a 10 mg/mL solution with 25% PEG 400(v/v), 5%
- Lutrol (w/v) in saline A solution of 6.67% Lutrol in saline was prepared by adding 933 ⁇ L of saline into a vial containing 66.7 mg of Lutrol, and mixed until the Lutrol dissolved in the saline.
- a solution of 40 mg/mL compound IR in PEG 400 was prepared by adding 973 ⁇ L of PEG 400 into a vial containing 40 mg of compound IR, vortexed and the vial was placed on shaker or rotator until IR was dissolved in PEG 400. The mixture can be heated occasionally to 5O 0 C to facilitate dissolution if needed.
- a solution of 10 mg/mL compound IR in 25% PEG 400 (v/v), 5% Lutrol (w/v) in saline was prepared by pipetting 750 ⁇ L of 6.67% Lutrol in saline into a vial containing 250 ⁇ L of 40 mg/mL compound IR in PEG 400, mixing while adding the saline solution.
- the solution was passed through 0.2 ⁇ m filter before injection.
- Compound IR also can be formulated as a 10 mg/mL solution in 7% PEG400/ 9%
- Compound IR also can be formulated as a 10 mg/mL solution in 10% cremophor/ 10% ethanol/ 80% saline, and used for IV injection.
- mice implanted with MXl human breast cancer tumors were treated with compound IR as described in EXAMPLE 11.
- Cisplatin was administered IP whereas doxorubicin was administered IV at the indicated doses.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Immunology (AREA)
- Dermatology (AREA)
- Hematology (AREA)
- Oncology (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Ophthalmology & Optometry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Pyrane Compounds (AREA)
- Medicinal Preparation (AREA)
Abstract
Description




































Claims







Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2007271743A AU2007271743B2 (en) | 2006-07-06 | 2007-07-06 | Substituted 4-aryl-chromene as activator of caspases and inducer of apoptosis and as antivascular agent and the use thereof |
| CA2656706A CA2656706C (en) | 2006-07-06 | 2007-07-06 | Substituted 4-aryl-chromene as activator of caspases and inducer of apoptosis and as antivascular agent and the use thereof |
| JP2009518401A JP2009542701A (en) | 2006-07-06 | 2007-07-06 | Substituted 4-aryl-chromenes as caspase activators and inducers of apoptosis and as anti-vascular agents and methods of use thereof |
| EP07810283A EP2049101A4 (en) | 2006-07-06 | 2007-07-06 | Substituted 4-aryl-chromene as activator of caspases and inducer of apoptosis and as antivascular agent and the use thereof |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US80667406P | 2006-07-06 | 2006-07-06 | |
| US60/806,674 | 2006-07-06 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2008005572A2 true WO2008005572A2 (en) | 2008-01-10 |
| WO2008005572A3 WO2008005572A3 (en) | 2008-03-20 |
Family
ID=38895260
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/015676 WO2008005572A2 (en) | 2006-07-06 | 2007-07-06 | Substituted 4-aryl-chromene as activator of caspases and inducer of apoptosis and as antivascular agent and the use thereof |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US7968595B2 (en) |
| EP (1) | EP2049101A4 (en) |
| JP (1) | JP2009542701A (en) |
| CN (1) | CN101657196A (en) |
| AU (1) | AU2007271743B2 (en) |
| CA (2) | CA2938241A1 (en) |
| WO (1) | WO2008005572A2 (en) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012062905A3 (en) * | 2010-11-12 | 2013-01-03 | Deutsches Krebsforschungszentrum (Dkfz) | Chromene derivatives and their analoga as wnt pathway antagonists |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2447010C (en) * | 2001-05-16 | 2011-08-02 | Cytovia, Inc. | Substituted 4h-chromenes and analogs as activators of caspases and inducers of apoptosis and the use thereof |
| ES2379242B1 (en) * | 2010-09-28 | 2013-03-04 | Consejo Superior De Investigaciones Científicas (Csic) | CHROME DERIVATIVES |
| CN103896899A (en) * | 2012-12-25 | 2014-07-02 | 韩冰 | Compounds for reducing intraocular pressure and preparation method and application thereof |
| CN104860915B (en) * | 2015-04-10 | 2016-11-23 | 昆明理工大学 | A kind of preparation method of 4H-4-aryl benzopyrans compounds |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE60008574T2 (en) | 1999-11-05 | 2009-10-01 | Cytovia, Inc., San Diego | Substituted 4H-chromenes and analogues as caspase activators and apoptosis inducers and their use |
| CA2447010C (en) * | 2001-05-16 | 2011-08-02 | Cytovia, Inc. | Substituted 4h-chromenes and analogs as activators of caspases and inducers of apoptosis and the use thereof |
| EP1392283A4 (en) | 2001-05-16 | 2004-10-20 | Cytovia Inc | Substituted coumarins and quinolines as caspase activators |
| US6858607B1 (en) | 2001-05-16 | 2005-02-22 | Cytovia, Inc. | 7,8-fused 4H-chromene and analogs as activators of caspases and inducers of apoptosis and the use thereof |
| EP1404329A4 (en) | 2001-06-04 | 2006-11-02 | Cytovia Inc | Substituted 4-aryl-3-(3-aryl-1-oxo-2-propenyl)-2(1h)-quinolinones and analogs as activators of caspases and inducers of apoptosis and the use thereof |
| AU2003241482A1 (en) | 2002-05-16 | 2003-12-02 | Cytovia, Inc. | Substituted 4h-chromenes, 2h-chromenes, chromans and analogs as activators of caspases and inducers of apoptosis and the use thereof |
| AU2003230411A1 (en) | 2002-05-16 | 2003-12-02 | Cytovia, Inc. | Substituted 4-aryl-4h-pyrrolo(2,3-h)chromenes and analogs as activators of caspases and inducers of apoptosis and the use thereof |
| US7135480B2 (en) | 2002-12-12 | 2006-11-14 | Cytovia, Inc. | Substituted 1-benzoyl-3-cyano-pyrrolo [1,2-a] quinolines and analogs as activators of caspases and inducers of apoptosis |
| KR20130081319A (en) | 2003-07-29 | 2013-07-16 | 시그너쳐 알 앤드 디 홀딩스, 엘엘씨 | Amino acid prodrugs |
-
2007
- 2007-07-06 CA CA2938241A patent/CA2938241A1/en not_active Abandoned
- 2007-07-06 AU AU2007271743A patent/AU2007271743B2/en not_active Ceased
- 2007-07-06 CN CN200780032539A patent/CN101657196A/en active Pending
- 2007-07-06 WO PCT/US2007/015676 patent/WO2008005572A2/en active Application Filing
- 2007-07-06 US US11/822,535 patent/US7968595B2/en not_active Expired - Fee Related
- 2007-07-06 JP JP2009518401A patent/JP2009542701A/en active Pending
- 2007-07-06 CA CA2656706A patent/CA2656706C/en not_active Expired - Fee Related
- 2007-07-06 EP EP07810283A patent/EP2049101A4/en not_active Ceased
Non-Patent Citations (3)
| Title |
|---|
| GREENWALD, J. MED. CHEM., vol. 42, 1999, pages 3657 - 3667 |
| LEU, J. MED. CHEM., vol. 42, 1999, pages 3623 - 3628 |
| See also references of EP2049101A4 |
Cited By (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012062905A3 (en) * | 2010-11-12 | 2013-01-03 | Deutsches Krebsforschungszentrum (Dkfz) | Chromene derivatives and their analoga as wnt pathway antagonists |
| US9371333B2 (en) | 2010-11-12 | 2016-06-21 | Deutsches Krebsforschungszentrum | Chromene derivatives and their analoga as Wnt pathway antagonists |
| US10065939B2 (en) | 2010-11-12 | 2018-09-04 | Deutsches Krebsforschungszentrum | Chromene derivatives and their analogs as Wnt pathway antagonists |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2009542701A (en) | 2009-12-03 |
| EP2049101A2 (en) | 2009-04-22 |
| WO2008005572A3 (en) | 2008-03-20 |
| AU2007271743B2 (en) | 2012-07-26 |
| US20080085328A1 (en) | 2008-04-10 |
| CN101657196A (en) | 2010-02-24 |
| US7968595B2 (en) | 2011-06-28 |
| AU2007271743A1 (en) | 2008-01-10 |
| CA2938241A1 (en) | 2008-01-10 |
| EP2049101A4 (en) | 2010-09-15 |
| CA2656706C (en) | 2016-09-20 |
| CA2656706A1 (en) | 2008-01-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2009200374B2 (en) | Substituted 4H-chromenes and analogs as activators of caspases and inducers of apoptosis and the use thereof | |
| US7476741B2 (en) | Substituted 4H-chromens, 2H-chromenes, chromans and analogs as activators of caspases and inducers of apoptosis and the use thereof | |
| AU781986B2 (en) | Substituted 4H-chromene and analogs as activators of caspases and inducers of apoptosis and the use thereof | |
| US7235674B2 (en) | Substituted coumarins and quinolines and analogs as activators of caspases and inducers of apoptosis and the use thereof | |
| AU2007271743B2 (en) | Substituted 4-aryl-chromene as activator of caspases and inducer of apoptosis and as antivascular agent and the use thereof | |
| US6747052B2 (en) | Substituted indole-2-carboxylic acid benzylidene-hydrazides and analogs as activators of caspases and inducers of apoptosis and the use thereof | |
| US7622497B2 (en) | Derivatives of gambogic acid and analogs as activators of caspases and inducers of apoptosis | |
| US7528164B2 (en) | Substituted 4-aryl-4h-pyrrolo[2,3-h]chromenes and analogs as activators of caspases and inducers of apoptosis and the use thereof | |
| US6858607B1 (en) | 7,8-fused 4H-chromene and analogs as activators of caspases and inducers of apoptosis and the use thereof | |
| EP1404329A1 (en) | Substituted 4-aryl-3-(3-aryl-1-oxo-2-propenyl)-2(1h)-quinolinones and analogs as activators of caspases and inducers of apoptosis and the use thereof | |
| EP1392294A1 (en) | 7,8-fused 4$i(h)-chromene and analogs as activators of caspases and inducers of apoptosis and the use thereof | |
| AU2019363148A1 (en) | Urea derivatives for treating and/or preventing cancer | |
| AU2002314781A1 (en) | Substituted 4H-chromenes and analogs as activators of caspases and inducers of apoptosis and the use thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780032539.3 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2656706 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009518401 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007271743 Country of ref document: AU |
|
| NENP | Non-entry into the national phase |
Ref country code: RU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007810283 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2007271743 Country of ref document: AU Date of ref document: 20070706 Kind code of ref document: A |