WO2007146824A2 - Quinoline compounds and methods of use - Google Patents
Quinoline compounds and methods of use Download PDFInfo
- Publication number
- WO2007146824A2 WO2007146824A2 PCT/US2007/070787 US2007070787W WO2007146824A2 WO 2007146824 A2 WO2007146824 A2 WO 2007146824A2 US 2007070787 W US2007070787 W US 2007070787W WO 2007146824 A2 WO2007146824 A2 WO 2007146824A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- fluorophenyl
- yloxy
- compound
- methoxyquinolin
- aryl
- Prior art date
Links
- 0 Cc(cc1)cc(C)c1C(NC*)=O Chemical compound Cc(cc1)cc(C)c1C(NC*)=O 0.000 description 35
- QDLRCGABZIHADX-UHFFFAOYSA-N CN1CCN(CCCOC)CC1 Chemical compound CN1CCN(CCCOC)CC1 QDLRCGABZIHADX-UHFFFAOYSA-N 0.000 description 2
- XFGCKLRUCQKWSV-UHFFFAOYSA-N CC#CCN(CC1)CCC1(C(F)(F)F)O Chemical compound CC#CCN(CC1)CCC1(C(F)(F)F)O XFGCKLRUCQKWSV-UHFFFAOYSA-N 0.000 description 1
- SLJPPDQJRFXTKR-UHFFFAOYSA-N CC(C)(C(CC1)C2)C1(C(Nc(cc1)cc(F)c1Oc1c(cc(c(OCCCN3CCOCC3)c3)OC)c3ncc1)=O)C2=O Chemical compound CC(C)(C(CC1)C2)C1(C(Nc(cc1)cc(F)c1Oc1c(cc(c(OCCCN3CCOCC3)c3)OC)c3ncc1)=O)C2=O SLJPPDQJRFXTKR-UHFFFAOYSA-N 0.000 description 1
- GZDWCJZYZWTVCL-UHFFFAOYSA-N CCN(CC)CC#CC Chemical compound CCN(CC)CC#CC GZDWCJZYZWTVCL-UHFFFAOYSA-N 0.000 description 1
- FFOYRWVKWXALDR-UHFFFAOYSA-N CCN1C(Nc2ccccc2)=NC=C(c(cc2)cc(F)c2[O](C)c2c(cc(c(OCCCN3CCOCC3)c3)OC)c3ncc2)C1=O Chemical compound CCN1C(Nc2ccccc2)=NC=C(c(cc2)cc(F)c2[O](C)c2c(cc(c(OCCCN3CCOCC3)c3)OC)c3ncc2)C1=O FFOYRWVKWXALDR-UHFFFAOYSA-N 0.000 description 1
- KHRMUOAOUDFSHG-UHFFFAOYSA-N CN(CCC1C(Nc(cc2)ccc2F)=O)C1=O Chemical compound CN(CCC1C(Nc(cc2)ccc2F)=O)C1=O KHRMUOAOUDFSHG-UHFFFAOYSA-N 0.000 description 1
- OBUNOEKDTMNWQK-UHFFFAOYSA-N CN(c1ccccc1)C(N1C)=NC=C(c(cc2)cc(F)c2Oc2c(cc(c(OCCCN3CCOCC3)c3)OC)c3ncc2)C1=O Chemical compound CN(c1ccccc1)C(N1C)=NC=C(c(cc2)cc(F)c2Oc2c(cc(c(OCCCN3CCOCC3)c3)OC)c3ncc2)C1=O OBUNOEKDTMNWQK-UHFFFAOYSA-N 0.000 description 1
- ZFELWKKEFVPBJQ-UHFFFAOYSA-N CN1CCN(CCOC)CC1 Chemical compound CN1CCN(CCOC)CC1 ZFELWKKEFVPBJQ-UHFFFAOYSA-N 0.000 description 1
- WMGKXFWDOJQJMK-UHFFFAOYSA-N CN1CCN(COC)CC1 Chemical compound CN1CCN(COC)CC1 WMGKXFWDOJQJMK-UHFFFAOYSA-N 0.000 description 1
- FHTGZDVYPCEHFQ-UHFFFAOYSA-N CNC1CCNCC1 Chemical compound CNC1CCNCC1 FHTGZDVYPCEHFQ-UHFFFAOYSA-N 0.000 description 1
- XUFJIJYJOOQTGK-UHFFFAOYSA-N CNc(nccc1)c1OCc1ccccc1 Chemical compound CNc(nccc1)c1OCc1ccccc1 XUFJIJYJOOQTGK-UHFFFAOYSA-N 0.000 description 1
- FITIAPUZOHEKLR-UHFFFAOYSA-N CNc1cccc(O)n1 Chemical compound CNc1cccc(O)n1 FITIAPUZOHEKLR-UHFFFAOYSA-N 0.000 description 1
- SVEUVITYHIHZQE-UHFFFAOYSA-N CNc1ncccc1 Chemical compound CNc1ncccc1 SVEUVITYHIHZQE-UHFFFAOYSA-N 0.000 description 1
- LTSVUPTVNGVMGM-UHFFFAOYSA-N CNc1ncccc1Oc1ccccn1 Chemical compound CNc1ncccc1Oc1ccccn1 LTSVUPTVNGVMGM-UHFFFAOYSA-N 0.000 description 1
- VGKOARVVEXZTPU-UHFFFAOYSA-N CNc1nccnc1 Chemical compound CNc1nccnc1 VGKOARVVEXZTPU-UHFFFAOYSA-N 0.000 description 1
- KNZJMAOIFHKXEK-UHFFFAOYSA-N COCCCN1CCOCC1 Chemical compound COCCCN1CCOCC1 KNZJMAOIFHKXEK-UHFFFAOYSA-N 0.000 description 1
- PTNHYVCQMCBKIS-UHFFFAOYSA-N COCCN1CCC(CO)CC1 Chemical compound COCCN1CCC(CO)CC1 PTNHYVCQMCBKIS-UHFFFAOYSA-N 0.000 description 1
- FPIOHADYDKAXRD-UHFFFAOYSA-N COCC[n]1cncc1 Chemical compound COCC[n]1cncc1 FPIOHADYDKAXRD-UHFFFAOYSA-N 0.000 description 1
- PKRABCJOENMWLW-UHFFFAOYSA-N COc(c(OC)cc1ncc2)cc1c2Oc(ccc(-c(cc1)ccc1Oc1ccccc1)c1)c1F Chemical compound COc(c(OC)cc1ncc2)cc1c2Oc(ccc(-c(cc1)ccc1Oc1ccccc1)c1)c1F PKRABCJOENMWLW-UHFFFAOYSA-N 0.000 description 1
- WIAJXGOOHAMRNL-UHFFFAOYSA-N COc(c(OC)cc1ncc2)cc1c2Oc(ccc(C1=CN=CN(Cc(ccc(F)c2)c2Cl)C1=O)c1)c1F Chemical compound COc(c(OC)cc1ncc2)cc1c2Oc(ccc(C1=CN=CN(Cc(ccc(F)c2)c2Cl)C1=O)c1)c1F WIAJXGOOHAMRNL-UHFFFAOYSA-N 0.000 description 1
- VIFJVGNINHQQMI-UHFFFAOYSA-N COc(c(OC)cc1ncc2)cc1c2Oc(ccc(C1=CN=CN(Cc2ccc(C(F)(F)F)cc2)C1=O)c1)c1F Chemical compound COc(c(OC)cc1ncc2)cc1c2Oc(ccc(C1=CN=CN(Cc2ccc(C(F)(F)F)cc2)C1=O)c1)c1F VIFJVGNINHQQMI-UHFFFAOYSA-N 0.000 description 1
- DYGVFOXTURBQHP-UHFFFAOYSA-N COc(c(OC)cc1ncc2)cc1c2Oc(ccc(N(C=CC(C(c1ccccc1)=O)=C1)C1=O)c1)c1F Chemical compound COc(c(OC)cc1ncc2)cc1c2Oc(ccc(N(C=CC(C(c1ccccc1)=O)=C1)C1=O)c1)c1F DYGVFOXTURBQHP-UHFFFAOYSA-N 0.000 description 1
- WTMYBXYGYLOQHJ-UHFFFAOYSA-N COc(c(OC)cc1ncc2)cc1c2Oc(ccc(N(C=CC=C1NC(c(cc2)ccc2F)=O)C1=O)c1)c1F Chemical compound COc(c(OC)cc1ncc2)cc1c2Oc(ccc(N(C=CC=C1NC(c(cc2)ccc2F)=O)C1=O)c1)c1F WTMYBXYGYLOQHJ-UHFFFAOYSA-N 0.000 description 1
- WCOUURYBEARRLP-UHFFFAOYSA-N COc(c(OCCCN1CCCCC1)cc1ncc2)cc1c2Oc(ccc(NC(C1=CC=NN(c(cc2)ccc2F)C1=O)=O)c1)c1F Chemical compound COc(c(OCCCN1CCCCC1)cc1ncc2)cc1c2Oc(ccc(NC(C1=CC=NN(c(cc2)ccc2F)C1=O)=O)c1)c1F WCOUURYBEARRLP-UHFFFAOYSA-N 0.000 description 1
- OIYYEFSPBRIBAB-UHFFFAOYSA-N COc(c(OCCCN1CCOCC1)cc1ncc2)cc1c2Oc(ccc(N(Cc(cccc1)c1N1)C1=O)c1)c1F Chemical compound COc(c(OCCCN1CCOCC1)cc1ncc2)cc1c2Oc(ccc(N(Cc(cccc1)c1N1)C1=O)c1)c1F OIYYEFSPBRIBAB-UHFFFAOYSA-N 0.000 description 1
- CYGULOBIQBPIGA-UHFFFAOYSA-N COc(c(OCCCN1CCOCC1)cc1ncc2)cc1c2Oc(ccc(NC(C1=CC=CN(Cc(cc2)ccc2Cl)C1=O)=O)c1)c1F Chemical compound COc(c(OCCCN1CCOCC1)cc1ncc2)cc1c2Oc(ccc(NC(C1=CC=CN(Cc(cc2)ccc2Cl)C1=O)=O)c1)c1F CYGULOBIQBPIGA-UHFFFAOYSA-N 0.000 description 1
- OSIQBRLXIPYVLY-UHFFFAOYSA-N COc(c(OCCCN1CCOCC1)cc1ncc2)cc1c2Oc(ccc(NC(C1=CC=NN(c(cc2)ccc2F)C1=O)=O)c1)c1F Chemical compound COc(c(OCCCN1CCOCC1)cc1ncc2)cc1c2Oc(ccc(NC(C1=CC=NN(c(cc2)ccc2F)C1=O)=O)c1)c1F OSIQBRLXIPYVLY-UHFFFAOYSA-N 0.000 description 1
- JQTSPNODCQKRBY-UHFFFAOYSA-N COc(c(OCC[n]1cncc1)cc1ncc2)cc1c2Oc(ccc(C1=CN=CN(Cc2ccccc2)C1=O)c1)c1F Chemical compound COc(c(OCC[n]1cncc1)cc1ncc2)cc1c2Oc(ccc(C1=CN=CN(Cc2ccccc2)C1=O)c1)c1F JQTSPNODCQKRBY-UHFFFAOYSA-N 0.000 description 1
- LDLQDZDYUPBUND-DHUJRADRSA-N C[C@](CCN1c(cc2)ccc2F)(C(Nc(cc2)cc(F)c2Oc2c(cc(c(OCCCN3CCOCC3)c3)OC)c3ncc2)=O)C1=O Chemical compound C[C@](CCN1c(cc2)ccc2F)(C(Nc(cc2)cc(F)c2Oc2c(cc(c(OCCCN3CCOCC3)c3)OC)c3ncc2)=O)C1=O LDLQDZDYUPBUND-DHUJRADRSA-N 0.000 description 1
- NAECWNOGCOGPLA-UHFFFAOYSA-N Cc1ccc(CN(C=NC=C2c(cc3)cc(F)c3Oc3c(cc(c(OC)c4)OC)c4ncc3)C2=O)cc1 Chemical compound Cc1ccc(CN(C=NC=C2c(cc3)cc(F)c3Oc3c(cc(c(OC)c4)OC)c4ncc3)C2=O)cc1 NAECWNOGCOGPLA-UHFFFAOYSA-N 0.000 description 1
- SIKXIUWKPGWBBF-UHFFFAOYSA-N Clc1nc(Cl)ncc1Br Chemical compound Clc1nc(Cl)ncc1Br SIKXIUWKPGWBBF-UHFFFAOYSA-N 0.000 description 1
- GIRDZQOHBHZJCC-UHFFFAOYSA-N O=C1NC(Cl)=NC=C1Br Chemical compound O=C1NC(Cl)=NC=C1Br GIRDZQOHBHZJCC-UHFFFAOYSA-N 0.000 description 1
- CLJIGLPOBRSBNU-UHFFFAOYSA-N O=C1NC=NC=C1Br Chemical compound O=C1NC=NC=C1Br CLJIGLPOBRSBNU-UHFFFAOYSA-N 0.000 description 1
- GLZGQYWJOQWBQB-UHFFFAOYSA-N OC(C1=NC=CN(c2ccccc2)C1=O)=O Chemical compound OC(C1=NC=CN(c2ccccc2)C1=O)=O GLZGQYWJOQWBQB-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/20—Oxygen atoms
- C07D215/22—Oxygen atoms attached in position 2 or 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the invention relates to quinoline compounds having protein tyrosine kinase activity.
- the quinoline compounds may be useful in the treatment of hyperproliferative disorders, such as cancer, in mammals.
- the invention also relates to pharmaceutical compositions and formulations, methods of synthesis, and methods of use such as treating hyperproliferative disorders.
- Met tyrosine kinase is a high-affinity transmembrane receptor for the hepatocyte growth factor (HGF, Bottaro et al (1991) Science 251:802-804). Met was cloned, named (Cooper et al (1984) 311:29-33) and identified as an oncogene (Park et al (1986) Cell 45:895-904). When deregulated by overexpression or mutations, Met receptor tyrosine kinase leads to tumor growth and invasion (Cristiani et al (2005) Biochem. 44:14110-14119).
- HGF also known as Scatter Factor
- Stimulation of Met by the ligand HGF initiates numerous physiological processes, including cell proliferation, scattering, morphogenic differentiation, angiogenesis, wound healing, tissue regeneration, and embryological development
- Receptor c-Met is rapidly internalized via clathrin-coated vesicles and traffics through an early endosomal compartment after hepatocyte growth factor stimulation.
- c-Met accumulates progressively in perinuclear compartments, which in part include the Golgi (Kermorgant et al (2003) J. of Biol. Chem. 278(31):28921-28929).
- c-Met Antibodies specific for c-Met have been expressed to block binding of HGF to c-Met (US 2005/0037431; US 2004/0166544). c-Met is also over-expressed in both non-small cell lung cancer and small cell lung cancer cells, in lung, breast, colon and prostate tumors (Herynk et al (2003) Cancer Res. 63(l l):2990-2996; Maulik et al (2002) Clin. Cancer Res. 8:620-627). Since c-Met appears to play an important role in oncogenesis of a variety of tumors, various inhibition strategies have been employed to therapeutically target this receptor tyrosine kinase.
- Protein kinases are enzymes that catalyze the phosphorylation of hydroxy groups on tyrosine, serine and threonine residues of proteins by transfer of the terminal (gamma) phosphate from ATP. Through signal transduction pathways, these enzymes modulate cell growth, differentiation and proliferation, i.e., virtually all aspects of cell life in one way or another depend on PK activity. Furthermore, abnormal PK activity has been related to a host of disorders, ranging from relatively non-life threatening diseases such as psoriasis to extremely virulent diseases such as glioblastoma (brain cancer). Protein kinases include two classes; protein tyrosine kinases (PTK) and serine-threonine kinases (STK).
- PTK protein tyrosine kinases
- STK serine-threonine kinases
- PTK activity is their involvement with growth factor receptors which are cell-surface proteins. When bound by a growth factor ligand, growth factor receptors are converted to an active form which interacts with proteins on the inner surface of a cell membrane. This leads to phosphorylation on tyrosine residues of the receptor and other proteins and to the formation inside the cell of complexes with a variety of cytoplasmic signaling 111-03-PCT - P2338R1 - 02120.004WO1
- HGF hepatocyte growth factor receptor tyrosine kinase
- hHGFR human hepatocyte growth factor receptor tyrosine kinase
- the zymogen-like form of HGF beta mutant was shown to bind Met with 14-fold lower affinity than the wild-type serine protease-like form, suggesting optimal interactions result from conformational changes upon cleavage of the single-chain form (US 2005/0037431).
- Extensive mutagenesis of the HGF beta region corresponding to the active site and activation domain of serine proteases showed that 17 of the 38 purified two-chain HGF mutants resulted in impaired cell migration or Met phosphorylation but no loss in Met binding.
- reduced biological activities were well correlated with reduced Met binding of corresponding mutants of HGF beta itself in assays eliminating dominant alpha-chain binding contributions.
- PTK Protein-tyrosine kinases
- RTK receptor tyrosine kinases
- NRTK non-receptor tyrosine kinases
- RTK span the plasma membrane and contain an extra-cellular domain, which binds ligand, and an intracellular portion, which possesses catalytic activity and regulatory sequences.
- Most RTK like the hepatocyte growth factor receptor c-Met, possess a single polypeptide chain and are monomeric in the absence of ligand.
- tyrosine autophosphorylation either stimulates the intrinsic catalytic kinase activity of the receptor or generates recruitment sites for downstream signaling 111-03-PCT- P2338R1 - 02120.004WO1
- SH2 Src homology 2
- PTB phosphotyrosine-binding
- PHA-665752 is a small molecule, ATP-competitive, active-site inhibitor of the catalytic activity of c-Met, as well as phenotypes such as cell growth, cell motility, invasion, and morphology of a variety of tumor cells (Ma et al (2005) Clin. Cancer Res. 11:2312-2319; Christensen et al (2003) Cancer Res. 63:7345-7355).
- the invention relates to quinoline compounds that are inhibitors of receptor tyrosine kinases (RTK), including c-Met.
- RTK receptor tyrosine kinases
- Certain hyperproliferative disorders are characterized by the overactivation of c-Met kinase function, for example by mutations or overexpression of the protein. Accordingly, the compounds of the invention are useful in the treatment of hyperproliferative disorders such as cancer.
- one aspect of the invention provides quinoline compounds of
- Another aspect of the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a quinoline compound of Formula I and a pharmaceutically acceptable carrier.
- the pharmaceutical composition may further comprise one or more additional therapeutic agents selected from anti-proliferative agents, anti-inflammatory agents, immunomodulatory agents, neurotropic factors, agents for treating cardiovascular disease, agents for treating liver disease, 111-03-PCT - P2338R1 - 02120.004WO1
- Another aspect of the invention provides methods of inhibiting c-Met kinase activity, comprising contacting a c-Met kinase with an effective inhibitory amount of a quinoline compound of Formula I, or a stereoisomer, geometric isomer, tautomer, solvate, metabolite, or pharmaceutically acceptable salt or prodrug thereof.
- Another aspect of the invention provides methods of preventing or treating a disease or disorder modulated by c-Met kinases, comprising administering to a mammal in need of such treatment an effective amount of a compound of Formula I, or a stereoisomer, geometric isomer, tautomer, solvate, metabolite, or pharmaceutically acceptable salt or prodrug thereof.
- diseases, conditions and disorders include, but are not limited to, hyperproliferative disorders (e.g., cancer, including melanoma and other cancers of the skin), neurodegeneration, cardiac hypertrophy, pain, migraine, neurotraumatic diseases, stroke, diabetes, hepatomegaly, cardiovascular disease, Alzheimer's disease, cystic fibrosis, viral diseases, autoimmune diseases, atherosclerosis, restenosis, psoriasis, allergic disorders, inflammation, neurological disorders, hormone-related diseases, conditions associated with organ transplantation, immunodeficiency disorders, destructive bone disorders, proliferative disorders, infectious diseases, conditions associated with cell death, thrombin-induced platelet aggregation, chronic myelogenous leukemia (CML), liver disease, pathologic immune conditions involving T cell activation, and CNS disorders.
- hyperproliferative disorders e.g., cancer, including melanoma and other cancers of the skin
- neurodegeneration e.g., cancer, including melanoma and other cancers of the skin
- Another aspect of the invention provides methods of preventing or treating a hyperproliferative disorder, comprising administering to a mammal in need of such treatment an effective amount of a compound of Formula I, or a stereoisomer, geometric isomer, tautomer, solvate, metabolite, or pharmaceutically acceptable salt or prodrug thereof, alone or in combination with one or more additional compounds having anti-hyperproliferative properties.
- the present invention provides a method of using a compound of this invention to treat a disease or condition modulated by c-Met in a mammal.
- An additional aspect of the invention is the use of a compound of this invention in the preparation of a medicament for the treatment or prevention of a disease or condition modulated by c-Met in a mammal.
- kits comprising a compound of Formula
- Another aspect of the invention includes methods of preparing, methods of separating, and methods of purifying compounds of Formula I.
- alkyl refers to a saturated linear or branched-chain monovalent hydrocarbon radical of one to twelve carbon atoms, wherein the alkyl radical may be optionally substituted independently with one or more substituents described below.
- alkyl groups include, but are not limited to, methyl (Me, -CH 3 ), ethyl (Et, -CH 2 CH 3 ), 1 -propyl (n- Pr, n-propyl, -CH 2 CH 2 CH 3 ), 2-propyl (i-Pr, i-propyl, -CH(CH 3 ) 2 ), 1 -butyl (n-Bu, n-butyl, -CH 2 CH 2 CH 2 CH 3 ), 2-methyl-l -propyl (i-Bu, i-butyl, -CH 2 CH(CH 3 ) 2 ), 2-butyl (s-Bu, s-butyl, -CH(CH 3 )CH 2 CH 3 ), 2-methyl-2-propyl (t-Bu, t-butyl, -C(CH 3 ) 3 ), 1-pentyl (n-pentyl, -CH 2 CH 2 CH 2 CH 2 CH 3 ), 2-pentyl (
- alkynyl refers to a linear or branched monovalent hydrocarbon radical of two to twelve carbon atoms with at least one site of unsaturation, i.e., a carbon-carbon, sp triple bond, wherein the alkynyl radical may be optionally substituted independently with one or more substituents described herein. Examples include, but are not limited to, ethynyl (-C ⁇ CH), propynyl (propargyl, -CH 2 C ⁇ CH), and the like.
- carrier refers to a monovalent or multivalent non-aromatic, saturated or partially unsaturated ring having 3 to 12 carbon atoms as a monocyclic ring or 7 to 12 carbon atoms as a bicyclic ring.
- Bicyclic carbocycles having 7 to 12 atoms can be arranged, for example, as a bicyclo [4,5], [5,5], [5,6] or [6,6] system, and bicyclic carbocycles having 9 or 10 ring atoms can be arranged as a bicyclo [5,6] or [6,6] system, or as bridged systems such as bicyclo[2.2.1]heptane, bicyclo[2.2.2]octane and bicyclo[3.2.2]nonane.
- monocyclic carbocycles include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, 1-cyclopent-l-enyl, l-cyclopent-2-enyl, l-cyclopent-3-enyl, cyclohexyl, 1-cyclohex-l-enyl, l-cyclohex-2-enyl, l-cyclohex-3-enyl, cyclohexadienyl, cycloheptyl, cyclooctyl, cyclononyl, cyclodecyl, cycloundecyl, cyclododecyl, and the like.
- Aryl means a monovalent or multivalent aromatic hydrocarbon radical of 6-20 carbon atoms derived by the removal of one or more hydrogen atoms from a carbon atom of a parent aromatic ring system. Some aryl groups are represented in the exemplary structures as "Ar”. Aryl includes bicyclic radicals comprising an aromatic ring fused to a saturated, partially unsaturated ring, or aromatic carbocyclic or heterocyclic ring.
- Typical aryl groups include, but are not limited to, radicals derived from benzene, substituted benzenes, naphthalene, anthracene, biphenyl, indenyl, indanyl, 1,2-dihydronapthalene, 1,2,3,4-tetrahydronapthyl, and the like.
- Examples of aryl fused to a heterocyclic ring include, but are not limited to, the structure: 111-03-PCT- P2338R1 - 02120.004WO1
- n 0, 1 or 2.
- aryl fused to a carbocylic ring include, but are not limited to, the structures:
- R 5 is as defined herein.
- heterocycle refers to a saturated or a partially unsaturated (i.e., having one or more double and/or triple bonds within the ring) carbocyclic, monovalent or multivalent radical of 3 to 20 carbon atoms, and in which at least one ring atom is a heteroatom selected from nitrogen, oxygen and sulfur, the remaining ring atoms being C, where one or more ring atoms is optionally substituted independently with one or more substituents described below.
- a heterocycle may be a monocycle having 3 to 7 ring members (2 to 6 carbon atoms and 1 to 3 heteroatoms selected from N, O, P, and S, wherein the S is optionally substituted with one or more oxo to provide the group SO or SO 2 ) or a bicycle having 7 to 10 ring members (4 to 9 carbon atoms and 1 to 3 heteroatoms selected from N, O, P, and S, wherein the S is optionally substituted with one or more oxo to provide the group SO or SO 2 ), for example: a bicyclo [4,5], [5,5], [5,6], or [6,6] system. Heterocycles are described in Paquette, Leo A.; "Principles of Modern Heterocyclic Chemistry" (W.A.
- heterocyclyl may be a carbon radical or heteroatom radical.
- heterocycle includes heterocycloalkoxy.
- Heterocyclyl also includes radicals where heterocycle radicals are fused with a saturated, partially unsaturated ring, or aromatic carbocyclic or heterocyclic ring. Examples of heterocyclic rings include, but are not limited to, pyrrolidinyl, 111-03-PCT - P2338R1 - 02120.004WO1
- the heterocycle groups herein are optionally substituted independently with one or more substituents described herein.
- heteroaryl refers to a monovalent or multivalent aromatic radical of 5-
- heteroaryl groups include fused ring systems (at least one of which is aromatic) of 1 to 20 carbon atoms, and containing one or more heteroatoms independently selected from nitrogen, oxygen, and sulfur.
- heteroaryl groups are pyridinyl (including, for example, 2-hydroxypyridinyl), imidazolyl, imidazopyridinyl, pyrimidinyl (including, for example, 4-hydroxypyrimidinyl), pyrazolyl, triazolyl, pyrazinyl, tetrazolyl, furyl, thienyl, isoxazolyl, thiazolyl, oxazolyl, isothiazolyl, pyrrolyl, quinolinyl, isoquinolinyl, indolyl, benzimidazolyl, benzofuranyl, cinnolinyl, indazolyl, indolizinyl, phthalazinyl, pyridaziny
- R 5 and R 10 are as defined herein.
- the heterocycle or heteroaryl groups may be C-attached or N-attached where such is possible.
- carbon bonded heterocycles or heteroaryls are bonded at position 2, 3, 4, 5, or 6 of a pyridine, position 3, 4, 5, or 6 of a pyridazine, position 2, 4, 5, or 6 of a pyrimidine, position 2, 3, 5, or 6 of a pyrazine, position 2, 3, 4, or 5 of a furan, tetrahydrofuran, thiofuran, thiophene, pyrrole or tetrahydropyrrole, position 2, 4, or 5 of an oxazole, imidazole or thiazole, position 3, 4, or 5 of an isoxazole, pyrazole, or 111-03-PCT- P2338R1 - 02120.004WO1
- nitrogen bonded heterocycles or heteroaryls are bonded at position 1 of an aziridine, azetidine, pyrrole, pyrrolidine, 2-pyrroline, 3- pyrroline, imidazole, imidazolidine, 2-imidazoline, 3-imidazoline, pyrazole, pyrazoline, 2- pyrazoline, 3-pyrazoline, piperidine, piperazine, indole, indoline, lH-indazole, position 2 of a isoindole, or isoindoline, position 4 of a morpholine, and position 9 of a carbazole, or ⁇ -carboline.
- Substituted alkyl mean alkyl, alkenyl, alkynyl, aryl, heteroaryl, heterocyclyl and cycloalkyl, respectively, in which one or more hydrogen atoms are each independently replaced with a substituent.
- Substituents may also be combinations of alkyl, alkenyl, alkynyl, carbocycle, aryl, and heteroaryl radicals, such as cyclopropylmethyl, cyclohexylethyl, benzyl, and N-ethylmorpholino, and substituted forms thereof.
- beneficial or desired clinical results include, but are not limited to, alleviation of symptoms, diminishment of extent of disease, stabilized (i.e., not worsening) state of disease, delay or slowing of disease progression, amelioration or palliation of the disease state, and remission (whether partial or total), whether detectable or undetectable.
- Treatment can also mean prolonging survival as compared to expected survival if not receiving treatment.
- Those in need of treatment include those already with the condition or disorder as well as those prone to have the condition or disorder or those in which the condition or disorder is to be prevented.
- the phrase "therapeutically effective amount” means an amount of a compound of the present invention that (i) treats or prevents the particular disease, condition, or disorder, (ii) attenuates, ameliorates, or eliminates one or more symptoms of the particular disease, condition, 111-03-PCT - P2338R1 - 02120.004WO1
- the therapeutically effective amount of the drug may reduce the number of cancer cells; reduce the tumor size; inhibit (i.e., slow to some extent and preferably stop) cancer cell infiltration into peripheral organs; inhibit (i.e., slow to some extent and preferably stop) tumor metastasis; inhibit, to some extent, tumor growth; and/or relieve to some extent one or more of the symptoms associated with the cancer.
- the drug may prevent growth and/or kill existing cancer cells, it may be cytostatic and/or cytotoxic.
- cancer therapy efficacy can be measured, for example, by assessing the time to disease progression (TTP) and/or determining the response rate (RR).
- TTP time to disease progression
- RR response rate
- cancer and “cancerous” refer to or describe the physiological condition in mammals that is typically characterized by unregulated cell growth.
- a “tumor” comprises one or more cancerous cells. Examples of cancer include, but are not limited to, carcinoma, lymphoma, blastoma, sarcoma, and leukemia or lymphoid malignancies.
- squamous cell cancer e.g., epithelial squamous cell cancer
- lung cancer including small- cell lung cancer, non-small cell lung cancer ("NSCLC"), adenocarcinoma of the lung and squamous carcinoma of the lung, cancer of the peritoneum, hepatocellular cancer, gastric or stomach cancer including gastrointestinal cancer, pancreatic cancer, glioblastoma, cervical cancer, ovarian cancer, liver cancer, bladder cancer, hepatoma, breast cancer, colon cancer, rectal cancer, colorectal cancer, endometrial or uterine carcinoma, salivary gland carcinoma, kidney or renal cancer, prostate cancer, vulval cancer, thyroid cancer, hepatic carcinoma, anal carcinoma, penile carcinoma, as well as head and neck cancer.
- NSCLC non-small cell lung cancer
- adenocarcinoma of the lung and squamous carcinoma of the lung cancer of the peritoneum, hepatocellular cancer, gastric or stomach cancer including gastrointestinal cancer
- a "chemotherapeutic agent” is a chemical compound useful in the treatment of cancer.
- chemotherapeutic agents include Erlotinib (TARCEVA®, Genentech/OSI Pharm.), Bortezomib (VELCADE®, Millennium Pharm.), Fulvestrant (FASLODEX®, AstraZeneca), Sutent (SUl 1248, Pfizer), Letrozole (FEMARA®, Novartis), Imatinib mesylate (GLEEVEC®, Novartis), PTK787/ZK 222584 (Novartis), Oxaliplatin (Eloxatin®, Sanofi), 5-FU (5-fluorouracil), Leucovorin, Rapamycin (Sirolimus, RAPAMUNE®, Wyeth), lapatinib (TYKERB®, GlaxoSmithKline PLC), Lonafarnib (SCH 66336), Sorafenib (BAY43-9006, Bayer Labs), and
- dynemicin including dynemicin A; bisphosphonates, such as clodronate; an esperamicin; as well as neocarzinostatin chromophore and related chromoprotein enediyne antibiotic chromophores), aclacinomysins, actinomycin, authramycin, azaserine, bleomycins, cactinomycin, carabicin, carminomycin, carzinophilin, chromomycinis, dactinomycin, daunorubicin, detorubicin, 6-diazo-5-oxo-L-norleucine, ADRIAMYCIN® (doxorubicin), morpholino-doxorubicin, cyanomorpholino-doxorubicin, 2-pyrrolino-doxorubicin and deoxydoxorubicin), epirubicin,
- chemotherapeutic agent also included in the definition of "chemotherapeutic agent” are: (i) anti -hormonal agents that act to regulate or inhibit hormone action on tumors such as anti-estrogens and selective estrogen receptor modulators (SERMs), including, for example, tamoxifen (including NOLVADEX®; tamoxifen citrate), raloxifene, droloxifene, 4-hydroxytamoxifen, trioxifene, keoxifene, LYl 17018, onapristone, and FARESTON® (toremifine citrate); (ii) aromatase inhibitors that inhibit the enzyme aromatase, which regulates estrogen production in the adrenal glands, such as, for example, 4(5)-imidazoles, aminoglutethimide, MEGASE® (megestrol acetate), AROMASIN® (exemestane; Pfizer), formestanie, fadrozole, RIVISOR® (vorozole), F
- prodrug refers to a precursor or derivative form of a compound of the invention that is less cytotoxic to cells compared to the parent compound or drug and is capable of being enzymatically or hydrolytically activated or converted into the more active parent form. See, e.g., Wilman, "Prodrugs in Cancer Chemotherapy” Biochemical Society Transactions, 14, pp. 375-382, 615th Meeting Harbor (1986) and Stella et al, "Prodrugs: A Chemical Approach to Targeted Drug Delivery,” Directed Drug Delivery, 111-03-PCT - P2338R1 - 02120.004WO1
- the prodrugs of this invention include, but are not limited to, phosphate-containing prodrugs, thiophosphate-containing prodrugs, sulfate-containing prodrugs, peptide-containing prodrugs, D-amino acid-modified prodrugs, glycosylated prodrugs, ⁇ -lactam-containing prodrugs, optionally substituted phenoxyacetamide- containing prodrugs, optionally substituted phenylacetamide-containing prodrugs, 5- fluorocytosine and other 5-fluorouridine prodrugs which can be converted into the more active cytotoxic free drug.
- cytotoxic drugs that can be derivatized into a prodrug form for use in this invention include, but are not limited to, compounds of the invention and chemotherapeutic agents such as described above.
- a "metabolite” is a product produced through metabolism in the body of a specified compound or salt thereof. Metabolites of a compound may be identified using routine techniques known in the art and their activities determined using tests such as those described herein. Such products may result for example from the oxidation, reduction, hydrolysis, amidation, deamidation, esterification, deesterification, enzymatic cleavage, and the like, of the administered compound. Accordingly, the invention includes metabolites of compounds of the invention, including compounds produced by a process comprising contacting a compound of this invention with a mammal for a period of time sufficient to yield a metabolic product thereof.
- a “liposome” is a small vesicle composed of various types of lipids, phospholipids and/or surfactant which is useful for delivery of a drug (such as the c-Met inhibitors disclosed herein and, optionally, a chemotherapeutic agent) to a mammal.
- a drug such as the c-Met inhibitors disclosed herein and, optionally, a chemotherapeutic agent
- the components of the liposome are commonly arranged in a bilayer formation, similar to the lipid arrangement of biological membranes.
- package insert is used to refer to instructions customarily included in commercial packages of therapeutic products, that contain information about the indications, usage, dosage, administration, contraindications and/or warnings concerning the use of such therapeutic products.
- chiral refers to molecules which have the property of non-superimposability of the mirror image partner, while the term “achiral” refers to molecules which are superimposable on their mirror image partner.
- stereoisomers refers to compounds which have identical chemical constitution, but differ with regard to the arrangement of the atoms or groups in space.
- “Diastereomer” refers to a stereoisomer with two or more centers of chirality and whose molecules are not mirror images of one another. Diastereomers have different physical properties, e.g. melting points, boiling points, spectral properties, and reactivities. Mixtures of 111-03-PCT - P2338R1 - 02120.004WO1
- 15 diastereomers may separate under high resolution analytical procedures such as electrophoresis and chromatography.
- Enantiomers refer to two stereoisomers of a compound which are non- superimposable mirror images of one another.
- the compounds of the invention may contain asymmetric or chiral centers, and therefore exist in different stereoisomeric forms. It is intended that all stereoisomeric forms of the compounds of the invention, including but not limited to, diastereomers, enantiomers and atropisomers, as well as mixtures thereof such as racemic mixtures, form part of the present invention.
- Many organic compounds exist in optically active forms, i.e., they have the ability to rotate the plane of plane-polarized light.
- the prefixes D and L, or R and S are used to denote the absolute configuration of the molecule about its chiral center(s).
- the prefixes d and 1 or (+) and (-) are employed to designate the sign of rotation of plane-polarized light by the compound, with (-) or 1 meaning that the compound is levorotatory.
- a compound prefixed with (+) or d is dextrorotatory.
- these stereoisomers are identical except that they are mirror images of one another.
- a specific stereoisomer may also be referred to as an enantiomer, and a mixture of such isomers is often called an enantiomeric mixture.
- a 50:50 mixture of enantiomers is referred to as a racemic mixture or a racemate, which may occur where there has been no stereoselection or stereospecificity in a chemical reaction or process.
- the terms “racemic mixture” and “racemate” refer to an equimolar mixture of two enantiomeric species, devoid of optical activity.
- the term “tautomer” or “tautomeric form” refers to structural isomers of different energies which are interconvertible via a low energy barrier.
- proton tautomers also known as prototropic tautomers
- Valence tautomers include interconversions by reorganization of some of the bonding electrons.
- a "salt,” as used herein, refers to organic or inorganic salts of a compound of the invention.
- Exemplary salts include, but are not limited, to sulfate, citrate, acetate, oxalate, chloride, bromide, iodide, nitrate, bisulfate, phosphate, acid phosphate, isonicotinate, lactate, salicylate, acid citrate, tartrate, oleate, tannate, pantothenate, bitartrate, ascorbate, succinate, maleate, gentisinate, fumarate, gluconate, glucuronate, saccharate, formate, benzoate, glutamate, methanesulfonate, ethanesulfonate, benzenesulfonate, /?-toluenesulfonate, and pamoate (i.e., 1,1'- 111-03-PCT - P2338R1
- a salt may involve the inclusion of another molecule such as an acetate ion, a succinate ion or other counter ion.
- the counter ion may be any organic or inorganic moiety that stabilizes the charge on the parent compound.
- a salt may have more than one charged atom in its structure. Instances where multiple charged atoms are part of the salt can have multiple counter ions. Hence, a salt can have one or more charged atoms and/or one or more counter ion.
- the desired salt may be prepared by any suitable method available in the art, for example, treatment of the free base with an inorganic acid, such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid and the like, or with an organic acid, such as acetic acid, maleic acid, succinic acid, mandelic acid, fumaric acid, malonic acid, pyruvic acid, oxalic acid, glycolic acid, salicylic acid, a pyranosidyl acid, such as glucuronic acid or galacturonic acid, an alpha hydroxy acid, such as citric acid or tartaric acid, an amino acid, such as aspartic acid or glutamic acid, an aromatic acid, such as benzoic acid or cinnamic acid, a sulfonic acid, such as p-toluenesulfonic acid or ethanesulfonic acid, or the like.
- an inorganic acid such as hydrochloric acid, hydrobro
- the desired salt may be prepared by any suitable method, for example, treatment of the free acid with an inorganic or organic base, such as an amine (primary, secondary or tertiary), an alkali metal hydroxide or alkaline earth metal hydroxide, or the like.
- suitable salts include, but are not limited to, organic salts derived from amino acids, such as glycine and arginine, ammonia, primary, secondary, and tertiary amines, and cyclic amines, such as piperidine, morpholine and piperazine, and inorganic salts derived from sodium, calcium, potassium, magnesium, manganese, iron, copper, zinc, aluminum and lithium.
- the salt is a pharmaceutically acceptable salt.
- pharmaceutically acceptable indicates that the substance or composition must be compatible chemically and/or toxicologically, with the other ingredients comprising a formulation, and/or the mammal being treated therewith.
- the compounds of Formula I also include salts of such compounds which are not necessarily pharmaceutically acceptable salts, and which may be useful as intermediates for preparing and/or purifying compounds of Formula I and/or for separating enantiomers of compounds of Formula I.
- a “solvate” refers to an association or complex of one or more solvent molecules and a compound of the invention.
- solvents that form solvates include, but are not 111-03-PCT - P2338R1 - 02120.004WO1
- protecting group refers to a substituent that is commonly employed to block or protect a particular functionality while reacting other functional groups on the compound.
- an "amino-protecting group” is a substituent attached to an amino group that blocks or protects the amino functionality in the compound.
- Suitable amino-protecting groups include acetyl, trifluoroacetyl, t-butoxycarbonyl (BOC), benzyloxycarbonyl (CBZ) and 9- fluorenylmethylenoxycarbonyl (Fmoc).
- a "hydroxy-protecting group” refers to a substituent of a hydroxy group that blocks or protects the hydroxy functionality.
- Suitable protecting groups include acetyl and silyl.
- a “carboxy-protecting group” refers to a substituent of the carboxy group that blocks or protects the carboxy functionality.
- Common carboxy-protecting groups include -CH 2 CH 2 SO 2 Ph, cyanoethyl, 2-(trimethylsilyl)ethyl, 2- (trimethylsilyl)ethoxymethyl, 2-(p-toluenesulfonyl) ethyl, 2-(p-nitrophenylsulfenyl)ethyl, 2- (diphenylphosphino)-ethyl, nitroethyl and the like.
- protecting groups and their use see T. W. Greene, Protective Groups in Organic Synthesis, John Wiley & Sons, New York, 1991.
- compound of this invention and “compounds of the present invention” and “compounds of Formula I” include compounds of Formula I and stereoisomers, geometric isomers, tautomers, solvates, metabolites, salts and pharmaceutically acceptable prodrugs thereof.
- mammal includes, but is not limited to, humans, dogs, cats, horses, cows, pigs, and sheep, and poultry.
- the present invention provides quinoline compounds, and pharmaceutical formulations thereof, that are potentially useful in the treatment of diseases, conditions and/or disorders modulated by c-Met. More specifically, the present invention provides compounds of Formula I:
- R 1 , R 2 and R 4 are independently selected from H, F, Cl, Br, I, CN,
- R 13 is H, Ci-C 6 alkyl, -(CR 14 R 15 ) n -cycloalkyl, -(CR 14 R 15 ) n -heterocyclyl,
- R 14 is null and R 10 and R 15 together with the atoms to which they are attached form a 5-6 membered heteroaryl ring,
- R 12 and R 14 together with the atoms to which they are attached form a saturated or partially unsaturated C 2 -Cj 2 heteorcyclic ring;
- R a and R b are independently H, C J -C J2 alkyl, C 2 -C 8 alkenyl, C 2 -C 8 alkynyl,
- Ci-C 12 alkyl C 2 -Cg alkenyl, C 2 -Cg alkynyl, C 6 -C 2O aryl, or C 1 -C 2 0 heteroaryl; [0077] Y, Y 1 and Y 2 are independently O or S;
- t is 1, 2, 3, 4, 5 or 6;
- n and m are independently 0, 1, 2, 3, 4, 5 or 6.
- R 1 and R 2 is -OR 10 where R 10 is Ci-Ci 2 alkyl.
- R 1 and R 2 are methoxy.
- R 10 is Ci-Ci 2 alkyl substituted with NR a R b or C 2 -C 2O heterocyclyl, wherein said heterocyclyl is optionally substituted with C 1 -C 6 alkyl, CH 2 OH or SO 2 Me.
- Exemplary embodiments include the structures:
- R 1 is methoxy and R 2 is selected from
- R 1 and R 2 is -OR 10 wherein R 10 is C]-Ci 2 alkyl substituted with Ci-C 20 heteroaryl, wherein said heteroaryl is optionally substituted with C]-C 6 alkyl.
- exemplary embodiments include the structures: 111-03-PCT - P2338R1 - 02120.004WO1
- R 1 and R 2 are independently selected from
- R 1 and R 2 are independently selected from optionally substituted aryl or heteroaryl, including the exemplary structures:
- R ! and R 2 is independently alkyl optionally substituted with one or more groups independently selected from OR 10 , NR 10 R 11 , heterocyclyl and heteroaryl. Examples include, but are not limited to, methyl, -CH 2 OH, -CH 2 CH 2 OH,
- R 1 and R 2 are independently -OR , including the exemplary structure:
- each R 4 is H.
- L-R 5 is (C 3 -Ci 2 carbocyclyO-R 5 , including the exemplary structures:
- L-R 5 is (C 2 -C 20 heterocyclyl)-R 5 wherein said heterocyclyl is optionally substituted, including the exemplary structures: 111-03-PCT- P2338R1 - 02120.004WO1
- L-R 5 is (C 6 -C 2 o aryl)-R 5 wherein said aryl is optionally substituted, including the exemplary structures:
- L-R 5 is (C 6 -C 2O aryl)-R 5 include the structures:
- L-R 5 is (C 6 -C 20 aryl)-R 5
- L-R 5 includes the structures: 111-03-PCT- P2338R1 - 02120.004WO1
- L-R 5 is (C 6 -C 2 O aryl)-R
- R 5 is (C 6 -C 2 O aryl)-R
- L-R 5 is (C 6 -C 20 aryl)-R 5 include the structures:
- L-R 5 is (Ci-C 20 heteroaryl)-R 5 .
- Exemplary embodiments include the structures:
- L-R 5 is (C]-C 2O heteroaryl)-R 5 also include the structures:
- R 13 is
- -(CR 14 R 15 ) n -cycloalkyl -(CR 14 R 15 ) n -aryl, -(CR 14 R 15 ) n -O-(CR 14 R 15 ) m -aryl, or -(CR 14 R 15 V heterocyclyl-(CR 14 R 15 ) r aryl, wherein said heterocyclyl portion is optionally substituted with SO 2 R 0 or C]-Ci 2 alkyl.
- Exemplary embodiments include the structures: 111-03-PCT - P2338R1 - 02120.004WO1
- R 10 is H or Ci-Ci 2 alkyl
- R 13 is H, Q-C 6 alkyl, -(CR 14 R 15 ) n -cycloalkyl, or -(CR 14 R 15 ) n -aryl, wherein said alkyl, cycloalkyl, and aryl portions are optionally substituted with F or 0R a .
- Exemplary embdodiments of R 5 include the structures:
- R 5 is -NR 10 R 13 .
- R 10 is H or
- Ci-Ci 2 alkyl, and R 13 is -(CR 14 R 15 ) n -heterocyclyl or -(CR 14 R 15 ) n -heteroaryl, wherein said heterocyclyl and heteroaryl are optionally substituted with OR a , SO 2 R 0 or O-(CH 2 )-aryl.
- Exemplary embdodiments of R 5 include the structures:
- R 15 and R 10 optionally together with the atoms to which they are attached form a 5-6 membered 111-03-PCT - P2338R1 - 02120.004WO1
- R 5 include the structures:
- exemplary embodiments of R 5 include the structures: 111-03-PCT - P2338R1 - 02120.004WO1
- R 11 is aryl or a Ci-Ci 2 alkyl substituted with aryl, wherein said aryl portions are optionally substituted.
- Particular embodiments include the structures:
- R 12 and R 14 together with the atoms to which they are attached form a 5-6 membered heterocyclic ring.
- exemplary embodiments include, but are not limited to 111-03-PCT - P2338R1 - 02120.004WO1
- a particular example includes
- R 13 is C 1 -
- Exemplary embodiments of R 5 include the structures:
- R 5 is -NR 12 SO 2 R 10 , including where R 10 is alkyl or optionally substituted aryl.
- exemplary embodiments include the structures: 111-03-PCT- P2338R1 - 02120.004WO1
- R 5 is an optionally substituted carbocyclyl, including the exemplary structures:
- R 5 is an optionallysubstituted heterocyclyl, including the exemplary structures:
- R 5 is an optionally substituted aryl, including the exemplary structures: 111-03-PCT - P2338R1 - 02120.004WO1
- R 5 is an optionally substituted heteroaryl, including the exemplary structures:
- R ⁇ is H, Ci-C 12 alkyl, C 3 -Ci 2 cycloalkyl, C 6 -C 20 aryl, or Ci-C 20 heteroaryl, and Ir 21 1 and
- R are independently selected from H or Ci-Ci 2 alkyl, wherein said alkyl, cycloalkyl, aryl, heteroaryl are optionally substituted with one or more groups independently selected from F, Cl, Br, I and Ci-Ci 2 alkyl; and where the wavy line indicates the point of attachment to L.
- R 20 is H. 111-03-PCT - P2338R1 - 02120.004WO1
- R 5 is an optionally substituted heteroaryl, including the exemplary structures:
- R 5 is an optionally substituted heteroaryl, including the exemplary structures: 111-03-PCT- P2338R1 - 02120.004WO1
- the quinoline compounds of the invention may contain asymmetric or chiral centers, and therefore exist in different stereoisomeric forms. It is intended that all stereoisomeric forms of the compounds of the invention, including but not limited to, diastereomers, enantiomers and atropisomers, as well as mixtures thereof such as racemic mixtures, form part of the present invention.
- the present invention embraces all geometric and positional isomers.
- a quinoline compound of the present invention incorporates a double bond or a fused ring
- the cis- and trans-forms, as well as mixtures thereof are embraced within the scope of the invention.
- Both the single positional isomers and mixture of positional isomers, e.g., resulting from the N-oxidation of the pyrimidine and pyrazine rings, are also within the scope of the present invention.
- stereochemistry of any particular chiral atom is not specified, then all stereoisomers are contemplated and included as the compounds of the invention. Where stereochemistry is specified by a solid wedge or dashed line representing a particular configuration, then that stereoisomer is so specified and defined.
- the compounds of the present invention may exist in unsolvated as well as solvated forms with pharmaceutically acceptable solvents such as water, ethanol, and the like, and it is intended that the invention embrace both solvated and unsolvated forms.
- tautomer or tautomeric form refers to structural isomers of different energies which are interconvertible via a low energy barrier.
- proton tautomers also known as prototropic tautomers
- Valence tautomers include interconversions by reorganization of some of the bonding electrons.
- the present invention also embraces isotopically-labeled compounds of the present invention which are identical to those recited herein, but for the fact that one or more atoms are replaced by an atom having an atomic mass or mass number different from the atomic 111-03-PCT - P2338R1 - 02120.004WO1
- isotopes of any particular atom or element as specified are contemplated within the scope of the compounds of the invention, and their uses.
- Exemplary isotopes that can be incorporated into compounds of the invention include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorus, sulfur, fluorine, chlorine and iodine, such as 2 H, 3 H, 11 C, 13 C, 14 C, 13 N, 15 N, 15 O, 17 O, 18 0, 32 P, 33 P, 35 S, 18 F, 36 Cl, 123 I and 125 I.
- Certain isotopically- labeled compounds of the present invention e.g., those labeled with 3 H and 14 C) are useful in compound and/or substrate tissue distribution assays.
- Tritiated ( 3 H) and carbon- 14 ( 14 C) isotopes are useful for their ease of preparation and detectability. Further, substitution with heavier isotopes such as deuterium (i.e., 2 H) may afford certain therapeutic advantages resulting from greater metabolic stability (e.g., increased in vivo half-life or reduced dosage requirements) and hence may be preferred in some circumstances.
- Positron emitting isotopes such as 15 O, 13 N, 11 C and 18 F are useful for positron emission tomography (PET) studies to examine substrate receptor occupancy.
- Isotopically labeled compounds of the present invention can generally be prepared by following procedures analogous to those disclosed in the Schemes and/or in the Examples herein below, by substituting an isotopically labeled reagent for a non-isotopically labeled reagent.
- Quinoline compounds of Formula I of the present invention may be synthesized by synthetic routes that include processes analogous to those well-known in the chemical arts, particularly in light of the description contained herein.
- the starting materials are generally available from commercial sources such as Aldrich Chemicals (Milwaukee, WI) or are readily prepared using methods well known to those skilled in the art (e.g., prepared by methods generally described in Louis F. Fieser and Mary Fieser, Reagents for Organic Synthesis, v. 1-19, Wiley, N.Y. (1967-1999 ed.), or Beilsteins Handbuch der organischen Chemie, 4, Aufl. ed. Springer- Verlag, Berlin, including supplements (also available via the Beilstein online database).
- compounds of Formula I may be readily prepared using procedures well-known to prepare quinoline compounds; and other heterocycles, which are described in: Comprehensive Heterocyclic Chemistry, Editors Katritzky and Rees, Pergamon Press, 1984; Klemm et al (1970) J. Hetero. Chem. 7(2):373-379; Klemm et al (1974) J. Hetero. Chem. 11(3): 355-361; Klemm et al (1976) J. Hetero. Chem. 13:273-275; Klemm et al (1985) J. Hetero. Chem. 22(5):1395-1396; Bisagni et al (1974) Bull. Soc. Chim. Fr.
- Compounds of Formula I may be prepared singly or as compound libraries comprising at least 2, for example 5 to 1,000 compounds, or 10 to 100 compounds.
- Libraries of compounds of Formula I may be prepared by a combinatorial 'split and mix' approach or by multiple parallel syntheses using either solution phase or solid phase chemistry, by procedures known to those skilled in the art.
- a compound library comprising at least 2 compounds, or pharmaceutically acceptable salts thereof.
- Schemes 1-20 show general methods for preparing the compounds of the present invention as well as key intermediates. For a more detailed description of the individual reaction steps, see the Examples section below. Those skilled in the art will appreciate that other synthetic routes may be used to synthesize the inventive compounds. Although specific starting materials and reagents are depicted in the Schemes and discussed below, other starting materials and reagents can be easily substituted to provide a variety of derivatives and/or reaction conditions. In addition, many of the compounds prepared by the methods described below can be further modified in light of this disclosure using conventional chemistry well known to those skilled in the art.
- Suitable amino-protecting groups include acetyl, trifluoroacetyl, t- butoxycarbonyl (BOC), benzyloxycarbonyl (CBz) and 9-fluorenylmethyleneoxycarbonyl (Fmoc).
- NH-Pg amino-protecting groups
- BOC t- butoxycarbonyl
- CBz benzyloxycarbonyl
- Fmoc 9-fluorenylmethyleneoxycarbonyl
- Scheme 1 shows a general method for the synthesis of intermediate compound 4, which is useful for the synthesis of compounds of Formula I. Syntheses of 4-phenoxy-6,7- dialkoxyquinolines have been previously reported in US 2004/0242603; US 2005/0002326; WO 2005/030140; J. Med. Chem. (2005) 48:1359-1366. As shown in Scheme 1, reaction of a 4- hydroxy-6,7-dialkoxyquinoline 1 with a variably substituted p-halonitroarene or heteroarene wherein X is F or Cl and Y is N or CH using an appropriate base (e.g. Cs 2 CO 3 , NaH, KOt-Bu, or the like) provides intermediate 2.
- an appropriate base e.g. Cs 2 CO 3 , NaH, KOt-Bu, or the like
- Scheme 2 shows a general synthetic route for the synthesis of compound 9, which is useful for the synthesis of compounds of Formula I.
- a 4-chloro-6,7-dialkoxyquinoline, such as compound 6, can be prepared by chlorination of the corresponding hydroxyquinoline (compound 1, Scheme 1) typically using POCl 3 , MeSO 2 Cl and the like.
- Compound 7 can then be reacted under basic conditions, typically DMAP in bromobenzene or Cs 2 C ⁇ 3 in DMF, with a functionalized phenol 8 (general schemes for synthesis of preferred functionalized phenols are shown below) to give rise to compound 9.
- a functionalized phenol 8 generally schemes for synthesis of preferred functionalized phenols are shown below
- Compound 9 can optionally be further manipulated depending on the phenol functionalization.
- PG exchange for R 11 can be omitted from the sequence and thus provide intermediate 9 with the 6,7-alkoxy substituents originally contained in intermediate 6.
- Scheme 3 shows a route for the preparation of phenol compound 14.
- Scheme 4 shows a route for the preparation of the 1 -substituted pyrimidinone intermediate 20 (wherein R 10 is independently selected from H, alkyl, aryl and heteroaryl).
- 5- bromo-2,4-dichloropyrimidine 15 is hydrolyzed with NaOH to give 5-bromo-2-chloropyrimidin- 4(3H)-one 16 as described in EP1506967 Al.
- Alkylation of compound 16 to provide the 1- substituted pyrimidinone can be accomplished with an alkylation agent (e.g. iodomethane, or the like) mediated by an appropriate base (e.g. sodium alkoxides, lithium or sodium hydride, or the like) providing a mixture of isomers 17 and 18.
- an alkylation agent e.g. iodomethane, or the like
- an appropriate base e.g. sodium alkoxides, lithium or sodium hydride, or the like
- Isomers 17 and 18 can be separated using purification techniques known to those skilled in the art (e.g. flash chromatography, reverse phase HPLC, or the like).
- Compound 18 is reacted with the appropriate zinc reagent and palladium catalyst to give 2-substituted intermediate 19.
- Suzuki coupling of compound 19 to an appropriate boronic acid followed by final deprotection of the phenol gives compound 20, which can be reacted with the appropriate core intermediate 7 as in Scheme 2 to provide compound 9.
- Scheme 5 shows a method for preparing phenol intermediate 27 (wherein R 10 is independently selected from H, alkyl, aryl and heteroaryl).
- Nucleophilic substitution of 2-chloro- 4-methoxypyrimidine 21 with a compound of the formula HY-R 10 , (wherein Y is O, N or S) can be accomplished in an appropriate solvent such as n-butanol, at refluxing temperature to give intermediate 22.
- Deprotection of the methoxypyrimidine with HBr in acetic acid provides 2- substituted pyrimidinone 23.
- Alkylation of 23 to provide the 1 -substituted pyrimidinone can be accomplished with an alkylation agent (e.g.
- iodomethane or the like
- an appropriate base e.g. sodium alkoxides, lithium or sodium hydride, or the like
- Isomers 24 and 25 can be separated using purification techniques known to those skilled in the art (e.g. flash chromatography, reverse phase HPLC, or the like).
- Bromination in the 5-position with a brominating agent such as Br 2 or NBS gives compound 26.
- Suzuki coupling of compound 26 to an appropriate boronic acid gives a bicyclic intermediate which after final deprotection of the phenol gives compound 27.
- Compound 27 can be reacted with the appropriate core intermediate 7 as in Scheme 2 to provide compound 9.
- Scheme 6 shows an alternative route to compound 27 (wherein R 10 is independently selected from H, alkyl, aryl and heteroaryl).
- Nucleophilic substitution of compound 28 with a compound of the formula HY-R 10 can be accomplished at elevated temperature with a base such as NaHCO 3 in an appropriate solvent such as n-butanol to give intermediate 26.
- Suzuki coupling of compound 26 to an appropriate boronic acid gives a bicyclic intermediate which after final deprotection of the phenol gives compound 27.
- Intermediate 27 can then be reacted with the appropriate core intermediate 7 as in Scheme 2 to give compound 9.
- Scheme 7 shows a route to phenol intermediate 32 (wherein R 10 is independently selected from H, alkyl, aryl and heteroaryl).
- Nucleophilic substitution of compound 29 with NaOMe can be accomplished at elevated temperature in an appropriate solvent such as methanol.
- Nucleophilic substitution of compound 30 with a compound of the formula HY-R 10 , (wherein Y is O, N or S), to form the 5-substituted pyrimidinone 31 can be accomplished at elevated temperature with a base such as NaHCO 3 in an appropriate solvent such as n-butanol. Under these reaction conditions, deprotection of the methoxypyrimidine to the pyrimidinone can also be achieved.
- deprotection of the methoxypyrimidine can be accomplished with HBr in acetic acid.
- Copper (I)-mediated coupling of compound 31 to an appropriate halide provides compound 32.
- the halide used in the coupling reaction contains a standard protecting group. In those cases, the protecting group can be removed by standard conditions known in the art.
- Compound 32 can then be reacted with the appropriate core intermediate 7 as in Scheme 2 to provide compound 9. 111-03-PCT - P2338R1 - 02120.004WO1
- Scheme 8 shows a route for the preparation of the 1 -substituted pyridone intermediate 38 (wherein R 10 is independently selected from H, alkyl, aryl and heteroaryl).
- Alkylation of 6-chloropyridin-2-ol 33 to provide the 1 -substituted pyridone 35 can be accomplished with an alkylation agent (e.g. iodomethane, or the like) mediated by an appropriate base (e.g. potassium carbonate, sodium alkoxides, lithium or sodium hydride, or the like) providing a mixture of isomers 34 and 35.
- alkylation agent e.g. iodomethane, or the like
- an appropriate base e.g. potassium carbonate, sodium alkoxides, lithium or sodium hydride, or the like
- Isomers 34 and 35 can be separated using purification techniques known to those skilled in the art (e.g.
- Scheme 9 shows a method for preparing phenol intermediate 43 (wherein R 10 is independently selected from H, alkyl, aryl and heteroaryl). 1 -Substituted pyridone 35, which can 111-03-PCT- P2338R1 - 02120.004WO1
- Scheme 10 shows a route for the preparation of the 6-acyl pyridin-2(lH)-one phenol compound 48.
- Base-mediated halogen exchange of the commercially available bromopyridine 44 followed by quenching with an aldehyde gives the secondary alcohol compound 45.
- Oxidation of the alcohol followed by demethylation gives compound 46.
- Bromination of compound 46 followed by a Suzuki coupling with an appropriate boronic acid gives a coupling compound 47.
- Final deprotection of the phenol gives compound 48, which can then be reacted with appropriate core intermediate 7 as in Scheme 2 to give compound 9.
- Sodium borohydride reduction of this compound gives compound 49, and acetylation of compound 49 gives intermediate 50.
- Compounds 49 and 50 can also be reacted with appropriate compound 7 as in Scheme 2 to provide compound 9.
- Scheme 12 shows a route for the preparation of the 5-benzyl-3-(4- hydroxyphenyl)pyrimidin-4(3H)-one phenol intermediate 58 which is useful for the synthesis of compounds of Formula I.
- Commercially available 4,6-dichloropyrimidine-5-carbaldehyde 54 is reacted with the appropriate substituted phenyl magnesium halide to give the secondary alcohol 55.
- Monobenzylation gives compound 56, which is subjected to hydrogenation to provide compound 57.
- Copper (I)-mediated coupling of compound 57 to an appropriate phenol provides the desired compound 58, which can be reacted with appropriate core intermediate 7 as in Scheme 2 to provide compound 9.
- the pyridazino carboxylic acid compound 62 can be prepared using methods described by McNab H. et al (1982) J. Chem. Soc. Perkin Trans. 1:1845 as depicted in Scheme 13. Substituted hydrazine 59 can be converted to hydrazono acetaldehyde 60 with standard dehydrating conditions such as acetic acid at room temperature.
- the carbonyl group condensation product 61 is prepared in a suitable organic solvent such as toluene, benzene or dioxane at room temperature using piperidinium acetate as catalyst.
- Carboxylic acid pyridazinone 62 is prepared from hydrazono ethylidene 61 by cyclization under basic conditions (sodium methoxide in methanol) at 7O 0 C.
- Compound 62 may then be used to acylate aniline intermediate 4 whose preparation is described in Scheme 1 to prepare compounds of Formula I.
- Scheme 14 shows a route for the preparation of oxo-4-phenyl-3,4- dihydropyrazine-2 -carboxylic acids.
- the pyrazine-2-carbonitrile 63 was prepared using methods described by Hoornaert, G., et al (1983) J. Heterocyclic Chem. 20:919 and Hoornaert, G., et al (1990) Tetrahedron 46:5715.
- the pyrazine-2-carboxylic acid 64 can be prepared by hydrolysis to the carboxylic acid followed by removal of the chloro group under hydrogenolysis conditions to 111-03-PCT - P2338R1 - 02120.004WO1
- Methyl 3-oxo-3,4-dihydropyrazine-2-carboxylate 65 can be converted to alkylpyrazino carboxylate 66 by standard basic alkylation conditions with alkyl halides. These conditions include but are not limited to treatment with K 2 CO 3 in acetone or DMF at room or elevated temperature, or NaH in THF at ambient or elevated temperature, followed by addition of the alkyl halide. In certain embodiments, this alkylation is achieved with LiH in DMF at O 0 C, followed by addition of alkyl chloride or alkyl bromide or alkyl iodide and warming to room temperature.
- Carboxylic acid 67 can then be prepared using standard saponification conditions such as LiOH or NaOH in standard mixed aqueous/organic solvent systems. The acid 67 can then be coupled via standard amide bond forming techniques to an aniline bearing core 4 constructed according to Scheme 1 to provide final compound 5.
- Scheme 16 shows a method for preparing pyrrolidin-2-one intermediate 70
- Scheme 17 shows a method for the preparation of the fused bicyclic cyclopropane lactam ester 74.
- An optionally substituted allylic amine 71 is acylated with a malonyl chloride ester under basic conditions to give the allylic amide intermediate 72.
- Cyclization under conditions which generate the malonyl carbine (preferably manganese III acetate catalyzed) provide the fused cyclopropyllactam compound 73.
- Deprotection under basic conditions typically LiOH or NaOH in an aqueous/organic solvent mixture
- the acid 74 can then be coupled via standard amide bond forming techniques to aniline bearing cores such as 4 constructed according to Scheme 1 to provide final compound 5.
- Scheme 18 shows a route for the preparation of (piperidin-l-yl)methanone phenol intermediate 78 which is useful for the synthesis of compounds of Formula I.
- Substituted and O- protected benzoyl chlorides of type 76 are reacted with an appropriate amine 75 to form the corresponding amide 77, which after final deprotection of the phenol gives compound 78.
- Compound 78 can then be reacted with the appropriate core intermediate 7 as in Scheme 2 to provide compound 9.
- Scheme 19 shows a method for the preparation of phenolic intermediate 82.
- 2,5- dibromopyridine 79 is treated with the appropriate boronic acid under Suzuki type reaction conditions to give selective coupling at the pyridine 2-position to provide compound 80.
- Buchwald type palladium coupling of compound 80 with an appropriate heteroatom bearing an R 10 group gives the protected compound 81.
- Final deprotection of compound 81 gives compound 82 which can be reacted with an appropriate core intermediate 7 as in Scheme 2 to provide compound 9.
- Scheme 20 shows a method for the preparation of phenolic intermediate 86. 2,5-
- Dibromopyrimidine 83 is treated with the appropriate heteroatom bearing an R 10 group with heating in an appropriate solvent such as 1-propanol. Reaction occurs selectively at the 2- position to give the bromopyrimidine intermediate 84. Suzuki coupling to the appropriately substituted boronic acid gives intermediate 85, which after deprotection gives the phenolic 111-03-PCT - P2338R1 - 02120.004WO1
- reaction products from one another and/or from starting materials.
- the desired products of each step or series of steps is separated and/or purified (hereinafter separated) to the desired degree of homogeneity by the techniques common in the art.
- separations involve multiphase extraction, crystallization from a solvent or solvent mixture, distillation, sublimation, or chromatography.
- Chromatography can involve any number of methods including, for example: reverse-phase and normal phase; size exclusion; ion exchange; high, medium and low pressure liquid chromatography methods and apparatus; small scale analytical; simulated moving bed (SMB) and preparative thin or thick layer chromatography, as well as techniques of small scale thin layer and flash chromatography.
- SMB simulated moving bed
- Another class of separation methods involves treatment of a mixture with a reagent selected to bind to or render otherwise separable a desired product, unreacted starting material, reaction by product, or the like.
- reagents include adsorbents or absorbents such as activated carbon, molecular sieves, ion exchange media, or the like.
- the reagents can be acids in the case of a basic material, bases in the case of an acidic material, binding reagents such as antibodies, binding proteins, selective chelators such as crown ethers, liquid/liquid ion extraction reagents (LIX), or the like.
- Diastereomeric mixtures can be separated into their individual diastereomers on the basis of their physical chemical differences by methods well known to those skilled in the art, such as by chromatography and/or fractional crystallization.
- Enantiomers can be separated by converting the enantiomeric mixture into a diastereomeric mixture by reaction with an appropriate optically active compound (e.g., chiral auxiliary such as a chiral alcohol or Mosher's acid chloride), separating the diastereomers and converting (e.g., hydrolyzing) the individual diastereoisomers to the corresponding pure enantiomers.
- an appropriate optically active compound e.g., chiral auxiliary such as a chiral alcohol or Mosher's acid chloride
- some of the compounds of the present invention may be atropisomers (e.g., substituted biaryls) and are considered as part of this invention. Enantiomers can also be separated by use of a chiral FIPLC column. 111-03-PCT- P2338R1 - 02120.004WO1
- a single stereoisomer e.g., an enantiomer, substantially free of its stereoisomer may be obtained by resolution of the racemic mixture using a method such as formation of diastereomers using optically active resolving agents (Eliel, E. and Wilen, S. "Stereochemistry of Organic Compounds,” John Wiley & Sons, Inc., New York, 1994; Lochmuller, C. H., (1975) J. Chromatogr., 113(3):283-302).
- Racemic mixtures of chiral compounds of the invention can be separated and isolated by any suitable method, including: (1) formation of ionic, diastereomeric salts with chiral compounds and separation by fractional crystallization or other methods, (2) formation of diastereomeric compounds with chiral derivatizing reagents, separation of the diastereomers, and conversion to the pure stereoisomers, and (3) separation of the substantially pure or enriched stereoisomers directly under chiral conditions. See: “Drug Stereochemistry, Analytical Methods and Pharmacology,” Irving W. Wainer, Ed., Marcel Dekker, Inc., New York (1993).
- diastereomeric salts can be formed by reaction of enantiomerically pure chiral bases such as brucine, quinine, ephedrine, strychnine, ⁇ -methyl- ⁇ - phenylethylamine (amphetamine), and the like with asymmetric compounds bearing acidic functionality, such as carboxylic acid and sulfonic acid.
- the diastereomeric salts may be induced to separate by fractional crystallization or ionic chromatography.
- the substrate to be resolved is reacted with one enantiomer of a chiral compound to form a diastereomeric pair
- a diastereomeric pair E. and Wilen, S. "Stereochemistry of Organic Compounds", John Wiley & Sons, Inc., 1994, p. 322
- Diastereomeric compounds can be formed by reacting asymmetric compounds with enantiomerically pure chiral derivatizing reagents, such as menthyl derivatives, followed by separation of the diastereomers and hydrolysis to yield the pure or enriched enantiomer.
- a method of determining optical purity involves making chiral esters, such as a menthyl ester, e.g., (-) menthyl chloroformate in the presence of base, or Mosher ester, oc-methoxy- ⁇ - (trifluoromethyl)phenyl acetate (Jacob III (1982) J. Org. Chem 47:4165), of the racemic mixture, and analyzing the 1 H NMR spectrum for the presence of the two atropisomeric enantiomers or diastereomers.
- chiral esters such as a menthyl ester, e.g., (-) menthyl chloroformate in the presence of base, or Mosher ester, oc-methoxy- ⁇ - (trifluoromethyl)phenyl acetate (Jacob III (1982) J. Org. Chem 47:4165), of the racemic mixture, and analyzing the 1 H NMR spectrum for the presence of the two atropisomeric
- Stable diastereomers of atropisomeric compounds can be separated and isolated by normal- and reverse-phase chromatography following methods for separation of atropisomeric naphthyl-isoquinolines (WO 96/15111).
- method (3) a racemic mixture of two enantiomers can be separated by chromatography using a chiral stationary phase ("Chiral Liquid 111-03-PCT - P2338R1 - 02120.004WO1
- Enriched or purified enantiomers can be distinguished by methods used to distinguish other chiral molecules with asymmetric carbon atoms, such as optical rotation and circular dichroism.
- Exemplary compounds of this invention include compounds 101-205 as described in Examples 1-105. BIOLOGICAL EVALUATION
- Determination of the activity of c-Met kinase activity of a compound of Formula I is possible by a number of direct and indirect detection methods.
- One example of an assay used for the determination of c-Met kinase activity is based on an enzyme linked immunosorbant assay (ELISA).
- the assay includes a compound of Formula I, c-Met (His-tagged recombinant human Met (amino acids 974-end), expressed by baculovirus), and ATP in assay buffer, as described in Example 106.
- Exemplary compounds described herein were prepared, characterized, and assayed for their c-Met binding activity and in vitro activity against tumor cells.
- the range of c- Met binding activities was less than 1 nM to about 10 ⁇ M.
- Certain exemplary compounds of the invention had c-Met binding activity IC 50 values less than 10 nM.
- Certain compounds of the invention had MKN45 cell-based activity IC 50 values less than 100 nM.
- the compounds of the invention may be administered by any route appropriate to the condition to be treated. Suitable routes include oral, parenteral (including subcutaneous, intramuscular, intravenous, intraarterial, intradermal, intrathecal and epidural), transdermal, rectal, nasal, topical (including buccal and sublingual), vaginal, intraperitoneal, intrapulmonary and intranasal.
- routes include oral, parenteral (including subcutaneous, intramuscular, intravenous, intraarterial, intradermal, intrathecal and epidural), transdermal, rectal, nasal, topical (including buccal and sublingual), vaginal, intraperitoneal, intrapulmonary and intranasal.
- the compounds may be administered by intralesional administration, including perfusing or otherwise contacting the graft with the inhibitor before transplantation. It will be appreciated that the preferred route may vary with for example the condition of the recipient. Where the compound is administered orally, it may be formulated as a pill, capsule, tablet, etc. with a pharmaceutically acceptable carrier or
- the compound may be formulated with a pharmaceutically acceptable parenteral vehicle and in a unit dosage injectable form, as detailed below.
- Compounds of the present invention are useful for treating diseases, conditions and/or disorders including, but not limited to, those characterized by over expression of receptor tyrosine kinases (RTK), e.g. c-Met kinase.
- RTK receptor tyrosine kinases
- another aspect of this invention includes methods of treating or preventing diseases or conditions that can be treated or prevented by inhibiting receptor tyrosine kinases (RTK), including c-Met.
- the method comprises administering to a mammal in need thereof a therapeutically effective amount of a compound of Formula I, or a stereoisomer, geometric isomer, tautomer, solvate, metabolite, or pharmaceutically acceptable salt or prodrug thereof.
- Diseases and conditions treatable according to the methods of this invention include, but are not limited to, cancer, stroke, diabetes, hepatomegaly, cardiovascular disease, Alzheimer's disease, cystic fibrosis, viral disease, autoimmune diseases, atherosclerosis, restenosis, psoriasis, allergic disorders, inflammation, neurological disorders, a hormone-related disease, conditions associated with organ transplantation, immunodeficiency disorders, destructive bone disorders, proliferative disorders, infectious diseases, conditions associated with cell death, thrombin-induced platelet aggregation, chronic myelogenous leukemia (CML), liver disease, pathologic immune conditions involving T cell activation, and CNS disorders in a patient.
- a human patient is treated with a compound of Formula I and a pharmaceutically acceptable carrier, adjuvant, or vehicle, wherein said compound of Formula I is present in an amount to detectably inhibit c-Met kinase activity.
- Cancers which can be treated according to the methods of this invention include, but are not limited to, breast, ovary, cervix, prostate, testis, genitourinary tract, esophagus, larynx, glioblastoma, neuroblastoma, stomach, skin, keratoacanthoma, lung, epidermoid carcinoma, large cell carcinoma, non-small cell lung carcinoma (NSCLC), small cell carcinoma, lung adenocarcinoma, bone, colon, adenoma, pancreas, adenocarcinoma, thyroid, follicular carcinoma, undifferentiated carcinoma, papillary carcinoma, seminoma, melanoma, sarcoma, bladder carcinoma, liver carcinoma and biliary passages, kidney carcinoma, myeloid disorders, lymphoid disorders, hairy cells, buccal cavity and pharynx (oral), lip, tongue, mouth, pharynx, small intestine, colon-rectum, large intestine,
- Cardiovascular diseases which can be treated according to the methods of this invention include, but are not limited to, restenosis, cardiomegaly, atherosclerosis, myocardial infarction, and congestive heart failure.
- Neurodegenerative disease which can be treated according to the methods of this invention include, but are not limited to, Alzheimer's disease, Parkinson's disease, amyotrophic 111-03-PCT - P2338R1 - 02120.004WO1
- Inflammatory diseases which can be treated according to the methods of this invention include, but are not limited to, rheumatoid arthritis, psoriasis, contact dermatitis, and delayed hypersensitivity reactions.
- Another aspect of this invention provides a compound of this invention for use in the treatment of the diseases or conditions described herein in a mammal, for example, a human, suffering from such disease or condition. Also provided is the use of a compound of this invention in the preparation of a medicament for the treatment of the diseases and conditions described herein in a warm-blooded animal, such as a mammal, for example a human, suffering from such disorder.
- a pharmaceutical composition comprising a compound of this invention in association with a pharmaceutically acceptable diluent or carrier.
- a typical formulation is prepared by mixing a compound of the present invention and a carrier, diluent or excipient.
- Suitable carriers, diluents and excipients are well known to those skilled in the art and include materials such as carbohydrates, waxes, water soluble and/or swellable polymers, hydrophilic or hydrophobic materials, gelatin, oils, solvents, water and the like.
- Solvents are generally selected based on solvents recognized by persons skilled in the art as safe (GRAS) to be administered to a mammal.
- safe solvents are non-toxic aqueous solvents such as water and other nontoxic solvents that are soluble or miscible in water.
- Suitable aqueous solvents include water, ethanol, propylene glycol, polyethylene glycols (e.g., PEG 400, PEG 300), etc. and mixtures thereof.
- the formulations may also include one or more buffers, stabilizing agents, surfactants, wetting agents, lubricating agents, emulsifiers, suspending agents, preservatives, antioxidants, opaquing agents, glidants, processing aids, colorants, sweeteners, perfuming agents, flavoring agents and other known additives to provide an elegant presentation of the drug (i.e., a compound of the present invention or pharmaceutical composition thereof) or aid in the manufacturing of the pharmaceutical product (i.e., medicament).
- buffers stabilizing agents, surfactants, wetting agents, lubricating agents, emulsifiers, suspending agents, preservatives, antioxidants, opaquing agents, glidants, processing aids, colorants, sweeteners, perfuming agents, flavoring agents and other known additives to provide an elegant presentation of the drug (i.e., a compound of the present invention or pharmaceutical composition thereof) or aid in the manufacturing of the pharmaceutical product (i.e., medicament).
- the formulations may be prepared using conventional dissolution and mixing procedures.
- the bulk drug substance i.e., compound of the present invention or stabilized form of the compound (e.g., complex with a cyclodextrin derivative or other known complexation agent) is dissolved in a suitable solvent in the presence of one or more of the excipients described above.
- the compound of the present invention is typically formulated into pharmaceutical dosage forms to provide an easily controllable dosage of the drug and to enable patient compliance with the prescribed regimen.
- the pharmaceutical composition (or formulation) for application may be packaged in a variety of ways depending upon the method used for administering the drug.
- an article for distribution includes a container having deposited therein the pharmaceutical formulation in an appropriate form.
- Suitable containers are well known to those skilled in the art and include materials such as bottles (plastic and glass), sachets, ampoules, plastic bags, metal cylinders, and the like.
- the container may also include a tamper-proof assemblage to prevent indiscreet access to the contents of the package.
- the container has deposited thereon a label that describes the contents of the container. The label may also include appropriate warnings.
- compositions of the compounds of the present invention may be prepared for various routes and types of administration.
- a compound of Formula I having the desired degree of purity may optionally be mixed with pharmaceutically acceptable diluents, carriers, excipients or stabilizers (Remington's Pharmaceutical Sciences (1980) 16th edition, Osol, A. Ed.), in the form of a lyophilized formulation, milled powder, or an aqueous solution.
- Formulation may be conducted by mixing at ambient temperature at the appropriate pH, and at the desired degree of purity, with physiologically acceptable carriers, i.e., carriers that are non-toxic to recipients at the dosages and concentrations employed.
- the pH of the formulation depends mainly on the particular use and the concentration of compound, but may range from about 3 to about 8.
- Formulation in an acetate buffer at pH 5 is a suitable embodiment.
- the compound of this invention for use herein is preferably sterile.
- formulations to be used for in vivo administration must be sterile. Such sterilization is readily accomplished by filtration through sterile filtration membranes.
- the compound ordinarily can be stored as a solid composition, a lyophilized formulation or as an aqueous solution.
- compositions of the invention will be formulated, dosed and administered in a fashion, i.e., amounts, concentrations, schedules, course, vehicles and route of administration, consistent with good medical practice.
- Factors for consideration in this context 111-03-PCT- P2338R1 - 0212 0 .004WOl
- the "therapeutically effective amount" of the compound to be administered will be governed by such considerations, and is the minimum amount necessary to prevent, ameliorate, or treat the coagulation factor mediated disorder. Such amount is preferably below the amount that is toxic to the host or renders the host significantly more susceptible to bleeding.
- the initial pharmaceutically effective amount of the inhibitor administered parenterally per dose will be in the range of about 0.01-100 mg/kg, namely about 0.1 to 20 mg/kg of patient body weight per day, with the typical initial range of compound used being 0.3 to 15 mg/kg/day.
- Acceptable diluents, carriers, excipients and stabilizers are nontoxic to recipients at the dosages and concentrations employed, and include buffers such as phosphate, citrate and other organic acids; antioxidants including ascorbic acid and methionine; preservatives (such as octadecyldimethylbenzyl ammonium chloride; hexamethonium chloride; benzalkonium chloride, benzethonium chloride; phenol, butyl or benzyl alcohol; alkyl parabens such as methyl or propyl paraben; catechol; resorcinol; cyclohexanol; 3-pentanol; and m-cresol); low molecular weight (less than about 10 residues) polypeptides; proteins, such as serum albumin, gelatin, or immunoglobulins; hydrophilic polymers such as polyvinylpyrrolidone; amino acids such as glycine, glutamine, aspara
- the active pharmaceutical ingredients may also be entrapped in microcapsules prepared, for example, by coacervation techniques or by interfacial polymerization, for example, hydroxymethylcellulose or gelatin-microcapsules and poly- (methylmethacylate) microcapsules, respectively, in colloidal drug delivery systems (for example, liposomes, albumin microspheres, microemulsions, nano-particles and nanocapsules) or in macroemulsions.
- colloidal drug delivery systems for example, liposomes, albumin microspheres, microemulsions, nano-particles and nanocapsules
- sustained-release preparations include semipermeable matrices of solid hydrophobic polymers containing a compound of Formula I, which matrices are in the form of shaped articles, e.g., films, or microcapsules.
- sustained-release matrices include 111-03-PCT - P2338R1 - 02120.004WO1
- polyesters for example, poly(2-hydroxyethyl-methacrylate), or poly(vinyl alcohol)
- polylactides U.S. Patent No. 3,773,919
- copolymers of L-glutamic acid and gamma-ethyl-L- glutamate non-degradable ethylene-vinyl acetate
- degradable lactic acid-glycolic acid copolymers such as the LUPRON DEPOT® (injectable microspheres composed of lactic acid- glycolic acid copolymer and leuprolide acetate) and poly-D-(-)-3-hydroxybutyric acid.
- the formulations include those suitable for the administration routes detailed herein.
- formulations may conveniently be presented in unit dosage form and may be prepared by any of the methods well known in the art of pharmacy. Techniques and formulations generally are found in Remington's Pharmaceutical Sciences (Mack Publishing Co., Easton, PA). Such methods include the step of bringing into association the active ingredient with the carrier which constitutes one or more accessory ingredients. In general the formulations are prepared by uniformly and intimately bringing into association the active ingredient with liquid carriers or finely divided solid carriers or both, and then, if necessary, shaping the product. [00178] Formulations of a compound of Formula I suitable for oral administration may be prepared as discrete units such as pills, capsules, cachets or tablets each containing a predetermined amount of a compound of Formula I.
- Compressed tablets may be prepared by compressing in a suitable machine the active ingredient in a free-flowing form such as a powder or granules, optionally mixed with a binder, lubricant, inert diluent, preservative, surface active or dispersing agent. Molded tablets may be made by molding in a suitable machine a mixture of the powdered active ingredient moistened with an inert liquid diluent. The tablets may optionally be coated or scored and optionally are formulated so as to provide slow or controlled release of the active ingredient therefrom.
- Tablets, troches, lozenges, aqueous or oil suspensions, dispersible powders or granules, emulsions, hard or soft capsules, e.g., gelatin capsules, syrups or elixirs may be prepared for oral use.
- Formulations of compounds of Formula I intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions and such compositions may contain one or more agents including sweetening agents, flavoring agents, coloring agents and preserving agents, in order to provide a palatable preparation. Tablets containing the active ingredient in admixture with non-toxic pharmaceutically acceptable excipient which are suitable for manufacture of tablets are acceptable.
- excipients may be, for example, inert diluents, such as calcium or sodium carbonate, lactose, calcium or sodium phosphate; granulating and disintegrating agents, such as maize starch, or alginic acid; binding agents, such as starch, gelatin or acacia; and lubricating 111-03-PCT - P2338R1 - 02120.004WO1
- 57 agents such as magnesium stearate, stearic acid or talc. Tablets may be uncoated or may be coated by known techniques including microencapsulation to delay disintegration and adsorption in the gastrointestinal tract and thereby provide a sustained action over a longer period.
- a time delay material such as glyceryl monostearate or glyceryl distearate alone or with a wax may be employed.
- the formulations are preferably applied as a topical ointment or cream containing the active ingredient(s) in an amount of, for example, 0.075 to 20% w/w.
- the active ingredients may be employed with either a paraffinic or a water-miscible ointment base.
- the active ingredients may be formulated in a cream with an oil-in-water cream base.
- the aqueous phase of the cream base may include a polyhydric alcohol, i.e., an alcohol having two or more hydroxyl groups such as propylene glycol, butane 1,3-diol, mannitol, sorbitol, glycerol and polyethylene glycol (including PEG 400) and mixtures thereof.
- the topical formulations may desirably include a compound which enhances absorption or penetration of the active ingredient through the skin or other affected areas. Examples of such dermal penetration enhancers include dimethyl sulfoxide and related analogs.
- the oily phase of the emulsions of this invention may be constituted from known ingredients in a known manner.
- the phase may comprise merely an emulsifier, it desirably comprises a mixture of at least one emulsifier with a fat or an oil or with both a fat and an oil.
- a hydrophilic emulsifier is included together with a lipophilic emulsifier which acts as a stabilizer. It is also preferred to include both an oil and a fat.
- the emulsifier(s) with or without stabilizer(s) make up the so-called emulsifying wax, and the wax together with the oil and fat make up the so-called emulsifying ointment base which forms the oily dispersed phase of the cream formulations.
- Emulsifiers and emulsion stabilizers suitable for use in the formulation of the invention include TWEEN® 60, Span® 80, cetostearyl alcohol, benzyl alcohol, myristyl alcohol, glyceryl mono-stearate and sodium lauryl sulfate.
- Aqueous suspensions of Formula I compounds contain the active materials in admixture with excipients suitable for the manufacture of aqueous suspensions.
- excipients include a suspending agent, such as sodium carboxymethylcellulose, croscarmellose, povidone, methylcellulose, hydroxypropyl methylcellulose, sodium alginate, polyvinylpyrrolidone, gum tragacanth and gum acacia, and dispersing or wetting agents such as a naturally occurring phosphatide (e.g., lecithin), a condensation product of an alkylene oxide with a fatty acid (e.g., polyoxyethylene stearate), a condensation product of ethylene oxide with a long chain aliphatic 111-03-PCT - P2338R1 - 02120.004WO1
- a suspending agent such as sodium carboxymethylcellulose, croscarmellose, povidone, methylcellulose, hydroxypropyl methylcellulose, sodium alginate, polyvinylpyr
- the aqueous suspension may also contain one or more preservatives such as ethyl or n-propyl p-hydroxybenzoate, one or more coloring agents, one or more flavoring agents and one or more sweetening agents, such as sucrose or saccharin.
- preservatives such as ethyl or n-propyl p-hydroxybenzoate, one or more coloring agents, one or more flavoring agents and one or more sweetening agents, such as sucrose or saccharin.
- compositions of compounds of Formula I may be in the form of a sterile injectable preparation, such as a sterile injectable aqueous or oleaginous suspension.
- a sterile injectable preparation such as a sterile injectable aqueous or oleaginous suspension.
- This suspension may be formulated according to the known art using those suitable dispersing or wetting agents and suspending agents which have been mentioned above.
- the sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally acceptable diluent or solvent, such as a solution in 1,3-butanediol or prepared as a lyophilized powder.
- a non-toxic parenterally acceptable diluent or solvent such as a solution in 1,3-butanediol or prepared as a lyophilized powder.
- acceptable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution.
- sterile fixed oils may conventionally be employed as a solvent or suspending medium.
- any bland fixed oil may be employed including synthetic mono- or diglycerides.
- fatty acids such as oleic acid may likewise be used in the preparation of injectables.
- a time-release formulation intended for oral administration to humans may contain approximately 1 to 1000 mg of active material compounded with an appropriate and convenient amount of carrier material which may vary from about 5 to about 95% of the total compositions (weight:weight).
- the pharmaceutical composition can be prepared to provide easily measurable amounts for administration.
- an aqueous solution intended for intravenous infusion may contain from about 3 to 500 ⁇ g of the active ingredient per milliliter of solution in order that infusion of a suitable volume at a rate of about 30 mL/hr can occur.
- Formulations suitable for parenteral administration include aqueous and nonaqueous sterile injection solutions which may contain anti-oxidants, buffers, bacteriostats and solutes which render the formulation isotonic with the blood of the intended recipient; and aqueous and non-aqueous sterile suspensions which may include suspending agents and thickening agents.
- Formulations suitable for topical administration to the eye also include eye drops wherein the active ingredient is dissolved or suspended in a suitable carrier, especially an aqueous solvent for the active ingredient.
- a suitable carrier especially an aqueous solvent for the active ingredient.
- the active ingredient is preferably present in such formulations 111-03-PCT - P2338R1 - 02120.004WO1
- 59 in a concentration of about 0.5 to 20% w/w, for example about 0.5 to 10% w/w, for example about 1.5% w/w.
- Formulations suitable for topical administration in the mouth include lozenges comprising the active ingredient in a flavored basis, usually sucrose and acacia or tragacanth; pastilles comprising the active ingredient in an inert basis such as gelatin and glycerin, or sucrose and acacia; and mouthwashes comprising the active ingredient in a suitable liquid carrier.
- Formulations for rectal administration may be presented as a suppository with a suitable base comprising for example cocoa butter or a salicylate.
- Formulations suitable for intrapulmonary or nasal administration have a particle size for example in the range of 0.1 to 500 microns (including particle sizes in a range between 0.1 and 500 microns in increments microns such as 0.5, 1, 30 microns, 35 microns, etc.), which is administered by rapid inhalation through the nasal passage or by inhalation through the mouth so as to reach the alveolar sacs.
- Suitable formulations include aqueous or oily solutions of the active ingredient.
- Formulations suitable for aerosol or dry powder administration may be prepared according to conventional methods and may be delivered with other therapeutic agents such as compounds heretofore used in the treatment or prophylaxis disorders as described below.
- Formulations suitable for vaginal administration may be presented as pessaries, tampons, creams, gels, pastes, foams or spray formulations containing in addition to the active ingredient such carriers as are known in the art to be appropriate.
- the formulations may be packaged in unit-dose or multi-dose containers, for example sealed ampoules and vials, and may be stored in a f ⁇ eeze-dried (lyophilized) condition requiring only the addition of the sterile liquid carrier, for example water, for injection immediately prior to use.
- sterile liquid carrier for example water
- Extemporaneous injection solutions and suspensions are prepared from sterile powders, granules and tablets of the kind previously described.
- Preferred unit dosage formulations are those containing a daily dose or unit daily sub-dose, as herein above recited, or an appropriate fraction thereof, of the active ingredient.
- the invention further provides veterinary compositions comprising at least one active ingredient as above defined together with a veterinary carrier therefore.
- Veterinary carriers are materials useful for the purpose of administering the composition and may be solid, liquid or gaseous materials which are otherwise inert or acceptable in the veterinary art and are compatible with the active ingredient. These veterinary compositions may be administered parenterally, orally or by any other desired route. COMBINATION THERAPY 111-03-PCT- P2338R1 - 02120.Q04WO1
- the compounds of Formula I may be employed alone or in combination with other therapeutic agents for the treatment of a disease or disorder described herein, such as a hyperproliferative disorder (e.g., cancer).
- a compound of Formula I is combined in a pharmaceutical combination formulation, or dosing regimen as combination therapy, with a second compound that has anti-hyperproliferative properties or that is useful for treating a hyperproliferative disorder (e.g., cancer).
- the second compound of the pharmaceutical combination formulation or dosing regimen preferably has complementary activities to the compound of Formula I such that they do not adversely affect each other.
- Such compounds are suitably present in combination in amounts that are effective for the purpose intended.
- a composition of this invention comprises a compound of Formula I, or a stereoisomer, geometric isomer, tautomer, solvate, metabolite, or pharmaceutically acceptable salt or prodrug thereof, in combination with a chemotherapeutic agent such as described herein.
- the combination therapy may be administered as a simultaneous or sequential regimen. When administered sequentially, the combination may be administered in two or more administrations.
- the combined administration includes coadministration, using separate formulations or a single pharmaceutical formulation, and consecutive administration in either order, wherein preferably there is a time period while both (or all) active agents simultaneously exert their biological activities.
- Suitable dosages for any of the above coadministered agents are those presently used and may be lowered due to the combined action (synergy) of the newly identified agent and other chemotherapeutic agents or treatments.
- the combination therapy may provide "synergy” and prove “synergistic", i.e., the effect achieved when the active ingredients used together is greater than the sum of the effects that results from using the compounds separately.
- a synergistic effect may be attained when the active ingredients are: (1) co-formulated and administered or delivered simultaneously in a combined, unit dosage formulation; (2) delivered by alternation or in parallel as separate formulations; or (3) by some other regimen.
- a synergistic effect may be attained when the compounds are administered or delivered sequentially, e.g., by different injections in separate syringes.
- an effective dosage of each active ingredient is administered sequentially, i.e., serially
- effective dosages of two or more active ingredients are administered together.
- a compound of Formula I or a stereoisomer, geometric isomer, tautomer, solvate, metabolite, or pharmaceutically acceptable 111-03-PCT - P2338R1 - 02120.004WO1
- Combination therapies may be combined with other chemotherapeutic, hormonal or antibody agents such as those described herein, as well as combined with surgical therapy and radiotherapy.
- Combination therapies according to the present invention thus comprise the administration of at least one compound of Formula I, or a stereoisomer, geometric isomer, tautomer, solvate, metabolite, or pharmaceutically acceptable salt or prodrug thereof, and the use of at least one other cancer treatment method.
- the amounts of the compound(s) of Formula I and the other pharmaceutically active chemotherapeutic agent(s) and the relative timings of administration will be selected in order to achieve the desired combined therapeutic effect.
- the invention includes metabolites of compounds of Formula I, including compounds produced by a process comprising contacting a compound of this invention with a mammal for a period of time sufficient to yield a metabolic product thereof.
- Metabolite products typically are identified by preparing a radiolabeled (e.g., 14 C or 3 H) isotope of a compound of the invention, administering it parenterally in a detectable dose (e.g., greater than about 0.5 mg/kg) to an animal such as rat, mouse, guinea pig, monkey, or to man, allowing sufficient time for metabolism to occur (typically about 30 seconds to 30 hours) and isolating its conversion products from the urine, blood or other biological samples.
- a detectable dose e.g., greater than about 0.5 mg/kg
- the metabolite structures are determined in conventional fashion, e.g., by MS, LC/MS or NMR analysis. In general, analysis of metabolites is done in the same way as conventional drug metabolism studies well known to those skilled in the art.
- the metabolite products so long as they are not otherwise found in vivo, are useful in diagnostic assays for therapeutic dosing of the compounds of the invention.
- kits containing materials useful for the treatment of the diseases and disorders described above.
- the kit comprises a container comprising a quinoline compound of Formula I, or a stereoisomer, geometric isomer, tautomer, solvate, metabolite, or pharmaceutically acceptable salt or prodrug thereof.
- the kit may further comprise a label or package insert on or associated with the container.
- package insert is used to refer to 111-03-PCT- P2338R1 - 02120.004WO1
- Suitable containers include, for example, bottles, vials, syringes, blister pack, etc.
- the container may be formed from a variety of materials such as glass or plastic.
- the container may hold a compound of Formula I or a formulation thereof which is effective for treating the condition and may have a sterile access port (for example, the container may be an intravenous solution bag or a vial having a stopper pierceable by a hypodermic injection needle).
- At least one active agent in the composition is a compound of Formula I.
- the label or package insert indicates that the composition is used for treating the condition of choice, such as cancer.
- the label or package insert may indicate that the patient to be treated is one having a disorder such as a hyperproliferative disorder, neurodegeneration, cardiac hypertrophy, pain, migraine or a neurotraumatic disease or event.
- the label or package inserts indicates that the composition comprising a compound of Formula I can be used to treat a disorder resulting from abnormal cell growth.
- the label or package insert may also indicate that the composition can be used to treat other disorders.
- the article of manufacture may further comprise a second container comprising a pharmaceutically acceptable buffer, such as bacteriostatic water for injection (BWFI), phosphate-buffered saline, Ringer's solution and dextrose solution. It may further include other materials desirable from a commercial and user standpoint, including other buffers, diluents, filters, needles, and syringes.
- BWFI bacteriostatic water for injection
- phosphate-buffered saline such as bacteriostatic water for injection (BWFI), phosphate-buffered saline, Ringer's solution and dextrose solution.
- BWFI bacteriostatic water for injection
- phosphate-buffered saline such as phosphate-buffered saline, Ringer's solution and dextrose solution.
- dextrose solution such as bacteriostatic water for injection (BWFI), phosphate-buffered saline, Ringer's solution and dext
- the kit may further comprise directions for the administration of the compound of
- kits are suitable for the delivery of solid oral forms of a compound of Formula I, such as tablets or capsules.
- a kit preferably includes a number of unit dosages.
- kits can include a card having the dosages oriented in the order of their intended use.
- An example of such a kit is a "blister pack". Blister packs are well known in the packaging industry and are widely used for packaging pharmaceutical unit dosage forms.
- a memory aid can be provided, for example in the form of numbers, letters, or other markings or with a calendar insert, designating the days in the treatment schedule in which the dosages can be administered.
- a kit may comprise (a) a first container with a compound of Formula I contained therein; and optionally (b) a second container with a second pharmaceutical formulation contained therein, wherein the second pharmaceutical formulation comprises a second compound with anti-hyperproliferative activity.
- the kit may further comprise a third container comprising a pharmaceutically- acceptable buffer, such as bacteriostatic water for injection (BWFI), phosphate-buffered saline, Ringer's solution and dextrose solution. It may further include other materials desirable from a commercial and user standpoint, including other buffers, diluents, filters, needles, and syringes.
- BWFI bacteriostatic water for injection
- phosphate-buffered saline such as bacteriostatic water for injection (BWFI), phosphate-buffered saline, Ringer's solution and dextrose solution. It may further include other materials desirable from a commercial and user standpoint, including other buffers, diluents,
- the kit may comprise a container for containing the separate compositions such as a divided bottle or a divided foil packet, however, the separate compositions may also be contained within a single, undivided container.
- the kit comprises directions for the administration of the separate components.
- the kit form is particularly advantageous when the separate components are preferably administered in different dosage forms (e.g., oral and parenteral), are administered at different dosage intervals, or when titration of the individual components of the combination is desired by the prescribing physician.
- Step A Preparation of 4-chloro-5-methyl-6-(2-methylbenzyl)pyrimidine: 2-
- Methylbenzylzinc chloride 25 ml of 0.5 M THF solution, 12 mmol was added to a solution of 4,6-dichloro-5-methylpyrimidine (2.0 g, 12 mmol) and bis(triphenylphosphine) palladium(II) chloride (0.4 g. 0.6 mmol) in THF (20 mL). The reaction mixture was heated to reflux for 2 hours, cooled to room temperature, and then poured onto water (10 mL). The reaction mixture was extracted with ethyl acetate, and the organic layer was washed with brine, dried over sodium sulfate, filtered, and concentrated.
- Step B Preparation of 4-(benzyloxy)-5-methyl-6-(2-methylbenzyl)pyrimidine:
- Step C Preparation of 5-methyl-6-(2-methylbenzyl)pyrimidin-4-ol: 4-
- Step D Preparation of 3-(3-fluoro-4-hydroxyphenyl)-5-methyl-6-(2- methylbenzyl)pyrimidin-4(3H)-one: Copper(I) iodide (90 mg, 0.5 mmol) was added to a solution of 5-methyl-6-(2-methylbenzyl)pyrimidin-4-ol (1.0 g, 5.0 mmol), 4-bromo-2-fluorophenol (0.90 g, 5.0 mmol), N,N'-dimethylethylenediamine (80 mg, 0.90 mmol) and potassium phosphate (2.0 g, 9.0 mmol).
- Step E Preparation of 3-(3-fluoro-4-(6-methoxy-7-(3-morpholino- propoxy)quinolin-4-yloxy)phenyl)-5-methyl-6-(2-methylbenzyl)pyrimidin-4(3H)-one: DMAP (0.75 mg, 0.0062 mmol) was added to a suspension of 3-(3-fluoro-4-hydroxyphenyl)-5-methyl-6- (2-methylbenzyl)pyrimidin-4(3H)-one (20 mg, 0.062 mmol) and 4-chloro-6-methoxy-7-(3- morpholinopropoxy)quinoline (prepared according to WO 01/55116, Example 2, 21 mg, 0.062 mmol).
- Step A Preparation of 4-benzyl-6-chloropyrimidine: Prepared from 4,6- dichloropyrimidine (2.0 g, 13 mmol) and benzyl zinc chloride (0.5 M solution in THF, 27 mL, 13 mmol) according to the procedure described for Example 1, Step A. The crude product was purified by silica gel flash column chromatography (1:10 Et 2 O/Hexane) to yield the product (1.3 g, 47%) as a yellow liquid.
- Step B Preparation of 4-benzyl-6-(benzyloxy)pyrimidine: Prepared from 4- benzyl-6-chloropyrimidine (1.1 g, 5.4 mmol) according to the procedure described for Example 1, Step B. The crude was purified by by silica gel flash column chromatography (1:1 Et 2 O/Hexane) to yield the product (1.3 g, 88%) as a colorless liquid.
- Step C Preparation of 6-benzylpyrimidin-4-ol: Prepared from 4-benzyl-6-
- Step D Preparation of 6-benzyl-3-(3-fluoro-4-hydroxyphenyl)pyrimidin-4(3H)- one: Prepared from 6-benzylpyrimidin-4-ol (0.50 g, 2.7 mmol) according to the procedure described for Example 1, Step D. The crude was purified by silica gel flash column chromatography (1:1 EtOAc/Hexane) to yield the product (0.50 g, 63%) as a white solid.
- Step E Preparation of 6-benzyl-3-(3-fluoro-4-(6-methoxy-7-(3- morpholinopropoxy)quinolin-4-yloxy)phenyl)pyrimidin-4(3H)-one: Prepared from 6-benzyl-3- (3-fiuoro-4-hydroxyphenyl)pyrimidin-4(3H)-one (18 mg, 0.059 mmol) according to the procedure described for Example 1, Step E. The crude product was purified by silica gel flash 111-03-PCT- P2338R1 - 02120.004WO1
- Step A Preparation of 4-benzyl-6-chloro-5-methylpyrimidine: Prepared from benzyl zinc bromide (0.5 M solution in THF, 25 mL, 12 mmol) and 4,6-dichloro-5- methylpyrimidine (2.0 g, 12 mmol) according to the procedure described for Example 1, Step A. The crude was purified by silica gel flash column chromatography (1:5 EtOAc/Hexane) to yield the product (0.86 g, 32%) as a colorless oil.
- 1 H NMR (CDCl 3 , 400 MHz) ⁇ 8.78 (s, IH), 7.28-7.33 (m, 2H), 7.18-7.26 (m, 3H), 4.19 (s, 2H), 2.35 (s, 3H).
- Step B Preparation of 4-benzyl-6-(benzyloxy)-5-methylpyrimidine: Prepared from 4-benzyl-6-chloro-5-methylpyrimidine (0.8 g, 4.0 mmol) according to the procedure described for Example 1, Step B. The crude product was purified by silica gel flash column chromatography (1:9 Et 2 O/Hexane) to yield the product (1.0 g, 94%) as a colorless oil.
- Step C Preparation of 6-benzyl-5-methylpyrimidin-4-ol: Prepared from 4- benzyl-6-(benzyloxy)-5-methylpyrimidine (1.0 g, 3.0 mmol) according to the procedure described for Example 1, Step C, to yield the product (0.50 g, 73%) as a white solid.
- Step D Preparation of 6-benzyl-3-(3-fluoro-4-hydroxyphenyl)-5- methylpyrimidin-4(3H)-one: Prepared from 6-benzyl-5-methylpyrimidin-4-ol (0.16 g, 0.80 mmol) according to the procedure described for Example 1, Step D. The crude product was purified by silica gel flash column chromatography (1 :1 EtOAc/Hexane) to yield the product (0.10 g, 64%) as a white solid.
- Step E Preparation of 6-benzyl-3-(3-fluoro-4-(6-methoxy-7-(3- morpholinopropoxy)quinolin-4-yloxy)phenyl)-5-methylpyrimidin-4(3H)-one: Prepared from 6- benzyl-3-(3-fluoro-4-hydroxyphenyl)-5-methylpyrimidin-4(3H)-one (18 mg, 0.06 mmol) according to the procedure described for Example 1, Step E. The crude product was purified by silica gel flash column chromatography (1:10 MeOH/EtOAc) to yield 103 (10 mg, 28%) as a white solid.
- Step A Preparation of (3-benzylpiperidin-l-yl)(3-fluoro-4- methoxyphenyl)methanone: Triethylamine (2 mL, 0.01 mol) was added into a solution of 3- fluoro-4-methoxybenzoyl chloride (500 mg, 2.65 mmol) and 3-benzylpiperidine hydrochloride (561 mg, 2.65 mmol) in CH 2 Cl 2 (20 mL). The reaction mixture was stirred for 30 minutes at room temperature and then poured into water (10 mL).
- Step B Preparation of (3-benzylpiperidin-l-yl)(3-fluoro-4- hydroxyphenyl)methanone: Boron tribromide (0.5 ml, 6.57 mmol) was added into a solution of (3-benzylpiperidin-l-yl)(3-fluoro-4-methoxyphenyl)methanone (0.86 g, 2.63 mmol) in CH 2 Cl 2 (2 mL) at O 0 C. The reaction mixture was stirred for 30 minutes at room temperature and then poured into water (10 mL).
- Step C Preparation of (3-benzylpiperidin-l-yl)(3-fluoro-4-(6-methoxy-7-(3- morpholinopropoxy)quinolin-4-yloxy)phenyl)methanone: Prepared from (3-benzylpiperidin-l- 111-03-PCT- P2338R1 - 02120.004WO1
- Step A Preparation of (4-benzylpiperidin-l-yl)(3-fluoro-4- methoxyphenyl)methanone: Prepared from 3-fluoro-4-methoxybenzoyl chloride (570 mg, 3.02 mmol) and 4-benzylpiperidine (530 mg, 3.02 mmol) according to the procedure described for Example 4, Step A, to yield the product (920 mg, 93%) as a white solid.
- Step B Preparation of (4-benzylpiperidin-l-yl)(3-fluoro-4- hydroxyphenyl)methanone: Prepared from (4-benzylpiperidin-l-yl)(3-fluoro-4- methoxyphenyl)methanone (130 mg, 0.40 mmol) according to the procedure described for Example 4, Step B, to yield the product (120 mg, 96%) as a white solid.
- Step C (4-benzylpiperidin-l-yl)(4-(6,7-dimethoxyquinolin-4-yloxy)-3- fluorophenyl)methanone: Prepared from 4-chloro-6,7-dimethoxyquinoline (for preparation see reference in Example 5) (79 mg, 0.35 mmol) and (4-benzylpiperidin-l-yl)(3-fluoro-4- hydroxyphenyl)methanone (110 g, 0.35 mmol) according to the procedure described for Example 1, Step E. The crude product was purified by silica gel flash column chromatography (1:10 MeOH/EtOAc) to yield 106 (20 mg, 11%) as a white solid.
- Step A Preparation of 3-benzyl-5-(4-(benzyloxy)-3-fluorophenyl)pyrimidin-
- Step B Preparation of 3-benzyl-5-(3-fiuoro-4-hydroxyphenyl)pyrimidin-4(3H)- one: Prepared from 3-benzyl-5-(4-(benzyloxy)-3-fluorophenyl)pyrimidin-4(3H)-one (0.3 g, 0.8 mmol) according to the procedure described for Example 1, Step C, to yield the product (0.2 g, 87%) as a white solid.
- Step C Preparation of 3-benzyl-5-(4-(7-(benzyloxy)-6-methoxyquinolin-4- yloxy)-3-fluorophenyi)pyrimidin-4(3H)-one: Prepared from 7-(benzyloxy)-4-chloro-6- methoxyquinoline (prepared according to WO2005030140, Example 32, 200 mg, 0.67 mmol) and 3-benzyl-5-(3-fluoro-4-hydroxyphenyl)pyrimidin-4(3H)-one (198 mg, 0.67 mmol) according to the procedure described for Example 1, Step E. The crude product was purified by silica gel flash column chromatography (1:10 MeOH/EtOAc) to yield the product (100 mg, 37%) as a white solid. LRMS (ESI pos) m/e 560 (M+l).
- Step D Preparation of 3-benzyl-5-(3-fluoro-4-(7-hydroxy-6-rnethoxyquinolin-4- yloxy)phenyl)pyrimidin-4(3H)-one: Prepared from 3-benzyl-5-(4-(7-(benzyloxy)-6- methoxyquinolin-4-yloxy)-3-fluorophenyl)pyrimidin-4(3H)-one (90 mg, 0.16 mmol) according to the procedure described for Example 1, Step C, to yield the product (50 mg, 66%) as a white solid.
- Step E Preparation of 3-benzyl-5-(3-fluoro-4-(6-methoxy-7-(3- morpholinopropoxy)quinolin-4-yloxy)phenyl)pyrimidin-4(3H)-one: Cesium carbonate (6.9 mg, 0.021 mmol) was added to a solution of 3-benzyl-5-(3-fluoro-4-(7-hydroxy-6-methoxyquinolin-4- yloxy)phenyl)pyrimidin-4(3H)-one (10 mg, 0.02 mmol) and 4-(3-chloropropyl)morpholine (3.5 mg, 0.021 mmol).
- reaction mixture was heated to 5O 0 C for 1 hour, and then poured onto water (1 mL).
- the reaction mixture was extracted with EtOAc, and the organic layer was washed with brine, dried over sodium sulfate, filtered, and concentrated.
- the residue was purified by silica gel flash column chromatography (1:10 MeOH/EtOAc) to yield 107 (6 mg, 47%) as a yellow solid.
- Step A Preparation of 5-bromo-3-(4-chlorobenzyl)pyrimidin-4(3H)-one: Sodium hydride (0.34 g, 8.6 mmol) was added into a solution of 5-bromopyrimidin-4(3H)-one (prepared according to Thomas J Kress (1985) J. Org. Chem. 50:3073-6, 1.5 g, 8.57 mmol) in THF (10 niL) and DMF (6 mL). The reaction was stirred for 10 minutes and 4-chlorobenzyl bromide (1.76 g, 8.57 mmol) was added.
- Step B Preparation of 5-(4-(benzyloxy)-3-fluorophenyl)-3-(4- chlorobenzyl)pyrimidin-4(3H)-one: Prepared from 5-bromo-3-(4-chlorobenzyl)pyrimidin-4(3H)- one (0.39 g, 1.3 mmol) according to the procedure described for Example 7, Step A. The crude was purified by silica gel flash column chromatography (1: 1 EtOAc/Hexane) to yield the product (0.17 g, 31%) as a white solid.
- Step C Preparation of 3-(4-chlorobenzyl)-5-(3-fluoro-4- hydroxyphenyl)pyrimidin-4(3H)-one: Prepared from 5-(4-(benzyloxy)-3-fluorophenyl)-3-(4- chlorobenzyl)pyrimidin-4(3H)-one (0.17 g, 0.40 mmol) according to the procedure described for Example 1, Step C, to yield the product (0.1 g, 75%) as a yellow solid.
- Step D Preparation of 3-(4-chlorobenzyl)-5-(3-fluoro-4-(6-methoxy-7-(3- morpholinopropoxy)quinolin-4-yloxy)phenyl)pyrimidin-4(3H)-one: Prepared from 3-(4- chlorobenzyl)-5-(3-fluoro-4-hydroxyphenyl)pyrimidin-4(3H)-one (27 mg, 0.08 mmol) according to the procedure described for Example 1, Step E. The crude was purified by silica gel flash column chromatography (1:10 MeOH/EtOAc) to yield 113 (10 mg, 36%) as a yellow solid.
- Step A Preparation of (6-(benzyloxy)pyridin-2-yl)(phenyl)methanol: nBuLi (2.5
- Step B Preparation of 6-benzylpyridin-2-ol: Palladium on carbon (10%, 1.5 g, 1.4 mmol) was added into a solution of (6-(benzyloxy)pyridin-2-yl)(phenyl)methanol (4 g, 14 mmol) in MeOH (20 mL). The reaction was pressurized with hydrogen using a balloon, stirred for 2 111-03-PCT- P2338R1 - 02120.004WO1
- Step C Preparation of 6-benzyl-3-bromopyridin-2-ol: Bromine (0.14 mL, 2.7 mmol) was added into a solution of 6-benzylpyridin-2-ol (0.5 g, 2.7 mmol) in CH 2 Cl 2 (5 mL). The reaction was stirred for 20 minutes at room temperature and then poured into 10% aqueous sodium bisulfite solution (10 mL). The reaction mixture was extracted with CH 2 Cl 2 and the organic layer was washed with brine, dried over sodium sulfate, filtered, and concentrated to yield the product (0.59 g, 82%) as a yellow solid. LRMS (ESI pos) m/e 263 (M+l).
- Step D Preparation of 6-benzyl-3-(4-(benzyloxy)-3-fluorophenyl)pyridin-2(lH)- one: Prepared from 6-benzyl-3-bromopyridin-2-ol (0.63 g, 2.39 mmol) according to the procedure described for Example 7, Step A. The crude product was purified by silica gel flash column chromatography (1:1 EtOAc/Hexane) to yield the product (200 mg, 22%) as a white solid.
- Step E Preparation of 6-benzyl-3-(3-fluoro-4-hydroxyphenyl)pyridin-2(lH)-one:
- Step F Preparation of 6-benzyl-3-(4-(6,7-dimethoxyquinolin-4-yloxy)-3- fluorophenyl)pyridin-2(lH)-one: Prepared from 4-chloro-6,7-dimethoxyquinoline (prepared according to reference in Example 5) (85 mg, 0.38 mmol) and 6-benzyl-3-(3-fluoro-4- hydroxyphenyl)pyridin-2(lH)-one (93 mg, 0.31 mmol) according to the procedure described for Example 1, Step E. The crude product was purified by silica gel flash column chromatography (1:10 MeOH/EtOAc) to yield 114 (50 mg, 33%) as a white solid.
- Example 14 (Example 14, Step E, 30 mg, 0.10 mmol) according to the procedure described for Example 1, Step E.
- the crude product was purified by silica gel flash column chromatography (1:10 MeOH/EtOAc) to yield 115 (32 mg, 53%) as a white solid.
- Step A Preparation of (2-(benzyloxy)-5-bromopyridin-4-yl)(phenyl)methanol:
- Step B Preparation of 4-benzylpyridin-2(lH)-one: Prepared from (2-(benzyloxy)-
- Step C Preparation of 4-benzyl-l-(3-fluoro-4-hydroxyphenyl)pyridin-2(lH)-one:
- Step D Preparation of 4-benzyl-l-(3-fluoro-4-(6-methoxy-7-(3- morpholinopropoxy)quinolin-4-yloxy)phenyl)pyridin-2(lH)-one: Prepared from 4-benzyl-l-(3- fluoro-4-hydroxyphenyl)pyridin-2(lH)-one (30 mg, 0.10 mmol) according to the procedure described for Example 1, Step E. The crude product was purified by silica gel flash column chromatography (1:10 MeOH/EtOAc) to yield 116 (41 mg, 68%) as a white solid.
- Step A Preparation of 5-bromo-3-(2-chlorobenzyl)pyrimidin-4(3H)-one:
- Step B Preparation ⁇ of 5-(4-(benzyloxy)-3-fluorophenyl)-3-(2- chlorobenzyl)pyrimidin-4(3H)-one: Prepared from 5-bromo-3-(2-chlorobenzyl)pyrimidin-4(3H)- one (0.37 g, 1.2 mmol) according to the procedure described for Example 7, Step A. The crude product was purified by silica gel flash column chromatography (1:1 EtOAc/Hexane) to yield the product (0.30 g, 58%) as a white solid.
- Step C Preparation of 3-(2-chlorobenzyl)-5-(3-fluoro-4- hydroxyphenyl)pyrimidin-4(3H)-one: Prepared from 5-(4-(benzyloxy)-3-fluorophenyl)-3-(2- chlorobenzyl)pyrimidin-4(3H)-one (0.4 g, 1.0 mmol) according to the procedure described for Example 1, Step C, to yield the product (0.20 g, 64%) as a white solid.
- Step D Preparation of 3-(2-chlorobenzyl)-5-(4-(6,7-dimethoxyquinolin-4-yloxy)-
- 3-fluorophenyl)pyrimidin-4(3H)-one Prepared from 4-chloro-6,7-dimethoxyquinoline (prepared according to reference procedure in Example 5) (60 mg, 0.27 mmol) and 3-(2-chlorobenzyl)-5-(3- fluoro-4-hydroxyphenyl)pyrimidin-4(3H)-one (89 mg, 0.27 mmol) according to the procedure described for Example 1, Step E. The crude product was purified by silica gel flash column chromatography (1:10 MeOH/EtOAc) to yield 119 (20 mg, 14%) as a white solid.
- Step A Preparation of 5-bromo-3-(2-methylbenzyl)pyrimidin-4(3H)-one:
- Step B Preparation of 5-(4-(benzyloxy)-3-fluorophenyl)-3-(2- methylbenzyl)pyrimidin-4(3H)-one: Prepared from 5-bromo-3-(2-chlorobenzyl)pyrimidin-4(3H)- one (280 mg, 1.0 mmol) according to the procedure described for Example 7, Step A. The crude product was purified by silica gel flash column chromatography (1:1 EtOAc/Hexane) to yield the product (0.31 g, 77%) as a white solid.
- Step C Preparation of 5-(3-fluoro-4-hydroxyphenyl)-3-(2- methylbenzyl)pyrimidin-4(3H)-one: Prepared frcm 5-(4-(benzyloxy)-3-fluorophenyl)-3-(2- methylbenzyl)pyrimidin-4(3H)-one (0.31 g, 0.77 mmol) according to the procedure described for Example 1, Step C, to yield the product (0.20 g, 84%) as a white solid.
- LRMS (ESI pos) m/e 331 (M+l).
- Step D Preparation of 5-(4-(6,7-dimethoxyquinolin-4-yloxy)-3-fluorophenyl)-3-
- (2-methylbenzyl)pyrimidin-4(3H)-one Prepared from 4-chloro-6,7-dimethoxyquinoline (prepared according to reference procedure in Example 5) (54 mg, 0.24 mmol) and 5-(3-fluoro-4- hydroxyphenyl)-3-(2-methylbenzyl)pyrimidin-4(3H)-one (75 mg, 0.24 mmol) according to the procedure described for Example 1, Step E.
- the crude product was purified by silica gel flash column chromatography (1:10 MeOH/EtOAc) to yield 120 (30 mg, 25%) as a white solid.
- Step A Preparation of 5-bromo-3-(3-chlorobenzyl)pyrimidin-4(3H)-one:
- Step B Preparation of 5-(4-(benzyloxy)-3-fluorophenyl)-3-(3- chlorobenzyl)pyrimidin-4(3H)-one: Prepared from 5-bromo-3-(3-chlorobenzyl)pyrimidin-4(3H)- one (0.41 g, 1.38 mmol) according to the procedure described for Example 7, Step A. The crude product was purified by silica gel flash column chromatography (1:1 EtOAc/Hexane) to yield the product (0.42 g, 72%) as a white solid. 1 H NMR (CDCl 3 , 400 MHz) ⁇ 8.15 (s, IH), 8.04 (s, IH), 111-03-PCT - P2338R1 - 02120.004WO1
- Step C Preparation of 3-(3-chlorobenzyl)-5-(3-fluoro-4- hydroxyphenyl)pyrimidin-4(3H)-one: Prepared from 5-(4-(benzyloxy)-3-fluorophenyl)-3-(3- chlorobenzyl)pyrimidin-4(3H)-one (0.42 g, 1.0 mmol) according to the procedure described for Example 1, Step C, to yield the product (0.20 g, 61%) as a white solid.
- LRMS (ESI pos) m/e 331 (M+l).
- Step D Preparation of 3-(3-chlorobenzyl)-5-(4-(6,7-dimethoxyquinolin-4-yloxy)-
- 3-fluorophenyl)pyrimidin-4(3H)-one Prepared from 4-chloro-6,7-dimethoxyquinoline (prepared according to reference procedure in Example 5) (64 mg, 0.29 mmol) and 3-(3-chlorobenzyl)-5-(3- fluoro-4-hydroxyphenyl)pyrimidin-4(3H)-one (95 mg, 0.30 mmol) according to the procedure described for Example 1, Step E.
- the crude product was purified by silica gel flash column chromatography (1:10 MeOH/EtOAc) to yield 121 (20 mg, 13%) as a white solid.
- Step A Preparation of 5-bromo-3-(2-fluorobenzyl)pyrimidin-4(3H)-one: Prepared from l-(bromomethyl)-2-fluorobenzene (1.58 g, 8.4 mmol) according to the procedure described for Example 13, Step A. The crude product was purified by silica gel flash column chromatography (1:1 EtOAc/Hexane) to yield the product (0.55 g, 23%) as a white solid. LRMS (ESI pos) m/e 283 (M+l).
- Step B Preparation of 5-(4-(benzyloxy)-3-fluorophenyl)-3-(3- chlorobenzyl)pyrimidin-4(3H)-one: Prepared from 5-bromo-3-(2-fluorobenzyl)pyrimidin-4(3H)- one (0.55 g, 1.9 mmol) according to the procedure described for Example 7, Step A. The crude product was purified by silica gel flash column chromatography (1:1 EtOAc/Hexane) to yield the product (0.35 g, 44%) as a white solid. LRMS (ESI pos) m/e 405 (M+l).
- Step C Preparation of 5-(3-fluoro-4-hydroxyphenyl)-3-(2- fluorobenzyl)pyrimidin-4(3H)-one: Prepared from 5-(4-(benzyloxy)-3-fluorophenyl)-3-(3- chlorobenzyl)pyrimidin-4(3H)-one (0.35 g, 8.6 mmol) according to the procedure described for Example 1, Step C, to yield the product (0.20 g, 74%) as a white solid.
- LRMS (ESI pos) m/e 315 (M+l).
- Step D Preparation of 5-(4-(6,7-dimethoxyquinolin-4-yloxy)-3-fluorophenyl)-3-
- (2-fluorobenzyl)pyrimidin-4(3H)-one Prepared from 4-chloro-6,7-dimethoxyquinoline (prepared according to reference procedure in Example 5) (100 mg, 0.45 mmol) and 3-(3-chlorobenzyl)-5- (3-fluoro-4-hydroxyphenyl)pyrimidin-4(3H)-one (92 mg, 0.29 mmol) according to the procedure described for Example 1, Step E. The crude product was purified by silica gel flash column chromatography (1:10 MeOH/EtOAc) to yield 123 (41 mg, 28%) as a white solid.
- Step A Preparation of 5-bromo-3-(4-fluorobenzyl)pyrimidin-4(3H)-one: Prepared from l-(bromomethyl)-4-fluorobenzene (1.58 g, 8.30 mmol) according to the procedure described for Example 13, Step A. The crude product was purified by silica gel flash column chromatography (1:1 EtOAc/Hexane) to yield the product (0.42 g, 17%) as a white solid. LRMS (ESI pos) m/e 283 (M+l).
- Step B Preparation of 5-(4-(benzyloxy)-3-fiuorophenyl)-3-(4- fluorobenzyl)pyrimidin-4(3H)-one: Prepared from 5-bromo-3-(4-fiuorobenzyl)pyrimidin-4(3H)- one (0.42 g, 1.5 mmol) according to the procedure described for Example 7, Step A. The crude product was purified by silica gel flash column chromatography (1:1 EtOAc/Hexane) to yield the product (0.33 g, 55%) as a white solid. LRMS (ESI pos) m/e 405 (M+l).
- Step C Preparation of 5-(3-fluoro-4-hydroxyphenyl)-3-(4- fluorobenzyl)pyrimidin-4(3H)-one: Prepared from 5-(4-(benzyloxy)-3-fluorophenyl)-3-(4- fluorobenzyl)pyrimidin-4(3H)-one (0.33 g, 0.82 mmol) according to the procedure described for Example 1, Step C, to yield the product (0.20 g, 78%) as a white solid.
- LRMS (ESI pos) m/e 315 (M+l).
- Step D Preparation of 5-(4-(6,7-dimethoxyquinolin-4-yloxy)-3-fluorophenyl)-3-
- (4-fluorobenzyl)pyrimidin-4(3H)-one Prepared from 4-chloro-6,7-dimethoxyquinoline (prepared according to reference procedure in Example 5) (105 mg, 0.47 mmol) and 5-(3-fluoro-4- hydroxyphenyl)-3-(4-fluorobenzyl)pyrimidin-4(3H)-one (90 mg, 0.29 mmol) according to the procedure described for Example 1, Step E. The crude product was purified by silica gel flash column chromatography (1:10 MeOH/EtOAc) to yield 124 (35 mg, 24 %) as a white solid.
- Step A Preparation of 5-bromo-3-(4-methylbenzyl)pyrimidin-4(3H)-one:
- Step B Preparation of 5-(4-(benzyloxy)-3-fluorophenyl)-3-(4- methylbenzyl)pyrimidin-4(3H)-one: Prepared from 5-bromo-3-(4-methylbenzyl)pyrimidin- 4(3H)-one (0.14 g, 0.5 mmol) according to the procedure described for Example 7, Step A. The crude product was purified by silica gel flash column chromatography (1 :1 EtOAc/Hexane) to yield the product (0.15 g, 75%) as a white solid. LRMS (ESI pos) m/e 401 (M+l).
- Step C Preparation of 5-(3-fluoro-4-hydroxyphenyl)-3-(4- methylbenzyl)pyrimidin-4(3H)-one: Prepared from 5-(4-(benzyloxy)-3-fluorophenyl)-3-(4- methylbenzyl)pyrimidin-4(3H)-one (0.15 g, 0.38 mmol) according to the procedure described for Example 1, Step C, to yield the product (0.10 g, 86%) as a white solid.
- LRMS (ESI pos) m/e 311 (M+l).
- Step D 5-(4-(6,7-dimethoxyquinolin-4-yloxy)-3-fluorophenyl)-3-(4- methylbenzyl)pyrimidin-4(3H)-one: Prepared from 4-chloro-6,7-dimethoxyquinoline (prepared according to reference procedure in Example 5) (105 mg, 0.50 mmol) and 5-(3-fluoro-4- hydroxyphenyl)-3-(4-methylbenzyl)pyrimidin-4(3H)-one (90 g, 0.29 mmol) according to the procedure described for Example 1, Step E.
- Step A Preparation of 5-bromo-3-(3,4-dichlorobenzyl)pyrimidin-4(3H)-one:
- Step B Preparation of 5-(4-(benzyloxy)-3-fluorophenyl)-3-(3,4- dichlorobenzyl)pyrimidin-4(3H)-one: Prepared from 5-bromo-3-(3,4-dichlorobenzyl)pyrimidin- 4(3H)-one (0.63 g, 0.20 mmol) according to the procedure described for Example 7, Step A. The crude product was purified by silica gel flash column chromatography (1:1 EtOAc/Hexane) to yield the product (0.11 g, 13%) as a white solid. LRMS (ESI pos) m/e 455 (M+l).
- Step C Preparation of 3-(3,4-dichlorobenzyl)-5-(3-fluoro-4- hydroxyphenyl)pyrimidin-4(3H)-one: Prepared from 5-(4-(benzyloxy)-3-fluorophenyl)-3-(3,4- dichlorobenzyl)pyrimidin-4(3H)-one (0.11 g, 0.24 mmol) according to the procedure described for Example 1, Step C, to yield the product (50 mg, 56%) as a white solid.
- Step D 3-(3,4-dichlorobenzyl)-5-(4-(6,7-dimethoxyquinolin-4-yloxy)-3- fluorophenyl)pyrimidin-4(3H)-one: Prepared from 4-chloro-6,7-dimethoxyquinoline (prepared according to reference procedure in Example 5) (60 mg, 0.14 mmol) and 3-(3,4-dichlorobenzyl)- 5-(3-fluoro-4-hydroxyphenyl)pyrimidin-4(3H)-one (50 mg, 0.1 mmol) according to the procedure described for Example 1, Step E.
- Step A Preparation of 5-bromo-3-(4-(trifluoromethyl)benzyl)pyrimidin-4(3H)- one: Prepared from l-(bromomethyl)-4-(trifluoromethyl)benzene (2.0 g, 8.3 mmol) according to the procedure described for Example 13, Step A. The crude product was purified by silica gel flash column chromatography (1:1 EtOAc/Hexane) to yield the product (0.28 g, 9.8%) as a white solid. LRMS (ESI pos) m/e 333 (M+l).
- Step B Preparation of 5-(4-(benzyloxy)-3-fluorophenyl)-3-(4-
- Step D Preparation of 5-(4-(6,7-dimethoxyquinolin-4-yloxy)-3-fluorophenyl)-3-
- (4-(trifluoromethyl)benzyl)pyrimidin-4(3H)-one Prepared from 4-chloro-6,7- dimethoxyquinoline (prepared according to reference procedure in Example 5) (100 mg, 0.45 mmol) and 5-(3-fluoro-4-hydroxyphenyl)-3-(4-(trifluoromethyl)benzyl)pyrimidin-4(3H)-one (100 mg, 0.22 mmol) according to the procedure described for Example 1, Step E. The crude product was purified by silica gel flash column chromatography (1:10 MeOH/EtOAc) to yield 127 (38 mg, 31 %) as a white solid.
- Step A Preparation of 5-bromo-3-(4-chloro-2-fluorobenzyl)pyrimidin-4(3H)-one:
- Step B Preparation of 5-(4-(benzyloxy)-3-fluorophenyl)-3-(4-chloro-2- fluorobenzyl)pyrimidin-4(3H)-one: Prepared from 5-bromo-3-(4-chloro-2- fluorobenzyl)pyrimidin-4(3H)-one (0.81 g, 2.5 mmol) according to the procedure described for Example 7, Step A. The crude product was purified by silica gel flash column chromatography (EtOAc) to yield the product (0.74 g, 66%) as a white solid. LRMS (ESI pos) m/e 439 (M+l).
- Step C Preparation of 3-(4-chloro-2-fluorobenzyl)-5-(3-fluoro-4- hydroxyphenyl)pyrimidin-4(3H)-one: Prepared from 5-(4-(benzyloxy)-3-fluorophenyl)-3-(4- chloro-2-fluorobenzyl)pyrimidin-4(3H)-one (0.74 g, 1.7 mmol) according to the procedure described for Example 1, Step C, to yield the product (0.5 g, 85%) as a white solid.
- LRMS (ESI pos) m/e 349 (M+l).
- Step D Preparation of 3-(4-chloro-2-fluorobenzyl)-5-(4-(6,7-dimethoxyquinolin-
- Step A Preparation of 5-bromo-3-(2-chloro-4-fluorobenzyl)pyrimidin-4(3H)-one:
- Step B Preparation of 5-(4-(benzyloxy)-3-fluorophenyl)-3-(2-chloro-4- fiuorobenzyl)pyrimidin-4(3H)-one: Prepared from 5-bromo-3-(2-chloro-4- fluorobenzyl)pyrimidin-4(3H)-one (0.66 g, 2.1 mmol) according to the procedure described for Example 7, Step A. The crude product was purified by silica gel flash column chromatography (1:1 EtOAc/Hexane) to yield the product (0.84 g, 92%) as a white solid. LRMS (ESI pos) m/e 439 (M+l).
- Step C Preparation of 3-(2-chloro-4-fluorobenzyl)-5-(3-fluoro-4- hydroxyphenyl)pyrimidin-4(3H)-one: Prepared from 5-(4-(benzyloxy)-3-fluorophenyl)-3-(2- chloro-4-fluorobenzyl)pyrimidin-4(3H)-one (0.84 g, 1.5 mmol) according to the procedure described for Example 1, Step C, to yield the product (0.5 g, 75%) as a white solid.
- Step D Preparation of 3-(2-chloro-4-fluorobenzyl)-5-(4-(6,7-dimethoxyquinolin-
- Example 30 Preparation of 3-(4-chloro-2,6-difluorobenzyl)-5-(4-(6,7- dimethoxyquinolin-4-yloxy)-3-fluorophenyl)pyrimidin-4(3H)-one 130 111-03-PCT - P2338R1 - 02120.0Q4WO1
- Step A Preparation of 5-bromo-3-(4-chloro-2,6-difluorobenzyl)pyrimidin-4(3H)- one: Prepared from 2-(bromomethyl)-5-chloro-l,3-difluorobenzene (2.0 g, 8.3 mmol) according to the procedure described for Example 13, Step A. The crude product was purified by silica gel flash column chromatography (1:1 EtOAc/Hexane) to yield the product (0.76 g, 26%) as a white solid. LRMS (ESI pos) m/e 335 (M+l).
- Step B Preparation of 5-(4-(benzyloxy)-3-fluorophenyl)-3-(4-chloro-2,6- difluorobenzyl)pyrimidin-4(3H)-one: Prepared from 5-bromo-3-(4-chloro-2,6- difluorobenzyl)pyrimidin-4(3H)-one (0.76 g, 2.2 mmol) according to the procedure described for Example 7, Step A. The crude product was purified by silica gel flash column chromatography (1:1 EtOAc/Hexane) to yield the product (0.52 g, 50%) as a white solid. LRMS (ESI pos) m/e 457 (M+l).
- Step C Preparation of 3-(4-chloro-2,6-difluorobenzyl)-5-(3-fluoro-4- hydroxyphenyl)pyrimidin-4(3H)-one: Prepared from 5-(4-(benzyloxy)-3-fluorophenyl)-3-(4- chloro-2,6-difluorobenzyl)pyrimidin-4(3H)-one (0.52 g, 1.14 mmol) according to the procedure described for Example 1, Step C, to yield the product (0.4 g, 97%) as a white solid.
- LRMS (ESI pos) m/e 367 (M+l).
- Step D Preparation of 3-(4-chloro-2,6-difluorobenzyl)-5-(4-(6,7- dimethoxyquinolin-4-yloxy)-3-fluorophenyl)pyrimidin-4(3H)-one: Prepared from 4-chloro-6,7- dimethoxyquinoline (prepared according to reference procedure in Example 5) (100 mg, 0.45 mmol) and 3-(4-chloro-2,6-difluorobenzyl)-5-(3-fluoro-4-hydroxyphenyl)pyrimidin-4(3H)-one (164 mg, 0.45 mmol) according to the procedure described for Example 1, Step E.
- Step A Preparation of 5-bromo-3-(3,4-dimethylbenzyl)pyrimidin-4(3H)-one:
- Step B Preparation of 5-(4-(benzyloxy)-3-fluorophenyl)-3-(3,4- dimethylbenzyl)pyrimidin-4(3H)-one: Prepared from 5-bromo-3-(3,4-dimethylbenzyl)pyrimidin- 4(3H)-one (0.68 g, 2.3 mmol) according to the procedure described for Example 7, Step A. The crude product was purified by silica gel flash column chromatography (1:1 EtOAc/Hexane) to yield the product (0.17 g, 17%) as a white solid. LRMS (ESI pos) m/e 415 (M+l).
- Step C Preparation of 3-(3,4-dimethylbenzyl)-5-(3-fluoro-4- hydroxyphenyl)pyrimidin-4(3H)-one: Prepared from 5-(4-(benzyloxy)-3-fluorophenyl)-3-(3,4- dimethylbenzyl)pyrimidin-4(3H)-one (0.17 g, 0.4 mmol) according to the procedure described for Example 1, Step C, to yield the product (0.1 g, 77%) as a white solid.
- LRMS (ESI pos) m/e 325 (M+l).
- Step D Preparation of 5-(4-(6,7-dimethoxyquinolin-4-yloxy)-3-fIuorophenyl)-3-
- (3,4-dimethylbenzyl)pyrimidin-4(3H)-one Prepared from 4-chloro-6,7-dimethoxyquinoline (prepared according to reference procedure in Example 5) (100 mg, 0.45 mmol) and 3-(3,4- dimethylbenzyl)-5-(3-fluoro-4-hydroxyphenyl)pyrimidin-4(3H)-one (145 mg, 0.45 mmol) according to the procedure described for Example 1, Step E. The crude product was purified by silica gel flash column chromatography (1:10 MeOH/EtOAc) to yield 131 (2 mg, 1%) as a white solid.
- Step A Preparation of 5-bromo-3-(4-fluoro-3-methylbenzyl)pyrimidin-4(3H)- one: Prepared from 4-(bromomethyl)-l-fluoro-2-methylbenzene (0.57 g, 2.8 mmol) according to the procedure described for Example 13, Step A. The crude product was purified by silica gel flash column chromatography (1:1 EtOAc/Hexane) to yield the product (0.46 g, 54%) as a white solid. LRMS (ESI pos) m/e 296 (M+l).
- Step B Preparation of 5-(4-(benzyloxy)-3-fluorophenyl)-3-(4-fluoro-3- methylbenzyl)pyrimidin-4(3H)-one: Prepared from 5-bromo-3-(4-fluoro-3- methylbenzyl)pyrimidin-4(3H)-one (0.46 g, 1.5 mmol) according to the procedure described for Example 7, Step A. The crude product was purified by silica gel flash column chromatography (1:1 EtOAc/Hexane) to yield the product (86 mg, 13%) as a white solid. LRMS (ESI pos) m/e 419 (M+l).
- Step C Preparation of 3-(4-fluoro-3-methylbenzyl)-5-(3-fluoro-4- hydroxyphenyl)pyrimidin-4(3H)-one: Prepared 5-(4-(benzyloxy)-3-fluorophenyl)-3-(4-fluoro-3- methylbenzyl)pyrimidin-4(3H)-one (86 mg, 0.2 mmol) according to the procedure described for Example 1, Step C, to yield the product (50 mg, 74%) as a white solid.
- Step D Preparation of 5-(4-(6,7-dimethoxyquinolin-4-yloxy)-3-fluorophenyl)-3-
- (4-fluoro-3-methylbenzyl)pyrimidin-4(3H)-one Prepared from 4-chloro-6,7-dimethoxyquinoline (prepared according to reference procedure in Example 5) (40 mg, 0.18 mmol) and 3-(4-fluoro- 3-methylbenzyl)-5-(3-fluoro-4-hydroxyphenyl)pyrimidin-4(3H)-one (59 mg, 0.18 mmol) according to the procedure described for Example 1, Step E. The crude product was purified by silica gel flash column chromatography (1:10 MeOH/EtOAc) to yield 132 (1.6 mg, 2%) as a white solid.
- Step A Preparation of (5-bromo-2-methoxypyridin-4-yl)(phenyl)methanol:
- Step B Preparation of (5-bromo-2-methoxypyridin-4-yl)(phenyl)methanone: PCC
- Step C Preparation of 4-benzoyl-5-bromopyridin-2(lH)-one: (5-bromo-2- methoxypyridin-4-yl)(phenyl)methanone (1.3 g, 4.5 mmol) and pyridine hydrochloride (2 g, 17 mmol) were heated at 150 0 C for 1 hour. CH 2 Cl 2 (30 mL) was added into the hot mixture. The mixture was cooled and the solvent was evaporated. The residue was purified by silica gel flash column chromatography (2:1 EtOAc/Hexane) to yield the product (0.3 g, 24%) as a white solid.
- Step D Preparation of 4-benzoyl-l-(3-fluoro-4-hydroxyphenyl)pyridin-2(lH)- one: Prepared from 4-benzoyl-5-bromopyridin-2(lH)-one (170 mg, 0.61 mmol) according to the procedure described for Example 1, Step D. The crude product was purified by silica gel flash column chromatography (EtOAc) to yield the product (170 mg, 90%) as a brown solid.
- EtOAc silica gel flash column chromatography
- Step E Preparation of 4-benzoyl-l-(4-(6,7-dimethoxyquinolin-4-yloxy)-3- fluorophenyl)pyridin-2(lH)-one: Prepared from 4-chloro-6,7-dimethoxyquinoline (prepared 111-03-PCT - P2338R1 - 02120.004WO1
- Example 34 60 mg, 0.16 mmol
- the crude product was purified by silica gel flash column chromatography (EtOAc) to yield 139 (40 mg, 53 %) as a white solid.
- Step A Preparation of l-(3-fluoro-4-hydroxyphenyl)-4-methylpyridin-2(lH)-one:
- Step B Preparation of l-(4-(6,7-dimethoxyquinolin-4-yloxy)-3-fluorophenyl)-4- methylpyridin-2(lH)-one: Prepared from l-(3-fluoro-4-hydroxyphenyl)-4-methylpyridin-2(lH)- one (0.1 g, 0.46 mmol) and 4-chloro-6,7-dimethoxyquinoline (prepared according to reference procedure in Example 5) (0.12 g, 0.55 mmol) using the procedure described for Example 1, Step E. The reaction mixture was purified by silica gel flash column chromatography (1:10 MeOH/EtOAc) to yield 140 (50 mg, 27%) as a white solid.
- Step A Preparation of 5-(4-(benzyloxy)-3-fluorophenyl)pyrimidin-4(3H)-one:
- Step B Preparation of 5-(3-fluoro-4-hydroxyphenyl)pyrimidin-4(3H)-one:
- Step C Preparation of 5-(4-(6,7-dimethoxyquinolin-4-yloxy)-3- fluorophenyl)pyrimidin-4(3H)-one: Prepared from 5-(3-fluoro-4-hydroxyphenyl)pyrimidin- 4(3H)-one (40 mg, 0.19 mmol) and 4-chloro-6,7-dimethoxyquinoline (prepared according to reference procedure in Example 5) (43 mg, 0.19 mmol) according to the procedure described for Example 1, Step E, to yield 143 (1 mg, 1%) as white solid.
- Step A Preparation of 2-amino-N-(3-fluoro-4-methoxyphenyl)benzamide: To a stirred suspension of isatoic anhydride (1.63 g, 10 mmol) in 15 mL dioxane at room temperature 111-03-PCT - P2338R1 - 02120.004WO1
- Step B Preparation of N-(2-aminobenzyl)-3-fluoro-4-methoxyaniline: To a stirred suspension of lithium aluminum hydride (121 mg, 3.2 mmol) in 2 mL dioxane at reflux under nitrogen was added 2-amino-N-(3-fluoro-4-methoxyphenyl)benzamide (260 mg, 1 mmol) as a solution in 2 mL dioxane. A vigorous reaction was evident. After refluxing overnight the reaction was cooled to room temperature and quenched by sequential treatment with H 2 O (150 uL), 15% NaOH (150 uL) and H 2 O (450 uL).
- Step D Preparation of 3-(3-fluoro-4-hydroxyphenyl)-3,4-dihydroquinazolin-
- Step E Preparation of 3-(3-fluoro-4-(6-methoxy-7-(3- morpholinopropoxy)quinolin-4-yloxy)phenyl)-3,4-dihydroquinazolin-2(lH)-one.
- Step E substituting 3-(3-fiuoro-4-hydroxyphenyl)-3,4- dihydroquinazolin-2( 1 H)-one for 3 -(3 -fluoro-4-hydroxyphenyl)-5 -methyl-6-(2- methylbenzyl)pyrimidin-4(3H)-one to give 145 after silica gel chromatography as a white foam (23 mg, 53% yield).
- Step A Preparation of 4-(2-fluoro-4-(piperazin-l-yl)phenoxy)-6,7- dimethoxyquinoline: Pd2(dba)3 (20 mg) was added into a suspension of 4-(4-bromo-2- fluorophenoxy)-6,7-dimethoxyquinoline (Example 34, 100 mg, 0.264 mmol), piperazine (228 mg, 2.64 mmol), Xanthphos (100 mg) and potassium phosphate (200 mg) in toluene (10 mL). The reaction mixture was heated to 100 0 C for 10 hours. The reaction mixture was filtered through a pad of silica gel.
- Step B Preparation of (4-(4-(6,7-dimethoxyquinolin-4-yloxy)-3- fluorophenyl)piperazin-l-yl)(phenyl)methanone: Benzoyl chloride (11 mg, 0.078 mmol) and triethylamine (0.5 mL) were added into a solution of 4-(2-fluoro-4-(piperazin-l-yl)phenoxy)-6,7- dimethoxyquinoline (30 mg, 0.078 mmol) in CH 2 Cl 2 . After a few minutes, water (1 mL) and CH 2 Cl 2 (2 mL) were added.
- Step A Preparation of 3-benzyl-5-(4-(benzyloxy)-3-fluorophenyl)pyrimidine-
- Step B 3-benzyl-5-(3-fluoro-4-hydroxyphenyl)pyrimidine-4(3H)-thione:
- Step C Preparation of 3-benzyl-5-(4-(6,7-dimethoxyquinolin-4-yloxy)-3- fluorophenyl)pyrimidine-4(3H)-thione: Prepared from 4-chloro-6,7-dimethoxyquinoline (prepared according to reference procedure in Example 5) (40 mg, 0.18 mmol) and 3-(4- chlorobenzyl)-5-(3-fluoro-4-hydroxyphenyl)pyrimidine-4(3H)-thione (62 mg, 0.18 mmol) according to the procedure described for Example 1, Step E to yield 148 (40 mg, 45%) as yellow solid.
- Step A Preparation of 3-benzyl-5-(4-(benzyloxy)-3-fluorophenyl)pyrimidin-
- the reaction mixture was heated at 100 0 C for 2 hours, cooled and poured into water (10 mL).
- the reaction mixture was extracted with ethyl acetate, and the organic layer was washed with brine, dried over sodium sulfate, filtered, and concentrated.
- the crude product was purified by silica gel flash column chromatography (2:1 EtOAc/Hexane) to yield the product (1.4 g, 32%) as a white solid.
- Step B Preparation of 3-benzyl-5-(3-fluoro-4-hydroxyphenyl)pyrimidin-4(3H)- one: Prepared from 3-benzyl-5-(4-(benzyloxy)-3-fluorophenyl)pyrimidin-4(3H)-one (0.3 g, 0.8 mmol) according to the procedure described for Example 1, Step C, to yield the product (0.2 g, 87%) as a white solid.
- LRMS (ESI pos) m/e 297 (M+l). 111 -03-PCT - P2338R 1 - 02120.004WO1
- Step C Preparation of 3-benzyl-5-(4-(7 ⁇ (benzyloxy)-6-methoxyquinolin-4- yloxy)-3-fluorophenyl)pyrimidin-4(3H)-one: Prepared from 7-(benzyloxy)-4-chloro-6- methoxyquinoline (prepared according to WO 2005/030140, Example32, 200 mg, 0.67 mmol) and 3-benzyl-5-(3-fluoro-4-hydroxyphenyl)pyrimidin-4(3H)-one (198 mg, 0.48 mmol) according to the procedure described for Example 1, Step E. The crude product was purified by silica gel flash column chromatography (1:10 MeOH/EtOAc) to yield 149 (100 mg, 27%) as a white solid. LRMS (ESI pos) m/e 560 (M+l).
- Step A Preparation of 5-bromo-3-(4-tert-butylbenzyl)pyrimidin-4(3H)-one:
- Step B Preparation of 5-(4-(benzyloxy)-3-fluorophenyl)-3-(4-tert- butylbenzyl)pyrimidin-4(3H)-one: Prepared from 5-bromo-3-(4-tert-butylbenzyl)pyrimidin- 4(3H)-one (0.65 mg, 2.0 mmol) according to the procedure described for Example 7, Step A. The crude product was purified by silica gel flash column chromatography (1:1 EtOAc/Hexane) to yield the product (0.50 g, 56%) as a white solid. LRMS (ESI pos) m/e 443 (M+l). 111-03-PCT - P2338R1 - 02120.004WO1
- Step C Preparation of 3-(4-tert-butylbenzyl)-5-(3-fluoro-4- hydroxyphenyl)pyrimidin-4(3H)-one: Prepared from 5-(4-(benzyloxy)-3-fluorophenyl)-3-(4-tert- butylbenzyl)pyrimidin-4(3H)-one (0.50 g, 1.1 mmol) according to the procedure described for Example I 5 Step C, to yield the product (0.30 g, 88%) as a white solid.
- Step D Preparation of 3-(4-tert-butylbenzyl)-5-(4-(6,7-dimethoxyquinolin-4- yloxy)-3-fluorophenyl)pyrimidin-4(3H)-one: Prepared from 4-chloro-6,7-dimethoxyquinoline (prepared according to reference procedure in Example 5) (100 mg, 0.46 mmol) and 3-(4-tert- butylbenzyl)-5-(3-fluoro-4-hydroxyphenyl)pyrimidin-4(3H)-one (130 mg, 0.30 mmol) according to the procedure described for Example 1, Step E.
- Step A Preparation of 5-bromo-3-(4-chloro-3-(trifluoromethyl)benzyl)pyrimidin-
- Step B Preparation of 5-(4-(benzyloxy)-3-fiuorophenyl)-3-(4-chloro-3-
- Step C Preparation of 3-(4-chloro-3-(trifiuoromethyl)benzyl)-5-(3-fluoro-4- hydroxyphenyl)pyrimidin-4(3H)-one: Prepared from 5-(4-(benzyloxy)-3-fluorophenyl)-3-(4- chloro-3-(trifluoromethyl)benzyl)pyrimidin-4(3H)-one (0.50 g, 1.0 mmol) according to the procedure described for Example 1, Step C, to yield the product (0.40 g, 86%) as a white solid.
- LRMS (ESI pos) m/e 331 (M+l).
- Step D Preparation of 3-(4-chloro-3-(trifluoromethyl)benzyl)-5-(4-(6,7- dimethoxyquinolin-4-yloxy)-3-fluorophenyl)pyrimidin-4(3H)-one: Prepared from 4-chloro-6,7- dimethoxyquinoline (prepared according to reference procedure in Example 5) (60 mg, 0.27 mmol) and 3-(4-chloro-3-(trifluoromethyl)benzyl)-5-(3-fluoro-4-hydroxyphenyl)pyrimidin- 4(3H)-one(107 mg, 0.27 mmol) according to the procedure described for Example 1, Step E.
- Step A Preparation of (4-benzylpiperazm-l-yl)(3-fiuoro-4- methoxyphenyl)methanone: Prepared from 1-benzylpiperazine (2.3 mg, 13 mmol) ) according to the procedure described for Example 4, Step A, to yield the product (2.6 g, 65%) as a white solid.
- Step B Preparation of (4-benzylpiperazin-l-yl)(3-fluoro-4- hydroxyphenyl)methanone: Prepared from (4-benzylpiperazin-l-yl)(3-fluoro-4- methoxyphenyl)methanone (2.0 g, 6.1 mmol) according to the procedure described for Example 4, Step B, to yield the product (1.0 g, 53%) as a white solid.
- Step C Preparation of 5 (4-benzylpiperazin-l-yl)(4-(6,7-dimethoxyquinolin-4- yloxy)-3-fluorophenyl)methanone: Prepared from (4-benzylpiperazin-l-yl)(3-fluoro-4- hydroxyphenyl)methanone (18 mg, 0.059 mmol) and 4-chloro-6-methoxy-7-(3- morpholinopropoxy)quinoline (20 mg, 0.059 mmol) according to the procedure described for Example 1, Step E, to yield 153 (40 mg, 18% yield) as a white solid.
- Step A Preparation of (E)-ethyl 3-amino-2-(4-(benzyloxy)phenylcarbamoyl)-4- phenylbut-2-enoate: A suspension of (E)-ethyl 3-amino-4-phenylbut-2-enoate (1 g, 4.9 mmol) and l-((4-isocyanatophenoxy)methyl)benzene (1.1 g, 4.9 mmol) was heated in DMF (20 mL) at 6O 0 C for 72 hours. The reaction mixture was poured into water (10 mL) and removed the solid by filtration.
- Step B Preparation of ethyl 4-benzyl-l-(4-(benzyloxy)phenyl)-6-oxo-l,6- dihydropyrimidine-5-carboxylate: Acetic anhydride (1 ml, 10.6 mmol) was added into a solution of (E)-ethyl 3-amino-2-((4-(benzyloxy)phenyl)carbamoyl)-4-phenylbut-2-enoate (1 g, 2.32 mmol) and triethyl orthroformate (5 ml, 30.1 mmol). The reaction was heated at HO 0 C for 16 hours, cooled and solvent was evaporated.
- Step C Preparation of 6-benzyl-3-(4-hydroxyphenyl)pyrimidin-4(3H)-one: Ethyl
- Step D Preparation of 6-benzyl-3-(4-(6-methoxy-7-(3- morpholinopropoxy)quinolin-4-yloxy)phenyl)pyrimidin-4(3H)-one: Prepared from 4-chloro-6- methoxy-7-(3-morpholinopropoxy)quinoline (34 mg, 0.10 mmol) and 6-benzyl-3-(4- hydroxyphenyl)pyrimidin-4(3H)-one (28 mg, 0.10 mmol) according to the procedure described for Example 1, Step E. The residue was purified by silica gel flash column chromatography column (1:10 MeOH/EtOAc) to yield 154 (20 mg, 34%) as a yellow solid.
- Step A preparation of (4,6-dichloropyrimidin-5-yl)(phenyl)methanol:
- Phenylmagnesium bromide (11 ml, 11 mmol) was added into a solution of 4,6-Dichloro-5- pyrimidinecarbaldehyde (2 g, 11 mmol) in THF (30 mL) at -78 0 C. The reaction was allowed to warm to room temperature and was poured into water (10 mL). The reaction mixture was extracted with EtOAc, and the organic layer was washed with brine, dried over sodium sulfate, filtered, and concentrated. The residue was purified by silica gel flash column chromatography (1:10 Et 2 O/Hexane) to yield the product (2.4 g, 83% yield) as a white solid.
- IH NMR (CDCl 3 , 400 MHz) ⁇ 8.76 (s, IH), 7.20-7.41 (m, 5H), 6.50 (d, IH), 3.00 (d, IH).
- Step B Preparation of (4-(benzyloxy)-6-chloropyrimidin-5-yl)(phenyl)methanol:
- Step C Preparation of 5-benzylpyrimidin-4-ol: Prepared from 5-benzylpyrimidin-
- Step D Preparation of 5-benzyl-3-(3-fluoro-4-hydroxyphenyl)pyrimidin-4(3H)- one: Prepared from 5-benzylpyrimidin-4-ol (0.1 g, 0.54 mmol) according to the procedure described for Example 1, Step D, to yield the product (20 mg, 13 % yield) as a brown solid.
- LRMS (ESI pos) m/e 279 (M+l).
- Step E Preparation of 5-benzyl-3-(3-fiuoro-4-(6-methoxy-7-(3- morpholinopropoxy)quinolin-4-yloxy)phenyl)pyrimidin-4(3H)-one: Prepared from 5-benzyl-3- (3-fluoro-4-hydroxyphenyl)pyrimidin-4(3H)-one (18 mg, 0.059 mmol) according to the procedure described for Example 1, Step E, to yield 155 (1 mg, 2.8% yield) as a light brown solid. LRMS (ESI pos) m/e 597 (M+l).
- Step A Preparation of 2-benzyl-4-methoxypyrimidine: A solution of 2-chloro-4- methoxypyrimidine (0.500 g, 3.46 mmol) and PdCl 2 (PPh 3 ) 2 (0.121 g, 0.173 mmol) in THF (10 mL) was sparged with N 2 . Benzylzinc(II) bromide (8.30 ml, 4.15 mmol; 0.5 M solution in THF) was added and the reaction mixture was stirred at reflux for 1 hour. The reaction mixture was cooled to room temperature and then partitioned between EtOAc and H 2 O. The phases were separated, and the aqueous phase was re-extracted with EtOAc (Ix).
- Step B Preparation of 2-benzylpyrimidin-4(3H)-one: To a solution of 2-benzyl-
- Step C Preparation of 2-benzyl-5-bromopyrimidin-4(3H)-one: To a solution of
- Step E Preparation of 2-benzyl-5-(3-fluoro-4-hydroxyphenyl)pyrimidin-4(3H)- one: A suspension of 2-benzyl-5-(4-(benzyloxy)-3-fluorophenyl)pyrimidin-4(3H)-one (0.284 g, 0.735 mmol) in trifluoroacetic acid (3 mL) was stirred at 40°C for 2 hours and then at room temperature for 16 hours. The reaction mixture was concentrated to dryness and then purified by silica gel flash column chromatography, eluting with 20:1 dichloromethane/MeOH. The desired product (0.177 g, 81%) was obtained as a white solid.
- Step F Preparation of 2-benzyl-5-(3-fluoro-4-(6-methoxy-7-(3- morpholinopropoxy)quinolin-4-yloxy)phenyl)pyrimidin-4(3H)-one: To a solution of 2-benzyl-5- (3-fluoro-4-hydroxyphenyl)pyrimidin-4(3H)-one (0.027 g; 0.091 mmol) in toluene (500 uL) in a microwave tube was added 4-(3-(4-chloro-6-methoxyquinolin-7-yloxy)propyl)morpholine (prepared according to the reference in Example 1, step E) (0.031 g, 0.091 mmol) and DMAP (0.011 g, 0.091 mmol).
- the reaction was stirred at 180°C in the microwave for 2 hours.
- the mixture was solubilized with a small amount of MeOH and purified by silica gel flash column chromatography, eluting with 9:1 EtOAc/MeOH.
- the desired product eluted along with residual 4-(3-(4-chloro-6-methoxyquinolin-7-yloxy)propyl)morpholine.
- the mixture was concentrated and repurified by silica gel flash column chromatography, eluting with a step gradient of CH 2 Cl 2 (150 mL), 2.5/97.5 MeOH/CH 2 Cl 2 (200 mL) and 5/95 MeOH/CH 2 Cl 2 (500 mL) to give 156 (0.022 g, 40%) as a tan foamy solid.
- Step A Preparation of 2-benzyl-5-(4-(benzyloxy)phenyl)pyrimidin-4(3H)-one:
- Step C Preparation of 2-benzyl-5-(4-(6-methoxy-7-(3- morpholinopropoxy)quinolin-4-yloxy)phenyl)pyrimidin-4(3H)-one: To a stirred suspension of 2- benzyl-5-(4-hydroxyphenyl)pyrimidin-4(3H)-one (0.021 g, 0.076 mmol) in bromobenzene (700 ⁇ L) at room temperature under N 2 was added 4-(3-(4-chloro-6-methoxyquinolin-7- yloxy)propyl)morpholine (prepared according to the referenced procedure in Example 1, step E) (0.028 g, 0.083 mmol) followed by DMAP (0.001 g, 0.008 mmol).
- Step A Preparation of 5-bromo-2-chloropyrimidin-4(3H)-one: Prepared from 5- bromo-2,4-dichloropyrimidine (10.00 g, 43.88 mmol) according to the procedure described in EP 1506967. The desired product (4.59 g, 50%) was obtained as a pale yellow solid.
- 1 H-NMR 400 MHz, DMSO-d 6 ) ⁇ 8.36 (s, IH).
- LRMS ESI neg
- m/e 207, 209 M-, Br pattern).
- Step B Preparation of 5-bromo-2-chloro-3-methylpyrimidin-4(3H)-one: To a solution of 5-bromo-2-chloropyrimidin-4(3H)-one (1.00 g, 4.78 mmol) in DME (12 mL)/DMF (3 mL) under N 2 at 0 0 C was added LiH (0.044 g, 5.25 mmol) and the reaction was stirred at room temperature for 15 minutes. Iodomethane (0.589 ml, 9.45 mmol) was then added and the reaction was stirred at room temperature for 30 minutes and then at 60 0 C for 1.5 hours.
- LiH 0.044 g, 5.25 mmol
- Step C Preparation of 2-benzyl-5-bromo-3-methylpyrimidin-4(3H)-one: A solution of 5-bromo-2-chloro-3-methylpyrimidin-4(3H)-one (0.100 g, 0.448 mmol) and PdCl 2 (PPh 3 ) 2 (0.016 g, 0.022 mmol) in THF (4 mL) was sparged with N 2 . Benzylzinc(II) bromide (0.904 ml, 0.452 mmol; 0.5 M solution in THF) was added and the reaction mixture was stirred at reflux for 30 minutes. The reaction mixture was cooled to room temperature and then partitioned between EtOAc and H 2 O.
- Step D Preparation of 2-benzyl-5-(4-(benzyloxy)-3-fluorophenyl)-3- methylpyrimidin-4(3H)-one: Prepared from 2-benzyl-5-bromo-3-methylpyrimidin-4(3H)-one (0.067 g, 0.240 mmol) according to the procedure described in Step D of Example 56. The crude product was purified by silica gel flash column chromatography, eluting with 10:1 dichloromethane/EtOAc. The desired product (0.082 g, 85%) was obtained as an off-white solid.
- 1 H-NMR 400 MHz, DMSOd 6 ) ⁇ 8.15 (s, IH), 7.66 (dd, IH), 7.51 (m, IH), 7.47 (m, 2H), 7.43-
- Step E Preparation of 2-benzyl-5-(3-fluoro-4-hydroxyphenyl)-3- methylpyrimidin-4(3H)-one: Prepared from 2-benzyl-5-(4-(benzyloxy)-3-fiuorophenyl)-3- methylpyrimidin-4(3H)-one (0.082 g, 0.20 mmol), according to the procedure described in Step E of Example 56. The desired product (0.064 g, 100%) was obtained as a pale yellow foamy solid.
- Step F Preparation of 2-benzyl-5-(3-fluoro-4-(6-methoxy-7-(3- morpholinopropoxy)quinolin-4-yloxy)phenyl)-3-methylpyrimidin-4(3H)-one: Prepared from 2- benzyl-5-(3-fluoro-4-hydroxyphenyl)-3-methylpyrimidin-4(3H)-one (0.025 g, 0.081 mmol), 4-(3- (4-chloro-6-methoxyquinolin-7-yloxy)propyl)morpholine (0.030 g, 0.089 mmol) and catalytic DMAP according to the procedure described in Step C of Example 58, to give 159 (0.006 g, 12%) as a yellow solid.
- Step A Preparation of 4-chloro-6-methoxy-5-methylpyrimidine: To a solution of 4,6-dichloro-5-methylpyrimidine (1.00 g, 6.13 mmol) in MeOH (50 mL) at 0°C was added solid sodium methoxide (0.348 g, 6.44 mmol) in portions. The reaction mixture was allowed to warm to room temperature and stirred at room temperature for 4 hours and then at 50 0 C for 12 hours. Additional sodium methoxide (0.348 g, 6.44 mmol) was added and the reaction mixture was stirred at 50°C for 4 hours.
- Step B Preparation of 5-methyl-6-(phenylamino)pyrimidin-4(3H)-one:
- a sealed tube was a mixture of 4-chloro-6-methoxy-5-methylpyrimidine (0.973 g, 6.14 mmol) and aniline (1.679 ml, 18.41 mmol) in n-BuOH (10 mL).
- the reaction mixture was stirred at reflux for 5 days and then at room temperature for 5 days.
- the reaction mixture was a purple suspension after cooling.
- the resulting solid was filtered and washed with Et 2 O, collected and dried under vacuum.
- the filtrate was concentrated, dried under vacuum and the trituration procedure was repeated with dichloromethane/Et 2 O.
- Step C Preparation of 3-(4-(benzyloxy)phenyl)-5-methyl-6-
- the secondary ligand Nl,N2-dimethylethane-l,2-diamine (0.0132 ml, 0.124 mmol) and additional l-(benzyloxy)-4-iodobenzene (0.020 g, 0.065 mmol) were added.
- the reaction mixture was then stirred at 110 0 C for an additional 16 hours.
- the reaction mixture was partitioned between EtOAc and saturated aqueous NaCl. The phases were separated, and the aqueous phase was re-extracted with EtOAc (Ix). The combined organic layers were dried (Na 2 SO 4 ), filtered and concentrated.
- Step D Preparation of 3-(4-hydroxyphenyl)-5-methyl-6-
- Step E Preparation of 3-(4-(6-methoxy-7-(3-morpholinopropoxy)quinolin-4- yloxy)phenyl)-5-methyl-6-(phenylamino)pyrimidin-4(3H)-one: Prepared from 3-(4- hydroxyphenyl)-5-methyl-6-(phenylamino)pyrimidin-4(3H)-one (0.019 g, 0.063 mmol), 4-(3-(4- chloro-6-methoxyquinolin-7-yloxy)propyl)morpholine (0.023 g, 0.069 mmol) and catalytic DMAP according to the procedure described in Step C of Example 58, to give 160 (0.026 g, 69%) as a pale yellow foamy solid.
- Step A Preparation of 3-(3-fluoro-4-hydroxyphenyl)-5-methyl-6-
- the mixture was flushed with N 2 and stirred at 11O 0 C for 16 hours.
- the reaction mixture was partitioned between EtOAc and saturated aqueous NaCl. The phases were separated, and the aqueous phase was re-extracted with EtOAc (2x). The combined organic layers were dried (Na 2 SO 4 ), filtered and concentrated.
- the crude product was purified by silica gel flash column chromatography, eluting with 20:1 dichloromethane/MeOH. The desired product (0.069 g, 45%) was obtained as a brown solid.
- Step B Preparation of 3-(3-fluoro-4-(6-methoxy-7-(3- morpholinopropoxy)quinolin-4-yloxy)phenyl)-5-methyl-6-(phenylamino)pyrimidin-4(3H)-one: Prepared from 3-(3-fluoro-4-hydroxyphenyl)-5-methyl-6-(phenylamino)pyrimidin-4(3H)-one (0.025 g, 0.0803 mmol), 4-(3-(4-chloro-6-methoxyquinolin-7-yloxy)propyl)morpholine (0.0298 g, 0.0883 mmol) and catalytic DMAP according to the procedure described in Step C of Example 58, to give 161 (0.035 g, 71%) as a pale yellow foamy solid.
- Step A Preparation of 4-methoxy-N-phenylpyrimidin-2-amine:
- 2-chloro-4-methoxypyrimidine (1.00 g, 6.92 mmol) in 2-propanol (5 mL).
- Aniline (0.757 ml, 8.30 mmol) and DIEA (1.45 ml, 8.30 mmol) were added and the reaction mixture was heated at 100°C until the reaction was complete by HPLC.
- the reaction mixture was cooled to room temperature.
- the resulting thick suspension was filtered, washed with ethanol, collected and dried under vacuum to yield the desired product (0.164 g) as a white solid.
- Step B Preparation of 2-(phenylamino)pyrimidin-4(3H)-one: To a solution of 4- methoxy-N-phenylpyrimidin-2 -amine (0.632 g, 3.14 mmol) in acetic acid (20 mL) was added HBr (2.132 ml, 18.84 mmol; 48 wt % in H 2 O). The reaction mixture was heated at 90-95°C for 3 hours. The reaction mixture was cooled to room temperature and diluted with H 2 O. The pH of the reaction mixture was adjusted to 5-6 with 6 M aqueous NaOH which resulted in the formation of a solid precipitate.
- Step C Preparation of 3-methyl-2-(phenylamino)pyrimidin-4(3H)-one.
- 2-(phenylamino)pyrimidin-4(3H)-one (0.250 g, 1.34 mmol) in DMF (10 mL) was added LiH (0.012 g, 1.47 mmol).
- iodomethane (0.166 ml, 2.67 mmol) was added.
- the reaction was stirred at room temperature for 18 hours.
- the reaction mixture was quenched with H 2 O and then partitioned between EtOAc and 111-03-PCT - P2338R1 - 02120.004WO1
- Step D Preparation of 5-bromo-3-methyl-2-(phenylamino)pyrimidin-4(3H)-one:
- Step F Preparation of 5-(3-fluoro-4-hydroxyphenyl)-3-methyl-2-
- Step G Preparation of 5-(3-fluoro-4-(6-methoxy-7-(3- morpholinopropoxy)quinolin-4-yloxy)phenyl)-3-methyl-2-(phenylamino)pyrimidin-4(3H)-one: Prepared from 5-(3-fluoro-4-hydroxyphenyl)-3-methyl-2-(phenylamino)pyrimidin-4(3H)-one (0.025 g, 0.0803 mmol), 4-(3-(4-chloro-6-methoxyquinolin-7-yloxy)propyl)morpholine (0.0298 g, 0.0883 mmol) and catalytic DMAP according to the procedure described in Step C of Example 58, to give 162 (0.029, 59%) as a pale yellow foamy solid.
- Step A Preparation of 5-bromo-2-(cyclopropylmethylamino)-3- methylpyrimidin-4(3H)-one: A mixture of 5-bromo-2-chloro-3-rnethylpyrimidin-4(3H)-one (0.100 g, 0.45 mmol; obtained from Example 59, Step B), cyclopropylmethanamine (0.051 ml, 0.58 mmol) and NaHCO 3 (0.150 g, 1.79 mmol) in n-BuOH (3 mL) was stirred at 60 0 C for 1 hour. The reaction mixture was cooled to room temperature and then diluted with EtOAc.
- Step C Preparation of 2-(cyclopropylmethylamino)-5-(3-fluoro-4- hydroxyphenyl)-3-methylpyrimidin-4(3H)-one: A solution of 5-(4-(benzyloxy)-3-fluorophenyl)- 2-(cyclopropylmethylamino)-3-methylpyrimidin-4(3H)-one (0.128 g, 0.337 mmol) in TFA (2 mL) was stirred at 40°C for 2 hours and 45 minutes. The reaction mixture was concentrated to dryness and then purified by silica gel flash column chromatography, eluting with 20:1 dichloromethane/MeOH.
- Step A Preparation of 5-bromo-2-(4-fluorophenylamino)-3-methylpyrimidin-
- Step B Preparation of 5-(4-(benzyloxy)-3-fluorophenyl)-2-(4- fluorophenylamino)-3-methylpyrimidin-4(3H)-one: Prepared from 5-bromo-2-(4- fluorophenylamino)-3-methylpyrimidin-4(3H)-one (0.130 g, 0.436 mmol) according to the procedure described in Step B of Example 63. The desired product (0.139 g, 76%) was obtained as a grey/white solid.
- Step C Preparation of 5-(3-fluoro-4-hydroxyphenyl)-2-(4-fluorophenylamino)-3- methylpyrimidin-4(3H)-one: Prepared from 5-(4-(benzyloxy)-3-fluorophenyl)-2-(4- fluorophenylamino)-3-methylpyrimidin-4(3H)-one (0.139 g, 0.331 mmol) according to the procedure described in Step C of Example 63. The desired product (0.089 g, 82%) was obtained as a white solid.
- Step D Preparation of 5-(3-fluoro-4-(6-methoxy-7-(3- mo ⁇ holinopropoxy)quinolin-4-yloxy)phenyl)-2-(4-fluorophenylamino)-3-methylpyrimidin- 4(3H)-one: Prepared from 5-(3-fluoro-4-hydroxyphenyl)-2-(4-fluorophenylamino)-3- methylpyrimidin-4(3H)-one (0.025 g, 0.0759 mmol), 4-(3-(4-chloro-6-methoxyquinolin-7- yloxy)propyl)morpholine (0.0281 g, 0.0835 mmol) and catalytic DMAP according to the procedure described in Step C of Example 58, to give 164 (0.026 g, 54%) as a pale yellow solid.
- Step A Preparation of 5-bromo-2-chloro-3-ethylpyrimidin-4(3H)-one: Prepared from 5-bromo-2-chloropyrimidin-4(3H)-one (1.00 g, 4.775 mmol; obtained from Example 59, Step A) according to the procedure described in Step B of Example 59, substituting iodoethane in place of iodomethane. The desired product (0.411 g, 36%) was obtained as a yellow crystalline waxy solid.
- 1 H-NMR 400 MHz, DMSO-d 6 ) ⁇ 8.26 (s, IH), 4.16 (q, 3H), 1.25 (t, 4H).
- LRMS (ESI pos) m/e 237, 239 (M+, Br pattern).
- Step B Preparation of 5-bromo-3-ethyl-2-(phenylamino)pyrimidin-4(3H)-one:
- Step E Preparation of 3-ethyl-5-(3-fluoro-4-(6-methoxy-7-(3- morpholinopropoxy)quinolin-4-yloxy)phenyl)-2-(phenylamino)pyrimidin-4(3H)-one: Prepared from 3-ethyl-5-(3-fluoro-4-hydroxyphenyl)-2-(phenylamino)pyrimidin-4(3H)-one (0.025 g, 0.0768 mmol), 4-(3-(4-chloro-6-methoxyquinolin-7-yloxy)propyl)mo ⁇ holine (0.0285 g, 0.0845 mmol) and catalytic DMAP according to the procedure described in Step C of Example 58, to give 165 (0.016 g, 33%) as a pale yellow solid.
Abstract
Description


































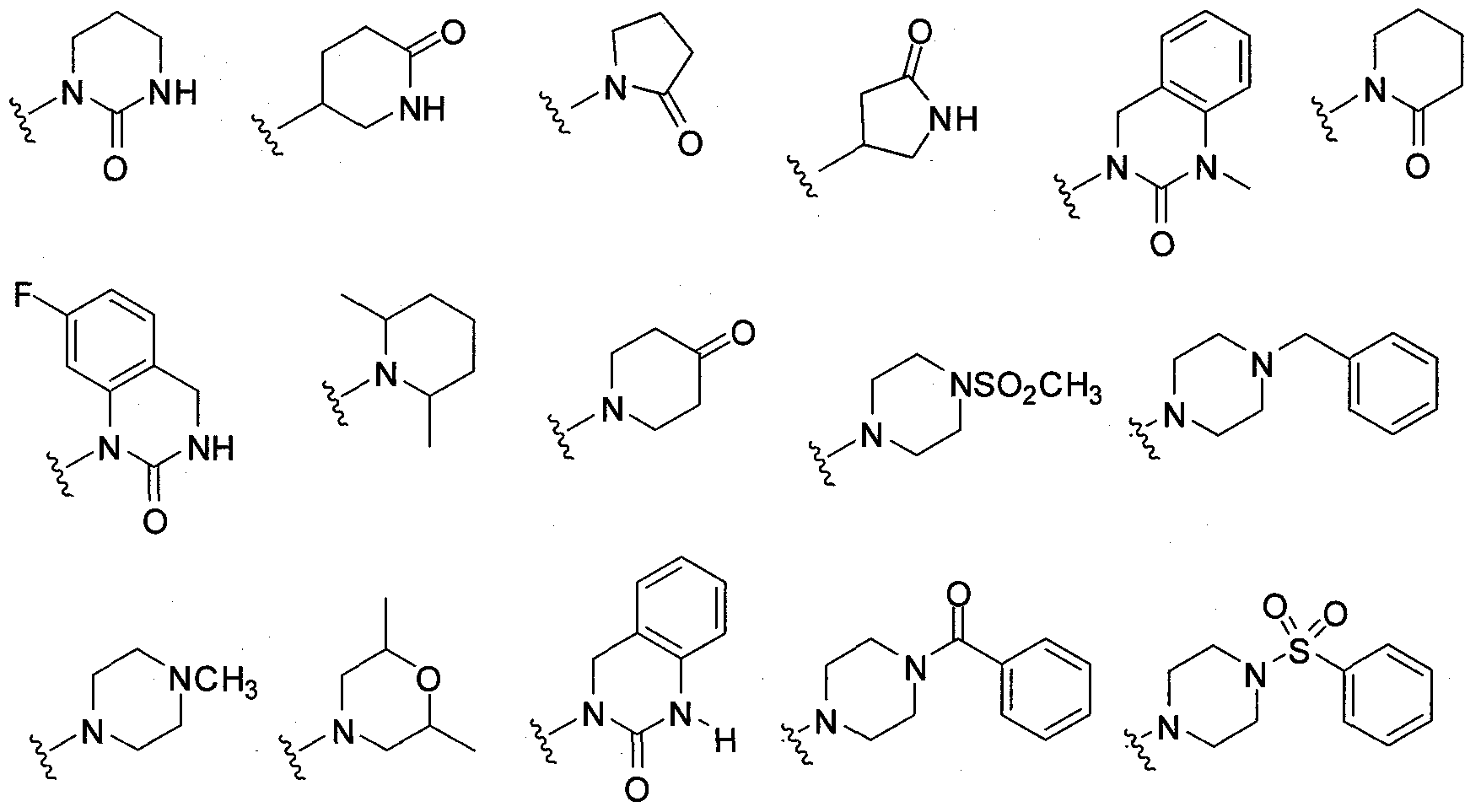
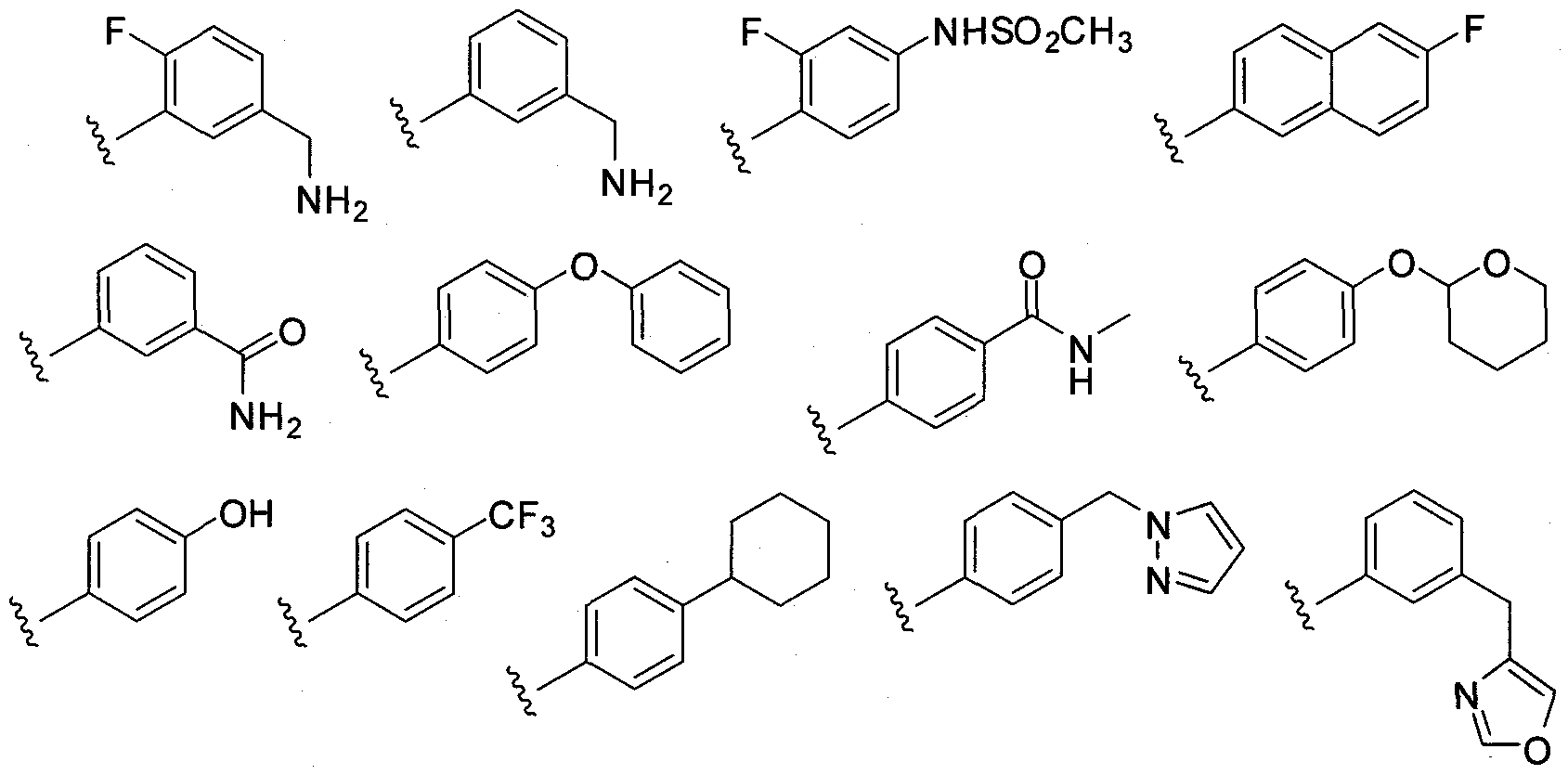

























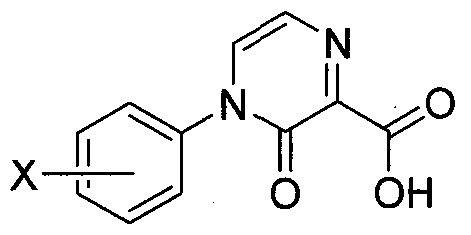
























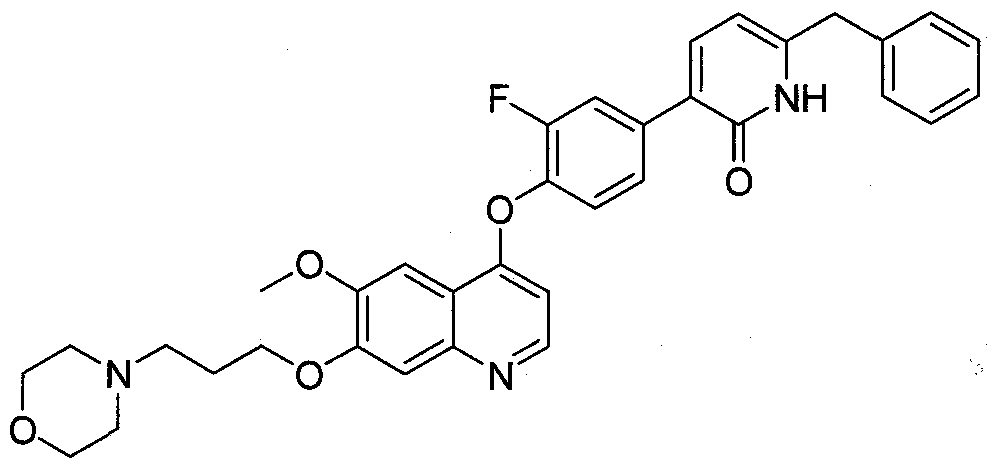







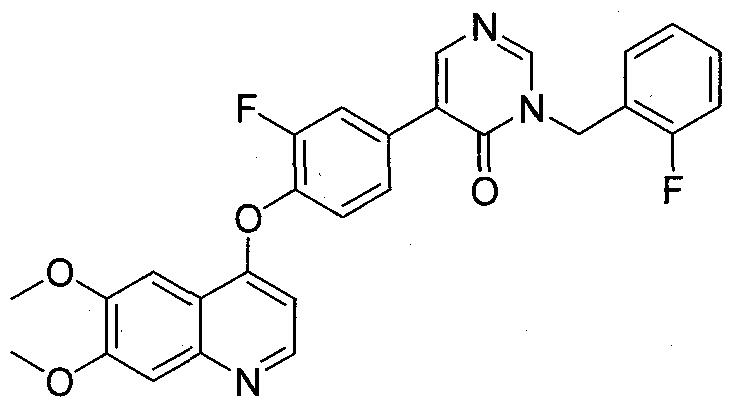



















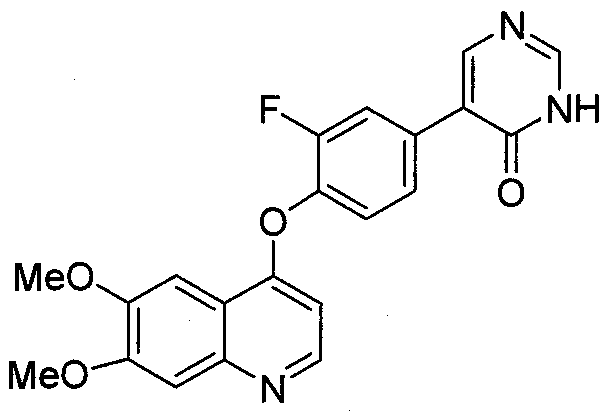

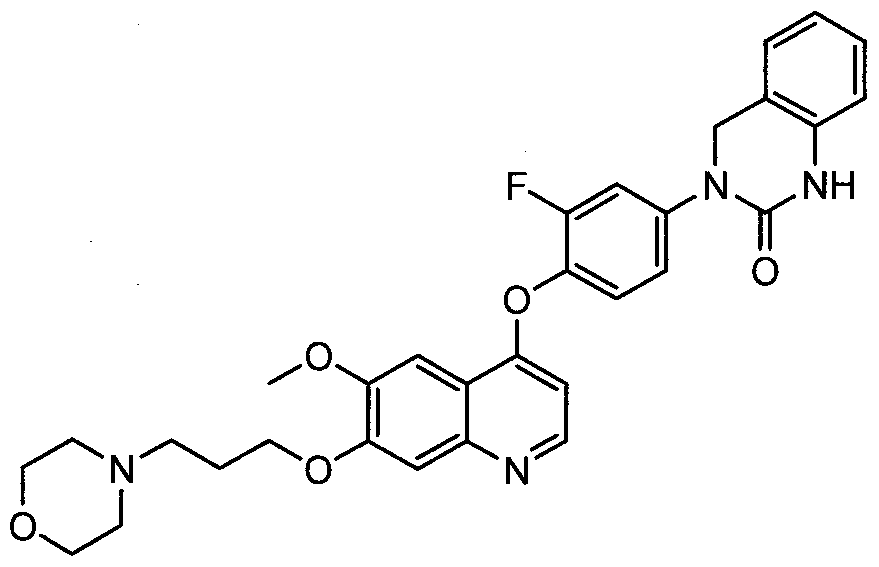








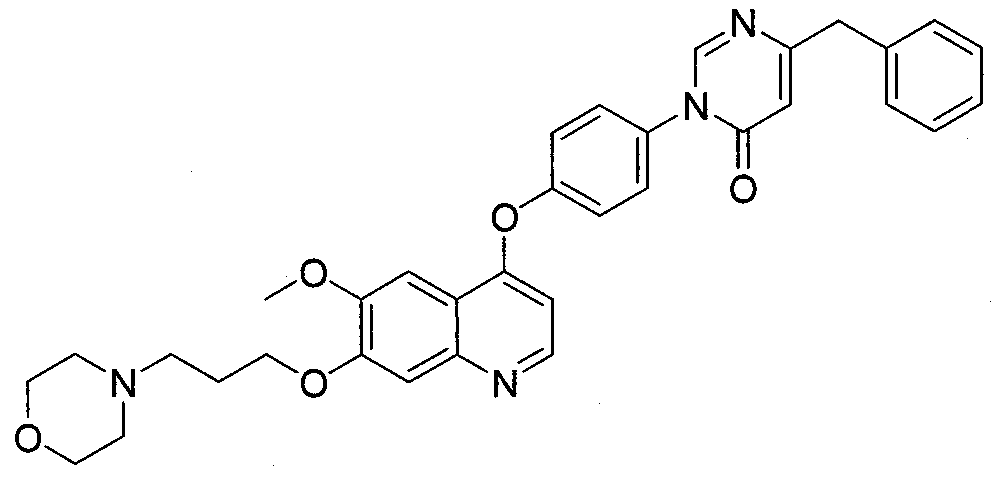







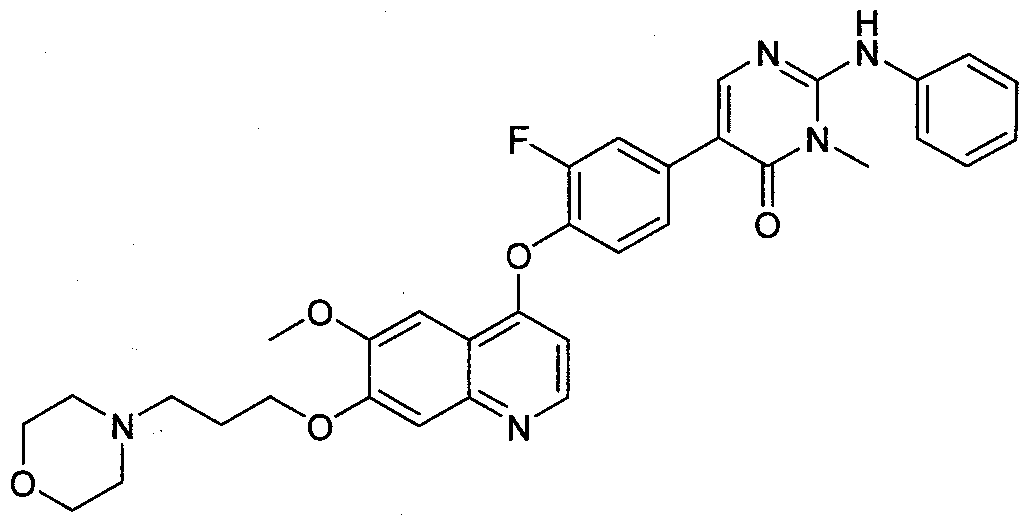








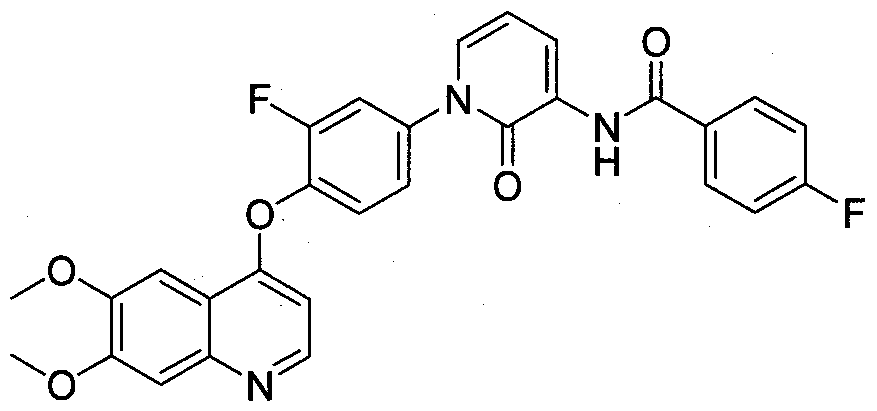















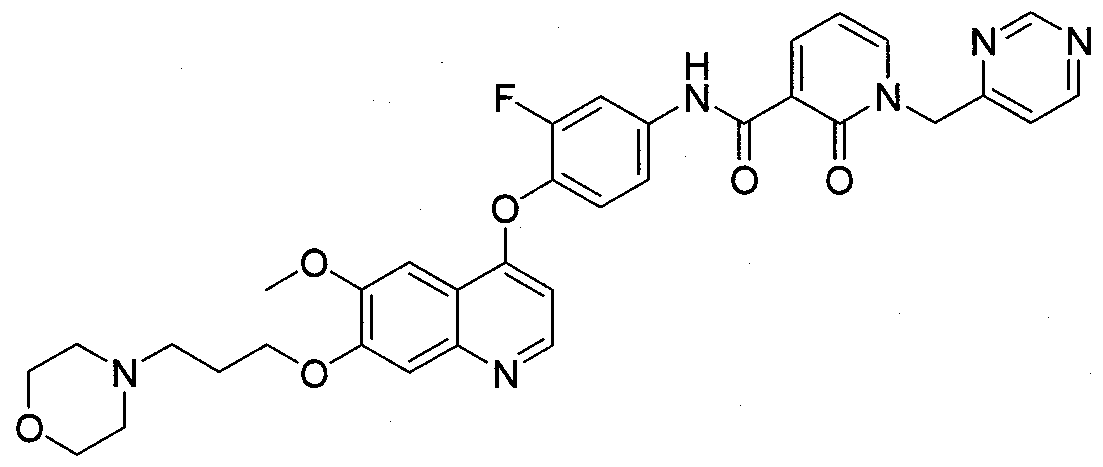










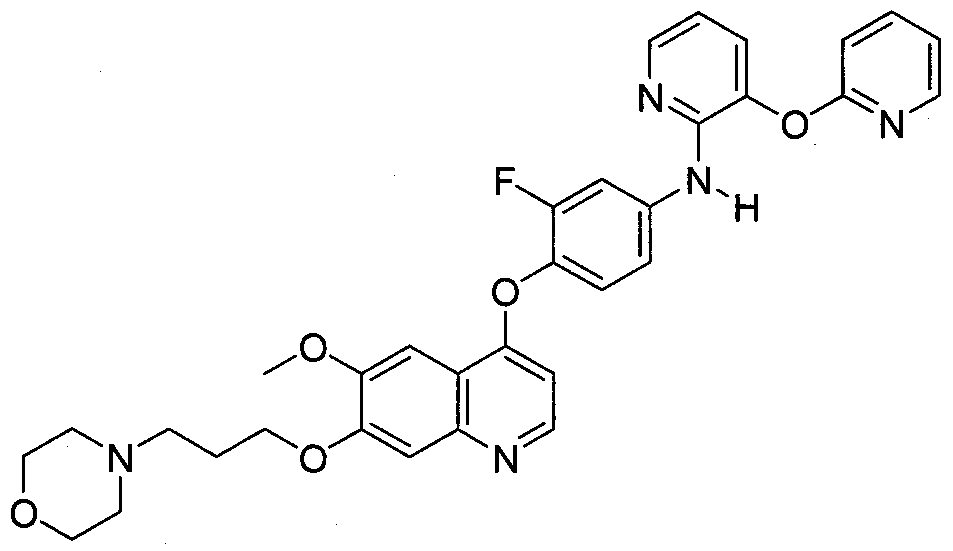





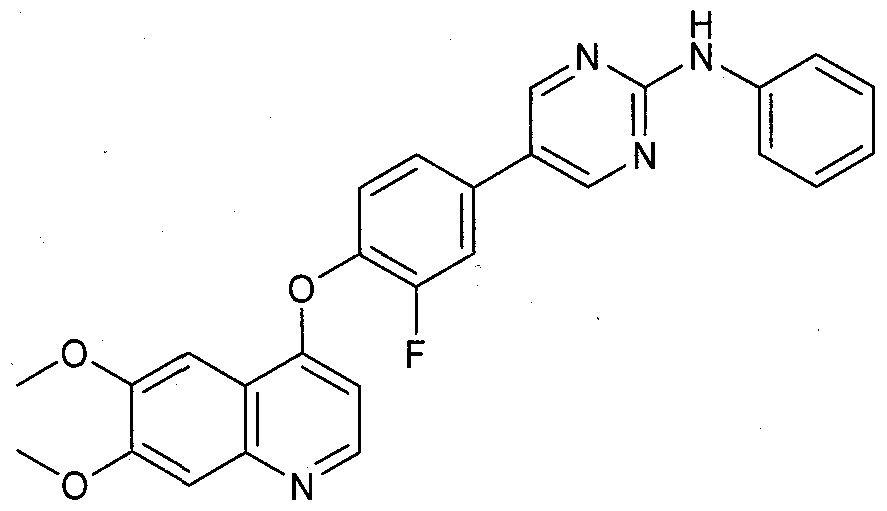

Claims







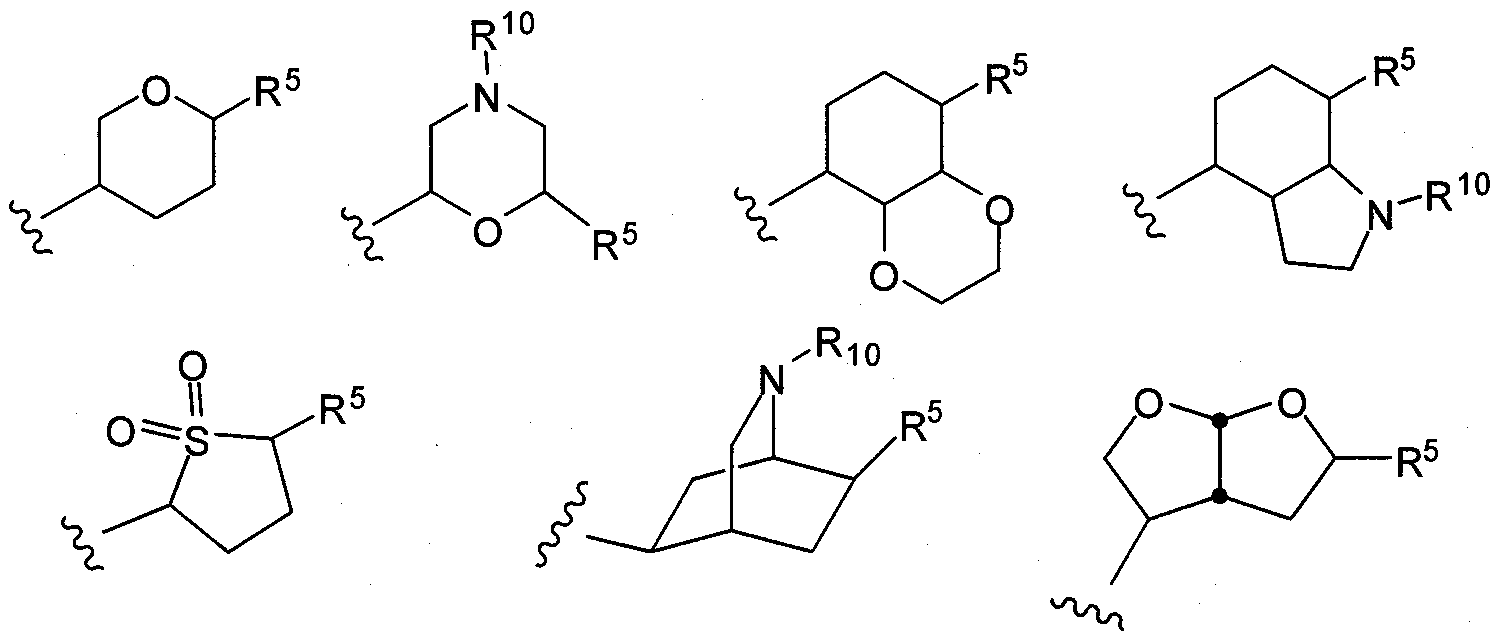































Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP07798333A EP2032538A2 (en) | 2006-06-08 | 2007-06-08 | Quinoline compounds and methods of use |
| CA002655128A CA2655128A1 (en) | 2006-06-08 | 2007-06-08 | Quinoline compounds and methods of use |
| US12/303,930 US20110053931A1 (en) | 2006-06-08 | 2007-06-08 | Quinoline compounds and methods of use |
| JP2009514553A JP2009539878A (en) | 2006-06-08 | 2007-06-08 | Quinoline compounds and methods of use |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US81190906P | 2006-06-08 | 2006-06-08 | |
| US60/811,909 | 2006-06-08 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007146824A2 true WO2007146824A2 (en) | 2007-12-21 |
| WO2007146824A3 WO2007146824A3 (en) | 2008-04-10 |
Family
ID=38658188
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/070787 WO2007146824A2 (en) | 2006-06-08 | 2007-06-08 | Quinoline compounds and methods of use |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20110053931A1 (en) |
| EP (1) | EP2032538A2 (en) |
| JP (1) | JP2009539878A (en) |
| CN (1) | CN101528702A (en) |
| CA (1) | CA2655128A1 (en) |
| WO (1) | WO2007146824A2 (en) |
Cited By (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2038272A2 (en) * | 2006-06-30 | 2009-03-25 | Sunesis Pharmaceuticals, Inc. | Pyridinonyl pdk1 inhibitors |
| FR2940289A1 (en) * | 2008-12-23 | 2010-06-25 | Biopharmed | AMINO HYDROXYQUINOLINE CLASS DERIVATIVES FOR THE TREATMENT OF PANCREATIC CANCER |
| WO2010056527A3 (en) * | 2008-10-30 | 2010-07-08 | Gilead Palo Alto, Inc. | B-biphenyl sulfonamide compounds as ion channel modulators |
| JP2011517689A (en) * | 2008-04-16 | 2011-06-16 | マックス プランク ゲゼルシャフト ツゥアー フェデルゥン デル ヴィッセンシャフテン エー フォー | Quinoline derivatives as AXL kinase inhibitors |
| EP2423208A1 (en) * | 2010-08-28 | 2012-02-29 | Lead Discovery Center GmbH | Pharmaceutically active compounds as Axl inhibitors |
| WO2012068096A2 (en) | 2010-11-15 | 2012-05-24 | Exelixis, Inc. | Benzoxazepines as inhibitors of pi3k/mtor and methods of their use and manufacture |
| US8404846B2 (en) | 2007-08-29 | 2013-03-26 | Methylgene Inc. | Inhibitors of protein tyrosine kinase activity |
| US8569319B2 (en) | 2010-04-29 | 2013-10-29 | Deciphera Pharmaceuticals, LLS | Pyridone amides and analogs exhibiting anti-cancer and anti-proliferative activities |
| WO2014001247A1 (en) | 2012-06-26 | 2014-01-03 | Bayer Pharma Aktiengesellschaft | N-[4-(Quinolin-4-yloxy)cyclohexyl(methyl)](hetero)arylcarboxamides as androgen receptor antagonists, production and use thereof as medicinal products |
| CN103739596A (en) * | 2013-12-10 | 2014-04-23 | 刘磊 | Quinazoline derivative used for cardio cerebrovascular diseases |
| WO2014194975A1 (en) * | 2013-06-06 | 2014-12-11 | Merck Patent Gmbh | A quinoline inhibitor of the macrophage stimulating 1 receptor mst1 r |
| US20140364431A1 (en) * | 2011-12-30 | 2014-12-11 | Shenyang Pharmaceutical University | Quinoline and cinnoline derivatives and their applications |
| WO2015012298A1 (en) | 2013-07-24 | 2015-01-29 | 小野薬品工業株式会社 | Quinoline derivative |
| EP2852388A4 (en) * | 2012-05-23 | 2016-01-13 | Univ Johns Hopkins | Compounds and methods of use thereof for treating neurodegenerative disorders |
| US9745283B2 (en) | 2011-11-14 | 2017-08-29 | Ignyta, Inc. | Uracil derivatives as AXL and c-MET kinase inhibitors |
| WO2017168448A1 (en) | 2016-03-30 | 2017-10-05 | Council Of Scientific & Industrial Research | Silicon incorporated quinolines with anti-malarial and anti-toxoplasmosis activity |
| AU2014219024B2 (en) * | 2013-02-20 | 2018-04-05 | KALA BIO, Inc. | Therapeutic compounds and uses thereof |
| WO2018108671A1 (en) | 2016-12-16 | 2018-06-21 | Basf Se | Pesticidal compounds |
| WO2018177781A1 (en) | 2017-03-28 | 2018-10-04 | Basf Se | Pesticidal compounds |
| US10208034B2 (en) | 2014-12-25 | 2019-02-19 | Ono Pharmaceutical Co., Ltd. | Quinoline derivative |
| WO2019121159A1 (en) | 2017-12-21 | 2019-06-27 | Basf Se | Pesticidal compounds |
| WO2019124548A1 (en) | 2017-12-22 | 2019-06-27 | 住友化学株式会社 | Heterocyclic compound and harmful arthropod controlling agent containing same |
| WO2019231942A1 (en) * | 2018-06-01 | 2019-12-05 | Rigel Pharmaceuticals, Inc. | Quinoline derivatives useful as tyrosine kinase inhibitors |
| WO2020027723A1 (en) * | 2018-08-01 | 2020-02-06 | Agency For Science, Technology And Research | Bicyclic compounds as kinase modulators, methods and uses thereof |
| WO2020058062A1 (en) | 2018-09-19 | 2020-03-26 | Bayer Aktiengesellschaft | Herbicidally active substituted phenylpyrimidine hydrazides |
| EP3643705A1 (en) | 2018-10-24 | 2020-04-29 | Basf Se | Pesticidal compounds |
| WO2020141470A1 (en) * | 2019-01-03 | 2020-07-09 | Array Biopharma Inc. | Quinoline compounds as inhibitors of tam and met kinases |
| WO2020154610A1 (en) * | 2019-01-25 | 2020-07-30 | Exelixis, Inc. | Compounds for the treatment of kinase-dependent disorders |
| US10836747B2 (en) | 2017-01-26 | 2020-11-17 | Ono Pharmaceutical Co., Ltd. | Ethane-sulfonate salt of quinoline derivative |
| EP3766879A1 (en) | 2019-07-19 | 2021-01-20 | Basf Se | Pesticidal pyrazole derivatives |
| WO2021025407A1 (en) * | 2019-08-02 | 2021-02-11 | 웰마커바이오 주식회사 | Oxo-pyridine fusion ring derivative and pharmaceutical composition comprising same |
| EP3795567A4 (en) * | 2018-05-15 | 2022-03-02 | Beijing InnoCare Pharma Tech Co., Ltd. | Indoline-1-formamide compound, preparation method therefor, and medical use thereof |
| US11753395B2 (en) | 2021-12-16 | 2023-09-12 | Kinnate Biopharma Inc. | Inhibitors of MET kinase |
| US11826363B2 (en) | 2017-10-13 | 2023-11-28 | Ono Pharmaceutical Co., Ltd. | Therapeutic agent for solid cancers, which comprises Axl inhibitor as active ingredient |
Families Citing this family (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2008236670B2 (en) * | 2007-04-05 | 2011-12-01 | Amgen Inc. | Aurora kinase modulators and method of use |
| SG188802A1 (en) * | 2008-03-06 | 2013-04-30 | Genentech Inc | Combination therapy with c-met and egfr antagonists |
| WO2012011548A1 (en) * | 2010-07-23 | 2012-01-26 | 国立大学法人 東京大学 | Nitrogen-containing heterocyclic derivative |
| CN102558147B (en) * | 2010-12-23 | 2014-09-17 | 江苏先声药物研究有限公司 | Compound, and preparation method and application thereof |
| ES2590778T3 (en) | 2011-02-28 | 2016-11-23 | Calitor Sciences, Llc | Quinoline Substituted Compounds |
| CN103965107B (en) * | 2013-02-06 | 2016-08-17 | 沈阳药科大学 | 2-aryl substituted quinoline derivatives and application thereof |
| AU2014216178B2 (en) | 2013-02-15 | 2018-06-28 | KALA BIO, Inc. | Therapeutic compounds and uses thereof |
| US9688688B2 (en) | 2013-02-20 | 2017-06-27 | Kala Pharmaceuticals, Inc. | Crystalline forms of 4-((4-((4-fluoro-2-methyl-1H-indol-5-yl)oxy)-6-methoxyquinazolin-7-yl)oxy)-1-(2-oxa-7-azaspiro[3.5]nonan-7-yl)butan-1-one and uses thereof |
| CN104072480B (en) * | 2013-03-27 | 2016-12-28 | 沈阳药科大学 | Quinolines and its preparation method and application |
| AU2014260011A1 (en) * | 2013-05-01 | 2015-11-12 | Celgene Corporation | Synthesis of 3-(5-amino-2-methyl-4-oxoquinazolin-3(4H)-yl)piperidine-2-6-dione |
| MX355330B (en) | 2013-11-01 | 2018-04-16 | Kala Pharmaceuticals Inc | CRYSTALLINE FORMS OF THERAPEUTIC COMPOUNDS and USES THEREOF. |
| US9890173B2 (en) | 2013-11-01 | 2018-02-13 | Kala Pharmaceuticals, Inc. | Crystalline forms of therapeutic compounds and uses thereof |
| MX2016012285A (en) | 2014-03-24 | 2017-01-23 | Genentech Inc | Cancer treatment with c-met antagonists and correlation of the latter with hgf expression. |
| CN104086529B (en) * | 2014-05-18 | 2015-12-30 | 马宝花 | A kind of quinazoline derivative, Its Preparation Method And Use |
| CN104817497B (en) * | 2015-03-20 | 2017-03-08 | 南京众睿缘生物科技有限公司 | A kind of alkynes is for quinoline and its production and use |
| CN104926789A (en) * | 2015-05-20 | 2015-09-23 | 沈阳药科大学 | 4-phenoxyl substituted quinoline compound containing imidazolone and application thereof |
| CN107474039B (en) * | 2016-06-08 | 2020-05-01 | 沈阳药科大学 | 4-phenoxy substituted quinoline compound containing triazazolone and imidazole and application thereof |
| EP3509421A4 (en) | 2016-09-08 | 2020-05-20 | Kala Pharmaceuticals, Inc. | Crystalline forms of therapeutic compounds and uses thereof |
| AU2017324716B2 (en) | 2016-09-08 | 2020-08-13 | KALA BIO, Inc. | Crystalline forms of therapeutic compounds and uses thereof |
| EP3509422A4 (en) | 2016-09-08 | 2020-05-20 | Kala Pharmaceuticals, Inc. | Crystalline forms of therapeutic compounds and uses thereof |
| CN108299292B (en) * | 2018-02-13 | 2021-03-26 | 安徽工业大学 | Application of 5, 7-dibromo-8- (methoxymethoxy) -2-methylquinoline or medicinal salt thereof in treating breast cancer |
| CN108707145B (en) * | 2018-07-05 | 2021-02-26 | 沈阳药科大学 | Quinoline compound containing five-membered heterocyclic structure and preparation and application thereof |
| EP3845527A4 (en) * | 2018-08-27 | 2022-06-08 | Beijing Yuezhikangtai Biomedicines Co., Ltd. | Multi-substituted pyridone derivatives and medical use thereof |
| CN111303024B (en) * | 2018-12-12 | 2023-03-28 | 安徽中科拓苒药物科学研究有限公司 | Quinoline-structured pan-KIT kinase inhibitor and application thereof |
| CN117362275A (en) * | 2022-06-29 | 2024-01-09 | 广州百霆医药科技有限公司 | Tyrosine protein kinase inhibitor and application thereof |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1548008A1 (en) * | 2002-08-23 | 2005-06-29 | Kirin Beer Kabushiki Kaisha | Compound having tgf-beta inhibitory activity and medicinal composition containing the same |
| WO2005070891A2 (en) * | 2004-01-23 | 2005-08-04 | Amgen Inc | Compounds and methods of use |
| WO2006060318A2 (en) * | 2004-11-30 | 2006-06-08 | Amgen Inc. | Quinolines and quinazoline analogs and their use as medicaments for treating cancer |
| WO2006117552A1 (en) * | 2005-05-05 | 2006-11-09 | Chroma Therapeutics Ltd | Quinoline and quinoxaline derivatives as inhibitors of kinase enzymatic activity |
Family Cites Families (50)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5880141A (en) * | 1995-06-07 | 1999-03-09 | Sugen, Inc. | Benzylidene-Z-indoline compounds for the treatment of disease |
| US20030162795A1 (en) * | 1998-10-22 | 2003-08-28 | Pfizer Inc. | Thienopyrimidine and thienopyridine derivatives useful as anticancer agents |
| GB9906566D0 (en) * | 1999-03-23 | 1999-05-19 | Zeneca Ltd | Chemical compounds |
| US6297238B1 (en) * | 1999-04-06 | 2001-10-02 | Basf Aktiengesellschaft | Therapeutic agents |
| US20020004511A1 (en) * | 2000-06-28 | 2002-01-10 | Luzzio Michael Joseph | Thiophene derivatives useful as anticancer agents |
| US6599902B2 (en) * | 2001-05-30 | 2003-07-29 | Sugen, Inc. | 5-aralkysufonyl-3-(pyrrol-2-ylmethylidene)-2-indolinone derivatives as kinase inhibitors |
| AU2002345792A1 (en) * | 2001-06-21 | 2003-01-08 | Pfizer Inc. | Thienopyridine and thienopyrimidine anticancer agents |
| EP1411046B1 (en) * | 2001-06-22 | 2009-09-16 | Kirin Pharma Kabushiki Kaisha | Quinoline derivative and quinazoline derivative inhibiting self-phosphorylation of hepatocytus proliferator receptor, and medicinal composition containing the same |
| US7495104B2 (en) * | 2001-10-17 | 2009-02-24 | Kirin Beer Kabushiki Kaisha | Quinoline or quinazoline derivatives inhibiting auto-phosphorylation of fibroblast growth factor receptors |
| EP1483268A2 (en) * | 2002-03-01 | 2004-12-08 | Pfizer Inc. | Indolyl-urea derivatives of thienopyridines useful as anti-angiogenic agents |
| US6790852B2 (en) * | 2002-04-18 | 2004-09-14 | Hoffmann-La Roche Inc. | 2-(2,6-dichlorophenyl)-diarylimidazoles |
| WO2004031184A1 (en) * | 2002-10-01 | 2004-04-15 | Johnson & Johnson Pharmaceutical Research & Development, Inc. | 4,6-diaminosubstituted-2-[oxy or aminoxy]-[1,3,5]triazines as protein tyrosine kinase inhibitors |
| CL2003002287A1 (en) * | 2002-11-25 | 2005-01-14 | Wyeth Corp | COMPOUNDS DERIVED FROM TIENO [3,2-b] -PIRIDINA-6-CARBONITRILOS AND TIENEO [2,3-b] -PIRIDINA-5-CARBONITRILS, PHARMACEUTICAL COMPOSITION, PROCEDURE OF PREPARATION AND INTERMEDIARY COMPOUNDS, AND THEIR USE IN THE TREATMENT OF CANCER, APOPLEJIA, OSTEOPOROSIS |
| US7276519B2 (en) * | 2002-11-25 | 2007-10-02 | Wyeth | Thieno[3,2-b]pyridine-6-carbonitriles and thieno[2,3-b]pyridine-5-carbonitriles as protein kinase inhibitors |
| MXPA05008521A (en) * | 2003-02-13 | 2005-10-20 | Pharmacia Corp | Antibodies to c-met for the treatment of cancers. |
| PL216368B1 (en) * | 2003-02-26 | 2014-03-31 | Sugen | Aminoheteroaryl compounds as protein kinase inhibitors |
| EP1615906A1 (en) * | 2003-04-03 | 2006-01-18 | Vertex Pharmaceuticals Incorporated | Compositions useful as inhibitors of protein kinases |
| KR20110117728A (en) * | 2003-06-06 | 2011-10-27 | 제넨테크, 인크. | Modulating the interaction between hgf beta chain and c-met |
| US20060009493A1 (en) * | 2003-07-02 | 2006-01-12 | Sugen, Inc. | Indolinone hydrazides as c-Met inhibitors |
| US7122548B2 (en) * | 2003-07-02 | 2006-10-17 | Sugen, Inc. | Triazolotriazine compounds and uses thereof |
| CA2536470A1 (en) * | 2003-08-15 | 2005-02-24 | Vertex Pharmaceuticals Incorporated | Pyrrole compositions useful as inhibitors of c-met |
| WO2005021554A1 (en) * | 2003-08-29 | 2005-03-10 | Pfizer Inc. | Thienopyridine-phenylacet amides and their derivatives useful as new anti-angiogenic agents |
| MXPA06002256A (en) * | 2003-08-29 | 2006-05-17 | Pfizer | Naphthalene carboxamides and their derivatives useful as new anti-angiogenic agents. |
| WO2005040345A2 (en) * | 2003-09-24 | 2005-05-06 | Vertex Pharmceuticals Incorporated | 4-azole substituted imidazole compositions useful as inhibitors or c-met receptor tyrosine kinase |
| EP2213661B1 (en) * | 2003-09-26 | 2011-07-20 | Exelixis Inc. | c-Met Modulators and Methods of Use |
| JP2007518823A (en) * | 2004-01-23 | 2007-07-12 | アムゲン インコーポレイテッド | Quinoline, quinazoline, pyridine, and pyrimidine compounds and their use in the treatment of inflammation, angiogenesis, and cancer |
| TW200538120A (en) * | 2004-02-20 | 2005-12-01 | Kirin Brewery | Compound having TGF-beta inhibitory activity and pharmaceutical composition containing same |
| US7459562B2 (en) * | 2004-04-23 | 2008-12-02 | Bristol-Myers Squibb Company | Monocyclic heterocycles as kinase inhibitors |
| TW200538453A (en) * | 2004-04-26 | 2005-12-01 | Bristol Myers Squibb Co | Bicyclic heterocycles as kinase inhibitors |
| AU2005245386B2 (en) * | 2004-05-07 | 2008-11-27 | Amgen Inc. | Nitrogenated heterocyclic derivatives as protein kinase modulators and use for the treatment of angiogenesis and cancer |
| EP1753428A4 (en) * | 2004-05-14 | 2010-09-15 | Abbott Lab | Kinase inhibitors as therapeutic agents |
| US7452887B2 (en) * | 2004-06-04 | 2008-11-18 | Amphora Discovery Corporation | Quinoline- and isoquinoline-based compounds exhibiting ATP-utilizing enzyme inhibitory activity, and compositions, and uses thereof |
| US7439246B2 (en) * | 2004-06-28 | 2008-10-21 | Bristol-Myers Squibb Company | Fused heterocyclic kinase inhibitors |
| US20050288290A1 (en) * | 2004-06-28 | 2005-12-29 | Borzilleri Robert M | Fused heterocyclic kinase inhibitors |
| JP5368701B2 (en) * | 2004-07-02 | 2013-12-18 | エクセリクシス、インコーポレイテッド | c-Met Modulator and Method of Use |
| EP1781664B1 (en) * | 2004-07-30 | 2013-09-04 | MethylGene Inc. | Inhibitors of vegf receptor and hgf receptor signaling |
| AU2006229343A1 (en) * | 2005-03-28 | 2006-10-05 | Kirin Pharma Kabushiki Kaisha | Thienopyridine derivative, or quinoline derivative, or quinazoline derivative, having c-Met autophosphorylation inhibiting potency |
| JP2008537748A (en) * | 2005-04-06 | 2008-09-25 | エクセリクシス、インコーポレイテッド | c-Met Modulator and Method of Use |
| US7470693B2 (en) * | 2005-04-21 | 2008-12-30 | Bristol-Myers Squibb Company | Oxalamide derivatives as kinase inhibitors |
| JO2787B1 (en) * | 2005-04-27 | 2014-03-15 | امجين إنك, | Substituted Amid derivatives & methods of use |
| BRPI0610382A2 (en) * | 2005-05-20 | 2010-06-15 | Methylgene Inc | vegf receptor and hgf receptor signaling inhibitors |
| CN101796055B (en) * | 2005-05-20 | 2013-09-04 | 梅特希尔基因公司 | Inhibitors of VEGF receptor and HGF receptor signaling |
| US7732613B2 (en) * | 2005-09-14 | 2010-06-08 | Bristol-Myers Squibb Company | Met kinase inhibitors |
| US7880004B2 (en) * | 2005-09-15 | 2011-02-01 | Bristol-Myers Squibb Company | Met kinase inhibitors |
| US7547782B2 (en) * | 2005-09-30 | 2009-06-16 | Bristol-Myers Squibb Company | Met kinase inhibitors |
| CN101522687A (en) * | 2006-01-30 | 2009-09-02 | 阿雷生物药品公司 | Heterobicyclic thiophene compounds and methods of use |
| US7723330B2 (en) * | 2006-03-07 | 2010-05-25 | Array Biopharma Inc. | Heterobicyclic pyrazole compounds and methods of use |
| ATE545417T1 (en) * | 2007-03-14 | 2012-03-15 | Ranbaxy Lab Ltd | PYRAZOLOA3,4-BUPYRIDINE DERIVATIVES AS PHOSPHODIESTERASE INHIBITORS |
| CN101918403A (en) * | 2007-09-06 | 2010-12-15 | 阵列生物制药公司 | Pyrazolo-pyridines as tyrosine kinase inhibitor |
| EP2225239B1 (en) * | 2007-12-21 | 2014-10-22 | F. Hoffmann-La Roche AG | Heterocyclic antiviral compounds |
-
2007
- 2007-06-08 EP EP07798333A patent/EP2032538A2/en not_active Withdrawn
- 2007-06-08 JP JP2009514553A patent/JP2009539878A/en active Pending
- 2007-06-08 CA CA002655128A patent/CA2655128A1/en not_active Abandoned
- 2007-06-08 CN CNA2007800294412A patent/CN101528702A/en active Pending
- 2007-06-08 WO PCT/US2007/070787 patent/WO2007146824A2/en active Application Filing
- 2007-06-08 US US12/303,930 patent/US20110053931A1/en not_active Abandoned
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1548008A1 (en) * | 2002-08-23 | 2005-06-29 | Kirin Beer Kabushiki Kaisha | Compound having tgf-beta inhibitory activity and medicinal composition containing the same |
| WO2005070891A2 (en) * | 2004-01-23 | 2005-08-04 | Amgen Inc | Compounds and methods of use |
| WO2006060318A2 (en) * | 2004-11-30 | 2006-06-08 | Amgen Inc. | Quinolines and quinazoline analogs and their use as medicaments for treating cancer |
| WO2006117552A1 (en) * | 2005-05-05 | 2006-11-09 | Chroma Therapeutics Ltd | Quinoline and quinoxaline derivatives as inhibitors of kinase enzymatic activity |
Cited By (69)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9873693B2 (en) | 2006-06-30 | 2018-01-23 | Sunesis Pharmaceuticals, Inc. | Methods of treatment using pyridinonyl PDK1 inhibitors |
| EP2038272A4 (en) * | 2006-06-30 | 2011-01-05 | Sunesis Pharmaceuticals Inc | Pyridinonyl pdk1 inhibitors |
| EP2038272A2 (en) * | 2006-06-30 | 2009-03-25 | Sunesis Pharmaceuticals, Inc. | Pyridinonyl pdk1 inhibitors |
| US8778977B2 (en) | 2006-06-30 | 2014-07-15 | Sunesis Pharmaceuticals, Inc. | Pyridinonyl PDK1 inhibitors |
| US8846927B2 (en) | 2007-08-29 | 2014-09-30 | Methylgene Inc. | Inhibitors of protein tyrosine kinase activity |
| US8404846B2 (en) | 2007-08-29 | 2013-03-26 | Methylgene Inc. | Inhibitors of protein tyrosine kinase activity |
| JP2011517689A (en) * | 2008-04-16 | 2011-06-16 | マックス プランク ゲゼルシャフト ツゥアー フェデルゥン デル ヴィッセンシャフテン エー フォー | Quinoline derivatives as AXL kinase inhibitors |
| WO2010056527A3 (en) * | 2008-10-30 | 2010-07-08 | Gilead Palo Alto, Inc. | B-biphenyl sulfonamide compounds as ion channel modulators |
| US8389500B2 (en) | 2008-10-30 | 2013-03-05 | Gilead Sciences, Inc. | Fused heterocyclic compounds as ion channel modulators |
| FR2940289A1 (en) * | 2008-12-23 | 2010-06-25 | Biopharmed | AMINO HYDROXYQUINOLINE CLASS DERIVATIVES FOR THE TREATMENT OF PANCREATIC CANCER |
| WO2010073235A1 (en) * | 2008-12-23 | 2010-07-01 | Biopharmed | Ppar agonist compositions and methods of use |
| US8569319B2 (en) | 2010-04-29 | 2013-10-29 | Deciphera Pharmaceuticals, LLS | Pyridone amides and analogs exhibiting anti-cancer and anti-proliferative activities |
| RU2573834C2 (en) * | 2010-08-28 | 2016-01-27 | Лид Дискавери Сентер Гмбх | PHARMACEUTICALLY ACTIVE COMPOUNDS AS Axl INHIBITORS |
| US8999982B2 (en) | 2010-08-28 | 2015-04-07 | Lead Discovery Center Gmbh | Pharmaceutically active compounds as Axl inhibitors |
| WO2012028332A1 (en) | 2010-08-28 | 2012-03-08 | Lead Discovery Center Gmbh | Pharmaceutically active compounds as axl inhibitors |
| EP2423208A1 (en) * | 2010-08-28 | 2012-02-29 | Lead Discovery Center GmbH | Pharmaceutically active compounds as Axl inhibitors |
| AU2011297889B2 (en) * | 2010-08-28 | 2015-12-24 | Lead Discovery Center Gmbh | Pharmaceutically active compounds as Axl inhibitors |
| WO2012068096A2 (en) | 2010-11-15 | 2012-05-24 | Exelixis, Inc. | Benzoxazepines as inhibitors of pi3k/mtor and methods of their use and manufacture |
| US10017496B2 (en) | 2011-11-14 | 2018-07-10 | Ignyta, Inc. | Uracil derivatives as AXL and c-MET kinase inhibitors |
| US9745283B2 (en) | 2011-11-14 | 2017-08-29 | Ignyta, Inc. | Uracil derivatives as AXL and c-MET kinase inhibitors |
| EP2799437A4 (en) * | 2011-12-30 | 2015-08-19 | Univ Shenyang Pharmaceutical | Quinoline and cinnoline compounds and use thereof |
| US20140364431A1 (en) * | 2011-12-30 | 2014-12-11 | Shenyang Pharmaceutical University | Quinoline and cinnoline derivatives and their applications |
| US9382232B2 (en) | 2011-12-30 | 2016-07-05 | Shenyang Pharmaceutical University | Quinoline and cinnoline derivatives and their applications |
| EP2852388A4 (en) * | 2012-05-23 | 2016-01-13 | Univ Johns Hopkins | Compounds and methods of use thereof for treating neurodegenerative disorders |
| US9539259B2 (en) | 2012-05-23 | 2017-01-10 | The Johns Hopkins University | Compounds and methods of use thereof for treating neurodegenerative disorders |
| US9428460B2 (en) | 2012-06-26 | 2016-08-30 | Bayer Pharma Aktiengesellschaft | N-[4-(quinolin-4-yloxy)cyclohexyl(methyl)](hetero)arylcarboxamides as androgen receptor antagonists, production and use thereof as medicinal products |
| WO2014001247A1 (en) | 2012-06-26 | 2014-01-03 | Bayer Pharma Aktiengesellschaft | N-[4-(Quinolin-4-yloxy)cyclohexyl(methyl)](hetero)arylcarboxamides as androgen receptor antagonists, production and use thereof as medicinal products |
| AU2014219024B2 (en) * | 2013-02-20 | 2018-04-05 | KALA BIO, Inc. | Therapeutic compounds and uses thereof |
| US9481668B2 (en) | 2013-06-06 | 2016-11-01 | Merck Patent Gmbh | Quinoline inhibitor of the macrophage stimulating 1 receptor MSTR1 |
| JP2016520627A (en) * | 2013-06-06 | 2016-07-14 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフツングMerck Patent Gesellschaft mit beschraenkter Haftung | Quinoline inhibitor of 1 receptor MSTR1 that stimulates macrophages |
| WO2014194975A1 (en) * | 2013-06-06 | 2014-12-11 | Merck Patent Gmbh | A quinoline inhibitor of the macrophage stimulating 1 receptor mst1 r |
| AU2014277262B2 (en) * | 2013-06-06 | 2019-10-03 | Lead Discovery Center Gmbh | A quinoline inhibitor of the macrophage stimulating 1 receptor MST1 R |
| JP2018024691A (en) * | 2013-07-24 | 2018-02-15 | 小野薬品工業株式会社 | Quinoline derivative |
| JPWO2015012298A1 (en) * | 2013-07-24 | 2017-03-02 | 小野薬品工業株式会社 | Quinoline derivatives |
| US9573935B2 (en) | 2013-07-24 | 2017-02-21 | Ono Pharmaceutical Co., Ltd. | Quinoline derivative |
| KR20160033707A (en) | 2013-07-24 | 2016-03-28 | 오노 야꾸힝 고교 가부시키가이샤 | Quinoline derivative |
| US9994549B2 (en) | 2013-07-24 | 2018-06-12 | Ono Pharmaceutical Co., Ltd. | Quinoline derivative |
| KR102251609B1 (en) | 2013-07-24 | 2021-05-13 | 오노 야꾸힝 고교 가부시키가이샤 | Quinoline derivative |
| WO2015012298A1 (en) | 2013-07-24 | 2015-01-29 | 小野薬品工業株式会社 | Quinoline derivative |
| US10676462B2 (en) | 2013-07-24 | 2020-06-09 | Ono Pharmaceutical Co., Ltd. | Quinoline derivative |
| EP3415501A1 (en) | 2013-07-24 | 2018-12-19 | ONO Pharmaceutical Co., Ltd. | Quinoline derivative |
| US10208022B2 (en) | 2013-07-24 | 2019-02-19 | Ono Pharmaceutical Co., Ltd. | Quinoline derivative |
| US10501442B2 (en) | 2013-07-24 | 2019-12-10 | Ono Pharmaceuticals Co., Ltd. | Quinoline derivative |
| CN103739596A (en) * | 2013-12-10 | 2014-04-23 | 刘磊 | Quinazoline derivative used for cardio cerebrovascular diseases |
| US10208034B2 (en) | 2014-12-25 | 2019-02-19 | Ono Pharmaceutical Co., Ltd. | Quinoline derivative |
| WO2017168448A1 (en) | 2016-03-30 | 2017-10-05 | Council Of Scientific & Industrial Research | Silicon incorporated quinolines with anti-malarial and anti-toxoplasmosis activity |
| US10550135B2 (en) | 2016-03-30 | 2020-02-04 | Council Of Scientific & Industrial Research | Silicon incorporated quinolines with anti-malarial and anti-toxoplasmosis activity |
| WO2018108671A1 (en) | 2016-12-16 | 2018-06-21 | Basf Se | Pesticidal compounds |
| US10836747B2 (en) | 2017-01-26 | 2020-11-17 | Ono Pharmaceutical Co., Ltd. | Ethane-sulfonate salt of quinoline derivative |
| WO2018177781A1 (en) | 2017-03-28 | 2018-10-04 | Basf Se | Pesticidal compounds |
| US11826363B2 (en) | 2017-10-13 | 2023-11-28 | Ono Pharmaceutical Co., Ltd. | Therapeutic agent for solid cancers, which comprises Axl inhibitor as active ingredient |
| WO2019121159A1 (en) | 2017-12-21 | 2019-06-27 | Basf Se | Pesticidal compounds |
| WO2019124548A1 (en) | 2017-12-22 | 2019-06-27 | 住友化学株式会社 | Heterocyclic compound and harmful arthropod controlling agent containing same |
| EP3795567A4 (en) * | 2018-05-15 | 2022-03-02 | Beijing InnoCare Pharma Tech Co., Ltd. | Indoline-1-formamide compound, preparation method therefor, and medical use thereof |
| WO2019231942A1 (en) * | 2018-06-01 | 2019-12-05 | Rigel Pharmaceuticals, Inc. | Quinoline derivatives useful as tyrosine kinase inhibitors |
| US10851093B2 (en) | 2018-06-01 | 2020-12-01 | Rigel Pharmaceuticals, Inc. | Tyrosine kinase inhibitors |
| WO2020027723A1 (en) * | 2018-08-01 | 2020-02-06 | Agency For Science, Technology And Research | Bicyclic compounds as kinase modulators, methods and uses thereof |
| US11702425B2 (en) | 2018-08-01 | 2023-07-18 | Agency For Science, Technology And Research | Bicyclic compounds as kinase modulators, methods and uses thereof |
| WO2020058062A1 (en) | 2018-09-19 | 2020-03-26 | Bayer Aktiengesellschaft | Herbicidally active substituted phenylpyrimidine hydrazides |
| WO2020083733A1 (en) | 2018-10-24 | 2020-04-30 | Basf Se | Pesticidal compounds |
| EP3643705A1 (en) | 2018-10-24 | 2020-04-29 | Basf Se | Pesticidal compounds |
| TWI745824B (en) * | 2019-01-03 | 2021-11-11 | 美商亞雷生物製藥股份有限公司 | Quinoline compounds as inhibitors of tam and met kinases |
| WO2020141470A1 (en) * | 2019-01-03 | 2020-07-09 | Array Biopharma Inc. | Quinoline compounds as inhibitors of tam and met kinases |
| WO2020154610A1 (en) * | 2019-01-25 | 2020-07-30 | Exelixis, Inc. | Compounds for the treatment of kinase-dependent disorders |
| CN113710322A (en) * | 2019-01-25 | 2021-11-26 | 埃克塞里艾克西斯公司 | Compounds for the treatment of kinase-dependent disorders |
| EP3766879A1 (en) | 2019-07-19 | 2021-01-20 | Basf Se | Pesticidal pyrazole derivatives |
| WO2021013561A1 (en) | 2019-07-19 | 2021-01-28 | Basf Se | Pesticidal pyrazole and triazole derivatives |
| WO2021025407A1 (en) * | 2019-08-02 | 2021-02-11 | 웰마커바이오 주식회사 | Oxo-pyridine fusion ring derivative and pharmaceutical composition comprising same |
| US11753395B2 (en) | 2021-12-16 | 2023-09-12 | Kinnate Biopharma Inc. | Inhibitors of MET kinase |
Also Published As
| Publication number | Publication date |
|---|---|
| US20110053931A1 (en) | 2011-03-03 |
| JP2009539878A (en) | 2009-11-19 |
| CA2655128A1 (en) | 2007-12-21 |
| EP2032538A2 (en) | 2009-03-11 |
| WO2007146824A3 (en) | 2008-04-10 |
| CN101528702A (en) | 2009-09-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20110053931A1 (en) | Quinoline compounds and methods of use | |
| US8003662B2 (en) | Heterobicyclic thiophene compounds and methods of use | |
| US7723330B2 (en) | Heterobicyclic pyrazole compounds and methods of use | |
| US20110130406A1 (en) | Pyrazolo-pyridines as tyrosine kinase inhibitors | |
| RU2592703C2 (en) | INHIBITORS OF TYPE ErbB INHIBITORS | |
| JP6026427B2 (en) | Substituted 6,6-fused nitrogen heterocyclic compounds and uses thereof | |
| JP5677945B2 (en) | Diazacarbazole and method of use thereof | |
| US20070049603A1 (en) | Raf inhibitor compounds and methods of use thereof | |
| JP5689069B2 (en) | Pyrazolopyridine PI3K inhibitor compounds and methods of use | |
| EP2440529A1 (en) | Janus kinase inhibitor compounds and methods | |
| JPWO2005080377A1 (en) | Compound having TGFβ inhibitory activity and pharmaceutical composition comprising the same | |
| TW202039490A (en) | Heterobicyclic inhibitors of mat2a and methods of use for treating cancer | |
| EP3319602B1 (en) | Fused quinoline compounds as pi3k/mtor inhibitors | |
| CN104822658A (en) | Fused tricyclic amide compounds as multiple kinase inhibitors | |
| TWI565698B (en) | Quinoxaline compounds, method for preparing the same and use thereof | |
| JP6943886B2 (en) | Condensed pyrimidinopiperidin derivative, and its production method and application |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780029441.2 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07798333 Country of ref document: EP Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009514553 Country of ref document: JP |
|
| ENP | Entry into the national phase |
Ref document number: 2655128 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 5043/KOLNP/2008 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007798333 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: RU |