WO2007144204A1 - Substituted aminopyrazolopyridines and salts thereof, their preparations and pharmaceutical compositions comprising them - Google Patents
Substituted aminopyrazolopyridines and salts thereof, their preparations and pharmaceutical compositions comprising them Download PDFInfo
- Publication number
- WO2007144204A1 WO2007144204A1 PCT/EP2007/005432 EP2007005432W WO2007144204A1 WO 2007144204 A1 WO2007144204 A1 WO 2007144204A1 EP 2007005432 W EP2007005432 W EP 2007005432W WO 2007144204 A1 WO2007144204 A1 WO 2007144204A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- alkyl
- phenyl
- preferably consisting
- heterocycloalkyl
- Prior art date
Links
- 150000003839 salts Chemical class 0.000 title claims abstract description 22
- 239000008194 pharmaceutical composition Substances 0.000 title claims abstract description 16
- 238000002360 preparation method Methods 0.000 title claims description 135
- MJEPPEQNLZSPAP-UHFFFAOYSA-N 1h-pyrazolo[4,3-b]pyridin-3-amine Chemical class C1=CN=C2C(N)=NNC2=C1 MJEPPEQNLZSPAP-UHFFFAOYSA-N 0.000 title description 8
- 150000001875 compounds Chemical class 0.000 claims abstract description 216
- 238000000034 method Methods 0.000 claims abstract description 83
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 82
- 201000010099 disease Diseases 0.000 claims abstract description 81
- 238000011282 treatment Methods 0.000 claims abstract description 32
- 230000006444 vascular growth Effects 0.000 claims abstract description 26
- 238000004519 manufacturing process Methods 0.000 claims abstract description 3
- 239000001257 hydrogen Substances 0.000 claims description 201
- 229910052739 hydrogen Inorganic materials 0.000 claims description 201
- -1 hydroxy, amino Chemical group 0.000 claims description 161
- 150000002431 hydrogen Chemical class 0.000 claims description 137
- 238000006243 chemical reaction Methods 0.000 claims description 118
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 110
- 229910052736 halogen Inorganic materials 0.000 claims description 89
- 150000002367 halogens Chemical class 0.000 claims description 89
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 61
- 125000000592 heterocycloalkyl group Chemical group 0.000 claims description 50
- 239000004202 carbamide Substances 0.000 claims description 40
- 206010028980 Neoplasm Diseases 0.000 claims description 36
- 125000003118 aryl group Chemical group 0.000 claims description 36
- 229910052760 oxygen Inorganic materials 0.000 claims description 31
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 30
- 229910052757 nitrogen Inorganic materials 0.000 claims description 30
- 239000001301 oxygen Substances 0.000 claims description 30
- 230000033115 angiogenesis Effects 0.000 claims description 29
- 125000001072 heteroaryl group Chemical group 0.000 claims description 28
- 125000000843 phenylene group Chemical group C1(=C(C=CC=C1)*)* 0.000 claims description 28
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 27
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 27
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 claims description 26
- 150000001412 amines Chemical class 0.000 claims description 23
- 230000009467 reduction Effects 0.000 claims description 23
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 22
- LPXPTNMVRIOKMN-UHFFFAOYSA-M sodium nitrite Chemical compound [Na+].[O-]N=O LPXPTNMVRIOKMN-UHFFFAOYSA-M 0.000 claims description 22
- OAKJQQAXSVQMHS-UHFFFAOYSA-N hydrazine Substances NN OAKJQQAXSVQMHS-UHFFFAOYSA-N 0.000 claims description 20
- 229910052799 carbon Inorganic materials 0.000 claims description 17
- 239000000460 chlorine Substances 0.000 claims description 17
- 206010030113 Oedema Diseases 0.000 claims description 16
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 claims description 15
- 229910052801 chlorine Inorganic materials 0.000 claims description 14
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 claims description 13
- 125000000217 alkyl group Chemical group 0.000 claims description 13
- 238000005859 coupling reaction Methods 0.000 claims description 13
- 125000004737 (C1-C6) haloalkoxy group Chemical group 0.000 claims description 12
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims description 12
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 claims description 12
- 229910052731 fluorine Inorganic materials 0.000 claims description 11
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 11
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 10
- 125000005549 heteroarylene group Chemical group 0.000 claims description 10
- 150000002429 hydrazines Chemical class 0.000 claims description 10
- 208000027866 inflammatory disease Diseases 0.000 claims description 10
- 208000014674 injury Diseases 0.000 claims description 10
- 125000006239 protecting group Chemical group 0.000 claims description 10
- 230000036573 scar formation Effects 0.000 claims description 10
- 239000001117 sulphuric acid Substances 0.000 claims description 10
- 235000011149 sulphuric acid Nutrition 0.000 claims description 10
- 230000008733 trauma Effects 0.000 claims description 10
- 206010027476 Metastases Diseases 0.000 claims description 9
- 201000004681 Psoriasis Diseases 0.000 claims description 9
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims description 9
- 239000002253 acid Substances 0.000 claims description 9
- 229910052804 chromium Inorganic materials 0.000 claims description 9
- 230000001419 dependent effect Effects 0.000 claims description 9
- 239000012948 isocyanate Substances 0.000 claims description 9
- 150000002513 isocyanates Chemical class 0.000 claims description 9
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims description 8
- 239000005864 Sulphur Substances 0.000 claims description 8
- 125000000732 arylene group Chemical group 0.000 claims description 8
- 125000001153 fluoro group Chemical group F* 0.000 claims description 8
- UCPYLLCMEDAXFR-UHFFFAOYSA-N triphosgene Chemical compound ClC(Cl)(Cl)OC(=O)OC(Cl)(Cl)Cl UCPYLLCMEDAXFR-UHFFFAOYSA-N 0.000 claims description 8
- 206010012442 Dermatitis contact Diseases 0.000 claims description 7
- 206010021143 Hypoxia Diseases 0.000 claims description 7
- 206010053613 Type IV hypersensitivity reaction Diseases 0.000 claims description 7
- 206010064930 age-related macular degeneration Diseases 0.000 claims description 7
- 208000006673 asthma Diseases 0.000 claims description 7
- 208000010247 contact dermatitis Diseases 0.000 claims description 7
- 208000002780 macular degeneration Diseases 0.000 claims description 7
- 201000006417 multiple sclerosis Diseases 0.000 claims description 7
- 208000002815 pulmonary hypertension Diseases 0.000 claims description 7
- 208000037803 restenosis Diseases 0.000 claims description 7
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 7
- 230000005951 type IV hypersensitivity Effects 0.000 claims description 7
- 208000027930 type IV hypersensitivity disease Diseases 0.000 claims description 7
- 125000006584 (C3-C10) heterocycloalkyl group Chemical group 0.000 claims description 6
- 208000003174 Brain Neoplasms Diseases 0.000 claims description 6
- 208000017442 Retinal disease Diseases 0.000 claims description 6
- 206010038923 Retinopathy Diseases 0.000 claims description 6
- 229910020008 S(O) Inorganic materials 0.000 claims description 6
- 206010052779 Transplant rejections Diseases 0.000 claims description 6
- 208000029078 coronary artery disease Diseases 0.000 claims description 6
- 230000007954 hypoxia Effects 0.000 claims description 6
- 208000030613 peripheral artery disease Diseases 0.000 claims description 6
- 230000002062 proliferating effect Effects 0.000 claims description 6
- 230000029663 wound healing Effects 0.000 claims description 6
- 206010001052 Acute respiratory distress syndrome Diseases 0.000 claims description 5
- 206010003445 Ascites Diseases 0.000 claims description 5
- 206010061692 Benign muscle neoplasm Diseases 0.000 claims description 5
- 206010004446 Benign prostatic hyperplasia Diseases 0.000 claims description 5
- 206010048962 Brain oedema Diseases 0.000 claims description 5
- 201000009273 Endometriosis Diseases 0.000 claims description 5
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 claims description 5
- 208000019693 Lung disease Diseases 0.000 claims description 5
- 206010025415 Macular oedema Diseases 0.000 claims description 5
- 201000004458 Myoma Diseases 0.000 claims description 5
- 150000001204 N-oxides Chemical class 0.000 claims description 5
- 206010033266 Ovarian Hyperstimulation Syndrome Diseases 0.000 claims description 5
- 206010055870 Postmenopausal haemorrhage Diseases 0.000 claims description 5
- 206010037423 Pulmonary oedema Diseases 0.000 claims description 5
- 208000013616 Respiratory Distress Syndrome Diseases 0.000 claims description 5
- 208000006011 Stroke Diseases 0.000 claims description 5
- 208000011341 adult acute respiratory distress syndrome Diseases 0.000 claims description 5
- 201000000028 adult respiratory distress syndrome Diseases 0.000 claims description 5
- 229910052794 bromium Inorganic materials 0.000 claims description 5
- 230000001684 chronic effect Effects 0.000 claims description 5
- 210000004087 cornea Anatomy 0.000 claims description 5
- 201000010230 macular retinal edema Diseases 0.000 claims description 5
- 210000005036 nerve Anatomy 0.000 claims description 5
- 201000011461 pre-eclampsia Diseases 0.000 claims description 5
- 230000008929 regeneration Effects 0.000 claims description 5
- 238000011069 regeneration method Methods 0.000 claims description 5
- 208000006386 Bone Resorption Diseases 0.000 claims description 4
- 230000024279 bone resorption Effects 0.000 claims description 4
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 4
- 230000009615 deamination Effects 0.000 claims description 4
- 238000006481 deamination reaction Methods 0.000 claims description 4
- 229910052701 rubidium Inorganic materials 0.000 claims description 4
- YGYAWVDWMABLBF-UHFFFAOYSA-N Phosgene Chemical compound ClC(Cl)=O YGYAWVDWMABLBF-UHFFFAOYSA-N 0.000 claims description 3
- XKJCHHZQLQNZHY-UHFFFAOYSA-N phthalimide Chemical compound C1=CC=C2C(=O)NC(=O)C2=C1 XKJCHHZQLQNZHY-UHFFFAOYSA-N 0.000 claims description 3
- SROYUQLUYMEPGP-UHFFFAOYSA-N 1-[4-(1,6-dimethylpyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-[2-fluoro-3-(trifluoromethyl)phenyl]urea Chemical compound C=12C=NN(C)C2=NC(C)=CC=1C(C=C1)=CC=C1NC(=O)NC1=CC=CC(C(F)(F)F)=C1F SROYUQLUYMEPGP-UHFFFAOYSA-N 0.000 claims description 2
- SDAXQKNDRGNTHA-UHFFFAOYSA-N 1-[4-(1,6-dimethylpyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-[2-fluoro-5-(trifluoromethyl)phenyl]urea Chemical compound C=12C=NN(C)C2=NC(C)=CC=1C(C=C1)=CC=C1NC(=O)NC1=CC(C(F)(F)F)=CC=C1F SDAXQKNDRGNTHA-UHFFFAOYSA-N 0.000 claims description 2
- YSLUKDOGCIIKHC-UHFFFAOYSA-N 1-[4-(1,6-dimethylpyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-[2-pyrrolidin-1-yl-5-(trifluoromethyl)phenyl]urea Chemical compound C=12C=NN(C)C2=NC(C)=CC=1C(C=C1)=CC=C1NC(=O)NC1=CC(C(F)(F)F)=CC=C1N1CCCC1 YSLUKDOGCIIKHC-UHFFFAOYSA-N 0.000 claims description 2
- MWEMKZUFOBTGSZ-UHFFFAOYSA-N 1-[4-(1,6-dimethylpyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound C=12C=NN(C)C2=NC(C)=CC=1C(C=C1)=CC=C1NC(=O)NC1=CC=CC(C(F)(F)F)=C1 MWEMKZUFOBTGSZ-UHFFFAOYSA-N 0.000 claims description 2
- KZRRCZBJCRGRBT-UHFFFAOYSA-N 1-[4-(1,6-dimethylpyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-[3-fluoro-5-(trifluoromethyl)phenyl]urea Chemical compound C=12C=NN(C)C2=NC(C)=CC=1C(C=C1)=CC=C1NC(=O)NC1=CC(F)=CC(C(F)(F)F)=C1 KZRRCZBJCRGRBT-UHFFFAOYSA-N 0.000 claims description 2
- JUNBZVWIJGYHTJ-UHFFFAOYSA-N 1-[4-(1,6-dimethylpyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-[4-fluoro-3-(trifluoromethyl)phenyl]urea Chemical compound C=12C=NN(C)C2=NC(C)=CC=1C(C=C1)=CC=C1NC(=O)NC1=CC=C(F)C(C(F)(F)F)=C1 JUNBZVWIJGYHTJ-UHFFFAOYSA-N 0.000 claims description 2
- XFNWQFGHTRDOQD-UHFFFAOYSA-N 1-[4-(1-methylpyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound C1=CN=C2N(C)N=CC2=C1C(C=C1)=CC=C1NC(=O)NC1=CC=CC(C(F)(F)F)=C1 XFNWQFGHTRDOQD-UHFFFAOYSA-N 0.000 claims description 2
- XGTVOKJZLRYVCR-UHFFFAOYSA-N 1-[4-(6-tert-butyl-1h-pyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-(3-ethylphenyl)urea Chemical compound CCC1=CC=CC(NC(=O)NC=2C=CC(=CC=2)C=2C=3C=NNC=3N=C(C=2)C(C)(C)C)=C1 XGTVOKJZLRYVCR-UHFFFAOYSA-N 0.000 claims description 2
- TWRUMWNCERKOAY-UHFFFAOYSA-N 1-[4-[1-methyl-6-(methylsulfonylmethyl)pyrazolo[3,4-b]pyridin-4-yl]phenyl]-3-[4-(trifluoromethyl)pyridin-2-yl]urea Chemical compound C1=C(CS(C)(=O)=O)N=C2N(C)N=CC2=C1C(C=C1)=CC=C1NC(=O)NC1=CC(C(F)(F)F)=CC=N1 TWRUMWNCERKOAY-UHFFFAOYSA-N 0.000 claims description 2
- PCLDEOPUBVOTMW-UHFFFAOYSA-N 1-[4-[1-methyl-6-(morpholin-4-ylmethyl)pyrazolo[3,4-b]pyridin-4-yl]phenyl]-3-(3-methylphenyl)urea Chemical compound CC1=CC=CC(NC(=O)NC=2C=CC(=CC=2)C=2C=3C=NN(C)C=3N=C(CN3CCOCC3)C=2)=C1 PCLDEOPUBVOTMW-UHFFFAOYSA-N 0.000 claims description 2
- 230000002378 acidificating effect Effects 0.000 claims description 2
- 229940113088 dimethylacetamide Drugs 0.000 claims description 2
- NGYKJTXBWGHOIZ-UHFFFAOYSA-N ethyl 1-[1-methyl-4-[4-[[3-(trifluoromethyl)phenyl]carbamoylamino]phenyl]pyrazolo[3,4-b]pyridin-6-yl]cyclopropane-1-carboxylate Chemical compound C=1C(C=2C=CC(NC(=O)NC=3C=C(C=CC=3)C(F)(F)F)=CC=2)=C2C=NN(C)C2=NC=1C1(C(=O)OCC)CC1 NGYKJTXBWGHOIZ-UHFFFAOYSA-N 0.000 claims description 2
- HXIOTKBQHXRMOX-UHFFFAOYSA-N ethyl 1-[4-[4-(phenylcarbamoylamino)phenyl]-1h-pyrazolo[3,4-b]pyridin-6-yl]cyclopropane-1-carboxylate Chemical compound C=1C(C=2C=CC(NC(=O)NC=3C=CC=CC=3)=CC=2)=C2C=NNC2=NC=1C1(C(=O)OCC)CC1 HXIOTKBQHXRMOX-UHFFFAOYSA-N 0.000 claims description 2
- MUXXBBUGTINDOI-UHFFFAOYSA-N 1-(2-fluoro-5-methylphenyl)-3-[4-(1-methylpyrazolo[3,4-b]pyridin-4-yl)phenyl]urea Chemical compound CC1=CC=C(F)C(NC(=O)NC=2C=CC(=CC=2)C=2C=3C=NN(C)C=3N=CC=2)=C1 MUXXBBUGTINDOI-UHFFFAOYSA-N 0.000 claims 1
- AJVQQDDBJULLHT-UHFFFAOYSA-N 1-(2-fluoro-5-methylphenyl)-3-[4-(1h-pyrazolo[3,4-b]pyridin-4-yl)phenyl]urea Chemical compound CC1=CC=C(F)C(NC(=O)NC=2C=CC(=CC=2)C=2C=3C=NNC=3N=CC=2)=C1 AJVQQDDBJULLHT-UHFFFAOYSA-N 0.000 claims 1
- KKSQNQFHTYAYMO-UHFFFAOYSA-N 1-(2-fluoro-5-methylphenyl)-3-[4-[1-methyl-6-(methylsulfonylmethyl)pyrazolo[3,4-b]pyridin-4-yl]phenyl]urea Chemical compound CC1=CC=C(F)C(NC(=O)NC=2C=CC(=CC=2)C=2C=3C=NN(C)C=3N=C(CS(C)(=O)=O)C=2)=C1 KKSQNQFHTYAYMO-UHFFFAOYSA-N 0.000 claims 1
- YBYWJWSXJXWZGJ-UHFFFAOYSA-N 1-(2-fluoro-5-methylphenyl)-3-[4-[1-methyl-6-(piperidin-1-ylmethyl)pyrazolo[3,4-b]pyridin-4-yl]phenyl]urea Chemical compound CC1=CC=C(F)C(NC(=O)NC=2C=CC(=CC=2)C=2C=3C=NN(C)C=3N=C(CN3CCCCC3)C=2)=C1 YBYWJWSXJXWZGJ-UHFFFAOYSA-N 0.000 claims 1
- GGPSYMHKBXFSEN-UHFFFAOYSA-N 1-(2-fluoro-5-methylphenyl)-3-[4-[1-methyl-6-[(3-oxopiperazin-1-yl)methyl]pyrazolo[3,4-b]pyridin-4-yl]phenyl]urea Chemical compound CC1=CC=C(F)C(NC(=O)NC=2C=CC(=CC=2)C=2C=3C=NN(C)C=3N=C(CN3CC(=O)NCC3)C=2)=C1 GGPSYMHKBXFSEN-UHFFFAOYSA-N 0.000 claims 1
- NHXXPINLNYZVMB-UHFFFAOYSA-N 1-(2-fluoro-5-methylphenyl)-3-[4-[1-methyl-6-[(4-methylpiperazin-1-yl)methyl]pyrazolo[3,4-b]pyridin-4-yl]phenyl]urea Chemical compound C1CN(C)CCN1CC1=CC(C=2C=CC(NC(=O)NC=3C(=CC=C(C)C=3)F)=CC=2)=C(C=NN2C)C2=N1 NHXXPINLNYZVMB-UHFFFAOYSA-N 0.000 claims 1
- BAVHPORQMZFSDD-UHFFFAOYSA-N 1-(2-fluoro-5-methylphenyl)-3-[4-[1-methyl-6-[1-(pyrrolidine-1-carbonyl)cyclopropyl]pyrazolo[3,4-b]pyridin-4-yl]phenyl]urea Chemical compound CC1=CC=C(F)C(NC(=O)NC=2C=CC(=CC=2)C=2C=3C=NN(C)C=3N=C(C=2)C2(CC2)C(=O)N2CCCC2)=C1 BAVHPORQMZFSDD-UHFFFAOYSA-N 0.000 claims 1
- AVGZPQAQXMDFTB-UHFFFAOYSA-N 1-(2-fluoro-5-methylphenyl)-3-[4-[6-(methoxymethyl)-1-methylpyrazolo[3,4-b]pyridin-4-yl]phenyl]urea Chemical compound C=12C=NN(C)C2=NC(COC)=CC=1C(C=C1)=CC=C1NC(=O)NC1=CC(C)=CC=C1F AVGZPQAQXMDFTB-UHFFFAOYSA-N 0.000 claims 1
- IPUYRXHKXNKBPD-UHFFFAOYSA-N 1-(2-fluoro-5-methylphenyl)-3-[4-[6-[(4-hydroxypiperidin-1-yl)methyl]-1-methylpyrazolo[3,4-b]pyridin-4-yl]phenyl]urea Chemical compound CC1=CC=C(F)C(NC(=O)NC=2C=CC(=CC=2)C=2C=3C=NN(C)C=3N=C(CN3CCC(O)CC3)C=2)=C1 IPUYRXHKXNKBPD-UHFFFAOYSA-N 0.000 claims 1
- NIAAGVAFAREREG-UHFFFAOYSA-N 1-[2-fluoro-5-(trifluoromethyl)phenyl]-3-[4-(1-methylpyrazolo[3,4-b]pyridin-4-yl)phenyl]urea Chemical compound C1=CN=C2N(C)N=CC2=C1C(C=C1)=CC=C1NC(=O)NC1=CC(C(F)(F)F)=CC=C1F NIAAGVAFAREREG-UHFFFAOYSA-N 0.000 claims 1
- VABXSELRSOOYNM-UHFFFAOYSA-N 1-[2-fluoro-5-(trifluoromethyl)phenyl]-3-[4-(1h-pyrazolo[3,4-b]pyridin-4-yl)phenyl]urea Chemical compound FC1=CC=C(C(F)(F)F)C=C1NC(=O)NC1=CC=C(C=2C=3C=NNC=3N=CC=2)C=C1 VABXSELRSOOYNM-UHFFFAOYSA-N 0.000 claims 1
- XTKQSZLRPSAAHE-UHFFFAOYSA-N 1-[3-methyl-4-[(4-methylpiperazin-1-yl)methyl]phenyl]-3-[4-(1-methylpyrazolo[3,4-b]pyridin-4-yl)phenyl]urea Chemical compound C1CN(C)CCN1CC(C(=C1)C)=CC=C1NC(=O)NC1=CC=C(C=2C=3C=NN(C)C=3N=CC=2)C=C1 XTKQSZLRPSAAHE-UHFFFAOYSA-N 0.000 claims 1
- YNTJLHUPRRHFKN-UHFFFAOYSA-N 1-[3-methyl-4-[(4-methylpiperazin-1-yl)methyl]phenyl]-3-[4-(1h-pyrazolo[3,4-b]pyridin-4-yl)phenyl]urea Chemical compound C1CN(C)CCN1CC(C(=C1)C)=CC=C1NC(=O)NC1=CC=C(C=2C=3C=NNC=3N=CC=2)C=C1 YNTJLHUPRRHFKN-UHFFFAOYSA-N 0.000 claims 1
- SNFMRIYKMBOUMP-UHFFFAOYSA-N 1-[4-(1-methylpiperidin-4-yl)oxy-3-(trifluoromethyl)phenyl]-3-[4-(1-methylpyrazolo[3,4-b]pyridin-4-yl)phenyl]urea Chemical compound C1CN(C)CCC1OC(C(=C1)C(F)(F)F)=CC=C1NC(=O)NC1=CC=C(C=2C=3C=NN(C)C=3N=CC=2)C=C1 SNFMRIYKMBOUMP-UHFFFAOYSA-N 0.000 claims 1
- VCBBDYQEYOVQBP-UHFFFAOYSA-N 1-[4-(1-methylpyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-phenylurea Chemical compound C1=CN=C2N(C)N=CC2=C1C(C=C1)=CC=C1NC(=O)NC1=CC=CC=C1 VCBBDYQEYOVQBP-UHFFFAOYSA-N 0.000 claims 1
- GCOCXYJUJVARQQ-UHFFFAOYSA-N 1-[4-(1h-pyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound FC(F)(F)C1=CC=CC(NC(=O)NC=2C=CC(=CC=2)C=2C=3C=NNC=3N=CC=2)=C1 GCOCXYJUJVARQQ-UHFFFAOYSA-N 0.000 claims 1
- UBXWQHJFLDNFNR-UHFFFAOYSA-N 1-[4-(6-cyclobutyl-1-methylpyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound N1=C2N(C)N=CC2=C(C=2C=CC(NC(=O)NC=3C=C(C=CC=3)C(F)(F)F)=CC=2)C=C1C1CCC1 UBXWQHJFLDNFNR-UHFFFAOYSA-N 0.000 claims 1
- QMUABAXRSAMJOL-UHFFFAOYSA-N 1-[4-(6-cyclohexyl-1-methylpyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound N1=C2N(C)N=CC2=C(C=2C=CC(NC(=O)NC=3C=C(C=CC=3)C(F)(F)F)=CC=2)C=C1C1CCCCC1 QMUABAXRSAMJOL-UHFFFAOYSA-N 0.000 claims 1
- HRJAFLDQXNTASV-UHFFFAOYSA-N 1-[4-(6-cyclopropyl-1-methylpyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound N1=C2N(C)N=CC2=C(C=2C=CC(NC(=O)NC=3C=C(C=CC=3)C(F)(F)F)=CC=2)C=C1C1CC1 HRJAFLDQXNTASV-UHFFFAOYSA-N 0.000 claims 1
- BUGQEVDWZBCYNJ-UHFFFAOYSA-N 1-[4-(6-cyclopropyl-1h-pyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound FC(F)(F)C1=CC=CC(NC(=O)NC=2C=CC(=CC=2)C=2C=3C=NNC=3N=C(C=2)C2CC2)=C1 BUGQEVDWZBCYNJ-UHFFFAOYSA-N 0.000 claims 1
- GHUZFSHGCIUHOO-UHFFFAOYSA-N 1-[4-(6-methyl-1h-pyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound C=12C=NNC2=NC(C)=CC=1C(C=C1)=CC=C1NC(=O)NC1=CC=CC(C(F)(F)F)=C1 GHUZFSHGCIUHOO-UHFFFAOYSA-N 0.000 claims 1
- LRHUSNHWNZSIBT-UHFFFAOYSA-N 1-[4-(6-tert-butyl-1-methylpyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-[2-[2-(4-methylpiperazin-1-yl)ethoxy]-5-(trifluoromethyl)phenyl]urea Chemical compound C1CN(C)CCN1CCOC1=CC=C(C(F)(F)F)C=C1NC(=O)NC1=CC=C(C=2C=3C=NN(C)C=3N=C(C=2)C(C)(C)C)C=C1 LRHUSNHWNZSIBT-UHFFFAOYSA-N 0.000 claims 1
- OTNNSSVSHLAAFI-UHFFFAOYSA-N 1-[4-(6-tert-butyl-1-methylpyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-[2-[2-(dimethylamino)ethoxy]-5-(trifluoromethyl)phenyl]urea Chemical compound CN(C)CCOC1=CC=C(C(F)(F)F)C=C1NC(=O)NC1=CC=C(C=2C=3C=NN(C)C=3N=C(C=2)C(C)(C)C)C=C1 OTNNSSVSHLAAFI-UHFFFAOYSA-N 0.000 claims 1
- FDNGYLCQEAECIB-UHFFFAOYSA-N 1-[4-(6-tert-butyl-1h-pyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-(2-methylphenyl)urea Chemical compound CC1=CC=CC=C1NC(=O)NC1=CC=C(C=2C=3C=NNC=3N=C(C=2)C(C)(C)C)C=C1 FDNGYLCQEAECIB-UHFFFAOYSA-N 0.000 claims 1
- CUCAMFXGWUKVLW-UHFFFAOYSA-N 1-[4-(6-tert-butyl-1h-pyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-(3,4-difluorophenyl)urea Chemical compound C=12C=NNC2=NC(C(C)(C)C)=CC=1C(C=C1)=CC=C1NC(=O)NC1=CC=C(F)C(F)=C1 CUCAMFXGWUKVLW-UHFFFAOYSA-N 0.000 claims 1
- HZTHQFPFMMNUOX-UHFFFAOYSA-N 1-[4-(6-tert-butyl-1h-pyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-(3-fluorophenyl)urea Chemical compound C=12C=NNC2=NC(C(C)(C)C)=CC=1C(C=C1)=CC=C1NC(=O)NC1=CC=CC(F)=C1 HZTHQFPFMMNUOX-UHFFFAOYSA-N 0.000 claims 1
- NKLXPIJJUOHMQD-UHFFFAOYSA-N 1-[4-(6-tert-butyl-1h-pyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-(3-methylphenyl)urea Chemical compound CC1=CC=CC(NC(=O)NC=2C=CC(=CC=2)C=2C=3C=NNC=3N=C(C=2)C(C)(C)C)=C1 NKLXPIJJUOHMQD-UHFFFAOYSA-N 0.000 claims 1
- QKEUOHYHRLHRGG-UHFFFAOYSA-N 1-[4-(6-tert-butyl-1h-pyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-(4-fluorophenyl)urea Chemical compound C=12C=NNC2=NC(C(C)(C)C)=CC=1C(C=C1)=CC=C1NC(=O)NC1=CC=C(F)C=C1 QKEUOHYHRLHRGG-UHFFFAOYSA-N 0.000 claims 1
- ZFMCEHCTDKYEPP-UHFFFAOYSA-N 1-[4-(6-tert-butyl-1h-pyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-[4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl]urea Chemical compound C1CN(C)CCN1CC(C(=C1)C(F)(F)F)=CC=C1NC(=O)NC1=CC=C(C=2C=3C=NNC=3N=C(C=2)C(C)(C)C)C=C1 ZFMCEHCTDKYEPP-UHFFFAOYSA-N 0.000 claims 1
- TWZPLBQPJDPWOG-UHFFFAOYSA-N 1-[4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl]-3-[4-(1-methylpyrazolo[3,4-b]pyridin-4-yl)phenyl]urea Chemical compound C1CN(C)CCN1CC(C(=C1)C(F)(F)F)=CC=C1NC(=O)NC1=CC=C(C=2C=3C=NN(C)C=3N=CC=2)C=C1 TWZPLBQPJDPWOG-UHFFFAOYSA-N 0.000 claims 1
- RQMUPHYTMHJIGB-UHFFFAOYSA-N 1-[4-[(4-methylpiperazin-1-yl)methyl]-3-(trifluoromethyl)phenyl]-3-[4-(1h-pyrazolo[3,4-b]pyridin-4-yl)phenyl]urea Chemical compound C1CN(C)CCN1CC(C(=C1)C(F)(F)F)=CC=C1NC(=O)NC1=CC=C(C=2C=3C=NNC=3N=CC=2)C=C1 RQMUPHYTMHJIGB-UHFFFAOYSA-N 0.000 claims 1
- QEBOTOXTUXYOLV-UHFFFAOYSA-N 1-[4-[1-methyl-6-(1,3-thiazol-2-yl)pyrazolo[3,4-b]pyridin-4-yl]phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound N1=C2N(C)N=CC2=C(C=2C=CC(NC(=O)NC=3C=C(C=CC=3)C(F)(F)F)=CC=2)C=C1C1=NC=CS1 QEBOTOXTUXYOLV-UHFFFAOYSA-N 0.000 claims 1
- QAJNZYMGNPARBR-UHFFFAOYSA-N 1-[4-[1-methyl-6-(2-phenylcyclopropyl)pyrazolo[3,4-b]pyridin-4-yl]phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound N1=C2N(C)N=CC2=C(C=2C=CC(NC(=O)NC=3C=C(C=CC=3)C(F)(F)F)=CC=2)C=C1C1CC1C1=CC=CC=C1 QAJNZYMGNPARBR-UHFFFAOYSA-N 0.000 claims 1
- BSNZWVMUAACBRR-UHFFFAOYSA-N 1-[4-[1-methyl-6-(methylsulfonylmethyl)pyrazolo[3,4-b]pyridin-4-yl]phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound C1=C(CS(C)(=O)=O)N=C2N(C)N=CC2=C1C(C=C1)=CC=C1NC(=O)NC1=CC=CC(C(F)(F)F)=C1 BSNZWVMUAACBRR-UHFFFAOYSA-N 0.000 claims 1
- WRRRCWADPATFAX-UHFFFAOYSA-N 1-[4-[1-methyl-6-[(3-oxopiperazin-1-yl)methyl]pyrazolo[3,4-b]pyridin-4-yl]phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound N1=C2N(C)N=CC2=C(C=2C=CC(NC(=O)NC=3C=C(C=CC=3)C(F)(F)F)=CC=2)C=C1CN1CCNC(=O)C1 WRRRCWADPATFAX-UHFFFAOYSA-N 0.000 claims 1
- RAFVVENBFDAWAI-UHFFFAOYSA-N 1-[4-[1-methyl-6-[(4-methylpiperazin-1-yl)methyl]pyrazolo[3,4-b]pyridin-4-yl]phenyl]-1-[3-(trifluoromethyl)phenyl]urea Chemical compound C1CN(C)CCN1CC1=CC(C=2C=CC(=CC=2)N(C(N)=O)C=2C=C(C=CC=2)C(F)(F)F)=C(C=NN2C)C2=N1 RAFVVENBFDAWAI-UHFFFAOYSA-N 0.000 claims 1
- QXBQIOODDHYQPM-UHFFFAOYSA-N 1-[4-[1-methyl-6-[(4-methylpiperazin-1-yl)methyl]pyrazolo[3,4-b]pyridin-4-yl]phenyl]-3-[4-(trifluoromethyl)pyridin-2-yl]urea Chemical compound C1CN(C)CCN1CC1=CC(C=2C=CC(NC(=O)NC=3N=CC=C(C=3)C(F)(F)F)=CC=2)=C(C=NN2C)C2=N1 QXBQIOODDHYQPM-UHFFFAOYSA-N 0.000 claims 1
- OBISVPJLRUMYSZ-UHFFFAOYSA-N 1-[4-[4-[(2-fluoro-5-methylphenyl)carbamoylamino]phenyl]-1-methylpyrazolo[3,4-b]pyridin-6-yl]-n,n-dimethylcyclopropane-1-carboxamide Chemical compound C=1C(C=2C=CC(NC(=O)NC=3C(=CC=C(C)C=3)F)=CC=2)=C2C=NN(C)C2=NC=1C1(C(=O)N(C)C)CC1 OBISVPJLRUMYSZ-UHFFFAOYSA-N 0.000 claims 1
- ANZNZKXMKQPXJC-UHFFFAOYSA-N 1-[4-[6-(methoxymethyl)-1-methylpyrazolo[3,4-b]pyridin-4-yl]phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound C=12C=NN(C)C2=NC(COC)=CC=1C(C=C1)=CC=C1NC(=O)NC1=CC=CC(C(F)(F)F)=C1 ANZNZKXMKQPXJC-UHFFFAOYSA-N 0.000 claims 1
- VCRHZIXCSHTRAA-UHFFFAOYSA-N 1-[4-[6-(methoxymethyl)-1-methylpyrazolo[3,4-b]pyridin-4-yl]phenyl]-3-[4-(trifluoromethyl)pyridin-2-yl]urea Chemical compound C=12C=NN(C)C2=NC(COC)=CC=1C(C=C1)=CC=C1NC(=O)NC1=CC(C(F)(F)F)=CC=N1 VCRHZIXCSHTRAA-UHFFFAOYSA-N 0.000 claims 1
- QFRZTVAXGJBUJA-UHFFFAOYSA-N 1-[4-[6-[(2,6-dimethylmorpholin-4-yl)methyl]-1-methylpyrazolo[3,4-b]pyridin-4-yl]phenyl]-3-(2-fluoro-5-methylphenyl)urea Chemical compound C1C(C)OC(C)CN1CC1=CC(C=2C=CC(NC(=O)NC=3C(=CC=C(C)C=3)F)=CC=2)=C(C=NN2C)C2=N1 QFRZTVAXGJBUJA-UHFFFAOYSA-N 0.000 claims 1
- BFMIVRGLXXLCDL-UHFFFAOYSA-N 1-[4-[6-[(2,6-dimethylmorpholin-4-yl)methyl]-1-methylpyrazolo[3,4-b]pyridin-4-yl]phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound C1C(C)OC(C)CN1CC1=CC(C=2C=CC(NC(=O)NC=3C=C(C=CC=3)C(F)(F)F)=CC=2)=C(C=NN2C)C2=N1 BFMIVRGLXXLCDL-UHFFFAOYSA-N 0.000 claims 1
- HDRLETXJEFXDRK-UHFFFAOYSA-N 1-[4-[6-[(4-hydroxypiperidin-1-yl)methyl]-1-methylpyrazolo[3,4-b]pyridin-4-yl]phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound N1=C2N(C)N=CC2=C(C=2C=CC(NC(=O)NC=3C=C(C=CC=3)C(F)(F)F)=CC=2)C=C1CN1CCC(O)CC1 HDRLETXJEFXDRK-UHFFFAOYSA-N 0.000 claims 1
- IITAVPXNKRUHIU-UHFFFAOYSA-N 1-[4-[6-[(dimethylamino)methyl]-1-methylpyrazolo[3,4-b]pyridin-4-yl]phenyl]-3-(2-fluoro-5-methylphenyl)urea Chemical compound C=12C=NN(C)C2=NC(CN(C)C)=CC=1C(C=C1)=CC=C1NC(=O)NC1=CC(C)=CC=C1F IITAVPXNKRUHIU-UHFFFAOYSA-N 0.000 claims 1
- RRHHMWFBKZHPMG-UHFFFAOYSA-N 1-[4-[6-[(dimethylamino)methyl]-1-methylpyrazolo[3,4-b]pyridin-4-yl]phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound C=12C=NN(C)C2=NC(CN(C)C)=CC=1C(C=C1)=CC=C1NC(=O)NC1=CC=CC(C(F)(F)F)=C1 RRHHMWFBKZHPMG-UHFFFAOYSA-N 0.000 claims 1
- ALRGJNXAQUPDCS-UHFFFAOYSA-N 1-[4-[6-[(dimethylamino)methyl]-1-methylpyrazolo[3,4-b]pyridin-4-yl]phenyl]-3-[4-(trifluoromethyl)pyridin-2-yl]urea Chemical compound C=12C=NN(C)C2=NC(CN(C)C)=CC=1C(C=C1)=CC=C1NC(=O)NC1=CC(C(F)(F)F)=CC=N1 ALRGJNXAQUPDCS-UHFFFAOYSA-N 0.000 claims 1
- YIOBPRAFJVROGN-UHFFFAOYSA-N 1-[4-[6-[[3-(dimethylamino)pyrrolidin-1-yl]methyl]-1-methylpyrazolo[3,4-b]pyridin-4-yl]phenyl]-3-(2-fluoro-5-methylphenyl)urea Chemical compound C1C(N(C)C)CCN1CC1=CC(C=2C=CC(NC(=O)NC=3C(=CC=C(C)C=3)F)=CC=2)=C(C=NN2C)C2=N1 YIOBPRAFJVROGN-UHFFFAOYSA-N 0.000 claims 1
- MMKHBLURYUEIJR-UHFFFAOYSA-N 1-phenyl-3-[4-(1h-pyrazolo[3,4-b]pyridin-4-yl)phenyl]urea Chemical compound C=1C=C(C=2C=3C=NNC=3N=CC=2)C=CC=1NC(=O)NC1=CC=CC=C1 MMKHBLURYUEIJR-UHFFFAOYSA-N 0.000 claims 1
- ASTAZOPDKJAWKA-UHFFFAOYSA-N 4-(6-methyl-1H-pyrazolo[3,4-b]pyridin-4-yl)aniline Chemical compound C=12C=NNC2=NC(C)=CC=1C1=CC=C(N)C=C1 ASTAZOPDKJAWKA-UHFFFAOYSA-N 0.000 claims 1
- 210000000988 bone and bone Anatomy 0.000 claims 1
- 239000003085 diluting agent Substances 0.000 claims 1
- MPRJUIORYHVVKA-UHFFFAOYSA-N n,n,2-trimethyl-2-[1-methyl-4-[4-[[3-(trifluoromethyl)phenyl]carbamoylamino]phenyl]pyrazolo[3,4-b]pyridin-6-yl]propanamide Chemical compound C=12C=NN(C)C2=NC(C(C)(C)C(=O)N(C)C)=CC=1C(C=C1)=CC=C1NC(=O)NC1=CC=CC(C(F)(F)F)=C1 MPRJUIORYHVVKA-UHFFFAOYSA-N 0.000 claims 1
- RMKMHVYKTDEDPU-UHFFFAOYSA-N n,n-diethyl-1-[1-methyl-4-[4-[[3-(trifluoromethyl)phenyl]carbamoylamino]phenyl]pyrazolo[3,4-b]pyridin-6-yl]cyclopropane-1-carboxamide Chemical compound C=1C(C=2C=CC(NC(=O)NC=3C=C(C=CC=3)C(F)(F)F)=CC=2)=C2C=NN(C)C2=NC=1C1(C(=O)N(CC)CC)CC1 RMKMHVYKTDEDPU-UHFFFAOYSA-N 0.000 claims 1
- SRLUMDYACGTJMZ-UHFFFAOYSA-N n-cyclopropyl-1-[4-[4-[(2-fluoro-5-methylphenyl)carbamoylamino]phenyl]-1-methylpyrazolo[3,4-b]pyridin-6-yl]cyclopropane-1-carboxamide Chemical compound CC1=CC=C(F)C(NC(=O)NC=2C=CC(=CC=2)C=2C=3C=NN(C)C=3N=C(C=2)C2(CC2)C(=O)NC2CC2)=C1 SRLUMDYACGTJMZ-UHFFFAOYSA-N 0.000 claims 1
- IQXPFERGRWPTCF-UHFFFAOYSA-N n-cyclopropyl-2-methyl-2-[1-methyl-4-[4-[[3-(trifluoromethyl)phenyl]carbamoylamino]phenyl]pyrazolo[3,4-b]pyridin-6-yl]propanamide Chemical compound N1=C2N(C)N=CC2=C(C=2C=CC(NC(=O)NC=3C=C(C=CC=3)C(F)(F)F)=CC=2)C=C1C(C)(C)C(=O)NC1CC1 IQXPFERGRWPTCF-UHFFFAOYSA-N 0.000 claims 1
- 101100481408 Danio rerio tie2 gene Proteins 0.000 abstract description 76
- 101100481410 Mus musculus Tek gene Proteins 0.000 abstract description 76
- 150000005229 pyrazolopyridines Chemical class 0.000 abstract description 20
- 230000011664 signaling Effects 0.000 abstract description 17
- 239000000543 intermediate Substances 0.000 description 233
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 116
- 238000005160 1H NMR spectroscopy Methods 0.000 description 110
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 85
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 78
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 68
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 50
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N DMSO Substances CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 49
- 238000000746 purification Methods 0.000 description 47
- 150000002148 esters Chemical class 0.000 description 46
- 239000000047 product Substances 0.000 description 46
- 235000013877 carbamide Nutrition 0.000 description 40
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 39
- PAQZWJGSJMLPMG-UHFFFAOYSA-N 2,4,6-tripropyl-1,3,5,2$l^{5},4$l^{5},6$l^{5}-trioxatriphosphinane 2,4,6-trioxide Chemical compound CCCP1(=O)OP(=O)(CCC)OP(=O)(CCC)O1 PAQZWJGSJMLPMG-UHFFFAOYSA-N 0.000 description 38
- 238000003818 flash chromatography Methods 0.000 description 36
- 239000000203 mixture Substances 0.000 description 35
- 102100034594 Angiopoietin-1 Human genes 0.000 description 34
- 101000924552 Homo sapiens Angiopoietin-1 Proteins 0.000 description 34
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 32
- 239000000243 solution Substances 0.000 description 32
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 32
- AMFYRKOUWBAGHV-UHFFFAOYSA-N 1h-pyrazolo[4,3-b]pyridine Chemical compound C1=CN=C2C=NNC2=C1 AMFYRKOUWBAGHV-UHFFFAOYSA-N 0.000 description 28
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 28
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 28
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 27
- 239000002904 solvent Substances 0.000 description 27
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 26
- 125000004432 carbon atom Chemical group C* 0.000 description 26
- 102100034608 Angiopoietin-2 Human genes 0.000 description 25
- 108091000080 Phosphotransferase Proteins 0.000 description 25
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 25
- 102000020233 phosphotransferase Human genes 0.000 description 25
- 101000924533 Homo sapiens Angiopoietin-2 Proteins 0.000 description 24
- 230000000694 effects Effects 0.000 description 23
- 239000003112 inhibitor Substances 0.000 description 23
- 239000011541 reaction mixture Substances 0.000 description 23
- 230000015572 biosynthetic process Effects 0.000 description 21
- 210000004027 cell Anatomy 0.000 description 21
- 238000003556 assay Methods 0.000 description 19
- 230000005764 inhibitory process Effects 0.000 description 19
- 210000002889 endothelial cell Anatomy 0.000 description 18
- HDZGCSFEDULWCS-UHFFFAOYSA-N monomethylhydrazine Chemical compound CNN HDZGCSFEDULWCS-UHFFFAOYSA-N 0.000 description 18
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 16
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 16
- 125000005842 heteroatom Chemical group 0.000 description 16
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 16
- 238000007363 ring formation reaction Methods 0.000 description 16
- 238000010626 work up procedure Methods 0.000 description 16
- 239000012044 organic layer Substances 0.000 description 15
- 239000000758 substrate Substances 0.000 description 15
- 238000009835 boiling Methods 0.000 description 14
- 238000002953 preparative HPLC Methods 0.000 description 14
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 14
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 13
- 230000014509 gene expression Effects 0.000 description 13
- 239000007787 solid Substances 0.000 description 13
- 239000005695 Ammonium acetate Substances 0.000 description 12
- 230000004913 activation Effects 0.000 description 12
- 235000019257 ammonium acetate Nutrition 0.000 description 12
- 229940043376 ammonium acetate Drugs 0.000 description 12
- 239000013522 chelant Substances 0.000 description 12
- 235000019439 ethyl acetate Nutrition 0.000 description 12
- 239000003446 ligand Substances 0.000 description 12
- 230000008569 process Effects 0.000 description 12
- 108090000765 processed proteins & peptides Proteins 0.000 description 12
- 230000009466 transformation Effects 0.000 description 12
- SINQIEAULQKUPD-UHFFFAOYSA-N 4-[4-(6-methoxy-2-naphthalenyl)-2-(4-methylsulfinylphenyl)-1H-imidazol-5-yl]pyridine Chemical compound C1=CC2=CC(OC)=CC=C2C=C1C=1N=C(C=2C=CC(=CC=2)S(C)=O)NC=1C1=CC=NC=C1 SINQIEAULQKUPD-UHFFFAOYSA-N 0.000 description 11
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 11
- 150000001408 amides Chemical class 0.000 description 11
- 230000001413 cellular effect Effects 0.000 description 11
- 230000008878 coupling Effects 0.000 description 10
- 238000010168 coupling process Methods 0.000 description 10
- 238000002868 homogeneous time resolved fluorescence Methods 0.000 description 10
- 239000002244 precipitate Substances 0.000 description 10
- 238000003756 stirring Methods 0.000 description 10
- 210000005166 vasculature Anatomy 0.000 description 10
- BXRFQSNOROATLV-UHFFFAOYSA-N 4-nitrobenzaldehyde Chemical compound [O-][N+](=O)C1=CC=C(C=O)C=C1 BXRFQSNOROATLV-UHFFFAOYSA-N 0.000 description 9
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 9
- ZKHQWZAMYRWXGA-KQYNXXCUSA-N Adenosine triphosphate Chemical compound C1=NC=2C(N)=NC=NC=2N1[C@@H]1O[C@H](COP(O)(=O)OP(O)(=O)OP(O)(O)=O)[C@@H](O)[C@H]1O ZKHQWZAMYRWXGA-KQYNXXCUSA-N 0.000 description 9
- ZKHQWZAMYRWXGA-UHFFFAOYSA-N Adenosine triphosphate Natural products C1=NC=2C(N)=NC=NC=2N1C1OC(COP(O)(=O)OP(O)(=O)OP(O)(O)=O)C(O)C1O ZKHQWZAMYRWXGA-UHFFFAOYSA-N 0.000 description 9
- 229960001456 adenosine triphosphate Drugs 0.000 description 9
- 239000002585 base Substances 0.000 description 9
- 201000011510 cancer Diseases 0.000 description 9
- 125000000753 cycloalkyl group Chemical group 0.000 description 9
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 9
- BDERNNFJNOPAEC-UHFFFAOYSA-N propan-1-ol Chemical compound CCCO BDERNNFJNOPAEC-UHFFFAOYSA-N 0.000 description 9
- COLRMVLTWJTLFJ-UHFFFAOYSA-N pyridin-2-yl trifluoromethanesulfonate Chemical class FC(F)(F)S(=O)(=O)OC1=CC=CC=N1 COLRMVLTWJTLFJ-UHFFFAOYSA-N 0.000 description 9
- 150000003384 small molecules Chemical class 0.000 description 9
- 235000010288 sodium nitrite Nutrition 0.000 description 9
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 8
- ZIUSEGSNTOUIPT-UHFFFAOYSA-N ethyl 2-cyanoacetate Chemical compound CCOC(=O)CC#N ZIUSEGSNTOUIPT-UHFFFAOYSA-N 0.000 description 8
- 102000005962 receptors Human genes 0.000 description 8
- 108020003175 receptors Proteins 0.000 description 8
- 238000001953 recrystallisation Methods 0.000 description 8
- 238000010992 reflux Methods 0.000 description 8
- 229940124530 sulfonamide Drugs 0.000 description 8
- 210000001519 tissue Anatomy 0.000 description 8
- WJKHJLXJJJATHN-UHFFFAOYSA-N triflic anhydride Chemical compound FC(F)(F)S(=O)(=O)OS(=O)(=O)C(F)(F)F WJKHJLXJJJATHN-UHFFFAOYSA-N 0.000 description 8
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 7
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 7
- 102000003746 Insulin Receptor Human genes 0.000 description 7
- 108010001127 Insulin Receptor Proteins 0.000 description 7
- 108010073929 Vascular Endothelial Growth Factor A Proteins 0.000 description 7
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 7
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 7
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 7
- 238000005804 alkylation reaction Methods 0.000 description 7
- 150000001448 anilines Chemical class 0.000 description 7
- 238000001514 detection method Methods 0.000 description 7
- UQLDLKMNUJERMK-UHFFFAOYSA-L di(octadecanoyloxy)lead Chemical compound [Pb+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O UQLDLKMNUJERMK-UHFFFAOYSA-L 0.000 description 7
- NSNHWTBQMQIDCF-UHFFFAOYSA-N dihydrate;hydrochloride Chemical compound O.O.Cl NSNHWTBQMQIDCF-UHFFFAOYSA-N 0.000 description 7
- 239000012039 electrophile Substances 0.000 description 7
- 238000001727 in vivo Methods 0.000 description 7
- 230000002401 inhibitory effect Effects 0.000 description 7
- 230000001404 mediated effect Effects 0.000 description 7
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 description 7
- 150000003456 sulfonamides Chemical class 0.000 description 7
- 238000000844 transformation Methods 0.000 description 7
- SXJYSIBLFGQAND-UHFFFAOYSA-N 1-isocyanato-3-(trifluoromethyl)benzene Chemical compound FC(F)(F)C1=CC=CC(N=C=O)=C1 SXJYSIBLFGQAND-UHFFFAOYSA-N 0.000 description 6
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 6
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 6
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 6
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 6
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 6
- 241000699670 Mus sp. Species 0.000 description 6
- 206010029113 Neovascularisation Diseases 0.000 description 6
- 108010090091 TIE-2 Receptor Proteins 0.000 description 6
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 6
- 238000010640 amide synthesis reaction Methods 0.000 description 6
- 238000011161 development Methods 0.000 description 6
- 230000018109 developmental process Effects 0.000 description 6
- 230000006870 function Effects 0.000 description 6
- 239000010410 layer Substances 0.000 description 6
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 6
- LVWZTYCIRDMTEY-UHFFFAOYSA-N metamizole Chemical compound O=C1C(N(CS(O)(=O)=O)C)=C(C)N(C)N1C1=CC=CC=C1 LVWZTYCIRDMTEY-UHFFFAOYSA-N 0.000 description 6
- 150000002825 nitriles Chemical class 0.000 description 6
- 125000001424 substituent group Chemical group 0.000 description 6
- 239000003826 tablet Substances 0.000 description 6
- 150000003672 ureas Chemical class 0.000 description 6
- JVVRJMXHNUAPHW-UHFFFAOYSA-N 1h-pyrazol-5-amine Chemical class NC=1C=CNN=1 JVVRJMXHNUAPHW-UHFFFAOYSA-N 0.000 description 5
- 125000004939 6-pyridyl group Chemical group N1=CC=CC=C1* 0.000 description 5
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 5
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 5
- 102000004190 Enzymes Human genes 0.000 description 5
- 108090000790 Enzymes Proteins 0.000 description 5
- 102000012753 TIE-2 Receptor Human genes 0.000 description 5
- 230000029936 alkylation Effects 0.000 description 5
- 125000004429 atom Chemical group 0.000 description 5
- 239000000872 buffer Substances 0.000 description 5
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Natural products OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 5
- SIPUZPBQZHNSDW-UHFFFAOYSA-N diisobutylaluminium hydride Substances CC(C)C[Al]CC(C)C SIPUZPBQZHNSDW-UHFFFAOYSA-N 0.000 description 5
- 239000003814 drug Substances 0.000 description 5
- 230000012010 growth Effects 0.000 description 5
- 239000012442 inert solvent Substances 0.000 description 5
- 230000003993 interaction Effects 0.000 description 5
- 238000005259 measurement Methods 0.000 description 5
- 230000004048 modification Effects 0.000 description 5
- 238000012986 modification Methods 0.000 description 5
- PSHKMPUSSFXUIA-UHFFFAOYSA-N n,n-dimethylpyridin-2-amine Chemical compound CN(C)C1=CC=CC=N1 PSHKMPUSSFXUIA-UHFFFAOYSA-N 0.000 description 5
- AICOOMRHRUFYCM-ZRRPKQBOSA-N oxazine, 1 Chemical compound C([C@@H]1[C@H](C(C[C@]2(C)[C@@H]([C@H](C)N(C)C)[C@H](O)C[C@]21C)=O)CC1=CC2)C[C@H]1[C@@]1(C)[C@H]2N=C(C(C)C)OC1 AICOOMRHRUFYCM-ZRRPKQBOSA-N 0.000 description 5
- 239000000725 suspension Substances 0.000 description 5
- 230000004614 tumor growth Effects 0.000 description 5
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 4
- XBPVIJSNIQYEST-UHFFFAOYSA-N 6-(bromomethyl)-1-methyl-4-(4-nitrophenyl)pyrazolo[3,4-b]pyridine Chemical compound C1=C(CBr)N=C2N(C)N=CC2=C1C1=CC=C([N+]([O-])=O)C=C1 XBPVIJSNIQYEST-UHFFFAOYSA-N 0.000 description 4
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 4
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 4
- 229910021380 Manganese Chloride Inorganic materials 0.000 description 4
- GLFNIEUTAYBVOC-UHFFFAOYSA-L Manganese chloride Chemical compound Cl[Mn]Cl GLFNIEUTAYBVOC-UHFFFAOYSA-L 0.000 description 4
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 4
- 102000038030 PI3Ks Human genes 0.000 description 4
- 108091007960 PI3Ks Proteins 0.000 description 4
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 description 4
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 4
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 4
- 125000003277 amino group Chemical group 0.000 description 4
- 239000012131 assay buffer Substances 0.000 description 4
- 125000005620 boronic acid group Chemical class 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 238000003776 cleavage reaction Methods 0.000 description 4
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 4
- VHJLVAABSRFDPM-QWWZWVQMSA-N dithiothreitol Chemical compound SC[C@@H](O)[C@H](O)CS VHJLVAABSRFDPM-QWWZWVQMSA-N 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- 239000000706 filtrate Substances 0.000 description 4
- 230000006698 induction Effects 0.000 description 4
- 230000002757 inflammatory effect Effects 0.000 description 4
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 4
- 230000003834 intracellular effect Effects 0.000 description 4
- 239000011565 manganese chloride Substances 0.000 description 4
- 230000000771 oncological effect Effects 0.000 description 4
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 4
- DGTNSSLYPYDJGL-UHFFFAOYSA-N phenyl isocyanate Chemical compound O=C=NC1=CC=CC=C1 DGTNSSLYPYDJGL-UHFFFAOYSA-N 0.000 description 4
- 230000003389 potentiating effect Effects 0.000 description 4
- 230000035755 proliferation Effects 0.000 description 4
- UBQKCCHYAOITMY-UHFFFAOYSA-N pyridin-2-ol Chemical class OC1=CC=CC=N1 UBQKCCHYAOITMY-UHFFFAOYSA-N 0.000 description 4
- 150000003222 pyridines Chemical class 0.000 description 4
- 230000007017 scission Effects 0.000 description 4
- 238000002560 therapeutic procedure Methods 0.000 description 4
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 3
- JNJRUQKIFNUNNE-UHFFFAOYSA-N 1-[4-(3-amino-6-propan-2-yl-2h-pyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound C=12C(N)=NNC2=NC(C(C)C)=CC=1C(C=C1)=CC=C1NC(=O)NC1=CC=CC(C(F)(F)F)=C1 JNJRUQKIFNUNNE-UHFFFAOYSA-N 0.000 description 3
- 229940044613 1-propanol Drugs 0.000 description 3
- OZAIFHULBGXAKX-UHFFFAOYSA-N 2-(2-cyanopropan-2-yldiazenyl)-2-methylpropanenitrile Chemical compound N#CC(C)(C)N=NC(C)(C)C#N OZAIFHULBGXAKX-UHFFFAOYSA-N 0.000 description 3
- KLSJWNVTNUYHDU-UHFFFAOYSA-N Amitrole Chemical compound NC1=NC=NN1 KLSJWNVTNUYHDU-UHFFFAOYSA-N 0.000 description 3
- 108010009906 Angiopoietins Proteins 0.000 description 3
- 102000009840 Angiopoietins Human genes 0.000 description 3
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 3
- 0 CC1(C*(*)C*(B*N(*)*)CCCC1)N Chemical compound CC1(C*(*)C*(B*N(*)*)CCCC1)N 0.000 description 3
- HTJDQJBWANPRPF-UHFFFAOYSA-N Cyclopropylamine Chemical compound NC1CC1 HTJDQJBWANPRPF-UHFFFAOYSA-N 0.000 description 3
- 238000002965 ELISA Methods 0.000 description 3
- 102000002254 Glycogen Synthase Kinase 3 Human genes 0.000 description 3
- 108010014905 Glycogen Synthase Kinase 3 Proteins 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- 101000805163 Mus musculus Docking protein 2 Proteins 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- 229910021626 Tin(II) chloride Inorganic materials 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- 125000003342 alkenyl group Chemical group 0.000 description 3
- 125000003545 alkoxy group Chemical group 0.000 description 3
- 125000002947 alkylene group Chemical group 0.000 description 3
- 125000000304 alkynyl group Chemical group 0.000 description 3
- 230000002491 angiogenic effect Effects 0.000 description 3
- 230000001772 anti-angiogenic effect Effects 0.000 description 3
- 239000002246 antineoplastic agent Substances 0.000 description 3
- 229940041181 antineoplastic drug Drugs 0.000 description 3
- 238000013459 approach Methods 0.000 description 3
- 230000035578 autophosphorylation Effects 0.000 description 3
- 125000005605 benzo group Chemical group 0.000 description 3
- 125000001743 benzylic group Chemical group 0.000 description 3
- 210000004204 blood vessel Anatomy 0.000 description 3
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical class OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 3
- 229940098773 bovine serum albumin Drugs 0.000 description 3
- 210000000481 breast Anatomy 0.000 description 3
- 239000012267 brine Substances 0.000 description 3
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 3
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 239000012043 crude product Substances 0.000 description 3
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 3
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 3
- 230000003247 decreasing effect Effects 0.000 description 3
- 230000007547 defect Effects 0.000 description 3
- 230000007783 downstream signaling Effects 0.000 description 3
- 238000006911 enzymatic reaction Methods 0.000 description 3
- 230000005284 excitation Effects 0.000 description 3
- 238000001914 filtration Methods 0.000 description 3
- 150000004820 halides Chemical class 0.000 description 3
- XLYOFNOQVPJJNP-ZSJDYOACSA-N heavy water Substances [2H]O[2H] XLYOFNOQVPJJNP-ZSJDYOACSA-N 0.000 description 3
- 125000000623 heterocyclic group Chemical group 0.000 description 3
- 238000011065 in-situ storage Methods 0.000 description 3
- 102000006495 integrins Human genes 0.000 description 3
- 108010044426 integrins Proteins 0.000 description 3
- 229910052740 iodine Inorganic materials 0.000 description 3
- 229940043355 kinase inhibitor Drugs 0.000 description 3
- 230000001926 lymphatic effect Effects 0.000 description 3
- 229910001629 magnesium chloride Inorganic materials 0.000 description 3
- 238000012423 maintenance Methods 0.000 description 3
- ANGDWNBGPBMQHW-UHFFFAOYSA-N methyl cyanoacetate Chemical compound COC(=O)CC#N ANGDWNBGPBMQHW-UHFFFAOYSA-N 0.000 description 3
- 125000002950 monocyclic group Chemical group 0.000 description 3
- 150000002828 nitro derivatives Chemical class 0.000 description 3
- 229910000069 nitrogen hydride Inorganic materials 0.000 description 3
- 230000001575 pathological effect Effects 0.000 description 3
- 230000037361 pathway Effects 0.000 description 3
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 3
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 3
- 125000005544 phthalimido group Chemical group 0.000 description 3
- 239000002798 polar solvent Substances 0.000 description 3
- 238000001556 precipitation Methods 0.000 description 3
- 239000000651 prodrug Substances 0.000 description 3
- 229940002612 prodrug Drugs 0.000 description 3
- 108090000623 proteins and genes Proteins 0.000 description 3
- 230000002829 reductive effect Effects 0.000 description 3
- 238000002165 resonance energy transfer Methods 0.000 description 3
- 125000006413 ring segment Chemical group 0.000 description 3
- 230000019491 signal transduction Effects 0.000 description 3
- 239000000741 silica gel Substances 0.000 description 3
- 229910002027 silica gel Inorganic materials 0.000 description 3
- 210000000329 smooth muscle myocyte Anatomy 0.000 description 3
- 229910000104 sodium hydride Inorganic materials 0.000 description 3
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 3
- 239000012453 solvate Substances 0.000 description 3
- 230000000638 stimulation Effects 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 238000006467 substitution reaction Methods 0.000 description 3
- 229910052717 sulfur Inorganic materials 0.000 description 3
- YBBRCQOCSYXUOC-UHFFFAOYSA-N sulfuryl dichloride Chemical compound ClS(Cl)(=O)=O YBBRCQOCSYXUOC-UHFFFAOYSA-N 0.000 description 3
- 230000001629 suppression Effects 0.000 description 3
- 238000003786 synthesis reaction Methods 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- AXZWODMDQAVCJE-UHFFFAOYSA-L tin(II) chloride (anhydrous) Chemical compound [Cl-].[Cl-].[Sn+2] AXZWODMDQAVCJE-UHFFFAOYSA-L 0.000 description 3
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 2
- 125000006376 (C3-C10) cycloalkyl group Chemical group 0.000 description 2
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 2
- LUBJCRLGQSPQNN-UHFFFAOYSA-N 1-Phenylurea Chemical compound NC(=O)NC1=CC=CC=C1 LUBJCRLGQSPQNN-UHFFFAOYSA-N 0.000 description 2
- FFIKXGYVHREKSV-UHFFFAOYSA-N 1-[4-(3-amino-1-methyl-6-propan-2-ylpyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound C=12C(N)=NN(C)C2=NC(C(C)C)=CC=1C(C=C1)=CC=C1NC(=O)NC1=CC=CC(C(F)(F)F)=C1 FFIKXGYVHREKSV-UHFFFAOYSA-N 0.000 description 2
- XFQIOKGHFOGODR-UHFFFAOYSA-N 1-[4-(3-amino-6-cyclobutyl-1-methylpyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound N1=C2N(C)N=C(N)C2=C(C=2C=CC(NC(=O)NC=3C=C(C=CC=3)C(F)(F)F)=CC=2)C=C1C1CCC1 XFQIOKGHFOGODR-UHFFFAOYSA-N 0.000 description 2
- AUFJZUKDKYLHMS-UHFFFAOYSA-N 1-[4-(3-amino-6-methyl-2h-pyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound C=12C(N)=NNC2=NC(C)=CC=1C(C=C1)=CC=C1NC(=O)NC1=CC=CC(C(F)(F)F)=C1 AUFJZUKDKYLHMS-UHFFFAOYSA-N 0.000 description 2
- GXZSFMBRSMZSCU-UHFFFAOYSA-N 1-[4-[3-amino-1-(2-hydroxyethyl)-6-propan-2-ylpyrazolo[3,4-b]pyridin-4-yl]phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound C=12C(N)=NN(CCO)C2=NC(C(C)C)=CC=1C(C=C1)=CC=C1NC(=O)NC1=CC=CC(C(F)(F)F)=C1 GXZSFMBRSMZSCU-UHFFFAOYSA-N 0.000 description 2
- IBYHHJPAARCAIE-UHFFFAOYSA-N 1-bromo-2-chloroethane Chemical compound ClCCBr IBYHHJPAARCAIE-UHFFFAOYSA-N 0.000 description 2
- HVCFCNAITDHQFX-UHFFFAOYSA-N 1-cyclopropylethanone Chemical compound CC(=O)C1CC1 HVCFCNAITDHQFX-UHFFFAOYSA-N 0.000 description 2
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide Chemical compound CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 2
- NAIKHCBDZGSGHH-UHFFFAOYSA-N 1-fluoro-2-isocyanato-4-(trifluoromethyl)benzene Chemical compound FC1=CC=C(C(F)(F)F)C=C1N=C=O NAIKHCBDZGSGHH-UHFFFAOYSA-N 0.000 description 2
- GLSUJZPVKMKUPJ-UHFFFAOYSA-N 1-fluoro-2-isocyanato-4-methylbenzene Chemical compound CC1=CC=C(F)C(N=C=O)=C1 GLSUJZPVKMKUPJ-UHFFFAOYSA-N 0.000 description 2
- OUQQNLNWNIFTFA-UHFFFAOYSA-N 1-methyl-6-(methylsulfonylmethyl)-4-(4-nitrophenyl)pyrazolo[3,4-b]pyridine Chemical compound C1=C(CS(C)(=O)=O)N=C2N(C)N=CC2=C1C1=CC=C([N+]([O-])=O)C=C1 OUQQNLNWNIFTFA-UHFFFAOYSA-N 0.000 description 2
- GKUOPERZLPMSEK-UHFFFAOYSA-N 1-methyl-6-[(4-methylpiperazin-1-yl)methyl]-4-(4-nitrophenyl)pyrazolo[3,4-b]pyridine Chemical compound C1CN(C)CCN1CC1=CC(C=2C=CC(=CC=2)[N+]([O-])=O)=C(C=NN2C)C2=N1 GKUOPERZLPMSEK-UHFFFAOYSA-N 0.000 description 2
- MOMFXATYAINJML-UHFFFAOYSA-N 2-Acetylthiazole Chemical compound CC(=O)C1=NC=CS1 MOMFXATYAINJML-UHFFFAOYSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- GBHCABUWWQUMAJ-UHFFFAOYSA-N 2-hydrazinoethanol Chemical compound NNCCO GBHCABUWWQUMAJ-UHFFFAOYSA-N 0.000 description 2
- SYBYTAAJFKOIEJ-UHFFFAOYSA-N 3-Methylbutan-2-one Chemical compound CC(C)C(C)=O SYBYTAAJFKOIEJ-UHFFFAOYSA-N 0.000 description 2
- MLDQJTXFUGDVEO-UHFFFAOYSA-N BAY-43-9006 Chemical compound C1=NC(C(=O)NC)=CC(OC=2C=CC(NC(=O)NC=3C=C(C(Cl)=CC=3)C(F)(F)F)=CC=2)=C1 MLDQJTXFUGDVEO-UHFFFAOYSA-N 0.000 description 2
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 2
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 2
- 238000012286 ELISA Assay Methods 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- 206010016654 Fibrosis Diseases 0.000 description 2
- 102000016621 Focal Adhesion Protein-Tyrosine Kinases Human genes 0.000 description 2
- 108010067715 Focal Adhesion Protein-Tyrosine Kinases Proteins 0.000 description 2
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- 206010051066 Gastrointestinal stromal tumour Diseases 0.000 description 2
- 241000238631 Hexapoda Species 0.000 description 2
- 102000038455 IGF Type 1 Receptor Human genes 0.000 description 2
- 108010031794 IGF Type 1 Receptor Proteins 0.000 description 2
- 239000002147 L01XE04 - Sunitinib Substances 0.000 description 2
- 239000005511 L01XE05 - Sorafenib Substances 0.000 description 2
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- 241001529936 Murinae Species 0.000 description 2
- 241000699660 Mus musculus Species 0.000 description 2
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 2
- 102000008052 Nitric Oxide Synthase Type III Human genes 0.000 description 2
- 108010075520 Nitric Oxide Synthase Type III Proteins 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 102000001253 Protein Kinase Human genes 0.000 description 2
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 2
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 2
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 description 2
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 2
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 2
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 2
- 229920002684 Sepharose Polymers 0.000 description 2
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 2
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 2
- 108010090089 TIE-1 Receptor Proteins 0.000 description 2
- 102000050000 TIE-1 Receptor Human genes 0.000 description 2
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- 108091008605 VEGF receptors Proteins 0.000 description 2
- AWTUVLBAXHNLAK-UHFFFAOYSA-N [1-methyl-4-(4-nitrophenyl)pyrazolo[3,4-b]pyridin-6-yl]methanol Chemical compound C1=C(CO)N=C2N(C)N=CC2=C1C1=CC=C([N+]([O-])=O)C=C1 AWTUVLBAXHNLAK-UHFFFAOYSA-N 0.000 description 2
- 230000001594 aberrant effect Effects 0.000 description 2
- 159000000021 acetate salts Chemical class 0.000 description 2
- 150000008065 acid anhydrides Chemical class 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 230000003213 activating effect Effects 0.000 description 2
- 230000010933 acylation Effects 0.000 description 2
- 238000005917 acylation reaction Methods 0.000 description 2
- 102000035181 adaptor proteins Human genes 0.000 description 2
- 108091005764 adaptor proteins Proteins 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 238000001042 affinity chromatography Methods 0.000 description 2
- 239000000556 agonist Substances 0.000 description 2
- 150000001299 aldehydes Chemical class 0.000 description 2
- 150000001351 alkyl iodides Chemical class 0.000 description 2
- 150000003863 ammonium salts Chemical class 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 239000005557 antagonist Substances 0.000 description 2
- 230000006907 apoptotic process Effects 0.000 description 2
- 239000012062 aqueous buffer Substances 0.000 description 2
- 229910052786 argon Inorganic materials 0.000 description 2
- 150000004982 aromatic amines Chemical class 0.000 description 2
- 230000003305 autocrine Effects 0.000 description 2
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 2
- 238000002306 biochemical method Methods 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 238000006795 borylation reaction Methods 0.000 description 2
- 210000004899 c-terminal region Anatomy 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 150000001733 carboxylic acid esters Chemical class 0.000 description 2
- 150000001735 carboxylic acids Chemical class 0.000 description 2
- 239000003054 catalyst Substances 0.000 description 2
- 239000006143 cell culture medium Substances 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 239000011248 coating agent Substances 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 210000004246 corpus luteum Anatomy 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 125000002993 cycloalkylene group Chemical group 0.000 description 2
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 2
- 125000006547 cyclononyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 2
- 125000000640 cyclooctyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C([H])([H])C1([H])[H] 0.000 description 2
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 125000000298 cyclopropenyl group Chemical group [H]C1=C([H])C1([H])* 0.000 description 2
- 210000000805 cytoplasm Anatomy 0.000 description 2
- 238000010511 deprotection reaction Methods 0.000 description 2
- BGRWYRAHAFMIBJ-UHFFFAOYSA-N diisopropylcarbodiimide Natural products CC(C)NC(=O)NC(C)C BGRWYRAHAFMIBJ-UHFFFAOYSA-N 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- 239000008298 dragée Substances 0.000 description 2
- 210000002257 embryonic structure Anatomy 0.000 description 2
- 150000002081 enamines Chemical class 0.000 description 2
- 230000003511 endothelial effect Effects 0.000 description 2
- ZBPJWOTYNPVILR-UHFFFAOYSA-N ethyl 1-[3-amino-1-methyl-4-[4-[[3-(trifluoromethyl)phenyl]carbamoylamino]phenyl]pyrazolo[3,4-b]pyridin-6-yl]cyclopropane-1-carboxylate Chemical compound C=1C(C=2C=CC(NC(=O)NC=3C=C(C=CC=3)C(F)(F)F)=CC=2)=C2C(N)=NN(C)C2=NC=1C1(C(=O)OCC)CC1 ZBPJWOTYNPVILR-UHFFFAOYSA-N 0.000 description 2
- DCSUGYIECPEZGQ-UHFFFAOYSA-N ethyl 2-[3-amino-1-methyl-4-[4-[[3-(trifluoromethyl)phenyl]carbamoylamino]phenyl]pyrazolo[3,4-b]pyridin-6-yl]-2-methylpropanoate Chemical compound C=12C(N)=NN(C)C2=NC(C(C)(C)C(=O)OCC)=CC=1C(C=C1)=CC=C1NC(=O)NC1=CC=CC(C(F)(F)F)=C1 DCSUGYIECPEZGQ-UHFFFAOYSA-N 0.000 description 2
- YVACJZRCOWZWPY-UHFFFAOYSA-N ethyl 2-cyano-3-(4-nitrophenyl)prop-2-enoate Chemical compound CCOC(=O)C(C#N)=CC1=CC=C([N+]([O-])=O)C=C1 YVACJZRCOWZWPY-UHFFFAOYSA-N 0.000 description 2
- ISJQKYRMMGFMTK-UHFFFAOYSA-N ethyl 3-amino-1-methyl-4-(4-nitrophenyl)pyrazolo[3,4-b]pyridine-6-carboxylate Chemical compound C=12C(N)=NN(C)C2=NC(C(=O)OCC)=CC=1C1=CC=C([N+]([O-])=O)C=C1 ISJQKYRMMGFMTK-UHFFFAOYSA-N 0.000 description 2
- VURXNMNYKYXFEK-UHFFFAOYSA-N ethyl 5-cyano-4-(4-nitrophenyl)-6-(trifluoromethylsulfonyloxy)pyridine-2-carboxylate Chemical compound FC(F)(F)S(=O)(=O)OC1=NC(C(=O)OCC)=CC(C=2C=CC(=CC=2)[N+]([O-])=O)=C1C#N VURXNMNYKYXFEK-UHFFFAOYSA-N 0.000 description 2
- JUTPJIZGWSVRJB-UHFFFAOYSA-N ethyl 5-cyano-4-(4-nitrophenyl)-6-oxo-1h-pyridine-2-carboxylate Chemical compound O=C1NC(C(=O)OCC)=CC(C=2C=CC(=CC=2)[N+]([O-])=O)=C1C#N JUTPJIZGWSVRJB-UHFFFAOYSA-N 0.000 description 2
- 239000011737 fluorine Substances 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 201000011243 gastrointestinal stromal tumor Diseases 0.000 description 2
- 125000001188 haloalkyl group Chemical group 0.000 description 2
- 125000005843 halogen group Chemical group 0.000 description 2
- 230000026030 halogenation Effects 0.000 description 2
- 238000005658 halogenation reaction Methods 0.000 description 2
- 125000004836 hexamethylene group Chemical group [H]C([H])([*:2])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[*:1] 0.000 description 2
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 238000002347 injection Methods 0.000 description 2
- 239000007924 injection Substances 0.000 description 2
- 229940125396 insulin Drugs 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- 210000004324 lymphatic system Anatomy 0.000 description 2
- 239000006166 lysate Substances 0.000 description 2
- 239000012139 lysis buffer Substances 0.000 description 2
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 2
- 239000000463 material Substances 0.000 description 2
- 201000001441 melanoma Diseases 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- 230000004060 metabolic process Effects 0.000 description 2
- 238000006263 metalation reaction Methods 0.000 description 2
- 230000005012 migration Effects 0.000 description 2
- 238000013508 migration Methods 0.000 description 2
- 229940043363 multi-kinase inhibitor Drugs 0.000 description 2
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 2
- 238000006396 nitration reaction Methods 0.000 description 2
- 238000010534 nucleophilic substitution reaction Methods 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 235000019198 oils Nutrition 0.000 description 2
- 238000006384 oligomerization reaction Methods 0.000 description 2
- 238000011275 oncology therapy Methods 0.000 description 2
- 230000002018 overexpression Effects 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- 230000003076 paracrine Effects 0.000 description 2
- 125000004817 pentamethylene group Chemical group [H]C([H])([*:2])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[*:1] 0.000 description 2
- 210000003668 pericyte Anatomy 0.000 description 2
- 230000035699 permeability Effects 0.000 description 2
- 230000002085 persistent effect Effects 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 2
- DCWXELXMIBXGTH-UHFFFAOYSA-N phosphotyrosine Chemical compound OC(=O)C(N)CC1=CC=C(OP(O)(O)=O)C=C1 DCWXELXMIBXGTH-UHFFFAOYSA-N 0.000 description 2
- IVDFJHOHABJVEH-UHFFFAOYSA-N pinacol Chemical compound CC(C)(O)C(C)(C)O IVDFJHOHABJVEH-UHFFFAOYSA-N 0.000 description 2
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- 230000001023 pro-angiogenic effect Effects 0.000 description 2
- GKMIMWCVLPIDLH-UHFFFAOYSA-N prop-1-en-2-yl n-[4-(trifluoromethyl)pyridin-2-yl]carbamate Chemical compound CC(=C)OC(=O)NC1=CC(C(F)(F)F)=CC=N1 GKMIMWCVLPIDLH-UHFFFAOYSA-N 0.000 description 2
- 108060006633 protein kinase Proteins 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 230000002685 pulmonary effect Effects 0.000 description 2
- 102000027426 receptor tyrosine kinases Human genes 0.000 description 2
- 108091008598 receptor tyrosine kinases Proteins 0.000 description 2
- 238000003118 sandwich ELISA Methods 0.000 description 2
- 238000007127 saponification reaction Methods 0.000 description 2
- FSYKKLYZXJSNPZ-UHFFFAOYSA-N sarcosine Chemical compound C[NH2+]CC([O-])=O FSYKKLYZXJSNPZ-UHFFFAOYSA-N 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 239000002002 slurry Substances 0.000 description 2
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 239000012312 sodium hydride Substances 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 125000000547 substituted alkyl group Chemical group 0.000 description 2
- 239000000454 talc Substances 0.000 description 2
- 235000012222 talc Nutrition 0.000 description 2
- 229910052623 talc Inorganic materials 0.000 description 2
- 125000000383 tetramethylene group Chemical group [H]C([H])([*:1])C([H])([H])C([H])([H])C([H])([H])[*:2] 0.000 description 2
- GZNAASVAJNXPPW-UHFFFAOYSA-M tin(4+) chloride dihydrate Chemical compound O.O.[Cl-].[Sn+4] GZNAASVAJNXPPW-UHFFFAOYSA-M 0.000 description 2
- FWPIDFUJEMBDLS-UHFFFAOYSA-L tin(II) chloride dihydrate Substances O.O.Cl[Sn]Cl FWPIDFUJEMBDLS-UHFFFAOYSA-L 0.000 description 2
- 238000011830 transgenic mouse model Methods 0.000 description 2
- 230000007704 transition Effects 0.000 description 2
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 description 2
- IHIXIJGXTJIKRB-UHFFFAOYSA-N trisodium vanadate Chemical compound [Na+].[Na+].[Na+].[O-][V]([O-])([O-])=O IHIXIJGXTJIKRB-UHFFFAOYSA-N 0.000 description 2
- 239000003643 water by type Substances 0.000 description 2
- HFVMEOPYDLEHBR-UHFFFAOYSA-N (2-fluorophenyl)-phenylmethanol Chemical compound C=1C=CC=C(F)C=1C(O)C1=CC=CC=C1 HFVMEOPYDLEHBR-UHFFFAOYSA-N 0.000 description 1
- CUGDYSSBTWBKII-LXGUWJNJSA-N (2r,3r,4r,5s)-6-(dimethylamino)hexane-1,2,3,4,5-pentol Chemical compound CN(C)C[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO CUGDYSSBTWBKII-LXGUWJNJSA-N 0.000 description 1
- IKXCHOUDIPZROZ-LXGUWJNJSA-N (2r,3r,4r,5s)-6-(ethylamino)hexane-1,2,3,4,5-pentol Chemical compound CCNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO IKXCHOUDIPZROZ-LXGUWJNJSA-N 0.000 description 1
- 125000006717 (C3-C10) cycloalkenyl group Chemical group 0.000 description 1
- CRPTXKKKIGGDBX-UHFFFAOYSA-N (z)-but-2-ene Chemical group [CH2]C=CC CRPTXKKKIGGDBX-UHFFFAOYSA-N 0.000 description 1
- PAAZPARNPHGIKF-UHFFFAOYSA-N 1,2-dibromoethane Chemical compound BrCCBr PAAZPARNPHGIKF-UHFFFAOYSA-N 0.000 description 1
- RVLWUWCBZVXWAZ-UHFFFAOYSA-N 1,6-dimethyl-4-(4-nitrophenyl)pyrazolo[3,4-b]pyridin-3-amine Chemical compound C=12C(N)=NN(C)C2=NC(C)=CC=1C1=CC=C([N+]([O-])=O)C=C1 RVLWUWCBZVXWAZ-UHFFFAOYSA-N 0.000 description 1
- BJHHDSPDNMCBGN-UHFFFAOYSA-N 1,6-dimethyl-4-(4-nitrophenyl)pyrazolo[3,4-b]pyridine Chemical compound C=12C=NN(C)C2=NC(C)=CC=1C1=CC=C([N+]([O-])=O)C=C1 BJHHDSPDNMCBGN-UHFFFAOYSA-N 0.000 description 1
- BUUYBOOVFSIJCN-UHFFFAOYSA-N 1-(2-fluoro-5-methylphenyl)-3-[4-[1-methyl-6-(morpholin-4-ylmethyl)pyrazolo[3,4-b]pyridin-4-yl]phenyl]urea Chemical compound CC1=CC=C(F)C(NC(=O)NC=2C=CC(=CC=2)C=2C=3C=NN(C)C=3N=C(CN3CCOCC3)C=2)=C1 BUUYBOOVFSIJCN-UHFFFAOYSA-N 0.000 description 1
- LFKRVDCVJRDOBG-UHFFFAOYSA-N 1-(2-phenylcyclopropyl)ethanone Chemical compound CC(=O)C1CC1C1=CC=CC=C1 LFKRVDCVJRDOBG-UHFFFAOYSA-N 0.000 description 1
- XTXFBKQCSKAZLS-UHFFFAOYSA-N 1-(3-ethylphenyl)-3-[4-[1-methyl-6-(morpholin-4-ylmethyl)pyrazolo[3,4-b]pyridin-4-yl]phenyl]urea Chemical compound CCC1=CC=CC(NC(=O)NC=2C=CC(=CC=2)C=2C=3C=NN(C)C=3N=C(CN3CCOCC3)C=2)=C1 XTXFBKQCSKAZLS-UHFFFAOYSA-N 0.000 description 1
- PVOAHINGSUIXLS-UHFFFAOYSA-N 1-Methylpiperazine Chemical compound CN1CCNCC1 PVOAHINGSUIXLS-UHFFFAOYSA-N 0.000 description 1
- USRPRZRMZCCBLZ-UHFFFAOYSA-N 1-[2-fluoro-5-(trifluoromethyl)phenyl]-3-[4-[1-methyl-6-(morpholin-4-ylmethyl)pyrazolo[3,4-b]pyridin-4-yl]phenyl]urea Chemical compound N1=C2N(C)N=CC2=C(C=2C=CC(NC(=O)NC=3C(=CC=C(C=3)C(F)(F)F)F)=CC=2)C=C1CN1CCOCC1 USRPRZRMZCCBLZ-UHFFFAOYSA-N 0.000 description 1
- RNYCLNYQRPSFRO-UHFFFAOYSA-N 1-[3-amino-1-methyl-4-[4-[[3-(trifluoromethyl)phenyl]carbamoylamino]phenyl]pyrazolo[3,4-b]pyridin-6-yl]-n-ethylcyclopropane-1-carboxamide Chemical compound C=1C(C=2C=CC(NC(=O)NC=3C=C(C=CC=3)C(F)(F)F)=CC=2)=C2C(N)=NN(C)C2=NC=1C1(C(=O)NCC)CC1 RNYCLNYQRPSFRO-UHFFFAOYSA-N 0.000 description 1
- XTYNHHMGHVCFJJ-UHFFFAOYSA-N 1-[3-amino-1-methyl-4-[4-[[3-(trifluoromethyl)phenyl]carbamoylamino]phenyl]pyrazolo[3,4-b]pyridin-6-yl]-n-methylcyclopropane-1-carboxamide Chemical compound C=1C(C=2C=CC(NC(=O)NC=3C=C(C=CC=3)C(F)(F)F)=CC=2)=C2C(N)=NN(C)C2=NC=1C1(C(=O)NC)CC1 XTYNHHMGHVCFJJ-UHFFFAOYSA-N 0.000 description 1
- HKJXXQWOKKGCLA-UHFFFAOYSA-N 1-[3-amino-1-methyl-4-[4-[[3-(trifluoromethyl)phenyl]carbamoylamino]phenyl]pyrazolo[3,4-b]pyridin-6-yl]-n-phenylcyclopropane-1-carboxamide Chemical compound N1=C2N(C)N=C(N)C2=C(C=2C=CC(NC(=O)NC=3C=C(C=CC=3)C(F)(F)F)=CC=2)C=C1C1(C(=O)NC=2C=CC=CC=2)CC1 HKJXXQWOKKGCLA-UHFFFAOYSA-N 0.000 description 1
- HSPQCNMTYSFXSV-UHFFFAOYSA-N 1-[3-amino-1-methyl-4-[4-[[3-(trifluoromethyl)phenyl]carbamoylamino]phenyl]pyrazolo[3,4-b]pyridin-6-yl]-n-propan-2-ylcyclopropane-1-carboxamide Chemical compound C=1C(C=2C=CC(NC(=O)NC=3C=C(C=CC=3)C(F)(F)F)=CC=2)=C2C(N)=NN(C)C2=NC=1C1(C(=O)NC(C)C)CC1 HSPQCNMTYSFXSV-UHFFFAOYSA-N 0.000 description 1
- VMWLTIKULXMSET-UHFFFAOYSA-N 1-[3-amino-1-methyl-4-[4-[[3-(trifluoromethyl)phenyl]carbamoylamino]phenyl]pyrazolo[3,4-b]pyridin-6-yl]cyclopropane-1-carboxamide Chemical compound N1=C2N(C)N=C(N)C2=C(C=2C=CC(NC(=O)NC=3C=C(C=CC=3)C(F)(F)F)=CC=2)C=C1C1(C(N)=O)CC1 VMWLTIKULXMSET-UHFFFAOYSA-N 0.000 description 1
- UVXWVPCBVHSSIU-UHFFFAOYSA-N 1-[3-amino-4-[4-[(2-fluoro-5-methylphenyl)carbamoylamino]phenyl]-1-methylpyrazolo[3,4-b]pyridin-6-yl]-n,n-diethylcyclopropane-1-carboxamide Chemical compound C=1C(C=2C=CC(NC(=O)NC=3C(=CC=C(C)C=3)F)=CC=2)=C2C(N)=NN(C)C2=NC=1C1(C(=O)N(CC)CC)CC1 UVXWVPCBVHSSIU-UHFFFAOYSA-N 0.000 description 1
- BYBSGHVXSJFZCS-UHFFFAOYSA-N 1-[4-(1,6-dimethylpyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-phenylurea Chemical compound C=12C=NN(C)C2=NC(C)=CC=1C(C=C1)=CC=C1NC(=O)NC1=CC=CC=C1 BYBSGHVXSJFZCS-UHFFFAOYSA-N 0.000 description 1
- XPBOSEJGZUCREQ-UHFFFAOYSA-N 1-[4-(3-amino-1,6-dimethylpyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-[2-fluoro-5-(trifluoromethyl)phenyl]urea Chemical compound C=12C(N)=NN(C)C2=NC(C)=CC=1C(C=C1)=CC=C1NC(=O)NC1=CC(C(F)(F)F)=CC=C1F XPBOSEJGZUCREQ-UHFFFAOYSA-N 0.000 description 1
- KROLCVQARUUHNV-UHFFFAOYSA-N 1-[4-(3-amino-1-methylpyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound C1=CN=C2N(C)N=C(N)C2=C1C(C=C1)=CC=C1NC(=O)NC1=CC=CC(C(F)(F)F)=C1 KROLCVQARUUHNV-UHFFFAOYSA-N 0.000 description 1
- LYHXSDWIWVVZPB-UHFFFAOYSA-N 1-[4-(3-amino-2h-pyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound C=12C(N)=NNC2=NC=CC=1C(C=C1)=CC=C1NC(=O)NC1=CC=CC(C(F)(F)F)=C1 LYHXSDWIWVVZPB-UHFFFAOYSA-N 0.000 description 1
- BARNJPJGHDEMQG-UHFFFAOYSA-N 1-[4-(3-amino-6-cyclohexyl-1-methylpyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound N1=C2N(C)N=C(N)C2=C(C=2C=CC(NC(=O)NC=3C=C(C=CC=3)C(F)(F)F)=CC=2)C=C1C1CCCCC1 BARNJPJGHDEMQG-UHFFFAOYSA-N 0.000 description 1
- UPVSBDANSROJQT-UHFFFAOYSA-N 1-[4-(3-amino-6-cyclopropyl-1-methylpyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound N1=C2N(C)N=C(N)C2=C(C=2C=CC(NC(=O)NC=3C=C(C=CC=3)C(F)(F)F)=CC=2)C=C1C1CC1 UPVSBDANSROJQT-UHFFFAOYSA-N 0.000 description 1
- VFLRJWMTKCLJHU-UHFFFAOYSA-N 1-[4-(3-amino-6-tert-butyl-1-methylpyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-(5-tert-butyl-1,2-oxazol-3-yl)urea Chemical compound C1=C(C(C)(C)C)N=C2N(C)N=C(N)C2=C1C(C=C1)=CC=C1NC(=O)NC=1C=C(C(C)(C)C)ON=1 VFLRJWMTKCLJHU-UHFFFAOYSA-N 0.000 description 1
- MTDGQRISQGFMAV-UHFFFAOYSA-N 1-[4-(3-amino-6-tert-butyl-1-methylpyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound C1=C(C(C)(C)C)N=C2N(C)N=C(N)C2=C1C(C=C1)=CC=C1NC(=O)NC1=CC=CC(C(F)(F)F)=C1 MTDGQRISQGFMAV-UHFFFAOYSA-N 0.000 description 1
- ZVBDPQBIRJLSIT-UHFFFAOYSA-N 1-[4-(3-amino-6-tert-butyl-2h-pyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-(3-ethylphenyl)urea Chemical compound CCC1=CC=CC(NC(=O)NC=2C=CC(=CC=2)C=2C=3C(N)=NNC=3N=C(C=2)C(C)(C)C)=C1 ZVBDPQBIRJLSIT-UHFFFAOYSA-N 0.000 description 1
- AIBILPUDAXOWDF-UHFFFAOYSA-N 1-[4-(3-amino-6-tert-butyl-2h-pyrazolo[3,4-b]pyridin-4-yl)phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound C=12C(N)=NNC2=NC(C(C)(C)C)=CC=1C(C=C1)=CC=C1NC(=O)NC1=CC=CC(C(F)(F)F)=C1 AIBILPUDAXOWDF-UHFFFAOYSA-N 0.000 description 1
- AQNUYFBTYHJFJV-UHFFFAOYSA-N 1-[4-[1-methyl-6-(morpholin-4-ylmethyl)pyrazolo[3,4-b]pyridin-4-yl]phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound N1=C2N(C)N=CC2=C(C=2C=CC(NC(=O)NC=3C=C(C=CC=3)C(F)(F)F)=CC=2)C=C1CN1CCOCC1 AQNUYFBTYHJFJV-UHFFFAOYSA-N 0.000 description 1
- WLWUADABAAOKCE-UHFFFAOYSA-N 1-[4-[1-methyl-6-(morpholin-4-ylmethyl)pyrazolo[3,4-b]pyridin-4-yl]phenyl]-3-phenylurea Chemical compound N1=C2N(C)N=CC2=C(C=2C=CC(NC(=O)NC=3C=CC=CC=3)=CC=2)C=C1CN1CCOCC1 WLWUADABAAOKCE-UHFFFAOYSA-N 0.000 description 1
- WHGUUFBYZLNGOQ-UHFFFAOYSA-N 1-[4-[3-amino-1-(2-morpholin-4-ylethyl)-6-propan-2-ylpyrazolo[3,4-b]pyridin-4-yl]phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound C=12C(N)=NN(CCN3CCOCC3)C2=NC(C(C)C)=CC=1C(C=C1)=CC=C1NC(=O)NC1=CC=CC(C(F)(F)F)=C1 WHGUUFBYZLNGOQ-UHFFFAOYSA-N 0.000 description 1
- BPXBWEXYRHPGHJ-UHFFFAOYSA-N 1-[4-[3-amino-1-methyl-6-(1,3-thiazol-2-yl)pyrazolo[3,4-b]pyridin-4-yl]phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound N1=C2N(C)N=C(N)C2=C(C=2C=CC(NC(=O)NC=3C=C(C=CC=3)C(F)(F)F)=CC=2)C=C1C1=NC=CS1 BPXBWEXYRHPGHJ-UHFFFAOYSA-N 0.000 description 1
- UQZQTUBNLPLHNI-UHFFFAOYSA-N 1-[4-[3-amino-1-methyl-6-(2-phenylcyclopropyl)pyrazolo[3,4-b]pyridin-4-yl]phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound N1=C2N(C)N=C(N)C2=C(C=2C=CC(NC(=O)NC=3C=C(C=CC=3)C(F)(F)F)=CC=2)C=C1C1CC1C1=CC=CC=C1 UQZQTUBNLPLHNI-UHFFFAOYSA-N 0.000 description 1
- ULOIWNJIAPUYOD-UHFFFAOYSA-N 1-[4-[3-amino-1-methyl-6-[1-(pyrrolidine-1-carbonyl)cyclopropyl]pyrazolo[3,4-b]pyridin-4-yl]phenyl]-3-(2-fluoro-5-methylphenyl)urea Chemical compound CC1=CC=C(F)C(NC(=O)NC=2C=CC(=CC=2)C=2C=3C(N)=NN(C)C=3N=C(C=2)C2(CC2)C(=O)N2CCCC2)=C1 ULOIWNJIAPUYOD-UHFFFAOYSA-N 0.000 description 1
- TUONPUQFQFTBDC-UHFFFAOYSA-N 1-[4-[3-amino-6-tert-butyl-1-(2-hydroxyethyl)pyrazolo[3,4-b]pyridin-4-yl]phenyl]-3-[3-(trifluoromethyl)phenyl]urea Chemical compound C=12C(N)=NN(CCO)C2=NC(C(C)(C)C)=CC=1C(C=C1)=CC=C1NC(=O)NC1=CC=CC(C(F)(F)F)=C1 TUONPUQFQFTBDC-UHFFFAOYSA-N 0.000 description 1
- ZGCHLAJIRWDGFE-UHFFFAOYSA-N 1-aminopropane-1,1-diol Chemical compound CCC(N)(O)O ZGCHLAJIRWDGFE-UHFFFAOYSA-N 0.000 description 1
- 125000004973 1-butenyl group Chemical group C(=CCC)* 0.000 description 1
- 125000004972 1-butynyl group Chemical group [H]C([H])([H])C([H])([H])C#C* 0.000 description 1
- JPJOOTWNILDNAW-UHFFFAOYSA-N 1-cyclobutylethanone Chemical compound CC(=O)C1CCC1 JPJOOTWNILDNAW-UHFFFAOYSA-N 0.000 description 1
- RIFKADJTWUGDOV-UHFFFAOYSA-N 1-cyclohexylethanone Chemical compound CC(=O)C1CCCCC1 RIFKADJTWUGDOV-UHFFFAOYSA-N 0.000 description 1
- DNFZCDLEGMEKMI-UHFFFAOYSA-N 1-ethyl-3-isocyanatobenzene Chemical compound CCC1=CC=CC(N=C=O)=C1 DNFZCDLEGMEKMI-UHFFFAOYSA-N 0.000 description 1
- WGSXKYXKAARAKD-UHFFFAOYSA-N 1-fluoro-3-isocyanato-5-(trifluoromethyl)benzene Chemical compound FC1=CC(N=C=O)=CC(C(F)(F)F)=C1 WGSXKYXKAARAKD-UHFFFAOYSA-N 0.000 description 1
- OPPYFFRLKJUEOS-UHFFFAOYSA-N 1-fluoro-4-isocyanato-2-(trifluoromethyl)benzene Chemical compound FC1=CC=C(N=C=O)C=C1C(F)(F)F OPPYFFRLKJUEOS-UHFFFAOYSA-N 0.000 description 1
- CPPGZWWUPFWALU-UHFFFAOYSA-N 1-isocyanato-3-methylbenzene Chemical compound CC1=CC=CC(N=C=O)=C1 CPPGZWWUPFWALU-UHFFFAOYSA-N 0.000 description 1
- MAJMPAMZRGVCDH-UHFFFAOYSA-N 1-methylpyrazolo[4,3-b]pyridine Chemical compound C1=CC=C2N(C)N=CC2=N1 MAJMPAMZRGVCDH-UHFFFAOYSA-N 0.000 description 1
- AVFZOVWCLRSYKC-UHFFFAOYSA-N 1-methylpyrrolidine Chemical compound CN1CCCC1 AVFZOVWCLRSYKC-UHFFFAOYSA-N 0.000 description 1
- ABEXEQSGABRUHS-UHFFFAOYSA-N 16-methylheptadecyl 16-methylheptadecanoate Chemical compound CC(C)CCCCCCCCCCCCCCCOC(=O)CCCCCCCCCCCCCCC(C)C ABEXEQSGABRUHS-UHFFFAOYSA-N 0.000 description 1
- YDTDKKULPWTHRV-UHFFFAOYSA-N 1H-indazol-3-amine Chemical class C1=CC=C2C(N)=NNC2=C1 YDTDKKULPWTHRV-UHFFFAOYSA-N 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- SIZDUQQDBXJXLQ-JGVFFNPUSA-N 2-[(1s,3s)-3-acetyl-2,2-dimethylcyclobutyl]acetic acid Chemical compound CC(=O)[C@H]1C[C@@H](CC(O)=O)C1(C)C SIZDUQQDBXJXLQ-JGVFFNPUSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- GHQHZMUTOOOZNX-UHFFFAOYSA-N 2-[3-[3-amino-1-methyl-4-[4-[[3-(trifluoromethyl)phenyl]carbamoylamino]phenyl]pyrazolo[3,4-b]pyridin-6-yl]-2,2-dimethylcyclobutyl]-n-cyclopropylacetamide Chemical compound N1=C2N(C)N=C(N)C2=C(C=2C=CC(NC(=O)NC=3C=C(C=CC=3)C(F)(F)F)=CC=2)C=C1C(C1(C)C)CC1CC(=O)NC1CC1 GHQHZMUTOOOZNX-UHFFFAOYSA-N 0.000 description 1
- RJWXVRUKDGLMBW-UHFFFAOYSA-N 2-[3-[3-amino-1-methyl-4-[4-[[3-(trifluoromethyl)phenyl]carbamoylamino]phenyl]pyrazolo[3,4-b]pyridin-6-yl]-2,2-dimethylcyclobutyl]acetic acid Chemical compound N1=C2N(C)N=C(N)C2=C(C=2C=CC(NC(=O)NC=3C=C(C=CC=3)C(F)(F)F)=CC=2)C=C1C1CC(CC(O)=O)C1(C)C RJWXVRUKDGLMBW-UHFFFAOYSA-N 0.000 description 1
- HCNSKLJHARORJF-UHFFFAOYSA-N 2-[3-amino-1-methyl-4-[4-[[3-(trifluoromethyl)phenyl]carbamoylamino]phenyl]pyrazolo[3,4-b]pyridin-6-yl]-2-methylpropanoic acid Chemical compound C1=C(C(C)(C)C(O)=O)N=C2N(C)N=C(N)C2=C1C(C=C1)=CC=C1NC(=O)NC1=CC=CC(C(F)(F)F)=C1 HCNSKLJHARORJF-UHFFFAOYSA-N 0.000 description 1
- WVDXIYVNRVAAJC-UHFFFAOYSA-N 2-[3-amino-1-methyl-4-[4-[[3-(trifluoromethyl)phenyl]carbamoylamino]phenyl]pyrazolo[3,4-b]pyridin-6-yl]-n,n,2-trimethylpropanamide Chemical compound C=12C(N)=NN(C)C2=NC(C(C)(C)C(=O)N(C)C)=CC=1C(C=C1)=CC=C1NC(=O)NC1=CC=CC(C(F)(F)F)=C1 WVDXIYVNRVAAJC-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-IVMDWMLBSA-N 2-amino-2-deoxy-D-glucopyranose Chemical compound N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O MSWZFWKMSRAUBD-IVMDWMLBSA-N 0.000 description 1
- KJJPLEZQSCZCKE-UHFFFAOYSA-N 2-aminopropane-1,3-diol Chemical compound OCC(N)CO KJJPLEZQSCZCKE-UHFFFAOYSA-N 0.000 description 1
- 125000004974 2-butenyl group Chemical group C(C=CC)* 0.000 description 1
- 125000000069 2-butynyl group Chemical group [H]C([H])([H])C#CC([H])([H])* 0.000 description 1
- XBTHHWYOEISJNF-UHFFFAOYSA-N 2-fluoro-1-isocyanato-3-(trifluoromethyl)benzene Chemical compound FC1=C(N=C=O)C=CC=C1C(F)(F)F XBTHHWYOEISJNF-UHFFFAOYSA-N 0.000 description 1
- VONGIOGTLFSXDE-UHFFFAOYSA-N 2-fluoro-4-iodopyridine-3-carbaldehyde Chemical compound FC1=NC=CC(I)=C1C=O VONGIOGTLFSXDE-UHFFFAOYSA-N 0.000 description 1
- RKUSRLUGUVDNKP-UHFFFAOYSA-N 2-methoxy-5-(trifluoromethyl)aniline Chemical compound COC1=CC=C(C(F)(F)F)C=C1N RKUSRLUGUVDNKP-UHFFFAOYSA-N 0.000 description 1
- BTERYDAAQMJMTD-UHFFFAOYSA-N 2-phenylcyclohexane-1-carboxylic acid Chemical compound OC(=O)C1CCCCC1C1=CC=CC=C1 BTERYDAAQMJMTD-UHFFFAOYSA-N 0.000 description 1
- HPDFKXPVMXSXGE-UHFFFAOYSA-N 2-pyrrolidin-1-yl-5-(trifluoromethyl)aniline Chemical compound NC1=CC(C(F)(F)F)=CC=C1N1CCCC1 HPDFKXPVMXSXGE-UHFFFAOYSA-N 0.000 description 1
- JMTMSDXUXJISAY-UHFFFAOYSA-N 2H-benzotriazol-4-ol Chemical compound OC1=CC=CC2=C1N=NN2 JMTMSDXUXJISAY-UHFFFAOYSA-N 0.000 description 1
- LUAQTQAFUORQHV-UHFFFAOYSA-N 2h-pyrazolo[3,4-b]pyridin-3-amine Chemical compound C1=CC=C2C(N)=NNC2=N1 LUAQTQAFUORQHV-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- 125000000474 3-butynyl group Chemical group [H]C#CC([H])([H])C([H])([H])* 0.000 description 1
- CSIQEOLAVILJSS-UHFFFAOYSA-N 4-(1,6-dimethylpyrazolo[3,4-b]pyridin-4-yl)aniline Chemical compound C=12C=NN(C)C2=NC(C)=CC=1C1=CC=C(N)C=C1 CSIQEOLAVILJSS-UHFFFAOYSA-N 0.000 description 1
- NBJHDLKSWUDGJG-UHFFFAOYSA-N 4-(2-chloroethyl)morpholin-4-ium;chloride Chemical compound Cl.ClCCN1CCOCC1 NBJHDLKSWUDGJG-UHFFFAOYSA-N 0.000 description 1
- DJJNIXWIUCTBBX-UHFFFAOYSA-N 4-(4-nitrophenyl)-2-oxo-1h-pyridine-3-carbonitrile Chemical compound C1=CC([N+](=O)[O-])=CC=C1C1=C(C#N)C(=O)NC=C1 DJJNIXWIUCTBBX-UHFFFAOYSA-N 0.000 description 1
- QMHZYJKTFCXYDO-UHFFFAOYSA-N 4-[1-methyl-6-(methylsulfonylmethyl)pyrazolo[3,4-b]pyridin-4-yl]aniline Chemical compound C1=C(CS(C)(=O)=O)N=C2N(C)N=CC2=C1C1=CC=C(N)C=C1 QMHZYJKTFCXYDO-UHFFFAOYSA-N 0.000 description 1
- FSAIMFTWBTYSCL-UHFFFAOYSA-N 4-[1-methyl-6-(morpholin-4-ylmethyl)pyrazolo[3,4-b]pyridin-4-yl]aniline Chemical compound N1=C2N(C)N=CC2=C(C=2C=CC(N)=CC=2)C=C1CN1CCOCC1 FSAIMFTWBTYSCL-UHFFFAOYSA-N 0.000 description 1
- DCSNGRLEEDFFSO-UHFFFAOYSA-N 4-[1-methyl-6-[(4-methylpiperazin-1-yl)methyl]pyrazolo[3,4-b]pyridin-4-yl]aniline Chemical compound C1CN(C)CCN1CC1=CC(C=2C=CC(N)=CC=2)=C(C=NN2C)C2=N1 DCSNGRLEEDFFSO-UHFFFAOYSA-N 0.000 description 1
- CGHYCVDODCGPIG-UHFFFAOYSA-N 4-[[1-methyl-4-(4-nitrophenyl)pyrazolo[3,4-b]pyridin-6-yl]methyl]morpholine Chemical compound N1=C2N(C)N=CC2=C(C=2C=CC(=CC=2)[N+]([O-])=O)C=C1CN1CCOCC1 CGHYCVDODCGPIG-UHFFFAOYSA-N 0.000 description 1
- WIFPJDJJFUSIFP-UHFFFAOYSA-N 4-aminobutane-1,2,3-triol Chemical compound NCC(O)C(O)CO WIFPJDJJFUSIFP-UHFFFAOYSA-N 0.000 description 1
- KVWLQZSOMITAJR-UHFFFAOYSA-N 4-hydroxypyrazolo[4,3-b]pyridine-5-carboxylic acid Chemical class ON1C(C(=O)O)=CC=C2N=NC=C21 KVWLQZSOMITAJR-UHFFFAOYSA-N 0.000 description 1
- YQYGPGKTNQNXMH-UHFFFAOYSA-N 4-nitroacetophenone Chemical compound CC(=O)C1=CC=C([N+]([O-])=O)C=C1 YQYGPGKTNQNXMH-UHFFFAOYSA-N 0.000 description 1
- PXRKCOCTEMYUEG-UHFFFAOYSA-N 5-aminoisoindole-1,3-dione Chemical compound NC1=CC=C2C(=O)NC(=O)C2=C1 PXRKCOCTEMYUEG-UHFFFAOYSA-N 0.000 description 1
- GGXGVZJHUKEJHO-UHFFFAOYSA-N 5-tert-butyl-1,2-oxazol-3-amine Chemical compound CC(C)(C)C1=CC(N)=NO1 GGXGVZJHUKEJHO-UHFFFAOYSA-N 0.000 description 1
- ZKXXIHFWZWPMMU-UHFFFAOYSA-N 6-(furan-2-yl)-4-(4-nitrophenyl)-2-oxo-1h-pyridine-3-carbonitrile Chemical compound C1=CC([N+](=O)[O-])=CC=C1C1=C(C#N)C(=O)NC(C=2OC=CC=2)=C1 ZKXXIHFWZWPMMU-UHFFFAOYSA-N 0.000 description 1
- HMGVHZFRXKQMCB-UHFFFAOYSA-N 6-cyclopropyl-4-(4-nitrophenyl)-2-oxo-1h-pyridine-3-carbonitrile Chemical compound C1=CC([N+](=O)[O-])=CC=C1C1=C(C#N)C(=O)NC(C2CC2)=C1 HMGVHZFRXKQMCB-UHFFFAOYSA-N 0.000 description 1
- FDNNBHGTFULOPT-UHFFFAOYSA-N 6-methyl-4-(4-nitrophenyl)-2-oxo-1h-pyridine-3-carbonitrile Chemical compound O=C1NC(C)=CC(C=2C=CC(=CC=2)[N+]([O-])=O)=C1C#N FDNNBHGTFULOPT-UHFFFAOYSA-N 0.000 description 1
- VCSRBPOZBMVVOZ-UHFFFAOYSA-N 6-tert-butyl-4-(4-nitrophenyl)-2-oxo-1h-pyridine-3-carbonitrile Chemical compound O=C1NC(C(C)(C)C)=CC(C=2C=CC(=CC=2)[N+]([O-])=O)=C1C#N VCSRBPOZBMVVOZ-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- OZAIFHULBGXAKX-VAWYXSNFSA-N AIBN Substances N#CC(C)(C)\N=N\C(C)(C)C#N OZAIFHULBGXAKX-VAWYXSNFSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 230000007730 Akt signaling Effects 0.000 description 1
- HJCMDXDYPOUFDY-WHFBIAKZSA-N Ala-Gln Chemical compound C[C@H](N)C(=O)N[C@H](C(O)=O)CCC(N)=O HJCMDXDYPOUFDY-WHFBIAKZSA-N 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 108010048036 Angiopoietin-2 Proteins 0.000 description 1
- 108020005544 Antisense RNA Proteins 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- 125000001313 C5-C10 heteroaryl group Chemical group 0.000 description 1
- BRUUWQWLYZGYCA-UHFFFAOYSA-N CC(C)(C)c1cc(-c(cc2)ccc2NC(Nc2cc(C(F)(F)F)ccc2)=O)c(cn[n]2CCO)c2n1 Chemical compound CC(C)(C)c1cc(-c(cc2)ccc2NC(Nc2cc(C(F)(F)F)ccc2)=O)c(cn[n]2CCO)c2n1 BRUUWQWLYZGYCA-UHFFFAOYSA-N 0.000 description 1
- JZYIFIZCRAWNHT-UHFFFAOYSA-N CC(C)(C)c1nc([nH]nc2)c2c(-c(cc2)ccc2NC(Nc(cc2)ccc2-c2ccccc2)=O)c1 Chemical compound CC(C)(C)c1nc([nH]nc2)c2c(-c(cc2)ccc2NC(Nc(cc2)ccc2-c2ccccc2)=O)c1 JZYIFIZCRAWNHT-UHFFFAOYSA-N 0.000 description 1
- LKVRTSYVMNIFSM-UHFFFAOYSA-N CC(C)(C)c1nc([nH]nc2)c2c(-c(cc2)ccc2NC(Nc2cc(C(F)(F)F)ccc2Cl)=O)c1 Chemical compound CC(C)(C)c1nc([nH]nc2)c2c(-c(cc2)ccc2NC(Nc2cc(C(F)(F)F)ccc2Cl)=O)c1 LKVRTSYVMNIFSM-UHFFFAOYSA-N 0.000 description 1
- ZNAQEXYFFOLJHK-UHFFFAOYSA-N CC(C)(C)c1nc([nH]nc2)c2c(-c(cc2)ccc2NC(Nc2cc(C(F)(F)F)ccc2F)=O)c1 Chemical compound CC(C)(C)c1nc([nH]nc2)c2c(-c(cc2)ccc2NC(Nc2cc(C(F)(F)F)ccc2F)=O)c1 ZNAQEXYFFOLJHK-UHFFFAOYSA-N 0.000 description 1
- ZWZKJJYQCVECLX-UHFFFAOYSA-N CC(C)(C)c1nc([nH]nc2)c2c(-c(cc2)ccc2NC(Nc2cc(C)cc(C)c2)=O)c1 Chemical compound CC(C)(C)c1nc([nH]nc2)c2c(-c(cc2)ccc2NC(Nc2cc(C)cc(C)c2)=O)c1 ZWZKJJYQCVECLX-UHFFFAOYSA-N 0.000 description 1
- XDNQPDDNMDQCLB-UHFFFAOYSA-N CC(C)(C)c1nc([n](C)nc2)c2c(-c(cc2)ccc2NC(C2(CC2)c2ccccc2)=O)c1 Chemical compound CC(C)(C)c1nc([n](C)nc2)c2c(-c(cc2)ccc2NC(C2(CC2)c2ccccc2)=O)c1 XDNQPDDNMDQCLB-UHFFFAOYSA-N 0.000 description 1
- MUNXRRQXDLOTSW-UHFFFAOYSA-N CC(C)(C)c1nc([n](C)nc2)c2c(-c(cc2)ccc2NC(Cc2ccccc2)=O)c1 Chemical compound CC(C)(C)c1nc([n](C)nc2)c2c(-c(cc2)ccc2NC(Cc2ccccc2)=O)c1 MUNXRRQXDLOTSW-UHFFFAOYSA-N 0.000 description 1
- IITIVPAADDCFAX-UHFFFAOYSA-N CC(C)(C)c1nc([n](C)nc2)c2c(-c(cc2)ccc2NC(Nc(cc2)cc(C(F)(F)F)c2OCCN(C)C)=O)c1 Chemical compound CC(C)(C)c1nc([n](C)nc2)c2c(-c(cc2)ccc2NC(Nc(cc2)cc(C(F)(F)F)c2OCCN(C)C)=O)c1 IITIVPAADDCFAX-UHFFFAOYSA-N 0.000 description 1
- CBOFKRUTYYTJFX-UHFFFAOYSA-N CC(C)(C)c1nc([n](C)nc2)c2c(-c(cc2)ccc2NC(Nc2cc(C(F)(F)F)c(CN3CCOCC3)cc2)=O)c1 Chemical compound CC(C)(C)c1nc([n](C)nc2)c2c(-c(cc2)ccc2NC(Nc2cc(C(F)(F)F)c(CN3CCOCC3)cc2)=O)c1 CBOFKRUTYYTJFX-UHFFFAOYSA-N 0.000 description 1
- MIRZCUUEWDGOJC-UHFFFAOYSA-N CC(C)(C)c1nc([n](C)nc2)c2c(-c(cc2)ccc2NC(Nc2cc(C(F)(F)F)ccc2)=O)c1 Chemical compound CC(C)(C)c1nc([n](C)nc2)c2c(-c(cc2)ccc2NC(Nc2cc(C(F)(F)F)ccc2)=O)c1 MIRZCUUEWDGOJC-UHFFFAOYSA-N 0.000 description 1
- SEIFRKJHZJHUQE-UHFFFAOYSA-N CC(C)(C)c1nc([n](C)nc2)c2c(-c(cc2)ccc2NC(Nc2cc(OC)c(CN3CCN(C)CC3)cc2)=O)c1 Chemical compound CC(C)(C)c1nc([n](C)nc2)c2c(-c(cc2)ccc2NC(Nc2cc(OC)c(CN3CCN(C)CC3)cc2)=O)c1 SEIFRKJHZJHUQE-UHFFFAOYSA-N 0.000 description 1
- HOXKOCVFJCDVDG-UHFFFAOYSA-N CC(C)(C)c1nc([n](C)nc2)c2c(-c(cc2)ccc2NS(Cc2ccccc2)(=O)=O)c1 Chemical compound CC(C)(C)c1nc([n](C)nc2)c2c(-c(cc2)ccc2NS(Cc2ccccc2)(=O)=O)c1 HOXKOCVFJCDVDG-UHFFFAOYSA-N 0.000 description 1
- RNMOLNKSDFKCBA-UHFFFAOYSA-O CC(NC1=O)=CC(c(cc2)ccc2[N+](O)=O)=C1C#N Chemical compound CC(NC1=O)=CC(c(cc2)ccc2[N+](O)=O)=C1C#N RNMOLNKSDFKCBA-UHFFFAOYSA-O 0.000 description 1
- LCFQKGADPMPYCE-UHFFFAOYSA-N CCN(CC)Cc(cc1)c(C(F)(F)F)cc1NC(Nc(cc1)ccc1-c1cc(C(C)(C)C)nc2c1cn[n]2C)=O Chemical compound CCN(CC)Cc(cc1)c(C(F)(F)F)cc1NC(Nc(cc1)ccc1-c1cc(C(C)(C)C)nc2c1cn[n]2C)=O LCFQKGADPMPYCE-UHFFFAOYSA-N 0.000 description 1
- MVHBUODNANHTIR-UHFFFAOYSA-N CCOC(C[n](c1nc(C(C)(C)C)c2)ncc1c2-c(cc1)ccc1NC(Nc1cc(C(F)(F)F)ccc1)=O)=O Chemical compound CCOC(C[n](c1nc(C(C)(C)C)c2)ncc1c2-c(cc1)ccc1NC(Nc1cc(C(F)(F)F)ccc1)=O)=O MVHBUODNANHTIR-UHFFFAOYSA-N 0.000 description 1
- MRAPLMNNFSKJTA-UHFFFAOYSA-N CCOc1cc(NC(Nc(cc2)ccc2-c2cc(C(C)(C)C)nc3c2cn[nH]3)=O)ccc1 Chemical compound CCOc1cc(NC(Nc(cc2)ccc2-c2cc(C(C)(C)C)nc3c2cn[nH]3)=O)ccc1 MRAPLMNNFSKJTA-UHFFFAOYSA-N 0.000 description 1
- ZXLYYQUMYFHCLQ-UHFFFAOYSA-N CN(C(c1ccccc11)=O)C1=O Chemical compound CN(C(c1ccccc11)=O)C1=O ZXLYYQUMYFHCLQ-UHFFFAOYSA-N 0.000 description 1
- 101100464197 Caenorhabditis elegans pak-1 gene Proteins 0.000 description 1
- 101100356682 Caenorhabditis elegans rho-1 gene Proteins 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- CKDWPUIZGOQOOM-UHFFFAOYSA-N Carbamyl chloride Chemical compound NC(Cl)=O CKDWPUIZGOQOOM-UHFFFAOYSA-N 0.000 description 1
- ZCBGGKZLOKHXNR-UHFFFAOYSA-N Cc1nc([nH]nc2)c2c(-c(cc2)ccc2NC(C2(CC2)c2cccc(C(F)(F)F)c2)=O)c1 Chemical compound Cc1nc([nH]nc2)c2c(-c(cc2)ccc2NC(C2(CC2)c2cccc(C(F)(F)F)c2)=O)c1 ZCBGGKZLOKHXNR-UHFFFAOYSA-N 0.000 description 1
- LFTVKVUHPQMEOD-UHFFFAOYSA-N Cc1nc([n](C)nc2)c2c(-c(cc2)ccc2NC(C2(CC2)c2cccc(OC)c2)=O)c1 Chemical compound Cc1nc([n](C)nc2)c2c(-c(cc2)ccc2NC(C2(CC2)c2cccc(OC)c2)=O)c1 LFTVKVUHPQMEOD-UHFFFAOYSA-N 0.000 description 1
- QIHDIGXNJCTSLB-UHFFFAOYSA-N Cc1nc([n](C)nc2)c2c(-c(cc2)ccc2NC(C2(CC2)c2ccccc2)=O)c1 Chemical compound Cc1nc([n](C)nc2)c2c(-c(cc2)ccc2NC(C2(CC2)c2ccccc2)=O)c1 QIHDIGXNJCTSLB-UHFFFAOYSA-N 0.000 description 1
- GXGJIOMUZAGVEH-UHFFFAOYSA-N Chamazulene Chemical group CCC1=CC=C(C)C2=CC=C(C)C2=C1 GXGJIOMUZAGVEH-UHFFFAOYSA-N 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 206010009944 Colon cancer Diseases 0.000 description 1
- 206010052360 Colorectal adenocarcinoma Diseases 0.000 description 1
- 206010052358 Colorectal cancer metastatic Diseases 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 208000011231 Crohn disease Diseases 0.000 description 1
- GSNUFIFRDBKVIE-UHFFFAOYSA-N DMF Natural products CC1=CC=C(C)O1 GSNUFIFRDBKVIE-UHFFFAOYSA-N 0.000 description 1
- 206010012689 Diabetic retinopathy Diseases 0.000 description 1
- 102100024746 Dihydrofolate reductase Human genes 0.000 description 1
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 1
- 101100015729 Drosophila melanogaster drk gene Proteins 0.000 description 1
- 102000009024 Epidermal Growth Factor Human genes 0.000 description 1
- 101800003838 Epidermal growth factor Proteins 0.000 description 1
- 208000019028 Epidermal thickening Diseases 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- XXRCUYVCPSWGCC-UHFFFAOYSA-N Ethyl pyruvate Chemical compound CCOC(=O)C(C)=O XXRCUYVCPSWGCC-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 1
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 1
- 108091006020 Fc-tagged proteins Proteins 0.000 description 1
- 108010073385 Fibrin Proteins 0.000 description 1
- 102000009123 Fibrin Human genes 0.000 description 1
- BWGVNKXGVNDBDI-UHFFFAOYSA-N Fibrin monomer Chemical group CNC(=O)CNC(=O)CN BWGVNKXGVNDBDI-UHFFFAOYSA-N 0.000 description 1
- 238000005863 Friedel-Crafts acylation reaction Methods 0.000 description 1
- 102000013446 GTP Phosphohydrolases Human genes 0.000 description 1
- 108091006109 GTPases Proteins 0.000 description 1
- 240000004260 Garcinia hombroniana Species 0.000 description 1
- 239000001828 Gelatine Substances 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 239000007821 HATU Substances 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 101001059454 Homo sapiens Serine/threonine-protein kinase MARK2 Proteins 0.000 description 1
- 101000851018 Homo sapiens Vascular endothelial growth factor receptor 1 Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 239000003458 I kappa b kinase inhibitor Substances 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 108060003951 Immunoglobulin Proteins 0.000 description 1
- 241000764238 Isis Species 0.000 description 1
- 208000007766 Kaposi sarcoma Diseases 0.000 description 1
- 208000008839 Kidney Neoplasms Diseases 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 101150018665 MAPK3 gene Proteins 0.000 description 1
- 108700029942 Megaloblastic Anemia due to Dihydrofolate Reductase Deficiency Proteins 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 102000002568 Multienzyme Complexes Human genes 0.000 description 1
- 108010093369 Multienzyme Complexes Proteins 0.000 description 1
- ZSXGLVDWWRXATF-UHFFFAOYSA-N N,N-dimethylformamide dimethyl acetal Chemical compound COC(OC)N(C)C ZSXGLVDWWRXATF-UHFFFAOYSA-N 0.000 description 1
- 230000006181 N-acylation Effects 0.000 description 1
- AHVYPIQETPWLSZ-UHFFFAOYSA-N N-methyl-pyrrolidine Natural products CN1CC=CC1 AHVYPIQETPWLSZ-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- QECVIPBZOPUTRD-UHFFFAOYSA-N N=S(=O)=O Chemical class N=S(=O)=O QECVIPBZOPUTRD-UHFFFAOYSA-N 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- 239000007832 Na2SO4 Substances 0.000 description 1
- 229910020889 NaBH3 Inorganic materials 0.000 description 1
- 102000007607 Non-Receptor Type 11 Protein Tyrosine Phosphatase Human genes 0.000 description 1
- 108010032107 Non-Receptor Type 11 Protein Tyrosine Phosphatase Proteins 0.000 description 1
- ZZQXQMMAJASHQH-UHFFFAOYSA-N O=C(Nc(cc1)ccc1-c1ccnc2c1cn[n]2CCN1CCOCC1)Nc1cc(C(F)(F)F)ccc1 Chemical compound O=C(Nc(cc1)ccc1-c1ccnc2c1cn[n]2CCN1CCOCC1)Nc1cc(C(F)(F)F)ccc1 ZZQXQMMAJASHQH-UHFFFAOYSA-N 0.000 description 1
- 208000022873 Ocular disease Diseases 0.000 description 1
- ZCQWOFVYLHDMMC-UHFFFAOYSA-N Oxazole Chemical compound C1=COC=N1 ZCQWOFVYLHDMMC-UHFFFAOYSA-N 0.000 description 1
- 239000012826 P38 inhibitor Substances 0.000 description 1
- 208000034038 Pathologic Neovascularization Diseases 0.000 description 1
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 1
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 1
- LGRFSURHDFAFJT-UHFFFAOYSA-N Phthalic anhydride Natural products C1=CC=C2C(=O)OC(=O)C2=C1 LGRFSURHDFAFJT-UHFFFAOYSA-N 0.000 description 1
- 108050001185 Pleckstrin homology domains Proteins 0.000 description 1
- 102000010995 Pleckstrin homology domains Human genes 0.000 description 1
- 229920002538 Polyethylene Glycol 20000 Polymers 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- WTKZEGDFNFYCGP-UHFFFAOYSA-N Pyrazole Chemical compound C=1C=NNC=1 WTKZEGDFNFYCGP-UHFFFAOYSA-N 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 101150058540 RAC1 gene Proteins 0.000 description 1
- 101150111584 RHOA gene Proteins 0.000 description 1
- 102100022122 Ras-related C3 botulinum toxin substrate 1 Human genes 0.000 description 1
- 208000006265 Renal cell carcinoma Diseases 0.000 description 1
- 102100022340 SHC-transforming protein 1 Human genes 0.000 description 1
- 108010077895 Sarcosine Proteins 0.000 description 1
- 102100028904 Serine/threonine-protein kinase MARK2 Human genes 0.000 description 1
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 description 1
- 208000006981 Skin Abnormalities Diseases 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 208000005718 Stomach Neoplasms Diseases 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 238000006069 Suzuki reaction reaction Methods 0.000 description 1
- 101710157310 Tegument protein UL47 homolog Proteins 0.000 description 1
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 description 1
- 108010040625 Transforming Protein 1 Src Homology 2 Domain-Containing Proteins 0.000 description 1
- 108700019146 Transgenes Proteins 0.000 description 1
- 108010053100 Vascular Endothelial Growth Factor Receptor-3 Proteins 0.000 description 1
- 208000032594 Vascular Remodeling Diseases 0.000 description 1
- 102100033178 Vascular endothelial growth factor receptor 1 Human genes 0.000 description 1
- 102100033179 Vascular endothelial growth factor receptor 3 Human genes 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000016383 Zea mays subsp huehuetenangensis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- VNRUCTBIUJOJSP-UHFFFAOYSA-N [3-cyano-4-(4-nitrophenyl)-6-propan-2-ylpyridin-2-yl] trifluoromethanesulfonate Chemical compound FC(F)(F)S(=O)(=O)OC1=NC(C(C)C)=CC(C=2C=CC(=CC=2)[N+]([O-])=O)=C1C#N VNRUCTBIUJOJSP-UHFFFAOYSA-N 0.000 description 1
- TZDZSLRQOLUVCX-UHFFFAOYSA-N [3-cyano-4-(4-nitrophenyl)pyridin-2-yl] trifluoromethanesulfonate Chemical compound C1=CC([N+](=O)[O-])=CC=C1C1=CC=NC(OS(=O)(=O)C(F)(F)F)=C1C#N TZDZSLRQOLUVCX-UHFFFAOYSA-N 0.000 description 1
- JTRIZJQAJGNVNX-UHFFFAOYSA-N [3-cyano-6-cyclopropyl-4-(4-nitrophenyl)pyridin-2-yl] trifluoromethanesulfonate Chemical compound C1=CC([N+](=O)[O-])=CC=C1C1=CC(C2CC2)=NC(OS(=O)(=O)C(F)(F)F)=C1C#N JTRIZJQAJGNVNX-UHFFFAOYSA-N 0.000 description 1
- RSJBTEWMKHTXFQ-UHFFFAOYSA-N [3-cyano-6-methyl-4-(4-nitrophenyl)pyridin-2-yl] trifluoromethanesulfonate Chemical compound FC(F)(F)S(=O)(=O)OC1=NC(C)=CC(C=2C=CC(=CC=2)[N+]([O-])=O)=C1C#N RSJBTEWMKHTXFQ-UHFFFAOYSA-N 0.000 description 1
- XISMTXALBRBWIM-UHFFFAOYSA-N [3-cyano-6-methyl-4-[4-[[3-(trifluoromethyl)phenyl]carbamoylamino]phenyl]pyridin-2-yl] trifluoromethanesulfonate Chemical compound FC(F)(F)S(=O)(=O)OC1=NC(C)=CC(C=2C=CC(NC(=O)NC=3C=C(C=CC=3)C(F)(F)F)=CC=2)=C1C#N XISMTXALBRBWIM-UHFFFAOYSA-N 0.000 description 1
- XZBVHYSUGSAZPB-UHFFFAOYSA-N [3-cyano-6-propan-2-yl-4-[4-[[3-(trifluoromethyl)phenyl]carbamoylamino]phenyl]pyridin-2-yl] trifluoromethanesulfonate Chemical compound FC(F)(F)S(=O)(=O)OC1=NC(C(C)C)=CC(C=2C=CC(NC(=O)NC=3C=C(C=CC=3)C(F)(F)F)=CC=2)=C1C#N XZBVHYSUGSAZPB-UHFFFAOYSA-N 0.000 description 1
- NWWWCYPMXRULPF-UHFFFAOYSA-N [4-(4-aminophenyl)-3-cyano-6-cyclopropylpyridin-2-yl] trifluoromethanesulfonate Chemical compound C1=CC(N)=CC=C1C1=CC(C2CC2)=NC(OS(=O)(=O)C(F)(F)F)=C1C#N NWWWCYPMXRULPF-UHFFFAOYSA-N 0.000 description 1
- SEMIOIFKSVFOGB-UHFFFAOYSA-N [4-(4-aminophenyl)-3-cyano-6-methylpyridin-2-yl] trifluoromethanesulfonate Chemical compound FC(F)(F)S(=O)(=O)OC1=NC(C)=CC(C=2C=CC(N)=CC=2)=C1C#N SEMIOIFKSVFOGB-UHFFFAOYSA-N 0.000 description 1
- SWKBSMXBIDAIMS-UHFFFAOYSA-N [4-(4-aminophenyl)-3-cyano-6-propan-2-ylpyridin-2-yl] trifluoromethanesulfonate Chemical compound FC(F)(F)S(=O)(=O)OC1=NC(C(C)C)=CC(C=2C=CC(N)=CC=2)=C1C#N SWKBSMXBIDAIMS-UHFFFAOYSA-N 0.000 description 1
- JRUYAWWZPYKWRG-UHFFFAOYSA-N [4-(4-aminophenyl)-3-cyanopyridin-2-yl] trifluoromethanesulfonate Chemical compound C1=CC(N)=CC=C1C1=CC=NC(OS(=O)(=O)C(F)(F)F)=C1C#N JRUYAWWZPYKWRG-UHFFFAOYSA-N 0.000 description 1
- QYRNBXGIUPTFJL-UHFFFAOYSA-N [4-(4-aminophenyl)-6-tert-butyl-3-cyanopyridin-2-yl] trifluoromethanesulfonate Chemical compound FC(F)(F)S(=O)(=O)OC1=NC(C(C)(C)C)=CC(C=2C=CC(N)=CC=2)=C1C#N QYRNBXGIUPTFJL-UHFFFAOYSA-N 0.000 description 1
- NKLCCMWGSJDVEP-UHFFFAOYSA-N [6-tert-butyl-3-cyano-4-(4-nitrophenyl)pyridin-2-yl] trifluoromethanesulfonate Chemical compound FC(F)(F)S(=O)(=O)OC1=NC(C(C)(C)C)=CC(C=2C=CC(=CC=2)[N+]([O-])=O)=C1C#N NKLCCMWGSJDVEP-UHFFFAOYSA-N 0.000 description 1
- QWFBPJKXJRUUFX-UHFFFAOYSA-N [6-tert-butyl-3-cyano-4-[4-[[3-(trifluoromethyl)phenyl]carbamoylamino]phenyl]pyridin-2-yl] trifluoromethanesulfonate Chemical compound FC(F)(F)S(=O)(=O)OC1=NC(C(C)(C)C)=CC(C=2C=CC(NC(=O)NC=3C=C(C=CC=3)C(F)(F)F)=CC=2)=C1C#N QWFBPJKXJRUUFX-UHFFFAOYSA-N 0.000 description 1
- ZVQOOHYFBIDMTQ-UHFFFAOYSA-N [methyl(oxido){1-[6-(trifluoromethyl)pyridin-3-yl]ethyl}-lambda(6)-sulfanylidene]cyanamide Chemical compound N#CN=S(C)(=O)C(C)C1=CC=C(C(F)(F)F)N=C1 ZVQOOHYFBIDMTQ-UHFFFAOYSA-N 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 125000005042 acyloxymethyl group Chemical group 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 208000037844 advanced solid tumor Diseases 0.000 description 1
- 230000001270 agonistic effect Effects 0.000 description 1
- 229910052783 alkali metal Inorganic materials 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 1
- 125000004849 alkoxymethyl group Chemical group 0.000 description 1
- 150000001347 alkyl bromides Chemical class 0.000 description 1
- 125000005907 alkyl ester group Chemical group 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- HSFWRNGVRCDJHI-UHFFFAOYSA-N alpha-acetylene Natural products C#C HSFWRNGVRCDJHI-UHFFFAOYSA-N 0.000 description 1
- 238000005576 amination reaction Methods 0.000 description 1
- 125000006242 amine protecting group Chemical group 0.000 description 1
- 125000006295 amino methylene group Chemical group [H]N(*)C([H])([H])* 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-N ammonia Natural products N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 1
- 210000000648 angioblast Anatomy 0.000 description 1
- 239000004037 angiogenesis inhibitor Substances 0.000 description 1
- 125000002178 anthracenyl group Chemical group C1(=CC=CC2=CC3=CC=CC=C3C=C12)* 0.000 description 1
- 230000003527 anti-angiogenesis Effects 0.000 description 1
- 230000002424 anti-apoptotic effect Effects 0.000 description 1
- 230000002682 anti-psoriatic effect Effects 0.000 description 1
- 229940045988 antineoplastic drug protein kinase inhibitors Drugs 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 230000004872 arterial blood pressure Effects 0.000 description 1
- 210000002565 arteriole Anatomy 0.000 description 1
- 125000005418 aryl aryl group Chemical group 0.000 description 1
- 150000001543 aryl boronic acids Chemical class 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 229940120638 avastin Drugs 0.000 description 1
- 210000003050 axon Anatomy 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 150000001540 azides Chemical class 0.000 description 1
- 125000004069 aziridinyl group Chemical group 0.000 description 1
- 125000004931 azocinyl group Chemical group N1=C(C=CC=CC=C1)* 0.000 description 1
- 125000003828 azulenyl group Chemical group 0.000 description 1
- 230000006420 basal activation Effects 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical class BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- NHOWLEZFTHYCTP-UHFFFAOYSA-N benzylhydrazine Chemical class NNCC1=CC=CC=C1 NHOWLEZFTHYCTP-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-UHFFFAOYSA-N beta-D-galactosamine Natural products NC1C(O)OC(CO)C(O)C1O MSWZFWKMSRAUBD-UHFFFAOYSA-N 0.000 description 1
- 229960000397 bevacizumab Drugs 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 210000000941 bile Anatomy 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 229960002685 biotin Drugs 0.000 description 1
- 235000020958 biotin Nutrition 0.000 description 1
- 239000011616 biotin Substances 0.000 description 1
- 230000002051 biphasic effect Effects 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- AZWXAPCAJCYGIA-UHFFFAOYSA-N bis(2-methylpropyl)alumane Chemical compound CC(C)C[AlH]CC(C)C AZWXAPCAJCYGIA-UHFFFAOYSA-N 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 150000001642 boronic acid derivatives Chemical class 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 201000008275 breast carcinoma Diseases 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229920005557 bromobutyl Polymers 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- 125000005510 but-1-en-2-yl group Chemical group 0.000 description 1
- JHIWVOJDXOSYLW-UHFFFAOYSA-N butyl 2,2-difluorocyclopropane-1-carboxylate Chemical compound CCCCOC(=O)C1CC1(F)F JHIWVOJDXOSYLW-UHFFFAOYSA-N 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 150000004657 carbamic acid derivatives Chemical class 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- 125000001589 carboacyl group Chemical group 0.000 description 1
- 150000001718 carbodiimides Chemical class 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 125000001721 carboxyacetyl group Chemical group 0.000 description 1
- 210000000748 cardiovascular system Anatomy 0.000 description 1
- 208000003295 carpal tunnel syndrome Diseases 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000003638 chemical reducing agent Substances 0.000 description 1
- 239000013626 chemical specie Substances 0.000 description 1
- 230000035605 chemotaxis Effects 0.000 description 1
- 125000004218 chloromethyl group Chemical group [H]C([H])(Cl)* 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 208000037976 chronic inflammation Diseases 0.000 description 1
- 208000037893 chronic inflammatory disorder Diseases 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 230000007882 cirrhosis Effects 0.000 description 1
- 208000019425 cirrhosis of liver Diseases 0.000 description 1
- 210000001072 colon Anatomy 0.000 description 1
- 238000004891 communication Methods 0.000 description 1
- 239000003184 complementary RNA Substances 0.000 description 1
- 239000003636 conditioned culture medium Substances 0.000 description 1
- 208000015806 constitutional megaloblastic anemia with severe neurologic disease Diseases 0.000 description 1
- 201000000159 corneal neovascularization Diseases 0.000 description 1
- 239000007819 coupling partner Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000001047 cyclobutenyl group Chemical group C1(=CCC1)* 0.000 description 1
- 125000001162 cycloheptenyl group Chemical group C1(=CCCCCC1)* 0.000 description 1
- 125000000596 cyclohexenyl group Chemical group C1(=CCCCC1)* 0.000 description 1
- 125000004090 cyclononenyl group Chemical group C1(=CCCCCCCC1)* 0.000 description 1
- 125000000522 cyclooctenyl group Chemical group C1(=CCCCCCC1)* 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 238000006114 decarboxylation reaction Methods 0.000 description 1
- 125000002704 decyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- KXGVEGMKQFWNSR-LLQZFEROSA-N deoxycholic acid Chemical compound C([C@H]1CC2)[C@H](O)CC[C@]1(C)[C@@H]1[C@@H]2[C@@H]2CC[C@H]([C@@H](CCC(O)=O)C)[C@@]2(C)[C@@H](O)C1 KXGVEGMKQFWNSR-LLQZFEROSA-N 0.000 description 1
- 229960003964 deoxycholic acid Drugs 0.000 description 1
- KXGVEGMKQFWNSR-UHFFFAOYSA-N deoxycholic acid Natural products C1CC2CC(O)CCC2(C)C2C1C1CCC(C(CCC(O)=O)C)C1(C)C(O)C2 KXGVEGMKQFWNSR-UHFFFAOYSA-N 0.000 description 1
- 239000005549 deoxyribonucleoside Substances 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 230000001687 destabilization Effects 0.000 description 1
- 125000005117 dialkylcarbamoyl group Chemical group 0.000 description 1
- 238000000502 dialysis Methods 0.000 description 1
- 230000009699 differential effect Effects 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 125000001028 difluoromethyl group Chemical group [H]C(F)(F)* 0.000 description 1
- 108020001096 dihydrofolate reductase Proteins 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 238000006073 displacement reaction Methods 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 125000005883 dithianyl group Chemical group 0.000 description 1
- 239000012636 effector Substances 0.000 description 1
- 238000003683 electrophilic halogenation reaction Methods 0.000 description 1
- 238000007336 electrophilic substitution reaction Methods 0.000 description 1
- 238000000132 electrospray ionisation Methods 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 230000013020 embryo development Effects 0.000 description 1
- 231100001129 embryonic lethality Toxicity 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 210000003038 endothelium Anatomy 0.000 description 1
- 210000003989 endothelium vascular Anatomy 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 229940116977 epidermal growth factor Drugs 0.000 description 1
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 1
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 1
- 230000032050 esterification Effects 0.000 description 1
- 238000005886 esterification reaction Methods 0.000 description 1
- 229940031098 ethanolamine Drugs 0.000 description 1
- AQSQLDOXNOVLRN-UHFFFAOYSA-N ethyl 1-[3-amino-4-(4-nitrophenyl)-2h-pyrazolo[3,4-b]pyridin-6-yl]cyclopropane-1-carboxylate Chemical compound C=1C(C=2C=CC(=CC=2)[N+]([O-])=O)=C2C(N)=NNC2=NC=1C1(C(=O)OCC)CC1 AQSQLDOXNOVLRN-UHFFFAOYSA-N 0.000 description 1
- SAKMKWYKBYRIEX-UHFFFAOYSA-N ethyl 1-[3-amino-4-[4-(phenylcarbamoylamino)phenyl]-2h-pyrazolo[3,4-b]pyridin-6-yl]cyclopropane-1-carboxylate Chemical compound C=1C(C=2C=CC(NC(=O)NC=3C=CC=CC=3)=CC=2)=C2C(N)=NNC2=NC=1C1(C(=O)OCC)CC1 SAKMKWYKBYRIEX-UHFFFAOYSA-N 0.000 description 1
- LSVOCJHADXQBJN-UHFFFAOYSA-N ethyl 1-[3-amino-4-[4-[(2-fluoro-5-methylphenyl)carbamoylamino]phenyl]-1-methylpyrazolo[3,4-b]pyridin-6-yl]cyclopropane-1-carboxylate Chemical compound C=1C(C=2C=CC(NC(=O)NC=3C(=CC=C(C)C=3)F)=CC=2)=C2C(N)=NN(C)C2=NC=1C1(C(=O)OCC)CC1 LSVOCJHADXQBJN-UHFFFAOYSA-N 0.000 description 1
- UHHQUPMIYZGTKO-UHFFFAOYSA-N ethyl 1-[4-(4-aminophenyl)-1h-pyrazolo[3,4-b]pyridin-6-yl]cyclopropane-1-carboxylate Chemical compound C=1C(C=2C=CC(N)=CC=2)=C2C=NNC2=NC=1C1(C(=O)OCC)CC1 UHHQUPMIYZGTKO-UHFFFAOYSA-N 0.000 description 1
- RRHSEWBWEGVNIB-UHFFFAOYSA-N ethyl 1-[4-(4-aminophenyl)-5-cyano-6-(trifluoromethylsulfonyloxy)pyridin-2-yl]cyclopropane-1-carboxylate Chemical compound C=1C(C=2C=CC(N)=CC=2)=C(C#N)C(OS(=O)(=O)C(F)(F)F)=NC=1C1(C(=O)OCC)CC1 RRHSEWBWEGVNIB-UHFFFAOYSA-N 0.000 description 1
- CBCYGUFNXAJDPR-UHFFFAOYSA-N ethyl 1-[4-(4-nitrophenyl)-1h-pyrazolo[3,4-b]pyridin-6-yl]cyclopropane-1-carboxylate Chemical compound C=1C(C=2C=CC(=CC=2)[N+]([O-])=O)=C2C=NNC2=NC=1C1(C(=O)OCC)CC1 CBCYGUFNXAJDPR-UHFFFAOYSA-N 0.000 description 1
- OBOBBYOPBNPSSL-UHFFFAOYSA-N ethyl 1-[5-cyano-4-(4-nitrophenyl)-6-(trifluoromethylsulfonyloxy)pyridin-2-yl]cyclopropane-1-carboxylate Chemical compound C=1C(C=2C=CC(=CC=2)[N+]([O-])=O)=C(C#N)C(OS(=O)(=O)C(F)(F)F)=NC=1C1(C(=O)OCC)CC1 OBOBBYOPBNPSSL-UHFFFAOYSA-N 0.000 description 1
- XAWAPJNFBJPHHD-UHFFFAOYSA-N ethyl 1-[5-cyano-4-(4-nitrophenyl)-6-oxo-1h-pyridin-2-yl]cyclopropane-1-carboxylate Chemical compound C=1C(C=2C=CC(=CC=2)[N+]([O-])=O)=C(C#N)C(=O)NC=1C1(C(=O)OCC)CC1 XAWAPJNFBJPHHD-UHFFFAOYSA-N 0.000 description 1
- UJALKFGYQCHQNI-UHFFFAOYSA-N ethyl 1-[5-cyano-4-[4-(phenylcarbamoylamino)phenyl]-6-(trifluoromethylsulfonyloxy)pyridin-2-yl]cyclopropane-1-carboxylate Chemical compound C=1C(C=2C=CC(NC(=O)NC=3C=CC=CC=3)=CC=2)=C(C#N)C(OS(=O)(=O)C(F)(F)F)=NC=1C1(C(=O)OCC)CC1 UJALKFGYQCHQNI-UHFFFAOYSA-N 0.000 description 1
- ZSCNVMUEVBTQEI-UHFFFAOYSA-N ethyl 1-[5-cyano-4-[4-[(2-fluoro-5-methylphenyl)carbamoylamino]phenyl]-6-(trifluoromethylsulfonyloxy)pyridin-2-yl]cyclopropane-1-carboxylate Chemical compound C=1C(C=2C=CC(NC(=O)NC=3C(=CC=C(C)C=3)F)=CC=2)=C(C#N)C(OS(=O)(=O)C(F)(F)F)=NC=1C1(C(=O)OCC)CC1 ZSCNVMUEVBTQEI-UHFFFAOYSA-N 0.000 description 1
- DISZFIFAESWGBI-UHFFFAOYSA-N ethyl 1-acetylcyclopropane-1-carboxylate Chemical compound CCOC(=O)C1(C(C)=O)CC1 DISZFIFAESWGBI-UHFFFAOYSA-N 0.000 description 1
- FNLJGRVHTNBZGV-UHFFFAOYSA-N ethyl 2-[3-amino-6-tert-butyl-4-[4-[[3-(trifluoromethyl)phenyl]carbamoylamino]phenyl]pyrazolo[3,4-b]pyridin-1-yl]acetate Chemical compound C1=C(C(C)(C)C)N=C2N(CC(=O)OCC)N=C(N)C2=C1C(C=C1)=CC=C1NC(=O)NC1=CC=CC(C(F)(F)F)=C1 FNLJGRVHTNBZGV-UHFFFAOYSA-N 0.000 description 1
- JQUGVWPBEIYCOQ-UHFFFAOYSA-N ethyl 2-[5-cyano-4-(4-nitrophenyl)-6-oxo-1h-pyridin-2-yl]-2-methylpropanoate Chemical compound O=C1NC(C(C)(C)C(=O)OCC)=CC(C=2C=CC(=CC=2)[N+]([O-])=O)=C1C#N JQUGVWPBEIYCOQ-UHFFFAOYSA-N 0.000 description 1
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 1
- 125000002534 ethynyl group Chemical group [H]C#C* 0.000 description 1
- GWQVMPWSEVRGPY-UHFFFAOYSA-N europium cryptate Chemical compound [Eu+3].N=1C2=CC=CC=1CN(CC=1N=C(C=CC=1)C=1N=C(C3)C=CC=1)CC(N=1)=CC(C(=O)NCCN)=CC=1C(N=1)=CC(C(=O)NCCN)=CC=1CN3CC1=CC=CC2=N1 GWQVMPWSEVRGPY-UHFFFAOYSA-N 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 210000002744 extracellular matrix Anatomy 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 229950003499 fibrin Drugs 0.000 description 1
- 230000004761 fibrosis Effects 0.000 description 1
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- 125000004216 fluoromethyl group Chemical group [H]C([H])(F)* 0.000 description 1
- 125000005816 fluoropropyl group Chemical group [H]C([H])(F)C([H])([H])C([H])([H])* 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 235000011389 fruit/vegetable juice Nutrition 0.000 description 1
- 238000007306 functionalization reaction Methods 0.000 description 1
- HYBBIBNJHNGZAN-UHFFFAOYSA-N furfural Chemical compound O=CC1=CC=CO1 HYBBIBNJHNGZAN-UHFFFAOYSA-N 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 108020001507 fusion proteins Proteins 0.000 description 1
- 102000037865 fusion proteins Human genes 0.000 description 1
- 206010017758 gastric cancer Diseases 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 238000001415 gene therapy Methods 0.000 description 1
- 229960002442 glucosamine Drugs 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 101150098203 grb2 gene Proteins 0.000 description 1
- 230000009036 growth inhibition Effects 0.000 description 1
- 125000004438 haloalkoxy group Chemical group 0.000 description 1
- 125000003187 heptyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 150000002390 heteroarenes Chemical class 0.000 description 1
- 125000006588 heterocycloalkylene group Chemical group 0.000 description 1
- 238000005734 heterodimerization reaction Methods 0.000 description 1
- 238000012766 histopathologic analysis Methods 0.000 description 1
- 239000000710 homodimer Substances 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 150000003840 hydrochlorides Chemical class 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 230000001146 hypoxic effect Effects 0.000 description 1
- 239000005457 ice water Substances 0.000 description 1
- 238000005417 image-selected in vivo spectroscopy Methods 0.000 description 1
- JBFYUZGYRGXSFL-UHFFFAOYSA-N imidazolide Chemical compound C1=C[N-]C=N1 JBFYUZGYRGXSFL-UHFFFAOYSA-N 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 238000002649 immunization Methods 0.000 description 1
- 230000003053 immunization Effects 0.000 description 1
- 102000018358 immunoglobulin Human genes 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 230000008595 infiltration Effects 0.000 description 1
- 238000001764 infiltration Methods 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 239000003999 initiator Substances 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 150000002485 inorganic esters Chemical class 0.000 description 1
- 239000012194 insect media Substances 0.000 description 1
- 238000012739 integrated shape imaging system Methods 0.000 description 1
- 230000031146 intracellular signal transduction Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 239000011630 iodine Substances 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 229940047889 isobutyramide Drugs 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical compound C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- MPVGZUGXCQEXTM-UHFFFAOYSA-N linifanib Chemical compound CC1=CC=C(F)C(NC(=O)NC=2C=CC(=CC=2)C=2C=3C(N)=NNC=3C=CC=2)=C1 MPVGZUGXCQEXTM-UHFFFAOYSA-N 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 238000004020 luminiscence type Methods 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 208000037841 lung tumor Diseases 0.000 description 1
- 210000002751 lymph Anatomy 0.000 description 1
- 230000035168 lymphangiogenesis Effects 0.000 description 1
- 210000004698 lymphocyte Anatomy 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- 235000009973 maize Nutrition 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- CUONGYYJJVDODC-UHFFFAOYSA-N malononitrile Chemical compound N#CCC#N CUONGYYJJVDODC-UHFFFAOYSA-N 0.000 description 1
- 239000003550 marker Substances 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 230000035800 maturation Effects 0.000 description 1
- 239000002609 medium Substances 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- 125000004184 methoxymethyl group Chemical group [H]C([H])([H])OC([H])([H])* 0.000 description 1
- DVAHCHCZPNEYNF-UHFFFAOYSA-N methyl 2-(3-acetyl-2,2-dimethylcyclobutyl)acetate Chemical compound COC(=O)CC1CC(C(C)=O)C1(C)C DVAHCHCZPNEYNF-UHFFFAOYSA-N 0.000 description 1
- OLOLHCNLFLXXMX-UHFFFAOYSA-N methyl 2-hydrazinylacetate;hydrochloride Chemical compound Cl.COC(=O)CNN OLOLHCNLFLXXMX-UHFFFAOYSA-N 0.000 description 1
- PYFSLJVSCGXYAJ-UHFFFAOYSA-N methyl 2-hydroxy-4-[[3-(2-hydroxyphenyl)phenyl]sulfonylamino]benzoate Chemical compound C1=C(O)C(C(=O)OC)=CC=C1NS(=O)(=O)C1=CC=CC(C=2C(=CC=CC=2)O)=C1 PYFSLJVSCGXYAJ-UHFFFAOYSA-N 0.000 description 1
- QSRRZKPKHJHIRB-UHFFFAOYSA-N methyl 4-[(2,5-dichloro-4-methylthiophen-3-yl)sulfonylamino]-2-hydroxybenzoate Chemical compound C1=C(O)C(C(=O)OC)=CC=C1NS(=O)(=O)C1=C(Cl)SC(Cl)=C1C QSRRZKPKHJHIRB-UHFFFAOYSA-N 0.000 description 1
- YGDGZDGRCWHDOU-UHFFFAOYSA-N methyl 4-[[5-chloro-4-(2-hydroxyphenyl)thiophen-2-yl]sulfonylamino]-2-hydroxybenzoate Chemical compound C1=C(O)C(C(=O)OC)=CC=C1NS(=O)(=O)C1=CC(C=2C(=CC=CC=2)O)=C(Cl)S1 YGDGZDGRCWHDOU-UHFFFAOYSA-N 0.000 description 1
- 230000011987 methylation Effects 0.000 description 1
- 238000007069 methylation reaction Methods 0.000 description 1
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 1
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 description 1
- 210000004088 microvessel Anatomy 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000003032 molecular docking Methods 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 230000004899 motility Effects 0.000 description 1
- 230000037257 muscle growth Effects 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- 230000000869 mutational effect Effects 0.000 description 1
- LBWFXVZLPYTWQI-IPOVEDGCSA-N n-[2-(diethylamino)ethyl]-5-[(z)-(5-fluoro-2-oxo-1h-indol-3-ylidene)methyl]-2,4-dimethyl-1h-pyrrole-3-carboxamide;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C LBWFXVZLPYTWQI-IPOVEDGCSA-N 0.000 description 1
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004957 naphthylene group Chemical group 0.000 description 1
- 230000001537 neural effect Effects 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 230000003472 neutralizing effect Effects 0.000 description 1
- 229940080607 nexavar Drugs 0.000 description 1
- 102000037979 non-receptor tyrosine kinases Human genes 0.000 description 1
- 108091008046 non-receptor tyrosine kinases Proteins 0.000 description 1
- 125000001400 nonyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000012038 nucleophile Substances 0.000 description 1
- 230000000269 nucleophilic effect Effects 0.000 description 1
- 235000015097 nutrients Nutrition 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 231100000590 oncogenic Toxicity 0.000 description 1
- 230000002246 oncogenic effect Effects 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 230000002611 ovarian Effects 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 150000003891 oxalate salts Chemical class 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000003585 oxepinyl group Chemical group 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- 230000001590 oxidative effect Effects 0.000 description 1
- 125000000466 oxiranyl group Chemical group 0.000 description 1
- 230000008506 pathogenesis Effects 0.000 description 1
- 230000009963 pathologic angiogenesis Effects 0.000 description 1
- 238000000059 patterning Methods 0.000 description 1
- 125000006340 pentafluoro ethyl group Chemical group FC(F)(F)C(F)(F)* 0.000 description 1
- RGSFGYAAUTVSQA-UHFFFAOYSA-N pentamethylene Natural products C1CCCC1 RGSFGYAAUTVSQA-UHFFFAOYSA-N 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 239000000546 pharmaceutical excipient Substances 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 229940127557 pharmaceutical product Drugs 0.000 description 1
- 239000003444 phase transfer catalyst Substances 0.000 description 1
- 125000001791 phenazinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3N=C12)* 0.000 description 1
- 125000001484 phenothiazinyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3NC12)* 0.000 description 1
- 125000001644 phenoxazinyl group Chemical group C1(=CC=CC=2OC3=CC=CC=C3NC12)* 0.000 description 1
- 125000003884 phenylalkyl group Chemical group 0.000 description 1
- 150000003904 phospholipids Chemical class 0.000 description 1
- 150000003014 phosphoric acid esters Chemical class 0.000 description 1
- 229910052698 phosphorus Inorganic materials 0.000 description 1
- 239000011574 phosphorus Substances 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 125000005633 phthalidyl group Chemical group 0.000 description 1
- PJGSXYOJTGTZAV-UHFFFAOYSA-N pinacolone Chemical compound CC(=O)C(C)(C)C PJGSXYOJTGTZAV-UHFFFAOYSA-N 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 229920001515 polyalkylene glycol Polymers 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 229920000136 polysorbate Polymers 0.000 description 1
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 1
- 229910000027 potassium carbonate Inorganic materials 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 238000012910 preclinical development Methods 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 238000002203 pretreatment Methods 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 150000003138 primary alcohols Chemical class 0.000 description 1
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 1
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 239000003909 protein kinase inhibitor Substances 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 239000008213 purified water Substances 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 150000003217 pyrazoles Chemical class 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 239000012066 reaction slurry Substances 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 108700015048 receptor decoy activity proteins Proteins 0.000 description 1
- 230000007115 recruitment Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 238000007634 remodeling Methods 0.000 description 1
- 238000009877 rendering Methods 0.000 description 1
- 230000008521 reorganization Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 239000002342 ribonucleoside Substances 0.000 description 1
- 229940043230 sarcosine Drugs 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 150000003333 secondary alcohols Chemical class 0.000 description 1
- 239000010703 silicon Substances 0.000 description 1
- 229910052710 silicon Inorganic materials 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- LYPGDCWPTHTUDO-UHFFFAOYSA-M sodium;methanesulfinate Chemical compound [Na+].CS([O-])=O LYPGDCWPTHTUDO-UHFFFAOYSA-M 0.000 description 1
- 229960003787 sorafenib Drugs 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- KXCAEQNNTZANTK-UHFFFAOYSA-N stannane Chemical class [SnH4] KXCAEQNNTZANTK-UHFFFAOYSA-N 0.000 description 1
- 229910000080 stannane Inorganic materials 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 239000011550 stock solution Substances 0.000 description 1
- 201000011549 stomach cancer Diseases 0.000 description 1
- 208000023516 stroke disease Diseases 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 125000005346 substituted cycloalkyl group Chemical group 0.000 description 1
- 239000001384 succinic acid Substances 0.000 description 1
- FDDDEECHVMSUSB-UHFFFAOYSA-N sulfanilamide Chemical compound NC1=CC=C(S(N)(=O)=O)C=C1 FDDDEECHVMSUSB-UHFFFAOYSA-N 0.000 description 1
- 230000006103 sulfonylation Effects 0.000 description 1
- 238000005694 sulfonylation reaction Methods 0.000 description 1
- 239000011593 sulfur Substances 0.000 description 1
- WINHZLLDWRZWRT-ATVHPVEESA-N sunitinib Chemical compound CCN(CC)CCNC(=O)C1=C(C)NC(\C=C/2C3=CC(F)=CC=C3NC\2=O)=C1C WINHZLLDWRZWRT-ATVHPVEESA-N 0.000 description 1
- 229960001796 sunitinib Drugs 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 230000002459 sustained effect Effects 0.000 description 1
- 229940034785 sutent Drugs 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 230000009897 systematic effect Effects 0.000 description 1
- 230000008685 targeting Effects 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 229910052723 transition metal Inorganic materials 0.000 description 1
- 238000007832 transition metal-catalyzed coupling reaction Methods 0.000 description 1
- 150000003624 transition metals Chemical class 0.000 description 1
- 108010075758 trebananib Proteins 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- 125000003866 trichloromethyl group Chemical group ClC(Cl)(Cl)* 0.000 description 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 1
- 125000001889 triflyl group Chemical group FC(F)(F)S(*)(=O)=O 0.000 description 1
- JLTRXTDYQLMHGR-UHFFFAOYSA-N trimethylaluminium Chemical compound C[Al](C)C JLTRXTDYQLMHGR-UHFFFAOYSA-N 0.000 description 1
- 125000003258 trimethylene group Chemical group [H]C([H])([*:2])C([H])([H])C([H])([H])[*:1] 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 125000005455 trithianyl group Chemical group 0.000 description 1
- 238000001665 trituration Methods 0.000 description 1
- 210000004881 tumor cell Anatomy 0.000 description 1
- 239000005483 tyrosine kinase inhibitor Substances 0.000 description 1
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 1
- 238000004704 ultra performance liquid chromatography Methods 0.000 description 1
- 241000701161 unidentified adenovirus Species 0.000 description 1
- VBEQCZHXXJYVRD-GACYYNSASA-N uroanthelone Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CS)C(=O)N[C@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)NCC(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)C(C)C)[C@@H](C)O)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CCSC)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CS)NC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC(N)=O)C(C)C)[C@@H](C)CC)C1=CC=C(O)C=C1 VBEQCZHXXJYVRD-GACYYNSASA-N 0.000 description 1
- LSGOVYNHVSXFFJ-UHFFFAOYSA-N vanadate(3-) Chemical compound [O-][V]([O-])([O-])=O LSGOVYNHVSXFFJ-UHFFFAOYSA-N 0.000 description 1
- 230000002792 vascular Effects 0.000 description 1
- 230000006496 vascular abnormality Effects 0.000 description 1
- 230000008728 vascular permeability Effects 0.000 description 1
- 230000004865 vascular response Effects 0.000 description 1
- 230000006426 vascular sprouting Effects 0.000 description 1
- 230000004862 vasculogenesis Effects 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 235000013311 vegetables Nutrition 0.000 description 1
- 201000009371 venous hemangioma Diseases 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 210000004269 weibel-palade body Anatomy 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 239000012224 working solution Substances 0.000 description 1
- 125000001834 xanthenyl group Chemical group C1=CC=CC=2OC3=CC=CC=C3C(C12)* 0.000 description 1
- 238000011911 α-alkylation Methods 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/78—Carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/84—Nitriles
- C07D213/85—Nitriles in position 3
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/06—Antiarrhythmics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Definitions
- the present invention relates to substituted pyrazolopyridine compounds of general formula (I) and salts thereof, to pharmaceutical compositions comprising said substituted pyrazolopyridine compounds, to methods of preparing said substituted pyrazolopyridines, as well as to uses thereof.
- Dysregulated vascular growth plays a critical role in a variety of inflammatory diseases, in particular psoriasis, delayed type hypersensitivity, contact dermatitis, asthma, multiple sclerosis, restenosis, rheumatoid arthritis and inflammatory bowl disease.
- Aberrant vascular growth is also involved in neovascular ocular diseases such as age-related macular degeneration and diabetic retinopathy.
- sustained vascular growth is accepted as one hallmark of cancer development (Hanahan, D.; Weinberg, R. A. Cell 2000, 100, 57).
- tumors initially grow either as an avascular mass or by co-opting existing host vessels, growth beyond a few mm 3 in size is depending on the induction of vessel neogrowth in order to sufficiently provide the tumor with oxygen and nutrients.
- Induction of angiogenesis is a prerequisite that the tumor surpasses a certain size (the so called angiogenic switch).
- An intricate signaling interaction network between cancer cells and the tumor microenvironment triggers the induction of vessel growth from existing vasculature.
- the dependence of tumors on neovascularization has led to a new treatment paradigm in cancer therapy (Ferrara et ⁇ /. Nature 2005, 438, 967; Carmeliet Nature 2005, 438, 932).
- Blocking tumor neovascularization by small molecule or antibody-mediated inhibition of relevant signal transduction pathways holds a great promise for extending currently available therapy options.
- angiogenesis The development of the cardiovascular system involves two basic stages. In the initial vasculogenesis stage, which only occurs during embryonal development, angioblasts differentiate into endothelial cells which subsequently form a primitive vessel network. The subsequent stage, termed angiogenesis, involves the remodeling of the initial vasculature and sprouting of new vessels (Risau, W. 2007 06 1 1
- angiogenesis occurs in wound healing, muscle growth, the female cycle and in the above mentioned disease states.
- receptor tyrosine kinases of the vascular endothelial growth factor (VEGF) family and the Tie (tyrosine kinase with immunoglobulin and epidermal growth factor homology domain) receptor tyrosine kinases are essential for both developmental and disease-associated angiogenesis (Ferrara et al Nat. Med. 2003, 9, 669; Dumont et al. Genes Dev. 1994, S, 1897; Sato et al. Nature 1995, 376, 70).
- Tie2 receptor tyrosine kinase is selectively expressed on endothelial cells (EC) of the adult vasculature (Schlaeger et al. Proc. Nat. Acad. Sci. USA 1997, 94, 3058). lmmunohistochemical analysis demonstrated the expression of Tie2 in adult rat tissues undergoing angiogenesis. During ovarian folliculogenesis, Tie2 is expressed in neovessels of the developing corpus luteum.
- angiopoietins 1 to 4 - have been identified for the type 1 transmembrane Tie2 (also named Tek) receptor, while no ligands have been identified so far for the Tie1 receptor. Binding of the extracellular Tie2 domain to the C-terminal fibrinogen-like domains of the various angiopoietins leads to significantly different cellular effects. In addition, heterodimerizations between Tie1 and Tie2 receptors have been postulated to influence ligand binding.
- Binding of Ang1 to Tie2 expressed on EC induces receptor cross-phosphorylation and kinase activation thus triggering various intracellular signaling pathways.
- the intracellular C-terminal tail of the Tie2 protein plays a crucial role in Tie2 signaling (Shewchuk et al. Structure 2000, 8, 1105).
- a conformational change is induced which removes the C-tail out of its inhibitory conformation thus allowing kinase activation by cross-phoshorylation of various Tyr residues in the C- tail, which subsequently function as docking sites for phosphotyrosine-binding (PTB) site possessing down-stream mediators.
- PTB phosphotyrosine-binding
- Ang1 activation of Tie2 includes inhibition of EC apoptosis, stimulation of EC migration and blood vessel reorganization, suppression of inflammatory gene expression and suppression of vascular permeability (Brindle et al. Circ. Res. 2006, 98, 1014). In 2007 06 11 contrast to VEGF-VEGFR signaling in EC, Ang1 activation of Tie2 does not stimulate EC proliferation in the majority of published assay settings.
- the anti-apoptotic effect of Tie2 signaling was shown to be mediated mainly by the PI3K-Akt signaling axis which is activated by binding of the regulatory p85 subunit of PI3K to Y1102 in the Tie2 C-tail (DeBusk et al. Exp. Cell. Res. 2004, 298, 167; Papapetropoulos et al. J. Biol. Chem. 2000, 275, 9102; Kim et al. Circ. Res. 2000, 86, 24).
- the chemotactic response downstream of the activated Tie2 receptor requires crosstalk between PI3K and the adaptor protein Dok-R.
- PI3K-mediated recruitment of the adaptor protein ShcA to Y1102 of the Tie2 C-tail is also believed to induce cellular sprouting and motility effects involving activation of endothelial nitric oxide synthase (eNOS), focal adhesion kinase (FAK) and the GTPases RhoA and Rac1.
- eNOS endothelial nitric oxide synthase
- FK focal adhesion kinase
- RhoA and Rac1 GTPases
- Other downstream mediators of Tie2 signaling include the adaptor protein Grb2, which mediates Erk1 /2 stimulation, and the SHP-2 phosphatase.
- basal activation of the Tie2 pathway by Ang1 is believed to maintain quiescence and integrity of the endothelium of the adult vasculature by providing a cell survival signal for ECs and by maintaining the integrity of the EC lining of blood vessels (Peters et al. Recent Prog. Horm. Res. 2004, 59, 51 ).
- Ang2 In contrast to Ang1 , Ang2 is not able to activate Tie2 on EC unless Ang2 is present in high concentration or for prolonged periods. However, Ang2 functions as a Tie2 agonist in non-endothelial cells transfected with Tie2. The structural basis for this context-dependence of the Ang2-Tie2 interaction is to date not understood.
- Ang2 functions as Tie2 antagonist and thus blocks the agonistic activity of Ang1 (Maisonpierre et al. Science 1997, 277, 55). Ang2 binding to Tie2 prevents Ang1 -mediated Tie2 activation which leads to vessel destabilization and results in vessel regression in the absence of pro-angiogenic stimuli such as VEGF. While Ang1 is widely expressed by periendothelial cells in 2007 06 11 quiescent vasculature such as pericytes or smooth muscle cells, Ang2 expression occurs in areas of ongoing angiogenesis. Ang2 can be stored in Weibel-Palade bodies in the cytoplasm of EC allowing for a quick vascular response upon stimulation.
- Ang1 and Ang2 are expressed in the corpus luteum, with Ang2 localizing to the leading edge of proliferating vessels and Ang1 localizing diffusively behind the leading edge.
- Ang2 expression is inter alia initiated by hypoxia (Pichiule et al. J. Biol. Chem. 2004, 279, 12171 ).
- Ang2 is upregulated in the tumor vasculature and represents one of the earliest tumor markers.
- Ang2 expression induces vessel permeability and - in the presence of e.g. pro-angiogenic VEGF - triggers angiogenesis.
- Tie2 RTK an attractive target for anti-angiogenesis therapy in diseases caused by or associated with dysregulated vascular growth.
- targeting the Tie2 pathway alone will be sufficient to achieve efficacious blockade of neovascularization. In certain diseases or disease subtypes it might be necessary or more efficacious to block several angiogenesis-relevant signaling pathways simultaneously.
- Integrin ⁇ 5 ⁇ l associates constitutively with Tie2 and increases the receptor's binding affinity for Ang1 resulting in initiation of downstream signaling at lower Ang1 effector concentrations in situations where integrin ⁇ 5 ⁇ l is present.
- the recently solved crystal structure of the Tie2-Ang2 complex suggests however that neither the oligomerization state nor a different binding mode causes the opposing cellular effects (Barton et al. Nat. Struc. MoI. Biol. 2006, advance online publication).
- Ang1 -Tie2 signaling plays also a role in the development of the lymphatic system and in lymphatic maintenance and sprouting (Tammela et al. Blood 2005, 105, 4642).
- An intimate cross-talk between Tie2 and VEGFR-3 signaling in lymphangiogenesis seems to equal the Tie2-KDR cross-talk in blood vessel angiogenesis.
- Tie2 signaling in the development and maintenance of the vasculature.
- Disruption of Tie2 function in Tie2 ⁇ transgenic mice leads to early embryonic lethality between days 9.5 and 12.5 as a consequence of vascular abnormalities.
- Tie2 " ⁇ embryos fail to develop the normal vessel hierachy suggesting a failure of vascular branching and differentiation.
- the heart and vessels in Tie2 A embryos show a decreased lining of EC and a loosened interaction between EC and underlying pericyte/smooth muscle cell matrix.
- Mice lacking functional Ang1 expression and mice overexpressing Ang2 display a phenotype reminiscent of the phenotype of Tie2 ⁇ mice (Suri et al.
- Ang2 ⁇ mice have profound defects in the growth and patterning of lymphatic vasculature and fail to remodel and regress the hyaloid vasculature of the neonatal lens (Gale et al. Dev. Cell 2002, 3, 411 ).
- Ang1 rescued the lymphatic defects, but not the vascular remodeling defects. Therefore, Ang2 might function as a Tie2 antagonist in blood vasculature but as a Tie2 agonist in developing lymph vasculature suggesting redundant roles of Ang1 and Ang2 in lymphatic development. 2007 06 11
- Tie2 pathway Aberrant activation of the Tie2 pathway is involved in various pathological settings. Activating Tie2 mutations leading to increased ligand-dependent and ligand- independent Tie2 kinase activity cause inherited venous malformations (Vikkula et al. Cell 1996, 87, 1181 ). Increased Ang1 mRNA and protein levels as well as increased Tie2 activation have been reported in patients with pulmonary hypertension (PH). Increased pulmonary arterial pressure in PH patients results from increased coverage of pulmonary arterioles with smooth muscle cells (Sullivan et al. Proc. Natl. Acad. Sci. USA 2003, 100, 12331 ).
- Tie2 and the ligands Ang1 and Ang2 are greatly upregulated in lesions, whereas a significant decrease in expression of Tie2 and ligands occur under anti-psoriatic treatment (Kuroda et al. J. Invest. Dermatol 2001 , 116, 713).
- Direct association of pathogenesis of disease with Tie2 expression has been demonstrated recently in transgenic mice overexpressing Tie2 (Voskas et al. Am. J. Pathol. 2005, 166, 843).
- a psoriasis-like phenotype such as epidermal thickening, rete ridges and lymphocyte infiltration.
- Tie2 expression was investigated in human breast cancer specimens and Tie2 expression was found in the vascular endothelium both in normal breast tissue as well as in tumor tissue. The proportion of Tie2-positive microvessels was increased in tumors as compared to normal breast tissue (Peters et al. Br. J. Cane. 1998, 77, 51 ). However, significant heterogeneity in endothelial Tie2 expression was observed in clinical specimen from a variety of human cancers (Fathers et al. Am. J. Path. 2005, 167, 1753).
- Tie2 and angiopoietins were found to be highly expressed in the cytoplasm of human colorectal adenocarcinoma cells indicating at the potential presence of an autocrine/ paracrine growth loop in certain cancers (Nakayama et al. World J. Gastroenterol. 2005, 11, 964).
- a similar autocrine/paracrine Ang1 -Ang2-Tie2 loop was postulated for certain human gastric cancer cell lines (Wang et al. Biochem. Biophys. Res. Comm. 2005, 337, 386).
- Ang1 -Tie2 signaling axis The relevance of the Ang1 -Tie2 signaling axis was challenged with various biochemical techniques. Inhibition of Ang1 expression by an antisense RNA 2007 06 11 approach resulted in decreased xenograft tumor growth (Shim et al. Int. J. Cane. 2001 , 94, 6 ; Shim et al. Exp. Cell Research 2002, 279, 299). However, other studies report that experimental overexpression of Ang1 in tumor models leads to decreased tumor growth (Hayes et al. Br. J. Cane. 2000, 83, 1154; Hawighorst et al. Am. J. Pathol. 2002, 160, 1381 ; Stoeltzing et al. Cancer Res.
- Gene therapy by adenoviral vector delivered sTie2 was capable of reducing tumor growth rates of a murine mammary carcinoma and a murine melanoma and resulted in reduction of metastasis formation (Lin et al. Proc. Natl. Acad. Sci. USA 1998, 95, 8829). Similar effects were observed with related sTie2 constructs (Sieffle et al. Cancer Res. 1999, 59, 3185) and a Tek- Fc construct (Fathers et al. Am. J. Path. 2005, 167, 1753).
- Adenovirus-delivered anti-Tie2 intrabodies were shown to inhibit growth of a human Kaposi's sarcoma and a human colon carcinoma upon peritumoral administration (Popkov et al. Cancer Res. 2005, 65, 972). Histopathologic analysis revealed a marked decrease in vessel density in treated vs. control tumors. Phenotypic simultaneous knockout of KDR and Tie2 by an adenovirus delivered intradiabody resulted in significantly higher growth inhibition of a human melanoma xenograft model than KDR knockout alone (Jendreyko et al. Proc. Natl. Acad. Sci. USA 2005, 102, 8293).
- small molecule inhibitors of the Tie2 kinase will be as efficacious in inhibiting angiogenesis as e.g. ligand antibodies, soluble decoy receptors or receptor intrabodies.
- angiogenesis e.g. ligand antibodies, soluble decoy receptors or receptor intrabodies.
- inhibition of Tie2 signaling alone might not be sufficient to induce an adequate antiangiogenic effect.
- Simultaneous inhibition of several angiogenesis relevant signaling pathways could overcome such inadequacies.
- novel chemotypes for small mocule inhibitors of the Tie2 kinase Fine tuning of additive anti-angiogenic activities as well as pharmacokinetic parameters such as e.g. solubility, membrane permeability, tissue distribution and metabolism will finally allow for chosing compounds of accurate profiles for various diseases caused by or associated with dysregulated vascular growth.
- Avastin (Bevacizumab), a VEGF neutralizing antibody, blocks KDR and VEGFR1 signaling and has been approved for first-line treatment of metastatic colorectal cancer.
- the small molecule multi-targeted kinase inhibitor Nexavar (Sorafenib) inhibits inter alia members of the VEGFR family and has been approved for the treatment of advanced renal cell carcinoma.
- Sutent (Sunitinib), another multi-targeted kinase inhibitor with activity vs. VEGFR 2007 06 11 family members, has been approved by the FDA for treatment of patients with gastrointestinal stromal tumors (GIST) or advanced kidney tumors.
- GIST gastrointestinal stromal tumors
- Several other small molecule inhibitors of angiogenesis-relevant targets are in clinical and preclinical development.
- AMG-386 an angiopoietin-targeting recombinant Fc fusion protein
- ABT-869, GW697465A and A- 422885.88 BSF466895
- Pyrazolopyridines have been disclosed as antimicrobiotic substances (e.g. Attaby et al., Phosphorus, Sulfur and Silicon and the related Elements 1999, 149, 49-64; Goda et al. Bioorg. Med. Chem. 2004, 12, 1845). US5478830 further discloses fused heterocycles for the treatment of atherosclerosis. Pyrazolopyridines have also been described as PDE4-lnhibitors (WO2006004188, US20060004003).
- WO 2001019828 discloses 125 templates, including 3-amino-1 H-pyrazolopyridines, as modulators of the activity of receptor and non-receptor tyrosine and serine/threonine kinases.
- WO 2004113304 discloses 3-amino-indazoles as inhibitors of protein tyrosine kinases, particularly as inhibitors as KDR kinase.
- WO 2006050109 discloses 3- aminopyrazolopyridines as tyrosine kinase inhibitors, particularly as KDR kinase inhibitors. 2007 06 11
- WO 2002024679 discloses tetrahydropyridine-substituted pyrazolopyridines as IKK inhibitors.
- WO 2004076450 further discloses 5-heteroaryl-pyrazolopyridines as p38 inhibitors.
- WO2005044181 discloses pyrazolopyridines as AbI kinase inhibitors.
- the insulin/IGF-1 receptor inhibitor NVP- ADW742 for example at concentrations which inhibit both the insulin and IGF-1 receptors strongly potentiated desoxycholic acid-induced apoptotic cell death, which as a consequence predicts strong liver toxic effects in case of impaired bile flow (Dent et al. Biochem. Pharmacol. 2005, 70, 1685). Even worse, inhibition of the neuronal insulin receptor causes Alzheimer-like disturbances in oxidative/energy brain metabolism (Hoyer et al. Ann. N. Y. Acad. Sci. 1999, 893, 301 ).
- compounds of the present invention which feature a pyrazole hinge binding moiety lacking the 3-amino group not only display potent activity as inhibitors of Tie2 kinase. Even more surprisingly, compounds of the present invention display a more selective inhibition of Tie2 kinase relative to not desired target kinases, particularly the insulin receptor kinase (InsR).
- InsR insulin receptor kinase
- Such a pharmacological profile is highly desirable not only for treating diseases of dysregulated vascular growth or diseases which are accompanied with dysregulated vascular growth, in particular solid tumors and metastases thereof, but for treating non-oncological diseases of dysregulated vascular growth or non-oncological diseases which are accompanied with dysregulated vascular growth, such as retinopathy, other angiogenesis dependent diseases of the eye, in particular cornea transplant rejection or age-related macular degeneration, rheumatoid arthritis, and other inflammatory diseases associated with angiogenesis, in particular psoriasis, delayed type hypersensitivity, contact dermatitis, asthma, multiple sclerosis, restenosis, pulmonary hypertension, stroke, and diseases of the bowel, diseases such as coronary and peripheral artery disease, wherein treatment of the said non-oncological diseases are preferably accomplished with less side-effects than in the treatment of oncological diseases.
- the invention thus relates to compounds of general formula (I) : 2007 06 1 1
- R 1 represents H or -C(O)R b , or is selected from the group comprising, preferably consisting of, CrC 6 -alkyl, C ⁇ -C ⁇ -alkenyl, C 2 -C 6 -alkynyl, C 3 - C 10 -cycloalkyl, C 3 -Cio-heterocycloalkyl, wherein said residues are unsubstituted or substituted one or more times, independently from each other, with R 6 ;
- R 2 stands for hydrogen, -NR d1 R d2 , -C(O)R b , or is selected from the group comprising, preferably consisting of, CrC 6 -alkyl, C 2 -C 6 -alkenyl, C 2 -C 6 - alkynyl, C 3 -Cio-cycloalkyl, C 3 -Cio-heterocycloalkyl, aryl, heteroaryl, wherein said residues are unsubstituted or singly or multiply substituted independently from each other with R 7 ;
- R s is selected from the group comprising, preferably consisting of, hydrogen, CrC 6 -alkyl, CrC 6 -alkoxy, CrC 6 -haloalkyl, Ci-C 6 -haloalkoxy, hydroxy, amino, halogen, cyano ;
- R 4 , R 5 , R 6 , R 7 , R 1 independently from each other, are selected from the group comprising, preferably consisting of, hydrogen, CrC 6 -alkyl, C 3 -C 10 - cycloalkyl, C 3 -Cio-heterocycloalkyl, CrC ⁇ -haloalkyl, Ci-C 6 -haloalkoxy, aryl, heteroaryl, hydroxy, amino, halogen, cyano, nitro, -C(O)R b , - S(O) 2 R b , -OR C , -NR d1 R d2 , -OP(O)(OR C ) 2 , wherein d-C 6 -alkyl, C 3 -C 10 - heterocycloalkyl and C 3 -Ci 0 -cycloalkyl of R 4 , R 5 , R 6 , and R 7 , are optionally substituted one or more times,
- R c is selected from the group comprising, preferably consisting of, hydrogen, -C(O)R 6 , C r C 6 -alkyl, C r C 6 -haloalkyl C 3 -Ci 0 -cycloalkyl, C 3 - C 10 -heterocycloalkyl, wherein CrC ⁇ -alkyl, Ci-C 6 -haloalkyl, C 3 -C 10 - cycloalkyl, C 3 -Cio-heterocycloalkyl are optionally substituted one or more times, in the same way or differently, with hydroxyl, halogen, aryl, or -NR d1 R d2 , and wherein C r C 6 -alkyl, C r C 6 -haloalkyl, C 3 -C 10 - cycloalkyl, C 3 -Ci 0 -heterocycloalkyl are optionally substituted once with -OR C ,
- R d1 and R d2 together with the nitrogen atom to which they are attached, form a 3 to 10 membered heterocycloalkyl ring, whereby the carbon backbone of this heterocycloalkyl ring is optionally interrupted one or more times, in the same way or differently, by a member of the group comprising, preferably consisting of, NH, NR d1 , oxygen or sulphur, and is optionally interrupted one or more times, in the same way or differently, with a -C(O)-, -S(O)- , and/or -S(O) 2 - group, and optionally contains one or more double bonds ;
- A is selected from the group comprising, preferably consisting of,
- B is a bond or a group selected from the group comprising, preferably consisting of CrC 6 -alkylene, C 3 -C 10 -cycloalkylene, C 3 -Ci 0 - heterocycloalkylene ;
- D, E are, independently from each other, arylene or heteroarylene ; and q represents an integer of 0, 1 , or 2 ;
- R 3 , R b , R c , R d1 , R d2 or R 8 when one or more of R 3 , R b , R c , R d1 , R d2 or R 8 is (are) present in one position in the molecule as well as in one or more further positions in the molecule, said R 3 , R b , R c , R d1 , R d2 or R 8 has (have), independently from each other, the same meanings as defined above in said first position in the molecule and in said second or further positions in the molecule, it being possible for the two or more occurrences of R 3 , R b , R c , R d1 , R d2 or R 8 within a single molecule to be identical or different.
- R a when R a is present twice in the molecule, then the meaning of the first R 3 may be H, for example, and the meaning of the second R a may be methyl, for example.
- the present invention relates to compounds of general formula (I), in which :
- R 1 represents H or -C(O)R b , or is selected from the group comprising, preferably consisting of, C r C 6 -alkyl, C 2 -C 6 -alkenyl, C 2 -C 6 -alkynyl, C 3 -
- R 2 stands for hydrogen, -NR d1 R d2 , -C(O)R b , or is selected from the group comprising, preferably consisting of, Ci-C 6 -alkyl, C 2 -C 6 -alkenyl, C 2 -C 6 - alkynyl, C 3 -Cio-cycloalkyl, C 3 -Cio-heterocycloalkyl, aryl, heteroaryl, wherein said residues are unsubstituted or singly or multiply substituted independently from each other with R 7 ; 2007 06 11
- R 3 is selected from the group comprising, preferably consisting of, hydrogen, Ci-C 6 -alkyl, Ci-C ⁇ -alkoxy, CrC ⁇ -haloalkyl, CrC 6 -haloalkoxy, hydroxy, amino, halogen ;
- R 4 , R 5 , R 6 , R 7 , R 8 independently from each other, are selected from the group comprising, preferably consisting of, hydrogen, C r C 6 -alkyl, C 3 -Ci 0 - cycloalkyl, C 3 -Cio-heterocycloalkyl, CrC ⁇ -haloalkyl, CrC 6 -haloalkoxy, hydroxy, amino, halogen, -C(O)R b , -S(O) 2 R b , OR C , NR d1 R d2 , OP(O)(OR C ) 2 , wherein CrC 6 -alkyl, C 3
- R c is selected from the group comprising, preferably consisting of, hydrogen, -C(O)R b , Ci-C 6 -alkyl, C r C 6 -haloalkyl C 3 -C 10 -cycloalkyl, C 3 - Cio-heterocycloalkyl, wherein CrC 6 -alkyl, CrC 6 -haloalkyl, C 3 -C 10 - cycloalkyl, C 3 -Cio-heterocycloalkyl are optionally substituted one or more times, in the same way or differently, with hydroxyl, halogen, aryl, or -NR d1 R d2 , and wherein C r C 6 -alkyl, C r C 6 -haloalkyl, C 3 -Ci 0 - cycloalkyl, C 3 -Ci 0 -heterocycloalkyl are optionally substituted once with -0R c
- R d1 and R d2 together with the nitrogen atom to which they are attached, form a 3 to 10 membered heterocycloalkyl ring, whereby the carbon backbone of this heterocycloalkyl ring is optionally interrupted one or more times, the same way or differently, by a member of the group comprising, preferably consisting of, NH, NR d1 , oxygen or sulphur, and is optionally interrupted one or more times, the same way or differently, with a -C(O)-, -S(O)- , and/or -S(O) 2 - group, and optionally contains one or more double bonds ;
- B is a bond or a group selected from the group comprising, preferably consisting of C r C 6 -alkylene, C 3 -C 10 -cycloalkylene, C 3 -Ci 0 - heterocycloalkylene ;
- D is phenylene ;
- E is phenylene or 5- or 6-membered heteroarylene ; and q represents an integer of 0 or 1 ;
- R a , R b , R c , R d1 , R d2 or R 8 when one or more of R a , R b , R c , R d1 , R d2 or R 8 is (are) present in one position in the molecule as well as in one or more further positions in the molecule, said R a , R b , R c , R d1 , R d2 or R 8 has (have), independently from each other, the same meanings as defined above in said first position in the molecule and in said second or further positions in the molecule, it being possible for the two or more occurrences of R 3 , R b , R c , R d1 , R d2 or R 8 within a single molecule to be identical or different.
- R a when R a is present twice in the molecule, then the meaning of the first R a may be H, for example, and the meaning of the second R 3 may be methyl, for example.
- the present invention relates to compounds of general formula (I), in which :
- R 1 represents H or -C(O)R b , or is selected from the group comprising, preferably consisting of, d-C 6 -alkyl, C 2 -C 6 -alkenyl, C 2 -C 6 -alkynyl, C 3 - 2007 06 11
- R 2 stands for hydrogen, -NR d1 R d2 , -C(O)R b , or is selected from the group comprising, preferably consisting of, CrC 6 -alkyl, C 2 -C 6 -alkenyl, C 2 -C 6 - alkynyl, C3-Cio-cycloalkyl, C3-Cio-heterocycloalkyl, aryl, heteroaryl, wherein said residues are unsubstituted or singly or multiply substituted independently from each other with R 7 ;
- R 3 is selected from the group comprising, preferably consisting of, hydrogen, d-C 6 -alkyl, CrC 6 -alkoxy, Ci-C 6 -haloalkyl, d-C 6 -haloalkoxy, hydroxy, amino, halogen ;
- R 8 independently from each other, are selected from the group comprising, preferably consisting of, hydrogen, d-C ⁇ -alkyl, C 3 -C 1O - cycloalkyl, C 3 -Cio-heterocycloalkyl, d-C 6 -haloalkyl, CrC 6 -haloalkoxy, hydroxy, amino, halogen, -C(O)R b , -S(O) 2 R 6 , OR C , NR d1 R d2 ,
- R a is selected from the group comprising, preferably consisting of, hydrogen or d-C 6 -alkyl ;
- R b is selected from the group comprising, preferably consisting of, hydroxyl, OR C , SR C , NR d1 R d2 ;
- R c is selected from the group comprising, preferably consisting of, hydrogen, -C(O)R b , C r C 6 -alkyl, C r C 6 -haloalkyl C 3 -C 10 -cycloalkyl, C 3 - do-heterocycloalkyl, wherein d-C ⁇ -alkyl, d-C 6 -haloalkyl, C 3 -C 1O - cycloalkyl, C 3 -do-heterocycloalkyl are optionally substituted one or more times with hydroxyl, halogen, aryl, or -NR d1 R d2 , and wherein C 1 -
- C 6 -alkyl, d-C 6 -haloalkyl, C 3 -C 1 o-cycloalkyl, Crdo-heterocycloalkyl are optionally substituted once with -0R c , or -OP(O)(OR C ) 2 ;
- R d1 , R d2 independently from each other are selected from the group comprising, preferably consisting of hydrogen, C r C 6 -alkyl, C 3 -C 1O - 2007 06 11 cycloalkyl, C3-Cio-heterocycloalkyl, C ⁇ -Cn-aryl, C 5 -C 10 -heteroaryl, or for a group -C(O)R 0 , -S(O) 2 R b , or C(O)NR d1 R d2 , wherein C r C 6 -alkyl, C 3 - Cio-cycloalkyl, C 3 -Ci 0 -heterocycloalkyl are optionally substituted one or more times, in the same way or differently, with halogen, hydroxy or an -0R c , -C(O)R b , -S(O) 2 R b , -OP(O)(OR C ) 2
- R d1 and R d2 together with the nitrogen atom to which they are attached, form a 3 to 10 membered heterocycloalkyl ring, whereby the carbon backbone of this heterocycloalkyl ring is optionally interrupted one or more times, the same way or differently, by a member of the group comprising, preferably consisting of, NH, NR d1 , oxygen or sulphur, and is optionally interrupted one or more times, the same way or differently, with a -C(O)-, -S(O)- , and/or -S(O) 2 - group, and optionally contains one or more double bonds ;
- A is selected from the group comprising, preferably consisting of,
- B is a bond or a group selected from the group comprising, preferably consisting of CrC 6 -alkylene, C 3 -Cio-cycloalkylene ;
- D is phenylene ;
- E is phenylene or 5- or 6-membered heteroarylene ; and q represents an integer of 0 or 1 ;
- R a , R b , R c , R d1 , R d2 or R 8 when one or more of R a , R b , R c , R d1 , R d2 or R 8 is (are) present in one position in the molecule as well as in one or more further positions in the molecule, said R a , R b , R c , R d1 , R d2 or R 8 has (have), independently from each other, the same meanings as defined above in said first position in the molecule and in said second or further positions in the molecule, it being possible for the two or more occurrences of R a , R b , R c , R d1 , R d2 or R 8 within a single molecule to be identical or different.
- R a when R a is present twice in the molecule, then the meaning of the first R a may be H, for example, and the meaning of the second R a may be methyl, for example.
- the present invention relates to compounds of general formula (I), in which :
- R 1 represents H or -C(O)R b , or is selected from the group comprising, preferably consisting of, d-C 6 -alkyl, C 3 -C 6 -cycloalkyl, C 3 -C 6 - heterocycloalkyl, wherein said residues are unsubstituted or substituted one or more times, independently from each other, with R 6 ;
- R 2 stands for hydrogen or is selected from the group comprising, preferably consisting of, Ci-C 6 -alkyl, C 3 -C 6 -cycloalkyl, C 3 -C 6 - heterocycloalkyl, aryl, heteroaryl, wherein said residues are unsubstituted or singly or multiply substituted independently from each other with R 7 ;
- R 3 is selected from the group comprising, preferably consisting of, hydrogen, CrC 6 -alkyl, CrC 6 -alkoxy, CrC ⁇ -haloalkyl, Ci-C 6 -haloalkoxy, hydroxy, amino, halogen ;
- R 4 is selected from the group comprising, preferably consisting of, hydrogen, Ci-C 6 -alkyl, CrC 6 -haloalkyl, CrC 6 -haloalkoxy, hydroxy, amino, halogen, 0R c , wherein Ci-C 6 -alkyl is optionally substituted one or more times with R 8 ;
- R 5 is selected from the group comprising, preferably consisting of, hydrogen, CrC 6 -alkyl, C 3 -C 6 -heterocycloalkyl, CrC 6 -haloalkyl, CrC 6 - haloalkoxy, hydroxy, amino, halogen, -C(O)R b , -S(O) 2 R 6 , OR C , NR d1 R d2 , wherein CrC ⁇ -alkyl and Cs-Q-heterocycloalkyl are optionally substituted one or more times with R 8 ;
- R 6 is selected from the group comprising, preferably consisting of, hydrogen, C 3 -C 6 -heterocycloalkyl, C r C 6 -haloalkoxy, hydroxy, amino, -C(O)R b , -S(O) 2 R", OR C , NR d1 R d2 , wherein C 3 -C 6 -heterocycloalkyl is optionally substituted one or more times with R 8 ;
- R 7 is selected from the group comprising, preferably consisting of, hydrogen, CrC 6 -alkyl, C 3 -C 6 -heterocycloalkyl, CrC 6 -haloalkyl, C 1 -C 6 - haloalkoxy, hydroxy, amino, -C(O)R b , -S(O) 2 R b , OR C , NR d1 R d2 , wherein Ci-C 6 -alkyl and C 3 -C 6
- R 8 is selected from the group comprising, preferably consisting of, Q-Ce- haloalkoxy, hydroxy, amino, halogen, -C(O)R b , -S(O) 2 R b , OR C , NR d1 R d2 ;
- R a is hydrogen
- R b is selected from the group comprising, preferably consisting of, OR C , NR d1 R d2 ;
- R c is selected from the group comprising, preferably consisting of, hydrogen, d-C ⁇ -alkyl, C 3 -C 6 -cycloalkyl, C 3 -C 6 -heterocycloalkyl, wherein d-C ⁇ -alkyl, C 3 -C 6 -cycloalkyl, C 3 -C 6 -heterocycloalkyl are optionally substituted one or more times, in the same way or differently, with -NR d1 R d2 , and wherein C r C 6 -alkyl, C 3 -C 6 -cycloalkyl,
- C 3 -C 6 -heterocycloalkyl are optionally substituted once with -OR C ;
- R d1 , R d2 independently from each other are selected from the group comprising, preferably consisting of hydrogen, CrC 6 -alkyl, C 3 -Ce- cycloalkyl, or for a -C(O)R C or C(O)NR d1 R d2 group, wherein C r C 6 -alkyl, and C 3 -C 6 -cycloalkyl are optionally substituted one or more times, in the same way or differently, with an -OR C , or -C(O)R b group, and wherein d-C ⁇ -alkyl, and C 3 -C 6 -cycloalkyl are optionally substituted once with an -NR d1 R d2 group ; or,
- R d1 and R d2 together with the nitrogen atom to which they are attached, form a 3 to 6 membered heterocycloalkyl ring, whereby the carbon backbone of this heterocycloalkyl ring is optionally interrupted one or more times, the same way or differently, by a member of the group comprising, preferably consisting of, NH, NR d1 , and oxygen ;
- A is selected from the group comprising, preferably consisting of, -C(O)-, -C(O)NR 3 -, -S(O) 2 - ;
- B is a bond or a group selected from the group comprising, preferably consisting of Ci-C 3 -alkylene, C 3 -cycloalkylene ;
- D and E are phenylene ; and q represents an integer of 0 ;
- R a , R b , R c , R d1 , R d2 or R 8 when one or more of R a , R b , R c , R d1 , R d2 or R 8 is (are) present in one position in the molecule as well as in one or more further positions in the molecule, said R a , R b , R c , R d1 , R d2 or R 8 has (have), independently from each 2007 06 11 other, the same meanings as defined above in said first position in the molecule and in said second or further positions in the molecule, it being possible for the two or more occurrences of R a , R b , R c , R d1 , R d2 or R 8 within a single molecule to be identical or different.
- R a when R a is present twice in the molecule, then the meaning of the first R a may be H, for example, and the meaning of the second R a may be methyl,
- the present invention relates to compounds of general formula (I), in which :
- R 1 represents H or -C(O)R b , or is selected from the group comprising, preferably consisting of, CrC 6 -alkyl, C 3 -C 6 -cycloalkyl, C 3 -C 6 - heterocycloalkyl, wherein said residues are unsubstituted or substituted one or more times, independently from each other, with R 6 ;
- R 2 stands for hydrogen or is selected from the group comprising, preferably consisting of, CrC ⁇ -alkyl, C 3 -C 6 -cycloalkyl, C 3 -C 6 - heterocycloalkyl, aryl, heteroaryl, wherein said residues are unsubstituted or singly or multiply substituted independently from each other with R 7 ;
- R 3 is selected from the group comprising, preferably consisting of, hydrogen, d-C ⁇ -alkyl, CrC ⁇ -alkoxy, CrC ⁇ -haloalkyl, d-C ⁇ -haloalkoxy, hydroxy, amino, halogen ;
- R 4 is selected from the group comprising, preferably consisting of, hydrogen, Ci-C 6 -alkyl, C r C 6 -haloalkyl, C r C 6 -haloalkoxy, hydroxy, amino, halogen, OR C , wherein CrC 6 -alkyl is optionally substituted one or more times with R 8 ;
- R 5 is selected from the group comprising, preferably consisting of, hydrogen, CrC 6 -alkyl, C 3 -C 6 -heterocycloalkyl, CrC 6 -haloalkyl, CrC 6 - haloalkoxy, hydroxy, amino, halogen, -C(O)R 6 , -S(O) 2 R b , 0R c , NR d1 R d2 , wherein CrC 6 -alkyl and C 3 -C 6 -heterocycloalkyl are optionally substituted one or more times with R 8 ;
- R 6 is selected from the group comprising, preferably consisting of, hydrogen, C3-C6-heterocycloalkyl, CrC ⁇ -haloalkoxy, hydroxy, amino, 2007 06 11
- R 7 is selected from the group comprising, preferably consisting of, hydrogen, Ci-C ⁇ -alkyi, C 3 -C 6 -heterocycloalkyl, Ci-C 6 -haloalkyl, d-C ⁇ - haloalkoxy, hydroxy, amino, -C(O)R 5 , -S(O) 2 R b , OR C , NR d1 R d2 , wherein
- CrC ⁇ -alkyl and C 3 -C 6 -heterocycloalkyl are optionally substituted one or more times with R 8 ;
- R 8 is selected from the group comprising, preferably consisting of, CrC ⁇ - haloalkoxy, hydroxy, amino, halogen, -C(O)R b , -S(O) 2 R b , 0R c , NR d1 R d2 ;
- R a is hydrogen ;
- R b is selected from the group comprising, preferably consisting of, OR C ,
- R c is selected from the group comprising, preferably consisting of, hydrogen, Ci-C 6 -alkyl, C 3 -C 6 -cycloalkyl, C 3 -C 6 -heterocycloalkyl, wherein C r C 6 -alkyl, C 3 -C 6 -cycloalkyl, C 3 -C 6 -heterocycloalkyl are optionally substituted one or more times with -NR d1 R d2 , and wherein d-C ⁇ -alkyl, Cs-C ⁇ -cycloalkyl, C 3 -C 6 -heterocycloalkyl are optionally substituted once with -OR C ;
- R d1 , R d2 independently from each other are selected from the group comprising, preferably consisting of hydrogen, Ci-Ce-alkyl, C 3 -C 6 - cycloalkyl, or for a -C(O)R C or C(O)NR d1 R d2 group, wherein C r C 6 -alkyl, and C 3 -C 6 -cycloalkyl are optionally substituted one or more times, in the same way or differently, with an -0R c , or -C(O)R b group, and wherein Ci-C 6 -alkyl, and C 3 -C 6 -cycloalkyl are optionally substituted once with an -NR d1 R d2 group ; or,
- R d1 and R d2 together with the nitrogen atom to which they are attached, form a 3 to 6 membered heterocycloalkyl ring, whereby the carbon backbone of this heterocycloalkyl ring is optionally interrupted one or more times, the same way or differently, by a member of the group comprising, preferably consisting of, NH, NR d1 , and oxygen ;
- A is -C(O)NR 3 - ;
- B is a bond or a group selected from the group comprising, preferably consisting of C r C 3 -alkylene, C 3 -cycloalkylene ;
- D and E are phenylene ; 2007 06 1 1 and q represents an integer of 0 ;
- R a , R b , R c , R d1 , R d2 or R 8 when one or more of R a , R b , R c , R d1 , R d2 or R 8 is (are) present in one position in the molecule as well as in one or more further positions in the molecule, said R a , R b , R c , R d1 , R d2 or R 8 has (have), independently from each other, the same meanings as defined above in said first position in the molecule and in said second or further positions in the molecule, it being possible for the two or more occurrences of R a , R b , R c , R d1 , R d2 or R 8 within a single molecule to be identical or different.
- R a when R a is present twice in the molecule, then the meaning of the first R a may be H, for example, and the meaning of the second
- R a may be methyl, for example.
- the first variant of the more particularly preferred embodiment, supra, of the present invention relates to compounds of general formula (I), in which :
- R 1 represents H or -C(O)R b , or is selected from the group comprising, preferably consisting of, CrC 6 -alkyl, C3-C6-cycloalkyl, C 3 -C 6 - heterocycloalkyl, wherein said residues are unsubstituted or substituted one or more times, independently from each other, with R 6 ;
- R 2 stands for hydrogen or is selected from the group comprising, preferably consisting of, d-C ⁇ -alkyl, C 3 -cycloalkyl ;
- R 3 is selected from the group comprising, preferably consisting of, hydrogen, methyl, fluoro, or chloro ;
- R 4 is selected from the group comprising, preferably consisting of, hydrogen, d-C ⁇ -alkyl, d-C 6 -haloalkyl, d-C 6 -haloalkoxy, hydroxy, amino, halogen, OR C , wherein d-C ⁇ -alkyl is optionally substituted one or more times with R 8 ;
- R 5 is selected from the group comprising, preferably consisting of, hydrogen, C r C 6 -alkyl, C 3 -C 6 -heterocydoalkyl, C r C 6 -haloalkyl, C 1 -C 6 - haloalkoxy, hydroxy, amino, halogen, -C(O)R b , -S(O) 2 R 6 , OR C , NR d1 R d2 , wherein C r C 6 -alkyl and C 3 -C 6 -heterocycloalkyl are optionally substituted one or
- R 6 is selected from the group comprising, preferably consisting of, hydrogen, C 3 -C 6 -heterocycloalkyl, Ci-C 6 -haloalkoxy, hydroxy, amino, -C(O)R b , -S(O) 2 R b , OR C , NR d1 R d2 , wherein C 3 -C 6 -heterocycloalkyl is optionally substituted one or more times with R 8 ; R 8 is selected from the group comprising, preferably consisting of, C 1 -C 6 - haloalkoxy, hydroxy, amino, halogen, -C(O)R b , -S(O) 2 R b , 0R c , NR d1 R d2 ;
- R a is hydrogen
- R b is selected from the group comprising, preferably consisting of, OR C ,
- R c is selected from the group comprising, preferably consisting of, hydrogen, C r C 6 -alkyl, C 3 -C 6 -cycloalkyl, C 3 -C 6 -heterocycloalkyl, wherein CrC 6 -alkyl, C 3 -C 6 -cycloalkyl, C 3 -C 6 -heterocycloalkyl are optionally substituted one or more times with -NR d1 R d2 , and wherein CrC ⁇ -alkyl, C 3 -C 6 -cycloalkyl, C 3 -C 6 -heterocycloalkyl are optionally substituted once with -OR C ;
- R d1 , R d2 independently from each other are selected from the group comprising, preferably consisting of hydrogen, CrC 6 -alkyl, C 3 -C 6 - cycloalkyl, or for a -C(O)R C or C(O)NR d1 R d2 group, wherein C r C 6 -alkyl, and C 3 -C 6 -cycloalkyl are optionally substituted one or more times, in the same way or differently, with an -0R c , or -C(O)R b group, and wherein CrC 6 -alkyl, and C 3 -C 6 -cycloalkyl are optionally substituted once with an -NR d1 R d2 group ; or,
- R d1 and R d2 together with the nitrogen atom to which they are attached, form a 3 to 6 membered heterocycloalkyl ring, whereby the carbon backbone of this heterocycloalkyl ring is optionally interrupted one or more times, the same way or differently, by a member of the group comprising, preferably consisting of, NH, NR d1 , and oxygen ;
- A is -C(O)NR 3 - ;
- B is a bond or a group selected from the group comprising, preferably consisting of Ci-C 3 -alkylene, C 3 -cycloalkylene ;
- D and E are phenylene ; and q represents an integer of 0 ; 2007 06 11 wherein, when one or more of R a , R b , R c , R d1 , R d2 or R 8 is (are) present in one position in the molecule as well as in one or more further positions in the molecule, said R a , R b , R c , R d1 , R d2 or R 8 has (have), independently from each other, the same meanings as defined above in said first position in the molecule and in said second or further positions in the molecule, it being possible for the two or more occurrences of R a , R b , R c , R d1 , R d2 or R 8 within a single molecule to be identical or different.
- R a is present twice in the molecule, then the meaning of the first R a may be H, for example, and the meaning of the second R a may be methyl, for example.
- the first variant of the more particularly preferred embodiment, supra, of the present invention relates to compounds of general formula (I), in which :
- R 1 represents H or CrC 6 -alkyl
- R 2 stands for hydrogen or is selected from the group comprising, preferably consisting of, CrC 6 -alkyl, C 3 -C 6 -cycloalkyl, C 3 -C 6 - heterocycloalkyl, aryl, heteroaryl, wherein said residues are unsubstituted or singly or multiply substituted independently from each other with R 7 ;
- R 3 is selected from the group comprising, preferably consisting of, hydrogen, methyl, fluor, chloro ;
- R 4 is selected from the group comprising, preferably consisting of, hydrogen, CrC 6 -alkyl, Ci-C 6 -haloalkyl, d-C 6 -haloalkoxy, hydroxy, amino, halogen, OR C , wherein C r C 6 -alkyl is optionally substituted one or more times with R 8 ;
- R 5 is selected from the group comprising, preferably consisting of, hydrogen, d-C 6 -alkyl, C 3 -C 6 -heterocycloalkyl, C r C 6 -haloalkyl, C 1 -C 6 - haloalkoxy, hydroxy, amino, halogen, -C(O)R b , -S(O) 2 R b , 0R c , NR d1 R d2 , wherein CrC ⁇ -alkyl and C 3 -C 6 -heterocycloalkyl are optionally substituted one or more times with R 8 ;
- R 7 is selected from the group comprising, preferably consisting of, hydrogen, CrC 6 -alkyl, C 3 -C 6 -heterocycloalkyl, C r C 6 -haloalkyl, C 1 -C 6 - haloalkoxy, hydroxy, amino, -C(O)R b , -S(O) 2 R b , OR C , NR d1 R d2 , wherein 2007 06 11
- CrC 6 -alkyl and C 3 -C 6 -heterocycloalkyl are optionally substituted one or more times with R 8 ;
- R 8 is selected from the group comprising, preferably consisting of, CrC 6 - haloalkoxy, hydroxy, amino, halogen, -C(O)R b , -S(O) 2 R b , 0R c , NR d1 R d2 ;
- R a is hydrogen ;
- R b is selected from the group comprising, preferably consisting of, OR C ,
- R c is selected from the group comprising, preferably consisting of, hydrogen, C r C 6 -alkyl, C 3 -C 6 -cycloalkyl, C 3 -C 6 -heterocycloalkyl, wherein CrC 6 -alkyl, C 3 -C 6 -cycloalkyl, C 3 -C 6 -heterocycloalkyl are optionally substituted one or more times with -NR d1 R d2 , and wherein C r C 6 -alkyl, C 3 -C 6 -cydoalkyl, C 3 -C 6 -heterocycloalkyl are optionally substituted once with -OR C ;
- R d1 , R d2 independently from each other are selected from the group comprising, preferably consisting of hydrogen, CrC 6 -alkyl, C 3 -C 6 - cycloalkyl, or for a -C(O)R C or C(O)NR d1 R d2 group, wherein C r C 6 -alkyl, and C 3 -C 6 -cycloalkyl are optionally substituted one or more times, in the same way or differently, with an -OR C , or -C(O)R b group, and wherein Ci-C 6 -alkyl, and C 3 -C 6 -cycloalkyl are optionally substituted once with an -NR d1 R d2 group ; or,
- R d1 and R d2 together with the nitrogen atom to which they are attached, form a 3 to 6 membered heterocycloalkyl ring, whereby the carbon backbone of this heterocycloalkyl ring is optionally interrupted one or more times, the same way or differently, by a member of the group comprising, preferably consisting of, NH, NR d1 , and oxygen ;
- A is -C(O)NR 3 - ;
- B is a bond or a group selected from the group comprising, preferably consisting of CrC 3 -alkylene, C 3 -cycloalkylene ;
- D and E are phenylene ; and q represents an integer of 0 ;
- R a , R b , R c , R d1 , R d2 or R 8 when one or more of R a , R b , R c , R d1 , R d2 or R 8 is (are) present in one position in the molecule as well as in one or more further positions in the 2007 06 11 molecule, said R a , R b , R c , R d1 , R d2 or R 8 has (have), independently from each other, the same meanings as defined above in said first position in the molecule and in said second or further positions in the molecule, it being possible for the two or more occurrences of R a , R b , R c , R d1 , R d2 or R 8 within a single molecule to be identical or different.
- R a when R a is present twice in the molecule, then the meaning of the first R a may be H, for example, and the meaning of the second R a may be methyl,
- the first variant of the more particularly preferred embodiment, supra, of the present invention relates to compounds of general formula (I), in which :
- R 1 represents H or CrC 6 -alkyl
- R 2 stands for hydrogen or is selected from the group comprising, preferably consisting of, CrC 6 -alkyl, C 3 -C 6 -heterocycloalkyl, aryl, heteroaryl, wherein said residues are unsubstituted or singly or multiply substituted independently from each other with R 7 ;
- R 3 is selected from the group comprising, preferably consisting of, hydrogen, methyl, fluor, chloro ;
- R 4 is selected from the group comprising, preferably consisting of, hydrogen, d-C 6 -alkyl, d-C 6 -haloalkyl, Ci-C 6 -haloalkoxy, hydroxy, amino, halogen, OR C , wherein Ci-C 6 -alkyl is optionally substituted one or more times with R 8 ;
- R 5 is selected from the group comprising, preferably consisting of, hydrogen, CrC 6 -alkyl, C 3 -C 6 -heterocycloalkyl, d-C ⁇ -haloalkyl, CrC 6 - haloalkoxy, hydroxy, amino, halogen, -C(O)R b , -S(O) 2 R 5 , 0R c , NR d1 R d2 , wherein d-C 6 -alkyl and C 3 -C 6 -heterocycloalkyl are optionally substituted one or more times with R 8 ;
- R 7 is selected from the group comprising, preferably consisting of, hydrogen, C r C 6 -alkyl, Cs-Q-heterocycloalkyl, d-C 6 -haloalkyl, C 1 -C 6 - haloalkoxy, hydroxy, amino, -C(O)R b , -S(O) 2 R b , OR C , NR d1 R d2 , wherein d-C 6 -alkyl and C 3 -C 6 -heterocycloalkyl are optionally substituted one or more times with R 8 ; 2007 06 11
- R 8 is selected from the group comprising, preferably consisting of, CrCo- haloalkoxy, hydroxy, amino, halogen, -C(O)R b , -S(O) 2 R b , OR C , NR d1 R d2 ;
- R a is hydrogen
- R b is selected from the group comprising, preferably consisting of, OR C , NR d1 R d2 ;
- R c is selected from the group comprising, preferably consisting of, hydrogen, d-C ⁇ -alkyl, C 3 -C 6 -cycloalkyl, C 3 -C 6 -heterocycloalkyl, wherein CrC 6 -alkyl, C 3 -C 6 -cycloalkyl, C 3 -C 6 -heterocycloalkyl are optionally substituted one or more times with -NR d1 R d2 , and wherein CrC 6 -alkyl, C 3 -C 6 -cycloalkyl, C 3 -C 6 -heterocycloalkyl are optionally substituted once with -OR C ;
- R d1 , R d2 independently from each other are selected from the group comprising, preferably consisting of hydrogen, CrC ⁇ -alkyl, C 3 -C 6 - cycloalkyl, or for a -C(O)R C or C(O)NR d1 R d2 group, wherein C r C 6 -alkyl and C 3 -C 6 -cycloalkyl are optionally substituted one or more times, in the same way or differently, with an -OR C or -C(O)R b group, and wherein CrC ⁇ -alkyl and C 3 -C 6 -cycloalkyl are optionally substituted once with an -NR d1 R d2 group ; or,
- R d1 and R d2 together with the nitrogen atom to which they are attached, form a 3 to 6 membered heterocycloalkyl ring, whereby the carbon backbone of this heterocycloalkyl ring is optionally interrupted one or more times, the same way or differently, by a member of the group comprising, preferably consisting of, NH, NR d1 , and oxygen ;
- A is -C(O)NR 3 - ;
- B is a bond or a group selected from the group comprising, preferably consisting of CrC 3 -alkylene, C 3 -cycloalkylene ;
- D and E are phenylene ; and q represents an integer of 0 ;
- R a , R b , R c , R d1 , R d2 or R 8 when one or more of R a , R b , R c , R d1 , R d2 or R 8 is (are) present in one position in the molecule as well as in one or more further positions in the molecule, said R a , R b , R c , R d1 , R d2 or R 8 has (have), independently from each other, the same meanings as defined above in said first position in the molecule 2007 06 11 and in said second or further positions in the molecule, it being possible for the two or more occurrences of R a , R b , R c , R d1 , R d2 or R 8 within a single molecule to be identical or different.
- the first variant of the more particularly preferred embodiment, supra, of the present invention relates to compounds of general formula (I), in which :
- R 1 is H or Ci-C 3 -alkyl
- R 2 stands for hydrogen or is selected from the group comprising, preferably consisting of, CrC 6 -alkyl, C 3 -cycloalkyl ;
- R 3 is selected from the group comprising, preferably consisting of, hydrogen, methyl, or fluoro;
- R 4 is selected from the group comprising, preferably consisting of, hydrogen, halogen, C r C 3 -alkyl, or Ci-C 3 -haloalkyl;
- R 5 is selected from the group comprising, preferably consisting of, hydrogen, Ci-C 3 -alkyl, CrC 3 -haloalkyl, CrC 3 -haloalkoxy, halogen, - OR C , -NR d1 R d2 , wherein C r C 3 -alkyl is optionally substituted by R 8 ;
- R 8 is selected from the group comprising, preferably consisting of, -OR C , and -NR d1 R d2 ;
- R a is hydrogen
- R c is selected from the group comprising, preferably consisting of, hydrogen, and Ci-C 3 -alkyl, wherein CrC 3 -alkyl is optionally substituted one or more times with -NR d1 R d2 , and wherein C r C 3 -alkyl is optionally substituted once with -OR C ;
- R d1 , R d2 independently from each other are selected from the group comprising, preferably consisting of hydrogen, Ci-C 3 -alkyl, wherein Cr
- C 3 -alkyl is optionally substituted one or more times, in the same way or differently, with an -0R c group, and wherein C r C 3 -alkyl is optionally substituted once with an -NR d1 R d2 group ; or,
- R d1 and R d2 together with the nitrogen atom to which they are attached, form a 6 membered heterocycloalkyl ring, whereby the carbon backbone of this heterocycloalkyl ring is optionally interrupted one time, by a 2007 06 11 member of the group comprising, preferably consisting of, NH, NR d1 , and oxygen ;
- A is -C(O)NR 3 - ;
- B is a bond
- D is para-phenylene
- E is phenylene ; q represents an integer of 0 ;
- R a , R b , R c , R d1 or R d2 when one or more of R a , R b , R c , R d1 or R d2 is (are) present in one position in the molecule as well as in one or more further positions in the molecule, said R a , R b , R c , R d1 or R d2 has (have), independently from each other, the same meanings as defined above in said first position in the molecule and in said second or further positions in the molecule, it being possible for the two or more occurrences of R a , R b , R c , R d1 or R d2 within a single molecule to be identical or different.
- R a when R a is present twice in the molecule, then the meaning of the first R a may be H, for example, and the meaning of the second R a may be methyl, for example.
- the present invention relates to compounds of general formula (I), in which :
- R 1 represents H or -C(O)R b , or is selected from the group comprising, preferably consisting of, C r C 6 -alkyl, C 3 -C 6 -cycloalkyl, C 3 -C 6 - heterocycloalkyl, wherein said residues are unsubstituted or substituted one or more times, independently from each other, with R 6 ;
- R 2 stands for hydrogen or is selected from the group comprising, preferably consisting of, C r C 6 -alkyl, C 3 -C 6 -cycloalkyl, C 3 -C 6 - heterocycloalkyl, aryl, heteroaryl, wherein said residues are unsubstituted or singly or multiply substituted independently from each other with R 7 ; 2007 06 11
- R 3 is selected from the group comprising, preferably consisting of, hydrogen, Ci-C 6 -alkyl, d-C 6 -alkoxy, d-C 6 -haloalkyl, d-C 6 -haloalkoxy, hydroxy, amino, halogen ;
- R 4 is selected from the group comprising, preferably consisting of, hydrogen, Ci-C 6 -alkyl, Ci-C ⁇ -haloalkyi, d-C 6 -haloalkoxy, hydroxy, amino, halogen, OR C , wherein CrC 6 -alkyl is optionally substituted one or more times with R 8 ;
- R 5 is selected from the group comprising, preferably consisting of, hydrogen, CrC 6 -alkyl, C 3 -C 6 -heterocycloalkyl, CrC 6 -haloalkyl, d-C 6 - haloalkoxy, hydroxy, amino, halogen, -C(O)R b , -S(O) 2 R b , OR C , NR d1 R d2 , wherein Ci-C 6 -alkyl and C 3 -C 6 -heterocycloalkyl are optionally substituted one or more times with R 8 ;
- R c is selected from the group comprising, preferably consisting of, hydrogen, Ci-C ⁇ -alkyl, C 3 -C ⁇ -cycloalkyl, C 3 -C 6 -heterocycloalkyl, wherein d-C 6 -alkyl, C 3 -C 6 -cycloalkyl, C 3 -C 6 -heterocycloalkyl are optionally substituted one or more times with -NR d1 R d2 , and wherein d-C 6 -alkyl, C 3 -C 6 -cycloalkyl, C 3 -C 6 -heterocycloalkyl are optionally substituted once with -OR C ; R d1 , R d2 independently from each other are selected from the group comprising, preferably consisting of hydrogen, d-C 6 -alkyl, C 3 -C 6 - 2007 06 11 cycloalkyl, or for
- R d1 and R d2 together with the nitrogen atom to which they are attached, form a 3 to 6 membered heterocycloalkyl ring, whereby the carbon backbone of this heterocycloalkyl ring is optionally interrupted one or more times, the same way or differently, by a member of the group comprising, preferably consisting of, NH, NR d1 , and oxygen ;
- A is -C(O)- or -S(O) 2 - ;
- B is a bond or a group selected from the group comprising, preferably consisting of C r C 3 -alkylene, C 3 -cycloalkylene ;
- D and E are phenylene ; and q represents an integer of 0 ;
- R a , R b , R c , R d1 , R d2 or R 8 when one or more of R a , R b , R c , R d1 , R d2 or R 8 is (are) present in one position in the molecule as well as in one or more further positions in the molecule, said R a , R b , R c , R d1 , R d2 or R 8 has (have), independently from each other, the same meanings as defined above in said first position in the molecule and in said second or further positions in the molecule, it being possible for the two or more occurrences of R a , R b , R c , R d1 , R d2 or R 8 within a single molecule to be identical or different.
- R a when R a is present twice in the molecule, then the meaning of the first R a may be H, for example, and the meaning of the second R a may be methyl, for example.
- the second variant of the more particularly preferred embodiment, supra, of the present invention relates to compounds of general formula (I), in which :
- R 1 represents H or -C(O)R b , or is selected from the group comprising, preferably consisting of, CrC 6 -alkyl, C 3 -C 6 -cycloalkyl, C 3 -C 6 - heterocycloalkyl, wherein said residues are unsubstituted or 2007 06 11 substituted one or more times, independently from each other, with R 6 ;
- R 2 stands for hydrogen or is selected from the group comprising, preferably consisting of, Ci-C 6 -alkyl, C 3 -cycloalkyl
- R 3 is selected from the group comprising, preferably consisting of, hydrogen, methyl, fluoro, or chloro ;
- R 4 is selected from the group comprising, preferably consisting of, hydrogen, CrC ⁇ -alkyl, d-C ⁇ -haloalkyl, CrC 6 -haloalkoxy, hydroxy, amino, halogen, 0R c , wherein CrC 6 -alkyl is optionally substituted one or more times with R 8 ;
- R 5 is selected from the group comprising, preferably consisting of, hydrogen, Ci-Ce-alkyl, C 3 -C 6 -heterocycloalkyl, CrC 6 -haloalkyl, Ci-C 6 - haloalkoxy, hydroxy, amino, halogen, -C(O)R b , -S(O) 2 R b , OR C , NR d1 R d2 , wherein d-C ⁇ -alkyl and C 3 -C 6 -heterocycloalkyl are optionally substituted one or more times with R 8 ;
- R 6 is selected from the group comprising, preferably consisting of, hydrogen, C 3 -C 6 -heterocycloalkyl, CrC 6 -haloalkoxy, hydroxy, amino, -C(O)R b , -S(O) 2 R b , OR C , NR d1 R d2 , wherein C 3 -C 6 -heterocycloalkyl is optionally substituted one or more times with R 8 ; R 8 is selected from the group comprising, preferably consisting of, C 1 -C 6 - haloalkoxy, hydroxy, amino, halogen, -C(O)R b , -S(O) 2 R b , 0R c , NR d1 R d2 ;
- R a is hydrogen
- R b is selected from the group comprising, preferably consisting of, OR C ,
- R c is selected from the group comprising, preferably consisting of, hydrogen, CrC 6 -alkyl, C 3 -C 6 -cycloalkyl, C 3 -C 6 -heterocycloalkyl, wherein CrC ⁇ -alkyl, C 3 -C ⁇ -cycloalkyl, C 3 -C 6 -heterocycloalkyl are optionally substituted one or more times with -NR d1 R d2 , and wherein d-C ⁇ -alkyl, C3-C6-cycloalkyl, C 3 -C 6 -heterocycloalkyl are optionally substituted once with -OR C ;
- R d1 , R d2 independently from each other are selected from the group comprising, preferably consisting of hydrogen, CrC ⁇ -alkyl, C 3 -C 6 - cycloalkyl, or for a -C(O)R C or C(O)NR d1 R d2 group, wherein C r C 6 -alkyl, and C 3 -C 6 -cycloalkyl are optionally substituted one or more times, in 2007 06 11 the same way or differently, with an -OR C , or -C(O)R b group, and wherein C r C 6 -alkyl, and C 3 -C 6 -cycloalkyl are optionally substituted once with an -NR d1 R d2 group ; or,
- R d1 and R d2 together with the nitrogen atom to which they are attached, form a 3 to 6 membered heterocycloalkyl ring, whereby the carbon backbone of this heterocycloalkyl ring is optionally interrupted one or more times, the same way or differently, by a member of the group comprising, preferably consisting of, NH, NR d1 , and oxygen ;
- A is -C(O)- or -S(O) 2 - ;
- B is a bond or a group selected from the group comprising, preferably consisting of C r C 3 -alkylene, C 3 -cycloalkylene ;
- D and E are phenylene ; and q represents an integer of 0 ;
- R a , R b , R c , R d1 , R d2 or R 8 when one or more of R a , R b , R c , R d1 , R d2 or R 8 is (are) present in one position in the molecule as well as in one or more further positions in the molecule, said R a , R b , R c , R d1 , R d2 or R 8 has (have), independently from each other, the same meanings as defined above in said first position in the molecule and in said second or further positions in the molecule, it being possible for the two or more occurrences of R a , R b , R c , R d1 , R d2 or R 8 within a single molecule to be identical or different.
- R a when R a is present twice in the molecule, then the meaning of the first R a may be H, for example, and the meaning of the second R a may be methyl, for example.
- the second variant of the more particularly preferred embodiment, supra, of the present invention relates to compounds of general formula (I), in which :
- R 1 represents H or d-C 6 -alkyl
- R 2 stands for hydrogen or is selected from the group comprising, preferably consisting of, CrC ⁇ -alkyl, C 3 -C 6 -cycloalkyl, C 3 -C 6 - heterocycloalkyl, aryl, heteroaryl, wherein said residues are 2007 06 11 unsubstituted or singly or multiply substituted independently from each other with R 7 ;
- R 3 is selected from the group comprising, preferably consisting of, hydrogen, methyl, fluor, chloro ;
- R 4 is selected from the group comprising, preferably consisting of, hydrogen, C r C 6 -alkyl, d-C 6 -haloalkyl, C r C 6 -haloalkoxy, hydroxy, amino, halogen, OR C , wherein CrC 6 -alkyl is optionally substituted one or more times with R 8 ;
- R 5 is selected from the group comprising, preferably consisting of, hydrogen, d-C ⁇ -alkyl, C 3 -C 6 -heterocycloalkyl, d-C ⁇ -haloalkyl, CrC 6 - haloalkoxy, hydroxy, amino, halogen, -C(O)R b , -S(O) 2 R b , OR C , NR d1 R d2 , wherein d-C 6 -alkyl and C 3 -C 6 -heterocycloalkyl are optionally substituted one or more times with R 8 ;
- R 7 is selected from the group comprising, preferably consisting of, hydrogen, d-Co-alkyl, C 3 -C 6 -heterocycloalkyl, CrC 6 -haloalkyl, C 1 -C 6 - haloalkoxy, hydroxy, amino, -C(O)R b , -S(O) 2 R b , OR C , NR d1 R d2 , wherein
- CrC ⁇ -alkyl and C 3 -C 6 -heterocycloalkyl are optionally substituted one or more times with R 8 ;
- R 8 is selected from the group comprising, preferably consisting of, d-C ⁇ - haloalkoxy, hydroxy, amino, halogen, -C(O)R b , -S(O) 2 R 5 , OR C , NR d1 R d2 ;
- R a is hydrogen
- R b is selected from the group comprising, preferably consisting of, OR C ,
- R c is selected from the group comprising, preferably consisting of, hydrogen, d-C 6 -alkyl, C 3 -C 6 -cycloalkyl, C 3 -C 6 -heterocycloalkyl, wherein d-C 6 -alkyl, C 3 -C 6 -cycloalkyl, C 3 -C 6 -heterocycloalkyl are optionally substituted one or more times with -NR d1 R d2 , and wherein d-C 6 -alkyl, C 3 -C 6 -cycloalkyl, C 3 -C 6 -heterocycloalkyl are optionally substituted once with -0R c ; R d1 , R d2 independently from each other are selected from the group comprising, preferably consisting of hydrogen, d-C 6 -alkyl, C 3 -C 6 - cycloalkyl, or for a -C(O)R C or C
- B is a bond or a group selected from the group comprising, preferably consisting of C r C 3 -alkylene, C 3 -cycloalkylene ;
- D and E are phenylene ; and q represents an integer of 0 ;
- R a , R b , R c , R d1 , R d2 or R 8 when one or more of R a , R b , R c , R d1 , R d2 or R 8 is (are) present in one position in the molecule as well as in one or more further positions in the molecule, said R a , R b , R c , R d1 , R d2 or R 8 has (have), independently from each other, the same meanings as defined above in said first position in the molecule and in said second or further positions in the molecule, it being possible for the two or more occurrences of R a , R b , R c , R d1 , R d2 or R 8 within a single molecule to be identical or different.
- R a when R a is present twice in the molecule, then the meaning of the first R a may be H, for example, and the meaning of the second R a may be methyl, for example.
- the second variant of the more particularly preferred embodiment, supra, of the present invention relates to compounds of general formula (I), in which :
- R 1 is H or Ci-C 3 -alkyl
- R 2 stands for hydrogen or is selected from the group comprising, preferably consisting of, C r C 6 -alkyl, C 3 -cycloalkyl
- R 3 is selected from the group comprising, preferably consisting of, hydrogen, methyl, or fluoro; 2007 06 11
- R 4 is selected from the group comprising, preferably consisting of, hydrogen, halogen, C r C 3 -alkyl, or Ci-C 3 -haloalkyl
- R 5 is selected from the group comprising, preferably consisting of, hydrogen, CrC 3 -alkyl, d-C 3 -haloalkyl, CrC 3 -haloalkoxy, halogen, - 0R c , -NR d1 R d2 , wherein C r C 3 -alkyl is optionally substituted by R 8 ;
- R 8 is selected from the group comprising, preferably consisting of, -0R c , and -NR d1 R d2 ;
- R a is hydrogen ;
- R c is selected from the group comprising, preferably consisting of, hydrogen, and C r C 3 -alkyl, wherein C r C 3 -alkyl is optionally substituted one or more times with -NR d1 R d2 , and wherein C r C 3 -alkyl is optionally substituted once with -OR C ;
- R d1 , R d2 independently from each other are selected from the group comprising, preferably consisting of hydrogen, Ci-C 3 -alkyl, wherein d- C 3 -alkyl is optionally substituted one or more times, with an -0R c group, and wherein C r C 3 -alkyl is optionally substituted once with an -
- NR d1 R d2 group or, R d1 and R d2 together with the nitrogen atom to which they are attached, form a 6 membered heterocycloalkyl ring, whereby the carbon backbone of this heterocycloalkyl ring is optionally interrupted one time, by a member of the group comprising, preferably consisting of, NH, NR d1 , and oxygen ;
- A is -C(O)- ;
- B is Ci-alkylene or C 3 -cycloalkylene ;
- D is para-phenylene ;
- E is phenylene ; q represents an integer of 0 ;
- R a , R b , R c , R d1 or R d2 when one or more of R a , R b , R c , R d1 or R d2 is (are) present in one position in the molecule as well as in one or more further positions in the molecule, said R a , R b , R c , R d1 or R d2 has (have), independently from each other, the same meanings as defined above in said first position in the molecule and in said second or further positions in the molecule, it being possible for the two or more occurrences of R a , R b , R c , R d1 or R d2 within a single molecule to be identical or 2007 06 11 different.
- R a when R a is present twice in the molecule, then the meaning of the first R a may be H, for example, and the meaning of the second R a may be methyl, for example.
- alkyl is to be understood as preferably meaning branched and unbranched alkyl, meaning e.g. methyl, ethyl, n-propyl, /so-propyl, n-butyl, iso- butyl, tert-butyl, sec-butyl, pentyl, /so-pentyl, hexyl, heptyl, octyl, nonyl and decyl and the isomers thereof.
- haloalkyl is to be understood as preferably meaning branched and unbranched alkyl, as defined supra, in which one or more of the hydrogen substituents is replaced in the same way or differently with halogen.
- said haloalkyl is, e.g. chloromethyl, fluoropropyl, fluoromethyl, difluoromethyl, trichloromethyl, 2,2,2-trifluoroethyl, pentafluoroethyl, bromobutyl, trifluoromethyl, iodoethyl, and isomers thereof.
- alkoxy is to be understood as preferably meaning branched and unbranched alkoxy, meaning e.g. methoxy, ethoxy, propyloxy, iso- propyloxy, butyloxy, /so-butyloxy, tert-butyloxy, sec-butyloxy, pentyloxy, iso- pentyloxy, hexyloxy, heptyloxy, octyloxy, nonyloxy, decyloxy, undecyloxy and dodecyloxy and the isomers thereof.
- haloalkoxy is to be understood as preferably meaning branched and unbranched alkoxy, as defined supra, in which one or more of the hydrogen substituents is replaced in the same way or differently with halogen, e.g. chloromethoxy, fluoromethoxy, pentafluoroethoxy, fluoropropyloxy, difluoromethyloxy, trichloromethoxy, 2,2,2-trifluoroethoxy, bromobutyloxy, trifluoromethoxy, iodoethoxy, and isomers thereof. 2007 06 11
- cycloalkyl is to be understood as preferably meaning a C 3 -C 10 cycloalkyl group, more particularly a saturated cycloalkyl group of the indicated ring size, meaning e.g. a cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclononyl, or cyclodecyl group ; and also as meaning an unsaturated cycloalkyl group containing one or more double bonds in the C-backbone, e.g.
- a C 3 - C 10 cycloalkenyl group such as, for example, a cyclopropenyl, cyclobutenyl, cyclopentenyl, cyclohexenyl, cycloheptenyl, cyclooctenyl, cyclononenyl, or cyclodecenyl group, wherein the linkage of said cyclolaklyl group to the rest of the molecule can be provided to the double or single bond.
- heterocycloalkyl is to be understood as preferably meaning a C 3 -C 10 cycloalkyl group, as defined supra, featuring the indicated number of ring atoms, wherein one or more ring atom(s) is (are) (a) heteroatom(s) such as NH, NR d1 , O, S, or (a) group(s) such as a C(O), S(O), S(O) 2 , or, otherwise stated, in a C n -cycloalkyl group, (wherein n is an integer of 3, 4, 5, 6, 7, 8, 9, or 10), one or more carbon atom(s) is (are) replaced by said heteroatom(s) or said group(s) to give such a C n cycloheteroalkyl group.
- said C n cycloheteroalkyl group refers, for example, to a three-membered heterocycloalkyl, expressed as C 3 -heterocycloalkyl, such as oxiranyl (C 3 ).
- heterocycloalkyls are oxetanyl (C 4 ), aziridinyl (C 3 ), azetidinyl (C 4 ), tetrahydrofuranyl (C 5 ), pyrrolidinyl (C 5 ), morpholinyl (C 6 ), dithianyl (C 6 ), thiomorpholinyl (C 6 ), piperidinyl (C 6 ), tetrahydropyranyl (C 6 ), piperazinyl (C 6 ), trithianyl (C 6 ) and chinuclidinyl (Cs).
- halogen or "Hal” is to be understood as preferably meaning fluorine, chlorine, bromine, or iodine.
- alkenyl is to be understood as preferably meaning branched and unbranched alkenyl, e.g. a vinyl, propen-1 -yl, propen-2-yl, but-1 -en-1-yl, but-1 -en- 2-yl, but-2-en-1 -yl, but-2-en-2-yl, but-1 -en-3-yl, 2-methyl-prop-2-en-1 -yl, or 2- methyl-prop-1 -en-1 -yl group.
- alkynyl is to be understood as preferably meaning branched and unbranched alkynyl, e.g. an ethynyl, prop-1 -yn-1 -yl, but-1 -yn-1 -yl, but-2-yn-1 -yl,or but-3-yn-1 -yl group. 2007 06 11
- aryl is defined in each case as having 3-14 carbon atoms, preferably 6-12 carbon atoms, such as, for example, cyclopropenyl, phenyl, tropyl, indenyl, naphthyl, azulenyl, biphenyl, fluorenyl, anthracenyl etc, phenyl being preferred.
- heteroaryl is understood as meaning an aromatic ring system which comprises 3-16 ring atoms, preferably 5 or 6 or 9 or 10 atoms, and which contains at least one heteroatom which may be identical or different, said heteroatom being such as oxygen, nitrogen or sulphur, and can be monocyclic, bicyclic, or tricyclic, and in addition in each case can be benzocondensed.
- heteroaryl is selected from thienyl, furanyl, pyrrolyl, oxazolyl, thiazolyl, imidazolyl, pyrazolyl, isoxazolyl, isothiazolyl, oxadiazolyl, triazolyl, thiadiazolyl, thia-4H-pyrazolyl etc., and benzo derivatives thereof, such as, e.g., benzofuranyl, benzothienyl, benzoxazolyl, benzimidazolyl, benzotriazolyl, indazolyl, indolyl, isoindolyl, etc.; or pyridyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl, etc., and benzo derivatives thereof, such as, for example, quinolinyl, isoquinolinyl, etc.; or azocinyl, indoliziny
- alkylene as used herein in the context of the compounds of general formula (I) is to be understood as meaning an optionally substituted alkyl chain or "tether", having 1 , 2, 3, 4, 5, or 6 carbon atoms, i.e. an optionally substituted - CH 2 - ("methylene” or “single membered tether” or e.g.
- alkylene tether is 1 ,
- cycloalkylene as used herein in the context of the compounds of general formula (I) is to be understood as meaning an optionally substituted cycloalkyl ring, having 3, 4, 5, 6, 7, 8, 9 or 10, preferably 3, 4, 5, or 6, carbon atoms, i.e. an optionally substituted cyclopropyl, cyclobutyl, cyclopenyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclononyl, or cyclodecyl ring, preferably a cyclopropyl, cyclobutyl, cyclopentyl or cyclohexyl ring.
- heterocycloalkylene as used herein in the context of the compounds of general formula (I) is to be understood as meaning a cycloalkylene ring, as defined supra, but which contains at least one heteroatom which may be identical or different, said heteroatom being such as 0, N, S, S(O) or S(O) 2 .
- arylene as used herein in the context of the compounds of general formula (I) which include the groups D and E, is to be understood as meaning an optionally substituted monocyclic or polycyclic arylene aromatic system e.g. arylene, naphthylene and biarylene, preferably an optionally substituted phenyl ring or "tether", having 6 or 10 carbon atoms. More preferably, said arylene tether is a ring having 6 carbon atoms. If the term "arylene” is used it is to be understood that the linking residues can be arranged to each other in ortho-, para- and meta- position, eg. an optionally substituted moiety of structure :
- heteroarylene as used herein in the context of the compounds of general formula (I) which include the groups D and E, is to be understood as meaning an optionally substituted monocyclic or polycyclic heteroarylene aromatic system, e.g. heteroarylene, benzoheteroarylene, preferably an optionally substituted 5-membered heterocycle, such as, for example, furan, pyrrole, thiazole, oxazole, isoxazole, or thiophene or "tether", or a 6-membered heterocycle, such as, for example, pyridine, pyrimidine, pyrazine, pyridazine.
- said heteroarylene tether is a ring having 6 carbon atoms, e.g. an optionally substituted structure as shown supra for the arylene moieties, but which contains at least one heteroatom which may be identical or different, said 2007 06 11 heteroatom being such as oxygen, nitrogen or sulphur. If the term "heteroarylene” is used it is to be understood that the linking residues can be arranged to each other in ortho-, para- and meta-position.
- C 1 -C 6 As used herein, the term "C 1 -C 6 ", as used throughout this text, e.g. in the context of the definition of "CrC 6 -alkyr, or “CrC ⁇ -alkoxy”, is to be understood as meaning an alkyl group having a finite number of carbon atoms of 1 to 6, i.e. 1 , 2, 3, 4, 5, or 6 carbon atoms. It is to be understood further that said term “CrC 6 " is to be interpreted as any sub-range comprised therein, e.g.
- C 2 -CO as used throughout this text, e.g. in the context of the definitions of "C 2 -C 6 -alkenyl” and “C 2 -C ⁇ -alkynyl”, is to be understood as meaning an alkenyl group or an alkynyl group having a finite number of carbon atoms of 2 to 6, i.e. 2, 3, 4, 5, or 6 carbon atoms. It is to be understood further that said term “C 2 -CO” is to be interpreted as any sub-range comprised therein, e.g. C 2 -C 6 , C 3 -C 5 , C 3 -C 4 , C 2 -C 3 , C 2 -C 4 , C 2 -C 5 ; preferably C 2 -C 3 .
- C 3 -Cio As used herein, the term "C 3 -Cio”, as used throughout this text, e.g. in the context of the definitions of "CrQo-cycloalkyl” or “C 3 -Cio-heterocycloalkyl”, is to be understood as meaning a cycloalkyl group having a finite number of carbon atoms of 3 to 10, i.e. 3, 4, 5, 6, 7, 8, 9 or 10 carbon atoms, preferably 3, 4, 5 or 6 carbon atoms. It is to be understood further that said term “C 3 -C 1O " is to be interpreted as any sub-range comprised therein, e.g. C 3 -C 1O , C 4 -Cg , C 5 -Cs , C 6 -C 7 ; preferably C 3 -Ce.
- C 3 -C O As used herein, the term "C 3 -C O ", as used throughout this text, e.g. in the context of the definitions of "C 3 -C 6 -cycloalkyl” or “C 3 -C 6 -heterocycloalkyl”, is to be understood as meaning a cycloalkyl group having a finite number of carbon atoms of 3 to 6, i.e. 3, 4, 5, or 6 carbon atoms. It is to be understood further that said term “C 3 -CO” is to be interpreted as any sub-range comprised therein, e.g. C 3 -C 4 , C 4 -C 6 , C 5 -C 6 . 2007 06 11
- C 6 -Cn As used herein, the term "C 6 -Cn", as used throughout this text, e.g. in the context of the definitions of "C 6 -C 1r aryr, is to be understood as meaning an aryl group having a finite number of carbon atoms of 5 to 11 , i.e. 5, 6, 7, 8, 9, 10 or 11 carbon atoms, preferably 5, 6, or 10 carbon atoms. It is to be understood further that said term “C 6 -Cn” is to be interpreted as any sub-range comprised therein, e.g. C 5 -Ci 0 , C 6 -C 9 , C 7 -C 8 ; preferably C 5 -C 6 .
- C 5 -C10 As used herein, the term "C 5 -C10", as used throughout this text, e.g. in the context of the definitions of "C 5 -Cio-heteroaryl", is to be understood as meaning a heteroaryl group having a finite number of carbon atoms of 5 to 10, in addition to the one or more heteroatoms present in the ring i.e. 5, 6, 7, 8, 9, or 10 carbon atoms, preferably 5, 6, or 10 carbon atoms. It is to be understood further that said term “C 5 -C 10 " is to be interpreted as any sub-range comprised therein, e.g. C 6 -Cg , C 7 -C 8 , C 7 -C 8 ; preferably C 5 -C 6 .
- C 1 -C 3 As used herein, the term "C 1 -C 3 ", as used throughout this text, e.g. in the context of the definitions of "CrC 3 -alkylene", is to be understood as meaning an alkylene group as defined supra having a finite number of carbon atoms of 1 to 3, i.e. 1 , 2, or 3. It is to be understood further that said term “C 1 -C 3 " is to be interpreted as any sub-range comprised therein, e.g. C 1 -C 2 , or C 2 -C 3 .
- the term "one or more times”, e.g. in the definition of the substituents of the compounds of the general formulae of the present invention, is understood as meaning “one, two, three, four or five times, particularly one, two, three or four tines, more particularly one, two or three times, more particularly one or two times”.
- isomers is to be understood as meaning chemical compounds with the same number and types of atoms as another chemical species. There are two main classes of isomers, constitutional isomers and stereoisomers.
- substitutional isomers is to be understood as meaning chemical compounds with the same number and types of atoms, but they are connected in 2007 06 11 differing sequences. There are functional isomers, structural isomers, tautomers or valence isomers.
- stereoisomers is to be understood as meaning chemical compounds having atoms which are connected sequentially in the same way, such that condensed formulae for two isomeric molecules are identical. The isomers differ, however, in the way the atoms are arranged in space. There are two major subclasses of stereoisomers : conformational isomers, which interconvert through rotations around single bonds, and configurational isomers, which are not readily interconvertable.
- Configurational isomers are, in turn, can be enantiomers and/or diastereomers.
- Enantiomers are stereoisomers which are related to each other as mirror images. Enantiomers can contain any number of stereogenic centers, as long as each center is the exact mirror image of the corresponding center in the other molecule. If one or more of these centers differs in configuration, the two molecules are no longer mirror images.
- Stereoisomers which are not enantiomers are called diastereomers.
- Diastereomers which still have a different constitution are another sub-class of diastereomers, the best known of which are simple cis - trans isomers.
- a suitably pharmaceutically acceptable salt of the pyrazolopyridines of the present invention may be, for example, an acid-addition salt of a pyrazolopyridine of the invention which is sufficiently basic, for example, an acid-addition salt with, for example, an inorganic or organic acid, for example hydrochloric, hydrobromic, sulphuric, phosphoric, trifluoroacetic, para-toluenesulphonic, methylsulphonic, citric, tartaric, succinic or maleic acid.
- an inorganic or organic acid for example hydrochloric, hydrobromic, sulphuric, phosphoric, trifluoroacetic, para-toluenesulphonic, methylsulphonic, citric, tartaric, succinic or maleic acid.
- an alkali metal salt for example a sodium or potassium salt
- an alkaline earth metal salt for example a calcium or magnesium salt
- an ammonium salt or a salt with an organic base which affords a physiologically 2007 06 11 acceptable cation, for example a salt with N-methyl-glucamine, dimethyl- glucamine, ethyl-glucamine, lysine, 1 ,6-hexadiamine, ethanolamine, glucosamine, sarcosine, serinol, tris-hydroxy-methyl-aminomethane, aminopropandiol, sovak- base, 1 -amino-2,3,4-butantriol.
- the compound according to Formula (I) can exist as N-oxides which are defined in that at least one nitrogen of the compounds of the general Formula (I) may be oxidized.
- the compound according to Formula (I) can exist as solvates, in particular as hydrate, wherein the compound according to Formula (I) may contain polar solvents, in particular water, as structural element of the crystal lattice of the compounds.
- the amount of polar solvents, in particular water may exist in a stoichiometric or unstoichiometric ratio.
- stoichiometric solvates e.g. hydrate
- the compounds of the present invention according to Formula (I) can exist as prodrugs, e.g. as in vivo hydrolysable esters.
- in vivo hydrolysable ester is understood as meaning an in vivo hydrolysable ester of a compound of formula (I) containing a carboxy or hydroxyl group, for example, a pharmaceutically acceptable ester which is hydrolysed in the human or animal body to produce the parent acid or alcohol.
- Suitable pharmaceutically acceptable esters for carboxy include for example alkyl, cycloalkyl and optionally substituted phenylalkyl, in particular benzyl esters, C r C 6 alkoxymethyl esters, e.g.
- Q -Ce alkanoyloxymethyl esters e.g. pivaloyloxymethyl, phthalidyl esters, C3-C8 cycloalkoxy-carbonyloxy-CrC ⁇ alkyl esters, e.g. 1 - cyclohexylcarbonyloxyethyl ; 1 ,3-dioxolen-2-onylmethyl esters, e.g. 5-methyl-1 ,3- dioxolen-2-onylmethyl ; and d-C ⁇ -alkoxycarbonyloxyethyl esters, e.g.
- An in vivo hydrolysable ester of a compound of formula (I) containing a hydroxyl group includes inorganic esters such as phosphate esters and [alpha] -acyloxyalkyl ethers and related compounds which as a result of the in vivo hydrolysis of the ester breakdown to give the parent hydroxyl group. 2007 06 11
- Examples of [alpha] -acyloxyalkyl ethers include acetoxymethoxy and 2,2- dimethylpropionyloxymethoxy.
- a selection of in vivo hydrolysable ester forming groups for hydroxyl include alkanoyl, benzoyl, phenylacetyl and substituted benzoyl and phenylacetyl, alkoxycarbonyl (to give alkyl carbonate esters), dialkylcarbamoyl and N-(dialkylaminoethyl)-N-alkylcarbamoyl (to give carbamates), dialkylaminoacetyl and carboxyacetyl.
- the compounds of the present invention according to Formula (I), or salts, N- oxides, or prodrugs thereof, may contain one or more asymmetric centers.
- Asymmetric carbon atoms may be present in the (R) or (S) configuration or (R,S) configuration.
- Substituents on a ring may also be present in either cis or trans form. It is intended that all such configurations (including enantiomers and diastereomers), are included within the scope of the present invention.
- Preferred stereoisomers are those with the configuration which produces the more desirable biological activity.
- Separated, pure or partially purified configurational isomers or racemic mixtures of the compounds of this invention are also included within the scope of the present invention. The purification of said isomers and the separation of said isomeric mixtures can be accomplished by standard techniques known in the art.
- the compounds of the present invention can be used in treating diseases of dysregulated vascular growth or diseases which are accompanied with dysregulated vascular growth. Especially, the compounds effectively interfere with Tie2 signalling. In addition, the compounds of the present invention allow for tunability of the inhibition of an additional kinase target according to the appropriate therapeutic needs.
- another aspect of the present invention is a use of the compound of general formula (I) described supra for manufacturing a pharmaceutical composition for the treatment of diseases of dysregulated vascular growth or of diseases which are accompanied with dysregulated vascular growth.
- the use is in the treatment of diseases, wherein the diseases are tumours and/or metastases thereof.
- diseases are retinopathy, other angiogenesis dependent diseases of the eye, in particular cornea transplant rejection or age-related macular degeneration, rheumatoid arthritis, and other inflammatory diseases associated with angiogenesis, in particular psoriasis, delayed type hypersensitivity, contact dermatitis, asthma, multiple sclerosis, restenosis, pulmonary hypertension, stroke, and diseases of the bowel. 2007 06 11
- a further use is in the treatment of diseases, wherein the diseases are coronary and peripheral artery disease.
- diseases are ascites, oedema such as brain tumour associated oedema, high altitude trauma, hypoxia induced cerebral oedema, pulmonary oedema and macular oedema or oedema following burns and trauma, chronic lung disease, adult respiratory distress syndrome, bone resorption and for benign proliferating diseases such as myoma, benign prostate hyperplasia and wound healing for the reduction of scar formation, reduction of scar formation during regeneration of damaged nerves, endometriosis, pre-eclampsia, postmenopausal bleeding and ovarian hyperstimulation.
- oedema such as brain tumour associated oedema, high altitude trauma, hypoxia induced cerebral oedema, pulmonary oedema and macular oedema or oedema following burns and trauma
- chronic lung disease such as myoma, benign prostate hyperplasia and wound healing for the reduction of scar formation, reduction of scar formation during regeneration of damaged nerves, endometri
- Yet another aspect of the invention is a method of treating a disease of dysregulated vascular growth or diseases which are accompanied with dysregulated vascular growth, by administering an effective amount of a compound of general formula (I) described supra.
- the diseases of said method is tumour and/or metastases thereof.
- the diseases of said method are retinopathy, other angiogenesis dependent diseases of the eye, in particular cornea transplant rejection or age-related macular degeneration, e.g. rheumatoid arthritis, and other inflammatory diseases associated with angiogenesis, in particular psoriasis, delayed type hypersensitivity, contact dermatitis, asthma, multiple sclerosis, restenosis, pulmonary hypertension, stroke, and diseases of the bowel.
- retinopathy other angiogenesis dependent diseases of the eye
- cornea transplant rejection or age-related macular degeneration e.g. rheumatoid arthritis
- other inflammatory diseases associated with angiogenesis in particular psoriasis, delayed type hypersensitivity, contact dermatitis, asthma, multiple sclerosis, restenosis, pulmonary hypertension, stroke, and diseases of the bowel.
- the disease of the method are coronary and peripheral artery disease.
- oedema such as brain tumour associated oedema, high altitude trauma, hypoxia induced cerebral oedema, pulmonary oedema and macular oedema or oedema following burns and trauma, chronic lung disease, adult respiratory distress syndrome, bone resorption and for benign proliferating diseases such as myoma, benign prostate hyperplasia and wound healing for the reduction of scar formation, reduction of scar formation during 2007 06 11 regeneration of damaged nerves, endometriosis, pre-eclampsia, postmenopausal bleeding and ovarian hyperstimulation.
- oedema such as brain tumour associated oedema, high altitude trauma, hypoxia induced cerebral oedema, pulmonary oedema and macular oedema or oedema following burns and trauma
- chronic lung disease such as myoma, benign prostate hyperplasia and wound healing for the reduction of scar formation, reduction of scar formation during 2007 06 11 regeneration of damaged nerves, endometri
- the compounds of the present invention can thus be applied for the treatment of diseases accompanied by neoangiogenesis.
- diseases accompanied by neoangiogenesis This holds principally for all solid tumours, e.g. breast, colon, renal, lung and/or brain tumours or metastases thereof and can be extended to a broad range of diseases, where pathologic angiogenesis is persistent.
- pathologic angiogenesis This applies for diseases with inflammatory association, diseases associated with oedema of various forms and diseases associated with stromal proliferation and pathologic stromal reactions broadly.
- Particularly suited is the treatment for gynaecological diseases where inhibition of angiogenic, inflammatory and stromal processes with pathologic character can be inhibited.
- the treatment is therefore an addition to the existing armament to treat diseases associated with neoangiogenesis.
- the compounds of the present invention can be used in particular in therapy and prevention of tumour growth and metastases, especially in solid tumours of all indications and stages with or without pre-treatment if the tumour growth is accompanied with persistent angiogenesis.
- tumour therapy is not restricted to tumour therapy but is also of great value for the treatment of other diseases with dysregulated vascular growth.
- This includes retinopathy and other angiogenesis dependent diseases of the eye (e.g. cornea transplant rejection, age-related macular degeneration), rheumatoid arthritis, and other inflammatory diseases associated with angiogenesis such as psoriasis, delayed type hypersensitivity, contact dermatitis, asthma, multiple sclerosis, restenosis, pulmonary hypertension, stroke and inflammatory diseases of the bowel, such as Crohn's disease.
- oedema such as brain tumour associated oedema, high altitude trauma, hypoxia induced cerebral oedema, pulmonary oedema and macular oedema or oedema following burns and trauma. Furthermore, it is useful for chronic lung disease, adult respiratory distress syndrome. Also for bone resorption and for benign proliferating diseases such as myoma, benign prostate hyperplasia and wound healing for the reduction of scar formation. It is therapeutically valuable for the treatment of diseases, where deposition of fibrin or extracellular matrix is an 2007 06 11 issue and stroma proliferation is accelerated (e.g. fibrosis, cirrhosis, carpal tunnel syndrome etc).
- Another aspect of the present invention is a pharmaceutical composition which contains a compound of Formula (I), or a pharmaceutically acceptable salt thereof, or an N-oxide thereof, or a pro-drug thereof, in admixture with one or more suitable excipients.
- This composition is particularly suited for the treatment of diseases of dysregulated vascular growth or of diseases which are accompanied with dysregulated vascular growth as explained above.
- the compounds or mixtures thereof may be provided in a pharmaceutical composition, which, as well as the compounds of the present invention for enteral, oral or parenteral application contain suitable pharmaceutically acceptable organic or inorganic inert base material, e.g. purified water, gelatine, gum Arabic, lactate, starch, magnesium stearate, talcum, vegetable oils, polyalkyleneglycol, etc.
- suitable pharmaceutically acceptable organic or inorganic inert base material e.g. purified water, gelatine, gum Arabic, lactate, starch, magnesium stearate, talcum, vegetable oils, polyalkyleneglycol, etc.
- compositions of the present invention may be provided in a solid form, e.g. as tablets, dragees, suppositories, capsules or in liquid form, e.g. as a solution, suspension or emulsion.
- the pharmaceutical composition may additionally contain auxiliary substances, e.g. preservatives, stabilisers, wetting agents or emulsifiers, salts for adjusting the osmotic pressure or buffers.
- sterile injection solutions or suspensions are preferred, especially aqueous solutions of the compounds in polyhydroxyethoxy containing castor oil.
- compositions of the present invention may further contain surface active agents, e.g. salts of gallenic acid, phospholipids of animal or vegetable origin, mixtures thereof and liposomes and parts thereof. 2007 06 11
- surface active agents e.g. salts of gallenic acid, phospholipids of animal or vegetable origin, mixtures thereof and liposomes and parts thereof. 2007 06 11
- dragees or capsules with talcum and/or hydrocarbon- containing carriers and binders are preferred.
- Further application in liquid form is possible, for example as juice, which contains sweetener if necessary.
- the dosage will necessarily be varied depending upon the route of administration, age, weight of the patient, the kind and severity of the illness being treated and similar factors.
- the daily dose is in the range of 0.5 to 1 ,500 mg.
- a dose can be administered as unit dose or in part thereof and distributed over the day. Accordingly the optimum dosage may be determined by the practitioner who is treating any particular patient.
- compounds of general formula(l) of the present invention can be used alone or, indeed in combination with one or more further drugs, particularly anti-cancer drugs or compositions thereof.
- said combination it is possible for said combination to be a single pharmaceutical composition entity, e.g. a single pharmaceutical formulation containing one or more compounds according to general formula(l)together with one or more further drugs, particularly anti-cancer drugs, or in a form, e.g. a "kit of parts", which comprises, for example, a first distinct part which contains one or more compounds according to general formula I, and one or more further distinct parts each containing one or more further drugs, particularly anti-cancer drugs. More particularly, said first distinct part may be used concomitantly with said one or more further distinct parts, or sequentially.
- Another aspect of the present invention is a method which may be used for preparing the compounds according to the present invention.
- the compounds may be purified by chromatography, particularly flash column chromatography, using for example prepacked silica gel cartridges, e.g. from Separtis such as Isolute® Flash silica gel or Isolute® Flash NH 2 silica gel in combination with a Flashmaster Il autopurifier (Argonaut/Biotage) and eluents such as gradients of hexane/EtOAc or DCM/ethanol.
- chromatography particularly flash column chromatography
- Separtis such as Isolute® Flash silica gel or Isolute® Flash NH 2 silica gel in combination with a Flashmaster Il autopurifier (Argonaut/Biotage) and eluents such as gradients of hexane/EtOAc or DCM/ethanol.
- the compounds may be purified by preparative HPLC using for example a Waters autopurifier equipped with a diode array detector and/or on-line electrospray ionization mass spectrometer in combination with a suitable prepacked reverse phase column and eluants such as gradients of water and acetonitrile which may contain additives such as trifluoroacetic acid or aqueous ammonia.
- a Waters autopurifier equipped with a diode array detector and/or on-line electrospray ionization mass spectrometer in combination with a suitable prepacked reverse phase column and eluants such as gradients of water and acetonitrile which may contain additives such as trifluoroacetic acid or aqueous ammonia.
- Reactions may be monitored and product purity may be analyzed by LC-MS analysis employing conditions such as, for example, the following specifications:
- 0.05% TFA can be replaced by, for example, 0.2% NH 3 .
- pyridones 4 are transformed into pyridines of general formula 5 carrying a leaving group X at the C2 position, wherein X stands for, but is not limited to, trifluoromethanesulfonyl (OTf), acetate (OAc), methoxy (OMe), Cl or F.
- X stands for Cl, even more preferably X stands for OTf.
- Conversion of intermediate compounds of general formula 4 into intermediates of general formula 5 may be achieved by a variety of methods, e.g.
- Intermediate compounds of general formula 7 are formed from intermediate compounds of general formula 6 by reaction with, for example, a suitably functionalized isocyanate (leading to ureas), a suitably functionalized sulfonyl chloride (leading to sulfonyl amides) or a suitably functionalized acid chloride (leading to carboxylic amides), in the presence of a suitable base as necessary, e.g. pyridine, which may also be used as solvent, optionally in the presence of an inert solvent, e.g. dichloromethane, acetonitrile, DMF or THF, at temperatures ranging from -20 °C to the boiling point of the solvent, whereby room temperature is preferred.
- a suitable base e.g. pyridine
- an inert solvent e.g. dichloromethane, acetonitrile, DMF or THF
- a variety of substituted hydrazine building blocks required for the conversion of pyridine 7 into intermediates 1 is commercially available, either in form of their free base or as various types of salts (e.g. hydrochlorides, oxalates), which can be transformed into their respective free bases by alkaline treatment either before the cyclization or in situ.
- salts e.g. hydrochlorides, oxalates
- substituted alkyl-, allyl-, and benzylhydrazines are accessible from the respective alkyl-, allyl- and benzylhalides, preferably the respective alkyl-, allyl- and benzylbromides, by nucleophilic substitution reaction with a protected hydrazine, such as BocNHNH ⁇ , in an inert solvent, preferably MeOH, in the presence of an amine promoter, e.g.
- the substituents R a , R 1 , R 2 , R 3 , R 4 , R 5 may be further modified on each step (general formula 1 to general formula 14) or in the last step (general formula I). These modifications can be such as the introduction of protecting groups, cleavage of protecting groups, reduction or oxidation of functional groups, substitution or other reactions. Appropiate protecting groups and their introduction and cleavage are well-known to the person skilled in the art (see for example T.W. Greene and P.G.M. Wuts in Protective Groups in Organic Synthesis, 3 rd edition, Wiley 1999).
- urea formation starting from amines of general formula 6 may be achieved by coupling with a second functionalized amine via in situ transformation of one of the reacting amines into the respective carbamoyl chloride, aryl- or alkenylcarbamate (see for example J. Org. Chem. 2005, 70, 6960 and references cited therein).
- This process may provide an alternative to the formation and isolation of the respective isocyanate derived from one of the starting amines (see for example Tetrahedron Lett. 2004, 45, 4769).
- ureas of formula 7a may be formed from two suitably functionalized amines and a suitable phosgene equivalent, preferably triphosgene, in an inert solvent, preferably acetonitrile, at temperatures ranging from -20 "C to room temperature, whereby room temperature is preferred.
- a suitable phosgene equivalent preferably triphosgene
- the corresponding acid which may be obtained from the corresponding ester by saponification, can be reacted with amines of general formula 6 in aprotic polar solvents, such as, for example, DMF, via an activated acid derivative, which is obtainable, for example, with hydroxybenzotriazole and a carbodiimide, such as, for example, diisopropylcarbodiimide (DIC), at temperatures of between 0 0 C and the boiling point of the solvent, preferably at 80 °C, or else with preformed reagents, such as, for example, O-(7-azabenzotriazol-1 -yl)-1 , 1 ,3,3- tetramethyluronium hexafluorophosphate (HATU) (see for example Chem.
- aprotic polar solvents such as, for example, DMF
- an activated acid derivative which is obtainable, for example, with hydroxybenzotriazole and a carbodiimide, such as, for example,
- DCC dicyclohexylcarbodiimide
- DMAP dimethylaminopyridine
- EDCI N-ethyl-N'- dimethylaminopropylcarbodiimide
- T3P T3P
- a suitable base such as N-methylmorpholine, for example, may be necessary.
- Amide formation may also be accomplished via the acid halide, mixed acid anhydride, imidazolide or azide.
- the carboxylic acids required for the above described amide coupling reactions are either commercially available or are accessible from commercially available carboxylic esters or nitriles.
- (hetero)aryls bearing a methylenenitrile substituent are easily accessible from the respective halides via a nucleophilic substitution reaction (e.g. KCN, cat. Kl, EtOH/H 2 O).
- Benzylic nitriles and esters can be efficiently alkylated at the benzylic position under basic conditions and subsequently hydrolyzed to the corresponding alkylated acids.
- Conditions for ⁇ -alkylations of nitriles and esters include, but are not limited to, the use of alkyl bromides or alkyl iodides as electrophiles under basic conditions in the presence or absence of a phase-transfer catalyst in a mono- or biphasic solvent system. Particularly, by using excess alkyl iodides as electrophilic species ⁇ , ⁇ -dialkylated nitriles are accessible.
- cycloalkyl moieties can be installed at the benzylic position of nitriles and esters (J. Med. Chem. 1975, 18,
- a cyclopropane ring can be installed at the benzylic position of a nitrile or ester.
- the hydrolysis of nitriles to yield carboxylic acids can be accomplished, as known to the person skilled in the art, under acid or base-mediated conditions.
- Suitable protecting groups for amino functions are well known to the person skilled in the art (see for example T. W. Greene and P. G. M. Wuts in Protective Groups in Organic Synthesis, 3 rd edition, Wiley 1999).
- the 3- amino group is protected by formation of the respective phthalimide.
- phthalimido protection of 3-aminopyrazoles can be achieved by reaction of the amine with phthalic anhydride in a suitable inert solvent, e.g. acetonitrile or dioxane, optionally in the presence of a basic mediator, e.g. Et 3 N, DIPEA or DMAP, at temperatures from room temperature up to the boiling point of the respective solvent.
- Nitro reduction yielding amino compounds of the general formula 10 and e.g. urea, sulfonamide, and amide formation to give compounds of general formula 11 are feasible as described above.
- the intermediates of formula 1 are accessible by deprotection of the amino group in compounds of the general formula 11.
- cleavage of the phthalimido group can be achieved, as known to the person skilled in the art, by reaction with hydrazine or hydrazine hydrate in solvents such as EtOH at temperatures from room temperature up to the boiling point of the respective solvent.
- anilines of general formula 8 can be transformed into ureas or sulfonamides or amides of general formula I as described above.
- anilines of formula 8" can be reacted with isocyanates of 2007 06 11 formula Ia' or anilines of formula Ia" in accordance to the before mentioned transformations to yield ureas of formula Ia.
- R 1 -substituents as present in intermediates of general formula 1 can be accomplished after formation of 1 H-pyrazolopyridines 12 by subsequent acylation or alkylation (Scheme 6). This process is of particular importance if appropriately substituted hydrazines are not readily available.
- 1 H- Pyrazolopyridines of general formula 12 are accessible from synthetic intermediates of formula 7 (which can be prepared as described above) by cyclization with hydrazine or more preferably with hydrazine hydrate in a suitable solvent, preferably 1 -propanol, at temperatures from room temperature up to the boiling point of the solvent, whereby in the case of 1 -PrOH 100 0 C is preferred.
- R 1 -groups to yield intermediates of general formula 1 can be achieved employing various conditions for introducing substituents to nitrogen atoms as known to the person skilled in the art. These conditions include, but are not limited to, alkylations under basic conditions employing alkyl-, allyl-, benzylhalides or ⁇ -halocarbonyl compounds as electrophiles (e.g. WO2005056532; Chem. Pharm. Bull. 1987, 35, 2292; J. Med. Chem. 2005, 48, 6843), alkylations under reductive conditions employing aldehydes as electrophiles and an 2007 06 1 1 appropriate reducing agent (e.g.
- Conditions for N1 -alkylation of 3-aminopyrazoles of the general formula 12 include, but are not limited to, treatment with an excess of the respective electrophile (e.g. alkyl-, allyl-, benzylhalides or ⁇ -halocarbonyl compounds) in the presence of a base, e.g. potassium carbonate or cesium carbonate, in DMF at temperatures from room temperature up to the boiling point of the solvent.
- a base e.g. potassium carbonate or cesium carbonate
- 1 H- pyrazoles of general formula 12 are deprotonated with sodium hydride in DMF at temperatures from 0 0 C up to 80 0 C followed by reaction with the respective electrophile (e.g. alkyl-, allyl-, benzylhalides or ⁇ -halocarbonyl compounds) in DMF at temperatures from room temperature up to the boiling point of the solvent.
- electrophile e.g. alkyl-, allyl-, benzylhalides or ⁇ -halocarbonyl compounds
- compounds of the present invention of general formula I are prepared by a transition metal catalyzed coupling of an appropriate halo precursor of general formula 15 and an appropriately substituted boronic acid or boronate esters. More particularly, compounds of the present invention can be prepared starting from a halogenated pyrazolopyridine (15) by Pd-catalyzed Suzuki-type coupling reactions with (hetero)aryl boronic acids (16) or even more particularly with their respective boronate esters (e.g. a pinacolate ester).
- Transition metal-catalyzed couplings of heteroaryl halides with (hetero)aryl boronic 2007 06 11 acids or aryl boronate esters are well known to the person skilled in the art.
- Various catalyst/ligand/base/solvent combinations have been published in the scientific literature ⁇ Tetrahedron 2005, 61, 5131 and references cited therein; Eur. J. Org. Chem. 2006, 1917 and references cited therein; Eur. J. Org. Chem. 2006, 2063 and references cited therein), which allow a fine-tuning of the required reaction conditions in order to allow for a broad set of additional functional groups on both coupling partners.
- the boronic acids of general formula 16 may be replaced by, for example, a suitably substituted stannane.
- Conditions for Stille-type couplings of aryl- or heteroaryl stannanes to aryl or heteroaryl halides employing a Pd catalyst and optionally a mediator are well known to the person skilled in the art.
- the respective halogenated pyrazolopyridines of general formula 15 can be synthesized according to literature procedures e.g. from 3-aminopyrazole (see for example J. Prakt. Chem. 1982, 324, 557) or from 5-carboxy-4- hydroxypyrazolopyridines (see for example J. Med. Chem. 1975, 18, 161 ) by decarboxylation followed by halogenation or from 2,4-dihalopyridine-3- carboxaldehydes (commercially available or synthesized e.g. according to US20040044040) by hydrazine cyclization.
- the boronic acids and their respective pinacolate esters (16) needed for the above mentioned coupling reactions can be prepared e.g.
- boronic acids or boronate esters can be introduced into aryl or heteroaryl compounds inter alia by substituting halogen atoms. This substitution can be accomplished by metalation followed by electrophilic borylation ⁇ Org. Biomol. Chem. 2004, 2, 852) or by direct Pd- or Cu-catalyzed borylation ⁇ Synlett 2003, 1204 and references cited therein; Org. Lett. 2006, S, 261 ). Interconversion of boronic acids into the respective esters (e.g. their pinacolate esters) can be accomplished under standard conditions (for example by treatment with pinacol in EtOH at r.t.). 2007 06 11
- the aniline (1.2 eq.) was dissolved in 10 mL acetonitrile and treated with triphosgene (0.2 mmol, 0.4 eq.) and stirred at room temperature for 1 h upon 2007 06 11 which the 2-pyridyltriflate was added and stirring was continued at r.t. for 16 h.
- the reaction mixture was concentrated and the crude urea used in the subsequent cyclization without additional purification.
- the aniline (1.2 eq.) was dissolved in 10 ml_ acetonitrile and treated with triphosgene (0.2 mmol, 0.4 eq.) and stirred at room temperature for 1 h upon which the 2-pyridyltriflate was added and stirring was continued at r.t. for 16 h.
- the reaction mixture was concentrated and the crude urea used in the subsequent cyclization without additional purification.
- the carboxylic acid ester was treated with EtOH and aqueous sodium hydroxide solution (1 M) and stirred for 3 hours at 80 0 C. To the cold solution was added the same volume of water. The mixture was acidified with a 20% solution of citric acid. The precipitate was filtered off, washed with water and dried to yield the carboxylic acid. 2007 06 11
- the carboxylic acid (1 eq.) was suspended in DCM and treated with the amine (1.3 eq.) and 4-methylmorpholine (5 eq.). The suspension was stirred for 10 minutes at room temperature and then cooled with ice. 2,4,6-tripropyl-[1 ,3,5,2,4,6]trioxa- triphosphinane 2,4,6-trioxide (T3P; 2 eq.) was added and the solution stirred over night at room temperature. The mixture was concentrated in vacuo, taken up in sodium bicarbonate solution and stirred. The precipitate was filtered off, washed with water and dried to yield the amide. In some cases additional purification steps (flash column chromatography and/or preparative HPLC) were necessary.
- reaction of 61.7 g ammonium acetate (800 mmol, 8 eq.), 10.73 ml ethyl cyanoacetate (100 mmol, 1 eq.), 12.55 ml 3,3-dimethylbutan-2-one (100 mmol, 1 eq.), and 15.12 g 4-nitrobenzaldehyde (100 mmol, 1 eq.) yielded 10.02 g product (34 % yield).
- reaction of 8.16 g ammonium acetate (106 mmol, 8 eq.), 1.41 ml ethyl cyanoacetate (13.23 mmol, 1 eq.), 0.98 ml dry acetone (13.23 mmol, 1 eq.), and 2 g 4-nitrobenzaldehyde (13.23 mmol, 1 eq.) yielded 1.56 g product (46 % yield).
- reaction of 1.85 g ammonium acetate (24 mmol, 8 eq.), 0.28 ml ethyl cyanoacetate (3 mmol, 1 eq.), 475 mg 2,2-dimethyl-3-oxo-butyric acid ethyl ester (3 mmol, 1 eq.), and 453 mg 4-nitrobenzaldehyde (3 mmol, 1 eq.) yielded 125 mg product (11 % yield).
- reaction of 1.85 g ammonium acetate (24 mmol, 8 eq.), 0.28 ml ethyl cyanoacetate (3 mmol, 1 eq.), 330 mg furan-2-carbaldehyde (3 mmol, 1 eq.), and 453 mg 4-nitrobenzaldehyde (3 mmol, 1 eq.) yielded 362 mg product (39 % yield).
- Step 3 The crude enamine from step 2 (13.0 g, 41.2 mmol) was dissolved in acetic acid (130 ml) containing 98% sulfuric acid (26 ml) and water (39 ml). The solution was refluxed for 2 h. After cooling, the precipitate was filtered and washed with water. Yield 6.8 g (28 mmol, 58%).
- step 1 reaction of 5.4 g Intermediate 3.2 (14 mmol, 1 eq.) with 3.15 g 1 -isocyanato-3-trifluoromethyl-benzene (16.82 mmol, 1.2 eq.) in 50 ml_ DCM yielded the crude urea in quantitative yield, which was used for subsequent cyclizations without further purification.
- step 1 reaction of 357.3 mg Intermediate 3.3 (1 mmol, 1 eq.) with 0.16 mL i -isocyanato-3-trifluoromethyl-benzene (1.2 mmol, 1.2 eq.) in 10 ml. DCM yielded the crude urea, which was used for subsequent cyclizations without further purification.
- step 1 reaction of 3.3 g Intermediate 3.5 (7.25 mmol, 1 eq.) with 1.63 g 1 -isocyanato-3-trifluoromethyl-benzene (8.7 mmol, 1.2 eq.) in 150 ml_ DCM yielded 3.78 g of the urea (5.9 mmol, 81 % yield), after purification by flash column chromatography.
- step 1 reaction of 0.95 g Intermediate 3.5 (2.1 mmol, 1 eq.) with 0.38 g 2-fluoro-5-methyl-1 -isocyanatobenzene (2.5 mmol, 1.2 eq.) in 25 ml_ DCM yielded 1.0 g of the urea (66 % yield), which was used without further purification.
- reaction of Intermediate 4.4 (420 mg, 0.73 mmol, 1 eq.) with with 110 ⁇ l_ 80% hydrazine hydrate (2.19 mmol, 3 eq.) in 20 mL 1 -PrOH yielded 242 mg of the 1 H-pyrazolopyridine (0.53 mmol, 72 % yield ).
- step 2 In a modification to GP 4a (step 2), Intermediate 4.1 (0.42 mmol) was dissolved in 7 ml. 1 -PrOH and treated with 195.8 mg hydrazino-acetic acid methyl ester hydrochloride (1.26 mmol, 3 eq.) and 0.17 ml_ Et 3 N (1.26 mmol, 3 eq.) yielding after typical work-up and product isolation 26 mg of the pyrazolopyridine (0.047 mmol, 11 % yield over 2 steps).
- reaction of Intermediate 4.2 (286,2 mg, 0.5 mmol) with 80 ⁇ l methyl hydrazine (1.5 mmol, 3 eq.) in 7.5 ml. 1 -PrOH yielded 183.1 mg of the pyrazolopyridine (0.39 mmol mmol, 78 % yield).
- step 2 reaction of 420 mg Intermediate 4.4 (0.73 mmol) with 120 ⁇ l methyl hydrazine (2.2 mmol, 3 eq.) in 20 mL 1 -PrOH yielded 219 mg of the pyrazolopyridine (0.46 mmol, 64 % yield).
- step 2 reaction of 3.78g Intermediate 4.5 (5.9 mmol) with 940 ⁇ l methyl hydrazine (17.7 mmol, 3 eq.) in 150 ml_ 1 -PrOH yielded 2.5 g of the pyrazolopyridine (4.7 mmol, 79 % yield).
- step 2 reaction of 1 g Intermediate 4.6 (1.65 mmol) with 0.26 ml methyl hydrazine (4.95 mmol, 3 eq.) in 50 ml_ 1 -PrOH yielded 0.7 g of the pyrazolopyridine (84 % yield).
- reaction of 1 -[4-(3-Amino-6-isopropyl-1H-pyrazolo[3,4- b]pyridin-4-yl)-phenyl]-3-(3-trifluoromethyl-phenyl)-urea (136 mg, 0.3 mmol, 1 eq.) with 39.3 mg NaH, 167 mg 4-(2-Chloro-ethyl)-morpholine hydrochloride (0.9 mmol, 3 eq.), 0.12 ml. triethylamine (0.9 mmol, 3 eq.) in 3+3 ml_ DMF yielded after HPLC purification 16.6 mg of the desired product (10% yield).
- the following intermediates 10.1 to 10.15 were prepared by applying GP 7 or GP 8 using the respective aniline intermediates, the respective carboxylic acid chlorides or sulfonyl chlorides and subsequently hydrazine hydrate or substituted hydrazines.
- reaction of 148 mg Intermediate 7.8 (0.27 mmol, 1 ,4 eq.) with 0.39 ml sodium methoxide solution (0.5 M in MeOH, 0.2 mmol, 1 eq.) and 42.44 mg formamide (0.94 mmol, 4.8 eq.) in 2 ml DMF yielded after purification with preparative HPLC 21 mg of the desired product (15 % yield).
- step 2 reaction of Intermediate 2.3 (2 g, 5.16 mmol, 1 eq.) with 270 ⁇ l_ methyl hydrazine (7.75 mmol, 1.5 eq.) in 90 ml_ 1 -PrOH at 100 0 C for 3h followed by concentration of the reaction mixture and filtration yielded 745 mg of the target compound (51 % yield ). Extractive work-up of the filtrate and subsequent flash column chromatography provided a second batch of the target compound.
- step 2 reaction of Intermediate 2.5 (1.1 g, 2.27 mmol, 1 eq.) with with 330 ⁇ l_ 80% hydrazine hydrate (6.8 mmol, 3 eq.) in 55 ml_ 1 -PrOH yielded 0.83 g of the 1 H-pyrazolopyridine (2.26 mmol, 99 % yield ).
- step 2 reaction of 755 mg Intermediate 16.5 (1.29 mmol) with 0.21 ml_ methyl hydrazine (3.88 mmol, 3 eq.) in 41 mL 1 -PrOH yielded 490 mg of the pyrazolopyridine (79 % yield).
- step 2 reaction of 2.04 g Intermediate 16.9 (3.33 mmol) with 0.53 ml. methyl hydrazine (9.99 mmol, 3 eq.) in 105 mL 1 -PrOH yielded 1.38 g of the pyrazolopyridine (81 % yield).
- step 2 reaction of 659 mg Intermediate 16.13 (1.02 mmol) with 0.16 mL methyl hydrazine (3.06 mmol, 3 eq.) in 32 mL 1 -PrOH yielded 415 mg of the pyrazolopyridine (75 % yield).
- step 2 reaction of 186 mg Intermediate 16.17 (0.3 mmol) with 0.048 ml. methyl hydrazine (0.91 mmol, 3 eq.) in 11 mL 1 -PrOH yielded 142 mg of the pyrazolopyridine (92 % yield).
- step 2 reaction of 1.85 g Intermediate 16.24 (2.7 mmol) with 0.44 ml methyl hydrazine (8.28 mmol, 3.06 eq.) in 100 mL 1 -PrOH yielded 1.13 g of the pyrazolopyridine (72 % yield).
- step 2 15.4 g of 5-cyano-4-(4-nitro-phenyl)-6- trifluoromethanesulfonyloxy-pyridine-2-carboxylic acid ethyl ester (Intermediate 17.3; 34.6 mmol, 1 eq.) were dissolved in 820 mL 1 -PrOH, treated with 3.22 mL methyl hydrazine (60.5 mmol, 1.75 eq.) and heated 110 °C for 2 h. The reaction mixture was cooled with ice and the precipitate filtered and washed with EtOH. The filtrate was concentrated, the newly formed solid was filtered and washed with ice-cold EtOH to provide a combined yield of 10.2 g of the 3- aminopyrazolopyridine (29.9 mmol, 86% yield).
- reaction of 171 mg of 1 -[4-(3-Amino-1 -methyl-1 H-pyrazolo[3, A- b]pyridin-4-yl)-phenyl]-3-(3-trifluoromethyl-phenyl)-urea (0.4 mmol, 1 eq.) with 63 ⁇ l_ concentrated sulphuric acid and 68 mg sodium nitrite (2.5 eq.) in 3.75 ml_ EtOH yielded the desired product.
- step 1 reaction of 83.4 mg of Intermediate 12.3 (0.35 mmol, 1 eq.) with 42 ⁇ l_ phenylisocyanate (0.38 mmol, 1.1 eq.) in 3.5 mL DCM yielded after extractive work-up and flash column chromatography 70 mg of Example Compound 2.1 as a beige solid (0.2 mmol, 56 % yield ).
- step 1 reaction of 83.4 mg of Intermediate 12.3 (0.35 mmol, 1 eq.) with 49 ⁇ l_ i -isocyanato-3-trifluoromethyl-benzene (0.38 mmol, 1.1 eq.) in 3.5 ml_ DCM yielded after extractive work-up and flash column chromatography 85 mg of Example Compound 2.2 as a beige solid (0.2 mmol, 58 % yield; mp 226 "C).
- step 1 reaction of 83.4 mg of Intermediate 12.3 (0.35 mmol, 1 eq.) with 56 ⁇ l_ i -fluoro ⁇ -isocyanaUM-trifluoromethyl-benzene (0.38 mmol, 1.1 eq.) in 3.5 mL DCM yielded after extractive work-up and flash column chromatography 64 mg of Example Compound 2.3 as a beige solid (0.144 mmol, 41 % yield; mp 225 0 C).
- step 1 reaction of 83.4 mg of Intermediate 12.3 (0.35 mmol, 1 eq.) with 79 mg 1 -fluoro-3-isocyanato-5-trifluoromethyl-benzene (0.38 mmol, 1.1 eq.) in 3.5 mL DCM yielded after extractive work-up and flash column chromatography 84 mg of Example Compound 2.4 as a beige solid (0.190 mmol, 54 % yield; mp 213 0 C).
- step 1 reaction of 83.4 mg of Intermediate 12.3 (0.35 mmol, 1 eq.) with 79 mg 1 -fluoro-4-isocyanato-2-trifluoromethyl-benzene (0.38 mmol, 1.1 eq.) in 3.5 ml_ DCM yielded after extractive work-up and flash column chromatography 93 mg of Example Compound 2.5 as a beige solid (0.21 mmol, 60 % yield; mp 232 0 C).
Abstract
Description






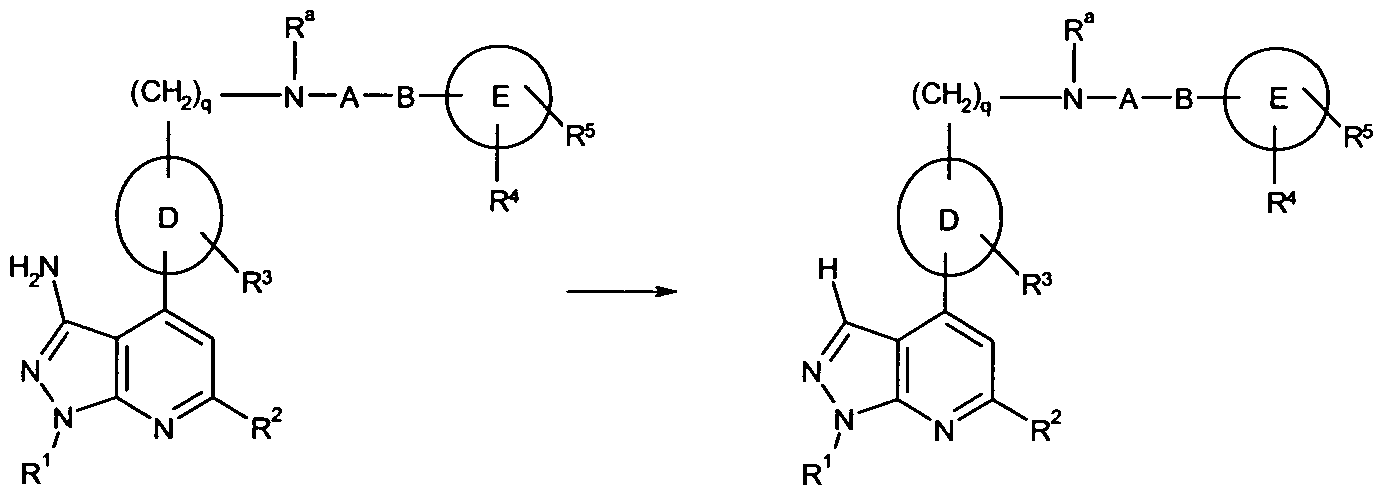




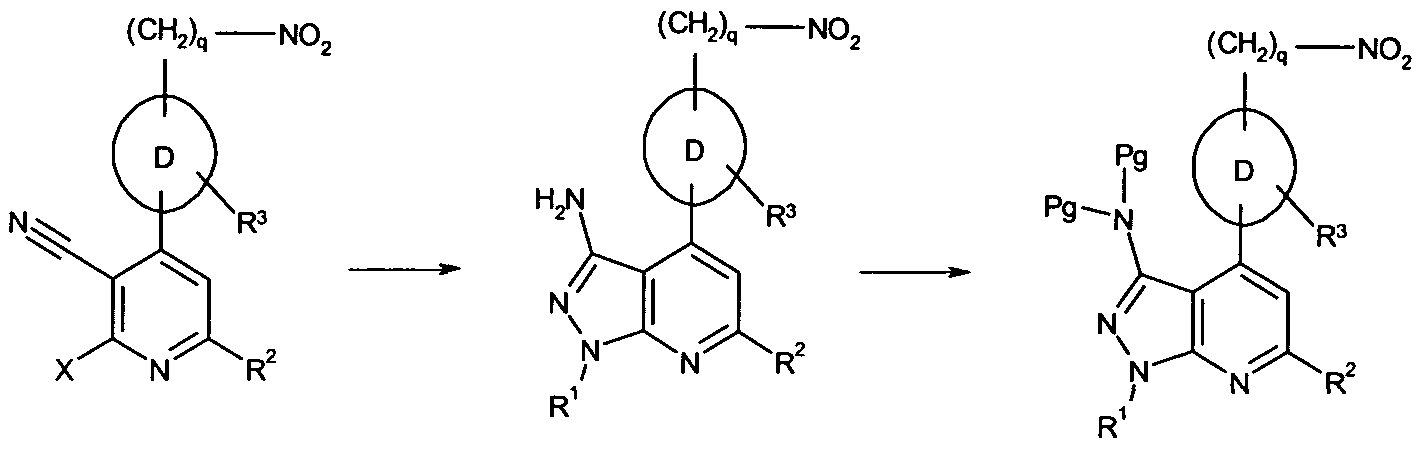

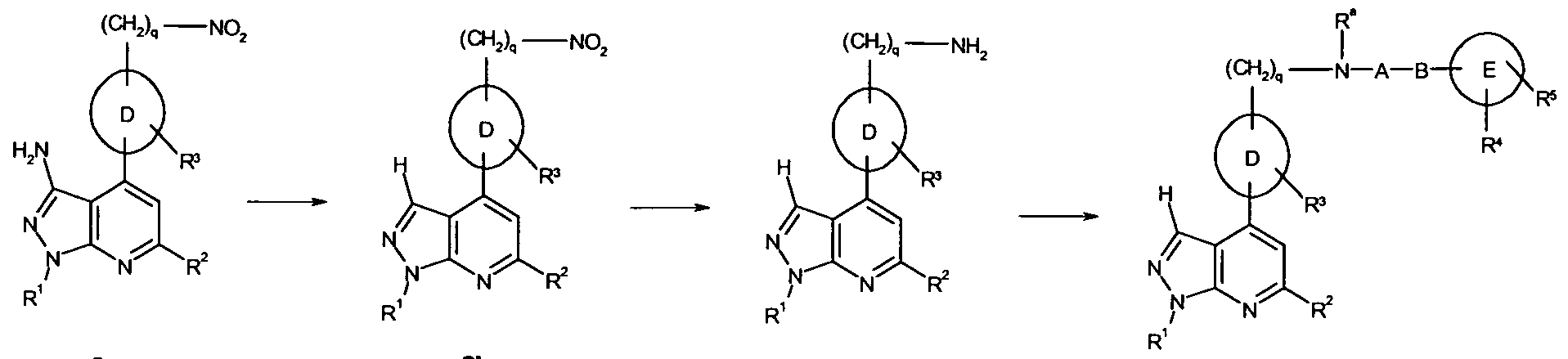



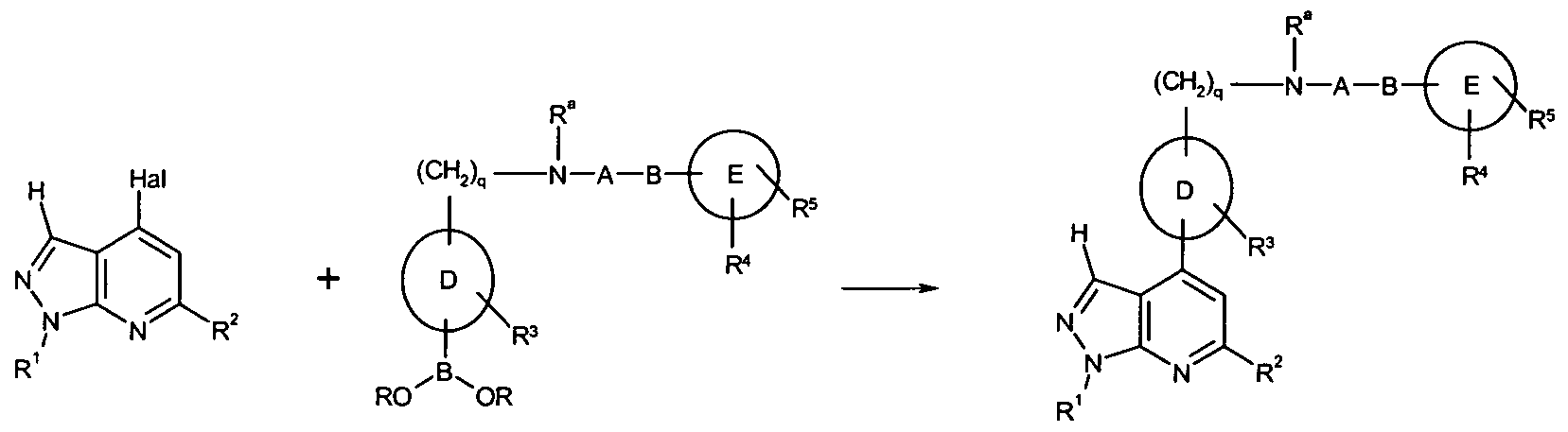



















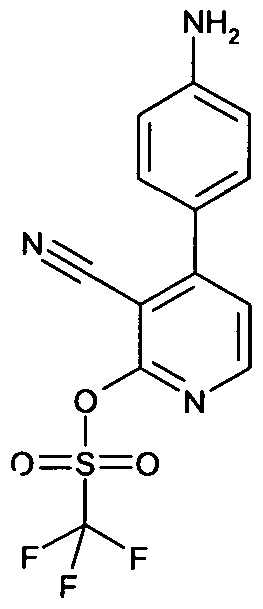
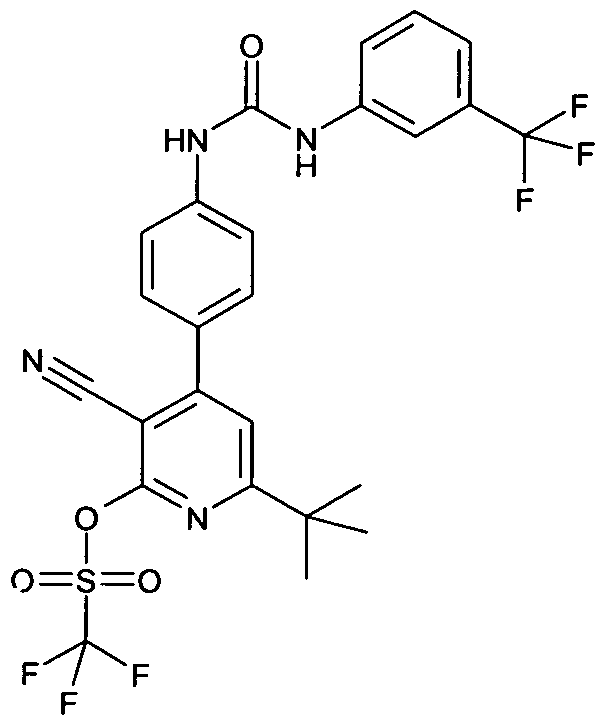






















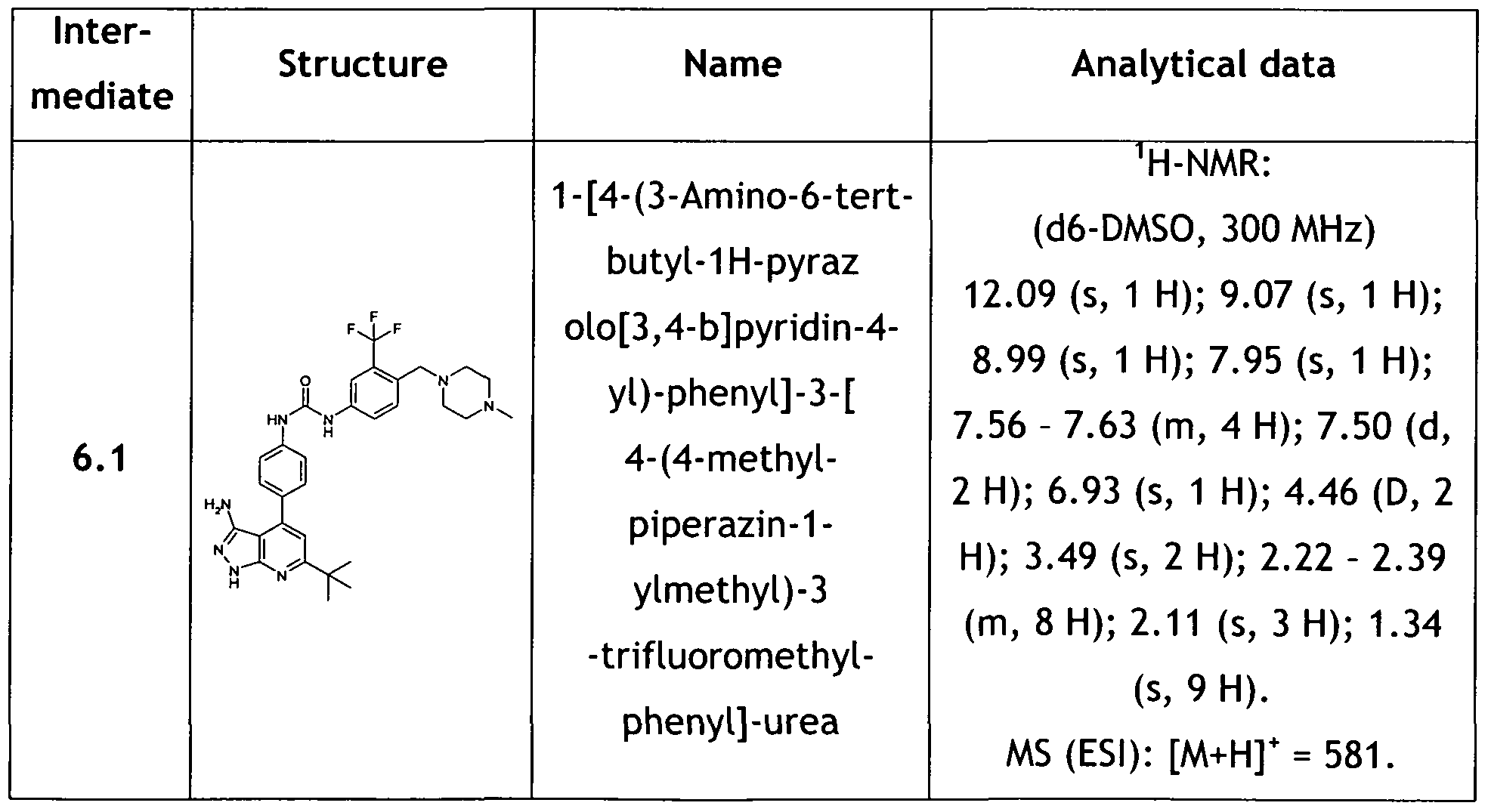












































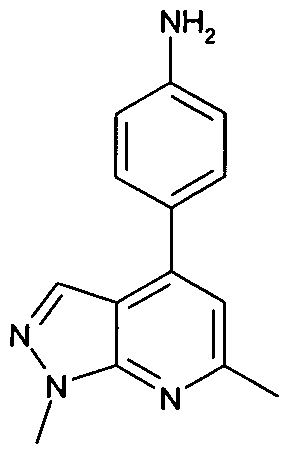





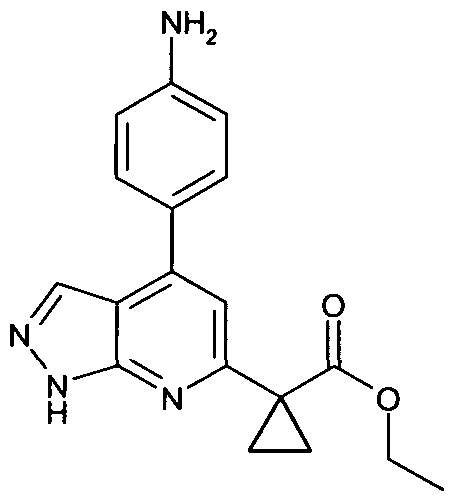







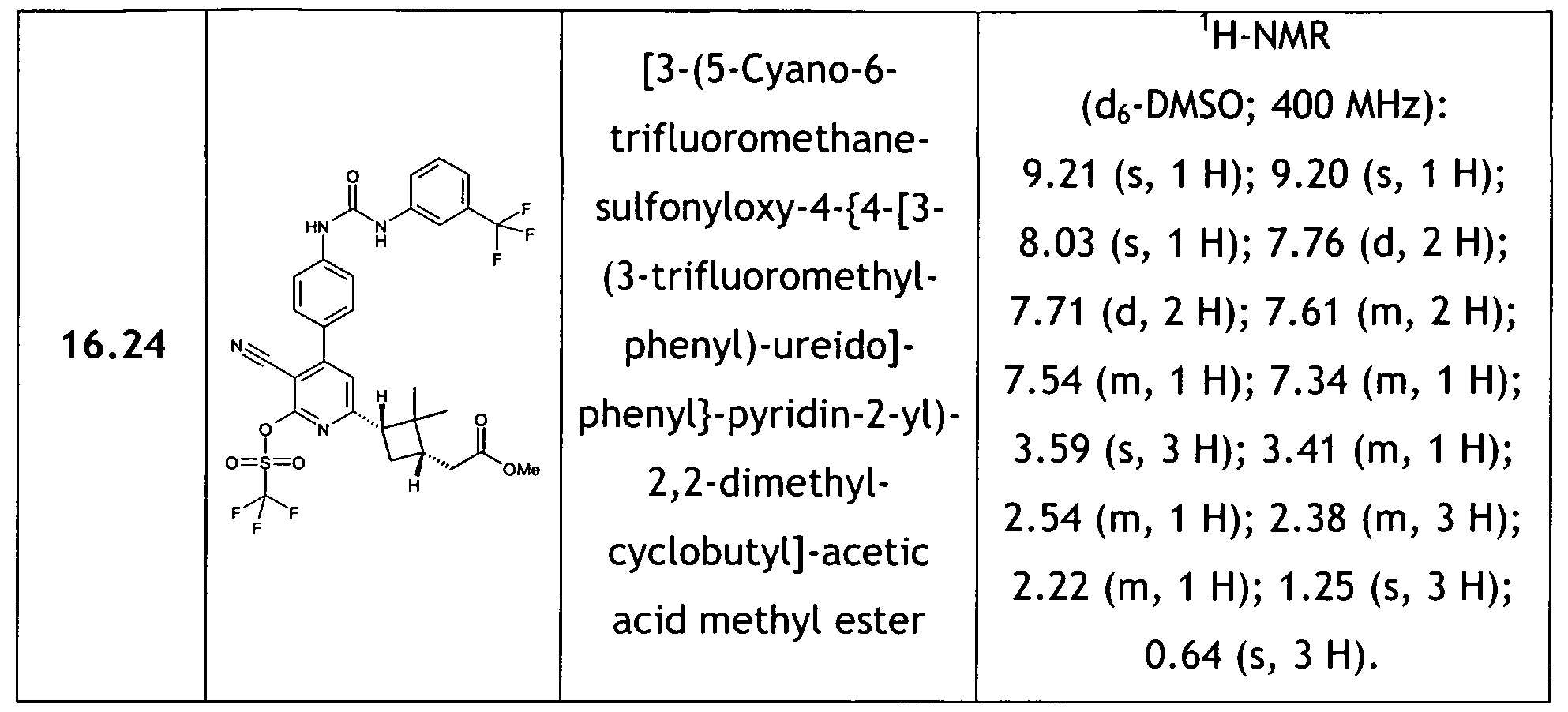








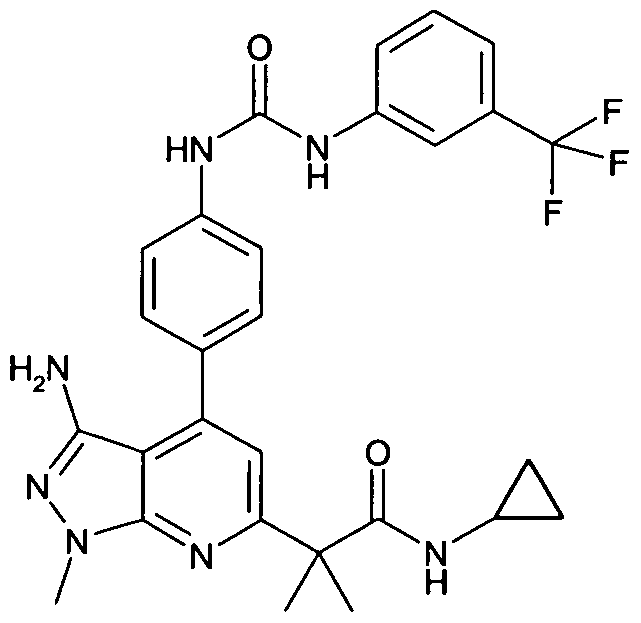
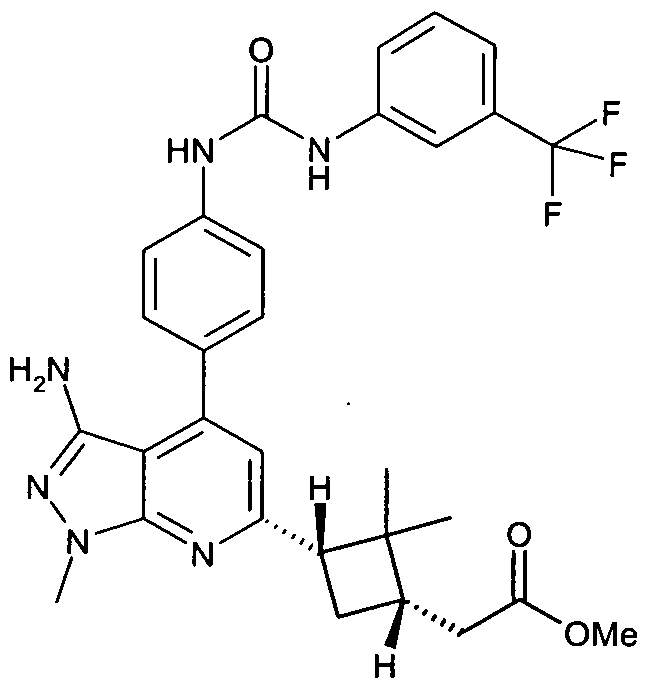


















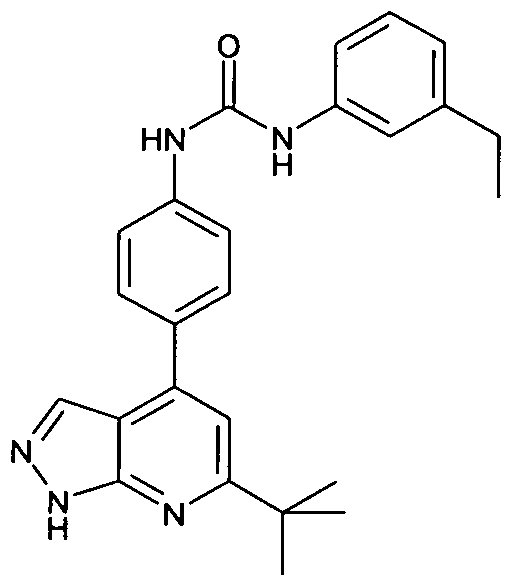











































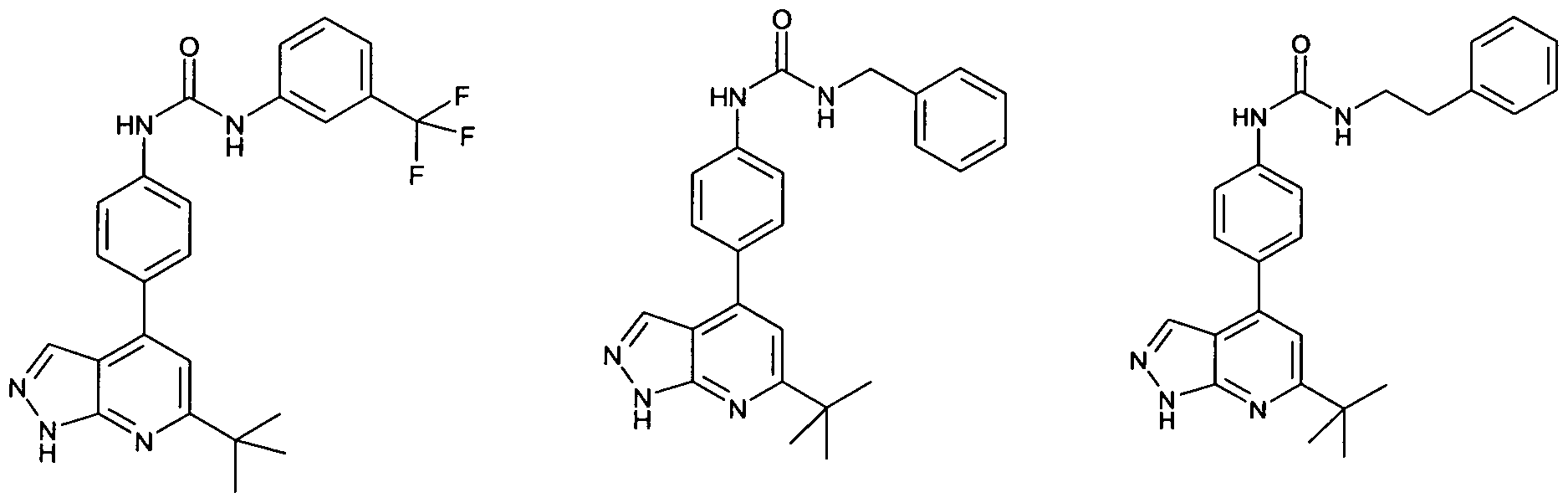













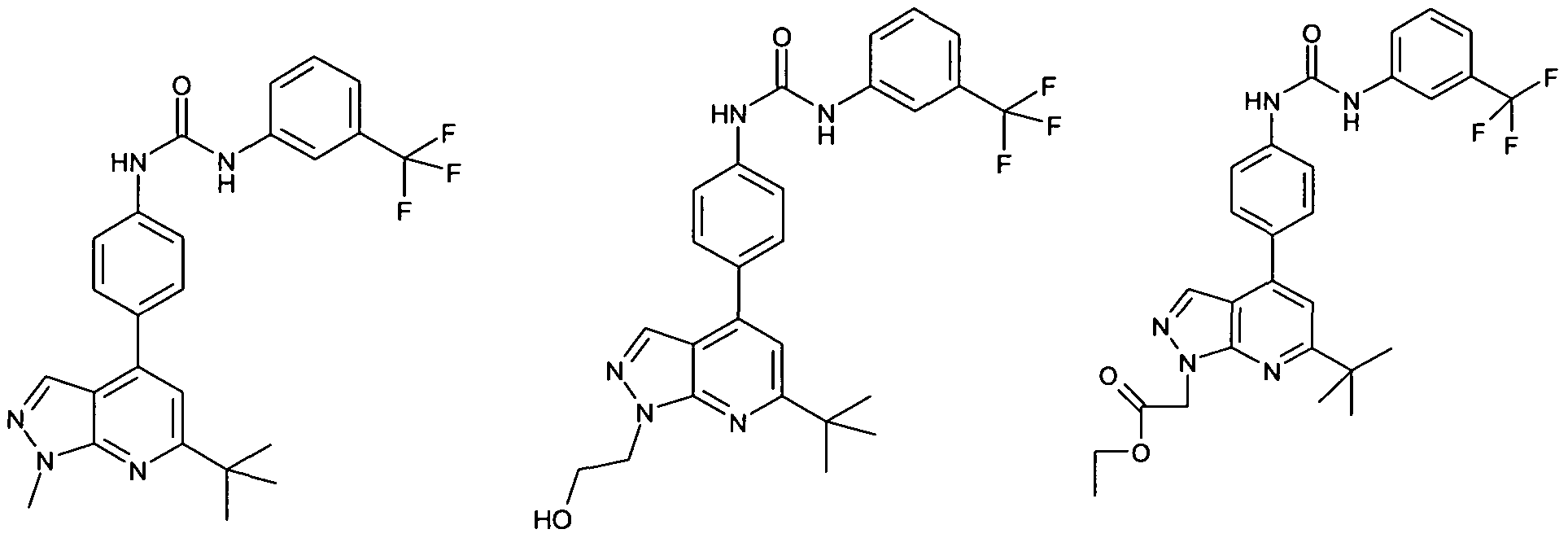













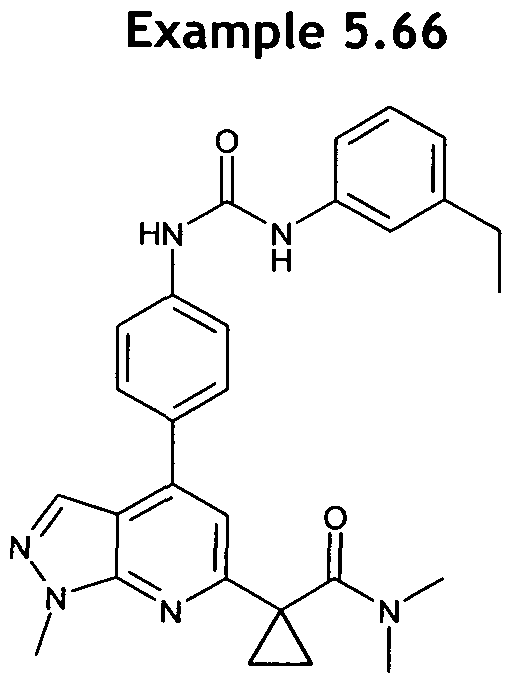







Claims


























Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ES07764753.5T ES2515117T3 (en) | 2006-06-13 | 2007-06-12 | Substituted aminopyrazolopyridines and their salts, their preparations and pharmaceutical compositions comprising them |
| EP07764753.5A EP2061790B1 (en) | 2006-06-13 | 2007-06-12 | Substituted aminopyrazolopyridines and salts thereof, their preparations and pharmaceutical compositions comprising them |
| JP2009514716A JP5475442B2 (en) | 2006-06-13 | 2007-06-12 | Substituted aminopyrazolopyrines and their salts, their preparation and pharmaceutical compositions comprising them |
| MX2008015955A MX2008015955A (en) | 2006-06-13 | 2007-06-12 | Substituted aminopyrazolopyridines and salts thereof, their preparations and pharmaceutical compositions comprising them. |
| EA200802407A EA200802407A1 (en) | 2006-06-13 | 2007-06-12 | SUBSTITUTED AMINOPYRAZOLOPIRIDINES AND THEIR SALTS, THEIR RECEIVES AND PHARMACEUTICAL COMPOSITIONS THAT CONTAIN THEM |
| AU2007260216A AU2007260216A1 (en) | 2006-06-13 | 2007-06-12 | Substituted aminopyrazolopyridines and salts thereof, their preparations and pharmaceutical compositions comprising them |
| CA2654789A CA2654789C (en) | 2006-06-13 | 2007-06-12 | Substituted aminopyrazolopyridines and salts thereof, their preparations and pharmaceutical compositions comprising them |
| BRPI0711961-5A BRPI0711961A2 (en) | 2006-06-13 | 2007-06-12 | substituted pyrazolopyridines and salts thereof, pharmaceutical compositions comprising them, methods of preparing them and uses thereof |
| IL195813A IL195813A0 (en) | 2006-06-13 | 2008-12-09 | Substituted aminopyrazolopyridines and salts thereof, their preparations and pharmaceutical compositions comprising them |
| NO20090171A NO20090171L (en) | 2006-06-13 | 2009-01-12 | Substituted aminopyrazole pyridines and their salts, processes for their preparation, and pharmaceutical preparations containing such |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP06090109A EP1867648A1 (en) | 2006-06-13 | 2006-06-13 | Substituted aminopyrazolopyridines and salts thereof, their preparations and pharmaceutical compositions comprising them. |
| EP06090109.7 | 2006-06-13 | ||
| EP07090020 | 2007-02-15 | ||
| EP07090020.4 | 2007-02-15 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007144204A1 true WO2007144204A1 (en) | 2007-12-21 |
Family
ID=38445661
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2007/005432 WO2007144204A1 (en) | 2006-06-13 | 2007-06-12 | Substituted aminopyrazolopyridines and salts thereof, their preparations and pharmaceutical compositions comprising them |
Country Status (21)
| Country | Link |
|---|---|
| US (1) | US7846928B2 (en) |
| EP (1) | EP2061790B1 (en) |
| JP (1) | JP5475442B2 (en) |
| KR (1) | KR20090018212A (en) |
| AR (1) | AR061346A1 (en) |
| AU (1) | AU2007260216A1 (en) |
| BR (1) | BRPI0711961A2 (en) |
| CA (1) | CA2654789C (en) |
| CL (1) | CL2007001711A1 (en) |
| CR (1) | CR10505A (en) |
| EA (1) | EA200802407A1 (en) |
| EC (1) | ECSP088963A (en) |
| ES (1) | ES2515117T3 (en) |
| GT (1) | GT200800285A (en) |
| IL (1) | IL195813A0 (en) |
| MX (1) | MX2008015955A (en) |
| NO (1) | NO20090171L (en) |
| PE (1) | PE20080205A1 (en) |
| TW (1) | TW200815438A (en) |
| UY (1) | UY30409A1 (en) |
| WO (1) | WO2007144204A1 (en) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009073300A1 (en) * | 2007-11-02 | 2009-06-11 | Vertex Pharmaceuticals Incorporated | [1h- pyrazolo [3, 4-b] pyridine-4-yl] -phenyle or -pyridin-2-yle derivatives as protein kinase c-theta |
| WO2011019780A1 (en) | 2009-08-11 | 2011-02-17 | Bristol-Myers Squibb Company | Azaindazoles as btk kinase modulators and use thereof |
| US8008322B2 (en) * | 2005-01-19 | 2011-08-30 | Aventis Pharma S.A. | Substituted pyrazolopyridines, compositions containing them, method for the production thereof, and their use |
| EP2671891A2 (en) | 2008-06-27 | 2013-12-11 | Amgen Inc. | Ang-2 inhibition to treat multiple sclerosis |
| US8653098B2 (en) | 2008-11-20 | 2014-02-18 | Genentech, Inc. | Pyrazolopyridine PI3K inhibitor compounds and methods of use |
| US8742106B2 (en) | 2009-12-21 | 2014-06-03 | Novartis Ag | Disubstituted heteroaryl-fused pyridines |
| JP2015524789A (en) * | 2012-03-26 | 2015-08-27 | スウェンソン,ロルフ,イー | Novel sphingosine 1-phosphate receptor antagonist |
| US10328053B2 (en) | 2016-08-26 | 2019-06-25 | Gilead Sciences, Inc. | Substituted pyrrolizine compounds and uses thereof |
| US10836769B2 (en) | 2018-02-26 | 2020-11-17 | Gilead Sciences, Inc. | Substituted pyrrolizine compounds and uses thereof |
| US11247987B2 (en) | 2017-10-06 | 2022-02-15 | Forma Therapeutics, Inc. | Inhibiting ubiquitin specific peptidase 30 |
| US11535618B2 (en) | 2018-10-05 | 2022-12-27 | Forma Therapeutics, Inc. | Fused pyrrolines which act as ubiquitin-specific protease 30 (USP30) inhibitors |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BRPI0912539A2 (en) * | 2008-05-05 | 2015-10-13 | Amgen Inc | compound, pharmaceutical composition, method for treating a disease, and use of the compound. |
| US20150164910A1 (en) * | 2012-06-15 | 2015-06-18 | The Regents Of The University Of Califonia | Antiviral compounds and methods of use |
| WO2017053604A1 (en) | 2015-09-23 | 2017-03-30 | The Regents Of The University Of California | Potent antiviral pyrazolopyridine compounds |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004113304A1 (en) * | 2003-05-22 | 2004-12-29 | Abbott Laboratories | Indazole, benzisoxazole, and benzisothiazole kinase inhibitors |
| WO2006050109A2 (en) * | 2004-10-29 | 2006-05-11 | Abbott Laboratories | Novel kinase inhibitors |
| WO2007000241A1 (en) * | 2005-06-27 | 2007-01-04 | Sanofi-Aventis | Pyrazolopyridine derivatives as inhibitors of beta-adrenergic receptor kinase 1 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6670364B2 (en) * | 2001-01-31 | 2003-12-30 | Telik, Inc. | Antagonists of MCP-1 function and methods of use thereof |
| EP1697369A1 (en) * | 2003-12-22 | 2006-09-06 | SB Pharmco Puerto Rico Inc | Crf receptor antagonists and methods relating thereto |
| FR2876691B1 (en) * | 2004-10-18 | 2006-12-29 | Sanofi Aventis Sa | PYRIDINE DERIVATIVES, THEIR PREPARATION, THEIR THERAPEUTIC APPLICATION |
| PE20061119A1 (en) * | 2005-01-19 | 2006-11-27 | Aventis Pharma Sa | PYRAZOLO PYRIDINES SUBSTITUTED AS INHIBITORS OF FAK, KDR AND Tie KINASES |
| EP1683796A1 (en) * | 2005-01-24 | 2006-07-26 | Schering Aktiengesellschaft | Pyrazolopyridines, their preparation and their medical use |
-
2007
- 2007-06-11 TW TW096121006A patent/TW200815438A/en unknown
- 2007-06-12 UY UY30409A patent/UY30409A1/en not_active Application Discontinuation
- 2007-06-12 BR BRPI0711961-5A patent/BRPI0711961A2/en not_active IP Right Cessation
- 2007-06-12 US US11/761,672 patent/US7846928B2/en not_active Expired - Fee Related
- 2007-06-12 EP EP07764753.5A patent/EP2061790B1/en active Active
- 2007-06-12 MX MX2008015955A patent/MX2008015955A/en not_active Application Discontinuation
- 2007-06-12 EA EA200802407A patent/EA200802407A1/en unknown
- 2007-06-12 WO PCT/EP2007/005432 patent/WO2007144204A1/en active Application Filing
- 2007-06-12 CL CL2007001711A patent/CL2007001711A1/en unknown
- 2007-06-12 AU AU2007260216A patent/AU2007260216A1/en not_active Abandoned
- 2007-06-12 PE PE2007000741A patent/PE20080205A1/en not_active Application Discontinuation
- 2007-06-12 JP JP2009514716A patent/JP5475442B2/en not_active Expired - Fee Related
- 2007-06-12 ES ES07764753.5T patent/ES2515117T3/en active Active
- 2007-06-12 CA CA2654789A patent/CA2654789C/en not_active Expired - Fee Related
- 2007-06-12 AR ARP070102552A patent/AR061346A1/en unknown
- 2007-06-12 KR KR1020097000562A patent/KR20090018212A/en not_active Application Discontinuation
-
2008
- 2008-12-09 IL IL195813A patent/IL195813A0/en unknown
- 2008-12-12 CR CR10505A patent/CR10505A/en not_active Application Discontinuation
- 2008-12-12 GT GT200800285A patent/GT200800285A/en unknown
- 2008-12-12 EC EC2008008963A patent/ECSP088963A/en unknown
-
2009
- 2009-01-12 NO NO20090171A patent/NO20090171L/en not_active Application Discontinuation
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004113304A1 (en) * | 2003-05-22 | 2004-12-29 | Abbott Laboratories | Indazole, benzisoxazole, and benzisothiazole kinase inhibitors |
| WO2006050109A2 (en) * | 2004-10-29 | 2006-05-11 | Abbott Laboratories | Novel kinase inhibitors |
| WO2007000241A1 (en) * | 2005-06-27 | 2007-01-04 | Sanofi-Aventis | Pyrazolopyridine derivatives as inhibitors of beta-adrenergic receptor kinase 1 |
Non-Patent Citations (1)
| Title |
|---|
| D.H. ALBERT ET AL.: "Preclinical activity of ABT-869, a multitargeted receptor tyrosine kinase inhibitor", MOLECULAR CANCER THERAPEUTICS, AMERICAN ASSOCIATION OF CANCER RESEARCH, US, vol. 5, no. 4, April 2006 (2006-04-01), pages 995 - 1006, XP002403181, ISSN: 1535-7163 * |
Cited By (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8008322B2 (en) * | 2005-01-19 | 2011-08-30 | Aventis Pharma S.A. | Substituted pyrazolopyridines, compositions containing them, method for the production thereof, and their use |
| JP2011502994A (en) * | 2007-11-02 | 2011-01-27 | バーテックス ファーマシューティカルズ インコーポレイテッド | [1H-pyrazolo [3,4-B] pyridin-4-yl] -phenylene or -pyridin-2-yl derivatives as protein kinase C-θ |
| US8173635B2 (en) | 2007-11-02 | 2012-05-08 | Vertex Pharmaceuticals Incorporated | Kinase inhibitors |
| US8367697B2 (en) | 2007-11-02 | 2013-02-05 | Vertex Pharmaceuticals Incorporated | Kinase inhibitors |
| WO2009073300A1 (en) * | 2007-11-02 | 2009-06-11 | Vertex Pharmaceuticals Incorporated | [1h- pyrazolo [3, 4-b] pyridine-4-yl] -phenyle or -pyridin-2-yle derivatives as protein kinase c-theta |
| EP2671891A2 (en) | 2008-06-27 | 2013-12-11 | Amgen Inc. | Ang-2 inhibition to treat multiple sclerosis |
| US8653098B2 (en) | 2008-11-20 | 2014-02-18 | Genentech, Inc. | Pyrazolopyridine PI3K inhibitor compounds and methods of use |
| EP2789615A1 (en) | 2009-08-11 | 2014-10-15 | Bristol-Myers Squibb Company | Azaindazoles as Btk kinase modulators and use thereof |
| WO2011019780A1 (en) | 2009-08-11 | 2011-02-17 | Bristol-Myers Squibb Company | Azaindazoles as btk kinase modulators and use thereof |
| US8846673B2 (en) | 2009-08-11 | 2014-09-30 | Bristol-Myers Squibb Company | Azaindazoles as kinase inhibitors and use thereof |
| US8742106B2 (en) | 2009-12-21 | 2014-06-03 | Novartis Ag | Disubstituted heteroaryl-fused pyridines |
| JP2015524789A (en) * | 2012-03-26 | 2015-08-27 | スウェンソン,ロルフ,イー | Novel sphingosine 1-phosphate receptor antagonist |
| US10328053B2 (en) | 2016-08-26 | 2019-06-25 | Gilead Sciences, Inc. | Substituted pyrrolizine compounds and uses thereof |
| US10874640B2 (en) | 2016-08-26 | 2020-12-29 | Gilead Sciences, Inc. | Substituted pyrrolizine compounds and uses thereof |
| US11247987B2 (en) | 2017-10-06 | 2022-02-15 | Forma Therapeutics, Inc. | Inhibiting ubiquitin specific peptidase 30 |
| US10836769B2 (en) | 2018-02-26 | 2020-11-17 | Gilead Sciences, Inc. | Substituted pyrrolizine compounds and uses thereof |
| US11420974B2 (en) | 2018-02-26 | 2022-08-23 | Gilead Sciences, Inc. | Substituted pyrrolizine compounds and uses thereof |
| US11535618B2 (en) | 2018-10-05 | 2022-12-27 | Forma Therapeutics, Inc. | Fused pyrrolines which act as ubiquitin-specific protease 30 (USP30) inhibitors |
| US11814386B2 (en) | 2018-10-05 | 2023-11-14 | Forma Therapeutics, Inc. | Fused pyrrolines which act as ubiquitin-specific protease 30 (USP30) inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2654789C (en) | 2014-08-05 |
| BRPI0711961A2 (en) | 2012-01-17 |
| CR10505A (en) | 2009-03-20 |
| TW200815438A (en) | 2008-04-01 |
| UY30409A1 (en) | 2008-01-31 |
| AR061346A1 (en) | 2008-08-20 |
| ECSP088963A (en) | 2009-01-30 |
| PE20080205A1 (en) | 2008-06-01 |
| EA200802407A1 (en) | 2009-06-30 |
| US20080058326A1 (en) | 2008-03-06 |
| CA2654789A1 (en) | 2007-12-21 |
| EP2061790B1 (en) | 2014-08-13 |
| US7846928B2 (en) | 2010-12-07 |
| KR20090018212A (en) | 2009-02-19 |
| JP2009539914A (en) | 2009-11-19 |
| CL2007001711A1 (en) | 2008-01-04 |
| ES2515117T3 (en) | 2014-10-29 |
| IL195813A0 (en) | 2009-09-22 |
| NO20090171L (en) | 2009-03-12 |
| JP5475442B2 (en) | 2014-04-16 |
| GT200800285A (en) | 2009-03-09 |
| MX2008015955A (en) | 2009-01-09 |
| AU2007260216A1 (en) | 2007-12-21 |
| EP2061790A1 (en) | 2009-05-27 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2061790B1 (en) | Substituted aminopyrazolopyridines and salts thereof, their preparations and pharmaceutical compositions comprising them | |
| EP2032572B1 (en) | Substituted aminopyrazolopyridines and salts thereof, their preparations and pharmaceutical compositions comprising them | |
| US20090062273A1 (en) | 3-H-pyrazolopyridines and salts thereof, pharmaceutical compositions comprising same, methods of preparing same and uses of same. | |
| EP2229389B1 (en) | Alkynylaryl compounds and salts thereof, pharmaceutical compositions comprising same, methods of preparing same and uses of same | |
| US7795264B2 (en) | Substituted arylpyrazolopyridines and salts thereof, pharmaceutical compositions comprising same, methods of preparing same and uses of same | |
| EP1867648A1 (en) | Substituted aminopyrazolopyridines and salts thereof, their preparations and pharmaceutical compositions comprising them. | |
| WO2007147574A1 (en) | Sulfonamido-macrocycles as tie2 inhibitors and salts thereof, pharmaceutical compositions comprising same, methods of preparing same and uses of same | |
| WO2007147575A2 (en) | Substituted sulfonamido-macrocycles as tie2 inhibitors and salts thereof, pharmaceutical compositions comprisings same, methods of preparing same and uses same | |
| EP1932845A1 (en) | 3-H-pyrazolopyridines and salts thereof, pharmaceutical compositions comprising same, methods of preparing same and uses of same | |
| EP1867647A1 (en) | Substituted aminopyrazolopyridines and salts thereof, their preparations and pharmaceutical compositions comprising them. | |
| EP2042500A1 (en) | N-[4-(1-H-pyrazolo[3,4-c]pyridin-4-yl)phenyl]-N'-[(hetero)aryl]-urea compounds, pharmaceutical compositions comprising them, their preparation and their use as Tie2-kinase inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780030210.3 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07764753 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2654789 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 195813 Country of ref document: IL Ref document number: 2007764753 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007260216 Country of ref document: AU Ref document number: 573511 Country of ref document: NZ Ref document number: 10214/DELNP/2008 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009514716 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12008502767 Country of ref document: PH Ref document number: 08131986 Country of ref document: CO Ref document number: CR2008-010505 Country of ref document: CR Ref document number: MX/A/2008/015955 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008121976 Country of ref document: EG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200802407 Country of ref document: EA |
|
| ENP | Entry into the national phase |
Ref document number: 2007260216 Country of ref document: AU Date of ref document: 20070612 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020097000562 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: PI0711961 Country of ref document: BR Kind code of ref document: A2 Effective date: 20081215 |