WO2007143597A2 - Organic compounds - Google Patents
Organic compounds Download PDFInfo
- Publication number
- WO2007143597A2 WO2007143597A2 PCT/US2007/070293 US2007070293W WO2007143597A2 WO 2007143597 A2 WO2007143597 A2 WO 2007143597A2 US 2007070293 W US2007070293 W US 2007070293W WO 2007143597 A2 WO2007143597 A2 WO 2007143597A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- carboxamide
- oxopyridin
- methyl
- group
- benzyl
- Prior art date
Links
- 150000002894 organic compounds Chemical class 0.000 title description 2
- 238000000034 method Methods 0.000 claims abstract description 192
- 125000000623 heterocyclic group Chemical group 0.000 claims abstract description 123
- 108010087894 Fatty acid desaturases Proteins 0.000 claims abstract description 91
- 230000000694 effects Effects 0.000 claims abstract description 50
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 33
- 102000016553 Stearoyl-CoA Desaturase Human genes 0.000 claims abstract 5
- 150000001875 compounds Chemical class 0.000 claims description 368
- 125000000217 alkyl group Chemical group 0.000 claims description 155
- 125000003118 aryl group Chemical group 0.000 claims description 135
- -1 ary] Chemical group 0.000 claims description 119
- 125000001072 heteroaryl group Chemical group 0.000 claims description 118
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 103
- 239000001257 hydrogen Substances 0.000 claims description 100
- 229910052739 hydrogen Inorganic materials 0.000 claims description 100
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 92
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 81
- 201000010099 disease Diseases 0.000 claims description 71
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 69
- 125000001316 cycloalkyl alkyl group Chemical group 0.000 claims description 65
- 125000004446 heteroarylalkyl group Chemical group 0.000 claims description 63
- 238000002360 preparation method Methods 0.000 claims description 63
- 125000001188 haloalkyl group Chemical group 0.000 claims description 61
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 61
- 125000004415 heterocyclylalkyl group Chemical group 0.000 claims description 57
- GGNIKGLUPSHSBV-UHFFFAOYSA-N thiazole-5-carboxamide Chemical compound NC(=O)C1=CN=CS1 GGNIKGLUPSHSBV-UHFFFAOYSA-N 0.000 claims description 52
- 125000002768 hydroxyalkyl group Chemical group 0.000 claims description 36
- 125000004432 carbon atom Chemical group C* 0.000 claims description 32
- 208000035475 disorder Diseases 0.000 claims description 32
- 125000004183 alkoxy alkyl group Chemical group 0.000 claims description 28
- 125000003545 alkoxy group Chemical group 0.000 claims description 26
- 230000001404 mediated effect Effects 0.000 claims description 26
- 125000003342 alkenyl group Chemical group 0.000 claims description 24
- 125000002947 alkylene group Chemical group 0.000 claims description 23
- 239000003112 inhibitor Substances 0.000 claims description 23
- 229910052757 nitrogen Inorganic materials 0.000 claims description 23
- 229940002612 prodrug Drugs 0.000 claims description 21
- 239000000651 prodrug Substances 0.000 claims description 21
- 241000124008 Mammalia Species 0.000 claims description 20
- 125000000304 alkynyl group Chemical group 0.000 claims description 20
- 238000011282 treatment Methods 0.000 claims description 20
- 239000003814 drug Substances 0.000 claims description 19
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 18
- 150000003839 salts Chemical class 0.000 claims description 18
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 17
- 208000008589 Obesity Diseases 0.000 claims description 15
- 235000020824 obesity Nutrition 0.000 claims description 14
- 125000004450 alkenylene group Chemical group 0.000 claims description 13
- 230000005764 inhibitory process Effects 0.000 claims description 12
- 229910052717 sulfur Inorganic materials 0.000 claims description 12
- 208000001072 type 2 diabetes mellitus Diseases 0.000 claims description 11
- 208000001145 Metabolic Syndrome Diseases 0.000 claims description 10
- 201000000690 abdominal obesity-metabolic syndrome Diseases 0.000 claims description 10
- 125000004419 alkynylene group Chemical group 0.000 claims description 10
- 239000003937 drug carrier Substances 0.000 claims description 10
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 claims description 10
- 229910052760 oxygen Inorganic materials 0.000 claims description 10
- YZVFSQQHQPPKNX-UHFFFAOYSA-N 1,3-thiazole-5-carboxylic acid Chemical compound OC(=O)C1=CN=CS1 YZVFSQQHQPPKNX-UHFFFAOYSA-N 0.000 claims description 9
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 9
- 206010012601 diabetes mellitus Diseases 0.000 claims description 9
- 230000002401 inhibitory effect Effects 0.000 claims description 9
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 9
- 208000032928 Dyslipidaemia Diseases 0.000 claims description 8
- 208000017170 Lipid metabolism disease Diseases 0.000 claims description 8
- 229910004749 OS(O)2 Inorganic materials 0.000 claims description 8
- SIARJEKBADXQJG-LFZQUHGESA-N stearoyl-CoA Chemical compound O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCSC(=O)CCCCCCCCCCCCCCCCC)O[C@H]1N1C2=NC=NC(N)=C2N=C1 SIARJEKBADXQJG-LFZQUHGESA-N 0.000 claims description 8
- 206010022489 Insulin Resistance Diseases 0.000 claims description 7
- DENPQNAWGQXKCU-UHFFFAOYSA-N thiophene-2-carboxamide Chemical compound NC(=O)C1=CC=CS1 DENPQNAWGQXKCU-UHFFFAOYSA-N 0.000 claims description 7
- PVNIIMVLHYAWGP-UHFFFAOYSA-N Niacin Chemical compound OC(=O)C1=CC=CN=C1 PVNIIMVLHYAWGP-UHFFFAOYSA-N 0.000 claims description 6
- 239000003446 ligand Substances 0.000 claims description 6
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 6
- 229910052727 yttrium Inorganic materials 0.000 claims description 5
- 208000002874 Acne Vulgaris Diseases 0.000 claims description 4
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 claims description 4
- 102000004877 Insulin Human genes 0.000 claims description 4
- 108090001061 Insulin Proteins 0.000 claims description 4
- 206010000496 acne Diseases 0.000 claims description 4
- 230000037396 body weight Effects 0.000 claims description 4
- 229940125396 insulin Drugs 0.000 claims description 4
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims description 4
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 4
- 206010020772 Hypertension Diseases 0.000 claims description 3
- 201000001431 Hyperuricemia Diseases 0.000 claims description 3
- 206010023330 Keloid scar Diseases 0.000 claims description 3
- 102000003728 Peroxisome Proliferator-Activated Receptors Human genes 0.000 claims description 3
- 108090000029 Peroxisome Proliferator-Activated Receptors Proteins 0.000 claims description 3
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 claims description 3
- 239000008103 glucose Substances 0.000 claims description 3
- 235000001968 nicotinic acid Nutrition 0.000 claims description 3
- 239000011664 nicotinic acid Substances 0.000 claims description 3
- 229960003512 nicotinic acid Drugs 0.000 claims description 3
- 230000036573 scar formation Effects 0.000 claims description 3
- 208000011580 syndromic disease Diseases 0.000 claims description 3
- 201000005665 thrombophilia Diseases 0.000 claims description 3
- ITMCEJHCFYSIIV-UHFFFAOYSA-M triflate Chemical compound [O-]S(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-M 0.000 claims description 3
- 208000016261 weight loss Diseases 0.000 claims description 3
- 230000004580 weight loss Effects 0.000 claims description 3
- 229940077274 Alpha glucosidase inhibitor Drugs 0.000 claims description 2
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 claims description 2
- 229940123208 Biguanide Drugs 0.000 claims description 2
- 206010006895 Cachexia Diseases 0.000 claims description 2
- 229920001268 Cholestyramine Polymers 0.000 claims description 2
- 201000004624 Dermatitis Diseases 0.000 claims description 2
- 208000002249 Diabetes Complications Diseases 0.000 claims description 2
- 206010012655 Diabetic complications Diseases 0.000 claims description 2
- 229940122355 Insulin sensitizer Drugs 0.000 claims description 2
- 102000016267 Leptin Human genes 0.000 claims description 2
- 108010092277 Leptin Proteins 0.000 claims description 2
- 206010033307 Overweight Diseases 0.000 claims description 2
- 201000004681 Psoriasis Diseases 0.000 claims description 2
- 102000018692 Sulfonylurea Receptors Human genes 0.000 claims description 2
- 108010091821 Sulfonylurea Receptors Proteins 0.000 claims description 2
- 241000534944 Thia Species 0.000 claims description 2
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 claims description 2
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 claims description 2
- 229960001138 acetylsalicylic acid Drugs 0.000 claims description 2
- 239000003888 alpha glucosidase inhibitor Chemical class 0.000 claims description 2
- 208000022531 anorexia Diseases 0.000 claims description 2
- 208000010668 atopic eczema Diseases 0.000 claims description 2
- 206010061428 decreased appetite Diseases 0.000 claims description 2
- 230000003247 decreasing effect Effects 0.000 claims description 2
- 229940125753 fibrate Drugs 0.000 claims description 2
- 230000002473 insulinotropic effect Effects 0.000 claims description 2
- NRYBAZVQPHGZNS-ZSOCWYAHSA-N leptin Chemical compound O=C([C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](N)CC(C)C)CCSC)N1CCC[C@H]1C(=O)NCC(=O)N[C@@H](CS)C(O)=O NRYBAZVQPHGZNS-ZSOCWYAHSA-N 0.000 claims description 2
- 229940039781 leptin Drugs 0.000 claims description 2
- 230000002265 prevention Effects 0.000 claims description 2
- 239000004059 squalene synthase inhibitor Substances 0.000 claims description 2
- 125000001475 halogen functional group Chemical group 0.000 claims 8
- 101000631826 Homo sapiens Stearoyl-CoA desaturase Proteins 0.000 claims 3
- 102000055981 human SCD1 Human genes 0.000 claims 3
- 101001032756 Rattus norvegicus Granzyme-like protein 1 Chemical class 0.000 claims 2
- 125000004438 haloalkoxy group Chemical group 0.000 claims 2
- 208000017520 skin disease Diseases 0.000 claims 2
- DAUYIKBTMNZABP-UHFFFAOYSA-N thiophene-3-carboxamide Chemical compound NC(=O)C=1C=CSC=1 DAUYIKBTMNZABP-UHFFFAOYSA-N 0.000 claims 2
- SMNDYUVBFMFKNZ-UHFFFAOYSA-M 2-furoate Chemical compound [O-]C(=O)C1=CC=CO1 SMNDYUVBFMFKNZ-UHFFFAOYSA-M 0.000 claims 1
- 125000003349 3-pyridyl group Chemical group N1=C([H])C([*])=C([H])C([H])=C1[H] 0.000 claims 1
- XNCOSPRUTUOJCJ-UHFFFAOYSA-N Biguanide Chemical class NC(N)=NC(N)=N XNCOSPRUTUOJCJ-UHFFFAOYSA-N 0.000 claims 1
- 102100025012 Dipeptidyl peptidase 4 Human genes 0.000 claims 1
- 229940121710 HMGCoA reductase inhibitor Drugs 0.000 claims 1
- 101000684208 Homo sapiens Prolyl endopeptidase FAP Proteins 0.000 claims 1
- OQIQSTLJSLGHID-WNWIJWBNSA-N aflatoxin B1 Chemical compound C=1([C@@H]2C=CO[C@@H]2OC=1C=C(C1=2)OC)C=2OC(=O)C2=C1CCC2=O OQIQSTLJSLGHID-WNWIJWBNSA-N 0.000 claims 1
- 239000002471 hydroxymethylglutaryl coenzyme A reductase inhibitor Substances 0.000 claims 1
- 239000004026 insulin derivative Substances 0.000 claims 1
- ILVXOBCQQYKLDS-UHFFFAOYSA-N pyridine N-oxide Chemical compound [O-][N+]1=CC=CC=C1 ILVXOBCQQYKLDS-UHFFFAOYSA-N 0.000 claims 1
- 125000004192 tetrahydrofuran-2-yl group Chemical group [H]C1([H])OC([H])(*)C([H])([H])C1([H])[H] 0.000 claims 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 234
- 239000007787 solid Substances 0.000 description 168
- 238000005160 1H NMR spectroscopy Methods 0.000 description 165
- 238000000524 positive electrospray ionisation mass spectrometry Methods 0.000 description 159
- 230000015572 biosynthetic process Effects 0.000 description 153
- 238000003786 synthesis reaction Methods 0.000 description 151
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 136
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 104
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 84
- 102100028897 Stearoyl-CoA desaturase Human genes 0.000 description 77
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 63
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 62
- 239000002253 acid Substances 0.000 description 60
- 239000000203 mixture Substances 0.000 description 55
- 239000011541 reaction mixture Substances 0.000 description 54
- 239000000243 solution Substances 0.000 description 51
- 150000002431 hydrogen Chemical group 0.000 description 33
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 32
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 31
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 30
- WGQKYBSKWIADBV-UHFFFAOYSA-N benzylamine Chemical compound NCC1=CC=CC=C1 WGQKYBSKWIADBV-UHFFFAOYSA-N 0.000 description 30
- 150000002632 lipids Chemical class 0.000 description 29
- 238000005481 NMR spectroscopy Methods 0.000 description 28
- 238000006243 chemical reaction Methods 0.000 description 28
- 150000003254 radicals Chemical class 0.000 description 27
- 241000282414 Homo sapiens Species 0.000 description 26
- 239000012044 organic layer Substances 0.000 description 25
- 238000004440 column chromatography Methods 0.000 description 23
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 22
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 20
- 239000002904 solvent Substances 0.000 description 20
- 239000000706 filtrate Substances 0.000 description 19
- 241001465754 Metazoa Species 0.000 description 18
- 239000012267 brine Substances 0.000 description 18
- 239000000194 fatty acid Substances 0.000 description 18
- 229920006395 saturated elastomer Polymers 0.000 description 18
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 18
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 17
- 125000005843 halogen group Chemical group 0.000 description 17
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 16
- 241000700605 Viruses Species 0.000 description 16
- 235000014113 dietary fatty acids Nutrition 0.000 description 16
- 229930195729 fatty acid Natural products 0.000 description 16
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 15
- 125000005605 benzo group Chemical group 0.000 description 15
- 150000004665 fatty acids Chemical class 0.000 description 15
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 15
- 235000017557 sodium bicarbonate Nutrition 0.000 description 15
- 238000012360 testing method Methods 0.000 description 15
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 14
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 13
- 229910052799 carbon Inorganic materials 0.000 description 13
- 229910052938 sodium sulfate Inorganic materials 0.000 description 13
- 235000011152 sodium sulphate Nutrition 0.000 description 13
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 12
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 12
- 125000006239 protecting group Chemical group 0.000 description 12
- 229940124597 therapeutic agent Drugs 0.000 description 11
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 10
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 10
- 241000699670 Mus sp. Species 0.000 description 10
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 10
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 10
- ZMXDDKWLCZADIW-UHFFFAOYSA-N Vilsmeier-Haack reagent Natural products CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 10
- 238000003556 assay Methods 0.000 description 10
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 10
- 229910000027 potassium carbonate Inorganic materials 0.000 description 10
- 241000699666 Mus <mouse, genus> Species 0.000 description 9
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 9
- 230000004071 biological effect Effects 0.000 description 9
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 9
- UFTFJSFQGQCHQW-UHFFFAOYSA-N triformin Chemical compound O=COCC(OC=O)COC=O UFTFJSFQGQCHQW-UHFFFAOYSA-N 0.000 description 9
- VEUMBMHMMCOFAG-UHFFFAOYSA-N 2,3-dihydrooxadiazole Chemical compound N1NC=CO1 VEUMBMHMMCOFAG-UHFFFAOYSA-N 0.000 description 8
- 102000004190 Enzymes Human genes 0.000 description 8
- 108090000790 Enzymes Proteins 0.000 description 8
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 8
- 150000001412 amines Chemical class 0.000 description 8
- 235000012000 cholesterol Nutrition 0.000 description 8
- 238000001914 filtration Methods 0.000 description 8
- 238000004519 manufacturing process Methods 0.000 description 8
- 239000012299 nitrogen atmosphere Substances 0.000 description 8
- WHIRHMIFXAFTLW-UHFFFAOYSA-N thiophen-2-ylmethanamine Chemical compound N[CH]C1=CC=CS1 WHIRHMIFXAFTLW-UHFFFAOYSA-N 0.000 description 8
- 150000003626 triacylglycerols Chemical class 0.000 description 8
- JVOCWIRGDNWGKH-UHFFFAOYSA-N 2-cyclopropylethyl 4-methylbenzenesulfonate Chemical compound C1=CC(C)=CC=C1S(=O)(=O)OCCC1CC1 JVOCWIRGDNWGKH-UHFFFAOYSA-N 0.000 description 7
- 0 CCO*C=C*(C(C)*)C1[C@](C)(*2)*2C(C)=*(C*C*(C)*(C)C*C2CC2)*1 Chemical compound CCO*C=C*(C(C)*)C1[C@](C)(*2)*2C(C)=*(C*C*(C)*(C)C*C2CC2)*1 0.000 description 7
- 101100041816 Homo sapiens SCD gene Proteins 0.000 description 7
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 7
- 235000005911 diet Nutrition 0.000 description 7
- 150000002430 hydrocarbons Chemical class 0.000 description 7
- 208000006575 hypertriglyceridemia Diseases 0.000 description 7
- 238000001727 in vivo Methods 0.000 description 7
- 230000037356 lipid metabolism Effects 0.000 description 7
- 210000001732 sebaceous gland Anatomy 0.000 description 7
- 208000024172 Cardiovascular disease Diseases 0.000 description 6
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 6
- 241000282412 Homo Species 0.000 description 6
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 6
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 6
- 108010062497 VLDL Lipoproteins Proteins 0.000 description 6
- 239000003795 chemical substances by application Substances 0.000 description 6
- 239000003085 diluting agent Substances 0.000 description 6
- 210000004185 liver Anatomy 0.000 description 6
- 238000010992 reflux Methods 0.000 description 6
- 238000003756 stirring Methods 0.000 description 6
- 239000000725 suspension Substances 0.000 description 6
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 5
- 101710159293 Acyl-CoA desaturase 1 Proteins 0.000 description 5
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical class [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 5
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 5
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 5
- 229960000583 acetic acid Drugs 0.000 description 5
- 238000010976 amide bond formation reaction Methods 0.000 description 5
- 150000005840 aryl radicals Chemical class 0.000 description 5
- 125000004429 atom Chemical group 0.000 description 5
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 5
- 239000000969 carrier Substances 0.000 description 5
- RGJOEKWQDUBAIZ-UHFFFAOYSA-N coenzime A Natural products OC1C(OP(O)(O)=O)C(COP(O)(=O)OP(O)(=O)OCC(C)(C)C(O)C(=O)NCCC(=O)NCCS)OC1N1C2=NC=NC(N)=C2N=C1 RGJOEKWQDUBAIZ-UHFFFAOYSA-N 0.000 description 5
- 239000005516 coenzyme A Substances 0.000 description 5
- 229940093530 coenzyme a Drugs 0.000 description 5
- 238000005859 coupling reaction Methods 0.000 description 5
- 125000004122 cyclic group Chemical group 0.000 description 5
- KDTSHFARGAKYJN-UHFFFAOYSA-N dephosphocoenzyme A Natural products OC1C(O)C(COP(O)(=O)OP(O)(=O)OCC(C)(C)C(O)C(=O)NCCC(=O)NCCS)OC1N1C2=NC=NC(N)=C2N=C1 KDTSHFARGAKYJN-UHFFFAOYSA-N 0.000 description 5
- 230000037213 diet Effects 0.000 description 5
- 239000007788 liquid Substances 0.000 description 5
- 229910052751 metal Inorganic materials 0.000 description 5
- 239000002184 metal Substances 0.000 description 5
- 230000008569 process Effects 0.000 description 5
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 5
- 239000007858 starting material Substances 0.000 description 5
- 239000000758 substrate Substances 0.000 description 5
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 4
- XVMSFILGAMDHEY-UHFFFAOYSA-N 6-(4-aminophenyl)sulfonylpyridin-3-amine Chemical compound C1=CC(N)=CC=C1S(=O)(=O)C1=CC=C(N)C=N1 XVMSFILGAMDHEY-UHFFFAOYSA-N 0.000 description 4
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 4
- 241000283690 Bos taurus Species 0.000 description 4
- RGJOEKWQDUBAIZ-IBOSZNHHSA-N CoASH Chemical compound O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCS)O[C@H]1N1C2=NC=NC(N)=C2N=C1 RGJOEKWQDUBAIZ-IBOSZNHHSA-N 0.000 description 4
- 206010014486 Elevated triglycerides Diseases 0.000 description 4
- 206010019708 Hepatic steatosis Diseases 0.000 description 4
- 208000035150 Hypercholesterolemia Diseases 0.000 description 4
- 206010028980 Neoplasm Diseases 0.000 description 4
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 4
- 235000021355 Stearic acid Nutrition 0.000 description 4
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzaldehyde Chemical compound O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 4
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 4
- AEILLAXRDHDKDY-UHFFFAOYSA-N bromomethylcyclopropane Chemical compound BrCC1CC1 AEILLAXRDHDKDY-UHFFFAOYSA-N 0.000 description 4
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 4
- 238000006555 catalytic reaction Methods 0.000 description 4
- 230000008859 change Effects 0.000 description 4
- 239000003153 chemical reaction reagent Substances 0.000 description 4
- 239000012043 crude product Substances 0.000 description 4
- 208000037765 diseases and disorders Diseases 0.000 description 4
- IPCSVZSSVZVIGE-UHFFFAOYSA-N hexadecanoic acid Chemical compound CCCCCCCCCCCCCCCC(O)=O IPCSVZSSVZVIGE-UHFFFAOYSA-N 0.000 description 4
- 230000007062 hydrolysis Effects 0.000 description 4
- 238000006460 hydrolysis reaction Methods 0.000 description 4
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 4
- XEEYBQQBJWHFJM-UHFFFAOYSA-N iron Substances [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 4
- 210000001853 liver microsome Anatomy 0.000 description 4
- 210000004379 membrane Anatomy 0.000 description 4
- 239000012528 membrane Substances 0.000 description 4
- 230000003228 microsomal effect Effects 0.000 description 4
- 125000004433 nitrogen atom Chemical group N* 0.000 description 4
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 4
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 4
- 150000003904 phospholipids Chemical class 0.000 description 4
- 239000008117 stearic acid Substances 0.000 description 4
- 125000001424 substituent group Chemical group 0.000 description 4
- 210000001519 tissue Anatomy 0.000 description 4
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 4
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 3
- YOETUEMZNOLGDB-UHFFFAOYSA-N 2-methylpropyl carbonochloridate Chemical compound CC(C)COC(Cl)=O YOETUEMZNOLGDB-UHFFFAOYSA-N 0.000 description 3
- LVQFHDAKZHGEAJ-UHFFFAOYSA-M 4-methylbenzenesulfonate Chemical compound [CH2]C1=CC=C(S([O-])(=O)=O)C=C1 LVQFHDAKZHGEAJ-UHFFFAOYSA-M 0.000 description 3
- DNUDULSSNHKPHP-UHFFFAOYSA-N 4-phenyl-1h-pyridin-2-one Chemical compound C1=NC(O)=CC(C=2C=CC=CC=2)=C1 DNUDULSSNHKPHP-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- 206010014476 Elevated cholesterol Diseases 0.000 description 3
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 3
- DTHNMHAUYICORS-KTKZVXAJSA-N Glucagon-like peptide 1 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1N=CNC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 DTHNMHAUYICORS-KTKZVXAJSA-N 0.000 description 3
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 3
- 108010010234 HDL Lipoproteins Proteins 0.000 description 3
- 241000725303 Human immunodeficiency virus Species 0.000 description 3
- WMFOQBRAJBCJND-UHFFFAOYSA-M Lithium hydroxide Chemical compound [Li+].[OH-] WMFOQBRAJBCJND-UHFFFAOYSA-M 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 3
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 3
- 239000004480 active ingredient Substances 0.000 description 3
- 150000001408 amides Chemical class 0.000 description 3
- 238000010171 animal model Methods 0.000 description 3
- 239000003524 antilipemic agent Substances 0.000 description 3
- 239000012298 atmosphere Substances 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 3
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- BVKZGUZCCUSVTD-UHFFFAOYSA-N carbonic acid Chemical compound OC(O)=O BVKZGUZCCUSVTD-UHFFFAOYSA-N 0.000 description 3
- 210000004027 cell Anatomy 0.000 description 3
- 150000001840 cholesterol esters Chemical class 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 239000010949 copper Substances 0.000 description 3
- 230000008878 coupling Effects 0.000 description 3
- 238000010168 coupling process Methods 0.000 description 3
- 230000007812 deficiency Effects 0.000 description 3
- 238000009472 formulation Methods 0.000 description 3
- 125000000524 functional group Chemical group 0.000 description 3
- ZZUFCTLCJUWOSV-UHFFFAOYSA-N furosemide Chemical group C1=C(Cl)C(S(=O)(=O)N)=CC(C(O)=O)=C1NCC1=CC=CO1 ZZUFCTLCJUWOSV-UHFFFAOYSA-N 0.000 description 3
- 208000006454 hepatitis Diseases 0.000 description 3
- 231100000283 hepatitis Toxicity 0.000 description 3
- 208000015181 infectious disease Diseases 0.000 description 3
- 239000004041 inotropic agent Substances 0.000 description 3
- INQOMBQAUSQDDS-UHFFFAOYSA-N iodomethane Chemical compound IC INQOMBQAUSQDDS-UHFFFAOYSA-N 0.000 description 3
- 239000010410 layer Substances 0.000 description 3
- 230000004060 metabolic process Effects 0.000 description 3
- 230000004048 modification Effects 0.000 description 3
- 238000012986 modification Methods 0.000 description 3
- 125000002950 monocyclic group Chemical group 0.000 description 3
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 3
- 150000007530 organic bases Chemical class 0.000 description 3
- 229910052763 palladium Inorganic materials 0.000 description 3
- 108090000623 proteins and genes Proteins 0.000 description 3
- HDOUGSFASVGDCS-UHFFFAOYSA-N pyridin-3-ylmethanamine Chemical compound NCC1=CC=CN=C1 HDOUGSFASVGDCS-UHFFFAOYSA-N 0.000 description 3
- 239000012312 sodium hydride Substances 0.000 description 3
- 229910000104 sodium hydride Inorganic materials 0.000 description 3
- 239000012453 solvate Substances 0.000 description 3
- 238000005556 structure-activity relationship Methods 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- PXQLVRUNWNTZOS-UHFFFAOYSA-N sulfanyl Chemical compound [SH] PXQLVRUNWNTZOS-UHFFFAOYSA-N 0.000 description 3
- 239000011593 sulfur Chemical group 0.000 description 3
- 208000024891 symptom Diseases 0.000 description 3
- 238000011287 therapeutic dose Methods 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- 230000000699 topical effect Effects 0.000 description 3
- ZLKZGKDNYHEPFV-UHFFFAOYSA-N 1,3-oxazol-2-ylmethanamine Chemical compound NCC1=NC=CO1 ZLKZGKDNYHEPFV-UHFFFAOYSA-N 0.000 description 2
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 2
- UUUHXMGGBIUAPW-UHFFFAOYSA-N 1-[1-[2-[[5-amino-2-[[1-[5-(diaminomethylideneamino)-2-[[1-[3-(1h-indol-3-yl)-2-[(5-oxopyrrolidine-2-carbonyl)amino]propanoyl]pyrrolidine-2-carbonyl]amino]pentanoyl]pyrrolidine-2-carbonyl]amino]-5-oxopentanoyl]amino]-3-methylpentanoyl]pyrrolidine-2-carbon Chemical compound C1CCC(C(=O)N2C(CCC2)C(O)=O)N1C(=O)C(C(C)CC)NC(=O)C(CCC(N)=O)NC(=O)C1CCCN1C(=O)C(CCCN=C(N)N)NC(=O)C1CCCN1C(=O)C(CC=1C2=CC=CC=C2NC=1)NC(=O)C1CCC(=O)N1 UUUHXMGGBIUAPW-UHFFFAOYSA-N 0.000 description 2
- 125000002941 2-furyl group Chemical group O1C([*])=C([H])C([H])=C1[H] 0.000 description 2
- WRMNZCZEMHIOCP-UHFFFAOYSA-N 2-phenylethanol Chemical compound OCCC1=CC=CC=C1 WRMNZCZEMHIOCP-UHFFFAOYSA-N 0.000 description 2
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 2
- 239000005725 8-Hydroxyquinoline Substances 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- JBYXPOFIGCOSSB-GOJKSUSPSA-N 9-cis,11-trans-octadecadienoic acid Chemical compound CCCCCC\C=C\C=C/CCCCCCCC(O)=O JBYXPOFIGCOSSB-GOJKSUSPSA-N 0.000 description 2
- 241000710929 Alphavirus Species 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- 239000005711 Benzoic acid Substances 0.000 description 2
- 102100038495 Bile acid receptor Human genes 0.000 description 2
- 239000004072 C09CA03 - Valsartan Substances 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- 102000012336 Cholesterol Ester Transfer Proteins Human genes 0.000 description 2
- 108010061846 Cholesterol Ester Transfer Proteins Proteins 0.000 description 2
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 2
- 241000711573 Coronaviridae Species 0.000 description 2
- RGHNJXZEOKUKBD-SQOUGZDYSA-N D-gluconic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C(O)=O RGHNJXZEOKUKBD-SQOUGZDYSA-N 0.000 description 2
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 2
- 208000004930 Fatty Liver Diseases 0.000 description 2
- 241000710781 Flaviviridae Species 0.000 description 2
- 102000001267 GSK3 Human genes 0.000 description 2
- DSLZVSRJTYRBFB-UHFFFAOYSA-N Galactaric acid Natural products OC(=O)C(O)C(O)C(O)C(O)C(O)=O DSLZVSRJTYRBFB-UHFFFAOYSA-N 0.000 description 2
- 101800000224 Glucagon-like peptide 1 Proteins 0.000 description 2
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 2
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 2
- 108010014905 Glycogen Synthase Kinase 3 Proteins 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- 241000711549 Hepacivirus C Species 0.000 description 2
- 206010019728 Hepatitis alcoholic Diseases 0.000 description 2
- 101000603876 Homo sapiens Bile acid receptor Proteins 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 2
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 2
- 108010007622 LDL Lipoproteins Proteins 0.000 description 2
- 102000007330 LDL Lipoproteins Human genes 0.000 description 2
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 208000021642 Muscular disease Diseases 0.000 description 2
- 201000009623 Myopathy Diseases 0.000 description 2
- 102000003729 Neprilysin Human genes 0.000 description 2
- 108090000028 Neprilysin Proteins 0.000 description 2
- YIKSCQDJHCMVMK-UHFFFAOYSA-N Oxamide Chemical compound NC(=O)C(N)=O YIKSCQDJHCMVMK-UHFFFAOYSA-N 0.000 description 2
- 235000021314 Palmitic acid Nutrition 0.000 description 2
- 102000004270 Peptidyl-Dipeptidase A Human genes 0.000 description 2
- 108090000882 Peptidyl-Dipeptidase A Proteins 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Chemical compound P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- 241000709664 Picornaviridae Species 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- 102100040918 Pro-glucagon Human genes 0.000 description 2
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- 241000283984 Rodentia Species 0.000 description 2
- YASAKCUCGLMORW-UHFFFAOYSA-N Rosiglitazone Chemical compound C=1C=CC=NC=1N(C)CCOC(C=C1)=CC=C1CC1SC(=O)NC1=O YASAKCUCGLMORW-UHFFFAOYSA-N 0.000 description 2
- 241000315672 SARS coronavirus Species 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- 102100033930 Stearoyl-CoA desaturase 5 Human genes 0.000 description 2
- 208000006011 Stroke Diseases 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 241000282898 Sus scrofa Species 0.000 description 2
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 description 2
- 241000710771 Tick-borne encephalitis virus Species 0.000 description 2
- 208000032109 Transient ischaemic attack Diseases 0.000 description 2
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 2
- OKJPEAGHQZHRQV-UHFFFAOYSA-N Triiodomethane Natural products IC(I)I OKJPEAGHQZHRQV-UHFFFAOYSA-N 0.000 description 2
- 239000004164 Wax ester Substances 0.000 description 2
- 241000710772 Yellow fever virus Species 0.000 description 2
- 230000001594 aberrant effect Effects 0.000 description 2
- WEVYAHXRMPXWCK-FIBGUPNXSA-N acetonitrile-d3 Chemical compound [2H]C([2H])([2H])C#N WEVYAHXRMPXWCK-FIBGUPNXSA-N 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- WNLRTRBMVRJNCN-UHFFFAOYSA-N adipic acid Chemical compound OC(=O)CCCCC(O)=O WNLRTRBMVRJNCN-UHFFFAOYSA-N 0.000 description 2
- 239000002671 adjuvant Substances 0.000 description 2
- 208000002353 alcoholic hepatitis Diseases 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 150000008064 anhydrides Chemical class 0.000 description 2
- 229940101006 anhydrous sodium sulfite Drugs 0.000 description 2
- 230000003178 anti-diabetic effect Effects 0.000 description 2
- 239000000883 anti-obesity agent Substances 0.000 description 2
- 229940030600 antihypertensive agent Drugs 0.000 description 2
- 239000002220 antihypertensive agent Substances 0.000 description 2
- 229940125710 antiobesity agent Drugs 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 230000004888 barrier function Effects 0.000 description 2
- 235000010233 benzoic acid Nutrition 0.000 description 2
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 2
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 2
- 125000002619 bicyclic group Chemical group 0.000 description 2
- XJHCXCQVJFPJIK-UHFFFAOYSA-M caesium fluoride Chemical compound [F-].[Cs+] XJHCXCQVJFPJIK-UHFFFAOYSA-M 0.000 description 2
- 229960001948 caffeine Drugs 0.000 description 2
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 201000011510 cancer Diseases 0.000 description 2
- 150000001720 carbohydrates Chemical class 0.000 description 2
- 235000014633 carbohydrates Nutrition 0.000 description 2
- 125000003636 chemical group Chemical group 0.000 description 2
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 2
- 229960001231 choline Drugs 0.000 description 2
- 239000003086 colorant Substances 0.000 description 2
- 239000012230 colorless oil Substances 0.000 description 2
- 229940108924 conjugated linoleic acid Drugs 0.000 description 2
- 229910052802 copper Inorganic materials 0.000 description 2
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- JXTHNDFMNIQAHM-UHFFFAOYSA-N dichloroacetic acid Chemical compound OC(=O)C(Cl)Cl JXTHNDFMNIQAHM-UHFFFAOYSA-N 0.000 description 2
- 230000000378 dietary effect Effects 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 230000006806 disease prevention Effects 0.000 description 2
- POULHZVOKOAJMA-UHFFFAOYSA-N dodecanoic acid Chemical compound CCCCCCCCCCCC(O)=O POULHZVOKOAJMA-UHFFFAOYSA-N 0.000 description 2
- 239000002552 dosage form Substances 0.000 description 2
- 229940079593 drug Drugs 0.000 description 2
- 239000003995 emulsifying agent Substances 0.000 description 2
- CFBIOWPDDZPIDP-UHFFFAOYSA-N ethyl 2-bromo-4-methyl-1,3-thiazole-5-carboxylate Chemical compound CCOC(=O)C=1SC(Br)=NC=1C CFBIOWPDDZPIDP-UHFFFAOYSA-N 0.000 description 2
- 208000010706 fatty liver disease Diseases 0.000 description 2
- 235000003599 food sweetener Nutrition 0.000 description 2
- DSLZVSRJTYRBFB-DUHBMQHGSA-N galactaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)C(O)=O DSLZVSRJTYRBFB-DUHBMQHGSA-N 0.000 description 2
- 239000012362 glacial acetic acid Substances 0.000 description 2
- KWIUHFFTVRNATP-UHFFFAOYSA-N glycine betaine Chemical compound C[N+](C)(C)CC([O-])=O KWIUHFFTVRNATP-UHFFFAOYSA-N 0.000 description 2
- 230000002440 hepatic effect Effects 0.000 description 2
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 2
- 210000003494 hepatocyte Anatomy 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- FUZZWVXGSFPDMH-UHFFFAOYSA-N hexanoic acid Chemical compound CCCCCC(O)=O FUZZWVXGSFPDMH-UHFFFAOYSA-N 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 2
- 230000001965 increasing effect Effects 0.000 description 2
- 238000011534 incubation Methods 0.000 description 2
- 150000007529 inorganic bases Chemical class 0.000 description 2
- 238000001990 intravenous administration Methods 0.000 description 2
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 2
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- OYHQOLUKZRVURQ-IXWMQOLASA-N linoleic acid Natural products CCCCC\C=C/C\C=C\CCCCCCCC(O)=O OYHQOLUKZRVURQ-IXWMQOLASA-N 0.000 description 2
- 102000004311 liver X receptors Human genes 0.000 description 2
- 108090000865 liver X receptors Proteins 0.000 description 2
- 239000011777 magnesium Substances 0.000 description 2
- 229910052749 magnesium Inorganic materials 0.000 description 2
- HPSSZFFAYWBIPY-UHFFFAOYSA-N malvalic acid Chemical compound CCCCCCCCC1=C(CCCCCCC(O)=O)C1 HPSSZFFAYWBIPY-UHFFFAOYSA-N 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 239000002609 medium Substances 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- 210000001589 microsome Anatomy 0.000 description 2
- 239000002480 mineral oil Substances 0.000 description 2
- 235000010446 mineral oil Nutrition 0.000 description 2
- 210000003205 muscle Anatomy 0.000 description 2
- WQEPLUUGTLDZJY-UHFFFAOYSA-N n-Pentadecanoic acid Natural products CCCCCCCCCCCCCCC(O)=O WQEPLUUGTLDZJY-UHFFFAOYSA-N 0.000 description 2
- 229930027945 nicotinamide-adenine dinucleotide Natural products 0.000 description 2
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 2
- 206010053219 non-alcoholic steatohepatitis Diseases 0.000 description 2
- WWZKQHOCKIZLMA-UHFFFAOYSA-N octanoic acid Chemical compound CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 description 2
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 2
- PXQPEWDEAKTCGB-UHFFFAOYSA-N orotic acid Chemical compound OC(=O)C1=CC(=O)NC(=O)N1 PXQPEWDEAKTCGB-UHFFFAOYSA-N 0.000 description 2
- 239000001301 oxygen Chemical group 0.000 description 2
- 229960003540 oxyquinoline Drugs 0.000 description 2
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 2
- SECPZKHBENQXJG-FPLPWBNLSA-N palmitoleic acid Chemical compound CCCCCC\C=C/CCCCCCCC(O)=O SECPZKHBENQXJG-FPLPWBNLSA-N 0.000 description 2
- MNBKLUUYKPBKDU-BBECNAHFSA-N palmitoyl-CoA Chemical compound O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCSC(=O)CCCCCCCCCCCCCCC)O[C@H]1N1C2=NC=NC(N)=C2N=C1 MNBKLUUYKPBKDU-BBECNAHFSA-N 0.000 description 2
- QNGNSVIICDLXHT-UHFFFAOYSA-N para-ethylbenzaldehyde Natural products CCC1=CC=C(C=O)C=C1 QNGNSVIICDLXHT-UHFFFAOYSA-N 0.000 description 2
- 229940124531 pharmaceutical excipient Drugs 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- VPRUMANMDWQMNF-UHFFFAOYSA-N phenylethane boronic acid Chemical compound OB(O)CCC1=CC=CC=C1 VPRUMANMDWQMNF-UHFFFAOYSA-N 0.000 description 2
- HYAFETHFCAUJAY-UHFFFAOYSA-N pioglitazone Chemical compound N1=CC(CC)=CC=C1CCOC(C=C1)=CC=C1CC1C(=O)NC(=O)S1 HYAFETHFCAUJAY-UHFFFAOYSA-N 0.000 description 2
- 235000020777 polyunsaturated fatty acids Nutrition 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- IOLCXVTUBQKXJR-UHFFFAOYSA-M potassium bromide Chemical compound [K+].[Br-] IOLCXVTUBQKXJR-UHFFFAOYSA-M 0.000 description 2
- 230000003389 potentiating effect Effects 0.000 description 2
- 239000002244 precipitate Substances 0.000 description 2
- 239000002243 precursor Substances 0.000 description 2
- 239000003755 preservative agent Substances 0.000 description 2
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 2
- AQHHHDLHHXJYJD-UHFFFAOYSA-N propranolol Chemical compound C1=CC=C2C(OCC(O)CNC(C)C)=CC=CC2=C1 AQHHHDLHHXJYJD-UHFFFAOYSA-N 0.000 description 2
- 235000018102 proteins Nutrition 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- MCJGNVYPOGVAJF-UHFFFAOYSA-N quinolin-8-ol Chemical compound C1=CN=C2C(O)=CC=CC2=C1 MCJGNVYPOGVAJF-UHFFFAOYSA-N 0.000 description 2
- JWEQRJSCTFBRSI-PCLIKHOPSA-N rboxylate Chemical compound COC(=O)C1C(N2C3=O)C4=CC=CC=C4OC1(C)N=C2S\C3=C\C(C=1)=CC=C(OC)C=1COC1=CC=CC=C1C JWEQRJSCTFBRSI-PCLIKHOPSA-N 0.000 description 2
- 238000001953 recrystallisation Methods 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 239000011347 resin Substances 0.000 description 2
- 229920005989 resin Polymers 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- CXMXRPHRNRROMY-UHFFFAOYSA-N sebacic acid Chemical compound OC(=O)CCCCCCCCC(O)=O CXMXRPHRNRROMY-UHFFFAOYSA-N 0.000 description 2
- 210000002966 serum Anatomy 0.000 description 2
- RYMZZMVNJRMUDD-HGQWONQESA-N simvastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)C(C)(C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 RYMZZMVNJRMUDD-HGQWONQESA-N 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 235000015424 sodium Nutrition 0.000 description 2
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 2
- 238000012453 sprague-dawley rat model Methods 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 231100000240 steatosis hepatitis Toxicity 0.000 description 2
- 125000003107 substituted aryl group Chemical group 0.000 description 2
- 238000006467 substitution reaction Methods 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 239000003765 sweetening agent Substances 0.000 description 2
- RMMXLENWKUUMAY-UHFFFAOYSA-N telmisartan Chemical compound CCCC1=NC2=C(C)C=C(C=3N(C4=CC=CC=C4N=3)C)C=C2N1CC(C=C1)=CC=C1C1=CC=CC=C1C(O)=O RMMXLENWKUUMAY-UHFFFAOYSA-N 0.000 description 2
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 2
- YAPQBXQYLJRXSA-UHFFFAOYSA-N theobromine Chemical compound CN1C(=O)NC(=O)C2=C1N=CN2C YAPQBXQYLJRXSA-UHFFFAOYSA-N 0.000 description 2
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- 201000010875 transient cerebral ischemia Diseases 0.000 description 2
- AQRLNPVMDITEJU-UHFFFAOYSA-N triethylsilane Chemical compound CC[SiH](CC)CC AQRLNPVMDITEJU-UHFFFAOYSA-N 0.000 description 2
- 125000004952 trihaloalkoxy group Chemical group 0.000 description 2
- CSRZQMIRAZTJOY-UHFFFAOYSA-N trimethylsilyl iodide Chemical compound C[Si](C)(C)I CSRZQMIRAZTJOY-UHFFFAOYSA-N 0.000 description 2
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 2
- 229960004699 valsartan Drugs 0.000 description 2
- SJSNUMAYCRRIOM-QFIPXVFZSA-N valsartan Chemical compound C1=CC(CN(C(=O)CCCC)[C@@H](C(C)C)C(O)=O)=CC=C1C1=CC=CC=C1C1=NN=N[N]1 SJSNUMAYCRRIOM-QFIPXVFZSA-N 0.000 description 2
- 229910052720 vanadium Inorganic materials 0.000 description 2
- SYOKIDBDQMKNDQ-XWTIBIIYSA-N vildagliptin Chemical compound C1C(O)(C2)CC(C3)CC1CC32NCC(=O)N1CCC[C@H]1C#N SYOKIDBDQMKNDQ-XWTIBIIYSA-N 0.000 description 2
- 235000019386 wax ester Nutrition 0.000 description 2
- 239000000080 wetting agent Substances 0.000 description 2
- 229940051021 yellow-fever virus Drugs 0.000 description 2
- 238000013293 zucker diabetic fatty rat Methods 0.000 description 2
- XINQFOMFQFGGCQ-UHFFFAOYSA-L (2-dodecoxy-2-oxoethyl)-[6-[(2-dodecoxy-2-oxoethyl)-dimethylazaniumyl]hexyl]-dimethylazanium;dichloride Chemical compound [Cl-].[Cl-].CCCCCCCCCCCCOC(=O)C[N+](C)(C)CCCCCC[N+](C)(C)CC(=O)OCCCCCCCCCCCC XINQFOMFQFGGCQ-UHFFFAOYSA-L 0.000 description 1
- ZCKAEFOHSOQKHN-UHFFFAOYSA-N (2-methyl-1,3-thiazol-4-yl)methanamine Chemical compound CC1=NC(CN)=CS1 ZCKAEFOHSOQKHN-UHFFFAOYSA-N 0.000 description 1
- XBHPFCIWRHJDCP-UHFFFAOYSA-N (2-trimethylsilylphenyl) trifluoromethanesulfonate Chemical compound C[Si](C)(C)C1=CC=CC=C1OS(=O)(=O)C(F)(F)F XBHPFCIWRHJDCP-UHFFFAOYSA-N 0.000 description 1
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- XUFXOAAUWZOOIT-SXARVLRPSA-N (2R,3R,4R,5S,6R)-5-[[(2R,3R,4R,5S,6R)-5-[[(2R,3R,4S,5S,6R)-3,4-dihydroxy-6-methyl-5-[[(1S,4R,5S,6S)-4,5,6-trihydroxy-3-(hydroxymethyl)-1-cyclohex-2-enyl]amino]-2-oxanyl]oxy]-3,4-dihydroxy-6-(hydroxymethyl)-2-oxanyl]oxy]-6-(hydroxymethyl)oxane-2,3,4-triol Chemical compound O([C@H]1O[C@H](CO)[C@H]([C@@H]([C@H]1O)O)O[C@H]1O[C@@H]([C@H]([C@H](O)[C@H]1O)N[C@@H]1[C@@H]([C@@H](O)[C@H](O)C(CO)=C1)O)C)[C@@H]1[C@@H](CO)O[C@@H](O)[C@H](O)[C@H]1O XUFXOAAUWZOOIT-SXARVLRPSA-N 0.000 description 1
- BOOOLEGQBVUTKC-NVQSDHBMSA-N (2e,4e)-3-methyl-5-[(1s,2s)-2-methyl-2-(5,5,8,8-tetramethyl-6,7-dihydronaphthalen-2-yl)cyclopropyl]penta-2,4-dienoic acid Chemical compound OC(=O)\C=C(/C)\C=C\[C@@H]1C[C@]1(C)C1=CC=C2C(C)(C)CCC(C)(C)C2=C1 BOOOLEGQBVUTKC-NVQSDHBMSA-N 0.000 description 1
- FONCZICQWCUXEB-RUZDIDTESA-N (2r)-2-[4-(9-bromo-2,3-dimethylbenzo[f][1]benzothiol-4-yl)-2,6-dimethylphenoxy]-3-phenylpropanoic acid Chemical compound C([C@@H](OC1=C(C)C=C(C=C1C)C=1C2=CC=CC=C2C(Br)=C2SC(=C(C2=1)C)C)C(O)=O)C1=CC=CC=C1 FONCZICQWCUXEB-RUZDIDTESA-N 0.000 description 1
- LPUDGHQMOAHMMF-JBACZVJFSA-N (2s)-2-[[[(2s)-6-amino-2-(methanesulfonamido)hexanoyl]amino]methyl]-3-[1-[[(1s)-1-carboxy-2-(4-hydroxyphenyl)ethyl]carbamoyl]cyclopentyl]propanoic acid Chemical compound N([C@@H](CC=1C=CC(O)=CC=1)C(O)=O)C(=O)C1(C[C@@H](CNC(=O)[C@H](CCCCN)NS(=O)(=O)C)C(O)=O)CCCC1 LPUDGHQMOAHMMF-JBACZVJFSA-N 0.000 description 1
- BIDNLKIUORFRQP-XYGFDPSESA-N (2s,4s)-4-cyclohexyl-1-[2-[[(1s)-2-methyl-1-propanoyloxypropoxy]-(4-phenylbutyl)phosphoryl]acetyl]pyrrolidine-2-carboxylic acid Chemical compound C([P@@](=O)(O[C@H](OC(=O)CC)C(C)C)CC(=O)N1[C@@H](C[C@H](C1)C1CCCCC1)C(O)=O)CCCC1=CC=CC=C1 BIDNLKIUORFRQP-XYGFDPSESA-N 0.000 description 1
- CABVTRNMFUVUDM-VRHQGPGLSA-N (3S)-3-hydroxy-3-methylglutaryl-CoA Chemical compound O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCSC(=O)C[C@@](O)(CC(O)=O)C)O[C@H]1N1C2=NC=NC(N)=C2N=C1 CABVTRNMFUVUDM-VRHQGPGLSA-N 0.000 description 1
- CAYQIZIAYYNFCS-UHFFFAOYSA-N (4-chlorophenyl)boronic acid Chemical compound OB(O)C1=CC=C(Cl)C=C1 CAYQIZIAYYNFCS-UHFFFAOYSA-N 0.000 description 1
- IIFVWLUQBAIPMJ-UHFFFAOYSA-N (4-fluorophenyl)methanamine Chemical compound NCC1=CC=C(F)C=C1 IIFVWLUQBAIPMJ-UHFFFAOYSA-N 0.000 description 1
- NRQHBNNTBIDSRK-YRNVUSSQSA-N (4e)-4-[(4-methoxyphenyl)methylidene]-2-methyl-1,3-oxazol-5-one Chemical compound C1=CC(OC)=CC=C1\C=C\1C(=O)OC(C)=N/1 NRQHBNNTBIDSRK-YRNVUSSQSA-N 0.000 description 1
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- METKIMKYRPQLGS-GFCCVEGCSA-N (R)-atenolol Chemical compound CC(C)NC[C@@H](O)COC1=CC=C(CC(N)=O)C=C1 METKIMKYRPQLGS-GFCCVEGCSA-N 0.000 description 1
- MIOPJNTWMNEORI-GMSGAONNSA-N (S)-camphorsulfonic acid Chemical compound C1C[C@@]2(CS(O)(=O)=O)C(=O)C[C@@H]1C2(C)C MIOPJNTWMNEORI-GMSGAONNSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- TWBNMYSKRDRHAT-RCWTXCDDSA-N (S)-timolol hemihydrate Chemical compound O.CC(C)(C)NC[C@H](O)COC1=NSN=C1N1CCOCC1.CC(C)(C)NC[C@H](O)COC1=NSN=C1N1CCOCC1 TWBNMYSKRDRHAT-RCWTXCDDSA-N 0.000 description 1
- 101150084750 1 gene Proteins 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- IFNMJBFIRKAQBT-UHFFFAOYSA-N 1-(bromomethyl)-4-(difluoromethoxy)benzene Chemical compound FC(F)OC1=CC=C(CBr)C=C1 IFNMJBFIRKAQBT-UHFFFAOYSA-N 0.000 description 1
- IKSNDOVDVVPSMA-UHFFFAOYSA-N 1-(bromomethyl)-4-(trifluoromethyl)benzene Chemical compound FC(F)(F)C1=CC=C(CBr)C=C1 IKSNDOVDVVPSMA-UHFFFAOYSA-N 0.000 description 1
- NVNPLEPBDPJYRZ-UHFFFAOYSA-N 1-(bromomethyl)-4-fluorobenzene Chemical compound FC1=CC=C(CBr)C=C1 NVNPLEPBDPJYRZ-UHFFFAOYSA-N 0.000 description 1
- VXNZUUAINFGPBY-UHFFFAOYSA-N 1-Butene Chemical group CCC=C VXNZUUAINFGPBY-UHFFFAOYSA-N 0.000 description 1
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 1
- 125000004973 1-butenyl group Chemical group C(=CCC)* 0.000 description 1
- BCMYXYHEMGPZJN-UHFFFAOYSA-N 1-chloro-2-isocyanatoethane Chemical compound ClCCN=C=O BCMYXYHEMGPZJN-UHFFFAOYSA-N 0.000 description 1
- BTQZKHUEUDPRST-UHFFFAOYSA-N 1-fluoro-3-methylbenzene Chemical compound CC1=CC=CC(F)=C1 BTQZKHUEUDPRST-UHFFFAOYSA-N 0.000 description 1
- RTVXQIMCVVFOGF-UHFFFAOYSA-N 1-methylthiophene-1-carboxylic acid Chemical compound OC(=O)S1(C)C=CC=C1 RTVXQIMCVVFOGF-UHFFFAOYSA-N 0.000 description 1
- VZOPVKZLLGMDDG-UHFFFAOYSA-N 1-oxido-4-phenylpyridin-1-ium Chemical compound C1=C[N+]([O-])=CC=C1C1=CC=CC=C1 VZOPVKZLLGMDDG-UHFFFAOYSA-N 0.000 description 1
- FRPZMMHWLSIFAZ-UHFFFAOYSA-N 10-undecenoic acid Chemical compound OC(=O)CCCCCCCCC=C FRPZMMHWLSIFAZ-UHFFFAOYSA-N 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Chemical group C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- 125000004206 2,2,2-trifluoroethyl group Chemical group [H]C([H])(*)C(F)(F)F 0.000 description 1
- SGTNSNPWRIOYBX-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-{[2-(3,4-dimethoxyphenyl)ethyl](methyl)amino}-2-(propan-2-yl)pentanenitrile Chemical compound C1=C(OC)C(OC)=CC=C1CCN(C)CCCC(C#N)(C(C)C)C1=CC=C(OC)C(OC)=C1 SGTNSNPWRIOYBX-UHFFFAOYSA-N 0.000 description 1
- WFLCAOGKZQTOIG-UHFFFAOYSA-N 2-(bromomethyl)-1,3-benzothiazole Chemical compound C1=CC=C2SC(CBr)=NC2=C1 WFLCAOGKZQTOIG-UHFFFAOYSA-N 0.000 description 1
- MHNWCBOXPOLLIB-UHFFFAOYSA-N 2-(bromomethyl)oxane Chemical compound BrCC1CCCCO1 MHNWCBOXPOLLIB-UHFFFAOYSA-N 0.000 description 1
- VOHILFSOWRNVJJ-UHFFFAOYSA-N 2-(bromomethyl)oxolane Chemical compound BrCC1CCCO1 VOHILFSOWRNVJJ-UHFFFAOYSA-N 0.000 description 1
- JQDNCGRNPYKRAO-UHFFFAOYSA-N 2-(bromomethyl)pyridine;hydron;bromide Chemical compound Br.BrCC1=CC=CC=N1 JQDNCGRNPYKRAO-UHFFFAOYSA-N 0.000 description 1
- WKAVKKUXZAWHDM-UHFFFAOYSA-N 2-acetamidopentanedioic acid;2-(dimethylamino)ethanol Chemical compound CN(C)CCO.CC(=O)NC(C(O)=O)CCC(O)=O WKAVKKUXZAWHDM-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-IVMDWMLBSA-N 2-amino-2-deoxy-D-glucopyranose Chemical compound N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O MSWZFWKMSRAUBD-IVMDWMLBSA-N 0.000 description 1
- HMSQZHBSTZZNGI-UHFFFAOYSA-N 2-bromo-4-methyl-1,3-thiazole-5-carboxylic acid Chemical compound CC=1N=C(Br)SC=1C(O)=O HMSQZHBSTZZNGI-UHFFFAOYSA-N 0.000 description 1
- SLOWTPCXWIWXFB-UHFFFAOYSA-N 2-chloro-4-(phenoxymethyl)pyridine Chemical compound C1=NC(Cl)=CC(COC=2C=CC=CC=2)=C1 SLOWTPCXWIWXFB-UHFFFAOYSA-N 0.000 description 1
- LUNMJRJMSXZSLC-UHFFFAOYSA-N 2-cyclopropylethanol Chemical compound OCCC1CC1 LUNMJRJMSXZSLC-UHFFFAOYSA-N 0.000 description 1
- ZYBUZXODDPRLDK-UHFFFAOYSA-N 2-methoxy-n-phenylpyridine-4-carboxamide Chemical compound C1=NC(OC)=CC(C(=O)NC=2C=CC=CC=2)=C1 ZYBUZXODDPRLDK-UHFFFAOYSA-N 0.000 description 1
- DPNDWFVORIGXQO-UHFFFAOYSA-N 2-methoxypyridine-4-carboxylic acid Chemical compound COC1=CC(C(O)=O)=CC=N1 DPNDWFVORIGXQO-UHFFFAOYSA-N 0.000 description 1
- 125000006088 2-oxoazepinyl group Chemical group 0.000 description 1
- KPGXRSRHYNQIFN-UHFFFAOYSA-N 2-oxoglutaric acid Chemical compound OC(=O)CCC(=O)C(O)=O KPGXRSRHYNQIFN-UHFFFAOYSA-N 0.000 description 1
- 125000004638 2-oxopiperazinyl group Chemical group O=C1N(CCNC1)* 0.000 description 1
- 125000004637 2-oxopiperidinyl group Chemical group O=C1N(CCCC1)* 0.000 description 1
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 description 1
- CZMRCDWAGMRECN-UHFFFAOYSA-N 2-{[3,4-dihydroxy-2,5-bis(hydroxymethyl)oxolan-2-yl]oxy}-6-(hydroxymethyl)oxane-3,4,5-triol Chemical compound OCC1OC(CO)(OC2OC(CO)C(O)C(O)C2O)C(O)C1O CZMRCDWAGMRECN-UHFFFAOYSA-N 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- JHDCDEHVUADNKQ-UHFFFAOYSA-N 3-(trifluoromethyl)-1h-pyridin-2-one Chemical compound OC1=NC=CC=C1C(F)(F)F JHDCDEHVUADNKQ-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- UOQHWNPVNXSDDO-UHFFFAOYSA-N 3-bromoimidazo[1,2-a]pyridine-6-carbonitrile Chemical compound C1=CC(C#N)=CN2C(Br)=CN=C21 UOQHWNPVNXSDDO-UHFFFAOYSA-N 0.000 description 1
- BNYIQEFWGSXIKQ-UHFFFAOYSA-N 3-methylfuran-2-carboxylic acid Chemical compound CC=1C=COC=1C(O)=O BNYIQEFWGSXIKQ-UHFFFAOYSA-N 0.000 description 1
- QZDTWJRYMXQXBX-UHFFFAOYSA-N 3-methylthiophene-2-carboxamide Chemical compound CC=1C=CSC=1C(N)=O QZDTWJRYMXQXBX-UHFFFAOYSA-N 0.000 description 1
- UIAGMCDKSXEBJQ-IBGZPJMESA-N 3-o-(2-methoxyethyl) 5-o-propan-2-yl (4s)-2,6-dimethyl-4-(3-nitrophenyl)-1,4-dihydropyridine-3,5-dicarboxylate Chemical compound COCCOC(=O)C1=C(C)NC(C)=C(C(=O)OC(C)C)[C@H]1C1=CC=CC([N+]([O-])=O)=C1 UIAGMCDKSXEBJQ-IBGZPJMESA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- NMAKJOWVEDTHOA-UHFFFAOYSA-N 4-(chloromethyl)-1,3-thiazol-2-amine;hydron;chloride Chemical compound Cl.NC1=NC(CCl)=CS1 NMAKJOWVEDTHOA-UHFFFAOYSA-N 0.000 description 1
- QKWSLYINUYKIRF-UHFFFAOYSA-N 4-(chloromethyl)-1,3-thiazole Chemical compound ClCC1=CSC=N1 QKWSLYINUYKIRF-UHFFFAOYSA-N 0.000 description 1
- YGTDRZIMYRYHHD-UHFFFAOYSA-N 4-(chloromethyl)-2-propan-2-yl-1,3-thiazole Chemical compound CC(C)C1=NC(CCl)=CS1 YGTDRZIMYRYHHD-UHFFFAOYSA-N 0.000 description 1
- SWLAMJPTOQZTAE-UHFFFAOYSA-N 4-[2-[(5-chloro-2-methoxybenzoyl)amino]ethyl]benzoic acid Chemical class COC1=CC=C(Cl)C=C1C(=O)NCCC1=CC=C(C(O)=O)C=C1 SWLAMJPTOQZTAE-UHFFFAOYSA-N 0.000 description 1
- SBQVQYPJWLJRQT-UHFFFAOYSA-N 4-amino-1h-pyridin-2-one Chemical compound NC=1C=CNC(=O)C=1 SBQVQYPJWLJRQT-UHFFFAOYSA-N 0.000 description 1
- 125000005986 4-piperidonyl group Chemical group 0.000 description 1
- AFKDNWKXTXSOTA-UHFFFAOYSA-N 5-bromo-n-[(4-fluorophenyl)methyl]-3-methylthiophene-2-carboxamide Chemical compound C1=C(Br)SC(C(=O)NCC=2C=CC(F)=CC=2)=C1C AFKDNWKXTXSOTA-UHFFFAOYSA-N 0.000 description 1
- SZFUWUOHDRMCKD-UHFFFAOYSA-N 5-chloro-1h-pyridin-2-one Chemical compound OC1=CC=C(Cl)C=N1 SZFUWUOHDRMCKD-UHFFFAOYSA-N 0.000 description 1
- RZTAMFZIAATZDJ-HNNXBMFYSA-N 5-o-ethyl 3-o-methyl (4s)-4-(2,3-dichlorophenyl)-2,6-dimethyl-1,4-dihydropyridine-3,5-dicarboxylate Chemical compound CCOC(=O)C1=C(C)NC(C)=C(C(=O)OC)[C@@H]1C1=CC=CC(Cl)=C1Cl RZTAMFZIAATZDJ-HNNXBMFYSA-N 0.000 description 1
- IYDVFMFKIKZKEK-UHFFFAOYSA-N 8-nonylsulfanyloctanoic acid Chemical compound CCCCCCCCCSCCCCCCCC(O)=O IYDVFMFKIKZKEK-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 101150067539 AMBP gene Proteins 0.000 description 1
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 1
- 102100034542 Acyl-CoA (8-3)-desaturase Human genes 0.000 description 1
- 102100034544 Acyl-CoA 6-desaturase Human genes 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 206010002091 Anaesthesia Diseases 0.000 description 1
- 229940123413 Angiotensin II antagonist Drugs 0.000 description 1
- 108010071619 Apolipoproteins Proteins 0.000 description 1
- 102000007592 Apolipoproteins Human genes 0.000 description 1
- 102000013918 Apolipoproteins E Human genes 0.000 description 1
- 108010025628 Apolipoproteins E Proteins 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 241001533362 Astroviridae Species 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- XUKUURHRXDUEBC-KAYWLYCHSA-N Atorvastatin Chemical compound C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CC[C@@H](O)C[C@@H](O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-KAYWLYCHSA-N 0.000 description 1
- XUKUURHRXDUEBC-UHFFFAOYSA-N Atorvastatin Natural products C=1C=CC=CC=1C1=C(C=2C=CC(F)=CC=2)N(CCC(O)CC(O)CC(O)=O)C(C(C)C)=C1C(=O)NC1=CC=CC=C1 XUKUURHRXDUEBC-UHFFFAOYSA-N 0.000 description 1
- 206010003694 Atrophy Diseases 0.000 description 1
- 241000271566 Aves Species 0.000 description 1
- XPCFTKFZXHTYIP-PMACEKPBSA-N Benazepril Chemical compound C([C@@H](C(=O)OCC)N[C@@H]1C(N(CC(O)=O)C2=CC=CC=C2CC1)=O)CC1=CC=CC=C1 XPCFTKFZXHTYIP-PMACEKPBSA-N 0.000 description 1
- 241000711515 Berne virus Species 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- 241001503592 Bovine calicivirus Species 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- 241000203231 Breda virus Species 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- 241001533357 Bymovirus Species 0.000 description 1
- 239000002083 C09CA01 - Losartan Substances 0.000 description 1
- 239000002947 C09CA04 - Irbesartan Substances 0.000 description 1
- 239000002053 C09CA06 - Candesartan Substances 0.000 description 1
- 239000005537 C09CA07 - Telmisartan Substances 0.000 description 1
- SQNAKTNYXOWYRK-UHFFFAOYSA-N CC1C=C(N(C=CC(OCc2ccccc2)=C2)C2=O)SC1C(NCc1ccccc1)=O Chemical compound CC1C=C(N(C=CC(OCc2ccccc2)=C2)C2=O)SC1C(NCc1ccccc1)=O SQNAKTNYXOWYRK-UHFFFAOYSA-N 0.000 description 1
- 101100337060 Caenorhabditis elegans glp-1 gene Proteins 0.000 description 1
- 229940127291 Calcium channel antagonist Drugs 0.000 description 1
- 241000714198 Caliciviridae Species 0.000 description 1
- LSPHULWDVZXLIL-UHFFFAOYSA-N Camphoric acid Natural products CC1(C)C(C(O)=O)CCC1(C)C(O)=O LSPHULWDVZXLIL-UHFFFAOYSA-N 0.000 description 1
- 241000711506 Canine coronavirus Species 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 239000005635 Caprylic acid (CAS 124-07-2) Substances 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 208000005623 Carcinogenesis Diseases 0.000 description 1
- 102000002666 Carnitine O-palmitoyltransferase Human genes 0.000 description 1
- 108010018424 Carnitine O-palmitoyltransferase Proteins 0.000 description 1
- 102100027943 Carnitine O-palmitoyltransferase 1, liver isoform Human genes 0.000 description 1
- 101710120614 Carnitine O-palmitoyltransferase 1, liver isoform Proteins 0.000 description 1
- 101710108984 Carnitine O-palmitoyltransferase 1, muscle isoform Proteins 0.000 description 1
- 201000002929 Carnitine palmitoyltransferase II deficiency Diseases 0.000 description 1
- 206010050215 Carnitine palmitoyltransferase deficiency Diseases 0.000 description 1
- SJSKOGVCMVNUJY-UHFFFAOYSA-N Cc1c(C(NCc2ccccc2)=O)[s]c(N(C(CCc2c3)=O)c2ccc3O)n1 Chemical compound Cc1c(C(NCc2ccccc2)=O)[s]c(N(C(CCc2c3)=O)c2ccc3O)n1 SJSKOGVCMVNUJY-UHFFFAOYSA-N 0.000 description 1
- RERBSGLAYHBULC-UHFFFAOYSA-N Cc1cc(COC(C=CN2c3nc(C)c(C(NCc4ccccc4)=O)[s]3)=CC2=O)n[o]1 Chemical compound Cc1cc(COC(C=CN2c3nc(C)c(C(NCc4ccccc4)=O)[s]3)=CC2=O)n[o]1 RERBSGLAYHBULC-UHFFFAOYSA-N 0.000 description 1
- 241000237074 Centris Species 0.000 description 1
- 208000008964 Chemical and Drug Induced Liver Injury Diseases 0.000 description 1
- 201000009182 Chikungunya Diseases 0.000 description 1
- 241000710777 Classical swine fever virus Species 0.000 description 1
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 1
- 241000709687 Coxsackievirus Species 0.000 description 1
- 241000699800 Cricetinae Species 0.000 description 1
- 201000003883 Cystic fibrosis Diseases 0.000 description 1
- 102100031655 Cytochrome b5 Human genes 0.000 description 1
- 102100033149 Cytochrome b5 reductase 4 Human genes 0.000 description 1
- 108030005700 Cytochrome-b5 reductases Proteins 0.000 description 1
- 108010007167 Cytochromes b5 Proteins 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- RGHNJXZEOKUKBD-UHFFFAOYSA-N D-gluconic acid Natural products OCC(O)C(O)C(O)C(O)C(O)=O RGHNJXZEOKUKBD-UHFFFAOYSA-N 0.000 description 1
- AEMOLEFTQBMNLQ-AQKNRBDQSA-N D-glucopyranuronic acid Chemical compound OC1O[C@H](C(O)=O)[C@@H](O)[C@H](O)[C@H]1O AEMOLEFTQBMNLQ-AQKNRBDQSA-N 0.000 description 1
- 108010073542 Delta-5 Fatty Acid Desaturase Proteins 0.000 description 1
- 208000001490 Dengue Diseases 0.000 description 1
- 206010012310 Dengue fever Diseases 0.000 description 1
- LTMHDMANZUZIPE-AMTYYWEZSA-N Digoxin Natural products O([C@H]1[C@H](C)O[C@H](O[C@@H]2C[C@@H]3[C@@](C)([C@@H]4[C@H]([C@]5(O)[C@](C)([C@H](O)C4)[C@H](C4=CC(=O)OC4)CC5)CC3)CC2)C[C@@H]1O)[C@H]1O[C@H](C)[C@@H](O[C@H]2O[C@@H](C)[C@H](O)[C@@H](O)C2)[C@@H](O)C1 LTMHDMANZUZIPE-AMTYYWEZSA-N 0.000 description 1
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 1
- 102000016622 Dipeptidyl Peptidase 4 Human genes 0.000 description 1
- 206010072268 Drug-induced liver injury Diseases 0.000 description 1
- 102100025027 E3 ubiquitin-protein ligase TRIM69 Human genes 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 241000710945 Eastern equine encephalitis virus Species 0.000 description 1
- 108010061435 Enalapril Proteins 0.000 description 1
- 206010014596 Encephalitis Japanese B Diseases 0.000 description 1
- 241000709661 Enterovirus Species 0.000 description 1
- 206010066919 Epidemic polyarthritis Diseases 0.000 description 1
- 241000283086 Equidae Species 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 108010011459 Exenatide Proteins 0.000 description 1
- 201000001376 Familial Combined Hyperlipidemia Diseases 0.000 description 1
- 102000009114 Fatty acid desaturases Human genes 0.000 description 1
- 241000711475 Feline infectious peritonitis virus Species 0.000 description 1
- 241000282326 Felis catus Species 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- IAJILQKETJEXLJ-UHFFFAOYSA-N Galacturonsaeure Natural products O=CC(O)C(O)C(O)C(O)C(O)=O IAJILQKETJEXLJ-UHFFFAOYSA-N 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 101000930822 Giardia intestinalis Dipeptidyl-peptidase 4 Proteins 0.000 description 1
- FAEKWTJYAYMJKF-QHCPKHFHSA-N GlucoNorm Chemical compound C1=C(C(O)=O)C(OCC)=CC(CC(=O)N[C@@H](CC(C)C)C=2C(=CC=CC=2)N2CCCCC2)=C1 FAEKWTJYAYMJKF-QHCPKHFHSA-N 0.000 description 1
- 208000002705 Glucose Intolerance Diseases 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 229920002527 Glycogen Polymers 0.000 description 1
- 208000036066 Hemophagocytic Lymphohistiocytosis Diseases 0.000 description 1
- 206010019668 Hepatic fibrosis Diseases 0.000 description 1
- 241000700721 Hepatitis B virus Species 0.000 description 1
- 206010019773 Hepatitis G Diseases 0.000 description 1
- 241000709721 Hepatovirus A Species 0.000 description 1
- 208000033981 Hereditary haemochromatosis Diseases 0.000 description 1
- 101000830203 Homo sapiens E3 ubiquitin-protein ligase TRIM69 Proteins 0.000 description 1
- 101100309604 Homo sapiens SCD5 gene Proteins 0.000 description 1
- 101000639987 Homo sapiens Stearoyl-CoA desaturase 5 Proteins 0.000 description 1
- 244000309467 Human Coronavirus Species 0.000 description 1
- 241001479210 Human astrovirus Species 0.000 description 1
- 241001428935 Human coronavirus OC43 Species 0.000 description 1
- 241000709701 Human poliovirus 1 Species 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 208000001021 Hyperlipoproteinemia Type I Diseases 0.000 description 1
- 201000004029 Immune dysregulation-polyendocrinopathy-enteropathy-X-linked syndrome Diseases 0.000 description 1
- 241000711450 Infectious bronchitis virus Species 0.000 description 1
- 206010065973 Iron Overload Diseases 0.000 description 1
- 102000008133 Iron-Binding Proteins Human genes 0.000 description 1
- 108010035210 Iron-Binding Proteins Proteins 0.000 description 1
- XRHGWAGWAHHFLF-UHFFFAOYSA-N Isazofos Chemical compound CCOP(=S)(OCC)OC=1N=C(Cl)N(C(C)C)N=1 XRHGWAGWAHHFLF-UHFFFAOYSA-N 0.000 description 1
- 208000032382 Ischaemic stroke Diseases 0.000 description 1
- 201000005807 Japanese encephalitis Diseases 0.000 description 1
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- 108010028554 LDL Cholesterol Proteins 0.000 description 1
- 238000008214 LDL Cholesterol Methods 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 239000005639 Lauric acid Substances 0.000 description 1
- 102000003960 Ligases Human genes 0.000 description 1
- 108090000364 Ligases Proteins 0.000 description 1
- 108010037138 Linoleoyl-CoA Desaturase Proteins 0.000 description 1
- 108010007859 Lisinopril Proteins 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 241000710769 Louping ill virus Species 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 241000608292 Mayaro virus Species 0.000 description 1
- 241000710185 Mengo virus Species 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- PCZOHLXUXFIOCF-UHFFFAOYSA-N Monacolin X Natural products C12C(OC(=O)C(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 PCZOHLXUXFIOCF-UHFFFAOYSA-N 0.000 description 1
- 241001529936 Murinae Species 0.000 description 1
- 241000710908 Murray Valley encephalitis virus Species 0.000 description 1
- 241000699660 Mus musculus Species 0.000 description 1
- 101100309601 Mus musculus Scd3 gene Proteins 0.000 description 1
- PCLIMKBDDGJMGD-UHFFFAOYSA-N N-bromosuccinimide Chemical compound BrN1C(=O)CCC1=O PCLIMKBDDGJMGD-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- ACFIXJIJDZMPPO-NNYOXOHSSA-N NADPH Chemical compound C1=CCC(C(=O)N)=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OC[C@@H]2[C@H]([C@@H](OP(O)(O)=O)[C@@H](O2)N2C3=NC=NC(N)=C3N=C2)O)O1 ACFIXJIJDZMPPO-NNYOXOHSSA-N 0.000 description 1
- 208000012902 Nervous system disease Diseases 0.000 description 1
- 208000025966 Neurological disease Diseases 0.000 description 1
- ZBBHBTPTTSWHBA-UHFFFAOYSA-N Nicardipine Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OCCN(C)CC=2C=CC=CC=2)C1C1=CC=CC([N+]([O-])=O)=C1 ZBBHBTPTTSWHBA-UHFFFAOYSA-N 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- JCXJVPUVTGWSNB-UHFFFAOYSA-N Nitrogen dioxide Chemical compound O=[N]=O JCXJVPUVTGWSNB-UHFFFAOYSA-N 0.000 description 1
- 241000714209 Norwalk virus Species 0.000 description 1
- TVCRAMGRUBUACO-UHFFFAOYSA-N O.OSO Chemical compound O.OSO TVCRAMGRUBUACO-UHFFFAOYSA-N 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- 241000725177 Omsk hemorrhagic fever virus Species 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- 102000004316 Oxidoreductases Human genes 0.000 description 1
- 108090000854 Oxidoreductases Proteins 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 108010016731 PPAR gamma Proteins 0.000 description 1
- 235000021319 Palmitoleic acid Nutrition 0.000 description 1
- 206010033645 Pancreatitis Diseases 0.000 description 1
- 208000002606 Paramyxoviridae Infections Diseases 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 208000018262 Peripheral vascular disease Diseases 0.000 description 1
- 102100038825 Peroxisome proliferator-activated receptor gamma Human genes 0.000 description 1
- 241000710778 Pestivirus Species 0.000 description 1
- 241001135549 Porcine epidemic diarrhea virus Species 0.000 description 1
- 241000156302 Porcine hemagglutinating encephalomyelitis virus Species 0.000 description 1
- 241000710078 Potyvirus Species 0.000 description 1
- 241000710884 Powassan virus Species 0.000 description 1
- TUZYXOIXSAXUGO-UHFFFAOYSA-N Pravastatin Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(O)C=C21 TUZYXOIXSAXUGO-UHFFFAOYSA-N 0.000 description 1
- 206010036618 Premenstrual syndrome Diseases 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 238000012228 RNA interference-mediated gene silencing Methods 0.000 description 1
- 241000903330 Rabbit coronavirus Species 0.000 description 1
- 241001428933 Rat coronavirus Species 0.000 description 1
- 208000017442 Retinal disease Diseases 0.000 description 1
- 206010038923 Retinopathy Diseases 0.000 description 1
- 241000710942 Ross River virus Species 0.000 description 1
- 241000710799 Rubella virus Species 0.000 description 1
- 241001533356 Rymovirus Species 0.000 description 1
- RYMZZMVNJRMUDD-UHFFFAOYSA-N SJ000286063 Natural products C12C(OC(=O)C(C)(C)CC)CC(C)C=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 RYMZZMVNJRMUDD-UHFFFAOYSA-N 0.000 description 1
- AJLFOPYRIVGYMJ-UHFFFAOYSA-N SJ000287055 Natural products C12C(OC(=O)C(C)CC)CCC=C2C=CC(C)C1CCC1CC(O)CC(=O)O1 AJLFOPYRIVGYMJ-UHFFFAOYSA-N 0.000 description 1
- 101100101423 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) UBI4 gene Proteins 0.000 description 1
- 101150042597 Scd2 gene Proteins 0.000 description 1
- 241000710961 Semliki Forest virus Species 0.000 description 1
- 241000710960 Sindbis virus Species 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 102100037202 Sodium/myo-inositol cotransporter 2 Human genes 0.000 description 1
- 101710090560 Sodium/myo-inositol cotransporter 2 Proteins 0.000 description 1
- 241000710888 St. Louis encephalitis virus Species 0.000 description 1
- 229910000831 Steel Inorganic materials 0.000 description 1
- PQRKPYLNZGDCFH-UHFFFAOYSA-N Sterculic-saeure Natural products CCCCCCCCC1=C(CCCCCCCC(O)=O)C1 PQRKPYLNZGDCFH-UHFFFAOYSA-N 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- 229940100389 Sulfonylurea Drugs 0.000 description 1
- 238000006069 Suzuki reaction reaction Methods 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 239000004809 Teflon Substances 0.000 description 1
- 229920006362 Teflon® Polymers 0.000 description 1
- 229940123464 Thiazolidinedione Drugs 0.000 description 1
- 208000004006 Tick-borne encephalitis Diseases 0.000 description 1
- NGBFQHCMQULJNZ-UHFFFAOYSA-N Torsemide Chemical compound CC(C)NC(=O)NS(=O)(=O)C1=CN=CC=C1NC1=CC=CC(C)=C1 NGBFQHCMQULJNZ-UHFFFAOYSA-N 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- VXFJYXUZANRPDJ-WTNASJBWSA-N Trandopril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](C[C@H]2CCCC[C@@H]21)C(O)=O)CC1=CC=CC=C1 VXFJYXUZANRPDJ-WTNASJBWSA-N 0.000 description 1
- 241000711484 Transmissible gastroenteritis virus Species 0.000 description 1
- GSEJCLTVZPLZKY-UHFFFAOYSA-N Triethanolamine Chemical compound OCCN(CCO)CCO GSEJCLTVZPLZKY-UHFFFAOYSA-N 0.000 description 1
- 241000711508 Turkey coronavirus Species 0.000 description 1
- 241000710959 Venezuelan equine encephalitis virus Species 0.000 description 1
- 241001494970 Vesicular exanthema of swine virus Species 0.000 description 1
- 206010051511 Viral diarrhoea Diseases 0.000 description 1
- 208000036142 Viral infection Diseases 0.000 description 1
- 239000003875 Wang resin Substances 0.000 description 1
- 241000710886 West Nile virus Species 0.000 description 1
- 241000710951 Western equine encephalitis virus Species 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- NERFNHBZJXXFGY-UHFFFAOYSA-N [4-[(4-methylphenyl)methoxy]phenyl]methanol Chemical compound C1=CC(C)=CC=C1COC1=CC=C(CO)C=C1 NERFNHBZJXXFGY-UHFFFAOYSA-N 0.000 description 1
- ROYYYTOVBUUPDX-UHFFFAOYSA-N [5-methyl-2-(trifluoromethyl)furan-3-yl]methanamine Chemical compound CC1=CC(CN)=C(C(F)(F)F)O1 ROYYYTOVBUUPDX-UHFFFAOYSA-N 0.000 description 1
- XPXVAYGVYBQKDE-UHFFFAOYSA-N [6-(trifluoromethyl)pyridin-3-yl]methanamine Chemical compound NCC1=CC=C(C(F)(F)F)N=C1 XPXVAYGVYBQKDE-UHFFFAOYSA-N 0.000 description 1
- 230000003187 abdominal effect Effects 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 229960002632 acarbose Drugs 0.000 description 1
- XUFXOAAUWZOOIT-UHFFFAOYSA-N acarviostatin I01 Natural products OC1C(O)C(NC2C(C(O)C(O)C(CO)=C2)O)C(C)OC1OC(C(C1O)O)C(CO)OC1OC1C(CO)OC(O)C(O)C1O XUFXOAAUWZOOIT-UHFFFAOYSA-N 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- GOEMGAFJFRBGGG-UHFFFAOYSA-N acebutolol Chemical compound CCCC(=O)NC1=CC=C(OCC(O)CNC(C)C)C(C(C)=O)=C1 GOEMGAFJFRBGGG-UHFFFAOYSA-N 0.000 description 1
- 229960002122 acebutolol Drugs 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 230000003044 adaptive effect Effects 0.000 description 1
- 239000001361 adipic acid Substances 0.000 description 1
- 235000011037 adipic acid Nutrition 0.000 description 1
- 229960000250 adipic acid Drugs 0.000 description 1
- 210000001789 adipocyte Anatomy 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- SNAAJJQQZSMGQD-UHFFFAOYSA-N aluminum magnesium Chemical compound [Mg].[Al] SNAAJJQQZSMGQD-UHFFFAOYSA-N 0.000 description 1
- 229940000806 amaryl Drugs 0.000 description 1
- 230000001668 ameliorated effect Effects 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- HTIQEAQVCYTUBX-UHFFFAOYSA-N amlodipine Chemical compound CCOC(=O)C1=C(COCCN)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1Cl HTIQEAQVCYTUBX-UHFFFAOYSA-N 0.000 description 1
- 229960000528 amlodipine Drugs 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- 230000037005 anaesthesia Effects 0.000 description 1
- 239000002333 angiotensin II receptor antagonist Substances 0.000 description 1
- 239000003472 antidiabetic agent Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 229960003121 arginine Drugs 0.000 description 1
- 208000037873 arthrodesis Diseases 0.000 description 1
- 125000004104 aryloxy group Chemical group 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 229960005261 aspartic acid Drugs 0.000 description 1
- 208000006673 asthma Diseases 0.000 description 1
- 244000309743 astrovirus Species 0.000 description 1
- 229960002274 atenolol Drugs 0.000 description 1
- 229960005370 atorvastatin Drugs 0.000 description 1
- 230000037444 atrophy Effects 0.000 description 1
- 125000005604 azodicarboxylate group Chemical group 0.000 description 1
- 229960004530 benazepril Drugs 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- UPABQMWFWCMOFV-UHFFFAOYSA-N benethamine Chemical compound C=1C=CC=CC=1CNCCC1=CC=CC=C1 UPABQMWFWCMOFV-UHFFFAOYSA-N 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000005870 benzindolyl group Chemical group 0.000 description 1
- 125000000928 benzodioxinyl group Chemical group O1C(=COC2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 229960004365 benzoic acid Drugs 0.000 description 1
- 150000001558 benzoic acid derivatives Chemical class 0.000 description 1
- 125000004619 benzopyranyl group Chemical group O1C(C=CC2=C1C=CC=C2)* 0.000 description 1
- 125000005874 benzothiadiazolyl group Chemical group 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 description 1
- PASDCCFISLVPSO-UHFFFAOYSA-N benzoyl chloride Chemical compound ClC(=O)C1=CC=CC=C1 PASDCCFISLVPSO-UHFFFAOYSA-N 0.000 description 1
- KKBIUAUSZKGNOA-HNAYVOBHSA-N benzyl (2s)-2-[[(2s)-2-(acetylsulfanylmethyl)-3-(1,3-benzodioxol-5-yl)propanoyl]amino]propanoate Chemical compound O=C([C@@H](NC(=O)[C@@H](CSC(C)=O)CC=1C=C2OCOC2=CC=1)C)OCC1=CC=CC=C1 KKBIUAUSZKGNOA-HNAYVOBHSA-N 0.000 description 1
- UIEATEWHFDRYRU-UHFFFAOYSA-N bepridil Chemical compound C1CCCN1C(COCC(C)C)CN(C=1C=CC=CC=1)CC1=CC=CC=C1 UIEATEWHFDRYRU-UHFFFAOYSA-N 0.000 description 1
- 229960003665 bepridil Drugs 0.000 description 1
- 102000012740 beta Adrenergic Receptors Human genes 0.000 description 1
- 108010079452 beta Adrenergic Receptors Proteins 0.000 description 1
- MSWZFWKMSRAUBD-UHFFFAOYSA-N beta-D-galactosamine Natural products NC1C(O)OC(CO)C(O)C1O MSWZFWKMSRAUBD-UHFFFAOYSA-N 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 229960003237 betaine Drugs 0.000 description 1
- 229960004324 betaxolol Drugs 0.000 description 1
- NWIUTZDMDHAVTP-UHFFFAOYSA-N betaxolol Chemical compound C1=CC(OCC(O)CNC(C)C)=CC=C1CCOCC1CC1 NWIUTZDMDHAVTP-UHFFFAOYSA-N 0.000 description 1
- 150000004283 biguanides Chemical class 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 229960002781 bisoprolol Drugs 0.000 description 1
- VHYCDWMUTMEGQY-UHFFFAOYSA-N bisoprolol Chemical compound CC(C)NCC(O)COC1=CC=C(COCCOC(C)C)C=C1 VHYCDWMUTMEGQY-UHFFFAOYSA-N 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- ZADPBFCGQRWHPN-UHFFFAOYSA-N boronic acid Chemical compound OBO ZADPBFCGQRWHPN-UHFFFAOYSA-N 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 125000001246 bromo group Chemical group Br* 0.000 description 1
- 239000007853 buffer solution Substances 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 125000005622 butynylene group Chemical group 0.000 description 1
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 1
- 229910000024 caesium carbonate Inorganic materials 0.000 description 1
- 239000000480 calcium channel blocker Substances 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- LSPHULWDVZXLIL-QUBYGPBYSA-N camphoric acid Chemical compound CC1(C)[C@H](C(O)=O)CC[C@]1(C)C(O)=O LSPHULWDVZXLIL-QUBYGPBYSA-N 0.000 description 1
- 230000036952 cancer formation Effects 0.000 description 1
- 229960000932 candesartan Drugs 0.000 description 1
- SGZAIDDFHDDFJU-UHFFFAOYSA-N candesartan Chemical compound CCOC1=NC2=CC=CC(C(O)=O)=C2N1CC(C=C1)=CC=C1C1=CC=CC=C1C1=NN=N[N]1 SGZAIDDFHDDFJU-UHFFFAOYSA-N 0.000 description 1
- KHAVLLBUVKBTBG-UHFFFAOYSA-N caproleic acid Natural products OC(=O)CCCCCCCC=C KHAVLLBUVKBTBG-UHFFFAOYSA-N 0.000 description 1
- 229960000830 captopril Drugs 0.000 description 1
- FAKRSMQSSFJEIM-RQJHMYQMSA-N captopril Chemical compound SC[C@@H](C)C(=O)N1CCC[C@H]1C(O)=O FAKRSMQSSFJEIM-RQJHMYQMSA-N 0.000 description 1
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 description 1
- 150000001721 carbon Chemical group 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 231100000504 carcinogenesis Toxicity 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 208000026106 cerebrovascular disease Diseases 0.000 description 1
- 229960005110 cerivastatin Drugs 0.000 description 1
- SEERZIQQUAZTOL-ANMDKAQQSA-N cerivastatin Chemical compound COCC1=C(C(C)C)N=C(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC(O)=O)=C1C1=CC=C(F)C=C1 SEERZIQQUAZTOL-ANMDKAQQSA-N 0.000 description 1
- GPUADMRJQVPIAS-QCVDVZFFSA-M cerivastatin sodium Chemical compound [Na+].COCC1=C(C(C)C)N=C(C(C)C)C(\C=C\[C@@H](O)C[C@@H](O)CC([O-])=O)=C1C1=CC=C(F)C=C1 GPUADMRJQVPIAS-QCVDVZFFSA-M 0.000 description 1
- 239000003610 charcoal Substances 0.000 description 1
- JUFFVKRROAPVBI-PVOYSMBESA-N chembl1210015 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(=O)N[C@H]1[C@@H]([C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O)[C@@H](O)[C@@H](CO[C@]3(O[C@@H](C[C@H](O)[C@H](O)CO)[C@H](NC(C)=O)[C@@H](O)C3)C(O)=O)O2)O)[C@@H](CO)O1)NC(C)=O)C(=O)NCC(=O)NCC(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 JUFFVKRROAPVBI-PVOYSMBESA-N 0.000 description 1
- 239000013043 chemical agent Substances 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- FZFAMSAMCHXGEF-UHFFFAOYSA-N chloro formate Chemical compound ClOC=O FZFAMSAMCHXGEF-UHFFFAOYSA-N 0.000 description 1
- 125000001309 chloro group Chemical group Cl* 0.000 description 1
- 208000019425 cirrhosis of liver Diseases 0.000 description 1
- SECPZKHBENQXJG-UHFFFAOYSA-N cis-palmitoleic acid Natural products CCCCCCC=CCCCCCCCC(O)=O SECPZKHBENQXJG-UHFFFAOYSA-N 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 230000006957 competitive inhibition Effects 0.000 description 1
- 239000012141 concentrate Substances 0.000 description 1
- 238000011109 contamination Methods 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- 208000029078 coronary artery disease Diseases 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 239000000625 cyclamic acid and its Na and Ca salt Substances 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- HCAJEUSONLESMK-UHFFFAOYSA-N cyclohexylsulfamic acid Chemical compound OS(=O)(=O)NC1CCCCC1 HCAJEUSONLESMK-UHFFFAOYSA-N 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000298 cyclopropenyl group Chemical group [H]C1=C([H])C1([H])* 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- WLVKDFJTYKELLQ-UHFFFAOYSA-N cyclopropylboronic acid Chemical compound OB(O)C1CC1 WLVKDFJTYKELLQ-UHFFFAOYSA-N 0.000 description 1
- 229960002887 deanol Drugs 0.000 description 1
- 125000004855 decalinyl group Chemical group C1(CCCC2CCCCC12)* 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 208000025729 dengue disease Diseases 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 238000003745 diagnosis Methods 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- LTMHDMANZUZIPE-PUGKRICDSA-N digoxin Chemical compound C1[C@H](O)[C@H](O)[C@@H](C)O[C@H]1O[C@@H]1[C@@H](C)O[C@@H](O[C@@H]2[C@H](O[C@@H](O[C@@H]3C[C@@H]4[C@]([C@@H]5[C@H]([C@]6(CC[C@@H]([C@@]6(C)[C@H](O)C5)C=5COC(=O)C=5)O)CC4)(C)CC3)C[C@@H]2O)C)C[C@@H]1O LTMHDMANZUZIPE-PUGKRICDSA-N 0.000 description 1
- 229960005156 digoxin Drugs 0.000 description 1
- LTMHDMANZUZIPE-UHFFFAOYSA-N digoxine Natural products C1C(O)C(O)C(C)OC1OC1C(C)OC(OC2C(OC(OC3CC4C(C5C(C6(CCC(C6(C)C(O)C5)C=5COC(=O)C=5)O)CC4)(C)CC3)CC2O)C)CC1O LTMHDMANZUZIPE-UHFFFAOYSA-N 0.000 description 1
- 150000004683 dihydrates Chemical class 0.000 description 1
- HSUGRBWQSSZJOP-RTWAWAEBSA-N diltiazem Chemical compound C1=CC(OC)=CC=C1[C@H]1[C@@H](OC(C)=O)C(=O)N(CCN(C)C)C2=CC=CC=C2S1 HSUGRBWQSSZJOP-RTWAWAEBSA-N 0.000 description 1
- 229960004166 diltiazem Drugs 0.000 description 1
- 125000005879 dioxolanyl group Chemical group 0.000 description 1
- 229910000396 dipotassium phosphate Inorganic materials 0.000 description 1
- 231100000676 disease causative agent Toxicity 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 201000008865 drug-induced hepatitis Diseases 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- GBXSMTUPTTWBMN-XIRDDKMYSA-N enalapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(O)=O)CC1=CC=CC=C1 GBXSMTUPTTWBMN-XIRDDKMYSA-N 0.000 description 1
- 229960000873 enalapril Drugs 0.000 description 1
- 206010014599 encephalitis Diseases 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 230000002255 enzymatic effect Effects 0.000 description 1
- 201000008220 erythropoietic protoporphyria Diseases 0.000 description 1
- AVOLMBLBETYQHX-UHFFFAOYSA-N etacrynic acid Chemical compound CCC(=C)C(=O)C1=CC=C(OCC(O)=O)C(Cl)=C1Cl AVOLMBLBETYQHX-UHFFFAOYSA-N 0.000 description 1
- 229960003199 etacrynic acid Drugs 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- 125000005678 ethenylene group Chemical group [H]C([*:1])=C([H])[*:2] 0.000 description 1
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 description 1
- FCZCIXQGZOUIDN-UHFFFAOYSA-N ethyl 2-diethoxyphosphinothioyloxyacetate Chemical compound CCOC(=O)COP(=S)(OCC)OCC FCZCIXQGZOUIDN-UHFFFAOYSA-N 0.000 description 1
- GSWZGDLAZFBBPB-UHFFFAOYSA-N ethyl 5-(aminomethyl)furan-2-carboxylate Chemical compound CCOC(=O)C1=CC=C(CN)O1 GSWZGDLAZFBBPB-UHFFFAOYSA-N 0.000 description 1
- QNZAHZICJQJSFJ-UHFFFAOYSA-N ethyl 5-bromo-3-methylthiophene-2-carboxylate Chemical compound CCOC(=O)C=1SC(Br)=CC=1C QNZAHZICJQJSFJ-UHFFFAOYSA-N 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 229960001519 exenatide Drugs 0.000 description 1
- 208000030533 eye disease Diseases 0.000 description 1
- 210000000744 eyelid Anatomy 0.000 description 1
- 208000029944 familial hemophagocytic lymphohistiocytosis type 1 Diseases 0.000 description 1
- 229950005203 fasidotril Drugs 0.000 description 1
- 230000004129 fatty acid metabolism Effects 0.000 description 1
- 229960003580 felodipine Drugs 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 235000019634 flavors Nutrition 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 235000019264 food flavour enhancer Nutrition 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 229960002490 fosinopril Drugs 0.000 description 1
- 125000003844 furanonyl group Chemical group 0.000 description 1
- 229960003883 furosemide Drugs 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 230000009368 gene silencing by RNA Effects 0.000 description 1
- 210000004907 gland Anatomy 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 229960004580 glibenclamide Drugs 0.000 description 1
- WIGIZIANZCJQQY-RUCARUNLSA-N glimepiride Chemical group O=C1C(CC)=C(C)CN1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)N[C@@H]2CC[C@@H](C)CC2)C=C1 WIGIZIANZCJQQY-RUCARUNLSA-N 0.000 description 1
- ZJJXGWJIGJFDTL-UHFFFAOYSA-N glipizide Chemical compound C1=NC(C)=CN=C1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)NC2CCCCC2)C=C1 ZJJXGWJIGJFDTL-UHFFFAOYSA-N 0.000 description 1
- 229960001381 glipizide Drugs 0.000 description 1
- 239000000174 gluconic acid Substances 0.000 description 1
- 235000012208 gluconic acid Nutrition 0.000 description 1
- 229960002442 glucosamine Drugs 0.000 description 1
- 229940097043 glucuronic acid Drugs 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- ZNNLBTZKUZBEKO-UHFFFAOYSA-N glyburide Chemical compound COC1=CC=C(Cl)C=C1C(=O)NCCC1=CC=C(S(=O)(=O)NC(=O)NC2CCCCC2)C=C1 ZNNLBTZKUZBEKO-UHFFFAOYSA-N 0.000 description 1
- 229940096919 glycogen Drugs 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 125000002795 guanidino group Chemical group C(N)(=N)N* 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- BCQZXOMGPXTTIC-UHFFFAOYSA-N halothane Chemical compound FC(F)(F)C(Cl)Br BCQZXOMGPXTTIC-UHFFFAOYSA-N 0.000 description 1
- 229960003132 halothane Drugs 0.000 description 1
- 208000019622 heart disease Diseases 0.000 description 1
- TZTFFIFDGZOKCS-UHFFFAOYSA-N heptadecanethioic s-acid Chemical compound CCCCCCCCCCCCCCCCC(O)=S TZTFFIFDGZOKCS-UHFFFAOYSA-N 0.000 description 1
- 125000004836 hexamethylene group Chemical group [H]C([H])([*:2])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[*:1] 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- 229960002885 histidine Drugs 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- DCPMPXBYPZGNDC-UHFFFAOYSA-N hydron;methanediimine;chloride Chemical compound Cl.N=C=N DCPMPXBYPZGNDC-UHFFFAOYSA-N 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 208000020346 hyperlipoproteinemia Diseases 0.000 description 1
- ZBNZAJFNDPPMDT-UHFFFAOYSA-N imidazole-4-carboxamide Natural products NC(=O)C1=CNC=N1 ZBNZAJFNDPPMDT-UHFFFAOYSA-N 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 208000026278 immune system disease Diseases 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 206010022000 influenza Diseases 0.000 description 1
- 239000007972 injectable composition Substances 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- 125000002346 iodo group Chemical group I* 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 229960002198 irbesartan Drugs 0.000 description 1
- YCPOHTHPUREGFM-UHFFFAOYSA-N irbesartan Chemical compound O=C1N(CC=2C=CC(=CC=2)C=2C(=CC=CC=2)C=2[N]N=NN=2)C(CCCC)=NC21CCCC2 YCPOHTHPUREGFM-UHFFFAOYSA-N 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 230000000302 ischemic effect Effects 0.000 description 1
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000004628 isothiazolidinyl group Chemical group S1N(CCC1)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 239000000644 isotonic solution Substances 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 229940099563 lactobionic acid Drugs 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 230000004322 lipid homeostasis Effects 0.000 description 1
- 230000008604 lipoprotein metabolism Effects 0.000 description 1
- 229960002394 lisinopril Drugs 0.000 description 1
- CZRQXSDBMCMPNJ-ZUIPZQNBSA-N lisinopril dihydrate Chemical compound O.O.C([C@H](N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(O)=O)C(O)=O)CC1=CC=CC=C1 CZRQXSDBMCMPNJ-ZUIPZQNBSA-N 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 239000002171 loop diuretic Substances 0.000 description 1
- 229960004773 losartan Drugs 0.000 description 1
- KJJZZJSZUJXYEA-UHFFFAOYSA-N losartan Chemical compound CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C=2[N]N=NN=2)C=C1 KJJZZJSZUJXYEA-UHFFFAOYSA-N 0.000 description 1
- 229960004844 lovastatin Drugs 0.000 description 1
- PCZOHLXUXFIOCF-BXMDZJJMSA-N lovastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=C[C@H](C)C[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 PCZOHLXUXFIOCF-BXMDZJJMSA-N 0.000 description 1
- QLJODMDSTUBWDW-UHFFFAOYSA-N lovastatin hydroxy acid Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CC(C)C=C21 QLJODMDSTUBWDW-UHFFFAOYSA-N 0.000 description 1
- 235000015263 low fat diet Nutrition 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 229960003646 lysine Drugs 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 230000036210 malignancy Effects 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 230000013011 mating Effects 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 235000012054 meals Nutrition 0.000 description 1
- 235000013622 meat product Nutrition 0.000 description 1
- 229950004994 meglitinide Drugs 0.000 description 1
- 210000004175 meibomian gland Anatomy 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- XZWYZXLIPXDOLR-UHFFFAOYSA-N metformin Chemical compound CN(C)C(=N)NC(N)=N XZWYZXLIPXDOLR-UHFFFAOYSA-N 0.000 description 1
- 229960003105 metformin Drugs 0.000 description 1
- TWXDDNPPQUTEOV-FVGYRXGTSA-N methamphetamine hydrochloride Chemical compound Cl.CN[C@@H](C)CC1=CC=CC=C1 TWXDDNPPQUTEOV-FVGYRXGTSA-N 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 229960002237 metoprolol Drugs 0.000 description 1
- IUBSYMUCCVWXPE-UHFFFAOYSA-N metoprolol Chemical compound COCCC1=CC=C(OCC(O)CNC(C)C)C=C1 IUBSYMUCCVWXPE-UHFFFAOYSA-N 0.000 description 1
- 229950009116 mevastatin Drugs 0.000 description 1
- AJLFOPYRIVGYMJ-INTXDZFKSA-N mevastatin Chemical compound C([C@H]1[C@@H](C)C=CC2=CCC[C@@H]([C@H]12)OC(=O)[C@@H](C)CC)C[C@@H]1C[C@@H](O)CC(=O)O1 AJLFOPYRIVGYMJ-INTXDZFKSA-N 0.000 description 1
- BOZILQFLQYBIIY-UHFFFAOYSA-N mevastatin hydroxy acid Natural products C1=CC(C)C(CCC(O)CC(O)CC(O)=O)C2C(OC(=O)C(C)CC)CCC=C21 BOZILQFLQYBIIY-UHFFFAOYSA-N 0.000 description 1
- PZRHRDRVRGEVNW-UHFFFAOYSA-N milrinone Chemical compound N1C(=O)C(C#N)=CC(C=2C=CN=CC=2)=C1C PZRHRDRVRGEVNW-UHFFFAOYSA-N 0.000 description 1
- 229960003574 milrinone Drugs 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 210000003470 mitochondria Anatomy 0.000 description 1
- 230000002438 mitochondrial effect Effects 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 150000004682 monohydrates Chemical class 0.000 description 1
- 235000021281 monounsaturated fatty acids Nutrition 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- 210000004400 mucous membrane Anatomy 0.000 description 1
- 201000006417 multiple sclerosis Diseases 0.000 description 1
- RTHZTQTZYJGDDM-UHFFFAOYSA-N n-benzyl-3-methylfuran-2-carboxamide Chemical compound C1=COC(C(=O)NCC=2C=CC=CC=2)=C1C RTHZTQTZYJGDDM-UHFFFAOYSA-N 0.000 description 1
- RIUZAWKIAJCXTF-UHFFFAOYSA-N n-benzyl-5-bromo-3-methylfuran-2-carboxamide Chemical compound C1=C(Br)OC(C(=O)NCC=2C=CC=CC=2)=C1C RIUZAWKIAJCXTF-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- XKVWVANEXDDABN-UHFFFAOYSA-N n-ethyl-3-methylthiophene-2-carboxamide Chemical compound CCNC(=O)C=1SC=CC=1C XKVWVANEXDDABN-UHFFFAOYSA-N 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- VWPOSFSPZNDTMJ-UCWKZMIHSA-N nadolol Chemical compound C1[C@@H](O)[C@@H](O)CC2=C1C=CC=C2OCC(O)CNC(C)(C)C VWPOSFSPZNDTMJ-UCWKZMIHSA-N 0.000 description 1
- 229960004255 nadolol Drugs 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical compound C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- OELFLUMRDSZNSF-BRWVUGGUSA-N nateglinide Chemical compound C1C[C@@H](C(C)C)CC[C@@H]1C(=O)N[C@@H](C(O)=O)CC1=CC=CC=C1 OELFLUMRDSZNSF-BRWVUGGUSA-N 0.000 description 1
- 229960000698 nateglinide Drugs 0.000 description 1
- 230000009826 neoplastic cell growth Effects 0.000 description 1
- PKWDZWYVIHVNKS-UHFFFAOYSA-N netoglitazone Chemical compound FC1=CC=CC=C1COC1=CC=C(C=C(CC2C(NC(=O)S2)=O)C=C2)C2=C1 PKWDZWYVIHVNKS-UHFFFAOYSA-N 0.000 description 1
- 229960001783 nicardipine Drugs 0.000 description 1
- BOPGDPNILDQYTO-NNYOXOHSSA-N nicotinamide-adenine dinucleotide Chemical compound C1=CCC(C(=O)N)=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OC[C@@H]2[C@H]([C@@H](O)[C@@H](O2)N2C3=NC=NC(N)=C3N=C2)O)O1 BOPGDPNILDQYTO-NNYOXOHSSA-N 0.000 description 1
- HYIMSNHJOBLJNT-UHFFFAOYSA-N nifedipine Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1[N+]([O-])=O HYIMSNHJOBLJNT-UHFFFAOYSA-N 0.000 description 1
- 229960001597 nifedipine Drugs 0.000 description 1
- 229960000715 nimodipine Drugs 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 208000008338 non-alcoholic fatty liver disease Diseases 0.000 description 1
- 238000013116 obese mouse model Methods 0.000 description 1
- 125000005060 octahydroindolyl group Chemical group N1(CCC2CCCCC12)* 0.000 description 1
- 125000005061 octahydroisoindolyl group Chemical group C1(NCC2CCCCC12)* 0.000 description 1
- 229960002446 octanoic acid Drugs 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 235000019198 oils Nutrition 0.000 description 1
- 229960002969 oleic acid Drugs 0.000 description 1
- XDUHQPOXLUAVEE-BPMMELMSSA-N oleoyl-CoA Chemical compound O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCSC(=O)CCCCCCC\C=C/CCCCCCCC)O[C@H]1N1C2=NC=NC(N)=C2N=C1 XDUHQPOXLUAVEE-BPMMELMSSA-N 0.000 description 1
- LVRLSYPNFFBYCZ-VGWMRTNUSA-N omapatrilat Chemical compound C([C@H](S)C(=O)N[C@H]1CCS[C@H]2CCC[C@H](N2C1=O)C(=O)O)C1=CC=CC=C1 LVRLSYPNFFBYCZ-VGWMRTNUSA-N 0.000 description 1
- 229950000973 omapatrilat Drugs 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 229960005010 orotic acid Drugs 0.000 description 1
- 230000003204 osmotic effect Effects 0.000 description 1
- 201000008482 osteoarthritis Diseases 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 229940116315 oxalic acid Drugs 0.000 description 1
- 125000000160 oxazolidinyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- PIBWKRNGBLPSSY-UHFFFAOYSA-L palladium(II) chloride Chemical compound Cl[Pd]Cl PIBWKRNGBLPSSY-UHFFFAOYSA-L 0.000 description 1
- 229940098695 palmitic acid Drugs 0.000 description 1
- QBYOCCWNZAOZTL-MDMKAECGSA-N palmitoleoyl-CoA Chemical compound O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCSC(=O)CCCCCCC\C=C/CCCCCC)O[C@H]1N1C2=NC=NC(N)=C2N=C1 QBYOCCWNZAOZTL-MDMKAECGSA-N 0.000 description 1
- WLJNZVDCPSBLRP-UHFFFAOYSA-N pamoic acid Chemical compound C1=CC=C2C(CC=3C4=CC=CC=C4C=C(C=3O)C(=O)O)=C(O)C(C(O)=O)=CC2=C1 WLJNZVDCPSBLRP-UHFFFAOYSA-N 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 239000006072 paste Substances 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 235000019371 penicillin G benzathine Nutrition 0.000 description 1
- 229940021222 peritoneal dialysis isotonic solution Drugs 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 239000008177 pharmaceutical agent Substances 0.000 description 1
- 125000001791 phenazinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3N=C12)* 0.000 description 1
- 125000001484 phenothiazinyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3NC12)* 0.000 description 1
- 125000001644 phenoxazinyl group Chemical group C1(=CC=CC=2OC3=CC=CC=C3NC12)* 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 229960005095 pioglitazone Drugs 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 229920000768 polyamine Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 229910000160 potassium phosphate Inorganic materials 0.000 description 1
- 239000008057 potassium phosphate buffer Substances 0.000 description 1
- 235000011009 potassium phosphates Nutrition 0.000 description 1
- 229960002965 pravastatin Drugs 0.000 description 1
- TUZYXOIXSAXUGO-PZAWKZKUSA-N pravastatin Chemical compound C1=C[C@H](C)[C@H](CC[C@@H](O)C[C@@H](O)CC(O)=O)[C@H]2[C@@H](OC(=O)[C@@H](C)CC)C[C@H](O)C=C21 TUZYXOIXSAXUGO-PZAWKZKUSA-N 0.000 description 1
- 201000009104 prediabetes syndrome Diseases 0.000 description 1
- 230000035935 pregnancy Effects 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- NPUSXSOBPNHOPH-UHFFFAOYSA-N propan-2-yl 4-(2-chlorophenyl)-1-ethyl-2-methyl-5-oxo-4,7-dihydrofuro[3,4-b]pyridine-3-carboxylate Chemical compound CC(C)OC(=O)C1=C(C)N(CC)C(COC2=O)=C2C1C1=CC=CC=C1Cl NPUSXSOBPNHOPH-UHFFFAOYSA-N 0.000 description 1
- 125000006410 propenylene group Chemical group 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 229960003712 propranolol Drugs 0.000 description 1
- 208000020016 psychiatric disease Diseases 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 150000003212 purines Chemical class 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000004307 pyrazin-2-yl group Chemical group [H]C1=C([H])N=C(*)C([H])=N1 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- GGOZGYRTNQBSSA-UHFFFAOYSA-N pyridine-2,3-diol Chemical compound OC1=CC=CN=C1O GGOZGYRTNQBSSA-UHFFFAOYSA-N 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 229940107700 pyruvic acid Drugs 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- JSDRRTOADPPCHY-HSQYWUDLSA-N quinapril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](CC2=CC=CC=C2C1)C(O)=O)CC1=CC=CC=C1 JSDRRTOADPPCHY-HSQYWUDLSA-N 0.000 description 1
- 229960001455 quinapril Drugs 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- HDACQVRGBOVJII-JBDAPHQKSA-N ramipril Chemical compound C([C@@H](C(=O)OCC)N[C@@H](C)C(=O)N1[C@@H](C[C@@H]2CCC[C@@H]21)C(O)=O)CC1=CC=CC=C1 HDACQVRGBOVJII-JBDAPHQKSA-N 0.000 description 1
- 229960003401 ramipril Drugs 0.000 description 1
- 229960002354 repaglinide Drugs 0.000 description 1
- 230000003362 replicative effect Effects 0.000 description 1
- 230000004141 reverse cholesterol transport Effects 0.000 description 1
- 230000006965 reversible inhibition Effects 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 229960004586 rosiglitazone Drugs 0.000 description 1
- 229960000672 rosuvastatin Drugs 0.000 description 1
- BPRHUIZQVSMCRT-VEUZHWNKSA-N rosuvastatin Chemical compound CC(C)C1=NC(N(C)S(C)(=O)=O)=NC(C=2C=CC(F)=CC=2)=C1\C=C\[C@@H](O)C[C@@H](O)CC(O)=O BPRHUIZQVSMCRT-VEUZHWNKSA-N 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 229950001780 sampatrilat Drugs 0.000 description 1
- 235000021003 saturated fats Nutrition 0.000 description 1
- 235000003441 saturated fatty acids Nutrition 0.000 description 1
- 150000004671 saturated fatty acids Chemical class 0.000 description 1
- 238000007423 screening assay Methods 0.000 description 1
- 210000002374 sebum Anatomy 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 229960002855 simvastatin Drugs 0.000 description 1
- 201000009890 sinusitis Diseases 0.000 description 1
- MFFMDFFZMYYVKS-SECBINFHSA-N sitagliptin Chemical compound C([C@H](CC(=O)N1CC=2N(C(=NN=2)C(F)(F)F)CC1)N)C1=CC(F)=C(F)C=C1F MFFMDFFZMYYVKS-SECBINFHSA-N 0.000 description 1
- 210000002027 skeletal muscle Anatomy 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- 238000003797 solvolysis reaction Methods 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- ZBMZVLHSJCTVON-UHFFFAOYSA-N sotalol Chemical compound CC(C)NCC(O)C1=CC=C(NS(C)(=O)=O)C=C1 ZBMZVLHSJCTVON-UHFFFAOYSA-N 0.000 description 1
- 229960002370 sotalol Drugs 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 125000003696 stearoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000010959 steel Substances 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 239000006228 supernatant Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229960005187 telmisartan Drugs 0.000 description 1
- 230000028016 temperature homeostasis Effects 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- ILMRJRBKQSSXGY-UHFFFAOYSA-N tert-butyl(dimethyl)silicon Chemical group C[Si](C)C(C)(C)C ILMRJRBKQSSXGY-UHFFFAOYSA-N 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 150000004685 tetrahydrates Chemical class 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 229960004559 theobromine Drugs 0.000 description 1
- 231100001274 therapeutic index Toxicity 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 150000001467 thiazolidinediones Chemical class 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 229930192474 thiophene Natural products 0.000 description 1
- 229960004605 timolol Drugs 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 239000012049 topical pharmaceutical composition Substances 0.000 description 1
- 229960005461 torasemide Drugs 0.000 description 1
- 231100000816 toxic dose Toxicity 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- 229960002051 trandolapril Drugs 0.000 description 1
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 1
- 238000011830 transgenic mouse model Methods 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 125000003866 trichloromethyl group Chemical group ClC(Cl)(Cl)* 0.000 description 1
- 125000004385 trihaloalkyl group Chemical group 0.000 description 1
- 150000004684 trihydrates Chemical class 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 125000005455 trithianyl group Chemical group 0.000 description 1
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 1
- 229960004418 trolamine Drugs 0.000 description 1
- 229960000281 trometamol Drugs 0.000 description 1
- 229910052721 tungsten Inorganic materials 0.000 description 1
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 1
- 229960002703 undecylenic acid Drugs 0.000 description 1
- 238000011144 upstream manufacturing Methods 0.000 description 1
- 229960001722 verapamil Drugs 0.000 description 1
- 229960001254 vildagliptin Drugs 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/14—Prodigestives, e.g. acids, enzymes, appetite stimulants, antidyspeptics, tonics, antiflatulents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4436—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a heterocyclic ring having sulfur as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4178—1,3-Diazoles not condensed 1,3-diazoles and containing further heterocyclic rings, e.g. pilocarpine, nitrofurantoin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/4439—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a five-membered ring with nitrogen as a ring hetero atom, e.g. omeprazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4427—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems
- A61K31/444—Non condensed pyridines; Hydrogenated derivatives thereof containing further heterocyclic ring systems containing a six-membered ring with nitrogen as a ring heteroatom, e.g. amrinone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/4965—Non-condensed pyrazines
- A61K31/497—Non-condensed pyrazines containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/02—Drugs for disorders of the urinary system of urine or of the urinary tract, e.g. urine acidifiers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/10—Anti-acne agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/06—Antigout agents, e.g. antihyperuricemic or uricosuric agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/48—Drugs for disorders of the endocrine system of the pancreatic hormones
- A61P5/50—Drugs for disorders of the endocrine system of the pancreatic hormones for increasing or potentiating the activity of insulin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/14—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F7/00—Compounds containing elements of Groups 4 or 14 of the Periodic Table
- C07F7/02—Silicon compounds
- C07F7/08—Compounds having one or more C—Si linkages
- C07F7/0834—Compounds having one or more O-Si linkage
Definitions
- the present invention relates generally to the field of inhibitors of stearoyl-CoA desaturase, such as heterocyclic derivatives, and uses for such compounds m treating and/or preventing various human diseases, including those mediated by stearoyl-CoA desaturase (SCD) enzymes, preferably SCDl, especially diseases related to elevated lipid levels, cardiovascular disease, diabetes, obesity, metabolic syndrome, d ⁇ ratological disorders and the like.
- SCD stearoyl-CoA desaturase
- Acyl desaturase enzymes catalyze the formation of a double bond in fatty acids derived from either dietary sources or de HOVD synthesis in the liver.
- fatty acids derived from either dietary sources or de HOVD synthesis in the liver.
- SCDs Stearoyl-CoA desaturases
- cofactors other agents
- other agents such as NADPH, cytochrome b5, cytochrome b5 reductase, Fe, and molecular O2 to introduce a double bond into the C9-C10 position (delta 9) of saturated fatty acids, when conjugated to Coenzyme A (CoA).
- the preferred substrates are palmitoyl-CoA (16:0) and stearoyl- CoA (18:0), which are converted to palmitoleoyl-CoA (16:1) and oleyl-CoA(18:l), respectively
- the resulting mono-unsaturated fatty acids are substrates foi further metabolism by fittty acid elongases or incorporation into phospholipids, triglycerides, and cholesterol esters.
- a number of mammalian SCD genes have been cloned For example, two genes have been identified in humans (h SCDl and hSCD5) and four SCD genes have been isolated from mouse (SCDl, SCD2, SCD3, and SCD4).
- hSCD 1 by Brownlie et al., FCT published patent application, WO 01/62954, the disclosure of which is hereby incorporated by reference in its entirety
- hSCD2 PCT published patent application, WO 02/26944, incorporated herein by reference in its entirety.
- sterculic acid 8-(2 octylcyclopropenyOoctanoic acid) and malvalic acid (7-(2-oclylcyclopropenyl)hepta ⁇ oic acid
- agents that may inhibit SCD activity include thia-fatty acids, such as 9-thiastearic acid (also called 8-nonylthiooctanoic acid) and other fatty acids with a thioestcr moiety
- the present invention solves this problem by presenting new drug-like classes of compounds that are useful in modulating SCD activity and regulating lipid levels, especially plasm, lipid levels, and which arc useful in the treatment of SCD-mediated diseases such as diseases related to dyslipidemia and disorders of lipid metabolism, especially diseases related to elevated lipid levels, cardiovascular disease, diabetes, obesity, metabolic syndrome and the like.
- the present invention provides heterocyclic derivatives that modulate the activity of ⁇ tearoyl-CoA de ⁇ atura ⁇ e. Methods of using such derivatives to modulate the activity of Stearoyl-CoA desaturase and pharmaceutics] compositions comprising such derivatives are also encompassed.
- the invention provides compounds of Formula (I):
- V is selected from -N(R 5 )C(O)-, -C(O)N(R*)-, -OC(O)N(R 5 )-, -N(R 5 )C(O)O-,
- W is selected fiom -N(R 5 )C(0>, -C(O)N(R 5 )-, -OC(O)N(R 5 )-, -N(R 5 )C(0)0-,
- X is selected from C(H) or N;
- Y is selected from S, O, N(H) or N(CH)); p is O, 1, 2, or 3; t is 1 or 2;
- R' is selected from the group consisting of lydrogen, alkyl, alkenyl, alkynyl,alkoxy, hydroxyalkyl, alkoxyalkyl, cycloalkyl, cycloalkylalkyl, aryl, aralk/L heterocyclyl, heteroeydylalkyl, heteroaryl, and hctcroarylalkyl; or R 1 is a multi-ring structure having 2 to 4 rings wherein the rings are independently selected from the group consisting of cycloalkyl, heterocyclyl, aryl Mid heteroaryl and where some or all of the rings may be fused to each other; R 2 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, alkoxy, hydro ⁇ yalkyl, alkoxyalkyl, cycloalkyl, cycloalkylalkyl, aryl, haloalkyl, aralkyl, heterocyclyl, hetero
- R 3 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkoxy, hydroxyalkyl, alkoxyalkyl, cycloalkyl, cycloalkylalkyl, heterocyclyl, aryl, aralkyl, heteroaryl, halo, haloalkyl, trihaloalkoxyl, cyano and -N(R 5 ) 2 j
- R 4 is selected from the group consisting of alkyl, hydroxyalkyl, cycloalkylalkyl, aralkyl, halo, haloalkyl, -OCF 3 , -OC(H)F 2 , and cyano; or two adjacent R 4 groups, together with the carbon atoms to which they are attached, may form a cycloalkyl, heterocyclyl, sryl or heteroaryl and the remaining R 4 groups, if present, are as described R is selected from the group consisting of hydrogen, alkyl, alkeny
- R ⁇ a is selected from the group consisting of hydrogen, alkyl, cycloalkylalkyl and cyano; a stereoisomer, enantiomer or tautomer thereof, a pharmaceutically acceptable salt thereof, a pharmaceutical composition thereof or a prodrug thereof.
- the invention provides methods of treating an SCD-mediated disease or condition in a mammal, preferably a human, wherein the methods comprise administering to the mammal in need thereof a therapeutically effective amount of a compound of the invention as set forth above.
- the invention provides compounds or pharmaceutical compositions useful in treating, preventing and/or diagnosing a disease or condition relating to SCD biological activity such as the diseases encompassed by cardiovascular disorders andior metabolic syndrome (including dyslipidemia, insulin resistance and obesity).
- a disease or condition relating to SCD biological activity such as the diseases encompassed by cardiovascular disorders andior metabolic syndrome (including dyslipidemia, insulin resistance and obesity).
- the invention provides methods of preventing or treating a disease or co ⁇ cM on related to elevated lipid levels, such as plasma lipid levels, especially elevated triglyceride or cholesterol levels, in a patient afflicted with such elevated levels, comprising administering to said patient a therapeutically or prophylacticaliy effective amount of a composition as disclosed herein.
- the present invention also relates to novel compounds having therapeutic ability to reduce lipid levels in an animal, especially triglyceride and cholesterol levels.
- the invention provides pharmaceutical compositions comprising
- the present invention relates to a pharmaceutical composition
- a pharmaceutical composition comprising a compound of the inye ⁇ tion in a pharmaceutically acceptable carrier and in an amount effective to modulate triglyceride level, or to treat diseases related to dyslipidemia and disorders of lipid metabolism, when administered to an animal, preferably a mammal, most preferably a human patient.
- the patient has an elevated lipid level, such as elevated plasma triglycerides or cholesterol, before administration of said compound and said compound is present in an amount effective to reduce said lipid level,
- the invention proridcs methods for treating a patient for, or protecting a patient from developing, a disease or condition mediated by stearoyl-CoA desaturase (SCD), which methods comprise administering to a patient afflicted with such disease or condition, or at risk of developing such disease or condition, a therapeutically effective amount of a compound that inhibits activity of SCD in a patient when administered [hereto.
- SCD stearoyl-CoA desaturase
- the invention provides methods for treating a range of diseases involving lipid metabolism and/or lipid homeostasis utilizing compounds identified by the methods disclosed herein.
- a range of compounds having said activity based on a screening assay for identifying, from a library of test compounds, a therapeutic agent which modulates the biological activity of said SCD and is useful in treating a human disorder or condition relating to serum levels of lipids, such as triglycerides, VLDL, HDL, LDL, and/or total cholesterol.
- the invention provides the use of the compounds of the invention, as set forth above, in the preparation of a medicament for the treatment of SCD-mediated disease or condition in a mammal, preferably a human.
- PCTFublished Patent Application WO 01/6045S; PCTFublished Patent Application, WO 01/60369; PCTFublished Patent Application, WO 94/26720; European Published Patent Application, 0438 230; European Published Patent Application, 1 184 442; CA 2,1 14,178; and US Patent No.5,334,328; US Patent No.5,310,499; and US Published Patent Application, 2003/0127627.
- C 7 -Ci 2 alkyl describes an alkyl group, as defined below, having a total of 7 to 12 carbon atoms
- C 4 -Ci 2 Cyclodk ⁇ la]kyl describes a cyclcalkylalkyl group, as defined below, having a total of X to 12 carbon atoms.
- the total number of carbons in the shorthand notation does not include carbons that may exist in substituents of the group described.
- Haldroxy refers to the -OH radical
- Niro refers to the -NO 2 radical
- Amino refers to the -NR 14 or NR 13 radical
- Trifluoromethyl refers to the — CFj radical
- Alkyl refers to a straight or branched hydrocarbon chain radical consisting solely of carbon and hydrogen atoms, containing no unsaturation, having from one to twelve carbon atoms, preferably one to eight carbon atoms or one to six caibon atoms,
- an alkyl group may be optionally substituted by one or more of the following groups: alkyl, alkenyl, halo, cyano, aryl, cycloalkyl, heterocyclyl, heteroaryl, -Si(CH 3 J 2 C(CHj) 3 , -OR 14 , -OC(O)-R 14 , - N(R 14 ) 2 , -C(O)R 14 , -C(O)OR", -C(O)N(R 14 J 2 , -N(R 14 )C(O)0R 16 , -N(R 14 )C(O)R 16 , - N(R 14 JS(O) 1 R 1 ', -S-, -S(O) 1 OR 16 , -S(O) 1 R 16 , and -S(OXN(R 14 J 2 and each R" is independently hydrogen, alkyl, haloalkyl, cycloalkyl, heterocyclyl
- Alkenyl refers to a straight or branched hydrocarbon chain radical group consisting solely of carbon and hydrogen atmns, containing at least one double bond, having from two to twelve carbon atoms, preferably two to eight carbon atoms or two to six carbon atoms and which is attached to the rest of the molecule by a single bond, e.g., ethenyl, prop.l.e ⁇ yl, but-1-enyl, pent-1-enyl, penta-l,4-dienyl, and the like.
- an alkenyl group may be optionally substituted by one or more of the following groups: alkyl, alkenyl, halo, haloalkyl, aryl, aralkyl, cycloalkyl, cycloalkylalkyl, heterocyclyl, heterocyclylalkyl, heteroaryl, heteroarylalkyl, - OR 14 , -OC(O)-K 14 -N(R 14 J 2 , -C(O)R 14 , -C(O)OR 14 , -C(O)N(R 14 J 2 , -N(R 14 JC(O)OR 16 , - N(R 14 JC(O)R 16 , -N(R 14 )S(0),R l ⁇ , -S-, -S(O] 1 OR 16 , -S(O) 1 R 16 , and -S(O) 1 N(R 14 J 2 and each R 14 , -OC(O)-K 14
- Alkynyl refers to a straight or branched hydrocarbon chain radical group consisting solely of carbon and hydrogen atoms, containing at least one triple bond, having from two to twelve carbon atoms, preferably two to eight carbon atoms or two to six carbon atoms and which is attached to the rest of the molecule by a single bond. Unless stated otherwise specifically in the specification, an alkynyl group may be optionally substituted by one or more of the following groups: alkyl, alkenyl, halo.
- Alkylene and “alkylene chain” refer to a straight or branched divalent hydrocarbon chain, linking the rest of the molecule to a radical group, consisting solely of carbon and hydrogen, containing no unsaturation and having from one to twelve carbon atoms, preferably having from one to eight carbons or one to six carbon atoms, e.g., methylene, ethylene, propylene, n-butylene, end the like.
- the alkylene chain may be attached to the rest of the molecule and to the radical group through one caibon within the chain or through any two carbons within the chain. Unless stated otherwise specifically in the specification, an alkylene chain may be optionally substituted by one or more of the following groups: alkyl. alkenyl.
- each R 14 is independently hydrogen, alkyl, haloalkyl, cycloalky], cycloalkylalkyl, aryl, aralkyl, heterocyclyl, heterocyclylalkyl, heteroaryl or heteroarylalkyl; and each R 16 is independently hydrogen, alkyl, haloalkyl, cycloalky], cycloalkylalkyl, aryl, aralkyl, heterocyclyl, heterocyclylalkyl, heteroaryl or heteroarylalkyl; and each R 16 is independently hydrogen, alkyl, haloalkyl, cycloalky], cycloalkylalkyl, aryl, aralkyl, heterocyclyl, heterocyclylalkyl, heteroaryl or heteroarylalkyl; and each R 16 is independently hydrogen, alkyl, haloalkyl, cycloalky], cycloalkylalkyl, aryl, aralkyl, heterocycly
- alkenylene and alkenylene chain refer to a straight or branched divalent hydrocarbon chain linking the rest of the molecule to a radical group, consisting solely of carbon and hydrogen, containing at least one double bond and having from two to twelve carbon atoms or two to six carbon atoms, e.g. ethenylene, propenylene, n-butenylene, and the like.
- an alkenylene chain may be optionally substituted by one or more of the following groups: alkyl, alkenyl, halo, cyano, aryl, cycloalkyl, heterocyclyl, heteroaryl, -OR 14 , -0C ⁇ 0)-R 14 , -T)(R 1 V - C(O)R 14 , -C(O)OR 14 , -C(O)N(R 14 ),, -N(R 14 )C(O)OR" ⁇ -N0l l4 )C(0)R le .
- Alkynylene and Alkynylene chain refer to a straight or branched divalent hydrocarbon chain linking the rest of the molecule to a radical group, consisting solely of carbon and hydrogen, containing at least one triple bond and having from two to twelve carbon atoms or two to six carbon atoms, e.g.
- an alkynylene chain may be optionally substituted by one or more of the following groups: alkyl, alk;n> I, halo, cyano, aryl, cycloalkyl, lictcrocyclyl, heteroaryl, -OR 14 , -OC(O)-R 1 ', -N(R I4 ) 2 , -C(O)R 14 , - C(O)OR 14 , -C(O)N(R 14 ) 2> -N(R l4 )C(0)0R", -N(R 14 JC(O)R 16 , -N(R 14 )S(O)iR 16 , -S-, - S(O) 1 OR 16 , -S(O) 1 R 16 , and -S(O),N(R 14 ) 2 , arid each R 14 is independently hydrogen,
- AIkOKy refers to a radical of the formula -OR, where R. is an alkyl radical as generally defined above.
- R. is an alkyl radical as generally defined above.
- the alkyl part of the alkoxy radical may be optionally substituted as defined above for an alkyl radical.
- Alkoityalkyl refers to a radical of ihe formula -R b -O-R 3 where Rj, is an alkylene chain as defined above and R, is an alkyl radical as defined above.
- the oxygen atom may be bonded to any carbon in the alkylene chair and in the alkyl radical.
- the allcyl part of the alkoxyalkyl radical may be optionally substituted as defined above for an alkyl group.
- the alkylene chain part of the alkoxyalkyl radical may be optionally substituted as defined above for an alkylene chain.
- Aryl refers to aromatic monocyclic O ⁇ multicyclic hydrocarbon ring system consisting only of hydrogen and carbon and containing from 6 to 19 carbon atoms, preferably 6 to 10 carbon atoms, where the ling system may be partially saturated.
- Aryl groups include, but are not limited to groups such as fluorenyl, phenyl, indenyl and naphthyl.
- the terra "aryl” or the prefix “ar-” (such as in “aralkyl”) is meant to include aryl radicals optionally substituted by one or more substitucnts selected from the group consisting of alkyl, aUcenyl, alkynyl,
- Aralkyl refers to a radical of the formula -R 1 R b where R, is an allcylene chain as defined above and R b is one or more aryl radicals as defined above, e.g., benzyl, diphenylmeihyl and the like.
- the aryl part of the aralkyl radical may be optionally substituted as described above For an aryl group.
- the alkylene chain part of the aralkyl radical may be optionally substituted as defined above for an alkyl group.
- Alkenyl refers to a radical of the formula -R a R b where R 2 is an alkenylene chain as defined above and R b i ⁇ one or more aryl radicals a ⁇ defined above, which may be optionally substituted as described above.
- the aryl part of the aralkenyl radical may be optionally substituted as described above for an aryl group.
- the alkenylene chain of the aralkenyl radical may be optionally substituted as defined above for an alkenyl group.
- Aryloxy refers to a radical of the formula -OR b where R b is an aryl group as defined above.
- R b is an aryl group as defined above.
- the aryl part of the aryloxy radical may be optionally substituted as defined above.
- Cycl ⁇ alkyl refers to a stable non-arcmatic monocyclic or polyeyeHc hydrocarbon radical consisting solely of carbon and hydrogen atoms, having from three to fifteen carbon atoms, preferably having from three to twelve carbon atoms or from three to six atoms, and which is saturated or unsaturated and attached to the rest of the molecule by a single bond, e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, decalinyl and the like.
- eycloalkyl is meant to include cycloalkyl radicals which are optionally substituted by one or more ⁇ ubstituent ⁇ selected from the group consisting of alkyl, alkenyl, halo, haloalkyl, cyano, aryl, aralkyl, cycloalkyl, cycloalkylalkyl, heterocyclyl,
- heterocydylalkyl heter ⁇ aryl, heteroarylalkyl, -R ls -0R 14 , -R 1 ⁇ OC(O)-R 1 ", -R 15 -N(R 14 ) 2 , - R ⁇ -C(O)R 1 ', -R IS -C(O)OR", -R 1S -C(0)N(R%, -R 15 -N(R 14 )C(O)OR 16 , -R 15 - N(R u )C(0)R 16 , -R IS -N(R U )S(O),R 16 , -S-, -R ls -S(O)OR 1 ⁇ , -R 1S -S(O),R 16 , and -R ]i - S(O),N(R 14 ) 2 and each R 14 is independently hydrogen, alky], haloalkyl, cycloalkyl, cycl ⁇ alkylal
- each R 1 ' is alkyl, tialoalkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocyclyl, heterocyclylalkyl, heteroaryl or heteroarylalkyl.
- Cycloalkylalkyl refers to a radical of the formula -R,Rj where R n is an alkylene chain as defined above and Rj is a cycloalkyl radical as defined above.
- the alkylene chain and the cycloalkyl radical may be optionally substituted as defined above.
- fused refers to any ring structure described herein which is fused to an existing ring structure in the compounds of the invention
- the fused ring is a heterocyclyl ring or a heteioaryl ring
- any carbon atom on the existing ring structure which becomes part of the fused heterocyclyl ring or the fused heteroaryl ring may be replaced with a nitrogen atom
- Halo refers to bromo, chloro, fluoro or iodo.
- Haloalkyl refers to an alkyl radical, as defined above, that is substituted by one or more halo radicals, as defined above, e.g., trifluoromethyl, difluororaethyl, trichloromethyl, 2,2,2-trifluoroethyl, l-fluoioinethyl-2-fluoroethyl, 3-bromo-2- fluoropropyl, 1 -bromomethyl-2-bromoethyl, and the like.
- the alkyl part of the haloalkyl radical may be optionally substituted as defined above for an alkyl group.
- Heteiocyclyl refers to a stable 3- to 18-membered non-aromatic ring radical which consists of carbon atoms and from ore to five heteroatoms selected from the group consisting of nitrogen, oxygen and sulfur.
- the heterocyclyl radical may be a monocyclic, bicyclic or tricyclic ring system, which may include fused or bridged ring systems; and the nitrogen, carbon or sulfur atoms in the het ⁇ rocyclyl radical may be optionally oxidized; the nitrogen atom may be optionally alkylated/substituted; and the heterocyclyl radical may be partially or fully saturated.
- aieh heterocyclyl radicals include, but are not limited to, dioxolanyl, decahydroisoquiriolyl, imidazolinyl, imidazolidinyl, isothiazolidinyl, lsomolidinyl,
- morpholinyl octahydroindolyl, octahydroisoindolyl, 2-oxopiperazinyl, 2-oxopiperidinyl, 2-oxopyrrolidirryl, oxazolidinyl, piperidinyl, piperazinyl, 4-piperidonyl, pyrrolidinyl, pyrazolidinyl, thiazolidi ⁇ yl, tetrahydrofuryl, trithianyl, tetrahydropyranyl, thiomorpholinyl, thiamorpholinyl, l-oxo-thiomorpholinyl, and 1,1-dioxc- thiomorpholinyl.
- heterocyclyl is meant to include heterocyclyl radicals as defined above which are optionally substituted by one or more substituents selected from the groupconsisting of alkyl, alkenyl, halo, haloalkyl, cyano, oxo, thioxo, aryl, aralkyl, cycloalkyl, cycloalkylallcyl, heterocyclyl. lieterocyclylalkyl.
- heteroaryl, heteroarylalkyl -R IS -OR 14 , -R I S -0C(0)-R", -R 15 -N(R 14 ) 2 , -R I5 -C(O)R U , -R.
- each R 14 is independently hydrogen, alkyl, haloalkyl, cyclcalkyl, cycloalkylalkyl, aryl, aialkyl, heterocycly], heterocyclylalkyl, heteroaryl or heteroarylalkyl; each R 15 is independently a direct bond or a straight or branched alkylene or alkenylene chain; and each R 16 is alkyl, haloalkyl, cyclcalkyl, cycloalkylallcyl, aryl, aralkyl, heterocyclyl, heterocyclylalkyl, heteroaryl or heteroarylalkyl.
- Heterocyclylalkyl refers to a radical of the formula -R a R c where R 3 is an alkylene chain as defined above and R* is a heterocyclyl radical as defined above, and if the heterocycly! is a nitrogen-containing heteiocyclyl, the heterocyclyl may be attached to the alkyl radical at the nitrogen atom.
- the alkyl part of the heterocyclylalkyl radical may be optionally substituted as defined above for an alkyl group.
- the heterocyclyl part of the heierocyclylalkyl radical may be optionally substituted as defined above for a heterocyclyl group.
- Heteroaryl refers to a 5- to 18-membered aromatic ring radical which consists of carbon atoms and from one to five heteroatoms selected from the group consisting of nitrogen, oxygen and sulfur.
- the heteroaryl radical may be a monocyclic, bicyclic or tricyclic ring system, which may include fused cr bridged ring systems; and the nitrogen, carbon or sulfur stems in the heteroaryl radical may be optionally oxidized; the nitrogen atom may be optionally alkylated/substituted Examples include, but are rot limited to, az ⁇ pi ⁇ yl, acridiriyl, benzimidazolyl, benzthiazolyl,
- Heteroarylalkyl refers to a radical of the formula -R 11 R f where R a is an alkylene chain as defined above and R f is a heteroaryl radical as defined above TTie heteroaryl part of the hetercarylallcyl radical may be optionally substituted as defined alove for a heteroaryl group.
- the alkyl part of the heterD-rylalkyl radical may be optionally substituted as defined above for an alkyl group
- Hydroxyalkyl refers to a radical of the formula -R 8 -OH where R 1 is an alkylene chain as defined above The hydroxy group may be attached to the alkyl radical on any carbon within the alkyl radical. The alkyl part of the hydroxyalkyl group may be optionally substituted as defined above for an alkyl group.
- Trihaloalkoxy refers to a radical of ttie formula -0R e where R 8 is a hsloalkyl group as defined above where three
- halo are substituted on an alky 1.
- the trihaloalkyl part of the trihaloalkoxy group may be optionally substituted as defined above for a tialoalkyl group.
- a multi-ring structure refers to a multicyclic ring system comprised of two to four rings wheiein the rings are independently selected from cycloalkyl, aryl, heterocyclyl or rieteroaryl as defined above
- Each cycloalkyl may be optionally substituted as defined above for a cycloalkyl group.
- Each aryl may be optionally substituted as defined above for an aryl group.
- Each heterocyclyl may be optionally substituted as defined above for a heterocyclyl group.
- Each heteroaryl may be optionally substituted as defined above for a heteroaryl sroup.
- the rings may be attached to each other through direct bonds or some or all of the rings may be fused to each other.
- prodrug is meant to indicate a compound that may be converted under physiological conditions or by solvolysis to a biologically active compound of the invention.
- prodrug refers to a metabolic precursor of a compound of the invention that is pharmaceutically acceptable.
- a prodrug may be inactive when administered to a subject in need thereof, but is converted in vivo to an active compound of the invention.
- Prodrugs are typically rapidly transformed m vivo to yield the parent compound of the invention, for example, by hydrolysis in blood or conversion in the gut or liver.
- the prodrug compound often offers advantages of solubility, tissue compatibility or delayed release in a mammalian organism (see, Bundgard, H., Design of Prodrugs (1985), pp.
- prodrugs are provided in Higuchi, T., et of., "Pro-drugs as Novel Delivery Systems," A.C.S. Symposium Series, Vol. 14, and in Bioreversible Carriers in Drug Design, ed. Edward B. Roche, Anglican Pharmaceutical Association arid Pergamon Press, 1987, both of which are incorporated in full by reference herein.
- prodrug is also meant to include any covalently bonded carriers which release the active compound of the invention in vivo when such prodrug is administered to a mammalian subject.
- Prodrugs of a compound of the invention may be prepared by modifying functional groups present in the compound of the invention in such a way that the modifications are cleaved, either in routine manipulation or in vivo, to the parent compound of the invention.
- Prodrugs include compounds of the invention wherein a hydroxy, amino or mcrcapto or acid group is bonded to any group that, when the prodrug
- prodrugs include, but are not limited to, acetate, formate and benzoate derivatives of alcohol or amides of amine functional groups in the compounds of the invention and the like.
- Solid compound and “stable structure” are meant to indicate a compound that is sufficiently robust to survive isolation to a useful degree of purity from areaction mixture, and formulation into an efficacious therapeutic agent.
- “Mammal” includes humans and domestic animals, such as cats, dogs, swine, cattle, sheep, goats, horses, rabbits, and the like.
- Optional or “optionally” means that the subsequently described event of circumstances may or may not occur, and tliat the description includes instances where said event or circumstance occurs and instances in which it does not.
- optionally substituted aryl means that the aryl radical may or may not be substituted and that the description includes both substituted aryl radicals and aryl radicals having no substitution
- “Pharmaceutically acceptable carrier, diluent or excipient” includes without limitation any adjuvant, carrier, excipient, glidant, sweetening agent, diluent, preservative, dye/colorant, flavor enhancer, surfactant, wetting agent, dispersing agent, suspending agent, stabilizer, isotonic agent, solvent, or emulsifier which has been approved by the United States Food and Drug Administration as being acceptable for use in humans or domestic animals.
- “Pharmaceutically acceptable salt” includes both acid and base addition salts
- “Pharmaceutically acceptable acid addition salt” refers to those salts which retain the biological effectiveness and properties cf the free bases, which are not biologically or otherwise undesirable, and which are formed with inorganic acids such as, but not limited to, hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid and the like, and organic acids such as, but not limited to, acetic acid, 2,2-dichloroacetic acid, adipic acid, alginic acid, ascorbic acid, aspartic acid, benzenesulfonic acid, benzoic acid, 4-acetamidcbenzoic acid, camphoric acid, camphor-10-sulfonic acid, capik acid, caproic acid, caprylic acid, carbonic acid, cin ⁇ amic acid, citric acid, cyclamic acid,
- dodecylsulfiiric acid ethane-l,2-disulfonic acid, ethanesulfonic acid, 2- hydroxyeihanesulfonlc acid, formic acid, njmarfc acid, galactaric acid, gent ⁇ sic acid, glu ⁇ hcptonic acid, gluconic acid, glucuronic acid, glutamic acid, glutatic acid, 2-oxo- glutaric acid, glycerophosphorirc acid, glycolic acid, hippuiic acid, isobutyric acid, lactic acid, lactobionic acid, lauric acid, maleic acid, malic acid, malonic acid, mandelic acid, methanesulfonic acid, mucic acid, naphthalene-], 5-disulfonic acid, naphthalene-2- sulfonic acid, l-hjdroxy-2-naphthoic acid, nicotinic acid, oleic acid
- “Pharmaceutically acceptable base addition salt” refers to those salts which retain the biological effectiveness and properties cf the free acids, which are not biologically or otherwise undesirable. These salts are prepared from addition of an inorganic base or an organic base to the free acid. Salts derived from inorganic bases include, but are not limited to, the sodium, potassium, lithium, ammonium, calcium, magnesium, iron, zinc, copper, manganese, aluminum salts and the like. Preferred inorganic salts are the ammonium, sodium, potassium, calcium, and magnesium salts.
- Salts derived from organic bases include, but are not limited to, salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines and basic ion exchange resins, such as ammonia, isopropylamine, trimethylamine, diethylamine,triethylamine, tripropylamirie, diethanolamine, ethanolamine, deanol, 2- dimethylaininoethanol, 2-diethylaminoethait ⁇ l, dicyclohexylamine, lysine, arginine, histidine, caffeine, procaine, hydrabami ⁇ e, choline, betaine, benethamine, benzathine, ethylenediamine, glucosamine, methylglucamine, theobromine, triethanolamine, tromethamine, purines, piperazine, piperidine, JV-ethylpiperidine, polyamine resins and the like. Particularly
- solvate refers to an aggregate that comprises one or more molecules of a compound of the invention with one or more molecules of solvent.
- solvent may be water, in which case the solv ⁇ te may be a hydrate.
- the solvent may be an organic solvent.
- the compounds of the present invention may exist as a hydrate, including a monohydrate, dihydrate, hemihydrate, sesquihydrate, trihydrate, tetrahydrate and the like, as well as the corresponding solvated forms.
- the compound of the invention may be true solvates, while in other cases, the compound of the invention may merely retain adventitious water or he a mixture of water plus some adventitious solvent.
- a “pharmaceutical composition” refers to a formulation of a compound of the invention and a medium generally accepted in the art for the delivery of the biologically active compound to mammals, e.g., hutnatis.
- a medium includes all pharmaceutically acceptable carriers, diluents orexcipients thereof.
- “Therapeutically effective amount” refers to that amount of a compound of the invention which, when administered to a mammal, preferably a human, is sufficient to effect treatment, as defined below, of an SCO-mediated disease or condition in the mammal, preferably a human.
- the amount of a compound of the invention which constitutes s. "therapeutically effective amount” will vary depending on the compound, the condition and its severity, and the age and body weight of the mammal to be treated, but can be determined routinely by one of ordinary skill in the art having regard to his own knowledge and to this disclosure.
- Treating covers the treatment of the disease or condition of interest in a mammal, preferably a human, having the disease or disorder of interest, and includes: (i) preventing the disease or condition from occurring in a mammal, in particular, when such mammal is predisposed to the condition but has not yet been diagnosed as having it; (H) inhibiting the disease or condition, i.e., airesting its development; (iii) relieving the disease or condition, i.e., causing regression of the disease or condition; or (iv) relieving the symptoms resulting from the disease or condition, i.e., relieving the symptoms without addressing the underlying disease or condition.
- the terms “disease” and “condition” may be used interchangeably or may be different in that the particular malady or condition may not have aUnown causative agent (so that etiology has not yet been worked out) and it is therefore not yet
- the compounds of the invention, or their pharmaceutically acceptable salts may contain one 01 more asymmetric centers and may thus give rise to enantiomers, diastereomers, and other stereo isomeric forms that may be defined, in terms of absolute stereochemistry, as [R)- or [S)- or, as (D)- or (L)- for amino acids.
- the present invention is meant to include all such possible isomers, as well as their racemic and optically pure forms.
- Optically active (+) and (-), (R)- and ⁇ S ⁇ -, or (D)- and (L)- isomers may be prepared using chiral synthon ⁇ or ch ⁇ ral reagents, or resolved using conventional techniques, such as HPLC using a chiral column
- the compounds described herein contain olef ⁇ nic double bonds or other centers of geometric asymmetry, and unless specified otherwise, it is intended that the compounds include both E and Z geometric isomers.
- all tautomeric Forms are also intended to be included
- stereoisomer refers to a compound made up of the same atoms bonded by the same bonds but having different three-dimensional structures, which are not interchangeable.
- the present invention contemplates various stereoisomers and mixtures thereof and includes “enantiomers”, which refers to two stereoisomers whose molecules are nonsuperimposeable mirror images of one another.
- a ' tautorner refers to a proton shift from one atom ⁇ f a molecule tc another atom of the same molecule.
- the present invention includes tautomers of any said compounds.
- One embodiment of the invention is the compounds of Formula -(I):
- V is selected from -N(R 5 )C(O)-, -C(O)N(R 3 )-, -OC(O)N(R 5 )-, -N(R 5 )C(O)O-,
- W is selected from -N(R 5 )C(0)-, -C(O)N(R 5 )-, -OC(O)N(R 5 )-, -N(R 5 JC(O)O-,
- X is selected from C(H) or N;
- Y is selected from S, O, N(H) or N(CHj); p is O, 1, 2, or 3; t is 1 or 2;
- R 1 is selected from the group consisting of hydrogen, alkyl, alke ⁇ yl, alkyryl, alkoxy, hydroxyalkyl, alkoxyalkyl, cycloalkyi, cycloalkylalkyl, aryl, aralkyl, heterocyclyl, hctcrocyclylalkyl, heteroaryl, and heteroarylalkyl, or R 1 is a multi-ring structure having 2 to 4 rings wherein the rings are independently selected from the group consisting of cycloalkyi, heterocyclyl, aryl and heteroaryl and where some or all of the rings may be fused to each other;
- R 2 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, alkoxy, hydro ⁇ yalkyl, alkoxyalkyl, cycloalkiyl, cycloalkylalkyl, aryl, haloalkyl, aralkyl, heterocyclyl, heterocyclylal
- K 3 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkoxy, hydroxyalkyl, alkoxyalkyl, cycloalkyl, cycloalkylalkyl, heterocyclyl, aryl, aralkyl, heteroaryl, halo, haloalkyl, haloalkoiyl, cyano and -N(R 5 > 2 ;
- R 4 is selected fiotn the group consisting of alkyl, hydroxyalkyl, cycloalkylalkyl, aralkyl, halo, haloalkyl, -OCF 3 , , -OC(H)F 3 , and cyano; or two adjacent R 4 groups, together with the carbon atoms to which they attached, may form a cycloalkyl, heterocyclyl, aryl or heteroaryl and the remaining R 4 groups, if present, are as
- R 5a is selected from the group consisting of hydrogen, alkyl, cycloalkylalkyl and cyano; a stereoisomer, enantiomer or tautomer thereof, a pharmaceutically acceptable salt thereof, a pharmaceutical composition thereof or a prodrug thereof.
- V is selected from -O- or a direct bond
- W is selected from -N(R 5 )C(0)-, -C(O)N(R 5 )-, -C(O)O- or a direct bond; p is 0, 1, 2, or 3;
- R 1 is selected from the group consisting of hydrogen, alkyl, aryl, aralkyl, heterocyclyl, neterocyclylalkyl, heteroaryl, and heteroarylalkyl;
- R 2 is selected from the group consisting of hydrogen, alkyl, alkynyl, hydroxyalkyl, alkoxy, alkoxyalkyl, cycloalkyl, cycloalkj/lalkyl, aryl, haloalkyl, aralkyl, heterocyclyl, heterocyclylalkyl, heteroaryl and heteroarylalkyl; or R 2 is a multi-ring structure having 2 to 4 rings wherein the rings are independently selected from the group consisting of cycloalkyl, heterocyclyl, aryl and heteroaryl and where some or all of the rings may be fused to each other;
- R 3 is selected from the group consisting of hydrogen and alky]
- R 4 is alkyl or haloalkyl
- R s is selacted from the group consisting of hydrogen or alkyl
- V is selected from -O- or a direct hold
- W is selected from -N(R 5 JC(O)-, -C(C)N(R S )-, -C(O)O- or a direct bond; p is O, I 1 2, or 3;
- R 1 is selected from the group consisting of hydrogen, alkyl, aryl, aralkyl, heterocycl yl, heterocyclylalkyl, heteroaryl, and heteroarylalkyl;
- R 2 is selected from the group consisting of hydrogen, alkyl, alkynyl, hydroxyalkyl,alkoxyalkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocyclyl, heterocyclylaflcyl, heteroaryl and heteroarylaHcyl; or R 3 is a multi-ring structure havlti£ 2 to 3 rings wherein the rings are independently selected from the group consisting of cycloalkyl, heterocydyl, aryl and heteroaryl and where some or all of the rings may be fused to each other;
- R 3 is selected from the group consisting of hydrogen and alkyl
- R 4 is alkyl or CF 3 ;
- R 5 is selected from the group consisting of hydrogen or alkyl.
- Another embodiment is a compound of Formula (I), wherein X is N and Y is N-CH 3 or NH;
- V is selected from -O- or a direct bond
- W is selected from -N(R s )C(0)-, -C(CI)N(R 5 )-, -OC(O)-, -C(O)O- oi a direct bond; p is O, 1, 2, or 3;
- R' isselected from the group consisting of hydrogen, alkyl, aryl, aralkyl, heterocycly], heterocyclylalkyl, heteroaryl, and heteroarylalkyl;
- R is selected from the group consisting of hydrogen, alkyl, alkynyl, hydroxyalkyl, alkoxy, alkoxyalkyl, cygloalkyl, cycloalkylalkyl, aryl, haloalkyl, aralkyl, heterocyclyl, heterocyclylalkyl, heteroaryl and heteroarylalkyl, or R 2 is a multi-ring structure having 2 to 4 rings wherein the rings are independently selected from the group consisting of cycloalkyl, heterocyclyl, aryl and heteroaryl and where some or all of the rings may be fused to each other;
- R ! Is selected from the group consisting of hydrogen and alkyl
- R' IS alkyl, halo and haloalkyl; or two adjacent R 4 groups, together with the carbon atoms to which they attached, may form a cycloalkyl, heterocyclyl, aryl or heteroaryl and the remaining R 4 groups, if present, are as described above;
- R 5 is selected from the group consisting of hydrogen or alkyl.
- Y is selected from S. O, N(H) or N(CHj):
- V is selected from -O- or a direct bond
- W is selected from -N(R 5 )C(0)-, -C(O)N(R 5 )-, -C(O)O- or a direct bond; p is 0, 1, 2, or 3;
- R 1 is selected from the group consisting of hydrogen, alkyl, aryl, aralkyl, alkoxy, heterocyclyl, heterocyclylalkyl, heteroaryl, and heteroarylalkyl;
- R 2 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, alkoxy, hydroxyalkyl, alkoxyalkyl, cycloalkyl, cycloalkylalkyl, aryl, haloallcyl, aralkyl, heterocyclyl, heterocyclylalkyl, heteroaryl and heteroarylalkyl; or R z is a multi-ring structure having 2 to 4 rings wherein the rings are independently selected from the group consisting of cycloalkyl, heterocyclyl, aryl and heteroaryl and where some or all of the rings may be fused to each other;
- R 3 is selected from the group consisting of hydrogen and alkyl
- R 4 is ⁇ lkyl or haloalkyl
- R s is selected from the group consisting of hydrogen or alkyl
- Another embodiment is a compound of Formula (1), wherein X is selected from CH or N;
- Y is selected from S, O, N(H) or N(CHj);
- V is selected from -O- or a direct bond
- W is selected from -N(R S )C(O)-, or -C(O)O-; p is 9, 1, 2, or 3;
- R 1 is selected from the group consisting of alkyl, aryl, aralkyl, heterocyclyl, heterocyclylalkyl, and heteroarylalkyl;
- R 2 is selected from the group consisting of hydrogen, alkyl, hydroxyalkyl, alkoxyalkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocyclyl, heterocyclylalkyl, heteroaryl and heteroarylalkyl; or R 1 is a multi-ring structure having 2 to 3 rings wherein the rings are independently selected from the group consisting of cycloalkyl, heterocyclyl, aryl and heteroaryl and where some or all of the rings may be fused to each other,
- R 3 is selected from the group consisting of hydrogen and alkyl
- R 1 is rialcalkl, or alkyl
- R 5 is selected from the group consisting of hydrogen or alkyl.
- Another embodiment is a compound of Formula (I), wherein X is selected from CH or N;
- Y is selected from S, O, N(H) or N(CHj);
- V is selected from -O- or a direct bond
- W is selected from -N(R S )C(O)-, or -C(O)O-; p is C, 1, 2, or 3;
- R' is selected from the group consisting of alkyl, aryl, aralkyl, heterocyclyl, heterocyclylalkyl, and heteroarylalkyl;
- R 2 is selected from the group consisting of hydrogen, alkyl, hydroiyalkyl, alkoxyalkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocyclyl, heterocyclylalkyl, heteroaryl and heteroarylalkyl; or R J is a multi-ring structure having 2 to 3 rings wherein the rings are independently selected from the group consisting of cycloalkyl, heterocyclyl, aryl and heteroaryl and where some or all of the rings may be fused to each other;
- R 3 is selected from the group consisting of hydrogen and alkyl
- R 4 ishal ⁇ alkyl, or alkyl
- R 5 is selected from the group consisting of hydrogen or alkyl.
- Another embodiment is a compound of Formula CI), wherein
- X is selected from CH or N;
- V is selected from S, O, NH or N-CH 3 ;
- V is selected from -O- or a direct bond
- W is selected from -N(R 5 )C(0)-, or -C(O)O-; p is 0, 1, 2, or 3;
- R' is selected from the group consisting of alkyl, aryl, aralkyl, heterocyclyl, heterocyclylalkyl, and heteroarylalkyl;
- R 2 is selected from the group consisting of hydrogen, alkyl, hydroxyalkyl, alkoxyalkyl, cycloalkyl, cyclDalkylalkyl, aryl, aralkyl, heterocyclyl, heterocyclylalkyl, heteroaryl and heteroarylalkyl; or R 2 is a multi-ring structure having 2 to 3 rings wherein the rings are independently selected from the group consisting of cycloalkyl, heterocyclyl, aryl and heteroaryl and where some or all of the rings may be fused to each other,
- R 3 is selected from the group consisting of hydrogen and alkyl
- R 4 is alkyl or haloalkyl
- R 5 is selected from the group consisting of hydrogen or alkyl
- Another embodiment is a compound of Formula (1), wherein
- V is selected from -O- or a direct bond
- W is selected from -N(R S )C(O)-, or -C(O)O-; p is O, I 1 2, or 3;
- R 1 is selected from the group consisting of alkyl, aryl, aralkyl, heterocyclyl, heterocyclylalkyl, and heteroarylalkyl;
- R J is selected from the group consisting of hydrogen, alkyl, hydroxyalkyl, alkoxyalkyl, cycloalkyl, cycloalkylalkyl, aryl, aralkyl, heterocyclyl, heterocyclylalkyl, heteroaryl and heteroarylalkyl; or R : is a multi-ring structure having 2 to 3 rings wherein the rings are independently selected from the group consisting of cycloalkyl, heterocyclyl, aryl and h ⁇ teroaryl and where some or all of the rings may be ftised to each other;
- R J is selected from the group consisting of hydrogen and alkyl
- R 4 is halcalkyl or alkyl
- R 5 is selected from the group consisting of hydrogen or alkyl.
- the methods of 1he invention are directed towards the treatment and/or prevention of diseases mediated by stearoyl-CoA desatiuase (SCD), especially human SCD (hSCD), preferably diseases related to dyslipidemiaand disorders of lipid metabolism, and especially a disease related to elevated plasma lipid levels, cardiovascular disease, diabetes, obesity, metabolic syndrome, dermatolog ⁇ cal disorders and the like by administering an effective amount of a compound of the invention.
- SCD stearoyl-CoA desatiuase
- hSCD human SCD
- diseases related to dyslipidemiaand disorders of lipid metabolism preferably diseases related to dyslipidemiaand disorders of lipid metabolism, and especially a disease related to elevated plasma lipid levels, cardiovascular disease, diabetes, obesity, metabolic syndrome, dermatolog ⁇ cal disorders and the like by administering an effective amount of a compound of the invention.
- the present invention also relates to pharmaceutical composition containing the compounds of tie Invention.
- the invention relates to 2 composition comprising compounds of the invention in a pharmaceutically acceptable carrier and in an amount effective to modulate triglyceride level or to treat diseases related to dyslipidemia and disorders of lipid metabolism, when administered to an animal, preferably a mammal, most preferably a human patient.
- the patient has an elevated lipid level, such as elevated triglycerides or cholesterol, before administration of said compound of the invention and the compound of the invention is present in an amount effective to reduce said lipid leveL
- the present invention relates to compounds, pharmaceutical compositions and methods of using the compounds and pharmaceutical compositions for the treatment and/or prevention of diseases mediated by stearoyl-CoA desaturase (SCD), especially human SCD (liSCD), preferably diseases related to dyslipidemia and disorders of lipid metabolism, and especially a disease related to elevated plasma lipid levels, especially cardiovascular disease, diabetes, obesity, metabolic syndrome, dermatologieal disorders and the like, by administering to a patient in need of such treatment an effective amount of an SCD modulating, especially inhibiting agent.
- SCD stearoyl-CoA desaturase
- liSCD human SCD
- diseases related to dyslipidemia and disorders of lipid metabolism preferably diseases related to dyslipidemia and disorders of lipid metabolism, and especially a disease related to elevated plasma lipid levels, especially cardiovascular disease, diabetes, obesity, metabolic syndrome, dermatologieal disorders and the like, by administering to a patient in need of such treatment an effective amount of an SCD modulating, especially inhibiting agent.
- the present invention provides a method for treating a patient for, or protecting a patient from developing, a disease related to dyslipidemia and/or a disorder oflipid metabolism, wherein lipid levels in an animal, especially a human being, are outside the normal range (i.e., abnormal lipid level, such as elevated plasma lipid levels),
- lipid-relaterj condition or disease is an SCD-mediated disease or condition, comprising administering to an animal, such as a mammal, especially a human patient, a therapeutically effective amount of a compound of the invention or a pharmaceutical composition comprising a compound of the invention wherein the compound modulates the activity of SCD, preferably human SCDl .
- the compounds of the invention modulate, preferably inhibit, the activity of human SCD enzymes, especially human SCDl.
- the general value of the compounds of the invention in modulating, especially inhibiting, the activity of SCD can be determined using the assay described below in Example 28.
- the general value of the compounds in treating disorders and diseases may be established in industry standard animal models for demonstrating the efficacy of compounds in treating obesity, diabetes or elevated triglyceride or cholesterol levels or for improving glucose tolerance.
- Such models include Zucker obese /a/fa rats (available from Harlan Sprague Dawley, Inc. (Indianapolis, Indiana)), or the Zucker diabetic fatty rat (ZDF/GmiCrl- ⁇ fe) (available from Charles River Laboratories (Montreal, Quebec)), and Sprague Dawley rate (Charles Rivers), as used in models for diet-induced obesity (Ohibaudi, L. etal., (2»C2), Obns. Res. Vol. 10, pp.956-963). Similar models have also been developed for mice.
- the compounds of the instant invention are inhibitors of delta- 0 de ⁇ aturaees and are useful for treating diseases and disorders in humans and other organisms, including all those human diseases and disorders which are the result of aberrant delta-9 desaturase biological activity or which may be amelicr-ted by modulation of delta-9 desaturase biological activity.
- an SCD-mediated disease or condition is defined as any disease or condition in which the activity of SCD is elevated and/or where inhibition of SCD activity can be demonstrated to bring about symptomatic improvements for the individual so treated.
- an SCD-mediated disease or condition includes
- dysl ⁇ pidemias including but not limited to disorders of serum levels of triglycerides, hypertriglyceridemia, VLDL, HDL, LDL, fatty acid Desaturation Index (e.g. the ratio of 18:1/18:0 fatty acids, or other fatty acids, as defined elsewhere herein), cholesterol, and total cholesterol, hypercholesterolemia, as well as cholesterol disorders (including disorders characterized by defective reverse cholesterol transport), familial combined hyperlipidemia, coronary artery disease, atherosclerosis, heart disease, cerebrovascular disease (including but not limited to stroke, ischemic stroke and transient ischemic attack (TIA)), peripheral vascular disease, and ischemic retinopathy.
- TIA transient ischemic attack
- An SCD-mediated disease or condition also includes metabolic syndrome (including bul not limited to dyslipidem ⁇ a, obesity and insulin resistance, hypertension, microalbuminem ⁇ a, hyperuricemia, and hypercoagulability), Syndrome X s diabetes, insulin resistance, decreased glucose tolerance, non -insulin-dependent diabetes mellitus, Type II diabetes, Type I diabetes, diabetic complications, body weight disorders (including bul not limited to obesity, overweight, cachexia and anorexia), weight loss, body mass index and leptin related diseases
- metabolic syndrome including bul not limited to dyslipidem ⁇ a, obesity and insulin resistance, hypertension, microalbuminem ⁇ a, hyperuricemia, and hypercoagulability
- Syndrome X s diabetes insulin resistance, decreased glucose tolerance, non -insulin-dependent diabetes mellitus, Type II diabetes, Type I diabetes, diabetic complications, body weight disorders (including bul not limited to obesity, overweight, cachexia and anorexia), weight loss, body mass index and leptin related diseases
- compounds of the invention will be used to treat diabetes me
- metabolic syndrome is a recognized clinical term used to describe a condition comprising combinations of Type Il diabetes, impaired glucose tolerance, insulin resistance, hypertension, obesity, increased abdominal girth, hypertriglyceridemia, low HDL, hyperuricemia, hypercoagulability and/or micro album ineinia.
- the American Heart Association has published guidelines for the diagnosis of metabolic syndrome, Grundy, S., et. aJ. f (3006) Cardiol. Rev. Vol. 13, No.6, pp. 322-327.
- An SCD-mediated disease or condition also includes fatty liver, hepatic steatosis, hepatitis, non-alcoholic hepatitis, non-alcoholic steatohepatitis (NASH) 5 alcoholic hepatitis, acute fatty liver, fatty liver of pregnancy, drug-induced hepatitis, erythrohepaiic protoporphyria, iron overload disorders, hereditary hemochromatosis, hepatic fibrosis, hepatic cirrhosis, hepatoma and conditions related thereto.
- NASH non-alcoholic steatohepatitis
- An SCD-mediated disease or condition also includes but is not limited to a disease or condition which is, or is related to primary hypertriglyceridemia, or
- hypertriglyceridemia secondary to another disorder or disease such as hyperlipoproteinemias, familial histiocytic reticulosis, lipoprotein lipase deficiency, apolipoprotein deficiency (such as ApoCII deficiency or ApoE deficiency), and the like, or hypertriglyceridemia of unknown or unspecified etiology.
- An SCD-ttiediated disease or condition also includes a disorder of polyunsaturated fatty acid (PUFA) disorder, ⁇ r a dermatoiogical disorder, including but not limited to eczema, acne, psoriasis, keloid scar formation or prevention, diseases related to production or secretions from mucous membranes, such as monounsaturated fasy acids, wax esters, and the like.
- PUFA polyunsaturated fatty acid
- ⁇ r a dermatoiogical disorder, including but not limited to eczema, acne, psoriasis, keloid scar formation or prevention, diseases related to production or secretions from mucous membranes, such as monounsaturated fasy acids, wax esters, and the like.
- the compounds of the invention inhibition of SCD acitivity can prevent or attenuate keloid scar formation by reduction of excessive sebum production that typically results in their Formation.
- An SCD-mediated disease or condition also includes inflammatica sinusitis, asthma, pancreatitis, osteoarthritis, rheumatoid arthritis, cystic fibrosis, and premenstrual syndrome.
- An SCD-mediated disease or condition also includes but is not limited to a disease or condition which is, or is related to cancer, neoplasia, malignancy, metastases, tumours (benign or malignant), carcinogenesis, hepatomas and the like.
- An SCD-mediated disease or condition also includes a condition where increasing lean body mass or lean muscle mass is desired, such as is desirable in enhancing performance through muscle building.
- Myopathies and lipid myopathies such as carnitine palmitoyltransferase deficiency (CPT I or CPT II) are also included herein.
- CPT I or CPT II carnitine palmitoyltransferase deficiency
- bovine, porcine or avian domestic animals or any other animal to reduce triglyceride production andJ ⁇ r provide leaner meat products and/or healthier animals.
- An SCD-mediated disease or condition also includes a disease or condition that is, or is related to, neurological diseases, psychiatric disorders, multiple sclerosis, immune disorders and eye diseases, including but not limited to, disorders characterized by excessive or inappropriate lipid production by meiobium glands..
- An SCD-mediated disease or condition also includes a disease or condition which is, or is related to, viral diseases or infections including but not limited to all positive strand RNA viiuses, c ⁇ ronaviruses, SARS virus, SARS-associated corona ⁇ irus, Togavlruses, Picornaviruses, Coxsackievirus, Yellow Fever virus, Flaviviridae, ALPHAVIRUS (TOGAVIRIDAE) including Rubella virus, Eastern equine encephalitis virus, Western equine encephalitis virus, Venezuelan equine encephalitis virus, Sindbis virus, Semliki forest virus, Chikungunya viius, O'nyong'nyong virus, Ross river virus, Mayaro virus, Alphaviruses; ASTROVIRIDAE including Astrovirus, Human Astroviruses; CALICIVIRIDAE including Vesicular exanthema of swine virus, Norwalk virus, CaliciviruE, Bovine calici
- Treatable viral infections include those where the virus employs an RNA intermediate as part of the replicative cycle (hepatitis or HIV); additionally it can be a disease or infection caused by or linked to RNA negative strand viruses such as influenza and parainfluenza viruses.
- the compounds identified in the instant specification inhibit the desaturation of various fatty acids (such as the C 9 -C 10 desaturation of stearoyl-CoA), which is accomplished by delta-9 desaturases, such as stearoyl-CoA desaturase I (SCDl). As such these compounds inhibit the formation of v ⁇ rious fatty acids and downstream metabolites thereof. This may lead to an accumulation of 5t ⁇ aroyl-CoA or palmitoyl-CoA and olher upstream precursors of various fatty acids; which may possibly result in anegative feedback loop causing an overall change in fatty acid metabolism. Any cf these consequences may ultimately be responsible for the overall therapeutic benefit provided by these compounds.
- a successful SCD inhibitory therapeutic agent will meet some or all of the following criteria.
- Oral availability should be at or above 20%
- Animal model efficacy is less than about 20 rng/Kg, 2 mg/Kg, 1 mg/Kg, or 0.5 mg/Kg and tile target human dose is between 10 and 250 mg/70 Kg, although doses outside of this range may be acceptable.
- mg/Kg means milligrams of compound per kilogram of body mass of the subject to whom it is being administered.
- the required dosage should preferably be no more than about once or twice a day or a! meal times.
- the therapeutic index (or ratio of toxic dose to therapeutic dose) should be greater than 10.
- the IC 50 is a measure of the amount of compound required to achieve 50% inhibition of SCD activity, over a specific time period, in an SCD biological activity assay. Any process for measuring the activity of SCD enzymes, preferably mouse or human SCD enzymes, may be utilized to assay the activity of the compounds useful in the methods of the invention in inhibiting said SCD activity.
- Compounds of the invention demonstrate an IC 50 ("Inhibitoty Concentration of 50%") in a 15 minute microsomal assay of preferably less than 10 ⁇ M, less than S ⁇ M, less than 2.5 ⁇ M, less than 1 ⁇ M, less than 750 nM, less than 50D nM, less than 250 nM, less than 100 nM, less than 50 ⁇ M, and most preferably less than 20 nM.
- IC 50 Inhibitoty Concentration of 50%
- SAR structure-activity relationship
- this is accomplished by administering said chemical agent to an animal afflicted withatriglyceride (TG)- or very low density lipoprotein (VLDL)-related disorder and subsequently detecting a change in plasma triglyceride level in said animal thereby identifying a therapeutic agent useful in treating a triglyceride (-T ( Jy. or very low density lipoprotein (VLDL)-related disorder.
- the animal may be a human, such as a human patient afflicted with such a disorder and in need of treatment of said disorder.
- said change in SCDl activity in said animal is a decrease in activity, preferably wherein said SCDl modulating agent d ⁇ es not substantially inhibit the biological activity of a delta-5 desaturase, delta-6 desaturase or fatty acid synthetase or other enzymes containing Fe at the active site.
- the model systems useful for compound evaluation may include, but are not
- liver microsomes such as from mice that have been maintained on a Mgh carbohydrate diet or from human donors, including persons suffering from obesity- Immortalized cell lines, such as HepG2 (from human liver), MCF-7 (ftom human breast cancer) and 3T3-L1 (from mouse adipocytes) may also be used.
- Primary cdl lines, such as mouse primary hepatocytes, are also useAil in testing the compounds of the invention.
- mice used as a source of primary hepatocyte cells may also be used wherein the mice have been maintained on a high carbohydrate diet to increase SCD activity in mirocrosomes and/or to elevate plasma triglyceride levels (i.e., the 1S:1/18:0 ratio); alternatively mice on anormal diet or mice with normal triglyceride levels may be used.
- Mouse models employing transgenic mice designed foi hypertriglyceridemia are also available. Ratbili and hamsters ate also useful as animal models, especially those expressing CETP (cholesterol ester transfer protein).
- Another suitable method for determining the in vivo efficacy of the compounds of the invention is to indirectly measure their impact on inhibition of SCD enzyme by measuring a subject's Desaturation Index after administration of the compound.
- “Desaturation Index” as employed in lhis specification means the ratio of the product over lhc substrate for the SCD enzyme as measured from a given tissue sample. This may be calculated using three different equations 18:ln-9/18:0 (oleic ⁇ cid over stearic acid); 16;ln-7/16:0 (palmitoleic acid over palmitic acid); and/or 16: ln-7 + 18:ln- 7/16:0 (measuring all reaction products of 16:0 desaturation over 16:0 substrate).
- Desatiiration Index is primarily measured in liver or plasma triglycerides, but may also be measured in other selected lipid fractions from a variety of tissues. Desaturation Index, generally sneaking, is a tool for plasma lipid proflling.
- a number of human diseases and disorders are the result of aberrant SCDI biological activity and may be ameliorated by modulation of SCDl biological activity using the therapeutic agents of the invention.
- Inhibition of SCD expression may also affect the fatty acid composition of membrane phospholipids, as well as production or levels of triglycerides and cholesterol esters.
- the fatty acid composition of phospholipids ultimately determines membrane fluidity, with a subsequent modulation of the activity of multiple enzymes present within the membrane, while the effects on the composition of triglycerides and cholesterol esters
- Another format can be used to measure the effect of SCD inhibition on sebaceous gland function.
- oral, intravenous or topical formulations of the SCD inhibitor are administered to a rodent for a period of 1 to 8 days.
- Skin samples are taken and prepared for histological assessment to determine sebaceous gland number, size, or lipid content
- a reduction of sebaceous gland size, number or function would indicate that the SCD inhibitor would have a beneficial impact on acne vulgaris, (Clark, S.B. et at "Pharmacological modulation of sebaceous gland activity: mechanisms and clinical applications", De/r ⁇ to/. Clin. (2007) Vol.25, ND 2, pp 137-46. Geiger, J.M , "Retinoids and sebaceous gland activity” Dermatology (1995), Vol. 191, No.4, pp 3C5-10).
- the present invention also relates to pharmaceutical composition containing the compounds of the invention disclosed herein.
- the present invention relates to a composition comprising compounds of the invention in a pharmaceutically acceptable carrier and in an amount effective to modulate triglyceride level or to treat diseases related to dyslipidemia and disorders of lipid metabolism, when administered to an animal, preferably a mammal, most preferably a human patient.
- the patient has an elevated lipid level, such as elevated triglycerides or
- cholesterol before administration of said compound of the invention and the compound of the invention is present in an amount effective to reduce said lipid level.
- compositions useful herein also contain a pharmaceutically acceptable carrier, including any suitable diluent or excipient, which includes any pharmaceutical agent that does not itself induce the production of antibodies harmful to the individual receiving the composition, and which may be administered without undue toxicity.
- Pharmaceutically acceptable carriers include, but are not limited to, liquids, such as water, saline, glycerol and ethanol, and the like.
- Therapeutic doses are generally identified through a dose ranging sludy in humans based on preliminary evidence derived frorn animal studies. Doses must be sufficient to result in a de&ited therapeutic benefit without causing unwanted ⁇ ide effects For the patient
- the preferred dosage range for an ⁇ nimal is 0.001 mg/Kg to 10,000 mg/Kg, including 0.5 mg/Kg, 1.0 mg/Kg, 2.0 mg/Kg 5,0 mg/Kg and 10 mg/Kg, though doses outside this range may be acceptable.
- the dosing schedule may be once or twice per day, although more often or less often may be satisfactory.
- the compounds ofthe invention can be used in in vitro or in vivo studies as exemplary agents for comparative purposes to find other compounds also useful in treatment of, or protection from, the various diseases disclosed herein.
- compositions according to the invention are those suitable for enteral, such as oral or rectal, transdermal, topical, and parenteral administration to mammals, including man, to inhibit stearo yl-CoA desaturase, and for the treatment of
- compositions comprise a therapeutically effective amount of a pharmacologically active compound of the instant invention, alone or In combination with one or more pharmaceutically acceptable carriers.
- the pharmacologically active compounds of the invention are useful in the manufacture of pharmaceutical compositions comprising a therapeutically effective amount thereof in conjunction or admixture with excipients or carriers suitable for either enteral or parenteral application.
- excipients or carriers suitable for either enteral or parenteral application.
- Such pharmaceutical compositions may comprise, for example, 1he active ingredient together with diluents (e.g., lactose, dextrose, sucrose, mannitol, sorbitol, cellulose and/or glycine), lubricants (e.g., silica, talcum, stearic acid, its magnesium or calcium salt and/or polyethvleneglycol), and for tablets also comprises binders (e.g , magnesium aluminum silicate, starch paste, gelatin, tragacanth, methylcellulose, sodium carboxymethylcellulose and/or polyvinylpyrrolidone) and disintegrants (eg-, starches, agar, alginie acid or its sodium salt) or effervescent mixtures and absorbanis, colorants, flavors and sweeteners.
- diluents e.g., lactose, dextrose, sucrose, mannitol, sorbitol, cellulose and/or gly
- the compounds may be in the form of injectable compositions, e.g. preferably aqueous isotonic solutions or suspensions, and suppositories, which can be advantageously prepared from fatty emulsions or suspensions.
- the compositions may be sterilized and/or contain adjuvants, such as preserving, stabilizing, wetting or emulsifying agents, solution promoters, salts for regulating the osmotic pressure and/or buffers. In addition, they may also contain other therapeutically valuable substances.
- the compositions may be prepared according to conventional mixing, granulating or coating methods, respectively, and contain about 0.1- 75%, preferably about 1-50%, of the active ingredient.
- transdermal devices are in the form of a bsntJage comprising a backing member, a reservoir containing the compound
- a rate controlling barrier to deliver the compound of the skin of the host at a controlled and pie-determined rate over a prolonged period of time, and means to secure the device to the skin.
- the compounds of the invention may be usefully combined with one or more other therapeutic agents for the treatment of SCD-mediated diseases and conditions
- the other therapeutic agent is selected from antidiabetics, hypolipidemic agents, anti-obesity agents, anti-hypertensive agents or inotropic agents.
- an additional aspect of the present invention concerns a pharmaceutical composition
- a pharmaceutical composition comprising a therapeutically effective amount of a compound of the invention in combination with one or more other therapeutic agents.
- the composition can be formulated to comprise a therapeutically effective amount of a compound of the invention as defined above, in combination with anothei therapeutic agent, each at an effective therapeutic dose as reported in the art.
- Such therapeutic agents may, for example, include insulin, insulin derivatives and mimetics; insulin secretogogues, such as the sulfonylureas, e.g., Glipizide, glyburide and Amaryl; insulinotropic sulfonylurea receptor ligands, such as meglitinides, e.g., nateglinide and repaglinide; PPAR ⁇ and/or PPARot (peroxisome proliferator-activated receptor) ligands such as MCC-555, MK767, L-165041, GW7282 or thiazolidinediones such as rosiglitazone, pioglitazone, troglttazone; insulin sensitizers, such as protein tyrosine phosphatase- IB (PTP-IB) inhibitors such as PTP-112; GSK3 (glycogen synthase kinase- 3) inhibitors such as SB
- coenzyme A HMG-CoA reductase inhibitors, e.g., lovastatin, p avastatin, simvastatin, pravastatin, cerivastatin, mevastatin, velostatin, fluvastat ⁇ n, dalva ⁇ tatin, atorvastatin, rosuvastatin, flmndostat ⁇ n and rivastatin, squalene synthase inhibitors or FXR (farnesoid X receptor) and LXR (liver X receptor) Ugands, cholestyramine, fibrates, nicotinic acid and aspirin; anti-obesity agents, such as orlisiat, anti-hypertensive agents, inotropic agents and hypolipidemic agents, e.g., loop diuretics, such as ethacrynic acid, furosemide and torsemide; angiotensin converting enzyme (ACE) inhibitor
- a compound of the present invention may be administered either simultaneously, before or after the other active ingredient, either separately by the same or different route of administration or together in the same pharmaceutical formulation.
- compositions as described above for production of a medicament for the treatment of SCD-mediated disease or conditions.
- compositions or combination as described above for the preparation of a medicament for the treatment of conditions associated with stearoyi-CoA desatruase activity.
- Suitable protecting groups include hydroxy, amino, mercapto and carboxylic add.
- Suitable protecting groups for hydroxy include trialkylsih/l or diarylalkylsilyl (e g., t-butyldimethylsilyl, t-butyldiphenylsilyl or trimethykilyl), tetrahydropyranyl. benzyl, and the like.
- Suitable protecting groups for amino, amidino and guanidino include t-butoxycarbonyl, ben ⁇ yloxycarbonyl, and the like.
- Suitable protecting groups for mercapto include -C(O)-R" (where R" is alkyl, aryl or aiylalkyl), p-methoxybenzyl, trityl and the like.
- Suitable protecting groups for ca ⁇ bo ⁇ ylic acid include alkyl, aryl or arylalkyl esters.
- the protecting group may also be a polymer re&in such as a Wang resin or a 2-chlorotrityl-cliloride resin
- starting components may be obtained from sources such as Sigma Aldricri, Lancaster Synthesis, Inc., Maybridge, Matrix Scientific, TCI, and Fluorochem USA, etc. or synthesized according to sources known to those skilled in the art (see, e.g., Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 5th edition (Wiley, December 2000)) or prepered as described in this invention.
- the compounds of Formula (I) of this invention can be synthesized following the general procedure as described it Reaction Scheme 1 where W is - N(R 5 JC(O)- and p, R 1 , R 2 , R 3 , R 4 , R ! , W , V, X and Y are defined as in the Specification unless specifically defined otherwise.
- R' is a protecting group.
- Compound (101) is coupled with compound (102) under metal catalyzed reaction conditions to generate compound (103) which undergoes a standard hydrolysis procedure known to one skilled in the art to generate the carboxylic acid (104). Coupling between compounds (1*4) and (105) under standard amide bond formation conditions known to th ⁇ one skilled in the art affords compounds of Formula (I) of the invention where W is - N(R 5 )C(O)-.
- the compounds of Formula (I) of this invention can be synthesized following the general procedure as described in Reaction Scheme 2 where W is - N(R 5 JC(O)-, ani p, X, Y, V, R 1 , R 2 , R 3 , R 4 ami R 5 are defined as in the Specification unless specifically defined otherwise.
- the starting compound (201) undergoes coupling reaction with amine (105) under standard amide bond formation conditions known to one skilled in the art Io afford compound (201).
- Compound (202) is then coupled with compound (102) under metal catalyzed reaction conditions to generate compounds of Formula (I) where W is - N(R 5 )C(O>.
- the compounds of Formula (I) of this invention can be synthesized following the general procedure as described in Reaction Scheme 3 where W is - N(R 5 )C(O>, V is -O- or direct bond and p, X, Y, R 1 , R 2 , R 3 , R 4 and R 5 are defined as in the Specification unless specifically defined otherwise. R' and R" are protecting groups.
- the compounds of Foiniula (I) of this invention can be synthesized following the general procedure as described in Reaction Scheme 4 where W is - N(R 5 )C(O)-, V is -N(H)C(O)- and p, X, Y, R 1 , R 2 , R 3 , R 4 and R 5 are defined as in the Specification unless specifically defined otherwise.
- R" is a protecting group.
- Compound (301) is coupled with compound (401) under metal catalyzed reaction conditions to generate compound (402) which undergoes a standard hydrolysis procedure known to one skilled in the art to generate the carboxylic acid (403). Coupling between compound (403) and amine (105) under standard amide bond formation conditions known to one skilled in the art affords the compound (404).
- Compound (404) is used as a key intermediate to generate compounds of Fo ⁇ nula (1) under the conditions of (a) reductive aminition or (b) amide bond formation.
- reaction mixture was stirred at ambient temperature for 1 hour, then diluted with chloroform (100 mL) and filtered through Celite. The filtrate was concentrated in vacuo. The residue was dissolved in a mixture of methanol/chloroform 1/10 (30 mL).
- TTie reaction mixture was stirred at ambient temperature for 5 hours and N,N-dimethylformarnide was removed in vacuo. The residue was dissolved in ethyl acetate (15 mL) and urashed with saturated aqueous sodium bicarbonate (2 x 5 mL) and brine (5 mL). The organic layer was dried over anhydrous
- the reaction mixture was kept stirring for 2 hours it ambient temperature, then another portion of benzaldehyde (0.10 mL, 0.98 mmol) and tricthylsilanc (0, 15 mL, 1.00 mmol) was added.
- the reaction mixture was ic «pt stirring for another 2 houis at ambient temperature.
- DMSO-A 8 167.9. 162.0. 151.6, 154.1, 150.2, 139.9, 132. «, 128.7, 127.7, 127.2, 123.6, 104.4, 98.0, 43.1, 17.5, MS (ES+) m/z 342.2 (M + 1).
- reaction mixture was heated at 100 0 C for 6 hours and then diluted with ethyl acetate (25 inL), washed with 14% aqueous ammonium hydroxide (2 x 7 mL) and brine (7 mL).
- ethyl acetate 25 inL
- 14% aqueous ammonium hydroxide 2 x 7 mL
- brine 7 mL
- the or ⁇ nic layer was dried over anhydrous sodium sulfate, filtered concentrated in vacuo to afford as a yellowish solid (0.45 g, 66%)- 'H NMROOO MHz.
- the reaction mixture was heated at reflux for 1 S hours, cooled to ambient temperature and ethyl acetate (30 mL) was added. The mixture was washed with saturated ammonium chloride (10 mL), dried over anhydrous sodium sulfate and filtered. The filtrate was concentrated m vacuo.
- dimethylfo ⁇ namide (8 mL) was added cesium carbonate (0.32 g, 0.99 mmol) and catalytic amount of w-tetrabutylammon jura iodide, followed by the addition of 2- (chloromethyl)-5-phenyl-l ,3,4-oxadiazole (0.15 g, 0.76 mmol).
- the reaction mixture was heated at 80 0 C for 20 hours and concentrated in vacuo, followed by the addition of dichloromethane (100 mL). The mixture was washed with water (70 mL). The organic layer was dried over anhydrous sodium sulfite and filtered. The solvent was concentrated in vacuo.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- General Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Diabetes (AREA)
- Epidemiology (AREA)
- Obesity (AREA)
- Dermatology (AREA)
- Hematology (AREA)
- Endocrinology (AREA)
- Heart & Thoracic Surgery (AREA)
- Child & Adolescent Psychology (AREA)
- Urology & Nephrology (AREA)
- Emergency Medicine (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Physical Education & Sports Medicine (AREA)
- Cardiology (AREA)
- Nutrition Science (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description

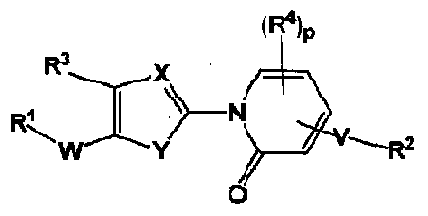













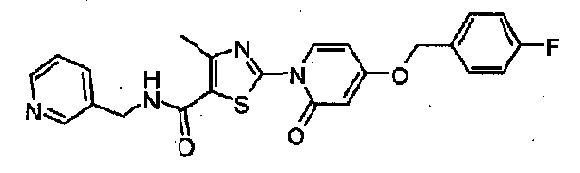

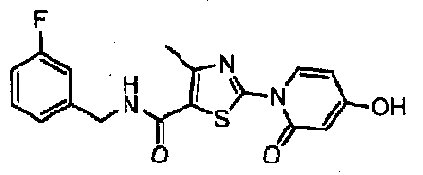















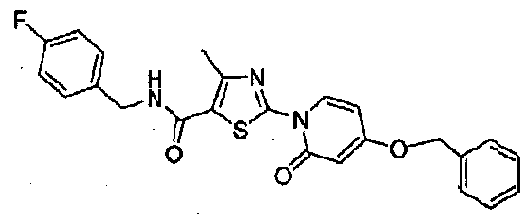

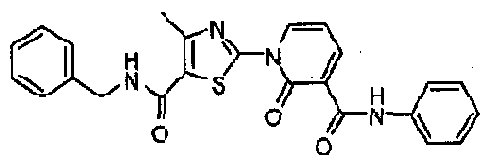



















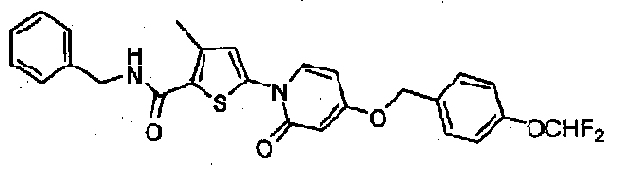








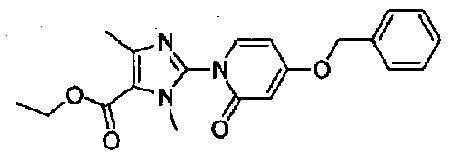










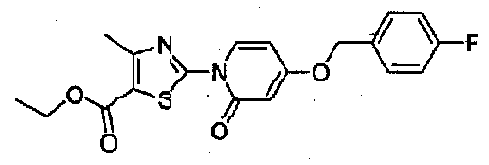

























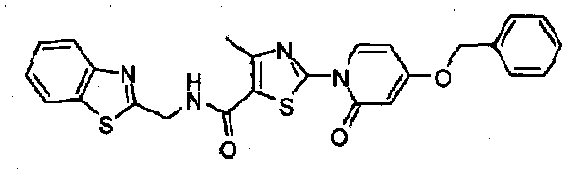









Claims


Priority Applications (14)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BRPI0712902A BRPI0712902A2 (en) | 2006-06-05 | 2007-06-04 | organic compounds |
| US12/303,490 US8501746B2 (en) | 2006-06-05 | 2007-06-04 | Organic compounds |
| AU2007256708A AU2007256708B2 (en) | 2006-06-05 | 2007-06-04 | Organic compounds |
| CN2007800204238A CN101460476B (en) | 2006-06-05 | 2007-06-04 | Organic compounds |
| AT07798049T ATE486862T1 (en) | 2006-06-05 | 2007-06-04 | ORGANIC COMPOUNDS |
| EP07798049A EP2029572B1 (en) | 2006-06-05 | 2007-06-04 | Organic compounds |
| MX2008015229A MX2008015229A (en) | 2006-06-05 | 2007-06-04 | Organic compounds. |
| DE602007010287T DE602007010287D1 (en) | 2006-06-05 | 2007-06-04 | ORGANIC CONNECTIONS |
| PL07798049T PL2029572T3 (en) | 2006-06-05 | 2007-06-04 | Organic compounds |
| KR1020087029648A KR101124070B1 (en) | 2006-06-05 | 2007-06-04 | Organic compounds |
| CA002653655A CA2653655A1 (en) | 2006-06-05 | 2007-06-04 | Organic compounds |
| JP2009514484A JP5043105B2 (en) | 2006-06-05 | 2007-06-04 | Organic compounds |
| RU2008151051/04A RU2491285C2 (en) | 2006-06-05 | 2007-06-04 | Organic compounds |
| US13/942,404 US20140024583A1 (en) | 2006-06-05 | 2013-07-15 | Organic compounds |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US81091506P | 2006-06-05 | 2006-06-05 | |
| US60/810,915 | 2006-06-05 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/303,490 A-371-Of-International US8501746B2 (en) | 2006-06-05 | 2007-06-04 | Organic compounds |
| US13/942,404 Division US20140024583A1 (en) | 2006-06-05 | 2013-07-15 | Organic compounds |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| WO2007143597A2 true WO2007143597A2 (en) | 2007-12-13 |
| WO2007143597A3 WO2007143597A3 (en) | 2008-04-10 |
| WO2007143597A9 WO2007143597A9 (en) | 2010-11-11 |
Family
ID=38802273
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/070293 WO2007143597A2 (en) | 2006-06-05 | 2007-06-04 | Organic compounds |
Country Status (20)
| Country | Link |
|---|---|
| US (2) | US8501746B2 (en) |
| EP (1) | EP2029572B1 (en) |
| JP (1) | JP5043105B2 (en) |
| KR (1) | KR101124070B1 (en) |
| CN (1) | CN101460476B (en) |
| AR (1) | AR061222A1 (en) |
| AT (1) | ATE486862T1 (en) |
| AU (1) | AU2007256708B2 (en) |
| BR (1) | BRPI0712902A2 (en) |
| CA (1) | CA2653655A1 (en) |
| CL (1) | CL2007001597A1 (en) |
| DE (1) | DE602007010287D1 (en) |
| ES (1) | ES2358297T3 (en) |
| MX (1) | MX2008015229A (en) |
| PE (1) | PE20080714A1 (en) |
| PL (1) | PL2029572T3 (en) |
| PT (1) | PT2029572E (en) |
| RU (1) | RU2491285C2 (en) |
| TW (1) | TW200815420A (en) |
| WO (1) | WO2007143597A2 (en) |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008128335A1 (en) * | 2007-04-20 | 2008-10-30 | Merck Frosst Canada Ltd. | Novel heteroaromatic compounds as inhibitors of stearoyl-coenzyme a delta-9 desaturase |
| KR20100117677A (en) * | 2008-02-20 | 2010-11-03 | 노파르티스 아게 | Heterocyclic inhibitors of stearoyl-coa desaturase |
| WO2011076725A1 (en) | 2009-12-21 | 2011-06-30 | Bayer Cropscience Ag | Thienylpyri (mi) dinylazole and their use for controlling phytopathogenic fungi |
| US8258160B2 (en) | 2006-12-20 | 2012-09-04 | Novartis Ag | SCD1 inhibitors triazole and tetrazole compounds |
| WO2013050437A1 (en) | 2011-10-06 | 2013-04-11 | Bayer Intellectual Property Gmbh | Heterocyclylpyri (mi) dinylpyrazole as fungicidals |
| WO2013056148A2 (en) | 2011-10-15 | 2013-04-18 | Genentech, Inc. | Methods of using scd1 antagonists |
| US9456998B2 (en) | 2012-05-22 | 2016-10-04 | Yissum Research Development Company Of The Hebrew University Of Jerusalem Ltd. | Selective inhibitors of undifferentiated cells |
| WO2018129403A1 (en) | 2017-01-06 | 2018-07-12 | Yumanity Therapeutics | Methods for the treatment of neurological disorders |
| WO2022167445A1 (en) * | 2021-02-02 | 2022-08-11 | Liminal Biosciences Limited | Gpr84 antagonists and uses thereof |
| WO2022167457A1 (en) * | 2021-02-02 | 2022-08-11 | Liminal Biosciences Limited | Gpr84 antagonists and uses thereof |
| US11873298B2 (en) | 2017-10-24 | 2024-01-16 | Janssen Pharmaceutica Nv | Compounds and uses thereof |
| US11970486B2 (en) | 2016-10-24 | 2024-04-30 | Janssen Pharmaceutica Nv | Compounds and uses thereof |
| US12098146B2 (en) | 2019-01-24 | 2024-09-24 | Janssen Pharmaceutica Nv | Compounds and uses thereof |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| RU2009109147A (en) * | 2006-08-15 | 2010-09-27 | Новартис АГ (CH) | ORGANIC COMPOUNDS |
| SI2178865T1 (en) | 2007-07-19 | 2015-11-30 | Lundbeck, H., A/S | 5-membered heterocyclic amides and related compounds |
| US9296719B2 (en) * | 2011-07-08 | 2016-03-29 | Indiana University Research And Technology Corporation | Compositions and methods for the treatment of norovirus infection |
| CN107074800B (en) * | 2014-01-14 | 2019-11-12 | 康内克斯生命科学私人有限公司 | The bicyclic heteroaryl compounds being substituted are as RXR agonist |
Citations (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001062954A2 (en) | 2000-02-24 | 2001-08-30 | Xenon Genetics, Inc. | Stearoyl-coa desaturase to identify triglyceride reducing therapeutic agents |
| WO2002026944A2 (en) | 2000-09-26 | 2002-04-04 | Xenon Genetics, Inc. | METHODS AND COMPOSITIONS EMPLOYING A NOVEL STEAROYL-CoA DESATURASE-hSCD5 |
| WO2005011653A2 (en) | 2003-07-30 | 2005-02-10 | Xenon Pharmaceuticals Inc. | Pyridazine derivatives and their use as therapeutic agents |
| WO2005011656A2 (en) | 2003-07-30 | 2005-02-10 | Xenon Pharmaceuticals Inc. | Pyridyl derivatives and their use as therapeutic agents |
| WO2005011655A2 (en) | 2003-07-30 | 2005-02-10 | Xenon Pharmaceuticals Inc. | Pyridazine derivatives and their use as therapeutic agents |
| WO2005011657A2 (en) | 2003-07-30 | 2005-02-10 | Xenon Pharmaceuticals Inc. | Piperazine derivatives and their use as therapeutic agents |
| WO2005011654A2 (en) | 2003-07-29 | 2005-02-10 | Xenon Pharmaceuticals Inc. | Pyridyl derivatives and their use as therapeutic agents |
| WO2006014168A1 (en) | 2004-07-06 | 2006-02-09 | Xenon Pharmaceuticals Inc. | Nicotinamide derivatives and their use as therapeutic agents |
| WO2006034279A1 (en) | 2004-09-20 | 2006-03-30 | Xenon Pharmaceuticals Inc. | Heterocyclic derivatives and their use as therapeutic agents |
| WO2006034338A1 (en) | 2004-09-20 | 2006-03-30 | Xenon Pharmaceuticals Inc. | Heterocyclic derivatives and their use as mediators of stearoyl-coa desaturase |
| WO2006034341A2 (en) | 2004-09-20 | 2006-03-30 | Xenon Pharmaceuticals Inc. | Pyridazine derivatives for inhibiting human stearoyl-coa-desaturase |
| WO2006034440A2 (en) | 2004-09-20 | 2006-03-30 | Xenon Pharmaceuticals Inc. | Heterocyclic derivatives and their use as stearoyl-coa desaturase inhibitors |
| WO2006034312A1 (en) | 2004-09-20 | 2006-03-30 | Xenon Pharmaceuticals Inc. | Bicyclic heterocyclic derivatives and their use as inhibitors of stearoyl-coa-desaturase (scd) |
| WO2006034446A2 (en) | 2004-09-20 | 2006-03-30 | Xenon Pharmaceuticals Inc. | Pyridine derivatives for inhibiting human stearoyl-coa-desaturase |
| WO2006034315A2 (en) | 2004-09-20 | 2006-03-30 | Xenon Pharmaceuticals Inc. | Heterocyclic derivatives for the treatment of diseases mediated by stearoyl-coa desaturase enzymes |
| WO2006034441A1 (en) | 2004-09-20 | 2006-03-30 | Xenon Pharmaceuticals Inc. | Heterocyclic derivatives and their use as stearoyl-coa desaturase inhibitors |
| WO2006130986A1 (en) | 2005-06-09 | 2006-12-14 | Merck Frosst Canada Ltd. | Azacyclohexane derivatives as inhibitors of stearoyl-coenzyme a delta-9 desaturase |
| WO2007009236A1 (en) | 2005-07-20 | 2007-01-25 | Merck Frosst Canada Ltd. | Heteroaromatic compounds as inhibitors of stearoyl-coenzyme a delta-9 desaturase |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3575991A (en) * | 1969-03-11 | 1971-04-20 | American Home Prod | 1-(4-aryl - 5 - carboxymethyl-2-thiazolyl)-1,6-dihydro -6 - oxonicotinic acids and esters thereof |
| US5716632A (en) * | 1989-11-22 | 1998-02-10 | Margolin; Solomon B. | Compositions and methods for reparation and prevention of fibrotic lesions |
| BR9406461A (en) * | 1993-05-07 | 1996-01-30 | Solomon Begelfor Margolin | Compositions and methods for the repair and prevention of fibrotic lesions |
| EP0813409B1 (en) * | 1995-03-03 | 2004-05-06 | MARGOLIN, Solomon B. | Treatment of cytokine growth factor caused disorders |
| US6114353A (en) * | 1995-03-03 | 2000-09-05 | Margolin; Solomon B. | Compositions and method for treatment of lymphomas, leukemias, and leiomyomas |
| WO1997010712A1 (en) * | 1995-09-19 | 1997-03-27 | Margolin Solomon B | Inhibition of tumor necrosis factor alpha |
| FR2754262B1 (en) * | 1996-10-08 | 1998-10-30 | Synthelabo | 1H-PYRIDO [3,4-B] INDOLE-4-CARBOXAMIDE DERIVATIVES, THEIR PREPARATION AND THEIR THERAPEUTIC APPLICATION |
| CN1395575A (en) * | 2000-01-20 | 2003-02-05 | 卫材株式会社 | Novel piperidine compounds and drugs containing the same |
| DE60028831D1 (en) * | 2000-02-21 | 2006-07-27 | Cymar Inc | COMPOSITIONS AND METHODS FOR TREATING EPILEPSY |
| EP1490064B1 (en) * | 2002-02-14 | 2009-11-18 | Pharmacia Corporation | Substituted pyridinones as modulators of p38 map kinase |
| IL163957A0 (en) * | 2002-03-14 | 2005-12-18 | Bayer Healthcare Ag | Monocyclic aroylpyridinones as antiinflammatory agents |
| NL1026826C2 (en) * | 2003-08-13 | 2007-01-04 | Pharmacia Corp | Substituted pyridinones. |
| EP1982978B1 (en) * | 2006-02-02 | 2014-08-27 | Kumiai Chemical Industry Co., Ltd. | Pyridone derivative and herbicide |
-
2007
- 2007-06-04 WO PCT/US2007/070293 patent/WO2007143597A2/en active Application Filing
- 2007-06-04 CA CA002653655A patent/CA2653655A1/en not_active Abandoned
- 2007-06-04 JP JP2009514484A patent/JP5043105B2/en not_active Expired - Fee Related
- 2007-06-04 TW TW096119972A patent/TW200815420A/en unknown
- 2007-06-04 PT PT07798049T patent/PT2029572E/en unknown
- 2007-06-04 DE DE602007010287T patent/DE602007010287D1/en active Active
- 2007-06-04 AU AU2007256708A patent/AU2007256708B2/en not_active Ceased
- 2007-06-04 KR KR1020087029648A patent/KR101124070B1/en not_active IP Right Cessation
- 2007-06-04 PE PE2007000690A patent/PE20080714A1/en not_active Application Discontinuation
- 2007-06-04 US US12/303,490 patent/US8501746B2/en not_active Expired - Fee Related
- 2007-06-04 AR ARP070102401A patent/AR061222A1/en unknown
- 2007-06-04 EP EP07798049A patent/EP2029572B1/en not_active Not-in-force
- 2007-06-04 MX MX2008015229A patent/MX2008015229A/en active IP Right Grant
- 2007-06-04 BR BRPI0712902A patent/BRPI0712902A2/en not_active IP Right Cessation
- 2007-06-04 RU RU2008151051/04A patent/RU2491285C2/en not_active IP Right Cessation
- 2007-06-04 PL PL07798049T patent/PL2029572T3/en unknown
- 2007-06-04 CN CN2007800204238A patent/CN101460476B/en not_active Expired - Fee Related
- 2007-06-04 CL CL200701597A patent/CL2007001597A1/en unknown
- 2007-06-04 AT AT07798049T patent/ATE486862T1/en active
- 2007-06-04 ES ES07798049T patent/ES2358297T3/en active Active
-
2013
- 2013-07-15 US US13/942,404 patent/US20140024583A1/en not_active Abandoned
Patent Citations (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001062954A2 (en) | 2000-02-24 | 2001-08-30 | Xenon Genetics, Inc. | Stearoyl-coa desaturase to identify triglyceride reducing therapeutic agents |
| WO2002026944A2 (en) | 2000-09-26 | 2002-04-04 | Xenon Genetics, Inc. | METHODS AND COMPOSITIONS EMPLOYING A NOVEL STEAROYL-CoA DESATURASE-hSCD5 |
| WO2005011654A2 (en) | 2003-07-29 | 2005-02-10 | Xenon Pharmaceuticals Inc. | Pyridyl derivatives and their use as therapeutic agents |
| WO2005011653A2 (en) | 2003-07-30 | 2005-02-10 | Xenon Pharmaceuticals Inc. | Pyridazine derivatives and their use as therapeutic agents |
| WO2005011656A2 (en) | 2003-07-30 | 2005-02-10 | Xenon Pharmaceuticals Inc. | Pyridyl derivatives and their use as therapeutic agents |
| WO2005011655A2 (en) | 2003-07-30 | 2005-02-10 | Xenon Pharmaceuticals Inc. | Pyridazine derivatives and their use as therapeutic agents |
| WO2005011657A2 (en) | 2003-07-30 | 2005-02-10 | Xenon Pharmaceuticals Inc. | Piperazine derivatives and their use as therapeutic agents |
| WO2006014168A1 (en) | 2004-07-06 | 2006-02-09 | Xenon Pharmaceuticals Inc. | Nicotinamide derivatives and their use as therapeutic agents |
| WO2006034279A1 (en) | 2004-09-20 | 2006-03-30 | Xenon Pharmaceuticals Inc. | Heterocyclic derivatives and their use as therapeutic agents |
| WO2006034338A1 (en) | 2004-09-20 | 2006-03-30 | Xenon Pharmaceuticals Inc. | Heterocyclic derivatives and their use as mediators of stearoyl-coa desaturase |
| WO2006034341A2 (en) | 2004-09-20 | 2006-03-30 | Xenon Pharmaceuticals Inc. | Pyridazine derivatives for inhibiting human stearoyl-coa-desaturase |
| WO2006034440A2 (en) | 2004-09-20 | 2006-03-30 | Xenon Pharmaceuticals Inc. | Heterocyclic derivatives and their use as stearoyl-coa desaturase inhibitors |
| WO2006034312A1 (en) | 2004-09-20 | 2006-03-30 | Xenon Pharmaceuticals Inc. | Bicyclic heterocyclic derivatives and their use as inhibitors of stearoyl-coa-desaturase (scd) |
| WO2006034446A2 (en) | 2004-09-20 | 2006-03-30 | Xenon Pharmaceuticals Inc. | Pyridine derivatives for inhibiting human stearoyl-coa-desaturase |
| WO2006034315A2 (en) | 2004-09-20 | 2006-03-30 | Xenon Pharmaceuticals Inc. | Heterocyclic derivatives for the treatment of diseases mediated by stearoyl-coa desaturase enzymes |
| WO2006034441A1 (en) | 2004-09-20 | 2006-03-30 | Xenon Pharmaceuticals Inc. | Heterocyclic derivatives and their use as stearoyl-coa desaturase inhibitors |
| WO2006130986A1 (en) | 2005-06-09 | 2006-12-14 | Merck Frosst Canada Ltd. | Azacyclohexane derivatives as inhibitors of stearoyl-coenzyme a delta-9 desaturase |
| WO2007009236A1 (en) | 2005-07-20 | 2007-01-25 | Merck Frosst Canada Ltd. | Heteroaromatic compounds as inhibitors of stearoyl-coenzyme a delta-9 desaturase |
Non-Patent Citations (25)
| Title |
|---|
| "Advanced Organic Chemistry: Reactions, Mechanisms, and Structure", 2000, WILEY |
| "Bioreversible Carriers in Drug Design", 1987, ANGLICAN PHARMACEUTICAL ASSOCIATION ARID PERGAMON PRESS |
| "REMINGTON'S PHARMACEUTICAL SCIENCES", MACK PUB. CO. |
| ATTIE, A.D.: "Relationship between stearoyl-CoA desaturase activity and plasma triglycerides in human and mouse hypertriglyceridemia", J. LIPID RES., vol. 43, no. 11, 2002, pages 1899 - 907 |
| BINCZEK, E.: "Obesity resistance of the stearoyl-CoA desaturase-deficient mouse results from disruption of the epidermal lipid barrier and adaptive thermoregulation", BIOL. CHEM., vol. 388, no. 4, 2007, pages 405 - 18 |
| BUNDGARD, H.: "Design of nrodrugs", 1985, ELSEVIER, pages: 7 - 9,21-24 |
| CLARK, S.B.: "Pharmacological modulation of sebaceous gland activity: mechanisms and clinical applications", DERMATOL. CLIN., vol. 25, no. 2, 2007, pages 137 - 46 |
| COHEN, P. ET AL.: "Role for stearoyl-CoA desaturase-1 in leptin mediated weight loss", SCIENCE, vol. 297, no. 5579, 2002, pages 240 - 3 |
| DE ANTUENO, R., LIPIDS, vol. 28, no. 4, 1993, pages 285 - 290 |
| DOBRZYN A.; DOBRZYN P.: "Stearoyl-CoA desaturase - a new player in skeletal muscle metabolism regulation", J PHYSIOL PHARMACOL., vol. 57, no. 10, 2006, pages 31 - 42 |
| GEIGER, J.M.: "Retinoids and sebaceous gland activity", DERMATOLOGY, vol. 191, no. 4, 1997, pages 305 - 10 |
| GHIBAUDI, L. ET AL., OBES. RES., vol. 10, 2002, pages 956 - 963 |
| GREENE, T.W.; P.G.M. WUTS: "Protective Groups in Organic Synthesis", 2006, WILEY |
| GRUNDY, S., CARDIOL. REV., vol. 13, no. 6, 2006, pages 322 - 327 |
| GUTIERREZ-JUAREZ, R. ET AL.: "Critical role of stearoyl CoA desaturase-1 (SCD1) in the onset of diet-induced hepatic insulin resistance", J. CLIN. INVEST., vol. 116, no. 6, 2006, pages 1686 - 95 |
| HIGUCHI, T. ET AL.: "Pro-drugs as Novel Delivery Systems", A.C.S. SYMPOSIUM SERIES, vol. 14, 1987 |
| JEFFCOAT, R. ET AL., EUR..L BIOCHEM., vol. 101, no. 2, 1979, pages 439 - 445 |
| MIYAZAKI, M.: "Targeted Disruption ofStearoyl-CoA Desaturasel Gene in Mice Causes Atrophy of Sebaceous and Meibomian Glands and Depletion of Wax Esters in the Eyelid", J. NUTR., vol. 131, 2001, pages 2260 - 68 |
| NTAMBI, J. M.: "Loss of stearoyl-CoA desaturase-1 function protects mice against adiposity", PROC. NATL. ACAD. SCI. U.S.A, vol. 99, no. 7, 2002, pages 11482 - 6 |
| PATEL MONA, EXPERT OPIN INVESTIG DRUGS., vol. 12, no. 4, 2003, pages 623 - 33 |
| SAMPATH, H.: "Stearoyl-CoA Desaturase-1 mediates the pro-lipogenic effects of dietary saturated fat", J. BIOL. CHEM., vol. 282, no. 4, 2007, pages 2483 - 93 |
| SHANKLIN J.; SUMMERVILLE C., PROC. NATL. ACAD. SCI. USA, vol. 88, 1991, pages 2510 - 2514 |
| XU H. ET AL.: "Hepatic knockdown of stearoyl-CoA desaturase 1 via RNA interference in obese mice decreases lipid content and change sfatty acid composition", FRONT. BIOSCI., vol. 12, 2007, pages 3781 - 94 |
| ZHAO ET AL., BIORGANIC AND MEDICINAL CHEMISTRY LETTERS, 2007 |
| ZHENG Y. ET AL.: "SCD1 is expressed in sebaceous glands and is disrupted in the asebia mouse", NAT. GENET., vol. 23, 1999, pages 268 - 270 |
Cited By (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8258160B2 (en) | 2006-12-20 | 2012-09-04 | Novartis Ag | SCD1 inhibitors triazole and tetrazole compounds |
| WO2008128335A1 (en) * | 2007-04-20 | 2008-10-30 | Merck Frosst Canada Ltd. | Novel heteroaromatic compounds as inhibitors of stearoyl-coenzyme a delta-9 desaturase |
| KR20100117677A (en) * | 2008-02-20 | 2010-11-03 | 노파르티스 아게 | Heterocyclic inhibitors of stearoyl-coa desaturase |
| KR101584826B1 (en) | 2008-02-20 | 2016-01-18 | 노파르티스 아게 | Heterocyclic inhibitors of stearoyl-coa desaturase |
| US8049016B2 (en) | 2008-02-20 | 2011-11-01 | Novartis Ag | Thiazole derivatives which inhibit stearoyl-CoA desaturase enzymes |
| US8318949B2 (en) | 2008-02-20 | 2012-11-27 | Novartis Ag | Organic compounds |
| US8685974B2 (en) | 2009-12-21 | 2014-04-01 | Bayer Cropscience Ag | Thienylpyri(mi)dinylazole |
| WO2011076725A1 (en) | 2009-12-21 | 2011-06-30 | Bayer Cropscience Ag | Thienylpyri (mi) dinylazole and their use for controlling phytopathogenic fungi |
| WO2013050437A1 (en) | 2011-10-06 | 2013-04-11 | Bayer Intellectual Property Gmbh | Heterocyclylpyri (mi) dinylpyrazole as fungicidals |
| WO2013056148A2 (en) | 2011-10-15 | 2013-04-18 | Genentech, Inc. | Methods of using scd1 antagonists |
| US9358250B2 (en) | 2011-10-15 | 2016-06-07 | Genentech, Inc. | Methods of using SCD1 antagonists |
| US9456998B2 (en) | 2012-05-22 | 2016-10-04 | Yissum Research Development Company Of The Hebrew University Of Jerusalem Ltd. | Selective inhibitors of undifferentiated cells |
| US11970486B2 (en) | 2016-10-24 | 2024-04-30 | Janssen Pharmaceutica Nv | Compounds and uses thereof |
| WO2018129403A1 (en) | 2017-01-06 | 2018-07-12 | Yumanity Therapeutics | Methods for the treatment of neurological disorders |
| US10973810B2 (en) | 2017-01-06 | 2021-04-13 | Yumanity Therapeutics, Inc. | Methods for the treatment of neurological disorders |
| US11873298B2 (en) | 2017-10-24 | 2024-01-16 | Janssen Pharmaceutica Nv | Compounds and uses thereof |
| US12098146B2 (en) | 2019-01-24 | 2024-09-24 | Janssen Pharmaceutica Nv | Compounds and uses thereof |
| WO2022167445A1 (en) * | 2021-02-02 | 2022-08-11 | Liminal Biosciences Limited | Gpr84 antagonists and uses thereof |
| WO2022167457A1 (en) * | 2021-02-02 | 2022-08-11 | Liminal Biosciences Limited | Gpr84 antagonists and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2007143597A9 (en) | 2010-11-11 |
| EP2029572A2 (en) | 2009-03-04 |
| DE602007010287D1 (en) | 2010-12-16 |
| EP2029572B1 (en) | 2010-11-03 |
| CL2007001597A1 (en) | 2008-05-16 |
| MX2008015229A (en) | 2008-12-12 |
| KR20090015107A (en) | 2009-02-11 |
| CN101460476B (en) | 2013-12-04 |
| AU2007256708B2 (en) | 2011-12-15 |
| AR061222A1 (en) | 2008-08-13 |
| US8501746B2 (en) | 2013-08-06 |
| CN101460476A (en) | 2009-06-17 |
| PE20080714A1 (en) | 2008-07-31 |
| US20090156615A1 (en) | 2009-06-18 |
| KR101124070B1 (en) | 2012-04-12 |
| PT2029572E (en) | 2011-02-09 |
| WO2007143597A3 (en) | 2008-04-10 |
| US20140024583A1 (en) | 2014-01-23 |
| RU2491285C2 (en) | 2013-08-27 |
| CA2653655A1 (en) | 2007-12-13 |
| ATE486862T1 (en) | 2010-11-15 |
| JP2009539868A (en) | 2009-11-19 |
| BRPI0712902A2 (en) | 2018-10-30 |
| TW200815420A (en) | 2008-04-01 |
| AU2007256708A1 (en) | 2007-12-13 |
| JP5043105B2 (en) | 2012-10-10 |
| PL2029572T3 (en) | 2011-05-31 |
| ES2358297T3 (en) | 2011-05-09 |
| RU2008151051A (en) | 2010-07-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2007143597A2 (en) | Organic compounds | |
| CA2729327C (en) | Heterocyclic derivatives that modulate the activity of stearoyl-coa desaturase | |
| CA2660201C (en) | Organic compounds | |
| US8258160B2 (en) | SCD1 inhibitors triazole and tetrazole compounds | |
| US8318949B2 (en) | Organic compounds | |
| AU2013206248A1 (en) | Organic compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780020423.8 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007256708 Country of ref document: AU Ref document number: 9405/DELNP/2008 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007798049 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2653655 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2008/015229 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2007256708 Country of ref document: AU Date of ref document: 20070604 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12303490 Country of ref document: US Ref document number: 2009514484 Country of ref document: JP Ref document number: 1020087029648 Country of ref document: KR Ref document number: KR |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07798049 Country of ref document: EP Kind code of ref document: A2 |
|
| ENP | Entry into the national phase |
Ref document number: 2008151051 Country of ref document: RU Kind code of ref document: A |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01E Ref document number: PI0712902 Country of ref document: BR Free format text: ESCLARECA A OMISSAO DE RAJENDER KAMBOJ DO QUADRO DE INVENTORES. |
|
| ENPW | Started to enter national phase and was withdrawn or failed for other reasons |
Ref document number: PI0712902 Country of ref document: BR Free format text: PEDIDO CONSIDERADO RETIRADO EM RELACAO AO BRASIL POR NAO TER CUMPRIDO EXIGENCIA PUBLICADA NA RPI NO 2188 DE 11/12/2012. |
|
| ENPZ | Former announcement of the withdrawal of the entry into the national phase was wrong |
Ref document number: PI0712902 Country of ref document: BR Kind code of ref document: A2 Free format text: A RETIRADA DO PEDIDO PUBLICADA NA RPI NO 2201 DE 12/03/2013 ESTA SENDO ANULADA EM RAZAO DE PARECER RECURSAL PUBLICADO NA RPI NO 2437, DE 19/09/2017. |
|
| ENP | Entry into the national phase |
Ref document number: PI0712902 Country of ref document: BR Kind code of ref document: A2 Effective date: 20081205 |