WO2007140222A2 - Pyrrolopyrimidine compounds and their uses - Google Patents
Pyrrolopyrimidine compounds and their uses Download PDFInfo
- Publication number
- WO2007140222A2 WO2007140222A2 PCT/US2007/069595 US2007069595W WO2007140222A2 WO 2007140222 A2 WO2007140222 A2 WO 2007140222A2 US 2007069595 W US2007069595 W US 2007069595W WO 2007140222 A2 WO2007140222 A2 WO 2007140222A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituted
- alkyl
- group
- cycloalkyl
- mmol
- Prior art date
Links
- NWLFTMLSFXSQKI-UHFFFAOYSA-N C(CC1)CC1[n]1c2nc(Nc(cc3)ncc3N3CCNCC3)ncc2cc1 Chemical compound C(CC1)CC1[n]1c2nc(Nc(cc3)ncc3N3CCNCC3)ncc2cc1 NWLFTMLSFXSQKI-UHFFFAOYSA-N 0.000 description 1
- QTHGFKIFWQPXGO-UHFFFAOYSA-N CC(C)(c1c(N2C3CCCC3)nc(Nc(cc3)ccc3N3CCNCC3)nc1)C2=O Chemical compound CC(C)(c1c(N2C3CCCC3)nc(Nc(cc3)ccc3N3CCNCC3)nc1)C2=O QTHGFKIFWQPXGO-UHFFFAOYSA-N 0.000 description 1
- DOBLBPCJWPDUOP-UHFFFAOYSA-N CC(N(CC1)CCN1c(cc1)ccc1Nc1ncc(c(C)c[n]2C3CCCC3)c2n1)=O Chemical compound CC(N(CC1)CCN1c(cc1)ccc1Nc1ncc(c(C)c[n]2C3CCCC3)c2n1)=O DOBLBPCJWPDUOP-UHFFFAOYSA-N 0.000 description 1
- LPUIZIRSCFAODM-UHFFFAOYSA-N CC(N(CC1)CCN1c(cc1)ccc1Nc1ncc(c(C)c[n]2C3CCCCC3)c2n1)=O Chemical compound CC(N(CC1)CCN1c(cc1)ccc1Nc1ncc(c(C)c[n]2C3CCCCC3)c2n1)=O LPUIZIRSCFAODM-UHFFFAOYSA-N 0.000 description 1
- PZDCVZQDCAJZPJ-UHFFFAOYSA-N CC(N(CC1)CCN1c(cc1)ccc1Nc1ncc(cc[n]2C3CCCC3)c2n1)=O Chemical compound CC(N(CC1)CCN1c(cc1)ccc1Nc1ncc(cc[n]2C3CCCC3)c2n1)=O PZDCVZQDCAJZPJ-UHFFFAOYSA-N 0.000 description 1
- AVPNJCGJPXIQOG-UHFFFAOYSA-N CC(N(CC1)CCN1c(cc1)cnc1N)=O Chemical compound CC(N(CC1)CCN1c(cc1)cnc1N)=O AVPNJCGJPXIQOG-UHFFFAOYSA-N 0.000 description 1
- ATZGMZJWAIXDJZ-UHFFFAOYSA-N CC(N(CC1)CCN1c(cc1)cnc1Nc1ncc(cc[n]2C3CCCC3)c2n1)=O Chemical compound CC(N(CC1)CCN1c(cc1)cnc1Nc1ncc(cc[n]2C3CCCC3)c2n1)=O ATZGMZJWAIXDJZ-UHFFFAOYSA-N 0.000 description 1
- XRKWOGZOQWSBCT-UHFFFAOYSA-N CC(N(CC1)CCN1c(cn1)ccc1[N+]([O-])=O)=O Chemical compound CC(N(CC1)CCN1c(cn1)ccc1[N+]([O-])=O)=O XRKWOGZOQWSBCT-UHFFFAOYSA-N 0.000 description 1
- ATPJIFCMUYJJTN-UHFFFAOYSA-N CC1(C=CC(Nc2ncc(c(C)c[n]3C4CC4)c3n2)=CC1)N(CC1)CCN1C(C)=O Chemical compound CC1(C=CC(Nc2ncc(c(C)c[n]3C4CC4)c3n2)=CC1)N(CC1)CCN1C(C)=O ATPJIFCMUYJJTN-UHFFFAOYSA-N 0.000 description 1
- KPLOIQVDAZAINB-UHFFFAOYSA-N CC12N(C3CCCC3)C=C(C)C1=CN=C(Nc(cc1)ccc1N1CCN(C)CC1)N2 Chemical compound CC12N(C3CCCC3)C=C(C)C1=CN=C(Nc(cc1)ccc1N1CCN(C)CC1)N2 KPLOIQVDAZAINB-UHFFFAOYSA-N 0.000 description 1
- RVWZDGBXGHOMKG-UHFFFAOYSA-N CCC(CC)N(C(C1)=O)c2c1cnc(Nc(cc1)ccc1N1CCNCC1)n2 Chemical compound CCC(CC)N(C(C1)=O)c2c1cnc(Nc(cc1)ccc1N1CCNCC1)n2 RVWZDGBXGHOMKG-UHFFFAOYSA-N 0.000 description 1
- RARLSEWFHXTVPD-UHFFFAOYSA-N CCC(CC)N(C(C1)=O)c2c1cnc(Nc(cc1)ncc1N1CCNCC1)n2 Chemical compound CCC(CC)N(C(C1)=O)c2c1cnc(Nc(cc1)ncc1N1CCNCC1)n2 RARLSEWFHXTVPD-UHFFFAOYSA-N 0.000 description 1
- UAJPTLDIFCCFJS-UHFFFAOYSA-O CCC(CC)N(c1nc(Cl)[nH+]cc1N1C)C1=O Chemical compound CCC(CC)N(c1nc(Cl)[nH+]cc1N1C)C1=O UAJPTLDIFCCFJS-UHFFFAOYSA-O 0.000 description 1
- JPNZFPOGAMFPDY-UHFFFAOYSA-N CCC(CC)Nc1nc(Cl)ncc1Br Chemical compound CCC(CC)Nc1nc(Cl)ncc1Br JPNZFPOGAMFPDY-UHFFFAOYSA-N 0.000 description 1
- LLYZJLNTGSIXEK-UHFFFAOYSA-N CCC(c1ccccc1)N1c2nc(N[AlH2])ncc2CC1=O Chemical compound CCC(c1ccccc1)N1c2nc(N[AlH2])ncc2CC1=O LLYZJLNTGSIXEK-UHFFFAOYSA-N 0.000 description 1
- MMDPMQBHVFPULC-UHFFFAOYSA-N CCN(CC)CCOc1cc(N)ccc1 Chemical compound CCN(CC)CCOc1cc(N)ccc1 MMDPMQBHVFPULC-UHFFFAOYSA-N 0.000 description 1
- SFEIACLAEPWULQ-UHFFFAOYSA-N CN(CC1)CCN1c(cc1)ccc1Nc1ncc(cc[n]2C3CCCC3)c2n1 Chemical compound CN(CC1)CCN1c(cc1)ccc1Nc1ncc(cc[n]2C3CCCC3)c2n1 SFEIACLAEPWULQ-UHFFFAOYSA-N 0.000 description 1
- OCLILGBSWCUZDJ-UHFFFAOYSA-N CN1CCN(CCOc2cccc(N)c2)CC1 Chemical compound CN1CCN(CCOc2cccc(N)c2)CC1 OCLILGBSWCUZDJ-UHFFFAOYSA-N 0.000 description 1
- XVCCHAMVUYNVAY-UHFFFAOYSA-N Cc(c1cnc(Nc(cc2)ncc2N2CCNCC2)nc11)c[n]1-c1ncccc1 Chemical compound Cc(c1cnc(Nc(cc2)ncc2N2CCNCC2)nc11)c[n]1-c1ncccc1 XVCCHAMVUYNVAY-UHFFFAOYSA-N 0.000 description 1
- MYBYPYDIECMJIG-UHFFFAOYSA-N Cc1c[n](C2CC2)c2nc(Nc(cc3)ccc3N3CCN(C)CC3)ncc12 Chemical compound Cc1c[n](C2CC2)c2nc(Nc(cc3)ccc3N3CCN(C)CC3)ncc12 MYBYPYDIECMJIG-UHFFFAOYSA-N 0.000 description 1
- AMPRRMMDLBKQON-UHFFFAOYSA-N Cc1c[n](C2CC2)c2nc(Nc(cc3)ccc3N3CCNCC3)ncc12 Chemical compound Cc1c[n](C2CC2)c2nc(Nc(cc3)ccc3N3CCNCC3)ncc12 AMPRRMMDLBKQON-UHFFFAOYSA-N 0.000 description 1
- BKOZEBTYQCGFBB-UHFFFAOYSA-N Cc1c[n](C2CCCC2)c2nc(Nc(cc3)ccc3N3CCNCC3)ncc12 Chemical compound Cc1c[n](C2CCCC2)c2nc(Nc(cc3)ccc3N3CCNCC3)ncc12 BKOZEBTYQCGFBB-UHFFFAOYSA-N 0.000 description 1
- FYPMCJASNFKOCG-UHFFFAOYSA-N Cc1c[n](C2CCCCC2)c2nc(Nc(cc3)ccc3N3CCN(C)CC3)ncc12 Chemical compound Cc1c[n](C2CCCCC2)c2nc(Nc(cc3)ccc3N3CCN(C)CC3)ncc12 FYPMCJASNFKOCG-UHFFFAOYSA-N 0.000 description 1
- XSHHFCAXOMRZKQ-UHFFFAOYSA-N Cc1c[n](C2CCCCC2)c2nc(Nc(cc3)ccc3N3CCNCC3)ncc12 Chemical compound Cc1c[n](C2CCCCC2)c2nc(Nc(cc3)ccc3N3CCNCC3)ncc12 XSHHFCAXOMRZKQ-UHFFFAOYSA-N 0.000 description 1
- KFOWCFUJSYGZMB-UHFFFAOYSA-N Nc1cc(OCCN2CCOCC2)ccc1 Chemical compound Nc1cc(OCCN2CCOCC2)ccc1 KFOWCFUJSYGZMB-UHFFFAOYSA-N 0.000 description 1
- YJDXLUSVVAGFFS-UHFFFAOYSA-N Nc1cccc(OCCN2CCCC2)c1 Chemical compound Nc1cccc(OCCN2CCCC2)c1 YJDXLUSVVAGFFS-UHFFFAOYSA-N 0.000 description 1
- VYGQEGNKBPMNCP-UHFFFAOYSA-N O=[N+2]c1cccc(NCCN2CCCC2)c1 Chemical compound O=[N+2]c1cccc(NCCN2CCCC2)c1 VYGQEGNKBPMNCP-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/02—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6
- C07D473/18—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6 one oxygen and one nitrogen atom, e.g. guanine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
- A61K31/52—Purines, e.g. adenine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
- A61K31/52—Purines, e.g. adenine
- A61K31/522—Purines, e.g. adenine having oxo groups directly attached to the heterocyclic ring, e.g. hypoxanthine, guanine, acyclovir
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/02—Stomatological preparations, e.g. drugs for caries, aphtae, periodontitis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/06—Antiabortive agents; Labour repressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/10—Anti-acne agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/14—Drugs for disorders of the endocrine system of the thyroid hormones, e.g. T3, T4
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/02—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6
- C07D473/24—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6 one nitrogen and one sulfur atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Definitions
- Protein kinases constitute a large family of structurally related enzymes that are responsible for the control of a variety of signal transduction processes within the cell. (Hardie, G. and Hanks, S. The Protein Kinase Facts Book, I and II, Academic Press, San Diego, Calif.: 1995). Protein kinases are thought to have evolved from a common ancestral gene due to the conservation of their structure and catalytic function. Almost all kinases contain a similar 250-300 amino acid catalytic domain. The kinases may be categorized into families by the substrates they phosphorylate ⁇ e.g., protein-tyrosine, protein-serine/threonine, lipids, etc.).
- protein kinases mediate intracellular signaling by affecting a phosphoryl transfer from a nucleoside triphosphate to a protein acceptor that is involved in a signaling pathway. These phosphorylation events act as molecular on/off switches that can modulate or regulate the target protein biological function. These phosphorylation events are ultimately triggered in response to a variety of extracellular and other stimuli.
- Examples of such stimuli include environmental and chemical stress signals ⁇ e.g., osmotic shock, heat shock, ultraviolet radiation, bacterial endotoxin, and H 2 O 2 ), cytokines ⁇ e.g., interleukin-1 (IL- 1) and tumor necrosis factor- ⁇ (TNF- ⁇ )), and growth factors ⁇ e.g., granulocyte macrophage- colony-stimulating factor (GM-CSF), and fibroblast growth factor (FGF)).
- An extracellular stimulus may affect one or more cellular responses related to cell growth, migration, differentiation, secretion of hormones, activation of transcription factors, muscle contraction, glucose metabolism, control of protein synthesis, and regulation of the cell cycle.
- diseases are associated with abnormal cellular responses triggered by protein kinase-mediated events as described above. These diseases include, but are not limited to, autoimmune diseases, inflammatory diseases, bone diseases, metabolic diseases, neurological and neurodegenerative diseases, cancer, cardiovascular diseases, allergies and asthma, Alzheimer's disease, and hormone-related diseases. Accordingly, there has been a substantial l effort in medicinal chemistry to find protein kinase inhibitors that are effective as therapeutic agents.
- the Janus kinases are a family of tyrosine kinases consisting of JAKl, JAK2, JAK3 and TYK2.
- the JAKs play a critical role in cytokine signaling.
- the down-stream substrates of the JAK family of kinases include the signal transducer and activator of transcription (STAT) proteins.
- STAT signal transducer and activator of transcription
- JAK/STAT signaling has been implicated in the mediation of many abnormal immune responses such as allergies, asthma, autoimmune diseases such as transplant rejection, rheumatoid arthritis, amyotrophic lateral sclerosis and multiple sclerosis as well as in solid and hematologic malignancies such as leukemias and lymphomas.
- the pharmaceutical intervention in the JAK/STAT pathway has been reviewed [Frank MoI. Med. 5: 432-456 (1999) & Seidel, et al, Oncogene 19: 2645-2656 (2000)].
- JAKl, JAK2, and TYK2 are ubiquitously expressed, while JAK3 is predominantly expressed in hematopoietic cells.
- JAK3 binds exclusively to the common cytokine receptor gamma chain ( ⁇ c ) and is activated by IL-2, IL-4, IL-7, IL-9, and IL-15.
- ⁇ c common cytokine receptor gamma chain
- the proliferation and survival of murine mast cells induced by IL-4 and IL-9 have, in fact, been shown to be dependent on JAK3- and 65 c -signaling [Suzuki et al, Blood 96: 2172-2180 (2000)].
- the JAK family of tyrosine kinases have also been shown to play a role in immunosuppression and allograft acceptance [Kirken, Transpl. Proc. 33: 3268-3270 (2001)], rheumatoid arthritis [Muller-Ladner, et al., J. Immunol. 164: 3894-3901 (2000)], Familial amyotrophic lateral sclerosis [Trieu, et al., Biochem. Biophys. Res. Commun. 267: 22-25 (2000)], and leukemia [Sudbeck, et al., Clin. Cancer Res. 5: 1569-1582 (1999)].
- CDK cyclin-dependent kinase
- CDKs The activity of CDKs is regulated post-translationally, by transitory associations with other proteins, and by alterations of their intracellular localization. Tumor development is closely associated with genetic alteration and deregulation of CDKs and their regulators, suggesting that inhibitors of CDKs may be useful anti-cancer therapeutics. Indeed, early results suggest that transformed and normal cells differ in their requirement for, e.g., cyclin A/CDK2 and that it may be possible to develop novel antineoplastic agents devoid of the general host toxicity observed with conventional cytotoxic and cytostatic drugs. While inhibition of cell cycle-related CDKs is clearly relevant in, e.g., oncology applications, this may not be the case for the inhibition of RNA polymerase-regulating CDKs.
- CDK9/cyclin T function was recently linked to prevention of HIV replication and the discovery of new CDK biology thus continues to open up new therapeutic indications for CDK inhibitors (Sausville, E. A. Trends Molec. Med. 2002, 8, S32-S37).
- the function of CDKs is to phosphorylate and thus activate or deactivate certain proteins, including e.g. retinoblastoma proteins, lamins, histone Hl, and components of the mitotic spindle.
- the catalytic step mediated by CDKs involves a phospho-transfer reaction from ATP to the macromolecular enzyme substrate.
- Several groups of compounds have been found to possess anti-proliferative properties by virtue of CDK-specific ATP antagonism.
- the invention provides a compound of Formula I:
- the protein kinase is a protein tyrosine kinase.
- the protein kinase is selected from the group consisting of abl, ATK, ber-abl, BIk, Brk, Btk, c-fms, e- kit, c- met, c-src, CDK, cRafl, CSFIR, CSK, EGFR, ErbB2, ErbB3, ErbB4, ERK, Fak, fes, FGFRI, 25 FGFR2, FGFR3, FGFR4, FGFR5, Fgr, FLK-4, flt-1, Fps, Frk, Fyn, GSK, Gst-Flkl, Hck, Her-2, Her-4, IGF- IR, INS-R, Jak, JNK, KDR, Lck, Lyn, MEK, p38, PANHER, PDGFR, PLK, PKC, PY
- the protein kinase is selected from the group consisting of CDKl, CDK2, CDK4, CDK5, CDK6, CDK7, CDK8 and CDK9.
- the protein kinase is selected from the group consisting of Jak 1, Jak2 and Jak3.
- the protein kinase is selected from the group consisting of Jak3 and CDK4.
- the protein kinase is in a cell culture. In still another aspect, the protein kinase is in a mammal.
- the invention provides a method of treating a protein kinase-associated disorder, wherein the method includes administering to a subject in need thereof a pharmaceutically acceptable amount of a compound of the Formula I, such that the protein kinase-associated disorder is treated.
- the protein kinase is selected from the group consisting of CDKl, CDK2, CDK4, CDK5, CDK6, CDK7, CDK8, CDK9, Jakl, Jak2 and Jak3.
- the protein kinase is selected from the group consisting of Jak3 and CDK4.
- the protein kinase-associated disorder is selected from the group consisting of blood vessel proliferative disorders, f ⁇ brotic disorders, mesangial cell proliferative disorders, metabolic disorders, allergies, asthma, thrombosis, nervous system diseases and cancer.
- the protein kinase-associated disorder is cancer.
- the cancer is selected from the group consisting of breast, stomach, ovary, colon, lung, brain, larynx, lymphatic system, genitourinary tract (including bladder and prostate), ovarian, gastric, bone, and pancreatic cancer.
- the protein kinase-associated disorder is selected from the group consisting of organ transplant rejection, xeno transplantation, lupus, multiple sclerosis, rheumatoid arthritis, psoriasis, Type 1 diabetes and complications from diabetes, cancer, asthma, atopic dermatitis, autoimmune thyroid disorders, ulcerative colitis, Crohn's disease, Alzheimer's disease and leukemia.
- the disease is selected from an immune response, an autoimmune disease, a neurodegenerative disease, or a solid or hematologic malignancy.
- the disease is selected from an allergic or type I hypersensitivity reaction, asthma, graft versus host disease, rheumatoid arthritis, amyotrophic lateral sclerosis, multiple sclerosis, Familial amyotrophic lateral sclerosis, leukemia, or lymphoma
- the invention provides a method of treating an autoimmune disease, wherein the treatment includes administering to a subject in need thereof a pharmaceutically acceptable amount of a compound of the Formula I, such that the autoimmune disease is treated.
- the autoimmune disease is selected from the group consisting of autoimmune hemolytic anemia, autoimmune neonatal thrombocytopenia, idiopathic thrombocytopenia purpura, autoimmunocytopenia, hemolytic anemia, antiphospholipid syndrome, dermatitis, allergic encephalomyelitis, myocarditis, relapsing polychondritis, rheumatic heart disease, glomerulonephritis, multiple sclerosis, neuritis, uveitis ophthalmia, polyendocrinopathies, purpura, Reiter's Disease, Stiff-Man Syndrome, autoimmune pulmonary inflammation, autism, Guillain-Barre Syndrome, insulin dependent diabetes mellitis, autoimmune inflammatory eye, autoimmune thyroiditis, hypo
- the invention provides a method of treating transplant rejection, wherein the treatment includes administering to a subject in need thereof a pharmaceutically acceptable amount of a compound of the Formula I such that the transplant rejection is treated.
- the transplant rejection is selected from the group consisting of graft versus host disease, rejection related to xeno transplantation, rejection related to organ transplant, rejection related to acute transplant, heterograft or homograft rejection and ischemic or reperfusion injury incurred during organ transplantation.
- the invention provides a method of treating cancer, wherein the method includes administering to a subject in need thereof a pharmaceutically acceptable amount of a compound of the Formula I such that the cancer disease or disorder is treated.
- the cancer is selected from the group consisting of bladder, head and neck, breast, stomach, ovary, colon, lung, brain, larynx, lymphatic system, genitourinary tract, gastrointestinal, ovarian, prostate, gastric, bone, small-cell lung, glioma, colorectal and pancreatic cancer.
- the Formula I or salt thereof is administered, simultaneously or sequentially, with an antiinflammatory, antiproliferative, chemotherapeutic agent, immunosuppressant, anti-cancer, cytotoxic agent or kinase inhibitor other than a compound of the Formula I or salt thereof.
- an antiinflammatory, antiproliferative, chemotherapeutic agent, immunosuppressant, anti-cancer, cytotoxic agent or kinase inhibitor other than a compound of the Formula I or salt thereof.
- Formula I or salt thereof is administered, simultaneously or sequentially, with one or more of a PTK inhibitor, cyclosporin A, CTLA4-Ig, antibodies selected from anti-ICAM-3, anti-IL-2 receptor, anti-CD45RB, anti-CD2, anti-CD3, anti-CD4, anti-CD80, anti-CD86, and monoclonal antibody OKT3, agents blocking the interaction between CD40 and gp39, fusion proteins constructed from CD40 and gp39, inhibitors of NF-kappa B function, non-steroidal antiinflammatory drugs, steroids, gold compounds, antiproliferative agents, FK506, mycophenolate mofetil, cytotoxic drugs, TNF- ⁇ inhibitors, anti-TNF antibodies or soluble TNF receptor, rapamycin, leflunimide, cyclooxygenase-2 inhibitors, paclitaxel, cisplatin, carboplatin, doxorubicin, carminomycin, daunorubicin, aminopterin, methotrexate,
- the invention provides a packaged protein kinase-associated disorder treatment, wherein the treatment includes a protein kinase-modulating compound of the Formula I, packaged with instructions for using an effective amount of the protein kinase- modulating compound to treat a protein kinase-associated disorder.
- This invention is directed to compounds, e.g., pyrrolopyrimidine compounds, and intermediates thereto, as well as pharmaceutical compositions containing the compounds for use in treatment of protein kinase-associated disorders.
- This invention is also directed to the compounds of the invention or compositions thereof as modulators of Jakl, Jak2 and Jak3, as well as CDKl, CDK2, CDK4, CDK5, CDK6, CDK7, CDK8 and CDK9.
- the present invention is also directed to methods of combination therapy for inhibiting protein kinase activity in cells, or for treating, preventing or ameliorating of one or more symptoms of cancer, transplant rejections, and autoimmune diseases in patients using the compounds of the invention or pharmaceutical compositions, or kits thereof.
- the invention provides compounds of the Formula I:
- A is N or CR 5 , wherein R 5 is hydrogen or CrQ-alkyl;
- R 2 and R 3 are each, independently, selected from the group consisting of hydrogen, hydroxyl, CrC 3 -alkyl, C 3 -C 8 -cycloalkyl, heterocyclyl, aryl, heteroaryl, substituted C 1 -C 3 - alkyl, substituted C 3 -Cg-cycloalkyl, substituted heterocyclyl, substituted aryl and substituted heteroaryl;
- R 6 and R 7 are each, independently selected from the group consisting of aryl, substituted aryl, heteroaryl, substituted heteroaryl, hydrogen, Ci-C 3 -alkyl, C 3 -C 8 -cycloalkyl, heterocyclyl, substituted alkyl, substituted cycloalkyl, and substituted heterocyclyl;
- R 8 is hydrogen, d-C 3 -alkyl, and C 3 -C 8 -cycloalkyl;
- R 9 and R 10 are each, independently, hydrogen, d-d-alkyl, or C 3 -C 8 -cycloalkyl;
- R 4 is branched or linear Cj-d-alkyl, wherein the branched C 1 - C 5 -alkyl group may be interrupted by one or more heteroatoms, and/or substituted with one or more heteroatoms, halogens, C 3 -Cg cycloalkyl groups, substituted C 3 -C 8 cycloalkyl groups, C 3 -Cg hetrocyclyl groups, aryl groups, heteroaryl groups, substituted aryl groups, or substituted heteroaryl groups.
- R 12 is not hydrogen
- R 4 is selected from the group consisting of hydrogen, d-Cg-alkyl, C 3 -Cg-cycloalkyl, C 3 -C 8 -substituted cycloalkyl, aryl, substituted aryl, heteroaryl, and substituted heteroaryl.
- R 12 is not hydrogen
- R 4 is branched or linear Ci-C 5 -alkyl, wherein the branched d-d-alkyl group may be interrupted by one or more heteroatoms, and/or substituted with one or more heteroatoms, halogens, C 3 -C 8 cycloalkyl groups, substituted C 3 -C 8 cycloalkyl groups, C 3 -Cg hetrocyclyl groups, aryl groups, heteroaryl groups, substituted aryl groups, or substituted heteroaryl groups.
- A is N.
- R 4 is selected from the group consisting of hydrogen, branched d-d-alkyl, branched CrC 5 -alkyl substituted by phenyl and d-C ⁇ -cycloalkyl.
- R 4 is C(H)(CH 2 CH 3 ) 2 , C(H)(CH 2 CH 3 )Ph, CH 2 CH 3 , cyclopropyl, cyclopentyl or cyclohexyl.
- the dashed line is a single bond
- the dashed line is a double bond
- X is CH, N, C- C(O)d-C 3 -alkyl or C-(Ci-C 3 -alkyl)
- R 2 is H.
- R 3 is an aryl group, which is further independently substituted one or more times by halogen, C 1 -C 4 -alkoxy, R 15 -amine, R 15 -heterocycle, or R 15 - heteroaryl, wherein R 15 is a bond, C(O), N(H)C(O), N(H)SO 2 , OC(O) or (CH 2 ) M , wherein the (CH 2 ) 1-4 group may be interrupted by O, N(CH 3 ) or N(H).
- the aryl group is phenyl
- the phenyl group is independently substituted one or more times with fluoro, methoxy, diethylamine, R 15 -piperazinyl, R 15 -morpholinyl, R 15 -piperidinyl, R 15 -triazolyl, R 15 -phenyl, R 15 -pyridinyl, R 15 -piperazinyl, R 15 -indazolyl, R 15 -pyrrolidinyl or R 15 -imidazolyl, wherein the piperazinyl, morpholinyl, piperidinyl, triazolyl, phenyl, pyridinyl, piperazinyl, indazolyl, pyrrolidinyl or imidazolyl groups may be further substituted with Ci-Oalkyl, C(O)Ci-C 4 -alkyl, S(O) 2 C rQ-alkyl, OH, C(O)(CH 2 )i -3 CN Or N
- the phenyl group is substituted by N(H)C(O)aryl, C(O)N(H)d-C 4 -alkyl, C(O)N(C r C 4 -alkyl) 2 or C(O)N(H)C 3 -C 6 -cycloalkyl.
- Preferred embodiments of Formula I are shown below in Table A, Table B, Table C and Table D, and are also considered to be “compounds of the invention.”
- the compounds of the invention are also referred to herein as "protein kinase inhibitors.”
- the compound of the present invention is further characterized as a modulator of a protein kinase, including, but not limited to, protein kinases selected from the group consisting of abl, ATK, ber-abl, BIk, Brk, Btk, c-fms, e- kit, c- met, c-src, CDK, cRafl, CSFIR, CSK, EGFR, ErbB2, ErbB3, ErbB4, ERK, Fak, fes, FGFRI, 25 FGFR2, FGFR3, FGFR4, FGFR5, Fgr, FLK-4, flt-1 , Fps, Frk, Fyn, GSK, Gst-Flkl, Hck, Her- 2, Her-4, IGF- IR, INS-R, Jak, JNK, KDR, Lck, Lyn, MEK, p38, PANHER, PDGFR, PLK, PKC,
- the protein kinase is selected from the group consisting of CDKl, CDK2, CDK4, CDK5, CDK6, CDK7, CDK8 and CDK9.
- the protein kinase is selected from the group consisting of Jakl, Jak2 and Jak3.
- the protein kinase is selected from the group consisting of Jak3 and CDK4.
- the compounds of the present invention are used for the treatment of protein kinase-associated disorders.
- protein kinase-associated disorder includes disorders and states (e.g., a disease state) that are associated with the activity of a protein kinase, e.g., CDK4 and Jak3.
- disorders and states e.g., a disease state
- Non-limiting examples of a protein kinase-associated disorder include blood vessel proliferative disorders, fibrotic disorders, mesangial cell proliferative disorders, metabolic disorders, allergies, asthma, thrombosis, nervous system diseases, organ transplant rejection, autoimmune diseases, and cancer.
- the compound of the present invention is further characterized as a modulator of a combination of protein kinases, e.g., Jak3 and CDK4.
- a compound of the present invention is used for protein kinase-associated diseases, and use of the compound of the present invention as an inhibitor of any one or more protein kinases. It is envisioned that a use can be a treatment of inhibiting one or more isoforms of protein kinases.
- the compounds of the invention are inhibitors of cyclin-dependent kinase enzymes (CDKs).
- CDKs cyclin-dependent kinase enzymes
- inhibition of the CDK4/cyclin Dl complex blocks phosphorylation of the Rb/inactive E2F complex, thereby preventing release of activated E2F and ultimately blocking E2F-dependent DNA transcription. This has the effect of inducing G 1 cell cycle arrest.
- the CDK4 pathway has been shown to have tumor-specific deregulation and cytotoxic effects.
- the compounds of this invention have the potential to block the expansion of auto- or alloreactive T cells, and thus have beneficial effects on autoimmune diseases, as well as transplant rejections.
- the present invention includes treatment of one or more symptoms of cancer, transplant rejections, and autoimmune diseases, as well as protein kinase-associated disorders, as described above, but the invention is not intended to be limited to the manner by which the compound performs its intended function of treatment of a disease.
- the present invention includes treatment of diseases described herein in any manner that allows treatment to occur, e.g., cancer, transplant rejections, and autoimmune diseases.
- the invention provides a pharmaceutical composition of any of the compounds of the present invention.
- the invention provides a pharmaceutical composition of any of the compounds of the present invention and a pharmaceutically acceptable carrier or excipient of any of these compounds.
- the invention includes the compounds as novel chemical entities.
- the invention includes a packaged protein kinase-associated disorder treatment.
- the packaged treatment includes a compound of the invention packaged with instructions for using an effective amount of the compound of the invention for an intended use.
- the compounds of the present invention are suitable as active agents in pharmaceutical compositions that are efficacious particularly for treating protein kinase- associated disorders, e.g., cancer, transplant rejections, and autoimmune diseases.
- the pharmaceutical composition in various embodiments has a pharmaceutically effective amount of the present active agent along with other pharmaceutically acceptable excipients, carriers, fillers, diluents and the like.
- phrases, "pharmaceutically effective amount” as used herein indicates an amount necessary to administer to a host, or to a cell, issue, or organ of a host, to achieve a therapeutic result, especially the regulating, modulating, or inhibiting protein kinase activity, e.g., inhibition of the activity of a protein kinase, or treatment of cancer, transplant rejections, or autoimmune diseases.
- the present invention provides a method for inhibiting the activity of a protein kinase.
- the method includes contacting a cell with any of the compounds of the present invention.
- the method further provides that the compound is present in an amount effective to selectively inhibit the activity of a protein kinase.
- the present invention provides a use of any of the compounds of the invention for manufacture of a medicament to treat cancer, transplant rejections, or autoimmune diseases in a subject.
- the invention provides a method of manufacture of a medicament, including formulating any of the compounds of the present invention for treatment of a subject.
- treat includes the diminishment or alleviation of at least one symptom associated or caused by the state, disorder or disease being treated.
- the treatment comprises the induction of a protein kinase-associated disorder, followed by the activation of the compound of the invention, which would in turn diminish or alleviate at least one symptom associated or caused by the protein kinase-associated disorder being treated.
- treatment can be diminishment of one or several symptoms of a disorder or complete eradication of a disorder.
- subject is intended to include organisms, e.g., prokaryotes and eukaryotes, which are capable of suffering from or afflicted with a disease, disorder or condition associated with the activity of a protein kinase.
- subjects include mammals, e.g., humans, dogs, cows, horses, pigs, sheep, goats, cats, mice, rabbits, rats, and transgenic non- human animals.
- the subject is a human, e.g., a human suffering from, at risk of suffering from, or potentially capable of suffering from cancer, transplant rejections, and autoimmune diseases, and for other diseases or conditions described herein.
- the subject is a cell.
- protein kinase-modulating compound refers to compounds that modulate, e.g., inhibit, or otherwise alter, the activity of a protein kinase.
- protein kinase-modulating compounds include compounds of Formula I, as well as Table A, Table B, Table C, Table D, Table E, and other examples as described herein (including pharmaceutically acceptable salts thereof, as well as enantiomers, stereoisomers, rotamers, tautomers, diastereomers, atropisomers or racemates thereof).
- a method of the invention includes administering to a subject an effective amount of a protein kinase-modulating compound of the invention, e.g., protein kinase-modulating compounds of Formula I, as well as Table A, Table B, Table C, Table D, Table E, and other examples as described herein (including pharmaceutically acceptable salts thereof, as well as enantiomers, stereoisomers, rotamers, tautomers, diastereomers, atropisomers or racemates thereof).
- a protein kinase-modulating compound of the invention e.g., protein kinase-modulating compounds of Formula I, as well as Table A, Table B, Table C, Table D, Table E, and other examples as described herein (including pharmaceutically acceptable salts thereof, as well as enantiomers, stereoisomers, rotamers, tautomers, diastereomers, atropisomers or racemates thereof).
- alkyl includes saturated aliphatic groups, including straight-chain alkyl groups (e.g., methyl, ethyl, propyl, butyl, pentyl, hexyl, heptyl, octyl, nonyl, decyl, etc.), branched-chain alkyl groups (isopropyl, tert-butyl, isobutyl, etc.), cycloalkyl (alicyclic) groups (cyclopropyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl), alkyl substituted cycloalkyl groups, and cycloalkyl substituted alkyl groups.
- straight-chain alkyl groups e.g., methyl, ethyl, propyl, butyl, pentyl, hexyl, heptyl, octyl, nonyl, decyl,
- alkyl also includes alkenyl groups and alkynyl groups.
- C x -C y -alkyl indicates a particular alkyl group (straight- or branched-chain) of a particular range of carbons.
- Q-Q-alkyl includes, but is not limited to, methyl, ethyl, propyl, butyl, isopropyl, tert-butyl and isobutyl.
- C 3 - 6 -cycloalkyl includes, but is not limited to, cyclopropyl, cyclopentyl, and cyclohexyl. As discussed below, these alkyl groups, as well as cycloalkyl groups, may be further substituted.
- halo as used herein means halogen, and includes fluorine, chlorine, bromine, or iodine, especially fluorine and chlorine.
- alkyl further includes alkyl groups which can further include oxygen, nitrogen, sulfur or phosphorous atoms replacing one or more carbons of the hydrocarbon backbone.
- a straight chain or branched chain alkyl has 10 or fewer carbon atoms in its backbone (e.g., Ci-C 10 for straight chain, C 3 -C 10 for branched chain), and more preferably 6 or fewer carbons.
- preferred cycloalkyls have from 4-7 carbon atoms in their ring structure, and more preferably have 5 or 6 carbons in the ring structure.
- alkyl e.g., methyl, ethyl, propyl, butyl, pentyl, hexyl, etc.
- alkyl include both
- unsubstituted alkyl and “substituted alkyl”, the latter of which refers to alkyl moieties having substituents replacing a hydrogen on one or more carbons of the hydrocarbon backbone, which allow the molecule to perform its intended function.
- substituted is intended to describe moieties having substituents replacing a hydrogen on one or more atoms, e.g. C, O or N, of a molecule.
- substituents can include, for example, oxo, alkyl, alkoxy, alkenyl, alkynyl, halogen, hydroxyl, alkylcarbonyloxy, arylcarbonyloxy, alkoxycarbonyloxy, aryloxycarbonyloxy, carboxylate, alkylcarbonyl, arylcarbonyl, alkoxycarbonyl, aminocarbonyl, alkylaminocarbonyl, dialkylaminocarbonyl, alkylthiocarbonyl, alkoxyl, phosphate, phosphonato, phosphinato, amino (including alkyl amino, dialkylamino, arylamino, diarylamino, and alkylarylamino), acylamino (including alkylcarbonylamino, ary
- substituents of the invention include moieties selected from straight or branched alkyl (preferably C 1 -C 5 ), cycloalkyl (preferably C 3 -Cg), alkoxy (preferably C 1 -C 6 ), thioalkyl (preferably C 1 -C 6 ), alkenyl (preferably C 2 -C 6 ), alkynyl (preferably C 2 -C 6 ), heterocyclic, carbocyclic, aryl (e.g., phenyl), aryloxy (e.g., phenoxy), aralkyl (e.g., benzyl), aryloxyalkyl (e.g., phenyloxyalkyl), arylacetamidoyl, alkylaryl, heteroaralkyl, alkylcarbonyl and arylcarbonyl or other such acyl group, heteroarylcarbonyl, or heteroaryl group, (CR'R")o- 3
- substituents can include, for example, halogen, hydroxyl, alkylcarbonyloxy, arylcarbonyloxy, alkoxycarbonyloxy, aryloxycarbonyloxy, carboxylate, alkylcarbonyl, alkoxycarbonyl, aminocarbonyl, alkylthiocarbonyl, alkoxyl, phosphate, phosphonato, phosphinato, cyano, amino (including alkyl amino, dialkylamino, arylamino, diarylamino, and alkylarylamino), acylamino (including alkylcarbonylamino, arylcarbonylamino, carbamoyl and ureido), amidino, imino, oxime, sulfhydryl, alkylthio, arylthio, thiocarboxylate, sulfates, sulfonato, sulfamoyl, sulfonamido, nitro,
- Cycloalkyls can be further substituted, e.g., with the substituents described above.
- An "aralkyl” moiety is an alkyl substituted with an aryl (e.g., phenylmethyl (i.e., benzyl)).
- alkenyl includes unsaturated aliphatic groups analogous in length and possible substitution to the alkyls described above, but which contain at least one double bond.
- alkenyl includes straight-chain alkenyl groups (e.g., ethenyl, propenyl, butenyl, pentenyl, hexenyl, heptenyl, octenyl, nonenyl, decenyl, etc.), branched- chain alkenyl groups, cycloalkenyl (alicyclic) groups (cyclopropenyl, cyclopentenyl, cyclohexenyl, cycloheptenyl, cyclooctenyl), alkyl or alkenyl substituted cycloalkenyl groups, and cycloalkyl or cycloalkenyl substituted alkenyl groups.
- alkenyl includes straight-chain alkenyl groups (e.g., ethenyl, propenyl, butenyl, pentenyl, hexenyl, heptenyl, octenyl, nonenyl, de
- alkenyl further includes alkenyl groups that include oxygen, nitrogen, sulfur or phosphorous atoms replacing one or more carbons of the hydrocarbon backbone.
- a straight chain or branched chain alkenyl group has 6 or fewer carbon atoms in its backbone (e.g., C 2 -C 6 for straight chain, C 3 -C 6 for branched chain).
- cycloalkenyl groups may have from 3-8 carbon atoms in their ring structure, and more preferably have 5 or 6 carbons in the ring structure.
- C 2 -C 6 includes alkenyl groups containing 2 to 6 carbon atoms.
- alkenyl includes both "unsubstituted alkenyls" and “substituted alkenyls”, the latter of which refers to alkenyl moieties having substituents replacing a hydrogen on one or more carbons of the hydrocarbon backbone.
- substituents can include, for example, alkyl groups, alkynyl groups, halogens, hydroxyl, alkylcarbonyloxy, arylcarbonyloxy, alkoxycarbonyloxy, aryloxycarbonyloxy, carboxylate, alkylcarbonyl, arylcarbonyl, alkoxycarbonyl, aminocarbonyl, alkylaminocarbonyl, dialkylaminocarbonyl, alkylthiocarbonyl, alkoxyl, phosphate, phosphonato, phosphinato, cyano, amino (including alkyl amino, dialkylamino, arylamino, diarylamino, and alkylarylamino), acylamino
- alkynyl includes unsaturated aliphatic groups analogous in length and possible substitution to the alkyls described above, but which contain at least one triple bond.
- alkynyl includes straight-chain alkynyl groups (e.g., ethynyl, propynyl, butynyl, pentynyl, hexynyl, heptynyl, octynyl, nonynyl, decynyl, etc.), branched- chain alkynyl groups, and cycloalkyl or cycloalkenyl substituted alkynyl groups.
- alkynyl further includes alkynyl groups that include oxygen, nitrogen, sulfur or phosphorous atoms replacing one or more carbons of the hydrocarbon backbone.
- a straight chain or branched chain alkynyl group has 6 or fewer carbon atoms in its backbone (e.g., C2-C 6 for straight chain, C 3 -C 6 for branched chain).
- C 2 -C 6 includes alkynyl groups containing 2 to 6 carbon atoms.
- alkynyl includes both "unsubstituted alkynyls" and “substituted alkynyls", the latter of which refers to alkynyl moieties having substituents replacing a hydrogen on one or more carbons of the hydrocarbon backbone.
- substituents can include, for example, alkyl groups, alkynyl groups, halogens, hydroxyl, alkylcarbonyloxy, arylcarbonyloxy, alkoxycarbonyloxy, aryloxycarbonyloxy, carboxylate, alkylcarbonyl, arylcarbonyl, alkoxycarbonyl, aminocarbonyl, alkylaminocarbonyl, dialkylaminocarbonyl, alkylthiocarbonyl, alkoxyl, phosphate, phosphonato, phosphinato, cyano, amino (including alkyl amino, dialkylamino, arylamino, diarylamino, and alkylarylamino), acylamino (including alkylcarbonylamino, arylcarbonylamino, carbamoyl and ureido), amidino, imino, sulfhydryl, alkylthio, arylthio, thiocarboxylate,
- amine or “amino” should be understood as being broadly applied to both a molecule, or a moiety or functional group, as generally understood in the art, and may be primary, secondary, or tertiary.
- amine or “amino” includes compounds where a nitrogen atom is covalently bonded to at least one carbon, hydrogen or heteroatom.
- alkylamino comprises groups and compounds wherein the nitrogen is bound to at least one additional alkyl group.
- dialkyl amino includes groups wherein the nitrogen atom is bound to at least two additional alkyl groups.
- arylamino and diarylamino include groups wherein the nitrogen is bound to at least one or two aryl groups, respectively.
- alkylarylamino refers to an amino group which is bound to at least one alkyl group and at least one aryl group.
- alkaminoalkyl refers to an alkyl, alkenyl, or alkynyl group bound to a nitrogen atom which is also bound to an alkyl group.
- amide includes compounds or moieties which contain a nitrogen atom which is bound to the carbon of a carbonyl or a thiocarbonyl group.
- the term includes "alkaminocarbonyl” or “alkylaminocarbonyl” groups which include alkyl, alkenyl, aryl or alkynyl groups bound to an amino group bound to a carbonyl group. It includes arylaminocarbonyl and arylcarbonylamino groups which include aryl or heteroaryl moieties bound to an amino group which is bound to the carbon of a carbonyl or thiocarbonyl group.
- alkylaminocarbonyl alkenylaminocarbonyl
- alkynylaminocarbonyl alkynylaminocarbonyl
- arylaminocarbonyl alkylcarbonylamino
- alkenylcarbonylamino alkynylcarbonylamino
- arylcarbonylamino alkylcarbonylamino
- alkenylcarbonylamino alkynylcarbonylamino
- arylcarbonylamino alkylcarbonylamino
- aryl includes groups, including 5- and 6-membered single-ring aromatic groups that may include from zero to four heteroatoms, for example, phenyl, pyrrole, furan, thiophene, thiazole, isothiaozole, imidazole, triazole, tetrazole, pyrazole, oxazole, isoxazole, pyridine, pyrazine, pyridazine, and pyrimidine, and the like.
- aryl includes multicyclic aryl groups, e.g., tricyclic, bicyclic, e.g., naphthalene, benzoxazole, benzodioxazole, benzothiazole, benzoimidazole, benzothiophene, methylenedioxyphenyl, quinoline, isoquinoline, anthryl, phenanthryl, napthridine, indole, benzofuran, purine, benzofuran, deazapurine, or indolizine.
- multicyclic aryl groups e.g., tricyclic, bicyclic, e.g., naphthalene, benzoxazole, benzodioxazole, benzothiazole, benzoimidazole, benzothiophene, methylenedioxyphenyl, quinoline, isoquinoline, anthryl, phenanthryl, napthridine, indole, benzofuran, purine,
- aryl groups having heteroatoms in the ring structure may also be referred to as "aryl heterocycles", “heterocycles,” “heteroaryls” or “heteroaromatics.”
- the aromatic ring can be substituted at one or more ring positions with such substituents as described above, as for example, alkyl, halogen, hydroxyl, alkoxy, alkylcarbonyloxy, arylcarbonyloxy, alkoxycarbonyloxy, aryloxycarbonyloxy, carboxylate, alkylcarbonyl, alkylaminoacarbonyl, aralkylaminocarbonyl, alkenylaminocarbonyl, alkylcarbonyl, arylcarbonyl, aralkylcarbonyl, alkenylcarbonyl, alkoxycarbonyl, aminocarbonyl, alkylthiocarbonyl, phosphate, phosphonato, phosphinato, cyano, amino (including alkyl amino,
- heteroaryl represents a stable monocyclic or bicyclic ring of up to 7 atoms in each ring, wherein at least one ring is aromatic and contains from 1 to 4 heteroatoms selected from the group consisting of O, N and S.
- Heteroaryl groups within the scope of this definition include but are not limited to: acridinyl, carbazolyl, cinnolinyl, quinoxalinyl, pyrrazolyl, indolyl, benzotriazolyl, furanyl, thienyl, benzothienyl, benzofuranyl, quinolinyl, isoquinolinyl, oxazolyl, isoxazolyl, indolyl, pyrazinyl, pyridazinyl, pyridinyl, pyrimidinyl, pyrrolyl, tetrahydroquinoline.
- heteroaryl is also understood to include the N-oxide derivative of any nitrogen-containing heteroaryl.
- heteroaryl substituent is bicyclic and one ring is non-aromatic or contains no heteroatoms, it is understood that attachment is via the aromatic ring or via the heteroatom containing ring, respectively.
- heterocycle or “heterocyclyl” as used herein is intended to mean a 5- to 10-membered aromatic or nonaromatic heterocycle containing from 1 to 4 heteroatoms selected from the group consisting of O, N and S, and includes bicyclic groups.
- Heterocyclyl therefore includes the above mentioned heteroaryls, as well as dihydro and tetrathydro analogs thereof. Further examples of “heterocyclyl” include, but are not limited to the following: benzoimidazolyl, benzofuranyl, benzofurazanyl, benzopyrazolyl, benzotriazolyl, benzothiophenyl, benzoxazolyl, carbazolyl, carbolinyl, cinnolinyl, furanyl, imidazolyl, indolinyl, indolyl, indolazinyl, indazolyl, isobenzofuranyl, isoindolyl, isoquinolyl, isothiazolyl, isoxazolyl, naphthpyridinyl, oxadiazolyl, oxazolyl, oxazoline, isoxazoline, oxetanyl, pyranyl, pyrazinyl,
- acyl includes compounds and moieties which contain the acyl radical (CH3CO-) or a carbonyl group.
- substituted acyl includes acyl groups where one or more of the hydrogen atoms are replaced by for example, alkyl groups, alkynyl groups, halogens, hydroxyl, alkylcarbonyloxy, arylcarbonyloxy, alkoxycarbonyloxy, aryloxycarbonyloxy, carboxylate, alkylcarbonyl, arylcarbonyl, alkoxycarbonyl, aminocarbonyl, alkylaminocarbonyl, dialkylaminocarbonyl, alkylthiocarbonyl, alkoxyl, phosphate, phosphonato, phosphinato, cyano, amino (including alkyl amino, dialkylamino, arylamino, diarylamino, and alkylarylamino), acylamino (including alkylcarbonylamino, ary
- acylamino includes moieties wherein an acyl moiety is bonded to an amino group.
- the term includes alkylcarbonylamino, arylcarbonylamino, carbamoyl and ureido groups.
- alkoxy includes substituted and unsubstituted alkyl, alkenyl, and alkynyl groups covalently linked to an oxygen atom. Examples of alkoxy groups include methoxy, ethoxy, isopropyloxy, propoxy, butoxy, and pentoxy groups and may include cyclic groups such as cyclopentoxy. Examples of substituted alkoxy groups include halogenated alkoxy groups.
- the alkoxy groups can be substituted with groups such as alkenyl, alkynyl, halogen, hydroxyl, alkylcarbonyloxy, arylcarbonyloxy, alkoxycarbonyloxy, aryloxycarbonyloxy, carboxylate, alkylcarbonyl, arylcarbonyl, alkoxycarbonyl, aminocarbonyl, alkylaminocarbonyl, dialkylaminocarbonyl, alkylthiocarbonyl, alkoxyl, phosphate, phosphonato, phosphinato, cyano, amino (including alkyl amino, dialkylamino, arylamino, diarylamino, and alkylarylamino), acylamino (including alkylcarbonylamino, arylcarbonylamino, carbamoyl and ureido), amidino, imino, sulfhydryl, alkylthio, arylthio, thiocarboxylate
- carbonyl or “carboxy” includes compounds and moieties which contain a carbon connected with a double bond to an oxygen atom, and tautomeric forms thereof.
- moieties that contain a carbonyl include aldehydes, ketones, carboxylic acids, amides, esters, anhydrides, etc.
- carboxy moiety refers to groups such as “alkylcarbonyl” groups wherein an alkyl group is covalently bound to a carbonyl group, "alkenylcarbonyl” groups wherein an alkenyl group is covalently bound to a carbonyl group, "alkynylcarbonyl” groups wherein an alkynyl group is covalently bound to a carbonyl group, “arylcarbonyl” groups wherein an aryl group is covalently attached to the carbonyl group.
- the term also refers to groups wherein one or more heteroatoms are covalently bonded to the carbonyl moiety.
- the term includes moieties such as, for example, aminocarbonyl moieties, (wherein a nitrogen atom is bound to the carbon of the carbonyl group, e.g., an amide), aminocarbonyloxy moieties, wherein an oxygen and a nitrogen atom are both bond to the carbon of the carbonyl group (e.g., also referred to as a "carbamate").
- aminocarbonylamino groups e.g., ureas
- heteroatom can be further substituted with one or more alkyl, alkenyl, alkynyl, aryl, aralkyl, acyl, etc. moieties.
- thiocarbonyl or “thiocarboxy” includes compounds and moieties which contain a carbon connected with a double bond to a sulfur atom.
- thiocarbonyl moiety includes moieties that are analogous to carbonyl moieties.
- thiocarbonyl moieties include aminothiocarbonyl, wherein an amino group is bound to the carbon atom of the thiocarbonyl group, furthermore other thiocarbonyl moieties include, oxythiocarbonyls (oxygen bound to the carbon atom), aminothiocarbonylamino groups, etc.
- ether includes compounds or moieties that contain an oxygen bonded to two different carbon atoms or heteroatoms.
- alkoxyalkyl which refers to an alkyl, alkenyl, or alkynyl group covalently bonded to an oxygen atom that is covalently bonded to another alkyl group.
- esters includes compounds and moieties that contain a carbon or a heteroatom bound to an oxygen atom that is bonded to the carbon of a carbonyl group.
- ester includes alkoxycarboxy groups such as methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl, butoxycarbonyl, pentoxycarbonyl, etc.
- alkyl, alkenyl, or alkynyl groups are as defined above.
- thioether includes compounds and moieties which contain a sulfur atom bonded to two different carbon or hetero atoms.
- examples of thioethers include, but are not limited to alkthioalkyls, alkthioalkenyls, and alkthioalkynyls.
- alkthioalkyls include compounds with an alkyl, alkenyl, or alkynyl group bonded to a sulfur atom that is bonded to an alkyl group.
- alkthioalkenyls and alkthioalkynyls refer to compounds or moieties wherein an alkyl, alkenyl, or alkynyl group is bonded to a sulfur atom which is covalently bonded to an alkynyl group.
- hydroxy or “hydroxyl” includes groups with an -OH or -O " .
- halogen includes fluorine, bromine, chlorine, iodine, etc.
- perhalogenated generally refers to a moiety wherein all hydrogens are replaced by halogen atoms.
- polycyclyl or “polycyclic radical” include moieties with two or more rings (e.g., cycloalkyls, cycloalkenyls, cycloalkynyls, aryls and/or heterocyclyls) in which two or more carbons are common to two adjoining rings, e.g., the rings are "fused rings".
- Rings that are joined through non-adjacent atoms are termed "bridged” rings.
- Each of the rings of the polycycle can be substituted with such substituents as described above, as for example, halogen, hydroxyl, alkylcarbonyloxy, arylcarbonyloxy, alkoxycarbonyloxy, aryloxycarbonyloxy, carboxylate, alkylcarbonyl, alkoxycarbonyl, alkylaminoacarbonyl, aralkylaminocarbonyl, alkenylaminocarbonyl, alkylcarbonyl, arylcarbonyl, aralkylcarbonyl, alkenylcarbonyl, aminocarbonyl, alkylthiocarbonyl, alkoxyl, phosphate, phosphonato, phosphinato, cyano, amino (including alkyl amino, dialkylamino, arylamino, diarylamino, and alkylarylamino), acylamino (including al
- Preferred heteroatoms are nitrogen, oxygen, sulfur and phosphorus.
- any combination thereof implies that any number of the listed functional groups and molecules may be combined to create a larger molecular architecture.
- bonds and/or hydrogen atoms are added to provide the following number of total bonds to each of the following types of atoms: carbon: four bonds; nitrogen: three bonds; oxygen: two bonds; and sulfur: two-six bonds.
- the structures of some of the compounds of this invention include asymmetric carbon atoms. It is to be understood accordingly that the isomers arising from such asymmetry (e.g., all enantiomers, stereoisomers, rotamers, tautomers, diastereomers, or racemates) are included within the scope of this invention. Such isomers can be obtained in substantially pure form by classical separation techniques and by stereochemically controlled synthesis. Furthermore, the structures and other compounds and moieties discussed in this application also include all tautomers thereof. Compounds described herein may be obtained through art recognized synthesis strategies.
- substituents of some of the compounds of this invention include isomeric cyclic structures. It is to be understood accordingly that constitutional isomers of particular substituents are included within the scope of this invention, unless indicated otherwise.
- tetrazole includes tetrazole, 2H-tetrazole, 3H- tetrazole, 4H-tetrazole and 5H-tetrazole.
- compounds of the present invention have valuable pharmacological properties and are useful in the treatment of diseases.
- compounds of the invention are useful in the treatment of a proliferative disease, or cancer.
- a proliferative disease is mainly a tumor disease (or cancer) (and/or any metastases).
- the inventive compounds are particularly useful for treating a tumor which is a breast cancer, genitourinary cancer, lung cancer, gastrointestinal cancer, epidermoid cancer, melanoma, ovarian cancer, pancreas cancer, neuroblastoma, head and/or neck cancer or bladder cancer, or in a broader sense renal, brain or gastric cancer; in particular (i) a breast tumor; an epidermoid tumor, such as an epidermoid head and/or neck tumor or a mouth tumor; a lung tumor, for example a small cell or non-small cell lung tumor; a gastrointestinal tumor, for example, a colorectal tumor; or a genitourinary tumor, for example, a prostate tumor (especially a hormone-refractory prostate tumor); or (ii) a proliferative disease that is refractory to the treatment with other chemotherapeutics; or (iii)
- a proliferative disease may furthermore be a hyperproliferative condition such as leukemias, hyperplasias, fibrosis (especially pulmonary, but also other types of fibrosis, such as renal fibrosis), angiogenesis, psoriasis, atherosclerosis and smooth muscle proliferation in the blood vessels, such as stenosis or restenosis following angioplasty.
- a hyperproliferative condition such as leukemias, hyperplasias, fibrosis (especially pulmonary, but also other types of fibrosis, such as renal fibrosis), angiogenesis, psoriasis, atherosclerosis and smooth muscle proliferation in the blood vessels, such as stenosis or restenosis following angioplasty.
- metastasis in the original organ or tissue and/or in any other location are implied alternatively or in addition, whatever the location of the tumor and/or metastasis.
- the inventive compound is selectively toxic or more toxic to rapidly proliferating cells than to normal cells, particularly in human cancer cells, e.g., cancerous tumors, the compound has significant antiproliferative effects and promotes differentiation, e.g., cell cycle arrest and apoptosis.
- compounds of the invention are useful in the treatment of transplant rejections.
- transplant rejections that may be treated by the compounds of the invention include, but are not limited to, graft versus host disease, rejection related to xeno transplantation, rejection related to organ transplant, rejection related to acute transplant, heterograft or homograft rejection and ischemic or reperfusion injury incurred during organ transplantation.
- compounds of the invention are useful in the treatment of autoimmune diseases.
- autoimmune diseases to be treated by the compounds of the invention include, but are not limited to, autoimmune hemolytic anemia, autoimmune neonatal thrombocytopenia, idiopathic thrombocytopenia purpura, autoimmunocytopenia, hemolytic anemia, antiphospholipid syndrome, dermatitis, allergic encephalomyelitis, myocarditis, relapsing polychondritis, rheumatic heart disease, glomerulonephritis, multiple sclerosis, neuritis, uveitis ophthalmia, polyendocrinopathies, purpura, Reiter's Disease, Stiff-Man Syndrome, autoimmune pulmonary inflammation, autism, Guillain-Barre Syndrome, insulin dependent diabetes mellitis, autoimmune inflammatory eye, autoimmune thyroiditis, hypothyroidism, systemic lupus erythematosus, Goodpasture's syndrome, Pemph
- use includes any one or more of the following embodiments of the invention, respectively: the use in the treatment of protein kinase-associated disorders; the use for the manufacture of pharmaceutical compositions for use in the treatment of these diseases, e.g., in the manufacture of a medicament; methods of use of compounds of the invention in the treatment of these diseases; pharmaceutical preparations having compounds of the invention for the treatment of these diseases; and compounds of the invention for use in the treatment of these diseases; as appropriate and expedient, if not stated otherwise.
- diseases to be treated and are thus preferred for use of a compound of the present invention are selected from cancer, transplant rejections, or autoimmune diseases, as well as those diseases that depend on the activity of protein kinases.
- compositions herein which bind to a protein kinase sufficiently to serve as tracers or labels, so that when coupled to a fluor or tag, or made radioactive, can be used as a research reagent or as a diagnostic or an imaging agent.
- Assays
- the inhibition of protein kinase activity by the compounds of the invention may be measured using a number of assays available in the art. Examples of such assays are described in the Exemplification section below.
- an effective amount of the compound is that amount necessary or sufficient to treat or prevent a protein kinase-associated disorder, e.g. prevent the various morphological and somatic symptoms of a protein kinase-associated disorder, and/or a disease or condition described herein.
- an effective amount of the compound of the invention is the amount sufficient to treat a protein kinase-associated disorder in a subject.
- the effective amount can vary depending on such factors as the size and weight of the subject, the type of illness, or the particular compound of the invention. For example, the choice of the compound of the invention can affect what constitutes an "effective amount.”
- One of ordinary skill in the art would be able to study the factors contained herein and make the determination regarding the effective amount of the compounds of the invention without undue experimentation.
- the regimen of administration can affect what constitutes an effective amount.
- the compound of the invention can be administered to the subject either prior to or after the onset of a protein kinase-associated disorder. Further, several divided dosages, as well as staggered dosages, can be administered daily or sequentially, or the dose can be continuously infused, or can be a bolus injection. Further, the dosages of the compound(s) of the invention can be proportionally increased or decreased as indicated by the exigencies of the therapeutic or prophylactic situation.
- Compounds of the invention may be used in the treatment of states, disorders or diseases as described herein, or for the manufacture of pharmaceutical compositions for use in the treatment of these diseases.
- compositions suitable for administration to mammals, e.g., humans.
- pharmaceutical compositions containing, for example, 0.1 to 99.5% (more preferably, 0.5 to 90%) of active ingredient in combination with a pharmaceutically acceptable carrier.
- pharmaceutically acceptable carrier is art recognized and includes a pharmaceutically acceptable material, composition or vehicle, suitable for administering compounds of the present invention to mammals.
- the carriers include liquid or solid filler, diluent, excipient, solvent or encapsulating material, involved in carrying or transporting the subject agent from one organ, or portion of the body, to another organ, or portion of the body.
- Each carrier must be "acceptable” in the sense of being compatible with the other ingredients of the formulation and not injurious to the patient.
- materials which can serve as pharmaceutically acceptable carriers include: sugars, such as lactose, glucose and sucrose; starches, such as corn starch and potato starch; cellulose, and its derivatives, such as sodium carboxymethyl cellulose, ethyl cellulose and cellulose acetate; powdered tragacanth; malt; gelatin; talc; excipients, such as cocoa butter and suppository waxes; oils, such as peanut oil, cottonseed oil, safflower oil, sesame oil, olive oil, corn oil and soybean oil; glycols, such as propylene glycol; polyols, such as glycerin, sorbitol, mannitol and polyethylene glycol; esters, such as ethyl oleate and ethyl laurate; agar; buffering agents, such as magnesium hydroxide and aluminum hydroxide; alginic acid; pyrogen-free water; isotonic saline; Ringer'
- wetting agents such as sodium lauryl sulfate and magnesium stearate, as well as coloring agents, release agents, coating agents, sweetening, flavoring and perfuming agents, preservatives and antioxidants can also be present in the compositions.
- antioxidants examples include: water soluble antioxidants, such as ascorbic acid, cysteine hydrochloride, sodium bisulfate, sodium metabisulfite, sodium sulfite and the like; oil-soluble antioxidants, such as ascorbyl palmitate, butylated hydroxyanisole (BHA), butylated hydroxytoluene (BHT), lecithin, propyl gallate, ⁇ -tocopherol, and the like; and metal chelating agents, such as citric acid, ethylenediamine tetraacetic acid (EDTA), sorbitol, tartaric acid, phosphoric acid, and the like.
- water soluble antioxidants such as ascorbic acid, cysteine hydrochloride, sodium bisulfate, sodium metabisulfite, sodium sulfite and the like
- oil-soluble antioxidants such as ascorbyl palmitate, butylated hydroxyanisole (BHA), butylated hydroxytoluene (BHT), lecithin
- Formulations of the present invention include those suitable for oral, nasal, topical, buccal, sublingual, rectal, vaginal and/or parenteral administration.
- the formulations may conveniently be presented in unit dosage form and may be prepared by any methods well known in the art of pharmacy.
- the amount of active ingredient that can be combined with a carrier material to produce a single dosage form will generally be that amount of the compound that produces a therapeutic effect. Generally, out of one hundred per cent, this amount will range from about 1 per cent to about ninety-nine percent of active ingredient, preferably from about 5 per cent to about 70 per cent, most preferably from about 10 per cent to about 30 per cent.
- Methods of preparing these formulations or compositions include the step of bringing into association a compound of the present invention with the carrier and, optionally, one or more accessory ingredients.
- the formulations are prepared by uniformly and intimately bringing into association a compound of the present invention with liquid carriers, or finely divided solid carriers, or both, and then, if necessary, shaping the product.
- Formulations of the invention suitable for oral administration may be in the form of capsules, cachets, pills, tablets, lozenges (using a flavored basis, usually sucrose and acacia or tragacanth), powders, granules, or as a solution or a suspension in an aqueous or non-aqueous liquid, or as an oil-in- water or water-in-oil liquid emulsion, or as an elixir or syrup, or as pastilles (using an inert base, such as gelatin and glycerin, or sucrose and acacia) and/or as mouth washes and the like, each containing a predetermined amount of a compound of the present invention as an active ingredient.
- a compound of the present invention may also be administered as a bolus, electuary or paste.
- the active ingredient is mixed with one or more pharmaceutically acceptable carriers, such as sodium citrate or dicalcium phosphate, and/or any of the following: fillers or extenders, such as starches, lactose, sucrose, glucose, mannitol, and/or silicic acid; binders, such as, for example, carboxymethylcellulose, alginates, gelatin, polyvinyl pyrrolidone, sucrose and/or acacia; humectants, such as glycerol; disintegrating agents, such as agar-agar, calcium carbonate, potato or tapioca starch, alginic acid, certain silicates, and sodium carbonate; solution retarding agents, such as paraffin; absorption accelerators, such as quaternary ammonium compounds; wetting agents, such as, for example, cetyl alcohol and glycerol monostea
- compositions may also comprise buffering agents.
- Solid compositions of a similar type may also be employed as fillers in soft and hard-filled gelatin capsules using such excipients as lactose or milk sugars, as well as high molecular weight polyethylene glycols and the like.
- a tablet may be made by compression or molding, optionally with one or more accessory ingredients.
- Compressed tablets may be prepared using binder (for example, gelatin or hydroxypropylmethyl cellulose), lubricant, inert diluent, preservative, disintegrant (for example, sodium starch glycolate or cross-linked sodium carboxymethyl cellulose), surface-active or dispersing agent.
- Molded tablets may be made by molding in a suitable machine a mixture of the powdered compound moistened with an inert liquid diluent.
- the tablets, and other solid dosage forms of the pharmaceutical compositions of the present invention may optionally be scored or prepared with coatings and shells, such as enteric coatings and other coatings well known in the pharmaceutical-formulating art. They may also be formulated so as to provide slow or controlled release of the active ingredient therein using, for example, hydroxypropylmethyl cellulose in varying proportions to provide the desired release profile, other polymer matrices, liposomes and/or microspheres.
- compositions may be sterilized by, for example, filtration through a bacteria-retaining filter, or by incorporating sterilizing agents in the form of sterile solid compositions that can be dissolved in sterile water, or some other sterile injectable medium immediately before use.
- These compositions may also optionally contain opacifying agents and may be of a composition that they release the active ingredient(s) only, or preferentially, in a certain portion of the gastrointestinal tract, optionally, in a delayed manner.
- embedding compositions that can be used include polymeric substances and waxes.
- the active ingredient can also be in micro-encapsulated form, if appropriate, with one or more of the above-described excipients.
- Liquid dosage forms for oral administration of the compounds of the invention include pharmaceutically acceptable emulsions, microemulsions, solutions, suspensions, syrups and elixirs.
- the liquid dosage forms may contain inert diluent commonly used in the art, such as, for example, water or other solvents, solubilizing agents and emulsifiers, such as ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene glycol, 1,3-butylene glycol, oils (in particular, cottonseed, groundnut, corn, germ, olive, castor and sesame oils), glycerol, tetrahydrofuryl alcohol, polyethylene glycols and fatty acid esters of sorbitan, and mixtures thereof.
- inert diluent commonly used in the art, such as, for example, water or other solvents, solubilizing agents and e
- the oral compositions can also include adjuvants such as wetting agents, emulsifying and suspending agents, sweetening, flavoring, coloring, perfuming and preservative agents.
- adjuvants such as wetting agents, emulsifying and suspending agents, sweetening, flavoring, coloring, perfuming and preservative agents.
- Suspensions in addition to the active compounds, may contain suspending agents as, for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar and tragacanth, and mixtures thereof.
- suspending agents as, for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar and tragacanth, and mixtures thereof.
- Formulations of the pharmaceutical compositions of the invention for rectal or vaginal administration may be presented as a suppository, which may be prepared by mixing one or more compounds of the invention with one or more suitable nonirritating excipients or carriers comprising, for example, cocoa butter, polyethylene glycol, a suppository wax or a salicylate, and which is solid at room temperature, but liquid at body temperature and, therefore, will melt in the rectum or vaginal cavity and release the active compound.
- suitable nonirritating excipients or carriers comprising, for example, cocoa butter, polyethylene glycol, a suppository wax or a salicylate, and which is solid at room temperature, but liquid at body temperature and, therefore, will melt in the rectum or vaginal cavity and release the active compound.
- Formulations of the present invention which are suitable for vaginal administration also include pessaries, tampons, creams, gels, pastes, foams or spray formulations containing such carriers as are known in the art to be appropriate.
- Dosage forms for the topical or transdermal administration of a compound of this invention include powders, sprays, ointments, pastes, creams, lotions, gels, solutions, patches and inhalants.
- the active compound may be mixed under sterile conditions with a pharmaceutically acceptable carrier, and with any preservatives, buffers, or propellants that may be required.
- the ointments, pastes, creams and gels may contain, in addition to an active compound of this invention, excipients, such as animal and vegetable fats, oils, waxes, paraffins, starch, tragacanth, cellulose derivatives, polyethylene glycols, silicones, bentonites, silicic acid, talc and zinc oxide, or mixtures thereof.
- excipients such as animal and vegetable fats, oils, waxes, paraffins, starch, tragacanth, cellulose derivatives, polyethylene glycols, silicones, bentonites, silicic acid, talc and zinc oxide, or mixtures thereof.
- Powders and sprays can contain, in addition to a compound of this invention, excipients such as lactose, talc, silicic acid, aluminum hydroxide, calcium silicates and polyamide powder, or mixtures of these substances.
- Sprays can additionally contain customary propellants, such as chlorofluorohydrocarbons and volatile unsubstituted hydrocarbons, such as butane and propane.
- Transdermal patches have the added advantage of providing controlled delivery of a compound of the present invention to the body.
- dosage forms can be made by dissolving or dispersing the compound in the proper medium.
- Absorption enhancers can also be used to increase the flux of the compound across the skin. The rate of such flux can be controlled by either providing a rate controlling membrane or dispersing the active compound in a polymer matrix or gel.
- Ophthalmic formulations, eye ointments, powders, solutions and the like, are also contemplated as being within the scope of this invention.
- compositions of this invention suitable for parenteral administration comprise one or more compounds of the invention in combination with one or more pharmaceutically acceptable sterile isotonic aqueous or nonaqueous solutions, dispersions, suspensions or emulsions, or sterile powders which may be reconstituted into sterile injectable solutions or dispersions just prior to use, which may contain antioxidants, buffers, bacteriostats, solutes which render the formulation isotonic with the blood of the intended recipient or suspending or thickening agents.
- aqueous and nonaqueous carriers examples include water, ethanol, polyols (such as glycerol, propylene glycol, polyethylene glycol, and the like), and suitable mixtures thereof, vegetable oils, such as olive oil, and injectable organic esters, such as ethyl oleate.
- polyols such as glycerol, propylene glycol, polyethylene glycol, and the like
- vegetable oils such as olive oil
- injectable organic esters such as ethyl oleate.
- Proper fluidity can be maintained, for example, by the use of coating materials, such as lecithin, by the maintenance of the required particle size in the case of dispersions, and by the use of surfactants.
- compositions may also contain adjuvants such as preservatives, wetting agents, emulsifying agents and dispersing agents. Prevention of the action of microorganisms may be ensured by the inclusion of various antibacterial and antifungal agents, for example, paraben, chlorobutanol, phenol sorbic acid, and the like. It may also be desirable to include isotonic agents, such as sugars, sodium chloride, and the like into the compositions. In addition, prolonged absorption of the injectable pharmaceutical form may be brought about by the inclusion of agents that delay absorption such as aluminum monostearate and gelatin. In some cases, in order to prolong the effect of a drug, it is desirable to slow the absorption of the drug from subcutaneous or intramuscular injection.
- adjuvants such as preservatives, wetting agents, emulsifying agents and dispersing agents.
- Prevention of the action of microorganisms may be ensured by the inclusion of various antibacterial and antifungal agents, for example, paraben, chlorobutan
- Injectable depot forms are made by forming microencapsule matrices of the subject compounds in biodegradable polymers such as polylactide-polyglycolide. Depending on the ratio of drug to polymer, and the nature of the particular polymer employed, the rate of drug release can be controlled. Examples of other biodegradable polymers include poly(orthoesters) and poly(anhydrides). Depot injectable formulations are also prepared by entrapping the drug in liposomes or microemulsions that are compatible with body tissue.
- the preparations of the present invention may be given orally, parenterally, topically, or rectally. They are of course given by forms suitable for each administration route. For example, they are administered in tablets or capsule form, by injection, inhalation, eye lotion, ointment, suppository, etc., administration by injection, infusion or inhalation; topical by lotion or ointment; and rectal by suppositories. Oral and/or IV administration is preferred.
- parenteral administration and “administered parenterally” as used herein means modes of administration other than enteral and topical administration, usually by injection, and includes, without limitation, intravenous, intramuscular, intraarterial, intrathecal, intracapsular, intraorbital, intracardiac, intradermal, intraperitoneal, transtracheal, subcutaneous, subcuticular, intraarticular, subcapsular, subarachnoid, intraspinal and intrasternal injection and infusion.
- systemic administration means the administration of a compound, drug or other material other than directly into the central nervous system, such that it enters the patient's system and, thus, is subject to metabolism and other like processes, for example, subcutaneous administration.
- These compounds may be administered to humans and other animals for therapy by any suitable route of administration, including orally, nasally, as by, for example, a spray, rectally, intravaginally, parenterally, intracisternally and topically, as by powders, ointments or drops, including buccally and sublingually.
- the compounds of the present invention which may be used in a suitable hydrated form, and/or the pharmaceutical compositions of the present invention, are formulated into pharmaceutically acceptable dosage forms by conventional methods known to those of skill in the art.
- Actual dosage levels of the active ingredients in the pharmaceutical compositions of this invention may be varied so as to obtain an amount of the active ingredient which is effective to achieve the desired therapeutic response for a particular patient, composition, and mode of administration, without being toxic to the patient.
- the selected dosage level will depend upon a variety of factors including the activity of the particular compound of the present invention employed, or the ester, salt or amide thereof, the route of administration, the time of administration, the rate of excretion of the particular compound being employed, the duration of the treatment, other drugs, compounds and/or materials used in combination with the particular compound employed, the age, sex, weight, condition, general health and prior medical history of the patient being treated, and like factors well known in the medical arts.
- a physician or veterinarian having ordinary skill in the art can readily determine and prescribe the effective amount of the pharmaceutical composition required.
- the physician or veterinarian could start doses of the compounds of the invention employed in the pharmaceutical composition at levels lower than that required in order to achieve the desired therapeutic effect and gradually increase the dosage until the desired effect is achieved.
- a suitable daily dose of a compound of the invention will be that amount of the compound that is the lowest dose effective to produce a therapeutic effect. Such an effective dose will generally depend upon the factors described above.
- intravenous and subcutaneous doses of the compounds of this invention for a patient when used for the indicated analgesic effects, will range from about 0.0001 to about 100 mg per kilogram of body weight per day, more preferably from about 0.01 to about 50 mg per kg per day, and still more preferably from about 1.0 to about 100 mg per kg per day.
- An effective amount is that amount treats a protein kinase-associated disorder.
- the effective daily dose of the active compound may be administered as two, three, four, five, six or more sub-doses administered separately at appropriate intervals throughout the day, optionally, in unit dosage forms. While it is possible for a compound of the present invention to be administered alone, it is preferable to administer the compound as a pharmaceutical composition.
- protecting group a readily removable group that is not a constituent of the particular desired end product of the compounds of the present invention.
- the protection of functional groups by such protecting groups, the protecting groups themselves, and their cleavage reactions are described for example in standard reference works, such as e.g., Science of Synthesis: Houben-Weyl Methods of Molecular Transformation. Georg Thieme Verlag, Stuttgart, Germany. 2005. 41627 pp. (URL: http://www.science-of-synthesis.com (Electronic Version, 48 Volumes)); J. F. W. McOmie, "Protective Groups in Organic Chemistry", Plenum Press, London and New York 1973, in T. W. Greene and P. G.
- Salts of compounds of the present invention having at least one salt-forming group may be prepared in a manner known per se.
- salts of compounds of the present invention having acid groups may be formed, for example, by treating the compounds with metal compounds, such as alkali metal salts of suitable organic carboxylic acids, e.g., the sodium salt of 2-ethylhexanoic acid, with organic alkali metal or alkaline earth metal compounds, such as the corresponding hydroxides, carbonates or hydrogen carbonates, such as sodium or potassium hydroxide, carbonate or hydrogen carbonate, with corresponding calcium compounds or with ammonia or a suitable organic amine, stoichiometric amounts or only a small excess of the salt-forming agent preferably being used.
- metal compounds such as alkali metal salts of suitable organic carboxylic acids, e.g., the sodium salt of 2-ethylhexanoic acid
- organic alkali metal or alkaline earth metal compounds such as the corresponding hydroxides, carbonates or hydrogen carbonates, such
- Acid addition salts of compounds of the present invention are obtained in customary manner, e.g., by treating the compounds with an acid or a suitable anion exchange reagent.
- Internal salts of compounds of the present invention containing acid and basic salt-forming groups, e.g., a free carboxy group and a free amino group, may be formed, e.g., by the neutralisation of salts, such as acid addition salts, to the isoelectric point, e.g., with weak bases, or by treatment with ion exchangers.
- Salts can be converted in customary manner into the free compounds; metal and ammonium salts can be converted, for example, by treatment with suitable acids, and acid addition salts, for example, by treatment with a suitable basic agent.
- diastereoisomers can be separated in a manner known per se into the individual isomers; diastereoisomers can be separated, for example, by partitioning between polyphasic solvent mixtures, recrystallisation and/or chromatographic separation, for example over silica gel or by, e.g., medium pressure liquid chromatography over a reversed phase column, and racemates can be separated, for example, by the formation of salts with optically pure salt-forming reagents and separation of the mixture of diastereoisomers so obtainable, for example by means of fractional crystallisation, or by chromatography over optically active column materials.
- Intermediates and final products can be worked up and/or purified according to standard methods, e.g., using chromatographic methods, distribution methods, (re-) crystallization, and the like.
- the process steps to synthesize the compounds of the invention can be carried out under reaction conditions that are known per se, including those mentioned specifically, in the absence or, customarily, in the presence of solvents or diluents, including, for example, solvents or diluents that are inert towards the reagents used and dissolve them, in the absence or presence of catalysts, condensation or neutralizing agents, for example ion exchangers, such as cation exchangers, e.g., in the H + form, depending on the nature of the reaction and/or of the reactants at reduced, normal or elevated temperature, for example in a temperature range of from about -100 0 C to about 19O 0 C, including, for example, from approximately - 8O 0 C to approximately 15O 0 C, for example at from -80 to -6O 0 C, at room temperature, at from -20 to 4O 0 C or at reflux temperature, under atmospheric pressure or in a closed vessel, where appropriate under pressure, and/or in an inert atmosphere
- mixtures of isomers that are formed can be separated into the individual isomers, for example diastereoisomers or enantiomers, or into any desired mixtures of isomers, for example racemates or mixtures of diastereoisomers, for example analogously to the methods described in Science of Synthesis: Houben-Weyl Methods of Molecular Transformation. Georg Thieme Verlag, Stuttgart, Germany. 2005.
- solvents from which those solvents that are suitable for any particular reaction may be selected include those mentioned specifically or, for example, water, esters, such as lower alkyl-lower alkanoates, for example ethyl acetate, ethers, such as aliphatic ethers, for example diethyl ether, or cyclic ethers, for example tetrahydrofuran or dioxane, liquid aromatic hydrocarbons, such as benzene or toluene, alcohols, such as methanol, ethanol or 1- or 2-propanol, nitriles, such as acetonitrile, halogenated hydrocarbons, such as methylene chloride or chloroform, acid amides, such as dimethylformamide or dimethyl acetamide, bases, such as heterocyclic nitrogen bases, for example pyridine or N-methylpyrrolidin-2- one, carboxylic acid anhydrides, such as lower alkanoic acid anhydrides, for example acetic anhydride,
- the compounds, including their salts, may also be obtained in the form of hydrates, or their crystals may, for example, include the solvent used for crystallization. Different crystalline forms may be present.
- the invention relates also to those forms of the process in which a compound obtainable as an intermediate at any stage of the process is used as starting material and the remaining process steps are carried out, or in which a starting material is formed under the reaction conditions or is used in the form of a derivative, for example in a protected form or in the form of a salt, or a compound obtainable by the process according to the invention is produced under the process conditions and processed further in situ.
- This invention also encompasses pharmaceutical compositions containing, and methods of treating protein kinase-associated disorders through administering, pharmaceutically acceptable prodrugs of compounds of the compounds of the invention.
- compounds of the invention having free amino, amido, hydroxy or carboxylic groups can be converted into prodrugs.
- Prodrugs include compounds wherein an amino acid residue, or a polypeptide chain of two or more ⁇ e.g., two, three or four) amino acid residues is covalently joined through an amide or ester bond to a free amino, hydroxy or carboxylic acid group of compounds of the invention.
- the amino acid residues include but are not limited to the 20 naturally occurring amino acids commonly designated by three letter symbols and also includes 4-hydroxyproline, hydroxylysine, demosine, isodemosine, 3-methylhistidine, norvalin, beta-alanine, gamma-aminobutyric acid, citrulline homocysteine, homoserine, ornithine and methionine sulfone. Additional types of prodrugs are also encompassed. For instance, free carboxyl groups can be derivatized as amides or alkyl esters.
- Free hydroxy groups may be derivatized using groups including but not limited to hemisuccinates, phosphate esters, dimethylaminoacetates, and phosphoryloxymethyloxycarbonyls, as outlined in Advanced Drug Delivery Reviews, 1996, 19, 115.
- Carbamate prodrugs of hydroxy and amino groups are also included, as are carbonate prodrugs, sulfonate esters and sulfate esters of hydroxy groups.
- acyl group may be an alkyl ester, optionally substituted with groups including but not limited to ether, amine and carboxylic acid functionalities, or where the acyl group is an amino acid ester as described above, are also encompassed.
- Prodrugs of this type are described in J. Med. Chem. 1996, 39, 10. Free amines can also be derivatized as amides, sulfonamides or phosphonamides. All of these prodrug moieties may incorporate groups including but not limited to ether, amine and carboxylic acid functionalities.
- a compound of the present invention may also be used in combination with other agents, e.g., an additional protein kinase inhibitor that is or is not a compound of the invention, for treatment of a protein kinase-associated disorder in a subject.
- agents e.g., an additional protein kinase inhibitor that is or is not a compound of the invention, for treatment of a protein kinase-associated disorder in a subject.
- combination is meant either a fixed combination in one dosage unit form, or a kit of parts for the combined administration where a compound of the present invention and a combination partner may be administered independently at the same time or separately within time intervals that especially allow that the combination partners show a cooperative, e.g., synergistic, effect, or any combination thereof.
- the compounds of the invention may be administered, simultaneously or sequentially, with an antiinflammatory, antiproliferative, chemotherapeutic agent, immunosuppressant, anti-cancer, cytotoxic agent or kinase inhibitor other than a compound of the Formula I or salt thereof.
- the compound of the invention and any additional agent may be formulated in separate dosage forms.
- the compound of the invention and any additional agent may be formulated together in any combination.
- the compound of the invention inhibitor may be formulated in one dosage form and the additional agent may be formulated together in another dosage form. Any separate dosage forms may be administered at the same time or different times.
- composition of this invention comprises an additional agent as described herein.
- Each component may be present in individual compositions, combination compositions, or in a single composition.
- Example 8 5 ,5-Dibromo-2-chloro-7-( 1 -ethyl-propyl)-5 ,7-dihydro-pyrrolo[2,3 -d]pyrimidin-6-one
- Example 30-33 By repeating the procedures described in example 29, using appropriate starting f materials, the following compounds are obtained.
- Example 135 2-[4-(4-Acetyl-piperazin- 1 -yl)-phenylamino]-7-( 1 -ethyl-propyl)-7H-pyrrolo[2,3- d]pyrimidine-6-carbaldehyde
- 1,4-dioxane (0.6 mL), Pd 2 (dba) 3 (12.2 mg, 0.013 mmol) and BINAP (16.6 mg, 0.026 mmol).
- the mixture is degassed, and heated at 100 0 C for 3 h.
- the mixture is cooled to room temperature, diluted with EtOAc, and filtered through a pad of Celite.
- the filtrate is concentrated under reduced pressure.
- the crude product is purified by preparative HPLC to give 84.9 mg of l-(l- ⁇ 4-[7-(l-ethyl-propyl)-7H-pyrrolo[2,3-d]pyrimidin-2-ylamino]- phenyl ⁇ -piperidin-4-yl)-ethanone as a pale white solid.
- 2-chloro-7-(cyclopentyl)-7H-pyrrolo[2,3-d]pyrimidine is prepared from cyclopentyl amine and 5-bromo-2,4-dichloropyrimidine using a method similar to that for the preparation of 2- chloro-7-(l-ethyl-propyl)-7H-pyrrolo[2,3-d]pyrimidine given in example 1.
- 2-Chloro-7-cyclopentyl-5-isopropyl-7H-pyrrolo[2,3-d]pyrimidine is prepared from 2-chloro- 7-(cyclopentyl)-7H-pyrrolo[2,3-d]pyrimidine and 2-chloropropane using a method similar to that for the preparation of l-(2-Chloro-7-cyclopentyl-7H-pyrrolo[2,3-d]pyrimidin-5-yl)- ethanone given in Example 325.
- 2-Chloro-7-cyclopentyl-6-methyl-7H-pyrrolo[2,3-d]pyrimidine is prepared from cyclopentyl amine and 5-bromo-2,4-dichloropyrimidine using a method similar to that for the preparation of2-chloro-7-(l-ethyl-propyl)-6-methyl-7H-pyrrolo[2,3-d]pyrimidine. given in example 328.
- Example 376 1 -(4- ⁇ 4-[7-(l -Ethyl-propyl)-6-(l -hydroxy-ethyl)-7H-pyrrolo[2,3-d]pyrimidin-2-ylamino]- phenyl ⁇ -piperazin- 1 -yl)-ethanone
- reaction mixture is stirred for 1 h, quenched with 10 % NaS 2 O 3 : saturated NaHCO 3 (1:1) aqueous solution, and extracted with CH 2 Cl 2 .
- the extracts are washed with water and brine, dried over Na 2 SO 4 , and concentrated in vacuo.
- the residue is purified by flash chromatography (SiO 2 , EtOAc/Hexane 1 :3) to afford 58 mg of l-[2-chloro-7-(l-ethyl- propyl)-7H-pyrrolo[2,3-d]pyrimidin-6-yl]-ethanone.
- (5-Bromo-2-chloro-pyrimidin-4-yl)cyclopentylamine is prepared from cyclopentyl amine and 5-bromo-2,4-dichloropyrimidine using a method similar to that for the preparation of (5- Bromo-2-chloro-pyrimidin-4-yl)-(l-ethylpropyl)amine given in Example 137.
- 2-chloro-7-cyclopentyl-6-methyl-7H-pyrrolo[2,3-d]pyrimidine-5-carboxylic acid methyl ester is prepared using the procedure shown for the preparation of its regio-isomer 2-chloro-7- cyclopentyl-5-methyl-7H-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid methyl ester given in the procedure for the synthesis of Example 384.
- Examples 387 - 408 are prepared using methods similar to those described in the syntheses of Examples 383-386 and standard synthetic methodology used in the synthesis of azole heterocycles with appropriate choice of starting materials.
- Phosphorylated STAT molecules dimerize and migrate to the nucleus where they bind to DNA and initiate transcription of responsive genes. Inhibitors of pathways downstream of cytokine/growth factor receptors have therapeutic potential for several indications.
- An enzymatic assay for JAK-3 and JAK-2 has been developed to identify T-cell selective inhibitors. GST fusion constructs of the kinase domains of both enzymes are used and a biotinylated, tyrosine containing peptide serves as substrate.
- Phosphorylation of this peptide by the respective kinase is quantified with an europium-labeled anti-phosphotyrosine antibody (Eu-PT66) as energy donor and a streptavidin-allophycocyanine conjugate (SA- APC) as energy acceptor.
- Eu-PT66 europium-labeled anti-phosphotyrosine antibody
- SA- APC streptavidin-allophycocyanine conjugate
- the assay has been established in a 384 well format.
- JAK LANCETM assay a biotinylated peptide is incubated together with compounds and ATP in buffer. The phosphorylation reaction starts after addition of JAK kinase. After incubation at RT the reaction is stopped with EDTA and the product detected by addition Streptavidin-Allophycocyanin and Europium-labeled antiphosphotyrosine antibody.
- the signal is measured using an EnVision Reader. Exc: 320nm, Em, Donor:
- Tables A and B Data acquired for the compounds of the invention using these assays are shown in Tables A and B.
- an ELISA based assay can be utilized, where the enzyme is a purified active CDK4/Cyclin-Dl kinase complex and the substrate is a purified Retinoblastoma (Rb) protein.
- the active CDK4/Cyclin-Dl kinase complex phosphorylates the Rb substrate at Serine780 residue, and then the phosphorylated Rb/S780 is detected via an antibody specific to the phosphorylated site.
- the compounds that inhibit the CDK4 kinase activity would inhibit the signal output of the assay.
- Data acquired for the compounds of the invention using this assay are shown in Table C.
- the CDK2 assay is a flourescence polarization assay, where the enzyme is a purified active CDK2/Cyclin-A kinase complex and the substrate is a synthesized peptide derived from Histone Hl .
- This assay utilizes the IMAP technology from Molecular Devices.
- the active CDK2/Cyclin-A complex phosphorylates the peptide substrate, which is conjugated with the TAMRA tag.
- the phosphorylated site is then recognized by a metal containing molecule that interacts with the TAMRA tag to induce a high flourescence polarization.
- the compounds that inhibit the CDK2 kinase activity would inhibit the flourescence output of the assay. Data acquired for the compounds of the invention using this assay are shown in Table C.
- Retinoblast Protein (pRb) antibody (4Hl Cell Signaling 9309L) diluted in DPBS (Phosphate Buffered Saline) overnight at 4 0 C.
- DPBS Phosphate Buffered Saline
- TBST Phosphate Buffered Saline
- Cells are plated at 50-60% confluency in a 96 well plate (Corning 3585) in lOOuL complete media (media containing fetal bovine serum (Gibco 1600-044), 2mM L-Glutamine (Gibco 25030), and 1% Penicillin/Streptomycin (Gibco 15140-122) and grown overnight in a humidified chamber at 37 0 C and 5% CO 2 .
- Compounds (in DMSO) are diluted in media to create a 7 point dilution series of compound with concentrations ranging from 1 lOuM to 0.027uM. lOul of the diluted compounds are added to the cells, with final concentrations on cells ranging from 1 OuM to 0.002uM. Cells are treated for 24 hrs in a humidified chamber at 37 0 C and 5% CO 2 .
- lOul of cell lysate and 5OuI lxPBS/10% Superblock (Gibco 10010 and Pierce 37535) is added to each well of the precoated and blocked Maxisorp plate and allowed to bind at room temperature for 2 hours on Oribtron Rotator II (Boekel Industries Model 260250). Plates are then washed 3x with Ix TBST (Teknova T9201) using Biotek platewasher equipped with a Biostack. The final wash is not aspirated. The final wash is removed by flicking off and tapping plate on paper towels.
- ppRbS780 antibody Cell Signaling 9307L
- lxPBS/10% Superblock Cell Signaling 9307L
- 50ul is added to each well. Plates are then incubated 1 hour on Oribtron Rotator II (Boekel Industries Model 260250). Plates are then washed as previously described.
- Goat anti-rabbit HRP Promega W401B
- Goat anti-rabbit HRP is diluted 1 :2500 lxPBS/10% Superblock (Gibco 10010 and Pierce 37535) and 50ul is added to each well. Plates are then incubated 30 minutes on Oribtron Rotator II. Plates are then washed as previously described.
- cells are plated in 96 well plates at 50-60% confluency in RMPI 1640 media. The next day, cells are treated with compounds at a desired concentration range and then incubated for 24 hrs in a humidified chamber at 37 0 C and 5% CO 2 . Following the protocol provided by the kit, cells are labeled with BrdU labeling agent for 2 hrs, and then fixed with 20OuL of
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Immunology (AREA)
- Diabetes (AREA)
- Rheumatology (AREA)
- Endocrinology (AREA)
- Physical Education & Sports Medicine (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Pulmonology (AREA)
- Hematology (AREA)
- Dermatology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Obesity (AREA)
- Reproductive Health (AREA)
- Cardiology (AREA)
- Pain & Pain Management (AREA)
- Hospice & Palliative Care (AREA)
- Psychiatry (AREA)
- Psychology (AREA)
- Heart & Thoracic Surgery (AREA)
- Oncology (AREA)
- Emergency Medicine (AREA)
- Gynecology & Obstetrics (AREA)
- Pregnancy & Childbirth (AREA)
Abstract
Description

















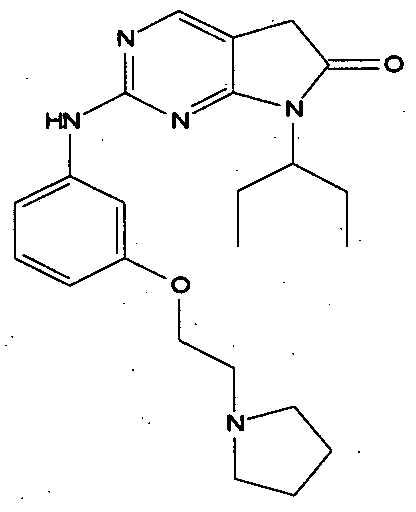










































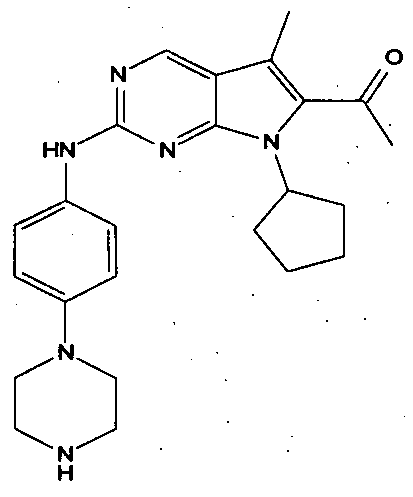






























































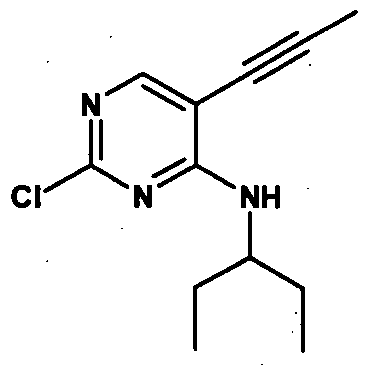























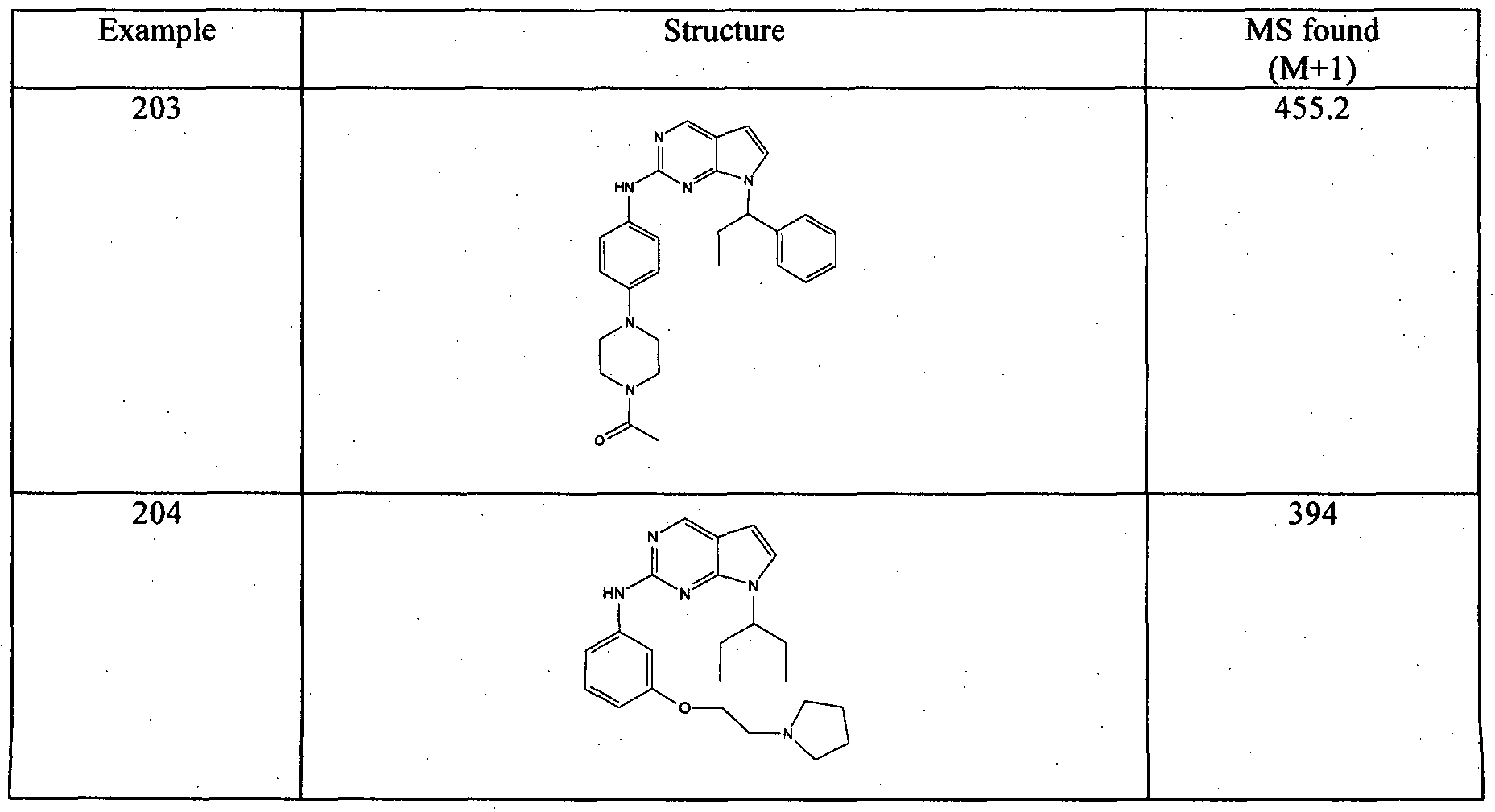









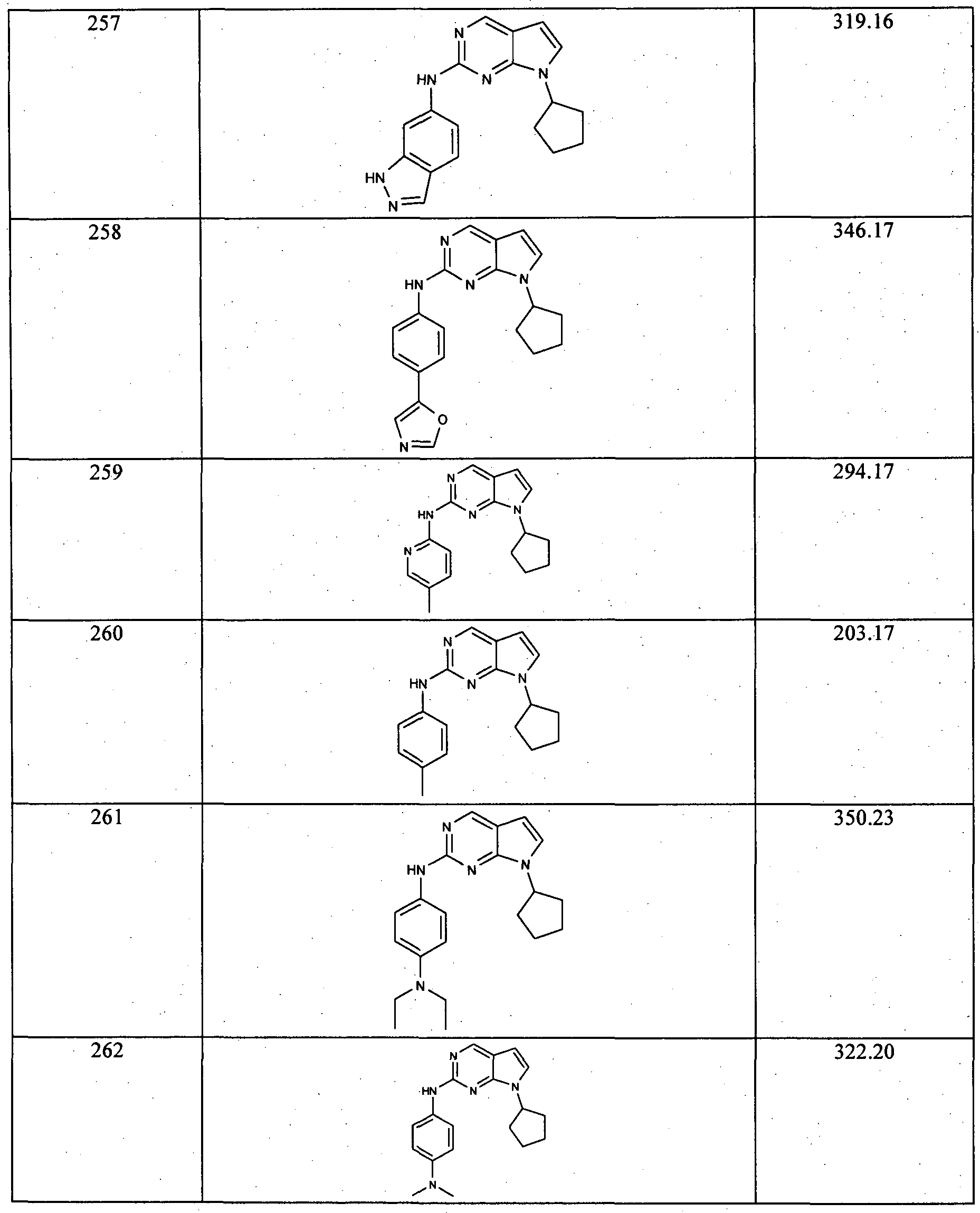












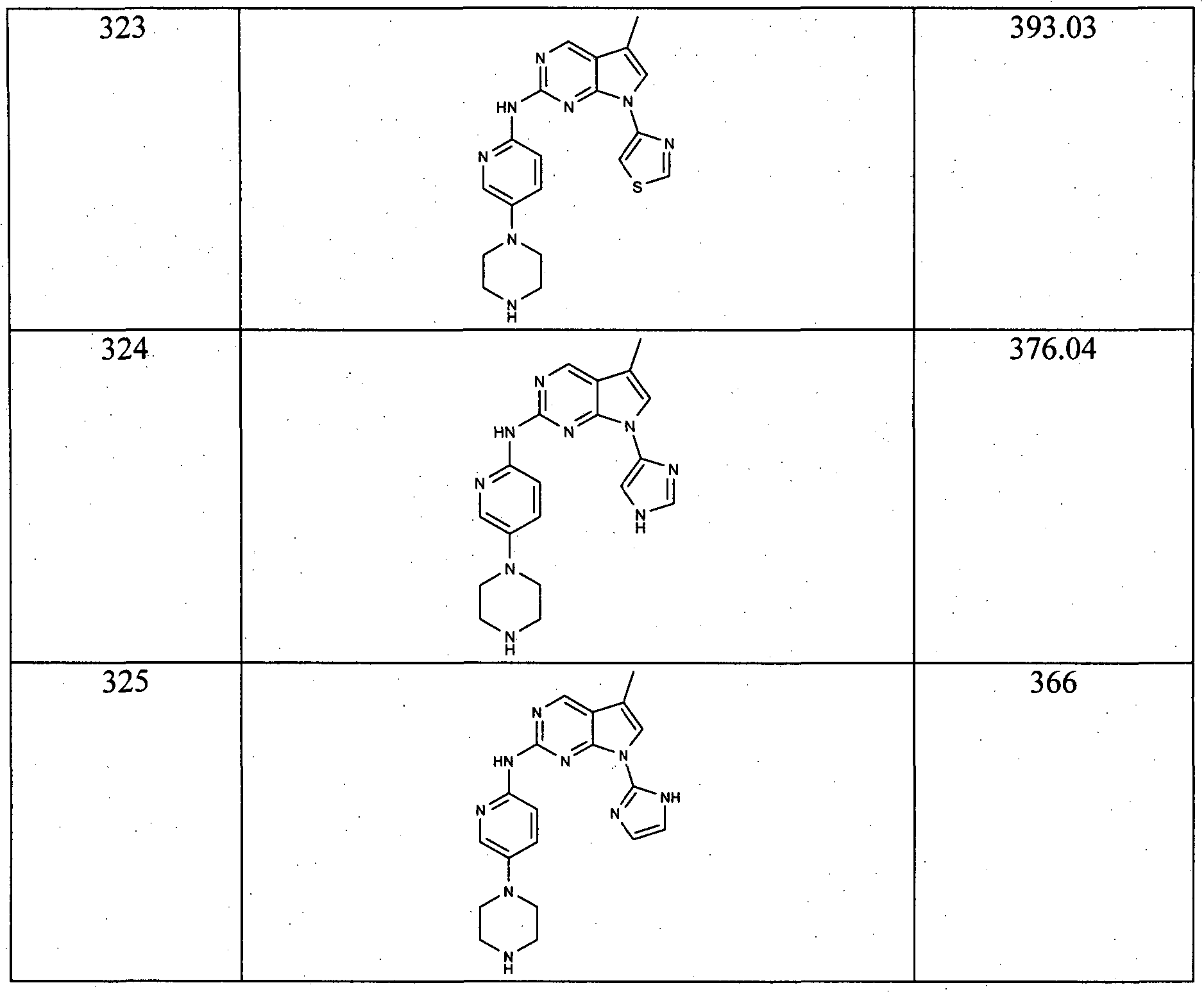



































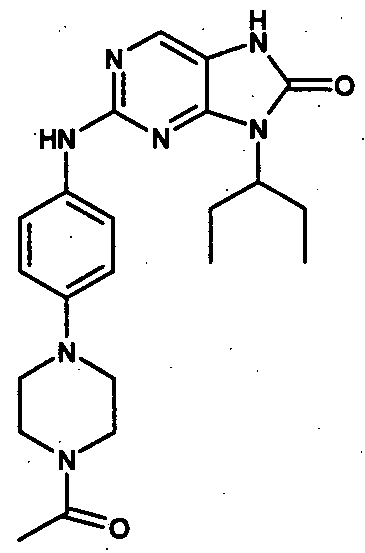





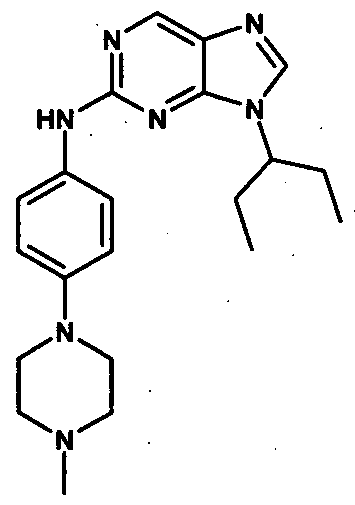


















Claims













Priority Applications (21)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BRPI0712816A BRPI0712816B8 (en) | 2006-05-26 | 2007-05-24 | pyrrolpyrimidine compounds and their uses |
| KR1020087031411A KR101466412B1 (en) | 2006-05-26 | 2007-05-24 | Pyrrolopyrimidine compounds and their uses |
| JP2009512291A JP2009538341A (en) | 2006-05-26 | 2007-05-24 | Pyrrolopyrimidine compounds and their use |
| CN2007800193572A CN101594871B (en) | 2006-05-26 | 2007-05-24 | Pyrrolopyrimidine compounds and their uses |
| EA200802332A EA016301B1 (en) | 2006-05-26 | 2007-05-24 | Pyrrolopyrimidine compounds and their uses |
| SM200800069T SMP200800069B (en) | 2006-05-26 | 2007-05-24 | Pyrrolopyrimidine compounds and their uses |
| ES07811927.8T ES2623133T3 (en) | 2006-05-26 | 2007-05-24 | Pyrrolopyrimidine compounds and their uses |
| NZ572549A NZ572549A (en) | 2006-05-26 | 2007-05-24 | Pyrrolopyrimidine compounds and their uses |
| CA2652044A CA2652044C (en) | 2006-05-26 | 2007-05-24 | Pyrrolopyrimidine compounds and their use as protein kinase inhibitors |
| MX2008015076A MX2008015076A (en) | 2006-05-26 | 2007-05-24 | Pyrrolopyrimidine compounds and their uses. |
| MEP-2008-766A ME00486B (en) | 2006-05-26 | 2007-05-24 | Pyrrolopyrimidine compounds and their uses |
| EP07811927.8A EP2029145B1 (en) | 2006-05-26 | 2007-05-24 | Pyrrolopyrimidine compounds and their uses |
| US12/302,223 US8324225B2 (en) | 2006-05-26 | 2007-05-24 | Pyrrolopyrimidine compounds and their uses |
| AU2007267645A AU2007267645C1 (en) | 2006-05-26 | 2007-05-24 | Pyrrolopyrimidine compounds and their uses |
| IL195086A IL195086A (en) | 2006-05-26 | 2008-11-03 | Pyrrolopyrimidine compounds and their uses |
| ZA2008/09382A ZA200809382B (en) | 2006-05-26 | 2008-11-03 | Pyrrolopyrimidine compounds and their uses |
| TNP2008000481A TNSN08481A1 (en) | 2006-05-26 | 2008-11-21 | Pyrrolopyrimidine compounds and their uses |
| CU2008000223A CU23831B1 (en) | 2006-05-26 | 2008-11-24 | PIRROLO-PYRIMIDINE COMPOUNDS |
| NO20085030A NO343182B1 (en) | 2006-05-26 | 2008-12-02 | Pyrrolopyrimidine Compounds and Their Uses |
| US13/452,100 US20120207763A1 (en) | 2006-05-26 | 2012-04-20 | Pyrrolopyrimidine compounds and their uses |
| HRP20170631TT HRP20170631T1 (en) | 2006-05-26 | 2017-04-21 | Pyrrolopyrimidine compounds and their uses |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US80860506P | 2006-05-26 | 2006-05-26 | |
| US60/808,605 | 2006-05-26 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US13/452,100 Division US20120207763A1 (en) | 2006-05-26 | 2012-04-20 | Pyrrolopyrimidine compounds and their uses |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007140222A2 true WO2007140222A2 (en) | 2007-12-06 |
| WO2007140222A3 WO2007140222A3 (en) | 2008-08-07 |
Family
ID=38779335
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/069595 WO2007140222A2 (en) | 2006-05-26 | 2007-05-24 | Pyrrolopyrimidine compounds and their uses |
Country Status (38)
| Country | Link |
|---|---|
| US (2) | US8324225B2 (en) |
| EP (1) | EP2029145B1 (en) |
| JP (2) | JP2009538341A (en) |
| KR (1) | KR101466412B1 (en) |
| CN (1) | CN101594871B (en) |
| AR (2) | AR061124A1 (en) |
| AU (1) | AU2007267645C1 (en) |
| BR (1) | BRPI0712816B8 (en) |
| CA (1) | CA2652044C (en) |
| CL (1) | CL2007001504A1 (en) |
| CR (1) | CR10433A (en) |
| CU (1) | CU23831B1 (en) |
| EA (1) | EA016301B1 (en) |
| EC (1) | ECSP088910A (en) |
| ES (1) | ES2623133T3 (en) |
| GE (1) | GEP20115283B (en) |
| GT (1) | GT200800258A (en) |
| HN (1) | HN2008001752A (en) |
| HR (1) | HRP20170631T1 (en) |
| IL (1) | IL195086A (en) |
| JO (1) | JO3235B1 (en) |
| MA (1) | MA30557B1 (en) |
| ME (1) | ME00486B (en) |
| MX (1) | MX2008015076A (en) |
| MY (1) | MY150650A (en) |
| NO (1) | NO343182B1 (en) |
| NZ (1) | NZ572549A (en) |
| PE (1) | PE20080263A1 (en) |
| PL (1) | PL2029145T3 (en) |
| PT (1) | PT2029145T (en) |
| SG (1) | SG172632A1 (en) |
| SM (1) | SMP200800069B (en) |
| TN (1) | TNSN08481A1 (en) |
| TW (1) | TWI398252B (en) |
| UA (1) | UA95632C2 (en) |
| UY (1) | UY30369A1 (en) |
| WO (1) | WO2007140222A2 (en) |
| ZA (1) | ZA200809382B (en) |
Cited By (127)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009024824A1 (en) * | 2007-08-23 | 2009-02-26 | Astrazeneca Ab | 2-anilinopurin-8-ones as inhibitors of ttk/mps1 for the treatment of proliferative disorders |
| WO2009085185A1 (en) * | 2007-12-19 | 2009-07-09 | Amgen Inc. | Fused pyridine, pyrimidine and triazine compounds as cell cycle inhibitors |
| WO2009110415A1 (en) * | 2008-03-03 | 2009-09-11 | 武田薬品工業株式会社 | Concomitant drug |
| WO2009124965A1 (en) | 2008-04-09 | 2009-10-15 | N.V. Organon | PYRROLO[2,3-d]PYRIMIDIN-2-YL-AMINE DERIVATIVES AS PKC-THETA INHIBITORS |
| WO2009152027A1 (en) * | 2008-06-12 | 2009-12-17 | Merck & Co., Inc. | 5,7-dihydro-6h-pyrrolo[2,3-d]pyrimidin-6-one derivatives for mark inhibition |
| WO2010020675A1 (en) * | 2008-08-22 | 2010-02-25 | Novartis Ag | Pyrrolopyrimidine compounds as cdk inhibitors |
| WO2010045451A1 (en) * | 2008-10-16 | 2010-04-22 | Glaxosmithkline Llc | Pyrrolopyrimidine compounds |
| WO2010100431A1 (en) | 2009-03-04 | 2010-09-10 | Medical Research Council Technology | Pyrrolopyrimidines used as kinase inhibitors |
| US7902187B2 (en) | 2006-10-04 | 2011-03-08 | Wyeth Llc | 6-substituted 2-(benzimidazolyl)purine and purinone derivatives for immunosuppression |
| US7915268B2 (en) | 2006-10-04 | 2011-03-29 | Wyeth Llc | 8-substituted 2-(benzimidazolyl)purine derivatives for immunosuppression |
| US7919490B2 (en) | 2006-10-04 | 2011-04-05 | Wyeth Llc | 6-substituted 2-(benzimidazolyl)purine and purinone derivatives for immunosuppression |
| JP2011518841A (en) * | 2008-04-24 | 2011-06-30 | ニューリンク ジェネティクス, インコーポレイテッド | IDO inhibitor |
| WO2011082400A2 (en) * | 2010-01-04 | 2011-07-07 | President And Fellows Of Harvard College | Modulators of immunoinhibitory receptor pd-1, and methods of use thereof |
| US7989459B2 (en) | 2006-02-17 | 2011-08-02 | Pharmacopeia, Llc | Purinones and 1H-imidazopyridinones as PKC-theta inhibitors |
| WO2011101409A1 (en) | 2010-02-19 | 2011-08-25 | Novartis Ag | Pyrrolopyrimidine compounds as inhibitors of cdk4/6 |
| WO2011101417A1 (en) | 2010-02-19 | 2011-08-25 | Novartis Ag | Deuterated pyrrolopyrimidine compounds as inhibitors of cdk4/6 |
| WO2011130232A1 (en) | 2010-04-13 | 2011-10-20 | Novartis Ag | Combination comprising a cyclin dependent kinase 4 or cyclin dependent kinase (cdk4/6) inhibitor and an mtor inhibitor for treating cancer |
| WO2012092880A1 (en) | 2011-01-07 | 2012-07-12 | Centaurus Biopharma Co., Ltd. | 2,4-DIAMINO-6,7-DIHYDRO-5H-PYRROLO[2,3]PYRIMIDINE DERIVATIVES AS FAK/Pyk2 INHIBITORS |
| WO2012110773A1 (en) | 2011-02-17 | 2012-08-23 | Cancer Therapeutics Crc Pty Limited | Fak inhibitors |
| WO2012110774A1 (en) | 2011-02-17 | 2012-08-23 | Cancer Therapeutics Crc Pty Limited | Selective fak inhibitors |
| WO2012127506A1 (en) | 2011-03-22 | 2012-09-27 | Advinus Therapeutics Limited | Substituted fused tricyclic compounds, compositions and medicinal applications thereof |
| WO2013006532A1 (en) | 2011-07-01 | 2013-01-10 | Novartis Ag | Combination therapy comprising a cdk4/6 inhibitor and a pi3k inhibitor for use in the treatment of cancer |
| WO2013006368A1 (en) | 2011-07-01 | 2013-01-10 | Novartis Ag | Combination therapy |
| WO2013017480A1 (en) * | 2011-07-29 | 2013-02-07 | Cellzome Limited | Pyrazolo[4,3-c]pyridine derivatives as jak inhibitors |
| WO2013017479A1 (en) * | 2011-07-29 | 2013-02-07 | Cellzome Limited | Pyrazolo[4,3-c]pyridine derivatives as jak inhibitors |
| US8389533B2 (en) | 2008-04-07 | 2013-03-05 | Amgen Inc. | Gem-disubstituted and spirocyclic amino pyridines/pyrimidines as cell cycle inhibitors |
| US8420657B2 (en) | 2008-02-06 | 2013-04-16 | Novartis Ag | Pyrrolo[2,3-D]pyrimidines and use thereof as tyrosine kinase inhibitors |
| US8501735B2 (en) | 2009-10-29 | 2013-08-06 | Palau Pharma, S.A. | N-containing heteroaryl derivatives as JAK3 kinase inhibitors |
| KR20130132433A (en) * | 2010-11-10 | 2013-12-04 | 노파르티스 아게 | Salt(s) of 7-cyclopentyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-7h-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid dimethylamide and processes of making thereof |
| WO2013188184A1 (en) * | 2012-06-14 | 2013-12-19 | Eli Lilly And Company | Inhibitor of jak1 and jak2 |
| US8623885B2 (en) | 2011-03-23 | 2014-01-07 | Amgen Inc. | Fused tricyclic dual inhibitors of CDK 4/6 and FLT3 |
| WO2014022830A2 (en) | 2012-08-03 | 2014-02-06 | Foundation Medicine, Inc. | Human papilloma virus as predictor of cancer prognosis |
| US8691807B2 (en) | 2011-06-20 | 2014-04-08 | Incyte Corporation | Azetidinyl phenyl, pyridyl or pyrazinyl carboxamide derivatives as JAK inhibitors |
| US8722693B2 (en) | 2007-06-13 | 2014-05-13 | Incyte Corporation | Salts of the Janus kinase inhibitor (R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| EP2742940A1 (en) | 2012-12-13 | 2014-06-18 | IP Gesellschaft für Management mbH | Salts of aza-bicyclo-di-aryl ethers for adminstration once daily, twice daily or thrice daily |
| WO2014130693A1 (en) * | 2013-02-25 | 2014-08-28 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| WO2014147573A2 (en) | 2013-03-21 | 2014-09-25 | Novartis Ag | Combination therapy |
| US8933086B2 (en) | 2005-12-13 | 2015-01-13 | Incyte Corporation | Heteroaryl substituted pyrrolo[2,3-B]pyridines and pyrrolo[2,3-B]pyrimidines as Janus kinase inhibitors |
| US8933085B2 (en) | 2010-11-19 | 2015-01-13 | Incyte Corporation | Cyclobutyl substituted pyrrolopyridine and pyrrolopyrimidine derivatives as JAK inhibitors |
| US8987443B2 (en) | 2013-03-06 | 2015-03-24 | Incyte Corporation | Processes and intermediates for making a JAK inhibitor |
| US9034884B2 (en) | 2010-11-19 | 2015-05-19 | Incyte Corporation | Heterocyclic-substituted pyrrolopyridines and pyrrolopyrimidines as JAK inhibitors |
| US9163021B2 (en) | 2012-10-04 | 2015-10-20 | Pfizer Limited | Pyrrolo[3,2-c]pyridine tropomyosin-related kinase inhibitors |
| WO2015160986A2 (en) | 2014-04-16 | 2015-10-22 | Infinity Pharmaceuticals, Inc. | Combination therapies |
| US20150313902A1 (en) * | 2012-12-20 | 2015-11-05 | Novartis Ag | Pharmaceutical combination comprising binimetinib |
| US9193733B2 (en) | 2012-05-18 | 2015-11-24 | Incyte Holdings Corporation | Piperidinylcyclobutyl substituted pyrrolopyridine and pyrrolopyrimidine derivatives as JAK inhibitors |
| WO2015181737A1 (en) | 2014-05-28 | 2015-12-03 | Piramal Enterprises Limited | Pharmaceutical combination for the treatment of cancer |
| US9216984B2 (en) | 2009-05-22 | 2015-12-22 | Incyte Corporation | 3-[4-(7H-pyrrolo[2,3-D]pyrimidin-4-yl)-1H-pyrazol-1-yl]octane—or heptane-nitrile as JAK inhibitors |
| US9233111B2 (en) | 2011-07-08 | 2016-01-12 | Novartis Ag | Pyrrolo pyrimidine derivatives |
| WO2016014904A1 (en) | 2014-07-24 | 2016-01-28 | Beta Pharma, Inc. | 2-h-indazole derivatives as cyclin-dependent kinase (cdk) inhibitors and therapeutic uses thereof |
| US9249145B2 (en) | 2009-09-01 | 2016-02-02 | Incyte Holdings Corporation | Heterocyclic derivatives of pyrazol-4-yl-pyrrolo[2,3-d]pyrimidines as janus kinase inhibitors |
| US9334286B2 (en) | 2012-09-07 | 2016-05-10 | Cancer Research Technology Limited | Pharmacologically active compounds |
| US9359358B2 (en) | 2011-08-18 | 2016-06-07 | Incyte Holdings Corporation | Cyclohexyl azetidine derivatives as JAK inhibitors |
| US9371319B2 (en) | 2011-03-14 | 2016-06-21 | Cancer Research Technology Limited | Pyrrolopyridineamino derivatives as MPS1 inhibitors |
| US9409907B2 (en) | 2012-09-07 | 2016-08-09 | Cancer Research Technology Limited | Inhibitor compounds |
| US9464088B2 (en) | 2010-03-10 | 2016-10-11 | Incyte Holdings Corporation | Piperidin-4-yl azetidine derivatives as JAK1 inhibitors |
| US9487521B2 (en) | 2011-09-07 | 2016-11-08 | Incyte Holdings Corporation | Processes and intermediates for making a JAK inhibitor |
| US9498467B2 (en) | 2014-05-30 | 2016-11-22 | Incyte Corporation | Treatment of chronic neutrophilic leukemia (CNL) and atypical chronic myeloid leukemia (aCML) by inhibitors of JAK1 |
| US9593115B2 (en) | 2012-09-21 | 2017-03-14 | Advinus Therapeutics Ltd. | Substituted fused tricyclic compounds, compositions, and medicinal applications thereof |
| US9616062B2 (en) | 2009-05-13 | 2017-04-11 | The University Of North Carolina At Chapel Hill | Cyclin dependent kinase inhibitors and methods of use |
| US9655854B2 (en) | 2013-08-07 | 2017-05-23 | Incyte Corporation | Sustained release dosage forms for a JAK1 inhibitor |
| WO2017114351A1 (en) * | 2015-12-27 | 2017-07-06 | Chongqing Fochon Pharmaceutical Co., Ltd. | Certain protein kinase inhibitors |
| US9808461B2 (en) | 2010-11-17 | 2017-11-07 | The University Of North Carolina At Chapel Hill | Protection of renal tissues from ischemia through inhibition of the proliferative kinases CDK4 and CDK6 |
| US9902721B2 (en) | 2014-02-28 | 2018-02-27 | Cancer Research Technology Limited | N2-phenyl-pyrido[3,4-d]pyrimidine-2, 8-diamine derivatives and their use as Mps1 inhibitors |
| WO2018039324A1 (en) | 2016-08-23 | 2018-03-01 | Eisai R&D Management Co., Ltd. | Combination therapies for the treatment of hepatocellular carcinoma |
| WO2018053437A1 (en) | 2016-09-19 | 2018-03-22 | Mei Pharma, Inc. | Combination therapy |
| WO2018073687A1 (en) | 2016-10-20 | 2018-04-26 | Pfizer Inc. | Anti-proliferative agents for treating pah |
| US10011874B2 (en) | 2013-02-25 | 2018-07-03 | Novartis Ag | Androgen receptor mutation |
| WO2018124001A1 (en) | 2016-12-27 | 2018-07-05 | 国立研究開発法人理化学研究所 | Bmp-signal-inhibiting compound |
| US20180244676A1 (en) * | 2015-02-13 | 2018-08-30 | Dana-Farber Cancer Institute, Inc. | Lrrk2 inhibitors and methods of making and using the same |
| EP3284746A4 (en) * | 2015-04-17 | 2018-09-12 | Changzhou Longthera Pharmaceuticals Inc | Preparation and use of kinase inhibitor |
| WO2018170447A1 (en) | 2017-03-16 | 2018-09-20 | Eisai R&D Management Co., Ltd. | Combination therapies for the treatment of breast cancer |
| WO2018232235A1 (en) | 2017-06-16 | 2018-12-20 | Beta Pharma, Inc. | Pharmaceutical formulations of n-(2-(2-(dimethylamino)ethoxy)-4-methoxy-5-((4-(1-methyl-1h-indol-3-yl)pyrimidin-2-yl)amino)phenyl)acrylamide and salts thereof |
| US10166191B2 (en) | 2012-11-15 | 2019-01-01 | Incyte Corporation | Sustained-release dosage forms of ruxolitinib |
| WO2019148161A1 (en) | 2018-01-29 | 2019-08-01 | Beta Pharma, Inc. | 2h-indazole derivatives as cdk4 and cdk6 inhibitors and therapeutic uses thereof |
| WO2019157020A1 (en) | 2018-02-06 | 2019-08-15 | The Board Of Trustees Of The University Of Illinois | Substituted benzothiophene analogs as selective estrogen receptor degraders |
| US10407446B2 (en) | 2016-12-20 | 2019-09-10 | Astrazeneca Ab | Amino-triazolopyridine compounds and their use in treating cancer |
| US10421747B2 (en) | 2014-03-26 | 2019-09-24 | Astex Therapeutics Ltd | Quinoxaline derivatives useful as FGFR kinase modulators |
| US10519137B2 (en) | 2010-04-30 | 2019-12-31 | Astex Therapeutics Ltd | Pyrazolyl quinoxaline kinase inhibitors |
| US10596161B2 (en) | 2017-12-08 | 2020-03-24 | Incyte Corporation | Low dose combination therapy for treatment of myeloproliferative neoplasms |
| US10662186B2 (en) | 2015-12-31 | 2020-05-26 | Shanghai Pharmaceuticals Holding Co., Ltd. | Substituted pyrimidines as cyclin-dependent kinase inhibitors |
| EP3560926A4 (en) * | 2016-12-21 | 2020-06-17 | Ono Pharmaceutical Co., Ltd. | Brk INHIBITORY COMPOUND |
| WO2020140052A1 (en) * | 2018-12-28 | 2020-07-02 | Spv Therapeutics Inc. | Cyclin-dependent kinase inhibitors |
| WO2020140054A1 (en) * | 2018-12-28 | 2020-07-02 | Spv Therapeutics Inc. | Cyclin-dependent kinase inhibitors |
| US10716787B2 (en) | 2014-03-26 | 2020-07-21 | Astex Therapeutics Ltd | Combinations |
| US10723739B2 (en) | 2018-05-14 | 2020-07-28 | Apotex Inc. | Processes for the preparation of Ribociclib and intermediates thereof |
| US10736900B2 (en) | 2014-03-26 | 2020-08-11 | Astex Therapeutics Ltd | Combinations of an FGFR inhibitor and an IGF1R inhibitor |
| WO2020168197A1 (en) * | 2019-02-15 | 2020-08-20 | Incyte Corporation | Pyrrolo[2,3-d]pyrimidinone compounds as cdk2 inhibitors |
| US10758543B2 (en) | 2010-05-21 | 2020-09-01 | Incyte Corporation | Topical formulation for a JAK inhibitor |
| WO2020205560A1 (en) * | 2019-03-29 | 2020-10-08 | Incyte Corporation | Sulfonylamide compounds as cdk2 inhibitors |
| US10899736B2 (en) | 2018-01-30 | 2021-01-26 | Incyte Corporation | Processes and intermediates for making a JAK inhibitor |
| WO2021017384A1 (en) * | 2019-07-30 | 2021-02-04 | 上海勋和医药科技有限公司 | Dihydro-pyrrolo-pyrimidine selective jak2 inhibitor |
| WO2021032582A1 (en) * | 2019-08-19 | 2021-02-25 | Galapagos Nv | Pyrazolo[4,3-c]pyridine derivatives and pharmaceutical compositions thereof for the treatment of inflammatory disorders |
| WO2021124106A1 (en) * | 2019-12-16 | 2021-06-24 | Lunella Biotech, Inc. | Selective cdk4/6 inhibitor cancer therapeutics |
| US11066404B2 (en) | 2018-10-11 | 2021-07-20 | Incyte Corporation | Dihydropyrido[2,3-d]pyrimidinone compounds as CDK2 inhibitors |
| US11155555B2 (en) | 2015-09-23 | 2021-10-26 | Janssen Pharmaceutica Nv | Compounds |
| US11207321B2 (en) | 2017-06-20 | 2021-12-28 | The Institute Of Cancer Research: Royal Cancer Hospital | Methods and medical uses |
| US11304949B2 (en) | 2018-03-30 | 2022-04-19 | Incyte Corporation | Treatment of hidradenitis suppurativa using JAK inhibitors |
| WO2022140231A1 (en) * | 2020-12-21 | 2022-06-30 | Incyte Corporation | Deazaguaine compounds as jak2 v617f inhibitors |
| US11414399B2 (en) | 2010-02-11 | 2022-08-16 | Celgene Corporation | Arylmethoxy isoindoline derivatives and compositions comprising and methods of using the same |
| US11427567B2 (en) | 2019-08-14 | 2022-08-30 | Incyte Corporation | Imidazolyl pyrimidinylamine compounds as CDK2 inhibitors |
| US11440914B2 (en) | 2019-05-01 | 2022-09-13 | Incyte Corporation | Tricyclic amine compounds as CDK2 inhibitors |
| US11447494B2 (en) | 2019-05-01 | 2022-09-20 | Incyte Corporation | Tricyclic amine compounds as CDK2 inhibitors |
| CZ309356B6 (en) * | 2020-09-15 | 2022-09-28 | Ústav experimentální botaniky AV ČR, v. v. i | Substituted purine compounds as protein kinase inhibitors, their use as medicaments and pharmaceutical preparations containing these derivatives |
| WO2022207788A2 (en) | 2021-04-01 | 2022-10-06 | Krka, D.D., Novo Mesto | Process for the preparation of ribociclib and pharmaceutically acceptable salts thereof |
| US11472791B2 (en) | 2019-03-05 | 2022-10-18 | Incyte Corporation | Pyrazolyl pyrimidinylamine compounds as CDK2 inhibitors |
| US11542247B2 (en) | 2015-09-23 | 2023-01-03 | Janssen Pharmaceutica Nv | Bi-heteroaryl substitute 1,4-benzodiazepines and uses thereof for the treatment of cancer |
| US11578067B2 (en) | 2017-01-30 | 2023-02-14 | Kyoto University | Compound, and method for producing regulatory T cells |
| US11661422B2 (en) | 2020-08-27 | 2023-05-30 | Incyte Corporation | Tricyclic urea compounds as JAK2 V617F inhibitors |
| EP3976624A4 (en) * | 2019-05-27 | 2023-06-14 | Dizal (Jiangsu) Pharmaceutical Co., Ltd. | Dna-dependent protein kinase inhibitor |
| US11684620B2 (en) | 2015-02-10 | 2023-06-27 | Astex Therapeutics Ltd | Pharmaceutical compositions comprising N-(3,5-dimethoxyphenyl)-N′-(1-methylethyl)-N-[3-(1-methyl-1H-pyrazol-4-yl)quinoxalin-6-yl]ethane-1,2-diamine |
| US11691971B2 (en) | 2020-06-19 | 2023-07-04 | Incyte Corporation | Naphthyridinone compounds as JAK2 V617F inhibitors |
| EP4009967A4 (en) * | 2019-08-08 | 2023-07-26 | Vimalan Biosciences, Inc. | Jak inhibitors |
| US11753413B2 (en) | 2020-06-19 | 2023-09-12 | Incyte Corporation | Substituted pyrrolo[2,1-f][1,2,4]triazine compounds as JAK2 V617F inhibitors |
| US11753476B2 (en) | 2018-04-08 | 2023-09-12 | Cothera Bioscience, Inc. | Combination therapy for cancers with BRAF mutation |
| US11767323B2 (en) | 2020-07-02 | 2023-09-26 | Incyte Corporation | Tricyclic pyridone compounds as JAK2 V617F inhibitors |
| US11780840B2 (en) | 2020-07-02 | 2023-10-10 | Incyte Corporation | Tricyclic urea compounds as JAK2 V617F inhibitors |
| EP4046999A4 (en) * | 2019-10-17 | 2023-11-22 | Cisen Pharmaceutical Co., Ltd. | Aminopyrimidine compound as cdk2/4/6 triple inhibitor |
| US11833155B2 (en) | 2020-06-03 | 2023-12-05 | Incyte Corporation | Combination therapy for treatment of myeloproliferative neoplasms |
| US11851426B2 (en) | 2019-10-11 | 2023-12-26 | Incyte Corporation | Bicyclic amines as CDK2 inhibitors |
| US11878968B2 (en) | 2021-07-09 | 2024-01-23 | Plexium, Inc. | Aryl compounds and pharmaceutical compositions that modulate IKZF2 |
| WO2024049926A1 (en) | 2022-08-31 | 2024-03-07 | Arvinas Operations, Inc. | Dosage regimens of estrogen receptor degraders |
| WO2024056090A1 (en) * | 2022-09-16 | 2024-03-21 | 华东师范大学 | Pyrrolopyrimidine derivative as rsk inhibitor and use thereof |
| US11958861B2 (en) | 2021-02-25 | 2024-04-16 | Incyte Corporation | Spirocyclic lactams as JAK2 V617F inhibitors |
| US11976073B2 (en) | 2021-12-10 | 2024-05-07 | Incyte Corporation | Bicyclic amines as CDK2 inhibitors |
| US11981671B2 (en) | 2021-06-21 | 2024-05-14 | Incyte Corporation | Bicyclic pyrazolyl amines as CDK2 inhibitors |
| US12076399B2 (en) | 2017-06-02 | 2024-09-03 | Cothera Bioscience, Inc. | Combination therapies for treating cancers |
| US12084430B2 (en) | 2022-03-17 | 2024-09-10 | Incyte Corporation | Tricyclic urea compounds as JAK2 V617F inhibitors |
Families Citing this family (54)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TW201100429A (en) | 2009-05-22 | 2011-01-01 | Incyte Corp | N-(hetero)aryl-pyrrolidine derivatives of pyrazol-4-yl-pyrrolo[2,3-d]pyrimidines and pyrrol-3-yl-pyrrolo[2,3-d]pyrimidines as janus kinase inhibitors |
| EP2571361A4 (en) | 2010-05-19 | 2013-11-13 | Univ North Carolina | Pyrazolopyrimidine compounds for the treatment of cancer |
| CN106967074A (en) * | 2010-10-25 | 2017-07-21 | G1治疗公司 | CDK inhibitor |
| CN103476776B (en) * | 2011-01-07 | 2016-09-28 | 北京赛林泰医药技术有限公司 | 2,4-diaminourea-6,7-dihydro-5H-pyrrolo-[2,3] pyrimidine derivatives as FAK/Pyk2 inhibitor |
| MX2013013331A (en) * | 2011-05-17 | 2014-10-17 | Principia Biopharma Inc | Azaindole derivatives as tyrosine kinase inhibitors. |
| JP2014532060A (en) | 2011-10-03 | 2014-12-04 | ザ・ユニヴァーシティ・オヴ・ノース・キャロライナ・アト・チャペル・ヒル | Pyrrolopyrimidine compounds for the treatment of cancer |
| WO2013059634A1 (en) | 2011-10-20 | 2013-04-25 | The Regents Of The University Of California | Use of cdk9 inhibitors to reduce cartilage degradation |
| CN104302627A (en) | 2012-05-22 | 2015-01-21 | 北卡罗来纳大学教堂山分校 | Pyrimidine compounds for the treatment of cancer |
| US9562047B2 (en) | 2012-10-17 | 2017-02-07 | The University Of North Carolina At Chapel Hill | Pyrazolopyrimidine compounds for the treatment of cancer |
| US9771330B2 (en) | 2012-11-27 | 2017-09-26 | The University Of North Carolina At Chapel Hill | Pyrimidine compounds for the treatment of cancer |
| EP2964648B1 (en) * | 2013-03-05 | 2016-11-16 | Merck Patent GmbH | 9-(aryl or heteroaryl)-2-(pyrazolyl, pyrrolidinyl or cyclopentyl)aminopurine derivatives as anticancer agents |
| AU2014253932B2 (en) | 2013-04-16 | 2020-04-30 | Memorial Sloan-Kettering Cancer Center | Companion diagnostic for CDK4 inhibitors |
| MX367918B (en) | 2013-04-25 | 2019-09-11 | Beigene Ltd | Fused heterocyclic compounds as protein kinase inhibitors. |
| US8895611B1 (en) | 2013-07-17 | 2014-11-25 | King Fahd University Of Petroleum And Minerals | Cytotoxic compounds for treating cancer |
| CN103408546A (en) * | 2013-08-22 | 2013-11-27 | 中国药科大学 | 2-phenylaminopurine PLK1 (Polo-like kinase 1) inhibitors and applications thereof |
| SI3702373T1 (en) | 2013-09-13 | 2022-11-30 | Beigene Switzerland Gmbh | Anti-pd1 antibodies and their use as therapeutics and diagnostics |
| WO2015157127A1 (en) | 2014-04-11 | 2015-10-15 | The University Of North Carolina At Chapel Hill | Therapuetic uses of selected pyrimidine compounds with anti-mer tyrosine kinase activity |
| JP2017516855A (en) * | 2014-05-28 | 2017-06-22 | シャンハイ フォチョン ファーマシューティカル カンパニー リミテッド | Certain protein kinase inhibitors |
| KR102003754B1 (en) | 2014-07-03 | 2019-07-25 | 베이진 엘티디 | Anti-PD-L1 Antibodies and Their Use as Therapeutics and Diagnostics |
| SG10202009598VA (en) | 2014-10-06 | 2020-10-29 | Signal Pharm Llc | Substituted aminopurine compounds, compositions thereof, and methods of treatment therewith |
| WO2016061144A1 (en) | 2014-10-14 | 2016-04-21 | The Regents Of The University Of California | Use of cdk9 and brd4 inhibitors to inhibit inflammation |
| CN105111215B (en) * | 2014-12-12 | 2019-06-18 | 苏州晶云药物科技股份有限公司 | A kind of crystal form and preparation method thereof of cyclin-dependent kinase inhibitor |
| CN104606197A (en) * | 2014-12-31 | 2015-05-13 | 芜湖杨燕制药有限公司 | Application of compound in tumor resistance |
| CN104610265A (en) * | 2014-12-31 | 2015-05-13 | 芜湖杨燕制药有限公司 | Compound and preparation method thereof |
| WO2017156263A1 (en) | 2016-03-09 | 2017-09-14 | Memorial Sloan-Kettering Cancer Center | Enigma and cdh18 as companion diagnostics for cdk4 inhibitors |
| US10709708B2 (en) | 2016-03-17 | 2020-07-14 | The University Of North Carolina At Chapel Hill | Method of treating cancer with a combination of MER tyrosine kinase inhibitor and an epidermal growth factor receptor (EGFR) inhibitor |
| EA039392B1 (en) | 2016-04-01 | 2022-01-21 | СИГНАЛ ФАРМАСЬЮТИКАЛЗ, ЭлЭлСи | Method of treating cancer using a substituted aminopurine compound |
| MX2018011970A (en) | 2016-04-01 | 2019-05-20 | Signal Pharm Llc | Solid forms of (1s,4s)-4-(2-(((3s4r)-3-fluorotetrahydro-2h-pyran- 4-yl) amino)-8-((2,4,6-trichlorophenyl) amino)-9h-purin-9-yl)-1-m ethylcyclohexane-1-carboxamide and methods of their use. |
| NZ749997A (en) | 2016-07-05 | 2022-11-25 | Beigene Ltd | Combination of a pd-l antagonist and a raf inhibitor for treating cancer |
| TW202233628A (en) | 2016-08-16 | 2022-09-01 | 英屬開曼群島商百濟神州有限公司 | Crystalline form of (s)-7-(1-acryloylpiperidin-4-yl)-2-(4-phenoxyphenyl)-4,5,6,7-tetrahydropyrazolo[1,5-a]pyrimidine-3-carboxamide, preparation, and uses thereof |
| ES2971881T3 (en) | 2016-08-19 | 2024-06-10 | Beigene Switzerland Gmbh | Combination of zanubrutinib with an anti-cd20 or anti-pd-1 antibody for use in cancer treatment |
| WO2018081211A1 (en) * | 2016-10-26 | 2018-05-03 | Li George Y | Deuterated 7-cyclopentyl-n, n-dimethyl-2-((5-(piperazin-1-yl)pyridin-2-yl)amino)-7h-pyrrolo[2,3-d]pyrimdine-6-carboxamide |
| JP2020502062A (en) * | 2016-11-17 | 2020-01-23 | ザ ユニバーシティ オブ ノース カロライナ アット チャペル ヒル | Alkylpyrrolopyrimidine analogs and methods of making and using same |
| EP3573989A4 (en) | 2017-01-25 | 2020-11-18 | Beigene, Ltd. | Crystalline forms of (s) -7- (1- (but-2-ynoyl) piperidin-4-yl) -2- (4-phenoxyphenyl) -4, 5, 6, 7-tetrahy dropyrazolo [1, 5-a]pyrimidine-3-carboxamide, preparation, and uses thereof |
| TW201906866A (en) | 2017-06-26 | 2019-02-16 | 英屬開曼群島商百濟神州有限公司 | Treatment of abnormal bone condition in patients with acid sphingomyelinase deficiency |
| WO2019034009A1 (en) | 2017-08-12 | 2019-02-21 | Beigene, Ltd. | Btk INHIBITORS WITH IMPROVED DUAL SELECTIVITY |
| JP6922085B2 (en) * | 2017-09-28 | 2021-08-18 | シャンハイ ハイヤン ファーマシューティカル テクノロジー カンパニー リミテッドShanghai Haiyan Pharmaceutical Technology Co., Ltd. | 4,6,7-Tri-substituted 1,2-dihydropyrrolo [3,4-c] pyridine / pyrimidine-3-one derivative and its use |
| KR20200061363A (en) | 2017-10-04 | 2020-06-02 | 셀진 코포레이션 | Cis-4-[2-{[(3S,4R)-3-fluorooxan-4-yl]amino}-8-(2,4,6-trichloroanilino)-9H-purin-9-yl ]-1-methylcyclohexane-1-carboxamide production process |
| BR122022005778B1 (en) | 2017-10-04 | 2024-01-09 | Celgene Corporation | CAPSULE COMPRISING CIS-4-[2-{[(3S,4R)-3-FLUORO-OXAN-4-IL]AMINO}-8-(2,4,6-TRICHLOROANILINO)-9H-PURIN-9-IL] -1-METHYLCYCLOHEXANE-1-CARBOXAMIDE |
| CN111801334B (en) | 2017-11-29 | 2023-06-09 | 百济神州瑞士有限责任公司 | Treatment of indolent or invasive B-cell lymphomas using combinations comprising BTK inhibitors |
| WO2019222524A1 (en) * | 2018-05-16 | 2019-11-21 | The University Of North Carolina At Chapel Hill | Alkyl pyrrolopyrimidines as pan-tam inhibitors and their application in cancer treatment |
| CN110577524B (en) * | 2018-06-07 | 2022-01-28 | 北京大学深圳研究生院 | Kinase selective inhibitor |
| CN109438447B (en) * | 2018-09-11 | 2020-10-16 | 北京工业大学 | Preparation method and application of 5, 7-dihydro-6H-pyrrolo [2,3-d ] pyrimidine-6-ketone derivative |
| US11136329B2 (en) | 2019-05-08 | 2021-10-05 | Vimalan Biosciences, Inc. | JAK inhibitors |
| BR112021022682A2 (en) | 2019-05-14 | 2022-02-22 | Provention Bio Inc | Methods and compositions for preventing type 1 diabetes |
| US20230024521A1 (en) * | 2019-09-25 | 2023-01-26 | Vimalan Biosciences, Inc. | Jak inhibitors |
| US20230357248A1 (en) * | 2019-11-13 | 2023-11-09 | Medshine Discovery Inc. | Pyrrolopyrimidine compound as btk inhibitor and use thereof |
| CA3182445A1 (en) | 2020-06-11 | 2021-12-16 | Francisco Leon | Methods and compositions for preventing type 1 diabetes |
| KR102409595B1 (en) * | 2020-06-29 | 2022-06-17 | 한국과학기술연구원 | Novel purinone derivatives as protein kinase CSF-1R inhibitor |
| CN112375081B (en) * | 2020-11-23 | 2022-04-12 | 中国医学科学院医药生物技术研究所 | Pyrrole [2,3-d ] pyrimidine derivative with CDK4, 6 or 9 inhibiting activity and preparation method and application thereof |
| CN116685323A (en) * | 2020-12-21 | 2023-09-01 | 江苏恒瑞医药股份有限公司 | Purinone derivative, preparation method and medical application thereof |
| WO2022226290A1 (en) * | 2021-04-22 | 2022-10-27 | H. Lee Moffitt Cancer Center And Research Institute, Inc. | 2-phenylamino pyrrolopyrimidines as ack1 inhibitors |
| US11786531B1 (en) | 2022-06-08 | 2023-10-17 | Beigene Switzerland Gmbh | Methods of treating B-cell proliferative disorder |
| WO2023241620A1 (en) * | 2022-06-14 | 2023-12-21 | Suzhou Keen Therapeutics Co., Ltd. | Biologically active compounds and methods thereof |
Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003074530A1 (en) * | 2002-03-07 | 2003-09-12 | F. Hoffman-La Roche Ag | Bicyclic pyridine and pyrimidine p38 kinase inhibitors |
| WO2005023761A2 (en) * | 2003-09-11 | 2005-03-17 | Kemia, Inc. | Cytokine inhibitors |
| WO2005080393A1 (en) * | 2004-02-14 | 2005-09-01 | Irm Llc | Compounds and compositions as protein kinase inhibitors |
| WO2005107760A1 (en) * | 2004-04-30 | 2005-11-17 | Irm Llc | Compounds and compositions as inducers of keratinocyte differentiation |
| WO2006045828A1 (en) * | 2004-10-29 | 2006-05-04 | Tibotec Pharmaceuticals Ltd. | Hiv inhibiting bicyclic pyrimidine derivatives |
| WO2006076595A1 (en) * | 2005-01-13 | 2006-07-20 | Signal Pharmaceuticals, Llc | Haloaryl substituted aminopurines, compositions thereof, and methods of treatment therewith |
| WO2006074985A1 (en) * | 2005-01-14 | 2006-07-20 | Janssen Pharmaceutica N.V. | 5-membered annelated heterocyclic pyrimidines as kinase inhibitors |
| WO2006091737A1 (en) * | 2005-02-24 | 2006-08-31 | Kemia, Inc. | Modulators of gsk-3 activity |
| JP2006241089A (en) * | 2005-03-04 | 2006-09-14 | Astellas Pharma Inc | Pyrroloprymidine derivative or its salt |
| WO2007030438A2 (en) * | 2005-09-06 | 2007-03-15 | Pharmacopeia, Inc. | Aminopurine derivatives for treating neurodegenerative diseases |
| WO2007058990A2 (en) * | 2005-11-14 | 2007-05-24 | Kemia, Inc. | Therapy using cytokine inhibitors |
| WO2007104053A2 (en) * | 2006-03-09 | 2007-09-13 | Pharmacopeia, Inc. | 8-heteroarylpurine mnk2 inhibitors for treating metabolic disorders |
| WO2007127382A1 (en) * | 2006-04-26 | 2007-11-08 | Signal Pharmaceuticals, Llc | Haloaryl substituted aminopurines, compositions thereof, and methods of treatment therewith |
Family Cites Families (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH05306226A (en) * | 1992-04-27 | 1993-11-19 | Takeda Chem Ind Ltd | Therapeutic agent for chronic immunological disease |
| EP1680424A2 (en) | 2003-09-05 | 2006-07-19 | Neurogen Corporation | Heteroaryl fused pyridines, pyrazines and pyrimidines as crf1 receptor ligands |
| US7319102B1 (en) * | 2003-12-09 | 2008-01-15 | The Procter & Gamble Company | Pyrrolo[2,3-d]pyrimidine cytokine inhibitors |
| EP1725562A1 (en) | 2004-03-05 | 2006-11-29 | Taisho Pharmaceutical Co., Ltd | Pyrrolopyrimidine derivatives |
| US7906528B2 (en) | 2004-10-05 | 2011-03-15 | Novartis International Pharmaceutical Ltd. | Pyrrolo-pyridine, pyrrolo-pyrimidine and related heterocyclic compounds |
| GB0520164D0 (en) | 2005-10-04 | 2005-11-09 | Novartis Ag | Organic compounds |
| GB0526246D0 (en) | 2005-12-22 | 2006-02-01 | Novartis Ag | Organic compounds |
| WO2007125405A2 (en) | 2006-05-01 | 2007-11-08 | Pfizer Products Inc. | Substituted 2-amino-fused heterocyclic compounds |
| EA017952B1 (en) | 2008-02-06 | 2013-04-30 | Новартис Аг | PYRROLO[2,3-d]PYRIDINES AND USE THEREOF AS TYROSINE KINASE INHIBITORS |
-
2007
- 2007-05-23 TW TW096118407A patent/TWI398252B/en active
- 2007-05-23 JO JOP/2007/0193A patent/JO3235B1/en active
- 2007-05-24 AU AU2007267645A patent/AU2007267645C1/en active Active
- 2007-05-24 CA CA2652044A patent/CA2652044C/en active Active
- 2007-05-24 SG SG2011037637A patent/SG172632A1/en unknown
- 2007-05-24 US US12/302,223 patent/US8324225B2/en active Active
- 2007-05-24 PL PL07811927T patent/PL2029145T3/en unknown
- 2007-05-24 EP EP07811927.8A patent/EP2029145B1/en active Active
- 2007-05-24 PE PE2007000646A patent/PE20080263A1/en active IP Right Grant
- 2007-05-24 AR ARP070102252A patent/AR061124A1/en active IP Right Grant
- 2007-05-24 NZ NZ572549A patent/NZ572549A/en unknown
- 2007-05-24 ES ES07811927.8T patent/ES2623133T3/en active Active
- 2007-05-24 SM SM200800069T patent/SMP200800069B/en unknown
- 2007-05-24 KR KR1020087031411A patent/KR101466412B1/en active IP Right Grant
- 2007-05-24 CN CN2007800193572A patent/CN101594871B/en active Active
- 2007-05-24 GE GEAP200710989A patent/GEP20115283B/en unknown
- 2007-05-24 JP JP2009512291A patent/JP2009538341A/en active Pending
- 2007-05-24 ME MEP-2008-766A patent/ME00486B/en unknown
- 2007-05-24 PT PT78119278T patent/PT2029145T/en unknown
- 2007-05-24 EA EA200802332A patent/EA016301B1/en not_active IP Right Cessation
- 2007-05-24 UA UAA200813340A patent/UA95632C2/en unknown
- 2007-05-24 WO PCT/US2007/069595 patent/WO2007140222A2/en active Application Filing
- 2007-05-24 MX MX2008015076A patent/MX2008015076A/en active IP Right Grant
- 2007-05-24 BR BRPI0712816A patent/BRPI0712816B8/en active IP Right Grant
- 2007-05-24 MY MYPI20084701 patent/MY150650A/en unknown
- 2007-05-25 CL CL200701504A patent/CL2007001504A1/en unknown
- 2007-05-25 UY UY30369A patent/UY30369A1/en active IP Right Grant
-
2008
- 2008-11-03 ZA ZA2008/09382A patent/ZA200809382B/en unknown
- 2008-11-03 IL IL195086A patent/IL195086A/en active IP Right Grant
- 2008-11-10 CR CR10433A patent/CR10433A/en unknown
- 2008-11-21 TN TNP2008000481A patent/TNSN08481A1/en unknown
- 2008-11-24 GT GT200800258A patent/GT200800258A/en unknown
- 2008-11-24 CU CU2008000223A patent/CU23831B1/en active IP Right Grant
- 2008-11-25 HN HN2008001752A patent/HN2008001752A/en unknown
- 2008-11-26 EC EC2008008910A patent/ECSP088910A/en unknown
- 2008-12-01 MA MA31431A patent/MA30557B1/en unknown
- 2008-12-02 NO NO20085030A patent/NO343182B1/en unknown
-
2012
- 2012-04-20 US US13/452,100 patent/US20120207763A1/en not_active Abandoned
-
2013
- 2013-01-23 JP JP2013010317A patent/JP5740417B2/en active Active
-
2017
- 2017-04-05 AR ARP170100874A patent/AR108179A2/en unknown
- 2017-04-21 HR HRP20170631TT patent/HRP20170631T1/en unknown
Patent Citations (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003074530A1 (en) * | 2002-03-07 | 2003-09-12 | F. Hoffman-La Roche Ag | Bicyclic pyridine and pyrimidine p38 kinase inhibitors |
| WO2005023761A2 (en) * | 2003-09-11 | 2005-03-17 | Kemia, Inc. | Cytokine inhibitors |
| WO2005080393A1 (en) * | 2004-02-14 | 2005-09-01 | Irm Llc | Compounds and compositions as protein kinase inhibitors |
| WO2005107760A1 (en) * | 2004-04-30 | 2005-11-17 | Irm Llc | Compounds and compositions as inducers of keratinocyte differentiation |
| WO2006045828A1 (en) * | 2004-10-29 | 2006-05-04 | Tibotec Pharmaceuticals Ltd. | Hiv inhibiting bicyclic pyrimidine derivatives |
| WO2006076595A1 (en) * | 2005-01-13 | 2006-07-20 | Signal Pharmaceuticals, Llc | Haloaryl substituted aminopurines, compositions thereof, and methods of treatment therewith |
| WO2006074985A1 (en) * | 2005-01-14 | 2006-07-20 | Janssen Pharmaceutica N.V. | 5-membered annelated heterocyclic pyrimidines as kinase inhibitors |
| WO2006091737A1 (en) * | 2005-02-24 | 2006-08-31 | Kemia, Inc. | Modulators of gsk-3 activity |
| JP2006241089A (en) * | 2005-03-04 | 2006-09-14 | Astellas Pharma Inc | Pyrroloprymidine derivative or its salt |
| WO2007030438A2 (en) * | 2005-09-06 | 2007-03-15 | Pharmacopeia, Inc. | Aminopurine derivatives for treating neurodegenerative diseases |
| WO2007058990A2 (en) * | 2005-11-14 | 2007-05-24 | Kemia, Inc. | Therapy using cytokine inhibitors |
| WO2007104053A2 (en) * | 2006-03-09 | 2007-09-13 | Pharmacopeia, Inc. | 8-heteroarylpurine mnk2 inhibitors for treating metabolic disorders |
| WO2007127382A1 (en) * | 2006-04-26 | 2007-11-08 | Signal Pharmaceuticals, Llc | Haloaryl substituted aminopurines, compositions thereof, and methods of treatment therewith |
Non-Patent Citations (3)
| Title |
|---|
| CHOI, HA-SOON ET AL: "Design and synthesis of 7H-pyrrolo[2,3-d]pyrimidines as focal adhesion kinase inhibitors. Part 1" BIOORGANIC & MEDICINAL CHEMISTRY LETTERS , 16(8), 2173-2176 CODEN: BMCLE8; ISSN: 0960-894X, 2006, XP002472293 * |
| GAULON C ET AL: "A General and Facile Route to New Trisubstituted Purin-8-ones" SYNTHESIS, GEORG THIEME VERLAG, STUTTGART, DE, vol. 13, 7 July 2005 (2005-07-07), pages 2227-2233, XP002413755 ISSN: 0039-7881 * |
| MORIARTY ET AL: "The synthesis and SAR of 2-amino-pyrrolo[2,3-d]pyrimidines: A new class of Aurora-A kinase inhibitors" BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, OXFORD, GB, vol. 16, no. 22, 30 October 2006 (2006-10-30), pages 5778-5783, XP005839098 ISSN: 0960-894X * |
Cited By (254)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10639310B2 (en) | 2005-12-13 | 2020-05-05 | Incyte Corporation | Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as Janus kinase inhibitors |
| US11331320B2 (en) | 2005-12-13 | 2022-05-17 | Incyte Holdings Corporation | Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as Janus kinase inhibitors |
| US9662335B2 (en) | 2005-12-13 | 2017-05-30 | Incyte Holdings Corporation | Heteroaryl substituted pyrrolo[2,3-B] pyridines and pyrrolo[2,3-B] pyrimidines as janus kinase inhibitors |
| US9974790B2 (en) | 2005-12-13 | 2018-05-22 | Incyte Corporation | Heteroaryl substituted pyrrolo[2,3-B] pyridines and pyrrolo[2,3-B] pyrimidines as janus kinase inhibitors |
| US11744832B2 (en) | 2005-12-13 | 2023-09-05 | Incyte Corporation | Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as Janus kinase inhibitors |
| US8946245B2 (en) | 2005-12-13 | 2015-02-03 | Incyte Corporation | Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as Janus kinase inhibitors |
| US8933086B2 (en) | 2005-12-13 | 2015-01-13 | Incyte Corporation | Heteroaryl substituted pyrrolo[2,3-B]pyridines and pyrrolo[2,3-B]pyrimidines as Janus kinase inhibitors |
| US10398699B2 (en) | 2005-12-13 | 2019-09-03 | Incyte Holdings Corporation | Heteroaryl substituted pyrrolo[2,3-b]pyridines and pyrrolo[2,3-b]pyrimidines as janus kinase inhibitors |
| US9079912B2 (en) | 2005-12-13 | 2015-07-14 | Incyte Corporation | Heteroaryl substituted pyrrolo[2,3-B] pyridines and pyrrolo[2,3-B] pyrimidines as Janus kinase inhibitors |
| US9814722B2 (en) | 2005-12-13 | 2017-11-14 | Incyte Holdings Corporation | Heteroaryl substituted pyrrolo[2,3-B] pyridines and pyrrolo[2,3-B] pyrimidines as janus kinase inhibitors |
| US9206187B2 (en) | 2005-12-13 | 2015-12-08 | Incyte Holdings Corporation | Heteroaryl substituted pyrrolo[2,3-B] pyridines and pyrrolo[2,3-B] pyrimidines as Janus kinase |
| US7989459B2 (en) | 2006-02-17 | 2011-08-02 | Pharmacopeia, Llc | Purinones and 1H-imidazopyridinones as PKC-theta inhibitors |
| US7902187B2 (en) | 2006-10-04 | 2011-03-08 | Wyeth Llc | 6-substituted 2-(benzimidazolyl)purine and purinone derivatives for immunosuppression |
| US7919490B2 (en) | 2006-10-04 | 2011-04-05 | Wyeth Llc | 6-substituted 2-(benzimidazolyl)purine and purinone derivatives for immunosuppression |
| US7915268B2 (en) | 2006-10-04 | 2011-03-29 | Wyeth Llc | 8-substituted 2-(benzimidazolyl)purine derivatives for immunosuppression |
| US10610530B2 (en) | 2007-06-13 | 2020-04-07 | Incyte Corporation | Salts of the Janus kinase inhibitor (R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| US8829013B1 (en) | 2007-06-13 | 2014-09-09 | Incyte Corporation | Salts of the Janus kinase inhibitor (R)-3-(4-(7H-pyrrolo[2,3-D]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| US10016429B2 (en) | 2007-06-13 | 2018-07-10 | Incyte Corporation | Salts of the janus kinase inhibitor (R)-3-(4-(7H-pyrrolo[2,3-D]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| US9376439B2 (en) | 2007-06-13 | 2016-06-28 | Incyte Corporation | Salts of the janus kinase inhibitor (R)-3(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| US8722693B2 (en) | 2007-06-13 | 2014-05-13 | Incyte Corporation | Salts of the Janus kinase inhibitor (R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| US8822481B1 (en) | 2007-06-13 | 2014-09-02 | Incyte Corporation | Salts of the janus kinase inhibitor (R)-3-(4-(7H-pyrrolo[2,3-d] pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| US11213528B2 (en) | 2007-06-13 | 2022-01-04 | Incyte Holdings Corporation | Salts of the janus kinase inhibitor (R)-3-(4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl)-3-cyclopentylpropanenitrile |
| WO2009024824A1 (en) * | 2007-08-23 | 2009-02-26 | Astrazeneca Ab | 2-anilinopurin-8-ones as inhibitors of ttk/mps1 for the treatment of proliferative disorders |
| WO2009085185A1 (en) * | 2007-12-19 | 2009-07-09 | Amgen Inc. | Fused pyridine, pyrimidine and triazine compounds as cell cycle inhibitors |
| US8841312B2 (en) | 2007-12-19 | 2014-09-23 | Amgen Inc. | Fused pyridine, pyrimidine and triazine compounds as cell cycle inhibitors |
| US8980903B2 (en) | 2007-12-19 | 2015-03-17 | Amgen Inc. | Fused pyridine, pyrimidine and triazine compounds as cell cycle inhibitors |
| JP2011507849A (en) * | 2007-12-19 | 2011-03-10 | アムジエン・インコーポレーテツド | Condensed pyridine, pyrimidine and triazine compounds as cell cycle inhibitors |
| AU2008343932B2 (en) * | 2007-12-19 | 2013-08-15 | Amgen Inc. | Fused pyridine, pyrimidine and triazine compounds as cell cycle inhibitors |
| US8420657B2 (en) | 2008-02-06 | 2013-04-16 | Novartis Ag | Pyrrolo[2,3-D]pyrimidines and use thereof as tyrosine kinase inhibitors |
| WO2009110415A1 (en) * | 2008-03-03 | 2009-09-11 | 武田薬品工業株式会社 | Concomitant drug |
| US8389533B2 (en) | 2008-04-07 | 2013-03-05 | Amgen Inc. | Gem-disubstituted and spirocyclic amino pyridines/pyrimidines as cell cycle inhibitors |
| WO2009124965A1 (en) | 2008-04-09 | 2009-10-15 | N.V. Organon | PYRROLO[2,3-d]PYRIMIDIN-2-YL-AMINE DERIVATIVES AS PKC-THETA INHIBITORS |
| CN101977913A (en) * | 2008-04-09 | 2011-02-16 | 欧加农股份有限公司 | Pyrrolo[2,3-d]pyrimidin-2-yl-amine derivatives as pkc-theta inhibitors |
| JP2011516522A (en) * | 2008-04-09 | 2011-05-26 | ナームローゼ・フエンノートチヤツプ・オルガノン | Pyrrolo [2,3-d] pyrimidin-2-yl-amine derivatives as PKC-theta inhibitors |
| US8748469B2 (en) | 2008-04-24 | 2014-06-10 | Newlink Genetics Corporation | IDO inhibitors |
| JP2011518841A (en) * | 2008-04-24 | 2011-06-30 | ニューリンク ジェネティクス, インコーポレイテッド | IDO inhibitor |
| US9174942B2 (en) | 2008-04-24 | 2015-11-03 | Newlink Genetics Corporation | IDO inhibitors |
| WO2009152027A1 (en) * | 2008-06-12 | 2009-12-17 | Merck & Co., Inc. | 5,7-dihydro-6h-pyrrolo[2,3-d]pyrimidin-6-one derivatives for mark inhibition |
| WO2010020675A1 (en) * | 2008-08-22 | 2010-02-25 | Novartis Ag | Pyrrolopyrimidine compounds as cdk inhibitors |
| JP2014129361A (en) * | 2008-08-22 | 2014-07-10 | Novartis Ag | Pyrrolopyrimidine compound as cdk inhibitor |
| AU2009284098B2 (en) * | 2008-08-22 | 2012-03-29 | Astex Therapeutics Ltd. | Pyrrolopyrimidine compounds as CDK inhibitors |
| CN102186856B (en) * | 2008-08-22 | 2014-09-24 | 诺华股份有限公司 | Pyrrolopyrimidine compounds as cdk inhibitors |
| CN102186856A (en) * | 2008-08-22 | 2011-09-14 | 诺瓦提斯公司 | Pyrrolopyrimidine compounds as cdk inhibitors |
| JP2012500785A (en) * | 2008-08-22 | 2012-01-12 | ノバルティス アーゲー | Pyrrolopyrimidine compounds as CDK inhibitors |
| KR101353857B1 (en) | 2008-08-22 | 2014-01-21 | 노파르티스 아게 | Pyrrolopyrimidine compounds as cdk inhibitors |
| EA019094B1 (en) * | 2008-08-22 | 2014-01-30 | Новартис Аг | Pyrrolopyrimidine compounds and use thereof |
| US8415355B2 (en) | 2008-08-22 | 2013-04-09 | Novartis Ag | Pyrrolopyrimidine compounds and their uses |
| TWI468409B (en) * | 2008-08-22 | 2015-01-11 | Novartis Ag | Pyrrolopyrimidine compounds and their uses |
| US8685980B2 (en) | 2008-08-22 | 2014-04-01 | Novartis Ag | Pyrrolopyrimidine compounds and their uses |
| US9416136B2 (en) | 2008-08-22 | 2016-08-16 | Novartis Ag | Pyrrolopyrimidine compounds and their uses |
| EP2716643A1 (en) | 2008-08-22 | 2014-04-09 | Novartis AG | Pyrrolopyrimidine compounds and their uses |
| US8962630B2 (en) | 2008-08-22 | 2015-02-24 | Novartis Ag | Pyrrolopyrimidine compounds and their uses |
| WO2010045451A1 (en) * | 2008-10-16 | 2010-04-22 | Glaxosmithkline Llc | Pyrrolopyrimidine compounds |
| WO2010100431A1 (en) | 2009-03-04 | 2010-09-10 | Medical Research Council Technology | Pyrrolopyrimidines used as kinase inhibitors |
| CN102341400A (en) * | 2009-03-04 | 2012-02-01 | 医学研究理事会技术公司 | Pyrrolopyrimidines used as kinase inhibitors |
| US9616062B2 (en) | 2009-05-13 | 2017-04-11 | The University Of North Carolina At Chapel Hill | Cyclin dependent kinase inhibitors and methods of use |
| US9216984B2 (en) | 2009-05-22 | 2015-12-22 | Incyte Corporation | 3-[4-(7H-pyrrolo[2,3-D]pyrimidin-4-yl)-1H-pyrazol-1-yl]octane—or heptane-nitrile as JAK inhibitors |
| US9623029B2 (en) | 2009-05-22 | 2017-04-18 | Incyte Holdings Corporation | 3-[4-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)-1H-pyrazol-1-yl]octane- or heptane-nitrile as JAK inhibitors |
| US9249145B2 (en) | 2009-09-01 | 2016-02-02 | Incyte Holdings Corporation | Heterocyclic derivatives of pyrazol-4-yl-pyrrolo[2,3-d]pyrimidines as janus kinase inhibitors |
| US8946257B2 (en) | 2009-10-29 | 2015-02-03 | Vectura Limited | N-containing heteroaryl derivatives as JAK3 kinase inhibitors |
| US8501735B2 (en) | 2009-10-29 | 2013-08-06 | Palau Pharma, S.A. | N-containing heteroaryl derivatives as JAK3 kinase inhibitors |
| WO2011082400A2 (en) * | 2010-01-04 | 2011-07-07 | President And Fellows Of Harvard College | Modulators of immunoinhibitory receptor pd-1, and methods of use thereof |
| WO2011082400A3 (en) * | 2010-01-04 | 2011-11-03 | President And Fellows Of Harvard College | Modulators of immunoinhibitory receptor pd-1, and methods of use thereof |
| US11414399B2 (en) | 2010-02-11 | 2022-08-16 | Celgene Corporation | Arylmethoxy isoindoline derivatives and compositions comprising and methods of using the same |
| TWI503322B (en) * | 2010-02-19 | 2015-10-11 | Novartis Ag | Pyrrolopyrimidine compounds as inhibitors of cdk4/6 |
| AU2011217199B2 (en) * | 2010-02-19 | 2014-05-15 | Novartis Ag | Deuterated pyrrolopyrimidine compounds as inhibitors of CDK4/6 |
| WO2011101409A1 (en) | 2010-02-19 | 2011-08-25 | Novartis Ag | Pyrrolopyrimidine compounds as inhibitors of cdk4/6 |
| WO2011101417A1 (en) | 2010-02-19 | 2011-08-25 | Novartis Ag | Deuterated pyrrolopyrimidine compounds as inhibitors of cdk4/6 |
| US9309252B2 (en) | 2010-02-19 | 2016-04-12 | Novartis Ag | Pyrrolopyrimidine compounds as inhibitors of CDK4/6 |
| AU2011217286B2 (en) * | 2010-02-19 | 2014-06-19 | Novartis Ag | Pyrrolopyrimidine compounds as inhibitors of CDK4/6 |
| EA022355B1 (en) * | 2010-02-19 | 2015-12-30 | Новартис Аг | Pyrrolopyrimidine compounds as inhibitors of cdk4/6 |
| CN102918043B (en) * | 2010-02-19 | 2015-07-29 | 诺华股份有限公司 | As the Pyrrolopyrimidine compounds of CDK4/6 inhibitor |
| KR101812357B1 (en) | 2010-02-19 | 2017-12-26 | 노파르티스 아게 | Pyrrolopyrimidine compounds as inhibitors of cdk4/6 |
| CN103003280A (en) * | 2010-02-19 | 2013-03-27 | 诺瓦提斯公司 | Deuterated pyrrolopyrimidine compounds as inhibitors of CDK4/6 |
| CN102918043A (en) * | 2010-02-19 | 2013-02-06 | 诺瓦提斯公司 | Pyrrolopyrimidine compounds as inhibitors of CDK4/6 |
| US11285140B2 (en) | 2010-03-10 | 2022-03-29 | Incyte Corporation | Piperidin-4-yl azetidine derivatives as JAK1 inhibitors |
| US10695337B2 (en) | 2010-03-10 | 2020-06-30 | Incyte Holdings Corporation | Piperidin-4-yl azetidine derivatives as JAK1 inhibitors |
| US9464088B2 (en) | 2010-03-10 | 2016-10-11 | Incyte Holdings Corporation | Piperidin-4-yl azetidine derivatives as JAK1 inhibitors |
| US9999619B2 (en) | 2010-03-10 | 2018-06-19 | Incyte Holdings Corporation | Piperidin-4-yl azetidine derivatives as JAK1 inhibitors |
| WO2011130232A1 (en) | 2010-04-13 | 2011-10-20 | Novartis Ag | Combination comprising a cyclin dependent kinase 4 or cyclin dependent kinase (cdk4/6) inhibitor and an mtor inhibitor for treating cancer |
| EP2558092B1 (en) | 2010-04-13 | 2018-06-27 | Novartis AG | Combination comprising a cyclin dependent kinase 4 or cyclin dependent kinase 6 (cdk4/6) inhibitor and an mtor inhibitor for treating cancer |
| AU2011240735B2 (en) * | 2010-04-13 | 2015-01-29 | Novartis Ag | Combination comprising a cyclin dependent kinase 4 or cyclin dependent kinase (CDK4/6) inhibitor and an mTOR inhibitor for treating cancer |
| RU2589696C2 (en) * | 2010-04-13 | 2016-07-10 | Новартис Аг | COMBINATION INCLUDING CYCLIN-DEPENDENT KINASE 4 INHIBITOR OR CYCLIN-DEPENDENT KINASE 6 INHIBITOR (CDK4/6) AND mTOR INHIBITOR FOR TREATING CANCER |
| US10519137B2 (en) | 2010-04-30 | 2019-12-31 | Astex Therapeutics Ltd | Pyrazolyl quinoxaline kinase inhibitors |
| US11219624B2 (en) | 2010-05-21 | 2022-01-11 | Incyte Holdings Corporation | Topical formulation for a JAK inhibitor |
| US11590136B2 (en) | 2010-05-21 | 2023-02-28 | Incyte Corporation | Topical formulation for a JAK inhibitor |
| US11571425B2 (en) | 2010-05-21 | 2023-02-07 | Incyte Corporation | Topical formulation for a JAK inhibitor |
| US10758543B2 (en) | 2010-05-21 | 2020-09-01 | Incyte Corporation | Topical formulation for a JAK inhibitor |
| US10869870B2 (en) | 2010-05-21 | 2020-12-22 | Incyte Corporation | Topical formulation for a JAK inhibitor |
| KR20130132433A (en) * | 2010-11-10 | 2013-12-04 | 노파르티스 아게 | Salt(s) of 7-cyclopentyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-7h-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid dimethylamide and processes of making thereof |
| TWI549953B (en) * | 2010-11-10 | 2016-09-21 | 諾華公司 | Salt(s) of 7-cyclopentyl-2-(5-piperazin-1-yl-pyridin-2-ylamino)-7h-pyrrolo[2,3-d]pyrimidine-6-carboxylic acid dimethylamide and processes of making thereof |
| US9808461B2 (en) | 2010-11-17 | 2017-11-07 | The University Of North Carolina At Chapel Hill | Protection of renal tissues from ischemia through inhibition of the proliferative kinases CDK4 and CDK6 |
| US9034884B2 (en) | 2010-11-19 | 2015-05-19 | Incyte Corporation | Heterocyclic-substituted pyrrolopyridines and pyrrolopyrimidines as JAK inhibitors |
| US8933085B2 (en) | 2010-11-19 | 2015-01-13 | Incyte Corporation | Cyclobutyl substituted pyrrolopyridine and pyrrolopyrimidine derivatives as JAK inhibitors |
| US10640506B2 (en) | 2010-11-19 | 2020-05-05 | Incyte Holdings Corporation | Cyclobutyl substituted pyrrolopyridine and pyrrolopyrimidines derivatives as JAK inhibitors |
| EP2661437A1 (en) * | 2011-01-07 | 2013-11-13 | Centaurus Biopharma Co., Ltd. | 2,4-DIAMINO-6,7-DIHYDRO-5H-PYRROLO[2,3]PYRIMIDINE DERIVATIVES AS FAK/Pyk2 INHIBITORS |
| AU2012204982B2 (en) * | 2011-01-07 | 2017-02-23 | Centaurus Biopharma Co., Ltd. | 2,4-diamino-6,7-dihydro-5H-pyrrolo[2,3]pyrimidine derivatives as FAK/Pyk2 inhibitors |
| WO2012092880A1 (en) | 2011-01-07 | 2012-07-12 | Centaurus Biopharma Co., Ltd. | 2,4-DIAMINO-6,7-DIHYDRO-5H-PYRROLO[2,3]PYRIMIDINE DERIVATIVES AS FAK/Pyk2 INHIBITORS |
| EP2661437A4 (en) * | 2011-01-07 | 2014-07-02 | Centaurus Biopharma Co Ltd | 2,4-DIAMINO-6,7-DIHYDRO-5H-PYRROLO[2,3]PYRIMIDINE DERIVATIVES AS FAK/Pyk2 INHIBITORS |
| US9428508B2 (en) | 2011-01-07 | 2016-08-30 | Centaurus Biopharma Co., Ltd. | 2,4-diamino-6,7-dihydro-5H-pyrrolo[2,3]pyrimidine derivatives as FAK/Pyk2 inhibitors |
| WO2012110774A1 (en) | 2011-02-17 | 2012-08-23 | Cancer Therapeutics Crc Pty Limited | Selective fak inhibitors |
| WO2012110773A1 (en) | 2011-02-17 | 2012-08-23 | Cancer Therapeutics Crc Pty Limited | Fak inhibitors |
| US9120761B2 (en) | 2011-02-17 | 2015-09-01 | Cancer Therapeutics Crc Pty Ltd | Selective FAK inhibitors |
| US9012461B2 (en) | 2011-02-17 | 2015-04-21 | Cancer Therapeutics Crc Pty Ltd | FAK inhibitors |
| US9421205B2 (en) | 2011-02-17 | 2016-08-23 | Cancer Therapeutics CRC Pty Ltd. | FAK inhibitors |
| US9174946B2 (en) | 2011-02-17 | 2015-11-03 | Cancer Therapeutics Crc Pty Ltd | Selective FAK inhibitors |
| US9371319B2 (en) | 2011-03-14 | 2016-06-21 | Cancer Research Technology Limited | Pyrrolopyridineamino derivatives as MPS1 inhibitors |
| WO2012127506A1 (en) | 2011-03-22 | 2012-09-27 | Advinus Therapeutics Limited | Substituted fused tricyclic compounds, compositions and medicinal applications thereof |
| US9359355B2 (en) | 2011-03-23 | 2016-06-07 | Amgen Inc. | Fused tricyclic dual inhibitors of CDK 4/6 and FLT3 |
| US8623885B2 (en) | 2011-03-23 | 2014-01-07 | Amgen Inc. | Fused tricyclic dual inhibitors of CDK 4/6 and FLT3 |
| US11214573B2 (en) | 2011-06-20 | 2022-01-04 | Incyte Holdings Corporation | Azetidinyl phenyl, pyridyl or pyrazinyl carboxamide derivatives as JAK inhibitors |
| US9023840B2 (en) | 2011-06-20 | 2015-05-05 | Incyte Corporation | Azetidinyl phenyl, pyridyl or pyrazinyl carboxamide derivatives as JAK inhibitors |
| US9611269B2 (en) | 2011-06-20 | 2017-04-04 | Incyte Corporation | Azetidinyl phenyl, pyridyl or pyrazinyl carboxamide derivatives as JAK inhibitors |
| US10513522B2 (en) | 2011-06-20 | 2019-12-24 | Incyte Corporation | Azetidinyl phenyl, pyridyl or pyrazinyl carboxamide derivatives as JAK inhibitors |
| US8691807B2 (en) | 2011-06-20 | 2014-04-08 | Incyte Corporation | Azetidinyl phenyl, pyridyl or pyrazinyl carboxamide derivatives as JAK inhibitors |
| WO2013006532A1 (en) | 2011-07-01 | 2013-01-10 | Novartis Ag | Combination therapy comprising a cdk4/6 inhibitor and a pi3k inhibitor for use in the treatment of cancer |
| WO2013006368A1 (en) | 2011-07-01 | 2013-01-10 | Novartis Ag | Combination therapy |
| US9233111B2 (en) | 2011-07-08 | 2016-01-12 | Novartis Ag | Pyrrolo pyrimidine derivatives |
| WO2013017480A1 (en) * | 2011-07-29 | 2013-02-07 | Cellzome Limited | Pyrazolo[4,3-c]pyridine derivatives as jak inhibitors |
| WO2013017479A1 (en) * | 2011-07-29 | 2013-02-07 | Cellzome Limited | Pyrazolo[4,3-c]pyridine derivatives as jak inhibitors |
| US9359358B2 (en) | 2011-08-18 | 2016-06-07 | Incyte Holdings Corporation | Cyclohexyl azetidine derivatives as JAK inhibitors |
| US9718834B2 (en) | 2011-09-07 | 2017-08-01 | Incyte Corporation | Processes and intermediates for making a JAK inhibitor |
| US9487521B2 (en) | 2011-09-07 | 2016-11-08 | Incyte Holdings Corporation | Processes and intermediates for making a JAK inhibitor |
| US9193733B2 (en) | 2012-05-18 | 2015-11-24 | Incyte Holdings Corporation | Piperidinylcyclobutyl substituted pyrrolopyridine and pyrrolopyrimidine derivatives as JAK inhibitors |
| US9062050B2 (en) | 2012-06-14 | 2015-06-23 | Eli Lilly And Company | Inhibitor of JAK1 and JAK2 |
| CN104349775A (en) * | 2012-06-14 | 2015-02-11 | 伊莱利利公司 | Inhibitor of jak1 and jak2 |
| WO2013188184A1 (en) * | 2012-06-14 | 2013-12-19 | Eli Lilly And Company | Inhibitor of jak1 and jak2 |
| AU2013274641B2 (en) * | 2012-06-14 | 2015-09-24 | Eli Lilly And Company | Inhibitor of JAK1 and JAK2 |
| US8673972B2 (en) | 2012-08-03 | 2014-03-18 | Foundation Medicine, Inc. | Human papilloma virus as predictor of cancer prognosis |
| WO2014022830A2 (en) | 2012-08-03 | 2014-02-06 | Foundation Medicine, Inc. | Human papilloma virus as predictor of cancer prognosis |
| EP4119676A1 (en) | 2012-08-03 | 2023-01-18 | Foundation Medicine, Inc. | Human papilloma virus as predictor of cancer prognosis |
| US11766439B2 (en) | 2012-08-03 | 2023-09-26 | Foundation Medicine, Inc. | Human papilloma virus as predictor of cancer prognosis |
| US9410954B2 (en) | 2012-08-03 | 2016-08-09 | Foundation Medicine, Inc. | Human papilloma virus as predictor of cancer prognosis |
| US11058687B2 (en) | 2012-08-03 | 2021-07-13 | Foundation Medicine, Inc. | Human papilloma virus as predictor of cancer prognosis |
| US9907798B2 (en) | 2012-08-03 | 2018-03-06 | Foundation Medicine, Inc. | Methods of treating head and neck cancer |
| US9834552B2 (en) | 2012-09-07 | 2017-12-05 | Cancer Research Technology Limited | Inhibitor compounds |
| US10188642B2 (en) | 2012-09-07 | 2019-01-29 | Cancer Research Technology Limited | Pharmacologically active compounds |
| US9409907B2 (en) | 2012-09-07 | 2016-08-09 | Cancer Research Technology Limited | Inhibitor compounds |
| US11897877B2 (en) | 2012-09-07 | 2024-02-13 | Cancer Research Technology Limited | Inhibitor compounds |
| US9334286B2 (en) | 2012-09-07 | 2016-05-10 | Cancer Research Technology Limited | Pharmacologically active compounds |
| US9895364B2 (en) | 2012-09-07 | 2018-02-20 | Cancer Research Technology Limited | Pharmacologically active compounds |
| US11046688B2 (en) | 2012-09-07 | 2021-06-29 | Cancer Research Technology Limited | Inhibitor compounds |
| US10479788B2 (en) | 2012-09-07 | 2019-11-19 | Cancer Research Technology Limited | Compounds that inhibit MPS1 kinase |
| US9890157B2 (en) | 2012-09-07 | 2018-02-13 | Cancer Research Technology Limited | Inhibitor compounds |
| US9593115B2 (en) | 2012-09-21 | 2017-03-14 | Advinus Therapeutics Ltd. | Substituted fused tricyclic compounds, compositions, and medicinal applications thereof |
| US9163021B2 (en) | 2012-10-04 | 2015-10-20 | Pfizer Limited | Pyrrolo[3,2-c]pyridine tropomyosin-related kinase inhibitors |
| US11576864B2 (en) | 2012-11-15 | 2023-02-14 | Incyte Corporation | Sustained-release dosage forms of ruxolitinib |
| US10166191B2 (en) | 2012-11-15 | 2019-01-01 | Incyte Corporation | Sustained-release dosage forms of ruxolitinib |
| US11337927B2 (en) | 2012-11-15 | 2022-05-24 | Incyte Holdings Corporation | Sustained-release dosage forms of ruxolitinib |
| US10874616B2 (en) | 2012-11-15 | 2020-12-29 | Incyte Corporation | Sustained-release dosage forms of ruxolitinib |
| US11576865B2 (en) | 2012-11-15 | 2023-02-14 | Incyte Corporation | Sustained-release dosage forms of ruxolitinib |
| US11896717B2 (en) | 2012-11-15 | 2024-02-13 | Incyte Holdings Corporation | Sustained-release dosage forms of ruxolitinib |
| EP2742940A1 (en) | 2012-12-13 | 2014-06-18 | IP Gesellschaft für Management mbH | Salts of aza-bicyclo-di-aryl ethers for adminstration once daily, twice daily or thrice daily |
| EP3251673A1 (en) | 2012-12-13 | 2017-12-06 | IP Gesellschaft für Management mbH | Combination therapy comprising a cdk4/6 inhibitor and a pi3k inhibitor for use in the treatment of cancer |
| US9867825B2 (en) * | 2012-12-20 | 2018-01-16 | Novartis Ag | Pharmaceutical combination comprising binimetinib |
| US20150313902A1 (en) * | 2012-12-20 | 2015-11-05 | Novartis Ag | Pharmaceutical combination comprising binimetinib |
| US10011874B2 (en) | 2013-02-25 | 2018-07-03 | Novartis Ag | Androgen receptor mutation |
| WO2014130693A1 (en) * | 2013-02-25 | 2014-08-28 | Pharmacyclics, Inc. | Inhibitors of bruton's tyrosine kinase |
| US9708326B2 (en) | 2013-02-25 | 2017-07-18 | Pharmacyclics Llc | Inhibitors of bruton's tyrosine kinase |
| US8987443B2 (en) | 2013-03-06 | 2015-03-24 | Incyte Corporation | Processes and intermediates for making a JAK inhibitor |
| US9221845B2 (en) | 2013-03-06 | 2015-12-29 | Incyte Holdings Corporation | Processes and intermediates for making a JAK inhibitor |
| US9714233B2 (en) | 2013-03-06 | 2017-07-25 | Incyte Corporation | Processes and intermediates for making a JAK inhibitor |
| WO2014147573A2 (en) | 2013-03-21 | 2014-09-25 | Novartis Ag | Combination therapy |
| US10561616B2 (en) | 2013-08-07 | 2020-02-18 | Incyte Corporation | Sustained release dosage forms for a JAK1 inhibitor |
| US9655854B2 (en) | 2013-08-07 | 2017-05-23 | Incyte Corporation | Sustained release dosage forms for a JAK1 inhibitor |
| US11045421B2 (en) | 2013-08-07 | 2021-06-29 | Incyte Corporation | Sustained release dosage forms for a JAK1 inhibitor |
| US9902721B2 (en) | 2014-02-28 | 2018-02-27 | Cancer Research Technology Limited | N2-phenyl-pyrido[3,4-d]pyrimidine-2, 8-diamine derivatives and their use as Mps1 inhibitors |
| US10399974B2 (en) | 2014-02-28 | 2019-09-03 | Cancer Research Technology Limited | N2-phenyl-pyrido[3,4-D]pyrimidine-2, 8-diamine derivatives and their use as Mps1 inhibitors |
| US10421747B2 (en) | 2014-03-26 | 2019-09-24 | Astex Therapeutics Ltd | Quinoxaline derivatives useful as FGFR kinase modulators |
| US10716787B2 (en) | 2014-03-26 | 2020-07-21 | Astex Therapeutics Ltd | Combinations |
| US11918576B2 (en) | 2014-03-26 | 2024-03-05 | Astex Therapeutics Ltd | Combination of an FGFR inhibitor and a CMET inhibitor |
| US10736900B2 (en) | 2014-03-26 | 2020-08-11 | Astex Therapeutics Ltd | Combinations of an FGFR inhibitor and an IGF1R inhibitor |
| WO2015160986A2 (en) | 2014-04-16 | 2015-10-22 | Infinity Pharmaceuticals, Inc. | Combination therapies |
| WO2015181737A1 (en) | 2014-05-28 | 2015-12-03 | Piramal Enterprises Limited | Pharmaceutical combination for the treatment of cancer |
| US9498467B2 (en) | 2014-05-30 | 2016-11-22 | Incyte Corporation | Treatment of chronic neutrophilic leukemia (CNL) and atypical chronic myeloid leukemia (aCML) by inhibitors of JAK1 |
| WO2016014904A1 (en) | 2014-07-24 | 2016-01-28 | Beta Pharma, Inc. | 2-h-indazole derivatives as cyclin-dependent kinase (cdk) inhibitors and therapeutic uses thereof |
| US11684620B2 (en) | 2015-02-10 | 2023-06-27 | Astex Therapeutics Ltd | Pharmaceutical compositions comprising N-(3,5-dimethoxyphenyl)-N′-(1-methylethyl)-N-[3-(1-methyl-1H-pyrazol-4-yl)quinoxalin-6-yl]ethane-1,2-diamine |
| US20180244676A1 (en) * | 2015-02-13 | 2018-08-30 | Dana-Farber Cancer Institute, Inc. | Lrrk2 inhibitors and methods of making and using the same |
| US10913744B2 (en) * | 2015-02-13 | 2021-02-09 | Dana-Farber Cancer Institute, Inc. | LRRK2 inhibitors and methods of making and using the same |
| EP3284746A4 (en) * | 2015-04-17 | 2018-09-12 | Changzhou Longthera Pharmaceuticals Inc | Preparation and use of kinase inhibitor |
| US11542247B2 (en) | 2015-09-23 | 2023-01-03 | Janssen Pharmaceutica Nv | Bi-heteroaryl substitute 1,4-benzodiazepines and uses thereof for the treatment of cancer |
| US11155555B2 (en) | 2015-09-23 | 2021-10-26 | Janssen Pharmaceutica Nv | Compounds |
| WO2017114351A1 (en) * | 2015-12-27 | 2017-07-06 | Chongqing Fochon Pharmaceutical Co., Ltd. | Certain protein kinase inhibitors |
| CN113549069A (en) * | 2015-12-27 | 2021-10-26 | 重庆复创医药研究有限公司 | Kinase inhibitor |
| CN108779117A (en) * | 2015-12-27 | 2018-11-09 | 重庆复创医药研究有限公司 | A kind of kinase inhibitor |
| US10662186B2 (en) | 2015-12-31 | 2020-05-26 | Shanghai Pharmaceuticals Holding Co., Ltd. | Substituted pyrimidines as cyclin-dependent kinase inhibitors |
| US10988476B2 (en) | 2015-12-31 | 2021-04-27 | Shanghai Pharmaceuticals Holding Co., Ltd. | Substituted pyrimidines as cyclin-dependent kinase inhibitors |
| WO2018039324A1 (en) | 2016-08-23 | 2018-03-01 | Eisai R&D Management Co., Ltd. | Combination therapies for the treatment of hepatocellular carcinoma |
| WO2018053437A1 (en) | 2016-09-19 | 2018-03-22 | Mei Pharma, Inc. | Combination therapy |
| WO2018073687A1 (en) | 2016-10-20 | 2018-04-26 | Pfizer Inc. | Anti-proliferative agents for treating pah |
| EP3804724A1 (en) | 2016-10-20 | 2021-04-14 | Pfizer Inc. | Cdk inhibitors for treating pah |
| US11746118B2 (en) | 2016-12-20 | 2023-09-05 | Astrazeneca Ab | Amino-triazolopyridine compounds and their use in treating cancer |
| US10407446B2 (en) | 2016-12-20 | 2019-09-10 | Astrazeneca Ab | Amino-triazolopyridine compounds and their use in treating cancer |
| US11136340B2 (en) | 2016-12-20 | 2021-10-05 | Astrazeneca Ab | Amino-triazolopyridine compounds and their use in treating cancer |
| US11052091B2 (en) | 2016-12-21 | 2021-07-06 | Ono Pharmaceutical Co., Ltd. | BRK inhibitory compound |
| EP3560926A4 (en) * | 2016-12-21 | 2020-06-17 | Ono Pharmaceutical Co., Ltd. | Brk INHIBITORY COMPOUND |
| WO2018124001A1 (en) | 2016-12-27 | 2018-07-05 | 国立研究開発法人理化学研究所 | Bmp-signal-inhibiting compound |
| US11578067B2 (en) | 2017-01-30 | 2023-02-14 | Kyoto University | Compound, and method for producing regulatory T cells |
| EP4218820A2 (en) | 2017-03-16 | 2023-08-02 | Eisai R&D Management Co., Ltd. | Combination therapies for the treatment of breast cancer |
| US11083722B2 (en) | 2017-03-16 | 2021-08-10 | Eisai R&D Management Co., Ltd. | Combination therapies for the treatment of breast cancer |
| WO2018170447A1 (en) | 2017-03-16 | 2018-09-20 | Eisai R&D Management Co., Ltd. | Combination therapies for the treatment of breast cancer |
| US12076399B2 (en) | 2017-06-02 | 2024-09-03 | Cothera Bioscience, Inc. | Combination therapies for treating cancers |
| WO2018232235A1 (en) | 2017-06-16 | 2018-12-20 | Beta Pharma, Inc. | Pharmaceutical formulations of n-(2-(2-(dimethylamino)ethoxy)-4-methoxy-5-((4-(1-methyl-1h-indol-3-yl)pyrimidin-2-yl)amino)phenyl)acrylamide and salts thereof |
| US11207321B2 (en) | 2017-06-20 | 2021-12-28 | The Institute Of Cancer Research: Royal Cancer Hospital | Methods and medical uses |
| US10596161B2 (en) | 2017-12-08 | 2020-03-24 | Incyte Corporation | Low dose combination therapy for treatment of myeloproliferative neoplasms |
| US11278541B2 (en) | 2017-12-08 | 2022-03-22 | Incyte Corporation | Low dose combination therapy for treatment of myeloproliferative neoplasms |
| US11352341B2 (en) | 2018-01-29 | 2022-06-07 | Beta Pharma, Inc. | 2H-indazole derivatives as CDK4 and CDK6 inhibitors and therapeutic uses thereof |
| WO2019148161A1 (en) | 2018-01-29 | 2019-08-01 | Beta Pharma, Inc. | 2h-indazole derivatives as cdk4 and cdk6 inhibitors and therapeutic uses thereof |
| US10899736B2 (en) | 2018-01-30 | 2021-01-26 | Incyte Corporation | Processes and intermediates for making a JAK inhibitor |
| WO2019157020A1 (en) | 2018-02-06 | 2019-08-15 | The Board Of Trustees Of The University Of Illinois | Substituted benzothiophene analogs as selective estrogen receptor degraders |
| US11304949B2 (en) | 2018-03-30 | 2022-04-19 | Incyte Corporation | Treatment of hidradenitis suppurativa using JAK inhibitors |
| US11753476B2 (en) | 2018-04-08 | 2023-09-12 | Cothera Bioscience, Inc. | Combination therapy for cancers with BRAF mutation |
| US10723739B2 (en) | 2018-05-14 | 2020-07-28 | Apotex Inc. | Processes for the preparation of Ribociclib and intermediates thereof |
| US11866432B2 (en) | 2018-10-11 | 2024-01-09 | Incyte Corporation | Dihydropyrido[2,3-d]pyrimidinone compounds as CDK2 inhibitors |
| US11066404B2 (en) | 2018-10-11 | 2021-07-20 | Incyte Corporation | Dihydropyrido[2,3-d]pyrimidinone compounds as CDK2 inhibitors |
| WO2020140054A1 (en) * | 2018-12-28 | 2020-07-02 | Spv Therapeutics Inc. | Cyclin-dependent kinase inhibitors |
| WO2020140052A1 (en) * | 2018-12-28 | 2020-07-02 | Spv Therapeutics Inc. | Cyclin-dependent kinase inhibitors |
| US11384083B2 (en) | 2019-02-15 | 2022-07-12 | Incyte Corporation | Substituted spiro[cyclopropane-1,5′-pyrrolo[2,3-d]pyrimidin]-6′(7′h)-ones as CDK2 inhibitors |
| WO2020168197A1 (en) * | 2019-02-15 | 2020-08-20 | Incyte Corporation | Pyrrolo[2,3-d]pyrimidinone compounds as cdk2 inhibitors |
| US11472791B2 (en) | 2019-03-05 | 2022-10-18 | Incyte Corporation | Pyrazolyl pyrimidinylamine compounds as CDK2 inhibitors |
| WO2020205560A1 (en) * | 2019-03-29 | 2020-10-08 | Incyte Corporation | Sulfonylamide compounds as cdk2 inhibitors |
| US11919904B2 (en) | 2019-03-29 | 2024-03-05 | Incyte Corporation | Sulfonylamide compounds as CDK2 inhibitors |
| US11447494B2 (en) | 2019-05-01 | 2022-09-20 | Incyte Corporation | Tricyclic amine compounds as CDK2 inhibitors |
| US11440914B2 (en) | 2019-05-01 | 2022-09-13 | Incyte Corporation | Tricyclic amine compounds as CDK2 inhibitors |
| EP3976624A4 (en) * | 2019-05-27 | 2023-06-14 | Dizal (Jiangsu) Pharmaceutical Co., Ltd. | Dna-dependent protein kinase inhibitor |
| JP2022540073A (en) * | 2019-07-30 | 2022-09-14 | シャンハイ シュンフェァ ファーマシューティカル テクノロジー カンパニー リミテッド | Dihydropyrrolopyrimidine-based selective JAK2 inhibitors |
| GB2603386B (en) * | 2019-07-30 | 2023-07-26 | Shanghai Xunhe Pharamceutical Tech Co Ltd | Selective dihydropyrrolopyrimidine JAK2 inhibitors |
| GB2603386A (en) * | 2019-07-30 | 2022-08-03 | Shanghai Xunhe Pharamceutical Tech Co Ltd | Dihydro-pyrrolo-pyrimidine selective JAK2 inhibitor |
| JP7335644B2 (en) | 2019-07-30 | 2023-08-30 | シャンハイ シュンフェァ ファーマシューティカル テクノロジー カンパニー リミテッド | Dihydropyrrolopyrimidine-based selective JAK2 inhibitor or pharmacologically acceptable salt thereof, method for producing the same, medicament, and composition |
| WO2021017384A1 (en) * | 2019-07-30 | 2021-02-04 | 上海勋和医药科技有限公司 | Dihydro-pyrrolo-pyrimidine selective jak2 inhibitor |
| EP4009967A4 (en) * | 2019-08-08 | 2023-07-26 | Vimalan Biosciences, Inc. | Jak inhibitors |
| US11427567B2 (en) | 2019-08-14 | 2022-08-30 | Incyte Corporation | Imidazolyl pyrimidinylamine compounds as CDK2 inhibitors |
| WO2021032582A1 (en) * | 2019-08-19 | 2021-02-25 | Galapagos Nv | Pyrazolo[4,3-c]pyridine derivatives and pharmaceutical compositions thereof for the treatment of inflammatory disorders |
| US11851426B2 (en) | 2019-10-11 | 2023-12-26 | Incyte Corporation | Bicyclic amines as CDK2 inhibitors |
| EP4046999A4 (en) * | 2019-10-17 | 2023-11-22 | Cisen Pharmaceutical Co., Ltd. | Aminopyrimidine compound as cdk2/4/6 triple inhibitor |
| WO2021124106A1 (en) * | 2019-12-16 | 2021-06-24 | Lunella Biotech, Inc. | Selective cdk4/6 inhibitor cancer therapeutics |
| CN114901666A (en) * | 2019-12-16 | 2022-08-12 | 卢内拉生物技术有限公司 | Selective CDK4/6 inhibitor type cancer therapeutic drug |
| US11833155B2 (en) | 2020-06-03 | 2023-12-05 | Incyte Corporation | Combination therapy for treatment of myeloproliferative neoplasms |
| US11691971B2 (en) | 2020-06-19 | 2023-07-04 | Incyte Corporation | Naphthyridinone compounds as JAK2 V617F inhibitors |
| US11753413B2 (en) | 2020-06-19 | 2023-09-12 | Incyte Corporation | Substituted pyrrolo[2,1-f][1,2,4]triazine compounds as JAK2 V617F inhibitors |
| US11767323B2 (en) | 2020-07-02 | 2023-09-26 | Incyte Corporation | Tricyclic pyridone compounds as JAK2 V617F inhibitors |
| US11780840B2 (en) | 2020-07-02 | 2023-10-10 | Incyte Corporation | Tricyclic urea compounds as JAK2 V617F inhibitors |
| US11661422B2 (en) | 2020-08-27 | 2023-05-30 | Incyte Corporation | Tricyclic urea compounds as JAK2 V617F inhibitors |
| CZ309356B6 (en) * | 2020-09-15 | 2022-09-28 | Ústav experimentální botaniky AV ČR, v. v. i | Substituted purine compounds as protein kinase inhibitors, their use as medicaments and pharmaceutical preparations containing these derivatives |
| US11919908B2 (en) | 2020-12-21 | 2024-03-05 | Incyte Corporation | Substituted pyrrolo[2,3-d]pyrimidine compounds as JAK2 V617F inhibitors |
| WO2022140231A1 (en) * | 2020-12-21 | 2022-06-30 | Incyte Corporation | Deazaguaine compounds as jak2 v617f inhibitors |
| US11958861B2 (en) | 2021-02-25 | 2024-04-16 | Incyte Corporation | Spirocyclic lactams as JAK2 V617F inhibitors |
| WO2022207788A2 (en) | 2021-04-01 | 2022-10-06 | Krka, D.D., Novo Mesto | Process for the preparation of ribociclib and pharmaceutically acceptable salts thereof |
| US11981671B2 (en) | 2021-06-21 | 2024-05-14 | Incyte Corporation | Bicyclic pyrazolyl amines as CDK2 inhibitors |
| US11878968B2 (en) | 2021-07-09 | 2024-01-23 | Plexium, Inc. | Aryl compounds and pharmaceutical compositions that modulate IKZF2 |
| US11976073B2 (en) | 2021-12-10 | 2024-05-07 | Incyte Corporation | Bicyclic amines as CDK2 inhibitors |
| US12084430B2 (en) | 2022-03-17 | 2024-09-10 | Incyte Corporation | Tricyclic urea compounds as JAK2 V617F inhibitors |
| WO2024049926A1 (en) | 2022-08-31 | 2024-03-07 | Arvinas Operations, Inc. | Dosage regimens of estrogen receptor degraders |
| WO2024056090A1 (en) * | 2022-09-16 | 2024-03-21 | 华东师范大学 | Pyrrolopyrimidine derivative as rsk inhibitor and use thereof |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2007267645B2 (en) | Pyrrolopyrimidine compounds and their uses | |
| ES2374480T3 (en) | PIRAZOL DERIVATIVES AND USE OF THE SAME AS INHIBITORS OF CYCLINE-DEPENDENT KINASES. | |
| US8076338B2 (en) | Kinase modulators and methods of use | |
| US20100093776A1 (en) | Organic Compounds and Their Uses | |
| EP2467382B1 (en) | Amino tetrahydro-pyridopyrimidine pde10 inhibitors | |
| EP2094682A2 (en) | Heteroaryl-heteroaryl compounds as cdk inhibitors for the treatment of cancer, inflammation and viral infections | |
| BRPI0917791A2 (en) | pyrrolopyrimidine compounds as cdk inhibitors | |
| US8957077B2 (en) | Pyrazolopyrimidine PDE 10 inhibitors | |
| CN104910137A (en) | CDK kinase inhibitor |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780019357.2 Country of ref document: CN |
|
| REEP | Request for entry into the european phase |
Ref document number: 2007811927 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12008502443 Country of ref document: PH Ref document number: 2007811927 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 572549 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007267645 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 9406/DELNP/2008 Country of ref document: IN Ref document number: CR2008-010433 Country of ref document: CR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2652044 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008111885 Country of ref document: EG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 08124716 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2009512291 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10989 Country of ref document: GE Ref document number: MX/A/2008/015076 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2007267645 Country of ref document: AU Date of ref document: 20070524 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200802332 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: DZP2008000754 Country of ref document: DZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: a200813340 Country of ref document: UA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12302223 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: PI0712816 Country of ref document: BR Kind code of ref document: A2 Effective date: 20081126 |