WO2007121918A2 - Use of 2-(purin-9-yl)-tetrahydofuran-3,4-diol derivatives as adenosine a2a receptor agonists - Google Patents
Use of 2-(purin-9-yl)-tetrahydofuran-3,4-diol derivatives as adenosine a2a receptor agonists Download PDFInfo
- Publication number
- WO2007121918A2 WO2007121918A2 PCT/EP2007/003433 EP2007003433W WO2007121918A2 WO 2007121918 A2 WO2007121918 A2 WO 2007121918A2 EP 2007003433 W EP2007003433 W EP 2007003433W WO 2007121918 A2 WO2007121918 A2 WO 2007121918A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- tetrahydro
- purin
- furan
- pyrrolidin
- ethylamino
- Prior art date
Links
- 0 CCCC=*N(*)c(nc1*)nc2c1nc[n]2 Chemical compound CCCC=*N(*)c(nc1*)nc2c1nc[n]2 0.000 description 1
- GQMPSVXWVRHKFY-HRDYMLBCSA-N CCNC([C@H]([C@H]1[O]#C)OC2(CC2)[C@@H]1[O]#C)=O Chemical compound CCNC([C@H]([C@H]1[O]#C)OC2(CC2)[C@@H]1[O]#C)=O GQMPSVXWVRHKFY-HRDYMLBCSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
- A61K31/52—Purines, e.g. adenine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/02—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6
- C07D473/16—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6 two nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
- C07H19/167—Purine radicals with ribosyl as the saccharide radical
Definitions
- This invention relates to organic compounds, their preparation and use as pharmaceuticals.
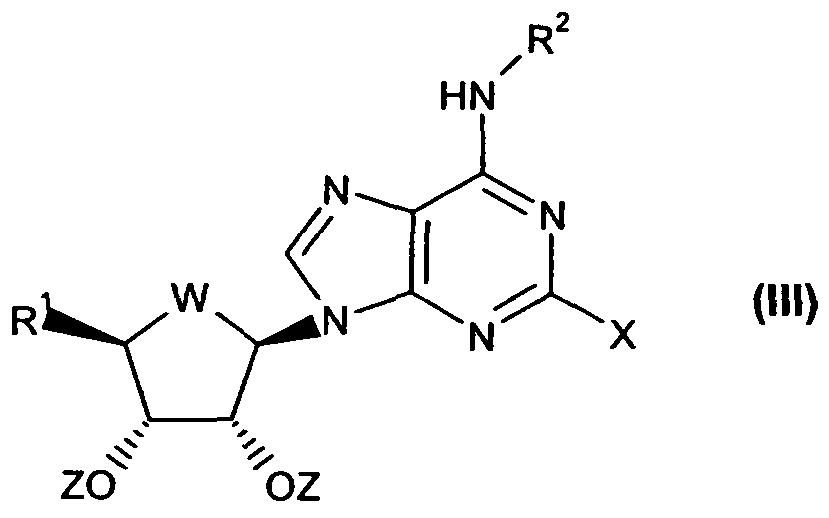
- the present invention provides for the use of compounds of formula (I)
- W is selected from CH 2 and O;
- R 1 is selected from CH 2 OH, CH 2 -O-C 1 -C 8 ⁇ IKyI, C(O)-O-C 1 -C 8 -BlRyI, C(O)NH 2 , C(O)-NH- d-C 8 -alkyl and a 3- to 10-membered heterocyclic ring containing at least one ring heteroatom selected from the group consisting of nitrogen, oxygen and sulphur, optionally substituted by d-C 8 -alkyl;
- R 2 is hydrogen or d-C 8 -alkyl optionally substituted by hydroxy or C 6 -Ci 0 -aryl;
- R 3 and R 4 together with the nitrogen atom to which they are attached, form a 3- to 10-membered heterocyclic group containing the indicated nitrogen atom as a ring heteroatom and optionally at least one ring heteroatom selected from the group consisting of nitrogen, oxygen and sulphur, optionally substituted by 0-3R 5 ;
- R 5 is selected from OH, d-C 8 -alkyl optionally substituted by OH, d-C 8 -alkoxy,
- C 7 -C 14 -aralkyl optionally substituted with OH, O-Ci-C 8 -alkyl, halogen C 6 -C 10 -aryl, or O-C 6 -C 10 -aryl, C r C 8 -alkoxy, C 6 -C 10 -aryl optionally substituted by OH, d-Ca-alkyl, O-d-C ⁇ -alkyl or -halogen, O-C 6 -C 10 -aryl optionally substituted by OH, d-C ⁇ -alkyl, O-CrC ⁇ -alkyl or -halogen, NR 5a R 5b , NHC(O)R 50 , NHS(O) 2 R 5 *, NHS(O) 2 R 56 , NR 5f C(O)NR 5g R 5h , NR 51 C(O)OR 5 ', d-C ⁇ -alkylcarbonyl, d-C 8 -alkoxycarbonyl, di
- R 6 is selected from OH, d-C 8 -alkyl optionally substituted by OH, C 7 -C 14 -aralkyl optionally substituted with OH, O-Ci-C 8 -alkyl, C 6 -C 10 -aryl, or O-C 6 -C 10 -aryl, C 1 -C 8 - alkoxy, C 6 -C 10 -aryl optionally substituted by OH, d-C 8 -alkyl, O-C r C 8 -alkyl or -halogen, O-C 6 -Ci 0 -aryl optionally substituted by OH, d-C 8 -alkyl, O-Ci-C 8 -alkyl or -halogen, NR 63 R 6 ", NHC(O)R 60 , NHS(O) 2 R 6 ", NHS(O) 2 R 66 , NR 6f C(O)NR 6g R 6h , NR 6i C(O)
- R 6k is H, d-C 8 -alkyl, C 6 -Ci 0 -aryl or a 3- to 10-membered heterocyclic group containing at least one ring heteroatom selected from the group consisting of nitrogen, oxygen and sulphur;
- R 6 ' is Ci-C 8 -alkyl, C 6 -C 10 -aryl or a 3- to 10-membered heterocyclic group containing at least one ring heteroatom selected from the group consisting of nitrogen, oxygen and sulphur, optionally substituted by COOR 10 ;
- R 7 is C00R 7a or a 3- to 10-membered heterocyclic group containing at least one ring heteroatom selected from the group consisting of nitrogen, oxygen and sulphur, optionally substituted by C00R 7b ;
- R 7a R 7b R 8 R 9 and R i o are se)ected from H d-C 8 -alkyl and C ⁇ -C 14 -aralkyl for the manufacture of a medicament for the treatment of a condition mediated by activation of the adenosine A 2A receptor, said condition mediated by activation of the adenosine A 2A receptor selected from the group consisting of cystic fibrosis, pulmonary hypertension, pulmonary fibrosis, inflammatory bowel syndrome, wound healing, diabetic nephropathy, reduction of inflammation in transplanted tissue, inflammatory diseases caused by pathogenic organisms, cardiovascular conditions, assessing the severity of coronary artery stenosis, imaging coronary activity in conjunction with radioactive imaging agents, adjunctive therapy with angioplasty, in combination with a protease inhibitor for treatment of organ ischaemia and reperfusion injury, wound healing in bronchial epithelial cells, and in combination with an integrin antagonist for treating platelet aggregation.
- Optionally substituted means the group referred to can be substituted at one or more positions by any one or any combination of the radicals listed thereafter.
- Halo or "halogen”, as used herein, may be fluorine, chlorine, bromine or iodine. Preferably halo is chlorine.
- Ci-C 8 -Alkyr denotes straight chain or branched alkyl having 1-8 carbon atoms.
- CVC ⁇ -alky! is d-C 4 -alkyl.
- Ci-C 8 -AIkOXy denotes straight chain or branched alkoxy having 1-8 carbon atoms.
- Ci-C 8 -alkoxy is C 1 -C ⁇ aIkOXy.
- C 3 -C 8 -Cycloalkyl denotes cycloalkyl having 3-8 ring carbon atoms, e.g., a monocyclic group, such as a cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl or cyclooctyl, any of which can be substituted by one or more, usually one or two, Ci-C 4 -alkyl groups; or a bicyclic group, such as bicycloheptyl or bicyclooctyl.
- C 3 -C 8 -cycloalkyl is C 3 -C 6 -cycloalkyl.
- Ci-C 8 -Alkylamino and di(C 1 -C 8 -alkyl)amino denote amino substituted respectively by one or two Ci-C 8 -alkyl groups as hereinbefore defined, which may be the same or different.
- d-C ⁇ -alkylamino and dKCrC ⁇ -alkylJamino are respectively C 1 -C 4 - alkylamino and di(C 1 -C 4 -alkyl)amino.
- Ci-C 8 -alkylcarbonyl and CrC ⁇ -alkoxycarbonyl are d-C 4 -alkylcarbonyl and C r C 4 -alkoxycarbonyl, respectively.
- C 3 -C 8 -Cycloalkylcarbonyr denotes C 3 -C 8 -cycloalkyl, as hereinbefore defined, attached by a carbon atom to a carbonyl group.
- C 3 -C 8 -cycloalkylcarbonyl is C 3 -C 5 -cycloalkylcarbonyl.
- C 3 -C8-Cycloalkylamino n denotes C 3 -C 8 -cycloalkyl, as hereinbefore defined, attached by a carbon atom to the nitrogen atom of an amino group.
- C 3 -C 8 - cycloalkylamino is C 3 -C 5 -cycloalkylamino.
- C 6 -CiO-ArVl denotes a monovalent carbocyclic aromatic group that contains 6-10 carbon atoms and which may be, e.g., a monocyclic group, such as phenyl; or a bicyclic group, such as naphthyl.
- C ⁇ -Cio-aryl is C 6 -C 8 -aryl, especially phenyl.
- C 7 -Ci 4 -Aralkyr denotes alkyl, e.g., Ci-C 4 -alkyl, as hereinbefore defined, substituted by C 6 -C 10 -aryl as hereinbefore defined.
- CyCu-aralkyl is C T -C 10 - aralkyl, such as phenyKVC-alkyl.
- Ci-C ⁇ -alkylaminocarbonyl and C 3 -C 8 -cycloalkylaminocarbonyl denote Ci-C 8 -alkylamino and C 3 -C 8 -cycloalkylamino, respectively, as hereinbefore defined, attached by a carbon atom to a carbonyl group.
- Ci-C ⁇ -alkylaminocarbonyl and C 3 -C 8 -cycloalkyl-aminocarbonyl are Ci-C 4 -alkylaminocarbonyl and C 3 -C 8 - cycloalkylaminocarbonyl, respectively.
- C 6 -C 10 -arylcarbonyl and C 7 -C 14 -arylkylcarbonyl are C 6 -C 8 - arylcarbonyl and Cz-Cio-arylkylcarbonyl, respectively.
- C 3 -C 15 -Carbocyclic group denotes a carbocyclic group having 3-15 ring carbon atoms, e.g., a monocyclic group, either aromatic or non-aromatic, such as a cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl or phenyl; or a bicyclic group, such as bicyclooctyl, bicyclononyl, bicyclodecyl, indanyl or indenyl, again any of which can be substituted by one or more, usually one or two, Ci-C 4 -alkyl groups.
- the C 3 -C 15 - carbocyclic group is a C 5 -C 10 -carbocyclic group, especially phenyl, cyclohexyl or indanyl.
- the C 5 -C 15 -carbocyclic group can unsubstituted or substituted.
- Preferred substituents on the heterocyclic ring include halo, cyano, hydroxy, carboxy, amino, aminocarbonyl, nitro, C 1 -Ci 0 - alkyl, C ⁇ C ⁇ -alkoxy and C 3 -C 10 -cycloalkyl, especially amino.
- 3- to 10-Membered heterocyclic ring containing at least one ring heteroatom selected from the group consisting of nitrogen, oxygen and sulphur may be, e.g., furan, pyrrole, pyrrolidine, pyrazole, imidazole, triazole, isotriazole, tetrazole, thiadiazole, isothiazole, oxadiazole, pyridine, piperidine, pyrazine, oxazole, isoxazole, pyrazine, pyridazine, pyrimidine, piperazine, pyrrolidine, morpholino, triazine, oxazine or thiazole.
- Preferred heterocyclic rings include piperazine, pyrrolidine, morpholino, imidazole, isotriazole, pyrazole, tetrazole, thiazole, thiadiazole, pyridine, piperidine, pyrazine, furan, oxazole, isoxazole, oxadiazole and azetidine.
- the 3- to-10-membered heterocyclic ring can be unsubstituted or substituted.
- Preferred substituents include halo, cyano, oxo, hydroxy, carboxy, nitro, Ci-C ⁇ -alkyl, CrCa-alkylcarbonyl, hydroxy-CrC ⁇ -alkyl, C ⁇ C ⁇ -haloalkyl, amino-CrC ⁇ -alkyl, amino(hydroxy)Ci-C 8 -alkyl and C 1 -C 8 - alkoxy optionally substituted by aminocarbonyl.
- substituents include halo, oxo, C r C 4 -alkyl, d-C ⁇ alkylcarbonyl, hydroxy-C 1 -C 4 -alkyl, C 1 -C 4 -haloalkyl, amino-C 1 -C 4 -alkyl and amino(hydroxy)C 1 -C 4 -alkyl.
- W is selected from CH 2 and O;
- R 1 is selected from CH 2 OH, CfOJ-NH-d-C ⁇ -alkyl and a 3- or10-membered heterocyclic ring containing at least one ring heteroatom selected from the group consisting of nitrogen, oxygen and sulphur optionally substituted by Ci-C 8 -alkyl;
- R 2 is hydrogen or C ⁇ C ⁇ -alkyl optionally substituted by C 6 -C 10 -aryl;
- R 3 and R 4 together with the nitrogen atom to which they are attached, form a 3- to 10-membered heterocyclic group containing the indicated nitrogen atom as a ring heteroatom and optionally at least one ring heteroatom selected from the group consisting of nitrogen, oxygen and sulphur, optionally substituted by 0-3R 5 ;
- R 5 is selected from OH, Ci-C 8 -alkyl optionally substituted by OH, or Ci-C 8 -alkoxy, C 7 -Ci 4 - aralkyl optionally substituted with OH, O-d-C 8 -alkyl, C 6 -C 10 -aryl, or O-C 6 -C 10 -aryl, CrC ⁇ -alkoxy, C 6 -C 10 -aryl optionally substituted by OH, C r C 8 -alkyl, O-C r C 8 -alkyl or halogen, O-C 6 -C 10 -aryl optionally substituted by OH, Ci-C 8 -alkyl, O-d-C 8 -alkyl or halogen, NR 53 R 5 ", NHC(O)R 50 , NHS(O) 2 R 5 ", NHS(O) 2 R 56 , NR 51 C(O)NR 59 R 511 , NR 5i C(O)OR 5
- R 53 , R 5b , R 50 , R 5f , R 5h and R 5i are, independently, H, d-C 8 -alkyl or C 6 -Ci 0 -aryl;
- R M , R 56 , R 59 and R 5 * are, independently, d-C 8 -alkyl or a 3- to 10-membered heterocyclic group containing at least one ring heteroatom selected from the group consisting of nitrogen, oxygen and sulphur, optionally substituted by 0-3R 6 ;
- R 5k is H, Ci-C 8 -alkyl, C 6 -C 10 -aryl or a 3- to 10-membered heterocyclic group containing at least one ring heteroatom selected from the group consisting of nitrogen, oxygen and sulphur;
- R 51 is CrC ⁇ -alkyl, C 6 -Ci 0 -aryl, NHR 7 or a 3- to 10-membered heterocyclic group containing at least one ring heteroatom selected from the group consisting of nitrogen, oxygen and sulphur;
- R 5m is H 1 CrC 8 -alkyl or Cr-Cu-aralkyl;
- R 6 is selected from OH, d-C 8 -alkyl optionally substituted by OH, C 7 -C 14 -aralkyl optionally substituted with OH, O-Ci-C 8 -alkyI, C 6 -C 10 -aryl or O-C 6 -C 10 -aryl, Ci-C 8 - alkoxy, C 6 -Ci 0 -aryl optionally substituted by OH, Ci-C 8 -alkyl, O-d-C 8 -alkyl or halogen, O-C 6 -Ci 0 -aryl optionally substituted by OH,
- R 6 ", R 68 , R 69 , R 6j and R 6 " 1 are, independently, d-C 8 -alkyl or a 3- to 10-membered heterocyclic group containing at least one ring heteroatom selected from the group consisting of nitrogen, oxygen and sulphur, optionally substituted by 0-3R 9 ;
- R 6k is H, Ci-C ⁇ -alkyl, C 6 -C 10 -aryl or a 3- to 10-membered heterocyclic group containing at least one ring heteroatom selected from the group consisting of nitrogen, oxygen and sulphur;
- R 61 is CrC ⁇ -alkyl, C 6 -Ci 0 -aryl or a 3- to 10-membered heterocyclic group containing at least one ring heteroatom selected from the group consisting of nitrogen, oxygen and sulphur, optionally substituted by COOR 10 ;
- R 7 isC00R 7a or a 3- to 10-membered heterocyclic group containing at least one
- Especially preferred compounds of the present invention include compounds of the formula (II) or stereoisomers or pharmaceutically acceptable salts thereof,
- R 1 is selected from CH 2 OH, C(O)-NH-Ci-C 4 -alkyl and a 3- to 10-membered heterocyclic ring containing at least one ring heteroatom selected from the group consisting of nitrogen, oxygen and sulphur optionally substituted by Ci-C 8 -alkyl;
- R 2 is hydrogen or Ci-C 4 -alkyl optionally substituted by C 6 -C 8 -aryl;
- R 3 and R 4 together with the nitrogen atom to which they are attached, form a 3- to 10-membered heterocyclic group containing the indicated nitrogen atom as a ring heteroatom and optionally at least one other ring nitrogen atom, optionally substituted by at least one heteroatom selected from the group consisting of nitrogen, oxygen and sulphur, optionally substituted by 0-3R 5 , the heterocyclic group being saturated or comprising a saturated heterocyclic ring fused to a carbocyclic ring or being a 5-membered unsaturated group;
- R 5 is selected from OH, d-C ⁇ alky! optionally substituted by OH, d-C 4 -alkoxy, C 6 -C 10 - aryl optionally substituted by halogen, O-C 6 -C 10 -aryl optionally substituted by halogen, NR 53 R 5 ", NHC(O)R 50 , NHS(O) 2 R 5 ", NHS(O) 2 R 58 , NR 91 C(O)NR 58 R 5 * 1 , NR 51 C(O)OR*.
- R 50 , R 56 , R 59 and R ⁇ are, independently, C 1 -C 4 -BlRyI or a 3- to 10-membered heterocyclic group containing at least one ring heteroatom selected from the group consisting of nitrogen, oxygen and sulphur, optionally substituted by 0-3R 6 ;
- R 5k is H, d-C 4 -alkyl or C 6 -C 10 -aryt
- R 51 is CrC 4 -alkyl, C 6 -C 10 -aryl, NHR 7 or a 3- to 10-membered heterocyclic group containing at least one ring heteroatom selected from the group consisting of nitrogen, oxygen and sulphur;
- R 5m is H, d-C ⁇ -alkyl or C 7 -C 14 -aralkyl
- R 6 is selected from OH, d-C 4 -alkyl optionally substituted by OH, C 6 -C 10 -aryl optionally substituted by OH, d-C 4 -alkyl, O-d-C 4 -alkyl or halogen, O-C 6 -C 10 -aryl optionally substituted by OH, d-C 4 -alkyl, O-d-C 4 -alkyl or halogen, NR 6a R 6b , NHC(O)R 6c , NHS(O) 2 R 60 , NHS(O) 2 R 68 , NR ⁇ C(O)NR 69 R 6 ", NR 6i C(O)OR 6j , d-C 4 -alkylcarbonyl, d-C ⁇ -alkoxycarbonyl, di(C 1 -C 4 -alkyl)aminocarbonyl, COOR 6k and C(O)R 61 , C(O)NHR 6 " 1
- R ⁇ a R 6b R 6c R ⁇ f R 6h an(j R a are i nde pendently, H, d-C 4 -alkyl or C 6 -C 10 -aryl;
- R 6d R 6 ⁇ R ⁇ g R 6j and R 6m are independently, d-C 4 -alkyl or a 3- to 10-membered heterocyclic group containing at least one ring heteroatom selected from the group consisting of nitrogen, oxygen and sulphur, optionally substituted by 0-3R 9 ;
- R 6k is H, d-C 4 -alkyl, C 6 -C 10 -aryl or a 3- to 10-membered heterocyclic group containing at least one ring heteroatom selected from the group consisting of nitrogen, oxygen and sulphur;
- R 61 is d-C 4 -alkyl, C 6 -C 10 -aryl or a 3- to 10-membered heterocyclic group containing at least one ring heteroatom selected from the group consisting of nitrogen, oxygen and sulphur, optionally substituted by COOR 10 ;
- R 7 is C00R 7a or a 3- to 10-membered heterocyclic group containing at least one ring heteroatom selected from the group consisting of nitrogen, oxygen and sulphur, optionally substituted by C00R 7a ;
- R ?a R 7b R 8 R 9 and R i o are selected from H, d-C 4 -alkyl and C 7 -C 14 -aralkyl.
- R ?a R 7b R 8 R 9 and R i o are selected from H, d-C 4 -alkyl and C 7 -C 14 -aralkyl.
- Especially preferred specific compounds of formula (I) are those described hereinafter in the Examples.
- the compounds represented by formula (I) may be capable of forming acid addition salts, particularly pharmaceutically acceptable acid addition salts.
- Pharmaceutically acceptable acid addition salts of the compound of formula (I) include those of inorganic acids, e.g., hydrohalic acids, such as hydrofluoric acid, hydrochloric acid, hydrobromic acid or hydroiodic acid, nitric acid, sulfuric acid or phosphoric acid; and organic acids, e.g., aliphatic monocarboxylic acids, such as formic acid, acetic acid, trifluoroacetic acid, propionic acid and butyric acid; aliphatic hydroxy acids, such as lactic acid, citric acid, tartaric acid or malic acid; dicarboxylic acids, such as maleic acid or succinic acid; aromatic carboxylic acids, such as benzoic acid, p-chlorobenzoic acid, diphenylacetic acid, para-biphenyl benzoic acid or triphenylacetic acid; aromatic hydroxy acids,
- Compounds of formula (I) which may contain acidic, e.g., carboxyl, groups, are also capable of forming salts with bases, in particular pharmaceutically acceptable bases, such as those well-known in the art; suitable such salts include metal salts, particularly alkali metal or alkaline earth metal salts, such as sodium, potassium, magnesium or calcium salts; or salts with ammonia or pharmaceutically acceptable organic amines or heterocyclic bases, such as ethanolamines, benzylamines or pyridine. These salts may be prepared from compounds of formula (Ia) by known salt-forming procedures.
- Stereoisomers are those compounds where there is an asymmetric carbon atom.
- the compounds exist in individual optically active isomeric forms or as mixtures thereof, e.g., as diastereomeric mixtures.
- the present invention embraces both individual optically active R and S isomers, as well as mixtures thereof.
- SYNTHESIS Another embodiment of the present invention, provides a process for the preparation of compounds of formula (I), in free or pharmaceutically acceptable salt form, which comprises the steps of:
- R 1 , R 2 and W are as defined in Claim 1;
- Z is H or a protecting group
- X is a leaving group, with a compound of formula (IV)
- R 3 and R 4 are as defined in Claim 1 ; and removing any protecting groups and recovering the resultant compound of formula (I), in free or pharmaceutically acceptable salt form.
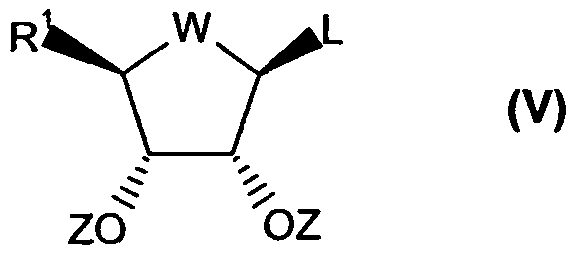
- the compound of formula (III) may be prepared by reacting a compound of formula (V)
- R 1 , Z and W are as defined in Claim 1;
- L represents a leaving group or a protected derivative thereof with a 2,6-dihalopurine, e.g., 2,6-dichloropurine, to provide a compound of formula (Vl)
- R 1 , Z and W are defined in Claim 1; and X and X 2 are halogen.
- the compounds of formula (I) can be prepared, e.g., using the reactions and techniques described below and in the Examples.
- the reactions may be performed in a solvent appropriate to the reagents and materials employed and suitable for the transformations being effected. It will be understood by those skilled in the art of organic synthesis that the functionality present on the molecule should be consistent with the transformations proposed. This will sometimes require a judgment to modify the order of the synthetic steps or to select one particular process scheme over another in order to obtain a desired compound of the invention.
- compounds of formula (I) can be prepared through two sequential nucleophilic aromatic substitution reactions to displace, e.g., chlorine atoms selectively and sequentially at the 6-position, to provide intermediate 2. Subsequent nucleophilic substitution at the 2-position with an appropriate amine provides compounds of formula (I). These reactions can be carried out either in the presence, or absence, of a base in addition to the reacting amine. A deprotection step may, or may not be necessary depending on the nature of the protecting group, if present.
- intermediate 3 or intermediate AD as referred to in the Examples is synthesized in accordance with the procedures outlined in the Examples, can be reacted with an amine through microwave or conventional heating described in the Examples to generate compound 4.
- purine derivative compounds with heterocyclic groups can be generated similar to the procedures outlined in Schemes 1-4 and the Examples.
- intermediate 9 where R 1 is a substituted tetrazole or a substituted isoxazole, such as ethyl substituted tetrazole or ethyl substituted isoxazole, can be generated according to the procedures outlined in WO 99/38877 and WO 98/28319.
- Intermediate 9 can then be reacted with an amine to provide compound 10.
- Compounds of formula (I), in free form, may be converted into salt form, and vice versa, in a conventional manner.
- the compounds in free or salt form can be obtained in the form of hydrates or solvates containing a solvent used for crystallisation.
- Compounds of formula (I) can be recovered from reaction mixtures and purified in a conventional manner. Isomers, such as stereoisomers, may be obtained in a conventional manner, e.g., by fractional crystallisation or asymmetric synthesis from correspondingly asymmetrically substituted, e.g., optically active, starting materials.
- Compounds of formula (I) and their pharmaceutically acceptable salts are useful as pharmaceuticals.
- they activate the adenosine A2A receptor, i.e., they act as A2A receptor agonists.
- Their properties as A2A agonists may be demonstrated using the method described by Murphree et a!., MoI Pharmacol, Vol. 61 , pp. 455-462 (2002).
- Ki values below 5.0 ⁇ M in the above assay For example, the compounds of Examples 2, 7, 9, 11, 13, 22, 24, 65, 77, 108, and 122 have Ki values of 0.61, 0.19, 0.16, 0.012, 0.054, 0.0005, 0.059, 0.002, 0.006, 0.005, and 0.004 ⁇ M respectively.
- agents of the invention are useful in the treatment of conditions which respond to the activation of the adenosine A2A receptor, particularly inflammatory or allergic conditions. Treatment in accordance with the invention may be symptomatic or prophylactic.
- agents of the invention are useful in the treatment of inflammatory or obstructive airways diseases, resulting, for example, in reduction of tissue damage, airways inflammation, bronchial hyperreactivity, remodelling or disease progression.
- Inflammatory or obstructive airways diseases and conditions to which the present invention is applicable include acute lung injury (ALI), adult/acute respiratory distress syndrome (ARDS), chronic obstructive pulmonary, airways or lung disease (COPD, COAD or COLD), including chronic bronchitis or dyspnea associated therewith, emphysema, as well as exacerbation of airways hyperreactivity consequent to other drug therapy, in particular other inhaled drug therapy.
- the invention is also applicable to the treatment of bronchitis of whatever type or genesis including, e.g., acute, arachidic, catarrhal, croupus, chronic or phthinoid bronchitis.
- bronchiectasis pneumoconiosis (an inflammatory, commonly occupational, disease of the lungs, frequently accompanied by airways obstruction, whether chronic or acute, and occasioned by repeated inhalation of dusts) of whatever type or genesis, including, for example, aluminosis, anthracosis, asbestosis, chalicosis, ptilosis, siderosis, silicosis, tabacosis and byssinosis.
- asthma inflammatory or obstructive airways diseases to which the present invention is applicable
- asthma of whatever type or genesis including both intrinsic (non-allergic) asthma and extrinsic (allergic) asthma, mild asthma, moderate asthma, severe asthma, bronchitic asthma, exercise-induced asthma, occupational asthma and asthma induced following bacterial infection.
- Treatment of asthma is also to be understood as embracing treatment of subjects, e.g. of less than 4 or 5 years of age, exhibiting wheezing symptoms and diagnosed or diagnosable as "whez infants", an established patient category of major medical concern and now often identified as incipient or early-phase asthmatics. (For convenience this particular asthmatic condition is referred to as "whez-infant syndrome".)
- Prophylactic efficacy in the treatment of asthma will be evidenced by reduced frequency or severity of symptomatic attack, e.g. of acute asthmatic or bronchoconstrictor attack, improvement in lung function or improved airways hyperreactivity. It may further be evidenced by reduced requirement for other, symptomatic therapy, i.e. therapy for or intended to restrict or abort symptomatic attack when it occurs, for example anti-inflammatory (e.g. cortico-steroid) or bronchodilatory.
- Prophylactic benefit in asthma may in particular be apparent in subjects prone to "morning dipping". "Morning dipping" is a recognised asthmatic syndrome, common to a substantial percentage of asthmatics and characterised by asthma attack, e.g. between the hours of about 4 to 6 am, i.e. at a time normally substantially distant from any previously administered symptomatic asthma therapy.
- agents of the invention are also useful in the treatment of eosinophil related disorders, e.g. eosinophilia, in particular eosinophil related disorders of the airways (e.g.
- eosinophilic infiltration of pulmonary tissues including hyper-eosinophilia as it effects the airways and/or lungs as well as, for example, eosinophil-related disorders of the airways consequential or concomitant to L ⁇ ffler's syndrome, eosinophilic pneumonia, parasitic (in particular metazoan) infestation (including tropical eosinophilia), bronchopulmonary aspergillosis, polyarteritis nodosa (including Churg-Strauss syndrome), eosinophilic granuloma and eosinophil-related disorders affecting the airways occasioned by drug-reaction.
- Agents of the invention are also useful in the treatment of inflammatory or allergic conditions of the skin, for example psoriasis, contact dermatitis, atopic dermatitis, alopecia areata, erythema multiforma, dermatitis herpetiformis, scleroderma, vitiligo, hypersensitivity angiitis, urticaria, bullous pemphigoid, lupus erythematosus, pemphisus, epidermolysis bullosa acquisita, and other inflammatory or allergic conditions of the skin.
- Agents of the invention may also be used for the treatment of other diseases or conditions, in particular diseases or conditions having an inflammatory component, for example, treatment of diseases and conditions of the eye such as conjunctivitis, keratoconjunctivitis sicca, and vernal conjunctivitis, diseases affecting the nose including allergic rhinitis, and inflammatory disease in which autoimmune reactions are implicated or having an autoimmune component or aetiology, including autoimmune haematological disorders (e.g.
- haemolytic anaemia haemolytic anaemia, aplastic anaemia, pure red cell anaemia and idiopathic thrombocytopenia
- systemic lupus erythematosus polychondritis, sclerodoma, Wegener granulamatosis, dermatomyositis, chronic active hepatitis, myasthenia gravis, Steven-Johnson syndrome, idiopathic sprue, autoimmune inflammatory bowel disease (e.g.
- ulcerative colitis and Crohn's disease endocrine opthalmopathy
- Grave's disease sarcoidosis, alveolitis, chronic hypersensitivity pneumonitis, multiple sclerosis, primary billiary cirrhosis, uveitis (anterior and posterior), keratoconjunct-ivitis sicca and vernal keratoconjunctivitis, interstitial lung fibrosis, psoriatic arthritis and glomerulonephritis (with and without nephrotic syndrome, e.g. including idiopathic nephrotic syndrome or minal change nephropathy).
- agents of the invention may also be used for the treatment of cystic fibrosis, pulmonary hypertension, pulmonary fibrosis, inflammatory bowel syndrome, wound healing, diabetic nephropathy as described in WO 05/107463, reduction of inflammation in transplanted tissue as described in US 2005/182018, inflammatory diseases caused by pathogenic organisms as described in WO 03/086408, and cardiovascular conditions as described in WO 03/029264.
- the agents of the invention may be used to assess the severity of coronary artery stenosis as described in WO 00/078774 and useful in conjunction with radioactive imaging agents to image coronary activity and useful in adjunctive therapy with angioplasty as described in WO 00/78779.
- Agents of the invention are also useful in combination with a protease inhibitor for prevention of organ ischaemia and reperfusion injury as described in WO 05/003150, and in combination with an integrin antagonist for treating platelet aggregation as described in WO 03/090733.
- Agents of the invention are also useful in promoting wound healing in bronchial epithelial cells as described in AJP-Lung 290: 849-855.
- diabetes e.g. diabetes mellitus type I Q ' uvenile diabetes
- diabetes mellitus type II diarrheal diseases
- ischemia/reperfusion injuries retinopathy, such as diabetic retinopathy or hyperbaric oxygen-induced retinopathy
- conditions characterised by elevated intraocular pressure or secretion of ocular aqueous humor such as glaucoma, ischemic tissue/organ damage from reperfusion, bedsores, as agents for promoting sleep, as agents for treating demyelinating diseases, eg multiple sclerosis and as neuroprotective agents for eg, cerebral haemorrhagic injury and spinal cord ischaemi-reperfusion injury.
- an agent of the invention in inhibiting inflammatory conditions, e.g., in inflammatory airways diseases, may be demonstrated in an animal model, e.g., a mouse or rat model, of airways inflammation or other inflammatory conditions, e.g., as described by Szarka et al., J Immunol Methods, Vol. 202, pp. 49-57 (1997); Renzi et al., Am Rev Respir Dis, Vol. 148, pp. 932-939 (1993); Tsuyuki et al., J Clin Invest, Vol. 96, pp. 2924-2931 (1995); Cernadas et al, Am J Respir Cell MoI Biol, Vol. 20, pp. 1-8 (1999); and Fozard et al., ErJ Pharmacol, Vol. 438, pp. 183-188 (2002).
- the agents of the invention are also useful as co-therapeutic agents for use in combination with other drug substances, such as anti-inflammatory, bronchodilatory, antihistamine or anti-tussive drug substances, particularly in the treatment of obstructive or inflammatory airways diseases, such as those mentioned hereinbefore, e.g., as potentiators of therapeutic activity of such drugs or as a means of reducing required dosaging or potential side effects of such drugs.
- An agent of the invention may be mixed with the other drug substance in a fixed pharmaceutical composition or it may be administered separately, before, simultaneously with or after the other drug substance.
- the invention includes a combination of an agent of the invention as hereinbefore described with an anti-inflammatory, bronchodilatory, antihistamine or anti-tussive drug substance, said agent of the invention and said drug substance being in the same or different pharmaceutical composition.
- Suitable anti-inflammatory drugs include steroids, in particular, glucocorticosteroids, such as budesonide, beclamethasone dipropionate, fluticasone propionate, ciclesonide or mometasone furoate; or steroids described in WO 02/88167, WO 02/12266, WO 02/100879, WO 02/00679 (especially those of Examples 3, 11, 14, 17, 19, 26, 34, 37, 39, 51 , 60, 67, 72, 73, 90, 99 and 101), WO 03/35668, WO 03/48181, WO 03/62259, WO 03/64445, WO 03/72592, WO 04/39827 and WO 04/66920; non-steroidal glucocorticoid receptor agonists, such as those described in DE 10261874, WO 00/00531, WO 02/10143, WO 03/82280, WO 03/82787, WO 03/86294, WO 03/104195, WO 03/10
- Suitable bronchodilatory drugs include anti-cholinergic or anti-muscarinic agents, in particular, ipratropium bromide, oxitropium bromide, tiotropium salts and CHF 4226 (Chiesi), and glycopyrrolate, but also those described in EP 424021 , US 3,714,357, US 5,171 ,744, WO 01/04118, WO 02/00652, WO 02/51841, WO 02/53564, WO 03/00840, WO 03/33495, WO 03/53966, WO 03/87094, WO 04/018422 and WO 04/05285.
- Suitable dual anti-inflammatory and bronchodilatory drugs include dual ⁇ -2 adrenoceptor agonist/muscarinic antagonists, such as those disclosed in US 2004/0167167, US 2004/0242622, US 2005/182092, WO 04/74246 WO 04/74812, WO 04/089892 and US 05/256114.
- Suitable anti-histamine drug substances include cetirizine hydrochloride, acetaminophen, clemastine fumarate, promethazine, loratidine, desloratidine, diphenhydramine and fexofenadine hydrochloride, activastine, astemizole, azelastine, ebastine, epinastine, mizolastine and tefenadine, as well as those disclosed in JP 2004107299, WO 03/099807 and WO 04/026841.
- agents of the invention with anti-inflammatory drugs are those with antagonists of chemokine receptors, e.g., CCR-1 , CCR-2, CCR-3, CCR-4, CCR-5, CCR-6, CCR-7, CCR-8, CCR-9 and CCR10, CXCR1, CXCR2, CXCR3, CXCR4, CXCR5, particularly CCR-5 antagonists, such as Schering-Plough antagonists SC-351125, SCH-55700 and SCH-D; Takeda antagonists, such as ⁇ /-[[4-[[[6,7-dihydro-2-(4-methylphenyl)-5H-benzo- cyclohepten-8-yl]carbonyl]amino]phenyl]-methyl]tetrahydro- ⁇ /, ⁇ /-dimethyl-2H-pyran-4-aminium chloride (TAK-770); and CCR-5 antagonists described in US 6,166,037 (particularly Claims 18 and 19), WO 00/665
- the invention also provides a method for the treatment of a condition responsive to activation of the adenosine A2A receptor, e.g., an inflammatory or allergic condition, particularly an inflammatory or obstructive airways disease, which comprises administering to a subject, particularly a human subject, in need thereof a compound of formula (I), in free form or in the form of a pharmaceutically acceptable salt.
- a compound of formula (I), in free form or in the form of a pharmaceutically acceptable salt for use in the manufacture of a medicament for the treatment of a condition responsive to activation of the adenosine A2A receptor, particularly an inflammatory or obstructive airways disease.
- the agents of the invention may be administered by any appropriate route, e.g., orally, e.g., in the form of a tablet or capsule; parenterally, e.g., intravenously; by inhalation, e.g., in the treatment of inflammatory or obstructive airways disease; intranasally, e.g., in the treatment of allergic rhinitis; topically to the skin, e.g., in the treatment of atopic dermatitis; or rectally, e.g., in the treatment of inflammatory bowel disease.
- routes e.g., orally, e.g., in the form of a tablet or capsule; parenterally, e.g., intravenously; by inhalation, e.g., in the treatment of inflammatory or obstructive airways disease; intranasally, e.g., in the treatment of allergic rhinitis; topically to the skin, e.g., in the treatment
- the invention also provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of formula (I), in free form or in the form of a pharmaceutically acceptable salt, optionally together with a pharmaceutically acceptable diluent or carrier therefor.
- the composition may contain a co-therapeutic agent, such as an anti-inflammatory, bronchodilatory, anti-histamine or anti-tussive drug, as hereinbefore described.
- a co-therapeutic agent such as an anti-inflammatory, bronchodilatory, anti-histamine or anti-tussive drug, as hereinbefore described.
- Such compositions may be prepared using conventional diluents or excipients and techniques known in the galenic art.
- oral dosage forms may include tablets and capsules.
- Formulations for topical administration may take the form of creams, ointments, gels or transdermal delivery systems, e.g., patches.
- Compositions for inhalation may comprise aerosol or other atomizable formulations or dry powder
- the composition comprises an aerosol formulation
- it preferably contains, e.g., a hydro-fluoro-alkane (HFA) propellant, such as HFA134a or HFA227 or a mixture of these, and may contain one or more co-solvents known in the art such as ethanol (up to 20% by weight); and/or one or more surfactants, such as oleic acid or sorbitan trioleate; and/or one or more bulking agents, such as lactose.
- HFA hydro-fluoro-alkane
- the composition comprises a dry powder formulation, it preferably contains, e.g., the compound of formula (I) having a particle diameter up to 10 microns, optionally together with a diluent or carrier, such as lactose, of the desired particle size distribution and a compound that helps to protect against product performance deterioration due to moisture, e.g., magnesium stearate.
- a diluent or carrier such as lactose
- the composition comprises a nebulised formulation, it preferably contains, e.g., the compound of formula (I) either dissolved, or suspended, in a vehicle containing water, a co-solvent, such as ethanol or propylene glycol and a stabiliser, which may be a surfactant.
- the invention includes: a) a compound of formula (I) in inhalable form, e.g., in an aerosol or other atomisable composition or in inhalable particulate, e.g., micronised, form; b) an inhalable medicament comprising a compound of formula (I) in inhalable form; c) a pharmaceutical product comprising a compound of formula (I) in inhalable form in association with an inhalation device; and d) an inhalation device containing a compound of formula (I) in inhalable form.
- Dosages of compounds of formula (I) employed in practising the present invention will of course vary depending, e.g., on the particular condition to be treated, the effect desired and the mode of administration.
- suitable daily dosages for administration by inhalation are of the order of 0.005-10 mg, while for oral administration suitable daily doses are of the order of 0.05-100 mg.
- the invention is illustrated by the following Examples.
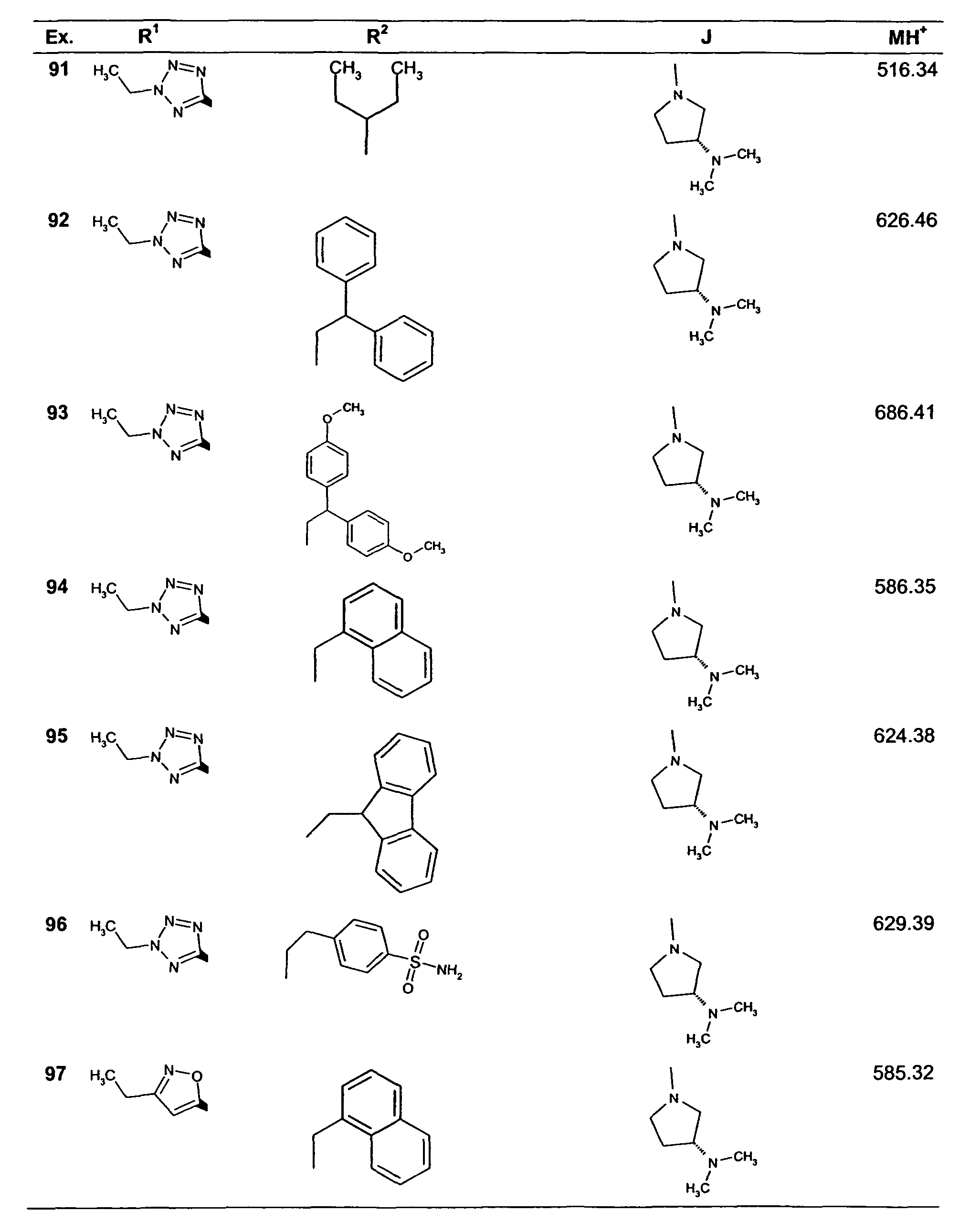
- Table 1 shows mass spectrometry, MH+ (ESI+), data.
- the title compound is prepared by the procedure of Preparation of Aminopurine- ⁇ -D- Ribofuranuronamide Derivatives as Antiinflammatories, Ayres et al., Glaxo Group Limited, UK, PCT Int. Appl., WO 96/02553, 49 pages (1996). - 46 -
- the title compound is prepared by the procedure of Preparation of 2-(purin-9-yl)- Tetrahydrofuran-3,4-diol Nucleosides as Antiinflammatory Agents and Agonists against Adenosine Receptors, Cox et al., Glaxo Group Ltd., UK, PCT Int. Appl. WO 98/28319 A1 , 118 pages (1998).
- Step AC1 Acetic acid (2f?,3f?,4f? t 5R)-4-acetoxy-5-acetoxymethyl-2-(6-chloro-2-nitro-purin-9- yl)-tetrahydro-furan-3-yl ester.
- the title compound is prepared by the procedure of Synthesis and Properties of 2-Nitrosoadenosine, Wanner, Koomen and Gerrit-Jan, Laboratory of Organic Chemistry, Institute of Molecular Chemistry, University of Amsterdam, Amsterdam, Neth., J Chem Soc, Perkin Transactions 1 (16), pp. 1908-1915 (2001).
- Step AC2 Acetic acid (2R,3R,4R,5R)-4-acetoxy-2-acetoxymethyl-5-(2-nitro-6-phenethylamino- purin-9-yl)-tetrahydro-furan-3-yl ester
- Step AEI Acetic acid (2f?,3f?,4/?,5S)-4-acetoxy-2-[6-((S)-1-benzyl-2-hydroxy-ethyl amino)-2- chloro-purin-9-yl]-5-(3-ethyl-isoxazol-5-yt)-tetrahydro-furan-3-yl ester hydrochloride
- Step AE2 (2/?,3R,4R,5S)-2-[6-((S)-1 -Benzyl-2-hydroxy-ethylamino)-2-chloro-purin-9-yl]-5-(3- ethyl-isoxazol-5-yl)-tetrahydro-furan-3,4-diol
- Step AH Acetic acid (2f?,3R,4/? l 5/?)-4-acetoxy-2-[6-((S)-1-benzyl-2-hydroxy-ethyl amino)-2- chloro-purin-9-yl]-5-(2-ethyl-2H-tetrazol-5-yl)-tetrahydro-furan-3-yl ester:
- Step AI2 (2R,3f?,4S,5R)-2-[6-((S)-1 -Benzyl-2-hydroxy-ethylamino)-2-chloro-purin-9-yl]-5-(2- ethyl-2H-tetrazol-5-yl)-tetrahydro-furan-3,4-diol
- Step AH analogously to (2R,3f?,4S,5S)-2-[6-((S)-1-benzyl-2-hydroxy-ethylamino)-2- chloro-purin-9-yl]-5-(3-ethyl-isoxazol-5-yl)-tetrahydro-furan-3,4-diol.
- Step BC1 4-Carbamoyl-3,4,5,6-tetrahydro-2H-[1,2 I ]bipyridinyl-5 I -carboxylic acid ethyl ester
- a stirred suspension comprising 6-chloro-nicotinic acid ethyl ester (1.86 g, 10.0 mmol), piperidine-4-carboxamide (1.54 g, 12.0 mmol) and DIPEA (2.1 mL, 12 mmol) in DMSO (7 mL) is heated to 90 0 C for 2 hours. MeOH (8 mL) is then added as the reaction mixture cools and the resulting precipitate is filtered, washed with water followed by ether and dried in vacuo (45°C) to yield the titled compound as a white powder.
- Step BC2 4-Amino-3,4,5,6-tetrahydro-2H-[1 ,2']bipyridinyl-5'-carboxylic acid ethyl ester
- a solution comprising 4-carbamoyl-3,4,5,6-tetrahydro-2H-[1 ⁇ 'Jbipyridinyl- ⁇ '-carboxylic acid ethyl ester (2.04 g, 7.36 mmol) and 6/s(trifluoroacetoxy) iodobenzene (3.80 g, 8.83 mmol) in acetonitrile (13 mL) is treated with water (5 mL) and heated to 65°C for 30 hours. The solvent is partially removed in vacuo and the resulting solution is acidified to pH 1 using 12 M HCI. The solution is extracted with EtOAc and this organic portion is discarded.
- the aqueous portion is basified to pH 8-9 using 2 M potassium carbonate solution and then extracted with EtOAc then DCM.
- the combined organic portions are washed with brine, dried (Na 2 SO 4 ) and concentrated in vacuo.
- the resulting residue is triturated with ether followed by ether/EtOAc (1 :1 , 5 x 0.7 mL) and dried in vacuo to yield the titled product as an off-white solid.
- Step BC3 4-[(lmidazole-1-carbonyl)-amino]-3,4,5,6-tetrahydro-2H-[1 ,2']bipyridinyl-5'-carboxylic acid ethyl ester
- Step BD1 1,3-/j/s-((R)-1-Benzyl-pyrrolidin-3-yl)-urea
- Step BD2 1 ,3-di(R)-Py ⁇ Olidin-3-yl-urea
- the title compound is prepared by the procedure of The Selective Reaction of Primary Carbonyl Imidazole Containing Compounds: Selective Amide and Carbamate Synthesis. Rannard and Davis, Org Lett, Vol. 2, No. 14, pp. 2117-2120 (2000).
- Step BH1 (R ⁇ - ⁇ -Benzyl-piperidine-i-carbonylJ-pyrrolidine-i-carboxylic acid benzyl ester
- Step BH2 (4-Benzyl-piperidin-1 -yl)-(R)-pyrrolidin-2-yl-methanone
- Step BH3 4-Benzyl-1-(/?)-1-pyrrolidin-2-ylmethyl-piperidine
- reaction mixture is stirred for 15 minutes at RT and then treated with 1 M HCI (520 mL). After stirring for 1.5 hours, the reaction mixture is extracted 3 times with DCM. The combined organic layers are dried (Na 2 SO 4 ) and concentrated in vacuo. The resulting crude is purified by flash chromatography on silica gel eluting with hexane.EtOAc (7:3) to afford the titled compound as a colourless oil.
- a cooled suspension comprising (2S,4R)-4-tert-butoxy-2-hydroxymethyl-pyrrolidine-1- carboxylic acid benzyl ester (19.2 g, 62.5 mmol), phthalimide (9.2 g, 62.5 mmol) and triphenylphosphine (6.7 g, 62.5 mmol) in THF (260 mL) is carefully treated dropwise with DEAD (3.7 mL, 62.46 mmol). After stirring at RT for 2 hours, further portions of phthalimide (0.92 g, 6.2 mmol), triphenylphosphine (0.67 g, 6.2 mmol) and DEAD (0.37 mL, 6.2 mmol) are added.
- Step BI3 (2S,4f?)-2-Aminomethyl-4-teAf-butoxy-pyrrolidine-1 -carboxylic acid benzyl ester
- Step BI4 (2S,4R)-4-tert-Butoxy-2-(fert-butoxycarbonylamino-methyl)-pyrrolidine-1 -carboxylic acid benzyl ester
- Step BI5 ((2S,4fi)-4-ferf-Butoxy-pyrrolidin-2-ylmethyl)-carbamic acid tert-butyl ester
- the title compound can be prepared by the procedure of Gregson, Michael; Ayres, Barry Edward; Ewan, George Blanch; Ellis, Frank; Knight, John. Preparation of diaminopurinylribofuranuronamide derivatives as antiinflammatories. (WO 94/17090)
- Step E2 (R)-[1 ,3 f ]Bipyrrolidinyl
- Step F1 6-((R)-1-Benzyl-pyrrolidin-3-ylamino)-nicotinonitrile
- Step F2 (5-Methyl-pyridin-2-yl)-(R)-pyrrolidin-3-yl-amine
- Step GV (RJ-S-KPyridine ⁇ -carbonyO-aminoj-pyrrolidine-i-carboxylic acid fert-butyl ester
- Step G2 (R)- ⁇ /-Pyrrolidin-3-yl-nicotinamide
- Step H2 (f?)-2-Pyrrolidin-3-yl-2,3-dihydro-1H-isoindole-5-carboxylic acid methyl ester
- Step 11 (RJ-S-KPyridine ⁇ -carbonyO-aminol-pyrrolidine-i-carboxylic acid te/f-butyl ester
- Racemic 3-(4-fluoro-phenyl)-pyrrolidine (696 g, 3.7 mol) is suspended in EtOH (11 L) and heated to 55-60 0 C to give a solution, whereupon a solution of (+)-di-O,O-p-tolyl tartaric acid (814 g, 2.1 mol) in EtOH (3 L) is added over 20 minutes. The solution is cooled to 0 0 C over 4 hours and stirred overnight to give an off-white suspension which is washed with two portions of cold EtOH (2 x 450 mL). The resulting solid is dissolved in EtOH (9 L) at 60 0 C and then cooled over 4 hours to 22°C. The resulting suspension is filtered and washed with two portions of EtOH (2 x 300 mL). The re-crystallisation was repeated twice more using EtOH (6.5 L) to afford the title product.
- Example 5 • ⁇ («)-1-[9-((2R,3R,4S I 5R)-3,4-dihydroxy-5-hydroxymethyl-tetrahydro-furan-2-yl)-6-(2 > 2- diphenyl-ethylamino)-9H-purin-2-yl]-pyrrolidin-3-yl ⁇ -carbamic acid terf-butyl ester trifluroacetate (Example 5), are prepared by an analogous procedure to Example 1 by replacing 4-(4-fluoro-phenyl)- piperidine with the appropriate amine.
- the titled compound is prepared analogously to Example 6 by replacing ⁇ (S)-1-[9-((2/?,3R,4S,5R)-3,4-dihydroxy-5-hydroxymethyl-tetrahydro-furan-2-yl)-6-(2 ) 2-diphenyl- ethylamino)-9H-purin-2-yl]-pyrrolidin-3-yl ⁇ -carbarnic acid terf-butyl ester trifluroacetate with ⁇ (R)-1-[9-((2f?,3R4S,5R)-3,4-dihydroxy-5-hydroxymethyl-tetrahydro-furan-2-yl)-6-(2,2-diphenyl- ethylamino)-9H-purin-2-yl]-pyrrolidin-3-yl ⁇ -carbamic acid terf-butyl ester trifluroacetate.
- Step i ⁇ (f?)-1-[9-((2f?,3R,4S,5R)-3,4-Dihydroxy-5-hydroxymethyl-tetrahydro-furan-2-yl)-6-(2 ) 2- diphenyl-ethylamino)-9H-purin-2-yl]-piperidin-3-yl ⁇ -carbamic acid terf-butyl ester
- the titled compound is prepared analogously to Example 1 by replacing 4-(4-fluoro- phenyl)-piperidine with (f?)-piperidin-3-yl-carbamic acid terf-butyl ester. - 59 -
- Step 2 (2R3R,4S,5fi)-2-[2-((/?)-3-Amino-piperidin-1 -yl)-6-(2,2-diphenyl-ethylamino)-purin-9-yl]- 5-hydroxymethyl-tetrahydro-furan-3,4-diol trifluroacetate
- the titled compound is prepared analogously to Example 6 by replacing ⁇ (S)-1-[9-((2/?,3/? > 4S,5R)-3,4-dihydroxy-5-hydroxymethyl-tetrahydro-furan-2-yl)-6-(2 I 2-diphenyl- ethylamino)-9H-purin-2-yl]-pyrrolidin-3-yl ⁇ -carbarnic acid tert-butyl ester trifluroacetate with ⁇ (/?)-1-[9-((2R,3R4S,5R)-3,4-dihydroxy-5-hydroxymethyl-tetrahydro-furan-2-yl)-6-(2,2-diphenyl- ethylamino)-9H-purin-2-yl]-piperidin-3-yl ⁇ -carbamic acid terf-butyl ester.
- Example 9 1 - ⁇ (ft)-1 -[9-((2/?,3/?,4S,5R)-3,4-Dihydroxy-5-hydroxymethyl-tetrahydro-furan- 2-yl)-6-(2 > 2-dihpenyl-ethylamino)-9H-purin-2-yl]-pyrrolidin-3-yl ⁇ -3-(3 f 4 > 5,6- tetrahydro-2H-[1 ,2']bipyridinyl-4-yl)-urea trifluoroacetate
- Example 10 4-(3- ⁇ (A?)-1-[9-((2/?,3/? > 4S,5/?)-3,4-Dihydroxy-5-hydromethyl-tetrahydro-furan- 2-yl)-6-(2,2-diphenyl-ethylamino)-9H-purin-2-yl]-pyrrolindin-3-yl ⁇ -ureido)- SAS.e-tetrahydro ⁇ W-II ⁇ 'lbipyridinyl-S'-carboxylic acid ethyl ester trifluoroacetate
- the titled compound is prepared by the same procedure as Example 9 by replacing the imidazole-1 -carboxylic acid (S ⁇ . ⁇ . ⁇ -tetrahydro ⁇ H-fi.ZJbipyridinyM-yO-amide with 4-[(imidazole-1 -carbonyl)-amino]-3,4,5,6-tetrahydro-2H-[1 ,21bipyridinyl-5'-carboxylic acid ethyl ester.
- Example 11 1 - ⁇ (/?)-1 -[9-((2/?,3f?,4S,5R)-3,4-Dihydroxy-5-hydroxymethyl-tetrahydro-f uran- 2-yl)-6-(2,2-diphenyl-ethylamino)-9H-purin-2-yl]-pyrrolidin-3-yl ⁇ -3-(R)- pyrrolidin-3-yl-urea trifluoroacetate
- Example 24 4- ⁇ [(/?)-3- ⁇ 3- ⁇ (R)-1-[9-((2/?,3/?,4S,5/?)-3,4-Dihydroxy-5-hydroxymethyl- tetrahydro-furan-2-yl)-6- ⁇ 2,2-diphenyl-ethylamino)-9H-purin-2-yl]- pyrrolidine-3-yl ⁇ -ureido)-pyrrolidine-1-carbonyl]-amino ⁇ -piperidine-1- carboxylic acid benzyl ester trifluoroacetate
- Example 25 4-(3- ⁇ (R)-1-[9-((2R,3R,4S,5R)-3,4-Dihydroxy-5-hydroxymethyl-tetrahydro- furan-2-yl)-6-(2,2-diphenyl-ethylamino)-9H-purin-2-yl]-pyrrolidin-3-yl ⁇ - ureido)-3,4,5 l 6-tetrahydro-2H-[1,2']bipyridinyl-5 I -carboxylic acid trifluoroacetate
- the titled compound is prepared by the same procedure as Example 11 by replacing (2R,3R,4S,5R)-2-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-5-ydroxymethyl-tetrahydro- - 62 -
- Step 1 (3aS,4S,6R,6aR)-6- ⁇ 6-Amino-2-[3-(3 I 4-dichloro-phenoxy)-azetidin-1 -yl]-purin-9-yl ⁇ -2,2- dimethyl-tetrahydro-furo[3,4-c(][1 ,3]dioxole-4-carboxylic acid ethylamide
- Step 2 (2/?,3R,4S,5R)-5- ⁇ 6-Amino-2-[3-(3,4-dichloro-phenoxy)-azetidin-1 -yl]-purin-9-yl ⁇ -3,4- dihydroxy-tetrahydro-furan-2-carboxylic acid ethylamide trifluoroacetate
- the titled compound is prepared by the same procedure as Example 33 by replacing 3-(3,4-dichloro-phenoxy)-azetidine with (R)-3-(4-fluoro-phenyl)-pyrrolidine.
- Step 1 Acetic acid (2R,3R4S f 5R)-3,4-diacetoxy-5- ⁇ 2-[3-(4-chloro-benzyl)-azetidin-1-yl]-6- phenethylamino-purin-9-yl ⁇ -tetrahydro-furan-2-ylmethyl ester
- the titled compound is prepared by the same procedure as Example 1 by replacing (2R,3f? ) 4S I 5R)-2-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-5-ydroxymethyl-tetrahydro- furan-3,4-diol with acetic acid (2R,3R,4S,5R)-4- a cetoxy-2-acetoxymethyl-5-(2-nitro-6- phenethylamino-purin-9-yl)-tetrahydro-furan-3-yl ester and by replacing 4-(4-fluoro-phenyl)- piperidine with 3-(4-chloro-benzyl)-azetidine (WO 2003/077907).
- Step 2 (2R,3R,4S,5R)-2- ⁇ 2-[3-(4-Chloro-benzyl)-azetidin-1-yl]-6-phenethylamino-purin-9-yl ⁇ -5- hydroxymethyl-tetrahydro-furan-3,4-diol
- Example 30 4- ⁇ 1-[9- ⁇ (2/?,3/?,4S,5/?)-3,4-Dihydroxy-5-hydroxymethyl-tetrahydro-furan-2- yl)-6-(2,2-diphenyl-ethylamino)-9W-purin-2-yl]-pyrrolidin-3-yl ⁇ -pipera2ine-1- carboxylic acid benzyl ester trifluoroacetate
- the titled compound is prepared analogously to Example 1 by replacing 4-(4-fluoro- phenyl)-piperidine with 4-pyrrolidin-3-yl-piperazine-1-carboxylic acid benzyl ester.
- Example 58 4-[(/?)-3-(3- ⁇ (/?)-1-[6-(2,2-Diphenyl-ethylamino)-9-((2/? I 3/? > 4S,5S)-5- ethylcarbamoyl-3,4-dihydroxy-tetrahydro-furan-2-yl)-9H-purin-2-yl]- pyrrolidin-3-yl ⁇ -ureido)-pyrrolidine-1-carbonyl]-benzoic acid methyl ester trifluoroacetate
- a suspension comprising (2S,3S,4R,5f?)-5- ⁇ 6-(2 I 2-diphenyl-ethylamino)-2-[(/?)-3-((R)-3- pyrrolidin-S-ylureidoJ-pyrrolidin-i-yll-purin- ⁇ -ylj-S ⁇ -dihydroxy-tetrahydro-furan ⁇ -carboxylic acid ethylamide hydrochloride (Example 57) (0.144 g, 0.2 mmol), methyl-4-chlorocarbonyl benzoate (0.059 g, 0.3 mmol) and TEA (83 ⁇ L, 0.6 mmol) in THF (2 ml_) and NMP (0.6 mL) is stirred at RT for 3 days. The solvent is removed in vacuo and purification by C-18 reverse phase column chromatography eluting with acetonitrile.water (0.1% TFA) (gradient of 0-100%
- Example 59 4-[(/?)-3-(3- ⁇ (/?)-1-[6-(2,2-Diphenyl-ethylamino)-9-((2/?,3/?,4S I 5S)-5- ethylcarbamoyl-3,4-dihydroxy-tetrahydro-furan-2-yl)-9H-purin-2-yl]- pyrrolidin-3-yl ⁇ -ureido)-pyrrolidine-1-carbonyl]-benzoic acid trifluoroacetate
- Example 64 • ⁇ /-((R)-1- ⁇ 6-(2,2-Diphenyl-ethylamino)-9-[(2R3R,4S,5R)-5-(2-ethyl-2H-tetrazol-5-yl)-3,4- dihydroxy-tetrahydro-furan-2-yl]-9H-purin-2-yl ⁇ -pyrrolidin-3-yl)-nicotinamide trifluoroacetate (Example 64), are prepared analogously to Example 11 by replacing (2R,3R,4S,5R)-2-[2-chloro-6-(2,2- diphenyl-ethylamino)-purin-9-yl]-5-ydroxymethyl-tetrahydro-furan-3,4-diol with (2/?,3R,4S,5R)-2- [2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-5-(2-eth
- Example 73 • ⁇ /-((R)-1- ⁇ 6-(2,2-Diphenyl-ethylamino)-9-[(2R3/?,4S,5S)-5-(3-ethyl-isoxazol-5-yl)-3,4- dihydroxy-tetrahydro-furan-2-yl]-9H-purin-2-yl ⁇ -pyrrolidin-3-yl)-nicotinamide trifluoroacetate (Example 73), are prepared analogously to Example 11 by replacing (2f?,3fi,4S,5R)-2-[2-chloro-6-(2,2- diphenyl-ethylamino)-purin-9-yl]-5-hydroxymethyl-tetrahydro-furan-3,4-diol with the appropriate intermediate (described herein) and by replacing 1,3-di(R)-pyrrolidin-3-yl-urea with the appropriate 3-(f?)-aminopyrrolidine derivative.
- Step 1 ⁇ /- ⁇ (/?)-1-[6-(2,2-Diphenyl-ethylamino)-9-((3aR > 4R > 6S,6aS)-6-ethylcarbamoyl-2,2- dimethyl-tetrahydro-furo[3,4-c(l[1,3]dioxol-4-yl)-9H-purin-2-yl]-pyrrolidin-3-yl ⁇ - isonicotinamide trifluoroacetate
- This compound is prepared analogously to Example 11 by replacing (2R,3R,4S,5f?)-2- [2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-5-hydroxymethyl-tetrahydro-furan-3,4-diol with (3aS,4S,6/ : ?,6af?)-6-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-2,2-dimethyl-tetrahydro- furo[3,4-d][1,3]dioxole-4-carboxylic acid ethylamide (WO 96/02553) and by replacing 1,3-di(f?)- pyrrolidin-3-yl-urea with (F?)- ⁇ /-pyrrolidin-3-yl-isonicotinamide (Intermediate I).
- Step 2 ⁇ /- ⁇ (R)-1-[6-(2,2-Diphenyl-ethylamino)-9-((2R I 3R,4S > 5S)-5-ethylcarbamoyl-3 > 4- dihydroxy-tetrahydro-furan-2-yl)-9H-purin-2-yl]-pyrrolidin-3-yl ⁇ -isonicotinamide
- Example 75 1 -((/?)-1 - ⁇ 6-(2,2-Diphenyl-ethylamino)-9-[(2/?,3/?,4S,5/?)-5- ⁇ 2-ethyl-2W- tetrazol-5-yl)-3,4-dihydroxy-tetrahydro-furan-2-yl]-9H-purin-2-yl ⁇ -pyrrolidin- 3-yl)-3-pyridin-3-yl-urea trifluoroacetate
- Step 1 (2R,3f?,4S,5R)-2-[2-((R)-3-Amino-pyrrolidin-1-yl)-6-(2,2-diphenyl-ethylannino)-purin-9- yl]-5-(2-ethyl-2f/-tetrazol-5-yl)-tetrahydro-furan-3,4-diol trifluoroacetate
- This compound is prepared analogously to Example 6 using ((R)-I - ⁇ 6-(2,2-diphenyl- ethylamino)-9-[(2R,3R,4S,5R)-5-(2-ethyl-2H-tetrazol-5-yl)-3,4-dihydroxy-tetrahydro-furan-2-yl]- 9H-purin-2-yl ⁇ -pyrrolidin-3-yl)-carbamic acid te/f-butyl ester which is prepared from Intermediate AL and (R)-pyrrolidin-3-yl-carbamic acid ferf-butyl ester.
- Step 2 1 -((R)-1 - ⁇ 6-(2,2-Diphenyl-ethylamino)-9-[(2R > 3/?,4S,5R)-5-(2-ethyl-2H-tetrazol-5-yl)-3,4- dihydroxy-tetrahydro-furan-2-yl]-9H-purin-2-yl ⁇ -pyrrolidin-3-yl)-3-pyridin-3-yl-urea trifluoroacetate
- Step 1 (2S,3S,4R,5R)-5-[2-((R)-3-Amino-pyrrolidin-1 -yl)-6-(2,2-diphenyl-ethylamino)-purin-9- yl]-3,4-dihydroxy-tetrahydro-furan-2-carboxylic acid ethylamide
- This compound is prepared from Intermediate J analogously to (2R,3R,4S,5R)-2-[2-((R)- 3-amino-pyrrolidin-1-yl)-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-5-(2-ethyl-2H-tetrazol-5-yl)- tetrahydro-furan-3,4-diol trifluoroacetate (Example 75, Step 1). - 70 -
- Step 2 ⁇ /- ⁇ (R)-1 -[6-(2,2-Diphenyl-ethylamino)-9-((2R3R,4S,5S)-5-ethylcarbamoyl-3,4- dihydroxy-tetrahydro-furan-2-yl)-9H-purin-2-yl]-pyrrolidin-3-yl ⁇ -6-mo ⁇ holin-4-yl- nicotinamide trifluoroacetate
- a reaction mixture comprising (2S,3S,4R,5R)-5-[2-((R)-3-amino-pyrrolidin-1-yl)-6-(2,2- diphenyl-ethylamino ⁇ purin- ⁇ -yll-S ⁇ -dihydroxy-tetrahydro-furan ⁇ -carboxylic acid ethylamide (Step 1) (30 mg, 0.052 mmol) and 6-morpholinonicotinyl chloride (35 mg, 0.156 mmol) in THF (1 mL) is treated with TE ⁇ A (134 ⁇ l_, 0.96 mmol) and stirred at room temperature for 5 days. The resulting mixture is diluted with THF (4 mL) and then filtered. The filtrate is concentrated in vacuo and then treated with DMSO (0.4 mL). The resulting suspension is filtered again and purified by preparative HPLC to afford the title compound.
- This compound is prepared analogously to Example 1 by replacing (2R ) 3R,4S,5R)-2-[2- chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-5-ydroxymethyl-tetrahydro-furan-3,4-diol (Intermediate AA) with (2R,3R,4S,5R)-2-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-5-(2- ethyl-2H-tetrazol-5-yl)-tetrahydro-furan-3,4-diol (Intermediate AL) and by replacing 4-(4-fluoro- phenyl)-piperidine (Intermediate BA) with (R)-3-(4-fluoro-phenyl)-pyrrolidine (Intermediate K). - 71 -
- a reaction mixture comprising (2S,3S,4R,5R)-5-[2-((/?)-3-amino-pyrrolidin-1-yl)-6-(2,2- diphenyl-ethylaminoJ-purin- ⁇ -yll-S ⁇ -dihydroxy-tetrahydro-furan ⁇ -carboxylic acid ethylamide (Example 76, Step 1) (19.6 mg, 0.03 mmol), pyridin-4-ylmethyl-carbamic acid phenyl ester (6.5 mg, 0.03 mmol) and DIPEA (18.3 mg, 0.14 mmol) in NMP (0.5 ml.) is heated to 110 0 C. Purification by C-18 reverse phase column chromatography eluting with acetonitrile:water (0.1% TFA) (gradient of 0-100% acetonitrile) affords the title compound.
- Example 86 are prepared analogously to Example 84 by replacing (2S,3S,4R,5R)-5-[2-((R)-3-amino- pyrrolidin-1-yl)-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-3,4-dihydroxy-tetrahydro-furan-2- carboxylic acid ethylamide (Example 76, Step 1) with the
- Example 110 are prepared analogously to Example 1 by replacing (2R,3f?,4S,5R)-2-[2-chloro-6-(2 > 2-diphenyl- ethylamino)-purin-9-yl]-5-ydroxymethyl-tetrahydro-furan-3,4-diol (Intermediate AA) with the appropriate intermediate (the preparations of which are described herein) and by replacing
- Example 111 1-((/?)-1- ⁇ 6-(2 > 2-Diphenyl-ethylamino)-9-[(2/?,3R,4S,5S)-5-(3-ethyl-isoxazol- 5-yl)-3,4-dihydroxy-tetrahydro-furan-2-yl]-9H-purin-2-yl ⁇ -pyrrolidin-3-yl)-3- pyrid i n-4-y I methyl-u rea trifl uoroactetate
- Step 1 (2R3f?,4S,5S)-2-[2-((f?)-3-Amino-pyrrolidin-1 -yl)-6-(2,2-diphenyl-ethylamino)-purin-9- yl]-5-(3-ethyl-isoxazol-5-yl)-tetrahydro-furan-3,4-diol trifluoroacetate
- This compound is prepared analogously to Example 6 using ((/?)-1- ⁇ 6-(2,2-diphenyl- ethylamino)-9-[(2/?,3R,4S,5S)-5-(3-ethyl-isoxazol-5-yl)-3,4-dihydroxy-tetrahydro-furan-2-yl]-9H- purin-2-yl ⁇ -pyrrolidin-3-yl)-carbamic acid te/f-butyl ester trifluoroacetate which is prepared from Intermediate AH and (f?)-pyrrolidin-3-yl-carbamic acid fe/f-butyl ester.
- Step 2 1-((R)-1- ⁇ 6-(2,2-Diphenyl-ethylamino)-9-[(2R3f?,4S,5S)-5-(3-ethyl-isoxazol-5-yl)-3,4- dihydroxy-tetrahydro-furan-2-ylI-9H-purin-2-yl ⁇ -pyrrolidin-3-yl)-3-pyridin-4-ylmethyl-urea trifl uoroactetate
- Example 112 1-((/?)-1- ⁇ 6-(2,2-Diphenyl-ethylamino)-9-[(2R,3R4S > 5/?)-5-(2-ethy
- This compound is prepared analogously to Example 111 by replacing (2f?,3f?,4S,5S)-2- [2-((/?)-3-amino-py ⁇ Olidin-1-yl)-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-5-(3-ethyl-isoxazol-5-yl)- tetrahydro-furan-3,4-diol trifluoroacetate with (2R,3R,4S,5/?)-2-[2-((R)-3-amino-pyrrolidin-1 -yl)-6- (2,2-diphenyl-ethylamino)-purin-9-yl]-5-(2-ethyl-2H-tetrazol-5-yl)-tetrahydro-furan-3,4-diol trifluoroacetate (Example 75, Step 1).
- Example 114 prepared analogously to Example 11 by replacing (2f?,3/?,4S,5R)-2-[2-chloro-6-(2,2-diphenyl- ethylamino)-purin-9-yl]-5-hydroxymethyl-tetrahydro-furan-3,4-diol with the appropriate intermediate (described herein) and by replacing 1 ,3-di(f?)-pyrrolidin-3-yl-urea with (R)- ⁇ /-pyrrolidin-3-yl-isonicot
- Example 115 1 -((R)-1 - ⁇ 6-(2,2-Diphenyl-ethylamino)-9-[(2R,3R,4S > 5S)-5-(3-ethyl-isoxazol- 5-yl)-3,4-dihydroxy-tetrahydro-furan-2-yl]-9H-purin-2-yl ⁇ -pyrrolidin-3-yl)-3- pyridin-3-yl-urea trifluoroacetate
- This compound is prepared analogously to Example 75 by replacing (2R,3/?,4S,5f?)-2- [2-((R)-3-amino-pyrrolidin-1-yl)-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-5-(2-ethyl-2H-tetrazol-5- yl)-tetrahydro-furan-3,4-diol trifluoroacetate with (2R,3f?,4S,5S)-2-[2-((f?)-3-amino-pyrrolidin-1- yl)-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-5-(3-ethyl-isoxazol-5-yl)-tetrahydro-furan-3,4-diol trifluoroacetate (Example 111, Step 1).
- Example 116 1 -((/?)-1 - ⁇ 6-(2,2-Diphenyl-ethylamino)-9-[(2/? f 3R,4S,5S)-5-(3-ethyl-isoxazol- 5-yl)-3,4-dihydroxy-tetrahydro-furan-2-yl]-9H-purln-2-yl ⁇ -pyrrolidin-3-yl)-3- pyridin-2-ylmethyl-urea trifluoroacetate - 76 -
- a reaction mixture comprising (2R,3f?,4S,5S)-2-[2-((/?)-3-amino-pyrrolidin-1-yl)-6-(2,2- diphenyl-ethylamino)-purin-9-yl]-5-(3-ethyl-isoxazol-5-yl)-tetrahydro-furan-3,4-diol trifluoroacetate (Example 111, Step 1) (20 mg, 0.034 mmol), phenyl chloroformate (10 mg, 0.069 mmol) and potassium carbonate (9 mg, 0.069 mmol) in THF (0.5 mL) is stirred at RT for 1 hour.
- Example 119 1 -((A?)-1 - ⁇ 6-[2,2-/>/s-(4-Hydroxy-phenyl)-ethylamino]-9-[(2/?,3/? I 4S,5R)-5-(2- ethyl-2W-tetrazol-5-yl)-3,4-dihydroxy-tetrahydro-furan-2-yl]-9H-purin-2-yl ⁇ - pyrrolidin-3-yl)-3-pyridin-3-yl-urea
- This compound is prepared analogously to Example 75 by replacing ((R)-I - ⁇ 6-(2, 2- diphenyl-ethylamino)-9-[(2R,3R,4S I 5R)-5-(2-ethyl-2H-tetrazol-5-yl)-3,4-dihydroxy-tetrahydro- furan-2-yl]-9/-/-purin-2-yl ⁇ -pyrrolidin-3-yl)-carbamic acid tert-butyl ester with (2R,3R,4S,5R)-2- ⁇ 2- ((R)-3-amino-pyrrolidin-1-yl)-6-[2 l 2-t>/s-(4-hydroxy-phenyl)-ethylamino]-purin-9-yl ⁇ -5-(2-ethyl-2H- - 77 -
- Example 120 1 -((/?)-1 - ⁇ 6-Amino-9-[(2/?,3/?,4S,5/?)-5- ⁇ 2-ethyl-2H-tetrazol-5-yl)-3,4- dihydroxy-tetrahydro-furan-2-yl]-9W-purin-2-yl ⁇ -pyrrolidin-3-yl)-3-(R)- pyrrolidin-3-yl-urea hydrochloride
- This compound is prepared analogously to Example 1 by replacing (2fi,3R,4S,5R)-2-[2- chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-5-ydroxymethyl-tetrahydro-furan-3,4-diol (Intermediate AA) with (2f?,3/?,4S,5R)-2-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-5-(2- ethyl-2/-/-tetrazol-5-yl)-tetrahydro-furan-3,4-diol (Intermediate AB) and by replacing 4-(4-fluoro- phenyl)-piperidine (Intermediate BA) with 1 ,3-di-(R)-pyrrolidin-3-yl-urea (Intermediate BD).
- Example 121 1 -((R)-1 - ⁇ 6-(2,2-Diphenyl-ethylamino)-9-[(2R > 3R > 4S,5R)-5-(2-ethyl-2H- tetrazol-5-yl)-3,4-dlhydroxy-tetrahydro-furan-2-yl]-9H-purin-2-yl ⁇ -pyrrolidin- 3-yl)- ⁇ /-cyano-2-phenyl-isourea
- Example 122 ⁇ /-((R)-1- ⁇ 6-(2 > 2-Dlphenyl-ethylamlno)-9-[(2R,3/?,4S,5/?)-5-(2-ethyl-2W- tetrazol-5-yl)-3,4-dihydroxy-tetrahydro-furan-2-yl]-9W-purin-2-yl ⁇ -pyrrolidin- 3-yl)- ⁇ f-cyano- ⁇ /"-pyridin-2-ylmethyl-guanidine
- a mixture comprising 1-((R)-1- ⁇ 6-(2,2-diphenyl-ethylamino)-9-[(2f?,3R,4S,5R)-5-(2-ethyl- 2H-tetrazol-5-yl)-3,4-dihydroxy-tetrahydro-furan-2-yl]-9H-purin-2-yl ⁇ -pyrrolidin-3-yl)- ⁇ /-cyano-2- phenyl-isourea (50 mg, 0.07 mmol) and 3-aminopyridine (7 mg, 0.07 mmol) in dry THF (2 ml.) and cat.
- DMAP is heated using microwave radiation in a Personal Chemistry EmrysTM Optimizer microwave reactor at 120 0 C for 1 hour.
- the solvent is removed in vacuo and the resulting crude product is partitioned between EtOAc and water.
- the organic portion is separated, dried (Na 2 SO 4 ) and concentrated in vacuo to afford a yellow oil. Purification of the oil by chromatography on silica eluting with EtOAc/iso-hexane (30-100% EtOAc) affords the title product as a yellow solid.
- This compound is prepared analogously to Example 123 by replacing 1-((f?)-1- ⁇ 6-(2,2- diphenyl-ethylamino)-9-[(2f?,3R,4S,5f?)-5-(2-ethyl-2H-tetrazol-5-yl)-3 ) 4-dihydroxy-tetrahydro- furan-2-yl]-9H-purin-2-yl ⁇ -pyrrolidin-3-yl)- ⁇ /-cyano-2-phenyl-isourea with (2R3R,4S,5/?)-2-[2- ((R)-3-amino-pyrrolidin-1-yl)-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-5-(2-ethyl-2H-tetrazol-5-yl)- tetrahydro-furan-3,4-diol trifluoroacetate and by replacing 3-aminopyridine with 3,4-dimethoxy-3-
- This compound is prepared analogously to Example 123 by replacing 1 -((R)- 1- ⁇ 6-(2,2- diphenyl-ethylamino)-9-[(2f?,3R,4S,5R)-5-(2-ethyl-2H-tetrazol-5-yl)-3,4-dihydroxy-tetrahydro- furan-2-yl]-9H-purin-2-yl ⁇ -pyrrolidin-3-yl)- ⁇ /-cyano-2-phenyl-isourea with (2R,3R,4S,5f?)-2-[2- ((R)-3-amino-pyrrolidin-1-yl)-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-5-(2-ethyl-2/-/-tetrazol-5-yl)- - 79 -
- Example 126 3-[/V-((/?)-1- ⁇ 6-(2,2-Diphenyl-ethylamino)-9-[(2/?,3/? > 4S,5/?)-5-(2-ethyl-2W- tetrazol-5-yl)-3,4-dihydroxy-tetrahydro-furan-2-yl]-9H-purin-2-yl ⁇ -pyrrolidin- 3-yl)- ⁇ /"-cyano-guanidino]-benzenesulfonamide
- This compound is prepared analogously to Example 123 by replacing 3-aminopyridine with 3-aminobenzene sulphonamide.
- Example 128 ⁇ /-((/?)-1- ⁇ 6-(2,2-Dlphenyl-ethylamino)-9-[(2/?,3/? f 4S f 5/?)-5-(2-ethyl-2H- tetrazol-5-yl)-3,4-dihydroxy-tetrahydro-furan-2-yl]-9H-purin-2-yl ⁇ -pyrrolidin- 3-yl)-oxalamic acid
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Engineering & Computer Science (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Diabetes (AREA)
- Heart & Thoracic Surgery (AREA)
- Epidemiology (AREA)
- Molecular Biology (AREA)
- Cardiology (AREA)
- Genetics & Genomics (AREA)
- Biotechnology (AREA)
- Biochemistry (AREA)
- Hematology (AREA)
- Urology & Nephrology (AREA)
- Emergency Medicine (AREA)
- Rheumatology (AREA)
- Pulmonology (AREA)
- Pain & Pain Management (AREA)
- Obesity (AREA)
- Endocrinology (AREA)
- Vascular Medicine (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Saccharide Compounds (AREA)
Abstract
Description










































Claims


Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BRPI0710816-8A BRPI0710816A2 (en) | 2006-04-21 | 2007-04-19 | organic compounds |
| EP07724370A EP2018381A2 (en) | 2006-04-21 | 2007-04-19 | Use of 2-(purin-9-yl)-tetrahydofuran-3,4-diol derivatives as adenosine a2a receptor agonists |
| CA002649205A CA2649205A1 (en) | 2006-04-21 | 2007-04-19 | Organic compounds |
| MX2008013523A MX2008013523A (en) | 2006-04-21 | 2007-04-19 | Use of 2-(purin-9-yl)-tetrahydofuran-3,4-diol derivatives as adenosine a2a receptor agonists. |
| AU2007241341A AU2007241341A1 (en) | 2006-04-21 | 2007-04-19 | Use of 2-(purin-9-yl)-tetrahydofuran-3,4-diol derivatives as adenosine A2A receptor agonists |
| US12/297,727 US20090240045A1 (en) | 2006-04-21 | 2007-04-19 | Organic Compounds |
| JP2009505777A JP2009534336A (en) | 2006-04-21 | 2007-04-19 | Use of 2- (purin-9-yl) -tetrahydrofuran-3,4-diol as an adenosine A2A receptor agonist |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0607951.1 | 2006-04-21 | ||
| GBGB0607951.1A GB0607951D0 (en) | 2006-04-21 | 2006-04-21 | Organic compounds |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007121918A2 true WO2007121918A2 (en) | 2007-11-01 |
| WO2007121918A3 WO2007121918A3 (en) | 2008-04-10 |
Family
ID=36581048
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2007/003433 WO2007121918A2 (en) | 2006-04-21 | 2007-04-19 | Use of 2-(purin-9-yl)-tetrahydofuran-3,4-diol derivatives as adenosine a2a receptor agonists |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US20090240045A1 (en) |
| EP (1) | EP2018381A2 (en) |
| JP (1) | JP2009534336A (en) |
| KR (1) | KR20080110836A (en) |
| CN (1) | CN101420959A (en) |
| AU (1) | AU2007241341A1 (en) |
| BR (1) | BRPI0710816A2 (en) |
| CA (1) | CA2649205A1 (en) |
| GB (1) | GB0607951D0 (en) |
| MX (1) | MX2008013523A (en) |
| RU (1) | RU2008145698A (en) |
| WO (1) | WO2007121918A2 (en) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| JP2011507907A (en) * | 2007-12-20 | 2011-03-10 | ピージーエックスヘルス、リミテッド、ライアビリティー、カンパニー | Substituted 4- {3- [6-amino-9- (3,4-dihydroxy-tetrahydro-furan-2-yl) -9H-purin-2-yl] -prop-2-ynyl} -piperidine as an A2AR agonist -1-carboxylic acid ester |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| EP2567959A1 (en) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2015086505A1 (en) * | 2013-12-09 | 2015-06-18 | Ucb Biopharma Sprl | Purine derivatives as modulators of tnf activity |
| US9296776B2 (en) | 2007-07-09 | 2016-03-29 | Eastern Virginia Medical School | Substituted nucleoside derivatives with antiviral and antimicrobial properties |
| US12084453B2 (en) | 2021-12-10 | 2024-09-10 | Incyte Corporation | Bicyclic amines as CDK12 inhibitors |
Families Citing this family (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GT200500281A (en) * | 2004-10-22 | 2006-04-24 | Novartis Ag | ORGANIC COMPOUNDS. |
| GB0500785D0 (en) | 2005-01-14 | 2005-02-23 | Novartis Ag | Organic compounds |
| GB0607944D0 (en) * | 2006-04-21 | 2006-05-31 | Novartis Ag | Organic compounds |
| KR20080110925A (en) | 2006-04-21 | 2008-12-19 | 노파르티스 아게 | Purine derivatives for use as adenosin a2a receptor agonists |
| GB0607950D0 (en) * | 2006-04-21 | 2006-05-31 | Novartis Ag | Organic compounds |
| EP1889846A1 (en) | 2006-07-13 | 2008-02-20 | Novartis AG | Purine derivatives as A2a agonists |
| EP1903044A1 (en) * | 2006-09-14 | 2008-03-26 | Novartis AG | Adenosine Derivatives as A2A Receptor Agonists |
| BRPI0718792A2 (en) * | 2006-11-10 | 2013-12-03 | Novartis Ag | ORGANIC COMPOUNDS |
| CA2930464A1 (en) * | 2013-12-10 | 2015-06-18 | Scinopharm Taiwan, Ltd. | A process for the preparation of regadenoson |
Family Cites Families (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7427606B2 (en) * | 1999-02-01 | 2008-09-23 | University Of Virginia Patent Foundation | Method to reduce inflammatory response in transplanted tissue |
| US6322771B1 (en) * | 1999-06-18 | 2001-11-27 | University Of Virginia Patent Foundation | Induction of pharmacological stress with adenosine receptor agonists |
| US6403567B1 (en) * | 1999-06-22 | 2002-06-11 | Cv Therapeutics, Inc. | N-pyrazole A2A adenosine receptor agonists |
| GB0003960D0 (en) * | 2000-02-18 | 2000-04-12 | Pfizer Ltd | Purine derivatives |
| ATE381336T1 (en) * | 2002-04-10 | 2008-01-15 | Univ Virginia | USE OF A2A ADENOSINE RECEPTOR AGONIST AND ANTIPATHOGENE CONTAINING COMBINATIONS FOR THE TREATMENT OF INFLAMMATORY DISEASES |
| US7396825B2 (en) * | 2004-05-03 | 2008-07-08 | University Of Virginia Patent Foundation | Agonists of A2A adenosine receptors for treatment of diabetic nephropathy |
| GB0500785D0 (en) * | 2005-01-14 | 2005-02-23 | Novartis Ag | Organic compounds |
| GB0505219D0 (en) * | 2005-03-14 | 2005-04-20 | Novartis Ag | Organic compounds |
-
2006
- 2006-04-21 GB GBGB0607951.1A patent/GB0607951D0/en not_active Ceased
-
2007
- 2007-04-19 JP JP2009505777A patent/JP2009534336A/en active Pending
- 2007-04-19 EP EP07724370A patent/EP2018381A2/en not_active Withdrawn
- 2007-04-19 CA CA002649205A patent/CA2649205A1/en not_active Abandoned
- 2007-04-19 AU AU2007241341A patent/AU2007241341A1/en not_active Abandoned
- 2007-04-19 US US12/297,727 patent/US20090240045A1/en not_active Abandoned
- 2007-04-19 RU RU2008145698/04A patent/RU2008145698A/en not_active Application Discontinuation
- 2007-04-19 MX MX2008013523A patent/MX2008013523A/en not_active Application Discontinuation
- 2007-04-19 CN CNA2007800134875A patent/CN101420959A/en active Pending
- 2007-04-19 BR BRPI0710816-8A patent/BRPI0710816A2/en not_active IP Right Cessation
- 2007-04-19 KR KR1020087025545A patent/KR20080110836A/en not_active Application Discontinuation
- 2007-04-19 WO PCT/EP2007/003433 patent/WO2007121918A2/en active Application Filing
Non-Patent Citations (1)
| Title |
|---|
| None |
Cited By (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9296776B2 (en) | 2007-07-09 | 2016-03-29 | Eastern Virginia Medical School | Substituted nucleoside derivatives with antiviral and antimicrobial properties |
| US9738678B2 (en) | 2007-07-09 | 2017-08-22 | Eastern Virginia Medical School | Substituted nucleoside derivatives with antiviral and antimicrobial properties |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| JP2011507907A (en) * | 2007-12-20 | 2011-03-10 | ピージーエックスヘルス、リミテッド、ライアビリティー、カンパニー | Substituted 4- {3- [6-amino-9- (3,4-dihydroxy-tetrahydro-furan-2-yl) -9H-purin-2-yl] -prop-2-ynyl} -piperidine as an A2AR agonist -1-carboxylic acid ester |
| WO2011157827A1 (en) | 2010-06-18 | 2011-12-22 | Sanofi | Azolopyridin-3-one derivatives as inhibitors of lipases and phospholipases |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| EP2567959A1 (en) | 2011-09-12 | 2013-03-13 | Sanofi | 6-(4-Hydroxy-phenyl)-3-styryl-1H-pyrazolo[3,4-b]pyridine-4-carboxylic acid amide derivatives as kinase inhibitors |
| WO2015086505A1 (en) * | 2013-12-09 | 2015-06-18 | Ucb Biopharma Sprl | Purine derivatives as modulators of tnf activity |
| CN105814056A (en) * | 2013-12-09 | 2016-07-27 | Ucb生物制药私人有限公司 | Purine derivatives as modulators of TNF activity |
| CN105814056B (en) * | 2013-12-09 | 2017-10-10 | Ucb生物制药私人有限公司 | Purine derivative as TNF active regulators |
| US9988383B2 (en) | 2013-12-09 | 2018-06-05 | Ucb Biopharma Sprl | Purine derivatives as modulators of TNF activity |
| RU2684644C1 (en) * | 2013-12-09 | 2019-04-11 | Юсб Байофарма Спрл | Purine derivatives as tnf activity modulators |
| US12084453B2 (en) | 2021-12-10 | 2024-09-10 | Incyte Corporation | Bicyclic amines as CDK12 inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2009534336A (en) | 2009-09-24 |
| KR20080110836A (en) | 2008-12-19 |
| RU2008145698A (en) | 2010-05-27 |
| BRPI0710816A2 (en) | 2011-08-23 |
| GB0607951D0 (en) | 2006-05-31 |
| AU2007241341A1 (en) | 2007-11-01 |
| CA2649205A1 (en) | 2007-11-01 |
| MX2008013523A (en) | 2009-01-16 |
| WO2007121918A3 (en) | 2008-04-10 |
| US20090240045A1 (en) | 2009-09-24 |
| EP2018381A2 (en) | 2009-01-28 |
| CN101420959A (en) | 2009-04-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP2018381A2 (en) | Use of 2-(purin-9-yl)-tetrahydofuran-3,4-diol derivatives as adenosine a2a receptor agonists | |
| US20080242683A1 (en) | Organic Compounds | |
| EP2066669B1 (en) | Adenosine derivatives as a2a receptor agonists | |
| EP2322525B1 (en) | Purine derivatives for use as adenosin A2A receptor agonists | |
| AU2006205878B2 (en) | Purine derivatives acting as A2A receptor agonists | |
| EP2044070B1 (en) | Purine derivatives as a2a agonists | |
| EP2013210A1 (en) | Purine derivatives with activity to the adenosine a2a receptor | |
| EP2018382A2 (en) | Purine derivatives as adenosine receptor activator |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07724370 Country of ref document: EP Kind code of ref document: A2 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007724370 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007241341 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780013487.5 Country of ref document: CN Ref document number: 2649205 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2007241341 Country of ref document: AU Date of ref document: 20070419 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12297727 Country of ref document: US Ref document number: 2009505777 Country of ref document: JP Ref document number: 1020087025545 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2008/013523 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 9440/DELNP/2008 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008145698 Country of ref document: RU |
|
| ENP | Entry into the national phase |
Ref document number: PI0710816 Country of ref document: BR Kind code of ref document: A2 Effective date: 20081020 |