WO2007114902A2 - Alkylthiobenzylpiperidine compounds - Google Patents
Alkylthiobenzylpiperidine compounds Download PDFInfo
- Publication number
- WO2007114902A2 WO2007114902A2 PCT/US2007/008142 US2007008142W WO2007114902A2 WO 2007114902 A2 WO2007114902 A2 WO 2007114902A2 US 2007008142 W US2007008142 W US 2007008142W WO 2007114902 A2 WO2007114902 A2 WO 2007114902A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phenyl
- compound
- methyl
- benzyl
- piperidin
- Prior art date
Links
- 0 *C(c(cc1)ccc1N*)=O Chemical compound *C(c(cc1)ccc1N*)=O 0.000 description 5
- BSKSYUHZZNIAIB-UHFFFAOYSA-N CC(c(cc1)ccc1N(C)C=C)=O Chemical compound CC(c(cc1)ccc1N(C)C=C)=O BSKSYUHZZNIAIB-UHFFFAOYSA-N 0.000 description 1
- CIDITTYDUGFFIH-UHFFFAOYSA-N CCCCSc1ccc(CN(CC2)CCC2c2cc(NC(C(C)C)=O)ccc2C)cc1 Chemical compound CCCCSc1ccc(CN(CC2)CCC2c2cc(NC(C(C)C)=O)ccc2C)cc1 CIDITTYDUGFFIH-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D211/26—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms with hydrocarbon radicals, substituted by nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- the present invention relates to compounds that are ligands at the MCHl receptor, and as such are useful to treat depression, anxiety or obesity.
- MCH Melanin-concentrating hormone
- MCHl The link between MCHl and the effects of MCH on feeding was suggested by reports on the phenotype of the MCHl knockout mice. Independent groups generated knock-out mice with the targeted deletion of the MCHl receptor. The phenotype of these mice was lean, hyperphag ⁇ c and hypermetabolic, with increased resistance to diet-induced obesity (D. J. Marsh, et al., Proc. Natl. Acad. ScL, 2002, 99, 3240-3245). These observations evidence that MCHl antagonists are useful to treat obesity.
- SNAP-7941 a selective MCHl small molecule antagonist
- Pharmacological blockade of the MCHl receptor with SNAP-7941 produced a profile similar to clinically used anti-depressants and anxiolytics in behavioral models of depression and/ or anxiety: the rat forced-swim, rat social interaction and guinea pig maternal-separation vocalization tests.
- the objective of the present invention is to provide compounds that are ligands at the MCHl receptor. Accordingly, the present invention relates to compounds of Formula I.
- each U 1 , U 2 , U 3 and U 4 is independently CR 4 or N, provided that if one U is N then the remaining U are each CR 4 ;
- each Y 1 , Y 2 , Y 3 and Y 4 is independently CR 5 or N, provided that if two Y are each N then the remaining Y are each CR 5 ;
- R 1 is straight chained or branched Ci-C 7 alkyl, C 3 -C 7 cycloalkyl or C 2 -C7 cyclic ether;
- R 2 is H or straight chained or branched C1-C7 alkyl
- R 3 is straight chained or branched Ci-C 7 alkyl, C3-C7 cycloalkyl or straight-chained or branched Ci-C 7 alkoxy;
- each R 4 is independently H, straight chained or branched Ci-C 7 alkyl, straight chained or branched Ci-C 7 fluoroalkyl, straight chained or branched Ci-C 7 alkoxy, F, Cl, Br or I;
- each R 5 is independently H, CH 3 , F, Cl or Br;
- n is an integer from 0 to 2 inclusive;
- the compound is selected from one of the specific compounds disclosed in the Experimental Section.
- the present invention provides a pharmaceutical composition comprising a therapeutically effective amount of a compound of Formula I and a pharmaceutically acceptable carrier.
- the present invention provides a method of treating a subject suffering from depression comprising administering to the subject a therapeutically effective amount of a compound of Formula I.
- the present invention further provides a method of treating a subject suffering from anxiety comprising administering to the subject a therapeutically effective amount of a compound of Formula I.
- the present invention further provides a method of treating a subject suffering from obesity comprising administering to the subject a therapeutically effective amount of a compound of Formula I.
- the present invention is directed to the use of a compound of Formula I for the manufacture of a medicament for treating a subject suffering from depression, anxiety or obesity.
- the term "straight chained or branched C 1 -C 7 alkyl” refers to a saturated hydrocarbon having from one to seven carbon atoms inclusive. Examples of such substituents include, but are not limited to, methyl, ethyl, 1-propyl, 2-propyl, 1-butyl, 2- butyl, 2-methyl-2-propy], 2-methyl-l -propyl and n-heptyl.
- the term “straight chained or branched Ci -C 4 alkyl” refers to a saturated hydrocarbon having from one to four carbon atoms inclusive.
- the term "straight chained or branched Ci-C 7 fluoroalkyl” refers to a saturated hydrocarbon having from one to seven carbon atoms inclusive substituted with one or more fluorine atoms. Examples of such substituents include, but are not limited to, trifluoromethyl, pentafluoroethyl, 1-fluoroethyl and 1 ,2-difluoroethyl and 2,3-difluoroheptyl. Similarly, the term “straight chained or branched Ci-C 4 fluoroalkyl” refers to a saturated hydrocarbon having from one to four carbon atoms inclusive substituted with one or more fluorine atoms.
- the term "straight chained or branched Ci-C 7 alkoxy” refers to a saturated alkoxy group having from one to seven carbon atoms inclusive with the open valency on the oxygen. Examples of such substituents include, but are not limited to, methoxy, ethoxy, n-butoxy, t- butoxy and n-heptyloxy. Similarly, the term “straight chained or branched Cj-C 4 alkoxy” refers to a saturated alkoxy group having from one to four carbon atoms inclusive with the open valency on the oxygen.
- C3-C 7 cycloalkyl refers to the group consisting of cyclopropane, cyclobutane, cyclopentane, cyclohexane and cycloheptane.
- C 2 -C 7 cyclic ether refers to the group consisting of oxirane, oxetane, tetrahydrofuran, tetrahydropyran, oxepane and oxecane with the open valency on a carbon of the cyclic ring.
- the aromatic ring containing Y 1 , Y 2 , Y 3 and Y 4 refers to the group consisting of phenyl, pyridyl, pyrimidinyl, pyrazinyl and pyridazinyl, in which the aromatic ring may be optionally substituted with one or more CH 3 , F, Cl or Br.
- the compound of example 2a has the following structure:
- This compound is constructed from Formula I wherein R 1 is tetrahydropyranyl; wherein R 2 is H; wherein R 3 is methoxy; wherein each U 1 , U 2 , U 3 and U 4 is CR 4 ; wherein R 4 is H; wherein each Y 1 , Y 2 , Y 3 and Y 4 is CR 5 ; wherein each R s is independently H or CH 3 ; and wherein n is 2.
- the present invention further provides certain embodiments that are described below.
- each Y 1 , Y 2 , Y 3 and Y 4 is independently CR 5 or N, provided that if one Y is N then the remaining Y are each CR 5 .
- each U 1 , U 2 , U 3 and U 4 is independently CR 4 ; and each R 4 is independently H, CH 3 , F 5 Cl, Br or I.
- R 2 is H or straight chained or branched C1-C4 alkyl.
- R 1 is C2-C 7 cyclic ether.
- R 1 is tetrahydropyranyl; and each R 4 is H.
- R 1 is C3-C 7 cycloalkyl.
- R 1 is straight chained or branched C 1 -C7 alkyl.
- R 1 is straight chained or branched Ci -C 4 alkyl; and each R 4 is H.
- R 3 is straight chained or branched Ci -C 4 alkoxy.
- R 3 is straight chained or branched C1-C4 alkyl.
- R 2 is straight chained or branched C1-C4 alkyl.
- R 2 is CH 3 ; each Y 1 , Y 2 , Y 3 and Y 4 is independently CR 5 ; and each R 5 is independently H, CH3 or F.
- R 2 is H.
- each Y 1 , Y 2 , Y 3 and Y 4 is independently CR 5 ; each R 5 is independently H, CH 3 or F; and n is 2.
- each Y 1 , Y 2 , Y 3 and Y 4 is independently CR 5 ; each R 5 is independently H, CH3 or F; and n is 0.
- the present invention is directed to the use of a compound as defined above for the manufacture of a medicament for treating a subject suffering from depression.
- the present invention is directed to the use of a compound as defined above for the manufacture of a medicament for treating a subject suffering from anxiety.
- the present invention is directed to the use of a compound as defined above for the manufacture of a medicament for treating a subject suffering from obesity.
- the present invention also comprises salts of the present compounds, typically, pharmaceutically acceptable salts.
- Such salts include pharmaceutically acceptable acid addition salts.
- Acid addition salts include salts of inorganic acids as well as organic acids.
- suitable inorganic acids include hydrochloric, hydrobromic, hydroiodic, phosphoric, sulfuric, sulfamic, nitric acids and the like.
- suitable organic acids include formic, acetic, trichloroacetic, trifluoroacetic, propionic, benzoic, cinnamic, citric, fumaric, glycolic, itaconic, lactic, methanesulfonic, maleic, malic, malonic, mandelic, oxalic, picric, pyruvic, salicylic, succinic, methane sulfonic, ethanesulfonic, tartaric, ascorbic, pamoic, bismethylene salicylic, ethanedisulfonic, gluconic, citraconic, aspartic, stearic, palmitic, EDTA, glycolic, p-aminobenzoic, glutamic, benzenesulfonic, p-tol
- inorganic or organic acid addition salts include the pharmaceutically acceptable salts listed in S. M. Berge, et al., J. Pharm. ScL, 1977, 66, 2, the contents of which are hereby incorporated by reference.
- the compounds of this invention may exist in unsolvated as well as in solvated forms with pharmaceutically acceptable solvents such as water, ethanol and the like. In general, the solvated forms are considered equivalent to the unsolvated forms for the purposes of this invention.
- Racemic forms may be. resolved into the optical antipodes by known methods, for example, by separation of diastereomeric salts thereof with an optically active acid, and liberating the optically active amine compound by treatment with a base. Separation • of such diastereomeric salts can be achieved, e.g. by fractional crystallization.
- the optically active acids suitable for this purpose may include, but are not limited to d- or 1- tartaric, madelic or camphorsulfonic acids. Another method for resolving racemates into the optical antipodes is based upon chromatography on an optically active matrix.
- the compounds of the present invention may also be resolved by the formation and chromatographic separation of diastereomeric derivatives from chiral derivatizing reagents, such as, e.g., chiral alkylating or acylating reagents, followed by cleavage of the chiral auxiliary. Any of the above methods may be applied either to resolve the optical antipodes of the compounds of the invention per se or to resolve the optical antipodes of synthetic intermediates, which can then be converted by methods described herein into the optically resolved final products which are the compound of the invention.
- chiral derivatizing reagents such as, e.g., chiral alkylating or acylating reagents
- Optically active compounds may also be prepared from optically active starting materials.
- the invention also encompasses prodrugs of the present compounds, which on administration undergo chemical conversion by metabolic processes before becoming pharmacologically active substances.
- prodrugs will be functional derivatives of the compounds of Formula I which are readily convertible in vivo into the required compound of Formula I.
- Conventional procedures for the selection and preparation of suitable prodrug derivatives are described in Design of Prodrugs, ed. H. Bundgaard, Elsevier, 1985.
- compositions The present invention further provides a pharmaceutical composition comprising a therapeutically effective amount of a compound of Formula I and a pharmaceutically acceptable carrier.
- the present invention also provides a pharmaceutical composition comprising a therapeutically effective amount of one of the specific compounds disclosed in the Experimental Section and a pharmaceutically acceptable carrier.
- the compounds of the invention may be administered alone or in combination with pharmaceutically acceptable carriers or excipients, in either single or multiple doses.
- the pharmaceutical compositions according to the invention may be formulated with pharmaceutically acceptable carriers or diluents as well as any other known adjuvants and excipients in accordance with conventional techniques such as those disclosed in Remington: The Science and Practice of Pharmacy, 19 th Edition, Gennaro, Ed., Mack Publishing Co., Easton, PA, 1995.
- compositions may be specifically formulated for administration by any suitable route such as oral, rectal, nasal, pulmonary, topical (including buccal and sublingual), transdermal, intracisternal, intraperitoneal, vaginal and parenteral (including subcutaneous, intramuscular, intrathecal, intravenous and intradermal) routes. It will be appreciated that the route will depend on the general condition and age of the subject to be treated, the nature of the condition to be treated and the active ingredient.
- compositions for oral administration include solid dosage forms such as capsules, tablets, dragees, pills, lozenges, powders and granules. Where appropriate, the compositions may be prepared with coatings such as enteric coatings or they may be formulated so as to provide controlled release of the active ingredient such as sustained or prolonged release according to methods well known in the art.
- Liquid dosage forms for oral administration include solutions, emulsions, suspensions, syrups and elixirs.
- compositions for parenteral administration include sterile aqueous and nonaqueous injectable solutions, dispersions, suspensions or emulsions as well as sterile powders to be reconstituted in sterile injectable solutions or dispersions prior to use.
- Suitable administration forms include, but are not limited to, suppositories, sprays, ointments, creams, gels, inhalants, dermal patches and implants.
- Typical oral dosages range from about 0.001 to about 100 mg/kg body weight per day. Typical oral dosages also range from about 0.01 to about 50 mg/kg body weight per day.
- Typical oral dosages further range from about 0.05 to about 10 mg/kg body weight per day.
- Oral dosages are usually administered in one or more dosages, typically, one to three dosages per day. The exact dosage will depend upon the frequency and mode of administration, the sex, age, weight and general condition of the subject treated, the nature and severity of the condition treated and any concomitant diseases to be treated and other factors evident to those skilled in the art.
- a typical unit dosage form for oral administration may contain from about 0.01 to about 1000 mg, from about 0.05 to about 500 mg, or from about 0.5 mg to about 200 mg.
- parenteral routes such as intravenous, intrathecal, intramuscular and similar administration
- typical doses are in the order of half the dose employed for oral administration.
- the present invention also provides a process for making a pharmaceutical composition
- a pharmaceutical composition comprising admixing a therapeutically effective amount of a compound of Formula I and a pharmaceutically acceptable carrier.
- the compound utilized in the aforementioned process is one of the specific compounds disclosed in the Experimental Section.
- the compounds of this invention are generally utilized as the free substance or as a pharmaceutically acceptable salt thereof.
- One example is an acid addition salt of a compound having the utility of a free base.
- a compound of Formula I contains a free base such salts are prepared in a conventional manner by treating a solution or suspension of a tree base of Formula I with a molar equivalent of a pharmaceutically acceptable acid.
- suitable organic and inorganic acids are described above.
- solutions of the compounds of Formula I in sterile aqueous solution, aqueous propylene glycol, aqueous vitamin E or sesame or peanut oil may be employed.
- aqueous solutions should be suitably buffered if necessary and the liquid diluent first rendered isotonic with sufficient saline or glucose.
- the aqueous solutions are particularly suitable for intravenous, intramuscular, subcutaneous and intraperitoneal administration.
- the compounds of Formula I may be readily incorporated into known sterile aqueous media using standard techniques known to those skilled in the art.
- Suitable pharmaceutical carriers include inert solid diluents or fillers, sterile aqueous solutions and various organic solvents.
- solid carriers include lactose, terra alba, sucrose, cyclodextrin, talc, gelatin, agar, pectin, acacia, magnesium stearate, stearic acid and lower alkyl ethers of cellulose.
- liquid carriers include, but are not limited to, syrup, peanut oil, olive oil, phospholipids, fatty acids, fatty acid amines, polyoxyethylene and water.
- the carrier or diluent may include any sustained release material known in the art, such as glyceryl monostearate or glyceryl distearate, alone or mixed with a wax.
- the pharmaceutical compositions formed by combining the compounds of Formula I and a pharmaceutically acceptable carrier are then readily administered in a variety of dosage forms suitable for the disclosed routes of administration.
- the formulations may conveniently be presented in unit dosage form by methods known in the art of pharmacy.
- Formulations of the present invention suitable for oral administration may be presented as discrete units such as capsules or tablets, each containing a predetermined amount of the active ingredient, and optionally a suitable excipient.
- the orally available formulations may be in the form of a powder or granules, a solution or suspension in an aqueous or non-aqueous liquid, or an oil-in-water or water-in-oil liquid emulsion.
- the preparation may be tabletted, placed in a hard gelatin capsule in powder or pellet form or it may be in the form of a troche or lozenge.
- the amount of solid carrier will vary widely but will range from about 25 mg to about 1 g per dosage unit.
- the preparation may be in the form of a syrup, emulsion, soft gelatin capsule or sterile injectable liquid such as an aqueous or non-aqueous liquid suspension or solution.
- the compounds of Formula I are ligands at the MCHl receptor.
- the present invention provides a method of treating a subject suffering from depression which comprises administering to the subject a therapeutically effective amount of a compound of this invention.
- This invention further provides a method of treating a subject suffering from anxiety which comprises administering to the subject a therapeutically effective amount of a compound of this invention.
- This invention also provides a method of treating a subject suffering from obesity which comprises administering to the subject a therapeutically effective amount of a compound of this invention.
- the subject is a human being.
- representative reagents such as bases or solvents.
- bases include but are not limited to K 2 CO 3 , Et 3 N or DIPEA (Diisopropylethylamine).
- TLC Thin-layer chromatography
- reagents used in the experimental section such as 2-propanethiol, 4- fluorobenzaldehyde, potassium carbonate, 3-chloroperoxybenzoic acid, 4- fluoroacetophenone, methyl magnesium bromide, methanesulfonyl chloride, methyl chloroformate, 4-chlorotetrahydropyran, N-bromosuccinimide, nitronium tetrafluoroborate were purchased from Aldrich Sigma. Cyclopentanethiol was purchased from TCI International.
- the compounds of Formula I may be synthesized according to the procedures described in Scheme 1.
- the compounds of Formula II and III are commercially available or may be synthesized by one skilled in the art.
- the aldehydes/ ketones of Formula IV are prepared via aromatic nucleophilic reaction of activated 4-halo-benzaldehydes/ ketones of Formula II and alkylthiols of Formula HI in the presence of base under reflux or microwave conditions.
- the corresponding sulfoxides IVb and sulfones IVc may be prepared via sequential oxidations of IVa by mCPBA.
- the compounds of Formula IV may be coupled with the amine of Formula VI to afford the compounds of the invention.
- the conversion of the aldehydes to ketones may be obtained via the Weinreb amide intermediate. For reaction conditions in connection with the Weinreb amide, see S. Nahm and S.M. Weinreb, Tetrahedron Lett., ⁇ 981 , 22, 3815-3818.
- aldehydes/ ketones of Formula IV may be prepared via Ullmann type reactions.
- Ullmann type reactions see T. Kondo, et al., Chem. Rev., 2000, 100, 3205-3220 and the references cited therein.
- the compounds of Formula I may be synthesized according to the procedures described in Scheme 2.
- the intermediates of Formula IV may be reduced to the alcohols of Formula V, which may be treated with mesylchloride in the presence of triethylamine in CH 2 Cl 2 to afford the mesylates of Formula VII.
- the mesylates may be coupled with the amines of Formula VI to afford the compounds of the invention.
- the compounds of Formula I may be synthesized according to the procedures described in Scheme 3.
- the compounds of Formula VIII which are commercially available or synthesized by those skilled in the art, may be treated with NBS to afford the bromides of Formula IX.
- the bromides may be coupled with the amines of Formula VI to afford the compounds of the invention.
- the amines of Formula VI may be prepared according to the procedures described in Scheme 4.
- the compounds of Formula X may be coupled with tert-buty ⁇ 4-(3- aminoaryl)piperidinecarboxy!ate of Formula XI to afford the N-Cbz protected intermediates of Formula XII.
- the Cbz group is removed to provide the compounds of Formula XV, which may be coupled with acid chlorides or chloroformates to yield the intermediates of Formula XVI.
- the Boc group is removed to afford the amines of Formula VI.
- the nitro compounds of Formula XIII may be used.
- N-Cbz bromo or iodo intermeditaes of Formula X may be prepared according to the procedures described in Scheme 5.
- the amino group of the commercially available starting materials of Formula XVII may be protected by treatment with benzyl chloroformate in the presence of base to afford the intermediates of Formula X.
- N-Cbz protected intermediates of Formula X may be prepared from the corresponding acids of Formula XVIII using diphenylphosphoryl azide via a Curtius type rearrangement, followed by trapping the isocyanates with benzyl alcohol to afford the compounds of Formula X.
- nitration (a) nitration, (b) bromination.
- the nitro-bromo or iodo intermediates of Formula XIII may be prepared according to the procedures described in Scheme 6.
- the 3-bromo or 3-iodo compounds of Formula XIII are available from commercial sources or may be prepared from the corresponding bromo or iodo compounds XIX by nitration methods or from the corresponding nitro intermediates of Formula XX by bromination methods.
- General information regarding aromatic nitration is described in the following references: J. G. Hoggett, et al. s Nitration and Aromatic Reactivity, Cambridge University Press, London, 1971; K. Schofield, Aromatic Nitration, Cambridge University Press, London, 1980; and G. A. Olah, et al., Nitration: Methods and Mechanism, (Ed.: H. Feuer), VCH Publishers, New York, 1989.
- the compounds of the invention may be prepared according to the procedures as outlined in Scheme 7.
- compounds of Formula XXI may be coupled with the mesylates of Formula VII or benzyl bromides of Formula IX in the presence of base to provide the advanced intermediates of Formula XXII.
- the advanced intermediates of Formula XXII may be treated with acid chlorides or chloroformates under standard coupling conditions to afford the compounds of Formula I. Preparation of intermediates
- Methanesulfonic acid l-[4-(propane-2-sulfonyl)-phenyl]-ethyl ester Methanesulfonyl chloride (2.00 eq, 3.06 mmol, 0.240 raL) was added to a solution of l-[4-(propane-2- sulfonyl)-phenyl]-ethanol (1.53 mmol, 0.350 g) and Et 3 N (3.00 eq, 4.59 mmol, 0.640 mL) in
- terf-butyl 4-(4,4,5,5-tetramethyM,3,2-dioxaborolan-2-yl)-l,2,5,6-tetrahydropyridine carboxylate of Formula XI was prepared according to the procedures described by P. R. Eastwood, Tetrahedron Lett., 2000, 41, 19, 3705-3708 and references cited therein.
- 2-Bromo-5-fluoro-4-nitro toluene 2-bromo-5-fluoro toluene (15.0 g, 10.0 mL, 79.0 mmol) was added to a solution of nitronium tetrafluoroborate (11.6 g, 87.0 mmol) in CH2CI2 (60.0 mL) over 5 min. After refluxing for 4.5 h, the mixture was cooled to rt and poured into ice water (150 mL). The mixture was extracted with CH 2 Cl 2 (3 X 50 mL). The combined organic layers were washed with brine (100 mL), dried over Na 2 SO 4 , filtered and concentrated in vacuo to give the crude product (18.3 g).
- Step 1 Bromine (21.1 mL, 0.393 mol) was added dropwise over ⁇ 20 min to a mixture of 2-hydroxy-5-nitropyridine (50.0 g, 0.358 mol) in water (7 L), which was warmed to 40 0 C. After stirring at 40 0 C for 2.5 h, the mixture was cooled to 10 0 C and the crude product was isolated by filtration.
- Step 2 To a cooled (0-5 0 C) mixture of 3-bromo-2-hydroxy-5-nitropyridine (47.0 g, 0.214 mol) and quinoline (13.7 g, 0.107 mol) was added POCl 3 (26.0 mL, 0.278 mol) dropwise over ⁇ 10 min (the mixture was difficult to stir initially but became less viscous as the reaction progressed and the mixture warmed). After stirring at 120 0 C for 3.5 h, the mixture was cooled to 100 0 C and water (90 mL) was added. The resulting mixture was stirred vigorously while cooling to 0-5 0 C.
- Benzyl 5-bromo-3-pyridinyl carbamate To a suspension of 5-bromonicotinic acid (20.0 g, 99.0 mmol) in toluene (200 mL) was added diphenylphosphoryl azide (25.6 mL, 118.8 mmol) and Et3N (16.6 mL, 118.8 mmol). After stirring at it for 30 min, benzyl alcohol (15.4 mL, 148.5 mmol) was added. The mixture was stirred at it for 1 h and refluxed overnight. After cooling to rt, the reaction mixture was washed with H 2 O, saturated aqueous NaHCO 3 and brine.
- Example Ia iV-(4-Methyl-3- ⁇ 1 -[4-(propane-2-sulfonyl)-benzyl]-piperidin-4-yl ⁇ -phenyl)- isobutyramide.
- Acetic acid (24.0 mg, 0.400 mmol) and sodium triacetoxyborohydride (169 mg, 0.800 mmol) were added to a solution of N-(4-methyl-3-piperidin-4-yl-phenyl)-isobutyramide (114 mg, 0.440 mmol) and 4-(propane-2-sulfonyl)-benzaldehyde (84.8 mg, 0.400 mmol) in CH 2 Cl 2 (5.00 mL) at rt. Stirring was continued under nitrogen at rt for 10 h. Saturated aqueous ⁇ aHC ⁇ 3 and CH 2 CI 2 (20 mL) were added. The phases were separated.
- Example Ib ⁇ 3-[l-(4-Isopropylsulfanyl-benzyl)-piperidin-4-yl]-4-methyl-phenyl ⁇ - carbamic acid methyl ester.
- Example Ic N- ⁇ 3-[ 1 -(4-Isopropylsulfanyl-benzy l)-piperid in-4-yl]-4-methyl-phenyl ⁇ - isobutyramide.
- Example Id N- ⁇ 3-[l -(4-Cyclopentanesulfonyl-benzyl)-piperidin-4-yl]-4-fluoro-phenyl ⁇ - isobutyramide.
- Example I (3- ⁇ t-[4-(Pro ⁇ ane-2-sulfonyl)-benzyl]-piperidin-4-yl ⁇ -phenyl)-carbamic acid methyl ester.
- Example 2a (4-Methyl-3- ⁇ 1 -[4-(tetrahydro-pyran-4-sulfonyl)-benzyl]-piperidin-4-yl ⁇ - phenyl)-carbamic acid methyl ester.
- Example 2b N-(4-Methyl-3- ⁇ 1 -[4-(tetrahydro-pyran-4-sulfonyl)-benzyl]-piperidin-4-yl ⁇ - phenyl)-isobutyramide.
- F Foorrmmuullaattiioonnss T Thhee pphhaa ⁇ rmaceutical formulations of the invention may be prepared by conventional methods in the art.
- tablets may be prepared by mixing the active ingredient with ordinary adjuvants and/or diluents and subsequently compressing the mixture in a conventional tabletting machine prepare tablets.
- adjuvants or diluents comprise: corn starch, potato starch, talcum, magnesium stearate, gelatine, lactose, gums, and the like. Any other adjuvants or additives usually used for such purposes such as colorings, flavorings, preservatives etc. may be used provided that they are compatible with the active ingredients.
- the affinity of the compounds was measured by their ability to displace tritiated SNAP-7941 from rat MCHl expressing membranes.
- the compound and radioligand were incubated with the membranes at 25 0 C for 90 min. Incubation was terminated by rapid vacuum filtration over GF/C glass fiber filters, presoaked in 5 % PEI using 50 nM Tris pH 7.4 as wash buffer. In all experiments, nonspecific binding was defined using 10 pM of tritiated SNAP-7941.
- MCHl receptor were determined to be 500 nM or less. For the majority of the compounds, the Ki values were determined to be 100 nM or less. For some compounds, the Ki values were determined to be 10 nM or less.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Hydrogenated Pyridines (AREA)
Abstract
This invention is directed to alkylthiobenzylpiperidine compounds which are ligands at the MCHl receptor. The invention provides a pharmaceutical composition comprising a therapeutically effective amount of a compound of the invention and a pharmaceutically acceptable carrier. This invention also provides a method of treating a subject suffering from depression comprising administering to the subject a therapeutically effective amount of a compound of the subject invention. This invention further provides a method of treating a subject suffering from anxiety comprising administering to the subject a therapeutically effective amount of a compound of the subject invention. This invention also provides a method of treating a subject suffering from obesity comprising administering to the subject a therapeutically effective amount of a compound of the subject invention. Furthermore, the present invention is directed to the use of a compound of the subject invention for the manufacture of a medicament for treating a subject suffering from depression, anxiety or obesity.
Description
ALKYLTHIOBENZYLPIPERIDINE COMPOUNDS
Field of the Invention The present invention relates to compounds that are ligands at the MCHl receptor, and as such are useful to treat depression, anxiety or obesity.
Background of the Invention
Throughout this application, various publications are referenced to in full citations. The disclosures of these publications are hereby incorporated by reference into this application to describe more fully the state of the art to which this invention pertains.
Melanin-concentrating hormone (MCH) is a cyclic 19-amϊno acid peptide produced by neurons in the lateral hypothalamus and zona incerta of the brain. Mammalian MCH is conserved between rat, mouse, and human, exhibiting 100 % amino acid homology, and the effects of MCH are mediated through receptors that belong in the rhodopsin superfamily of G protein-coupled receptors. Presently, two receptor subtypes for MCH have been identified in humans, MCHl and MCH2.
The link between MCHl and the effects of MCH on feeding was suggested by reports on the phenotype of the MCHl knockout mice. Independent groups generated knock-out mice with the targeted deletion of the MCHl receptor. The phenotype of these mice was lean, hyperphagϊc and hypermetabolic, with increased resistance to diet-induced obesity (D. J. Marsh, et al., Proc. Natl. Acad. ScL, 2002, 99, 3240-3245). These observations evidence that MCHl antagonists are useful to treat obesity.
To further assess the physiological role of the MCHl receptor, SNAP-7941, a selective MCHl small molecule antagonist, was evaluated in several animal models (B. Borowsky, et al., Nature Medicine, 2002, 8, 825-830). Pharmacological blockade of the MCHl receptor with SNAP-7941 produced a profile similar to clinically used anti-depressants and anxiolytics in behavioral models of depression and/ or anxiety: the rat forced-swim, rat social interaction and guinea pig maternal-separation vocalization tests. These observations evidence that MCHl antagonists are useful to treat depression and anxiety.
Current treatments for depression, anxiety and obesity are on the market. However, numerous patients do not respond to current treatments. Hence, there remains the need for alternative methods of treatment.
Summary of the Invention
The objective of the present invention is to provide compounds that are ligands at the MCHl receptor. Accordingly, the present invention relates to compounds of Formula I.

Formula I wherein each U1, U2, U3 and U4 is independently CR4 or N, provided that if one U is N then the remaining U are each CR4;
wherein each Y1, Y2, Y3 and Y4 is independently CR5 or N, provided that if two Y are each N then the remaining Y are each CR5;
wherein R1 is straight chained or branched Ci-C7 alkyl, C3-C7 cycloalkyl or C2-C7 cyclic ether;
wherein R2 is H or straight chained or branched C1-C7 alkyl;
wherein R3 is straight chained or branched Ci-C7 alkyl, C3-C7 cycloalkyl or straight-chained or branched Ci-C7 alkoxy;
wherein each R4 is independently H, straight chained or branched Ci-C7 alkyl, straight chained or branched Ci-C7 fluoroalkyl, straight chained or branched Ci-C7 alkoxy, F, Cl, Br or I;
wherein each R5 is independently H, CH3, F, Cl or Br; and
wherein n is an integer from 0 to 2 inclusive;
or a pharmaceutically acceptable salt thereof.
In separate embodiments of the invention, the compound is selected from one of the specific compounds disclosed in the Experimental Section.
Furthermore, the present invention provides a pharmaceutical composition comprising a therapeutically effective amount of a compound of Formula I and a pharmaceutically acceptable carrier.
Moreover, the present invention provides a method of treating a subject suffering from depression comprising administering to the subject a therapeutically effective amount of a compound of Formula I. The present invention further provides a method of treating a subject suffering from anxiety comprising administering to the subject a therapeutically effective amount of a compound of Formula I. The present invention further provides a method of treating a subject suffering from obesity comprising administering to the subject a therapeutically effective amount of a compound of Formula I.
Furthermore, the present invention is directed to the use of a compound of Formula I for the manufacture of a medicament for treating a subject suffering from depression, anxiety or obesity.
Detailed Description of the Invention
Definitions
In the present invention, the term "straight chained or branched C1-C7 alkyl" refers to a saturated hydrocarbon having from one to seven carbon atoms inclusive. Examples of such substituents include, but are not limited to, methyl, ethyl, 1-propyl, 2-propyl, 1-butyl, 2- butyl, 2-methyl-2-propy], 2-methyl-l -propyl and n-heptyl. Similarly, the term "straight chained or branched Ci -C4 alkyl" refers to a saturated hydrocarbon having from one to four carbon atoms inclusive.
The term "straight chained or branched Ci-C7 fluoroalkyl" refers to a saturated hydrocarbon having from one to seven carbon atoms inclusive substituted with one or more fluorine atoms. Examples of such substituents include, but are not limited to, trifluoromethyl, pentafluoroethyl, 1-fluoroethyl and 1 ,2-difluoroethyl and 2,3-difluoroheptyl. Similarly, the term "straight chained or branched Ci-C4 fluoroalkyl" refers to a saturated hydrocarbon having from one to four carbon atoms inclusive substituted with one or more fluorine atoms.
The term "straight chained or branched Ci-C7 alkoxy" refers to a saturated alkoxy group having from one to seven carbon atoms inclusive with the open valency on the oxygen. Examples of such substituents include, but are not limited to, methoxy, ethoxy, n-butoxy, t- butoxy and n-heptyloxy. Similarly, the term "straight chained or branched Cj-C4 alkoxy" refers to a saturated alkoxy group having from one to four carbon atoms inclusive with the open valency on the oxygen.
The term "C3-C7 cycloalkyl" refers to the group consisting of cyclopropane, cyclobutane, cyclopentane, cyclohexane and cycloheptane. The term "C2-C7 cyclic ether" refers to the group consisting of oxirane, oxetane, tetrahydrofuran, tetrahydropyran, oxepane and oxecane with the open valency on a carbon of the cyclic ring.
The aromatic ring containing Y1, Y2, Y3 and Y4 refers to the group consisting of phenyl, pyridyl, pyrimidinyl, pyrazinyl and pyridazinyl, in which the aromatic ring may be optionally substituted with one or more CH3, F, Cl or Br.
For illustrative purposes, and without limiting the invention, the compound of example 2a has the following structure:

This compound is constructed from Formula I wherein R1 is tetrahydropyranyl; wherein R2 is H; wherein R3 is methoxy; wherein each U1, U2, U3 and U4 is CR4; wherein R4 is H; wherein each Y1, Y2, Y3 and Y4 is CR5; wherein each Rs is independently H or CH3; and wherein n is 2.
Additionally, the present invention further provides certain embodiments that are described below.
In one embodiment, each Y1, Y2, Y3 and Y4 is independently CR5 or N, provided that if one Y is N then the remaining Y are each CR5.
In another embodiment, each U1, U2, U3 and U4 is independently CR4; and each R4 is independently H, CH3, F5 Cl, Br or I.
In one embodiment, R2 is H or straight chained or branched C1-C4 alkyl.
In yet another embodiment, R1 is C2-C7 cyclic ether.
In one embodiment, R1 is tetrahydropyranyl; and each R4 is H.
In one embodiment, R1 is C3-C7 cycloalkyl.
In another embodiment, R1 is straight chained or branched C1-C7 alkyl.
In one embodiment, R1 is straight chained or branched Ci -C4 alkyl; and each R4 is H.
In an embodiment of the present invention, R3 is straight chained or branched Ci -C4 alkoxy.
In one embodiment, R3 is straight chained or branched C1-C4 alkyl.
In yet another embodiment, R2 is straight chained or branched C1-C4 alkyl.
In one embodiment, R2 is CH3; each Y1, Y2, Y3 and Y4 is independently CR5; and each R5 is independently H, CH3 or F.
In one embodiment, R2 is H.
In one embodiment, each Y1, Y2, Y3 and Y4 is independently CR5; each R5 is independently H, CH3 or F; and n is 2.
In another embodiment, each Y1, Y2, Y3 and Y4 is independently CR5; each R5 is independently H, CH3 or F; and n is 0.
Furthermore, the present invention is directed to the use of a compound as defined above for the manufacture of a medicament for treating a subject suffering from depression.
In one embodiment, the present invention is directed to the use of a compound as defined above for the manufacture of a medicament for treating a subject suffering from anxiety.
In a separate embodiment, the present invention is directed to the use of a compound as defined above for the manufacture of a medicament for treating a subject suffering from obesity.
Pharmaceutically Acceptable Salts
The present invention also comprises salts of the present compounds, typically, pharmaceutically acceptable salts. Such salts include pharmaceutically acceptable acid addition salts. Acid addition salts include salts of inorganic acids as well as organic acids.
Representative examples of suitable inorganic acids include hydrochloric, hydrobromic, hydroiodic, phosphoric, sulfuric, sulfamic, nitric acids and the like. Representative examples of suitable organic acids include formic, acetic, trichloroacetic, trifluoroacetic, propionic, benzoic, cinnamic, citric, fumaric, glycolic, itaconic, lactic, methanesulfonic, maleic, malic, malonic, mandelic, oxalic, picric, pyruvic, salicylic, succinic, methane sulfonic, ethanesulfonic, tartaric, ascorbic, pamoic, bismethylene salicylic, ethanedisulfonic, gluconic, citraconic, aspartic, stearic, palmitic, EDTA, glycolic, p-aminobenzoic, glutamic, benzenesulfonic, p-toluenesulfonic acids, theophylline acetic acids, as well as the 8- halotheophyllines, for example 8-bromotheophylline and the like. Further examples of pharmaceutically acceptable inorganic or organic acid addition salts include the pharmaceutically acceptable salts listed in S. M. Berge, et al., J. Pharm. ScL, 1977, 66, 2, the contents of which are hereby incorporated by reference.
Furthermore, the compounds of this invention may exist in unsolvated as well as in solvated forms with pharmaceutically acceptable solvents such as water, ethanol and the like. In general, the solvated forms are considered equivalent to the unsolvated forms for the purposes of this invention.
Racemic forms may be. resolved into the optical antipodes by known methods, for example, by separation of diastereomeric salts thereof with an optically active acid, and liberating the optically active amine compound by treatment with a base. Separation • of such diastereomeric salts can be achieved, e.g. by fractional crystallization. The optically active acids suitable for this purpose may include, but are not limited to d- or 1- tartaric, madelic or camphorsulfonic acids. Another method for resolving racemates into the optical antipodes is based upon chromatography on an optically active matrix. The compounds of the present invention may also be resolved by the formation and chromatographic separation of diastereomeric derivatives from chiral derivatizing reagents, such as, e.g., chiral alkylating or acylating reagents, followed by cleavage of the chiral auxiliary. Any of the above methods may be applied either to resolve the optical antipodes of the compounds of the invention per se or to resolve the optical antipodes of synthetic intermediates, which can then be converted by methods described herein into the optically resolved final products which are the compound of the invention.
Additional methods for the resolution of optical isomers, known to those skilled in the art, may be used. Such methods include those discussed by J. Jaques, A. Collet and S. Wilen in Enantiomers, Racemates, and Resolutions, John Wiley and Sons, New York 1981. Optically active compounds may also be prepared from optically active starting materials.
The invention also encompasses prodrugs of the present compounds, which on administration undergo chemical conversion by metabolic processes before becoming pharmacologically active substances. In general, such prodrugs will be functional derivatives of the compounds of Formula I which are readily convertible in vivo into the required compound of Formula I. Conventional procedures for the selection and preparation of suitable prodrug derivatives are described in Design of Prodrugs, ed. H. Bundgaard, Elsevier, 1985.
Pharmaceutical compositions The present invention further provides a pharmaceutical composition comprising a therapeutically effective amount of a compound of Formula I and a pharmaceutically
acceptable carrier. The present invention also provides a pharmaceutical composition comprising a therapeutically effective amount of one of the specific compounds disclosed in the Experimental Section and a pharmaceutically acceptable carrier.
The compounds of the invention may be administered alone or in combination with pharmaceutically acceptable carriers or excipients, in either single or multiple doses. The pharmaceutical compositions according to the invention may be formulated with pharmaceutically acceptable carriers or diluents as well as any other known adjuvants and excipients in accordance with conventional techniques such as those disclosed in Remington: The Science and Practice of Pharmacy, 19th Edition, Gennaro, Ed., Mack Publishing Co., Easton, PA, 1995.
The pharmaceutical compositions may be specifically formulated for administration by any suitable route such as oral, rectal, nasal, pulmonary, topical (including buccal and sublingual), transdermal, intracisternal, intraperitoneal, vaginal and parenteral (including subcutaneous, intramuscular, intrathecal, intravenous and intradermal) routes. It will be appreciated that the route will depend on the general condition and age of the subject to be treated, the nature of the condition to be treated and the active ingredient.
Pharmaceutical compositions for oral administration include solid dosage forms such as capsules, tablets, dragees, pills, lozenges, powders and granules. Where appropriate, the compositions may be prepared with coatings such as enteric coatings or they may be formulated so as to provide controlled release of the active ingredient such as sustained or prolonged release according to methods well known in the art. Liquid dosage forms for oral administration include solutions, emulsions, suspensions, syrups and elixirs.
Pharmaceutical compositions for parenteral administration include sterile aqueous and nonaqueous injectable solutions, dispersions, suspensions or emulsions as well as sterile powders to be reconstituted in sterile injectable solutions or dispersions prior to use.
Other suitable administration forms include, but are not limited to, suppositories, sprays, ointments, creams, gels, inhalants, dermal patches and implants.
Typical oral dosages range from about 0.001 to about 100 mg/kg body weight per day. Typical oral dosages also range from about 0.01 to about 50 mg/kg body weight per day.
Typical oral dosages further range from about 0.05 to about 10 mg/kg body weight per day.
Oral dosages are usually administered in one or more dosages, typically, one to three dosages per day. The exact dosage will depend upon the frequency and mode of administration, the sex, age, weight and general condition of the subject treated, the nature and severity of the condition treated and any concomitant diseases to be treated and other factors evident to those skilled in the art.
The formulations may also be presented in a unit dosage form by methods known to those skilled in the art. For illustrative purposes, a typical unit dosage form for oral administration may contain from about 0.01 to about 1000 mg, from about 0.05 to about 500 mg, or from about 0.5 mg to about 200 mg.
For parenteral routes such as intravenous, intrathecal, intramuscular and similar administration, typical doses are in the order of half the dose employed for oral administration.
The present invention also provides a process for making a pharmaceutical composition comprising admixing a therapeutically effective amount of a compound of Formula I and a pharmaceutically acceptable carrier. In an embodiment of the present invention the compound utilized in the aforementioned process is one of the specific compounds disclosed in the Experimental Section.
The compounds of this invention are generally utilized as the free substance or as a pharmaceutically acceptable salt thereof. One example is an acid addition salt of a compound having the utility of a free base. When a compound of Formula I contains a free base such salts are prepared in a conventional manner by treating a solution or suspension of a tree base of Formula I with a molar equivalent of a pharmaceutically acceptable acid. Representative examples of suitable organic and inorganic acids are described above.
For parenteral administration, solutions of the compounds of Formula I in sterile aqueous solution, aqueous propylene glycol, aqueous vitamin E or sesame or peanut oil may be employed. Such aqueous solutions should be suitably buffered if necessary and the liquid diluent first rendered isotonic with sufficient saline or glucose. The aqueous solutions are particularly suitable for intravenous, intramuscular, subcutaneous and intraperitoneal administration. The compounds of Formula I may be readily incorporated into known sterile aqueous media using standard techniques known to those skilled in the art.
Suitable pharmaceutical carriers include inert solid diluents or fillers, sterile aqueous solutions and various organic solvents. Examples of solid carriers include lactose, terra alba, sucrose, cyclodextrin, talc, gelatin, agar, pectin, acacia, magnesium stearate, stearic acid and lower alkyl ethers of cellulose. Examples of liquid carriers include, but are not limited to, syrup, peanut oil, olive oil, phospholipids, fatty acids, fatty acid amines, polyoxyethylene and water. Similarly, the carrier or diluent may include any sustained release material known in the art, such as glyceryl monostearate or glyceryl distearate, alone or mixed with a wax. The pharmaceutical compositions formed by combining the compounds of Formula I and a pharmaceutically acceptable carrier are then readily administered in a variety of dosage forms suitable for the disclosed routes of administration. The formulations may conveniently be presented in unit dosage form by methods known in the art of pharmacy.
Formulations of the present invention suitable for oral administration may be presented as discrete units such as capsules or tablets, each containing a predetermined amount of the active ingredient, and optionally a suitable excipient. Furthermore, the orally available formulations may be in the form of a powder or granules, a solution or suspension in an aqueous or non-aqueous liquid, or an oil-in-water or water-in-oil liquid emulsion.
If a solid carrier is used for oral administration, the preparation may be tabletted, placed in a hard gelatin capsule in powder or pellet form or it may be in the form of a troche or lozenge. The amount of solid carrier will vary widely but will range from about 25 mg to about 1 g per dosage unit.
If a liquid carrier is used, the preparation may be in the form of a syrup, emulsion, soft gelatin capsule or sterile injectable liquid such as an aqueous or non-aqueous liquid suspension or solution.
Treatment of Disorders
As mentioned above, the compounds of Formula I are ligands at the MCHl receptor. The present invention provides a method of treating a subject suffering from depression which comprises administering to the subject a therapeutically effective amount of a compound of this invention. This invention further provides a method of treating a subject suffering from anxiety which comprises administering to the subject a therapeutically effective amount of a compound of this invention. This invention also provides a method of treating a subject suffering from obesity which comprises administering to the subject a therapeutically
effective amount of a compound of this invention. In an embodiment of this invention, the subject is a human being.
The invention will be better . understood from the Experimental Details which follow. However, one skilled in the art will readily appreciate that the specific methods and results discussed therein are merely illustrative of the invention as described more fully in the claims which follow thereafter. Furthermore, the variables depicted in Schemes 1-7 are consistent with the variables recited in the Summary of the Invention. For clarity purposes, the variables U1, U2, U3 and U4 are designated as variable U in the experimental schemes. Moreover, the variables Y1, Y2, Y3 and Y4 are designated as variable Y in the experimental schemes.
In the Experimental Section, standard acronyms are used. Examples of such acronyms include, but are not limited to, AIBN (2,2'-Azobisisobutyronitrile); DMF (N,N- Dimethylformamide); DMSO (Dimethylsulfoxide); NBS (N-Bromosuccinimide); MTBE (Methyl t-butyl ether); HATU (O-(7-Azabenzotriazol-l-yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate); mCPBA (3-Chloroperoxybenzoic acid); CbzCl (Benzyl chloroformate); and BOC (fert-Butoxycarbonyl). Furthermore in certain instances, the methods of preparing the compounds of the invention are described generally by referring to representative reagents such as bases or solvents. The particular reagent identified is representative but is not inclusive or does not limit the invention in any way. For example, representative bases include but are not limited to K2CO3, Et3N or DIPEA (Diisopropylethylamine).
Experimental Section
General Methods: All reactions were performed under a nitrogen atmosphere and the reagents, neat or in appropriate solvents, were transferred to the reaction vessel via syringe and cannula techniques. Anhydrous solvents were purchased from the Aldrich Chemical Company and used as received. The NMR spectra were recorded on a Bruker Avance (400 MHz) or GE QEPlus300 in CDCl3, MeOH-d4 or DMSOd6 as solvent with tetramethylsilane as the internal standard unless otherwise noted. Chemical shifts (δ) are expressed in ppm, coupling constants (J) are expressed in Hz, and splitting patterns are described as follows: s - singlet; d = doublet; t = triplet; q = quartet; quintet; sextet; septet; br = broad; m = multiplet; dd = doublet of doublets; dt = doublet of triplets; td = triplet of doublets; dm = doublet of multiplets; ddd = doublet of doublet of doublets. Unless otherwise noted, mass spectra were obtained using electrospray ionization (ESMS, Micromass Platform II or Quattro Micro) and (M+H)+ is reported. Thin-layer chromatography (TLC) was carried out on glass plates pre-coated with silica gel 60 F254 (0.25 mm, EM Separations Tech.). Preparative TLC was carried out on glass sheets pre-coated with silica gel GF (2 mm, Analtech). Flash column chromatography was performed on Merck silica gel 60 (230-400 mesh). Microwave experiments were carried out using a Biotage Emyrs Optimizer or Smithcreator.
List and source of chemicals
Most of the reagents used in the experimental section such as 2-propanethiol, 4- fluorobenzaldehyde, potassium carbonate, 3-chloroperoxybenzoic acid, 4- fluoroacetophenone, methyl magnesium bromide, methanesulfonyl chloride, methyl chloroformate, 4-chlorotetrahydropyran, N-bromosuccinimide, nitronium tetrafluoroborate were purchased from Aldrich Sigma. Cyclopentanethiol was purchased from TCI International.

Il III IVa IVb IVc

IV (R2 = H or C1-C7 alky) Vl
(a) K2CO3/ DMF or DMSO/ 90 0C5 10 h or microwave/ 250 0C, 5 min. (b) mCPBA (leq)/ CH2Cl2/ 5 0C, 10 min. (c) mCPBA (1.5 eq)/ CH2Cl2/ 5 0C, 15 min. (d) Mgalkylbromide. (e) Dess-Martin reagent (f) (R2=H)/ NaBH(OAc)3/ HOAc/ CH2Cl2 or 1,2-dichIoroethane or (R2= Ci-C7 alkyl)/ Ti(OiPr)4/ THF/ rt, 12 h, then NaBH4/ EtOH/ rt, 1 h.
The compounds of Formula I may be synthesized according to the procedures described in Scheme 1. The compounds of Formula II and III are commercially available or may be synthesized by one skilled in the art. In summary, the aldehydes/ ketones of Formula IV are prepared via aromatic nucleophilic reaction of activated 4-halo-benzaldehydes/ ketones of Formula II and alkylthiols of Formula HI in the presence of base under reflux or microwave conditions. The corresponding sulfoxides IVb and sulfones IVc may be prepared via sequential oxidations of IVa by mCPBA.
Alternatively, the ketones of Formula IV (R2=Ci-C7 alkyl) may be prepared from the corresponding aldehydes of Formula IV (R2=H) by the addition of a Grignard reagent such as magnesiummethylbromide to the aldehydes of Formula IV (R2=H) to afford the alcohols of Formula V. The alcohols may be oxidized to the corresponding ketones of Formula IV (R2=C|-C7 alkyl). The compounds of Formula IV may be coupled with the amine of Formula VI to afford the compounds of the invention. Additionally, the conversion of the
aldehydes to ketones may be obtained via the Weinreb amide intermediate. For reaction conditions in connection with the Weinreb amide, see S. Nahm and S.M. Weinreb, Tetrahedron Lett., \ 981 , 22, 3815-3818.
Alternatively, the aldehydes/ ketones of Formula IV may be prepared via Ullmann type reactions. For reaction conditions in connection with Ullmann type reactions, see T. Kondo, et al., Chem. Rev., 2000, 100, 3205-3220 and the references cited therein.
Scheme 2

IV V VII

(a) NaBH4/ MeOH/ rt, 30 min. (b) MsCl/ TEA/ CH2Cl2/ rt, 30 min. (c) TEA/ THF/ rt.
Alternatively, the compounds of Formula I may be synthesized according to the procedures described in Scheme 2. The intermediates of Formula IV may be reduced to the alcohols of Formula V, which may be treated with mesylchloride in the presence of triethylamine in CH2Cl2 to afford the mesylates of Formula VII. The mesylates may be coupled with the amines of Formula VI to afford the compounds of the invention.
Scheme 3

VIII IX
(a) NBS/ AIBN/ CCl4/ refluxing, 12 h. (b) Amine of Formula VI; K2CO3/ DMF/ 90 0C5 5h or K2CO3/ EtOH/ rt/ 10 h.
Alternatively, the compounds of Formula I may be synthesized according to the procedures described in Scheme 3. The compounds of Formula VIII, which are commercially available or synthesized by those skilled in the art, may be treated with NBS to afford the bromides of Formula IX. The bromides may be coupled with the amines of Formula VI to afford the compounds of the invention.
Scheme 4

XIII Xl XIV
(a) K2CO3/ PdCbdppf/ DMF/ 60-80 0C 12 h. (b) 10% Pd/C/ H2 (50-60 psi)/ EtOH/ rt, 24-72 h. (c) Acid chloride or chloroformate/ Pyridine/ CH2Cl2. (d) 4M HCl in 1,4-dioxane/ rt, 1 h or TFA/ CH2Cl2/ rt, 10 min.
The amines of Formula VI may be prepared according to the procedures described in Scheme 4. In summary, the compounds of Formula X may be coupled with tert-butyϊ 4-(3- aminoaryl)piperidinecarboxy!ate of Formula XI to afford the N-Cbz protected intermediates
of Formula XII. The Cbz group is removed to provide the compounds of Formula XV, which may be coupled with acid chlorides or chloroformates to yield the intermediates of Formula XVI. The Boc group is removed to afford the amines of Formula VI. Alternatively, the nitro compounds of Formula XIII may be used. These compounds are also coupled with tert-butyl 4-(3-aminoaryl)piperidinecarboxylate of Formula XI to afford the compounds of Formula XIV. The nitro group and the double bond are simultaneously reduced to provide the compounds of Formula XV which may be converted to the amines of Formula VI.
For representative reaction conditions in connection with Suzuki couplings and hydrogenation reactions see A. Suzuki et al., Chem. Rev. 1995, 95, 2457-2483; A. Suzuki, J. Organomet. Chem. 1999, 576, 147-168 and the references cited therein; and P. N. Rylander, Hydrogenation Methods (Best Synthetic Methods Series), Academic Press, 1990. tert-Butyl 4-(454,5,5-tetramethyl- 1 ,3,2-dioxaborolan-2-yl)- 1 ,2,5,6-tetrahydropyridine carboxylate of Formula XI may be prepared according to the procedures described in P. R. Eastwood, Tetrahedron Lett., 2000, 41, 3705-3708 and references cited therein.
Scheme 5

XVII X XVI" (a) CbzCl/ K2CO3/ THF. (b) Curtius reaction.
The N-Cbz bromo or iodo intermeditaes of Formula X may be prepared according to the procedures described in Scheme 5. The amino group of the commercially available starting materials of Formula XVII may be protected by treatment with benzyl chloroformate in the presence of base to afford the intermediates of Formula X. Alternatively, N-Cbz protected intermediates of Formula X may be prepared from the corresponding acids of Formula XVIII using diphenylphosphoryl azide via a Curtius type rearrangement, followed by trapping the isocyanates with benzyl alcohol to afford the compounds of Formula X. For reaction conditions in connection with the Curtius reaction, see Yamada et al., Tetrahedron 1974, 30, 2151-2157.
Scheme 6
γγ Br (γor I) γγ Brγ (or I) O
XIX XlII xx
(a) nitration, (b) bromination. The nitro-bromo or iodo intermediates of Formula XIII may be prepared according to the procedures described in Scheme 6. The 3-bromo or 3-iodo compounds of Formula XIII are available from commercial sources or may be prepared from the corresponding bromo or iodo compounds XIX by nitration methods or from the corresponding nitro intermediates of Formula XX by bromination methods. General information regarding aromatic nitration is described in the following references: J. G. Hoggett, et al.s Nitration and Aromatic Reactivity, Cambridge University Press, London, 1971; K. Schofield, Aromatic Nitration, Cambridge University Press, London, 1980; and G. A. Olah, et al., Nitration: Methods and Mechanism, (Ed.: H. Feuer), VCH Publishers, New York, 1989.
Scheme 7

(a) IV, (R1HHy NaBH(OAc)3/ HOAc/ CH2Cl2 or 1,2-dichloroethane (b) IX/ K2CO3/ DMF/ 90 0C, 5h or IX/ K2CO3/ EtOH/ it, 1O h. (c) VII/ TEA/ THF/ rt. (d) Acid chloride or chloroformate/ Pyridine/ CH2CI2/ rt.
Alternatively the compounds of the invention may be prepared according to the procedures as outlined in Scheme 7. The compounds of Formula XXI may be coupled with the benzaldehydes or benzophenones of Formula IV (R2=H) in the presence of NaBH(OA 0)3 to provide the advanced intermediates of Formula XXII. Likewise, compounds of Formula XXI may be coupled with the mesylates of Formula VII or benzyl bromides of Formula IX in the presence of base to provide the advanced intermediates of Formula XXII. The advanced intermediates of Formula XXII may be treated with acid chlorides or chloroformates under standard coupling conditions to afford the compounds of Formula I.
Preparation of intermediates
Representative intermediates of the compounds of the Formula I were synthesized as follows:
5
Intermediate of Formula IVa (R2 = H)
4-lsopropyIsuIfanyI-benzaldehyde: A mixture of 4-fiuorobenzaldehyde (1.57 g, 12.6 mmol) and propane-2-thiol (0.958 g, 12.6 mmol), K2CO3 (2.09 g, 15.1 mmol) in DMF (5.00 raL) was heated at 90 0C for 10 h. After cooling to rt, the reaction mixture was poured into a
10 separatory funnel containing water (100 mL). The phases were separated and the aqueous layer was extracted with CH2Cl2 (3 x 50 mL). The organic layers were combined, washed with water (2 x 100 mL), brine (50 mL), and dried over MgSO4. Removal of the solvents in vacuo gave a light yellow liquid. Purification by silica gel flash chromatography (5 % EtOAc in Hexane) provided 4-isopropylsulfanyl-benzaldehyde (2.01 g, 88.2 % yield) as a
15 light yellow liquid.
Intermediate of Formula IVb (R2 = H)
4-(Propane-2-suIfinyl)-benzaldehyde: A mixture of 4-isopropylsulfanyl-benzaldehyde (653 mg, 3.63 mmol) and 3-chloroperoxybenzoic acid (max 77 %, 813 mg, 3.63 mmol) in
20 CH2CI2 (5.00 mL) was stirred at 0-5 0C for 10 min. The reaction mixture was poured into a separatory funnel containing 5 % aqueous KOH solution (10 mL). The phases were separated and the aqueous layer was extracted with CH2CI2 (3 x 10 mL). The combined organic extracts were washed with water (10 mL), brine (50 mL) and dried over MgSO4. Removal of solvents in vacuo gave a light yellow liquid. Purification by silica gel flash
25 chromatography (5 % EtOAc in Hexane) provided 4-(propane-2-sulfinyl)-benzaldehyde (400 mg, 56.2 % yield) as a light yellow solid.
Intermediate of Formula IVc (R2 = H)
4-(Propane-2-sulfonyl)-benzaldehyde: A mixture of 4-isopropylsulfanyl-benzaldehyde (653
30 mg, 3.63 mmol) and 3-chloroperoxybenzoic acid (max 77%, 2.44 g, 10.9 mmol) in CH2Cl2 (5.00 mL) was stirred at 0-5 0C for 15 min. The reaction mixture poured into a separatory funnel containing 5 % aqueous KOH solution (20 mL). The phases were separated and the aqueous layer was extracted with CH2Cl2 (3 x 10 mL). The combined organic layers were washed with water (10 mL), brine (50 mL) and dried over MgSO4. Removal of solvents in
35. vacuo gave a light yellow liquid. Purification by silica gel flash chromatography (5 %
EtOAc in Hexane) provided 4-(propane-2-sulfonyl)-benzaldehyde (700 mg, 91.0 % yield) as a light yellow solid. 1H NMR (CDCl3) δ 10.14 (s, IH)5 8.08 (m, 4H), 3.32 (m5 IH), 1.32 (d, J=6.87Hz, 6H).
Intermediate of Formula IV (R2 = Ct -C7 alkyl)
4-(isopropylthio)acetophenone: 2-propanethiol (152 mg, 2.0 mmol) was added to a stirred solution of 4'-fluoroacetophenone (276 mg, 2.0 mmol) and K2CO3 (276 mg, 2.0 mmol) in N,N-dimethylformamide (3 mL). The mixture was stirred in a sealed glass tube at 9O0C for 18 h. After cooling, the reaction was filtered and the solvent was removed in vacuo to produce a yellow oil which was purified by silica gel flash column chromatography (2-15 % EtOAc in Hexane) to furnish the title compound (293 mg, 76 % yield) as a clear oil. 1H NMR (CDCl3, 400 MHz)5 δ 7.85(d, J=8.5Hz, 2H), 7.35 (d, J=8.5Hz, 2H), 3.56 (m, IH), 2.57 (s, 3H)5 1.36 (d, J-6.7HZ, 6H).
Conversion of Alkylbenzaldehydes to Alkylphenylketones: Intermediate of Formula IV (R2 = Ci-C7 alkyl) l-[4-(Propane-2-sulfonyl)-phenyl]-ethanol: MeMgBr (1.4 M in toluene, 2.0 eq, 4.72 mmol, 3.37 mL) was added to a solution of 4-(propane-2-sulfonyl)-benzaldehyde (0.500 g, 2.36 mmol,) in THF (20 mL) at -78 0C. The resulting mixture was allowed to warm to rt and quenched with saturated aqueous NH4Cl solution. The resultant aqueous solution was extracted with EtOAc (3 X 50 mL). The organic layers were combined, washed with brine, dried over MgSO4 and concentrated in vacuo. The crude material was purified by silica gel flash chromatography (0 to 60% EtOAc in Hexane) to afford the desired alcohol (0.520 g, 98.1%).
Intermediate of Formula IV (R2 = C1-C7 alkyl) l-[4-(Propane-2-sulfonyl)-phenyl]-ethanone: Dess-Martin reagent (1.28 g, 1.5 eq) was added to a solution of l-[4-(propane-2-sulfonyl)-phenyl]-ethanol (0.520 g, 2.28 mmol) in CH2CI2 (20 mL). The resultant mixture was stirred overnight at rt and quenched with saturated aqueous NaHCθ3 solution. The aqueous solution was extracted with CH2CI2 (3 X 50 mL). The organic layers were combined, dried over MgSO4 and concentrated in vacuo. The product was purified by silica gel flash chromatography (0 to 60% ethyl acetate:Hexane) to afford the ketone (0.481 g, 92.1 % yield).
Conversion to Mesylate Intermediate of Formula VII
Methanesulfonic acid l-[4-(propane-2-sulfonyl)-phenyl]-ethyl ester: Methanesulfonyl chloride (2.00 eq, 3.06 mmol, 0.240 raL) was added to a solution of l-[4-(propane-2- sulfonyl)-phenyl]-ethanol (1.53 mmol, 0.350 g) and Et3N (3.00 eq, 4.59 mmol, 0.640 mL) in
CH2CI2 (50 mL). The resultant mixture was stirred at rt for 2 h. The mixture was diluted with CH2CI2 and washed with 0.5 N aqueous HCl, saturated aqueous NaHCO3, H2O and brine. The phases were separated. The organic layers were combined, dried over MgSO4 and concentrated in vacuo to afford the mesylate (quantitative yield). This compound was used without further purification.
Conversion to Benzyl Bromide Intermediate of Formula IX
4-(4-Bromomethyl-benzenesulfonyl)-tetrahydro-pyran: A mixture of 4-(toluene-4- sulfonyl)-tetrahydro-pyran (1.100 g, 4.20 mmol), N-bromosuccinimide (1.05 g, 4.41 mmol) and 2,2'-azobis-(2-methylpropionitrile) (68.9 mg, 0.420 mmol) in CCl4 (25.0 mL) was stirred for 5 min at rt and heated at reflux for 12 h. The reaction mixture was cooled to rt and filtered. The solvents were removed in vacuo to afford the desired product which was used without any further purification (1.05 g, 78.6 %).
terf-butyl 4-(4,4,5,5-tetramethyM,3,2-dioxaborolan-2-yl)-l,2,5,6-tetrahydropyridine carboxylate of Formula XI was prepared according to the procedures described by P. R. Eastwood, Tetrahedron Lett., 2000, 41, 19, 3705-3708 and references cited therein.
Intermediates of Formula Xiπ l-Bromo-2,4-difluoro-5-nitrobenzene: HΝO3 (68.0 mL) was added to a 0 0C solution of 1- bromo-2,4-difluorobenzene (20.0 g, 11.7 mL, 0.100 mol) and H2SO4 (76.8 mL) over 45 min at such a rate that the internal temperature remained < 7 0C. The resulting mixture was stirred for 1 h at 0 0C and poured into ice water (400 mL). This reaction was vigorously stirred for 2-3 min and extracted with CH2CI2 (400 mL). The organic layers were combined and washed with brine (1 X 500 mL), dried over Na2SO4, filtered and concentrated in vacuo to give the product as a yellow oil (23.5 g, 95 % yield). 1H NMR (CDCl3) δ 8.39 (t, J=7.2Hz, IH), 7.14 (ddd, J=0.3, 7.8, 9.9Hz, IH).
The following compound was prepared analogously: l-Bromo-3-nitro-2,4,6-trifluorobenzene: 1H NMR (CDCl3) δ 7.01 (ddd, J=2.4, 7.8, 9.3Hz5 IH); 19F NMR (CDCl3) δ -116.20 to -116.10, -107.73 to -107.71, -93.80 to - 93.70.
2-Bromo-5-fluoro-4-nitro toluene: 2-bromo-5-fluoro toluene (15.0 g, 10.0 mL, 79.0 mmol) was added to a solution of nitronium tetrafluoroborate (11.6 g, 87.0 mmol) in CH2CI2 (60.0 mL) over 5 min. After refluxing for 4.5 h, the mixture was cooled to rt and poured into ice water (150 mL). The mixture was extracted with CH2Cl2 (3 X 50 mL). The combined organic layers were washed with brine (100 mL), dried over Na2SO4, filtered and concentrated in vacuo to give the crude product (18.3 g). The crude product was treated with Hexane and cooled to —70 0C. The Hexane was decanted away from the resulting solid to give the desired product as a semi-solid (9.77 g, 53 % yield). The mother liquors were combined and evaporated. Additional product was purified by silica gel flash column chromatography (2 % EtOAc in Hexane) to give the desired product (1.0 g). 1H NMR (CDCl3) δ 8.26 (d, J=6.9Hz, IH), 7.20 (d, J=I 1.7Hz, IH), 2.48 (s, 3H).
The following compound of Formula XIII was prepared via a series of functional group transformations that are known to one skilled in the art:
3-Bromo-2-methyl-5-nitropyridine: Step 1 : Bromine (21.1 mL, 0.393 mol) was added dropwise over ~20 min to a mixture of 2-hydroxy-5-nitropyridine (50.0 g, 0.358 mol) in water (7 L), which was warmed to 40 0C. After stirring at 40 0C for 2.5 h, the mixture was cooled to 10 0C and the crude product was isolated by filtration. The solid was washed with water and dried in vacuo to give 3-bromo-2-hydroxy-5-nitropyridine as a solid (70.0 g, 90 % yield), mp 212-214 0C (with decomposition); 1H NMR (CD3OD) δ 8.66 (d, J=2.9Hz, IH), 8.64 (d, J=2.9Hz, IH).
Step 2: To a cooled (0-5 0C) mixture of 3-bromo-2-hydroxy-5-nitropyridine (47.0 g, 0.214 mol) and quinoline (13.7 g, 0.107 mol) was added POCl3 (26.0 mL, 0.278 mol) dropwise over ~10 min (the mixture was difficult to stir initially but became less viscous as the reaction progressed and the mixture warmed). After stirring at 120 0C for 3.5 h, the mixture was cooled to 100 0C and water (90 mL) was added. The resulting mixture was stirred vigorously while cooling to 0-5 0C. The product was collected by filtration, washed with water and dried in vacuo at 45 0C to give 3-bromo-2-chloro-5-nitropyridine (42.0 g, 82 %). 1H NMR (CD3OD) δ 9.19 (d, J=2.4Hz, 1 H)5 8.93 (d, J=2.4Hz, 1 H).
Step 3: NaH (as a 60 % dispersion in oil, 2.32 g, 58.0 mmol) was added over 5 min to a cooled (15 0C) solution of diethyl malonate (8.8 mL; 58.0 mmol) in diethyl ether (110 mL). 3-Bromo-2-chloro-5-nitropyridine (12.5 g, 52.6 mmol) was added in four portions over —15 min (an exotherm to 26 0C was observed), followed by removal of diethyl ether in vacuo to give a red oil. After stirring the resulting red oil at 1140C for 1 h 15 min, H2SO4 (6M, 67.0 mL) was added. The resulting mixture was heated at reflux for 8 h then cooled to 0 0C and the pH value was adjusted to 7 with 25 % KOH aqueous solution (135 mL). The resulting mixture was stirred in an ice bath for 25 min and the crude product was collected and washed with water (50 mL) by filtration. The crude product was stirred in CH2CI2 (350 mL) for 30 min and the impurity was removed by filtration. The organic layer was dried over Na2SO4, filtered and concentrated to give the impure product as red oil. The red oil was dissolved in CH2Cl2 (100 mL) and Hexane (200 mL). The resulting mixture was filtered and the organic portion was concentrated to give 3-bromo-2~methyl-5-nitropyridine as an orange crystalline solid (9.30 g, 81 % yield). 1H NMR (CDCl3) δ 9.25 (d, J=2.3Hz, IH), 8.61 (d, J=2.3Hz, IH), 2.80 (s, 3H).
Intermediate of Formula X
Benzyl 5-bromo-3-pyridinyl carbamate: To a suspension of 5-bromonicotinic acid (20.0 g, 99.0 mmol) in toluene (200 mL) was added diphenylphosphoryl azide (25.6 mL, 118.8 mmol) and Et3N (16.6 mL, 118.8 mmol). After stirring at it for 30 min, benzyl alcohol (15.4 mL, 148.5 mmol) was added. The mixture was stirred at it for 1 h and refluxed overnight. After cooling to rt, the reaction mixture was washed with H2O, saturated aqueous NaHCO3 and brine. The organic layers were combined, dried over MgSO4 and concentrated in vacuo. Purification by silica gel flash chromatography (15-50 % EtOAc in Hexane) provided benzyl 5-bromo-3-pyridinylcarbamate (22.2 g, 72.5 mmol, 73 % yield): 1H NMR (CDCl3) δ 8.39-8.32 (m, 2H), 8.29 (s, IH), 7.45-7.32 (m, 5H), 6.94 (s, IH), 5.22 (s, 2H); ESMS m/e: 307.0 (M + H)+.
Intermediates of Formula XV
/erZ-Butyl 4-(3-aminophenyl)piperidine carboxylate, tert-butyl 4-(3-amino-4-fluorophenyl) piperidine carboxylate and tert-butyl 4-(3-amino-4,6-difluorophenyl)piperidine carboxylate were prepared according to the procedures described by M. R. Marzabadi et ah, PCT International Publication No. WO 2004/005257 (pp. 48-82).
The following intermediates were prepared analogously:
tert-Buty\ 4-(3-amino-6-methylphenyl)piperidinecarboxylate
1H NMR (CDCl3) δ 6.93 (d, J=8.1Hz, IH), 6.53 (d, J=2.4Hz, IH)5 6.47 (dd, J=2.4, 8.1Hz, IH), 4.30-4.18 (m, 2H), 3.53 (br s, 2H), 2.86-2.51 (m, 3H), 2.23 (s, 3H), 1.77-1.68 (m, 2H), 1.50-1.63 (m, 2H), 1.49 (s, 9H).
tert-Butyl 4-(3-amino-6-fluorophenyl)piperidinecarboxylate
1H NMR (CDCl3) δ 6.85 -6.76 (m, IH), 6.51-6.44 (m, 2H), 4.30-4.15 (m, 2H)5 3.51 (br s, 2H), 2.98-2.73 (m, 3H), 1.82 -1.73 (m, 2H), 1.66 -1.50 (m, 2H), 1.48 (s, 9H).
tert-Butyl 4-(3-amino-4-fluoro-6-methylphenyl)piperidinecarboxylate
1H NMR (CDCl3) δ 6.77 (d, J=12.0Hz, IH), 6.60 (d, J=9.0Hz, IH), 4.32-4.16 (m, 2H)5 3.86-
3.52 (br, 2H), 2.86-2.67 (m, 3H), 2.22 (s, 3H), 1.69 (m, 2H), 1.60-1.43 (m, 1 IH).
terf-Butyl 4-(3-arnino-2,4,6-trifluorophenyl)piperidinecarboxylate
1HNMR (CDCI3) δ 6.67-6.54 (m, IH), 4.32-4.15 (m, 2H), 3.60-3.48 (m, 2H), 3.10-2.97 (m, IH), 2.84-2.68 (m, 2H), 2.06-1.88 (m, 2H), 1.70-1.60 (m, 2H), 1.46 (s, 9H).
tert-Butyl 4-(5-amino-3-pyridyl) piperidinecarboxylate
1H NMR (CDCl3) δ 8.01-7.95 (m, IH)5 7.89 (s, IH), 6.83 (s, IH), 4.39-4.09 (br, 2H), 3.90- 3.50 (br, 2H), 2.88-2.68 (m, 2H), 2.67-2.52 (m, IH), 1.88-1.71 (m, 2H), 1.68-1.49 (m, 2H), 1.48 (s, 9H); ESMS m/e: 278.3 (M + H)+.
tert-Butyl 4-(5-amino-2-methyl-3-pyridyl)piperidinecarboxylate
1H NMR (CDCl3) δ 7.87 (d, J=2.7Hz, I H), 6.80 (d, J=2.7Hz, I H), 4.33-4.17 (m, 2H), 3.57- 3.50 (br, 2H), 2.88-2.70 (m, 3H), 2.46 (s, 3H), 1.79-1.70 (m, 2H), 1.61-1.43 (m, 11H).
Compounds of the Invention The following compounds of the invention were synthesized according to the procedures described in Scheme 1 :
Example Ia iV-(4-Methyl-3-{ 1 -[4-(propane-2-sulfonyl)-benzyl]-piperidin-4-yl} -phenyl)- isobutyramide.

The following compounds were prepared analogously:
Example Ib {3-[l-(4-Isopropylsulfanyl-benzyl)-piperidin-4-yl]-4-methyl-phenyl}- carbamic acid methyl ester.

Prepared from 4-isopropylsulfanyl-benzaldehyde and (4-methyl-3-piperidin-4-yl-phenyl)- carbamic acid methyl ester. Yield: 84 %. ESMS m/e: 413.2 (M+H)+.
Example Ic N- {3-[ 1 -(4-Isopropylsulfanyl-benzy l)-piperid in-4-yl]-4-methyl-phenyl } - isobutyramide.

Example Id N-{3-[l -(4-Cyclopentanesulfonyl-benzyl)-piperidin-4-yl]-4-fluoro-phenyl}- isobutyramide.

Prepared from 4-cyclopentanesulfonyI-benzaldehyde and N-(4-fIuoro-3-piperidin-4-yl- phenyl)-isobutyramide. Yield: 78.5 %. ESMS m/e: 487.0 (MB-H)+.
Example Ie N-{4-Methyl-3-[l-(4-methylsulfanyl-benzyl)-piperidin-4-yl]-phenyl}- isobutyramide.

Prepared from 4-methylsulfanyl-benzaldehyde and N-(4-methyl-3-piperidin-4-yl-phenyl)- isobutyramide. Yield: 85.2 %. ESMS m/e: 397.1 (M+H)+.
Example If {3-[l -^-Ethanesulfonyl-benzyO-piperidin^-yl^-methyl-phenyty-carbamic acid methyl ester.

Prepared from 4-ethanesulfonyl-benzaldehyde and (4-methyl-3-piperidin-4-yl-phenyl)- carbamic acid methyl ester. Yield: 34.5 %. ESMS m/e: 431.1 (M+H)+.
Example Ig Cyclobutanecarboxylic acid {2-methyl-l'-[4-(propane-2-sulfonyl)-benzyl]- 11,2',3t,4'55',6I-hexahydro-[3,4']bipyridinyl-5-yl} -amide.
Example Ih N-{3-[l -(4-sec-Butylsulfanyl-benzyl)-piperidin-4-yl]-4-methyl-phenyl}- isobutyramide.

Example Ii N-(4-Methyl-3-{ 1 -[4-(2-methyI-propane-l -sulfonyl)-benzyl]-piperidin-4-yl} - phenyl)-isobutyramide.

Prepared from 4-(2-methyl-propane-l-sulfonyl)-benzaldehyde and N-(4-methyl-3-piperidin- 4-yl-phenyl)-isobutyramide. Yield: 52.4 %. ESMS m/e: 471.4 (M+H)+.
Example Ij Cyclobutanecarboxylic acid (4-methyl-3-{l-[4-(propane-l-sulfonyl)-benzyl]- piperidin-4-yl}-phenyl)-amide.

Prepared from 4-(propane-l-sulfonyl)-benzaldehyde and cyclobutanecarboxylic acid (4- methyl-3-piperidin-4-yl-phenyl)-amide. Yield: 84.2 %. ESMS m/e: 469.2 (M+H)+.
Example Ik Cyclobutanecarboxylic acid (4-methyl-3-{l-[4-(propane-2-sulfonyl)-benzyl]- piperidin-4-y 1 } -phenyl)-amide.
Prepared from 4-(propane-2-sulfonyl)-benzaldehyde and cyclobutanecarboxylic acid (4- methyl-3-piperidin-4-y!-phenyl)-amide. Yield: 46.6 %. ESMS m/e: 469.5 (M+H)+.
Example 11 Cyclopropanecarboxylic acid (4-fluoro-3-{ l-[4-(propane-2-sulfonyl)-benzyl]- piperidin-4-yl}-phenyl)-amide.
Example Im N-{2-Methyl-ll-[4-(propane-2-sulfonyl)-benzyl]-r,2>,3'>4',5I,6t-hexahydro- [3,4']bipyridinyl-5-yl}-isobutyramide.

Prepared from 4-(propane-2-sulfonyl)-benzaldehyde and N-(2-methyl-r,2',3',4',5',6'- hexahydro-[3,4']bipyridinyl-5-yl)-isobutyramide. Yield: 31.2 %. ESMS m/e: 458.4 (M+H)+.
Example In N-(3-{ l-[4-(Propane-2-sulfonyl)-benzyl]-piperidin-4-yl}-phenyl)-acetamide:

Prepared from 4-(propane-2-sulfonyl)-benzaldehyde and N-(3-piperidin-4-yl-phenyl)- acetamide. Yield: 16.6 %. ESMS m/e: 415.4 (M+H)+.
Example Io Cyclopropanecarboxylic acid (4-methyl-3-{1-[4-(propane-2-sulfonyl)-benzyl]- piperidin-4-yl}-phenyl)-amide.

Prepared from 4-(propane-2-sulfonyl)-benzaldehyde and cyclopropanecarboxylic acid (4- methyl-3-piperidin-4-yl-phenyl)-amide. Yield: 22.8 %. ESMS m/e: 455.5 (M+H)+.
Example Ip N-(2,4,6-Trifluoro-3-{ 1 -[4-(propane-2-sulfonyl)-benzyl]-piperidin-4-yl}- pheny l)-isobutyram ide .

Example Iq N-(2,4-Difluoro-5-{ l-[4-(propane-2-sulfonyl)-benzyl]-piperidin-4-yl}- phenyl)-isobutyr amide.

Prepared from 4-(propane-2-sulfonyl)-benzaldehyde and N-(2,4-difluoro-5-piperidin-4-yl- phenyO-isobutyramide. Yield: 29.3 %. ESMS m/e: 479.4 (M+H)+.
Example Ir N-(2-Fluoro-4-methyl-5 -{ 1 -[4-(propane-2-sulfonyl)-benzylj-piperidin-4-yl} - phenyl)-isobutyramide.
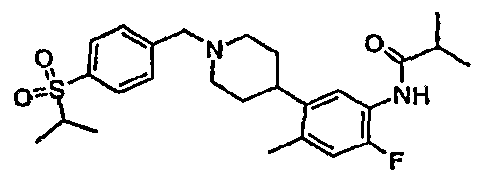
Prepared from 4-(propane-2-sulfonyl)-benzaldehyde and N-(2-fiuoro-4-methyl-5-piperidin- 4-yl-phenyl)-isobutyramide. Yield: 17.8 %. ESMS m/e: 475.5 (M+H)+.
Example Is (3-{t-[4-(Proρane-2-sulfonyl)-benzyl]-piperidin-4-yl}-phenyl)-carbamic acid methyl ester.

Example It N-{3-[l-(4-ButylsulfanyI-benzyl)-piperidin-4-yl]-4-methyl-phenyl}- isobutyramide.

Prepared from 4-butylsulfanyl-benzaldehyde and N-(4-methyl-3-piperidin-4-yl-phenyl)- isobutyramide. Yield: 99.0 %. ESMS m/e: 439.4 (M+H)+.
Example Iu N-(3-{l-[l-(4-Isopropylsulfanyl-phenyl)-ethyl]-piperidin-4-yl}-4-methyl- phenyl)-isobutyramide.

Prepared from N-(4-methyl-3-(piperidin-4-yl)phenyl)isobutyrarnide and 4-(isopropylthio) acteophenone using titanium (IV) isopropoxide and NaBH4. Yield: 52 %. ESMS m/e: 439.4 (M+H)+.
Example Iv N-(4-Methyl-3-{l-[l-(4-methylsulfanyl-phenyl)-ethyl]-piperidin-4-yl}- phenyl)-isobutyramide.

Prepared from 4-(methylthio)acteophenone and N-(4-methyl-3-(piperidin-4- yl)phenyl)isobutyramide using titanium (IV) isopropoxide and NaBH4. Yield: 58 %. ESMS m/e: 411.3 (M+H)+.
The following compounds of the invention were synthesized according to the procedures described in Scheme 3:
Example 2a (4-Methyl-3-{ 1 -[4-(tetrahydro-pyran-4-sulfonyl)-benzyl]-piperidin-4-yl}- phenyl)-carbamic acid methyl ester.

Into a vial was added 4-(bromomethyl)-l-[(4-(tetrahydro-pyran-4-sulfonyl]benzene (100 mg, 0.320 mmol), (4-methyl-3-piperidin-4-yl-phenyl)-carbamic acid methyl ester (47.2 mg, 0.190 mmol), potassium carbonate (60.0 mg, 0.430 mmol), NaI (30.0 mg, 0.200 mmol) and DMF (2.50 mL). The mixture was stirred for 2-3 min at 25 0C and heated to 90 0C in an oil bath. After stirring for 12 h at 90 0C, the mixture was allowed to cool to 25 0C and was diluted with EtOAc (50 mL). The reaction solution was washed with water (3 x 30 mL) and the aqueous solution was extracted with EtOAc (30 mL). The organic layers were combined and dried over MgSO4. Removal of solvent in vacuo gave crude product that was purified by silica gel flash chromatography (97 % EtOAc:3 % methanol 2 M ammonia to afford (4- methyl-3- { 1 -[4-(tetrahydro-pyran-4-sulfonyl)-benzyl]-piperidin-4-yl} -phenyl)-carbamic acid methyl ester (36.0 mg, 39.1 % yield). 1HNMR (CDCl3) δ 7.89-7.79 (m, 2H), 7.66-7.55 (m, 2H), 7.32 (br s, IH), 7.18-7.01 (m, 2H), 6.80 (br s, IH), 4.15-4.00 (m, 2H), 3.76 (s, 3H), 3.63 (s 2H), 3.37-3.28 (m, 2H), 3.21-3.10 (m, IH), 3.04-2.95 (m, 2H), 2.75-2.65 (m, IH), 2.27 (s, 3H), 2.21-2.13 (m, 2H), 1.97-1.66 (m, 8H). ESMS m/e: 487.2 (M+H)+.
The following compound was prepared analogously:
Example 2b N-(4-Methyl-3-{ 1 -[4-(tetrahydro-pyran-4-sulfonyl)-benzyl]-piperidin-4-yl} - phenyl)-isobutyramide.

Prepared from 4-(bromomethyl)-l-[(4-(tetrahydro-pyran-4-sulfonyl]benzene and N-(4- methyl-3-pϊperidin-4-yI-phenyl)-isobutyramide. Yield: 30.5 %. ESMS m/e: 499.1 (M+H)+.
F Foorrmmuullaattiioonnss T Thhee pphhaaπrmaceutical formulations of the invention may be prepared by conventional methods in the art.
For example, tablets may be prepared by mixing the active ingredient with ordinary adjuvants and/or diluents and subsequently compressing the mixture in a conventional tabletting machine prepare tablets. Examples of adjuvants or diluents comprise: corn starch, potato starch, talcum, magnesium stearate, gelatine, lactose, gums, and the like. Any other adjuvants or additives usually used for such purposes such as colorings, flavorings, preservatives etc. may be used provided that they are compatible with the active ingredients.
1) Tablets containing 5.0 mg of Compound 2a calculated as the free base:
Compound 2a 5.0 mg
Lactose 60 mg
Maize starch 30 mg
Hydroxypropylcellulose 2.4 mg
Microcrystalline cellulose 19.2 mg
Croscarmellose Sodium Type A 2.4 mg
Magnesium stearate 0.84 mg
lets containing 0.5 mg of Compound 2a calcula
Compound 2a 0.5 mg
Lactose 46.9 mg
Maize starch 23.5 mg
Povidone 1.8 mg
Microcrystalline cellulose 14.4 mg
Croscarmellose Sodium Type A 1.8 mg
Magnesium stearate 0.63 mg
3) Syrup containing 25 mg of Compound 2a per milliliter:
Compound 2a 25 mg
Sorbitol 500 mg
Hydroxypropylcellulose 15 mg
Glycerol 50 mg
Methyl-paraben 1 mg
Propyl-paraben 0.1 mg
Ethanol 0.005 mL
Flavor 0.05 mg
Saccharin 0.5 mg
Water 1 mL
In Vitro Methods
The pharmacological properties of the compounds of the present invention were evaluated at the cloned rat MCHl receptor using the protocols disclosed in U.S. Patent No. 6,727,264, the contents of which are hereby incorporated by reference.
Using this protocol, the inhibition by the compound of the binding of a radiolabeled ligand (tritiated SNAP-7941) to membranes harvested from CHO cells expressing cloned rat MCHl receptors was determined in vitro. The radiochemical synthesis of tritiated SNAP- 7941 was performed by Amersham Pharmacia Biotech, Cardiff, Wales.
Briefly, the affinity of the compounds was measured by their ability to displace tritiated SNAP-7941 from rat MCHl expressing membranes. The compound and radioligand were incubated with the membranes at 25 0C for 90 min. Incubation was terminated by rapid vacuum filtration over GF/C glass fiber filters, presoaked in 5 % PEI using 50 nM Tris pH 7.4 as wash buffer. In all experiments, nonspecific binding was defined using 10 pM of tritiated SNAP-7941.
The binding affinities for the compounds of the present invention, described herein, at the
MCHl receptor were determined to be 500 nM or less. For the majority of the compounds, the Ki values were determined to be 100 nM or less. For some compounds, the Ki values were determined to be 10 nM or less.
Claims
Claim 1. A compound having the structure:

wherein each Y1, Y2, Y3 and Y4 is independently CR5 or N, provided that if two Y are each N then the remaining Y are each CR5;
wherein R1 is straight chained or branched C1-C7 alkyl, C3-C7 cycloalkyl or C2-C7 cyclic ether;
wherein R2 is H or straight chained or branched C1-C7 alkyl;
wherein R3 is straight chained or branched Ci-C7 alkyl, C3-C7 cycloalkyl or straight-chained or branched C1-C7 alkoxy;
wherein each R4 is independently H, straight chained or branched Ci -C7 alkyl, straight chained or branched C1-C7 fluoroalkyU straight chained or branched Ci-C7 alkoxy, F, Cl, Br or I;
wherein each R5 is independently H5 CH3, F, Cl or Br; and
wherein n is an integer from 0 to 2 inclusive; or a pharmaceutically acceptable salt thereof.
Claim 2. The compound of claim 1, wherein each Y1, Y2, Y3 and Y4 is independently
CR5 or N, provided that if one Y is N then the remaining Y are each CR5.
Claim 3. The compound of claim 2, wherein each U1, JJ2, U3 and U4 is independently CR4; and wherein each R4 is independently H, CH3, F, Cl, Br or I.
Claim 4. The compound of claim 3, wherein R2 is H or straight chained or branched Q-C4 alkyl.
Claim 5. The compound of claim 4, wherein R1 is C2-C7 cyclic ether.
Claim 6. . The compound of claim 5, wherein R1 is tetrahydropyranyl; and wherein each R4 is H.
Claim 7. The compound of claim 4, wherein R1 is C3-C7 cycloalkyl.
Claim 8. The compound of claim 4, wherein R1 is straight chained or branched Ci-C7 alkyl.
Claim 9. The compound of claim 8, wherein R1 is straight chained or branched C1-C4 alkyl; and wherein each R4 is H.
Claim 10. The compound of claim 9, wherein R3 is straight chained or branched Q-C4 alkoxy.
Claim 1 1. The compound of claim 9, wherein R3 is straight chained or branched C1-C4 alkyl.
Claim 12. The compound of claim 11, wherein R2 is straight chained or branched C1-C4 alkyl.
Claim 13. The compound of claim 12, wherein R2 is CH3; wherein each Y1, Y2, Y3 and Y4 is independently CR5; and wherein each R5 is independently H, CH3 or F.
Claim 14. The compound of claim 11, wherein R2 is H.
Claim 15. The compound of claim 14, wherein each Y1, Y2, Y3 and Y4 is independently CR5; wherein each R5 is independently H, CH3 or F; and wherein n is 2.
Claim 16. The compound of claim 14, wherein each Y1, Y2, Y3 and Y4 is independently
CR5; wherein each R5 is independently H, CH3 or F; and wherein n is 0.
Claim 17. The compound of claim 1, wherein the compound is selected from the group consisting of N-(4-methyl-3-{l-[4-(propane-2-sulfonyl)-benzyl]-piperidin-4-yl}-phenyl)- isobutyramide; {3-[l-(4-isopropylsulfanyl-ben2yl)-piperidin-4-yl]-4-methyl-phenyl}- carbamic acid methyl ester; N-{3-[l-(4-isopropylsulfanyl-benzyl)-piperidin-4-yl]-4-methyl- phenyl}-isobutyramide; N-{3-[l-(4-cyclopentanesulfonyl-benzyl)-piperidin-4-yl]-4-fluoro- phenyl}-isobutyramide; N-{4-methyl-3-[l-(4-methylsuIfanyI-benzyl)-piperidin-4-yl].phenyl}- isobutyramide; {3-[l-(4-ethanesυlfonyl-benzyi)-piperidiπ-4-yl]-4-methyl-phenyl} -carbamic acid methyl ester; cyclobutanecarboxylic acid {2-methyl-r-[4-(propane-2-sulfonyl)-benzyl]- l',2l,31,4',5l !,6l-hexahydrθ"[3,4l]bipyridinyl-5-y]}-amide; N-{3-[l-(4-sec-butylsulfanyl-benzyl)- piperidin-4-yl]-4-methyl-phenyl} -isobutyramide; N-(4-methyl-3-{ 1 -[4-(2~methyl-propane-l - sulfonyl)-benzyl]-piperidin-4-yl}-phenyl)-isobutyraraide; cyclobutanecarboxylic acid (4- methyl-3-{l-[4-(propane-l-sulfonyl)-benzyl]-pipeπdin-4-yl}-phenyl)-amide; cyclobutanecarboxylic acid (4-methyl-3-{ 1 -[4-(propane-2-sulfonyl)-benzyl]-piperidin-4-yl}- phenyl)-amide; cyclopropanecarboxylic acid (4-fluoro-3-{l-[4-(propane-2-sulfonyl)-ber_zyl]- ρiρeridin-4-yl}-phenyi)-amide; N-{2"methyI-r-[4'(propane-2"Su!fonyl)-benzyl]-r,2l,3',4',5l,6'- hexahydro-[3,4']bipyridinyl-5-y l}-isobutyramide; N-(3-{ I -[4-(propane-2-sulfonyl)-benzyI]- piperidin-4-yl}-phenyl)-acetamide; cyclopropanecarboxylic acid (4-methyl-3-{l-[4-(proρane>2- sulfonyl)-benzyl]-ρiρeridin4-yl}-phenyl)-amide; N-(2,4,6-trifluoro-3-{ 1 -[4-(propane-2- sulfonyl)-benzyl]-piperidin-4-yl} -phenyl)-isobutyramide; N-(2j4-difluoro-5-{ l-[4-(propane-2- sulfonyl)-benzyl]-piperidin-4-yl} -phenyl)-isobufyramide; N-(2-fluoro-4-methyl-5-{ 1 -[4-
(propane-2-sulfonyl)-benzyl]-piperidin4-yl}-phenyl)-isobutyramide; (3-{l-[4-(propane-2- sulfonyl)-benzyl3-piperidin-4-yl}-phenyl)"Caitatnic acid methyl ester; N-{3-[I-(4-butylsulfanyI- benzyl)-piperidin-4-yl]-4-metiiyl-phenyl}-isobutyramide; N-(3-{l-[l-(4-isopropyIsulianyl- phenyl)-ethyl]-piperidin-4'yl}-4-methyl-phenyl)-isobutyramide; N-(4-methyl-3-{l-[l-(4- methylsulfanyl-phenyl)-ethyl]-piperidin4-yi} -phenyl)-isobutyramide; (4-methy 1-3- { 1 -[4- (tetrahydro-pyran4-sulfonyl)-benzyl]-piperidin-4-yl}-phenyl)-carbamic acid methyl ester and N-(4-methyl-3-{l-[4-(tetrahydro-pyran4-sulfonyl)-benzyl]-piperidin4-yl}-phenyl)- isobutyramide.
Claim 18. A pharmaceutical composition comprising a therapeutically effective amount of a compound of claim 1 and a pharmaceutically acceptable carrier,
Claim 19, Use of a compound as defined in claim 1 for the manufacture of a medicament useful for treating a subject suffering from depression.
Claim 20. Use of a compound as defined in claim 1 for the manufacture of a medicament useful for treating a subject suffering from anxiety.
Claim 21. Use of a compound as defined in claim 1 for the manufacture of a medicament useful for treating a subject suffering from obesity.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US78927406P | 2006-04-04 | 2006-04-04 | |
| US60/789,274 | 2006-04-04 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007114902A2 true WO2007114902A2 (en) | 2007-10-11 |
| WO2007114902A3 WO2007114902A3 (en) | 2008-07-03 |
Family
ID=38564129
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/008142 WO2007114902A2 (en) | 2006-04-04 | 2007-04-02 | Alkylthiobenzylpiperidine compounds |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2007114902A2 (en) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| CN103848767A (en) * | 2014-01-27 | 2014-06-11 | 华东师范大学 | Synthetic method of aryl thioether compound |
| CN114539073A (en) * | 2022-02-18 | 2022-05-27 | 郑州萃智医药科技有限公司 | Synthesis method of 3-bromo-2-chloro-4, 6-difluoroaniline |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6727264B1 (en) * | 2001-07-05 | 2004-04-27 | Synaptic Pharmaceutical Corporation | Substituted anilinic piperidines as MCH selective antagonists |
-
2007
- 2007-04-02 WO PCT/US2007/008142 patent/WO2007114902A2/en active Application Filing
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6727264B1 (en) * | 2001-07-05 | 2004-04-27 | Synaptic Pharmaceutical Corporation | Substituted anilinic piperidines as MCH selective antagonists |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| CN103848767A (en) * | 2014-01-27 | 2014-06-11 | 华东师范大学 | Synthetic method of aryl thioether compound |
| CN114539073A (en) * | 2022-02-18 | 2022-05-27 | 郑州萃智医药科技有限公司 | Synthesis method of 3-bromo-2-chloro-4, 6-difluoroaniline |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2007114902A3 (en) | 2008-07-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU757747B2 (en) | 4-aroyl-piperidin-CCR-3 receptor antagonists III | |
| US7205319B2 (en) | N-[phenyl (piperidin-2-yl) methyl]benzamide derivatives, preparation thereof, and use thereof in therapy | |
| US5387593A (en) | Piperazinyl-and piperidinyl-cyclohexanols | |
| AU2005254800B2 (en) | Benzimidazolone carboxylic acid derivatives | |
| US6020345A (en) | Pyridin-2-yl-methylamine derivatives, method of preparing and application as medicine | |
| WO2007114902A2 (en) | Alkylthiobenzylpiperidine compounds | |
| US20100173925A1 (en) | Oxyindole derivatives | |
| US6867220B2 (en) | Phenoxypropanolamines, method for producing them and pharmaceutical compositions containing them | |
| MXPA01012450A (en) | Metalloprotease inhibitors. | |
| JPH04364164A (en) | Psychotropic n-aralkylpiperidine derivative | |
| RU2464268C2 (en) | 4-alkoxypyridazine derivatives as fast dissociating dopamine 2 receptor antagonists | |
| CA2344807A1 (en) | Naphthalenecarboxamides as tachykinin receptor antagonists | |
| CA2468691C (en) | 4-piperidinyl alkylamine derivatives as muscarinic receptor antagonists | |
| HUE025163T2 (en) | Novel compounds as histamine h3 receptor ligands | |
| WO2007114916A2 (en) | Arylbenzylpiperidine compounds | |
| EP1799223A2 (en) | Arylthiobenzylpiperidine derivates | |
| EP1804796A2 (en) | Amino substituted aryloxybenzylpiperidine derivatives | |
| US7329656B2 (en) | Arylthiobenzylpiperidine derivatives | |
| JP4129445B2 (en) | Benzimidazolonecarboxylic acid derivatives | |
| US4518713A (en) | Analgesic substituted-1-aminoalkylamino-4-aryloxypiperidines | |
| CA2525849A1 (en) | 4-arylsulphonylpiperidine derivatives for antagonism of the 5-ht2a receptor | |
| US7446204B2 (en) | Amino substituted aryloxybenzylpiperidine derivatives | |
| HUT77311A (en) | Phenyl-oxo-alkyl-(4-piperidinyl)benzoate derivatives, pharmaceutical compositions containing them, process for producing them and their use | |
| MXPA01002485A (en) | 4,4-biarylpiperidine derivatives with opioid receptor activity | |
| ZA200102658B (en) | Naphthalenecarboxamides as tachykinin receptor antagonists. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 07754639 Country of ref document: EP Kind code of ref document: A2 |
|
| NENP | Non-entry into the national phase in: |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 07754639 Country of ref document: EP Kind code of ref document: A2 |