WO2007103839A2 - Pyrrolotriazine aniline prodrug compounds useful as kinase inhibitors - Google Patents
Pyrrolotriazine aniline prodrug compounds useful as kinase inhibitors Download PDFInfo
- Publication number
- WO2007103839A2 WO2007103839A2 PCT/US2007/063250 US2007063250W WO2007103839A2 WO 2007103839 A2 WO2007103839 A2 WO 2007103839A2 US 2007063250 W US2007063250 W US 2007063250W WO 2007103839 A2 WO2007103839 A2 WO 2007103839A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituted
- optionally
- compound
- methyl
- alkyl
- Prior art date
Links
- 0 C*1=CC=C(C=CC(O2)=O)C2=CC1 Chemical compound C*1=CC=C(C=CC(O2)=O)C2=CC1 0.000 description 2
- GTKNRXQSCVVESA-UHFFFAOYSA-N C1N=Nc2c1cccc2 Chemical compound C1N=Nc2c1cccc2 GTKNRXQSCVVESA-UHFFFAOYSA-N 0.000 description 1
- RWGFKTVRMDUZSP-UHFFFAOYSA-N CC(C)c1ccccc1 Chemical compound CC(C)c1ccccc1 RWGFKTVRMDUZSP-UHFFFAOYSA-N 0.000 description 1
- DXLSVPUSCYKDND-UHFFFAOYSA-N CCCN(C(c(c(C)c12)c[n]1ncnc2Nc1cc(C(NC2CC2)=O)ccc1C)=O)C(OCOC(c1cnccc1)=O)=O Chemical compound CCCN(C(c(c(C)c12)c[n]1ncnc2Nc1cc(C(NC2CC2)=O)ccc1C)=O)C(OCOC(c1cnccc1)=O)=O DXLSVPUSCYKDND-UHFFFAOYSA-N 0.000 description 1
- KONVXCZBTUVWLG-MDZDMXLPSA-N CCCNC(c(c(C)c12)c[n]1ncnc2N(C(OCOC(/C=C/C(O)=O)=O)=O)c1cc(C(NC2CC2)=O)ccc1C)=O Chemical compound CCCNC(c(c(C)c12)c[n]1ncnc2N(C(OCOC(/C=C/C(O)=O)=O)=O)c1cc(C(NC2CC2)=O)ccc1C)=O KONVXCZBTUVWLG-MDZDMXLPSA-N 0.000 description 1
- XLLPAWJXOXUTFM-SFHVURJKSA-N CCCNC(c(c(C)c12)c[n]1ncnc2N(C(OCOC([C@H](C)NC=O)=O)=O)c1cc(C(NC2CC2)=O)ccc1C)=O Chemical compound CCCNC(c(c(C)c12)c[n]1ncnc2N(C(OCOC([C@H](C)NC=O)=O)=O)c1cc(C(NC2CC2)=O)ccc1C)=O XLLPAWJXOXUTFM-SFHVURJKSA-N 0.000 description 1
- HFVQYQZMXLYNPF-UHFFFAOYSA-N CCCNC(c(c(C)c12)c[n]1ncnc2N(C(OCOP(OCc1ccccc1)(OCc1ccccc1)=O)=O)c1cc(C(NC2CC2)=O)ccc1C)=O Chemical compound CCCNC(c(c(C)c12)c[n]1ncnc2N(C(OCOP(OCc1ccccc1)(OCc1ccccc1)=O)=O)c1cc(C(NC2CC2)=O)ccc1C)=O HFVQYQZMXLYNPF-UHFFFAOYSA-N 0.000 description 1
- NDZRYTPYNNVMFL-UAYVFMOQSA-N CCCNC(c1c[nH]c(/C(/N(C(OCOC(/C=C/C(O)=O)=O)=O)c2c(C)ccc(C(NCCC)=O)c2)=N\C=N)c1C)=O Chemical compound CCCNC(c1c[nH]c(/C(/N(C(OCOC(/C=C/C(O)=O)=O)=O)c2c(C)ccc(C(NCCC)=O)c2)=N\C=N)c1C)=O NDZRYTPYNNVMFL-UAYVFMOQSA-N 0.000 description 1
- YESYBFGCAORBGP-WFWCCXBYSA-N CCCNC(c1c[nH]c(/C(/N(C(OCOC(/C=C/C(O)=O)=O)=O)c2cc(C(NCC)=O)ccc2C)=N\C=N)c1C)=O Chemical compound CCCNC(c1c[nH]c(/C(/N(C(OCOC(/C=C/C(O)=O)=O)=O)c2cc(C(NCC)=O)ccc2C)=N\C=N)c1C)=O YESYBFGCAORBGP-WFWCCXBYSA-N 0.000 description 1
- XZXQYROXECZVBY-WFWCCXBYSA-N CCCNC(c1ccc(C)c(N(/C(/c2c(C)c(C(NCC)=O)c[nH]2)=N/C=N)C(OCOC(/C=C/C(O)=O)=O)=O)c1)=O Chemical compound CCCNC(c1ccc(C)c(N(/C(/c2c(C)c(C(NCC)=O)c[nH]2)=N/C=N)C(OCOC(/C=C/C(O)=O)=O)=O)c1)=O XZXQYROXECZVBY-WFWCCXBYSA-N 0.000 description 1
- SALZNTRWWDLXCF-RYBRMRFGSA-N CCNC(c1c[nH]c(/C(/N(C(OCOC(/C=C/C(O)=O)=O)=O)C2=CC(C(NCC)=O)=CCC2(C)C)=N\C=N)c1C)=O Chemical compound CCNC(c1c[nH]c(/C(/N(C(OCOC(/C=C/C(O)=O)=O)=O)C2=CC(C(NCC)=O)=CCC2(C)C)=N\C=N)c1C)=O SALZNTRWWDLXCF-RYBRMRFGSA-N 0.000 description 1
- MFGIBHUOBYNQDB-HPKGBOGASA-O CCNC(c1c[nH]c(/C(/N(C(OCOC(/C=C/C(O)=O)=O)=O)c2c(C)ccc(C(Nc3n[o]cc3)=O)c2)=N\C=[NH2+])c1C)=O Chemical compound CCNC(c1c[nH]c(/C(/N(C(OCOC(/C=C/C(O)=O)=O)=O)c2c(C)ccc(C(Nc3n[o]cc3)=O)c2)=N\C=[NH2+])c1C)=O MFGIBHUOBYNQDB-HPKGBOGASA-O 0.000 description 1
- BUIPHDGKYGEMCY-XSHMTDCFSA-N CC[NH+](Cc1c[nH]c(/C(/N(C(OCOC(/C=C/C(O)=O)=O)=O)c2cc(C(NC3CC3)=O)ccc2C)=N\C=N)c1C)[O-] Chemical compound CC[NH+](Cc1c[nH]c(/C(/N(C(OCOC(/C=C/C(O)=O)=O)=O)c2cc(C(NC3CC3)=O)ccc2C)=N\C=N)c1C)[O-] BUIPHDGKYGEMCY-XSHMTDCFSA-N 0.000 description 1
- FXJCNYQETUPKJU-UHFFFAOYSA-N Cc(ccc(C(N(C1CC1)C(OCCl)=O)=O)c1)c1N Chemical compound Cc(ccc(C(N(C1CC1)C(OCCl)=O)=O)c1)c1N FXJCNYQETUPKJU-UHFFFAOYSA-N 0.000 description 1
- RZVHIXYEVGDQDX-UHFFFAOYSA-N O=C(c1ccccc11)c(cccc2)c2C1=O Chemical compound O=C(c1ccccc11)c(cccc2)c2C1=O RZVHIXYEVGDQDX-UHFFFAOYSA-N 0.000 description 1
- SIKJAQJRHWYJAI-UHFFFAOYSA-N c1c[nH]c2c1cccc2 Chemical compound c1c[nH]c2c1cccc2 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 1
- AWBOSXFRPFZLOP-UHFFFAOYSA-N c1cc2n[o]nc2cc1 Chemical compound c1cc2n[o]nc2cc1 AWBOSXFRPFZLOP-UHFFFAOYSA-N 0.000 description 1
- BAXOFTOLAUCFNW-UHFFFAOYSA-N c1n[nH]c2c1cccc2 Chemical compound c1n[nH]c2c1cccc2 BAXOFTOLAUCFNW-UHFFFAOYSA-N 0.000 description 1
- HYZJCKYKOHLVJF-UHFFFAOYSA-N c1nc(cccc2)c2[nH]1 Chemical compound c1nc(cccc2)c2[nH]1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 description 1
- BCMCBBGGLRIHSE-UHFFFAOYSA-N c1nc(cccc2)c2[o]1 Chemical compound c1nc(cccc2)c2[o]1 BCMCBBGGLRIHSE-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/53—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with three nitrogens as the only ring hetero atoms, e.g. chlorazanil, melamine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/02—Drugs for disorders of the urinary system of urine or of the urinary tract, e.g. urine acidifiers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/06—Antigout agents, e.g. antihyperuricemic or uricosuric agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/04—Drugs for disorders of the muscular or neuromuscular system for myasthenia gravis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
- A61P31/06—Antibacterial agents for tuberculosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/16—Antivirals for RNA viruses for influenza or rhinoviruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/02—Antiprotozoals, e.g. for leishmaniasis, trichomoniasis, toxoplasmosis
- A61P33/06—Antimalarials
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/06—Antianaemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C11/00—Aliphatic unsaturated hydrocarbons
Definitions
- This invention relates to pyrrolotriazine compounds, more particularly, to prodrugs of pyrrolotriazine aniline compounds useful for treating p38 kinase- associated conditions.
- the invention further pertains to pharmaceutical compositions containing at least one compound according to the invention useful for treating p38 kinase-associated conditions and methods of inhibiting the activity of p38 kinase in a mammal.
- cytokines participate in the inflammatory response, including IL-I, IL-6, IL-8 and TNF- ⁇ .
- Overproduction of cytokines such as IL-I and TNF- ⁇ are implicated in a wide variety of diseases, including inflammatory bowel disease, rheumatoid arthritis, psoriasis, multiple sclerosis, endotoxin shock, osteoporosis, Alzheimer's disease, and congestive heart failure, among others [Henry et al, Drugs FuL, 24: 1345-1354 (1999); Salituro et al, Curr. Med. Chem., 6:807-823 (1999)].
- TNF- ⁇ monoclonal antibody to TNF- ⁇
- Etanercept soluble TNF- ⁇ receptor-Fc fusion protein
- the biosynthesis of TNF- ⁇ occurs in many cell types in response to an external stimulus, such as, for example, a mitogen, an infectious organism, or trauma.
- MAP mitogen-activated protein
- p38 mitogen-activated protein
- p38- ⁇ There are four known isoforms of p38, i.e., p38- ⁇ , p38 ⁇ , p38 ⁇ , and p38 ⁇ .
- the ⁇ and ⁇ isoforms are expressed in inflammatory cells and are key mediators of TNF- ⁇ production. Inhibiting the p38 ⁇ and ⁇ enzymes in cells results in reduced levels of TNF- ⁇ expression. Also, administering p38 ⁇ and ⁇ inhibitors in animal models of inflammatory disease has proven that such inhibitors are effective in treating those diseases. Accordingly, the p38 enzymes serve an important role in inflammatory processes mediated by IL-I and TNF- ⁇ .
- the present invention provides certain prodrugs of pyrrolotriazine compounds, particularly, pyrrolotriazine aniline compounds useful as kinase inhibitors, particularly kinases p38 ⁇ and ⁇ .
- pyrrolotriazine compounds useful as tyrosine kinase inhibitors are disclosed in US patent application Serial No.
- Prodrug strategies or methodologies can be used to markedly enhance properties of a drug or to overcome an inherent deficiency in the pharmaceutical or pharmacokinetic properties of a drug.
- Prodrugs are new chemical entities which, upon administration to the patient, regenerates the parent molecule within the body.
- a myriad of prodrug strategies exist which provide choices in modulating the conditions for regeneration of the parent drug, the physical, pharmaceutic, or pharmacokinetic properties of the prodrug, and the functionality to which the prodrug modifications may be attached.
- none of the existing technologies teaches or suggests the specific prodrugs of the present disclosure. The identification of prodrugs with desired properties is often difficult and non straightforward.
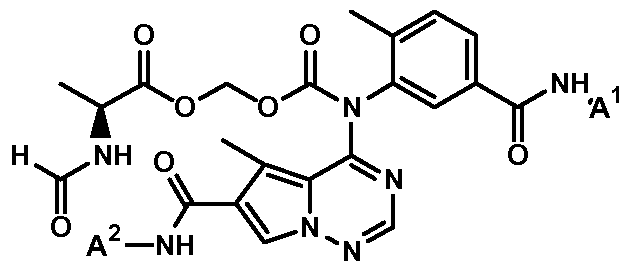
- the instant invention pertains to compounds of Formula I:
- a 1 and A 2 are each independently selected from optionally-substituted alkyl, optionally-substituted cycloalkyl, optionally-substituted aryl, optionally-substituted aralkyl, optionally-substituted heterocyclo and optionally-substituted heteroaryl;
- R is independently selected from hydrogen and optionally-substituted alkyl
- R 4 is independently selected from:
- R 6 is attached to any available carbon atom of the phenyl ring and at each occurrence is independently selected from optionally-substituted alkyl, halogen, trifluoromethoxy, trifluoromethyl, hydroxy, alkoxy, alkanoyl, alkanoyloxy, thiol, alkylthio, ureido, nitro, cyano, carboxy, carboxyalkyl, carbamyl, alkoxycarbonyl, alkylthiono, arylthiono, arylsulfonylamine, alkylsulfonylamine, sulfonic acid, alkysulfonyl, sulfonamido, phenyl, benzyl, aryloxy and benzyloxy, wherein each R 6 group in turn may be further substituted by one to two R 18 ;
- the invention further pertains to pharmaceutical compositions containing compounds of Formula I, and to methods of treating conditions associated with the activity of p38 kinase ( ⁇ and ⁇ ), comprising administering to a mammal a pharmaceutically effective amount of a compound of Formula I, including pharmaceutically acceptable salts thereof, and one or more pharmaceutically acceptable carriers, excipients or diluents.
- the present case is for a method of treating an inflammatory disorder comprising administering to a patient in need of such treatment a pharmaceutical composition as specified herein.
- the instant invention is directed to the use of a carbamate substituted pyrrolotriazine compound of Formula I as a prodrug for releasing a parent drug containing the substituted pyrrolotriazine compound after removal of the carbamate moiety in animals or humans.
- the instant invention is for a compound having the Formula II:
- R 7 is independently selected from:
- R 8 and R 9 are independently selected from optionally-substituted alkyl, optionally-substituted cycloalkyl, optionally-substituted aryl, optionally-substituted aralkyl, optionally-substituted heterocyclo and optionally-substituted heteroaryl, or R 8 and R 9 can be taken together to be optionally substituted lactam; and n is 0 or 1.
- This disclosure relates to a prodrug approach which enhances the maximum exposure and/or the ability to increase exposure multiples (i.e., multiples of drug exposure greater than EC50 or EC90) upon dose escalation of efficacious members of a previously disclosed class of p38 kinase inhibitors.
- the improvements offered by the prodrug are beneficial, for they allow drug levels in the body to be increased, which provides greater efficacy.
- the compounds of the present invention may possess asymmetric centers and, therefore, occur as mixtures of diastereomers and enantiomers, the present invention includes the individual diastereoisomeric and enantiomeric forms of the compounds of Formula I, Formula II and Formula III, in addition to the mixtures thereof.
- this invention relates to a carbamate prodrug of a pyrrolotriazine compound.
- the carbamate moiety increases the utility of the parent compounds by increasing the absorption of the compounds in the body.
- this disclosure describes prodrugs of amides which are effective at improving the oral utility of the parent molecules in the body.
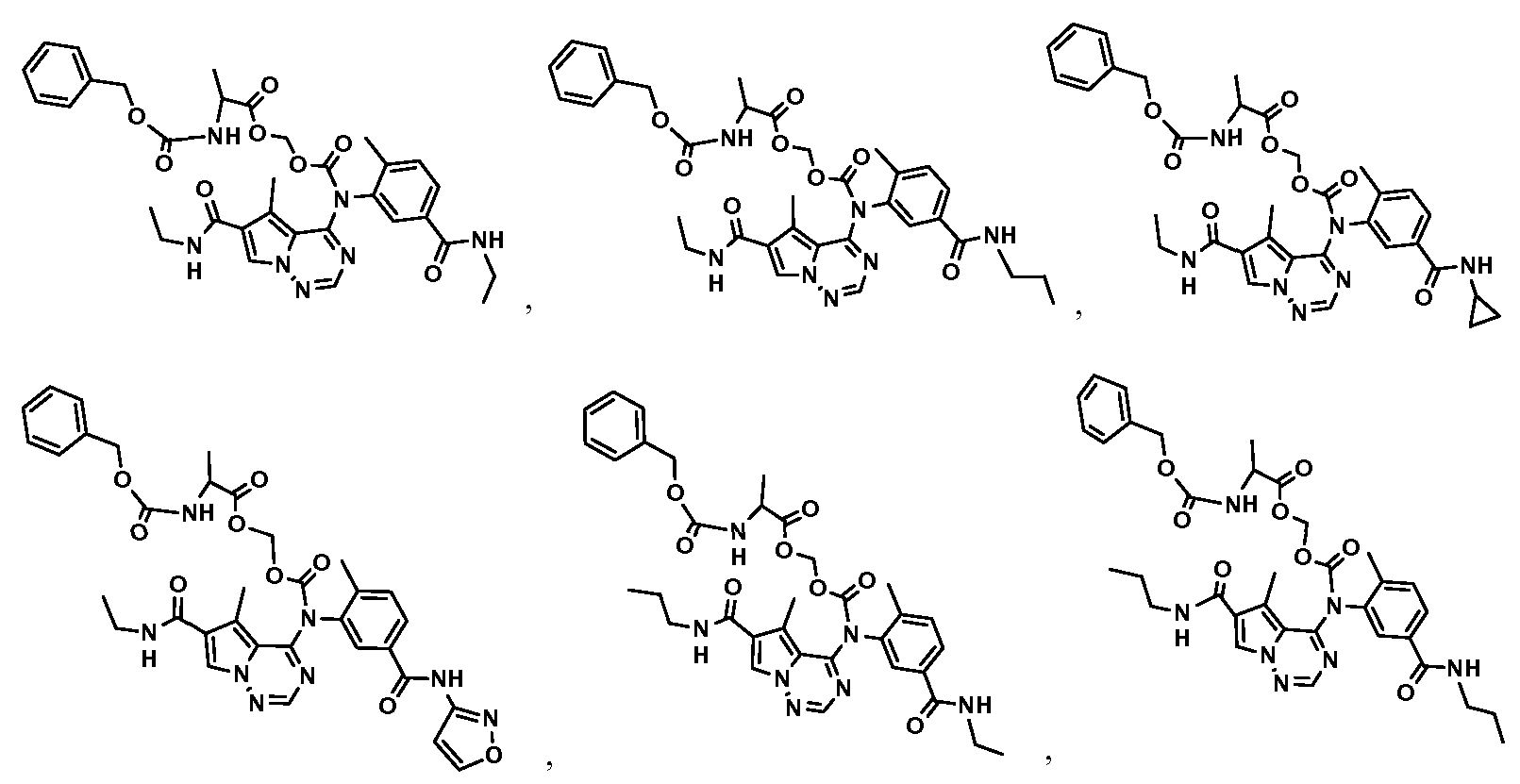
- An embodiment of the present invention is for compounds, and salts thereof, selected from the group consisting of:
- An embodiment of the present invention is for compounds, and salts thereof, selected from the group consisting of:
- [ t 001 present invention is for compounds, and salts thereof, selected from the ; g prrnonunp n cnonnsskisttiinngp o off-:
- An embodiment of the present invention is for compounds, and salts thereof, selected from the group consisting of:
- An embodiment of the present invention is for compounds, and salts thereof, selected from the group consisting of:
- An embodiment of the present invention is for compounds, and salts thereof, selected from the group consisting of:
- An embodiment of the present invention is for compounds, and salts thereof, selected from the group consisting of:
- An embodiment of the present invention is for compounds, , and salts thereof, selected from * t*h • e" g ⁇ -ro ⁇ up consisting of:
- An embodiment of the present invention is for compounds, and salts thereof, selected from the group consisting of:
- An embodiment of the present invention is for compounds, and salts thereof, selected from the group consisting of:
- An embodiment of the present invention is for compounds, and salts thereof, selected from the group consisting of:
- An embodiment of the present invention is for compounds, and salts thereof, selected from the group consisting of:
- An embodiment of the present invention is for compounds, and salts thereof, selected from the group consisting of:
- prodrug of the present disclosure more of the drug will be absorbed and reach the target, and pill burden, cost to the patient and dosing intervals could be reduced.
- the following discussion will show that the prodrugs described in this invention work surprisingly well. They release parent drug quickly and efficiently and enhance the exposure to levels which are higher than reported for many prodrugs.
- a successful prodrug strategy requires that a chemically reactive site in a molecule be modified via addition of the prodrug moiety and that later, under the desired conditions in the patients, the prodrug moiety will unmask and release parent drug.
- the prodrug molecule must have suitable stability in an acceptable dosage form prior to dosing.
- the release mechanism must allow the prodrug to regenerate parent drug efficiently and with kinetics that provide therapeutic levels of parent drug at the disease target.
- alkyl refers to straight or branched chain unsubstituted hydrocarbon groups of 1 to 20 carbon atoms, preferably 1 to 7 carbon atoms.
- lower alkyl refers to unsubstituted alkyl groups of 1 to 4 carbon atoms.
- the subscript refers to the number of carbon atoms that the group may contain.
- Co- 4 alkyl includes a bond and alkyl groups of 1 to 4 carbon atoms.
- the substituent on the alkyl optionally in turn may be further substituted, in which case it will be with substituted one or more of Ci- 4 alkyl, C 2 - 4 alkenyl, halogen, haloalkyl, haloalkoxy, cyano, nitro, amino, hydroxy, hydroxyCi_ 4 alkyl, alkoxy, alkylthio, phenyl, benzyl, phenyloxy, and/or benzyloxy.
- alkenyl refers to straight or branched chain hydrocarbon groups of 2 to 20 carbon atoms, preferably 2 to 15 carbon atoms, and most preferably 2 to 8 carbon atoms, having at least one double bond, and depending on the number of carbon atoms, up to four double bonds.
- substituted alkenyl refers to an alkenyl group substituted by one to two substituents selected from those recited above for substituted alkyl groups.
- alkynyl refers to straight or branched chain hydrocarbon groups of 2 to 20 carbon atoms, preferably 2 to 15 carbon atoms, and most preferably 2 to 8 carbon atoms, having at least one triple bond, and depending on the number of carbon atoms, up to four triple bonds.
- substituted alkynyl refers to an alkynyl group substituted by one to two substituents selected from those recited above for alkyl groups.
- alkyl is used in connection with another group, as in heterocycloalkyl or cycloalkylalkyi, this means the identified (first named) group is bonded directly through an alkyl group which may be branched or straight chain (e.g., cyclopropylCi_ 4 alkyl means a cyclopropyl group bonded through a straight or branched chain alkyl group having one to four carbon atoms.).
- substituted cycloalkylalkyi the alkyl portion of the group, besides being branched or straight chain, may be substituted as recited above for substituted alkyl groups and/or the first named group (e.g., cycloalkyl) may be substituted as recited herein for that group.
- halogen refers to fluorine, chlorine, bromine and iodine
- halo refers to fluoro, choro, bromo and iodo
- aryl refers to monocyclic or bicyclic aromatic substituted or unsubstituted hydrocarbon groups having 6 to 12 carbon atoms in the ring portion, such as phenyl, naphthyl, and biphenyl groups. Aryl groups may optionally include one to three additional rings (either cycloalkyl, heterocyclo or heteroaryl) fused thereto.
- Examples include:
- Each ring of the aryl may be optionally substituted with one to three R c groups, wherein R c at each occurrence is selected from alkyl, substituted alkyl, halogen, trifluoromethoxy, trifluoromethyl, -SR, -OR, -NRR', -NRSO 2 R', -SO 2 R, -SO 2 NRR', -CO 2 R', -C(O)R', -C(O)NRR', -OC(O)R', -OC(O)NRR', -NRC(O)R', -NRCO 2 R', phenyl, C 3 -?
- each R and R' is selected from hydrogen, alkyl, substituted alkyl, alkenyl, substituted alkenyl, phenyl, C 3 _ 7 cycloalkyl, and five-to-six membered heterocyclo or heteroaryl, except in the case of a sulfonyl group, then R is not going to be hydrogen.
- Each substituent R c optionally in turn may be further substituted by one or more (preferably O to 2) Ra groups, wherein Ra is selected from Ci_ 6 alkyl, C 2 _ 6 alkenyl, halogen, haloalkyl, haloalkoxy, cyano, nitro, amino, Ci-4alkylamino, aminoCi-4alkyl, hydroxy, hydroxyCi-4alkyl, alkoxy, alkylthio, phenyl, benzyl, phenylethyl, phenyloxy, and benzyloxy.
- Ra is selected from Ci_ 6 alkyl, C 2 _ 6 alkenyl, halogen, haloalkyl, haloalkoxy, cyano, nitro, amino, Ci-4alkylamino, aminoCi-4alkyl, hydroxy, hydroxyCi-4alkyl, alkoxy, alkylthio, phenyl, benzyl, phenylethyl, phen
- aralkyl refers to an aryl group bonded directly through an alkyl group, such as benzyl, wherein the alkyl group may be branched or straight chain.
- alkyl group such as benzyl
- substituted aralkyl the alkyl portion of the group besides being branched or straight chain, may be substituted as recited above for substituted alkyl groups and/or the aryl portion may be substituted as recited herein for aryl.
- each R group may be hydrogen or may also be selected from R c as defined above, in turn optionally substituted with one or more Ra. At least two of these "R" groups should be hydrogen and preferably at least five of the "R” groups is hydrogen.
- a preferred benzyl group involves the alkyl-portion being branched to define
- heteroaryl refers to a substituted or unsubstituted aromatic group for example, which is a 4 to 7 membered monocyclic, 7 to 11 membered bicyclic, or 10 to 15 membered tricyclic ring system, which has at least one heteroatom and at least one carbon atom-containing ring.
- Each ring of the heteroaryl group containing a heteroatom can contain one or two oxygen or sulfur atoms and/or from one to four nitrogen atoms, provided that the total number of heteroatoms in each ring is four or less and each ring has at least one carbon atom.
- the fused rings completing the bicyclic and tricyclic groups may contain only carbon atoms and may be saturated, partially saturated, or unsaturated.
- the nitrogen and sulfur atoms may optionally be oxidized and the nitrogen atoms may optionally be quaternized.
- Heteroaryl groups which are bicyclic or tricyclic must include at least one fully aromatic ring but the other fused ring or rings may be aromatic or non-aromatic.
- the heteroaryl group may be attached at any available nitrogen or carbon atom of any ring. It may optionally be substituted with one to three (preferably 0 to 2) R c groups, as defined above for aryl, which in turn may be substituted with one or more (preferably o to 2) Ra groups, also as recited above.
- Exemplary monocyclic heteroaryl groups include pyrrolyl, pyrazolyl,
- pyrazolinyl imidazolyl, oxazolyl, isoxazolyl, thiazolyl ⁇ i.e., N ), thiadiazolyl, isothiazolyl, furanyl, thienyl, oxadiazolyl, pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, triazinyl and the like.
- Exemplary bicyclic heteroaryl groups include indolyl, benzothiazolyl, benzodioxolyl, benzoxaxolyl, benzothienyl, quinolinyl, tetrahydroisoquinolinyl, isoquinolinyl, benzimidazolyl, benzopyranyl, indolizinyl, benzofuranyl, chromonyl, coumarinyl, benzopyranyl, cinnolinyl, quinoxalinyl, indazolyl, pyrrolopyridyl, furopyridinyl, dihydroisoindolyl, tetrahydroquinolinyl and the like.
- exemplary tricyclic heteroaryl groups include carbazolyl, benzidolyl, phenanthrollinyl, acridinyl, phenanthridinyl, xanthenyl and the like.
- cycloalkyl refers to a saturated or partially unsaturated non- aromatic cyclic hydrocarbon ring system, preferably containing 1 to 3 rings and 3 to 7 carbon atoms per ring, which may be substituted or unsubstituted and/or which may be fused with a C3-C7 carbocylic ring, a heterocyclic ring, or which may have a bridge of 3 to 4 carbon atoms.
- cycloalkyl groups including any available carbon or nitrogen atoms on any fused or bridged rings optionally may have 0 to 3 (preferably 0-2) substituents selected from R c groups, as recited above, and/or from keto (where appropriate) which in turn may be substituted with one to three Ra groups, also as recited above.
- a carbon-carbon bridge may be optionally substituted
- the carbon atoms in the bridged ring optionally may be substituted with an R c group, which preferably is seleted from Ci- 4 alkyl, C 2 - 4 alkenyl, halogen, haloalkyl, haloalkoxy, cyano, amino, hydroxy, and Exemplary cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, bicycloheptane, cycloctyl, cyclodecyl, cyclododecyl, and adamantyl.
- heterocycle each refer to a fully saturated or partially unsaturated nonaromatic cyclic group, which may be substituted or unsubstituted, for example, which is a 4 to 7 membered monocyclic, 7 to 11 membered bicyclic, or 10 to 15 membered tricyclic ring system, which has at least one heteroatom in at least one carbon atom-containing ring.
- Each ring of the heterocyclic group containing a heteroatom may have 1 , 2 or 3 heteroatoms selected from nitrogen, oxygen, and sulfur atoms, where the nitrogen and sulfur heteroatoms also optionally may be oxidized and the nitrogen heteroatoms also optionally may be quaternized.
- heterocyclic group may be attached at any nitrogen or carbon atom.
- Exemplary monocyclic heterocyclic groups include pyrrolidinyl, pyrrolyl, indolyl, pyrazolyl, oxetanyl, pyrazolinyl, imidazolyl, imidazolinyl, imidazolidinyl, oxazolyl, oxazolidinyl, isoxazolinyl, isoxazolyl, thiazolyl, thiadiazolyl, thiazolidinyl, isothiazolyl, isothiazolidinyl, furyl, tetrahydrofuryl, thienyl, oxadiazolyl, piperidinyl, piperazinyl, 2-oxopiperazinyl, 2-oxopiperidinyl, 2-oxopyrrolidinyl, 2-oxazepinyl, azepinyl, 4-piperidonyl, pyridyl, N-oxo-pyridyl, N
- Exemplary bicyclic hetrocyclic groups include 2,3-dihydro-2-oxo-lH- indolyl, benzothiazolyl, benzoxazolyl, benzothienyl, quinuclidinyl, quinolinyl, quinolinyl-N-oxide, tetrahydroisoquinolinyl, isoquinolinyl, benzimidazolyl, benzopyranyl, indolizinyl, benzofuryl, chromonyl, coumarinyl, cinnolinyl, quinoxalinyl, indazolyl, pyrrolopyridyl, furopyridinyl (such as furo[2,3-c]pyridinyl, furo[3,l-b]pyridinyl] or furo[2,3-b]pyridinyl), dihydroisoindolyl, dihydroquinazolinyl (such as 3,4-
- heterocyclos such as epoxides and aziridines.
- aryl e.g., phenyl
- cycloalkyl e.g., cyclohexyl
- heterocyclo e.g., pyrrolidinyl
- heteroaryl e.g., indolyl
- the reference is intended to include rings having 0 to 3, preferably 0-2, substituents selected from those recited above for the the aryl, cycloalkyl, heterocyclo and/or heteroaryl groups, as appropriate.
- isoquinoline refers to isoquinoline and tetrahydroisoquinoline.
- heteroatoms shall include oxygen, sulfur and nitrogen.
- haloalkyl means an alkyl having one or more halo substituents.
- perfluoromethyl means a methyl group substituted by one, two, or three fluoro atoms, i.e., CH 2 F, CHF 2 and CF3.
- perfluoroalkyl means an alkyl group having from one to five fluoro atoms, such as pentafluoroethyl.
- haloalkoxy means an alkoxy group having one or more halo substituents. For example, “haloalkoxy” includes -OCF3.
- carbocyclic means a saturated or unsaturated monocyclic or bicyclic ring in which all atoms of all rings are carbon. Thus, the term includes cycloalkyl and aryl rings. The carbocyclic ring may be substituted in which case the substituents are selected from those recited above for cycloalkyl and aryl groups.
- unsaturated is used herein to refer to a ring or group, the ring or group may be fully unsaturated or partially unsaturated.
- alkoxy is -OR e
- aryloxy is -OAr
- amino is -NH 2
- alkylamino is -NHR e or -N(R e ) 2
- arylamino is -NHAr or -NR 6 Ar
- aralkylamino is -NH-R f -Ar
- thiol is thiol
- NH arylsulfonylamine is -NHSO( q )Ar
- guanidino is — N — C — NH 2 '
- ureido is
- R e is alkyl or substituted alkyl as defined above
- R f is H alkylene or substituted alkylene as defined above
- R s and R are selected from alkyl, substituted alkyl, aryl, aralkyl, cycloalkyl, heterocyclo, and heteraryl
- Ar is an aryl as defined above
- q is 2 or 3.
- the compounds of the present invention may form salts with alkali metal hydroxides such as sodium hydroxide, potassium hydroxide and lithium hydroxide; with alkaline earth metal hydroxides such as calcium hydroxide and magnesium hydroxide; with organic bases such as triethanolamine, tributylamine and pyridine triethanolamine; and with amino acids such as arginine, lysine and the like.
- alkali metal hydroxides such as sodium hydroxide, potassium hydroxide and lithium hydroxide
- alkaline earth metal hydroxides such as calcium hydroxide and magnesium hydroxide
- organic bases such as triethanolamine, tributylamine and pyridine triethanolamine
- amino acids such as arginine, lysine and the like.
- Such salts can be formed as known to those skilled in the art.
- the compounds of the present invention may form salts with a variety of organic and inorganic acids and bases.
- Such salts include those formed with hydrogen chloride, hydrogen bromide, methanesulfonic acid, sulfuric acid, acetic acid, trifluoroacetic acid, oxalic acid, maleic acid, benzenesulfonic acid, toluenesulfonic acid and various others (e.g., nitrates, phosphates, borates, tartrates, citrates, succinates, benzoates, ascorbates, salicylates and the like).
- Such salts can be formed as known to those skilled in the art. Salt forms of the compounds may be advantageous for improving the compound dissolution rate and oral bioavailability.
- zwitterions inner salts
- All stereoisomers of the compounds of the instant invention are contemplated, either in a mixture or in pure or substantially pure form.
- the definition of compounds according to the invention embraces all the possible stereoisomers and their mixtures. It embraces the racemic forms and the isolated optical isomers having the specified activity.
- the racemic forms can be resolved by physical methods, such as, for example, fractional crystallization, separation or crystallization of diastereomeric derivatives or separation by chiral column chromatography.
- the individual optical isomers can be obtained from the racemates from the conventional methods, such as, for example, salt formation with an optically active acid followed by crystallization.
- prodrugs are well known in the art.
- prodrug derivatives see: a) Design of Prodrugs, edited by H. Bundgaard, (Elsevier, 1985) and Methods in Enzymology, Vol. 112, pp. 309-396, edited by K. Widder, et al.
- solvates e.g., hydrates
- Methods of solvation are generally known in the art.
- the compounds of the invention are prodrugs that release selective inhibitors of p38 kinase activity, and in particular, isoforms p38 ⁇ and p38 ⁇ . Accordingly, compounds of formula (I) have utility in treating conditions associated with p38 kinase activity. Such conditions include diseases in which cytokine levels are modulated as a consequence of intracellular signaling via p38, and in particular, diseases that are associated with an overproduction of cytokines IL-I, IL-4, IL-8, and TNF- ⁇ .
- the terms “treating” or “treatment” encompass either or both responsive and prophylaxis measures, e.g., measures designed to inhibit or delay the onset of the disease or disorder, achieve a full or partial reduction of the symptoms or disease state, and/or to alleviate, ameliorate, lessen, or cure the disease or disorder and/or its symptoms.
- responsive and prophylaxis measures e.g., measures designed to inhibit or delay the onset of the disease or disorder, achieve a full or partial reduction of the symptoms or disease state, and/or to alleviate, ameliorate, lessen, or cure the disease or disorder and/or its symptoms.
- inhibition of p-38 ⁇ kinase this means that either p38 ⁇ and/or p38 ⁇ kinase are inhibited.
- reference to an IC50 value for inhibiting p-38 ⁇ / ⁇ kinase means that the compound has such effectiveness for inhibiting at least one of, or both of, p38 ⁇ and p38 ⁇ kinases.
- the compounds of Formula (I) are useful in treating p-38 associated conditions including, but not limited to, inflammatory diseases, autoimmune diseases, destructive bone disorders, proliferative disorders, angiogenic disorders, infectious diseases, neurodegenerative diseases, and viral diseases.
- the specific conditions or diseases that may be treated with the inventive compounds include, without limitation, pancreatitis (acute or chronic), asthma, allergies, adult respiratory distress syndrome, chronic obstructive pulmonary disease, glomerulonephritis, rheumatoid arthritis, systemic lupus erythematosis, scleroderma, chronic thyroiditis, Graves' disease, autoimmune gastritis, diabetes, autoimmune hemolytic anemia, autoimmune neutropenia, thrombocytopenia, atopic dermatitis, chronic active hepatitis, myasthenia gravis, multiple sclerosis, inflammatory bowel disease, ulcerative colitis, Crohn's disease, psoriasis, graft vs.
- p38 inhibitors of this invention inhibit the expression of inducible pro-inflammatory proteins such as prostaglandin endoperoxide synthase-2 (PGHS-2), also referred to as cyclooxygenase-2 (COX-2).
- PGHS-2 prostaglandin endoperoxide synthase-2
- COX-2 cyclooxygenase-2
- additional p38-associated conditions include edema, analgesia, fever and pain, such as neuromuscular pain, headache, pain caused by cancer, dental pain and arthritis pain.
- inventive compounds also may be used to treat veterinary viral infections, such as lentivirus infections, including, but not limited to equine infectious anemia virus; or retro virus infections, including feline immunodeficiency virus, bovine immunodeficiency virus, and canine immunodeficiency virus.
- p38 associated condition or "p38 associated disease or disorder” are used herein, each is intended to encompass all of the conditions identified above as if repeated at length, as well as any other condition that is affected by p38 kinase activity.
- the present invention thus provides methods for treating such conditions, comprising administering to a subject in need thereof an effective amount of at least one compound of Formula (I) or a salt thereof.
- the methods of treating p38 kinase- associated conditions may comprise administering compounds of Formula (I) alone or in combination with each other and/or other suitable therapeutic agents useful in treating such conditions.
- Such other therapeutic agents include corticosteroids, rolipram, calphostin, CSAIDs, 4-substituted imidazo [1,2- A]quinoxalines as disclosed in U.S. Pat. No. 4,200,750; Interleukin-10, glucocorticoids, salicylates, nitric oxide, and other immunosuppressants; nuclear translocation inhibitors, such as deoxyspergualin (DSG); non-steroidal antiinflammatory drugs (NSAIDs) such as ibuprofen, celecoxib and rofecoxib; steroids such as prednisone or dexamethasone; antiviral agents such as abacavir; antiproliferative agents such as methotrexate, leflunomide, FK506 (tacrolimus, Prograf); cytotoxic drugs such as azathiprine and cyclophosphamide; TNF- ⁇ inhibitors such as tenidap, anti-TNF antibodies or soluble TNF-
- the above other therapeutic agents when employed in combination with the compounds of the present invention, may be used, for example, in those amounts indicated in the Physicians' Desk Reference (PDR) or as otherwise determined by one of ordinary skill in the art. In the methods of the present invention, such other therapeutic agent(s) may be administered prior to, simultaneously with, or following the administration of the inventive compounds.
- the present invention also provides pharmaceutical compositions capable of treating p38-kinase associated conditions, including TNF- ⁇ , IL-I, and/or IL-8 mediated conditions, as described above.
- inventive compositions may contain other therapeutic agents as described above and may be formulated, for example, by employing conventional solid or liquid vehicles or diluents, as well as pharmaceutical additives of a type appropriate to the mode of desired administration (e.g., excipients, binders, preservatives, stabilizers, flavors, etc.) according to techniques such as those well known in the art of pharmaceutical formulation.
- pharmaceutical additives e.g., excipients, binders, preservatives, stabilizers, flavors, etc.
- the compounds of Formula (I) may be administered by any means suitable for the condition to be treated, which may depend on the need for site- specific treatment or quantity of drug to be delivered. Topical administration is generally preferred for skin-related diseases, and systematic treatment preferred for cancerous or pre-cancerous conditions, although other modes of delivery are contemplated.
- the compounds may be delivered orally, such as in the form of tablets, capsules, granules, powders, or liquid formulations including syrups; topically, such as in the form of solutions, suspensions, gels or ointments; sublingually; bucally; parenterally, such as by subcutaneous, intravenous, intramuscular or intrasternal injection or infusion techniques (e.g., as sterile injectable aq.
- compositions for topical administration include a topical carrier such as PLASTIBASE ® (mineral oil gelled with polyethylene).
- compositions for oral administration include suspensions which may contain, for example, microcrystalline cellulose for imparting bulk, alginic acid or sodium alginate as a suspending agent, methylcellulose as a viscosity enhancer, and sweeteners or flavoring agents such as those known in the art; and immediate release tablets which may contain, for example, microcrystalline cellulose, dicalcium phosphate, starch, magnesium stearate and/or lactose and/or other excipients, binders, extenders, disintegrants, diluents and lubricants such as those known in the art.
- the inventive compounds may also be orally delivered by sublingual and/or buccal administration, e.g., with molded, compressed, or freeze- dried tablets.
- compositions may include fast-dissolving diluents such as mannitol, lactose, sucrose, and/or cyclodextrins.
- fast-dissolving diluents such as mannitol, lactose, sucrose, and/or cyclodextrins.
- high molecular weight excipients such as celluloses (AVICEL ® ) or polyethylene glycols (PEG); an excipient to aid mucosal adhesion such as hydroxypropyl cellulose (HPC), hydroxypropyl methyl cellulose (HPMC), sodium carboxymethyl cellulose (SCMC), and/or maleic anhydride copolymer (e.g.,
- compositions for nasal aerosol or inhalation administration include solutions which may contain, for example, benzyl alcohol or other suitable preservatives, absorption promoters to enhance absorption and/or bioavailability, and/or other solubilizing or dispersing agents such as those known in the art.
- compositions for parenteral administration include injectable solutions or suspensions which may contain, for example, suitable non-toxic, parenterally acceptable diluents or solvents, such as mannitol, 1,3-butanediol, water, Ringer's solution, an isotonic sodium chloride solution, or other suitable dispersing or wetting and suspending agents, including synthetic mono- or diglycerides, and fatty acids, including oleic acid.
- suitable non-toxic, parenterally acceptable diluents or solvents such as mannitol, 1,3-butanediol, water, Ringer's solution, an isotonic sodium chloride solution, or other suitable dispersing or wetting and suspending agents, including synthetic mono- or diglycerides, and fatty acids, including oleic acid.
- compositions for rectal administration include suppositories which may contain, for example, suitable non-irritating excipients, such as cocoa butter, synthetic glyceride esters or polyethylene glycols, which are solid at ordinary temperatures but liquefy and/or dissolve in the rectal cavity to release the drug.
- suitable non-irritating excipients such as cocoa butter, synthetic glyceride esters or polyethylene glycols, which are solid at ordinary temperatures but liquefy and/or dissolve in the rectal cavity to release the drug.
- the effective amount of a compound of the present invention may be determined by one of ordinary skill in the art, and includes exemplary dosage amounts for a mammal of from about 0.05 to 100 mg/kg of body weight of active compound per day, which may be administered in a single dose or in the form of individual divided doses, such as from 1 to 4 times per day.
- the specific dose level and frequency of dosage for any particular subject may be varied and will depend upon a variety of factors, including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the species, age, body weight, general health, sex and diet of the subject, the mode and time of administration, rate of excretion, drug combination, and severity of the particular condition.
- Preferred subjects for treatment include animals, most preferably mammalian species such as humans, and domestic animals such as dogs, cats, horses, and the like.
- this term is intended to include all subjects, most preferably mammalian species, that are affected by mediation of p38 enzyme levels.
- p38 Kinases [0087] cDNAs of human p38 ⁇ , ⁇ and ⁇ isozymes were cloned by PCR. These cDNAs were subcloned in the pGEX expression vector (Pharmacia). GST-p38 fusion protein was expressed in E. CoIi and purified from bacterial pellets by affinity chromatography using glutathione agarose. p38 fusion protein was activated by incubating with constitutively active MKK6. Active p38 was separated from MKK6 by affinity chromatography. Constitutively active MKK6 was generated according to Raingeaud et al. [MoL Cell. Biol., 1247-1255 (1996)].
- PBMCs Peripheral blood mononuclear cells
- assay medium RPMI medium containing 10% fetal bovine serum
- 50 ul of cell suspension was incubated with 50 ul of test compound (4X concentration in assay medium containing 0.2% DMSO) in 96-well tissue culture plates for 5 minutes at RT.
- TNF- ⁇ concentration in the medium was quantified using a standard ELISA kit (Pharmingen-San Diego, CA). Concentrations of TNF- ⁇ and IC50 values for test compounds (concentration of compound that inhibited LPS-stimulated TNF- ⁇ production by 50%) were calculated by linear regression analysis.
- the assays were performed in V-bottomed 96-well plates. The final assay volume was 60 ⁇ prepared from three 20 ⁇ additions of enzyme, substrates (MBP and ATP) and test compounds in assay buffer (50 mM Tris pH 7.5, 10 mM MgCl 2 , 50 mM NaCl and 1 mM DTT). Bacterially expressed, activated p38 was pre-incubated with test compounds for 10 min. prior to initiation of reaction with substrates. The reaction was incubated at 25°C for 45 min. and terminated by adding 5 ⁇ of 0.5 M EDTA to each sample.
- assay buffer 50 mM Tris pH 7.5, 10 mM MgCl 2 , 50 mM NaCl and 1 mM DTT.
- Bacterially expressed, activated p38 was pre-incubated with test compounds for 10 min. prior to initiation of reaction with substrates. The reaction was incubated at 25°C for 45 min. and terminated by adding
- the reaction mixture was aspirated onto a pre-wet f ⁇ ltermat using a Skatron Micro96 Cell Harvester (Skatron, Inc.), then washed with PBS.
- the filtermat was then dried in a microwave oven for 1 min., treated with MeltilLex A scintillation wax (Wallac), and counted on a Microbeta scintillation counter Model 1450 (Wallac). Inhibition data were analyzed by nonlinear least-squares regression using Prizm (GraphPadSoftware).
- the final concentration of reagents in the assays are ATP, 1 /M; [ ⁇ - 33 P]ATP, 3 nM; MBP (Sigma, #M1891), 2 ⁇ g/well; p38, 10 nM; and DMSO, 0.3%.
- LPS lipopolysaccharide
- mice were sedated by C ⁇ 2 : ⁇ 2 inhalation and a blood sample was obtained. Serum was separated and analyzed for TNF-alpha concentrations by commercial ELISA assay per the manufacturer's instructions (R&D Systems, Minneapolis, MN).
- Test compounds were administered orally at various times before LPS injection. The compounds were dosed either as suspensions or as solutions in various vehicles or solubilizing agents.
- Boc tert-butyloxycarbonyl
- Bz benzyl
- DIPEA diisopropylethylamine
- EDC or EDCI l-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride
- HATU O-(7-Azabenzotriazol-l-yl-N,N,N',N'-tetramethyluronim hexafluorophosphate
- HOBt 1-hydroxybenzotriazole hydrate
- K 2 CO 3 potassium carbonate
- KOH potassium hydroxide
- LC/MS high performance liquid chromatography/mass spectrometry
- m-CPBA m-chloroperbenzoic acid
- NaH sodium hydride
- NaOH sodium hydroxide
- TFA trifluoroacetic acid
- THF tetrahydrofuran
- TLC thin layer chromatography
- Compounds of Formula I may generally be prepared according to the following schemes and the knowledge of one skilled in the art, and/or the methods described in US patent applications Serial Nos. 10/036,293 and/or 09/573,829, incorporated herein by reference.
- the groups A 1 and A 2 are as described herein for compounds of Formula (I), and said groups are also further exemplified in the Examples described hereinbelow.
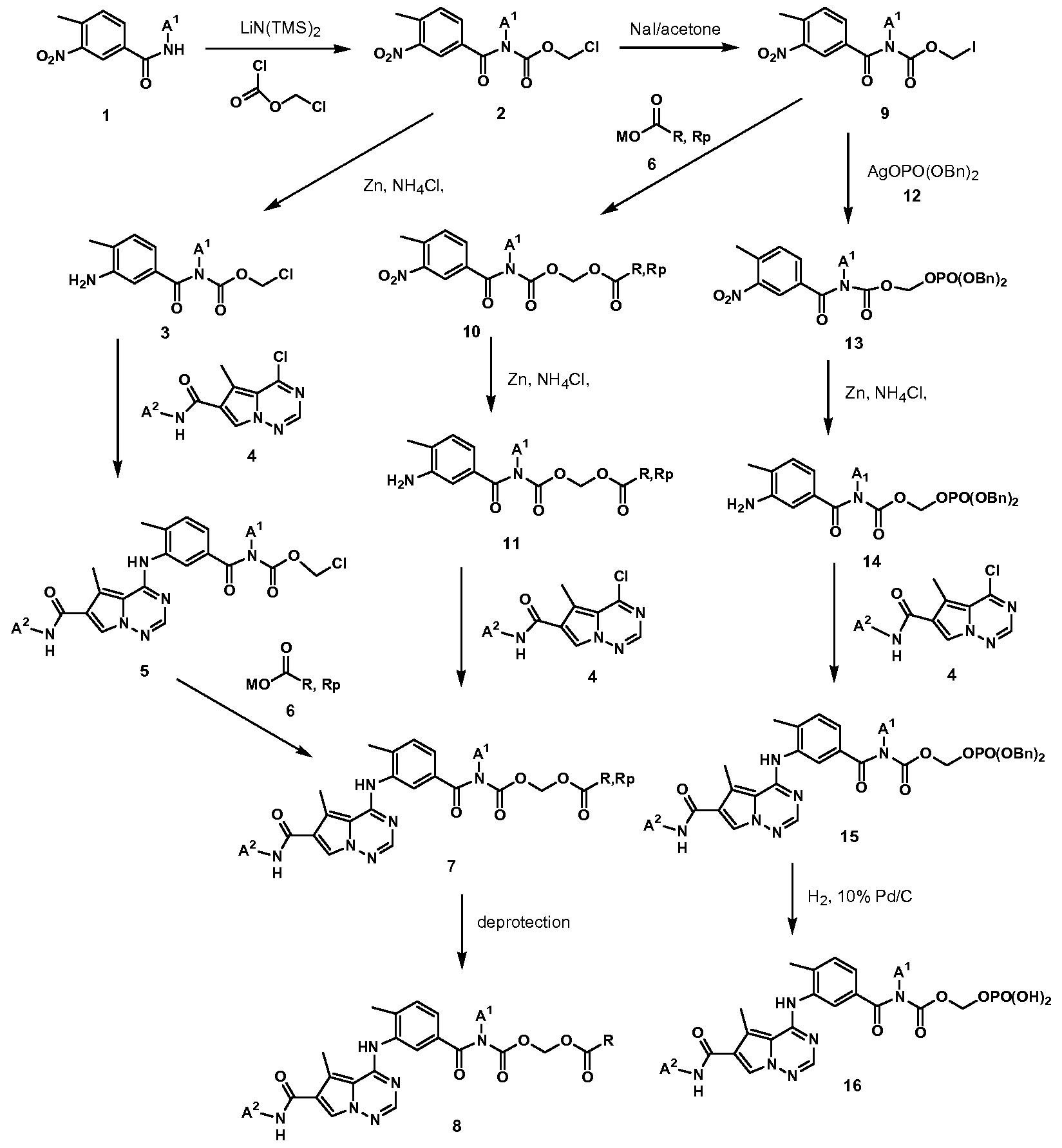
- Prodrug 7 and 8 can also be prepared via an alternative route where 0-chloromethyl carbamate 2 is converted into O-iodomethyl carbamate 9, followed by treatment with carboxylate 6, nitro reduction, and reaction with 4.
- treatment of 9 with the silver salt of dibenzyl hydrogen phosphate 12 supplies phosphate 13.
- Reduction of the nitro into amino (14) and subsequent reaction with 4 gives compound 15, which is hydrogenated to prodrug 16.
- Scheme 2 illustrates the preparation of the carbamoyl prodrugs where the carbamoyl is built on the pyrrolo[l,2-fJ[l,2,4]triazine-6-carboxamido nitrogen.
- Scheme 3 shows the preparation of the carbamoyl prodrugs where the carbamoyl is built on the anilino nitrogen. Reaction of the parent drug 17 with chloro formic acid chloromethyl ester gives carbamate 24. The rest of the transformations are similar to those in Scheme 1 and 2.
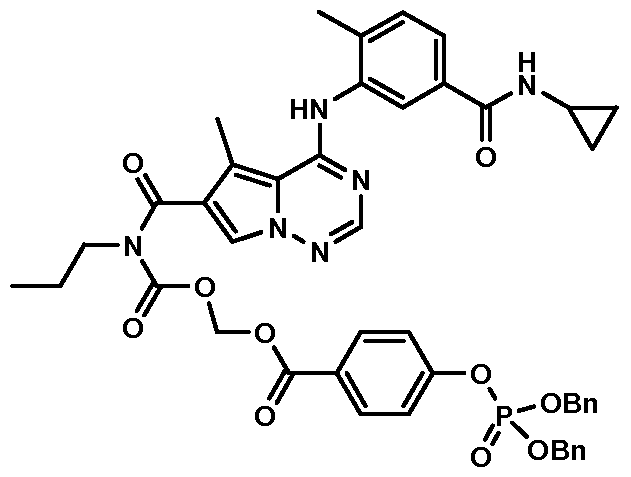
- step 2 in Ex. 14 (0.300 g, 0.501 mmol) and 2-(4- (benzyloxy)phenyl)acetic acid silver salt (0.350 g, 1.00 mmol) in toluene (25 mL) was heated at reflux for 16hr. Additional 2-(4-(benzyloxy)phenyl)acetic acid silver salt (0.200 g, 0.573 mmol) was added, and the mixture was continued to be heated for additional 24 hr. The insoluble material was removed by suction filtration through Celite ® 545. The filtrate was concentrated under vacuum to dryness.
Abstract
Description





















































































































































Claims



Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2007223342A AU2007223342A1 (en) | 2006-03-07 | 2007-03-05 | Pyrrolotriazine aniline prodrug compounds useful as kinase inhibitors |
| CA002645031A CA2645031A1 (en) | 2006-03-07 | 2007-03-05 | Pyrrolotriazine aniline prodrug compounds useful as kinase inhibitors |
| BRPI0708644-0A BRPI0708644A2 (en) | 2006-03-07 | 2007-03-05 | pyrrolotriazine aniline prodrug compounds useful as kinase inhibitors |
| MX2008011136A MX2008011136A (en) | 2006-03-07 | 2007-03-05 | Pyrrolotriazine aniline prodrug compounds useful as kinase inhibitors. |
| JP2008558486A JP2009535295A (en) | 2006-03-07 | 2007-03-05 | Pyrrolotriazine aniline prodrug compounds useful as kinase inhibitors |
| EA200801945A EA200801945A1 (en) | 2006-03-07 | 2007-03-05 | PYRROLOTRIAZINANILINUM MEDICAL AND MEDICINAL COMPOUNDS, USEFUL AS KINASE INHIBITORS |
| EP07757860A EP2001886A2 (en) | 2006-03-07 | 2007-03-05 | Pyrrolotriazine aniline prodrug compounds useful as kinase inhibitors |
| IL193653A IL193653A0 (en) | 2006-03-07 | 2008-08-24 | Pyrrolotriazine aniline prodrug compounds useful as kinase inhibitors |
| NO20083717A NO20083717L (en) | 2006-03-07 | 2008-08-29 | Pyrrolotriazine-aniline prodrugs useful as kinase inhibitors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US77985106P | 2006-03-07 | 2006-03-07 | |
| US60/779,851 | 2006-03-07 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007103839A2 true WO2007103839A2 (en) | 2007-09-13 |
| WO2007103839A3 WO2007103839A3 (en) | 2008-06-05 |
Family
ID=38475749
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/063250 WO2007103839A2 (en) | 2006-03-07 | 2007-03-05 | Pyrrolotriazine aniline prodrug compounds useful as kinase inhibitors |
Country Status (17)
| Country | Link |
|---|---|
| US (2) | US7572795B2 (en) |
| EP (1) | EP2001886A2 (en) |
| JP (1) | JP2009535295A (en) |
| KR (1) | KR20080107408A (en) |
| CN (1) | CN101395158A (en) |
| AR (1) | AR059778A1 (en) |
| AU (1) | AU2007223342A1 (en) |
| BR (1) | BRPI0708644A2 (en) |
| CA (1) | CA2645031A1 (en) |
| EA (1) | EA200801945A1 (en) |
| IL (1) | IL193653A0 (en) |
| MX (1) | MX2008011136A (en) |
| NO (1) | NO20083717L (en) |
| PE (1) | PE20080139A1 (en) |
| TW (1) | TW200804390A (en) |
| WO (1) | WO2007103839A2 (en) |
| ZA (1) | ZA200807459B (en) |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2011038572A1 (en) * | 2009-09-29 | 2011-04-07 | Glaxo Group Limited | Novel compounds |
| CN102596921A (en) * | 2009-09-29 | 2012-07-18 | 葛兰素集团有限公司 | Novel compounds |
| US8471005B2 (en) | 2008-12-19 | 2013-06-25 | Cephalon, Inc. | Pyrrolotriazines as ALK and JAK2 inhibitors |
| US8791257B2 (en) | 2010-03-31 | 2014-07-29 | Bristol-Myers Squibb Company | Substituted pyrrolotriazines as protein kinase inhibitors |
| US9050345B2 (en) | 2013-03-11 | 2015-06-09 | Bristol-Myers Squibb Company | Pyrrolotriazines as potassium ion channel inhibitors |
| WO2016091825A1 (en) * | 2014-12-09 | 2016-06-16 | Bayer Pharma Aktiengesellschaft | Compounds for the treatment of cancer |
| US10342786B2 (en) | 2017-10-05 | 2019-07-09 | Fulcrum Therapeutics, Inc. | P38 kinase inhibitors reduce DUX4 and downstream gene expression for the treatment of FSHD |
| US11254695B2 (en) | 2016-04-04 | 2022-02-22 | Chemocentryx, Inc. | Soluble C5aR antagonists |
| US11291659B2 (en) | 2017-10-05 | 2022-04-05 | Fulcrum Therapeutics, Inc. | P38 kinase inhibitors reduce DUX4 and downstream gene expression for the treatment of FSHD |
Families Citing this family (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2289894A3 (en) * | 2002-04-23 | 2011-07-20 | Bristol-Myers Squibb Company | Pyrrolo-triazine compounds useful as kinase inhibitors |
| KR20080107408A (en) * | 2006-03-07 | 2008-12-10 | 브리스톨-마이어스 스큅 컴퍼니 | Pyrrolotriazine aniline prodrug compounds useful as kinase inhibitors |
| WO2009158450A1 (en) * | 2008-06-25 | 2009-12-30 | Bristol-Myers Squibb Company | Crystalline forms of ((((4-((5-(cyclopropylcarbamoyl)-2-methylphenyl)amino)-5-methylpyrrolo[2,1-f][1,2,4]triazin-6-yl)carbonyl)(propyl)carbamoyl)oxy)methyl (4-(phosphonooxy)phenyl)acetate, preparation and use thereof |
| WO2009158446A2 (en) * | 2008-06-25 | 2009-12-30 | Bristol-Myers Squibb Company | Crystalline forms of ((((4-((5-(cyclopropylcarbamoyl)-2-methylphenyl)amino)-5-methylpyrrolo[2,1-f][1,2,4]triazin-6-yl)carbonyl)(propyl)carbamoyl)oxy)methyl (4-(phosphonooxy)phenyl)acetate, method of preparation and use thereof |
| ES2477884T3 (en) | 2008-08-19 | 2014-07-18 | Xenoport, Inc. | Methyl hydrogen hydrogen fumarate prodrugs, pharmaceutical compositions thereof and methods of use |
| US8686009B2 (en) | 2009-06-25 | 2014-04-01 | Alkermes Pharma Ireland Limited | Prodrugs of NH-acidic compounds |
| WO2012031057A1 (en) | 2010-09-01 | 2012-03-08 | Bristol-Myers Squibb Company | Bms- 582949 for the treatment of resistant rheumatic disease |
| CN102153558B (en) * | 2011-02-23 | 2012-11-21 | 扬州永济医药新技术有限公司 | Derivative of multi-target antitumor inhibitor 2-aminopyrrole-triazine and synthesis method thereof |
| US8901305B2 (en) * | 2012-07-31 | 2014-12-02 | Bristol-Myers Squibb Company | Aryl lactam kinase inhibitors |
| WO2014031897A1 (en) | 2012-08-22 | 2014-02-27 | Xenoport, Inc. | Oral dosage forms having a high loading of (n,n- diethylcarbamoyl)methyl methyl|(2e)but-2-ene-1,4-dioate |
| WO2014160633A1 (en) | 2013-03-24 | 2014-10-02 | Xenoport, Inc. | Pharmaceutical compositions of dimethyl fumarate |
| WO2014197860A1 (en) | 2013-06-07 | 2014-12-11 | Xenoport, Inc. | Method of making monomethyl fumarate |
| US9421182B2 (en) | 2013-06-21 | 2016-08-23 | Xenoport, Inc. | Cocrystals of dimethyl fumarate |
| TW201516020A (en) | 2013-09-06 | 2015-05-01 | Xenoport Inc | Crystalline forms of (N,N-diethylcarbamoyl)methyl methyl (2E)but-2-ene-1,4-dioate, methods of synthesis and use |
| US9999672B2 (en) | 2014-03-24 | 2018-06-19 | Xenoport, Inc. | Pharmaceutical compositions of fumaric acid esters |
| WO2017070135A1 (en) * | 2015-10-20 | 2017-04-27 | Bristol-Myers Squibb Company | Prodrugs of 2-(4-(3-((4-amino-7-cyano-imidazo[2,1-f][1,2,4]triazin-2-yl)amino)phenyl)piperaz in-1-yl)propanamide derivatives as ck2 inhibitors for the treatment of cancer |
| EP3761983A1 (en) | 2018-03-05 | 2021-01-13 | Alkermes Pharma Ireland Limited | Aripiprazole dosing strategy |
| CN110590839B (en) * | 2018-06-13 | 2022-04-05 | 四川海思科制药有限公司 | Levatinib derivative and preparation method and application thereof |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3629301A (en) | 1969-10-24 | 1971-12-21 | Du Pont | 3 3-difluoro-2-substituted steroids and their preparation |
| US4200750A (en) | 1977-01-07 | 1980-04-29 | Westwood Pharmaceuticals Inc. | 4-Substituted imidazo [1,2-a]quinoxalines |
| WO2001014378A1 (en) | 1999-08-23 | 2001-03-01 | Shionogi & Co., Ltd. | PYRROLOTRIAZINE DERIVATIVES HAVING sPLA2-INHIBITORY ACTIVITIES |
Family Cites Families (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5658903A (en) * | 1995-06-07 | 1997-08-19 | Smithkline Beecham Corporation | Imidazole compounds, compositions and use |
| NZ327044A (en) * | 1996-01-11 | 2000-01-28 | Smithkline Beecham Corp | Substituted imidazole compounds |
| US5945418A (en) * | 1996-12-18 | 1999-08-31 | Vertex Pharmaceuticals Incorporated | Inhibitors of p38 |
| US6147080A (en) * | 1996-12-18 | 2000-11-14 | Vertex Pharmaceuticals Incorporated | Inhibitors of p38 |
| US6087496A (en) * | 1998-05-22 | 2000-07-11 | G. D. Searle & Co. | Substituted pyrazoles suitable as p38 kinase inhibitors |
| WO1998052937A2 (en) * | 1997-05-22 | 1998-11-26 | G.D. Searle And Co. | 4-aryl-3(5)-heteroaryl substituted pyrazoles as p38 kinase |
| WO1999001452A1 (en) * | 1997-07-02 | 1999-01-14 | Smithkline Beecham Corporation | Novel cycloalkyl substituted imidazoles |
| US6130235A (en) * | 1998-05-22 | 2000-10-10 | Scios Inc. | Compounds and methods to treat cardiac failure and other disorders |
| AU772477B2 (en) | 1998-08-28 | 2004-04-29 | Scios Inc. | Inhibitors of p38-alpha kinase |
| US6184226B1 (en) | 1998-08-28 | 2001-02-06 | Scios Inc. | Quinazoline derivatives as inhibitors of P-38 α |
| GB9906566D0 (en) | 1999-03-23 | 1999-05-19 | Zeneca Ltd | Chemical compounds |
| US6982265B1 (en) * | 1999-05-21 | 2006-01-03 | Bristol Myers Squibb Company | Pyrrolotriazine inhibitors of kinases |
| GB9924092D0 (en) | 1999-10-13 | 1999-12-15 | Zeneca Ltd | Pyrimidine derivatives |
| NZ512957A (en) | 1999-11-10 | 2005-01-28 | Ortho Mcneil Pharm Inc | Substituted 2-aryl-3-(heteroaryl)-imidazo[1,2-a] pyrimidines and their use in inhibiting in vitro secretion of TNF-alpha and IL-1beta to treat neurodegenerative diseases |
| AU1304801A (en) | 1999-11-15 | 2001-05-30 | Shionogi & Co., Ltd. | Tricyclic azaindolizine derivatives having an SPLA2- inhibiting effect |
| US6376548B1 (en) | 2000-01-28 | 2002-04-23 | Rohm And Haas Company | Enhanced propertied pesticides |
| US6670357B2 (en) * | 2000-11-17 | 2003-12-30 | Bristol-Myers Squibb Company | Methods of treating p38 kinase-associated conditions and pyrrolotriazine compounds useful as kinase inhibitors |
| EP2289894A3 (en) | 2002-04-23 | 2011-07-20 | Bristol-Myers Squibb Company | Pyrrolo-triazine compounds useful as kinase inhibitors |
| WO2003091229A1 (en) | 2002-04-23 | 2003-11-06 | Bristol-Myers Squibb Company | Aryl ketone pyrrolo-triazine compounds useful as kinase inhibitors |
| US7388009B2 (en) * | 2002-04-23 | 2008-06-17 | Bristol-Myers Squibb Company | Heteroaryl-substituted pyrrolo-triazine compounds useful as kinase inhibitors |
| TWI329112B (en) | 2002-07-19 | 2010-08-21 | Bristol Myers Squibb Co | Novel inhibitors of kinases |
| US6951859B2 (en) | 2002-08-02 | 2005-10-04 | Bristol-Myers Squibb Company | Pyrrolotriazine kinase inhibitors |
| TW200420565A (en) | 2002-12-13 | 2004-10-16 | Bristol Myers Squibb Co | C-6 modified indazolylpyrrolotriazines |
| MXPA05008183A (en) * | 2003-02-05 | 2005-10-05 | Bristol Myers Squibb Co | Process for preparing pyrrolotriazine kinase inhibitors. |
| US7265140B2 (en) | 2003-09-23 | 2007-09-04 | Pfizer Inc | Acyloxymethylcarbamate prodrugs of oxazolidinones |
| WO2007138381A2 (en) | 2005-10-14 | 2007-12-06 | Targanta Therapeutics Inc. | Phosphonated oxazolidinones and uses thereof for the prevention and treatment of bone and joint infections |
| KR20080107408A (en) * | 2006-03-07 | 2008-12-10 | 브리스톨-마이어스 스큅 컴퍼니 | Pyrrolotriazine aniline prodrug compounds useful as kinase inhibitors |
-
2007
- 2007-03-05 KR KR1020087021851A patent/KR20080107408A/en not_active Application Discontinuation
- 2007-03-05 BR BRPI0708644-0A patent/BRPI0708644A2/en not_active IP Right Cessation
- 2007-03-05 MX MX2008011136A patent/MX2008011136A/en not_active Application Discontinuation
- 2007-03-05 JP JP2008558486A patent/JP2009535295A/en active Pending
- 2007-03-05 AU AU2007223342A patent/AU2007223342A1/en not_active Abandoned
- 2007-03-05 CN CNA2007800079168A patent/CN101395158A/en active Pending
- 2007-03-05 WO PCT/US2007/063250 patent/WO2007103839A2/en active Application Filing
- 2007-03-05 CA CA002645031A patent/CA2645031A1/en not_active Abandoned
- 2007-03-05 EA EA200801945A patent/EA200801945A1/en unknown
- 2007-03-05 EP EP07757860A patent/EP2001886A2/en not_active Withdrawn
- 2007-03-06 PE PE2007000242A patent/PE20080139A1/en not_active Application Discontinuation
- 2007-03-06 US US11/682,331 patent/US7572795B2/en active Active
- 2007-03-07 AR ARP070100944A patent/AR059778A1/en unknown
- 2007-03-07 TW TW096107889A patent/TW200804390A/en unknown
-
2008
- 2008-08-24 IL IL193653A patent/IL193653A0/en unknown
- 2008-08-29 NO NO20083717A patent/NO20083717L/en not_active Application Discontinuation
- 2008-08-29 ZA ZA200807459A patent/ZA200807459B/en unknown
-
2009
- 2009-02-12 US US12/370,081 patent/US20090156555A1/en not_active Abandoned
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3629301A (en) | 1969-10-24 | 1971-12-21 | Du Pont | 3 3-difluoro-2-substituted steroids and their preparation |
| US4200750A (en) | 1977-01-07 | 1980-04-29 | Westwood Pharmaceuticals Inc. | 4-Substituted imidazo [1,2-a]quinoxalines |
| WO2001014378A1 (en) | 1999-08-23 | 2001-03-01 | Shionogi & Co., Ltd. | PYRROLOTRIAZINE DERIVATIVES HAVING sPLA2-INHIBITORY ACTIVITIES |
Non-Patent Citations (8)
| Title |
|---|
| "Design of Prodrugs", 1985, ELSEVIER |
| "Methods in Enzymology", vol. 112, 1985, ACADEMIC PRESS, pages: 309 - 396 |
| H. BUNDGAARD, ADVANCED DRUG DELIVERY REVIEWS, vol. 8, 1992, pages 1 - 38 |
| H. BUNDGAARD: "A Textbook of Drug Design and Development", 1991, article "Design and Application of Prodrugs", pages: 113 - 191 |
| HENRY ET AL., DRUGS FUT., vol. 24, 1999, pages 1345 - 1354 |
| MORELAND ET AL., ANN. INTERN. MED., vol. 130, 1999, pages 478 - 486 |
| RANKIN ET AL., BR. .1 RHEUMATOL., vol. 34, 1995, pages 334 - 342 |
| SALITURO ET AL., CURR. MED. CHEM., vol. 6, 1999, pages 807 - 823 |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8471005B2 (en) | 2008-12-19 | 2013-06-25 | Cephalon, Inc. | Pyrrolotriazines as ALK and JAK2 inhibitors |
| CN102596921B (en) * | 2009-09-29 | 2015-04-29 | 葛兰素集团有限公司 | Novel compounds |
| EA021437B1 (en) * | 2009-09-29 | 2015-06-30 | Глэксо Груп Лимитед | Compounds inhibiting lrrk2 kinase activity |
| CN102596921A (en) * | 2009-09-29 | 2012-07-18 | 葛兰素集团有限公司 | Novel compounds |
| JP2013505969A (en) * | 2009-09-29 | 2013-02-21 | グラクソ グループ リミテッド | New compounds |
| CN104292154A (en) * | 2009-09-29 | 2015-01-21 | 葛兰素集团有限公司 | Novel compounds |
| WO2011038572A1 (en) * | 2009-09-29 | 2011-04-07 | Glaxo Group Limited | Novel compounds |
| US8791257B2 (en) | 2010-03-31 | 2014-07-29 | Bristol-Myers Squibb Company | Substituted pyrrolotriazines as protein kinase inhibitors |
| US9050345B2 (en) | 2013-03-11 | 2015-06-09 | Bristol-Myers Squibb Company | Pyrrolotriazines as potassium ion channel inhibitors |
| WO2016091825A1 (en) * | 2014-12-09 | 2016-06-16 | Bayer Pharma Aktiengesellschaft | Compounds for the treatment of cancer |
| US11254695B2 (en) | 2016-04-04 | 2022-02-22 | Chemocentryx, Inc. | Soluble C5aR antagonists |
| US10342786B2 (en) | 2017-10-05 | 2019-07-09 | Fulcrum Therapeutics, Inc. | P38 kinase inhibitors reduce DUX4 and downstream gene expression for the treatment of FSHD |
| US10537560B2 (en) | 2017-10-05 | 2020-01-21 | Fulcrum Therapeutics. Inc. | P38 kinase inhibitors reduce DUX4 and downstream gene expression for the treatment of FSHD |
| US11291659B2 (en) | 2017-10-05 | 2022-04-05 | Fulcrum Therapeutics, Inc. | P38 kinase inhibitors reduce DUX4 and downstream gene expression for the treatment of FSHD |
| US11479770B2 (en) | 2017-10-05 | 2022-10-25 | Fulcrum Therapeutics, Inc. | Use of p38 inhibitors to reduce expression of DUX4 |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2007223342A1 (en) | 2007-09-13 |
| KR20080107408A (en) | 2008-12-10 |
| JP2009535295A (en) | 2009-10-01 |
| NO20083717L (en) | 2008-10-06 |
| IL193653A0 (en) | 2009-08-03 |
| MX2008011136A (en) | 2008-09-08 |
| US7572795B2 (en) | 2009-08-11 |
| TW200804390A (en) | 2008-01-16 |
| EP2001886A2 (en) | 2008-12-17 |
| US20090156555A1 (en) | 2009-06-18 |
| PE20080139A1 (en) | 2008-03-10 |
| AR059778A1 (en) | 2008-04-30 |
| EA200801945A1 (en) | 2009-02-27 |
| ZA200807459B (en) | 2009-11-25 |
| BRPI0708644A2 (en) | 2011-06-07 |
| CN101395158A (en) | 2009-03-25 |
| CA2645031A1 (en) | 2007-09-13 |
| US20070213300A1 (en) | 2007-09-13 |
| WO2007103839A3 (en) | 2008-06-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2007103839A2 (en) | Pyrrolotriazine aniline prodrug compounds useful as kinase inhibitors | |
| EP1363910B1 (en) | Methods of treating p38 kinase-associated conditions and pyrrolotriazine compounds useful as kinase inhibitors | |
| EP1497019B1 (en) | Pyrrolo-triazine aniline compounds useful as kinase inhibitors | |
| US6670357B2 (en) | Methods of treating p38 kinase-associated conditions and pyrrolotriazine compounds useful as kinase inhibitors | |
| EP1503996B1 (en) | Aryl ketone pyrrolo-triazine compounds useful as kinase inhibitors | |
| US7419978B2 (en) | Phenyl-aniline substituted bicyclic compounds useful as kinase inhibitors | |
| AU2002232760A1 (en) | Methods of treating p38 kinase-associated conditions and pyrrolotriazine compounds useful as kinase inhibitors | |
| EP2307411B1 (en) | Triazolopyridine compounds useful as kinase inhibitors | |
| US7388009B2 (en) | Heteroaryl-substituted pyrrolo-triazine compounds useful as kinase inhibitors | |
| ZA200303786B (en) | Methods of treating p38 kinase-associated conditions and pyrrolotriazine compounds useful as kinase inhibitors. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 193653 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 7337/DELNP/2008 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 570885 Country of ref document: NZ Ref document number: MX/a/2008/011136 Country of ref document: MX Ref document number: 2007223342 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 08093116 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200780007916.8 Country of ref document: CN Ref document number: 2645031 Country of ref document: CA Ref document number: 2008558486 Country of ref document: JP Ref document number: 1020087021851 Country of ref document: KR |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2007223342 Country of ref document: AU Date of ref document: 20070305 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007757860 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200801945 Country of ref document: EA |
|
| ENP | Entry into the national phase |
Ref document number: PI0708644 Country of ref document: BR Kind code of ref document: A2 Effective date: 20080908 |