New primary amines
The present invention was made as a result of activities undertaken within the scope of a research collaboration agreement between Merck & Co., Inc., Actelion Pharmaceuticals Ltd, and Actelion Ltd. The agreement was executed on December 4, 2003.
The invention relates to novel compounds of the formula (I). The invention also concerns related aspects including processes for the preparation of the compounds, pharmaceutical compositions containing one or more compounds of formula (I) and especially their use as renin inhibitors in cardiovascular events and renal insufficiency.
In the renin-angiotensin system (RAS) the biologically active angiotensin II (Ang II) is generated by a two-step mechanism. The highly specific enzyme renin cleaves angiotensinogen to angiotensin I (Ang I), which is then further processed to Ang II by the less specific angiotensin-converting enzyme (ACE). Ang II is known to work on at least two receptor subtypes called ATi and AT2. Whereas ATi seems to transmit most of the known functions of Ang II, the role of AT2 is still unknown.
Modulation of the RAS represents a major advance in the treatment of cardiovascular diseases. ACE inhibitors and ATi blockers have been accepted to treat hypertension (Waeber B. et al., "The renin-angiotensin system: role in experimental and human hypertension", in Birkenhager W. H., Reid J. L. (eds): Hypertension, Amsterdam, Elsevier Science Publishing Co, 1986, 489-519; Weber M. A., Am. J. Hypertens., 1992, 5, 247S). In addition, ACE inhibitors are used for renal protection (Rosenberg M. E. et al., Kidney International, 1994, 45, 403; Breyer J. A. et al, Kidney International, 1994, 45, S156), in the prevention of congestive heart failure (Vaughan D. E. et al., Cardiovasc. Res., 1994, 28, 159; Fouad-Tarazi F. et al., Am. J. Med., 1988, 84 (Suppl. 3A), 83) and myocardial infarction (Pfeffer M. A. et al., N. Engl. J. Med., 1992, 327, 669).
The rationale to develop renin inhibitors is the specificity of renin (Kleinert H. D., Cardiovasc. Drugs, 1995, 9, 645). The only substrate known for renin is angiotensinogen,
which can only be processed (under physiological conditions) by renin. In contrast, ACE can also cleave bradykinin besides Ang I and can be by-passed by chymase, a serine protease (Husain A., J. Hypertens., 1993, 11, 1155). In patients inhibition of ACE thus leads to bradykinin accumulation causing cough (5-20%) and potentially life-threatening angioneurotic edema (0.1-0.2%) (Israili Z. H. et al., Annals of Internal Medicine, 1992, 117, 234). ACE inhibitors do not inhibit Chymase. Therefore, the formation of Ang II is still possible in patients treated with ACE inhibitors. Blockade of the ATi receptor (e.g. by losartan) on the other hand overexposes other AT -receptor subtypes (e.g. AT2) to Ang II, whose concentration is significantly increased by the blockade of ATi receptors. In summary, renin inhibitors are expected to demonstrate a different pharmaceutical profile than ACE inhibitors and ATi blockers with regard to efficacy in blocking the RAS and in safety aspects.
Only limited clinical experience (Azizi M. et al., J. Hypertens., 1994, 12, 419; Neutel J. M. et ah, Am. Heart, 1991, 122, 1094) has been created with renin inhibitors because of their insufficient oral activity due to their peptidomimetic character (Kleinert H. D., Cardiovasc. Drugs, 1995, 9, 645). The clinical development of several compounds has been stopped because of this problem together with the high cost of goods. Only one compound containing four chiral centers has entered clinical trials (Rahuel J. et al, Chem. Biol, 2000, 7, 493; Mealy N. E., Drugs of the Future, 2001, 26, 1139). Thus, renin inhibitors with good oral bioavailability and long duration of action are required. Recently, the first non-peptide renin inhibitors were described which show high in vitro activity (Oefner C. et al., Chem. Biol, 1999, 6, 127; Patent Application WO 97/09311; Marki H. P. et al., 11 Farmaco, 2001, 56, 21). However, the development status of these compounds is not known.
The present invention relates to renin inhibitors of a non-peptidic nature and of low molecular weight. Described are orally active renin inhibitors of formula (I) which have a long duration of action and which are active in indications beyond blood pressure regulation where the tissular renin-chymase system may be activated leading to pathophysiologically altered local functions such as renal, cardiac and vascular remodelling, atherosclerosis, and possibly restenosis. So, the present invention describes these non-peptidic renin inhibitors of formula (I).
In particular, the present invention relates to novel compounds of the formula (I)
Formula (I)
wherein
X represents CH, N, or N+-O";
W represents a/?αra-substituted pyridinyl or a thiazolyl, such as especially:
V represents -CH
2CH
2CH
2-, -CH
2CH
2-A-, -CH
2-A-CH
2-, -A-CH
2CH
2-, -CH
2CH
2CH
2CH
2- , -A-CH
2CH
2CH
2-, -CH
2-A-CH
2CH
2-, -CH
2CH
2-A-CH
2-, -CH
2CH
2CH
2-A-, -A-CH
2CH
2- B- (preferred), -CH
2CH
2CH
2CH
2CH
2-, -A-CH
2CH
2CH
2CH
2-, -CH
2-A-CH
2CH
2CH
2-, -CH
2CH
2-A-CH
2CH
2-, -CH
2CH
2CH
2-A-CH
2-, -CH
2CH
2CH
2CH
2-A-, -A-CH
2CH
2CH
2-B-, -CH
2-A-CH
2CH
2-B-, -A-CH
2CH
2-B-CH
2-, -A-CH
2CH
2CH
2-B-CH
2-, -CH
2-A- CH
2CH
2CH
2-B-, -0-CH
2-Q-, wherein Q is bound to the group U of formula (I), or preferably a pyrrolidinyl of the formula:
U represents unsubstituted aryl, especially phenyl; mono-, di-, tri- or tetra-substituted aryl (especially mono- di-, tri-, or tetra-substituted phenyl), wherein the substituents are independently selected from the group consisting of Ci_7-alkyl (such as especially methyl), -CF3, halogen, and hydroxy-Ci_7-alkyl (such as especially CH3CH(OH)-); or a five- membered heteroaryl with two heteroatoms independently selected from nitrogen, oxygen and sulphur (preferably pyrazolyl or isoxazolyl), wherein said heteroaryl radical is optionally mono-, di- or tri-substituted, wherein the substitutents are independently selected from the group consisting of Ci_7-alkyl, Ci_7-alkoxy, -CF3, -OCF3, and halogen;
Q represents a fϊve-membered heteroaryl with two or three heteroatoms independently selected from O and N;
A and B represent independently from each others -O- or -S-, especially -O-;
R1 represents Ci_7-alkyl or cycloalkyl, preferably cycloalkyl such as especially cyclopropyl;
R2 represents halogen or Ci_7-alkyl, preferably chloro or methyl;
R represents hydrogen, halogen (such as especially chloro), Ci_7-alkyl (such as especially methyl), Ci_7-alkoxy, or -CF3; and
R4 represents hydrogen; Ci_7-alkyl-0-(CH2)o-4-CH2-, such as especially CH3-O-(CH2)lj2- CH2-; CF3-O-(CH2V4-CH2-; R'2N-(CH2)o.4-CH2-, wherein R' is independently selected from the group consisting of hydrogen, Ci_7-alkyl (optionally but preferably substituted by one to three fluorine), cyclopropyl (optionally substituted by one to three fluorine), cyclopropyl-Ci_7-alkyl (optionally but preferably substituted by one to three fluorine), and -C(=O)-R" wherein R" is C1-4-alkyl, Ci_4-alkoxy, -CF3, -CH2-CF3, or cyclopropyl; or R5- C(=0)-(0)o-i-(CH2)o_4-, wherein R5 is Ci_4-alkyl, Ci_4-alkoxy, or cyclopropyl; wherein R' and R' ' preferably do not both simultaneously represent hydrogen;
and salts thereof.
The general terms used hereinbefore and hereinafter preferably have, within this disclosure, the following meanings, unless otherwise indicated:
Where the plural form is used for compounds, salts, pharmaceutical compositions, diseases and the like, this is intended to mean also a single compound, salt, or the like.
Any reference to a compound of formula (I) is to be understood as referring also to salts (especially pharmaceutically acceptable salts) of a compound of formula (I), as appropriate and expedient.
The term Ci-7-alkyl, alone or in combination with other groups, means saturated, straight or branched chain groups with one to seven carbon atoms, preferably one to four carbon atoms, i.e. Ci_4-alkyl. Examples of Ci_7-alkyl groups are methyl, ethyl, n-propyl, iso- propyl, n-butyl, iso-butyl, sec-butyl, tert-butyl, pentyl, hexyl and heptyl. The methyl, ethyl and isopropyl groups are preferred, especially the methyl and ethyl groups.
The term Ci-7-alkoxy, alone or in combination with other groups, refers to an R-O- group, wherein R is a Ci_7-alkyl group. Examples of Ci_7-alkoxy groups are methoxy, ethoxy, propoxy, iso-propoxy, iso-butoxy, sec-butoxy and tert-butoxy.
The term hydroxy-Ci-7-alkyl, alone or in combination with other groups, refers to an HO- R- group, wherein R is a Ci_7-alkyl group. Examples of hydroxy-Ci_7-alkyl groups are HO-CH2-, HO-CH2CH2-, HO-CH2CH2CH2- and CH3CH(OH)-.
The term halogen means fluorine, chlorine, bromine or iodine, preferably fluorine, chlorine or bromine. In a more preferred embodiment of the invention the term halogen means fluorine or chlorine.
The term cycloalkyl, alone or in combination with other groups, means a saturated cyclic hydrocarbon ring system with 3 to 7 carbon atoms, i.e. cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl or cycloheptyl, preferably cyclopropyl.
The term aryl, alone or in combination, refers to a phenyl, naphthyl or indanyl group, preferably a phenyl group.
The expression pharmaceutically acceptable salts encompasses either salts with inorganic acids or organic acids like hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, sulfamic acid, phosphoric acid, nitric acid, phosphorous acid, nitrous acid, citric acid, formic acid, acetic acid, oxalic acid, maleic acid, lactic acid, tartaric acid, fumaric acid, benzoic acid, mandelic acid, cinnamic acid, palmoic acid, stearic acid, glutamic acid, aspartic acid, methanesulfonic acid, ethanesulfonic acid, ethanedisulfonic acid, /?-toluenesulfonic acid, salicylic acid, succinic acid, trifluoroacetic acid, and the like that are non toxic to living organisms or in case the compound of formula (I) is acidic in nature with an inorganic base like an alkali or earth alkali base, e.g. sodium hydroxide, potassium hydroxide, calcium hydroxide and the like. For other examples of pharmaceutically acceptable salts, reference can be made to "Salt selection for basic drugs", Int. J. Pharm. (1986), 33, 201-217.
The compounds of the formula (I) may contain asymmetric carbon atoms. Substituents at a double bond or a ring may be present in cis- (= Z-) or trans (= E-) form unless indicated otherwise. The compounds of formula (I) may thus be present as mixtures of stereoisomers or preferably as pure stereoisomers. Mixtures of stereoisomers may be separated in a manner known per se, e.g. by column chromatography, thin layer chromatography, HPLC or crystallization.
Compounds of the invention also include nitrosated compounds of formula (I) that have been nitrosated through one or more sites such as oxygen (hydroxyl condensation), sulfur
(sulfydryl condensation) and/or nitrogen. The nitrosated compounds of the present invention can be prepared using conventional methods known to one skilled in the art. For example, known methods for nitrosating compounds are described in U.S. Pat. Nos.
5,380,758, 5,703,073, 5,994,294, 6,242,432 and 6,218,417; WO 98/19672; and Oae et al, Org. Prep. Proc. Int., 15(3): 165-198 (1983).
A preferred embodiment of the present invention relates to a compound of formula (I), wherein
X respresents CH or N; and
R4 represents hydrogen; Ci_7-alkyl-0-(CH2)o-4-CH2-; CF3-O-(CH2V4-CH2-; or R'2N- (CH2)o-4-CH2-, wherein R' is independently selected from the group consisting of
hydrogen, Ci_7-alkyl (optionally substituted by one to three fluorine), cyclopropyl (optionally substituted by one to three fluorine), cyclopropyl-Ci_7-alkyl (optionally substituted by one to three fluorine), and -C(=O)-R" wherein R" is Ci_4-alkyl, -CF3, -CH2- CF3, or cyclopropyl.
A preferred embodiment of the present invention relates to a compound of formula (I), wherein X represents CH or N+-O".
A preferred embodiment of the present invention relates to a compound of formula (I), wherein A and B both represent -O- .
A preferred embodiment of the present invention relates to a compound of formula (I), wherein R1 represents cyclopropyl.
A preferred embodiment of the present invention relates to a compound of formula (I), wherein W represents
V
A preferred embodiment of the present invention relates to a compound of formula (I), wherein V represents -0-CH2CH2-O-, -CH2-CH2-O- wherein the -CH2 part of -CH2-CH2- O- is bound to the group W of formula (I), -0-CH2-Q-, or
A preferred embodiment of the present invention relates to a compound of formula (I), wherein V represents -0-CH2CH2-O- or -0-CH2-Q-.
A preferred embodiment of the present invention relates to a compound of formula (I), wherein V-W represents:
A preferred embodiment of the present invention relates to a compound of formula (I), wherein U represents
A preferred embodiment of the present invention relates to a compound of formula (I), wherein U represents
A preferred embodiment of the present invention relates to a compound of formula (I), wherein Q represents an isoxazolyl or an oxadiazolyl.
A preferred embodiment of the present invention relates to a compound of formula (I), wherein Q represents an isoxazolyl, especially an isoxazolyl that is connected to the rest of the molecule of formula (I) as follows:
A preferred embodiment of the present invention relates to a compound of formula (I), wherein R
2 represents Cl, and R
3 represents hydrogen.
A preferred embodiment of the present invention relates to a compound of formula (I), wherein R4 represents CH3-O-(CH2)2_3- or CH3-C(=O)-NH-CH2-CH2-.
A preferred embodiment of the present invention relates to a compound of formula (I), wherein R4 represents -CH2CH2CH2-O-CH3 or -CH2CH2-O-CH3.
A preferred embodiment of the present invention relates to a compound of formula (I), wherein R4 represents -CH2CH2-O-CH3.
A preferred embodiment of the present invention relates to a compound of formula (I), wherein the moiety
represents one of the following possibilities:
In an especially preferred embodiment, the present invention relates to a compound of formula (I), wherein X represents CH or N;
W represents a/?αra-substituted pyridinyl or a thiazolyl; V represents -0-CH
2CH
2-O- or a pyrrolidinyl of the formula:
U represents di-, tri-, or tetra-substituted phenyl, wherein the substituents are independently selected from the group consisting of C1-7-alkyl, halogen and hydroxy-Ci_7-alkyl; R1 represents cyclopropyl; R2 represents halogen or Ci_7-alkyl;
R represents hydrogen, halogen, or Ci_7-alkyl; and R4 represents hydrogen or Ci_7-alkyl-0-(CH2)o-4-CH2-.
The present invention also relates to compounds of formula (I) wherein the meanings of one or more of the substituents and symbols as defined for formula (I), or a preferred embodiment of formula (I), are replaced by their preferred meanings as defined herein, such as those defined for the above-given preferred embodiments.
A very preferred embodiment of the present invention relates to a compound of formula (I) selected from the group consisting of:
(i?)-3-amino-Λ/-cyclopropyl-2-{2-[2-(2,6-dichloro-4-methyl-phenoxy)-ethoxy]-thiazol-5- ylmethyl}-Λ/-(2,3-dimethyl-benzyl)-propionamide,
(i?)-3-amino-2-{2-[2-(2-chloro-3,6-difluoro-phenoxy)-ethoxy]-thiazol-5-ylmethyl}-Λ/- cyclopropyl-Λ/-(2,3-dimethyl-benzyl)-propionamide, (i?)-3-amino-Λ/-cyclopropyl-2-{2-[2-(2,6-dichloro-phenoxy)-ethoxy]-thiazol-5-ylmethyl}- N-(2,3-dimethyl-benzyl)-propionamide,
(i?)-3-amino-Λ/-cyclopropyl-2-{2-[2-(2,6-dichloro-3,4-dimethyl-phenoxy)-ethoxy]-thiazol- 5-ylmethyl}-Λ/-(2,3-dimethyl-benzyl)-propionamide,
(i?)-3-amino-2-{2-[2-(2-chloro-6-fluoro-3-methyl-phenoxy)-ethoxy]-thiazol-5-ylmethyl}- Λ/-cyclopropyl-Λ/-(2,3-dimethyl-benzyl)-propionamide,
(i?)-3-amino-iV-cyclopropyl-2-(2- {2-[2,6-dichloro-4-(l -hydroxy-ethyl)-phenoxy]-ethoxy} - thiazol-5-ylmethyl)-Λ/-(2,3-dimethyl-benzyl)-propionamide,
(i?)-3-amino-2-{2-[2-(3-chloro-2,6-difluoro-phenoxy)-ethoxy]-thiazol-5-ylmethyl}-Λ/- cyclopropyl-Λ/-(2,3-dimethyl-benzyl)-propionamide,
(i?)-3-amino-Λ/-cyclopropyl-2-{2-[2-(2,6-dichloro-4-fluoro-phenoxy)-ethoxy]-thiazol-5- ylmethyl}-Λ/-(2,3-dimethyl-benzyl)-propionamide, (i?)-2-aminomethyl-N-cyclopropyl-N-(2,3-dichloro-benzyl)-3-{2-[(i?)-3-(2,6-dichloro-4- methyl-phenoxy)-pyrrolidin- 1 -yl]-thiazol-5-yl} -propionamide,
(i?)-2-aminomethyl-N-cyclopropyl-N-(2,3-dichloro-benzyl)-3-{2-[(i?)-3-(2,6-dichloro- phenoxy)-pyrrolidin- 1 -yl] -thiazol-5 -yl} -propionamide,
(i?)-2-aminomethyl-N-cyclopropyl-N-(2,3-dichloro-benzyl)-3-{2-[(i?)-3-(2,6-dichloro-3,4- dimethyl-phenoxy)-pyrrolidin- 1 -yl] -thiazol-5 -yl} -propionamide,
(i?)-2-aminomethyl-3-{2-[(i?)-3-(2-chloro-6-fluoro-3-methyl-phenoxy)-pyrrolidin-l-yl]- thiazol-5-yl}-Λ/-cyclopropyl-Λ/-(2,3-dichloro-benzyl)-propionamide,
(i?)-2-aminomethyl-N-cyclopropyl-N-(2,3-dichloro-benzyl)-3-(2-{(i?)-3-[(i?)-2,6-dichloro- 4-(l -hydroxy-ethyl)-phenoxy]-pyrrolidin- 1 -yl} -thiazol-5 -yl)-propionamide, (i?)-2-aminomethyl-N-cyclopropyl-N-(2,3-dichloro-benzyl)-3-(2-{(i?)-3-[(5)-2,6-dichloro- 4-(l -hydroxy-ethyl)-phenoxy]-pyrrolidin- 1 -yl} -thiazol-5 -yl)-propionamide,
(i?)-2-aminomethyl-3- {2-[(i?)-3-(3-chloro-2,6-difluoro-phenoxy)-pyrrolidin- 1 -yl]-thiazol- 5-yl}-Λ/-cyclopropyl-Λ/-(2,3-dichloro-benzyl)-propionamide,
(i?)-2-aminomethyl-N-cyclopropyl-N-(2,3-dichloro-benzyl)-3-{2-[(i?)-3-(2,6-dichloro-4- fluoro-phenoxy)-pyrrolidin- 1 -yl]-thiazol-5-yl} -propionamide,
(i?)-2-aminomethyl-Λ/-[2-chloro-5-(3-methoxy-propyl)-benzyl]-N-cyclopropyl-3-{6-[(i?)-3- (2,6-dichloro-4-methyl-phenoxy)-pyrrolidin- 1 -yl]-pyridin-3-yl} -propionamide, and
(i?)-2-aminomethyl-Λ/-[5-chloro-2-(3-methoxy-propyl)-pyridin-4-ylmethyl]-Λ/-cyclopropyl- 3- {6-[(i?)-3-(2,6-dichloro-4-methyl-phenoxy)-pyrrolidin- 1 -yl]-pyridin-3-yl} -propionamide.
A further very preferred embodiment of the present invention relates to a compound of formula (I) selected from the group consisting of:
(i?)-2-aminomethyl-Λ/-[2-chloro-5-(2-methoxy-ethyl)-benzyl]-Λ/-cyclopropyl-3-{6-[2-(2,6- dichloro-4-methyl-phenoxy)-ethoxy]-pyridin-3-yl}-propionamide,
(i?)-2-aminomethyl-Λ/-[2-chloro-5-(3-methoxy-propyl)-benzyl]-N-cyclopropyl-3-{6-[2- (2,6-dichloro-4-methyl-phenoxy)-ethoxy]-pyridin-3-yl}-propionamide, (i?)-2-aminomethyl-Λ/-[2-chloro-5-(2-methoxy-ethyl)-benzyl]-Λ/-cyclopropyl-3-{6-[(R)-3- (2,6-dichloro-4-methyl-phenoxy)-pyrrolidin- 1 -yl]-pyridin-3-yl} -propionamide,
(i?)-2-aminomethyl-Λ/-[5-chloro-2-(3-methoxy-propyl)-pyridin-4-ylmethyl]-Λ/-cyclopropyl- 3- {6-[2-(2,6-dichloro-4-methyl-phenoxy)-ethoxy]-pyridin-3-yl} -propionamide, and
(i?)-2-aminomethyl-Λ/-[2-chloro-5-(2-methoxy-ethyl)-benzyl]-Λ/-cyclopropyl-3-{6-[(5)-3- (2,6-dichloro-4-methyl-phenoxy)-pyrrolidin-l-yl]-pyridin-3-yl} -propionamide.
The compounds of formula (I) are useful for the treatment and/or prophylaxis of diseases such as or related to hypertension, congestive heart failure, pulmonary hypertension, renal insufficiency, renal ischemia, renal failure, renal fibrosis, cardiac insufficiency, cardiac hypertrophy, cardiac fibrosis, myocardial ischemia, cardiomyopathy, glomerulonephritis, renal colic, complications resulting from diabetes such as nephropathy, vasculopathy and neuropathy, glaucoma, elevated intra-ocular pressure, atherosclerosis, restenosis post angioplasty, complications following vascular or cardiac surgery, erectile dysfunction, hyperaldosteronism, lung fibrosis, scleroderma, anxiety, cognitive disorders, complications of treatments with immunosuppressive agents, and other diseases related to the renin- angiotensin system.
The compounds of formula (I) are especially useful for the treatment and/or prophylaxis of hypertension, congestive heart failure, pulmonary hypertension, renal insufficiency, renal ischemia, renal failure, renal fibrosis, cardiac insufficiency, cardiac hypertrophy, cardiac fibrosis, myocardial ischemia, cardiomyopathy, complications resulting from diabetes such as nephropathy, vasculopathy and neuropathy.
In one embodiment, the invention relates to a method for the treatment and/or prophylaxis of diseases, which are associated with a dysregulation of the renin-angiotensin system, in particular to a method for the treatment and/or prophylaxis of the above-mentioned
diseases, said methods comprising administering to a patient a pharmaceutically active amount of a compound of formula (I).
A further aspect of the present invention relates to pharmaceutical compositions comprising a compound of formula (I) and a pharmaceutically acceptable carrier material.
These pharmaceutical compositions may be used for the treatment and/or prophylaxis of the above-mentioned diseases. The pharmaceutical compositions can be used for enteral, parenteral, or topical administration. They can be administered, for example, perorally, e.g. in the form of tablets, coated tablets, dragees, hard and soft gelatine capsules, solutions, emulsions or suspensions, rectally, e.g. in the form of suppositories, parenterally, e.g. in the form of injection solutions or infusion solutions, or topically, e.g. in the form of ointments, creams or oils.
The invention also relates to the use of a compound of formula (I) for the preparation of pharmaceutical compositions for the treatment and/or prophylaxis of the above-mentioned diseases.
The production of the pharmaceutical compositions can be effected in a manner which will be familiar to any person skilled in the art (see for example Mark Gibson, Editor, Pharmaceutical Preformulation and Formulation, IHS Health Group, Englewood, CO, USA, 2001; Remington, The Science and Practice of Pharmacy, 20th Edition, Philadelphia College of Pharmacy and Science) by bringing the described compounds of formula (I) or their pharmaceutically acceptable salts, optionally in combination with other therapeutically valuable substances, into a galenical administration form together with suitable, non-toxic, inert, therapeutically compatible solid or liquid carrier materials and, if desired, usual pharmaceutical adjuvants.
Compounds of formula (I) or the above-mentioned pharmaceutical compositions are also of use in combination with other pharmacologically active compounds such as ACE- inhibitors, neutral endopeptidase inhibitors, aldosterone antagonists, angiotensin II receptor antagonists, endothelin receptors antagonists, vasodilators, calcium antagonists, potassium activators, diuretics, sympatholitics, beta-adrenergic antagonists, alpha-adrenergic
antagonists, 1 lbeta-hydroxysteroid dehydrogenase type 1 inhibitors, soluble guanylate cyclase activators and/or other drugs beneficial for the prevention or the treatment of the above-mentioned diseases.
The present invention also relates to pro-drugs of a compound of formula (I) that convert in vivo to the compound of formula (I) as such. Any reference to a compound of formula (I) is therefore to be understood as referring also to the corresponding pro-drugs of the compound of formula (I), as appropriate and expedient.
The compounds of formula (I) can be manufactured by the methods outlined below, by the methods described in the examples or by analogous methods.
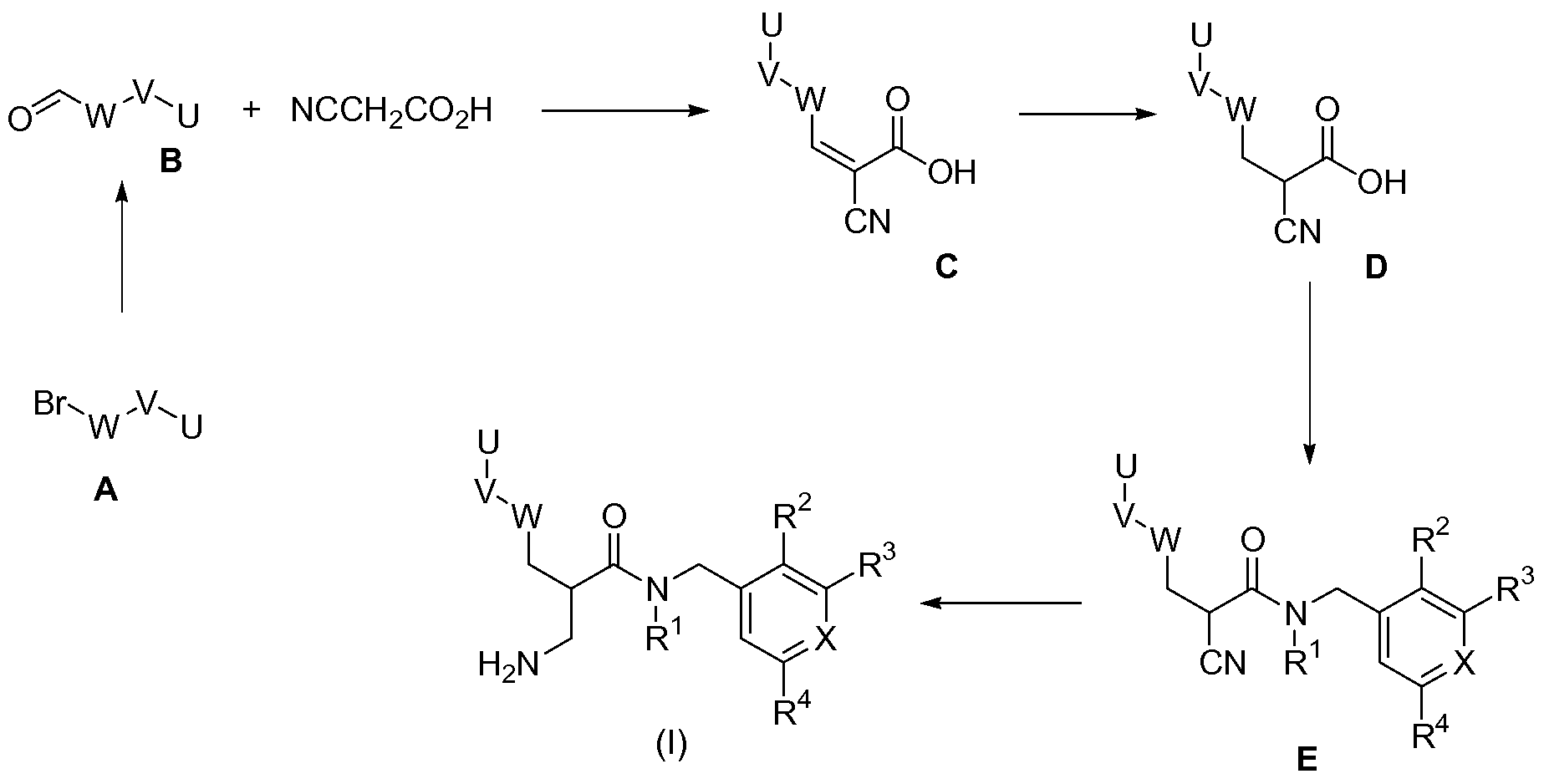
An aryl bromide or heteroaryl bromide of type A, wherein U, V and W are as defined for formula (I), can be converted into the corresponding aldehyde of type B, as described in Scheme 1. A Knoevenhagel condensation yields a compound of type C. Reduction of the double bond yields a compound of type D. An amide coupling yields a compound of type E, and final reduction of the nitrile leads a compound of formula (I).
Scheme 1
Sometimes the U-V-W-Br fragment cannot be prepared as such, or is not suitable for the subsequent chemistry. In this case, a fragment V
a-W-Br of type F, as described in Scheme 2, can be prepared, wherein V
a stands for a precursor of the V-substituent. The V
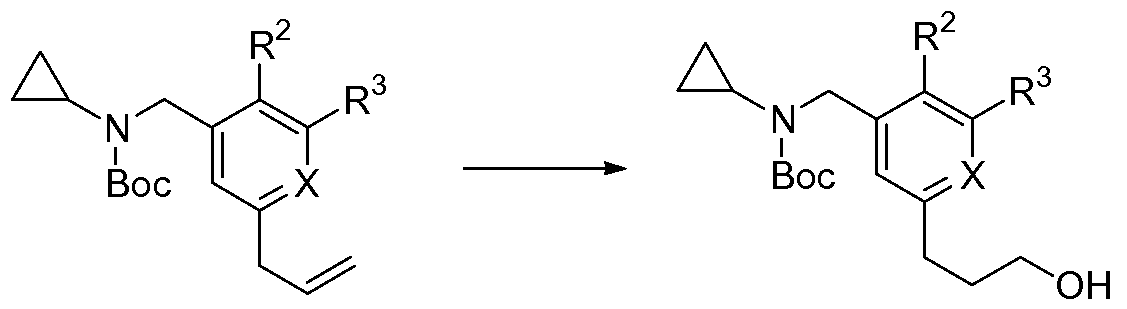
a- substituent can then be modified along the synthesis. The same chemistry as described in Scheme 1 leads to the compounds of types G, H, J, and K, respectively. Completion of the U-V-W-fragment leads to a compound of type E.
Scheme 2
B-w'va
Also, a compound of type K can be reduced into a compound of type L, as represented in Scheme 3. Protection with a protecting group PG leads to a compound of type M. Completion of the U-V-W fragment leads to a compound of type N. Final deprotection leads to a compound of formula (I).
Scheme 3
The U-V-W- or Va- W- fragments that are used to prepare a compound of type A or F have to be prepared separately. The preparation of several such substituents is described in the patent applications WO 2003/093267, WO 2004/002957, WO 2004/096769, WO 2004/096803, WO 2004/096799, and WO 2004/096366. Otherwise a pyrrolidine substituent can be attached to an aromatic ring by a copper- or palladium-catalysed coupling as described in Scheme 4. Under certain circumstances a transition metal is not necessary to catalyse this reaction. A protected pyrrolidine derivative, wherein PG' stands for a suitable protecting group, will be transformed into a compound of type F, wherein X' stands for N. If W in formula (I) represents a thiazolyl, the same chemistry can be applied as well.
Scheme 4
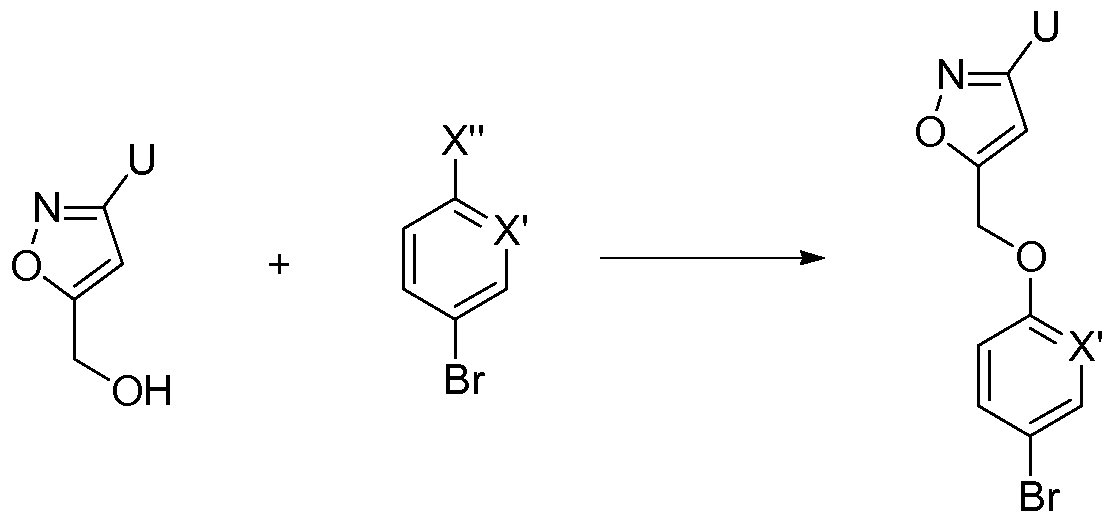
If V represents -0-CH2-Q-, the isoxazolyl moiety is prepared by cycloaddition. This cycloaddition can be realized on the W- Va- fragment in a compound of type K, leading to a compound of type E as described in Scheme 2. Otherwise the cycloaddition can be
performed separately as, for instance, described in Scheme 5. Cycloaddition on a compound of type F with an often commercially available aldehyde leads to a compound of type A. Of course the aldehyde moiety can be built on the W-Va-fragment, and a compound of the form U-CCH can be constructed, to give after cycloaddition another isoxazolyl moiety. The same principles can be used to prepare oxadiazolyl moieties, using methodologies described in the literature.
Scheme 5
Also a hydroxymethyl isoxazole (Scheme 6) can be prepared from the aldehyde mentioned in Scheme 3 and propargyl alcohol. Coupling to a phenyl or heteroaryl derivative, wherein X" typically stands for -OH, -Br, or -I, leads to a compound of type A.
Scheme 6
The amines used for such amide couplings have to be prepared separately, as described specifically in the examples, vide infra.
Enantiomerically pure compounds can always be obtained e.g. by chromatographic separation of the corresponding racemate, using a chiral solid support.
The following examples serve to illustrate the present invention in more details. They are, however, not intended to limit its scope in any manner.
Experimental Part
Abbreviations (as used herein):
AcOH acetic acid
ADDP azodicarboxylic dipiperidide
Ang angiotensin aq. aqueous
Boc te/t-butyloxycarbonyl
BSA bovine serum albumine
Bu butyl
BuLi n-butyllithium
Cy cyclohexyl dba dibenzylidene acetone
DDQ 2,3-dichloro-5,6-dicyano-l,4-benzoquinone
DIPEA diisopropylethylamine
DMAP 4-Λ/,Λ/-dimethylaminopyridine
DMF Λ/,Λ/-dimethylformamide
DMSO dimethylsulfoxide dppp 1 ,3 -bis(diphenylphosphino)propane
EDC HCl ethyl-Λ/,Λ/-dimethylaminopropylcarbodiimide hydrochloride
EIA enzyme immunoassay
ELSD evaporative light scattering detection eq. equivalent(s)
ES electrospray
ES+ electrospray, positive ionization
Et ethyl
EtOAc ethyl acetate
EtOH ethanol
FC flash chromatography h hour(s)
HATU O-(7-azabenzotriazol- 1 -yl)-N,N,N',N'-tetramethyluronium hexafluorophosphate
HOBt hydroxybenzotriazol
HPLC high performance liquid chromatography
LC-MS liquid chromatography - mass spectrometry
Me methyl
MeOH methanol min minute(s)
MS mass spectrometry org. organic
P para
PG protecting group rt room temperature sat. saturated sol. solution
TBAF tetra-n-butylammonium fluoride
TBME tert-butyl methyl ether
TBDMS te/t-butyldimethylsilyl tBu tert-butyi
TFA trifluoroacetic acid
THF tetrahydrofuran
TLC thin layer chromatography tR retention time (in LC-MS or HPLC) given in minutes
UV ultra violet
Vis visible xantphos 4,5-bis(diphenylphosphino)-9,9-dimethylxanthene
HPLC- or LC-MS-conditions (if not indicated otherwise):
Analytic: Zorbax 59 SB Aqua column, 4.6 x 50 mm from Agilent Technologies. Eluents: A: acetonitrile; B: H2O + 0.5% TFA. Gradient: 90% B → 5% B over 2 min. Flow: 1 niL/min. Detection: UV/Vis + MS. Preparative: Zorbax SB Aqua column, 20 x 500 mm from Agilent Technologies. Eluent: A: Acetonitrile; B: H2O + 0.05% ammonium hydroxide (25% aq.). Gradient: 80% B → 10% B over 6 min. Flow: 40 mL/min. Detection: UV + MS, or UV + ELSD. Chiral, analytic: a) Regis Whelk column, 4.6 x 250 mm, 10 μm. Eluent A: EtOH + 0.05% Et3N. Eluent B: hexane. Flow 1 mL/min. b) ChiralPak AD, 4.6x250 mm, 5 μm. Eluent A: EtOH + 0.05% Et3N. Eluent B: hexane. Flow 1 mL/min. c) ChiralCel OD, 4.6x250 mm, 10 μm. Eluent A: EtOH + 0.1% Et3N. Eluent B: hexane. Flow 0.8 mL/min. Chiral, preparative: a) Regis Whelk 01 column, 50x250 mm and a flow of 100 mL/min. Eluent A: EtOH + 0.05% Et3N. Eluent B: hexane. b) ChiralCel OD, 20 μm, 50 mm x 250 mm, flow 100 mL/min. Eluent A: EtOH + 0.1% Et3N. Eluent B: hexane.
General conditions for a Mitsunobu coupling (General procedure A)
A mixture of the desired alcohol (0.1 mmol), of the desired phenol (0.12 mmol), ADDP (0.2 mmol) and PBu3 (0.4 mmol) in toluene (2 mL) was prepared at 0 0C. The mixture was stirred for 2 h at rt, for 3 h at reflux, and then overnight at rt again. The solvents were removed under reduced pressure, and the residue was purified by HPLC.
General conditions for the cleavage of a Boc-protecting group (General procedure B)
HCl (4M in dioxane, 1 mL) was added to a sol. of the starting material in CH2Cl2 (1 mL) at 0 0C. The mixture was stirred for 1 h at 0 0C, and aq. IM NaOH (4 mL) was added. The mixture was filtered on Isolute®, and the org. layer was evaporated under reduced pressure. Purification of the crude by HPLC yielded the title compound.
l-Bromo-S-chloro-pyridine^-carbaldehyde
To a stirred sol. of diisopropylamine (20.9 mL, 148 mmol) in dry THF (350 mL) at -5 °C was added dropwise BuLi (1.6M in hexane, 89.5 mL, 143 mmol), and the resulting sol. was stirred for 30 min at -5 0C. The sol. was allowed to cool to -70 0C, and a sol. of 2- bromo-5-chloropyridine (25.0 g, 130 mmol) in THF ( 100 mL) was added dropwise at -70 0C over 15 min, such that the internal temperature did not exceed -65 0C. The mixture was stirred at -70 0C for 30 min. DMF (10.52 mL, 136 mmol) was added dropwise over 20 min such that the internal temperature did not exceed -70 0C. The orange mixture was stirred at -70 0C for 40 min. The mixture was allowed to warm up to rt, and was poured onto a mixture of water (200 mL) and aq. IM NaOH (50 mL). The mixture was extracted with EtOAc (2x), and the combined org. extracts were washed back with aq. IM NaOH (2x). The org. extracts were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the crude by FC (EtO Ac/heptane 1 :9 — > 1 :8 — > 1 :6 — > 1 :4 → 1 :2 → 1 :1) yielded the title compound (21.55 g, 72 %). LC-MS: tR = 0.74 min; ES+: 295.01.
l-Bromo-S-chloro^-dimethoxymethyl-pyridine
To a sol. of 2-bromo-5-chloro-pyridine-4-carbaldehyde (43.9 g, 199 mmol) in MeOH (800 mL) were successively added at rt trimethyl ortho formate (65.3 mL, 597 mmol) and p- toluenesulfonic acid monohydrate (1.90 g, 10.0 mmol). This reaction mixture was then heated to reflux for 3 h. The mixture was allowed to cool to rt and was concentrated under reduced pressure. The residue was dissolved in CH2Cl2 and this mixture was washed with aq. 10% K2CO3. The org. layer was dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Drying under high vacuum yielded the title compound
(51.7 g, 97 %). LC-MS: tR = 0.92 min; ES+: 309.06.
5-Chloro-4-dimethoxymethyl-2-(3-methoxy-propyl)-pyridine
To a suspension of Mg (911 mg, 37.5 mmol) and of iodine (one crystal) in dry THF (30 mL) was added dropwise 5% of the total amount of l-bromo-3-methoxypropane (4.59 g,
30.0 mmol). The mixture was heated to reflux with the help of a heat gun until the
Grignard formation had started. The rest of the l-bromo-3-methoxypropane was added
slowly, while an exothermic reaction proceeded. After the end of the addition, the reaction mixture was stirred under reflux for 20 min, and was allowed to cool to rt. This Grignard sol. (IM in THF, 23.5 mL, 23.5 mmol) was added dropwise to a mixture of 2-bromo-5- chloro-4-dimethoxymethyl-pyridine (2.50 g, 9.38 mmol) and Ni(dppp)Cl2 (495 mg, 0.938 mmol) in THF (50 mL) at 0 0C. The reaction mixture was stirred at rt for 30 min, and was then heated to reflux for 2 h. The mixture was allowed to cool to rt, and was dissolved with EtOAc. This mixture was washed with aq. sat. NaHCO3. The org. layer was dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the residue by FC (heptane — > EtO Ac/heptane 1 :1) yielded the title compound (1.51 g, 62%). LC-MS: tR = 0.80 min; ES+: 260.15.
5-Chloro-2-(3-methoxy-propyl)-pyridine-4-carbaldehyde
5-Chloro-4-dimethoxymethyl-2-(3-methoxy-propyl)-pyridine (25.5 g, 98.2 mmol) was dissolved in aq. IM HCl (500 mL), and the mixture was heated to 80 0C for 2 h. The mixture was allowed to cool to rt, and EtOAc was added. The mixture was cooled to 0 0C, and was basified with aq. 2.5M NaOH until a pH = 10 was reached. The layers were separated, and the org. layer was dried over MgSO4, filtered, and concentrated under reduced pressure. Drying the residue under high vacuum yielded the crude title compound (98.1 mmol, 99%) that was used further without purification. LC-MS: tR = 0.62 min; ES+: 246.12.
[5-Chloro-2-(3-methoxy-propyl)-pyridin-4-ylmethyl]-cyclopropyl-amine
A mixture of 5-chloro-2-(3-methoxy-propyl)-pyridine-4-carbaldehyde (21.0 g, 98.2 mmol) and cyclopropylamine (13.8 mL, 196 mmol) in MeOH (450 mL) was stirred at rt overnight. NaBH4 (4.83 g, 128 mmol) was added at 0 0C, and the mixture was stirred at rt overnight. Ice was added, and the mixture was concentrated under reduced pressure. The crude product was dissolved in EtOAc, and this mixture was washed with aq. IM NaOH. The aq. layer was extracted back with EtOAc. The combined org. extracts were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the crude by FC (EtO Ac/heptane 1 :5 → 1 :4 → 1 :3 → 1 :1 → 3:1 → EtOAc) yielded the title compound (11.8 g) and [5-chloro-2-(3-methoxy-propyl)-pyridin-4-ylmethylene]- cyclopropyl-amine (10.7 g). This unreacted imine was dissolved in MeOH (20 mL), and
this sol. was cooled to 0 0C. NaBH4 (3.20 g, 84.6 mmol) was added, and the mixture was stirred at rt overnight. NaBH4 (3.20 g, 84.6 mmol) was added again, and the mixture was stirred for 3 days. Ice was added to the reaction mixture, and the mixture was concentrated under reduced pressure. The crude product was dissolved in EtOAc and the resulting mixture was washed with aq. IM NaOH. The aq. phase was extracted back with EtOAc. The combined org. extracts were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the crude by FC (EtO Ac/heptane 1 :3 — > 1 :2 — > 1 :1 — > EtOAc) yielded the title compound (9.4 g). The fractions of the title compound were mixed together (21.2 g, 85%). LC-MS: tR = 0.55 min; ES+: 296.16.
S-Bromo-l-chloro-TV-cyclopropylbenzamide
Into a flame-dried 250 mL round-bottom flask equipped with a magnetic stir bar and under N2 were added 5-bromo-2-chlorobenzoic acid (10.0 g, 42.5 mmol) and DMF (3.9 mL, 51.0 mmol) in toluene (80 mL). The sol. was cooled to 0 0C, and oxalyl chloride (4.4 mL, 51.0 mmol) was added dropwise over 1 h. The resulting mixture was stirred at 0 0C for 2 h and then the volatiles were removed. The resulting crude reaction mixture was dissolved in CH2Cl2 (100 mL) and cooled to 0 0C in an ice bath. Cyclopropylamine (4.5 mL, 63.7 mmol) was added dropwise over 1 h followed by addition of DIPEA (11.8 mL, 85.0 mmol). The resulting sol. was stirred at rt for 16 h. The reaction mixture was poured into a 1 L separatory funnel containing IM aq. HCl (600 mL). The mixture was extracted with CH2Cl2 (6 x 250 mL). The combined org. layers were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The product was crystallized from hexanes/CH2Cl2 and isolated by filtration to give the title compound (8.24 g, 71%).
7V-(5-bromo-2-chlorobenzyl)cyclopropylamine
A sol. of 5-bromo-2-chloro-Λ/-cyclopropylbenzamide (12.0 g, 43.7 mmol) in THF (100 mL) was placed into a 250 mL round-bottom flask, equipped with a magnetic stir bar and under N2. The sol. was treated with dropwise addition OfBH3-Me2S (13.1 mL, 131 mmol), and the resulting suspension was stirred at rt for 1 h. The mixture was heated to reflux for 1 h, cooled to rt, and slowly quenched with dropwise addition of IM aq. HCl (25 mL). The suspension was again refluxed for 1 h, cooled to rt, and basified to pH = 10-11 with IM aq. NaOH. The mixture was poured into a 500 mL separatory funnel containing IM aq. NaOH
(350 niL). The mixture was extracted with EtOAc (3 x 100 mL). The combined org. layers were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. The crude amine was used directly in the next step.
General procedure for the reductive amination of substituted benzaldehydes with cyclopropylamine:
Y = halogen
A sol. of substituted benzaldehyde (17.8 mmol, 1.0 eq.), cyclopropylamine (3.13 mL, 44.5 mmol, 2.5 eq.) and sodium cyanoborohydride (1.34 g, 21.4 mmol, 1.2 eq.) in MeOH (100 mL) was treated with dropwise addition of glacial AcOH (3.06 mL, 53.4 mmol, 3.0 eq.). The resulting sol. was stirred at rt for 16 h overnight. The reaction mixture was quenched with dropwise addition of sat. aq. NaHCO3, and concentrated under reduced pressure to remove the MeOH. The crude residue was poured into a 250 mL separatory funnel containing sat. aq. NaHCO3 (150 mL), and extracted with EtOAc (3 x 50 mL). The combined org. layers were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. Purification by FC yielded the benzamine product.
General procedure for the Boc-protection of cyclopropylbenzamines:
Y = halogen
A sol. of the cyclopropylbenzamine (43.7 mmol, 1.0 eq.) in a biphasic mixture Of CH
2Cl
2 (50 mL) and IM aq. NaOH (50 mL) was treated with BoC
2O (15.1 mL, 65.6 mmol, 1.5 eq.). The mixture was stirred at rt vigorously for 16 h. The mixture was poured into a 500 mL separatory funnel containing H
2O (300 mL), and extracted with CH
2Cl
2 (3 x 100 mL). The combined org. layers were washed with brine, dried over MgSO
4, filtered and concentrated under reduced pressure. Purification by FC yielded the Boc-protected amine.
General procedure for the allylation ofBoc-protected cyclopropylbenzamines:
Y = halogen
Into a flame-dried round-bottom flask or Schlenk tube, under N2 was added Pd[PCy3]2 (0.05 eq.), CsF (2.0 eq.) and the corresponding aryl bromide (1.0 eq.). If the aryl chloride was being used as a starting material, the (Pd[P?Bu3]Br)2 dimer (0.025 eq.) was used in place of the Pd[PCy3]2 catalyst. The flask was evacuated under reduced pressure (0.1 mm Hg) and backfilled with N2 (repeated 3 times). The resulting solids were dissolved in anhydrous THF or dioxane (0.15 M sol.) and tri-n-butyl allyltin (1.5 eq.) was added and the resulting mixture was refluxed for 8-16 h, until TLC shows complete consumption of starting material. The reaction mixture was cooled to rt, and filtered through a pad of silica gel on a sintered glass funnel, washing with Et2O. The filtrate was concentrated and purified by FC to give the corresponding allylbenzamide derivative.
General procedure for the hydroboration/oxidation ofallylbenzamines:
Into a flame-dried round-bottom flask equipped with a magnetic stir bar was added the allylbenzamine (1.0 eq.) and anhydrous THF (0.3 M sol.). The sol. was cooled to 0 0C and
BH3*Me2S (1.1 eq.) was added dropwise over 20 min. The sol. was stirred at 0 0C for 1 h, then allowed to warm to rt, and stirred for an additional 2 h. The sol. was cooled to 0 0C and IM aq. NaOH was added dropwise (CAUTION - EXOTHERMIC REACTION), followed by dropwise addition of 30% aq. H2O2. The mixture was allowed to warm to rt, and stirred for 2 h. The mixture was poured into a separatory funnel containing H2O and extracted with Et2O (3 times). The combined org. layers were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. Purification by FC yielded the desired alcohol product.
General procedure for the oxidative cleavage/reduction of allylbenzamines:
A sol. of allylbenzamine (1.0 eq.) in CH2Cl2 (0.4 M sol.) was cooled to -78 0C and O3 gas was introduced into the sol. using a gas dispersion tube. The ozone gas was introduced until all of the starting material had been consumed, as determined by TLC, and the reaction mixture maintained a slight blue colour. The reaction was stirred at -78 0C for 20 min, then EtOH (0.5 M sol.) and NaBH4 (2.5 eq.) were added. The mixture was allowed to warm to rt overnight (16 h). The reaction mixture was quenched with dropwise addition of sat. aq. NH4Cl (5 mL), and poured into a separatory funnel containing sat. aq. NH4Cl. The mixture was extracted with Et2O (3 times). The combined org. layers were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. Purification by FC yielded the desired alcohol.
General procedure for the etherification of aromatic primary alcohols with methyl iodide:
A suspension of the primary alcohol (1.0 eq.) in THF (0.25 M sol.) was cooled to 0 0C and treated with NaH (60% in oil, 2.0 eq.). The resulting mixture was stirred at 0 0C for 30 min and then at rt for another 30 min. The suspension was re-cooled to 0 0C and then MeI (8.0 eq.) was added in a single portion. The reaction mixture was stirred at 0 0C for 30 min, at rt for 30 min, and then heated to reflux for 4 h until all of the starting material was consumed as determined by TLC. The cooled reaction mixture was quenched with dropwise addition of sat. aq. NH4Cl and poured into a separatory funnel containing sat. aq. NH4Cl, and extracted with EtOAc (3 times). The combined org. layers were washed with brine, dried over MgSO4, filtered and concentrated under reduced pressure. Purification by FC yielded the methyl ether.
General procedure for the deprotection of Boc-protected cyclopropylbenzamines:
To a sol. of Boc-protected cyclopropylbenzamine (1.0 eq.) in CH2Cl2 (0.1-0.5 M sol.) was added 4 M HCl in dioxane (5.0 eq.). The resulting mixture was stirred at rt for 8-16 h until TLC shows complete conversion of starting material. The reaction was poured into a separatory funnel containing IM aq. NaOH, and extracted with CH2Cl2 (3 times). Purification by FC yielded the corresponding free amine.
2,6-Dichloro-4-hydroxymethylphenol
BH3 (IM in THF, 250 mL, 250 mmol) was added dropwise to a cooled sol. of 3,5- dichloro-4-hydroxybenzoic acid (20 g, 96.6 mmol) in THF (200 mL) at 0 0C. The resulting mixture was stirred at 0 0C for 15 min, and then at rt for 13 h. The milky mixture was cooled to 0 0C and MeOH (150 mL), then water (100 mL), were added dropwise. The mixture was further stirred at 0 0C for 15 min, and then at rt for 5 h. The mixture was then partially concentrated under reduced pressure. EtOAc (200 mL) and water (50 mL) were added to the residue, and the phases were shaken and separated. The aq. phase was further extracted with EtOAc. The combined org. extracts were washed with brine, dried over MgSO4, filtered, and concentrated under reduced pressure. Purification by FC (CH2C12/CH3OH, 100:1) led to the title compound as a slightly beige solid (17.86 g, 96%). LC-MS: tR = 0.69 min.
3,5-Dichloro-4-hydroxybenzaldehyde 2,6-Dichloro-4-hydroxymethylphenol (3.56 g, 18.4 mmol) was dissolved in dioxane, and DDQ (4.19 g, 18.4 mmol) was added. The reaction mixture was stirred at rt overnight. The solvents were removed under reduced pressure. The residue was diluted with CH2Cl2, and the mixture was filtered. The filtrate was dried over MgSO4, filtered, and the solvents
were removed under reduced pressure. Crystallization from EtOAc yielded the title compound (0.77 g, 22%). LC-MS: tR = 0.82 min.
(rac.)-2,6-Dichloro-4-(l-hydroxyethyl)phenol A sol. of 3,5-dichloro-4-hydroxybenzaldehyde (1.635 g, 8.56 mmol) in Et2O (30 mL) was cooled to -78 0C. MeMgBr (3M in Et2O, 7.15 mL, 21.5 mmol) was added dropwise to the cooled reaction mixture over 18 min. Et2O (20 mL) was added again during the addition of MeMgBr. Stirring was continued at -78 0C for 1 h, and then the reaction mixture was allowed to warm up to rt over 1 h. The mixture was cooled to 0 0C, and aq. sat. NH4Cl (10 mL) was added dropwise. The mixture was allowed to warm up to rt, and additional aq. sat. NH4Cl (35 mL) and water (35 mL) were added. The phases were then separated and the aq. phase was extracted with Et2O. The combined org. extracts were then washed with brine (50 mL), dried over MgSO4, filtered, and concentrated under reduced pressure. Purification by FC (EtO Ac/heptane, 1 :1) yielded the title compound (1.68 g, 95%). LC- MS: tR = 0.74 min.
(rac.)-2-(tert-Butyldimethylsilanyloxy)-5-[l-(tert-butyldimethylsilanyloxy)ethyl]-l,3- dichlorobenzene
To a sol. of (rac.)-2,6-dichloro-4-(l-hydroxyethyl)phenol (100 mg, 0.483 mmol) in DMF (5.5 mL) were added TBDMS-Cl (175 mg, 1.16 mmol), and imidazole (145 mg, 2.42 mmol). The sol. was stirred at rt overnight. The sol. was cooled to 0 0C, and aq. sat. NH4Cl was added. The mixture was extracted with heptane/Et2O (1/1, 4x). The combined org. extracts were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification by FC (CH2Cl2) yielded the title compound (188 mg, (90%).
(rac.)-4-[l-(tert-Butyldimethylsilanyloxy)ethyl]-2,6-dichlorophenol
A sol. of (rαc.)-2-(tert-butyldimethylsilanyloxy)-5-[l-(tert-butyldimethylsilanyloxy)ethyl]- 1,3-dichlorobenzene (188 mg, 0.432 mmol) and Cs2CO3 (76.2 mg, 0.126 mmol) in DMF (0.50 mL) and water (50 μL) was stirred at rt overnight. Et2O (75 mL) was added. The sol. was washed with brine, dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification by FC (CH2Cl2) yielded the title compound (122 mg, 88%). LC-MS: tR = 1.15 min.
2,6-Dichloro-3,4-dimethylphenol
To a sol. of 3,4-dimethylphenol (3.00 g, 24.6 mmol) in CH2Cl2 (5 niL) was added SO2Cl2 (4.98 niL, 61.3 mmol). The resulting sol. was heated to 50 0C for 4 h. The mixture was poured onto ice-water. CH2Cl2 (200 mL) was added, the layers were separated, and the org. layer was washed with water, then with aq. sat. NaHCO3. The org. layer was dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification by FC (EtO Ac/heptane 1 :4) yielded the title compound (1.17 g , 25%).
2-[2-(før*-Butyldimethylsilanyloxy)ethoxy]thiazole
NaH (50% suspension in oil, 2.98 g, 62.1 mmol) was suspended in hexane and washed twice with hexane. THF (20 mL) was then added followed by a sol. of 2-{tert- butyldimethylsilanyloxy)ethanol (McDougal, P. G., Rico, J. G., Oh, Y. L, Condon, B. D., J. Org. Chem., 1986, 51, 3388, 9.49 g, 53.8 mmol) in THF (30 mL) over 30 min. The mixture was then stirred for 2 h at rt. 2-Bromothiazole (6.79 g, 41.4 mmol) was then added dropwise and the reaction mixture was then stirred at reflux for 20 h. Aq. sat. NH4Cl was added carefully and the product was extracted with Et2O (3x). The combined org. extracts were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the residue by FC (Et2O/hexane 5:95) yielded the title compound (3.80 g, 35%).
(5)-l-Thiazol-2-yl-pyrrolidin-3-ol
A mixture of 2,5-dibromothiazole (15.0 g, 59.9 mmol), (5)-3-hydroxypyrrolidine (6.00 mL, 71.9 mmol) and DIPEA (13.3 mL, 77.9 mmol) in dioxane (875 mL) was stirred at 80 0C for 17 h. The solvents were removed under reduced pressure. Aq. sat. NaHCO3 was added, and the mixture was extracted with CH2Cl2 (2x). The combined org. extracts were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. The crude title product (12.2 g) was used in the next reaction without purification. LC-MS: tR = 0.54 min; ES+: 249.06.
(S)-2-[3-(tert-Butyl-dimethyl-silanyloxy)-pyrrolidin-l-yl]-thiazole
A mixture of (5)-l-thiazol-2-yl-pyrrolidin-3-ol (12.2 g, 48.9 mmol), TBDMS-Cl (8.84 g, 58.7 mmol) and imidazole (8.30 g, 122 mmol) in DMF (150 mL) was stirred at rt for 30 min. Water (150 mL) was added, and the mixture was extracted with heptane (3x). The combined org. extracts were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the residue by FC (heptane/EtOAc 14:1 — > 13:1 — > 12: l→ 10:1) yielded the title compound (11.3 g, 52% over 2 steps). LC-MS: tR = 1.10 min; ES+: 363.14.
2-Cyano-7V-cyclopropyl-7V-(2,3-dimethyl-benzyl)-acetamide HATU (6.51 g, 17.1 mmol) was added to a sol. of cyano-acetic acid (1.46 g, 17.1 mmol), cyclopropyl-(2,3-dimethyl-benzyl)-amine (prepared from 2,3-dimethyl-benzaldehyde and cyclopropylamine by reductive amination; 3.00 g, 17.1 mmol) and DIPEA (5.86 mL, 34.2 mmol) in DMF (15 mL) at 0 0C. The mixture was stirred for 1.5 h at 0 0C, and was diluted with EtOAc. The resulting mixture was washed with aq. IM HCl (2x) and aq. sat. NaHCO3 (Ix). The combined aq. phases were extracted back with EtOAc (Ix). The combined org. extracts were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the residue by FC (EtO Ac/heptane 1 :3 → 1 :2 → 1 :1) yielded the title compound (3.36 g, 81%). LC-MS: tR = 0.93 min; ES+: 243.22.
2-Cyano-7V-cyclopropyl-7V-(2,3-dichloro-benzyl)-acetamide
HATU (4.47 g, 11.8 mmol) was added to a sol. of cyano-acetic acid (1.00 g, 11.8 mmol), cyclopropyl-(2,3-dichloro-benzyl)-amine (prepared from 2,3-dichloro-benzaldehyde and cyclopropylamine by reductive amination; 2.54 g, 11.8 mmol) and DIPEA (4.03 mL, 23.5 mmol) in DMF (10 mL) at 0 0C. The mixture was stirred for 1 h at 0 0C, and for 1.5 h at rt. EtOAc was added, and the mixture was washed with aq. IM HCl (2x), and aq. sat. NaHCO3 (Ix). The combined aq. phases were extracted back with EtOAc (Ix). The combined org. extracts were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the crude by FC (EtO Ac/heptane 1 :1) yielded the title compound (2.58 g, 78%). LC-MS: tR = 0.94 min; ES+: 324.09.
(JE)-3-{2-[2-(tert-Butyl-dimethyl-silanyloxy)-ethoxy]-thiazol-5-yl}-2-cyano-7V- cyclopropyl-7V-(2,3-dimethyl-benzyl)-acrylamide
A mixture of 2-cyano-Λ/-cyclopropyl-Λ/-(2,3-dimethyl-benzyl)-acetamide (3.36 g, 13.9 mmol), compound Bl (3.99 g, 13.9 mmol) and pyridine (5 droplets) in toluene (100 mL) was heated to 120 0C for 2 days. The mixture was allowed to cool to rt, and was evaporated under reduced pressure. The residue was dried under high vacuum. The resulting yellow crystals were triturated with heptane, and were filtered. Drying these crystals under high vacuum yielded the title compound (4.86 g, 69%). LC-MS: tR = 1.21 min; ES+: 512.37.
(^-(^^-{l-IS-^ert-Butyl-dimethyl-silanyloxyJ-pyrrolidin-l-yll-thiazol-S-ylJ-l-cyano- 7V-cyclopropyl-7V-(2,3-dichloro-benzyl)-acrylamide
A mixture of 2-cyano-7V-cyclopropyl-iV-(2,3-dichloro-benzyl)-acetamide (2.13 g, 7.52 mmol), compound B2 (2.35 g, 7.52 mmol) and pyridine (5 droplets) in toluene (100 mL) was heated to 120 0C for 2 days. The mixture was allowed to cool to rt, and the solvents were removed under reduced pressure. The residue was dried under high vacuum, and was dissolved in a mixture of EtOAc and heptane. The solvents were removed under reduced pressure, and the resulting solid was triturated with heptane. Drying the solid under high vacuum yielded the title compound (3.76 g, 86%). LC-MS: tR = 1.24 min; ES+: 577.02.
(rac.)-2-{2-[2-(tert-Butyl-dimethyl-silanyloxy)-ethoxy]-thiazol-5-ylmethyl}-2-cyano-7V- cyclopropyl-7V-(2,3-dimethyl-benzyl)-acetamide
(E)-3-{2-[2-(tert-Butyl-dimethyl-silanyloxy)-ethoxy]-thiazol-5-yl}-2-cyano-N- cyclopropyl-Λ/-(2,3-dimethyl-benzyl)-acrylamide (4.17 g, 8.15 mmol) was suspended in MeOH (50 mL), and THF was added until a clear sol. was obtained (about 80 mL). The mixture was cooled to 0 0C, and CeCl3-7H2θ (6.20 g, 16.3 mmol) was added. NaBH4 (1.61 g, 40.7 mmol) was added carefully in portions. The mixture was stirred for 30 min at
0 0C, and CH2Cl2 (100 mL) was added. Aq. IM NaOH was added, and the mixture was stirred efficiently for 10 min. The phases were separated, and the aq. phase was extracted with CH2Cl2. The combined org. extracts were washed with brine, dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Drying the residue under high vacuum yielded the title crude compound (4.47 g, quantitative) that was used further without purification. LC-MS: tR = 1.20 min; ES+: 514.43.
Mixture of (2R)-3-{2-[(3S)-3-(før*-Butyl-dimethyl-silanyloxy)-pyrrolidin-l-yl]-thiazol- 5-yl}-2-cyano-7V-cyclopropyl-7V-(2,3-dichloro-benzyl)-propionamide and (2S)-3-{2- [(3S)-3-(tert-Butyl-dimethyl-silanyloxy)-pyrrolidin-l-yl]-thiazol-5-yl}-2-cyano-7V- cyclopropyl-7V-(2,3-dichloro-benzyl)-propionamide (E)-(5)-3-{2-[3-(tert-Butyl-dimethyl-silanyloxy)-pyrrolidin-l-yl]-thiazol-5-yl}-2-cyano-Λ/- cyclopropyl-iV-(2,3-dichloro-benzyl)-acrylamide (4.35 g, 7.53 mmol) was suspended in MeOH (65 niL), and THF was added until a clear sol. was obtained (about 30 rnL). The mixture was cooled to 0 0C, and CeCl3-VH2O (5.70 g, 15.1 mmol) was added. NaBH4 (2.97 g, 75.3 mmol) was added carefully in portions. The mixture was stirred for 2 h at 0 0C. CH2Cl2 (300 rnL), MeOH (15 mL), and aq. IM NaOH (200 mL) were added, and the mixture was stirred efficiently for 2 h. The phases were separated, and the org. layer was washed with aq. IM NaOH and brine. The org. layer was dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Drying the residue under high vacuum yielded the crude title compounds mixture (4.50 g, quantitative yield) that was used without further purification. LC-MS: tR = 1.01 min; ES+: 579.20.
2-(2,6-Dichloro-4-methyl-phenoxy)-ethanol
In a three-necked flask equipped with a gas droplet counter and an efficient cooling system, a mixture of 2,6-dichloro-/?-cresol (20.0 g, 113 mmol), [l,3]dioxolan-2-one (9.95 g, 113 mmol) and imidazole (115 mg, 1.70 mmol) was heated to 160 0C for 25 h. The mixture was allowed to cool to rt. Purification by FC (Et2O/heptane 1 :1) yielded the title compound (18.7 g, 75%). LC-MS: tR = 0.88 min.
(R)-l-(5-Bromo-pyridin-2-yl)-pyrrolidin-3-ol 2,5-Dibromopyridine (4.00 g, 16.4 mmol) and (i?)-pyrrolidine-3-ol (1.11 g, 12.6 mmol) were dissolved in toluene. tBuONa (1.87 g, 18.9 mmol), Pd2(dba)3 (231 mg, 0.252 mmol) and xantphos (451 mg, 0.756 mmol) were added, and the mixture was degassed with nitrogen. The mixture was stirred at 95 0C for 4 h, and was allowed to cool to rt. EtOAc was added, and the mixture was washed with water (2x). The combined org. layers were extracted back with EtOAc (Ix). The combined org. extracts were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the crude
by FC (MeOH/CH2Cl2 1 :30 with 1% Et3N) yielded the title compound (2.57 g, 84%). LC- MS: tR = 0.44 min; ES+: 243.07.
(R)-5-Bromo-2-[3-(2,6-dichloro-4-methyl-phenoxy)-pyrrolidin-l-yl]-pyridine (Al) A mixture of compound Fl (3.38 g, 13.9 mmol), 2,6-dichloro-/?-cresol (3.69 g, 20.9 mmol), ADDP (5.26 g, 20.9 mmol) and PBu3 (85%, 10.3 mL, 35.4 mmol) in toluene (200 mL) was heated to reflux for 2 h. The mixture was allowed to cool to rt, and was diluted with heptane. The mixture was filtered, washed with heptane, and the filtrate was evaporated under reduced pressure. The residue was diluted with CH2Cl2, and this mixture was washed with aq. IM NaOH (2x). The org. layer was dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the residue by FC (CH2Cl2/heptane 4:1 → CH2Cl2) yielded the title compound (5.46g, 98%). LC-MS: tR = 0.91 min; ES+: 403.00.
5-Bromo-2- [2-(2,6-dichloro-4-methyl-phenoxy)-ethoxy] -pyridine (A2)
A sol. of 2-(2,6-dichloro-4-methyl-phenoxy)-ethanol (18.6 g, 84 mmol) in THF (360 mL) was cooled to 0 0C. NaH (about 55% in oil, 6.60 g, about 153 mmol) was added in portions, and the mixture was stirred at rt for 30 min. A sol. of 2,5-dibrompyridine (18.0 g, 76.3 mmol) in THF (60 mL) was added dropwise, and the mixture was heated to reflux for 90 min. The mixture was allowed to cool to rt, and ice was added carefully. The solvents were partially removed under reduced pressure, and the residue was diluted with EtOAc. This mixture was washed with aq. sat. NH4Cl. The aq. layer was extracted back with EtOAc (2x). The combined org. extracts were washed with brine, dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the crude by FC (EtOAc/heptane 3:97) yielded the title compound (22.7 g, 79%). LC-MS: tR = 1.13 min; ES+: 378.08.
(S)-5-Bromo-2-[3-(2,6-dichloro-4-methyl-phenoxy)-pyrrolidin-l-yl]-pyridine (A3)
A mixture of (i?)-l-(5-bromo-pyridin-2-yl)-pyrrolidin-3-ol (2.56 g, 10.5 mmol), 2,6- dichloro-/?-cresol (3.88 g, 21.1 mmol), ADDP (4.07 g, 15.8 mmol) and PBu3 (85%, 9.17 mL, 26.8 mmol) in toluene (150 mL) was stirred at 80 0C for 1 h. The mixture was allowed to cool to rt, and was diluted with heptane. The precipitate was washed with
heptane thoroughly, and the filtrate was evaporated under reduced pressure. The residue was diluted with CH2Cl2, and the resulting mixture was washed with aq. IM NaOH (2x). The org. layer was dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the crude by FC (EtO Ac/heptane 1 :7) yielded the title compound (2.88 g, 68%). LC-MS: tR = 0.93 min; ES+: 402.89.
2-[2-(før^Butyl-dimethyl-silanyloxy)-ethoxy]-thiazole-5-carbaldehyde (Bl)
BuLi (9.95 mL, 15.6 mmol) was added to a sol. of 2-[2-(tert- butyldimethylsilanyloxy)ethoxy]thiazole (4.00 g, 15.4 mmol) in THF (25 mL) at -78 0C. DMF (1.29 mL, 16.7 mmol) was added, and the mixture was stirred for 4 h, while being allowed to warm up to rt. Water was added, and the mixture was extracted with EtOAc (2x). The combined org. extracts were washed with brine (Ix), dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Drying the residue under high vacuum yielded the crude title compound (4.35g, 98%) that was used further without purification. LC-MS: tR = 1.08 min; ES+: 288.22.
(S)-2-[3-(tert-Butyl-dimethyl-silanyloxy)-pyrrolidin-l-yl]-thiazole-5-carbaldehyde (B2)
BuLi (1.6M in hexane, 2.27 mL, 3.63 mmol) was added to a sol. of (S)-2-[3-(tert-butyl- dimethyl-silanyloxy)-pyrrolidin-l-yl]-thiazole (1.00 g, 3.52 mmol) in THF (6.00 mL) at
-78 0C. The mixture was stirred for 10 min at -78 0C, and DMF (0.294 mL, 3.81 mmol) was added. The mixture was stirred for 2 h while being allowed to warm up to rt. Water was added, and the mixture was extracted with EtOAc (2x). The combined org. extracts were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. The crude title compound (1.10 g, quantitative yield) was used without further purification.
LC-MS: tR = 1.08 min; ES+: 313.18.
(if)-6-[3-(2,6-Dichloro-4-methyl-phenoxy)-pyrrolidin-l-yl]-pyridine-3-carbaldehyde (B3) BuLi (1.6M in hexane, 9.80 mL, 16.3 mmol) was added to a sol. of compound Al (5.46 g, 13.6 mmol) in THF (280 mL) at -78 0C. The mixture was stirred for 20 min at -78 0C, and DMF (1.57 mL, 20.3 mmol) was added. The mixture was stirred for 2.5 h at -78 0C, and
aq. sat. NH4Cl (120 niL) was added. The mixture was allowed to warm up to rt, and was extracted with TBME (2x). The combined org. extracts were washed with aq. sat. NH4Cl, dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the crude by FC (EtO Ac/heptane 1 :4 — > 2:3) yielded the title compound (2.97 g, 62%). LC-MS: tR = 0.89 min; ES+: 351.12.
6- [2-(2,6-Dichloro-4-methyl-phenoxy)-ethoxy] -pyridine-3-carbaldehyde (B4)
BuLi (1.6M in hexane, 9.53 mL, 15.3 mmol) was added to a sol. of compound A2 (5.00 g, 13.3 mmol) in THF (280 mL) at -78 0C. The mixture was stirred for 1 h at -78 0C, and DMF (1.54 mL, 19.9 mmol) was added. The mixture was stirred for 2.5 h at -78 0C, and DMF (1.00 mL, 12.9 mmol) was added again. The mixture was stirred overnight while warming up to rt. Aq. sat. NH4Cl (120 mL) was added, and the mixture was extracted with TBME (2x). The combined org. extracts were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the crude by FC (EtO Ac/heptane 1 :4) yielded the title compound (1.94 g, 45%). LC-MS: tR = 1.06 min; ES+: 326.02.
(S)-6-[3-(2,6-Dichloro-4-methyl-phenoxy)-pyrrolidin-l-yl]-pyridine-3-carbaldehyde (B5)
BuLi (1.6M in hexane, 5.15 mL, 8.24 mmol) was added to a sol. of compound A3 (2.88 g, 7.16 mmol) in THF (150 mL) at -78 0C. The mixture was stirred for 10 min at -78 0C, and
DMF (0.832 mL, 10.7 mmol) was added. The mixture was stirred for 2.5 h at -78 0C, and aq. sat. NH4Cl (120 mL) was added. The mixture was allowed to warm up to rt, and was extracted with TBME (2x). The combined org. extracts were washed with aq. sat. NH4Cl, dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the crude by FC (EtO Ac/heptane 1 :4 — > 2:3) yielded the title compound
(1.55 g, 61%). LC-MS: tR = 0.90 min; ES+: 351.14.
(£)-2-Cyano-3-{6-[(R)-3-(2,6-dichloro-4-methyl-phenoxy)-pyrrolidin-l-yl]-pyridin-3- yl}-acrylic acid (Cl) Compound B3 (2.97 g, 8.46 mmol) and cyanoacetic acid (719 mg, 8.46 mmol) were dissolved in toluene (85 mL), and piperidine (20 droplets) was added. The mixture was heated to reflux for 4 h, and was allowed to cool to rt. The stirring was stopped, and
crystals formed slowly overnight. Crystallization continued at 0 0C. The crystals were filtered, and washed with cold heptane. A second crop was obtained from the mother liquors from little CH2Cl2/heptane. Drying both crops under high vacuum yielded the title compound (4.06 g, 95%). LC-MS: tR = 0.93 min; ES+: 418.09.
(£)-2-Cyano-3-{6-[2-(2,6-dichloro-4-methyl-phenoxy)-ethoxy]-pyridin-3-yl}-acrylic acid (C2)
A mixture of compound B4 (3.40 g, 10.4 mmol), cyanoacetic acid (887 mg, 10.4 mmol) and piperidine (20 drops) in toluene (96 mL) was heated to reflux for 3 h. The mixture was allowed to cool to rt, while the product precipitated. Filtration, washing with cold heptane and drying this precipitate yielded the title compound (4.01 g, 98%). LC-MS: tR = 1.03 min; ES+: 393.07.
(JE)-2-Cyano-3-{6-[(S)-3-(2,6-dichloro-4-methyl-phenoxy)-pyrrolidin-l-yl]-pyridin-3- yl}-acrylic acid (C3)
Compound B5 (1.55 g, 4.40 mmol) and cyanoacetic acid (374 mg, 4.40 mmol) were dissolved in toluene (40 mL), and piperidine (20 droplets) was added. The mixture was heated to reflux overnight, and was allowed to cool to rt. The solvents were removed under reduced pressure. Purification of the crude by FC (CH2Cl2/MeOH/AcOH=20:0.5:0.05) yielded the title compound (1.24 g, 67%). LC-MS: tR = 0.94 min; ES+: 418.14.
Mixture of (R)-2-cyano-3-{6-[(R)-3-(2,6-dichloro-4-methyl-phenoxy)-pyrrolidin-l-yl]- pyridin-3-yl}-propionic acid and (S)-2-cyano-3-{6-[(R)-3-(2,6-dichloro-4-methyl- phenoxy)-pyrrolidin-l-yl]-pyridin-3-yl}-propionic acid (Dl)
Compound Cl (3.32 g, 7.94 mmol) was dissolved in MeOH (156 mL), and water (78 mL) and NaHCθ3 (567 mg, 5.56 mmol) were added. The mixture was cooled to 0 0C, and NaBH4 (1.80 g, 47.7 mmol) was added in portions over 30 min. The mixture was allowed to warm to rt, and aq. IM HCl was added till a pH=4 was reached. The solvents were partially removed under reduced pressure, and the residue was extracted with CH2Cl2 (3x). The combined org. extracts were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Drying under high vacuum yielded the crude title
compounds mixture (2.1O g, 63%) that was used without further purification. LC-MS: tR = 0.80 min; ES+: 420.10.
(rac.)-2-Cyano-3-{6-[2-(2,6-dichloro-4-methyl-phenoxy)-ethoxy]-pyridin-3-yl}- propionic acid (D2)
NaHCO3 (599 mg, 7.13 mmol) and water (100 mL) were added to a sol. of compound C2 (4.01 g, 10.9 mmol) in MeOH (200 mL). The mixture was cooled to 0 0C, and NaBH4 (2.31 g, 61.1 mmol) was added. The mixture was stirred for 75 min at 0 0C, and for 4.5 h at rt. Aq. IM HCl was added to pH 4, and the solvents were partially removed under reduced pressure. The aq. residue was extracted with CH2Cl2 (3x). The combined org. extracts were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Azeotropic removal of the solvents with toluene yielded the crude title compound (2.60 g, 65%) that was used without further purification. LC-MS: tR = 0.97 min; ES+: 395.09.
Mixture of (R)-2-cyano-3-{6-[(S)-3-(2,6-dichloro-4-methyl-phenoxy)-pyrrolidin-l-yl]- pyridin-3-yl}-propionic acid and (S)-2-cyano-3-{6-[(S)-3-(2,6-dichloro-4-methyl- phenoxy)-pyrrolidin-l-yl]-pyridin-3-yl}-propionic acid (D3)
Compound C3 (1.24 g, 2.95 mmol) was dissolved in MeOH (100 mL), and water (37 mL) and NaHCO3 (322 mg, 3.84 mmol) were added. The mixture was cooled to 0 0C, and
NaBH4 (1.78 g, 45.2 mmol) was added in portions over 4 h. The mixture was allowed to warm to rt, and aq. IM HCl was added till a pH=4 was reached. The solvents were partially removed under reduced pressure, and the residue was extracted with CH2Cl2 (3x).
The combined org. extracts were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Drying under high vacuum yielded the crude title compounds mixture (1.18 g, 95%) that was used without further purification. LC-MS: tR =
0.78 min; ES+: 420.10.
Mixture of (R)-7V-[2-Chloro-5-(3-methoxy-propyl)-benzyl]-2-cyano-7V-cyclopropyl-3- {6-[(R)-3-(2,6-dichloro-4-methyl-phenoxy)-pyrrolidin-l-yl]-pyridin-3-yl}- propionamide and (S)-N- [2-Chloro-5-(3-methoxy-propyl)-benzyl]-2-cyano-7V-
cyclopropyl-3-{6-[(R)-3-(2,6-dichloro-4-methyl-phenoxy)-pyrrolidin-l-yl]-pyridin-3- yl}-propionamide (El)
A mixture of compounds Dl (700 mg, 1.67 mmol), DMAP (50.8 mg, 0.416 mmol), HOBt
(270 mg, 2.00 mmol), DIPEA (1.14 mL, 6.66 mmol) and EDC-HCl (798 mg, 4.16 mmol) in CH2Cl2 (35 mL) was stirred for 75 min at rt. [2-Chloro-5-(3-methoxy-propyl)-benzyl]- cyclopropyl-amine (634 mg, 2.50 mmol) was added, and the mixture was stirred for 3 days.
The mixture was dissolved with CHCI3, and the resulting mixture was washed with aq. IM
HCl (2x). The combined aq. layers were extracted back with CHCI3, and the combined org. extracts were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the crude by FC (MeOH/CH2Cl2 1 :99 with 0.1% Et3N) yielded the title compounds mixture (436 mg, 40%). LC-MS: tR = 0.98 min; ES+: 656.96.
Mixture of (R)-N- [5-chloro-2-(3-methoxy-propyl)-pyridin-4-ylm ethyl] -2-cyano-7V- cyclopropyl-3-{6-[(R)-3-(2,6-dichloro-4-methyl-phenoxy)-pyrrolidin-l-yl]-pyridin-3- yl}-propionamide and (S)-N- [5-chloro-2-(3-methoxy-propyl)-pyridin-4-ylmethyl] -2- cyano-7V-cyclopropyl-3-{6-[(R)-3-(2,6-dichloro-4-methyl-phenoxy)-pyrrolidin-l-yl]- pyridin-3-yl}-propionamide (E2) A mixture of compounds Dl (700 mg, 1.67 mmol), DMAP (50.8 mg, 0.416 mmol), HOBt
(270 mg, 2.00 mmol), DIPEA (1.14 mL, 6.66 mmol) and EDC-HCl (798 mg, 4.16 mmol) in CH2Cl2 (35 mL) was stirred for 45 min at rt. [5-Chloro-2-(3-methoxy-propyl)-pyridin-
4-ylmethyl]-cyclopropyl-amine (637 mg, 2.50 mmol) was added, and the mixture was stirred for 3 days. CHCI3 was added, and the mixture was washed with aq. IM HCl (2x).
The combined aq. phases were extracted back with CHCI3. The combined org. extracts were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the residue by FC (CH2Cl2/Me0H/Et3N 20:0.4:0.04) yielded the title compounds mixture (796 mg, 73%). LC-MS: tR = 0.91 min; ES+: 657.97.
(rac.)-jV-[2-Chloro-5-(2-methoxy-ethyl)-benzyl]-2-cyano-7V-cyclopropyl-3-{6-[2-(2,6- dichloro-4-methyl-phenoxy)-ethoxy]-pyridin-3-yl}-propionamide (E3) A mixture of compound D2 (600 mg, 1.52 mmol), DMAP (46.4 mg, 0.380 mmol), HOBt
(246 mg, 1.82 mmol), DIPEA (1.04 mL, 6.07 mmol) and EDC-HCl (727 mg, 3.08 mmol) in CH2Cl2 (26 mL) was stirred for 45 min at rt. [2-Chloro-5-(3-methoxy-ethyl)-benzyl]-
cyclopropyl-amine (546 mg, 2.28 mmol) was added, and the mixture was stirred for 3 days. CHCI3 was added, and the mixture was washed with aq. IM HCl (2x). The combined aq. phases were extracted back with CHCI3. The combined org. extracts were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the residue by FC (CH2Cl2/Me0H/Et3N 25:0.2:0.05) yielded the title compound (550 mg, 59%). LC-MS: tR = 1.19 min; ES+: 616.46.
(rac.)-7V-[2-Chloro-5-(3-methoxy-propyl)-benzyl]-2-cyano-7V-cyclopropyl-3-{6-[2-(2,6- dichloro-4-methyl-phenoxy)-ethoxy]-pyridin-3-yl}-propionamide (E4) A mixture of compound D2 (800 mg, 2.02 mmol), DMAP (61.8 mg, 0.506 mmol), HOBt
(328 mg, 2.43 mmol), DIPEA (1.39 mL, 8.10 mmol) and EDC-HCl (970 mg, 5.10 mmol) in CH2Cl2 (35 mL) was stirred for 45 min at rt. [2-Chloro-5-(3-methoxy-propyl)-benzyl]- cyclopropyl-amine (771 mg, 3.04 mmol) was added, and the mixture was stirred for 3 days. CHCI3 was added, and the mixture was washed with aq. IM HCl (2x). The combined aq. phases were extracted back with CHCI3. The combined org. extracts were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the residue by FC (CH2Cl2/Me0H/Et3N 25:0.2:0.02) yielded the title compound (860 mg, 67%). LC-MS: tR = 1.09 min; ES+: 630.35.
Mixture of (R)-7V-[2-chloro-5-(2-methoxy-ethyl)-benzyl]-2-cyano-7V-cyclopropyl-3-{6- [(R)-3-(2,6-dichloro-4-methyl-phenoxy)-pyrrolidin-l-yl]-pyridin-3-yl}-propionamide and (S)-N- [2-chloro-5-(2-methoxy-ethyl)-benzyl]-2-cyano-7V-cyclopropyl-3- {6- [(R)-3- (2,6-dichloro-4-methyl-phenoxy)-pyrrolidin-l-yl]-pyridin-3-yl}-propionamide (E5)
A mixture of compounds Dl (350 mg, 0.833 mmol), DMAP (25.4 mg, 0.208 mmol), HOBt (135 mg, 1.00 mmol), DIPEA (0.570 mL, 3.33 mmol) and EDC-HCl (399 mg, 2.08 mmol) in CH2Cl2 (18 mL) was stirred for 45 min at rt. [2-Chloro-5-(3-methoxy-ethyl)-benzyl]- cyclopropyl-amine (300 mg, 1.25 mmol) was added, and the mixture was stirred for 3 days. CHCI3 was added, and the mixture was washed with aq. IM HCl (2x). The combined aq. phases were extracted back with CHCI3. The combined org. extracts were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the residue by FC (CH2Cl2/Me0H/Et3N 20:0.3:0.05) yielded the title compounds mixture (461 mg, 86%). LC-MS: tR = 0.99 min; ES+: 642.93.
(rac.)-7V-[5-Chloro-2-(3-methoxy-propyl)-pyridin-4-ylmethyl]-2-cyano-7V-cyclopropyl- 3-{6-[2-(2,6-dichloro-4-methyl-phenoxy)-ethoxy]-pyridin-3-yl}-propionamide (E6)
A mixture of compound D2 (800 mg, 2.02 mmol), DMAP (61.8 mg, 0.506 mmol), HOBt (328 mg, 2.43 mmol), DIPEA (1.39 mL, 8.10 mmol) and EDC-HCl (970 mg, 5.10 mmol) in CH2Cl2 (35 mL) was stirred for 45 min at rt. [5-Chloro-2-(3-methoxy-propyl)-pyridin- 4-ylmethyl]-cyclopropyl-amine (515 mg, 2.02 mmol) was added, and the mixture was stirred for 3 days. CHCI3 was added, and the mixture was washed with aq. IM HCl (2x). The combined aq. phases were extracted back with CHCI3. The combined org. extracts were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the residue by FC (CH2Cl2/Me0H/Et3N 20:0.3:0.02) yielded the title compound (680 mg, 53%). LC-MS: tR = 1.11 min; ES+: 632.97.
Mixture of (R)-7V-[2-Chloro-5-(2-methoxy-ethyl)-benzyl]-2-cyano-7V-cyclopropyl-3-{6- [(S)-3-(2,6-dichloro-4-methyl-phenoxy)-pyrrolidin-l-yl]-pyridin-3-yl}-propionamide and (S)-7V-[2-Chloro-5-(2-methoxy-ethyl)-benzyl]-2-cyano-7V-cyclopropyl-3-{6-[(S)-3- (2,6-dichloro-4-methyl-phenoxy)-pyrrolidin-l-yl]-pyridin-3-yl}-propionamide (E7)
A mixture of compounds D3 (540 mg, 1.29 mmol), DMAP (39.2 mg, 0.321 mmol), HOBt
(208 mg, 1.54 mmol), EDC-HCl (616 mg, 3.21 mmol) and DIPEA (1.10 mL, 6.43 mmol) in CH2Cl2 (27 mL) was stirred at rt for 45 min. [5-Chloro-2-(3-methoxy-ethyl)-pyridin-4- ylmethyl]-cyclopropyl-amine (532 mg, 1.93 mmol) was added, and the mixture was stirred overnight. CH2Cl2 was added, and the mixture was washed with aq. IM HCl. The org. layer was dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the crude by FC (CH2Cl2/MeOH/Et3N=20:0.3:0.05) yielded the title compounds mixture (483 mg, 59%). LC-MS: tR = 0.99 min; ES+: 641.16.
(S)-l-(5-Bromo-pyridin-2-yl)-pyrrolidin-3-ol (Fl)
A mixture of 2,5-dibromopyridine (28.6 g, 121 mmol) and (5)-3-hydroxypyrolidine (10.0 g, 115 mmol) in dry toluene (150 mL) was stirred under reflux for 20 h. The mixture was allowed to cool to rt, and the solvents were removed under reduced pressure. The residue was dissolved with EtOAc, and the resulting mixture was washed with aq. 10% K2CO3. The org. layer was dried over MgSO4, filtered, and the solvents were concentrated under
reduced pressure. Purification of the residue by FC (CH2Cl2/Me0H 99:1 → 98:2 → 97:3 → 96:4 → 95:5 → 94:6 → 93:7) yielded the title compound (15.39 g, 55 %). LC-MS: tR = 0.45 min; ES+: 245.11.
(rac.J-S-Amino-l-ll-^-^rt-butyl-dimethyl-silanyloxyJ-ethoxyl-thiazol-S-ylmethylJ-TV- cyclopropyl-7V-(2,3-dimethyl-benzyl)-propionamide (Ll)
A mixture of (rαc.)-2-{2-[2-(tert-butyl-dimethyl-silanyloxy)-ethoxy]-thiazol-5-ylmethyl}- 2-cyano-Λ/-cyclopropyl-Λ/-(2,3-dimethyl-benzyl)-acetamide (4.19 g, 8.15 mmol) and CoCl2 (108 mg, 0.815 mmol) in MeOH (90 mL) was cooled to 0 0C, and NaBH4 (1.28 g, 32.6 mmol) was added in portions. The mixture was stirred at 0 0C for 30 min, and NaBH4 (642 mg, 16.3 mmol) was added again. The mixture was stirred at 0 0C for 30 min, and NaBH4 (642 mg, 16.3 mmol) was added again. The mixture was stirred at 0 0C for 2 h, and NaBH4 (642 mg, 16.3 mmol) and MeOH (30 mL) were added. The mixture was stirred for 2 h at 0 0C, and was filtered over celite. The filtrate was evaporated under reduced pressure, and the residue was partitioned between CH2Cl2 and aq. IM NaOH. The org. layer was dried over Na2SO4, filtered, and the solvents were removed under reduced pressure. Drying the residue under high vacuum yielded the crude title compound (4.18 g, 99%) that was used without further purification. LC-MS: tR = 0.95 min; ES+: 518.46.
Mixture of (if)-2-aminomethyl-3-{2-[(S)-3-(tert-butyl-dimethyl-silanyloxy)-pyrrolidin- l-yl]-thiazol-5-yl}-7V-cyclopropyl-7V-(2,3-dichloro-benzyl)-propionamide and (S)-2- aminomethyl-3-{2-[(S)-3-(tert-butyl-dimethyl-silanyloxy)-pyrrolidin-l-yl]-thiazol-5- yl}-7V-cyclopropyl-7V-(2,3-dichloro-benzyl)-propionamide (L2)
A mixture of (i?)-3-{2-[(5)-3-(tert-butyl-dimethyl-silanyloxy)-pyrrolidin-l-yl]-thiazol-5- yl}-2-cyano-N-cyclopropyl-N-(2,3-dichloro-benzyl)-propionamide and (S)-3-{2-[(S)-3-
(tert-butyl-dimethyl-silanyloxy)-pyrrolidin- 1 -yl]-thiazol-5-yl} -2-cyano-N-cyclopropyl-N-
(2,3-dichloro-benzyl)-propionamide (3.66 g, 6.31 mmol) and CoCl2 (83.6 mg, 0.631 mmol) in MeOH (50 mL) was cooled to 0 0C, and NaBH4 (746 mg, 18.9 mmol) was added in portions. The mixture was stirred for a total time of 6 h at 0 0C, whereas NaBH4 (498 mg, 12.6 mmol) was added every 2 h. After the last addition, the mixture was stirred for 1 h at rt, and was filtered over celite. The filtrate was reduced under reduced pressure, and
partitioned between CH2Cl2 and aq. IM NaOH. The org. layer was dried over Na2SO4, filtered, and the solvents were removed under reduced pressure. Purification of the crude by FC (MeOH/CH2Cl2 1 :15) yielded the title compounds mixture (1.71 g, 46%). LC-MS: tR = 0.79 min; ES+: 583.31.
(rac.)-{3-{2-[2-(tert-Butyl-dimethyl-silanyloxy)-ethoxy]-thiazol-5-yl}-2-[cyclopropyl- (2,3-dimethyl-benzyl)-carbamoyl]-propyl}-carbamic acid tert-buty\ ester (Ml)
BoC2O (2.70 g, 12.1 mmol) was added to a sol. of compound Ll (4.18 g, 8.07 mmol) and DIPEA (2.82 mL, 16.1 mmol) in CH2Cl2 (150 mL) at 0 0C. The mixture was stirred for 3 days, while warming up to rt. The mixture was cooled to 0 0C, and was washed with aq. IM HCl (2x) and aq. sat. NaHCO3 (Ix). The combined aq. phases were extracted back with CH2Cl2 (Ix). The combined org. extracts were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the crude by FC (MeOH/CH2Cl2 1 :66) yielded the title compound (2.14 g, 43%). LC-MS: tR = 1.20 min; ES+: 618.56.
(rac.)-{2-[Cyclopropyl-(2,3-dimethyl-benzyl)-carbamoyl]-3-[2-(2-hydroxy-ethoxy)- thiazol-5-yl]-propyl}-carbamic acid tert-buty\ ester (M2)
TBAF (IM in THF, 6.90 mL; 6.90 mmol) was added to a sol. of compound Ml (2.14 g, 3.46 mmol) in THF (50 mL) at 0 0C. The mixture was stirred for 60 min at 0 0C, and aq. sat. NH4Cl was added. The mixture was extracted with Et2O (2x). The combined org. extracts were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the crude by FC (MeOH/CH2Cl2 1 :45 → 1 :40 → 1 :35) yielded the title compound (1.18 g, 68%). LC-MS: tR = 0.95 min; ES+: 504.44.
Mixture of (R)-{3-{2-[(5)-3-(tert-Butyl-dimethyl-silanyloxy)-pyrrolidin-l-yl]-thiazol-5- yl}-2-[cyclopropyl-(2,3-dichloro-benzyl)-carbamoyl]-propyl}-carbamic acid tert-buty\ ester and (S)-{3-{2-[(S)-3-(tert-Butyl-dimethyl-silanyloxy)-pyrrolidin-l-yl]-thiazol-5- yl}-2-[cyclopropyl-(2,3-dichloro-benzyl)-carbamoyl]-propyl}-carbamic acid tert-buty\ ester (M3)
BoC2O (979 mg, 4.39 mmol) was added to a sol. of compounds L2 (1.71 g, 2.93 mmol) and DIPEA (1.02 mL, 5.86 mmol) in CH2Cl2 (50 mL) at 0 0C. The mixture was stirred
overnight, while warming up to rt. Ice was added, and the mixture was washed with cold aq. IM HCl (2x) and aq. sat. NaHCO3 (Ix). The combined aq. layers were extracted back with CH2Cl2 (Ix). The combined org. extracts were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the crude by FC (MeOH/CH2Cl2 1 :19) yielded the title compounds mixture (1.85 g, 93%). LC-MS: tR = 1.03 min; ES+: 683.37.
Mixture of (R)-{2- [cyclopropyl-(2,3-dichloro-benzyl)-carbamoyl] -3- [2-((S)-3-hydroxy- pyrrolidin-l-yl)-thiazol-5-yl]-propyl}-carbamic acid tert-buty\ ester and (S)-{2- [cyclopropyl-(2,3-dichloro-benzyl)-carbamoyl]-3-[2-((S)-3-hydroxy-pyrrolidin-l-yl)- thiazol-5-yl]-propyl}-carbamic acid tert-butyl ester (M4)
TBAF (IM in THF, 5.4 mL; 5.4 mmol) was added to a sol. of compounds M3 (1.85 g, 2.71 mmol) in THF (40 mL) at 0 0C. The mixture was stirred for 60 min at 0 0C, and aq. sat. NH4Cl was added. The mixture was extracted with Et2O (2x). The combined org. extracts were dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the crude by FC (MeOH/CH2Cl2 1 :45 → 1 :40 → 1 :35) yielded the title compounds mixture (1.48 g, 96%). LC-MS: tR = 0.85 min; ES+: 569.37.
(rac. )-(2- [Cyclopr opyl-(2,3-dimethyl-benzyl)-carbamoyl] -3- {2- [2-(2,6-dichloro-4- methyl-phenoxy)-ethoxy]-thiazol-5-yl}-propyl)-carbamic acid tert-butyl ester (Nl)
According to General procedure A, from compound M2 and 2,6-dichloro-p-cresol. LC- MS: tR = 1.20 min; ES+: 662.36.
(rac.)-{3-{2-[2-(2-Chloro-3,6-difluoro-phenoxy)-ethoxy]-thiazol-5-yl}-2-[cyclopropyl- (2,3-dimethyl-benzyl)-carbamoyl]-propyl}-carbamic acid tert-butyl ester (N2)
According to General procedure A, from compound M2 and 2-chloro-3,6-difluorophenol. LC-MS: tR = 1.13 min; ES+: 650.41.
(rac. )-(2- [Cyclopr opyl-(2,3-dimethyl-benzyl)-carbamoyl] -3- {2- [2-(2,6-dichloro- phenoxy)-ethoxy]-thiazol-5-yl}-propyl)-carbamic acid tert-butyl ester (N3)
According to General procedure A, from compound M2 and 2,6-dichlorophenol. LC-MS: tR = 1.17 min; ES+: 648.33.
(rac. )-(2- [Cyclopr opyl-(2,3-dimethyl-benzyl)-carbamoyl] -3- {2- [2-(2,6-dichloro-3,4- dimethyl-phenoxy)-ethoxy]-thiazol-5-yl}-propyl)-carbamic acid tert-butyl ester (N4)
According to General procedure A, from compound M2 and 2,6-dichloro-3,4- dimethylphenol. LC-MS: tR = 1.22 min; ES+: 676.39.
(rac.)-{3-{2-[2-(2-Chloro-6-fluoro-3-methyl-phenoxy)-ethoxy]-thiazol-5-yl}-2- [cyclopropyl-(2,3-dimethyl-benzyl)-carbamoyl]-propyl}-carbamic acid tert-butyl ester
(N5) According to General procedure A, from compound M2 and 2-chloro-6-fluoro-3- methylphenol. LC-MS: tR = 1.18 min; ES+: 646.40.
Mixture of (rac.)-(R*)-{3-[2-(2-{(R*)-4-[l-(tert-butyl-dimethyl-silanyloxy)-ethyl]-2,6- dichloro-phenoxy}-ethoxy)-thiazol-5-yl]-2-[cyclopropyl-(2,3-dimethyl-benzyl)- carbamoylj-propylj-carbamic acid tert-butyl ester and (røc.)-(R*)-{3-[2-(2-{(S*)-4-[l- (tert-butyl-dimethyl-silanyloxy)-ethyl]-2,6-dichloro-phenoxy}-ethoxy)-thiazol-5-yl]-2- [cyclopropyl-(2,3-dimethyl-benzyl)-carbamoyl]-propyl}-carbamic acid tert-butyl ester (N6) According to General procedure A, from compound M2 and (rac.)-4-[\-(tert- butyldimethylsilanyloxy)ethyl]-2,6-dichlorophenol. LC-MS: tR = 1.31 min; ES+: 808.47.
(rac.)-{3-{2-[2-(3-Chloro-2,6-difluoro-phenoxy)-ethoxy]-thiazol-5-yl}-2-[cyclopropyl- (2,3-dimethyl-benzyl)-carbamoyl]-propyl}-carbamic acid tert-butyl ester (N7)
According to General procedure A, from compound M2 and 3-chloro-2,6-difluorophenol. LC-MS: tR = 1.16 min; ES+: 650.39.
(rac. )-(2- [Cyclopr opyl-(2,3-dimethyl-benzyl)-carbamoyl] -3- {2- [2-(2,6-dichloro-4- fluoro-phenoxy)-ethoxy]-thiazol-5-yl}-propyl)-carbamic acid tert-butyl ester (N 8)
According to General procedure A, from compound M2 and 2,6-dichloro-4-fluorophenol. LC-MS: tR = 1.18 min; ES+: 666.38.
Mixture of ((R)-2- [cyclopropyl-(2,3-dichloro-benzyl)-carbamoyl] -3-{2- [(R)-3-(2,6- dichloro-^methyl-phenoxyJ-pyrrolidin-l-ylJ-thiazol-S-ylJ-propylJ-carbamic acid tert- butyl ester and ((S)-2-[cyclopropyl-(2,3-dichloro-benzyl)-carbamoyl]-3-{2-[(R)-3-(2,6- dichloro-^methyl-phenoxyJ-pyrrolidin-l-yll-thiazol-S-ylJ-propylJ-carbamic acid tert- butyl ester (N9)
According to General procedure A, from compounds M4 and 2,6-dichloro-/?-cresol. LC- MS: tR = 1.04 min; ES+: 729.18.
Mixture of ((R)-2- [cyclopropyl-(2,3-dichloro-benzyl)-carbamoyl] -3-{2- [(R)-3-(2,6- dichloro-phenoxyJ-pyrrolidin-l-ylJ-thiazol-S-ylJ-propylJ-carbamic acid tert-bntyλ ester and ((S)-I- [cyclopr opyl-(2,3-dichlor o-benzyl)-carbamoyl] -3- {2- [(R)-3-(2,6- dichloro-phenoxyJ-pyrrolidin-l-yll-thiazol-S-ylJ-propylJ-carbamic acid tert-buty\ ester (NlO)
According to General procedure A, from compounds M4 and 2,6-dichlorophenol. LC-MS: tR = 1.02 min; ES+: 715.28.
Mixture of ((R)-2- [cyclopropyl-(2,3-dichloro-benzyl)-carbamoyl] -3-{2- [(R)-3-(2,6- dichloro-S^-dimethyl-phenoxyJ-pyrrolidin-l-ylJ-thiazol-S-ylJ-propylJ-carbamic acid tert-buty\ ester and ((S)-2-[cyclopropyl-(2,3-dichloro-benzyl)-carbamoyl]-3-{2-[(R)-3- (2,6-dichloro-3,4-dimethyl-phenoxy)-pyrrolidin-l-yl]-thiazol-5-yl}-propyl)-carbamic acid tert-buty\ ester (Nil)
According to General procedure A, from compounds M4 and 2,6-dichloro-3,4- dimethylphenol. LC-MS: tR = 1.06 min; ES+: 741.37.
Mixture of {(R)-3-{2-[(/f)-3-(2-chloro-6-fluoro-3-methyl-phenoxy)-pyrrolidin-l-yl]- thiazol-5-yl}-2- [cyclopropyl-(2,3-dichloro-benzyl)-carbamoyl] -propyl}-carbamic acid tert-buty\ ester and {(S)-3-{2-[(R)-3-(2-chloro-6-fluoro-3-methyl-phenoxy)-pyrrolidin- l-yl]-thiazol-5-yl}-2-[cyclopropyl-(2,3-dichloro-benzyl)-carbamoyl]-propyl}-carbamic acid tert-buty\ ester (N12) According to General procedure A, from compounds M4 and 2-chloro-6-fluoro-3- methylphenol. LC-MS: tR = 1.02 min; ES+: 713.32.
Mixture of {(R)-3-[2-((R)-3-{(R)-4-[l-(tert-butyl-dimethyl-silanyloxy)-ethyl]-2,6- dichloro-phenoxy}-pyrrolidin-l-yl)-thiazol-5-yl]-2-[cyclopropyl-(2,3-dichloro-benzyl)- carbamoyl]-propyl}-carbamic acid tert-buty\ ester, {(R)-3-[2-((R)-3-{(S)-4-[l-(tert- butyl-dimethyl-silanyloxy)-ethyl]-2,6-dichloro-phenoxy}-pyrrolidin-l-yl)-thiazol-5- yl]-2-[cyclopropyl-(2,3-dichloro-benzyl)-carbamoyl]-propyl}-carbamic acid tert-buty\ ester, {(5)-3-[2-((R)-3-{(R)-4-[l-(tert-butyl-dimethyl-silanyloxy)-ethyl]-2,6-dichloro- phenoxy}-pyrrolidin-l-yl)-thiazol-5-yl]-2-[cyclopropyl-(2,3-dichloro-benzyl)- carbamoyl]-propyl}-carbamic acid tert-buty\ ester, and {(S)-3-[2-((R)-3-{(S)-4-[l-(tert- butyl-dimethyl-silanyloxy)-ethyl]-2,6-dichloro-phenoxy}-pyrrolidin-l-yl)-thiazol-5- yl]-2-[cyclopropyl-(2,3-dichloro-benzyl)-carbamoyl]-propyl}-carbamic acid tert-buty\ ester (N13)
According to General procedure A, from compounds M4 and (rac.)-4-[\-(tert- butyldimethylsilanyloxy)ethyl]-2,6-dichlorophenol. LC-MS: tR = 1.15 min; ES+: 873.44.
Mixture of {(R)-3-{2-[(R)-3-(3-chloro-2,6-difluoro-phenoxy)-pyrrolidin-l-yl]-thiazol- 5-yl}-2-[cyclopropyl-(2,3-dichloro-benzyl)-carbamoyl]-propyl}-carbamic acid tert- butyl ester and {(S)-3-{2-[(R)-3-(3-chloro-2,6-difluoro-phenoxy)-pyrrolidin-l-yl]- thiazol-5-yl}-2- [cyclopropyl-(2,3-dichloro-benzyl)-carbamoyl] -propyl}-carbamic acid tert-buty\ ester (N14) According to General procedure A, from compounds M4 and 3-chloro-2,6-difluorophenol. LC-MS: tR = 1.01 min; ES+: 717.35.
Mixture of ((R)-I- [cyclopropyl-(2,3-dichloro-benzyl)-carbamoyl] -3-{2- [(R)-3-(2,6- dichloro^-fluoro-phenoxyJ-pyrrolidin-l-yll-thiazol-S-ylJ-propylJ-carbamic acid tert- butyl ester and ((S)-2-[cyclopropyl-(2,3-dichloro-benzyl)-carbamoyl]-3-{2-[(R)-3-(2,6- dichloro^-fluoro-phenoxyJ-pyrrolidin-l-ylJ-thiazol-S-ylJ-propylJ-carbamic acid tert- butyl ester (N15)
According to General procedure A, from compounds M4 and 2,6-dichloro-4-fluorophenol. LC-MS: tR = 1.02 min; ES+: 733.32.
Examples
Example 1
(rac.)-3-Amino-7V-cyclopropyl-2-{2-[2-(2,6-dichloro-4-methyl-phenoxy)-ethoxy]- thiazol-5-ylmethyl}-7V-(2,3-dimethyl-benzyl)-propionamide According to General procedure B, from compound Nl. LC-MS: tR = 0.94 min; ES+: 562.33.
Example 2
(rac.)-3-Amino-2-{2-[2-(2-chloro-3,6-difluoro-phenoxy)-ethoxy]-thiazol-5-ylmethyl}- 7V-cyclopropyl-7V-(2,3-dimethyl-benzyl)-pr opionamide
According to General procedure B, from compound N2. LC-MS: tR = 0.89 min; ES+: 550.30.
Example 3 (rac.)-3-Amino-7V-cyclopropyl-2-{2-[2-(2,6-dichloro-phenoxy)-ethoxy]-thiazol-5- ylmethyl}-7V-(2,3-dimethyl-benzyl)-propionamide
According to General procedure B, from compound N3. LC-MS: tR = 0.92 min; ES+: 550.26.
Example 4
(rac.)-3-Amino-7V-cyclopropyl-2-{2-[2-(2,6-dichloro-3,4-dimethyl-phenoxy)-ethoxy]- thiazol-5-ylmethyl}-7V-(2,3-dimethyl-benzyl)-propionamide
According to General procedure B, from compound N4. LC-MS: tR = 0.97 min; ES+:
576.32.
Example 5
(me. )-3- Amino-2- {2- [2-(2-chloro-6-fluor o-3-methyl-phenoxy)-ethoxy] -thiazol-5- ylmethyl}-7V-cyclopropyl-7V-(2,3-dimethyl-benzyl)-propionamide
According to General procedure B, from compound N5. LC-MS: tR = 0.92 min; ES+: 546.26.
Example 6
(rac.)-(R*)-3-Amino-7V-cyclopropyl-2-(2-{2-[2,6-dichloro-4-((R*)-l-hydroxy-ethyl)- phenoxy]-ethoxy}-thiazol-5-ylmethyl)-7V-(2,3-dimethyl-benzyl)-propionamide and (rac.)-(R*)-3-amino-7V-cyclopropyl-2-(2-{2-[2,6-dichloro-4-((S*)-l-hydroxy-ethyl)- phenoxy]-ethoxy}-thiazol-5-ylmethyl)-7V-(2,3-dimethyl-benzyl)-propionamide
According to General procedure B, from compound N6. LC-MS: tR = 0.87 min; ES+: 592.31.
Example 7 (me. )-3- Amino-2- {2- [2-(3-chloro-2,6-difluoro-phenoxy)-ethoxy] -thiazol-5-ylmethyl}- 7V-cyclopropyl-7V-(2,3-dimethyl-benzyl)-propionamide
According to General procedure B, from compound N7. LC-MS: tR = 0.92 min; ES+: 550.25.
Example 8
(rac.)-3-Amino-7V-cyclopropyl-2-{2-[2-(2,6-dichloro-4-fluoro-phenoxy)-ethoxy]- thiazol-5-ylmethyl}-7V-(2,3-dimethyl-benzyl)-propionamide
According to General procedure B, from compound N8. LC-MS: tR = 0.93 min; ES+: 566.36.
Example 9
Mixture of (R)-2-aminomethyl-7V-cyclopropyl-7V-(2,3-dichloro-benzyl)-3-{2-[(R)-3- (2,6-dichloro-4-methyl-phenoxy)-pyrrolidin-l-yl]-thiazol-5-yl}-propionamide and (S)- 2-aminomethyl-7V-cyclopropyl-7V-(2,3-dichloro-benzyl)-3-{2-[(R)-3-(2,6-dichloro-4- methyl-phenoxy)-pyrrolidin-l-yl]-thiazol-5-yl}-propionamide
According to General procedure B, from compounds N9. LC-MS: tR = 0.84 min; ES+: 629.18.
Example 10 Mixture of (R)-2-aminomethyl-7V-cyclopropyl-7V-(2,3-dichloro-benzyl)-3-{2-[(R)-3- (2,6-dichloro-phenoxy)-pyrrolidin-l-yl]-thiazol-5-yl}-propionamide and (S)-2-
aminomethyl-7V-cyclopropyl-7V-(2,3-dichloro-benzyl)-3-{2-[(R)-3-(2,6-dichloro- phenoxy)-pyrrolidin-l-yl]-thiazol-5-yl}-propionamide
According to General procedure B, from compounds NlO. LC-MS: tR = 0.82 min; ES+: 613.19.
Example 11
Mixture of (R)-2-aminomethyl-7V-cyclopropyl-7V-(2,3-dichloro-benzyl)-3-{2-[(R)-3- (2,6-dichloro-3,4-dimethyl-phenoxy)-pyrrolidin-l-yl]-thiazol-5-yl}-propionamide and (S)-2-aminomethyl-7V-cyclopropyl-7V-(2,3-dichloro-benzyl)-3-{2-[(R)-3-(2,6-dichloro- 3,4-dimethyl-phenoxy)-pyrrolidin-l-yl]-thiazol-5-yl}-propionamide
According to General procedure B, from compounds Nil. LC-MS: tR = 0.86 min; ES+: 643.09.
Example 12 Mixture of (R)-2-aminomethyl-3-{2-[(R)-3-(2-chloro-6-fluoro-3-methyl-phenoxy)- pyrrolidin-l-yl]-thiazol-5-yl}-7V-cyclopropyl-7V-(2,3-dichloro-benzyl)-propionamide and (S)-2-aminomethyl-3-{2-[(/f)-3-(2-chloro-6-fluoro-3-methyl-phenoxy)-pyrrolidin- l-yl]-thiazol-5-yl}-7V-cyclopropyl-7V-(2,3-dichloro-benzyl)-propionamide According to General procedure B, from compounds N12. LC-MS: tR = 0.82 min; ES+: 611.21.
Example 13
Mixture of (R)-2-aminomethyl-7V-cyclopropyl-7V-(2,3-dichloro-benzyl)-3-(2-{(R)-3-
[(R)-2,6-dichloro-4-(l-hydroxy-ethyl)-phenoxy]-pyrrolidin-l-yl}-thiazol-5-yl)- propionamide, (S)-2-aminomethyl-7V-cyclopropyl-7V-(2,3-dichloro-benzyl)-3-(2-{(R)-3- [(/f)-2,6-dichloro-4-(l-hydroxy-ethyl)-phenoxy]-pyrrolidin-l-yl}-thiazol-5-yl)- propionamide, (R)-2-aminomethyl-7V-cyclopropyl-7V-(2,3-dichloro-benzyl)-3-(2-{(/f)-3- [(S)-2,6-dichloro-4-(l-hydroxy-ethyl)-phenoxy]-pyrrolidin-l-yl}-thiazol-5-yl)- propionamide, and (S)-2-aminomethyl-7V-cyclopropyl-7V-(2,3-dichloro-benzyl)-3-(2- {(R)-3-[(S)-2,6-dichloro-4-(l-hydroxy-ethyl)-phenoxy]-pyrrolidin-l-yl}-thiazol-5-yl)- propionamide
According to General procedure B, from compounds N13. LC-MS: tR = 0.78 min; ES+: 659.23.
Example 14 Mixture of (R)-2-aminomethyl-3-{2-[(R)-3-(3-chloro-2,6-difluoro-phenoxy)- pyrrolidin-l-yl]-thiazol-5-yl}-7V-cyclopropyl-7V-(2,3-dichloro-benzyl)-propionamide and (S)-2-aminomethyl-3-{2-[(R)-3-(3-chloro-2,6-difluoro-phenoxy)-pyrrolidin-l-yl]- thiazol-5-yl}-7V-cyclopropyl-7V-(2,3-dichloro-benzyl)-propionamide According to General procedure B, from compounds N14. LC-MS: tR = 0.82 min; ES+: 617.24.
Example 15
Mixture of (R)-2-aminomethyl-7V-cyclopropyl-7V-(2,3-dichloro-benzyl)-3-{2-[(R)-3- (2,6-dichloro-4-fluoro-phenoxy)-pyrrolidin-l-yl]-thiazol-5-yl}-propionamide and (S)- 2-aminomethyl-7V-cyclopropyl-7V-(2,3-dichloro-benzyl)-3-{2-[(R)-3-(2,6-dichloro-4- fluoro-phenoxy)-pyrrolidin-l-yl]-thiazol-5-yl}-propionamide
According to General procedure B, from compounds N15. LC-MS: tR = 0.83 min; ES+: 633.22.
Example 16
(R)-2-Aminomethyl-7V-[2-chloro-5-(3-methoxy-propyl)-benzyl]-7V-cyclopropyl-3-{6- [(R)-3-(2,6-dichloro-4-methyl-phenoxy)-pyrrolidin-l-yl]-pyridin-3-yl}-propionamide
CoCl2 (17.3 mg, 0.133 mmol) was added to a sol. of compounds El (436 mg, 0.665 mmol) in MeOH (8.00 mL) at 0 0C. NaBH4 (101 mg, 2.66 mmol) was added in portions. The mixture was stirred for 90 min, and CH2Cl2 and aq. IM NaOH were added. The mixture was filtered through celite, and the phases were separated. The org. phase was washed with aq. IM NaOH, water, and brine. The org. layer was dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the crude by FC (MeOH/CH2Cl2 1 :20 with 0.1% Et3N) yielded the title compound mixed with its corresponding (S, i?)-diastereoisomer (241 mg, 55%). Separation of this mixture by HPLC using a chiral stationary phase (Regis Whelk column, 10 μm, 50 m x 250 mm, 120 niL/min, gradient from EtOH/hexane 25:75 with 0.15% Et3N to EtOH/hexane 70:30 with
0.15% Et3N over 30 min) yielded the title compound. LC-MS: tR = 0.98 min; ES+: 656.22. Chiral, preparative Regis Whelk 01 column: tR = 25.9 min.
Example 17 (R)-2-Aminomethyl-7V-[5-chloro-2-(3-methoxy-propyl)-pyridin-4-ylmethyl]-7V- cyclopropyl-3-{6-[(R)-3-(2,6-dichloro-4-methyl-phenoxy)-pyrrolidin-l-yl]-pyridin-3- yl}-propionamide
CoCl2 (41.9 mg, 0.323 mmol) was added to a sol. of compounds E2 (796 mg, 1.21 mmol) in MeOH (13 mL). NaBH4 (183 mg, 4.85 mmol) was added in portions, and the mixture was stirred for 4 h at 0 0C. The mixture was diluted with CH2Cl2 and aq. IM NaOH. The mixture was filtered over celite, and the phases were separated. The org. layer was washed with aq. IM NaOH and brine, was dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the residue by FC (CH2Cl2/Me0H/Et3N 25:1.0:0.1) yielded the title compound mixed with its corresponding (S, R)- diastereoisomer. Separation of this mixture by HPLC using a chiral stationary phase (Regis Whelk column, 10 μm, 50 m x 250 mm, 120 mL/min, gradient from EtOH/hexane 25:75 with 0.15% Et3N to EtOH/hexane 70:30 with 0.15% Et3N over 30 min) yielded the title compound. LC-MS: tR = 0.78 min; ES+: 660.21. Chiral, preparative Regis Whelk 01 column: tR = 27.9 min.
Example 18
(R)-2-Aminomethyl-7V-[2-chloro-5-(2-methoxy-ethyl)-benzyl]-7V-cyclopropyl-3-{6-[2-
(2,6-dichloro-4-methyl-phenoxy)-ethoxy]-pyridin-3-yl}-propionamide
CoCl2 (23.1 mg, 0.178 mmol) was added to a sol. of compound E3 (550 mg, 0.891 mmol) in MeOH (11 mL) at 0 0C. NaBH4 (135 mg, 3.57 mmol) was added in portions, and the mixture was stirred for 30 min. CH2Cl2 was added, and the mixture was washed with aq.
IM NaOH. The org. layer was dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the crude by FC (CH2Cl2/Me0H/Et3N=25: 1 :0.1) yielded the racemic title compound (236 mg, 43%). Separation of this mixture by HPLC using a chiral stationary phase (Regis Whelk column, 10 μm, 50 m x 250 mm, 120 mL/min, gradient from EtOH/hexane 25:75 with 0.15% Et3N to EtOH/hexane 70:30 with
0.15% Et3N over 30 min) yielded the title compound (73 mg, 37%). LC-MS: tR = 0.93 min; ES+: 619.94. Chiral, preparative Regis Whelk 01 column: tR = 16.6 min.
Example 19 (R)-2-Aminomethyl-7V-[2-chloro-5-(3-methoxy-propyl)-benzyl]-7V-cyclopropyl-3-{6-[2- (2,6-dichloro-4-methyl-phenoxy)-ethoxy]-pyridin-3-yl}-propionamide
CoCl2 (35.4 mg, 0.273 mmol) was added to a sol. of compound E4 (860 mg, 1.36 mmol) in MeOH (15 mL) at 0 0C. NaBH4 (306 mg, 5.45 mmol) was added in portions, and the mixture was stirred for 30 min. CH2Cl2 was added, and the mixture was washed with aq. IM NaOH. The org. layer was dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the crude by FC (CH2Cl2/Me0H/Et3N=25: 1 :0.1) yielded the racemic title compound (453 mg, 52%). Separation of this mixture by HPLC using a chiral stationary phase (Regis Whelk column, 10 μm, 50 m x 250 mm, 120 mL/min, isocratic conditions EtOH/hexane 40:60 with 0.1% Et3N) yielded the title compound (100 mg, 23%). LC-MS: tR = 0.96 min; ES+: 636.43. Chiral, preparative Regis Whelk 01 column: tR = 14.3 min.
Example 20 (R)-2-Aminomethyl-7V-[2-chloro-5-(2-methoxy-ethyl)-benzyl]-7V-cyclopropyl-3-{6-[(/f)- 3-(2,6-dichloro-4-methyl-phenoxy)-pyrrolidin-l-yl]-pyridin-3-yl}-propionamide
CoCl2 (18.7 mg, 0.144 mmol) was added to a sol. of compounds E5 (461 mg, 0.718 mmol) in MeOH (8.6 mL) at 0 0C. NaBH4 (109 mg, 2.87 mmol) was added in portions, and the mixture was stirred for 30 min. CH2Cl2 was added, and the mixture was washed with aq. IM NaOH. The org. layer was dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the crude by FC (CH2Cl2/Me0H/Et3N=25: 1 :0.1) yielded the title compound mixed with its diastereoisomer from compounds E5 (229 mg, 50%). Separation of this mixture by HPLC using a chiral stationary phase (Regis Whelk column, 10 μm, 50 m x 250 mm, 120 mL/min, gradient from EtOH/hexane 25:75 with 0.15% Et3N to EtOH/hexane 70:30 with 0.15% Et3N over 30 min) yielded the title compound (62 mg, 27%). LC-MS: tR = 0.96 min; ES+: 636.43. Chiral, preparative Regis Whelk 01 column: tR = 25.9 min.
Example 21
(R)-2-Aminomethyl-7V-[5-chloro-2-(3-methoxy-propyl)-pyridin-4-ylmethyl]-7V- cyclopropyl-3-{6-[2-(2,6-dichloro-4-methyl-phenoxy)-ethoxy]-pyridin-3-yl}- propionamide CoCl2 (20.0 mg, 0.154 mmol) was added to a sol. of compound E6 (470 mg, 0.744 mmol) in MeOH (8.0 niL) at 0 0C. NaBH4 (200 mg, 3.17 mmol) was added in portions, and the mixture was stirred for 30 min. CH2Cl2 was added, and the mixture was washed with aq. IM NaOH. The org. layer was dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the crude by FC (CH2Cl2/MeOH/Et3N=20: 1 :0.1) yielded the racemic title compound (275 mg, 58%). Separation of this mixture by HPLC using a chiral stationary phase (Regis Whelk column, 10 μm, 50 m x 250 mm, 120 mL/min, isocratic conditions EtOH/hexane 40:60 with 0.1% Et3N) yielded the title compound (93 mg, 28%). LC-MS: tR = 0.87 min; ES+: 636.99. Chiral, preparative Regis Whelk 01 column: tR = 17.0 min.
Example 22
(R)-2-Aminomethyl-7V-[2-chloro-5-(2-methoxy-ethyl)-benzyl]-7V-cyclopropyl-3-{6-[(S)-
3-(2,6-dichloro-4-methyl-phenoxy)-pyrrolidin-l-yl]-pyridin-3-yl}-propionamide
CoCl2 (19.6 mg, 0.151 mmol) was added to a sol. of compounds E7 (483 mg, 0.753 mmol) in MeOH (9.3 mL) at 0 0C. NaBH4 (114 mg, 3.02 mmol) was added in portions, and the mixture was stirred for 30 min. CH2Cl2 was added, and the mixture was washed with aq. IM NaOH. The org. layer was dried over MgSO4, filtered, and the solvents were removed under reduced pressure. Purification of the crude by FC (CH2Cl2/MeOH/Et3N=20: 1 :0.1) yielded the title compound together with its (S, 5)-diastereoisomer (300 mg, 62%). Separation of this mixture by HPLC using a chiral stationary phase (Regis Whelk column, 10 μm, 50 m x 250 mm, 120 mL/min, isocratic conditions EtOH/hexane 40:60 with 0.1% Et3N) yielded the title compound (75 mg, 25%). LC-MS: tR = 0.82 min; ES+: 645.20. Chiral, preparative Regis Whelk 01 column: tR = 29.1 min.
Biological Assays
1. Enzyme immuno assay (EIA) to estimate Angl accumulation and renin inhibition
1.1 Preparation of Angl-BSA conjugate
1.3 mg (1 μmol) of Angl [1-10 (Bachem, H-1680)] and 17 mg (0.26 μmol) of BSA (Fluka, 05475) were dissolved in 4 rnL of 0.1M phosphate buffer, pH 7.4, after which 2 mL of a 1 :100 dilution of glutaraldehyde in H2O (Sigma G-5882) was added dropwise. The mixture was incubated overnight at 4 0C, then dialyzed against 2 liters of 0.9% NaCl, twice for 4 h at rt, followed by dialysis against 2 liters of PBS IX overnight at rt. The solution was then filtered with a Syringe filter, 0.45 μm (Nalgene, Cat. No. 194-2545). The conjugate can be stored in polypropylene tubes in 0.05% sodium azide at 4 0C for at least 12 months.
1.2 Preparation of BS A- Angl coated MTP
Microtiter plates (MPT384, MaxiSorpTM^ Nunc) were incubated overnight at 4 0C with 80 μl of Angl (l-10)/BSA conjugate, diluted l :100'000 in PBS IX in a teflon beaker (exact dilution dependent on batch of conjugate), emptied, filled with 90 μl of blocking solution [0.5% BSA (Sigma A-2153) in PBS IX, 0.02% NaN3], and incubated for at least 2 h at rt, or overnight at 4 0C. 96 well MTP (MaxiSorp™, Nunc) were coated with 200 μl conjugate and blocked with 250 μl blocking solution as above, except that the blocking solution contained 3% BSA. The plates can be stored in blocking solution at 4 0C for 1 month.
1.3 Angl-EIA in 384 well MTP The Angl (l-10)/BSA coated MTP were washed 3 times with wash buffer (PBS IX, 0.01% Tween 20) and filled with 75 μl of primary antibody solution (anti-Angl antiserum, pre- diluted 1 :10 in horse serum), diluted to a final concentration of 1 :100 OOO in assay buffer (PBS IX, ImM EDTA, 0.1% BSA, pH 7.4). 5 μl of the renin reaction (or standards in assay buffer) (see below) were added to the primary antibody solution and the plates were incubated overnight at 4 0C. After the incubation the plates were washed 3 times with wash buffer and incubated with secondary antibody [anti-rabbit IgG, linked to horseradish peroxidase (Amersham Bioscience, NA 934V), diluted 1 :2 OOO in wash buffer] for 2 h at
rt. The plates were washed 3 times with wash buffer and then incubated for 1 h at rt with substrate solution [1.89mM ABTS (2.2'-azino-di-(3-ethyl-benzthiazolinsulfonate)] (Roche Diagnostics, 102 946) and 2.36mM H2O2 [30%, (Fluka, 95300] in substrate buffer (0.1M sodium acetate, 0.05M sodium dihydrogen phosphate, pH 4.2). The OD of the plate was read at 405 nm in a microplate reader (FLUOStar Optima from BMG). The production of Angl during the renin reaction was quantified by comparing the OD of the sample with the OD of a standard curve of Angl(l-lO), measured in parallel.
2. Primary renin inhibition assay: IC50 in buffer, 384 well MTP The renin assay was adapted from an assay described before (Fischli W. et ah, Hypertension, 1991, 18:22-31) and consists of two steps: in the first step, recombinant human renin is incubated with its substrate (commercial human tetradecapeptide renin substrate) to create the product Angiotensin I (Angl). In the second step, the accumulated Angl is measured by an immunological assay (enzyme immuno assay, EIA). The detailed description of this assay is found below. The EIA is very sensitive and well suited for renin activity measurements in buffer or in plasma. Due to the low concentration of renin used in this assay (2 fmol per assay tube or 10 pM) it is possible to measure inhibitor affinities in this primary assay down to low pM concentration.
2.1 Methodology
Recombinant human renin (3 pg/μl) in assay buffer (PBS IX, ImM EDTA, 0.1% BSA, pH 7.4), human tetradecapeptide (1-14) substrate (Bachem, M-1120) [5 μM in 10 mM HCl], hydroxyquinoline sulfate (Fluka, 55100) [30 mM in H2O] and assay buffer were premixed at 4 0C at a ratio of 100:30:10:145. 47.5 μl per well of this premix was transferred into polypropylene plates (MTP384, Nunc). Test compounds were dissolved and diluted in 100% DMSO and 2.5 μl added to the premix, then incubated at 37 0C for 3 h. At the end of the incubation period, 5 μl of the renin reaction (or standards in assay buffer) were transferred into EIA assays (as described above) and Angl produced by renin was quantified. The percentage of renin inhibition (Angl decrease) was calculated for each concentration of compound and the concentration of renin inhibition was determined that inhibited the enzyme activity by 50% (IC50). The ICso-values of the compounds of the
Examples are below 100 nM. Moreover, the compounds exhibit a very good bioavailability and are metabolically more stable than prior art compounds.
Examples of inhibition: