WO2007094962A2 - Pyrazoles for the treatment of obesity and other cns disorders - Google Patents
Pyrazoles for the treatment of obesity and other cns disorders Download PDFInfo
- Publication number
- WO2007094962A2 WO2007094962A2 PCT/US2007/002547 US2007002547W WO2007094962A2 WO 2007094962 A2 WO2007094962 A2 WO 2007094962A2 US 2007002547 W US2007002547 W US 2007002547W WO 2007094962 A2 WO2007094962 A2 WO 2007094962A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phenyl
- pyrazole
- methyl
- ylpropoxy
- pyrrolidin
- Prior art date
Links
- 0 CCC(CC1)C*(C)c2c1[n](C)nc2* Chemical compound CCC(CC1)C*(C)c2c1[n](C)nc2* 0.000 description 3
- LZSYGJNFCREHMD-UHFFFAOYSA-N BrCc1ccccc1Br Chemical compound BrCc1ccccc1Br LZSYGJNFCREHMD-UHFFFAOYSA-N 0.000 description 1
- VGCLOPCQODEGOA-UHFFFAOYSA-N C(CN1CCCC1)COc(cc1)ccc1-[n]1nc(cccc2)c2c1 Chemical compound C(CN1CCCC1)COc(cc1)ccc1-[n]1nc(cccc2)c2c1 VGCLOPCQODEGOA-UHFFFAOYSA-N 0.000 description 1
- OXTKSKZTTWYFJM-NZZSEEIWSA-N CC/C(/CCCC/C(/C=C/c1ccccc1)=C\CI)=C/C(C)CCCN1CCCC1 Chemical compound CC/C(/CCCC/C(/C=C/c1ccccc1)=C\CI)=C/C(C)CCCN1CCCC1 OXTKSKZTTWYFJM-NZZSEEIWSA-N 0.000 description 1
- STSCVKRWJPWALQ-UHFFFAOYSA-N CCOC(C(F)(F)F)=O Chemical compound CCOC(C(F)(F)F)=O STSCVKRWJPWALQ-UHFFFAOYSA-N 0.000 description 1
- FOWZCNMPRRZSDV-BQYQJAHWSA-N COc(cc1)ccc1-[n]1c(/C=C/c2ccccc2)cc(C(F)(F)F)c1 Chemical compound COc(cc1)ccc1-[n]1c(/C=C/c2ccccc2)cc(C(F)(F)F)c1 FOWZCNMPRRZSDV-BQYQJAHWSA-N 0.000 description 1
- BLEBOOVJJNMSCV-UHFFFAOYSA-N COc(cc1)ccc1-[n]1nc(cccc2)c2c1 Chemical compound COc(cc1)ccc1-[n]1nc(cccc2)c2c1 BLEBOOVJJNMSCV-UHFFFAOYSA-N 0.000 description 1
- BHAAPTBBJKJZER-UHFFFAOYSA-N COc(cc1)ccc1N Chemical compound COc(cc1)ccc1N BHAAPTBBJKJZER-UHFFFAOYSA-N 0.000 description 1
- ATYLMPAHHHRNDD-UHFFFAOYSA-N COc(cc1)ccc1N(Cc(cccc1)c1Br)N Chemical compound COc(cc1)ccc1N(Cc(cccc1)c1Br)N ATYLMPAHHHRNDD-UHFFFAOYSA-N 0.000 description 1
- CSOPVKUECMSWBR-VOTSOKGWSA-N O=C(CC(/C=C/c1ccccc1)=O)C(F)(F)F Chemical compound O=C(CC(/C=C/c1ccccc1)=O)C(F)(F)F CSOPVKUECMSWBR-VOTSOKGWSA-N 0.000 description 1
- JHFUZEPROLIAPX-UHFFFAOYSA-N Oc(cc1)ccc1-[n]1nc(cccc2)c2c1 Chemical compound Oc(cc1)ccc1-[n]1nc(cccc2)c2c1 JHFUZEPROLIAPX-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/54—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings condensed with carbocyclic rings or ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/06—Antimigraine agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/54—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings condensed with carbocyclic rings or ring systems
- C07D231/56—Benzopyrazoles; Hydrogenated benzopyrazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
Definitions
- This invention relates to compounds having pharmacological activity, to compositions containing these compounds, and to a method of treatment employing the compounds and compositions. More particularly, this invention concerns certain pyrazole derivatives and their salts and solvates. These compounds alter H 3 histamine receptor activity. This invention also relates to pharmaceutical compositions containing these compounds and to a method of treating disorders in which histamine H 3 receptor modulation is beneficial.
- Histamine is. a chemical messenger involved in various complex biological actions. When released, histamine interacts with specific macromolecular receptors on the cell surface or within a target cell to elicit changes in many different bodily functions.
- Various cell types including smooth muscle, blood cells, cells of the immune system, endocrine and exocrine col is as well as neurons respond to histamine by modulating the formation of intracellular signals, including of phosphatidylinositoL or adenylate cyclase.
- Evidence that histami ne plays a role as a neurotransmitter was established by the mid-to-late 197CPs (Schwartz, 1 ⁇ 75 ) Life. Sci. 17:503-518.
- histamine H 1 and H 2 Two histamine receptors (H 1 and H 2 ) were reported to mediate the biochemical actions of histamine on neurons. More recently, studies have demonstrated the existence of a third subtype of histamine receptor, the histamine H 3 receptor (Schwartz et al., 1986) TIPS 8: 24-28. Various studies have now demonstrated that histamine H 3 receptors are found on the histaminergic nerve terminals in the brains of several species, including man (Arrang et al., 1 983) Nature 102: 832-837. The H 3 receptor found on the histaminergic nerve terminal was defined as an autoreceptor and could intimately control the amount of histamine released from the neurons.
- H ;i - receptors have been identified on cholinergic, serotonergic, glutamaterg ⁇ c and monoamine nerve terminals in the peripheral nervous system (PNS) and central nervous system including the cerebral cortex and cerebral vessels.
- CNS histaminergic cell bodies are found in the magnocel l ular nuclei of the hypothalamic mammillary region and these neurons project diffusely to large areas of the forebra ⁇ n.
- the presence of histaminerg ⁇ c cell bodies in the tuberomammillury nucleus of the posterior hypothalamus, a brain area involved in the maintenance of wakefulness, and their projections to the cerebral cortex suggest a role in modulating the ar ⁇ usul .-.tatu ⁇ >r sleep- wake cycle.
- the histaminergic projection to many limbic structures such as the hippocanipal formation and the amygdaloid complex suggest roles in functions such as ⁇ ut-jnomic regulation, control of emotions and motivated behaviors, and memory processes.
- histamine is important for the state of arousal, as v sjgested by the location of histaminergic pathways, is supported by other types of evidence Lesions of the posterior hypothalamus are well known to produce sleep. Neuroehci.i.v.al and electrophysiological- studies have also indicated that the activity of histaminergic neurons is maximal during periods of wakefulness and is suppressed by barbiturates and ⁇ ther h> pnotics. Intraventricular histamine induces the appearances of an arousal EEG pattern i n rabbits and increased spontaneous locomotor activity, grooming and exploratory behavior in b ⁇ .-;i ⁇ saline and pentobarbital-treated rats.
- Thioperamide has also been shown to increase wakefulness and decrease slow-wave and IUZM sleep in rats. These findings are consistent with in vivo studies demonstrating that thioperamide caused an increase in synthesis and release of histamine. Together, these data demonstrate that selective H 3 antagonists or inverse agonists may be useful in the treatment of arousal states and sleep disorders. - ⁇
- H 3 receptor has been demonstrated to regulate the release of each of these neurotransmitters.
- An H 3 receptor antagonist or inverse agonist would therefore be expected to increase the release of these neurotransmitters in the brain. Since histamine has been demonstrated to be important in arousal and vigilance, Hj receptor antagonists or inverse agonists might enhance arousal and vigilance via increasing levels of neurotransmitter release and thereby improve cognition.
- Hj receptor antagonists or inverse agonists might enhance arousal and vigilance via increasing levels of neurotransmitter release and thereby improve cognition.
- H 3 receptor agonist may be useful in treating several other CNS disorders. It has been suggested that histamine may be involved in cerebral circulation, energy metabolism, and hypothalmic hormone secretion. For example, H 3 receptor agonists, antagonists or inverse agonists have been demonstrated to affect food intake and body weight gain in rodents. Recent evidence has indicated the possible use of H 3 agonists, antagonists or inverse agonists in the treatment of epilepsy. Work has demonstrated an inverse correlation between the duration of clonic convulsions and brain histamine levels. Thioperamide was also shown to significantly and dose-dependently decrease the durations of every convulsive phase after electrically-induced convulsions and increase the electroconvulsive threshold. For examples of therapeutical uses of H 3 receptor agonists, inverse agonists or antagonists, sec CJS Patent No. 6,316,475 or WO 03050099A1.
- an H 3 receptor binding compound may be useful in the treatment of diseases and conditions such as asthma, rhinitis, airway congestion, inflammation, hyper and hypo motility and acid secretion of the gastrointestinal tract.
- H 3 receptors may also contribute to changes in blood pressure, heart rate and cardiovascular output and could be used in the treatment of cardiovascular diseases, and in the treatment of diseases or conditions such as obesity, migraine, inflammation, motion sickness, pain, ADHD, dementia, depression, Parkinson's disease, schizophrenia, epilepsy, narcolepsy, acute myocardial infarction and asthma.
- Various pyrazole derivatives are discfosed in WO 03/024935; WO 03/095430; WO 89/03385; WO 93/23036; EP 01 78035; and EP 0647629.
- both WO 03/024935 and WO 03/095430 disclose certain substituted pyrazolyl compounds for treatment of inflammation.
- WO 00/19994 and WO 98/27061 disclose various cyclic compounds which may " have a spectrum of agontst/ ' antagontst properties.
- the present invention provides, in its principal aspect, compounds of the general formulae:
- X is O or NR 7 ; y is 0, I or 2; n is 0 or 1 q is O, I 5 or 2;
- Ri and R 2 are independently selected from the group consisting of (Ci-Cs)alkyl and (C r C( 5 )cycloalkyl; or where X is O, Rt and R 2 taken together with the nitrogen to which they are attached form a 5- 7 member heterocyclic ring system with 0 or. 1 additional hetero atoms selected from O, S, and NR O5 wherein the resulting ring may optionally be substituted with 1 -3 (C]-C 5 )alkyl or (C 3 -C 6 )cycloalkyl groups;
- R 3 is 0-2 of groups selected from halogen, (Ci-Cg)alkyl, (C t -C 8 )alkoxy, (C 3 -C 7 )cycloalkyl, (C 3 -C7)cycloalkyl-(C
- R 4 and R 6 are independently selected from (Ci-C 8 )alkyl, (Ci-Cs)alkoxy, (C 3 -C 7 )cycloalkyl, (C 3 -C 7 )cycloalkyl-(Ci-C 6 )aIkyI, heterocycloalkyl containing 1-3 hetero atoms selected from O 3 S, N, (C r Cs)alkyl-O-(Ci-C 5 )alkyl, amide, (C 1 -C 5 )alkyl-aryl 3 and CF 3 ;
- Rs is selected from the group consisting of hydrogen, (C t -Cg)alkyl, aryl, (Ci-C 5 )alkyl-O-(Cj- C 5 )alkyi J and (C r C 5 )alkyI-aryI,
- R 5 and R 6 and the atoms to which they are attached form a fused 5-6 member saturated carbocyclic ring or a fused 10 member bi-cycl ⁇ c ring system, such as
- R > and R 6 and the atoms to which they are attached form a fused benzothiophene or fused be ⁇ zofuran ring system, such as
- X is NR7, R7 and R 2 taken together are -(CH2CH2)- to form a two nitrogen containing ring where y is 0 (piperazine) or y is 1 (homopiperazine), and wherein Ri is as defined previously, and the pharmaceutically acceptable salts thereof.
- compositions comprising compounds of formulae 1 -3, pharmaceutically acceptable salts, solvates, or formulations thereof, and pharmaceutically acceptable carriers in combination with an effective amount of at least one compound of formulae 1-3.
- he present invention also provides a method of treating conditions in which modulation of histamine f I 3 receptors may be of therapeutic importance such as inflammation, migraine, motion sickness, pain, Parkinson's Disease, epilepsy, cardiovascular disease (i.e. hyper or hypotension, acute myocardial infarction), gastrointestinal disorders (acid secretion, motility) and CNS disorders involving attention or cognitive- disorders (i.e., Alzheimer's, Attention Deficit Disorder, age-related memory dysfunction, stroke, etc.), psychiatric disorders (i.e., depression, schizophrenia, obsessive-compulsive disorders, etc.); sleep disorders (i.e.
- narcolepsy sleep apnea, insomnia, disturbed biological and circadian rhythms, hyper and hypsomnolence), and disorders such as obesity, anorexia/bulimia, thermoregulation, hormone release
- administering an effective amount of a compound of formulae 1 -3 to a patient in need of such treatment.
- Presently preferred compounds include:
- Certain compounds of the invention may exist in different isomeric (e.g. enantiomers and distereoisomers) forms.
- the invention contemplates all such isomers both in pure form and in a mixture, including racemic mixtures. Enol and tautomeric forms are also included.
- the compounds of the invention can exist in u ⁇ solvaled as well as solvated forms, including hydrated forms, e.g., hemt-hydrate.
- solvated forms including hydrated forms, e.g., hemt-hydrate.
- pharmaceutically acceptable solvents such as water, ethanol, and the like are equivalent to the ⁇ nsolvated forms for the purposes of the invention.
- Certain compounds of the invention also form pharmaceutically acceptable salts, e.g., acid addition salts.
- the nitrogen atoms may form salts with acids.
- suitable acids for salt formation are hydrochloric, sulfuric, phosphoric, acetic, citric, oxalic, mal ⁇ nic, salicylic, malic, fumaric, succinic, ascorbic, maleic, methanesulfonic and other mineral carboxylic acids well known to those in the art.
- the salts are prepared by contacting the free base form with a sufficient amount of the desired acid to produce a salt in the conventional manner
- the free base forms may be regenerated by treating the salt with a suitable dilute aqueous base solution such as dilute aqueous hydroxide, potassium carbonate, ammonia, and sodium bicarbonate.
- the free base forms differ from their respective salt forms somewhat in certain physical properties, such as solubility in polar solvents, but the acid salts are equivalent to their respective free base forms for purposes of the invention (See, for example S M. Berge, et ai., "Pharmaceutical Salts,” J. Pharm. Sci., 66: 1-19 (1977) which is incorporated herein by reference.
- alkyl refers to straight or branched chain radicals derived from saturated hydrocarbons by the removal of one hydrogen atom.
- Representative examples of alkyl groups include methyl, ethyl, n-propyl, iso-propyl, n-butyl, sec-butyl, iso- butyl, tert-butyl, and the like.
- ⁇ cycloaikyl refers to an aliphatic ring system having 3 to 10 carbon atoms and I to 3 rings, including, but not limited to cyclopropyl, cyclopentyl, cycl ⁇ hexyl, norbornyl, and adamantly among others.
- Cycloalkyi groups can be unsubstituted or substituted with one, two or three substituents independently selected from lower alkyl, haloalkyl, alkoxy, thioalkoxy, amino, alkylarntno, d ⁇ alkylamino, hydroxy ⁇ halo, mercapto, nitro, carboxaldehyde, carboxy, alkoxycarbonyl and carboximide.
- Cycloalkyi includes cis or trans forms. Furthermore, the substituents may either be in endo or exo positions in the bridged btcyclic systems.
- halo or halogen' ' as used herein refers to I, Br, Cl or F.
- heteroatom refers to at least one N, O or S atom.
- heterocycloalkyl refers to a non-aromatic 3- to 10- membered ring containing at least one endocyclic N, O, or S atom
- the heterocycle may be optionally aryl-fused.
- the heterocycle may also optionally be substituted with at least one substituent which is independently selected from the group consisting of hydrogen, halogen, hydroxy!, amino, nitro, ttiflouromethyl, trifluoromethoxy, alkyl, aralkyt, alkenyl, alkynyl, aryl, cyano, carboxy, carboalkoxy, carboxyalkyl, oxo, arylsulfonyl and aralkylaminocarbonyl among others.
- substituent is independently selected from the group consisting of hydrogen, halogen, hydroxy!, amino, nitro, ttiflouromethyl, trifluoromethoxy, alkyl, aralkyt, alkenyl, alkynyl, aryl, cyano, carboxy, carboalkoxy, carboxyalkyl, oxo, arylsulfonyl and aralkylaminocarbonyl among others.
- composition is intended to encompass a
- the compounds of the present invention can be used in the form of pharmaceutically acceptable salts derived from inorganic or organic acids.
- pharmaceutically acceptable salt means those salts which are. within the scope of sound medical judgment, suitable for use in contact with the tissues uf humans and lower animals without undue toxicity, irritation, allergic response and the like, and are commensurate with a reasonable benefit/risk, ratio.
- Pharmaceutically acceptable salts are well-known in the art. For example, S. M. Berge et al. describe pharmaceutically acceptable salts in detail in J.
- the salts can be prepared in situ during the final isolation and purification of the compounds of the in ⁇ entiun or separately by reacting a free base function with a suitable organic acid.
- Representative acid addition salts include, but are not limited to acetate, adipate, alginate, citrate, aspartate, benzoate, benzenesulfonate, bisulfate, butyrate, camphorate, camphorsulfonate. digluconate, glycerophosphate, hemisulfate, heptanoate, hexanoate, fumarate. hydrochloride, hydrobromide.
- halide such as methyl, ethyl, propyl, and butyl chlorides, bromides and iodides; dialkyt sulfates like dimethyl, diethyl, dibutyl and diamyl sulfates; long chain hal ⁇ des such as decyl, Iauryl. myristyl and stearyl chlorides, bromides and iodides; arylalkyl halides like benzyl and phenethyl bromides and others. Water or oil-soluble or dispersible products are thereby obtained.
- acids which can be employed to form pharmaceutically acceptable acid addition salts include such inorganic acids as hydrochloric acid, hydrobromic acid, sulphuric acid and phosphoric acid and sueh organic acids as oxalic acid, maleic acid, succinic acid and citric acid
- Basic addition salts can be prepared in situ during the final isolation and purification of compounds of this invention by reacting a carboxylic acid-containing moiety with a suitable base such as the hydroxide, carbonate or bicarbonate of a pharmaceutically acceptable metal cation or with ammonia or an organic primary, secondary or tertiary amine.
- Pharmaceutically acceptable salts include, but are not limited to. cations based on alkali metals or alkaline earth metals such as lithium, sodium, potassium, calcium, magnesium and aluminum salts and the tike and- nontoxic quaternary ammonia and amine cations including ammonium, tetramethylammon ⁇ um, tetraethylammontum.
- methylammonium methylammonium, dimethylammontum, trimethyl ammonium, triethylammonium, diethylammonium, and ethylammonium among others.
- Other representative organic amines useful for the formation of base addition salts include ethylenediamine, ethanoiamtne, d ⁇ ethanolamine, piperidine, piperazine and the like.
- Dosage forms for topical administration of a compound of this invention include powders, sprays, ointments and inhalants.
- the active compound is mixed under sterile conditions with a pharmaceutically acceptable carrier and any needed preservatives, buffers or propellants which can be required.
- Opthalm ⁇ c Formulations, eye ointments, powders and solutions are also contemplated as being within the scope of this invention.
- Actual dosage levels of active ingredients in the pharmaceutical compositions of this invention can be varied so as to obtain an amount of the active compound(s) which is effective to achieve the desired therapeutic response for a particular patient, compositions and mode of administration.
- the selected dosage level will depend upon the activity of the particular compound, the route of administration, the seventy of the condition being treated and the condition and prior medical history of the patient being treated. However, it is within the skill of the art to start doses of the compound at levels lower than required to achieve the desired therapeutic effect and to gradually increase the dosage until the desired effect is achieved.
- a therapeutically effective amount of one of the compounds of the present invention can be employed in pure form or, where such forms exist, in pharmaceutically acceptable salt, ester or prodrug form.
- the compound can be administered as a pharmaceutical composition containing the compound of interest in combination with one or more pharmaceutically acceptable excipients.
- therapeutically effective amount means a sufficient amount of the compound to treat disorders, at a reasonable benefit/risk ratio applicable to any medical treatment. It will be understood, however, that the total daily usage of the compounds and compositions of the present invention will be decided by the attending physician within the scope of sound medical judgment.
- the specific therapeutically effective dose level for any particular patient will depend upon a variety of factors including the disorder being treated and the severity of the disorder; activity of the specific compound employed; the specific composition employed; the age, body weight, general health, sex and diet of the patient the time of administration, route of administration, and rate of excretion of the speci fic compound employed; the duration of the treatment; drugs used in combination- or coincidental w ith the specific compound employed, and like factors well known in the " ' medical arts
- the total daily dose of the compound-, or this invention administered to a human or lower animal may range from about - 0 0001 to about HX ) O mg/kg/day.
- more preferable doses ". can be in the range o t from about 0.001 to about 5 mg/ky/day.
- the effective daily ' dose can ⁇ c div ided into multiple doses for purposes of administration; consequently, singleriff dose compositions may contain such amounts or submultiples thereof to make up the daily ⁇ dose
- present invention also provides pharmaceutical compositions that .- compr.se compounds ot the present invention formulated together with one or more non-toxic pharmaceutic al l v acceptable carriers.
- the pharmaceutical compositions can be specially formulated tor oral administration in solid or liquid form lor parenteral injection or for rectal - administration
- compositions of this invention can be administered to humans ant! other mammals orally, rectally, parenteral Iy , intracisternaUy, intravaginally, intraperi toneal! ⁇ topically (as by powders, ointments or drops), bucally or as an oral or nasal - sprav
- parenterally refers to modes of administration which ' include i ntrav e nous intramuscular, intraperitoneal, 1 utrastemal, subcutaneous and intraarticular mic tion and infusion.
- the present invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a component of the present invention and a physiologically tolerable - diluent T
- he present invention includes one or more compounds as described above formulated into compositions together with one or more non-toxic physiologically tolerable * or acceptable di luents carriers, adjuvants or vehicles that are collectively referred to herein as ".
- diluents for parenteral injection, for intranasal delivery, for oral administration in solid or liquid form, for rectal or topical administration, among others.
- I he compositions can also be delivered through a catheter for local delivery at a target site via an mtracoronary stent (a tubular device composed of a fine wire mesh), or v/(/ a biodegradable polymer.
- the compounds may also be complexed to tigands, such as .v.' . i iodies, for targeted delivery. - ⁇
- compositions suitable for parenteral injection may comprise physiologically ⁇ jcr ⁇ table, sterile aqueous or nonaqueous solutions, dispersions, suspensions or emulsions cif i> 1 sterile powders for reconstitution into sterile injectable solutions or dispersions.
- * t ⁇ a..- iiples of suitable aqueous and nonaqueous carriers, diluents, solvents or vehicles include witi--, ethanol, polyols (propytenegiycol, polyethyleneglycol, glycerol, and the like), ⁇ . cfc-:able oils (such as olive oil), injectable organic esters such as ethyl ⁇ leate, and suitable ⁇ : i ⁇ : .» ires thereof.
- compositions can also contain adjuvants such as preserving, wetting, cini Uifying, and dispensing agents.
- adjuvants such as preserving, wetting, cini Uifying, and dispensing agents.
- Prevention of the action of microorganisms can be ff. --.i-ed by various antibacterial and antifungal agents, for example, parabens, chlorobutanol, ⁇ '-.- ⁇ ol, sorbic acid, and the like. It may also be desirable to include isotonic agents, for ex. rnple sugars, sodium chloride and the like.
- Prolonged absorption of the injectable phar maceutical form can be brought about by the use of agents delaying absorption, for c:- . ⁇ :rple, aluminum monostearate and gelatin.
- Suspensions in addition to the active compounds, may contain suspending :' ⁇ r.-' fs, as for example, ethoxylated isostearyi alcohols, polyoxyethylene sorbitol and sorbitan '.- ⁇ IVr-., microcrystatline cellulose, aluminum metahydroxide, bentonite. agar-agar and f r .! • • ..• ⁇ anth, or mixtures of these substances, and the like.
- suspending :' ⁇ r.-' fs, as for example, ethoxylated isostearyi alcohols, polyoxyethylene sorbitol and sorbitan '.- ⁇ IVr-., microcrystatline cellulose, aluminum metahydroxide, bentonite. agar-agar and f r .! • • ..• ⁇ anth, or mixtures of these substances, and the like.
- Injectable depot forms are made by forming microencapsule matrices of the f iri!;'. in biodegradable polymers such as polylactide-polyglycolide. Depending upon the ratio oi J ⁇ ig to polymer and the nature of the particular polymer employed, the rate of drug release •j.ifi :>e controlled. Examples of other biodegradable polymers include poly(orthoesters) and pi • ' :-' ⁇ anhydrides). Depot injectable formulations are also prepared by entrapping the drug in Mr-. nines or microemuls ⁇ o ⁇ s which are compatible with body tissues.
- the injectable formulations can be sterilized, for example, by filtration through a bacterial-retaining filter or by incorporating sterilizing agents in the form of sterile solid compositions which can be dissolved or dispersed in sterile water or other sterile injectable medium just prior to use.
- Solid dosage forms for oral administration include capsules, tablets, pills, powders and granules, tn such solid dosage forms, the active compound may be mixed with at least one inert, pharmaceutically acceptable excipient or carrier, such as sodium citrate or dicalciurn phosphate atid ' oi ai fillers or extenders such as starches, lactose, sucrose, glucose, mann ⁇ tol and silicic acid, b) binders such as carboxymeLhy [cellulose, alginates, gelatin, polyvinylpyrrolidone sucn ⁇ e and acacia; c) humectants such as glycerol; d) disintegrating agents such as agar-agar calcium carbonate, potato or tapioca starch, alginic acid, certain silicates and sodium LarKmare; e) solution retarding agents such as paraffin; (f) absorption accelerators such as quaternary ammonium compounds; g) wetting agents such as cet
- Solid compositions of a similar type may also be employed as fillers in soft and hard-filled gelatin capsules using such excipients as lactose or milk sugar as well as high molecular weight p ⁇ Kcthv icr.c glycols and the like.
- he solid d ⁇ ⁇ a ⁇ e forms of tablets, dragees, capsules, pills and granules can be prepared with coatings and .-hells such as enteric coatings and other coatings well-known in the pharmaceutical formulating art. They may optionally contain opacifying agents and may also be of a composition MR h that they release the active ingredie ⁇ t(s) only, or preferentially, in a certain part of the intestinal tract, optionally, in a delayed manner. Examples of embedding compositions which can be used include polymeric substances and waxes.
- the active compounds can also be in micro-encapsulated form, if appropriate, with one or more of the above-mentioned excipients.
- Liquid d ⁇ sa ⁇ e forms for oral administration include pharmaceutically acceptable emulsions, solutions, suspensions, syrups and elixirs.
- the liquid dosaue forms may contain inert diluents commonly used in the art such as, for example, uatcr or other solvents, sohibtlizing agents and emulsifiers such as ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene gKcol.
- oils in particular, cottonseed, groundnut, corn, germ, olive, castor and sesame oils
- glycerol tetrahydrofurfuryl alcohol
- polyethylene glycols polyethylene glycols and fatty -acid esters of sorbitan and mixtures thereof.
- the oral compositions may also include adjuvants such as wetting agents, emulsifying and suspending agents, sweetening, flavoring and perfuming agents.
- adjuvants such as wetting agents, emulsifying and suspending agents, sweetening, flavoring and perfuming agents.
- compositions for rectal or vaginal administration are preferably suppositories which can be prepared by mixing the compounds of this invention with suitable non-irritating excipients or carriers such as cocoa butter, polyethylene glycol or a suppository wax which are solid at room temperature but liquid at body temperature and therefore melt in the tectum or vaginal cavity and release the active compound.
- suitable non-irritating excipients or carriers such as cocoa butter, polyethylene glycol or a suppository wax which are solid at room temperature but liquid at body temperature and therefore melt in the tectum or vaginal cavity and release the active compound.
- Liposomes are generally derived from phospholipids or other lipid substances. Liposomes are formed by mono- or multi-lameltar hydrated liquid crystals which are dispersed in an aqueous medium. Any non-toxic, physiologically acceptable and metabolizable lipid capable of forming liposomes can be used.
- the present compositions in liposome form can contain, in addition to a compound of the present invention, stabilizers, preservatives, excipients and the like.
- the preferred lipids are natural and synthetic phospholipids and phosphatidyl cholines (lecithins) used separately or together.
- prodrugs of the compounds of the present invention represents those prodrugs of the compounds of the present invention "which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of humans and lower animals without undue toxicity, irritation, allergic response, and the like, commensuiate with a reasonable benefit/risk ratio, and effective for their intended use, as well as the zw ⁇ tterionic forms, where possible, of the compounds of the invention.
- Prodrugs of the present invention may be rapidly transformed in vivo to the parent compound of the above formula, for example, by hydrolysis in blood. A thorough discussion is provided in T. Higuchi and V. Stella, Pro-d ⁇ ms as Novel Delivery Systems, V. 14 of the A. CS.
- Stereoisomers include enant ⁇ omers and d ⁇ astereomers, and mixtures of enantiomers or diastereomers.
- Individual stereoisomers of compounds of the present invention may be prepared synthetically from commercially available starting materials which contain asymmetric or chirai centers or by preparation of racemic mixtures followed by resolution well-known to those of ordinary skill in the art.
- the compounds of the invention can exist in unsolvated as well as soivated forms, including hydrated forms, such as hemi-hydrates.
- soivated forms with pharmaceutically acceptable solvents such as water and ethanol among others are equivalent to the unsolvated forms for the purposes of the invention.
- l-(4-Methoxyphe ⁇ yl)-5-styryl-3-trifluoromethyl- ⁇ /7-p ⁇ ra/ole l,l,l-Trifluoro-6- phenyIhex-5-ene-2,4-dione (2.28 mmol) and 4-meth ⁇ xyphen> I hydrazine hydrochloride (435 mg, 2.5 mmol) were heated in ethanol (7 mL) at 70° C ⁇ vern ⁇ ht. The solution was diluted with water and extracted with ethyl acetate. The ethyl acetate extracts were washed with 1 N HCl 5 saturated sodium bicarbonate solution, and brine, then dried over MgSC> 4 and concentrated.
- Acetyloctahydronaphthalen-I-one (0.96 g, 4.95 mmol) was dissolved in absolute ethanol (10 mL), and 4-methoxyphenyihydrazine hydrochloride (0.95 g, 5.44 mmol ) was added. The reaction was stirred overnight at 70° C. The mixture was diluted with water and extracted with ethyl acetate. The organic extracts were washed with 1 N FICl. saturated sodium bicarbonate solution, and brine, then dried over MgS ⁇ 4 and concentrated.
- Benzyloxyphenyl)-8-methoxy-3-methyl-4,5-dihydro-lH-benzo[g]indazole (42 mg, 0.1 mmol) was dissolved in methanol/tetrahydrofuran (2/1, v/v, 1.5 mL) and a catalytic amount of 10% Pd/C (wet) was added The flask was purged with nitrogen and hydrogen, then stirred under 1 atmosphere of hydrogen for 2 hours. The mixture was filtered through Celite and washed with methanol, and the filtrate was concentrated. The reaction was assumed to be quantitative. LC-MS (Ci 9 H, S N Z O 2 calculated 306) m/z 307 (M+H).
- Methoxyphenylhydrazine hydrochloride (6.05 g, 34.8 mmol) and ethyl 2,4-dioxovalerate (4.45 mL, 31.6 mmol) were stirred overnight at 80° C in cthanol (SO mL). After the solution was allowed to cool to room temperature, it was diluted with water and extracted with ethyl acetate. The organic layer was washed with water. 1 N " HCl, and saturated sodium bicarbonate.
- 2-(4-Methoxyphenyl)-5-methyl-2jy-pyrazole-3-carboxylic acid cyclohexylamide 2-(4- Methoxyphenyl)-5-methyl-2/f-pyrazoIe-3-carboxyIic acid (752 mg, 3.24 mmol) was suspended in methylene chloride (25 mL) and DMSO (3 drops) was added. After the reaction was stirred at room temperature for 1 hour, it was concentrated in vacuo. The residue was diluted twice with methylene chloride (60 mL) and concentrated to dryness.
- Example 12 2-[4-(l-Isopropylpiperidi ⁇ -4-yIoxy)phenyl]-5-methyI-2H-pyra7.ole-3-earbo ⁇ > lie nc ⁇ d cyclohexylamide
- ToIuene-4-sulfonfc acid l,4-dioxa-spiro[4.5]dec-8-yl ester The alcohol prepared previously. 1 ,4-dioxa-spiro[4.5]decan-8-ol (4.92g, 31. 1 mmol) was dissolved in pyridine (15 mL) at 0 C followed by addition of p-toluenesulfonyl chloride (6.1 g, 32.1 rnmol). The mixture was stirred at 0 C for 2 hours and allowed to warm to room temperature overnight. The reaction was diluted with water (15 mL) and stirred for 30 minutes.
- Cis-4- (3,5-diisopropyl-pyrazol-l-yl)-cyclohexanol (17.5 mg, 0.068 mmul) was dissolved in dry DMF (2 mL) followed by addition of NaH (excess, 60% in oil) and stirred at room temperature until hydrogen evolution stopped.
- To the reaction was added l -(3-Chloro- propyl)-pyrrolidine (20 mg) and catalytic NaI followed by heating to 80 ° C overnight.
- Trans-4-(3,5-ditsopropyI-pyrazol-l-yl)-cyclohexanol was prepared by the same method starting with trans-4-(3,5-d ⁇ isopropyl-pyrazol-l-yl)-cycIohexanol.
- the human histamine H i receptor was stably expressed in HTlOSO cells containing the chimeric G-protein, GqQo ( Coward et ah, Anal Biochem 1999; 270:242-8).
- HT1080- Gq ⁇ i5 cells were grown in alpha-moditkd MBM containing 10% fetal bovine serum and 7 ⁇ g/ml blasticidin at 37° C m 5% C ⁇ 2 ' ⁇ S% atmosphere.
- Cells (4.8xlO 9 ) were irradiated with 50 rads from a 137 Cs source and the nFG8-HH3 RAGE (Random Activation of Gene Expression; see Harrington et a! . ⁇ attire Biotechnology.
- the RAGE vector pFG8-HH3 contained cDN ⁇ strqueoce coding for the first exon (83 amino acids) of human H3 receptor.
- the culture medium was replaced 48 hrs after electroporation with alpha-modified MEM 3 10% fetal ho vine serum, 500 ⁇ g/ml hygromycin B and 3 ⁇ g/ml puromycin. Medium was replaced everv tour days during cell expansion.
- RAGE activated cells expressing the H3 receptor pools of approximately 10,000 colonies (5x 10 7 - 1.5x10 8 cells total) were screened by PCR for the desired gene product (using primers specific to the RAGE vector and exon 2 of the H3 receptor). Pools that were found to contain the appropriate transcript, as confirmed by sequencing, were subcloned into pools of 100 cells/well. Positive 100 cells/well pools were identified by PCR, confirmed by sequencing, and subsequently subcloned to 0.8 cells/well. Once clones expressing the H3 receptor were identified by PCR analysis, assays (FLIPR or radioligand binding) were performed to confirm that the activated gene produced functional protein.
- the protein expression in the initial clones obtained from the RAGE library was increased by growth in the presence ⁇ i " methotrexate. Since the integrated RAGE vector contains the DHFR gene, such treatment selects for cells that have amplified the genetic locus containing the RAGE insert. Subclones obtained after methotrexate amplification were tested for functional activity in FLiPR assays to identify the clone that was most suitable for HTS.
- the final HT1080-Gq ⁇ i5 RAGF clone (RAGE-H3) expressing the human histamine H3 receptor was grown in alpha-mud ⁇ fied MEM containing 10% fetal bovine serum, 3 ⁇ g/ml puromycin, 500 ⁇ g/mt hygromycin B, 1.2 ⁇ M methotrexate at 37° C in 5% CO 2 /95% atmosphere.
- RAGE-H3 cells (IO 9 ) were washed twice with cold PBS, scraped off the plates, and centrifuged at 1000 x g for 5 minutes. Cells were resuspended in ice-cold 10 niM Tm HCl, pH 7.4, containing 5 raM EDTA and protease inhibitor cocktail tablets (Roche Molecular Biochem ⁇ cals). After incubating on ice for 10 min, the cells were homogenized with a dounce homogenizer or a polytron tissue grinder, and centrifuged at 1000 x g for 10 min at 4° C. T he resulting supernatant was centrifuged at 32, 000 x g for 30 min at 4° C. The membrane pellets were resuspended in 50 mM Tris HCI, pH 7.4, and stored at — 80° C until use. Protein concentration was determined by the Bradford method (Bio-Rad Laboratories, CA)-
- Binding assays were carried out in 96- well polypropylene plates in 50 mM Tris HCL pH 7.4, containing 1 mM EDTA. Reaction mixtures contained 100 ⁇ l of membrane > suspension, 50 ⁇ l of 4% DMSO, and 50 ⁇ l of increasing amounts of [ I25 I]iodoproxyfan (final concentration 0.0005-1.8 nM for human H3 receptor saturation binding assay). Nonspecific binding was defined by adding 10 ⁇ M clobenpropit to the reaction mixtures. Competition binding assays were performed in a reaction mixture containing 100 ⁇ l of membrane suspension ( ⁇ 20 ⁇ g of protein/well), 50 ⁇ l of [ l25 I]iodoproxyfan (final concentration of ⁇
Abstract
This invention relates to compounds having pharmacological activity, to compositions containing these compounds, and to a method of treatment employing the compounds and compositions. More particularly, this invention concerns certain pyrazole derivatives, their salts and solvates. These compounds have H3 histamine receptor binding activity. This invention also relates to pharmaceutical compositions containing these compounds and to a method of treating disorders in which histamine H3 receptor modulation is beneficial.
Description
PYRAZOLES FOR THE TREATMENT OF OBESITY AJND OTHER CNS
DISORDERS
TECHNICAL FIELD
This invention relates to compounds having pharmacological activity, to compositions containing these compounds, and to a method of treatment employing the compounds and compositions. More particularly, this invention concerns certain pyrazole derivatives and their salts and solvates. These compounds alter H3 histamine receptor activity. This invention also relates to pharmaceutical compositions containing these compounds and to a method of treating disorders in which histamine H3 receptor modulation is beneficial.
BACKGROUND O F THE INVENTION
Histamine is. a chemical messenger involved in various complex biological actions. When released, histamine interacts with specific macromolecular receptors on the cell surface or within a target cell to elicit changes in many different bodily functions. Various cell types including smooth muscle, blood cells, cells of the immune system, endocrine and exocrine col is as well as neurons respond to histamine by modulating the formation of intracellular signals, including of phosphatidylinositoL or adenylate cyclase. Evidence that histami ne plays a role as a neurotransmitter was established by the mid-to-late 197CPs (Schwartz, 1 ^75 ) Life. Sci. 17:503-518. Immunohistochemical studies identified histaminergϊc cell bodies in the tuberornammϊllary nucleus of the posterior hypothalamus with widespread projections in the dicencephalon and telencephalon (Inagaki et al., 1998) J. Camp. Neurol. 273 :283-300.
Two histamine receptors (H 1 and H2) were reported to mediate the biochemical actions of histamine on neurons. More recently, studies have demonstrated the existence of a third subtype of histamine receptor, the histamine H3 receptor (Schwartz et al., 1986) TIPS 8: 24-28. Various studies have now demonstrated that histamine H3 receptors are found on the histaminergic nerve terminals in the brains of several species, including man (Arrang et al., 1 983) Nature 102: 832-837. The H3 receptor found on the histaminergic nerve terminal was defined as an autoreceptor and could intimately control the amount of histamine released from the neurons. 1 Hstamϊne, the natural compound, was capable of stimulating this
autoreceptor but testing of known Hi and H2 receptor agonists and antagonists suggested that the H3 receptor has a distinct pharmacological profile. Further, H;i- receptors have been identified on cholinergic, serotonergic, glutamatergϊc and monoamine nerve terminals in the peripheral nervous system (PNS) and central nervous system including the cerebral cortex and cerebral vessels. These observations suggest that Hj receptors are uniquely located to modulate histamine as well as other neurotransmitter release, and compounds th.it bind H3 receptors could be important mediators of neuronal activity.
As stated, CNS histaminergic cell bodies are found in the magnocel l ular nuclei of the hypothalamic mammillary region and these neurons project diffusely to large areas of the forebraϊn. The presence of histaminergϊc cell bodies in the tuberomammillury nucleus of the posterior hypothalamus, a brain area involved in the maintenance of wakefulness, and their projections to the cerebral cortex suggest a role in modulating the arυusul .-.tatu κ>r sleep- wake cycle. The histaminergic projection to many limbic structures such as the hippocanipal formation and the amygdaloid complex suggest roles in functions such as αut-jnomic regulation, control of emotions and motivated behaviors, and memory processes.
The concept that histamine is important for the state of arousal, as v sjgested by the location of histaminergic pathways, is supported by other types of evidence Lesions of the posterior hypothalamus are well known to produce sleep. Neuroehci.i.v.al and electrophysiological- studies have also indicated that the activity of histaminergic neurons is maximal during periods of wakefulness and is suppressed by barbiturates and υther h> pnotics. Intraventricular histamine induces the appearances of an arousal EEG pattern i n rabbits and increased spontaneous locomotor activity, grooming and exploratory behavior in b<.-;iι saline and pentobarbital-treated rats.
In contrast, a highly selective inhibitor of histidine decarboxylase, -.he sole enzyme responsible for histamine synthesis, has been shown to impair waking in t at-.. These data support the hypothesis that histamine may function in modulating behavioi ai arousal. The role of the H3 receptor in sleep- waking parameters has been demonstrated ( I i n et al., 1990) Brain Rex. 592: 325-330. Oral administration of RAMHA, a I h agonist, caused a significant increase in deep slow wave sleep in the cat. Conversely, thioperamide, a H3 antagonist/inverse agonist, enhanced wakefulness in a dose-dependent fashion. Thioperamide has also been shown to increase wakefulness and decrease slow-wave and IUZM sleep in rats. These findings are consistent with in vivo studies demonstrating that thioperamide caused an increase in synthesis and release of histamine. Together, these data
demonstrate that selective H3 antagonists or inverse agonists may be useful in the treatment of arousal states and sleep disorders. - ■
Serotonin,, histamine, and acetylcholine have all been demonstrated to be diminished in the Alzheimer's (AP) brain. The histamine H3 receptor has been demonstrated to regulate the release of each of these neurotransmitters. An H3 receptor antagonist or inverse agonist would therefore be expected to increase the release of these neurotransmitters in the brain. Since histamine has been demonstrated to be important in arousal and vigilance, Hj receptor antagonists or inverse agonists might enhance arousal and vigilance via increasing levels of neurotransmitter release and thereby improve cognition. Thus, the use of compounds that bind the use uf I (3 receptor in AD, attention deficit disorders (ADD), age- related memory dysfunction and other cognitive disorders would be supported.'
H3 receptor agonist:,, antagonists or inverse agonists may be useful in treating several other CNS disorders. It has been suggested that histamine may be involved in cerebral circulation, energy metabolism, and hypothalmic hormone secretion. For example, H3 receptor agonists, antagonists or inverse agonists have been demonstrated to affect food intake and body weight gain in rodents. Recent evidence has indicated the possible use of H3 agonists, antagonists or inverse agonists in the treatment of epilepsy. Work has demonstrated an inverse correlation between the duration of clonic convulsions and brain histamine levels. Thioperamide was also shown to significantly and dose-dependently decrease the durations of every convulsive phase after electrically-induced convulsions and increase the electroconvulsive threshold. For examples of therapeutical uses of H3 receptor agonists, inverse agonists or antagonists, sec CJS Patent No. 6,316,475 or WO 03050099A1.
In spite of their 1O\Λ density, H3 receptor binding sites can be detected outside the brain. Several studies have revealed the presence of H3 hetero receptors in the gastrointestinal tract, as well as upon neurons of the respitory tract. Accordingly, an H3 receptor binding compound may be useful in the treatment of diseases and conditions such as asthma, rhinitis, airway congestion, inflammation, hyper and hypo motility and acid secretion of the gastrointestinal tract. Peripheral or central blockage of H3 receptors may also contribute to changes in blood pressure, heart rate and cardiovascular output and could be used in the treatment of cardiovascular diseases, and in the treatment of diseases or conditions such as obesity, migraine, inflammation, motion sickness, pain, ADHD, dementia, depression, Parkinson's disease, schizophrenia, epilepsy, narcolepsy, acute myocardial infarction and asthma.
Various pyrazole derivatives are discfosed in WO 03/024935; WO 03/095430; WO 89/03385; WO 93/23036; EP 01 78035; and EP 0647629. For example, both WO 03/024935 and WO 03/095430 disclose certain substituted pyrazolyl compounds for treatment of inflammation. WO 00/19994 and WO 98/27061 disclose various cyclic compounds which may "have a spectrum of agontst/'antagontst properties.
SUMMARY OF THE INVENTION
The present invention provides, in its principal aspect, compounds of the general formulae:

where
X is O or NR7; y is 0, I or 2; n is 0 or 1 q is O, I 5 or 2;
Ri and R2 are independently selected from the group consisting of (Ci-Cs)alkyl and (C r C(5)cycloalkyl;
or where X is O, Rt and R2 taken together with the nitrogen to which they are attached form a 5- 7 member heterocyclic ring system with 0 or. 1 additional hetero atoms selected from O, S, and NRO5 wherein the resulting ring may optionally be substituted with 1 -3 (C]-C5)alkyl or (C3-C6)cycloalkyl groups;
R3 is 0-2 of groups selected from halogen, (Ci-Cg)alkyl, (Ct-C8)alkoxy, (C3-C7)cycloalkyl, (C3-C7)cycloalkyl-(C|-C6)aikyl; heterocycloalkyl containing 1-3 hetero atoms selected from O7 S, and (C1-C5)alkyl-O-(CrC5)aIkyl;
R4 and R6 are independently selected from (Ci-C8)alkyl, (Ci-Cs)alkoxy, (C3-C7)cycloalkyl, (C3-C7)cycloalkyl-(Ci-C6)aIkyI, heterocycloalkyl containing 1-3 hetero atoms selected from O3 S, N, (CrCs)alkyl-O-(Ci-C5)alkyl, amide, (C1-C5)alkyl-aryl3 and CF3;
Rs is selected from the group consisting of hydrogen, (Ct-Cg)alkyl, aryl, (Ci-C5)alkyl-O-(Cj- C5)alkyiJ and (CrC5)alkyI-aryI,
or
Rs and R4 and the atoms to which they are attached form a fused 5-6 member saturated carbocyclic ring or a fused 10 member bi-cyclic ring system, such as

R5 and R6 and the atoms to which they are attached form a fused 5-6 member saturated carbocyclic ring or a fused 10 member bi-cyclϊc ring system, such as
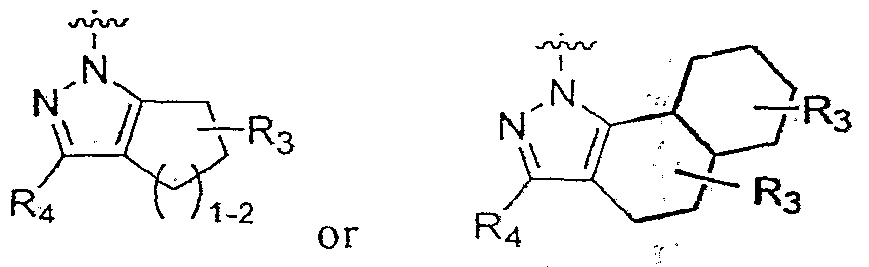
Rs -and R4 and the atoms to which they are attached form a fused 5-6 member saturated carbocyclic ring to which a 6 member aromatic ring is fused, such as

Rs and Rt, and the atoms to which they are attached form a fused 5-6 member saturated carbocyclic ring to which a 6 member aromatic ring is fused, such as

R> and R6 and the atoms to which they are attached form a fused benzothiophene or fused beπzofuran ring system, such as

where X is NR7, R7 and R2 taken together are -(CH2CH2)- to form a two nitrogen containing ring where y is 0 (piperazine) or y is 1 (homopiperazine), and wherein Ri is as defined previously, and the pharmaceutically acceptable salts thereof.
This invention also provides pharmaceutical compositions comprising compounds of formulae 1 -3, pharmaceutically acceptable salts, solvates, or formulations thereof, and pharmaceutically acceptable carriers in combination with an effective amount of at least one compound of formulae 1-3.
1 he present invention also provides a method of treating conditions in which modulation of histamine f I3 receptors may be of therapeutic importance such as inflammation, migraine, motion sickness, pain, Parkinson's Disease, epilepsy, cardiovascular disease (i.e.
hyper or hypotension, acute myocardial infarction), gastrointestinal disorders (acid secretion, motility) and CNS disorders involving attention or cognitive- disorders (i.e., Alzheimer's, Attention Deficit Disorder, age-related memory dysfunction, stroke, etc.), psychiatric disorders (i.e., depression, schizophrenia, obsessive-compulsive disorders, etc.); sleep disorders (i.e. narcolepsy, sleep apnea, insomnia, disturbed biological and circadian rhythms, hyper and hypsomnolence), and disorders such as obesity, anorexia/bulimia, thermoregulation, hormone release) comprising administering an effective amount of a compound of formulae 1 -3 to a patient in need of such treatment.
DETAILED DESCRIPTION OF THE INVENTION
Presently preferred compounds include:
3-Methyt-l-[4-(3-pyrrolidin-l-ylpropoxy)phenyl]-4,5-dihydro-lH-benzo[g]inda7.ole;
3-Methyl-I-{4-[ 3-(2/?-methylpyrrolidin-l -yI)propoxy]phenyl}-4,5-dihydro-l H- benzo[g-] indazo 1 e :
3-Methyl- 1 -[4-(3-morpholϊn- 1 -yIpropoxy)phenyl]-4,5-dihydro-l H-benzo[g]Indazole; l-[4-(3-PyrroIidin-l -ylpropoxy)phenyl]-5-styryl-3-trifluoromethyl-l //-pyra7olc;
3-Methyl-l-[4-(3-pyrroIidin-l -ylpropoxy)phenyl]-415,5a,6,7,8,9,9a-octahydro-} H- benzo[g]indazole;
3-Methyl-2-[4-(3-pyrτolidin- l -ylpropoxy)phenyl]-435,5a:>6:>7,8,9,9a-octahydro-2Λr- benzo [g] i n d azo I e ;
8-Methoxy-3-methyl-l -[4-(3-pyrrolidin- l-ylpropoxy)phenyl]-4,5-dihydro-I H- benzofg-jindayolc:
7-Methoxy-3-methyl- l-[4-(3-pyrrolidin-l -ylpropoxy)phenyl]-4,5-dihydro-I H- benzo[g-]indazole;
6-Methoxy-3-methyl-l -[4-(3-pyrrolidin-l-yIpropoxy)phenyl]-4,5-dihydro-IH- benzo[^]indazole;
2-[4-(l-Cyclopentyl-piperidin-4-yIoxy)phenylJ-5-methyl-2H-pyrazole-3-carboxyIic acid cyclohexylamide:
2-[4-(l-CycIohexytptperidIn-4-yloxy)phenyI]-5-methyl-2i?-pyrazole-3-carboxylic acid cyclohexylamide;
2-[4-(l-IsopropyIpiperidin-4-yloxy)phenyl]-5-methyl-2iϊ"-pyrazole-3-carboxylic acid cyclohexylamide;
2-[4-(l-CycIobutylpiperidin-4-ytoxy)phenyl]-5-methyl-2/Y-pyra7.ole-3-carboxylic acid cyclohexylamide; - • - •
{5-Methyl-2-[4-(3-pyrroIidIπ- 1 -ylρropoxy)phenyl]-2H-pyrazol-3-yl \ methanol; 5-CyclopentyIoxymethyl-3-methyI-l-[4-(3-pyrroIidin-l -yl-propoxy)phenyl]-l//-pyrazole; 5-Cyclopentyloxymethyl-3-ineLhyl- l-[4-(3-pyrrolidin-l-ylpropoxy)phenyl]-lH-pyrazole; 5-Isopropy!oxymethy!-3 -methyl- 1 -[4-(3-pyrrolidin-l-ylpiOpoxy)phenyl]-lHr-pyrazole; 2-[4-(3-Pyrrolidin-l -yIpropoxy)phcnyl]-2//-ϊndazole;
4-(4-Methoxyphenyl)-3,5-dimethyI-l-[4~(3-pyrrolidin-l -yIpropoxy)phenyl]-lH'-pyrazoIe; 1 -[4-(3-PyrroIidiri-I -ylpropoxy)phenyl]-l/f-indazote; 3,5-DiethyI- 1 -[4-(3-pyrτo 1 ϊdtn- 1 -y lpropoxy)phenyl]- 1 H-pyrazo Ie; 3,5-DiethyI-l-[4-(3-piperidm-l-yIpropoxy)pheπyl]-lH-pyrazσIe: 3,5-Diethyl-l-[4-(3-morphotin-l -ytpropoxy)pheny!]-lH-pyrazoIe: 3,5-DiisopropyI- 1 -[4-(3-pyrrolidin-l -yl-propoxy)-phenylj-l//-pyrazole; 3,5-DiϊsopropyI- 1 -[4-(3 -piperidin- 1 -yl-propoxy)-phenyl]- 1 H-pyrazo Ie; 3-tert-ButyI-5-methyl-l-[4-(3-pyrrofidin-l-ylpropoxy)-phenyl]- l //-pyrazαle; 3-ferf-Butyl-5-methyI- 1 -[4-(3-pϊperidin-l-ylpropoxy)-phenyI]- 1 H-pyrazole; 5-IsobutyI-3-methyt-l -[4-(3-pyrτolidin-l-yIpropoxy)-phenyI]-l//-pyrazole; 5-Isobutyl-3 -methyl- 1 -[4-(3-piperϊdiα-l-ylpropoxy)-phenylJ- 1 H-pyrazole; 5-Isobutyl-3-methy!-l -[4-(3-pipeπ'diα-l-ylpropoxy)-phenyl]-2H-pyrazole; 5-IsobutyI-3 -methyl- 1 -[4-(3 -pyrrol idiπ-l-ylpropoxy)-phenyl]-2//-pyrazole; l -Cyclobutyl-4-[4-(3,5-diisopropylpyrazoI-l-yl)phenoxy]pipcridinc: 5 -tert-Butyϊ-3 -methyl- 1 -[4-(3-pyrroIidin-l-ylpropoxy)-phenyl]- 1 /-/-pyrazole; 5-ter/-ButyI-3-methyl- 1 -[4-(3-pϊperidin-l-ylpropoxy)-phenyl]-l H-pyrazole; 3,5-Dimethyl-I -[4-(3-pyrrolidin- 1 -yIpropoxy)phenyl]-lH-pyrazυlc; 3,4,5-Trϊmethyl- 1 -[4-(3-pyrroiidin- l-ylpropoxy)phenylJ-l H-pyrazole; 4-Ethyl-3,5-dimethyl-l -[4-(3-pyrrolidin-l-ylpropoxy)ρhcnyl]- 1 H-pyrazole; 4-Butyl-3J5-dimethyt-l-[4-(3-pyrroIidin-l-ylpropoxy)pheayI]-l H-pyrazole; 4-Phenyl-3,5-dimethyl-1 -[4-(3-pyrrolidin-l-ylpropoxy)phenylJ- 1 H-pyrazole; 5-Methyl-3-phenyl-l -[4-(3-pyrrolidin-l-ylpropoxy)phenyl]-I H-pyrazole; 5-Methyl-3-phenyl-l -[4-(3-pyrrolidiα-l-ylpropoxy)phenylJ-2H-pyrazole; 3-/β/'t-ButyI-5-phenyI- 1 -[4-(3 -pyrrol idin-l-ylpropoxy)pheny I]- 1 //-pyrazole; 3-Phenyl-l -[4-(3-pyrrolidϊn- 1 -ylpropoxy)phenyl]-4,5,6,7-tetrahydro- 1 H-indazole; 3-Phenyl-l-[4-(3-pyrrolidin-l-ylpropoxy)phenyl]-4,5,6)7-tetrahydrø-2JH:-indazole; 5-Furan-2-yI-3-methyl- 1 -[4-(3-pyrrolidin-l-ylpropoxy)phenyl]- l H-pyrazole;
3-Diflυoromethyl-5-furan-2-yl-l-[4-(3-pyrrolidin-l-yIpropoxy)phenyl]-li/-pyrazole;
3-TrifluoromethyI-5- furan-2-yl-I -[4-(3-pyrrolidin- 1 -ylprαpoxy)phenyl)-l /-/-pyrazole;
3-Trifluoromethyl-5-thiophen-2-yl- 1 -[4-(3-pyrrolidin-l -ylpropoxy)phenyl]- 1 //-pyrazole;
3-Difluoromethyl-5-phenyl-l-[4-(3-pyrτolidϊn-I-yIpropoxy)pheπyl]-lH-pyrazolc;
5-Phenyl-l-[4-(3-pyrrolidin-l-yl-propoxy)-pheny}]-3-trifluoromethyl-l //-pyrazole; l-{4-[3-(2-(R)-Mcthyl-pyrrolidin-I-yl)-propoxy]-phcnyl}-5-phenyI-3-trifluoromethyl-l H- pyrazole;
DimethyI-(l -{3-[4-(5-phenyl-3-trifIuoromethyl-pyrazoI-i-yl)-phenoxy]-propyI}-pyrrotidiπ-3- yl)-amine;
4-{3-[4-(5-Phenyl-3-trifluoromethyI-pyrazol-l-yl)-phenoxy]-propyI}-morpholinc; l-{3-[4-(5-Phenyi-3-trifluoromethyl-pyrazol-l-y])-phenoxy]-propyl}-piperidine;
3-Methyl- 1 -[4-(3-pγrrolidin-l -yI-propoxy)-phenyl]-435,6,7-tetrahydro-l Η-indazo!e;
3-Methyl-2-[4-(3-pyrrolidin-l-yI-propoxy)-phenyl]-4,5ϊ6,7-tetrahydro-2H-indazoIe;
3-Methyl- 1 -[4-(3-pyτrolidin- 1 -yl-propoxy)-pheny I]- 1 ,4,5, ό-tetrahydro-cyclopentapyrazole;
3-Methyl-2-[4-(3-pyrrolidin-l-yl-propoxy)-phenyl]-2.435,6-tetrahydro-cyclopentapyra2ole;
3-Methyl- 1 -[4-(3 -pi peridin-1 -yl-propoxy)-phenyl]-l ,4,5,6-tetrahydro-cycIopentapyrazole;
3-Methyl-2-[4-(3-piperidin-l-yl-propoxy)-phenyI]-2,4,5J6-tetrahydro-cycIopentapyrazolc;
3,5-DiisopropyI- 1 -[ 2-methyl-4-(3-pyrτoIidin-l -ylpropoxy)phenyl]-l//-pyrazole;
3,5-Diisopropyl- l -[2-methyl-4-(3-piperidin-l-yIpropoxy)phen.yl]-l/:/'-pyrazole;
5-Benzofuran-2-yl-l -[4-(3-pyrroIidiπ-l -ylpropoxy)phenyl]-3-trifluoromethyt-I //-pyrazole:
3-Methyl-l-[4-(3-pyrrolidin-I -yl-propoxy)-phenyI]-lH-bcnzo[4,5]thieno[3,2-c]pyrazole;
3-Methyl- l-{4-[3-(2-methyl-pyrrolϊdin-l-yl)-propoxy]-pheαyl}-lH-benzo[4,5]thienof 3,2- c]pyrazole;
3-[4-(3-Pyrrolϊdin- 1 -yl-propoxy)-phenyl]-l -trifluoromethyl-3H-8-oxa-2,3-diaza- cyclopenta[a] indene,
3-{4-[3-(2-Methy!-pyrτolidin-l-yl)-propoxy]-phenyl}-l-trifluoromethyI-3H-8-oxa-2.3-diaza- eye lopenta[a] indene;
Dimethyl-(1 - { 3-[4-( 1 -trifluoromethyl-δ-oxa^p-diaza-cyclopentafaJinden-S-yO-phenox.y]- propyl}-pyrτolidin-3-yl)-amme;
1 -[4-trans-(3 ,5 -Diisopropyl-pyrazol- 1 -yl)-cycIohexyI]-4-isopropyl-piperazine;
1 -[4-cis-(3 ,5-Diisopropyl-pyrazol-l -yl)-cyclohexyl]-4-isαpropyl-piperazine;
3,5-Diisopropyl- I -[trans-4-(3-pyrrolϊdin-l-yi-propoxy)-cyclohexyl]-lH-pyrazoIe;
3,5-Diisopropyl- 1 -[cis-4-(3-pyrrolidin- 1 -yI-propoxy)-cyclϋhexyl]-lH-pyrazoIe;
5-Methyl-2-[4-(3-pyrτolidin-l-yl-propoxy)-phenyl]-2H-pyrazole-3-carbυxylic acid cyclohexylamide - - - - •
5-Methyi-2-{4-[3-(2i?-methy lpyrrol idϊn-1 -yl)propoxy]phenyl}-2//-pyrazole-3-carboxylic acid cyclohexylamide;
5-Methyl-2-[4-(3-piperidin-l -yIpropoxy)phenylj-2//-pyrazole-3-carboxylic acid cyclohexylamide;
5-Methyl-2-[4-(3-morpholin-4-yIpropoxy)phenyl]-2//-pyrazolc-3-carboxylϊc acid cyclohexylamide;
5-Methyl-2-{4-[2-(l-methylpyrτolidin-2-yl)ethoxy]phenyl}-2//-pyrazoIe-3-carboxyIic acid cycl ohexylamide;
{5-Methyl-2-[4-(3-pyrroIidin-I-ylpropoxv)phenyπ-2//-pyrazol-3-yI}pyrrolidin-l- ylmeLhanone;
5-Methyl-2-[4-(3-pyrτolidin-l -yIpropoxy)phenyl]-2//-pyrazole-3-carboxylic acid cyclohexylmethylamide;
 cyclobutylamide;
cyclobutylamide;
5-Methyl-2-[4-(3-pyrrolidin-l-ylpropoxy)phenylj-2//-pyrazole-3-carboxylic acid phenyl amide;
5-Methyl-2-[4-(octahydroquinoltzin- I-ylmethoxy)phenyI]-2//-pyrazole-3-carboxylic acid cycfohexylamide; and
5-Methyl-l-[4-(3-pyrrolidin-I -ylpropoxy)pheayl]-l //-pyrazole-3-carboxylic acid cyclohexylamide.
Certain compounds of the invention may exist in different isomeric (e.g. enantiomers and distereoisomers) forms. The invention contemplates all such isomers both in pure form and in a mixture, including racemic mixtures. Enol and tautomeric forms are also included.
The compounds of the invention can exist in uπsolvaled as well as solvated forms, including hydrated forms, e.g., hemt-hydrate. In general, the solvated forms, with pharmaceutically acceptable solvents such as water, ethanol, and the like are equivalent to the υnsolvated forms for the purposes of the invention.
Certain compounds of the invention also form pharmaceutically acceptable salts, e.g., acid addition salts. For example, the nitrogen atoms may form salts with acids. Examples of suitable acids for salt formation are hydrochloric, sulfuric, phosphoric, acetic,
citric, oxalic, malυnic, salicylic, malic, fumaric, succinic, ascorbic, maleic, methanesulfonic and other mineral carboxylic acids well known to those in the art.- The salts are prepared by contacting the free base form with a sufficient amount of the desired acid to produce a salt in the conventional manner The free base forms may be regenerated by treating the salt with a suitable dilute aqueous base solution such as dilute aqueous hydroxide, potassium carbonate, ammonia, and sodium bicarbonate. The free base forms differ from their respective salt forms somewhat in certain physical properties, such as solubility in polar solvents, but the acid salts are equivalent to their respective free base forms for purposes of the invention (See, for example S M. Berge, et ai., "Pharmaceutical Salts," J. Pharm. Sci., 66: 1-19 (1977) which is incorporated herein by reference.
As throughout this specification and appended claims, the following terms have the meanings ascribed to them:
The term "alkyl" as used herein refers to straight or branched chain radicals derived from saturated hydrocarbons by the removal of one hydrogen atom. Representative examples of alkyl groups include methyl, ethyl, n-propyl, iso-propyl, n-butyl, sec-butyl, iso- butyl, tert-butyl, and the like.
The term ^cycloaikyl" as used herein refers to an aliphatic ring system having 3 to 10 carbon atoms and I to 3 rings, including, but not limited to cyclopropyl, cyclopentyl, cyclσhexyl, norbornyl, and adamantly among others. Cycloalkyi groups can be unsubstituted or substituted with one, two or three substituents independently selected from lower alkyl, haloalkyl, alkoxy, thioalkoxy, amino, alkylarntno, dϊalkylamino, hydroxy^ halo, mercapto, nitro, carboxaldehyde, carboxy, alkoxycarbonyl and carboximide.
"Cycloalkyi" includes cis or trans forms. Furthermore, the substituents may either be in endo or exo positions in the bridged btcyclic systems.
The term "'halo" or "halogen'' as used herein refers to I, Br, Cl or F.
The term "heteroatom" as used herein refers to at least one N, O or S atom.
The term "heterocycloalkyl" as used herein, alone or in combination, refers to a non-aromatic 3- to 10- membered ring containing at least one endocyclic N, O, or S atom The heterocycle may be optionally aryl-fused. The heterocycle may also optionally be substituted with at least one substituent which is independently selected from the group consisting of hydrogen, halogen, hydroxy!, amino, nitro, ttiflouromethyl, trifluoromethoxy, alkyl, aralkyt, alkenyl, alkynyl, aryl, cyano, carboxy, carboalkoxy, carboxyalkyl, oxo, arylsulfonyl and aralkylaminocarbonyl among others.
As used herein, the term "composition" is intended to encompass a product comprising the specified ingredients in the specified amounts, as well as any product which results, directly or indirectly, from a combination of the specified ingredients in the specified amounts.
The compounds of the present invention can be used in the form of pharmaceutically acceptable salts derived from inorganic or organic acids. The phrase "pharmaceutically acceptable salt" means those salts which are. within the scope of sound medical judgment, suitable for use in contact with the tissues uf humans and lower animals without undue toxicity, irritation, allergic response and the like, and are commensurate with a reasonable benefit/risk, ratio. Pharmaceutically acceptable salts are well-known in the art. For example, S. M. Berge et al. describe pharmaceutically acceptable salts in detail in J. Pharmaceutical Sciences, 1977, 66: I et seq The salts can be prepared in situ during the final isolation and purification of the compounds of the in\ entiun or separately by reacting a free base function with a suitable organic acid. Representative acid addition salts include, but are not limited to acetate, adipate, alginate, citrate, aspartate, benzoate, benzenesulfonate, bisulfate, butyrate, camphorate, camphorsulfonate. digluconate, glycerophosphate, hemisulfate, heptanoate, hexanoate, fumarate. hydrochloride, hydrobromide. hydroϊodide, 2- hydroxyethansulfonate (isothionate), lactate, maleate. rnethanesulfonate, nieυtinate, 2- naphthalenesulfonate, oxalate, palmitoate, pectinate, persutfate, 3-phenylpropϊonate, picrate, pivalate, propionate, succinate, tartrate, thiocyanate, phosphate, glutamate. bicarbonate, p- toluenesulfonate and. undecanoate. Also, the basic nitrogen-containing groups can be quatemized with such agents as lower alky! halide.s such as methyl, ethyl, propyl, and butyl chlorides, bromides and iodides; dialkyt sulfates like dimethyl, diethyl, dibutyl and diamyl sulfates; long chain halϊdes such as decyl, Iauryl. myristyl and stearyl chlorides, bromides and iodides; arylalkyl halides like benzyl and phenethyl bromides and others. Water or oil-soluble or dispersible products are thereby obtained. Examples of acids which can be employed to form pharmaceutically acceptable acid addition salts include such inorganic acids as hydrochloric acid, hydrobromic acid, sulphuric acid and phosphoric acid and sueh organic acids as oxalic acid, maleic acid, succinic acid and citric acid
Basic addition salts can be prepared in situ during the final isolation and purification of compounds of this invention by reacting a carboxylic acid-containing moiety with a suitable base such as the hydroxide, carbonate or bicarbonate of a pharmaceutically acceptable metal cation or with ammonia or an organic primary, secondary or tertiary amine. Pharmaceutically acceptable salts include, but are not limited to. cations based on alkali
metals or alkaline earth metals such as lithium, sodium, potassium, calcium, magnesium and aluminum salts and the tike and- nontoxic quaternary ammonia and amine cations including ammonium, tetramethylammonϊum, tetraethylammontum. methylammonium, dimethylammontum, trimethyl ammonium, triethylammonium, diethylammonium, and ethylammonium among others. Other representative organic amines useful for the formation of base addition salts include ethylenediamine, ethanoiamtne, dϊethanolamine, piperidine, piperazine and the like.
Dosage forms for topical administration of a compound of this invention include powders, sprays, ointments and inhalants. The active compound is mixed under sterile conditions with a pharmaceutically acceptable carrier and any needed preservatives, buffers or propellants which can be required. Opthalmϊc Formulations, eye ointments, powders and solutions are also contemplated as being within the scope of this invention.
Actual dosage levels of active ingredients in the pharmaceutical compositions of this invention can be varied so as to obtain an amount of the active compound(s) which is effective to achieve the desired therapeutic response for a particular patient, compositions and mode of administration. The selected dosage level will depend upon the activity of the particular compound, the route of administration, the seventy of the condition being treated and the condition and prior medical history of the patient being treated. However, it is within the skill of the art to start doses of the compound at levels lower than required to achieve the desired therapeutic effect and to gradually increase the dosage until the desired effect is achieved.
When used in the above or other treatments, a therapeutically effective amount of one of the compounds of the present invention can be employed in pure form or, where such forms exist, in pharmaceutically acceptable salt, ester or prodrug form. Alternatively, the compound can be administered as a pharmaceutical composition containing the compound of interest in combination with one or more pharmaceutically acceptable excipients. The phrase "therapeutically effective amount" of the compound of the invention means a sufficient amount of the compound to treat disorders, at a reasonable benefit/risk ratio applicable to any medical treatment. It will be understood, however, that the total daily usage of the compounds and compositions of the present invention will be decided by the attending physician within the scope of sound medical judgment. The specific therapeutically effective dose level for any particular patient will depend upon a variety of factors including the disorder being treated and the severity of the disorder; activity of the specific compound employed; the specific composition employed; the age, body weight, general health, sex and
diet of the patient the time of administration, route of administration, and rate of excretion of the speci fic compound employed; the duration of the treatment; drugs used in combination- or coincidental w ith the specific compound employed, and like factors well known in the "' medical arts For example, it is well within the skill of the art to start doses of the compound at levek lower than lequired to achieve the desired therapeutic effect and to gradually increase the dosage until the desired effect is achieved t hese compounds have been tested in vitro and in vivo and have been shown to be potent and se lective inhibitors of H3 receptor activation. The total daily dose of the compound-, or this invention administered to a human or lower animal may range from about - 0 0001 to about HX)O mg/kg/day. For purposes of oral administration, more preferable doses ". can be in the range o t from about 0.001 to about 5 mg/ky/day. If desired, the effective daily ' dose can ^c div ided into multiple doses for purposes of administration; consequently, single „ dose compositions may contain such amounts or submultiples thereof to make up the daily \ dose
1 1'v, present invention also provides pharmaceutical compositions that .- compr.se compounds ot the present invention formulated together with one or more non-toxic pharmaceutic al l v acceptable carriers. The pharmaceutical compositions can be specially formulated tor oral administration in solid or liquid form lor parenteral injection or for rectal - administration
1 he pharmaceutical compositions of this invention can be administered to humans ant! other mammals orally, rectally, parenteral Iy , intracisternaUy, intravaginally, intraperi toneal! ^ topically (as by powders, ointments or drops), bucally or as an oral or nasal - sprav The term "parenterally," as used herein, refers to modes of administration which ' include i ntrav e nous intramuscular, intraperitoneal, 1 utrastemal, subcutaneous and intraarticular mic tion and infusion.
Ip another aspect, the present invention provides a pharmaceutical composition comprising a component of the present invention and a physiologically tolerable - diluent T he present invention includes one or more compounds as described above formulated into compositions together with one or more non-toxic physiologically tolerable * or acceptable di luents carriers, adjuvants or vehicles that are collectively referred to herein as ". diluents, for parenteral injection, for intranasal delivery, for oral administration in solid or liquid form, for rectal or topical administration, among others.
I he compositions can also be delivered through a catheter for local delivery at a target site via an mtracoronary stent (a tubular device composed of a fine wire mesh), or
v/(/ a biodegradable polymer. The compounds may also be complexed to tigands, such as .v.' .i iodies, for targeted delivery. - ■
Compositions suitable for parenteral injection may comprise physiologically αjcrμtable, sterile aqueous or nonaqueous solutions, dispersions, suspensions or emulsions cif i> 1 sterile powders for reconstitution into sterile injectable solutions or dispersions.* t: \a..- iiples of suitable aqueous and nonaqueous carriers, diluents, solvents or vehicles include witi--, ethanol, polyols (propytenegiycol, polyethyleneglycol, glycerol, and the like), \. cfc-:able oils (such as olive oil), injectable organic esters such as ethyl υleate, and suitable π: i \:.» ires thereof.
These compositions can also contain adjuvants such as preserving, wetting, cini Uifying, and dispensing agents. Prevention of the action of microorganisms can be ff. --.i-ed by various antibacterial and antifungal agents, for example, parabens, chlorobutanol, ι^'-.-πol, sorbic acid, and the like. It may also be desirable to include isotonic agents, for ex. rnple sugars, sodium chloride and the like. Prolonged absorption of the injectable phar maceutical form can be brought about by the use of agents delaying absorption, for c:- .ι:rple, aluminum monostearate and gelatin.
Suspensions, in addition to the active compounds, may contain suspending :'Λ r.-' fs, as for example, ethoxylated isostearyi alcohols, polyoxyethylene sorbitol and sorbitan '.-^IVr-., microcrystatline cellulose, aluminum metahydroxide, bentonite. agar-agar and f r .!• •..• αanth, or mixtures of these substances, and the like.
In some cases, in order to prolong the effect of the drug, it is desirable to slow i}'.-.: ..'.bsorptxon of the drug from subcutaneous or intramuscular injection. This can be LiC-'.. ifiplished by the use of a liquid suspension of crystalline or amorphous material with r-i .r-r water solubility. The rate of absorption of the drug then depends upon its rate of J ; ..-.: lution which, in turn, may depend upon crystal size and crystalline form. Alternatively, del,;', ed absorption of a parenterally administered drug form is accomplished by dissolving or SU -pending the drug in an oil vehicle.
Injectable depot forms are made by forming microencapsule matrices of the f iri!;'. in biodegradable polymers such as polylactide-polyglycolide. Depending upon the ratio oi J πig to polymer and the nature of the particular polymer employed, the rate of drug release •j.ifi :>e controlled. Examples of other biodegradable polymers include poly(orthoesters) and pi •' :-'{ anhydrides). Depot injectable formulations are also prepared by entrapping the drug in Mr-. nines or microemulsϊoπs which are compatible with body tissues.
The injectable formulations can be sterilized, for example, by filtration through a bacterial-retaining filter or by incorporating sterilizing agents in the form of sterile solid compositions which can be dissolved or dispersed in sterile water or other sterile injectable medium just prior to use.
Solid dosage forms for oral administration include capsules, tablets, pills, powders and granules, tn such solid dosage forms, the active compound may be mixed with at least one inert, pharmaceutically acceptable excipient or carrier, such as sodium citrate or dicalciurn phosphate atid 'oi ai fillers or extenders such as starches, lactose, sucrose, glucose, mannϊtol and silicic acid, b) binders such as carboxymeLhy [cellulose, alginates, gelatin, polyvinylpyrrolidone sucn^e and acacia; c) humectants such as glycerol; d) disintegrating agents such as agar-agar calcium carbonate, potato or tapioca starch, alginic acid, certain silicates and sodium LarKmare; e) solution retarding agents such as paraffin; (f) absorption accelerators such as quaternary ammonium compounds; g) wetting agents such as cetyf alcohol and glycerol rnunostcarate; h) absorbents such as kaolin and bentonite clay and i) lubricants such as talc, calcium stearate, magnesium stearatc. solid polyethylene glycols, sodium lauryl sul fate and mixtures thereof. In the case of capsules, tablets and pills, the dosage form may also comprise buffering agents.
Solid compositions of a similar type may also be employed as fillers in soft and hard-filled gelatin capsules using such excipients as lactose or milk sugar as well as high molecular weight pυKcthv icr.c glycols and the like.
'[ he solid dυ^aμe forms of tablets, dragees, capsules, pills and granules can be prepared with coatings and .-hells such as enteric coatings and other coatings well-known in the pharmaceutical formulating art. They may optionally contain opacifying agents and may also be of a composition MR h that they release the active ingredieαt(s) only, or preferentially, in a certain part of the intestinal tract, optionally, in a delayed manner. Examples of embedding compositions which can be used include polymeric substances and waxes.
The active compounds can also be in micro-encapsulated form, if appropriate, with one or more of the above-mentioned excipients.
Liquid dυsaμe forms for oral administration include pharmaceutically acceptable emulsions, solutions, suspensions, syrups and elixirs. In addition to the active compounds, the liquid dosaue forms may contain inert diluents commonly used in the art such as, for example, uatcr or other solvents, sohibtlizing agents and emulsifiers such as ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propylene gKcol. i ,3-butylene glycol, dimethyl formamide, oils (in particular,
cottonseed, groundnut, corn, germ, olive, castor and sesame oils), glycerol, tetrahydrofurfuryl alcohol, polyethylene glycols and fatty -acid esters of sorbitan and mixtures thereof.
Besides inert diluents, the oral compositions may also include adjuvants such as wetting agents, emulsifying and suspending agents, sweetening, flavoring and perfuming agents.
Compositions for rectal or vaginal administration are preferably suppositories which can be prepared by mixing the compounds of this invention with suitable non-irritating excipients or carriers such as cocoa butter, polyethylene glycol or a suppository wax which are solid at room temperature but liquid at body temperature and therefore melt in the tectum or vaginal cavity and release the active compound.
Compounds of the present invention can also be administered in the form of liposomes. As is known in the art, liposomes are generally derived from phospholipids or other lipid substances. Liposomes are formed by mono- or multi-lameltar hydrated liquid crystals which are dispersed in an aqueous medium. Any non-toxic, physiologically acceptable and metabolizable lipid capable of forming liposomes can be used. The present compositions in liposome form can contain, in addition to a compound of the present invention, stabilizers, preservatives, excipients and the like. The preferred lipids are natural and synthetic phospholipids and phosphatidyl cholines (lecithins) used separately or together.
Methods to form liposomes are known in the art. See, for example, Prescott, Ed., Methods in Cell Biology.. Volume XIV, Academic Press, New York, N. Y. ( 1 076 ). p. 33 et seq.
The term "pharmaceutically acceptable prodrugs" as used herein represents those prodrugs of the compounds of the present invention "which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of humans and lower animals without undue toxicity, irritation, allergic response, and the like, commensuiate with a reasonable benefit/risk ratio, and effective for their intended use, as well as the zwϊtterionic forms, where possible, of the compounds of the invention. Prodrugs of the present invention may be rapidly transformed in vivo to the parent compound of the above formula, for example, by hydrolysis in blood. A thorough discussion is provided in T. Higuchi and V. Stella, Pro-dαms as Novel Delivery Systems, V. 14 of the A. CS. Symposium Series, and in Edward B. Roche, ed., Bioreversible Carriers in Drug Design, American Pharmaceutical Association and Pergamon Press (1987), hereby incorporated by reference.
Compounds of the present invention that are formed by in vivo conversion of a different compound that was administered to a mammal are intended to be included within the scope of the present invention.
Compounds of the present invention may exist as stereoisomers wherein asymmetric or chirai centers are present. These stereoisomers are "R." or "S" depending on the configuration of substituents around the chirai carbon atom. The present invention contemplates various stereoisomers and mixtures thereof. Stereoisomers include enantϊomers and dϊastereomers, and mixtures of enantiomers or diastereomers. Individual stereoisomers of compounds of the present invention may be prepared synthetically from commercially available starting materials which contain asymmetric or chirai centers or by preparation of racemic mixtures followed by resolution well-known to those of ordinary skill in the art. These methods of resolution are exemplified by (1) attachment of a mixture of enantiomers to a chiral auxiliary, separation of the resulting mixture of diastereomers by recrystallizatϊon or chromatography and liberation of the optically pure product from the auxiliary or (2) direct separation of the mixture of optical enantiomers on chirai chromatographic columns.
The compounds of the invention can exist in unsolvated as well as soivated forms, including hydrated forms, such as hemi-hydrates. In general, the soivated forms, with pharmaceutically acceptable solvents such as water and ethanol among others are equivalent to the unsolvated forms for the purposes of the invention.
The invention ma\ be illustrated by the following representative schemes and examples.


3-Methyl-l-[4-(3-pyrroIidin-l-yIpropoxy)pheπylj-4,5-dihydro-l//-benzo|^linda-/.o!e

l-(4-Methoxy-pheπyl)-3-raethyI-4,5-dihydro-lH-benzo[g]indazole. To a solution of ?. acetyl- 1 -tetralone (329 rag, 1.75 mmol) in ethano! (12 mL) was added 4-metho.xypb.cn> ϊ hydrazine hydrochloride. The stirred suspension was heated to SO0 C overnight. The reaction was cooled to room temperature and diluted with water (100 mL) and extracted with EtO Ac (2x 50 mL). The combined organic layers were washed with water, 10% NaOH, 10% HCl. brine and dried over Na2SC*4- The solvent was removed under reduced pressure and the- residue purified on silica gel using a 5% EtOAc to 25% EtOAc in hexane gradient (vidd 3^6 mg). LC-MS (C19H18N2O calculated 290) m/z 291 (M÷H).
4-(3-Methyl-4,5-dihydrobenzo[grJindazol-l-yl)phenol. I-(4-Methoxyphenyl)-3-methyl-4,ς - dihydro-I //-benzo[g-]indazole (200 mg, 0.69 mmol} was dissolved in dichloromethane (2 m l ) under N2 and cooled to — 400 C. Boron tribromϊde (2.07 mL, 1 M in dichloromethane, 2.O7 mmol) was added dropwise and the solution was stirred for 4 hours, warming to room temperature. The reaction mixture was carefully diluted with saturated NaHCO.* solution The mixture was extracted with dichloromethane, and the extracts were dried over MgSC >j and concentrated. SiO2 chromatography with 20-80% ethyl acetate/hexanes gave 144 mu of the desired product (76% yield). LC-MS (C18Hi6N2O calculated 276) m/z 277 (M+H).
3-Methyl-l-[4-(3-pyrrolidin-l-ylpropoxy)phenylJ-4,5-dihydro-l /f-benzo[»]inda7.oIe. 4- (3-Methyi-4,5-dihydrobenzo[g-]indazol-l-yl)phenol (48 mg, 0.174 mmol) was dissolved in jV,N-dimethylformamide (1 mL), and l -(3-chloropropyl)pyrrolidine (31 mg, 0.21 mmol,). sodium hydride (8 mg, 60% dispersion in mineral oil, 0.21 mmol) and sodium iodide (32 mg. 0.21 mmol) were added. The reaction was heated at 70° C for 1.5 hours, then cooled to room temperature. Saturated sodium bicarbonate solution was added, and the mixture was extracted
with ethyl acetate. The extracts were dried over MgSO4 and concentrated. S1O2 chromatography with ethyl acetate, then 2% triethyIamine/10% methanol/ethyl acetate gave 27.4 mg of the desired product (40% yield). LC-MS (C25H29N3O calculated 387) m/z 388 (M+H); 1H NMR (300 MHz, CDCl3) δ 7.39 - 7.34 (m, 2H), 7.27 (d, J = 7.5 Hz, IH), 7.12 (t, J = 7.5 Hz, IH), 7.01 - 6.93 (m, 3H), 6.81 (d, J= 7.8 Hz, IH), 4.09 (t, J = 6.3 Hz, 2H), 2.98 (t, J = 6.9 Hz, 2H), 2.74 - 2.60 (m, 8 H). 2.30 (s, 3H), 2.14 - 2.04 (m, 2H), 1.86 - 1.82 (m, 4H).
Example 2 3-MethyI-l-{4-[3-(2i?-methylpyrrolidin-l-yl)propoxy}phenyl}-4,5-dihydro-ljHr- benzυ [gjϊndazole

3 -M ethyl -1 - {4-[3-(2ft-methylpyrrolidin- 1 -y I ipropoxyjphenyl } -4,5-dihydro- 1 H- benzo[g]indazole was synthesized by a method analogous to that used for Example L LC- MS (C26H31N3O calculated 401) m/z 402 (M+H).
Example 3 3-Methyl-l-[4-(3-morpholin-l-ylpropoxy)phenyI]-4,5-dihydro-li!/-benzo[^]inda2oIe

3-M ethyl- 1 -[4-(3-morpholin-l -ylpropoxy)phcnyl]-435-dϊhydro-l H-benzo[g]indazole was synthesized by a method analogous to that used for Example 1. LC-MS (C25H298N3O2 calculated 403) m/z 404 (M+H); 1H NMR (100 MHz, CDCI3) δ 7.40 (m, 2H), 7.27 (d, ./= 6.9 Hz, 1 H)3 7.12 (dt, J = 7.5 Hz3 1.2 Hz3 1 H), 7 01 - 6.93 (m. 3H), 6.81 (d3 J = 6.9 Hz, IH), 4.09 (t, J= 6.3 Hz, 2H), 3.85 - 3.77 (m, 4H). 2 98 (t, J= 6.9 Hz, 2H), 2.74 - 2.60 (m, 8H), 2.30 (s, 3H), 2.13 - 2.04 (m, 2H).

Exam ple 4 l-j4-(3-PyrroIidin-l-yIpropoxy)phenyil-5-stv r3 l-3-trϊfl u{)ruπ(iethyHi?:-pyrazoIe

l,I,l-Trifluor&-6-phenylhex-5-ene-2,4~dione. Sodium h> dmL- i ^47 mg, 60% dispersion in mineral oil, 13.68 mmol) was added to ethyl trifluυroaceicue (1 .6i rαL, 13.68 mmol). (Note: Carefulness is required. The reaction caught fire upon ,ιdJmy sodium hydride to ethyl trifluoroacctate.) 7r<my-4-Phenyl-3-buten-2-υne (' 1 U1. 6.X-5 rπrnol) was added, and the reaction was stirred for 3 hours at 40° C. The reaction
 αMld to room temperature and quenched with water, then diluted with I N F lC'!. The inr.\t>.!;c was extracted with ethyl acetate, dried over MgSθ4, and concentrated. The reaction w:t^ assumed to be quantitative. LC-MS (C12H9F3O2 calculated 242) m/z 243 (M+H).
αMld to room temperature and quenched with water, then diluted with I N F lC'!. The inr.\t>.!;c was extracted with ethyl acetate, dried over MgSθ4, and concentrated. The reaction w:t^ assumed to be quantitative. LC-MS (C12H9F3O2 calculated 242) m/z 243 (M+H).
l-(4-Methoxypheπyl)-5-styryl-3-trifluoromethyl-ϊ/7-p\ ra/ole. l,l,l-Trifluoro-6- phenyIhex-5-ene-2,4-dione (2.28 mmol) and 4-methυxyphen> I hydrazine hydrochloride (435 mg, 2.5 mmol) were heated in ethanol (7 mL) at 70° C υvernϊμht. The solution was diluted with water and extracted with ethyl acetate. The ethyl acetate extracts were washed with 1 N HCl5 saturated sodium bicarbonate solution, and brine, then dried over MgSC>4 and concentrated. SiO2 chromatography with 5-20% ethyl acctate.'hcxanes gave 0.14 g of the desired product, along with many mixed fractions. Oπ!> - he clean fractions were carried on. LC-MS (Ci9Hi5F3N2O calculated 344) m/z 345 (M+H ): : H NM R ( 100 MHz, CDCI3) δ 7.43 -
7.30 (m, 7H), 7.12 (d, J = 16.2 Hz, I H), 7.05 - 7.00 (m, 2H), 6.88 (s, IH), 6.78 (d, J = 16.2 Hz, I H), 3.89 (s, 3 H).
4-(5-Styryl-3-trifluoromethylpyrazol-I-yl)phenoL l-(4-Methoxyphenyl)-5-styryl-3- trifluQromethyl- l /7r-pyrazole (0.14 g, 0.4 mmol) was dissolved in dichloromethane (1.2 ml) and cooled to —10" C. Boron tribromide (1 .2 ml. 1 M in dichloromethane, 1 .2 mmol) was added, and the reaction was stirred overnight, warming up to room temperature. Saturated sodium bicarbonate solution was added, and the mixture was extracted with ethyl acetate. The extracts were dried over MgSO4 and concentrated. The reaction was assumed to be quantitative. LC-MS (Ci8HuF-SN2O calculated 330) m/z 33l (M+H).
l-[4-(3-Pyrro[idϊn-I-γlpropoxy)phenyI]-5-styryl-3-trifluoromethyI-l//-pyrazole. 4-(5- StyryI-3-friπuoromethylpyrazol-l-yl)phenol (U.2 mmol) was dissolved in MtN- dimcthylhbπnamtde ( L mL), and l -(3-chloropropyl)pyrrolidine (32 mg, 0.22 mmol), sodium hydride (9 mg. 60"Zo dispersion in mineral oil, 0.22 mmol) and sodium iodide (33 rag, 0.22 mmol) were added. The reaction was heated at 70° C overnight, then cooled to room temperature. Saturated sodium bicarbonate solution was added, and the mixture was extracted with ethyl acetate. The extracts were dried over MgSO4 and concentrated. Semi-prep LC- MS purification gave 20.8 mg of the desired product. LC-MS (C25H26F3N3O calculated 441) m/∑ 442 (M f f I): 1 H NMR (300 MHz3 CDCI3) δ. 7.42 - 7.30 (m, 7H), 7.12 (d, J = 16.2 Hz, IH), 7 00 (d, J = 8.7 Hz, 2H), 6.88 (s, IH), 6.77 (d, J = 16.5 Hz, IH), 4.10 (t, J - 6 Hz, 2H), 2.93 - 2.86 (m. 6H), 2.21 - 2.12 (m, 2H), 1.94 - 1 .90 (m, 4H).


2-AcetyIoctahydrGnaphthaIeπ-l-oπe. Boron trifiuoride-acetie acid complex (5.2 mL, 37.5 mmol) was cooled to 00C. /V-αw-l-Decalone (3.82 g, 25 mmol) in acetic anhydride (4.7 mL, 50 mrnol) was added, and the reaction was stirred at room temperature for 3.5 hours. Saturated ammonium chloride solution (95 mL) was added, and the mixture was heated to 80° C for 45 minutes. After cooling to room temperature, the mixture was extracted with ethyl acetate. The extracts were dried over MgSO4 and concentrated SiOj chromatography with 10-40% ethyl acetate/hexanes gave 0.96 g of the desired product
2-(4-Methoxyphenyl)-3-methyI-4,5,5a,6,7,8,9,9a-ocrahydro-2H'-benzc>[(§r| ϊndazole and 1- (4-Methoxyphenyl)-3-methyl-4,5,5a,6,7,8,9,9a-oetahydro-lH-benzo[^]indazole. 2-
Acetyloctahydronaphthalen-I-one (0.96 g, 4.95 mmol) was dissolved in absolute ethanol (10 mL), and 4-methoxyphenyihydrazine hydrochloride (0.95 g, 5.44 mmol ) was added. The reaction was stirred overnight at 70° C. The mixture was diluted with water and extracted with ethyl acetate. The organic extracts were washed with 1 N FICl. saturated sodium bicarbonate solution, and brine, then dried over MgSθ4 and concentrated. S1O2 chromatography with 5-40% ethyl acetate/hexanes gave 0.15 g of 2-(4-methoxyphenyI)-3- methyl-4,5,5^6,7J8,9,9a-octahydro-2/τr-benzo[g]indazole and 0.66 g of 1 -(4-methoxyphenyl)- 3-methyl-4,5-5a,6,7,8,9ϊ9a-octahydro-lH-benzo[^]indazoIe. Regiochemistry was determined by x-ray crystal structure of l-(4-methθϊ£yphenyl)-3-methyl-4.5,5a.6,7,8,Q,9a-octahydro-l//- benzυ[g-]indazole. LC-MS (C19H24N2O calculated 296) m/z 297 (M+H).
4-(3-MethyI-4,5,5a,6r7,8,9,9a-octahydrobenzofg]indazoI-l-yI)phenϋI. l-(4-
Methoxyphenyl)-3-methyl-4,5,5a,6,73839,9a-octahydro-lH-benzo[^Jinda2oIe (0.44 g, 1.49 mmol) was dissolved in dichloromethane (4 mL) and cooled to -40° C, and boron tribromide (0.5 mL) was added. The reaction was stirred for 2 hours, then carefully quenched with
saturated sodium bicarbonate solution. The mixture was extracted with dichloromethane. The organic extracts were washed with brine, dried over MgSO4, and concentrated. The reaction was assumed to be quantitative. LC-MS (C18.H22N2O calculated 282) m/z 283
(M+H).
3-Methyl-l-[4-(3-pyrrolidin-l-yIpropoxy)phenyl]-4,5,5a,6,7,8,9,9a-octahydro-lHr- benzo[g]indazole. 4-(3-Me&yl-4,5,5a,6,7,8,9,9a-octahydrobenzo[g]indazol-l-yl)phenol
(1.49 mmol) vvas dissolved in ΛζTV'-dimethylforrnamide (6 mL), and l -(3- chloropropyl)pyrrolidιne (0.22 g, 1.49 mmol), sodium hydride (72 mg, 60% dispersion in mineral oil, 1.79 mmol) and a catalytic amount of sodium iodide were added. The reaction was heated at 70° C for 3 hours. The solution was diluted with saturated sodium bicarbonate and extracted with ethyl acetate. The extracts were dried over MgSO4 and concentrated. SiO2 chromatograph} with 2% triethylamine/10% methanol/ethyl acetate gave 0.268 g of the desired product [ .C-MS (C25H35N3O calc'd 393) m/z 394 (M+H). 1H NMR (300 MHz, CDCl3) δ 7.27 (d, J - 8.7 Hz, 2H)3 6.91 (d, J= 9 Hz, 2H), 4.05 (t, J= 6.3 Hz, 2H), 2.64 (t, J = 7.2 Hz, 2H), 2 58 - 2 39 (m, 7H)5 2.20 (s, 3H), 2.07 - 2.00 (m, 2H)1 1.82 - 1.10 (m, 14H), 0.91 - 0.79 (m, I H).
Example 6 3-Methyl-2-[4-(3-pyrrolidin-l-ylpropoxy)phenyϊ]-4,5,5a,6,7,8,9,9a-octahydro-2/f- beπzo [g] ϊ π d azo 1 e


K x ample 7 8-Methoxy-3-.methvl-l-[4-( Vp\ rrolidin-l-ylpropoxy)phenyl)-4,5-dihydro-UΪ-

2-AcetyI-7-methαxy-3,4-dihydro-2//-πaphthaIen-l-one. Boron trifluoride etherate (0.53 mL) was added dropwise to a stirred mixture of 7-methoxytetraIone (176 mgτ 1 mmol) in acetic anhydride (1.8 mL). f he reaction was stirred at room temperature for 2 hours, then poured into ice-water and stirred tor 1 hour. The mixture was extracted with ether, and the ether extracts were evaporated. The residue was diluted with methanol (12 mL) and saturated sodium acetate (S mL) and stirred at reflux for 4 hours. After cooling to room temperature, the solution was extracted with diuhlorornethane. The organic extracts were washed with brine, dried over MgSO1J. and concentrated. SiO2 chromatography with 5-20% ethyl acetate/hexanes gave 72.5 rng of the desired product. LC-MS (CI3HI 4O3 calculated 218) m/z 217 (M-H).
l-(4-BenzyloxyphenyJ)-8-methoxv-3-methyl-4,S-dihydro-l/f-benzo[^]indazale. 2-Acetyl-
7-methoxy-3>4-dihydro-2/-/r-naphthaien- 1 -one (72.5 mg, . 0.33 mmol) and 4- benzyloxyphenythydrazine hydrochloride (90 mg, 0.36 mmol) were heated in elhanol at 70° C for 2.5 days. The reaction was diluted with water and extracted with ethyl acetate. The
organic extracts were washed with 1 N HCl, saturated sodium bicarbonate solution, and brine, dried over MgSO4, and concentrated. SiO2 chromatography with 5-20% ethyl acetate/hexanes gave 42 mg of the desired product. LC-MS (C26H24N2O2 calculated 396) m/z 397 (M+H).
4-(8-Methoxy-3-methyI-4,5-dihydrGbenzo[g]rndazol-l-yl)-phenol. l-(4-
Benzyloxyphenyl)-8-methoxy-3-methyl-4,5-dihydro-lH-benzo[g]indazole (42 mg, 0.1 mmol) was dissolved in methanol/tetrahydrofuran (2/1, v/v, 1.5 mL) and a catalytic amount of 10% Pd/C (wet) was added The flask was purged with nitrogen and hydrogen, then stirred under 1 atmosphere of hydrogen for 2 hours. The mixture was filtered through Celite and washed with methanol, and the filtrate was concentrated. The reaction was assumed to be quantitative. LC-MS (Ci9H, SNZO2 calculated 306) m/z 307 (M+H).
8-Methoxy-3-nτethyl-l-[4-(3-pyrrolidin-l-yIpropoxy)phenyI]-4r5-dihydro-lHr- benzo[g]indazole. 4-(8-Methoxy-3-methyl-4,5-dihydrobenzo[gjindazol-l-yl)-phenol (0 1 mmol) was dissolved in /V,/V-dimethylfoτmamide (I mL), and -(3-chloropropyl)pyrrolidine (15 mg, 0.1 mmol), sodium hydride (4 mg, 60% dispersion in mineral oil, 0.1 mmol) and sodium iodide (15 mg, 0.1 mmol) were added. The reaction was heated at 700 C for 3 hours. The solution was diluted with saturated sodium bicarbonate and extracted with ethyl acetate. The extracts were dried over MgSO4 and concentrated. SiO2 chromatography with 2% triethyIamine/10% methanol/ethyl acetate gave 8.6 mg of the desired product. LC-MS (C25H31N3O2 calculated 417) m/= 418 (M+H). 1H NMR (300 MHz, CDCl3) δ 7.41 - 7.36 (m, 2H), 7.17 (d, J = S.7 Hz1 IH), 6.99 - 6.94 (m, 2H), 6.67 (dd, J - 8.7 Hz, 2.4 Hz, IH), 6.40 (d, J = 2.4 Hz, I H), 4.07 (t, J = 6.3 Hz, 2H), 3.52 (s, 3H), 2.91 (m, 2H), 2.72 - 2.58 (m, 8H)5 2.30 (s, 3H), 2.12 - 2.04 (m, 2Fi), 1 .86 - 1.81 (m, 4H).
Example 8 7-Methoxy-3-methyI-l-(4-(3-pyrrolidin-l-ylpropoxy)phenyl]-4,5-dihydro-lJΪ- benzofgjindazole

7-Methoxy-3-methyl-l-[4-(3-pyrrolidin-l-yIpropoxy)pheαyl]-4,5-dihydro-l//- benzofgjindazole was synthesized by a method analogous to that used for Example 7. LC- MS (C26H31N3O2 calc'd 417) m/z 418 (M+H). 1H NMR (300 MHz, CDCi3) δ 7.39 - 7.34 (m, 2H), 6.95 - 6.90 (m, 2H)3 6.83 (d, J= 2.7 Hz3 IH), 6.73 (d, J= 8.7 Hz, IH), 6.53 (dd, J- 8.7 Hz, 2.7 Hz, IH), 4.08 (t, J= 6 Hz, 2H)5 3.77 (s, 3H), 3.12 - 3.07 (m, 6H), 2.95 (m, 2H), 2.65 (m, 2H), 2.29 (s, 3H), 2.25 - 2.18 (m, 2H), 2.05 - 1.96 (m, 4H).
Example 9 6-Methoxy-3-methyI-l-[4-(3-pyrrolidin-l-yIpropoxy)phenylJ-4,5-dihydro-1 /1/- benzo[g\[indazole

6-Methoxy-3-methyl-l-[4-(3-pyτrolidin-l-ylpropoxy)phenyI]-4,5-dihydro-l//- benzo[,g]indazole was synthesized by a method analogous to that used for Example 7. LC- MS (C26H31N3O2 calc'd 417) m/z 418 (M+H). 1H NMR (300 MHz, CDCI3) δ 7.37 - 7.32 (m, 2H), 6.98 - 6.91 (ra, 3H), 6.75 (d, J= 8.1 Hz, IH), 6.45 (d, J= 7.8 Hz, IH), 4.08 (t, J = 6 Hz, 2H), 3.85 (s, 3FI), 3.02 - 2.80 (m, 8H), 2.65 - 2.60 (m, 2H), 2.30 (s, 3H), 2.19 - 2.09 (m, 2H), 1.95 - 1.88 (m, 4H).

Example 10
2-[4-(l-CycIopentyI-piperidin-4-yloxy)phenyl]-5-methyI-2//-pyrazoIe-3-carboxylic acid cyclohexylamlde

2-(4-MethoxyphenyI)-5-methyl-2//-pyrazole-3-carboxyIic acid ethyl . ester. 4-
Methoxyphenylhydrazine hydrochloride (6.05 g, 34.8 mmol) and ethyl 2,4-dioxovalerate (4.45 mL, 31.6 mmol) were stirred overnight at 80° C in cthanol (SO mL). After the solution
was allowed to cool to room temperature, it was diluted with water and extracted with ethyl acetate. The organic layer was washed with water. 1 N" HCl, and saturated sodium bicarbonate. Concentration, and SiC«2 chromatography with 20-30% ethyl acetate/hexanes gave 3.1 g of the title compound along with 2.9 g of l-(4-methoxyphenyi)-5-methyl-l/f- pyrazole-3-carboxylic acid ethyl ester.
2-(4-Merhoxyphenyl)-5-methyl-2//-pyrazoIe~3-carboxylic acid. To a solution of 2-(4- methoxyphenyt)-5-methyl-2JT-pyrazole-3-carboxylic acid ethyl ester (1.00 g, 3.84 mmol) in ethanol (20 ml) and water (20 mL) was added a sodium hydroxide solution (50% in water, 1 mL). The solution was stirred at 45° C overnight. After the solution was allowed to cool to room temperature. 1 N HCl was added until the solution was acidic to pH paper. The solution was extracted with ethyl acetate (3x). The combined organic layers were dried (MgSO<ι) and concentrated to give 761 mg of the title compound, uhieh was used in the next reaction without further purification. LC-MS (CJ2HI2N2O3 calculated 232) m/z 233 (M+H).
2-(4-Methoxyphenyl)-5-methyl-2jy-pyrazole-3-carboxylic acid cyclohexylamide. 2-(4- Methoxyphenyl)-5-methyl-2/f-pyrazoIe-3-carboxyIic acid (752 mg, 3.24 mmol) was suspended in methylene chloride (25 mL) and DMSO (3 drops) was added. After the reaction was stirred at room temperature for 1 hour, it was concentrated in vacuo. The residue was diluted twice with methylene chloride (60 mL) and concentrated to dryness. The residue was diluted again w ith methylene chloride (45 mL), cyclohexv famine (650 μL, 5.68 mmol) was added, and the reaction was stirred at room temperature for I hour. The reaction was diluted further with methylene chloride, washed with NaOH (10%) and 1 N HCl, then dried over Na^SO.}. Concentration gave 630 mg of the title compound, which was used in the next reaction without further purification. LC-MS (Ci8H2JN3O2 calculated 313) m/z 314 (M+H).
2-(4-HydroxyphenyI)-5-raethyI-2//-pyrazole-3-carbox> lic acid cyclohexylamide. To a solution of 2-(4-methσxyphenyl)-5-methyl-2i/~pyrazole-3-carboxyIic acid cyclohexylamide -(630 mg, 2.01 mmol) in methylene chloride (30 mL) at -40° C was added boron tribromide (6 mL, 1 M in DCM, 6 mmol). After the reaction was stirred at -40° C for 10 minutes and at room temperature for 2 hours, it was quenched with saturated sodium bicarbonate. Ethyl acetate was added, and the mixture was stirred for 1 hour The layers were separated, and the aqueous layer was extracted an additional time with ethyl acetate. The combined organic
layers were washed with water and brine, dried (MgSO4), and concentrated to give 600 mg of the title compound which was used in the next reaction without further purification. LC-MS (CnH21N3O2 calculated 299} m/z 300 (M+H).
4-[4-(5-CyclohexyIcarbamoyl-3-methyipyrazol-l-yI)phenoxyJpipeπdϊne-l-carboxyltc acid tert-huty\ ester. To a solution of 2-(4-hydroxyphenyl)-5-methyl-2//-pyrazole-3- carboxylic acid cyclohexylamide (300 mg, LOt) mmol) and triphenylphosphine (291 mg, 1.1 1 mmol) in tetrahydrofuran (5 mL) was added l -ter/-butoxycarbonyl-4-hydrσxypiperidme (221 mg, 1.10 mmol) followed by dropwtse addition of diisopropylazodicarboxylate (216 μL, 1.10 mmol). The solution was stirred at room temperature overnight, concentrated and dissolved in ethyl acetate. The ethyl acetate was washed with water, dried (MgS Oa) and concentrated to give the crude product in quantitative yield. LC-MS (C27HSgN4O4 calculated 482) m/z 4S3 (M+H).
5-Metbyl-2-[4-(piperidin-4-yloxy)phenyl]-2/f-pyrazoIe-3-carboxyIic acid
I cyclohexylamide. To a solution of 4-[4-(5-cyclohexyIcarbamoyl-3-rnethylpyra2;oI-l- yl)phenoxy}piperidine-l -carboxylic acid tert-bwty\ ester (482 mg, 1.00 mmol) in methylene chloride (10 mL) was added trifluoro acetic acid (2 mL). After the reaction was stirred at room temperature for 2 hours, it was quenched with saturated sodium bicarbonate and the aqueous layer was extracted with methylene chloride. The combined organic layers were i dried (MgSO4) and concentrated to give the crude product in quantitative yield. LC-MS (C22H30N4O2 calculated 382) m/z 383 (M+H).
2-[4-(l-CyclopentyIpiperidin-4-yIoxy)phenylj-5-methyI-2//-pyrazole-3-carbϋxylic acid cyclohexylamide. To a solution of 5-methyl-2-[4-(piperidϊn-4-yIoxy)phenyl]-2H-pyrazoie-
► 3-carboxylIc acid cyclohexylamide (60 mg, 0.157 mmol) in methylene chloride (9 mL) was added cyclopentanone (21 μL, 0.24 mmol) and acetic acid (150 μL). After 1 hour at room temperature, sodium triaeetoxyborohydride (51 mg, 0.24 mmol) was added and the reaction was allowed to stir for an additional 4 hours. The reaction was quenched with 10% NaOH and extracted with methylene chloride. The methylene chloride solution was dried (MgSO4) i and concentrated. The- residue was purified by semi-prep LC-MS to give 2.4 mg of the desired product. LC-MS (C27H38N4O2 calculated 450) m/z 451 (M+H). 1 H NMR (300 MHz,
CDCl3) δ 7.34-7.31 (m, 2H), 7.01 -6.98 (m, 21 1), 6.78 (d; J = 8.4 Hz, I H), 6.70 (s, I H), 4.42
(m, IH), 3.95-3.93 (m, IH), 2.87-2.83 (m, 2H), 2.6S-2.54 (m, 3H). 2.23 (s, 3 H), 2 15 - 1 . 14 (m, 22H).
Example 11 2-[4-(l-CyclohexyIpiperidin-4-ytoxy)phenylj-5-methyl-2H-pyraz(jle-3-carbo\> lic acid cyclohexylamide
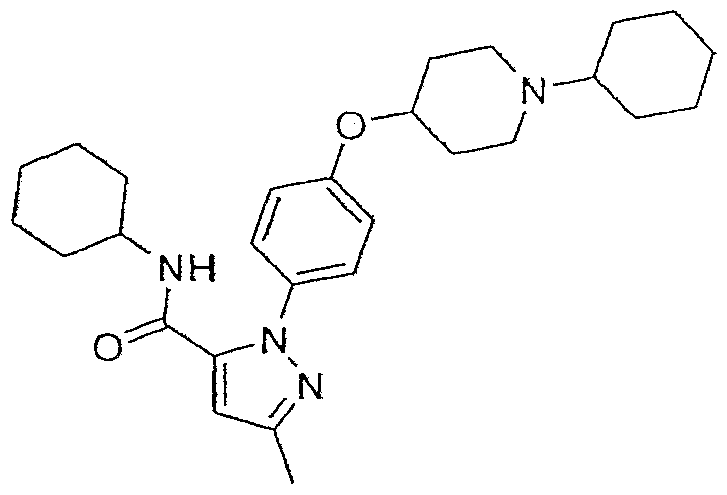
2-[4-(l-CyclohexyIpiperidin-4-yloxy)phenyl]-5-methyl-2//-pyrazole-3-carbo\'\hc acid cyclohexylamide was synthesized by a method analogous to that used fuϊ Px amp Ib 10 LC- MS (C2SH40N4O2 calculated 464) m/z 465 (M+H).
Example 12 2-[4-(l-Isopropylpiperidiπ-4-yIoxy)phenyl]-5-methyI-2H-pyra7.ole-3-earbo\> lie ncϊd cyclohexylamide

2-[4-(l-Isopropylpiperidin-4-yloxy)phenyI]-5-methyl-2H-pyrazole-3-carbox>lic actd cyclohexylamide was synthesized by a method analogous to that used for txample 10 LC- MS (C25H36N4O2 calculated 424) m/z 425 (M+H).
Example 13 2-[4-(l-CyclobutyIpϊperidiπ-4-yloxy)phenyI]-5-methyI-2//-pyra/.oIe-3-carbox> lic acid cyclohexylamide
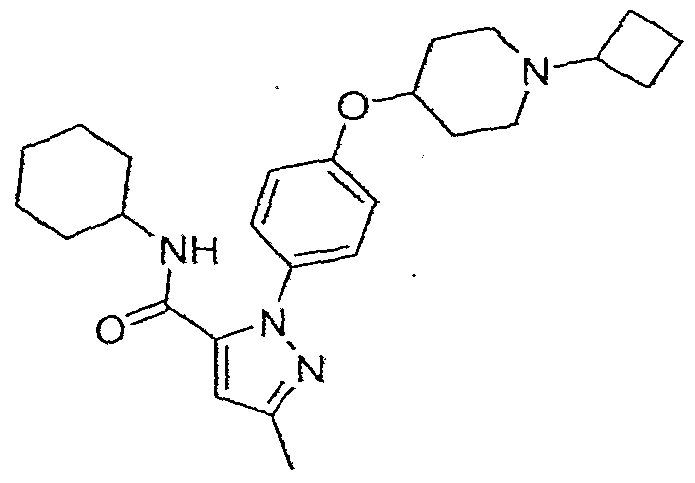
2-[4-(l-CyclobutyIpiρeridin-4-yIoxy)phenyl]-5-methyl-2//-pyrazole-3-carboxylic acid cyclohexyiamide was synthesized by a method analogous to that used for Example 10. LC- MS (C26H36N4O2 calculated 436) mfz 437 (M+H).

Example 14 {5-Methyl-2-|4-(3-pyrroIidin-l-ylpropoxy)phenylJ-2/f-pyrazol-3-yi} methanol

4-(5-JHydroxymethyI-3-methyI-pyrazαt-l-yl)-pheπol. 2-(4-Hydroxyphenyl)-5-methyl-2H- pyrazole-3-carboxylϊc acid ethyl ester (233 mg, 0.946 mmol) was dissolved in tetrahydrofuran (20 mL). Lithium aluminum hydride (1.42 mL, 1 M in THF, 1.42 mmol) was added, and the reaction was stirred overnight. The reaction was quenched with water and extracted with ethyl acetate. The ethyl acetate solution was dried (MgSO4) and concentrated to gϊve the crude product in quantitative yield. LC-MS (CnHi2N2O2 calculated 204) m/z 205 (M+H).
{5-Methyl-2-[4-(3-pyrrolidin-l~yIpropoxy)phenyl]-2jHr-pyrazoI-3-yl}methanoI. To a solution of 4-(5-hydroxymethyl-3-methylpyrazol-l-yl)phenol (326 mg, 1 60 mrπol) in 2- butanone (8 mL) was added potassium carbonate (243 mg, 1.76 mmol) and I-(3- chloropropyl)pyrrolidine (260 mg, 1 76 mmol). The reaction was heated overnight at 80° C. After the reaction, was diluted with water and extracted with methylene chloride, the organic layer was dried (MgSO4) and concentrated to give 315 mg of the desired product. LC-MS (C18H25N3O2 calculated 315) m/z 316 (M+H).
Example 15
5-CyclopentyloxymethyI-3-methyl-l-[4-(3-pyrroIidin-l-yl-propoxy)phenylJ-ljHr-pyrazole

Example 16
5-CycIopentyIoxymethyl-3-πaethyl-l-[4-(3-pyrroHdin-l-ylpropoxy)pheny I]-IH- pyrazole

5-CycIohexyIoxymethyl-3-methyI-l-[4-(3-pyrrolidin-l-ylpropoxy)pheny!]-li7-pyrazoie was synthesized by a method analogous to that used for example 15. LC-MS (C24H35N3O2 calculated 397) m/z 39S (M+H).
Example 17 5-ϊsopropyloxymefhy{-3-methyl-J-[4-(3-pyrroIidrn-l-ylpropoxy)phenyl]-l//-pyrazoFe

5-Cyclohexyloxymethyl-3-methyl-l-[4-(3-pyrrolidin-l-ylpropoxy)phenyl]-l//-pyrazole was synthesized by a method analogous to that used for Example 15. LC-MS
 calculated 357) m/z 358 (M+H).
calculated 357) m/z 358 (M+H).

Example 18 2-[4-(3-PyrroIidm-l-ylpropoxy)phenylJ-2f/-indazole

/Y-(2-BromαbenzyI)-iV-(4-methoxypheπyl)hvdrazine. To a suspension of 2-brornobenzyl bromide (500 mg, 2.00 mmol) and 4-methυxyphenylhydrazine hydrochloride (348 mg, 2.00 mmol) in ΛyV-dirnethylformarnide (8 nil.) was added potassium carbonate (1.38 g, 10.0 mmol). The reaction mixture was heated at L>0° C for 4 hours. The reaction was partitioned between water and methylene chloride. The methylene chloride was dried (MgSCλj), concentrated and purified by SiOi chromatography with 10-50% ethyl acetate/hexanes to give 298 mg of the title compound. LC-MS ( C I4Hi5BrN2O calculated 306) m/z 307 (M+H).
Z-^-Methoxyphenyty-lH-indazole. 2-(4-Methoxyphenyl)-2H:-ϊndazoIe was prepared according to Song and Yee (Org. Lett. 2000, 2, 519). To a solution of iV-(2-bromobenzyl)-iV- (4-methoxyphenyI)hydrazine (298 rng, 0.97 mmol) in toluene (3.5 mL) were added palladium acetate (11 mg, 0.05 mmol), l ,l '-bis(diphenylphosphino)ferrocene (46 mg, 0.075 mmol) and sodium /ert-butoxide (140 mg, 1.46 mmol). The vial was capped and the reaction was stirred at .90° C overnight. After the reaction was allowed to cool, it was filtered through a pad of silica and concentrated to give the desired product. The reaction was assumed to be quantitative. LC-MS (Ci4H12N2O calculated 224) m/z 225 (M4-H).
4-Indazol-2-ylpheπol. To a solution of 2-(4-methoxyphenyl)-2/^-indazoIe. (50 mg, 0.22 mmol) in methylene chloride (3 mL) at -78° C was added boron tribromide (62 μL, 0.66 mmol). The reaction was stirred at -78° C for 1 hour and room temperature for 1 hour. The reaction was quenched with saturated sodium bicarbonate solution. After the aqueous layer was extracted with methylene chloride, the combined organic layers were dried (MgSO4) and concencentrated to give 28.4 mg of the desired indazole. LC-MS (C13H10N2O calculated 210) m/z 21 \ (M+H).
2-[4-(3-PyrroHdiπ-l-ytpropoxy)pheπyI]-2JE/-indazoIe. To a solution of 4-Indazol-2- ylphenol (28 mg, 0.13 mmol) and sodium iodide (6 mg, 0.04 mmol) in N1N- dimethylforrnamide (3 mL) was added sodium hydride (8.0 mg, 60% in mineral oil, 0.2 mmol) followed by l -(3-chloropropyl)pyrrolidine (30 mg, 0.20 mmol). After the reaction was heated at 85° C overnight and allowed to cool, it was partitioned between water and methylene chloride. The organic layer was dried (MgSO4), concentrated and purified by semi-prep LC-MS to give 5.0 mg of the desired indazole. LC-MS (C20H23N3O calc'd 321) m/z 322 (M+H); 1H NMR (300 MHz3 CDCI3) δ 8.33 (s, I H), 7.81-7.77 (m, 3H), 7.71 (d, J = 8.7 Hz, I H), 7.34-7.29 (m, IH), 7.14-7.09 (m, IH)3 7.05-7.02 (m, 2H), 4.10 (t, J = 6.6 Hz, 2H), 2.72 (t, J = 7.2 Hz, 2H), 2.63 (m, 4H), 2.08 (quint, J= 6.3 Hz, 2H), 1.86-1.81 (m, 4H).


l-(4-Benzyloxγphenyl)-4-(4-metboxyphenyl)-3r5-dimethyl-lfl-pyrazole. A solution of 3- (4-methoxyphenyI)pentane-2,4-dione (50 mg, 0.24 mrαol, prepared according to Ghosh et al, Bioorg. Med. Chem. 2003, 11, 629) and (4-benzyloxyphenyl)hydrazine hydrochloride (61 mg, 0.24 mmol) in ethanol (3 mL) was heated at 80° C overnight. The reaction was diluted with water and extracted with ethyl acetate. The organic layer was washed with 1 N ΗCI (2x) and saturated sodium bicarbonate (Ix), dried (MgSO-t), and concentrated to give 57 mg of the crude pyrazole. LC-MS (C25H24N2O2 calculated 384) m/z 385 (M+H).
4-[4-(4-Methoxyphenyl)-3,5-dimethylpyrazol-l-ylJphenoL A solution of l-(4- ben7.yIoxyphenyl)-4-(4-methoxyphenyI)-3}5-dimethyl-l/-r-pyra2θIe (57 mg, 0.15 mmol) in methanol (2 ml ,} and tetrahydrofuran (3 mL) was flushed with nitrogen. A catalytic amount of palladium on carbon (10% wet) was added, and the reaction was again flushed with nitrogen followed by hydrogen. After the reaction was allowed to stir at room temperature for 1 hour, it was filtered through a pad of Celite and concentrated to give 37 mg of the desired phenol. LC-MS (Ct8Hi8N2O2 calculated 294) m/z 295 (M+H).
4-(4-Methoxyphenyl)-3,5-dimethyI-l-[4-(3-pyrroIidin-l-yIpropoxy)phenyI]-lH-pyrazole.
To a solution of 4-[4-(4-methoxyphenyl)-3J5-dimethylpyrazoI-l-yl]phenol (37 mg, 0.13 mmol) and sodium iodide (6 mg, 0.04 mmol) in /V,yV-dimethylformamide (3 mL) was added sodium hydride (8.0 mg, 60% in mineral oil, 0.2 mmol) followed by l -(3- chloropropyl)pyrrolidine (30 mg, 0.20 mmol). After the reaction was heated at 85° C overnight and allowed to cool, it was partitioned between water and methylene chloride. The organic layer was dried (MgSO4), concentrated and purified by semi-prep LC-MS to give 2.4 mg of the desired iαdazole. LC-MS (C25H31N3O2 calculated 405) m/z 406 (M+H); 1H NMR
(300 MHz3 CDCl3) δ 7.37-734 (m, 2H), 7.26-7.22 (m, 2H), 6.99-6.96 (m, 4H), 4.07 (t, J = 6.3 Hz, 2H), 3.85 (s, 3H)3 2.70 (t, J = 7.2 -Hz3 2H-), 2.61 (m, 4H), 2.31 (s, 3H), 2.23 (s, 3H), 2.06 (quint, J= 6.3 Hz3 2H), 1.85-1.80 (m, 4H).

Example 20
I-[4-(3-Pyrrolidin-l-ylpropoxy)phenyI}-liT-indazoIe

4-IndazoI-l-yl-pheπol. l-(4-Methoxyphenyl)-lH-indazole (30 mg, 0. 1 34 mmol) was dissolved in dich!oromethane (0.4 mL) and cooled to -400 C. Boron tribromidc (0.4 mL, 1 M solution in dichloromethane, 0.4 mmol) was added, and the reaction was warmed to rt and stirred for 5 hours. Saturated sodium bicarbonate solution was added, and the mixture was extracted with dichloromethane. The extracts were dried over MgSC>4 and concentrated to give the desired product. LC-MS (CI3HI0N2O calculated 210) m/z 21 1 (M-I H).
l-[4-(3-PyrroHdin-l-ylpropoxy)phenyI]-lJζT-indazole. 4-Indazol-l -yl-phenol (0.07 mmol) was dissolved in N,N'dimethylformamide (0.5 mL), and sodium hydride (3 mg, 60%
dispersion in mineral oil, 0.085 mmol), l-(3-chlσropropyl)pyτrolidine (10 mg, 0.07 mmol) and a catalytic amount of sodium iodide were added. The reaction was heated at 70° C for 2.5 hours. Saturated sodium bicarbonate solution was added, and the mixture was extracted with ethyl acetate. The organic extracts were dried over MgSO4, concentrated, and purified by semi-prep LC-MS to give 3.5 mg of the desired product. LC-MS (C2OH23N3O calculated 321) m/z 322 (M+H).


2-butanσne. 80 0C


Example 21 3,5-Dϊethyl-1-[4-(3-pyrroIidin-l-yIpropoxy)phenyl]-lH-pyrazole
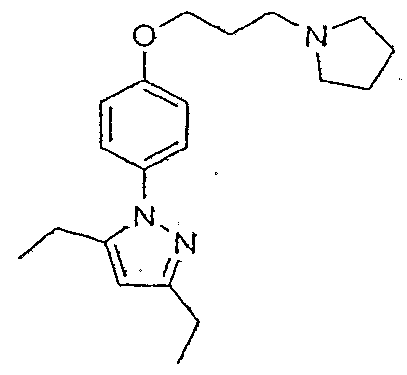
3,5-DiethyI-l-(4-methoxyphenyl)-I.H-pyrazole. A solution of 3,5-heptanedione (1.06 mL, 7.80 mmol) and 4-methoxyphenylhydrazine hydrochloride (1.49 g, 8.58 mmol) in ethanol (25 mL) was stirred at 60° C overnight. After the reaction was concentrated, the residue was partitioned between 1 N HCl and ethyl acetate. The ethyl acetate layer was washed two times with 1 N HCl. The organic layer was dried (MgSO4) and concentrated to give 893 mg of the title compound. LC-MS (Ci4H18N2O calculated 230) m/z 231 (M+H).
4-(3,5-Diethylpyrazol-l-yI)phenoI. To a solution of 3,5-diethyl-l-(4-methoxyphenyl)-l/f~ pyrazoie (893 mg, 3.88 mmol) in methylene chloride (50 mL) at -78° C was added boron
tribromide (1.10 mL, 11.63 ramol). The reaction, was stirred at -78° C for 1 hour and at room temperature -for- an additional 1 hour. The reaction was quenched with saturated sodium bicarbonate. After the aqueous layer was extracted with methylene chloride, the combined organic layers were dried (MgSO4) and concentrated to give the desired product. The reaction was assumed to be quantitative. LC-MS (Ci3HIeN2O calculated 216) m/z 217 (M+H).
3,5-DiethyI-l-[4-(3-pyrroIidin-I-yIpropoxy)phenyI)-liϊr-pyrazole. To a solution of 4-(3,5- diethylpyrazol-l-yl)phenol (50 mg, 0.23 mmol) in 2-butanone (2 mL) was added potassium carbonate (35 mg, 0.25 mmol) and I-(3-chloropropyl)pyrroIidine (37 mg, 0.25 rϊimol). The vial was capped and the reaction was heated overnight at 80° C. After the reaction was diluted with water and extracted with methylene chloride, the organic layer was dried (MgSO4) and concentrated. The residue was purified by semi-prep LC-MS to give 7.0 mg of the desired product. LC-MS (C20H29N3O calculated 327) m/z 328 (M M l); 1H NMR (300 MHz, CDCl3) δ 7.29 (d, J = 9.0 I Iz, 211), 6.94 (d, J = 8.7 Hz, 2H), 6.02 (s, 1 H). 4.05 (t, J = 6.3 Hz, 2H)3 2.73-2.54 (m, I Of T), 2.06 (quint, J = 6.3 Hz, 2H)3 1.83 (s, 411), 1 .28 (t, J = 7.5 Hz, 3H), 1.19 (t, J= 7.5 Hz, 3H).
The following compounds were synthesized according to the procedure for Example 21 :




Acetone
2M HCI (aq)


Examples 69 and 70 l-f^trans^S-S-Diisopropyl-pyrazol-l-y^-cycIohexylJ^-isopropyl-piperazine and l-[4- cis-(3,5-Dϊisopropyl-pyrazoI-l-yl)-cyclohexyI]-4-isopropyI-piperazinc

l ,4-Dioxa-spiro[4.5]decan-8-oL To a methanol (50 mL) solution of 1 ,4-dioxa- spiro[4 5jdecan-8-one (Sg, 32 mraol) at 0°C was added NaBH.-, ( 1.34g, 35.3 ramol) in 4 portions 1 he solution was stirred at 0°C for 15 min followed by stirring at room temperature, tor 1 hour The methanol was evaporated and the residue partitioned between ether and water. The organic layer was separated, dried over Na2SO4 and concentrated to give the desired alcohol as a colorless oil (4.92 g). LC-MS (C8Hi4O3 calculated 158) m/z 159 (M+H); 1H NMR (300 MHz3 CDCl3) 53.90 (s, 4 H), 3.77 (m, 1 H), 1.82 (m, 4 H), 1 .55 (m, 4 H).
ToIuene-4-sulfonfc acid l,4-dioxa-spiro[4.5]dec-8-yl ester. The alcohol prepared previously. 1 ,4-dioxa-spiro[4.5]decan-8-ol (4.92g, 31. 1 mmol) was dissolved in pyridine (15 mL) at 0 C followed by addition of p-toluenesulfonyl chloride (6.1 g, 32.1 rnmol). The mixture was stirred at 0 C for 2 hours and allowed to warm to room temperature overnight. The reaction was diluted with water (15 mL) and stirred for 30 minutes. The reaction was filtered and the precipitate washed with water and recrystallizcd from hexanes to give the tobvlate as an off white solid (yield 7.3Ig). LC-MS (C15H20O5S calculated 312) m/z 313 (M+H ).
l-(l,4-Dioxa-spiro[4.5]dec-8-yl)-3,5-diisopropyI-l/f-pyrazoIe. Sodium hydride (66 mg, 60% suspension in oil) was added to a solution of 2.5-diisopropylpyrazole (0.25g, 1 64 mmol) in dry DMF (5ml). The solution was stirred at room temperature until hydrogen evolution stopped and the solution cooled on ice for 5 minutes. To the reaction was added a solution of totuene-4-sulfonϊc acid l ,4-dioxa-spiro[4.5]dec-8-yI ester (0.467g, 1.5 mmol) in DMF ( 1 mL) and stirred at 0 C for 5 minutes. The reaction was then heated to 60 C overnight. 1 he reaction was cooled, quenched with water and partitioned between ethyl
acetate and water. The organic layer was separated, washed with water, dried over Na2SO4 and purified on silica gel (2:1 Hexane.ΕtOAc) to give the desired product (yield 4-lmg). LC- MS (Ci7H28N2O2 calculated 292) m/z 293 (M+H).
4-(3,5-Dϊisoprσpyl-pyrazoI-l-yl)-cycIohexanone. To a solution of l-(l,4-dioxa- spiro[4.5]dec-8-yI)-3,5-diisopropyl~l ff-pyrazole (40 mg) in acetone (20 ml) was added HCl (20 mL, 2M aq). The reaction was heated to reflux overnight. The reaction was cooled, concentrated and extracted with EtOAc. The organic fractions were washed with water, saturated NaHCO3 and dried over Na2SOa. The solution was concentrated and used without further purification (Yield 30.6 mg). LC-MS (C15H24N2O calculated 248) m/z 249 (M+H).
l-[4-trans-(3,5-DiisopropyI-pyrazol-l-yi)-«ϊycIohexyl]-4-isopropyI-piperazϊne and l-[4- cis-(3,5-DiϊsopropyI-pyrazoI-l-yl)-cyct&hexyI]-4-isopropyϊ-pϊpera7.ine. To a solution of 4-
(3,5-diisopropyl-pyrazol-l-yl)-cycIohexanone (30 mg, 0.121 rπmol) , and 1- isopropylptperazine (23.3 mg, 26 uL) in dtchloroethane (2 mL) was added acetic acid (100 uL). The solution was stirred at room temperature for 2 hours followed by addition of NaBH(OAc)3. The reaction was concentrated under reduced pressure and the products separated by reverse phase HPLC to give l-[4-cis-(3,5-D)isopropyl-pyrazol-l-yl)- cycIohexyl]-4-isopropyl-pϊperazme (yield 13.2 mg) and l-[4-trans-(3,5-Diisopropyl-pyrazol- l-yl)-cycIohexyl]-4-isopropyl-piperazine (yield 6.1 mg). LC-MS (C20H40N4 calculated 360) m/z 361 (M→ H).
Examples 71 and 72
SjS-Diisopropyl-l-ltrans^^-pyrrolidin-l-yl-propoxy^cycIohexyH-lH-pyrazoIe and 3,5- Diisopropyl-l-[cis-4-(3-pyrrolidin-l-yl-propoxy)-cycIohexyIJ-lH-pyrazole

3,5-Diϊsopropyl-l-[cis-4-(3-pyrroHditt-l-yl-propoxy)-cyclohexyl}-l i/-pyrazole. Cis-4- (3,5-diisopropyl-pyrazol-l-yl)-cyclohexanol (17.5 mg, 0.068 mmul) was dissolved in dry DMF (2 mL) followed by addition of NaH (excess, 60% in oil) and stirred at room temperature until hydrogen evolution stopped. To the reaction was added l -(3-Chloro- propyl)-pyrrolidine (20 mg) and catalytic NaI followed by heating to 80° C overnight. The reaction was cooled, quenched with methanol and concentrated under reduced pressure. The residue was purified by prep HPLC to give the desired product (yield 10.6 mg). LC-MS (C22II39N3O calculated 361) m/z 362 (M+H).
Trans-4-(3,5-ditsopropyI-pyrazol-l-yl)-cyclohexanol was prepared by the same method starting with trans-4-(3,5-dϊisopropyl-pyrazol-l-yl)-cycIohexanol.

Example 73
5-Methyl-2-f4-(3-pyrroHdin-l-yI-propoxy)-phenyl}-2H-py rii7θle-3-carboxylϊc acid cyclohexylamide
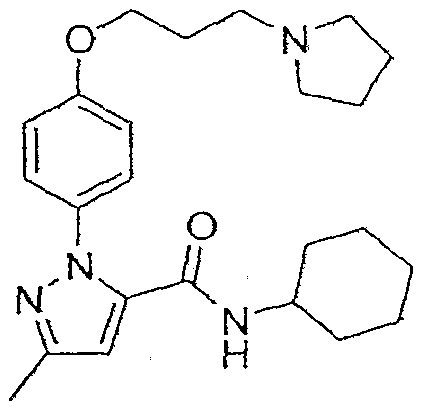
2-(4-Methoxy-phenyl)-5-methyl-2H-pyrazole-3-carboxyfic acid trth> I ester Cthyi-2S4- dioxovalerate (0.555g, 3.5 mmol) and 4-methoxyphenylhydrazine hydrochloride (0.67g, 3.8mmol) were mixed in ethanoi and heated to 800C overnight. The rcactiutι was cooled, diluted with water and extracted with EtOAc. Organic layers were combined and washed with water, 10% HCl, sat. NaHCO3, dried over MgSO4 and concentrated under reduced
pressure. The residue was purified on silica gel (20% EtOAc in hexanes to 30% EtOAc in hexanes). Two regioisomers were isolated, -the title regioisomer being the more polar (yield 492 mg). LC-MS (Ci4H16N5O. calculated 260) m/z 261 (M+H).
2-(4-Methoxy-phenyl)-5-methyl-211-pyrazole-3-carboxylϊc acid. To a solution of 2-(4- Methoxy-pheπyI)-5-methyl-2H-pyra7.ole-3-carboxylic acid ethyl ester (400 mg) in a 1 :1 : 1 mixture of MeOH:THF:water was added 10% NaOH (20 mL). The mixture was heated to reflux and the reaction monitored by TLC and HPLC until hydrolysis was complete. The solution was cooled and concentrated under reduced pressure. The residue was acidified with 10% HCl and extracted with RtOAc. The organic fractions were combined and dried over MgSO4 and concentrated under reduced pressure. The resulting solid was used, without further purification (Yield 242 uiμ). LC-MS (C12Hj2N2O3 calculated 232) m/z 231 (M-H).
2-(4-Methoxy-phenyl)-5-methvl-2H-pyrazoIe-3-carboxyIic acid cyclohexyl amide. The acid prepared above, 2-(4-Methoxy-phenyl)-5-methyl-2H-pyrazole-3-carboxylic acid (234 mg, 1 mmol) was suspended in DCM (8 mL). To this suspension was added oxalyl chloride (1 15 uL, 166 mg, 1.3 mmol) FuI lowed by one drop of DMSO. The reaction was stirred at room temperature for I hour. The reaction was concentrated to dryness, diluted with DCM (20 mL) and concentrated under reduced pressure a second time to ensure removal of any excess oxalyl chloride, ['he intermediate acid chloride was dissolved in DCM (15 mL) followed by drop wise addition of" cyclohexylamiπe (0.22g, 2.2 mmol) at room temperature, for 1 hour. The reaction was diluted with DCM and extracted with water, 10% NaOH, 10% HCl and dried over Na2SO1U The solvent was removed and the product used without further purification (Yield 0.167g). LC-MS (C18H23N4O2 calculated 313) m/z 3 14 (M+H).
2-(4-Methoxy-phenyl)-5-methyl-2H-pyrazole-3-carboxylic acid cyctohexyl amide was converted into the final product by the method described in Example 21 ..
The following compounds were synthesized according to the procedure for Example 73:

Representative compounds of the present invention that were prepared by the procedures of Examples 1-83 were evaluated in binding assays against cells expressing human H3 receptor by the following procedure.
Cell culture Materials
[125I]iodoproxyfan (2000 Ci/mmol) was obtained from Amershara Bioscience (Piscataway, NJ). All other chemicals were either from- Sigma-Aldrich (St. Louis, MO) or Tocris Cookson Inc. (EUisville, MO)
RAGE methodology
The human histamine H i receptor was stably expressed in HTlOSO cells containing the chimeric G-protein, GqQo ( Coward et ah, Anal Biochem 1999; 270:242-8). HT1080- Gqαi5 cells were grown in alpha-moditkd MBM containing 10% fetal bovine serum and 7 μg/ml blasticidin at 37° C m 5% Cϋ2'^S% atmosphere. Cells (4.8xlO9) were irradiated with 50 rads from a 137Cs source and the nFG8-HH3 RAGE (Random Activation of Gene Expression; see Harrington et a! . \ attire Biotechnology. 2001; 19:440-45) vector was subsequently integrated into the cells v ia electroporation (250V, 600μF, 50Ω). The RAGE vector pFG8-HH3 contained cDNΛ strqueoce coding for the first exon (83 amino acids) of human H3 receptor. After electropoπuioα, cells were plated in T75 flasks and grown in alpha-modified MEM. The culture medium was replaced 48 hrs after electroporation with alpha-modified MEM3 10% fetal ho vine serum, 500 μg/ml hygromycin B and 3 μg/ml puromycin. Medium was replaced everv tour days during cell expansion. To identify RAGE activated cells expressing the H3 receptor, pools of approximately 10,000 colonies (5x 107- 1.5x108 cells total) were screened by PCR for the desired gene product (using primers specific to the RAGE vector and exon 2 of the H3 receptor). Pools that were found to contain the appropriate transcript, as confirmed by sequencing, were subcloned into pools of 100 cells/well. Positive 100 cells/well pools were identified by PCR, confirmed by sequencing, and subsequently subcloned to 0.8 cells/well. Once clones expressing the H3 receptor were
identified by PCR analysis, assays (FLIPR or radioligand binding) were performed to confirm that the activated gene produced functional protein. The protein expression in the initial clones obtained from the RAGE library was increased by growth in the presence υi" methotrexate. Since the integrated RAGE vector contains the DHFR gene, such treatment selects for cells that have amplified the genetic locus containing the RAGE insert. Subclones obtained after methotrexate amplification were tested for functional activity in FLiPR assays to identify the clone that was most suitable for HTS. The final HT1080-Gqαi5 RAGF clone (RAGE-H3) expressing the human histamine H3 receptor was grown in alpha-mudϊ fied MEM containing 10% fetal bovine serum, 3 μg/ml puromycin, 500 μg/mt hygromycin B, 1.2 μM methotrexate at 37° C in 5% CO2/95% atmosphere.
Membrane preparation
RAGE-H3 cells (IO9) were washed twice with cold PBS, scraped off the plates, and centrifuged at 1000 x g for 5 minutes. Cells were resuspended in ice-cold 10 niM Tm HCl, pH 7.4, containing 5 raM EDTA and protease inhibitor cocktail tablets (Roche Molecular Biochemϊcals). After incubating on ice for 10 min, the cells were homogenized with a dounce homogenizer or a polytron tissue grinder, and centrifuged at 1000 x g for 10 min at 4° C. T he resulting supernatant was centrifuged at 32, 000 x g for 30 min at 4° C. The membrane pellets were resuspended in 50 mM Tris HCI, pH 7.4, and stored at — 80° C until use. Protein concentration was determined by the Bradford method (Bio-Rad Laboratories, CA)-
Radioligand binding assays
Binding assays were carried out in 96- well polypropylene plates in 50 mM Tris HCL pH 7.4, containing 1 mM EDTA. Reaction mixtures contained 100 μl of membrane
> suspension, 50 μl of 4% DMSO, and 50 μl of increasing amounts of [I25I]iodoproxyfan (final concentration 0.0005-1.8 nM for human H3 receptor saturation binding assay). Nonspecific binding was defined by adding 10 μM clobenpropit to the reaction mixtures. Competition binding assays were performed in a reaction mixture containing 100 μl of membrane suspension (~ 20 μg of protein/well), 50 μl of [l25I]iodoproxyfan (final concentration of ~
► 0.15 nM) and 50 μl of test compound. Compounds were dissolved in DMSO and then diluted with 4% DMSO; the final maximal DMSO concentration in the binding assays was 1%. Incubations were performed for 1.5 hrs at room temperature and reactions were terminated by rapid filtration over glass Fiber GF/C filters (Perkin Elmer, MA) using a B rand el cell harvester. The filters were presoaked in Q.3% polyethyleπeϊmine for 30 minutes and were i washed with 500 ml of ice-cold 50 mM Tπs I ICl, pH 7.4. The filters were dried, impregnated with Meltilex wax scintillate (Perkin Elmer, MA) and counted with a Betaplate scintillation counter (Perkin Elmer, MA).
Data analysis i All data were analyzed by nonlinear least squares curve fitting using Prism 4.0 software. The KD and Bmax for [l2itjiodoproxyfan were derived from the equation RL=RtL /(KQ +L), where RL is concentration of receptor-bound lϊgand at equilibrium, L is the free ligand concentration, and Rt is the total receptor concentration (i.e., Bmax)- For competition binding experiments, ICso values (the concentration of compound producing 50% inhibition of specific binding) and Hill Coefficients (nf/) were derived from fitting the data to a 4- parameter logistic equation. Apparent K1 values were calculated using the Cheπg-Prussof equation of K1- = ICSQ/(1 +(L/KD)), where I. is the ligand concentration.
Selected Examples

53

54

55




Claims
1. A compound of the formulae:

1 2 3
where
X is O or NR7;
y is O, 1 or 2;
n is 0 or 1 ;
q is 0, I , or 2;
Ri and R2 are independently selected from the group consisting of (C[-C5)alkyl and (C3-
C6>cycloalkyl;
or
where X is O, Ri and R2 taken together with the nitrogen to which they are attached form a 5- 7 member heterocyclic ring system with 0 or 1 additional hetero atoms selected from O3 S, and NR^, wherein the resulting ring may optionally be substituted with 1-3 (C1-Cs)alkyl or (C3-C6)cycloalkyl groups; R3 Is 0-2 of groups selected from halogen, (Ci-C8)alkyl, (Ci-Cs)alkoxy, (C3-C7)cycloalkyl, (C3-C7)eycIoaIkyl-(Cι-C(i)aIkyI, heterocycloalkyl containing 1-3 hetero atoms selected from O, S, and (Ct-C5)aIkyL-O-(Ci-C5)alkyl;
R4 and R6 are independently selected from (Ci-Cg)alkyl, (C|-C8)aikoxy, (C3-C7)cycioalkyi, (C3-C7)cycloalkyl-(Ct-C6)alkyl, heterocycloalkyl containing 1-3 hetero atoms selected from O, S, N, (Ci-C5)aLkyl-O-(Gi-C5)alkyl, amide, (C1-C3)alkyI-aryl, and CF3;
R5 is selected from the group consisting of hydrogen, (Ci-Cg)alkyl, aryl, (Ci-Cs)alkyl-O-(Ci- C5)alkyl, and (C1-C5)alkyl-aryl;
or
R 5 and K4 and the atoms to which they are attached form a fused 5-6 member saturated carbocyclic ring or a fused 10 member bi-cyclϊc ring system;
or
R5 and R6 and the atoms to which they are attached form a fused 5-6 member saturated carbocyclic ring or a fused 10 member bi-cyclic ring system;
or
Rs and R4 and the atoms to which they are attached form a fused 5-6 member saturated carbocyclic ring to which a 6 member aromatic ring Is fused;
or
R5 and R6 and the atoms to which they are attached form a fused 5-6 member saturated carbocyclic ring to which a 6 member aromatic ring is fused;
or Rs and R6 and the atoms to which they are attached form a fused benzothiophene or fused benzofuran ring system;.
where X is NR7, R7 and R2 taken together arc -(CH2CH2)- to form a two nitrogen containing ring where y is 0 or I , and wherein Ri is as defined previously, and the pharmaceutically acceptable salts thereof.
2. A compound as in claim 1 where X is O, y is 1, and q is 1.
3. A compound of claim I selected from the group consisting of" 3-MethyI-l-[4-(3-pyrτolidin-l-ylpropo>cy)phenyl]-4,5-dihydro-l/f-benzo[g]indazole; 3-Methyl-l-{4-[3-(27?-methylpyrrolidin-l-yl)propoxy]phenyl}-4,5-dihydro-l/:f- benzo[g]indazole;
3-MethyI-I -[4-(3-moφholin-l-ylpropoxy)phenyl]-4,5-dihydro-l//-benzo[g]indazole: l-[4-(3-PyrroIidin-l-ylpropoxy)phenyi]-5-styryl-3-trifluoromethyl-l H-pyrazole; 3-Methyl-l-[4-(3-pyrrolidin-l-ylpropoxy)phenyl]-4,5,5aJ6,7,S,9,9a-octahydro-lH- benzo [gjm' dazole;
3-Methyl-2-[4-(3-pyrrolidin-l-ylpropoxy)phenyl]-4;,5,5a,6:,73839,9a-octahydro-2H- benzo[g)indazole,
8-Methoxy-3-methyl-l-[4-(3-pyrrolidin-l-ylpropoxy)phenyl]-4,5-dihydro-l//- benzo[g]indazole;
7-Methoxy-3-methyl-l-[4-(3-pyrrolidin-I-ylpropoxy)phenyl]-4,5-dihydro- l/f- benzo[g]indazote,
6-Methoxy-3-methyl-l-[4-(3-pyrrolidin-l-ylpropoxy)phenyl]-435-dihydro-I//- benzo[g]indazole;
2-[4-(l-CycIopentyl-piperidin-4-yloxy)phenyl]-5-methyl-2H-pyrazole-3-carboxylic acid cyclohexylamide;
2-[4-(l-Cyclohexylpiperidin-4-yloxy)phenyl)-5-methyl-2i/-pyrazole-3-carboxylic acid cyclohexylamide;
2-[4-(I-Isopropylpiperidin-4-yioxy)phenyl}-5-methyl-2icf-pyrazole-3-carboxylic acid cyclohexylamide;
2-[4-(l-Cyclobutylpiperidin-4-yloxy)phenyl]-5-methyl-2H'-pyrazole-3-carboxylic acid cyclohexylamide; {5-Methyl-2-[4-(3-pyrroIidin-l-ylpropoxy)phenyI]-2i/-pyrazϋl-3-yl}methanol; 5-CyclopentyloxymethyI-3-methyl-l-[4-(3-pyrrolidin-l -yl-propoxy)phenyl]-liϊ"-pyjazole; 5-Cyclopentyloxymethyl-3-methyl-l-[4-(3-pyrrolιdin-l-yIpropQxy)phenyl]-li:/-pyrazole; 5-IsopropyIoxymethyl-3-methyl-l-[4-(3-pyrrolidin-l -yIpropoxy)phenyI]-lH-pyrazole; -[4-(3-Pyrrolidm- 1 -ylpropoxy)phenyl]-2H-indazole; -(4-Methoxyphenyl)-3,5-dimethyl-l-[4-(3-pyrrolidin- l-yIpropo Ky)PlIeHyI]-Ii1J-PyTaZoIe; l-[4-(3-PyrroItdin--l -ylpropoxy)ρhenyl]-lH-indazole; 3,5-Dϊethyl-l -[4-(3-pyrrolϊdin-l-ylpropoxy)phenyl}-lH-pyrazυle; 3,5-Diethyl-l -[4-(3-piperidin-l-ylpropoxy)phenyl]-l H-pyrazole; ,5-DiethyI-l-[4-(3-mθφhoiin-l-ylpropoxy)phenyl]-l//-pyrazole; 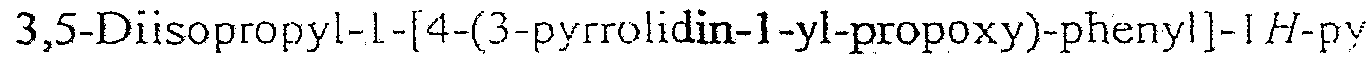 razole; 3,5-DϊisopropyI- 1 -|4-(3-pφeridin-l-yI-propoxy)-pheπyIl-l H-pyrazole; 3-tert-Butyl-5-methyI - 1 -[4-(3-pyrrolidin-l-ylpropoxy)-pheαy 11- 1 H-pyrazole; 3-ϊer/-Butyl-5-methyl- 1 -[4-(3-piperidm-l-ylpropoxy)-phenyl]- 1 //-pyrazole; 5-IsobutyI-3-methyl- 1 -[4-(_3-pyrroIidin-l-ylpropoxy)-phenylJ-l //-pyrazole; 5-Isobutyl-3-methyl- I-[4-(3-pϊperidin-l-yIpropoxy)-phenylJ- 1 H- pyrazole; 5-Isobutyl-3-methyI-I -f4-(3-ptperidin-l-ylpropoxy)-phenyl)-2H-pyrazoIe; 5-Isobuty 1-3 -methy I - 1 -[4 - (3 -pyrτolidin-1 -ylpropoxy)-pheny 1] -2 H-pyrazole; l-Cyclobutyl-4-[4-(3,5-dϊisopropylpyrazol-l-yl)phenoxy]piperidiαe; 5-;ert-ButyI-3-methy[-I-[4-(3-pyrrolidin-l-ylpropoxy)-phenyIj- 1 H-pyrazole; 5-/er/-ButyI-3-methyl- 1 -f4-(3-piperidin-l-yLpropoxy)-phenyl]- 1 H-pyrazole; 3,5-Dimethyl-l-[4-(3-pyrrolidin-l-ylpropoxy)phenyIJ-l H-pyrazole; 3,4,5-Trimethyl- l -[4-(':i-pyrrolidin.-l-yIpropoxy)phenyIJ-l //-pyrazole; 4-Ethyl-3,5~dimethyl- l -[4-(3-pyrrolidin-l-ylpropoxy)phenyl |- l //-pyrazole; 4-Butyl-3,5-dimethyl-l-[4-(3-pyrroIidin-l-ylpropoxy)phcnyl]- l /"/-pyrazole; 4-Phenyl-3,5-dimethyI- 1 -[4-(3-pyirolidin-l-ylpropoxy)phenyI |- 1 H-pyrazole; 5-MethyI-3-phenyI-l -[4-(3-pyrrolidin-l-ylpropoxy)phenyI]-lH pyrazole; 5-Methyl-3-phenyl-l -[4-(3-pyrrolidin-l-ylpropoxy)phenyl]-2H-pyrazole; 3-/err-Butyl-5-phenyl-l -f4-(3-pyrrolidin-l-ylpropoxy)phenyl]- 1 H-pyrazole; 3-PhenyI-l-f4-{3-pyrrolidin-l-ylpropoxy)phenyI]-4,5,6,7-telrahydro-li:ir-indazole; 3-PhenyI-l-[4-(3-pyrrolidin-l -ylpropoxy)phenyl]-435J6,7-tetrahydro-2ϋ-r-inda2θle; 5-Furan-2-yl-3-methyl-1 -[4-(3-pyrrolidin-l-yIpropoxy)phenyIj- l /f-pyrazole; 3-Difluoromethyl-5-furan-2-yl-l-[4-(3-pyrroIidin-l-yIpropoxy)phenyl]-l H-pyrazole; 3-Trifluoromethyl-5-furan-2-yl-l-[4-(3-pyτroΗdin-l-ylpropoxy)phenyl]-lH-pyrazole; 3-TrijQuoromet.hyl-5-thiophen-2-yl-l-[4-(3-pyrrolidin-l-ylpropoxy)phenyI]-l//-pyrazole;
razole; 3,5-DϊisopropyI- 1 -|4-(3-pφeridin-l-yI-propoxy)-pheπyIl-l H-pyrazole; 3-tert-Butyl-5-methyI - 1 -[4-(3-pyrrolidin-l-ylpropoxy)-pheαy 11- 1 H-pyrazole; 3-ϊer/-Butyl-5-methyl- 1 -[4-(3-piperidm-l-ylpropoxy)-phenyl]- 1 //-pyrazole; 5-IsobutyI-3-methyl- 1 -[4-(_3-pyrroIidin-l-ylpropoxy)-phenylJ-l //-pyrazole; 5-Isobutyl-3-methyl- I-[4-(3-pϊperidin-l-yIpropoxy)-phenylJ- 1 H- pyrazole; 5-Isobutyl-3-methyI-I -f4-(3-ptperidin-l-ylpropoxy)-phenyl)-2H-pyrazoIe; 5-Isobuty 1-3 -methy I - 1 -[4 - (3 -pyrτolidin-1 -ylpropoxy)-pheny 1] -2 H-pyrazole; l-Cyclobutyl-4-[4-(3,5-dϊisopropylpyrazol-l-yl)phenoxy]piperidiαe; 5-;ert-ButyI-3-methy[-I-[4-(3-pyrrolidin-l-ylpropoxy)-phenyIj- 1 H-pyrazole; 5-/er/-ButyI-3-methyl- 1 -f4-(3-piperidin-l-yLpropoxy)-phenyl]- 1 H-pyrazole; 3,5-Dimethyl-l-[4-(3-pyrrolidin-l-ylpropoxy)phenyIJ-l H-pyrazole; 3,4,5-Trimethyl- l -[4-(':i-pyrrolidin.-l-yIpropoxy)phenyIJ-l //-pyrazole; 4-Ethyl-3,5~dimethyl- l -[4-(3-pyrrolidin-l-ylpropoxy)phenyl |- l //-pyrazole; 4-Butyl-3,5-dimethyl-l-[4-(3-pyrroIidin-l-ylpropoxy)phcnyl]- l /"/-pyrazole; 4-Phenyl-3,5-dimethyI- 1 -[4-(3-pyirolidin-l-ylpropoxy)phenyI |- 1 H-pyrazole; 5-MethyI-3-phenyI-l -[4-(3-pyrrolidin-l-ylpropoxy)phenyI]-lH pyrazole; 5-Methyl-3-phenyl-l -[4-(3-pyrrolidin-l-ylpropoxy)phenyl]-2H-pyrazole; 3-/err-Butyl-5-phenyl-l -f4-(3-pyrrolidin-l-ylpropoxy)phenyl]- 1 H-pyrazole; 3-PhenyI-l-f4-{3-pyrrolidin-l-ylpropoxy)phenyI]-4,5,6,7-telrahydro-li:ir-indazole; 3-PhenyI-l-[4-(3-pyrrolidin-l -ylpropoxy)phenyl]-435J6,7-tetrahydro-2ϋ-r-inda2θle; 5-Furan-2-yl-3-methyl-1 -[4-(3-pyrrolidin-l-yIpropoxy)phenyIj- l /f-pyrazole; 3-Difluoromethyl-5-furan-2-yl-l-[4-(3-pyrroIidin-l-yIpropoxy)phenyl]-l H-pyrazole; 3-Trifluoromethyl-5-furan-2-yl-l-[4-(3-pyτroΗdin-l-ylpropoxy)phenyl]-lH-pyrazole; 3-TrijQuoromet.hyl-5-thiophen-2-yl-l-[4-(3-pyrrolidin-l-ylpropoxy)phenyI]-l//-pyrazole;
3-Difluoromethyl-5-phenyl-l-[4-(3-pyrrolidin-l-yIpropoxy)phenyI3-l//-pyrazole;
5-Phenyl- t -f4-(3-pyrrolidin-l-yl-propoxy)-phenyl}-3-trifluoromethyl-l/^-pyrazole; l-{4-[3-(2-(R)-Methyl-pyrroIidin-l-yl)-propoxy]-pheαyl}-5-phenyl-3-trifluorornethyI-l /f- pyrazole;
Dimcthyl-(1 -{3-[4-(5-phenyl-3-trifluoromethyI-pyτazol-l-yl)-phenoxy]-propyI}-pyrrolidin-3- yl)-amine;
4-{3-[4-(5-Phenyl-3-trifluoromethyl-pyrazol-l-yl)-phenoxy]-propyl}-morpholiπe; l-{3-[4-(5-Phenyt-3-trifluoromethyl-pyrazol-l-yl)-phen.oxy]-propyl}-piperidine:
3 -Methyl -1 -[4-(3 -pyrrolidin-l-yl-propoxy)-phenyll-4,5,6,7-tetrahydro-l H-indazolc;
3-Methy]-2-[4-(3-pyrrolidin-l-yl-propoxy)-phenyl]-4,5,6,7-tetrahydro-2H-indazole;
S-Methyl-l-^-fj-pyrroIidin-l-yl-propoxy^phenylj-l ^.S.β-tetrahydro-cyclopentapyrazole;
3-Methyl-2-[4-(3-pyrroIidin-l-yl-propoxy}-phenyl]-2,4,5,6-tetrahydro-cyclopentapyrazole;
3-Methyi-l -[4-(3-piperidϊn-l-yl-propoxy)-phenyIJ- 1 ,4,5,6-tetrahydro-cyclopentapyrazole;
3-Methyl-2- [4-( ">-piperidin-l -yl-propoxy)-phenyl]-2,4,5,6-tetrahydro-cycIopentapyrazole;
3,5-DiisopropyI- 1 -[2-methyI-4-(3-pyrrolidin-l-ylpropoxy)phenyI]-l H-pyrazole;
3,5-Diisopropyl- 1 -[2-methyl-4-(3-piperidin-l -ylpropoxy)phenyl]-l/f-pyτazole;
5-Benzofuran-2-yI-l-[4-(3-pyrτoIidin-l-ylpropoxy)phenyl]-3-trifluoromethyl-l /-/-pyrazole;
3-Methyl-l -[4-(3 -pyrroIidin-l-yl-propoxy)-phenyl]-l H-benzo[4,5]thieno[3,2-c]pyrazole;
3-Methyl-l -{4-[3-(2-raethyl-pyrrolidin-I-yI)-propoxy]-phenyl}-lH-benzo[4,5Jthieno[3,2- cjpyrazole; ;.
3-[4-(3-Pyrrolidirι-l-yI-propoxy)-phenyI]-l-trifIuororaethyl-3H-8-oxa-2J3-diaza- cyclopenta[ajiπclcne;
3-{4-[3-(2-Merhyt-pyτrolldin-l -yI)-propoxy]-phenyI}-l-trifluoromethyl-3H-8-oxa-2)3-diaza- cyclopenta[a}indene;
Dimethyl-( 1 - {3 [4-(l-trifluoromethyl-8-oxa-2,3-diaza-cyclopenta[a]indcn-3-yI)-phenoxy]- propyl}-pyrrolidin-3-yl)-amine;
1 - [4 - trans-( 3 ,5-Di isopropyl-pyrazo 1- 1 -yl)-cy clohexyl] -4-ϊsopropy 1-piperazi ne ;
1 -[4-cis-(3 ,5 -Dii sopropyl-pyrazol-1 -yl)rcycIohexyI]-4-isopropyl-piperazine;
3,5-DiisoρropyI-l -[trans-4-(3-pyrτolidin-l-yl-propoxy)-cyclohexyl]-l/f-pyrazoIe;
3,5-Diisoρropy [- \ -[cis-4-(3-pyrrolidin-l ^1-PrOpOXy)-CyCIoIIeXyI]-IH-PyFaZoIe;
5-Methyl-2-[4-(3-pyrrolidin-l-yl-propoxy)-phenyl]-2H-pyrazole-3-carboxylic acid cyclohexylamide 5-lyfethyl-2-{4-[3-(2i?-methylpyτrolidiQ-l-yl)propoxy]phenyl}-2H-pyra2θIe-3-carboxylic acid cyclohexylamide;
5-Methyl-2-[4-(3-piperidin-l-ylpropoxy)phenyl]-2H-pyrazole-3-carboxyIic acid eye lohexy lamide;
5-MTethyl-2-[4-(3-mθφholin-4-yIpropoxy)phenyl]-2H-pyra2ole-3-carboxylic acid cyclohexylamide;
5-Methyl-2-{4-[2-(l-methylp)ττolidϊn-2-yl)ethoxy]phenyl}-2H'-pyrazole-3-carboxyric acid cyclohexylamide;
{5-Methy]-2-[4-(3-pyrroIidϊn- 1 -y lpropoxy)phenylJ-2H-pyrazoI-3-yl }pyrrolidin- 1 - ylmethanoπe;
5-Methyl-2-[4-(3-pyrroiidin- 1 -ylpropoxy)phenyl]-2H-pyrazole-3-carboxy lie acid cyclohexylmethylamide;
5-Methyl-2-[4-(3-pyrroIidin- 1 -yIpropoxy)phenyl]-2H-pyrazαIe-3-carboxyltc acid cyclobutylamide;
5-Methyl-2-[4-(3-pyrro[idin-I-yIpropoxy}phenyl]-2H;-pyτazole-3-carboxylic acid phenylamide;
5-Methyl-2-[4-(octahydroquinolizin-l-ylinethoxy)phenyl]-2//-pyτazoIe-3-caiboxylic acid cyciohexylamide; and
5-Methyl-l-[4-(3-pyiToIidin-l-ylpropoxy)pIienyl]-lH-pyrazole-3-carboxylic acid cyclohexylamide.
4. A pharmaceutical composition comprising at least one compound of any one of claims 1-3 in combination with a pharmaceutically acceptable carrier.
5. A method of treating a condition in a patient in which modulation of histamine Η3 receptors is of therapeutic importance comprising administering an effective amount of at least one compound of any one of claims.1-3 to a patient in need of such treatment
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002641441A CA2641441A1 (en) | 2006-02-09 | 2007-01-30 | Pyrazoles for the treatment of obesity and other cns disorders |
| AT07749543T ATE525072T1 (en) | 2006-02-09 | 2007-01-30 | PYRAZOLES FOR THE TREATMENT OF OBESITY AND OTHER CNS DISEASES |
| JP2008554271A JP5207983B2 (en) | 2006-02-09 | 2007-01-30 | Pyrazoles for treating obesity and other CNS disorders |
| EP07749543A EP1988777B1 (en) | 2006-02-09 | 2007-01-30 | Pyrazoles for the treatment of obesity and other cns disorders |
| DK07749543.0T DK1988777T3 (en) | 2006-02-09 | 2007-01-30 | Pyrazoles for the treatment of obesity and other cns disorders |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US77176806P | 2006-02-09 | 2006-02-09 | |
| US60/771,768 | 2006-02-09 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007094962A2 true WO2007094962A2 (en) | 2007-08-23 |
| WO2007094962A3 WO2007094962A3 (en) | 2008-01-31 |
Family
ID=38371978
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2007/002547 WO2007094962A2 (en) | 2006-02-09 | 2007-01-30 | Pyrazoles for the treatment of obesity and other cns disorders |
Country Status (9)
| Country | Link |
|---|---|
| US (2) | US7897634B2 (en) |
| EP (1) | EP1988777B1 (en) |
| JP (1) | JP5207983B2 (en) |
| AR (1) | AR059428A1 (en) |
| AT (1) | ATE525072T1 (en) |
| CA (1) | CA2641441A1 (en) |
| DK (1) | DK1988777T3 (en) |
| TW (1) | TWI386403B (en) |
| WO (1) | WO2007094962A2 (en) |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008019302A1 (en) * | 2006-08-04 | 2008-02-14 | Decode Genetics Ehf | Pyrazolylphenyl and pyrrolylphenyl inhibitors of lta4h for treating inflammation |
| WO2008072724A1 (en) * | 2006-12-14 | 2008-06-19 | Taisho Pharmaceutical Co., Ltd. | Pyrazole derivative |
| WO2008137087A1 (en) | 2007-05-03 | 2008-11-13 | Cephalon, Inc. | Processes for preparing (r)-2-methylpyrrolidine and (s)-2-methylpyrrolidine and tartrate salts thereof |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| WO2009063953A1 (en) | 2007-11-13 | 2009-05-22 | Taisho Pharmaceutical Co., Ltd. | Phenylpyrazole derivatives |
| JP2010285423A (en) * | 2009-05-12 | 2010-12-24 | Taisho Pharmaceutical Co Ltd | Phenylpyrazole derivative |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2013085018A1 (en) | 2011-12-08 | 2013-06-13 | 大正製薬株式会社 | Phenylpyrrole derivative |
| WO2013100054A1 (en) | 2011-12-27 | 2013-07-04 | 大正製薬株式会社 | Phenyltriazole derivative |
| WO2015134998A1 (en) | 2014-03-07 | 2015-09-11 | Biocryst Pharmaceuticals, Inc. | Human plasma kallikrein inhibitors |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11189975B1 (en) | 2017-05-07 | 2021-11-30 | Jeffrey P. Baldwin | Powered wall plate |
| US11509102B1 (en) | 2017-05-07 | 2022-11-22 | Jeffrey P. Baldwin | Powered wall plate with plug prongs |
| US11489280B1 (en) | 2019-06-04 | 2022-11-01 | Jeffrey P. Baldwin | Powered wall plate with keyed interface |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3940418A (en) * | 1972-04-07 | 1976-02-24 | G. D. Searle & Co. | Esters and amides of 4,5-dihydrobenz[g]indazole-3-carboxylic acids and related compounds |
| FR2711140B1 (en) * | 1993-10-12 | 1996-01-05 | Sanofi Sa | 1-Naphtylpyrazole-3-substituted carboxamides active on neurotensin, their preparation, pharmaceutical compositions containing them. |
| DK1212327T3 (en) * | 1999-09-17 | 2003-12-15 | Abbott Gmbh & Co Kg | Pyrazolopyrimidines as therapeutic agents |
| AU2002341729A1 (en) * | 2001-09-19 | 2003-04-01 | Pharmacia Corporation | Substituted pyrazolyl compounds for the treatment of inflammation |
| US7202257B2 (en) * | 2003-12-24 | 2007-04-10 | Deciphera Pharmaceuticals, Llc | Anti-inflammatory medicaments |
| WO2005016877A2 (en) * | 2003-08-07 | 2005-02-24 | Merck & Co., Inc. | Pyrazole carboxamides as inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 |
| EP1670460B1 (en) * | 2003-10-10 | 2014-11-26 | Bristol-Myers Squibb Company | Pyrazole derivatives as cannabinoid receptor modulators |
| EP1751139B1 (en) * | 2004-04-30 | 2011-07-27 | Bayer HealthCare LLC | Substituted pyrazolyl urea derivatives useful in the treatment of cancer |
| JP4857267B2 (en) * | 2004-06-30 | 2012-01-18 | アサーシス, インク. | Non-imidazole tertiary amines that are histamine 3 receptor inhibitors for the treatment of cognitive impairment, sleep disorders, obesity and other CNS disorders |
| WO2007073405A1 (en) * | 2005-12-21 | 2007-06-28 | Decode Genetics Ehf | N-linked aryl heteroaryl inhibitors of lta4h for treating inflammation |
| US8115005B2 (en) * | 2006-08-04 | 2012-02-14 | Decode Genetics Ehf. | Pyrazolylphenyl and pyrrolylphenyl inhibitors of LTA4H for treating inflammation |
-
2007
- 2007-01-30 CA CA002641441A patent/CA2641441A1/en not_active Abandoned
- 2007-01-30 US US11/699,662 patent/US7897634B2/en not_active Expired - Fee Related
- 2007-01-30 JP JP2008554271A patent/JP5207983B2/en not_active Expired - Fee Related
- 2007-01-30 EP EP07749543A patent/EP1988777B1/en not_active Not-in-force
- 2007-01-30 WO PCT/US2007/002547 patent/WO2007094962A2/en active Application Filing
- 2007-01-30 AT AT07749543T patent/ATE525072T1/en not_active IP Right Cessation
- 2007-01-30 DK DK07749543.0T patent/DK1988777T3/en active
- 2007-02-09 TW TW096104902A patent/TWI386403B/en not_active IP Right Cessation
- 2007-02-09 AR ARP070100559A patent/AR059428A1/en unknown
-
2011
- 2011-01-13 US US13/006,161 patent/US7981896B2/en not_active Expired - Fee Related
Non-Patent Citations (1)
| Title |
|---|
| See references of EP1988777A4 * |
Cited By (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8115005B2 (en) | 2006-08-04 | 2012-02-14 | Decode Genetics Ehf. | Pyrazolylphenyl and pyrrolylphenyl inhibitors of LTA4H for treating inflammation |
| WO2008019302A1 (en) * | 2006-08-04 | 2008-02-14 | Decode Genetics Ehf | Pyrazolylphenyl and pyrrolylphenyl inhibitors of lta4h for treating inflammation |
| WO2008072724A1 (en) * | 2006-12-14 | 2008-06-19 | Taisho Pharmaceutical Co., Ltd. | Pyrazole derivative |
| WO2008137087A1 (en) | 2007-05-03 | 2008-11-13 | Cephalon, Inc. | Processes for preparing (r)-2-methylpyrrolidine and (s)-2-methylpyrrolidine and tartrate salts thereof |
| WO2009021740A2 (en) | 2007-08-15 | 2009-02-19 | Sanofis-Aventis | Substituted tetrahydronaphthalenes, process for the preparation thereof and the use thereof as medicaments |
| US8193176B2 (en) | 2007-11-13 | 2012-06-05 | Taisho Pharmaceutical Co., Ltd | Phenylpyrazole derivatives |
| EP2221298A1 (en) * | 2007-11-13 | 2010-08-25 | Taisho Pharmaceutical Co., Ltd | Phenylpyrazole derivatives |
| JP4543344B2 (en) * | 2007-11-13 | 2010-09-15 | 大正製薬株式会社 | Phenylpyrazole derivatives |
| US7888354B2 (en) | 2007-11-13 | 2011-02-15 | Taisho Pharmaceutical Co., Ltd | Phenylpyrazole derivatives |
| JPWO2009063953A1 (en) * | 2007-11-13 | 2011-03-31 | 大正製薬株式会社 | Phenylpyrazole derivatives |
| EP2221298A4 (en) * | 2007-11-13 | 2011-09-21 | Taisho Pharmaceutical Co Ltd | Phenylpyrazole derivatives |
| JP2010143943A (en) * | 2007-11-13 | 2010-07-01 | Taisho Pharmaceutical Co Ltd | Phenylpyrazole derivative |
| US8183387B2 (en) | 2007-11-13 | 2012-05-22 | Taisho Pharmaceutical Co., Ltd | Phenylpyrazole derivatives |
| WO2009063953A1 (en) | 2007-11-13 | 2009-05-22 | Taisho Pharmaceutical Co., Ltd. | Phenylpyrazole derivatives |
| AU2008321823B2 (en) * | 2007-11-13 | 2013-03-07 | Taisho Pharmaceutical Co., Ltd. | Phenylpyrazole derivatives |
| JP2010285423A (en) * | 2009-05-12 | 2010-12-24 | Taisho Pharmaceutical Co Ltd | Phenylpyrazole derivative |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2013085018A1 (en) | 2011-12-08 | 2013-06-13 | 大正製薬株式会社 | Phenylpyrrole derivative |
| CN103958499A (en) * | 2011-12-08 | 2014-07-30 | 大正制药株式会社 | Phenylpyrrole derivative |
| JPWO2013085018A1 (en) * | 2011-12-08 | 2015-04-27 | 大正製薬株式会社 | Phenylpyrrole derivatives |
| US9284324B2 (en) | 2011-12-08 | 2016-03-15 | Taisho Pharmaceutical Co., Ltd | Phenylpyrrole derivative |
| WO2013100054A1 (en) | 2011-12-27 | 2013-07-04 | 大正製薬株式会社 | Phenyltriazole derivative |
| WO2015134998A1 (en) | 2014-03-07 | 2015-09-11 | Biocryst Pharmaceuticals, Inc. | Human plasma kallikrein inhibitors |
| EP4180424A1 (en) | 2014-03-07 | 2023-05-17 | BioCryst Pharmaceuticals, Inc. | Substituted pyrazoles as human plasma kallikrein inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1988777B1 (en) | 2011-09-21 |
| EP1988777A4 (en) | 2010-08-25 |
| DK1988777T3 (en) | 2012-01-16 |
| AR059428A1 (en) | 2008-04-09 |
| US20110112088A1 (en) | 2011-05-12 |
| JP5207983B2 (en) | 2013-06-12 |
| US7897634B2 (en) | 2011-03-01 |
| US7981896B2 (en) | 2011-07-19 |
| TWI386403B (en) | 2013-02-21 |
| EP1988777A2 (en) | 2008-11-12 |
| WO2007094962A3 (en) | 2008-01-31 |
| US20070197526A1 (en) | 2007-08-23 |
| JP2009526054A (en) | 2009-07-16 |
| TW200745092A (en) | 2007-12-16 |
| CA2641441A1 (en) | 2007-08-23 |
| ATE525072T1 (en) | 2011-10-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2007094962A2 (en) | Pyrazoles for the treatment of obesity and other cns disorders | |
| EP1221440B1 (en) | Benzoheterocyclic derivatives useful as vasopressin or oxytocin modulators | |
| US7319103B2 (en) | Non-imidazole tertiary amines as histamine 3 receptor inhibitors for the treatment of cognitive and sleep disorders, obesity and other CNS disorders | |
| US20070197506A1 (en) | Modulators of 11-beta hydroxyl steroid dehydrogenase type 1, pharmaceutical compositions thereof, and methods of using the same | |
| JP2005255685A (en) | New pyrazole analog acting on cannabinoid receptor | |
| MX2008011791A (en) | N-substituted-azacyclylamines as histamine-3 antagonists. | |
| JP2010532748A (en) | 2-Substituted indole derivatives as calcium channel blockers | |
| JP2010505851A (en) | N-substituted azacyclylamines as histamine-3 antagonists | |
| AU2007343063B2 (en) | 5-(Heterocyclyl)alkyl-N-(arylsulfonyl)indole compounds and their use as 5-HT6 ligands | |
| JP2010540628A (en) | N-substituted oxyindoline derivatives as calcium channel blockers | |
| CA2683124C (en) | Aminoalkoxy aryl sulfonamide compounds and their use as 5-ht6 ligands | |
| CA2626028C (en) | Indole derivatives as histamine 3 receptor inhibitors for the treatment of cognitive and sleep disorders, obesity and other cns disorders | |
| EP2121602B1 (en) | 4-(heterocyclyl)alkyl-n-(arylsulfonyl) indole compounds and their use as 5-ht6 ligands | |
| JP2010540631A (en) | N-substituted oxyindoline derivatives as calcium channel blockers | |
| KR100824506B1 (en) | Quinazolinone Derivative | |
| WO2008144299A1 (en) | 5- (aminoazacyclyl) -3-sulfonyl-lh- indazoles as 5-hydroxytryptamine- 6 ligands for the treatment of cns disorders |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 2641441 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008554271 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007749543 Country of ref document: EP |