WO2007075852A2 - Calcium channel antagonists - Google Patents
Calcium channel antagonists Download PDFInfo
- Publication number
- WO2007075852A2 WO2007075852A2 PCT/US2006/048722 US2006048722W WO2007075852A2 WO 2007075852 A2 WO2007075852 A2 WO 2007075852A2 US 2006048722 W US2006048722 W US 2006048722W WO 2007075852 A2 WO2007075852 A2 WO 2007075852A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituted
- unsubstituted
- compound
- aryl
- heteroaryl
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/54—Nitrogen and either oxygen or sulfur atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/02—Muscle relaxants, e.g. for tetanus or cramps
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/04—Drugs for disorders of the muscular or neuromuscular system for myasthenia gravis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/30—Drugs for disorders of the nervous system for treating abuse or dependence
- A61P25/36—Opioid-abuse
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
- A61P29/02—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID] without antiinflammatory effect
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/06—Antiarrhythmics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/38—Nitrogen atoms
- C07D277/44—Acylated amino or imino radicals
- C07D277/46—Acylated amino or imino radicals by carboxylic acids, or sulfur or nitrogen analogues thereof
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D277/00—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings
- C07D277/02—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings
- C07D277/20—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D277/32—Heterocyclic compounds containing 1,3-thiazole or hydrogenated 1,3-thiazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D277/38—Nitrogen atoms
- C07D277/44—Acylated amino or imino radicals
- C07D277/48—Acylated amino or imino radicals by radicals derived from carbonic acid, or sulfur or nitrogen analogues thereof, e.g. carbonylguanidines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
Definitions
- Calcium is an important signaling molecule for many normal physiological processes in the human body. These include electrical signaling in the nervous system, as well as controlling heart and smooth muscle contraction, and hormone release. The entry of calcium into cells is regulated by a diverse set of proteins called calcium channels.
- a fundamental role of Ca2+ channels is to translate an electrical signal on the surface membrane into a chemical signal within the cytoplasm, which, in turn, activates many important intracellular processes including contraction, secretion, neurotransmission and regulation of enzymatic activities and gene expression.
- Continuing studies have revealed that there are multiple types of Ca2+ currents as defined by physiological and pharmacological criteria. See, e. g., Catterall, W.A., (2000) Annul Rev. Cell Dev.
- Voltage dependent calcium channels have been classified by their electrophysiological and pharmacological properties (McCleskey, E. W. et al. Curr Topics Membr (1991) 39:295-326, and Dunlap, K. et al. Trends Neurosci (1995) 18:89-98).
- Voltage-gated calcium channels can be divided into Low Voltage Activated calcium channels (LVA), that are activated at a lower voltage, and High Voltage Activated (HVA) calcium channels, that areactivated at a higher voltage with respect to typical resting membrane potentials.
- LVA Low Voltage Activated calcium channels
- HVA High Voltage Activated calcium channels
- T-type calcium channels activate at more positive potentials (high voltage activated) and display diverse kinetics and voltage-dependent properties.
- T-type calcium channels only one class of low-threshold calcium channels is known, the T-type calcium channels. These channels are so called because they carry a transient current with a low voltage of activation and rapid inactivation. (Ertel and Ertel (1997) Trends Pharmacol. Sci. 18:37-42.).
- T- type calcium channels are involved in the generation of low threshold spikes to produce burst firing (Huguenard, J.R., Annul Rev. Physiol., 329-348, 1996).
- CACNAlG alphalG, Cav3.1
- CACNAlH alphalH, Cav3.2
- CACNAlI alphalL Cav3.3
- T-type calcium channels are located in the nervous system, cardiac & vascular smooth muscle; as well as a variety of endocrine cell types (see Perez-Reyes, Physiol Rev. 2003 83:117-61). Generally, T-type channels are believed to be involved in electrical pacemaker activity, low-threshold calcium spikes, neuronal oscillations and resonance (Perez-Reyes, Physiol Rev. 2003 83: 117-61). The functional roles for T-type calcium channels in neurons include, membrane depolarization, calcium entry and burst firing. (White et al. (1989) Proc. Natl. Acad. Sci. USA 86:6802-6806). Functionally unique calcium channels allow for temporal and spatial control of intracellular calcium and support regulation of cellular activity.
- T-type calcium channels have more negative activation ranges and inactivate more rapidly than other calcium channels.
- T-type calcium channels can undergo rapid cycling between open, inactivated, and closed states, giving rise to continuous calcium influx in a range of negative membrane potentials where HVA channels are not normally activated.
- the membrane depolarizing influence of T-type calcium channel activation can become regenerative and produce calcium action potentials and oscillations.
- changes to calcium influx into neuronal cells may be implicated in conditions such as epilepsy, stroke, brain trauma, Alzheimer's disease, multiinfarct dementia, other classes of dementia, Korsakoff s disease, neuropathy caused by a viral infection of the brain or spinal cord (e.g., human immunodeficiency viruses, etc.), amyotrophic lateral sclerosis, convulsions, seizures, Huntington's disease, amnesia, pain transmission, cardiac pacemaker activity or damage to the nervous system resulting from reduced oxygen supply, poison or other toxic substances (Goldin et al., US Pat. No. 5,312,928).
- Other pathological conditions associated with elevated intracellular free calcium levels include muscular dystrophy and hypertension (Steinhardt et al., US Pat. No. 5,559,004).
- T- type calcium currents are prominent in neurons from inferior olive, thalamus, hippocampus, lateral habenular cells, dorsal horn neurons, sensory neurons (DRG, nodose), cholinergic forebrain neurons, hippocampal intraneurons, CAl , CA3 dentate gyros pyramidal cells, basal forebrain neurons, amygdala neurons (Talley et al., J. Neurosci., 19: 1895-1911, 1999) and neurons in the thalamus (Suzaki and Rogawski, Proc. Natl. Acad. Sci. USA 86:7228-7232, 1998).
- T-type channels are prominent in the some and dendrites of neurons that reveal robust Ca dependent burst firing behaviors such as the thalamic relay neurons and cerebellar Purkinje cells (Huguenard, J.R., Annul Rev. Physiol., 329-348, 1996). Consequently, improper functioning of these T-type calcium channels has been implicated in arrhythmias, chronic peripheral pain, inappropriate pain transmission in the central nervous system.
- T-type calcium channel antagonists mibefradil and/or ethosuximide
- T-type calcium channels promote oscillatory behavior, which has important consequences for epilepsy.
- the ability of a cell to fire low threshold spikes is critical in the genesis of oscillatory behavior and increased burst firing (groups of action potentials separated by about 50-100 ms).
- T-type calcium channels are believed to play a vital role in absence epilepsy, a type of generalized non-convulsive seizure.
- the evidence that voltage- gated calcium currents contribute to the epileptogenic discharge, including seizure maintenance and propagation includes: 1) a specific enhancement of T-type currents in the reticular thalamic (nRT) neurons which are hypothesized to be involved in the genesis of epileptic seizures in a rat genetic model for absence epilepsy (Tsakiridou et al., J. Neurosci., 15: 3110- 3117, 1995); 2) anti epileptics against absence petit mal epilepsy (ethosuximide and dimethadione) have been shown at physiologically relevant doses to partially depress T-type currents in thalamic neurons (Courter et al., Ann. Neurol., 25:582-93, 1989; U.S.
- T-type calcium channels underlie the intrinsic bursting properties of particular neurons that are hypothesized to be involved in epilepsy (nRT, thalamic relay and hippocampal pyramidal cells) (Huguenard).
- T-type calcium channels have been implicated in thalamic oscillations and cortical synchrony, and their involvement has been directly implicated in the generation of cortical spike waves that are thought to underlie absence epilepsy and the onset of sleep (McCormick and BaI, Annul Rev. Neurosci., 20: 185-215, 1997). Oscillations of neural networks are critical in normal brain function such during sleep-wave cycles. It is widely recognized that the thalamus is intimately involved in cortical rhythniogenesis.
- Thalamic neurons most frequently exhibit tonic firing (regularly spaced spontaneous firing) in awake animals, whereas phasic burst firing is typical of slow- wave sleep and may account for the accompanying spindling in the cortical EEG.
- the shift to burst firing occurs as a result of activation of a low threshold Ca2+ spike which is stimulated by synaptically mediated inhibition (i.e., activated upon hyperpolarization of the RP).
- synaptically mediated inhibition i.e., activated upon hyperpolarization of the RP.
- the reciprocal connections between pyramidal neurons in deeper layers of the neocortex, cortical relay neurons in the thalamus, and their respective inhibitory interneurons are believed to form the elementary pacemaking circuit.
- Tremor can be controlled through the basal ganglia and the thalamus, regions in which T-type calcium channels are strongly expressed (Talley et al J Neurosci. 1999 19:1895-911). T-type calcium channels have been implicated in the pathophysiology of tremor since the anti-epileptic drug ethosuximide is used for treating tremor, in particular, tremor associated with Parkinson's disease, essential tremor, or cerebellar disease (U.S. Pat. No. 4,981,867; D. A. Prince).
- Cortisol is the precursor for glucocorticoids and prolonged exposure to glucocorticoids causes breakdown of peripheral tissue protein, increased glucose production by the liver and mobilization of lipid from the fat depots. Furthermore, individuals suffering from anxiety and stress produce abnormally high levels of glucocorticoids. Consequently, drugs that would regulate these levels would aid in the treatment of stress disorders. In this regard, the observations (Enyeart et al., MoI. Endocrinol., 7:1031-1040, 1993) that T-type channels in adrenal zone fasciculata cells of the adrenal cortex modulate Cortisol secretion will greatly aid in the identification of such a therapeutic candidate.
- T-type calcium channels may also be involved sperm production.
- Sertoli cells secrete a number of proteins including transport proteins, hormones and growth factors, enzymes which regulate germinal cell development and other biological processes related to reproduction (Griswold, Int. Rev. Cytol., 133-156, 1988). While the role of T-type calcium channels remains to be fully elucidated, it is believed that they may be important in the release of nutrients, inhibin B, and/or plasminogen activator and thus may impact sperm production. According to researchers, the inhibition of T-type calcium channels in sperm during gamete interaction inhibits zona pellucida-dependent Ca2+ elevations and inhibits acrosome reactions, thus directly linking sperm T- type calcium channels to fertilization.
- T-type calcium channel function is very important and therapeutic moieties capable of modulating T-type currents may find utility in the practice of medicine, i.e., calcium channel blockers for the treatment of pain, epilepsy, hypertension, and angina pectoris etc.
- Compounds identified thereby may be candidates for use in the treatment of disorders and conditions associated with T-channel activity in humans and animals. Such activities include, but are not.
- cardiovascular disorders e.g. myocardial infarct, cardiac arrhythmia, heart failure and angina pectoris
- neurological disorders e.g. epilepsy, pain, schizophrenia, depression and sleep
- peripheral muscle disorders e.g. respiratory disorders; and endocrine disorders.
- the present invention meets these and other needs in the art.
- substituted 5-membered nitrogen-containing heteroaryls may be used to antagonize calcium channels.
- the calcium channel antagonist of the present invention has the formula:
- X 1 is -S-, -O-, or -N(R 3 )-.
- R 1 is substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocycloalkyl, substituted or unsubstituted aryl, or substituted or unsubstituted heteroaryl.
- R 2 is substituted or unsubstituted heteroaryl, substituted or unsubstituted heterocycloalkyl, substituted or unsubstituted cycloalkyl, or substituted or unsubstituted aryl.
- L 1 and L 2 are independently a bond, substituted or unsubstituted alkylene, substituted or unsubstituted heteroalkylene, -S(O) n , -N(R 4 )-, -N(R 4 )C(O)-, -N(R 4 )C(O)O-, -N(R 4 )C(O)N(R 4 )-, -C(O)-, -O-, -C(O)N(R 4 )-, -CH(OR 4 )-, -CH 2 -, -CH 2 O-.
- n represents an integer from 0 to 2.
- R 3 and R 4 are independently hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted heteroalkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocycloalkyl, substituted or unsubstituted aryl, or substituted or unsubstituted heteroaryl.
- the present invention provides pharmaceutical compositions comprising a pharmaceutically acceptable carrier and an antagonist of the present invention (e.g. a compound of the present invention or a complex of the present invention).
- an antagonist of the present invention e.g. a compound of the present invention or a complex of the present invention.
- the present invention provides a method for decreasing ion flow through a voltage-dependant calcium channel in a cell.
- the method includes contacting the cell with a calcium channel-closing amount of an antagonist of the present invention.
- the present invention provides a method for treating a disease through antagonizing calcium ion flow through calcium channels.
- alkyl by itself or as part of another substituent, means, unless otherwise stated, a straight or branched chain, or combination thereof, which may be fully saturated, mono- or polyunsaturated and can include di- and multivalent radicals, having the number of carbon atoms designated ⁇ i.e. C 1 -C 1O or 1- to 10- membered means one to ten carbons).
- saturated hydrocarbon radicals include, but are not limited to, groups such as methyl, ethyl, n-propyl, isopropyl, n-butyl, t-butyl, isobutyl, sec-butyl, cyclohexyl, (cyclohexyl)methyl, cyclopropylmethyl, homo logs and isomers of, for example, n-pentyl, n- hexyl, n-heptyl, n-octyl, and the like.
- An unsaturated alkyl group is one having one or more double bonds or triple bonds.
- unsaturated alkyl groups include, but are not limited to, vinyl, 2-propenyl, crotyl, 2-isopentenyl, 2-(butadienyl), 2,4-pentadienyl, 3-(l,4- pentadienyl), ethynyl, 1- and 3-propynyl, 3-butynyl, and the higher homologs and isomers.
- alkylene by itself or as part of another substituent means a divalent radical derived from an alkyl, as exemplified, but not limited, by -CHzCH 2 CH 2 CHi-, and further includes those groups described below as “heteroalkylene.”
- an alkyl (or alkylene) group will have from 1 to 24 carbon atoms, with those groups having 10 or fewer carbon atoms being preferred in the present invention.
- a “lower alkyl” or “lower alkylene” is a shorter chain alkyl or alkylene group, generally having eight or fewer carbon atoms.
- alkenylene is an alkylene that includes at least one carbon-carbon double bond
- alkynylene is an alkylene that includes at least one carbon-carbon triple bond
- a homoalkylene is an alkylene that does not include carbon-carbon double bonds or triple bonds.
- heteroalkyl by itself or in combination with another term, means, unless otherwise stated, a stable straight or branched chain, or cyclic hydrocarbon radical, or combinations thereof, consisting of the stated number of carbon atoms and at least one heteroatom selected from the group consisting of O, N, and S, and wherein the nitrogen and sulfur atoms may optionally be oxidized and the nitrogen heteroatom may optionally be quaternized.
- the heteroatom(s) O, N and S may be placed at any interior position of the heteroalkyl group or at the position at which the alkyl group is attached to the remainder of the molecule.
- heteroalkylene by itself or as part of another substituent means a divalent radical derived from heteroalkyl, as exemplified, but not limited by, -CH 2 -CH 2 -S-CH 2 -CH 2 - and -CH 2 -S- CH2-CH2-NH-CH2-.
- heteroatoms can also occupy either or both of the chain termini (e.g., alkyleneoxy, alkylenedioxy, alkyleneamino, alkylenediamino, and the like).
- cycloalkyl and “heterocycloalkyl”, by themselves or in combination with other terms, represent, unless otherwise stated, cyclic versions of “alkyl” and “heteroalkyl”, respectively.
- a cycloalkyl or heterocycloalkyl include saturated and unsaturated ring linkages.
- a heteroatom can occupy the position at which the heterocycle is attached to the remainder of the molecule.
- Examples of cycloalkyl include, but are not limited to, cyclopentyl, cyclohexyl, 1-cyclohexenyl, 3- cyclohexenyl, cycloheptyl, and the like.
- heterocycloalkyl examples include, but are not limited to, 1— (1,2,5,6-tetrahydropyridyl), 1-piperidinyl, 2-piperidinyl, 3-piperidinyl, 4- morpholinyl, 3-morpholinyl, tetrahydrofuran-2-yl, tetrahydrofuran-3-yl, tetrahydrothien-2-yl, tetrahydrothien-3-yl, 1— piperazinyl, 2-piperazinyl, and the like.
- haloalkyl are meant to include monohaloalkyl and polyhaloalkyl.
- 1IaIo(C 1 -C 4 )alkyl is mean to include, but not be limited to, trifluoromethyl, 2,2,2-trifluoroethyl, 4-chlorobutyl, 3-bromopropyl, and the like.
- aryl means, unless otherwise stated, a polyunsaturated, aromatic, hydrocarbon substituent which can be a single ring or multiple rings (preferably from 1 to 3 rings) which are fused together or linked covalently.
- heteroaryl refers to aryl groups (or rings) that contain from one to four heteroatoms selected from N, O, and S, wherein the nitrogen and sulfur atoms are optionally oxidized, and the nitrogen atom(s) are optionally quaternized.
- a heteroaryl group can be attached to the remainder of the molecule through a heteroatom.
- Non-limiting examples of aryl and heteroaryl groups include phenyl, 1-na ⁇ hthyl, 2-naphthyl, 4-biphenyl, 1-pyrrolyl, 2-pyrrolyl, 3-pyrrolyl, 3-pyrazolyl, 2- imidazolyl, 4-imidazolyl, pyrazinyl, 2-oxazolyl, 4-oxazolyl, 2-phenyl-4-oxazolyl, 5-oxazolyl, 3-isoxazolyl, 4-isoxazolyl, 5-isoxazolyl, 2-thiazolyl, 4-thiazolyl, 5-thiazolyl, 2-furyl, 3-furyl, 2-thienyl, 3-thienyl, 2-pyridinyl, 3-pyridinyl, 4-pyridinyl, 2-pyrimidyl, 4-pyrimidyl, 5- benzothiazolyl, purinyl, 2-benzimidazolyl, 5-indolyl, 1-isoquinoly
- aryl when used in combination with other terms (e.g., aryloxy, arylthioxy, arylalkyl) includes both aryl and heteroaryl rings as defined above.
- arylalkyl is meant to include those radicals in which an aryl group is attached to an alkyl group (e.g., benzyl, phenethyl, pyridylmethyl and the like) including those alkyl groups in which a carbon atom (e.g., a methylene group) has been replaced by, for example, an oxygen atom (e.g., phenoxymethyl, 2-pyridyloxymethyl, 3-(l- naphthyloxy)propyl, and the like).
- alkyl group e.g., benzyl, phenethyl, pyridylmethyl and the like
- an oxygen atom e.g., phenoxymethyl, 2-pyridyloxymethyl, 3-(l- naphthyloxy
- oxo as used herein means an oxygen that is double bonded to a carbon atom.
- Oxa as used herein, means an oxygen that is bonded to two carbon atoms.
- Thia as used herein, means a sulfur that is bonded to two carbon atoms.
- R', R", R" 1 and R 11 each preferably independently refer to hydrogen, substituted or unsubstituted heteroalkyl, substituted or unsubstituted aryl, e.g., aryl substituted with 1 to 3 halogens, substituted or unsubstituted alkyl, alkoxy or thioalkoxy groups, or arylalkyl groups.
- each of the R groups is independently selected as are each R 1 , R", R'" and R"" groups when more than one of these groups is present.
- R 1 and R" When R 1 and R" are attached to the same nitrogen atom, they can be combined with the nitrogen atom to form a 4-,5-, 6-, or 7-membered ring.
- - NR 1 R" is meant to include, but not be limited to, 1-pyrrolidinyl and 4-morpholinyl.
- alkyl is meant to include groups including carbon atoms bound to groups other than hydrogen groups, such as haloalkyl (e.g., -CF 3 and -CH 2 CF 3 ) and acyl ⁇ e.g., -C(O)CH 3 , -C(O)CF 3 , - C(O)CH 2 OCH 3 , and the like).
- haloalkyl e.g., -CF 3 and -CH 2 CF 3
- acyl ⁇ e.g., -C(O)CH 3 , -C(O)CF 3 , - C(O)CH 2 OCH 3 , and the like.
- Two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula — T-C(O)-(CRR') q -U-, wherein T and U are independently -NR-, -O-, -CRR'- or a single bond, and q is an integer of from 0 to 3.
- two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula — A-(CH2)r-B-, wherein A and B are independently -CRR'-, -O-, -NR-, -S-, -S(O)-, -S(O) 2 -, -S(O) 2 NR'- or a single bond, and r is an integer of from 1 to 4.
- One of the single bonds of the new ring so formed may optionally be replaced with a double bond.
- two of the substituents on adjacent atoms of the aryl or heteroaryl ring may optionally be replaced with a substituent of the formula -(CRR l ) s -X-(CR"R'")d-, where s and d are independently integers of from 0 to 3, and X is -O-, -C(O)-,-NR'-, -S-, -S(O)-, -S(O) 2 -, or -S(O) 2 NR'-.
- the substituents R, R', R" and R'" are preferably independently selected from hydrogen or substituted or unsubstituted (C 1 -C 6 )HIlCyI.
- heteroatom is meant to include oxygen (O), nitrogen (N), and sulfur (S).
- a "substituent group,” as used herein, means a group selected from the following moieties:
- a “size-limited substituent” or “size-limited substituent group,” as used herein means a group selected from all of the substituents described above for a “substituent group,” wherein each substituted or unsubstituted alkyl is a substituted or unsubstituted Ci-C 2 O alkyl, each substituted or unsubstituted heteroalkyl is a substituted or unsubstituted 2- to 20- membered heteroalkyl, each substituted or unsubstituted cycloalkyl is a substituted or unsubstituted C 3 -Cg cycloalkyl, and each substituted or unsubstituted heterocycloalkyl is a substituted or unsubstituted 3 to 8 membered heterocycloalkyl.
- a “lower substituent” or “lower substituent group,” as used herein means a group selected from all of the substituents described above for a “substituent group,” wherein each substituted or unsubstituted alkyl is a substituted or unsubstituted Ci-Cs alkyl, each substituted or unsubstituted heteroalkyl is a substituted or unsubstituted 2 to 8 membered heteroalkyl, each substituted or unsubstituted cycloalkyl is a substituted or unsubstituted C 5 - C 7 cycloalkyl, and each substituted or unsubstituted heterocycloalkyl is a substituted or unsubstituted 5 to 7 membered heterocycloalkyl.
- salts of the active antagonists which are prepared with relatively nontoxic acids or bases, depending on the particular substituents found on the antagonists described herein.
- base addition salts can be obtained by contacting the neutral form of such antagonists with a sufficient amount of the desired base, either neat or in a suitable inert solvent.
- pharmaceutically acceptable base addition salts include sodium, potassium, calcium, ammonium, organic amino, or magnesium salt, or a similar salt.
- acid addition salts can be obtained by contacting the neutral form of such antagonists with a sufficient amount of the desired acid, either neat or in a suitable inert solvent.
- Examples of pharmaceutically acceptable acid addition salts include those derived from inorganic acids like hydrochloric, hydrobromic, nitric, carbonic, monohydrogencarbonic, phosphoric, monohydrogenphosphoric, dihydrogenphosphoric, sulfuric, monohydrogensulfuric, hydriodic, or phosphorous acids and the like, as well as the salts derived from relatively nontoxic organic acids like acetic, propionic, isobutyric, maleic, tnalonic, benzoic, succinic, suberic, fumaric, lactic, mandelic, phthalic, benzenesulfonic, p- tolylsulfonic, citric, tartaric, methanesulfoiiic, and the like.
- inorganic acids like hydrochloric, hydrobromic, nitric, carbonic, monohydrogencarbonic, phosphoric, monohydrogenphosphoric, dihydrogenphosphoric, sulfuric, monohydrogensulfuric, hydriodic,
- salts of amino acids such as arginate and the like, and salts of organic acids like glucuronic or galactunoric acids and the like (see, for example, Berge et al, "Pharmaceutical Salts", Journal of Pharmaceutical Science 66: 1-19 (1977)).
- Certain specific antagonists of the present invention contain both basic and acidic functionalities that allow the antagonists to be converted into either base or acid addition salts.
- the neutral forms of the antagonists are preferably regenerated by contacting the salt with a base or acid and isolating the parent antagonist in the conventional manner.
- the parent form of the antagonist differs from the various salt forms in certain physical properties, such as solubility in polar solvents.
- the present invention provides antagonists, which are in a prodrug form.
- Prodrugs of the antagonists described herein are those compounds or complexes that readily undergo chemical changes under physiological conditions, in vivo, to provide the antagonists of the present invention.
- prodrugs can be converted to the antagonists of the present invention by chemical or biochemical methods in an ex vivo environment. For example, prodrugs can be slowly converted to the antagonists of the present invention when placed in a transdermal patch reservoir with a suitable enzyme or chemical reagent.
- ring as used herein means a substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocycloalkyl, substituted or unsubstituted aryl, or substituted or unsubstituted heteroaryl.
- a ring includes fused ring moities. The number of atoms in a ring are typically defined by the number of members in the ring. For example, a "5- to 7- membered ring” means there are 5-7 atoms in the encircling arrangement. The ring optionally includes a heteroatom.
- the term “5- to 7- membered ring” includes, for example pyridinyl, piperidinyl and thiazolyl rings.
- poly as used herein means at least 2.
- a polyvalent metal ion is a metal ion having a valency of at least 2.
- “Moiety” refers to the radical of a molecule that is attached to another moiety.
- the symbol / vv ⁇ ⁇ whether utilized as a bond or displayed perpendicular to a bond indicates the point at which the displayed moiety is attached to the remainder of the molecule.
- Certain antagonists of the present invention can exist in unsolvated forms as well as solvated forms, including hydrated forms. In general, the solvated forms are equivalent to unsolvated forms and are encompassed within the scope of the present invention. Certain antagonists of the present invention may exist in multiple crystalline or amorphous forms. In general, all physical forms are equivalent for the uses contemplated by the present invention and are intended to be within the scope of the present invention.
- the antagonists of the present invention may also contain unnatural proportions of atomic isotopes at one or more of the atoms that constitute such antagonists.
- the antagonists may be radiolabeled with radioactive isotopes, such as for example tritium
- NaI sodium iodide
- Hz Hertz
- NaOH sodium hydroxide
- RP reverse phase
- TFAA trifluoroacetic anhydride
- THF tetrahydrofuran
- CHCI 3 chloroform
- NaCl sodium chloride
- HI hydrogen iodide
- Pd-C palladium on charcoal
- LCMS liquid chromatography couple mass spectrometry
- the calcium channel antagonist of the present invention has the formula:
- X 1 is -S-, -O-, or -N(R 3 )-.
- R 1 is substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocycloalkyl, substituted or unsubstituted aryl, or substituted or unsubstituted heteroaryl.
- R 2 is substituted or unsubstituted heteroaryl, substituted or unsubstituted heterocycloalkyl, substituted or unsubstituted cycloalkyl, or substituted or unsubstituted aryl.
- L 1 and L 2 are independently a bond, substituted or unsubstituted alkylene, substituted or unsubstituted heteroalkylene, - S(O) n , -N(R 4 )-, -N(R 4 )C(O)-, -N(R 4 )C(O)O-, -N(R 4 )C(O)N(R 4 )-, -C(O)-, -O-, -C(O)N(R 4 )-, - CH(OR 4 )-, -CH 2 -, -CH 2 O-.
- n represents an integer from O to 2.
- R 3 and R 4 are independently hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted heteroalkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocycloalkyl, substituted or unsubstituted aryl, or substituted or unsubstituted heteroaryl.
- R 1 is substituted or unsubstituted aryl (e.g. substituted or unsubstituted phenyl).
- R 2 may be substituted or unsubstituted aryl (e.g. substituted or unsubstituted phenyl) or substituted or unsubstituted heteroaryl (e.g. substituted or unsubstituted pyridinyl, or substituted or unsubstituted pyrimidinyl).
- X 1 is -S-; R 1 is substituted or unsubstituted aryl, or substituted or unsubstituted heteroaryl; L 1 is substituted or unsubstituted alkylene, -S(O) 2 -, -N(R 4 )-, or -O-; and L 2 is -S(O) 2 -, -N(R 4 )-, -O-, or -C(O)N(R 4 )-.
- X 1 is -S-; R 1 is substituted aryl, or unsubstituted heteroaryl; R 2 is substituted aryl, or substituted or unsubstituted heteroaryl; L 1 is -S(O) 2 -; L 2 is -N(R 4 )-; and R 4 is hydrogen, substituted or unsubstituted alkyl, or substituted or unsubstituted aryl.
- R 2 may also have the formula
- R 5 and R 6 are independently hydrogen, halogen, -CF 3 , - L 3 -R 7 ,-L 3 -OR 7 , -L 3 -NR 8 R 9 , substituted or unsubstituted alkyl, substituted or unsubstituted heteroalkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocycloalkyl, substituted or unsubstituted aryl, or substituted or unsubstituted heteroaryl.
- L 3 is a bond, substituted or unsubstituted alkylene (e.g.
- R 7 , R 8 , and R 9 are independently hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted heteroalkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocycloalkyl, substituted or unsubstituted aryl, or substituted or unsubstituted heteroaryl.
- R 8 and an R 9 are optionally joined with the nitrogen to which they are attached to form a substituted or unsubstituted heterocycloalkyl, or substituted or unsubstituted heteroaryl.
- Two R 7 substituents are optionally joined with the oxygen to which they are attached to form a substituted or unsubstituted heterocycloalkyl (e.g. furanyl).
- R 8 and an R 7 are optionally joined with the nitrogen and oxygen to which they are attached, respectively, to form a substituted or unsubstituted heterocycloalkyl, or substituted or unsubstituted heteroaryl.
- L 3 is a bond, or unsubstituted alkylene.
- L 3 may also be a bond, or unsubstituted C 1 -Cs alkylene (e.g. a Ci-C 8 alkylene comprising an alkynylene linking moiety).
- R 6 may be -L 3 -NR 8 R 9 .
- L 3 is a bond, R 9 is hydrogen; and R 8 is substituted or unsubstituted alkyl, or substituted or unsubstituted heteroalkyl.
- X 1 is -S-; R 1 is substituted or unsubstituted aryl, or unsubstituted heteroaryl; L 1 is -S(O) 2 -; L 2 is -N(R 4 )-; and R 5 is hydrogen, halogen, substituted or unsubstituted alkyl, or substituted or unsubstituted aryl.
- R 4 may be hydrogen, unsubstituted alkyl, or unsubstituted heterocycloalkyl.
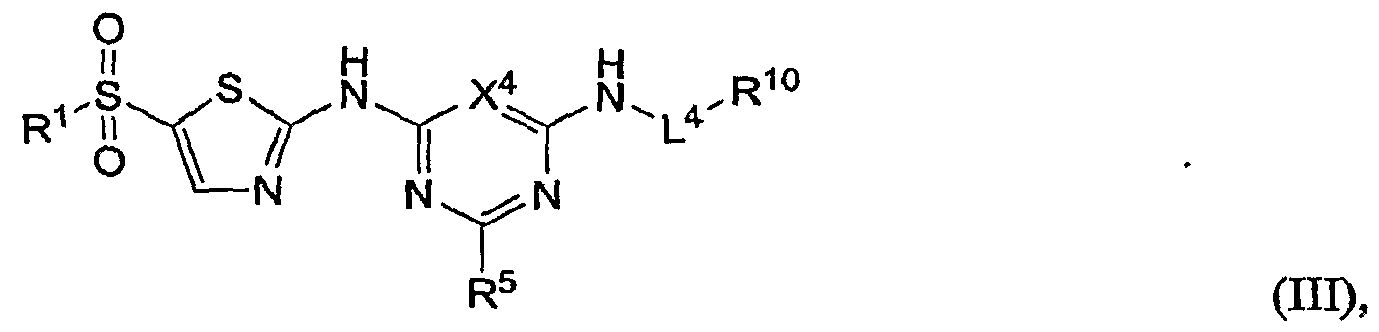
- the calcium channel antagonist has the formula:
- X 4 is as defined above.
- L 4 is a bond or substituted or unsubstituted alkylene.
- R 10 is hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted heteroalkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocycloalkyl, substituted or unsubstituted aryl, or substituted or unsubstituted heteroaryl.
- L 4 is unsubstituted alkylene.
- R 1 may be substituted or unsubstituted phenyl.
- each substituted group described above in the compound of Formulae (T), (II) and/or (HI) is substituted with at least one substituent group. More specifically, in some embodiments, each substituted alkyl, substituted heteroalkyl, substituted cycloalkyl, substituted heterocycloalkyl, substituted aryl, substituted heteroaryl, substituted alkylene, and/or substituted heteroalkylene, described above in the compounds of Formulae (J), (II) and/or (III) is substituted with at least one substituent group. In other embodiments, at least one or all of these groups are substituted with at least one size-limited substituent group. Alternatively, at least one or all of these groups are substituted with at least one lower substituent group.
- each substituted or unsubstituted alkyl is a substituted or unsubstituted > Ci-C2o alkyl
- each substituted or unsubstituted heteroalkyl is a substituted or unsubstituted 2 to 20 membered heteroalkyl
- each substituted or unsubstituted cycloalkyl is a substituted or u ⁇ substituted C4- C 8 cycloalkyl
- each substituted or unsubstituted heterocycloalkyl is a substituted or unsubstituted 4 to 8 membered heterocycloalkyl
- each substituted or unsubstituted alkylene is a substituted or unsubstituted C 1 -C 20 alkylene
- each substituted or unsubstituted heteroalkylene is a substituted or unsubstituted 2 to 20 membered heteroalky
- each substituted or unsubstituted alkyl is a substituted or unsubstituted C 1 -Cg alkyl
- each substituted or unsubstituted heteroalkyl is a substituted or unsubstituted 2 to 8 membered heteroalkyl
- each substituted or unsubstituted cycloalkyl is a substituted or unsubstituted Cs-C 7 cycloalkyl
- each substituted or unsubstituted heterocycloalkyl is a substituted or unsubstituted 5 to 7 membered heterocycloalkyl
- each substituted or unsubstituted alkylene is a substituted or unsubstituted C 1 -C 8 alkylene
- each substituted or unsubstituted heteroalkylene is a substituted or unsubstituted 2 to 8 membered heteroalkylene.
- the compounds of the present invention include the compounds of Tables 1-10, Examples 1-36, and/or Table A below.
- the following exemplary schemes illustrate methods of preparing the calcium channel antagonists of the present invention. These methods are not limited to producing the compounds shown, but can be used to prepare a variety of antagonists such as the compounds and complexes described above.
- the antagonists of the invention can also be produced by methods not explicitly illustrated in the schemes but are well within the skill of one in the art.
- the antagonists can be prepared using readily available starting materials or known intermediates. It is understood that protecting groups for sensitive or reactive functional groups may be employed where necessary in accordance with general principles of chemistry. Protecting groups are manipulated according to standard methods of organic synthesis (T. W. Green and P. G. M. Wuts (1991) Protecting Groups in Oreanic Synthesis. John Wiley & Sons).
- Acylation of the amino group may be accomplished via reaction with an acyl halide or another activated carbonyl compound, such as a mixed anhydride, generated in situ if necessary, and an added base in an appropriate solvent, such as CH2CI 2 , to provide 5.
- amine 4 may react with agents such as isocyanates or chloroformates to deliver 5.
- the reaction may optionally be heated, in an aprotic solvent such as DMF, to temperatures between 50 0 C and 150 0 C, typically 100 0 C.
- Oxidation of the sulfide for example with Oxone®, provides the corresponding sulfonyl subunit and compounds of general structure 5.
- Chlorination typically using phosphorous oxychloride, provides the dichloro analogs (12) that may be reacted with ammonia (using either ammonium hydroxide or a solution of ammonia in an appropriate alcoholic solvent) or amines to obtain chJoropyrimidines (8).
- ammonia using either ammonium hydroxide or a solution of ammonia in an appropriate alcoholic solvent
- amines to obtain chJoropyrimidines (8).
- conversion of the dichloropyrimidine to the corresponding di-iodidopyrimidine (13) followed by introduction of the amino group may be used to generate iodopyrimidines (14).
- the base is preferably a trialkylamine, such as DIEA, or an alkali metal hydride, such as NaH.
- T-type calcium channels can be assessed using a variety of in vitro assays, including, but not limited to, measuring changes in cellular cation flux, transmembrane potential, and/or cellular electrical currents. Measurement of ionic fluxes can be accomplished by measuring changes in the concentration of the permeant species using, for example, calcium sensitive fluorescent dyes (e.g. FLUO-4), or by tracking the movement of small amounts of an appropriately permeant radioactive tracer (e.g. 45-calcium).
- FLUO-4 calcium sensitive fluorescent dyes
- an appropriately permeant radioactive tracer e.g. 45-calcium
- a preferred means to determine changes in cellular polarization is by measuring changes in current or voltage with the voltage-clamp and patch-clamp techniques, using the "cell- attached” mode, the "inside-out” mode, the “outside-out” mode, the “perforated patch” mode, the "whole cell” mode, or other means of controlling or measuring changes in transmembrane potential (see, e.g., Ackerman et al., New Engl. J. Med., 336: 1575-1595 (1997)).
- Whole cell currents are conveniently determined using the standard methodology (see, e.g., Hamill et al., Pflugers. Archiv. 391: 85 (1981). Functional consequences of the test compound on ion flux can be quite varied.
- any suitable physiological change can be used to assess the influence of a test compound on the channels of this invention.
- the effects of a test compound can be measured by a toxin-binding assay.
- the functional consequences are determined using intact cells or animals, one can also measure a variety of effects such as transmitter release, hormone release, transcriptional changes to both known and uncharacterized genetic markers, changes in cell metabolism such as cell growth or pH changes, and changes in intracellular second messengers such as Ca2+, or cyclic nucleotides.
- Antagonists of T-type calcium channels can be tested using recombinant channels, or by examining cells that express native T-type calcium currents (i.e. dorsal ganglion neurons, Todorovic SM, et al (2001) Neuron. 31:75-85).
- Recombinant T-type calcium channels can be transiently or stably expressed in a host cell which can be mammalian in origin (for example, human embryonic kidney (HEK-293) or Chinese Hamster Ovary (CHO) cells) or in other cell systems like amphibian oocytes or insect cells.
- Assays for compounds capable of inhibiting or increasing divalent cation flux through T-type calcium channel proteins can be performed by application of the compounds to a bath solution containing cells expressing functional T-type calcium channels. The compounds are then allowed to contact the cells in the bath. Samples or assays that are treated with a potential T-type calcium channel antagonist are compared to control samples without the test compound, to examine the extent of modulation. Control samples (untreated with inhibitors) are assigned a relative calcium channel activity value of 100.
- Inhibition of T- type calcium channels is achieved when the calcium channel activity value relative to the control is less than 70%, preferably less than 40%, and still more preferably less than 30% at a concentration of 100 ⁇ M, preferably less than 10 ⁇ M, and still more preferably less than 1 ⁇ M.
- the compounds to be tested are present in the range from about 1 nM to about 100 mM, preferably from about 1 nM to about 30 ⁇ M. hi some embodiments, the compounds to be tested are present in the range from about 1 nM to about 3 ⁇ M.
- the present invention provides pharmaceutical compositions comprising a pharmaceutically acceptable carrier and an antagonist of the present invention (e.g. a compound of the present invention or a complex of the present invention).
- an antagonist of the present invention e.g. a compound of the present invention or a complex of the present invention.
- the antagonists of the present invention can be prepared and administered in a wide variety of oral, parenteral arid topical dosage forms.
- the antagonists of the present invention can be administered by injection, that is, intravenously, intramuscularly, intracutaneously. subcutaneously, intraduodenally, or intraperitoneally.
- the antagonists described herein can be administered by inhalation, for example, intranasally.
- the antagonists of the present invention can be administered transdermally.
- the present invention also provides pharmaceutical compositions comprising a pharmaceutically acceptable carrier and either an antagonist, or a pharmaceutically acceptable salt of an antagonist.
- pharmaceutically acceptable carriers can be either solid or liquid.
- Solid form preparations include powders, tablets, pills, capsules, cachets, suppositories, and dispersible granules.
- a solid carrier can be one or more substances, which may also act as diluents, flavoring agents, binders, preservatives, tablet disintegrating agents, or an encapsulating material.
- the carrier is a finely divided solid, which is in a mixture with the finely divided active component.
- the active component is mixed with the carrier having the necessary binding properties in suitable proportions and compacted in the shape and size desired.
- the powders and tablets preferably contain from 5% or 10% to 70% of the active antagonist.
- Suitable carriers are magnesium carbonate, magnesium stearate, talc, sugar, lactose, pectin, dextrin, starch, gelatin, tragacanth, methylcellulose, sodium carboxymethylcellulose, a low melting wax, cocoa butter, and the like.
- the term "preparation" is intended to include the formulation of the active antagonist with encapsulating material as a carrier providing a capsule in which the active component with or without other carriers, is surrounded by a carrier, which is thus in association with it.
- cachets and lozenges are included. Tablets, powders, capsules, pills, cachets, and lozenges can be used as solid dosage forms suitable for oral administration.
- a low melting wax such as a mixture of fatty acid glycerides or cocoa butter
- the active component is dispersed homogeneously therein, as by stirring.
- the molten homogeneous mixture is then poured into convenient sized molds, allowed to cool, and thereby to solidify.
- Liquid form preparations include solutions, suspensions, and emulsions, for example, water or water/propylene glycol solutions.
- liquid preparations can be formulated in solution in aqueous polyethylene glycol solution.
- Aqueous solutions suitable for oral use can be prepared by dissolving the active component in water and adding suitable colorants, flavors, stabilizers, and thickening agents as desired.
- Aqueous suspensions suitable for oral use can be made by dispersing the finely divided active component in water with viscous material, such as natural or synthetic gums, resins, methylcellulose, sodium carboxymethylcellulose, and other well-known suspending agents.
- solid form preparations which are intended to be converted, shortly before use, to liquid form preparations for oral administration.
- liquid forms include solutions, suspensions, and emulsions.
- These preparations may contain, in addition to the active component, colorants, flavors, stabilizers, buffers, artificial and natural sweeteners, dispersants, thickeners, solubilizing agents, and the like.
- the pharmaceutical preparation is preferably in unit dosage form. In such form the preparation is subdivided into unit doses containing appropriate quantities of the active component.
- the unit dosage form can be a packaged preparation, the package containing discrete quantities of preparation, such as packeted tablets, capsules, and powders in vials or ampoules.
- the unit dosage form can be a capsule, tablet, cachet, or lozenge itself, or it can be the appropriate number of any of these in packaged form.
- the quantity of active component in a unit dose preparation may be varied or adjusted from 0.1 mg to 10000 mg, more typically 1.0 mg to 1000 mg, most typically 10 mg to 500 mg, according to the particular application and the potency of the active component.
- the composition can, if desired, also contain other compatible therapeutic agents.
- the present invention provides a method for decreasing ion flow through a voltage-dependant calcium channel in a cell.
- the method includes contacting the cell with a calcium channel-closing amount of an antagonist of the present invention.
- the voltage-dependent calcium channel is a T- type calcium channel.
- the present invention provides a method for treating a disease through antagonizing calcium ion flow through calcium channels.
- An "antagonist,” as used herein, means a compound capable of decreasing the flow of ions in a calcium channel relative to the absence of the antagonist.
- the antagonists are useful in the treatment of epilepsy, stroke, anxiety, stress-related disorders, brain trauma, Alzheimer's disease, multi-infarct dementia, Korsakoff s disease, neuropathy caused by a viral infection of the brain or spinal cord, amyotrophic lateral sclerosis, convulsions, seizures, Huntington's disease, amnesia, pain transmission, damage to the nervous system resulting from reduced oxygen supply, poison or other toxic substances, muscular dystrophy, hypertension, cardiac arrhythmia, or low sperm count.
- This method involves administering, to a patient, an effective amount (e.g. a therapeutically effective amount) of an antagonist of the present invention (a compound or complex of the present invention).
- the antagonists provided herein find therapeutic utility via antagonism of calcium channels in the treatment of diseases or conditions.
- methods include contacting the cell with a calcium channel-closing amount of an antagonist of the present invention.
- the calcium channel is a T-type calcium channel.
- the cell may be isolated or form part of a organ or organism (e.g. a mammal such as a human).
- the antagonists utilized in the pharmaceutical method of the invention are administered at the initial dosage of about 0.001 mg/kg to about 1000 mg/kg daily.
- a daily dose range of about 0.1 mg/kg to about 100 mg/kg is more typical.
- the dosages may be varied depending upon the requirements of the patient, the severity of the condition being treated, and the antagonist being employed. Determination of the proper dosage for a particular situation is within the skill of the practitioner. Generally, treatment is initiated with smaller dosages, which are less than the optimum dose of the antagonist. Thereafter, the dosage is increased by small increments until the optimum effect under the circumstances is reached. For convenience, the total daily dosage may be divided and administered in portions during the day.
- Part B Copper (II) bromide (12.6 g, 57.0 mmol) was added to a mixture of 5-(3- ethoxy-phenylsulfanyl)-thiazol-2-ylamine (13.0 g, 52.0 mol) and acetonitrile (500 mL). The reaction mixture was cooled to 0 0 C and /-butyl nitrite (9.80 mL, 82.0 mmol) was added dropwise. The reaction mixture was stirred at 0 0 C for 2 hours and was allowed to warm to RT overnight. The reaction mixture was concentrated under reduced pressure.
- Part A A mixture of 4,6-dichloro-2-methylpyrimidine (1.63 g, 10.0 mmol) in
- Part B A mixture of 4-amino-6-chloro-2-methylpyrimidine (1.54 g, 10.8 mmol) and NaH (60% dispersion in mineral oil, 540 mg, 13.5 mmol) in THF (120 mL) was stirred, under Ar, at 0 0 C for 30 min. A solution of Intl-A (2.61 g, 7.49 mmol) in THF (30 mL) was added. The reaction mixture was heated at reflux overnight and was allowed to cool to RT. The reaction mixture was quenched with water, acidified with IN aqueous HCl and partitioned with 10% MeOH/CHCl3. The organic phase was separated, dried (Na 2 SO 4 ), and concentrated under reduced pressure.
- Part C A mixture of 4,6-dichloro-5-fluoro-2-methylpyrimidine (1.55 g, 8.56 mmol), ammonium hydroxide (35%, 10.0 mL, 257 mmol), and MeOH (1.00 mL) was heated, in a sealed tube, at 70 0 C for 2h. The reaction mixture was cooled to RT, and a precipitate was formed. The reaction mixture was diluted with water (ca. 10 mL) and was stirred 30 min. The solids were collected by suction filtration, washed with water and air-dried to give 4-amino-6-chloro-5-fluoro-2-methylpyrimidine (845 mg, 61%) as a tan solid. LCMS (m/z): 162,164 (M+H) +
- Part D A mixture of 4-amino-6-chloro-5-fluoro-2-methylpyrimidine (840 mg, 5.20 mmol) and NaH (60% dispersion in mineral oil, 229 mg, 5.73 mmol) in DMF (20.0 mL) was stirred, under Ar, at RT for 15 min. A solution of ⁇ ntl-A (1.81 g, 5.20 mmol) in DMF (5.0 mL) was added, and the reaction mixture was stirred at RT 15 min. Additional NaH (60% dispersion in mineral oil, 210 mg, 5.25 mmol) was added and the reaction mixture was heated at 60 0 C for 30 min.
- Part A Hydrogen iodide (3.5 M in water, 30.0 mL) was added to a solution of 4,6- dichloro-2-methylpyrimidine (5.00 g, 0.03 mol) and sodium iodide (23.0 g, 0.15 mol) in acetone (150 mL) at RT for 2 h. The reaction mixture was stirred at RT for 16 h, poured onto ice:water [(ca. 1:1) approx. 250 mL] and allowed to warm to RT. The solids were collected by suction filtration, washed with water, and air-dried to give 4,6-diodo-2-methylpyrimidine (9.80 g, 92%) as an off-white solid.
- Part B A suspension of 4,6-diodo-2-methylpy ⁇ midme (1.83 g, 5.29 mmol) in ammonia (2 M solution in EtOH, 10 mL) was heated, in a sealed tube, at 100 °C for 18 h. The reaction mixture was cooled to RT and concentrated under reduced pressure. The solid residue was washed with EtOAc and the filtrate was concentrated under reduced pressure to give 4-amino-6-diodo-2-methylpyrimidine (1.05 g, 84%) as a pale yellow solid.
- N-(2-Pyrrolidin-l-ethy])-N'-[5-(3-ethoxy-benzenesulfonyl)-thiazoI-2-yI]- pyrimidine-4,6-diamine A mixture of Int-2 (250 mg, 0.63 mmol), N-(2- aminoethyl)pyrrolidine (0.40 mL, 3.0 mmol) and Et 3 N (0.19 mL, 1.40 mmol) in 1,4-Dioxane (4 mL) was heated at 90 0 C overnight. The reaction mixture was concentrated under reduced pressure.
- N-Cl-Amino-l-methyl-propyO-N'-IS-CS-ethoxy-benzenesulfony ⁇ -thiazol-l-yll-Z- methyl-pyrimidine-4,6-diamine.TFA salt A mixture of Int-3 (600 mg, 1.3 mmol), 1,2- Diamino-2-methylpropane (0.30 mL, 3.0 mmol) and N,iV-Dusopropylethylamine (0.51 mL, 2.9 mmol) in 1,4-Dioxane (9 mL) was heated, in a sealed tube, at 100 °C overnight.
- T-type calcium channel inhibitory activity of some compounds of the invention was evaluated using both fluorometric as well as electrophysiological measurement methodologies, which are known to those skilled in the art.
- Electrophysiological measurements of test compound induced changes in T-type calcium channel activity were assessed as follows. Native cells natively expressing T-type channels or HEK-293 cells transiently or stably expressing recombinant mammalian T-type calcium channels were grown in DMEM/High glucose, Hyclone, Fetal Bovine Serum (10%), 2 mM sodium pyruvate 2 mM (and for cells lines recombinantly expressing T-type calcium channels, G418 @ 400 mg/liter) on glass coverslips in 35 mm tissue culture dishes.
- Test compound effects were typically assessed under conditions in which approximately half of the available channels were inactivated either by an 8 second conditioning depolarization from a holding potential of -100 mV to a potential ranging from -70 mV to -60 mV or by continually holding the membrane potential at -70 mV. Test compounds were assessed for their ability to reduce the amplitude of the inward T-type calcium current elicited by a 100 ms step depolarization -20 or -30 mV.
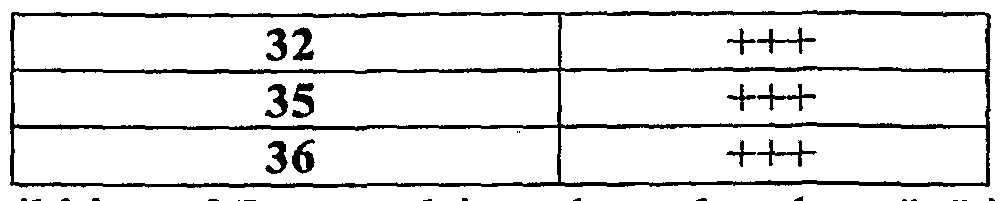
- Activity refers to inhibition of T-type calcium channels, where "+” is 10 ⁇ M ⁇ IC50 ⁇ 1- mM; "++” is 1 ⁇ M ⁇ IC50 ⁇ 10 ⁇ M; and “+++” is 1 nM ⁇ IC50 ⁇ 1 ⁇ M.
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Pain & Pain Management (AREA)
- Psychiatry (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Physical Education & Sports Medicine (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Addiction (AREA)
- Hospice & Palliative Care (AREA)
- Urology & Nephrology (AREA)
- Vascular Medicine (AREA)
- Communicable Diseases (AREA)
- Rheumatology (AREA)
- Psychology (AREA)
- Endocrinology (AREA)
- Reproductive Health (AREA)
- Virology (AREA)
- Oncology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description



























Claims


Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002634442A CA2634442A1 (en) | 2005-12-22 | 2006-12-20 | Calcium channel antagonists |
| AU2006331656A AU2006331656B2 (en) | 2005-12-22 | 2006-12-20 | Calcium channel antagonists |
| EP06847881A EP1968596A4 (en) | 2005-12-22 | 2006-12-20 | Calcium channel antagonists |
| JP2008547529A JP2009521461A (en) | 2005-12-22 | 2006-12-20 | Calcium channel antagonist |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US75343905P | 2005-12-22 | 2005-12-22 | |
| US60/753,439 | 2005-12-22 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007075852A2 true WO2007075852A2 (en) | 2007-07-05 |
| WO2007075852A3 WO2007075852A3 (en) | 2008-10-23 |
Family
ID=38218592
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2006/048722 WO2007075852A2 (en) | 2005-12-22 | 2006-12-20 | Calcium channel antagonists |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US8034954B2 (en) |
| EP (1) | EP1968596A4 (en) |
| JP (1) | JP2009521461A (en) |
| AU (1) | AU2006331656B2 (en) |
| CA (1) | CA2634442A1 (en) |
| WO (1) | WO2007075852A2 (en) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2014021383A1 (en) | 2012-07-31 | 2014-02-06 | 協和発酵キリン株式会社 | Condensed ring heterocyclic compound |
| US9000186B2 (en) | 2011-02-01 | 2015-04-07 | Kyowa Hakko Kirin Co., Ltd. | Ring-fused heterocyclic derivative |
| US9856250B2 (en) | 2014-05-28 | 2018-01-02 | Toa Eiyo Ltd. | Substituted tropane derivatives |
| US11427540B2 (en) | 2019-07-11 | 2022-08-30 | Praxis Precision Medicines, Inc. | Formulations of T-type calcium channel modulators and methods of use thereof |
| CN115244044A (en) * | 2020-03-25 | 2022-10-25 | 优迈特株式会社 | Fluorine-containing pyrimidine compound and method for producing same |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1968956A2 (en) * | 2005-12-22 | 2008-09-17 | Icagen, Inc. | Calcium channel antagonists |
| WO2007120729A2 (en) * | 2006-04-12 | 2007-10-25 | Merck & Co., Inc. | Pyridyl amide t-type calcium channel antagonists |
| AU2008317352A1 (en) * | 2007-10-24 | 2009-04-30 | Merck Sharp & Dohme Corp. | Heterocycle amide T-type calcium channel antagonists |
| AU2008317353B2 (en) * | 2007-10-24 | 2014-08-07 | Merck Sharp & Dohme Llc | Heterocycle phenyl amide T-type calcium channel antagonists |
| KR100958286B1 (en) * | 2007-11-15 | 2010-05-19 | 한국과학기술원 | A Method for the prevention and treatment of essential tremor by regulating a1G T?type calcium channel or by T?type calcium channel blockers |
| CA2800521A1 (en) | 2010-05-24 | 2011-12-01 | Toa Eiyo Ltd. | Fused imidazole derivative |
| WO2012118935A1 (en) | 2011-03-03 | 2012-09-07 | Proteotech Inc | Compounds for the treatment of neurodegenerative diseases |
| US9199975B2 (en) | 2011-09-30 | 2015-12-01 | Asana Biosciences, Llc | Biaryl imidazole derivatives for regulating CYP17 |
| EP2831071B1 (en) * | 2012-03-29 | 2018-11-14 | Merck Sharp & Dohme Corp. | Imidazolyl methyl piperidine t-type calcium channel antagonists |
| JP2016506248A (en) * | 2013-01-10 | 2016-03-03 | ティーエーユー・セラピューティクス・エルエルシー | T-type calcium channel inhibitors for the treatment of cancer |
Family Cites Families (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4210762A (en) * | 1978-08-17 | 1980-07-01 | Monsanto Company | 2[5-(3-Trifluoromethylphenyl)-1,3,4-oxadiazol-2-yl] benzoates |
| CN100384825C (en) * | 1994-06-15 | 2008-04-30 | 大塚制药株式会社 | Benzheterocyclic derivatives |
| EP0912562A1 (en) * | 1996-07-19 | 1999-05-06 | Takeda Chemical Industries, Ltd. | Heterocyclic compounds, their production and use |
| ES2237919T4 (en) * | 1998-06-18 | 2007-05-01 | Bristol-Myers Squibb Company | AMINOTIAZOL INHIBITORS REPLACED WITH CARBON OF CYCLINE DEPENDENT KINASES. |
| DE19858192A1 (en) * | 1998-12-17 | 2000-06-21 | Aventis Cropscience Gmbh | 4-Trifluoromethyl-3-oxazolylpyridines, process for their preparation, compositions containing them and their use as pesticides |
| AU2935200A (en) * | 1999-04-30 | 2000-11-17 | Pfizer Products Inc. | Compounds for the treatment of obesity |
| US20020137755A1 (en) * | 2000-12-04 | 2002-09-26 | Bilodeau Mark T. | Tyrosine kinase inhibitors |
| AU2002231139B2 (en) * | 2000-12-21 | 2007-03-22 | Bristol-Myers Squibb Company | Thiazolyl inhibitors of tec family tyrosine kinases |
| US20030199523A1 (en) * | 2002-02-28 | 2003-10-23 | Snutch Terrance P. | Heterocyclic calcium in channel blockers |
| AU2003253364B2 (en) * | 2002-08-07 | 2007-06-07 | F. Hoffmann-La Roche Ag | Thiazole derivatives |
| JP2006507302A (en) * | 2002-10-30 | 2006-03-02 | メルク エンド カムパニー インコーポレーテッド | Kinase inhibitor |
| MXPA06001570A (en) * | 2003-08-12 | 2006-05-15 | Hoffmann La Roche | 2-amino-5-benzoylthiazole npy antagonists. |
| KR20060037436A (en) * | 2003-08-12 | 2006-05-03 | 에프. 호프만-라 로슈 아게 | Thiazole derivatives as npy antagonists |
| ES2344007T3 (en) * | 2003-10-14 | 2010-08-16 | The Arizona Board Of Regents On Behalf Of The University Of Arizona | PROTEIN QUINASE INHIBITORS. |
| WO2005051932A1 (en) | 2003-11-28 | 2005-06-09 | Nippon Soda Co., Ltd. | Arylheterocycle derivative and agricultural or horticulrual bactericide and insecticide |
| TW200526635A (en) * | 2003-12-22 | 2005-08-16 | Shionogi & Co | Hydroxypyrimidinone derivative having HIV integrase inhibitory activity |
| US20060019284A1 (en) * | 2004-06-30 | 2006-01-26 | Fei Huang | Identification of polynucleotides for predicting activity of compounds that interact with and/or modulate protein tyrosine kinases and/or protein tyrosine kinase pathways in lung cancer cells |
| US7786132B2 (en) * | 2004-12-17 | 2010-08-31 | Amgen Inc. | Aminopyrimidine compounds and methods of use |
| NZ561000A (en) * | 2005-02-28 | 2010-01-29 | Japan Tobacco Inc | Novel aminopyridine compound with Syk inhibitory activity |
-
2006
- 2006-12-20 CA CA002634442A patent/CA2634442A1/en not_active Abandoned
- 2006-12-20 EP EP06847881A patent/EP1968596A4/en not_active Withdrawn
- 2006-12-20 US US11/613,872 patent/US8034954B2/en not_active Expired - Fee Related
- 2006-12-20 AU AU2006331656A patent/AU2006331656B2/en not_active Expired - Fee Related
- 2006-12-20 JP JP2008547529A patent/JP2009521461A/en not_active Withdrawn
- 2006-12-20 WO PCT/US2006/048722 patent/WO2007075852A2/en active Application Filing
Non-Patent Citations (1)
| Title |
|---|
| See references of EP1968596A4 * |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9000186B2 (en) | 2011-02-01 | 2015-04-07 | Kyowa Hakko Kirin Co., Ltd. | Ring-fused heterocyclic derivative |
| WO2014021383A1 (en) | 2012-07-31 | 2014-02-06 | 協和発酵キリン株式会社 | Condensed ring heterocyclic compound |
| US9856250B2 (en) | 2014-05-28 | 2018-01-02 | Toa Eiyo Ltd. | Substituted tropane derivatives |
| US11427540B2 (en) | 2019-07-11 | 2022-08-30 | Praxis Precision Medicines, Inc. | Formulations of T-type calcium channel modulators and methods of use thereof |
| US11649207B2 (en) | 2019-07-11 | 2023-05-16 | Praxis Precision Medicines, Inc. | Formulations of T-type calcium channel modulators and methods of use thereof |
| US12077502B2 (en) | 2019-07-11 | 2024-09-03 | Praxis Precision Medicines, Inc. | Formulations of T-type calcium channel modulators and methods of use thereof |
| CN115244044A (en) * | 2020-03-25 | 2022-10-25 | 优迈特株式会社 | Fluorine-containing pyrimidine compound and method for producing same |
Also Published As
| Publication number | Publication date |
|---|---|
| US20070173504A1 (en) | 2007-07-26 |
| WO2007075852A3 (en) | 2008-10-23 |
| EP1968596A2 (en) | 2008-09-17 |
| CA2634442A1 (en) | 2007-07-05 |
| EP1968596A4 (en) | 2010-12-01 |
| US8034954B2 (en) | 2011-10-11 |
| JP2009521461A (en) | 2009-06-04 |
| AU2006331656A1 (en) | 2007-07-05 |
| AU2006331656B2 (en) | 2012-08-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2006331656B2 (en) | Calcium channel antagonists | |
| US20070197523A1 (en) | Calcium channel antagonists | |
| CA2620179C (en) | Inhibitors of voltage-gated sodium channels | |
| AU775166B2 (en) | Selective NPY (Y5) antagonists | |
| CA2600531A1 (en) | Pyrimidine compounds and methods of use | |
| WO2003037274A2 (en) | Pyrazole-amides and-sulfonamides | |
| WO2005082871A2 (en) | Guanidine compounds, and use thereof as binding partners for 5-ht5 receptors | |
| US20100267697A1 (en) | Ion channel modulators | |
| MXPA06005736A (en) | Quinazolinone compounds with reduced bioaccumulation. | |
| DE102005005395A1 (en) | New thiazolidinone compounds are polo-like kinase inhibitors, useful for treating e.g. cancer, autoimmune diseases, cardiovascular diseases, infectious diseases, nephrological diseases and viral diseases | |
| US8338608B2 (en) | Inhibitors of ion channels | |
| MX2008008200A (en) | New phenanthridine derivatives as bradykinin antagonists. | |
| AU2004253447A1 (en) | Asymmetric benzimidazoles and related compounds as potassium channel modulators | |
| US6989379B1 (en) | Selective NPY (Y5) antagonists | |
| WO1992016526A1 (en) | Thiazole derivatives |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2008547529 Country of ref document: JP Ref document number: 2634442 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006331656 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006847881 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2006331656 Country of ref document: AU Date of ref document: 20061220 Kind code of ref document: A |