WO2007054741A1 - Tetracyclic indole derivatives as antiviral agents - Google Patents
Tetracyclic indole derivatives as antiviral agents Download PDFInfo
- Publication number
- WO2007054741A1 WO2007054741A1 PCT/GB2006/050378 GB2006050378W WO2007054741A1 WO 2007054741 A1 WO2007054741 A1 WO 2007054741A1 GB 2006050378 W GB2006050378 W GB 2006050378W WO 2007054741 A1 WO2007054741 A1 WO 2007054741A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- carboxylic acid
- fluorocyclohexyl
- tetrahydroindolo
- benzodiazocine
- ethyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CCCCC(****C1=CC2=*C(*)=*)C1=C(C1C(*)CCCC1)C2=** Chemical compound CCCCC(****C1=CC2=*C(*)=*)C1=C(C1C(*)CCCC1)C2=** 0.000 description 4
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Definitions
- the present invention relates to fluorinated tetracyclic indole compounds, to pharmaceutical compositions containing them, to their use in the prevention and treatment of hepatitis C infections and to methods of preparation of such compounds and compositions.
- HCV Hepatitis C
- A, Z, Ri, R 2 , R 3 , R 4 and n are defined therein, as useful in compositions and methods for treating psychiatric and neurological disorders.
- this document does not disclose the use of tetracyclic indole derivatives in treating or preventing viral infections.
- A, B, R 1 , R 2 , R 3 and n are defined therein, and their use in treating hepatitis C.
- R 1 , R 2 , A, Ar, W, X, Y, and Z are defined therein, useful for the treatment or prevention of infection by hepatitis C virus.
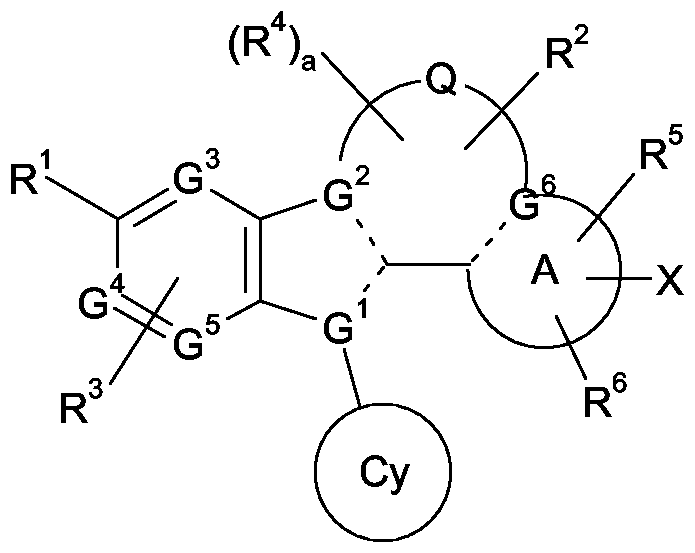
- Ar is a moiety containing at least one aromatic ring and possesses 5-, 6-, 9- or 10-ring atoms optionally containing 1, 2 or 3 heteroatoms independently selected from N, O and S, which ring is optionally substituted at any substitutable position by groups Q 1 and Q 2 ;
- Q 1 is halogen, hydroxy, Ci -6 alkyl, Ci -6 alkoxy, aryl, heteroaryl, CONR°R d , (CH 2 V 3 NR 0 R *1 , 0(CH 2 )o -3 C 3-8 cycloalkyl, 0(CH 2 ) J-3 NR 0 R 1 , O(CH 2 ) 0-3 CONR°R d , O(CH 2 ) 0-3 aryl, OCH(CH 3 )aryl, 0(CH 2 )o -3 heteroaryl, OCH(CH 3 )heteroaryl or OCHR 0 R ⁇
- R c and R d are each independently selected from hydrogen, Ci -4 alkyl and C(O)Ci -4 alkyl; or R c , R d and the nitrogen atom to which they are attached form a heteroaliphatic ring of 4 to 7 ring atoms, where said ring is optionally substituted by halogen, hydroxy,
- R e and R f are each independently selected from hydrogen and Ci -4 alkoxy; or R e and R f are linked by a heteroatom selected from N, O and S to form a heteroaliphatic ring of 4 to 7 ring atoms, where said ring is optionally substituted by halogen, hydroxy, or Ci ⁇ alkoxy; and wherein said and aryl groups are optionally substituted by halogen or hydroxy;
- Q 2 is halogen, hydroxy, where said Ci -4 alkyl and Ci -4 alkoxy groups are optionally substituted by halogen or hydroxy; or Q 1 and Q 2 may be linked by a bond or a heteroatom selected from N, O and S to form a ring of
- D 1 is N or CR a ;
- D 2 is N or CR 1 ;
- D 3 is N or CR 2 ;
- D 4 is N or CR b ; with the proviso that D 2 and D 3 are not both N;
- R a and R b are each independently selected from hydrogen, fluorine, chlorine, Ci -4 alkyl, C 2-4 alkenyl or where said Ci -4 alkyl, C 2-4 alkenyl and Ci -4 alkoxy groups are optionally substituted by hydroxy or fluorine;
- one of R 1 or R 2 is hydrogen, halogen, Ci -4 alkyl, Ci -4 alkoxy, CN, CO 2 H, aryl, heteroaryl or C(O)NR 3 R 4 , where said aryl and heteroaryl groups are optionally substituted by hydroxy or fluorine;
- R 3 is hydrogen or Ci -4 alkyl
- R 4 is hydrogen, Ci -4 alkyl, C 2-4 alkenyl, (CH 2 V 3 R 5 , SO 2 R 6 Or -L-CO 2 R 20 ;
- R 5 is NRV, OR h , aryl, heteroaryl or Het;
- R h and R 1 are each independently selected from hydrogen and Ci -4 alkyl;
- Het is a heteroaliphatic ring of 4 to 7 ring atoms, which ring may contain 1, 2 or 3 heteroatoms selected from N, O or S or a group S(O), S(O) 2 , NH or NCi -4 alkyl;
- R 6 is C 2-4 alkenyl or (CH 2 V 3 R 7 ;
- R 20 is hydrogen or Ci -6 alkyl
- R 21 and R 22 are independently selected from hydrogen, halogen, Ci -4 alkyl, C 2-4 alkenyl or
- R 21 and R 22 are linked to form a C 3-8 cycloalkyl group
- B is aryl, heteroaryl or CONR 23 R 24 , optionally substituted by halogen, C 2-4 alkenyl or
- R 23 is hydrogen or Ci -6 alkyl; or R 23 is linked to R 21 and/or R 22 to form a 5- to 10-membered ring, where said ring may be saturated, partially saturated or unsaturated, and where said ring is optionally substituted by halogen, C 2-4 alkenyl, C 2 - 4 alkynyl or Ci -4 alkoxy;
- R 24 is aryl or heteroaryl; or R 23 , R 24 and the nitrogen atom to which they are attached form a 5- to 10-membered mono- or bi-cyclic ring system, where said ring may be saturated, partially saturated or unsaturated, and where said ring is optionally substituted by halogen, C 2-4 alkenyl, C 2-4 alkynyl or D is a bond, Ci -6 alkylene, C 2-6 alkenylene, C 2-6 alkynylene, aryl or heteroaryl, where said aryl or heteroaryl is optionally substituted by halogen, or C 2-4 alkenyl;
- R 10 , R 11 , R 12 , R 13 , R 14 and R 15 are each independently selected from hydrogen, hydroxy, Ci -6 alkyl, C 2-6 alkenyl, Ci -6 alkoxy, C(O)Ci -6 alkyl, (CH 2 V 3 C 3-8 CyClOaIlCyI, (CH 2 ) 0-3 heteroaryl, (CH 2 ) 0-3 Het, (CH 2 ) 0-3 C(0)(CH 2 )o -3 Het, (CH 2 )Q -3 NR 16 R 17 , (CH 2 ) O-3 C(O)(CH 2 ) O-3 NR 16 R 17 and NHC(O)(CH 2 ) 0-3 NR 16 R 17 ;
- R 16 and R 17 are independently selected from hydrogen, Ci -6 alkyl and (CH 2 ) 0-4 NR 18 R 19 ; or R 16 , R 17 and the nitrogen atom to which they are attached form a heteroaliphatic ring of 4 to 7 ring atoms, which ring may optionally contain 1 or 2 more heteroatoms selected from O or S or a group S(O), S(O) 2 , NH or and which ring is optionally substituted by halogen, hydroxy, Ci -4 alkyl or
- R 18 and R 19 are independently selected from hydrogen and Ci -6 alkyl; or R 18 , R 19 and the nitrogen atom to which they are attached form a heteroaliphatic ring of 4 to 7 ring atoms, which ring may optionally contain 1 or 2 more heteroatoms selected from O or S or a group S(O),
- Ar is a moiety containing at least one aromatic ring and possesses 5-, 6-, 9- or 10-ring atoms optionally containing 1, 2 or 3 heteroatoms independently selected from N, O and S, which ring is optionally substituted at any substitutable position by groups Q 1 and Q 2 ;
- Q 1 is halogen, hydroxy, Ci -6 alkyl, Ci -6 alkoxy, aryl, heteroaryl, CONR°R d , (CH 2 V 3 NR 0 R *1 , 0(CH 2 ) O-3 C 3-8 CyClOaIlCyI, O(CH 2 ) 1-3 NR°R d , O(CH 2 ) 0-3 CONR°R d , O(CH 2 ) 0-3 aryl, O(CH 2 ) 0-3 heteroaryl, OCHR 6 R ⁇ R c and R d are each independently selected from hydrogen, Ci -4 alkyl and C(O)Ci -4 alkyl; or R c , R d and the nitrogen atom to which they are attached form a heteroaliphatic ring of 4 to 7 ring atoms, where said ring is optionally substituted by halogen, hydroxy,
- R e and R f are each independently selected from hydrogen and Ci -4 alkoxy; or R e and R f are linked by a heteroatom selected from N, O and S to form a heteroaliphatic ring of 4 to 7 ring atoms, where said ring is optionally substituted by halogen, hydroxy, and wherein said and aryl groups are optionally substituted by halogen or hydroxy;
- Q 2 is halogen, hydroxy, where said Ci -4 alkyl and Ci -4 alkoxy groups are optionally substituted by halogen or hydroxy; or Q 1 and Q 2 may be linked by a bond or a heteroatom selected from N, O and S to form a ring of
- D 1 is N or CR a ;
- D 2 is N or CR 1 ;
- D 3 is N or CR 2 ;
- D 4 is N or CR b ; with the proviso that D 2 and D 3 are not both N;
- R a and R b are each independently selected from hydrogen, fluorine, chlorine, Ci -4 alkyl, C 2-4 alkenyl or where said Ci -4 alkyl, C 2-4 alkenyl and Ci -4 alkoxy groups are optionally substituted by hydroxy or fluorine; - -
- R 1 or R 2 is hydrogen, halogen, Ci -4 alkyl, Ci -4 alkoxy, CN, CO 2 H, aryl, heteroaryl or C(O)NR 3 R 4 , where said aryl and heteroaryl groups are optionally substituted by hydroxy or fluorine;
- R 3 is hydrogen or R 4 is hydrogen, Ci -4 alkyl, C 2-4 alkenyl, (CH 2 V 3 R 5 or SO 2 R 6 ;
- W, X, Y and Z are as defined in relation to formula (I); and pharmaceutically acceptable salts thereof.
- Ar is a five- or six-membered aromatic ring optionally containing 1, 2 or 3 heteroatoms independently selected from N, O and S, and which ring is optionally substituted by groups Q 1 and Q 2 as hereinbefore defined.
- Ar is a five- or six-membered aromatic ring optionally containing a heteroatom selected from N, O or S such as phenyl, pyridyl, pyrrolyl, furanyl and thienyl, which ring is optionally substituted by groups Q 1 and Q 2 as hereinbefore defined. More preferably, Ar is phenyl or 2-thienyl, particularly phenyl, optionally substituted by groups Q 1 and Q 2 as hereinbefore defined.
- Q 1 is halogen, hydroxy, Ci -6 alkyl or Ci -6 alkoxy, O(CH 2 ) 0-3 C 3 . 8 cycloalkyl, O(CH 2 ) 1-3 NR°R d , 0(CH 2 )o -3 aryl, OCH(CH 3 )aryl, O(CH 2 ) 0-3 heteroaryl or OCH(CH 3 )heteroaryl, where R c and R d are as hereinbefore defined.
- Q 1 is halogen, 0(CH 2 )o -3 C 3-6 cycloalkyl, O(CH 2 ) 1-2 NR°R d , O(CH 2 ) 0-3 ⁇ henyl, OCH(CH 3 )phenyl, O(CH 2 ) 0-2 heteroaryl or OCH(CH 3 )heteroaryl, where R c and R d are independently selected from hydrogen and and heteroaryl is a 5- or 6-membered heteroaromatic ring containing 1, 2 or 3 heteroatoms selected from N, O and S.
- Q 1 is halogen, Ci -2 alkyl, Ci -3 alkoxy, O(CH 2 )C 3-6 cycloalkyl, O(CH 2 )i -2 N(Ci -4 alkyl) 2 , O(CH 2 ) 0- iphenyl, OCH(CH 3 )phenyl, O(CH 2 ) 0- iheteroaryl or OCH(CH 3 )heteroaryl, where heteroaryl is a 5- or 6-membered heteroaromatic ring containing 1 or 2 heteroatoms selected from N and S.
- Q 1 groups examples include fluorine, chlorine, methyl, methoxy, ethoxy, n-propoxy,
- Q 2 is absent.
- D 1 is CR a wherein R a is as hereinbefore defined.
- R a is hydrogen, fluorine, methyl or trifluoromethyl. More preferably, R a is hydrogen.
- D 4 is CR b wherein R b is as hereinbefore defined.
- R b is hydrogen, fluorine, methyl, methoxy or trifluoromethyl. More preferably, R b is hydrogen.
- D 2 is CR 1 wherein R 1 is as hereinbefore defined.
- R 1 is CO 2 H, CO 2 Ci. 4 alkyl, heteroaryl or C(O)NR 3 R 4 , where R 3 and R 4 are as hereinbefore defined. More preferably, R 1 is CO 2 H, heteroaryl or C(O)NHR 4 , where heteroaryl is a 5- or 6-membered heteroaromatic ring containing 1 to 4 nitrogen atoms, and R 4 is as hereinbefore defined.
- R 1 is CO 2 H, a 5-membered heteroaromatic ring containing 1 to 4 nitrogen atoms, C(O)NH(d -4 alkyl), C(O)NHSO 2 R 6 or C(O)NH-L-CO 2 R 20 , where R 6 , L and R 20 are as hereinbefore defined.
- suitable R 1 groups include CO 2 H, tetrazolyl, C(O)NH(CH 3 ), C(O)NHSO 2 (CH 2 CH 3 ), C(O)NHSO 2 N(CH 3 ) 2 and
- D 3 is CR 2 wherein R 2 is as hereinbefore defined.
- R 2 is hydrogen, fluorine, chlorine, C 2-4 alkenyl or Ci -4 alkoxy, where said Ci -4 alkyl, C 2-4 alkenyl and groups are optionally substituted by hydroxy or fluorine. More preferably, R 2 is hydrogen or Most preferably, R 2 is hydrogen.
- W is -(CR 10 R 11 MCR 12 R 13 Vi-, such as -CH 2 -,
- W is -CH 2 - or -CH 2 CH 2 -. Most preferably, W is -CH 2 -. - -
- Z is a bond, O or -CCR 10 R 11 HCR 12 R 13 Vi-. More preferably, Z is a bond O or -CH 2 -.
- Y is -CR 14 R 15 - or NR 14 where R 14 and R 15 are as hereinbefore defined.
- Y is -CH 2 - or NR 14 where R 14 is hydrogen, Ci -6 alkyl, (CH 2 ) 0-3 C 3-8 cycloalkyl, (CH 2 ) 0-3 C(0)(CH 2 )o -3 Het, (CH 2 ) 1-3 NR 16 R 17 , (CH 2 ) 0-3 heteroaryl or (CH 2 ) 0-3 C(O)(CH 2 ) 0-3 NR 16 R 17 , where R 16 and R 17 are as hereinbefore defined.
- Y is -CH 2 - or NR 14 where R 14 is hydrogen, C 1-4 alkyl, C 3-6 cycloalkyl, C(O)Het, (CH 2 ) 2 NR 16 R 17 , (CH 2 ) 0-3 pyridyl or (CH 2 ) 0- iC(O)(CH 2 ) 0- iNR 16 R 17 , where R 16 and R 17 are independently selected from hydrogen and Ci -4 alkyl, or R 16 and R 17 , together with the nitrogen atom to which they are attached, form a heteroaliphatic ring of 5 or 6 ring atoms, which ring may optionally contain one oxygen atom and/or a NH or group.
- R 14 is hydrogen, C 1-4 alkyl, C 3-6 cycloalkyl, C(O)Het, (CH 2 ) 2 NR 16 R 17 , (CH 2 ) 0-3 pyridyl or (CH 2 ) 0- iC(O)(CH 2
- R groups include hydrogen, methyl, ethyl, n-propyl, i-propyl, cyclopropyl,
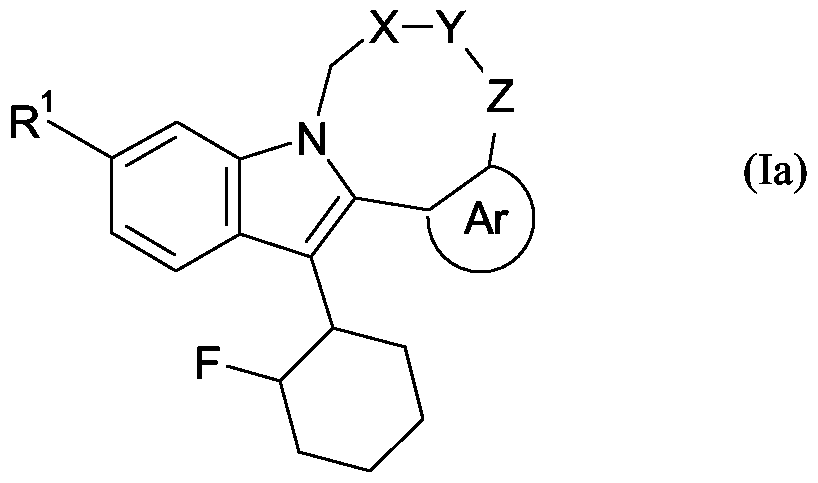
- the compound of formula (I) as hereinbefore described has the following relative stereochemical configuration: - -
- the compound of formula (Ia) as hereinbefore described has the following relative stereochemical configuration:
- alkyl or "alkoxy" as a group or part of a group means that the group is straight or branched.
- suitable alkyl groups include methyl, ethyl, n-propyl, i-propyl, n-butyl, s-butyl and t-butyl.
- suitable alkoxy groups include methoxy, ethoxy, n-propoxy, i-propoxy, n-butoxy, s-butoxy and t-butoxy.
- cycloalkyl groups referred to herein may represent, for example, cyclopropyl, cyclobutyl, cyclopentyl or cyclohexyl.
- a suitable cycloalkylalkyl group may be, for example, cyclopropylmethyl.
- alkenyl as a group or part of a group means that the group is straight or branched. Examples of suitable alkenyl groups include vinyl and allyl.
- halogen means fluorine, chlorine, bromine and iodine.
- aryl as a group or part of a group means a carbocyclic aromatic ring.
- suitable aryl groups include phenyl and naphthyl.
- heteroaryl as a group or part of a group means a 5- to 10-membered heteroaromatic ring system containing 1 to 4 heteroatoms selected from N, O and S.
- groups include pyrrolyl, furanyl, thienyl, pyridyl, pyrazolyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, pyrazinyl, pyrimidinyl, pyridazinyl, triazolyl, oxadiazolyl, thiadiazolyl, triazinyl, tetrazolyl, indolyl, benzothienyl, benzimidazolyl and quinolinyl.
- Het as a group or part of a group means a heteroaliphatic ring of 4 to 7 atoms, which ring may contain 1, 2 or 3 heteroatoms selected from N, O and S or a group S(O), S(O)2, NH ⁇ r NCi_4alkyl.
- substituents may be present.
- Optional substituents may be attached to the compounds or groups which they substitute in a variety of ways, either directly or through a connecting group of which the following are examples: amine, amide, ester, ether, thioether, sulfonamide, sulfamide, sulfoxide, urea, thiourea and urethane.
- an optional substituent may itself be substituted by another substituent, the latter being connected directly to the former or through a connecting group such as those exemplified above.
- the salts of the compounds of formula (I) will be non-toxic pharmaceutically acceptable salts.
- Other salts may, however, be useful in the preparation of the compounds according to the invention or of their non-toxic pharmaceutically acceptable salts.
- Suitable pharmaceutically acceptable salts of the compounds of this invention include acid addition salts which may, for example, be formed by mixing a solution of the compound according to the invention with a solution of a pharmaceutically acceptable acid such as hydrochloric acid, fumaric acid, p-toluenesulfonic acid, maleic acid, succinic acid, acetic acid, citric acid, tartaric acid, carbonic acid, phosphoric acid or sulfuric acid.
- a pharmaceutically acceptable acid such as hydrochloric acid, fumaric acid, p-toluenesulfonic acid, maleic acid, succinic acid, acetic acid, citric acid, tartaric acid, carbonic acid, phosphoric acid or sulfuric acid.
- Salts of amine groups may also comprise quaternary ammonium salts in which the amino nitrogen atom carries a suitable organic group such as an alkyl, alkenyl, alkynyl or aralkyl moiety.
- suitable pharmaceutically acceptable salts thereof may include metal salts such as alkali metal salts, e.g. sodium or potassium salts; and alkaline earth metal salts, e.g. calcium or magnesium salts.
- the salts may be formed by conventional means, such as by reacting the free base form of the product with one or more equivalents of the appropriate acid in a solvent or medium in which the salt is insoluble, or in a solvent such as water which is removed in vacuo or by freeze drying or by exchanging the anions of an existing salt for another anion on a suitable ion exchange resin.
- the present invention includes within its scope prodrugs of the compounds of formula (I) above.
- prodrugs will be functional derivatives of the compounds of formula (I) which are readily convertible in vivo into the required compound of formula (I).
- Conventional procedures for the selection and preparation of suitable prodrug derivatives are described, for example, in "Design of Prodrugs", ed. H. Bundgaard, Elsevier, 1985.
- a prodrug may be a pharmacologically inactive derivative of a biologically active substance (the "parent drug” or “parent molecule”) that requires transformation within the body in order to release the active drug, and that has improved delivery properties over the parent drug molecule.
- the transformation in vivo may be, for example, as the result of some metabolic process, such as chemical or enzymatic hydrolysis of a carboxylic, phosphoric or sulfate ester, or reduction or oxidation of a susceptible functionality.
- the present invention includes within its scope solvates of the compounds of formula (I) and salts thereof, for example, hydrates.
- the present invention also includes within its scope N-oxides of the compounds of formula (I).
- the present invention also includes within its scope any enantiomers, diastereomers, geometric isomers and tautomers of the compounds of formula (I). It is to be understood that all such isomers and mixtures thereof are encompassed within the scope of the invention.
- the present invention further provides a compound of formula (I) or a pharmaceutically acceptable salt thereof for use in therapy.
- the invention provides the use of a compound of formula (I) as defined above, or a pharmaceutically acceptable salt thereof, for the manufacture of a medicament for treatment or prevention of infection by hepatitis C virus in a human or animal.
- a further aspect of the invention provides a pharmaceutical composition comprising a compound of formula (I) as defined above, or a pharmaceutically acceptable salt thereof, in association with a pharmaceutically acceptable carrier.
- the composition may be in any suitable form, depending on the intended method of administration. It may for example be in the form of a tablet, capsule or liquid for oral administration, or of a solution or suspension for administration parenterally.
- the pharmaceutical compositions optionally also include one or more other agents for the treatment of viral infections such as an antiviral agent, or an immunomodulatory agent such as ⁇ -, ⁇ - or ⁇ - interferon.
- the invention provides a method of inhibiting hepatitis C virus polymerase and/or of treating or preventing an illness due to hepatitis C virus, the method involving administering to a human or animal (preferably mammalian) subject suffering from the condition a therapeutically or prophylactically effective amount of the pharmaceutical composition described above or of a compound of formula (I) as defined above, or a pharmaceutically acceptable salt thereof.
- Effective amount means - -
- the dosage rate at which the compound is administered will depend on a variety of factors including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the age of the patient, body weight, general health, sex, diet, mode and time of administration, rate of excretion, drug combination, the severity of the particular condition and the host undergoing therapy. Suitable dosage levels may be of the order of 0.02 to 5 or 10 g per day, with oral dosages two to five times higher. For instance, administration of from 1 to 20 or 50 mg of the compound per kg of body weight from one to three times per day may be in order. Appropriate values are selectable by routine testing. The compound may be administered alone or in combination with other treatments, either simultaneously or sequentially.
- it may be administered in combination with effective amounts of antiviral agents, immunomodulators, anti-infectives or vaccines known to those of ordinary skill in the art. It may be administered by any suitable route, including orally, intravenously, cutaneously and subcutaneously. It may be administered directly to a suitable site or in a manner in which it targets a particular site, such as a certain type of cell. Suitable targeting methods are already known.
- An additional aspect of the invention provides a method of preparation of a pharmaceutical composition, involving admixing at least one compound of formula (I) as defined above, or a pharmaceutically acceptable salt thereof, with one or more pharmaceutically acceptable adjuvants, diluents or carriers and/or with one or more other therapeutically or prophylactically active agents.
- the present invention also provides a process for the preparation of compounds of formula (I).
- compounds of formula (I) where Y is NR 14 may be prepared by internal ring closure of a compound of formula (II):
- R 14 , D 1 , D 2 , D 3 , D 4 , Ar, W, X and Z are defined in relation to formula (I).
- the reaction is conveniently performed in the presence of a coupling reagent, such as HATU, and a base, such as diisopropylethylamine, in a solvent.
- a coupling reagent such as HATU
- a base such as diisopropylethylamine
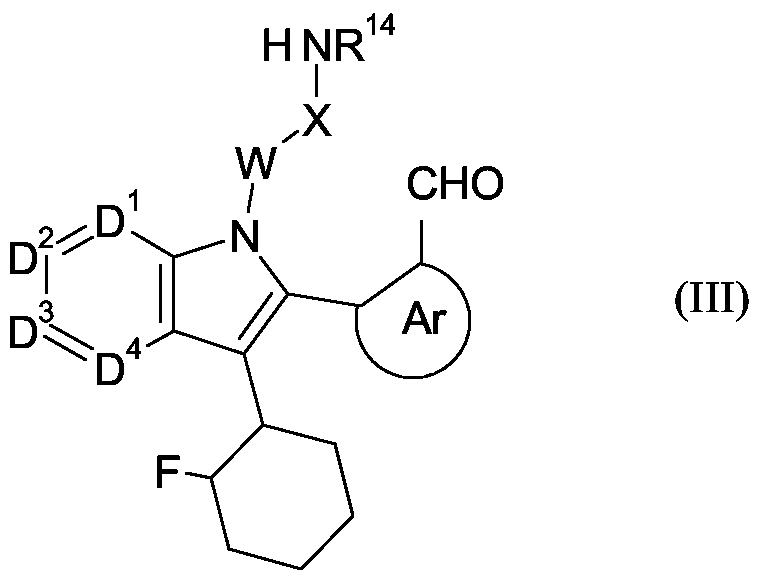
- compounds of formula (I) where Y is NR 14 and Z is -CH 2 - may be prepared by reduction and internal ring closure of a compound of formula (III): - -
- R 14 , D 1 , D 2 , D 3 , D 4 , Ar, W and X are as defined in relation to formula (I).
- the reduction is conveniently performed in the presence of a mild reducing agent, such as sodium cyanoborohydride, in a suitable solvent, such as methanol.
- a suitable solvent such as methanol.
- the ring closure is conveniently performed in the presence of a coupling reagent, such as HATU, and a base, such as diisopropylethylamine, in a solvent.
- Suitable solvents include dichloromethane.
- compounds of formula (I) may be prepared by internal ring closure of a compound of formula (FV):
- R 1 , R 2 , A, Ar, Y and Z are as defined in relation to formula (I) and X' is X as defined in relation to formula (I) or is converted to X during or after the cyclisation reaction, and W is W as defined in relation to formula (I) or is converted to W during or after the cyclisation reaction.
- W and X' may be suitable activated precursors of groups W and X respectively which can be converted into Wand X respectively during the ring closure or after it using methods described in the accompanying Schemes and Examples or known to the person skilled in the art.
- W may be CH 2 -halogen or W and X' together may be an epoxide or aziridine group.
- the compound of formula (I) where D 2 is CCO 2 alkyl may be converted into the compound of formula (I) where D 2 is CCO 2 H by conversion of the ester to the carboxylic acid, for example, by treatment with with with KOH or NaOH in a suitable solvent, such as dioxane (optionally mixed with water), THF and/or methanol.
- a suitable solvent such as dioxane (optionally mixed with water), THF and/or methanol.
- the compound of formula (I) where D 2 is CCO 2 H may be converted into the compound of formula (I) where D 2 is CC(O)NR 3 R 4 by reacting the carboxylic acid with HNR 3 R 4 in the presence of a coupling reagent, such as HATU, and a base, such as diisopropylethylamine, in a solvent.
- a coupling reagent such as HATU
- a base such as diisopropylethylamine
- a borane reagent such as BH 3 -THF
- the compound of formula (I) where Q 1 is OH may be converted into the compound of formula (T) where Q 1 is O(CH 2 ) 0-3 heteroaryl, O(CH 2 ) 0-3 aryl or O(CH 2 ) 0-3 C 3 . 8 cycloalkyl by reacting the hydroxy group with hal-(CH 2 ) 0-3 heteroaryl, hal-(CH 2 ) 0-3 aryl or hal-(CH 2 ) 0-3 C 3 . 8 cycloalkyl respectively, where hal is a suitable halogen atom such as bromine or chlorine.
- the reaction is conveniently carried out in the presence of a base, such as sodium hydride, in a suitable solvent, such as DMF.
- 2-Bromoindole intermediate (prepared as described in Example 1) was functionalised on the indole nitrogen to introduce precursor functionality W'/X' to either or both of the elements W/X of the tether.
- Pd mediated cross-coupling methodology e.g., Suzuki, Stille, etc.
- Functional group manipulation followed by ring closure afforded the tetracyclic system.
- Ester deprotection then yielded the target indole carboxylic acids, with the C2 aromatic tethered to the indole nitrogen.
- the skilled person will readily understand that the order of the reactions may be reversed (i.e. Pd-mediated coupling followed by functionalisation on the indole nitrogen).
- the C2 aromatic was introduced at the outset via Pd mediated cross-coupling methodology (Suzuki, Stille, etc.).
- the tether was then built up, with cyclisation onto the indole nitrogen finally closing the ring. Ester deprotection then yielded the target indole carboxylic acids, with the C2 aromatic tethered to the indole nitrogen.
- Tethered indole carboxylic acids arising from Methods A-D were further derivatised through manipulation of the carboxylate functionality to give compounds bearing a carboxylate replacement or carboxamide.
- any of the above synthetic sequences it may be necessary and/or desirable to protect sensitive or reactive groups on any of the molecules concerned. This may be achieved by means of conventional protecting groups, such as those described in Protective Groups in Organic Chemistry, ed. J.F.W. McOmie, Plenum Press, 1973; and T.W. Greene & P.G.M. Wuts, Protective Groups in Organic Synthesis, John Wiley & Sons, 3rd edition, 1999.
- the protecting groups may be removed at a convenient subsequent stage using methods known from the art.
- the compounds of the invention were tested for inhibitory activity against the HCV RNA dependent RNA polymerase (NS5B) in an enzyme inhibition assay (example i)) and in a cell based sub- genomic replication assay (example U)).
- the compounds have IC50's below 5 ⁇ M in the enzyme assay and several examples have EC50's below 2 ⁇ M in the cell based assay. - -
- the standard reaction (50 ⁇ l) was carried out in a buffer containing 20 mM tris/HCl pH 7.5, 5 mM MgCl 2 , 1 mM DTT, 50 mM NaCl, 0.03% N-octylglucoside, 1 ⁇ Ci [ 3 H]-UTP (40 Ci/mmol, NEN), 10 ⁇ M UTP and 10 ⁇ g/ml poly(A) or 5 ⁇ M NTPs and 5 ⁇ g/ml heteropolymeric template. Oligo(U)i 2 (1 ⁇ g/ml, Genset) was added as a primer in the assay working on PoIy(A) template. The final NS5B enzyme concentration was 5 nM.
- the order of assembly was: 1) compound, 2) enzyme, 3) template/primer, 4) NTP. After 1 h incubation at 22 0 C the reaction was stopped by adding 50 ⁇ l of 20% TCA and applying samples to DE81 filters. The filters were washed thoroughly with 5% TCA containing IM Na 2 HPOVNaH 2 PO 4 , pH 7.0, rinsed with water and then ethanol, air dried, and the filter-bound radioactivity was measured in the scintillation counter. Carrying out this reaction in the presence of various concentrations of each compound set out above allowed determination of IC 50 values by utilising the formula:
- Ai absorbance value of HBIlO cells supplemented with the indicated inhibitor concentration.
- a 0 absorbance value of HBIlO cells incubated without inhibitor.
- Reagents were usually obtained directly from commercial suppliers (and used as supplied) but a limited number of compounds from in-house corporate collections were utilised. In the latter case the reagents were readily accessible using routine synthetic steps that are either reported in the scientific literature or are known to those skilled in the art.
- 1 H NMR spectra were recorded on Bruker AM series spectrometers operating at (reported) frequencies between 300 and 600 MHz. Chemical shifts ( ⁇ ) for signals corresponding to non- exchangeable protons (and exchangeable protons where visible) are recorded in parts per million (ppm) relative to tetramethylsilane and are measured using the residual solvent peak as reference.
- Example 1 14-[(IR,2S) or (LS ⁇ J ⁇ -fluorocyclohexyll- ⁇ -isopropyl-S- ⁇ yridiii ⁇ -ylmethoxyJ-Sj ⁇ jVjS- tctrahydroindolo[2,l - ⁇ ] [2,5]bcnzodiazocinc-l 1 -carboxylic acid
- Step 1 methyl S-cyclohex-l-en-l-yl-lH-indole- ⁇ -carboxylate
- Step 3 ( ⁇ )-methyl 3- ⁇ (trans)-2-iluorocyclohex ⁇ l-lH-indole-6-carboxylate
- Step 4 ( ⁇ Ymethyl 2-bromo-3-f(trans)-2-fluorocvclohexylJ-lH-indole-6-carboxylate A solution (0.16 M) of the foregoing material in CH 2 Cl 2 was treated with NBS (1.1 eq) over 2 h. The resulting mixture was stirred for 4 h then diluted with aqueous Na 2 S 2 O 3 (1 N) and stirred for 12 h. The organic phase was separated and washed with aqueous Na 2 S 2 O 3 (1 N) and brine.
- Step 5 methyl 2-bromo-3-r(lR,2S)-2-fluorocvclohexyll-lH-indole-6-carboxylate and methyl 2-bromo-3- [(lS,2R)-2- ⁇ uorocvclohexyn-lH-indole-6-carboxylate
- the preceding material was dissolved in MeOH and the enantiomers were separated by SFC chromatography (stationary phase: Chiralcel OJ-H 250 x 10 mm; mobile phase: 25% MeOH containing 0.2% diethylamine/CO 2 ; flow rate 10 mL/min; column pressure: 100 bar; column temperature: 35 0 C; detection UV 254 nm).
- Step 6 methyl 2-bromo-l-(2-tert-butoxy-2-oxoethyl)-3- ⁇ (lR,2S) or (lS,2R)-2-fluorocvclohexyll-lH- indole-6-carboxylate
- Step 7 methyl 2-F4-(benzyloxy)-2-formylphenyll-l-(2-tert-butoxy-2-oxoethyl)-3-F(lR,2S) or (lS,2R)-2- fluorocvclohexylJ-lH-indole- ⁇ -carboxylate
- a pyrex tube was charged with a solution (0.2 M) of the foregoing material in toluene.
- the solution was degassed then treated with [4-(benzyloxy)-2-formylphenyl]boronic acid (1.5 eq), 2- dicyclohexylphosphino-2',6'-dimethoxybiphenyl (0.06 eq), K 3 PO 4 (2 eq) and Pd 2 (dba) 3 (0.02 eq).
- the tube was sealed and heated at 100 0 C for 4.5 h, then the mixture was cooled and diluted with EtOAc and H 2 O. The organic phase was separated then washed with brine and dried.
- Step 8 methyl 2- ⁇ 4-(benzyloxy)-2-r(isoyrovylamino)methyllvhenyl ⁇ -l-(2-tert-butoxy-2-oxoethyl)-3- FHRJS) or (lSJR ⁇ -fluorocvclohexyll-lH-indole- ⁇ -carboxylate
- Step 9 methyl 3-(benzyloxy)-14-F(lR,2S) or (lS,2R)-2-fluorocvclohexyll-6-isopropyl-7-oxo-5,6,7,8- tetrahvdroindolo[2,l-al [2,51benzodiazocine-l 1-carboxylate
- the foregoing material was treated with a 1 : 1 mixture of CH 2 C1 2 :TFA and the resulting solution (0.05 M) was stirred for 6 h. The volatiles were removed in vacuo to afford a residue that was diluted with CH 2 Cl 2 .
- Step 10 methyl 3-(benzyloxy)-14-[(lR,2S) or (1 S,2R)-2-fluorocvclohexyl] -6-isopropyl-5, 6, 7,8- tetrahydroindolo[2,l-al [2,51benzodiazocine-l 1-carboxylate
- a solution (0.04 M) of the foregoing material in THF was treated with BH 3 THF (1 M solution in THF; 5 eq) and stirred for 2 h. The mixture was cooled to 0 0 C, diluted to 0.02 M by dropwise addition of MeOH then treated with methanolic HCl (1.25 M, 3 eq). The solution was heated to 65 0 C for 2 h then cooled to - -
- Stev 11 methyl 14-FHRJS) or (lS,2R)-2-fluorocvclohexyll-3-hydroxy-6-isoyroyyl-5,6,7,8- tetrahydroindolo[2,l-al [2,51benzodiazocine-l 1-carboxylate
- Stev 12 methyl 14- ⁇ (1R,2S) or aSJR ⁇ -fluorocvclohexylJ- ⁇ -isoyrovyl-S-f ⁇ yridin ⁇ -ylmethoxy ⁇ -S. ⁇ J.S- tetrahydroindolo[2,l-al [2,51benzodiazocine-l 1-carboxylate
- a solution (0.03 M) of the foregoing material in a 1: 1 mixture of dioxane:H 2 O was treated with aqueous KOH (5 N, 2 eq) and heated at 40 0 C for 48 h. The volatiles were removed in vacuo and the residue was acidified with aqueous HCl (1 N).
- Example 2 7-[[2-(dimethylamino)ethyl](methyl)amino]-14-[(/ J R,2 1 S> or f/S ⁇ -fluorocyclohexyl]- 7,8-dihydro-6//-indolo[l,2-e] [l ⁇ Jbcnzoxazocinc-l 1-carboxylic acid
- Stev 1 methyl 3-[(lR,2S) or (lS,2R)-2- ⁇ uorocvclohexyn-2-(2-hydroxwhenyl)-lH-indole-6-c ⁇ rboxyl ⁇ te
- Step 4 treatment of a solution (0.16 M) of (-)-methyl 2- bromo-3-[(tr ⁇ ns)-2-fluorocyclohexyl]-lH-indole-6-c ⁇ rboxyl ⁇ te (Isomer A, from Example 1, Step 5) with 2-hydroxyphenylboronic acid (1.8 eq), aqueous Na 2 CO 3 (2 N, 4.6 eq) and Pd(PPh 3 ) 4 (0.1 eq) afforded a - -
- Step 2 methyl 3-F(lR,2S) or (lS,2R)-2-fluorocvclohexyll-2- ⁇ 2-F(2S)-oxiran-2-ylmethoxylvhenyl ⁇ -lH- indole-6-carboxylate
- Step 6 methyl (7R)-14-r(lR.2S)-2-fluorocyclohexyll-7-(methylamino)-7,8-dihydro-6H-indolori.2- el [1 ,51benzoxazocine-l 1-carboxylate
- Stev 7 methyl (7R)-7- ⁇ (2-aminoethyl)(methyl)aminol-14- ⁇ (lR.2S) or (lS.2R)-2-fluorocvclohexyll-7.8- dihydro-6H-indolo[l ,2-el [1 ,51benzoxazocine-l 1-carboxylate
- AcOH 1.6 eq
- tert-butyl(2- oxoethyl)carbamate 2.5 eq
- NaBH 3 CN 2 eq
- Stev 8 7- ⁇ 2-(dimethylamino)ethyll(methyl)aminol-14- ⁇ (lR,2S) or aS.2R)-2-fluorocvclohexyll-7.8- dihydro-6H-indolo[l ,2-el fl ,51benzoxazocine-l 1-carboxylic acid
- Example 4 6-[2-(dimethylamino)ethyl]-14-[(l 1 S',2 ⁇ )-2-fluorocyclohexyl]-3-methoxy-5,6,7,8- tctrahydroindolo[2,l -a] [2,5]bcnzodiazocinc-ll -carboxylic acid and 6-[2-(dimethylamino)ethyl]-14- [(l/?,25)-2-fluorocyclohcxyl]-3-mcthoxy-5,6,7,8-tctrahydroindolo[2,1-a][2,5]bcnzodiazocinc-ll- carboxylic acid
- Step 1 methyl l-(2-tert-butoxy-2-oxoethyl)-3-F(lR,2S) or (ISJR) -2-fluorocvclohexyll-2-(2-formyl-4- methoxyphenyl)-lH-indole-6-carboxylate
- Step 2 methyl 6- ⁇ 2-(dimethylamino)ethy ⁇ l-14- ⁇ (lR,2S) or (1S,2R) -l-fluorocyclohexyllS-methoxy-J-oxo- 5,6,7,8-tetrahvdroindolof2,l-a]f2,5]benzodiazocine-ll-carboxylate
- Step 3 6-F2-(dimethylamino)ethyll-14-F(lR,2S) or (lS,2R)-2-fluorocvclohexyll-3-methoxy-5,6,7,8- tetrahydroindolo[2,l-al J2,5Jbenzodiazocine-l 1-carboxylic acid
- Step 1 6-[2-(dimethylamino)ethyll-14-[(trans-2-fluorocyclohexyll-5,6, 7,8-tetrahydroindolo[2,l- a] J2,5 Jbenzodiazocine-11-carboxylic acid
- Example 6 6-ethyl-14-[(/ ⁇ ,2S> or (75,2/?>2-fluorocyclohexyl]-3-(pyridiii-2-ylmethoxy)-5,6,7,8- tctrahydroindolo[2,l- ⁇ ] [2,5]bcnzodiazocinc-l 1-carboxylic acid
- Step 1 methyl 3-(benzyloxy)-6-ethyl-14-F(lR,2S) or (lS,2R)-2-fluorocvclohexyll-7-oxo-5,6J,8- tetrahvdroindolo[2,l-al [2,51benzodiazocine-l 1-carboxylate
- Step 2 methyl 6-ethyl-14-[(lR.2S) or (lS.2R)-2-fluorocvclohexyll-3-hvdroxy-5.6.7.8- tetrahvdroindolo[2,l-al [2,51benzodiazocine-l 1-carboxylate
- Example 7 14-[(1R,2S) or ('/.S' ⁇ -fluorocyclohexyll- ⁇ -isopropyl-S- ⁇ yridiii-S-ylmethoxyJ-Sj ⁇ jVjS- tctrahydroindolo[2,l - ⁇ ] [2,5]bcnzodiazocinc-l 1-carboxylic acid
- Step 2 3-(Benzyloxy)-4-bromovhenyl 4-methylbenzenesulfonate
- a solution (0.5 M) of the preceding compound in DMF was cooled to 0 0 C then treated with NaH (60% in mineral oil, 1.8 eq). After 1 h, (bromomethyl)benzene (1.2 eq) was added and the reaction was stirred for 12 h before being diluted with EtOAc and washed with aqueous HCl(I N) and brine. The organic layer was dried and concentrated to give a residue that was triturated with hexane/Et 2 O to give the title compound (88%) as a white solid.
- Step 4 Methyl 2-(2-(benzyloxy)-4- ⁇ [(4-methylvhenyl)sulfonvnoxy ⁇ yhenyl)-3-[(trans)-2- fluorocyclohexyli-lH-indole-o-carboxylate A solution (0.06 M) of (-)-methyl 2-bromo-3-[(trans)-2-fluorocyclohexyl]-lH-indole-6-carboxylate
- Step 5 Methyl 3-FHRJS) or (1S.2R) -2-fluorocvclohexy ⁇ -2- ⁇ 4-m4-methylvhenyl)sulfonylJoxy ⁇ -2-raS)- oxiran-2-ylmethoxyJphenyl ⁇ -lH-indole-6-carboxylate
- Step 6 Methyl (7S)-14- ⁇ (1R,2S) or(lS,2R)-2-fluorocvclohexyll-7-hvdroxy-3- ⁇ r(4- methylvhenvtisulfonylloxyl-J.S-dihvdro- ⁇ H-indoloriJ-eiri.Slbenzoxazocine-ll-carboxylate
- a solution (0.02 M) of the preceding material in DMF was treated with NaHMDS (1.2 eq) for 1 h.
- the mixture was diluted with EtOAc and washed with aqueous HCl (1 N), s.s. NaHCO 3 , and brine.
- the dried organics were concentrated to give a residue that was purified by flash chromatography to afford the title compound (60%) as a solid; MS (ES + ) m/z 594 (M+H) + .
- Stev 8 Methyl (7R)-7-azido-14-f(lR.2S ) or (1S.2R) -2-nuorocvclohexy ⁇ l-3-(mridin-3-ylmethoxy)-7,8- dihvdro-6H-indolo[l ,2-el [1 ,51benzoxazocine-l 1-carboxylate
- Stev 1 Methyl 2-f2-(benzyloxy)-4-hvdroxwhenyll-3-f(lR.2S) or (lS.2R)-2-fluorocvclohexyll-lH-indole-
- Step 4 Methyl 3-[(lR,2S) or (lS,2R)-2-fluorocvclohexyl]-2- ⁇ 4-methoxy-2-[(2S)-oxiran-2- ylmethoxylvhenvU-lH-indole-6-carboxylate
- CsF 3.0 eq
- (25)-oxiran-2- ylmethyl 3-nitrobenzenesulfonate 1.3 eq.
- the mixture was stirred at 20 0 C for 12 h, then diluted with EtOAc and washed with H 2 O and brine.
- the dried organic layer was concentrated in vacuo to give a - -
- Step 5 Methyl 14-F(1R,2S) or (lSJR ⁇ -fluorocvclohexyll-S-methoxy ⁇ -oxo ⁇ .S-dihydro- ⁇ H-indoloFlJ- el [l,51benzoxazocine-l 1-carboxylate
- Step 6 7R and 7S- ⁇ 2-(dimethylamino)ethy ⁇ l(methyl)aminol-14- ⁇ (lR,2S) or (lS.2R)-2- fluorocvclohexyl]-3-methoxy-7,8-dihydro-6H-indolo ⁇ ,2-el [1 ,51benzoxazocine-l 1-carboxylic acid
- a solution (0.05 M) of the preceding material in 1,2-DCE was treated with N.N-dimethylethane-1,2- diamine (5 eq), Na(OAc) 3 BH (1.5 eq) and AcOH (2 eq). After 12 h the mixture was diluted with EtOAc and washed with s.s.
- Step 1 ethyl (2E)-3-(4- ⁇ f(l-aminocvclopentyl)carbonylJamino ⁇ phenyl)acrylate trifluoroacetate l- ⁇ [(benzyloxy)carbonyl]amino ⁇ cyclopentanecarboxylic acid was dissolved in DMF (0.2 M). HATU (1 eq) and triethylamine (3 eq) were added, followed by ethyl (2u)-3-(4-aminophenyl)acrylate (0.95 eq). The resulting mixture was stirred for 48 h at 40 0 C.
- Step 2 (2E)-3- ⁇ 4-r( ⁇ l-r( ⁇ 14-r(trans)-2-fluorocvclohexyll-6-isopropyl-3-methoxy-5.6.7.8- tetrahvdroindolo[2,l-a][2,5]benzodiazocin-ll-yl ⁇ carbonyl)amino]cvclopentyl ⁇ carbonyl)amino] yhenvUacrylic acid
- Example 12 IR and 7£-[2-(dimethylamino)ethyl]-14-[(LS',2 J R) or (LR ⁇ -fluorocyclohexylJ-S- (pyridin-S-ylmethoxyJ-VjS-dihydro- ⁇ /T-indoloIl ⁇ - ⁇ IljSlbeiizoxazocine-ll-carboxylic acid
- Step 2 (2-(methoxymethoxy)-4- ⁇ f(4-methylphenyl)sulfonylloxy ⁇ phenyl)boronic acid
- Step 3 methyl 3-fflR,2S) or dS,2R)-2-fluorocvclohexylJ-2-( ' 2-fmethoxymethoxy)-4- ⁇ ff4-methylphenyl) sulfonyl]oxy ⁇ phenyl)-lH-indole-6-carboxylate
- Step 4 methyl 3-FHRJS) or (lS.2R)-2-nuorocvclohexyll-2-(2-hvdroxy-4-U(4-methylphenyl) sulfonyl]oxylphenyl)-lH-indole-6-carboxylate
- Stev 7 methyl 14-FHRJS) or (lSJR)-2-fluorocvclohexyll-3- ⁇ F(4-methylvhenyl)sulfonylloxy ⁇ -7R and 7S- ( ⁇ F(4-methylvhenyl)sulfonvnoxy ⁇ methyl)-7,8-dihydro-6H-indoloFlJ-elF1.51benzoxazocine-ll- carboxylate
- Stev 8 methyl 7R and 7S-(cyanomethyl)-14-FHR.2S or (lSJR)-2-fluorocvclohexyll-3- ⁇ F(4-methylvhenyl) sulfonyl]oxy ⁇ -7,8-dihvdro-6H-indoloFl J-e] Fl ,5]benzoxazocine-l 1-carboxylate
- a solution (0.04 M) of the preceding material in DMF was treated with NaCN (1.2 eq) then stirred for 18 h. After dilution with EtOAc the organics were washed with s.s. NaHCO 3 , and brine. The organic layer was dried and concentrated in vacuo. The residue was purified by flash chromatography (EtOAc : PE 25:75) to give the title compound as a colourless oil (55%); MS (ES + ) m/z 617 (M+H) + .
- Stev 10 methyl 7R and 7S-(2-aminoethyl)-14-F(lR.2S) or (lS,2R)-2-fluorocvclohexyll-3-hvdroxy-7,8- dihydro-6H-indolo[l ,2-el Fl ,5]benzoxazocine-l 1-carboxylate
- Stev 11 methyl 7R and 7S-F2-(dimethylamino)ethyll-14-F(lR,2S) or (lS,2R)-2-fluorocvclohexyll-3- hydroxy ⁇ .S-dihydro- ⁇ H-indoloFU-elFLSlbenzoxazocine-ll-carboxylate
- Stev 12 methyl 7R and 7S-F2-(dimethylamino)ethyll-14-F(lR.2S) or (lS,2R)-2-fluorocvclohexyll-3- (yyridin-3-ylmethoxy)-7 ⁇ 8-dihydro-6H-indolo[l ,2-el Fl ,5]benzoxazocine-l 1-carboxylate
- Step 13 7R and 7S -F2-(dimethylamino)ethyll-14-F(lR,2S) or (lS,2R)-2-fluorocvclohexyll-3-(vyridin-3- ylmethoxy)-7,8-dihydro-6H-indoloFl ,2-el Fl ,5Jbenzoxazocine-l 1-carboxylic acid
- Stev 1 methyl 7R and 7S-(cvanomethyl)-14- ⁇ (lR.2S) or (lS,2R)-2-fluorocvclohexyll-3-methoxy-7,8- dihydro-6H-indolo[l ,2-el [1 ,51benzoxazocine-l 1-carboxylate
- Step 2 methyl 7R and 7S-F2-(dimethylamino)ethyll-14-F(lR,2S) or (lS,2R)-2-fluorocvclohexyll-3- methoxy ⁇ .S-dihydro- ⁇ H-indoloflJ-eHl.Slbenzoxazocine-ll-carboxylate
- Example 14 7R and 7S-[(dimethylamino)methyl]-14-[(lR,2S) or (lS,2R)-2-fluorocydohexyl]-3- (pyridazin-S-ylmethoxyJ ⁇ jS-dihydro- ⁇ H-indoloIl ⁇ -ellljSlbenzoxazocine-ll-carboxylic acid
- Stev 1 methyl 7R and 7S- ⁇ (dimethylamino)methyll-14- ⁇ (1R,2S) or (lS,2R)-2-nuorocvclohexy ⁇ l-3-U(4- methylphenyl)sulfonyl]oxy ⁇ -7,8-dihydro-6H-indolo[l ,2-e] [1 ,5]benzoxazocine-l 1-carboxylate
- Step 2 methyl 7R and 7S- ⁇ (dimethylamino)methv ⁇ l-14- ⁇ (1R,2S) or (lS,2R)-2-fluorocvclohexyll-3- hvdroxy-7,8-dihvdro-6H-indolofl,2-e]fl,5]benzoxazocine-ll-carboxylate
- Stev 3 methyl 7R and 7S-f(dimethylamino)methyll-14-f(lR.2S) or (lS.2R)-2-fluorocvclohexyll-3- (vyridazin-3-ylmethoxy)-7 ⁇ 8-dihvdro-6H-indolo[l ,2-el l [l ,51benzoxazocine-l 1-carboxylate
- Step 4 7R and 7S- ⁇ (dimethylamino)methv ⁇ l-14- ⁇ (1R,2S) or (lS,2R)-2-fluorocvclohexyll-3-(Oyridazin-3- ylmethoxy)-?
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Oncology (AREA)
- Virology (AREA)
- Communicable Diseases (AREA)
- Molecular Biology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description






































Claims








Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US12/084,846 US20090136449A1 (en) | 2005-11-10 | 2006-11-09 | Tetracyclic Indole Derivatives as Antiviral Agents |
| JP2008539509A JP2009515865A (en) | 2005-11-10 | 2006-11-09 | Tetracyclic indole derivatives as antiviral agents |
| CA002628937A CA2628937A1 (en) | 2005-11-10 | 2006-11-09 | Tetracyclic indole derivatives as antiviral agents |
| EP06808742A EP1948660A1 (en) | 2005-11-10 | 2006-11-09 | Tetracyclic indole derivatives as antiviral agents |
| AU2006313539A AU2006313539A1 (en) | 2005-11-10 | 2006-11-09 | Tetracyclic indole derivatives as antiviral agents |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB0522881.2A GB0522881D0 (en) | 2005-11-10 | 2005-11-10 | Therapeutic compounds |
| GB0522881.2 | 2005-11-10 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007054741A1 true WO2007054741A1 (en) | 2007-05-18 |
Family
ID=35516651
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2006/050378 Ceased WO2007054741A1 (en) | 2005-11-10 | 2006-11-09 | Tetracyclic indole derivatives as antiviral agents |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US20090136449A1 (en) |
| EP (1) | EP1948660A1 (en) |
| JP (1) | JP2009515865A (en) |
| CN (1) | CN101305008A (en) |
| AU (1) | AU2006313539A1 (en) |
| CA (1) | CA2628937A1 (en) |
| GB (1) | GB0522881D0 (en) |
| WO (1) | WO2007054741A1 (en) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007129119A1 (en) * | 2006-05-08 | 2007-11-15 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Pentacyclic indole derivatives as antiviral agents |
| WO2008011521A3 (en) * | 2006-07-20 | 2008-06-26 | Genelabs Tech Inc | Polycyclic viral inhibitors |
| GB2451184A (en) * | 2007-07-17 | 2009-01-21 | Angeletti P Ist Richerche Bio | Macrocyclic hepatitis C virus NS3 protease inhibitors having a 6-(N-sulphamoyl(carbamoyl))-[7-aza-]indole moiety within the macrocyclic ring path |
| WO2009076747A1 (en) | 2007-12-19 | 2009-06-25 | Boehringer Ingelheim International Gmbh | Viral polymerase inhibitors |
| US7659263B2 (en) | 2004-11-12 | 2010-02-09 | Japan Tobacco Inc. | Thienopyrrole compound and use thereof as HCV polymerase inhibitor |
| WO2010080874A1 (en) | 2009-01-07 | 2010-07-15 | Scynexis, Inc. | Cyclosporine derivative for use in the treatment of hcv and hiv infection |
| WO2010082050A1 (en) | 2009-01-16 | 2010-07-22 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti S.P.A. | Macrocyclic and 7-aminoalkyl-substituted benzoxazocines for treatment of hepatitis c infections |
| US7977331B1 (en) | 2004-02-24 | 2011-07-12 | Japan Tobacco Inc. | Tetracyclic fused heterocyclic compound and use thereof as HCV polymerase inhibitor |
| US8921355B2 (en) | 2008-07-08 | 2014-12-30 | Janssen R & D Ireland | Macrocyclic indole derivatives useful as hepatitis C virus inhibitors |
| US9127010B2 (en) | 2010-06-24 | 2015-09-08 | Janssen Sciences Ireland Uc | Preparation of 13-cyclohexyl-3-methoxy-6-[methyl-(2-{2-[methyl-(sulphamoyl)-amino]-ethoxy}-ethyl)-carbamoyl]-7H-indolo-[2,1-a]-[2]-benzazepine-10-carboxylic acid |
| US9334282B2 (en) | 2007-12-24 | 2016-05-10 | Janssen Sciences Ireland Uc | Macrocyclic indoles as hepatitis C virus inhibitors |
| US12274700B1 (en) | 2020-10-30 | 2025-04-15 | Accencio LLC | Methods of treating symptoms of coronavirus infection with RNA polymerase inhibitors |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB0518390D0 (en) * | 2005-09-09 | 2005-10-19 | Angeletti P Ist Richerche Bio | Therapeutic compounds |
| SG11201708622UA (en) | 2015-02-02 | 2017-11-29 | Forma Therapeutics Inc | 3-aryl-4-amido-bicyclic [4,5,0] hydroxamic acids as hdac inhibitors |
| WO2016126726A1 (en) | 2015-02-02 | 2016-08-11 | Forma Therapeutics, Inc. | Bicyclic [4,6,0] hydroxamic acids as hdac6 inhibitors |
| EP3472131B1 (en) | 2016-06-17 | 2020-02-19 | Forma Therapeutics, Inc. | 2-spiro-5- and 6-hydroxamic acid indanes as hdac inhibitors |
| UY38705A (en) * | 2019-05-23 | 2020-12-31 | Irbm S P A | TRICYCLIC INHIBITORS SUBSTITUTED WITH HEPATITIS B VIRUS OXALAMIDE |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005080399A1 (en) * | 2004-02-24 | 2005-09-01 | Japan Tobacco Inc. | Fused heterotetracyclic compounds and use tehreof as hcv polymerase inhibitor |
| WO2006046030A2 (en) * | 2004-10-26 | 2006-05-04 | Istituto Di Ricerche Di Biologia Molecolare P Angeletti Spa | Tetracyclic indole derivatives as antiviral agents |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7795247B2 (en) * | 2004-10-26 | 2010-09-14 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Tetracyclic indole derivatives as antiviral agents |
| GB0518390D0 (en) * | 2005-09-09 | 2005-10-19 | Angeletti P Ist Richerche Bio | Therapeutic compounds |
-
2005
- 2005-11-10 GB GBGB0522881.2A patent/GB0522881D0/en not_active Ceased
-
2006
- 2006-11-09 AU AU2006313539A patent/AU2006313539A1/en not_active Abandoned
- 2006-11-09 CA CA002628937A patent/CA2628937A1/en not_active Abandoned
- 2006-11-09 US US12/084,846 patent/US20090136449A1/en not_active Abandoned
- 2006-11-09 EP EP06808742A patent/EP1948660A1/en not_active Withdrawn
- 2006-11-09 CN CNA2006800417723A patent/CN101305008A/en active Pending
- 2006-11-09 JP JP2008539509A patent/JP2009515865A/en not_active Withdrawn
- 2006-11-09 WO PCT/GB2006/050378 patent/WO2007054741A1/en not_active Ceased
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005080399A1 (en) * | 2004-02-24 | 2005-09-01 | Japan Tobacco Inc. | Fused heterotetracyclic compounds and use tehreof as hcv polymerase inhibitor |
| EP1719773A1 (en) * | 2004-02-24 | 2006-11-08 | Japan Tobacco, Inc. | Fused heterotetracyclic compounds and use tehreof as hcv polymerase inhibitor |
| WO2006046030A2 (en) * | 2004-10-26 | 2006-05-04 | Istituto Di Ricerche Di Biologia Molecolare P Angeletti Spa | Tetracyclic indole derivatives as antiviral agents |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7977331B1 (en) | 2004-02-24 | 2011-07-12 | Japan Tobacco Inc. | Tetracyclic fused heterocyclic compound and use thereof as HCV polymerase inhibitor |
| US7659263B2 (en) | 2004-11-12 | 2010-02-09 | Japan Tobacco Inc. | Thienopyrrole compound and use thereof as HCV polymerase inhibitor |
| WO2007129119A1 (en) * | 2006-05-08 | 2007-11-15 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Pentacyclic indole derivatives as antiviral agents |
| US8232390B2 (en) | 2006-05-08 | 2012-07-31 | Istituto Di Richerche Di Biologia Molecolare P. Angeletti Spa | Pentacyclic indole derivatives as antiviral agents |
| AU2007246850B2 (en) * | 2006-05-08 | 2012-07-05 | Istituto Di Richerche Di Biologia Molecolare P. Angeletti Spa | Pentacyclic indole derivatives as antiviral agents |
| WO2008011521A3 (en) * | 2006-07-20 | 2008-06-26 | Genelabs Tech Inc | Polycyclic viral inhibitors |
| GB2451184B (en) * | 2007-07-17 | 2011-09-21 | Angeletti P Ist Richerche Bio | Novel anti-viral macrocyclic indole compounds |
| GB2451184A (en) * | 2007-07-17 | 2009-01-21 | Angeletti P Ist Richerche Bio | Macrocyclic hepatitis C virus NS3 protease inhibitors having a 6-(N-sulphamoyl(carbamoyl))-[7-aza-]indole moiety within the macrocyclic ring path |
| WO2009010783A1 (en) * | 2007-07-17 | 2009-01-22 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Macrocyclic indole derivatives for the treatment of hepatitis c infections |
| WO2009010785A1 (en) * | 2007-07-17 | 2009-01-22 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Macrocyclic indole derivatives for the treatment of hepatitis c infections |
| JP2010533699A (en) * | 2007-07-17 | 2010-10-28 | イステイチユート・デイ・リチエルケ・デイ・ビオロジア・モレコラーレ・ピ・アンジエレツテイ・エツセ・ピー・アー | Macrocyclic indole derivatives for the treatment of hepatitis C infection |
| US7989438B2 (en) | 2007-07-17 | 2011-08-02 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Therapeutic compounds |
| WO2009076747A1 (en) | 2007-12-19 | 2009-06-25 | Boehringer Ingelheim International Gmbh | Viral polymerase inhibitors |
| US9334282B2 (en) | 2007-12-24 | 2016-05-10 | Janssen Sciences Ireland Uc | Macrocyclic indoles as hepatitis C virus inhibitors |
| US8921355B2 (en) | 2008-07-08 | 2014-12-30 | Janssen R & D Ireland | Macrocyclic indole derivatives useful as hepatitis C virus inhibitors |
| US9427440B2 (en) | 2008-07-08 | 2016-08-30 | Janssen Sciences Ireland Uc | Macrocyclic indole derivatives useful as hepatitis C virus inhibitors |
| WO2010080874A1 (en) | 2009-01-07 | 2010-07-15 | Scynexis, Inc. | Cyclosporine derivative for use in the treatment of hcv and hiv infection |
| WO2010082050A1 (en) | 2009-01-16 | 2010-07-22 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti S.P.A. | Macrocyclic and 7-aminoalkyl-substituted benzoxazocines for treatment of hepatitis c infections |
| US9127010B2 (en) | 2010-06-24 | 2015-09-08 | Janssen Sciences Ireland Uc | Preparation of 13-cyclohexyl-3-methoxy-6-[methyl-(2-{2-[methyl-(sulphamoyl)-amino]-ethoxy}-ethyl)-carbamoyl]-7H-indolo-[2,1-a]-[2]-benzazepine-10-carboxylic acid |
| US12274700B1 (en) | 2020-10-30 | 2025-04-15 | Accencio LLC | Methods of treating symptoms of coronavirus infection with RNA polymerase inhibitors |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1948660A1 (en) | 2008-07-30 |
| AU2006313539A1 (en) | 2007-05-18 |
| CN101305008A (en) | 2008-11-12 |
| GB0522881D0 (en) | 2005-12-21 |
| US20090136449A1 (en) | 2009-05-28 |
| CA2628937A1 (en) | 2007-05-18 |
| JP2009515865A (en) | 2009-04-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7662809B2 (en) | Tetracyclic indole derivatives as antiviral agents | |
| US8232390B2 (en) | Pentacyclic indole derivatives as antiviral agents | |
| US8101595B2 (en) | Antiviral indoles | |
| WO2007054741A1 (en) | Tetracyclic indole derivatives as antiviral agents | |
| US7989438B2 (en) | Therapeutic compounds | |
| US20090149526A1 (en) | Tetracyclic Indole Derivatives as Antiviral Agents | |
| US20070167447A1 (en) | Indole acetamides as inhibitors of the hepatitis c virus ns5b polymerase | |
| US7973026B2 (en) | Thienopyrroles as antiviral agents | |
| US7795247B2 (en) | Tetracyclic indole derivatives as antiviral agents | |
| US7767660B2 (en) | Antiviral indoles |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200680041772.3 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2006313539 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 2006313539 Country of ref document: AU Date of ref document: 20061109 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006313539 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2628937 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2008539509 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006808742 Country of ref document: EP Ref document number: 12084846 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 4493/DELNP/2008 Country of ref document: IN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006808742 Country of ref document: EP |