WO2007045989A1 - Pyridyl derivatives useful as h3 ligands - Google Patents
Pyridyl derivatives useful as h3 ligands Download PDFInfo
- Publication number
- WO2007045989A1 WO2007045989A1 PCT/IB2006/002951 IB2006002951W WO2007045989A1 WO 2007045989 A1 WO2007045989 A1 WO 2007045989A1 IB 2006002951 W IB2006002951 W IB 2006002951W WO 2007045989 A1 WO2007045989 A1 WO 2007045989A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- pharmaceutically acceptable
- general formula
- alkyl
- inhibitors
- Prior art date
Links
- SKCNYHLTRZIINA-UHFFFAOYSA-N ClCc(cn1)ccc1Cl Chemical compound ClCc(cn1)ccc1Cl SKCNYHLTRZIINA-UHFFFAOYSA-N 0.000 description 1
- SZTLDEFMPWLFOE-UHFFFAOYSA-N N#CC1(CCOCC1)c(cn1)ccc1Cl Chemical compound N#CC1(CCOCC1)c(cn1)ccc1Cl SZTLDEFMPWLFOE-UHFFFAOYSA-N 0.000 description 1
- BEYUBHYVZOYVBK-UHFFFAOYSA-N N#CC1(CCOCC1)c(cn1)ccc1OCCCN1CCCC1 Chemical compound N#CC1(CCOCC1)c(cn1)ccc1OCCCN1CCCC1 BEYUBHYVZOYVBK-UHFFFAOYSA-N 0.000 description 1
- PMCPJUYOAKGHTO-UHFFFAOYSA-N N#CC1(CCOCC1)c1ccc[n+]([O-])c1 Chemical compound N#CC1(CCOCC1)c1ccc[n+]([O-])c1 PMCPJUYOAKGHTO-UHFFFAOYSA-N 0.000 description 1
- GFDQXRQBDKIUEK-UHFFFAOYSA-N N#CC1(CCOCC1)c1cccnc1 Chemical compound N#CC1(CCOCC1)c1cccnc1 GFDQXRQBDKIUEK-UHFFFAOYSA-N 0.000 description 1
- BLGUCBUETMYJTB-UHFFFAOYSA-N N#CCc(cn1)ccc1Cl Chemical compound N#CCc(cn1)ccc1Cl BLGUCBUETMYJTB-UHFFFAOYSA-N 0.000 description 1
- OIPHWUPMXHQWLR-UHFFFAOYSA-N N#CCc1cccnc1 Chemical compound N#CCc1cccnc1 OIPHWUPMXHQWLR-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
Definitions
- the present invention relates to pyridyl derivatives of general formula:
- R and X have the meanings indicated below, and to processes for the preparation of, intermediates used in the preparation of, compositions containing and the uses of, such derivatives.
- Histamine H 3 receptors are found inter alia on presynaptic terminals of peripheral nerves, where they modulate autonomic neurotransmission and modulate a variety of end organ responses under control of the autonomic nervous system. They are also heteroreceptors, modulating the release of numerous other neurotransmitters such as dopamine, glutamate, noradrenaline, serotonin, GABA, acetylcholine, some peptides and co-transmitters.
- Histamine H 3 receptor ligands Recently numerous histamine H 3 receptor ligands have been developed. An overview of the current advance in H 3 ligand research and patenting is given in Expert Opin. Ther. Patents (2003) 13(6). Examples of Histamine H 3 receptor ligands can be found in WO02/76925, WO00/06254, WO02/12190, WO02/12214 and WO02/06223.
- H 3 receptor ligands are believed to be suitable for the treatment of various diseases including both disorders of the central nervous system and inflammatory disorders.
- diseases where treatment with H 3 ligands is believed to be useful are obesity, inflammatory bowel disease, Crohn's disease, colitis ulcerosa, sleep disorders, migraine, dyskinesia, stress-induced anxiety, psychotic disorders, epilepsy, Cognition deficiency diseases such as Alzheimer's disease or mild coginitive impairment, depression, mood disorders, schizophrenia, anxiety disorders, attention-deficit hyperactivity disorder (ADHD), psychotic disorders, obesity, dizziness, epilepsy, motion sickness, vertigo, respiratory diseases such as adult respiratory distress syndrome, acute respiratory distress syndrome, bronchitis, chronic bronchitis, chronic obstructive pulmonary disease, cystic fibrosis, asthma, emphysema, rhinitis, chronic sinusitis, allergy, allergy-induced airway responses, allergic rhinitis, viral rhinitis, non-allergic
- H 3 ligands are known there is still a need to provide new H 3 ligands that are good drug candidates.
- preferred compounds should bind potently to the histamine H 3 receptor whilst showing little affinity for other receptors. They should be well absorbed from the gastrointestinal tract, be metabolically stable and possess favourable pharmacokinetic properties. They should be non-toxic and demonstrate few side-effects.
- the present invention concerns new pyridyl derivatives of general formula (1) : i or a pharmaceutically acceptable salt and/or solvate (including hydrate) thereof wherein :
- X is selected from -CN, -CH 2 NR 1 R 2 , -C(O)NR 3 R 4 , -CH 2 -het 1 and net 2 , the groups net 1 and het 2 being optionally substituted by one or two substituents independently selected from halo, cyano, (Ci-C 4 )alkyl and (C r C 4 )alkoxy;
- R 1 and R 2 are each independently selected from hydrogen, (Ci-C 4 )alkyl and (C 3 -C 6 )cycloalkyl, said (C r C 4 )alkyl and (C 3 -C 6 )cycloalkyl being optionally substituted by one group selected from (C r C 4 )alkoxy and (C 3 -C 6 )cycloalkyl, or alternatively R 1 and R 2 together with the nitrogen atom to which they are attached form a A-, 5-, 6- or 7-membered saturated heterocycle wherein one carbon atom may be optionally replaced by a nitrogen or an oxygen atom, and wherein said saturated heterocycle is optionally substituted by one or two groups independently selected from (C r C 4 )alkyl, (C r C 4 )alkoxy or -C(O)(C r C 4 )alkyl;
- R 3 and R 4 are each independently selected from hydrogen, (C r C 4 )alkyl and (C 3 -C 6 )cycloalkyl, said (C 1 - C 4 )alkyl and (C 3 -C 6 )cycloalkyl being optionally substituted by one group selected from (C r C 4 )alkoxy and (C 3 -C 6 )cycloalkyl, or alternatively R 3 and R 4 together with the nitrogen atom to which they are attached form a A-, 5-, 6- or 7-membered saturated heterocycle wherein one carbon atom may be optionally replaced by a nitrogen or an oxygen atom, and wherein said saturated heterocycle is optionally substituted by one or two groups independently selected from (C r C 4 )alkyl, (C r C 4 )alkoxy or -C(O)(C r C 4 )alkyl;
- het 1 is N-linked and is selected from monocyclic or bicyclic heteroaromatic groups having 5, 6, 7, 8 or 9 ring members, which contain 1, 2, 3 or 4 heteroatom(s) selected from nitrogen, oxygen and sulphur;
- het 2' is C-linked and is selected from monocyclic or bicyclic heteroaromatic groups having 5, 6, 7, 8, 9 or 10 ring members, which contain 1 , 2, 3 or 4 heteroatom(s) selected from nitrogen, oxygen and sulphur;
- R is either a group of formula :
- * ⁇ represents the attachment point to the oxygen atom
- the N-containing ring is a 4-, 5-, 6- or 7- membered saturated heterocycle
- n is an integer equal to 0, 1 or 2
- R 7 represents a substituent selected from hydrogen, (CrC 4 )alkyl, and (C 3 -C 6 )cycloalkyl.
- H 3 ligands are H 3 ligands and are thus particularly useful for the treatment of H 3 -related diseases such as neurologic disorders, or inflammatory, respiratory and allergic diseases, disorders and conditions.
- Halo denotes a halogen atom selected from the group consisting of fluoro, chloro, bromo and iodo.
- (C 1 -C 4 )all ⁇ yl denotes a saturated, straight-chain or branched hydrocarbon group having from 1 to 4 carbon atoms and includes for example methyl, ethyl, propyl, iso-propyl, n-butyl, iso-butyl, sec-butyl and tert-butyl. This also applies if the alkyl group carries substituents or is a substituent for another group, e.g. in (C r C 4 )alkoxy and -C(O)(C r C 4 )alkyl.
- (C r C 4 )alkoxy denotes straight-chain and branched alkoxy groups and includes for example methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, iso-butoxy, sec-butoxy and tert-butoxy.
- (C 2 -C 6 )alkylene denotes a divalent radical derived from straight-chain or branched alkane containing from 2 to 6 carbon atoms.
- Examples of (C 2 -C 6 )alkylene radicals are methylene, ethylene (1,2-ethylene or 1,1 -ethylene), trimethylene (1,3-propylene), tetramethylene (1 ,4-butylene), pentamethylene and hexamethylene.
- (C 3 -C 6 )cycloalkyl denotes a saturated monocyclic carbocyclic group having 3, 4, 5 or 6 carbon atoms and includes for example cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl.
- saturated heterocycle denotes a saturated monocyclic group having 4, 5, 6 or 7 ring members, which contains one nitrogen atom.
- saturated heterocycles are azetidinyl, pyrrolidinyl and piperidinyl.
- Het 1 and het 2 are monocyclic or bicyclic heteroaromatic groups having 5, 6, 7, 8 or 9 and 5, 6, 7, 8, 9 or 10 ring members respectively, which contain 1 , 2, 3 or 4 heteroatom(s) selected from nitrogen, oxygen and sulphur.
- Het 1 is N-linked, which means that the group is linked to the adjacent atom by a ring nitrogen atom.
- Het 2 is C-linked, which means that the group is linked to the adjacent atom by a ring carbon atom.
- the heteroaromatic group can be unsubstituted, monosubstituted or disubstituted, as indicated in the definition of X hereabove for general formula (1) according to the present invention.
- heteroaryl groups include, but are not limited to thiophenyl, furanyl, pyrrolyl, pyrazolyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, triazolyl, oxadiazolyl, thiadiazolyl, tetrazolyl, pyranyl, pyridinyl, pyrazinyl, pyrimidinyl, pyridazinyl, triazinyl, thiadiazinyl, isobenzofuranyl, benzofuranyl, chromenyl, indolizinyl, isoindolyl, indolyl, indazolyl, purinyl, quinolinyl, isoquinolyl, cinnolinyl, phthalazinyl, naphthyridinyl, quinazolinyl, quinoxalinyl, benzoxazolyl, be
- X is selected from -CN, -CH 2 NR 1 R 2 , -CH 2 -het 1 and het 2 , het 2 being optionally substituted once by (Ci-C 4 )alkyl, wherein R 1 , R 2 , het 1 and het 2 are as previously defined.
- X is selected from -CN, -CH 2 NR 1 R 2 , -CH 2 -het 1 and het 2 , het 2 being optionally substituted once by (Ci-C 4 )alkyl, wherein:
- R 1 and R 2 are both hydrogen atoms or alternatively R 1 and R 2 together with the nitrogen atom to which they are attached form a A-, 5-, 6- or 7-membered saturated heterocycle wherein one carbon atom may be optionally replaced by a nitrogen or an oxygen atom; het 1 is a N-linked bicyclic heteroaromatic group having 9 ring members which contain a total of two nitrogen atoms; and het 2 is a C-linked monocyclic heteroaromatic group having 5 ring members which contain one nitrogen atom and one sulphur atom.
- X is selected from -CN, -CH 2 -NH 2 , -CH 2 -morpholine, -CH 2 -benzimidazole and 1,3- thiazol-2-yl optionally substituted by a (C r C 4 )alkyl group, preferably a methyl group.
- R is a group of formula
- R 5 and R s together with the nitrogen atom to which they are attached form a 5- or 6- membered saturated heterocycle.
- the saturated heterocycle is a pyrrolidinyl group.
- L is propylene.
- Particularly preferred compounds of the invention include those in which each variable in formula (1) is selected from the suitable and/or preferred groups for each variable. Even more preferable compounds of the invention include those where each variable in formula (1) is selected from the more preferred or most preferred groups for each variable.
- the compounds of formula (1) are selected from:
- Another embodiment of the invention is an intermediate as described herein.
- Pharmaceutically acceptable salts of the compounds of formula (1) include the acid addition and base salts thereof.
- Suitable acid addition salts are formed from acids which form non-toxic salts. Examples include the acetate, aspartate, benzoate, besylate, bicarbonate/carbonate, bisulphate/sulphate, borate, camsylate, citrate, edisylate, esylate, formate, fumarate, gluceptate, gluconate, glucuronate, hexafluorophosphate, hibenzate, hydrochloride/chloride, hydrobromide/bromide, hydroiodide/iodide, isethionate, lactate, malate, maleate, malonate, mesylate, methylsulphate, naphthylate, 2-napsylate, nicotinate, nitrate, orotate, oxalate, palmitate, pamoate, phosphate/hydrogen phosphate/dihydrogen phosphate, saccharate, stearate, succinate, tartrate, tosylate and trifluor
- Suitable base salts are formed from bases which form non-toxic salts. Examples include the aluminium, arginine, benzathine, calcium, choline, diethylamine, diolamine, glycine, lysine, magnesium, meglumine, olamine, potassium, sodium, tromethamine and zinc salts.
- Hemisalts of acids and bases may also be formed, for example, hemisulphate and hemicalcium salts.
- salts of compounds of formula (1) may be prepared by one or more of three methods:
- the resulting salt may precipitate out and be collected by filtration or may be recovered by evaporation of the solvent.
- the degree of ionisation in the resulting salt may vary from completely ionised to almost non-ionised.
- the compounds of the invention may exist in both unsolvated and solvated forms.
- 'solvate' is used herein to describe a molecular complex comprising the compound of the invention and a stoichiometric amount of one or more pharmaceutically acceptable solvent molecules, for example, ethanol.
- solvent molecules for example, ethanol.
- 'hydrate' is employed when said solvent is water.
- complexes such as clathrates, drug-host inclusion complexes wherein, in contrast to the aforementioned solvates, the drug and host are present in stoichiometric or non-stoichiometric amounts.
- complexes of the drug containing two or more organic and/or inorganic components which may be in stoichiometric or non-stoichiometric amounts.
- the resulting complexes may be ionised, partially ionised, or non-ionised.
- references to compounds of formula (1) include references to salts, solvates and complexes thereof and to solvates and complexes of salts thereof.
- the compounds of the invention include compounds of formula (1) as hereinbefore defined, including all polymorphs and crystal habits thereof, prodrugs and isomers thereof (including optical, geometric and tautomeric isomers) as hereinafter defined and isotopically-labeled compounds of formula (1).
- 'pro-drugs' of the compounds of formula (1) are also within the scope of the invention.
- certain derivatives of compounds of formula (1) which may have little or no pharmacological activity themselves can, when administered into or onto the body, be converted into compounds of formula (1) having the desired activity, for example, by hydrolytic cleavage.
- Such derivatives are referred to as 'prodrugs'.
- Further information on the use of prodrugs may be found in 'Prodrugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series (T. Higuchi and W. Stella) and 'Bioreversible Carriers in Drug Design', Pergamon Press, 1987 (ed. E. B Roche, American Pharmaceutical Association).
- Prodrugs in accordance with the invention can, for example, be produced by replacing appropriate functionalities present in the compounds of formula (1) with certain moieties known to those skilled in the art as 'pro-moieties' as described, for example, in "Design of Prodrugs" by H. Bundgaard (Elsevier, 1985).
- prodrugs in accordance with the invention include, where the compound of general formula (1) contains a primary or secondary amino functionality (-NH 2 or -NHR where R ⁇ H), an amide thereof, for example, a compound wherein, as the case may be, one or both hydrogens of the amino functionality of the compound of formula (1) is/are replaced by (C r Ci 0 )alkanoyl.
- metabolites of compounds of formula (1) that is, compounds formed in vivo upon administration of the drug.
- Some examples of metabolites in accordance with the invention include : (i) where the compound of formula (1) contains a methyl group, an hydroxymethyl derivative thereof
- stereoisomers, geometric isomers and tautomeric forms of the compounds of formula (1) including compounds exhibiting more than one type of isomerism, and mixtures of one or more thereof.
- acid addition or base salts wherein the counterion is optically active for example, d-lactate or /-lysine, or racemic, for example, d/-tartrate or dl- arginine.
- Conventional techniques for the preparation/isolation of individual enantiomers include chiral synthesis from a suitable optically pure precursor or resolution of the racemate (or the racemate of a salt or derivative) using, for example, chiral high pressure liquid chromatography (HPLC),
- the racemate (or a racemic precursor) may be reacted with a suitable optically active compound, for example, an alcohol, or, in the case where the compound of formula (1) contains an acidic or basic moiety, an acid or base such as tartaric acid or 1-phenylethylamine.
- a suitable optically active compound for example, an alcohol, or, in the case where the compound of formula (1) contains an acidic or basic moiety, an acid or base such as tartaric acid or 1-phenylethylamine.
- the resulting diastereomeric mixture may be separated by chromatography and/or fractional crystallization and one or both of the diastereoisomers converted to the corresponding pure enantiomer(s) by means well known to a skilled person.
- Chiral compounds of the invention may be obtained in enantiomerically- enriched form using chromatography, typically HPLC, on an asymmetric resin with a mobile phase consisting of a hydrocarbon, typically heptane or hexane, containing from 0 to 50% by volume of isopropanol, typically from 2% to 20%, and from 0 to 5% by volume of an alkylamine, typically 0.1% diethylamine. Concentration of the eluate affords the enriched mixture.
- chromatography typically HPLC
- a mobile phase consisting of a hydrocarbon, typically heptane or hexane, containing from 0 to 50% by volume of isopropanol, typically from 2% to 20%, and from 0 to 5% by volume of an alkylamine, typically 0.1% diethylamine.
- Stereoisomeric conglomerates may be separated by conventional techniques known to those skilled in the art - see, for example, "Stereochemistry of Organic Compounds" by E. L. Eliel (Wiley, New York, 1994).
- the present invention includes all pharmaceutically acceptable isotopically-labelled compounds of formula (1) wherein one or more atoms are replaced by atoms having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number which predominates in nature.
- isotopes suitable for inclusion in the compounds of the invention include isotopes of hydrogen, such as 2 H and 3 H, carbon, such as 11 C, 13 C and 14 C, chlorine, such as 36 CI, fluorine, such as 18 F, iodine, such as 123 I and 125 I, nitrogen, such as 13 N and 15 N, oxygen, such as 15 O, 17 O and 18 O, phosphorus, such as 32 P, and sulphur, such as 35 S.
- isotopically-labelled compounds of formula (1) for example, those incorporating a radioactive isotope, are useful in drug and/or substrate tissue distribution studies.
- substitution with heavier isotopes such as deuterium, i.e. 2 H, may afford certain therapeutic advantages resulting from greater metabolic stability, for example, increased in vivo half-life or reduced dosage requirements, and hence may be preferred in some circumstances.
- Isotopically-labeled compounds of formula (1) can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described in the accompanying Examples and Preparations using an appropriate isotopically-labeled reagents in place of the non-labeled reagent previously employed.
- solvates in accordance with the invention include those wherein the solvent of crystallization may be isotopically substituted, e.g. D 2 O 1 d 6 -acetone, d s -DMSO.
- Compounds of general formula (1) wherein X is -CN may be prepared in step (vi) by reacting 4-(6- chloropyridin-3-yl)tetrahydro-2H-pyran-4-carbonitrile with ROH and a suitable base (e.g. NaH or KOtBu) in a suitable solvent (e.g. THF) at temperatures between room temperature and reflux, typically at room temperature.
- ROH a suitable base
- THF suitable solvent
- 4-(6-Chloropyridin-3-yl)tetrahydro-2H-pyran-4-carbonitrile may be prepared by several methods, two of which are illustrated in scheme 1: Sequential tetrahydro-2H-pyran construction ((i) e.g. with a 2-haloethyl ether such as 2-bromoethylether and a base such as NaH, optionally with Kl, in a suitable solvent such as THF at temperatures between O 0 C room temperature), N-oxidation ((H) e.g.
- a suitable oxidising agent such as oxone and a base such as sodium bicarbonate in a suitable solvent such as aqueous acetone at temperatures between 0°C and room temperature
- halogenation (Hi) e.g. with a chlorinating agent such as POCI 3 optionally with a co-solvent such as acetonitrile at temperatures between room temperature and reflux).
- sequential cyanation (iv) e.g. with NaCN in a suitable solvent such as aqueous EtOH at temperatures between room temperature and reflux)
- tetrahydro-2H-pyran construction (v) e.g. with a 2-haloethyl ether such as 2-bromoethylether and a base such as NaH, optionally with Kl, in a suitable solvent such as THF at temperatures between O 0 C and room temperature).
- pyridyl derivatives of general formula (1) wherein R is as previsouly defined in general formula (1) and X is not -CN may be prepared from analogous pyridyl derivatives of formula (1) wherein X is -CN (preparation described above) using conventional procedures exemplified but not limited by those illustrated in scheme 2:
- X is het 2 , and het 2 is ally substituted with
- Compounds of general formula (1) wherein X is C(O)NR 3 R 4 may be prepared from compounds of general formula (2) in step (viii) by using standard techniques for amide coupling.
- Conditions for step (viii) include sequential treatment of compounds of general formula (2) with a reagent capable of activating a carboxylic acid (e.g. thionyl chloride) and then with the amino derivative of formula HNR 3 R 4 , in a suitable solvent (e.g. dichloromethane) at between O 0 C and reflux.
- a suitable solvent e.g. dichloromethane
- the acid may be treated with the amine of formula HNR 3 R 4 , in the presence of a coupling agent (e.g.
- TBTU O-benzotriazol-1-yl-N,N,N',N'- tetramethyluronium hexafluorophosphate, 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride), optionally in the presence of a suitable additive (e.g. 1-hydroxybenzotriazole) and base (e.g. triethylamine) in a suitable solvent (e.g. dichloromethane, N,N-dimethylformamide) at room temperature (about 20 0 C).
- a suitable additive e.g. 1-hydroxybenzotriazole
- base e.g. triethylamine
- a suitable solvent e.g. dichloromethane, N,N-dimethylformamide
- Compounds of general formula (2) may be prepared from compounds of general formula (1) wherein X is -CN in step (vii) by using standard techniques for nitrile hydrolysis. Conditions for step (vii) include treatment of the nitrile compound with hydrochloric acid, optionally in the presence of a co-solvent such as dioxane, at temperatures between room temperature and reflux.
- a co-solvent such as dioxane
- Compounds of general formula (1) wherein X is net 2 may be prepared from compounds of general formula (1) wherein X is -CN using multistep procedures known to those skilled in the art such as those described in general heterocyclic texts such as Heterocyclic Chemistry, JA Joule and K. Mills, 4 th Edition, Blackwell publishing, 2000 or Comprehensive Heterocyclic chemistry I and II, Pergamon Press.
- This is exemplified in scheme 2 by the formation of compounds of general formula (1) wherein X is het 2 and het 2 is thiazole optionally substituted with (CrC ⁇ alkyl:
- These compounds can be prepared from compounds of general formula (3) in step (x) by treatment with a suitable reagent (e.g. bromoacetaldehyde dimethyl acetal or chloroacetone) optionally with an acid (e.g. HBr or HCI) in a suitable solvent (e.g. EtOH) at temperatures between room temperature and reflux.
- a suitable reagent e
- Compounds of general formula (3) may be prepared from compounds of general formula (1) wherein X is -CN in step (ix) by treatment with diethyldithiophosphate and water at temperatures between room temperature and reflux, typically at 60 0 C.
- alkylations with alkyl halides and a base e.g. potassium carbonate, potassium ferf-butoxide
- a suitable solvent e.g. acetonitrile, tetrahydrofuran
- Compounds of general formula (1) wherein X is -CH 2 NH 2 can be prepared from compounds of general formula (1) wherein X is -CN in step (xi) by using a suitable reducing agent (e.g. LiAIH 4 or hydrogen gas in the presence of a catalyst such as PtO 2 ) in a suitable solvent (e.g. Et 2 O, dichloromethane, tetrahydrofuran or isopropanol) at temperatures between room temperature and reflux.
- a suitable reducing agent e.g. LiAIH 4 or hydrogen gas in the presence of a catalyst such as PtO 2
- a suitable solvent e.g. Et 2 O, dichloromethane, tetrahydrofuran or isopropanol
- Compounds of general formula (4) can be prepared from compounds of general formula (1) wherein X is -CH 2 NH 2 in step (xiii) by reaction with 2-halo-nitrobenzene (e.g. 2-fluoro-nitrobenzene) and a base (e.g. potassium carbonate) in a suitable solvent (e.g. tetrahydrofuran) at temperatures between room temperature and reflux.
- 2-halo-nitrobenzene e.g. 2-fluoro-nitrobenzene
- a base e.g. potassium carbonate
- a suitable solvent e.g. tetrahydrofuran
- Suitable protecting groups for hydroxyl include trialkylsilyl and diarylalkylsilyl (e.g. tert-butyldimethylsilyl, ferf-butyldiphenylsilyl or trimethylsilyl), alkyl (e.g. methyl or methoxyethyl) and tetrahydropyranyl.
- Suitable protecting groups for amino include fert-butyloxycarbonyl, 9-fluorenylmethoxycarbony! or benzyloxycarbonyl.
- Suitable protecting groups for carboxylic acid include (C r C 4 )alkyl or benzyl esters.
- the compounds of formula (1) as well as intermediates for the preparation thereof can be purified according to various well-known methods such as recrystallisation and chromatography.
- the compounds of formula (1) are valuable pharmaceutically active compounds, which are suitable for the therapy and prophylaxis of numerous disorders in which the histamine H 3 receptor is involved or in which agonism or antagonism of this receptor may induce benefit.
- Compounds of the invention intended for pharmaceutical use may be administered as crystalline or amorphous products. They may be obtained, for example, as solid plugs, powders, or films by methods such as precipitation, crystallization, freeze drying, spray drying, or evaporative drying. Microwave or radio frequency drying may be used for this purpose. They may be administered alone or in combination with one or more other compounds of the invention or in combination with one or more other drugs (or as any combination thereof). Generally, they will be administered as a formulation in association with one or more pharmaceutically acceptable excipients.
- the term 'excipient' is used herein to describe any ingredient other than the compound(s) of the invention. The choice of excipient will to a large extent depend on factors such as the particular mode of administration, the effect of the excipient on solubility and stability, and the nature of the dosage form.
- a pharmaceutical composition including a compound of the formula (1) or a pharmaceutically acceptable salt and/or solvate thereof, as defined in any one of the preceding claims, together with a pharmaceutically acceptable excipient.
- compositions suitable for the delivery of compounds of the present invention and methods for their preparation will be readily apparent to those skilled in the art. Such compositions and methods for their preparation may be found, for example, in Remington's Pharmaceutical Sciences. 19th Edition (Mack Publishing Company, 1995).
- the compounds of the invention may be administered orally.
- Oral administration may involve swallowing, so that the compound enters the gastrointestinal tract, or buccal or sublingual administration may be employed by which the compound enters the blood stream directly from the mouth.
- Formulations suitable for oral administration include solid formulations such as tablets, capsules containing particulates, liquids, or powders, lozenges (including liquid-filled), chews, multi- and nano-particulates, gels, solid solution, liposome, films, ovules, sprays and liquid formulations.
- Liquid formulations include suspensions, solutions, syrups and elixirs. Such formulations may be employed as fillers in soft or hard capsules and typically comprise a carrier, for example, water, ethanol, polyethylene glycol, propylene glycol, methylcellulose, or a suitable oil, and one or more emulsifying agents and/or suspending agents. Liquid formulations may also be prepared by the reconstitution of a solid, for example, from a sachet.
- the compounds of the invention may also be used in fast-dissolving, fast-disintegrating dosage forms such as those described in Expert Opinion in Therapeutic Patents, H (6), 981-986, by Liang and Chen (2001).
- the drug may make up from 1 weight % to 80 weight % of the dosage form, more typically from 5 weight % to 60 weight % of the dosage form.
- tablets generally contain a disintegrant.
- disintegrants include sodium starch glycolate, sodium carboxymethyl cellulose, calcium carboxymethyl cellulose, croscarmellose sodium, crospovidone, polyvinylpyrrolidone, methyl cellulose, microcrystalline cellulose, lower alkyl-substituted hydroxypropyl cellulose, starch, pregelatinised starch and sodium alginate.
- the disintegrant will comprise from 1 weight % to 25 weight %, preferably from 5 weight % to 20 weight % of the dosage form.
- Binders are generally used to impart cohesive qualities to a tablet formulation. Suitable binders include microcrystalline cellulose, gelatin, sugars, polyethylene glycol, natural and synthetic gums, polyvinylpyrrolidone, pregelatinised starch, hydroxypropyl cellulose and hydroxypropyl methylcellulose. Tablets may also contain diluents, such as lactose (monohydrate, spray-dried monohydrate, anhydrous and the like), mannitol, xylitol, dextrose, sucrose, sorbitol, microcrystalline cellulose, starch and dibasic calcium phosphate dihydrate.
- lactose monohydrate, spray-dried monohydrate, anhydrous and the like
- mannitol xylitol
- dextrose sucrose
- sorbitol microcrystalline cellulose
- starch dibasic calcium phosphate dihydrate
- Tablets may also optionally comprise surface active agents, such as sodium lauryl sulfate and polysorbate 80, and glidants such as silicon dioxide and talc.
- surface active agents such as sodium lauryl sulfate and polysorbate 80
- glidants such as silicon dioxide and talc.
- surface active agents may comprise from 0.2 weight % to 5 weight % of the tablet, and glidants may comprise from 0.2 weight % to 1 weight % of the tablet.
- Tablets also generally contain lubricants such as magnesium stearate, calcium stearate, zinc stearate, sodium stearyl fumarate, and mixtures of magnesium stearate with sodium lauryl sulphate.
- Lubricants generally comprise from 0.25 weight % to 10 weight %, preferably from 0.5 weight % to 3 weight % of the tablet.
- ingredients include anti-oxidants, colourants, flavouring agents, preservatives and taste- masking agents.
- Exemplary tablets contain up to about 80% drug, from about 10 weight % to about 90 weight % binder, from about 0 weight % to about 85 weight % diluent, from about 2 weight % to about 10 weight % disintegrant, and from about 0.25 weight % to about 10 weight % lubricant.
- Tablet blends may be compressed directly or by roller to form tablets. Tablet blends or portions of blends may alternatively be wet-, dry-, or melt-granulated, melt congealed, or extruded before tabletting.
- the final formulation may comprise one or more layers and may be coated or uncoated; it may even be encapsulated.
- Consumable oral films for human or veterinary use are typically pliable water-soluble or water-swellable thin film dosage forms which may be rapidly dissolving or mucoadhesive and typically comprise a compound of formula (1), a film-forming polymer, a binder, a solvent, a humectant, a plasticiser, a stabiliser or emulsifier, a viscosity-modifying agent and a solvent. Some components of the formulation may perform more than one function.
- the compound of formula (1) may be water-soluble or insoluble.
- a water-soluble compound typically comprises from 1 weight % to 80 weight %, more typically from 20 weight % to 50 weight %, of the solutes. Less soluble compounds may comprise a greater proportion of the composition, typically up to 88 weight % of the solutes.
- the compound of formula (1) may be in the form of multiparticulate beads.
- the film-forming polymer may be selected from natural polysaccharides, proteins, or synthetic hydrocolloids and is typically present in the range 0.01 to 99 weight %, more typically in the range 30 to 80 weight %.
- ingredients include anti-oxidants, colorants, flavourings and flavour enhancers, preservatives, salivary stimulating agents, cooling agents, co-solvents (including oils), emollients, bulking agents, anti-foaming agents, surfactants and taste-masking agents.
- Films in accordance with the invention are typically prepared by evaporative drying of thin aqueous films coated onto a peelable backing support or paper. This may be done in a drying oven or tunnel, typically a combined coater dryer, or by freeze-drying or vacuuming.
- Solid formulations for oral administration may be formulated to be immediate and/or modified release.
- Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
- Suitable modified release formulations for the purposes of the invention are described in US Patent No. 6,106,864. Details of other suitable release technologies such as high energy dispersions and osmotic and coated particles are to be found in Pharmaceutical Technology On-line. 25(2), 1-14, by Verma et a/ (2001). The use of chewing gum to achieve controlled release is described in WO 00/35298.
- the compounds of the invention may also be administered directly into the blood stream, into muscle, or into an internal organ.
- Suitable means for parenteral administration include intravenous, intraarterial, intraperitoneal, intrathecal, intraventricular, intraurethral, intrastemal, intracranial, intramuscular and subcutaneous.
- Suitable devices for parenteral administration include needle (including microneedle) injectors, needle-free injectors and infusion techniques.
- Parenteral formulations are typically aqueous solutions which may contain excipients such as salts, carbohydrates and buffering agents (preferably to a pH of from 3 to 9), but, for some applications, they may be more suitably formulated as a sterile non-aqueous solution or as a dried form to be used in conjunction with a suitable vehicle such as sterile, pyrogen-free water.
- excipients such as salts, carbohydrates and buffering agents (preferably to a pH of from 3 to 9)
- a suitable vehicle such as sterile, pyrogen-free water.
- parenteral formulations under sterile conditions may readily be accomplished using standard pharmaceutical techniques well known to those skilled in the art.
- solubility of compounds of formula (1) used in the preparation of parenteral solutions may be increased by the use of appropriate formulation techniques, such as the incorporation of solubility- enhancing agents.
- Formulations for parenteral administration may be formulated to be immediate and/or modified release.
- Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
- compounds of the invention may be formulated as a solid, semi-solid, or thixotropic liquid for administration as an implanted depot providing modified release of the active compound.
- examples of such formulations include drug-coated stents and poly(cf/-lactic-coglycolic)acid (PGLA) microspheres.
- the compounds of the invention may also be administered topically to the skin or mucosa, that is, dermally or transdermally.
- Typical formulations for this purpose include gels, hydrogels, lotions, solutions, creams, ointments, dusting powders, dressings, foams, films, skin patches, wafers, implants, sponges, fibres, bandages and microemulsions. Liposomes may also be used.
- Typical carriers include alcohol, water, mineral oil, liquid petrolatum, white petrolatum, glycerin, polyethylene glycol and propylene glycol. Penetration enhancers may be incorporated - see, for example, J Pharm Sci, 88 (10), 955-958, by Finnin and Morgan (October 1999).
- topical administration include delivery by electroporation, iontophoresis, phonophoresis, sonophoresis and microneedle or needle-free (e.g. PowderjectTM, BiojectTM, etc.) injection.
- Formulations for topical administration may be formulated to be immediate and/or modified release.
- Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
- the compounds of the invention can also be administered intranasally or by inhalation, typically in the form of a dry powder (either alone, as a mixture, for example, in a dry blend with lactose, or as a mixed component particle, for example, mixed with phospholipids, such as phosphatidylcholine) from a dry powder inhaler or as an aerosol spray from a pressurised container, pump, spray, atomiser (preferably an atomiser using electrohydrodynamics to produce a fine mist), or nebuliser, with or without the use of a suitable propellant, such as 1,1 ,1 ,2-tetrafluoroethane or 1 ,1,1,2,3,3,3-heptafluoropropane.
- the powder may comprise a bioadhesive agent, for example, chitosan or cyclodextrin.
- the pressurised container, pump, spray, atomizer, or nebuliser contains a solution or suspension of the compound(s) of the invention comprising, for example, ethanol, aqueous ethanol, or a suitable alternative agent for dispersing, solubilising, or extending release of the active, a propellant(s) as solvent and an optional surfactant, such as sorbitan trioleate, oleic acid, or an oligolactic acid.
- a solution or suspension of the compound(s) of the invention comprising, for example, ethanol, aqueous ethanol, or a suitable alternative agent for dispersing, solubilising, or extending release of the active, a propellant(s) as solvent and an optional surfactant, such as sorbitan trioleate, oleic acid, or an oligolactic acid.
- the drug product Prior to use in a dry powder or suspension formulation, the drug product is micronised to a size suitable for delivery by inhalation (typically less than 5 microns). This may be achieved by any appropriate comminuting method, such as spiral jet milling, fluid bed jet milling, supercritical fluid processing to form nanoparticles, high pressure homogenisation, or spray drying.
- Capsules (made, for example, from gelatin or hydroxypropylmethylcellulose), blisters and cartridges for use in an inhaler or insufflator may be formulated to contain a powder mix of the compound of the invention, a suitable powder base such as lactose or starch and a performance modifier such as /-leucine, mannitol, or magnesium stearate.
- the lactose may be anhydrous or in the form of the monohydrate, preferably the latter.
- suitable excipients include dextran, glucose, maltose, sorbitol, xylitol, fructose, sucrose and trehalose.
- a suitable solution formulation for use in an atomiser using electrohydrodynamics to produce a fine mist may contain from 1 ⁇ g to 20mg of the compound of the invention per actuation and the actuation volume may vary from 1 ⁇ l to 100 ⁇ l.
- a typical formulation may comprise a compound of formula (1), propylene glycol, sterile water, ethanol and sodium chloride.
- Alternative solvents which may be used instead of propylene glycol include glycerol and polyethylene glycol.
- Suitable flavours such as menthol and levomenthol, or sweeteners, such as saccharin or saccharin sodium, may be added to those formulations of the invention intended for inhaled/intranasal administration.
- Formulations for inhaled/intranasal administration may be formulated to be immediate and/or modified release using, for example, PGLA.
- Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
- the dosage unit is determined by means of a valve which delivers a metered amount.
- Units in accordance with the invention are typically arranged to administer a metered dose or "puff' containing from 1 ⁇ g to 4000 ⁇ g of the compound of formula (1).
- the overall daily dose will typically be in the range 1 ⁇ g to 20 mg which may be administered in a single dose or, more usually, as divided doses throughout the day.
- the compounds of the invention may be administered rectally or vaginally, for example, in the form of a suppository, pessary, or enema.
- Cocoa butter is a traditional suppository base, but various alternatives may be used as appropriate.
- Formulations for rectal/vaginal administration may be formulated to be immediate and/or modified release.
- Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
- the compounds of the invention may also be administered directly to the eye or ear, typically in the form of drops of a micronised suspension or solution in isotonic, pH-adjusted, sterile saline.
- Other formulations suitable for ocular and aural administration include ointments, biodegradable (e.g. absorbable gel sponges, collagen) and non-biodegradable (e.g. silicone) implants, wafers, lenses and particulate or vesicular systems, such as niosomes or liposomes.
- a polymer such as crossed-linked polyacrylic acid, polyvinylalcohol, hyaluronic acid, a cellulosic polymer, for example, hydroxypropylmethylcellulose, hydroxyethylcellulose, or methyl cellulose, or a heteropolysaccharide polymer, for example, gelan gum, may be incorporated together with a preservative, such as benzalkonium chloride.
- a preservative such as benzalkonium chloride.
- Such formulations may also be delivered by iontophoresis.
- Formulations for ocular/aural administration may be formulated to be immediate and/or modified release.
- Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted, or programmed release.
- the compounds of the invention may be combined with soluble macromolecular entities, such as cyclodextrin and suitable derivatives thereof or polyethylene glycol-containing polymers, in order to improve their solubility, dissolution rate, taste-masking, bioavailability and/or stability for use in any of the aforementioned modes of administration.
- soluble macromolecular entities such as cyclodextrin and suitable derivatives thereof or polyethylene glycol-containing polymers
- Drug-cyclodextrin complexes are found to be generally useful for most dosage forms and administration routes. Both inclusion and non-inclusion complexes may be used.
- the cyclodextrin may be used as an auxiliary additive, i.e. as a carrier, diluent, or solubiliser. Most commonly used for these purposes are alpha-, beta- and gamma- cyclodextrins, examples of which may be found in International Patent Applications Nos. WO 91/11172, WO 94/02518 and WO 98/55148.
- compositions may conveniently be combined in the form of a kit suitable for coadministration of the compositions.
- the kit of the invention comprises two or more separate pharmaceutical compositions, at least one of which contains a compound of formula . (1) in accordance with the invention, and means for separately retaining said compositions, such as a container, divided bottle, or divided foil packet.
- a container, divided bottle, or divided foil packet An example of such a kit is the familiar blister pack used for the packaging of tablets, capsules and the like.
- the kit of the invention is particularly suitable for administering different dosage forms, for example, oral and parenteral, for administering the separate compositions at different dosage intervals, or for titrating the separate compositions against one another.
- the kit typically comprises directions for administration and may be provided with a so-called memory aid.
- the total daily dose of the compounds of the invention is typically in the 0.001 mg to 2000 mg depending, of course, on the mode of administration.
- oral administration may require a total daily dose of from 1 mg to 2000 mg, while an intravenous dose may only require from 0.01 mg to 100 mg.
- the total daily dose may be administered in single or divided doses and may, at the physician's discretion, fall outside of the typical range given herein. These dosages are based on an average human subject having a weight of about 60kg to 70kg. The physician will readily be able to determine doses for subjects whose weight falls outside this range, such as infants and the elderly.
- references herein to "treatment” include references to curative, palliative and prophylactic treatment.
- the compounds of the formula (1), or pharmaceutically acceptable salts, derived forms or compositions thereof can also be used as a combination with one or more additional therapeutic agents to be co-administered to a patient to obtain some particularly desired therapeutic end result.
- the second and more additional therapeutic agents may also be a compound of the formula (1), or a pharmaceutically acceptable salt, derived forms or compositions thereof, or one or more histamine H 3 receptor ligands known in the art. More typically, the second and more therapeutic agents will be selected from a different class of therapeutic agents.
- co-administration As used herein, the terms “co-administration”, “co-administered” and “in combination with”, referring to the compounds of formula (1) and one or more other therapeutic agents, is intended to mean, and does refer to and include the following:
- Suitable examples of other therapeutic agents which may be used in combination with the compound(s) of formula (1), or pharmaceutically acceptable salts, derived forms or compositions thereof, include, but are by no means limited to :
- Histamine H 1 receptor antagonists for instance loratidine, desloratidine, fexofenadine and cetirizine;
- Leukotriene antagonists including antagonists of LTB 4 , LTC 4 , LTD 4 , and LTE 4 , in particular Montelukast;
- Phosphodiesterase inhibitors such as PDE4 inhibitors or PDE5 inhibitors
- neurotransmitter re-uptake inhibitors for instance fluoxetine, sertraline, paroxetine, ziprasidone;
- Anti-tumor necrosis factor (anti-TNF- ⁇ ) agents Anti-TNF- ⁇ agents
- Adhesion molecule inhibitors including VLA-4 antagonists
- MMPs matrix metalloproteases
- Modulators of the NFic ⁇ pathway e.g. IKK inhibitors
- Agents that can be classed as mucolytics or anti-tussive Agents that can be classed as mucolytics or anti-tussive
- cytokine signalling pathways such as p38 MAP kinase, syk kinase or JAK kinase inhibitors
- HDAC histone deacetylase
- Histamine H 1 receptor antagonists for instance loratidine, desloratidine, fexofenadine and cetirizine
- Leukotriene antagonists including antagonists of LTB 4 , LTC 4 , LTD 4 , and LTE 4 , in particular Montelukast • Phosphodiesterase PDE4 inhibitors
- neurotransmitter re-uptake inhibitors for instance fluoxetine, sertraline, paroxetine, ziprasidone
- H 3 ligands are meant to include H 3 receptor antagonists, agonists and inverse agonists.
- H 3 antagonists are believed to be most suitable.
- a further aspect of the present invention relates to the compounds of formula (1), or pharmaceutically acceptable salts, derived forms or compositions thereof, for use in the treatment of diseases, disorders, and conditions in which the H 3 receptor is involved. More specifically, the present invention also concerns the compounds of formula (1), or pharmaceutically acceptable salts, derived forms or compositions thereof, for use in the treatment of diseases, disorders, and conditions selected from the group consisting of :
- diseases of the central nervous system sleep disorders, migraine, dyskinesia, stress-induced anxiety, psychotic disorders, epilepsy, Cognition deficiency diseases such as Alzheimer's disease or mild cognitive impairment, depression, mood disorders, schizophrenia, anxiety disorders, attention-deficit hyperactivity disorder (ADHD), psychotic disorders, obesity, dizziness, vertigo, epilepsy, motion sickness
- respiratory diseases adult respiratory distress syndrome, acute respiratory distress syndrome, bronchitis, chronic bronchitis, chronic obstructive pulmonary disease, cystic fibrosis, asthma, emphysema, rhinitis, chronic sinusitis
- allergy allergy-induced airway responses
- allergic rhinitis allergic rhinitis
- viral rhinitis non-allergic rhinitis
- perennial and seasonal rhinitis nasal congestion
- allergic congestion allergic congestion
- Female sexual dysfunction including hypoactive sexual desire disorder, sexual arousal disorder, orgasmic disorder and sexual pain disorder
- Male sexual dysfunction including male desire disorders, male erectile dysfunction, male orgasmic disorders such as premature ejaculation
- cardiac dysfunctions such as myocardial ischaemia and arrythmia
- the compounds of formula (1 ) of the invention are particularly suitable for the treatment of allergy, allergy- induced airway responses, allergic rhinitis, viral rhinitis, non-allergic rhinitis, perennial and seasonal rhinitis, nasal congestion, allergic congestion.
- a stiil further aspect of the present invention also relates to the use of the compounds of formula (1), or pharmaceutically acceptable salts, derived forms or compositions thereof, for the manufacture of a drug being a H 3 ligand.
- the present inventions concerns the use of the compounds of formula (1), or pharmaceutically acceptable salts, derived forms or compositions thereof, for the manufacture of a drug for the treatment of H 3 -mediated diseases and/or conditions, in particular the diseases and/or conditions listed above.
- the present invention provides a particularly interesting method to treat a mammal, including a human being, with an effective amount of a compound of formula (1), or a pharmaceutically acceptable salt, derived form or composition thereof. More precisely, the present invention provides a particularly interesting method for the treatment of a H 3 -mediated diseases and/or conditions in a mammal, including a human being, in particular the diseases and/or conditions listed above, comprising administering to said mammal an effective amount of a compound of formula (1), its pharmaceutically acceptable salts and/or derived forms.
- the crude material was purified by column chromatography using an ISCO ® silica cartridge eluting with dichloromethane: methanol: ammonia (99:1:0.1 to 95:5:0.5).
- the title compound was obtained as a yellow oil (310mg, 0.70mmol, 56%).
- the mixture was extracted from a saturated aqueous solution of sodium hydrogen carbonate (200ml) into ethyl acetate (250ml), dried and the solvent evaporated in vacuo.
- the crude material was purified by column chromatography over silica gel eluting with dichloromethane: methanol: 0.880 ammonia (90:10:1).
- the title compound was obtained as a pale yellow oil (1.46g, 4.18mmol, 66%).
- the mixture was diluted with diethyl ether (200ml), washed with water (100ml), brine (100ml), dried over magnesium sulphate, filtered and evaporated in vacuo.
- the crude material was purified by column chromatography using a Combiflash ® silica cartridge eluting with dichloromethane to dichloromethane: methanol: ammonia (80:20:2). The title compound was obtained as an orange oil (13.55g, 43mmol, 64%).
- the crude material was purified by column chromatography using a Combifiash ® silica cartridge eluting with dichloromethane: methanol: ammonia (95:5:0.5 to 85:15:1.5), to yield the title compound (0.9g, 2.8mmol, 44%).
- the crude material was purified by column chromatography using an ISCO ® silica cartridge eluting with dichloromethane: methanol: ammonia (99:1:0.1 to 90:10:1) to give the title compound (26mg, 0.07mmol, 5%).
- the crude material was purified by column chromatography using an ISCO ® silica cartridge eluting with dichloromethane: methanol: ammonia (99:1:0.1 to 90:10:1).
- the title compound was obtained as a colourless oil (352mg, 0.91 mmol, 65%).
- Compounds were evaluated using a cell based functional assay measuring cAMP through ⁇ -lactamase reporter gene activity.
- a stable cell line was generated from HEK-293 cells expressing a CRE ⁇ -lactamase reporter gene and transfected with human histamine H 3 receptor cDNA. Cells were seeded at a density of 500,000 cells/ml, and grown overnight in MEM (Invitrogen) supplemented with 1% dialysed FBS (Sigma), 2mM glutamine (Sigma), 1mM sodium pyruvate (Sigma), 0.1mM non essential amino acids (Invitrogen) and 25mM HEPES (Sigma) in poly D lysine coated 384 well plates (BD Biosciences).
- H 3 receptor agonist imetit dose dependently inhibited 10 ⁇ M forskolin (Calbiochem) stimulated synthesis of cAMP measured after 4.5hours by ⁇ -lactamase cleavage of CCF4-AM dye (Invitrogen).
- test compounds were prepared in PBS (Sigma) and DMSO (Sigma) at a dose response of 5x10 "10 to 5x10 "5 M with a final DMSO concentration in the assay of 0.5%. Cells were incubated for 15 minutes plus/minus compound and their ability to permit 10 ⁇ M forskolin-stimulated cAMP synthesis in the presence of 1 nM imetit was measured as described above.
Abstract
The invention relates to pyridyl derivatives of general formula (1) and to processes for the preparation of, intermediates used in the preparation of, compositions containing and the uses of, such derivatives. Said pyridyl derivatives are H3 ligands and are useful in numerous diseases, disorders and conditions, in particular inflammatory, allergic and respiratory diseases, disorders and conditions.
Description
PYRIDYL DERIVATIVES USEFUL AS H3 LIGANDS
The present invention relates to pyridyl derivatives of general formula:

Histamine H3 receptors are found inter alia on presynaptic terminals of peripheral nerves, where they modulate autonomic neurotransmission and modulate a variety of end organ responses under control of the autonomic nervous system. They are also heteroreceptors, modulating the release of numerous other neurotransmitters such as dopamine, glutamate, noradrenaline, serotonin, GABA, acetylcholine, some peptides and co-transmitters.
Recently numerous histamine H3 receptor ligands have been developed. An overview of the current advance in H3 ligand research and patenting is given in Expert Opin. Ther. Patents (2003) 13(6). Examples of Histamine H3 receptor ligands can be found in WO02/76925, WO00/06254, WO02/12190, WO02/12214 and WO02/06223.
H3 receptor ligands are believed to be suitable for the treatment of various diseases including both disorders of the central nervous system and inflammatory disorders. Examples of diseases where treatment with H3 ligands is believed to be useful are obesity, inflammatory bowel disease, Crohn's disease, colitis ulcerosa, sleep disorders, migraine, dyskinesia, stress-induced anxiety, psychotic disorders, epilepsy, Cognition deficiency diseases such as Alzheimer's disease or mild coginitive impairment, depression, mood disorders, schizophrenia, anxiety disorders, attention-deficit hyperactivity disorder (ADHD), psychotic disorders, obesity, dizziness, epilepsy, motion sickness, vertigo, respiratory diseases such as adult respiratory distress syndrome, acute respiratory distress syndrome, bronchitis, chronic bronchitis, chronic obstructive pulmonary disease, cystic fibrosis, asthma, emphysema, rhinitis, chronic sinusitis, allergy, allergy-induced airway responses, allergic rhinitis, viral rhinitis, non-allergic rhinitis, perennial and seasonal rhinitis, nasal congestion and allergic congestion.
Although H3 ligands are known there is still a need to provide new H3 ligands that are good drug candidates. In particular, preferred compounds should bind potently to the histamine H3 receptor whilst showing little affinity for other receptors. They should be well absorbed from the gastrointestinal tract, be metabolically stable and possess favourable pharmacokinetic properties. They should be non-toxic and demonstrate few side-effects.
In this context, the present invention concerns new pyridyl derivatives of general formula (1) : i
 or a pharmaceutically acceptable salt and/or solvate (including hydrate) thereof wherein :
or a pharmaceutically acceptable salt and/or solvate (including hydrate) thereof wherein :
X is selected from -CN, -CH2NR1R2, -C(O)NR3R4, -CH2-het1 and net2, the groups net1 and het2 being optionally substituted by one or two substituents independently selected from halo, cyano, (Ci-C4)alkyl and (CrC4)alkoxy;
R1 and R2 are each independently selected from hydrogen, (Ci-C4)alkyl and (C3-C6)cycloalkyl, said (Cr C4)alkyl and (C3-C6)cycloalkyl being optionally substituted by one group selected from (CrC4)alkoxy and (C3-C6)cycloalkyl, or alternatively R1 and R2 together with the nitrogen atom to which they are attached form a A-, 5-, 6- or 7-membered saturated heterocycle wherein one carbon atom may be optionally replaced by a nitrogen or an oxygen atom, and wherein said saturated heterocycle is optionally substituted by one or two groups independently selected from (CrC4)alkyl, (CrC4)alkoxy or -C(O)(CrC4)alkyl;
R3 and R4 are each independently selected from hydrogen, (CrC4)alkyl and (C3-C6)cycloalkyl, said (C1- C4)alkyl and (C3-C6)cycloalkyl being optionally substituted by one group selected from (CrC4)alkoxy and (C3-C6)cycloalkyl, or alternatively R3 and R4 together with the nitrogen atom to which they are attached form a A-, 5-, 6- or 7-membered saturated heterocycle wherein one carbon atom may be optionally replaced by a nitrogen or an oxygen atom, and wherein said saturated heterocycle is optionally substituted by one or two groups independently selected from (CrC4)alkyl, (CrC4)alkoxy or -C(O)(CrC4)alkyl;
het1 is N-linked and is selected from monocyclic or bicyclic heteroaromatic groups having 5, 6, 7, 8 or 9 ring members, which contain 1, 2, 3 or 4 heteroatom(s) selected from nitrogen, oxygen and sulphur;
het2' is C-linked and is selected from monocyclic or bicyclic heteroaromatic groups having 5, 6, 7, 8, 9 or 10 ring members, which contain 1 , 2, 3 or 4 heteroatom(s) selected from nitrogen, oxygen and sulphur;
and R is either a group of formula :
R5 \ N-L — *
R6/ wherein * represents the attachment point to the oxygen atom, L is a straight chain or branched (C2- C6)alkylene and R5 and R6 are each independently selected from hydrogen, (CrC4)alkyl or (C3- C6)cycloalkyl, or alternatively R5 and R6 together with the nitrogen atom to which they are attached form a A-, 5-, 6- or 7-membered saturated heterocycle wherein said saturated heterocycle is optionally substituted by one or two groups independently selected from (CrC4)alkyl and (CrC4)alkoxy,
or alternatively R is a group of formula:

It has been found that these compounds are H3 ligands and are thus particularly useful for the treatment of H3-related diseases such as neurologic disorders, or inflammatory, respiratory and allergic diseases, disorders and conditions.
In the present description the following definitions are used, unless otherwise specified:
"Halo" denotes a halogen atom selected from the group consisting of fluoro, chloro, bromo and iodo.
"(C1-C4)all<yl" denotes a saturated, straight-chain or branched hydrocarbon group having from 1 to 4 carbon atoms and includes for example methyl, ethyl, propyl, iso-propyl, n-butyl, iso-butyl, sec-butyl and tert-butyl. This also applies if the alkyl group carries substituents or is a substituent for another group, e.g. in (CrC4)alkoxy and -C(O)(CrC4)alkyl.
"(CrC4)alkoxy" denotes straight-chain and branched alkoxy groups and includes for example methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, iso-butoxy, sec-butoxy and tert-butoxy.
"(C2-C6)alkylene" denotes a divalent radical derived from straight-chain or branched alkane containing from 2 to 6 carbon atoms. Examples of (C2-C6)alkylene radicals are methylene, ethylene (1,2-ethylene or 1,1 -ethylene), trimethylene (1,3-propylene), tetramethylene (1 ,4-butylene), pentamethylene and hexamethylene.
"(C3-C6)cycloalkyl" denotes a saturated monocyclic carbocyclic group having 3, 4, 5 or 6 carbon atoms and includes for example cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl.
"Saturated heterocycle" denotes a saturated monocyclic group having 4, 5, 6 or 7 ring members, which contains one nitrogen atom. Examples of saturated heterocycles are azetidinyl, pyrrolidinyl and piperidinyl.
Het1 and het2 are monocyclic or bicyclic heteroaromatic groups having 5, 6, 7, 8 or 9 and 5, 6, 7, 8, 9 or 10 ring members respectively, which contain 1 , 2, 3 or 4 heteroatom(s) selected from nitrogen, oxygen and sulphur. Het1 is N-linked, which means that the group is linked to the adjacent atom by a ring nitrogen atom. Het2 is C-linked, which means that the group is linked to the adjacent atom by a ring carbon atom. The heteroaromatic group can be unsubstituted, monosubstituted or disubstituted, as indicated in the
definition of X hereabove for general formula (1) according to the present invention. Examples of heteroaryl groups include, but are not limited to thiophenyl, furanyl, pyrrolyl, pyrazolyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, triazolyl, oxadiazolyl, thiadiazolyl, tetrazolyl, pyranyl, pyridinyl, pyrazinyl, pyrimidinyl, pyridazinyl, triazinyl, thiadiazinyl, isobenzofuranyl, benzofuranyl, chromenyl, indolizinyl, isoindolyl, indolyl, indazolyl, purinyl, quinolinyl, isoquinolyl, cinnolinyl, phthalazinyl, naphthyridinyl, quinazolinyl, quinoxalinyl, benzoxazolyl, benzothiazolyl, benzimidazolyl, benzofuranyl, benzothiophenyl, pyrrolopyrazinyl, pyrrolopyridinyl, and imidazopyridinyl. Preferred definitions for het1 and net2 will follow hereafter.
In the general formula (1) according to the present invention, when a radical is mono- or poly-substituted, said substituent(s) can be located at any desired position(s), unless otherwise stated. Also, when a radical is polysubstituted, said substituents can be identical or different, unless otherwise stated.
According to a preferred aspect of the invention, X is selected from -CN, -CH2NR1R2, -CH2-het1 and het2, het2 being optionally substituted once by (Ci-C4)alkyl, wherein R1, R2, het1 and het2 are as previously defined.
According to a more preferred aspect, X is selected from -CN, -CH2NR1R2, -CH2-het1 and het2, het2 being optionally substituted once by (Ci-C4)alkyl, wherein:
R1 and R2 are both hydrogen atoms or alternatively R1 and R2 together with the nitrogen atom to which they are attached form a A-, 5-, 6- or 7-membered saturated heterocycle wherein one carbon atom may be optionally replaced by a nitrogen or an oxygen atom; het1 is a N-linked bicyclic heteroaromatic group having 9 ring members which contain a total of two nitrogen atoms; and het2 is a C-linked monocyclic heteroaromatic group having 5 ring members which contain one nitrogen atom and one sulphur atom.
Still more preferably, X is selected from -CN, -CH2-NH2, -CH2-morpholine, -CH2-benzimidazole and 1,3- thiazol-2-yl optionally substituted by a (CrC4)alkyl group, preferably a methyl group.
In another preferred aspect of the invention R is a group of formula
R
\
N-L —
R6/ wherein * represents the attachment point to the oxygen atom, L is a straight chain or branched (C2- C6)alkylene and R5 and R6 together with the nitrogen atom to which they are attached form a 4-, 5-, 6- or 7-membered saturated heterocycle wherein said saturated heterocycle is optionally substituted by one or two groups independently selected from (CrC4)alkyl and (CrC4)alkoxy.
More preferably, R5 and Rs together with the nitrogen atom to which they are attached form a 5- or 6- membered saturated heterocycle. Even more preferably, the saturated heterocycle is a pyrrolidinyl group.
Preferably, L is propylene.
When one of the groups in the compound of formula (1) is substituted by halo, fluoro or chloro are preferred. Fluoro is most preferred.
Particularly preferred compounds of the invention include those in which each variable in formula (1) is selected from the suitable and/or preferred groups for each variable. Even more preferable compounds of the invention include those where each variable in formula (1) is selected from the more preferred or most preferred groups for each variable.
Most preferably, the compounds of formula (1) are selected from:
4-[6-(3-pyrrolidin-1-ylpropoxy)pyridin-3-yl]tetrahydro-2H-pyran-4-carbonitrile;
1-{4-[6-(3-pyrrolidin-1-ylpropoxy)pyridin-3-yl]tetrahydro-2H-pyran-4-yl}methanamine;
4_({4_[6-(3-pyrrolidin-1-ylpropoxy)pyridin-3-yl]tetrahydro-2H-pyran-4-yl}methyl)morpholine;
1-({4-[6-(3-pyrrolidin-1-ylpropoxy)pyridin-3-yl]tetrahydro-2H-pyran-4-yl}methyl)-1H-benzimidazole;
2-(3-pyrrolidin-1 -ylpropoxy)-5-[4-(1 ,3-thiazol-2-yl)tetrahydro-2H-pyran-4-yl]pyridine and
5-[4-(4-methyl-1 ,3-thiazol-2-yl)tetrahydro-2H-pyran-4-yl]-2-(3-pyrrolidin-1-ylpropoxy)pyridine.
Another embodiment of the invention is an intermediate as described herein.
Pharmaceutically acceptable salts of the compounds of formula (1) include the acid addition and base salts thereof.
Suitable acid addition salts are formed from acids which form non-toxic salts. Examples include the acetate, aspartate, benzoate, besylate, bicarbonate/carbonate, bisulphate/sulphate, borate, camsylate, citrate, edisylate, esylate, formate, fumarate, gluceptate, gluconate, glucuronate, hexafluorophosphate, hibenzate, hydrochloride/chloride, hydrobromide/bromide, hydroiodide/iodide, isethionate, lactate, malate, maleate, malonate, mesylate, methylsulphate, naphthylate, 2-napsylate, nicotinate, nitrate, orotate, oxalate, palmitate, pamoate, phosphate/hydrogen phosphate/dihydrogen phosphate, saccharate, stearate, succinate, tartrate, tosylate and trifluoroacetate salts.
Suitable base salts are formed from bases which form non-toxic salts. Examples include the aluminium, arginine, benzathine, calcium, choline, diethylamine, diolamine, glycine, lysine, magnesium, meglumine, olamine, potassium, sodium, tromethamine and zinc salts.
Hemisalts of acids and bases may also be formed, for example, hemisulphate and hemicalcium salts.
For a review on suitable salts, see "Handbook of Pharmaceutical Salts: Properties, Selection, and Use" by Stahl and Wermuth (Wiley-VCH, Weinheim, Germany, 2002).
Pharmaceutically acceptable salts of compounds of formula (1) may be prepared by one or more of three methods:
(i) by reacting the compound of formula (1 ) with the desired acid or base;
(ii) by removing an acid- or base-labile protecting group from a suitable precursor of the compound of formula (1) or by ring-opening a suitable cyclic precursor, for example, a lactone or lactam, using the desired acid or base; or (iii) by converting one salt of the compound of formula (1) to another by reaction with an appropriate acid or base or by means of a suitable ion exchange column.
All three reactions are typically carried out in solution. The resulting salt may precipitate out and be collected by filtration or may be recovered by evaporation of the solvent. The degree of ionisation in the resulting salt may vary from completely ionised to almost non-ionised.
The compounds of the invention may exist in both unsolvated and solvated forms. The term 'solvate' is used herein to describe a molecular complex comprising the compound of the invention and a stoichiometric amount of one or more pharmaceutically acceptable solvent molecules, for example, ethanol. The term 'hydrate' is employed when said solvent is water.
Included within the scope of the invention are complexes such as clathrates, drug-host inclusion complexes wherein, in contrast to the aforementioned solvates, the drug and host are present in stoichiometric or non-stoichiometric amounts. Also included are complexes of the drug containing two or more organic and/or inorganic components which may be in stoichiometric or non-stoichiometric amounts. The resulting complexes may be ionised, partially ionised, or non-ionised. For a review of such complexes, see J Pharm Sci, 64 (8), 1269-1288 by Haleblian (August 1975).
Hereinafter all references to compounds of formula (1) include references to salts, solvates and complexes thereof and to solvates and complexes of salts thereof.
The compounds of the invention include compounds of formula (1) as hereinbefore defined, including all polymorphs and crystal habits thereof, prodrugs and isomers thereof (including optical, geometric and tautomeric isomers) as hereinafter defined and isotopically-labeled compounds of formula (1).
As indicated, so-called 'pro-drugs' of the compounds of formula (1) are also within the scope of the invention. Thus certain derivatives of compounds of formula (1) which may have little or no pharmacological activity themselves can, when administered into or onto the body, be converted into compounds of formula (1) having the desired activity, for example, by hydrolytic cleavage. Such derivatives are referred to as 'prodrugs'. Further information on the use of prodrugs may be found in 'Prodrugs as Novel Delivery Systems, Vol. 14, ACS Symposium Series (T. Higuchi and W. Stella) and 'Bioreversible Carriers in Drug Design', Pergamon Press, 1987 (ed. E. B Roche, American Pharmaceutical Association).
Prodrugs in accordance with the invention can, for example, be produced by replacing appropriate functionalities present in the compounds of formula (1) with certain moieties known to those skilled in the art as 'pro-moieties' as described, for example, in "Design of Prodrugs" by H. Bundgaard (Elsevier, 1985).
Some examples of prodrugs in accordance with the invention include, where the compound of general formula (1) contains a primary or secondary amino functionality (-NH2 or -NHR where R ≠ H), an amide thereof, for example, a compound wherein, as the case may be, one or both hydrogens of the amino functionality of the compound of formula (1) is/are replaced by (CrCi0)alkanoyl.
Further examples of replacement groups in accordance with the foregoing examples and examples of other prodrug types may be found in the aforementioned references.
Moreover, certain compounds of formula (1 ) may themselves act as prodrugs of other compounds of formula (1).
Also included within the scope of the invention are metabolites of compounds of formula (1), that is, compounds formed in vivo upon administration of the drug. Some examples of metabolites in accordance with the invention include : (i) where the compound of formula (1) contains a methyl group, an hydroxymethyl derivative thereof
(-CH3 → -CH2OH): (ii) where the compound of formula (1) contains an alkoxy group, an hydroxy derivative thereof (-OR
→ -OH); (iii) where the compound of formula (1) contains a tertiary amino group, a secondary amino derivative thereof (-NRaRb → -NHRa or -NHRb); (iv) where the compound of formula (1) contains a secondary amino group, a primary derivative thereof (-NHRa→ -NH2); (v) where the compound of formula (1) contains an amide group, a carboxylic acid derivative thereof
(-CONR°Rd → COOH).
Compounds of formula (1) containing one or more asymmetric carbon atoms_can exist as two or more stereoisomers. Where structural isomers are interconvertible via a low energy barrier, tautomeric isomerism ('tautomerism') can occur. This can take the form of proton tautomerism in compounds of formula (1) containing, for example, an imino, keto, or oxime group, or so-called valence tautomerism in compounds which contain an aromatic moiety. It follows that a single compound may exhibit more than one type of isomerism.
Included within the scope of the present invention are all stereoisomers, geometric isomers and tautomeric forms of the compounds of formula (1), including compounds exhibiting more than one type of isomerism, and mixtures of one or more thereof. Also included are acid addition or base salts wherein the counterion is optically active, for example, d-lactate or /-lysine, or racemic, for example, d/-tartrate or dl- arginine.
Conventional techniques for the preparation/isolation of individual enantiomers include chiral synthesis from a suitable optically pure precursor or resolution of the racemate (or the racemate of a salt or derivative) using, for example, chiral high pressure liquid chromatography (HPLC),
Alternatively, the racemate (or a racemic precursor) may be reacted with a suitable optically active compound, for example, an alcohol, or, in the case where the compound of formula (1) contains an acidic or basic moiety, an acid or base such as tartaric acid or 1-phenylethylamine. The resulting diastereomeric mixture may be separated by chromatography and/or fractional crystallization and one or both of the diastereoisomers converted to the corresponding pure enantiomer(s) by means well known to a skilled person.
Chiral compounds of the invention (and chiral precursors thereof) may be obtained in enantiomerically- enriched form using chromatography, typically HPLC, on an asymmetric resin with a mobile phase consisting of a hydrocarbon, typically heptane or hexane, containing from 0 to 50% by volume of isopropanol, typically from 2% to 20%, and from 0 to 5% by volume of an alkylamine, typically 0.1% diethylamine. Concentration of the eluate affords the enriched mixture.
Stereoisomeric conglomerates may be separated by conventional techniques known to those skilled in the art - see, for example, "Stereochemistry of Organic Compounds" by E. L. Eliel (Wiley, New York, 1994).
The present invention includes all pharmaceutically acceptable isotopically-labelled compounds of formula (1) wherein one or more atoms are replaced by atoms having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number which predominates in nature.
Examples of isotopes suitable for inclusion in the compounds of the invention include isotopes of hydrogen, such as 2H and 3H, carbon, such as 11C, 13C and 14C, chlorine, such as 36CI, fluorine, such as 18F, iodine, such as 123I and 125I, nitrogen, such as 13N and 15N, oxygen, such as 15O, 17O and 18O, phosphorus, such as 32P, and sulphur, such as 35S.
Certain isotopically-labelled compounds of formula (1), for example, those incorporating a radioactive isotope, are useful in drug and/or substrate tissue distribution studies. The radioactive isotopes tritium, i.e. 3H, and carbon-14, i.e. 14C, are particularly useful for this purpose in view of their ease of incorporation and ready means of detection.
Substitution with heavier isotopes such as deuterium, i.e. 2H, may afford certain therapeutic advantages resulting from greater metabolic stability, for example, increased in vivo half-life or reduced dosage requirements, and hence may be preferred in some circumstances.
Substitution with positron emitting isotopes, such as 11C, 18F, 15O and 13N, can be useful in Positron Emission Topography (PET) studies for examining substrate receptor occupancy.
Isotopically-labeled compounds of formula (1) can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described in the accompanying Examples and Preparations using an appropriate isotopically-labeled reagents in place of the non-labeled reagent previously employed.
Pharmaceutically acceptable solvates in accordance with the invention include those wherein the solvent of crystallization may be isotopically substituted, e.g. D2O1 d6-acetone, ds-DMSO.
The pyridyl derivatives of general formula (1) wherein R is as previously defined in general formula (1) above and X is -CN may be prepared using conventional procedures exemplified but not limited by those illustrated in scheme 1:

+ ROH (vi)

Scheme 1
Compounds of general formula (1) wherein X is -CN may be prepared in step (vi) by reacting 4-(6- chloropyridin-3-yl)tetrahydro-2H-pyran-4-carbonitrile with ROH and a suitable base (e.g. NaH or KOtBu) in a suitable solvent (e.g. THF) at temperatures between room temperature and reflux, typically at room temperature.
Compounds of general formula ROH wherein R is as defined previously are either commercial or can be made using procedures known to the skilled person.
4-(6-Chloropyridin-3-yl)tetrahydro-2H-pyran-4-carbonitrile may be prepared by several methods, two of which are illustrated in scheme 1: Sequential tetrahydro-2H-pyran construction ((i) e.g. with a 2-haloethyl ether such as 2-bromoethylether and a base such as NaH, optionally with Kl, in a suitable solvent such as THF at temperatures between O0C room temperature), N-oxidation ((H) e.g. with a suitable oxidising agent such as oxone and a base such as sodium bicarbonate in a suitable solvent such as aqueous acetone at temperatures between 0°C and room temperature) and halogenation ((Hi) e.g. with a chlorinating agent such as POCI3 optionally with a co-solvent such as acetonitrile at temperatures between room temperature and reflux). Alternatively, sequential cyanation ((iv) e.g. with NaCN in a suitable solvent such as aqueous EtOH at temperatures between room temperature and reflux) and tetrahydro-2H-pyran construction ((v) e.g. with a 2-haloethyl ether such as 2-bromoethylether and a base such as NaH, optionally with Kl, in a suitable solvent such as THF at temperatures between O0C and room temperature).
The pyridyl derivatives of general formula (1) wherein R is as previsouly defined in general formula (1) and X is not -CN may be prepared from analogous pyridyl derivatives of formula (1) wherein X is -CN (preparation described above) using conventional procedures exemplified but not limited by those illustrated in scheme 2:

(1) wherein X is -CN (2) (1) wherein X is -C(O)NFW
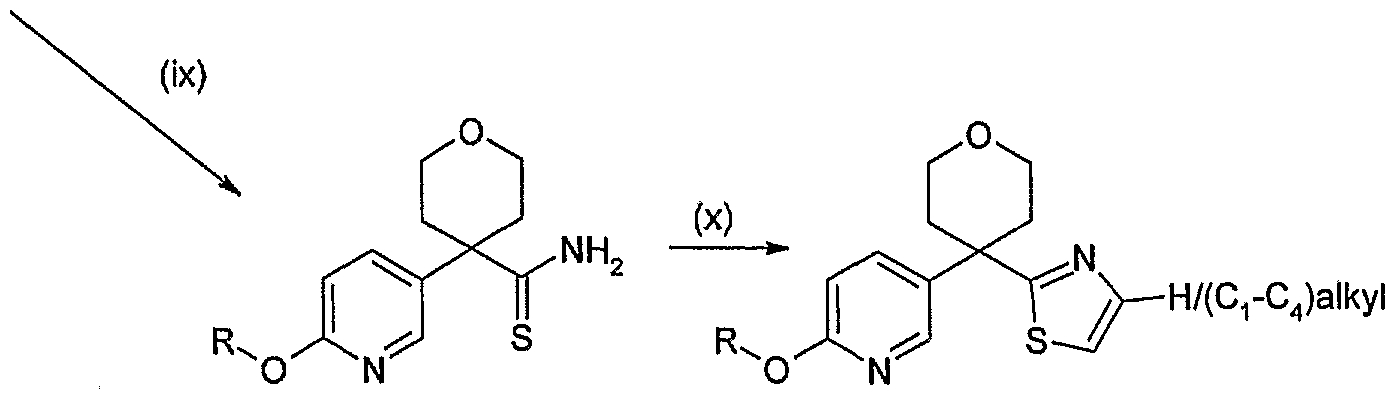


(1) wherein X is -NH2 (1) wherein X is -CH2NR1R2

Scheme 2
Compounds of general formula (1) wherein X is C(O)NR3R4 may be prepared from compounds of general formula (2) in step (viii) by using standard techniques for amide coupling. Conditions for step (viii) include sequential treatment of compounds of general formula (2) with a reagent capable of activating a carboxylic acid (e.g. thionyl chloride) and then with the amino derivative of formula HNR3R4, in a suitable solvent (e.g. dichloromethane) at between O0C and reflux. Alternatively the acid may be treated with the amine of formula HNR3R4, in the presence of a coupling agent (e.g. TBTU, O-benzotriazol-1-yl-N,N,N',N'- tetramethyluronium hexafluorophosphate, 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride), optionally in the presence of a suitable additive (e.g. 1-hydroxybenzotriazole) and base (e.g. triethylamine) in a suitable solvent (e.g. dichloromethane, N,N-dimethylformamide) at room temperature (about 200C).
Compounds of general formula (2) may be prepared from compounds of general formula (1) wherein X is -CN in step (vii) by using standard techniques for nitrile hydrolysis. Conditions for step (vii) include
treatment of the nitrile compound with hydrochloric acid, optionally in the presence of a co-solvent such as dioxane, at temperatures between room temperature and reflux.
Compounds of general formula (1) wherein X is net2 may be prepared from compounds of general formula (1) wherein X is -CN using multistep procedures known to those skilled in the art such as those described in general heterocyclic texts such as Heterocyclic Chemistry, JA Joule and K. Mills, 4th Edition, Blackwell publishing, 2000 or Comprehensive Heterocyclic chemistry I and II, Pergamon Press. This is exemplified in scheme 2 by the formation of compounds of general formula (1) wherein X is het2 and het2 is thiazole optionally substituted with (CrC^alkyl: These compounds can be prepared from compounds of general formula (3) in step (x) by treatment with a suitable reagent (e.g. bromoacetaldehyde dimethyl acetal or chloroacetone) optionally with an acid (e.g. HBr or HCI) in a suitable solvent (e.g. EtOH) at temperatures between room temperature and reflux.
Compounds of general formula (3) may be prepared from compounds of general formula (1) wherein X is -CN in step (ix) by treatment with diethyldithiophosphate and water at temperatures between room temperature and reflux, typically at 600C.
Compounds of general formula (1) wherein X is -CH2NR1R2 can be prepared from compounds of general formula (1) wherein X is NH2 in step (xii) using standard techniques for functional group interconversion known to those skilled in the art, for example as described in Comprehensive Organic Transformations, R.C. Larock, 1st Edition, 1989 VCH Publishers Inc. These techniques include:
1) Reductive alkylation with aldehydes or ketones with a reducing agent (e.g. formic acid or NaHB(OAc)3) in a suitable solvent (e.g. dichloromethane, tetrahydrofuran), optionally with the addition of acetic acid, at temperatures between room temperature and reflux;
2) Alkylations with alkyl halides and a base (e.g. potassium carbonate, potassium ferf-butoxide) in a suitable solvent (e.g. acetonitrile, tetrahydrofuran) at temperatures between room temperature and reflux.
Compounds of general formula (1) wherein X is -CH2NH2 can be prepared from compounds of general formula (1) wherein X is -CN in step (xi) by using a suitable reducing agent (e.g. LiAIH4 or hydrogen gas in the presence of a catalyst such as PtO2) in a suitable solvent (e.g. Et2O, dichloromethane, tetrahydrofuran or isopropanol) at temperatures between room temperature and reflux.
Compounds of general formula (1) wherein X is -CH2het1 can be prepared from compounds of general formula (1) wherein X is -CH2NH2 using multistep methods known to those skilled in the art, such as those described in general heterocyclic texts such as Heterocyclic Chemistry, J.A. Joule and K. Mills, 4th Edition, Blackwell publishing, 2000 or Comprehensive Heterocyclic chemistry I and II, Pergamon Press. This is exemplified in scheme 2 wherein het1 is benzimidazole: These compounds can be prepared from compounds of general formula (4) in step (xiv) by sequential reduction with a suitable reducing agent (e.g. hydrogen gas in the presence of a catalyst such as palladium on carbon) in a suitable solvent (e.g. formic acid) at temperatures between room temperature and 600C followed by cyclisation with an orthoester (e.g.
trimethylorthoformate) in a suitable solvent (e.g. formic acid) at temperatures between room temperature and reflux.
Compounds of general formula (4) can be prepared from compounds of general formula (1) wherein X is -CH2NH2 in step (xiii) by reaction with 2-halo-nitrobenzene (e.g. 2-fluoro-nitrobenzene) and a base (e.g. potassium carbonate) in a suitable solvent (e.g. tetrahydrofuran) at temperatures between room temperature and reflux.
It will be appreciated by those skilled in the art that, certain compounds of general formula (1) may be converted to alternative compounds of general formula (1) using standard chemical transformations. Those skilled in the art will also appreciate that compounds of general formula (1) may also be prepared by reordering the steps illustrated in schemes 1 and 2.
It will be appreciated by those skilled in the art that, in the course of carrying out the processes described above, the functional groups of intermediate compounds may need to be protected by protecting groups.
These functional groups include hydroxyl, amino and carboxylic acid. Suitable protecting groups for hydroxyl include trialkylsilyl and diarylalkylsilyl (e.g. tert-butyldimethylsilyl, ferf-butyldiphenylsilyl or trimethylsilyl), alkyl (e.g. methyl or methoxyethyl) and tetrahydropyranyl. Suitable protecting groups for amino include fert-butyloxycarbonyl, 9-fluorenylmethoxycarbony! or benzyloxycarbonyl. Suitable protecting groups for carboxylic acid include (CrC4)alkyl or benzyl esters.
The protection and deprotection of functional groups may take place before or after any of the reaction steps described hereinbefore.
The introduction and removal of protecting groups is fully described in Protective Groups in Organic Chemistry, edited by J.W.F. McOmie, Plenum Press (1973), Protective Groups in Organic Synthesis, 2nd edition, T.W. Greene & P.G.M. Wutz, Wiley-lnterscience Publication, 1991) and Protecting Groups, P.J. Kocienski, Thieme, 1994.
Also, the compounds of formula (1) as well as intermediates for the preparation thereof, can be purified according to various well-known methods such as recrystallisation and chromatography.
The compounds of formula (1), their pharmaceutically acceptable salts and/or derived forms, are valuable pharmaceutically active compounds, which are suitable for the therapy and prophylaxis of numerous disorders in which the histamine H3 receptor is involved or in which agonism or antagonism of this receptor may induce benefit.
Compounds of the invention intended for pharmaceutical use may be administered as crystalline or amorphous products. They may be obtained, for example, as solid plugs, powders, or films by methods such as precipitation, crystallization, freeze drying, spray drying, or evaporative drying. Microwave or radio frequency drying may be used for this purpose.
They may be administered alone or in combination with one or more other compounds of the invention or in combination with one or more other drugs (or as any combination thereof). Generally, they will be administered as a formulation in association with one or more pharmaceutically acceptable excipients. The term 'excipient' is used herein to describe any ingredient other than the compound(s) of the invention. The choice of excipient will to a large extent depend on factors such as the particular mode of administration, the effect of the excipient on solubility and stability, and the nature of the dosage form.
According to another aspect of the invention, there is provided a pharmaceutical composition including a compound of the formula (1) or a pharmaceutically acceptable salt and/or solvate thereof, as defined in any one of the preceding claims, together with a pharmaceutically acceptable excipient.
Pharmaceutical compositions suitable for the delivery of compounds of the present invention and methods for their preparation will be readily apparent to those skilled in the art. Such compositions and methods for their preparation may be found, for example, in Remington's Pharmaceutical Sciences. 19th Edition (Mack Publishing Company, 1995).
The compounds of the invention may be administered orally. Oral administration may involve swallowing, so that the compound enters the gastrointestinal tract, or buccal or sublingual administration may be employed by which the compound enters the blood stream directly from the mouth.
Formulations suitable for oral administration include solid formulations such as tablets, capsules containing particulates, liquids, or powders, lozenges (including liquid-filled), chews, multi- and nano-particulates, gels, solid solution, liposome, films, ovules, sprays and liquid formulations.
Liquid formulations include suspensions, solutions, syrups and elixirs. Such formulations may be employed as fillers in soft or hard capsules and typically comprise a carrier, for example, water, ethanol, polyethylene glycol, propylene glycol, methylcellulose, or a suitable oil, and one or more emulsifying agents and/or suspending agents. Liquid formulations may also be prepared by the reconstitution of a solid, for example, from a sachet.
The compounds of the invention may also be used in fast-dissolving, fast-disintegrating dosage forms such as those described in Expert Opinion in Therapeutic Patents, H (6), 981-986, by Liang and Chen (2001).
For tablet dosage forms, depending on dose, the drug may make up from 1 weight % to 80 weight % of the dosage form, more typically from 5 weight % to 60 weight % of the dosage form. In addition to the drug, tablets generally contain a disintegrant. Examples of disintegrants include sodium starch glycolate, sodium carboxymethyl cellulose, calcium carboxymethyl cellulose, croscarmellose sodium, crospovidone, polyvinylpyrrolidone, methyl cellulose, microcrystalline cellulose, lower alkyl-substituted hydroxypropyl
cellulose, starch, pregelatinised starch and sodium alginate. Generally, the disintegrant will comprise from 1 weight % to 25 weight %, preferably from 5 weight % to 20 weight % of the dosage form.
Binders are generally used to impart cohesive qualities to a tablet formulation. Suitable binders include microcrystalline cellulose, gelatin, sugars, polyethylene glycol, natural and synthetic gums, polyvinylpyrrolidone, pregelatinised starch, hydroxypropyl cellulose and hydroxypropyl methylcellulose. Tablets may also contain diluents, such as lactose (monohydrate, spray-dried monohydrate, anhydrous and the like), mannitol, xylitol, dextrose, sucrose, sorbitol, microcrystalline cellulose, starch and dibasic calcium phosphate dihydrate.
Tablets may also optionally comprise surface active agents, such as sodium lauryl sulfate and polysorbate 80, and glidants such as silicon dioxide and talc. When present, surface active agents may comprise from 0.2 weight % to 5 weight % of the tablet, and glidants may comprise from 0.2 weight % to 1 weight % of the tablet.
Tablets also generally contain lubricants such as magnesium stearate, calcium stearate, zinc stearate, sodium stearyl fumarate, and mixtures of magnesium stearate with sodium lauryl sulphate. Lubricants generally comprise from 0.25 weight % to 10 weight %, preferably from 0.5 weight % to 3 weight % of the tablet.
Other possible ingredients include anti-oxidants, colourants, flavouring agents, preservatives and taste- masking agents.
Exemplary tablets contain up to about 80% drug, from about 10 weight % to about 90 weight % binder, from about 0 weight % to about 85 weight % diluent, from about 2 weight % to about 10 weight % disintegrant, and from about 0.25 weight % to about 10 weight % lubricant.
Tablet blends may be compressed directly or by roller to form tablets. Tablet blends or portions of blends may alternatively be wet-, dry-, or melt-granulated, melt congealed, or extruded before tabletting. The final formulation may comprise one or more layers and may be coated or uncoated; it may even be encapsulated.
The formulation of tablets is discussed in Pharmaceutical Dosaαe Forms: Tablets. Vol. 1 , by H. Lieberman and L. Lachman (Marcel Dekker, New York, 1980).
Consumable oral films for human or veterinary use are typically pliable water-soluble or water-swellable thin film dosage forms which may be rapidly dissolving or mucoadhesive and typically comprise a compound of formula (1), a film-forming polymer, a binder, a solvent, a humectant, a plasticiser, a stabiliser or emulsifier, a viscosity-modifying agent and a solvent. Some components of the formulation may perform more than one function.
The compound of formula (1) may be water-soluble or insoluble. A water-soluble compound typically comprises from 1 weight % to 80 weight %, more typically from 20 weight % to 50 weight %, of the solutes. Less soluble compounds may comprise a greater proportion of the composition, typically up to 88 weight % of the solutes. Alternatively, the compound of formula (1) may be in the form of multiparticulate beads.
The film-forming polymer may be selected from natural polysaccharides, proteins, or synthetic hydrocolloids and is typically present in the range 0.01 to 99 weight %, more typically in the range 30 to 80 weight %.
Other possible ingredients include anti-oxidants, colorants, flavourings and flavour enhancers, preservatives, salivary stimulating agents, cooling agents, co-solvents (including oils), emollients, bulking agents, anti-foaming agents, surfactants and taste-masking agents.
Films in accordance with the invention are typically prepared by evaporative drying of thin aqueous films coated onto a peelable backing support or paper. This may be done in a drying oven or tunnel, typically a combined coater dryer, or by freeze-drying or vacuuming.
Solid formulations for oral administration may be formulated to be immediate and/or modified release. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
Suitable modified release formulations for the purposes of the invention are described in US Patent No. 6,106,864. Details of other suitable release technologies such as high energy dispersions and osmotic and coated particles are to be found in Pharmaceutical Technology On-line. 25(2), 1-14, by Verma et a/ (2001). The use of chewing gum to achieve controlled release is described in WO 00/35298.
The compounds of the invention may also be administered directly into the blood stream, into muscle, or into an internal organ. Suitable means for parenteral administration include intravenous, intraarterial, intraperitoneal, intrathecal, intraventricular, intraurethral, intrastemal, intracranial, intramuscular and subcutaneous. Suitable devices for parenteral administration include needle (including microneedle) injectors, needle-free injectors and infusion techniques.
Parenteral formulations are typically aqueous solutions which may contain excipients such as salts, carbohydrates and buffering agents (preferably to a pH of from 3 to 9), but, for some applications, they may be more suitably formulated as a sterile non-aqueous solution or as a dried form to be used in conjunction with a suitable vehicle such as sterile, pyrogen-free water.
The preparation of parenteral formulations under sterile conditions, for example, by lyophilisation, may readily be accomplished using standard pharmaceutical techniques well known to those skilled in the art.
The solubility of compounds of formula (1) used in the preparation of parenteral solutions may be increased by the use of appropriate formulation techniques, such as the incorporation of solubility- enhancing agents.
Formulations for parenteral administration may be formulated to be immediate and/or modified release. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release. Thus compounds of the invention may be formulated as a solid, semi-solid, or thixotropic liquid for administration as an implanted depot providing modified release of the active compound. Examples of such formulations include drug-coated stents and poly(cf/-lactic-coglycolic)acid (PGLA) microspheres.
The compounds of the invention may also be administered topically to the skin or mucosa, that is, dermally or transdermally. Typical formulations for this purpose include gels, hydrogels, lotions, solutions, creams, ointments, dusting powders, dressings, foams, films, skin patches, wafers, implants, sponges, fibres, bandages and microemulsions. Liposomes may also be used. Typical carriers include alcohol, water, mineral oil, liquid petrolatum, white petrolatum, glycerin, polyethylene glycol and propylene glycol. Penetration enhancers may be incorporated - see, for example, J Pharm Sci, 88 (10), 955-958, by Finnin and Morgan (October 1999).
Other means of topical administration include delivery by electroporation, iontophoresis, phonophoresis, sonophoresis and microneedle or needle-free (e.g. Powderject™, Bioject™, etc.) injection.
Formulations for topical administration may be formulated to be immediate and/or modified release. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
The compounds of the invention can also be administered intranasally or by inhalation, typically in the form of a dry powder (either alone, as a mixture, for example, in a dry blend with lactose, or as a mixed component particle, for example, mixed with phospholipids, such as phosphatidylcholine) from a dry powder inhaler or as an aerosol spray from a pressurised container, pump, spray, atomiser (preferably an atomiser using electrohydrodynamics to produce a fine mist), or nebuliser, with or without the use of a suitable propellant, such as 1,1 ,1 ,2-tetrafluoroethane or 1 ,1,1,2,3,3,3-heptafluoropropane. For intranasal use, the powder may comprise a bioadhesive agent, for example, chitosan or cyclodextrin.
The pressurised container, pump, spray, atomizer, or nebuliser contains a solution or suspension of the compound(s) of the invention comprising, for example, ethanol, aqueous ethanol, or a suitable alternative agent for dispersing, solubilising, or extending release of the active, a propellant(s) as solvent and an optional surfactant, such as sorbitan trioleate, oleic acid, or an oligolactic acid.
Prior to use in a dry powder or suspension formulation, the drug product is micronised to a size suitable for delivery by inhalation (typically less than 5 microns). This may be achieved by any appropriate comminuting method, such as spiral jet milling, fluid bed jet milling, supercritical fluid processing to form nanoparticles, high pressure homogenisation, or spray drying.
Capsules (made, for example, from gelatin or hydroxypropylmethylcellulose), blisters and cartridges for use in an inhaler or insufflator may be formulated to contain a powder mix of the compound of the invention, a suitable powder base such as lactose or starch and a performance modifier such as /-leucine, mannitol, or magnesium stearate. The lactose may be anhydrous or in the form of the monohydrate, preferably the latter. Other suitable excipients include dextran, glucose, maltose, sorbitol, xylitol, fructose, sucrose and trehalose.
A suitable solution formulation for use in an atomiser using electrohydrodynamics to produce a fine mist may contain from 1μg to 20mg of the compound of the invention per actuation and the actuation volume may vary from 1μl to 100μl. A typical formulation may comprise a compound of formula (1), propylene glycol, sterile water, ethanol and sodium chloride. Alternative solvents which may be used instead of propylene glycol include glycerol and polyethylene glycol.
Suitable flavours, such as menthol and levomenthol, or sweeteners, such as saccharin or saccharin sodium, may be added to those formulations of the invention intended for inhaled/intranasal administration.
Formulations for inhaled/intranasal administration may be formulated to be immediate and/or modified release using, for example, PGLA. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
In the case of dry powder inhalers and aerosols, the dosage unit is determined by means of a valve which delivers a metered amount. Units in accordance with the invention are typically arranged to administer a metered dose or "puff' containing from 1 μg to 4000 μg of the compound of formula (1). The overall daily dose will typically be in the range 1 μg to 20 mg which may be administered in a single dose or, more usually, as divided doses throughout the day. •
The compounds of the invention may be administered rectally or vaginally, for example, in the form of a suppository, pessary, or enema. Cocoa butter is a traditional suppository base, but various alternatives may be used as appropriate.
Formulations for rectal/vaginal administration may be formulated to be immediate and/or modified release. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
The compounds of the invention may also be administered directly to the eye or ear, typically in the form of drops of a micronised suspension or solution in isotonic, pH-adjusted, sterile saline. Other formulations suitable for ocular and aural administration include ointments, biodegradable (e.g. absorbable gel sponges, collagen) and non-biodegradable (e.g. silicone) implants, wafers, lenses and particulate or vesicular systems, such as niosomes or liposomes. A polymer such as crossed-linked polyacrylic acid, polyvinylalcohol, hyaluronic acid, a cellulosic polymer, for example, hydroxypropylmethylcellulose, hydroxyethylcellulose, or methyl cellulose, or a heteropolysaccharide polymer, for
example, gelan gum, may be incorporated together with a preservative, such as benzalkonium chloride. Such formulations may also be delivered by iontophoresis.
Formulations for ocular/aural administration may be formulated to be immediate and/or modified release. Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted, or programmed release. „
The compounds of the invention may be combined with soluble macromolecular entities, such as cyclodextrin and suitable derivatives thereof or polyethylene glycol-containing polymers, in order to improve their solubility, dissolution rate, taste-masking, bioavailability and/or stability for use in any of the aforementioned modes of administration.
Drug-cyclodextrin complexes, for example, are found to be generally useful for most dosage forms and administration routes. Both inclusion and non-inclusion complexes may be used. As an alternative to direct complexation with the drug, the cyclodextrin may be used as an auxiliary additive, i.e. as a carrier, diluent, or solubiliser. Most commonly used for these purposes are alpha-, beta- and gamma- cyclodextrins, examples of which may be found in International Patent Applications Nos. WO 91/11172, WO 94/02518 and WO 98/55148.
Inasmuch as it may desirable to administer a combination of active compounds, for example, for the purpose of treating a particular disease or condition, it is within the scope of the present invention that two or more pharmaceutical compositions, at least one of which contains a compound in accordance with the invention, may conveniently be combined in the form of a kit suitable for coadministration of the compositions.
Thus the kit of the invention comprises two or more separate pharmaceutical compositions, at least one of which contains a compound of formula . (1) in accordance with the invention, and means for separately retaining said compositions, such as a container, divided bottle, or divided foil packet. An example of such a kit is the familiar blister pack used for the packaging of tablets, capsules and the like.
The kit of the invention is particularly suitable for administering different dosage forms, for example, oral and parenteral, for administering the separate compositions at different dosage intervals, or for titrating the separate compositions against one another. To assist compliance, the kit typically comprises directions for administration and may be provided with a so-called memory aid.
For administration to human patients, the total daily dose of the compounds of the invention is typically in the 0.001 mg to 2000 mg depending, of course, on the mode of administration. For example, oral administration may require a total daily dose of from 1 mg to 2000 mg, while an intravenous dose may only require from 0.01 mg to 100 mg. The total daily dose may be administered in single or divided doses and may, at the physician's discretion, fall outside of the typical range given herein.
These dosages are based on an average human subject having a weight of about 60kg to 70kg. The physician will readily be able to determine doses for subjects whose weight falls outside this range, such as infants and the elderly.
For the avoidance of doubt, references herein to "treatment" include references to curative, palliative and prophylactic treatment.
According to another embodiment of the present invention, the compounds of the formula (1), or pharmaceutically acceptable salts, derived forms or compositions thereof, can also be used as a combination with one or more additional therapeutic agents to be co-administered to a patient to obtain some particularly desired therapeutic end result. The second and more additional therapeutic agents may also be a compound of the formula (1), or a pharmaceutically acceptable salt, derived forms or compositions thereof, or one or more histamine H3 receptor ligands known in the art. More typically, the second and more therapeutic agents will be selected from a different class of therapeutic agents.
As used herein, the terms "co-administration", "co-administered" and "in combination with", referring to the compounds of formula (1) and one or more other therapeutic agents, is intended to mean, and does refer to and include the following:
• simultaneous administration of such combination of compound(s) of formula (1) and therapeutic agent(s) to a patient in need of treatment, when such components are formulated together into a single dosage form which releases said components at substantially the same time to said patient,
• substantially simultaneous administration of such combination of compound(s) of formula (1) and therapeutic agent(s) to a patient in need of treatment, when such components are formulated apart from each other into separate dosage forms which are taken at substantially the same time by said patient, whereupon said components are released at substantially the same time to said patient,
• sequential administration of such combination compound(s) of formula (1) and therapeutic agent(s) to a patient in need of treatment, when such components are formulated apart from each other into separate dosage forms which are taken at consecutive times by said patient with a significant time interval between each administration, whereupon said components are released at substantially different times to said patient; and
• sequential administration of such combination of compound(s) of formula (1) and therapeutic agent(s) to a patient in need of treatment, when such components are formulated together into a single dosage form which releases said components in a controlled manner whereupon they are concurrently, consecutively, and/or overlapingly administered at the same and/or different times by said patient, where each part may be administered by either the same or different route.
Suitable examples of other therapeutic agents which may be used in combination with the compound(s) of formula (1), or pharmaceutically acceptable salts, derived forms or compositions thereof, include, but are by no means limited to :
• Histamine H1 receptor antagonists, for instance loratidine, desloratidine, fexofenadine and cetirizine;
• Histamine H4 receptor antagonists;
• Histamine H2 receptor antagonists;
• Leukotriene antagonists, including antagonists of LTB4, LTC4, LTD4, and LTE4, in particular Montelukast;
• Phosphodiesterase inhibitors such as PDE4 inhibitors or PDE5 inhibitors;
• neurotransmitter re-uptake inhibitors, for instance fluoxetine, sertraline, paroxetine, ziprasidone;
• 5-Lipoxygenase (5-LO) inhibitors or 5-lipoxygenase activating protein (FLAP) antagonists;
• <xr and α2-adrenoceptor agonist vasoconstrictor sympathomimetic agents for decongestant use;
• Muscarinic M3 receptor antagonists or anticholinergic agents;
• β2-adrenoceptor agonists;
• Theophylline;
• Sodium cromoglycate;
• COX-1 inhibitors (NSAIDs) and COX-2 selective inhibitors;
• Oral or inhaled Glucocorticosteroids;
• Monoclonal antibodies active against endogenous inflammatory entities;
• Anti-tumor necrosis factor (anti-TNF-α) agents;
• Adhesion molecule inhibitors including VLA-4 antagonists;
• KJnJn-B1 - and B2 -receptor antagonists;
• Immunosuppressive agents,;
• Inhibitors of matrix metalloproteases (MMPs);
• Tachykinin NKi, NK2 and NK3 receptor antagonists;
• Elastase inhibitors;
• , Adenosine A2a receptor agonists;
• Inhibitors of urokinase;
• Compounds that act on dopamine receptors, e.g. D2 agonists;
• Modulators of the NFicβ pathway, e.g. IKK inhibitors;
• Agents that can be classed as mucolytics or anti-tussive;
• Antibiotics;
• modulators of cytokine signalling pathways such as p38 MAP kinase, syk kinase or JAK kinase inhibitors;
• DP1 receptor antagonists;
• prostaglandin CRTH2 receptor antagonists
• HDAC (histone deacetylase) inhibitors and
• PI3 kinase inhibitors.
According to the present invention, combination of the compounds of formula (1) with :
• Histamine H1 receptor antagonists, for instance loratidine, desloratidine, fexofenadine and cetirizine
• Histamine H4 receptor antagonists
• Histamine H2 receptor antagonists
• Leukotriene antagonists, including antagonists of LTB4, LTC4, LTD4, and LTE4, in particular Montelukast
• Phosphodiesterase PDE4 inhibitors
• neurotransmitter re-uptake inhibitors, for instance fluoxetine, sertraline, paroxetine, ziprasidone
• DP1 receptor antagonists and
• prostaglandin CRTH2 receptor antagonists are preferred.
The compounds of formula (1) have the ability to interact with the H3 receptor and thereby have a wide range of therapeutic applications, as described further below, because of the essential role which the H3 receptor plays in the physiology of all mammals. According to this invention H3 ligands are meant to include H3 receptor antagonists, agonists and inverse agonists. For the preferred indications to be treated according to the invention, H3 antagonists are believed to be most suitable.
In another aspect of the invention there is provided a compound of the formula (1) as defined in herein, or a pharmaceutically acceptable salt and/or solvate thereof, for use as a medicament.
A further aspect of the present invention relates to the compounds of formula (1), or pharmaceutically acceptable salts, derived forms or compositions thereof, for use in the treatment of diseases, disorders, and conditions in which the H3 receptor is involved. More specifically, the present invention also concerns the compounds of formula (1), or pharmaceutically acceptable salts, derived forms or compositions thereof, for use in the treatment of diseases, disorders, and conditions selected from the group consisting of :
• diseases of the central nervous system: sleep disorders, migraine, dyskinesia, stress-induced anxiety, psychotic disorders, epilepsy, Cognition deficiency diseases such as Alzheimer's disease or mild cognitive impairment, depression, mood disorders, schizophrenia, anxiety disorders, attention-deficit hyperactivity disorder (ADHD), psychotic disorders, obesity, dizziness, vertigo, epilepsy, motion sickness
• inflammatory diseases
• respiratory diseases (adult respiratory distress syndrome, acute respiratory distress syndrome, bronchitis, chronic bronchitis, chronic obstructive pulmonary disease, cystic fibrosis, asthma, emphysema, rhinitis, chronic sinusitis), allergy, allergy-induced airway responses, allergic rhinitis, viral rhinitis, non-allergic rhinitis, perennial and seasonal rhinitis, nasal congestion, allergic congestion
• Female sexual dysfunction including hypoactive sexual desire disorder, sexual arousal disorder, orgasmic disorder and sexual pain disorder
• Male sexual dysfunction including male desire disorders, male erectile dysfunction, male orgasmic disorders such as premature ejaculation
• cardiac dysfunctions such as myocardial ischaemia and arrythmia
• diseases of the gastrointestinal tract such as inflammatory bowel disease, Crohn's disease and colitis ulcerosa
• cancer
• hypotension
• obesity;
• pain and
• overactive bladder conditions.
The compounds of formula (1 ) of the invention are particularly suitable for the treatment of allergy, allergy- induced airway responses, allergic rhinitis, viral rhinitis, non-allergic rhinitis, perennial and seasonal rhinitis, nasal congestion, allergic congestion.
A stiil further aspect of the present invention also relates to the use of the compounds of formula (1), or pharmaceutically acceptable salts, derived forms or compositions thereof, for the manufacture of a drug being a H3 ligand. In particular, the present inventions concerns the use of the compounds of formula (1), or pharmaceutically acceptable salts, derived forms or compositions thereof, for the manufacture of a drug for the treatment of H3-mediated diseases and/or conditions, in particular the diseases and/or conditions listed above.
As a consequence, the present invention provides a particularly interesting method to treat a mammal, including a human being, with an effective amount of a compound of formula (1), or a pharmaceutically acceptable salt, derived form or composition thereof. More precisely, the present invention provides a particularly interesting method for the treatment of a H3-mediated diseases and/or conditions in a mammal, including a human being, in particular the diseases and/or conditions listed above, comprising administering to said mammal an effective amount of a compound of formula (1), its pharmaceutically acceptable salts and/or derived forms.
EXAMPLES
Glossary
APCI atmospheric pressure chemical ionisation br broad δ chemical shift (in ppm) d doublet
LRMS low resolution mass spectrum m muliplet m/z mass charge ratio
NMR nuclear magnetic resonance q quartet
S singlet t triplet
The following examples illustrate the preparation of pyridyl derivatives of general formula (1)
Intermediate 1: (θ-Chloropyridin-S-yDacetonitrile
To a solution of 2-chloro-5-(chlofomethyl)pyridine (75g, 0.46mol) in ethanol (120ml) stirring at O0C, was added a solution of sodium cyanide (24.5Og, 0.50mol) in water (40ml). The reaction mixture heated at 10O0C for 2 hours then stirred at room temperature for a further 18 hours. The solvent was evaporated in vacuo and the residue extracted from water into dichloromethane (500ml), washed with brine, dried over sodium sulphate, filtered and evaporated in vacuo. The crude material was purified by column chromatography over silica gel eluting with dichloromethane. The title compound was obtained as a brown oil which solidified on standing (67g, 0.44mol, 96%). 1H-NMR (CDCI3, 400MHz); δ 3.7 (s, 2H), 7.4 (d, 1H), 7.65 (d, 1H), 8.4 (s, 1H).
Intermediate 2: 4-(6-Chloropyridin-3-yl)tetrahvdro-2H-pyran-4-carbonitrile
Sodium hydride (60% in mineral oil) (38.65g, 0.97mol) was added portionwise to an ice cooled solution of (6-chIoropyridin-3-yl)acetonitrile (67g, 0.44mol) in tetrahydrofuran, maintaining the reaction temperature below 2O0C. The reaction mixture was stirred for 20 minutes after which time 2-bromoethyl ether (61ml, 0.48mol) was added dropwise, maintaining the reaction temperature below 3O0C. Potassium iodide (8Og, 0.48mol) was added and the reaction mixture stirred at room temperature for 3 hours. The mixture was poured into water, extracted into diethyl ether (3x1000ml), dried over sodium sulphate, filtered and evaporated in vacuo. The crude material was purified by column chromatography over silica gel eluting with 1:4 to 1:1 ethyl acetate: pentane. The material was recrystallised from ethyl acetate: pentane to yield the title compound as a pale yellow solid (48g, 0.22mol, 50%). 1H-NMR (CDCI3, 400MHz); δ 2.1 (m, 4H), 3.9 (m, 2H), 4.1 (m, 2H)1 7.4 (d, 1H), 7.8 (d, 1H), 8.6 (s, 1H).
Intermediate 3: 2-Nitro-N-(f4f6-(3-pyrrolidin-1-yl propoxy)pyridine-3-vntetrahvdro-2H-Pyran-4- yllmethvPaniline
1-{4-[6-(3-Pyrrolidin-1-ylpropoxy)pyridin-3-yl]tetrahydro-2H-pyran-4-yl}methanamine (400mg, 1.25mmol), 2-fluoronitrobenzene (0.145ml, 1.37mmol) and potassium carbonate (190mg, 1.37mmol) were combined in tetrahydrofuran (8ml) and the reaction mixture stirred at room temperature for 3 hours. The solvent was evaporated in vacuo and the residue redissolved in dimethylformamide then stirred at room temperature for 18 hours. The mixture was diluted with ethyl acetate and washed with water, brine, dried over sodium sulphate, filtered and evaporated in vacuo. The crude material was purified by column chromatography using an ISCO® silica cartridge eluting with dichloromethane: methanol: ammonia (99:1:0.1 to 95:5:0.5). The title compound was obtained as a yellow oil (310mg, 0.70mmol, 56%).
1H-NMR (CDCI3, 400MHz); jS 1.75 (m, 4H), 2.0 (m, 4H), 2.3 (m, 2H), 2.5 (m, 4H), 2.6 (t, 2H), 3.4 (d, 2H), 3.6 (m, 2H), 3.8 (m, 2H), 4.3 (t, 2H), 6.6 (t, 1H), 6.7 (d, 1 H), 6.75 (d, 1 H), 7.3 (m, 1 H), 7.6 (m, 1 H), 7.9 (br t, 1H), 8.1 (d, 1H), 8.2 (s, 1H).
Intermediate 4: 4-rβ-f3-Pyrrolidin-1 -yl propoxy)pyridine-3-ylltetrahvdro-2H-pyran-4-carbothioamide
Diethyl dithiophosphate (5.9g, 31.7mmol) was stirred at O0C, 4-[6-(3-pyrrolidin-1-ylpropoxy)pyridin-3- y|]tetrahydro-2H-pyran-4-carbonitrile (2.Og, 6.35mmol) was added dropwise and the solution stirred at O0C for 1 hour. The mixture was allowed to warm to room temperature, water (1.2ml) was added and the reaction mixture heated to 800C for 30 minutes then stirred at room temperature for 18 hours. The mixture was extracted from a saturated aqueous solution of sodium hydrogen carbonate (200ml) into ethyl acetate (250ml), dried and the solvent evaporated in vacuo. The crude material was purified by column chromatography over silica gel eluting with dichloromethane: methanol: 0.880 ammonia (90:10:1). The title compound was obtained as a pale yellow oil (1.46g, 4.18mmol, 66%).
1H-NMR (CDCI3, 400MHz); .δ 2.1 (m, 2H), 2.25 (m, 4H), 2.4 (m, 2H), 2.65 (m, 2H), 2.9 (m, 2H), 3.4 (m, 2H), 3.6 (m, 2H), 3.85 (m, 2H)1 4.1 (m, 2H), 4.4 (t, 2H), 6.75 (d, 1H), 7.3 (br s, 1 H), 7.65 (br s, 1H), 7.7 (m, 1 H)1 8.3 (m, 1 H).
Example 1 : 4-r6-(3-Pyrrolidin-1 -ylpropoxy)pyridin-3-vntetrahvdro-2H-pyran-4-carbonitrile

Potassium f-butoxide (11.35g, 101.3mmol) was added portionwise to a solution of 3-pyrrolidin-1-ylpropan- 1-ol {Australian Journal of Chemistry, 52, 12, 1131-1138, 1999) (13.05g, 101.3mmol) in tetrahydrofuran (150ml), maintaining the reaction temperature below 3O0C. The reaction mixture was stirred at room temperature for 30 minutes. 4-(6-Chloropyridin-3-yl)tetrahydro-2H-pyran-4-carbonitrile (15g, 67.4mmol) was added portionwise and the reaction mixture stirred at room temperature for 20 hours. The mixture was diluted with diethyl ether (200ml), washed with water (100ml), brine (100ml), dried over magnesium sulphate, filtered and evaporated in vacuo. The crude material was purified by column chromatography using a Combiflash® silica cartridge eluting with dichloromethane to dichloromethane: methanol: ammonia (80:20:2). The title compound was obtained as an orange oil (13.55g, 43mmol, 64%). 1H-NMR (CDCI3, 400MHz); .δ 1.8 (m, 4H), 2.0 (m, 6H)1 2.5 (m, 4H), 2.6 (m, 2H), 3.9 (m, 2H), 4.1 (m, 2H), 4.35 (t, 2H), 6.8 (d, 1 H), 7.6 (d, 1 H), 8.25 (s, 1 H).
Example 2: 1-(4-r6-(3-Pyrrolidin-1-ylpropoxy)pyridin-3-vπtetrahvdro-2H-pyran-4-yl)methanamine

4-[6-(3-Pyrrolidin-1 -ylpropoxy)pyridin-3-yl]tetrahydro-2H-pyran-4-carbonitrile (2.Og1 6.34mmol) was dissolved in ethanol (80ml), platinum oxide (1.0g) and a 1N solution of hydrochloric acid in diethyl ether (6.34ml, 6.34mmol) were added and the reaction mixture stirred at room temperature, under 4
atmospheres of hydrogen for 5 hours. The reaction mixture was filtered through arbocel® and the filtrate evaporated in vacuo. The crude material was purified by column chromatography using a Combifiash® silica cartridge eluting with dichloromethane: methanol: ammonia (95:5:0.5 to 85:15:1.5), to yield the title compound (0.9g, 2.8mmol, 44%).
1H-NMR (CDCI3, 400MHz): j5 1.8 (m, 6H), 2.0 (m, 4H), 2.5 (m, 4H), 2.6 (m, 2H), 2.8 (s, 2H), 3.5 (m, 2H)1 3.8 (m, 2H), 4.4 (t, 2H), 6.8 (d, 1H), 7.5 (d, 1H), 8.1 (s, 1 H).
Example 3: 4-^4-r6-f3-Pyrrolidin-1-ylpropoxy)pyridin-3-yl1tetrahvdro-2H-pyran-4-yl}methyl)morpholine

1 -{4-[6-(3-Pyrrolidin-1 -ylpropoxy)pyridin-3-yl]tetrahydro-2H-pyran-4-yl}methanamine (2.3Og, 7.20mmol) and potassium carbonate (2.19g, 15.84mmol) were suspended in acetonitrile, 2-bromoethyl ether (0.3ml, 2.64mmol) was added and the reaction mixture heated at 6O0C for 30 minutes. 2-bromoethyl ether (0.3ml, 2.64mmol) was added and the reaction mixture heated at 6O0C for a further 30 minutes before the addition of the final portion of 2-bromoethyl ether (0.3ml, 2.64mmol). The mixture was heated at 6O0C for 4 hours. The solvent was evaporated in vacuo and the residue extracted from water into ethyl acetate, dried over sodium sulphate, filtered and evaporated in vacuo. The crude material was purified by column chromatography over silica gel eluting with dichloromethane: methanol: ammonia (94:6:0.6 to 90:10:1) to yield the title compound (730mg, 1.87mmol, 26%).
1H-NMR (CDCI3, 400MHz); δ 1.8 (m, 6H), 2.05 (m, 4H), 2.2 (m, 4H), 2.4 (s, 2H), 2.7 (m, 6H), 3.5 (m, 6H), 3.7 (m, 2H), 4.3 (t, 2H)1 6.7 (d, 1 H), 7.6 (d, 1H), 8.05 (s, 1H).
Example 4: 1-({4-f6-(3-Pyrrolidin-1-ylpropoxy)pyridin-3-yl1tetrahvdro-2H-pyran-4-yl>methyl)-1 H- benzimidazole

2-Nitro-N-({4[6-(3-pyrrolidin-1-yl propoxy)pyridine-3-yl]tetrahydro-2H-pyran-4-yl}methyl)aniline (300mg, 0.69mmol), formic acid (0.13ml, 3.45mmol), trimethylorthoformate (1.5ml, 13.7mmol) and 10% palladium on carbon (30mg) were combined in ethanol (20ml) and stirred at room temperature under 3.4 atmospheres of hydrogen for 5 hours. The reaction mixture was filtered through arbocel® and the filtrate evaporated in vacuo. The residue was dissolved in ethyl acetate, washed with an aqueous solution of sodium carbonate, brine, dried over sodium sulphate, filtered and evaporated in vacuo. The crude material
was purified by column chromatography using an ISCO® silica cartridge eluting with dichloromethane: methanol: ammonia (98:2:0.2 to 95:5:0.5). The title compound was obtained as a white powder (210mg, O.δmmol, 72%).
1H-NMR (CDCI3, 400MHz); δ 1.8 (m, 4H), 2.0 (m, 4H), 2.2 (m, 2H), 2.5 (m, 4H), 2.6 (t, 2H), 3.4 (m, 2H), 3.8 (m, 2H), 4.2 (s, 2H), 4.3 (t, 2H), 6.6 (d, 1H), 7.0 (d, 1H), 7.2 (m, 4H)1 7.7 (d, 1H), 7.9 (s, 1 H).
Example 5: 2-(3-Pyrrolidin-1-yl propoxy)-5-[4-(1 ,3-thiazol-2-yl)tetrahydro-2H-pyran-4-vπpyridine

4-[6-(3-Pyrrolidin-1-yl propoxy)pyridine-3-yl]tetrahydro-2H-pyran-4-carbothioamide (500mg, 1.4mmol), bromoacetaldehyde diethyl acetal (280mg, 1.4mmol) and a 48% aqueous solution of hydrogen bromide (0.3ml) were combined in ethanol (2ml) and heated to reflux for 4 hours. The solvent was evaporated in vacuo and the residue redissolved in dichloromethane (50ml), washed with a saturated aqueous solution of sodium hydrogen carbonate (2x25ml), dried over magnesium sulphate, filtered and evaporated in vacuo. The crude material was purified by column chromatography using an ISCO® silica cartridge eluting with dichloromethane: methanol: ammonia (99:1:0.1 to 90:10:1) to give the title compound (26mg, 0.07mmol, 5%).
1H-NMR (CDCI3, 400MHz); δ 1.75 (m, 4H), 2.0 (m, 2H), 2.4 (m, 2H), 2.55 (m, 4H), 2.6 (m, 4H), 3.7 (m, 2H), 3.85 (m, 2H), 4.3 (t, 2H), 6.65 (d, 1H), 7.25 (m, 1H), 7.5 (m, 1H), 7.7 (d, 1H), 8.1 (m, 1H). '
Example 6: 5-r4-(methyl-1 ,3-thiazol-2-yl)tetrahvdro-2H-pyran-4-vπ-2-(3-pyrrolidin-1-yl propoxy)pyridine

4-[6-(3-Pyrrolidin-1-yl propoxy)pyridine-3-yl]tetrahydro-2H-pyran-4-carbothioamide (500mg, 1.4mmol) and chloroacetone (129mg, 1.4mmol) were combined in ethanol (2ml) and the reaction mixture heated at reflux for 4 hours. The solvent was evaporated in vacuo and the residue dissolved in dichloromethane, washed with a saturated aqueous solution of sodium hydrogen carbonate, dried over magnesium sulphate, filtered and evaporated in vacuo. The crude material was purified by column chromatography using an ISCO® silica cartridge eluting with dichloromethane: methanol: ammonia (99:1:0.1 to 90:10:1). The title compound was obtained as a colourless oil (352mg, 0.91 mmol, 65%).
1H-NMR (CDCI3, 400MHz); δ 1.8 (m, 4H), 2.0 (m, 2H), 2.35 (m, 2H), 2.4 (s, 3H)1 2.5 (m, 4H), 2.6 (m, 4H), 3.7 (m, 2H), 3.8 (m, 2H)1 4.3 (t, 2H), 6.6 (d, 1H), 6.8 (s, 1H), 7.5 (m, 1H), 8.05 (s, 1 H).
H5 Cell Based Functional Assay
Compounds were evaluated using a cell based functional assay measuring cAMP through β-lactamase reporter gene activity. A stable cell line was generated from HEK-293 cells expressing a CRE β-lactamase reporter gene and transfected with human histamine H3 receptor cDNA. Cells were seeded at a density of 500,000 cells/ml, and grown overnight in MEM (Invitrogen) supplemented with 1% dialysed FBS (Sigma), 2mM glutamine (Sigma), 1mM sodium pyruvate (Sigma), 0.1mM non essential amino acids (Invitrogen) and 25mM HEPES (Sigma) in poly D lysine coated 384 well plates (BD Biosciences). H3 receptor agonist imetit (Tocris) dose dependently inhibited 10μM forskolin (Calbiochem) stimulated synthesis of cAMP measured after 4.5hours by β-lactamase cleavage of CCF4-AM dye (Invitrogen). For IC50 determination, test compounds were prepared in PBS (Sigma) and DMSO (Sigma) at a dose response of 5x10"10 to 5x10"5M with a final DMSO concentration in the assay of 0.5%. Cells were incubated for 15 minutes plus/minus compound and their ability to permit 10μM forskolin-stimulated cAMP synthesis in the presence of 1 nM imetit was measured as described above. Functional K1 values were calculated from the IC50 of compounds tested as antagonists based on an experimentally determined imetit EC50 (represented in the equation as Kd) of 35OpM, and an imetit concentration [L] of 1 nM, according to the Cheng-Prussoff equation where K, = (IC50)/(1+([L]/Kd)).
All the examples were tested in the assay described above and they have found to have a K1 value of less than 1 μM. The data for examples 1-5 are given below by way of example:

Claims
1. A compound of general formula (1 ) :

X is selected from -CN, -CH2NR1R2, -C(O)NR3R4, -CH2-het1 and het2, the groups net1 and het2 being optionally substituted by one or two substituents independently selected from halo, cyano, (Ci-C4)alkyl and (C1-C4JaIkOXy;
R1 and R2 are each independently selected from hydrogen, (CrC4)alkyl and (C3-C6)cycloalkyl, said (C1- C4)alkyl and (C3-C6)cycloalkyl being optionally substituted by one group selected from (C1-C4JaIkOXy and (C3-C6)cycloalkyl, or alternatively R1 and R2 together with the nitrogen atom to which they are attached form a 4-, 5-, 6- or 7-membered saturated heterocycle wherein one carbon atom may be optionally replaced by a nitrogen or an oxygen atom, and wherein said saturated heterocycle is optionally substituted by one or two groups independently selected from (CrC4)alkyl, (CrC4)alkoxy or-C(0)(CrC4)alkyl;
R3 and R4 are each independently selected from hydrogen, (CrC4)alkyl and (C3-C6)cycloalkyl, said (C1- C4)alkyl and (C3-C6)cycloalkyl being optionally substituted by one group selected from (CrC4)alkoxy and (C3-C6)cycloalkyl, or alternatively R3 and R4 together with the nitrogen atom to which they are attached form a 4-, 5-, 6- or 7-membered saturated heterocycle wherein one carbon atom may be optionally replaced by a nitrogen or an oxygen atom, and wherein said saturated heterocycle is optionally substituted by one or two groups independently selected from (CrC4)alkyl, (C-ι-C4)alkoxy or -C(O)(Ci-C4)alkyl;
het1 is N-linked and is selected from monocyclic or bicyclic heteroaromatic groups having 5, 6, 7, 8 or 9 ring members, which contain 1, 2, 3 or 4 heteroatom(s) selected from nitrogen, oxygen and sulphur;
het2 is C-linked and is selected from monocyclic or bicyclic heteroaromatic groups having 5, 6, 7, 8, 9 or 10 ring members, which contain 1, 2, 3 or 4 heteroatom(s) selected from nitrogen, oxygen and sulphur;
and R is either a group of formula :
R \
N-L —
R6/ wherein * represents the attachment point to the oxygen atom, L is a straight chain or branched (C2- C6)alkylene and R5 and R6 are each independently selected from hydrogen, (CrC4)alkyl or (C3- C6)cycloalkyl, or alternatively R5 and R6 together with the nitrogen atom to which they are attached form a 4-, 5-, 6- or 7-membered saturated heterocycle wherein said .saturated heterocycle is optionally substituted by one or two groups independently selected from (CrC4)alkyl and (C1-C4JaIkOXy, or alternatively R is a group of formula:

2. A compound of general formula (1) according to claim 1 or a pharmaceutically acceptable salt and/or solvate thereof wherein:
X is selected from -CN, -CH2NR1R2, -CH2-het1 and het2, net2 being optionally substituted once by (C1-
C4)alkyl;
R1 and R2 are both hydrogen atoms or alternatively R1 and R2 together with the nitrogen atom to which they are attached form a 4-, 5-, 6- or 7-membered saturated heterocycle wherein one carbon atom may be optionally replaced by a nitrogen or an oxygen atom; het1 is a N-linked bicyclic heteroaromatic group having 9 ring members which contains a total of two nitrogen atoms; and het2 is a C-linked monocyclic heteroaromatic group having 5 ring members which contains one nitrogen atom and one sulphur atom.
3. A compound of general formula (1) according to claim 1 or a pharmaceutically acceptable salt and/or solvate thereof wherein X selected from -CN, -CH2-NH2, -CH2-morpholine, -CH2-benzimidazole, 1,3- thiazol-2-yl and 1,3-thiazol-2-yl substituted by a (CrC4)alkyl group.
4. A compound of general formula (1) according to any one of claims 1 to 3 or a pharmaceutically acceptable salt and/or solvate thereof wherein R is a group of formula :
R\
N-L — *
R6/ wherein * represents the attachment point to the oxygen atom, L is a straight chain or branched (C2- C6)alkylene and R5 and R6 together with the nitrogen atom to which they are attached form a 4-, 5-, 6- or 7-membered saturated heterocycle wherein said saturated heterocycle is optionally substituted by one or two groups independently selected from (CrC4)alkyl and (CrC4)alkoxy.
5. A compound of general formula (1) according to claim 4 or a pharmaceutically acceptable salt and/or solvate thereof wherein R5 and R6 together with the nitrogen atom to which they are attached form a 5- or 6-membered saturated heterocycle.
6. A compound of general formula (1) according to claim 4 or 5 or a pharmaceutically acceptable salt aanndd//oorr ssoollvvaattee tthheerreeof wherein R5 and R6 together with the nitrogen atom to which they are attached form a pyrrolidinyl group.
7. A compound of general formula (1) according to any one of claims 4 to 6 or a pharmaceutically acceptable salt and/or solvate thereof wherein L is propylene.
8. A compound of general formula (1) according to claim 1 or a pharmaceutically acceptable salt and/or solvate thereof selected from:
4-[6-(3-pyrrolidin-1-ylpropoxy)pyridin-3-yl]tetrahydro-2H-pyran-4-carbonitrile; 1-{4-[6-(3-pyrrolidin-1-ylpropoxy)pyridin-3-yl]tetrahydro-2H-pyran-4-yl}methanamine; 4-({4-[6-(3-pyrrolidin-1-ylpropoxy)pyridin-3-yl]tetrahydro-2H-pyran-4-yl}methyl)morpholine; 1-({4-[6-(3-pyrrolidin-1-ylpropoxy)pyridin-3-yl]tetrahydro-2H-pyran-4-yl}methyl)-1 H-benzimidazole; 2-(3-pyrrolidin-1 -ylpropoxy)-5-[4-(1 ,3-thiazol-2-yl)tetrahydro-2H-pyran-4-yl]pyridine and 5-[4-(4-methyl-1 ,3-thiazol-2-yl)tetrahydro-2H-pyran-4-yl]-2-(3-pyrrolidin-1-ylpropoxy)pyridine.
9. A pharmaceutical composition including a compound of general formula (1) or a pharmaceutically acceptable salt and/or solvate thereof, as defined in any one of claims 1 to 8, together with a pharmaceutically acceptable excipient.
10. A compound of general formula (1) as defined in any one of claims 1 to 8 or a pharmaceutically acceptable salt and/or solvate thereof, for use as a medicament.
11. The use of a compound of general formula (1) as defined in any one of claims 1 to 8 or of a pharmaceutically acceptable salt and/or solvate thereof, for the treatment of a disease for which a H3 ligand is indicated.
12. The use of a compound of general formula (1) according to claim 11 or a pharmaceutically acceptable salt and/or solvate thereof wherein said disease is selected from:
• diseases of the central nervous system: sleep disorders, migraine, dyskinesia, stress-induced anxiety, psychotic disorders, epilepsy, Cognition deficiency diseases such as Alzheimer's disease or mild cognitive impairment, depression, mood disorders, schizophrenia, anxiety disorders, attention-deficit hyperactivity disorder (ADHD), psychotic disorders, obesity, dizziness, vertigo, epilepsy, motion sickness
• inflammatory diseases
• respiratory diseases (adult respiratory distress syndrome, acute respiratory distress syndrome, bronchitis, chronic bronchitis, chronic obstructive pulmonary disease, cystic fibrosis, asthma, emphysema, rhinitis, chronic sinusitis), allergy, allergy-induced airway responses, allergic rhinitis, viral rhinitis, non-allergic rhinitis, perennial and seasonal rhinitis, nasal congestion, allergic congestion
• Female sexual dysfunction including hypoactive sexual desire disorder, sexual arousal disorder, orgasmic disorder and sexual pain disorder • Male sexual dysfunction including male desire disorders, male erectile dysfunction, male orgasmic disorders such as premature ejaculation
• cardiac dysfunctions such as myocardial ischaemia and arrythmia
• diseases of the gastrointestinal tract such as inflammatory bowel disease, Crohn's disease and colitis ulcerosa
• cancer
• hypotension
• obesity
• pain and
• overactive bladder conditions.
13. The use of a compound of general formula (1) as defined in any one of claims 1 to 8 or of a pharmaceutically acceptable salt and/or solvate thereof, for the manufacture of a medicament to treat a disease for which a H3 ligand is indicated.
14. The use of a compound of general formula (1) or a pharmaceutically acceptable salt and/or solvate thereof according to claim 13 wherein said disease is selected from those of claim 12.
15. A method of treatment of a mammal, including a human being, suffering from a disease for which a H3 ligand is indicated, comprising administering to said mammal an effective amount of a compound of the formula (1) as defined in any one of claims 1 to 8 or a pharmaceutically acceptable salt and/or solvate thereof, or composition thereof.
16. A method of treatment of a mammal according to claim 15 wherein said disease is selected from those of claim 12.
17. A combination of a compound of general formula (1) as defined in any one of claims 1 to 8 or a pharmaceutically acceptable salt and/or solvate thereof with another pharmacologically active agent.
18. A combination according to claim 17 wherein said other pharmacologically active agent is selected from:
• Histamine H1 receptor antagonists, for instance loratidine, desloratidine, fexofenadine and cetirizine
• Histamine H4 receptor antagonists
• Histamine H2 receptor antagonists
• Leukotriene antagonists, including antagonists of LTB4, LTC4, LTD4, and LTE4, in particular Montelukast
• Phosphodiesterase inhibitors such as PDE4 inhibitors or PDE5 inhibitors,
• neurotransmitter re-uptake inhibitors, for instance fluoxetine, sertraline, paroxetine, ziprasidone
• 5-Lipoxygenase (5-LO) inhibitors or 5-lipoxygenase activating protein (FLAP) antagonists,
• OC1- and α2-adrenoceptor agonist vasoconstrictor sympathomimetic agents for decongestant use,
• Muscarinic M3 receptor antagonists or anticholinergic agents, β2-adrenoceptor agonists, .
Theophylline,
Sodium cromoglycate,
COX-1 inhibitors (NSAIDs) and COX-2 selective inhibitors,
Oral or inhaled Glucocorticosteroids,
Monoclonal antibodies active against endogenous inflammatory entities,
Anti-tumor necrosis factor (anti-TNF-α) agents,
Adhesion molecule inhibitors including VLA-4 antagonists,
KJnJn-B1 - and B2 -receptor antagonists,
Immunosuppressive agents,
Inhibitors of matrix metalloproteases (MMPs),
Tachykinin NK1, NK2 and NK3 receptor antagonists,
Elastase inhibitors,
Adenosine A2a receptor agonists,
Inhibitors of urokinase,
Compounds that act on dopamine receptors, e.g. D2 agonists,
Modulators of the NFxβ pathway, e.g. IKK inhibitors,
Agents that can be classed as mucolytics or anti-tussive, antibiotics, modulators of cytokine signalling pathways such as p38 MAP kinase, syk kinase or JAK kinase inhibitors,
DP1 receptor antagonists, prostaglandin CRTH2 receptor antagonists,
HDAC (histone deacetylase) inhibitors and
PI3 kinase inhibitors.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US72914805P | 2005-10-20 | 2005-10-20 | |
| US60/729,148 | 2005-10-20 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007045989A1 true WO2007045989A1 (en) | 2007-04-26 |
Family
ID=37670745
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2006/002951 WO2007045989A1 (en) | 2005-10-20 | 2006-10-12 | Pyridyl derivatives useful as h3 ligands |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2007045989A1 (en) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013064462A1 (en) | 2011-10-31 | 2013-05-10 | Bayer Intellectual Property Gmbh | Substituted 4-cyano-3-phenyl-4-(pyridin-3-yl)butanoates, processes for preparation thereof and use thereof as herbicides and plant growth regulators |
| WO2013092500A1 (en) | 2011-12-19 | 2013-06-27 | Bayer Intellectual Property Gmbh | Substituted 4-cyan-3-phenyl-4-(pyridine-3-yl)butanoates, processes for preparation thereof and use thereof as herbicides and plant growth regulators. |
| WO2014024195A1 (en) * | 2012-08-09 | 2014-02-13 | Can-Fite Biopharma Ltd. | A3 adenosine receptor ligands for use in treatment of a sexual dysfunction |
| WO2014063642A1 (en) | 2012-10-25 | 2014-05-01 | 中国中化股份有限公司 | Substituted pyrimidine compound and uses thereof |
| WO2015085935A1 (en) | 2013-12-13 | 2015-06-18 | 中国中化股份有限公司 | Pyrazolyl pyrimidinamine compound and application thereof |
| WO2018167677A1 (en) | 2017-03-15 | 2018-09-20 | Oat & Iil India Laboratories Private Limited | Novel dithiolane compound or a salt or an n-oxide and use thereof |
| US10342786B2 (en) | 2017-10-05 | 2019-07-09 | Fulcrum Therapeutics, Inc. | P38 kinase inhibitors reduce DUX4 and downstream gene expression for the treatment of FSHD |
| US11291659B2 (en) | 2017-10-05 | 2022-04-05 | Fulcrum Therapeutics, Inc. | P38 kinase inhibitors reduce DUX4 and downstream gene expression for the treatment of FSHD |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002012214A2 (en) * | 2000-08-08 | 2002-02-14 | Ortho Mcneil Pharmaceutical Inc. | Non-imidazole aryloxyalkylamines as h3 receptor ligands |
| WO2004056369A1 (en) * | 2002-12-20 | 2004-07-08 | Glaxo Group Limited | Benzo ‘ d!azepine derivatives for the treatment of neurological disorders |
| WO2005108384A1 (en) * | 2004-05-07 | 2005-11-17 | Warner-Lambert Company Llc | 3- or 4-monosubstituted phenol and thiophenol derivatives useful as h3 ligands |
-
2006
- 2006-10-12 WO PCT/IB2006/002951 patent/WO2007045989A1/en active Application Filing
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2002012214A2 (en) * | 2000-08-08 | 2002-02-14 | Ortho Mcneil Pharmaceutical Inc. | Non-imidazole aryloxyalkylamines as h3 receptor ligands |
| WO2004056369A1 (en) * | 2002-12-20 | 2004-07-08 | Glaxo Group Limited | Benzo ‘ d!azepine derivatives for the treatment of neurological disorders |
| WO2005108384A1 (en) * | 2004-05-07 | 2005-11-17 | Warner-Lambert Company Llc | 3- or 4-monosubstituted phenol and thiophenol derivatives useful as h3 ligands |
Non-Patent Citations (1)
| Title |
|---|
| STARK H: "Recent advances in histamine H3/H4 receptor ligands", EXPERT OPINION ON THERAPEUTIC PATENTS, ASHLEY PUBLICATIONS, GB, vol. 13, no. 6, 2003, pages 851 - 865, XP002353598, ISSN: 1354-3776 * |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2013064462A1 (en) | 2011-10-31 | 2013-05-10 | Bayer Intellectual Property Gmbh | Substituted 4-cyano-3-phenyl-4-(pyridin-3-yl)butanoates, processes for preparation thereof and use thereof as herbicides and plant growth regulators |
| WO2013092500A1 (en) | 2011-12-19 | 2013-06-27 | Bayer Intellectual Property Gmbh | Substituted 4-cyan-3-phenyl-4-(pyridine-3-yl)butanoates, processes for preparation thereof and use thereof as herbicides and plant growth regulators. |
| WO2014024195A1 (en) * | 2012-08-09 | 2014-02-13 | Can-Fite Biopharma Ltd. | A3 adenosine receptor ligands for use in treatment of a sexual dysfunction |
| EP3530273A3 (en) * | 2012-08-09 | 2020-02-26 | Can-Fite Biopharma Ltd. | A3 adenosine receptor ligands for use in treatment of a sexual dysfunction |
| US9549943B2 (en) | 2012-08-09 | 2017-01-24 | Can-Fite Biopharma Ltd. | A3 adenosine receptor ligands for use in treatment of a sexual dysfunction |
| WO2014063642A1 (en) | 2012-10-25 | 2014-05-01 | 中国中化股份有限公司 | Substituted pyrimidine compound and uses thereof |
| US9682962B2 (en) | 2013-12-13 | 2017-06-20 | Shenyang Sinochem Agrochemicals R&D Co., Ltd. | Pyrazolyl pyrimidinamine compound and application thereof |
| WO2015085935A1 (en) | 2013-12-13 | 2015-06-18 | 中国中化股份有限公司 | Pyrazolyl pyrimidinamine compound and application thereof |
| WO2018167677A1 (en) | 2017-03-15 | 2018-09-20 | Oat & Iil India Laboratories Private Limited | Novel dithiolane compound or a salt or an n-oxide and use thereof |
| US10342786B2 (en) | 2017-10-05 | 2019-07-09 | Fulcrum Therapeutics, Inc. | P38 kinase inhibitors reduce DUX4 and downstream gene expression for the treatment of FSHD |
| US10537560B2 (en) | 2017-10-05 | 2020-01-21 | Fulcrum Therapeutics. Inc. | P38 kinase inhibitors reduce DUX4 and downstream gene expression for the treatment of FSHD |
| US11291659B2 (en) | 2017-10-05 | 2022-04-05 | Fulcrum Therapeutics, Inc. | P38 kinase inhibitors reduce DUX4 and downstream gene expression for the treatment of FSHD |
| US11479770B2 (en) | 2017-10-05 | 2022-10-25 | Fulcrum Therapeutics, Inc. | Use of p38 inhibitors to reduce expression of DUX4 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP4084410B2 (en) | Tetrahydronaphthylidine derivatives useful as histamine H3 receptor ligands | |
| JP6728208B2 (en) | Benzazepine dicarboxamide compound | |
| US7456164B2 (en) | 3- or 4-monosubtituted phenol and thiophenol derivatives useful as H3 ligands | |
| JP4173191B2 (en) | 3- or 4-monosubstituted phenol and thiophenol derivatives useful as H3 ligands | |
| EP1893609B1 (en) | N-(pyridin-2-yl)-sulfonamide derivatives | |
| EP2297112B1 (en) | Pyrazole compounds as ccr1 antagonists | |
| US20070179175A1 (en) | Tetrahydronaphthyridine Derivative | |
| US20060111416A1 (en) | Octahydropyrrolo[3,4-C]pyrrole derivatives | |
| US10344026B2 (en) | Compositions and methods of targeting mutant K-ras | |
| WO2007045989A1 (en) | Pyridyl derivatives useful as h3 ligands | |
| CA2592092A1 (en) | Nonnucleoside inhibitors of hiv-1 reverse transcriptase | |
| WO2007034279A2 (en) | C3a antagonists and pharmaceutical compositions thereof | |
| EP1846409A1 (en) | Octahydropyrrolo[3,4-c]pyrrole derivatives | |
| ZA200505957B (en) | Triazole compounds useful in therapy | |
| EP1680423A1 (en) | Azabenzodiazepines as phosphodiesterase-4 inhibitors | |
| EP1671972A1 (en) | Octahydropyrrolo[3,4-c]pyrrole derivatives | |
| EP1593679A1 (en) | 3- Or 4-monosubstituted phenol derivatives useful as H3 ligands |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 06809088 Country of ref document: EP Kind code of ref document: A1 |