WO2007034312A2 - Pyridinaminosulfonyl substituted benzamides as inhibitors of cytochrome p450 3a4 (cyp3a4) - Google Patents
Pyridinaminosulfonyl substituted benzamides as inhibitors of cytochrome p450 3a4 (cyp3a4) Download PDFInfo
- Publication number
- WO2007034312A2 WO2007034312A2 PCT/IB2006/002639 IB2006002639W WO2007034312A2 WO 2007034312 A2 WO2007034312 A2 WO 2007034312A2 IB 2006002639 W IB2006002639 W IB 2006002639W WO 2007034312 A2 WO2007034312 A2 WO 2007034312A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- sulfonyl
- hydrogen
- compound
- amino
- Prior art date
Links
- 239000003112 inhibitor Substances 0.000 title description 15
- 102000004328 Cytochrome P-450 CYP3A Human genes 0.000 title description 10
- 108010081668 Cytochrome P-450 CYP3A Proteins 0.000 title description 10
- 229940054066 benzamide antipsychotics Drugs 0.000 title 1
- 150000003936 benzamides Chemical class 0.000 title 1
- 150000001875 compounds Chemical class 0.000 claims abstract description 389
- 150000003839 salts Chemical class 0.000 claims abstract description 104
- 239000012453 solvate Substances 0.000 claims abstract description 75
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 28
- 238000002360 preparation method Methods 0.000 claims abstract description 17
- -1 -O(CR12aR12b) Chemical group 0.000 claims description 154
- 229910052739 hydrogen Inorganic materials 0.000 claims description 143
- 239000001257 hydrogen Substances 0.000 claims description 134
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 133
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 73
- 125000003118 aryl group Chemical group 0.000 claims description 67
- 125000006694 (C2-C10) heterocyclyl group Chemical group 0.000 claims description 61
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 59
- 125000000041 C6-C10 aryl group Chemical group 0.000 claims description 58
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 56
- 241000124008 Mammalia Species 0.000 claims description 53
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 53
- 125000000623 heterocyclic group Chemical group 0.000 claims description 45
- 125000001072 heteroaryl group Chemical group 0.000 claims description 43
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 42
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 42
- 229910052736 halogen Inorganic materials 0.000 claims description 40
- 150000002367 halogens Chemical class 0.000 claims description 40
- QWAXKHKRTORLEM-UGJKXSETSA-N saquinavir Chemical compound C([C@@H]([C@H](O)CN1C[C@H]2CCCC[C@H]2C[C@H]1C(=O)NC(C)(C)C)NC(=O)[C@H](CC(N)=O)NC(=O)C=1N=C2C=CC=CC2=CC=1)C1=CC=CC=C1 QWAXKHKRTORLEM-UGJKXSETSA-N 0.000 claims description 38
- 125000004429 atom Chemical group 0.000 claims description 29
- 108010019625 Atazanavir Sulfate Proteins 0.000 claims description 28
- 150000002431 hydrogen Chemical group 0.000 claims description 28
- KJHKTHWMRKYKJE-SUGCFTRWSA-N Kaletra Chemical compound N1([C@@H](C(C)C)C(=O)N[C@H](C[C@H](O)[C@H](CC=2C=CC=CC=2)NC(=O)COC=2C(=CC=CC=2C)C)CC=2C=CC=CC=2)CCCNC1=O KJHKTHWMRKYKJE-SUGCFTRWSA-N 0.000 claims description 27
- 125000000217 alkyl group Chemical group 0.000 claims description 27
- YMARZQAQMVYCKC-OEMFJLHTSA-N amprenavir Chemical compound C([C@@H]([C@H](O)CN(CC(C)C)S(=O)(=O)C=1C=CC(N)=CC=1)NC(=O)O[C@@H]1COCC1)C1=CC=CC=C1 YMARZQAQMVYCKC-OEMFJLHTSA-N 0.000 claims description 27
- AXRYRYVKAWYZBR-GASGPIRDSA-N atazanavir Chemical compound C([C@H](NC(=O)[C@@H](NC(=O)OC)C(C)(C)C)[C@@H](O)CN(CC=1C=CC(=CC=1)C=1N=CC=CC=1)NC(=O)[C@@H](NC(=O)OC)C(C)(C)C)C1=CC=CC=C1 AXRYRYVKAWYZBR-GASGPIRDSA-N 0.000 claims description 27
- SUJUHGSWHZTSEU-FYBSXPHGSA-N tipranavir Chemical compound C([C@@]1(CCC)OC(=O)C([C@H](CC)C=2C=C(NS(=O)(=O)C=3N=CC(=CC=3)C(F)(F)F)C=CC=2)=C(O)C1)CC1=CC=CC=C1 SUJUHGSWHZTSEU-FYBSXPHGSA-N 0.000 claims description 24
- 238000011282 treatment Methods 0.000 claims description 24
- NCDNCNXCDXHOMX-UHFFFAOYSA-N Ritonavir Natural products C=1C=CC=CC=1CC(NC(=O)OCC=1SC=NC=1)C(O)CC(CC=1C=CC=CC=1)NC(=O)C(C(C)C)NC(=O)N(C)CC1=CSC(C(C)C)=N1 NCDNCNXCDXHOMX-UHFFFAOYSA-N 0.000 claims description 23
- 229960000311 ritonavir Drugs 0.000 claims description 23
- NCDNCNXCDXHOMX-XGKFQTDJSA-N ritonavir Chemical compound N([C@@H](C(C)C)C(=O)N[C@H](C[C@H](O)[C@H](CC=1C=CC=CC=1)NC(=O)OCC=1SC=NC=1)CC=1C=CC=CC=1)C(=O)N(C)CC1=CSC(C(C)C)=N1 NCDNCNXCDXHOMX-XGKFQTDJSA-N 0.000 claims description 23
- 125000000876 trifluoromethoxy group Chemical group FC(F)(F)O* 0.000 claims description 23
- 239000004030 hiv protease inhibitor Substances 0.000 claims description 22
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 claims description 22
- JORVRJNILJXMMG-OLNQLETPSA-N brecanavir Chemical compound C([C@@H]([C@H](O)CN(CC(C)C)S(=O)(=O)C=1C=C2OCOC2=CC=1)NC(=O)O[C@@H]1[C@@H]2CCO[C@@H]2OC1)C(C=C1)=CC=C1OCC1=CSC(C)=N1 JORVRJNILJXMMG-OLNQLETPSA-N 0.000 claims description 21
- 229940122440 HIV protease inhibitor Drugs 0.000 claims description 20
- 229950009079 brecanavir Drugs 0.000 claims description 20
- PMDQGYMGQKTCSX-HQROKSDRSA-L calcium;[(2r,3s)-1-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-[[(3s)-oxolan-3-yl]oxycarbonylamino]-4-phenylbutan-2-yl] phosphate Chemical compound [Ca+2].C([C@@H]([C@H](OP([O-])([O-])=O)CN(CC(C)C)S(=O)(=O)C=1C=CC(N)=CC=1)NC(=O)O[C@@H]1COCC1)C1=CC=CC=C1 PMDQGYMGQKTCSX-HQROKSDRSA-L 0.000 claims description 20
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 20
- AXRYRYVKAWYZBR-UHFFFAOYSA-N Atazanavir Natural products C=1C=C(C=2N=CC=CC=2)C=CC=1CN(NC(=O)C(NC(=O)OC)C(C)(C)C)CC(O)C(NC(=O)C(NC(=O)OC)C(C)(C)C)CC1=CC=CC=C1 AXRYRYVKAWYZBR-UHFFFAOYSA-N 0.000 claims description 19
- QAGYKUNXZHXKMR-UHFFFAOYSA-N CPD000469186 Natural products CC1=C(O)C=CC=C1C(=O)NC(C(O)CN1C(CC2CCCCC2C1)C(=O)NC(C)(C)C)CSC1=CC=CC=C1 QAGYKUNXZHXKMR-UHFFFAOYSA-N 0.000 claims description 19
- SUJUHGSWHZTSEU-UHFFFAOYSA-N Tipranavir Natural products C1C(O)=C(C(CC)C=2C=C(NS(=O)(=O)C=3N=CC(=CC=3)C(F)(F)F)C=CC=2)C(=O)OC1(CCC)CCC1=CC=CC=C1 SUJUHGSWHZTSEU-UHFFFAOYSA-N 0.000 claims description 19
- 229960001830 amprenavir Drugs 0.000 claims description 19
- 230000036436 anti-hiv Effects 0.000 claims description 19
- 229960003277 atazanavir Drugs 0.000 claims description 19
- CJBJHOAVZSMMDJ-HEXNFIEUSA-N darunavir Chemical compound C([C@@H]([C@H](O)CN(CC(C)C)S(=O)(=O)C=1C=CC(N)=CC=1)NC(=O)O[C@@H]1[C@@H]2CCO[C@@H]2OC1)C1=CC=CC=C1 CJBJHOAVZSMMDJ-HEXNFIEUSA-N 0.000 claims description 19
- 229960005107 darunavir Drugs 0.000 claims description 19
- 229960002933 fosamprenavir calcium Drugs 0.000 claims description 19
- 229960001936 indinavir Drugs 0.000 claims description 19
- CBVCZFGXHXORBI-PXQQMZJSSA-N indinavir Chemical compound C([C@H](N(CC1)C[C@@H](O)C[C@@H](CC=2C=CC=CC=2)C(=O)N[C@H]2C3=CC=CC=C3C[C@H]2O)C(=O)NC(C)(C)C)N1CC1=CC=CN=C1 CBVCZFGXHXORBI-PXQQMZJSSA-N 0.000 claims description 19
- 229940088976 invirase Drugs 0.000 claims description 19
- 229960004525 lopinavir Drugs 0.000 claims description 19
- QAGYKUNXZHXKMR-HKWSIXNMSA-N nelfinavir Chemical compound CC1=C(O)C=CC=C1C(=O)N[C@H]([C@H](O)CN1[C@@H](C[C@@H]2CCCC[C@@H]2C1)C(=O)NC(C)(C)C)CSC1=CC=CC=C1 QAGYKUNXZHXKMR-HKWSIXNMSA-N 0.000 claims description 19
- 229960000884 nelfinavir Drugs 0.000 claims description 19
- RXBWRFDZXRAEJT-SZNOJMITSA-N palinavir Chemical compound C([C@H](NC(=O)[C@@H](NC(=O)C=1N=C2C=CC=CC2=CC=1)C(C)C)[C@H](O)CN1[C@@H](C[C@@H](CC1)OCC=1C=CN=CC=1)C(=O)NC(C)(C)C)C1=CC=CC=C1 RXBWRFDZXRAEJT-SZNOJMITSA-N 0.000 claims description 19
- 229950006460 palinavir Drugs 0.000 claims description 19
- 229960001852 saquinavir Drugs 0.000 claims description 19
- 229960000838 tipranavir Drugs 0.000 claims description 19
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 18
- 125000005843 halogen group Chemical group 0.000 claims description 17
- 239000003814 drug Substances 0.000 claims description 15
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 13
- DLFVBJFMPXGRIB-UHFFFAOYSA-N thioacetamide Natural products CC(N)=O DLFVBJFMPXGRIB-UHFFFAOYSA-N 0.000 claims description 11
- 239000003937 drug carrier Substances 0.000 claims description 8
- 125000000475 sulfinyl group Chemical group [*:2]S([*:1])=O 0.000 claims description 6
- 208000031886 HIV Infections Diseases 0.000 claims description 2
- 208000037357 HIV infectious disease Diseases 0.000 claims description 2
- 208000033519 human immunodeficiency virus infectious disease Diseases 0.000 claims description 2
- 238000000034 method Methods 0.000 abstract description 68
- 239000000203 mixture Substances 0.000 description 74
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 54
- 241000725303 Human immunodeficiency virus Species 0.000 description 49
- KXDAEFPNCMNJSK-UHFFFAOYSA-N Benzamide Chemical compound NC(=O)C1=CC=CC=C1 KXDAEFPNCMNJSK-UHFFFAOYSA-N 0.000 description 47
- 239000003795 chemical substances by application Substances 0.000 description 43
- 239000000243 solution Substances 0.000 description 41
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 36
- 238000009472 formulation Methods 0.000 description 30
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 27
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 21
- 239000002904 solvent Substances 0.000 description 21
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 21
- 102000002004 Cytochrome P-450 Enzyme System Human genes 0.000 description 20
- 108010015742 Cytochrome P-450 Enzyme System Proteins 0.000 description 20
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 20
- 239000007787 solid Substances 0.000 description 20
- BEUUJDAEPJZWHM-COROXYKFSA-N tert-butyl n-[(2s,3s,5r)-3-hydroxy-6-[[(2s)-1-(2-methoxyethylamino)-3-methyl-1-oxobutan-2-yl]amino]-6-oxo-1-phenyl-5-[(2,3,4-trimethoxyphenyl)methyl]hexan-2-yl]carbamate Chemical compound C([C@@H]([C@@H](O)C[C@H](C(=O)N[C@H](C(=O)NCCOC)C(C)C)CC=1C(=C(OC)C(OC)=CC=1)OC)NC(=O)OC(C)(C)C)C1=CC=CC=C1 BEUUJDAEPJZWHM-COROXYKFSA-N 0.000 description 19
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 18
- 239000002585 base Substances 0.000 description 17
- 230000005764 inhibitory process Effects 0.000 description 17
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 16
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 16
- 230000002401 inhibitory effect Effects 0.000 description 16
- HINZVVDZPLARRP-YSVIXOAZSA-N (4r,5s,6s,7r)-1,3-bis[(3-aminophenyl)methyl]-4,7-dibenzyl-5,6-dihydroxy-1,3-diazepan-2-one;methanesulfonic acid Chemical compound CS(O)(=O)=O.CS(O)(=O)=O.NC1=CC=CC(CN2C(N(CC=3C=C(N)C=CC=3)[C@H](CC=3C=CC=CC=3)[C@H](O)[C@@H](O)[C@H]2CC=2C=CC=CC=2)=O)=C1 HINZVVDZPLARRP-YSVIXOAZSA-N 0.000 description 15
- 125000004432 carbon atom Chemical group C* 0.000 description 15
- 201000010099 disease Diseases 0.000 description 14
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 14
- 235000019439 ethyl acetate Nutrition 0.000 description 14
- 239000002253 acid Substances 0.000 description 13
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 12
- 102000004190 Enzymes Human genes 0.000 description 12
- 108090000790 Enzymes Proteins 0.000 description 12
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 12
- 238000006243 chemical reaction Methods 0.000 description 12
- 230000000694 effects Effects 0.000 description 12
- 239000000546 pharmaceutical excipient Substances 0.000 description 12
- 238000005160 1H NMR spectroscopy Methods 0.000 description 11
- 239000002259 anti human immunodeficiency virus agent Substances 0.000 description 11
- 229940124411 anti-hiv antiviral agent Drugs 0.000 description 11
- 229910002092 carbon dioxide Inorganic materials 0.000 description 11
- CUFQBQOBLVLKRF-RZDMPUFOSA-N (4r)-3-[(2s,3s)-2-hydroxy-3-[(3-hydroxy-2-methylbenzoyl)amino]-4-phenylbutanoyl]-5,5-dimethyl-n-[(2-methylphenyl)methyl]-1,3-thiazolidine-4-carboxamide Chemical compound CC1=CC=CC=C1CNC(=O)[C@@H]1C(C)(C)SCN1C(=O)[C@@H](O)[C@@H](NC(=O)C=1C(=C(O)C=CC=1)C)CC1=CC=CC=C1 CUFQBQOBLVLKRF-RZDMPUFOSA-N 0.000 description 10
- 101100352919 Caenorhabditis elegans ppm-2 gene Proteins 0.000 description 10
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 10
- NJBBLOIWMSYVCQ-VZTVMPNDSA-N Kynostatin 272 Chemical compound C([C@H](NC(=O)[C@@H](NC(=O)COC=1C2=CC=NC=C2C=CC=1)CSC)[C@H](O)C(=O)N1[C@@H](CSC1)C(=O)NC(C)(C)C)C1=CC=CC=C1 NJBBLOIWMSYVCQ-VZTVMPNDSA-N 0.000 description 10
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 10
- 239000000969 carrier Substances 0.000 description 10
- 239000006184 cosolvent Substances 0.000 description 10
- 108010075606 kynostatin 272 Proteins 0.000 description 10
- 239000012044 organic layer Substances 0.000 description 10
- JFKIHXVQWVFSNI-FCJVTMLMSA-N (2r,3r,4r,5r,6r,7r)-3,6-bis[(3-aminophenyl)methoxy]-2,7-dibenzyl-1,1-dioxothiepane-4,5-diol Chemical compound NC1=CC=CC(CO[C@H]2[C@H](S(=O)(=O)[C@H](CC=3C=CC=CC=3)[C@H](OCC=3C=C(N)C=CC=3)[C@H](O)[C@H]2O)CC=2C=CC=CC=2)=C1 JFKIHXVQWVFSNI-FCJVTMLMSA-N 0.000 description 9
- IXZYCIFRVZKVRJ-RKKDRKJOSA-N (2s)-n-[(2s,3r)-4-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]-2-[[2-[(3-fluorophenyl)methylamino]acetyl]amino]-3,3-dimethylbutanamide Chemical compound C([C@@H]([C@H](O)CN(CC(C)C)S(=O)(=O)C=1C=CC(N)=CC=1)NC(=O)[C@@H](NC(=O)CNCC=1C=C(F)C=CC=1)C(C)(C)C)C1=CC=CC=C1 IXZYCIFRVZKVRJ-RKKDRKJOSA-N 0.000 description 9
- 239000007832 Na2SO4 Substances 0.000 description 9
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 9
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 9
- BINXAIIXOUQUKC-UIPNDDLNSA-N [(3as,4r,6ar)-2,3,3a,4,5,6a-hexahydrofuro[2,3-b]furan-4-yl] n-[(2s,3r)-3-hydroxy-4-[(4-methoxyphenyl)sulfonyl-(2-methylpropyl)amino]-1-phenylbutan-2-yl]carbamate Chemical compound C1=CC(OC)=CC=C1S(=O)(=O)N(CC(C)C)C[C@@H](O)[C@@H](NC(=O)O[C@@H]1[C@@H]2CCO[C@@H]2OC1)CC1=CC=CC=C1 BINXAIIXOUQUKC-UIPNDDLNSA-N 0.000 description 9
- 150000001412 amines Chemical class 0.000 description 9
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 9
- 229940079593 drug Drugs 0.000 description 9
- 208000015181 infectious disease Diseases 0.000 description 9
- 230000010076 replication Effects 0.000 description 9
- 229910052938 sodium sulfate Inorganic materials 0.000 description 9
- 125000001424 substituent group Chemical group 0.000 description 9
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 8
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 8
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 8
- 239000012043 crude product Substances 0.000 description 8
- 238000010438 heat treatment Methods 0.000 description 8
- 239000007924 injection Substances 0.000 description 8
- 238000002347 injection Methods 0.000 description 8
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 8
- ITAXXEIRIFKXHI-UHFFFAOYSA-N methyl 2-(pyridin-3-ylsulfamoyl)benzoate Chemical compound COC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 ITAXXEIRIFKXHI-UHFFFAOYSA-N 0.000 description 8
- 229920001223 polyethylene glycol Polymers 0.000 description 8
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 8
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 8
- 239000000725 suspension Substances 0.000 description 8
- PNIFFZXGBAYVMQ-RKKDRKJOSA-N (2s)-n-[(2s,3r)-4-[(3-aminophenyl)sulfonyl-(2-methylpropyl)amino]-3-hydroxy-1-phenylbutan-2-yl]-2-[[2-[(3-fluorophenyl)methylamino]acetyl]amino]-3,3-dimethylbutanamide Chemical compound C([C@@H]([C@H](O)CN(CC(C)C)S(=O)(=O)C=1C=C(N)C=CC=1)NC(=O)[C@@H](NC(=O)CNCC=1C=C(F)C=CC=1)C(C)(C)C)C1=CC=CC=C1 PNIFFZXGBAYVMQ-RKKDRKJOSA-N 0.000 description 7
- 108010010803 Gelatin Proteins 0.000 description 7
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 7
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 7
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 7
- 239000004480 active ingredient Substances 0.000 description 7
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 7
- 239000002775 capsule Substances 0.000 description 7
- 239000008273 gelatin Substances 0.000 description 7
- 229920000159 gelatin Polymers 0.000 description 7
- 235000019322 gelatine Nutrition 0.000 description 7
- 235000011852 gelatine desserts Nutrition 0.000 description 7
- 239000007788 liquid Substances 0.000 description 7
- KVPBBXNZIREVEW-UHFFFAOYSA-N n-[(3,4-difluorophenyl)methyl]-2-[(4-methoxypyridin-3-yl)sulfamoyl]-n-methylbenzamide Chemical compound COC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)N(C)CC1=CC=C(F)C(F)=C1 KVPBBXNZIREVEW-UHFFFAOYSA-N 0.000 description 7
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 7
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 7
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 6
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 6
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- 229920002472 Starch Polymers 0.000 description 6
- MUMGGOZAMZWBJJ-DYKIIFRCSA-N Testostosterone Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 MUMGGOZAMZWBJJ-DYKIIFRCSA-N 0.000 description 6
- 238000004458 analytical method Methods 0.000 description 6
- 229910052799 carbon Inorganic materials 0.000 description 6
- 239000001768 carboxy methyl cellulose Substances 0.000 description 6
- 210000004027 cell Anatomy 0.000 description 6
- 239000003153 chemical reaction reagent Substances 0.000 description 6
- 238000004440 column chromatography Methods 0.000 description 6
- 239000003085 diluting agent Substances 0.000 description 6
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 6
- 230000004060 metabolic process Effects 0.000 description 6
- 150000007522 mineralic acids Chemical class 0.000 description 6
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 6
- 229910052757 nitrogen Inorganic materials 0.000 description 6
- 150000007524 organic acids Chemical class 0.000 description 6
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 6
- 239000011541 reaction mixture Substances 0.000 description 6
- 230000002829 reductive effect Effects 0.000 description 6
- 229920006395 saturated elastomer Polymers 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- 239000003826 tablet Substances 0.000 description 6
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 6
- WFDIJRYMOXRFFG-UHFFFAOYSA-N Acetic anhydride Chemical compound CC(=O)OC(C)=O WFDIJRYMOXRFFG-UHFFFAOYSA-N 0.000 description 5
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 5
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 5
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 5
- 239000002202 Polyethylene glycol Substances 0.000 description 5
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 5
- 150000007513 acids Chemical class 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 210000004369 blood Anatomy 0.000 description 5
- 239000008280 blood Substances 0.000 description 5
- 238000011260 co-administration Methods 0.000 description 5
- 229910052681 coesite Inorganic materials 0.000 description 5
- 229910052906 cristobalite Inorganic materials 0.000 description 5
- 238000001704 evaporation Methods 0.000 description 5
- 238000003818 flash chromatography Methods 0.000 description 5
- 239000012458 free base Substances 0.000 description 5
- 230000002209 hydrophobic effect Effects 0.000 description 5
- 238000000338 in vitro Methods 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 239000002207 metabolite Substances 0.000 description 5
- NYGRNTSORGSTJK-UHFFFAOYSA-N n-[(3,4-difluorophenyl)methyl]-2-[(4-ethoxypyridin-3-yl)sulfamoyl]-n-methylbenzamide Chemical compound CCOC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)N(C)CC1=CC=C(F)C(F)=C1 NYGRNTSORGSTJK-UHFFFAOYSA-N 0.000 description 5
- WZPOEIYGBPZWFU-UHFFFAOYSA-N n-[(3,4-difluorophenyl)methyl]-2-[(4-ethoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound CCOC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)NCC1=CC=C(F)C(F)=C1 WZPOEIYGBPZWFU-UHFFFAOYSA-N 0.000 description 5
- CYFWKTRCWDFGNM-UHFFFAOYSA-N n-[[4-fluoro-3-(trifluoromethyl)phenyl]methyl]-2-[(4-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)NCC1=CC=C(F)C(C(F)(F)F)=C1 CYFWKTRCWDFGNM-UHFFFAOYSA-N 0.000 description 5
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 5
- 239000000843 powder Substances 0.000 description 5
- 238000000746 purification Methods 0.000 description 5
- 239000000377 silicon dioxide Substances 0.000 description 5
- 235000019698 starch Nutrition 0.000 description 5
- 229910052682 stishovite Inorganic materials 0.000 description 5
- 239000000758 substrate Substances 0.000 description 5
- KDYFGRWQOYBRFD-UHFFFAOYSA-N succinic acid Chemical compound OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 5
- 235000000346 sugar Nutrition 0.000 description 5
- 238000003786 synthesis reaction Methods 0.000 description 5
- 238000004809 thin layer chromatography Methods 0.000 description 5
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 5
- 229910052905 tridymite Inorganic materials 0.000 description 5
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 4
- AYJGIISEEPZUAU-UHFFFAOYSA-N 2-(pyridin-3-ylmethylsulfanyl)benzoic acid Chemical compound OC(=O)C1=CC=CC=C1SCC1=CC=CN=C1 AYJGIISEEPZUAU-UHFFFAOYSA-N 0.000 description 4
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 4
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 4
- 206010001513 AIDS related complex Diseases 0.000 description 4
- UXCAQJAQSWSNPQ-XLPZGREQSA-N Alovudine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](F)C1 UXCAQJAQSWSNPQ-XLPZGREQSA-N 0.000 description 4
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 4
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 4
- IAJILQKETJEXLJ-UHFFFAOYSA-N Galacturonsaeure Natural products O=CC(O)C(O)C(O)C(O)C(O)=O IAJILQKETJEXLJ-UHFFFAOYSA-N 0.000 description 4
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 4
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 4
- 108010078851 HIV Reverse Transcriptase Proteins 0.000 description 4
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 4
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 4
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 4
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 4
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 4
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 4
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 4
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 4
- WQDUMFSSJAZKTM-UHFFFAOYSA-N Sodium methoxide Chemical compound [Na+].[O-]C WQDUMFSSJAZKTM-UHFFFAOYSA-N 0.000 description 4
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 4
- 239000008186 active pharmaceutical agent Substances 0.000 description 4
- 235000001014 amino acid Nutrition 0.000 description 4
- 150000001413 amino acids Chemical class 0.000 description 4
- 239000007864 aqueous solution Substances 0.000 description 4
- 239000012267 brine Substances 0.000 description 4
- 239000002552 dosage form Substances 0.000 description 4
- 239000008298 dragée Substances 0.000 description 4
- 239000000839 emulsion Substances 0.000 description 4
- 235000019253 formic acid Nutrition 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 238000011534 incubation Methods 0.000 description 4
- 239000008101 lactose Substances 0.000 description 4
- ZCSHNCUQKCANBX-UHFFFAOYSA-N lithium diisopropylamide Chemical compound [Li+].CC(C)[N-]C(C)C ZCSHNCUQKCANBX-UHFFFAOYSA-N 0.000 description 4
- KLPBXNJPVWIQKI-UHFFFAOYSA-N methyl 2-[(4-methoxypyridin-3-yl)sulfamoyl]benzoate Chemical compound COC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CN=CC=C1OC KLPBXNJPVWIQKI-UHFFFAOYSA-N 0.000 description 4
- HERGORGVVPQEFP-UHFFFAOYSA-N methyl 2-[(5-cyanopyridin-3-yl)sulfamoyl]benzoate Chemical compound COC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CN=CC(C#N)=C1 HERGORGVVPQEFP-UHFFFAOYSA-N 0.000 description 4
- ZNFNYMJJSRSQKF-UHFFFAOYSA-N methyl 2-[(5-methoxypyridin-3-yl)sulfamoyl]benzoate Chemical compound COC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CN=CC(OC)=C1 ZNFNYMJJSRSQKF-UHFFFAOYSA-N 0.000 description 4
- HUNUAFNLLYVTQD-UHFFFAOYSA-N methyl 2-chlorosulfonylbenzoate Chemical compound COC(=O)C1=CC=CC=C1S(Cl)(=O)=O HUNUAFNLLYVTQD-UHFFFAOYSA-N 0.000 description 4
- AIXZEHAFOAWCBI-UHFFFAOYSA-N n-[(3,4-difluorophenyl)methyl]-2-[(4-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)NCC1=CC=C(F)C(F)=C1 AIXZEHAFOAWCBI-UHFFFAOYSA-N 0.000 description 4
- 239000003921 oil Substances 0.000 description 4
- 235000019198 oils Nutrition 0.000 description 4
- 239000003960 organic solvent Substances 0.000 description 4
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 4
- 239000000825 pharmaceutical preparation Substances 0.000 description 4
- 230000036470 plasma concentration Effects 0.000 description 4
- 229910000027 potassium carbonate Inorganic materials 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 4
- 210000002966 serum Anatomy 0.000 description 4
- 239000011734 sodium Substances 0.000 description 4
- 229910052708 sodium Inorganic materials 0.000 description 4
- 229910000029 sodium carbonate Inorganic materials 0.000 description 4
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 4
- 239000003381 stabilizer Substances 0.000 description 4
- 239000007858 starting material Substances 0.000 description 4
- 150000008163 sugars Chemical class 0.000 description 4
- 229910052717 sulfur Inorganic materials 0.000 description 4
- 125000004434 sulfur atom Chemical group 0.000 description 4
- 239000003981 vehicle Substances 0.000 description 4
- 230000003612 virological effect Effects 0.000 description 4
- ILAYIAGXTHKHNT-UHFFFAOYSA-N 4-[4-(2,4,6-trimethyl-phenylamino)-pyrimidin-2-ylamino]-benzonitrile Chemical compound CC1=CC(C)=CC(C)=C1NC1=CC=NC(NC=2C=CC(=CC=2)C#N)=N1 ILAYIAGXTHKHNT-UHFFFAOYSA-N 0.000 description 3
- CTQPCFFQBYXOAJ-UHFFFAOYSA-N 5-methoxypyridin-3-amine Chemical compound COC1=CN=CC(N)=C1 CTQPCFFQBYXOAJ-UHFFFAOYSA-N 0.000 description 3
- WVKLERKKJXUPIK-UHFFFAOYSA-N 7-phenylmethoxy-4-(trifluoromethyl)chromen-2-one Chemical compound C1=CC=2C(C(F)(F)F)=CC(=O)OC=2C=C1OCC1=CC=CC=C1 WVKLERKKJXUPIK-UHFFFAOYSA-N 0.000 description 3
- 229920001817 Agar Polymers 0.000 description 3
- LSNNMFCWUKXFEE-UHFFFAOYSA-M Bisulfite Chemical compound OS([O-])=O LSNNMFCWUKXFEE-UHFFFAOYSA-M 0.000 description 3
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 3
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 3
- 102000018832 Cytochromes Human genes 0.000 description 3
- 108010052832 Cytochromes Proteins 0.000 description 3
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 3
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 3
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 3
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 3
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 3
- 108010002459 HIV Integrase Proteins 0.000 description 3
- 108010050904 Interferons Proteins 0.000 description 3
- 102000014150 Interferons Human genes 0.000 description 3
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 3
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 3
- BZLVMXJERCGZMT-UHFFFAOYSA-N Methyl tert-butyl ether Chemical compound COC(C)(C)C BZLVMXJERCGZMT-UHFFFAOYSA-N 0.000 description 3
- ACFIXJIJDZMPPO-NNYOXOHSSA-N NADPH Chemical compound C1=CCC(C(=O)N)=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](COP(O)(=O)OP(O)(=O)OC[C@@H]2[C@H]([C@@H](OP(O)(O)=O)[C@@H](O2)N2C3=NC=NC(N)=C3N=C2)O)O1 ACFIXJIJDZMPPO-NNYOXOHSSA-N 0.000 description 3
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 3
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 3
- 239000013543 active substance Substances 0.000 description 3
- 235000010419 agar Nutrition 0.000 description 3
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 3
- 230000002924 anti-infective effect Effects 0.000 description 3
- 229940124522 antiretrovirals Drugs 0.000 description 3
- 238000003556 assay Methods 0.000 description 3
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 3
- 230000031018 biological processes and functions Effects 0.000 description 3
- 229910052791 calcium Inorganic materials 0.000 description 3
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 3
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 3
- 229940105329 carboxymethylcellulose Drugs 0.000 description 3
- 150000001768 cations Chemical class 0.000 description 3
- 229920002678 cellulose Polymers 0.000 description 3
- 239000001913 cellulose Substances 0.000 description 3
- 235000010980 cellulose Nutrition 0.000 description 3
- 229940125782 compound 2 Drugs 0.000 description 3
- 239000013078 crystal Substances 0.000 description 3
- 230000003247 decreasing effect Effects 0.000 description 3
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 3
- 230000008020 evaporation Effects 0.000 description 3
- 239000000945 filler Substances 0.000 description 3
- 239000007903 gelatin capsule Substances 0.000 description 3
- 235000011187 glycerol Nutrition 0.000 description 3
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 150000007529 inorganic bases Chemical class 0.000 description 3
- 229940079322 interferon Drugs 0.000 description 3
- JMMWKPVZQRWMSS-UHFFFAOYSA-N isopropyl acetate Chemical compound CC(C)OC(C)=O JMMWKPVZQRWMSS-UHFFFAOYSA-N 0.000 description 3
- 238000001294 liquid chromatography-tandem mass spectrometry Methods 0.000 description 3
- 239000007937 lozenge Substances 0.000 description 3
- 239000011777 magnesium Substances 0.000 description 3
- 229910052749 magnesium Inorganic materials 0.000 description 3
- 235000019359 magnesium stearate Nutrition 0.000 description 3
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 3
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 3
- 230000001404 mediated effect Effects 0.000 description 3
- MXRUQSWIPRERAN-UHFFFAOYSA-N methyl 2-[(5-methylpyridin-3-yl)sulfamoyl]benzoate Chemical compound COC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CN=CC(C)=C1 MXRUQSWIPRERAN-UHFFFAOYSA-N 0.000 description 3
- DDLIGBOFAVUZHB-UHFFFAOYSA-N midazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NC=C2CN=C1C1=CC=CC=C1F DDLIGBOFAVUZHB-UHFFFAOYSA-N 0.000 description 3
- 229960003793 midazolam Drugs 0.000 description 3
- RIEHXZKHLIMPRG-UHFFFAOYSA-N n-[(3,4-dichlorophenyl)methyl]-2-[(5-ethoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound CCOC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCC=2C=C(Cl)C(Cl)=CC=2)=C1 RIEHXZKHLIMPRG-UHFFFAOYSA-N 0.000 description 3
- ZWRFIJHLXJKXTN-UHFFFAOYSA-N n-[(3,4-dichlorophenyl)methyl]-2-[(5-methylpyridin-3-yl)sulfamoyl]benzamide Chemical compound CC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCC=2C=C(Cl)C(Cl)=CC=2)=C1 ZWRFIJHLXJKXTN-UHFFFAOYSA-N 0.000 description 3
- QWCBQIYPSKLQGZ-UHFFFAOYSA-N n-[(3,4-difluorophenyl)methyl]-2-[(5-ethoxypyridin-3-yl)sulfamoyl]-n-methylbenzamide Chemical compound CCOC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)N(C)CC=2C=C(F)C(F)=CC=2)=C1 QWCBQIYPSKLQGZ-UHFFFAOYSA-N 0.000 description 3
- BKNCIMAPPAXVNR-UHFFFAOYSA-N n-[(3,4-difluorophenyl)methyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCC=2C=C(F)C(F)=CC=2)=C1 BKNCIMAPPAXVNR-UHFFFAOYSA-N 0.000 description 3
- QVOWRNNWQKOAJN-UHFFFAOYSA-N n-[(3,5-dichlorophenyl)methyl]-2-[(5-methylpyridin-3-yl)sulfamoyl]benzamide Chemical compound CC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCC=2C=C(Cl)C=C(Cl)C=2)=C1 QVOWRNNWQKOAJN-UHFFFAOYSA-N 0.000 description 3
- UCEXMYNDLQJYOM-UHFFFAOYSA-N n-[2-(2-fluorophenyl)ethyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCCC=2C(=CC=CC=2)F)=C1 UCEXMYNDLQJYOM-UHFFFAOYSA-N 0.000 description 3
- LFXBOBVVGLBSHE-UHFFFAOYSA-N n-[2-(2-methylphenyl)ethyl]-2-[(5-methylpyridin-3-yl)sulfamoyl]benzamide Chemical compound CC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCCC=2C(=CC=CC=2)C)=C1 LFXBOBVVGLBSHE-UHFFFAOYSA-N 0.000 description 3
- YFHUJXZHIOMPFO-UHFFFAOYSA-N n-[[4-fluoro-3-(trifluoromethyl)phenyl]methyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCC=2C=C(C(F)=CC=2)C(F)(F)F)=C1 YFHUJXZHIOMPFO-UHFFFAOYSA-N 0.000 description 3
- NKERIASEZLPHHZ-UHFFFAOYSA-N n-[[4-fluoro-3-(trifluoromethyl)phenyl]methyl]-2-[(5-methylpyridin-3-yl)sulfamoyl]benzamide Chemical compound CC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCC=2C=C(C(F)=CC=2)C(F)(F)F)=C1 NKERIASEZLPHHZ-UHFFFAOYSA-N 0.000 description 3
- 229930027945 nicotinamide-adenine dinucleotide Natural products 0.000 description 3
- 150000007530 organic bases Chemical class 0.000 description 3
- 125000004430 oxygen atom Chemical group O* 0.000 description 3
- 239000012071 phase Substances 0.000 description 3
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 3
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 description 3
- 229940068968 polysorbate 80 Drugs 0.000 description 3
- 229920000053 polysorbate 80 Polymers 0.000 description 3
- 238000012545 processing Methods 0.000 description 3
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 3
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 3
- 239000000600 sorbitol Substances 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 229940032147 starch Drugs 0.000 description 3
- 239000004094 surface-active agent Substances 0.000 description 3
- 238000013268 sustained release Methods 0.000 description 3
- 239000012730 sustained-release form Substances 0.000 description 3
- 239000006188 syrup Substances 0.000 description 3
- 235000020357 syrup Nutrition 0.000 description 3
- 239000000454 talc Substances 0.000 description 3
- 229910052623 talc Inorganic materials 0.000 description 3
- 235000012222 talc Nutrition 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- 229960003604 testosterone Drugs 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- 229940086542 triethylamine Drugs 0.000 description 3
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 2
- YDEDQHURUUATAT-PIZZNKLWSA-N (4r)-3-[(2s,3s)-3-[(3-amino-2-chlorobenzoyl)amino]-2-hydroxy-4-(3-methoxyphenyl)butanoyl]-n-[(2,6-dimethylphenyl)methyl]-5,5-dimethyl-1,3-thiazolidine-4-carboxamide Chemical compound COC1=CC=CC(C[C@H](NC(=O)C=2C(=C(N)C=CC=2)Cl)[C@H](O)C(=O)N2[C@@H](C(C)(C)SC2)C(=O)NCC=2C(=CC=CC=2C)C)=C1 YDEDQHURUUATAT-PIZZNKLWSA-N 0.000 description 2
- WBYWAXJHAXSJNI-VOTSOKGWSA-M .beta-Phenylacrylic acid Natural products [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 2
- BQLUNWRBACRWBA-UHFFFAOYSA-N 2-(2,7-diazaspiro[3.5]nonane-2-carbonyl)-n-pyridin-3-ylbenzenesulfonamide Chemical compound C=1C=CC=C(S(=O)(=O)NC=2C=NC=CC=2)C=1C(=O)N(C1)CC21CCNCC2 BQLUNWRBACRWBA-UHFFFAOYSA-N 0.000 description 2
- PXYHTCLWTKVRAB-UHFFFAOYSA-N 2-(2,7-diazaspiro[3.5]nonane-7-carbonyl)-n-pyridin-3-ylbenzenesulfonamide Chemical compound C=1C=CC=C(S(=O)(=O)NC=2C=NC=CC=2)C=1C(=O)N(CC1)CCC21CNC2 PXYHTCLWTKVRAB-UHFFFAOYSA-N 0.000 description 2
- HIEZDQFLOSUONH-UHFFFAOYSA-N 2-[(4-ethoxypyridin-3-yl)sulfamoyl]-n-[(2-fluorophenyl)methyl]-n-methylbenzamide Chemical compound CCOC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)N(C)CC1=CC=CC=C1F HIEZDQFLOSUONH-UHFFFAOYSA-N 0.000 description 2
- LJFJIBGKXPGKKY-UHFFFAOYSA-N 2-[(4-ethoxypyridin-3-yl)sulfamoyl]-n-[(3-fluorophenyl)methyl]-n-methylbenzamide Chemical compound CCOC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)N(C)CC1=CC=CC(F)=C1 LJFJIBGKXPGKKY-UHFFFAOYSA-N 0.000 description 2
- ZCRMIBSPUMHJIU-UHFFFAOYSA-N 2-[(4-ethoxypyridin-3-yl)sulfamoyl]-n-[(4-fluorophenyl)methyl]-n-methylbenzamide Chemical compound CCOC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)N(C)CC1=CC=C(F)C=C1 ZCRMIBSPUMHJIU-UHFFFAOYSA-N 0.000 description 2
- FEVBOALFQRJYOG-UHFFFAOYSA-N 2-[(4-ethoxypyridin-3-yl)sulfamoyl]-n-[2-(2-methylphenyl)ethyl]benzamide Chemical compound CCOC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)NCCC1=CC=CC=C1C FEVBOALFQRJYOG-UHFFFAOYSA-N 0.000 description 2
- ZOCLBCDMJXZMHF-UHFFFAOYSA-N 2-[(4-ethoxypyridin-3-yl)sulfamoyl]-n-[[4-fluoro-3-(trifluoromethyl)phenyl]methyl]benzamide Chemical compound CCOC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)NCC1=CC=C(F)C(C(F)(F)F)=C1 ZOCLBCDMJXZMHF-UHFFFAOYSA-N 0.000 description 2
- QEHCVWOCKYDNAS-UHFFFAOYSA-N 2-[(4-ethoxypyridin-3-yl)sulfamoyl]-n-methyl-n-[[4-(trifluoromethyl)phenyl]methyl]benzamide Chemical compound CCOC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)N(C)CC1=CC=C(C(F)(F)F)C=C1 QEHCVWOCKYDNAS-UHFFFAOYSA-N 0.000 description 2
- ZDSCIYPGWDXEEH-UHFFFAOYSA-N 2-[(4-methoxypyridin-3-yl)sulfamoyl]-n-[2-(2-methylphenyl)ethyl]benzamide Chemical compound COC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)NCCC1=CC=CC=C1C ZDSCIYPGWDXEEH-UHFFFAOYSA-N 0.000 description 2
- VJFLKSJZFAKBOU-UHFFFAOYSA-N 2-[(4-methoxypyridin-3-yl)sulfamoyl]-n-[2-[2-(trifluoromethoxy)phenyl]ethyl]benzamide Chemical compound COC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)NCCC1=CC=CC=C1OC(F)(F)F VJFLKSJZFAKBOU-UHFFFAOYSA-N 0.000 description 2
- AAXJQLKAJKWMNQ-UHFFFAOYSA-N 2-[(4-methoxypyridin-3-yl)sulfamoyl]-n-methyl-n-[[4-(trifluoromethyl)phenyl]methyl]benzamide Chemical compound COC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)N(C)CC1=CC=C(C(F)(F)F)C=C1 AAXJQLKAJKWMNQ-UHFFFAOYSA-N 0.000 description 2
- HCKHZGMYCRMGNN-UHFFFAOYSA-N 2-[(5-ethoxypyridin-3-yl)sulfamoyl]-n-[2-(2-methylphenyl)ethyl]benzamide Chemical compound CCOC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCCC=2C(=CC=CC=2)C)=C1 HCKHZGMYCRMGNN-UHFFFAOYSA-N 0.000 description 2
- CNXACKFWYRYUCW-UHFFFAOYSA-N 2-[(5-ethoxypyridin-3-yl)sulfamoyl]-n-[[4-fluoro-3-(trifluoromethyl)phenyl]methyl]benzamide Chemical compound CCOC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCC=2C=C(C(F)=CC=2)C(F)(F)F)=C1 CNXACKFWYRYUCW-UHFFFAOYSA-N 0.000 description 2
- YNUUXHDSDKOWNS-UHFFFAOYSA-N 2-[(5-methoxypyridin-3-yl)sulfamoyl]-n-[2-(2-methylphenyl)ethyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCCC=2C(=CC=CC=2)C)=C1 YNUUXHDSDKOWNS-UHFFFAOYSA-N 0.000 description 2
- XFQVZVMSFLRCHS-UHFFFAOYSA-N 2-[(5-methylpyridin-3-yl)sulfamoyl]-n-[2-[2-(trifluoromethoxy)phenyl]ethyl]benzamide Chemical compound CC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCCC=2C(=CC=CC=2)OC(F)(F)F)=C1 XFQVZVMSFLRCHS-UHFFFAOYSA-N 0.000 description 2
- IZHVBANLECCAGF-UHFFFAOYSA-N 2-hydroxy-3-(octadecanoyloxy)propyl octadecanoate Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)COC(=O)CCCCCCCCCCCCCCCCC IZHVBANLECCAGF-UHFFFAOYSA-N 0.000 description 2
- CUYKNJBYIJFRCU-UHFFFAOYSA-N 3-aminopyridine Chemical compound NC1=CC=CN=C1 CUYKNJBYIJFRCU-UHFFFAOYSA-N 0.000 description 2
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 2
- FZWUIWQMJFAWJW-UHFFFAOYSA-N 3-bromo-5-methoxypyridine Chemical compound COC1=CN=CC(Br)=C1 FZWUIWQMJFAWJW-UHFFFAOYSA-N 0.000 description 2
- GWNOTCOIYUNTQP-FQLXRVMXSA-N 4-[4-[[(3r)-1-butyl-3-[(r)-cyclohexyl(hydroxy)methyl]-2,5-dioxo-1,4,9-triazaspiro[5.5]undecan-9-yl]methyl]phenoxy]benzoic acid Chemical compound N([C@@H](C(=O)N1CCCC)[C@H](O)C2CCCCC2)C(=O)C1(CC1)CCN1CC(C=C1)=CC=C1OC1=CC=C(C(O)=O)C=C1 GWNOTCOIYUNTQP-FQLXRVMXSA-N 0.000 description 2
- HSBKFSPNDWWPSL-VDTYLAMSSA-N 4-amino-5-fluoro-1-[(2s,5r)-5-(hydroxymethyl)-2,5-dihydrofuran-2-yl]pyrimidin-2-one Chemical compound C1=C(F)C(N)=NC(=O)N1[C@@H]1C=C[C@H](CO)O1 HSBKFSPNDWWPSL-VDTYLAMSSA-N 0.000 description 2
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 2
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical compound [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 2
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 2
- 208000030507 AIDS Diseases 0.000 description 2
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 2
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 2
- 239000004475 Arginine Substances 0.000 description 2
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 239000005711 Benzoic acid Substances 0.000 description 2
- 102100035875 C-C chemokine receptor type 5 Human genes 0.000 description 2
- 101710149870 C-C chemokine receptor type 5 Proteins 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- VWFCHDSQECPREK-LURJTMIESA-N Cidofovir Chemical compound NC=1C=CN(C[C@@H](CO)OCP(O)(O)=O)C(=O)N=1 VWFCHDSQECPREK-LURJTMIESA-N 0.000 description 2
- WBYWAXJHAXSJNI-SREVYHEPSA-N Cinnamic acid Chemical compound OC(=O)\C=C/C1=CC=CC=C1 WBYWAXJHAXSJNI-SREVYHEPSA-N 0.000 description 2
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 2
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 2
- XQSPYNMVSIKCOC-NTSWFWBYSA-N Emtricitabine Chemical compound C1=C(F)C(N)=NC(=O)N1[C@H]1O[C@@H](CO)SC1 XQSPYNMVSIKCOC-NTSWFWBYSA-N 0.000 description 2
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 2
- 239000004471 Glycine Substances 0.000 description 2
- 108010010369 HIV Protease Proteins 0.000 description 2
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 2
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 2
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 2
- 229930195725 Mannitol Natural products 0.000 description 2
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 2
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 2
- IWUCXVSUMQZMFG-AFCXAGJDSA-N Ribavirin Chemical compound N1=C(C(=O)N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 IWUCXVSUMQZMFG-AFCXAGJDSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 2
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 2
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 2
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 2
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 2
- 241000700605 Viruses Species 0.000 description 2
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 2
- RLAHNGKRJJEIJL-RFZPGFLSSA-N [(2r,4r)-4-(2,6-diaminopurin-9-yl)-1,3-dioxolan-2-yl]methanol Chemical compound C12=NC(N)=NC(N)=C2N=CN1[C@H]1CO[C@@H](CO)O1 RLAHNGKRJJEIJL-RFZPGFLSSA-N 0.000 description 2
- BHIIGRBMZRSDRI-UHFFFAOYSA-N [chloro(phenoxy)phosphoryl]oxybenzene Chemical compound C=1C=CC=CC=1OP(=O)(Cl)OC1=CC=CC=C1 BHIIGRBMZRSDRI-UHFFFAOYSA-N 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- WOZSCQDILHKSGG-UHFFFAOYSA-N adefovir depivoxil Chemical compound N1=CN=C2N(CCOCP(=O)(OCOC(=O)C(C)(C)C)OCOC(=O)C(C)(C)C)C=NC2=C1N WOZSCQDILHKSGG-UHFFFAOYSA-N 0.000 description 2
- 239000000443 aerosol Substances 0.000 description 2
- 239000008272 agar Substances 0.000 description 2
- IAJILQKETJEXLJ-RSJOWCBRSA-N aldehydo-D-galacturonic acid Chemical compound O=C[C@H](O)[C@@H](O)[C@@H](O)[C@H](O)C(O)=O IAJILQKETJEXLJ-RSJOWCBRSA-N 0.000 description 2
- IAJILQKETJEXLJ-QTBDOELSSA-N aldehydo-D-glucuronic acid Chemical compound O=C[C@H](O)[C@@H](O)[C@H](O)[C@H](O)C(O)=O IAJILQKETJEXLJ-QTBDOELSSA-N 0.000 description 2
- 235000010443 alginic acid Nutrition 0.000 description 2
- 229920000615 alginic acid Polymers 0.000 description 2
- 229910052783 alkali metal Inorganic materials 0.000 description 2
- 150000001340 alkali metals Chemical class 0.000 description 2
- 229910001860 alkaline earth metal hydroxide Inorganic materials 0.000 description 2
- 229950004424 alovudine Drugs 0.000 description 2
- 229940061720 alpha hydroxy acid Drugs 0.000 description 2
- 150000001280 alpha hydroxy acids Chemical class 0.000 description 2
- 229910052782 aluminium Inorganic materials 0.000 description 2
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 2
- 229910021529 ammonia Inorganic materials 0.000 description 2
- 230000000798 anti-retroviral effect Effects 0.000 description 2
- 230000000840 anti-viral effect Effects 0.000 description 2
- 229960005475 antiinfective agent Drugs 0.000 description 2
- 239000000010 aprotic solvent Substances 0.000 description 2
- 239000008135 aqueous vehicle Substances 0.000 description 2
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 2
- 125000006615 aromatic heterocyclic group Chemical group 0.000 description 2
- 235000003704 aspartic acid Nutrition 0.000 description 2
- 125000002393 azetidinyl group Chemical group 0.000 description 2
- 235000010233 benzoic acid Nutrition 0.000 description 2
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910000019 calcium carbonate Inorganic materials 0.000 description 2
- 235000010216 calcium carbonate Nutrition 0.000 description 2
- 150000001721 carbon Chemical group 0.000 description 2
- 235000013985 cinnamic acid Nutrition 0.000 description 2
- 229930016911 cinnamic acid Natural products 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 229940126214 compound 3 Drugs 0.000 description 2
- 229910052802 copper Inorganic materials 0.000 description 2
- 239000010949 copper Substances 0.000 description 2
- ZYGHJZDHTFUPRJ-UHFFFAOYSA-N coumarin Chemical compound C1=CC=C2OC(=O)C=CC2=C1 ZYGHJZDHTFUPRJ-UHFFFAOYSA-N 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 2
- 229960000975 daunorubicin Drugs 0.000 description 2
- 230000007423 decrease Effects 0.000 description 2
- WHBIGIKBNXZKFE-UHFFFAOYSA-N delavirdine Chemical compound CC(C)NC1=CC=CN=C1N1CCN(C(=O)C=2NC3=CC=C(NS(C)(=O)=O)C=C3C=2)CC1 WHBIGIKBNXZKFE-UHFFFAOYSA-N 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 239000008121 dextrose Substances 0.000 description 2
- LMBWSYZSUOEYSN-UHFFFAOYSA-N diethyldithiocarbamic acid Chemical compound CCN(CC)C(S)=S LMBWSYZSUOEYSN-UHFFFAOYSA-N 0.000 description 2
- 125000004852 dihydrofuranyl group Chemical group O1C(CC=C1)* 0.000 description 2
- 125000005043 dihydropyranyl group Chemical group O1C(CCC=C1)* 0.000 description 2
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 2
- 235000011180 diphosphates Nutrition 0.000 description 2
- VLCYCQAOQCDTCN-UHFFFAOYSA-N eflornithine Chemical group NCCCC(N)(C(F)F)C(O)=O VLCYCQAOQCDTCN-UHFFFAOYSA-N 0.000 description 2
- 229960002759 eflornithine Drugs 0.000 description 2
- PEASPLKKXBYDKL-FXEVSJAOSA-N enfuvirtide Chemical compound C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(N)=O)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(C)=O)[C@@H](C)O)[C@@H](C)CC)C1=CN=CN1 PEASPLKKXBYDKL-FXEVSJAOSA-N 0.000 description 2
- 238000005516 engineering process Methods 0.000 description 2
- 229960002049 etravirine Drugs 0.000 description 2
- PYGWGZALEOIKDF-UHFFFAOYSA-N etravirine Chemical compound CC1=CC(C#N)=CC(C)=C1OC1=NC(NC=2C=CC(=CC=2)C#N)=NC(N)=C1Br PYGWGZALEOIKDF-UHFFFAOYSA-N 0.000 description 2
- 239000010685 fatty oil Substances 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 239000001530 fumaric acid Substances 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 229940097043 glucuronic acid Drugs 0.000 description 2
- 239000004220 glutamic acid Substances 0.000 description 2
- 235000013922 glutamic acid Nutrition 0.000 description 2
- 229960004275 glycolic acid Drugs 0.000 description 2
- 125000005842 heteroatom Chemical group 0.000 description 2
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 2
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 2
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 2
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 2
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 2
- 239000002955 immunomodulating agent Substances 0.000 description 2
- 229940121354 immunomodulator Drugs 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- 229910052500 inorganic mineral Inorganic materials 0.000 description 2
- 239000011630 iodine Substances 0.000 description 2
- 229910052740 iodine Inorganic materials 0.000 description 2
- 239000003456 ion exchange resin Substances 0.000 description 2
- 229920003303 ion-exchange polymer Polymers 0.000 description 2
- 229910052742 iron Inorganic materials 0.000 description 2
- 239000002502 liposome Substances 0.000 description 2
- 229910052744 lithium Inorganic materials 0.000 description 2
- 210000001853 liver microsome Anatomy 0.000 description 2
- 231100000053 low toxicity Toxicity 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 239000011976 maleic acid Substances 0.000 description 2
- 229960002510 mandelic acid Drugs 0.000 description 2
- WPBNNNQJVZRUHP-UHFFFAOYSA-L manganese(2+);methyl n-[[2-(methoxycarbonylcarbamothioylamino)phenyl]carbamothioyl]carbamate;n-[2-(sulfidocarbothioylamino)ethyl]carbamodithioate Chemical compound [Mn+2].[S-]C(=S)NCCNC([S-])=S.COC(=O)NC(=S)NC1=CC=CC=C1NC(=S)NC(=O)OC WPBNNNQJVZRUHP-UHFFFAOYSA-L 0.000 description 2
- 239000000594 mannitol Substances 0.000 description 2
- 235000010355 mannitol Nutrition 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 238000005259 measurement Methods 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- FJYBYEOOHROJMA-UHFFFAOYSA-N methyl 2-(pyridin-3-ylmethylsulfanyl)benzoate Chemical compound COC(=O)C1=CC=CC=C1SCC1=CC=CN=C1 FJYBYEOOHROJMA-UHFFFAOYSA-N 0.000 description 2
- BAQGCWNPCFABAY-UHFFFAOYSA-N methyl 2-sulfanylbenzoate Chemical compound COC(=O)C1=CC=CC=C1S BAQGCWNPCFABAY-UHFFFAOYSA-N 0.000 description 2
- QPJVMBTYPHYUOC-UHFFFAOYSA-N methyl benzoate Chemical compound COC(=O)C1=CC=CC=C1 QPJVMBTYPHYUOC-UHFFFAOYSA-N 0.000 description 2
- WBYWAXJHAXSJNI-UHFFFAOYSA-N methyl p-hydroxycinnamate Natural products OC(=O)C=CC1=CC=CC=C1 WBYWAXJHAXSJNI-UHFFFAOYSA-N 0.000 description 2
- 235000010755 mineral Nutrition 0.000 description 2
- 239000011707 mineral Substances 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 description 2
- 125000002950 monocyclic group Chemical group 0.000 description 2
- BELBLPSOLXIMFJ-UHFFFAOYSA-N n-(2-phenylethyl)-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound C=1C=CC=C(S(=O)(=O)NC=2C=NC=CC=2)C=1C(=O)NCCC1=CC=CC=C1 BELBLPSOLXIMFJ-UHFFFAOYSA-N 0.000 description 2
- RGWFZLVNZLOKOJ-UHFFFAOYSA-N n-(4-methoxypyridin-3-yl)-2-(piperidine-1-carbonyl)benzenesulfonamide Chemical compound COC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)N1CCCCC1 RGWFZLVNZLOKOJ-UHFFFAOYSA-N 0.000 description 2
- UFJXNMMVTRLRIJ-UHFFFAOYSA-N n-[(2,3-dichlorophenyl)methyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCC=2C(=C(Cl)C=CC=2)Cl)=C1 UFJXNMMVTRLRIJ-UHFFFAOYSA-N 0.000 description 2
- BSGJWRBUGYBDHK-UHFFFAOYSA-N n-[(2,4-difluorophenyl)methyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCC=2C(=CC(F)=CC=2)F)=C1 BSGJWRBUGYBDHK-UHFFFAOYSA-N 0.000 description 2
- VOIJRRHNYDZHMB-UHFFFAOYSA-N n-[(2-chloro-6-fluorophenyl)methyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCC=2C(=CC=CC=2F)Cl)=C1 VOIJRRHNYDZHMB-UHFFFAOYSA-N 0.000 description 2
- HAKGGFRXAALSRL-UHFFFAOYSA-N n-[(2-fluorophenyl)methyl]-2-[(4-methoxypyridin-3-yl)sulfamoyl]-n-methylbenzamide Chemical compound COC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)N(C)CC1=CC=CC=C1F HAKGGFRXAALSRL-UHFFFAOYSA-N 0.000 description 2
- HDKZLYYQCUODOF-UHFFFAOYSA-N n-[(2-fluorophenyl)methyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]-n-methylbenzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)N(C)CC=2C(=CC=CC=2)F)=C1 HDKZLYYQCUODOF-UHFFFAOYSA-N 0.000 description 2
- LLOVTLAOQUZYAI-UHFFFAOYSA-N n-[(3,4-dichlorophenyl)methyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCC=2C=C(Cl)C(Cl)=CC=2)=C1 LLOVTLAOQUZYAI-UHFFFAOYSA-N 0.000 description 2
- BWEQTNKWSRHHBB-UHFFFAOYSA-N n-[(3,4-difluorophenyl)methyl]-2-[(5-ethoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound CCOC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCC=2C=C(F)C(F)=CC=2)=C1 BWEQTNKWSRHHBB-UHFFFAOYSA-N 0.000 description 2
- BAJVGLYEMVHBDU-UHFFFAOYSA-N n-[(3,4-difluorophenyl)methyl]-2-[(5-methylpyridin-3-yl)sulfamoyl]benzamide Chemical compound CC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCC=2C=C(F)C(F)=CC=2)=C1 BAJVGLYEMVHBDU-UHFFFAOYSA-N 0.000 description 2
- CKTYJCWBRORZQJ-UHFFFAOYSA-N n-[(3,4-difluorophenyl)methyl]-n-methyl-2-[(5-methylpyridin-3-yl)sulfamoyl]benzamide Chemical compound C=1C=CC=C(S(=O)(=O)NC=2C=C(C)C=NC=2)C=1C(=O)N(C)CC1=CC=C(F)C(F)=C1 CKTYJCWBRORZQJ-UHFFFAOYSA-N 0.000 description 2
- HGWNRXIUMRGXJG-UHFFFAOYSA-N n-[(3-chloro-2-fluorophenyl)methyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCC=2C(=C(Cl)C=CC=2)F)=C1 HGWNRXIUMRGXJG-UHFFFAOYSA-N 0.000 description 2
- VKLFHKFXCIIWSR-UHFFFAOYSA-N n-[(3-chloro-2-methylphenyl)methyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCC=2C(=C(Cl)C=CC=2)C)=C1 VKLFHKFXCIIWSR-UHFFFAOYSA-N 0.000 description 2
- JDNMGMXTNUPZEB-UHFFFAOYSA-N n-[(3-chloro-4-fluorophenyl)methyl]-2-[(5-methylpyridin-3-yl)sulfamoyl]benzamide Chemical compound CC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCC=2C=C(Cl)C(F)=CC=2)=C1 JDNMGMXTNUPZEB-UHFFFAOYSA-N 0.000 description 2
- FJMUWXSITDDMKV-UHFFFAOYSA-N n-[(3-chloro-4-methylphenyl)methyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCC=2C=C(Cl)C(C)=CC=2)=C1 FJMUWXSITDDMKV-UHFFFAOYSA-N 0.000 description 2
- QFESGTJXYSELGZ-UHFFFAOYSA-N n-[(3-chlorophenyl)methyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCC=2C=C(Cl)C=CC=2)=C1 QFESGTJXYSELGZ-UHFFFAOYSA-N 0.000 description 2
- GWYDCYZEPASTRK-UHFFFAOYSA-N n-[(3-fluoro-4-methylphenyl)methyl]-2-[(5-methylpyridin-3-yl)sulfamoyl]benzamide Chemical compound CC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCC=2C=C(F)C(C)=CC=2)=C1 GWYDCYZEPASTRK-UHFFFAOYSA-N 0.000 description 2
- TXMAHYZRZXTSDE-UHFFFAOYSA-N n-[(4-chloro-2-methylphenyl)methyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCC=2C(=CC(Cl)=CC=2)C)=C1 TXMAHYZRZXTSDE-UHFFFAOYSA-N 0.000 description 2
- JRZIWWDYNKTTLW-UHFFFAOYSA-N n-[(4-chloro-3-fluorophenyl)methyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCC=2C=C(F)C(Cl)=CC=2)=C1 JRZIWWDYNKTTLW-UHFFFAOYSA-N 0.000 description 2
- XKPXPHGUKFYMKJ-UHFFFAOYSA-N n-[(4-fluorophenyl)methyl]-2-[(4-methoxypyridin-3-yl)sulfamoyl]-n-methylbenzamide Chemical compound COC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)N(C)CC1=CC=C(F)C=C1 XKPXPHGUKFYMKJ-UHFFFAOYSA-N 0.000 description 2
- SZOMYOBZUIOLGG-UHFFFAOYSA-N n-[(4-fluorophenyl)methyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]-5-(2-morpholin-4-ylethoxy)benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC(OCCN3CCOCC3)=CC=2)C(=O)NCC=2C=CC(F)=CC=2)=C1 SZOMYOBZUIOLGG-UHFFFAOYSA-N 0.000 description 2
- LHTHLXKPPFKMEA-UHFFFAOYSA-N n-[(4-fluorophenyl)methyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]-5-piperidin-4-yloxybenzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC(OC3CCNCC3)=CC=2)C(=O)NCC=2C=CC(F)=CC=2)=C1 LHTHLXKPPFKMEA-UHFFFAOYSA-N 0.000 description 2
- IWWGHFQZUHGVOQ-UHFFFAOYSA-N n-[1-(3,4-difluorophenyl)propan-2-yl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NC(C)CC=2C=C(F)C(F)=CC=2)=C1 IWWGHFQZUHGVOQ-UHFFFAOYSA-N 0.000 description 2
- GDWVAORSNCTOSP-UHFFFAOYSA-N n-[1-(4-chlorophenyl)-3-hydroxypropan-2-yl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NC(CO)CC=2C=CC(Cl)=CC=2)=C1 GDWVAORSNCTOSP-UHFFFAOYSA-N 0.000 description 2
- DHXZDPBYPLFVER-UHFFFAOYSA-N n-[1-(4-chlorophenyl)ethyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NC(C)C=2C=CC(Cl)=CC=2)=C1 DHXZDPBYPLFVER-UHFFFAOYSA-N 0.000 description 2
- OXQFXVFRKBMTFG-UHFFFAOYSA-N n-[2-(2-chlorophenyl)ethyl]-2-[(4-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)NCCC1=CC=CC=C1Cl OXQFXVFRKBMTFG-UHFFFAOYSA-N 0.000 description 2
- WYDFZTPBMVKRGJ-UHFFFAOYSA-N n-[2-(2-chlorophenyl)ethyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCCC=2C(=CC=CC=2)Cl)=C1 WYDFZTPBMVKRGJ-UHFFFAOYSA-N 0.000 description 2
- CDTPNUKUWNVTNH-UHFFFAOYSA-N n-[2-(2-chlorophenyl)ethyl]-2-[(5-methylpyridin-3-yl)sulfamoyl]benzamide Chemical compound CC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCCC=2C(=CC=CC=2)Cl)=C1 CDTPNUKUWNVTNH-UHFFFAOYSA-N 0.000 description 2
- AEADHDKNHFTDMD-UHFFFAOYSA-N n-[2-(2-fluorophenyl)ethyl]-2-[(4-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)NCCC1=CC=CC=C1F AEADHDKNHFTDMD-UHFFFAOYSA-N 0.000 description 2
- YZVIXULBKRGWKM-UHFFFAOYSA-N n-[2-(2-methylphenyl)ethyl]-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound CC1=CC=CC=C1CCNC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 YZVIXULBKRGWKM-UHFFFAOYSA-N 0.000 description 2
- JLASHBATSXBRLI-UHFFFAOYSA-N n-[2-(3,4-difluorophenyl)propan-2-yl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NC(C)(C)C=2C=C(F)C(F)=CC=2)=C1 JLASHBATSXBRLI-UHFFFAOYSA-N 0.000 description 2
- SBUZYNMNIUYJOA-UHFFFAOYSA-N n-[2-(3-chlorophenyl)ethyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCCC=2C=C(Cl)C=CC=2)=C1 SBUZYNMNIUYJOA-UHFFFAOYSA-N 0.000 description 2
- XGXLERLENSUUJC-UHFFFAOYSA-N n-[2-(3-methoxyphenyl)ethyl]-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound COC1=CC=CC(CCNC(=O)C=2C(=CC=CC=2)S(=O)(=O)NC=2C=NC=CC=2)=C1 XGXLERLENSUUJC-UHFFFAOYSA-N 0.000 description 2
- SMFQLEUJOFYQQY-UHFFFAOYSA-N n-[2-(3-methylphenyl)ethyl]-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound CC1=CC=CC(CCNC(=O)C=2C(=CC=CC=2)S(=O)(=O)NC=2C=NC=CC=2)=C1 SMFQLEUJOFYQQY-UHFFFAOYSA-N 0.000 description 2
- UXGYZEAZGGCZNG-UHFFFAOYSA-N n-butyl-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound CCCCNC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 UXGYZEAZGGCZNG-UHFFFAOYSA-N 0.000 description 2
- WPCKOJUACWQXJV-UHFFFAOYSA-N n-butyl-2-[(4-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound CCCCNC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CN=CC=C1OC WPCKOJUACWQXJV-UHFFFAOYSA-N 0.000 description 2
- JABWAVANHSNLFE-UHFFFAOYSA-N n-methyl-2-[(5-methylpyridin-3-yl)sulfamoyl]-n-[[4-(trifluoromethyl)phenyl]methyl]benzamide Chemical compound C=1C=CC=C(S(=O)(=O)NC=2C=C(C)C=NC=2)C=1C(=O)N(C)CC1=CC=C(C(F)(F)F)C=C1 JABWAVANHSNLFE-UHFFFAOYSA-N 0.000 description 2
- ILXITCKQRIYFEG-UHFFFAOYSA-N n-methyl-n-[2-(2-methylphenyl)ethyl]-2-[(5-methylpyridin-3-yl)sulfamoyl]benzamide Chemical compound C=1C=CC=C(S(=O)(=O)NC=2C=C(C)C=NC=2)C=1C(=O)N(C)CCC1=CC=CC=C1C ILXITCKQRIYFEG-UHFFFAOYSA-N 0.000 description 2
- NQDJXKOVJZTUJA-UHFFFAOYSA-N nevirapine Chemical compound C12=NC=CC=C2C(=O)NC=2C(C)=CC=NC=2N1C1CC1 NQDJXKOVJZTUJA-UHFFFAOYSA-N 0.000 description 2
- 229910017604 nitric acid Inorganic materials 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 239000002777 nucleoside Substances 0.000 description 2
- 150000003833 nucleoside derivatives Chemical class 0.000 description 2
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 2
- 229940116315 oxalic acid Drugs 0.000 description 2
- 235000006408 oxalic acid Nutrition 0.000 description 2
- 230000003647 oxidation Effects 0.000 description 2
- 238000007254 oxidation reaction Methods 0.000 description 2
- 229910052760 oxygen Inorganic materials 0.000 description 2
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 230000036961 partial effect Effects 0.000 description 2
- 239000008188 pellet Substances 0.000 description 2
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- 125000004193 piperazinyl group Chemical group 0.000 description 2
- 125000003367 polycyclic group Chemical group 0.000 description 2
- 229940068886 polyethylene glycol 300 Drugs 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 239000008057 potassium phosphate buffer Substances 0.000 description 2
- 150000003141 primary amines Chemical class 0.000 description 2
- 229960004063 propylene glycol Drugs 0.000 description 2
- 235000013772 propylene glycol Nutrition 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 2
- 229940107700 pyruvic acid Drugs 0.000 description 2
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 2
- 108010043277 recombinant soluble CD4 Proteins 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 239000011347 resin Substances 0.000 description 2
- 229920005989 resin Polymers 0.000 description 2
- ATEBXHFBFRCZMA-VXTBVIBXSA-N rifabutin Chemical compound O([C@](C1=O)(C)O/C=C/[C@@H]([C@H]([C@@H](OC(C)=O)[C@H](C)[C@H](O)[C@H](C)[C@@H](O)[C@@H](C)\C=C\C=C(C)/C(=O)NC(=C2N3)C(=O)C=4C(O)=C5C)C)OC)C5=C1C=4C2=NC13CCN(CC(C)C)CC1 ATEBXHFBFRCZMA-VXTBVIBXSA-N 0.000 description 2
- 229960000885 rifabutin Drugs 0.000 description 2
- 239000003419 rna directed dna polymerase inhibitor Substances 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 2
- 229960004889 salicylic acid Drugs 0.000 description 2
- 150000003335 secondary amines Chemical class 0.000 description 2
- 238000000926 separation method Methods 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 239000001384 succinic acid Substances 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 230000009885 systemic effect Effects 0.000 description 2
- 239000011975 tartaric acid Substances 0.000 description 2
- 235000002906 tartaric acid Nutrition 0.000 description 2
- VCMJCVGFSROFHV-WZGZYPNHSA-N tenofovir disoproxil fumarate Chemical compound OC(=O)\C=C\C(O)=O.N1=CN=C2N(C[C@@H](C)OCP(=O)(OCOC(=O)OC(C)C)OCOC(=O)OC(C)C)C=NC2=C1N VCMJCVGFSROFHV-WZGZYPNHSA-N 0.000 description 2
- 150000003512 tertiary amines Chemical class 0.000 description 2
- 125000000335 thiazolyl group Chemical group 0.000 description 2
- FYSNRJHAOHDILO-UHFFFAOYSA-N thionyl chloride Chemical compound ClS(Cl)=O FYSNRJHAOHDILO-UHFFFAOYSA-N 0.000 description 2
- 231100000419 toxicity Toxicity 0.000 description 2
- 230000001988 toxicity Effects 0.000 description 2
- 102000003390 tumor necrosis factor Human genes 0.000 description 2
- 239000011701 zinc Substances 0.000 description 2
- 229910052725 zinc Inorganic materials 0.000 description 2
- NIDRYBLTWYFCFV-UHFFFAOYSA-N (-)-calanolide b Chemical compound C1=CC(C)(C)OC2=C1C(OC(C)C(C)C1O)=C1C1=C2C(CCC)=CC(=O)O1 NIDRYBLTWYFCFV-UHFFFAOYSA-N 0.000 description 1
- AOSZTAHDEDLTLQ-AZKQZHLXSA-N (1S,2S,4R,8S,9S,11S,12R,13S,19S)-6-[(3-chlorophenyl)methyl]-12,19-difluoro-11-hydroxy-8-(2-hydroxyacetyl)-9,13-dimethyl-6-azapentacyclo[10.8.0.02,9.04,8.013,18]icosa-14,17-dien-16-one Chemical compound C([C@@H]1C[C@H]2[C@H]3[C@]([C@]4(C=CC(=O)C=C4[C@@H](F)C3)C)(F)[C@@H](O)C[C@@]2([C@@]1(C1)C(=O)CO)C)N1CC1=CC=CC(Cl)=C1 AOSZTAHDEDLTLQ-AZKQZHLXSA-N 0.000 description 1
- XMAYWYJOQHXEEK-OZXSUGGESA-N (2R,4S)-ketoconazole Chemical compound C1CN(C(=O)C)CCN1C(C=C1)=CC=C1OC[C@@H]1O[C@@](CN2C=NC=C2)(C=2C(=CC(Cl)=CC=2)Cl)OC1 XMAYWYJOQHXEEK-OZXSUGGESA-N 0.000 description 1
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- GTXSRFUZSLTDFX-HRCADAONSA-N (2s)-n-[(2s)-3,3-dimethyl-1-(methylamino)-1-oxobutan-2-yl]-4-methyl-2-[[(2s)-2-sulfanyl-4-(3,4,4-trimethyl-2,5-dioxoimidazolidin-1-yl)butanoyl]amino]pentanamide Chemical compound CNC(=O)[C@H](C(C)(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](S)CCN1C(=O)N(C)C(C)(C)C1=O GTXSRFUZSLTDFX-HRCADAONSA-N 0.000 description 1
- QPVWMQXBTCSLCB-BYAJYZPISA-N (2s)-n-[(2s,3s,4r,5s)-3,4-dihydroxy-5-[[(2s)-3-methyl-2-[[methyl(pyridin-2-ylmethyl)carbamoyl]amino]butanoyl]amino]-1,6-diphenylhexan-2-yl]-3-methyl-2-[[methyl(pyridin-2-ylmethyl)carbamoyl]amino]butanamide Chemical compound N([C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)[C@H](O)[C@H](O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)N(C)CC=1N=CC=CC=1)C(C)C)C(=O)N(C)CC1=CC=CC=N1 QPVWMQXBTCSLCB-BYAJYZPISA-N 0.000 description 1
- QVSVMNXRLWSNGS-UHFFFAOYSA-N (3-fluorophenyl)methanamine Chemical compound NCC1=CC=CC(F)=C1 QVSVMNXRLWSNGS-UHFFFAOYSA-N 0.000 description 1
- NTWKGLJNQAZSHF-ZRTHHSRSSA-N (4r,5s,6s,7r)-1-[(3-amino-1h-indazol-5-yl)methyl]-3,4,7-tribenzyl-5,6-dihydroxy-1,3-diazepan-2-one Chemical compound C([C@H]1N(C(N(CC=2C=CC=CC=2)[C@H](CC=2C=CC=CC=2)[C@H](O)[C@H]1O)=O)CC1=CC=C2NN=C(C2=C1)N)C1=CC=CC=C1 NTWKGLJNQAZSHF-ZRTHHSRSSA-N 0.000 description 1
- JJWJSIAJLBEMEN-ZDUSSCGKSA-N (4s)-6-chloro-4-(2-cyclopropylethynyl)-4-(trifluoromethyl)-1,3-dihydroquinazolin-2-one Chemical compound C([C@]1(C2=CC(Cl)=CC=C2NC(=O)N1)C(F)(F)F)#CC1CC1 JJWJSIAJLBEMEN-ZDUSSCGKSA-N 0.000 description 1
- UXDWYQAXEGVSPS-GFUIURDCSA-N (4s)-6-chloro-4-[(e)-2-cyclopropylethenyl]-4-(trifluoromethyl)-1,3-dihydroquinazolin-2-one Chemical compound C(/[C@]1(C2=CC(Cl)=CC=C2NC(=O)N1)C(F)(F)F)=C\C1CC1 UXDWYQAXEGVSPS-GFUIURDCSA-N 0.000 description 1
- GHOKWGTUZJEAQD-ZETCQYMHSA-N (D)-(+)-Pantothenic acid Chemical compound OCC(C)(C)[C@@H](O)C(=O)NCCC(O)=O GHOKWGTUZJEAQD-ZETCQYMHSA-N 0.000 description 1
- GWKIPRVERALPRD-ZDUSSCGKSA-N (s)-4-isopropoxycarbonyl-6-methoxy-3-methylthiomethyl-3,4-dihydroquinoxalin-2(1h)-thione Chemical compound N1C(=S)[C@H](CSC)N(C(=O)OC(C)C)C2=CC(OC)=CC=C21 GWKIPRVERALPRD-ZDUSSCGKSA-N 0.000 description 1
- SWMNGXODFOCPKQ-BTJKTKAUSA-N (z)-but-2-enedioic acid;n'-methoxy-4-[5-[4-[(z)-n'-methoxycarbamimidoyl]phenyl]furan-2-yl]benzenecarboximidamide Chemical compound OC(=O)\C=C/C(O)=O.C1=CC(C(=N)NOC)=CC=C1C1=CC=C(C=2C=CC(=CC=2)C(=N)NOC)O1 SWMNGXODFOCPKQ-BTJKTKAUSA-N 0.000 description 1
- NWUYHJFMYQTDRP-UHFFFAOYSA-N 1,2-bis(ethenyl)benzene;1-ethenyl-2-ethylbenzene;styrene Chemical compound C=CC1=CC=CC=C1.CCC1=CC=CC=C1C=C.C=CC1=CC=CC=C1C=C NWUYHJFMYQTDRP-UHFFFAOYSA-N 0.000 description 1
- DDMOUSALMHHKOS-UHFFFAOYSA-N 1,2-dichloro-1,1,2,2-tetrafluoroethane Chemical compound FC(F)(Cl)C(F)(F)Cl DDMOUSALMHHKOS-UHFFFAOYSA-N 0.000 description 1
- BDNKZNFMNDZQMI-UHFFFAOYSA-N 1,3-diisopropylcarbodiimide Chemical compound CC(C)N=C=NC(C)C BDNKZNFMNDZQMI-UHFFFAOYSA-N 0.000 description 1
- JPRPJUMQRZTTED-UHFFFAOYSA-N 1,3-dioxolanyl Chemical group [CH]1OCCO1 JPRPJUMQRZTTED-UHFFFAOYSA-N 0.000 description 1
- WXNZNIQENJBHBC-UHFFFAOYSA-N 1,5-naphthyridine-3-carboxamide Chemical class C1=CC=NC2=CC(C(=O)N)=CN=C21 WXNZNIQENJBHBC-UHFFFAOYSA-N 0.000 description 1
- LMDZBCPBFSXMTL-UHFFFAOYSA-N 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide Substances CCN=C=NCCCN(C)C LMDZBCPBFSXMTL-UHFFFAOYSA-N 0.000 description 1
- OKGPFTLYBPQBIX-CQSZACIVSA-N 1-[(2r)-4-benzoyl-2-methylpiperazin-1-yl]-2-(4-methoxy-1h-pyrrolo[2,3-b]pyridin-3-yl)ethane-1,2-dione Chemical compound C1=2C(OC)=CC=NC=2NC=C1C(=O)C(=O)N([C@@H](C1)C)CCN1C(=O)C1=CC=CC=C1 OKGPFTLYBPQBIX-CQSZACIVSA-N 0.000 description 1
- ONBQEOIKXPHGMB-VBSBHUPXSA-N 1-[2-[(2s,3r,4s,5r)-3,4-dihydroxy-5-(hydroxymethyl)oxolan-2-yl]oxy-4,6-dihydroxyphenyl]-3-(4-hydroxyphenyl)propan-1-one Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1OC1=CC(O)=CC(O)=C1C(=O)CCC1=CC=C(O)C=C1 ONBQEOIKXPHGMB-VBSBHUPXSA-N 0.000 description 1
- USMXIXZBRZZOQH-UHFFFAOYSA-N 1-[[4-(1,4,8,11-tetrazacyclotetradec-1-ylmethyl)phenyl]methyl]-1,4,8,11-tetrazacyclotetradecane;dihydrate;octahydrochloride Chemical compound O.O.Cl.Cl.Cl.Cl.Cl.Cl.Cl.Cl.C=1C=C(CN2CCNCCCNCCNCCC2)C=CC=1CN1CCCNCCNCCCNCC1 USMXIXZBRZZOQH-UHFFFAOYSA-N 0.000 description 1
- QHSMEGADRFZVNE-UHFFFAOYSA-N 1-hydroxymidazolam Chemical compound C12=CC(Cl)=CC=C2N2C(CO)=NC=C2CN=C1C1=CC=CC=C1F QHSMEGADRFZVNE-UHFFFAOYSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- RZRNAYUHWVFMIP-KTKRTIGZSA-N 1-oleoylglycerol Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC(O)CO RZRNAYUHWVFMIP-KTKRTIGZSA-N 0.000 description 1
- 125000001462 1-pyrrolyl group Chemical group [*]N1C([H])=C([H])C([H])=C1[H] 0.000 description 1
- HCSBTDBGTNZOAB-UHFFFAOYSA-N 2,3-dinitrobenzoic acid Chemical compound OC(=O)C1=CC=CC([N+]([O-])=O)=C1[N+]([O-])=O HCSBTDBGTNZOAB-UHFFFAOYSA-N 0.000 description 1
- RQOLVVSZHICZQK-UHFFFAOYSA-N 2-(1,3-dihydroisoindole-2-carbonyl)-n-pyridin-3-ylbenzenesulfonamide Chemical compound C1C2=CC=CC=C2CN1C(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 RQOLVVSZHICZQK-UHFFFAOYSA-N 0.000 description 1
- QRRQEXYHWSGFFB-UHFFFAOYSA-N 2-(3,3-dimethylpiperidine-1-carbonyl)-n-pyridin-3-ylbenzenesulfonamide Chemical compound C1C(C)(C)CCCN1C(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 QRRQEXYHWSGFFB-UHFFFAOYSA-N 0.000 description 1
- XBJRDVOMBNOGII-UHFFFAOYSA-N 2-(3,5-dimethylpiperidine-1-carbonyl)-n-pyridin-3-ylbenzenesulfonamide Chemical compound C1C(C)CC(C)CN1C(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 XBJRDVOMBNOGII-UHFFFAOYSA-N 0.000 description 1
- KVZDNSCGJQLVBU-UHFFFAOYSA-N 2-(4-hydroxypiperidine-1-carbonyl)-n-pyridin-3-ylbenzenesulfonamide Chemical compound C1CC(O)CCN1C(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 KVZDNSCGJQLVBU-UHFFFAOYSA-N 0.000 description 1
- BURFYGDOBPEGQD-UHFFFAOYSA-N 2-(4-methoxypiperidine-1-carbonyl)-n-pyridin-3-ylbenzenesulfonamide Chemical compound C1CC(OC)CCN1C(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 BURFYGDOBPEGQD-UHFFFAOYSA-N 0.000 description 1
- SBYGPIWXUUNLEP-UHFFFAOYSA-N 2-(4-methylpiperazine-1-carbonyl)-n-pyridin-3-ylbenzenesulfonamide Chemical compound C1CN(C)CCN1C(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 SBYGPIWXUUNLEP-UHFFFAOYSA-N 0.000 description 1
- RDAKOFVWKNIKNS-UHFFFAOYSA-N 2-(4-methylpiperidine-1-carbonyl)-n-pyridin-3-ylbenzenesulfonamide Chemical compound C1CC(C)CCN1C(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 RDAKOFVWKNIKNS-UHFFFAOYSA-N 0.000 description 1
- YVCLDTKIKNEEAR-UHFFFAOYSA-N 2-(phenylsulfamoyl)-n-(quinolin-6-ylmethyl)benzamide Chemical compound C=1C=C2N=CC=CC2=CC=1CNC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CC=C1 YVCLDTKIKNEEAR-UHFFFAOYSA-N 0.000 description 1
- UHAXVBFSTFIHFJ-UHFFFAOYSA-N 2-(piperidine-1-carbonyl)-n-pyridin-3-ylbenzenesulfonamide Chemical compound C=1C=CC=C(S(=O)(=O)NC=2C=NC=CC=2)C=1C(=O)N1CCCCC1 UHAXVBFSTFIHFJ-UHFFFAOYSA-N 0.000 description 1
- TZBFNTDQLQKPAZ-UHFFFAOYSA-N 2-(piperidine-1-carbonyl)benzenesulfonamide Chemical compound NS(=O)(=O)C1=CC=CC=C1C(=O)N1CCCCC1 TZBFNTDQLQKPAZ-UHFFFAOYSA-N 0.000 description 1
- NZOUXUIBJTZWDL-UHFFFAOYSA-N 2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound NC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 NZOUXUIBJTZWDL-UHFFFAOYSA-N 0.000 description 1
- VHVPQPYKVGDNFY-DFMJLFEVSA-N 2-[(2r)-butan-2-yl]-4-[4-[4-[4-[[(2r,4s)-2-(2,4-dichlorophenyl)-2-(1,2,4-triazol-1-ylmethyl)-1,3-dioxolan-4-yl]methoxy]phenyl]piperazin-1-yl]phenyl]-1,2,4-triazol-3-one Chemical compound O=C1N([C@H](C)CC)N=CN1C1=CC=C(N2CCN(CC2)C=2C=CC(OC[C@@H]3O[C@](CN4N=CN=C4)(OC3)C=3C(=CC(Cl)=CC=3)Cl)=CC=2)C=C1 VHVPQPYKVGDNFY-DFMJLFEVSA-N 0.000 description 1
- NYKUMWAPRICTSX-UHFFFAOYSA-N 2-[(4-ethoxypyridin-3-yl)sulfamoyl]-n-(2-methoxyethyl)-n-methylbenzamide Chemical compound CCOC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)N(C)CCOC NYKUMWAPRICTSX-UHFFFAOYSA-N 0.000 description 1
- FOJSTUKBOKAIOO-UHFFFAOYSA-N 2-[(4-ethoxypyridin-3-yl)sulfamoyl]-n-(2-methoxyethyl)-n-propan-2-ylbenzamide Chemical compound CCOC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)N(CCOC)C(C)C FOJSTUKBOKAIOO-UHFFFAOYSA-N 0.000 description 1
- UUMROOFDNHEROD-UHFFFAOYSA-N 2-[(4-ethoxypyridin-3-yl)sulfamoyl]-n-(2-methoxyethyl)benzamide Chemical compound CCOC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)NCCOC UUMROOFDNHEROD-UHFFFAOYSA-N 0.000 description 1
- IEXMSSINFFHJMR-UHFFFAOYSA-N 2-[(4-ethoxypyridin-3-yl)sulfamoyl]-n-(2-methylbutan-2-yl)benzamide Chemical compound CCOC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)NC(C)(C)CC IEXMSSINFFHJMR-UHFFFAOYSA-N 0.000 description 1
- LVKFFHHAWCZXPY-UHFFFAOYSA-N 2-[(4-ethoxypyridin-3-yl)sulfamoyl]-n-(2-methylbutyl)benzamide Chemical compound CCOC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)NCC(C)CC LVKFFHHAWCZXPY-UHFFFAOYSA-N 0.000 description 1
- QNKZNOITBHKCNK-UHFFFAOYSA-N 2-[(4-ethoxypyridin-3-yl)sulfamoyl]-n-(2-phenoxyethyl)benzamide Chemical compound CCOC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)NCCOC1=CC=CC=C1 QNKZNOITBHKCNK-UHFFFAOYSA-N 0.000 description 1
- CQLVVCFVTZIBSI-UHFFFAOYSA-N 2-[(4-ethoxypyridin-3-yl)sulfamoyl]-n-(3-methylbutyl)benzamide Chemical compound CCOC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)NCCC(C)C CQLVVCFVTZIBSI-UHFFFAOYSA-N 0.000 description 1
- ZNVPFVAJEGXMCQ-CQSZACIVSA-N 2-[(4-ethoxypyridin-3-yl)sulfamoyl]-n-[(2r)-3-methylbutan-2-yl]benzamide Chemical compound CCOC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)N[C@H](C)C(C)C ZNVPFVAJEGXMCQ-CQSZACIVSA-N 0.000 description 1
- ZNVPFVAJEGXMCQ-AWEZNQCLSA-N 2-[(4-ethoxypyridin-3-yl)sulfamoyl]-n-[(2s)-3-methylbutan-2-yl]benzamide Chemical compound CCOC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)N[C@@H](C)C(C)C ZNVPFVAJEGXMCQ-AWEZNQCLSA-N 0.000 description 1
- CXMXQJHTIKAJFA-UHFFFAOYSA-N 2-[(4-ethoxypyridin-3-yl)sulfamoyl]-n-methyl-n-pentylbenzamide Chemical compound CCCCCN(C)C(=O)C1=CC=CC=C1S(=O)(=O)NC1=CN=CC=C1OCC CXMXQJHTIKAJFA-UHFFFAOYSA-N 0.000 description 1
- GJQJDSSELKFYDO-UHFFFAOYSA-N 2-[(4-ethoxypyridin-3-yl)sulfamoyl]-n-methyl-n-propan-2-ylbenzamide Chemical compound CCOC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)N(C)C(C)C GJQJDSSELKFYDO-UHFFFAOYSA-N 0.000 description 1
- GGASDZBISMAILC-UHFFFAOYSA-N 2-[(4-methoxypyridin-3-yl)sulfamoyl]-n-(2-methylbutan-2-yl)benzamide Chemical compound CCC(C)(C)NC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CN=CC=C1OC GGASDZBISMAILC-UHFFFAOYSA-N 0.000 description 1
- ROQHUMUSZRNSJM-UHFFFAOYSA-N 2-[(4-methoxypyridin-3-yl)sulfamoyl]-n-(2-methylbutyl)benzamide Chemical compound CCC(C)CNC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CN=CC=C1OC ROQHUMUSZRNSJM-UHFFFAOYSA-N 0.000 description 1
- KUGAVCFFQGVMDA-UHFFFAOYSA-N 2-[(4-methoxypyridin-3-yl)sulfamoyl]-n-(2-phenoxyethyl)benzamide Chemical compound COC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)NCCOC1=CC=CC=C1 KUGAVCFFQGVMDA-UHFFFAOYSA-N 0.000 description 1
- OBVNLPPUVGSUCY-UHFFFAOYSA-N 2-[(4-methoxypyridin-3-yl)sulfamoyl]-n-methyl-n-pentylbenzamide Chemical compound CCCCCN(C)C(=O)C1=CC=CC=C1S(=O)(=O)NC1=CN=CC=C1OC OBVNLPPUVGSUCY-UHFFFAOYSA-N 0.000 description 1
- BLVMZXNGPQUESD-UHFFFAOYSA-N 2-[(4-methoxypyridin-3-yl)sulfamoyl]-n-methyl-n-propan-2-ylbenzamide Chemical compound COC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)N(C)C(C)C BLVMZXNGPQUESD-UHFFFAOYSA-N 0.000 description 1
- ACTOXUHEUCPTEW-BWHGAVFKSA-N 2-[(4r,5s,6s,7r,9r,10r,11e,13e,16r)-6-[(2s,3r,4r,5s,6r)-5-[(2s,4r,5s,6s)-4,5-dihydroxy-4,6-dimethyloxan-2-yl]oxy-4-(dimethylamino)-3-hydroxy-6-methyloxan-2-yl]oxy-10-[(2s,5s,6r)-5-(dimethylamino)-6-methyloxan-2-yl]oxy-4-hydroxy-5-methoxy-9,16-dimethyl-2-o Chemical compound O([C@H]1/C=C/C=C/C[C@@H](C)OC(=O)C[C@@H](O)[C@@H]([C@H]([C@@H](CC=O)C[C@H]1C)O[C@H]1[C@@H]([C@H]([C@H](O[C@@H]2O[C@@H](C)[C@H](O)[C@](C)(O)C2)[C@@H](C)O1)N(C)C)O)OC)[C@@H]1CC[C@H](N(C)C)[C@@H](C)O1 ACTOXUHEUCPTEW-BWHGAVFKSA-N 0.000 description 1
- ZOLIBEKXICARLI-UHFFFAOYSA-N 2-[(5-ethoxypyridin-3-yl)sulfamoyl]-n-(3-methylbutyl)benzamide Chemical compound CCOC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCCC(C)C)=C1 ZOLIBEKXICARLI-UHFFFAOYSA-N 0.000 description 1
- JEKULYLYGIPSAU-UHFFFAOYSA-N 2-[(5-methoxypyridin-3-yl)sulfamoyl]-n-methyl-5-piperidin-4-yloxy-n-propan-2-ylbenzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC(OC3CCNCC3)=CC=2)C(=O)N(C)C(C)C)=C1 JEKULYLYGIPSAU-UHFFFAOYSA-N 0.000 description 1
- SFFQYKBIMWQWGC-UHFFFAOYSA-N 2-[(5-methoxypyridin-3-yl)sulfamoyl]-n-methyl-n-(2-phenylethyl)benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)N(C)CCC=2C=CC=CC=2)=C1 SFFQYKBIMWQWGC-UHFFFAOYSA-N 0.000 description 1
- GXWYPVFLJMINEG-UHFFFAOYSA-N 2-[(5-methoxypyridin-3-yl)sulfamoyl]-n-methyl-n-(3-methylbutyl)benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)N(C)CCC(C)C)=C1 GXWYPVFLJMINEG-UHFFFAOYSA-N 0.000 description 1
- DVURLAWPVFJXTO-UHFFFAOYSA-N 2-[(5-methoxypyridin-3-yl)sulfamoyl]-n-methyl-n-pentylbenzamide Chemical compound CCCCCN(C)C(=O)C1=CC=CC=C1S(=O)(=O)NC1=CN=CC(OC)=C1 DVURLAWPVFJXTO-UHFFFAOYSA-N 0.000 description 1
- KVXRWCVCDDTQQA-UHFFFAOYSA-N 2-[(5-methoxypyridin-3-yl)sulfamoyl]-n-pentan-3-ylbenzamide Chemical compound CCC(CC)NC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CN=CC(OC)=C1 KVXRWCVCDDTQQA-UHFFFAOYSA-N 0.000 description 1
- OIQOAYVCKAHSEJ-UHFFFAOYSA-N 2-[2,3-bis(2-hydroxyethoxy)propoxy]ethanol;hexadecanoic acid;octadecanoic acid Chemical compound OCCOCC(OCCO)COCCO.CCCCCCCCCCCCCCCC(O)=O.CCCCCCCCCCCCCCCCCC(O)=O OIQOAYVCKAHSEJ-UHFFFAOYSA-N 0.000 description 1
- BNHYMYBGHFACHA-UHFFFAOYSA-N 2-[4-(4-fluorophenyl)piperazine-1-carbonyl]-n-pyridin-3-ylbenzenesulfonamide Chemical compound C1=CC(F)=CC=C1N1CCN(C(=O)C=2C(=CC=CC=2)S(=O)(=O)NC=2C=NC=CC=2)CC1 BNHYMYBGHFACHA-UHFFFAOYSA-N 0.000 description 1
- BTOMIMSUTLPSHA-UHFFFAOYSA-N 2-[4-chloro-2-(3-chloro-5-cyanobenzoyl)phenoxy]-n-(2-methyl-4-sulfamoylphenyl)acetamide Chemical compound CC1=CC(S(N)(=O)=O)=CC=C1NC(=O)COC1=CC=C(Cl)C=C1C(=O)C1=CC(Cl)=CC(C#N)=C1 BTOMIMSUTLPSHA-UHFFFAOYSA-N 0.000 description 1
- MDCGTBRYRLWFJI-WDCKKOMHSA-N 2-amino-9-(2-hydroxyethoxymethyl)-3h-purin-6-one;(8s,9s,10r,11s,13s,14s,17r)-11,17-dihydroxy-17-(2-hydroxyacetyl)-10,13-dimethyl-2,6,7,8,9,11,12,14,15,16-decahydro-1h-cyclopenta[a]phenanthren-3-one Chemical compound N1C(N)=NC(=O)C2=C1N(COCCO)C=N2.O=C1CC[C@]2(C)[C@H]3[C@@H](O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 MDCGTBRYRLWFJI-WDCKKOMHSA-N 0.000 description 1
- GWFOVSGRNGAGDL-FSDSQADBSA-N 2-amino-9-[(1r,2r,3s)-2,3-bis(hydroxymethyl)cyclobutyl]-3h-purin-6-one Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1C[C@H](CO)[C@H]1CO GWFOVSGRNGAGDL-FSDSQADBSA-N 0.000 description 1
- BCSLVBZWCFTDPK-UDJQAZALSA-N 2-amino-9-[(2r,3r,4s)-3,4-bis(hydroxymethyl)oxetan-2-yl]-3h-purin-6-one Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@@H]1O[C@H](CO)[C@H]1CO BCSLVBZWCFTDPK-UDJQAZALSA-N 0.000 description 1
- RTJUXLYUUDBAJN-KVQBGUIXSA-N 2-amino-9-[(2r,4s,5r)-4-fluoro-5-(hydroxymethyl)oxolan-2-yl]-3h-purin-6-one Chemical compound C1=2NC(N)=NC(=O)C=2N=CN1[C@H]1C[C@H](F)[C@@H](CO)O1 RTJUXLYUUDBAJN-KVQBGUIXSA-N 0.000 description 1
- SLVAPEZTBDBAPI-UHFFFAOYSA-N 2-cyclopentyl-2-[2-(2,6-diethylpyridin-4-yl)ethyl]-5-[(5,7-dimethyl-[1,2,4]triazolo[1,5-a]pyrimidin-2-yl)methyl]-4-hydroxy-3h-pyran-6-one Chemical compound CCC1=NC(CC)=CC(CCC2(OC(=O)C(CC3=NN4C(C)=CC(C)=NC4=N3)=C(O)C2)C2CCCC2)=C1 SLVAPEZTBDBAPI-UHFFFAOYSA-N 0.000 description 1
- HZLCGUXUOFWCCN-UHFFFAOYSA-N 2-hydroxynonadecane-1,2,3-tricarboxylic acid Chemical compound CCCCCCCCCCCCCCCCC(C(O)=O)C(O)(C(O)=O)CC(O)=O HZLCGUXUOFWCCN-UHFFFAOYSA-N 0.000 description 1
- JMTMSDXUXJISAY-UHFFFAOYSA-N 2H-benzotriazol-4-ol Chemical compound OC1=CC=CC2=C1N=NN2 JMTMSDXUXJISAY-UHFFFAOYSA-N 0.000 description 1
- 125000001698 2H-pyranyl group Chemical group O1C(C=CC=C1)* 0.000 description 1
- SOSPMXMEOFGPIM-UHFFFAOYSA-N 3,5-dibromopyridine Chemical compound BrC1=CN=CC(Br)=C1 SOSPMXMEOFGPIM-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- UZFPOOOQHWICKY-UHFFFAOYSA-N 3-[13-[1-[1-[8,12-bis(2-carboxyethyl)-17-(1-hydroxyethyl)-3,7,13,18-tetramethyl-21,24-dihydroporphyrin-2-yl]ethoxy]ethyl]-18-(2-carboxyethyl)-8-(1-hydroxyethyl)-3,7,12,17-tetramethyl-22,23-dihydroporphyrin-2-yl]propanoic acid Chemical compound N1C(C=C2C(=C(CCC(O)=O)C(C=C3C(=C(C)C(C=C4N5)=N3)CCC(O)=O)=N2)C)=C(C)C(C(C)O)=C1C=C5C(C)=C4C(C)OC(C)C1=C(N2)C=C(N3)C(C)=C(C(O)C)C3=CC(C(C)=C3CCC(O)=O)=NC3=CC(C(CCC(O)=O)=C3C)=NC3=CC2=C1C UZFPOOOQHWICKY-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-M 3-carboxy-2,3-dihydroxypropanoate Chemical compound OC(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-M 0.000 description 1
- 125000004364 3-pyrrolinyl group Chemical group [H]C1=C([H])C([H])([H])N(*)C1([H])[H] 0.000 description 1
- 125000001397 3-pyrrolyl group Chemical group [H]N1C([H])=C([*])C([H])=C1[H] 0.000 description 1
- 125000001963 4 membered heterocyclic group Chemical group 0.000 description 1
- XEDONBRPTABQFB-UHFFFAOYSA-N 4-[(2-formyl-3-hydroxyphenoxy)methyl]benzoic acid Chemical compound C1=CC(C(=O)O)=CC=C1COC1=CC=CC(O)=C1C=O XEDONBRPTABQFB-UHFFFAOYSA-N 0.000 description 1
- HSBKFSPNDWWPSL-CAHLUQPWSA-N 4-amino-5-fluoro-1-[(2r,5s)-5-(hydroxymethyl)-2,5-dihydrofuran-2-yl]pyrimidin-2-one Chemical compound C1=C(F)C(N)=NC(=O)N1[C@H]1C=C[C@@H](CO)O1 HSBKFSPNDWWPSL-CAHLUQPWSA-N 0.000 description 1
- BIUDHHGROGJSHN-UHFFFAOYSA-N 4-fluoro-3-(trifluoromethyl)benzaldehyde Chemical group FC1=CC=C(C=O)C=C1C(F)(F)F BIUDHHGROGJSHN-UHFFFAOYSA-N 0.000 description 1
- SJZRECIVHVDYJC-UHFFFAOYSA-N 4-hydroxybutyric acid Chemical compound OCCCC(O)=O SJZRECIVHVDYJC-UHFFFAOYSA-N 0.000 description 1
- BZPVREXVOZITPF-UHFFFAOYSA-N 4-methoxy-3-nitropyridine Chemical compound COC1=CC=NC=C1[N+]([O-])=O BZPVREXVOZITPF-UHFFFAOYSA-N 0.000 description 1
- STWMPIWLSQKHSJ-UHFFFAOYSA-N 4-methoxypyridin-3-amine Chemical compound COC1=CC=NC=C1N STWMPIWLSQKHSJ-UHFFFAOYSA-N 0.000 description 1
- OBKXEAXTFZPCHS-UHFFFAOYSA-N 4-phenylbutyric acid Chemical compound OC(=O)CCCC1=CC=CC=C1 OBKXEAXTFZPCHS-UHFFFAOYSA-N 0.000 description 1
- 125000001826 4H-pyranyl group Chemical group O1C(=CCC=C1)* 0.000 description 1
- 125000002373 5 membered heterocyclic group Chemical group 0.000 description 1
- GJOHLWZHWQUKAU-UHFFFAOYSA-N 5-azaniumylpentan-2-yl-(6-methoxyquinolin-8-yl)azanium;dihydrogen phosphate Chemical compound OP(O)(O)=O.OP(O)(O)=O.N1=CC=CC2=CC(OC)=CC(NC(C)CCCN)=C21 GJOHLWZHWQUKAU-UHFFFAOYSA-N 0.000 description 1
- BEUMBMWUTPTRGO-UHFFFAOYSA-N 5-hydroxypyrimidine-4-carboxamide Chemical class NC(=O)C1=NC=NC=C1O BEUMBMWUTPTRGO-UHFFFAOYSA-N 0.000 description 1
- JXUWZXFVCBODAN-UHFFFAOYSA-N 5-methylpyridin-3-amine Chemical compound CC1=CN=CC(N)=C1 JXUWZXFVCBODAN-UHFFFAOYSA-N 0.000 description 1
- XSEGWEUVSZRCBC-UHFFFAOYSA-N 6beta-Hydroxytestosterone Natural products O=C1CCC2(C)C3CCC(C)(C(CC4)O)C4C3CC(O)C2=C1 XSEGWEUVSZRCBC-UHFFFAOYSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- NIXOWILDQLNWCW-UHFFFAOYSA-M Acrylate Chemical compound [O-]C(=O)C=C NIXOWILDQLNWCW-UHFFFAOYSA-M 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 108010064760 Anidulafungin Proteins 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- BTBUEUYNUDRHOZ-UHFFFAOYSA-N Borate Chemical compound [O-]B([O-])[O-] BTBUEUYNUDRHOZ-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 1
- CIUUIPMOFZIWIZ-UHFFFAOYSA-N Bropirimine Chemical compound NC1=NC(O)=C(Br)C(C=2C=CC=CC=2)=N1 CIUUIPMOFZIWIZ-UHFFFAOYSA-N 0.000 description 1
- 0 CC(C)(*)OC(N1CCC(C2)(CN2C(c2ccccc2S(Nc2cccnc2)(=O)=O)=O)CC1)=O Chemical compound CC(C)(*)OC(N1CCC(C2)(CN2C(c2ccccc2S(Nc2cccnc2)(=O)=O)=O)CC1)=O 0.000 description 1
- 108010036239 CD4-IgG(2) Proteins 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 108010020326 Caspofungin Proteins 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- HZZVJAQRINQKSD-UHFFFAOYSA-N Clavulanic acid Natural products OC(=O)C1C(=CCO)OC2CC(=O)N21 HZZVJAQRINQKSD-UHFFFAOYSA-N 0.000 description 1
- 229940126657 Compound 17 Drugs 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 239000001879 Curdlan Substances 0.000 description 1
- 229920002558 Curdlan Polymers 0.000 description 1
- 208000009011 Cytochrome P-450 CYP3A Inhibitors Diseases 0.000 description 1
- 241000701022 Cytomegalovirus Species 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 206010012735 Diarrhoea Diseases 0.000 description 1
- 239000004338 Dichlorodifluoromethane Substances 0.000 description 1
- BXZVVICBKDXVGW-NKWVEPMBSA-N Didanosine Chemical compound O1[C@H](CO)CC[C@@H]1N1C(NC=NC2=O)=C2N=C1 BXZVVICBKDXVGW-NKWVEPMBSA-N 0.000 description 1
- DKMROQRQHGEIOW-UHFFFAOYSA-N Diethyl succinate Chemical compound CCOC(=O)CCC(=O)OCC DKMROQRQHGEIOW-UHFFFAOYSA-N 0.000 description 1
- 208000032928 Dyslipidaemia Diseases 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- XPOQHMRABVBWPR-UHFFFAOYSA-N Efavirenz Natural products O1C(=O)NC2=CC=C(Cl)C=C2C1(C(F)(F)F)C#CC1CC1 XPOQHMRABVBWPR-UHFFFAOYSA-N 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 108010032976 Enfuvirtide Proteins 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- 239000004606 Fillers/Extenders Substances 0.000 description 1
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 description 1
- BDAGIHXWWSANSR-UHFFFAOYSA-M Formate Chemical compound [O-]C=O BDAGIHXWWSANSR-UHFFFAOYSA-M 0.000 description 1
- 238000005033 Fourier transform infrared spectroscopy Methods 0.000 description 1
- 241000206672 Gelidium Species 0.000 description 1
- 102000004457 Granulocyte-Macrophage Colony-Stimulating Factor Human genes 0.000 description 1
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 239000012981 Hank's balanced salt solution Substances 0.000 description 1
- 101100005713 Homo sapiens CD4 gene Proteins 0.000 description 1
- 101000987586 Homo sapiens Eosinophil peroxidase Proteins 0.000 description 1
- 101000920686 Homo sapiens Erythropoietin Proteins 0.000 description 1
- 101001054334 Homo sapiens Interferon beta Proteins 0.000 description 1
- 102000002265 Human Growth Hormone Human genes 0.000 description 1
- 108010000521 Human Growth Hormone Proteins 0.000 description 1
- 239000000854 Human Growth Hormone Substances 0.000 description 1
- 241000713772 Human immunodeficiency virus 1 Species 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 108010054710 IMREG-1 Proteins 0.000 description 1
- 108060003951 Immunoglobulin Proteins 0.000 description 1
- 102100034343 Integrase Human genes 0.000 description 1
- 108010061833 Integrases Proteins 0.000 description 1
- 108010054698 Interferon Alfa-n3 Proteins 0.000 description 1
- 102000008070 Interferon-gamma Human genes 0.000 description 1
- 108010074328 Interferon-gamma Proteins 0.000 description 1
- 108010065805 Interleukin-12 Proteins 0.000 description 1
- 102000013462 Interleukin-12 Human genes 0.000 description 1
- 108010002350 Interleukin-2 Proteins 0.000 description 1
- 102000000588 Interleukin-2 Human genes 0.000 description 1
- 208000007766 Kaposi sarcoma Diseases 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 240000007472 Leucaena leucocephala Species 0.000 description 1
- 235000010643 Leucaena leucocephala Nutrition 0.000 description 1
- 208000017170 Lipid metabolism disease Diseases 0.000 description 1
- 235000019759 Maize starch Nutrition 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-L Malonate Chemical compound [O-]C(=O)CC([O-])=O OFOBLEOULBTSOW-UHFFFAOYSA-L 0.000 description 1
- YFGBQHOOROIVKG-FKBYEOEOSA-N Met-enkephalin Chemical compound C([C@@H](C(=O)N[C@@H](CCSC)C(O)=O)NC(=O)CNC(=O)CNC(=O)[C@@H](N)CC=1C=CC(O)=CC=1)C1=CC=CC=C1 YFGBQHOOROIVKG-FKBYEOEOSA-N 0.000 description 1
- 241001465754 Metazoa Species 0.000 description 1
- 108010042237 Methionine Enkephalin Proteins 0.000 description 1
- VVQNEPGJFQJSBK-UHFFFAOYSA-N Methyl methacrylate Chemical compound COC(=O)C(C)=C VVQNEPGJFQJSBK-UHFFFAOYSA-N 0.000 description 1
- 108010021062 Micafungin Proteins 0.000 description 1
- WHNWPMSKXPGLAX-UHFFFAOYSA-N N-Vinyl-2-pyrrolidone Chemical compound C=CN1CCCC1=O WHNWPMSKXPGLAX-UHFFFAOYSA-N 0.000 description 1
- 238000005481 NMR spectroscopy Methods 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 206010028813 Nausea Diseases 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 229930012538 Paclitaxel Natural products 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- JNTOCHDNEULJHD-UHFFFAOYSA-N Penciclovir Chemical compound N1C(N)=NC(=O)C2=C1N(CCC(CO)CO)C=N2 JNTOCHDNEULJHD-UHFFFAOYSA-N 0.000 description 1
- 108091005804 Peptidases Proteins 0.000 description 1
- 102000035195 Peptidases Human genes 0.000 description 1
- 108010071384 Peptide T Proteins 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-L Phosphate ion(2-) Chemical compound OP([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-L 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- 229920002642 Polysorbate 65 Polymers 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 description 1
- 239000004365 Protease Substances 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- 229920002125 Sokalan® Polymers 0.000 description 1
- 239000004187 Spiramycin Substances 0.000 description 1
- XNKLLVCARDGLGL-JGVFFNPUSA-N Stavudine Chemical compound O=C1NC(=O)C(C)=CN1[C@H]1C=C[C@@H](CO)O1 XNKLLVCARDGLGL-JGVFFNPUSA-N 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- LSNNMFCWUKXFEE-UHFFFAOYSA-N Sulfurous acid Chemical compound OS(O)=O LSNNMFCWUKXFEE-UHFFFAOYSA-N 0.000 description 1
- PZBFGYYEXUXCOF-UHFFFAOYSA-N TCEP Chemical compound OC(=O)CCP(CCC(O)=O)CCC(O)=O PZBFGYYEXUXCOF-UHFFFAOYSA-N 0.000 description 1
- 229920002253 Tannate Polymers 0.000 description 1
- HIINQLBHPIQYHN-JTQLQIEISA-N Tyr-Gly-Gly Chemical compound OC(=O)CNC(=O)CNC(=O)[C@@H](N)CC1=CC=C(O)C=C1 HIINQLBHPIQYHN-JTQLQIEISA-N 0.000 description 1
- HDOVUKNUBWVHOX-QMMMGPOBSA-N Valacyclovir Chemical compound N1C(N)=NC(=O)C2=C1N(COCCOC(=O)[C@@H](N)C(C)C)C=N2 HDOVUKNUBWVHOX-QMMMGPOBSA-N 0.000 description 1
- WPVFJKSGQUFQAP-GKAPJAKFSA-N Valcyte Chemical compound N1C(N)=NC(=O)C2=C1N(COC(CO)COC(=O)[C@@H](N)C(C)C)C=N2 WPVFJKSGQUFQAP-GKAPJAKFSA-N 0.000 description 1
- 108010067390 Viral Proteins Proteins 0.000 description 1
- 208000036142 Viral infection Diseases 0.000 description 1
- 206010047700 Vomiting Diseases 0.000 description 1
- WREGKURFCTUGRC-POYBYMJQSA-N Zalcitabine Chemical compound O=C1N=C(N)C=CN1[C@@H]1O[C@H](CO)CC1 WREGKURFCTUGRC-POYBYMJQSA-N 0.000 description 1
- ATSMZGDYBPOXMZ-HTQZYQBOSA-N [(2r,4r)-4-[2-amino-6-(cyclopropylamino)purin-9-yl]-1,3-dioxolan-2-yl]methanol Chemical compound C=12N=CN([C@@H]3O[C@H](CO)OC3)C2=NC(N)=NC=1NC1CC1 ATSMZGDYBPOXMZ-HTQZYQBOSA-N 0.000 description 1
- ZNOCHZVUAKTEPH-YDWIMUEDSA-N [(2s,3s)-1-[(4r)-4-(tert-butylcarbamoyl)-5,5-dimethyl-1,3-thiazolidin-3-yl]-3-[[2-(2,6-dimethylphenoxy)acetyl]amino]-1-oxo-4-phenylbutan-2-yl] 5-[[2-[[(2s,3s,5r)-3-azido-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy]-2-oxoethyl]amino]-5-oxopenta Chemical compound CC1=CC=CC(C)=C1OCC(=O)N[C@H]([C@H](OC(=O)CCCC(=O)NCC(=O)OC[C@@H]1[C@H](C[C@@H](O1)N1C(NC(=O)C(C)=C1)=O)N=[N+]=[N-])C(=O)N1[C@@H](C(C)(C)SC1)C(=O)NC(C)(C)C)CC1=CC=CC=C1 ZNOCHZVUAKTEPH-YDWIMUEDSA-N 0.000 description 1
- ZWBTYMGEBZUQTK-PVLSIAFMSA-N [(7S,9E,11S,12R,13S,14R,15R,16R,17S,18S,19E,21Z)-2,15,17,32-tetrahydroxy-11-methoxy-3,7,12,14,16,18,22-heptamethyl-1'-(2-methylpropyl)-6,23-dioxospiro[8,33-dioxa-24,27,29-triazapentacyclo[23.6.1.14,7.05,31.026,30]tritriaconta-1(32),2,4,9,19,21,24,26,30-nonaene-28,4'-piperidine]-13-yl] acetate Chemical compound CO[C@H]1\C=C\O[C@@]2(C)Oc3c(C2=O)c2c4NC5(CCN(CC(C)C)CC5)N=c4c(=NC(=O)\C(C)=C/C=C/[C@H](C)[C@H](O)[C@@H](C)[C@@H](O)[C@@H](C)[C@H](OC(C)=O)[C@@H]1C)c(O)c2c(O)c3C ZWBTYMGEBZUQTK-PVLSIAFMSA-N 0.000 description 1
- ZGDKVKUWTCGYOA-URGPHPNLSA-N [4-[4-[(z)-c-(4-bromophenyl)-n-ethoxycarbonimidoyl]piperidin-1-yl]-4-methylpiperidin-1-yl]-(2,4-dimethyl-1-oxidopyridin-1-ium-3-yl)methanone Chemical compound C=1C=C(Br)C=CC=1C(=N/OCC)\C(CC1)CCN1C(CC1)(C)CCN1C(=O)C1=C(C)C=C[N+]([O-])=C1C ZGDKVKUWTCGYOA-URGPHPNLSA-N 0.000 description 1
- NYRAVIYBIHCEGB-UHFFFAOYSA-N [K].[Ca] Chemical compound [K].[Ca] NYRAVIYBIHCEGB-UHFFFAOYSA-N 0.000 description 1
- 229960004748 abacavir Drugs 0.000 description 1
- MCGSCOLBFJQGHM-SCZZXKLOSA-N abacavir Chemical compound C=12N=CN([C@H]3C=C[C@@H](CO)C3)C2=NC(N)=NC=1NC1CC1 MCGSCOLBFJQGHM-SCZZXKLOSA-N 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000000205 acacia gum Substances 0.000 description 1
- XOYXESIZZFUVRD-UVSAJTFZSA-M acemannan Chemical compound CC(=O)O[C@@H]1[C@H](O)[C@@H](OC)O[C@H](CO)[C@H]1O[C@@H]1[C@@H](O)[C@@H](OC(C)=O)[C@H](O[C@@H]2[C@H]([C@@H](OC(C)=O)[C@H](O[C@@H]3[C@H]([C@@H](O)[C@H](O[C@@H]4[C@H]([C@@H](OC(C)=O)[C@H](O[C@@H]5[C@H]([C@@H](OC(C)=O)[C@H](O[C@@H]6[C@H]([C@@H](OC(C)=O)[C@H](O[C@@H]7[C@H]([C@@H](OC(C)=O)[C@H](OC)[C@@H](CO)O7)O)[C@@H](CO)O6)O)[C@H](O5)C([O-])=O)O)[C@@H](CO)O4)O)[C@@H](CO)O3)NC(C)=O)[C@@H](CO)O2)O)[C@@H](CO)O1 XOYXESIZZFUVRD-UVSAJTFZSA-M 0.000 description 1
- 229960005327 acemannan Drugs 0.000 description 1
- WETWJCDKMRHUPV-UHFFFAOYSA-N acetyl chloride Chemical compound CC(Cl)=O WETWJCDKMRHUPV-UHFFFAOYSA-N 0.000 description 1
- 239000012346 acetyl chloride Substances 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- YTIVTFGABIZHHX-UHFFFAOYSA-L acetylenedicarboxylate(2-) Chemical compound [O-]C(=O)C#CC([O-])=O YTIVTFGABIZHHX-UHFFFAOYSA-L 0.000 description 1
- 229960004150 aciclovir Drugs 0.000 description 1
- MKUXAQIIEYXACX-UHFFFAOYSA-N aciclovir Chemical compound N1C(N)=NC(=O)C2=C1N(COCCO)C=N2 MKUXAQIIEYXACX-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- 229960003205 adefovir dipivoxil Drugs 0.000 description 1
- 230000000274 adsorptive effect Effects 0.000 description 1
- 229940040563 agaric acid Drugs 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 150000008044 alkali metal hydroxides Chemical class 0.000 description 1
- 229910000272 alkali metal oxide Inorganic materials 0.000 description 1
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 229950005846 amdoxovir Drugs 0.000 description 1
- 239000003708 ampul Substances 0.000 description 1
- 229960003348 anidulafungin Drugs 0.000 description 1
- JHVAMHSQVVQIOT-MFAJLEFUSA-N anidulafungin Chemical compound C1=CC(OCCCCC)=CC=C1C1=CC=C(C=2C=CC(=CC=2)C(=O)N[C@@H]2C(N[C@H](C(=O)N3C[C@H](O)C[C@H]3C(=O)N[C@H](C(=O)N[C@H](C(=O)N3C[C@H](C)[C@H](O)[C@H]3C(=O)N[C@H](O)[C@H](O)C2)[C@@H](C)O)[C@H](O)[C@@H](O)C=2C=CC(O)=CC=2)[C@@H](C)O)=O)C=C1 JHVAMHSQVVQIOT-MFAJLEFUSA-N 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 230000001028 anti-proliverative effect Effects 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000003903 antiretrovirus agent Substances 0.000 description 1
- 239000003443 antiviral agent Substances 0.000 description 1
- 229940121357 antivirals Drugs 0.000 description 1
- RYMCFYKJDVMSIR-RNFRBKRXSA-N apricitabine Chemical compound O=C1N=C(N)C=CN1[C@@H]1S[C@H](CO)OC1 RYMCFYKJDVMSIR-RNFRBKRXSA-N 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 229910052786 argon Inorganic materials 0.000 description 1
- 150000004945 aromatic hydrocarbons Chemical class 0.000 description 1
- 239000012131 assay buffer Substances 0.000 description 1
- 229960003159 atovaquone Drugs 0.000 description 1
- KUCQYCKVKVOKAY-CTYIDZIISA-N atovaquone Chemical compound C1([C@H]2CC[C@@H](CC2)C2=C(C(C3=CC=CC=C3C2=O)=O)O)=CC=C(Cl)C=C1 KUCQYCKVKVOKAY-CTYIDZIISA-N 0.000 description 1
- DXZFFLRJVDZCMT-UHFFFAOYSA-N azane 2,2,2-trichloro-1,3,2lambda6-dioxatellurolane Chemical compound Cl[TeH]1(OCCO1)(Cl)Cl.N DXZFFLRJVDZCMT-UHFFFAOYSA-N 0.000 description 1
- HONIICLYMWZJFZ-UHFFFAOYSA-N azetidine Chemical compound C1CNC1 HONIICLYMWZJFZ-UHFFFAOYSA-N 0.000 description 1
- 229960004099 azithromycin Drugs 0.000 description 1
- MQTOSJVFKKJCRP-BICOPXKESA-N azithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)N(C)C[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 MQTOSJVFKKJCRP-BICOPXKESA-N 0.000 description 1
- 230000004888 barrier function Effects 0.000 description 1
- 230000008901 benefit Effects 0.000 description 1
- 239000000440 bentonite Substances 0.000 description 1
- 229910000278 bentonite Inorganic materials 0.000 description 1
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 1
- 150000008331 benzenesulfonamides Chemical class 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004601 benzofurazanyl group Chemical group N1=C2C(=NO1)C(=CC=C2)* 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- AGEZXYOZHKGVCM-UHFFFAOYSA-N benzyl bromide Chemical compound BrCC1=CC=CC=C1 AGEZXYOZHKGVCM-UHFFFAOYSA-N 0.000 description 1
- 229960000397 bevacizumab Drugs 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 229920000249 biocompatible polymer Polymers 0.000 description 1
- 230000036983 biotransformation Effects 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 229950009494 bropirimine Drugs 0.000 description 1
- HQABUPZFAYXKJW-UHFFFAOYSA-N butan-1-amine Chemical compound CCCCN HQABUPZFAYXKJW-UHFFFAOYSA-N 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 229930184135 calanolide Natural products 0.000 description 1
- 239000001506 calcium phosphate Chemical class 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- MIOPJNTWMNEORI-UHFFFAOYSA-N camphorsulfonic acid Chemical compound C1CC2(CS(O)(=O)=O)C(=O)CC1C2(C)C MIOPJNTWMNEORI-UHFFFAOYSA-N 0.000 description 1
- YQXCVAGCMNFUMQ-UHFFFAOYSA-N capravirine Chemical compound C=1C(Cl)=CC(Cl)=CC=1SC1=C(C(C)C)N=C(COC(N)=O)N1CC1=CC=NC=C1 YQXCVAGCMNFUMQ-UHFFFAOYSA-N 0.000 description 1
- 229950008230 capravirine Drugs 0.000 description 1
- 239000007963 capsule composition Substances 0.000 description 1
- 239000001569 carbon dioxide Substances 0.000 description 1
- 229960004424 carbon dioxide Drugs 0.000 description 1
- 238000001460 carbon-13 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- JYIKNQVWKBUSNH-WVDDFWQHSA-N caspofungin Chemical compound C1([C@H](O)[C@@H](O)[C@H]2C(=O)N[C@H](C(=O)N3CC[C@H](O)[C@H]3C(=O)N[C@H](NCCN)[C@H](O)C[C@@H](C(N[C@H](C(=O)N3C[C@H](O)C[C@H]3C(=O)N2)[C@@H](C)O)=O)NC(=O)CCCCCCCC[C@@H](C)C[C@@H](C)CC)[C@H](O)CCN)=CC=C(O)C=C1 JYIKNQVWKBUSNH-WVDDFWQHSA-N 0.000 description 1
- 229960003034 caspofungin Drugs 0.000 description 1
- 229960000541 cetyl alcohol Drugs 0.000 description 1
- ZXFCRFYULUUSDW-OWXODZSWSA-N chembl2104970 Chemical compound C([C@H]1C2)C3=CC=CC(O)=C3C(=O)C1=C(O)[C@@]1(O)[C@@H]2CC(O)=C(C(=O)N)C1=O ZXFCRFYULUUSDW-OWXODZSWSA-N 0.000 description 1
- 239000000460 chlorine Substances 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- KVSASDOGYIBWTA-UHFFFAOYSA-N chloro benzoate Chemical compound ClOC(=O)C1=CC=CC=C1 KVSASDOGYIBWTA-UHFFFAOYSA-N 0.000 description 1
- 229960000724 cidofovir Drugs 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 229960002626 clarithromycin Drugs 0.000 description 1
- AGOYDEPGAOXOCK-KCBOHYOISA-N clarithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@](C)([C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)OC)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 AGOYDEPGAOXOCK-KCBOHYOISA-N 0.000 description 1
- 229940090805 clavulanate Drugs 0.000 description 1
- HZZVJAQRINQKSD-PBFISZAISA-N clavulanic acid Chemical compound OC(=O)[C@H]1C(=C/CO)/O[C@@H]2CC(=O)N21 HZZVJAQRINQKSD-PBFISZAISA-N 0.000 description 1
- KDLRVYVGXIQJDK-AWPVFWJPSA-N clindamycin Chemical compound CN1C[C@H](CCC)C[C@H]1C(=O)N[C@H]([C@H](C)Cl)[C@@H]1[C@H](O)[C@H](O)[C@@H](O)[C@@H](SC)O1 KDLRVYVGXIQJDK-AWPVFWJPSA-N 0.000 description 1
- 229960002227 clindamycin Drugs 0.000 description 1
- 230000002281 colonystimulating effect Effects 0.000 description 1
- 230000002860 competitive effect Effects 0.000 description 1
- 229940126142 compound 16 Drugs 0.000 description 1
- 238000007796 conventional method Methods 0.000 description 1
- ARUVKPQLZAKDPS-UHFFFAOYSA-L copper(II) sulfate Chemical compound [Cu+2].[O-][S+2]([O-])([O-])[O-] ARUVKPQLZAKDPS-UHFFFAOYSA-L 0.000 description 1
- 229910000366 copper(II) sulfate Inorganic materials 0.000 description 1
- 235000001671 coumarin Nutrition 0.000 description 1
- 229960000956 coumarin Drugs 0.000 description 1
- 230000008878 coupling Effects 0.000 description 1
- 238000010168 coupling process Methods 0.000 description 1
- 238000005859 coupling reaction Methods 0.000 description 1
- 235000019316 curdlan Nutrition 0.000 description 1
- 229940078035 curdlan Drugs 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000002433 cyclopentenyl group Chemical group C1(=CCCC1)* 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 229940087451 cytovene Drugs 0.000 description 1
- 229950006497 dapivirine Drugs 0.000 description 1
- GHVNFZFCNZKVNT-UHFFFAOYSA-N decanoic acid Chemical compound CCCCCCCCCC(O)=O GHVNFZFCNZKVNT-UHFFFAOYSA-N 0.000 description 1
- 229960005319 delavirdine Drugs 0.000 description 1
- 239000003405 delayed action preparation Substances 0.000 description 1
- 230000002939 deleterious effect Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000010511 deprotection reaction Methods 0.000 description 1
- 239000007933 dermal patch Substances 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 229960002086 dextran Drugs 0.000 description 1
- 229960000633 dextran sulfate Drugs 0.000 description 1
- 125000002576 diazepinyl group Chemical group N1N=C(C=CC=C1)* 0.000 description 1
- ACYGYJFTZSAZKR-UHFFFAOYSA-J dicalcium;2-[2-[bis(carboxylatomethyl)amino]ethyl-(carboxylatomethyl)amino]acetate Chemical compound [Ca+2].[Ca+2].[O-]C(=O)CN(CC([O-])=O)CCN(CC([O-])=O)CC([O-])=O ACYGYJFTZSAZKR-UHFFFAOYSA-J 0.000 description 1
- PXBRQCKWGAHEHS-UHFFFAOYSA-N dichlorodifluoromethane Chemical compound FC(F)(Cl)Cl PXBRQCKWGAHEHS-UHFFFAOYSA-N 0.000 description 1
- 235000019404 dichlorodifluoromethane Nutrition 0.000 description 1
- 229940042935 dichlorodifluoromethane Drugs 0.000 description 1
- 229940087091 dichlorotetrafluoroethane Drugs 0.000 description 1
- 229960002656 didanosine Drugs 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- 229940116901 diethyldithiocarbamate Drugs 0.000 description 1
- XXJWXESWEXIICW-UHFFFAOYSA-N diethylene glycol monoethyl ether Chemical compound CCOCCOCCO XXJWXESWEXIICW-UHFFFAOYSA-N 0.000 description 1
- 150000004683 dihydrates Chemical class 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-M dihydrogenphosphate Chemical compound OP(O)([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-M 0.000 description 1
- 125000005057 dihydrothienyl group Chemical group S1C(CC=C1)* 0.000 description 1
- IJKVHSBPTUYDLN-UHFFFAOYSA-N dihydroxy(oxo)silane Chemical compound O[Si](O)=O IJKVHSBPTUYDLN-UHFFFAOYSA-N 0.000 description 1
- BGRWYRAHAFMIBJ-UHFFFAOYSA-N diisopropylcarbodiimide Natural products CC(C)NC(=O)NC(C)C BGRWYRAHAFMIBJ-UHFFFAOYSA-N 0.000 description 1
- SPCNPOWOBZQWJK-UHFFFAOYSA-N dimethoxy-(2-propan-2-ylsulfanylethylsulfanyl)-sulfanylidene-$l^{5}-phosphane Chemical compound COP(=S)(OC)SCCSC(C)C SPCNPOWOBZQWJK-UHFFFAOYSA-N 0.000 description 1
- AYXBAIULRDEVAS-UHFFFAOYSA-N dimethyl-[[4-[[3-(4-methylphenyl)-8,9-dihydro-7h-benzo[7]annulene-6-carbonyl]amino]phenyl]methyl]-(oxan-4-yl)azanium;iodide Chemical compound [I-].C1=CC(C)=CC=C1C1=CC=C(CCCC(=C2)C(=O)NC=3C=CC(C[N+](C)(C)C4CCOCC4)=CC=3)C2=C1 AYXBAIULRDEVAS-UHFFFAOYSA-N 0.000 description 1
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 1
- 125000000532 dioxanyl group Chemical group 0.000 description 1
- 239000001177 diphosphate Substances 0.000 description 1
- 239000002270 dispersing agent Substances 0.000 description 1
- 238000004090 dissolution Methods 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 125000005883 dithianyl group Chemical group 0.000 description 1
- 125000005411 dithiolanyl group Chemical group S1SC(CC1)* 0.000 description 1
- POULHZVOKOAJMA-UHFFFAOYSA-M dodecanoate Chemical compound CCCCCCCCCCCC([O-])=O POULHZVOKOAJMA-UHFFFAOYSA-M 0.000 description 1
- 230000036267 drug metabolism Effects 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 229940009662 edetate Drugs 0.000 description 1
- XPOQHMRABVBWPR-ZDUSSCGKSA-N efavirenz Chemical compound C([C@]1(C2=CC(Cl)=CC=C2NC(=O)O1)C(F)(F)F)#CC1CC1 XPOQHMRABVBWPR-ZDUSSCGKSA-N 0.000 description 1
- 229960003804 efavirenz Drugs 0.000 description 1
- 238000000921 elemental analysis Methods 0.000 description 1
- 229950005627 embonate Drugs 0.000 description 1
- 229960000366 emtricitabine Drugs 0.000 description 1
- 229960002062 enfuvirtide Drugs 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 229960000980 entecavir Drugs 0.000 description 1
- YXPVEXCTPGULBZ-WQYNNSOESA-N entecavir hydrate Chemical compound O.C1=NC=2C(=O)NC(N)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)C1=C YXPVEXCTPGULBZ-WQYNNSOESA-N 0.000 description 1
- 229950000206 estolate Drugs 0.000 description 1
- KMCVEQSRKFYOIH-BSRZVIQJSA-N ethyl (2s)-2-[2-[[4-[(2s,3r)-2-[[(3as,4r,6ar)-2,3,3a,4,5,6a-hexahydrofuro[2,3-b]furan-4-yl]oxycarbonylamino]-3-hydroxy-4-[(4-methoxyphenyl)sulfonyl-(2-methylpropyl)amino]butyl]phenyl]methylamino]ethyl-phenoxyphosphoryl]oxypropanoate Chemical compound C([C@@H](O)[C@@H](NC(=O)O[C@@H]1[C@@H]2CCO[C@@H]2OC1)CC1=CC=C(C=C1)CNCCP(=O)(O[C@@H](C)C(=O)OCC)OC=1C=CC=CC=1)N(CC(C)C)S(=O)(=O)C1=CC=C(OC)C=C1 KMCVEQSRKFYOIH-BSRZVIQJSA-N 0.000 description 1
- GLLOSPJJYDPIRO-UHFFFAOYSA-N ethyl 4-[2-(pyridin-3-ylsulfamoyl)benzoyl]piperazine-1-carboxylate Chemical compound C1CN(C(=O)OCC)CCN1C(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 GLLOSPJJYDPIRO-UHFFFAOYSA-N 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- 229960004396 famciclovir Drugs 0.000 description 1
- GGXKWVWZWMLJEH-UHFFFAOYSA-N famcyclovir Chemical compound N1=C(N)N=C2N(CCC(COC(=O)C)COC(C)=O)C=NC2=C1 GGXKWVWZWMLJEH-UHFFFAOYSA-N 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- LLYJISDUHFXOHK-GOCONZMPSA-N ferroptocide Chemical compound C[C@@H]1CC[C@@]23C[C@@H](C(=O)[C@]2([C@@]1([C@@H](C[C@H]([C@@H]3C)C4=CCN5C(=O)N(C(=O)N5C4)C6=CC=CC=C6)OC(=O)CCl)C)O)O LLYJISDUHFXOHK-GOCONZMPSA-N 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- RFHAOTPXVQNOHP-UHFFFAOYSA-N fluconazole Chemical compound C1=NC=NN1CC(C=1C(=CC(F)=CC=1)F)(O)CN1C=NC=N1 RFHAOTPXVQNOHP-UHFFFAOYSA-N 0.000 description 1
- 229960004884 fluconazole Drugs 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 229910052731 fluorine Inorganic materials 0.000 description 1
- 229960001447 fomivirsen Drugs 0.000 description 1
- XCWFZHPEARLXJI-UHFFFAOYSA-N fomivirsen Chemical compound C1C(N2C3=C(C(NC(N)=N3)=O)N=C2)OC(CO)C1OP(O)(=S)OCC1OC(N(C)C(=O)\N=C(\N)C=C)CC1OP(O)(=S)OCC1OC(N2C3=C(C(NC(N)=N3)=O)N=C2)CC1OP(O)(=S)OCC1OC(N2C(NC(=O)C(C)=C2)=O)CC1OP(O)(=S)OCC1OC(N2C(NC(=O)C(C)=C2)=O)CC1OP(O)(=S)OCC1OC(N2C(NC(=O)C(C)=C2)=O)CC1OP(O)(=S)OCC1OC(N2C3=C(C(NC(N)=N3)=O)N=C2)CC1OP(O)(=S)OCC1OC(N2C(N=C(N)C=C2)=O)CC1OP(O)(=S)OCC(C(C1)OP(S)(=O)OCC2C(CC(O2)N2C(N=C(N)C=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C(NC(=O)C(C)=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C(NC(=O)C(C)=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C(N=C(N)C=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C(NC(=O)C(C)=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C(NC(=O)C(C)=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C(N=C(N)C=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C(NC(=O)C(C)=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C(NC(=O)C(C)=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C3=C(C(NC(N)=N3)=O)N=C2)OP(O)(=S)OCC2C(CC(O2)N2C(N=C(N)C=C2)=O)OP(O)(=S)OCC2C(CC(O2)N2C3=C(C(NC(N)=N3)=O)N=C2)O)OC1N1C=C(C)C(=O)NC1=O XCWFZHPEARLXJI-UHFFFAOYSA-N 0.000 description 1
- 230000037406 food intake Effects 0.000 description 1
- 229940044170 formate Drugs 0.000 description 1
- 229960000848 foscarnet sodium Drugs 0.000 description 1
- 229940050411 fumarate Drugs 0.000 description 1
- 125000003838 furazanyl group Chemical group 0.000 description 1
- 125000004612 furopyridinyl group Chemical group O1C(=CC2=C1C=CC=N2)* 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 230000004927 fusion Effects 0.000 description 1
- 229940044627 gamma-interferon Drugs 0.000 description 1
- 239000007789 gas Substances 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- 238000007429 general method Methods 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 102000018146 globin Human genes 0.000 description 1
- 108060003196 globin Proteins 0.000 description 1
- 229960001731 gluceptate Drugs 0.000 description 1
- KWMLJOLKUYYJFJ-VFUOTHLCSA-N glucoheptonic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O)C(O)=O KWMLJOLKUYYJFJ-VFUOTHLCSA-N 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 229940049906 glutamate Drugs 0.000 description 1
- YQEMORVAKMFKLG-UHFFFAOYSA-N glycerine monostearate Natural products CCCCCCCCCCCCCCCCCC(=O)OC(CO)CO YQEMORVAKMFKLG-UHFFFAOYSA-N 0.000 description 1
- 229960005150 glycerol Drugs 0.000 description 1
- RZRNAYUHWVFMIP-HXUWFJFHSA-N glycerol monolinoleate Natural products CCCCCCCCC=CCCCCCCCC(=O)OC[C@H](O)CO RZRNAYUHWVFMIP-HXUWFJFHSA-N 0.000 description 1
- SVUQHVRAGMNPLW-UHFFFAOYSA-N glycerol monostearate Natural products CCCCCCCCCCCCCCCCC(=O)OCC(O)CO SVUQHVRAGMNPLW-UHFFFAOYSA-N 0.000 description 1
- 229940074045 glyceryl distearate Drugs 0.000 description 1
- 229940075507 glyceryl monostearate Drugs 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 210000003714 granulocyte Anatomy 0.000 description 1
- 238000000227 grinding Methods 0.000 description 1
- 229920000591 gum Polymers 0.000 description 1
- LVASCWIMLIKXLA-LSDHHAIUSA-N halofuginone Chemical compound O[C@@H]1CCCN[C@H]1CC(=O)CN1C(=O)C2=CC(Cl)=C(Br)C=C2N=C1 LVASCWIMLIKXLA-LSDHHAIUSA-N 0.000 description 1
- 229950010152 halofuginone Drugs 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- MNWFXJYAOYHMED-UHFFFAOYSA-N heptanoic acid Chemical compound CCCCCCC(O)=O MNWFXJYAOYHMED-UHFFFAOYSA-N 0.000 description 1
- KKLGDUSGQMHBPB-UHFFFAOYSA-N hex-2-ynedioic acid Chemical compound OC(=O)CCC#CC(O)=O KKLGDUSGQMHBPB-UHFFFAOYSA-N 0.000 description 1
- IPCSVZSSVZVIGE-UHFFFAOYSA-M hexadecanoate Chemical compound CCCCCCCCCCCCCCCC([O-])=O IPCSVZSSVZVIGE-UHFFFAOYSA-M 0.000 description 1
- FUZZWVXGSFPDMH-UHFFFAOYSA-M hexanoate Chemical compound CCCCCC([O-])=O FUZZWVXGSFPDMH-UHFFFAOYSA-M 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 239000002835 hiv fusion inhibitor Substances 0.000 description 1
- 102000044890 human EPO Human genes 0.000 description 1
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical compound C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-M hydrogensulfate Chemical compound OS([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-M 0.000 description 1
- 229920001600 hydrophobic polymer Polymers 0.000 description 1
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 description 1
- BTXNYTINYBABQR-UHFFFAOYSA-N hypericin Chemical compound C12=C(O)C=C(O)C(C(C=3C(O)=CC(C)=C4C=33)=O)=C2C3=C2C3=C4C(C)=CC(O)=C3C(=O)C3=C(O)C=C(O)C1=C32 BTXNYTINYBABQR-UHFFFAOYSA-N 0.000 description 1
- 229940005608 hypericin Drugs 0.000 description 1
- PHOKTTKFQUYZPI-UHFFFAOYSA-N hypericin Natural products Cc1cc(O)c2c3C(=O)C(=Cc4c(O)c5c(O)cc(O)c6c7C(=O)C(=Cc8c(C)c1c2c(c78)c(c34)c56)O)O PHOKTTKFQUYZPI-UHFFFAOYSA-N 0.000 description 1
- 229950010245 ibalizumab Drugs 0.000 description 1
- 125000002962 imidazol-1-yl group Chemical group [*]N1C([H])=NC([H])=C1[H] 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 102000018358 immunoglobulin Human genes 0.000 description 1
- 238000002513 implantation Methods 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 230000028709 inflammatory response Effects 0.000 description 1
- 238000002329 infrared spectrum Methods 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 229940102223 injectable solution Drugs 0.000 description 1
- 229940102213 injectable suspension Drugs 0.000 description 1
- 230000002452 interceptive effect Effects 0.000 description 1
- 229940109242 interferon alfa-n3 Drugs 0.000 description 1
- 229940117681 interleukin-12 Drugs 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- KQNPFQTWMSNSAP-UHFFFAOYSA-N isobutyric acid Chemical compound CC(C)C(O)=O KQNPFQTWMSNSAP-UHFFFAOYSA-N 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- FZWBNHMXJMCXLU-BLAUPYHCSA-N isomaltotriose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1OC[C@@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@@H](OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O)O1 FZWBNHMXJMCXLU-BLAUPYHCSA-N 0.000 description 1
- BMFVGAAISNGQNM-UHFFFAOYSA-N isopentylamine Chemical compound CC(C)CCN BMFVGAAISNGQNM-UHFFFAOYSA-N 0.000 description 1
- 229940011051 isopropyl acetate Drugs 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- GWYFCOCPABKNJV-UHFFFAOYSA-N isovaleric acid Chemical compound CC(C)CC(O)=O GWYFCOCPABKNJV-UHFFFAOYSA-N 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 229960004130 itraconazole Drugs 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 229960004125 ketoconazole Drugs 0.000 description 1
- 239000004922 lacquer Substances 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 229940099584 lactobionate Drugs 0.000 description 1
- JYTUSYBCFIZPBE-AMTLMPIISA-N lactobionic acid Chemical compound OC(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O JYTUSYBCFIZPBE-AMTLMPIISA-N 0.000 description 1
- JTEGQNOMFQHVDC-NKWVEPMBSA-N lamivudine Chemical compound O=C1N=C(N)C=CN1[C@H]1O[C@@H](CO)SC1 JTEGQNOMFQHVDC-NKWVEPMBSA-N 0.000 description 1
- 229960001627 lamivudine Drugs 0.000 description 1
- 229940070765 laurate Drugs 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 229940113354 lexiva Drugs 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 229940057995 liquid paraffin Drugs 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- 229950005339 lobucavir Drugs 0.000 description 1
- 235000019341 magnesium sulphate Nutrition 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 238000007726 management method Methods 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical compound [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 229960004296 megestrol acetate Drugs 0.000 description 1
- RQZAXGRLVPAYTJ-GQFGMJRRSA-N megestrol acetate Chemical compound C1=C(C)C2=CC(=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@@](C(C)=O)(OC(=O)C)[C@@]1(C)CC2 RQZAXGRLVPAYTJ-GQFGMJRRSA-N 0.000 description 1
- 238000002844 melting Methods 0.000 description 1
- 230000008018 melting Effects 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 125000005341 metaphosphate group Chemical group 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- IZYBEMGNIUSSAX-UHFFFAOYSA-N methyl benzenecarboperoxoate Chemical compound COOC(=O)C1=CC=CC=C1 IZYBEMGNIUSSAX-UHFFFAOYSA-N 0.000 description 1
- 229940095102 methyl benzoate Drugs 0.000 description 1
- 229920000609 methyl cellulose Polymers 0.000 description 1
- QAHLFXYLXBBCPS-IZEXYCQBSA-N methyl n-[(2s)-1-[[(5s)-5-[(4-aminophenyl)sulfonyl-(2-methylpropyl)amino]-6-hydroxyhexyl]amino]-1-oxo-3,3-diphenylpropan-2-yl]carbamate Chemical compound C=1C=CC=CC=1C([C@H](NC(=O)OC)C(=O)NCCCC[C@@H](CO)N(CC(C)C)S(=O)(=O)C=1C=CC(N)=CC=1)C1=CC=CC=C1 QAHLFXYLXBBCPS-IZEXYCQBSA-N 0.000 description 1
- JZMJDSHXVKJFKW-UHFFFAOYSA-M methyl sulfate(1-) Chemical compound COS([O-])(=O)=O JZMJDSHXVKJFKW-UHFFFAOYSA-M 0.000 description 1
- 235000010981 methylcellulose Nutrition 0.000 description 1
- 239000001923 methylcellulose Substances 0.000 description 1
- 229960002159 micafungin Drugs 0.000 description 1
- PIEUQSKUWLMALL-YABMTYFHSA-N micafungin Chemical compound C1=CC(OCCCCC)=CC=C1C1=CC(C=2C=CC(=CC=2)C(=O)N[C@@H]2C(N[C@H](C(=O)N3C[C@H](O)C[C@H]3C(=O)N[C@H](C(=O)N[C@H](C(=O)N3C[C@H](C)[C@H](O)[C@H]3C(=O)N[C@H](O)[C@H](O)C2)[C@H](O)CC(N)=O)[C@H](O)[C@@H](O)C=2C=C(OS(O)(=O)=O)C(O)=CC=2)[C@@H](C)O)=O)=NO1 PIEUQSKUWLMALL-YABMTYFHSA-N 0.000 description 1
- 229960005225 mifamurtide Drugs 0.000 description 1
- JMUHBNWAORSSBD-WKYWBUFDSA-N mifamurtide Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@@H](OC(=O)CCCCCCCCCCCCCCC)COP(O)(=O)OCCNC(=O)[C@H](C)NC(=O)CC[C@H](C(N)=O)NC(=O)[C@H](C)NC(=O)[C@@H](C)O[C@H]1[C@H](O)[C@@H](CO)OC(O)[C@@H]1NC(C)=O JMUHBNWAORSSBD-WKYWBUFDSA-N 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 230000003020 moisturizing effect Effects 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- KMCBHFNNVRCAAH-UHFFFAOYSA-N n,n-dimethyldodecan-1-amine oxide;2-[dodecyl(dimethyl)azaniumyl]acetate Chemical compound CCCCCCCCCCCC[N+](C)(C)[O-].CCCCCCCCCCCC[N+](C)(C)CC([O-])=O KMCBHFNNVRCAAH-UHFFFAOYSA-N 0.000 description 1
- AMWSIDBIPRPFCD-UHFFFAOYSA-N n-(1-cyclopropylethyl)-2-[(4-ethoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound CCOC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)NC(C)C1CC1 AMWSIDBIPRPFCD-UHFFFAOYSA-N 0.000 description 1
- NLIIEAPQRWWUBY-UHFFFAOYSA-N n-(1-cyclopropylethyl)-2-[(4-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)NC(C)C1CC1 NLIIEAPQRWWUBY-UHFFFAOYSA-N 0.000 description 1
- YWXRVHFSQALKIX-UHFFFAOYSA-N n-(1-hydroxy-2-methylpropan-2-yl)-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound OCC(C)(C)NC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 YWXRVHFSQALKIX-UHFFFAOYSA-N 0.000 description 1
- QZGGQPHSCMHMRN-UHFFFAOYSA-N n-(2,2-dimethylpropyl)-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound CC(C)(C)CNC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 QZGGQPHSCMHMRN-UHFFFAOYSA-N 0.000 description 1
- ITFXJSDGFWKYSN-UHFFFAOYSA-N n-(2,3-dihydro-1-benzofuran-5-ylmethyl)-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound C=1C=C2OCCC2=CC=1CNC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 ITFXJSDGFWKYSN-UHFFFAOYSA-N 0.000 description 1
- VZVQFHWKYLWVDI-UHFFFAOYSA-N n-(2,3-dihydro-1h-inden-2-yl)-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound C1C2=CC=CC=C2CC1NC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 VZVQFHWKYLWVDI-UHFFFAOYSA-N 0.000 description 1
- VDGVEXDMLJRBIZ-UHFFFAOYSA-N n-(2,4-dimethylpentan-3-yl)-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NC(C(C)C)C(C)C)=C1 VDGVEXDMLJRBIZ-UHFFFAOYSA-N 0.000 description 1
- FEUWXJZZJONHOS-UHFFFAOYSA-N n-(2-adamantyl)-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NC2C3CC4CC(C3)CC2C4)=C1 FEUWXJZZJONHOS-UHFFFAOYSA-N 0.000 description 1
- ZHFAIGZYRXXORW-UHFFFAOYSA-N n-(2-cyclopentylethyl)-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCCC2CCCC2)=C1 ZHFAIGZYRXXORW-UHFFFAOYSA-N 0.000 description 1
- ZTSCYQWFXQFOEE-UHFFFAOYSA-N n-(2-cyclopropylpropan-2-yl)-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound C1CC1C(C)(C)NC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 ZTSCYQWFXQFOEE-UHFFFAOYSA-N 0.000 description 1
- OGBVQLHLLYHTRZ-UHFFFAOYSA-N n-(2-ethoxyethyl)-2-[(4-ethoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound CCOCCNC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CN=CC=C1OCC OGBVQLHLLYHTRZ-UHFFFAOYSA-N 0.000 description 1
- DJOFZBOMTDDUAU-UHFFFAOYSA-N n-(2-ethoxyethyl)-2-[(4-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound CCOCCNC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CN=CC=C1OC DJOFZBOMTDDUAU-UHFFFAOYSA-N 0.000 description 1
- LAVQEDXCJSKOAA-UHFFFAOYSA-N n-(2-methoxyethyl)-2-[(4-methoxypyridin-3-yl)sulfamoyl]-n-methylbenzamide Chemical compound COCCN(C)C(=O)C1=CC=CC=C1S(=O)(=O)NC1=CN=CC=C1OC LAVQEDXCJSKOAA-UHFFFAOYSA-N 0.000 description 1
- WWHCETIWDUIDFW-UHFFFAOYSA-N n-(2-methoxyethyl)-2-[(4-methoxypyridin-3-yl)sulfamoyl]-n-propan-2-ylbenzamide Chemical compound COCCN(C(C)C)C(=O)C1=CC=CC=C1S(=O)(=O)NC1=CN=CC=C1OC WWHCETIWDUIDFW-UHFFFAOYSA-N 0.000 description 1
- IATJZHRITCCTNH-UHFFFAOYSA-N n-(2-methoxyethyl)-2-[(5-methoxypyridin-3-yl)sulfamoyl]-n-propan-2-ylbenzamide Chemical compound COCCN(C(C)C)C(=O)C1=CC=CC=C1S(=O)(=O)NC1=CN=CC(OC)=C1 IATJZHRITCCTNH-UHFFFAOYSA-N 0.000 description 1
- CVKQAAIPXODBFM-UHFFFAOYSA-N n-(2-methyl-1-morpholin-4-ylpropan-2-yl)-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound C=1C=CC=C(S(=O)(=O)NC=2C=NC=CC=2)C=1C(=O)NC(C)(C)CN1CCOCC1 CVKQAAIPXODBFM-UHFFFAOYSA-N 0.000 description 1
- GUJYBDOPAYGNER-UHFFFAOYSA-N n-(2-methyl-1-phenylmethoxypropan-2-yl)-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound C=1C=CC=C(S(=O)(=O)NC=2C=NC=CC=2)C=1C(=O)NC(C)(C)COCC1=CC=CC=C1 GUJYBDOPAYGNER-UHFFFAOYSA-N 0.000 description 1
- AUEDMNCSRHBIFW-UHFFFAOYSA-N n-(2-methyl-1-piperidin-1-ylpropan-2-yl)-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound C=1C=CC=C(S(=O)(=O)NC=2C=NC=CC=2)C=1C(=O)NC(C)(C)CN1CCCCC1 AUEDMNCSRHBIFW-UHFFFAOYSA-N 0.000 description 1
- GCDAFEUMGHHHPM-UHFFFAOYSA-N n-(2-methyl-1-pyrrolidin-1-ylpropan-2-yl)-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound C=1C=CC=C(S(=O)(=O)NC=2C=NC=CC=2)C=1C(=O)NC(C)(C)CN1CCCC1 GCDAFEUMGHHHPM-UHFFFAOYSA-N 0.000 description 1
- YVSPZFFGYXSLIC-UHFFFAOYSA-N n-(2-methyl-2-morpholin-4-ylpropyl)-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound C1COCCN1C(C)(C)CNC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 YVSPZFFGYXSLIC-UHFFFAOYSA-N 0.000 description 1
- XXBFLTPZBBNITI-UHFFFAOYSA-N n-(2-methylbutan-2-yl)-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound CCC(C)(C)NC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 XXBFLTPZBBNITI-UHFFFAOYSA-N 0.000 description 1
- WBPRTFNBODFHDD-UHFFFAOYSA-N n-(2-phenoxyethyl)-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound C=1C=CC=C(S(=O)(=O)NC=2C=NC=CC=2)C=1C(=O)NCCOC1=CC=CC=C1 WBPRTFNBODFHDD-UHFFFAOYSA-N 0.000 description 1
- DFTSCRDWLSGSDD-UHFFFAOYSA-N n-(3-butoxypropyl)-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound CCCCOCCCNC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 DFTSCRDWLSGSDD-UHFFFAOYSA-N 0.000 description 1
- OYNSGAOTPPALPA-UHFFFAOYSA-N n-(3-methylbutyl)-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound CC(C)CCNC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 OYNSGAOTPPALPA-UHFFFAOYSA-N 0.000 description 1
- OACKUIGDTKXUKU-UHFFFAOYSA-N n-(3-pyridin-2-ylpropyl)-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound C=1C=CC=C(S(=O)(=O)NC=2C=NC=CC=2)C=1C(=O)NCCCC1=CC=CC=N1 OACKUIGDTKXUKU-UHFFFAOYSA-N 0.000 description 1
- HJSBJTRVRVLCQO-UHFFFAOYSA-N n-(5-cyanopyridin-3-yl)-2-(piperidine-1-carbonyl)benzenesulfonamide Chemical compound C=1C=CC=C(S(=O)(=O)NC=2C=C(C=NC=2)C#N)C=1C(=O)N1CCCCC1 HJSBJTRVRVLCQO-UHFFFAOYSA-N 0.000 description 1
- XXUVMKBKKNXDGR-UHFFFAOYSA-N n-(5-methylpyridin-3-yl)-2-(piperidine-1-carbonyl)benzenesulfonamide Chemical compound CC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)N2CCCCC2)=C1 XXUVMKBKKNXDGR-UHFFFAOYSA-N 0.000 description 1
- FUCBWSKXZHNEAK-UHFFFAOYSA-N n-(cyclohexylmethyl)-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCC2CCCCC2)=C1 FUCBWSKXZHNEAK-UHFFFAOYSA-N 0.000 description 1
- FZSCTNCHHTYMKG-UHFFFAOYSA-N n-(cyclopentylmethyl)-n-methyl-2-[(5-methylpyridin-3-yl)sulfamoyl]benzamide Chemical compound C=1C=CC=C(S(=O)(=O)NC=2C=C(C)C=NC=2)C=1C(=O)N(C)CC1CCCC1 FZSCTNCHHTYMKG-UHFFFAOYSA-N 0.000 description 1
- OSAPNIHGXDXMGK-UHFFFAOYSA-N n-(cyclopropylmethyl)-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound C=1C=CC=C(S(=O)(=O)NC=2C=NC=CC=2)C=1C(=O)NCC1CC1 OSAPNIHGXDXMGK-UHFFFAOYSA-N 0.000 description 1
- AXZZPZQPKXHVCC-UHFFFAOYSA-N n-(cyclopropylmethyl)-n-propyl-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound C=1C=CC=C(S(=O)(=O)NC=2C=NC=CC=2)C=1C(=O)N(CCC)CC1CC1 AXZZPZQPKXHVCC-UHFFFAOYSA-N 0.000 description 1
- WZELQYXDGTUXRE-UHFFFAOYSA-N n-[(2,3-difluorophenyl)methyl]-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound FC1=CC=CC(CNC(=O)C=2C(=CC=CC=2)S(=O)(=O)NC=2C=NC=CC=2)=C1F WZELQYXDGTUXRE-UHFFFAOYSA-N 0.000 description 1
- SNVJDQWYGGTMCO-UHFFFAOYSA-N n-[(2,4-dimethoxyphenyl)methyl]-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound COC1=CC(OC)=CC=C1CNC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 SNVJDQWYGGTMCO-UHFFFAOYSA-N 0.000 description 1
- FANBITMJHONWIQ-UHFFFAOYSA-N n-[(2,6-dimethoxyphenyl)methyl]-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound COC1=CC=CC(OC)=C1CNC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 FANBITMJHONWIQ-UHFFFAOYSA-N 0.000 description 1
- YWUDQZCQZMXFSP-UHFFFAOYSA-N n-[(2-chloro-4-fluorophenyl)methyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCC=2C(=CC(F)=CC=2)Cl)=C1 YWUDQZCQZMXFSP-UHFFFAOYSA-N 0.000 description 1
- FXZBTOZIQRWFJA-UHFFFAOYSA-N n-[(2-ethoxyphenyl)methyl]-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound CCOC1=CC=CC=C1CNC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 FXZBTOZIQRWFJA-UHFFFAOYSA-N 0.000 description 1
- CTFBPFOCFXBVDG-UHFFFAOYSA-N n-[(2-fluorophenyl)methyl]-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound FC1=CC=CC=C1CNC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 CTFBPFOCFXBVDG-UHFFFAOYSA-N 0.000 description 1
- YBLUJFSABNPTHO-UHFFFAOYSA-N n-[(2-hydroxy-2-adamantyl)methyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCC2(O)C3CC4CC(C3)CC2C4)=C1 YBLUJFSABNPTHO-UHFFFAOYSA-N 0.000 description 1
- KPTIZVKMCRPXKC-UHFFFAOYSA-N n-[(2-methylphenyl)methyl]-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound CC1=CC=CC=C1CNC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 KPTIZVKMCRPXKC-UHFFFAOYSA-N 0.000 description 1
- MTWNTUVFMLOHRI-UHFFFAOYSA-N n-[(3,4-difluorophenyl)methyl]-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound C1=C(F)C(F)=CC=C1CNC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 MTWNTUVFMLOHRI-UHFFFAOYSA-N 0.000 description 1
- RKMDAEWYMUTXPQ-UHFFFAOYSA-N n-[(3,4-difluorophenyl)methyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]-n-methylbenzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)N(C)CC=2C=C(F)C(F)=CC=2)=C1 RKMDAEWYMUTXPQ-UHFFFAOYSA-N 0.000 description 1
- IRLJRPIJCHDLTA-UHFFFAOYSA-N n-[(3,5-dimethoxyphenyl)methyl]-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound COC1=CC(OC)=CC(CNC(=O)C=2C(=CC=CC=2)S(=O)(=O)NC=2C=NC=CC=2)=C1 IRLJRPIJCHDLTA-UHFFFAOYSA-N 0.000 description 1
- SUUFZSBHKMCVAX-UHFFFAOYSA-N n-[(3-chloro-4-fluorophenyl)methyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCC=2C=C(Cl)C(F)=CC=2)=C1 SUUFZSBHKMCVAX-UHFFFAOYSA-N 0.000 description 1
- GGHKDGMFOORARP-UHFFFAOYSA-N n-[(3-fluorophenyl)methyl]-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound FC1=CC=CC(CNC(=O)C=2C(=CC=CC=2)S(=O)(=O)NC=2C=NC=CC=2)=C1 GGHKDGMFOORARP-UHFFFAOYSA-N 0.000 description 1
- JCYKDOHFVTXJPE-UHFFFAOYSA-N n-[(3-fluorophenyl)methyl]-2-[(4-methoxypyridin-3-yl)sulfamoyl]-n-methylbenzamide Chemical compound COC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)N(C)CC1=CC=CC(F)=C1 JCYKDOHFVTXJPE-UHFFFAOYSA-N 0.000 description 1
- XMRMLVIOSLLRBM-UHFFFAOYSA-N n-[(3-methoxyphenyl)methyl]-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound COC1=CC=CC(CNC(=O)C=2C(=CC=CC=2)S(=O)(=O)NC=2C=NC=CC=2)=C1 XMRMLVIOSLLRBM-UHFFFAOYSA-N 0.000 description 1
- KKHAUHJOQPPGSL-UHFFFAOYSA-N n-[(4-fluorophenyl)methyl]-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound C1=CC(F)=CC=C1CNC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 KKHAUHJOQPPGSL-UHFFFAOYSA-N 0.000 description 1
- KOVBCRJVKWBPDG-UHFFFAOYSA-N n-[(4-fluorophenyl)methyl]-5-methoxy-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound C=1C(OC)=CC=C(S(=O)(=O)NC=2C=C(OC)C=NC=2)C=1C(=O)NCC1=CC=C(F)C=C1 KOVBCRJVKWBPDG-UHFFFAOYSA-N 0.000 description 1
- XOKMABJSBHUADT-UHFFFAOYSA-N n-[(4-methoxyphenyl)methyl]-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound C1=CC(OC)=CC=C1CNC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 XOKMABJSBHUADT-UHFFFAOYSA-N 0.000 description 1
- VZKIXKREDXYDMB-UHFFFAOYSA-N n-[(4-methylphenyl)methyl]-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound C1=CC(C)=CC=C1CNC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 VZKIXKREDXYDMB-UHFFFAOYSA-N 0.000 description 1
- ZFDCCKLPSGJYBO-UHFFFAOYSA-N n-[1-(hydroxymethyl)cyclopentyl]-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound C=1C=CC=C(S(=O)(=O)NC=2C=NC=CC=2)C=1C(=O)NC1(CO)CCCC1 ZFDCCKLPSGJYBO-UHFFFAOYSA-N 0.000 description 1
- OMISWPXZIWPQGF-UHFFFAOYSA-N n-[2-(2,3-difluoro-4-methylphenyl)ethyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCCC=2C(=C(F)C(C)=CC=2)F)=C1 OMISWPXZIWPQGF-UHFFFAOYSA-N 0.000 description 1
- DQVITSNBPXNMEX-UHFFFAOYSA-N n-[2-(2,4-difluorophenyl)ethyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCCC=2C(=CC(F)=CC=2)F)=C1 DQVITSNBPXNMEX-UHFFFAOYSA-N 0.000 description 1
- BKFRLCHKKFLSFV-UHFFFAOYSA-N n-[2-(2,5-difluorophenyl)ethyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCCC=2C(=CC=C(F)C=2)F)=C1 BKFRLCHKKFLSFV-UHFFFAOYSA-N 0.000 description 1
- ZKSCOYLWGBEIHC-UHFFFAOYSA-N n-[2-(2-fluorophenyl)ethyl]-2-[(5-methylpyridin-3-yl)sulfamoyl]benzamide Chemical compound CC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCCC=2C(=CC=CC=2)F)=C1 ZKSCOYLWGBEIHC-UHFFFAOYSA-N 0.000 description 1
- ITLSJVSTDRVSPQ-UHFFFAOYSA-N n-[2-(2-methoxyphenyl)ethyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCCC=2C(=CC=CC=2)OC)=C1 ITLSJVSTDRVSPQ-UHFFFAOYSA-N 0.000 description 1
- XIASXGXRVIMZEI-UHFFFAOYSA-N n-[2-(2-methylphenyl)ethyl]-2-[(4-methylpyridin-3-yl)sulfamoyl]benzamide Chemical compound CC1=CC=CC=C1CCNC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CN=CC=C1C XIASXGXRVIMZEI-UHFFFAOYSA-N 0.000 description 1
- DSZPGAREUFRJFP-UHFFFAOYSA-N n-[2-(2-methylphenyl)ethyl]-2-[methyl(pyridin-3-yl)sulfamoyl]benzamide Chemical compound C=1C=CC=C(C(=O)NCCC=2C(=CC=CC=2)C)C=1S(=O)(=O)N(C)C1=CC=CN=C1 DSZPGAREUFRJFP-UHFFFAOYSA-N 0.000 description 1
- LHBGHAXUFKQUTB-UHFFFAOYSA-N n-[2-(3,4-difluorophenyl)ethyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCCC=2C=C(F)C(F)=CC=2)=C1 LHBGHAXUFKQUTB-UHFFFAOYSA-N 0.000 description 1
- MJSOSBJFWOAXIZ-UHFFFAOYSA-N n-[2-(dimethylamino)ethyl]-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound CN(C)CCNC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 MJSOSBJFWOAXIZ-UHFFFAOYSA-N 0.000 description 1
- HZWCLQYTKPKKAM-UHFFFAOYSA-N n-[2-[3-fluoro-4-(trifluoromethyl)phenyl]ethyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCCC=2C=C(F)C(=CC=2)C(F)(F)F)=C1 HZWCLQYTKPKKAM-UHFFFAOYSA-N 0.000 description 1
- LRRSQNXXBYYUNM-UHFFFAOYSA-N n-[2-methyl-1-(4-methylpiperidin-1-yl)propan-2-yl]-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound C1CC(C)CCN1CC(C)(C)NC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 LRRSQNXXBYYUNM-UHFFFAOYSA-N 0.000 description 1
- UBEWJBQYUDNOEP-UHFFFAOYSA-N n-[3-(2-methoxyphenyl)propyl]-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CN=CC(NS(=O)(=O)C=2C(=CC=CC=2)C(=O)NCCCC=2C(=CC=CC=2)OC)=C1 UBEWJBQYUDNOEP-UHFFFAOYSA-N 0.000 description 1
- GAQZNFUDILDDDI-UHFFFAOYSA-N n-[4-[[2-[4-chloro-2-(3-chloro-5-cyanobenzoyl)phenoxy]acetyl]amino]-3-methylphenyl]sulfonylpropanamide Chemical compound CC1=CC(S(=O)(=O)NC(=O)CC)=CC=C1NC(=O)COC1=CC=C(Cl)C=C1C(=O)C1=CC(Cl)=CC(C#N)=C1 GAQZNFUDILDDDI-UHFFFAOYSA-N 0.000 description 1
- FHQOKEKEXHYXKO-UHFFFAOYSA-N n-[[3-(difluoromethoxy)phenyl]methyl]-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound FC(F)OC1=CC=CC(CNC(=O)C=2C(=CC=CC=2)S(=O)(=O)NC=2C=NC=CC=2)=C1 FHQOKEKEXHYXKO-UHFFFAOYSA-N 0.000 description 1
- LGOFYTSUXWGTBO-UHFFFAOYSA-N n-[[4-(methylcarbamoyl)phenyl]methyl]-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound C1=CC(C(=O)NC)=CC=C1CNC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 LGOFYTSUXWGTBO-UHFFFAOYSA-N 0.000 description 1
- WVEJJEVZJIVRAR-UHFFFAOYSA-N n-benzyl-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound C=1C=CC=C(S(=O)(=O)NC=2C=NC=CC=2)C=1C(=O)NCC1=CC=CC=C1 WVEJJEVZJIVRAR-UHFFFAOYSA-N 0.000 description 1
- JHWGAHLPJDDMBN-UHFFFAOYSA-N n-benzyl-n-ethyl-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound C=1C=CC=C(S(=O)(=O)NC=2C=NC=CC=2)C=1C(=O)N(CC)CC1=CC=CC=C1 JHWGAHLPJDDMBN-UHFFFAOYSA-N 0.000 description 1
- SPGMMGAQYGORJQ-UHFFFAOYSA-N n-benzyl-n-methyl-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound C=1C=CC=C(S(=O)(=O)NC=2C=NC=CC=2)C=1C(=O)N(C)CC1=CC=CC=C1 SPGMMGAQYGORJQ-UHFFFAOYSA-N 0.000 description 1
- WUEPAUUIMWUKCQ-UHFFFAOYSA-N n-butan-2-yl-2-[(4-ethoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound CCOC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)NC(C)CC WUEPAUUIMWUKCQ-UHFFFAOYSA-N 0.000 description 1
- VRTBKQLKMHSVBW-UHFFFAOYSA-N n-butan-2-yl-2-[(4-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound CCC(C)NC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CN=CC=C1OC VRTBKQLKMHSVBW-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- YAKNUNDUTNFTOY-UHFFFAOYSA-N n-butyl-2-[(4-ethoxypyridin-3-yl)sulfamoyl]-n-methylbenzamide Chemical compound CCCCN(C)C(=O)C1=CC=CC=C1S(=O)(=O)NC1=CN=CC=C1OCC YAKNUNDUTNFTOY-UHFFFAOYSA-N 0.000 description 1
- KPCWIONSYPSRFG-UHFFFAOYSA-N n-butyl-2-[(5-cyanopyridin-3-yl)sulfamoyl]benzamide Chemical compound CCCCNC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CN=CC(C#N)=C1 KPCWIONSYPSRFG-UHFFFAOYSA-N 0.000 description 1
- CGJWSHMACKWUEU-UHFFFAOYSA-N n-butyl-2-[(5-methoxypyridin-3-yl)sulfamoyl]-n-methylbenzamide Chemical compound CCCCN(C)C(=O)C1=CC=CC=C1S(=O)(=O)NC1=CN=CC(OC)=C1 CGJWSHMACKWUEU-UHFFFAOYSA-N 0.000 description 1
- HLHFNFQIKRHGQR-UHFFFAOYSA-N n-butyl-2-[(5-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound CCCCNC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CN=CC(OC)=C1 HLHFNFQIKRHGQR-UHFFFAOYSA-N 0.000 description 1
- OCUBLIXVGLWZLN-UHFFFAOYSA-N n-butyl-2-[(5-methylpyridin-3-yl)sulfamoyl]benzamide Chemical compound CCCCNC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CN=CC(C)=C1 OCUBLIXVGLWZLN-UHFFFAOYSA-N 0.000 description 1
- CUACXIWPWUDTDD-UHFFFAOYSA-N n-cyclobutyl-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound C=1C=CC=C(S(=O)(=O)NC=2C=NC=CC=2)C=1C(=O)NC1CCC1 CUACXIWPWUDTDD-UHFFFAOYSA-N 0.000 description 1
- FREGWFQAUVJGCN-UHFFFAOYSA-N n-cyclohexyl-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound C=1C=CC=C(S(=O)(=O)NC=2C=NC=CC=2)C=1C(=O)NC1CCCCC1 FREGWFQAUVJGCN-UHFFFAOYSA-N 0.000 description 1
- AXUXYOAJWPWUMV-UHFFFAOYSA-N n-ethyl-n-(2-pyrazol-1-ylethyl)-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound C=1C=CC=C(S(=O)(=O)NC=2C=NC=CC=2)C=1C(=O)N(CC)CCN1C=CC=N1 AXUXYOAJWPWUMV-UHFFFAOYSA-N 0.000 description 1
- CUFSQQUDQKPSLL-UHFFFAOYSA-N n-methyl-2-(piperidine-1-carbonyl)-n-pyridin-3-ylbenzenesulfonamide Chemical compound C=1C=CC=C(C(=O)N2CCCCC2)C=1S(=O)(=O)N(C)C1=CC=CN=C1 CUFSQQUDQKPSLL-UHFFFAOYSA-N 0.000 description 1
- VSYZHJOACOQBEK-UHFFFAOYSA-N n-methyl-2-[(5-methylpyridin-3-yl)sulfamoyl]-n-(2-phenylethyl)benzamide Chemical compound C=1C=CC=C(S(=O)(=O)NC=2C=C(C)C=NC=2)C=1C(=O)N(C)CCC1=CC=CC=C1 VSYZHJOACOQBEK-UHFFFAOYSA-N 0.000 description 1
- JMJNGGPGGOZKSK-UHFFFAOYSA-N n-methyl-2-[(5-methylpyridin-3-yl)sulfamoyl]-n-pentylbenzamide Chemical compound CCCCCN(C)C(=O)C1=CC=CC=C1S(=O)(=O)NC1=CN=CC(C)=C1 JMJNGGPGGOZKSK-UHFFFAOYSA-N 0.000 description 1
- HBZUDXWOAKGQHD-UHFFFAOYSA-N n-methyl-n-(2-phenylethyl)-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound C=1C=CC=C(S(=O)(=O)NC=2C=NC=CC=2)C=1C(=O)N(C)CCC1=CC=CC=C1 HBZUDXWOAKGQHD-UHFFFAOYSA-N 0.000 description 1
- ZPGNYQSLYVVQIL-UHFFFAOYSA-N n-methyl-n-pentyl-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound CCCCCN(C)C(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 ZPGNYQSLYVVQIL-UHFFFAOYSA-N 0.000 description 1
- FVSAMYKBPQMQSY-UHFFFAOYSA-N n-pentyl-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound CCCCCNC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 FVSAMYKBPQMQSY-UHFFFAOYSA-N 0.000 description 1
- FUFHSCYHWXYDRP-UHFFFAOYSA-N n-propan-2-yl-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound CC(C)NC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 FUFHSCYHWXYDRP-UHFFFAOYSA-N 0.000 description 1
- QDSPTCBZQDWTPG-UHFFFAOYSA-N n-propyl-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound CCCNC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 QDSPTCBZQDWTPG-UHFFFAOYSA-N 0.000 description 1
- PIRHGTRRBZHYDG-UHFFFAOYSA-N n-pyridin-3-yl-2-(4-pyridin-2-ylpiperazine-1-carbonyl)benzenesulfonamide Chemical compound C=1C=CC=C(S(=O)(=O)NC=2C=NC=CC=2)C=1C(=O)N(CC1)CCN1C1=CC=CC=N1 PIRHGTRRBZHYDG-UHFFFAOYSA-N 0.000 description 1
- HFWVOZLXAXWTNO-UHFFFAOYSA-N n-pyridin-3-yl-2-(pyrrolidine-1-carbonyl)benzenesulfonamide Chemical compound C=1C=CC=C(S(=O)(=O)NC=2C=NC=CC=2)C=1C(=O)N1CCCC1 HFWVOZLXAXWTNO-UHFFFAOYSA-N 0.000 description 1
- OBKCUFCRJMULAX-UHFFFAOYSA-N n-tert-butyl-2-(pyridin-3-ylsulfamoyl)benzamide Chemical compound CC(C)(C)NC(=O)C1=CC=CC=C1S(=O)(=O)NC1=CC=CN=C1 OBKCUFCRJMULAX-UHFFFAOYSA-N 0.000 description 1
- GJENYNRYMORIRF-UHFFFAOYSA-N n-tert-butyl-2-[(4-ethoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound CCOC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)NC(C)(C)C GJENYNRYMORIRF-UHFFFAOYSA-N 0.000 description 1
- WZJFHJYTSUEFEU-UHFFFAOYSA-N n-tert-butyl-2-[(4-methoxypyridin-3-yl)sulfamoyl]benzamide Chemical compound COC1=CC=NC=C1NS(=O)(=O)C1=CC=CC=C1C(=O)NC(C)(C)C WZJFHJYTSUEFEU-UHFFFAOYSA-N 0.000 description 1
- PSZYNBSKGUBXEH-UHFFFAOYSA-N naphthalene-1-sulfonic acid Chemical compound C1=CC=C2C(S(=O)(=O)O)=CC=CC2=C1 PSZYNBSKGUBXEH-UHFFFAOYSA-N 0.000 description 1
- KVBGVZZKJNLNJU-UHFFFAOYSA-N naphthalene-2-sulfonic acid Chemical compound C1=CC=CC2=CC(S(=O)(=O)O)=CC=C21 KVBGVZZKJNLNJU-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 230000008693 nausea Effects 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 229960000689 nevirapine Drugs 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 230000036963 noncompetitive effect Effects 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- 235000016709 nutrition Nutrition 0.000 description 1
- 230000035764 nutrition Effects 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- WWZKQHOCKIZLMA-UHFFFAOYSA-M octanoate Chemical compound CCCCCCCC([O-])=O WWZKQHOCKIZLMA-UHFFFAOYSA-M 0.000 description 1
- 229940049964 oleate Drugs 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 229920000620 organic polymer Polymers 0.000 description 1
- 239000003791 organic solvent mixture Substances 0.000 description 1
- 125000002524 organometallic group Chemical group 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000003551 oxepanyl group Chemical group 0.000 description 1
- 125000003566 oxetanyl group Chemical group 0.000 description 1
- 229960001592 paclitaxel Drugs 0.000 description 1
- 229940014662 pantothenate Drugs 0.000 description 1
- 235000019161 pantothenic acid Nutrition 0.000 description 1
- 239000011713 pantothenic acid Substances 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 235000010603 pastilles Nutrition 0.000 description 1
- 230000001575 pathological effect Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 235000010987 pectin Nutrition 0.000 description 1
- 229920001277 pectin Polymers 0.000 description 1
- 239000001814 pectin Substances 0.000 description 1
- 229960001179 penciclovir Drugs 0.000 description 1
- XDRYMKDFEDOLFX-UHFFFAOYSA-N pentamidine Chemical compound C1=CC(C(=N)N)=CC=C1OCCCCCOC1=CC=C(C(N)=N)C=C1 XDRYMKDFEDOLFX-UHFFFAOYSA-N 0.000 description 1
- 229960004448 pentamidine Drugs 0.000 description 1
- 239000000575 pesticide Substances 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- DYUMLJSJISTVPV-UHFFFAOYSA-N phenyl propanoate Chemical compound CCC(=O)OC1=CC=CC=C1 DYUMLJSJISTVPV-UHFFFAOYSA-N 0.000 description 1
- WLJVXDMOQOGPHL-UHFFFAOYSA-N phenylacetic acid Chemical class OC(=O)CC1=CC=CC=C1 WLJVXDMOQOGPHL-UHFFFAOYSA-N 0.000 description 1
- 229950009215 phenylbutanoic acid Drugs 0.000 description 1
- DHRLEVQXOMLTIM-UHFFFAOYSA-N phosphoric acid;trioxomolybdenum Chemical compound O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.O=[Mo](=O)=O.OP(O)(O)=O DHRLEVQXOMLTIM-UHFFFAOYSA-N 0.000 description 1
- XNGIFLGASWRNHJ-UHFFFAOYSA-L phthalate(2-) Chemical compound [O-]C(=O)C1=CC=CC=C1C([O-])=O XNGIFLGASWRNHJ-UHFFFAOYSA-L 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 239000002504 physiological saline solution Substances 0.000 description 1
- SIOXPEMLGUPBBT-UHFFFAOYSA-N picolinic acid Chemical compound OC(=O)C1=CC=CC=N1 SIOXPEMLGUPBBT-UHFFFAOYSA-N 0.000 description 1
- 239000000049 pigment Substances 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 239000004014 plasticizer Substances 0.000 description 1
- 229960002169 plerixafor Drugs 0.000 description 1
- 229920001467 poly(styrenesulfonates) Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 235000010988 polyoxyethylene sorbitan tristearate Nutrition 0.000 description 1
- 239000001816 polyoxyethylene sorbitan tristearate Substances 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 150000004804 polysaccharides Chemical class 0.000 description 1
- 229940099511 polysorbate 65 Drugs 0.000 description 1
- 229960004293 porfimer sodium Drugs 0.000 description 1
- 229960001589 posaconazole Drugs 0.000 description 1
- RAGOYPUPXAKGKH-XAKZXMRKSA-N posaconazole Chemical compound O=C1N([C@H]([C@H](C)O)CC)N=CN1C1=CC=C(N2CCN(CC2)C=2C=CC(OC[C@H]3C[C@@](CN4N=CN=C4)(OC3)C=3C(=CC(F)=CC=3)F)=CC=2)C=C1 RAGOYPUPXAKGKH-XAKZXMRKSA-N 0.000 description 1
- 159000000001 potassium salts Chemical class 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 229940069328 povidone Drugs 0.000 description 1
- 238000011533 pre-incubation Methods 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 229960005179 primaquine Drugs 0.000 description 1
- FYPMFJGVHOHGLL-UHFFFAOYSA-N probucol Chemical compound C=1C(C(C)(C)C)=C(O)C(C(C)(C)C)=CC=1SC(C)(C)SC1=CC(C(C)(C)C)=C(O)C(C(C)(C)C)=C1 FYPMFJGVHOHGLL-UHFFFAOYSA-N 0.000 description 1
- 229960003912 probucol Drugs 0.000 description 1
- KCXFHTAICRTXLI-UHFFFAOYSA-N propane-1-sulfonic acid Chemical compound CCCS(O)(=O)=O KCXFHTAICRTXLI-UHFFFAOYSA-N 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 238000011321 prophylaxis Methods 0.000 description 1
- UORVCLMRJXCDCP-UHFFFAOYSA-M propynoate Chemical compound [O-]C(=O)C#C UORVCLMRJXCDCP-UHFFFAOYSA-M 0.000 description 1
- 230000029983 protein stabilization Effects 0.000 description 1
- 235000018102 proteins Nutrition 0.000 description 1
- 102000004169 proteins and genes Human genes 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- 238000000425 proton nuclear magnetic resonance spectrum Methods 0.000 description 1
- SSKVDVBQSWQEGJ-UHFFFAOYSA-N pseudohypericin Natural products C12=C(O)C=C(O)C(C(C=3C(O)=CC(O)=C4C=33)=O)=C2C3=C2C3=C4C(C)=CC(O)=C3C(=O)C3=C(O)C=C(O)C1=C32 SSKVDVBQSWQEGJ-UHFFFAOYSA-N 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- 125000002755 pyrazolinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- WKSAUQYGYAYLPV-UHFFFAOYSA-N pyrimethamine Chemical compound CCC1=NC(N)=NC(N)=C1C1=CC=C(Cl)C=C1 WKSAUQYGYAYLPV-UHFFFAOYSA-N 0.000 description 1
- 229960000611 pyrimethamine Drugs 0.000 description 1
- VTGOHKSTWXHQJK-UHFFFAOYSA-N pyrimidin-2-ol Chemical class OC1=NC=CC=N1 VTGOHKSTWXHQJK-UHFFFAOYSA-N 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 150000003856 quaternary ammonium compounds Chemical class 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 229950005950 rebimastat Drugs 0.000 description 1
- 238000010992 reflux Methods 0.000 description 1
- 230000000979 retarding effect Effects 0.000 description 1
- 229960000329 ribavirin Drugs 0.000 description 1
- HZCAHMRRMINHDJ-DBRKOABJSA-N ribavirin Natural products O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1N=CN=C1 HZCAHMRRMINHDJ-DBRKOABJSA-N 0.000 description 1
- 229940100486 rice starch Drugs 0.000 description 1
- 229960004641 rituximab Drugs 0.000 description 1
- 102200046712 rs752492870 Human genes 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 239000000741 silica gel Substances 0.000 description 1
- 229910002027 silica gel Inorganic materials 0.000 description 1
- 238000010898 silica gel chromatography Methods 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 239000012064 sodium phosphate buffer Substances 0.000 description 1
- 239000008109 sodium starch glycolate Substances 0.000 description 1
- 229920003109 sodium starch glycolate Polymers 0.000 description 1
- 229940079832 sodium starch glycolate Drugs 0.000 description 1
- JJICLMJFIKGAAU-UHFFFAOYSA-M sodium;2-amino-9-(1,3-dihydroxypropan-2-yloxymethyl)purin-6-olate Chemical compound [Na+].NC1=NC([O-])=C2N=CN(COC(CO)CO)C2=N1 JJICLMJFIKGAAU-UHFFFAOYSA-M 0.000 description 1
- 239000007901 soft capsule Substances 0.000 description 1
- 239000012439 solid excipient Substances 0.000 description 1
- 229960002920 sorbitol Drugs 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 229960001294 spiramycin Drugs 0.000 description 1
- 229930191512 spiramycin Natural products 0.000 description 1
- 235000019372 spiramycin Nutrition 0.000 description 1
- 125000003003 spiro group Chemical group 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 230000000087 stabilizing effect Effects 0.000 description 1
- 229960001203 stavudine Drugs 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 239000012058 sterile packaged powder Substances 0.000 description 1
- TYFQFVWCELRYAO-UHFFFAOYSA-L suberate(2-) Chemical compound [O-]C(=O)CCCCCCC([O-])=O TYFQFVWCELRYAO-UHFFFAOYSA-L 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- BDHFUVZGWQCTTF-UHFFFAOYSA-M sulfonate Chemical compound [O-]S(=O)=O BDHFUVZGWQCTTF-UHFFFAOYSA-M 0.000 description 1
- 150000003457 sulfones Chemical class 0.000 description 1
- 150000003462 sulfoxides Chemical class 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 238000007910 systemic administration Methods 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical compound O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 1
- 229960004556 tenofovir Drugs 0.000 description 1
- LDEKQSIMHVQZJK-CAQYMETFSA-N tenofovir alafenamide Chemical compound O([P@@](=O)(CO[C@H](C)CN1C2=NC=NC(N)=C2N=C1)N[C@@H](C)C(=O)OC(C)C)C1=CC=CC=C1 LDEKQSIMHVQZJK-CAQYMETFSA-N 0.000 description 1
- 229960004946 tenofovir alafenamide Drugs 0.000 description 1
- 229960004693 tenofovir disoproxil fumarate Drugs 0.000 description 1
- 229950002757 teoclate Drugs 0.000 description 1
- CZDOFRBRPKDZPS-UHFFFAOYSA-N tert-butyl 2-[2-(pyridin-3-ylsulfamoyl)benzoyl]-2,7-diazaspiro[3.5]nonane-7-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCC11CN(C(=O)C=2C(=CC=CC=2)S(=O)(=O)NC=2C=NC=CC=2)C1 CZDOFRBRPKDZPS-UHFFFAOYSA-N 0.000 description 1
- VBCPZLRADGTWFZ-UHFFFAOYSA-N tert-butyl 7-[2-(pyridin-3-ylsulfamoyl)benzoyl]-2,7-diazaspiro[3.5]nonane-2-carboxylate Chemical compound C1N(C(=O)OC(C)(C)C)CC21CCN(C(=O)C=1C(=CC=CC=1)S(=O)(=O)NC=1C=NC=CC=1)CC2 VBCPZLRADGTWFZ-UHFFFAOYSA-N 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- OQHZMGOXOOOFEE-SYQUUIDJSA-N tert-butyl n-[(2s,3r)-3-hydroxy-4-[[(2r,3s)-2-hydroxy-3-[(2-methylpropan-2-yl)oxycarbonylamino]-4-[4-(2-morpholin-4-yl-2-oxoethoxy)phenyl]butyl]amino]-1-phenylbutan-2-yl]carbamate Chemical compound C([C@H](NC(=O)OC(C)(C)C)[C@H](O)CNC[C@@H](O)[C@H](CC=1C=CC(OCC(=O)N2CCOCC2)=CC=1)NC(=O)OC(C)(C)C)C1=CC=CC=C1 OQHZMGOXOOOFEE-SYQUUIDJSA-N 0.000 description 1
- KKRYDPVDJYCEER-QEGGNFSNSA-N tert-butyl n-[(2s,3r)-3-hydroxy-4-[[(2r,3s)-2-hydroxy-3-[(2-methylpropan-2-yl)oxycarbonylamino]-4-phenylbutyl]amino]-1-phenylbutan-2-yl]carbamate Chemical compound C([C@H](NC(=O)OC(C)(C)C)[C@H](O)CNC[C@@H](O)[C@H](CC=1C=CC=CC=1)NC(=O)OC(C)(C)C)C1=CC=CC=C1 KKRYDPVDJYCEER-QEGGNFSNSA-N 0.000 description 1
- 125000006169 tetracyclic group Chemical group 0.000 description 1
- HWCKGOZZJDHMNC-UHFFFAOYSA-M tetraethylammonium bromide Chemical compound [Br-].CC[N+](CC)(CC)CC HWCKGOZZJDHMNC-UHFFFAOYSA-M 0.000 description 1
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 125000005958 tetrahydrothienyl group Chemical group 0.000 description 1
- 125000004632 tetrahydrothiopyranyl group Chemical group S1C(CCCC1)* 0.000 description 1
- 125000003831 tetrazolyl group Chemical group 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 229940126585 therapeutic drug Drugs 0.000 description 1
- 238000002560 therapeutic procedure Methods 0.000 description 1
- 238000011285 therapeutic regimen Methods 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000005308 thiazepinyl group Chemical group S1N=C(C=CC=C1)* 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 125000001583 thiepanyl group Chemical group 0.000 description 1
- 125000002053 thietanyl group Chemical group 0.000 description 1
- ZFEAMMNVDPDEGE-LGRGJMMZSA-N tifuvirtide Chemical compound C([C@H](NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(C)=O)[C@@H](C)CC)[C@@H](C)O)[C@@H](C)CC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(N)=O)C1=CC=C(O)C=C1 ZFEAMMNVDPDEGE-LGRGJMMZSA-N 0.000 description 1
- 230000036964 tight binding Effects 0.000 description 1
- 239000004408 titanium dioxide Substances 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 150000003626 triacylglycerols Chemical class 0.000 description 1
- 125000004306 triazinyl group Chemical group 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical class [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- CYRMSUTZVYGINF-UHFFFAOYSA-N trichlorofluoromethane Chemical compound FC(Cl)(Cl)Cl CYRMSUTZVYGINF-UHFFFAOYSA-N 0.000 description 1
- 229940029284 trichlorofluoromethane Drugs 0.000 description 1
- 125000006168 tricyclic group Chemical group 0.000 description 1
- IEDVJHCEMCRBQM-UHFFFAOYSA-N trimethoprim Chemical compound COC1=C(OC)C(OC)=CC(CC=2C(=NC(N)=NC=2)N)=C1 IEDVJHCEMCRBQM-UHFFFAOYSA-N 0.000 description 1
- 229960001082 trimethoprim Drugs 0.000 description 1
- NOYPYLRCIDNJJB-UHFFFAOYSA-N trimetrexate Chemical compound COC1=C(OC)C(OC)=CC(NCC=2C(=C3C(N)=NC(N)=NC3=CC=2)C)=C1 NOYPYLRCIDNJJB-UHFFFAOYSA-N 0.000 description 1
- 229960001099 trimetrexate Drugs 0.000 description 1
- DFHAXXVZCFXGOQ-UHFFFAOYSA-K trisodium phosphonoformate Chemical compound [Na+].[Na+].[Na+].[O-]C(=O)P([O-])([O-])=O DFHAXXVZCFXGOQ-UHFFFAOYSA-K 0.000 description 1
- YFNGWGVTFYSJHE-UHFFFAOYSA-K trisodium;phosphonoformate Chemical compound [Na+].[Na+].[Na+].OP(O)(=O)C([O-])=O.OP(O)(=O)C([O-])=O.OP(O)(=O)C([O-])=O YFNGWGVTFYSJHE-UHFFFAOYSA-K 0.000 description 1
- WVPSKSLAZQPAKQ-CDMJZVDBSA-N trovafloxacin Chemical compound C([C@H]1[C@@H]([C@H]1C1)N)N1C(C(=CC=1C(=O)C(C(O)=O)=C2)F)=NC=1N2C1=CC=C(F)C=C1F WVPSKSLAZQPAKQ-CDMJZVDBSA-N 0.000 description 1
- 229960000497 trovafloxacin Drugs 0.000 description 1
- 229950009795 tucaresol Drugs 0.000 description 1
- 230000036967 uncompetitive effect Effects 0.000 description 1
- 241001529453 unidentified herpesvirus Species 0.000 description 1
- 229940093257 valacyclovir Drugs 0.000 description 1
- NQPDZGIKBAWPEJ-UHFFFAOYSA-N valeric acid Chemical class CCCCC(O)=O NQPDZGIKBAWPEJ-UHFFFAOYSA-N 0.000 description 1
- 229960002149 valganciclovir Drugs 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
- 229940100050 virazole Drugs 0.000 description 1
- BCEHBSKCWLPMDN-MGPLVRAMSA-N voriconazole Chemical compound C1([C@H](C)[C@](O)(CN2N=CN=C2)C=2C(=CC(F)=CC=2)F)=NC=NC=C1F BCEHBSKCWLPMDN-MGPLVRAMSA-N 0.000 description 1
- 229960004740 voriconazole Drugs 0.000 description 1
- 239000001993 wax Substances 0.000 description 1
- 229940100445 wheat starch Drugs 0.000 description 1
- 229960000523 zalcitabine Drugs 0.000 description 1
- 229960002555 zidovudine Drugs 0.000 description 1
- HBOMLICNUCNMMY-XLPZGREQSA-N zidovudine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](N=[N+]=[N-])C1 HBOMLICNUCNMMY-XLPZGREQSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/76—Nitrogen atoms to which a second hetero atom is attached
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/165—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide
- A61K31/166—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide having the carbon of a carboxamide group directly attached to the aromatic ring, e.g. procainamide, procarbazine, metoclopramide, labetalol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4406—Non condensed pyridines; Hydrogenated derivatives thereof only substituted in position 3, e.g. zimeldine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P39/00—General protective or antinoxious agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/10—Spiro-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/08—Bridged systems
Definitions
- the cytochrome P450 (CYP450) enzyme system is responsible for the biotransformation of drugs from active to inactive metabolites that are readily excreted by the body. Furthermore, the rapid metabolism of certain drugs by the CYP450 enzyme system can markedly alter their pharmacokinetic (PK) profile and can result in sub-therapeutic plasma levels of those drugs over time. In the area of anti-infective therapy, such as treating viral infections such as human immunodeficiency virus (HIV) infections, such sub-therapeutic drug plasma levels can lead to an increase in resistance of the virus.
- HIV human immunodeficiency virus
- Ritonavir is a marketed HlV protease inhibitor (Pl) that, due to its ability to inhibit the cytochrome P450 3A4 enzyme, is also used to "boost" the pharmacokinetic exposure of many coadministered anti-retrovirals.
- Pl HlV protease inhibitor
- Administering low doses of a compound with potent antiviral activity may also contribute to the selection of drug- resistant strains of HIV.
- a novel CYP3A4 inhibitor capable of boosting antivirals as effectively as RTV but devoid of antiviral activity and significant side-effects would offer significant advantages and therapeutic value in the treatment of those suffering from infection with the HIV virus.
- the present invention discloses compounds that are useful in the inhibition of the CYP450 enzyme system and may be used to boost the pharmacokinetic exposure of co-administered drugs, including anti-retrovirals. It also discloses pharmaceutical formulations comprising such compounds, methods of making them, and methods of using them.
- R 1 , R 2 , R 3 , R 4 are independently selected from hydrogen, C 1 -C 6 alkyl, -(CR 12a R 12b ) t halo,
- R 5 is hydrogen or C 1 -C 6 alkyl
- R 6 , R 7 , R 8 , and R 9 are independently selected from hydrogen, C 1 -C 6 alkyl, -(CR 12a R 12b ) t halo, -(CR 12a R 12b ) t CN, -(CR 12a R 12b ) t CF 3 , -(CR 12a R 12b ) t OR 12a , -(CR 12a R 12b ) t N(R 12a R 12b ), -(CR 12a R 12b ) t R 12a , -(CR 123 R 1215 XC 3 -C 11 cycloalkyl, -(CR 12a R 12b ) t C 6 -C 10 aryl, -(CR 12a R 12b ) t C 2 -C 10 heterocyclyl, -(CR 12a R 12b ) t heteroaryl, -O(CR 12a R 12
- R 10 and R 11 are independently selected from hydrogen, -(CR 12a R 12b ) t C r C 6 alkyl, -(CR 123 R 1 ⁇ )A-C 11 cycloalkyl, -(CR 12a R 12b ) t C 6 -C 10 aryl, -(CR 12a R 12b ),C 2 -C 10 heterocyclyl, and -(CR 12a R 12b ) t heteroaryl, wherein each of said C 1 -C 6 alkyl, C 3 -C 11 cycloalkyl, C 6 -C 10 aryl, C 2 -C 10 heterocyclyl, and heteroaryl groups is optionally substituted with one or more R 13 ; or
- R 10 and R 11 together with the atom to which they are attached, form a C 2 -C 10 heterocyclyl group, optionally substituted with one or more R 13 ; each R 12a and R 12b is independently selected from hydrogen and C 1 -C 6 alkyl; or
- R 12a and R 12b together with the atom to which they are attached, form a C 3 -C 11 cycloalkyl or a C 2 -C 10 heterocyclyl group; each R 13 is independently selected from C 1 -C 6 alkyl, halogen, -(CR 12a R 12b ) t CN, -(CR 12a R 12b ) t CF 3 , -(CR 12a R 12b ) t OCF 3 , -(CR 123 R 12 ⁇ C 3 -C 11 cycloalkyl, -(CR 12a R 12b ),C 6 -C 10 aryl, -(CR 12a R 12b ) t C 2 -C 10 heterocyclyl, -(CR 12a R 12b ) t heteroaryl, -O(CR 12a R 12b ) t R 12a , -O(CR 12a R 12b ) t C 6 -C 10 aryl, -O(CR
- R 1 , R 2 , R 3 , R 4 are independently selected from hydrogen, C 1 -C 6 alkyl, -(CR 12a R 12b ) t halo, -(CR 12a R 12b ),CN, -(CR 12a R 12b ),OR 123 , -(CR 12a R 12b ),N(R 12a R 12b ), and -(CR 12a R 12b ) t CF 3 ;
- R 5 is hydrogen or C 1 -C 6 alkyl;
- R 6 , R 7 , R 8 , and R 9 are independently selected from hydrogen, C 1 -C 6 alkyl, -(CR 12a R 12b ) t halo, -(CR 12a R 12b ) t CN, -(CR 12a R 12b ) t CF 3l -(CR 12a R 12b ) t OR 12a , -(CR 12a R 12b ) t N(R 12a R 12b ),
- R 10 and R 11 are independently selected from hydrogen, -(CR 12a R 12b ) t C r C 6 alkyl, -(CR 123 R 1 ⁇ ) 1 C 3 -C 11 cycloalkyl, -(CR 12a R 12b ) ( C 6 -C 10 aryl, -(CR 12a R 12b ) t C 2 -C 10 heterocyclyl, and -(CR 12a R 12b ) t heteroaryl, wherein each of said C 1 -C 6 alkyl, C 3 -C 11 cycloal
- R 12a and R 12b together with the atom to which they are attached, form a C 3 -C 11 cycloalkyl or a C 2 -C 10 heterocyclyl group; each R 13 is independently selected from C 1 -C 6 alkyl, halogen, -(CR 12a R 12b ) t CN, -(CR 12a R 12b ) t CF 3 , -(CR 12a R 12b ) t OCF 3 , -(CR 123 R 1213 XC 3 -C 11 cycloalkyl, -(CR 12a R 12b ),C 6 -C 10 aryl, -(CR 12a R 12b ) t C 2 -C 10 heterocyclyl, -(CR 12a R 12b ),heteroaryl, -O(CR 12a R 12b ) t R 12a , -O(CR 12a R 12b ) t C 6 -C 10 aryl, -Oary
- R 1 , R 2 , R 3 , R 4 are independently selected from hydrogen, C 1 -C 6 alkyl, halo, -CN, -OR 12a , and -CF 3 ;
- R 5 is hydrogen
- R 6 , R 7 , R 8 , and R 9 are independently selected from hydrogen, C 1 -C 6 alkyl, -(CR 12a R 12b ) t halo, -(CR 12a R 12b ) t CN, -(CR 12a R 12b ) t CF 3 , -(CR 12a R 12b ) t OR 12a , -(CR 12a R 12b ) t N(R 12a R 12b ),
- R 10 and R 11 are independently selected from hydrogen, -(CR 12a R 12b ) t C r C 6 alkyl, -(CR 123 R 1213 XC 3 -C 11 cycloalkyl, -(CR 12a R 12b ) t C 6 -C 10 aryl, -(CR 12a R 12b ),C 2 -C 10 heterocyclyl, and -(CR 12a R 12b ) t heteroaryl, wherein each of said C 1 -C 6 alkyl, C 3 -C 11 cycloalkyl, C 6 -C 10 aryl, C 2 -C 10 heterocyclyl, and heteroaryl groups is optionally substituted with one or more R 13 ; each R 12a and R 12b is independently selected from hydrogen and C 1 -C 6 alkyl; or
- R 12a and R 12b together with the atom to which they are attached, form a C 3 -Ci 1 cycloalkyl or a C 2 -C 10 heterocyclyl group; each R 13 is independently selected from C 1 -C 6 alkyl, halogen, -(CR 12a R 12b ) t CN, -(CR 123 R 12b ) t CF 3 , -(CR 12a R 12b ) t OCF 3 , -(CR 123 R 1213 XC 3 -C 11 cycloalkyl, -(CR 12a R 12b ),C 6 -C 10 aryl,
- Still another embodiment provides compounds of formula (I), wherein:
- R 1 , R 2 , R 3 , R 4 are independently selected from hydrogen, C 1 -C 6 alkyl, halo, -CN, -OR 12a , and -CF 3 ;
- R 5 is hydrogen
- R 6 , R 7 , R 8 , and R 9 are independently selected from hydrogen, Ci-C 6 alkyl, -(CR 12a R 12b ) t halo, -(CR 12a R 12b ) t CN, -(CR 12a R 12b )»CF 3 , -(CR 12a R 12b ) t OR 12a , -(CR 12a R 12b ) t N(R 12a R 12b ),
- R 10 is hydrogen or C 1 -C 6 alkyl
- R 11 is selected from -(CR 12a R 12b ) t C r C 6 alkyl, -(CR 123 R 12 ⁇ C 3 -C 11 cycloalkyl, - (CR 123 R 12 VVC 10 aryl, -(CR 12a R 12b ) ( C 2 -Ci 0 heterocyclyl, and -(CR 12a R 12b ) t heteroaryl, wherein each of said C 1 -C 6 alkyl, C 3 -Ci 1 cycloalkyl, C 6 -C 10 aryl, C 2 -C 10 heterocyclyl, and heteroaryl groups is optionally substituted with one or more R 13 ; each R 12a and R 12b is independently selected from hydrogen and C 1 -C 6 alkyl; or R 12a and R 12b , together with the atom to which they are attached, form a C 3 -Cn cycloalkyl or a C 2 -Ci 0 heterocyclyl group; each
- R 1 , R 2 , R 3 , R 4 are independently selected from hydrogen, C 1 -C 6 alkyl, halo, -CN, -OR 12a , and -CF 3 ;
- R 5 is hydrogen;
- R 6 , R 7 , R 8 , and R 9 are hydrogen;
- R 10 is hydrogen or C 1 -C 6 alkyl
- R 11 is selected from -(CR 12a R 12b ) t C r C 6 alkyl, cycloalkyl, - (CR 12a R 12b ) t C 6 -C 10 aryl, -(CR 12a R 12b ) t C 2 -C 10 heterocyclyl, and -(CR 12a R 12b ) t heteroaryl, wherein each of said C 1 -C 6 alkyl, C 3 -C 11 cycloalkyl, C 6 -C 10 aryl, C 2 -C 10 heterocyclyl, and heteroaryl groups is optionally substituted with one or more R 13 ; each R 12a and R 12b is independently selected from hydrogen and C 1 -C 6 alkyl; or
- R 12a and R 12b together with the atom to which they are attached, form a C 3 -C 11 cycloalkyl or a C 2 -C 10 heterocyclyl group; each R 13 is independently selected from C 1 -C 6 alkyl, halogen, -(CR 12a R 12b ) t CN, -(CR 12a R 12b ) t CF 3 , -(CR 12a R 12b ) t OCF 3 , -(CR 123 R 1 ⁇ ) 1 C 3 -C 11 cycloalkyl, -(CR 12a R 12b ) t C 6 -C 10 aryl,
- each of said C 1 -C 6 alkyl, C 3 -C 11 cycloalkyl, C 6 -C 10 aryl, C 2 - Ci 0 heterocyclyl, and heteroaryl groups is optionally substituted with one or more R 14 ; each R 14 is independently selected from C 1 -C 6 alkyl, halogen, -CN, -CF 3 , and -OR 12a ; and each t is independently selected from 0, 1 , 2, 3, 4, 5, and 6; or a pharmaceutically acceptable salt or solvate thereof.
- Still another embodiment affords compounds of formula (I), wherein:
- R 1 , R 2 , R 3 , R 4 are independently selected from hydrogen, C 1 -C 6 alkyl, halo, -CN, -OR 12a , and -CF 3 ;
- R 5 is hydrogen;
- R 6 , R 7 , R 8 , and R 9 are hydrogen;
- R 10 is hydrogen or C 1 -C 6 alkyl
- R 11 is -(CR 12a R 12b ) t C 6 -C 10 aryl, wherein said C 6 -C 10 aryl is optionally substituted with one or more R 13 ; each R 12a and R 12b is independently selected from hydrogen and C 1 -C 6 alkyl; or R 12a and R 12b , together with the atom to which they are attached, form a C 3 -C 11 cycloalkyl or a C 2 -C 10 heterocyclyl group; each R 13 is independently selected from C 1 -C 6 alkyl, halogen, -(CR 12a R 12b ) t CN,
- each of said C 1 -C 6 alkyl, C 3 -C 11 cycloalkyl, C 6 -C 10 aryl, C 2 - C 10 heterocyclyl, and heteroaryl groups is optionally substituted with one or more R 14 ; each R 14 is independently selected from C 1 -C 6 alkyl, halogen, -CN, -CF 3 , and -OR 12a ; and each t is independently selected from 0, 1 , 2, 3, 4, 5, and 6; or a pharmaceutically acceptable salt or solvate thereof.
- R 1 , R 2 , R 3 , R 4 are independently selected from hydrogen, C 1 -C 6 alkyl, halo, -CN, -OR 12a , and -CF 3 ;
- R 5 is hydrogen
- R 6 , R 7 , R 8 , and R 9 are hydrogen;
- R 10 is hydrogen or C 1 -C 6 alkyl;
- R 11 is -(CR 12a R 12b )C 6 -C 10 aryl or -(CR 12a R 12b ) 2 C 6 -C 10 aryl, wherein said C 6 -C 10 aryl is optionally substituted with one or more R 13 ; each R 12a and R 12b is independently selected from hydrogen and C 1 -C 6 alkyl; or R 12a and R 12b , together with the atom to which they are attached, form a C 3 -Ci 1 cycloalkyl or a C 2 -C 10 heterocyclyl group; each R 13 is independently selected from C 1 -C 6 alkyl, halogen, -(CR 123 R 12b ) t CN, -(CR 12a R 12b ) t CF 3 , -(CR 12a R 12b ) t OCF 3 , -(CR 123 R 1 ⁇ ) 1 C 3 -C 11 cycloalkyl, -(CR 12a R 12b
- each of said C 1 -C 6 alkyl, C 3 -C 11 cycloalkyl, C 6 -C 10 aryl, C 2 - C 10 heterocyclyl, and heteroaryl groups is optionally substituted with one or more R 14 ; each R 14 is independently selected from C 1 -C 6 alkyl, halogen, -CN, -CF 3 , and -OR 12a ; and each t is independently selected from 0, 1 , 2, 3, 4, 5, and 6; or a pharmaceutically acceptable salt or solvate thereof.
- Still another embodiment provides compounds of formula (I), wherein: R 1 , R 2 , R 3 , R 4 are independently selected from hydrogen, C r C ⁇ alkyl, halo, -CN, -OR 12a , and -CF 3 ;
- R 5 is hydrogen;
- R 6 , R 7 , R 8 , and R 9 are hydrogen;
- R 10 is hydrogen or C 1 -C 6 alkyl
- R 11 is -(CH 2 )C 6 -C 10 aryl or -(CH 2 ) 2 C 6 -C 10 aryl, wherein said C 6 -C 10 aryl is optionally substituted with one or more R 13 ;
- R 12a is hydrogen or C 1 -C 6 alkyl; and each R 13 is independently selected from C 1 -C 6 alkyl, halogen, -CN, -CF 3 , and -OCF 3 ; or a pharmaceutically acceptable salt or solvate thereof.
- a further embodiment provides compounds of formula (I), wherein: R 1 , R 3 , and R 4 are hydrogen; R 2 is C 1 -C 6 alky!, halo, -CN, -OR 12a , or -CF 3 ; R 5 is hydrogen;
- R 6 , R 7 , R 8 , and R 9 are hydrogen;
- R 10 is hydrogen or CrC 6 alkyl;
- R 11 is -(CH 2 )C 6 -Ci 0 aryl or -(CH 2 J 2 C 6 -Ci 0 aryl, wherein said C 6 -C 10 aryl is optionally substituted with one or more R 13 ;
- R 12a is hydrogen or C 1 -C 6 alkyl; and each R 13 is independently selected from C 1 -C 6 alkyl, halogen, -CN, -CF 3 , and -OCF 3 ; or a pharmaceutically acceptable salt or solvate thereof.
- R 2 is C 1 -C 6 alkyl, -Cl, -F, -CN, -OCH 3 , -OCH 2 CH 3 , Or -CF 3 ;
- R 5 is hydrogen
- R 6 , R 7 , R 8 , and R 9 are hydrogen
- R 10 is hydrogen or -CH 3 ;
- R 11 is -(CH 2 )C 6 -C 10 aryl or -(CH 2 ) 2 C 6 -Ci 0 aryl, wherein said C 6 -C 10 aryl is optionally substituted with one or more R 13 ; and each R 13 is independently selected from C 1 -C 6 alkyl, -Cl, -F, -CN, -CF 3 , and -OCF 3 ; or a pharmaceutically acceptable salt or solvate thereof.
- R 1 , R 3 , and R 4 are hydrogen
- R 2 is C 1 -C 6 alkyl, -OCH 3 , or -OCH 2 CH 3 ;
- R 5 is hydrogen
- R 6 , R 7 , R 8 , and R 9 are hydrogen;
- R 10 is hydrogen or -CH 3 ;
- R 11 is -(CH 2 )C 6 -C 10 aryl or -(CH 2 J 2 C 6 -C 10 aryl, wherein said C 6 -C 10 aryl is optionally substituted with one or more R 13 ; and each R 13 is independently selected from C 1 -C 6 alkyl, -Cl, -F, -CN, -CF 3 , and -OCF 3 ; or a pharmaceutically acceptable salt or solvate thereof.
- R 1 , R 3 , and R 4 are hydrogen
- R 2 is -CH 3 , -OCH 3 , Or -OCH 2 CH 3 ;
- R 5 is hydrogen;
- R 6 , R 7 , R 8 , and R 9 are hydrogen;
- R 10 is hydrogen Or -CH 3 ;
- R 11 is -(CH 2 )C 6 -Ci 0 aryl or -(CH 2 J 2 C 6 -Ci 0 aryl, wherein said C 6 -C 10 aryl is optionally substituted with one or more R 13 ; and each R 13 is independently selected from C 1 -C 6 alkyl, -Cl, -F, -CN, -CF 3 , and -OCF 3 ; or a pharmaceutically acceptable salt or solvate thereof.
- R 1 , R 2 , and R 4 are hydrogen
- R 3 is C 1 -C 6 alkyl, -Cl, -F, -CN, -OCH 3 , -OCH 2 CH 3 , or -CF 3 ;
- R 5 is hydrogen;
- R 6 , R 7 , R 8 , and R 9 are hydrogen
- R 10 is hydrogen Or -CH 3 ;
- R 11 is -(CH 2 )C 6 -C 10 aryl or -(CH 2 ) 2 C 6 -Ci 0 aryl, wherein said C 6 -C 10 aryl is optionally substituted with one or more R 13 ; and each R 13 is independently selected from C 1 -C 6 alkyl, -Cl, -F, -CN, -CF 3 , and -OCF 3 ; or a pharmaceutically acceptable salt or solvate thereof.
- R 1 , R 2 , and R 4 are hydrogen;
- R 3 is -CH 3 , -OCH 3 or -OCH 2 CH 3 ;
- R 5 is hydrogen
- R 6 , R 7 , R 8 , and R 9 are hydrogen
- R 10 is hydrogen or -CH 3 ;
- R 11 is -(CH 2 )C 6 -C 10 aryl or -(CH 2 J 2 C 6 -C 10 aryl, wherein said C 6 -Ci 0 aryl is optionally substituted with one or more R 13 ; and each R 13 is independently selected from C 1 -C 6 alkyl, -Cl, -F, -CN, -CF 3 , and -OCF 3 ; or a pharmaceutically acceptable salt or solvate thereof.
- R 3 is -OCH 3 or -OCH 2 CH 3 ;
- R 5 is hydrogen
- R 6 , R 7 , R 8 , and R 9 are hydrogen
- R 10 is hydrogen or -CH 3 ;
- R 11 is -(CH 2 )C 6 -C 10 aryl or -(CH 2 J 2 C 6 -C 10 aryl, wherein said C 6 -C 10 aryl is optionally substituted with one or more substituents independently selected from -Cl and -F; or a pharmaceutically acceptable salt or solvate thereof.
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , and R 9 are hydrogen;
- R 10 is hydrogen or -CH 3 ;
- R 11 is -(CH 2 )C 6 -Ci 0 aryl or -(CH 2 )2C 6 -Ci 0 aryl, wherein said C 6 -C 10 aryl is optionally substituted with one or more R 13 ; and each R 13 is independently selected from C 1 -C 6 alky!, halogen, -CN, -CF 3 , and -OCF 3 ; or a pharmaceutically acceptable salt or solvate thereof.
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , R 9 , and R 10 are hydrogen;
- R 11 is -(CH 2 )C 6 -Ci 0 aryl or -(CH 2 JaC 6 -C 10 aryl, wherein said C 6 -C 10 aryl is optionally substituted with one or more R 13 ; and each R 13 is independently selected from C 1 -C 6 alkyl, halogen, -CN, -CF 3 , and -OCF 3 ; or a pharmaceutically acceptable salt or solvate thereof.
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , and R 9 are hydrogen; R 10 -CH 3 ;
- R 11 is -(CH 2 )C 6 -C 10 aryl or -(CHa) 2 C 6 -C 10 aryl, wherein said C 6 -C 10 aryl is optionally substituted with one or more R 13 ; and each R 13 is independently selected from C 1 -C 6 alkyl, halogen, -CN, -CF 3 , and
- R 1 , R 2 , R 3 , R 4 are independently selected from hydrogen, C 1 -C 6 alkyl, halo, -CN, -OR 12a , and -CF 3 ;
- R 5 is hydrogen
- R 6 , R 7 , R 8 , and R 9 are independently selected from hydrogen, Ci-C 6 alkyl,
- R 1 , R 2 , R 3 , R 4 are independently selected from hydrogen, C 1 -C 6 alkyl, halo, -CN, -OR 12a , and -CF 3 ;
- R 5 is hydrogen
- R 6 , R 7 , R 8 , and R 9 are hydrogen
- R 10 and R 11 together with the atom to which they are attached, form a C 2 -Ci 0 heterocyclyl group, optionally substituted with one or more R 13 ; each R 12a and R 12b is independently selected from hydrogen and C r C 6 alkyl; or
- R 12a and R 12b together with the atom to which they are attached, form a C 3 -C 1 - I cycloalkyl or a C 2 -C 10 heterocyclyl group; each R 13 is independently selected from C 1 -C 6 alkyl, halogen, -(CR 12a R 12b ) t CN, -(CR 12a R 12b )tCF 3 , -(CR 12a R 12b )t0CF 3 , -(CR 123 R 1 ⁇ ) 1 C 3 -C 11 cycloalkyl, -(CR 12a R 12b ) t C 6 -C 10 aryl, -(CR 12a R 12b ) t C 2 -C 10 heterocyclyl, -(CR 12a R 12b ),heteroaryl, -O(CR 12a R 12b ) t R 12a , -O(CR 12a R 12b ) t C 6 -C 10 aryl,
- R 1 , R 2 , R 3 , R 4 are independently selected from hydrogen, C 1 -C 6 alkyl, halo, -CN, -OR 12a , and -CF 3 ;
- R 5 is hydrogen
- R 6 , R 7 , R 8 , and R 9 are hydrogen; R 10 and R 11 , together with the atom to which they are attached, form a C 2 -C 10 heterocyclyl group, optionally substituted with one or more R 13 ; R 12a is hydrogen or C r C 6 alkyl; and each R 13 is independently selected from C 1 -C 6 alkyl, halogen, -CN, -CF 3 , and -OCF 3 ; or a pharmaceutically acceptable salt or solvate thereof.
- R 1 , R 3 , and R 4 are hydrogen
- R 2 is C 1 -C 6 alkyl, halo, -CN, -OR 12a , or -CF 3 ;
- R 5 is hydrogen
- R 6 , R 7 , R 8 , and R 9 are hydrogen; R 10 and R 11 , together with the atom to which they are attached, form a C 2 -C 10 heterocyclyl group, optionally substituted with one or more R 13 ;
- R 12a is hydrogen or C 1 -C 6 alkyl; and each R 13 is independently selected from C 1 -C 6 alkyl, halogen, -CN, -CF 3 , and -OCF 3 ; or a pharmaceutically acceptable salt or solvate thereof.
- R 1 , R 3 , and R 4 are hydrogen
- R 2 is C 1 -C 6 alkyl, -Cl, -F, -CN, -OCH 3 , -OCH 2 CH 3 , or -CF 3 ;
- R 5 is hydrogen;
- R 6 , R 7 , R 8 , and R 9 are hydrogen;
- R 10 and R 11 together with the atom to which they are attached, form a C 2 -C 10 heterocyclyl group, optionally substituted with one or more R 13 ; and each R 13 is independently selected from C 1 -C 6 alkyl, halogen, -CN, -CF 3 , and -OCF 3 ; or a pharmaceutically acceptable salt or solvate thereof.
- R 1 , R 2 , and R 4 are hydrogen
- R 3 is C 1 -C 6 alkyl, -Cl, -F, -CN, -OCH 3 , -OCH 2 CH 3 , or -CF 3 ;
- R 5 is hydrogen;
- R 6 , R 7 , R 8 , and R 9 are hydrogen;
- R 10 and R 11 together with th ee aattoomm ttoo wwhhiicchh they are attached, form a C 2 -C 10 heterocyclyl group, optionally substituted with one or more R 13 ; and eeaacchh RR 1133 iiss iinnddeeppeennddeennttllyy sseelleecctteedd ffrroomm CC 11 --CC 66 aallkkyyll,, r halogen, -CN, -CF 3 , and -OCF 3 ; or a pharmaceutically acceptable salt or solvate thereof.
- R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , and R 9 are hydrogen;
- R 10 and R 11 together with the atom to which they are attached, form a C 2 -C 10 heterocyclyl group, optionally substituted with one or more R 13 ; and each R 13 is independently selected from C 1 -C 6 alkyl, halogen, -CN, -CF 3 , and -OCF 3 ; or a pharmaceutically acceptable salt or solvate thereof.
- R 13 is independently selected from C 1 -C 6 alkyl, halogen, -CN, -CF 3 , and -OCF 3 ; or a pharmaceutically acceptable salt or solvate thereof.
- R 1 , R 2 , R 3 , R 4 are independently selected from hydrogen, C 1 -C 6 alkyl, -(CR 12a R 12b ) t halo,
- R 5 is hydrogen or Ci-C 6 alkyl
- R 6 , R 7 , R 8 , and R 9 are independently selected from hydrogen, C 1 -C 6 alkyl, -(CR 12a R 12b ) t halo, -(CR 12a R 12b ) t CN, -(CR 12a R 12b ),OR 12a , and -(CR 12a R 12b ) t N(R 12a R 12b );
- R 10 and R 11 are independently selected from hydrogen, alkyl,
- each R 10 and R 11 together with the atom to which they are attached, form a C 2 -C 10 heterocyclyl group, optionally substituted with at least one R 13 ;
- each R 12a and R 12b is independently selected from hydrogen and C 1 -C 6 alkyl; or R 12a and R 12b , together with the atom to which they are attached, form a C 3 -C 11 cycloalkyl group;
- each R 13 is independently selected from C 1 -C 6 alkyl, halogen, -O(CR 12a R 12b ) t R 12a , -O(CR 12a R 12b ) t C 6 -C 10 aryl, -O(CR 12a R 12b ) ,C 2 -C 10 heterocyclyl, -O(CR 12a R 12b ) t heteroaryl, -CO 2 (C 1 -
- C 1 -C 6 alkyl, Ce-C 10 aryl, C 2 -C 10 heterocyclyl, and heteroaryl groups is optionally substituted with at least one R 14 ; each R 14 is independently selected from C 1 -C 6 alkyl, halogen, and -OR 12a ; and each t is independently selected from 0, 1, 2, 3, 4, 5, and 6; or a pharmaceutically acceptable salt or solvate thereof.
- R 1 , R 2 , R 3 , R 4 are independently selected from hydrogen, C 1 -C 6 alkyl, -(CR 12a R 12b ) t halo, -(CR 12a R 12b ) t CN, -(CR 12a R 12b ) t OR 12a , and -(CR 12a R 12b ) t N(R 12a R 12b );
- R 5 is hydrogen or C 1 -C 6 alkyl;
- R 6 , R 7 , R 8 , and R 9 are independently selected from hydrogen, C 1 -C 6 alkyl, -(CR 12a R 12b ) t halo, -(CR 12a R 12b ),CN, -(CR 12a R 12b ) t OR 12a , and -(CR 12a R 12b ) t N(R 12a R 12b );
- R 10 and R 11 are independently selected from hydrogen, -(CR 12a R 12b )tC r C 6 alkyl, -(CR 1Za R 12b )tC ⁇ -Cii cycloalkyl, -(CR 12a R 12b ) t C 6 -C 10 aryl, -(C R 12a R 1 ⁇ ) 1 C 2 -C 10 heterocyclyl, and -(CR 12a R 12b ) t heteroaryl, wherein each of said C 1 -C 6 alkyl, C 3 -C 11 cycloalky
- each of said C 1 -C 6 alkyl, C 6 -C 10 aryl, C 2 -C 10 heterocyclyl, and heteroaryl groups is optionally substituted with at least one R 14 ; each R 14 is independently selected from C r C 6 alkyl, halogen, and -OR 12a ; and each t is independently selected from 0, 1 , 2, 3, 4, 5, and 6; or a pharmaceutically acceptable salt or solvate thereof.
- R 1 , R 2 , R 3 , R 4 , R 6 , R 7 , R 8 , and R 9 are hydrogen.
- R 5 is hydrogen.
- R 10 is hydrogen and R 11 is -(CR 12a R 12b ) t C r C 6 alkyl, -(CR 123 R 12 ⁇ C 3 -C 11 cycloalkyl, -(CR 12a R 12b ) t C 6 -C 10 aryl, -(CR 12a R 12b ) t C 2 -C 10 heterocyciyl, and -(CR 12a R 12b ) t heteroaryl, wherein each of said C 1 -C 6 alkyl, C 3 -C- I i cycloalkyl, C 6 -C 10 aryl, C 2 -C 10 heterocyclyl, and heteroaryl groups is optionally substituted with at least one R 13 .
- R 11 is selected from -(CR 12a R 12b ) t C r C 6 alkyl, -(CR 123 R 1 ⁇ ) 1 C 3 -C 11 cycloalkyl,
- R 11 is C 1 -C 6 alkyl and -(CH 2 )C 6 -C 10 aryl, wherein said C 1 -C 6 and aryl groups are optionally substituted with at least one substituent independently selected from C r C 6 alkyl, halogen, and -OR 12a .
- R 11 is C 1 -C 6 alkyl.
- R 10 and R 11 together with the atom to which they are attached, form a C 2 -Ci 0 heterocyclyl group, optionally substituted with at least one R 13 .
- compounds of formula (I) wherein said C 2 -C 10 heterocyclyl group is optionally substituted with at least one substituent independently selected from C 1 -C 6 alkyl, C 6 -C 10 aryl, -OH, -OCH 3 , heteroaryl, and -CO 2 (C 1 -C 6 alkyl), wherein said C 6 -
- C 10 aryl and heteroaryl groups are optionally substituted with at least one halogen or C 1 -C 6 alkyl group.
- compounds of formula (I), wherein said C 2 -C 10 heterocyclyl group is selected from 2,7-diazaspiro[3.5]non-7-yl, 2,7-diazaspiro[3.5]non-2-yl, piperazinyl, piperidinyl, morpholinyl, and pyrrolidinyl.
- compounds of formula (I) selected from N-[1,1- dimethyl-2-(4-methylpiperidin-1-yl)ethyl]-2-[(pyridin-3-ylamino)sulfonyl]benzamide; N-(1 ,1- dimethyl-2-piperidin-1-ylethyl)-2-[(pyridin-3-ylamino)sulfonyl]benzamide; N-(1,1-dimethyl-2- pyrrolidin-1-ylethyl)-2-[(pyridin-3-ylamino)sulfonyl]benzamide; N-(1-cyclopropyl-1-methylethyl)-2- [(pyridin-3-ylamino)sulfonyl]benzamide; N-(2-fluorobenzyl)-2-[(pyridin-3- ylamino)sulfonyl]benzamide; 2-(2,7-diazaspiro[3.5]non-7
- Another embodiment provides a compound selected from N-(3,4-difluorobenzyl)-2- ⁇ [(5- methoxypyridin-3-yl)amino]sulfonyl ⁇ benzamide; N-(3,4-difluorobenzyl)-2- ⁇ [(5-methylpyridin-3- yl)amino]sulfonyl ⁇ benzamide; N-(3,4-dichlorobenzyl)-2- ⁇ [(5-methylpyridin-3- yl)amino]sulfonyl ⁇ benzamide; N-[4-fluoro-3-(trifluoromethyl)benzyl]-2- ⁇ [(5-methylpyridin-3- yl)amino]sulfonyl ⁇ benzamide; N-(3,4-dichlorobenzyl)-2- ⁇ [(5-methoxypyridin-3- yl)amino]sulfonyl ⁇ benzamide; N-[4-fluoro-3
- a further embodiment provides a compound selected from N-(3,4-difluorobenzyl)-2- ⁇ [(4- methoxypyridin-3-yl)amino]sulfonyl ⁇ benzamide; N-(3,4-difluorobenzyl)-2- ⁇ [(4-methoxypyridin-3- yl)amino]sulfonyl ⁇ -N-methylbenzamide; N-[4-fluoro-3-(trifluoromethyl)benzyl]-2- ⁇ [(4- methoxypyridin-3-yl)amino]sulfonyl ⁇ benzamide; N-(3,4-difluorobenzyl)-2- ⁇ [(4-ethoxypyridin-3- yl)amino]sulfonyl ⁇ benzamide; N-(3,4-difluorobenzyl)-2- ⁇ [(4-ethoxypyridin-3-yl)amino]sulfonyl ⁇ benz
- Still another embodiment provides a compound selected from N-butyl-2- ⁇ [(4- methoxypyridin-3-yl)amino]sulfonyl ⁇ benzamide; N-(4-methoxypyridin-3-yl)-2-(piperidin-1- ylcarbonyl)benzenesulfonamide; N-butyl-2- ⁇ t(4-methoxypyridin-3-yl)amino]sulfonyl ⁇ -N- methylbenzamide; N-isopropyl-2- ⁇ [(4-methoxypyridin-3-yl)amino]sulfonyl ⁇ -N-methylbenzamide; 2- ⁇ [(4-methoxypyridin-3-yl)amino]sulfonyl ⁇ -N-methyl-N-pentylbenzamide; 2- ⁇ t(4-methoxypyridin-3- yl)amino]sulfonyl ⁇ -N-(3-methylbutyl)
- a still further embodiment provides a compound selected from 2- ⁇ [(5-methoxypyridin-3- yl)amino]sulfonyl ⁇ -N-[2-(2-methylphenyl)ethyl]benzamide; N-(3,4-difluorobenzyl)-2- ⁇ [(5- methoxypyridin-3-yl)amino]sulfonyl ⁇ benzamide; N-butyl-2- ⁇ [(5-methoxypyridin-3- yl)amino]sulfonyl ⁇ -N-methylbenzamide; 2- ⁇ [(5-methoxypyridin-3-yl)amino]sulfonyl ⁇ -N-methyl-N- pentylbenzamide; 2- ⁇ [(5-methoxypyridin-3-yl)amino]sulfonyl ⁇ -N-methyl-N-(3- methylbutyl)benzamide; N-(cyc!opropylmethyl)-2- ⁇ [
- Another embodiment provides a compound selected from N-(3,4-difluorobenzyl)-2- ⁇ [(5- methoxypyridin-3-yl)amino]sulfonyl ⁇ benzamide; N-[2-(2-methylphenyl)ethyl]-2- ⁇ [(5-methylpyridin- 3-yl)amino]sulfonyl ⁇ benzamide; N-(3,4-dichlorobenzyl)-2- ⁇ [(5-methylpyridin-3- yl)amino]sulfonyl ⁇ benzamide; N-[4-fluoro-3-(trifluoromethyl)benzyl]-2- ⁇ [(5-methylpyridin-3- yl)amino]sulfonyl ⁇ benzamide; N-[4-fluoro-3-(trifluoromethyl)benzyl]-2- ⁇ [(5-methoxypyridin-3- yl)amino]sulfonyl ⁇ benzamide; 2- ⁇
- Another embodiment provides N-(3,4-difluorobenzyl)-2- ⁇ [(4-methoxypyridin-3- yl)amino]sulfonyl ⁇ benzamide, or a pharmaceutically acceptable salt or solvate thereof.
- Another embodiment provides N-[2-(2-fluorophenyl)ethyl]-2- ⁇ [(5-methoxypyridin-3- yl)amino]sulfonyl ⁇ benzamide, or a pharmaceutically acceptable salt or solvate thereof.
- Another embodiment provides N-[4-fluoro-3-(trifluoromethyl)benzyl]-2- ⁇ [(4-methoxypyridin-3- yl)amino]sulfonyl ⁇ benzamide, or a pharmaceutically acceptable salt or solvate thereof.
- Another embodiment provides N-(3,4- difluorobenzyl)-2- ⁇ [(4-ethoxypyridin-3-yl)amino]sulfonyl ⁇ -N-methylbenzamide, or a pharmaceutically acceptable salt or solvate thereof.
- compositions comprising an effective amount of at least one compound of formula (I), and a pharmaceutically acceptable carrier.
- pharmaceutical compositions further comprising an effective amount of at least one compound that is metabolized by cytochrome P4503A4 enzyme.
- pharmaceutical compositions wherein said at least one compound that is metabolized by cytochrome P4503A4 enzyme is an anti-HIV compound.
- compositions comprising an effective amount of at least one compound that is metabolized by cytochrome P450 enzyme and an effective amount of any of the compounds of formula (I).
- at least one anti-HIV compound is an HIV protease inhibitor.
- HIV protease inhibitor is selected from amprenavir, CGP-73547, CGP-61755, DMP-450, nelfinavir, ritonavir, saquinavir, invirase, lopinavir, TMC-126, atazanavir, palinavir, GS-3333, KN 1-413, KNI-272, LG-71350, CGP-61755, PD 173606, PD 177298, PD 178390, PD 178392, U- 140690, ABT-378, DMP-450, AG-1776, MK-944, VX-478, indinavir, tipranavir, darunavir, brecanavir, DPC-681, DPC-684, (4R)-N-allyl-3- ⁇ (2S,3S)-2-hydroxy-3-[(3-hydroxy-2- methylbenzoyl)amino]-4-phenylbutanoyl ⁇
- HIV protease inhibitor is selected from amprenavir, nelfinavir, ritonavir, saquinavir, invirase, lopinavir, atazanavir, palinavir, indinavir, tipranavir, darunavir, brecanavir, brecanavir, (4R)-N-allyl-3- ⁇ (2S,3S)-2-hydroxy-3-[(3-hydroxy-2-methylbenzoyl)amino]-4-phenylbutanoyl ⁇ -5,5- dimethyl-1 ,3-thiazolidine-4-carboxamide, and fosamprenavir calcium.
- a further embodiment provides such pharmaceutical compositions, wherein said at least one additional compound is (4R)-N-allyl-3- ⁇ (2S,3S)-2-hydroxy-3-[(3-hydroxy-2-methylbenzoyl)amino]-4-phenylbutanoyl ⁇ -5,5- dimethyl-1 ,3-thiazolidine-4-carboxamide.
- compositions comprising an effective amount of (N- ⁇ (1 S)-3-[3-isopropyl-5-methyl-4H-1 ,2,4-triazole-4-yl]-exo-8-azabicyclo[3.2.1]oct-8- yl ⁇ -1-phenylpropyl)-4,4-difluorocyclohexanecarboxamide), ethyl 1-e ⁇ do- ⁇ 8-[(3S)-3-(acetylamino)- 3-(3-fluorophenyl)propyl]-8-azabicyclo[3.2.1]oct-3-yl ⁇ -2-methyl-4,5,6,7-tetrahydro-1H-imidazo[4,5- c]pyridine-5-carboxylate, or ⁇ /- ⁇ (1 S)-3-[3-encfo-(5-lsobutyryl-2-methyl-4,5,6,7-tetrahydro-1 H- imidazo[4,5-c]pyridin
- compositions comprising an effective amount of (N- ⁇ (1S)-3-[3- isopropyl-S-methyMH-i ⁇ -triazole ⁇ -yll-exo-S-azabicyclop ⁇ .iloct- ⁇ -ylJ-i-phenylpropylH ⁇ - difluorocyclohexanecarboxamide), at least compound of formula (I), and a pharmaceutically acceptable carrier.
- compositions comprising at least one compound of formula (I), and an anti-HIV compound as a combined preparation for simultaneous, separate, or sequential administration to an HIV-infected mammal for the treatment of HIV in said mammal.
- methods of inhibiting the metabolism of a first compound that is metabolized by cytochrome P4503A4 enzyme comprising administering said first compound and an effective amount of a second compound to said mammal, wherein said second compound is selected from those of formula (I).
- said first compound is 6-cyclopentyl-6-[2-(2,6-diethylpyridin-4-yl)ethyl]-3-[(5,7- dimethyl[1 ,2,4]triazolo[1 ,5-a]pyrimidin-2-yl)methyl]-4-hydroxy-5,6-dihydro-2H-pyran-2-one, or a pharmaceutically acceptable salt thereof.
- said first compound is (N- ⁇ (1S)-3-[3-isopropyl-5-methyl-4H-1,2,4-triazole-4-yl]-exo-8- azabicyclo[3.2.1]oct-8-yl ⁇ -1-phenylpropyl)-4,4-difluorocyclohexanecarboxamide), ethyl 1-endo- ⁇ 8- [(3S)-3-(acetylamino)-3-(3-fluorophenyl)propyl]-8-azabicyclo[3.2.1]oct-3-yl ⁇ -2-methyl-4,5,6,7- tetrahydro-1 H-imidazo[4,5-c]pyridine-5-carboxylate, or ⁇ /- ⁇ (1 S)-3-[3-encto-(5-lsobutyryl-2-methyl- 4,5,6,7-tetrahydro-1H-imidazo[4,5-c]pyridin-1-y
- said first compound is (N- ⁇ (1S)-3-[3-isopropyl-5-methyl-4H-1,2,4-triazole-4-yl]-exo-8-azabicyclo[3.2.1]oct- ⁇ -ylj-i-phenylpropyl ⁇ -difluorocyclohexanecarboxamide).
- said first compound is an anti-HIV compound.
- said anti-HIV compound is an HIV protease inhibitor.
- HIV protease inhibitor is selected from amprenavir, CGP-73547, CGP-61755, DMP-450, nelfinavir, ritonavir, saquinavir, invirase, lopinavir, TMC-126, atazanavir, palinavir, GS-3333, KN 1-413, KNI-272, LG-71350, CGP-61755, PD 173606, PD 177298, PD 178390, PD 178392, U-140690, ABT-378, DMP-450, AG-1776, MK- 944, VX-478, indinavir, tipranavir, darunavir, brecanavir, DPC-681 , DPC-684, (4R)-N-allyl-3- ⁇ (2S,3S)-2-hydroxy-3-[(3-hydroxy-2-methylbenzoyl)amino]-4-phenylbutanoyl ⁇ -5
- HIV protease inhibitor is selected from amprenavir, nelfinavir, ritonavir, saquinavir, invirase, lopinavir, atazanavir, palinavir, indinavir, tipranavir, darunavir, brecanavir, (4R)-N-allyl-3- ⁇ (2S,3S)-2-hydroxy-3-[(3-hydroxy-2-methylbenzoyl)amino]-4- phenylbutanoyl ⁇ -5,5-dimethyl-1,3-thiazolidine-4-carboxamide and fosamprenavir calcium.
- said first compound is (N- ⁇ (1 S)-3-[3- isopropyl-S-methyl ⁇ H-i ⁇ -triazole ⁇ -ylJ-exo- ⁇ -azabicyclop ⁇ .iJoct- ⁇ -ylJ-i-phenylpropyl) ⁇ - difluorocyclohexanecarboxamide).
- methods of improving the pharmacokinetics in a mammal of a first compound comprising administering said first compound and an effective amount of a second compound to said mammal, wherein said second compound is selected from those of formula (I).
- Another embodiment provides such methods, wherein said first compound is 6-cyclopentyl-6-[2-(2,6-diethylpyridin-4-yl)ethyl]-3-t(5,7-dimethyl[1,2,4]triazolo[1 ,5-a]pyrimidin- 2-yl)methyl]-4-hydroxy-5,6-dihydro-2H-pyran-2-one, or a pharmaceutically acceptable salt thereof.
- said first compound is (N- ⁇ (1S)-3-[3- isopropyl- ⁇ -methyMH-i ⁇ -triazole ⁇ -yll-exo- ⁇ -azabicyclofS ⁇ .iloct-S-ylJ-i-phenylpropylH ⁇ - difluorocyclohexanecarboxamide), ethyl 1-endo- ⁇ 8-[(3S)-3-(acetylamino)-3-(3- fluorophenyl)propyl]- ⁇ -azabicyclo[3.2.1]oct-3-yl ⁇ -2-methyl-4,5,6,7-tetrahydro-1/-/-imidazo[4,5- c]pyridine-5-carboxylate, or ⁇ /- ⁇ (1S)-3-[3-endo-(5-lsobutyryl-2-methyl-4,5,6,7-tetrahydro-1H- imidazo[4,5-c]pyridin-1-
- a further embodiment provides such methods, wherein said first compound is (N- ⁇ (1S)-3-[3- isopropyl- ⁇ -methyl ⁇ H-i ⁇ -triazole ⁇ -yll-exo- ⁇ -azabicyclop ⁇ . ⁇ oct- ⁇ -ylJ-i-phenylpropylH ⁇ - difluorocyclohexanecarboxamide).
- said first compound is an anti-HIV compound.
- said anti-HIV compound is an HIV protease inhibitor.
- HIV protease inhibitor is selected from amprenavir, CGP-73547, CGP-61755, DMP-450, nelfinavir, ritonavir, saquinavir, invirase, lopinavir, TMC-126, atazanavir, palinavir, GS-3333, KN 1-413, KNI-272, LG-71350, CGP-61755, PD 173606, PD 177296, PD 176390, PD 176392, U-140690, ABT-378, DMP-450, AG-1776, MK-944, VX-47 ⁇ , indinavir, tipranavir, darunavir, brecanavir, DPC-661, DPC-684, (4R)-N-allyl-3- ⁇ (2S,3S)-2- hydroxy-3-[(3-hydroxy-2-methylbenzoyl)amino]-4-phenylbutanoyl ⁇ -5,
- HIV protease inhibitor is selected from amprenavir, nelfinavir, ritonavir, saquinavir, invirase, lopinavir, atazanavir, palinavir, indinavir, tipranavir, darunavir, brecanavir, (4R)-N-allyl-3- ⁇ (2S,3S)-2-hydroxy-3-[(3-hydroxy-2-methylbenzoyl)amino]-4-phenylbutanoyl ⁇ -5,5-dimethyl-1 ,3- thiazolidine-4-carboxamide and fosamprenavir calcium.
- HIV protease inhibitor is (4R)-N-allyl-3- ⁇ (2S,3S)-2-hydroxy-3-[(3- hydroxy-2-methylbenzoyl)amino]-4-phenylbutanoyl ⁇ -5,5-dimethyl-1 ,3-thiazolidine-4-carboxamide.
- Another embodiment provides methods of inhibiting HIV replication in an HIV-infected mammal, comprising administering to said mammal an effective amount of a first compound and an effective amount of a second compound, wherein said first compound is an HIV replication- inhibiting compound and said second compound is selected from those of formula (I).
- Still another embodiment provides such methods, wherein said first compound is (N- ⁇ (1S)-3-[3- isopropyl-5-methyl-4H-1,2,4-triazole-4-yl]-exo-8-azabicyclo[3.2.1]oct-8-yl ⁇ -1-phenylpropyl)-4,4- difluorocyclohexanecarboxamide), ethyl 1-enc/o- ⁇ 8-[(3S)-3-(acetylamino)-3-(3- fluorophenyl)propyl]-8-azabicyclo[3.2.1]oct-3-yl ⁇ -2-methyl-4,5,6,7-tetrahydro-1H-imidazo[4,5- c]pyridine-5-carboxylate, or ⁇ /- ⁇ (1 S)-3-[3-endo-(5-lsobutyryl-2-methyl-4,5,6,7-tetrahydro-1 H- imidazo[4,5-c]pyridin-1-yl
- Yet another embodiment provides such methods, wherein said first compound is (N- ⁇ (1S)-3-[3- isopropyl-S-methyl ⁇ H-i ⁇ -triazole ⁇ -yll-exo-S-azabicyclop ⁇ .iJoct- ⁇ -ylJ-i-phenylpropylH ⁇ - difluorocyclohexanecarboxamide). Still another embodiment affords such methods, wherein said anti-HIV compound is an HIV protease inhibitor.
- a further embodiment provides such methods, wherein said HIV protease inhibitor is selected from amprenavir, CGP-73547, CGP-61755, DMP- 450, nelfinavir, ritonavir, saquinavir, invirase, lopinavir, TMC-126, atazanavir, palinavir, GS-3333, KN 1-413, KNI-272, LG-71350, CGP-61755, PD 173606, PD 177298, PD 178390, PD 178392, U- 140690, ABT-378, DMP-450, AG-1776, MK-944, VX-478, indinavir, tipranavir, darunavir, brecanavir, DPC-681, DPC-684, (4R)-N-allyl-3- ⁇ (2S,3S)-2-hydroxy-3-[(3-hydroxy-2- methylbenzoyl)amino]-4-phenylbutanoyl ⁇ -5,
- HIV protease inhibitor is selected from amprenavir, nelfinavir, ritonavir, saquinavir, invirase, lopinavir, atazanavir, palinavir, indinavir, tipranavir, darunavir, brecanavir, (4R)-N-allyl-3- ⁇ (2S,3S)-2- hydroxy-3-[(3-hydroxy-2-methylbenzoyl)amino]-4-phenylbutanoyl ⁇ -5,5-dimethyl-1 ,3-thiazolidine- 4-carboxamide, and fosamprenavir calcium.
- a further embodiment provides such methods, wherein said first compound is (4R)-N-allyl-3- ⁇ (2S,3S)-2-hydroxy-3-[(3-hydroxy-2- methylbenzoyl)amino]-4-phenylbutanoyl ⁇ -5,5-dimethyl-1 ,3-thiazolidine-4-carboxamide.
- Fruther provided herein are such uses, wherein said second compound is (N- ⁇ ISJ-S-P-isopropyl- ⁇ -methyMH-i ⁇ -triazole ⁇ -ylj-exo- ⁇ -azabicycloP ⁇ .IJoct- ⁇ -ylJ-i- phenylpropyl)-4,4-difluorocyclohexanecarboxamide), ethyl 1-encto- ⁇ 8-[(3S)-3-(acetylamino)-3-(3- fluorophenyl)propyl]-8-azabicyclo[3.2.1]oct-3-yl ⁇ -2-methyl-4,5,6,7-tetrahydro-1f/-imidazo[4,5- c]py ⁇ dine-5-carboxylate, or ⁇ /- ⁇ (1 S)-3-[3-encfo-(5-lsobutyryl-2-methyl-4,5,6,7-tetrahydro-1 H- imid
- a further embodiment affords such methods, wherein said second compound is (N- ⁇ (1S)-3-[3- isopropyl-S-methyMH-i ⁇ -triazole ⁇ -yll-exo- ⁇ -azabicyclop ⁇ .iJoct-S-ylJ-i-phenylpropylH ⁇ - difluorocyclohexanecarboxamide).
- said second compound is an HIV protease inhibitor.
- a further embodiment affords such uses, wherein said second compound is selected from amprenavir, CGP-73547, CGP-61755, DMP- 450, nelfinavir, ritonavir, saquinavir, invirase, lopinavir, TMC-126, atazanavir, palinavir, GS-3333, KN 1-413, KNI-272, LG-71350, CGP-61755, PD 173606, PD 177298, PD 178390, PD 178392, U- 140690, ABT-378, DMP-450, AG-1776, MK-944, VX-478, indinavir, tipranavir, darunavir, brecanavir, DPC-681 , DPC-684, (4R)-N-allyl-3- ⁇ (2S,3S)-2-hydroxy-3-[(3-hydroxy-2- methylbenzoyl)amino]-4-phenylbutanoyl ⁇ -5,5-
- Another embodiment provides such uses, wherein said second compound is selected from amprenavir, nelfinavir, ritonavir, saquinavir, invirase, lopinavir, atazanavir, palinavir, indinavir, tipranavir, darunavir, brecanavir, (4R)-N-allyl-3- ⁇ (2S,3S)-2- hydroxy-3-[(3-hydroxy-2-methylbenzoyl)amino]-4-phenylbutanoyl ⁇ -5,5-dimethyl-1,3-thiazolidine- 4-carboxamide, and fosamprenavir calcium.
- said second compound is selected from amprenavir, nelfinavir, ritonavir, saquinavir, invirase, lopinavir, atazanavir, palinavir, indinavir, tipranavir, darunavir, brecanavir, (4R)-N-allyl-3- ⁇ (2S
- a further embodiment provides such uses, wherein said second compound is (4R)-N-allyl-3- ⁇ (2S,3S)-2-hydroxy-3-[(3-hydroxy-2- methylbenzoyl)amino]-4-phenylbutanoyl ⁇ -5,5-dimethyl-1,3-thiazolidine-4-carboxamide. Further embodiments provide such uses, wherein said medicament is for simultaneous, separate, or sequential administration to said mammal for the treatment of HIV.
- said second compound is (N- ⁇ (1S)- S-p-isopropyl- ⁇ -methyMH-i ⁇ -triazole ⁇ -yll-exo- ⁇ -azabicyclotS ⁇ .iloct-S-ylJ-i-phenylpropyl)- 4,4-difluorocyclohexanecarboxamide), ethyl 1-e ⁇ c/o- ⁇ 8-[(3S)-3-(acetylamino)-3-(3- fluorophenyOpropyll- ⁇ -azabicyclotS ⁇ . ⁇ oct-S-ylJ ⁇ -methyM.S.ey-tetrahydro-IH-imidazo ⁇ .S- c]pyridine-5-carboxylate, or ⁇ /- ⁇ (1S)-3-[3-e ⁇ do-(5-lsobutyryl-2-methyl-4,5,6,7-tetrahydro-1H- imidazo[
- said second compound is (N- ⁇ (1S)-3-[3-isopropyl-5-methyl-4H- i ⁇ -triazole ⁇ -yll-exo- ⁇ -azabicyclo ⁇ .iJoct- ⁇ -ylJ-i-phenylpropyO-fM- difluorocyclohexanecarboxamide). Further provided are such uses, wherein said second compound is an HIV protease inhibitor.
- Another embodiment affords such uses, wherein said second compound is selected from amprenavir, CGP-73547, CGP-61755, DMP-450, nelfinavir, ritonavir, saquinavir, invirase, lopinavir, TMC-126, atazanavir, palinavir, GS-3333, KN 1-413, KNI- 272, LG-71350, CGP-61755, PD 173606, PD 177293, PD 17 ⁇ 390, PD 176392, U-140690, ABT- 378, DMP-450, AG-1776, MK-944, VX-47 ⁇ , indinavir, tipranavir, darunavir, brecanavir, DPC-661 , DPC-684, (4R)-N-allyl-3- ⁇ (2S,3S)-2-hydroxy-3-[(3-hydroxy-2-methylbenzoyl)amino]-4- phenylbutanoyl ⁇ -5,5-
- said second compound is selected from amprenavir, nelfinavir, ritonavir, saquinavir, invirase, lopinavir, atazanavir, palinavir, indinavir, tipranavir, darunavir, brecanavir, (4R)-N-allyl-3- ⁇ (2S,3S)-2-hydroxy-3-[(3-hydroxy-2-methyIbenzoyl)amino]- 4-phenylbutanoyl ⁇ -5,5-dimethyl-1 ,3-thiazolidine-4-carboxamide, and fosamprenavir calcium.
- Another embodiment provides such uses, wherein said second compound is (4R)-N-allyl-3- ⁇ (2S,3S)-2-hydroxy-3-[(3-hydroxy-2-methylbenzoyl)amino]-4-phenylbutanoyl ⁇ -5,5-dimethyl-1 ,3- thiazolidine-4-carboxamide.
- a first compound in the preparation of a medicament for improving the pharmacokinetics in a mammal of a second compound, wherein said first compound is selected from those according of formula (I), and said second compound is metabolized by cytochrome P450.
- said second compound is (N- ⁇ (1S)-3-t3-isopropyl-5-methyl-4H-1 ,2,4-triazole-4-yl]-exo-8-azabicyclo[3.2.1]oct- ⁇ -ylj-i-phenylpropyl ⁇ -difluorocyclohexanecarboxamide), ethyl 1-enofo- ⁇ 8-[(3S)-3-
- said second compound is (N- ⁇ (1S)-3-[3-isopropyl-5-methyl-4H-1 ,2,4-triazole-4-yl]-exo-8-azabicyclo[3.2.1]oct-8-yl ⁇ -1- phenylpropyl)-4,4-difluorocyclohexanecarboxamide). Further provided are such uses, wherein said second compound is an HlV protease inhibitor.
- Another embodiment affords such uses, wherein said second compound is selected from amprenavir, CGP-73547, CGP-61755, DMP- 450, nelfinavir, ritonavir, saquinavir, invirase, lopinavir, TMC-126, atazanavir, palinavir, GS-3333, KN 1-413, KNI-272, LG-71350, CGP-61755, PD 173606, PD 177298, PD 178390, PD 178392, U- 140690, ABT-378, DMP-450, AG-1776, MK-944, VX-478, indinavir, tipranavir, darunavir, brecanavir, DPC-681, DPC-684, (4R)-N-allyl-3- ⁇ (2S,3S)-2-hydroxy-3-[(3-hydroxy-2- methylbenzoyl)amino]-4-phenyIbutanoyl ⁇ -5,5-di
- said second compound is selected from amprenavir, nelfinavir, ritonavir, saquinavir, invirase, lopinavir, atazanavir, palinavir, indinavir, tipranavir, darunavir, brecanavir, (4R)-N-allyl-3- ⁇ (2S,3S)-2-hydroxy-3-[(3- hydroxy-2-methylbenzoyl)amino]-4-phenylbutanoyl ⁇ -5,5-dimethyl-1 ,3-thiazolidine-4-carboxamide, and fosamprenavir calcium.
- Another embodiment provides such uses, wherein said second compound is (4R)-N-allyl-3- ⁇ (2S,3S)-2-hydroxy-3-[(3-hydroxy-2-methylbenzoyl)amino]-4- phenylbutanoyl ⁇ -5,5-dimethyl-1 ,3-thiazolidine-4-carboxamide.
- methods of treating HIV in an HIV-infected mammal comprising administering to said mammal an effective amount of an anti-HIV compound and an effective amount of a compound according to formula (I).
- said anti-HIV compound is an HIV protease inhibitor.
- HIV protease inhibitor is selected from amprenavir, CGP-73547, CGP-61755, DMP-450, nelfinavir, ritonavir, saquinavir, invirase, lopinavir, TMC-126, atazanavir, palinavir, GS-3333, KN 1-413, KNI-272, LG-71350, CGP-61755, PD 173606, PD 177298, PD 178390, PD 178392, U-140690, ABT-378, DMP-450, AG-1776, MK- 944, VX-478, indinavir, tipranavir, darunavir, brecanavir, DPC-681 , DPC-684, (4R)-N-allyl-3- ⁇ (2S,3S)-2-hydroxy-3-[(3-hydroxy-2-methylbenzoyl)amino]-4-phenylbutanoyl ⁇ -5
- HIV protease inhibitor is selected from amprenavir, nelfinavir, ritonavir, saquinavir, invirase, lopinavir, atazanavir, palinavir, indinavir, tipranavir, darunavir, brecanavir, (4R)-N-allyl-3- ⁇ (2S,3S)-2-hydroxy-3-[(3-hydroxy-2-methylbenzoyl)amino]-4- phenylbutanoyl ⁇ -5,5-dimethyl-1 ,3-thiazolidine-4-carboxamide, and fosamprenavir calcium.
- HIV protease inhibitor is (4R)-N-allyl- 3- ⁇ (2S,3S)-2-hydroxy-3-[(3-hydroxy-2-methylbenzoyl)amino]-4-phenylbutanoyl ⁇ -5,5-dimethyl-1,3- thiazolidine-4-carboxamide.
- the anti-HlV compound is (N- ⁇ (1S)-3-[3-isopropyl-5-methyl-4H-1,2,4-triazole-4-yl]-exo-8- azabicyclo[3.2.1]oct-8-yl ⁇ -1-phenylpropyl)-4,4-difluorocyclohexanecarboxamide), ethyl 1-enc/o- ⁇ 8- [(3S)-3-(acetylamino)-3-(3-fluorophenyl)propyl]-8-azabicyclo[3.2.1]oct-3-yl ⁇ -2-methyl-4,5,6,7- tetrahydro-1 H-imidazo[4,5-c]pyridine-5-carboxylate, or ⁇ /- ⁇ (1 S)-3-[3-endo-(5-Isobutyryl-2-methyl- 4,5,6,7-tetrahydro-1f/-imidazo[4,5-
- anti-HIV compound is (N- ⁇ (1S)-3-[3-isopropyl-5-methyl-4H-1 ,2,4-triazole-4-yl]-exo-8- azabicyclo[3.2.1]oct-8-yl ⁇ -1-phenylpropyl)-4,4-difluorocyclohexanecarboxamide).
- the first compound is 6- cyclopentyl-6-[2-(2,6-diethylpyridin-4-yl)ethyl]-3-[(5,7-dimethyl[1,2,4]triazolo[1 ,5-a]pyrimidin-2- y!methyl]-4-hydroxy-5,6-dihydro-2H-pyran-2-one.
- HIV Human Immunodeficiency Virus.
- HIV integrase means the Human Immunodeficiency Virus integrase enzyme.
- C 1 -C 6 alkyl means saturated monovalent hydrocarbon radicals having straight or branched moieties and containing from 1 to 6 carbon atoms. Examples of such groups include, but are not limited to, methyl, ethyl, propyl, iso-propyl, n-butyl, iso-butyl, and tert-butyl.
- C 3 -C 1 1 cycloalkyl means a saturated, monocyclic, fused, or spiro, polycyclic ring structure having a total of from 3 to 11 carbon ring atoms.
- Examples of such groups include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclopentenyl, cyclohexyl, cycloheptyl, and adamantyl.
- C 6 -Ci 0 ar yl means a group derived from an aromatic hydrocarbon containing from 6 to 10 carbon atoms. Examples of such groups include, but are not limited to, phenyl or naphthyl.
- C 2 -Ci 0 heterocyclyl as used herein, means a non-aromatic, monocyclic, bicyclic, tricyclic, or tetracyclic group having a total of from 4 to 10 atoms in its ring system, and containing from 2 to 10 carbon atoms and from one to four heteroatoms each independently selected from O, S and N, and with the proviso that the ring of said group does not contain two adjacent O atoms or two adjacent S atoms.
- C 2 -C 10 heterocyclyl groups may comprise polycyclic, spiro ring systems. Also, such groups may be optionally benzofused.
- C 2 -Ci 0 heterocyclyl groups may contain an oxo substituent at any available atom that will result in a stable compound.
- a group may contain an oxo atom at an available carbon or nitrogen atom.
- Such a group may contain more than one oxo substituent if chemically feasible.
- An example of a 4-membered heterocyclic group is azetidinyl (derived from azetidine).
- C 2 -Ci 0 heterocyclyl groups include, but are not limited to, pyrrolidinyl, tetrahydrofuranyl, dihydrofuranyl, tetrahydrothienyl, tetrahydropyranyl, dihydropyranyl, tetrahydrothiopyranyl, piperidino, morpholino, thiomorpholino, thioxanyl, piperazinyl, azetidinyl, oxetanyl, thietanyl, homopiperidinyl, oxepanyl, thiepanyl, oxazepinyl, diazepinyl, thiazepinyl, 1 ,2,3,6-tetrahydropyridinyl, 2-pyrrolinyl,
- heteroaryl means an aromatic heterocyclic group having a total of from 5 to 10 atoms in its ring, and containing from 2 to 9 carbon atoms and from one to four heteroatoms each independently selected from O, S and N, and with the proviso that the ring of said group does not contain two adjacent O atoms or two adjacent S atoms.
- the heterocyclic groups include benzo-fused ring systems.
- aromatic heterocyclic groups are pyridinyl, imidazolyl, pyrimidinyl, pyrazolyl, triazolyl, pyrazinyl, tetrazolyl, furyl, thienyl, isoxazolyl, thiazolyl, oxazolyl, isothiazolyl, pyrrolyl, quinolinyl, isoquinolinyl, indolyl, benzimidazolyl, benzofuranyl, cinnolinyl, indazolyl, indolizinyl, phthalazinyl, pyridazinyl, triazinyl, isoindolyl, pteridinyl, purinyl, oxadiazolyl, thiadiazolyl, furazanyl, benzofurazanyl, benzothiophenyl, benzothiazolyl, benzoxazolyl, quinazolinyl, quinox
- heteroaryl groups may be C-attached or N-attached where such is possible.
- a group derived from pyrrole may be pyrrol-1-yl (N-attached) or pyrrol-3-yl (C-attached).
- a group derived from imidazole may be imidazol-1-yl (N-attached) or imidazol-3-yl (C-attached).
- halogen and "halo,” as used herein, mean fluorine, chlorine, bromine or iodine.
- substituted means that the specified group or moiety bears one or more substituents.
- unsubstituted means that the specified group bears no substituents.
- optionally substituted means that the specified group is unsubstituted or substituted by one or more substituents. It is to be understood that in the compounds of the present invention when a group is said to be “unsubstituted,” or is “substituted” with fewer groups than would fill the valencies of all the atoms in the compound, the remaining valencies on such a group are filled by hydrogen.
- a C 6 aryl group also called “phenyl” herein
- phenyl is substituted with one additional substituent
- one of ordinary skill in the art would understand that such a group has 4 open positions left on carbon atoms of the C 6 aryl ring (6 initial positions, minus one to which the remainder of the compound of the present invention is bonded, minus an additional substituent, to leave 4). In such cases, the remaining 4 carbon atoms are each bound to one hydrogen atom to fill their valencies.
- a C 6 aryl group in the present compounds is said to be "disubstituted," one of ordinary skill in the art would understand it to mean that the C 6 aryl has 3 carbon atoms remaining that are unsubstituted. Those three unsubstituted carbon atoms are each bound to one hydrogen atom to fill their valencies.
- solvate means a pharmaceutically acceptable solvate form of a compound of the present invention that retains the biological effectiveness of such compound.
- solvates include, but are not limited to, compounds of the invention in combination with water, isopropanol, ethanol, methanol, dimethylsulfoxide (DMSO), ethyl acetate, acetic acid, ethanolamine, or mixtures thereof.
- DMSO dimethylsulfoxide
- ethyl acetate acetic acid
- ethanolamine or mixtures thereof.
- one solvent molecule can be associated with one molecule of the compounds of the present invention, such as a hydrate.
- more than one solvent molecule may be associated with one molecule of the compounds of the present invention, such as a dihydrate.
- solvates of the present invention are contemplated as solvates of compounds of the present invention that retain the biological effectiveness of the non-hydrate form of the compounds.
- cytochrome P450-inhibiting amount and “cytochrome P450 enzyme activity-inhibiting amount,” as used herein, refer to an amount of a compound required to decrease the activity of cytochrome P450 enzymes or a particular cytochrome P450 enzyme isoform in the presence of such compound. Whether a particular compound of decreases cytochrome P450 enzyme activity, and the amount of such a compound required to do so, can be determined by methods known to those of ordinary skill in the art and the methods described herein.
- inhibitors refer to decreasing the activity of a cytochrome P450 enzyme or enzymes using an agent that is capable of decreasing such activity either in vitro or in vivo after administration to a mammal, such as a human. Such inhibition may take place by the compound binding directly to the cytochrome P450 enzyme or enzymes. In addition, the activity of such cytochrome P450 enzymes may be decreased in the presence of such a compound when such direct binding between the enzyme and the compound does not take place. Furthermore, such inhibition may be competitive, non-competitive, or uncompetitive, as described in T.F. Woolf, Handbook of Drug Metabolism, Marcel Dekker, Inc., New York, 1999. Such inhibition may be determined using in vitro or in vivo systems, or a combination of both, using methods known to those of ordinary skill in the art.
- bioavailability refers to the systemic availability of a given amount of a chemical compound administered to a mammal. Bioavailability can be assessed by measuring the area under the curve (AUC) or the maximum serum or plasma concentration (C max ) of the unchanged form of a compound following administration of the compound to a mammal. AUC is a determination of the Area Under the Curve plotting the serum or plasma concentration of a compound along the ordinate (Y-axis) against time along the abscissa (X- axis). Generally, the AUC for a particular compound can be calculated using methods known to those of ordinary skill in the art and as described in G.S. Banker, Modern Pharmaceutics. Drugs and the Pharmaceutical Sciences, v.
- the C max value is defined as the maximum concentration of the compound achieved in the serum or plasma of a mammal following administration of the compound to the mammal.
- the C max value of a particular compound can be measured using methods known to those of ordinary skill in the art.
- the phrases "increasing bioavailability" or “improving the pharmacokinetics,” as used herein mean that the systemic availability of a first compound, measured as AUC, C max , or C min in a mammal is greater when co-administered with a compound of the present invention than when such coadministration does not take place.
- administration refers to the delivery of a compound, or a pharmaceutically acceptable salt or solvate thereof, or of a pharmaceutical composition containing the compound, or a pharmaceutically acceptable salt or solvate thereof, to a mammal such that the compound is absorbed into the serum or plasma of the mammal.
- co-administration refers to the administration of a combination of a first compound and a compound of the present invention, or a pharmaceutically acceptable salt or solvate thereof. Such co-administration can be performed such that the first compound and the compound of the present invention are part of the same composition or part of the same unitary dosage form. Co-administration also includes administering a first compound and a compound of the present invention separately, but as part of the same therapeutic regimen. The two components, if administered separately, need not necessarily be administered at essentially the same time, although they can be if so desired. Thus co-administration includes, for example, administering a first compound and a compound of the present invention as separate dosages or dosage forms, but at the same time. Coadministration also includes separate administration at different times and in any order.
- pharmaceutically acceptable formulation or “pharmaceutical composition,” as used herein, mean a combination of a compound of the invention, or a pharmaceutically acceptable salt or solvate thereof, and a carrier, diluent, and/or excipients that are compatible with a compound of the present invention, and is not deleterious to the recipient thereof.
- Pharmaceutical formulations can be prepared by procedures known to those of ordinary skill in the art.
- the compounds of the present invention can be formulated with common excipients, diluents, or carriers, and formed into tablets, capsules, and the like.
- excipients, diluents, and carriers that are suitable for such formulations include the following: fillers and extenders such as starch, sugars, mannitol, and silicic derivatives; binding agents such as carboxymethyl cellulose and other cellulose derivatives, alginates, gelatin, and polyvinyl pyrrolidone; moisturizing agents such as glycerol; disintegrating agents such as povidone, sodium starch glycolate, sodium carboxymethylcellulose, agar agar, calcium carbonate, and sodium bicarbonate; agents for retarding dissolution such as paraffin; resorption accelerators such as quaternary ammonium compounds; surfacelactive agents such as cetyl alcohol, glycerol monostearate; adsorptive carriers such as kaolin and bentonite; and lubricants such as talc, calcium and magnesium stearate and solid polyethylene glycols.
- fillers and extenders such as starch, sugars, mannitol, and silicic derivatives
- Final pharmaceutical forms may be pills, tablets, powders, lozenges, saches, cachets, or sterile packaged powders, and the like, depending on the type of excipient used. Additionally, it is specifically contemplated that pharmaceutically acceptable formulations of the present invention can contain more than one active ingredient. For example, such formulations may contain more than one compound according to the present invention. Alternatively, such formulations may contain one or more compounds of the present invention and one or more additional anti-HIV agents.
- inhibiting HIV replication means inhibiting human immunodeficiency virus (HIV) replication in a ceil.
- a cell may be present in vitro, or it may be present in vivo, such as in a mammal, such as a human.
- Such inhibition may be accomplished by administering one or more anti-HIV compounds to the cell, such as in a mammal, in an HIV-inhibiting amount.
- the quantification of inhibition of HlV replication in a cell, such as in a mammal can be measured using methods known to those of ordinary skill in the art.
- an anti-HIV compound may be administered to a mammal, either alone or as part of a pharmaceutically acceptable formulation.
- Blood samples may then be withdrawn from the mammal and the amount of HIV virus in the sample may be quantified using methods known to those of ordinary skill in the art.
- a reduction in the amount of HIV virus in the sample compared to the amount found in the blood before administration of one or more anti-HIV compounds would represent inhibition of the replication of HIV virus in the mammal.
- the administration of one or more anti-HIV compounds to the cell, such as in a mammal may be in the form of single dose or a series of doses. In the case of more than one dose, the doses may be administered in one day or they may be administered over more than one day.
- anti-HIV compound and "HIV-inhibiting agent,” as used herein, mean a compound or combination of compounds capable of inhibiting the replication of HIV in a cell, such as a cell in a mammal. Such compounds may inhibit the replication of HIV through any mechanism known to those of ordinary skill in the art.
- human immunodeficiency virus-inhibiting amount refers to the amount of an anti-HIV compound, or a pharmaceutically acceptable salt of solvate thereof, required to inhibit replication of the human immunodeficiency virus (HIV) in vivo, such as in a mammal, or in vitro.
- the amount of such compounds required to cause such inhibition can be determined without undue experimentation using methods described herein and those known to those of ordinary skill in the art.
- therapeutically effective amount or “effective amount,” as used herein, means an amount of a compound that, when administered to a mammal in need of such treatment, is sufficient to effect treatment, as defined herein.
- a therapeutically effective amount or effective amount of a compound is a quantity sufficient to modulate or inhibit the activity of a particular enzyme target or biological process such that a disease condition that is mediated by activity of such an enzyme target or biological process is reduced or alleviated.
- modulation or inhibition of a particular target enzyme target or biological process include, but are not limited to, inhibition of the HIV protease enzyme and inhibition of the CYP450 enzymes, such as the CYP3A4 enzyme.
- treat refers to any treatment of any disease or condition in a mammal, particularly a human, and include: (i) preventing the disease or condition from occurring in a subject which may be predisposed to the condition, such that the treatment constitutes prophylactic treatment for the pathologic condition; (ii) modulating or inhibiting the disease or condition, i.e., arresting its development; (iii) relieving the disease or condition, i.e., causing regression of the disease or condition; or (iv) relieving and/or alleviating the disease or condition or the symptoms resulting from the disease or condition, e.g., relieving an inflammatory response without addressing the underlying disease or condition.
- resistant refers to HIV virus demonstrating a reduction in sensitivity to a particular drug.
- a mammal infected with HIV that is resistant to a particular anti-HIV agent or combination of agents usually manifests an increase in HIV viral load despite continued administration of the agent or agents.
- Resistance may be either genotypic, meaning that a mutation in the HIV genetic make-up has occurred, or phenotypic, meaning that resistance is discovered by successfully growing laboratory cultures of HIV virus in the presence of an anti-HIV agent or a combination of such agents.
- protease inhibitor refers to compounds or combinations of compounds that interfere with the proper functioning of the HIV protease enzyme that is responsible for cleaving long strands of viral protein into the separate proteins making up the viral core.
- HIV load and "HIV viral load,” as used herein, mean the amount of HIV in the circulating blood of a mammal, such as a human.
- the amount of HIV virus in the blood of mammal can be determined by measuring the quantity of HIV RNA in the blood using methods known to those of ordinary skill in the art.
- compound of the present invention refers to any compound of formula (I), including those in the Examples that follow, and also include those generically described or those described as species.
- the term also refers to pharmaceutically acceptable salts or solvates of these compounds.
- the compounds of the present invention may be administered to a mammal, such as a human, in combination with an additional compound so that there is an increase of the exposure, or an increase in the bioavailability of the additional compound, or an improvement in the pharmacokinetics, of the additional compound in the mammal.
- exposure refers to the concentration of an additional or second compound in the plasma of a mammal as measured over a period of time.
- An increase of the exposure of a mammal to an additional or second compound can be measured by first administering the additional or second compound to a mammal in an appropriate form and in the absence of the administration of a compound of the invention, withdrawing plasma samples at predetermined times, and measuring the amount of the compound in the plasma using an appropriate analytical technique, such as liquid chromatography or liquid chromatography-tandem mass spectroscopy. The same study is then repeated, except that a compound of the present invention is co-administered with the additional or second compound. The amount of the additional or second compound present in the plasma at a certain time is determined and the concentration and time data from all the samples are plotted to afford a curve. The area under this curve is calculated and affords the exposure of the mammal to the compound.
- Such co-administration to a mammal of a compound of the present invention and a second or additional compound, as described above, may occur such that a compound or compounds of the present invention are present in the same formulation as the additional agents described above.
- a combination may be administered such that the compound or compounds of the present invention are present in a formulation that is separate from the formulation in which the additional agent is found. If the compound or compounds of the present invention are administered separately from the additional agent, such administration may take place at the same or sequentially with an appropriate period of time in between.
- the choice of whether to include the compound or compounds of the present invention in the same formulation as the additional agent or agents is within the knowledge of one of ordinary skill in the art.
- ⁇ represents a methyl group
- ⁇ CH 3 represents an ethyl group
- stereoisomers refers to compounds that have identical chemical constitution, but differ with regard to the arrangement of their atoms or groups in space.
- enantiomers refers to two stereoisomers of a compound that are non-superimposable mirror images of one another.
- racemic or “racemic mixture,” as used herein, refer to a 1 :1 mixture of enantiomers of a particular compound.
- diastereomers refers to the relationship between a pair of stereoisomers that comprise two or more asymmetric centers and are not mirror images of one another.
- the compounds of the present invention may have asymmetric carbon atoms.
- the bonds of the compounds of the present invention may be depicted herein using a solid line ( ), a solid wedge ( """ ⁇ - ), or a dotted wedge ( ""HI ).
- the use of a solid line to depict bonds from asymmetric carbon atoms is meant to indicate that all possible stereoisomers at that carbon atom are included.
- the use of either a solid or dotted wedge to depict bonds from asymmetric carbon atoms is meant to indicate that only the stereoisomer shown is meant to be included. It is possible that compounds of the invention may contain more than one asymmetric carbon atom.
- a solid line to depict bonds from asymmetric carbon atoms is meant to indicate that all possible stereoisomers are meant to be included.
- the use of a solid line to depict bonds from one or more asymmetric carbon atoms in a compound of the invention and the use of a solid or dotted wedge to depict bonds from other asymmetric carbon atoms in the same compound is meant to indicate that a mixture of diastereomers is present.
- a desired salt may be prepared by any suitable method known to the art, including treatment of the free base with an inorganic acid, such as hydrochloric acid; hydrobromic acid; sulfuric acid; nitric acid; phosphoric acid; and the like, or with an organic acid, such as acetic acid; maleic acid; succinic acid; mandelic acid; fumaric acid; malonic acid; pyruvic acid; oxalic acid; glycolic acid; salicylic acid; pyranosidyl acid, such as glucuronic acid or galacturonic acid; alpha-hydroxy acid, such as citric acid or tartaric acid; amino acid, such as aspartic acid or glutamic acid; aromatic acid, such as benzoic acid or cinnamic acid; sulfonic acid, such as p-toluenesulfonic acid or ethanesulfonic acid; and the like.
- an inorganic acid such as hydrochloric acid; hydrobromic acid; sulfuric acid;
- a desired salt may be prepared by any suitable method known to the art, including treatment of the free acid with an inorganic or organic base, such as an amine (primary, secondary, or tertiary); an alkali metal or alkaline earth metal hydroxide; or the like.
- suitable salts include organic salts derived from amino acids such as glycine and arginine; ammonia; primary, secondary, and tertiary amines; and cyclic amines, such as piperidine, morpholine, and piperazine; as well as inorganic salts derived from sodium, calcium, potassium, magnesium, manganese, iron, copper, zinc, aluminum, and lithium.
- solvate is intended to mean a pharmaceutically acceptable solvate form of a specified compound that retains the biological effectiveness of such compound.
- solvates include, but are not limited to, compounds of the invention in combination with water, isopropanol, ethanol, methanol, dimethylsulfoxide (DMSO), N,N-dimethylformamide (DMF), ethyl acetate, acetic acid, ethanolamine, or mixtures thereof.
- a "pharmaceutically acceptable salt” is intended to mean a salt that retains the biological effectiveness of the free acids and bases of the specified derivative, containing pharmacologically acceptable anions, and is not biologically or otherwise undesirable.
- pharmaceutically acceptable salts include, but are not limited to, acetate, acrylate, benzenesulfonate, benzoate (such as chlorobenzoate, methylbenzoate, dinitrobenzoate, hydroxybenzoate, and methoxybenzoate), bicarbonate, bisulfate, bisulfite, bitartrate, borate, bromide, butyne-1 ,4-dioate, calcium edetate, camsylate, carbonate, chloride, caproate, caprylate, clavulanate, citrate, decanoate, dihydrochloride, dihydrogenphosphate, edetate, edislyate, estolate, esylate, ethylsuccinate, formate, fumarate, glu
- the compounds of the present invention that are basic in nature are capable of forming a wide variety of different salts with various inorganic and organic acids. Although such salts must be pharmaceutically acceptable for administration to animals, it is often desirable in practice to initially isolate the compound of the present invention from the reaction mixture as a pharmaceutically unacceptable salt and then simply convert the latter back to the free base compound by treatment with an alkaline reagent and subsequently convert the latter free base to a pharmaceutically acceptable acid addition salt.
- the acid addition salts of the base compounds of this invention can be prepared by treating the base compound with a substantially equivalent amount of the selected mineral or organic acid in an aqueous solvent medium or in a suitable organic solvent, such as methanol or ethanol. Upon evaporation of the solvent, the desired solid salt is obtained.
- the desired acid salt can also be precipitated from a solution of the free base in an organic solvent by adding an appropriate mineral or organic acid to the solution.
- Those compounds of the present invention that are acidic in nature are capable of forming base salts with various pharmacologically acceptable cations.
- such salts include the alkali metal or alkaline-earth metal salts and particularly, the sodium and potassium salts. These salts are all prepared by conventional techniques.
- the chemical bases which are used as reagents to prepare the pharmaceutically acceptable base salts of this invention are those which form nontoxic base salts with the acidic compounds of the present invention.
- Such non-toxic base salts include those derived from such pharmacologically acceptable cations as sodium, potassium calcium and magnesium, etc.
- salts can be prepared by treating the corresponding acidic compounds with an aqueous solution containing the desired pharmacologically acceptable cations, and then evaporating the resulting solution to dryness, preferably under reduced pressure.
- they may also be prepared by mixing lower alkanolic solutions of the acidic compounds and the desired alkali metal alkoxide together, and then evaporating the resulting solution to dryness in the same manner as before.
- stoichiometric quantities of reagents are preferably employed in order to ensure completeness of reaction and maximum yields of the desired final product.
- the desired pharmaceutically acceptable salt may be prepared by any suitable method available in the art, for example, treatment of the free base with an inorganic acid, such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid and the like, or with an organic acid, such as acetic acid, maleic acid, succinic acid, mandelic acid, fumaric acid, malonic acid, pyruvic acid, oxalic acid, glycolic acid, salicylic acid, a pyranosidyl acid, such as glucuronic acid or galacturonic acid, an alpha-hydroxy acid, such as citric acid or tartaric acid, an amino acid, such as aspartic acid or glutamic acid, an aromatic acid, such as benzoic acid or cinnamic acid, a sulfonic acid, such as p-toluenesulfonic acid or ethanesulfonic acid, or the like.
- an inorganic acid such as hydrochloric acid
- the desired pharmaceutically acceptable salt may be prepared by any suitable method, for example, treatment of the free acid with an inorganic or organic base, such as an amine (primary, secondary or tertiary), an alkali metal hydroxide or alkaline earth metal hydroxide, or the like.
- suitable salts include organic salts derived from amino acids, such as glycine and arginine, ammonia, primary, secondary, and tertiary amines, and cyclic amines, such as piperidine, morpholine and piperazine, and inorganic salts derived from sodium, calcium, potassium, magnesium, manganese, iron, copper, zinc, aluminum and lithium.
- compositions of the invention comprise a therapeutically effective amount of at least one compound of the present invention and an inert, pharmaceutically acceptable carrier or diluent.
- the pharmaceutical carriers employed may be either solid or liquid.
- Exemplary solid carriers are lactose, sucrose, talc, gelatin, agar, pectin, acacia, magnesium stearate, stearic acid and the like.
- Exemplary liquid carriers are syrup, peanut oil, olive oil, water and the like.
- the inventive compositions may include time-delay or time-release material known in the art, such as glyceryl monostearate or glyceryl distearate alone or with a wax, ethylcellulose, hydroxypropylmethylcellulose, methylmethacrylate or the like. Further additives or excipients may be added to achieve the desired formulation properties.
- a bioavailability enhancer such as Labrasol ® , Gelucire ® or the like, or formulator, such as CMC (carboxy- methylcellulose), PG (propyleneglycol), or PEG (polyethyleneglycol), may be added.
- CMC carboxy- methylcellulose
- PG propyleneglycol
- PEG polyethyleneglycol
- a semi-solid vehicle that protects active ingredients from light, moisture and oxidation may be added, e.g., when preparing a capsule formulation.
- the preparation can be tableted, placed in a hard gelatin capsule in powder or pellet form, or formed into a troche or lozenge.
- the amount of solid carrier may vary, but generally will be from about 25 mg to about 1 g.
- the preparation may be in the form of syrup, emulsion, soft gelatin capsule, sterile injectable solution or suspension in an ampoule or vial or non-aqueous liquid suspension.
- a semi-solid carrier is used, the preparation may be in the form of hard and soft gelatin capsule formulations.
- the inventive compositions are prepared in unit-dosage form appropriate for the mode of administration, e.g., parenteral or oral administration.
- a pharmaceutically acceptable salt of a compound of the present invention may be dissolved in an aqueous solution of an organic or inorganic acid, such as 0.3 M solution of succinic acid or citric acid.
- the agent may be dissolved in a suitable cosolvent or combinations of cosolvents.
- suitable cosolvents include alcohol, propylene glycol, polyethylene glycol 300, polysorbate 80, glycerin and the like in concentrations ranging from 0-60% of the total volume.
- a compound of Formula I is dissolved in DMSO and diluted with water.
- the composition may also be in the form of a solution of a salt form of the active ingredient in an appropriate aqueous vehicle such .as water or isotonic saline or dextrose solution.
- the agents of the compounds of the present invention may be formulated into aqueous solutions, preferably in physiologically compatible buffers such as Hanks solution, Ringer's solution, or physiological saline buffer.
- physiologically compatible buffers such as Hanks solution, Ringer's solution, or physiological saline buffer.
- penetrants appropriate to the barrier to be permeated are used in the formulation. Such penetrants are generally known in the art.
- the compounds can be formulated by combining the active compounds with pharmaceutically acceptable carriers known in the art.
- Such carriers enable the compounds of the invention to be formulated as tablets, pills, dragees, capsules, liquids, gels, syrups, slurries, suspensions and the like, for oral ingestion by a subject to be treated.
- Pharmaceutical preparations for oral use can be obtained using a solid excipient in admixture with the active ingredient (agent), optionally grinding the resulting mixture, and processing the mixture of granules after adding suitable auxiliaries, if desired, to obtain tablets or dragee cores.
- Suitable excipients include: fillers such as sugars, including lactose, sucrose, mannitol, or sorbitol; and cellulose preparations, for example, maize starch, wheat starch, rice starch, potato starch, gelatin, gum, methyl cellulose, hydroxypropylmethyl-cellulose, sodium carboxymethylcellulose, or polyvinylpyrrolidone (PVP).
- disintegrating agents may be added, such as crosslinked polyvinyl pyrrolidone, agar, or alginic acid or a salt thereof such as sodium alginate.
- Dragee cores are provided with suitable coatings.
- concentrated sugar solutions may be used, which may optionally contain gum arabic, polyvinyl pyrrolidone, Carbopol gel, polyethylene glycol, and/or titanium dioxide, lacquer solutions, and suitable organic solvents or solvent mixtures.
- Dyestuffs or pigments may be added to the tablets or dragee coatings for identification or to characterize different combinations of active agents.
- compositions that can be used orally include push-fit capsules made of gelatin, as well as soft, sealed capsules made of gelatin and a plasticizer, such as glycerol or sorbitol.
- the push-fit capsules can contain the active ingredients in admixture with fillers such as lactose, binders such as starches, and/or lubricants such as talc or magnesium stearate, and, optionally, stabilizers.
- the active agents may be dissolved or suspended in suitable liquids, such as fatty oils, liquid paraffin, or liquid polyethylene glycols.
- stabilizers may be added. All formulations for oral administration should be in dosages suitable for such administration.
- the compositions may take the form of tablets or lozenges formulated in conventional manner.
- the compounds for use according to the present invention may be conveniently delivered in the form of an aerosol spray presentation from pressurized packs or a nebuliser, with the use of a suitable propellant, e.g., dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide, or other suitable gas.
- a suitable propellant e.g., dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide, or other suitable gas.
- a suitable propellant e.g., dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane, carbon dioxide, or other suitable gas.
- a suitable propellant e.g., dichlorodifluoromethane, trichlorofluoromethane, dichlorotetrafluoroethane
- the compounds may be formulated for parenteral administration by injection, e.g., by bolus injection or continuous infusion.
- Formulations for injection may be presented in unit- dosage form, e.g., in ampoules or in multi-dose containers, with an added preservative.
- the compositions may take such forms as suspensions, solutions or emulsions in oily or aqueous vehicles, and may contain formulatory agents such as suspending, stabilizing and/or dispersing agents.
- compositions for parenteral administration include aqueous solutions of the active compounds in water-soluble form.
- suspensions of the active agents may be prepared as appropriate oily injection suspensions.
- Suitable lipophilic solvents or vehicles include fatty oils such as sesame oil, or synthetic fatty acid esters, such as ethyl oleate or triglycerides, or liposomes.
- Aqueous injection suspensions may contain substances that increase the viscosity of the suspension, such as sodium carboxymethyl cellulose, sorbitol, or dextran.
- the suspension may also contain suitable stabilizers or agents that increase the solubility of the compounds to allow for the preparation of highly concentrated solutions.
- the active ingredient may be in powder form for constitution with a suitable vehicle, e.g., sterile pyrogen-free water, before use.
- the compounds of the present invention may also be formulated as a depot preparation.
- Such long-acting formulations may be administered by implantation (for example, subcutaneously or intramuscularly) or by intramuscular injection.
- the compounds may be formulated with suitable polymeric or hydrophobic materials (for example, as an emulsion in an acceptable oil) or ion- exchange resins, or as sparingly soluble derivatives, for example, as a sparingly soluble salt.
- a pharmaceutical carrier for hydrophobic compounds is a cosolvent system comprising benzyl alcohol, a nonpolar surfactant, a water-miscible organic polymer, and an aqueous phase.
- the cosolvent system may be a VPD co-solvent system.
- VPD is a solution of 3% w/v benzyl alcohol, 8% w/v of the nonpolar surfactant polysorbate 80, and 65% w/v polyethylene glycol 300, made up to volume in absolute ethanol.
- the VPD co-solvent system (VPD: 5W) contains VPD diluted 1 :1 with a 5% dextrose in water solution. This co-solvent system dissolves hydrophobic compounds well, and itself produces low toxicity upon systemic administration.
- the proportions of a co-solvent system may be suitably varied without destroying its solubility and toxicity characteristics.
- co-solvent components may be varied: for example, other low-toxicity nonpolar surfactants may be used instead of polysorbate 80; the fraction size of polyethylene glycol may be varied; other biocompatible polymers may replace polyethylene glycol, e.g. polyvinyl pyrrolidone; and other sugars or polysaccharides may be substituted for dextrose.
- other delivery systems for hydrophobic pharmaceutical compounds may be employed. Liposomes and emulsions are known examples of delivery vehicles or carriers for hydrophobic drugs. Certain organic solvents such as dimethylsulfoxide also may be employed, although usually at the cost of greater toxicity due to the toxic nature of DMSO.
- the compounds may be delivered using a sustained-release system, such as semipermeable matrices of solid hydrophobic polymers containing the therapeutic agent.
- sustained-release materials have been established and are known by those skilled in the art.
- Sustained- release capsules may, depending on their chemical nature, release the compounds for a few weeks up to over 100 days.
- additional strategies for protein stabilization may be employed.
- the pharmaceutical compositions also may comprise suitable solid- or gel-phase carriers or excipients. These carriers and excipients may provide marked improvement in the bioavailability of poorly soluble drugs.
- Such carriers or excipients include calcium carbonate, calcium phosphate, sugars, starches, cellulose derivatives, gelatin, and polymers such as polyethylene glycols. Furthermore, additives or excipients such as Gelucire®, Capryol®, Labrafil®, Labrasol®, Lauroglycol®, Plurol®, Peceol® Transcutol® and the like may be used. Further, the pharmaceutical composition may be incorporated into a skin patch for delivery of the drug directly onto the skin.
- an exemplary daily dose generally employed will be from about 0.001 to about 1000 mg/kg of body weight, with courses of treatment repeated at appropriate intervals.
- the pharmaceutically acceptable formulations of the present invention may contain a compound of the present invention, or a pharmaceutically acceptable salt or solvate thereof, in an amount of about 10 mg to about 2000 mg, or from about 10 mg to about 1500 mg, or from about 10 mg to about 1000 mg, or from about 10 mg to about 750 mg, or from about 10 mg to about 500 mg, or from about 25 mg to about 500 mg, or from about 50 to about 500 mg, or from about 100 mg to about 500mg.
- the pharmaceutically acceptable formulations of the present invention may contain a compound of the present invention, or a pharmaceutically acceptable salt or solvate thereof, in an amount from about 0.5 w/w% to about 95 w/w%, or from about 1 w/w% to about 95 w/w%, or from about 1 w/w% to about 75 w/w%, or from about 5 w/w% to about 75 w/w%, or from about 10 w/w% to about 75 w/w%, or from about 10 w/w% to about 50 w/w%.
- a pharmaceutical composition of the invention in a suitable formulation is administered in combination with at least one anti-HIV agent.
- a combined formulation of a compound of the present invention and an at least anti-HIV agent may be prepared by combining a therapeutically effective amount (i.e., a cytochrome P450-inhibiting amount) of at least one compound of the present invention (as an active ingredient) with one or more anti-HIV agents and at least one pharmaceutically suitable carrier, which may be selected, for example, from diluents, excipients and auxiliaries that facilitate processing of the active compounds into the final pharmaceutical preparations.
- a therapeutically effective amount i.e., a cytochrome P450-inhibiting amount
- at least one compound of the present invention as an active ingredient
- at least one pharmaceutically suitable carrier which may be selected, for example, from diluents, excipients and auxiliaries that facilitate processing of the active compounds into the final pharmaceutical preparations.
- a pharmaceutical composition of the invention in a suitable formulation is administered at the same time as at least one anti-HIV agent that is in a separate, pharmaceutically acceptable formulation.
- a dosing regimen may be designed such that a compound of the present invention is administered to an HIV-infected mammal prior to, at the same time as, or after the administration of the pharmaceutical formulation containing at least one anti-HIV agent.
- the pharmaceutically acceptable formulation of the compound of the present invention may be prepared by combining the compound and at least one pharmaceutically suitable carrier, which may be selected, for example, from diluents, excipients and auxiliaries that facilitate processing of the active compounds into the final pharmaceutical preparations.
- the compounds of the present invention may be administered to a mammal suffering from infection with HIV, such as a human, either alone or as part of a pharmaceutically acceptable formulation, once a day, twice a day, or three times a day in combination with an anti-HIV agent.
- the compounds of the present invention may be administered in combination with an additional agent or agents for the treatment of a mammal, such as a human, that is suffering from an infection with the HIV virus, AIDS, AIDS-related complex (ARC), or any other disease or condition which is related to infection with the HIV virus.
- a mammal such as a human
- AIDS AIDS-related complex
- ARC AIDS-related complex
- agents that may be used in combination with the compounds of the present invention include, but are not limited to, those useful as HIV protease inhibitors, HIV reverse transcriptase inhibitors, non-nucleoside HIV reverse transcriptase inhibitors, inhibitors of HIV integrase, CCR5 inhibitors, HIV fusion inhibitors, compounds useful as immunomodulators, compounds that inhibit the HIV virus by an unknown mechanism, compounds useful for the treatment of herpes viruses, compounds useful as anti- infectives, and others as described below.
- Compounds useful as HIV protease inhibitors that may be used in combination with the compounds of the present invention include, but are not limited to, 141 W94 (amprenavir), CGP- 73547, CGP-61755, DMP-450, nelfinavir, ritonavir, saquinavir (invirase), lopinavir, TMC-126, atazanavir, palinavir, GS-3333, KN 1-413, KNI-272, LG-71350, CGP-61755, PD 173606, PD 177298, PD 178390, PD 178392, U-140690, ABT-378, DMP-450, AG-1776, MK-944, VX-478, indinavir, tipranavir, darunavir, brecanavir, DPC-681, DPC-684, fosamprenavir calcium (Lexiva), benzenesulfonamide derivatives disclosed in WO 03053435, R-944,
- Compounds useful as inhibitors of the HIV reverse transcriptase enzyme that may be used in combination with the compounds of the present invention include, but are not limited to, abacavir, FTC, GS-840, lamivudine, adefovir dipivoxil, beta-fluoro-ddA, zalcitabine, didanosine, stavudine, zidovudine, tenofovir, amdoxovir, SPD-754, SPD-756, racivir, reverset (DPC-817), MIV-210 (FLG), beta-L-Fd4C (ACH-126443), MIV-310 (alovudine, FLT), dOTC, DAPD, entecavir, GS-7340, emtricitabine, and alovudine.
- abacavir FTC
- GS-840 lamivudine
- adefovir dipivoxil beta-fluoro-ddA
- Compounds useful as non-nucleoside inhibitors of the HIV reverse transcriptase enzyme that may be used in combination with the compounds of the present invention include, but are not limited to, efavirenz, HBY-097, nevirapine, TMC-120 (dapivirine), TMC-125, etravirine, delavirdine, DPC-083, DPC-961, TMC-120, capravirine, GW-678248, GW-695634, calanolide, and tricyclic pyrimidinone derivatives as disclosed in WO 03062238.
- Compounds useful as CCR5 inhibitors that may be used in combination with the compounds of the present invention include, but are not limited to, TAK-779, SC-351125, SCH- D, (N-ftiSJ-S-fS-isopropyl- ⁇ -methyl ⁇ H-i ⁇ -triazole ⁇ -ylJ-exo-S-azabicycloP ⁇ .IJoct- ⁇ -ylJ-i- phenylpropyl)-4,4-difluorocyclohexanecarboxamide), ethyl 1-e/7oto- ⁇ 8-[(3S)-3-(acetylamino)-3-(3- fluorophenyl)propyl]-8-azabicyclo[3.2.1]oct-3-yl ⁇ -2-methyl-4,5,6,7-tetrahydro-1/-/-imidazo[4,5- c]pyridine-5-carboxylate, ⁇ /- ⁇ (1 S)-3-[3-encf
- Compounds useful as inhibitors of HIV integrase enzyme that may be used in combination with the compounds of the present invention include, but are not limited to, GW- 810781, 1 ,5-naphthyridine-3-carboxamide derivatives disclosed in WO 03062204, compounds disclosed in WO 03047564, compounds disclosed in WO 03049690, and 5-hydroxypyrimidine-4- carboxamide derivatives disclosed in WO 03035076.
- Fusion inhibitors for the treatment of HIV include, but are not limited to enfuvirtide (T-20), T-1249, AMD-3100, and fused tricyclic compounds disclosed in JP 2003171381.
- compositions of the present invention include, but are not limited to, Soluble CD4, TNX-355, PRO-542, BMS-806, tenofovir disoproxil fumarate, and compounds disclosed in JP 2003119137.
- Compounds useful in the treatment or management of infection from viruses other than HIV that may be used in combination with the compounds of the present invention include, but are not limited to, acyclovir, fomivirsen, penciclovir, HPMPC, oxetanocin G, AL-721, cidofovir, cytomegalovirus immune globin, cytovene, fomivganciclovir, famciclovir, foscarnet sodium, lsis 2922, KNI-272, valacyclovir, virazole ribavirin, valganciclovir, ME-609, PCL-016
- Compounds that act as immunomodulators and may be used in combination with the compounds of the present invention include, but are not limited to, AD-439, AD-519, Alpha
- Interferon AS-101, bropirimine, acemannan, CL246,738, EL10, FP-21399, gamma interferon, granulocyte macrophage colony stimulating factor, IL-2, immune globulin intravenous, IMREG-1 ,
- IMREG-2 imuthiol diethyl dithio carbamate, alpha-2 interferon, methionine-enkephalin, MTP-PE, granulocyte colony stimulating sactor, remune, rCD4, recombinant soluble human CD4, interferon alfa-2, SK&F106528, soluble T4 yhymopentin, tumor necrosis factor (TNF), tucaresol, recombinant human interferon beta, and interferon alfa n-3.
- TNF tumor necrosis factor
- Anti-infectives that may be used in combination with the compounds of the present invention include, but are not limited to, atovaquone, azithromycin, clarithromycin, trimethoprim, trovafloxacin, pyrimethamine, daunorubicin, clindamycin with primaquine, fluconazole, pastill, ornidyl, eflornithine pentamidine, rifabutin, spiramycin, intraconazole-R51211 , trimetrexate, daunorubicin, recombinant human erythropoietin, recombinant human growth hormone, megestrol acetate, testerone, and total enteral nutrition.
- Antifungals that may be used in combination with the compounds of the present invention include, but are not limited to, anidulafungin, C31G, caspofungin, DB-289, fluconzaole, itraconazole, ketoconazole, micafungin, posaconazole, and voriconazole.
- BMS-234475 BMS-234475, CI-1012, curdlan sulfate, dextran sulfate, STOCRINE EL10, hypericin, lobucavir, novapren, peptide T octabpeptide sequence, trisodium phosphonoformate, probucol, and RBC- CD4.
- the compounds of the present invention may be used in combination with anti-proliferative agents for the treatment of conditions such as Kaposi's sarcoma.
- agents include, but are not limited to, inhibitors of metallo-matrix proteases, A-007, bevacizumab, BMS-
- an additional agent or agents will depend on a number of factors that include, but are not limited to, the condition of the mammal being treated, the particular condition or conditions being treated, the identity of the compound or compounds of the present invention and the additional agent or agents, and the identity of any additional compounds that are being used to treat the mammal.
- the particular choice of the compound or compounds of the invention and the additional agent or agents is within the knowledge of one of ordinary skill in the art and can be made without undue experimentation.
- the compounds of the present invention may be administered in combination with any of the above additional agents for the treatment of a mammal, such as a human, that is suffering from an infection with the HIV virus, AIDS, AIDS-related complex (ARC), or any other disease or condition which is related to infection with the HIV virus.
- a mammal such as a human
- Such a combination may be administered to a mammal such that a compound or compounds of the present invention are present in the same formulation as the additional agents described above.
- such a combination may be administered to a mammal suffering from infection with the HIV virus such that the compound or compounds of the present invention are present in a formulation that is separate from the formulation in which the additional agent is found.
- the compound or compounds of the present invention are administered separately from the additional agent, such administration may take place concomitantly or sequentially with an appropriate period of time in between.
- the choice of whether to include the compound or compounds of the present invention in the same formulation as the additional agent or agents is within the knowledge of one of ordinary skill in the art.
- inventive agents may be prepared using the reaction routes and synthesis schemes as described below, employing the techniques available in the art using starting materials that are readily available.
- the preparation of certain embodiments of the present invention are described in detail in the following examples, but those of ordinary skill in the art will recognize that the preparations described may be readily adapted to prepare other embodiments of the present invention.
- the synthesis of non-exemplified compounds according to the invention may be performed by modifications apparent to those skilled in the art, e.g., by appropriately protecting interfering groups, by changing to other suitable reagents known in the art, or by making routine modifications of reaction conditions.
- other reactions disclosed herein or known in the art will be recognized as having adaptability for preparing other compounds of the invention.
- the compounds of Formula (I) can be prepared from compounds of formulae 3 and 4, as shown below in Scheme 1.
- compounds of formula 3, wherein R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , and R 9 are as hereinbefore defined, and R 15 is C 1 -Ci 0 alkyl may be allowed to react with compounds of formula 4, wherein R 10 and R 11 are as hereinbefore defined.
- Such reactions may be performed in an aprotic solvent, such as acetonitrile for example, at a temperature in the range of from about 50 0 C to about 150 0 C, for example about 110 0 C, and optionally in the presence of microwave energy.
- compounds of formula (I) may be prepared using methyl 2-[(pyridin-3-ylamino)sulfonyl]benzoate by preparing a mixture of methyl 2-[(pyridin-3- ylamino)sulfonyl]benzoate (1 eq) and an appropriate compound of formula 4 (1.5 eq) in CH 3 CN (0.35 M) and heating the resulting mixture to about 110 0 C for 30 minutes in a microwave. The reaction mixtures are then allowed to cool to about room temperature and the solvents are removed by evaporation. The resulting crude products may be further purified by methods known to those of ordinary skill in the art, such as the use of silica gel chromatography using an appropriate solvent or mixture of solvents as the eluant.
- Compounds of formula 3 may be prepared from compounds of formulae 1 and 2, wherein R 1 , R 2 , R 3 , R 4 , R 5 , R 6 , R 7 , R 8 , and R 9 are as hereinbefore defined, and R 15 is C 1 -Ci 0 alkyl, as shown below in Scheme 2.
- these reactions may be performed in an aprotic solvent, such as acetonitrile for example, at a temperature in the range of from about 0 0 C to about 75 0 C, for example about 25 0 C, and in the presence of a base.
- Suitable bases include, but are not limited to, inorganic bases, such as potassium carbonate or sodium carbonate for example, or organic bases, such as triethyl amine or N,N-4,4-dimethylaminopyridine for example.
- a second equivalent of the compound of formula 2 may be used as a suitable base. The choice of whether to use an additional base or an additional equivalent of compound 2 as the base is within the knowledge of one of ordinary skill in the art and can be determined without undue experimentation.
- Compounds of formula 1 are commercially available or may be prepared by those of ordinary skill in the art without undue experimentation using methods found in the literature, such as those found in Kim et al., Synthesis (1992), (12), 1203-4; Bastide et al., Pesticide Science (1994), 40(4), 293-7; and Diehr et al., United States Patent No. 4,743,294.
- Compounds of formula 2 are either commercially available, may be prepared by those of ordinary skill in the art without undue experimentation using methods found in the literature, or may be prepared using methods described herein.
- the inhibition of the cytochrome P450 enzyme system by a compound of the present invention can be determined according to methods known to those of skill in the art and the methods described herein. For example, see Morrison, J. F., Biochim Biophys Acta., 1969,185:
- NMR spectra were recorded on a Bruker instrument operating at 300 MHz and 13 C-NMR spectra were recorded at 75 MHz.
- NMR spectra were obtained as DMSO-d6 or CDCI 3 solutions (reported in ppm), using chloroform as the reference standard (7.25 ppm and 77.00 ppm) or DMSO-d6 ((2.50 ppm and 39.52 ppm)). Other NMR solvents were used as needed.
- Infrared spectra were recorded on a Perkin-Elmer FT-IR Spectrometer as neat oils, as KBr pellets, or as CDCI 3 solutions, and when reported are in wave numbers (cm "1 ). The mass spectra were obtained using LC/MS or APCI. All melting points are uncorrected.
- Step 3 methyl 2- ⁇ [(5-methoxypyridin-3-yl)amino]sulfonyl ⁇ benzoate
- Example D Preparation of methyl 2- ⁇ [(4-methoxypyridin-3-yl)amino]sulfonyl ⁇ benzoate Step 1 : 4-methoxypyridin-3-amine
- Methyl 2- ⁇ [(5-cyanopyridin-3-yl)amino]sulfonyl ⁇ benzoate was prepared following the procedure published in Schareina, T. et al., J. Organometallic Chemistry 2004, 689, 4576.
- Step 1 Methyl 2-mercaptobenzoate (2): Acid 1 (170.0 g, 1.1 mol), H 2 SO 4 (20 mL, 0.37 mol) and anhydrous MeOH (300 mL) were refluxed for 2 days. The solvent was evaporated under reduced pressure. Water was added and the mixture was extracted with CH 2 CI 2 (3*1000 mL). The organic layers were combined, washed with aq. NaHCO 3 (2x300 mL), water (2*250 mL), brine (500 mL) and dried over Na 2 SO 4 to afford crude compound 2 (165.0 g, 89.6%). This was used without further purification.
- Step 2 Methyl 2-(pyridin-3-ylmethylthio)benzoate (3): A mixture of compound 2 (70.0 g, 0.4167 mol), 1-(bromomethyl)benzene (82.8 g, 0.5 mol), K 2 CO 3 (350 g, 2.49 mol) and CH 3 CN (500 mL) was refluxed overnight. The mixture was allowed to cool to room temperature and filtered. The filtrate was concentrated under reduced pressure. After addition of water, the mixture was extracted with CH 2 CI 2 (3x500 mL). The organic layers were combined, washed with aq. Na 2 CO 3 (2*200 mL), water (2*300 mL), brine (300 mL) and dried over Na 2 SO 4 .
- Step 3 2-(pyridin-3-ylmethylthio)benzoic acid (4): To a solution of compound 3 (55.0 g, 0.212 mol) in MeOH/H 2 O (300 mL / 50 mL)was added LiOH- H 2 O (17.8 g, 0.425 mol) in portions at 0 0 C. The reaction mixture was stirred at room temperature overnight and TLC showed the starting material was consumed. The solvent was removed under reduced pressure. The residue was diluted with water, and extracted with Et 2 O (2x1 L) to remove neutral impurities. The aqueous layer was adjusted to pH 3-4 with 1 N aq.
- Step 4 N-(2-methyl-1-morpholinopropan-2-yl)-2-(pyridin-3-ylmethylthio)benzamide(5): To a solution of acid 4 (2.75 g, 11.2 mmol) and 2-rnethyl-1-morpholinopropan-2-amine (1.12 mL, 7.4 mmol) ) in anhydrous DMF/DMSO were added EDCI (2.58 g, 13.32 mmol), HOBt (0.182 g, 13.32 mmol) and NMM (3.35 mL, 29.6 mmol) and the mixture was stirred at room temperature overnight. The solvent was removed in vacuo. After addition of water (10 mL), the mixture was extracted with CH 2 CI 2 .
- Step 5 N-(2-methyl-1-morpholinopropan-2-yl)-2-(pyridin-3-ylmethy)sulfinyl)benzamide (6): To a solution of intermediate 5 (0.143, 0.370 mmol) in CHCI 3 (6 ml) were added Et 4 NBr (0.004 g, 0.018 mmol) and IBX (0.142 g, 0.507 mmol) and the reaction mixture was stirred at room temperature for 1 hour. The mixture was diluted with saturated aqueous NaHCO 3 and extracted with CH 2 CI 2 . The organic layer was dried over Na 2 SO 4 , filtered and concentrated.
- the title compound was prepared according to procedures described above, using methyl 2-[(pyridin-3-ylamino)sulfonyl]benzoate and 3-methylbutylamine.
- the title compound was further purified by dissolving 2.0 g in ethyl acetate (20 ml_) and heating to reflux. When compound had fully dissolved the resultant solution was left to cool very slowly to room temperature. The resulting crystals were left to stand at RT for 30 min, after which time they were filtered using a funnel, washed with cold ethyl acetate (20 ml_), and dried under high vacuum for
- CDCI 3 ⁇ 8.64 (s, 1 H) 8.26 - 8.43 (m, 2 H) 7.60 - 7.68 (m, 2 H) 7.45 - 7.59 (m, 2 H) 7.39 (t, 1 H) 7.12 - 7.21 (m, 1 H) 6.08 - 6.17 (m, 1 H) 3.52 (q, 2 H) 1.67 - 1.79 (m, 1 H) 1.57 (q, 2 H) 0.98 (d, 6
- the title compound was prepared according to procedures described above, using methyl 2-[(pyridin-3-ylamino)sulfonyl]benzoate and 3-fluorobenzylamine.
- the title compound was further purified by dissolving 25 g in EtOH (300 mL) and gently heating using water bath to 80 0 C. The resulting solids material were then slowly dissolved and more EtOH (50 mL) was added to completely dissolve all the solids. The solution left at room temperature to crystallize for 1 h. The resulting crystals were collected and dried under high vacuum for 16 h to afford the title compound as a crystalline solid (21 g, 84 %).
- the title compound was prepared according to procedures described above, using methyl 2-[(pyridin-3-ylamino)sulfonyl]benzoate and n-butylamine.
- the title compound was further purified by dissolving 2.Og in ethyl acetate (10 mL). Hexane was then added dropwise to the solution until it became cloudy. A few drops of ethyl acetate were then added to obtain a clear solution. The resulting solution was left at rt for approximately 48 h.
- Example G In vitro activity data for Examples 1. Determination of compound K Iapp
- a determination of the K ia pp of the compounds of the invention against recombinant CYP3A4 enzyme was performed as follows. The assay was performed in a 10OmM sodium phosphate buffer pH 7.0, 5 mM TCEP and containing 2 % dimethylformamide (final concentration) upon addition of substrate and inhibitor. A typical reaction for the determination of Kiapp values was carried at room temperature in a solid black Costar u-bottom 96-well polypropylene plate. In each well, recombinant CYP3A4 enzyme (5.5 nM or 8 nM, final concentration depending on the commercial source of the enzyme) was pre-incubated in the presence of the inhibitor for at least 30 minutes in the assay buffer.
- the initial reaction velocities were measured during the first 5 min of the reaction when the release of the fluorescent product is linear with time, in the absence and in the presence of various concentrations of inhibitors.
- the Ki app values were determined by using the equation for tight-binding inhibitor developed by Morrison, JF (Morrison JF. Biochim Biophys Acta. 1969, 185: 269-86) and by Szedlaseck SE et al. (Szedlascek, S.E., Ostafe, V., Serban, M., and Vlad, M.O. Biochem. J. 1988, 254:311-312), respectively.
- the fluorescent substrate BFC was purchased from Sigma (St Louis, MO). Two commercial sources of recombinant enzymes were used in this study: recombinant CYP3A4 - b5 enzyme (Baculosomes ® ) was purchased from Invitrogen (Carlsbad, CA) and the recombinant CYP3A4 + b5 enzyme (Supersomes ® ) was purchased from BD Biosciences (Woburn, MA). 2. Determination of IC 50 against CYP3A4 by measurement of inhibition of testosterone metabolism
- IC50 values are calculated from the percent inhibition determined for each test compound at 6 concentrations (for example: 750, 250, 83.3, 27.8, 9.3 and 3.1 nM).
- the incubation substrate mix contains 25 ⁇ M testosterone, 0.1 mg/mL human liver microsomes, 1 mM NADPH, and potassium phosphate buffer (100 mM, pH 7.4).
- Quantitation of metabolite peak area ratio against an internal standard is determined by LC-MS/MS analysis.
- the production of 6- ⁇ -OH-testosterone from testosterone metabolism is determined after incubation for eight minutes by comparison to a standard curve generated for the metabolite
- the sample injection volume was 10 ⁇ L and flow was split post-column with 0.4 mL/min going to the mass spectrometer. Analysis was performed using the following API 3000 mass spectrometer settings: Instrument settings
- IC 50 against CYP3A4 by measurement of inhibition of midazolam metabolism This assay was performed using a standard 96 well plate design. IC50s are calculated from the percent inhibition determined for each test compound at 6 concentrations (for example: 750, 250, 83.3, 27.8, 9.3 and 3.1 nM).
- the incubation substrate mix contains 2 ⁇ M midazolam, 0.1 mg/mL human liver microsomes, 1 mM NADPH, and potassium phosphate buffer (100 mM, pH 7.4). Quantitation of metabolite peak area ratio against internal standard is determined by LC-MS/MS analysis. The production of 1-hydroxymidazolam from midazolam metabolism is determined after incubation for eight minutes by comparison to a standard curve generated for the metabolite.
- the sample injection volume was 10 ⁇ L and flow was split post-column with 0.4 mL/min going to the mass spectrometer. Analysis was performed using the following API 3000 mass spectrometer settings: Instrument settings
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Virology (AREA)
- Epidemiology (AREA)
- Toxicology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Tropical Medicine & Parasitology (AREA)
- Molecular Biology (AREA)
- Engineering & Computer Science (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- AIDS & HIV (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Pyridine Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Description








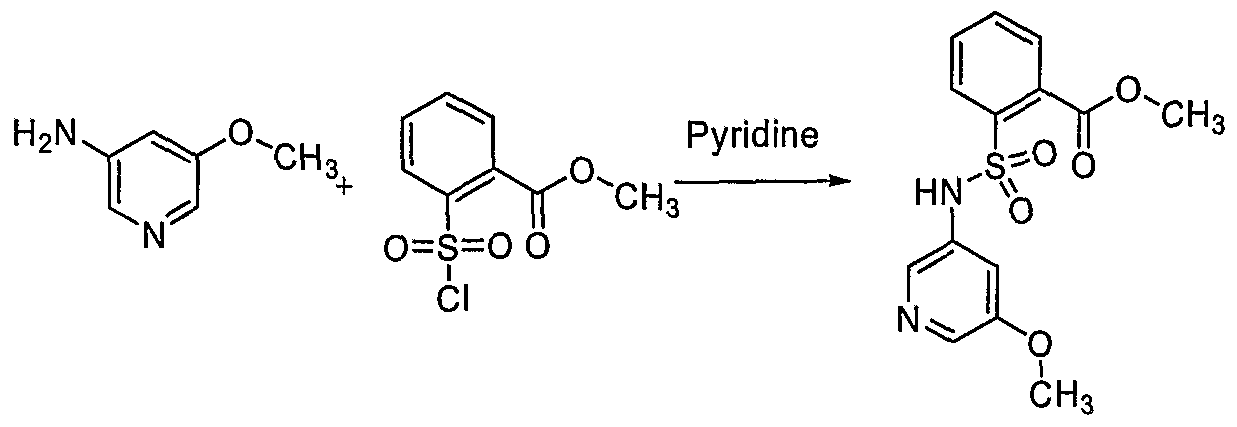

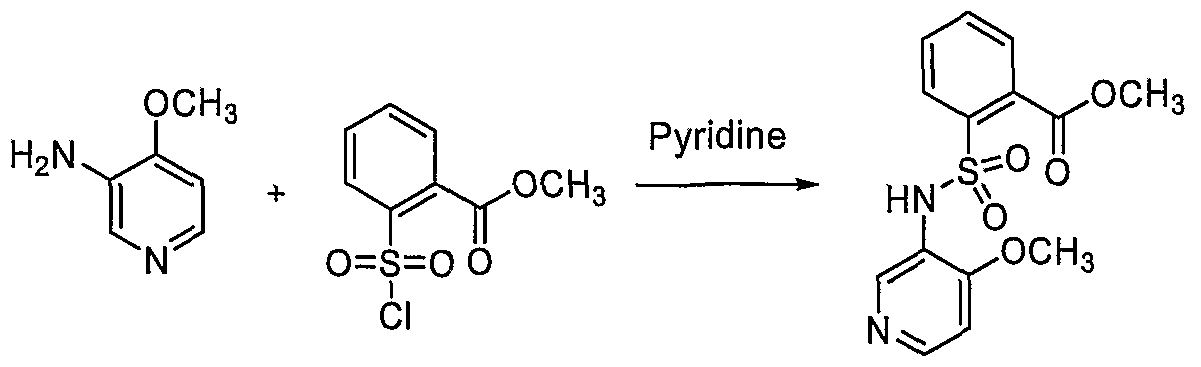



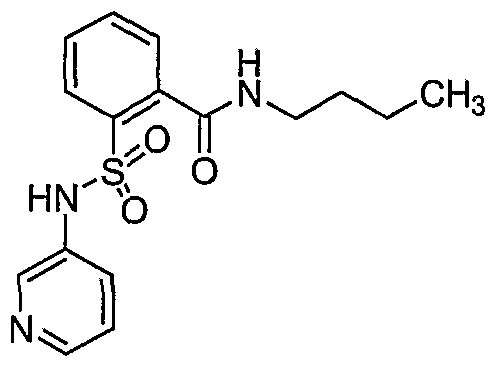








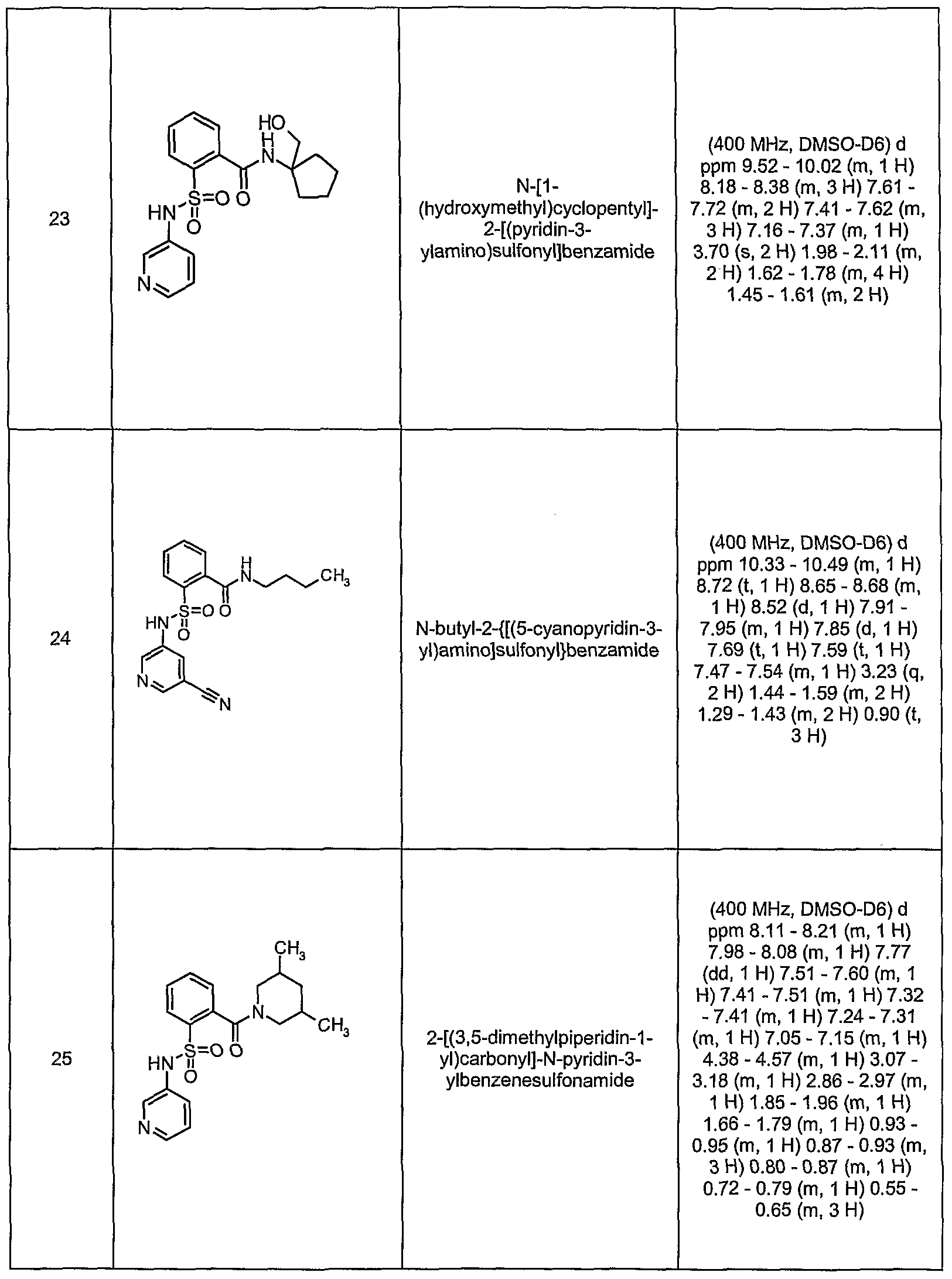








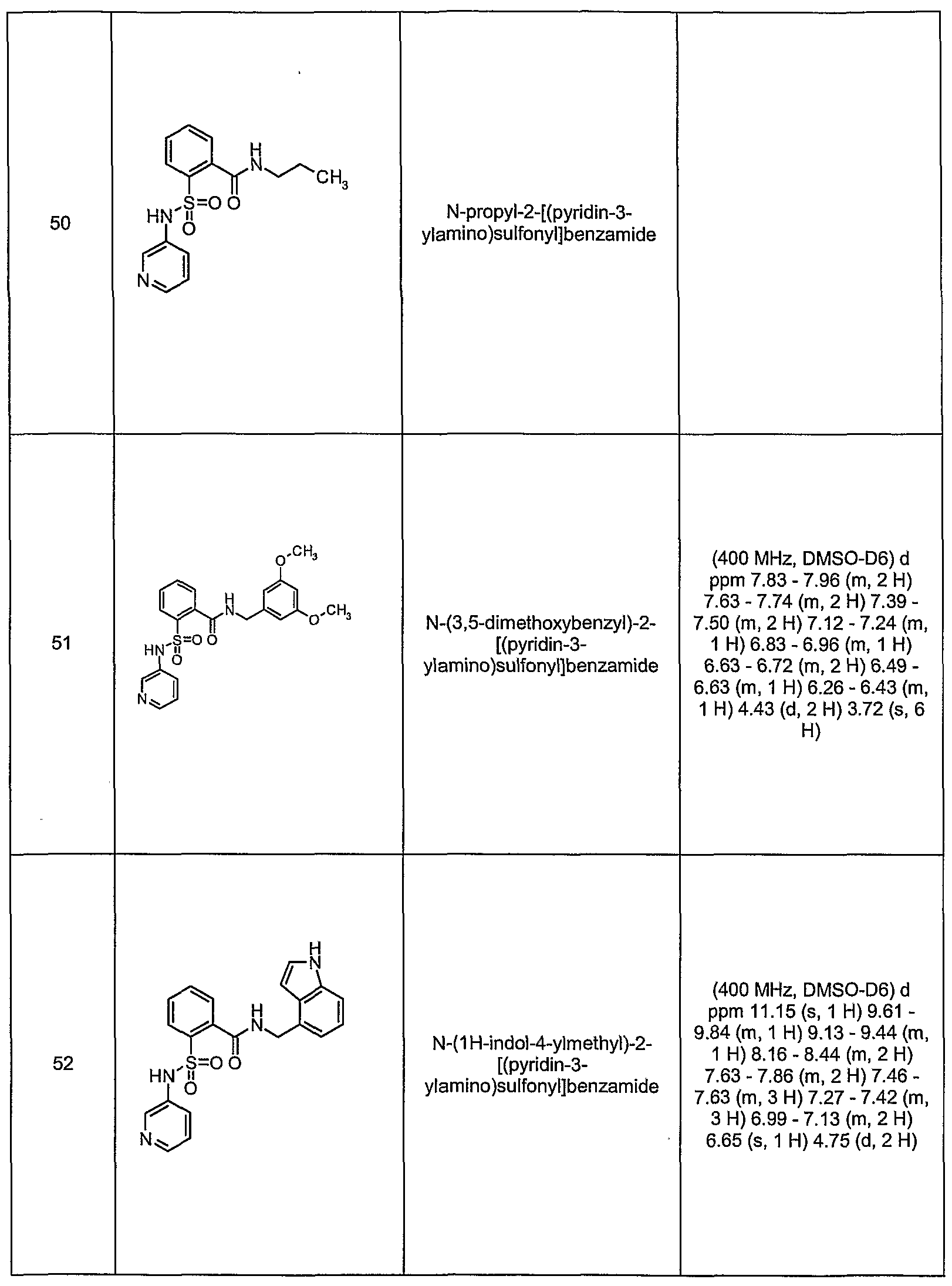





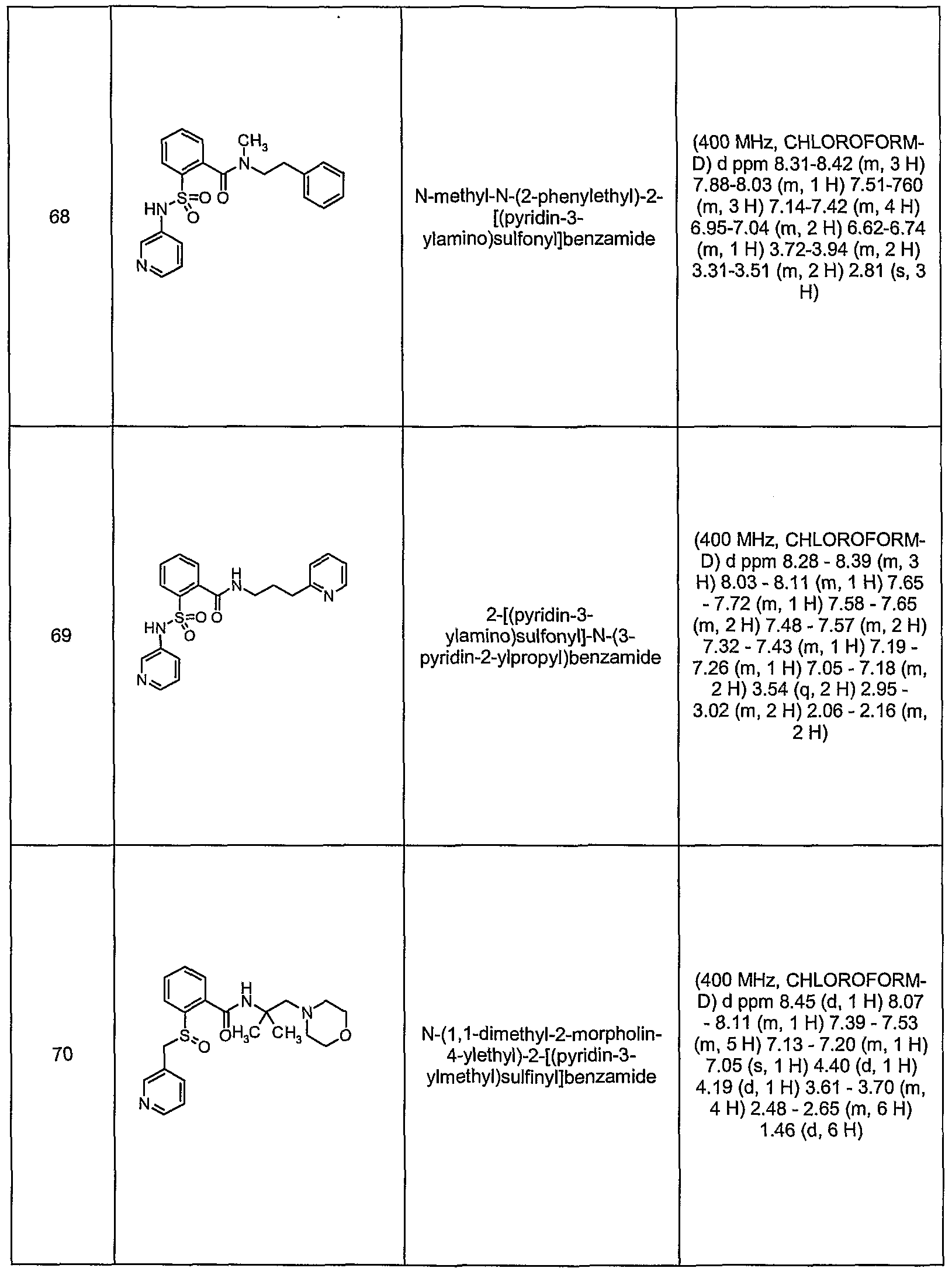





























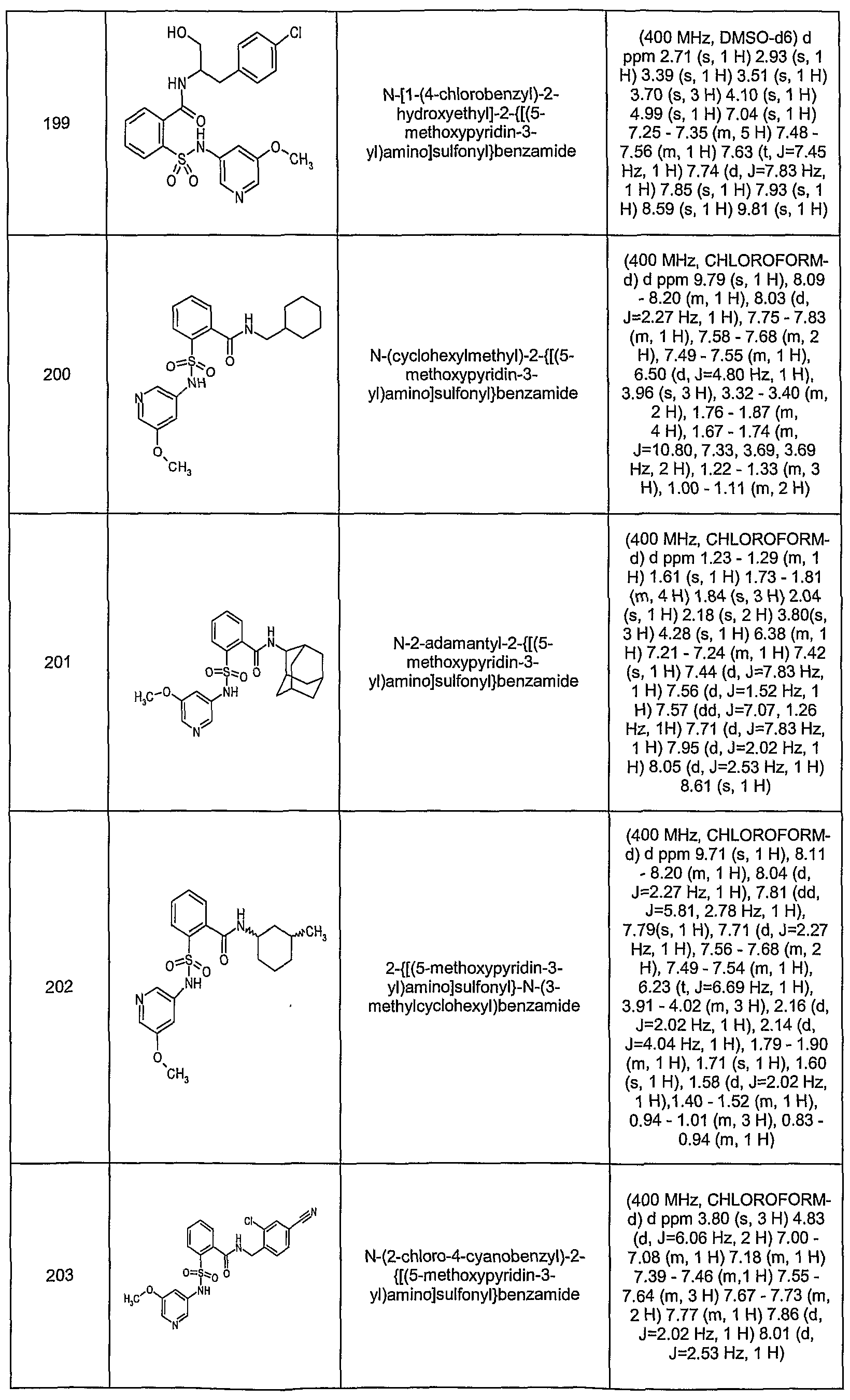



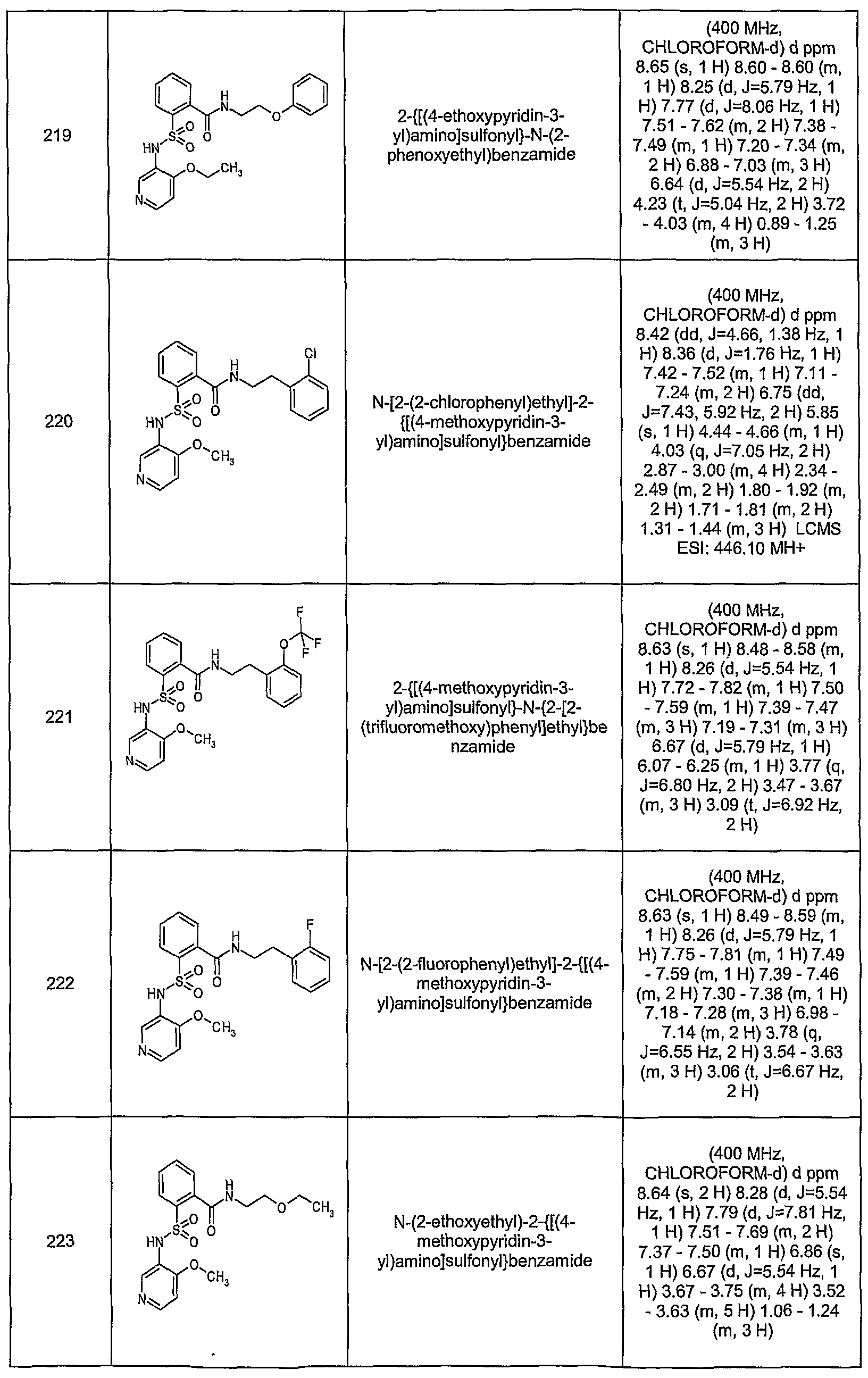

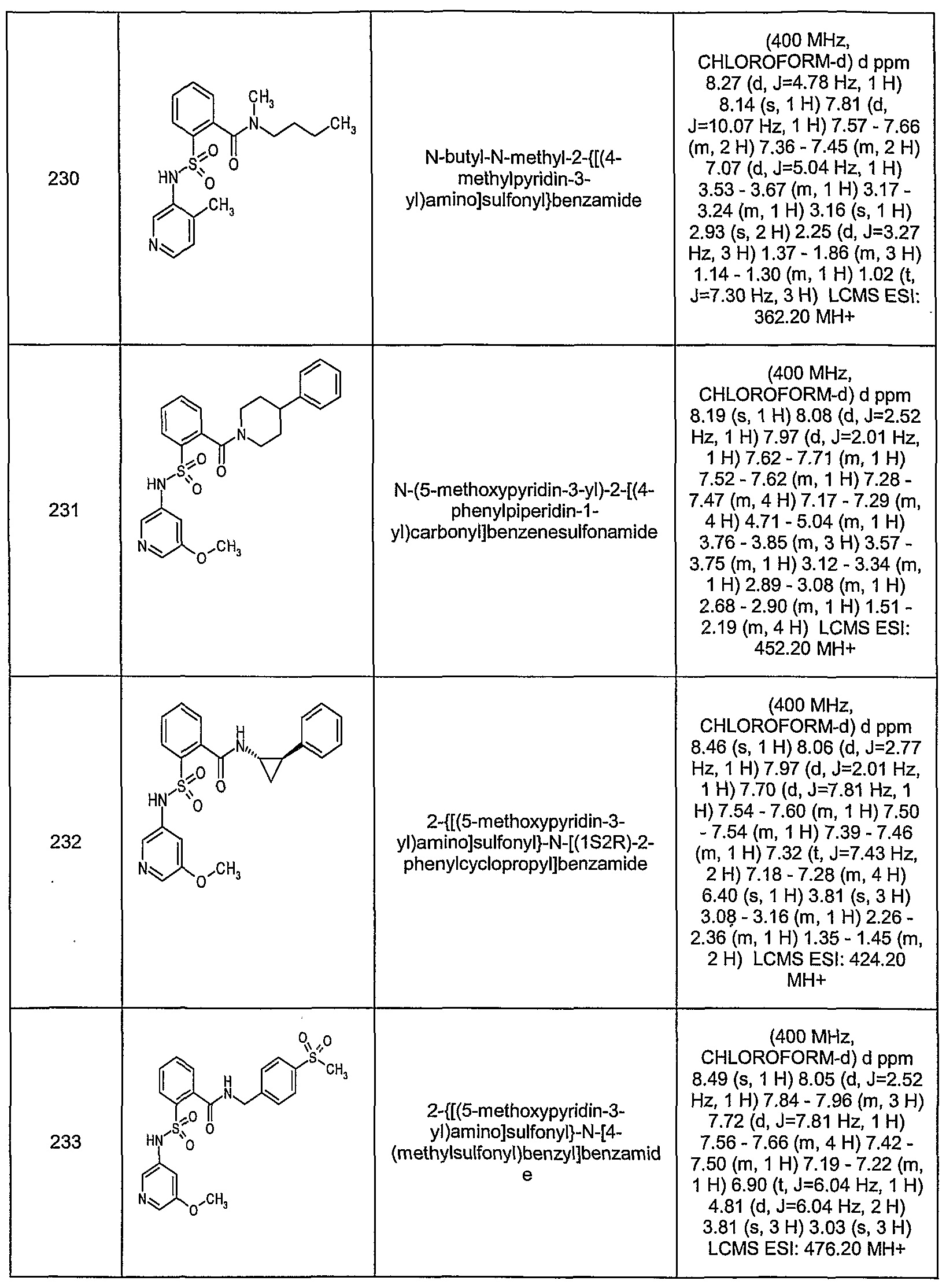








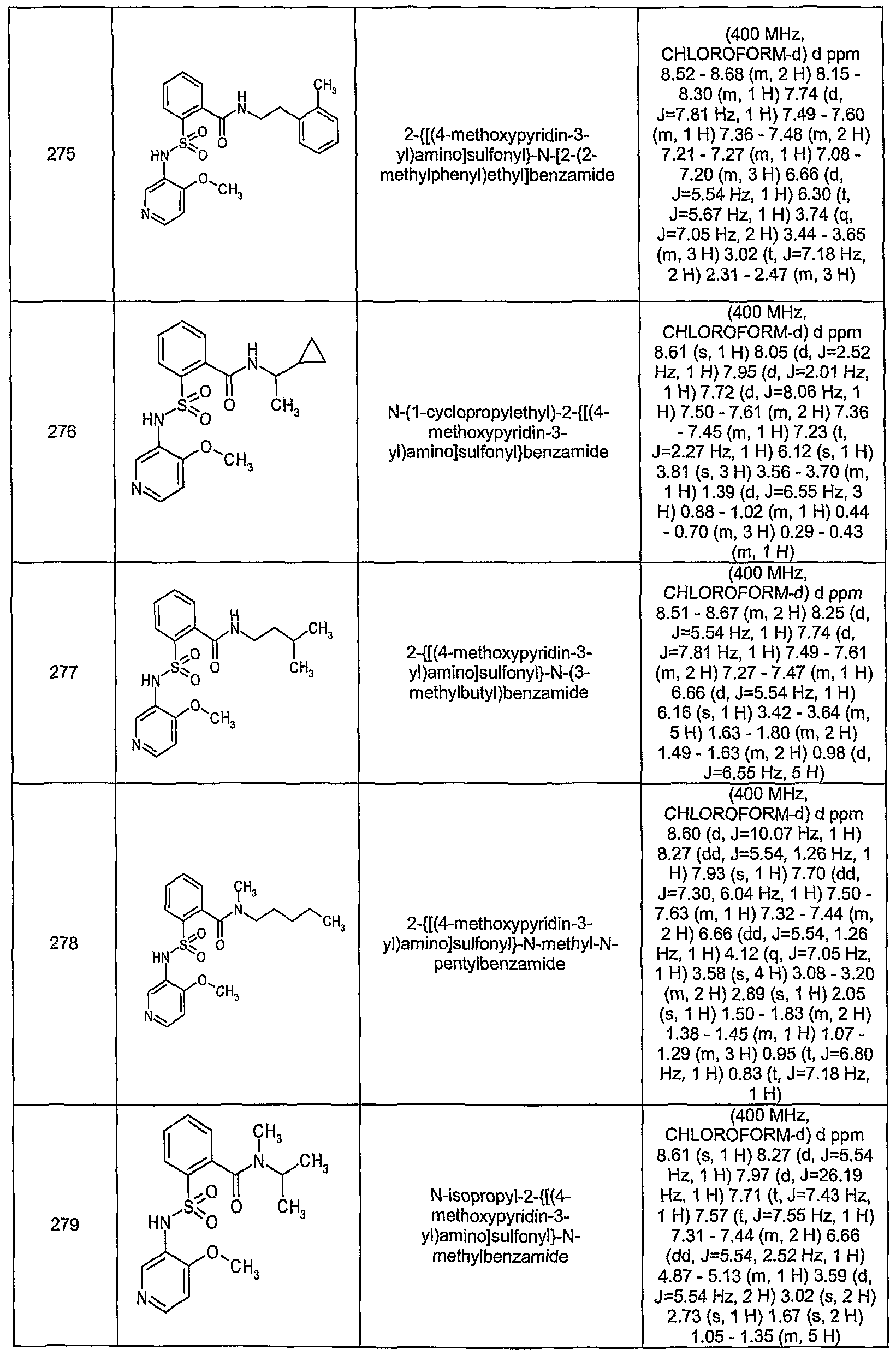

































Claims

Priority Applications (21)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2006293605A AU2006293605B2 (en) | 2005-09-23 | 2006-09-11 | Pyridinaminosulfonyl substituted benzamides as inhibitors of cytochrome P450 3A4 (CYP3A4) |
| PL06795544T PL1937639T3 (en) | 2005-09-23 | 2006-09-11 | Pyridinaminosulfonyl substituted benzamides as inhibitors of cytochrome p450 3a4 (cyp3a4) |
| AT06795544T ATE459603T1 (en) | 2005-09-23 | 2006-09-11 | PYRIDINEMINOSULFONYL SUBSTITUTED BENZAMIDES AS INHIBITORS OF CYTOCHROME P450 3A4 (CYP3A4) |
| JP2008531812A JP4336738B2 (en) | 2005-09-23 | 2006-09-11 | Pyridineaminosulfonyl substituted benzamides as inhibitors of cytochrome P4503A4 (CYP3A4) |
| SI200630666T SI1937639T1 (en) | 2005-09-23 | 2006-09-11 | Pyridinaminosulfonyl substituted benzamides as inhibitors of cytochrome p450 3a4 (cyp3a4) |
| DK06795544.3T DK1937639T3 (en) | 2005-09-23 | 2006-09-11 | Pyridinaminosulfonyl-substituted benzamides as inhibitors of cytochrome P450 3A4 (CYP3A4) |
| ES06795544T ES2340200T3 (en) | 2005-09-23 | 2006-09-11 | BENZAMIDS REPLACED WITH PYRIDINAMINOSULFONIL AS CITROMO INHIBITORS P450 3A4 (CYP3A4). |
| DE602006012713T DE602006012713D1 (en) | 2005-09-23 | 2006-09-11 | PYRIDINAMINOSULFONYL SUBSTITUTED BENZAMIDES AS INHIBITORS OF CYTOCHROME P450 3A4 (CYP3A4) |
| RSP-2008/0118A RS20080118A (en) | 2005-09-23 | 2006-09-11 | Pyridinaminosulfonyl substituted benzamides as inhibitors of cytochrome p450 3a4 (cyp3a4) |
| CA2621805A CA2621805C (en) | 2005-09-23 | 2006-09-11 | Pyridinaminosulfonyl substituted benzamides as inhibitors of cytochrome p450 3a4 (cyp3a4) |
| CN2006800348278A CN101287708B (en) | 2005-09-23 | 2006-09-11 | Pyridinaminosulfonyl substituted benzamides as inhibitors of cytochrome p450 3a4 (cyp3a4) |
| NZ566136A NZ566136A (en) | 2005-09-23 | 2006-09-11 | Pyridinaminosulfonyl substituted benzamides as inhibitors of cytochrome p450 3a4 (cyp3a4) |
| EP06795544A EP1937639B1 (en) | 2005-09-23 | 2006-09-11 | Pyridinaminosulfonyl substituted benzamides as inhibitors of cytochrome p450 3a4 (cyp3a4) |
| EA200800542A EA014329B1 (en) | 2005-09-23 | 2006-09-11 | Pyridinaminosulphonyl substituted benzamides as inhibitors of cytochrome p450 3a4 (cyp3a4) |
| BRPI0616156-1A BRPI0616156A2 (en) | 2005-09-23 | 2006-09-11 | therapeutic compounds, pharmaceutical composition comprising them and use |
| US11/621,410 US7868026B2 (en) | 2005-09-23 | 2007-01-09 | Therapeutic compounds |
| IL189666A IL189666A (en) | 2005-09-23 | 2008-02-21 | Pyridinaminosulfonyl substituted benzamides as inhibitors of cytochrome p450 3a4 (cyp3a4) |
| TNP2008000139A TNSN08139A1 (en) | 2005-09-23 | 2008-03-19 | THERAPEUTIC COMPOUNDS |
| NO20081844A NO20081844L (en) | 2005-09-23 | 2008-04-16 | Therapeutic compounds |
| HK08113572.1A HK1120506A1 (en) | 2005-09-23 | 2008-12-15 | Pyridinaminosulfonyl substituted benzamides as inhibitors of cytochrome p450 3a4 (cyp3a4) |
| HR20100272T HRP20100272T2 (en) | 2005-09-23 | 2010-05-14 | Pyridinaminosulfonyl substituted benzamides as inhibitors of cytochrome p450 3a4 (cyp3a4) |
Applications Claiming Priority (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US72015105P | 2005-09-23 | 2005-09-23 | |
| US60/720,151 | 2005-09-23 | ||
| US72311505P | 2005-10-03 | 2005-10-03 | |
| US60/723,115 | 2005-10-03 | ||
| US72546905P | 2005-10-11 | 2005-10-11 | |
| US60/725,469 | 2005-10-11 | ||
| US76225606P | 2006-01-25 | 2006-01-25 | |
| US60/762,256 | 2006-01-25 | ||
| US82166406P | 2006-08-07 | 2006-08-07 | |
| US60/821,664 | 2006-08-07 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US11/621,410 Continuation US7868026B2 (en) | 2005-09-23 | 2007-01-09 | Therapeutic compounds |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2007034312A2 true WO2007034312A2 (en) | 2007-03-29 |
| WO2007034312A3 WO2007034312A3 (en) | 2007-08-23 |
Family
ID=37889174
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2006/002639 WO2007034312A2 (en) | 2005-09-23 | 2006-09-11 | Pyridinaminosulfonyl substituted benzamides as inhibitors of cytochrome p450 3a4 (cyp3a4) |
Country Status (29)
| Country | Link |
|---|---|
| US (1) | US7868026B2 (en) |
| EP (1) | EP1937639B1 (en) |
| JP (1) | JP4336738B2 (en) |
| KR (1) | KR100975448B1 (en) |
| CN (1) | CN101287708B (en) |
| AT (1) | ATE459603T1 (en) |
| AU (1) | AU2006293605B2 (en) |
| BR (1) | BRPI0616156A2 (en) |
| CA (1) | CA2621805C (en) |
| CY (1) | CY1110010T1 (en) |
| DE (1) | DE602006012713D1 (en) |
| DK (1) | DK1937639T3 (en) |
| EA (1) | EA014329B1 (en) |
| EC (1) | ECSP088296A (en) |
| ES (1) | ES2340200T3 (en) |
| GE (1) | GEP20115230B (en) |
| HK (1) | HK1120506A1 (en) |
| HR (1) | HRP20100272T2 (en) |
| IL (1) | IL189666A (en) |
| MA (1) | MA29791B1 (en) |
| MY (1) | MY143391A (en) |
| NO (1) | NO20081844L (en) |
| NZ (1) | NZ566136A (en) |
| PL (1) | PL1937639T3 (en) |
| PT (1) | PT1937639E (en) |
| RS (2) | RS20080118A (en) |
| SI (1) | SI1937639T1 (en) |
| TN (1) | TNSN08139A1 (en) |
| WO (1) | WO2007034312A2 (en) |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2010125831A1 (en) * | 2009-05-01 | 2010-11-04 | Raqualia Pharma Inc. | Sulfamoyl benzoic acid derivatives as trpm8 antagonists |
| EP2349279A1 (en) * | 2008-10-28 | 2011-08-03 | The Board of Trustees of The Leland Stanford Junior University | Modulators of aldehyde dehydrogenase and methods of use thereof |
| WO2014000178A1 (en) * | 2012-06-27 | 2014-01-03 | Merck Sharp & Dohme Corp. | Sulfonamide derivatives and methods of use thereof for improving the pharmacokinetics of a drug |
| US8906942B2 (en) | 2008-09-08 | 2014-12-09 | The Board Of Trustees Of The Leland Stanford Junior University | Modulators of aldhehyde dehydrogenase activity and methods of use thereof |
| WO2014197345A2 (en) | 2013-06-07 | 2014-12-11 | Merck Sharp & Dohme Corp. | Imidazole derivatives and methods of use thereof for improving the pharmacokinetics of a drug |
| US20150105456A1 (en) | 2007-03-08 | 2015-04-16 | The Board Of Trustees Of The Leland Stanford Junior University | Mitochondrial Aldehyde Dehydrogenase-2 Modulators and Methods of Use Thereof |
| WO2015073310A1 (en) | 2013-11-12 | 2015-05-21 | Merck Sharp & Dohme Corp. | Piperidine or piperazine linked imidazole and triazole derivatives and methods of use thereof for improving the pharmacokinetics of a drug |
| US9067922B2 (en) | 2013-04-19 | 2015-06-30 | Pfizer Limited | Chemical compounds |
| US9670162B2 (en) | 2013-03-14 | 2017-06-06 | The Board Of Trustees Of The Leland Stanford Junio | Mitochondrial aldehyde dehyrogenase-2 modulators and methods of use thereof |
| US9975888B2 (en) | 2013-11-12 | 2018-05-22 | Merck Sharp & Dohme Corp. | Aryl linked imidazole and triazole derivatives and methods of use thereof for improving the pharmacokinetics of a drug |
| US10457659B2 (en) | 2011-04-29 | 2019-10-29 | The Board Of Trustees Of The Leland Stanford Junior University | Compositions and methods for increasing proliferation of adult salivary stem cells |
| US11098048B2 (en) | 2014-03-26 | 2021-08-24 | Hoffmann-La Roche Inc. | Bicyclic compounds as autotaxin (ATX) and lysophosphatidic acid (LPA) production inhibitors |
| US11352330B2 (en) | 2015-09-04 | 2022-06-07 | Hoffmann-La Roche Inc. | Phenoxymethyl derivatives |
| US11673888B2 (en) | 2017-03-16 | 2023-06-13 | Hoffmann-La Roche Inc. | Bicyclic compounds as ATX inhibitors |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NZ574207A (en) * | 2006-07-05 | 2010-10-29 | Pfizer Prod Inc | Pyrazole derivatives as cytochrome p450 inhibitors |
| AU2007343726A1 (en) * | 2006-12-26 | 2008-07-24 | Amgen Inc. | N-cyclohexyl benzamides and benzeneacetamides as inhibitors of 11-beta-hydroxysteroid dehydrogenases |
| AU2008307195B2 (en) * | 2007-10-04 | 2012-11-22 | F. Hoffmann-La Roche Ag | Cyclopropyl aryl amide derivatives and uses thereof |
| PE20142099A1 (en) | 2009-01-12 | 2014-12-13 | Icagen Inc | SULFONAMIDE DERIVATIVES |
| EP2590972B1 (en) | 2010-07-09 | 2015-01-21 | Pfizer Limited | N-sulfonylbenzamides as inhibitors of voltage-gated sodium channels |
| CN104540813A (en) * | 2012-06-27 | 2015-04-22 | 默沙东公司 | Sulfonamide derivatives and methods of use thereof for improving the pharmacokinetics of a drug |
| WO2016101118A1 (en) * | 2014-12-23 | 2016-06-30 | Merck Sharp & Dohme Corp. | Amidoethyl azole orexin receptor antagonists |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20030166584A1 (en) * | 2002-02-22 | 2003-09-04 | Hu Oliver Yoa-Pu | Cytochrome P450 3A inhibitors and enhancers |
-
2006
- 2006-09-11 ES ES06795544T patent/ES2340200T3/en active Active
- 2006-09-11 AU AU2006293605A patent/AU2006293605B2/en not_active Ceased
- 2006-09-11 NZ NZ566136A patent/NZ566136A/en not_active IP Right Cessation
- 2006-09-11 PL PL06795544T patent/PL1937639T3/en unknown
- 2006-09-11 EA EA200800542A patent/EA014329B1/en not_active IP Right Cessation
- 2006-09-11 CN CN2006800348278A patent/CN101287708B/en not_active Expired - Fee Related
- 2006-09-11 RS RSP-2008/0118A patent/RS20080118A/en unknown
- 2006-09-11 WO PCT/IB2006/002639 patent/WO2007034312A2/en active Application Filing
- 2006-09-11 JP JP2008531812A patent/JP4336738B2/en not_active Expired - Fee Related
- 2006-09-11 AT AT06795544T patent/ATE459603T1/en active
- 2006-09-11 BR BRPI0616156-1A patent/BRPI0616156A2/en not_active IP Right Cessation
- 2006-09-11 GE GEAP200610575A patent/GEP20115230B/en unknown
- 2006-09-11 PT PT06795544T patent/PT1937639E/en unknown
- 2006-09-11 DE DE602006012713T patent/DE602006012713D1/en active Active
- 2006-09-11 DK DK06795544.3T patent/DK1937639T3/en active
- 2006-09-11 KR KR1020087006967A patent/KR100975448B1/en not_active IP Right Cessation
- 2006-09-11 RS RSP-2010/0237A patent/RS51349B/en unknown
- 2006-09-11 EP EP06795544A patent/EP1937639B1/en active Active
- 2006-09-11 CA CA2621805A patent/CA2621805C/en not_active Expired - Fee Related
- 2006-09-11 SI SI200630666T patent/SI1937639T1/en unknown
-
2007
- 2007-01-09 US US11/621,410 patent/US7868026B2/en not_active Expired - Fee Related
-
2008
- 2008-02-21 IL IL189666A patent/IL189666A/en not_active IP Right Cessation
- 2008-03-19 MA MA30764A patent/MA29791B1/en unknown
- 2008-03-19 EC EC2008008296A patent/ECSP088296A/en unknown
- 2008-03-19 TN TNP2008000139A patent/TNSN08139A1/en unknown
- 2008-03-21 MY MYPI20080779A patent/MY143391A/en unknown
- 2008-04-16 NO NO20081844A patent/NO20081844L/en not_active Application Discontinuation
- 2008-12-15 HK HK08113572.1A patent/HK1120506A1/en not_active IP Right Cessation
-
2010
- 2010-04-20 CY CY20101100359T patent/CY1110010T1/en unknown
- 2010-05-14 HR HR20100272T patent/HRP20100272T2/en unknown
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20030166584A1 (en) * | 2002-02-22 | 2003-09-04 | Hu Oliver Yoa-Pu | Cytochrome P450 3A inhibitors and enhancers |
Non-Patent Citations (1)
| Title |
|---|
| DRESSER G K ET AL: "PHARMACOKINETIC-PHARMACODYNAMIC CONSEQUENCES AND CLINICAL RELEVANCEOF CYTOCHROME P450 3A4 INHIBITION" CLINICAL PHARMACOKINETICS, LEA & FEBIGER, PHILADELPHIA, PA, US, vol. 38, no. 1, January 2000 (2000-01), pages 41-57, XP000952582 * |
Cited By (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20150105456A1 (en) | 2007-03-08 | 2015-04-16 | The Board Of Trustees Of The Leland Stanford Junior University | Mitochondrial Aldehyde Dehydrogenase-2 Modulators and Methods of Use Thereof |
| US9315484B2 (en) | 2007-03-08 | 2016-04-19 | The Board of Trustees—Leland Stanford Junior University | Mitochondrial aldehyde dehydrogenase-2 modulators and methods of use thereof |
| US9102651B2 (en) | 2007-03-08 | 2015-08-11 | The Board of Trustees-Leland Stanford Junior University | Mitochondrial aldehyde dehydrogenase-2 modulators and methods of use thereof |
| US8906942B2 (en) | 2008-09-08 | 2014-12-09 | The Board Of Trustees Of The Leland Stanford Junior University | Modulators of aldhehyde dehydrogenase activity and methods of use thereof |
| US9345693B2 (en) | 2008-09-08 | 2016-05-24 | The Board of Trustees-Leland Stanford Junior University | Modulators of aldehyde dehydrogenase activity and methods of use thereof |
| EP2349279A4 (en) * | 2008-10-28 | 2013-12-25 | Univ Leland Stanford Junior | Modulators of aldehyde dehydrogenase and methods of use thereof |
| US8772295B2 (en) | 2008-10-28 | 2014-07-08 | The Board Of Trustees Of The Leland Stanford Junior University | Modulators of aldehyde dehydrogenase and methods of use thereof |
| EP2349279A1 (en) * | 2008-10-28 | 2011-08-03 | The Board of Trustees of The Leland Stanford Junior University | Modulators of aldehyde dehydrogenase and methods of use thereof |
| WO2010125831A1 (en) * | 2009-05-01 | 2010-11-04 | Raqualia Pharma Inc. | Sulfamoyl benzoic acid derivatives as trpm8 antagonists |
| US10457659B2 (en) | 2011-04-29 | 2019-10-29 | The Board Of Trustees Of The Leland Stanford Junior University | Compositions and methods for increasing proliferation of adult salivary stem cells |
| US9556202B2 (en) | 2012-06-27 | 2017-01-31 | Merck Sharp & Dohme Corp. | Sulfonamide derivatives and methods of use thereof for improving the pharmacokinetics of a drug |
| WO2014000178A1 (en) * | 2012-06-27 | 2014-01-03 | Merck Sharp & Dohme Corp. | Sulfonamide derivatives and methods of use thereof for improving the pharmacokinetics of a drug |
| WO2014004416A1 (en) | 2012-06-27 | 2014-01-03 | Merck Sharp & Dohme Corp. | Sulfonamide derivatives and methods of use thereof for improving the pharmacokinetics of a drug |
| US10227304B2 (en) | 2013-03-14 | 2019-03-12 | The Board Of Trustees Of The Leland Stanford Junior University | Mitochondrial aldehyde dehydrogenase-2 modulators and methods of use thereof |
| US9670162B2 (en) | 2013-03-14 | 2017-06-06 | The Board Of Trustees Of The Leland Stanford Junio | Mitochondrial aldehyde dehyrogenase-2 modulators and methods of use thereof |
| US9067922B2 (en) | 2013-04-19 | 2015-06-30 | Pfizer Limited | Chemical compounds |
| US9981969B2 (en) | 2013-06-07 | 2018-05-29 | Merck Sharp & Dohme Corp. | Imidazole derivatives and methods of use thereof for improving the pharmacokinetics of a drug |
| WO2014197345A2 (en) | 2013-06-07 | 2014-12-11 | Merck Sharp & Dohme Corp. | Imidazole derivatives and methods of use thereof for improving the pharmacokinetics of a drug |
| US9975888B2 (en) | 2013-11-12 | 2018-05-22 | Merck Sharp & Dohme Corp. | Aryl linked imidazole and triazole derivatives and methods of use thereof for improving the pharmacokinetics of a drug |
| WO2015073310A1 (en) | 2013-11-12 | 2015-05-21 | Merck Sharp & Dohme Corp. | Piperidine or piperazine linked imidazole and triazole derivatives and methods of use thereof for improving the pharmacokinetics of a drug |
| EP3587411A1 (en) | 2013-11-12 | 2020-01-01 | Merck Sharp & Dohme Corp. | Piperidine or thiomorpholine linked imidazole and triazole derivatives and uses thereof for improving the pharmacokinetics of a drug |
| US10745377B2 (en) | 2013-11-12 | 2020-08-18 | Merck Sharp & Dohme Corp. | Piperidine or piperazine linked imidazole and triazole derivatives and methods of use |
| US11098048B2 (en) | 2014-03-26 | 2021-08-24 | Hoffmann-La Roche Inc. | Bicyclic compounds as autotaxin (ATX) and lysophosphatidic acid (LPA) production inhibitors |
| US11352330B2 (en) | 2015-09-04 | 2022-06-07 | Hoffmann-La Roche Inc. | Phenoxymethyl derivatives |
| US11673888B2 (en) | 2017-03-16 | 2023-06-13 | Hoffmann-La Roche Inc. | Bicyclic compounds as ATX inhibitors |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2007034312A2 (en) | Pyridinaminosulfonyl substituted benzamides as inhibitors of cytochrome p450 3a4 (cyp3a4) | |
| US7919488B2 (en) | Therapeutic compounds | |
| CA2647448A1 (en) | Pyrrolidine derivatives as modulators of chemokine ccr5 receptors | |
| US20080161311A1 (en) | Nitrogen-containing fused ring compound and use thereof as HIV integrase inhibitor | |
| CA2595574A1 (en) | Chemical compounds | |
| AU2014234906B2 (en) | Cycloalkyl nitrile pyrazolo pyridones as Janus kinase inhibitors | |
| CN115942972A (en) | LPA receptor antagonists and uses thereof | |
| CA2901766A1 (en) | N-(2-cyano heterocyclyl)pyrazolo pyridones as janus kinase inhibitors | |
| WO2008004100A2 (en) | Therapeutic compounds | |
| EP3068391B1 (en) | Aryl linked imidazole and triazole derivatives and methods of use thereof for improving the pharmacokinetics of a drug | |
| CA2594602A1 (en) | 8-aza-bicyclo (3.2.1) octane derivatives with an activity on chemokine ccr5 receptors | |
| JP2023551272A (en) | Heterocyclic compounds and their uses as diacylglycerol kinase inhibitors | |
| JP2009511463A (en) | Inhibitor of HIV integrase enzyme | |
| EP3587411B1 (en) | 1-(alpha-methylbenzyl)-5-(piperidinomethyl)imidazole derivatives and uses thereof for improving the pharmacokinetics of a drug | |
| WO2006136917A1 (en) | Triazolylpiperidine derivatives and use thereof in therapy | |
| WO2006085199A1 (en) | Piperazine derivatives | |
| WO2006097848A1 (en) | Hiv-1 virion maturation inhibitors, compositions and treatments using the same |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200680034827.8 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11621410 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 11621410 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 566136 Country of ref document: NZ Ref document number: 189666 Country of ref document: IL Ref document number: 2006293605 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: AP/P/2008/004370 Country of ref document: AP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006795544 Country of ref document: EP Ref document number: 1720/DELNP/2008 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2008/002924 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200800542 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2621805 Country of ref document: CA Ref document number: 08024643 Country of ref document: CO |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008030411 Country of ref document: EG |
|
| WWE | Wipo information: entry into national phase |
Ref document number: CR2008-009817 Country of ref document: CR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12008500709 Country of ref document: PH |
|
| ENP | Entry into the national phase |
Ref document number: 2006293605 Country of ref document: AU Date of ref document: 20060911 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006293605 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10575 Country of ref document: GE Ref document number: P-2008/0118 Country of ref document: RS Ref document number: 1020087006967 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: DZP2008000180 Country of ref document: DZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008531812 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 06795544 Country of ref document: EP Kind code of ref document: A2 |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006795544 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P-2010/0237 Country of ref document: RS |
|
| ENP | Entry into the national phase |
Ref document number: PI0616156 Country of ref document: BR Kind code of ref document: A2 Effective date: 20080319 |