WO2007009681A1 - 1 , 1-DIOXID0-2 , 3-DIHYDRO-l , 2-BENZISOTHIAZ0L-6-YL-1H-INDAZOL-4-YL-2 , 4-PYRIMIDINEDI AMINE DERIVATIVES - Google Patents
1 , 1-DIOXID0-2 , 3-DIHYDRO-l , 2-BENZISOTHIAZ0L-6-YL-1H-INDAZOL-4-YL-2 , 4-PYRIMIDINEDI AMINE DERIVATIVES Download PDFInfo
- Publication number
- WO2007009681A1 WO2007009681A1 PCT/EP2006/006953 EP2006006953W WO2007009681A1 WO 2007009681 A1 WO2007009681 A1 WO 2007009681A1 EP 2006006953 W EP2006006953 W EP 2006006953W WO 2007009681 A1 WO2007009681 A1 WO 2007009681A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- fluoro
- indazol
- benzisothiazol
- ethyl
- Prior art date
Links
- 0 Cc(c(Nc1nc(I)ncc1*)ccc1)c1N Chemical compound Cc(c(Nc1nc(I)ncc1*)ccc1)c1N 0.000 description 2
- CXNWVKMEFQKJGQ-UHFFFAOYSA-N CC(Nc1c(C)c(N(CCCOC)c2nc(C)ncc2F)ccc1)=O Chemical compound CC(Nc1c(C)c(N(CCCOC)c2nc(C)ncc2F)ccc1)=O CXNWVKMEFQKJGQ-UHFFFAOYSA-N 0.000 description 1
- MDCQESIFFYEQPN-UHFFFAOYSA-N CC(Nc1c(C)c(Nc(nc(nc2)Cl)c2F)ccc1)=O Chemical compound CC(Nc1c(C)c(Nc(nc(nc2)Cl)c2F)ccc1)=O MDCQESIFFYEQPN-UHFFFAOYSA-N 0.000 description 1
- SVTBIYUFZHUTBS-UHFFFAOYSA-N CCN(c1c(cn[nH]2)c2ccc1)c1nc(Cl)ncc1F Chemical compound CCN(c1c(cn[nH]2)c2ccc1)c1nc(Cl)ncc1F SVTBIYUFZHUTBS-UHFFFAOYSA-N 0.000 description 1
- IVLMQSOUUCWUTG-UHFFFAOYSA-N CN(Cc(cc1)c2cc1N)S2(=O)=O Chemical compound CN(Cc(cc1)c2cc1N)S2(=O)=O IVLMQSOUUCWUTG-UHFFFAOYSA-N 0.000 description 1
- MJRBDTGIFOARNW-UHFFFAOYSA-N Nc1ccc(CNS2(=O)=O)c2c1 Chemical compound Nc1ccc(CNS2(=O)=O)c2c1 MJRBDTGIFOARNW-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing three or more hetero rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
Definitions
- the present invention relates to novel chemical compounds which have activity against the spleen tyrosine kinase (Syk kinase), processes for their preparation, pharmaceutically acceptable formulations containing them and their use in therapy.
- Syk kinase spleen tyrosine kinase
- Allergic rhinitis and asthma are diseases associated with hypersensitivity reactions and inflammatory events involving a multitude of cell types including mast cells, eosinophils, T cells and dendritic cells.
- high affinity immunoglobulin receptors for IgE (Fc ⁇ RI) and IgG (Fc ⁇ RI) become cross-linked and activate downstream processes in mast cells and other cell types leading to the release of pro-inflammatory mediators and airway spasmogens.
- IgE receptor cross-linking by allergen leads to release of mediators including histamine from pre-formed granules, as well as the synthesis and release of newly synthesised lipid mediators including prostaglandins and leukotrienes.
- Syk kinase is a non-receptor linked tyrosine kinase which is important in transducing the downstream cellular signals associated with cross-linking Fc ⁇ RI and or Fc ⁇ RI receptors, and is positioned early in the signalling cascade.
- the early sequence of Fc ⁇ RI signalling following allergen cross-linking of receptor-lgE complexes involves first Lyn (a Src family tyrosine kinase) and then Syk kinase.
- Inhibitors of Syk kinase activity would therefore be expected to inhibit all downstream signalling cascades thereby alleviating the immediate allergic response and adverse events initiated by the release of pro-inflammatory mediators and spasmogens (Wong, B., Grossbard, E.B. Payan, D.G & Masuda, E. S. Expert Opin. Investig. Drugs (2004) 13 (7) 743-762).
- Rheumatoid Arthritis is an auto-immune disease affecting approximately 1 % of the population. It is characterised by inflammation of articular joints leading to debilitating destruction of bone and cartilage.
- Recent clinical studies with Rituximab, which causes a reversible B cell depletion, (J. CW. Edwards et al 2004, New Eng. J. Med. 350: 2572-2581 ) have shown that targeting B cell function is an appropriate therapeutic strategy in auto-immune diseases such as RA.
- Clinical benefit correlates with a reduction in auto-reactive antibodies (or Rheumatoid Factor) and these studies suggest that B cell function and indeed auto-antibody production are central to the ongoing pathology in the disease.
- WO 03/057695 (Boehringer lngelheim Pharmaceuticals, Inc.) describes substituted [1 ,6]-naphthyridines that inhibit Syk kinase.
- WO2003/063794, WO2004/014382, WO2005/012294 and WO2005/16893 describes a series of 2,4-pyrimidinediamine compounds which inhibit Syk kinase, for use in treating autoimmune diseases.
- WO2005/026158 (Novartis AG) describes 2,4-di (hetero)-arylamino-pyrimidine derivatives which have ZAP-70 and/or Syk inhibitory activities.
- WO 04/035604 discloses the structural co-ordinates of the human Syk protein.
- the present invention provides a compound of formula (I): in which:
- R 1 and R 2 is each independently hydrogen, C 1-6 alkyl or CH 2 C 3-7 cycloalkyl; R 3 is C 1-3 alkyl or C 2-3 alkyl substituted by hydroxy or Ci. 3 alkoxy; and X is halogen; or a salt or solvate, preferably a pharmaceutically acceptable salt or solvate, thereof.
- Compounds of the present invention are useful as inhibitors of Syk kinase and thus potentially of use in treating diseases resulting from inappropriate mast cell activation, for instance allergic and inflammatory diseases.
- R 1 include hydrogen, methyl, ethyl, 2-methylpropyl, cyclopropylmethyl, cyclobutylmethyl, and cyclohexylmethyl.
- R 2 include hydrogen.
- R 3 include methyl, ethyl, propyl, -(CH 2 ) n OH and -(CH 2 ) n 0C 1-3 alkyl where n is 3.
- R 3 is methyl, ethyl, propyl, -(CH 2 ) 3 OH and -(CH 2 ) 3 OCH 3 .
- X include fluoro, chloro and bromo.
- X is fluoro
- the present invention provides for a compound of formula (IB):
- R 1 is hydrogen, C 1-6 alkyl or CH 2 C 3-7 cycloalkyl
- R 3 is C 1 . 3 alkyl
- X is halogen; or a salt or solvate, preferably a pharmaceutically acceptable salt or solvate, thereof.
- the present invention provides for a compound of formula (IC):
- R 3 is C 2-3 alkyl substituted by hydroxy or C 1-3 alkoxy
- X is halogen; or a salt or solvate, preferably a pharmaceutically acceptable salt or solvate, thereof.
- Representative examples of compounds of formula (I) include:
- examples of compounds of formula (I) include: / ⁇ -(i .i-dioxido ⁇ .S-dihydro-i ⁇ -benzisothiazol- ⁇ -yO-S-fluoro- ⁇ -IH-indazol ⁇ -yl- ⁇ / 4 - propyl-2,4-pyrimidinediamine; and
- references to alkyl include references to both straight chain and branched chain aliphatic isomers of the corresponding alkyl. It will be appreciated that references to alkylene and alkoxy shall be interpreted similarly.
- references to C 3-7 cycloalkyl include references to all alicyclic (including branched) isomers of the corresponding alkyl.
- the term “pharmaceutically acceptable” refers to those compounds, materials, compositions, and dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, or other problem or complication, commensurate with a reasonable benefit/risk ratio.
- pharmaceutically acceptable salts of the compound of the present invention may be prepared.
- pharmaceutically acceptable salts refers to salts that retain the desired biological activity of the subject compound and exhibit minimal undesired toxicological effects.
- compositions may be prepared in situ during the final isolation and purification of the compound, or by separately reacting the purified compound in its free acid or free base form with a suitable base or acid, respectively. Indeed, in certain embodiments of the invention, pharmaceutically acceptable salts may be preferred over the respective free base or free acid because such salts impart greater stability or solubility to the molecule thereby facilitating formulation into a dosage form.
- the compound of the present invention may contain one or more acidic functional groups.
- suitable pharmaceutically acceptable salts include salts of such acidic functional groups.
- Representative salts include pharmaceutically acceptable metal salts such as sodium, potassium, lithium, calcium, magnesium, aluminum, and zinc salts; carbonates and bicarbonates of a phamnaceutically- acceptable metal cation such as sodium, potassium, lithium, calcium, magnesium, aluminum, and zinc; pharmaceutically acceptable organic primary, secondary, and tertiary amines including aliphatic amines, aromatic amines, aliphatic diamines, and hydroxy alkylamines such as methylamine, ethylamine, 2-hydroxyethylamine, diethylamine, triethylamine, ethylenediamine, ethanolamine, diethanolamine, and cyclohexylamine.
- Compounds of the present invention are basic and accordingly generally capable of forming pharmaceutically acceptable acid addition salts by treatment with a suitable acid.
- Suitable acids include pharmaceutically acceptable inorganic acids and pharmaceutically acceptable organic acids.
- Representative pharmaceutically acceptable acid addition salts include hydrochloride, hydrobromide, nitrate, methylnitrate, sulfate, bisulfate, sulfamate, phosphat ⁇ i acetate, hydroxyacetate, phenylacetate, propionate, butyrate, isobutyrate, valerate, maleate, hydroxymaleate, acrylate, fumarate, malate, tartrate, citrate, salicylate, p-aminosalicyclate, glycollate, lactate, heptanoate, phthalate, oxalate, succinate, benzoate, o-acetoxybenzoate, chlorobenzoate, methylbenzoate, dinitrobenzoate, hydroxybenzoate, methoxybenzoate, mandelate, t
- the term “compound” refers to one or more compounds.
- the term “a compound of the present invention” refers to one or more compounds of the present invention.
- the compound of the present invention may exist in solid or liquid form.
- the compound of the present invention may exist in crystalline or noncrystalline form, or as a mixture thereof.
- pharmaceutically acceptable solvates may be formed wherein solvent molecules are incorporated into the crystalline lattice during crystallization.
- Solvates may involve non-aqueous solvents such as, but not limited to, ethanol, isopropanol, n-butanol, i-butanol, acetone, tetrahydrofuran, dioxane, DMSO, acetic acid, ethanolamine, and ethyl acetate, or they may involve water as the solvent that is incorporated into the crystalline lattice. Solvates wherein water is the solvent incorporated into the crystalline lattice are typically referred to as "hydrates.” Hydrates include stoichiometric hydrates as well as compositions containing variable amounts of water. The invention includes all such solvates.
- polymorphs may exhibit polymorphism (i.e. the capacity to occur in different crystalline structures). These different crystalline forms are typically known as "polymorphs.”
- the invention includes all such polymorphs. Polymorphs have the same chemical composition but differ in packing, geometrical arrangement, and other descriptive properties of the crystalline solid state. Polymorphs, therefore, may have different physical properties such as shape, density, hardness, deformability, stability, and dissolution properties. Polymorphs typically exhibit different melting points, IR spectra, and X-ray powder diffraction patterns, which may be used for identification.
- polymorphs may be produced, for example, by changing or adjusting the reaction conditions or reagents, used in making the compound. For example, changes in temperature, pressure, or solvent may result in polymorphs. In addition, one polymorph may spontaneously convert to another polymorph under certain conditions.
- the compound of the present invention may contain one or more asymmetric centres (also referred to as a chiral centre) and may, therefore, exist as individual enantiomers, diastereoisomers, or other stereoisomeric forms, or as mixtures thereof.
- Chiral centres such as chiral carbon atoms, may also be present in a substituent such as an alkyl group.
- the stereochemistry of a chiral centre is not specified the structure is intended to encompass any stereoisomer and all mixtures thereof.
- the compound of the present invention containing one or more chiral centres may be used as racemic mixtures, enantiomerically enriched mixtures, or as enantiomerically pure individual stereoisomers.
- Individual stereoisomers of a compound according to formula (I) which contain one or more asymmetric centre may be resolved by methods known to those skilled in the art. For example, such resolution may be carried out (1 ) by formation of diastereoisomeric salts, complexes or other derivatives; (2) by selective reaction with a stereoisomer-specific reagent, for example by enzymatic oxidation or reduction; or (3) by gas-liquid or liquid chromatography in a chiral environment, for example, on a chiral support such as silica with a bound chiral ligand or in the presence of a chiral solvent.
- stereoisomers may be synthesized by asymmetric synthesis using optically active reagents, substrates, catalysts or solvents, or by converting one enantiomer to the other by asymmetric transformation.
- the compound of the present invention may also contain double bonds or other centres of geometric asymmetry. Where the stereochemistry of a centre of geometric asymmetry is not specified, the structure is intended to encompass the trans (E) geometric isomer, the cis (Z) geometric isomer, and all mixtures thereof. Likewise, all tautomeric forms are also included the compound of the present invention whether such tautomers exist in equilibrium or predominately in one form.
- the present invention provides a process for preparing a compound of formula (I), or a salt or solvate thereof, which process comprises: reacting a compound of formula (II):
- the process may be performed in the presence of a solvent (for example, aqueous acetone or propan-2-ol), in the presence of concentrated hydrochloric acid, at a suitable temperature, preferably in the range of 0-170° C.
- a solvent for example, aqueous acetone or propan-2-ol
- concentrated hydrochloric acid at a suitable temperature, preferably in the range of 0-170° C.
- the process may be performed in the presence of a solvent such as 2-propanol, and hydrogen chloride in ether, at a suitable temperature, preferably in the range of 0-100°C, preferably under a nitrogen atmosphere.
- a solvent such as 2-propanol, and hydrogen chloride in ether
- the process may be performed in the presence of a solvent such as 2-propanol, and a dilute acid such as dilute hydrochloric acid, at a suitable temperature, preferably in the range of 0- 100°C.
- a solvent such as 2-propanol
- a dilute acid such as dilute hydrochloric acid
- Suitable leaving groups (L 1 ) include a halide such as chloride, bromide or iodide.
- Other leaving groups include, but are not restricted to, an alkyl and aryl sulfide, an alkyl and aryl sulfinyl, an alkyl and aryl sulfonyl, and an alcohol derived leaving group (such as triflate, mesylate, methylsulfonate or tosylate).
- the process may be performed in the presence of a non-nucleophilic base such as triethylamine or di-isopropylethylamine, as a melt, at a suitable temperature, preferably in the range of 0-180°C.
- a non-nucleophilic base such as triethylamine or di-isopropylethylamine
- R 1 in which R 1 is hydrogen, may be converted into further compounds of formula (I) in which R 1 is C 1-6 alkyl or CH 2 C 3 . 7 cycloalkyl by reaction with an appropriate alkylating agent, for instance an alkyl or cycloalkyl halide R 1 L 3 , in the presence of a mild base such as cesium carbonate, in a solvent such as DMF.
- an appropriate alkylating agent for instance an alkyl or cycloalkyl halide R 1 L 3
- the starting compound of formula (I) may be treated with the base, in a preliminary step, prior to addition of the alkylating agent. Examples of protecting groups and the means for their removal can be found in T. W. Greene 'Protective Groups in Organic Synthesis' (J. Wiley and Sons, 1991).
- Suitable amine protecting groups include, but are not restricted to, sulphonyl (such as tosyl), acyl (such as benzyloxycarbonyl or t-butoxycarbonyl) and arylalkyl (such as benzyl), which may be removed by hydrolysis or hydrogenolysis as appropriate.
- Other suitable amine protecting groups include trifluoroacetyl (-C(O)CF 3 ), which may be removed by base catalysed hydrolysis, or a solid phase resin bound benzyl group, such as a Merrifield resin bound 2,6-dimethoxybenzyl group (Ellman linker) which may be removed by acid catalysed hydrolysis (using, for example, trifluoroacetic acid).
- L 1 , and X are as hereinbefore defined, or a protected derivative thereof, by reacting with a sodium hydride in DMF, followed by adding alkyl iodide R 3 I, at a temperature of about O 0 C, under a nitrogen atmosphere.
- L 2 is a leaving group such as chloro and L 1 and X are as hereinbefore defined; in a solvent such as propan-2-ol or acetone in the presence or a base such as di-isopropylethylamine, and at a temperature in the range 120-180 0 C.
- Compounds of formula (II) may also be prepared by reacting a compound of formula (Vl): in which R 3 , L 1 and X are as hereinbefore defined, or a protected derivative thereof, with an organic nitrite derivative, such as, but not limited to, tert-butyl nitrite. This reaction is preferably performed in the presence of an acid, such as acetic acid or hydrochloric acid.
- the aforementioned reaction may be performed in the presence of acetic anhydride to yield a product in which an indazolyl nitrogen atom is protected.
- L 1 and X are as hereinbefore defined, or a protected derivative thereof, with an alkyl iodide R 3 I in the presence of a base such as cesium carbonate, in an aprotic solvent such as DMF.
- L 1 , L 2 and X are as hereinbefore defined, in aqueous methanol, at a temperature of about 7O 0 C.
- the compound of formula (II) is the following N-acetyl protected derivative:
- L 1 , X, and R 3 are as defined above. This may be prepared from the corresponding N-acetyl compounds of formula (Vl), (VII) and (VIII), as hereinbefore described.
- Compounds of the present invention are useful as inhibitors of Syk and thus potentially of use in treating diseases resulting from inappropriate mast cell activation, for instance allergic and inflammatory diseases.
- the present invention provides for a compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof, for use in therapy.
- the present invention provides for a method of treating inappropriate mast cell activation which method comprises administering to a patient in need thereof an effective compound of formula I, or a or a pharmaceutically acceptable salt or solvate thereof.
- the present invention provides a method comprising administering to a patient in need thereof an effective compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof, to inhibit a Syk kinase.
- the present invention provides a method of treating an inflammatory disease which comprises administering to a patient in need thereof an effective compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof.
- the present invention provides a method of treating an allergic disorder which comprises administering to a patient in need thereof an effective compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof.
- Syk kinase diseases and pathological conditions thought to be mediated by Syk kinase include inflammatory and allergic disorders involving mast cell activation, such as chronic obstructive pulmonary disease (COPD), adult respiratory distress syndrome (ARDS), asthma, ulcerative colitis, Crohn's Disease, bronchitis, conjunctivitis, psoriasis, sclerodoma, urticaria, dermatitis, and allergic rhinitis. They also include inflammatory conditions which involve B cells, for instance lupus and rheumatoid arthritis.
- COPD chronic obstructive pulmonary disease
- ARDS adult respiratory distress syndrome
- asthma ulcerative colitis
- Crohn's Disease bronchitis
- conjunctivitis conjunctivitis
- psoriasis psoriasis
- sclerodoma urticaria
- dermatitis dermatitis
- allergic rhinitis allergic r
- Compounds of the present invention may also be used in combination with other classes of therapeutic agents which are known in the art.
- Representative classes of agents for use in such combinations include, for treating asthma, anti-inflammatory steroids (in particular corticosteroids), topical glucocorticoid agonists, PDE4 inhibitors, IKK2 inhibitors, A2a agonists, ⁇ 2 -adrenoreceptor agonists (including both slow acting and long acting ⁇ 2 -adrenoreceptor agonists), alpha 4 integrin inhibitors, and anti-muscarinics, and, for treating allergies, the foregoing agents, as well as H1 and H1/H3 antagonists.
- Representative agents for use in combination therapy for treating severe asthma include topically acting p38 inhibitors, and IKK2 inhibitors.
- Anti-inflammatory corticosteroids are well known in the art. Representative examples include fluticasone propionate (e.g. see US patent 4,335,121), beclomethasone 17- propionate ester, beclomethasone 17,21-dipropionate ester, dexamethasone or an ester thereof, mometasone or an ester thereof (e.g. mometasone furoate), ciclesonide, budesonide, and flunisolide.
- fluticasone propionate e.g. see US patent 4,335,121
- beclomethasone 17- propionate ester beclomethasone 17,21-dipropionate ester
- dexamethasone or an ester thereof dexamethasone or an ester thereof
- mometasone or an ester thereof e.g. mometasone furoate
- ciclesonide e.g. mometasone furoate
- ciclesonide esonide
- anti-inflammatory corticosteroids are described in WO 02/12266 A1 (Glaxo Group Ltd), in particular, the compounds of Example 1 ( 6 ⁇ ,9 ⁇ -difluoro-17 ⁇ -[(2-furanylcarbonyl)oxy]-11 ⁇ -hydroxy- 16 ⁇ -methyl-3-oxo-androsta-1 ,4-diene-17 ⁇ -carbothioic acid S-fluoromethyl ester) and Example 41 (6 ⁇ ,9 ⁇ -difluoro-11 ⁇ -hydroxy-16 ⁇ -methyl-17 ⁇ -[(4-methyl-1 ,3-thiazole-5- carbonyl)oxy]-3-oxo-androsta-1 ,4-diene-17 ⁇ -carbothioic acid S-fluoromethyl ester), or a pharmaceutically acceptable salt thereof.
- ⁇ 2 -adrenoreceptor agonists examples include salmeterol (e.g. as racemate or a single enantiomer such as the R-enantiomer), salbutamol, formoterol, salmefamol, fenoterol or terbutaline and salts thereof, for example the xinafoate salt of salmeterol, the sulphate salt or free base of salbutamol or the fumarate salt of formoterol.
- Long- acting ⁇ 2 -adrenoreceptor agonists are preferred, especially those having a therapeutic effect over a 24 hour period such as salmeterol or formoterol.
- anti-histamines examples include methapyrilene, or loratadine, cetirizine, desloratadine or fexofenadine.
- anticholinergic compounds include muscarinic (M) receptor antagonists, in particular M-
- muscarinic M3 antagonists include ipratropium bromide, oxitropium bromide or tiotropium bromide.
- PDE4 or mixed PDE3/4 inhibitors that may be used in combination with compounds of the invention include AWD-12-281 (Elbion), PD-168787 (Pfizer), roflumilast, and cilomilast (GlaxoSmithKline). Further examples of PDE4 inhibitors are described in WO 2004/103998 (Glaxo Group Ltd).
- the present invention also provides for so-called "triple combination" therapy, comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof together with ⁇ 2 -adrenoreceptor agonist and an anti-inflammatory corticosteroid.
- this combination is for treatment and/or prophylaxis of asthma, COPD or allergic rhinitis.
- the ⁇ 2 -adrenoreceptor agonist and/or the anti-inflammatory corticosteroid can be as described above and/or as described in WO 03/030939 A1.
- a representative example of such a "triple” combination comprises a compound of formula (I) or a pharmaceutically acceptable salt thereof, salmeterol or a pharmaceutically acceptable salt thereof (e.g. salmeterol xinafoate) and fluticasone propionate.
- Preferred compounds of formula (I) for use as inhibitors of Syk kinase are those which exhibit selectivity for the Syk kinase against other key kinases such as Aurora A, Aurora B, Zap 70, JNK3, for instance at least 10x (based on either pKi or plCso values for the enzymes)
- the compound of the present invention will normally, but not necessarily, be formulated into pharmaceutical compositions prior to administration to a patient. Accordingly, in another aspect the invention is directed to pharmaceutical compositions comprising a compound of the invention and one or more pharmaceutically acceptable excipient.
- compositions of the invention may be prepared and packaged in bulk form wherein a safe and effective amount of a compound of the invention can be extracted and then given to the patient, such as with powders or syrups.
- the pharmaceutical compositions of the invention may be prepared and packaged in unit dosage form wherein each physically discrete unit contains a safe and effective amount of a compound of the invention.
- the pharmaceutical compositions of the invention typically contain from about 0.1 to 99.9 wt.%, depending on the nature of the formulation.
- compositions of the invention typically contain one compound of the invention. However, in certain embodiments, the pharmaceutical compositions of the invention contain more than one compound of the invention. For example, in certain embodiments the pharmaceutical compositions of the invention contain two compounds of the invention. In addition, the pharmaceutical compositions of the invention may optionally further comprise one or more additional pharmaceutically active compounds.
- pharmaceutically acceptable excipient means a pharmaceutically acceptable material, composition or vehicle involved in giving form or consistency to the pharmaceutical composition.
- Each excipient must be compatible with the other ingredients of the pharmaceutical composition when commingled, such that interactions which would substantially reduce the efficacy of the compound of the invention when administered to a patient and would result in pharmaceutically unacceptable compositions are avoided.
- each excipient must of course be of sufficiently high purity to render it pharmaceutically acceptable.
- compositions of the present invention comprising a compound of the invention and the pharmaceutically acceptable excipient or excipients will typically be provided as a dosage form adapted for administration to the patient by the desired route of administration.
- dosage forms include those adapted for (1) inhalation, such as aerosols and solutions; and (2) intranasal administration, such as solutions or sprays.
- Suitable pharmaceutically acceptable excipients will vary depending upon the particular dosage form chosen.
- suitable pharmaceutically acceptable excipients may be chosen for a particular function that they may serve in the composition.
- certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the production of uniform dosage forms.
- Certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the production of stable dosage forms.
- Certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the carrying or transporting the compound of the present invention once administered to the patient from one organ, or portion of the body, to another organ, or portion of the body.
- Certain pharmaceutically acceptable excipients may be chosen for their ability to enhance patient compliance.
- Suitable pharmaceutically acceptable excipients include the following types of excipients: Diluents, fillers, lubricants, glidants, granulating agents, wetting agents, solvents, co-solvents, suspending agents, emulsifiers, anticaking agents, chelating agents, viscosity increasing agents, antioxidants, preservatives, stabilizers, surfactants, and buffering agents.
- Diluents Diluents, fillers, lubricants, glidants, granulating agents, wetting agents, solvents, co-solvents, suspending agents, emulsifiers, anticaking agents, chelating agents, viscosity increasing agents, antioxidants, preservatives, stabilizers, surfactants, and buffering agents.
- certain pharmaceutically acceptable excipients may serve more than one function and may serve alternative functions depending on how much of the excipient is present in the formulation and what other ingredients are present in the formulation.
- compositions of the invention are prepared using techniques and methods known to those skilled in the art. Some of the methods commonly used in the art are described in Remington's Pharmaceutical Sciences (Mack Publishing Company).
- Dosage forms for nasal or inhaled administration may conveniently be formulated as aerosols, solutions, drops, gels or dry powders.
- Dosage forms for topical administration to the nasal cavity include pressurised aerosol formulations and aqueous formulations administered to the nose by pressurised pump.
- Formulations which are non-pressurised and adapted for nasal administration are of particular interest. Suitable formulations contain water as the diluent or carrier for this purpose.
- Aqueous formulations for administration to the nose may be provided with conventional excipients such as buffering agents, tonicity modifying agents and the like. Aqueous formulations may also be administered to the nose by nebulisation.
- dosage forms for nasal administration are provided in a metered dose device.
- the dosage form may be provided as a fluid formulation for delivery from a fluid dispenser having a dispensing nozzle or dispensing orifice through which a metered dose of the fluid formulation is dispensed upon the application of a user-applied force to a pump mechanism of the fluid dispenser.
- Such fluid dispensers are generally provided with a reservoir of multiple metered doses of the fluid formulation, the doses being dispensable upon sequential pump actuations.
- the dispensing nozzle or orifice may be configured for insertion into the nostrils of the user for spray dispensing of the fluid formulation into the nasal cavity.
- the fluid dispenser is of the general type described and illustrated in WO-A-2005/044354.
- the dispenser has a housing which houses a fluid discharge device having a compression pump mounted on a container for containing a fluid formulation.
- the housing has at least one finger-operable side lever which is movable inwardly with respect to the housing to cam the container upwardly in the housing to cause the pump to compress and pump a metered dose of the formulation out of a pump stem through a nasal nozzle of the housing.
- a particularly preferred fluid dispenser is of the general type illustrated in Figures 30-40 of WO-A- 2005/044354.
- the compound or salt of formula (I) is in a particle-size-reduced form, and more preferably the size-reduced form is obtained or obtainable by micronisation.
- the preferable particle size of the size-reduced (e.g. micronised) compound or salt or solvate is defined by a D50 value of about 0.5 to about 10 microns (for example as measured using laser diffraction).
- Aerosol compositions can comprise a solution or fine suspension of the active substance in a pharmaceutically acceptable aqueous or non-aqueous solvent. Aerosol formulations can be presented in single or multidose quantities in sterile form in a sealed container, which can take the form of a cartridge or refill for use with an atomising device or inhaler. Alternatively the sealed container may be a unitary dispensing device such as a single dose nasal inhaler or an aerosol dispenser fitted with a metering valve (metered dose inhaler) which is intended for disposal once the contents of the container have been exhausted.
- a metering valve metered dose inhaler
- the dosage form comprises an aerosol dispenser
- it preferably contains a suitable propellant under pressure such as compressed air, carbon dioxide or an organic propellant such as a hydrofluorocarbon (HFC).
- suitable HFC propellants include 1 ,1 ,1 ,2,3,3,3-heptafluoropropane and 1 ,1 ,1 ,2-tetrafluoroethane.
- the aerosol dosage forms can also take the form of a pump-atomiser.
- the pressurised aerosol may contain a solution or a suspension of the active compound. This may require the incorporation of additional excipients e.g. co-solvents and/or surfactants to improve the dispersion characteristics and homogeneity of suspension formulations. Solution formulations may also require the addition of co-solvents such as ethanol.
- Other excipient modifiers may also be incorporated to improve, for example, the stability and/or taste and/or fine particle mass characteristics (amount and/or profile) of the formulation.
- the pharmaceutical composition is a dry powder inhalable composition.
- a dry powder inhalable composition can comprise a powder base such as lactose, glucose, trehalose, mannitol or starch, the compound of formula (I) or salt or solvate thereof (preferably in particle-size-reduced form, e.g. in micronised form), and optionally a performance modifier such as L-leucine or another amino acid, cellobiose octaacetate and/or metals salts of stearic acid such as magnesium or calcium stearate.
- the dry powder inhalable composition comprises a dry powder blend of lactose and the compound of formula (! or salt thereof.
- the lactose is preferably lactose hydrate e.g. lactose monohydrate and/or is preferably inhalation- grade and/or fine-grade lactose.
- the particle size of the lactose is defined by 90% or more (by weight or by volume) of the lactose particles being less than 1000 microns (micrometres) (e.g. 10-1000 microns e.g. 30-1000 microns) in diameter, and/or 50% or more of the lactose particles being less than 500 microns (e.g. 10-500 microns) in diameter. More preferably, the particle size of the lactose is defined by 90% or more of the lactose particles being less than 300 microns (e.g.
- the particle size of the lactose is defined by 90% or more of the lactose particles being less than 100-200 microns in diameter, and/or 50% or more of the lactose particles being less than 40- 70 microns in diameter.
- a suitable inhalation-grade lactose is E9334 lactose (10% fines) (Borculo Domo Ingredients, Hanzeplein 25, 8017 JD Zwolle, Netherlands).
- a pharmaceutical composition for inhaled administration can be incorporated into a plurality of sealed dose containers (e.g. containing the dry powder composition) mounted longitudinally in a strip or ribbon inside a suitable inhalation device.
- the container is rupturable or peel-openable on demand and the dose of e.g. the dry powder composition can be administered by inhalation via the device such as the DISKUS TM device, marketed by GlaxoSmithKline.
- the DISKUS TM inhalation device is for example described in GB 2242134 A, and in such a device at least one container for the pharmaceutical composition in powder form (the container or containers preferably being a plurality of sealed dose containers mounted longitudinally in a strip or ribbon) is defined between two members peelably secured to one another; the device comprises: a means of defining an opening station for the said container or containers; a means for peeling the members apart at the opening station to open the container; and an outlet, communicating with the opened container, through which a user can inhale the pharmaceutical composition in powder form from the opened container.
- a composition of the present invention, for intranasal administration may also be adapted for dosing by insufflation, as a dry powder formulation.
- the compound of the present invention when administered in combination with other therapeutic agents normally administered by the inhaled, intravenous, oral or intranasal route, that the resultant pharmaceutical composition may be administered by the same routes.
- the compound of the present invention may conveniently be administered in amounts of, for example, 1 ⁇ g to 100mg.
- the precise dose will of course depend on the age and condition of the patient and the particular route of administration chosen.
- Recombinant human Syk was expressed as a His-tagged protein * .
- the activity of Syk was assessed using a time-resolved fluorescence resonance energy transfer (TR-FRET) assay.
- TR-FRET time-resolved fluorescence resonance energy transfer
- the reaction was incubated for 40min at room temperature, then terminated by the addition of 3 ⁇ l of read reagent containing 60 mM EDTA, 15OmM NaCI, 5OnM Streptavidin APC (Prozyme, San Leandro, California, USA), 0.5nM antiphosphotyrosine antibody labelled with W-1024 europium chelate (Wallac OY, Turku, Finland) in 4OmM HEPES pH 7.4, 0.03% BSA. The reaction was further incubated for 60min at room temperature.
- the degree of phosphorylation of Biotin- AAAEEIYGEI was measured using a BMG Rubystar plate reader (BMG LabTechnologies Ltd, Aylesbury, UK) as a ratio of specific 665 nm energy transfer signal to reference europium 620 nm signal.
- Version B - Syk was pre-activated at room temperature for 30 mins in the presence of 16.6mM MgCI 2 , 8.3mM ATP and then diluted to 4nM in 4OmM Hepes pH 7.4, 0.01% BSA.
- 3 ⁇ l of substrate reagent containing biotinylated peptide, Biotin- AAAEEIYGEI (0.5 ⁇ M final), ATP (30 ⁇ M final) and MgCI 2 (1OmM final) in 4OmM HEPES pH 7.4, 0.01% BSA were added to wells containing 0.1 ⁇ l of various concentrations of compound or DMSO vehicle (1.7% final) in Greiner low volume 384 well black plate.
- the reaction was initiated by the addition of 3 ⁇ l of diluted Syk (2nM final). The reaction was incubated for 60min at room temperature, then terminated by the addition of 3 ⁇ l of read reagent containing 60 mM EDTA, 15OmM NaCI, 5OnM Streptavidin APC (Prozyme, San Leandro, California, USA), 0.5nM antiphosphotyrosine antibody labelled with W-1024 europium chelate (Wallac OY, Turku, Finland) in 4OmM HEPES pH 7.4, 0.03% BSA. The reaction was further incubated for 45min at room temperature.
- the degree of phosphorylation of Biotin- AAAEEIYGEI was measured using a BMG Rubystar plate reader (BMG LabTechnologies Ltd, Aylesbury, UK) as a ratio of specific 665 nm energy transfer signal to reference europium 620 nm signal.
- Compounds according to the present invention were assayed in this, or a similar Time-resolved fluorescence resonance energy transfer kinase assay, and gave IC 50 values less than 10 ⁇ M.
- the 30OmM Imidazole fractions were pooled buffer exchanged using G25M (Amersham Biosciences, Buckinghamshire, UK) into 2OmM MES pH 6.0, 2OmM NaCI, 1OmM ⁇ McEtOH,10% glycerol.
- the buffer exchanged 6His-Syk was loaded onto a Source15S column (Amersham Biosciences, Buckinghamshire, UK) and the column eluted using a NaCI gradient 0-50OmM over 50 column volumes.
- the 6His-Syk containing fractions were pooled and concentrated by ultra-filtration. The identity of 6His-Syk was confirmed by peptide mass finger printing and intact LC-MS.
- Cells of the mouse fibroblast cell line NIH-3T3 are stably transfected with a cFms- SYK chimera.
- Addition of the ligand (MCSF) produces dimerisation of the chimera resulting in autophosphorylation of the SYK kinase domain.
- MCSF ligand
- Cells are plated at a density of 1x10 5 /well in a volume of 200 ⁇ l growth medium (DMEM containing 10% heat inactivated foetal calf serum, 1% L-glutamine, 400 ⁇ g/ml geneticin and 400 ⁇ g/ml zeocin) in 96 well Collagen 1 coated tissue culture plates. Following incubation at 37°C, 10% CO 2 , for 2Oh, the cell supernatant is removed and replaced with 200 ⁇ l DMEM containing 1% penicillin/streptomycin (serum free DMEM). The cells are incubated for one hour under the conditions described above. The medium is removed, 50 ⁇ l appropriately diluted compound solution added and the plate incubated for a further hour.
- DMEM containing 10% heat inactivated foetal calf serum, 1% L-glutamine, 400 ⁇ g/ml geneticin and 400 ⁇ g/ml zeocin
- Cells are stimulated with 25 ⁇ l MCSF (0.66 ⁇ g/ml final) for 20min at 37 0 C. After removal of the supernatant, the cells are washed with cold PBS and lysed with 100 ⁇ l lysis buffer for 4h at 4°C.
- Cells are plated at a density of 1x105/well in a volume of 200 ⁇ l growth medium (DMEM containing 10% heat inactivated foetal calf serum, 1% L-glutamine, 400 ⁇ g/ml geneticin and 400 ⁇ g/ml zeocin) in 96 well Collagen 1 coated tissue culture plates. Following incubation at 37°C, 10% CO2, for 2Oh the cell supernatant is removed and 50 ⁇ l appropriately diluted compound solution added and the plate incubated for an hour. Cells are stimulated with 25 ⁇ l MCSF (0.66 ⁇ g/ml final) for 20min at 37°C. After removal of the supernatant, the cells are washed with cold PBS and lysed with 100 ⁇ l lysis buffer for 4h at 4°C.
- DMEM containing 10% heat inactivated foetal calf serum, 1% L-glutamine, 400 ⁇ g/ml geneticin and 400 ⁇ g/ml zeocin
- 85 ⁇ l cell lysate is transferred to a 96 well ELISA plate coated with goat anti human M-CSF R capture antibody and incubated for 16 hours at 4°C.
- the plate is washed and a biotinylated anti-phosphotyrosine detection antibody added (100 ⁇ l/well) for 2h at room temperature. This is removed and replaced with 100 ⁇ l Streptavidin-HRP for 30min. Captured phosphorylated SYK is visualised using 100 ⁇ l TMB substrate. The reaction is terminated with 50 ⁇ l 1 M sulphuric acid and the absorbance measured at 450nm.
- Compound Preparation Compound is prepared as a 1OmM stock in DMSO and a dilution series prepared in DMSO using 9 successive 5-fold dilutions. This dilution series is diluted a further 1 :333 with serum free DMEM to give the concentration range to be tested of 1x10 5 to 1.54x10 "11 M. Compound dilutions are prepared using the Biomek 2000 or Biomek Nx automated robotic pipetting systems.
- the population of B cells observed in this assay are the naive mature IgM/lgD expressing population. These form at least 70% of the purified B cell population (the rest being isotype switched memory B cells) and are the only cells that proliferate as the cells are stimulated with anti-lgM.
- Anti-lgM drives signalling through the B cell receptor which is Syk dependant. Proliferation is a functional measure of B cell signalling that can be measured by observing the incorporation of tritiated methyl thymidine into the cells.
- Protocol Purified human tonsillar B cells are resuspended in Buckleys * medium at a concentration of 1.25 x 10 6 ml.
- 160 ⁇ l of cells re-suspended in Buckley's medium is added to the compound and control wells of a 96 well plate.
- the control wells are located on column 11 and 12 of the 96 well plate.
- the background wells are located in column 12 and 20 ⁇ l of 10 ⁇ M control is added to provide an appropriate background control.
- 20 ⁇ l of 1% DMSO is added to the wells in column 1 1 for the stimulated control.
- the compound titrations are located between columns 1 and 10. Three compounds are run in duplicate on each plate and row A and B are used for the control compound titration.
- the final concentration of DMSO is 0.1% in the assay.
- the cells are left for 45min, after 45min the proliferative stimulus is added to the first 11 wells of the 96 well plate and 20 ⁇ l of medium is added to column 12.
- F(ab')2 fragments of a polyclonal goat anti-sera raised to human IgM is used at a final concentration of 15 ⁇ g/ ml to stimulate the cells. (Biosource. Cat no: AMI 4601 ).
- Tritiated methyl thymidine is added to the cells at a concentration of 1 ⁇ Ci per well. (Amersham, TRK 758). The radioactivity is added 65 hours after the initial stimulus and is left on the cells for 6 to 8 hours. After pulsing with methyl thymidine the cells are harvested on a Skatron 96 well cell harvester onto glass fibre mats. Once these have dried these are counted on a Wallac 1450 Microbeta scintillation counter.
- Data is downloaded as an XL file and IC50's determined using Activity base.
- Buckleys Medium 450 ml Iscoves (Sigma I 3390), 50ml FCS, 2.5 g BSA, 5ml Pen/ strep, 5ml Glutamine (20OmM), 500 ⁇ l Apo transferrin (50mg/ml) Sigma (T 1147), 100 ⁇ l Bovine Insulin (10mg/ml) Sigma (I 1882).
- Compound is prepared as a 1OmM stock in DMSO and a dilution series prepared in DMSO using 9 successive 3-fold dilutions.
- This dilution series is diluted a further 1 :100 with Buckleys medium to give the concentration range to be tested of 100 ⁇ M to 5nM.
- This is added as 20 ⁇ l to 96 well plates in duplicate to generate two IC50's for each compound tested. Each plate is run in the presence of a control compound, which acts as an internal standard. .
- LAD2 is a stem cell factor (SCF)-dependent human mast cell line that was established by the NIH from bone marrow aspirates from a patient with mast cell sarcoma/leukaemia.
- SCF stem cell factor
- LAD2 cells resemble CD34+-derived human mast cells and express functional Fc ⁇ RI.
- the Fc ⁇ RI is up-regulated in the presence of IL-4, SCF and IgE, subsequent cross linking of cell-bound IgE results in degranulation which can be measured as hexosaminidase release.
- LAD2 cells are re-suspended at 1x10 5 /ml in complete stem pro-34SFM (Gibco Cat 10640-019 media containing Stem Pro-34 nutrient supplement (1 :40), glutamine
- LAD2 cells Activation of LAD2 cells with anti-lgE Version A Primed LAD2 cells are centrifuged (30Og, 5min), the supernatant discarded and the cell pellet re-suspended at 1x10 4 cells/ml in RPMI supplemented with glutamine (2mM). Following a further centrifugation (30Og, 5min) the cells are re-suspended in fresh RPMI with glutamine (2mM), adjusted to a density of 2.85x10 5 /ml, and pipetted into sterile V-well plates (70 ⁇ l/well; Greiner) containing 20 ⁇ l diluted compound (prepared as detailed above).
- Cells are then incubated for 1 h (37°C, 5% CO 2 in a humidified atmosphere) before activating with a sub-maximal concentration of anti- lgE (10 ⁇ l volume to give a final assay dilution of 1 :2700; Sigma).
- a sub-maximal concentration of anti- lgE 10 ⁇ l volume to give a final assay dilution of 1 :2700; Sigma.
- plates are centrifuged (120Og, 10min, 4 C C) and the supernatant removed for hexosaminidase assay.
- the cell pellet is lysed in 100 ⁇ l/well triton-X (0.5% in RPMI 2mM glutamine) at 37°C for 30min.
- Primed LAD2 cells are centrifuged (40Og, 5min), the supernatant discarded and the cell pellet re-suspended at 1x10 4 cells/ml in RPMI supplemented with glutamine (2mM). Following a further centrifugation (40Og, 5min) the cells are re-suspended in fresh RPMI with glutamine (2mM), adjusted to a density of 5.7 x10 5 AnI, and pipetted into sterile V-well plates (70 ⁇ l/well; Greiner) containing 20 ⁇ l diluted compound (prepared as detailed above).
- Cells are then incubated for 1 h (37°C, 5% CO 2 in a humidified atmosphere) before activating with a sub-maximal concentration of anti- IgE (10 ⁇ l volume to give a final assay dilution of 1 :2700; Sigma).
- a sub-maximal concentration of anti- IgE 10 ⁇ l volume to give a final assay dilution of 1 :2700; Sigma.
- plates are centrifuged (120Og, 10min, 4 0 C) and the supernatant removed for hexosaminidase assay.
- the cell pellet is lysed in 100 ⁇ l/well triton-X (0.5% in RPMI 2mM glutamine) at 37°C for 30min.
- Beta-hexosaminidase activity is measured by the conversion of 4-methylumbelliferyl N-acetyl- ⁇ -D glucosaminide (Sigma) to a fluorescent product.
- a useful screening strategy comprises assay 1 (enzyme assay (pKi), assay 2 and then assay 3 (B Cell Proliferation) or assay 4 (LAD2).
- DCM dichloromethane DMSO refers to dimethylsulfoxide.
- DMF refers to ⁇ /, ⁇ /-dimethylformamide IPA refers to propan-2-ol
- THF refers to tetrahydrofuran.
- HPLC refers to high performance liquid chromatography.
- SPE refers to solid phase extraction cartridges marketed by lsolute
- LC/MS was conducted on a Supelcosil LCABZ+PLUS column (3.3 cm x 4.6 mm ID) eluting with 0.1% HCO2H and 0.01 M ammonium acetate in water (solvent A) and 0.05% HCO 2 H 5% water in acetonitrile (solvent B), using the following elution gradient 0.0-7min 0%B, 0.7-4.2 min 100%B, 4.2-5.3 min 0%B, 5.3-5.5min 0%B at a flow rate of 3ml/min.
- the mass spectra were recorded on a Fisons VG Platform spectrometer using electrospray positive and negative mode (ES+ve and ES-ve).
- Mass directed autoprep / "preparative mass directed HPLC” was conducted on a system such as; a Waters FractionLynx system comprising of a Waters 600 pump with extended pump heads, Waters 2700 autosampler, Waters 996 diode array and Gilson 202 fraction collector on a 10 cm 2.54 cm ID ABZ+ column, eluting with either 0.1% formic acid or trifluoroacetic acid in water (solvent A) and 0.1% formic or trifluoroacetic acid in acetonitrile (solvent B) using the appropriate elution gradient.
- Mass spectra were recorded on Micromass ZMD mass spectrometer using electrospray positive and negative mode, alternate scans. The software used was MassLynx 3.5 with OpenLynx and FractionLynx optio or using equivalent alternative systems.
- “Hydrophobic frits” refers to filtration tubes sold by Whatman. SPE (solid phase extraction) refers to the use of cartridges sold by International Sorbent Technology Ltd.
- the Flashmaster Il is an automated multi-user flash chromatography system, available from Argonaut Technologies Ltd, which utilises disposable, normal phase, SPE cartridges (2 g to 100 g). It provides quaternary on-line solvent mixing to enable gradient methods to be run. Samples are queued using the multi-functional open access software, which manages solvents, flow-rates, gradient profile and collection conditions.
- the system is equipped with a Knauer variable wavelength uv-detector and two Gilson FC204 fraction-collectors enabling automated peak cutting, collection and tracking.
- Silica chromatography techniques include either automated (Flashmaster) techniques or manual chromatography on pre-packed cartridges (SPE) or manually- packed flash columns.
- Microwave chemistry was typically performed in sealed vessels, irradiating with a suitable microwave reactor system, such as a Biotage InitiatorTM Microwave Synthesiser.
- a microwave vessel was charged with the crude ⁇ /-(2-chloro-5-fluoro-4-pyrimidinyl)- ⁇ /-ethyl-1-( ⁇ [2-(trimethylsilyl)ethyl]oxy ⁇ methyl)-1H-indazol-4-amine dissolved in IPA (3ml) and hydrochloric acid (2M, 0.2ml). 2,3-Dihydro-1 ,2-benzisothiazol-6-amine 1 ,1- dioxide (30mg) was added and the mixture was heated in the microwave at 150°C for 30min.
- a microwave vessel was charged with the crude ⁇ /-(2-chloro-5-chloro-4-pyrimidinyl)- ⁇ /-ethyl-1-( ⁇ [2-(trimethylsilyl)ethyl]oxy ⁇ methyl)-1H-indazol-4-amine dissolved in IPA (3ml) and hydrochloric acid (2M, 0.2ml). 2,3-Dihydro-1 ,2-benzisothiazol-6-amine 1 ,1- dioxide (30mg) was added and the mixture was heated in the microwave at 150 0 C for 30min.
- a suspension of ethyl 6-nitro-1 ,2-benzisothiazole-2(3H)-carboxylate 1 ,1-dioxide (10.Og), Pd/C (5%, 1.0g, 50%wet) and methanesulfonic acid (4.5ml) in IPA (100ml) was hydrogenated under hydrogen (2bar) at 50 0 C for ⁇ 6min.
- the mixture was purged with nitrogen, cooled to 2O 0 C and treated with a mixture of sodium hydroxide (2M, 90ml) and sodium hydroxide (10M, 10ml).
- the reaction was stirred for 2h at ambient temperature and filtered through a filter pad, washing the pad with water (20ml).
- the second portion was absorbed onto a 75Og silica cartridge before being purified using a CombiFlash ® CompanionTM system eluting with a gradient of ethyl acetate in cyclohexane (0-100%).
- the required pure fractions were combined and the solvent was evaporated in vacuo to give a further quantity of the ⁇ /-(2-chloro-5-fluoro-4- pyrimidinyl)-1-( ⁇ [2-(trimethylsilyl)ethyl]oxy ⁇ methyl)-1/-/-indazol-4-amine as an orange/brown solid (10.96g).
- Example 3 formic acid - 5-bromo-/vVi .1-dioxido-2,3-dihvdro-1 ,2-benzisothiazol-6- vD- ⁇ -ethyl- ⁇ -i /-/-indazol-4-yl-2,4-pyrimidinediamine (1 :1)
- a microwave vessel was charged with ⁇ /-(2-chloro-5-fluoro-4-pyrimidinyl)- ⁇ /-ethyl-1- ( ⁇ [2-(trimethylsilyl)ethyl]oxy ⁇ methyl)-1H-indazol-4-amine (0.05g) and 2,3-dihydro-1 ,2- benzisothiazol-6-amine 1 ,1 -dioxide (0.025g), followed by and a solution of acetone/water/conc. hydrochloric acid (150:100:1 , 3ml) and N-methyl pyrrolidinone (0.1ml). The mixture was heated by microwave irradiation in a sealed vessel at 150 0 C for 60min.
- 2,3-Dihydro-1 ,2-benzisothiazol-6-amine 1 ,1-dioxide (185mg) was dissolved in dry methanol (15ml). Benzaldehyde (138 ⁇ l) was added and the mixture stirred at room temperature under nitrogen for 4h. The solvent was removed in vacuo and the residue re-dissolved in dry methanol (10ml) and benzaldehyde (100 ⁇ l) added. The reaction mixture was stirred for a further 3h, filtered, the solid washed with a minimal amount of methanol and sucked dry on the sinter.
- Example 6 - ⁇ / ⁇ d .i-dioxido ⁇ .S-dihydro-i ⁇ -benzisothiazol-e-vn- ⁇ -fluoro- ⁇ -i /-/- indazol-4-yl- ⁇ / 4 -methyl-2.4-pyrimidinediamine trifluoroacetate
- N-(2,5-dichloro-4-pyrimidinyl)-N-ethyl-1 -( ⁇ [2-(trimethylsilyl)ethyl]oxy ⁇ methyl)-1 H- indazol-4-amine (520mg) in 5M hydrochloric acid / IPA (2:1 ; 15ml) was heated at 55°C for 6.5h. The cooled mixture was then evaporated to dryness in vacuo and the crude residue was left at room temperature overnight. The resultant residue was basified with saturated sodium carbonate solution (20ml) and extracted with ethyl acetate (3x 30ml). The combined organic extracts were washed with water (2x 20ml), dried (MgSO 4 ) and the solvent evaporated in vacuo.
- the residue was purified by chromatography on a silica cartridge (5Og) eluting with an ethyl acetate / cyclohexane gradient (0-50%) over 40min. The product containing fractions were combined and reduced to dryness in vacuo. The residue was purified by mass directed autoprep to give, after evaporation of the solvents, ⁇ /-(2,5-dichloro-4-pyrimidinyl)- ⁇ /-ethyl-1H- indazol-4-amine (79mg). LC/MS; Rt 3.33min. MH + 308, 310.
- N-(2,5-dichloro-4-pyrimidinyl)-1-( ⁇ [2-(trimethylsilyl)ethyl]oxy ⁇ methyl)-1 H-indazol-4- amine 870mg was dissolved in DMF (20ml) at room temperature under nitrogen. Cesium carbonate (760mg) was added and the mixture stirred for 30min. Ethyl iodide (255 ⁇ l) was added and the mixture was left to stir at 60 0 C for 1.5h. The solvent was evaporated in vacuo and the residue was partitioned between ethyl acetate and water. The organic extract was washed with water (3x 15ml), dried (MgSO 4 ) and reduced to dryness.
- a fourth reaction used 1-acetyl- ⁇ /-(2-chloro-5-fluoro-4- pyrimidinyl)- ⁇ /-ethyl-1H-indazol-4-amine (2.Og) and 2,3-dihydro-1 ,2-benzisothiazol-6- amine 1 ,1-dioxide (1.21g) in 15ml of the solvent mix.
- Example 8 - / 1 Z-(I , i-dioxido ⁇ .S-dihvdro-i ⁇ -benzisothiazol-e-v ⁇ - ⁇ -ethyl- ⁇ -fluoro- ⁇ i H-indazol-4- ⁇ l-2,4-pyrimidinediamine
- the mixture was seeded, aged at 40 0 C for 1h, then cooled to 15°C over 1h and aged for 1h.
- the product was isolated by filtration, washed with methanol (2x 130ml) and dried in vacuo at 50°C to give the title compound as a beige solid (30.7g) containing some residual solvents.
- the orange mixture was treated cautiously with aqueous potassium carbonate (1 M, 259ml) over 20min, stirred for a further 30min and the layers were separated.
- the organic layer was washed with water (150ml) and the organic layer was concentrated to ca 110ml under reduced pressure.
- the residue was diluted by addition of propan-1-ol (296ml) and the organic layer was concentrated to ca 110ml under reduced pressure. Further propan-1-ol (222ml) was added and the organic layer was concentrated to ca 110ml under reduced pressure.
- the residue was diluted with further propan-1-ol (110ml), warmed to 40-50 0 C, and treated with water (222ml) over 20min.
- the slurry was aged at ca 50 0 C for 15min, then cooled to 5°C over 3h.
- the product was isolated by filtration, washed with cold water / propan-1-ol (1 :1 , 2x 75ml), and dried in vacuo to give the title compound as a yellow solid (34.91 g).
- the mixture was seeded with ⁇ /- ⁇ 3-[(2-chloro-5-fluoro-4-pyrimidinyl)(ethyl)amino]-2-methylphenyl ⁇ acetamide, aged for 30min at 60 0 C and treated with further water (100ml) over 30min.
- the yellow suspension was aged for 1 h, cooled to 10 0 C and aged for 2h.
- the product was isolated by filtration, washed with water / DMF (2:1 , 60ml) and then water (2x 60ml). The product was dried in vacuo at 55-60 0 C to give the title product as a yellow solid (20.1g).
- the mixture was further cooled to 5-10 0 C, aged for 2h and the solid isolated by filtration.
- the product was washed with water / IPA (3:1 , 100ml), then water (2x 100ml), and dried in vacuo to give the title product as a white solid (35.15g).
- Example 10 - ⁇ / 4 -ethyl- ⁇ / 2 -(2-ethyl-1.1-dioxido-2.3-dihvdro-1 ,2-benzisothiazol-6-yl)-5- fluoro- ⁇ / 4 -1/-/-indazol-4-yl-2,4-pyrimidinediamine trifluoroacetate
- Example 14 - ⁇ -d .i-dioxido ⁇ .S-dihvdro-i ⁇ -benzisothiazol- ⁇ -vD- ⁇ -fluoro- ⁇ -i /-/- indazoM-yl- ⁇ propyl ⁇ -pyrimidinediamine
- the liquor was decanted from the oily residue, the oily solid re-dissolved in IPA ( ⁇ 20ml) and water ( ⁇ 35ml) added; product precipitated as an oily residue.
- the decanted filtrates and the oily solid were recombined in methanol and the organic solvent removed in vacuo.
- the resultant solid was filtered, washed sequentially with water and dichloromethane to give the hydrochloride salt of the title compound as light brown solid (4.Og).
- Example 15 - ⁇ -d .i-dioxido ⁇ .S-dihvdro-i ⁇ -benzisothiazol-e-vn- ⁇ -fluoro- ⁇ -I H- indazol-4-yl- ⁇ / 4 -f3-(methyloxy)propyll-2.4-Pyrimidinediamine trifluoroacetate
- the reaction mixture was concentrated and the residue purified using SCX SPE (500mg) by loading the residue in methanol and eluting with methanol then ammonia in methanol.
- the ammonia fraction was concentrated and the residue dissolved in DMSO / methanol (1:1) and purified using Mass Directed HPLC.
- the product fractions were concentrated and purified again using SCX SPE (500mg) loading in methanol and eluting with methanol then ammonia in methanol.
- the ammonia fraction was concentrated and the residue dissolved in DMSO / methanol (1 :1 ) and re-purified using Mass Directed HPLC.
- the fractions containing product were evaporated to dryness to give the title compound (9.2mg): LC/MS; Rt 2.84min, MH + 484.
- the reaction was treated with 2,3-dihydro-1 ,2-benzisothiazol-6-amine 1 ,1-dioxide (92mg, Manchester Organics) and irradiated in a Biotage microwave at 16O 0 C for a further 2x 30min.
- the reaction was concentrated and applied to an SCX SPE (10g), eluting with methanol (50ml) and ammonia in methanol (50ml).
- the ammonia fraction was concentrated and absorbed onto florisil (3g).
- the solid was applied to a silica cartridge (2Og) and the cartridge eluted with an ethyl acetate / cyclohexane gradient (0-100%) over 30min.
- the product fractions were evaporated to dryness to give the title compound. (125mg). LC/MS; Rt 2.88min, MH + 484.
- 2,3-Dihydro-1 ,2-benzisothiazol-6-amine 1 ,1 -dioxide 13.11g was added to a mixture of 1 -acetyl- ⁇ /-(2-chloro-5-fluoro-4-pyrimidinyl)- ⁇ /-propyl-1 /-/-indazol-4-amine (22.46g) in IPA (125ml,) stirring at room temperature under nitrogen.
- the resulting suspension was treated with hydrochloric acid (2M, 25.4ml) and the mixture heated to reflux and maintained at that temperature for ⁇ 24h.
- the solid was divided into 1.5g portions, each portion dissolved in DMF (10ml) and purified by preparative HPLC on a 2" x 25cm column packed with Kromasil C8 10 ⁇ m RP and eluting with a gradient of 10 -100% of (70:30 acetonitrile / 0.1 M ammonium dihydrogenphosphate, adjusted to pH 2.6 with phosphoric acid) in (0.1 M ammonium dihydrogenphosphate, adjusted to pH 2.6 with phosphoric acid) over 21.5mins (flow rate 100ml/min). Material eluting in a broad peak at ⁇ 15mins was collected from each run, the fractions from the separate runs combined and the acetonitrile evaporated in vacuo.
- Example 20 - /v ⁇ -d .i-dioxido ⁇ .S-dihydro-i ⁇ -benzisothiazol-e-vD-S-fluoro- ⁇ -i/-/- indazol-4-yl- ⁇ / 4 -r3-(methyloxy)propyll-2,4-pyrimidinediamine
- the solid was then dried in vacuo at 45°C, before suspending in methanol (460ml). To the stirring mixture was added water (153ml), the resulting mixture was heated to 65°C and treated with sodium hydroxide (2M, 30ml). The solution was allowed to cool to room temperature, the resulting precipitate collected by filtration, and washed with methanol / water (1 :1), then water. The solid was dried in vacuo at 40 0 C.
- the solid was divided into 1.5g portions, each portion dissolved in DMF (10ml) and purified by preparative HPLC on a 2" x 25cm column packed with Kromasil C8 10 ⁇ m RP and eluting with a gradient of 10 -100% of (70:30 acetonitrile / 0.1 M ammonium dihydrogenphosphate, adjusted to pH 2.6 with phosphoric acid) in (0.1 M ammonium dihydrogenphosphate, adjusted to pH 2.6 with phosphoric acid) over 21.5mins (flow rate 100ml/min). Material eluting in a broad peak at ⁇ 13mins was collected from each run, the fractions from the separate runs combined and the acetonitrile evaporated in vacuo.
- Triphenyl phosphine (138.7g) was added to a solution of 3-methoxy-1-propanol (47.6g) in anhydrous DCM (500ml). The resulting mixture was stirred under nitrogen and cooled to 0 0 C, N-bromosuccinimide (94.13g) was added portion-wise keeping the internal temperature under 2O 0 C during the addition. The mixture was stirred overnight at room temperature, then, quenched by addition of sodium metabisulfite solution (5%). The phases were separated, the organic phase washed sequentially with sodium hydroxide (0.5M) then brine and dried (magnesium sulphate).
- Example 24 2-naphthalenesulfonic acid - /v ⁇ -d .i-dioxido ⁇ .S-dihydro-i ⁇ - benzisothiazol-S-vD-Zv ⁇ -ethyl-S-fluoro-A ⁇ -IH-indazol ⁇ -yl ⁇ -pyrimidinediamine
- a suspension of / ⁇ -(i .i-dioxido ⁇ .S-dihydro-i ⁇ -benzisothiazol- ⁇ -yO- ⁇ -ethyl-S- fluoro- ⁇ /MH-indazol-4-yl-2,4-pyrimidinediamine (as prepared in Example 8, 1.0g) in water / IPA (1 :5, 12ml) was warmed to reflux and the suspension was treated with an aqueous solution of 2-naphthalenenesulfonic acid monohydrate (0.58g) in water (3ml) to give a solution and the residual acid rinsed in with water (2x 2ml). The solution was cooled to ambient temperature, stirred for 2h, the precipitated solid filtered off, washed with water (6x 1 ml), and dried in vacuo at 70 0 C to give the title compound as a pale yellow solid (1.23g).
- the solid was divided into 2 portions.
- the first portion (284.13g) was suspended in methanol (4260ml) and water (1420ml), the mixture was heated to 65°C and treated with sodium hydroxide (2M, 290ml).
- the resulting solution was seeded with a pure sample of / ⁇ -(i.i-dioxido ⁇ .S-dihydro-i ⁇ -benzisothiazol- ⁇ -yO- ⁇ -fluoro- ⁇ i/-/- indazol-4-yl- ⁇ / 4 -[3-(methyloxy)propyl]-2,4-pyrimidinediamine and cooled to 40 0 C and maintained at 40 0 C for 1 h.
- the second portion (274.71 g) was suspended in methanol (4120ml) and water (1370ml), the mixture was heated to 65°C and treated with sodium hydroxide (2M, 280ml). The resulting solution was seeded with a pure sample of ⁇ / 2 -(1 ,1-dioxido-2,3- dihydro-1 ⁇ -benzisothiazol- ⁇ -yO- ⁇ -fluoro- ⁇ i /-/-indazol-4-yl- ⁇ / 4 -[3-(methyloxy)propyl]- 2,4-pyrimidinediamine and cooled to 40°C and maintained at 40 0 C for 1h.
- the reaction was cooled to room temperature and aqueous potassium carbonate (1M, 2100ml) added portionwise. The phases were separated, the aqueous extracted with chloroform, and the combined organic phases washed with water, brine and dried (magnesium sulphate). The solvent was evaporated in vacuo, the residual oil dissolved in ether (200ml) and cyclohexane (700ml) added to the solution in 100ml portions. Scratching of the flask initialled precipitation. The precipitate was isolated by filtration, washed with ether / cyclohexane (1 :4) and dried in vacuo at 40 0 C to give the title compound (246.28g).
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Immunology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pulmonology (AREA)
- Pain & Pain Management (AREA)
- Rheumatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
A compound of formula (I): in which: R1 and R2 is each independently hydrogen, C1-6alkyl or CH2C3-7cycloalkyl; R3 is C1-3alkyl or C2-3alkyl substituted by hydroxy or C1-3alkoxy; and X is halogen; or a salt or solvate, preferably a pharmaceutically acceptable salt or solvate, thereof; useful as inhibitors of Syk kinase and thus useful in treating diseases resulting from inappropriate mast cell activation, for instance allergic and inflammatory diseases.
Description
1 , l-DIOXIDO-2 , 3-DIHYDRO-l , 2-BENZISOTHIAZ0L-6-YL-1H-INDAZOL-4-YL-2 , 4-PYRIMIDINEDI
AMINE
DERIVATIVES
The present invention relates to novel chemical compounds which have activity against the spleen tyrosine kinase (Syk kinase), processes for their preparation, pharmaceutically acceptable formulations containing them and their use in therapy.
Allergic rhinitis and asthma are diseases associated with hypersensitivity reactions and inflammatory events involving a multitude of cell types including mast cells, eosinophils, T cells and dendritic cells. Following exposure to allergen, high affinity immunoglobulin receptors for IgE (FcεRI) and IgG (FcγRI) become cross-linked and activate downstream processes in mast cells and other cell types leading to the release of pro-inflammatory mediators and airway spasmogens. In the mast cell, for example, IgE receptor cross-linking by allergen leads to release of mediators including histamine from pre-formed granules, as well as the synthesis and release of newly synthesised lipid mediators including prostaglandins and leukotrienes.
Syk kinase is a non-receptor linked tyrosine kinase which is important in transducing the downstream cellular signals associated with cross-linking FcεRI and or FcγRI receptors, and is positioned early in the signalling cascade. In mast cells, for example, the early sequence of FcεRI signalling following allergen cross-linking of receptor-lgE complexes involves first Lyn (a Src family tyrosine kinase) and then Syk kinase. Inhibitors of Syk kinase activity would therefore be expected to inhibit all downstream signalling cascades thereby alleviating the immediate allergic response and adverse events initiated by the release of pro-inflammatory mediators and spasmogens (Wong, B., Grossbard, E.B. Payan, D.G & Masuda, E. S. Expert Opin. Investig. Drugs (2004) 13 (7) 743-762).
Recently, it has been shown that the Syk kinase inhibitor R112 (Rigel), dosed intranasally in a phase I/I I study for the treatment of allergic rhinitis, gave a statistically significant decrease in PGD2, a key immune mediator that is highly correlated with improvements in allergic rhinorrhea, as well as being safe across a range of indicators, thus providing the first evidence for the clinical safety and efficacy of a topical Syk kinase inhibitor. Meltzer, EIi O.; Berkowitz, Robert B.; Grossbard, Elliott B. An intranasal Syk -kinase inhibitor ( R112 ) improves the symptoms of seasonal allergic rhinitis in a park environment. Journal of Allergy and
Clinical Immunology (2005), 115(4), 791-796. In a more recent phase Il clinical trial for allergic rhinitis (Clinical Trials.gov Identifier NCT0015089), R112 was however shown as having a lack of efficacy versus placebo.
Rheumatoid Arthritis (RA) is an auto-immune disease affecting approximately 1 % of the population. It is characterised by inflammation of articular joints leading to debilitating destruction of bone and cartilage. Recent clinical studies with Rituximab,
which causes a reversible B cell depletion, (J. CW. Edwards et al 2004, New Eng. J. Med. 350: 2572-2581 ) have shown that targeting B cell function is an appropriate therapeutic strategy in auto-immune diseases such as RA. Clinical benefit correlates with a reduction in auto-reactive antibodies (or Rheumatoid Factor) and these studies suggest that B cell function and indeed auto-antibody production are central to the ongoing pathology in the disease.
Studies using cells from mice deficient in the Syk kinase have demonstrated a non- redundant role of this kinase in B cell function. The deficiency in Syk kinase is characterised by a block in B cell development (M. Turner et al 1995 Nature 379: 298-302 and Cheng et al 1995, Nature 378: 303-306). These studies, along with studies on mature B cells deficient in Syk kinase (Kurasaki et al 2000, Immunol. Rev. 176:19-29), demonstrate that Syk kinase is required for the differentiation and activation of B cells. Hence, inhibition of Syk kinase in RA patients is likely to block B cell function and hence to reduce Rheumatoid Factor production. In addition to the role of Syk kinase in B cell function, of relevance to the treatment of RA, is the requirement for Syk kinase activity in Fc receptor (FcR) signalling. FcR activation by immune complexes in RA has been suggested to contribute to the release of multiple pro-inflammatory mediators.
The contribution of Syk kinase dependent processes to the pathology of RA has been reviewed in Wong et al (2004, ibid).
WO 03/057695 (Boehringer lngelheim Pharmaceuticals, Inc.) describes substituted [1 ,6]-naphthyridines that inhibit Syk kinase.
WO2003/063794, WO2004/014382, WO2005/012294 and WO2005/16893 (Rigel Pharmaceuticals, Inc) describes a series of 2,4-pyrimidinediamine compounds which inhibit Syk kinase, for use in treating autoimmune diseases.
WO2005/026158 (Novartis AG) describes 2,4-di (hetero)-arylamino-pyrimidine derivatives which have ZAP-70 and/or Syk inhibitory activities.
WO 04/035604 discloses the structural co-ordinates of the human Syk protein.
There remains however the need to identify further compounds which are inhibitors of Syk kinase.
Thus, in a first aspect invention, the present invention provides a compound of formula (I):
 in which:
in which:
R1 and R2 is each independently hydrogen, C1-6alkyl or CH2C3-7cycloalkyl; R3 is C1-3 alkyl or C2-3alkyl substituted by hydroxy or Ci.3alkoxy; and X is halogen; or a salt or solvate, preferably a pharmaceutically acceptable salt or solvate, thereof.
Compounds of the present invention are useful as inhibitors of Syk kinase and thus potentially of use in treating diseases resulting from inappropriate mast cell activation, for instance allergic and inflammatory diseases.
Representative examples of R1 include hydrogen, methyl, ethyl, 2-methylpropyl, cyclopropylmethyl, cyclobutylmethyl, and cyclohexylmethyl.
Representative examples of R2 include hydrogen.
Representative examples of R3 include methyl, ethyl, propyl, -(CH2)nOH and -(CH2)n0C1-3alkyl where n is 3.
In a further embodiment, R3 is methyl, ethyl, propyl, -(CH2)3OH and -(CH2)3OCH3.
In a further embodiment, representative examples of X include fluoro, chloro and bromo.
In a further embodiment, X is fluoro.
In a further aspect, the present invention provides for a compound of formula (IB):

R1 is hydrogen, C1-6alkyl or CH2C3-7cycloalkyl;
R3 is C1.3alkyl; and
X is halogen; or a salt or solvate, preferably a pharmaceutically acceptable salt or solvate, thereof.
In a further aspect, the present invention provides for a compound of formula (IC):

R3 is C2-3alkyl substituted by hydroxy or C1-3alkoxy; and
X is halogen; or a salt or solvate, preferably a pharmaceutically acceptable salt or solvate, thereof.
Representative examples of compounds of formula (I) include:
Λ/^i .i-dioxido^.S-dihydro-i ^-benzisothiazol-β-yO-Λ^-ethyl-δ-fluoro-Λ^-IH-indazol-
4-yl-2,4-pyrimidinediamine;
5-chloro-Λ/2-(1 ,1-dioxido-2,3-dihydro-1 ,2-benzisothiazol-6-yl)-Λ/4-ethyl-/v4-1H-indazol-
4-yl-2,4-pyrimidinediamine; 5-bromo-/V2-(1 , 1 -dioxido-2,3-dihydro-1 ^-benzisothiazol-θ-yO-ΛΛethyl-Λ/4-! H-indazol-
4-yl-2,4-pyrimidinediamine;
Λ/4-Ethyl-5-fluoro-Λ/4-1H-indazol-4-yl-Λ/2-(2-methyl-1 ,1-dioxido-2,3-dihydro-1 ,2- benzisothiazol-6-yl)-2,4-pyrimidinediamine;
Λ/2-(1 ,1-dioxido-2,3-dihydro-1 ,2-benzisothiazol-6-yl)-5-fluoro-AT*-1 H-indazol-4-yl-Λ/4- methyl-2,4-pyrimidinediamine;
/^-^-(cyclopropylmethyO-i ,1 -dioxido-2,3-dihydro-1 ^-benzisothiazol-θ-ylJ-Λ^-ethyl-δ- fluoro-/V*-1 H-indazol-4-yl-2,4-pyrimidinediamine;
Λ/4-ethyl-Λ/2-(2-ethyl-1 ,1-dioxido-2,3-dihydro-1 ^-benzisothiazol-θ-yO-S-fluoro-A/M H- indazol-4-yl-2,4-pyrimidinediamine; Λ^-ethyl-δ-fluoro-Λ/4-! H-indazol-4-yl-Λ/2-[2-(2-methylpropyl)-1 ,1 -dioxido-2,3-dihydro-
1 ,2-benzisothiazol-6-yl]-2,4-pyrimidinediamine;
Λ/2-[2-(cyclobutylmethyl)-1 , 1 -dioxido-2,3-dihydro-1 ^-benzisothiazol-β-yll-Λ^-ethyl-δ- fluoro-Λ^-IH-indazoM-yl^^-pyrimidinediamine;
/^-^-(cyclohexylmethyO-i , 1 -dioxido-2,3-dihydro-1 ^-benzisothiazol-δ-yll-Λ^-ethyl-S- fluoro-Λ/4-! H-indazol-4-yl-2,4-pyrimidinediamine;
Af-(1 , 1-dioxido-2,3-dihydro-1 ,2-benzisothiazol-6-yl)-5-fluoro-Λ/4-1H-indazol-4-yl-/V4- propyl-2,4-pyrimidinediamine;
Af-(1 , 1-dioxido-2,3-dihydro-1 ,2-benzisothiazol-6-yl)-5-fluoro-/v4-1H-indazol-4-yl-/V*-[3-
(methyloxy)propyl]-2,4-pyrimidinediamine; and 3-[{2-[(1 ,1-dioxido-2,3-dihydro-1 ,2-benzisothiazol-6-yl)amino]-5-fluoro-4- pyrimidinyl}(1 /-/-indazol-4-yl)amino]-1 -propanol; or salt or solvate thereof, in particular, a pharmaceutically acceptable salt or solvate thereof.
In a further embodiment, examples of compounds of formula (I) include:
/^-(i .i-dioxido^.S-dihydro-i ^-benzisothiazol-θ-yO-S-fluoro-Λ^-IH-indazol^-yl-Λ/4- propyl-2,4-pyrimidinediamine; and
Λf-0 ,1 -dioxido-2,3-dihydro-1 ^-benzisothiazol-θ-yO-S-fluoro-Λ^-i H-indazol-4-yl-ΛΛ[3- (methyloxy)propyl]-2,4-pyrimidinediamine; or a salt or solvate thereof, in particular, a pharmaceutically acceptable salt or solvate thereof.
References to alkyl include references to both straight chain and branched chain aliphatic isomers of the corresponding alkyl. It will be appreciated that references to alkylene and alkoxy shall be interpreted similarly.
References to C3-7 cycloalkyl include references to all alicyclic (including branched) isomers of the corresponding alkyl.
When used herein, the term "pharmaceutically acceptable" refers to those compounds, materials, compositions, and dosage forms which are, within the scope of sound medical judgment, suitable for use in contact with the tissues of human beings and animals without excessive toxicity, irritation, or other problem or complication, commensurate with a reasonable benefit/risk ratio. The skilled artisan will appreciate that pharmaceutically acceptable salts of the compound of the present invention may be prepared. As used herein, the term "pharmaceutically acceptable salts" refers to salts that retain the desired biological activity of the subject compound and exhibit minimal undesired toxicological effects. These pharmaceutically acceptable salts may be prepared in situ during the final isolation and purification of the compound, or by separately reacting the purified compound in its free acid or free base form with a suitable base or acid, respectively. Indeed, in certain embodiments of the invention, pharmaceutically acceptable salts may be preferred over the respective free base or free acid because such salts impart greater stability or solubility to the molecule thereby facilitating formulation into a dosage form.
In certain embodiments, the compound of the present invention may contain one or more acidic functional groups. Suitable pharmaceutically acceptable salts include salts of such acidic functional groups. Representative salts include pharmaceutically acceptable metal salts such as sodium, potassium, lithium, calcium, magnesium, aluminum, and zinc salts; carbonates and bicarbonates of a phamnaceutically- acceptable metal cation such as sodium, potassium, lithium, calcium, magnesium, aluminum, and zinc; pharmaceutically acceptable organic primary, secondary, and tertiary amines including aliphatic amines, aromatic amines, aliphatic diamines, and hydroxy alkylamines such as methylamine, ethylamine, 2-hydroxyethylamine, diethylamine, triethylamine, ethylenediamine, ethanolamine, diethanolamine, and cyclohexylamine.
Compounds of the present invention are basic and accordingly generally capable of forming pharmaceutically acceptable acid addition salts by treatment with a suitable acid. Suitable acids include pharmaceutically acceptable inorganic acids and pharmaceutically acceptable organic acids. Representative pharmaceutically acceptable acid addition salts include hydrochloride, hydrobromide, nitrate, methylnitrate, sulfate, bisulfate, sulfamate, phosphatβi acetate, hydroxyacetate, phenylacetate, propionate, butyrate, isobutyrate, valerate, maleate, hydroxymaleate, acrylate, fumarate, malate, tartrate, citrate, salicylate, p-aminosalicyclate, glycollate, lactate, heptanoate, phthalate, oxalate, succinate, benzoate, o-acetoxybenzoate, chlorobenzoate, methylbenzoate, dinitrobenzoate, hydroxybenzoate, methoxybenzoate, mandelate, tannate, formate, stearate, ascorbate, palmitate, oleate, pyruvate, pamoate, malonate, laurate, glutarate, glutamate, estolate, methanesulfonate (mesylate), ethanesulfonate (esylate), 2-hydroxyethanesulfonate, benzenesulfonate (besylate), p-aminobenzenesulfonate, p-toluenesulfonate (tosylate), and napthalene-2-sulfonate.
As used herein, the term "compound" refers to one or more compounds. Similarly, the term "a compound of the present invention" refers to one or more compounds of the present invention.
The compound of the present invention may exist in solid or liquid form. In the solid state, the compound of the present invention may exist in crystalline or noncrystalline form, or as a mixture thereof. For a compound of the present invention that is in crystalline form, the skilled artisan will appreciate that pharmaceutically acceptable solvates may be formed wherein solvent molecules are incorporated into the crystalline lattice during crystallization. Solvates may involve non-aqueous solvents such as, but not limited to, ethanol, isopropanol, n-butanol, i-butanol, acetone, tetrahydrofuran, dioxane, DMSO, acetic acid, ethanolamine, and ethyl acetate, or they may involve water as the solvent that is incorporated into the crystalline lattice. Solvates wherein water is the solvent incorporated into the crystalline lattice are typically referred to as "hydrates." Hydrates include stoichiometric hydrates as well as compositions containing variable amounts of water. The invention includes all such solvates.
The skilled artisan will further appreciate that certain compounds of the invention that exist in crystalline form, including the various solvates thereof, may exhibit polymorphism (i.e. the capacity to occur in different crystalline structures). These different crystalline forms are typically known as "polymorphs." The invention includes all such polymorphs. Polymorphs have the same chemical composition but differ in packing, geometrical arrangement, and other descriptive properties of the crystalline solid state. Polymorphs, therefore, may have different physical properties such as shape, density, hardness, deformability, stability, and dissolution properties.
Polymorphs typically exhibit different melting points, IR spectra, and X-ray powder diffraction patterns, which may be used for identification. The skilled artisan will appreciate that different polymorphs may be produced, for example, by changing or adjusting the reaction conditions or reagents, used in making the compound. For example, changes in temperature, pressure, or solvent may result in polymorphs. In addition, one polymorph may spontaneously convert to another polymorph under certain conditions.
The compound of the present invention may contain one or more asymmetric centres (also referred to as a chiral centre) and may, therefore, exist as individual enantiomers, diastereoisomers, or other stereoisomeric forms, or as mixtures thereof. Chiral centres, such as chiral carbon atoms, may also be present in a substituent such as an alkyl group. Where the stereochemistry of a chiral centre is not specified the structure is intended to encompass any stereoisomer and all mixtures thereof. Thus, the compound of the present invention containing one or more chiral centres may be used as racemic mixtures, enantiomerically enriched mixtures, or as enantiomerically pure individual stereoisomers. Generally it is preferred to use a compound of formula (I) in the form of a purified single enantiomer.
Individual stereoisomers of a compound according to formula (I) which contain one or more asymmetric centre may be resolved by methods known to those skilled in the art. For example, such resolution may be carried out (1 ) by formation of diastereoisomeric salts, complexes or other derivatives; (2) by selective reaction with a stereoisomer-specific reagent, for example by enzymatic oxidation or reduction; or (3) by gas-liquid or liquid chromatography in a chiral environment, for example, on a chiral support such as silica with a bound chiral ligand or in the presence of a chiral solvent. The skilled artisan will appreciate that where the desired stereoisomer is converted into another chemical entity by one of the separation procedures described above, a further step is required to liberate the desired form. Alternatively, specific stereoisomers may be synthesized by asymmetric synthesis using optically active reagents, substrates, catalysts or solvents, or by converting one enantiomer to the other by asymmetric transformation.
The compound of the present invention may also contain double bonds or other centres of geometric asymmetry. Where the stereochemistry of a centre of geometric asymmetry is not specified, the structure is intended to encompass the trans (E) geometric isomer, the cis (Z) geometric isomer, and all mixtures thereof. Likewise, all tautomeric forms are also included the compound of the present invention whether such tautomers exist in equilibrium or predominately in one form.
Compounds of the present invention, as well as salts and solvates thereof, may be prepared by one or more of the general synthetic schemes described hereinafter.
Scheme 1A

(I)
Scheme 1B

Scheme 1C

in which schemes R1, R2, R3 and X are as hereinbefore defined and Pr is a protecting group such as 2-(trimethylsilyl)ethoxymethyl or 1 ,1-dimethylethylcarboxylate.
Accordingly, in a further aspect, the present invention provides a process for preparing a compound of formula (I), or a salt or solvate thereof, which process comprises: reacting a compound of formula (II):

or a protected derivative thereof wherein L1 represents a suitable leaving group, with a compound of formula (III)

or a protected derivative thereof, wherein X, R1, R2 and R3 are as defined above.
The process may be performed in the presence of a solvent (for example, aqueous acetone or propan-2-ol), in the presence of concentrated hydrochloric acid, at a suitable temperature, preferably in the range of 0-170° C.
Alternatively, with a 2-(trimethylsilyl)ethoxymethyl protecting group, the process may be performed in the presence of a solvent such as 2-propanol, and hydrogen chloride in ether, at a suitable temperature, preferably in the range of 0-100°C, preferably under a nitrogen atmosphere.
Alternatively, with a 1,1-dimethylethylcarboxylate protecting group, the process may be performed in the presence of a solvent such as 2-propanol, and a dilute acid such as dilute hydrochloric acid, at a suitable temperature, preferably in the range of 0- 100°C.
Examples of suitable leaving groups (L1) include a halide such as chloride, bromide or iodide. Other leaving groups include, but are not restricted to, an alkyl and aryl sulfide, an alkyl and aryl sulfinyl, an alkyl and aryl sulfonyl, and an alcohol derived leaving group (such as triflate, mesylate, methylsulfonate or tosylate).
Alternatively the process may be performed in the presence of a non-nucleophilic base such as triethylamine or di-isopropylethylamine, as a melt, at a suitable temperature, preferably in the range of 0-180°C.
Compounds of formula (I) having a [1 ,1-dioxido-2,3-dihydro-1 ,2-benzisothiazol-6-yl] moiety:

Compounds of formula (II) may be prepared from the corresponding compound of formula (IV):

Compounds of formula (IV) may be prepared by reacting a corresponding (protected) indazole-4-amine with a pyrimidine compound of formula (V):

Compounds of formula (II) may also be prepared by reacting a compound of formula (Vl):
 in which R3, L1 and X are as hereinbefore defined, or a protected derivative thereof, with an organic nitrite derivative, such as, but not limited to, tert-butyl nitrite. This reaction is preferably performed in the presence of an acid, such as acetic acid or hydrochloric acid.
in which R3, L1 and X are as hereinbefore defined, or a protected derivative thereof, with an organic nitrite derivative, such as, but not limited to, tert-butyl nitrite. This reaction is preferably performed in the presence of an acid, such as acetic acid or hydrochloric acid.
The aforementioned reaction may be performed in the presence of acetic anhydride to yield a product in which an indazolyl nitrogen atom is protected.
Compounds of formula (Vl) may be prepared from the corresponding compound of formula (VII):

in which L1 and X are as hereinbefore defined, or a protected derivative thereof, with an alkyl iodide R3I in the presence of a base such as cesium carbonate, in an aprotic solvent such as DMF.
Compounds of formula (VII) may be prepared by reacting the corresponding optionally protected aniline of formula (VIII):

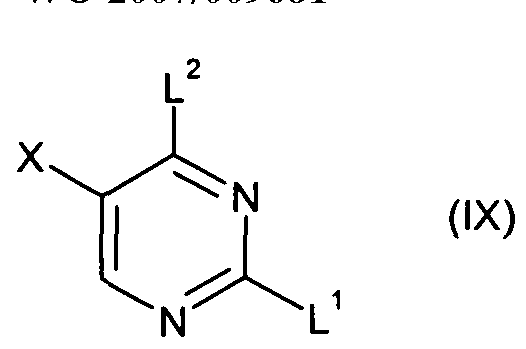
in which L1, L2 and X are as hereinbefore defined, in aqueous methanol, at a temperature of about 7O0C.
In a further embodiment, the compound of formula (II) is the following N-acetyl protected derivative:

Compounds of formula (III) are either obtainable from commercial sources, known in the art or may be prepared from the corresponding nitro compound using conventional reducing conditions for such a transformation, for example sodium dithionite, hydrogen gas in the presence of palladium, or iron metal with hydrochloric acid.
Compounds of formula (II) in which the leaving group represents other than halogen, may be obtained via the corresponding halogen by conventional methods.
Certain compounds of formula (II) are new and form a further aspect of the invention.
Compounds of the present invention are useful as inhibitors of Syk and thus potentially of use in treating diseases resulting from inappropriate mast cell activation, for instance allergic and inflammatory diseases.
Thus, in a further aspect, the present invention provides for a compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof, for use in therapy.
In a further embodiment, the present invention provides for a method of treating inappropriate mast cell activation which method comprises administering to a patient in need thereof an effective compound of formula I, or a or a pharmaceutically acceptable salt or solvate thereof.
In a further aspect, the present invention provides a method comprising administering to a patient in need thereof an effective compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof, to inhibit a Syk kinase.
In a further aspect, the present invention provides a method of treating an inflammatory disease which comprises administering to a patient in need thereof an effective compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof.
In a further aspect, the present invention provides a method of treating an allergic disorder which comprises administering to a patient in need thereof an effective compound of formula (I), or a pharmaceutically acceptable salt or solvate thereof.
Diseases and pathological conditions thought to be mediated by Syk kinase include inflammatory and allergic disorders involving mast cell activation, such as chronic obstructive pulmonary disease (COPD), adult respiratory distress syndrome (ARDS), asthma, ulcerative colitis, Crohn's Disease, bronchitis, conjunctivitis, psoriasis, sclerodoma, urticaria, dermatitis, and allergic rhinitis. They also include inflammatory conditions which involve B cells, for instance lupus and rheumatoid arthritis.
Compounds of the present invention may also be used in combination with other classes of therapeutic agents which are known in the art. Representative classes of agents for use in such combinations include, for treating asthma, anti-inflammatory steroids (in particular corticosteroids), topical glucocorticoid agonists, PDE4 inhibitors, IKK2 inhibitors, A2a agonists, β2-adrenoreceptor agonists (including both slow acting and long acting β2-adrenoreceptor agonists), alpha 4 integrin inhibitors, and anti-muscarinics, and, for treating allergies, the foregoing agents, as well as H1 and H1/H3 antagonists. Representative agents for use in combination therapy for treating severe asthma include topically acting p38 inhibitors, and IKK2 inhibitors.
Anti-inflammatory corticosteroids are well known in the art. Representative examples include fluticasone propionate (e.g. see US patent 4,335,121), beclomethasone 17- propionate ester, beclomethasone 17,21-dipropionate ester, dexamethasone or an ester thereof, mometasone or an ester thereof (e.g. mometasone furoate), ciclesonide, budesonide, and flunisolide. Further examples of anti-inflammatory corticosteroids are described in WO 02/12266 A1 (Glaxo Group Ltd), in particular, the compounds of Example 1 ( 6α,9α-difluoro-17α-[(2-furanylcarbonyl)oxy]-11 β-hydroxy- 16α-methyl-3-oxo-androsta-1 ,4-diene-17β-carbothioic acid S-fluoromethyl ester) and
Example 41 (6α,9α-difluoro-11β-hydroxy-16α-methyl-17α-[(4-methyl-1 ,3-thiazole-5- carbonyl)oxy]-3-oxo-androsta-1 ,4-diene-17β-carbothioic acid S-fluoromethyl ester), or a pharmaceutically acceptable salt thereof.
Examples of β2-adrenoreceptor agonists include salmeterol (e.g. as racemate or a single enantiomer such as the R-enantiomer), salbutamol, formoterol, salmefamol, fenoterol or terbutaline and salts thereof, for example the xinafoate salt of salmeterol, the sulphate salt or free base of salbutamol or the fumarate salt of formoterol. Long- acting β2-adrenoreceptor agonists are preferred, especially those having a therapeutic effect over a 24 hour period such as salmeterol or formoterol.
Examples of anti-histamines include methapyrilene, or loratadine, cetirizine, desloratadine or fexofenadine.
Examples of anticholinergic compounds include muscarinic (M) receptor antagonists, in particular M-| , Iv^, M1/M2, or M3 receptor antagonists, in particular a (selective)
M3 receptor antagonist. Examples of anticholinergic compounds are described in
WO 03/011274 A2 and WO 02/069945 A2 / US 2002/0193393 A1 and US 2002/052312 A1. Examples of muscarinic M3 antagonists include ipratropium bromide, oxitropium bromide or tiotropium bromide.
Representative PDE4 or mixed PDE3/4 inhibitors that may be used in combination with compounds of the invention include AWD-12-281 (Elbion), PD-168787 (Pfizer), roflumilast, and cilomilast (GlaxoSmithKline). Further examples of PDE4 inhibitors are described in WO 2004/103998 (Glaxo Group Ltd).
The present invention also provides for so-called "triple combination" therapy, comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof together with β2-adrenoreceptor agonist and an anti-inflammatory corticosteroid. Preferably this combination is for treatment and/or prophylaxis of asthma, COPD or allergic rhinitis. The β2-adrenoreceptor agonist and/or the anti-inflammatory corticosteroid can be as described above and/or as described in WO 03/030939 A1. A representative example of such a "triple" combination comprises a compound of formula (I) or a pharmaceutically acceptable salt thereof, salmeterol or a pharmaceutically acceptable salt thereof (e.g. salmeterol xinafoate) and fluticasone propionate.
Preferred compounds of formula (I) for use as inhibitors of Syk kinase are those which exhibit selectivity for the Syk kinase against other key kinases such as Aurora A, Aurora B, Zap 70, JNK3, for instance at least 10x (based on either pKi or plCso values for the enzymes)
The compound of the present invention will normally, but not necessarily, be formulated into pharmaceutical compositions prior to administration to a patient.
Accordingly, in another aspect the invention is directed to pharmaceutical compositions comprising a compound of the invention and one or more pharmaceutically acceptable excipient.
The pharmaceutical compositions of the invention may be prepared and packaged in bulk form wherein a safe and effective amount of a compound of the invention can be extracted and then given to the patient, such as with powders or syrups. Alternatively, the pharmaceutical compositions of the invention may be prepared and packaged in unit dosage form wherein each physically discrete unit contains a safe and effective amount of a compound of the invention. When prepared in unit dosage form, the pharmaceutical compositions of the invention typically contain from about 0.1 to 99.9 wt.%, depending on the nature of the formulation.
The pharmaceutical compositions of the invention typically contain one compound of the invention. However, in certain embodiments, the pharmaceutical compositions of the invention contain more than one compound of the invention. For example, in certain embodiments the pharmaceutical compositions of the invention contain two compounds of the invention. In addition, the pharmaceutical compositions of the invention may optionally further comprise one or more additional pharmaceutically active compounds.
As used herein, "pharmaceutically acceptable excipient" means a pharmaceutically acceptable material, composition or vehicle involved in giving form or consistency to the pharmaceutical composition. Each excipient must be compatible with the other ingredients of the pharmaceutical composition when commingled, such that interactions which would substantially reduce the efficacy of the compound of the invention when administered to a patient and would result in pharmaceutically unacceptable compositions are avoided. In addition, each excipient must of course be of sufficiently high purity to render it pharmaceutically acceptable.
Compositions of the present invention comprising a compound of the invention and the pharmaceutically acceptable excipient or excipients will typically be provided as a dosage form adapted for administration to the patient by the desired route of administration. For example, dosage forms include those adapted for (1) inhalation, such as aerosols and solutions; and (2) intranasal administration, such as solutions or sprays.
Suitable pharmaceutically acceptable excipients will vary depending upon the particular dosage form chosen. In addition, suitable pharmaceutically acceptable excipients may be chosen for a particular function that they may serve in the composition. For example, certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the production of uniform dosage forms. Certain pharmaceutically acceptable excipients may be chosen for their ability to facilitate the production of stable dosage forms. Certain pharmaceutically acceptable excipients
may be chosen for their ability to facilitate the carrying or transporting the compound of the present invention once administered to the patient from one organ, or portion of the body, to another organ, or portion of the body. Certain pharmaceutically acceptable excipients may be chosen for their ability to enhance patient compliance.
Suitable pharmaceutically acceptable excipients include the following types of excipients: Diluents, fillers, lubricants, glidants, granulating agents, wetting agents, solvents, co-solvents, suspending agents, emulsifiers, anticaking agents, chelating agents, viscosity increasing agents, antioxidants, preservatives, stabilizers, surfactants, and buffering agents. The skilled artisan will appreciate that certain pharmaceutically acceptable excipients may serve more than one function and may serve alternative functions depending on how much of the excipient is present in the formulation and what other ingredients are present in the formulation.
Skilled artisans possess the knowledge and skill in the art to enable them to select suitable pharmaceutically acceptable excipients in appropriate amounts for use in the invention. In addition, there are a number of resources that are available to the skilled artisan which describe pharmaceutically acceptable excipients and may be useful in selecting suitable pharmaceutically acceptable excipients. Examples include Remington's Pharmaceutical Sciences (Mack Publishing Company), Remington: The Science and Practice of Pharmacy, (Lippincott Williams & Wilkins), The Handbook of Pharmaceutical Additives (Gower Publishing Limited), and The Handbook of Pharmaceutical Excipients (the American Pharmaceutical Association and the Pharmaceutical Press).
The pharmaceutical compositions of the invention are prepared using techniques and methods known to those skilled in the art. Some of the methods commonly used in the art are described in Remington's Pharmaceutical Sciences (Mack Publishing Company).
Dosage forms for nasal or inhaled administration may conveniently be formulated as aerosols, solutions, drops, gels or dry powders.
Dosage forms for topical administration to the nasal cavity (nasal administration) include pressurised aerosol formulations and aqueous formulations administered to the nose by pressurised pump. Formulations which are non-pressurised and adapted for nasal administration are of particular interest. Suitable formulations contain water as the diluent or carrier for this purpose. Aqueous formulations for administration to the nose may be provided with conventional excipients such as buffering agents, tonicity modifying agents and the like. Aqueous formulations may also be administered to the nose by nebulisation.
In a further embodiment, dosage forms for nasal administration are provided in a metered dose device. The dosage form may be provided as a fluid formulation for
delivery from a fluid dispenser having a dispensing nozzle or dispensing orifice through which a metered dose of the fluid formulation is dispensed upon the application of a user-applied force to a pump mechanism of the fluid dispenser. Such fluid dispensers are generally provided with a reservoir of multiple metered doses of the fluid formulation, the doses being dispensable upon sequential pump actuations. The dispensing nozzle or orifice may be configured for insertion into the nostrils of the user for spray dispensing of the fluid formulation into the nasal cavity. In one embodiment, the fluid dispenser is of the general type described and illustrated in WO-A-2005/044354. The dispenser has a housing which houses a fluid discharge device having a compression pump mounted on a container for containing a fluid formulation. The housing has at least one finger-operable side lever which is movable inwardly with respect to the housing to cam the container upwardly in the housing to cause the pump to compress and pump a metered dose of the formulation out of a pump stem through a nasal nozzle of the housing. A particularly preferred fluid dispenser is of the general type illustrated in Figures 30-40 of WO-A- 2005/044354.
For compositions suitable and/or adapted for inhaled administration, it is preferred that the compound or salt of formula (I) is in a particle-size-reduced form, and more preferably the size-reduced form is obtained or obtainable by micronisation. The preferable particle size of the size-reduced (e.g. micronised) compound or salt or solvate is defined by a D50 value of about 0.5 to about 10 microns (for example as measured using laser diffraction).
Aerosol compositions, e.g. for inhaled administration, can comprise a solution or fine suspension of the active substance in a pharmaceutically acceptable aqueous or non-aqueous solvent. Aerosol formulations can be presented in single or multidose quantities in sterile form in a sealed container, which can take the form of a cartridge or refill for use with an atomising device or inhaler. Alternatively the sealed container may be a unitary dispensing device such as a single dose nasal inhaler or an aerosol dispenser fitted with a metering valve (metered dose inhaler) which is intended for disposal once the contents of the container have been exhausted.
Where the dosage form comprises an aerosol dispenser, it preferably contains a suitable propellant under pressure such as compressed air, carbon dioxide or an organic propellant such as a hydrofluorocarbon (HFC). Suitable HFC propellants include 1 ,1 ,1 ,2,3,3,3-heptafluoropropane and 1 ,1 ,1 ,2-tetrafluoroethane. The aerosol dosage forms can also take the form of a pump-atomiser. The pressurised aerosol may contain a solution or a suspension of the active compound. This may require the incorporation of additional excipients e.g. co-solvents and/or surfactants to improve the dispersion characteristics and homogeneity of suspension formulations. Solution formulations may also require the addition of co-solvents such as ethanol. Other excipient modifiers may also be incorporated to improve, for example, the stability
and/or taste and/or fine particle mass characteristics (amount and/or profile) of the formulation.
For pharmaceutical compositions suitable and/or adapted for inhaled administration, it is preferred that the pharmaceutical composition is a dry powder inhalable composition. Such a composition can comprise a powder base such as lactose, glucose, trehalose, mannitol or starch, the compound of formula (I) or salt or solvate thereof (preferably in particle-size-reduced form, e.g. in micronised form), and optionally a performance modifier such as L-leucine or another amino acid, cellobiose octaacetate and/or metals salts of stearic acid such as magnesium or calcium stearate. Preferably, the dry powder inhalable composition comprises a dry powder blend of lactose and the compound of formula (!) or salt thereof. The lactose is preferably lactose hydrate e.g. lactose monohydrate and/or is preferably inhalation- grade and/or fine-grade lactose. Preferably, the particle size of the lactose is defined by 90% or more (by weight or by volume) of the lactose particles being less than 1000 microns (micrometres) (e.g. 10-1000 microns e.g. 30-1000 microns) in diameter, and/or 50% or more of the lactose particles being less than 500 microns (e.g. 10-500 microns) in diameter. More preferably, the particle size of the lactose is defined by 90% or more of the lactose particles being less than 300 microns (e.g. 10- 300 microns e.g. 50-300 microns) in diameter, and/or 50% or more of the lactose particles being less than 100 microns in diameter. Optionally, the particle size of the lactose is defined by 90% or more of the lactose particles being less than 100-200 microns in diameter, and/or 50% or more of the lactose particles being less than 40- 70 microns in diameter. Most importantly, it is preferable that about 3 to about 30% (e.g. about 10%) (by weight or by volume) of the particles are less than 50 microns or less than 20 microns in diameter. For example, without limitation, a suitable inhalation-grade lactose is E9334 lactose (10% fines) (Borculo Domo Ingredients, Hanzeplein 25, 8017 JD Zwolle, Netherlands).
Optionally, in particular for dry powder inhalable compositions, a pharmaceutical composition for inhaled administration can be incorporated into a plurality of sealed dose containers (e.g. containing the dry powder composition) mounted longitudinally in a strip or ribbon inside a suitable inhalation device. The container is rupturable or peel-openable on demand and the dose of e.g. the dry powder composition can be administered by inhalation via the device such as the DISKUS TM device, marketed by GlaxoSmithKline. The DISKUS ™ inhalation device is for example described in GB 2242134 A, and in such a device at least one container for the pharmaceutical composition in powder form (the container or containers preferably being a plurality of sealed dose containers mounted longitudinally in a strip or ribbon) is defined between two members peelably secured to one another; the device comprises: a means of defining an opening station for the said container or containers; a means for peeling the members apart at the opening station to open the container; and an
outlet, communicating with the opened container, through which a user can inhale the pharmaceutical composition in powder form from the opened container.
A composition of the present invention, for intranasal administration, may also be adapted for dosing by insufflation, as a dry powder formulation.
It will be appreciated that when the compound of the present invention is administered in combination with other therapeutic agents normally administered by the inhaled, intravenous, oral or intranasal route, that the resultant pharmaceutical composition may be administered by the same routes.
The compound of the present invention may conveniently be administered in amounts of, for example, 1 μg to 100mg. The precise dose will of course depend on the age and condition of the patient and the particular route of administration chosen.
Biological test methods
Compounds of the invention may be tested for in vitro activity in accordance with the following assays:
1. Enzyme Assay - Time-resolved fluorescence resonance energy transfer kinase assay
Recombinant human Syk was expressed as a His-tagged protein*. The activity of Syk was assessed using a time-resolved fluorescence resonance energy transfer (TR-FRET) assay.
Version A - 3μl of substrate reagent containing biotinylated peptide, Biotin- AAAEEIYGEI (0.5μM final), ATP (30μM final) and MgCI2 (1OmM final) in HEPES pH 7.4, (4OmM final), were added to wells containing 0.2μl of various concentrations of compound or DMSO vehicle (3.3% final) in Greiner low volume 384 well black plate. The reaction was initiated by the addition of 3μl of Syk (2OnM final) in HEPES pH 7.4 (4OmM final). The reaction was incubated for 40min at room temperature, then terminated by the addition of 3μl of read reagent containing 60 mM EDTA, 15OmM NaCI, 5OnM Streptavidin APC (Prozyme, San Leandro, California, USA), 0.5nM antiphosphotyrosine antibody labelled with W-1024 europium chelate (Wallac OY, Turku, Finland) in 4OmM HEPES pH 7.4, 0.03% BSA. The reaction was further incubated for 60min at room temperature. The degree of phosphorylation of Biotin- AAAEEIYGEI was measured using a BMG Rubystar plate reader (BMG LabTechnologies Ltd, Aylesbury, UK) as a ratio of specific 665 nm energy transfer signal to reference europium 620 nm signal.
Version B - Syk was pre-activated at room temperature for 30 mins in the presence of 16.6mM MgCI2, 8.3mM ATP and then diluted to 4nM in 4OmM Hepes pH 7.4, 0.01% BSA. 3μl of substrate reagent containing biotinylated peptide, Biotin- AAAEEIYGEI (0.5μM final), ATP (30μM final) and MgCI2 (1OmM final) in 4OmM HEPES pH 7.4, 0.01% BSA, were added to wells containing 0.1 μl of various concentrations of compound or DMSO vehicle (1.7% final) in Greiner low volume 384 well black plate. The reaction was initiated by the addition of 3μl of diluted Syk (2nM final). The reaction was incubated for 60min at room temperature, then terminated by the addition of 3μl of read reagent containing 60 mM EDTA, 15OmM NaCI, 5OnM Streptavidin APC (Prozyme, San Leandro, California, USA), 0.5nM antiphosphotyrosine antibody labelled with W-1024 europium chelate (Wallac OY, Turku, Finland) in 4OmM HEPES pH 7.4, 0.03% BSA. The reaction was further incubated for 45min at room temperature. The degree of phosphorylation of Biotin- AAAEEIYGEI was measured using a BMG Rubystar plate reader (BMG LabTechnologies Ltd, Aylesbury, UK) as a ratio of specific 665 nm energy transfer signal to reference europium 620 nm signal.
Compounds according to the present invention were assayed in this, or a similar Time-resolved fluorescence resonance energy transfer kinase assay, and gave IC50 values less than 10μM.
Compounds according to the present invention were assayed in this, or a similar Time-resolved fluorescence resonance energy transfer kinase assay, and gave IC50 values less than 10μM.
* Preparation of Recombinant Human Full Length Spleen Tyrosine Kinase (Svk)
Full length human Syk was expressed with a 6His tag on the N-terminal using the baculovirus system (Invitrogen, Paisley, Scotland). The cells were disrupted by dounce homogenisation, the debris removed by centrifugation and the lysate contacted with NiNTA Superflow (Qiagen, Crawley, UK). The NiNTA was packed into a column and eluted using 10 column volumes each of buffer (2OmM Tris pH8.0, 30OmM NaCI, 1OmM βMcEtOH, 10% glycerol), buffer + 1 M NaCI, buffer + 2OmM Imidazole and buffer + 30OmM imidazole. The 30OmM Imidazole fractions were pooled buffer exchanged using G25M (Amersham Biosciences, Buckinghamshire, UK) into 2OmM MES pH 6.0, 2OmM NaCI, 1OmM βMcEtOH,10% glycerol. The buffer exchanged 6His-Syk was loaded onto a Source15S column (Amersham Biosciences, Buckinghamshire, UK) and the column eluted using a NaCI gradient 0-50OmM over 50 column volumes. The 6His-Syk containing fractions were pooled and concentrated by ultra-filtration. The identity of 6His-Syk was confirmed by peptide mass finger printing and intact LC-MS.
2. Whole Cell Assay - cFms assay
Principle of the assay
Cells of the mouse fibroblast cell line NIH-3T3 are stably transfected with a cFms- SYK chimera. Addition of the ligand (MCSF) produces dimerisation of the chimera resulting in autophosphorylation of the SYK kinase domain. Following cell lysis phosphorylated SYK is detected by ELISA.
Stimulation of cFms-SYK cells with MCSF Version A
Cells are plated at a density of 1x105/well in a volume of 200μl growth medium (DMEM containing 10% heat inactivated foetal calf serum, 1% L-glutamine, 400μg/ml geneticin and 400μg/ml zeocin) in 96 well Collagen 1 coated tissue culture plates. Following incubation at 37°C, 10% CO2, for 2Oh, the cell supernatant is removed and replaced with 200μl DMEM containing 1% penicillin/streptomycin (serum free DMEM). The cells are incubated for one hour under the conditions described above. The medium is removed, 50μl appropriately diluted compound solution added and
the plate incubated for a further hour. Cells are stimulated with 25μl MCSF (0.66μg/ml final) for 20min at 370C. After removal of the supernatant, the cells are washed with cold PBS and lysed with 100μl lysis buffer for 4h at 4°C.
Stimulation of cFms-SYK cells with MCSF Version B
Cells are plated at a density of 1x105/well in a volume of 200μl growth medium (DMEM containing 10% heat inactivated foetal calf serum, 1% L-glutamine, 400μg/ml geneticin and 400μg/ml zeocin) in 96 well Collagen 1 coated tissue culture plates. Following incubation at 37°C, 10% CO2, for 2Oh the cell supernatant is removed and 50μl appropriately diluted compound solution added and the plate incubated for an hour. Cells are stimulated with 25μl MCSF (0.66μg/ml final) for 20min at 37°C. After removal of the supernatant, the cells are washed with cold PBS and lysed with 100μl lysis buffer for 4h at 4°C.
cFms ELISA
85μl cell lysate is transferred to a 96 well ELISA plate coated with goat anti human M-CSF R capture antibody and incubated for 16 hours at 4°C. The plate is washed and a biotinylated anti-phosphotyrosine detection antibody added (100μl/well) for 2h at room temperature. This is removed and replaced with 100μl Streptavidin-HRP for 30min. Captured phosphorylated SYK is visualised using 100μl TMB substrate. The reaction is terminated with 50μl 1 M sulphuric acid and the absorbance measured at 450nm.
Compound Preparation Compound is prepared as a 1OmM stock in DMSO and a dilution series prepared in DMSO using 9 successive 5-fold dilutions. This dilution series is diluted a further 1 :333 with serum free DMEM to give the concentration range to be tested of 1x105 to 1.54x10"11M. Compound dilutions are prepared using the Biomek 2000 or Biomek Nx automated robotic pipetting systems.
3. B Cell Proliferation Assay
Background The population of B cells observed in this assay are the naive mature IgM/lgD expressing population. These form at least 70% of the purified B cell population (the rest being isotype switched memory B cells) and are the only cells that proliferate as the cells are stimulated with anti-lgM.
Anti-lgM drives signalling through the B cell receptor which is Syk dependant. Proliferation is a functional measure of B cell signalling that can be measured by observing the incorporation of tritiated methyl thymidine into the cells.
Protocol
Purified human tonsillar B cells are resuspended in Buckleys* medium at a concentration of 1.25 x 106 ml.
160μl of cells re-suspended in Buckley's medium is added to the compound and control wells of a 96 well plate. The control wells are located on column 11 and 12 of the 96 well plate. The background wells are located in column 12 and 20μl of 10μM control is added to provide an appropriate background control. 20μl of 1% DMSO is added to the wells in column 1 1 for the stimulated control.
The compound titrations are located between columns 1 and 10. Three compounds are run in duplicate on each plate and row A and B are used for the control compound titration.
The final concentration of DMSO is 0.1% in the assay. The cells are left for 45min, after 45min the proliferative stimulus is added to the first 11 wells of the 96 well plate and 20μl of medium is added to column 12. F(ab')2 fragments of a polyclonal goat anti-sera raised to human IgM is used at a final concentration of 15μg/ ml to stimulate the cells. (Biosource. Cat no: AMI 4601 ).
Tritiated methyl thymidine is added to the cells at a concentration of 1μCi per well. (Amersham, TRK 758). The radioactivity is added 65 hours after the initial stimulus and is left on the cells for 6 to 8 hours. After pulsing with methyl thymidine the cells are harvested on a Skatron 96 well cell harvester onto glass fibre mats. Once these have dried these are counted on a Wallac 1450 Microbeta scintillation counter.
Data is downloaded as an XL file and IC50's determined using Activity base.
* Buckleys Medium: 450 ml Iscoves (Sigma I 3390), 50ml FCS, 2.5 g BSA, 5ml Pen/ strep, 5ml Glutamine (20OmM), 500μl Apo transferrin (50mg/ml) Sigma (T 1147), 100μl Bovine Insulin (10mg/ml) Sigma (I 1882).
Compound Preparation
Compound is prepared as a 1OmM stock in DMSO and a dilution series prepared in DMSO using 9 successive 3-fold dilutions. This dilution series is diluted a further 1 :100 with Buckleys medium to give the concentration range to be tested of 100μM to 5nM. This is added as 20μl to 96 well plates in duplicate to generate two IC50's for each compound tested. Each plate is run in the presence of a control compound, which acts as an internal standard. .
4. LAD2 Assay
Principle of the assay
LAD2 is a stem cell factor (SCF)-dependent human mast cell line that was established by the NIH from bone marrow aspirates from a patient with mast cell sarcoma/leukaemia. LAD2 cells resemble CD34+-derived human mast cells and express functional FcεRI. The FcεRI is up-regulated in the presence of IL-4, SCF and IgE, subsequent cross linking of cell-bound IgE results in degranulation which can be measured as hexosaminidase release.
Priming LAD2 cells to up-regulate FcεRI
LAD2 cells are re-suspended at 1x105/ml in complete stem pro-34SFM (Gibco Cat 10640-019 media containing Stem Pro-34 nutrient supplement (1 :40), glutamine
(2mM), penicillin (100μg/ml), streptomycin (100μg/ml)) with additional supplements of human recombinant SCF (100ng/ml; R&D systems), human recombinant Interleukin-
4 (6ng/ml; R&D Systems) and IgE (100μg/ml; Calbiochem). Cells are then maintained for 5 days at 37°C, 5% CO2 in a humidified atmosphere.
Compound Preparation
Compounds are titrated from a 2mM stock in 100% DMSO to give 9 successive 1 :3 dilutions (V 96-well Nunc; Biomek 2000). From this master plate 3μl is dispensed into a daughter plate (flat 96-well NuncBiomek Fx) which, is then diluted 1 :40 in RPMI with 2mM glutamine, and 20μl of the diluted compound transferred into the Greiner cell plate. Therefore the final compound concentration range is 1x10"5M to 5x10"10M in a constant 0.5% DMSO. Control wells are treated with 0.5% DMSO.
Activation of LAD2 cells with anti-lgE Version A Primed LAD2 cells are centrifuged (30Og, 5min), the supernatant discarded and the cell pellet re-suspended at 1x104 cells/ml in RPMI supplemented with glutamine (2mM). Following a further centrifugation (30Og, 5min) the cells are re-suspended in fresh RPMI with glutamine (2mM), adjusted to a density of 2.85x105/ml, and pipetted into sterile V-well plates (70μl/well; Greiner) containing 20μl diluted compound (prepared as detailed above). Cells are then incubated for 1 h (37°C, 5% CO2 in a humidified atmosphere) before activating with a sub-maximal concentration of anti- lgE (10μl volume to give a final assay dilution of 1 :2700; Sigma). Following a 40min incubation (37°C, 5% CO2 in a humidified atmosphere), plates are centrifuged (120Og, 10min, 4CC) and the supernatant removed for hexosaminidase assay. The cell pellet is lysed in 100μl/well triton-X (0.5% in RPMI 2mM glutamine) at 37°C for 30min.
Activation of LAD2 cells with anti-lgE Version B
Primed LAD2 cells are centrifuged (40Og, 5min), the supernatant discarded and the cell pellet re-suspended at 1x104 cells/ml in RPMI supplemented with glutamine (2mM). Following a further centrifugation (40Og, 5min) the cells are re-suspended in fresh RPMI with glutamine (2mM), adjusted to a density of 5.7 x105AnI, and pipetted into sterile V-well plates (70μl/well; Greiner) containing 20μl diluted compound (prepared as detailed above). Cells are then incubated for 1 h (37°C, 5% CO2 in a
humidified atmosphere) before activating with a sub-maximal concentration of anti- IgE (10μl volume to give a final assay dilution of 1 :2700; Sigma). Following a 40min incubation (370C, 5% CO2 in a humidified atmosphere), plates are centrifuged (120Og, 10min, 40C) and the supernatant removed for hexosaminidase assay. The cell pellet is lysed in 100μl/well triton-X (0.5% in RPMI 2mM glutamine) at 37°C for 30min.
Beta-hexosaminidase assay
Beta-hexosaminidase activity is measured by the conversion of 4-methylumbelliferyl N-acetyl-ε-D glucosaminide (Sigma) to a fluorescent product.
Supernatant or lysate (25μl) is incubated with an equal volume of 4- methylumbelliferyl N-acetyl-ε-D glucosaminide (500μM in 0.2M sodium citrate buffer, pH 4.5) in black 96-well plate (Nunc) for 1 h at 370C. The reaction is then terminated by addition of Trizma pH9 (90μl) and the fluorescent product measured using excitation 356nm and emission 450nm (Tecan Safire)
A useful screening strategy comprises assay 1 (enzyme assay (pKi), assay 2 and then assay 3 (B Cell Proliferation) or assay 4 (LAD2).
Intermediates and Examples
General
All temperatures are in 0C.
DCM refers to dichloromethane DMSO refers to dimethylsulfoxide. DMF refers to Λ/,Λ/-dimethylformamide IPA refers to propan-2-ol THF refers to tetrahydrofuran.
HPLC refers to high performance liquid chromatography.
SPE refers to solid phase extraction cartridges marketed by lsolute
1H NMR spectra were recorded using a Bruker DPX 400MHz, referenced to tetramethylsilane.
LC/MS was conducted on a Supelcosil LCABZ+PLUS column (3.3 cm x 4.6 mm ID) eluting with 0.1% HCO2H and 0.01 M ammonium acetate in water (solvent A) and 0.05% HCO2H 5% water in acetonitrile (solvent B), using the following elution gradient 0.0-7min 0%B, 0.7-4.2 min 100%B, 4.2-5.3 min 0%B, 5.3-5.5min 0%B at a flow rate of 3ml/min. The mass spectra were recorded on a Fisons VG Platform spectrometer using electrospray positive and negative mode (ES+ve and ES-ve).
"Mass directed autoprep" / "preparative mass directed HPLC" was conducted on a system such as; a Waters FractionLynx system comprising of a Waters 600 pump with extended pump heads, Waters 2700 autosampler, Waters 996 diode array and Gilson 202 fraction collector on a 10 cm 2.54 cm ID ABZ+ column, eluting with either 0.1% formic acid or trifluoroacetic acid in water (solvent A) and 0.1% formic or trifluoroacetic acid in acetonitrile (solvent B) using the appropriate elution gradient. Mass spectra were recorded on Micromass ZMD mass spectrometer using electrospray positive and negative mode, alternate scans. The software used was MassLynx 3.5 with OpenLynx and FractionLynx optio or using equivalent alternative systems.
"Hydrophobic frits" refers to filtration tubes sold by Whatman. SPE (solid phase extraction) refers to the use of cartridges sold by International Sorbent Technology Ltd.
The Flashmaster Il is an automated multi-user flash chromatography system, available from Argonaut Technologies Ltd, which utilises disposable, normal phase, SPE cartridges (2 g to 100 g). It provides quaternary on-line solvent mixing to enable gradient methods to be run. Samples are queued using the multi-functional open access software, which manages solvents, flow-rates, gradient profile and collection conditions. The system is equipped with a Knauer variable wavelength uv-detector
and two Gilson FC204 fraction-collectors enabling automated peak cutting, collection and tracking.
Silica chromatography techniques include either automated (Flashmaster) techniques or manual chromatography on pre-packed cartridges (SPE) or manually- packed flash columns.
Microwave chemistry was typically performed in sealed vessels, irradiating with a suitable microwave reactor system, such as a Biotage Initiator™ Microwave Synthesiser.
When the name of a commercial supplier is given after the name of a compound or a reagent, for instance "compound X (Aldrich)" or "compound X / Aldrich", this means that compound X is obtainable from a commercial supplier, such as the commercial supplier named.
Similarly, when a literature or a patent reference is given after the name of a compound, for instance compound Y (EP 0 123 456), this means that the preparation of the compound is described in the named reference.
The names of the above mentioned Examples have been obtained using the compound naming programme "ACD Name Pro 6.02".
Example 1 - Λ/2-(1.1-dioxido-2.3-dihvdro-1 ,2-benzisothiazol-6-yl)-Λ/4-ethyl-5-fluoro-Λ/4- 1H-indazol-4-yl-2.4-pyrimidinediamine

To a solution of Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-1-({[2-
(trimethylsilyl)ethyl]oxy}methyl)-1 /-/-indazol-4-amine (50mg) in DMF (5ml) was added sodium hydride (60% in mineral oil, 5.3mg). The reaction was stirred for 15min before the addition of ethyl iodide (0.02ml). The reaction was stirred at room temperature overnight. The reaction was quenched with IPA (1ml) and concentrated in vacuo to
afford crude Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-ethyl-1-({[2- (trimethylsilyl)ethyl]oxy}methyl)-1H-indazol-4-amine which was used without further purification. LC/MS Rt 3.92 min, MH+ 422, 424.
A microwave vessel was charged with the crude Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)- Λ/-ethyl-1-({[2-(trimethylsilyl)ethyl]oxy}methyl)-1H-indazol-4-amine dissolved in IPA (3ml) and hydrochloric acid (2M, 0.2ml). 2,3-Dihydro-1 ,2-benzisothiazol-6-amine 1 ,1- dioxide (30mg) was added and the mixture was heated in the microwave at 150°C for 30min. The crude material was concentrated in vacuo, then purified by mass directed autoprep to afford /^-(i .i-dioxido^.S-dihydro-i ^-benzisothiazol-δ-yO-ΛΛethyl-δ- fluoro-/V*-1/-/-indazol-4-yl-2,4-pyrimidinediamine (4.2 mg) LC/MS; Rt 2.93 min, MH+ 440.
Example 2 - δ-chloro-Λ^-d .i-dioxido^.S-dihydro-i ^-benzisothiazol-β-vn-Λ^-ethyl- Λ^-IH-indazol^-yl^^-pyrimidinediamine

To a solution of Λ/-(2-chloro-5-chloro-4-pyrimidinyl)-1-({[2- (trimethylsilyl)ethyl]oxy}methyl)-1H-indazol-4-amine (50mg) in DMF (5ml) was added sodium hydride (60% in mineral oil, 5.3mg). The reaction was stirred for 15 minutes before the addition of ethyl iodide (0.02ml). The reactions were then stirred at room temperature overnight. The reaction was quenched with IPA (1ml) and concentrated to afford crude Λ/-(2-chloro-5-chloro-4-pyrimidinyl)-Λ/-ethyl-1-({[2- (trimethylsilyl)ethyl]oxy}methyl)-1H-indazol-4-amine which was used without further purification. LC/MS; Rt 4.07 min, MH+ 438, 440, 442.
A microwave vessel was charged with the crude Λ/-(2-chloro-5-chloro-4-pyrimidinyl)- Λ/-ethyl-1-({[2-(trimethylsilyl)ethyl]oxy}methyl)-1H-indazol-4-amine dissolved in IPA (3ml) and hydrochloric acid (2M, 0.2ml). 2,3-Dihydro-1 ,2-benzisothiazol-6-amine 1 ,1- dioxide (30mg) was added and the mixture was heated in the microwave at 1500C for 30min. The crude material was concentrated in vacuo, then purified by mass directed autoprep to afford 5-chloro-Λ/2-(1 ,1-dioxido-2,3-dihydro-1 ,2-benzisothiazol-6-yl)-Λ/4-
ethyl-A/MH-indazol^-yl^-pyrimidinediamine (2.4 mg) LC/MS; Rt 3.11min, MH+ 455, 457.
Intermediate 1 - 2,3-dihvdro-1 ,2-benzisothiazol-6-amine 1 ,1-dioxide

To a suspension of 6-amino-2-benzisothiazol-3(2H)-one 1 ,1-dioxide (6.37g, Bull Chem Soc Jpn 1982 55(12) 3824-7) in cone, hydrochloric acid (79.6ml) was added zinc dust (18.9g) portionwise over half an hour and cooling with an ice bath when necessary (maximum temperature 700C). The reaction was stirred at room temperature for 3h. The mixture was basified with a solution of saturated NaHCO3 (50ml), then with solid NaHCO3. The mixture was filtered and the filtrate was extracted with ethyl acetate (3x 150ml). The organic phases were combined, dried (brine and magnesium sulphate), filtered and concentrated under vacuum to give a pale yellow solid (1.548g). The residue from filtration of the basified reaction was triturated with ethyl acetate (500ml) and filtered. The orange filtrate was concentrated under vacuum to give an orange solid (1.789g). The residue obtained from filtration was triturated again with ethyl acetate (250ml), filtered and concentrated under vacuum to give a pale orange solid (0.357g). All the solids were dissolved in methanol, combined and then concentrated to give the desired product as a beige solid (3.151g). LC/MS; Rt 0.83min.
Intermediate 2 - ethyl 1 ,2-benzisothiazole-2(3/-/)-carboxylate 1.1 -dioxide

Ethyl chloroformate (136ml) was added dropwise to a chilled solution of 2,3-dihydro- 1 ,2-benzisothiazol-1 ,1 -dioxide (68.Og) in pyridine (340ml) over 1 h, maintaining the
temperature below 18°C. The slurry was stirred for 10min at 200C, diluted dropwise with water (1000ml) over 75min and stirred for a further 30min. The mixture was chilled to 0-50C, aged for 1 h and isolated by filtration. The solid was washed with cold water (2x 250ml), and dried in vacuo to give the title compound as a white solid
(72.35g).
NMR; [D6-DMSO] δH 1.30,(3H1 1, J=7.1 Hz), 4.32,(2H, q, J=7.1 Hz), 5.01 ,(2H, s), 7.64-
7.70,(2H1 m), 7.81 ,(1 H, td, J=7.6Hz J=LOHz), 7.98,(1 H d, J=8.0Hz).
Intermediate 3 - ethyl 6-nitro-1 ,2-benzisothiazole-2(3H)-carboxylate 1 ,1-dioxide

A solution of ethyl 1 ,2-benzisothiazole-2(3H)-carboxylate 1 ,1-dioxide (68.Og) in concentrated sulphuric acid (180ml) was cooled to 0-50C and treated dropwise with fuming nitric acid (25ml) over 45min. The mixture was treated further with fuming nitric acid (15ml) over 30min, aged for 40min and quenched into water (700ml) at 15- 200C over 71 min in the presence of seed. The suspension was aged for 45min, filtered and washed with water (3x150ml). The product was dried in vacuo at 50°C to give the title compound as a mixture of regioisomers.
This was suspended in acetonitrile (400ml) and water (320ml) at almost reflux, treated with further acetonitrile (40ml), and aged for 1 h. The mixture was cooled slowly to ambient temperature over 3h, cooled further to 0-50C and aged for 30min. The product was isolated by filtration, washed with 1 :1 water / acetonitrile (140ml), then water (140ml), and dried in vacuo at 50°C to give the title compound as an off- white solid (64.5g). NMR; [D6-DMSO] δH 1.31 ,(3H, t, J=7.1 Hz), 4.35,(2H, q J=7.1 Hz), 5.16,(2H, s), 7.96,(1 H, dq J=8.6Hz J=0.5Hz), 8.61 ,(1 H, dd, J=8.6Hz J=2.2Hz), 8.90,(1 H, d, J=2.1Hz).
Intermediate 4 - 2,3-dihvdro-1 ,2-benzisothiazol-6-amine 1.1-dioxide

A suspension of ethyl 6-nitro-1 ,2-benzisothiazole-2(3H)-carboxylate 1 ,1-dioxide (10.Og), Pd/C (5%, 1.0g, 50%wet) and methanesulfonic acid (4.5ml) in IPA (100ml) was hydrogenated under hydrogen (2bar) at 500C for ~6min. The mixture was purged with nitrogen, cooled to 2O0C and treated with a mixture of sodium hydroxide (2M, 90ml) and sodium hydroxide (10M, 10ml). The reaction was stirred for 2h at ambient temperature and filtered through a filter pad, washing the pad with water (20ml). Hydrochloric acid (5M, 45ml) was added to the filtrate to adjust to pH7. The resultant slurry was stirred at ambient temperature for 2h, cooled to 0-50C and aged for a further 1 h. The product was isolated by filtration, washed with cold water / IPA (2:1, 30ml) then water (2x 30ml). The solid was dried in vacuo at 500C to give the title compound as a light grey solid (5.23g). NMR; [D6-DMSO] δH 4.18, (2H, s), 5.59,(2H1 s), 6.79,(1 H, d, J=2.1 Hz), 6.83,(1 H, dd, J=8.3Hz J=2.1Hz), 7.14,(1 H, d, J=8.3Hz), 7.52,(1H, br s).
Intermediate 5 - Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-ethyl-1-({r2- (trimethylsilyl)ethvπoxy)methyl)-1H-indazol-4-amine

To a flask charged with Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-1-({[2- (trimethylsilyl)ethyl]oxy}methyl)-1H-indazol-4-amine (10.84g) in DMF (400ml) was added potassium carbonate (4.98g) portionwise. The mixture was stirred for 30min prior to the addition of ethyl iodide (2.63ml). The reaction mixture was stirred overnight. The solvent was removed in vacuo and the residue partitioned with water (3x 300ml) and DCM (3x 300ml). The dried organic extracts (MgSO4) were filtered and concentrated. The crude residue was purified by chromatography on silica cartridges (2x 7Og) eluting with 20% ethylacetate in cyclohexane to afford, after
evaporation of the solvents, Λ/-(2-chloro-5-fluoro-4-pyhmidinyl)-Λ/-ethyl-1-({[2- (trimethylsilyl)ethyl]oxy}methyl)-1H-indazol-4-amine as a yellow gum which solidified on cooling (6.25g). LC/MS Rt 3.88min, MH+ 422, 424.
Intermediate 6 - Λ/-(5-bromo-2-chloro-4-pyrimidinyl)-Λ/-ethyl-1-((r2- (trimethylsilyl)ethvHoxy)methyl)-1H-indazol-4-amine

To a flask charged with Λ/-(2-chloro-5-bromo-4-pyrimidinyl)-1 -({[2-
(trimethylsilyl)ethyl]oxy}methyl)-1H-indazol-4-amine (2.Og) in DMF (100ml) was added potassium carbonate (0.79g) portionwise. The mixture was stirred for 30min prior to the addition of ethyl iodide (0.42ml). The reaction mixture was stirred at 23°C for 16h after which time a further aliquot of ethyl iodide (0.42ml) was added and the reaction stirred for 6h. At this time an additional aliquot of ethyl iodide (0.42ml) was added and the reaction stirred for 16h. The reaction was concentrated in vacuo and the residue partitioned with water (50ml) and DCM (3x 50ml). The dried organic extracts (hydrophobic frit) were concentrated and the crude residue purified by chromatography (2Og cartridge) eluting with an ethyl acetate / cyclohexane gradient. Evaporation of the solvents gave Λ/-(5-bromo-2-chloro-4-pyrimidinyl)-Λ/-ethyl-1 -({[2- (trimethylsilyl)ethyl]oxy}methyl)-1/-/-indazol-4-amine as a pale yellow oil. (1.0g). LC/MS; Rt 4.17min, MH+ 482, 484, 486.
Intermediate 7 - Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-1-((r2- (trimethylsilyl)ethyl1oxy)methyl)-1H-indazol-4-amine

The second portion was absorbed onto a 75Og silica cartridge before being purified using a CombiFlash® Companion™ system eluting with a gradient of ethyl acetate in cyclohexane (0-100%). The required pure fractions were combined and the solvent was evaporated in vacuo to give a further quantity of the Λ/-(2-chloro-5-fluoro-4- pyrimidinyl)-1-({[2-(trimethylsilyl)ethyl]oxy}methyl)-1/-/-indazol-4-amine as an orange/brown solid (10.96g). LC/MS; Rt 3.57min, MH+ 394,396.
Intermediate 8 - 1-((f2-(trimethylsilyl)ethylloxy>methyl)-1 H-indazol-4-amine mixture with 2-({f2-(trimethylsilyl)ethylloxy>methyl)-2H-indazol-4-amine

Two vigorously stirred solutions, each containing 4-nitro-1-({[2- (trimethylsilyl)ethyl]oxy}methyl)-1 H-indazole mixture with 4-nitro-2-({[2- (trimethylsilyl)ethyl]oxy}methyl)-2H-indazole (21.17g) in ethanol (150ml), were hydrogenated at room temperature and 1atm. of pressure using 10% palladium on carbon catalyst (2.65g) for 1.5h. The mixtures were both filtered through Celite and the filter cakes were washed with portions of ethanol. The combined ethanol filtrates and washings from both reactions were evaporated in vacuo to give 1-({[2-
(trimethylsilyl)ethyl]oxy}methyl)-1 H-indazol-4-amine mixture with 2-({[2- (trimethylsilyl)ethyl]oxy}methyl)-2H-indazol-4-amine as a brown oil (36.83g). LC/MS; Rt 3.14 and 3.35min, MH+ 264.
Intermediate 9 - 4-nitro-1-((r2-(trimethylsilyl)ethvπoxy)methyl)-1H-indazole mixture with 4-nitro-2-((r2-(trimethylsilyl)ethylloxy>methyl)-2H-indazole

To a stirred solution of 4-nitroindazole (24.Og, Bionet Research Intermediates Catalogue) in dry tetrahydrofuran (380ml), stirred under nitrogen and cooled using an ice bath was added portion wise sodium-terf-butoxide (14.85g) followed after 5min by 2-(trimethylsilyl)ethoxymethylchloride (28.2ml). After removal of the cooling bath, the mixture was stirred at room temperature for 3h. Methanol (240ml) was added to the mixture and the solvent was evaporated in vacuo. The residue was partitioned between chloroform (300ml) and water (200ml), the phases were separated and the aqueous phase was extracted with chloroform (3x 100ml). The combined organic phases were dried (MgSO4), filtered and the solvent evaporated in vacuo to give 4- nitro-1-({[2-(trimethylsilyl)ethyl]oxy}methyl)-1 H-indazole mixture with 4-nitro-2-({[2- (trimethylsilyl)ethyl]oxy}methyl)-2H-indazole as a orange/brown oil (42.34g). LC/MS; Rt 3.68 and 3.77min, MH+ 294.
Intermediate 10 - Λ/-(5-bromo-2-chloro-4-pyrimidinyl)-1-(([2- (trimethylsily0ethvπoxy)methyl)-1H-indazol-4-amine

Example 3 - formic acid - 5-bromo-/vVi .1-dioxido-2,3-dihvdro-1 ,2-benzisothiazol-6- vD-Λ^-ethyl-Λ^-i /-/-indazol-4-yl-2,4-pyrimidinediamine (1 :1)

A flask was charged with Λ/-(5-bromo-2-chloro-4-pyrimidinyl)-Λ/-ethyl-1-({[2- (trimethylsilyl)ethyl]oxy}methyl)-1H-indazol-4-amine (0.05g), 2,3-dihydro-i ,2- benzisothiazol-6-amine 1 ,1 -dioxide (0.042g), and a solution of acetone / water / cone. hydrochloric acid (150:100:1 , 4ml) The solution was heated at reflux (ca. 900C) for 16h. At this time a further aliquot of cone, hydrochloric acid was added (1 ml) and the reaction heated at reflux for a further 16h. The crude material was concentrated in vacuo, then purified by preparative mass directed HPLC to afford the title compound as a pink solid (4.0mg) LC/MS; Rt 3.13min, MH+ 500, 502.
Example 4 - Λ/2-(1.1-dioxido-2,3-dihvdro-1 ,2-benzisothiazol-6-yl)-Λ/4-ethyl-5-fluoro-/V4- 1/-/-indazol-4-yl-2.4-pyrimidinediamine

A microwave vessel was charged with Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-ethyl-1- ({[2-(trimethylsilyl)ethyl]oxy}methyl)-1H-indazol-4-amine (0.05g) and 2,3-dihydro-1 ,2- benzisothiazol-6-amine 1 ,1 -dioxide (0.025g), followed by and a solution of acetone/water/conc. hydrochloric acid (150:100:1 , 3ml) and N-methyl pyrrolidinone (0.1ml). The mixture was heated by microwave irradiation in a sealed vessel at 1500C for 60min. The crude material was added to an SCX cartridge (10g), primed with methanol. The cartridge was eluted with 3x column volumes of methanol and then flushed with 2x column volumes ammonia in methanol solution (2M). The basic fractions were combined and concentrated and the residue purified by preparative mass directed HPLC to afford the title compound (31.3mg) LC/MS; Rt 2.92min, MH+ 440.
Intermediate 11 - N-(2-chloro-5-fluoro-4-pyrimidinyl)-N-ethyl-1 H-indazol-4-amine

A solution of Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-ethyl-1-({[2-
(trimethylsilyl)ethyl]oxy}methyl)-1H-indazol-4-amine (4.2g) in IPA (20ml) and hydrochloric acid (5M, 60ml) was stirred with heating at 550C for 4h. After allowing to cool, the solvents were evaporated in vacuo, the residue was basified with saturated aqueous sodium carbonate solution and extracted with ethyl acetate (3x 100ml). The combined organic phases were washed with water (3x 100ml), dried (MgSO4), filtered, and the solvent evaporated in vacuo. The residue was absorbed onto a silica SPE cartridge (100g) which was then eluted with an ethyl acetate / cyclohexane
gradient (0-100%) over 40min, followed by a gradient of methanol in ethyl acetate (0- 20%). The required fractions were combined and the solvent was evaporated in vacuo to give the N-(2-chloro-5-fluoro-4-pyrimidinyl)-N-ethyl-1 H-indazol-4-amine as a yellow oil (1.55g). LC/MS; Rt 3.05min, MH+ 292, 294.
Example 5 - /v4-Ethyl-5-fluoro-Λ/4-1H-indazol-4-yl-Λ/2-(2-methyl-1.1-dioxido-2.3- dihvdro-1.2-benzisothiazol-6-yl)-2.4-pyrimidinediamine

2-Methyl-2,3-dihydro-1 ,2-benzisothiazol-6-amine 1 ,1-dioxide (40mg) and Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-ethyl-1 H-indazol-4-amine (50mg) were dissolved in a mixture of acetone / water / cone, hydrochloric acid (1.2ml, 150:100:1 ). The solution was heated at 160°C by microwave irradiation in a sealed vessel for 30min. The reaction was then evaporated to dryness and the crude residue was purified by mass directed autoprep to give the title compound (36mg). LC/MS; Rt 3.17min, MH+ 454.
Intermediate 12 - 2-Methyl-2,3-dihydro-1 ,2-benzisothiazol-6-amine 1 ,1-dioxide

2,3-Dihydro-1 ,2-benzisothiazol-6-amine 1 ,1-dioxide (185mg) was dissolved in dry methanol (15ml). Benzaldehyde (138μl) was added and the mixture stirred at room temperature under nitrogen for 4h. The solvent was removed in vacuo and the residue re-dissolved in dry methanol (10ml) and benzaldehyde (100μl) added. The reaction mixture was stirred for a further 3h, filtered, the solid washed with a minimal amount of methanol and sucked dry on the sinter. The resulting solid (137mg) was dissolved in dry DMF (1 ml) and added dropwise to a mixture of sodium hydride (60% in mineral oil; 24mg) in dry DMF (2ml) under nitrogen. The mixture was stirred at
room temperature for 15min and methyl iodide (100μl) added. After stirring for 2h, hydrochloric acid (2N, 2ml) was added and the mixture stirred for 2h and left standing for 18h. The solvent was removed in vacuo, the residue dissolved in methanol and applied to an SPE cartridge (SCX-2, 7Og) eluting with ammonia in methanol (2N). The appropriate fractions were combined and concentrated to give the title compound (89mg). LC/MS; MH+ 199, Rt 1.93min.
Example 6 - Λ/^d .i-dioxido^.S-dihydro-i ^-benzisothiazol-e-vn-δ-fluoro-Λ^-i /-/- indazol-4-yl-Λ/4-methyl-2.4-pyrimidinediamine trifluoroacetate

A mixture of Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-N-methyl-1 H-indazol-4-amine (28mg) and 2,3-dihydro-1 ,2-benzisothiazol-6-amine 1 ,1 -dioxide (0.15mmol) in acetone / water / cone, hydrochloric acid (150:100:1 , 1.5ml) was heated at 800C for 2 days. The reaction was then evaporated to dryness (genevac) and the crude residue was dissolved in methanol and was loaded on to an SCX-2 SPE cartridge (500mg), which was washed with methanol and product eluted with 10% ammonia/methanol. The methanolic ammonia was evaporated under a stream of nitrogen, and the residue was further purified by mass directed autoprep to give the title compound (11.6mg). LC/MS: MH+ 426, Rt 2.79.
Intermediate 13 - Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-methyl-1 H-indazol-4-amine

Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-methyl-1-({[2-(trimethylsilyl)ethyl]oxy}methyl)- 1 H-indazol-4-amine (2.12g) in 5M hydrochloric acid / IPA (2:1 , 60ml) was heated at 550C for 3.5h. The cooled reaction was then evaporated to dryness. The resultant residue was basified with saturated sodium carbonate solution (70ml) and extracted
with ethyl acetate (3x 70ml). The combined organic extracts were washed with water (3x 70ml), dried (MgSO4) and the solvent evaporated. The residue was purified by chromatography on a silica cartridge (10Og) eluting with an ethyl acetate / cyclohexane gradient (0-50%) over 40min to give, after evaporation of the solvents Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-methyl-1 H-indazol-4-amine (285mg). LC/MS; Rt 2.97min, MH+ 278.
Intermediate 14 - Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-methyl-1-((r2- (trimethylsilyl)ethylloxy)rnethyl)-1 H-indazol-4-amine

Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-1-({[2-(trimethylsilyl)ethyl]oxy}methyl)-1 H-indazol- 4-amine (1.938g) was dissolved in DMF (20ml) and cooled to 00C in an ice-bath under nitrogen. Sodium hydride (60% in mineral oil, 236mg) was added and the mixture stirred for 15min. Methyl iodide (459μl) was added and the mixture was allowed to warm at room temperature and stirred for 3h. Saturated ammonium chloride (5ml) was added and the mixture reduced to dryness. The crude residue was partitioned between ethyl acetate (25ml) and water (15ml). The organic extract was washed with water (3x 15ml), dried (MgSO4) and the solvent evaporated in vacuo to give Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-methyl-1-({[2-
(trimethylsilyl)ethyl]oxy}methyl)-1 H-indazol-4-amine (2.12g). LC/MS; Rt 3.82min, MH+ 408.
Intermediate 15 - Λ/-(2,5-dichloro-4-pyrirnidinyl)-Λ/-ethyl-1 H-indazol-4-amine

N-(2,5-dichloro-4-pyrimidinyl)-N-ethyl-1 -({[2-(trimethylsilyl)ethyl]oxy}methyl)-1 H- indazol-4-amine (520mg) in 5M hydrochloric acid / IPA (2:1 ; 15ml) was heated at
55°C for 6.5h. The cooled mixture was then evaporated to dryness in vacuo and the crude residue was left at room temperature overnight. The resultant residue was basified with saturated sodium carbonate solution (20ml) and extracted with ethyl acetate (3x 30ml). The combined organic extracts were washed with water (2x 20ml), dried (MgSO4) and the solvent evaporated in vacuo. The residue was purified by chromatography on a silica cartridge (5Og) eluting with an ethyl acetate / cyclohexane gradient (0-50%) over 40min. The product containing fractions were combined and reduced to dryness in vacuo. The residue was purified by mass directed autoprep to give, after evaporation of the solvents, Λ/-(2,5-dichloro-4-pyrimidinyl)-Λ/-ethyl-1H- indazol-4-amine (79mg). LC/MS; Rt 3.33min. MH+ 308, 310.
Intermediate 16 - N-(2,5-dichloro-4-pyrimidinyl)-N-ethyl-1-((r2- (trimethylsilyl)ethylloxy)methyl)-1 H-indazol-4-amine

N-(2,5-dichloro-4-pyrimidinyl)-1-({[2-(trimethylsilyl)ethyl]oxy}methyl)-1 H-indazol-4- amine (870mg) was dissolved in DMF (20ml) at room temperature under nitrogen. Cesium carbonate (760mg) was added and the mixture stirred for 30min. Ethyl iodide (255μl) was added and the mixture was left to stir at 600C for 1.5h. The solvent was evaporated in vacuo and the residue was partitioned between ethyl acetate and water. The organic extract was washed with water (3x 15ml), dried (MgSO4) and reduced to dryness. The residue was purified by chromatography on silica cartridges (5Og) eluting with an ethyl acetate / cyclohexane gradient (0-50%) over 40min to give N-(2,5-dichloro-4-pyrimidinyl)-N-ethyl-1-({[2-(trimethylsilyl)ethyl]oxy}methyl)-1 H- indazol-4-amine (520mg). LC/MS; Rt 4.08min. MH+ 438, 440.
Intermediate 17 - N-(2.5-dichloro-4-pyrimidinyl)-1-((r2- (trimethylsilyl)ethvπoxy)methyl)-1 H-indazol-4-amine
A mixture of 1-({[2-(trimethylsilyl)ethyl]oxy}methyl)-1 H-indazol-4-amine and 2-({[2- (trimethylsilyl)ethyl]oxy}methyl)-2H-indazol-4-amine (5g) was mixed with 2,4,5- trichloropyrimidine (2.17ml, Aldrich) in N,N-diisopropylethylamine (40ml) was heated at 1500C for 5h. The reaction was then evaporated in vacuo and the residue was purified by chromatography on silica cartridges (2x 100g) eluting with an ethyl acetate / cyclohexane gradient (0-100%) over 40min to give after evaporation of the solvents N-(2,5-dichloro-4-pyrimidinyl)-1 -({[2-(trimethylsilyl)ethyl]oxy}methyl)-1 H- indazol-4-amine (945mg). LC/MS; Rt 3.80min. MH+ 410.
Example 7 - Λ/^d .i-dioxido^.S-dihvdro-i ^-benzisothiazol-θ-vD-ΛΛ-ethyl-δ-fluoro-Λ/4- 1H-indazol-4-yl-2.4-pyrimidinediamine

A mixture of 1-acetyl-Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-ethyl-1H-indazol-4-amine (1.0g) and 2,3-dihydro-1 ,2-benzisothiazol-6-amine 1 ,1-dioxide (1.1g) in acetone : water : concentrated hydrochloric acid mixture (150:100:1 , 15ml) at heated at 1600C in a Biotage Initiator microwave apparatus for 60min. The reaction was repeated using 1-acetyl-Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-ethyl-1/-/-indazol-4-amine (1.Og) and 2,3-dihydro-1 ,2-benzisothiazol-6-amine 1 ,1-dioxide (0.825g). A third reaction was carried out using 1-acetyl-Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-ethyl-1H-indazol- 4-amine (1.0g) and 2,3-dihydro-1 ,2-benzisothiazol-6-amine 1 ,1-dioxide (605mg) in 10ml of the solvent mix. A fourth reaction used 1-acetyl-Λ/-(2-chloro-5-fluoro-4- pyrimidinyl)-Λ/-ethyl-1H-indazol-4-amine (2.Og) and 2,3-dihydro-1 ,2-benzisothiazol-6- amine 1 ,1-dioxide (1.21g) in 15ml of the solvent mix. The reaction was finally
repeated twice more using 1-acetyl-Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-ethyl-1H- indazol-4-amine (2.58g) and 2,3-dihydro-1 ,2-benzisothiazol-6-amine 1 ,1-dioxide (1.56g) in 15ml of the solvent mix. All the six reaction mixtures were combined and concentrated in vacuo to remove the acetone. The resulting suspension was diluted with water (200ml) and the mixture stirred vigorously for several hours. The solid was filtered and dried in vacuo at 400C resulting in 12.1g of the title compound. This solid was dissolved in methanol (~100ml) and applied to a 5Og aminopropyl SPE cartridge initially under gravity then under pressure. The column was eluted with further methanol (150ml). The eluant was concentrated slightly and then filtered to give the title compound as a beige solid (5.3Og). The filtrate was concentrated to ca 100ml volume and a second crop (1.79g) of the title compound was obtained by filtration. A third crop of the title compound was obtained after standing the filtrate overnight (0.36g). The first and 1.59g of the second crop were combined to give one batch of the title compound. LC/MS; Rt 2.86min, MH+ 440.
Λ/2-(1 ,1-dioxido-2,3-dihydro-1 ,2-benzisothiazol-6-yl)-Λ/4-ethyl-5-fluoro-Λ/4-1H-indazol- 4-yl-2,4-pyrimidinediamine (3.4g) was dissolved in hot ethyl acetate (~750ml), the solution filtered and reduced in vacuo to approx. 600ml. The suspension was reheated and refiltered and the filtrate once again heated to boiling. The solution was allowed to cool to room temperature and then further cooled for 5 days at ca. 4°C. The crystals thus formed were isolated by filtration, washed with a little ethyl acetate and sucked dry on the sinter to give Λ/2-(1 ,1-dioxido-2,3-dihydro-1 ,2- benzisothiazol-6-yl)-Λ/4-ethyl-5-fluoro-Λ/4-1H-indazol-4-yl-2,4-pyrimidinediamine as beige crystals containing 1 equivalent of ethyl acetate (3.26g). NMR; [D6-DMSO] δH 13.29,(1 H, bs), 9.82,(1 H, s), 8.48,(1 H, d), 8.00,(1 H, d), 7.94,(1 H, s), 7.74,(2H, m), 7.51 ,(1 H, d), 7.41-7.35,(2H, m). 7.05,(1 H, d), 4.32,(2H, s), 4.11 ,(2H, q), 4.03,(2H, q, EtOAc), 1.99,(3H1 s, EtOAc), 1.23,(3H, t), 1.17,(3H, t, EtOAc).
Example 8 - /1Z-(I , i-dioxido^.S-dihvdro-i ^-benzisothiazol-e-vπ-Λ^-ethyl-δ-fluoro- ΛΛi H-indazol-4-γl-2,4-pyrimidinediamine

A mixture of 1-acetyl-Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-ethyl-1/-/-indazol-4-amine (33.3g) and 2,3-dihydro-1 ,2-benzisothiazol-6-amine 1 ,1-dioxide (20.Og) in IPA (80ml) and hydrochloric acid (2M, 40ml) was heated to reflux and stirred for 21 h. The mixture was cooled to 6O0C, diluted with DMSO (82ml) and then methanol (235ml). The mixture was treated with sodium hydroxide (2M, 130ml) to pH8 and cooled to 400C. The mixture was seeded, aged at 400C for 1h, then cooled to 15°C over 1h and aged for 1h. The product was isolated by filtration, washed with methanol (2x 130ml) and dried in vacuo at 50°C to give the title compound as a beige solid (30.7g) containing some residual solvents.
NMR; [D6-DMSO] δH 1.24,(3H, t, J=7.1 Hz), 4.12,(2H, q, J=7.0Hz), 4.32,(2H, s), 7.05,(1H, d, J=7.3Hz), 7.35-7.42,(2H, m), 7.51,(1 H, d, J=8.4Hz), 7.70-7.80, (2H, dd + br s), 7.95,(1 H, s), 8.00,(1 H, d, J=5.5Hz), 8.49,(1 H, d, J=1.9Hz), 9.83,(1 H, s), 13.30,(1 H, br s).
Intermediate 18 - 1-acetyl-Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-ethyl-1 /-/-indazol-4- amine
Intermediate 19 - 1-acetyl-Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-ethyl-1/-/-indazol-4- amine

Intermediate 20 - Λ/-(3-r(2-chloro-5-fluoro-4-pyrimidinyl)(ethv0aminol-2- methylphenvDacetamide

A mixture of Λ/-{3-[(2-chloro-5-fluoro-4-pyrimidinyl)amino]-2-methylphenyl}acetamide (6.9g) in anhydrous DMF (50ml) was stirred with potassium carbonate (3.6g) and ethyl iodide (2.06ml). The reaction mixture was stirred for approximately 5.5h and the solvent removed in vacuo. The residue was partitioned between ethyl acetate (50ml) and water (25ml). The organic layer was separated and washed with water (2x 25ml) and dried with brine (25ml) and over magnesium sulphate. The solvent was removed in vacuo to give the title compound (7.2g). LC/MS; Rt 2.8min, MH+ 323, 325.
Intermediate 21 - Λ/-{3-r(2-chloro-5-fluoro-4-pyrimidinyl)(ethyl)aminol-2- methylphenvDacetamide

Intermediate 22 - Λ/-(3-[(2-chloro-5-fluoro-4-pyrimidinyl)amino1-2- methylphenvDacetamide

A mixture of 3-acetylamino-2-methylaniline (EP425345A1 , 7.72g) and 2,4-dichloro-5- fluoropyrimidine (7.84g) in methanol / water (1 :3, 188ml) was stirred at 700C for 1 h. The reaction mixture was cooled and then filtered. The residue was washed with ice cold methanol / water (1 :3) and the solid dried in vacuo at 50°C and then at 65°C to give the title compound as a white solid (6.9g). LC/MS; Rt 2.2min, MH+ 295, 297.
Intermediate 23 - Λ/-{3-r(2-chloro-5-fluoro-4-pyrimidinyl)amino1-2- methylphenyllacetamide

A mixture of 2,4-dichloro-5-fluoropyrimidine (23.Og), 2-acetylamino-6-aminotoluene (23.71 g) and sodium acetate (22.52g) was slurried in IPA (115ml) and water (345ml), and warmed to 7O0C. The mixture was seeded with Λ/-{3-[(2-chloro-5-fluoro-4- pyrimidinyl)amino]-2-methylphenyl}acetamide after 10min at 7O0C; the mixture was
stirred at 7O0C for 5h, then allowed to cool to ambient temperature overnight. The mixture was further cooled to 5-100C, aged for 2h and the solid isolated by filtration. The product was washed with water / IPA (3:1 , 100ml), then water (2x 100ml), and dried in vacuo to give the title product as a white solid (35.15g).
Example 9 - Λ/2-r2-(cvclopropylmethyl)-1.1-dioxido-2.3-dihvdro-1.2-benzisothiazol-6- yll-Λ/4-ethyl-5-fluoro-Λ/4-1/-/-indazol-4-yl-2.4-pyrimidinediamine trifluoroacetate

A mixture of /V2-(1 J-dioxido^.S-dihydro-i ^-benzisothiazol-θ-yO-ΛΛethyl-S-fluoro-Λ^- IH-indazol^-yl^^-pyrimidinediamine (10mg) and cesium carbonate (10mg) in DMF (2ml) was stirred at room temperature under nitrogen for 10min. Bromomethylcyclopropane (3μl) was then added and the reaction mixture was stirred at room temperature under nitrogen for 16h. Saturated sodium chloride solution (0.5ml) was added and the solvent was removed under vacuum. The residue was then purified by preparative HPLC to give the title compound as a white solid (4.5mg). LC/MS; Rt 3.34min, MH+ 494.
Example 10 - Λ/4-ethyl-Λ/2-(2-ethyl-1.1-dioxido-2.3-dihvdro-1 ,2-benzisothiazol-6-yl)-5- fluoro-Λ/4-1/-/-indazol-4-yl-2,4-pyrimidinediamine trifluoroacetate

A mixture of ^-(i .i-dioxido^.S-dihydro-i ^-benzisothiazol-S-yO-Λ^-ethyl-δ-fluoro-Λ/4- 1 H-indazol-4-yl-2,4-pyrimidinediamine (35mg), bromoethane (7μl) and cesium carbonate (31 mg) in dry DMF (2ml) was stirred at room temperature under nitrogen for 16h. Saturated ammonium chloride solution (0.5ml) was added and the solvent
was evaporated under a stream of nitrogen. The residue was partitioned between ethyl acetate and water. The organic phase was separated / dried (hydrophobic frit) and the solvent removed by evaporation. The product was then purified by preparative HPLC to give the title compound (16mg). LC/MS; Rt 3.17min, MH+ 468.
The following compounds were prepared in a similar manner;
O

Example 14 - Λ^-d .i-dioxido^.S-dihvdro-i ^-benzisothiazol-θ-vD-δ-fluoro-Λ^-i /-/- indazoM-yl-ΛΛpropyl^Λ-pyrimidinediamine

1 -Acetyl-Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-propyl-1 /-/-indazol-4-amine (3.54g, 10.2mmol ) was suspended in IPA (20ml), 2,3-dihydro-1 ,2-benzisothiazol-6-amine 1 ,1-dioxide (2.06g, 11.2mmol) and hydrochloric acid (4ml, 2M) were added and the mixture heated under reflux for 21 h. Water (35ml) was added and the solution cooled, an oily precipitate formed. The liquor was decanted from the oily residue, the oily solid re-dissolved in IPA (~20ml) and water (~35ml) added; product precipitated as an oily residue. The decanted filtrates and the oily solid were recombined in methanol and the organic solvent removed in vacuo. The resultant solid was filtered, washed sequentially with water and dichloromethane to give the hydrochloride salt of the title compound as light brown solid (4.Og).
The hydrochloride salt (3.45g) was dissolved in methanol and saturated aqueous sodium bicarbonate added until the pH=8.4. The solid was isolated by filtration, washed sequentially with methanol and water. A portion of this material was divided into appropriate portions and purified by mass directed autoprep to give, after recombination of the purified material and evaporation of the solvents, the title compound as a pale yellow solid, 1.04g. LC/MS; Rt 3.00min, MH+ 453.9. NMR; [D6- DMSO] δH 13.28,(NH, bs), 9.74,(1 H, s), 8.29,(1 H, s), 7.99,(1 H, d), 7.92,(1 H, s), 7.83,(1 H, d), 7.70,(NH, bs), 7.50,(1 H, d), 7.40-7.35,(2H, m), 7.06,(1 H, d), 4.32,(2H1 s), 4.03,(2H, t), 1.65.(2H1 m), 0.89.(3H1 1).
Intermediate 24 - 1-acetyl-Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-propyl-1H-indazol-4- amine

Λ/-{3-[(2-chloro-5-fluoro-4-pyrimidinyl)(propyl)amino]-2-methylphenyl}acetamide (4.72g) was dissolved in chloroform (30ml), potassium acetate (1.44g), acetic acid (0.9ml) and acetic anhydride (1.34ml) were added followed by the addition of tert- butylnitrite (3.43ml) over 20min. The mixture was stirred at ambient temperature for 30mins then at 55-6O0C for 22h. The reaction mixture was cooled and aqueous potassium carbonate (30ml, 1 M) was added portionwise. The aqueous phase was removed and extracted with chloroform. The combined organic phases were washed with water and brine before drying over sodium sulphate and the solvent removed in vacuo to give a brown oil which was triturated with diethyl ether to give the desired product as a pale yellow solid (2.9g). The ethereal filtrate was evaporated and the residue purified by column chromatography on silica eluting with 10% ethyl acetate / hexane. The purified fractions were combined with the material previously isolated, the solvent evaporated and the residue triturated with methanol to give a pale yellow solid which was dried at 450C in vacuo to give the title compound (3.57g, 73%). NMR; [CDCI3] δH 8.45,(1 H, d), 7.94,(1 H1 s), 7.86,(1 H, d), 7.58,(1 H, t), 7.20,(1 H, d), 4.01 ,(2H, t), 2.80,(3H, s), 1.73-1.67,(2H, m), 0.94,(3H1 1).
Intermediate 25 - Λ/-(3-f(2-chloro-5-fluoro-4-pyrimidinyl)(propyl)amino1-2- methylphenvDacetamide

Λ/-{3-[(2-chloro-5-fluoro-4-pyrimidinyl)amino]-2-methylphenyl}acetamide (4.81 g) and potassium carbonate (2.95g) were added to DMF (24ml) and heated to 5O0C. n- Propyliodide (3.33g) was added and the suspension heated at 55°C for 4.25h. Water
(250ml) was added at 55°C to give an oily precipitation. The mixture was cooled and the liquor decanted from the oily precipitate. The oily precipitate was dissolved in diethyl ether and extracted with brine. The organic phase was dried over sodium sulphate and the solvent removed in vacuo to give a yellow solid which was washed with water, dried at 450C in vacuo to give the title compound, 4.74g (86%). NMR; [D6-DMSO] δH 9.40,(NH, bs), 8.11 ,(1 H, d), 7.47,(1 H, d), 7.22,(1 H, t), 7.12,(1 H, d), 3.99,(1 H, m), 3.41 ,(1 H, m), 2.06,(3H, s), 2.02,(3H, s), 1.60,(2H, m), 0.87,(3H, t).
Example 15 - Λ^-d .i-dioxido^.S-dihvdro-i ^-benzisothiazol-e-vn-δ-fluoro-Λ^-I H- indazol-4-yl-Λ/4-f3-(methyloxy)propyll-2.4-Pyrimidinediamine trifluoroacetate

Λ/-(2-Chloro-5-fluoro-4-pyrimidinyl)-Λ/-[3-(methyloxy)propyl]-1H-indazol-4-amine (33.5mg) was taken up in water (1.5ml), acetone (1 ml) and hydrochloric acid (20μl, 2N) the mixture was placed in a microwave vial and treated with 2,3-dihydro-1 ,2- benzisothiazol-6-amine 1 ,1-dioxide (27.6mg). The vial was sealed and the reaction irradiated in a biotage microwave at 16O0C for 2x 30min. The reaction mixture was concentrated and the residue purified using SCX SPE (500mg) by loading the residue in methanol and eluting with methanol then ammonia in methanol. The ammonia fraction was concentrated and the residue dissolved in DMSO / methanol (1:1) and purified using Mass Directed HPLC. The product fractions were concentrated and purified again using SCX SPE (500mg) loading in methanol and eluting with methanol then ammonia in methanol. The ammonia fraction was concentrated and the residue dissolved in DMSO / methanol (1 :1 ) and re-purified using Mass Directed HPLC. The fractions containing product were evaporated to dryness to give the title compound (9.2mg): LC/MS; Rt 2.84min, MH+ 484.
Intermediate 26 - Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-r3-(methyloxy)propyll-1H- indazol-4-amine

Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-[3-(methyloxy)propyl]-1-({[2- (trimethylsilyl)ethyl]oxy}methyl)-1/-/-indazol-4-amine (1.87 g) was treated with a mixture of hydrochloric acid (5N) and IPA (2:1 , 45ml). The mixture was stirred at 55°C for 5.5h, allowed to cool to room temperature and the solvents evaporated in vacuo. The residue was basified with saturated sodium carbonate solution (50ml) and extracted with ethyl acetate (3x 50ml). The combined organic extracts were dried (MgSO4), filtered and the solvent evaporated in vacuo. The residue was purified by chromatography on a silica cartridge (100g) eluting with a methanol / DCM gradient (0-50%) over 60min. Fractions containing the product were combined and reduced to dryness in vacuo. The residue was dissolved in IPA, treated with hydrochloric acid (5N, 9ml) and concentrated in vacuo. The residue was left to stand at room temperature for 3 days, basified with saturated sodium carbonate (25ml) and extracted with ethyl acetate (2x 25ml). The combined organic phases were dried (MgSO4) and concentrated in vacuo. The residue was purified by chromatography on a silica cartridge (100g) eluting with an ethyl acetate / cyclohexane gradient (0- 100%) over 60min to afford the title compound (220mg). LC/MS; Rt 2.95min, MH+ 336, 338. Additional impure product fractions were further purified by chromatography on silica, eluting with an ethyl acetate / cyclohexane gradient (0 - 50%) to afford another batch of the title compound (140mg). LC/MS; Rt 2.94min, MH+ 336. The remaining impure product fractions were purified by mass-directed autoprep to afford the title compound (129mg). LC/MS; Rt 3.04min, MH+ 335.99.
Intermediate 27 - Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-r3-(methyloxy)propyll-1-({[2- (trimethylsilyl)ethyl1oxy}methyl)-1 H-indazol-4-amine

Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-1-({[2-(trimethylsilyl)ethyl]oxy}methyl)-1/-/-indazol- 4-amine (2g) was dissolved in DMF (20ml) and the mixture cooled to 0cC in an ice- water bath, under nitrogen. Sodium hydride (60% in mineral oil, 244mg) was added and stirring continued for 15min. 1 -Bromo-3-(methyloxy)propane (0.93g) was added and the reaction mixture allowed to warm to room temperature and stirred under a nitrogen atmosphere for 3 days. Saturated ammonium chloride solution (5ml) was added to the reaction. The mixture was transferred to a larger flask using a minimum of chloroform and concentrated in vacuo. The residue was partitioned between ethyl acetate (25ml) and water (15ml). The organic phase was washed with water (3x 15ml) and concentrated in vacuo. The residue was purified by chromatography on a silica cartridge (100g), eluting with an ethyl acetate / cyclohexane gradient (0-50%) over 60min to afford, after evaporation of the solvents, the title compound (1.87g). LC/MS; Rt 3.91 min, MH+ 466.
Example 16 - Λ^-d .i-dioxido^.S-dihydro-i ^-benzisothiazol-e-vπ-δ-fluoro-Λ^-I H- indazol-4-yl-Λ/4-[3-(methyloxy)propyn-2.4-pyrimidinediamine

Λ/-(2-Chloro-5-fluoro-4-pyrimidinyl)-Λ/-[3-(methyloxy)propyl]-1/-/-indazol-4-amine (167.5mg) was taken up in water (3ml), acetone (2ml) and hydrochloric acid (40μl, 2N), the mixture was treated with 2,3-dihydro-1 ,2-benzisothiazol-6-amine 1 ,1 -dioxide (138mg, Manchester Organics). The mixture was placed into a microwave vial, the vial sealed and the reaction irradiated in a Biotage microwave at 1600C for 1 h. The reaction was treated with 2,3-dihydro-1 ,2-benzisothiazol-6-amine 1 ,1-dioxide (92mg,
Manchester Organics) and irradiated in a Biotage microwave at 16O0C for a further 2x 30min. The reaction was concentrated and applied to an SCX SPE (10g), eluting with methanol (50ml) and ammonia in methanol (50ml). The ammonia fraction was concentrated and absorbed onto florisil (3g). The solid was applied to a silica cartridge (2Og) and the cartridge eluted with an ethyl acetate / cyclohexane gradient (0-100%) over 30min. The product fractions were evaporated to dryness to give the title compound. (125mg). LC/MS; Rt 2.88min, MH+ 484.
Example 17 - Λ^-d .i-dioxido^.S-dihvdro-i ^-benzisothiazol-β-vD-δ-fluoro-Λ^-IH-

A^-(1 ,1 -dioxido-2,3-dihydro-1 ^-benzisothiazol-θ-yO-δ-fluoro-Λ^-i H-indazol^-yl-Λ/4-^- (methyloxy)propyl]-2,4-pyrimidinediamine (50mg) was taken up in a minimal amount of methanol and treated with 1 M HCI in ethanol (520μl) the mixture was allowed to stir at room temperature for IOmins and evaporated to dryness to give title compound (53mg). LC/MS; Rt 2.92min, MH+ 484. NMR; [D6-DMSO] δH 10.04,(1 H, bs), 8.22,(1 H, d), 8.06,(1 H, d), 7.99,(1 H, d), 7.83,(1 H, dd), 7.53,(1 H, d), 7.43-7.36,(2H, m), 7.10,(1 H, d), 4.34,(2H, s), 4.11 ,(2H, m), 3.34,(2H1 1), 3.08,(3H, s), 1.88,(2H, m).
Example 18 - Λ/^d .i-dioxido^.S-dihvdro-i ^-benzisothiazol-e-vπ-δ-fluoro-Λ^-I H- indazol-4-yl-Λ/V3-(methyloxy)propyll-2Λ-pyrimidinediamine

To a mixture of 1-acetyl-Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-[3-(methyloxy)propyl]- 1H-indazol-4-amine (6.23 g) in IPA (33ml) was added 2,3-dihydro-1 ,2-benzisothiazol- 6-amine 1 ,1-dioxide (3.41g) and hydrochloric acid (2M, 6.6ml). The resulting mixture was heated at reflux for ~25h. The IPA was evaporated in vacuo and water (30ml) and ethyl acetate (50ml) added to the residue. Saturated aqueous solution of sodium
hydrogen carbonate was added and the resulting mixture was further diluted with water and ethyl acetate / DCM (1 :1 ) so that 250ml of aqueous layer and 250ml of organic layer were obtained. The phases were separated and the aqueous phase was extracted with DCM (4x 100ml). The combined organics, which contained some of brown solid, were concentrated in vacuo to give a yellow-brown solid (8.175g). 0.985g of this residue was adsorbed onto florisil and purified by chromatography on a silica cartridge, eluting with a (methanol + 1% triethylamine) / DCM gradient (0-30%), to give, after evaporation of the solvents in vacuo the desired product as a yellow- orange solid (0.365g). LC/MS; Rt 2.90min, MH+ 484 (76% purity by HPLC).
A further 5.87g of the residue was dissolved in methanol and applied to an SCX SPE cartridge. The cartridge was washed with methanol and the product was eluted from the column with 2M ammonia in methanol. The appropriate fractions were combined and the solvent was evaporated in vacuo to leave a yellow-orange residue. The residue was adsorbed onto florisil and purified by chromatography on a silica cartridge, eluting with a (methanol + 1% triethylamine) / DCM gradient (0-30%), to give, after evaporation of the solvents in vacuo, the title compound as a orange solid (2.87g). LC/MS; Rt 2.86min, MH+ 484 (77% purity by HPLC).
The combined solids (3.235g) were applied to a silica column (100g), and the column eluted with a mixture of methanol, DCM and triethylamine (5:94:1) and then methanol. Appropriate fractions were combined and evaporated in vacuo. Portions of approximately 130mg, 250mg and 5 x 500mg were each dissolved in DMF (5ml) and trifluoroacetic acid (0.5ml) and injected in turn onto the HPLC column of 7micron Kromasil C8 (25 x 5cm), with a gradient of 0 to 40%B (where A is water +0.25% TFA and B is acetonitrile +0.25% TFA), at a flow of 80ml/min. The peaks between retention times 38.5 - 41.5min of mass 484 (MH+) were bulked from all the chromatographic separations. The bulked fractions were divided into 3 portions and diluted with water (2 volumes). Each portion was applied to an Amberchrom CG 161 column (25cm x 2cm) to adsorb the compound. The columns were washed in turn with ammonium hydroxide (0.2M, 300ml), water (500ml), and then the compound eluted with acetone (250ml). The acetone elution samples were bulked and dried to afford the title compound (1.8g). LC/MS; Rt 2.82min, MH+ 484. NMR; [D6-DMSO] δH 13.29,(br. s, 1 H), 9.76,(s, 1 H), 8.26,(s, 1 H), 8.00,(d, 1 H), 7.93,(s, 1 H), 7.85,(dd, 1 H), 7.76,(m, 1 H), 7.51 , (d, 1 H), 7.38,(m, 2H), 7.07,(d, 1 H), 4.32,(m, 2H), 4.11 ,(m, 2H), 3.37,(t, 2H), 3.09,(s, 3H), 1.90,(m, 2H)
Intermediate 28 - 1-acetyl-Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-r3-(methyloxy)propyll- 1 H-indazol-4-amine

To a solution of Λ/-(3-{(2-chloro-5-fluoro-4-pyrimidinyl)[3-(methyloxy)propyl]amino}-2- methylphenyl)acetamide (6.045g) in chloroform (50ml) were added potassium acetate (1.7Og), acetic acid (1.09g, 1.04ml) and acetic anhydride (1.7Og, 1.57ml). tert-Butylnitrite (90% w/w, 4.48ml) was added slowly to the reaction mixture. The resulting yellow solution was heated at 600C under nitrogen overnight. The reaction mixture was allowed to cool to room temperature, aqueous potassium carbonate (1 M, 100ml) was added to the mixture, followed by chloroform (70ml). The phases were separated and the aqueous phase extracted with chloroform (2x 30ml). The combined organic phases were dried (hydrophobic frit) and concentrated in vacuo to give the title compound (6.23 g). LC/MS; Rt 3.26min, MH+ 378.
Intermediate 29 - Λ/-(3-((2-chloro-5-fluoro-4-pyrimidinyl)f3-(methyloxy)propyllamino)- 2-methylphenyl)acetamide

To a stirred solution of Λ/-{3-[(2-chloro-5-fluoro-4-pyrimidinyl)amino]-2- methylphenyl}acetamide (5.Og), in dry DMF (50ml) under nitrogen, was added potassium carbonate (2.82g) and 1-bromo-3-(methyloxy)propane (2.86g). The resulting mixture was heated at 500C under nitrogen for 30min and then stirred at room temperature under nitrogen overnight. 1-Bromo-3-(methyloxy)propane (520mg) was added and the resulting mixture was stirred at room temperature under nitrogen
for 1 h, then heated at 400C under nitrogen for 1h. Further 1-bromo-3- (methyloxy)propane (520mg) was added and the resulting mixture was heated at 6O0C under nitrogen for 90min. The reaction mixture was allowed to cool to room temperature and diluted with water (50ml). The solvents were evaporated in vacuo and the residue partitioned between water (50ml) and ethyl acetate (50ml). The aqueous phase was extracted with ethyl acetate, the combined organic phases were dried (hydrophobic frit) and concentrated in vacuo to give the title compound as a yellow solid (6.045g). LC/MS; Rt 2.78min, MH+ 367.
Example 19 - /V2-H J-dioxido-2.3-dihvdro-1 ,2-benzisothiazol-6-yl)-5-fluoro-Λ/4-1/-/- indazol-4-yl-/V*-propyl-2.4-pyrimidinediannine

2,3-Dihydro-1 ,2-benzisothiazol-6-amine 1 ,1 -dioxide (13.11g) was added to a mixture of 1 -acetyl-Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-propyl-1 /-/-indazol-4-amine (22.46g) in IPA (125ml,) stirring at room temperature under nitrogen. The resulting suspension was treated with hydrochloric acid (2M, 25.4ml) and the mixture heated to reflux and maintained at that temperature for ~24h. A further portion of 2,3-dihydro-1 ,2- benzisothiazol-6-amine 1 ,1-dioxide (1.19g) was then added and stirring at reflux was maintained for a further ~20h. The reaction mixture was then cooled to room temperature and a precipitate formed. The mixture was diluted with IPA then the solid was collected by filtration. The solid was washed with IPA and dried in vacuo at 45°C (27.42g).
1g of this solid was dissolved in methanol (15ml) with warming to 400C. The resulting solution was then treated with sodium hydroxide (2M, 1.02ml) followed by water (0.2ml) and the resulting precipitate collected by filtration and washed with water. The filtrate also contained a precipitate that was collected by filtration and washed with water. The solids were combined and dried in vacuo at 400C for 2h.
The remaining portion of solid (26.42g) was suspended in methanol (400ml). The stirring suspension was heated to 400C, water (26ml) added and the resulting solution treated with sodium hydroxide (2M, 27ml) followed by addition of the seed
crystals from the small scale conversion. The suspension was diluted with water (80ml) and cooled to room temperature. The resulting solid was collected by filtration, washed with water and dried in vacuo at 400C overnight.
The solid was divided into 1.5g portions, each portion dissolved in DMF (10ml) and purified by preparative HPLC on a 2" x 25cm column packed with Kromasil C8 10μm RP and eluting with a gradient of 10 -100% of (70:30 acetonitrile / 0.1 M ammonium dihydrogenphosphate, adjusted to pH 2.6 with phosphoric acid) in (0.1 M ammonium dihydrogenphosphate, adjusted to pH 2.6 with phosphoric acid) over 21.5mins (flow rate 100ml/min). Material eluting in a broad peak at ~15mins was collected from each run, the fractions from the separate runs combined and the acetonitrile evaporated in vacuo. The residual aqueous was basified to pH 8.5 with 0.880 ammonia and the precipitated solid isolated by filtration. The residue was washed with water, then resuspended in water and freeze-dried to yield the title compound (16.9g). LC/MS; MH+ 454, Rt 3.00min.
Intermediate 30 - 1-acetyl-Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-propyl-1/-/-indazol-4- amine

To Λ/-{3-[(2-chloro-5-fluoro-4-pyrimidinyl)(propyl)amino]-2-methylphenyl}acetamide (29.42g) dissolved in anhydrous chloroform (210ml) was added potassium acetate (9.02g) followed by glacial acetic acid (5.61 ml) and acetic anhydride (8.35ml). The resulting mixture was stirred under a nitrogen atmosphere at room temperature and t- butyl nitrite (21.45ml) added drop-wise over 10min. Once the addition was complete the mixture was heated to 600C and maintained at 6O0C overnight. The mixture was allowed to cool to room temperature, potassium carbonate (1 M, 250ml) added and the phases separated. The aqueous phase was extracted with chloroform, the combined organics were washed with water then brine and dried (magnesium sulphate). The solvent was evaporated in vacuo to leave a brown oil, which was
triturated with diethyl ether to yield the title compound as a peach coloured solid (22.88g). LC/MS; MH+ 348, 350, Rt 3.45min.
Intermediate 31 - /V-(3-r(2-chloro-5-fluoro-4-pyrimidinyl)(propyDaminol-2- methylphenyllacetamide

To Λ/-{3-[(2-chloro-5-fluoro-4-pyrimidinyl)amino]-2-methylphenyl}acetamide (15g) in DMF (75ml) was added potassium carbonate (8.44g). The resulting mixture was then heated to 5O0C and treated with iodopropane (5.46ml) dropwise over 10min. The resulting mixture was then stirred overnight at 500C. A further portion of iodopropane (0.248ml) was added and stirring continued for 4h. The mixture was cooled to room temperature and poured into water (90ml) stirring at 5O0C. The mixture was diluted with a further portion of water (60ml) and the resulting suspension was stirred at 5O0C. The solid was collected by filtration, washed with water and dried in vacuo at 400C to yield the title compound as a cream coloured solid (15.63g). LC/MS; MH+ 337,339, Rt 3.00min.
Example 20 - /v^-d .i-dioxido^.S-dihydro-i ^-benzisothiazol-e-vD-S-fluoro-Λ^-i/-/- indazol-4-yl-Λ/4-r3-(methyloxy)propyll-2,4-pyrimidinediamine

To 1 -acetyl-Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-[3-(methyloxy)propyl]-1 H-indazol-4- amine (25.14g) in IPA (140ml) stirring at room temperature under nitrogen was
added 2,3-dihydro-1,2-benzisothiazol-6-amine 1 ,1 -dioxide (13.51g). The resulting mixture was treated with hydrochloric acid (2M, 26ml). The mixture was heated to reflux and maintained at reflux overnight. The suspension was cooled to room temperature and the solid was collected by filtration, and washed with IPA. The solid was then dried in vacuo at 45°C, before suspending in methanol (460ml). To the stirring mixture was added water (153ml), the resulting mixture was heated to 65°C and treated with sodium hydroxide (2M, 30ml). The solution was allowed to cool to room temperature, the resulting precipitate collected by filtration, and washed with methanol / water (1 :1), then water. The solid was dried in vacuo at 400C. The solid was divided into 1.5g portions, each portion dissolved in DMF (10ml) and purified by preparative HPLC on a 2" x 25cm column packed with Kromasil C8 10μm RP and eluting with a gradient of 10 -100% of (70:30 acetonitrile / 0.1 M ammonium dihydrogenphosphate, adjusted to pH 2.6 with phosphoric acid) in (0.1 M ammonium dihydrogenphosphate, adjusted to pH 2.6 with phosphoric acid) over 21.5mins (flow rate 100ml/min). Material eluting in a broad peak at ~13mins was collected from each run, the fractions from the separate runs combined and the acetonitrile evaporated in vacuo. The residual aqueous was basified to pH 8.5 with 0.880 ammonia and the precipitated solid isolated by filtration. The residue was washed with water, then resuspended in water and freeze-dried to yield the title compound (17g). LC/MS; MH+ 484, Rt 2.85min.
Example 21 - Λ/2-M J-dioxido-2.3-dihvdro-1 ,2-benzisothiazol-6-yl)-5-fluoro-Λ/4-1 H- indazol-4-yl-/v4-^3-(methyloxy)propyl1-2Λ-pyrimidinediamine

Λf*-(1 , i-dioxido^.S-dihydro-i ^-benzisothiazol-θ-yO-S-fluoro-Λ^-IH-indazol^-yl-Λ^-tS-
(methyloxy)propyl]-2,4-pyrimidinediamine (0.5g) was dissolved in DMSO (0.5g), filtered, and methanol (10g) was added to the filtrate. The solution was seeded with
/^-(i .i-dioxido^.S-dihydro-i ^-benzisothiazol-e-yO-δ-fluoro-Λ^-IH-indazol^-yl-Λ/4-^- (methyloxy)propyl]-2,4-pyrimidinediamine (1mg) and allowed to crystallise overnight
at 50C. The crystallised solid was filtered off, washed with methanol (4x 0.5ml), water (2x 0.5ml) and dried for 16h under vacuum at 9O0C, to give the title compound (0.43g) as a tan solid. LC/MS; MH+ 484.
Intermediate 32 - 1-acetyl-Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-r3-(methyloxy)propyll- 1 H-indazol-4-amine

To Λ/-(3-{(2-chloro-5-fluoro-4-pyrimidinyl)[3-(methyloxy)propyl]amino}-2- methylphenyl)acetamide (34.11g) dissolved in anhydrous chloroform (250ml) was added potassium acetate (9.6Og), followed by glacial acetic acid (5.97ml) and acetic anhydride (8.89ml). The resulting mixture was stirred under a nitrogen atmosphere at room temperature and t-butyl nitrite (22.83ml) added drop-wise over 10min. The mixture was heated to 60°C and maintained at 600C overnight. The mixture was allowed to cool to room temperature, potassium carbonate (1 M, 250ml) added and the phases separated. The aqueous phase was extracted with chloroform, the combined organics washed with water then brine and dried (magnesium sulphate).
The solvent was evaporated, the resulting brown oil was dissolved in ether (50ml) and cyclohexane was added (150ml) to the solution. The resulting solid precipitate was collected by filtration and was washed with cyclohexane / ether (3:1). The solid was dried in vacuo at 35°C to yield the title compound (26.4g). LC/MS; MH+ 378,
380, Rt 3.22min.
Intermediate 33 - Λ/-(3-((2-chloro-5-fluoro-4-pyrimidinyl)r3-(methyloxy)propyl1amino}- 2-methylphenyl)acetamide

To /V-{3-[(2-chloro-5-fluoro-4-pyrimidinyl)amino]-2-methylphenyl}acetamide (3Og) in DMF (150ml) was added potassium carbonate (16.8g). The resulting mixture was then heated to 500C under nitrogen and 1 -bromo-3-(methyloxy)propane (17.1g) added. The reaction was stirred overnight at 50°C under nitrogen, poured onto ice/water (450ml) and extracted with methyl-tert-butyl ether. The aqueous was re- extracted with methyl-terf-butyl ether, the combined organics were washed sequentially with lithium chloride solution (10%) and brine then dried (magnesium sulphate). The solvent was evaporated to yield the title compound as a yellow foam (34.11g) LC/MS; MH+ 367, 369, Rt 2.75min.
Intermediate 34 - 1-bromo-3-(methyloxy)propane
Triphenyl phosphine (138.7g) was added to a solution of 3-methoxy-1-propanol (47.6g) in anhydrous DCM (500ml). The resulting mixture was stirred under nitrogen and cooled to 00C, N-bromosuccinimide (94.13g) was added portion-wise keeping the internal temperature under 2O0C during the addition. The mixture was stirred overnight at room temperature, then, quenched by addition of sodium metabisulfite solution (5%). The phases were separated, the organic phase washed sequentially with sodium hydroxide (0.5M) then brine and dried (magnesium sulphate). The resulting green residue was triturated with hexane and the resulting solid collected by filtration and washed with hexane. The filtrate contained solid so it was re-filtered to remove the solid. The filtrate was concentrated to leave a pale brown oil that was distilled under reduced pressure (~112mbar). The second boiling fraction (bp=68°C) was collected to give the title compound as a colourless oil (33.66g). NMR; [CDCI3] δH, 3.52-3.49,(4H, m), 3.35,(3H1 s), 2.11-2.08,(2H, m).
Example 22 - 3-r(2-r(1.1-dioxido-2.3-dihvdro-1.2-benzisothiazol-6-yl)aminol-5-fluoro- 4-pyrimidinylH1 H-indazol-4-yl)aminol-1 -propanol

A mixture of 1-acetyl-Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-(3-{[(1 ,1- dimethylethyl)(dimethyl)silyl]oxy}propyl)-1H-indazol-4-amine (1.35g), hydrochloric acid (2N, 1.14ml), 2,3-dihydro-1 ,2-benzisothiazol-6-amine 1 ,1-dioxide (1.04g) in IPA (15ml) was heated in a sealed vial by microwave irradiation for 90min at 1200C. The reaction mixture was poured into aqueous sodium bicarbonate solution (100ml), extracted with ethyl acetate (2x 100ml). The combined organic extracts were dried (hydrophobic frit) and concentrated in vacuo. The residue was dissolved in ethyl acetate and purified by chromatography on a silica cartridge (5Og) eluting with an ethyl acetate / ether gradient (50-100%) and then with 10% methanol in ethyl acetate. Solvent was evaporated from the desired fractions in vacuo to give the title compound as a pink/white solid (0.38g). LC/MS; Rt 2.72min, MH+ 469.9.
The solid was further purified by warming in ethyl acetate, the mixture allowed to cool and the solid isolated by filtration. The resulting yellow solid was dried at 450C for 4h to give 3-[{2-[(1 ,1-dioxido-2,3-dihydro-1 ,2-benzisothiazol-6-yl)amino]-5-fluoro-4- pyrimidinyl}(1H-indazol-4-yl)amino]-1 -propanol (299mg). LC/MS; MH+ 469.9.
Intermediate 35 - 1-acetyl-Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-(3-(r(1.1- dimethylethyl)(dimethv0silyl1oxy)propyl)-1H-indazol-4-amine

To a mixture of Λ/-{3-[(2-chloro-5-fluoro-4-pyrimidinyl)(3-{[(1 ,1- dimethylethyl)(dimethyl)silyl]oxy}propyl)amino]-2-methylphenyl}acetamide (7.705g), in chloroform (50ml) was added potassium acetate (1.7Og), acetic acid (1.09g, 1.04ml) acetic anhydride (1.7Og, 1.57ml) and tert-butylnitrite (3.88g, 90% w/w). The resulting yellow suspension was heated at 600C under nitrogen overnight. The reaction mixture was allowed to cool to room temperature. Aqueous potassium carbonate solution (1 M, 100ml) was added to the mixture followed by chloroform (50ml). The phases were separated and the aqueous phase extracted with chloroform (2x 50ml). The combined organic phases were dried (hydrophobic frit) and concentrated in vacuo to give the title compound as a brown oil (7.93g). LC/MS; Rt 4.19min, MH+ 478.
Intermediate 36 - Λ/-(3-r(2-chloro-5-fluoro-4-pyrimidinvn(3-(r(1.1- dimethylethyl)(dimethyl)silylloxy>propyl)aminol-2-methylphenyl>acetamide

To a stirred solution of of Λ/-{3-[(2-chloro-5-fluoro-4-pyrimidinyl)amino]-2- methylphenyl}acetamide (5.Og), in DMF (50ml), was added potassium carbonate (2.82g) and [(3-bromopropyl)oxy](1,1-dimethylethyl)dimethylsilane (4.74g, Aldrich). The resulting mixture was heated at 500C under nitrogen for 90min and then at 600C under nitrogen overnight. The reaction mixture was allowed to cool to room temperature and concentrated under vacuum. The resulting residue was partitioned between ethyl acetate (50ml) and water (50ml). The aqueous phase was extracted with ethyl acetate (2x 25ml). The combined organic phases were dried (hydrophobic
frit) and concentrated in vacuo. The resulting residue was dried in vacuo, to give title compound (7.705g). LC/MS; Rt 3.75min, MH+ 467.
Example 23 - Λ^-M J-dioxido^.S-dihvdro-i ^-benzisothiazol-e-vO-ΛΛethyl-S-fluoro- Λ^-IH-indazoM-yl^Λ-pyrimidinediamine 4-methylbenzenesulfonate

A suspension of /N^-(1 , 1-dioxido-2, 3-dihydro-1 , 2-benzisothiazol-6-yl)-Λ/4-ethyl-5- fluoro-Λ^-I H-indazoM-yl^^-pyrimidinediarnine (as prepared in Example 8, 1.13g) in water / acetone (1 :9, 20ml) was warmed to reflux and the suspension was treated with p-toluenesulfonic acid (0.73g) to give a solution. This was cooled to 40°C at which point the mixture spontaneously crystallised. The slurry was heated to reflux, aged for 1 h and cooled to ambient temperature over 2h. The mixture was cooled further to 0-50C, filtered and the solid washed with water / acetone (1:9, 2x 4ml), and dried in vacuo to give the title compound as a white solid (1.106g).
NMR; [D6-DMSO] δH 1.23,(3H, t, J=7.1 Hz), 2.29,(3H, s), 4.11 ,(2H, q, J=7.1 Hz),
4.35,(2H, s), 7.09,(1 H, d, J=7.3Hz), 7.12,(2H, d, J=8.0Hz), 7.39,(1 H, dd, J=8.2Hz
J=7.5Hz), 7.45,(1 H, d, J=8.5Hz), 7.49,(2H, d, J=8.0Hz), 7.55,(1 H, d, J=8.4Hz), 7.72,(1 H, dd, J=8.4Hz J=1.9Hz), 8.01 ,(1 H, s), 8.07,(1 H, d, J=6.2Hz), 8.39,(1 H, d,
J=1.4Hz), 10.04,(1 H, s).
Example 24 - 2-naphthalenesulfonic acid - /v^-d .i-dioxido^.S-dihydro-i ^- benzisothiazol-S-vD-Zv^-ethyl-S-fluoro-A^-IH-indazol^-yl^^-pyrimidinediamine

A suspension of /^-(i .i-dioxido^.S-dihydro-i^-benzisothiazol-θ-yO-Λ^-ethyl-S- fluoro-Λ/MH-indazol-4-yl-2,4-pyrimidinediamine (as prepared in Example 8, 1.0g) in water / IPA (1 :5, 12ml) was warmed to reflux and the suspension was treated with an aqueous solution of 2-naphthalenenesulfonic acid monohydrate (0.58g) in water (3ml) to give a solution and the residual acid rinsed in with water (2x 2ml). The solution was cooled to ambient temperature, stirred for 2h, the precipitated solid filtered off, washed with water (6x 1 ml), and dried in vacuo at 700C to give the title compound as a pale yellow solid (1.23g).
NMR; [D6-DMSO] δH 1.22,(3H, t, J=7.1 Hz), 4.10,(2H, q, J=7.1 Hz), 4.36,(2H, s), 7.11 ,(1 H, d, J=7.2Hz), 7.40,(1 H, dd, J=8.3Hz J=7.4Hz), 7.47,(1 H, d, J=8.5Hz), 7.50- 7.55,(2H, m), 7.58,(1 H, d, J=8.5Hz), 7.70,(1 H, dd, J=8.6Hz J=2.0Hz), 7.73,(1 H, dd, J=8.5Hz J=1.7Hz), 7.87,(1 H, d, J=8.4Hz), 7.90,(1 H, m), 7.97,(1 H, m), 8.04,(1 H, d J=LOHz), 8.11 ,(1 H1Cl1 J=6.4Hz), 8.17,(1 H, s), 8.36,(1 H, d, J=1.7Hz), 10.16,(1 H, s).
Example 25 - /V^1 J-dioxido-2.3-dihvdro-1.2-benzisothiazol-6-yl)-5-fluoro-ΛΛi /-/- indazol-4-yl-Λ/4-r3-(methyloxy)propyll-2,4-pyrimidinediamine

The solid was divided into 2 portions. The first portion (284.13g) was suspended in methanol (4260ml) and water (1420ml), the mixture was heated to 65°C and treated with sodium hydroxide (2M, 290ml). The resulting solution was seeded with a pure sample of /^-(i.i-dioxido^.S-dihydro-i ^-benzisothiazol-θ-yO-δ-fluoro-ΛΛi/-/- indazol-4-yl-Λ/4-[3-(methyloxy)propyl]-2,4-pyrimidinediamine and cooled to 400C and maintained at 400C for 1 h. The precipitate was collected by filtration, washed with methanol / water (1 :1 ) and the solid dried in vacuo at 400C to give the title compound (201.59g). NMR; [D6-DMSO] δH 13.28,(1 H, bs), 9.75,(1 H, s), 8.26,(1 H, s), 8.00,(1 H, d), 7.93,(1 H, s), 7.85,(1 H, d), 7.74,(1 H, bs), 7.51 ,(1 H, d), 7.39-7.35,(2H, m), 7.07,(1 H, d), 4.32,(2H, s), 4.11 ,(2H, t), 3.39-3.34(2H, obscured by water), 3.09,(3H, s), 1.90,(2H, m).
The second portion (274.71 g) was suspended in methanol (4120ml) and water (1370ml), the mixture was heated to 65°C and treated with sodium hydroxide (2M, 280ml). The resulting solution was seeded with a pure sample of Λ/2-(1 ,1-dioxido-2,3- dihydro-1 ^-benzisothiazol-θ-yO-δ-fluoro-ΛΛi /-/-indazol-4-yl-Λ/4-[3-(methyloxy)propyl]- 2,4-pyrimidinediamine and cooled to 40°C and maintained at 400C for 1h. The precipitate was collected by filtration, washed with methanol / water (1 :1 ) and the solid dried in vacuo at 40°C to give the title compound (189.58g). NMR; [D6-DMSO] δH 13.28,(1 H1 bs), 9.75,(1 H, s), 8.27,(1 H, s), 8.00,(1 H, d), 7.93,(1 H, s), 7.86,(1 H, d), 7.74,(1 H, bs), 7.51 , (1 H, d), 7.39-7.35,(2H, m), 7.07,(1 H, d), 4.33,(2H, s), 4.12,(2H, t), 3.39-3.36(2H, t), 3.10,(3H, s), 1.91 ,(2H, m).
Λ/2-(1 ,1 -dioxido-2,3-dihydro-1 ^-benzisothiazol-e-yO-δ-fluoro-Λ^-i H-indazol^-yl-Λ/4-^- (methyloxy)propyl]-2,4-pyrimidinediamine (380.28g, from combining the product from the above two portions) was further purified by suspending in DMSO (380ml) and heating to 400C. Methanol (4940ml) was added over 35min, maintaining the temperature at 40±2°C. The mixture was heated to reflux, maintained at this
temperature for 1h, then cooled to 60°C and filtered under reduced pressure. The residue was washed with methanol (3800ml) and dried in vacuo at 500C to room temperature overnight and then for a further 3 days under vacuum at room temperature to give the title compound (333.9g).
Intermediate 37 - 1-acetyl-Λ/-(2-chloro-5-fluoro-4-pyrimidinyl)-Λ/-[3-(methyloxy)propyll- 1 H-indazol-4-amine

To Λ/-(3-{(2-chloro-5-fluoro-4-pyhmidinyl)[3-(methyloxy)propyl]amino}-2- methylphenyl)acetamide (297.81 g) dissolved in anhydrous chloroform (2100ml) was added potassium acetate (83.8g), followed by glacial acetic acid (52ml) and acetic anhydride (77.6ml). The mixture was stirred under a nitrogen atmosphere at room temperature and t-butyl nitrite (199ml) added drop-wise over 10min. The mixture was heated to 600C and maintained at 60°C overnight. The reaction was cooled to room temperature and aqueous potassium carbonate (1M, 2100ml) added portionwise. The phases were separated, the aqueous extracted with chloroform, and the combined organic phases washed with water, brine and dried (magnesium sulphate). The solvent was evaporated in vacuo, the residual oil dissolved in ether (200ml) and cyclohexane (700ml) added to the solution in 100ml portions. Scratching of the flask initialled precipitation. The precipitate was isolated by filtration, washed with ether / cyclohexane (1 :4) and dried in vacuo at 400C to give the title compound (246.28g). NMR; [D6-DMSO] δH 8.51 ,(1 H, s), 8.31 , (1 H, d), 8.18,(1 H, d), 7.68,(1 H, t), 7.46,(1 H1 d), 4.08,(2H, t), 3.35-3.32,(2H, obscured by water), 3.14,(3H, s), 2.73,(3H, s), 1.82,(2H, m).
Intermediate 38 - Λ/-(3-{(2-chloro-5-fluoro-4-pyrimidinyl)f3-(methyloxy)propyllamino)- 2-methylphenyl)acetamide

To Λ/-{3-[(2-chloro-5-fluoro-4-pyrimidinyl)amino]-2-methylphenyl}acetamide (278.65g) in DMF (1393ml) was added potassium carbonate (157g). The mixture was heated to 500C and 1-bromo-3-(methyloxy)propane (159g) added. The reaction was stirred overnight at 5O0C, poured into water (4179ml) and extracted with methyl-tert-butyl ether. The aqueous was re-extracted with methyl-fe/t-butyl ether, the combined organics washed sequentially with aqueous lithium chloride solution (10%, 500ml) and brine (500ml) and dried (magnesium sulphate). The solvent was evaporated to yield the title compound as a yellow solid (298.72g). NMR; [D6-DMSO] δH 9.41 ,(1 H, s), 8.12,(1 H, d), 7.47,(1 H, d), 7.22,(1 H1 1), 7.14,(1H, d), 4.14-4.06,(1 H, m), 3.53- 3.47,(1 H, m), 3.38-3.31 , (2H, partially obscured by water), 3.19,(3H, s), 2.06,(3H, s), 2.02,(3H, s), 1.92-1.74,(2H1 m).
Claims
1. A compound

R1 and R2 is each independently hydrogen,  or CH2C3-7cycloalkyl;
or CH2C3-7cycloalkyl;
R3 is C1-3alkyl or C2.3alkyl substituted by hydroxy or C1-3alkoxy; and
X is halogen; or a salt or solvate, preferably a pharmaceutically acceptable salt or solvate, thereof.
2. A compound as claimed in claim 1 in which R1 is hydrogen.
3. A compound as claimed in claim 1 or 2 in which R2 is hydrogen.
4. A compound as claimed in any one of claims 1 to 3 in which R3 is ethyl, propyl, -(CH2)3OH and -(CH2)3OCH3.
5. A compound as claimed in any one of claims 1 to 4 in which X is fluoro.
6. A compound as claimed in any one of claims 1 to 5 of formula (IB):

R1 is hydrogen, C1-6alkyl or CH2C3-7cycloalkyl; R3 is C1-3alkyl; and X is halogen; or a salt or solvate, preferably a pharmaceutically acceptable salt or solvate, thereof.
7. A compound as claimed in any one of claims 1 to 5 of formula (IC):  in which:
in which:
R3 is C2.3alkyl substituted by hydroxy or C1-3alkoxy; and X is halogen; or a salt or solvate, preferably a pharmaceutically acceptable salt or solvate, thereof.
8. A compound of formula (I) or a salt or solvate thereof, selected from the group consisting of:
Λ/2-(1 ,1 -dioxido-2,3-dihydro-1 ^-benzisothiazol-θ-yO-Λ^-ethyl-δ-fluoro-Λ/4-! H-indazol- 4-yl-2,4-pyrimidinediamine;
S-chloro-^i .i-dioxido^.S-dihydro-i ^-benzisothiazol-θ-yO-Λ^-ethyl-Λ^-IH-indazol-
4-yl-2,4-pyrimidinediamine;
5-bromo-Λ/2-(1 )1-dioxido-2,3-dihydro-1 >2-benzisothiazol-6-yl)-Λ/4-ethyl-Λ/4-1H-indazol-
4-yl-2,4-pyrimidinediamine; Λ/4-Ethyl-5-fluoro-Λ/4-1 H-indazol-4-yl-Λ/2-(2-methyl-1 ,1 -dioxido-2,3-dihydro-1 ,2- benzisothiazol-6-yl)-2,4-pyrimidinediamine;
/N^-(1 ,1-dioxido-2,3-dihydro-1 ^-benzisothiazol-θ-yO-δ-fluoro-ΛΛi H-indazol-4-yl-Λ/4- methyl-2,4-pyrimidinediamine;
^-^-(cyclopropylmethyO-i .i-dioxido^.S-dihydro-i^-benzisothiazol-θ-yll-Λ^-ethyl-δ- fluoro-/V*-1 H-indazol-4-yl-2,4-pyrimidinediamine;
Λ/4-ethyl-Λ/2-(2-ethyl-1 ,1-dioxido-2,3-dihydro-1 ^-benzisothiazol-β-yO-S-fluoro-Λ/4-! H- indazol-4-yl-2,4-pyrimidinediamine;
ΛΛethyl-δ-fluoro-Λ/M H-indazol-4-yl-Λ/2-[2-(2-methylpropyl)-1 , 1 -dioxido-2,3-dihydro-
1 ,2-benzisothiazol-6-yl]-2,4-pyrimidinediamine; Λ/2-[2-(cyclobutylmethyl)-1 ,1-dioxido-2,3-dihydro-1 ^-benzisothiazol-e-yll-Λ^-ethyl-δ- fluoro-ΛΛiH-indazoM-yl^^-pyrimidinediamine;
/^-^-(cyclohexylmethyO-i .i-dioxido^.S-dihydro-i ^-benzisothiazol-β-yll-Λ^-ethyl-S- fluoro-Λ^-IH-indazol^-yl^^-pyrimidinediamine;
A^-(I , i-dioxido^.S-dihydro-i ^-benzisothiazol-θ-yO-S-fluoro-Λ^-IH-indazol^-yl-Λ/4- propyl-2,4-pyrimidinediaminei
A^-(I , i-dioxido^.S-dihydro-i ^-benzisothiazol-θ-yO-S-fluoro-Λ^-IH-indazoM-yl-Λ/4-^-
(methyloxy)propyl]-2,4-pyrimidinediamine; and
3-[{2-[(1 ,1-dioxido-2,3-dihydro-1 ,2-benzisothiazol-6-yl)amino]-5-fluoro-4- pyrimidinyl}(1H-indazol-4-yl)amino]-1-propanol; or a salt or solvate thereof, in particular, a pharmaceutically acceptable salt or solvate thereof.
9. A compound of formula (I) selected from the group consisting of:
Λ/2-(1 ,1-dioxido-2,3-dihydro-1 ^-benzisothiazol-θ-yO-ΛΛethyl-δ-fluoro-ΛΛi H-indazol- 4-yl-2,4-pyrimidinediamine; or a salt or solvate thereof, in particular, a pharmaceutically acceptable salt or solvate thereof.
10. A compound of formula (I) selected from the group consisting of:
/V^-(1 , 1-dioxido-2, 3-dihydro-1 , 2-benzisothiazol-6-yl)-5-fluoro-Λ/4-1H-indazol-4-yl-Λ/4- propyl-2,4-pyrimidinediamine; or a salt or solvate thereof, in particular, a pharmaceutically acceptable salt or solvate thereof.
11. A compound of formula (I) selected from the group consisting of: Λ/^CI .I-dioxido^.S-dihydro-i ^-benzisothiazol-θ-yO-S-fluoro-Λ^-IH-indazoM-yl-Λ/4-^- (methyloxy)propyl]-2,4-pyrimidinediamine; or a salt or solvate thereof, in particular, a pharmaceutically acceptable salt or solvate thereof.
12. A process for preparing a compound of formula (I), or a salt or solvate thereof, as defined in any one of claims 1 to 11 , which process comprises reacting a compound of formula (II):

or a protected derivative thereof wherein L1 represents a suitable leaving group, with a compound of formula (III)

or a protected derivative thereof, wherein X, R1, R2, R3 and R4 are as defined for formula (I) in claim 1.
13. A pharmaceutical formulation comprising a compound of formula (I), or a salt or solvate thereof, as defined in any one of claims 1 to 11 and pharmaceutically acceptable excipients.
14. A compound of formula (I) as defined in any one of claims 1 to 11 for use in therapy.
15. The use of a compound of formula (I) or a salt or solvate thereof, as defined in any one of claims 1 to 11 , in the manufacture of a medicament for treating a disease associated with inappropriate mast cell activation
16. The use of a compound of formula (I) or a salt or solvate thereof, as defined in any one of claims 1 to 11 , in the manufacture of a medicament to inhibit a Syk kinase.
17. The use of a compound of formula (I) or a salt or solvate thereof, as defined in any one of claims 1 to 11 , in the manufacture of a medicament for of treating an inflammatory disease
18. The use of a compound of formula (I) or a salt or solvate thereof, as defined in any one of claims 1 to 11 , in the manufacture of a medicament for treating an allergic disorder.
19. The use of a compound of formula (I) or a salt or solvate thereof, as defined in any one of claims 1 to 11 , in the manufacture of a medicament for treating rhinitis.
Applications Claiming Priority (14)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0514580A GB0514580D0 (en) | 2005-07-15 | 2005-07-15 | Novel compounds |
| GB0514580.0 | 2005-07-15 | ||
| GB0524785A GB0524785D0 (en) | 2005-12-05 | 2005-12-05 | Novel compounds |
| GB0524785.3 | 2005-12-05 | ||
| GB0601728A GB0601728D0 (en) | 2006-01-27 | 2006-01-27 | Novel Compounds |
| GB0601728.9 | 2006-01-27 | ||
| GB0606763.1 | 2006-04-03 | ||
| GB0606763A GB0606763D0 (en) | 2006-04-03 | 2006-04-03 | Novel compounds |
| GB0610513.4 | 2006-05-26 | ||
| GB0610513A GB0610513D0 (en) | 2006-05-26 | 2006-05-26 | Novel compounds |
| GB0610924.3 | 2006-06-02 | ||
| GB0610924A GB0610924D0 (en) | 2006-06-02 | 2006-06-02 | Novel compounds |
| GB0611488A GB0611488D0 (en) | 2006-06-09 | 2006-06-09 | Novel compounds |
| GB0611488.8 | 2006-06-09 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2007009681A1 true WO2007009681A1 (en) | 2007-01-25 |
Family
ID=37022880
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2006/006953 WO2007009681A1 (en) | 2005-07-15 | 2006-07-13 | 1 , 1-DIOXID0-2 , 3-DIHYDRO-l , 2-BENZISOTHIAZ0L-6-YL-1H-INDAZOL-4-YL-2 , 4-PYRIMIDINEDI AMINE DERIVATIVES |
Country Status (4)
| Country | Link |
|---|---|
| AR (1) | AR054834A1 (en) |
| PE (1) | PE20070362A1 (en) |
| TW (1) | TW200740805A (en) |
| WO (1) | WO2007009681A1 (en) |
Cited By (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007085540A1 (en) * | 2006-01-27 | 2007-08-02 | Glaxo Group Limited | 1h-indaz0l-4-yl-2 , 4-pyrimidinediamine derivatives |
| US7718653B2 (en) | 2007-07-16 | 2010-05-18 | Astrazeneca Ab | Pyrimidine derivatives for inhibiting Eph receptors |
| EP2489663A1 (en) | 2011-02-16 | 2012-08-22 | Almirall, S.A. | Compounds as syk kinase inhibitors |
| WO2012123311A1 (en) * | 2011-03-11 | 2012-09-20 | Glaxo Group Limited | Pyridinyl- and pyrazinyl -methyloxy - aryl derivatives useful as inhibitors of spleen tyrosine kinase (syk) |
| CN103864764A (en) * | 2012-12-11 | 2014-06-18 | 齐鲁制药有限公司 | Indazole-substituted pyrimidinamine derivative, and preparation method and use thereof |
| US8987456B2 (en) | 2011-10-05 | 2015-03-24 | Merck Sharp & Dohme Corp. | 3-pyridyl carboxamide-containing spleen tyrosine kinase (SYK) inhibitors |
| US9006444B2 (en) | 2011-10-05 | 2015-04-14 | Merck Sharp & Dohme Corp. | Phenyl carboxamide-containing spleen tyrosine kinase (SYK) inhibitors |
| US9120785B2 (en) | 2011-05-10 | 2015-09-01 | Merck Sharp & Dohme Corp. | Pyridyl aminopyridines as Syk inhibitors |
| US9145391B2 (en) | 2011-05-10 | 2015-09-29 | Merck Sharp & Dohme Corp. | Bipyridylaminopyridines as Syk inhibitors |
| US9216173B2 (en) | 2011-10-05 | 2015-12-22 | Merck Sharp & Dohme Corp. | 2-Pyridyl carboxamide-containing spleen tyrosine kinase (SYK) inhibitors |
| US9242984B2 (en) | 2012-06-20 | 2016-01-26 | Merck Sharp & Dohme Corp. | Pyrazolyl derivatives as Syk inhibitors |
| US9290490B2 (en) | 2011-05-10 | 2016-03-22 | Merck Sharp & Dohme Corp. | Aminopyrimidines as Syk inhibitors |
| US9353066B2 (en) | 2012-08-20 | 2016-05-31 | Merck Sharp & Dohme Corp. | Substituted phenyl-Spleen Tyrosine Kinase (Syk) inhibitors |
| US9376418B2 (en) | 2012-06-22 | 2016-06-28 | Merck Sharp & Dohme Corp. | Substituted pyridine spleen tyrosine kinase (SYK) inhibitors |
| US9416111B2 (en) | 2012-06-22 | 2016-08-16 | Merck Sharp & Dohme Corp. | Substituted diazine and triazine spleen tyrosine kinease (Syk) inhibitors |
| US9487504B2 (en) | 2012-06-20 | 2016-11-08 | Merck Sharp & Dohme Corp. | Imidazolyl analogs as syk inhibitors |
| US9499534B2 (en) | 2013-04-26 | 2016-11-22 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminopyrimidines as spleen tyrosine kinase inhibitors |
| US9586931B2 (en) | 2012-09-28 | 2017-03-07 | Merck Sharp & Dohme Corp. | Triazolyl derivatives as Syk inhibitors |
| US9598405B2 (en) | 2012-12-21 | 2017-03-21 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminopyridines as spleen tyrosine kinase inhibitors |
| US9624210B2 (en) | 2012-12-12 | 2017-04-18 | Merck Sharp & Dohme Corp. | Amino-pyrimidine-containing spleen tyrosine kinase (Syk) inhibitors |
| US9670196B2 (en) | 2013-12-20 | 2017-06-06 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminoheteroaryls as Spleen Tyrosine Kinase inhibitors |
| US9745295B2 (en) | 2013-04-26 | 2017-08-29 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminoheteroaryls as spleen tyrosine kinase inhibitors |
| US9775839B2 (en) | 2014-03-13 | 2017-10-03 | Merck Sharp & Dohme Corp. | 2-pyrazine carboxamides as spleen tyrosine kinase inhibitors |
| US9783531B2 (en) | 2013-12-20 | 2017-10-10 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminoheteroaryls as spleen tyrosine kinase inhibitors |
| US9822107B2 (en) | 2013-12-20 | 2017-11-21 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminoheteroaryls as spleen tyrosine kinase inhibitors |
| WO2020188015A1 (en) | 2019-03-21 | 2020-09-24 | Onxeo | A dbait molecule in combination with kinase inhibitor for the treatment of cancer |
| WO2021089791A1 (en) | 2019-11-08 | 2021-05-14 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for the treatment of cancers that have acquired resistance to kinase inhibitors |
| WO2021148581A1 (en) | 2020-01-22 | 2021-07-29 | Onxeo | Novel dbait molecule and its use |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1054004A1 (en) * | 1997-12-15 | 2000-11-22 | Yamanouchi Pharmaceutical Co. Ltd. | Novel pyrimidine-5-carboxamide derivatives |
| WO2001060816A1 (en) * | 2000-02-17 | 2001-08-23 | Amgen Inc. | Kinase inhibitors |
| WO2003030909A1 (en) * | 2001-09-25 | 2003-04-17 | Bayer Pharmaceuticals Corporation | 2- and 4-aminopyrimidines n-substtituded by a bicyclic ring for use as kinase inhibitors in the treatment of cancer |
| WO2003063794A2 (en) * | 2002-02-01 | 2003-08-07 | Rigel Pharmaceuticals, Inc. | 2,4-pyrimidinediamine compounds and their uses |
| WO2004014382A1 (en) * | 2002-07-29 | 2004-02-19 | Rigel Pharmaceuticals | Methods of treating or preventing autoimmune diseases with 2,4-pyrimidinediamine compounds |
| WO2004080980A1 (en) * | 2003-03-14 | 2004-09-23 | Novartis Ag | 2, 4- di (phenylamino) pyrimidines useful in the treatment of neoplastic diseases, inflammatory and immune system disorders |
| WO2005016894A1 (en) * | 2003-08-15 | 2005-02-24 | Novartis Ag | 2, 4-pyrimidinediamines useful in the treatment of neoplastic diseases, inflammatory and immune system disorders |
-
2006
- 2006-07-13 WO PCT/EP2006/006953 patent/WO2007009681A1/en active Application Filing
- 2006-07-13 TW TW095125594A patent/TW200740805A/en unknown
- 2006-07-13 AR ARP060103014A patent/AR054834A1/en not_active Application Discontinuation
- 2006-07-13 PE PE2006000835A patent/PE20070362A1/en not_active Application Discontinuation
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1054004A1 (en) * | 1997-12-15 | 2000-11-22 | Yamanouchi Pharmaceutical Co. Ltd. | Novel pyrimidine-5-carboxamide derivatives |
| WO2001060816A1 (en) * | 2000-02-17 | 2001-08-23 | Amgen Inc. | Kinase inhibitors |
| WO2003030909A1 (en) * | 2001-09-25 | 2003-04-17 | Bayer Pharmaceuticals Corporation | 2- and 4-aminopyrimidines n-substtituded by a bicyclic ring for use as kinase inhibitors in the treatment of cancer |
| WO2003063794A2 (en) * | 2002-02-01 | 2003-08-07 | Rigel Pharmaceuticals, Inc. | 2,4-pyrimidinediamine compounds and their uses |
| WO2004014382A1 (en) * | 2002-07-29 | 2004-02-19 | Rigel Pharmaceuticals | Methods of treating or preventing autoimmune diseases with 2,4-pyrimidinediamine compounds |
| WO2004080980A1 (en) * | 2003-03-14 | 2004-09-23 | Novartis Ag | 2, 4- di (phenylamino) pyrimidines useful in the treatment of neoplastic diseases, inflammatory and immune system disorders |
| WO2005016894A1 (en) * | 2003-08-15 | 2005-02-24 | Novartis Ag | 2, 4-pyrimidinediamines useful in the treatment of neoplastic diseases, inflammatory and immune system disorders |
Cited By (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007085540A1 (en) * | 2006-01-27 | 2007-08-02 | Glaxo Group Limited | 1h-indaz0l-4-yl-2 , 4-pyrimidinediamine derivatives |
| US7718653B2 (en) | 2007-07-16 | 2010-05-18 | Astrazeneca Ab | Pyrimidine derivatives for inhibiting Eph receptors |
| EP2489663A1 (en) | 2011-02-16 | 2012-08-22 | Almirall, S.A. | Compounds as syk kinase inhibitors |
| CN103502239B (en) * | 2011-03-11 | 2015-07-01 | 葛兰素集团有限公司 | Pyridinyl- and pyrazinyl-methyloxy-aryl derivatives useful as inhibitors of spleen tyrosine kinase (SKY) |
| EP2937344A1 (en) * | 2011-03-11 | 2015-10-28 | Glaxo Group Limited | Pyridinyl- and pyrazinyl -methyloxy - aryl derivatives useful as inhibitors of spleen tyrosine kinase (syk) |
| JP2014507457A (en) * | 2011-03-11 | 2014-03-27 | グラクソ グループ リミテッド | Pyridinyl and pyrazinylmethyloxyaryl derivatives useful as inhibitors of spleen tyrosine kinase (Syk) |
| AU2012228439C1 (en) * | 2011-03-11 | 2016-11-03 | Glaxo Group Limited | Pyridinyl- and pyrazinyl -methyloxy - aryl derivatives useful as inhibitors of spleen tyrosine kinase (SYK) |
| WO2012123311A1 (en) * | 2011-03-11 | 2012-09-20 | Glaxo Group Limited | Pyridinyl- and pyrazinyl -methyloxy - aryl derivatives useful as inhibitors of spleen tyrosine kinase (syk) |
| US8993560B2 (en) | 2011-03-11 | 2015-03-31 | Glaxo Group Limited | Compounds |
| EA022437B1 (en) * | 2011-03-11 | 2015-12-30 | Глаксо Груп Лимитед | Pyridinyl- and pyrazinyl-methyloxy-aryl derivatives useful as inhibitors of spleen tyrosine kinase (syk) |
| US9447074B2 (en) | 2011-03-11 | 2016-09-20 | Glaxo Group Limited | Compounds |
| AU2012228439B2 (en) * | 2011-03-11 | 2015-11-26 | Glaxo Group Limited | Pyridinyl- and pyrazinyl -methyloxy - aryl derivatives useful as inhibitors of spleen tyrosine kinase (SYK) |
| CN103502239A (en) * | 2011-03-11 | 2014-01-08 | 葛兰素集团有限公司 | Pyridinyl- and pyrazinyl-methyloxy-aryl derivatives useful as inhibitors of spleen tyrosine kinase (SKY) |
| KR101564007B1 (en) | 2011-03-11 | 2015-10-28 | 글락소 그룹 리미티드 | Pyridinyl- and pyrazinyl-methyloxy-aryl derivatives useful as inhibitors of spleen tyrosine kinase(syk) |
| US9145391B2 (en) | 2011-05-10 | 2015-09-29 | Merck Sharp & Dohme Corp. | Bipyridylaminopyridines as Syk inhibitors |
| US9120785B2 (en) | 2011-05-10 | 2015-09-01 | Merck Sharp & Dohme Corp. | Pyridyl aminopyridines as Syk inhibitors |
| US9290490B2 (en) | 2011-05-10 | 2016-03-22 | Merck Sharp & Dohme Corp. | Aminopyrimidines as Syk inhibitors |
| US9216173B2 (en) | 2011-10-05 | 2015-12-22 | Merck Sharp & Dohme Corp. | 2-Pyridyl carboxamide-containing spleen tyrosine kinase (SYK) inhibitors |
| US9006444B2 (en) | 2011-10-05 | 2015-04-14 | Merck Sharp & Dohme Corp. | Phenyl carboxamide-containing spleen tyrosine kinase (SYK) inhibitors |
| US8987456B2 (en) | 2011-10-05 | 2015-03-24 | Merck Sharp & Dohme Corp. | 3-pyridyl carboxamide-containing spleen tyrosine kinase (SYK) inhibitors |
| US9242984B2 (en) | 2012-06-20 | 2016-01-26 | Merck Sharp & Dohme Corp. | Pyrazolyl derivatives as Syk inhibitors |
| US9487504B2 (en) | 2012-06-20 | 2016-11-08 | Merck Sharp & Dohme Corp. | Imidazolyl analogs as syk inhibitors |
| US9416111B2 (en) | 2012-06-22 | 2016-08-16 | Merck Sharp & Dohme Corp. | Substituted diazine and triazine spleen tyrosine kinease (Syk) inhibitors |
| US9376418B2 (en) | 2012-06-22 | 2016-06-28 | Merck Sharp & Dohme Corp. | Substituted pyridine spleen tyrosine kinase (SYK) inhibitors |
| US9353066B2 (en) | 2012-08-20 | 2016-05-31 | Merck Sharp & Dohme Corp. | Substituted phenyl-Spleen Tyrosine Kinase (Syk) inhibitors |
| US9586931B2 (en) | 2012-09-28 | 2017-03-07 | Merck Sharp & Dohme Corp. | Triazolyl derivatives as Syk inhibitors |
| CN103864764A (en) * | 2012-12-11 | 2014-06-18 | 齐鲁制药有限公司 | Indazole-substituted pyrimidinamine derivative, and preparation method and use thereof |
| US9624210B2 (en) | 2012-12-12 | 2017-04-18 | Merck Sharp & Dohme Corp. | Amino-pyrimidine-containing spleen tyrosine kinase (Syk) inhibitors |
| US9598405B2 (en) | 2012-12-21 | 2017-03-21 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminopyridines as spleen tyrosine kinase inhibitors |
| US9499534B2 (en) | 2013-04-26 | 2016-11-22 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminopyrimidines as spleen tyrosine kinase inhibitors |
| US9745295B2 (en) | 2013-04-26 | 2017-08-29 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminoheteroaryls as spleen tyrosine kinase inhibitors |
| US9670196B2 (en) | 2013-12-20 | 2017-06-06 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminoheteroaryls as Spleen Tyrosine Kinase inhibitors |
| US9783531B2 (en) | 2013-12-20 | 2017-10-10 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminoheteroaryls as spleen tyrosine kinase inhibitors |
| US9822107B2 (en) | 2013-12-20 | 2017-11-21 | Merck Sharp & Dohme Corp. | Thiazole-substituted aminoheteroaryls as spleen tyrosine kinase inhibitors |
| US9775839B2 (en) | 2014-03-13 | 2017-10-03 | Merck Sharp & Dohme Corp. | 2-pyrazine carboxamides as spleen tyrosine kinase inhibitors |
| WO2020188015A1 (en) | 2019-03-21 | 2020-09-24 | Onxeo | A dbait molecule in combination with kinase inhibitor for the treatment of cancer |
| WO2021089791A1 (en) | 2019-11-08 | 2021-05-14 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Methods for the treatment of cancers that have acquired resistance to kinase inhibitors |
| WO2021148581A1 (en) | 2020-01-22 | 2021-07-29 | Onxeo | Novel dbait molecule and its use |
Also Published As
| Publication number | Publication date |
|---|---|
| PE20070362A1 (en) | 2007-04-23 |
| TW200740805A (en) | 2007-11-01 |
| AR054834A1 (en) | 2007-07-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2007009681A1 (en) | 1 , 1-DIOXID0-2 , 3-DIHYDRO-l , 2-BENZISOTHIAZ0L-6-YL-1H-INDAZOL-4-YL-2 , 4-PYRIMIDINEDI AMINE DERIVATIVES | |
| WO2007028445A1 (en) | 6-indolyl-4-yl-amino-5-halogeno-2-pyrimidinyl-amino derivatives | |
| EP2376481B1 (en) | Pyrimidinecarboxamide derivatives as inhibitors of syk kinase | |
| WO2007085540A1 (en) | 1h-indaz0l-4-yl-2 , 4-pyrimidinediamine derivatives | |
| RU2682245C1 (en) | Indazolie wnt signal pathway inhibitors and their therapeutic applications | |
| WO2006129100A1 (en) | Novel compounds | |
| US9533988B2 (en) | Ring-fused bicyclic pyridyl derivatives as FGFR4 inhibitors | |
| EP2705032B1 (en) | Dihydroquinoline derivatives as bromodomain inhibitors | |
| JP6663857B2 (en) | Pyrazolopyridine and pyrazolopyrimidine | |
| EP3842420B1 (en) | Heteroaryl inhibitors of pde4 | |
| EP2613781B1 (en) | Indazole derivatives for use in the treatment of influenza virus infection | |
| EP2699573B1 (en) | 7-(3,5-dimethyl-4-isoxazolyl)-8-(methyloxy)-1h-imidazo[4,5-c]quinoline derivatives | |
| EP3433254B1 (en) | Naphthyridines as integrin antagonists | |
| JP2009511527A (en) | Pyrrolopyrimidine derivatives as Syk inhibitors | |
| JP5520969B2 (en) | Pyrazole derivatives for use as CCR4 receptor antagonists | |
| JP6412102B2 (en) | Cycloalkylnitrile pyrazolopyridones as Janus kinase inhibitors | |
| JP2009512669A (en) | Cinnoline compounds as inhibitors of type IV phosphodiesterase (PDE4) | |
| US11273146B2 (en) | Benzofuran derivatives and their use as bromodomain inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Country of ref document: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 06776251 Country of ref document: EP Kind code of ref document: A1 |