WO2006116412A2 - Fused heterocyclic compounds - Google Patents
Fused heterocyclic compounds Download PDFInfo
- Publication number
- WO2006116412A2 WO2006116412A2 PCT/US2006/015646 US2006015646W WO2006116412A2 WO 2006116412 A2 WO2006116412 A2 WO 2006116412A2 US 2006015646 W US2006015646 W US 2006015646W WO 2006116412 A2 WO2006116412 A2 WO 2006116412A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- optionally substituted
- compound
- mmol
- chloro
- Prior art date
Links
- 0 CC(*)[C@](C(N*)=**)N Chemical compound CC(*)[C@](C(N*)=**)N 0.000 description 4
- URVFXOCXGUHMQM-UHFFFAOYSA-N CC(C)(C)C(c(cc1)c2[n](C)c(Nc(c(OC)c3)c(C)cc3Cl)nc2c1Cl)=O Chemical compound CC(C)(C)C(c(cc1)c2[n](C)c(Nc(c(OC)c3)c(C)cc3Cl)nc2c1Cl)=O URVFXOCXGUHMQM-UHFFFAOYSA-N 0.000 description 1
- ARVHJNDOTXEBSY-UHFFFAOYSA-N CC(CN1)NC1O Chemical compound CC(CN1)NC1O ARVHJNDOTXEBSY-UHFFFAOYSA-N 0.000 description 1
- BMWVVWVDBHYAOY-UHFFFAOYSA-N CCC(CC)c(cc1)c2[n](C)c(Nc(c(Cl)cc(OC(F)(F)F)c3)c3Cl)nc2c1Cl Chemical compound CCC(CC)c(cc1)c2[n](C)c(Nc(c(Cl)cc(OC(F)(F)F)c3)c3Cl)nc2c1Cl BMWVVWVDBHYAOY-UHFFFAOYSA-N 0.000 description 1
- WMIUKSJJCFREED-UHFFFAOYSA-N CCC(CC)c(cc1)c2[n](C)c(Oc(c(Br)c3)ccc3Cl)nc2c1Cl Chemical compound CCC(CC)c(cc1)c2[n](C)c(Oc(c(Br)c3)ccc3Cl)nc2c1Cl WMIUKSJJCFREED-UHFFFAOYSA-N 0.000 description 1
- ZHTLMSDKPHVCMF-UHFFFAOYSA-N CCC(CC)c1c2[n](C)c(Nc3c(C)cc(C)cc3OC)nc2c(C(F)(F)F)cc1 Chemical compound CCC(CC)c1c2[n](C)c(Nc3c(C)cc(C)cc3OC)nc2c(C(F)(F)F)cc1 ZHTLMSDKPHVCMF-UHFFFAOYSA-N 0.000 description 1
- HLDVBPIPQBAEBP-UHFFFAOYSA-N CCC(CC)c1c2[n](C)c(Oc3c(C)cc(C)cc3C)nc2c(C)cc1 Chemical compound CCC(CC)c1c2[n](C)c(Oc3c(C)cc(C)cc3C)nc2c(C)cc1 HLDVBPIPQBAEBP-UHFFFAOYSA-N 0.000 description 1
- TYFIVKKDDRAAHJ-UHFFFAOYSA-N CCCC(CCC)c(cc1)c2[n](C)c(Nc(cc(C(F)(F)F)c(O)c3)c3Cl)nc2c1Cl Chemical compound CCCC(CCC)c(cc1)c2[n](C)c(Nc(cc(C(F)(F)F)c(O)c3)c3Cl)nc2c1Cl TYFIVKKDDRAAHJ-UHFFFAOYSA-N 0.000 description 1
- FOKRIXVMQJMZND-UHFFFAOYSA-N CCCC(CCC)c(cc1)c2[n](C)c(Nc(cc(C(F)(F)F)cc3)c3OC)nc2c1Cl Chemical compound CCCC(CCC)c(cc1)c2[n](C)c(Nc(cc(C(F)(F)F)cc3)c3OC)nc2c1Cl FOKRIXVMQJMZND-UHFFFAOYSA-N 0.000 description 1
- MSWYOUMZUWYJFJ-UHFFFAOYSA-N CCCC(CCC)c(cc1)c2[n](C)c(Oc(c(Cl)c3)ccc3Br)nc2c1Cl Chemical compound CCCC(CCC)c(cc1)c2[n](C)c(Oc(c(Cl)c3)ccc3Br)nc2c1Cl MSWYOUMZUWYJFJ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/24—Benzimidazoles; Hydrogenated benzimidazoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached in position 2
- C07D235/26—Oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/24—Benzimidazoles; Hydrogenated benzimidazoles with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached in position 2
- C07D235/30—Nitrogen atoms not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D335/00—Heterocyclic compounds containing six-membered rings having one sulfur atom as the only ring hetero atom
- C07D335/02—Heterocyclic compounds containing six-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/04—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/10—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/06—Peri-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D495/04—Ortho-condensed systems
Definitions
- the present invention relates to novel nitrogen- containing fused heterocyclic compounds having CRF (corticotropin releasing factor) antagonistic activity and pharmaceutical compositions containing them.
- CRF corticotropin releasing factor
- Corticotropin-releasing factor (hereinafter, abbreviated as "CRF”) is a neuropeptide composed of 41 amino acids, and was isolated and purified as a peptide promoting release of adrenocorticotropic hormone (ACTH) from pituitary gland.
- ACTH adrenocorticotropic hormone
- the structure thereof was determined from sheep hypothalamus and, thereafter, the presence thereof was confirmed also in a rat or a human, and the structure thereof was determined [Science, 213, 1394(1981); Proc. Natl. Acad. Sci USA, 80, 4851(1983); EMBO J. 5, 775(1983)].
- An amino acid sequence is the same in a human and a rat, but differed in 7 amino acids in ovine.
- CRF is synthesized as a carboxy-terminal of prepro CRF, cut and secreted.
- the CRF peptide and a mRNA thereof are present at the largest amount in hypothalamus and pituitary gland, and are widely distributed in a brain such as cerebral cortex, cerebellum, hippocampus and corpus amygdaloideum.
- a brain such as cerebral cortex, cerebellum, hippocampus and corpus amygdaloideum.
- peripheral tissues the existence has been confirmed in placenta, adrenal gland, lung, liver, pancreas, skin and digestive tract [J. Clin. Endocrinol. Metab., 65, 176(1987); J. Clin. Endocrinol. Metab., 67, 768(1988); Regul .
- a CRF receptor is a 7- transmembrane G protein-coupled receptor, and two subtypes of CRFl and CRF2 are present. It is reported that CRFl is present mainly in cerebral cortex, cerebellum, olfactory bulb, pituitary gland and tonsil nucleus. On the other hand, the CRF2 receptor has two subtypes of CRF2 ⁇ and CRF2 ⁇ .
- the CRF2 ⁇ receptor is distributed much in hypothalamus, septal area and choroids plexus, and the CRF2 ⁇ receptor is present mainly in peripheral tissues such as skeletal muscle and is distributed in a blood vessel in a brain [J. Neurosci. 15, 6340(1995); Endocrinology, 137, 72(1996); Biochim. Biophys . Acta, 1352, 129(1997)]. Since each receptor differs in distribution in a living body, it is suggested that a role thereof is also different [Trends. Pharmacol. Sci. 23, 71(2002)].
- CRF As a physiological action of CRF, the action on the endocrine system is known in which CRF is produced and secreted in response to stress in hypothalamus and acts on pituitary gland to promote the release of ACTH [Recent Prog. Horm. Res., 39, 245(1983)].
- CRF acts as a neurotransmitter or a neuroregulating factor in a brain, and integrates electrophysiology, autonomic nerve and conducts to stress [Brain Res. Rev., 15, 71(1990); Pharmacol. Rev., 43, 425(1991)].
- ⁇ - helical CRF (9-41) of a peptidergic CRF receptor antagonist exerts an anti-anxiety action in an animal model [Brain Res., 509, 80(1990); J. Neurosci., 14, 2579(1994)].
- a blood pressure, a heart rate and a body temperature of a rat are increased by stress or CRF administration, but the ⁇ -helical CRF (9-41) of a peptidergic CRF antagonist inhibits the increase in a blood pressure, a heart rate and a body temperature due to stress [J. Physiol., 460, 221(1993)].
- the ⁇ -helical CRF (9-41) of a peptidergic CRF receptor antagonist inhibits abnormal conducts due to withdrawal of a dependent drug such as an alcohol and a cocaine [Psychopharmacology, 103, 227(1991); Pharmacol. Rev.53, 209(2001)].
- a dependent drug such as an alcohol and a cocaine
- learning and memory are promoted by CRF administration in a rat [Nature, 375, 284(1995); Neuroendocrinology, 57, 1071(1993); Eur. J. Pharmacol., 405, 225(2000)].
- CRF is associated with stress response in a living body
- the CRF concentration in a cerebrospinal fluid of a depression patient is higher as compared with that of a normal person [Am. J. Psychiatry, 144, 873(1987)]
- the mRNA level of CRF in hypothalamus of a depression patient is increased as compared with that of a normal person [Am. J. Psychiatry, 152, 1372(1995)].
- a CRF binding site of cerebral cortex of a patient who suicided by depression is decreased [Arch. Gen. Psychiatry, 45, 577(1988)].
- the increase in the plasma ACTH concentration due to CRF administration is small in a depression patient [N. Engl.
- CRF In a CRF- overexpressing mouse, excessive secretions of ACTH and adrenal cortex steroid occur, and abnormalities analogous to Gushing' s syndrome such as atrophy of muscle, alopecia, infertility and the like are observed [Endorcrinology, 130, 3378(1992)].
- CRF inhibits ingestion in an experimental animal such as a rat [Life Sci., 31, 363 (1982); Neurophamacology, 22, 337(1983)].
- ⁇ -helical CRF (9-41) of a peptidergic CRF antagonist inhibited decrease of ingestion due to stress loading in an experimental model [Brain Res. Bull., 17, 285(1986)].
- CRF inhibited weight gain in a hereditary obesity animal [Physiol. Behav.
- CRF is centrally or peripherally associated with the digestive tract movement involved in stress or inflammation [Am. J. Physiol. Gastrointest . Liver Physiol. 280,
- CRF acts centrally or peripherally, weakens the shrinkablity of stomach, and decreases the gastric excreting ability [Regulatory Peptides, 21, 173(1988); Am. J. Physiol., 253, G241(1987)].
- ⁇ -helical CRF (9-41) of a peptidergic CRF antagonist has a restoring action for hypofunction of stomach by abdominal operation [Am. J. Physiol., 258, G152(1990)].
- CRF inhibits secretion of a bicarbonate ion in stomach, decreases gastric acid secretion and inhibits ulcer due to cold restriction stress [Am. J. Physiol., 258, G152(1990)].
- ⁇ -helical CRF (9-41) of a peptidergic CRF antagonist shows the inhibitory action on gastric acid secretion decrease, gastric excretion decrease, small intestinal transport decrease and large intestinal transport enhancement due to restriction stress [Gastroenterology, 95, 1510(1988)].
- CRF decreases a threshold of discomfort [Gastroenterology, 109, 1772(1995); Neurogastroenterol . Mot., 8, 9 [1996].
- a irritable bowel syndrome patient large intestinal , movement is excessively enhanced by CRF administration as compared with a healthy person [Gut, 42, 845(1998)].
- CRF In an inflammatory site of an experimental animal and in a joint fluid of a rheumatoid arthritis patient, production of CRF is topically increased [Science, 254, 421(1991); J. Clin. Invest., 90, 2555(1992); J. Immunol., 151, 1587(1993)].
- CRF induces degranulation of a mast cell and enhances the blood vessel permeability [Endocrinology, 139, 403(1998); J.Pharmacol. Exp. Ther., 288, 1349(1999)].
- CRF can be detected also in a thyroid gland of autoimmune thyroiditis patient [Am. J. Pathol. 145, 1159(1994)].
- CRF receptor antagonistic compound would exert an excellent effect for treating or preventing various diseases in which CRF is involved.
- a CRF antagonist for example, peptide CRF receptor antagonists are reported in which a part of an amino acid sequence of CRF or associated peptides of a human or other mammals is altered or deleted, and they are reported to show a pharmacological action such as ACTH release- inhibiting action and anti-anxiety action [Science, 224, 889(1984); J. Pharmacol. Exp. Ther., 269, 564(1994); Brain Res. Rev., 15, 71(1990)].
- peptide derivatives have a low utility value as a medicine.
- R 1 is an optionally substituted hydrocarbyl, an_ optionally substituted C-linked heterocyclic group, an optionally substituted N-linked heteroaryl group, or an acyl, provided that methyl, and trifluoromethyl are excluded;
- R 2 is an optionally substituted cyclic hydrocarbyl or an optionally substituted heterocyclic group, provided that 2- [2- (1, l-dimethylethyl)phenyloxy] -3-pyridinyl is excluded;
- X is oxygen, sulfur or -NR 3 - (wherein R 3 is a hydrogen, an optionally substituted hydrocarbyl or an acyl) ;
- Y 1 , Y 2 and Y 3 are each an optionally substituted carbon or a nitrogen, provided that one or less of Y 1 , Y 2 and Y 3 is nitrogen; and Z is a bond, -CO-, oxygen, sulfur, -SO-, -SO 2 -, -NR 4 -, -NR 4 - alk-, -CONR 4 - or -NR 4 CO- (wherein alk is an optionally substituted C 1 - 4 alkylene and R 4 is a hydrogen, an optionally substituted hydrocarbyl or an acyl) ; provided that (i) the compound wherein X is -NH-, and R 2 is an optionally substituted thiophene ring, (ii) the compound wherein R 1 is cyano, Y 3 is carbon which is substituted with methyl substituted with three substituents, one of which is acyl, and other two of which may form a ring,
- a process for producing the compound according to the above-mentioned (1) which comprises reacting a compound represented by the formula: wherein L represents a leaving group selected from halogen atom, sulfonyloxy group and acyloxy group, and other symbols are as defined in the above-mentioned (1), with a compound represented by the formula:
- a pharmaceutical composition which comprises the compound according to the above-mentioned (1) ;
- a CRF receptor antagonist which is the compound represented by the formula (I 1 ) :
- R 1 is an optionally substituted hydrocarbyl, an optionally substituted C-linked heterocyclic group, an optionally substituted N-linked heteroaryl group, a cyano or an acyl;
- R 2 is an optionally substituted cyclic hydrocarbyl or an optionally substituted heterocyclic group
- X is oxygen, sulfur or -NR - (wherein R is a hydrogen, an optionally substituted hydrocarbyl or an acyl)
- Y 1 , Y 2 and Y 3 are each an optionally substituted carbon or a nitrogen, provided that one or less of Y 1 , Y 2 and Y 3 is nitrogen;
- Z is a bond, -CO-, oxygen, sulfur, -SO-, -SO 2 -, -NR 4 -, -NR 4 - alk-, -CONR 4 - or -NR 4 CO- (wherein alk is an optionally substituted C ⁇ - 4 alkylene and R 4 is a hydrogen, an optionally substituted hydrocarbyl or an acyl) ; or a salt thereof;
- a method for treating or preventing a disease wherein a CRF receptor is implicated which comprises administering to a subject in need thereof an effective amount of the CRF receptor antagonist according to the above-mentioned (19); (21) The method according to the above-mentioned (20) wherein the disease being treated or prevented is selected from affective disorder, depression or anxiety;
- composition according to the above- mentioned (24) wherein the disease being treated or prevented is selected from affective disorder, depression or anxiety; and the like.
- hydrocarbyl means a univalent group containing only carbon and hydrogen.
- X represents an oxygen, a sulfur or -NR 3 - (wherein R 3 is a hydrogen, an optionally substituted hydrocarbyl or an acyl) . That is, examples of the 5-membered ring in the formula (I) and (I 1 ) include an oxazole ring, a thiazole ring and an imidazole ring.
- Examples of the "hydrocarbyl" of the “optionally substituted hydrocarbyl” represented by R 3 of the formula: -NR 3 - include an optionally substituted aliphatic hydrocarbon group, an optionally substituted alicyclic hydrocarbon group, an optionally substituted alicyclic- aliphatic hydrocarbon group, an optionally substituted alicyclic-alicyclic hydrocarbon group, an optionally substituted aromatic hydrocarbon group, an optionally substituted aromatic-aliphatic hydrocarbon group (an aralkyl group) , and the like.
- aliphatic hydrocarbon group examples include a saturated aliphatic hydrocarbon group having 1-8 carbon atoms (e.g., alkyl group) such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, sec-butyl, tert-butyl, pentyl, isopentyl, neopentyl, tert-pentyl, hexyl, isohexyl, heptyl, octyl, etc.; and an unsaturated aliphatic hydrocarbon group having 2-8 carbon atoms (e.g., alkenyl group, alkynyl group, alkadienyl group, alkadiynyl group, etc.) such as vinyl, allyl, 1-propenyl,- 2-methyl-l-propenyl, 1-butenyl, 2- butenyl, 3-butenyl, 3-methyl-2-butenyl, 1-penten
- alicyclic hydrocarbon group examples include a saturated alicyclic hydrocarbon group having 3-7 carbon atoms (e.g., cycloalkyl group, etc.) such as cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, and the like; an unsaturated alicyclic hydrocarbon group having 3-7 carbon atoms (e.g., cycloalkenyl group, cycloalkadienyl group, etc.) such as 1-cyclopentenyl, 2-cyclopentenyl, 3- cyclopentenyl, 1-cyclohexenyl, 2-cyclohexenyl, 3- cyclohexenyl, 1-cycloheptenyl, 2-cycloheptenyl, 3- cycloheptenyl, 2, 4-cycloheptadienyl, etc.; a partly saturated and fused bicyclic hydrocarbon group [preferably, Cg-io partly saturated and fused
- Said alicyclic hydrocarbon group may be cross-linked.
- alicyclic-aliphatic hydrocarbon group examples include those where the above-mentioned alicyclic hydrocarbon group and the above-mentioned aliphatic hydrocarbon group are combined, for example, those having 4-14 carbon atoms such as cyclopropylmethyl, cyclopropylethyl, cyclobutylmethyl, cyclobutylethyl, cyclopentylmethyl, 2-cyclopentenylmethyl, 3- cyclopentenylmethyl, cyclopentylethyl, cyclohexylmethyl, 2- cyclohexenylmethyl, 3-cyclohexenylmethyl, cyclohexylethyl, cycloheptylmethyl, cycloheptylethyl, 2- (3, 4-dihydro-2- naphtyl) ethyl, 2- (1, 2, 3, 4-tetrahydro-2-naphtyl) ethyl, 2- (3, 4-dihydro-2-na
- C 3 _ 7 cycloalkyl- Ci_ 4 alkyl group e.g., C 3 _ 7 cycloalkyl- Ci_ 4 alkyl group, C 3 - 7 cycloalkenyl-Ci- 4 alkyl group, C3- 7 cycloalkyl-C2-4 alkenyl group, C3_ 7 cycloalkenyl-C2-4 alkenyl group, C 9 - 10 partly saturated and fused bicyclic hydrocarbon-Ci- 4 alkyl group, C 9 -. 3.0 partly saturated and fused bicyclic hydrocarbon-C2-4 alkenyl groups, etc.).
- alicyclic-alicyclic hydrocarbon group examples include a C 1 - 4 alkyl group substituted with two C 3 - 7 cycloalkyls selected from the above-mentioned alicyclic hydrocarbon group, for example, those represented by the formula:
- aromatic hydrocarbon group examples include an aryl group having 6-10 carbon atoms (including that where a 5- to ⁇ -membered non-aromatic hydrocarbon ring is fused with phenyl group) such as phenyl, ⁇ -naphthyl, ⁇ -naphthyl, 4-indenyl, 5-indenyl, 4-indanyl, 5-indanyl, 5,6,7,8- tetrahydro-1-naphthyl, 5, 6, 7, 8-tetrahydro-2-naphthyl, 5,6- dihydro-1-naphthyl, 5, 6-dihydro-2-naphthyl, 5, 6-dihydro-3- naphthyl, 5, 6-dihydro-4-naphthyl, etc.; and the like.
- aryl group having 6-10 carbon atoms including that where a 5- to ⁇ -membered non-aromatic hydrocarbon ring is fused with
- aromatic-aliphatic hydrocarbon group examples include an aralkyl group having 7-14 carbon atoms (C ⁇ -io aryl-Ci-4 alkyl group) such as phenyl-Ci_ 4 alkyl group, e.g., benzyl, phenethyl, 1-phenylethyl, 1-phenylpropyl, 2- phenylpropyl, 3-phenylpropyl, etc.; naphthyl-Ci_4 alkyl group such as ⁇ -naphthylmethyl, ⁇ -naphthylethyl, ⁇ - naphthylmethyl, ⁇ -naphthylethyl, etc.; C 6 -I 0 aryl-C2-4 alkenyl group such as phenyl-C 2 - 4 alkenyl group, e.g., styryl, cinnamyl, etc.; and the like.
- hydrocarbyl group may have a substituent at a substitutable position.
- substituents include a halogen, nitro, cyano, oxo, (1) an optionally substituted heterocyclic group, (2) an optionally substituted sulfinyl group, (3) an optionally substituted sulfonyl group, (4) optionally substituted hydroxyl group, (5) optionally substituted thiol group, (6) an optionally substituted amino group, (7) an acyl group,
- Examples of the substituent of above-mentioned (2) an optionally substituted sulfinyl group, (3) an optionally substituted sulfonyl group, (4) optionally substituted hydroxyl group, (5) optionally substituted thiol group and (6) an optionally substituted amino group include an optionally substituted hydrocarbyl.
- hydrocarbyl of such optionally substituted hydrocarbyl include those exemplified above. Such hydrocarbyl may be substituted by one or more substituents at a substitutable position. Examples of the substituent group of the optionally substituted hydrocarbyl as a substituent group include halogen, nitro, cyano, hydroxyl, thiol, amino and carboxyl .
- Ci_ 6 alkylsulfinyl e.g., methylsulfinyl, ethylsulfinyl, propylsulfinyl, butylsulfinyl etc.
- C ⁇ -io arylsulfinyl e.g., phenylsulfinyl, naphthylsulfinyl etc.
- Ci_ 6 alkylsulfonyl e.g., methylsulfonyl, ethylsulfonyl, propylsulfonyl, butylsulfonyl etc.
- C ⁇ -io arylsulfonyl e.g., phenylsulfonyl, naphthylsulfonyl etc.
- hydroxyl, Ci_ 6 alkoxy e.g., methoxy, ethoxy, n-propoxy, isopropoxy, n-butoxy, isobutoxy, sec-butoxy, t-butoxy, n-pentyloxy, isopentyloxy, neopentyloxy, etc.
- C ⁇ -io aryloxy e.g., phenoxy, naphthoxy, etc.
- thiol As the optionally substituted thiol group of above- mentioned (5), specifically, thiol, C 1 - S alkylthio (e.g., methylthio, ethylthio, propylthio, etc.) and C ⁇ -IO arylthio (e.g., phenylthio, naphthylthio etc.) are exemplified.
- C 1 - S alkylthio e.g., methylthio, ethylthio, propylthio, etc.
- C ⁇ IO arylthio e.g., phenylthio, naphthylthio etc.
- amino, mono-Ci_ 6 alkylamino e.g., methylamino, ethylamino, propylamino, isopropylamino, butylamino etc.
- di-Ci- 6 alkylamino e.g., dimethylamino, diethylamino, ethylmethylamino, dipropylamino, diisopropylamino, dibutylamino etc.
- Examples of the acyl group of above-mentioned (7) include the same group as the acyl for R 3 .
- ester group or amide group of the optionally esterified or amidated carboxyl group of above- mentioned (8) examples include ester group with the same optionally substituted hydrocarbyl as the substituent of optionally substituted hydroxyl group of above-mentioned (4) or amide group with optionally substituted amino group of above- mentioned (6) .
- carboxyl specifically, carboxyl, Ci_ 6 alkoxy-carbonyl (e.g., methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl, tert- butoxycarbonyl etc.), C ⁇ -io aryloxy-carbonyl (e.g., phenoxycarbonyl etc.), C 7 - I6 aralkyloxy-carbonyl (e.g., benzyloxycarbonyl, • phenetyloxycarbonyl etc.), and the like are exemplified.
- carboxyl Ci_ 6 alkoxy-carbonyl (e.g., methoxycarbonyl, ethoxycarbonyl, propoxycarbonyl, tert- butoxycarbonyl etc.)
- C ⁇ -io aryloxy-carbonyl e.g., phenoxycarbonyl etc.
- C 7 - I6 aralkyloxy-carbonyl e.g., benz
- carbamoyl mono-Ci_ 6 alkyl-carbamoyl (e.g., methylcarbamoyl, ethylcarbamoyl etc.), di-C ⁇ - 6 alkyl- carbamoyl (e.g., dimethylcarbamoyl, diethylcarbamoyl, ethylmethylcarbamoyl etc.), Ce-io aryl-carbamoyl (e.g., phenylcarbamoyl, 1-naphthylcarbamoyl, 2-naphthylcarbamoyl etc.), 5- to 6-membered heterocyclic carbamoyl (e.g., 2- pyridylcarbamoyl, 3-pyridylcarbamoyl, 4-pyridylcarbamoyl, 2-thienylcarbamoyl,
- acyl represented by R 3 of the formula: -NR 3 - include a formyl and a group where the carbonyl group is combined with a Ci_io alkyl group, a C 2 - 10 alkenyl group, a C 2 -io alkynyl group, a C 3 -.7 cycloalkyl group, a C 5 - 7 cycloalkenyl group or an aromatic group (e.g., phenyl group, pyridyl group, etc.) (e.g., acetyl, propionyl, butyryl, isobytyryl, valeryl, isovaleryl, pivaloyl, hexanoyl, heptanoyl, octanoyl, cyclobutanecarbonyl, cyclopentanecarbonyl, cyclohexanecarbonyl, cycloheptanecarbonyl, crotonyl, 2-
- R 3 is preferably hydrogen, Ci_io alkyl, C 2 -io alkenyl, C2-10 alkynyl, and more preferably hydrogen, Ci- 10 alkyl.
- R 3 methyl, ethyl, hydroxyethyl and the like are preferred.
- R 1 in the formula (I) and (I 1 ) is an optionally substituted hydrocarbyl, an optionally substituted C-linked heterocyclic group, an optionally substituted N-linked heteroaryl group, a cyano or an acyl.
- C- linked of said “optionally substituted C-linked heterocyclic group” means that R 1 is linked via a carbon atom of the heterocyclic group of R 1 to the fused bicyclic ring represented by the formula (I) .
- N- linked of the "optionally substituted N-linked heteroaryl group” means that R 1 is linked via a nitrogen atom of the heteroaryl group of R 1 to the fused bicyclic ring represented by the formula (I) .
- Examples of the "optionally substituted hydrocarbyl” for R 1 include the same groups as those exemplified with respect to the optionally substituted hydrocarbyl of R 3 .
- Examples of the "optionally substituted heterocyclic group" in the “optionally substituted C-linked heterocyclic group” for R 1 include the same groups as those exemplified below with respect to the optionally substituted heterocyclic group of R 2 .
- heteroaryl group in the "optionally substituted N-linked heteroaryl group” for R 1 examples include a 5- to 10-membered aromatic heterocyclic group optionally containing 1 to 3 hetero atoms selected from a nitrogen atom, a sulfur atom and an oxygen atom in addition to one nitrogen atom (e.g., pyrrolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, imidazolyl, pyrazolyl, 1,2,3- oxadiazolyl, 1, 2, 4-oxadiazolyl, 1, 3, 4-oxadiazolyl, furazanyl, 1, 2, 3-thiadiazolyl, 1, 2, 4-thiadiazolyl, 1,3,4- thiadiazolyl, 1, 2, 3-triazolyl, 1, 2, 4-triazolyl, tetrazolyl, pyridyl, pyrimidinyl, pyridazinyl, pyrazinyl, triaziny
- Said heteroaryl group may be substituted with 1 to 3 substituents selected from the group consisting of halogen, Ci- 6 alkyl, C 2 -6 alkenyl, Ci- 6 alkoxy-Ci- 6 alkyl, C 5 _ 7 cycloalkyl, C ⁇ -io aryl (said aryl may have 1 or 2 substituents selected from halogen, Ci- 6 alkyl, halogeno Ci- 6 alkyl and Ci_ 6 alkoxy) , C 7 - I4 aralkyl (said aralkyl may have 1 or 2 substituents selected from halogen, Ci-e alkyl, halogeno Ci_ 6 alkyl and Ci_ 6 alkoxy) , hydroxy, hydroxy-Ci_ 6 alkyl, C ⁇ -io aryloxy (said aryloxy may have 1 or 2 substituents selected from halogen, Ci- 6 alkyl, halogeno Ci_6 alkyl and Ci_ 6 al
- acyl examples include the same groups as those exemplified with respect to the acyl of R 3 .
- R 1 in the formula (I) and (I 1 ) is preferably an optionally substituted acyclic branched C 3 _ 14 hydrocarbyl (preferably acyclic branched C 3 _ 7 hydrocarbyl such as 2-propyl, 3-hexyl, 3-pentyl, 4-heptyl, etc. ) , an optionally substituted C ⁇ -io aryl, an optionally substituted C-linked 5- to 14-membered heterocyclic group or N-linked 5- to 10-membered heteroaryl group.
- R 2 in the formula (I) and (I 1 ) is an optionally substituted cyclic hydrocarbyl or an optionally substituted heterocyclic group.
- Examples of the "cyclic hydrocarbyl" of the "optionally substituted cyclic hydrocarbyl” for R 2 include a C 3 - 7 cycloalkyl group (e.g., cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, etc) , a C 3 - 7 cycloalkenyl group (e.g., 1-cyclopentenyl, 2-cyclopentenyl, 3-cyclopentenyl, 1-cyclohexenyl, 2-cyclohexenyl, 3- cyclohexenyl, 1-cycloheptenyl, 2-cycloheptenyl, 3- cycloheptenyl, etc) , an aryl group having 6-10 carbon atoms
- phenyl such as phenyl, ⁇ -naphthyl, ⁇ -naphthyl, 4-indenyl, 5-indenyl, 4-indanyl, 5- indanyl, 5, 6, 7 , 8-tetrahydro-l-naphthyl, 5, 6, 7, 8-tetrahydro- 2-naphthyl, 5, 6-dihydro-l-naphthyl, 5, 6-dihydro-2-naphthyl, 5, 6-dihydro-3-naphthyl, 5, 6-dihydro-4-naphthyl, etc.; and the like.
- each of the heterocyclic groups exemplified in (i) to (iv) may be a saturated or unsaturated heterocyclic group and the unsaturated heterocyclic group may be either aromatic or non-aromatic.
- heterocyclic group for an optionally substituted heterocyclic group of R 2 examples include an aromatic monocyclic heterocyclic group and a non-aromatic heterocyclic group.
- heterocyclic group for an optionally substituted heterocyclic group include (i) an aromatic monocyclic heterocyclic group (e.g., furyl, thienyl, pyrrolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, imidazolyl, pyrazolyl, 1, 2, 3-oxadiazolyl, 1, 2, 4-oxadiazolyl, 1, 3, 4-oxadiazolyl, furazanyl, 1,2,3- thiadiazolyl, 1, 2, 4-thiadiazolyl, 1, 3, 4-thiadiazolyl, 1, 2, 3-triazolyl, 1, 2, 4-triazolyl, tetrazolyl, pyridyl, pyrimidinyl, pyridazinyl, pyrazinyl, triazinyl, etc.) and (ii) a non-aromatic, heterocyclic group (e.g., oxiranyl, azetidiny
- cycloalkyl may have the same substituent as those exemplified with respect to the optionally substituted hydrocarbyl group of R 3 and further may have the same group as optionally substituted hydrocarbyl group of R 3 as their substituent.
- two of the substituents of the "cyclic hydrocarbyl” in the “optionally substituted cyclic hydrocarbyl” or “heterocyclic group” in the “optionally substituted heterocyclic group” for R 2 may be combined each other to form a fused ring with the cyclic hydrocarbyl or heterocyclic group.
- fused ring examples include, for example, an aromatic fused heterocyclic group such as 8- to 12-membered aromatic fused heterocyclic group (preferably, heterocyclic group consisting of the above-mentioned 5- or 6-membered aromatic monocyclic heterocyclic group fused with a benzene ring or heterocyclic group consisting of the above-mentioned 5- or 6-membered aromatic monocyclic heterocyclic group fused with the same or different above- mentioned 5- or ⁇ -membered aromatic monocyclic heterocyclic group), etc. (e.g.
- substituent of the "cyclic hydrocarbyl” in the “optionally substituted cyclic hydrocarbyl” or “heterocyclic group” in the “optionally substituted heterocyclic group” for R 2 may be combined together with the substituent: R 4 of -NR 4 -, -NR 4 -alk-, - CONR 4 - or -NR 4 CO- in Z of formula (I) or (I 1 ) to form a nitrogen-containing fused ring with the cyclic hydrocarbyl or heterocyclic group of R 2 .
- fused ring examples include, for example, 8- to 12-membered bicyclic heterocyclic group formed by a fusion of a benzene ring with a saturated monocyclic heterocyclic group containing one nitrogen atom such as 1, 2, 3, 4-tetrahydroquinolyl, 2, 3, 4, 5-tetrahydro-lH-l-benzazepinyl, and the like.
- fused ring and “nitrogen- containing fused ring” may further have one to three substituents selected from an acyl (e.g., acetyl, propionyl, etc.), an amide (e.g., dimethylaminocarbonyl, methylaminocarbonyl, etc.), an amine (e.g., dimethylamino, methylamino, amino, etc.), a halogen (e.g., fluorine, chlorine, bromine, etc.), a lower alkyl (e.g., methyl, ethyl, trifluoromethyl, etc.) and a lower alkoxy (e.g., methoxy, ethoxy, trifluoromethoxy, etc.), each of which may be substituted.
- an acyl e.g., acetyl, propionyl, etc.
- an amide e.g., dimethylaminocarbonyl, methylaminocarbonyl,
- R 2 is preferably an optionally- substituted Ce-io aryl (more preferably phenyl) or an optionally substituted 5- to 8-membered (more preferably 5- to 6-membered) heterocyclic group (more preferably pyridyl) .
- R 2 is more preferably phenyl which is 2, 4-disubstituted, 2, 4, 6-trisubstituted or 2, 4, 5-trisubstituted with two or three substituents, or pyridyl which is disubstituted or trisubstituted with two or three substituents.
- the substituents for the phenyl and pyridyl may be the same or different and examples thereof include an acyl (e.g., acetyl, propionyl, etc.), an ' amide (e.g., dimethylaminocarbonyl, methylaminocarbonyl, etc.), an amine (e.g., dimethylamino, methylamino, amino, etc.), a halogen (e.g., fluorine, chlorine, bromine, etc.), a lower alkyl (e.g., methyl, ethyl, trifluoromethyl, etc.) and a lower alkoxy (e.g., methoxy, ethoxy, trifluoromethoxy, etc.), each of which may be substituted.
- an acyl e.g., acetyl, propionyl, etc.
- an ' amide e.g., dimethylaminocarbonyl, methylamino
- Y 1 is CR 3a or a nitrogen
- Y 2 is CR 3b or a nitrogen
- Y 3 is CR 3c or a nitrogen
- R 3a , R 3b and R 3c are independently a hydrogen, a halogen, a nitro, a cyano, an optionally substituted hydrocarbyl, an optionally substituted hydrocarbyloxy, an optionally substituted hydrocarbylthio, an optionally substituted amino or an acyl
- the 6-membered ring with Y 1 , Y 2 and Y 3 of the formula (I) and (I 1 ) is a ring containing one or less nitrogen atom such as benzene ring and pyridine ring.
- halogen include fluorine, chlorine, bromine, iodine, and the like, preferably, chlorine and bromine .
- Examples of the "optionally substituted hydrocarbyl" in R 3a , R 3b and R 3c include the same groups as those exemplified with respect to the optionally substituted hydrocarbyl of R 3 .
- an optionally substituted Ci_ 3 alkyl is preferred, and an unsubstituted Ci- 3 alkyl, C 1 - 3 alkyl substituted with hydroxy and C 1 - 3 alkyl substituted with an amine (e.g., dimethylamino, methylamino, pyrrolidine, etc.) are more preferred.
- hydrocarbyl for said "optionally substituted hydrocarbyloxy" and “optionally substituted hydrocarbylthio" of R 3a , R 3b and R 3c include the same groups as those exemplified with respect to the optionally substituted hydrocarbyl of R 3 .
- hydrocarbyl having 1 to 4 carbon atoms is preferred.
- an optionally substituted C 1S alkoxy is preferred, and an substituted C 1 -. 3 alkoxy and halogenated substituted C1- 3 alkoxy are more preferred, and in particular, methoxy, difluoromethoxy and trifluoromethoxy are preferred.
- the "optionally substituted amino" for R 3a , R 3b and R 3c include amino group, an N-mono- substituted amino group, and an N, N-di-substituted amino group.
- substituted amino groups include that having one or two substituents of an optionally substituted hydrocarbyl group (e.g., a Ci_ 8 alkyl group, a C 3 _ 7 cycloalkyl group, a C 2 - 8 alkenyl group, a C 2 -8 alkynyl group, a C 3 - 7 cycloalkenyl group, a C 6 - I0 aryl group that may have a C 1 - 4 alkyl group, etc.), an optionally substituted heterocyclic group (e.g., the same group as an optionally substituted heterocyclic group of R 2 ), or the formula: COR 3d (wherein R 3d represents hydrogen atom or an optionally substituted hydrocarbyl group or an optionally substituted heterocyclic group.
- an optionally substituted hydrocarbyl group e.g., a Ci_ 8 alkyl group, a C 3 _ 7 cycloalkyl group, a C 2 - 8 alkenyl
- the hydrocarbyl group” or “the heterocyclic group” in “an optionally substituted hydrocarbyl group” or “an optionally substituted heterocyclic group” of R 3d may have the same substituent as that of "the hydrocarbyl group” or “the heterocyclic group” in “an optionally substituted hydrocarbyl” of R 3 or “an optionally substituted heterocyclic group” of R 2 ) , preferably a Ci- 10 acyl group (e.g., a C 2 - 7 alkanoyl, benzoyl, nicotinoyl, etc.).
- a Ci- 10 acyl group e.g., a C 2 - 7 alkanoyl, benzoyl, nicotinoyl, etc.
- methylamino dimethylamino, ethylamino, diethylamino, dipropylamino, dibutylamino, diallylamino, cyclohexylamino, phenylamino, N-methyl-N-phenylamino, acetylamino, propionylamino, benzoylamino, nicotinoylamino, and the like.
- the two groups in said substituted amino groups may be combined to form a nitrogen-containing 5- to
- 7-membered ring e.g., piperidino, piperazino, morpholino, thiomorpholino, etc.
- acyl for R 3a , R 3b and R 3c examples include the same groups as those exemplified with respect to the acyl for R 3 .
- an acyl having 2 to 4 carbon atoms is preferred.
- Y 1 , Y 2 and Y 3 are preferably CR 3a , CR 3b and CR 3c respectively, or one of Y 1 , Y 2 and Y 3 is nitrogen.
- R 3a , R 3b and R 3c are preferably hydrogen, halogen, cyano, acyl, C 1 - 4 alkyl optionally substituted by hydroxy (for example, methyl, ethyl, hydroxymethyl) , amino, and C 1 - 4 alkoxy (for example, methoxy, ethoxy) .
- R 3a chlorine, bromine, methoxy and methyl are more preferred.
- Z is a bond, -CO-, an oxygen (-0-) , a sulfur (-S-), -SO-, -SO 2 -, -NR 4 -, -NR 4 -alk-,
- alk is an optionally substituted C 1 -4 alkylene such as methylene, ethylene, propylene, butylene and the like.
- R 4 is a hydrogen, an optionally substituted hydrocarbyl or an acyl.
- the "optionally substituted hydrocarbyl” and “acyl” for R 4 include the same groups as those exemplified with respect to the optionally substituted hydrocarbyl group and acyl for R 3 .
- R 4 may be combined together with the substituent of the cyclic hydrocarbyl or heterocyclic group in the optionally substituted hydrocarbyl or optionally substituted heterocyclic group of R 2 to form a ring.
- Examples of said ring include the same rings as those exemplified with respect to the rings formed by the two substituents of R 2 mentioned above. Provided that (i) the compound wherein X is -NH-, and
- R 2 is an optionally substituted thiophene ring, (ii) the compound wherein R 1 is cyano, R 3c is methyl substituted with three substituents, one of which is acyl, and other two of which may form a ring, (iii) a) 6-amino-2- [ (2, ⁇ -dichlorophenyl) amino] -1-methyl-lH- benzimidazole-7-carbonitrile, b) N- ⁇ 7-cyano-2- [ (2, ⁇ -dichlorophenyl) amino] -1-methyl-IH- benzimidazol- ⁇ -yl ⁇ acetamide, c) ⁇ -amino-2- [ (2, ⁇ -dichlorophenyl) amino] -1-methyl-lH- benzimidazole-7-carboxamide, and d) 6- ⁇ [ (allylamino) carbonothioyl] amino ⁇ -2- [ (2, 6- dichlorophenyl) amino]
- a compound wherein X is NR 3 (wherein R 3 is preferably methyl, ethyl, hydroxyethyl, etc.); Y 1 is CR 3a (wherein R 3a is preferably H, Me, halogen (eg. F, Cl, Br), cyano, acyl, alkoxy, etc.), Y 2 is CR 3b (wherein R 3b is preferably H, Me, halogen (eg. F, Cl, Br), etc.) or nitrogen and Y 3 is CR 3c (wherein R 3c is preferably H, Me, halogen (eg.
- Z is NR 4 (wherein R 4 is preferably H, Ci_ 4 alkyl, etc.) or oxygen;
- R 1 is an optionally substituted acyclic branched C 3 _ 7 hydrocarbyl (in particular, 2-propyl, 3-hexyl, 3-pentyl, 4-heptyl) ;
- R 2 is an optionally substituted C ⁇ -io aryl (in particular, phenyl, more preferably di- or tri-substituted phenyl) or an optionally substituted pyridyl (in particular, pyridyl, more preferably di- or tri-substituted pyridyl) is exemplified.
- R 1 is 3-pentyl, 3-hexyl or 4-heptyl
- X is NR 3
- R 3 is methyl, ethyl or hydroxyethyl
- Y 1 is CR 3a
- Y 2 is CR 3b
- Y 3 is CR 3c
- R 3a is chloro, bromo, methoxy or methyl
- R 3b is a hydrogen
- R 3c is a hydrogen
- R 2 is phenyl which is 2, 4, 6-trisubstituted, 2, 4, 5-trisubstituted or 2, 4-disubstituted with substituents or pyridyl which is trisubstituted or disubstituted with substituents.
- substituents for the phenyl and pyridyl include an acyl such as acetyl and propionyl, an amide such as dimethylaminocarbonyl and methylaminocarbonyl, an amine such as dimethylamino, methylamino and amino, a halogen such as fluoro, chloro and bromo, a lower alkyl such as methyl, ethyl and trifluoromethyl and a lower alkoxy such as methoxy, ethoxy and trifluoromethoxy, each of which may be substituted.
- an acyl such as acetyl and propionyl
- an amide such as dimethylaminocarbonyl and methylaminocarbonyl
- an amine such as dimethylamino, methylamino and amino
- a halogen such as fluoro, chloro and bromo
- a lower alkyl such as methyl, ethyl and trifluoromethyl
- Compound (I) or (I 1 ) may be in the form of a prodrug thereof.
- the prodrug of compound (I) or (I') refers to a compound that is converted into compound (I) or (I 1 ) by a reaction with an enzyme, gastric acid, or the like under a physiological condition in the living body, namely, (i) a compound that is converted into compound (I) or (I') by an enzymatic oxidation, reduction, hydrolysis, or the like, and (ii) a compound that is converted into compound (I) or (I 1 ) by hydrolysis with gastric acid or the like.
- Examples of a prodrug of compound (I) or (!') to be used include a compound or its salt wherein hydroxyl group in compound (I) or (I 1 ) is acylated, alkylated, phosphorylated, or converted into borate (e.g., a compound or its salt wherein hydroxyl group in compound (I) or (I 1 ) is converted into acetyloxy, palmitoyloxy, propanoyloxy, pivaloyloxy, succinyloxy, fumaryloxy, alanyloxy, dimethylaminomethylcarbonyloxy, etc.), a compound or its salt wherein carboxyl group in compound (I) or (I 1 ) is esterified or amidated (e.g., a compound or its salt wherein carboxyl group in compound (I) or (I 1 ) is subjected to ethyl esterification, phenyl esterification, carboxyoxymethyl esterification, dimethylaminomethyl esterification,
- prodrugs can be produced according to a per se known method or its modified method.
- a prodrug of compound (I) or (I') may be a compound or its salt that is converted into compound (I) or (I 1 ) under physiological conditions as described in "Development of Drugs", Volume 7, Molecular Design, Hirokawa Shoten, 1990; pages 163-198.
- General synthetic method
- R la is an optionally substituted hydrocarbyl, an optionally substituted C-linked heterocyclic, an optionally substituted N-linked heteroaryl and acyl
- Ar is an optionally substituted aryl
- L 1 is a leaving group (for example, halogen atom such as chlorine, bromine and iodine, etc, sulfonyloxy group such as p-toluenesulfonyloxy group, methanesulfonyloxy group and trifluoromethanesulfonyloxy group, etc., and acyloxy group such as acetyloxy group and benzoyloxy group, etc.) and each of other symbols has a meaning defined above.
- Compound (III) or a salt thereof can be prepared by halogenation, sulfonylation or acylation of compound (II) or a salt thereof with a halogenation agent, sulfonylation agent or acylation agent, respectively.
- a halogenation agent include phosphorous oxychloride, phosphorous oxybromide, phosphorous trichloride, phosphorous tribromide, phosphorous pentachloride, chlorine, bromine and thionyl chloride.
- the halogenation agent is employed in an amount of 1 moles to excess per 1 mole of compound (II) or as a solvent.
- solvent having no adverse effect on the reaction examples include aromatic hydrocarbons such as benzene, toluene and xylene, ethers such as diethyl ether, dioxane and tetrahydrofuran, esters such as ethyl acetate, nitriles such as acetonitrile, halogenated hydrocarbon such as chloroform and dichloromethane, amides such as N,N- dimethlformamide, W ⁇ iV-dimethylacetamide and l-methyl-2- pirrolidinone, ketones such as acetone and 2-butanone and sulfoxides such as dimethylsulfoxide. These solvents may be used by mixing at an appropriate ratio.
- aromatic hydrocarbons such as benzene, toluene and xylene
- ethers such as diethyl ether, dioxane and tetrahydrofuran
- esters such as ethyl acetate
- reaction temperature may vary depending on compound (II) or a salt thereof employed as well as other conditions, it is 0 to 200 0 C, preferably 20 to 150 0 C.
- the reaction time is 10 minutes to 24 hours, preferably 30 minutes to 12 hours.
- the thus obtained compound (III) can be isolated and purified by the known isolating and purifying methods, for example, concentration under reduced pressure, extraction with solvent, crystallization, recrystallization, transfer dissolution and chromatography.
- compound (III) or a salt thereof can be prepared by reacting compound (II) with a sulfonylation agent or an acylation agent after base treatment of compound (II) .
- a base may for example be an alkaline metal hydroxide such as sodium hydroxide and potassium hydroxide, etc., an alkaline metal hydrogen carbonate such as sodium hydrogen carbonate and potassium hydrogen carbonate, etc., an alkaline metal carbonate such as sodium carbonate and potassium carbonate, etc., a cesium salt such as cesium carbonate, etc., an alkaline metal hydride such as sodium hydride and potassium hydride, etc., sodium amide, an alkoxide such as sodium methoxide and sodium ethoxide, etc., an amine such as trimethylamine, triethylamine and diisopropylethylamine, etc., a cyclic amine such as pyridine, etc.
- a sulfonylation agent examples include p- toluenesulfonyl chloride, methanesulfonylchloride, trifluoromethanesulfonylchloride, etc.
- the sulfonylation agent is employed in an amount of 1 to 10 moles, preferably 1 to 5 moles per 1 mole of compound (II) .
- an acylation agent examples include acetylchloride, benzoyl chloride, etc.
- the acylation agent is employed in an amount of 1 to 10 moles, preferably 1 to 5 moles per 1 mole of compound (II) .
- Examples of the solvent having no adverse effect on the reaction include water, alcohols such as methanol and ethanol, ethers such as diethyl ether, dioxane and tetrahydrofuran, aromatic hydrocarbons such as benzene, toluene and xylene, esters such as ethyl acetate, halogenated hydrocarbons such as chloroform and dichloromethane, nitriles such as acetonitrile, amides such as W ⁇ i ⁇ -dimethylformamide and N ⁇ iV-dimethylacetamide, and sulfoxides such as dimethylsulfoxide. These solvents may be used by mixing at an appropriate ratio.
- reaction temperature may vary depending on compound (II) or a salt thereof employed as well as other conditions, it is 0 to 200 0 C, preferably 0 to 150 0 C.
- the reaction time is 10 minutes to 24 hours, preferably 30 minutes to 12 hours.
- the thus obtained compound (III) can be isolated and purified by the known isolating and purifying methods, for example, concentration under reduced pressure, extraction with solvent, crystallization, recrystallization, transfer dissolution and chromatography.
- Compound (Ia) or a salt thereof, which is encompassed within compound (I) of the invention, can be prepared by reacting compound (III) with ArZH. In this step, 1 to 20 moles, preferably 1 to 10 moles of a compound represented by ArZH or a salt thereof are employed per 1 mole of compound (III) or a salt thereof.
- a base may for example be an alkaline metal hydroxide such as sodium hydroxide and potassium hydroxide, etc., an alkaline metal hydrogen carbonate such as sodium hydrogen carbonate and potassium hydrogen carbonate, etc., an alkaline metal carbonate such as sodium carbonate and potassium carbonate, etc., a cesium salt such as cesium carbonate, etc., an alkaline metal hydride such as sodium hydride and potassium hydride, etc., sodium amide, an alkaline metal alkoxide such as sodium methoxide, sodium ethoxide, sodium tert-butoxide and potassium tert-butoxide, etc., an amine such as trimethylamine, triethylamine and diisopropylethylamine, etc., a cyclic amine such as pyridine, etc.
- an alkaline metal hydroxide such as sodium hydroxide and potassium hydroxide, etc.
- an alkaline metal hydrogen carbonate such as sodium hydrogen carbonate and potassium hydrogen carbonate, etc.
- solvent having no adverse effect on the reaction examples include water, alcohols such as methanol and ethanol, ethers such as diethyl ether, dioxane and tetrahydrofuran, aromatic hydrocarbons such as benzene, toluene and xylene, esters such as ethyl acetate, halogenated hydrocarbons such as chloroform and dichloromethane, nitriles such as acetonitrile, amides such as I ⁇ N-dimethylformamide, N,N-dimethylacetamide and 1- methyl-2-pyrrolidinone, ketones such as acetone and 2- butanone and sulfoxides such as dimethylsulfoxide. These solvents may be used by mixing at an appropriate ratio, or may not be used.
- reaction temperature may vary depending on compound (III) or a salt thereof employed as well as other reaction conditions, it is 0 to 200 °C, preferably 20 to 150 °C, or the reaction may be heated by microwave irradiation.
- the reaction time is 5 minutes to 48 hours, preferably 5 minutes to 24 hours.
- the thus obtained compound (Ia) can be isolated and purified by the known isolating and purifying methods, for example, concentration, concentration under reduced pressure, extraction with solvent, crystallization, recrystallization, transfer dissolution and chromatography.
- R 5 is hydrogen and an optionally substituted hydrocarbyl
- R 3 and R 5 may be combined each other to form a ring
- R 6 and R 7 are optionally substituted hydrocarbyl
- L 2 and L 3 are halogen atoms such as chlorine, bromine and iodine and each of other symbols has a meaning defined above .
- Compound (Ha) or a salt thereof can be prepared by treatment of compound (IV) with 1, 1' -carbonyl diimidazole, phosgene, triphosgene, alkyl haloformate such as ethyl chloroformate, phenyl haloformate such as phenyl chloroformate or urea, etc.
- Compound (IV) or a salt thereof is mainly commercially available or can be prepared from the nitro derivatives corresponded to compound (IV) .
- solvent having no adverse effect on the reaction examples include water, alcohols such as methanol and ethanol, ethers such as diethyl ether, dioxane and tetrahydrofuran, aromatic hydrocarbons such as benzene, toluene and xylene, esters such as ethyl acetate, halogenated hydrocarbons such as chloroform and dichloromethane, nitriles such as acetonitrile, amides such as N ⁇ iV-dimethylformamide, I ⁇ F,W-dimethylacetamide and 1- methyl-2-pyrrolidinone, ketones such as acetone and 2- butanone and sulfoxides such as dimethylsulfoxide. These solvents may be used by mixing at an appropriate ratio, or may not be used.
- reaction temperature may vary depending on compound (IV) or a salt thereof employed as well as other reaction conditions, it is 0 to 150 °C, preferably 20 to
- reaction time is 5 minutes to 48 hours, preferably 5 minutes to 24 hours.
- compound (Ha) can be isolated and purified by the known isolating and purifying methods, for example, concentration, concentration under reduced pressure, extraction with solvent, crystallization, recrystallization, transfer dissolution and chromatography.
- Compound (lib) or a salt thereof can be prepared by Grinard reaction of compound (Ha) or a salt thereof with R 6 MgL 2 and R 7 MgL 3 .
- R 6 MgL 2 may be used in this step.
- the Grinard reactions may be performed stepwise by R 6 MgL 2 and R 7 MgL 3 in this step. In this step, 1 to 20 moles, preferably 1 to 10 moles of a compound represented by R 6 MgL 2 and R 7 MgL 3 or a salt thereof are employed per 1 mole of compound (Ha) or a salt thereof.
- solvent having no adverse effect on the reaction examples include ethers such as diethyl ether, dioxane and tetrahydrofuran, aromatic hydrocarbons such as benzene, toluene and xylene, halogenated hydrocarbons such as chloroform and dichloromethane, ketones such as acetone and 2-butanone and sulfoxides such as dimethylsulfoxide .
- ethers such as diethyl ether, dioxane and tetrahydrofuran
- aromatic hydrocarbons such as benzene, toluene and xylene
- halogenated hydrocarbons such as chloroform and dichloromethane
- ketones such as acetone and 2-butanone
- sulfoxides such as dimethylsulfoxide .
- reaction temperature may vary depending on compound (Ha) or a salt thereof employed as well as other reaction conditions, it is -20 to 150 0 C, preferably 0 to 100 °C.
- the reaction time is 5 minutes to 24 hours, preferably 5 minutes to 12 hours.
- the thus obtained compound (lib) can be isolated and purified by the known isolating and purifying methods, for example, concentration, concentration under reduced pressure, extraction with solvent, crystallization, recrystallization, transfer dissolution and chromatography.
- Compound (lie) or a salt thereof can be prepared by dehydration of compound (lib) or a salt thereof with an acid, and the olefine is then reduced by an appropriate reducing agent or catalytic hydrogenation.
- An acid may for example be an inorganic acid such as hydrochloric acid, sulfuric acid, nitric acid and thionyl chloride, etc., and an ordinary organic acid such as formic acid, acetic acid, trifluoroacetic acid and methanesulfonic acid, etc. as well as a Lewis acid.
- an inorganic acid such as hydrochloric acid, sulfuric acid, nitric acid and thionyl chloride, etc.
- an ordinary organic acid such as formic acid, acetic acid, trifluoroacetic acid and methanesulfonic acid, etc. as well as a Lewis acid.
- 1 mole to excess of an acid is employed per 1 mole of compound (lib) or a salt thereof or an acid may be employed as a solvent.
- solvent having no adverse effect on the reaction examples include water, alcohols such as methanol and ethanol, ethers such as diethyl ether, dioxane and tetrahydrofuran, aromatic hydrocarbons such as benzene, toluene and xylene, esters such as ethyl acetate, halogenated hydrocarbons such as chloroform and dichloromethane, nitriles such as acetonitrile, amides such as 2V, ⁇ T-dimethylformamide, I ⁇ 7V ⁇ 7-dimethylacetamide and 1- methyl-2-pyrrolidinone, ketones such as acetone and 2- butanone and sulfoxides such as dimethylsulfoxide. These solvents may be used by mixing at an appropriate ratio, or may not be used.
- reaction temperature may vary depending on compound (lib) or a salt thereof employed as well as other reaction conditions, it is 0 to 200 °C, preferably 20 to 150 °C.
- the reaction time is 5 minutes to 48 hours, preferably 5 minutes to 24 hours.
- a reducing agent is preferably sodium borohydride, lithium borohydride, sodium cyanoborohydride and sodium triacetoxyborohydride.
- Catalytic hydrogenation may be performed in this step.
- a catalyst include a palladium catalyst such as palladium black, palladium oxide, palladium barium sulfate, palladium on carbon, palladium hydroxide, a platinum catalyst such as platinum black, platinum oxide and platinum on carbon, or nickel catalyst such as reduced nickel, oxidized nickel, and Raney nickel.
- solvent having no adverse effect on the reaction examples include water, alcohols such as methanol and ethanol, ethers such as diethyl ether, dioxane and tetrahydrofuran, aromatic hydrocarbons such as benzene, toluene and xylene, esters such as ethyl acetate, halogenated hydrocarbons such as chloroform and dichloromethane, nitriles such as acetonitrile, amides such as N,N ⁇ dimethylfor ⁇ a ⁇ ide, IV,I ⁇ /-dimethylacetamide and 1- methyl-2-pyrrolidinone, ketones such as acetone and 2- butanone and sulfoxides such as dimethylsulfoxide.
- alcohols such as methanol and ethanol
- ethers such as diethyl ether, dioxane and tetrahydrofuran
- aromatic hydrocarbons such as benzene, toluene and xylene
- esters
- reaction temperature may vary depending on the olefine or a salt thereof employed as well as other reaction conditions, it is 0 to 150 °C, preferably 0 to 100 °C.
- the reaction time is 5 minutes to 48 hours, preferably 5 minutes to 24 hours.
- the thus obtained compound (lie) can be isolated and purified by the known isolating and purifying methods, for example, concentration, concentration under reduced pressure, extraction with solvent, crystallization, recrystallization, transfer dissolution and chromatography.
- R lb is an optionally substituted hydrocarbyl and an optionally substituted C-linked heterocyclic
- R lc is an optionally substituted hydrocarbyl, an optionally substituted C-linked heterocyclic and an optionally substituted N-linked heteroaryl
- R 8 and R 9 are independently hydrogen, optionally substituted hydrocarbyl, hydroxy and optionally substituted alkoxy and may be combined each
- L 4 is a halogen atom such as chlorine, bromine and iodine each of other symbols has a meaning defined above.
- Compound (VI) or a salt thereof can be prepared by halogenation of compound (V) or a salt thereof with a halogenation agent.
- halogenation agent examples include chlorine, bromine, iodine, thionyl chloride, copper (I) chloride, copper (II) chloride, copper (I) bromide, copper (II) bromide, sodium chloride, sodium bromide, sodium iodide, potassium iodide, etc.
- the halogenation agent is employed in an amount of 0.5 moles to 10 moles, preferably 0.5 moles to 5 moles, per 1 mole of compound (V) .
- the diazonium type compound may be produced before introduction of a halogen atom.
- an agent to produce the diazonium type compound include sodium nitrite, potassium nitrite and tert-butyl nitrite, etc.
- the agent is employed in an amount of 1 mole to 10 moles, preferably 1 mole to 5 moles, per 1 mole of compound (V) .
- This reaction can be carried out under an acidic condition.
- an acid include an inorganic acid such as hydrochloric acid, hydrobromic acid, hydroiodic acid, sulfuric acid, a nitric acid and copper sulfate, etc., as well as Lewis acid.
- An acid is employed in an amount of 2 moles to excess per 1 mole of compound (V) .
- solvent having no adverse effect on the reaction examples include water, alcohols such as methanol and ethanol, ethers such as diethyl ether, dioxane and tetrahydrofuran, aromatic hydrocarbons such as benzene, toluene and xylene, esters such as ethyl acetate, halogenated hydrocarbons such as chloroform and dichloromethane, nitriles such as acetonitrile, amides such as NV-V-dimethylformamide, I ⁇ J>N-dimethylacetamide and 1- methyl-2-pyrrolidinone, ketones such as acetone and 2- butanone and sulfoxides such as dimethylsulfoxide. These solvents may be used by mixing at an appropriate ratio.
- reaction temperature may vary depending on compound (V) or a salt thereof employed as well as other conditions, it is -20 to 150 0 C, preferably 0 to 100 0 C.
- the reaction time is 10 minutes to 24 hours, preferably 30 minutes to 12 hours.
- the thus obtained compound (VI) can be isolated and purified by the known isolating and purifying methods, for example, concentration under reduced pressure, extraction with solvent, crystallization, recrystallization, transfer dissolution and chromatography.
- R lc is an optionally substituted hydrocarbyl
- an optionally substituted C-linked heterocyclic in compound (lie) or a salt thereof compound (lie) or a salt thereof can be prepared by reacting compound (VI) with R lb BR 8 R 9 or a salt thereof in the presence of a palladium catalyst, preferably tetrakis (triphenylphosphine) palladium(O) and tris (dibenzylidenacetate) dipalladium(O) , a catalytic amount of a phosphine ligand, preferably 2-
- a palladium catalyst preferably palladium (II) acetate or a copper agent, preferably copper (II) acetate.
- a catalytic amount of a phosphine ligand preferably 2- (dicyclohexylphosphino) biphenyl, may be employed. This reaction can be carried out according to the procedure of Buchwald et al. (J. Am. Chem. Soc. 1998, 120, 9722) and Lam et. al. (Tetrahedron Lett., 1998, 39, 2941) and the modified methods.
- Compound (VII) or a salt thereof can be prepared by reaction of compound (VI) or a salt thereof via lithiation or coupling reaction with a boron agent.
- examples of a boron agent include trialkyl borate, preferably triisopropyl borate.
- a lithiation agent may for example be alkyl lithium, preferably ⁇ -butyl lithium, sec-butyl lithium and tert- butyl lithium and is employed in an amount of 1 to 5 moles, preferably 1 to 3 moles per 1 mole of compound (VI) or a salt thereof.
- solvent having no adverse effect on the reaction examples include aromatic hydrocarbons such as benzene, toluene and xylene, ethers such as diethyl ether, dioxane and tetrahydrofuran, and halogenated hydrocarbon such as chloroform and dichloromethane . These solvents may be used by mixing at an appropriate ratio.
- reaction temperature may vary depending on compound (VI) or a salt thereof employed as well as other conditions, it is -100 to 100 0 C, preferably -80 to 50 0 C.
- the reaction time is 10 minutes to 24 hours, preferably 30 minutes to 12 hours.
- examples of a boron agent include 4,4,5,5- tetramethyl-1, 3, 2-dioxaborolane and bis (pinacolato) diborane.
- 1 to 10 moles, preferably 1 to 5 moles of a boron agent are employed per 1 mole of compound (VI) or a salt thereof.
- a palladium catalyst preferably palladium (II) diacetate, a catalytic amount of a phosphine ligand, preferably 2- (dicyclohexylphosphino) biphenyl and a base can be employed according to the procedure described in J. Org.
- solvent having no adverse effect on the reaction examples include water, alcohols such as methanol and ethanol, ethers such as diethyl ether, dioxane and tetrahydrofuran, aromatic hydrocarbons such as benzene, toluene and xylene, esters such as ethyl acetate, halogenated hydrocarbons such as chloroform and dichloromethane, nitriles such as acetonitrile, amides such as A ⁇ N-dimethylformamide, N,N-dimethylacetamide and 1- methyl-2-pyrrolidinone, ketones such as acetone and 2- butanone and sulfoxides such as dimethylsulfoxide . These solvents may be used by mixing at an appropriate ratio, or may not be used.
- reaction temperature may vary depending on compound (VI) or a salt thereof employed as well as other conditions, it is 0 to 200 0 C, preferably 20 to 150 0 C.
- the reaction time is 10 minutes to 48 hours, preferably 30 minutes to 24 hours.
- the thus obtained compound (VII) can be isolated and purified by the known isolating and purifying methods, for example, concentration under reduced pressure, extraction with solvent, crystallization, recrystallization, transfer dissolution and chromatography.
- Preparation of compound (VIII) or a salt thereof from compound (VI) or a salt thereof can be carried out similar to preparation of compound (III) or a salt thereof in Scheme 1.
- Preparation of compound (IX) or a salt thereof from compound (VIII) or a salt thereof can be carried out similar to preparation of compound (Ia) or a salt thereof in Scheme 1.
- Compound (X) or a salt thereof can be prepared by reaction of compound (V) or a salt thereof with sodium nitrite, and the obtained diazonium salt is reduced by an appropriate reducing agent.
- Examples of a reducing agent include alkaline metal borohydride, preferably sodium borohydride, lithium borohydride, sodium cyanoborohydride and sodium triacetoxyborohydride, metal, preferably Fe, Zn, Sn and
- a palladium catalyst such as palladium black, palladium oxide, palladium barium sulfate, palladium on carbon, palladium hydroxide, a platinum catalyst such as platinum black, platinum oxide and platinum on carbon, or nickel catalyst such as reduced nickel, oxidized nickel, and Raney nickel.
- the reducing agent is employed in an amount of catalytic amount to excess per 1 mole of compound (V) .
- solvent having no adverse effect on the reaction examples include water, alcohols such as methanol and ethanol, ethers such as diethyl ether, dioxane and tetrahydrofuran, aromatic hydrocarbons such as benzene, toluene and xylene, esters such as ethyl acetate, halogenated hydrocarbons such as chloroform and dichloromethane, nitriles such as acetonitrile, amides such as _V,_V-dimethylformamide, N,N-dimethylacetamide and 1- methyl-2-pyrrolidinone, and sulfoxides such as dimethylsulfoxide . These solvents may be used by mixing at an appropriate ratio, or may not be used.
- reaction temperature may vary depending on compound (V) or a salt thereof employed as well as other conditions, it is -20 to 150 0 C, preferably 0 to 100 0 C.
- the reaction time is 10 minutes to 24 hours, preferably 30 minutes to 12 hours.
- compound (X) can be isolated and purified by the known isolating and purifying methods, for example, concentration under reduced pressure, extraction with solvent, crystallization, recrystallization, transfer dissolution and chromatography.
- Compound (Hf) or a salt thereof can be prepared by reaction of compound (X) or a salt thereof with compound (XI) or a salt thereof.
- An acid may be used and for example be an inorganic acid such as hydrochloric acid, sulfuric acid and nitric acid, etc., and an ordinary organic acid such as formic acid, acetic acid, p-toluenesulfonic acid, trifluoroacetic acid and methanesulfonic acid, etc. as well as a Lewis acid.
- an inorganic acid such as hydrochloric acid, sulfuric acid and nitric acid, etc.
- an ordinary organic acid such as formic acid, acetic acid, p-toluenesulfonic acid, trifluoroacetic acid and methanesulfonic acid, etc. as well as a Lewis acid.
- solvent having no adverse effect on the reaction examples include water, alcohols such as methanol and ethanol, ethers such as diethyl ether, dioxane and tetrahydrofuran, aromatic hydrocarbons such as benzene, toluene and xylene, esters such as ethyl acetate, halogenated hydrocarbons such as chloroform and dichloromethane, nitriles such as acetonitrile, amides such as IV,I ⁇ 7-dimethylformamide, WVN-dimethylacetamide and 1- methyl-2-pyrrolidinone, acids such as formic acid and acetic acid and sulfoxides such as dimethylsulfoxide . These solvents may be used by mixing at an appropriate ratio.
- reaction temperature may vary depending on compound (V) or a salt thereof employed as well as other conditions, it is 0 to 200 0 C, preferably 20 to 150 0 C.
- the reaction time is 10 minutes to 24 hours, preferably 30 minutes to 12 hours.
- the thus obtained compound (Hf) can be isolated and purified by the known isolating and purifying methods, for example, concentration under reduced pressure, extraction with solvent, crystallization, recrystallization, transfer dissolution and chromatography.
- phenylcarbonyl for example. formyl, methylcarbonyl and ethylcarbonyl, etc.
- phenylcarbonyl for example, a Ci- 6 alkyloxycarbonyl (for example, methoxycarbonyl, ethoxycarbonyl and tert-butoxycarbonyl, etc.), phenyloxycarbonyl (for example, benzoxycarbonyl) , C 7 - IO aralkylcarbonyl (for example, benzyloxycarbonyl) , C 7 -io aralkyl (for example, benzyl and 4-methoxybenzyl, etc.), trityl, phthaloyl, etc.
- a substituent on each of the groups listed above may be a halogen atom (for example, fluorine, chlorine, bromine and iodine, etc.), a Ci_ 6 alkylcarbonyl (for example, methylcarbonyl, ethylcarbonyl and butylcarbonyl, etc.) and a nitro group, and each of other symbols has a meaning defined above.
- Compound (Ha) or a salt thereof can be also prepared by the procedure as shown in scheme 11.
- Compound (XIV) or a salt thereof can be prepared by nitration of compound (XIII) or a salt thereof with a nitration agent.
- Compound (XIII) or a salt thereof is mainly commercially available or can be prepared from the aniline derivatives corresponded to compound (XIII) by a usual acetylation method.
- nitration agent examples include nitric acids (for example, fuming nitric acid, a solution of nitric acid and sulfuric acid etc. ) , nitrates (for example, sodium nitrate, potassium nitrate, silver nitrate, ammonium nitrate, benzoyl nitrate, benzyltriphenylphosphonium nitrate, bismuth subnitrate, etc) .
- the nitration agent is employed in an amount of 0.5 moles to 50 moles, or may be employed as a solvent, preferably 0.5 moles to 30 moles, per 1 mole of compound (XIII) .
- additives include anhydrides (for example, acetic anhydride, trifluoroacetic anhydride, methanesulfonic anhydride, etc) , acid chlorides (for example, thionyl chloride, etc) , acids
- the additives are employed in an amount of 0.5 moles to 50 moles, preferably 0.5 moles to 30 moles, per 1 mole of compound (XIII) .
- solvent having no adverse effect on the reaction examples include water, acetic acid, halogenated hydrocarbons such as chloroform and dichloromethane, 1,2- dichloroethane, etc. These solvents may be used by mixing at an appropriate ratio, or may not be used.
- the reaction temperature may vary depending on compound (XIII) or a salt thereof employed as well as other reaction conditions, it is -20 to 150 0 C, preferably 0 to 100 °C
- the reaction time is 5 minutes to 48 hours, preferably 5 minutes to 24 hours.
- The' thus obtained compound (XIV) can be isolated and purified by the known isolating and purifying methods, for example, concentration, concentration under reduced pressure, extraction with solvent, crystallization, recrystallization, transfer dissolution and chromatography.
- Compound (XV) or a salt thereof can be prepared by deacetylation of compound (XIV) or a salt thereof with an acid or base, and then reduction of the nitro group by an appropriate reducing agent or catalytic hydrogenation.
- An acid may for example be an inorganic acid such as hydrochloric acid, sulfuric acid, nitric acid and thionyl chloride, etc., and an ordinary organic acid such as formic acid, acetic acid, trifluoroacetic acid and methanesulfonic acid, etc. as well as a Lewis acid.
- an inorganic acid such as hydrochloric acid, sulfuric acid, nitric acid and thionyl chloride, etc.
- an ordinary organic acid such as formic acid, acetic acid, trifluoroacetic acid and methanesulfonic acid, etc. as well as a Lewis acid.
- a base may for example be an alkaline metal hydroxide such as sodium hydroxide and potassium hydroxide, etc., an alkaline metal hydrogen carbonate such as sodium hydrogen carbonate and potassium hydrogen carbonate, etc., an alkaline metal carbonate such as sodium carbonate and potassium carbonate, etc., a cesium salt such as cesium carbonate, etc. , an alkaline metal hydride such as sodium hydride and potassium hydride, etc., sodium amide, an alkaline metal alkoxide such as sodium methoxide, sodium ethoxide, sodium tert-butoxide and potassium tert-butoxide, etc.
- solvent having no adverse effect on the reaction include water, alcohols such as methanol and ethanol, ethers such as diethyl ether, dioxane and tetrahydrofuran, aromatic hydrocarbons such as benzene, toluene and xylene, esters such as ethyl acetate, halogenated hydrocarbons such as chloroform and dichloromethane, nitriles such as acetonitrile, amides such as I ⁇ 7V.W-dimethylformamide, iV ⁇ iV-dimethylacetamide and 1- methyl-2-pyrrolidinone, ketones such as acetone and 2- butanone and sulfoxides such as dimethylsulfoxide. These solvents may be used by mixing at an appropriate ratio, or may not be used.
- reaction temperature may vary depending on compound (XIV) or a salt thereof employed as well as other reaction conditions, it is 0 to 200 °C, preferably 20 to 150 °C.
- the reaction time is 5 minutes to 48 hours, preferably 5 minutes to 24 hours.
- nitro compounds can be isolated and purified by the known isolating and purifying methods, for example, concentration, concentration under reduced pressure, extraction with solvent, crystallization, recrystallization, transfer dissolution and chromatography.
- a reducing agent is preferably sodium borohydride, lithium borohydride, sodium cyanoborohydride and sodium triacetoxyborohydride .
- Catalytic hydrogenation may be performed in this step.
- a catalyst include a palladium catalyst such as palladium black, palladium oxide, palladium barium sulfate, palladium on carbon, palladium hydroxide, a platinum catalyst such as platinum black, platinum oxide and platinum on carbon, or nickel catalyst such as reduced nickel, oxidized nickel, and Raney nickel.
- solvent having no adverse effect on the reaction include water, alcohols such as methanol and ethanol, ethers such as diethyl ether, dioxane and tetrahydrofuran, aromatic hydrocarbons such as benzene, toluene and xylene, esters such as ethyl acetate, halogenated hydrocarbons such as chloroform and dichloromethane, nitriles such as acetonitrile, amides such as A ⁇ AT-dimethylformamide, N,iV-dimethylacetamide and 1- methyl-2-pyrrolidinone, ketones such as acetone and 2- butanone and sulfoxides such as dimethylsulfoxide. These solvents may be used by mixing at an appropriate ratio.
- reaction temperature may vary depending on compound (XIV) or a salt thereof employed as well as other reaction conditions, it is 0 to 150 0 C, preferably 0 to 100
- reaction time is 5 minutes to 48 hours, preferably 5 minutes to 24 hours.
- compound (XV) can be isolated and purified by the known isolating and purifying methods, for example, concentration, concentration under reduced pressure, extraction with solvent, crystallization, recrystallization, transfer dissolution and chromatography.
- Preparation of compound (XVI) or a salt thereof from compound (XV) or a salt thereof can be carried out similar to preparation of compound (Ha) in Scheme 2.
- Compound (XVII) or a salt thereof can be prepared by reacting compound (XVI) with R 12 -L 2 or anhydride (R 12 ) 2 O.
- a base may for example be an alkaline metal hydroxide such as sodium hydroxide and potassium hydroxide, etc., an alkaline metal hydrogen carbonate such as sodium hydrogen carbonate and potassium hydrogen carbonate, etc., an alkaline metal carbonate such as sodium carbonate and potassium carbonate, etc., a cesium salt such as cesium carbonate, etc., an alkaline metal hydride such as sodium hydride and potassium hydride, etc., sodium amide, an alkaline metal alkoxide such as sodium methoxide, sodium ethoxide, sodium tert-butoxide and potassium tert-butoxide, etc., an amine such as trimethylamine, triethylamine and diisopropylethylamine, etc., a cyclic amine such as pyridine, 4-dimethylaminopyridine, DBU, etc.
- an alkaline metal hydroxide such as sodium hydroxide and potassium hydroxide, etc.
- an alkaline metal hydrogen carbonate such as sodium
- solvent having no adverse effect on the reaction examples include water, alcohols such as methanol and ethanol, ethers such as diethyl ether, dioxane and tetrahydrofuran, aromatic hydrocarbons such as benzene, toluene and xylene, esters such as ethyl acetate, halogenated hydrocarbons such as chloroform and dichloromethane, nitriles such as acetonitrile, amides such as WVN-dimethylformamide, N,W-dimethylacetamide and 1- methyl-2-pyrrolidinone, ketones such as acetone and 2- butanone and sulfoxides such as dimethylsulfoxide. These solvents may be used by mixing at an appropriate ratio, or may not be used. While the reaction temperature may vary depending on compound (XVI) or a salt thereof employed as well as other reaction conditions, it is 0 to 200 0 C, preferably 20 to
- reaction 150 0 C or the reaction may be heated by microwave irradiation.
- the reaction time is 5 minutes to 48 hours, preferably 5 minutes to 24 hours.
- compound (XVII) can be isolated and purified by the known isolating and purifying methods, for example, concentration, concentration under reduced pressure, extraction with solvent, crystallization, recrystallization, transfer dissolution and chromatography.
- Preparation of compound (XVIII) or a salt thereof from compound (XVII) or a salt thereof can be carried out similar to preparation of compound (XVII) .
- Compound (Ha) or a salt thereof can be prepared by deprotection of compound (XVIII) or a salt thereof with an acid or a base, or catalytic hydrogenation.
- An acid may for example be an inorganic acid such as hydrochloric acid, sulfuric acid, nitric acid and thionyl chloride, etc., and an ordinary organic acid such as formic acid, acetic acid, trifluoroacetic acid and methanesulfonic acid, etc. as well as a Lewis acid.
- an inorganic acid such as hydrochloric acid, sulfuric acid, nitric acid and thionyl chloride, etc.
- an ordinary organic acid such as formic acid, acetic acid, trifluoroacetic acid and methanesulfonic acid, etc. as well as a Lewis acid.
- a base may for example be an alkaline metal hydroxide such as sodium hydroxide and potassium hydroxide, etc., an alkaline metal hydrogen carbonate such as sodium hydrogen carbonate and potassium hydrogen carbonate, etc., an alkaline metal carbonate such as sodium carbonate and potassium carbonate, etc., a cesium salt such as cesium carbonate, etc., an alkaline metal hydride such as sodium hydride and potassium hydride, etc., sodium amide, an alkaline metal alkoxide such as sodium methoxide, sodium ethoxide, sodium tert-butoxide and potassium tert-butoxide, etc.
- Catalytic hydrogenation may be performed in this step.
- a catalyst include a palladium catalyst such as palladium black, palladium oxide, palladium barium sulfate, palladium on carbon, palladium hydroxide, a platinum catalyst such as platinum black, platinum oxide and platinum on carbon, or nickel catalyst such as reduced nickel, oxidized nickel, and Raney nickel.
- solvent having no adverse effect on the reaction examples include water, alcohols such as methanol and ethanol, ethers such as diethyl ether, dioxane and tetrahydrofuran, aromatic hydrocarbons such as benzene, toluene and xylene, esters such as ethyl acetate, halogenated hydrocarbons such as chloroform and dichloromethane, nitriles such as acetonitrile, amides such as i ⁇ 7,I ⁇ J-dimethylformamide, N,W-dimethylacetamide and 1- methyl-2-pyrrolidinone, ketones such as acetone and 2- butanone and sulfoxides such as dimethylsulfoxide. These solvents may be used by mixing at an appropriate ratio, or may not be used.
- reaction temperature may vary depending on compound (XVIII) or a salt thereof employed as well as other reaction conditions, it is 0 to 200 0 C, preferably 20 to 150 °C.
- the reaction time is 5 minutes to 48 hours, preferably 5 minutes to 24 hours.
- the thus obtained compounds can be isolated and purified by the known isolating and purifying methods, for example, concentration, concentration under reduced pressure, extraction with solvent, crystallization, recrystallization, transfer dissolution and chromatography.
- a starting compound for compound (I) according to the invention may be in a form of a salt, including a salt with an inorganic acid (for example, hydrochloric acid, phosphoric acid, hydrobromic acid and sulfuric acid, etc.) and a salt with an organic acid (for example, acetic acid, formic acid, propionic acid, fumaric acid, maleic acid, succinic acid, tartaric acid, citric acid, malic acid, oxalic acid, benzoic acid, methanesulfonic acid and benzenesulfonic acid, etc.).
- an inorganic acid for example, hydrochloric acid, phosphoric acid, hydrobromic acid and sulfuric acid, etc.
- an organic acid for example, acetic acid, formic acid, propionic acid, fumaric acid, maleic acid, succinic acid, tartaric acid, citric acid, malic acid, oxalic acid, benzoic acid, methanesulfonic acid and benzenes
- a salt with an inorganic base for example, an alkaline metal or an alkaline earth metal such as sodium, potassium, calcium and magnesium, ammonia, etc.
- an organic base for example, tri-Ci_ 3 alkylamine such as triethylamine, etc.
- a starting compound when carries as a substituent an amino group, an amide group, a hydrozino group, a urea group, a carboxyl group or a hydroxyl group, then such group may be derivatized with a protective group employed ordinarily in peptide chemistry, which is cleaved after a reaction if desired to yield an intended compound.
- a protective group for an amino group, an amide group and a urea group may for example be an optionally substituted Ci_ 6 alkylcarbonyl (for example. formyl, methylcarbonyl and ethylcarbonyl, etc.), phenylcarbonyl, a Ci- 6 alkyloxycarbonyl (for example, methoxycarbonyl, ethoxycarbonyl and tert-butoxycarbonyl, etc.), phenyloxycarbonyl (for example, benzoxycarbonyl) , C 7 -I 0 aralkylcarbonyl (for example, benzyloxycarbonyl) , C 7 -I 0 aralkyl (for example, benzyl and 4-methoxybenzyl, etc.), trityl, phthaloyl, etc.
- Ci_ 6 alkylcarbonyl for example. formyl, methylcarbonyl and ethylcarbonyl, etc.
- a substituent on each of the groups listed above may be a halogen atom (for example, fluorine, chlorine, bromine and iodine, etc.), a C ⁇ - 6 alkylcarbonyl (for example, methylcarbonyl, ethylcarbonyl and butylcarbonyl, etc.) and a nitro group, which may occur 1 to about 3 times .
- a halogen atom for example, fluorine, chlorine, bromine and iodine, etc.
- a C ⁇ - 6 alkylcarbonyl for example, methylcarbonyl, ethylcarbonyl and butylcarbonyl, etc.
- a nitro group which may occur 1 to about 3 times .
- a protective group for a carboxyl group may for example be an optionally substituted Ci_ 6 alkyl (for example, methyl, ethyl, n-propyl, i-propyl, n-butyl and t-butyl, etc.), phenyl, trityl and silyl, etc.
- a substituent on each of the groups listed above may be a halogen atom (for example, fluorine, chlorine, bromine and iodine, etc.), a Ci_ 6 alkylcarbonyl (for example, formyl, methylcarbonyl, ethylcarbonyl and butylcarbonyl, etc.) and a nitro group, which may occur 1 to about 3 times.
- a protective group for a hydroxyl group may for example be an optionally substituted Ci- 6 alkyl (for example, methyl, ethyl, n-propyl, i-propyl, n-butyl and tert-butyl, etc.), phenyl, a C 7 - I0 aralkyl (for example, benzyl, etc.), a Ci_6 alkylcarbonyl (for example, formyl, methylcarbonyl and ethylcarbonyl, etc.), phenyloxycarbonyl (for example, benzoxycarbonyl, etc.), C 7 - I0 aralkylcarbonyl (for example, benzyloxycarbonyl, etc.), pyranyl, furanyl, silyl, etc.
- Ci- 6 alkyl for example, methyl, ethyl, n-propyl, i-propyl, n-butyl and tert-butyl, etc
- a substituent on each of the groups listed above may be a halogen atom (for example, fluorine, chlorine, bromine and iodine, etc.), a Ci_ 6 alkyl, phenyl, a C 7 -io aralkyl, nitro, etc., which may occur 1 to about 4 times.
- a method for cleaving a protective group is a method known per se or an analogous method, such as a treatment for example with an acid, a base, a reduction, UV light, hydrazine, phenylhydrazine, sodium N-methyldithiocarbamate, tetrabutylammonium fluoride, palladium acetate, etc.
- the pharmaceutical composition containing compound (I) or (I 1 ) of the present invention is expected to be useful in the treatment and prevention of diseases, in which CRF is involved, such as depression, major depression, bipolar depression, dysthymia, seasonal affective disorder, recurrent depression, postpartum depression, suppression symptom, mania, anxiety, generalized anxiety disorder, anxiety syndrome, panic disorder, phobia, social phobia, obsessive-compulsive disorder, posttraumatic stress disorder, stress-induced insomnia, post psychic trauma stress disorder, Tourette' s syndrome, autism, passion disorder, adjustment disorder, dysthymic disorder, sleep disorder, insomnia, bipolar disorder, circulatory disease, neurosis, schizophrenia, digestive ulcer, irritable bowl syndrome, inflammatory bowel disease, ulcerative colitis, Crohn's disease, stress-induced gastrointestinal disorder, nervous emesis, peptic ulcer, diarrhea, constipation, postoperative ileus, gastrointestine dysfunction and nervous vomiting associated with stress, Alzheimer's disease, Alzheimer's type senile dementia, nervous degenerated disease such
- Compound (I) or (I 1 ) of the present invention can be formulated with a pharmaceutically acceptable carrier and can be orally or parenterally administered as solid formulations such as tablets, capsules, granules, powders, or the like; or liquid formulations such as syrups, injections, or the like. Also, there can be prepared formulations for transdermal administration such as patchings, cataplasms, ointments (including creams) , plasters, tapes, lotions, liquids and solutions, suspensions, emulsions, sprays, and the like.
- a variety of organic or inorganic carrier substances which have been conventionally employed as formulation materials, is used and compounded as a bulking agent, a lubricant, a binding agent, and a disintegrator in solid formulations; a vehicle, a solubilizing agent, a suspending agent, an isotonicity agent, a buffering agent, and an analgesic in liquid formulations.
- formulation excipients such as a preservative, an antioxidant, a stabilizer, a coloring agent, a sweetening agent, and the like can be used.
- Preferred examples of the bulking agent include lactose, sucrose, D-mannitol, starch, crystalline cellulose, light anhydrous silicic acid, and the like.
- Preferred examples of the lubricant include magnesium stearate, potassium stearate, talc, colloidal silica, and the like.
- Preferred examples of the binding agent include crystalline cellulose, ⁇ -starch, sucrose, D-mannitol, dextrin, hydroxypropyl cellulose, hydroxypropylmethyl cellulose, polyvinyl pyrrolidone, and the like.
- Preferred examples of the disintegrator include starch, carboxymethyl cellulose, calcium carboxymethyl cellulose, croscarmellose sodium, sodium carboxymethyl starch, low-substituted hydroxypropyl cellulose, and the like.
- Preferred examples of the vehicle include water for injection, alcohol, propylene glycol, macrogol, sesame oil, corn oil, and the like. If necessary, for the purpose of taste masking, enteric coating, or prolonged action, oral formulations can be prepared by coating by a per se known method.
- this coating agent examples include hydroxypropylmethyl cellulose, ethyl cellulose, hydroxymethyl cellulose, hydroxypropyl cellulose, polyoxyethylene glycol, Tween 80, Pluronic F68 [polyoxyethylene (160) polyoxypropylene (30) glycol], cellulose acetate phthalate, hydroxypropylmethyl cellulose phthalate, hydroxymethyl cellulose acetate phthalate, Eudragit (manufactured by Rohm Company, methacrylic acid- acrylic acid copolymer), and the like.
- Preferred examples of the solubilizing agent include polyethylene glycol, propylene glycol, benzyl benzoate, ethanol, trisamiomethane, cholesterol, triethanolamine, sodium carbonate, sodium citrate, and the like.
- Preferred examples of the suspending agent include surface active agents such as stearyltriethanolamine, sodium lauryl sulfate, laurylaminopropionic acid, lecithin, benzalkonium chloride, benzethonium chloride, glycerin monostearate, and the like; hydrophilic, high molecular substances such as polyvinyl alcohol, polyvinyl pyrrolidone, sodium carboxymethyl cellulose, methyl cellulose, hydroxymethyl cellulose, hydroxyethyl cellulose, hydroxypropyl cellulose, and the like; and so on.
- Preferred examples of the isotonicity agent include sodium chloride, glycerin, D- mannitol, and the like.
- Preferred examples of the buffering agent include buffer solutions of a phosphate, an acetate, a carbonate, a citrate, or the like.
- Preferable examples of the analgesic include benzyl alcohol and the like.
- Preferred examples of the preservative include paraoxybenzoic acid esters, chlorobutanol, benzyl alcohol, phenethyl alcohol, dehydroacetic acid, sorbic acid, and the like.
- Preferred examples of the antioxidant include sulfites, ascorbic acid, and the like.
- the aqueous mixture was extracted with 50 mL of diethyl ether and twice with 50 mL of dichloromethane. The organics were combined, dried over sodium sulfate, filtered, and concentrated in vacuo to a residue. This residue was dissolved in 50 mL of ethanol and 10 mL of 6N aqueous hydrochloric acid was added. The mixture was heated at 75 0 C for 2 h and then concentrated in vacuo. The resulting residue was dissolved in dichloromethane and washed with aqueous sodium bicarbonate.
- Examples 16-54 in Table 4 were prepared as trifluoroacetic acid salt and examples 55-60 were prepared as free base in the similar method described in Example 15. Table 4
- the mixture was alkalified by 12 N sodium hydroxide, followed by addition of ethyl acetate to the suspension. After addition of 24.6 mL of di-tert-butyl dicarbonate (107 mmol), the mixture was stirred at room temperature for 15 hours. The aqueous layer was separated and extracted with ethyl acetate (Xl) . The organic layer was washed with brine (Xl) , dried over sodium sulfate and concentrated in vacuo. The residual solids were washed with hexane to give 8.53 g of the Boc derivative of the title compound as yellow crystals.
- 2-chloro-7- (2, 5- dimethyl-li ⁇ -pyrrol-1-yl) -1-methyl-lH-benzimidazole and 225 mg (1.04 mmol) of 4-bromo-2-methoxy-6-methylaniline was stirred at 110 0 C for 15 hours. After cooling, the reaction mixture was neutralized by saturated aqueous sodium hydrogen carbonate and extracted with ethyl acetate (X2) .
- Example 70 and 71 in Table 7 were prepared in the similar method described in Example 15. Table 7
- This compound was prepared in a similar manner described in Example 66.
- reaction was allowed to cool to room temperature and concentrated in vacuo. The residue was dissolved in ethyl acetate, washed with aqueous sodium bicarbonate and brine.
- N- (4-Bromo-2-methoxy-6-methylphenyl) -4-chloro-7- (1- ethylpropyl) -1-methyl-lH-benzimidazol-2-amine A mixture of 2, 4-dichloro-7- (1-ethylpropyl) -1-methyl- lff-benzimidazole (140 mg, 0.52 iranol) , 4-bromo-2-methoxy-6- methylaniline (335 mg, 1.55 mmol) and l-methyl-2- pyrrolidone (5 drops) was stirred at 130 0 C for 2 days under nitrogen atmosphere.
- Examples 78 - 96 were prepared in the similar method described in Example 77.
- Examples 98 - 99 were prepared in the similar method described in Example 97.
- N- (4-Chloro-2-methoxy- ⁇ -methylphenyl) -7- (1-ethylpropyl) - 1, 4-dimethyl-lH-benzimidazol-2-amine A mixture of 2-chloro-7- (1-ethylpropyl) -1, 4-dimethyl- lH-benzimidazole (100 mg, 0.399 mmol), 4 ⁇ chloro-2-methoxy- 6-r ⁇ ethylaniline (205 mg, 1.20 mmol) and l-methyl-2- pyrrolidone (0.2 mL) was stirred at 120 0 C for 2 days under nitrogen atmosphere. The mixture was diluted with water and extracted with ethyl acetate, washed with brine.
- the reaction mixture was diluted with saturated aqueous sodium hydrogen carbonate and extracted with dichloromethane (X2) .
- the combined organic layer was washed with water (Xl) and brine (Xl), dried over sodium sulfate and concentrated in vacuo.
- the residue was purified by silica gel column chromatography eluting with a 10-50% ethyl acetate/hexane gradient mixture to give 3.83 g (11.3 mmol, 65.0%) of the title compound as a colorless solid.
- Example 109 was prepared in the similar method described in Example 108. mp. 166-168 0 C.
- Example 110-124 were prepared in the similar method described in Example 77.
- Example 124-144 were prepared in the similar method described in Example 97.
- Example 140 4-Chloro-2- [2, 6-dichloro-4- (trifluoromethoxy) phenoxy] -7- (1- ethylpropyl) -1-methyl-lH-benzimidazole
- Example 146 was prepared in the similar method described in Example 145. mp 106-107 0 C.
- Example 151 was ⁇ prepared in the similar method described in Example 150. rap 192-192 0 C.
Abstract
Description






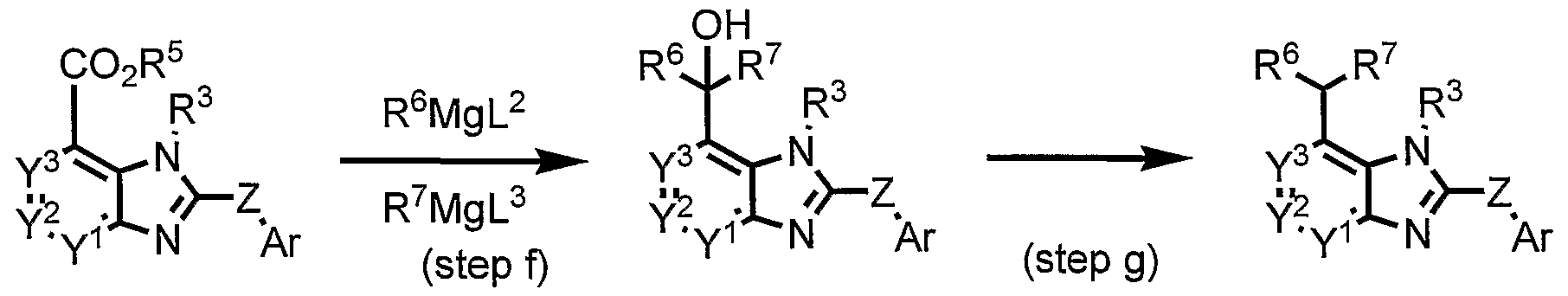






























































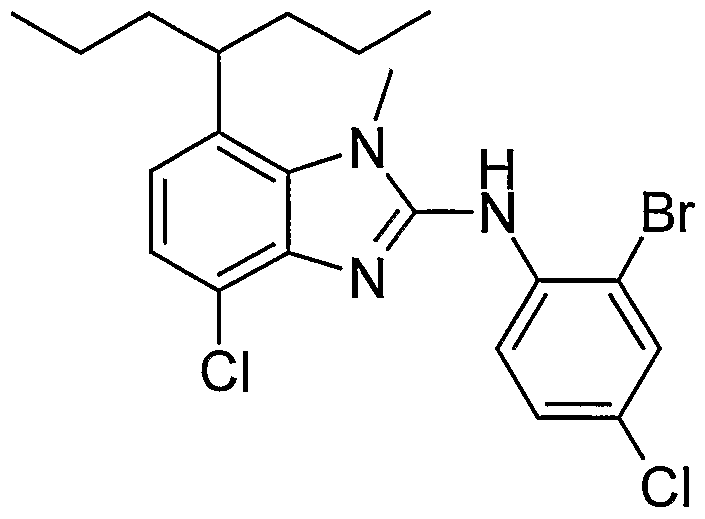





















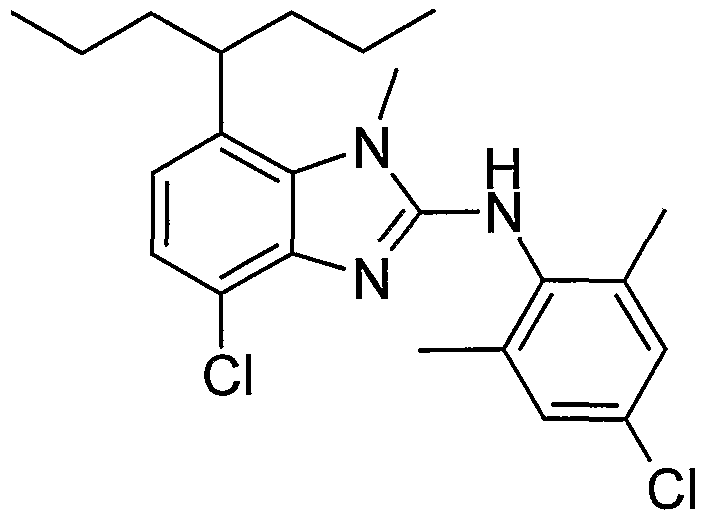



















































Claims



Priority Applications (12)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002605453A CA2605453A1 (en) | 2005-04-27 | 2006-04-26 | Novel benzimidazole compounds having crf antagonistic activity and pharmaceutical compositions containing them |
| MX2007013154A MX2007013154A (en) | 2005-04-27 | 2006-04-26 | Fused heterocyclic compounds. |
| CN2006800147137A CN101166729B (en) | 2005-04-27 | 2006-04-26 | Fused heterocyclic compounds |
| EP06751379A EP1874736A4 (en) | 2005-04-27 | 2006-04-26 | Fused heterocyclic compounds |
| KR1020077024873A KR101368037B1 (en) | 2005-04-27 | 2006-04-26 | Fused heterocyclic compounds |
| US11/919,435 US8163935B2 (en) | 2005-04-27 | 2006-04-26 | Fused heterocyclic compounds |
| BRPI0610150-0A BRPI0610150A2 (en) | 2005-04-27 | 2006-04-26 | compound, prodrug, process for producing the compound, pharmaceutical composition, crf receptor antagonist, and use of crf receptor antagonist |
| JP2008509045A JP5061097B2 (en) | 2005-04-27 | 2006-04-26 | Fused heterocyclic compounds |
| AU2006241210A AU2006241210B2 (en) | 2005-04-27 | 2006-04-26 | Fused heterocyclic compounds |
| NZ562235A NZ562235A (en) | 2005-04-27 | 2006-04-26 | Benzimidazole and pyridoimidazole derivatives as CRF receptor antagonists, for treating affective disorder, depression, or anxiety |
| IL186407A IL186407A (en) | 2005-04-27 | 2007-10-07 | Benzimidazole and imidazo[4,5-c]pyridine derivatives as crf receptor antagonists, production thereof, pharmaceutical compositions comprising them and use thereof for manufacturing medicaments |
| NO20075936A NO20075936L (en) | 2005-04-27 | 2007-11-19 | Fused heterocyclic compounds |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US67511305P | 2005-04-27 | 2005-04-27 | |
| US60/675,113 | 2005-04-27 | ||
| US74210105P | 2005-12-02 | 2005-12-02 | |
| US60/742,101 | 2005-12-02 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| WO2006116412A2 true WO2006116412A2 (en) | 2006-11-02 |
| WO2006116412A8 WO2006116412A8 (en) | 2007-03-01 |
| WO2006116412A3 WO2006116412A3 (en) | 2007-06-28 |
Family
ID=37215421
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2006/015646 WO2006116412A2 (en) | 2005-04-27 | 2006-04-26 | Fused heterocyclic compounds |
Country Status (19)
| Country | Link |
|---|---|
| US (1) | US8163935B2 (en) |
| EP (1) | EP1874736A4 (en) |
| JP (1) | JP5061097B2 (en) |
| KR (1) | KR101368037B1 (en) |
| AR (1) | AR053859A1 (en) |
| AU (1) | AU2006241210B2 (en) |
| BR (1) | BRPI0610150A2 (en) |
| CA (1) | CA2605453A1 (en) |
| CR (1) | CR9421A (en) |
| GE (1) | GEP20104962B (en) |
| IL (1) | IL186407A (en) |
| MA (1) | MA29486B1 (en) |
| MX (1) | MX2007013154A (en) |
| NO (1) | NO20075936L (en) |
| NZ (1) | NZ562235A (en) |
| PE (1) | PE20061377A1 (en) |
| RU (1) | RU2408586C2 (en) |
| TW (1) | TWI370820B (en) |
| WO (1) | WO2006116412A2 (en) |
Cited By (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008051533A2 (en) | 2006-10-25 | 2008-05-02 | Takeda Pharmaceutical Company Limited | Benzimidazole compounds |
| WO2008082003A1 (en) * | 2006-12-29 | 2008-07-10 | Takeda Pharmaceutical Company Limited | Fused heterocyclic compounds having crf antagonistic activity |
| WO2009093747A1 (en) * | 2008-01-22 | 2009-07-30 | Takeda Pharmaceutical Company Limited | Tricyclic compounds having corticotropin-releasing factor antagonistic activity and pharmaceutical compositions containing them |
| WO2010018874A1 (en) * | 2008-08-12 | 2010-02-18 | Takeda Pharmaceutical Company Limited | Amide compound |
| WO2010032461A1 (en) * | 2008-09-17 | 2010-03-25 | 武田薬品工業株式会社 | Nitrogen-containing fused ring compound |
| US7812013B2 (en) | 2005-06-14 | 2010-10-12 | Schering Corporation | Macrocyclic heterocyclic aspartyl protease inhibitors |
| WO2011078360A1 (en) | 2009-12-24 | 2011-06-30 | 武田薬品工業株式会社 | Amide compound |
| US8329914B2 (en) | 2008-10-31 | 2012-12-11 | Merck Sharp & Dohme Corp | Cyclic benzimidazole derivatives useful as anti-diabetic agents |
| WO2013024898A1 (en) | 2011-08-18 | 2013-02-21 | 日本新薬株式会社 | Heterocyclic derivative and pharmaceutical drug |
| US8394969B2 (en) | 2008-09-26 | 2013-03-12 | Merck Sharp & Dohme Corp. | Cyclic benzimidazole derivatives useful as anti-diabetic agents |
| US8410284B2 (en) | 2008-10-22 | 2013-04-02 | Merck Sharp & Dohme Corp | Cyclic benzimidazole derivatives useful as anti-diabetic agents |
| US8563746B2 (en) | 2008-10-29 | 2013-10-22 | Merck Sharp & Dohme Corp | Cyclic benzimidazole derivatives useful as anti-diabetic agents |
| US8895596B2 (en) | 2010-02-25 | 2014-11-25 | Merck Sharp & Dohme Corp | Cyclic benzimidazole derivatives useful as anti-diabetic agents |
| US8987250B2 (en) | 2012-04-20 | 2015-03-24 | Gilead Sciences, Inc. | Therapeutic compounds |
| US9006229B2 (en) | 2011-04-21 | 2015-04-14 | Gilead Sciences, Inc. | Benzothiazole compounds and their pharmaceutical use |
| WO2015112642A1 (en) | 2014-01-21 | 2015-07-30 | Neurocrine Biosciences, Inc. | Crf1 receptor antagonists for the treatment of congenital adrenal hyperplasia |
| US9102614B2 (en) | 2010-07-02 | 2015-08-11 | Gilead Sciences, Inc. | Naphth-2-ylacetic acid derivatives to treat AIDS |
| US9284323B2 (en) | 2012-01-04 | 2016-03-15 | Gilead Sciences, Inc. | Naphthalene acetic acid derivatives against HIV infection |
| US9296758B2 (en) | 2010-07-02 | 2016-03-29 | Gilead Sciences, Inc. | 2-quinolinyl-acetic acid derivatives as HIV antiviral compounds |
| US9376392B2 (en) | 2012-01-04 | 2016-06-28 | Gilead Sciences, Inc. | 2-(tert-butoxy)-2-(7-methylquinolin-6-yl) acetic acid derivatives for treating AIDS |
| WO2016174183A1 (en) | 2015-04-30 | 2016-11-03 | Bayer Pharma Aktiengesellschaft | Combinations of inhibitors of irak4 with inhibitors of btk |
| EP3195865A1 (en) | 2016-01-25 | 2017-07-26 | Bayer Pharma Aktiengesellschaft | Combinations of inhibitors of irak4 with inhibitors of btk |
| EP3219329A1 (en) | 2016-03-17 | 2017-09-20 | Bayer Pharma Aktiengesellschaft | Combinations of copanlisib |
| WO2017207386A1 (en) | 2016-06-01 | 2017-12-07 | Bayer Pharma Aktiengesellschaft | Use of 2-substituted indazoles for the treatment and prophylaxis of autoimmune diseases |
| WO2017207481A1 (en) | 2016-06-01 | 2017-12-07 | Bayer Animal Health Gmbh | Substituted indazoles ueful for treatment and prevention of allergic and/or inflammatory diseases in animals |
| WO2018095953A1 (en) | 2016-11-23 | 2018-05-31 | Bayer Cropscience Aktiengesellschaft | 2-[3-(alkylsulfonyl)-2h-indazol-2-yl]-3h-imidazo[4,5-b]pyridine derivatives and similar compounds as pesticides |
| US10308634B2 (en) | 2014-11-26 | 2019-06-04 | Bayer Pharma Aktiengesellschaft | Substituted indazoles, methods for the production thereof, pharmaceutical preparations that contain said new substituted indazoles, and use of said new substituted indazoles to produce drugs |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017155052A1 (en) * | 2016-03-09 | 2017-09-14 | 日本曹達株式会社 | Pyridine compound and use thereof |
Family Cites Families (43)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4373497B2 (en) | 1996-06-19 | 2009-11-25 | ローン−プーラン・ロレ・リミテツド | Substituted azabicyclo compounds and their use as inhibitors of TNF and cyclic AMP phosphodiesterase production |
| AU3899097A (en) * | 1996-08-05 | 1998-02-25 | Rhone-Poulenc Rorer Pharmaceuticals Inc. | Substituted aromatic compounds |
| US6124463A (en) | 1998-07-02 | 2000-09-26 | Dupont Pharmaceuticals | Benzimidazoles as corticotropin release factor antagonists |
| US6365589B1 (en) | 1998-07-02 | 2002-04-02 | Bristol-Myers Squibb Pharma Company | Imidazo-pyridines, -pyridazines, and -triazines as corticotropin releasing factor antagonists |
| US6376664B1 (en) * | 1999-03-17 | 2002-04-23 | The Ohio State University | Cyclic bis-benzimidazole ligands and metal complexes thereof |
| MXPA01012283A (en) | 1999-06-23 | 2002-07-30 | Aventis Pharma Gmbh | Substituted benzimidazole. |
| EA004939B1 (en) | 1999-06-28 | 2004-10-28 | Янссен Фармацевтика Н.В. | Respiratory syncytial virus replication inhibitors |
| AU7314200A (en) * | 1999-09-17 | 2001-04-24 | Yamanouchi Pharmaceutical Co., Ltd. | Benzimidazole derivatives |
| WO2001021160A2 (en) | 1999-09-23 | 2001-03-29 | Axxima Pharmaceuticals Aktiengesellschaft | Carboxymide and aniline derivatives as selective inhibitors of pathogens |
| US6506769B2 (en) | 1999-10-06 | 2003-01-14 | Boehringer Ingelheim Pharmaceuticals, Inc. | Heterocyclic compounds useful as inhibitors of tyrosine kinases |
| JP5036112B2 (en) | 1999-10-06 | 2012-09-26 | ベーリンガー インゲルハイム ファーマシューティカルズ インコーポレイテッド | Heterocyclic compounds useful as inhibitors of tyrosine kinases |
| KR100526487B1 (en) | 2000-06-21 | 2005-11-08 | 에프. 호프만-라 로슈 아게 | Benzothiazole derivatives |
| JP5000068B2 (en) | 2000-08-11 | 2012-08-15 | ベーリンガー インゲルハイム ファーマシューティカルズ インコーポレイテッド | Heterocyclic compounds useful as inhibitors of tyrosine kinases |
| DE10060292A1 (en) | 2000-12-05 | 2002-06-20 | Aventis Pharma Gmbh | Use of substituted benzimidazoles for the manufacture of a medicament for the treatment of diseases which can be influenced by inhibition of the Na + / H + exchanger and medicament containing them |
| US20020146469A1 (en) | 2000-12-20 | 2002-10-10 | Benjamin Wiegand | Methods for reducing chronic stress in mammals |
| CN1531530A (en) * | 2001-02-05 | 2004-09-22 | 罗奇维生素股份公司 | Novel 2-benzoxazolyl benzene derivatives and their use as UV screening agents |
| US7030150B2 (en) | 2001-05-11 | 2006-04-18 | Trimeris, Inc. | Benzimidazole compounds and antiviral uses thereof |
| US6734179B2 (en) | 2001-12-12 | 2004-05-11 | Hoffmann-La Roche Inc. | Benzothiazoles |
| GB0203045D0 (en) | 2002-02-08 | 2002-03-27 | Johnson & Johnson Consumer | Method of afefecting sleep and sleep-related behaviours |
| PL402389A1 (en) | 2002-03-29 | 2013-04-02 | Novartis Ag | Method for inhibiting Raf kinase activity in humans or inanimals |
| US8299108B2 (en) | 2002-03-29 | 2012-10-30 | Novartis Ag | Substituted benzazoles and methods of their use as inhibitors of raf kinase |
| US7049333B2 (en) | 2002-06-04 | 2006-05-23 | Sanofi-Aventis Deutschland Gmbh | Substituted thiophenes: compositions, processes of making, and uses in disease treatment and diagnosis |
| JP2006505570A (en) * | 2002-10-17 | 2006-02-16 | アムジエン・インコーポレーテツド | Benzimidazole derivatives and their use as vanilloid receptor ligands |
| WO2004043913A2 (en) | 2002-11-08 | 2004-05-27 | Trimeris, Inc. | Hetero-substituted benzimidazole compounds and antiviral uses thereof |
| CA2524352A1 (en) | 2003-05-09 | 2004-11-18 | Pharmacia & Upjohn Company Llc | Substituted pyrimidine derivatives |
| US20040242560A1 (en) | 2003-05-22 | 2004-12-02 | Aventis Pharma Deutschland Gmbh | Process for synthesizing heterocyclic compounds |
| JP2007521296A (en) | 2003-07-01 | 2007-08-02 | メルク エンド カムパニー インコーポレーテッド | Ophthalmic composition for the treatment of high intraocular pressure |
| US20050042212A1 (en) | 2003-07-24 | 2005-02-24 | Nanda Steven A. | Method of reducing CRF receptor mRNA |
| DE10337942A1 (en) | 2003-08-18 | 2005-03-17 | Merck Patent Gmbh | aminobenzimidazole derivatives |
| DE10349587A1 (en) | 2003-10-24 | 2005-05-25 | Merck Patent Gmbh | Benzimidazolylderivate |
| JP4842829B2 (en) * | 2003-10-31 | 2011-12-21 | 武田薬品工業株式会社 | Nitrogen-containing fused heterocyclic compounds |
| AR046959A1 (en) | 2003-12-18 | 2006-01-04 | Tibotec Pharm Ltd | MORFOLINILO CONTAINING BENCIMIDAZOLS AS INHIBITORS OF THE REPLICATION OF RESPIRATORY SYNTHETIC VIRUSES |
| ES2326813T3 (en) | 2003-12-18 | 2009-10-20 | Tibotec Pharmaceuticals Ltd. | DERIVATIVES OF AMINO-BENCIMIDAZOLES AS INHIBITORS OF THE REPLICATION OF THE SYNTHETIC RESPIRATORY VIRUS. |
| ATE501135T1 (en) | 2003-12-18 | 2011-03-15 | Tibotec Pharm Ltd | PIPERIDINAMINO-BENZIMIDAZOLE DERIVATIVES AL RESPIRATORY SYNCYTIALVIRUS REPLICATION INHIBITORS |
| AU2004312001B2 (en) | 2003-12-19 | 2009-08-27 | Merck & Co., Inc. | Cyclic guanidines, compositions containing such compounds and methods of use |
| US7470712B2 (en) | 2004-01-21 | 2008-12-30 | Bristol-Myers Squibb Company | Amino-benzazoles as P2Y1 receptor inhibitors |
| WO2005077936A1 (en) | 2004-02-11 | 2005-08-25 | Ulkar Kimya Sanayii Ve Ticaret A.S. | Pyridine benzimidazole sulfoxides with high purity |
| US7429608B2 (en) | 2005-01-20 | 2008-09-30 | Amgen Inc. | Benzo[d]imidazol analogs as vanilloid receptor ligands and their use in treatments |
| CA2600570C (en) | 2005-03-14 | 2011-12-06 | Transtech Pharma, Inc. | Benzazole derivatives, compositions, and methods of use as .beta.-secretase inhibitors |
| WO2008013270A1 (en) | 2006-07-28 | 2008-01-31 | Ono Pharmaceutical Co., Ltd. | Method for diagnosis of neuropsychiatric disease |
| WO2008082003A1 (en) * | 2006-12-29 | 2008-07-10 | Takeda Pharmaceutical Company Limited | Fused heterocyclic compounds having crf antagonistic activity |
| MX2010008039A (en) * | 2008-01-22 | 2010-08-10 | Takeda Pharmaceutical | Tricyclic compounds having corticotropin-releasing factor antagonistic activity and pharmaceutical compositions containing them. |
| UY32045A (en) * | 2008-08-12 | 2010-03-26 | Takeda Pharmaceutical | AMIDA COMPOUND |
-
2006
- 2006-04-21 TW TW095114290A patent/TWI370820B/en not_active IP Right Cessation
- 2006-04-26 EP EP06751379A patent/EP1874736A4/en not_active Withdrawn
- 2006-04-26 WO PCT/US2006/015646 patent/WO2006116412A2/en active Application Filing
- 2006-04-26 CA CA002605453A patent/CA2605453A1/en not_active Abandoned
- 2006-04-26 MX MX2007013154A patent/MX2007013154A/en active IP Right Grant
- 2006-04-26 AR ARP060101661A patent/AR053859A1/en unknown
- 2006-04-26 GE GEAP200610339A patent/GEP20104962B/en unknown
- 2006-04-26 JP JP2008509045A patent/JP5061097B2/en not_active Expired - Fee Related
- 2006-04-26 KR KR1020077024873A patent/KR101368037B1/en not_active IP Right Cessation
- 2006-04-26 NZ NZ562235A patent/NZ562235A/en not_active IP Right Cessation
- 2006-04-26 US US11/919,435 patent/US8163935B2/en not_active Expired - Fee Related
- 2006-04-26 RU RU2007143966/04A patent/RU2408586C2/en not_active IP Right Cessation
- 2006-04-26 AU AU2006241210A patent/AU2006241210B2/en not_active Ceased
- 2006-04-26 BR BRPI0610150-0A patent/BRPI0610150A2/en not_active IP Right Cessation
- 2006-04-26 PE PE2006000437A patent/PE20061377A1/en not_active Application Discontinuation
-
2007
- 2007-10-07 IL IL186407A patent/IL186407A/en not_active IP Right Cessation
- 2007-10-08 CR CR9421A patent/CR9421A/en unknown
- 2007-11-19 NO NO20075936A patent/NO20075936L/en not_active Application Discontinuation
- 2007-11-26 MA MA30420A patent/MA29486B1/en unknown
Non-Patent Citations (1)
| Title |
|---|
| None |
Cited By (43)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8557798B2 (en) | 2005-06-14 | 2013-10-15 | Merck Sharp & Dohme Corp. | Macrocyclic heterocyclic aspartyl protease inhibitors |
| US7812013B2 (en) | 2005-06-14 | 2010-10-12 | Schering Corporation | Macrocyclic heterocyclic aspartyl protease inhibitors |
| EP2088861A2 (en) * | 2006-10-25 | 2009-08-19 | Takeda Pharmaceutical Company Limited | Benzimidazole compounds |
| EP2088861A4 (en) * | 2006-10-25 | 2010-07-07 | Takeda Pharmaceutical | Benzimidazole compounds |
| WO2008051533A2 (en) | 2006-10-25 | 2008-05-02 | Takeda Pharmaceutical Company Limited | Benzimidazole compounds |
| WO2008082003A1 (en) * | 2006-12-29 | 2008-07-10 | Takeda Pharmaceutical Company Limited | Fused heterocyclic compounds having crf antagonistic activity |
| JP2010514672A (en) * | 2006-12-29 | 2010-05-06 | 武田薬品工業株式会社 | Fused heterocyclic compounds having CRF antagonist activity |
| US8039500B2 (en) | 2006-12-29 | 2011-10-18 | Takeda Pharmaceutical Company Limited | Fused heterocyclic compounds |
| US8901141B2 (en) | 2008-01-22 | 2014-12-02 | Takeda Pharmaceutical Company, Limited | Tricyclic compounds having corticotropin-releasing factor antagonistic activity and pharmaceutical compositions containing them |
| WO2009093747A1 (en) * | 2008-01-22 | 2009-07-30 | Takeda Pharmaceutical Company Limited | Tricyclic compounds having corticotropin-releasing factor antagonistic activity and pharmaceutical compositions containing them |
| US8785460B2 (en) | 2008-01-22 | 2014-07-22 | Takeda Pharmaceutical Company Limited | Tricyclic compounds and use thereof |
| JP2011509923A (en) * | 2008-01-22 | 2011-03-31 | 武田薬品工業株式会社 | Tricyclic compounds having antagonism of corticotropin releasing factor and pharmaceutical compositions containing them |
| WO2010018874A1 (en) * | 2008-08-12 | 2010-02-18 | Takeda Pharmaceutical Company Limited | Amide compound |
| WO2010032461A1 (en) * | 2008-09-17 | 2010-03-25 | 武田薬品工業株式会社 | Nitrogen-containing fused ring compound |
| US8394969B2 (en) | 2008-09-26 | 2013-03-12 | Merck Sharp & Dohme Corp. | Cyclic benzimidazole derivatives useful as anti-diabetic agents |
| US8410284B2 (en) | 2008-10-22 | 2013-04-02 | Merck Sharp & Dohme Corp | Cyclic benzimidazole derivatives useful as anti-diabetic agents |
| US8563746B2 (en) | 2008-10-29 | 2013-10-22 | Merck Sharp & Dohme Corp | Cyclic benzimidazole derivatives useful as anti-diabetic agents |
| US8329914B2 (en) | 2008-10-31 | 2012-12-11 | Merck Sharp & Dohme Corp | Cyclic benzimidazole derivatives useful as anti-diabetic agents |
| WO2011078360A1 (en) | 2009-12-24 | 2011-06-30 | 武田薬品工業株式会社 | Amide compound |
| US8895596B2 (en) | 2010-02-25 | 2014-11-25 | Merck Sharp & Dohme Corp | Cyclic benzimidazole derivatives useful as anti-diabetic agents |
| US9296758B2 (en) | 2010-07-02 | 2016-03-29 | Gilead Sciences, Inc. | 2-quinolinyl-acetic acid derivatives as HIV antiviral compounds |
| US9102614B2 (en) | 2010-07-02 | 2015-08-11 | Gilead Sciences, Inc. | Naphth-2-ylacetic acid derivatives to treat AIDS |
| US9006229B2 (en) | 2011-04-21 | 2015-04-14 | Gilead Sciences, Inc. | Benzothiazole compounds and their pharmaceutical use |
| WO2013024898A1 (en) | 2011-08-18 | 2013-02-21 | 日本新薬株式会社 | Heterocyclic derivative and pharmaceutical drug |
| US9284323B2 (en) | 2012-01-04 | 2016-03-15 | Gilead Sciences, Inc. | Naphthalene acetic acid derivatives against HIV infection |
| US9376392B2 (en) | 2012-01-04 | 2016-06-28 | Gilead Sciences, Inc. | 2-(tert-butoxy)-2-(7-methylquinolin-6-yl) acetic acid derivatives for treating AIDS |
| US9096586B2 (en) | 2012-04-20 | 2015-08-04 | Gilead Sciences, Inc. | Therapeutic compounds |
| US8987250B2 (en) | 2012-04-20 | 2015-03-24 | Gilead Sciences, Inc. | Therapeutic compounds |
| WO2015112642A1 (en) | 2014-01-21 | 2015-07-30 | Neurocrine Biosciences, Inc. | Crf1 receptor antagonists for the treatment of congenital adrenal hyperplasia |
| US11730739B2 (en) | 2014-01-21 | 2023-08-22 | Neurocrine Biosciences, Inc. | Treatment of congenital adrenal hyperplasia |
| US11311544B2 (en) | 2014-01-21 | 2022-04-26 | Neurocrine Biosciences, Inc. | Treatment of congenital adrenal hyperplasia |
| US10905690B2 (en) | 2014-01-21 | 2021-02-02 | Neurocrine Biosciences, Inc. | Treatment of congenital adrenal hyperplasia |
| EP3674298A1 (en) | 2014-11-26 | 2020-07-01 | Bayer Pharma Aktiengesellschaft | Substituted indazoles, process for their preparation, pharmaceutical formulations containing them, and their use for the preparation of medicaments |
| US10308634B2 (en) | 2014-11-26 | 2019-06-04 | Bayer Pharma Aktiengesellschaft | Substituted indazoles, methods for the production thereof, pharmaceutical preparations that contain said new substituted indazoles, and use of said new substituted indazoles to produce drugs |
| US10793545B2 (en) | 2014-11-26 | 2020-10-06 | Bayer Pharma Aktiengesellschaft | Substituted indazoles, methods for the production thereof, pharmaceutical preparations that contain said new substituted indazoles, and use of said new substituted indazoles to produce drugs |
| EP4260909A2 (en) | 2014-11-26 | 2023-10-18 | Bayer Pharma Aktiengesellschaft | Substituted indazoles, process for their preparation, pharmaceutical preparations containing them and their use in the preparation of drugs |
| WO2016174183A1 (en) | 2015-04-30 | 2016-11-03 | Bayer Pharma Aktiengesellschaft | Combinations of inhibitors of irak4 with inhibitors of btk |
| EP3195865A1 (en) | 2016-01-25 | 2017-07-26 | Bayer Pharma Aktiengesellschaft | Combinations of inhibitors of irak4 with inhibitors of btk |
| WO2017157792A1 (en) | 2016-03-17 | 2017-09-21 | Bayer Pharma Aktiengesellschaft | Combinations of copanlisib |
| EP3219329A1 (en) | 2016-03-17 | 2017-09-20 | Bayer Pharma Aktiengesellschaft | Combinations of copanlisib |
| WO2017207386A1 (en) | 2016-06-01 | 2017-12-07 | Bayer Pharma Aktiengesellschaft | Use of 2-substituted indazoles for the treatment and prophylaxis of autoimmune diseases |
| WO2017207481A1 (en) | 2016-06-01 | 2017-12-07 | Bayer Animal Health Gmbh | Substituted indazoles ueful for treatment and prevention of allergic and/or inflammatory diseases in animals |
| WO2018095953A1 (en) | 2016-11-23 | 2018-05-31 | Bayer Cropscience Aktiengesellschaft | 2-[3-(alkylsulfonyl)-2h-indazol-2-yl]-3h-imidazo[4,5-b]pyridine derivatives and similar compounds as pesticides |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1874736A4 (en) | 2008-08-20 |
| IL186407A (en) | 2014-04-30 |
| BRPI0610150A2 (en) | 2012-09-25 |
| IL186407A0 (en) | 2008-01-20 |
| RU2408586C2 (en) | 2011-01-10 |
| NO20075936L (en) | 2007-11-19 |
| TW200716639A (en) | 2007-05-01 |
| CA2605453A1 (en) | 2006-11-02 |
| GEP20104962B (en) | 2010-04-26 |
| AU2006241210B2 (en) | 2011-11-17 |
| US8163935B2 (en) | 2012-04-24 |
| KR20080000625A (en) | 2008-01-02 |
| JP2008539247A (en) | 2008-11-13 |
| WO2006116412A3 (en) | 2007-06-28 |
| EP1874736A2 (en) | 2008-01-09 |
| US20090312383A1 (en) | 2009-12-17 |
| AU2006241210A1 (en) | 2006-11-02 |
| TWI370820B (en) | 2012-08-21 |
| AR053859A1 (en) | 2007-05-23 |
| JP5061097B2 (en) | 2012-10-31 |
| PE20061377A1 (en) | 2007-01-18 |
| MX2007013154A (en) | 2008-01-21 |
| WO2006116412A8 (en) | 2007-03-01 |
| NZ562235A (en) | 2010-03-26 |
| MA29486B1 (en) | 2008-05-02 |
| KR101368037B1 (en) | 2014-02-26 |
| RU2007143966A (en) | 2009-06-10 |
| CR9421A (en) | 2007-12-12 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US8163935B2 (en) | Fused heterocyclic compounds | |
| KR102228764B1 (en) | Heterocyclic amides as kinase inhibitors | |
| CA2543707A1 (en) | Nitrogen-containing fused heterocyclic compounds | |
| EP2088861A2 (en) | Benzimidazole compounds | |
| CA3203080A1 (en) | Preparation and application method of heterocyclic compound as kras inhibitor | |
| HRP20040311A2 (en) | Spiro-hydantoin compounds useful as anti-inflammatory agents | |
| CA2873921A1 (en) | 1,3-dihydro-2h-benzimidazol-2-one derivatives substituted with benzimidazoles as respiratory syncytial virus antiviral agents | |
| KR20050084170A (en) | 4-substituted benzimidazoles and their use as inhibitors of gastric secretion | |
| KR20010049859A (en) | NEW β-CARBOLINE COMPOUNDS, A PROCESS FOR THEIR PREPARATION AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM | |
| AU2007264791A1 (en) | Derivatives of ureas of piperidine or pyrrolidine, their preparation and their therapeutical use | |
| EP0682021A1 (en) | Benzimidazoles, medicines containing them and process for their preparation | |
| JPH06145170A (en) | Heterocyclic compound, its preparation and medicinal composition for treatment of hypertension and congestive heart failure | |
| JPWO2010035745A1 (en) | Heterocyclic biaryl derivatives and PDE inhibitors containing them as active ingredients | |
| JP2011516398A (en) | Benzimidazole derivatives having heterospiro-decane residues as NPY-Y5 antagonists | |
| EP2125753A1 (en) | Fused heterocyclic compounds having crf antagonistic activity | |
| CA2100927A1 (en) | Benzimidazoles, pharmaceutical compositions containing these compounds and processes for preparing them | |
| SK279409B6 (en) | Benzimidazoles, pharmaceutical compositions containing them and process for their preparation | |
| JP2012509249A (en) | Glycogen synthase kinase-3 beta inhibitor comprising 7-hydroxy-benzimidazol-4-yl-methanone derivative | |
| CN101166729B (en) | Fused heterocyclic compounds | |
| EP2197872A1 (en) | Phenylisoquinoline and phenylquinazoline derivatives for the treatment of bone diseases | |
| SK282473B6 (en) | Benzimidazole compound, its production method, stable crystal, pharmaceutical preparation and intermediate |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200680014713.7 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 562235 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 186407 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3812/KOLNP/2007 Country of ref document: IN Ref document number: CR2007-009421 Country of ref document: CR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006241210 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 2605453 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2007/013154 Country of ref document: MX Ref document number: 2006751379 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12007502388 Country of ref document: PH |
|
| ENP | Entry into the national phase |
Ref document number: 2008509045 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10339 Country of ref document: GE Ref document number: 07113630 Country of ref document: CO Ref document number: 1020077024873 Country of ref document: KR |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2006241210 Country of ref document: AU Date of ref document: 20060426 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007143966 Country of ref document: RU Ref document number: A20071447 Country of ref document: BY |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11919435 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: PI0610150 Country of ref document: BR Kind code of ref document: A2 Effective date: 20071026 |