WO2006106052A1 - Pyrazoles - Google Patents
Pyrazoles Download PDFInfo
- Publication number
- WO2006106052A1 WO2006106052A1 PCT/EP2006/061057 EP2006061057W WO2006106052A1 WO 2006106052 A1 WO2006106052 A1 WO 2006106052A1 EP 2006061057 W EP2006061057 W EP 2006061057W WO 2006106052 A1 WO2006106052 A1 WO 2006106052A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- methyl
- pyrazol
- octahydro
- phenyl
- methanone
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/14—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing three or more hetero rings
Definitions
- the invention relates to inhibitors of ll ⁇ -hydroxysteroid dehydrogenase.
- the inhibitors include, for example, pyrazoles and derivatives thereof and are useful for the treatment of diseases such as type II diabetes mellitus and metabolic syndrome.
- the compounds according to the present invention can be characterized by formula I:
- R 1 or R 2 is hydrogen or alkyl and the other is lower alkyl or (CH 2 ) P Y, wherein Y is a substituted or unsubstituted, saturated, partially unsaturated, or unsaturated mono-, bi- or tri-cyclic 5-10 membered cycloalkyl ring and p is 0 or 1, and wherein substituents on Y are lower alkyl, lower alkoxy, hydroxy, hydroxy-alkyl, alkyl-phenyl, phenyl-alkyl, pyridine or halogen,
- R 1 and R 2 together with the N atom to which they are attached, form a substituted or unsubstituted ring Z, wherein Z is a 5- to 7-membered monocyclic or 7- to 10-membered bicyclic saturated, partially unsaturated or unsaturated substituted or unsubstituted heterocyclic ring which contains the N atom to which R 1 and R 2 are attached, and optionally an- other hetero atom which is selected from N, O and S, wherein the substituted heterocyclic ring is mono- or di- substituted with lower alkyl, hydroxy, hydroxy-alkyl, alkyl-phenyl, phenyl-alkyl, pyridine or halogen;
- R 3 is an aromatic ring system selected from the group consisting of [2,2']bithiophenyl, 1- methyl-indole, 2,3-dihydro-benzo[l,4]dioxin, benzo[l,3]dioxole, benzo[
- R 3 is:
- Ar is a carbocyclic or heterocyclic aryl group which may be unsubstituted or substituted with one or more groups selected from the group consisting of halogen, lower alkyl, lower alkoxy, trifluoromethyl, cyano and nitro; and
- R 4 is lower alkyl
- Diabetes mellitus is a serious illness that affects an increasing number of people across the world. Its incidence is increasing along with the increasing trend to obesity in many countries. The serious consequences of the disease include increased risk of stroke, heart dis- ease, kidney damage, blindness, and amputation. Diabetes is characterized by decreased insulin secretion and/or an impaired ability of peripheral tissues to respond to insulin, resulting in increased plasma glucose levels. There are two forms of diabetes: insulin- dependent and non- insulin-dependent, with the great majority of diabetics suffering from the non- insulin-dependent form of the disease, known as type 2 diabetes or non-insulin- dependent diabetes mellitus (NIDDM). Because of the serious consequences, there is an urgent need to control diabetes.
- NIDDM non-insulin- dependent diabetes mellitus
- NIDDM NIDDM-induced diabetes fibrosis .
- Treatment of NIDDM generally starts with weight loss, a healthy diet and an exercise program. These factors are especially important in addressing the increased cardiovascular risks associated with diabetes, but they are generally ineffective in controlling the disease itself.
- drug treatments available including insulin, metformin, sul- fonylureas, acarbose, and thiazolidinediones.
- each of these treatments has disadvantages, and there is an ongoing need for new drugs to treat diabetes.
- Metformin is an effective agent that reduces fasting plasma glucose levels and enhances the insulin sensitivity of peripheral tissue. Metformin has a number of effects in vivo, in- eluding an increase in the synthesis of glycogen, the polymeric form in which glucose is stored [R. A. De Fronzo Drugs 1999, 58 Suppl. 1, 29]. Metformin also has beneficial effects on lipid profile, with favorable results on cardiovascular health — treatment with metformin leads to reductions in the levels of LDL cholesterol and triglycerides [S. E. Inzucchi JAMA 2002, 287, 360]. However, over a period of years, metformin loses its effectiveness [R. C. Turner et al. JAMA 1999, 281, 2005] and there is consequently a need for new treatments for diabetes.
- Thiazolidinediones are activators of the nuclear receptor peroxisome-proliferator activated receptor-gamma. They are effective in reducing blood glucose levels, and their efficacy has been attributed primarily to decreasing insulin resistance in skeletal muscle [M. Tadayyon and S. A. Smith Expert Opin. Investig. Drugs 2003, 12, 307].
- One disadvantage associated with the use of thiazolidinediones is weight gain.
- Sulfonylureas bind to the sulfonylurea receptor on pancreatic beta cells, stimulate insulin secretion, and consequently reduce blood glucose levels. Weight gain is also associated with the use of sulfonylureas [S. E. Inzucchi JAMA 2002, 287, 360] and, like metformin, efficacy decreases over time [R. C. Turner et al. JAMA 1999, 281, 2005].
- a further problem often encountered in patients treated with sulfonylureas is hypoglycemia [M. Salas and J. J. Caro Adv. Drug React. Tox. Rev. 2002, 21, 205-217].
- Acarbose is an inhibitor of the enzyme alpha-glucosidase, which breaks down disaccha- rides and complex carbohydrates in the intestine. It has lower efficacy than metformin or the sulfonylureas, and it causes intestinal discomfort and diarrhea which often lead to the discontinuation of its use [S. E. Inzucchi JAMA 2002, 287, 360] - A -
- the metabolic syndrome is a condition where patients exhibit more than two of the following symptoms: obesity, hypertriglyceridemia, low levels of HDL-cholesterol, high blood pressure, and elevated fasting glucose levels.
- This syndrome is often a precursor of type 2 diabetes, and has high prevalence in the United States with an estimated prevalence of 24% (E. S. Ford et al. JAMA 2002, 287, 356).
- a therapeutic agent that ameliorates the metabolic syndrome would be useful in potentially slowing or stopping the progression to type 2 diabetes.
- glucose is produced by two different processes: gluconeogenesis, where new glucose is generated in a series of enzymatic reactions from pyruvate, and glycolysis, where glucose is generated by the breakdown of the polymer glycogen.
- PEPCK phosphoenolpyruvate car- boxykinase
- G6Pase glucose-6-phosphatase
- both PEPCK and G6Pase are upregulated, allowing the rate of gluconeogenesis to increase.
- the levels of these enzymes are controlled in part by the corticosteroid hormones (Cortisol in human and corticosterone in mouse).
- corticosteroid hormones Cortisol in human and corticosterone in mouse.
- corticosteroid receptor When the corticosteroid binds to the corticosteroid receptor, a signaling cascade is triggered which results in the upregulation of these enzymes.
- corticosteroid hormones are found in the body along with their oxidized 11-dehydro counterparts (cortisone and 11-dehydrocorticosterone in human and mouse, respectively), which do not have activity at the glucocorticoid receptor.
- the actions of the hormone depend on the local concentration in the tissue where the corticosteroid receptors are expressed. This local concentration can differ from the circulating levels of the hormone in plasma, because of the actions of redox enzymes in the tissues.
- the enzymes that modify the oxidation state of the hormones are 1 lbeta-hydroxysteroid dehydrogenases forms I and II.
- Form I (ll ⁇ -HSDl) is responsible for the reduction of cortisone to Cortisol in vivo, while form II (ll ⁇ -HSD2) is responsible for the oxidation of Cortisol to cortisone.
- the enzymes have low homology and are expressed in different tissues.
- ll ⁇ -HSDl is highly expressed in a number of tissues including liver, adipose tissue, and brain, while ll ⁇ -HSD2 is highly expressed in mineralocorticoid target tissues, such as kidney and colon, ll ⁇ -
- HSD2 prevents the binding of Cortisol to the mineralocorticoid receptor, and defects in this enzyme have been found to be associated with the syndrome of apparent mineralocorticoid excess (AME).
- mice demonstrate that modulation of the activity of ll ⁇ -HSDl could have beneficial therapeutic effects in diabetes and in the metabolic syndrome.
- the ll ⁇ -HSDl gene is knocked out in mice, fasting does not lead to the normal increase in levels of G6Pase and PEPCK, and the animals are not susceptible to stress- or obesity-related hyperglycemia.
- knockout animals which are rendered obese on a high-fat diet have significantly lower fasting glucose levels than weight- matched controls (Y. Kotolevtsev et al. Proc. Natl. Acad. ScL USA 1997, 94, 14924).
- mice have also been found to have improved lipid profile, insulin sensitiv- ity, and glucose tolerance (N. M. Morton et al. J. Biol. Chem. 2001, 276, 41293).
- the effect of overexpressing the ll ⁇ -HSDl gene in mice has also been studied.
- These transgenic mice displayed increased ll ⁇ -HSDl activity in adipose tissue and exhibited visceral obesity which is associated with the metabolic syndrome. Levels of the corticosterone were increased in adipose tissue, but not in serum, and the mice had increased levels of obesity, especially when on a high-fat diet. Mice fed on low-fat diets were hyperglycemic and hy- perinsulinemic, and also showed glucose intolerance and insulin resistance (H. Masuzaki et al. Science, 2001, 294, 2166).
- carbe- noxolone was found to lead to an increase in whole body insulin sensitivity, and this increase was attributed to a decrease in hepatic glucose production (B. R. Walker et al. J. Clin. Endocrinol. Metab. 1995, 80, 3155).
- carbenoxolone was found to improve cognitive function in healthy elderly men and also in type 2 diabetics (T. C. Sandeep et al. Proc. Natl. Acad. Sci USA 2004, 101, 6734).
- a number of non-specific inhibitors of ll ⁇ -HSDl and ll ⁇ -HSD2 have been identified, including glycyrrhetinic acid, abietic acid, and carbenoxolone.
- a number of selective inhibitors of ll ⁇ -HSDl have been found, including chenodeoxycholic acid, fla- vanone and 2'-hydroxyflavanone (S. Diederich et al. Eur. J. Endocrinol. 2000, 142, 200 and R. A. S. Schweizer et al. MoI. Cell. Endocrinol. 2003, 212, 41).
- WO 2004089470, WO 2004089416 and WO 2004089415 (Novo Nordisk A/S); and WO 0190090, WO 0190091, WO 0190092, WO 0190093, WO 03043999, WO 0190094, WO 03044000, WO 03044009, and WO 2004103980 (Biovitrum AB) disclose compounds as inhibitors of ll ⁇ -HSDl. These compounds are different in structure from the compounds of the current invention.
- WO 2004112781 and WO 2004112782 disclose the method of use of some of these compounds for the promotion of wound healing.
- WO 03065983, WO 03075660, WO 03104208, WO 03104207, US20040133011, WO 2004058741, WO2005016877 and WO 2004106294 disclose com- pounds as inhibitors of ll ⁇ -HSDl. These compounds are different in structure from the compounds of the current invention.
- US2004122033 discloses the combination of an appetite suppressant with inhibitors of ll ⁇ -HSDl for the treatment of obesity, and obesity-related disorders.
- WO 2004065351 discloses compounds as inhibitors of ll ⁇ -HSDl. These compounds are different in structure from the compounds of the current invention.
- WO 2004089415 (Novo Nordisk A/S) discloses the use of an inhibitor of ll ⁇ -HSDl in combination with an agonist of the glucocorticoid receptor for the treatment of diseases including cancer and diseases involving inflammation.
- ll ⁇ - HSDl inhibitors include amino-ketones, benzimidazoles, carboxamides, 2,3-dihydrobenzofuran-7-carboxamides, indoles, methylenedioxyphenyl-carboxamides, oxazole-4-carboxamides, oxazole-5-carboxamides pyrazolo[l,5-a]pyrimidines, pyrazole-4- carboxamides, thiazole-4-carboxamides, thiazole-5-carboxamides, and 1,2,4-triazoles.
- WO 2004089416 (Novo Nordisk A/S) discloses the use of an inhibitor of ll ⁇ -HSDl in combination with an antihypertensive agent for the treatment of diseases including insulin resistance, dyslipidemia and obesity.
- WO 2004089470 (Novo Nordisk A/S) discloses substituted amides as inhibitors of ll ⁇ -HSDl .
- WO 2004089471 (Novo Nordisk A/S) discloses pyrazolo[l,5-a]pyrimidines as inhibitors of ll ⁇ -HSDl.
- WO 2004089896 (Novo Nordisk A/S) discloses compounds as inhibitors of ll ⁇ -HSDl. These compounds are different in structure from the compounds of the current invention.
- WO 2004011410, WO 2004033427, and WO 2004041264 (AstraZeneca UK Limited) dis- close compounds as inhibitors of ll ⁇ -HSDl. These compounds are different in structure from the compounds of the current invention.
- WO 02076435A2 (The University of Edinburgh) claims the use of an agent which lowers levels of ll ⁇ -HSDl in the manufacture of a composition for the promotion of an athero- protective lipid profile.
- Agents mentioned as inhibitors of ll ⁇ -HSDl include carbe- noxolone, 11-oxoprogesterone, 3 ⁇ ,17,21-trihydroxy-5 ⁇ -pregnan-3-one, 21 -hydro xy-pregn- 4-ene-3,ll,20-trione, androst-4-ene-3,ll,20-trione and 3 ⁇ -hydroxyandrost-5-en-17-one. None of these compounds is similar in structure to the compounds of the current invention.
- WO 03059267 (Rhode Island Hospital) claims a method for treating a glucocorticoid- associated state by the administration of a ll ⁇ -HSDl inhibitor such as 11-keto testosterone, 11-keto-androsterone, 11-keto-pregnenolone, 11-keto-dehydro-epiandrostenedione, 3 ⁇ ,5 ⁇ - reduced-11-ketoprogesterone, 3 ⁇ ,5 ⁇ -reduced-l 1-keto testosterone, 3 ⁇ ,5 ⁇ -reduced-l 1-keto- androstenedione, or 3 ⁇ ,5 ⁇ -tetrahydro-ll ⁇ -dehydro-corticosterone. None of these com- pounds is similar in structure to the compounds of the current invention.
- WO 2001070671 discloses compounds as insecticides. These compounds are different in structure from the compounds of the current invention.
- EP 360701 Rhone-Poulenc Agrochimie discloses compounds as agrochemical fungicides.
- aryl is used to mean a mono- or polycyclic aromatic ring system, in which the rings may be carbocyclic or may contain one or more atoms selected from O, S, and N.
- aryl groups are phenyl, pyridyl, benzimidazolyl, benzofu- ranyl, benzothiazolyl, benzothiophenyl, cinnolinyl, furyl, imidazo[4,5-c]pyridinyl, imida- zolyl, indolyl, isoquinolinyl, isoxazolyl, naphthyl, [l,7]naphthyridinyl, oxadiazolyl, oxa- zolyl, phthalazinyl, purinyl, pyidazinyl, pyrazolyl, pyrido[2,3-d]pyrimidinyl, pyrimidinyl, pyrimidimide,
- alkyl means, for example, a branched or unbranched, cyclic or acyclic, saturated or unsaturated (e.g. alkenyl or alkynyl) hydrocarbyl radical which may be substituted or unsubstituted.
- the alkyl group is preferably C 3 to C 12 , more preferably C 5 to C 1 O, more preferably C 5 to C 7 .
- the alkyl group is preferably C 1 to C 1 O, more preferably C 1 to C 6 , more preferably methyl, ethyl, propyl (n-propyl or isopropyl), butyl (n- butyl, isobutyl or tertiary-butyl) or pentyl (including n-pentyl and isopentyl), more preferably methyl.
- alkyl as used herein includes alkyl (branched or unbranched), substituted alkyl (branched or un- branched), alkenyl (branched or unbranched), substituted alkenyl (branched or un- branched), alkynyl (branched or unbranched), substituted alkynyl (branched or unbranched), cycloalkyl, substituted cycloalkyl, cycloalkenyl, substituted cycloalkenyl, cycloalkynyl and substituted cycloalkynyl. Saturated acyclic, branched or unbranches groups are preferred alkyl groups.
- lower alkyl means, for example, a branched or unbranched, cyclic or acyclic, saturated or unsaturated (e.g. alkenyl or alkynyl) hydrocarbyl radical wherein said cyclic lower alkyl group is C 5 , C 6 or C 7 , and wherein said acyclic lower alkyl group is C 1 , C 2 , C 3 or C 4 , and is preferably selected from methyl, ethyl, propyl (n-propyl or isopropyl) or butyl (n- butyl, sec-butyl, isobutyl or tertiary- butyl).
- lower alkyl as used herein includes lower alkyl (branched or unbranched), lower alkenyl (branched or unbranched), lower alkynyl (branched or unbranched), cycloloweralkyl, cycloloweralkenyl and cycloloweralkynyl. Saturated acyclic, branched or unbranches groups are preferred lower alkyl groups.
- alkyl and aryl groups may be substituted or unsubstituted. Where substituted, there will generally be, for example, 1 to 3 substituents present, preferably 1 substituent.
- Sub- stituents may include, for example: carbon-containing groups such as alkyl, aryl, arylalkyl (e.g. substituted and unsubstituted phenyl, substituted and unsubstituted benzyl); halogen atoms and halogen-containing groups such as haloalkyl (e.g. trifluoro methyl); oxygen- containing groups such as alcohols (e.g. hydroxyl, hydroxyalkyl, aryl(hydroxyl) alkyl), ethers (e.g.
- aminocarbonyl mono- or di-alkylaminocarbonyl, aminocarbonylalkyl, mono-or di- alkylaminocarbonylalkyl, arylaminocarbonyl
- carbamates e.g. alkoxycarbonylamino, ar- loxycarbonylamino, aminocarbonyloxy, mono-or di-alkylaminocarbonyloxy, arylamino- carbonyloxy
- ureas e.g. mono- or di- alkylaminocarbonylamino or arylaminocarbon- ylamino
- nitrogen-containing groups such as amines (e.g.
- the lower alkyl groups may be substituted or unsubstituted, preferably unsubstituted. Where substituted, there will generally be, for example, 1 to 3 substitutents present, preferably 1 substituent.
- alkoxy means, for example, alkyl-O- and "alkoyl” means, for example, alkyl-CO-.
- Alkoxy substituent groups or alkoxy-containing substituent groups may be substituted by, for example, one or more alkyl groups.
- lower alkoxy means, for example, lower alkyl-O-.
- halogen means, for example, a fluorine, chlorine, bromine or iodine radical, preferably a fluorine, chlorine or bromine radical, and more preferably a fluorine or chlorine radical.
- salts may be prepared from pharmaceutically acceptable non-toxic acids and bases including inorganic and organic acids and bases.
- acids include, for example, acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethenesulfonic, dichloroacetic, formic, fumaric, gluconic, glutamic, hippuric, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, nitric, oxalic, pamoic, pantothenic, phosphoric, succinic, sulfuric, tartaric, oxalic, p- toluenesulfonic and the like.
- Acceptable base salts include alkali metal (e.g. sodium, potassium), alkaline earth metal (e.g. calcium, magnesium) and aluminum salts.
- alkali metal e.g. sodium, potassium
- alkaline earth metal e.g. calcium, magnesium
- aluminum salts e.g. aluminum salts.
- R 1 or R 2 is hydrogen or alkyl and the other is lower alkyl or (CH 2 ) P Y, wherein Y is a substituted or unsubstituted, saturated, partially unsaturated, or unsaturated mono-, bi- or tri-cyclic 5-10 membered cycloalkyl ring and p is 0 or 1, and wherein substituents on Y are lower alkyl, lower alkoxy, hydroxy, hydroxy-alkyl, alkyl-phenyl, phenyl-alkyl, pyridine or halogen,
- R 1 and R 2 together with the N atom to which they are attached, form a substituted or unsubstituted ring Z, wherein Z is a 5- to 7-membered monocyclic or 7- to 10-membered bicyclic saturated, partially unsaturated or unsaturated substituted or unsubstituted heterocyclic ring which contains the N atom to which R 1 and R 2 are attached, and optionally another hetero atom which is selected from N, O and S, wherein the substituted heterocyclic ring is mono- or di- substituted with lower alkyl, hydroxy, hydroxy-alkyl, alkyl-phenyl, phenyl-alkyl, pyridine or halogen;
- R 3 is an aromatic ring system selected from the group consisting of [2,2']bithiophenyl, 1- methyl- indole, 2,3-dihydro-benzo[l,4]dioxin, benzo[l,3]dioxole, benzothiophene, diben- zofuran, furane, naphthalene, phenyl, biphenyl, quinoline, thianthrene and thiophene, wherein said aromatic ring may be unsubstituted or substituted with one or more amino, cyano, formyl, halo, hydroxy, hydroxymethyl, lower-acyl, lower-acyl-amino, lower-alkoxy, lower-alkoxy-carbonyl, 2-(lower-alkoxy-carbonyl)-ethenyl, lower-alkyl, lower-alkyl-thio, nitro, trifluoromethoxy or trifluoromethyl, wherein said pheny
- R 3 is:
- Ar is a carbocyclic or heterocyclic aryl group which may be unsubstituted or substituted with one or more groups selected from the group consisting of halogen, lower alkyl, lower alkoxy, trifluoro methyl, cyano and nitro; and
- R 4 is lower alkyl
- Preferred compounds of formula I as defined above are those, wherein R 1 is hydrogen and R 2 is a substituted 6-8 membered cycloalkyl ring. Saturated monocyclic cycloalkyl rings are preferred in this context.
- Other preferred compounds as defined above are those wherein R 2 is l,7,7-trimethyl-bicyclo[2.2.1]hept-2-yl, 2,6,6-trimethyl-bicyclo[3.1.1]hept-3- yl , 3-noradamantyl, adamantan-1-yl, adamantan-1-yl- methyl, adamantan-2-yl, 1,2,3,4- tetrahydronaphthyl, cyclohexyl, cyclooctyl, or cycoheptyl.
- Z is selected from the group consisting of 2- ethyl-piperidine, 3-phenyl-pyrrolidine, 3-(pyridin-3-yl)-pyrrolidine, 4-chloro-decahydro- quinoline, 4a-bromo-decahydro-isoquinoline, 6-bromo-octahydro-isoquinoline, 3- cyclohexyl-piperidine, 3-benzyl-piperidine, decahydro-quinoline and decahydro- isoquinoline.
- R 3 is substituted or unsubstituted benzothiophene or phenyl. More preferably, R 3 is substituted with one or more halogen, lower-alkoxy or lower-alkyl.
- Preferred compounds of formula I as defined above are those selected from the group consisting of
- the compounds of formula (I) can have one or more asymmetric C atoms and can therefore exist as an enantiomeric mixture, diastereomeric mixture or as optically pure compounds.
- the compounds of general formula (I) in this invention may be derivatised at functional groups to provide derivatives which are capable of conversion back to the parent compound in vivo.
- novel compounds of the present invention have been found to inhibit ll ⁇ -hydroxysteroid dehydrogenase. They can therefore be used in the treatment and prophylaxis of diseases which are modulated by ll ⁇ -hydroxysteroid dehydrogenase inhibitors, preferably a metabolic disorder. Such diseases include type II diabetes, obesity and metabolic syndrome.
- the invention therefore also relates to pharmaceutical compositions comprising a compound as defined above and a pharmaceutically acceptable carrier and/or adjuvant.
- the invention likewise embraces compounds as described above for use as therapeutically active substances, especially as therapeutically active substances for the treatment and/or prophylaxis of diseases which are modulated by 11 ⁇ -hydroxysteroid dehydrogenase inhibitors, particularly as therapeutically active substances for the treatment and/or prophylaxis of type II diabetes or metabolic syndrome.
- the invention relates to a method for the therapeutic and/or prophylactic treatment of diseases which are modulated by ll ⁇ -hydroxysteroid de- hydro genase inhibitors, particularly for the therapeutic and/or prophylactic treatment of type II diabetesobesity or metabolic syndrome, which method comprises administering a compound as defined above to a human being or animal.
- said therapeutically effective amount is about 10 to about 1000 mg per day.
- the invention also embraces the use of compounds as defined above for the therapeutic and/or prophylactic treatment of diseases which are modulated by 11 ⁇ -hydroxysteroid dehydrogenase inhibitors, particularly for the therapeutic and/or prophylactic treatment of type II diabetes, obesity or metabolic syndrome.
- the invention also relates to the use of compounds as described above for the preparation of medicaments for the therapeutic and/or prophylactic treatment of diseases which are modulated by 11 ⁇ -hydroxysteroid dehydrogenase inhibitors, particularly for the therapeutic and/or prophylactic treatment of type II diabetes, obesity or metabolic syndrome.
- medicaments comprise a compound as described above.
- Type II diabetes is particularly preferred.
- the present invention refers to a process for the preparation of a compound as defined above, which process comprises reacting a compound of formula II
- Another embodiment of the present invention refers to compounds as defined above, when prepared by a process as defined above.
- the compounds according to the present invention can be prepared according to the following general synthetic methods given below, by the methods given in the specific examples, or in analogy thereto.
- a ⁇ -keto-ester of formula 2 is converted to a compound of formula 3 where X represents dialkylamino (such as dimethylamino) or lower-alkoxy (such as ethoxy) and then the compound of formula 3 is reacted with a hydrazine to give the compound of formula 4.
- the ester protective group in the compound of formula 2 is then cleaved and the resulting carboxylic acid is coupled with an amine of formula HNR 1 R 2 to give the desired compound of formula 1.
- the reaction of a compound of formula 2 to give a compound of formula 3 can be carried out using conditions that are well known in the art.
- the com- pound of formula 3 can be prepared by treating a compound of formula 2 with N,N- dimethylformamide dimethyl acetal in an inert solvent such as an aromatic hydrocarbon (for example, toluene) at a temperature between about 50 0 C and about 100 0 C.
- an inert solvent such as an aromatic hydrocarbon (for example, toluene)
- Examples of conditions for this reaction can be found in the literature, for example, in H. H. Wasser- mann et al. Tetrahedron Lett. 1984, 25, 3743-3746, in S. Gelin et al. Synthesis 1983, 566- 568, and in J. Svete et al. Synthesis 1990, 70-72.
- the compound of formula 3 can be prepared by treating a compound of formula 2 with triethy- lorthoformate in the presence of acetic anhydride at the reflux temperature.
- Examples of conditions for this reaction can be found in the literature, for example, in L. Claisen Lie- bigs Ann. Chem. 1897, 297, 1-18; in L. Crombie et al. J. Chem. Soc. Perkin Trans. 1 1979, 464-471; in M. S. S. Palanki et al. J. Med. Chem. 2000, 43, 3995-4004; and in M. T. Herrero et al. Tetrahedron 2002, 58, 8581-8589.
- the reaction of the compound of formula 3 with a hydrazine can be carried out under a va- riety of conditions.
- the compound of formula 3 can be reacted with a hydrazine or the acid addition salt of a hydrazine in an inert solvent such as an alcohol (for example, ethanol).
- an acid addition salt of the hydrazine is used, then the reaction is carried out in the additional presence of a base such as a tertiary alkylamine (for example, triethylamine or diisopropylethylamine).
- a base such as a tertiary alkylamine (for example, triethylamine or diisopropylethylamine).
- the reaction is conveniently carried out at a temperature between about -20 0 C and about 80 0 C. Examples of conditions for this reaction can be found in the literature, for example, in J.
- reaction conditions that are well known in the field of organic synthesis, many of which are outlined in "Protective Groups in Organic Synthesis” [T. W. Greene and P. G. M. Wuts, 2nd Edition, John Wiley & Sons, N. Y. 1991].
- R 4 represents methyl or ethyl
- the reaction can be conveniently effected by treating the compound with one equivalent of an alkali metal hydroxide, such as potassium hydroxide, sodium hydroxide, or lithium hydroxide, preferably lithium hydroxide, in a suitable solvent, such as a mixture of tetrahydrofuran, methanol, and water.
- an alkali metal hydroxide such as potassium hydroxide, sodium hydroxide, or lithium hydroxide, preferably lithium hydroxide
- the reaction can be carried out at a temperature between about 0 0 C and about room temperature, preferably at about room temperature.
- the ester may be treated with a strong inorganic acid, for example a hydrohalic acid such as hydrogen chloride or hydro- gen bromide, or a strong organic acid, for example a halogenated alkane carboxylic acid such as trifluoro acetic acid and the like.
- the reaction is conveniently carried out in the presence of an inert organic solvent (such as dichloromethane) and at a temperature between about 0 0 C and about room temperature, preferably at about room temperature.
- the reaction may be carried out by hydrogenation in the presence of a noble metal catalyst such as palladium-on-carbon in the presence of an inert solvent (for example, an alcohol such as ethanol) at about room temperature and under atmospheric pressure.
- a noble metal catalyst such as palladium-on-carbon
- an inert solvent for example, an alcohol such as ethanol
- the coupling of a carboxylic acid of structure 4 where R 4 represents hydrogen with an amine of structure HNR 1 R 2 can be achieved using methods well known to one of ordinary skill in the art.
- the transformation can be carried out by reaction of a carboxylic acid of structure 4 where R 4 represents hydrogen or of an appropriate derivative thereof such as an activated ester, with an amine of structure HNR 1 R 2 or a corresponding acid addition salt (e.g., the hydrochloride salt) in the presence, if necessary, of a coupling agent, many examples of which are well known per se in peptide chemistry.
- the reaction is conveniently carried out by treating the carboxylic acid of structure 4 where R 4 represents hydrogen with the hydrochloride of the amine of structure HNR 1 R 2 in the presence of an appropriate base, such as diisopropylethylamine, a coupling agent such as O-(benzotriazol-l-yl)-l,l,3,3-tetramethyluronium hexafluorophosphate, and in the optional additional presence of a substance that increases the rate of the reaction, such as 1 -hydro xybenzotriazole or 1 -hydro xy-7-azabenzotriazole, in an inert solvent, such as a chlorinated hydrocarbon (e.g., dichloromethane) or N,N-dimethylformamide or N- methylpyrrolidinone, at a temperature between about 0 0 C and about room temperature, preferably at about room temperature.
- an appropriate base such as diisopropylethylamine
- a coupling agent such as
- reaction can be carried out by converting the carboxylic acid of formula 4 where R 4 represents hydrogen to an activated ester derivative, such as the N-hydroxysuccinimide ester, and subsequently reacting this with the amine of structure HNR 1 R 2 or a corresponding acid addition salt.
- This reaction sequence can be carried out by reacting the carboxylic acid of formula 4 where R 4 represents hydrogen with N-hydroxysuccinimide in the presence of a coupling agent such as N,N'-dicyclohexylcarbodiimide in an inert solvent such as tetrahydrofuran at a temperature between about 0 0 C and about room temperature.
- N-hydroxysuccinimide es- ter is then treated with the amine of structure HNR1R2 or a corresponding acid addition salt, in the presence of a base, such as an organic base (e.g., triethylamine or diisopro- pylethylamine or the like) in a suitable inert solvent such as N,N-dimethylformamide at around room temperature.
- a base such as an organic base (e.g., triethylamine or diisopro- pylethylamine or the like) in a suitable inert solvent such as N,N-dimethylformamide at around room temperature.
- the reaction sequence shown in Scheme 1 can also be carried out using solid-phase synthesis, in the case where X represents a polymer-bound amino group.
- the compound of formula 2 is treated with N-formylimidazole dimethyl acetal and a polymer-bound amine such as an aniline-functionalized cellulose derivative (for example, 4-amino-phenyl-sulfonyl-ethoxy-cellulose, which is available from Iontosorb, Usti nad La- bem, Czech Republic) in the presence of an acid catalyst such as camphor- sulfonic acid in an inert solvent, such as N,N-dimethylformamide at a temperature around 80 0 C, to give a compound of formula 3 where X represents a polymer-bound aniline.
- an aniline-functionalized cellulose derivative for example, 4-amino-phenyl-sulfonyl-ethoxy-cellulose, which is available from Iontosorb, Usti nad La- bem, Czech Republic
- the compound of formula 3 is then converted into the compound of formula 4 by treatment with a hydrazine in an inert solvent such as an alcohol (for example, isopropanol) at a temperature around the boiling point of the solvent.
- an inert solvent such as an alcohol (for example, isopropanol)
- Examples of conditions for this reaction can be found in the literature, for example, in L. De Luca et al. J. Comb. Chem. 2003, 5, 465-471.
- a pyrazole-4-carboxamide of formula 1 can be prepared according to Scheme 2, where a ⁇ -keto-amide of formula 5 is converted to a compound of formula 6 where X represents dialkylamino (such as dimethylamino) or lower-alkoxy (such as ethoxy) and then the com- pound of formula 6 reacts with a hydrazine to give the compound of formula 1.
- the reaction of a compound of formula 5 to give a compound of formula 6 can be carried out using conditions that are well known in the art.
- the compound of formula 6 can be prepared by treating a compound of formula 5 with N,N-dimethylformamide dimethyl acetal in an inert solvent such as an aromatic hydrocarbon (for example, toluene) at a temperature between about 50 0 C and about 100 0 C.
- an inert solvent such as an aromatic hydrocarbon (for example, toluene)
- Examples of conditions for this reaction can be found in the literature, for example, in R. Zupet et al. J. Heterocycl. Chem. 1991, 28, 1731-1740; in D. E. Seitz et al. Tetrahedron Lett. 1995, 36, 1413-1416; in A. V. Rama Rao et al. Tetrahedron Lett.
- the compound of formula 6 can be prepared by treating a compound of formula 5 with triethylortho formate in the presence of acetic anhydride at the reflux temperature. Examples of conditions for this reaction can be found in the literature, for example, in J. H. Dewar et al. J. Chem. Soc. 1961, 3254-3260.
- the reaction of the compound of formula 6 with a hydrazine can be carried out under a va- riety of conditions.
- the compound of formula 6 can be reacted with a hydrazine or the acid addition salt of a hydrazine in an inert solvent such as an alcohol (for example, ethanol).
- an acid addition salt of the hydrazine is used, then the reaction is carried out in the additional presence of a base such as a tertiary alkylamine (for example, triethylamine or diisopropylethylamine).
- a base such as a tertiary alkylamine (for example, triethylamine or diisopropylethylamine).
- the reaction is conveniently carried out at a temperature between about -20 0 C and about 80 0 C. Examples of conditions for this reaction can be found in the literature, for example, in A.
- a compound of formula 6 can be prepared from a compound of formula 5 by treatment with an N-(alkoxymethylene)-aniline, in the optional presence of an inert solvent such as kerosene, at elevated temperature such as between about 125 0 C and about 140 0 C.
- an inert solvent such as kerosene
- Examples of conditions for this reaction can be found in the literature, for example, in F. B. Dains Chem. Ber. 1902, 35, 2496-2500; in F. B. Dains et al. J. Am. Chem. Soc. 1909, 31, 1148-1157; in F. B. Dains et al. J. Am. Chem. Soc.
- the compound of formula 6 can then be converted to the compound of formula 1 by treatment with a hydrazine in an inert solvent such as ethanol at a temperature around the reflux temperature of the solvent.
- an inert solvent such as ethanol
- Examples of conditions for this reaction can be found in the literature, for example, in F. B. Dains et al. J. Am. Chem. Soc. 1909, 31, 1148-1157; in F. B. Dains et al. J. Am. Chem. Soc. 1916, 38, 1515; in F. B. Dains et al. J. Am. Chem. Soc. 1918, 40, 562-569; and in A. N. Borisevich et al. Ukrainskii Khimicheskii Zhurnal 1986, 52, 641-7 Chemical Abstracts AN 1987:458919.
- a l-alkyl-5-pyrrolyl-pyrazole-4-carboxylic acid derivative of formula 9 can be prepared starting from a 3-alkoxy-2-cyano-acrylic acid ester of formula 7 by reaction with a hydrazine of formula RNHNH2 to give an intermediate 5-amino-pyrazole of formula 8, which can then be reacted with 2,5-dimethoxy-tetrahydrofuran to give the 5- pyrrolyl-pyrazole of formula 9.
- This can be converted to a carboxamide of the invention by reactions analogous to those discussed above with reference to Scheme 1.
- the pyrazole-forming annulation reaction can be conveniently carried out by treating a 3-alkoxy-2- cyano-acrylic acid ester of formula 7 (such as 3-ethoxy-2-cyano-acrylic acid ethyl ester) with a hydrazine of formula RNHNH2 in an inert solvent such as ethanol at the reflux temperature.
- the subsequent annulation to form the pyrrole ring is conveniently carried out by heating the intermediate 5-amino-pyrazole with 2,5-dimethoxy-tetrahydrofuran in an organic acid such as acetic acid at a temperature of around 100 0 C.
- An example of conditions suitable for this process can be found in the literature, for example, in M. Kopp et al. J. Heterocycl.
- the carboxylate ester of formula 9 can then be hydrolyzed to the corresponding carboxylic acid and coupled with an amine of formula HNR1R2 using procedures analogous to those described above for the conversion of a carboxylate ester of formula 4 to a compound of the invention of formula 1.
- a l-alkyl-5-pyrrolyl-pyrazole-4-carboxylic acid derivative of formula 13 can be prepared starting from a 5-amino-pyrazole-4-carboxylate ester of formula 10 by diazotization of the amino group in the presence of a brominating agent such as cop- per(II) bromide.
- the reaction is conveniently carried out by treating the compound of formula 10 with an alkyl nitrite such as tert-butyl nitrite or isoamyl nitrite in an inert solvent such as a halogenated hydrocarbon (for example, carbon tetrachloride) at a temperature around 50 0 C, in the presence of a bromine source such as bromine, copper(II) bromide, dibromomethane, or bromoform.
- an alkyl nitrite such as tert-butyl nitrite or isoamyl nitrite in an inert solvent such as a halogenated hydrocarbon (for example, carbon tetrachloride) at a temperature around 50 0 C
- a bromine source such as bromine, copper(II) bromide, dibromomethane, or bromoform.
- the conversion of the ester of formula 11 to an amide of formula 12 is analogous to the conversion of a compound of formula 4 to a compound of formula 1 as discussed above, and can be carried out using similar reactions.
- the conversion of a compound of formula 12 to a compound of the invention of formula 13 can be carried out using a Suzuki reaction with an organoboron intermediate such as an aryl-boronic acid or an ester thereof, a reaction that is well known to one of average skill in the art.
- the reaction can be conveniently carried out by reacting a compound of formula 12 with an aryl-boronic acid in a convenient inert solvent such as a polar aprotic solvent (e.g., N,N- dimethylformamide) or an ether (e.g., dioxane) or water, in the presence of a catalytic amount of a palladium(O) complex (e.g., tetrakis(triphenylphosphine)palladium(0)) or a compound which can be reduced in situ to give palladium(O) (for example, palladium(II) acetate or bis(triphenylphosphine)-palladium(II) chloride), in the optional additional presence of a catalytic amount of a phosphine ligand, for example tri-o-tolylphosphine or tri- tert-butylphosphine, or alternatively in the presence of a preformed complex of palla- dium(0) with a
- the starting material of formula 10 can be made from a 3-alkoxy-2- cyano-acrylic acid ester of formula 7 by reaction with an alkyl-hydrazine by reactions analogous to those described above for the preparation of a compound of formula 8.
- Conditions appropriate for this reaction can be found in the literature, for example in F. Bon- davalli et al. J. Med. Chem. 2002, 45, 4875-4887; in S. Schenone et al. Bioorg. Med. Chem. Lett. 2001, 11, 2529-2531; in M. Kopp et al. J. Heterocycl. Chem. 2001, 38, 1045-1050; and in P. Seneci et al. Synth. Commun. 1999, 29, 311-341.
- a compound of formula 1 in which R 1 represents lower alkyl can be prepared from a compound of formula 1 in which R 1 represents hydrogen, by reaction with a strong base (such as sodium hydride) in an inert solvent (such as dimethylforma- mide) at room temperature to give the corresponding anion. This is then reacted without isolation with a lower-alkyl halide of formula RlX, again at room temperature, to give the desired compound of formula 1 in which R 1 represents lower alkyl.
- a strong base such as sodium hydride
- an inert solvent such as dimethylforma- mide
- the resulting intermediate of formula 15 is then heated with an alcohol of formula HOR 4 , either using the alcohol as solvent (for example in the case where the alcohol is methanol or ethanol), or in an inert solvent such as benzene (for example in the case where the alcohol is benzyl alcohol or tert-butyl alcohol).
- the reaction is conveniently carried out at a temperature between about 60 0 C and about 80 0 C. Condi- tions suitable for this reaction can be found in the literature, for example in Y. Oikawa et al. J. Org. Chem. 1978, 43, 2087-2088.
- ⁇ -Keto-amides of formula 5 can be prepared from the intermediate of formula 15 by treatment with a stoichiometric amount of an amine of formula HNR 1 R 2 in a suitable inert solvent such as toluene at the refluxing temperature.
- a suitable inert solvent such as toluene at the refluxing temperature.
- trans'-decahydroquinorine can be prepared by the dissolving metal reduction of A ll9 -octahydroquinoline which is in turn prepared in a multistep sequence from N-1-cyclohexenylpyrrolidine and acrylonitrile. Conditions for these reactions can be found in F. W. Vierhapper and E. L. Eliel J. Org. Chem. 1975, 40, 2734-2742 and in L. A. Cohen and B. Witkop / Am. Chem. Soc. 1955, 77, 6595-6600.
- synthesis of an amine of formula HNR 1 R 2 1 -hydro xyadamantan-
- a secondary amine can be prepared by making use of a process called reductive amination, which is well known to one of average skill in the art of organic synthesis, whereby an amine is treated with a ketone to give an imine which is reduced by one of a number of reducing agents.
- reductive amination which is well known to one of average skill in the art of organic synthesis, whereby an amine is treated with a ketone to give an imine which is reduced by one of a number of reducing agents.
- Many examples of conditions that can be used for this reaction are enumerated in "Comprehensive Organic Transformations: A Guide to Functional Group Preparations” [R. C. Larock, VCH Publishers, Inc. New York, 1989] on pages 421-423.
- the amine and ketone can be treated with a reducing agent such as tetrabutylammo- nium cyanoborohydride in an inert solvent such as a halogenated hydrocarbon (e.g., di- chloro methane) in the presence of methanolic HCl at about room temperature.
- a reducing agent such as tetrabutylammo- nium cyanoborohydride
- an inert solvent such as a halogenated hydrocarbon (e.g., di- chloro methane) in the presence of methanolic HCl at about room temperature.
- a halogenated hydrocarbon e.g., di- chloro methane
- an effective amount of any one of the compounds of this invention or a combination of any of the compounds of this invention or a pharmaceutically acceptable salt thereof is administered via any of the usual and acceptable methods known in the art, either singly or in combination.
- the compounds or compositions can thus be administered orally (e.g., buccal cavity), sublingually, parenter- ally (e.g., intramuscularly, intravenously, or subcutaneously), rectally (e.g., by suppositories or washings), transdermally (e.g., skin electroporation) or by inhalation (e.g., by aerosol), and in the form or solid, liquid or gaseous dosages, including tablets and suspensions.
- buccal cavity e.g., buccal cavity
- parenter- ally e.g., intramuscularly, intravenously, or subcutaneously
- rectally e.g., by suppositories or washings
- transdermally e.g., skin electroporation
- the administration can be conducted in a single unit dosage form with continuous therapy or in a single dose therapy ad libitum.
- the therapeutic composition can also be in the form of an oil emulsion or dispersion in conjunction with a lipophilic salt such as pamoic acid, or in the form of a biodegradable sustained-release composition for subcutaneous or intramuscular administration.
- Useful pharmaceutical carriers for the preparation of the compositions hereof can be solids, liquids or gases; thus, the compositions can take the form of tablets, pills, capsules, suppositories, powders, enterically coated or other protected formulations (e.g. binding on ion- exchange resins or packaging in lipid-protein vesicles), sustained release formulations, solutions, suspensions, elixirs, aerosols, and the like.
- the carrier can be selected from the various oils including those of petroleum, animal, vegetable or synthetic origin, e.g., peanut oil, soybean oil, mineral oil, sesame oil, and the like.
- formulations for intravenous administration comprise sterile aqueous solutions of the active ingredient(s) which are prepared by dissolving solid ac- tive ingredient(s) in water to produce an aqueous solution, and rendering the solution sterile.
- Suitable pharmaceutical excipients include starch, cellulose, talc, glucose, lactose, gelatin, malt, rice, flour, chalk, silica, magnesium stearate, sodium stearate, glycerol monostearate, sodium chloride, dried skim milk, glycerol, propylene glycol, water, ethanol, and the like.
- the compositions may be subjected to conventional pharmaceutical additives such as preservatives, stabilizing agents, wetting or emulsifying agents, salts for adjusting osmotic pressure, buffers and the like.
- Suitable pharmaceutical carriers and their formulation are described in Remington's Pharmaceutical Sciences by E. W. Martin. Such compositions will, in any event, contain an effective amount of the active compound together with a suitable carrier so as to prepare the proper dosage form for proper administration to the recipient.
- the dose of a compound of the present invention depends on a number of factors, such as, for example, the manner of administration, the age and the body weight of the subject, and the condition of the subject to be treated, and ultimately will be decided by the attending physician or veterinarian.
- Such an amount of the active compound as determined by the attending physician or veterinarian is referred to herein, and in the claims, as an "effective amount".
- the dose of a compound of the present invention is typically in the range of about 10 to about 1000 mg per day.
- LC/MS liquid chromatography/mass spectroscopy
- the simultaneous chromatographic separation was achieved with the following HPLC system: ES Industries Chromegabond WR C- 18 3u 120A (3.2 x 30mm) column cartridge; Mobile Phase A: Water (0.02% TFA) and Phase B: Acetonitrile (0.02% TFA); gradient 10% B to 90% B in 3 minutes; equilibration time of 1 minute; flow rate of 2 mL/minute.
- Step 2 (5-Bromo-l-methyl-lH-pyrazol-4-yl)-(octahydro-quinolin-l-yl)-methanone
- 5-bromo-l-methyl-lH-pyrazole-4-carboxylic acid ethyl ester (6.9 g, 29.6 mmol) in CH 3 OH (25 mL) and water (25 mL) was added LiOH (0.78 g, 32.6 mmol).
- the reaction mixture was stirred at reflux for 4 h, and then the solution was concentrated under reduced pressure to remove the methanol.
- the residue was diluted with water and the solu- tion was acidified to pH 2 with concentrated HCl ( ⁇ 3 mL).
- the resulting mixture was then extracted with ethyl acetate.
- the combined organic extracts were concentrated in vacuo to give 5-bromo-l-methyl-lH-pyrazole-4-carboxylic acid, which was used without further purification.
- reaction mixture was cooled to room temperature and then filtered through celite and a silica plug.
- the eluant was then partitioned between ethyl acetate and water and the water layer was extracted three times with ethyl acetate.
- the organic layers were combined, concentrated in vacuo and the desired product was obtained after purification by C- 18 reversed phase HPLC with a gradient of 10-100% Acetonitrile/Water.
- Example numbers 1-107 below were prepared by one of the three methods described above.
- Example 108 l-Methyl-S-phenyl-lH-pyrazole- ⁇ carboxylic acid methyl-((lR,2S,4R)- l,7,7-trimethyl-bicyclo[2.2.1]hept-2-yl)-amide
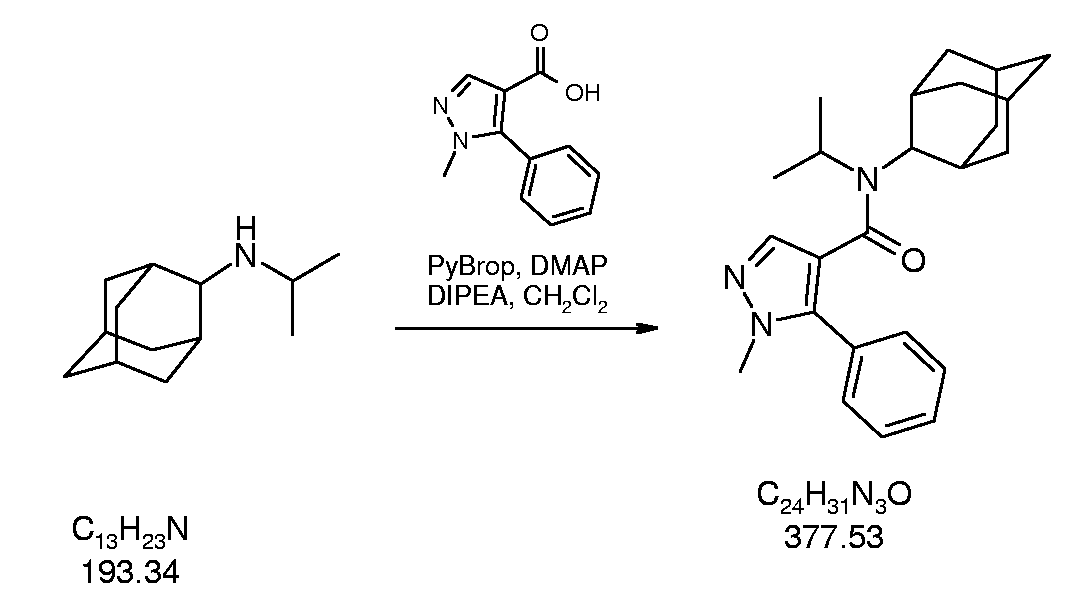
- Example 109 l-Methyl-S-phenyl-lH-pyrazole- ⁇ carboxylic acid adamantan-2-yl- isopropyl-amide
- Methyl-5-phenyl-lH-pyrazole-4-carboxylic acid adamantan-2-yl-isopropyl- amide is prepared from adamantan-2-yl-isopropyl- amine (of intermediate 3) and l-methyl-5-phenyl- lH-pyrazole-4-carboxylic acid (Maybridge pic, Cornwall, UK) according to general procedure C.
- Purified human HSDl was diluted in 50 mM Tris-HCl, 100 mM NaCl, 0.1 mg/ml BSA, 0.02% Lubrol, 20 mM MgC12, 10 mM glucose 6-phosphate, 0.4 mM NADPH, 60 U/ml glucose 6-phosphate dehydrogenase to a concentration of 1.5 ug/ml (Enzyme Solution).
- Cortisone (100 uM) in DMSO was diluted to 1 uM with 50 mM Tris-HCl, 100 mM NaCl (Substrate Solution).
- Testing compounds (40 uM) in DMSO were diluted 3 fold in series in DMSO and further diluted 20 fold in Substrate Solution.
- Enzyme Solution (10 ul/ well) was added into 384 well microtiter plates followed by diluted compound solutions (10 ul/well) and mixed well. Samples were then incubated at 37 0 C for 30 min.
- EDTA/biotin- cortisol solution (10 ul/well) in 28 mM EDTA, 100 nM biotin-cortisol, 50 mM Tris-HCl, 100 mM NaCl was then added followed by 5 ul/well of anti-cortisol antibody (3.2 ug/ml) in 50 mM Tris-HCl, 100 mM NaCl, 0.1 mg/ml BSA and the solution was incubated at 37 0 C for 30 min.
- % Inhibition 100* [l-(Fs-Fb)/(Ft-Fb)], where:
- Fs is the fluorescence signal of the sample which included the agent
- Fb is the fluorescence signal in the absence of HSDl and agent
- Ft is the fluorescence signal in the presence of HSDl, but no agent.
- the inhibitory activities of test compounds were determined by the IC 5 oS, or the concentration of compound that gave 50% inhibition.
- the compounds of the present invention preferably exhibit IC50 values below 10 ⁇ M, preferably in the range between 10 ⁇ M and 1 nM, more preferably between 2 ⁇ M and 5 nM.
- the results of the in vitro inhibition of ll ⁇ -HSDl by representative compounds of the present invention are shown in the following Table:
- Example 111 Testing of Compounds of the Invention in vivo
- the compound of the invention is formulated in 7.5% Modified Gelatin in water and is administered IP at 100 mg/kg to mice (male C57B1/6J, age -97 Days). After 30 minutes, cortisone formulated in gelatin is administered by s.c. injection at 1 mg/kg. After a further 40 minutes, blood samples are taken from the mice and are analyzed using LC-MS for the concentrations of cortisone, Cortisol, and drug.
- Percent inhibition of HSDl activity by the inhibitor is calculated by the following formula:
- C V eh is the conversion of cortisone to Cortisol when the animal is dosed with vehicle
- Film coated tablets containing the following ingredients can be manufactured in a conventional manner:
- the active ingredient is sieved and mixed with microcristalline cellulose and the mixture is granulated with a solution of polyvinylpyrrolidon in water.
- the granulate is mixed with sodium starch glycolate and magesiumstearate and compressed to yield kernels of 120 or 350 mg respectively.
- the kernels are lacquered with an aqueous solution / suspension of the above mentioned film coat.
- Capsules containing the following ingredients can be manufactured in a conventional manner:
- the components are sieved and mixed and filled into capsules of size 2.
- Injection solutions can have the following composition:
- the active ingredient is dissolved in a mixture of Polyethylene Glycol 400 and water for injection (part).
- the pH is adjusted to 5.0 by Acetic Acid.
- the volume is adjusted to 1.0 ml by addition of the residual amount of water.
- the solution is filtered, filled into vials using an appropriate overage and sterilized.
- Soft gelatin capsules containing the following ingredients can be manufactured in a conventional manner:
- the active ingredient is dissolved in a warm melting of the other ingredients and the mixture is filled into soft gelatin capsules of appropriate size.
- the filled soft gelatin capsules are treated according to the usual procedures.
- Sachets containing the following ingredients can be manufactured in a conventional manner:
- Microcristalline cellulose (AVICEL PH 102) 1400.0 mg
- Flavoring additives 1.0 mg
- the active ingredient is mixed with lactose, microcristalline cellulose and sodium carboxymethyl cellulose and granulated with a mixture of polyvinylpyrrolidon in water.
- the granulate is mixed with magnesiumstearate and the flavouring additives and filled into sachets.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Diabetes (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Endocrinology (AREA)
- Emergency Medicine (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Child & Adolescent Psychology (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description









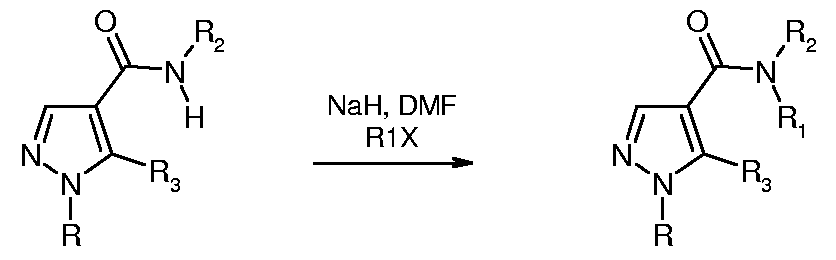




































Claims



Priority Applications (13)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AT06725326T ATE519744T1 (en) | 2005-04-05 | 2006-03-27 | 1H-PYRAZOLE-4-CARBONIC ACID AMIDES, THEIR PREPARATION AND USE AS 11-BETA-HYDROXYSTEROID DEHYDROGENASE INHIBITORS |
| EP06725326A EP1928840B1 (en) | 2005-04-05 | 2006-03-27 | 1H-Pyrazole-4-carboxamides, their preparation and their use as 11-beta-hydroxysteroid dehydrogenase inhibitors |
| CA2602781A CA2602781C (en) | 2005-04-05 | 2006-03-27 | 1h-pyrazole 4-carboxylamides, their preparation and their use as 11beta-hydroxysteroid dehydrogenase |
| AU2006232660A AU2006232660B2 (en) | 2005-04-05 | 2006-03-27 | 1H-Pyrazole 4-Carboxylamides, their preparation and their use as 11Beta-hydroxysteroid dehydrogenase |
| MX2007012212A MX2007012212A (en) | 2005-04-05 | 2006-03-27 | Pyrazoles. |
| DK06725326.0T DK1928840T3 (en) | 2005-04-05 | 2006-03-27 | 1H-pyrazole-4-carboxamides, their preparation and their use as 11-beta-hydroxysteroid dehydrogenase inhibitors |
| PL06725326T PL1928840T3 (en) | 2005-04-05 | 2006-03-27 | 1H-Pyrazole-4-carboxamides, their preparation and their use as 11-beta-hydroxysteroid dehydrogenase inhibitors |
| KR1020077022649A KR100931411B1 (en) | 2005-04-05 | 2006-03-27 | 1H-pyrazole 4-carboxyamide, process for its preparation, and use thereof as 11beta-hydroxysteroid dehydrogenase |
| BRPI0610459-2A BRPI0610459A2 (en) | 2005-04-05 | 2006-03-27 | compound, process for its preparation, pharmaceutical compositions comprising it, method for the therapeutic and / or prophylactic treatment of diseases that are modulated by hydroxysteroid-11b dehydrogenase inhibitors and use of the compound |
| CN2006800111775A CN101155783B (en) | 2005-04-05 | 2006-03-27 | Pyrazoles |
| JP2008504736A JP4880671B2 (en) | 2005-04-05 | 2006-03-27 | 1H-pyrazole 4-carboxylamido process for its preparation and its use as 11β-hydroxysteroid dehydrogenase |
| IL186118A IL186118A0 (en) | 2005-04-05 | 2007-09-20 | 1h-pyrazole 4-carboxylamides, their preparation and their use as 11beta- hydroxysteroid dehydrogenase |
| NO20074872A NO20074872L (en) | 2005-04-05 | 2007-09-25 | pyrazoles |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US66836705P | 2005-04-05 | 2005-04-05 | |
| US60/668,367 | 2005-04-05 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006106052A1 true WO2006106052A1 (en) | 2006-10-12 |
Family
ID=36514279
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2006/061057 WO2006106052A1 (en) | 2005-04-05 | 2006-03-27 | Pyrazoles |
Country Status (21)
| Country | Link |
|---|---|
| US (1) | US7345058B2 (en) |
| EP (1) | EP1928840B1 (en) |
| JP (1) | JP4880671B2 (en) |
| KR (1) | KR100931411B1 (en) |
| CN (1) | CN101155783B (en) |
| AR (1) | AR053203A1 (en) |
| AT (1) | ATE519744T1 (en) |
| AU (1) | AU2006232660B2 (en) |
| BR (1) | BRPI0610459A2 (en) |
| CA (1) | CA2602781C (en) |
| DK (1) | DK1928840T3 (en) |
| ES (1) | ES2369389T3 (en) |
| IL (1) | IL186118A0 (en) |
| MX (1) | MX2007012212A (en) |
| NO (1) | NO20074872L (en) |
| PL (1) | PL1928840T3 (en) |
| PT (1) | PT1928840E (en) |
| RU (1) | RU2381217C2 (en) |
| TW (1) | TWI309645B (en) |
| WO (1) | WO2006106052A1 (en) |
| ZA (1) | ZA200708419B (en) |
Cited By (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007058346A1 (en) * | 2005-11-21 | 2007-05-24 | Shionogi & Co., Ltd. | HETEROCYCLIC COMPOUND HAVING INHIBITORY ACTIVITY ON 11-β-HYDROXYSTEROID DEHYDROGENASE TYPE I |
| WO2007107470A2 (en) * | 2006-03-22 | 2007-09-27 | F. Hoffmann-La Roche Ag | Pyrazoles as 11-beta-hsd-1 |
| WO2007128761A2 (en) | 2006-05-04 | 2007-11-15 | Boehringer Ingelheim International Gmbh | Uses of dpp-iv inhibitors |
| WO2008120653A1 (en) * | 2007-04-02 | 2008-10-09 | Banyu Pharmaceutical Co., Ltd. | Indoledione derivative |
| WO2008142986A1 (en) | 2007-05-18 | 2008-11-27 | Shionogi & Co., Ltd. | NITROGEN-CONTAINING HETEROCYCLIC DERIVATIVE HAVING 11 β-HYDROXYSTEROID DEHYDROGENASE TYPE I INHIBITORY ACTIVITY |
| WO2009010416A2 (en) * | 2007-07-17 | 2009-01-22 | F. Hoffmann-La Roche Ag | Inhibitors of 11b-hydroxysteroid dehydrogenase |
| CN114560811A (en) * | 2022-03-11 | 2022-05-31 | 中国药科大学 | 1,3, 5-trisubstituted-pyrazole-4 carboxylic acid derivative and preparation method and application thereof |
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2659155A1 (en) * | 2006-07-20 | 2008-01-24 | Amgen Inc. | Substituted azole aromatic heterocycles as inhibitors of 11.beta.-hsd-1 |
| TW200827346A (en) * | 2006-11-03 | 2008-07-01 | Astrazeneca Ab | Chemical compounds |
| TW200836719A (en) * | 2007-02-12 | 2008-09-16 | Astrazeneca Ab | Chemical compounds |
| FR2924931B1 (en) * | 2007-12-12 | 2010-01-15 | Oreal | USES OF 1- (1H-PYRAZOL-4-Y) HETEROCYCLIC DERIVATIVES FOR STIMULATING OR INDUCING THE PUSH OF KERATIN FIBERS AND / OR BRAKING THEIR FALL; NOVEL COMPOUNDS; COMPOSITIONS CONTAINING THEM. |
| WO2009098501A1 (en) * | 2008-02-04 | 2009-08-13 | Astrazeneca Ab | Novel crystalline forms of 4- [4- (2-adamantylcarbam0yl) -5-tert-butyl-pyrazol-1-yl] benzoic acid |
| US20120041211A1 (en) * | 2009-01-30 | 2012-02-16 | Sythana Suresh Kumar | Novel process for preparing carboxy-containing pyrazoleamido compounds 597 |
| AR078887A1 (en) * | 2009-11-06 | 2011-12-07 | Boehringer Ingelheim Int | ARILO AND HETEROARILCARBONILO DERIVATIVES OF HEXAHYDROINDENOPIRIDINE AND OCTAHYDROBENZOQUINOLINE AND PHARMACEUTICAL COMPOSITIONS CONTAINING THEM. |
| CN104284898A (en) | 2012-06-20 | 2015-01-14 | 霍夫曼-拉罗奇有限公司 | Pyranopyridone inhibitors of tankyrase |
| DK3074011T3 (en) * | 2013-11-25 | 2019-09-30 | Corcept Therapeutics Inc | Octahydro-condensed azadecaline glucocorticoid receptor modulators |
| US9273254B2 (en) * | 2013-12-20 | 2016-03-01 | Ecolab Usa Inc. | Amino acetals and ketals as hydrogen sulfide and mercaptan scavengers |
| CN103772282B (en) * | 2014-02-26 | 2015-09-23 | 上海毕得医药科技有限公司 | A kind of preparation method of the 3-tertiary butyl-1H-pyrazoles-4-formaldehyde |
| US11234971B2 (en) | 2018-12-19 | 2022-02-01 | Corcept Therapeutics Incorporated | Methods of treating cancer comprising administration of a glucocorticoid receptor modulator and a cancer chemotherapy agent |
| US11389432B2 (en) | 2018-12-19 | 2022-07-19 | Corcept Therapeutics Incorporated | Methods of treating cancer comprising administration of a glucocorticoid receptor modulator and a cancer chemotherapy agent |
| AU2020226863B2 (en) | 2019-02-22 | 2023-04-06 | Corcept Therapeutics Incorporated | Therapeutic uses of relacorilant, a heteroaryl-ketone fused azadecalin glucocorticoid receptor modulator |
| UA128573C2 (en) | 2019-12-11 | 2024-08-14 | Корсепт Терапьютікс Інкорпорейтед | Methods of treating antipsychotic-induced weight gain with miricorilant |
| AU2021282256A1 (en) * | 2020-05-27 | 2022-12-15 | Corcept Therapeutics Incorporated | Concomitant administration of glucocorticoid receptor modulator relacorilant and CYP2C8 substrates |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2612155A1 (en) * | 1975-03-25 | 1976-10-14 | Byk Gulden Lomberg Chem Fab | NITROFURYL PYRAZOLE DERIVATIVES, METHOD FOR MANUFACTURING THEREOF AND MEDICINAL PRODUCTS CONTAINING THEM |
| DE3713774A1 (en) * | 1986-04-24 | 1987-10-29 | Mitsui Toatsu Chemicals | NEW PYRAZOLE DERIVATIVES, METHOD FOR THEIR PRODUCTION AND FUNGICIDES FOR AGRICULTURE AND GARDENING, WHICH CONTAIN THESE COMPOUNDS |
| EP0360701A1 (en) * | 1988-09-01 | 1990-03-28 | Rhone-Poulenc Agrochimie | Antifungal compounds containing amides and phenyl groups |
| WO2004089470A2 (en) * | 2003-04-11 | 2004-10-21 | Novo Nordisk A/S | New amide derivatives and pharmaceutical use thereof |
| WO2005016877A2 (en) * | 2003-08-07 | 2005-02-24 | Merck & Co., Inc. | Pyrazole carboxamides as inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 |
| US20050080087A1 (en) * | 2003-10-10 | 2005-04-14 | Annapurna Pendri | Pyrazole derivatives as cannabinoid receptor modulators |
Family Cites Families (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4620865A (en) * | 1983-11-07 | 1986-11-04 | Eli Lilly And Company | Herbicidal and algicidal 1,5-disubstituted-1H-pyrazole-4-carboxamides |
| JPH0768220B2 (en) * | 1986-04-24 | 1995-07-26 | 三井東圧化学株式会社 | Novel pyrazole derivative, production method thereof, and agricultural / horticultural fungicide containing them |
| MY138097A (en) | 2000-03-22 | 2009-04-30 | Du Pont | Insecticidal anthranilamides |
| SE0001899D0 (en) | 2000-05-22 | 2000-05-22 | Pharmacia & Upjohn Ab | New compounds |
| JP2002003410A (en) | 2000-06-27 | 2002-01-09 | Fuji Photo Film Co Ltd | Method for producing aromatic halogen compound |
| WO2002044133A1 (en) | 2000-11-28 | 2002-06-06 | Pfizer Products Inc. | Preparation of sodium-hydrogen exchanger type-1 inhibitors |
| GB0107383D0 (en) | 2001-03-23 | 2001-05-16 | Univ Edinburgh | Lipid profile modulation |
| WO2003044000A1 (en) | 2001-11-22 | 2003-05-30 | Biovitrum Ab | Inhibitors of 11-beta-hydroxy steroid dehydrogenase type 1 |
| CN1633428A (en) | 2001-11-22 | 2005-06-29 | 比奥维特罗姆股份公司 | Inhibitors of 11-beta-hydroxy steroid dehydrogenase type 1 |
| PL370111A1 (en) | 2001-11-22 | 2005-05-16 | Biovitrum Ab | Inhibitors of 11-beta-hydroxy steroid dehydrogenase type 1 |
| CA2469305A1 (en) | 2001-12-19 | 2003-06-26 | Frank Robert Busch | Methods for preparing sodium-hydrogen exchanger type-1 inhibitors |
| AU2002361861A1 (en) | 2001-12-21 | 2003-07-30 | Rhode Island Hospital | SELECTIVE 11Beta-HSD INHIBITORS AND METHODS FOR USE THEREOF |
| EP1474139B1 (en) | 2002-02-01 | 2007-11-21 | Merck & Co., Inc. | 11-beta-hydroxysteroid dehydrogenase 1 inhibitors useful for the treatment of diabetes, obesity and dyslipidemia |
| WO2003075660A1 (en) | 2002-03-06 | 2003-09-18 | Merck & Co., Inc. | Method of treatment or prevention of obesity |
| AR040241A1 (en) | 2002-06-10 | 2005-03-23 | Merck & Co Inc | INHIBITORS OF 11-BETA-HYDROXIESTEROID DEHYDROGRENASE 1 FOR THE TREATMENT OF DIABETES OBESITY AND DISLIPIDEMIA |
| CA2494668A1 (en) | 2002-07-27 | 2004-02-05 | Astrazeneca Ab | Chemical compounds |
| AU2003269242A1 (en) | 2002-10-11 | 2004-05-04 | Astrazeneca Ab | 1,4-disubstituted piperidine derivatives and their use as 11-betahsd1 inhibitors |
| AU2003276458A1 (en) | 2002-11-07 | 2004-06-07 | Astrazeneca Ab | 2-oxo-ethanesulfonamide derivates |
| US20040122033A1 (en) * | 2002-12-10 | 2004-06-24 | Nargund Ravi P. | Combination therapy for the treatment of obesity |
| JO2397B1 (en) * | 2002-12-20 | 2007-06-17 | ميرك شارب اند دوم كوربوريشن | Triazole Derivatives As Inhibitors Of 11-Beta -Hydroxysteriod Dehydrogenase-1 |
| TW200503994A (en) | 2003-01-24 | 2005-02-01 | Novartis Ag | Organic compounds |
| EP1615697A2 (en) | 2003-04-11 | 2006-01-18 | Novo Nordisk A/S | New pyrazolo[1,5-a] pyrimidine derivatives and pharmaceutical use thereof |
| JP2006522750A (en) | 2003-04-11 | 2006-10-05 | ノボ ノルディスク アクティーゼルスカブ | Combination therapy using 11β-hydroxysteroid dehydrogenase type 1 inhibitors and antihypertensive agents to treat metabolic syndrome and related diseases and disorders |
| WO2004089415A2 (en) | 2003-04-11 | 2004-10-21 | Novo Nordisk A/S | COMBINATIONS OF AN 11β-HYDROXYSTEROID DEHYDROGENASE TYPE 1 INHIBITOR AND A GLUCOCORTICOID RECEPTOR AGONIST |
| EP1631558A1 (en) | 2003-05-21 | 2006-03-08 | Biovitrum Ab | Inhibitors of 11-beta-hydroxy steroid dehydrogenase type i |
| CA2526712A1 (en) | 2003-05-29 | 2004-12-09 | Merck & Co., Inc. | Triazole derivatives as inhibitors of 11-beta hydroxysteroid dehydrogenase-1 |
| SE0301886D0 (en) | 2003-06-25 | 2003-06-25 | Biovitrum Ab | New use V |
| SE0301883D0 (en) | 2003-06-25 | 2003-06-25 | Biovitrum Ab | New use II |
-
2006
- 2006-03-27 AU AU2006232660A patent/AU2006232660B2/en not_active Ceased
- 2006-03-27 KR KR1020077022649A patent/KR100931411B1/en not_active IP Right Cessation
- 2006-03-27 PL PL06725326T patent/PL1928840T3/en unknown
- 2006-03-27 CA CA2602781A patent/CA2602781C/en not_active Expired - Fee Related
- 2006-03-27 PT PT06725326T patent/PT1928840E/en unknown
- 2006-03-27 CN CN2006800111775A patent/CN101155783B/en not_active Expired - Fee Related
- 2006-03-27 DK DK06725326.0T patent/DK1928840T3/en active
- 2006-03-27 RU RU2007140737/04A patent/RU2381217C2/en not_active IP Right Cessation
- 2006-03-27 JP JP2008504736A patent/JP4880671B2/en not_active Expired - Fee Related
- 2006-03-27 MX MX2007012212A patent/MX2007012212A/en active IP Right Grant
- 2006-03-27 AT AT06725326T patent/ATE519744T1/en active
- 2006-03-27 ES ES06725326T patent/ES2369389T3/en active Active
- 2006-03-27 EP EP06725326A patent/EP1928840B1/en not_active Not-in-force
- 2006-03-27 WO PCT/EP2006/061057 patent/WO2006106052A1/en active Application Filing
- 2006-03-27 BR BRPI0610459-2A patent/BRPI0610459A2/en not_active IP Right Cessation
- 2006-03-31 US US11/395,763 patent/US7345058B2/en not_active Expired - Fee Related
- 2006-04-03 TW TW095111719A patent/TWI309645B/en not_active IP Right Cessation
- 2006-04-03 AR ARP060101312A patent/AR053203A1/en unknown
-
2007
- 2007-09-20 IL IL186118A patent/IL186118A0/en unknown
- 2007-09-25 NO NO20074872A patent/NO20074872L/en not_active Application Discontinuation
- 2007-10-02 ZA ZA200708419A patent/ZA200708419B/en unknown
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2612155A1 (en) * | 1975-03-25 | 1976-10-14 | Byk Gulden Lomberg Chem Fab | NITROFURYL PYRAZOLE DERIVATIVES, METHOD FOR MANUFACTURING THEREOF AND MEDICINAL PRODUCTS CONTAINING THEM |
| DE3713774A1 (en) * | 1986-04-24 | 1987-10-29 | Mitsui Toatsu Chemicals | NEW PYRAZOLE DERIVATIVES, METHOD FOR THEIR PRODUCTION AND FUNGICIDES FOR AGRICULTURE AND GARDENING, WHICH CONTAIN THESE COMPOUNDS |
| EP0360701A1 (en) * | 1988-09-01 | 1990-03-28 | Rhone-Poulenc Agrochimie | Antifungal compounds containing amides and phenyl groups |
| WO2004089470A2 (en) * | 2003-04-11 | 2004-10-21 | Novo Nordisk A/S | New amide derivatives and pharmaceutical use thereof |
| WO2005016877A2 (en) * | 2003-08-07 | 2005-02-24 | Merck & Co., Inc. | Pyrazole carboxamides as inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 |
| US20050080087A1 (en) * | 2003-10-10 | 2005-04-14 | Annapurna Pendri | Pyrazole derivatives as cannabinoid receptor modulators |
Cited By (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007058346A1 (en) * | 2005-11-21 | 2007-05-24 | Shionogi & Co., Ltd. | HETEROCYCLIC COMPOUND HAVING INHIBITORY ACTIVITY ON 11-β-HYDROXYSTEROID DEHYDROGENASE TYPE I |
| US8324265B2 (en) | 2005-11-21 | 2012-12-04 | Shionogi & Co., Ltd. | Heterocyclic compounds having type I 11β hydroxysteroid dehydrogenase inhibitory activity |
| EP2295411A1 (en) * | 2006-03-22 | 2011-03-16 | F. Hoffmann-La Roche AG | Pyrazoles as 11-beta-hsd-1 |
| WO2007107470A3 (en) * | 2006-03-22 | 2007-11-08 | Hoffmann La Roche | Pyrazoles as 11-beta-hsd-1 |
| CN101405270B (en) * | 2006-03-22 | 2013-01-16 | 霍夫曼-拉罗奇有限公司 | Pyrazoles as 11-beta-HSD-1 |
| US7728029B2 (en) | 2006-03-22 | 2010-06-01 | Hoffmann-La Roche Inc. | Adamantyl-pyrazole carboxamides as inhibitors of 11β-hdroxysteroid dehydrogenase |
| WO2007107470A2 (en) * | 2006-03-22 | 2007-09-27 | F. Hoffmann-La Roche Ag | Pyrazoles as 11-beta-hsd-1 |
| WO2007128761A2 (en) | 2006-05-04 | 2007-11-15 | Boehringer Ingelheim International Gmbh | Uses of dpp-iv inhibitors |
| EP2351568A2 (en) | 2006-05-04 | 2011-08-03 | Boehringer Ingelheim International GmbH | Uses of dpp-iv inhibitors |
| JP5319518B2 (en) * | 2007-04-02 | 2013-10-16 | Msd株式会社 | Indoledione derivative |
| US8106086B2 (en) | 2007-04-02 | 2012-01-31 | Msd K.K. | Indoledione derivative |
| WO2008120653A1 (en) * | 2007-04-02 | 2008-10-09 | Banyu Pharmaceutical Co., Ltd. | Indoledione derivative |
| CN103288738A (en) * | 2007-05-18 | 2013-09-11 | 盐野义制药株式会社 | Nitrogen-containing heterocyclic derivative having 11 beta-hydroxysteroid dehydrogenase type I inhibitory activity |
| JP2011098974A (en) * | 2007-05-18 | 2011-05-19 | Shionogi & Co Ltd | NITROGEN-CONTAINING HETEROCYCLIC DERIVATIVE HAVING 11beta-HYDROXYSTEROID DEHYDROGENASE TYPE I-INHIBITORY ACTIVITY |
| CN103288738B (en) * | 2007-05-18 | 2016-03-16 | 盐野义制药株式会社 | There is the nitogen-contained heterocycle derivant of 11beta-Hydroxysteroid dehydrogenase I type inhibit activities |
| CN101754954B (en) * | 2007-05-18 | 2015-11-25 | 盐野义制药株式会社 | There is the nitogen-contained heterocycle derivant of 11beta-Hydroxysteroid dehydrogenase I type inhibit activities |
| JPWO2008142986A1 (en) * | 2007-05-18 | 2010-08-05 | 塩野義製薬株式会社 | Nitrogen-containing heterocyclic derivatives having type I 11β-hydroxysteroid dehydrogenase inhibitory activity |
| JP4671369B2 (en) * | 2007-05-18 | 2011-04-13 | 塩野義製薬株式会社 | Nitrogen-containing heterocyclic derivatives having type I 11β-hydroxysteroid dehydrogenase inhibitory activity |
| WO2008142986A1 (en) | 2007-05-18 | 2008-11-27 | Shionogi & Co., Ltd. | NITROGEN-CONTAINING HETEROCYCLIC DERIVATIVE HAVING 11 β-HYDROXYSTEROID DEHYDROGENASE TYPE I INHIBITORY ACTIVITY |
| US8383622B2 (en) | 2007-05-18 | 2013-02-26 | Shionogi & Co., Ltd. | Nitrogen-containing heterocyclic derivative having 11β-hydroxysteroid dehydrogenase type I inhibitory activity |
| WO2009010416A3 (en) * | 2007-07-17 | 2009-03-05 | Hoffmann La Roche | Inhibitors of 11b-hydroxysteroid dehydrogenase |
| US7790711B2 (en) | 2007-07-17 | 2010-09-07 | Hoffmann-La Roche Inc. | Inhibitors of 11β-Hydroxysteroid Dehydrogenase |
| KR101158191B1 (en) | 2007-07-17 | 2012-06-19 | 에프. 호프만-라 로슈 아게 | Inhibitors of 11b-hydroxysteroid dehydrogenase |
| WO2009010416A2 (en) * | 2007-07-17 | 2009-01-22 | F. Hoffmann-La Roche Ag | Inhibitors of 11b-hydroxysteroid dehydrogenase |
| JP2010533670A (en) * | 2007-07-17 | 2010-10-28 | エフ.ホフマン−ラ ロシュ アーゲー | Inhibitors of 11β-hydroxysteroid dehydrogenase |
| CN114560811A (en) * | 2022-03-11 | 2022-05-31 | 中国药科大学 | 1,3, 5-trisubstituted-pyrazole-4 carboxylic acid derivative and preparation method and application thereof |
| CN114560811B (en) * | 2022-03-11 | 2023-09-01 | 上海立森印迹医药技术有限公司 | 1,3, 5-trisubstituted-pyrazole-4 carboxylic acid derivative, preparation method and application thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| MX2007012212A (en) | 2007-12-07 |
| RU2007140737A (en) | 2009-05-20 |
| CA2602781A1 (en) | 2006-10-12 |
| ES2369389T3 (en) | 2011-11-30 |
| EP1928840B1 (en) | 2011-08-10 |
| US20060223852A1 (en) | 2006-10-05 |
| DK1928840T3 (en) | 2011-09-12 |
| JP2008534647A (en) | 2008-08-28 |
| CN101155783A (en) | 2008-04-02 |
| JP4880671B2 (en) | 2012-02-22 |
| AU2006232660B2 (en) | 2010-03-25 |
| NO20074872L (en) | 2007-10-30 |
| KR100931411B1 (en) | 2009-12-10 |
| ATE519744T1 (en) | 2011-08-15 |
| RU2381217C2 (en) | 2010-02-10 |
| CA2602781C (en) | 2011-02-08 |
| US7345058B2 (en) | 2008-03-18 |
| TWI309645B (en) | 2009-05-11 |
| BRPI0610459A2 (en) | 2010-06-22 |
| PL1928840T3 (en) | 2012-01-31 |
| ZA200708419B (en) | 2008-11-26 |
| EP1928840A1 (en) | 2008-06-11 |
| KR20070108937A (en) | 2007-11-13 |
| TW200643013A (en) | 2006-12-16 |
| AU2006232660A1 (en) | 2006-10-12 |
| CN101155783B (en) | 2012-01-04 |
| IL186118A0 (en) | 2008-01-20 |
| AR053203A1 (en) | 2007-04-25 |
| PT1928840E (en) | 2011-10-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1928840B1 (en) | 1H-Pyrazole-4-carboxamides, their preparation and their use as 11-beta-hydroxysteroid dehydrogenase inhibitors | |
| US7728029B2 (en) | Adamantyl-pyrazole carboxamides as inhibitors of 11β-hdroxysteroid dehydrogenase | |
| CN101743226B (en) | Inhibitors of 11beta-hydroxysteroid dehydrogenase | |
| JP4839377B2 (en) | 11-β-hydroxysteroid dehydrogenase-1-inhibitor 2-1 type diabetes |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DPE2 | Request for preliminary examination filed before expiration of 19th month from priority date (pct application filed from 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2006725326 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 186118 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2602781 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2007/012212 Country of ref document: MX Ref document number: 12007502164 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006232660 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020077022649 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2008504736 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200680011177.5 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 7775/DELNP/2007 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 2006232660 Country of ref document: AU Date of ref document: 20060327 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006232660 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007140737 Country of ref document: RU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006725326 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: PI0610459 Country of ref document: BR Kind code of ref document: A2 |