WO2006100310A1 - Heterobicylic inhibitors of hcv - Google Patents
Heterobicylic inhibitors of hcv Download PDFInfo
- Publication number
- WO2006100310A1 WO2006100310A1 PCT/EP2006/061070 EP2006061070W WO2006100310A1 WO 2006100310 A1 WO2006100310 A1 WO 2006100310A1 EP 2006061070 W EP2006061070 W EP 2006061070W WO 2006100310 A1 WO2006100310 A1 WO 2006100310A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- ring
- alkyl
- optionally substituted
- compounds
- Prior art date
Links
- 0 *N(*)C(c(cncc1)c1N)=O Chemical compound *N(*)C(c(cncc1)c1N)=O 0.000 description 1
- DNQRMIMPLQPNQV-UHFFFAOYSA-N CN(C)CCNc(cc1)nc2c1c(O)nc(-c(cc(cc1)Cl)c1F)n2 Chemical compound CN(C)CCNc(cc1)nc2c1c(O)nc(-c(cc(cc1)Cl)c1F)n2 DNQRMIMPLQPNQV-UHFFFAOYSA-N 0.000 description 1
- GJNJDELEHIGPKJ-UHFFFAOYSA-N N#Cc(cc(cc1)Cl)c1F Chemical compound N#Cc(cc(cc1)Cl)c1F GJNJDELEHIGPKJ-UHFFFAOYSA-N 0.000 description 1
- SROQEVHFERCATR-UHFFFAOYSA-N NC(c(cc(cc1)Cl)c1F)=N Chemical compound NC(c(cc(cc1)Cl)c1F)=N SROQEVHFERCATR-UHFFFAOYSA-N 0.000 description 1
- AQTMUXWHODGBHI-UHFFFAOYSA-N NC(c(cncc1)c1Nc1nc(-c(cc(cc2)Cl)c2F)nc2c1nccn2)=O Chemical compound NC(c(cncc1)c1Nc1nc(-c(cc(cc2)Cl)c2F)nc2c1nccn2)=O AQTMUXWHODGBHI-UHFFFAOYSA-N 0.000 description 1
- OBTWVHJAUQPKKE-UHFFFAOYSA-N Oc1nc(-c(cc(cc2)Cl)c2F)nc2c1ccc(F)n2 Chemical compound Oc1nc(-c(cc(cc2)Cl)c2F)nc2c1ccc(F)n2 OBTWVHJAUQPKKE-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Definitions
- the invention relates to methods of treating disorders associated with hepatitis C infection. More specifically, it concerns certain fused bicyclic pyrimidine compounds that have an amide-substituted 4-pyridylamine group on the pyrimidine ring that are useful in these methods.
- TGF ⁇ Transforming growth factor-beta
- TGF ⁇ denotes a superfamily of proteins that includes, for example, TGF ⁇ l, TGF ⁇ 2, and TGF ⁇ 3, which are pleiotropic modulators of cell growth and differentiation, embryonic and bone development, extracellular matrix formation, hematopoiesis, and immune and inflammatory responses (Roberts and Sporn Handbook of Experimental Pharmacology (1990) 95:419-58; Massague, et al, Ann. Rev. Cell. Biol. (1990) 6:597-646).
- Other members of this superfamily include activin, inhibin, bone morphogenic protein, and Mullerian inhibiting substance.
- fibroproliferative diseases include kidney disorders associated with unregulated TGF ⁇ activity and excessive fibrosis including glomerulonephritis (GN), such as mesangial proliferative GN, immune GN, and crescentic GN.
- GN glomerulonephritis
- renal conditions include diabetic nephropathy, renal interstitial fibrosis, renal fibrosis in transplant patients receiving cyclosporin, and HFV-associated nephropathy.
- Collagen vascular disorders include progressive systemic sclerosis, polymyositis, scleroderma, dermatomyositis, eosinophilic fascitis, morphea, or those associated with the occurrence of Raynaud's syndrome.
- Lung fibroses resulting from excessive TGF ⁇ activity include adult respiratory distress syndrome, chronic obstructive pulmonary disease (COPD), idiopathic pulmonary fibrosis, and interstitial pulmonary fibrosis often associated with autoimmune disorders, such as systemic lupus erythematosus and scleroderma, chemical contact, or allergies.
- COPD chronic obstructive pulmonary disease
- Another autoimmune disorder associated with fibroproliferative characteristics is rheumatoid arthritis.
- Fibroproliferative conditions can be associated with surgical eye procedures. Such procedures include retinal reattachment surgery accompanying proliferative vitreoretinopathy, cataract extraction with intraocular lens implantation, and post glaucoma drainage surgery.
- TGF ⁇ l inhibits the formation of tumors, probably by inhibition of the proliferation of nontransformed cells.
- TGF ⁇ l promotes the growth of the tumor.
- N. Dumont and CL. Arteaga Breast Cancer Res., Vol. 2, 125-132 (2000).
- inhibitors of the TGF ⁇ pathway are also useful for the treatment of many forms of cancer, such as lung cancer, skin cancer, and colorectal cancer.
- the compounds of the invention herein are derivatives of pyrimidine having an additional ring fused onto the pyrimidine.
- PCT publication WOO 1/47921 describes pyrimidine and triazine compounds that are inhibitors of kinase activities associated with various inflammatory conditions, as opposed to the treatment of fibroproliferative disorders described herein.
- the above mentioned PCT publication describes the use of the disclosed compounds only for treatment of the inflammatory aspects of certain autoimmune diseases.
- the compounds described differ from those described herein by virtue of the substitutions required on the pyrimidine nucleus; among other distinctions, the compounds disclosed in the PCT publication do not include phenyl bound directly to the pyrimidine ring.
- 6,476,031 also discloses compounds containing a quinazoline ring, which cam be a fused bicyclic derivative of a pyrimidine; it includes compounds where the quinazoline ring is linked to an aryl group at C-4 of the quinazoline.
- the compounds are reported to act at the TGF ⁇ site, and the compounds can include a 4- pyridylamine group as the aryl group linked to the quinazoline at C-4.
- the invention is directed to methods, compositions, and novel compounds useful in treating conditions that are characterized by excessive TGF ⁇ activity. These conditions are, most prominently, fibroproliferative diseases, such as conditions associated with hepatitis C virus infection, and certain cancers. However, the conditions for which the compounds and methods are useful include any medical condition characterized by an undesirably high level of TGF ⁇ activity.
- the compounds of the invention have been found to inhibit TGF ⁇ and are thus useful in treating diseases mediated by the activity of this family of iactors.
- the compounds of the invention are of the formula (I):
- R 1 represents H or OH, or an optionally substituted alkyl, alkoxy, heteroalkyl, amino, acyl, heteroacyl, aryl, arylalkyl, heteroaryl, or heteroarylalkyl group;
- R 2 represents H or optionally substituted alkyl, heteroalkyl, acyl, heteroacyl, aryl, heteroaryl, arylalkyl, or heteroarylalkyl;
- B represents H or a C1-C8 acyl group that may be substituted or unsubstituted; each of W, X, Y and Z is independently C-H, C-J or N, provided that not more than two of W, X, Y and Z represent N;
- Ar represents an optionally substituted phenyl ring; each J independently represents halo, OH, SH, or optionally substituted alkyl, alkenyl, alkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, aryl, acyl, heteroacyl, or heteroaryl, or NR 1 R 2 , NO 2 , CN, CF 3 , COOR, CONR 2 , or SO 2 R, wherein each R is independently H or an optionally substituted alkyl, alkenyl, alkynyl, acyl, aryl, heteroalkyl, heteroalkenyl, heteroalkynyl, heteroacyl or heteroaryl group, R 1 and R 2 of any NR 1 R 2 can cyclize to form a 3-8 membered ring that can be saturated, unsaturated, or aromatic, and that contains 1-3 heteroatoms selected from N, O and S as ring members, and is optionally substituted; and n is 0-3;
- the invention is also directed to pharmaceutical compositions containing one or more compounds of formula (I) or their pharmaceutically acceptable salts, or prodrug forms thereof, as active ingredients and to methods of treating conditions characterized by an excessive level of TGF ⁇ activity, particularly fibroproliferative conditions, using compounds of formula (I) or compositions containing such compounds.
- TGF ⁇ refers to the superfamily which includes TGF ⁇ l, TGF ⁇ 2, and TGF ⁇ 3 as well as other members of the family known or which become known in the art such as inhibin, bone morphogenic protein, and the like. One or more of these family members may be more active than desired in the conditions which the compounds of the invention are designed to ameliorate or prevent.
- Conditions "characterized by an excessive level of TGF ⁇ activity” include those wherein TGF ⁇ synthesis is stimulated so that TGF ⁇ is present in enhanced amount, and those wherein TGF ⁇ latent protein is undesirably activated or converted to active TGF ⁇ protein, and those wherein TGF ⁇ receptors are upregulated, and those wherein the
- TGF ⁇ protein shows enhanced binding to cells or extracellular matrix in the location of the disease.
- "excessive level of TGF ⁇ activity” refers to any condition wherein the activity of TGF ⁇ is undesirably high, regardless of the cause and regardless of whether the actual amount or activity of TGF ⁇ present is within a 'normal' range.
- the compounds useful in the invention are fused bicyclic derivatives of pyrimidine containing mandatory substituents at positions corresponding to the 2- and 4-positions of the pyrimidine ring.
- the bicyclic pyrimidines further have another aromatic ring fused onto the pyrimidine at positions 5 and 6 of the pyrimidine ring. They further include a 4-pyridylamine group at position 4 of the pyrimidine ring and a phenyl group at position 2 of the pyrimidine ring.
- the 4-pyridyl group may be a pyridine- N-oxide.
- the compounds further include an amide group that is attached at position 3 of the pyridyl ring through its carbonyl carbon.
- Other substituents may also be included on the pyrimidine, pyridine and phenyl rings and on the aromatic ring fursed onto the pyrimidine.
- alkyl straight-chain, branched-chain and cyclic monovalent hydrocarbyl radicals, and combinations of these, which contain only C and H when they are unsubstituted. Examples include methyl, ethyl, isobutyl, cyclohexyl, cyclopentylethyl, 2-propenyl, 3-butynyl, and the like.
- the total number of carbon atoms in each such group is sometimes described herein, e.g., either as 1 -1OC or as Cl-ClO when the group can contain up to ten carbon atoms.
- heteroatoms N, O and S typically
- the numbers describing the group represent the sum of the number of carbon atoms in the group plus the number of such heteroatoms that are included as replacements for carbon atoms.
- the alkyl, alkenyl and alkynyl substituents of the invention contain 1-lOC (alkyl) or 2- 1OC (alkenyl or alkynyl). Preferably they contain 1-8C (alkyl) or 2-8C (alkenyl or alkynyl). Sometimes they contain 1-4C (alkyl) or 2-4C (alkenyl or alkynyl).
- a single group can include more than one type of multiple bond, or more than one multiple bond; such groups are included within the definition of the term "alkenyl” when they contain at least one carbon-carbon double bond, and are included within the term "alkynyl" when they contain at least one carbon-carbon triple bond.
- Heteroalkyl “heteroalkenyl”, and “heteroalkynyl” are defined similarly to the corresponding hydrocarbyl (alkyl, alkenyl and alkynyl) groups, but the 'hetero' terms refer to groups that contain 1-3 O, S or N heteroatoms or combinations thereof within the backbone residue; thus at least one carbon atom of a corresponding alkyl, alkenyl, or alkynyl group is replaced by one of the specified heteroatoms to form a heteroalkyl, heteroalkenyl, or heteroalkynyl group.
- heteroforms of alkyl, alkenyl and alkynyl groups are the same as for the corresponding hydrocarbyl groups, and the substituents that may be present on the heteroforms are the same as those described above for the hydrocarbyl groups.
- substituents that may be present on the heteroforms are the same as those described above for the hydrocarbyl groups.
- such groups do not include more than two contiguous heteroatoms except where an oxo group is present on N or S as in a nitro or sulfonyl group.
- alkyl as used herein includes cycloalkyl and cycloalkylalkyl groups
- cycloalkyl may be used herein to describe a carbocyclic non-aromatic group that is typically connected via a ring carbon atom
- cycloalkylalkyl may be used to describe a carbocyclic non-aromatic group that is connected to the molecule through an alkyl linker.
- heterocyclyl may be used to describe a non-aromatic cyclic group that contains at least one heteroatom as a ring member and that is typically connected to the molecule via a ring atom, which may be C or N; and “heterocyclylalkyl” may be used to describe such a group that is connected to another molecule through a linker.
- the sizes and substituents that are suitable for the cycloalkyl, cycloalkylalkyl, heterocyclyl, and heterocyclylalkyl groups are the same as those described above for alkyl groups As used herein, these terms also include rings that contain a double bond or two, as long as the ring is not aromatic.
- acyl encompasses groups comprising an alkyl, alkenyl, alkynyl, aryl or arylalkyl radical attached at one of the two available valence positions of a carbonyl carbon atom
- heteroacyl refers to the corresponding groups wherein at least one carbon other than the carbonyl carbon has been replaced by a heteroatom chosen from N, O and S.
- Acyl and heteroacyl groups are bonded to any group or molecule to which they are attached through the open valence of the carbonyl carbon atom. Typically, they are C1-C8 acyl groups, which include formyl, acetyl, pivaloyl, and benzoyl, and C2-C8 heteroacyl groups, which include methoxyacetyl, ethoxycarbonyl, and 4-pyridinoyl.
- the hydrocarbyl groups, aryl groups, and heteroforms of such groups that comprise an acyl or heteroacyl group can be substituted with the substituents described herein as generally suitable substituents for each of the corresponding components of the acyl or heteroacyl group.
- Aromaatic moiety or aryl moiety refers to a monocyclic or fused bicyclic moiety having the well-known characteristics of aromaticity; examples include phenyl and naphthyl.
- heteroaryl refers to such monocyclic or fused bicyclic ring systems which contain as ring members one or more heteroatoms selected from O, S and N. The inclusion of a heteroatom permits aromaticity in 5-membered rings as well as 6-membered rings.
- Typical heteroaromatic systems include monocyclic C5-C6 aromatic groups such as pyridyl, pyrimidyl, pyrazinyl, thienyl, iuranyl, pyrrolyl, pyrazolyl, thiazolyl, oxazolyl, and imidazolyl and the fused bicyclic moieties formed by fusing one of these monocyclic groups with a phenyl ring or with any of the heteroaromatic monocyclic groups to form a C8-C10 bicyclic group such as indolyl, benzimidazolyl, indazolyl, benzotriazolyl, isoquinolyl, quinolyl, benzothiazolyl, benzofuranyl, pyrazolopyridyl, quinazolinyl, quinoxalinyl, cinnolinyl, and the like.
- monocyclic C5-C6 aromatic groups such as pyridyl, pyrimi
- any monocyclic or fused ring bicyclic system which has the characteristics of aromaticity in terms of electron distribution throughout the ring system is included in this definition. It also includes bicyclic groups where at least the ring which is directly attached to the remainder of the molecule has the characteristics of aromaticity.
- the ring systems contain 5-12 ring member atoms.
- the monocyclic heteroaryls contain 5-6 ring members, and the bicyclic heteroaryls contain 8-10 ring members.
- Aryl and heteroaryl moieties may be substituted with a variety of substituents including halo, C1-C8 alkyl, C2-C8 alkenyl, C2-C8 alkynyl, OR, NR 2 , SR, SO 2 R, SO 2 NR 2 , NRSO 2 R, NRCONR 2 , NRCOOR, NRCOR, CN, COOR, CONR 2 , OOCR, COR, and NO 2 , wherein each R is independently H, Cl -C8 alkyl, C2-C8 heteroalkyl, C2-C8 alkenyl, C2-C8 heteroalkenyl, C2-C8 alkynyl, C2-C8 heteroalkynyl, C6-C10 aryl, C5-C10 heteroaryl, C7-C12 arylalkyl, or C6-C12 heteroarylalkyl, and each R is optionally substituted as described above for alkyl groups.
- arylalkyl and “heteroarylalkyl” refer to aromatic and heteroaromatic ring systems which are bonded to their attachment point through a linking group such as an alkylene, including substituted or unsubstituted, saturated or unsaturated, cyclic or acyclic linkers.
- the linker is C1-C8 alkyl or a hetero form thereof.
- These linkers may also include a carbonyl group, thus making them able to provide substituents as an acyl or heteroacyl moiety.
- An aryl or heteroaryl ring in an arylalkyl or heteroarylalkyl group may be substituted with the same substituents described above for aryl groups.
- an arylalkyl group includes a phenyl ring optionally substituted with the groups defined above for aryl groups and a C1-C4 alkylene that is unsubstituted or is substituted with one or two C1-C4 alkyl groups or heteroalkyl groups, where the alkyl or heteroalkyl groups can optionally cyclize to form a ring such as cyclopropane, dioxolane, or oxacyclopentane.
- a heteroarylalkyl group preferably includes a C5-C6 monocyclic heteroaryl group that is optionally substituted with the groups described above as substituents typical on aryl groups and a C1-C4 alkylene that is unsubstituted or is substituted with one or two C1-C4 alkyl groups or heteroalkyl groups, or it includes an optionally substituted phenyl ring or C5-C6 monocyclic heteroaryl and a C1-C4 heteroalkylene that is unsubstituted or is substituted with one or two C1-C4 alkyl or heteroalkyl groups, where the alkyl or heteroalkyl groups can optionally cyclize to form a ring such as cyclopropane, dioxolane, or oxacyclopentane.
- substituents may be on either the alkyl or heteroalkyl portion or on the aryl or heteroaryl portion of the group.
- the substituents optionally present on the alkyl or heteroalkyl portion are the same as those described above for alkyl groups generally; the substituents optionally present on the aryl or heteroaryl portion are the same as those described above for aryl groups generally.
- Arylalkyl groups as used herein are hydrocarbyl groups if they are unsubstituted, and are described by the total number of carbon atoms in the ring and alkylene or similar linker.
- a benzyl group is a C7-arylalkyl group
- phenylethyl is a C8-arylalkyl.
- Heteroarylalkyl refers to a moiety comprising an aryl group that is attached through a linking group, and differs from “arylalkyl” in that at least one ring atom of the aryl moiety or one atom in the linking group is a heteroatom selected from N, O and S.
- heteroarylalkyl groups are described herein according to the total number of atoms in the ring and linker combined, and they include aryl groups linked through a heteroalkyl linker; heteroaryl groups linked through a hydrocarbyl linker such as an alkylene; and heteroaryl groups linked through a heteroalkyl linker.
- C7 -heteroarylalkyl would include pyridylmethyl, phenoxy, and N-pyrrolylmethoxy.
- Alkylene refers to a divalent hydrocarbyl group; because it is divalent, it can link two other groups together.
- n 1-8 and preferably n is 1-4
- an alkylene can also be substituted by other groups, and can be of other lengths, and the open valences need not be at opposite ends of a chain.
- -CH(Me)- and -C(Me) 2 - may also be referred to as alkylenes, as can a cyclic group such as cyclopropan-l,l-diyl.
- the substituents include those typically present on alkyl groups as described herein.
- any alkyl, alkenyl, alkynyl, acyl, or aryl or arylalkyl group or any heteroform of one of these groups that is contained in a substituent may itself optionally be substituted by additional substituents.
- the nature of these substituents is similar to those recited with regard to the primary substituents themselves if the substituents are not otherwise described.
- R 7 is alkyl
- this alkyl may optionally be substituted by the remaining substituents listed as embodiments for R 7 where this makes chemical sense, and where this does not undermine the size limit provided for the alkyl per se; e.g.
- alkyl substituted by alkyl or by alkenyl would simply extend the upper limit of carbon atoms for these embodiments, and is not included.
- each such alkyl, alkenyl, alkynyl, acyl, or aryl group may be substituted with a number of substituents according to its available valences; in particular, any of these groups may be substituted with fluorine atoms at any or all of its available valences, for example.
- Heteroform refers to a derivative of a group such as an alkyl, aryl, or acyl, wherein at least one carbon atom of the designated carbocyclic group has been replaced by a heteroatom selected from N, O and S.
- heteroforms of alkyl, alkenyl, alkynyl, acyl, aryl, and arylalkyl are heteroalkyl, heteroalkenyl, heteroalkynyl, heteroacyl, heteroaryl, and heteroarylalkyl, respectively. It is understood that no more than two N, O or S atoms are ordinarily connected sequentially, except where an oxo group is attached to N or S to form a nitro or sulfonyl group. "Optionally substituted" as used herein indicates that the particular group or groups being described may have no non-hydrogen substituents, or the group or groups may have one or more non-hydrogen substituents.
- Halo as used herein includes fluoro, chloro, bromo and iodo. Fluoro and chloro are often preferred.
- Amino refers to NH 2 , but where an amino is described as “substituted” or “optionally substituted”, the term includes NR'R" wherein each R' and R" is independently H, or is an alkyl, alkenyl, alkynyl, acyl, aryl, or arylalkyl group or a heteroform of one of these groups, and each of the alkyl, alkenyl, alkynyl, acyl, aryl, or arylalkyl groups or heteroforms of one of these groups is optionally substituted with the substituents described herein as suitable for the corresponding group.
- R' and R" are linked together to form a 3-8 membered ring which may be saturated, unsaturated or aromatic and which contains 1-3 heteroatoms independently selected from N, O and S as ring members, and which is optionally substituted with the substituents described as suitable for alkyl groups or, if NR'R" is an aromatic group, it is optionally substituted with the substituents described as typical for heteroaryl groups.
- the compounds of the invention include a pyrimidine ring, and another six-membered aromatic ring is fused onto the C5 and C6 positions of the pyrimidine.
- the C2 position of the pyrimidine is occupied by an optionally substituted phenyl group referred to in formula (I) as Ar.
- the C4 position of the pyrimidine is linked by a nitrogen linker to the C-4 carbon of a pyridine ring.
- Substituents J may be present on the pyridine ring in formula (I) at any or all of the positions not otherwise expressly occupied.
- n in formula (I) can be 0-3.
- n is 0; in some embodiments n is 1 or 2.
- Typical embodiments of J in formula (I) include the substituents described herein as substituents for an aryl group generally.
- Preferred embodiments for J include CF 3 and CN, as well as halo, C1-C4 alkyl, OR, SR, and NR 2 , wherein each R is independently H or C1-C4 alkyl or C1-C4 heteroalkyl, where each alkyl or heteroalkyl is optionally substituted with the substituents described above for alkyl groups, and where two R groups on N can optionally cyclize to form a 3-8 membered ring containing one or two heteroatoms selected from N, O and S as ring members.
- Halo, methyl, methoxy and CF 3 are often preferred for each J present.
- Ar represents phenyl which may be unsubstituted, but is typically substituted with at least one and preferably two or more substituents selected from the group consisting of halo, C1-C4 alkyl, CN, CF 3 , OR, NO 2 , COOR, CONR 2 , SO 2 R, NR 2 , and C1-C8 acyl, where each R is independently H, C1-C4 alkyl, C1-C8 acyl, or C2-C8 heteroacyl.
- Ar is substituted with one or two substituents.
- the substituents on Ar may be at any available position on the phenyl ring, but frequently one substituent occupies a ring position adjacent to the atom through which Ar is linked to the pyrimidine ring.
- position 1 the position of the phenyl ring that is attached to the pyrimidine ring in formula (I) is referred to as position 1, and other positions on the phenyl ring are numbered relative to that position.
- Preferred embodiments often have Ar as a phenyl ring that is substituted by at least one halo substituent, which may be at position 2 of that phenyl.
- a preferred embodiment includes a phenyl ring substituted with two groups, which may both be halo. 2,5-dihalo phenyl is sometimes specifically preferred, particularly where each halo is F or Cl; and 2-fluoro-5-chlorophenyl is especially preferred.
- R 1 and R 2 attaches substituents R 1 and R 2 to the pyridyl ring specifically at the 3 -position.
- the selection of R 1 and R 2 is important for its effect on the intrinsic activity of the TGF ⁇ inhibitor compounds, and also can strongly influence their properties related to bioavailability.
- R 1 is H, OH, or NH 2 ; in other embodiments, R 1 is an optionally substituted alkyl, heteroalkyl, alkoxy, amino, acyl, heteroacyl, aryl, arylalkyl, heteroaryl, or heteroarylalkyl group.
- R 1 is C1-C8 alkoxy, amino, C1-C8 alkyl, C2-C8 heteroalkyl, C6-C10 aryl, C5-C10 heteroaryl, C7-C12-arylalkyl, or C6-C12 heteroarylalkyl, where each of the foregoing groups except H is optionally substituted by the substituents described herein as suitable substituents for such groups.
- Each R in these substituents is independently H, C1-C8 alkyl, C2-C8 heteroalkyl, C6-C10 aryl, C5-C10 heteroaryl, C1-C8 acyl or C2-C8 heteroacyl.
- Preferred embodiments of R 1 include H, C1-C8 alkoxy, NH 2 , C1-C8 alkyl and C2-C8 heteroalkyl, wherein each alkyl or heteroalkyl is optionally substituted as just described.
- R 1 and R 2 is H, so in many embodiments the amide is a secondary or tertiary amide.
- R 2 is H, or an optionally substituted alkyl, acyl, heteroacyl, aryl, heteroaryl, arylalkyl, or heteroarylalkyl group.
- R 2 is H or a C1-C8 alkyl group, and in others it is a C1-C8 acyl or C2-C8 heteroacyl group or a C7-C12 arylalkyl or C6-C12 heteroarylalkyl group; in each of these embodiments where R 2 is other than H, the group represented by R 2 is optionally substituted with the substituents described above for R 1 .
- Preferred substituents for R 2 when R 2 is other than H include halo, OR, NR 2 , COOR, and CONR 2 , where each R is independently H, C1-C4 alkyl, or Cl -C4 heteroalkyl.
- R 1 and R 2 Of-C(O)NR 1 R 2 R 1 can cyclize to form a 3-8 membered ring that can be saturated, unsaturated, or aromatic, and can contain 1-3 heteroatoms selected from N, O and S as ring members, and can be substituted.
- R 1 and R 2 cyclize to form a 3 to 6 membered ring that is saturated or unsaturated and contains either O or 1 heteroatom in addition to the N to which R 1 and R 2 are attached.
- R 1 and R 2 cyclize to form a saturated 6-membered ring containing one heteroatom that is either O or N in addition to the N to which R 1 and R 2 are attached.
- any ring that is formed by linking R 1 and R 2 OfNR 1 R 2 is optionally substituted by the substituents that are described herein as suitable substituents for alkyl groups if the ring so formed is non-aromatic, or by the substituents described above for aryl groups if the ring formed by linking R 1 and R 2 is aromatic.
- R 1 and/or R 2 examples include ethers, amines, alcohols, esters, amides, carbamates, ketones, sulfones, sulfonamides, phosphate esters, polyhydroxylated alkyl or cycloalkyl groups including monosaccharide derivatives, amidines, oximes, guanidines, cyanoguanidines, and the like. In certain embodiments, at least one and preferably two of such polar groups are included in compounds of formula (1).
- B in formula (1) can be H or a C1-C8 optionally substituted acyl group. In certain embodiments, B is H. Where B is an acyl group, the compound may serve as a prodrug to release a compound wherein B is H upon metabolic or chemical hydrolysis to cleave off the acyl group.
- Each of W, X, Y and Z in formula (I) is independently CH, CJ or N, provided that no more than two of W, X, Y and Z represent N.
- the combination of W, X, Y and Z, together with the pyrimidine-ring carbon atoms to which W and Z are attached forms a six membered ring that is aromatic.
- at least one of W, X, Y and Z is N, and in some fo these, at least one of W, Z is N.
- Z is N
- W, X and Y each independently represent CH or CJ
- W and Z are each N and X and Y each represent CH or CJ.
- Some embodiments have W, X, Y and Z each independently representing CH or C-J, thus forming a carbocyclic ring that, taken with the pyrimidine ring, forms a quinazoline nucleus.
- Each embodiment of the ring containing W, X, Y and Z is optionally substituted as described herein.
- Preferred embodiments include those in which the fused ring containing W, X and Z is phenyl or pyridyl, each of which is optionally substituted as defined above. Pyridyl is sometimes more preferred for this ring, especially when either Z or W represents the pyridyl ring nitrogen.
- fused ring containing W, X, Y and Z is a pyrazine wherein W and Z are both N, and X and Y each represent CH or CJ.
- the preferred aromatic fused rings mentioned are substituted by at least one group such as halo, optionally substituted C1-C8 alkyl, COOR, CONR 2 , OR, or NR 2 , wherein each R is independently H, C1-C8 alkyl or C2-C8 heteroalkyl, and each alkyl or heteroalkyl comprising R is optionally substituted with the substituents defined above for alkyl groups.
- At least one of W, X, Y and Z represents C-J, while the others represent N or CH.
- J comprises NH; and in certain embodiments, the NH that J comprises is directly linked to the carbon atom of the group C-J.
- Y represents C-J, where J comprises an amine, amide or carbamate group. Especially when Z represents N, Y is often C-J, i.e. a substituted carbon. While J in such embodiments can be any of the groups provided herein as suitable substituents for an aromatic ring, in many embodiments, and especially when Z represents N, Y represents C-J wherein J is an amine or a substituted amine group.
- Typical examples include NH 2 , C1-C4 monoalkyl amines where the alkyl group may be substituted with, for example, one or two C1-C4 alkoxy, amino, C1-C4 alkylamino or di-(Cl-C4)-alkylamino groups.
- a dialkylamine can be present, it can represent a cyclic group such as a pyrrolidine, piperidine, morpholine, and the like, which may be substituted.
- J when Y represents C-J, J can be an arylalkylamine group such as a benzylamino substituent; and the benzyl group can be substituted with the groups that are described herein as typical for an aryl ring if on the phenyl portion, or with any of the groups suitable for an alkyl group if substitution is on the alkylene portion of the arylalkyl group.
- Preferred substituents for the phenyl ring of a benzyl in such embodiments include halo, CF 3 , C1-C4 alkyl, and C1-C4 alkoxy.
- any aryl, alkyl, heteroaryl, heteroalkyl, acyl, heteroacyl, arylalkyl, or heteroarylalkyl group included within a substituent may itself be substituted with the substituents described above as typical for such aryl, alkyl, acyl, or arylalkyl groups. These substituents may occupy all available positions of the group, preferably 1-2 positions, or more preferably only one position. Where two substituents are present on a single atom, such as but not limited to NR 2 of an amine or amide, the two substituents may be linked together to form a ring where this is chemically reasonable.
- Such rings may be saturated or unsaturated and may be further substituted if substitution is permitted for the substituents linked to form the ring. It is specifically contemplated that R 1 and R 2 or any two R groups on one N can cyclize to form a 3-8 membered ring that may be saturated or unsaturated, and may include 1-3 heteroatoms selected from N, O and S, and which may be optionally substituted as described for the substituents or R groups being linked to form the ring.
- any of the aryl or cyclic moieties can optionally contain at least two substituents, if those substituents occupy adjacent positions on a ring or they are on a single atom, they may also be linked together to form a 5-7 membered carbocyclic ring or a heterocyclic ring containing 1-3 heteroatoms selected from N, O and S.
- substituents include a dioxolane fused to a phenyl ring; oxazole fused to a pyridine ring; an acetonide of a 1,2-diol or a 1,3-diol; and a cyclic ketal.
- An embodiment of the present invention relates to the pyrido[2,3-d]pyrimidine compounds of formula (II),
- R 1 represents H or OH, or an optionally substituted alkyl, alkoxy, heteroalkyl, amino, acyl, heteroacyl, aryl, arylalkyl, heteroaryl, or heteroarylalkyl group;
- R 2 represents H or optionally substituted alkyl, heteroalkyl, acyl, heteroacyl, aryl, heteroaryl, arylalkyl, or heteroarylalkyl;
- B represents H or a C1-C8 acyl group that may be substituted or unsubstituted;
- Y is C-H, or C-J;
- Ar represents an optionally substituted phenyl ring; each J independently represents halo, OH, SH, or optionally substituted alkyl, alkenyl, alkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, aryl, acyl, heteroacyl, or heteroaryl, or NR 1 R 2 , NO 2 , CN, CF 3 , COOR, CONR 2 , or SO 2 R, wherein each R is independently H or an optionally substituted alkyl, alkenyl, alkynyl, acyl, aryl, heteroalkyl, heteroalken
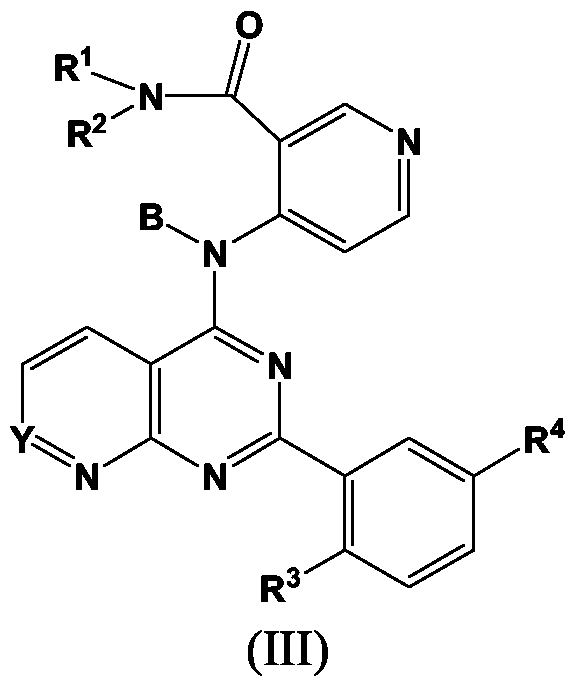
- a further embodiment of the present invention relates to the pyrido[2,3-d]pyrimidine compounds of formula (III),
- R 1 represents H or OH, or an optionally substituted alkyl, alkoxy, heteroalkyl, amino, acyl, heteroacyl, aryl, arylalkyl, heteroaryl, or heteroarylalkyl group;
- R 2 represents H or optionally substituted alkyl, heteroalkyl, acyl, heteroacyl, aryl, heteroaryl, arylalkyl, or heteroarylalkyl;
- B represents H or a C1-C8 acyl group that may be substituted or unsubstituted
- Y is C-H, or C-J
- R 3 represents H, or halo;
- R 4 represents halo; each J independently represents halo, OH, SH, or optionally substituted alkyl, alkenyl, alkynyl, heteroalkyl, heteroalkenyl, heteroalkynyl, aryl, acyl, heteroacyl, or heteroaryl, or NR 1 R 2 , NO 2 , CN, CF 3 , COOR, CONR 2 , or SO 2 R, wherein each R is independently H or an optionally substituted alkyl, alkenyl, alkynyl, acyl, aryl, heteroalkyl, heteroalkenyl, heteroalkynyl, heteroacyl or heteroaryl group.
- the compounds of the present invention may be supplied in the form of their pharmaceutically acceptable acid-addition salts including salts of inorganic acids such as hydrochloric, sulfuric, hydrobromic, or phosphoric acid or salts of organic acids such as acetic, tartaric, succinic, benzoic, salicylic, citric, alkylsulfonic, arylsulfonic, and glucuronic acids and the like. If a carboxyl moiety is present on the compounds of the present invention, the compound may also be supplied as a salt with a pharmaceutically acceptable cation, such as sodium, potassium, or an ammonium salt.
- a pharmaceutically acceptable cation such as sodium, potassium, or an ammonium salt.
- the compounds of the present invention may also be supplied in the form of a "prodrug" which is designed to release the compounds the present invention when administered to a subject.
- Prodrug designs are well known in the art, and depend on the substituents contained in the compounds of the present invention.
- a substituent containing sulfhydryl could be coupled to a carrier which renders the compound biologically inactive until removed by endogenous enzymes or, for example, by enzymes targeted to a particular receptor or location in the subject.
- ester and amide linkages may be employed to mask hydroxyl, amino, or carboxyl groups on an active molecule within the scope of the invention, and such groups may be enzymatically cleaved in vivo to release the active molecule.
- B can represent an acyl group that is selected for its ability to hydro lyze at a suitable rate in vivo; thus B could be acetyl or formyl, or B-N in formula (1) can be an amide formed from the carboxylate of an amino acid or a dipeptide, each of which would readily hydrolyze from the nitrogen flanked by two heteroaryl rings in formula (1). Accordingly, such amides wherein B is an acyl group are suitable as prodrugs for delivering a compound of formula (1) wherein B is H.
- the invention includes each stereoisomeric form thereof, both as an isolated stereoisomer and as a component of a mixture of these stereoisomeric forms.
- Such mixtures of stereoisomers may be racemic or may be enriched in one enantiomer of a pair of enantiomers where a single chiral center is present.
- the invention includes mixtures wherein either, neither or each center is enriched in one stereoisomeric form.
- a number of synthetic routes may be employed to produce the compounds of the invention. In general, they may be synthesized from conventional starting materials using reactions known in the art. Specific routes and reactions suitable for synthesis of many of the compounds of the invention are described in U.S. Patent No. 6,476,031, and in published PCT application WO 2004/024159, and in published US application US 2005/0004143 Al, and in published PCT application US2004/032430, each of which is incorporated by reference specifically for its disclosure of such methods.
- the fused ring system is constructed from an aryl ring that corresponds to the ring in formula (1) containing W, X, Y and Z; that aryl ring would having an acylating group adjacent to an amine or a leaving group that can be used to introduce an amine.
- the acylating group of the aromatic ring is used to acylate a phenyl amidine, whose phenyl group corresponds to Ar in formula (1).
- Cyclization is then effected under known conditions to produce a fused ring system with a 4-hydroxypyrimidine.
- One example of this condensation is illustrated in Scheme 5 below.
- the hydroxyl group is then converted to a halo (e.g., Cl or I), which is displaced with a 4-aminopyridine derivative, as shown in Scheme 1.
- Scheme 1 shows how a 4-hydroxy pyrimidine can be converted into a 4-halo pyrimidine, which is then coupled to a 4-aminopyridine.
- the coupling is done using a palladium catalyst, and may be done with the 4-chloro pyrimidine derivative in some cases, but was done with the 4-iodo derivative in some cases.
- the requisite 3-carboxamide group may be present on the 4-aminopyridine when the pyridine is added to the pyrimidine, or the pyridyl group may contain an ester at the 3 -position as illustrated in Scheme 1.
- the ester can be hydro lyzed with base to form a carboxylic acid after the pyridine group is installed.
- This carboxylic acid is readily coupled to a wide variety of amine groups by methods well known in the art for forming amide bonds as illustrated in Scheme 2. Because of the wide variety of amines that are available and the generality of this amide formation reaction, this method provides access to a tremendous variety of compounds of the present invention.
- Schcmc Converting an ester to a carboxamide of formula (I).
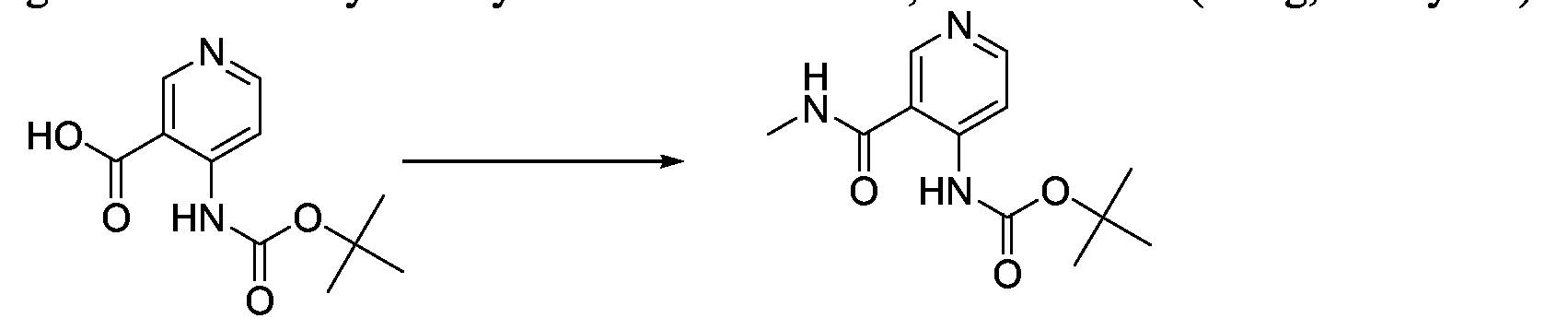
- the amide can be formed on the pyridine ring before it is coupled to the pyrimidine.
- Preparation of such 3-carboxamide-4-amino pyridines is shown in Schemes 3a and 3b.
- Scheme 3a provides a route to prepare the pyridyl nucleus and further substitution thereon.
- R substituent is exemplified as hydrogen or methyl in the above scheme, it may also include the other substituents as listed under the definitions of R 1 and R 2 .
- Scheme 5 depicts an overall sequence wherein a fused ring compound of formula (1) wherein Z represents N can be prepared from a suitable pyridine derivative and a phenyl amidine. It further illustrates how a suitably substituted compound of this type can be further modified after it has been synthesized to provide other compounds of formula (1).
- R 2 NH is bis-(p-methoxy- benzyl)amine
- R 2 N in Scheme 5 represents a bis(p-methoxybenzyl)amine
- the /7-methoxybenzyl groups can be cleaved by well-known methods such as reduction or treatment with a strong acid, leaving NH 2 , which can be derivatized by methods well known in the art.
- Schcmc 6 Preparation of pyrido[2,3-d]pyrimidines
- Methyl 2-amino-3-pyridinecarboxylate (6a) is reacted with an aroyl chloride in the presence of a suitable solvent such as chloroform or pyridine to afford
- 2-aroylaminopyridin-3-carboxylates (6b) The latter carboxylates (6b) are converted into 2-acylaminopyridin-3 -amides (6d), for example by reacting the starting carboxylates with ammonia.
- 2-acylaminopyridin-3 -amides (6d) may be obtained directly by aroylation of a 2-amino-3-pyridineamide (6c).
- the 2-acylaminopyridin-3 -amides (6d) are then cyclized by the addition of a base to form pyrido[2,3-d]pyrimidin-4-ol derivates of formula (6e).
- the alcohol group in the latter may then be replaced by a halogen with the help of a halogenating agent such as thionyl chloride in a suitable solvent like chloroform, dichloroethane or tetrahydroiuran (THF), preferably in the presence of a catalytic amount of dimethylformamide (DMF).
- a halogenating agent such as thionyl chloride in a suitable solvent like chloroform, dichloroethane or tetrahydroiuran (THF)
- THF tetrahydroiuran
- DMF dimethylformamide
- formula 5 preferably in the presence of a suitable base, e.g. a tertiary amine such as TEA or DIPEA, in an organic solvent such as DCM, THF or DMF.
- a suitable base e.g. a tertiary amine such as TEA or DIPEA
- the 2-aroylaminopyridin-3 -amides (6e) may be converted in a one-pot procedure into the pyrido[2,3-d]pyrimidines of formula (II) by reacting (6e) with an aminopyridinamide as specified in the previous paragraph, with a suitable base, in particular a tertiary amine such as TEA or DIPEA, in the presence of benzotriazo Ie-I- yl-oxy-tris-pyrrolidino-phosphonium hexafluorophosphate (PyBOP).
- the pyrimidine amine in this reaction may be a 4-aminonicotinic acid alkyl ester such as the methyl ester, which is converted after the substitution reaction to the corresponding acid (7d) and then condensed with an amine HNR 1 R 2 using an amide forming agent such as a carbodiimide or PyBOP.
- an amide forming agent such as a carbodiimide or PyBOP.
- the pyridine compounds of the present invention can be oxidized to N-oxides using commonly known oxidation reagents such as, for example, meta-chloroperoxy benzoic acid or peracetic acid.
- the compounds of the invention are useful in treating conditions associated with conditions characterized by excessive TGF ⁇ activity such as fibroproliferation.
- the compounds of the invention or their pharmaceutically acceptable salts or prodrug forms are also useful for the manufacture of a medicament for prophylactic or therapeutic treatment of mammals, including humans, in respect of conditions characterized by excessive activity of TGF ⁇ .
- TGF ⁇ inhibition activity is useful in treating fibroproliferative diseases, treating collagen vascular disorders, treating eye diseases associated with a fibroproliferative condition, venting excessive scarring, treating neurological conditions and other conditions that are targets for TGF ⁇ inhibitors and in preventing excessive scarring that elicits and accompanies restenosis following coronary angioplasty, cardiac fibrosis occurring after infarction and progressive heart failure, and in hypertensive vasculopathy, and keloid formation or hypertrophic scars occurring during the healing of wounds including surgical wounds and traumatic lacerations.
- Neurological conditions characterized by TGF ⁇ production include CNS injury after traumatic and hypoxic insults, Alzheimer's disease, and Parkinson's disease.
- Other conditions that are potential clinical targets for TGF ⁇ inhibitors include myelofibrosis, tissue thickening resulting from radiation treatment, nasal polyposis, polyp surgery, liver cirrhosis, and osteoporosis.
- Cardiovascular diseases such as congestive heart failure, dilated cardiomyopathy, myocarditis, or vascular stenosis associated with atherosclerosis, angioplasty treatment, or surgical incisions or mechanical trauma
- kidney diseases associated with fibrosis and/or sclerosis including glomerulonephritis of all etiologies, diabetic nephropathy, and all causes of renal interstitial fibrosis, including hypertension, complications of drug exposure, such as cyclosporin, HIV-associated nephropathy, transplant nephropathy, chronic ureteral obstruction
- hepatic diseases associated with excessive scarring and progressive sclerosis including cirrhosis due to all etiologies, disorders of the biliary tree, and hepatic dysfunction attributable to infections such as hepatitis virus or parasites
- the invention provides the compounds of the invention for use as a medicament, in particular for use as a medicament for treating conditions associated with HCV infection.
- the invention moreover relates to the use for the manufacture of a medicament for the prophylactic or therapeutic treatment of mammals, including humans, running the risk of developing or suffering from conditions associated with hepatitis C virus.
- TGF ⁇ The modulation of the immune and inflammation systems by TGF ⁇ (Wahl, et al, Immunol.
- TGF ⁇ is a stimulator for the excess production of extracellular matrix proteins, including fibronectin and collagen. It also inhibits the production of enzymes that degrade these matrix proteins. The net effect is the accumulation of fibrous tissue which is the hallmark of fibroproliferative diseases.
- TGF ⁇ is active as a homodimer, but is synthesized and secreted from cells as an inactive latent complex of the mature homodimer and proregions, called latency associated protein (LAP). These proteins bind to each other through noncovalent interactions (Lyons and Moses, Eur. J. Biochem. (1990) 187:467). LAP is often disulfide-linked to separate gene products, called latent TGF ⁇ binding proteins or LTBP's. These latent forms provide stability for the mature cytokine and a means for targeting it to the extracellular matrix and cell surfaces (Lawrence, Eur. Cytokine Network (1996) 7:363-74).
- Activation of the latent complex occurs after secretion from cells and is believed to result from the action of proteases, such as plasmin (Munger, et al., Kidney Intl. (1997) 51:1376-82), on LAP, thrombospondin-1 binding (Crawford, et al., Cell (1998) 93:1159-70), and binding to the integrin v6 (Munger, et al., Cell (1999) 319-28).
- proteases such as plasmin (Munger, et al., Kidney Intl. (1997) 51:1376-82), on LAP, thrombospondin-1 binding (Crawford, et al., Cell (1998) 93:1159-70), and binding to the integrin v6 (Munger, et al., Cell (1999) 319-28).
- proteases such as plasmin (Munger, et al., Kidney Intl. (1997) 51:
- Type IV is present only in the pituitary gland while the others are ubiquitous.
- the binding affinities among the three isoforms for the type I and II receptors differ such that these two receptors bind TGF ⁇ l and TGF ⁇ 3 more tightly than TGF ⁇ 2 (Massague, Cell (1992) 69:1067-70).
- the type IV receptor or endoglin has a similar isoform binding profile in contrast to the type III receptor, betaglycan, which binds equally well to all three isoforms (Wang, et al, Cell (1991) 67:797-805; Lopez-Casillas, Cell (1991) 67:785-95).
- the type V receptor binds to IGFBP-3 and is thought to have an active kinase domain similar to the type I and II receptors.
- the bound receptor then recruits type I receptor into a multimeric membrane complex, whereupon the constitutively active type II receptor kinase phosphorylates and activates type I receptor kinase.
- the function of the type I receptor kinase is to phosphorylate a receptor-associated co-transcription factor, smad-2/3, thereby releasing it into the cytoplasm where it binds to smad-4.
- This smad complex translocates into the nucleus, associates with a DNA-binding cofactor, such as Fast-1, binds to enhancer regions of specific genes, and activates transcription.
- the expression of these genes leads to the synthesis of cell cycle regulators that control proliferative responses or extracellular matrix proteins that mediate outside-in cell signaling, cell adhesion, migration, and intercellular communication.
- the manner of administration and formulation of the compounds useful in the invention and their related compounds will depend on the nature of the condition, the severity of the condition, the particular subject to be treated, and the judgment of the practitioner; formulation will depend on mode of administration.
- the compounds of the invention are small molecules, they are conveniently administered by oral administration by compounding them with one or more suitable pharmaceutical excipients so as to provide tablets, capsules, syrups, and the like.
- suitable formulations for oral administration may also include minor components such as buffers, flavoring agents and the like.
- the amount of active ingredient in the formulations will be in the range of 5%-95% of the total formulation, but wide variation is permitted depending on the carrier.
- Suitable carriers include sucrose, pectin, magnesium stearate, lactose, peanut oil, olive oil, water, and the like.
- the compounds useful in the invention may also be administered through suppositories or other transmucosal vehicles.
- such formulations will include excipients that facilitate the passage of the compound through the mucosa such as pharmaceutically acceptable detergents.
- the compounds may also be administered topically, for topical conditions such as psoriasis, or in formulation intended to penetrate the skin. These include lotions, creams, ointments and the like which can be formulated by known methods.
- the compounds may also be administered by injection, including intravenous, intramuscular, subcutaneous or intraperitoneal injection. Typical formulations for such use are liquid formulations in isotonic vehicles such as Hank's solution or Ringer's solution.
- Alternative formulations include nasal sprays, liposomal formulations, slow-release formulations, and the like, as are known in the art. Any suitable formulation may be used. A compendium of art-known formulations is found in Remington's Pharmaceutical Sciences, latest edition, Mack Publishing Company, Easton, PA. Reference to this manual is routine in the art.
- the dosages of the compounds of the invention will depend on a number of factors which will vary from patient to patient. However, it is believed that generally, the routine oral dosage will utilize 0.001-100 mg/kg total body weight, preferably from 0.01-50 mg/kg and more preferably about 0.01 mg/kg-10 mg/kg. Dosages will typically be administered at least once per day, but the dose regimen will vary, depending on the conditions being treated and the judgment of the practitioner. For some uses, the compounds or compositions may be administered several times per day and for other uses they may be administered less frequently than once per day.
- the compounds of the present invention can be administered as individual active ingredients, or as mixtures of several embodiments of this formula.
- the compounds of the invention may be used as single therapeutic agents or in combination with other therapeutic agents.
- Drugs that could be usefully combined with these compounds include natural or synthetic corticosteroids, particularly prednisone and its derivatives, monoclonal antibodies targeting cells of the immune system, antibodies or soluble receptors or receptor fusion proteins targeting immune or nonimmune cytokines, and small molecule inhibitors of cell division, protein synthesis, or mRNA transcription or translation, or inhibitors of immune cell differentiation or activation.
- the compounds of the invention may be used in humans, they are also available for veterinary use in treating animal subjects.
- Compounds of the invention show anti- viral properties and in particular are active against HCV.
- Compounds of the invention therefore are useful in the treatment of individuals infected by HCV and for the prophylaxic treatment of individuals at risk of being infected.
- Compounds of the present invention may also find use in the treatment of warm-blooded animals infected with flaviviruses.
- Conditions which may be prevented or treated with compounds of the present invention are conditions associated with HCV and other pathogenic flaviviruses, such as Yellow fever, Dengue fever (types 1-4), haemorraghic fever, encephalitis (St.
- HCV human encephalitis
- Japanese encephalitis Japanese encephalitis
- Murray valley encephalitis West Nile virus
- Kunjin virus Conditions associated with HCV include progressive liver fibrosis, inflammation and necrosis leading to cirrhosis, end-stage liver disease, and HCC.
- the present invention provides a method of treating HCV infection in a warm-blood animal, in particular a human, said method comprising the administration of an effective amount of a compound of formula (I), and in particular a compound of formula (II) or (III), as specified herein.
- this invention provides a method for treating a warm-blooded animal, in particular a human, from conditions associated with HCV infection said method comprising the administration of an effective amount of a compound of formula (I) and in particular a compound of formula (II) or (III), as specified herein.
- Compounds of the invention and in particular compounds of formula (II) or (III) or any subgroup thereof, may therefore be used as medicines against the above-mentioned conditions.
- Said use as a medicine or method of treatment comprises the systemic administration to HCV-infected subjects of an amount effective to combat the conditions associated with HCV and other pathogenic flaviviruses. Consequently, the compounds of the present invention can be used in the manufacture of a medicament useful for treating conditions associated with HCV and other pathogenic flaviviruses.
- the invention relates to the use of a compound of the invention and in particular a compound of formula (II) or (III) or any subgroup thereof as defined herein in the manufacture of a medicament for treating or combating infection or disease associated with HCV infection in a mammal.
- the invention also relates to a method of treating a flaviviral infection, or a disease associated with flavivirus infection comprising administering to a mammal in need thereof an effective amount of a compound of the invention and in particular of a compound of formula (II) or (III) or a subgroup thereof as defined herein.
- the present invention relates to the use of a compound of the invention and in particular a compound formula (II) or (III) or any subgroup thereof as defined herein, for the manufacture of a medicament useful for inhibiting viral activity in a mammal infected with flaviviruses, in particular with HCV.
- the present invention relates to the use of formula (II) or (III) or any subgroup thereof as defined herein for the manufacture of a medicament useful for inhibiting viral activity in a mammal infected with flaviviruses, or in particular infected with HCV, wherein said flaviviruses or HCV is inhibited in their or its replication.
- the invention furthermore relates to combinations of a compound of this invention, in particular a compound of formula (II) or (III) as specified herein, and another anti- HCV compound.
- the invention also provides methods of treating warm-blooded animals, in particular humans, suffering from HIV infection or conditions associated with HCV infection, as mentioned above, said methods comprising the administration of a combination of a compound of this invention, in particular a compound of formula (II) or (III) as specified herein, and another anti-HCV compound.
- Anti-HCV compounds comprise, for instance, interferon- ⁇ (IFN- ⁇ ), pegylated interferon- ⁇ and/or ribavirin.
- IFN- ⁇ interferon- ⁇
- the combinations of a compound of the invention and in particular of a compound of formula (II) or (III), with another anti-HCV compound can be used as a medicine in a combination therapy.
- combination therapy relates to a product containing (a) a compound of the invention, in particular a compound of formula (II) or (III), and (b) another anti-HCV compound, as a combined preparation for simultaneous, separate or sequential use in treatment of HCV infections, in particular, in the treatment of infections with HCV type 1.
- the compounds of the invention may be co-administered in combination with for instance, interferon- ⁇ (IFN- ⁇ ), pegylated interferon- ⁇ and/or ribavirin, as well as therapeutics based on antibodies targeted against HCV epitopes, small interfering RNA (Si RNA), ribozymes, DNAzymes, antisense RNA, small molecule antagonists of for instance NS3 protease, NS3 helicase and NS5B polymerase.
- IFN- ⁇ interferon- ⁇
- Si RNA small interfering RNA
- ribozymes DNAzymes
- antisense RNA small molecule antagonists of for instance NS3 protease, NS3 helicase and NS5B polymerase.
- the present invention relates to the use of a compound of the invention, in particular a compound of formula (II) or (III) or any subgroup thereof as defined above, for the manufacture of a medicament useful for inhibiting HCV activity in a mammal infected with HCV viruses, wherein said medicament is used in a combination therapy, said combination therapy preferably comprising a compound of formula (II) or (III) and (pegylated) IFN- ⁇ and/or ribavirin, and possibly an anti-HIV compound.
- Appropriate cell types can be equipped by stable transfection with a luciferase reporter gene whose expression is dependent on a constitutively active gene promoter, and such cells can be used as a counter-screen to eliminate non-selective inhibitors.
- a luciferase reporter gene whose expression is dependent on a constitutively active gene promoter
- Such cells can be used as a counter-screen to eliminate non-selective inhibitors.
- the following examples are intended to illustrate, but not to limit, the invention.
- 2,6-Difluoro-nicotinic acid To a solution of anhydrous THF (50 mL) and diisopropyl amine (14.02 mL) cooled to-78°C was added n-BuLi (2M, 50 mL). The mixture was allowed to warm to O 0 C for 30 min and was cooled to -78 0 C. 2,6-Di- fluoropyridine (11.5 g) dissolved in THF (200 mL) was added to the LDA mixture at -78 0 C. The mixture stirred at -78 0 C for 2h, the ice bath was removed and the mixture stirred at O 0 C for 10 min.
- 2,6-Difluoro-nicotinoyl chloride A mixture of 2,6-difluoronicotinic acid (6.2 g), thionyl chloride (15 mL) and CH 2 Cl 2 (100 mL) was heated to reflux for 3 h. The mixture was evaporated to dryness, CH 2 Cl 2 , was added and evaporated to dryness to afford 1.1 g of the 2,6-difluoronicotinic acid chloride. This material used without further purification.
- the 2-(5-Chloro-2-fluoro-phenyl)-7-(2-di- methylamino-ethylamino)-pyrido[2,3-d]pyrimidin-4-ol (0.18 g) was dissolved in P(O)Cl 3 (10 mL) and heated to reflux for 2 hr. The mixture was reduced in volume and NaHCO 3 (sat aq) was added.
- reaction mixture was evaporated to dryness and purified by silica gel chromatography (dicholoromethane/ EtOAc gradient 95/5 to 5/95) to afford 4-[7-Amino-2-(5-chloro-2-fluoro-phenyl)-pyrido[2,3-d]pyrimidin-4-ylamino]- N-methyl-nicotinamide (0.78 g).
- TGF ⁇ R 1 autophosphorylation protocol The compounds of the invention were tested for their ability to inhibit TGF ⁇ by a TGF ⁇ R 1 autophosphorylation protocol. This was conducted as follows: Compound dilutions and reagents were prepared fresh daily. Compounds were diluted from DMSO stock solutions to 2 times the desired assay concentration, keeping final DMSO concentration in the assay less than or equal to 1%. TGF ⁇ Rl was diluted to 4 times the desired assay concentration in buffer + DTT. ATP was diluted into 4x reaction buffer, and gamma- 33 P-ATP was added at 60uCi/mL.
- the assay was performed by adding lOul of the enzyme to 20ul of the compound solution.
- the reaction was initiated by the addition of lOul of ATP mix.
- Final assay conditions included lOuM ATP, 17OnM TGF ⁇ Rl, and IM DTT in 2OmM MOPS, pH7.
- the reactions were incubated at room temperature for 20 minutes.
- the reactions were stopped by transferring 23ul of reaction mixture onto a phosphocellulose 96-well filter plate, which had been pre-wetted with 15ul of 0.25M H 3 PO 4 per well. After 5 minutes, the wells were washed 4x with 75mM H 3 PO 4 and once with 95% ethanol.
- the plate was dried, scintillation cocktail was added to each well, and the wells were counted in a Packard TopCount microplate scintillation counter.
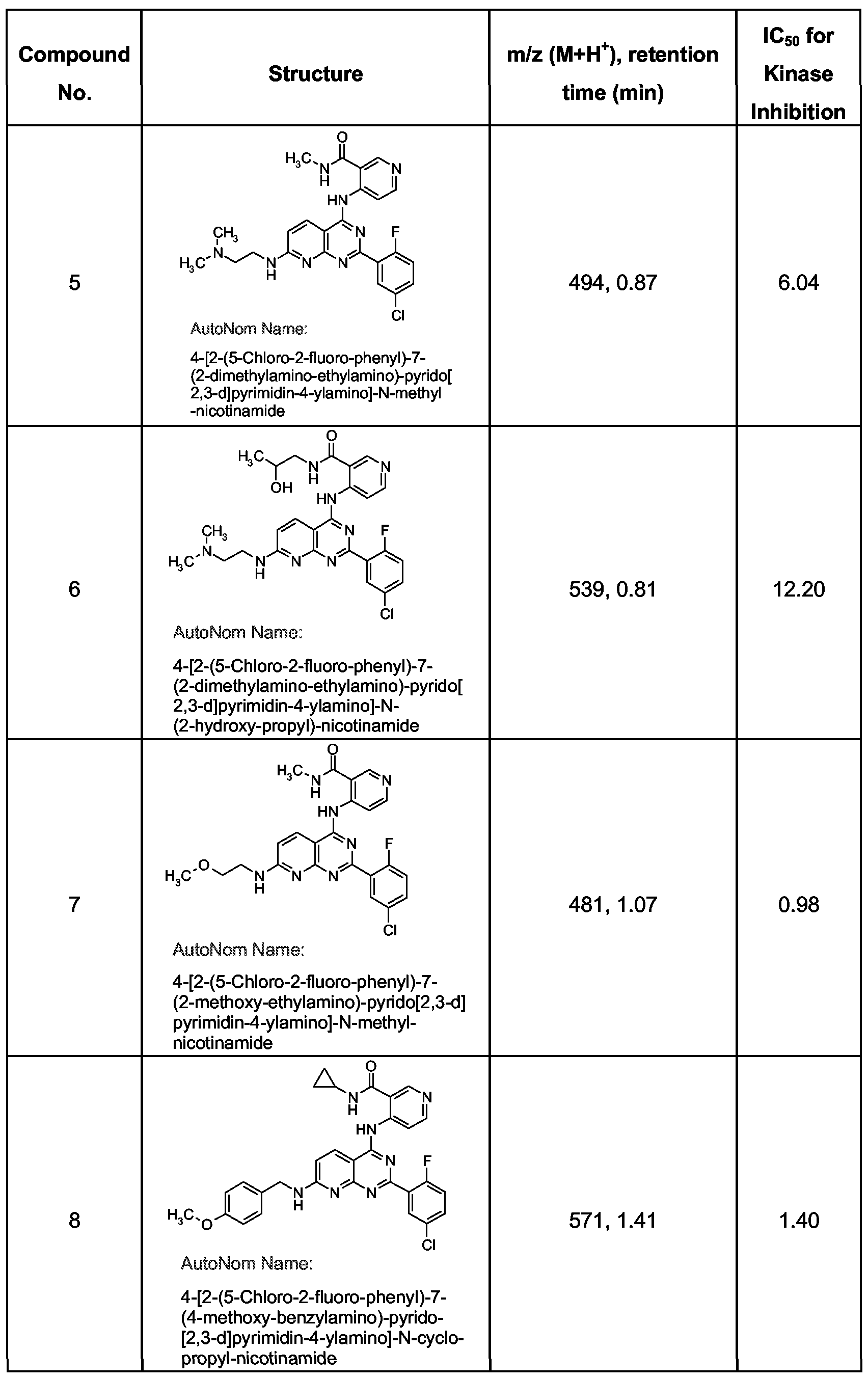
- the compounds in Table 1 were prepared by the methods set forth herein. The compounds were characterized at least by LC-mass spectrometry. For each compound in the Table, the product observed by LC (liquid chromatography) provided the molecular ion expected for the desired product; the characteristic ion is listed in Table 1 for each compound, along with the retention time from the LC. These compounds provide, in this assay, IC 50 values in the range of 0.01-12 micromolar. Table 1
- HPLC solvents A: water with 0.1% trifluoroacetic acid.
- the pyrido[2,3-d]pyrimidine compounds of the present invention were examined for activity in the inhibition of HCV RNA replication in a cellular assay.
- the assay demonstrated that the tested compounds exhibit activity against HCV replicons functional in a cell culture.
- the cellular assay was based on a bicistronic expression construct, as described by Lohmann et al. (1999) Science vol. 285 pp. 110-113 with modifications described by Krieger et al. (2001) Journal of Virology 75: 4614-4624, in a multi-target screening strategy. In essence, the method was as follows.
- the assay utilized the stably transfected cell line Huh-7 luc/neo (hereafter referred to as Huh-Luc).
- This cell line harbored an RNA encoding a bicistronic expression construct comprising the wild type NS3-NS5B regions of HCV type Ib translated from an Internal Ribosome Entry Site (IRES) from encephalomyocarditis virus (EMCV), preceded by a reporter portion (FfL- luciferase), and a selectable marker portion (neo R , neomycine phosphotransferase). The construct was bordered by 5' and 3' NTRs (non- translated regions) from HCV type Ib.
- G418 neo R
- the stably transfected replicon cells that expressed HCV RNA which replicated autonomously and to high levels, encoding inter alia luciferase, were used for screening the antiviral compounds.
- the replicon cells were plated in 384-well plates in the presence of the test and control compounds which were added in various concentrations. Following an incubation of three days, HCV replication was measured by assaying luciferase activity (using standard luciferase assay substrates and reagents and a Perkin Elmer ViewLux Tm ultraHTS microplate imager). Replicon cells in the control cultures had high luciferase expression in the absence of any inhibitor. The inhibitory activity of the compound on luciferase activity was monitored on the Huh-Luc cells, enabling a dose-response curve for each test compound. EC 50 values were then calculated, which value represents the amount of the compound required to decrease by 50% the level of detected luciferase activity, or more specifically, the ability of the genetically linked HCV replicon RNA to replicate.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Virology (AREA)
- Epidemiology (AREA)
- Molecular Biology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description





























Claims


Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP06708816A EP1869037B1 (en) | 2005-03-25 | 2006-03-27 | Heterobicylic inhibitors of hvc |
| DK06708816.1T DK1869037T3 (en) | 2005-03-25 | 2006-03-27 | Heterobicyclic inhibitors of HVC |
| AT06708816T ATE517897T1 (en) | 2005-03-25 | 2006-03-27 | HETEROBICYCLIC INHIBITORS OF HVC |
| JP2008502426A JP2008534479A (en) | 2005-03-25 | 2006-03-27 | Heterobicyclic inhibitors of HCV |
| AU2006226322A AU2006226322B2 (en) | 2005-03-25 | 2006-03-27 | Heterobicylic inhibitors of HCV |
| CA2602294A CA2602294C (en) | 2005-03-25 | 2006-03-27 | Heterobicylic inhibitors of hvc |
| US11/909,118 US8030318B2 (en) | 2005-03-25 | 2006-03-27 | Fused bicyclic inhibitors of HCV |
| MX2007011850A MX2007011850A (en) | 2005-03-25 | 2006-03-27 | Heterobicylic inhibitors of hcv. |
| CN2006800091593A CN101189234B (en) | 2005-03-25 | 2006-03-27 | Heterobicyclic inhibitors of HCV |
| BRPI0609650-6A BRPI0609650A2 (en) | 2005-03-25 | 2006-03-27 | heterobicolytic hvc inhibitors |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US66515105P | 2005-03-25 | 2005-03-25 | |
| US60/665,151 | 2005-03-25 | ||
| US68040505P | 2005-05-12 | 2005-05-12 | |
| US60/680,405 | 2005-05-12 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006100310A1 true WO2006100310A1 (en) | 2006-09-28 |
Family
ID=36572091
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2006/061070 WO2006100310A1 (en) | 2005-03-25 | 2006-03-27 | Heterobicylic inhibitors of hcv |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US8030318B2 (en) |
| EP (1) | EP1869037B1 (en) |
| JP (1) | JP2008534479A (en) |
| KR (1) | KR20070122194A (en) |
| AT (1) | ATE517897T1 (en) |
| AU (1) | AU2006226322B2 (en) |
| BR (1) | BRPI0609650A2 (en) |
| CA (1) | CA2602294C (en) |
| DK (1) | DK1869037T3 (en) |
| MX (1) | MX2007011850A (en) |
| RU (1) | RU2405783C2 (en) |
| WO (1) | WO2006100310A1 (en) |
Cited By (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006120252A2 (en) * | 2005-05-12 | 2006-11-16 | Tibotec Pharmaceuticals Ltd. | Pyrido[2,3-d]pyrimidines useful as hcv inhibitors, and methods for the preparation thereof |
| WO2008121634A2 (en) | 2007-03-30 | 2008-10-09 | Pharmasset, Inc. | Nucleoside phosphoramidate prodrugs |
| JP2009521479A (en) * | 2005-12-21 | 2009-06-04 | アボット・ラボラトリーズ | Antiviral compounds |
| WO2010002779A2 (en) * | 2008-07-03 | 2010-01-07 | Merck Serono S.A. | Naphthyridininones as aurora kinase inhibitors |
| JP2010505797A (en) * | 2006-10-04 | 2010-02-25 | テイボテク・フアーマシユーチカルズ・リミテツド | Carboxamide 4-[(4-pyridyl) amino] pyrimidine useful as an HCV inhibitor |
| WO2010075549A2 (en) | 2008-12-23 | 2010-07-01 | Pharmasset, Inc. | Nucleoside phosphoramidates |
| WO2010075554A1 (en) | 2008-12-23 | 2010-07-01 | Pharmasset, Inc. | Synthesis of purine nucleosides |
| WO2010075517A2 (en) | 2008-12-23 | 2010-07-01 | Pharmasset, Inc. | Nucleoside analogs |
| WO2010135569A1 (en) | 2009-05-20 | 2010-11-25 | Pharmasset, Inc. | N- [ (2 ' r) -2 ' -deoxy-2 ' -fluoro-2 ' -methyl-p-phenyl-5 ' -uridylyl] -l-alanine 1-methylethyl ester and process for its production |
| US7915411B2 (en) | 2005-12-21 | 2011-03-29 | Abbott Laboratories | Anti-viral compounds |
| US7977342B2 (en) | 2004-09-30 | 2011-07-12 | Tibotec-Virco Virology Bvba | HCV inhibiting bi-cyclic pyrimidines |
| US8030318B2 (en) | 2005-03-25 | 2011-10-04 | Tibotec Pharmaceuticals Ltd. | Fused bicyclic inhibitors of HCV |
| WO2011123645A2 (en) | 2010-03-31 | 2011-10-06 | Pharmasset, Inc. | Nucleoside phosphoramidates |
| WO2011123672A1 (en) | 2010-03-31 | 2011-10-06 | Pharmasset, Inc. | Purine nucleoside phosphoramidate |
| US8236950B2 (en) | 2006-12-20 | 2012-08-07 | Abbott Laboratories | Anti-viral compounds |
| US8575184B2 (en) | 2009-09-03 | 2013-11-05 | Bristol-Myers Squibb Company | Quinazolines as potassium ion channel inhibitors |
| US8618076B2 (en) | 2009-05-20 | 2013-12-31 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US8729079B2 (en) | 2011-08-23 | 2014-05-20 | Endo Pharmaceuticals Inc. | Pyrimido-pyridazinone compounds and methods of use thereof |
| US8759510B2 (en) | 2008-06-11 | 2014-06-24 | Gilead Pharmasset Llc | Nucleoside cyclicphosphates |
| US8841275B2 (en) | 2010-11-30 | 2014-09-23 | Gilead Pharmasset Llc | 2′-spiro-nucleosides and derivatives thereof useful for treating hepatitis C virus and dengue virus infections |
| US8889159B2 (en) | 2011-11-29 | 2014-11-18 | Gilead Pharmasset Llc | Compositions and methods for treating hepatitis C virus |
| US9290502B2 (en) | 2005-05-12 | 2016-03-22 | Janssen Sciences Ireland Uc | Pteridines useful as HCV inhibitors and methods for the preparation thereof |
| US10456414B2 (en) | 2011-09-16 | 2019-10-29 | Gilead Pharmasset Llc | Methods for treating HCV |
| US11116783B2 (en) | 2013-08-27 | 2021-09-14 | Gilead Pharmasset Llc | Combination formulation of two antiviral compounds |
| US12036224B2 (en) | 2017-04-28 | 2024-07-16 | Libertas Bio, Inc. | Formulations, methods, kits, and dosage forms for treating atopic dermatitis and for improved stability of an active pharmaceutical ingredient |
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TW201639852A (en) * | 2008-12-09 | 2016-11-16 | 吉李德科學股份有限公司 | Intermediate compounds for preparation of compounds useful as modulators of toll-like receptors |
| MX2012006877A (en) | 2009-12-18 | 2012-08-31 | Idenix Pharmaceuticals Inc | 5,5-fused arylene or heteroarylene hepatitis c virus inhibitors. |
| US8563530B2 (en) | 2010-03-31 | 2013-10-22 | Gilead Pharmassel LLC | Purine nucleoside phosphoramidate |
| US8940742B2 (en) | 2012-04-10 | 2015-01-27 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| SG11201506021XA (en) | 2013-01-31 | 2015-08-28 | Gilead Pharmasset Llc | Combination formulation of two antiviral compounds |
| WO2015051241A1 (en) | 2013-10-04 | 2015-04-09 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| MX2016004340A (en) | 2013-10-04 | 2016-08-08 | Infinity Pharmaceuticals Inc | Heterocyclic compounds and uses thereof. |
| ES2813875T3 (en) | 2014-01-01 | 2021-03-25 | Medivation Tech Llc | Compounds and procedures for use |
| PT3119397T (en) | 2014-03-19 | 2022-04-11 | Infinity Pharmaceuticals Inc | Heterocyclic compounds for use in the treatment of pi3k-gamma mediated disorders |
| MX2017000026A (en) | 2014-07-11 | 2017-05-01 | Gilead Sciences Inc | Modulators of toll-like receptors for the treatment of hiv. |
| SI3194401T1 (en) | 2014-09-16 | 2020-12-31 | Gilead Sciences, Inc. | Solid forms of a toll-like receptor modulator |
| US9708348B2 (en) | 2014-10-03 | 2017-07-18 | Infinity Pharmaceuticals, Inc. | Trisubstituted bicyclic heterocyclic compounds with kinase activities and uses thereof |
| WO2016067099A1 (en) | 2014-10-29 | 2016-05-06 | King Abdullah University Of Science And Technology | 3-alkyl pyridinium compound from red sea sponge with potent antiviral activity |
| US10160761B2 (en) | 2015-09-14 | 2018-12-25 | Infinity Pharmaceuticals, Inc. | Solid forms of isoquinolinones, and process of making, composition comprising, and methods of using the same |
| WO2017161116A1 (en) | 2016-03-17 | 2017-09-21 | Infinity Pharmaceuticals, Inc. | Isotopologues of isoquinolinone and quinazolinone compounds and uses thereof as pi3k kinase inhibitors |
| US10919914B2 (en) | 2016-06-08 | 2021-02-16 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001047921A1 (en) | 1999-12-28 | 2001-07-05 | Pharmacopeia, Inc. | Pyrimidine and triazine kinase inhibitors |
| US6476031B1 (en) | 1998-08-28 | 2002-11-05 | Scios, Inc. | Quinazoline derivatives as medicaments |
| WO2003097615A1 (en) | 2002-05-17 | 2003-11-27 | Scios, Inc. | TREATMENT OF FIBROPROLIFERATIVE DISORDERS USING TGF-β INHIBITORS |
| US20040032430A1 (en) | 2002-06-04 | 2004-02-19 | Kai Yung | System and method for generating user interfaces for different instrument types |
| WO2004024159A1 (en) | 2002-09-10 | 2004-03-25 | Scios Inc. | INHIBITORS OF TFGβ |
| WO2004047818A2 (en) | 2002-11-22 | 2004-06-10 | Scios, Inc. | USE OF TFG-β INHIBITORSTO COUNTERACT PATHOLOGIC CHANGES IN THE LEVEL OR FUNCTION OF STEROID/THYROID RECEPTORS |
| US20040132159A1 (en) | 1999-05-13 | 2004-07-08 | Ziyang Zhong | Novel beta-secretase and modulation of beta-secretase activity |
| US20050004143A1 (en) | 2003-03-28 | 2005-01-06 | Sundeep Dugar | Bi-cyclic pyrimidine inhibitors of TGFbeta |
| WO2005032481A2 (en) * | 2003-09-30 | 2005-04-14 | Scios Inc. | Quinazoline derivatives as medicaments |
Family Cites Families (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BE789496A (en) | 1971-10-05 | 1973-03-29 | S M B Anciens Ets J Muelberger | |
| IL112249A (en) | 1994-01-25 | 2001-11-25 | Warner Lambert Co | Pharmaceutical compositions containing di and tricyclic pyrimidine derivatives for inhibiting tyrosine kinases of the epidermal growth factor receptor family and some new such compounds |
| GB2295387A (en) | 1994-11-23 | 1996-05-29 | Glaxo Inc | Quinazoline antagonists of alpha 1c adrenergic receptors |
| US6638926B2 (en) | 2000-09-15 | 2003-10-28 | Vertex Pharmaceuticals Incorporated | Pyrazole compounds useful as protein kinase inhibitors |
| CA2441492C (en) | 2001-03-23 | 2011-08-09 | Bayer Corporation | Rho-kinase inhibitors |
| GB0127433D0 (en) * | 2001-11-15 | 2002-01-09 | Smithkline Beecham Corp | Compounds |
| GB0127430D0 (en) * | 2001-11-15 | 2002-01-09 | Smithkline Beecham Corp | Compounds |
| ES2305435T3 (en) | 2002-01-10 | 2008-11-01 | Bayer Healthcare Ag | INHIBITORS OF RHO-QUINASA. |
| JP2003321472A (en) | 2002-02-26 | 2003-11-11 | Takeda Chem Ind Ltd | Grk inhibitor |
| EP1485380B1 (en) | 2002-03-15 | 2010-05-19 | Vertex Pharmaceuticals Incorporated | Azolylaminoazines as inhibitors of protein kinases |
| ATE466580T1 (en) | 2002-03-15 | 2010-05-15 | Vertex Pharma | AZOLYLAMINOAZINE AS PROTEIN KINASE INHIBITORS |
| ES2289279T3 (en) | 2002-03-15 | 2008-02-01 | Vertex Pharmaceuticals Incorporated | USEFUL COMPOSITIONS AS PROTEINQUINASE INHIBITORS. |
| AU2003225800A1 (en) | 2002-03-15 | 2003-09-29 | Hayley Binch | Azolylaminoazine as inhibitors of protein kinases |
| US7202249B2 (en) | 2002-08-27 | 2007-04-10 | Bristol-Myers Squibb Company | Antagonists of chemokine receptors |
| JP2006524633A (en) | 2002-11-22 | 2006-11-02 | サイオス・インコーポレーテツド | Methods for counteracting pathological changes in the β-adrenergic pathway |
| WO2004065392A1 (en) | 2003-01-24 | 2004-08-05 | Smithkline Beecham Corporation | Condensed pyridines and pyrimidines and their use as alk-5 receptor ligands |
| US7148226B2 (en) | 2003-02-21 | 2006-12-12 | Agouron Pharmaceuticals, Inc. | Inhibitors of hepatitis C virus RNA-dependent RNA polymerase, and compositions and treatments using the same |
| TW200626157A (en) | 2004-09-30 | 2006-08-01 | Tibotec Pharm Ltd | HCV inhibiting bi-cyclic pyrimidines |
| US20070066632A1 (en) | 2005-03-25 | 2007-03-22 | Scios, Inc. | Fused bicyclic inhibitors of TGFbeta |
| ATE517897T1 (en) | 2005-03-25 | 2011-08-15 | Tibotec Pharm Ltd | HETEROBICYCLIC INHIBITORS OF HVC |
| AR054122A1 (en) | 2005-05-12 | 2007-06-06 | Tibotec Pharm Ltd | PIRIDO [2,3-D] USEFUL PYRIMIDES AS HCV INHIBITORS, AND METHODS FOR THE PREPARATION OF THE SAME |
| AR056347A1 (en) | 2005-05-12 | 2007-10-03 | Tibotec Pharm Ltd | USE OF PTERIDINE COMPOUNDS TO MANUFACTURE PHARMACEUTICAL MEDICINES AND COMPOSITIONS |
-
2006
- 2006-03-27 AT AT06708816T patent/ATE517897T1/en not_active IP Right Cessation
- 2006-03-27 WO PCT/EP2006/061070 patent/WO2006100310A1/en active Application Filing
- 2006-03-27 BR BRPI0609650-6A patent/BRPI0609650A2/en not_active IP Right Cessation
- 2006-03-27 RU RU2007139453/04A patent/RU2405783C2/en not_active IP Right Cessation
- 2006-03-27 DK DK06708816.1T patent/DK1869037T3/en active
- 2006-03-27 JP JP2008502426A patent/JP2008534479A/en active Pending
- 2006-03-27 EP EP06708816A patent/EP1869037B1/en active Active
- 2006-03-27 KR KR1020077018260A patent/KR20070122194A/en not_active Application Discontinuation
- 2006-03-27 CA CA2602294A patent/CA2602294C/en not_active Expired - Fee Related
- 2006-03-27 AU AU2006226322A patent/AU2006226322B2/en not_active Ceased
- 2006-03-27 MX MX2007011850A patent/MX2007011850A/en active IP Right Grant
- 2006-03-27 US US11/909,118 patent/US8030318B2/en not_active Expired - Fee Related
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6476031B1 (en) | 1998-08-28 | 2002-11-05 | Scios, Inc. | Quinazoline derivatives as medicaments |
| US20040132159A1 (en) | 1999-05-13 | 2004-07-08 | Ziyang Zhong | Novel beta-secretase and modulation of beta-secretase activity |
| WO2001047921A1 (en) | 1999-12-28 | 2001-07-05 | Pharmacopeia, Inc. | Pyrimidine and triazine kinase inhibitors |
| WO2003097615A1 (en) | 2002-05-17 | 2003-11-27 | Scios, Inc. | TREATMENT OF FIBROPROLIFERATIVE DISORDERS USING TGF-β INHIBITORS |
| US20040032430A1 (en) | 2002-06-04 | 2004-02-19 | Kai Yung | System and method for generating user interfaces for different instrument types |
| WO2004024159A1 (en) | 2002-09-10 | 2004-03-25 | Scios Inc. | INHIBITORS OF TFGβ |
| WO2004047818A2 (en) | 2002-11-22 | 2004-06-10 | Scios, Inc. | USE OF TFG-β INHIBITORSTO COUNTERACT PATHOLOGIC CHANGES IN THE LEVEL OR FUNCTION OF STEROID/THYROID RECEPTORS |
| US20050004143A1 (en) | 2003-03-28 | 2005-01-06 | Sundeep Dugar | Bi-cyclic pyrimidine inhibitors of TGFbeta |
| WO2005032481A2 (en) * | 2003-09-30 | 2005-04-14 | Scios Inc. | Quinazoline derivatives as medicaments |
Non-Patent Citations (7)
| Title |
|---|
| "Remington's Pharmaceutical Sciences", MACK PUBLISHING COMPANY |
| KRIEGER ET AL., JOURNAL OF VIROLOGY, vol. 75, 2001, pages 4614 - 4624 |
| LOHMANN ET AL., SCIENCE, vol. 285, 1999, pages 110 - 113 |
| M.P. DE CAESTECKER; E. PIEK; A.B. ROBERTS, J. NATIONAL CANCER INST., vol. 92, no. 17, 2000, pages 1388 - 1402 |
| MASSAGUE ET AL., ANN. REV. CELL. BIOL., vol. 6, 1990, pages 597 - 646 |
| N. DUMONT; C.L. ARTEAGA, BREAST CANCER RES., vol. 2, 2000, pages 125 - 132 |
| ROBERTS; SPORN, HANDBOOK OF EXPERIMENTAL PHARMACOLOGY, vol. 95, 1990, pages 419 - 458 |
Cited By (76)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7977342B2 (en) | 2004-09-30 | 2011-07-12 | Tibotec-Virco Virology Bvba | HCV inhibiting bi-cyclic pyrimidines |
| US8030318B2 (en) | 2005-03-25 | 2011-10-04 | Tibotec Pharmaceuticals Ltd. | Fused bicyclic inhibitors of HCV |
| US9708328B2 (en) | 2005-05-12 | 2017-07-18 | Janssen Sciences Ireland Uc | Pteridines useful as HCV inhibitors and methods for the preparation thereof |
| WO2006120252A2 (en) * | 2005-05-12 | 2006-11-16 | Tibotec Pharmaceuticals Ltd. | Pyrido[2,3-d]pyrimidines useful as hcv inhibitors, and methods for the preparation thereof |
| US9290502B2 (en) | 2005-05-12 | 2016-03-22 | Janssen Sciences Ireland Uc | Pteridines useful as HCV inhibitors and methods for the preparation thereof |
| US9951075B2 (en) | 2005-05-12 | 2018-04-24 | Janssen Sciences Ireland Uc | Pteridines useful as HCV inhibitors and methods for the preparation thereof |
| US8022077B2 (en) | 2005-05-12 | 2011-09-20 | Tibotec Pharmaceuticals Ltd. | Pyrido[2,3-d]pyrimidines useful as HCV inhibitors, and methods for the preparation thereof |
| WO2006120252A3 (en) * | 2005-05-12 | 2007-02-22 | Tibotec Pharm Ltd | Pyrido[2,3-d]pyrimidines useful as hcv inhibitors, and methods for the preparation thereof |
| JP2009521479A (en) * | 2005-12-21 | 2009-06-04 | アボット・ラボラトリーズ | Antiviral compounds |
| US7910595B2 (en) * | 2005-12-21 | 2011-03-22 | Abbott Laboratories | Anti-viral compounds |
| US7915411B2 (en) | 2005-12-21 | 2011-03-29 | Abbott Laboratories | Anti-viral compounds |
| JP2010505797A (en) * | 2006-10-04 | 2010-02-25 | テイボテク・フアーマシユーチカルズ・リミテツド | Carboxamide 4-[(4-pyridyl) amino] pyrimidine useful as an HCV inhibitor |
| US9987277B2 (en) | 2006-10-04 | 2018-06-05 | Janssen Sciences Ireland Uc | Carboxamide 4-[(4-pyridyl)amino] pryimidines for the treatment of hepatitis C |
| US8236950B2 (en) | 2006-12-20 | 2012-08-07 | Abbott Laboratories | Anti-viral compounds |
| US10183037B2 (en) | 2007-03-30 | 2019-01-22 | Gilead Pharmasset Llc | Nucleoside phosphoramidate prodrugs |
| DE202008018643U1 (en) | 2007-03-30 | 2017-03-16 | Gilead Pharmasset Llc | Nucleosidphosphoramidat prodrugs |
| US9585906B2 (en) | 2007-03-30 | 2017-03-07 | Gilead Pharmasset Llc | Nucleoside phosphoramidate prodrugs |
| US8580765B2 (en) | 2007-03-30 | 2013-11-12 | Gilead Pharmasset Llc | Nucleoside phosphoramidate prodrugs |
| US9085573B2 (en) | 2007-03-30 | 2015-07-21 | Gilead Pharmasset Llc | Nucleoside phosphoramidate prodrugs |
| US11642361B2 (en) | 2007-03-30 | 2023-05-09 | Gilead Sciences, Inc. | Nucleoside phosphoramidate prodrugs |
| WO2008121634A2 (en) | 2007-03-30 | 2008-10-09 | Pharmasset, Inc. | Nucleoside phosphoramidate prodrugs |
| US8957046B2 (en) | 2007-03-30 | 2015-02-17 | Gilead Pharmasset Llc | Nucleoside phosphoramidate prodrugs |
| US8906880B2 (en) | 2007-03-30 | 2014-12-09 | Gilead Pharmasset Llc | Nucleoside phosphoramidate prodrugs |
| US8735372B2 (en) | 2007-03-30 | 2014-05-27 | Gilead Pharmasset Llc | Nucleoside phosphoramidate prodrugs |
| US8759510B2 (en) | 2008-06-11 | 2014-06-24 | Gilead Pharmasset Llc | Nucleoside cyclicphosphates |
| JP2011526912A (en) * | 2008-07-03 | 2011-10-20 | メルク パテント ゲゼルシャフト ミット ベシュレンクテル ハフツング | Naphthyridinone as a protein kinase inhibitor |
| AU2009267161B2 (en) * | 2008-07-03 | 2014-11-06 | Merck Patent Gmbh | Naphthyridininones as Aurora kinase inhibitors |
| WO2010002779A2 (en) * | 2008-07-03 | 2010-01-07 | Merck Serono S.A. | Naphthyridininones as aurora kinase inhibitors |
| WO2010002779A3 (en) * | 2008-07-03 | 2011-03-03 | Merck Patent Gmbh | Naphthyridininones as aurora kinase inhibitors |
| WO2010075554A1 (en) | 2008-12-23 | 2010-07-01 | Pharmasset, Inc. | Synthesis of purine nucleosides |
| EP2671888A1 (en) | 2008-12-23 | 2013-12-11 | Gilead Pharmasset LLC | 3',5'-cyclic nucleoside phosphate analogues |
| US8716263B2 (en) | 2008-12-23 | 2014-05-06 | Gilead Pharmasset Llc | Synthesis of purine nucleosides |
| US8716262B2 (en) | 2008-12-23 | 2014-05-06 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| WO2010075517A2 (en) | 2008-12-23 | 2010-07-01 | Pharmasset, Inc. | Nucleoside analogs |
| EP3222628A1 (en) | 2008-12-23 | 2017-09-27 | Gilead Pharmasset LLC | Nucleoside phosphoramidates |
| WO2010075549A2 (en) | 2008-12-23 | 2010-07-01 | Pharmasset, Inc. | Nucleoside phosphoramidates |
| US8551973B2 (en) | 2008-12-23 | 2013-10-08 | Gilead Pharmasset Llc | Nucleoside analogs |
| US9045520B2 (en) | 2008-12-23 | 2015-06-02 | Gilead Pharmasset Llc | Synthesis of purine nucleosides |
| US8957045B2 (en) | 2008-12-23 | 2015-02-17 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| EP3321275A1 (en) | 2009-05-20 | 2018-05-16 | Gilead Pharmasset LLC | Crystalline form of sofosbuvir |
| WO2010135569A1 (en) | 2009-05-20 | 2010-11-25 | Pharmasset, Inc. | N- [ (2 ' r) -2 ' -deoxy-2 ' -fluoro-2 ' -methyl-p-phenyl-5 ' -uridylyl] -l-alanine 1-methylethyl ester and process for its production |
| US8618076B2 (en) | 2009-05-20 | 2013-12-31 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US8629263B2 (en) | 2009-05-20 | 2014-01-14 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| EP2610264A2 (en) | 2009-05-20 | 2013-07-03 | Gilead Pharmasset LLC | N-[(2'r)-2'-deoxy-2'-fluoro-2'-methyl-p-phenyl-5'-uridylyl]-l-alanine 1-methylethyl ester and process for its production |
| US8633309B2 (en) | 2009-05-20 | 2014-01-21 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US8642756B2 (en) | 2009-05-20 | 2014-02-04 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US8735569B2 (en) | 2009-05-20 | 2014-05-27 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| EP2910562A1 (en) | 2009-05-20 | 2015-08-26 | Gilead Pharmasset LLC | N-[(2'r)-2'-deoxy-2 '-fluoro-2'-methyl-p-phenyl-5 '-uridylyl]-l-alanine 1-methylethyl ester in crystalline form |
| EP2913337A1 (en) | 2009-05-20 | 2015-09-02 | Gilead Pharmasset LLC | N-[(2'r)-2'-deoxy-2'-fluoro-2'-methyl-p-phenyl-5'-uridylyl]-l-alanine 1-methylethyl ester and process for its production |
| US9206217B2 (en) | 2009-05-20 | 2015-12-08 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US9284342B2 (en) | 2009-05-20 | 2016-03-15 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US9637512B2 (en) | 2009-05-20 | 2017-05-02 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US9822096B2 (en) | 2009-09-03 | 2017-11-21 | Bristol-Myers Squibb Company | Quinazolines as potassium ion channel inhibitors |
| US9458114B2 (en) | 2009-09-03 | 2016-10-04 | Bristol-Myers Squibb Company | Quinazolines as potassium ion channel inhibitors |
| US11008306B2 (en) | 2009-09-03 | 2021-05-18 | Bristol-Myers Squibb Company | Quinazolines as potassium ion channel inhibitors |
| US10676460B2 (en) | 2009-09-03 | 2020-06-09 | Bristol-Myers Squibb Company | Quinazolines as potassium ion channel inhibitors |
| US8575184B2 (en) | 2009-09-03 | 2013-11-05 | Bristol-Myers Squibb Company | Quinazolines as potassium ion channel inhibitors |
| US10214511B2 (en) | 2009-09-03 | 2019-02-26 | Bristol-Myers Squibb Company | Quinazolines as potassium ion channel inhibitors |
| US8859756B2 (en) | 2010-03-31 | 2014-10-14 | Gilead Pharmasset Llc | Stereoselective synthesis of phosphorus containing actives |
| WO2011123672A1 (en) | 2010-03-31 | 2011-10-06 | Pharmasset, Inc. | Purine nucleoside phosphoramidate |
| WO2011123645A2 (en) | 2010-03-31 | 2011-10-06 | Pharmasset, Inc. | Nucleoside phosphoramidates |
| EP3290428A1 (en) | 2010-03-31 | 2018-03-07 | Gilead Pharmasset LLC | Tablet comprising crystalline (s)-isopropyl 2-(((s)-(((2r,3r,4r,5r)-5-(2,4-dioxo-3,4-dihydropyrimidin-1 (2h)-yl)-4-fluoro-3-hydroxy-4-methyltetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)propanoate |
| WO2011123668A2 (en) | 2010-03-31 | 2011-10-06 | Pharmasset, Inc. | Stereoselective synthesis of phosphorus containing actives |
| EP2752422A1 (en) | 2010-03-31 | 2014-07-09 | Gilead Pharmasset LLC | Stereoselective synthesis of phosphorus containing actives |
| EP2609923A2 (en) | 2010-03-31 | 2013-07-03 | Gilead Pharmasset LLC | Nucleoside Phosphoramidates |
| US8841275B2 (en) | 2010-11-30 | 2014-09-23 | Gilead Pharmasset Llc | 2′-spiro-nucleosides and derivatives thereof useful for treating hepatitis C virus and dengue virus infections |
| US10183944B2 (en) | 2011-08-23 | 2019-01-22 | Asana Biosciences, Llc | Pyrimido-pyridazinone compounds and methods of use thereof |
| US8729079B2 (en) | 2011-08-23 | 2014-05-20 | Endo Pharmaceuticals Inc. | Pyrimido-pyridazinone compounds and methods of use thereof |
| US10647720B2 (en) | 2011-08-23 | 2020-05-12 | Asan BioSciences, LLC | Pyrimido-pyridazinone compounds and methods of use thereof |
| US9382277B2 (en) | 2011-08-23 | 2016-07-05 | Asana Biosciences, Llc | Pyrimido-pyridazinone compounds and methods of use thereof |
| US10456414B2 (en) | 2011-09-16 | 2019-10-29 | Gilead Pharmasset Llc | Methods for treating HCV |
| US9549941B2 (en) | 2011-11-29 | 2017-01-24 | Gilead Pharmasset Llc | Compositions and methods for treating hepatitis C virus |
| US8889159B2 (en) | 2011-11-29 | 2014-11-18 | Gilead Pharmasset Llc | Compositions and methods for treating hepatitis C virus |
| US11116783B2 (en) | 2013-08-27 | 2021-09-14 | Gilead Pharmasset Llc | Combination formulation of two antiviral compounds |
| US11707479B2 (en) | 2013-08-27 | 2023-07-25 | Gilead Sciences, Inc. | Combination formulation of two antiviral compounds |
| US12036224B2 (en) | 2017-04-28 | 2024-07-16 | Libertas Bio, Inc. | Formulations, methods, kits, and dosage forms for treating atopic dermatitis and for improved stability of an active pharmaceutical ingredient |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1869037A1 (en) | 2007-12-26 |
| DK1869037T3 (en) | 2011-10-24 |
| CA2602294C (en) | 2015-02-24 |
| KR20070122194A (en) | 2007-12-28 |
| MX2007011850A (en) | 2007-10-03 |
| CA2602294A1 (en) | 2006-09-28 |
| US8030318B2 (en) | 2011-10-04 |
| BRPI0609650A2 (en) | 2010-04-20 |
| ATE517897T1 (en) | 2011-08-15 |
| RU2007139453A (en) | 2009-04-27 |
| EP1869037B1 (en) | 2011-07-27 |
| US20080182863A1 (en) | 2008-07-31 |
| RU2405783C2 (en) | 2010-12-10 |
| AU2006226322A1 (en) | 2006-09-28 |
| AU2006226322B2 (en) | 2012-03-29 |
| JP2008534479A (en) | 2008-08-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA2602294C (en) | Heterobicylic inhibitors of hvc | |
| US20070066632A1 (en) | Fused bicyclic inhibitors of TGFbeta | |
| JP5046942B2 (en) | HCV-inhibiting bicyclic pyrimidines | |
| US7345045B2 (en) | Pyrido-pyrimidine compounds as medicaments | |
| US7232824B2 (en) | Quinazoline derivatives as medicaments | |
| JP2023508054A (en) | Substituted Heteroaryl Compounds Useful as T Cell Activators | |
| US20060281763A1 (en) | Carboxamide inhibitors of TGFbeta | |
| US9987277B2 (en) | Carboxamide 4-[(4-pyridyl)amino] pryimidines for the treatment of hepatitis C | |
| CA3042960A1 (en) | Fgfr4 inhibitor, preparation method therefor and pharmaceutical use thereof | |
| JP2008543888A (en) | Pyrido (3,2-d) pyrimidine and pharmaceutical compositions useful for treating hepatitis C | |
| AU2002315006A1 (en) | Urea substituted imidazopyridines | |
| US20040132730A1 (en) | Inhibitors of TGFbeta | |
| CN109790158A (en) | As JAK inhibitor heterocyclic compound, the salt and its therapeutical uses of the compound | |
| KR20210093293A (en) | Pyridine-sulfonamide compounds for treating conditions associated with interleukin 1 beta | |
| JP2024517867A (en) | WDR5 inhibitors and modulators |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2006226322 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020077018260 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: 2006226322 Country of ref document: AU Date of ref document: 20060327 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006226322 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2602294 Country of ref document: CA Ref document number: 2008502426 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11909118 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200680009159.3 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2007/011850 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 7779/DELNP/2007 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006708816 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007139453 Country of ref document: RU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2006708816 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: PI0609650 Country of ref document: BR Kind code of ref document: A2 |