WO2006092692A1 - Use of combinations of pde7 inhibitors and alpha-2-delty ligands for the treatment of neuropathic pain - Google Patents
Use of combinations of pde7 inhibitors and alpha-2-delty ligands for the treatment of neuropathic pain Download PDFInfo
- Publication number
- WO2006092692A1 WO2006092692A1 PCT/IB2006/000385 IB2006000385W WO2006092692A1 WO 2006092692 A1 WO2006092692 A1 WO 2006092692A1 IB 2006000385 W IB2006000385 W IB 2006000385W WO 2006092692 A1 WO2006092692 A1 WO 2006092692A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- acid
- pain
- alpha
- compound
- methyl
- Prior art date
Links
- 0 C*1C(*)CC(CC(O)=O)(CN)C1 Chemical compound C*1C(*)CC(CC(O)=O)(CN)C1 0.000 description 7
- XPGQIHGEQGZUFD-UHFFFAOYSA-N CC1C(CC(O)=O)(CN)CCC2C1C2 Chemical compound CC1C(CC(O)=O)(CN)CCC2C1C2 XPGQIHGEQGZUFD-UHFFFAOYSA-N 0.000 description 1
- BZFBCJQSBQZUFQ-VIFPVBQESA-N CCC(CC)=CC[C@@H](CN)C(O)=O Chemical compound CCC(CC)=CC[C@@H](CN)C(O)=O BZFBCJQSBQZUFQ-VIFPVBQESA-N 0.000 description 1
- NASNEFQXPOULDZ-AEFFLSMTSA-N CCC(CC)CC[C@@](C)(C(OC1)=O)N[C@H]1c1ccccc1 Chemical compound CCC(CC)CC[C@@](C)(C(OC1)=O)N[C@H]1c1ccccc1 NASNEFQXPOULDZ-AEFFLSMTSA-N 0.000 description 1
- LEKQUDBVQFBMTG-UHFFFAOYSA-N NCC1(CC(O)=O)C(C2)C2CC1 Chemical compound NCC1(CC(O)=O)C(C2)C2CC1 LEKQUDBVQFBMTG-UHFFFAOYSA-N 0.000 description 1
- IOYHERBFPMDMPG-UHFFFAOYSA-N NCC1(CC(O)=O)CC(C2)C2CC1 Chemical compound NCC1(CC(O)=O)CC(C2)C2CC1 IOYHERBFPMDMPG-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
- C07D239/72—Quinazolines; Hydrogenated quinazolines
- C07D239/78—Quinazolines; Hydrogenated quinazolines with hetero atoms directly attached in position 2
- C07D239/80—Oxygen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/195—Carboxylic acids, e.g. valproic acid having an amino group
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/185—Acids; Anhydrides, halides or salts thereof, e.g. sulfur acids, imidic, hydrazonic or hydroximic acids
- A61K31/19—Carboxylic acids, e.g. valproic acid
- A61K31/195—Carboxylic acids, e.g. valproic acid having an amino group
- A61K31/197—Carboxylic acids, e.g. valproic acid having an amino group the amino and the carboxyl groups being attached to the same acyclic carbon chain, e.g. gamma-aminobutyric acid [GABA], beta-alanine, epsilon-aminocaproic acid, pantothenic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/357—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having two or more oxygen atoms in the same ring, e.g. crown ethers, guanadrel
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/401—Proline; Derivatives thereof, e.g. captopril
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/527—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim spiro-condensed
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/537—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines spiro-condensed or forming part of bridged ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/54—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one sulfur as the ring hetero atoms, e.g. sulthiame
- A61K31/547—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one sulfur as the ring hetero atoms, e.g. sulthiame spiro-condensed or forming part of bridged ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
Definitions
- PDE7 inhibitor The invention also relates to the use of a combination of an alpha-2-delta ligand and a phosphodiesterase 7 (PDE7) inhibitor for the manufacture of a medicament for the treatment of pain, particularly inflammatory, neuropathic, visceral and nociceptive pain. It also relates to a method for treating pain through the use of effective amounts of a combination of an 0 alpha-2-delta ligand and a phosphodiesterase 7 (PDE7) inhibitor.
- Alpha-2-delta receptor ligands may also be known as GABA analogs.
- Alpha-2-delta ligands have been described for the treatment of a number of indications, including 5 epilepsy and pain.
- Phosphodiesterases are a family of enzymes which affect various cellular signaling processes by the process of hydrolyzing the second messenger molecules cAMP and cGMP to the corresponding inactive 5'-monophosphate nucleotides and thereby regulating their 0 physiological level.
- the secondary messengers cAMP and cGMP are responsible for the regulation of numerous intracellular processes.
- PDE7 is one member of the PDE family and comprises 2 subclass members PDE7 A and B.
- the mRNA of PDE7 is expressed in various tissues and cell types known to be important in the 5 pathogenesis of several diseases such as Tcell related disorders, in particular PDE7A and its splice variants are upregulated in activated Tcells, [L. Li, C. Yee and J.A. Beavo. Science 283 (1999), pp. 848-851], and in B-lymphocytes. [R. Lee, S. Wolda, E. Moon, J. Esselstyn, C. Hertel and A. Lerner. Ce//. Signal 14 (2002), pp. 277-284], autoimmune disease . [L. Li, C.
- PDE7A mRNA is found to be widely distributed in rat brain in both neuronal and non-neuronal cell populations. The highest levels are observed in the olfactory bulb, olfactory tubercle, hippocampus, cerebellum, medial habenula nucleus, pineal gland, area postrema, and choroid plexus. PDE7A mRNA is also widely detected in other non brain tissue. These results are consistent with PDE7A being involved in the regulation of cAMP signaling in many brain functions and suggests that PDE7A could have an effect on memory, depression, and emesis [X. Mir ⁇ , S. Perez-Torres , J. M. Palacios , P. Puigdomenech , G. Mengod 1 Synapse
- PDE7A has been isolated from yeast [Michaeli, T., et al J. Biol. Chem. 268 1993 12925 - 12932] , human [Han, P., Xiaoyan, Z., Tamar, M., Journ. Biol. Chem 272 26 1997 16152 - 16157], mouse [Bloom, T., Beavo, JA., proc. Natl. Acad. Sci. USA 93 1996 14188 - 14192] and mouse, and upregulation of PDE7A levels is seen in human T lymphocytes [lchimura, M., Kase, H. Biochem. Biophys. Res. Commun 193, 1993 985 - 990].
- PDE7B the second member of the PDE7 family, shares 70% amino acid homology with PDE7A in the C-terminal catalytic domain (N terminal domain is the regulatory domain containing the phosphorylation site which is conserved across the PDE family].
- PDE7B is cAMP specific and has been cloned from mouse [accession number - AJ251858] and human [accession number - AJ251860] sources [C. Gardner, N. Robas, D. Cawkill and M. Fidock. Biochem. Biophys. Res. Commun. 272 (2000), pp. 186-192].
- PDE7B has also been shown to discriminate among several general PDE inhibitors [J. M. Hetman, S. H. Soderling, N.A. Glavas and J.A. Beavo. PNAS 97 (2000), pp. 472-476], many standard PDE inhibitors, zaprinast, rolipram, milrinone do not specifically inhibit PDE7B.
- amino acid and nucleotide sequences that encode PDE7 of various species are known to those skilled in the art and can be found in GenBank under accession numbers AB057409, U77880, AB038040, L12052, AK035385, AY007702.
- Inhibitors of PDE7 are known as is their use in the treatment of various PDE7 related diseases.
- the patent application EP-A-1348701 discloses pharmaceutical compositions comprising phosphodiesterase 7 inhibitors.
- EP-A-1348701 addresses the problem of providing a means of alleviating visceral pain using such compositions.
- Visceral pain is known to be a particular and narrow class of nociceptive pain. It is known that there are 2 fundamental and different types of pain: nociceptive pain and neuropathic pain. It is further known that nociceptive and neuropathic pain are clinically and mechanistically distinct from each other.
- the clinical characteristics of nociceptive pain are determined by excessive and/or prolonged activation of specific sensory neurones A ⁇ and C fibers. These may be activated by a mechanical, chemical, or thermal stimulus and become sensitised in chronic inflammatory conditions.
- Neuropathic pain however is defined as pain which arises as a result of damage to or dysfunction of the nervous system.
- the clinical characteristics of neuropathic pain are therefore determined predominantly by the mechanisms, location, and severity of the neuropathology process itself and arises from neurons that have themselves been damaged.
- Neuropathic pain has important elements which are mediated via activitiy in sensory nerves which do not normally convey pain, the A ⁇ neurones.
- neuropathic pain is notoriously difficult to treat; it responds very poorly or not at all to standard analgesic therapies which are effective in the treatment of nociceptive pain such as nonsteroidal anti-inflammatory drugs and acetaminophen; and responds less predictably and less robustly to opioids than do nociceptive pain conditions.
- Effective treatments for nociceptive pain are not expected to extend to neuropathic pain.
- medicaments such as gabapentin, pregabalin and amitriptyline, which provide some relief to neuropathic pain, are often not effective in the treatment of nociceptive pain.
- combination therapy with an alpha-2-delta ligand and a PDE7 inhibitor when administered simultaneously, sequentially or separately, results in improvement in the treatment of pain, particularly inflammatory, neuropathic, visceral and nociceptive pain, most preferably neuropathic pain.
- the alpha-2-delta ligand and PDE7 inhibitor can interact in a synergistic manner to control pain. This synergy allows a reduction in the dose req ⁇ ired of each compound, leading to a reduction in the side effects and enhancement of the clinical utility of the compounds.
- the invention provides, as a first aspect, a combination of an alpha-2-delta ligand and a PDE7 inhibitor.
- the invention further provides a combination of an alpha-2-delta ligand and a PDE7 inhibitor for the treatment of pain, particularly inflammatory, neuropathic, visceral and nociceptive pain, most preferably neuropathic pain.
- the invention further provides the use of a combination of an alpha-2-delta ligand and a PDE7 inhibitor for the manufacture of a medicament for the treatment of pain, particularly inflammatory, neuropathic, visceral and nociceptive pain, most preferably neuropathic pain.
- the invention further provides a method for treating pain, particularly inflammatory, neuropathic, visceral and nociceptive pain, most preferably neuropathic pain, through the use of effective amounts of a combination of an alpha-2-delta ligand and a PDE7 inhibitor.
- a second alpha-2-delta ligand, pregabalin, (S)-(+)-4-amino-3-(2-methylpropyl)butanoic acid is described in European Patent Application Number EP-A-0641330 as an anti-convulsant treatment useful in the treatment of epilepsy and in EP0934061 for the treatment of pain.
- alpha-2-delta ligands are described in the following documents.
- n is an integer of from 1 to 4.
- each center may be independently R or S, preferred compounds being those of Formulae I-IV above in which n is an integer of from 2 to 4.
- WO-A-02/85839 describes alpha-2-delta ligands of the following formulae:
- Ri and R 2 together with the carbon to which they are attached, form a three to six membered cycloalkyl ring;
- R 3 is (d-CeJalkyl, (C 3 -C 6 )cycloalkyl, (C 3 -C 6 )cycloalkyl-(C 1 -C 3 )alkyl, phenyl, phenyl-(C r C 3 )alkyl, pyridyl, pyridyl-(Ci-C 3 )alkyl, phenyl-N(H)-, or pyridyl-N(H)- , wherein each of the foregoing alkyl moieties can be optionally substituted with from one to five fluorine atoms, preferably with from zero to three fluorine atoms, and wherein said phenyl and said pyridyl and the phenyl and pyridyl moieties of said phenyl-(C r C 3 )alkyl and said pyridyl-(C r C 3 )alkyl, respectively, can be optionally substitute
- R 4 is hydrogen or (CrC 6 )alkyl optionally substituted with from one to five fluorine atoms;
- R 6 is hydrogen or (C r C 6 )alkyl; or a pharmaceutically acceptable salt thereof.
- R is a 3-12 membered cycloalkyl, 4-12 membered heterocycloalkyl, aryl or heteroaryl, where any ring may be optionally substituted with one or more substituents independently selected from halogen, hydroxy, cyano, nitro, amino, hydroxycarbonyl, CrC 6 alkyl, Ci-C 6 alkenyl, C 1 -C 6 alkynyl,
- C r C 6 acyl C r C 6 acyl, C r C 6 acyloxy, CrC 6 acyloxyC r C 6 alkyl, C 1 -C 6 acylamino, C 1 -C 6 alkylthio, C 1 -C 6 alkylthiocarbonyl, C 1 -C 6 alkylthioxo, C 1 -C 6 alkoxycarbonyl, C 1 -C 6 alkylsulfonyl, C 1 -C 6 alkylsulfonylamino, aminosulfonyl, C 1 -C 6 alkylaminosulfonyl, di-C r C 6 alkylaminosulfonyl,
- R 1 is C 1 -C 6 alkyl, said C 1 -C 6 alkyl being optionally substituted by one or more halo, -R 5 , -OR 5 or -
- R 2 is methyl, optionally substituted by one or more fluoro groups
- R 3 and R 4 are each independently H or a group which is converted to H following administration of the compound to a mammal;
- R 5 is C 1 -C 6 alkyl, C 1 -C 6 haloalkyl, C 3 -C 8 cycloalkyl or aryl;
- aryl is phenyl or naphthyl, each optionally substituted by one or more substituents selected from halo, -NR 6 R 6 , C 1 -C 6 alkyl, C 3 -C 8 cycloalkyl, C 1 -C 6 alkoxy and cyano; and
- R 6 is H, C 1 -C 6 alkyl or C 3 -C 8 cycloalkyl.
- alpha-2-delta ligands for use in the present invention are those compounds, or pharmaceutically acceptable salts thereof, generally or specifically disclosed in US4024175, particularly gabapentin, EP641330, particularly pregabalin, US5563175, WO-A-97/33858, WO-A- 97/33859, WO-A-99/31057, WO-A-99/31074, WO-A-97/29101, WO-A-02/085839, particularly [(1R,5R,6S)-6-(aminomethyl)bicyclo[3.2.0]hept-6-yl]acetic acid, WO-A-99/31075, particularly 3-(1- aminomethyl-cyclohexylmethyl)-4H-[1 ,2,4]oxadiazol-5-one and C-[1-(1 H-tetrazol-5-ylmethyl)- cyclohepty
- EP1178034 EP1201240, WO-A-99/31074, WO-A ⁇ 03/000642, WO-A-02/22568, WO-A-02/30871 , WO-A-02/30881 WO-A-02/100392, WO-A-02/100347, WO-A-02/42414, WO-A-02/32736 and WO-A-02/28881, US Provisional Patent Application Number 60/676025 (unpublished at the filing date of the present application) , particularly (2S)-2-amino-4-ethyl-2-methylhexanoic acid and US Provisional Patent Application Number 60/733591 (unpublished at the filing date of the present application), particularly (2S)-2-aminomethyl-5-ethyl-heptanoic acid, all of which are incorporated herein by reference.
- Preferred alpha-2-delta ligands for use in the combination of the present invention include: gabapentin, pregabalin, [(1R,5R,6S)-6-(aminomethyl)bicyclo[3.2.0]hept-6-yl]acetic acid, 3-(1- aminomethyl-cyclohexylmethyl)-4H-[1 ,2,4]oxadiazol-5-one, C-[1-(1H-tetrazol-5-ylmethyl)- cycloheptyl]-methylamine, (3S,4S)-(1-aminomethyl-3,4-dimethyl-cyclopentyl)-acetic acid, (1 ⁇ ,3 ⁇ ,5 ⁇ )(3-amino-methyl-bicyclo[3.2.0]hept-3-yl)-acetic acid, (3S,5R)-3-aminomethyl-5-methyl- octanoic acid, (3S,5R)-3-amino-5-methyl-heptanoic acid, (3
- alpha-2-delta ligands for use in the combination of the present invention are (3S,5R)-3-amino-5-methyloctanoic acid, (3S,5R)-3-amino-5-methylnonanoic acid, (3R,4R,5R)-3- amino-4,5-dimethylheptanoic acid and (3R,4R,5R)-3-amino-4,5-dimethyloctanoic acid, and the pharmaceutically acceptable salts thereof.
- alpha-2-delta ligands for use in the combination of the present invention are selected from gabapentin, pregabalin, (1 ⁇ ,3 ⁇ ,5 ⁇ )(3-amino-methyl-bicyclo[3.2.0]hept-3-yl)-acetic acid, (2S,4S)-4-(3-chlorophenoxy)proline, (2S,4S)-4-(3-fluorobenzyl)proline, (2S)-2-amino-4- ethyl-2-methylhexanoic acid, and (2S)-2-aminomethyl-5-ethyl-heptanoic acid, or pharmaceutically acceptable salts thereof.
- Suitable PDE7 inhibitors for use in the invention include those compounds generally or specifically described in the patent applications WO-A-2002/074754 (Warner Lambert) and WO-A- 2002/076953 (Warner Lambert), which describe Spiroquinazolinones which are PDE7 inhibitors (also described in the publication, Bioorganic and Medicinal Chemistry Letters 2004, 14, 4623- 4626. Bioorganic and Medicinal Chemistry Letters 2004, 14, 4627-4631).
- PDE7 inhibitors according to WO-A-2002/074754 and WO-A-2002/076953 have the following formula (I), (II) or (III),
- X 1 , X 2 , X 3 and X 4 are the same or different and are selected from:
- N provided that not more than two of the groups Xi, X 2 , X3 and X 4 simultaneously represent a nitrogen atom, or,
- R 1 is selected from: - Q1 , or lower alkyl, lower alkenyl or lower alkynyl, these groups being unsubstituted or substituted with one or several groups Q2; - the group X 5 -R 5 in which,
- n is an integer selected from O, 1 , 2 and 3
- R 1 and R" together with the nitrogen atom to which they are linked, can form a 4- to 8-membered heterocyclic ring, which may contain one or two heteroatoms selected from O, S or N; or, - when X 1 and X 2 both represent C-R 1 , the 2 substituents R 1 may form together with the carbon atoms to which they are attached, a 5-membered heterocyclic ring comprising a nitrogen atom and optionally a second heteroatom selected from O, S or N;
- X is O or NR 9 , in which R 9 is selected from,
- Y is selected from O, S or N-R 12 , in which R 12 is selected from:
- R 14 and R 15 being independently selected from hydrogen or lower alkyl, or, R 14 and R 15 , together with the nitrogen atom to which they are linked, can form a 4- to 8-membered heterocyclic ring which may contain one or two heteroatoms chosen from O, S or N, and which may be substituted with a lower alkyl, or,
- R 14 and R 15 being chosen from hydrogen or lower alkyl
- R 14 and R 15 , and/or, R 16 and R 17 , together with the nitrogen atom to which they are linked, can form a 4- to 8-membered heterocyclic ring which may contain one or two heteroatoms chosen from O, S or N, and which may be substituted with a lower alkyl;
- A is a cycle chosen from:
- R 19 and R 20 , and/or, R 21 and R 22 , together with the nitrogen atom to which they are linked, can form a 4- to 8-membered heterocyclic ring;
- 2 atoms of the cycle A which are not adjacent, may be linked by a 2, 3 or 4 carbon atom chain which may be interrupted with 1 heteroatom chosen from O, S or N; provided that: - not more than two of the groups A 1 , A 2 , A 3 , A 4 , A 5 and A 6 simultaneously represent a heteroatom; the cycle A does not contain more than 2 carbon atoms in an sp 2 hybridization state;
- Halogen includes fluoro, chloro, bromo, and iodo. Preferred halogens are F and Cl.
- Lower alkyl includes straight and branched carbon chains having from 1 to 6 carbon atoms.
- alkyl groups include methyl, ethyl, isopropyl, tert-butyl and the like.
- Lower alkenyl includes straight and branched hydrocarbon radicals having from 2 to 6 carbon atoms and at least one double bond. Examples of such alkenyl groups are ethenyl, 3-buten-1-yl,
- Lower alkynyl includes straight and branched hydrocarbon radicals having from 2 to 6 carbon atoms and at least one triple bond.
- alkynyl groups are ethynyl, 3-butyn-1-yl, propynyl, 2-butyn-1-yl, 3-pentyn-1-yl, and the like.
- Lower haloalkyl includes a lower alkyl as defined above, substituted with one or several halogens.
- a preferred haloalkyl is trifluoromethyl.
- Aryl is understood to refer to an aromatic carbocycle containing between 6 and 10, preferably 6, carbon atoms.
- a preferred aryl group is phenyl.
- Heteroaryl includes aromatic cycles which have from 5 to 10 ring atoms, from 1 to 4 of which are independently selected from the group consisting of O, S, and N.
- Preferred heteroaryl groups have 1 , 2, 3 or 4 heteroatoms in a 5- or 6-membered aromatic ring. Examples of such groups are tetrazole, pyridyl, thienyl and the like.
- Preferred cycloalkyl contain from 3 to 8 carbon atoms. Examples of such groups are cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and cyclooctyl.
- interrupted means that in a backbone chain, a carbon atom is replaced by an heteroatom or a group as defined herein.
- Bicyclic substituents refer to two cycles, which are the same or different and which are chosen from aryl, heterocyclic ring, cycloalkyl or cycloalkenyl, fused together to form said bicyclic substituents.
- a preferred bicyclic substituent is indolyl.
- WO-A-2002/028847 descibes compounds of the following formula (I) as PDE7 inhibitors:
- R2 is: lower alkyl, C 2 -C 10 alkenyl, C 4 -Ci 0 alkynyl, cycloalkyl, cycloalkenyl, heterocycle, aryl; each optionally substituted with one or several groups which are the same or different and which are selected from:
- - R'3 is: cycloalkyl, cycloalkenyl, aryl, heterocycle, or a polycyclic group; each optionally substituted with one or several groups X 3 -Ri 7 , identical or different, in which: - X 3 is: a single bond, lower alkylene, C 2 -C 6 alkenylene, C 2 -C 6 alkynylene, cycloalkylene, arylene, divalent heterocycle or a divalent polycyclic group, and,
- heterocycle optionally substituted with one or several groups R 5 ;
- R 5 and R 6 are the same or different and are selected from : - H 1 - - lower alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl;
- X 5 is a single bond or lower alkylene and R 18 , Ri 9 and R 20 are the same or different and are selected from H or lower alkyl; - X 6 -heterocycle, X 6 -aryl, X 6 -cycloalkyl, X 6 -cycloalkenyl, X 6 -polycyclic group in which X 6 is selected from a single bond or lower alkylene, these groups being optionally substituted with one or several groups, identical or different, selected from halogens, COOR 21 , OR 21 , or (CH 2 J n NR 21 R 22 in which n is O, 1 or 2 and R 21 and R 22 are the same or different and are selected from H or lower alkyl; R 9 is selected from H, CN, OH, lower alkyl, O-lower alkyl, aryl, heterocycle, SO 2 NH 2 or
- Ri9 in which X 5 is a single bond or lower alkylene and R 18 and R 19 are the same or different and are selected from H or lower alkyl;
- Rio is selected from hydrogen, lower alkyl, cyclopropyl or heterocycle; or a pharmaceutically acceptable derivative thereof, with the proviso that,
- - aryl is understood to refer to an unsaturated carbocycle, exclusively comprising carbon atoms in the cyclic structure, the number of which is between 5 and 10, including phenyl, naphthyl or tetrahydronaphthyl;
- heterocycle is understood to refer to a non-saturated or saturated monocycle containing between 1 and 7 carbon atoms in the cyclic structure and at least one heteroatom in the cyclic structure, such as nitrogen, oxygen, or sulfur, preferably from 1 to 4 heteroatoms, identical or different, selected from nitrogen, sulfur and oxygen atoms.
- Suitable heterocycles include morpholinyl, piperazinyl, pyrrolidinyl, piperidinyl, pyrimidinyl, 2- and 3-furanyl, 2- and 3-thienyl, 2- pyridyl, 2- and 3-pyranyl, hydroxypyridyl, pyrazolyl, isoxazolyl, tetrazole, imidazole, triazole and the like;
- polycyclic groups include at least two cycles, identical or different, selected from aryl, heterocycle, cycloalkyl, cycloalkenyl groups fused together to form said polycyclic group such as 2- and 3-benzothienyl, 2- and 3-benzofuranyl, 2-indolyl, 2- and 3-quinolinyl, acridinyl, quinazolinyl, indolyl benzo[1 ,3]dioxolyl and 9-thioxantanyl;
- Preferred polycyclic groups include 2 or 3 cycles as defined above. More preferred polycyclic groups include 2 cycles (bicyclic substituents) as defined above.
- bicyclic groups refer to two cycles, which are the same or different and which are chosen from aryl, heterocycle, cycloalkyl or cycloalkenyl, fused together to form said bicyclic groups;
- - halogen is understood to refer to fluorine, chlorine, bromine or iodine
- - lower alkyl is understood to mean that the alkyl is linear or branched and contains 1 to 6 carbon atoms; Examples of lower alkyl groups include methyl, ethyl, propyl, butyl, isopropyl, tert-butyl, isobutyl, n-butyl, pentyl, hexyl and the like.
- - alkenyl is understood to refer to a linear or branched unsaturated carbon atom chain, comprising one or several double bonds, preferably one or two double bonds. Preferred alkenyls comprise from 3 to 6 carbon atoms and one double bonds.
- alkynyl is understood to refer to a linear or branched unsaturated carbon atom chain, comprising one or several triple bonds, preferably one or two triple bonds.
- Preferred alkynyls comprise from 3 to 6 carbon atoms and one triple bonds.
- lower haloalkyl are understood to refer to a lower alkyl substituted with one or several halogens;
- Preferred lower haloalkyl groups include perhaloalkyl groups such as CF 3 .
- - cycloalkyl is understood to refer to saturated monocarbocyle containing from 3 to 10 carbon atoms; preferred cycloalkyl groups comprise cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl and cycloheptyl.
- - cycloalkenyl is understood to refer to unsaturated monocarbocyle containing from 3 to 10 carbon atoms.
- Preferred cyloalkenyl groups contain 1 or 2 double bonds.
- suitable cycloalkenyl are 3-cyclohexene, 3-cycloheptene or the like.
- carboxylic acid bioisostere has the classical meaning; common carboxylic acid bioisostere are tetrazole, hydroxamic acid, isoxazole, hydroxythiadiazole, sulfonamide, sulfonylcarboxamide, phosphonates, phosphonamides, phosphinates, sulfonates, acyl sulfonamide, mercaptoazole, acyl cyanamides.
- WO-A-2004/026818 describes compounds of the following formula (I) as PDE7 inhibitors:
- • m is 1 , 2 or 3, and, • R 1 is selected from CH 3 , Cl, Br and F and,
- R 2 is selected from,
- ⁇ Q 1 is a single bond or a linear or branched (CrC ⁇ Jalkylene group;
- ⁇ Q 2 is a saturated 4 to 6-membered heterocycle comprising one or two heteroatoms selected from O or N;
- ⁇ Q 3 is a linear or branched (C r C 6 )alkylene group
- R 5 is selected from R 4 , H and (C r C 6 )alkyl; or,
- R 9 is selected from H, CN, OH, OCH 3 , SO 2 CH 3 , SO 2 NH 2 and (C 1 -C 6 )alkyl, and,
- m 0, 1 or 2;
- X is O, S or N-CN
- R is F, Cl or CN
- A is a C3-6 cycloalkylene group optionally substituted with a C 1 ⁇ alkyl group
- B is a single bond or a C 1-2 alkylene group; or a pharmaceutically acceptable salt, solvate or
- suitable PDE7 inhibitors for use in the invention include those compounds generally or specifically disclosed in the patent application WO-A-01/098274 (CellTech Chiroscience Ltd), M-substituted phenyl-N-phenylsulfonamides particularly N-phenyl-3- benzoxazol-2-ylphenylsulfonamide and N-phenyl-3-benzimidazol-2-ylphenylsulfonamide derivatives.
- Patent application WO-A-01 /098274 Celltech Chiroscience discloses further examples of suitable PDE7 inhibitors which are sulfonamides and suitable for use in the invention.
- patent application WO-A-01/074786 discloses further examples of PDE7 inhibitors suitable for use in the invention and which are a series of heterobiarylsulphonamides. Particularly suitable are the N-aryl-3- benzimidazolylbenzenesulfonamides.
- Patent application WO-A-00/068230 discloses further suitable PDE7 inhibitors, 9-(1 ,2,3,4-Tetrahydronapthalen-1-yl)-1 ,9- dihydropurin-6-one derivatives (also published in, Bioorganic and Medicinal Chemistry Letters 2001 , 1081-1083).
- Patent applications WO-A-01/029049 (Merck), WO-A-01/036425 (Merck) and DE 19954707 (Merck) disclose imidazole derivatives, WO-A-01 /032175 (Merck) and DE 19953024 (Merck) disclose isoxazole derivatives, WO-A-01/032618 (Merck) and DE 19953025 (Merck) disclose pyrrole derivatives, DE19953414 (Merck) discloses imidazo[4,5-c]pyridine derivatives, all of which are further examples of PDE7 inhibitors and suitable for use in the invention.
- WO-A-2002/074754 particularly 5'-carboxypropoxy-8'-chloro-spiro[cyclohexane-1 -4'-(3',4'- dihydro)quinazolin]-2'(1 'H)-one;
- Preferred PDE7 inhibitors for use with the present invention are selected from:
- a combination comprising 5'- carboxypropoxy-8'-chloro-spiro[cyclohexane-1-4'-(3',4'-dihydro)quinazolin]-2'(1'H)-one, or a pharmaceutically acceptable salt thereof, and an alpha-2-delta ligand selected from gabapentin, pregabalin, [(1R,5R,6S)-6-(aminomethyl)bicyclo[3.2.0]hept-6-yl]acetic acid, 3-(1-aminomethyl- cyclohexylmethyl)-4H-[1 ,2,4]oxadiazol-5-one, C-[1-(1 H-tetrazol-5-ylmethyl)-cycloheptyl]- methylamine, (3S,4S)-(1-aminomethyl-3,4-dimethyl-cyclopentyl)-acetic acid, (1 ⁇ ,3 ⁇ ,5 ⁇ )(
- a combination comprising c/s-3-[(8'- chloro-2'-oxo-2',3'-dihydro-1'H-spiro[cyclohexane-1 ,4'-quinazolin]-5'-yl)oxy]cyclobutanecarboxylic acid, or a pharmaceutically acceptable salt thereof, and an alpha-2-delta ligand selected from gabapentin, pregabalin, [(1 R,5R,6S)-6-(aminomethyl)bicyclo[3.2.0]hept-6-yl]acetic acid, 3-(1- aminomethyl-cyclohexylmethyl)-4H-[1,2,4]oxadiazol-5-one, C-[1-(1H-tetrazol-5-ylmethyl)- cycloheptyl]-methylamine, (3S,4S)-(1-aminomethyl-3,4-dimethyl-cyclopentyl)-ace
- a combination comprising trans-3- [( ⁇ '-chloro ⁇ '-oxo ⁇ '.S'-dihydro-i'H-spirotcyclohexane-i ⁇ '-quinazolinl- ⁇ 1 - yl)oxy]cyclobutanecarboxylic acid, or a pharmaceutically acceptable salt thereof, and an alpha-2- delta ligand selected from gabapentin, pregabalin, [(1R,5R,6S)-6- (aminomethyl)bicyclo[3.2.0]hept-6-yl]acetic acid, 3-(1 -aminomethyl-cyclohexylmethyl)-4H- [1 ,2,4]oxadiazol-5-one, C-[1-(1 H-tetrazol-S-ylmethyO-cycloheptyll-methylamine, (3S,4S)-(1- aminomethyl-3,4-dimethyl-cyclopentyl)
- the combination is selected from:
- (2S)-2-aminomethyl-5-ethyl-heptanoic acid or pharmaceutically acceptable salts or solvates of either or both components of any such combination.
- the combination is selected from: c/s-3-[(8'-chloro-2'-oxo-2',3'-dihydro-1'H-spiro[cyclohexane-1,4'-quinazolin]-5'- yl)oxy]cyclobutanecarboxylic acid and gabapentin; c/s-3-[(8'-chloro-2'-oxo-2',3'-dihydro-1'H-spiro[cyclohexane-1 ,4'-quinazolin]-5'- yl)oxy]cyclobutanecarboxylic acid and pregabalin; c/s-3-[(8'-chloro-2'-oxo-2',3'-dihydro-1'H-spiro[cyclohexane-1,4'-quinazolin]-5'- yl)oxy]cyclobutanecarboxylic acid and gabapentin; c/s-3
- the combination is selected from: frans-3-[(8'-chloro-2'-oxo-2',3'-dihydro-1'H-spiro[cyclohexane-1,4'-quinazolin]-5'- yl)oxy]cyclobutanecarboxylic acid and gabapentin; frans-3-[(8'-chloro-2'-oxo-2',3'-dihydro-1'H-spiro[cyclohexane-1,4'-quinazolin]-5'- yl)oxy]cyclobutanecarboxylic acid and pregabalin; fra ⁇ s-3-[(8'-chloro-2' ⁇ »co-2 ⁇ 3'-dihydro-1 ⁇ -spiro[cyclohexane-1 ,4'-quinazolin]-5'- yl)oxy]cyclobutanecarboxylic acid and (1 ⁇ ,3 ⁇ ,
- Particularly preferred combinations of the invention include those in which each variable of the combination is selected from the suitable parameters for each variable. Even more preferable combinations of the invention include those where each variable of the combination is selected from the more preferred or more preferred parameters for each variable.
- Pharmaceutically acceptable salts of PDE7 inhibitors and alpha-2-delta ligands include the acid addition and base salts thereof.
- Suitable acid addition salts are formed from acids which form non-toxic salts. Examples include the acetate, adipate, aspartate, benzoate, besylate, bicarbonate/carbonate, bisulphate/sulphate, borate, camsylate, citrate, cyclamate, edisylate, esylate, formate, fumarate, gluceptate, gluconate, glucuronate, hexafluorophosphate, hibenzate, hydrochloride/chloride, hydrobromide/bromide, hydroiodide/iodide, isethionate, lactate, malate, maleate, malonate, mesylate, methylsulphate, naphthylate, 2-napsylate, nicotinate, nitrate, orotate, oxalate, palmitate, pamoate, phosphate/hydrogen phosphate/dihydrogen phosphate, pyroglutamate, saccharate, ste
- Suitable base salts are formed from bases which form non-toxic salts. Examples include the aluminium, arginine, benzathine, calcium, choline, diethylamine, diolamine, glycine, lysine, magnesium, meglumine, olamine, potassium, sodium, tromethamine and zinc salts.
- Hemisalts of acids and bases may also be formed, for example, hemisulphate and hemicalcium salts.
- a suitable cyclic precursor for example, a lactone or lactam
- the resulting salt may precipitate out and be collected by filtration or may be recovered by evaporation of the solvent.
- the degree of ionisation in the resulting salt may vary from completely ionised to almost non-ionised.
- the components of the combination of the present invention may exist in a continuum of solid states ranging from fully amorphous to fully crystalline.
- the term 'amorphous' refers to a state in which the material lacks long range order at the molecular level and, depending upon temperature, may exhibit the physical properties of a solid or a liquid. Typically such materials do not give distinctive X-ray diffraction patterns and, while exhibiting the properties of a solid, are more formally described as a liquid.
- a change from solid to liquid properties occurs which is characterised by a change of state, typically second order ('glass transition').
- 'crystalline' refers to a solid phase in which the material has a regular ordered internal structure at the molecular level and gives a distinctive X-ray diffraction pattern with defined peaks. Such materials when heated sufficiently will also exhibit the properties of a liquid, but the change from solid to liquid is characterised by a phase change, typically first order ('melting point').
- the components of the combination of the invention may also exist in unsolvated and solvated forms.
- 'solvate' is used herein to describe a molecular complex comprising the PDE7 inhibitor or alpha-2-delta ligand and one or more pharmaceutically acceptable solvent molecules, for example, ethanol.
- solvent molecules for example, ethanol.
- 'hydrate' is employed when said solvent is water.
- Isolated site hydrates are ones in which the water molecules are isolated from direct contact with each other by intervening organic molecules.
- channel hydrates the water molecules lie in lattice channels where they are next to other water molecules.
- metal-ion coordinated hydrates the water molecules are bonded to the metal ion.
- the complex When the solvent or water is tightly bound, the complex will have a well-defined stoichiometry independent of humidity. When, however, the solvent or water is weakly bound, as in channel solvates and hygroscopic compounds, the water/solvent content will be dependent on humidity and drying conditions. In such cases, non-stoichiometry will be the norm.
- multi-component complexes other than salts and solvates
- Complexes of this type include clathrates (drug-host inclusion complexes) and co-crystals.
- the latter are typically defined as crystalline complexes of neutral molecular constituents which are bound together through non-covalent interactions, but could also be a complex of a neutral molecule with a salt.
- Co-crystals may be prepared by melt crystallisation, by recrystallisation from solvents, or by physically grinding the components together - see Chem Commun, .17, 1889-1896, by O. Almarsson and M. J. Zaworotko (2004).
- the components of the combination of the invention may also exist in a mesomorphic state (mesophase or liquid crystal) when subjected to suitable conditions.
- the mesomorphic state is intermediate between the true crystalline state and the true liquid state (either melt or solution).
- Mesomorphism arising as the result of a change in temperature is described as 'thermotropic' and that resulting from the addition of a second component, such as water or another solvent, is described as 'lyotropic'.
- references to PDE7 inhibitors include references to salts, solvates, multi- component complexes and liquid crystals thereof and to solvates, multi-component complexes and liquid crystals of salts thereof.
- references to alpha-2-delta ligands include references to salts, solvates, multi- component complexes and liquid crystals thereof and to solvates, multi-component complexes and liquid crystals of salts thereof.
- a number of the alpha-2-delta ligands of the combination of the present invention are amino acids. Since amino acids are amphoteric, pharmacologically compatible salts can be salts of appropriate non-toxic inorganic or organic acids or bases. Salts with quaternary ammonium ions can also be prepared with, for example, the tetramethyl-ammonium ion.
- the alpha-2-delta ligands of the combination of the invention may also be formed as a zwitterion.
- a suitable salt for amino acid compounds of the present invention is the hydrochloride salt.
- PDE7 inhibitor' includes PDE7 inhibitors as hereinbefore defined, including all polymorphs and crystal habits thereof, prodrugs and isomers thereof (including optical, geometric and tautomeric isomers) as hereinafter defined and isotopically-labeled PDE7 inhibitors.
- alpha-2-delta ligand' includes alpha-2-delta ligands as hereinbefore defined, including all polymorphs and crystal habits thereof, prodrugs and isomers thereof (including optical, geometric and tautomeric isomers) as hereinafter defined and isotopically-labeled alpha-2-delta ligands.
- so-called 'prodrugs' of the components of the combination are also within the scope of the invention.
- certain derivatives of PDE7 inhibitors or alpha-2-delta ligands which may have little or no pharmacological activity themselves can, when administered into or onto the body, be converted into PDE7 inhibitors or alpha-2-delta ligands having the desired activity, for example, by hydrolytic cleavage.
- Such derivatives are referred to as 'prodrugs'.
- Further information on the use of prodrugs may be found in Pro-drugs as Novel Delivery Systems. Vol. 14, ACS Symposium Series (T. Higuchi and W. Stella) and Bioreversible Carriers in Drug Design, Pergamon Press, 1987 (Ed. E. B. Roche, American Pharmaceutical Association).
- Prodrugs in accordance with the invention can, for example, be produced by replacing appropriate functionalities present in the PDE7 inhibitor or alpha-2-delta ligand with certain moieties known to those skilled in the art as 'pro-moieties' as described, for example, in Design of Prodrugs by H. Bundgaard (Elsevier, 1985).
- prodrugs in accordance with the invention include
- the PDE7 inhibitor or alpha-2-delta ligand contains a carboxylic acid functionality (- COOH), an ester thereof, for example, a compound wherein the hydrogen of the carboxylic acid functionality of the PDE7 inhibitor or alpha-2-delta ligand is replaced by (C r C 8 )alkyl;
- the PDE7 inhibitor or alpha-2-delta ligand contains an alcohol functionality (-OH), an ether thereof, for example, a compound wherein the hydrogen of the alcohol functionality of the PDE7 inhibitor or alpha-2-delta ligand is replaced by (C 1 -C 6 )alkanoyloxymethyl; and
- the PDE7 inhibitor or alpha-2-delta ligand contains a primary or secondary amino functionality (-NH 2 Or -NHR where R ⁇ H), an amide thereof, for example, a compound wherein, as the case may be, one or both hydrogens of the amino functionality of the PDE7 inhibitor or alpha- 2-delta
- PDE7 inhibitors or alpha-2-delta ligands may themselves act as prodrugs of other PDE7 inhibitors or alpha-2-delta ligands.
- Aminoacyl-glycolic and -lactic esters are known as prodrugs of amino acids (Wermuth CG. , Chemistry and Industry, 1980:433-435).
- the carbonyl group of the amino acids can be esterified by known means.
- Prodrugs and soft drugs are known in the art (Palomino E., Drugs of the Future, 1990;15(4):361-368). The last two citations are hereby incorporated by reference.
- metabolites of the components of the combination that is, compounds formed in vivo upon administration of the PDE7 inhibitor or alpha-2-delta ligand.
- Some examples of metabolites in accordance with the invention include
- the PDE7 inhibitor or alpha-2-delta ligand contains a tertiary amino group, a secondary amino derivative thereof (-NR 1 R 2 -> -NHR 1 or -NHR 2 );
- a PDE7 inhibitor or alpha-2-delta ligand containing one or more asymmetric carbon atoms can exist as two or more stereoisomers. Where a PDE7 inhibitor or alpha-2-delta ligand contains an alkenyl or alkenylene group, geometric cisltrans (or Z/E) isomers are possible. Where structural isomers are interconvertible via a low energy barrier, tautomeric isomerism ('tautomerism') can occur. This can take the form of proton tautomerism in PDE7 inhibitors or alpha-2-delta ligands containing, for example, an imino, keto, or oxime group, or so-called valence tautomerism in compounds which contain an aromatic moiety. It follows that a single compound may exhibit more than one type of isomerism.
- Cisltrans isomers may be separated by conventional techniques well known to those skilled in the art, for example, chromatography and fractional crystallisation.
- the racemate (or a racemic precursor) may be reacted with a suitable optically active compound, for example, an alcohol, or, in the case where the PDE7 inhibitor or alpha-2-delta ligand contains an acidic or basic moiety, a base or acid such as 1-phenylethylamine or tartaric acid.
- a suitable optically active compound for example, an alcohol, or, in the case where the PDE7 inhibitor or alpha-2-delta ligand contains an acidic or basic moiety, a base or acid such as 1-phenylethylamine or tartaric acid.
- the resulting diastereomeric mixture may be separated by chromatography and/or fractional crystallization and one or both of the diastereoisomers converted to the corresponding pure enantiomer(s) by means well known to a skilled person.
- Chiral PDE7 inhibitors or alpha-2-delta ligands may be obtained in enantiomerically-enriched form using chromatography, typically HPLC, on an asymmetric resin with a mobile phase consisting of a hydrocarbon, typically heptane or hexane, containing from 0 to 50% by volume of isopropanol, typically from 2% to 20%, and from 0 to 5% by volume of an alkylamine, typically 0.1% diethylamine. Concentration of the eluate affords the enriched mixture.
- chromatography typically HPLC
- a mobile phase consisting of a hydrocarbon, typically heptane or hexane, containing from 0 to 50% by volume of isopropanol, typically from 2% to 20%, and from 0 to 5% by volume of an alkylamine, typically 0.1% diethylamine.
- Racemic mixtures may be separated by conventional techniques known to those skilled in the art - see, for example, Stereochemistry of Organic Compounds by E. L. Eliel and S. H. Wilen (Wiley, 1994).
- the present invention includes all pharmaceutically acceptable isotopically-labelled PDE7 inhibitors or alpha-2-delta ligands wherein one or more atoms are replaced by atoms having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number which predominates in nature.
- isotopes suitable for inclusion in the PDE7 inhibitors or alpha-2-delta ligands include isotopes of hydrogen, such as 2 H and 3 H, carbon, such as 11 C, 13 C and 14 C, chlorine, such as 36 CI, fluorine, such as 18 F, iodine, such as 123 I and 125 I, nitrogen, such as 13 N and 15 N, oxygen, such as 15 O, 17 O and 18 O, phosphorus, such as 32 P, and sulphur, such as 35 S.
- Certain isotopically-labelled PDE7 inhibitors or alpha-2-delta ligands are useful in drug and/or substrate tissue distribution studies.
- the radioactive isotopes tritium, i.e. 3 H, and carbon-14, i.e. 14 C, are particularly useful for this purpose in view of their ease of incorporation and ready means of detection.
- substitution with heavier isotopes such as deuterium, i.e. 2 H, may afford certain therapeutic advantages resulting from greater metabolic stability, for example, increased in vivo half-life or reduced dosage requirements, and hence may be preferred in some circumstances.
- Isotopically-labeled PDE7 inhibitors or alpha-2-delta ligands can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described in the accompanying Examples and Preparations using an appropriate isotopically- labeled reagent in place of the non-labeled reagent previously employed.
- solvates in accordance with the invention include those wherein the solvent of crystallization may be isotopically substituted, e.g. D 2 O, d 6 -acetone, d 6 -DMSO.
- a method for the treatment of pain comprising simultaneous, sequential or separate administration of a therapeutically effective amount of an alpha-2-delta ligand and a PDE7 inhibitor, to a mammal in need of said treatment.
- a method for the treatment of pain comprising simultaneous, sequential or separate administration of a therapeutically synergistic amount of an alpha-2-delta ligand and PDE7 inhibitor, to a mammal in need of said treatment.
- Physiological pain is an important protective mechanism designed to warn of danger from potentially injurious stimuli from the external environment.
- the system operates through a specific set of primary sensory neurones and is activated by noxious stimuli via peripheral transducing mechanisms (see Millan, 1999, Prog. Neurobiol., 57, 1-164 for a review).
- These sensory fibres are known as nociceptors and are characteristically small diameter axons with slow conduction velocities. Nociceptors encode the intensity, duration and quality of noxious stimulus and by virtue of their topographically organised projection to the spinal cord, the location of the stimulus.
- nociceptive nerve fibres of which there are two main types, A-delta fibres (myelinated) and C fibres (non-myelinated).
- A-delta fibres myelinated
- C fibres non-myelinated.
- the activity generated by nociceptor input is transferred, after complex processing in the dorsal horn, either directly, or via brain stem relay nuclei, to the ventrobasal thalamus and then on to the cortex, where the sensation of pain is generated.
- Clinical pain is present when discomfort and abnormal sensitivity feature among the patient's symptoms. Patients tend to be quite heterogeneous and may present with various pain symptoms. Such symptoms include: 1) spontaneous pain which may be dull, burning, or stabbing; 2) exaggerated pain responses to noxious stimuli (hyperalgesia); and 3) pain produced by normally innocuous stimuli (allodynia - Meyer et al., 1994, Textbook of Pain, 13-44). Although patients suffering from various forms of acute and chronic pain may have similar symptoms, the underlying mechanisms may be different and may, therefore, require different treatment strategies. Pain can also therefore be divided into a number of different subtypes according to differing pathophysiology, including nociceptive, inflammatory and neuropathic pain.
- nociceptors activates two types of afferent nerve fibres.
- Myelinated A-delta fibres transmit rapidly and are responsible for sharp and stabbing pain sensations, whilst unmyelinated C fibres transmit at a slower rate and convey a dull or aching pain.
- Moderate to severe acute nociceptive pain is a prominent feature of pain from central nervous system trauma, strains/sprains, burns, myocardial infarction and acute pancreatitis, post-operative pain (pain following any type of surgical procedure), posttraumatic pain, renal colic, cancer pain and back pain.
- Cancer pain may be chronic pain such as tumour related pain (e.g. bone pain, headache, facial pain or visceral pain) or pain associated with cancer therapy (e.g.
- Neuropathic pain is currently defined as pain initiated or caused by a primary lesion or dysfunction in the nervous system. Nerve damage can be caused by trauma and disease and thus the term 'neuropathic pain' encompasses many disorders with diverse aetiologies.
- peripheral neuropathy include, but are not limited to, peripheral neuropathy, diabetic neuropathy, post herpetic neuralgia, trigeminal neuralgia, back pain, cancer neuropathy, HIV neuropathy, phantom limb pain, carpal tunnel syndrome, central post-stroke pain and pain associated with chronic alcoholism, hypothyroidism, uremia, multiple sclerosis, spinal cord injury, Parkinson's disease, epilepsy and vitamin deficiency.
- Neuropathic pain is pathological as it has no protective role. It is often present well after the original cause has dissipated, commonly lasting for years, significantly decreasing a patient's quality of life (Woolf and Mannion, 1999, Lancet, 353, 1959-1964).
- neuropathic pain The symptoms of neuropathic pain are difficult to treat, as they are often heterogeneous even between patients with the same disease (Woolf & Decosterd, 1999, Pain Supp, 6, S141-S147; Woolf and Mannion, 1999, Lancet, 353, 1959-1964). They include spontaneous pain, which can be continuous, and paroxysmal or abnormal evoked pain, such as hyperalgesia (increased sensitivity to a noxious stimulus) and allodynia (sensitivity to a normally innocuous stimulus).
- the inflammatory process is a complex series of biochemical and cellular events, activated in response to tissue injury or the presence of foreign substances, which results in swelling and pain (Levine and Taiwo, 1994, Textbook of Pain, 45-56).
- Arthritic pain is the most common inflammatory pain.
- Rheumatoid disease is one of the commonest chronic inflammatory conditions in developed countries and rheumatoid arthritis is a common cause of disability. The exact aetiology of rheumatoid arthritis is unknown, but current hypotheses suggest that both genetic and microbiological factors may be important (Grennan & Jayson, 1994, Textbook of Pain, 397-407).
- Visceral pain is pain associated with the viscera, which encompass the organs of the abdominal cavity. These organs include the sex organs, spleen and part of the digestive system. Pain associated with the viscera can be divided into digestive visceral pain and non-digestive visceral pain.
- Gl gastrointestinal
- FBD functional bowel disorder
- IBD inflammatory bowel disease
- Gl disorders include a wide range of disease states that are currently only moderately controlled, including, in respect of FBD, gastro-esophageal reflux, dyspepsia, irritable bowel syndrome (IBS) and functional abdominal pain syndrome (FAPS), and, in respect of IBD, Crohn's disease, ileitis and ulcerative colitis, all of which regularly produce visceral pain.
- visceral pain include the pain associated with dysmenorrhea, cystitis and pancreatitis and pelvic pain.
- heart and vascular pain including pain caused by angina, myocardical infarction, mitral stenosis, pericarditis, Raynaud's phenomenon, scleredoma and skeletal muscle ischemia;
- head pain such as migraine (including migraine with aura and migraine without aura), cluster headache, tension-type headache mixed headache and headache associated with vascular disorders; and
- orofacial pain including dental pain, otic pain, burning mouth syndrome and temporomandibular myofascial pain.
- R 1 , R 2 , R 3 and R 4 are as previously defined for a compound of formula (I) unless otherwise stated.
- a compound of formula (I), wherein R 3 and R 4 are H, may be prepared by the hydrogenolytic deprotection of a compound of formula (III)
- R 1 and R 2 are as defined above.
- the hydrogenation is typically carried out using a source of hydrogen such as hydrogen gas, cyclohexadiene or ammonium formate (preferably hydrogen gas) and a transition metal catalyst such as a palladium, platinum or rhodium catalyst (preferably a palladium catalyst).
- a source of hydrogen such as hydrogen gas, cyclohexadiene or ammonium formate (preferably hydrogen gas) and a transition metal catalyst such as a palladium, platinum or rhodium catalyst (preferably a palladium catalyst).
- An acid such as hydrochloric or trifluoroacetic acid, may also be used to increase the rate of reaction.
- a solution of the compound of formula (III) in a suitable solvent, such as ethanol is treated with palladium on carbon and hydrochloric acid hydrogenated at about 414 kPa (60 psi).
- a compound of formula (III) may be prepared by treating an imine of formula (IV):
- R 1 is as defined above, with a compound of formula:
- R 2 is as defined above and M 1 is a suitable metal, optionally bearing one or more further ligands; or by treating an imine of formula (Vl):
- R 2 is as defined above, with a compound of formula:
- organometallic reagent of formula (V) or (VII) is typically an organolithium or an organomagnesium derivative.
- the reaction is carried out in a suitable inert solvent such as tetrahydrofuran or diethyl ether at low temperature, typically between 0 and -78 0 C.
- a solution of the compound of formula (IV) or (Vl) in a suitable solvent, such as tetrahydrofuran, is treated with a suitable Grignard reagent of formula (V) or (VII), respectively, at -5O 0 C and in the presence of boron trifluoride etherate.
- R 1 is as defined above and X is Ci-C ⁇ alkyl; or a compound of formula (X):
- R 2 is as defined above and X is C 1 -C 6 alkyl.
- the condensation may be carried out under basic, neutral or acidic conditions and generally requires elevated temperatures and/or prolonged reaction times.
- a solution of the compound of formula (VIII) and the compound of formula (IX) or (X), in a suitable solvent, such as trifluoroethanol, is heated at about 8O 0 C in the presence of a dehydrating agent such as 4A molecular sieves.
- a compound of formula (I), wherein R 3 and R 4 are both H, may alternatively be prepared by the hydrolysis of a nitrile of formula (Xl):
- R 1 and R 2 are as defined above.
- the hydrolysis is typically accomplished with acidic or basic catalysis in an aqueous solvent at an elevated temperature.
- a solution of the compound of formula (Xl) in water is treated with 6 molar hydrochloric acid and heated to about 100 0 C.
- a compound of formula (Xl) may be prepared by the addition of cyanide to a compound of formula (XII):
- a preferred source of cyanide for the addition is a compound of formula M 2 CN wherein M 2 is a metal cation, optionally bearing other ligands. Most preferred is a dialkylaluminium cyanide such as diethylaluminium cyanide.
- the reaction is carried out as a solution in a suitable inert solvent such as tetrahydrofuran, dichloromethane or diethyl ether.
- a solution of a compound of formula (XII) in a mixture of isoproanol and tetrahydrofuran is treated with diethylaluminium cyanide at a temperature of between -78 and -2O 0 C.
- a compound of formula (XII) may be prepared by the reaction of a compound of formula (XIII)
- R 1 and R 2 are as defined above, under dehydrating conditions.
- the reaction is catalysed by a Lewis acid (e.g. titanium tetraethoxide).
- a Lewis acid e.g. titanium tetraethoxide
- a solution of the compound of formula (XIII) and the compound of formula (XIV) in a suitable solvent such as tetrahydrofuran is treated with titanium tetraethoxide at a temperature of about 5O 0 C.
- a compound of formula (I), wherein R 3 and R 4 are both H, may alternatively be prepared by the hydrolysis of an ester of formula (XV):
- R 1 and R 2 are as defined above.
- the hydrolysis may be carried out under acidic or basic conditions. In a typical procedure, a solution of a compound of formula (XV) in aqueous hydrochloric acid is heated under reflux for 16 hours.
- a compound of formula (XV) may be prepared by the methanolysis of a compound of formula (XVI):
- R 1 and R 2 are as defined above and Y 1 and Y 2 are each selected from Ci-C 6 alkyl.
- the reaction may be carried out with acid or base catalysis.
- a solution of a compound of formula (XVI) in methanolic hydrochloric acid is stirred at room temperature for about 72 hours.
- a compound of formula (XVI) may be prepared by the alkylation of a compound of formula (XVII):
- R 1 , Y 1 and Y 2 are as defined above, with a compound of formula R 2 L 1 , wherein R 2 is as defined above and L 1 is a suitable leaving group.
- L 1 is preferably halo (particularly bromo), trifluoromethanesulphonate or methanesulphonate.
- the compound of formula (XVII) is deprotonated with a base, such as butyl lithium, in an inert solvent such as diethyl ether or tetrahydrofuran, at low temperature (usually in the range -78 to -2O 0 C). A solution of the alkylating agent in an inert solvent is then added.
- a solution of the compound of formula (XVII) in tetrahydrofuran is treated with n-butyl lithium at -78 0 C and an excess of the alkylating agent is then added.
- a compound of formula (XVII) may be prepared by the double alkylation of a compound of formula (XVIII):
- the compound of formula (XVIII) is deprotonated using a base (e.g. potassium fert-butoxide, potassium hexamethyldisilazide or sodium hydride) in an inert solvent, such as tetrahydrofuran or diethyl ether.
- a base e.g. potassium fert-butoxide, potassium hexamethyldisilazide or sodium hydride
- an inert solvent such as tetrahydrofuran or diethyl ether.
- a suitable alkylating agent such as an alkyl halide (particularly an alkyl bromide) or an alkyl sulphonate ester (e.g. an alkyl mesylate) is then added at a temperature of from -2O 0 C to room temperature.
- a compound of formula (XVIII) may be prepared by the cyclisation of a compound of formula (XIX):
- R 1 is as defined above.
- the reduction is typically accomplished using hydrogen and a hydrogenation catalyst such as a palladium, platinum or rhodium catalyst.
- a solution of the compound of formula (XX) in a suitable solvent, such as aqueous ethanolic hydrochloric acid is treated with hydrogen at room temperature.
- a compound of formula (XX) may be prepared by coupling an amine of formula (XXI):
- the acid is first activated, either by conversion to the corresponding acid chloride or by treatment with a suitable peptide coupling agent. If the acid chloride is used it is preformed and then reacted with the amine as a solution in a suitable inert solvent (such as dichloromethane or tetrahydrofuran) in the presence of a base (such as triethylamine). Alternatively, as solution of the acid and the amine in a suitable solvent (such as dichloromethane or tetrahydrofuran) is treated with a base (such as triethylamine) and a coupling agent (such as a carbodiimide).
- a suitable inert solvent such as dichloromethane or tetrahydrofuran
- a base such as triethylamine
- a coupling agent such as a carbodiimide
- Compounds of formula (I) can also be prepared by using the reactions described above to construct a compound wherein R 1 or R 2 are partially formed and then completing the synthesis by functional group manipulation. For instance, a group may be carried through the synthesis in a protected form and deprotected in a final step.
- Suitable protecting groups are described in 'Protective Groups in Organic Synthesis' by Theodora Greene and Peter Wuts (third edition, 1999, John Wiley and Sons).
- Suitable functional group transformations are described in 'Comprehensive Organic Transformations' by Richard Larock (1999, VCH Publishers Inc.).
- (2S)-2-aminomethyl-5-ethyl-heptanoic acid (the compound of formula (I)) may be prepared from a compound of formula (IV) as shown in scheme 1 , below:
- the compound of formula (II) may be obtained as described in Organic Letters, 2000; 2(22); 3527-3529.
- the reaction is typically achieved by the treatment of (II) with a strong base, for example, lithium diisopropylamide (LDA), lithium hexamethyldisilylazide (LHMDS), or sodium hexamethyldisilylazide (NaHMDS), optionally in the presence of an additive, (e.g. lithium chloride (LiCI)) in a suitable solvent (e.g.
- a strong base for example, lithium diisopropylamide (LDA), lithium hexamethyldisilylazide (LHMDS), or sodium hexamethyldisilylazide (NaHMDS
- an additive e.g. lithium chloride (LiCI)
- a suitable solvent e.g.
- tetrahydrofuran (THF), ether) at low temperature, for example at a temperature of from -1O 0 C to O 0 C for about 1 hour, followed by the quenching of the resulting anion with the alkyl bromide (Vl).
- THF tetrahydrofuran
- ether a solution of 1 equivalent of the compound of formula (II) in THF is treated with 3.2 equivalents LHMDS and 4 equivalents LiCI, at a temperature of from -5 0 C to O 0 C for about 1 hour followed by treatment with 1 equivalent of the alkenyl bromide (Vl) at O 0 C and the reaction is allowed to warm to room temperature over 18 hours.
- the alkenyl bromide (Vl) may be prepared from commercially available starting materials using standard chemical transformations as exemplified in Preparations 1 and 2.
- a suitable catalyst for example 10% palladium on charcoal or platinum oxide.
- compound (IV) may be prepared in one step by replacing the alkenyl bromide (Vl) with an alkyl halide (Via) in step (a) as in scheme 1.
- the alkly bromide may be prepared according to the processes described by Bestmann ef al. (Liebigs Ann. Chem. 1979, 1189-1204) and Pinazzi ef al. (Bull. Soc. Chim., 1975, 1-2, 201-205).
- the alkyl iodide may be prepared by analogous processes.
- the compound of formula (I) may be prepared by changing the order of steps (b) and (c) as shown in Scheme 2.
- (2S)-2-aminomethyl-5-ethyl-heptanoic acid (compound (I)) may be prepared from a compound of formula (IX) as shown in scheme 3.
- the reaction may be achieved by treatment of the compound of formula (VII) with a strong base, for example, LDA, LHMDS or NaHMDS, in a suitable solvent (e.g. THF, ether) at a temperature of from -78 0 C to -6O 0 C, for example -78 0 C, for about 1 hour, followed by quenching of the resulting anion with the alkyl bromide, at a temperature of from -78 0 C to about room temperature.
- a strong base for example, LDA, LHMDS or NaHMDS
- a suitable solvent e.g. THF, ether
- Step (g): The compound of formula (IX) may be prepared by treatment of the compound of formula (VIII) with a suitable base, and quenching of the resulting anion with aqueous acid.
- the reaction may be achieved by treatment of the compound of formula (VIII) with a strong base, for example, LDA, LHMDS or NaHMDS, in a suitable solvent (e.g. THF, ether) at very low temperature, for example at a temperature of from about -78° to about -6O 0 C for about 3 hours, followed by quenching of the resulting anion with aqueous acid.
- a strong base for example, LDA, LHMDS or NaHMDS
- a suitable solvent e.g. THF, ether
- This reaction may be achieved under basic or acidic catalysis, but is typically carried out under aqueous acidic conditions (e.g. hydrochloric acid (HCI) or sulphuric acid (H 2 SO 4 )), optionally in the presence of a suitable solvent (e.g. THF) at reflux for about 24 hours.
- aqueous acidic conditions e.g. hydrochloric acid (HCI) or sulphuric acid (H 2 SO 4 )
- a suitable solvent e.g. THF
- R is a suitable protecting group such as (CrC ⁇ Jalkyl.
- the aldehyde (XIV) is described in Tetrahedron 1988, 44(4)1091-1106.
- Step (j): Reduction of a compound of formula (XV) to a compound of formula (XVI) may be carried out by hydrogenation in a suitable solvent, typically ethanol, in the presence of a metal catalyst (e.g. platinum oxide, palladium on charcoal).
- a metal catalyst e.g. platinum oxide, palladium on charcoal
- Step (k): Resolution of a compound of formula (XVI) may be achieved by formation of a chiral salt (e.g. the (+) di-o-tolyl tartrate or (L)-dibenzoyl tartrate salt) and recrystallisation, followed by reformation of the free amine.
- Step (m): The compound of formula (I) may be prepared by hydrolysis of a compound of formula (XVII) under acidic or basic conditions, typically using HCI, in dioxan, at a temperature of 8O 0 C, for about 18 hours.
- Compound (I) may alternatively be prepared by stereospecific hydrolysis of compound (XVI) with an enzyme (e.g. pig liver esterase, lipase).
- an enzyme e.g. pig liver esterase, lipase
- the compound of formula (I) may be prepared from a compound of formula (XVII) according to scheme 6.
- a base e.g. potassium carbonate
- a solvent e.g. dimethylforamide
- Step (o): Reduction of a compound of formula (XVIII) to a compound of formula (XVI) may be carried out by hydrogenation in a suitable solvent, typically ethanol, in the presence of a metal catalyst (e.g. platinum oxide or palladium on charcoal).
- a metal catalyst e.g. platinum oxide or palladium on charcoal
- the compound of formula (I) may also prepared by analogy with the methods described in WO-A- 2003/082807 and references therein, and also by analogy with the method of Lavielle et al in European Journal of Organic Chemistry, 2000(1), 83-89.
- DMF dimethylformamide
- DMSO dimethyl sulphoxide
- TEMPO 2,2,6,6-tetramethylpiperidine-N-oxide
- THF tetrahydrofuran
- P represents a hydroxy-protecting group, suitable examples of which are described in "Protective Groups in Organic Synthesis" by T. W. Greene and P. Wuts, Wiley and Sons, 1991
- LG represents a suitable leaving group, such as halogen or sulphonate (eg methanesulphonate, p-toluenesulphonate or trifluoromethanesulphonate).
- P is benzyl and LG is p-toluenesulphonate.
- the compound of formula (III) may be prepared from compound (II) and an appropriate agent capable of converting a hydroxy group into a leaving group, typically a sulphonylating reagent (eg methanesulphonyl chloride or p-toluenesulphonyl chloride) in the presence of a base (eg triethylamine or pyridine) in a suitable solvent (eg pyridine or dichloromethane) at O 0 C to room temperature for 15 minutes to 24 hours.
- a base eg triethylamine or pyridine
- suitable solvent eg pyridine or dichloromethane
- Preferred conditions are: 1eq compound (II) in dichloromethane, 1.2eq p-toluenesulphonyl chloride, 2 eq pyridine at room temperature for 18 hours.
- Step (b): The compound of formula (IV) may be prepared from compound (III) and the hydroxy compound of formula (Vl) in a suitable solvent (eg DMF, DMSO) in the presence of a suitable base (eg Cs 2 CO 3 , K 2 CO 3 ), optionally in the presence of a crown ether (eg 18-crown-6) at 50- 12O 0 C overnight.
- a suitable solvent eg DMF, DMSO
- a suitable base eg Cs 2 CO 3 , K 2 CO 3
- a crown ether eg 18-crown-6
- a deprotecting agent in a suitable solvent to yield the compound of formula (V).
- suitable reagents and methods are described in "Protective Groups in Organic Synthesis" (referred to above).
- P is benzyl
- suitable reagents include boron trichloride or iron (III) chloride.
- Preferred conditions are: 1eq compound (IV) in dichloromethane, 4 eq BCI 3 at room temperature for 18 hours.
- the compound of formula (I) may be prepared by oxidation of the compound of formula (V) using an oxidising agent in a suitable solvent.
- Typical reagents and conditions include catalytic chromium trioxide and periodic acid (H 5 IO 6 ) in a solvent such as acetonitrile at room temperature to 5O 0 C for 18 to 36 hours, or alternatively NaOCI plus NaCIO 2 in the presence of catalytic TEMPO in a solvent such as acetonitrile at O 0 C to room temperature for 18 to 36 hours.
- Preferred conditions are: 1eq compound (V), 2.5 eq periodic acid, 0.02 eq CrO 3 , in 0.75% aqueous acetonitrile, 24 hours at 4O 0 C.
- the compounds of formula (I) may alternatively be prepared by oxidation of compounds of formula (V) in a two-step procedure via the aldehydes of formula (VII) as shown in Scheme 2.
- a suitable solvent eg acetonitrile, acetone at O 0 C to room temperature for 2- 18 hours
- sulphur trioxide- pyridine complex with DMSO in a solvent such as THF at O 0 C to room temperature for 2-18 hours.
- a solvent such as aqueous t-butanol
- catalytic TEMPO eg acetone or acetonitrile
- Trans compounds (II) and (X) may be obtained from cis compounds (II) and (X) respectively by inversion using Mitsunobu chemistry analogous to that described in Synthesis, (1981), 1.
- R a is an ester residue, suitable examples of which are described in "Protective Groups in Organic Synthesis” (referred to above) (eg (C ⁇ alkyl, benzyl or (+) or (-)-menthyl), and LG is a leaving group such as halogen or sulphonate (eg methanesulphonate, p- toluenesulphonate or trifluoromethanesulphonate).
- Preferred conditions are: 1eq compound (VIII), 1.1 eq. 1 ,1'-carbonyl diimidazole, in ethyl acetate at reflux for 1 hour followed by 1eq R 3 OH at room temperature for 4 hours.
- Step (b): Reduction of compound (IX) to the alcohol (X) may be carried out using a suitable reducing agent, eg sodium borohydride or L-Selectride ® , in a suitable solvent such as THF.
- a suitable reducing agent eg sodium borohydride or L-Selectride ®
- THF a suitable solvent
- Preferred conditions are: 1eq compound (IX), 0.5 eq NaBH 4 in 20:1 THF:methanol at O 0 C for 20 minutes.
- Step (c): The compound of formula (Xl) may be prepared from compound (X) using reagents and conditions similar to those described in Scheme 1 , step (a). Preferred conditions are: 1eq compound (X) 1 1.05 eq p-toluenesulphonyl chloride in pyridine at O 0 C to room temperature.
- the components of the combination of the invention should be assessed for their biopharmaceutical properties, such as solubility and solution stability (across pH), permeability, efc, in order to select the most appropriate dosage form and route of administration for treatment of the proposed indication.
- biopharmaceutical properties such as solubility and solution stability (across pH), permeability, efc, in order to select the most appropriate dosage form and route of administration for treatment of the proposed indication.
- the components of the combiantion of the invention intended for pharmaceutical use may be administered as crystalline or amorphous products. They may be obtained, for example, as solid plugs, powders, or films by methods such as precipitation, crystallization, freeze drying, or spray drying, or evaporative drying. Microwave or radio frequency drying may be used for this purpose.
- compositions suitable for the delivery of combinations of the present invention and methods for their preparation will be readily apparent to those skilled in the art. Such compositions and methods for their preparation may be found, for example, in Remington's Pharmaceutical Sciences, 19th Edition (Mack Publishing Company, 1995).
- Oral administration may involve swallowing, so that the compound enters the gastrointestinal tract, and/or buccal, lingual, or sublingual administration by which the compound enters the blood stream directly from the mouth.
- Liquid formulations include suspensions, solutions, syrups and elixirs. Such formulations may be employed as fillers in soft or hard capsules (made, for example, from gelatin or hydroxypropylmethylcellulose) and typically comprise a carrier, for example, water, ethanol, polyethylene glycol, propylene glycol, methylcellulose, or a suitable oil, and one or more emulsifying agents and/or suspending agents. Liquid formulations may also be prepared by the reconstitution of a solid, for example, from a sachet. The components of the combination of the invention may also be used in fast-dissolving, fast- disintegrating dosage forms such as those described in Expert Opinion in Therapeutic Patents, H (6), 981-986, by Liang and Chen (2001).
- the drug may make up from 1 weight % to 80 weight % of the dosage form, more typically from 5 weight % to 60 weight % of the dosage form.
- tablets generally contain a disintegrant.
- disintegrants include sodium starch glycolate, sodium carboxymethyl cellulose, calcium carboxymethyl cellulose, croscarmellose sodium, crospovidone, polyvinylpyrrolidone, methyl cellulose, microcrystalline cellulose, lower alkyl-substituted hydroxypropyl cellulose, starch, pregelatinised starch and sodium alginate.
- the disintegrant will comprise from 1 weight % to 25 weight %, preferably from 5 weight % to 20 weight % of the dosage form.
- Binders are generally used to impart cohesive qualities to a tablet formulation. Suitable binders include microcrystalline cellulose, gelatin, sugars, polyethylene glycol, natural and synthetic gums, polyvinylpyrrolidone, pregelatinised starch, hydroxypropyl cellulose and hydroxypropyl methylcellulose. Tablets may also contain diluents, such as lactose (monohydrate, spray-dried monohydrate, anhydrous and the like), mannitol, xylitol, dextrose, sucrose, sorbitol, microcrystalline cellulose, starch and dibasic calcium phosphate dihydrate.
- lactose monohydrate, spray-dried monohydrate, anhydrous and the like
- mannitol xylitol
- dextrose sucrose
- sorbitol microcrystalline cellulose
- starch dibasic calcium phosphate dihydrate
- Tablets may also optionally comprise surface active agents, such as sodium lauryl sulfate and polysorbate 80, and glidants such as silicon dioxide and talc.
- surface active agents such as sodium lauryl sulfate and polysorbate 80
- glidants such as silicon dioxide and talc.
- surface active agents may comprise from 0.2 weight % to 5 weight % of the tablet, and glidants may comprise from 0.2 weight % to 1 weight % of the tablet.
- Tablets also generally contain lubricants such as magnesium stearate, calcium stearate, zinc stearate, sodium stearyl fumarate, and mixtures of magnesium stearate with sodium lauryl sulphate.
- Lubricants generally comprise from 0.25 weight % to 10 weight %, preferably from 0.5 weight % to 3 weight % of the tablet.
- ingredients include anti-oxidants, colourants, flavouring agents, preservatives and taste-masking agents.
- Exemplary tablets contain up to about 80% drug, from about 10 weight % to about 90 weight % binder, from about 0 weight % to about 85 weight % diluent, from about 2 weight % to about 10 weight % disintegrant, and from about 0.25 weight % to about 10 weight % lubricant.
- Tablet blends may be compressed directly or by roller to form tablets. Tablet blends or portions of blends may alternatively be wet-, dry-, or melt-granulated, melt congealed, or extruded before tabletting.
- the final formulation may comprise one or more layers and may be coated or uncoated; it may even be encapsulated.
- the formulation of tablets is discussed in Pharmaceutical Dosage Forms: Tablets. Vol. 1, by H. Lieberman and L. Lachman (Marcel Dekker, New York, 1980).
- Consumable oral films for human or veterinary use are typically pliable water-soluble or water- swellable thin film dosage forms which may be rapidly dissolving or mucoadhesive and typically comprise a compound of formula I 1 a film-forming polymer, a binder, a solvent, a humectant, a plasticiser, a stabiliser or emulsifier, a viscosity-modifying agent and a solvent. Some components of the formulation may perform more than one function.
- the components of the combination may be water-soluble or insoluble.
- a water-soluble compound typically comprises from 1 weight % to 80 weight %, more typically from 20 weight % to 50 weight %, of the solutes. Less soluble compounds may comprise a greater proportion of the composition, typically up to 88 weight % of the solutes.
- the components of the combination may be in the form of multiparticulate beads.
- the film-forming polymer may be selected from natural polysaccharides, proteins, or synthetic hydrocolloids and is typically present in the range 0.01 to 99 weight %, more typically in the range 30 to 80 weight %.
- ingredients include anti-oxidants, colorants, flavourings and flavour enhancers, preservatives, salivary stimulating agents, cooling agents, co-solvents (including oils), emollients, bulking agents, anti-foaming agents, surfactants and taste-masking agents.
- Films in accordance with the invention are typically prepared by evaporative drying of thin aqueous films coated onto a peelable backing support or paper. This may be done in a drying oven or tunnel, typically a combined coater dryer, or by freeze-drying or vacuuming.
- Solid formulations for oral administration may be formulated to be immediate and/or modified controlled release.
- Modified release formulations include delayed-, sustained-, pulsed-, controlled- , targeted and programmed release.
- the components of the combination of the invention may also be administered directly into the blood stream, into muscle, or into an internal organ.
- Suitable means for parenteral administration include .intravenous, intraarterial, intraperitoneal, intrathecal, intraventricular, intraurethral, intrasternal, intracranial, intramuscular, intrasynovial and subcutaneous.
- Suitable devices for parenteral administration include needle (including microneedle) injectors, needle-free injectors and infusion techniques.
- parenteral formulations under sterile conditions may readily be accomplished using standard pharmaceutical techniques well known to those skilled in the art.
- Formulations for parenteral administration may be formulated to be immediate and/or modified controlled release.
- Modified release formulations include delayed-, sustained-, pulsed-, controlled- , targeted and programmed release.
- the components of the combination of the invention may be formulated as a suspension or as a solid, semi-solid, or thixotropic liquid for administration as an implanted depot providing modified release of the active compound.
- examples of such formulations include drug-coated stents and semi-solids and suspensions comprising drug-loaded poly(d/-!actic-coglycolic)acid (PGLA) microspheres.
- PGLA poly(d/-!actic-coglycolic)acid
- the components of the combination of the invention may also be administered topically, (intra)dermally, or transdermal ⁇ to the skin or mucosa.
- Typical formulations for this purpose include gels, hydrogels, lotions, solutions, creams, ointments, dusting powders, dressings, foams, films, skin patches, wafers, implants, sponges, fibres, bandages and microemulsions. Liposomes may also be used.
- Typical carriers include alcohol, water, mineral oil, liquid petrolatum, white petrolatum, glycerin, polyethylene glycol and propylene glycol. Penetration enhancers may be incorporated - see, for example, J Pharm Sci, 88 (10), 955-958, by Finnin and Morgan (October 1999).
- a suitable solution formulation for use in an atomiser using electrohydrodynamics to produce a fine mist may contain from 1 ⁇ g to 20mg of the compound of the invention per actuation and the actuation volume may vary from 1 ⁇ l to 100 ⁇ l.
- a typical formulation may comprise a compound of formula I 1 propylene glycol, sterile water, ethanol and sodium chloride.
- Alternative solvents which may be used instead of propylene glycol include glycerol and polyethylene glycol.
- Suitable flavours such as menthol and levomenthol, or sweeteners, such as saccharin or saccharin sodium, may be added to those formulations of the invention intended for inhaled/intranasal administration.
- Formulations for inhaled/intranasal administration may be formulated to be immediate and/or modified controlled release using, for example, PGLA.
- Controlled release formulations include Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
- the dosage unit is determined by means of a valve which delivers a metered amount.
- Units in accordance with the invention are typically arranged to administer a metered dose.
- the overall daily dose may be administered in a single dose or, more usually, as divided doses throughout the day.
- the compounds of the invention may be administered rectally or vaginally, for example, in the form of a suppository, pessary, or enema.
- Cocoa butter is a traditional suppository base, but various alternatives may be used as appropriate.
- Formulations for rectal/vaginal administration may be formulated to be immediate and/or modified controlled release.
- Controlled release formulations include Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
- the components of the combination of the invention may also be administered directly to the eye or ear, typically in the form of drops of a micronised suspension or solution in isotonic, pH- adjusted, sterile saline.
- Other formulations suitable for ocular and aural administration include ointments, gels, biodegradable (e.g. absorbable gel sponges, collagen) and non-biodegradable (e.g. silicone) implants, wafers, lenses and particulate or vesicular systems, such as niosomes or liposomes.
- the components of the combination of the invention may be combined with soluble macromolecular entities, such as cyclodextrin and suitable derivatives thereof or polyethylene glycol-containing polymers, in order to improve their solubility, dissolution rate, taste-masking, bioavailability and/or stability for use in any of the aforementioned modes of administration.
- soluble macromolecular entities such as cyclodextrin and suitable derivatives thereof or polyethylene glycol-containing polymers
- Drug-cyclodextrin complexes are found to be generally useful for most dosage forms and administration routes. Both inclusion and non-inclusion complexes may be used.
- the cyclodextrin may be used as an auxiliary additive, i.e. as a carrier, diluent, or solubiliser. Most commonly used for these purposes are alpha-, beta- and gamma-cyclodextrins, examples of which may be found in International Patent Applications Nos. WO 91/11172, WO 94/02518 and WO 98/55148.
- These dosages are based on an average human subject having a weight of about 60kg to 70kg. The physician will readily be able to determine doses for subjects whose weight falls outside this range, such as infants and the elderly.
- references herein to "treatment” include references to curative, palliative and prophylactic treatment.
- a pharmaceutical composition comprising an alpha-2-delta ligand, a PDE7 inhibitor, or pharmaceutically acceptable salts thereof, and one or more suitable excipients.
- the composition is suitable for use in the treatment of pain, particularly neuropathic pain.
- a pharmaceutical composition comprising a synergistic combination comprising an alpha-2-delta ligand, a PDE7 inhibitor, or pharmaceutically acceptable salts thereof, and one or more suitable excipients.
- the composition is suitable for use in the treatment of pain, particularly neuropathic pain.
- the combination of the present invention may be usefully combined with another pharmacologically active compound, or with two or more other pharmacologically active compounds, particularly in the treatment of pain.
- the combination of the present invention, or pharmaceutically acceptable salts or solvates thereof, as defined above may be administered simultaneously, sequentially or separately in combination with one or more agents selected from:
- an opioid analgesic e.g. morphine, heroin, hydromorphone, oxymorphone, levorphanol, levallorphan, methadone, meperidine, fentanyl, cocaine, codeine, dihydrocodeine, oxycodone, hydrocodone, propoxyphene, nalmefene, nalorphine, naloxone, naltrexone, buprenorphine, butorphanol, nalbuphine or pentazocine;
- NSAID nonsteroidal antiinflammatory drug
- NSAID nonsteroidal antiinflammatory drug
- diclofenac diflusinal, etodolac
- fenbufen fenoprofen
- flufenisal flurbiprofen
- ibuprofen indomethacin
- ketoprofen ketorolac
- meclofenamic acid mefenamic acid
- meloxicam nabumetone, naproxen, nimesulide, nitroflurbiprofen, olsalazine, oxaprozin, phenylbutazone, piroxicam, sulfasalazine, sulindac, tolmetin or zomepirac
- NSAID nonsteroidal antiinflammatory drug
- a benzodiazepine having a sedative action e.g. chlordiazepoxide, clorazepate, diazepam, flurazepam, lorazepam, oxazepam, temazepam or triazolam;
- an Hi antagonist having a sedative action e.g. diphenhydramine, pyrilamine, promethazine, chlorpheniramine or chlorcyclizine; • a sedative such as glutethimide, meprobamate, methaqualone or dichloralphenazone;
- a skeletal muscle relaxant e.g. baclofen, carisoprodol, chlorzoxazone, cyclobenzaprine, methocarbamol or orphrenadine;
- an NMDA receptor antagonist e.g. dextromethorphan ((+)-3-hydroxy-N- methylmorphinan) or its metabolite dextrorphan ((+)-3-hydroxy-N-methylmorphinan), ketamine, memantine, pyrroloquinoline quinine, cis-4-(phosphonomethyl)-2- piperidinecarboxylic acid, budipine, EN-3231 (MorphiDex®, a combination formulation of morphine and dextromethorphan), topiramate, neramexane or perzinfotel including an NR2B antagonist, e.g.
- an NMDA receptor antagonist e.g. dextromethorphan ((+)-3-hydroxy-N- methylmorphinan) or its metabolite dextrorphan ((+)-3-hydroxy-N-methylmorphinan), ketamine, memantine, pyrroloquinoline quinine, cis-4-(phosphonomethyl
- an alpha-adrenergic e.g. doxazosin, tamsulosin, clonidine, guanfacine, dexmetatomidine, modafinil, or 4-amino-6,7-dimethoxy-2-(5-methane-sulfonamido- 1 ,2,3,4-tetrahydroisoquinol-2-yl)-5-(2-pyridyl) quinazoline;
- a tricyclic antidepressant e.g. desipramine, imipramine, amitriptyline or nortriptyline
- an anticonvulsant e.g. carbamazepine, lamotrigine, topiratmate or valproate
- a tachykinin (NK) antagonist particularly an NK-3, NK-2 or NK-1 antagonist, e.g. ( ⁇ R,9R)-7-[3, 5-bis(trifluoromethyl)benzyl]-8,9, 10,11 -tetrahydro-9-methyl-5-(4- methylphenyl)-7H-[1 ,4]diazocino[2, 1 -g][1 ,7]-naphthyridine-6-13-dione (TAK-637), 5- [[(2R,3S)-2-[( 1 R)-1 -[3,5-bis(trifluoromethyl)phenyl]ethoxy-3-(4-fluorophenyl)-4- morpholinyl]-methyl]-1 ,2-dihydro-3H-1 ,2,4-triazol-3-one (MK-869), aprepitant, lanepitant, dapitant or 3-[[2-methoxy-5-(trifluoromethoxy)phen
- a muscarinic antagonist e.g oxybutynin, tolterodine, propiverine, tropsium chloride, darifenacin, solifenacin, temiverine and ipratropium;
- COX-2 selective inhibitor e.g. celecoxib, rofecoxib, parecoxib, valdecoxib, deracoxib, etoricoxib, or lumiracoxib;
- a neuroleptic such as droperidol, chlorpromazine, haloperidol, perphenazine, thioridazine, mesoridazine, trifluoperazine, fluphenazine, clozapine, olanzapine, risperidone, ziprasidone, quetiapine, sertindole, aripiprazole, sonepiprazole, blonanserin, iloperidone, perospirone, raclopride, zotepine, bifeprunox, asenapine, lurasidone, amisulpride, balaperidone, palindore, eplivanserin, osanetant, rimonabant, meclinertant, Miraxion® or sarizotan; • a vanilloid receptor agonist (e.g. resinferatoxin) or antagonist (e.g. capsazepine);
- a beta-adrenergic such as propranolol
- a corticosteroid such as dexamethasone
- a cholinergic (nicotinic) analgesic such as ispronicline (TC-1734), (E)-N-methyl- 4-(3-pyridinyl)-3-buten-1 -amine (RJR-2403), (R)-5-(2-azetidinylmethoxy)-2-chloropyridine (ABT-594) or nicotine;
- a PDEV inhibitor such as 5-[2-ethoxy-5-(4-methyl-1-piperazinyl- sulphonyl)phenyl]-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (sildenafil), (6R,12aR)-2,3,6,7,12,12a-hexahydro-2-methyl-6-(3,4-methylenedioxyphenyl)- pyrazino[2',1':6,1]-pyrido[3,4-b]indole-1 ,4-dione (IC-351 or tadalafil), 2-[2-ethoxy-5-(4- ethyl-piperazin-1 -yl-1 -sulphonyl)-phenyl]-5-methyl-7-propyl-3H-imidazo[5, 1 -f][1 ,2,4]triazin- 4-one (vardenafil),
- mGluRI metabotropic glutamate subtype 1 receptor
- a noradrenaline (norepinephrine) reuptake inhibitor such as maprotiline, lofepramine, mirtazepine, oxaprotiline, fezolamine, tomoxetine, mianserin, buproprion, bupropion metabolite hydroxybuproprion, nomifensine and viloxazine (Vivalan®), especially a selective noradrenaline reuptake inhibitor such as reboxetine, in particular
- a dual serotonin-noradrenaline reuptake inhibitor such as venlafaxine, venlafaxine metabolite O-desmethylvenlafaxine, clomipramine, clomipramine metabolite desmethylclomipramine, duloxetine, milnacipran and imipramine; • an inducible nitric oxide synthase (iNOS) inhibitor such as S-[2-[(1- iminoethyl)amino]ethyl]-L-homocysteine, S-[2-[(1-iminoethyl)-amino]ethyl]-4,4-dioxo-L- cysteine, S-[2-[(1-iminoethyl)amino]ethyl]-2-methyl-L-cysteine, (2S,5Z)-2-amino-2-methyl- 7-[(1-iminoethyl)amino]-5-he
- an acetylcholinesterase inhibitor such as donepezil
- a prostaglandin E 2 subtype 4 (EP4) antagonist such as ⁇ /-[( ⁇ 2-[4-(2-ethyl-4,6- dimethyl-I H-imidazo ⁇ . ⁇ -clpyridin-i-yOphenylJethylJaminoJ-carbonylH- methylbenzenesulfonamide or 4-[(1 S)-1-( ⁇ [5-chloro-2-(3-fluorophenoxy)pyridin-3- yl]carbonyl ⁇ amino)ethyl]benzoic acid;
- a leukotriene B4 antagonist such as 1-(3-biphenyl-4-ylmethyl-4-hydroxy- chroman-7-yl)-cyclopentanecarboxylic acid (CP-105696), 5-[2-(2-Carboxyethyl)-3-[6-(4- methoxyphenyl)-5E- hexenyl]oxyphenoxy]-valeric acid (ONO-4057) or DPC-11870, • a 5-lipoxygenase inhibitor, such as zileuton, 6-[(3-fluoro-5-[4-methoxy-3,4,5,6- tetrahydro-2H-pyran-4-yl])phenoxy-methyl]-1-methyl-2-quinolone (ZD-2138), or 2,3,5- trimethyl-6-(3-pyridylmethyl),1 ,4-benzoquinone (CV-6504);
- a leukotriene B4 antagonist such as 1-(3-biphenyl-4-yl
- a sodium channel blocker such as lidocaine
- a 5-HT3 antagonist such as ondansetron
- the present invention extends to a product comprising an alpha-2-delta ligand, an PDE7 inhibitor and one or more other therapeutic agents, such as those listed above, for simultaneous, separate or sequential use in the curative, prophylactic treatment of pain, particularly inflammatory, neuropathic, visceral or nociceptive pain.
- the kit of the invention is particularly suitable for administering different dosage forms, for example, oral and parenteral, for administering the separate compositions at different dosage intervals, or for titrating the separate compositions against one another.
- the kit typically comprises directions for administration and may be provided with a so-called memory aid.
- the biological activity of the alpha-2-delta ligands of the invention may be measured in a radioligand binding assay using [3H]gabapentin and the ⁇ subunit derived from porcine brain tissue based on the method given in J. Biol. Chem., 1996, 271(10), 5768-5776). This assay is reproduced below.
- membranes are pelleted as above twice more by centrifugation with Buffer B, before a final re-suspension in approximately 3 volumes of storage buffer (1.25 mM EDTA/1.25 mM EGTA/25% Glycerol/12.5 mM Hepes/KOH, pH 7.4) to give a concentration of about 3 milligrams of protein per millilitre. Aliquots are stored at -8O 0 C until required.
- Binding of [ 3 H]gabapentin to pig cerebral cortex membranes is carried out at 22°C in 10 mM Hepes/KOH, pH 7.4 for 60 minutes.
- Non-specific binding (nsb) is defined as the binding obtained in the presence of 10 ⁇ M pregabalin.
- An assay volume of 250 ⁇ l is employed, comprising 200 ⁇ l of membranes, 25 ⁇ l test compound/buffer/nsb, 25 ⁇ l [ 3 H]gabapentin (final assay ' concentration ⁇ 10nM).
- Comparatve assays according to the methods of Gee et al. may be performed between different alpha-2-delta subunit subtypes to determine selectivity.
- the biological activity of PDE7 inhibitors may be measured using the following methods:
- Reactions were initiated with enzyme, incubated for 30-60min at 3O 0 C to give ⁇ 30% substrate turnover and terminated with 50 ⁇ l yttrium silicate SPA beads (containing 3mM of the respective unlabelled cyclic nucleotide for PDEs 9 and 11). Plates were re-sealed and shaken for 20min, after which the beads were allowed to settle for 30min in the dark and then counted on a TopCount plate reader (Packard, Meriden, CT) Radioactivity units were converted to % activity of an uninhibited control (100%), plotted against inhibitor concentration and inhibitor IC 50 values obtained using the 'Fit Curve' Microsoft Excel extension.
- TopCount plate reader Packard, Meriden, CT
- PDE7 ligands and inhibitors can be identified, for example by screening a compound library and by employing a variety of screening techniques against PDE7. Methods of identifying ligands and inhibitors of the enzyme are known and examples of these are presented below:
- the binding may take place in solution or on a solid surface.
- the test compound is previously labelled for detection.
- Any detectable group may be used for labelling, such as but not limited to, a luminescent, fluorescent, or radioactive isotope or group containing same, or a nonisotopic label, such as an enzyme or dye.
- the reaction is exposed to conditions and manipulations that remove excess or non-specifically bound test compound. Typically, this involves washing with an appropriate buffer. Finally, the presence a PDE7-test compound complex is detected.
- binding interactions can be detected by measuring changes in changes in fluoresence on ligand displacement from the enzyme.change in protein fluorescence or molecular tumbling rate or molecular sedimentation in solution of the enzyme in the presence of test compound.
- the binding assay is carried out with one or more components immobilized on a solid surface.
- the solid support could be, but is not restricted to, polycarbonate, polystyrene, polypropylene, polyethylene, glass, nitrocellulose, dextran, nylon, polyacrylamide and agarose.
- the support configuration can include beads, membranes, microparticles, the interior surface of a reaction vessel such as a microtitre plate, test tube or other reaction vessel.
- the immobilization of PDE7, or other component can be achieved through covalent or non-covalent attachments.
- the attachment may be indirect, i.e. through an attached antibody.
- PDE7 is tagged with an epitope, such as glutatione S-transferase (GST) so that the attachment to the solid surface can be mediated by a commercially available antibody such as anti-GST (Santa Cruz Biotechnology).
- GST glutatione S-transferase
- an affinity binding assay may be performed using a PDE7 which is immobilized to a solid support.
- the non-immobilized component of the binding reaction in this case the test compound, is labelled to enable detection.
- labelling methods are available and may be used, such as detection of luminescent, chromophoric, fluorescent, or radioactive isotopes or groups, or detection of nonisotopic labels, such as enzymes or dyes.
- the test compound is labelled with a fluorophore such as fluorescein isothiocyanate (FITC, available from Sigma Chemicals, St. Louis).
- FITC fluorescein isothiocyanate
- the labelled test compound is then allowed to contact with the solid support with the immobilised PDE7, under conditions that allow specific binding to occur. After the binding reaction has taken place, unbound and non-specifically bound test compounds are separated by means of washing the surface.
- Attachment of the binding partner to the solid phase can be accomplished in various ways known to those skilled in the art, including but not limited to chemical cross-linking, non-specific adhesion to a plastic surface, interaction with an antibody attached to the solid phase, interaction between a ligand attached to the binding partner (such as biotin) and a ligand-binding protein (such as avidin or streptavidin) attached to the solid phase, and the like.
- the label remaining on the solid surface may be detected by any detection method known in the art. For example, if the test compound is labelled with a fluorophore, a fluorimeter may be used to detect complexes. Alternatively, the binding reaction may be carried out in solution.
- the labelled component is allowed to interact with its binding partner(s) in solution. If the size differences between the labelled component and its binding partner(s) permit such a separation, the separation can be achieved by passing the products of the binding reaction through an ultrafilter whose pores allow passage of unbound labelled component but not of its binding partner(s) or of labelled component bound to its partner(s) to determine levels of bound vs free ligand. Separation can also be achieved using any reagent capable of capturing a binding partner of the labelled component from solution, such as an antibody against the binding partner, a ligand- binding protein which can interact with a ligand previously attached to the binding partner, and so on.
- Effects of a test compound on the catalytic activity of a PDE7 can be most easily determined by standard competitive binding experiments between PDE inhibitors and cAMP on enzyme activity for which known amounts of cAMP substrate and fixed amounts of enzyme are incubated together with various amounts of inhibitor substance for fixed periods of time, after which the reaction is stopped and the residual amount of unhydrolysed cAMP is measured.
- This may be done for any test sample by use of a scintillation proximity based assay (SPA) designed to measure the competition between cAMP in the test sample and a known amount of radiolabeled cAMP for binding to a cAMP-specific antibody attached to scintillant beads (Hancock, A. A., Vodenlich, A.
- SPA scintillation proximity based assay
- Identification of inhibitor activity can be judged using a standard SPA (scintillation proximity assay) assay with a PDE7 enzyme.
- the PDE7 enzyme can be for example recombinant mouse, human or yeast or can be derived from a whole cell lysate of Hut78 Tcell line as a surrogate for the use of a recombinant PDE7A according to the method of Pitts, WJ., et al Biorg. Med. Chem. Lett 14 2004 2955 - 2958.
- IC50 values of ⁇ 1 micromolar in the presence of inhibitor are indicative of good inhibition.
- a binding assay can be performed as follows:
- Phosphodiesterase activity of PDE7 can be measured using the phosphodiesterase Scintillation Proximity Assay (SPA) (Amersham) according to the manufacturer's protocol, for convenience the assays can be done in triplicate in 96 well format. Reaction times and enzyme dilution are optimised so that the lowest substrate concentration gives no more than 30% conversion of substrate to product to ensure linearity.
- the reactions can contain for example 25 ⁇ l of the appropriately diluted enzyme, 25 ⁇ l buffer (20 mM Tris with 5 mM MgCL2.6H2O, pH 7.4 plus 2 mg/ml BSA) and initiated by the addition of 50 ⁇ l of either cAMP or cGMP to give a total reaction volume of 100 ⁇ l.
- SPA phosphodiesterase Scintillation Proximity Assay
- [ 3 H]-CAMP (Amersham Cat. No. TRK304 B70, 24.Ci/mmol) or [ 3 H]-CGMP (Amersham Cat. No. TRK392 B37, 10.7 Ci/mmol) is mixed with the corresponding cold cyclic nucleotide to give a final concentration range of 1 ⁇ M-0.002 ⁇ M. This is achieved by performing doubling dilutions across a 96 well plate. Following a 40 min incubation at 3O 0 C, the plates are immediately centrifuged at 2000 rpm for 5 min and then counted on TopCount. Background levels for each cAMP concentration were determined using a Scintillation Counter.
- a binding assay can be performed as follows: Inhibition of PDE activity can be determined using Hut78 cell lysate (Hut78 is a Tcell line which expresses PDE7) and an SPA specific for cAMP (Amersham Pharmacia Biotech, Buckinghamshire, UK) according to the manufacturers instructions with minor modifications. Enzyme assays are performed at room temperature in the presence of 5OmM Tris-HCI, pH7.5, containing 8.3mM MgCI 2 , 1.7mM EGTA, and 0.5mg/mL BSA.
- Each assay is performed in a 100 ⁇ L reaction volume in 96 well microtitre plates containing the above buffer, 0.3 ⁇ L of Hut78 cell lysate treated with 2 ⁇ M Zardaverine to inhibit PDE3 and PDE4, 0.05 ⁇ Ci of [5 ,8- 3 H] Adenosine 3 ,5-cyclic phosphate as an ammonium salt for 20min.
- Inhibitors are included at a concentration range of 0.5-300 ⁇ M for each inhibitor is used and cAMP concentration is kept constant, the assay blank contains all reagents minus the enzyme. The reaction was terminated by the addition of 50 ⁇ L
- PDE SPA beads (1mg) water with 1OmM cold cAMP (Sigma, St. Louis MO). The reaction mix was allowed to settle for 20min before counting in a Top Count-NXT scintillation counter (Packard BioScience, Meriden, CT). For selectivity studies, the assay is essentially unchanged except that 3 H-cyclic GMP is used as the substrate for PDE1 , PDE5, and PDE6.
- the following PDEs/activators and enzyme sources are used: PDE1 , bovine (Sigma St. Louis), calmodulin; PDE2, rat kidney, cGMP; PDE3, human platelet; PDE4, rat kidney; PDE5, human platelet, and PDE6, bovine retina.
- the PDE7 inhibitors for use in the combination of the invention are preferably potent PDE7 inhibitors. These compounds have low IC 50 values for PDE7, typically at less than 10OnM, preferably less than 10 nM, more preferably less thani nM.
- the PDE7 inhibitors for use in the combination of the invention are preferably selective PDE7 inhibitors.
- the selectivity of PDE7 inhibitor is preferably at least 10 fold selective for PDE7 over other PDEs 1 preferably it should be at least 100 fold selective and further preferably at least 1000 fold selective.
- Selectivity in general represents the relative potency of a compound between two enzyme subtypes for the appropriate ligand or inhibitor for the enzyme of interest.
- a PDE7 ligand or inhibitor can be tested for selectivity for the PDE7 in comparison with another PDE such as for example PDE4.
- the capacity of each test compound to compete with binding of labelled-cAMP is measured at both the PDE7 and PDE4 enzymes, and an IC 50 value (in ⁇ M) is determined.
- Any of the above mentioned binding assay procedures can be used.
- test compounds are assayed for their ability to disrupt the binding and hydrolysis of cAMP by PDE7.
- Labelled cAMP may be mixed with PDE7 or a fragment or derivative thereof, and placed under conditions in which the interaction between them would normally occur, either with or without the addition of the test compound.
- the amount of labelled cAMP that binds and is hydrolysed by PDE7 or PDE4 may be compared to the amount bound and hydrolysed in the presence or absence of test compound, thus the level of inhibition of the process can be determined for any test compound addition at either PDE and compared.
- the potency of a PDE7 inhibitor (based on IC50 potency which can be defined as the concentration of inhibitor that gives a halving of the value of the functional activity of a enzyme in a functional assay as described below) is preferably at least 10OnM IC50 at the human enzyme (recombinant and/or native), more preferably less than 1OnM and further preferably less than 1 nM.
- IC50 is the molar concentration of an inhibitor that inhibits by 50% the maximal activity of the human PDE7 for example in response to cAMP.
- IC50 is the molar concentration of an inhibitor that displaces 50% of the specific binding of labelled cAMP or other appropriate ligand or the moalr concentration at which the test compound occupies half of the available PDE7 binding sites.
- Functional assay methods are known for identifying compounds that are inhibitors of PDE7.
- the methods generally include the steps comprising: a) contacting a PDE7-expressing cell with a test compound optionaly in the presence of cAMP or another PDE7 substrate ligand; and b) measuring the resultant level of a PDE7 activity, or the level of expression of PDE7 in the cell, such that if said level of measured activity or expression differs from that measured in the absence of the test compound, then a compound that modulates a PDE7-cAMP-mediated process is identified.
- the PDE7 activity measured can be the ability to interact with cAMP or by a change in cAMP / AMP levels in the cell or the response of the cell to cAMP for example by alterations in gene transcription or protein activity.
- Example protocols for functional assays are provided below.
- the key advantage of functional cell based assays is that they facilitate early and direct pharmacological characterization of compounds by high-throughput quantification and allow identification of compounds that act both at the binding site of the PDE or on a modulatory binding site on a PDE that is topographically distinct from the binding site.
- the most common systems of functional cell based assays are based on cyclic AMP detection and are reviewed in Williams, C, Nature Reviews Drug Discovery 3 2004 125-135.
- Cell-based assays in HTS provides the advantage of having the ability to identify inhibitor compounds and to obtain additional information about the mode of action of the compound.
- HTS-compatible accumulation assays for cAMP measurement follow a general principle, with changes in intracellular cAMP being detected by the competition between cellular cAMP and a labelled form of cAMP for binding to an anti-cAMP sequestering antibody or directly to the PDE. Protocols for these assays differ markedly and include: radiometric assays, fluorescence polarization cAMP assays, time-resolved fluorescence assays, assays which detect alterations in gene transcription or protein activity for example via initiation of phosphorylation events that regulate target enzymes and transcription factors, enzymatic assays, assays to determine binding to protein kinases within the cell.
- Homogeneous radiometric assays such as scintillation proximity assays (SPA.Amersham Biosciences) and Flashplate technology (NEN/Perkin Elmer) enable the direct detection of [125I]- labelled cAMP once it is inclose proximity to a solid scintillant surface [Amersham Life Science. High throughput screening forcAMP formation by scintillation proximityradioimmunoassay. Proximity News Issue No. 23. (1996).&. NEN Life Science Products. A novel adenylyl cyclaseactivation assay on FlashPlate (Flasplate File #1 , ApplicationNote). (NEN Life Science Products Inc., Boston, Massachusetts, 1998).18. Kariv, I. I. et al. High throughput quantitation of cAMPproduction mediated by activation of seven transmembranedomain receptors. J. Biomol. Screen. 4, 27-32 (1999)].
- Fluorescence polarization cAMP assays (available in kit form from companies such as Perkin Elmer and Amersham Biosciences) monitor the light emitted from a fluorescently tagged cAMP molecule following excitation with a polarized light source, the assays is based on a decrease in the extent of molecular rotation of a fluorescently labelled cAMP that occurs following binding to the larger anti-cAMP antibody.
- dyes such as Bodipy-TMR,MR121 ,Alexa, Cy3 and Cy5 have been used in FP binding assays.
- the HTRF (homogeneous time-resolved fluorescence) technology uses anti-cAMP antibodies labelled with europium cryptate and cAMP that is labelled with a modifiedallophyocyanin (see the CIS Bio International HTRF web site). In the absence of cellular cAMP, these two fluorescent molecules are in close proximity, FRET occurs and longlifetime fluorescence is emitted at two different wavelengths. When the two molecules are separated by competition with cellularcAMP, no FRET occurs and only emission from the europium is detected. This technique has been successfully applied to high-throughput screening with whole cells in miniaturized formats. [Claret E, Roux P, Ouled-Diaf J, Preaudat C, Drexler C, Grepin C, Seguin P.
- Reporter-gene assays for cAMP detection follow a general principle.where by receptor-mediated changes in intracellular cAMP con-centrations are detected via changes in the expression level of a particular gene (the reporter), the transcrip-tion of which is regulated by the transcription factorcAMP response-element binding protein (CREB) binding to upstream cAMP response elements (CREs).
- CREB transcription factorcAMP response-element binding protein
- CREs upstream cAMP response elements
- reporter genes have been used in in vitro and in vivo studies, including ⁇ -galactosidase, green fluorescent protein(GFP),luciferase and ⁇ -lactamase 28- 31.
- the reporter-gene method is compatible with screening for activity in live cells or enabling transfected cell popula-tions.
- Cell lines commonly used inreporter-gene assays are for example Chinese hamster ovary cells (CHO) and human embryonic kidney cells.
- the first of these ALPHAScreen (amplified luminescent proximity homogeneous assay; PackardBioscience/Perkin Elmer) — is a homogeneous assay format using chemiluminscent readout.
- the second system an enzyme complementation technology from DiscoveRx(Fremont,California) — uses a cAMP molecule tagged with an inactive ⁇ -galactosidase component and uses fluorescent or luminescent readout.
- the third system uses electrochemiluminescence detection and is a technology available from Meso ScaleDiscovery (Gaithersburg, Maryland). In this case, the cAMP, is tagged with a ruthenium derivative, which results in the production of light from the labelled cAMP (see Meso Scale Discovery web site).
- the analgesic effect of PDE7 inhibitors and alpha-2-delta ligands may be determined in vivo using animal models of selected pain conditions. Several models of pain conditions are known and specific procedures that can be used to determine the analgesic effect of PDE7 inhibitors and alpha-2-delta ligands are presented below.
- An alternative pain model is the streptozocin induced diabetic model of neuropathic pain in rats. This procedure involves administration of streptozocin (50mg/kg, i.p.) in a single dose to animals such as Charles River Sprague dawley rats (225 - 25Og) to induce diabetes. Animals are evaluated 2 weeks following administration using static and dynamic allodynia tests and if neuropathic pain is confirmed they are used to further evaluate compounds for their effect on neuropathic pain.
- the chronic constrictive injury (CCI) model of neuropathic pain in rats involves the tying of loose ligatures around the sciatic nerve Charles River male Sprague dawley rats (175-20Og) are placed in an anaesthetic chamber and anaesthetised with a 2% isofluorane O 2 mixture. The right hind thigh is shaved and swabbed with 1 % iodine. Animals are then transferred to a homeothermic blanket for the duration of the procedure and anaesthesia maintained during surgery via a nose cone. The skin is cut along the line of the thigh bone. The common sciatic nerve is exposed at the middle of the thigh by blunt dissection through biceps femoris.
- Proximal to the sciatic trifurcation about 7mm of nerve is freed by inserting forceps under the nerve and the nerve gently lifted out of the thigh. The forceps are gently opened and closed several times to aid clearance of the fascia from the nerve. Suture is pulled under the nerve using forceps and tied in a simple knot until slight resistance is felt and then double knotted. The procedure is repeated until 4 ligatures (4-0 silk) are tied loosely around the nerve with approx 1 mm spacing. The incision is closed in layers. Fourteen days following surgery, animals are assessed for static allodynia, dynamic allodynia or weight bearing deficit.
- 'Alternative animal models of neuropathic pain conditions include the Seltzer model, partial tight ligation of the sciatic nerve (Seltzer, Z. (1995). Sem. Neurosci, 8: pp. 34-39) or Chung's model, tight ligation of one of the two spinal nerves of the sciatic nerve. (Kim SH, Chung JM. Pain (1992); 50: pp. 355-63) or of the Chronic Constrictive Injury model (CCI) (Bennett GJ, Xie Y-K. Pain (1988); 33: pp. 87-107).
- neuropathic pain conditions may involve selection of an animal that naturally possesses a painful disease condition providing neuropathic pain and its symptoms such as HIV or Herpes or cancer or diabetes.
- the animal may be arranged to experience a pain condition by modification of the animal to possess a pain inducing disease condition such as arthritis or HIV or Herpes or cancer or diabetes.
- Animals may be modified to possess a pain condition due to a disease in a variety of ways for example by administration of Streptozocin to induce a diabetic neuropathy (Courteix.C, Eschalier.A., Lavarenne.J., Pain, 53 (1993) pp. 81-88.) or by administration of viral proteins to cause HIV related neuropathic pain (Herzberg U.
- Dynamic allodynia can be assessed by lightly stroking the plantar surface of the hind paw of the animal with a cotton bud. Care is taken to perform this procedure in fully habituated rats that are not active, to avoid recording general motor activity. At least two measurements are taken at each time point, the mean of which represents the paw withdrawal latency (PWL). If no reaction is exhibited within 15s the procedure is terminated and animals are assigned this withdrawal time. Thus, 15s effectively represents no withdrawal. A withdrawal response is often accompanied with repeated flinching or licking of the paw. Dynamic allodynia is considered to be present if animals responded to the cotton stimulus within 8s of commencing stroking.
- animals can be administered compounds for analgesic assessment by one of the following routes, oral administration, subcutaneous., intra-peritoneal., intra-venous or intra-thecal.
- the PWL is re-evaluated at some or all of the following time points, 30 min, 1h, 2h, 3h, 4h, 5h, 6h, 7h, 24h.
- Animals are assigned randomly to each compound group according to their baseline values.
- the mean and standard error mean are calculated for each compound group at each time point. Measures of dynamic allodynia are compared to their respective controls using a one way ANOVA followed by a Dunnett's t-test comparing vehicle to compound at each time point.
- the minimum number of animals per group is 6.
- Static allodynia can be evaluated by application of von Frey hairs (Stoelting, Wood Dale, Illinois, USA) in ascending order of force (0.6, 1 , 1.4, 2, 4, 6, 8, 10, 15 and 26 grams) to the plantar surface of hind paws. Animals are habituated to wire bottom test cages prior to the assessment of allodynia. Each von Frey hair is applied to the paw for a maximum of 6 seconds, or until a withdrawal response occurs. Once a withdrawal response to a von Frey hair is established, the paw is re-tested, starting with the filament below the one that produces a withdrawal, and subsequently with the remaining filaments in descending force sequence until no withdrawal occurs.
- paw withdrawal threshold PWT
- Static allodynia is defined as present if animals responded to a stimulus of, or less than, 4g, which is innocuous in normal rats. Following baseline evaluation, animals are administered compounds for analgesic assessment by one of the following routes, orally, subcutaneous, intra-peritoneal., intra-venous or intra-thecal.
- Thermal hyperalgesia is assessed using the rat plantar test (Ugo Basile, Italy) following a modified method of Hargreaves et al., (1988) Pain 32:77-88. Rats are habituated to the apparatus that consists of three individual perspex boxes on an elevated glass table. A mobile radiant heat source is located under the table and focused onto the hind paw and paw withdrawal latencies • (PWL) are recorded. There is an automatic cut off point of 22.5 s to prevent tissue damage. PWL are taken 2-3 times for both hind paws of each animal, the mean of which represented baselines for right and left hind paws. The apparatus is calibrated to give a PWL of approximately 10 s. PWL are reassessed 2h following administration of carrageenan.
- PWL's are reassessed hourly for up to 6 hours.
- PWL's of compound groups are compared to their respective controls using a one way ANOVA followed by a Dunnett's t-test. The minimum number of animals per group will be 6.
- Weight bearing deficit can be measured according to the method of Bove SE, et: al. Weight bearing as a measure of disease progression and efficacy of anti-inflammatory compounds in a model of monosodium iodoacetate-induced osteoarthritis. Osteoarthritis Cartilage. 2003
- Open field test can be carried out according to the method of Prut L and Belzung.C. The open field as a paradigm to measure the effects of compounds on anxiety-like behaviors: a review. Eur J Pharmacol. 2003;463::3-33.
- the locomotor test can be carried out according to the method of Salmi P and Ahlenius S- Sedative effects of the dopamine D1 enzyme agonist A 68930 on rat open-field behavior. Neuroreport. 2000 Apr 27; 11(6): 1269-72.
- Suitable alpha-2-delta ligand compounds of the present invention may be prepared as described herein below or in the aforementioned patent literature references, which are illustrated by the following non-limiting examples and intermediates.
- reaction mixture was stirred for a further 75 minutes at -78°C then allowed to warm to -20 0 C and quenched with saturated aqueous ammonium chloride (100ml). More tetrahydrofuran (100ml) was added and the organic layer was separated from the aqueous layer. The organic layer was dried (MgSO 4 ) and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel using an elution gradient of pentane to pentane:ether (30:70) to afford the title compound as a white solid (3.21 g).
- the title compound was prepared from (5S)-3-methyl-5-phenyl-5,6-dihydro-2H-1 ,4-oxazin-2-one (0.454g, 2.39mmol, see WO-A-02/051983) and 1-bromo-3-ethylpentane (see Bull. Soc. Chim.
- the title compound was prepared from (5S)-3-methyl-5-phenyl-5,6-dihydro-2H-1,4-oxazin-2-one (1g, 5.28mmol, see WO-A-02/051983) and (bromomethyl)cyclobutane according to the procedure outlined in Preparation 1. The total amount of compound synthesised was 0.445g.
- Dicyclohexyl carbodiimide (9.51g, 46.1mmol), N,N'-dimethyl-4-aminopyridine (1.13g, 9.2mmol) and triethylamine (6.4ml, 46.1mmol) were added to a solution of Meldrum's acid (6.64g, 46.1mmol) in dichloromethane (150ml).
- (4R)-4-Methylhexanoic acid (6g, 46.1mmol) was added and the reaction mixture was stirred overnight.
- the reaction mixture was filtered and the solid was washed with dichloromethane (2 x 100ml). The filtrate and washings were combined and evaporated under reduced pressure to give an orange oil.
- lsopropanol (0.63ml, 8.2mmol) was added to a mixture of a 1 molar solution of diethylaluminium cyanide in toluene (12.3ml) and tetrahydrofuran (5ml). The mixture was stirred at room temperature for 10 min and then cooled to -78 0 C. A solution of the compound of Preparation 7 (1.9g, 8.2mmol) in tetrahydrofuran (20ml) was added dropwise over 2 minutes. The reaction was then stirred at -78 0 C for 5 minutes and at room temperature for 90 minutes.
- the amide of Preparation 5 (8.6g, 0.026mol) was dissolved in 1 ,4-dioxane (100ml), water (100ml) was added and the reaction mixture heated at reflux for 116 hours. The mixture was filtered through arbocel ® and the filtrate evaporated in vacuo. The crude material was triturated with acetonitrile then recrystalised from water to yield the title compound as a white solid (2.9g, 0.0156mol, 60%).
- Zinc dust (6.54g, 0.1 mol) was suspended in water (30ml) and argon bubbled through the suspension for 15 minutes before the addition of copper (II) sulphate (780mg, 3.1mmol).
- the reaction mixture was stirred at room temperature, under argon for 30 minutes.
- the mixture was filtered under a stream of argon and the solid washed with water (100ml), acetone (100ml) and dried in vacuo for 4 hours.
- the resultant zinc/ copper couple was suspended in diethyl ether: 1 ,2- dimethoxyethane (70ml: 10ml) under argon and allyl benzyl ether (4.6ml, 30mmol) added.
Abstract
The instant invention relates to a combination of a PDE7 inhibitor and an alpha-2-delta ligand, and pharmaceutically acceptable salts thereof, pharmaceutical compositions thereof and their use in the treatment of pain, particularly inflammatory, neuropathic, visceral and nociceptive pain.
Description
SE OF COMBINATIONS OF PDE7 INHIBITORS AND ALPHA-2-DELTY LIGANDS FOR THE TREATMENT OF NEUROPATHIC PAIN
FIELD OF THE INVENTION
5 This invention relates to a combination of an alpha-2-delta ligand and a phosphodiesterase 7
(PDE7) inhibitor. The invention also relates to the use of a combination of an alpha-2-delta ligand and a phosphodiesterase 7 (PDE7) inhibitor for the manufacture of a medicament for the treatment of pain, particularly inflammatory, neuropathic, visceral and nociceptive pain. It also relates to a method for treating pain through the use of effective amounts of a combination of an 0 alpha-2-delta ligand and a phosphodiesterase 7 (PDE7) inhibitor.
BACKGROUND TO THE INVENTION
An alpha-2-delta receptor ligand is any molecule which binds to any sub-type of the human 5 calcium channel alpha-2-delta sub-unit. The calcium channel alpha-2-delta sub-unit comprises a number of receptor sub-types which have been described in the literature (e.g. N. S. Gee, J. P.
Brown, V. U. Dissanayake, J. Offord, R. Thurlow, and G. N. Woodruff, J-Biol-Chem 271
(10):5768-76, 1996, (type 1); Gong, J. Hang, W. Kohler, Z. Li, and T-Z. Su, J.Membr.Biol. 184
(1):35-43, 2001 , (types 2 and 3); E. Marais, N. Klugbauer, and F. Hofmann, MoI. Pharmacol. 59 0 (5): 1243-1248, 2001. (types 2 and 3); and N. Qin, S. Yagel, M. L. Momplaisir, E. E. Codd, and M.
R. D'Andrea. MoI. Pharmacol. 62 (3):485-496, 2002, (type 4)). Alpha-2-delta receptor ligands may also be known as GABA analogs.
Alpha-2-delta ligands have been described for the treatment of a number of indications, including 5 epilepsy and pain.
Phosphodiesterases (PDEs) are a family of enzymes which affect various cellular signaling processes by the process of hydrolyzing the second messenger molecules cAMP and cGMP to the corresponding inactive 5'-monophosphate nucleotides and thereby regulating their 0 physiological level. The secondary messengers cAMP and cGMP are responsible for the regulation of numerous intracellular processes. There are at least 11 families of PDE's, some (PDE3, 4, 7, 8) being specific for cAMP, and others for cGMP (PDE5, 6, and 9).
PDE7 is one member of the PDE family and comprises 2 subclass members PDE7 A and B. The mRNA of PDE7 is expressed in various tissues and cell types known to be important in the 5 pathogenesis of several diseases such as Tcell related disorders, in particular PDE7A and its splice variants are upregulated in activated Tcells, [L. Li, C. Yee and J.A. Beavo. Science 283 (1999), pp. 848-851], and in B-lymphocytes. [R. Lee, S. Wolda, E. Moon, J. Esselstyn, C. Hertel and A. Lerner. Ce//. Signal 14 (2002), pp. 277-284], autoimmune disease . [L. Li, C. Yee and J.A. Beavo. Science 283 (1999), pp. 848-851], and airway disease [Smith SJ, et al Am. J. Physiol. 0 Lung. Cell. MoI. Physiol 2003, 284, L279-L289]. Consequently it is expected that selective
inhibitors of PDE7 will have broad application as both immunosuppressants and treatment for respiratory conditions, for example chronic obstructive pulmonary disease and asthma. [N.A. Glavas, C. Ostenson, J. B. Schaefer, V. Vasta and J.A. Beavo. PNAS 98 (2001), pp. 6319-6324.]
Studies in rat have shown that PDE7A mRNA is found to be widely distributed in rat brain in both neuronal and non-neuronal cell populations. The highest levels are observed in the olfactory bulb, olfactory tubercle, hippocampus, cerebellum, medial habenula nucleus, pineal gland, area postrema, and choroid plexus. PDE7A mRNA is also widely detected in other non brain tissue. These results are consistent with PDE7A being involved in the regulation of cAMP signaling in many brain functions and suggests that PDE7A could have an effect on memory, depression, and emesis [X. Mirό , S. Perez-Torres , J. M. Palacios , P. Puigdomenech , G. Mengod1 Synapse
40:201-214, 2001] a link to Alzheimers disease is also suggested [S. Perez Torres R, Cortes M, Tolnay A., Probst J. M., Palaciosand G. Mengod, Experimental Neurology, 182,2, August 2003, Pages 322-334]. Additionally PDE7 has also been implicated in both fertility disorders [WO 0183772] and leukemia [Lee r, et. al. Cell Signalling 2002, 14, 277-284].
PDE7A has been isolated from yeast [Michaeli, T., et al J. Biol. Chem. 268 1993 12925 - 12932] , human [Han, P., Xiaoyan, Z., Tamar, M., Journ. Biol. Chem 272 26 1997 16152 - 16157], mouse [Bloom, T., Beavo, JA., proc. Natl. Acad. Sci. USA 93 1996 14188 - 14192] and mouse, and upregulation of PDE7A levels is seen in human T lymphocytes [lchimura, M., Kase, H. Biochem. Biophys. Res. Commun 193, 1993 985 - 990]. PDE7B, the second member of the PDE7 family, shares 70% amino acid homology with PDE7A in the C-terminal catalytic domain (N terminal domain is the regulatory domain containing the phosphorylation site which is conserved across the PDE family]. PDE7B is cAMP specific and has been cloned from mouse [accession number - AJ251858] and human [accession number - AJ251860] sources [C. Gardner, N. Robas, D. Cawkill and M. Fidock. Biochem. Biophys. Res. Commun. 272 (2000), pp. 186-192]. It has been shown to be expressed in a wide variety of tissues: the caudate nucleus, putamen and occipital lobe of the brain and peripherally in the heart, ovary and pituriary gland, kidney and liver small intestine and thymus, additionally in skeletal muscle, colon, bladder, uterus, prostate, stomach adrenal gland and thyroid gland. PDE7B has also been shown to discriminate among several general PDE inhibitors [J. M. Hetman, S. H. Soderling, N.A. Glavas and J.A. Beavo. PNAS 97 (2000), pp. 472-476], many standard PDE inhibitors, zaprinast, rolipram, milrinone do not specifically inhibit PDE7B.
The amino acid and nucleotide sequences that encode PDE7 of various species are known to those skilled in the art and can be found in GenBank under accession numbers AB057409, U77880, AB038040, L12052, AK035385, AY007702.
Inhibitors of PDE7 are known as is their use in the treatment of various PDE7 related diseases. The patent application EP-A-1348701 discloses pharmaceutical compositions comprising phosphodiesterase 7 inhibitors. EP-A-1348701 addresses the problem of providing a means of
alleviating visceral pain using such compositions. Visceral pain is known to be a particular and narrow class of nociceptive pain. It is known that there are 2 fundamental and different types of pain: nociceptive pain and neuropathic pain. It is further known that nociceptive and neuropathic pain are clinically and mechanistically distinct from each other.
The clinical characteristics of nociceptive pain are determined by excessive and/or prolonged activation of specific sensory neurones Aδ and C fibers. These may be activated by a mechanical, chemical, or thermal stimulus and become sensitised in chronic inflammatory conditions.
Neuropathic pain however is defined as pain which arises as a result of damage to or dysfunction of the nervous system. The clinical characteristics of neuropathic pain are therefore determined predominantly by the mechanisms, location, and severity of the neuropathology process itself and arises from neurons that have themselves been damaged. Neuropathic pain has important elements which are mediated via activitiy in sensory nerves which do not normally convey pain, the Aβ neurones.
Additionally, in contrast to nociceptive pain, neuropathic pain is notoriously difficult to treat; it responds very poorly or not at all to standard analgesic therapies which are effective in the treatment of nociceptive pain such as nonsteroidal anti-inflammatory drugs and acetaminophen; and responds less predictably and less robustly to opioids than do nociceptive pain conditions. Effective treatments for nociceptive pain are not expected to extend to neuropathic pain. In addition, medicaments such as gabapentin, pregabalin and amitriptyline, which provide some relief to neuropathic pain, are often not effective in the treatment of nociceptive pain. Thus for these reasons: difference in clinical characteristics, difference in mechanism and difference in amenability to treatment, neuropathic pain is clearly distinguished as different from nociceptive pain.
There is a medical need to develop new combinations of analgesic compounds which in combination either act synergistically to avert neuropathic pain or in combination treat different symptoms of neuropathic pain.
SUMMARY OF THE INVENTION
It has now been found that combination therapy with an alpha-2-delta ligand and a PDE7 inhibitor, when administered simultaneously, sequentially or separately, results in improvement in the treatment of pain, particularly inflammatory, neuropathic, visceral and nociceptive pain, most preferably neuropathic pain. Advantageously, the alpha-2-delta ligand and PDE7 inhibitor can interact in a synergistic manner to control pain. This synergy allows a reduction in the dose
reqυired of each compound, leading to a reduction in the side effects and enhancement of the clinical utility of the compounds.
Accordingly, the invention provides, as a first aspect, a combination of an alpha-2-delta ligand and a PDE7 inhibitor.
The invention further provides a combination of an alpha-2-delta ligand and a PDE7 inhibitor for the treatment of pain, particularly inflammatory, neuropathic, visceral and nociceptive pain, most preferably neuropathic pain.
The invention further provides the use of a combination of an alpha-2-delta ligand and a PDE7 inhibitor for the manufacture of a medicament for the treatment of pain, particularly inflammatory, neuropathic, visceral and nociceptive pain, most preferably neuropathic pain.
The invention further provides a method for treating pain, particularly inflammatory, neuropathic, visceral and nociceptive pain, most preferably neuropathic pain, through the use of effective amounts of a combination of an alpha-2-delta ligand and a PDE7 inhibitor.
The best known alpha-2-delta ligand, gabapentin (Neurontin®), 1-(aminomethyl)-cyclohexylacetic acid, was first described in the patent literature in the patent family comprising US Patent Number US4024175. The compound is approved for the treatment of epilepsy and neuropathic pain.
A second alpha-2-delta ligand, pregabalin, (S)-(+)-4-amino-3-(2-methylpropyl)butanoic acid, is described in European Patent Application Number EP-A-0641330 as an anti-convulsant treatment useful in the treatment of epilepsy and in EP0934061 for the treatment of pain.
Further suitable alpha-2-delta ligands are described in the following documents.
International Patent Application Publication No. WO-A-01/28978, describes a series of novel bicyclic amino acids, their pharmaceutically acceptable salts, and their prodrugs of formula:

I II III iv wherein n is an integer of from 1 to 4. Where there are stereocentres, each center may be independently R or S, preferred compounds being those of Formulae I-IV above in which n is an integer of from 2 to 4.
WO-A-02/85839 describes alpha-2-delta ligands of the following formulae:

(I) (H) (III) (IV)
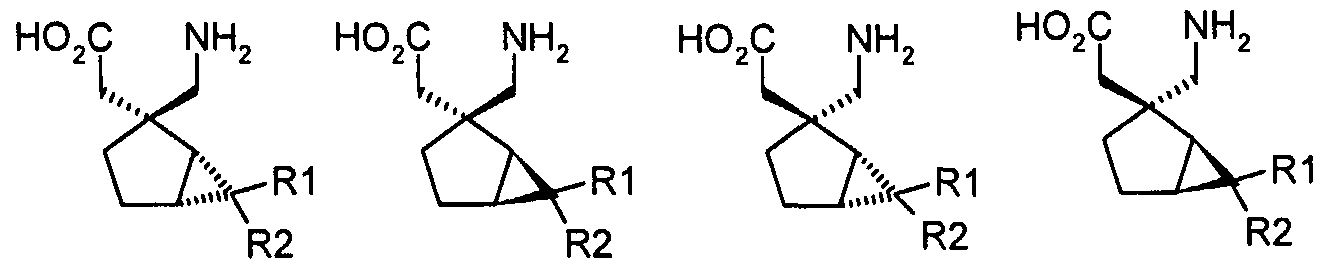
(V) (Vl) (VII) (VIIi)

(X) (Xl) (XIl)

XXH XXiIi xxiv xxv wherein R1 and R2 are each independently selected from H, straight or branched alkyl of 1-6 carbon atoms, cycloalkyl of from 3-6 carbon atoms, phenyl and benzyl, subject to the proviso that, except in the case of a tricyclooctane compound of formula (XVII), R1 and R2 are not simultaneously hydrogen; for use in the treatment of a number of indications, including pain.
lnternational Patent Application Publication No. WO-A-03/082807, describes compounds of the formula I, below:

Ri and R2, together with the carbon to which they are attached, form a three to six membered cycloalkyl ring;
R3 is (d-CeJalkyl, (C3-C6)cycloalkyl, (C3-C6)cycloalkyl-(C1-C3)alkyl, phenyl, phenyl-(CrC3)alkyl, pyridyl, pyridyl-(Ci-C3)alkyl, phenyl-N(H)-, or pyridyl-N(H)- , wherein each of the foregoing alkyl moieties can be optionally substituted with from one to five fluorine atoms, preferably with from zero to three fluorine atoms, and wherein said phenyl and said pyridyl and the phenyl and pyridyl moieties of said phenyl-(CrC3)alkyl and said pyridyl-(CrC3)alkyl, respectively, can be optionally substituted with from one to three substituents, preferably with from zero to two substituents, independently selected from chloro, fluoro, amino, nitro, cyano, (CrC3)alkylamino, (CrC3)alkyl optionally substituted with from one to three fluorine atoms and (C-ι-C3)alkoxy optionally substituted with from one to three fluorine atoms;
R4 is hydrogen or (CrC6)alkyl optionally substituted with from one to five fluorine atoms;
R5 is hydrogen or (CrC6)alkyl optionally substituted with from one to five fluorine atoms; and
R6 is hydrogen or (CrC6)alkyl; or a pharmaceutically acceptable salt thereof.
International Patent Application No. WO-A-2004/039367 describes compounds of the formula (I), below:

C1-C6 alkoxy, hydroxyCrC6 alkyl, C1-C6 alkoxyCrC6 alkyl, perfluoro C1-C6 alkyl, perfluorod-Ce alkoxy,
CrC6 alkylamino, di- C1-C6 alkylamino, aminoCi-C6 alkyl, C1-C6 alkylaminoCrCe alkyl, di-CrC6 alkylaminoCrCe alkyl,
CrC6acyl, CrC6acyloxy, CrC6acyloxyCrC6 alkyl, C1-C6 acylamino, C1-C6 alkylthio, C1-C6 alkylthiocarbonyl, C1-C6 alkylthioxo, C1-C6 alkoxycarbonyl, C1-C6 alkylsulfonyl, C1-C6 alkylsulfonylamino, aminosulfonyl, C1-C6 alkylaminosulfonyl, di-CrC6 alkylaminosulfonyl,
3-8 membered cycloalkyl, 4-8 membered heterocycloalkyl, phenyl and monocyclic heteroaryl; or a pharmaceutically acceptable salt thereof.
US Provisional Patent Application Number 60/676025 (unpublished at the filing date of the present application) describes compounds of the formula (I) below:

or a pharmaceutically acceptable salt or solvate thereof, wherein R1 is C1-C6 alkyl, said C1-C6 alkyl being optionally substituted by one or more halo, -R5, -OR5 or -
SR5 groups;
R2 is methyl, optionally substituted by one or more fluoro groups;
R3 and R4 are each independently H or a group which is converted to H following administration of the compound to a mammal; R5 is C1-C6 alkyl, C1-C6 haloalkyl, C3-C8 cycloalkyl or aryl; aryl is phenyl or naphthyl, each optionally substituted by one or more substituents selected from halo, -NR6R6, C1-C6 alkyl, C3-C8 cycloalkyl, C1-C6 alkoxy and cyano; and
R6 is H, C1-C6 alkyl or C3-C8 cycloalkyl.
US Provisional Patent Application Number 60/733591 (unpublished at the filing date of the present application) desribes the comound (2S)-2-aminomethyl-5-ethyl-heptanoic acid, or a pharmaceutically acceptable salt or solvate thereof.
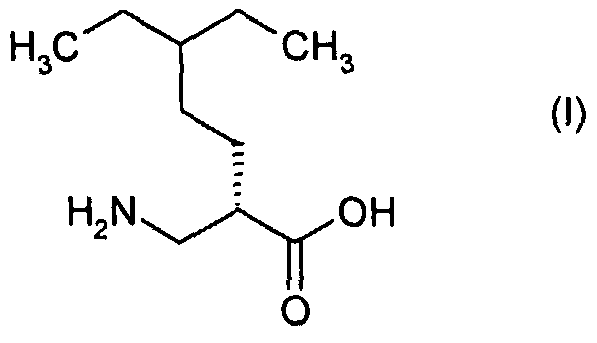
(2S)-2-Aminomethyl-5-ethyl-heptanoic acid
Examples of alpha-2-delta ligands for use in the present invention are those compounds, or pharmaceutically acceptable salts thereof, generally or specifically disclosed in US4024175, particularly gabapentin, EP641330, particularly pregabalin, US5563175, WO-A-97/33858, WO-A- 97/33859, WO-A-99/31057, WO-A-99/31074, WO-A-97/29101, WO-A-02/085839, particularly [(1R,5R,6S)-6-(aminomethyl)bicyclo[3.2.0]hept-6-yl]acetic acid, WO-A-99/31075, particularly 3-(1- aminomethyl-cyclohexylmethyl)-4H-[1 ,2,4]oxadiazol-5-one and C-[1-(1 H-tetrazol-5-ylmethyl)- cycloheptyl]-methylamine, WO-A-99/21824, particularly (3S,4S)-(1-aminomethyl-3,4-dimethyl- cyclopentyl)-acetic acid, WO-A-01/90052, WO-A-01 /28978, particularly (1α,3α,5α)(3-amino- methyl-bicyclo[3.2.0]hept-3-yl)-acetic acid , EP0641330, WO-A-98/17627, WO-A-00/76958, particularly (3S,5R)-3-aminomethyl-5-methyl-octanoic acid, WO-A-03/082807, particularly (3S,5R)-3-amino-5-methyl-heptanoic acid, (3S,5R)-3-amino-5-methyl-nonanoic acid and (3S.5R)- 3-amino-5-methyl-octanoic acid, WO-A-2004/039367, particularly (2S,4S)-4-(3-fluoro- phenoxymethyl)-pyrrolidine-2-carboxylic acid, (2S,4S)-4-(2,3-difluoro-benzyl)-pyrrolidine-2- carboxylic acid, (2S,4S)-4-(3-chlorophenoxy)proline and (2S,4S)-4-(3-fluorobenzyl)proline,
EP1178034, EP1201240, WO-A-99/31074, WO-Aτ03/000642, WO-A-02/22568, WO-A-02/30871 , WO-A-02/30881 WO-A-02/100392, WO-A-02/100347, WO-A-02/42414, WO-A-02/32736 and WO-A-02/28881, US Provisional Patent Application Number 60/676025 (unpublished at the filing date of the present application) , particularly (2S)-2-amino-4-ethyl-2-methylhexanoic acid and US Provisional Patent Application Number 60/733591 (unpublished at the filing date of the present application), particularly (2S)-2-aminomethyl-5-ethyl-heptanoic acid, all of which are incorporated herein by reference.
Preferred alpha-2-delta ligands for use in the combination of the present invention include: gabapentin, pregabalin, [(1R,5R,6S)-6-(aminomethyl)bicyclo[3.2.0]hept-6-yl]acetic acid, 3-(1- aminomethyl-cyclohexylmethyl)-4H-[1 ,2,4]oxadiazol-5-one, C-[1-(1H-tetrazol-5-ylmethyl)- cycloheptyl]-methylamine, (3S,4S)-(1-aminomethyl-3,4-dimethyl-cyclopentyl)-acetic acid, (1α,3α,5α)(3-amino-methyl-bicyclo[3.2.0]hept-3-yl)-acetic acid, (3S,5R)-3-aminomethyl-5-methyl- octanoic acid, (3S,5R)-3-amino-5-methyl-heptanoic acid, (3S,5R)-3-amino-5-methyl-nonanoic acid, (3S,5R)-3-amino-5-methyl-octanoic acid, (2S,4S)-4-(3-chlorophenoxy)proline, (2S,4S)-4-(3- fluorobenzyl)proline, (2S)-2-amino-4-ethyl-2-methylhexanoic acid, and (2S)-2-aminomethyl-5- ethyl-heptanoic acid or pharmaceutically acceptable salts thereof.
Further preferred alpha-2-delta ligands for use in the combination of the present invention are (3S,5R)-3-amino-5-methyloctanoic acid, (3S,5R)-3-amino-5-methylnonanoic acid, (3R,4R,5R)-3- amino-4,5-dimethylheptanoic acid and (3R,4R,5R)-3-amino-4,5-dimethyloctanoic acid, and the pharmaceutically acceptable salts thereof.
Particularly preferred alpha-2-delta ligands for use in the combination of the present invention are selected from gabapentin, pregabalin, (1α,3α,5α)(3-amino-methyl-bicyclo[3.2.0]hept-3-yl)-acetic
acid, (2S,4S)-4-(3-chlorophenoxy)proline, (2S,4S)-4-(3-fluorobenzyl)proline, (2S)-2-amino-4- ethyl-2-methylhexanoic acid, and (2S)-2-aminomethyl-5-ethyl-heptanoic acid, or pharmaceutically acceptable salts thereof.
Suitable PDE7 inhbiitors are described in the following documents.
Suitable PDE7 inhibitors for use in the invention include those compounds generally or specifically described in the patent applications WO-A-2002/074754 (Warner Lambert) and WO-A- 2002/076953 (Warner Lambert), which describe Spiroquinazolinones which are PDE7 inhibitors (also described in the publication, Bioorganic and Medicinal Chemistry Letters 2004, 14, 4623- 4626. Bioorganic and Medicinal Chemistry Letters 2004, 14, 4627-4631).
PDE7 inhibitors according to WO-A-2002/074754 and WO-A-2002/076953 have the following formula (I), (II) or (III),

(l) (H) ("I)
in which, a) X1, X2, X3 and X4 are the same or different and are selected from:
N, provided that not more than two of the groups Xi, X2, X3 and X4 simultaneously represent a nitrogen atom, or,
C-R1, in which R1 is selected from: - Q1 , or lower alkyl, lower alkenyl or lower alkynyl, these groups being unsubstituted or substituted with one or several groups Q2; - the group X5-R5 in which,
X5 is selected from : - a single bond, lower alkylene, lower alkenylene or lower alkynylene, optionally interrupted with 1 or 2 heteroatoms chosen from O, S, S(=O), SO2 or N, , the carbon atoms of these groups being unsubstituted or substituted with one or several groups, identical or different, selected from SR6, OR6, NR6R7, =0, =S or =N-R6 in which R6 and R7 are the same or different and are selected from hydrogen or lower alkyl, and,
- R5 is selected from aryl, heteroaryl, cycloalkyl optionally interrupted with C(=O) or with 1 , 2, or 3 heteroatoms chosen from O, S, S(=O), SO2 or N1 cycloalkenyl optionally interrupted with C(=O) or with 1 , 2, or 3 heteroatoms chosen from O, S, S(=0), SO2 or N, or a bicyclic group, these groups being unsubstituted or substituted with one or several groups selected from Q3, heteroaryl or lower alkyl optionally substituted with Q3; in which Q1 , Q2, Q3 are the same or different and are selected from
- hydrogen, halogen, CN1 NO2, SO3H1 P(=O)(OH)2
- OR2, 0C(=0)R2, C(=O)OR2, SR2, S(=O)R2, C(=O)-NH-SO2-CH3, NR3R4, Q-R2, Q-NR3R4, NR2-Q-NR3R4 or NR3-Q-R2 in which Q is selected from C(=NR), C(=0), C(=S) or SO2, R is selected from hydrogen, CN, SO2NH2 or lower alkyl and R2, R3 and R4 are the same or different and are selected from: hydrogen, lower alkyl optionally interrupted with C(=0), Q4-aryl, Q4-heteroaryl, Q4-cycloalkyl optionally interrupted with C(=0) or with 1 or 2 heteroatoms chosen from O, S, S(=0), SO2 or
N, or Q4-cycloalkenyl optionally interrupted with C(=0) or with 1 or 2 heteroatoms chosen from O, S, S(=0), SO2 or N, in which
Q4 is selected from (CH2)n, lower alkyl interrupted with one heteroatom selected from O, S or N, lower alkenyl or lower alkynyl, these groups being optionally substituted with lower alkyl, OR' or NR1R" in which R' and R" are the same or different and are selected from hydrogen or lower alkyl; n is an integer selected from O, 1 , 2, 3 or 4; these groups being unsubstituted or substituted with one or several groups selected from lower alkyl, halogen, CN1 CH3, SO3H, SO2CH3, C(=O)-NH-SO2-CH3, CF3, OR6, COOR6, C(=0)R6, NR6R7, NR6C(=O)R7, C(=O)NR6R7 or SO2NR6R7, in which R6 and R7 are the same or different and are selected from hydrogen or lower alkyl optionally substituted with one or two groups selected from OR, COOR or NRR8 in which R and R8 are hydrogen or lower alkyl, and,
R6 and R7, and/or, R3 and R4, together with the nitrogen atom to which they are linked, can form a 4- to 8-membered heterocyclic ring, which may contain one or two heteroatoms selected from O, S, S(=0), SO2, or N, and which may be substituted with,
(CH2)n-Q5, in which n is an integer selected from O, 1 , 2 and 3, and Q5 is a 4- to 8- membered heterocyclic ring which may contain one or two heteroatoms selected from O, S or N and which may be substituted with a lower alkyl, or, a lower alkyl optionally substituted with OR1, NR'R", C(=O)NR'R" or COOR' in which R' and R" are the same or different and are selected from,
- H, or, lower alkyl optionally substituted with OR or COOR in which R is hydrogen or lower alkyl and,
R1 and R" together with the nitrogen atom to which they are linked, can form a 4- to 8-membered heterocyclic ring, which may contain one or two heteroatoms selected from O, S or N; or,
- when X1 and X2 both represent C-R1, the 2 substituents R1 may form together with the carbon atoms to which they are attached, a 5-membered heterocyclic ring comprising a nitrogen atom and optionally a second heteroatom selected from O, S or N;
b) X is O or NR9, in which R9 is selected from,
- hydrogen, CN, OH, NH2, lower alkyl, lower alkenyl or lower alkynyl, these groups being unsubstituted or substituted with cycloalkyl optionally interrupted with 1 or 2 heteroatoms chosen from O, S, S(=O), SO2 or N, cycloalkenyl optionally interrupted with 1 or 2 heteroatoms chosen from O, S, S(=0), SO2 or N, aryl, heteroaryl, OR10, COOR10 or NR10R11 in which R10 and R11 are the same or different and are selected from hydrogen or lower alkyl;
c) Y is selected from O, S or N-R12, in which R12 is selected from:
- hydrogen, CN, OH, NH2, - lower alkyl, lower alkenyl or lower alkynyl, these groups being unsubstituted or substituted with, cycloalkyl optionally interrupted with 1 or 2 heteroatoms chosen from O, S, S(=0), SO2 or N, cycloalkenyl optionally interrupted with 1 or 2 heteroatoms chosen from O, S, S(=0), SO2 or N, aryl, heteroaryl, OR10, COOR10 or NR10R11 in which R10 and R11 are the same or different and are selected from hydrogen or lower alkyl;
d) Z is chosen from CH-NO2, O, S or NR13 in which R13 is selected from: hydrogen, CN, OH1 NH2, aryl, heteroaryl, cycloalkyl optionally interrupted with one or several heteroatoms chosen from O, S, S(=0), SO2 or N, cycloalkenyl optionally interrupted with one or several heteroatoms chosen from O, S, S(=0), SO2 or N, C(=O)R14, C(=O)NR14R15, OR14, or, lower alkyl, unsubstituted or substituted with one or several groups which are the same or different and which are selected OR14, COOR10 or NR14R15;
R14 and R15 being independently selected from hydrogen or lower alkyl, or, R14 and R15, together with the nitrogen atom to which they are linked, can form a 4- to 8-membered heterocyclic ring which may contain one or two heteroatoms chosen from O, S or N, and which may be substituted with a lower alkyl, or,
- when Y is N-R12 and Z is N-R13, may form together a -CH=N- group or a -C=C- group,
- when X is N-R9 and Z is N-R13, R9 and R13 may form together a -CH=N- group or a -C=C- group; e) Z1 is chosen from H, CH3 or NR16R17 in which R16 and R17 are the same or different and are selected from: hydrogen, CN, aryl, heteroaryl, cycloalkyl optionally interrupted with one or several heteroatoms chosen from O, S, S(=0), SO2 or N, cycloalkenyl optionally interrupted with one or several heteroatoms chosen from O, S1 S(=0), SO2 or N, C(=O)R14, C(=O)NR14R15, OR14, or,
- lower alkyl unsubstituted or substituted with one or several groups selected from OR14,
COOR14 or NR14R15,
R14 and R15 being chosen from hydrogen or lower alkyl, and,
R14 and R15, and/or, R16 and R17, together with the nitrogen atom to which they are linked, can form a 4- to 8-membered heterocyclic ring which may contain one or two heteroatoms chosen from O, S or N, and which may be substituted with a lower alkyl;
f) A is a cycle chosen from:

, or, in which,
A1, A2, A4, A5 and A6 are the same or different and are selected from O, S, C, C(=0), SO, SO2 or N-R18 in which R18 is selected from: hydrogen, aryl, heteroaryl, cycloalkyl optionally interrupted with one or several heteroatoms chosen from O, S, S(=0), SO2 or N, cycloalkenyl optionally interrupted with one or several heteroatoms chosen from O, S, S(=0), SO2 or N, lower alkyl unsubstituted or substituted with aryl, heteroaryl, cycloalkyl optionally interrupted with one or several heteroatoms chosen from O, S, S(=0), SO2 or N, cycloalkenyl optionally interrupted with one or several heteroatoms chosen from O, S, S(=0), SO2 or N, CN1 NR19R20, C(=O)NR19R20, OR19, C(=O)R19 or C(=O)OR19 in which R19 and R20 are identical or different and are selected from hydrogen or lower alkyl;
- A3 is selected from O, S, C, C(=0), SO or SO2, or N-R18 when A1 and/or A2 are C(=0) or when Y is O or S, wherein R18 is as defined above;
* represents the carbon atom which is shared between the cycle A and the backbone cycle containing X and/or Y; - each carbon atom of the cycle A is unsubstituted or substituted with 1 or 2 groups, identical or different, selected from lower alkyl optionally substituted with OR21, NR21R22, COOR21 or CONR21R22, lower haloalkyl, CN, F, =0, SO2NR19R20, OR19, SR19, C(=0)0R19, C(=O)NR19R20 or NR19R20 in which R19 and R20 are identical or different and are selected from hydrogen or lower alkyl optionally substituted with OR21, NR21R22, COOR21 or CONR21R22 in which R21 and R22 are identical or different and are selected from hydrogen or lower alkyl, and,
R19 and R20, and/or, R21 and R22, together with the nitrogen atom to which they are linked, can form a 4- to 8-membered heterocyclic ring;
2 atoms of the cycle A, which are not adjacent, may be linked by a 2, 3 or 4 carbon atom chain which may be interrupted with 1 heteroatom chosen from O, S or N; provided that:
- not more than two of the groups A1, A2, A3, A4, A5 and A6 simultaneously represent a heteroatom; the cycle A does not contain more than 2 carbon atoms in an sp2 hybridization state;
- when X is O, X2 is not C-R1 in which R1 is - a thienyl substituted with CN or with CN and CH3, a phenyl substituted with CN1 Cl, NO2 or CN and F,
- Br
- F; or their tautomeric forms, their racemic forms or their isomers and their pharmaceutically acceptable derivatives.
The following definitions apply to the formulae described in WO-A-2002/074754 and WO-A-
2002/076953:
Halogen includes fluoro, chloro, bromo, and iodo. Preferred halogens are F and Cl.
Lower alkyl includes straight and branched carbon chains having from 1 to 6 carbon atoms. Examples of such alkyl groups include methyl, ethyl, isopropyl, tert-butyl and the like.
Lower alkenyl includes straight and branched hydrocarbon radicals having from 2 to 6 carbon atoms and at least one double bond. Examples of such alkenyl groups are ethenyl, 3-buten-1-yl,
2-ethenylbutyl, 3-hexen-1-yl, and the like.
Lower alkynyl includes straight and branched hydrocarbon radicals having from 2 to 6 carbon atoms and at least one triple bond. Examples of such alkynyl groups are ethynyl, 3-butyn-1-yl, propynyl, 2-butyn-1-yl, 3-pentyn-1-yl, and the like.
Lower haloalkyl includes a lower alkyl as defined above, substituted with one or several halogens.
A preferred haloalkyl is trifluoromethyl.
Aryl is understood to refer to an aromatic carbocycle containing between 6 and 10, preferably 6, carbon atoms. A preferred aryl group is phenyl.
Heteroaryl includes aromatic cycles which have from 5 to 10 ring atoms, from 1 to 4 of which are independently selected from the group consisting of O, S, and N. Preferred heteroaryl groups have 1 , 2, 3 or 4 heteroatoms in a 5- or 6-membered aromatic ring. Examples of such groups are tetrazole, pyridyl, thienyl and the like. Preferred cycloalkyl contain from 3 to 8 carbon atoms. Examples of such groups are cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and cyclooctyl.
The term "interrupted" means that in a backbone chain, a carbon atom is replaced by an heteroatom or a group as defined herein. For example, in "cycloalkyl or cycloalkenyl optionally interrupted with C(=O) or with 1 heteroatom chosen from O, S, S(=O), SO2 or N", the term "interrupted" means that C(=O) or a heteroatom can replace a carbon atom of the ring. Examples of such groups are morpholine or piperazine.
Cycloalkenyl includes 3- to 10- membered cycloalkyl containing at least one double bond.
Heterocyclic ring include heteroaryl as defined above and cycloalkyl or cycloalkenyl, as defined above, interrupted with 1 , 2 or 3 heteroatoms chosen from O, S, S(=O), SO2, or N.
Bicyclic substituents refer to two cycles, which are the same or different and which are chosen from aryl, heterocyclic ring, cycloalkyl or cycloalkenyl, fused together to form said bicyclic substituents. A preferred bicyclic substituent is indolyl.
WO-A-2002/028847 descibes compounds of the following formula (I) as PDE7 inhibitors:

in which
- Y is O or S;
- R1 is: C4-C10 alkyl,
C2-C10 alkenyl, C2-C10 alkynyl, cycloalkyl, cycloalkenyl, heterocycle, aryl, or a bicyclic group; each optionally substituted with one or several groups X1-R4, identical or different, in which: - X1 is: a single bond, lower alkylene, C2-C6 alkenylene, cycloalkylene, arylene or divalent heterocycle, and, R4 is:
1) H, =0, NO2, CN, halogen, lower haloalkyl, lower alkyl, carboxylic acid bioisostere,
2) COOR5, C(=O)R5, C(=S)R5, SO2R5, SOR5, SO3R5, SR5, OR5,
3) C(=O)NR7R8, C(=S)NR7R8, C(=CH-NO2)NR7R8, C(=N-CN)NR7R8, C(=N- SO2NH2)NR7R8, C(=NR7)NHR8, C(=NR7)R8, C(=NR9)NHR8, C(=NR9)R8, SO2NR7R8 or NR7R8 in which R7 and R8 are the same or different and are selected from OH, R5, R6, C(=O)NR5R6, C(=O)R5, SO2R5, C(=NR9)NHR10,
C(=NR9)R10, C(=CH-NO2)NR9R10, C(=N-SO2NH2)NR9R10, C(=N-CN)NR9R10 or C(=S)NR9R10;
- R2 is: lower alkyl,
C2-C10 alkenyl, C4-Ci0 alkynyl, cycloalkyl, cycloalkenyl, heterocycle, aryl; each optionally substituted with one or several groups which are the same or different and which are selected from:
1) H, carboxylic acid bioisostere, lower haloalkyl, halogen, 2) COOR5, OR5, SO2R5,
3) SO2NRi1R12, Cl=O)NR11R12 or NR11R12 in which R11 and R12 are the same or different and are selected from OH, R5, R6, C(=O)NR5R6, C(=O)R5, SO2R5, C(=S)NR9R10, C(=CH-NO2)NR9R10, C(=N-CN)NR9R10, C(=N-SO2NH2)NR9R10, C(=NR9)NHR10 or C(=NR9)R10;
- R3 is X2-R'3 wherein:
X2 is a single bond or, a group selected from C1-C4 alkylene, C2-C6 alkenylene, C2-C6 alkynylene, each optionally substituted with one or several groups which are the same or different and which are selected from:
1) H, C1-C3 alkyl, C3-C4 cycloalkyl, aryl, heterocycle, =0, CN,
2) OR5, =NR5 or,
3) NR13Ri4 in which R13 and R14 are the same or different and are selected from R5, R6, C(=O)NR5R6, C(=0)R5, SO2R5, C(=S)NR9R10, C(=CH-NO2)NR9R10, C(=NR9)NHR10 or C(=NR9)R10;
- R'3 is: cycloalkyl, cycloalkenyl, aryl, heterocycle, or a polycyclic group; each optionally substituted with one or several groups X3-Ri7, identical or different, in which: - X3 is: a single bond, lower alkylene, C2-C6 alkenylene, C2-C6 alkynylene, cycloalkylene, arylene, divalent heterocycle or a divalent polycyclic group, and,
Ri7 is:
1) H, =0, NO2, CN, lower haloalkyl, halogen, cycloalkyl, 2) COOR5, C(=O)R5, C(=S)R5, SO2R5, SOR5, SO3R5, SR5, OR5,
3) C(=O)NR15R16, C(=S)NR15Ri6, C(=N-CN)NR15Ri6, C(=N-Sθ2NH2)NR15R16, C(=CH- NO2)NR15R16, SO2NR15R16, C(=NR15)NHR16, C(=NR15)Ri6, C(=NR9)NHR16, C(=NR9)R16 or
NR15R16 in which R15 and R16 are the same or different and are selected from OH, R5, R6, C(=O)NR5R6, C(=0)R5, SO2R5, C(=S)NR9R10, C(=CH-NO2)NR9R10, C(=N-CN)NR9R10, C(=N-SO2NH2)NR9R10, C(=NR9)NHR10 or C(=NR9)R10
4) heterocycle optionally substituted with one or several groups R5;
R5 and R6 are the same or different and are selected from : - H1 - - lower alkyl, C2-C6 alkenyl, C2-C6 alkynyl;
- X4-cycloalkyl, X^cycloalkenyl, X4-aryl, X4-heterocycle or X4-polycyclic group, in which X4 is a single bond, lower alkylene or C2-C6 alkenylene; each optionally substituted with one or several groups which are the same or different and which are selected from: - halogen, =0, COOR20, CN, OR20, lower alkyl optionally substituted with OR20, O- lower alkyl optionally sustituted with OR20, C(=O)-lower alkyl, lower haloalkyl,
X5-N-"1*18 p
19 in which X5 is a single bond or lower alkylene and R18, Ri9 and R20 are the same or different and are selected from H or lower alkyl; - X6-heterocycle, X6-aryl, X6-cycloalkyl, X6-cycloalkenyl, X6-polycyclic group in which X6 is selected from a single bond or lower alkylene, these groups being optionally substituted with one or several groups, identical or different, selected from halogens, COOR21, OR21, or (CH2JnNR21R22 in which n is O, 1 or 2 and R21 and R22 are the same or different and are selected from H or lower alkyl; R9 is selected from H, CN, OH, lower alkyl, O-lower alkyl, aryl, heterocycle, SO2NH2 or
Ri9 in which X5 is a single bond or lower alkylene and R18 and R19 are the same or different and are selected from H or lower alkyl;
Rio is selected from hydrogen, lower alkyl, cyclopropyl or heterocycle; or a pharmaceutically acceptable derivative thereof, with the proviso that,
- when R1 is phenyl, it bears at least one substituent other than H,
- when X2 is a single bond and both R1 and R'3 are phenyl, each of R1 and R'3 bear at least one substituent other than H,
- when X2 is a single bond and R'3 is phenyl, R'3 is not substituted by an ester or a carboxylic acid in the ortho position,
- the atom of R3 which is linked to the thiadiazole group is a carbon atom, with the exclusion of the following compounds,
1-Phenyl-1-[4-phenyl-5-(5-trifluoromethyl-2H-[1,2,4]triazol-3-ylimino)-4,5-dihydro-
[1 ,3,4]thiadiazol-2-yl]-methanone,
1-[4-Phenyl-5-(5-trifluoromethyl-2H-[1,2,4]triazol-3-ylimino)-4,5-dihydro-[1 ,3,4]thiadiazol-2-yl]-1- thiophen-2-yl-methanone, 1-Phenyl-1-(4-phenyl-5-p-tolylimino-4,5-dihydro-[1 ,3,4]thiadiazol-2-yl)-methanone,
Cyclohexyl-[3-(2,4,6-trichloro-phenyl)-5-(2,3,3-trimethyl-cyclopent-1-enylmethyl)-3H-
[1 ,3,4]thiadiazol-2-ylidene]-amine,
2-(3,5-Diphenyl-3H-[1 ,3,4]thiadiazol-2-ylideneamino)-1 ,4-diphenyl-but-2-ene-1 ,4-dione,
2-[3-Phenyl-5-( 1 -phenyl-methanoyl)-3/-/-[1 , 3,4]thiadiazol-2-ylideneamiπo]-but-2-enedioic acid dimethyl ester,
2-[5-(1-Phenyl-methanoyl)-3-p-tolyl-3H-[1,3,4]thiadiazol-2-ylideneamino]-but-2-enedioic acid dimethyl ester, and,
2-[3-(4-Chloro-phenyl)-5-(1-phenyl-methanoyl)-3H-[1 ,3,4]thiadiazol-2-ylideneamino]-but-2- enedioic acid dimethyl ester.
The following definitions apply to the compounds of the formula (I) described in WO-A-
2002/028847:
- aryl is understood to refer to an unsaturated carbocycle, exclusively comprising carbon atoms in the cyclic structure, the number of which is between 5 and 10, including phenyl, naphthyl or tetrahydronaphthyl;
- heterocycle is understood to refer to a non-saturated or saturated monocycle containing between 1 and 7 carbon atoms in the cyclic structure and at least one heteroatom in the cyclic structure, such as nitrogen, oxygen, or sulfur, preferably from 1 to 4 heteroatoms, identical or different, selected from nitrogen, sulfur and oxygen atoms. Suitable heterocycles include morpholinyl, piperazinyl, pyrrolidinyl, piperidinyl, pyrimidinyl, 2- and 3-furanyl, 2- and 3-thienyl, 2- pyridyl, 2- and 3-pyranyl, hydroxypyridyl, pyrazolyl, isoxazolyl, tetrazole, imidazole, triazole and the like;
- polycyclic groups include at least two cycles, identical or different, selected from aryl, heterocycle, cycloalkyl, cycloalkenyl groups fused together to form said polycyclic group such as 2- and 3-benzothienyl, 2- and 3-benzofuranyl, 2-indolyl, 2- and 3-quinolinyl, acridinyl, quinazolinyl, indolyl benzo[1 ,3]dioxolyl and 9-thioxantanyl; Preferred polycyclic groups include 2 or 3 cycles as defined above. More preferred polycyclic groups include 2 cycles (bicyclic substituents) as defined above.
- bicyclic groups refer to two cycles, which are the same or different and which are chosen from aryl, heterocycle, cycloalkyl or cycloalkenyl, fused together to form said bicyclic groups;
- halogen is understood to refer to fluorine, chlorine, bromine or iodine;
- lower alkyl is understood to mean that the alkyl is linear or branched and contains 1 to 6 carbon atoms; Examples of lower alkyl groups include methyl, ethyl, propyl, butyl, isopropyl, tert-butyl, isobutyl, n-butyl, pentyl, hexyl and the like.
- alkenyl is understood to refer to a linear or branched unsaturated carbon atom chain, comprising one or several double bonds, preferably one or two double bonds. Preferred alkenyls comprise from 3 to 6 carbon atoms and one double bonds.
- alkynyl is understood to refer to a linear or branched unsaturated carbon atom chain, comprising one or several triple bonds, preferably one or two triple bonds. Preferred alkynyls comprise from 3 to 6 carbon atoms and one triple bonds.
- lower haloalkyl are understood to refer to a lower alkyl substituted with one or several halogens; Preferred lower haloalkyl groups include perhaloalkyl groups such as CF3.
- cycloalkyl is understood to refer to saturated monocarbocyle containing from 3 to 10 carbon atoms; preferred cycloalkyl groups comprise cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl and cycloheptyl.
- cycloalkenyl is understood to refer to unsaturated monocarbocyle containing from 3 to 10 carbon atoms. Preferred cyloalkenyl groups contain 1 or 2 double bonds. Examples of suitable cycloalkenyl are 3-cyclohexene, 3-cycloheptene or the like. - carboxylic acid bioisostere has the classical meaning; common carboxylic acid bioisostere are tetrazole, hydroxamic acid, isoxazole, hydroxythiadiazole, sulfonamide, sulfonylcarboxamide, phosphonates, phosphonamides, phosphinates, sulfonates, acyl sulfonamide, mercaptoazole, acyl cyanamides.
WO-A-2004/026818 describes compounds of the following formula (I) as PDE7 inhibitors:

• m is 1 , 2 or 3, and, • R1 is selected from CH3, Cl, Br and F and,
• R2 is selected from,
o Q1-Q2-Q3-Q4 wherein,
■ Q1 is a single bond or a linear or branched (CrCβJalkylene group; ■ Q2 is a saturated 4 to 6-membered heterocycle comprising one or two heteroatoms selected from O or N;
■ Q3 is a linear or branched (CrC6)alkylene group;
■ Q4 is a 4 to 8-membered, aromatic or non aromatic, heterocycle comprising 1 to 4 heteroatoms selected from O, S, S(=O), SO2 and N, said heterocycle being optionally substituted with one or several groups selected from OR, NRR', CN and
(CrC6)alkyl, wherein R and R' are the same or different and are selected from H and (CrC6)alkyl;
■the atom of Q2 bound to Q1 is a carbon atom, and, •the atom of Q4 bound to Q3 is a carbon atom.
o (C-CβJalkyl,
■ said alkyl group being substituted with 1 to 3 groups, preferably 1 , selected from OR4, COOR4, NR4R5, NRC(=0)R4, C(=O)NR4R5 and SO2NR4R5, wherein,
• R is H or ^rCeJalkyl; » R4 is (Ci-C6)alkyl substituted with one or several groups, preferably 1 to
3, selected from F, CN, S(=O)R6, SO3H, SO2R6, SR7, C(=O)-NH-SO2-CH3, C(=O)R7, NR C(=O)R7, NR SO2R6, C(=O)NR7R8, O-C(=O)NR7R8 and SO2NR7R8, wherein R' is H or (CrC6)alkyl, R6 is (C1-C6)alkyl optionally substituted with one or two groups OR" wherein R" is selected from H and (CrC6)alkyl and R7 and R8 are the same or different and are selected from
H and R6;
• R5 is selected from R4, H and (CrC6)alkyl; or,
■ said alkyl group being
1) substituted with 1 to 3 groups, preferably 1 , selected from OC(=O)R4, SR4, S(=0)R3, C(=NR9)R4, C(=NR9)-NR4R5, NR-C(=NR9)-NR4R5, NRCOOR4,
NR-C(=O)-NR4R5, NR-SO2-NR4R5, NR-C(=NR9)-R4 and NR-SO2-R3 and,
2) optionally substituted with 1 or 2 groups selected from OR4, COOR4, C(=O)-R4, NR4R5, NRC(=O)R4, C(=O)NR4R5 and SO2NR4R5; wherein, • R is selected from H and (CrC6)alkyl;
• R9 is selected from H, CN, OH, OCH3, SO2CH3, SO2NH2 and (C1-C6)alkyl, and,
• R3 is (CrC6)alkyl, unsubstituted or substituted with one or several groups, preferably 1 to 3, selected from F, CN, S(=O)R6, SO3H, SO2R6, C(=O)-NH-SO2-CH3, OR7, SR7, COOR7, C(=0)R7, O-C(=O)NR7R8, NR7R8,
NRC(=O)R7, NR1SO2R6, C(=O)NR7R8 and SO2NR7R8, wherein R1 is H or (CrC6)alkyl, R6 is (CrC6)alkyl optionally substituted with one or two groups OR", wherein R" is selected from H and (CrC6)alkyl and R7 and R8 are the same or different and are selected from H and R6; • R4 and R5 are the same or different and are selected from H and R3.
US Provisional Patent Application No. 60/741854 (unpublished at the filing date of the present application) describes compounds of the formula (I) below:
,
- 20 -

X is O, S or N-CN;
R is F, Cl or CN;
A is a C3-6 cycloalkylene group optionally substituted with a C1^ alkyl group; and
B is a single bond or a C 1-2 alkylene group; or a pharmaceutically acceptable salt, solvate or | prodrug thereof.
Examples of suitable PDE7 inhibitors for use in the invention include those compounds generally or specifically disclosed in the publication of A. Castro, M.I. Abasolo, C. Gil, V. Segarra and A. Martinez. Eur. J. Med. Chem. 36 (2001), pp. 333-338 in particular the compounds which are benzyl derivatives of 2,1,3-benzo [3,2-a] thiadiazine 2,2-dioxides and 2,1,3- benzothieno[3,2- ajthiadiazine 2,2-dioxides and pharmaceutically acceptable salts and solvates thereof.
Further examples of suitable PDE7 inhibitors for use in the invention include those compounds generally or specifically disclosed in the publication of Barnes Mj, Cooper N, Davenport RJ, Biorg. Med. Chem. Lett. (2001) 23 (8): 1081 - 1083, 338 in particular the compounds which are guanine analogues, the 8-bromo-9-substitued compounds being the most preferred, and pharmaceutically acceptable salts and solvates thereof.
Further examples of suitable PDE7 inhibitors for use in the invention include those compounds generally or specifically disclosed in the publication of Pitts, WJ., et al Biorg. Med. Chem. Lett 14 2004 2955 - 2958, particularly the compounds which are purine based compounds and pharmaceutically acceptable salts and solvates thereof.
Further examples of suitable PDE7 inhibitors for use in the invention include those compounds generally or specifically disclosed in the publication of lorthiois, E., et al Biorg. Med. Chem. Lett, 14 2004 4623 - 4626 particularly the compounds which are spiroquinazolinones and pharmaceutically acceptable salts and solvates thereof.
Further examples of suitable PDE7 inhibitors for use in the invention include those compounds generally or specifically disclosed in the publication of Bernardelli, P., et al Bioorg. Med. Chem. Lett, 14 2004 4627 - 4631 , particularly the compounds which are 5, 8-disubstituted spirocyclohexane-quinazolinones particularly 5 substituted 8-chloro-spirocyclohexane-
quinazolinones derivatives such as 5-alkoxy-8 chloro-quinazolinone, and pharmaceutically acceptable salts and solvates thereof.
Further examples of suitable PDE7 inhibitors for use in the invention include those compounds generally or specifically disclosed in the publication of Vergne, F., et al Bioorg. Med. Chem. Lett, 2004, 14, 4607 - 461 & Vergne, F., et al Bioorg. Med. Chem. Lett, 2004, 14, 4615 - 4621 , particularly the compounds which are thiadiazoles and pharmaceutically acceptable salts and solvates thereof.
Further examples of suitable PDE7 inhibitors for use in the invention include those compounds generally or specifically disclosed in the patent application WO-A-01/098274 (CellTech Chiroscience Ltd), M-substituted phenyl-N-phenylsulfonamides particularly N-phenyl-3- benzoxazol-2-ylphenylsulfonamide and N-phenyl-3-benzimidazol-2-ylphenylsulfonamide derivatives. Patent application WO-A-01 /098274 (Celltech Chiroscience) discloses further examples of suitable PDE7 inhibitors which are sulfonamides and suitable for use in the invention.
In addition, patent application WO-A-01/074786 (Darwin Discovery Ltd) discloses further examples of PDE7 inhibitors suitable for use in the invention and which are a series of heterobiarylsulphonamides. Particularly suitable are the N-aryl-3- benzimidazolylbenzenesulfonamides. Patent application WO-A-00/068230 (Darwin Discovery Ltd) discloses further suitable PDE7 inhibitors, 9-(1 ,2,3,4-Tetrahydronapthalen-1-yl)-1 ,9- dihydropurin-6-one derivatives (also published in, Bioorganic and Medicinal Chemistry Letters 2001 , 1081-1083).
Patent applications WO-A-01/029049 (Merck), WO-A-01/036425 (Merck) and DE 19954707 (Merck) disclose imidazole derivatives, WO-A-01 /032175 (Merck) and DE 19953024 (Merck) disclose isoxazole derivatives, WO-A-01/032618 (Merck) and DE 19953025 (Merck) disclose pyrrole derivatives, DE19953414 (Merck) discloses imidazo[4,5-c]pyridine derivatives, all of which are further examples of PDE7 inhibitors and suitable for use in the invention.
Examples of suitable PDE7 inhibitors for use with the present invention are those compounds, or pharmaceutically acceptable salts thereof, generally or specifically disclosed in: WO-A-2002/028847, particularly
S^δ-cyclohexylimino^-methyM.S-dihydroli .S^lthiadiazol^-yO^-methoxy-benzene-I .S-diol; compound with trifluoro-methanesulfonic acid, δ^S-cyclohexylimino^-methyM.δ-dihydroII .SΛlthiadiazol^-yO^.S-dimethoxy-phenol; compound with trifluoro-methanesulfonic acid,
2-chloro-5-(5-cyclohexylimino-4-methyl-4,5-dihydro-[1 ,3,4]thiadiazol-2-yl)-benzenesulfonamide,
2-chloro-5-(5-cyclohexylimino-4-methyl-4,5-dihydro[1,3,4]thiadiazo!-2-yl)-N,N-diethyl- benzenesulfonamide,
{S-^-chloro-S^-methyl-piperazine-i-sulfonylJ-phenyll-S-methyl-SH-ti.a^lthiadiazol-a-ylidene}- cyclohexyl-amine,
2-chloro-5-(5-cyclohexylimino-4-methyl-4,5-dihydro-[1 ,3,4]thiadiazol-2-yl)-N-pyridin-4-ylmethyl- benzenesulfonamide,
2-chloro-5-(5-cyclohexylimino-4-methyl-4,5-dihydro-[1 ,3,4]thiadiazol-2-yl)-N-(2-morpholin-4-yl- ethyl)-benzenesulfonamide,
2-chloro-5-(5-cyclohexylimino-4-methyl-4,5-dihydro-[1,3,4]thiadiazol-2-yl)-N-ethyl- benzenesulfonamide,
2-chloro-5-(5-cyclohexylimino-4-methyl-4,5-dihydro-[1,3,4]thiadiazol-2-yl)-N-ethyl-N-(2- morpholin-4-yl-ethyl)-benzenesulfonamide,
2-chloro-5-(5-cyclohexylimino-4-methyl-4,5-dihydro-[1 ,3,4]thiadiazol-2-yl)-N-isopropyl-N-(2- morpholin-4-yl-ethyl)-benzenesulfonamide,
2-chloro-5-(5-cyclohexylimino-4-methyl-4,5-dihydro-[.1 ,3,4]thiadiazol-2-yl)-N-ethyl-N-[2-(2-methoxy- ethoxy)-ethyl]-benzenesulfonamide,
2-chloro-5-(cyclohexylimino-methyl-4,5-dihydro-[1,3,4]thiadiazol-2-yl)-N-(3-dimethylamino-2- hydroxy-propyl)-N-ethyl-benzenesulfonamide,
2-chloro-5-(5-cyclohexylimino-4-methyl-4,5-dihydro-[1,3,4]thiadiazol-2-yl)-N-(2,3-dihydroxy-propyl)-
N-ethyl-benzenesulfonamide,
2-chloro-5-(5-cyclohexylimino-4-methyl-4,5-dihydro-[1 ,3,4]thiadiazol-2-yl)-N-ethyl-N-(2-hydroxy-
3-pyrrolidin-1-yl-propyl)-benzenesulfonamide,
S-Cδ-cyclohexylimino^-methyM.δ-dihydro-ti .S^lthiadiazol^-yO-benzamide,
4-(5-cyclohexylimino-4-methyl-4,5-dihydro-[1,3,4]thiadiazol-2-yl)-benzamide,
4-(5-cyclohexylimino-4-methyl-4,5-dihydro[1,3,4]thiadiazol-2-yl)-N-quinolin-8-yl-benzamide,
4-(5-cyclohexylimino-4-methyl-4,5-dihydro[1 ,3,4]thiadiazol-2-yl)-N-(2,6-dimethoxy-pyridin-3-yl)- benzamide,
4-(5-cyclohexylimino-4-methyl-4,5-dihydro[1 ,3,4]thiadiazol-2-yl)-N-isopropyl-benzamide,
4-(5-cyclohexylimino-4-methyl-4,5-dihydro[1 ,3,4]thiadiazol-2-yl)-N-ethyl-benzamide,
4-(5-cyclohexylimino-4-methyl-4,5-dihydro[1,3,4]thiadiazol-2-yl)-N-(2-dimethylamino-ethyl)- benzamide,
4-(5-cyclohexylimino-4-methyl-4,5-dihydro[1 ,3,4]thiadiazol-2-yl)-N-pyridin-4-ylmethyl- benzamide,
4-(5-cyclohexylimino-4-methyl-4,5-dihydro-[1 ,3,4]thiadiazol-2-yl)-N-methyl-N-(1-methyl- piperidin-4-yl)-benzamide,
4-(5-cyclohexylimino-4-methyi-4,5-dihydro-[1 ,3,4]thiadiazol-2-yl)-N-methyl-benzamide,
2-[4-(5-cyclohexylimino-4-methyl-4,5-dihydro-[1,3,4]thiadiazol-2-yl)-benzoylamino]-3-(4- hydroxy-phenyl)-propionic acid tert-butyl ester,
(S)-2-[4-(5-cyclohexylimino-4-methyl-4,5-dihydro-[1 ,3,4]thiadiazol-2-yl)-benzoylamino]-3-(4-
hydroxy-phenyl)-propionic acid; compound with 2,2,2-trifluoro-acetic acid,
^(S-cyclohexylimino^-methyl^.S-dihydro-ti.a^lthiadiazol-Z-yO-N^S^.δ-trimethoxy-benzyl)- benzamide,
4-(5-cyclohexylimino-4-methyl-4,5-dihydro-[1 ,3,4]thiadiazol-2-yl)-N-[3-(4-methyl-piperazin-1-yl)- propyl]-beπzamide,
4-(5-cyclohexylimino-4-methyl-4,5-dihydro-[1 ,3,4]thiadiazol-2-yl)-N-pyridin-3-ylmethyl- benzamide,
N^I-benzyl-piperidin^-yO^S-cyclohexylimino^-methyM.δ-dihydro-ti .S^lthiadiazol^-yl)- benzamide,
4-(5-cyclohexylimino-4-methyl-4,5-dihydro-[1 ,3,4]thiadiazol-2-yl)-N-(2-ethyl-2H-pyrazol-3-yl)- benzamide,
4-(5-cyclohexylimino-4-methyl-4,5-dihydro-[1 ,3,4]thiadiazol-2-yl)-N-(2-morpholin-4-yl-ethyl)- benzamide,
4-(5-cyclohexylimino-4-methyl-4,5-dihydro-[1 ,3,4]thiadiazol-2-yl)-N-(2-pyrrolidin-1-yl-ethyl)- benzamide,
3-[5-(4-carbamoyl-phenyl)-3-methyl-3H-[1,3,4]thiadiazol-2-ylideneamino]-benzoic acid,
[5-(4-chloro-phenyl)-3-methyl-3H-[1,3,4]thiadiazol-2-ylidene]-[3-(1H-tetrazol-5-yl)-phenyl]- amine,
2-amino-4-(5-cyclohexylimino-4-methyl-4,5-dihydro-[1 ,3,4]thiadiazol-2-yl)-benzoic acid methyl ester,
2-amino-4-(5-cyclohexylimino-4-methyl-4,5-dihydro-[1 ,3,4]thiadiazol-2-yl)-benzamide,
7-(5-cyclohexylimino-4-methyl-4,5-dihydro-[1,3,4]thiadiazol-2-yl)-3H<|uinazolin^<>ne,
7-(5-cyclohexylimino-4-methyl-4,5-dihydro-[1,3,4]thiadiazol-2-yl)-quinazolin-4-ylamine,
N-^-lδ-cyclohexylimino^-methyl^.S-dihydro-li.S^lthiadiazol^-yO-phenyll-acetamide,
1-[4-(cyclohexylimino-methyl-4,5-dihydro-[1 ,3,4]thiadiazol-2-yl)-phenyl]-3-(2-dimethylamino- ethyl)-urea,
5'-(2-[(2-amino-2-oxoethyl)amino]ethoxy)-8'-chloro-1'H-spiro[cyclohexane-1 ,4'-quinazolin]- 2'(3'H)-one, δ'-chloro-δ'^lmethylsulfinyllmethoxyJ-i'H-spiroIcyclohexane-i^'-quinazolinl^XSΗ^one, 5'-(2-{[2-(acetylamino)ethyl]amino}ethoxy)-8'-chloro-1Η-spiro[cyclohexane-1,4'-quinazolin]- 2'(3Η)-one,
8'-fluoro-5'-[3-(methylsulfinyl)propoxy]-1'H-spiro[cyclohexane-1 ,4'-quinazolin]-2'(3'H)-one, δ'-fluoro-δ'^tmethylsulfinyllmethoxyVI'H-spirolcyclohexane-i ^'-quinazolinl-ZXS'H^one, and, 8'-fluoro-5'-(2-{[1-(1 H-pyrazol-3-ylmethyl)azetidin-3-yl]oxy}1'H-spiro[cyclohexane-1 ,4'- quinazolin]-2'(3'H)-one;
WO-A-2002/074754, particularly 5'-carboxypropoxy-8'-chloro-spiro[cyclohexane-1 -4'-(3',4'- dihydro)quinazolin]-2'(1 'H)-one; and
US Provisional Patent Application Number 60/741854, particularly c/s-3-[(8'-chloro-2'-oxo-2',3'- dihydro-1'H-spiro[cyclohexane-1 ,4'-quinazolin]-5'-yl)oxy]cyclobutanecarboxylic acid and trans-3-
^ B2006/000385
[(δ'-chloro-Z'-oxo^'.S'-dihydro-IΗ-spirolcyclohexane-i^'-quinazolinl-δ'- yl)oxy]cyclobutanecarboxylic acid; all of which are incorporated by reference.
Preferred PDE7 inhibitors for use with the present invention are selected from:
5'-carboxypropoxy-8'-chloro-spiro[cyclohexane-1-4'-(3',4'-dihydro)quinazolin]-2l(1'H)-one; c/s-3-[(8'-chloro-2'-oxo-2\3'-dihydro-1'H-spiro[cyclohexane-1 ,4'-quinazolin]-5'- yl)oxy]cyclobutanecarboxylic acid; and frans-S-^δ'-chloro^'-oxo-a'.S'-dihydro-IΗ-spiroIcyclohexane-i^'-quinazolinl-δ1- yl)oxy]cyclobutanecarboxylic acid; and their pharmaceutically acceptable salts.
As a further aspect of the present invention, there is provided a combination comprising 5'- carboxypropoxy-8'-chloro-spiro[cyclohexane-1-4'-(3',4'-dihydro)quinazolin]-2'(1'H)-one, or a pharmaceutically acceptable salt thereof, and an alpha-2-delta ligand selected from gabapentin, pregabalin, [(1R,5R,6S)-6-(aminomethyl)bicyclo[3.2.0]hept-6-yl]acetic acid, 3-(1-aminomethyl- cyclohexylmethyl)-4H-[1 ,2,4]oxadiazol-5-one, C-[1-(1 H-tetrazol-5-ylmethyl)-cycloheptyl]- methylamine, (3S,4S)-(1-aminomethyl-3,4-dimethyl-cyclopentyl)-acetic acid, (1α,3α,5α)(3-amino- methyl-bicyclo[3.2.0]hept-3-yl)-acetic acid, (3S,5R)-3-aminomethyl-5-methyl-octanoic acid, (3S,5R)-3-amino-5-methyl-heptanoic acid, (3S,5R)-3-amino-5-methyl-nonanoic acid, (3S.5R)- 3-Amino-5-methyl-octanoic acid, (2S,4S)-4-(3-chlorophenoxy)proline, (2S,4S)-4-(3- fluorobenzyl)proline, (2S)-2-amino-4-ethyl-2-methylhexanoic acid, and (2S)-2-aminomethyl-5- ethyl-heptanoic acid or a pharmaceutically acceptable salt thereof.
As a further aspect of the present invention, there is provided a combination comprising c/s-3-[(8'- chloro-2'-oxo-2',3'-dihydro-1'H-spiro[cyclohexane-1 ,4'-quinazolin]-5'-yl)oxy]cyclobutanecarboxylic acid, or a pharmaceutically acceptable salt thereof, and an alpha-2-delta ligand selected from gabapentin, pregabalin, [(1 R,5R,6S)-6-(aminomethyl)bicyclo[3.2.0]hept-6-yl]acetic acid, 3-(1- aminomethyl-cyclohexylmethyl)-4H-[1,2,4]oxadiazol-5-one, C-[1-(1H-tetrazol-5-ylmethyl)- cycloheptyl]-methylamine, (3S,4S)-(1-aminomethyl-3,4-dimethyl-cyclopentyl)-acetic acid,
(1α,3α,5α)(3-amino-methyl-bicyclo[3.2.0]hept-3-yl)-acetic acid, (3S,5R)-3-aminomethyl-5-methyl- octanoic acid, (3S,5R)-3-amino-5-methyl-heptanoic acid, (3S,5R)-3-amino-5-methyl-nonanoic acid, (3S,5R)-3-amino-5-methy!-octanoic acid, (2S,4S)-4-(3-chlorophenoxy)proline, (2S,4S)-4-(3- fluorobenzyOproline, (2S)-2-amino-4-ethyl-2-methylhexanoic acid, and (2S)-2-aminomethyl-5- ethyl-heptanoic acid or a pharmaceutically acceptable salt thereof.
As a further aspect of the present invention, there is provided a combination comprising trans-3- [(δ'-chloro^'-oxo^'.S'-dihydro-i'H-spirotcyclohexane-i^'-quinazolinl-δ1- yl)oxy]cyclobutanecarboxylic acid, or a pharmaceutically acceptable salt thereof, and an alpha-2- delta ligand selected from gabapentin, pregabalin, [(1R,5R,6S)-6-
(aminomethyl)bicyclo[3.2.0]hept-6-yl]acetic acid, 3-(1 -aminomethyl-cyclohexylmethyl)-4H- [1 ,2,4]oxadiazol-5-one, C-[1-(1 H-tetrazol-S-ylmethyO-cycloheptyll-methylamine, (3S,4S)-(1- aminomethyl-3,4-dimethyl-cyclopentyl)-acetic acid, (1α,3α,5α)(3-amino-methyl-bicyclo[3.2.0]hept- 3-yl)-acetic acid, (3S,5R)-3-aminomethyl-5-methyl-octanoic acid, (3S,5R)-3-amino-5-methyl- heptanoic acid, (3S,5R)-3-amino-5-methyl-nonanoic acid, (3S,5R)-3-amino-5-methyl-octanoic acid, (2S,4S)-4-(3-chlorophenoxy)proline, (2S,4S)-4-(3-fluorobenzyl)proline, (2S)-2-amino-4- ethyl-2-methylhexanoic acid, and (2S)-2-aminomethyl-5-ethyl-heptanoic acid or a pharmaceutically acceptable salt thereof.
As a yet further preferred aspect of the present invention, the combination is selected from:
5'-carboxypropoxy-8'-chloro-spiro[cyclohexane-1-4'-(3',4'-dihydro)quinazolin]-2'(1'H)-one and gabapentin;
5'-carboxypropoxy-8'-chloro-spiro[cyclohexane-1-4'-(3',4'-dihydro)quinazolin]-2'(1'H)-one and pregabaliπ; 5'-carboxypropoxy-8'-chloro-spiro[cyclohexane-1-4'-(3',4'-dihydro)quinazolin]-2'(1'H)-one and
(1α,3α,5α)(3-amino-methyl-bicyclo[3.2.0]hept-3-yl)-acetic acid;
5'-carboxypropoxy-8'-chloro-spiro[cyclohexane-1-4'-(3',4'-dihydro)quinazolin]-2'(1'H)-one and
[(1R,5R,6S)-6-(aminomethyl)bicyclo[3.2.0]hept-6-yl]acetic acid;
5'-carboxypropoxy-8'-chloro-spiro[cyclohexane-1-4'-(3',4'-dihydro)quinazolin]-2'(1'H)-one and (3S,4S)-(1-aminomethyl-3,4-dimethyl-cyclopentyl)-acetic acid;
5'-carboxypropoxy-8'-chloro-spiro[cyclohexane-1-4'-(3',4'-dihydro)quinazolin]-2'(1'H)-one and
(2S,4S)-4-(3-fluorobenzyl)proline;
5'-carboxypropoxy-8'-chloro-spiro[cyclohexane-1-4'-(3',4'-dihydro)quinazolin]-2'(1'H)-one and
(2S)-2-amino-4-ethyl-2-methylhexanoic acid; and δ'-carboxypropoxy-δ'-chloro-spiroIcyclohexane-i^'-CS'^'-dihyd^quinazolinl^XI'H^one and
(2S)-2-aminomethyl-5-ethyl-heptanoic acid; or pharmaceutically acceptable salts or solvates of either or both components of any such combination.
As a yet further preferred aspect of the present invention, the combination is selected from: c/s-3-[(8'-chloro-2'-oxo-2',3'-dihydro-1'H-spiro[cyclohexane-1,4'-quinazolin]-5'- yl)oxy]cyclobutanecarboxylic acid and gabapentin; c/s-3-[(8'-chloro-2'-oxo-2',3'-dihydro-1'H-spiro[cyclohexane-1 ,4'-quinazolin]-5'- yl)oxy]cyclobutanecarboxylic acid and pregabalin; c/s-3-[(8'-chloro-2'-oxo-2',3'-dihydro-1'H-spiro[cyclohexane-1,4'-quinazolin]-5'- yl)oxy]cyclobutanecarboxylic acid and (1 α,3α,5α)(3-amino-methyl-bicyclo[3.2.0]hept-3-yl)-acetic acid; c/s-3-[(8'-chloro-2'-oxo-2\3'-dihydro-1Η-spiro[cyclohexane-1,4'-quinazolin]-5'- yl)oxy]cyclobutanecarboxylic acid and [(1 R,5R,6S)-6-(aminomethyl)bicyclo[3.2.0]hept-6-yl]acetic acid;
c/s-3-[(8'-chloro-2'-oxo-2\3'-dihydro-1Η-spiro[cyclohexane-1 ,4'-quinazolin]-5'- yl)oxy]cyclobutanecarboxylic acid and (3S,4S)-(1-aminomethyl-3,4-dimethyl-cyclopentyl)-acetic acid; c/s-3-[(8'-chloro-2l-oxo-2l l3'-dihydro-1'H-spiro[cyclohexane-1 ,4'-quinazolin]-5'- yl)oxy]cyclobutanecarboxylic acid and (2S,4S)-4-(3-flucτobenzyl)proline; c/s-3-[(8'-chloro-2'-oxo-2',3'-dihydro-1'H-spiro[cyclohexane-1 ,4'-quinazolin]-5'- yl)oxy]cyclobutanecarboxylic acid and (2S)-2-amino-4-ethyl-2-methylhexanoic acid; and c/s-3-[(8'-chloro-2'<>xo-2\3'-dihydro-1Η-spiro[cyclohexane-1 ,4'-quinazolin]-5'- yl)oxy]cyclobutanecarboxylic acid and (2S)-2-aminomethyl-5-ethyl-heptanoic acid or pharmaceutically acceptable salts or solvates of either or both components of any such combination.
As a yet further preferred aspect of the present invention, the combination is selected from: frans-3-[(8'-chloro-2'-oxo-2',3'-dihydro-1'H-spiro[cyclohexane-1,4'-quinazolin]-5'- yl)oxy]cyclobutanecarboxylic acid and gabapentin; frans-3-[(8'-chloro-2'-oxo-2',3'-dihydro-1'H-spiro[cyclohexane-1,4'-quinazolin]-5'- yl)oxy]cyclobutanecarboxylic acid and pregabalin; fraπs-3-[(8'-chloro-2'<»co-2\3'-dihydro-1Η-spiro[cyclohexane-1 ,4'-quinazolin]-5'- yl)oxy]cyclobutanecarboxylic acid and (1α,3α,5α)(3-amino-methyl-bicyclo[3.2.0]hept-3-yl)-acetic acid; frans-3-[(8'-chloro-2'-oxo-2',3'-dihydro-1'H-spiro[cyclohexane-1 ,4'-quinazolin]-5'- yl)oxy]cyclobutanecarboxylic acid and [(1R,5R,6S)-6-(aminomethyl)bicyclo[3.2.0]hept-6-yl]acetic acid; frans-3-[(8'-chloro-2'-oxo-2l,3'-dihydro-1Η-spiro[cyclohexane-1 ,4'-quinazolin]-51- yl)oxy]cyclobutanecarboxylic acid and (3S,4S)-(1-aminomethyl-3,4-dimethyl-cyclopentyl)-acetic acid; fra/7s-3-[(8'-chloro-2'-oxo-2',3'-dihydro-1'H-spiro[cyclohexane-1,4'-quinazolin]-5'- yl)oxy]cyclobutanecarboxylic acid and (2S,4S)-4-(3-fluorobenzyl)proline; fraA7S-3-[(8'-chloro-2'-oxo-2',3'-dihydro-1'H-spiro[cyclohexane-1 ,4'-quinazolin]-5'- yl)oxy]cyclobutanecarboxylic acid and (2S)-2-amino-4-ethyl-2-methylhexanoic acid; and frans-S-KB'-chloro^'-oxo^S'-dihydro-IΗ-spiroIcyclohexane-i^'-quinazolinl-δ1- yl)oxy]cyclobutanecarboxylic acid and (2S)-2-aminomethyl-5-ethyl-heptanoic acid or pharmaceutically acceptable salts or solvates of either or both components of any such combination.
Particularly preferred combinations of the invention include those in which each variable of the combination is selected from the suitable parameters for each variable. Even more preferable combinations of the invention include those where each variable of the combination is selected from the more preferred or more preferred parameters for each variable.
Pharmaceutically acceptable salts of PDE7 inhibitors and alpha-2-delta ligands include the acid addition and base salts thereof.
Suitable acid addition salts are formed from acids which form non-toxic salts. Examples include the acetate, adipate, aspartate, benzoate, besylate, bicarbonate/carbonate, bisulphate/sulphate, borate, camsylate, citrate, cyclamate, edisylate, esylate, formate, fumarate, gluceptate, gluconate, glucuronate, hexafluorophosphate, hibenzate, hydrochloride/chloride, hydrobromide/bromide, hydroiodide/iodide, isethionate, lactate, malate, maleate, malonate, mesylate, methylsulphate, naphthylate, 2-napsylate, nicotinate, nitrate, orotate, oxalate, palmitate, pamoate, phosphate/hydrogen phosphate/dihydrogen phosphate, pyroglutamate, saccharate, stearate, succinate, tannate, tartrate, tosylate, trifluoroacetate and xinofoate salts.
Suitable base salts are formed from bases which form non-toxic salts. Examples include the aluminium, arginine, benzathine, calcium, choline, diethylamine, diolamine, glycine, lysine, magnesium, meglumine, olamine, potassium, sodium, tromethamine and zinc salts.
Hemisalts of acids and bases may also be formed, for example, hemisulphate and hemicalcium salts.
For a review on suitable salts, see Handbook of Pharmaceutical Salts: Properties. Selection, and Use by Stahl and Wermuth (Wiley-VCH, 2002).
Pharmaceutically acceptable salts of PDE7 inhibitors and alpha-2-delta ligands may be prepared by one or more of three methods:
(i) by reacting the PDE7 inhibitor or alpha-2-delta ligand with the desired acid or base;
(ii) by removing an acid- or base-labile protecting group from a suitable precursor of the
PDE7 inhibitor or alpha-2-delta ligand or by ring-opening a suitable cyclic precursor, for example, a lactone or lactam, using the desired acid or base; or (iii) by converting one salt of the PDE7 inhibitor or alpha-2-delta ligand to another by reaction with an appropriate acid or base or by means of a suitable ion exchange column.
All three reactions are typically carried out in solution. The resulting salt may precipitate out and be collected by filtration or may be recovered by evaporation of the solvent. The degree of ionisation in the resulting salt may vary from completely ionised to almost non-ionised.
The components of the combination of the present invention may exist in a continuum of solid states ranging from fully amorphous to fully crystalline. The term 'amorphous' refers to a state in which the material lacks long range order at the molecular level and, depending upon temperature, may exhibit the physical properties of a solid or a liquid. Typically such materials do not give distinctive X-ray diffraction patterns and, while exhibiting the properties of a solid, are
more formally described as a liquid. Upon heating, a change from solid to liquid properties occurs which is characterised by a change of state, typically second order ('glass transition'). The term 'crystalline' refers to a solid phase in which the material has a regular ordered internal structure at the molecular level and gives a distinctive X-ray diffraction pattern with defined peaks. Such materials when heated sufficiently will also exhibit the properties of a liquid, but the change from solid to liquid is characterised by a phase change, typically first order ('melting point').
The components of the combination of the invention may also exist in unsolvated and solvated forms. The term 'solvate' is used herein to describe a molecular complex comprising the PDE7 inhibitor or alpha-2-delta ligand and one or more pharmaceutically acceptable solvent molecules, for example, ethanol. The term 'hydrate' is employed when said solvent is water.
A currently accepted classification system for organic hydrates is one that defines isolated site, channel, or metal-ion coordinated hydrates - see Polymorphism in Pharmaceutical Solids by K. R. Morris (Ed. H. G. Brittain, Marcel Dekker, 1995). Isolated site hydrates are ones in which the water molecules are isolated from direct contact with each other by intervening organic molecules. In channel hydrates, the water molecules lie in lattice channels where they are next to other water molecules. In metal-ion coordinated hydrates, the water molecules are bonded to the metal ion.
When the solvent or water is tightly bound, the complex will have a well-defined stoichiometry independent of humidity. When, however, the solvent or water is weakly bound, as in channel solvates and hygroscopic compounds, the water/solvent content will be dependent on humidity and drying conditions. In such cases, non-stoichiometry will be the norm.
Also included within the scope of the invention are multi-component complexes (other than salts and solvates) wherein the drug and at least one other component are present in stoichiometric or non-stoichiometric amounts. Complexes of this type include clathrates (drug-host inclusion complexes) and co-crystals. The latter are typically defined as crystalline complexes of neutral molecular constituents which are bound together through non-covalent interactions, but could also be a complex of a neutral molecule with a salt. Co-crystals may be prepared by melt crystallisation, by recrystallisation from solvents, or by physically grinding the components together - see Chem Commun, .17, 1889-1896, by O. Almarsson and M. J. Zaworotko (2004). For a general review of multi-component complexes, see J Pharm Sci, 64 (8), 1269-1288, by Haleblian (August 1975).
The components of the combination of the invention may also exist in a mesomorphic state (mesophase or liquid crystal) when subjected to suitable conditions. The mesomorphic state is intermediate between the true crystalline state and the true liquid state (either melt or solution). Mesomorphism arising as the result of a change in temperature is described as 'thermotropic' and that resulting from the addition of a second component, such as water or another solvent, is described as 'lyotropic'. Compounds that have the potential to form lyotropic mesophases are
described as 'amphiphilic' and consist of molecules which possess an ionic (such as -COO"Na+, -COO'K+, or -SO3 "Na+) or non-ionic (such as -N~N+(CH3)3) polar head group. For more information, see Crystals and the Polarizing Microscope by N. H. Hartshorne and A. Stuart, 4th Edition (Edward Arnold, 1970).
Hereinafter all references to PDE7 inhibitors include references to salts, solvates, multi- component complexes and liquid crystals thereof and to solvates, multi-component complexes and liquid crystals of salts thereof.
Hereinafter all references to alpha-2-delta ligands include references to salts, solvates, multi- component complexes and liquid crystals thereof and to solvates, multi-component complexes and liquid crystals of salts thereof.
A number of the alpha-2-delta ligands of the combination of the present invention are amino acids. Since amino acids are amphoteric, pharmacologically compatible salts can be salts of appropriate non-toxic inorganic or organic acids or bases. Salts with quaternary ammonium ions can also be prepared with, for example, the tetramethyl-ammonium ion. The alpha-2-delta ligands of the combination of the invention may also be formed as a zwitterion.
A suitable salt for amino acid compounds of the present invention is the hydrochloride salt.
The term 'PDE7 inhibitor' includes PDE7 inhibitors as hereinbefore defined, including all polymorphs and crystal habits thereof, prodrugs and isomers thereof (including optical, geometric and tautomeric isomers) as hereinafter defined and isotopically-labeled PDE7 inhibitors.
The term 'alpha-2-delta ligand' includes alpha-2-delta ligands as hereinbefore defined, including all polymorphs and crystal habits thereof, prodrugs and isomers thereof (including optical, geometric and tautomeric isomers) as hereinafter defined and isotopically-labeled alpha-2-delta ligands.
As indicated, so-called 'prodrugs' of the components of the combination are also within the scope of the invention. Thus certain derivatives of PDE7 inhibitors or alpha-2-delta ligands which may have little or no pharmacological activity themselves can, when administered into or onto the body, be converted into PDE7 inhibitors or alpha-2-delta ligands having the desired activity, for example, by hydrolytic cleavage. Such derivatives are referred to as 'prodrugs'. Further information on the use of prodrugs may be found in Pro-drugs as Novel Delivery Systems. Vol. 14, ACS Symposium Series (T. Higuchi and W. Stella) and Bioreversible Carriers in Drug Design, Pergamon Press, 1987 (Ed. E. B. Roche, American Pharmaceutical Association).
Prodrugs in accordance with the invention can, for example, be produced by replacing appropriate functionalities present in the PDE7 inhibitor or alpha-2-delta ligand with certain moieties known to
those skilled in the art as 'pro-moieties' as described, for example, in Design of Prodrugs by H. Bundgaard (Elsevier, 1985).
Some examples of prodrugs in accordance with the invention include
(i) where the PDE7 inhibitor or alpha-2-delta ligand contains a carboxylic acid functionality (- COOH), an ester thereof, for example, a compound wherein the hydrogen of the carboxylic acid functionality of the PDE7 inhibitor or alpha-2-delta ligand is replaced by (CrC8)alkyl; (ii) where the PDE7 inhibitor or alpha-2-delta ligand contains an alcohol functionality (-OH), an ether thereof, for example, a compound wherein the hydrogen of the alcohol functionality of the PDE7 inhibitor or alpha-2-delta ligand is replaced by (C1-C6)alkanoyloxymethyl; and (iii) where the PDE7 inhibitor or alpha-2-delta ligand contains a primary or secondary amino functionality (-NH2 Or -NHR where R ≠ H), an amide thereof, for example, a compound wherein, as the case may be, one or both hydrogens of the amino functionality of the PDE7 inhibitor or alpha- 2-delta ligand is/are replaced by (Ci-Cio)alkanoyl.
Further examples of replacement groups in accordance with the foregoing examples and examples of other prodrug types may be found in the aforementioned references.
Moreover, certain PDE7 inhibitors or alpha-2-delta ligands may themselves act as prodrugs of other PDE7 inhibitors or alpha-2-delta ligands.
Aminoacyl-glycolic and -lactic esters are known as prodrugs of amino acids (Wermuth CG. , Chemistry and Industry, 1980:433-435). The carbonyl group of the amino acids can be esterified by known means. Prodrugs and soft drugs are known in the art (Palomino E., Drugs of the Future, 1990;15(4):361-368). The last two citations are hereby incorporated by reference.
Also included within the scope of the invention are metabolites of the components of the combination, that is, compounds formed in vivo upon administration of the PDE7 inhibitor or alpha-2-delta ligand. Some examples of metabolites in accordance with the invention include
(i) where the PDE7 inhibitor or alpha-2-delta ligand contains a methyl group, an hydroxymethyl derivative thereof (-CH3 -> -CH2OH):
(ii) where the PDE7 inhibitor or alpha-2-delta ligand contains an alkoxy group, an hydroxy derivative thereof (-OR -> -OH);
(iii) where the PDE7 inhibitor or alpha-2-delta ligand contains a tertiary amino group, a secondary amino derivative thereof (-NR1R2 -> -NHR1 or -NHR2);
(iv) where the PDE7 inhibitor or alpha-2-delta ligand contains a secondary amino group, a primary derivative thereof (-NHR1 -> -NH2); (v) where the PDE7 inhibitor or alpha-2-delta ligand contains a phenyl moiety, a phenol derivative thereof (-Ph -> -PhOH); and
(vi) where the PDE7 inhibitor or alpha-2-delta ligand contains a carboxamide group, a carboxylic acid derivative thereof (-CONH2 -> COOH).
A PDE7 inhibitor or alpha-2-delta ligand containing one or more asymmetric carbon atoms can exist as two or more stereoisomers. Where a PDE7 inhibitor or alpha-2-delta ligand contains an alkenyl or alkenylene group, geometric cisltrans (or Z/E) isomers are possible. Where structural isomers are interconvertible via a low energy barrier, tautomeric isomerism ('tautomerism') can occur. This can take the form of proton tautomerism in PDE7 inhibitors or alpha-2-delta ligands containing, for example, an imino, keto, or oxime group, or so-called valence tautomerism in compounds which contain an aromatic moiety. It follows that a single compound may exhibit more than one type of isomerism.
Included within the scope of the present invention are all stereoisomers, geometric isomers and tautomeric forms of the components of the combination of the invention, including compounds exhibiting more than one type of isomerism, and mixtures of one or more thereof. Also included are acid addition or base salts wherein the counterion is optically active, for example, d-lactate or /-lysine, or racemic, for example, (//-tartrate or d/-arginine.
Cisltrans isomers may be separated by conventional techniques well known to those skilled in the art, for example, chromatography and fractional crystallisation.
Conventional techniques for the preparation/isolation of individual enantiomers include chiral synthesis from a suitable optically pure precursor or resolution of the racemate (or the racemate of a salt or derivative) using, for example, chiral high pressure liquid chromatography (HPLC).
Alternatively, the racemate (or a racemic precursor) may be reacted with a suitable optically active compound, for example, an alcohol, or, in the case where the PDE7 inhibitor or alpha-2-delta ligand contains an acidic or basic moiety, a base or acid such as 1-phenylethylamine or tartaric acid. The resulting diastereomeric mixture may be separated by chromatography and/or fractional crystallization and one or both of the diastereoisomers converted to the corresponding pure enantiomer(s) by means well known to a skilled person.
Chiral PDE7 inhibitors or alpha-2-delta ligands (and chiral precursors thereof) may be obtained in enantiomerically-enriched form using chromatography, typically HPLC, on an asymmetric resin with a mobile phase consisting of a hydrocarbon, typically heptane or hexane, containing from 0 to 50% by volume of isopropanol, typically from 2% to 20%, and from 0 to 5% by volume of an alkylamine, typically 0.1% diethylamine. Concentration of the eluate affords the enriched mixture.
When any racemate crystallises, crystals of two different types are possible. The first type is the racemic compound (true racemate) referred to above wherein one homogeneous form of crystal is produced containing both enantiomers in equimolar amounts. The second type is the racemic
mixture or conglomerate wherein two forms of crystal are produced in equimolar amounts each comprising a single enantiomer.
While both of the crystal forms present in a racemic mixture have identical physical properties, they may have different physical properties compared to the true racemate. Racemic mixtures may be separated by conventional techniques known to those skilled in the art - see, for example, Stereochemistry of Organic Compounds by E. L. Eliel and S. H. Wilen (Wiley, 1994).
The present invention includes all pharmaceutically acceptable isotopically-labelled PDE7 inhibitors or alpha-2-delta ligands wherein one or more atoms are replaced by atoms having the same atomic number, but an atomic mass or mass number different from the atomic mass or mass number which predominates in nature.
Examples of isotopes suitable for inclusion in the PDE7 inhibitors or alpha-2-delta ligands include isotopes of hydrogen, such as 2H and 3H, carbon, such as 11C, 13C and 14C, chlorine, such as 36CI, fluorine, such as 18F, iodine, such as 123I and 125I, nitrogen, such as 13N and 15N, oxygen, such as 15O, 17O and 18O, phosphorus, such as 32P, and sulphur, such as 35S.
Certain isotopically-labelled PDE7 inhibitors or alpha-2-delta ligands, for example, those incorporating a radioactive isotope, are useful in drug and/or substrate tissue distribution studies. The radioactive isotopes tritium, i.e. 3H, and carbon-14, i.e. 14C, are particularly useful for this purpose in view of their ease of incorporation and ready means of detection.
Substitution with heavier isotopes such as deuterium, i.e. 2H, may afford certain therapeutic advantages resulting from greater metabolic stability, for example, increased in vivo half-life or reduced dosage requirements, and hence may be preferred in some circumstances.
Substitution with positron emitting isotopes, such as 11C, 18F, 15O and 13N, can be useful in Positron Emission Topography (PET) studies for examining substrate receptor occupancy.
Isotopically-labeled PDE7 inhibitors or alpha-2-delta ligands can generally be prepared by conventional techniques known to those skilled in the art or by processes analogous to those described in the accompanying Examples and Preparations using an appropriate isotopically- labeled reagent in place of the non-labeled reagent previously employed.
Pharmaceutically acceptable solvates in accordance with the invention include those wherein the solvent of crystallization may be isotopically substituted, e.g. D2O, d6-acetone, d6-DMSO.
The combination of the present invention is useful for the treatment of pain, inflammatory, neuropathic, visceral and nociceptive pain, more particularly neuropathic pain.
As a further aspect of the invention, there is provided the use of a PDE7 inhibitor and an alpha- 2-delta ligand in the manufacture of a medicament for the treatment of pain, particularly inflammatory, neuropathic, visceral and nociceptive pain, more particularly neuropathic pain.
As an alternative feature, the invention provides the use of a synergistic effective amount of a PDE7 inhibitor and an alpha-2-delta ligand in the manufacture of a medicament for the curative, prophylactic or palliative treatment of pain, particularly inflammatory, neuropathic, visceral and nociceptive pain, more particularly neuropathic pain.
As an alternative aspect, there is provided a method for the treatment of pain, particularly inflammatory, neuropathic, visceral and nociceptive pain, more particularly neuropathic pain, comprising simultaneous, sequential or separate administration of a therapeutically effective amount of an alpha-2-delta ligand and a PDE7 inhibitor, to a mammal in need of said treatment.
As an alternative feature, there is provided a method for the treatment of pain, particularly inflammatory, neuropathic, visceral and nociceptive pain, more particularly neuropathic pain, comprising simultaneous, sequential or separate administration of a therapeutically synergistic amount of an alpha-2-delta ligand and PDE7 inhibitor, to a mammal in need of said treatment.
Physiological pain is an important protective mechanism designed to warn of danger from potentially injurious stimuli from the external environment. The system operates through a specific set of primary sensory neurones and is activated by noxious stimuli via peripheral transducing mechanisms (see Millan, 1999, Prog. Neurobiol., 57, 1-164 for a review). These sensory fibres are known as nociceptors and are characteristically small diameter axons with slow conduction velocities. Nociceptors encode the intensity, duration and quality of noxious stimulus and by virtue of their topographically organised projection to the spinal cord, the location of the stimulus. The nociceptors are found on nociceptive nerve fibres of which there are two main types, A-delta fibres (myelinated) and C fibres (non-myelinated). The activity generated by nociceptor input is transferred, after complex processing in the dorsal horn, either directly, or via brain stem relay nuclei, to the ventrobasal thalamus and then on to the cortex, where the sensation of pain is generated.
Pain may generally be classified as acute or chronic. Acute pain begins suddenly and is shortlived (usually twelve weeks or less). It is usually associated with a specific cause such as a specific injury and is often sharp and severe. It is the kind of pain that can occur after specific injuries resulting from surgery, dental work, a strain or a sprain. Acute pain does not generally result in any persistent psychological response. In contrast, chronic pain is long-term pain, typically persisting for more than three months and leading to significant psychological and emotional problems. Common examples of chronic pain are neuropathic pain (e.g. painful diabetic neuropathy, postherpetic neuralgia), carpal tunnel syndrome, back pain, headache, cancer pain, arthritic pain and chronic post-surgical pain.
When a substantial injury occurs to body tissue, via disease or trauma, the characteristics of nociceptor activation are altered and there is sensitisation in the periphery, locally around the injury and centrally where the nociceptors terminate. These effects lead to a hightened sensation of pain. In acute pain these mechanisms can be useful, in promoting protective behaviours which may better enable repair processes to take place. The normal expectation would be that sensitivity returns to normal once the injury has healed. However, in many chronic pain states, the hypersensitivity far outlasts the healing process and is often due to nervous system injury. This injury often leads to abnormalities in sensory nerve fibres associated with maladaptation and aberrant activity (Woolf & Salter, 2000, Science, 288, 1765-1768).
Clinical pain is present when discomfort and abnormal sensitivity feature among the patient's symptoms. Patients tend to be quite heterogeneous and may present with various pain symptoms. Such symptoms include: 1) spontaneous pain which may be dull, burning, or stabbing; 2) exaggerated pain responses to noxious stimuli (hyperalgesia); and 3) pain produced by normally innocuous stimuli (allodynia - Meyer et al., 1994, Textbook of Pain, 13-44). Although patients suffering from various forms of acute and chronic pain may have similar symptoms, the underlying mechanisms may be different and may, therefore, require different treatment strategies. Pain can also therefore be divided into a number of different subtypes according to differing pathophysiology, including nociceptive, inflammatory and neuropathic pain.
Nociceptive pain is induced by tissue injury or by intense stimuli with the potential to cause injury. Pain afferents are activated by transduction of stimuli by nociceptors at the site of injury and activate neurons in the spinal cord at the level of their termination. This is then relayed up the spinal tracts to the brain where pain is perceived (Meyer et al., 1994, Textbook of Pain, 13-44).
The activation of nociceptors activates two types of afferent nerve fibres. Myelinated A-delta fibres transmit rapidly and are responsible for sharp and stabbing pain sensations, whilst unmyelinated C fibres transmit at a slower rate and convey a dull or aching pain. Moderate to severe acute nociceptive pain is a prominent feature of pain from central nervous system trauma, strains/sprains, burns, myocardial infarction and acute pancreatitis, post-operative pain (pain following any type of surgical procedure), posttraumatic pain, renal colic, cancer pain and back pain. Cancer pain may be chronic pain such as tumour related pain (e.g. bone pain, headache, facial pain or visceral pain) or pain associated with cancer therapy (e.g. postchemotherapy syndrome, chronic postsurgical pain syndrome or post radiation syndrome). Cancer pain may also occur in response to chemotherapy, immunotherapy, hormonal therapy or radiotherapy. Back pain may be due to herniated or ruptured intervertebral discs or abnormalities of the lumber facet joints, sacroiliac joints, paraspinal muscles or the posterior longitudinal ligament. Back pain may resolve naturally but in some patients, where it lasts over 12 weeks, it becomes a chronic condition which can be particularly debilitating.
Neuropathic pain is currently defined as pain initiated or caused by a primary lesion or dysfunction in the nervous system. Nerve damage can be caused by trauma and disease and thus the term 'neuropathic pain' encompasses many disorders with diverse aetiologies. These include, but are not limited to, peripheral neuropathy, diabetic neuropathy, post herpetic neuralgia, trigeminal neuralgia, back pain, cancer neuropathy, HIV neuropathy, phantom limb pain, carpal tunnel syndrome, central post-stroke pain and pain associated with chronic alcoholism, hypothyroidism, uremia, multiple sclerosis, spinal cord injury, Parkinson's disease, epilepsy and vitamin deficiency. Neuropathic pain is pathological as it has no protective role. It is often present well after the original cause has dissipated, commonly lasting for years, significantly decreasing a patient's quality of life (Woolf and Mannion, 1999, Lancet, 353, 1959-1964). The symptoms of neuropathic pain are difficult to treat, as they are often heterogeneous even between patients with the same disease (Woolf & Decosterd, 1999, Pain Supp, 6, S141-S147; Woolf and Mannion, 1999, Lancet, 353, 1959-1964). They include spontaneous pain, which can be continuous, and paroxysmal or abnormal evoked pain, such as hyperalgesia (increased sensitivity to a noxious stimulus) and allodynia (sensitivity to a normally innocuous stimulus).
The inflammatory process is a complex series of biochemical and cellular events, activated in response to tissue injury or the presence of foreign substances, which results in swelling and pain (Levine and Taiwo, 1994, Textbook of Pain, 45-56). Arthritic pain is the most common inflammatory pain. Rheumatoid disease is one of the commonest chronic inflammatory conditions in developed countries and rheumatoid arthritis is a common cause of disability. The exact aetiology of rheumatoid arthritis is unknown, but current hypotheses suggest that both genetic and microbiological factors may be important (Grennan & Jayson, 1994, Textbook of Pain, 397-407). It has been estimated that almost 16 million Americans have symptomatic osteoarthritis (OA) or degenerative joint disease, most of whom are over 60 years of age, and this is expected to increase to 40 million as the age of the population increases, making this a public health problem of enormous magnitude (Houge & Mersfelder, 2002, Ann Pharmacother., 36, 679-686; McCarthy et al., 1994, Textbook of Pain, 387-395). Most patients with osteoarthritis seek medical attention because of the associated pain. Arthritis has a significant impact on psychosocial and physical function and is known to be the leading cause of disability in later life. Ankylosing spondylitis is also a rheumatic disease that causes arthritis of the spine and sacroiliac joints. It varies from intermittent episodes of back pain that occur throughout life to a severe chronic disease that attacks the spine, peripheral joints and other body organs.
Another type of inflammatory pain is visceral pain which includes pain associated with inflammatory bowel disease (IBD). Visceral pain is pain associated with the viscera, which encompass the organs of the abdominal cavity. These organs include the sex organs, spleen and part of the digestive system. Pain associated with the viscera can be divided into digestive visceral pain and non-digestive visceral pain. Commonly encountered gastrointestinal (Gl) disorders that cause pain include functional bowel disorder (FBD) and inflammatory bowel disease (IBD). These Gl disorders include a wide range of disease states that are currently only
moderately controlled, including, in respect of FBD, gastro-esophageal reflux, dyspepsia, irritable bowel syndrome (IBS) and functional abdominal pain syndrome (FAPS), and, in respect of IBD, Crohn's disease, ileitis and ulcerative colitis, all of which regularly produce visceral pain.
Other types of visceral pain include the pain associated with dysmenorrhea, cystitis and pancreatitis and pelvic pain.
It should be noted that some types of pain have multiple aetiologies and thus can be classified in more than one area,' e.g. back pain and cancer pain have both nociceptive and neuropathic components.
Other types of pain include:
• pain resulting from musculoskeletal disorders, including myalgia, fibromyalgia, spondylitis, sero-negative (non-rheumatoid) arthropathies, non-articular rheumatism, dystrophinopathy, glycogenosis, polymyositis and pyomyositis;
• heart and vascular pain, including pain caused by angina, myocardical infarction, mitral stenosis, pericarditis, Raynaud's phenomenon, scleredoma and skeletal muscle ischemia;
• head pain, such as migraine (including migraine with aura and migraine without aura), cluster headache, tension-type headache mixed headache and headache associated with vascular disorders; and
• orofacial pain, including dental pain, otic pain, burning mouth syndrome and temporomandibular myofascial pain.
DETAILED DESCRIPTION OF THE INVENTION
The components of the combination of the present invention are prepared by methods well known to those skilled in the art. Specifically, the patents, patent applications and publications, mentioned hereinabove, each of which is hereby incorporated herein by reference, exemplify compounds which can be used in the combinations, pharmaceutical compositions, methods and kits in accord with the present invention, and refer to methods of preparing those compounds.
The following experimental procedures illustrate the preparation of certain preferred alpha-2-delta ligands described in US Provisional Patent Application Number 60/676025 (unpublished at the filing date of the present invention):
In the following general methods, R1, R2, R3 and R4 are as previously defined for a compound of formula (I) unless otherwise stated.
A compound of formula (I), wherein R3 and R4 are H, may be prepared by the hydrogenolytic deprotection of a compound of formula (III)

wherein R1 and R2 are as defined above. The hydrogenation is typically carried out using a source of hydrogen such as hydrogen gas, cyclohexadiene or ammonium formate (preferably hydrogen gas) and a transition metal catalyst such as a palladium, platinum or rhodium catalyst (preferably a palladium catalyst). An acid, such as hydrochloric or trifluoroacetic acid, may also be used to increase the rate of reaction. In a preferred procedure, a solution of the compound of formula (III) in a suitable solvent, such as ethanol, is treated with palladium on carbon and hydrochloric acid hydrogenated at about 414 kPa (60 psi).
A compound of formula (III) may be prepared by treating an imine of formula (IV):

R2M1 (V)
wherein R2 is as defined above and M1 is a suitable metal, optionally bearing one or more further ligands; or by treating an imine of formula (Vl):

wherein R2 is as defined above, with a compound of formula:
R1M1 (VII)
wherein R1 and M1are as defined above. In such an imine addition reaction the organometallic reagent of formula (V) or (VII) is typically an organolithium or an organomagnesium derivative. An organomagensium (Grignard) reagent, wherein M1 is MgX, X being a halide, is preferred. The reaction is carried out in a suitable inert solvent such as tetrahydrofuran or diethyl ether at low temperature, typically between 0 and -780C. In a preferred procedure, a solution of the compound of formula (IV) or (Vl) in a suitable solvent, such as tetrahydrofuran, is treated with a suitable Grignard reagent of formula (V) or (VII), respectively, at -5O0C and in the presence of boron trifluoride etherate.
Compounds of formula (IV) and (Vl) can be prepared by the condensation of a compound of formula:

with, respectively, a compound of formula:

wherein R1 is as defined above and X is Ci-Cε alkyl; or a compound of formula (X):
O xo JL π R2 n ° (X)
wherein R2 is as defined above and X is C1-C6 alkyl. The condensation may be carried out under basic, neutral or acidic conditions and generally requires elevated temperatures and/or prolonged reaction times. In a typical procedure, a solution of the compound of formula (VIII) and the compound of formula (IX) or (X), in a suitable solvent, such as trifluoroethanol, is heated at about 8O0C in the presence of a dehydrating agent such as 4A molecular sieves.
Compounds of formula (X), (IX) and (VIII) are either commercially available or easily prepared by standard methods well known to the skilled person, either as a result of common general knowledge (e.g. see 'Comprehensive Organic Transformations' by Richard Larock (1999, VCH Publishers Inc.) or by reference to specific published procedures.
Compounds of formula (I) wherein R3 and/or R4 are not H may be prepared from compounds of formula (I) wherein R3 and/or R4 are H by simple chemical transformations well known to the skilled man. Suitable conditions for such amide and ester forming reactions may be found in
Comprehensive Organic Transformations referenced above.
A compound of formula (I), wherein R3 and R4 are both H, may alternatively be prepared by the hydrolysis of a nitrile of formula (Xl):

wherein R1 and R2 are as defined above. The hydrolysis is typically accomplished with acidic or basic catalysis in an aqueous solvent at an elevated temperature. In a typical procedure, a solution of the compound of formula (Xl) in water is treated with 6 molar hydrochloric acid and heated to about 1000C.
A compound of formula (Xl) may be prepared by the addition of cyanide to a compound of formula (XII):

wherein R1 and R2 are as defined above. A preferred source of cyanide for the addition is a compound of formula M2CN wherein M2 is a metal cation, optionally bearing other ligands. Most preferred is a dialkylaluminium cyanide such as diethylaluminium cyanide. The reaction is carried out as a solution in a suitable inert solvent such as tetrahydrofuran, dichloromethane or diethyl ether. In a preferred procedure a solution of a compound of formula (XII) in a mixture of isoproanol and tetrahydrofuran is treated with diethylaluminium cyanide at a temperature of between -78 and -2O0C.
A compound of formula (XII) may be prepared by the reaction of a compound of formula (XIII)

with a compound of formula (XIV):
RV Il R2 (XiV) O
wherein R1 and R2 are as defined above, under dehydrating conditions. Typically the reaction is catalysed by a Lewis acid (e.g. titanium tetraethoxide). In a preferred procedure, a solution of the compound of formula (XIII) and the compound of formula (XIV) in a suitable solvent (such as tetrahydrofuran) is treated with titanium tetraethoxide at a temperature of about 5O0C.
Compounds of formula (XIII) and (XIV) are either commercially available or easily prepared by standard methods well known to the skilled person, either as a result of common general knowledge (e.g. see 'Comprehensive Organic Transformations' by Richard Larock (1999, VCH Publishers Inc.) or by reference to specific published procedures.
A compound of formula (I), wherein R3 and R4 are both H, may alternatively be prepared by the hydrolysis of an ester of formula (XV):

wherein R1 and R2 are as defined above. The hydrolysis may be carried out under acidic or basic conditions. In a typical procedure, a solution of a compound of formula (XV) in aqueous hydrochloric acid is heated under reflux for 16 hours.
A compound of formula (XV) may be prepared by the methanolysis of a compound of formula (XVI):
(XVI)

A compound of formula (XVI) may be prepared by the alkylation of a compound of formula (XVII):

wherein R1, Y1 and Y2 are as defined above, with a compound of formula R2L1, wherein R2 is as defined above and L1 is a suitable leaving group. L1 is preferably halo (particularly bromo), trifluoromethanesulphonate or methanesulphonate. Typically, the compound of formula (XVII) is deprotonated with a base, such as butyl lithium, in an inert solvent such as diethyl ether or tetrahydrofuran, at low temperature (usually in the range -78 to -2O0C). A solution of the alkylating agent in an inert solvent is then added. In a preferred procedure, a solution of the compound of formula (XVII) in tetrahydrofuran is treated with n-butyl lithium at -780C and an excess of the alkylating agent is then added.
A compound of formula (XVII) may be prepared by the double alkylation of a compound of formula (XVIII):

wherein R1 is as defined above. Typically, the compound of formula (XVIII) is deprotonated using a base (e.g. potassium fert-butoxide, potassium hexamethyldisilazide or sodium hydride) in an inert solvent, such as tetrahydrofuran or diethyl ether. A suitable alkylating agent, such as an alkyl halide (particularly an alkyl bromide) or an alkyl sulphonate ester (e.g. an alkyl mesylate) is then added at a temperature of from -2O0C to room temperature. In a preferred procedure, an excess of trimethoxonium tetrafluoroborate in is used as the alkylating agent.
A compound of formula (XVIII) may be prepared by the cyclisation of a compound of formula (XIX):

wherein R1 is as defined above. In a typical procedure, a solution of a compound of formula (XIX) in a suitable solvent, such as toluene, is heated under reflux.
A compound of formula (XIX) may be prepared by the reduction of a compound of formula (XX):

wherein R1 is as defined above. The reduction is typically accomplished using hydrogen and a hydrogenation catalyst such as a palladium, platinum or rhodium catalyst. In a preferred procedure, a solution of the compound of formula (XX) in a suitable solvent, such as aqueous ethanolic hydrochloric acid, is treated with hydrogen at room temperature.
A compound of formula (XX) may be prepared by coupling an amine of formula (XXI):

with an acid of formula (XXII):

Compounds of formula (XXI) and (XXII) are either commercially available or easily prepared by standard methods well known to the skilled person, either as a result of common general knowledge (e.g. see 'Comprehensive Organic Transformations' by Richard Larock (1999, VCH Publishers Inc.) or by reference to specific published procedures.
Compounds of formula (I) can also be prepared by using the reactions described above to construct a compound wherein R1 or R2 are partially formed and then completing the synthesis by functional group manipulation. For instance, a group may be carried through the synthesis in a protected form and deprotected in a final step. Suitable protecting groups are described in 'Protective Groups in Organic Synthesis' by Theodora Greene and Peter Wuts (third edition, 1999, John Wiley and Sons). Suitable functional group transformations are described in 'Comprehensive Organic Transformations' by Richard Larock (1999, VCH Publishers Inc.).
The following experimental procedures illustrate the preparation of , (2S)-2-aminomethyl-5-ethyl- heptanoic acid, described in US Provisional Patent Application Number 60/733591 (unpublished at the filing date of the present invention):
Diverse methods exist for the preparation of chiral and racemic beta-amino acids. Such methods can be found in "Enantioselective Synthesis of β-Amino Acids", Juaristi, Eusebio; Editor. USA, 1997, Wiley-VCH, New York, N.Y.
The methods described below are illustrative of methods that can be used for the preparation of the compound but are not limiting.
According to a first process, (2S)-2-aminomethyl-5-ethyl-heptanoic acid (the compound of formula (I)) may be prepared from a compound of formula (IV) as shown in scheme 1 , below:

Scheme 1
The compound of formula (II) may be obtained as described in Organic Letters, 2000; 2(22); 3527-3529.
Step (a): The compound of formula (II) is de-protonated using a suitable base, and the resulting anion quenched by addition of the alkyating agent, typically the bromide (Vl), to provide the compound of formula (III). The reaction is typically achieved by the treatment of (II) with a strong base, for example, lithium diisopropylamide (LDA), lithium hexamethyldisilylazide (LHMDS), or sodium hexamethyldisilylazide (NaHMDS), optionally in the presence of an additive, (e.g. lithium chloride (LiCI)) in a suitable solvent (e.g. tetrahydrofuran (THF), ether) at low temperature, for example at a temperature of from -1O0C to O0C for about 1 hour, followed by the quenching of the resulting anion with the alkyl bromide (Vl). In a preferred procedure, a solution of 1 equivalent of the compound of formula (II) in THF is treated with 3.2 equivalents LHMDS and 4 equivalents LiCI, at a temperature of from -50C to O0C for about 1 hour followed by treatment with 1 equivalent of the alkenyl bromide (Vl) at O0C and the reaction is allowed to warm to room temperature over 18 hours.

Step (b): The compound of formula (III) may be reduced to provide the compound of formula (IV). This may be achieved with hydrogen and a suitable catalyst, (for example 10% palladium on charcoal or platinum oxide).
In a preferred procedure, a solution of the amide of formula (III) in ethyl acetate is reduced using 4 atmospheres of H2, in the presence of 10% palladium on charcoal, at room temperature for 72 hours.
Alternatively compound (IV) may be prepared in one step by replacing the alkenyl bromide (Vl) with an alkyl halide (Via) in step (a) as in scheme 1. The alkly bromide may be prepared according to the processes described by Bestmann ef al. (Liebigs Ann. Chem. 1979, 1189-1204) and Pinazzi ef al. (Bull. Soc. Chim., 1975, 1-2, 201-205). The alkyl iodide may be prepared by analogous processes.

X = Br, I
Step (c): The compound of formula (IV) may be hydrolysed to provide the compound of formula (I). This reaction may be achieved under neutral, basic or acidic catalysis, but typically under neutral conditions. The reaction is performed in water, optionally in the presence of a co-solvent (e.g. dioxan, THF) at reflux for about 4 days. In a preferred procedure, a solution of the amide of formula (IV) in waterdioxan (1:1 by volume) is heated at reflux for 112 hours.
Alternatively, the compound of formula (I), may be prepared by changing the order of steps (b) and (c) as shown in Scheme 2.
H

Scheme 2 (D
According to a second process, (2S)-2-aminomethyl-5-ethyl-heptanoic acid (compound (I)), may be prepared from a compound of formula (IX) as shown in scheme 3.

(IX) Scheme 3
Compounds of formula (VII) may be obtained according to the methods of Juaristi et al. Tetrahedron Asymmetry, 1996, 2233-46.
Step (T): The compound of formula (VII) is treated with a suitable base, and the resulting anion is quenched by addition of an alkylating agent of formula (Via) to provide the compound of formula (VIII). The reaction may be achieved by treatment of the compound of formula (VII) with a strong base, for example, LDA, LHMDS or NaHMDS, in a suitable solvent (e.g. THF, ether) at a temperature of from -780C to -6O0C, for example -780C, for about 1 hour, followed by quenching of the resulting anion with the alkyl bromide, at a temperature of from -780C to about room temperature.
Step (g): The compound of formula (IX) may be prepared by treatment of the compound of formula (VIII) with a suitable base, and quenching of the resulting anion with aqueous acid. The reaction may be achieved by treatment of the compound of formula (VIII) with a strong base, for example, LDA, LHMDS or NaHMDS, in a suitable solvent (e.g. THF, ether) at very low temperature, for example at a temperature of from about -78° to about -6O0C for about 3 hours, followed by quenching of the resulting anion with aqueous acid.
Step (h): Hydrolysis of the compound of formula (IX) provides the compound of formula (I). This reaction may be achieved under basic or acidic catalysis, but is typically carried out under aqueous acidic conditions (e.g. hydrochloric acid (HCI) or sulphuric acid (H2SO4)), optionally in the presence of a suitable solvent (e.g. THF) at reflux for about 24 hours.
According to a third process, the compound of formula (I) may be prepared from a compound of formula (XIII) according to scheme 4.

Scheme 4
Steps (f) and (h) are as described above for scheme 3.
According to a fourth process, the compound of formula (I) may be prepared from a compound of formula (XVII) by the method outlined in scheme 5.

(XVII) (I)
Scheme 5
R is a suitable protecting group such as (CrCβJalkyl.
Step (i): The aldehyde (XIV) may undergo Knoevenagel condensation with a cyanoacetate derivative to give the compound of formula (XV) in an analogous manner to that described in Journal of Organic Chemistry, 1961 , 26, 2738-2740. The aldehyde (XIV) is described in Tetrahedron 1988, 44(4)1091-1106.
Step (j): Reduction of a compound of formula (XV) to a compound of formula (XVI) may be carried out by hydrogenation in a suitable solvent, typically ethanol, in the presence of a metal catalyst (e.g. platinum oxide, palladium on charcoal).
Step (k): Resolution of a compound of formula (XVI) may be achieved by formation of a chiral salt (e.g. the (+) di-o-tolyl tartrate or (L)-dibenzoyl tartrate salt) and recrystallisation, followed by reformation of the free amine.
Step (m): The compound of formula (I) may be prepared by hydrolysis of a compound of formula (XVII) under acidic or basic conditions, typically using HCI, in dioxan, at a temperature of 8O0C, for about 18 hours.
Compound (I) may alternatively be prepared by stereospecific hydrolysis of compound (XVI) with an enzyme (e.g. pig liver esterase, lipase).
According to a fifth process, the compound of formula (I) may be prepared from a compound of formula (XVII) according to scheme 6.

(XVII) (I)
Scheme 6
Step (n): The alkenyl bromide (Vl), as exemplified in Preparations 1 and 2, may be used to alkylate a cyanoacetate derivative in the presence of a base (e.g. potassium carbonate) and a solvent (e.g. dimethylforamide) to give a compound of formula (XVIII).
Step (o): Reduction of a compound of formula (XVIII) to a compound of formula (XVI) may be carried out by hydrogenation in a suitable solvent, typically ethanol, in the presence of a metal catalyst (e.g. platinum oxide or palladium on charcoal).
Steps (k) and (m) are as described above for scheme 5.
The compound of formula (I) may also prepared by analogy with the methods described in WO-A- 2003/082807 and references therein, and also by analogy with the method of Lavielle et al in European Journal of Organic Chemistry, 2000(1), 83-89.
The following experimental procedures illustrate the preparation of certain preferred PDE7 inhibitors described in US Provisional Patent Application Number 60/741854 (unpublished at the filing date of the present invention):
The following abbreviations are used:
DMF = dimethylformamide DMSO = dimethyl sulphoxide TEMPO = 2,2,6,6-tetramethylpiperidine-N-oxide THF = tetrahydrofuran
The compounds of formula (I) may be prepared as shown in Scheme 1 below.


(V)
(I)
Scheme 1
In Scheme 1 , P represents a hydroxy-protecting group, suitable examples of which are described in "Protective Groups in Organic Synthesis" by T. W. Greene and P. Wuts, Wiley and Sons, 1991 , and LG represents a suitable leaving group, such as halogen or sulphonate (eg methanesulphonate, p-toluenesulphonate or trifluoromethanesulphonate). Preferably P is benzyl and LG is p-toluenesulphonate.
Step (a): The compound of formula (III) may be prepared from compound (II) and an appropriate agent capable of converting a hydroxy group into a leaving group, typically a sulphonylating reagent (eg methanesulphonyl chloride or p-toluenesulphonyl chloride) in the presence of a base (eg triethylamine or pyridine) in a suitable solvent (eg pyridine or dichloromethane) at O0C to room temperature for 15 minutes to 24 hours. Preferred conditions are: 1eq compound (II) in dichloromethane, 1.2eq p-toluenesulphonyl chloride, 2 eq pyridine at room temperature for 18 hours.
Step (b): The compound of formula (IV) may be prepared from compound (III) and the hydroxy compound of formula (Vl) in a suitable solvent (eg DMF, DMSO) in the presence of a suitable base (eg Cs2CO3, K2CO3), optionally in the presence of a crown ether (eg 18-crown-6) at 50- 12O0C overnight.
Preferred conditions are: 1eq compound (Vl), 1.1eq compound (III), 1.2 eq Cs2CO3, in DMF at 8O0C for 24 hours.
Compounds of formula (Vl) are generally described in WO 02/074754. Specific compounds of formula (Vl) wherein X is O, m is 1 and R is Cl may be prepared as described in Bioorg. Med. Chem. Lett., (2004), 14 (18), 4627-32.
Step (c): The compound of formula (IV) may be deprotected by reaction with a deprotecting agent in a suitable solvent to yield the compound of formula (V). Suitable reagents and methods are described in "Protective Groups in Organic Synthesis" (referred to above). When P is benzyl, examples of suitable reagents include boron trichloride or iron (III) chloride. Preferred conditions are: 1eq compound (IV) in dichloromethane, 4 eq BCI3 at room temperature for 18 hours.
Step (d): The compound of formula (I) may be prepared by oxidation of the compound of formula (V) using an oxidising agent in a suitable solvent. Typical reagents and conditions include catalytic chromium trioxide and periodic acid (H5IO6) in a solvent such as acetonitrile at room temperature to 5O0C for 18 to 36 hours, or alternatively NaOCI plus NaCIO2 in the presence of catalytic TEMPO in a solvent such as acetonitrile at O0C to room temperature for 18 to 36 hours.
Preferred conditions are: 1eq compound (V), 2.5 eq periodic acid, 0.02 eq CrO3, in 0.75% aqueous acetonitrile, 24 hours at 4O0C.
The compounds of formula (I) may alternatively be prepared by oxidation of compounds of formula (V) in a two-step procedure via the aldehydes of formula (VII) as shown in Scheme 2.

Step (a): Oxidation of the alcohol (V) to the aldehyde (VII) is typically carried out using NaOCI with catalytic TEMPO in a suitable solvent, eg acetonitrile, acetone at O0C to room temperature for 2- 18 hours, or alternatively using sulphur trioxide- pyridine complex with DMSO in a solvent such as THF at O0C to room temperature for 2-18 hours.
Step (b): Further oxidation of the aldehyde (VII) to the acid (I) with is typically carried out using NaCIO2 in the presence of potassium phosphate in a solvent such as aqueous t-butanol at O0C to room temperature for 2-18 hours, or alternatively using trichloroisocyanuric acid with catalytic TEMPO in a suitable solvent, eg acetone or acetonitrile, at O0C to room temperature for 2-18 hours.
Compounds of formula (II) are known in the literature. For example, compounds of formula (II) wherein A is a c/s-1 ,3-cyclobutylene group and B is a single bond may be prepared as described in J. Chem. Soc, Perkin Trans. 1, (1995), 18, 2281-7.
Alternatively compounds of formula (Ib), which are compounds of formula (I) wherein A is a cis- or frans-1 ,3-cyclobutylene group and B is a single bond may be prepared from compound (VIII) or compound (IX) by standard methods, such as shown in Scheme 3.
Trans compounds (II) and (X) may be obtained from cis compounds (II) and (X) respectively by inversion using Mitsunobu chemistry analogous to that described in Synthesis, (1981), 1.

(Ib)
Scheme 3
In Scheme 3, Ra is an ester residue, suitable examples of which are described in "Protective Groups in Organic Synthesis" (referred to above) (eg (C^alkyl, benzyl or (+) or (-)-menthyl), and LG is a leaving group such as halogen or sulphonate (eg methanesulphonate, p- toluenesulphonate or trifluoromethanesulphonate).
Step (a): The compound of formula (IX) may be prepared by reaction of compound (VIII) with a suitable alcohol of formula R3OH (eg methanol, t-butanol, (-) menthol) under a variety of conditions, suitable examples of which are described in "Protective Groups in Organic Synthesis"
(referred to above). Preferred conditions are: 1eq compound (VIII), 1.1 eq. 1 ,1'-carbonyl diimidazole, in ethyl acetate at reflux for 1 hour followed by 1eq R3OH at room temperature for 4 hours.
Step (b): Reduction of compound (IX) to the alcohol (X) may be carried out using a suitable reducing agent, eg sodium borohydride or L-Selectride®, in a suitable solvent such as THF. Preferred conditions are: 1eq compound (IX), 0.5 eq NaBH4 in 20:1 THF:methanol at O0C for 20 minutes.
Step (c): The compound of formula (Xl) may be prepared from compound (X) using reagents and conditions similar to those described in Scheme 1 , step (a). Preferred conditions are: 1eq compound (X)1 1.05 eq p-toluenesulphonyl chloride in pyridine at O0C to room temperature.
Step (d): The compound of formula (Ia) may be prepared from compound (Xl) and the hydroxy compound of formula (Vl) using reagents and conditions similar to those described in Scheme 1, step (b).
Preferred conditions are: 1.2 eq compound (Xl), 1.0 eq compound (Vl), 1.5 eq Cs2CO3 in DMF at 8O0C for 18 hours.
Step (e): The compound of formula (Ia) may be hydrolysed to provide the compound of formula (Ib). This reaction may be achieved under a variety of conditions, suitable examples of which are described in "Protective Groups in Organic Synthesis" (referred to above). Preferred conditions are: compound (Ia), 2 eq NaOH in 1 :1 ethanol:water at 6O0C for 2 hours.
Compound (VIII) is described in J. Org. Chem., (1981), 53, 3841-43 and compound (IX) is described in J. Org. Chem., (1994), 59, 2132-34.

(VIM) (IX)
The components of the combination of the invention should be assessed for their biopharmaceutical properties, such as solubility and solution stability (across pH), permeability,
efc, in order to select the most appropriate dosage form and route of administration for treatment of the proposed indication.
The components of the combiantion of the invention intended for pharmaceutical use may be administered as crystalline or amorphous products. They may be obtained, for example, as solid plugs, powders, or films by methods such as precipitation, crystallization, freeze drying, or spray drying, or evaporative drying. Microwave or radio frequency drying may be used for this purpose.
They may be administered alone or in combination with one or more other drugs (or as any combination thereof). Generally, they will be administered as a formulation in association with one or more pharmaceutically acceptable excipients. The term 'excipient' is used herein to describe any ingredient other than the components of the combination of the invention. The choice of excipient will to a large extent depend on factors such as the particular mode of administration, the effect of the excipient on solubility and stability, and the nature of the dosage form.
Pharmaceutical compositions suitable for the delivery of combinations of the present invention and methods for their preparation will be readily apparent to those skilled in the art. Such compositions and methods for their preparation may be found, for example, in Remington's Pharmaceutical Sciences, 19th Edition (Mack Publishing Company, 1995).
ORAL ADMINISTRATION
The components of the combination of the invention may be administered orally. Oral administration may involve swallowing, so that the compound enters the gastrointestinal tract, and/or buccal, lingual, or sublingual administration by which the compound enters the blood stream directly from the mouth.
Formulations suitable for oral administration include solid, semi-solid and liquid systems such as tablets; soft or hard capsules containing multi- or nano-particulates, liquids, or powders; lozenges (including liquid-filled); chews; gels; fast dispersing dosage forms; films; ovules; sprays; and and buccal/mucoadhesive patches.
Liquid formulations include suspensions, solutions, syrups and elixirs. Such formulations may be employed as fillers in soft or hard capsules (made, for example, from gelatin or hydroxypropylmethylcellulose) and typically comprise a carrier, for example, water, ethanol, polyethylene glycol, propylene glycol, methylcellulose, or a suitable oil, and one or more emulsifying agents and/or suspending agents. Liquid formulations may also be prepared by the reconstitution of a solid, for example, from a sachet.
The components of the combination of the invention may also be used in fast-dissolving, fast- disintegrating dosage forms such as those described in Expert Opinion in Therapeutic Patents, H (6), 981-986, by Liang and Chen (2001).
For tablet dosage forms, depending on dose, the drug may make up from 1 weight % to 80 weight % of the dosage form, more typically from 5 weight % to 60 weight % of the dosage form. In addition to the drug, tablets generally contain a disintegrant. Examples of disintegrants include sodium starch glycolate, sodium carboxymethyl cellulose, calcium carboxymethyl cellulose, croscarmellose sodium, crospovidone, polyvinylpyrrolidone, methyl cellulose, microcrystalline cellulose, lower alkyl-substituted hydroxypropyl cellulose, starch, pregelatinised starch and sodium alginate. Generally, the disintegrant will comprise from 1 weight % to 25 weight %, preferably from 5 weight % to 20 weight % of the dosage form.
Binders are generally used to impart cohesive qualities to a tablet formulation. Suitable binders include microcrystalline cellulose, gelatin, sugars, polyethylene glycol, natural and synthetic gums, polyvinylpyrrolidone, pregelatinised starch, hydroxypropyl cellulose and hydroxypropyl methylcellulose. Tablets may also contain diluents, such as lactose (monohydrate, spray-dried monohydrate, anhydrous and the like), mannitol, xylitol, dextrose, sucrose, sorbitol, microcrystalline cellulose, starch and dibasic calcium phosphate dihydrate.
Tablets may also optionally comprise surface active agents, such as sodium lauryl sulfate and polysorbate 80, and glidants such as silicon dioxide and talc. When present, surface active agents may comprise from 0.2 weight % to 5 weight % of the tablet, and glidants may comprise from 0.2 weight % to 1 weight % of the tablet.
Tablets also generally contain lubricants such as magnesium stearate, calcium stearate, zinc stearate, sodium stearyl fumarate, and mixtures of magnesium stearate with sodium lauryl sulphate. Lubricants generally comprise from 0.25 weight % to 10 weight %, preferably from 0.5 weight % to 3 weight % of the tablet.
Other possible ingredients include anti-oxidants, colourants, flavouring agents, preservatives and taste-masking agents.
Exemplary tablets contain up to about 80% drug, from about 10 weight % to about 90 weight % binder, from about 0 weight % to about 85 weight % diluent, from about 2 weight % to about 10 weight % disintegrant, and from about 0.25 weight % to about 10 weight % lubricant.
Tablet blends may be compressed directly or by roller to form tablets. Tablet blends or portions of blends may alternatively be wet-, dry-, or melt-granulated, melt congealed, or extruded before tabletting. The final formulation may comprise one or more layers and may be coated or uncoated; it may even be encapsulated.
The formulation of tablets is discussed in Pharmaceutical Dosage Forms: Tablets. Vol. 1, by H. Lieberman and L. Lachman (Marcel Dekker, New York, 1980).
Consumable oral films for human or veterinary use are typically pliable water-soluble or water- swellable thin film dosage forms which may be rapidly dissolving or mucoadhesive and typically comprise a compound of formula I1 a film-forming polymer, a binder, a solvent, a humectant, a plasticiser, a stabiliser or emulsifier, a viscosity-modifying agent and a solvent. Some components of the formulation may perform more than one function.
The components of the combination may be water-soluble or insoluble. A water-soluble compound typically comprises from 1 weight % to 80 weight %, more typically from 20 weight % to 50 weight %, of the solutes. Less soluble compounds may comprise a greater proportion of the composition, typically up to 88 weight % of the solutes. Alternatively, the components of the combination may be in the form of multiparticulate beads.
The film-forming polymer may be selected from natural polysaccharides, proteins, or synthetic hydrocolloids and is typically present in the range 0.01 to 99 weight %, more typically in the range 30 to 80 weight %.
Other possible ingredients include anti-oxidants, colorants, flavourings and flavour enhancers, preservatives, salivary stimulating agents, cooling agents, co-solvents (including oils), emollients, bulking agents, anti-foaming agents, surfactants and taste-masking agents.
Films in accordance with the invention are typically prepared by evaporative drying of thin aqueous films coated onto a peelable backing support or paper. This may be done in a drying oven or tunnel, typically a combined coater dryer, or by freeze-drying or vacuuming.
Solid formulations for oral administration may be formulated to be immediate and/or modified controlled release. Modified release formulations include delayed-, sustained-, pulsed-, controlled- , targeted and programmed release.
Suitable modified release formulations for the purposes of the invention are described in US Patent No. 6,106,864. Details of other suitable release technologies such as high energy dispersions and osmotic and coated particles are to be found in Pharmaceutical Technology Online. 25(2), 1-14, by Verma ef a/ (2001). The use of chewing gum to achieve controlled release is described in WO 00/35298.
PARENTERAL ADMINISTRATION
The components of the combination of the invention may also be administered directly into the blood stream, into muscle, or into an internal organ. Suitable means for parenteral administration include .intravenous, intraarterial, intraperitoneal, intrathecal, intraventricular, intraurethral, intrasternal, intracranial, intramuscular, intrasynovial and subcutaneous. Suitable devices for parenteral administration include needle (including microneedle) injectors, needle-free injectors and infusion techniques.
Parenteral formulations are typically aqueous solutions which may contain excipients such as salts, carbohydrates and buffering agents (preferably, to a pH of from 3 to 9), but, for some applications, they may be more suitably formulated as a sterile non-aqueous solution or as powdered a dried form to be used in conjunction with a suitable vehicle such as sterile, pyrogen- free water.
The preparation of parenteral formulations under sterile conditions, for example, by lyophilisation, may readily be accomplished using standard pharmaceutical techniques well known to those skilled in the art.
The solubility of the components of the combination used in the preparation of parenteral solutions may be increased by the use of appropriate formulation techniques, such as the incorporation of solubility-enhancing agents. Formulations for use with needle-free injection administration comprise a compound of the invention in powdered form in conjunction with a suitable vehicle such as sterile, pyogen-free water.
Formulations for parenteral administration may be formulated to be immediate and/or modified controlled release. Modified release formulations include delayed-, sustained-, pulsed-, controlled- , targeted and programmed release. Thus the components of the combination of the invention may be formulated as a suspension or as a solid, semi-solid, or thixotropic liquid for administration as an implanted depot providing modified release of the active compound. Examples of such formulations include drug-coated stents and semi-solids and suspensions comprising drug-loaded poly(d/-!actic-coglycolic)acid (PGLA) microspheres.
TOPICAL ADMINISTRATION
The components of the combination of the invention may also be administered topically, (intra)dermally, or transdermal^ to the skin or mucosa. Typical formulations for this purpose include gels, hydrogels, lotions, solutions, creams, ointments, dusting powders, dressings, foams, films, skin patches, wafers, implants, sponges, fibres, bandages and microemulsions. Liposomes may also be used. Typical carriers include alcohol, water, mineral oil, liquid petrolatum, white petrolatum, glycerin, polyethylene glycol and propylene glycol. Penetration enhancers may be incorporated - see, for example, J Pharm Sci, 88 (10), 955-958, by Finnin and Morgan (October 1999).
Other means of topical administration include delivery by electroporation, iontophoresis, phonophoresis, sonophoresis and microneedle or needle-free (e.g. Powderject™, Bioject™, ete.) injection.Topical administration may also be achieved using a patch, such as a transdernal iontophoretic patch.
Formulations for topical administration may be formulated to be immediate and/or modified controlled release.. Controlled release formulations include Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
INHALED/INTRANASAL ADMINISTRATION
The components of the combination of the invention can also be administered intranasally or by inhalation, typically in the form of a dry powder (either alone, as a mixture, for example, in a dry blend with lactose, or as a mixed component particle, for example, mixed with phospholipids, such as phosphatidylcholine) from a dry powder inhaler, as an aerosol spray from a pressurised container, pump, spray, atomiser (preferably an atomiser using electrohydrodynamics to produce a fine mist), or nebuliser, with or without the use of a suitable propellant, such as 1 , 1 , 1 ,2- tetrafluoroethane or 1 ,1 ,1 ,2,3,3,3-heptafluoropropane, or as nasal drops. For intranasal use, the powder may comprise a bioadhesive agent, for example, chitosan or cyclodextrin.
The pressurised container, pump, spray, atomizer, or nebuliser contains a solution or suspension of the components of the combination of the invention comprising, for example, ethanol, aqueous ethanol, or a suitable alternative agent for dispersing, solubilising, or extending release of the active, a propellant(s) as solvent and an optional surfactant, such as sorbitan trioleate, oleic acid, or an oligolactic acid.
Prior to use in a dry powder or suspension formulation, the drug product is micronised to a size suitable for delivery by inhalation (typically less than 5 microns). This may be achieved by any appropriate comminuting method, such as spiral jet milling, fluid bed jet milling, supercritical fluid processing to form nanoparticles, high pressure homogenisation, or spray drying.
Capsules (made, for example, from gelatin or hydroxypropylmethylcellulose), blisters and cartridges for use in an inhaler or insufflator may be formulated to contain a powder mix of the compound of the invention, a suitable powder base such as lactose or starch and a performance modifier such as /-leucine, mannitol, or magnesium stearate. The lactose may be anhydrous or in the form of the monohydrate, preferably the latter. Other suitable excipients include dextran, glucose, maltose, sorbitol, xylitol, fructose, sucrose and trehalose.
A suitable solution formulation for use in an atomiser using electrohydrodynamics to produce a fine mist may contain from 1 μg to 20mg of the compound of the invention per actuation and the
actuation volume may vary from 1μl to 100μl. A typical formulation may comprise a compound of formula I1 propylene glycol, sterile water, ethanol and sodium chloride. Alternative solvents which may be used instead of propylene glycol include glycerol and polyethylene glycol.
Suitable flavours, such as menthol and levomenthol, or sweeteners, such as saccharin or saccharin sodium, may be added to those formulations of the invention intended for inhaled/intranasal administration.
Formulations for inhaled/intranasal administration may be formulated to be immediate and/or modified controlled release using, for example, PGLA.. Controlled release formulations include Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
In the case of dry powder inhalers and aerosols, the dosage unit is determined by means of a valve which delivers a metered amount. Units in accordance with the invention are typically arranged to administer a metered dose. The overall daily dose may be administered in a single dose or, more usually, as divided doses throughout the day.
RECTAL/INTRAVAGINAL ADMINISTRATION
The compounds of the invention may be administered rectally or vaginally, for example, in the form of a suppository, pessary, or enema. Cocoa butter is a traditional suppository base, but various alternatives may be used as appropriate.
Formulations for rectal/vaginal administration may be formulated to be immediate and/or modified controlled release. Controlled release formulations include Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted and programmed release.
OCULAR/AURAL ADMINISTRATION
The components of the combination of the invention may also be administered directly to the eye or ear, typically in the form of drops of a micronised suspension or solution in isotonic, pH- adjusted, sterile saline. Other formulations suitable for ocular and aural administration include ointments, gels, biodegradable (e.g. absorbable gel sponges, collagen) and non-biodegradable (e.g. silicone) implants, wafers, lenses and particulate or vesicular systems, such as niosomes or liposomes. A polymer such as crossed-linked polyacrylic acid, polyvinylalcohol, hyaluronic acid, a cellulosic polymer, for example, hydroxypropylmethylcellulose, hydroxyethylcellulose, or methyl cellulose, or a heteropolysaccharide polymer, for example, gelan gum, may be incorporated together with a preservative, such as benzalkonium chloride. Such formulations may also be delivered by iontophoresis.
Formulations for ocular/aural administration may be formulated to be immediate and/or modified controlled release. Controlled release formulations include Modified release formulations include delayed-, sustained-, pulsed-, controlled-, targeted, or programmed release.
OTHER TECHNOLOGIES
The components of the combination of the invention may be combined with soluble macromolecular entities, such as cyclodextrin and suitable derivatives thereof or polyethylene glycol-containing polymers, in order to improve their solubility, dissolution rate, taste-masking, bioavailability and/or stability for use in any of the aforementioned modes of administration.
Drug-cyclodextrin complexes, for example, are found to be generally useful for most dosage forms and administration routes. Both inclusion and non-inclusion complexes may be used. As an alternative to direct complexation with the drug, the cyclodextrin may be used as an auxiliary additive, i.e. as a carrier, diluent, or solubiliser. Most commonly used for these purposes are alpha-, beta- and gamma-cyclodextrins, examples of which may be found in International Patent Applications Nos. WO 91/11172, WO 94/02518 and WO 98/55148.
DOSAGE
For administration to human patients, the total daily dose of the components of the combination of the invention depends on the mode of administration. The total daily dose may be administered in single or divided doses and may, at the physician's discretion, fall outside of the typical range given herein.
These dosages are based on an average human subject having a weight of about 60kg to 70kg. The physician will readily be able to determine doses for subjects whose weight falls outside this range, such as infants and the elderly.
For the avoidance of doubt, references herein to "treatment" include references to curative, palliative and prophylactic treatment.
Thus, as a further aspect of the present invention, there is provided a pharmaceutical composition comprising an alpha-2-delta ligand, a PDE7 inhibitor, or pharmaceutically acceptable salts thereof, and one or more suitable excipients. The composition is suitable for use in the treatment of pain, particularly neuropathic pain.
As an alternative aspect of the present invention, there is provided a pharmaceutical composition comprising a synergistic combination comprising an alpha-2-delta ligand, a PDE7 inhibitor, or pharmaceutically acceptable salts thereof, and one or more suitable excipients. The composition is suitable for use in the treatment of pain, particularly neuropathic pain.
The combination of the present invention may be usefully combined with another pharmacologically active compound, or with two or more other pharmacologically active compounds, particularly in the treatment of pain. For example, the combination of the present invention, or pharmaceutically acceptable salts or solvates thereof, as defined above, may be administered simultaneously, sequentially or separately in combination with one or more agents selected from:
• an opioid analgesic, e.g. morphine, heroin, hydromorphone, oxymorphone, levorphanol, levallorphan, methadone, meperidine, fentanyl, cocaine, codeine, dihydrocodeine, oxycodone, hydrocodone, propoxyphene, nalmefene, nalorphine, naloxone, naltrexone, buprenorphine, butorphanol, nalbuphine or pentazocine;
• a nonsteroidal antiinflammatory drug (NSAID), e.g. aspirin, diclofenac, diflusinal, etodolac, fenbufen, fenoprofen, flufenisal, flurbiprofen, ibuprofen, indomethacin, ketoprofen, ketorolac, meclofenamic acid, mefenamic acid, meloxicam, nabumetone, naproxen, nimesulide, nitroflurbiprofen, olsalazine, oxaprozin, phenylbutazone, piroxicam, sulfasalazine, sulindac, tolmetin or zomepirac;
• a barbiturate sedative, e.g. amobarbital, aprobarbital, butabarbital, butabital, mephobarbital, metharbital, methohexital, pentobarbital, phenobartital, secobarbital, talbutal, theamylal or thiopental;
• a benzodiazepine having a sedative action, e.g. chlordiazepoxide, clorazepate, diazepam, flurazepam, lorazepam, oxazepam, temazepam or triazolam;
• an Hi antagonist having a sedative action, e.g. diphenhydramine, pyrilamine, promethazine, chlorpheniramine or chlorcyclizine; • a sedative such as glutethimide, meprobamate, methaqualone or dichloralphenazone;
• a skeletal muscle relaxant, e.g. baclofen, carisoprodol, chlorzoxazone, cyclobenzaprine, methocarbamol or orphrenadine;
• an NMDA receptor antagonist, e.g. dextromethorphan ((+)-3-hydroxy-N- methylmorphinan) or its metabolite dextrorphan ((+)-3-hydroxy-N-methylmorphinan), ketamine, memantine, pyrroloquinoline quinine, cis-4-(phosphonomethyl)-2- piperidinecarboxylic acid, budipine, EN-3231 (MorphiDex®, a combination formulation of morphine and dextromethorphan), topiramate, neramexane or perzinfotel including an NR2B antagonist, e.g. ifenprodil, traxoprodil or (-)-(R)-6-{2-[4-(3-fluorophenyl)-4-hydroxy- 1-piperidinyl]-1-hydroxyethyl-3,4-dihydro-2(1 H)-quinolinone;
• an alpha-adrenergic, e.g. doxazosin, tamsulosin, clonidine, guanfacine, dexmetatomidine, modafinil, or 4-amino-6,7-dimethoxy-2-(5-methane-sulfonamido- 1 ,2,3,4-tetrahydroisoquinol-2-yl)-5-(2-pyridyl) quinazoline;
• a tricyclic antidepressant, e.g. desipramine, imipramine, amitriptyline or nortriptyline;
• an anticonvulsant, e.g. carbamazepine, lamotrigine, topiratmate or valproate;
• a tachykinin (NK) antagonist, particularly an NK-3, NK-2 or NK-1 antagonist, e.g. (αR,9R)-7-[3, 5-bis(trifluoromethyl)benzyl]-8,9, 10,11 -tetrahydro-9-methyl-5-(4- methylphenyl)-7H-[1 ,4]diazocino[2, 1 -g][1 ,7]-naphthyridine-6-13-dione (TAK-637), 5- [[(2R,3S)-2-[( 1 R)-1 -[3,5-bis(trifluoromethyl)phenyl]ethoxy-3-(4-fluorophenyl)-4- morpholinyl]-methyl]-1 ,2-dihydro-3H-1 ,2,4-triazol-3-one (MK-869), aprepitant, lanepitant, dapitant or 3-[[2-methoxy-5-(trifluoromethoxy)phenyl]-methylamino]-2-phenylpiperidine
(2S.3S);
• a muscarinic antagonist, e.g oxybutynin, tolterodine, propiverine, tropsium chloride, darifenacin, solifenacin, temiverine and ipratropium;
• a COX-2 selective inhibitor, e.g. celecoxib, rofecoxib, parecoxib, valdecoxib, deracoxib, etoricoxib, or lumiracoxib;
• a coal-tar analgesic, in particular paracetamol;
• a neuroleptic such as droperidol, chlorpromazine, haloperidol, perphenazine, thioridazine, mesoridazine, trifluoperazine, fluphenazine, clozapine, olanzapine, risperidone, ziprasidone, quetiapine, sertindole, aripiprazole, sonepiprazole, blonanserin, iloperidone, perospirone, raclopride, zotepine, bifeprunox, asenapine, lurasidone, amisulpride, balaperidone, palindore, eplivanserin, osanetant, rimonabant, meclinertant, Miraxion® or sarizotan; • a vanilloid receptor agonist (e.g. resinferatoxin) or antagonist (e.g. capsazepine);
• a beta-adrenergic such as propranolol;
• a local anaesthetic such as mexiletine;
• a corticosteroid such as dexamethasone;
• a 5-HT receptor agonist or antagonist, particularly a 5-HT1B/ID agonist such as eletriptan, sumatriptan, naratriptan, zolmitriptan or rizatriptan;
• a 5-HT2A receptor antagonist such as R(+)-alpha-(2,3-dimethoxy-phenyl)-1-[2-(4- fluorophenylethyl)]-4-piperidinemethanol (MDL-100907);
• a cholinergic (nicotinic) analgesic, such as ispronicline (TC-1734), (E)-N-methyl- 4-(3-pyridinyl)-3-buten-1 -amine (RJR-2403), (R)-5-(2-azetidinylmethoxy)-2-chloropyridine (ABT-594) or nicotine;
• Tramadol®;
• a PDEV inhibitor, such as 5-[2-ethoxy-5-(4-methyl-1-piperazinyl- sulphonyl)phenyl]-1-methyl-3-n-propyl-1,6-dihydro-7H-pyrazolo[4,3-d]pyrimidin-7-one (sildenafil), (6R,12aR)-2,3,6,7,12,12a-hexahydro-2-methyl-6-(3,4-methylenedioxyphenyl)- pyrazino[2',1':6,1]-pyrido[3,4-b]indole-1 ,4-dione (IC-351 or tadalafil), 2-[2-ethoxy-5-(4- ethyl-piperazin-1 -yl-1 -sulphonyl)-phenyl]-5-methyl-7-propyl-3H-imidazo[5, 1 -f][1 ,2,4]triazin- 4-one (vardenafil), 5-(5-acetyl-2-butoxy-3-pyridinyl)-3-ethyl-2-(1-ethyl-3-azetidinyl)-2,6- dihydro-7/-/-pyrazolo[4,3-d]pyrimidin-7-one, 5-(5-acetyl-2-propoxy-3-pyridinyl)-3-ethyl-2- (1-isopropyl-3-azetidinyl)-2,6-dihydro-7H-pyrazolo[4,3-c/]pyrimidin-7-one, 5-[2-ethoxy-5-(4- ethylpiperazin-1-ylsulphonyl)pyridin-3-yl]-3-ethyl-2-[2-methoxyethyl]-2,6-dihydro-7H-
pyrazolo[4,3-d]pyrimidin-7-one, 4-[(3-chloro-4-methoxybenzyl)amino]-2-[(2S)-2- (hydroxymethyl)pyrrolidin-1-yl]-N-(pyrimidin-2-ylmethyl)pyrimidine-5-carboxamide, 3-(1- methyl-7-oxo-3-propyl-6,7-dihydro-1 H-pyrazolo[4,3-d]pyrimidin-5-yl)-N-[2-(1- methylpyrrolidin-2-yl)ethyl]-4-propoxybenzenesulfonamide; • a cannabiπoid;
• metabotropic glutamate subtype 1 receptor (mGluRI) antagonist;
• a serotonin reuptake inhibitor such as sertraline, sertraline metabolite demethylsertraline, fluoxetine, norfluoxetine (fluoxetine desmethyl metabolite), fluvoxamine, paroxetine, citalopram, citalopram metabolite desmethylcitalopram, escitalopram, d,l-fenfluramine, femoxetine, ifoxetine, cyanodothiepin, litoxetine, dapoxetine, nefazodone, cericlamine and trazodone;
• a noradrenaline (norepinephrine) reuptake inhibitor, such as maprotiline, lofepramine, mirtazepine, oxaprotiline, fezolamine, tomoxetine, mianserin, buproprion, bupropion metabolite hydroxybuproprion, nomifensine and viloxazine (Vivalan®), especially a selective noradrenaline reuptake inhibitor such as reboxetine, in particular
(S.S)-reboxetine;
• a dual serotonin-noradrenaline reuptake inhibitor, such as venlafaxine, venlafaxine metabolite O-desmethylvenlafaxine, clomipramine, clomipramine metabolite desmethylclomipramine, duloxetine, milnacipran and imipramine; • an inducible nitric oxide synthase (iNOS) inhibitor such as S-[2-[(1- iminoethyl)amino]ethyl]-L-homocysteine, S-[2-[(1-iminoethyl)-amino]ethyl]-4,4-dioxo-L- cysteine, S-[2-[(1-iminoethyl)amino]ethyl]-2-methyl-L-cysteine, (2S,5Z)-2-amino-2-methyl- 7-[(1-iminoethyl)amino]-5-heptenoic acid, 2-[[(1 R,3S)-3-amino-4- hydroxy-1-(5-thiazolyl)- butyl]thio]-5-chloro-3-pyridinecarbonitrile; 2-[[(1 R,3S)-3-amino-4-hydroxy-1 -(5- thiazolyl)butyl]thio]-4-chlorobenzonitrile, (2S,4R)-2-amino-4-[[2-chloro-5-
(trifluoromethyl)phenyl]thio]-5-thiazolebutanol,
2-[[(1 R,3S)-3-amino-4-hydroxy-1-(5-thiazolyl) butyl]thio]-6-(trifluoromethyl)-3 pyridinecarbonitrile, 2-[[(1 R,3S)-3- amino-4-hydroxy- 1 -(5-thiazolyl)butyl]thio]-5- chlorobenzonitrile, N-[4-[2-(3-chlorobenzylamino)ethyl]phenyl]thiophene-2-carboxamidine, or guanidinoethyldisulfide;
• an acetylcholinesterase inhibitor such as donepezil;
• a prostaglandin E2 subtype 4 (EP4) antagonist such as Λ/-[({2-[4-(2-ethyl-4,6- dimethyl-I H-imidazo^.δ-clpyridin-i-yOphenylJethylJaminoJ-carbonylH- methylbenzenesulfonamide or 4-[(1 S)-1-({[5-chloro-2-(3-fluorophenoxy)pyridin-3- yl]carbonyl}amino)ethyl]benzoic acid;
• a leukotriene B4 antagonist; such as 1-(3-biphenyl-4-ylmethyl-4-hydroxy- chroman-7-yl)-cyclopentanecarboxylic acid (CP-105696), 5-[2-(2-Carboxyethyl)-3-[6-(4- methoxyphenyl)-5E- hexenyl]oxyphenoxy]-valeric acid (ONO-4057) or DPC-11870,
• a 5-lipoxygenase inhibitor, such as zileuton, 6-[(3-fluoro-5-[4-methoxy-3,4,5,6- tetrahydro-2H-pyran-4-yl])phenoxy-methyl]-1-methyl-2-quinolone (ZD-2138), or 2,3,5- trimethyl-6-(3-pyridylmethyl),1 ,4-benzoquinone (CV-6504);
• a sodium channel blocker, such as lidocaine; • a 5-HT3 antagonist, such as ondansetron;
and the pharmaceutically acceptable salts and solvates thereof.
The present invention extends to a product comprising an alpha-2-delta ligand, an PDE7 inhibitor and one or more other therapeutic agents, such as those listed above, for simultaneous, separate or sequential use in the curative, prophylactic treatment of pain, particularly inflammatory, neuropathic, visceral or nociceptive pain.
KIT-OF-PARTS
Inasmuch as it may desirable to administer a combination of active compounds, for example, for the purpose of treating a particular disease or condition, it is within the scope of the present invention that two or more pharmaceutical compositions, at least one of which contains a component of the combination in accordance with the invention, may conveniently be combined in the form of a kit suitable for coadministration of the compositions.
Thus the kit of the invention comprises two or more separate pharmaceutical compositions, at least one of which contains a component of the combination in accordance with the invention, and means for separately retaining said compositions, such as a container, divided bottle, or divided foil packet. An example of such a kit is the familiar blister pack used for the packaging of tablets, capsules and the like.
The kit of the invention is particularly suitable for administering different dosage forms, for example, oral and parenteral, for administering the separate compositions at different dosage intervals, or for titrating the separate compositions against one another. To assist compliance, the kit typically comprises directions for administration and may be provided with a so-called memory aid.
BIOLOGY EXAMPLES METHODS
The biological activity of the alpha-2-delta ligands of the invention may be measured in a radioligand binding assay using [3H]gabapentin and the σ^δ subunit derived from porcine brain
tissue based on the method given in J. Biol. Chem., 1996, 271(10), 5768-5776). This assay is reproduced below.
[3H]Gabapentin binding assay
Preparation of brain membranes
All solutions are maintained at 40C throughout. Pig brain cortex (up to 50 g) (fresh or frozen) is homogenised in 10 volumes of Buffer A (0.32 M Sucrose/1 mM EDT A/1 mM EGTA/10 mM Hepes/KOH, pH 7.4) by six strokes of a glass/teflon homogeniser at 600 r.p.m. After removal of the 1000 g x 10 minute pellet, the supernatant is centrifuged at 40,000 g for 20 minutes and the resulting pellet is resuspended in 10 volumes of Buffer B (1 mM EDTA/1 mM EGTA/10 mM Hepes/KOH, pH 7.4). Following 30 minutes of continuous stirring, membranes are pelleted as above twice more by centrifugation with Buffer B, before a final re-suspension in approximately 3 volumes of storage buffer (1.25 mM EDTA/1.25 mM EGTA/25% Glycerol/12.5 mM Hepes/KOH, pH 7.4) to give a concentration of about 3 milligrams of protein per millilitre. Aliquots are stored at -8O0C until required.
Binding assay protocol:
Binding of [3H]gabapentin to pig cerebral cortex membranes is carried out at 22°C in 10 mM Hepes/KOH, pH 7.4 for 60 minutes. Non-specific binding (nsb) is defined as the binding obtained in the presence of 10μM pregabalin. An assay volume of 250μl is employed, comprising 200μl of membranes, 25μl test compound/buffer/nsb, 25μl [3H]gabapentin (final assay' concentration ~10nM). Separation of unbound radioligand is effected by rapid filtration under vacuum through cold 50 mM Tris/HCI, pH 7.4-dipped GF/B unifilter plates, using 2 x 1ml of cold 50 mM Tris/HCI, pH 7.4. Plates are left to dry before addition of 50μl/well microscint-40 and the amount of radioactivity bound determined using a TopCount scintillation counter. Results may be expressed as an IC50 in terms of μM or nM.
The binding affinity of an alpha-2-delta ligand is preferably less than 20OnM IC50, more preferably less than 10OnM, more preferably less than 1OnM and most preferably less than 1 nM.
Comparatve assays according to the methods of Gee et al. may be performed between different alpha-2-delta subunit subtypes to determine selectivity.
The biological activity of PDE7 inhibitors may be measured using the following methods:
PRIMARY BINDING ASSAYS
In vitro PDE inhibitory activities against cyclic guanosine 3',5'-monophosphate (cGMP) and cyclic adenosine 3',5'-monophosphate (cAMP) phosphodiesterases can be determined by measurement of their IC50 values (the concentration of compound required for 50% inhibition of enzyme activity).
The required PDE enzymes can be isolated from a variety of sources, including human corpus cavemosum, human and rabbit platelets, human cardiac ventricle, human skeletal muscle and bovine retina, essentially by a modification of the method of Thompson WJ and Appleman MM; Biochemistry 10(2),311-316, 1971 , as described by Ballard SA et al.; J. Urology 159(6), 2164- 2171 , 1998. In particular, cGMP-specific PDE5 and cGMP-inhibited cAMP PDE3 can be obtained from human corpus cavemosum tissue, human platelets or rabbit platelets; cGMP-stimulated PDE2 was obtained from human corpus cavemosum; calcium/calmodulin (Ca/CAM)-dependent PDE1 from human cardiac ventricle; cAMP-specific PDE4 from human skeletal muscle; and photoreceptor PDE6 from bovine retina. Phosphodiesterases 7-11 can be generated from full length human recombinant clones transfected into SF9 cells.
Assays can be performed either using a modification of the "batch" method of Thompson, WJ et a/.; Biochemistry 18(23), 5228-5237, 1979, essentially as described by Ballard SA et al.; J. Urology 159(6), 2164-2171 , 1998 or using a scintillation proximity assay for the direct detection of [3H]-labelled AMP/GMP using a modification of the protocol described by Amersham pic under product code TRKQ7090/7100. In summary, for the scintillation proximity assay the effect of PDE inhibitors was investigated by assaying a fixed amount of enzyme in the presence of varying inhibitor concentrations and low substrate, (cGMP or cAMP in a 3:1 ratio unlabelled to [3H]- labeled at a concentration of -1/3 Km or less) such that IC50 ≡ K1. The final assay volume was made up to 100μl with assay buffer [2OmM Tris-HCI pH 7.4, 5mM MgCI2, 1 mg/ml bovine serum albumin]. Reactions were initiated with enzyme, incubated for 30-60min at 3O0C to give <30% substrate turnover and terminated with 50μl yttrium silicate SPA beads (containing 3mM of the respective unlabelled cyclic nucleotide for PDEs 9 and 11). Plates were re-sealed and shaken for 20min, after which the beads were allowed to settle for 30min in the dark and then counted on a TopCount plate reader (Packard, Meriden, CT) Radioactivity units were converted to % activity of an uninhibited control (100%), plotted against inhibitor concentration and inhibitor IC50 values obtained using the 'Fit Curve' Microsoft Excel extension.
PDE7 ligands and inhibitors can be identified, for example by screening a compound library and by employing a variety of screening techniques against PDE7. Methods of identifying ligands and inhibitors of the enzyme are known and examples of these are presented below:
The identification of test compounds as ligands of PDE7 and the affinity with which a test compound binds to the PDE7 may be determined through use of labelled ligand binding assays, for example standard radioligand binding assays, although othe modes of labelling are available, wherein the test compound is labelled to detect binding, for example by radiolabelling, and incubated with a preparation of the target PDE7 enzyme. Such an enzyme preparation may be obtained from cells transfected with and expressing a recombinant PDE7 enzyme or chosen from a cell lysate of a cell line known to naturally express PDE7.
In a direct binding assay, PDE7 is contacted with a test compound under conditions that allow binding of the test compound to the PDE7. The binding may take place in solution or on a solid surface. Preferably, the test compound is previously labelled for detection. Any detectable group may be used for labelling, such as but not limited to, a luminescent, fluorescent, or radioactive isotope or group containing same, or a nonisotopic label, such as an enzyme or dye. After a period of incubation sufficient for binding to take place, the reaction is exposed to conditions and manipulations that remove excess or non-specifically bound test compound. Typically, this involves washing with an appropriate buffer. Finally, the presence a PDE7-test compound complex is detected. Alternatively binding interactions can be detected by measuring changes in changes in fluoresence on ligand displacement from the enzyme.change in protein fluorescence or molecular tumbling rate or molecular sedimentation in solution of the enzyme in the presence of test compound.
In a preferred embodiment of the direct binding assay, to facilitate complex formation and detection, the binding assay is carried out with one or more components immobilized on a solid surface. In various embodiments, the solid support could be, but is not restricted to, polycarbonate, polystyrene, polypropylene, polyethylene, glass, nitrocellulose, dextran, nylon, polyacrylamide and agarose. The support configuration can include beads, membranes, microparticles, the interior surface of a reaction vessel such as a microtitre plate, test tube or other reaction vessel. The immobilization of PDE7, or other component, can be achieved through covalent or non-covalent attachments. In one embodiment, the attachment may be indirect, i.e. through an attached antibody. In another embodiment, PDE7 is tagged with an epitope, such as glutatione S-transferase (GST) so that the attachment to the solid surface can be mediated by a commercially available antibody such as anti-GST (Santa Cruz Biotechnology). For example, such an affinity binding assay may be performed using a PDE7 which is immobilized to a solid support. Typically, the non-immobilized component of the binding reaction, in this case the test compound, is labelled to enable detection. A variety of labelling methods are available and may be used, such as detection of luminescent, chromophoric, fluorescent, or radioactive isotopes or groups, or detection of nonisotopic labels, such as enzymes or dyes. In one preferred embodiment, the test compound is labelled with a fluorophore such as fluorescein isothiocyanate (FITC, available from Sigma Chemicals, St. Louis). The labelled test compound, is then allowed to contact with the solid support with the immobilised PDE7, under conditions that allow specific binding to occur. After the binding reaction has taken place, unbound and non-specifically bound test compounds are separated by means of washing the surface. Attachment of the binding partner to the solid phase can be accomplished in various ways known to those skilled in the art, including but not limited to chemical cross-linking, non-specific adhesion to a plastic surface, interaction with an antibody attached to the solid phase, interaction between a ligand attached to the binding partner (such as biotin) and a ligand-binding protein (such as avidin or streptavidin) attached to the solid phase, and the like. Finally, the label remaining on the solid surface may be detected by any detection method known in the art. For example, if the test compound is labelled with a fluorophore, a fluorimeter may be used to detect complexes.
Alternatively, the binding reaction may be carried out in solution. In this assay, the labelled component is allowed to interact with its binding partner(s) in solution. If the size differences between the labelled component and its binding partner(s) permit such a separation, the separation can be achieved by passing the products of the binding reaction through an ultrafilter whose pores allow passage of unbound labelled component but not of its binding partner(s) or of labelled component bound to its partner(s) to determine levels of bound vs free ligand. Separation can also be achieved using any reagent capable of capturing a binding partner of the labelled component from solution, such as an antibody against the binding partner, a ligand- binding protein which can interact with a ligand previously attached to the binding partner, and so on.
Effects of a test compound on the catalytic activity of a PDE7 can be most easily determined by standard competitive binding experiments between PDE inhibitors and cAMP on enzyme activity for which known amounts of cAMP substrate and fixed amounts of enzyme are incubated together with various amounts of inhibitor substance for fixed periods of time, after which the reaction is stopped and the residual amount of unhydrolysed cAMP is measured. This may be done for any test sample by use of a scintillation proximity based assay (SPA) designed to measure the competition between cAMP in the test sample and a known amount of radiolabeled cAMP for binding to a cAMP-specific antibody attached to scintillant beads (Hancock, A. A., Vodenlich, A. D., Maldonado, C, Janis, R. (1995) a2-adrenergic agonist-induced inhibition of cyclic AMP formation in transfected cell lines using a microtiter-based Scintillation Proximity Assay. J. of Receptor and Signal Transduction research 15:557-579). The assay is read in a scintillation counter where the counts per sample are inversely related to the amount of cAMP present in the test sample. SPA kits for measurement of cAMP are available from Amersham Pharmacia Biotech (Amersham, UK).
Identification of inhibitor activity can be judged using a standard SPA (scintillation proximity assay) assay with a PDE7 enzyme. The PDE7 enzyme can be for example recombinant mouse, human or yeast or can be derived from a whole cell lysate of Hut78 Tcell line as a surrogate for the use of a recombinant PDE7A according to the method of Pitts, WJ., et al Biorg. Med. Chem. Lett 14 2004 2955 - 2958. IC50 values of <1 micromolar in the presence of inhibitor are indicative of good inhibition.
In a preferred embodiment, a binding assay can be performed as follows:
Phosphodiesterase activity of PDE7 can be measured using the phosphodiesterase Scintillation Proximity Assay (SPA) (Amersham) according to the manufacturer's protocol, for convenience the assays can be done in triplicate in 96 well format. Reaction times and enzyme dilution are optimised so that the lowest substrate concentration gives no more than 30% conversion of substrate to product to ensure linearity. The reactions can contain for example 25 μl of the appropriately diluted enzyme, 25 μl buffer (20 mM Tris with 5 mM MgCL2.6H2O, pH 7.4 plus 2
mg/ml BSA) and initiated by the addition of 50 μl of either cAMP or cGMP to give a total reaction volume of 100 μl. [3H]-CAMP (Amersham Cat. No. TRK304 B70, 24.Ci/mmol) or [3H]-CGMP (Amersham Cat. No. TRK392 B37, 10.7 Ci/mmol) is mixed with the corresponding cold cyclic nucleotide to give a final concentration range of 1 μM-0.002 μM. This is achieved by performing doubling dilutions across a 96 well plate. Following a 40 min incubation at 3O0C, the plates are immediately centrifuged at 2000 rpm for 5 min and then counted on TopCount. Background levels for each cAMP concentration were determined using a Scintillation Counter. Average counts of triplicate results for each assay are determined and the corresponding background subtracted. Counts per min for each assay are converted into pmol of cAMP hydrolysed per min per ml of enzyme and plotted against cAMP concentration (μM). For inhibitor profiling a concentration range of 0.5-300 μM in 1% dimethyl sulphoxide for each inhibitor is used and cAMP concentration is kept constant at 1/3 Km. The assay blank contains all reagents minus the enzyme. Values for K01 and IC50 were determined using the computer package GraFit4.
According to an altenative preferred embodiment, a binding assay can be performed as follows: Inhibition of PDE activity can be determined using Hut78 cell lysate (Hut78 is a Tcell line which expresses PDE7) and an SPA specific for cAMP (Amersham Pharmacia Biotech, Buckinghamshire, UK) according to the manufacturers instructions with minor modifications. Enzyme assays are performed at room temperature in the presence of 5OmM Tris-HCI, pH7.5, containing 8.3mM MgCI2, 1.7mM EGTA, and 0.5mg/mL BSA. Each assay is performed in a 100μL reaction volume in 96 well microtitre plates containing the above buffer, 0.3μL of Hut78 cell lysate treated with 2μM Zardaverine to inhibit PDE3 and PDE4, 0.05μCi of [5 ,8-3H] Adenosine 3 ,5-cyclic phosphate as an ammonium salt for 20min. Inhibitors are included at a concentration range of 0.5-300 μM for each inhibitor is used and cAMP concentration is kept constant, the assay blank contains all reagents minus the enzyme. The reaction was terminated by the addition of 50μL
PDE SPA beads (1mg) water with 1OmM cold cAMP (Sigma, St. Louis MO). The reaction mix was allowed to settle for 20min before counting in a Top Count-NXT scintillation counter (Packard BioScience, Meriden, CT). For selectivity studies, the assay is essentially unchanged except that 3H-cyclic GMP is used as the substrate for PDE1 , PDE5, and PDE6. The following PDEs/activators and enzyme sources are used: PDE1 , bovine (Sigma St. Louis), calmodulin; PDE2, rat kidney, cGMP; PDE3, human platelet; PDE4, rat kidney; PDE5, human platelet, and PDE6, bovine retina.
SELECTIVITY OF INHIBITORS The PDE7 inhibitors for use in the combination of the invention are preferably potent PDE7 inhibitors. These compounds have low IC50 values for PDE7, typically at less than 10OnM, preferably less than 10 nM, more preferably less thani nM.
The PDE7 inhibitors for use in the combination of the invention are preferably selective PDE7 inhibitors. The selectivity of PDE7 inhibitor is preferably at least 10 fold selective for PDE7 over
other PDEs1 preferably it should be at least 100 fold selective and further preferably at least 1000 fold selective. Selectivity in general represents the relative potency of a compound between two enzyme subtypes for the appropriate ligand or inhibitor for the enzyme of interest.
A PDE7 ligand or inhibitor, can be tested for selectivity for the PDE7 in comparison with another PDE such as for example PDE4. In the assay, the capacity of each test compound to compete with binding of labelled-cAMP is measured at both the PDE7 and PDE4 enzymes, and an IC50 value (in μM) is determined. Any of the above mentioned binding assay procedures can be used. For example in an inhibition assay, test compounds are assayed for their ability to disrupt the binding and hydrolysis of cAMP by PDE7. Labelled cAMP may be mixed with PDE7 or a fragment or derivative thereof, and placed under conditions in which the interaction between them would normally occur, either with or without the addition of the test compound. The amount of labelled cAMP that binds and is hydrolysed by PDE7 or PDE4 may be compared to the amount bound and hydrolysed in the presence or absence of test compound, thus the level of inhibition of the process can be determined for any test compound addition at either PDE and compared.
The potency of a PDE7 inhibitor (based on IC50 potency which can be defined as the concentration of inhibitor that gives a halving of the value of the functional activity of a enzyme in a functional assay as described below) is preferably at least 10OnM IC50 at the human enzyme (recombinant and/or native), more preferably less than 1OnM and further preferably less than 1 nM. For instance in a functional cell based assay, IC50 is the molar concentration of an inhibitor that inhibits by 50% the maximal activity of the human PDE7 for example in response to cAMP. In a binding assay, IC50 is the molar concentration of an inhibitor that displaces 50% of the specific binding of labelled cAMP or other appropriate ligand or the moalr concentration at which the test compound occupies half of the available PDE7 binding sites.
FUNCTIONAL ASSAYS
Functional assay methods are known for identifying compounds that are inhibitors of PDE7. The methods generally include the steps comprising: a) contacting a PDE7-expressing cell with a test compound optionaly in the presence of cAMP or another PDE7 substrate ligand; and b) measuring the resultant level of a PDE7 activity, or the level of expression of PDE7 in the cell, such that if said level of measured activity or expression differs from that measured in the absence of the test compound, then a compound that modulates a PDE7-cAMP-mediated process is identified. The PDE7 activity measured can be the ability to interact with cAMP or by a change in cAMP / AMP levels in the cell or the response of the cell to cAMP for example by alterations in gene transcription or protein activity. Example protocols for functional assays are provided below.
The key advantage of functional cell based assays is that they facilitate early and direct pharmacological characterization of compounds by high-throughput quantification and allow identification of compounds that act both at the binding site of the PDE or on a modulatory binding
site on a PDE that is topographically distinct from the binding site. The most common systems of functional cell based assays are based on cyclic AMP detection and are reviewed in Williams, C, Nature Reviews Drug Discovery 3 2004 125-135. Cell-based assays in HTS provides the advantage of having the ability to identify inhibitor compounds and to obtain additional information about the mode of action of the compound.
HTS-compatible accumulation assays for cAMP measurement follow a general principle, with changes in intracellular cAMP being detected by the competition between cellular cAMP and a labelled form of cAMP for binding to an anti-cAMP sequestering antibody or directly to the PDE. Protocols for these assays differ markedly and include: radiometric assays, fluorescence polarization cAMP assays, time-resolved fluorescence assays, assays which detect alterations in gene transcription or protein activity for example via initiation of phosphorylation events that regulate target enzymes and transcription factors, enzymatic assays, assays to determine binding to protein kinases within the cell.
Homogeneous radiometric assays, such as scintillation proximity assays (SPA.Amersham Biosciences) and Flashplate technology (NEN/Perkin Elmer) enable the direct detection of [125I]- labelled cAMP once it is inclose proximity to a solid scintillant surface [Amersham Life Science. High throughput screening forcAMP formation by scintillation proximityradioimmunoassay. Proximity News Issue No. 23. (1996).&. NEN Life Science Products. A novel adenylyl cyclaseactivation assay on FlashPlate (Flasplate File #1 , ApplicationNote). (NEN Life Science Products Inc., Boston, Massachusetts, 1998).18. Kariv, I. I. et al. High throughput quantitation of cAMPproduction mediated by activation of seven transmembranedomain receptors. J. Biomol. Screen. 4, 27-32 (1999)].
Fluorescence polarization cAMP assays (available in kit form from companies such as Perkin Elmer and Amersham Biosciences) monitor the light emitted from a fluorescently tagged cAMP molecule following excitation with a polarized light source, the assays is based on a decrease in the extent of molecular rotation of a fluorescently labelled cAMP that occurs following binding to the larger anti-cAMP antibody. Alternatively, dyes such as Bodipy-TMR,MR121 ,Alexa, Cy3 and Cy5 have been used in FP binding assays.
The HTRF (homogeneous time-resolved fluorescence) technology uses anti-cAMP antibodies labelled with europium cryptate and cAMP that is labelled with a modifiedallophyocyanin (see the CIS Bio International HTRF web site). In the absence of cellular cAMP, these two fluorescent molecules are in close proximity, FRET occurs and longlifetime fluorescence is emitted at two different wavelengths. When the two molecules are separated by competition with cellularcAMP, no FRET occurs and only emission from the europium is detected. This technique has been successfully applied to high-throughput screening with whole cells in miniaturized formats. [Claret E, Roux P, Ouled-Diaf J, Preaudat C, Drexler C, Grepin C, Seguin P. Phosphodiesterase assays with HTRF (R)10th SBS annual conference. September 2004, Orlando, US. Cisbio]
Additionally changes in the intracellular levels of cAMP produce alterations in gene transcription or protein activity and result in the observed functional response of the cell; these events can be measured via transcription factors such as NFAT (nuclear factor activated in T-cells) or CREB (cAMP response element binding protein) and reporter genes under the control of appropriate upstream elements [Hill, S. J. et al. Reporter-gene systems for the study of G-protein-coupled receptors. Curr. Opin. Pharmacol. 1 ,526-532 (2001 ).29. Wood, K. V. Marker proteins for gene expression. Curr. Opin.Biotechnol. 6, 50-58 (1995).3O. Southward, C. M. & Surett, M. G. The dynamic microbe:green fluorescen.
Reporter-gene assays for cAMP detection Reporter-gene assays follow a general principle.where by receptor-mediated changes in intracellular cAMP con-centrations are detected via changes in the expression level of a particular gene (the reporter), the transcrip-tion of which is regulated by the transcription factorcAMP response-element binding protein (CREB) binding to upstream cAMP response elements (CREs). Various reporter genes have been used in in vitro and in vivo studies, including β-galactosidase, green fluorescent protein(GFP),luciferase and β-lactamase 28- 31. The reporter-gene method is compatible with screening for activity in live cells or enabling transfected cell popula-tions. Cell lines commonly used inreporter-gene assays are for example Chinese hamster ovary cells (CHO) and human embryonic kidney cells.
Recently.three innovative technologies have emerged that also aim to provide non-radiometric high-sensitivity assays of cAMP accumulation. The first of these — ALPHAScreen (amplified luminescent proximity homogeneous assay; PackardBioscience/Perkin Elmer) — is a homogeneous assay format using chemiluminscent readout. The second system — an enzyme complementation technology from DiscoveRx(Fremont,California) — uses a cAMP molecule tagged with an inactive β-galactosidase component and uses fluorescent or luminescent readout. The third system uses electrochemiluminescence detection and is a technology available from Meso ScaleDiscovery (Gaithersburg, Maryland). In this case, the cAMP, is tagged with a ruthenium derivative, which results in the production of light from the labelled cAMP (see Meso Scale Discovery web site).
IN VIVO PROCEDURES
The analgesic effect of PDE7 inhibitors and alpha-2-delta ligands may be determined in vivo using animal models of selected pain conditions. Several models of pain conditions are known and specific procedures that can be used to determine the analgesic effect of PDE7 inhibitors and alpha-2-delta ligands are presented below.
An alternative pain model is the streptozocin induced diabetic model of neuropathic pain in rats. This procedure involves administration of streptozocin (50mg/kg, i.p.) in a single dose to animals such as Charles River Sprague dawley rats (225 - 25Og) to induce diabetes. Animals are
evaluated 2 weeks following administration using static and dynamic allodynia tests and if neuropathic pain is confirmed they are used to further evaluate compounds for their effect on neuropathic pain.
The chronic constrictive injury (CCI) model of neuropathic pain in rats involves the tying of loose ligatures around the sciatic nerve Charles River male Sprague dawley rats (175-20Og) are placed in an anaesthetic chamber and anaesthetised with a 2% isofluorane O2 mixture. The right hind thigh is shaved and swabbed with 1 % iodine. Animals are then transferred to a homeothermic blanket for the duration of the procedure and anaesthesia maintained during surgery via a nose cone. The skin is cut along the line of the thigh bone. The common sciatic nerve is exposed at the middle of the thigh by blunt dissection through biceps femoris. Proximal to the sciatic trifurcation, about 7mm of nerve is freed by inserting forceps under the nerve and the nerve gently lifted out of the thigh. The forceps are gently opened and closed several times to aid clearance of the fascia from the nerve. Suture is pulled under the nerve using forceps and tied in a simple knot until slight resistance is felt and then double knotted. The procedure is repeated until 4 ligatures (4-0 silk) are tied loosely around the nerve with approx 1 mm spacing. The incision is closed in layers. Fourteen days following surgery, animals are assessed for static allodynia, dynamic allodynia or weight bearing deficit.
'Alternative animal models of neuropathic pain conditions include the Seltzer model, partial tight ligation of the sciatic nerve (Seltzer, Z. (1995). Sem. Neurosci, 8: pp. 34-39) or Chung's model, tight ligation of one of the two spinal nerves of the sciatic nerve. (Kim SH, Chung JM. Pain (1992); 50: pp. 355-63) or of the Chronic Constrictive Injury model (CCI) (Bennett GJ, Xie Y-K. Pain (1988); 33: pp. 87-107).
Alternative animal models of neuropathic pain conditions may involve selection of an animal that naturally possesses a painful disease condition providing neuropathic pain and its symptoms such as HIV or Herpes or cancer or diabetes. Alternatively the animal may be arranged to experience a pain condition by modification of the animal to possess a pain inducing disease condition such as arthritis or HIV or Herpes or cancer or diabetes. Animals may be modified to possess a pain condition due to a disease in a variety of ways for example by administration of Streptozocin to induce a diabetic neuropathy (Courteix.C, Eschalier.A., Lavarenne.J., Pain, 53 (1993) pp. 81-88.) or by administration of viral proteins to cause HIV related neuropathic pain (Herzberg U. Sagen J., Journal of Neuroimmunology. (2001 May 1), 116(1): pp. 29-39) or adminstration of varicella ■ zoster virus to cause Herpes and post herpatic neuralgia (Fleetwood-Walker SM. Quiπn JP. Wallace C. Blackburn-Munro G. Kelly BG. Fiskerstrand CE. Nash AA. Dalziel RG., Journal of General Virology. 80 ( Pt 9):2433-6, 1999 Sep.) or adminstration of a carcinogen or of cancer cells to an animal to cause cancer (Shimoyama M. Tanaka K. Hasue F. Shimoyama N, Pain. 99(1-2): pp. 167-74, 2002 Sep).
Dynamic allodynia can be assessed by lightly stroking the plantar surface of the hind paw of the animal with a cotton bud. Care is taken to perform this procedure in fully habituated rats that are not active, to avoid recording general motor activity. At least two measurements are taken at each time point, the mean of which represents the paw withdrawal latency (PWL). If no reaction is exhibited within 15s the procedure is terminated and animals are assigned this withdrawal time. Thus, 15s effectively represents no withdrawal. A withdrawal response is often accompanied with repeated flinching or licking of the paw. Dynamic allodynia is considered to be present if animals responded to the cotton stimulus within 8s of commencing stroking. Following baseline evaluation, animals can be administered compounds for analgesic assessment by one of the following routes, oral administration, subcutaneous., intra-peritoneal., intra-venous or intra-thecal. The PWL is re-evaluated at some or all of the following time points, 30 min, 1h, 2h, 3h, 4h, 5h, 6h, 7h, 24h. Animals are assigned randomly to each compound group according to their baseline values. The mean and standard error mean are calculated for each compound group at each time point. Measures of dynamic allodynia are compared to their respective controls using a one way ANOVA followed by a Dunnett's t-test comparing vehicle to compound at each time point. The minimum number of animals per group is 6.
Static allodynia can be evaluated by application of von Frey hairs (Stoelting, Wood Dale, Illinois, USA) in ascending order of force (0.6, 1 , 1.4, 2, 4, 6, 8, 10, 15 and 26 grams) to the plantar surface of hind paws. Animals are habituated to wire bottom test cages prior to the assessment of allodynia. Each von Frey hair is applied to the paw for a maximum of 6 seconds, or until a withdrawal response occurs. Once a withdrawal response to a von Frey hair is established, the paw is re-tested, starting with the filament below the one that produces a withdrawal, and subsequently with the remaining filaments in descending force sequence until no withdrawal occurs. The highest force of 26g lifts the paw as well as eliciting a response, thus representing the cut off point. Each animal has both hind paws tested in this manner. The lowest amount of force required to elicit a response is recorded as paw withdrawal threshold (PWT) in grams. Static allodynia is defined as present if animals responded to a stimulus of, or less than, 4g, which is innocuous in normal rats. Following baseline evaluation, animals are administered compounds for analgesic assessment by one of the following routes, orally, subcutaneous, intra-peritoneal., intra-venous or intra-thecal. and the PWT re-evaluated at some or all of the following time points, 30 min, 1h, 2h, 3h, 4h, 5h, 6h, 7h, 24h. Static allodynia measurements are analysed using a Kruskall-Wallis test for non- parametric results, followed by Mann-Whitney's U test vs vehicle group. The minimum number of animals per group is 6.
Thermal hyperalgesia is assessed using the rat plantar test (Ugo Basile, Italy) following a modified method of Hargreaves et al., (1988) Pain 32:77-88. Rats are habituated to the apparatus that consists of three individual perspex boxes on an elevated glass table. A mobile radiant heat source is located under the table and focused onto the hind paw and paw withdrawal latencies • (PWL) are recorded. There is an automatic cut off point of 22.5 s to prevent tissue damage. PWL
are taken 2-3 times for both hind paws of each animal, the mean of which represented baselines for right and left hind paws. The apparatus is calibrated to give a PWL of approximately 10 s. PWL are reassessed 2h following administration of carrageenan. Following administration of compounds for analgesic assessment PWL's are reassessed hourly for up to 6 hours. PWL's of compound groups are compared to their respective controls using a one way ANOVA followed by a Dunnett's t-test. The minimum number of animals per group will be 6.
Weight bearing deficit can be measured according to the method of Bove SE, et: al. Weight bearing as a measure of disease progression and efficacy of anti-inflammatory compounds in a model of monosodium iodoacetate-induced osteoarthritis. Osteoarthritis Cartilage. 2003
Nov;11 (11):821-30. Open field test can be carried out according to the method of Prut L and Belzung.C. The open field as a paradigm to measure the effects of compounds on anxiety-like behaviors: a review. Eur J Pharmacol. 2003;463::3-33. The locomotor test can be carried out according to the method of Salmi P and Ahlenius S- Sedative effects of the dopamine D1 enzyme agonist A 68930 on rat open-field behavior. Neuroreport. 2000 Apr 27; 11(6): 1269-72.
The ability of compounds to act as PDE7 inhibitors or alpha-2-delta ligands can be measured according to established procedures, particularly those described in the documents mentioned hereinabove.
Suitable alpha-2-delta ligand compounds of the present invention may be prepared as described herein below or in the aforementioned patent literature references, which are illustrated by the following non-limiting examples and intermediates.
The following examples and preparations illustrate the preparation of alpha-2-delta ligands disclosed in US Provisional Patent Application Number 60/676025 (unpublished at the filing date of the present application):
Example 1 (2S)-2-Amino-4-ethyl-2-methylhexanoic acid

Pearlman's catalyst (0.15g) was added to a solution of (3S,5S)-3-(2-ethylbutyl)-3-methyl-5- phenylmorpholin-2-one (0.15g, 0.54mmol, Preparation 1) in ethanol (5ml) and 1 molar aqueous hydrochloric acid (1 ml). The reaction was stirred under hydrogen gas (414 kPa, 60psi) at room temperature for 24 hours. The reaction mixture was filtered through arbocel and washed with ethanol (20ml). The liquor was evaporated under reduced pressure and the residue was partitioned between dichloromethane (50ml) and water (20ml). The organic layer was removed and the aqueous phase was extracted with more dichloromethane (50ml). The aqueous phase
was evaporated under reduced pressure to give a yellow solid. The material was purified by ion exchange chromatography on Dowex 50 WX8 resin, eluting with 0.88 aqueous ammonia:water (2:98 to 14:86) to give the title compound as a white crystalline solid (30mg).
1HNMR (CD3OD, 400MHz) δ: 0.83-0.89 (m, 6H), 1.29-1.43 (m, 8H), 1.58-1.62 (dd, 1 H), 1.81-1.86 (dd, 1 H). LRMS (ESI): m/z 174 [M+H+], 172 [M-H"].
Example 2 2.5.5-Trimethyl-L-norleucine

The title compound was prepared according to the procedure outlined in Example 1 from (3S.5S)- 3-(3,3-dimethylbutyl)-3-methyl-5-phenylmorpholin-2-one (0.394g, 1.43mmol, Preparation 2). The product (0.138g) was obtained as a white crystalline solid. 1HNMR (CD3OD, 400MHz) δ: 0.91(s, 9H), 1.17-1.25 (m, 1 H), 1.28-1.36 (m, 1 H), 1.44 (s, 3H), 1.59-1.67 (m, 1H), 1.83-1.91 (m, 1H). LRMS (ESI): m/z 174 [M+H+], 172 [M-H ].
Example 3 (2S)-2-Amino-3-cvclopentyl-2-methylpropanoic acid

The title compound was prepared according to the procedure outlined in Example 1 from (3S.5S)- 3-(cyclopentylmethyl)-3-methyl-5-phenylmorpholin-2-one (0.426g, 1.56mmol, Preparation 3). The product (0.157g) was obtained as a white crystalline solid. 1HNMR (CD3OD, 400MHz) δ: 1.08-1.22 (m, 2H), 1.44 (s, 3H), 1.48-1.68 (m, 4H), 1.74-2.00 (m, 5H). LRMS (ESI): m/z 174 [M+H+], 172 [M-H'].
Example 4 (2S)-2-Amino-5-ethyl-2-methylheptanoic acid

1HNMR (CD3OD, 400MHz) δ: 0.85-0.92 (m, 6H), 1.16-1.48 (m, 7H), 1.56 (s, 3H), 1.72-1.99 (m,
2H).
LRMS (ESI): m/z 188 [M+H+], 186 [M-H"].
Example 5
(2S)-2-Amino-3-cvclobutyl-2-methylpropanoic acid

Pearlman's catalyst (0.5g) was added to a solution of (3S,5S)-3-(cyclobutylmethyl)-3-methyl-5- phenylmorpholin-2-one (0.445g, Ummol, Preparation 5) in ethanol (15ml), water (2ml) and trifluoroacetic acid (0.5ml). The reaction was stirred under hydrogen gas (414 kPa, 60psi) at room temperature for 24 hours. The reaction mixture was filtered through Arbocel® and washed with ethanol (20ml). The liquor was evaporated under reduced pressure and the residue was partitioned between dichloromethane (50ml) and water (50ml). The organic layer was removed and the aqueous phase was extracted with more dichloromethane (50ml). The water layer was evaporated under reduced pressure to give a yellow solid. The product was purified by ion exchange chromatography on Dowex 50 WX8 resin, eluting with 0.88 aqueous ammonia:water (2:98 to 14:86) to give the title compound (0.034g) as a white solid.
1HNMR (CD3OD, 400MHz) δ: 1.40 (s, 3H), 1.69-1.84 (m, 6H), 2.04-2.17 (m, 2H), 2.41-2.54 (m,
1 H).
LRMS (ESI): m/z 158 [M+H+], 156 [M-H"].
Example 6
(2S.5R)-2-Amino-2.5-dimethylheptanoic acid

A solution of the compound of Preparation 8 (1.1Og) in dioxane (3ml) and 6N aqueous hydrochloric acid (15ml) was heated under reflux for 16 hours. The solution was then allowed to cool to room temperature, the solvent was evaporated and the residue was redissolved in 2ml of water. A column of DOWEX-50X8-200 (25g) was washed with 250ml of deionised water. The crude product was then loaded and the column was eluted with 250ml of deionised water and then 250ml of 10% aqueous ammonia. The basic fractions were evaporated to give the title compound (0.18g) as a white solid.
1HNMR (CD3OD, 400MHz) δ: 0.89 (βH, m), 1.08-1.24 (2H1 m), 1.26-1.49 (6H, m), 1.56-1.65 (1 H, m), 1.87-1.96 (1 H, m). LRMS (APCI): m/z 174 [M+H+].
Example 7
(4S)-2.4-Dimethyl-L-norleucine

The title compound was prepared according to the procedure outlined in Example 1 from (3S.5S)- 3-methyl-3-[(2S)-2-methylbutyl]-5-phenylmorpholin-2-one (0.586g, 2.24mmol, Preparation 9). The product (0.018g) was obtained as a white crystalline solid.
1HNMR (CD3OD, 400MHz) δ: 0.90-0.92 (m, 3H), 0.94-0.95 (m, 3H), 1.17-1.40 (m, 2H), 1.45 (s, 3H), 1.50-1.58 (bs, 1 H), 1.69-1.81 (m, 1H), 1.70-1.83 (m, 1 H). LRMS (ESI): m/z 262 [M+H+].
The following preparations show how intermediates used in the preparation of the Examples described above may themselves be synthesised.
Preparation 1
(3S.5S)-3-(2-Ethylbutyl)-3-methyl-5-phenylmorpholin-2-one

Boron trifluoride etherate (5.85ml, 46mmol) was added slowly to a solution of (5S)-3-methyl-5- phenyl-5,6-dihydro-2H-1 ,4-oxazin-2-one (4.35g, 23mmol, see WO-A-02/051983) in tetrahydrofuran at -780C. The solution was stirred for 50 minutes and then a solution of the Grignard reagent prepared from 3-(bromomethyl)pentane (10.9g, 66mmol) and magnesium turnings (2.5g, 99mmol) in ether (250ml) was added over 40 minutes. The reaction mixture was stirred for a further 75 minutes at -78°C then allowed to warm to -200C and quenched with saturated aqueous ammonium chloride (100ml). More tetrahydrofuran (100ml) was added and the organic layer was separated from the aqueous layer. The organic layer was dried (MgSO4) and concentrated under reduced pressure. The residue was purified by column chromatography on silica gel using an elution gradient of pentane to pentane:ether (30:70) to afford the title compound as a white solid (3.21 g).
1HNMR (CDCI3, 400MHz) δ: 0.79 (q, 6H)1 1.26-1.35 (m, 5H), 1.41 (s, 3H), 1.63 (dd, 1H), 1.91 (dd, 1H), 4.17-4.26 (m, 1 H)1 4.28-4.35 (m, 2H)1 7.25-7.36 (m, 5H). LRMS (ESI): m/z 276 [M+H+].
Preparation 2
(3S.5S)-3-(3.3-Dimethylbutyl)-3-methyl-5-phenylmorpholin-2-one

A suspension of Rieke Magnesium (343mg, 14.1 mmol) in tetrahydrofuran (13.7ml) was added to a solution of 1-iodo-3,3-dimethylbutane in diethylether (50ml) over a period of twenty minutes and the reaction was stirred for 40 minutes at room temperature. The Grignard reagent solution and
(5S)-3-methyl-5-phenyl-5,6-dihydro-2H-1 ,4-oxazin-2-one (1g, 5.28mmol, see WO-A-02/051983) were used according to the method of Preparation 1 to generate the title compound. The total amount of compound synthesised was 0.394g.
1HNMR (CDCI3, 400MHz) δ: 0.91 (s, 9H), 1.13-1.31 (m, 3H), 1.42 (s, 3H), 1.67-1.77 (bs, 1H)1 2.02-2.10 (m, 1H)1 4.31-4.43 (m, 3H), 7.34-7.46 (m, 5H).
LRMS (ESI): m/z 276 [M+H+].
Preparation 3
(3S.5S)-3-(Cvclopentylmethyl)-3-methyl-5-phenylmorpholin-2-one

The compound was prepared according to the procedure outlined in Preparation 1 using (5S)-3- methyl-5-phenyl-5,6-dihydro-2H-1,4-oxazin-2-one (1g, 5.28mmol, see WO-A-02/051983) and (iodomethyl)cyclopentane. The total amount of compound synthesised was 0.426g. 1HNMR (CDCI3, 400MHz) δ: 1.12-1.22 (m, 2H)1 1.43-1.65 (m, 8H)1 1.78-1.94 (m, 4H), 2.13-2.18 (m, 1H), 4.23-4.42 (m, 3H), 7.31-7.45 (m, 5H). LRMS (ESI): m/z274 [M+H+].
Preparation 4
(3S,5S)-3-(3-Ethylpentvπ-3-methyl-5-phenylmorpholin-2-one

Fr, 1975, 201-205) according to the procedure outlined in Preparation 1. The total amount of compound synthesised was 0.07g. 1HNMR (CDCI3, 400MHz) δ: 0.91 (s, 9H), 1.13-1.31 (m, 3H), 1.42 (s, 3H), 1.67-1.77 (bs, 1 H),
2.02-2.10 (m, 1 H)1 4.31-4.43 (m, 3H), 7.34-7.46 (m, 5H).
LRMS (ESI): m/z 276 [M+H+].
Preparation 5 (3S,5S)-3-(Cvclobutylmethyl)-3-methyl-5-phenylmorpholin-2-one

The title compound was prepared from (5S)-3-methyl-5-phenyl-5,6-dihydro-2H-1,4-oxazin-2-one (1g, 5.28mmol, see WO-A-02/051983) and (bromomethyl)cyclobutane according to the procedure outlined in Preparation 1. The total amount of compound synthesised was 0.445g. 1HNMR (CDCI3, 400MHz) δ: 1.43 (s, 3H), 1.73-1.82 (m, 2H), 1.86-1.92 (m, 2H), 2.02-2.12 (m, 2H), 2.14-2.19 (m, 1 H), 2.43-2.52 (m, 1H), 4.22-4.27 (m, 1 H), 4.33-4.37 (m, 2H), 7.31-7.43 (m, 5H). LRMS (ESI): m/z 260 [M+H+].
Preparation 6 (5R)-5-Methylheptan-2-one

Dicyclohexyl carbodiimide (9.51g, 46.1mmol), N,N'-dimethyl-4-aminopyridine (1.13g, 9.2mmol) and triethylamine (6.4ml, 46.1mmol) were added to a solution of Meldrum's acid (6.64g, 46.1mmol) in dichloromethane (150ml). (4R)-4-Methylhexanoic acid (6g, 46.1mmol) was added and the reaction mixture was stirred overnight. The reaction mixture was filtered and the solid was washed with dichloromethane (2 x 100ml). The filtrate and washings were combined and evaporated under reduced pressure to give an orange oil. Acetic acid (50ml) and water (50ml) were added and the reaction was heated under reflux overnight. After the solution had cooled to room temperature it was extracted with pentane (100ml). The organic extract was dried (MgSO4) and evaporated to give a residue that was purified by column chromatography on silica gel using an elution gradient of pentane to pentane:diethyl ether 19:1 to give the title compound (2.8g) as a pale yellow oil.
1HNMR (CDCI3, 400MHz) δ: 0.82-0.88 (6H, m), 1.07-1.19 (1 H, m), 1.25-1.41 (3H, m), 1.54-1.64 (1 H, m), 2.12 (3H, s), 2.34-2.47 (2H, m).
Preparation 7
N-[(1 E.4R)-1.4-dimethylhexylidenel-(S)-2-methylpropane-2-sulfinamide

Preparation 8
N-r(1S.4R)-1-cvano-1.4-dimethylhexyll-2-methylpropane-(S)-2-sulfinamide

1HNMR (CDCI3, 400MHz) δ: 0.86-0.93 (6H, m), 1.13-1.26 (10H, m), 1.29-1.44 (3H, m), 1.48-1.57 (2H, m), 1.64 (3H, s), 1.81-1.98 (2H, m), 3.41 (1H, s). LRMS (APCI): m/z 259 [M+H+].
Preparation 9
(3S.5S)-3-Methvl-3-[(2SV2-methylbutvll-5-phenvlmorpholin-2-one

The following examples and preparations illustrate the preparation.of (2S)-2-(Aminomethyl)-5- ethylheptanoic acid disclosed in US Provisional Patent Application Number 60/733591 (unpublished at the filing date of the present application):
Example 1
(2S)-2-(Aminomethvl)-5-ethvlheDtanoic acid

Method A
10% Palladium on carbon (600mg) was added to a solution of the alkene of Preparation 4 (6.7Og, 0.04mol) in 10% aqueous acetic acid (130ml) and the reaction mixture stirred at room temperature, under 4.08 atmosheres of hydrogen for 4 hours. The reaction mixture was filtered through arbocel® and the filtrate evaporated in vacuo. The crude material was recrystalised from water to yield the title compound as a white solid (5.41 g, 0.029mol, 73%). 1H-NMR (CD3OD, 400MHz): $ 0.86 (t, 6H), 1.15-1.23 (m, 1 H), 1.29-1.36 (m, 6H), 1.44-1.53 (m, 1H), 1.64-1.73 (m, 1H), 2.33-2.40 (m, 1H), 2.93-3.03 (m, 2H). LRMS m/z APCI 188 [MH]+
Method B
The amide of Preparation 5 (8.6g, 0.026mol) was dissolved in 1 ,4-dioxane (100ml), water (100ml) was added and the reaction mixture heated at reflux for 116 hours. The mixture was filtered through arbocel® and the filtrate evaporated in vacuo. The crude material was triturated with
acetonitrile then recrystalised from water to yield the title compound as a white solid (2.9g, 0.0156mol, 60%).
Preparation 1
3-Ethylpent-1-en-3-ol

To a 1M solution of vinyl magnesium bromide in tetrahydrofuran (400ml, 0.4mol) stirring at O0C, was added dropwise a solution of 3-pentanone (34.7ml, 0.33mol) in tetrahydrofuran (30ml), maintaining the temperature between 5-150C. The reaction mixture was allowed to warm to room temperature overnight then quenched with a saturated aqueous solution of ammonium chloride (20ml). The mixture was diluted with diethyl ether (400ml), washed with water (2x100ml), dried over magnesium sulphate, filtered and evaporated in vacuo. The title compound was obtained as a yellow oil (30.7g, 0.269mol, 82%). 1H-NMR (CDCI3, 400MHz): δ 0.85 (t, 6H), 1.5-1.6 (m, 4H), 5.10-5.25 (m, 2H), 5.75-5.85 (m, 1H).
Preparation 2 1-Bromo-3-ethylpent-2-ene

48% aqueous Hydrobromic acid (74.58g, 0.44mol) was added to a solution of the alkene of Preparation 1 (55.8g, 49mmol) in pentane (950ml) and the reaction mixture stirred at room temperature for 5 hours then left to stand for 18 hours. The layers were separated and the organic layer washed with water (2x200ml), dried over magnesium sulphate, filtered and evaporated in vacuo. The crude material was returned to the reaction conditions for a further 2 hours, left to stir at room temperature overnight, then worked up as previously described. The crude material was purified by fractional distillation. The title compound was obtained as a colourless oil with a boiling point of 47-5O0C at 10 millibar (47.2g, 0.27mol, 55%).
1H-NMR (CDCI3, 400MHz): δ 1.0 (m, 6H), 2.1-2.2 (m, 4H), 4.05 (d, 2H), 5.5 (t, 1 H).
Preparation 3
(2SV2-(Aminomethvn-5-ethyl-N-r(1 S.2SV2-hvdroxy-1-methyl-2-phenylethvn-N-methylhept-4- enamide

To a suspension of N-[(1S,2S)-2-hydroxy-1-methyl-2-phenylethyl]-N-methyl-beta-alaninamide (Org. Lett, 2, 22, 2000, 3527-3530) (36.9g, 0.156mol) and lithium chloride (26.49g, 0.625mol) in tetrahydrofuran (400ml) stirring at O0C1 was added a 2M solution of 1 ,1 , 1 ,3, 3,3- hexamethyldisilazane lithium salt (500ml, 0.5mol), maintaining the reaction temperature below 50C. The reaction mixture was stirred at O0C for 1 hour. A solution of the allyl bromide of Preparation 2 (30.6g, 0.172mol) in tetrahydrofuran (30ml) was added dropwise, maintaining the reaction temperature at O0C. The reaction mixture was allowed to warm to room temperature over 18 hours then quenched with water (40ml) and the solvent evaporated in vacuo. The residue was extracted from water (400ml) into ethyl acetate (2x400ml) and the combined organic phases washed with brine (400ml), dried over magnesium sulphate, filtered and evaporated in vacuo. The crude material was triturated with pentane then recrystalised from diethyl ether: pentane to yield the title compound as a white solid (14.76g, 0.044mol, 28%). The mother liquors were evaporated in vacuo and the crude material purified by column chromatography over silica gel eluting with dichloromethane to dichloromethane: methanol: 0.880 ammonia (90: 10: 1). The material was recrystalised from diethyl ether to yield the title compound (4.Og, 0.012mol, 8%). 1H-NMR (CDCI3, 400MHz): δ 0.92-1.00 (m, 9H), 1.96-2.06 (m, 4H), 2.08-2.17 (m, 1H), 2.20-2.27 (m, 1 H), 2.80-2.88 (m, 2H), 2.91-2.93 (m, 1 H), 2.95 (m, 3H), 4.52 (d, 1 H), 4.81-4.85 (m, 1 H), 5.00 (t, 1 H), 7.24-7.39 (m, 5H). LRMS m/z APCI 333 [MH]+
Preparation 4
(2S)-2-(Aminomethyl)-5-ethylhept-4-enoic acid

The amide of Preparation 3 (14.03g, 0.042mol) was dissolved in 1 ,4-dioxane (150ml), water (150ml) was added and the reaction mixture heated to reflux for 112 hours. The solvent was evaporated in vacuo and the residue triturated with acetonitrile to yield the title compound as a white solid (3.83g, 0.021 mol, 49%).
1H-NMR (CDCI3, 400MHz): δ 1.00 (m, 6H), 2.00-2.15 (m, 4H), 2.20-2.35 (m, 1 H), 2.40-2.55 (m, 2H), 2.90-3.00 (m, 2H), 5.15 (t, 1 H). LRMS m/z APCI 186 [MH]+
Preparation 5
(2S)-2-(Aminomethyl)-5-ethyl-N-r(1S.2SV2-hvdroxy-1-methyl-2-phenylethvn-N- methylheptanamide

Suitable PDE7 inhibitor compounds of the present invention may be prepared as described herein below or in the aforementioned patent literature references, which are illustrated by the following non-limiting examples and intermediates.
The following examples and preparations illustrate the preparation of certain preferred PDE7 inhibitors described in US Provisional Patent Application Number 60/741854 (unpublished at the filing date of the present application):
1H Nuclear magnetic resonance (NMR) spectra were in all cases consistent with the proposed structures. Characteristic chemical shifts (δ) are given in parts per million (ppm) downfietd from tetramethylsilane using conventional abbreviations for designation of major peaks: e.g. s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad. The mass spectra (m/z) were recorded using either electrospray ionisation (ESI) or atmospheric pressure chemical ionisation (APCI). The following abbreviations have been used for common solvents: CDCI3, deuterochloroform; D6- DMSO, hexadeuterodimethylsulphoxide.
Example 1
C/s-3-[(8'-chloro-2'-oxo-2',3'-dihvdro-1'H-spiro[cvclohexane-1.4'-αuinazolinl-5'- vDoxylcvclobutanecarboxylic acid

To a solution of the alcohol of Preparation 8 (50mg, 0.14mmol) in 99.25: 0.75 acetonitrile: water (2ml) was added a solution of periodic acid (82mg, 0.359mmol) and chromium (Vl) oxide (1.6mg, 0.016mmol) in 99.25: 0.75 acetonitrile: water (2ml), maintaining the reaction temperature below 50C. The reaction mixture was stirred at room temperature for 18 hours. The reaction mixture was filtered and the residue washed with 99.25: 0.75 acetonitrile: water, 2N hydrochloric acid: methanol (5:1), water and methanol. The residue was dried in vacuo to yield the title compound as a white solid (28mg, 0.077mmol, 55%). 1H-NMR (D6-DMSO, 400MHz): δ 1.17 (m, 1 H), 1.40-1.65 (m, 5H), 1.79 (m, 2H), 2.16 (m, 2H), 2.48 (m, 2H), 2.72 (m, 3H), 4.64 (m, 1 H), 6.43 (d, 1H), 7.0 (s, 1 H), 7.21 (d, 1 H), 7.90 (s, 1 H), 12.26 (bs, 1 H). LRMS m/z (APCI): 365[M+H]+, 406[M+CH3CN+H]+
Example 2 Trans-S-ffδ'-chloro^'-oxo^'.S'-dihvdro-i'H-spirofcvclohexane-i ^'-quinazolini-δ'- vDoxylcvclobutanecarboxvlic acid

To a solution of the alcohol of Preparation 11 (2.05g, 5.84mmol) in acetonitrile containing 0.75% water (50ml) was added a solution of chromium (Vl) oxide (12mg, 0.11mmol) and periodic acid (3.33g, 14.6mmol) and the reaction mixture stirred at 40°C for 96 hours. Water (100ml) was added and the suspension stirred for 2 hours. The resulting precipitate was collected by filtration, washed with water and dried in vacuo to yield the title compound (1.9Og, 5.2mmol, 89%). 1H-NMR (D6-DMSO, 400MHz): δ 1.2 (m, 1 H), 1.2 (m, 2H), 1.6 (m, 2H), 1.8 (m, 2H), 2.3 (m, 2H), 2.6 (m, 2H), 3.1 (m, 1 H), 3.2 (s, 1H), 4.0 (bs, 1 H), 4.8 (m, 1H)1 6.4 (d, 1 H), 7.0 (s, 1 H), 7.2 (d, 1 H), 7.9 (S, 1 H). LRMS m/z (APCI) 365 [MH]+
Preparation 1 3-f(Benzyloxy)methyll-2.2-dichlorocvclobutanone

Zinc dust (6.54g, 0.1 mol) was suspended in water (30ml) and argon bubbled through the suspension for 15 minutes before the addition of copper (II) sulphate (780mg, 3.1mmol). The reaction mixture was stirred at room temperature, under argon for 30 minutes. The mixture was filtered under a stream of argon and the solid washed with water (100ml), acetone (100ml) and dried in vacuo for 4 hours. The resultant zinc/ copper couple was suspended in diethyl ether: 1 ,2- dimethoxyethane (70ml: 10ml) under argon and allyl benzyl ether (4.6ml, 30mmol) added. A solution of trichloroacetyl chloride (9ml, 81 mmol) in diethyl ether: 1 ,2-dimethoxyethane (58ml: 7ml) was added dropwise over 45 minutes and the reaction mixture heated to reflux for 48 hours. The reaction mixture was filtered through Celite®and the salts washed with diethyl ether (3x70ml). The filtrate was evaporated in vacuo and the residue redissolved in hexane (150ml). The remaining solids were removed by filtration and the filtrate washed with a saturated aqueous solution of sodium hydrogen carbonate (2x100ml), brine (80ml), dried over magnesium sulphate, filtered and evaporated in vacuo. The crude material was purified by column chromatography over silica gel eluting with 10-25% hexane: diethyl ether. The title compound was obtained as a yellow oil (7.03g, 27.3mmol, 91%).
1H-NMR (CDCI3, 400MHz): δ 3.11-3.21 (m, 2H), 3.48 (m, 1 H), 3.70 (m, 1H), 3.85 (m, 1H), 7.35 (m, 5H), 4.58 (s, 2H).
Preparation 2 3-[(Benzyloxy)methyllcvclobutanone
To a solution of the dichlorocyclobutanone of Preparation 1 (5.98g, 23.08mmol) in methanol saturated with ammonium chloride (90ml) was added zinc powder (9.25g, 142mmol) and the reaction mixture stirred at room temperature for 2 hours. Ammonium chloride was added and the reaction mixture stirred at room temperature for a further 6 hours. The mixture was filtered through Celite® and the salts washed with diethyl ether (50ml). The filtrate was concentrated in vacuo and the residue partitioned between diethyl ether (200ml) and water (100ml). The mixture was filtered and the organic phase washed with water, dried over magnesium sulphate, filtered and evaporated in vacuo. The title compound was obtained as a yellow oil (3.7g, 19.5mmol, 84%). 1H-NMR (CDCI3, 400MHz): $ 2.69 (m, 1 H), 2.90 (m, 2H), 3.11 (m, 2H), 3.60 (d, 2H), 4.56 (s, 2H), 7.34 (m, 5H).
Preparation 3
C/s-3-[(benzyloxy)methvHcvclob_ιtanol

To a solution of the cyclobutanone of Preparation 2 (1.166g, 6.13mmol) in tetrahydrofuran stirring at -7O0C, was added dropwise a 1M solution of lithium tri-sec-butylborohydride in tetrahydrofuran (40ml), maintaining the reaction temperature below -650C. The reaction was allowed to warm to room temperature over 18 hours. The reaction mixture was quenched with a saturated aqueous solution of sodium hydrogen carbonate (25ml) then cooled to 50C. 30% Aqueous hydrogen peroxide (4ml) was added dropwise, maintaining the reaction temperature below 1O0C. The mixture was extracted from water into ethyl acetate (50ml) and the combined organic phases washed with brine (30ml), dried over magnesium sulphate, filtered and evaporated in vacuo. The crude material was purified by column chromatography over silica gel eluting with 25-50% ethyl acetate: pentane to yield a colourless oil (1.05g, 5.5mmol, 89%). 1H-NMR indicated that a 15:1 ratio of cis: trans isomers had been obtained.
1H-NMR (CDCI3, 400MHz): δ 1.70 (m, 2H), 2.10 (m, 1H), 2.46 (m, 2H), 3.45 (d, 2H), 4.15 (q, 1H)1 4.52 (S, 2H), 7.33 (m, 5H).
Preparation 4 7rans-3-r(benzyloxy)methyllcvclobutyl 4-nitrobenzoate

A solution of diethyl azodicarboxylate (2g, 11.δmmol) in tetrahydrofuran (5ml) was added dropwise to a stirred solution of the cyclobutyl alcohol of Preparation 3 (1.05g, 5.47mmol), 4- nitrobenzoic acid (1.82g, 10.9mmol) and triphenylphosphine (3.016g, 11.5mmol) in tetrahydrofuran (20ml) at O0C. The reaction mixture was stirred at room temperature for 18 hours.
The solvent was evaporated in vacuo and the residue redissolved in diethyl ether (30ml). The remaining solid was removed by filtration and the filtrate evaporated in vacuo. The crude material was purified by column chromatography over silica gel eluting with 1:10 to 1 :3 ethyl acetate: pentane to yield a colourless oil (1.64g, 4. δmmol, 88%). 1H-NMR indicated that a 15:1 ratio of trans: cis isomers had been obtained.
1H-NMR (CDCI3, 400MHz): δ 2.40 (m, 4H), 2.67 (m, 1 H), 3.53 (d, 2H), 4.57 (s, 2H), 5.36 (q, 1 H),
7.37 (m, 5H), 8.20 (d, 2H), 8.29 (d, 2H).
Preparation 5 rrans-3-r(benzyloxy)methyllcvclobutanol
To a solution of the p-nitroester of Preparation 4 (1.64g, 4.8mmol) in 1,4-dioxane (35ml) was added a solution of sodium hydroxide (385mg, 9.6mmol) in water (25ml) and the reaction mixture stirred at room temperature for 30 minutes. Acetic acid (0.4ml, 7mmol) was added and the mixture concentrated in vacuo. The residue was extracted from a saturated aqueous solution of sodium hydrogen carbonate into ethyl acetate (20ml), dried over magnesium sulphate, filtered and evaporated in vacuo. The title compound was obtained as a yellow oil (850mg, 4.4mmol, 92%). 1H-NMR (CDCI3, 400MHz): δ 2.08 (m, 2H), 2.20 (m, 2H), 2.47 (m, 1 H), 3.47 (d, 2H), 4.39 (q, 1H), 4.52 (s, 2H), 7.34 (m, 5H).
Preparation 6
7rans-3-[(benzyloxy)methyllcvclobutyl p-toluenesulphonate

p-Toluenesulphonyl chloride (1.18g, 6.2mmol) was added portionwise to a stirred solution of the cyclobutanol of Preparation 5 (850mg, 4.42mmol) in pyridine (5ml) at O0C and the reaction mixture stirred at room temperature for 18 hours. The solvent was concentrated in vacuo and the residue redissolved in ethyl acetate (30ml), washed with 2N hydrochloric acid, (30ml) a saturated aqueous solution of sodium hydrogen carbonate (30ml), brine (30ml), dried over magnesium sulphate, filtered and evaporated in vacuo. The crude material was purified by column chromatography over silica gel eluting with dichloromethane. The title compound was obtained as a colourless oil (1.53g, 4.4mmol).
1H-NMR (CDCI3, 400MHz): $ 2.15 (m, 2H), 2.31 (m, 2H), 2.44 (s, 3H), 2.49 (m, 1 H), 3.4 (d, 2H), 4.49 (s, 2H), 4.93 (q, 1 H), 7.32 (m, 7H), 7.75 (d, 2H).
Preparation 7
5'-({C>s-3-[(benzyloxy)methyl1cvclobutyl}oxy)-8'-chloro-1'H-spirorcvclohexane-1.4'-quinazolin1-
2'(3'HVone
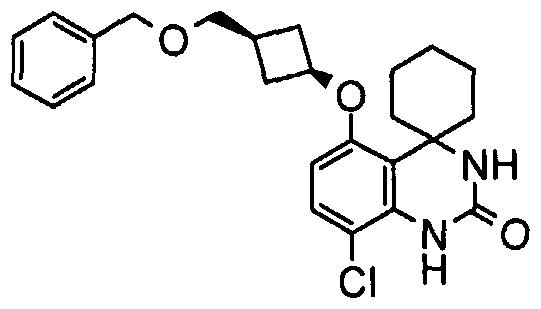
8M2hloro-5'-hydroxy-1Η-spiro[cyclohexane-1 ,4'-quinazolin]-2'(3Η)-one (prepared as described in Bioorg. Med. Chem. Lett, (2004), 14 (18), 4627-4632) (640mg, 2.4mmol), potassium carbonate (400mg, 2.9mmol) and 18-crown-6 (767mg, 2.9mmol) were combined in dimethylformamide (8ml) and the reaction mixture heated to 8O0C. A solution of the tosylate of Preparation 6 (1g, 2.9mmol) in dimethylformamide was added in 3 portions and the mixture heated at 8O0C for a further 18 hours. The reaction mixture was partitioned between ethyl acetate (100ml) and water (150ml) and the solid collected by filtration. The phases were separated and the aqueous phase reextracted with ethyl acetate, diluted with brine and again extracted into ethyl acetate. The combined organic phases were concentrated in vacuo and the residue triturated with water and methanol. The combined crude products were purified by column chromatography over silica gel eluting with dichloromethane to dichloromethane: ethyl acetate (1 :1) to obtain the title compound as an off- white solid (685mg, 1.156mmol, 64%). 1H-NMR (D6-DIvISO1 400MHz): δ 1.1 (m, 1 H), 1.4 (m, 2H), 1.6 (m, 3H), 1.7 (m, 2H), 1.8 (m, 2H), 2.3 (m, 1 H), 2.5 (m, 4H), 3.4 (s, 2H), 4.4 (s, 2H), 4.6 (m, 1 H), 6.4 (d, 1 H), 7.0 (s, 1 H), 7.2 (d, 1 H), 7.3 (m, 5H), 7.8 (s, 1 H).
Preparation 8 8'-Chloro-5'-lfc/s-3-(hvdroxymethyl)cvclobutvnoxy)-1'H-spiro[cvclohexane-1.4'-quinazolin1-2'(3'H)- one

A 2M solution of boron trichloride-dimethyl sulfide complex in dichloromethane (1.8ml, 3.6mmol) was added to a suspension of the benzyl alcohol of Preparation 7 (400mg, 0.9mmol) in dichloromethane (10ml) and the reaction mixture stirred at room temperature overnight. A saturated aqueous solution of sodium hydrogen carbonate (10ml) was added and the mixture stirred for 5 minutes. Dichloromethane and water were added and the resultant solid collected by filtration. The title compound was obtained as a white solid (230mg, 0.657mmol, 73%). 1H-NMR (D6-DMSO, 400MHz): δ 1.17 (m, 1 H), 1.42 (m, 2H), 1.57 (m, 3H), 1.82 (m, 4H), 2.05 (m, 1H), 2.45 (m, 4H), 3.38 (t, 2H), 4.58 (m, 2H), 6.41 (d, 1 H), 6.99 (s, 1H), 7.20 (d, 1H), 7.86 (s, 1H). LRMS m/z (APCI) 351 [MH]+
Preparation 9
C/s-3-f(benzyloxy)methyl1cvclobutyl p-toluenesulphonate

Pyridine (14.3ml, 176mmol) and p-toluenesulphonyl chloride (20.2g, 105.9mmol) were added to a solution of the alcohol of Preparation 3 (17g, 88.4mmol) in dichloromethane (90ml) stirring at 50C and the reaction mixture was stirred at room temperature for 18 hours. The reaction mixture was diluted with dichloromethane (50ml), washed with 2N hydrochloric acid (50ml), a saturated aqueous solution of sodium hydrogen carbonate (50ml), dried over magnesium sulphate, filtered and evaporated in vacuo. The crude material was purified by column chromatography over silica gel eluting with pentane: ethyl acetate (19:1 , 9:1 , 4:1). The title compound was obtained as a colourless oil (24.8g, 71.6mmol, 81%).
1H-NMR (CDCI3, 400MHz): δ 1.95 (m, 2H), 2.1 (m, 1H)1 2.35 (m, 2H), 2.45 (s, 3H), 3.4 (m, 2H), 4.5 (s, 2H), 4.7 (m, 1 H), 7.3 (m, 7H), 7.8 (m, 2H). LRMS m/z (ESI) 347 [MH]+
Preparation 10
5'-((rrans-3-f(benzyloxy)methvncyclobutyl)oxy)-8'-chloro-1'H-spirofcyclohexane-1.4'-αuinazolin1-
2Y3'H)-one

Method A
Caesium carbonate (730mg, 2.24mmol) was added to a stirred suspension of 8'-chloro-5'- hydroxy-1'H-spiro[cyclohexane-1 ,4'-quinazolin]-2'(3'H)-one (500mg, 1.87mmol) in dimethylformamide (2ml) and the reaction mixture heated to 8O0C. After 5 minutes a solution of the tosylate of Preparation 9 (710mg, 2.05mmol) in dimethylformamide (1 ml) was added and the reaction mixture heated at 8O0C for 18 hours. The mixture was extracted from brine (60ml) into ethyl acetate (1x80ml, 2x30ml), washed with brine (3x100ml), dried over magnesium sulphate, filtered and evaporated in vacuo. The title compound was obtained as a slightly impure cream solid (800mg, 0.96mmol, 96%).
Method B
To a solution of S'-chloro-δ'-hydroxy-IΗ-spiroIcyclohexane-i ^'-quinazolin^XS'HJ-one (950mg, 3.56mmol) in dimethylformamide (12ml) stirring at 8O0C was added potassium carbonate (590mg, 4.27mmol) and 18-crown-6 (1.1g, 4.27mmol). The reaction mixture was stirred for 10 minutes before the addition of a solution of the tosylate of Preparation 9 (1.48g, 4.27mmol) in dimethylformamide (3ml). The reaction mixture was heated at 8O0C for 24 hours. The mixture was poured onto water: methanol (75ml: 25ml), stirred for 10 minutes and the resulting precipitate collected by filtration and washed with methanol. The solid was dissolved in dichloromethane, filtered through Celite®and the resulting filtrate evaporated in vacuo to yield the title compound as a 9:1 mixture of trans: cis isomers (887mg, 2.0mmol, 56%).
1H-NMR (CDCI3, 400MHz): δ 1.3 (m, 1H), 1.5-1.9 (m, 9H), 2.4 (m, 3H), 2.6 (m, 2H), 3.5 (d, 2H), 4.6 (s, 2H), 4.75 (m, 1 H), 5.85 (bs, 1H), 6.25 (d, 1 H), 7.05 (bs, 1 H), 7.1 (d, 1 H), 7.3-7.4 (m, 5H). LRMS m/z (ESI) 441 [MH]+
Preparation 11
8'-Chloro-5'-(ffra/?s-3-(hvdroxymethyl)cvclobutyl]oxy}-1'H-spiro[cvclohexane-1.4'-αuinazolinl-
2'(3'm-one

A 2M solution of boron trichloride-dimethyl sulfide complex in dichloromethane (15ml) was added dropwise to a solution of the benzyl ether of Preparation 10 (3.5g, 7.9mmol) in dichloromethane (80ml) and the reaction mixture stirred at room temperature for 18 hours. The mixture was poured into a saturated aqueous solution of sodium hydrogen carbonate (200ml) and stirred until the effervescence ceased. The mixture was extracted into dichloromethane (1x200ml, 2x100ml), washed with brine (50ml), dried over magnesium sulphate, filtered and evaporated in vacuo. The crude material was recrystalised from acetonitrile to yield the title compound as a 91:9 ratio of trans: cis products (2.33g, 6.65mmol, 84%). 1H-NMR (CDCI3, 400MHz): δ 1.3 (m, 1 H), 1.5 (m, 2H), 1.8 (m, 5H), 2.4 (m, 4H), 2.6 (m, 3H), 3.8 (d, 2H), 4.8 (m, 1H), 5.7 (bs, 1H), 6.25 (d, 1 H), 7.0 (bs, 1 H), 7.1 (d, 1 H). LRMS m/z (ESI) 351 [MH]+
Claims
1. A combination including a PDE7 inhibitor and an alpha-2-delta ligand.
2. A combination according to claim 1 , wherein the PDE7 inhibitor is selected from: 5'-carboxypropoxy-8'-chloro-spiro[cyclohexane-1-4'-(3',4l-dihydro)quinazolin]-2'(1'H)-one; c/s-3-[(8'-chloro-2l-oxo-2',3'-dihydro-1'H-spiro[cyclohexane-1 ,4'-quinazolin]-5'- yl)oxy]cyclobutanecarboxylic acid; and frans-3-[(8'-chloro-2'-oxo-2',3'-dihydro-1'H-spiro[cyclohexane-1 ,4'-quinazolin]-5'- yl)oxy]cyclobutanecarboxylic acid; or a pharmaceutically acceptable salt or solvate thereof.
3. A combination according to claim 1 or claim 2 wherein the PDE7 inhibitor is 5'- carboxypropoxy-8'-chloro-spiro[cyclohexane-1 -4'-(3',4'-dihydro)quinazolin]-2'(1 Η)-one or a pharmaceutically acceptable salt or solvate thereof.
4. A combination according to claim 1 or claim 2, wherein the PDE7 inhibitor is c/s-3-[(8'- chloro^'-oxo^'.S'-dihydro-IΗ-spiroIcyclohexane-i ^'-quinazolinl-δ'-yOoxylcyclobutanecarboxylic acid or a pharmaceutically acceptable salt or solvate thereof.
5. A combination according to claim 1 or claim 2 wherein the PDE7 inhibitor is trans-Z-[(8'- chloro-2'-oxo-2',3'-dihydro-1'H-spiro[cyclohexane-1 ,4'-quinazolin]-5'-yl)oxy]cyclobutanecarboxylic acid or a pharmaceutically acceptable salt or solvate thereof.
6. A combination according to any one of claims 1 to 5, wherein the alpha-2-delta ligand is selected from gabapentin, pregabalin, [(1R,5R,6S)-6-(aminomethyl)bicyclo[3.2.0]hept-6-yl]acetic acid, 3-(1-aminomethyl-cyclohexylmethyl)-4H-[1 ,2,4]oxadiazol-5-one, C-[1-(1 H-tetrazol-5- ylmethyl)-cycloheptyl]-methylamine, (3S,4S)-(1-aminomethyl-3,4-dimethyl-cyclopentyl)-acetic acid, (1 α,3α,5α)(3-amino-methyl-bicyclo[3.2.0]hept-3-yl)-acetic acid, (3S,5R)-3-aminomethyl- 5-methyl-octanoic acid, (3S,5R)-3-amino-5-methyl-heptanoic acid, (3S,5R)-3-amino-5-methyl- nonanoic acid , (SS.δRJ-S-amino-δ-methyl-octanoic acid, (2S,4S)-4-(3-chlorophenoxy)proline, (2S,4S)-4-(3-fluorobenzyl)proline, (2S)-2-amino-4-ethyl-2-methylhexanoic acid, and (2S)-2- aminomethyl-5-ethyl-heptanoic acid, and pharmaceutically acceptable salts and solvates thereof.
7. A combination according to any one of claims 1 to 6, wherein the alpha-2-delta ligand is gabapentin or a pharmaceutically acceptable salt or solvate thereof.
8. A combination according to any one of claims 1 to 6, wherein the alpha-2-delta ligand is pregabalin or a pharmaceutically acceptable salt or solvate thereof.
9. A combination according to any one of claims 1 to 6, wherein the alpha-2-delta ligand is (2S,4S)-4-(3-fluorobenzyl)proline or a pharmaceutically acceptable salt or solvate thereof.
10. A combination according to any one of claims 1 to 6, wherein the alpha-2-delta ligand is (2S)-2-amino-4-ethyl-2-methylhexanoic acid or a pharmaceutically acceptable salt or solvate thereof..
I
11. A combination according to any one of claims 1 to 6, wherein the alpha-2-delta ligand is
(2S)-2-aminomethyl-5-ethyl-heptanoic acid or a pharmaceutically acceptable salt or solvate thereof.
12. A pharmaceutical composition including a combination according to any one of claims 1 to 11 , aud a pharmaceutically acceptable excipient.
13. A pharmaceutical composition for the treatment of pain, including a therapeutically effective amount of a combination according to any one of claims 1 to 11 , and a pharmaceutically acceptable excipient.
14. A combination of a PDE7 inhibitor and an alpha-2-delta ligand according to any one of claims 1 to 11 for use as a medicament.
15. Use of a combination of a PDE7 inhibitor and an alpha-2-delta ligand according to any one of claims 1 to 11 in the manufacture of a medicament for the treatment of pain.
16. Use according to claim 15 wherein the pain is neuropathic pain.
17. A method for the treatment of pain, including simultaneous, sequential or separate administration, in combination, of therapeutically effective amounts of a PDE7 inhibitor and an alpha-2-delta ligand, to a mammal in need of said treatment.
18. The method according to claim 17 wherein the pain is neuropathic pain.
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0504209A GB0504209D0 (en) | 2005-03-01 | 2005-03-01 | New use of PDE7 inhibitors |
| GB0504209.8 | 2005-03-01 | ||
| US67576105P | 2005-04-27 | 2005-04-27 | |
| US60/675,761 | 2005-04-27 | ||
| US74185405P | 2005-12-02 | 2005-12-02 | |
| US60/741,854 | 2005-12-02 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006092692A1 true WO2006092692A1 (en) | 2006-09-08 |
Family
ID=36190470
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2006/000385 WO2006092692A1 (en) | 2005-03-01 | 2006-02-16 | Use of combinations of pde7 inhibitors and alpha-2-delty ligands for the treatment of neuropathic pain |
Country Status (1)
| Country | Link |
|---|---|
| WO (1) | WO2006092692A1 (en) |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007052134A1 (en) * | 2005-11-04 | 2007-05-10 | Pfizer Limited | (2s)-2-aminomethyl-5-ethyl heptanoic acid its pharmaceutical use |
| NL2000336C2 (en) * | 2005-12-02 | 2007-08-07 | Pfizer Ltd | Spirocyclic derivatives. |
| WO2008119057A2 (en) | 2007-03-27 | 2008-10-02 | Omeros Corporation | The use of pde7 inhibitors for the treatment of movement disorders |
| USRE41920E1 (en) | 1996-07-24 | 2010-11-09 | Warner-Lambert Company Llc | Isobutylgaba and its derivatives for the treatment of pain |
| WO2012064667A2 (en) | 2010-11-08 | 2012-05-18 | Omeros Corporation | Treatment of addiction and impulse-control disorders using pde7 inhibitors |
| US8242126B2 (en) | 2007-10-03 | 2012-08-14 | Sanofi | Quinazolinedione derivatives, preparation thereof and therapeutic uses thereof |
| EP2558101A1 (en) * | 2010-04-15 | 2013-02-20 | The Royal Institution for the Advancement of Learning / McGill University | Topical treatments for pain |
| US8637528B2 (en) | 2007-03-27 | 2014-01-28 | Omeros Corporation | Use of PDE7 inhibitors for the treatment of movement disorders |
| US8722659B2 (en) | 2009-04-09 | 2014-05-13 | Sanofi | Quinazolinedione derivatives, preparation thereof and various therapeutic uses thereof |
| US8846654B2 (en) | 2009-04-09 | 2014-09-30 | Sanofi | Therapeutic applications in the cardiovascular field of quinazolinedione derivatives |
| US9220715B2 (en) | 2010-11-08 | 2015-12-29 | Omeros Corporation | Treatment of addiction and impulse-control disorders using PDE7 inhibitors |
| WO2024038089A1 (en) | 2022-08-18 | 2024-02-22 | Mitodicure Gmbh | Use of a therapeutic agent with phosphodiesterase-7 inhibitory activity for the treatment and prevention of diseases associated with chronic fatigue, exhaustion and/or exertional intolerance |
Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999021824A1 (en) * | 1997-10-27 | 1999-05-06 | Warner-Lambert Company | Cyclic amino acids and derivatives thereof useful as pharmaceutical agents |
| WO2001028978A1 (en) * | 1999-10-20 | 2001-04-26 | Warner-Lambert Company | Bicyclic amino acids as pharmaceutical agents |
| WO2002074754A1 (en) * | 2001-03-21 | 2002-09-26 | Warner-Lambert Company Llc | New spirotricyclic derivatives and their use as phosphodiesterase-7 inhibitors |
| WO2003082807A2 (en) * | 2002-03-28 | 2003-10-09 | Warner-Lambert Company Llc | Amino acids with affinity for the alpha-2-delta-protein |
| WO2004016259A1 (en) * | 2002-08-15 | 2004-02-26 | Pfizer Limited | Synergistic combination of an alpha-2-delta ligand and a pdev inhibitor for use in the treatment of pain |
| EP1400244A1 (en) * | 2002-09-17 | 2004-03-24 | Warner-Lambert Company LLC | New spirocondensed quinazolinones and their use as phosphodiesterase inhibitors |
| WO2004039367A1 (en) * | 2002-10-31 | 2004-05-13 | Pfizer Limited | Proline derivatives having affinity for the calcium channel alpha-2-delta subunit |
| US6774132B1 (en) * | 1999-07-21 | 2004-08-10 | Astrazeneca Ab | Spirooxindole derivatives that act as analgesics |
| US20050277659A1 (en) * | 2004-03-29 | 2005-12-15 | Yoshinobu Hashizume | Alpha aryl or heteroaryl methyl beta piperidino propanoic acid compounds as ORL1-receptor antagonists |
-
2006
- 2006-02-16 WO PCT/IB2006/000385 patent/WO2006092692A1/en not_active Application Discontinuation
Patent Citations (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999021824A1 (en) * | 1997-10-27 | 1999-05-06 | Warner-Lambert Company | Cyclic amino acids and derivatives thereof useful as pharmaceutical agents |
| US6774132B1 (en) * | 1999-07-21 | 2004-08-10 | Astrazeneca Ab | Spirooxindole derivatives that act as analgesics |
| WO2001028978A1 (en) * | 1999-10-20 | 2001-04-26 | Warner-Lambert Company | Bicyclic amino acids as pharmaceutical agents |
| WO2002074754A1 (en) * | 2001-03-21 | 2002-09-26 | Warner-Lambert Company Llc | New spirotricyclic derivatives and their use as phosphodiesterase-7 inhibitors |
| WO2003082807A2 (en) * | 2002-03-28 | 2003-10-09 | Warner-Lambert Company Llc | Amino acids with affinity for the alpha-2-delta-protein |
| WO2004016259A1 (en) * | 2002-08-15 | 2004-02-26 | Pfizer Limited | Synergistic combination of an alpha-2-delta ligand and a pdev inhibitor for use in the treatment of pain |
| EP1400244A1 (en) * | 2002-09-17 | 2004-03-24 | Warner-Lambert Company LLC | New spirocondensed quinazolinones and their use as phosphodiesterase inhibitors |
| WO2004039367A1 (en) * | 2002-10-31 | 2004-05-13 | Pfizer Limited | Proline derivatives having affinity for the calcium channel alpha-2-delta subunit |
| US20050277659A1 (en) * | 2004-03-29 | 2005-12-15 | Yoshinobu Hashizume | Alpha aryl or heteroaryl methyl beta piperidino propanoic acid compounds as ORL1-receptor antagonists |
Non-Patent Citations (5)
| Title |
|---|
| BERNARDELLI, PATRICK ET AL: "Spiroquinazolinones as novel, potent, and selective PDE7 inhibitors. Part 2: optimization of 5,8-disubstituted derivatives", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS , 14(18), 4627-4631 CODEN: BMCLE8; ISSN: 0960-894X, 2004, XP002378324 * |
| EUGENE G ET AL: "SELECTIVITY OF SILDENAFIL AND OTHER PHOSPHODIESTERASE TYPE 5 (PDE5) INHIBITORS AGAINST ALL HUMAN PHOSPHODIESTERASE FAMILES", EUROPEAN UROLOGY SUPPLEMENTS, vol. 1, no. 1, January 2002 (2002-01-01), pages 63, XP001157408, ISSN: 1569-9056 * |
| GUAY ET AL: "Pregabalin in neuropathic pain: A more ''pharmaceutically elegant'' gabapentin?", THE AMERICAN JOURNAL OF GERIATRIC PHARMACOTHERAPY, EXCERPTA MEDICA, vol. 3, no. 4, December 2005 (2005-12-01), pages 274 - 287, XP005304524, ISSN: 1543-5946 * |
| LAIRD M A ET AL: "USE OF GABAPENTIN IN THE TREATMENT OF NEUROPATHIC PAIN", ANNALS OF PHARMACOTHERAPY, vol. 34, no. 6, June 2000 (2000-06-01), pages 802 - 807, XP008036307, ISSN: 1060-0280 * |
| LORTHIOIS, EDWIGE ET AL: "Spiroquinazolinones as novel, potent, and selective PDE7 inhibitors. Part 1", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS , 14(18), 4623-4626 CODEN: BMCLE8; ISSN: 0960-894X, 2004, XP002378323 * |
Cited By (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| USRE41920E1 (en) | 1996-07-24 | 2010-11-09 | Warner-Lambert Company Llc | Isobutylgaba and its derivatives for the treatment of pain |
| WO2007052134A1 (en) * | 2005-11-04 | 2007-05-10 | Pfizer Limited | (2s)-2-aminomethyl-5-ethyl heptanoic acid its pharmaceutical use |
| NL2000336C2 (en) * | 2005-12-02 | 2007-08-07 | Pfizer Ltd | Spirocyclic derivatives. |
| WO2007063391A3 (en) * | 2005-12-02 | 2007-09-13 | Pfizer Ltd | Spirocyclic quinazoline derivatives as pde7 inhibitors |
| US7507742B2 (en) | 2005-12-02 | 2009-03-24 | Pfizer Inc. | Spirocyclic derivatives |
| US8637528B2 (en) | 2007-03-27 | 2014-01-28 | Omeros Corporation | Use of PDE7 inhibitors for the treatment of movement disorders |
| WO2008119057A2 (en) | 2007-03-27 | 2008-10-02 | Omeros Corporation | The use of pde7 inhibitors for the treatment of movement disorders |
| US9119822B2 (en) | 2007-03-27 | 2015-09-01 | Omeros Corporation | Use of PDE7 inhibitors for the treatment of movement disorders |
| US8242126B2 (en) | 2007-10-03 | 2012-08-14 | Sanofi | Quinazolinedione derivatives, preparation thereof and therapeutic uses thereof |
| US8846654B2 (en) | 2009-04-09 | 2014-09-30 | Sanofi | Therapeutic applications in the cardiovascular field of quinazolinedione derivatives |
| US8722659B2 (en) | 2009-04-09 | 2014-05-13 | Sanofi | Quinazolinedione derivatives, preparation thereof and various therapeutic uses thereof |
| EP2558101A4 (en) * | 2010-04-15 | 2013-09-18 | Univ Mcgill | Topical treatments for pain |
| EP2558101A1 (en) * | 2010-04-15 | 2013-02-20 | The Royal Institution for the Advancement of Learning / McGill University | Topical treatments for pain |
| WO2012064667A2 (en) | 2010-11-08 | 2012-05-18 | Omeros Corporation | Treatment of addiction and impulse-control disorders using pde7 inhibitors |
| US9220715B2 (en) | 2010-11-08 | 2015-12-29 | Omeros Corporation | Treatment of addiction and impulse-control disorders using PDE7 inhibitors |
| US11207275B2 (en) | 2010-11-08 | 2021-12-28 | Omeros Corporation | Treatment of addiction and impulse-control disorders using PDE7 inhibitors |
| US11464785B2 (en) | 2010-11-08 | 2022-10-11 | Omeros Corporation | Treatment of addiction and impulse-control disorders using PDE7 inhibitors |
| EP4275752A2 (en) | 2010-11-08 | 2023-11-15 | Omeros Corporation | Treatment of addiction and impulse-control disorders using pde7 inhibitors |
| WO2024038089A1 (en) | 2022-08-18 | 2024-02-22 | Mitodicure Gmbh | Use of a therapeutic agent with phosphodiesterase-7 inhibitory activity for the treatment and prevention of diseases associated with chronic fatigue, exhaustion and/or exertional intolerance |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP3625214B1 (en) | Deuterated pyridone amides and prodrugs thereof as modulators of sodium channels | |
| WO2006092692A1 (en) | Use of combinations of pde7 inhibitors and alpha-2-delty ligands for the treatment of neuropathic pain | |
| CA2574600C (en) | Pyridine derivatives | |
| US20090111837A1 (en) | Use of pde7 inhibitors for the treatment of neuropathic pain | |
| CA2631535C (en) | Spirocyclic quinazoline derivatives as pde7 inhibitors | |
| EP3022175A1 (en) | Sulfonamides as modulators of sodium channels | |
| WO2012007877A2 (en) | Chemical compounds | |
| WO2007129188A1 (en) | Cyclopropanecarboxamide compound | |
| US20100216823A1 (en) | Spirocyclic Derivatives | |
| US20090227680A1 (en) | Amino Acid Derivatives | |
| JP4512052B2 (en) | New uses of PDE7 inhibitors | |
| WO2006056874A1 (en) | Salt form | |
| WO2013093688A1 (en) | Sulfonamide derivatives and use thereof as vgsc inhibitors | |
| JP2006241158A (en) | CONCOMITANT MEDICINE CONTAINING alpha-2-delta-LIGAND | |
| US20080293746A1 (en) | Combinations comprising pregabalin | |
| WO2007052134A1 (en) | (2s)-2-aminomethyl-5-ethyl heptanoic acid its pharmaceutical use |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DPE2 | Request for preliminary examination filed before expiration of 19th month from priority date (pct application filed from 20040101) | ||
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| NENP | Non-entry into the national phase |
Ref country code: RU |
|
| WWW | Wipo information: withdrawn in national office |
Country of ref document: RU |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 06710445 Country of ref document: EP Kind code of ref document: A1 |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 6710445 Country of ref document: EP |