WO2006074330A2 - Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme - Google Patents
Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme Download PDFInfo
- Publication number
- WO2006074330A2 WO2006074330A2 PCT/US2006/000402 US2006000402W WO2006074330A2 WO 2006074330 A2 WO2006074330 A2 WO 2006074330A2 US 2006000402 W US2006000402 W US 2006000402W WO 2006074330 A2 WO2006074330 A2 WO 2006074330A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- hydrogen
- adamantane
- methyl
- amino
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/18—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member
- C07D207/22—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/24—Oxygen or sulfur atoms
- C07D207/26—2-Pyrrolidones
- C07D207/273—2-Pyrrolidones with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to other ring carbon atoms
- C07D207/277—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/165—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/48—Drugs for disorders of the endocrine system of the pancreatic hormones
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/01—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C233/02—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having nitrogen atoms of carboxamide groups bound to hydrogen atoms or to carbon atoms of unsubstituted hydrocarbon radicals
- C07C233/11—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having nitrogen atoms of carboxamide groups bound to hydrogen atoms or to carbon atoms of unsubstituted hydrocarbon radicals with carbon atoms of carboxamide groups bound to carbon atoms of an unsaturated carbon skeleton containing six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/01—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C233/45—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by carboxyl groups
- C07C233/52—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by carboxyl groups with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by a carbon atom of a ring other than a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/57—Carboxylic acid amides having carbon atoms of carboxamide groups bound to carbon atoms of rings other than six-membered aromatic rings
- C07C233/58—Carboxylic acid amides having carbon atoms of carboxamide groups bound to carbon atoms of rings other than six-membered aromatic rings having the nitrogen atoms of the carboxamide groups bound to hydrogen atoms or to carbon atoms of unsubstituted hydrocarbon radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/57—Carboxylic acid amides having carbon atoms of carboxamide groups bound to carbon atoms of rings other than six-membered aromatic rings
- C07C233/63—Carboxylic acid amides having carbon atoms of carboxamide groups bound to carbon atoms of rings other than six-membered aromatic rings having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by carboxyl groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C237/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups
- C07C237/24—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atom of at least one of the carboxamide groups bound to a carbon atom of a ring other than a six-membered aromatic ring of the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/68—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member
- C07D211/72—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member with hetero atoms or with carbon atoms having three bonds to hetero atoms, with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D211/78—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/54—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/56—Amides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/02—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings
- C07D231/10—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members
- C07D231/12—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings not condensed with other rings having two or three double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D261/00—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings
- C07D261/02—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings
- C07D261/06—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members
- C07D261/08—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D333/00—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom
- C07D333/02—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings
- C07D333/04—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom
- C07D333/06—Heterocyclic compounds containing five-membered rings having one sulfur atom as the only ring hetero atom not condensed with other rings not substituted on the ring sulphur atom with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to the ring carbon atoms
- C07D333/24—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/02—Systems containing only non-condensed rings with a three-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/04—Systems containing only non-condensed rings with a four-membered ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/06—Systems containing only non-condensed rings with a five-membered ring
- C07C2601/08—Systems containing only non-condensed rings with a five-membered ring the ring being saturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/12—Systems containing only non-condensed rings with a six-membered ring
- C07C2601/14—The ring being saturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2603/00—Systems containing at least three condensed rings
- C07C2603/56—Ring systems containing bridged rings
- C07C2603/58—Ring systems containing bridged rings containing three rings
- C07C2603/70—Ring systems containing bridged rings containing three rings containing only six-membered rings
- C07C2603/74—Adamantanes
Definitions
- the present invention relates to compounds that are inhibitors of the 11-beta- hydroxysteroid dehydrogenase Type 1 enzyme.
- the present invention further relates to the use of inhibitors of 11-beta-hydroxysteroid dehydrogenase Type 1 enzyme for the treatment of non-insulin dependent type 2 diabetes, insulin resistance, obesity, lipid disorders, metabolic syndrome and other diseases and conditions that are mediated by excessive glucocorticoid action.
- Insulin is a hormone that modulates glucose and lipid metabolism. Impaired action of insulin (i.e., insulin resistance) results in reduced insulin-induced glucose uptake, oxidation and storage, reduced insulin-dependent suppression of fatty acid release from adipose tissue (i.e., lipolysis) and reduced insulin-mediated suppression of hepatic glucose production and secretion. Insulin resistance frequently occurs in diseases that lead to increased and premature morbidity and mortality.
- Diabetes mellitus is characterized by an elevation of plasma glucose levels (hyperglycemia) in the fasting state or after administration of glucose during a glucose tolerance test. While this disease may be caused by several underlying factors, it is generally grouped into two categories, Type 1 and Type 2 diabetes.
- Type 1 diabetes also referred to as Insulin Dependent Diabetes Mellitus (“IDDM”)
- IDDM Insulin Dependent Diabetes Mellitus
- type 2 diabetes also referred to as non-insulin dependent diabetes mellitus, or NIDDM
- insulin resistance is a significant pathogenic factor in the development of hyperglycemia.
- the insulin levels in type 2 diabetes patients are elevated (i.e., hyperinsulinemia), but this compensatory increase is not sufficient to overcome the insulin resistance.
- Persistent or uncontrolled hyperglycemia in both type 1 and type 2 diabetes mellitus is associated with increased incidence of macro vascular and/or microvascular complications including atherosclerosis, coronary heart disease, peripheral vascular disease, stroke, nephropathy, neuropathy and retinopathy.
- Insulin resistance is a component of the metabolic syndrome.
- diagnostic criteria for metabolic syndrome have been established. To qualify a patient as having metabolic syndrome, three out of the five following criteria must be met: elevated blood pressure above 130/85 mmHg, fasting blood glucose above 110 mg/dl, abdominal obesity above 40" (men) or 35" (women) waist circumference and blood lipid changes as defined by an increase in triglycerides above 150 mg/dl or decreased HDL cholesterol below 40 mg/dl (men) or 50 mg/dl (women). It is currently estimated that 50 million adults, in the US alone, fulfill these criteria. That population, whether or not they develop overt diabetes mellitus, are at increased risk of developing the macrovascular and microvascular complications of type 2 diabetes listed above.
- Type 2 diabetes Available treatments for type 2 diabetes have recognized limitations. Diet and physical exercise can have profound beneficial effects in type 2 diabetes patients, but compliance is poor. Even in patients having good compliance, other forms of therapy may be required to further improve glucose and lipid metabolism.
- One therapeutic strategy is to increase insulin levels to overcome insulin resistance. This maybe achieved through direct injection of insulin or through stimulation of the endogenous insulin secretion in pancreatic beta cells.
- Sulfonylureas e.g., tolbutamide and glipizide
- meglitinide are examples of drugs that stimulate insulin secretion (i.e., insulin secretagogues) thereby increasing circulating insulin concentrations high enough to stimulate insulin-resistant tissue.
- insulin and insulin secretagogues may lead to dangerously low glucose concentrations (i.e., hypoglycemia).
- insulin secretagogues frequently lose therapeutic potency over time.
- Alpha-glucosidase inhibitors e.g., acarbose
- acarbose may delay carbohydrate absorption from the gut after meals, which may in turn lower blood glucose levels, particularly in the postprandial period.
- these compounds may also cause gastrointestinal side effects.
- Glitazones i.e., 5-benzylthiazolidine-2,4-diones
- Glitazones are a newer class of compounds used in the treatment of type 2 diabetes. These agents may reduce insulin resistance in multiple tissues, thus lowering blood glucose. The risk of hypoglycemia may also be avoided.
- Glitazones modify the activity of the Peroxisome Proliferator Activated Receptor ("PPAR") gamma subtype. PPAR is currently believed to be the primary therapeutic target for the main mechanism of action for the beneficial effects of these compounds.
- Other modulators of the PPAR family of proteins are currently in development for the treatment of type 2 diabetes and/or dyslipidemia. Marketed glitazones suffer from side effects including bodyweight gain and peripheral edema.
- GLP-I Glucagon-Like Peptide 1
- DPP-IV Dipeptidyl Peptidase IV
- GLP-I Glucagon-Like Peptide 1
- DPP-IV Dipeptidyl Peptidase IV
- Other examples include: Inhibitors of key enzymes involved in the hepatic glucose production and secretion (e.g., fructose- 1,6-bisphosphatase inhibitors) and direct modulation of enzymes involved in insulin signaling (e.g., Protein Tyrosine Phosphatase-1B, or "PTP-IB").
- Another method of treating or prophylactically treating diabetes mellitus includes using inhibitors of 11- ⁇ -hydroxysteroid dehydrogenase Type 1 (1 l ⁇ -HSDl). Such methods are discussed in J.R. Seckl et al., Endocrinology, 142: 1371-1376, 2001 and references cited therein.
- Glucocorticoids are steroid hormones that are potent regulators of glucose and lipid metabolism. Excessive glucocorticoid action may lead to insulin resistance, type 2 diabetes, dyslipidemia, increased abdominal obesity and hypertension. Glucocorticoids circulate in the blood in an active form (i.e., Cortisol in humans) and an inactive form (i.e., cortisone in humans).
- 11 ⁇ -HSD 1 which is highly expressed in liver and adipose tissue, converts cortisone to Cortisol leading to higher local concentration of Cortisol. Inhibition of 1 l ⁇ -HSDl prevents or decreases the tissue specific amplification of glucocorticoid action thus imparting beneficial effects on blood pressure and glucose- and lipid-metabolism.
- One aspect of the present invention is directed toward a compound of formula (I), or a pharmaceutically acceptable salt, prodrug, salt of a prodrug, or a combination thereof
- a 1 , A 2 , A 3 and A 4 is selected from the group consisting of alkyl-NH-alkyl, alkylcarbonyl, alkylsulfonyl, cycloalkyl, cycloalkylcarbonyl, cycloalkylsulfonyl, arylcarbonyl, arylsulfonyl, heteroaryl, heteroarylalkyl, heteroaryloxyalkyl, heterocyclecarbonyl, heterocyclesulfonyl, aryl 1 , arylalkyl, aryloxyalkyl, carboxyalkyl, carboxycycloalkyl, haloalkyl, heterocyclealkyl, heterocycleoxyalkyl, -S(O) 2 -N(R 5 R 6 ), -NR 7 - [C(R 8 R 9 )] n -C(O)-R 10 , -OR 14a , -N(R 15 R 16 ),
- D is selected from the group consisting of a bond, -C(R 27 R 28 )-X- and -C(R 27 R 28 )-
- E is selected from the group consisting of a cycloalkyl, alkyl, aryl, heteroaryl and heterocycle, wherein the heteroaryl and the heterocycle are appended to the parent molecular moiety through an available carbon atom, or R 4 and E together with the atoms to which they are attached form a ring selected from the group consisting of cycloalkyl and heterocycle;
- X is selected from the group consisting of a bond, -N(R 31 )-, -O-, -S-, -S(O)- and - S(O) 2 -;
- R 1 is selected from the group consisting of hydrogen and alkyl;
- R 2 is selected from the group consisting of hydrogen, alkyl and cycloalkyl
- R 3 and R 4 are each independently selected from the group consisting of hydrogen, alkyl, carboxyalkyl, carboxycycloalkyl, cycloalkyl, haloalkyl, aryl, arylalkyl, heteroaryl, heteroarylalkyl, heterocycle and heterocyclealkyl, or R 3 and R 4 together with the atom to which they are attached form a ring selected from the group consisting of cycloalkyl and heterocycle;
- R 5 and R 6 are each independently selected from the group consisting of hydrogen, alkoxy, alkyl, alkylcarbonyl, alkylsufonyl, carboxy, carboxyalkyl, carboxycycloalkyl, cycloalkyl, cycloalkyloxy, cycloalkylsulfonyl, aryl, arylalkyl, arylalkylcarbonyl, arylcarbonyl, aryloxy, aryloxyalkyl, arylsulfonyl, heteroaryl, heteroarylalkyl, heteroarylalkylcarbonyl, heteroarylcarbonyl, heteroaryloxyalkyl, heteroarylsulfonyl, heterocycle, heterocyclealkyl, heterocyclealkylcarbonyl, heterocyclecarbonyl, heterocyclecarbonyl, heterocycleoxyalkyl, heterocycleoxy, heterocyclesulfonyl and hydroxy, or R 5 and R 6 together with the atom to which they are attached form
- R 8 and R 9 are each independently selected from the group consisting of hydrogen and alkyl, or R and R taken together with the atom to which they are attached form a ring selected from the group consisting of cycloalkyl and heterocycle;
- R 10 is selected from the group consisting of hydrogen, alkyl, carboxyalkyl, cycloalkyl, carboxycycloalkyl, aryl, arylalkyl, aryloxy, aryloxyalkyl, hydroxy, alkoxy, cycloalkyloxy, heteroaryl, heteroarylalkyl, heteroaryloxy, heteroaryloxyalkyl, heterocycle, heterocyclealkyl, heterocycleoxy, heterocycleoxyalkyl and -N(R R ); R 11 and R 12 are each independently selected from the group consisting of hydrogen and alkyl or R 11 and R 12 taken together with the atom to which they are attached form a ring selected from the group consisting of cycloalkyl and heterocycle; R 13 is selected from the group consisting of hydroxy and -N(R 34 R 35 ); R 14a is selected from the group consisting of carboxyalkyl, cycloalkyl, carboxycycloalkyl, aryl, arylalkyl,
- R 15 and R 16 are each independently selected from the group consisting of hydrogen, alkyl, alkylcarbonyl, carboxyalkyl, cycloalkyl, carboxycycloalkyl, aryl, arylalkyl, arylalkylcarbonyl, arylcarbonyl, aryloxyalkyl, heteroaryl, heteroarylalkyl, heteroarylalkylcarbonyl, heteroarylcarbonyl, heteroaryloxyalkyl, heteroarylsulfonyl, heterocycle, heterocyclealkyl, heterocyclecarbonyl, heterocycleoxyalkyl, heterocyclesulfonyl, alkylsufon
- R 18 and R 19 are each independently selected from the group consisting of hydrogen, alkoxy, alkyl, alkylsufonyl, carboxy, carboxyalkyl, carboxycycloalkyl, cycloalkyl, cycloalkyloxy, cycloalkylsulfonyl, aryl, arylalkyl, aryloxy, aryloxyalkyl, arylsulfonyl, heteroaryl, heteroarylalkyl, heteroaryloxyalkyl, heteroarylsulfonyl, heterocycle, heterocyclealkyl, heterocycleoxyalkyl, heterocycleoxy, heterocyclesulfonyl and hydroxy, or R 18 and R 19 together with the atom to which they are attached form a heterocycle;
- R , R and R are each independently selected from the group consisting of hydrogen, alkyl, aryl, arylalkyl, carboxyalkyl, carboxycycloalkyl, cycloalkyl, haloalkyl, heteroaryl, heteroarylalkyl, heterocycle and heterocyclealkyl;
- R 23 and R 24 are each independently selected from the group consisting of hydrogen, alkyl, alkylcarbonyl, alkylsulfonyl, aryl, arylcarbonyl, arylsulfonyl, carboxyalkyl, carboxycycloalkyl, cycloalkyl, cycloalkylcarbonyl, cycloalkylsulfonyl, heteroaryl, heteroarylcarbonyl, heteroarylsulfonyl, heterocycle, heterocyclecarbonyl and heterocyclesulfonyl;
- R 25 and R 26 are each independently selected from the group consisting of hydrogen, alkoxy, alkyl, alkylcarbonyl, alkylsulfonyl, aryl, arylcarbonyl, aryloxy, arylsulfonyl, carboxyalkyl, carboxycycloalkyl, cycloalkyl, cycloalkylcarbonyl, cycloalkyloxy, cycloalkylsulfonyl, heteroaryl, heteroarylcarbonyl, heteroaryloxy, heteroarylsulfonyl, heterocycle, heterocyclecarbonyl, heterocycleoxy, heterocyclesulfonyl and hydroxy, or R 25 and R 26 together with the nitrogen to which they are attached form a ring selected from the group consisting of heteroaryl and heterocycle;
- R 27 and R 28 are each independently selected from the group consisting of hydrogen, alkyl, aryl, cycloalkyl, heteroaryl and heterocycle or R 27 and R 28 together with the atom to which they are attached form a ring selected from the group consisting of cycloalkyl and heterocycle, or R 27 and R 29 together with the atoms to which they are attached form a ring selected from the group consisting of cycloalkyl and heterocycle, or R 28 and R 4 together with the atoms to which they are attached form a ring selected from the group consisting of cycloalkyl and heterocycle;
- R 29 and R 3O are each independently selected from the group consisting of hydrogen, alkyl, alkoxy, aryl, aryloxy, cycloalkyl, cycloalkyloxy, heteroaryl, heterocycle, and - N(R 36 R 37 ), or R 29 and R 30 together with the atom to which they are attached form a ring selected from the group consisting of cycloalkyl and heterocycle, or R 29 and R 4 together with the atoms to which they are attached form a ring selected from the group consisting of cycloalkyl and heterocycle, or R 29 and E together with the atoms to which they are attached form a ring selected from the group consisting of cycloalkyl and heterocycle;
- R 31 is selected from the group consisting of hydrogen, alkyl, aryl, cycloalkyl, heterocycle and heteroaryl, or R 31 and E together with the atom to which they are attached form a ring selected from the group consisting of heteroaryl and heterocycle, or R 31 and R 4 together with the atoms to which they are attached form a heterocycle;
- R and R are each independently selected from the group consisting of hydrogen, alkyl, carboxy, carboxyalkyl, cycloalkyl, cycloalkyloxy, carboxycycloalkyl, aryl, arylalkyl, aryloxy, aryloxyalkyl, heterocycle, heterocyclealkyl, heterocycleoxyalkyl, heterocycleoxy, hydroxy, alkoxy, alkylsufonyl, cycloalkylsulfonyl, arylsulfonyl, and heterocyclesulfonyl, or R 32 and R 33 together with the atom to which they are attached
- R 36 and R 37 are each independently selected from the group consisting of hydrogen, alkyl and aryl.
- a further aspect of the present invention encompasses the use of the compounds of formula (I) for the treatment of disorders that are mediated by 11-beta-hydroxysteroid dehydrogenase Type 1 enzyme, such as non-insulin dependent type 2 diabetes, insulin resistance, obesity, lipid disorders, metabolic syndrome and other diseases and conditions that are mediated by excessive glucocorticoid action, comprising administering a therapeutically effective amount of a compound of formula (I) .
- the present invention is directed to a pharmaceutical composition
- a pharmaceutical composition comprising a therapeutically effective amount of a compound of formula (I) in combination with a pharmaceutically suitable carrier.
- One aspect of the present invention is directed toward a compound of formula (I), or a pharmaceutically acceptable salt, prodrug, salt of a prodrug, or a combination thereof
- a 1 , A 2 , A 3 and A 4 is selected from the group consisting of alkyl-NH-alkyl, alkylcarbonyl, alkylsulfonyl, cycloalkyl, cycloalkylcarbonyl, cycloalkylsulfonyl, arylcarbonyl, arylsulfonyl, heteroaryl, heteroarylalkyl, heteroaryloxyalkyl, heterocyclecarbonyl, heterocyclesulfonyl, aryl 1 , arylalkyl, aryloxyalkyl, carboxyalkyl, carboxycycloalkyl, haloalkyl, heterocyclealkyl, heterocycleoxyalkyl, -S(O) 2 -N(R 5 R 6 ), -NR 7 - [C(R 8 R 9 )] n -C(O)-R 10 , -O-[C(R ⁇ R 12 )] p
- D is selected from the group consisting of a bond, -C(R 27 R 28 )-X- and -C(R 27 R 28 )-
- E is selected from the group consisting of a cycloalkyl, alkyl, aryl, heteroaryl and heterocycle, wherein the heteroaryl and the heterocycle are appended to the parent molecular moiety through an available carbon atom, or R 4 and E together with the atoms to which they are attached form a ring selected from the group consisting of cycloalkyl and heterocycle;
- X is selected from the group consisting of a bond, -N(R 31 )-, -O-, -S-, -S(O)- and -
- R 1 is selected from the group consisting of hydrogen and alkyl
- R 2 is selected from the group consisting of hydrogen, alkyl and cycloalkyl
- R 3 and R 4 are each independently selected from the group consisting of hydrogen, alkyl, carboxyalkyl, carboxycycloalkyl, cycloalkyl, haloalkyl, aryl, arylalkyl, heteroaryl, heteroarylalkyl, heterocycle and heterocyclealkyl, or R 3 and R 4 together with the atom to which they are attached form a ring selected from the group consisting of cycloalkyl and heterocycle;
- R 5 and R 6 are each independently selected from the group consisting of hydrogen, alkoxy, alkyl, alkylcarbonyl, alkylsufonyl, carboxy, carboxyalkyl, carboxycycloalkyl, cycloalkyl, cycloalkyloxy, cycloalkylsulfonyl, aryl, arylalkyl, arylalkylcarbonyl, arylcarbonyl, aryloxy, aryloxyalkyl, arylsulfonyl, heteroaryl, heteroarylalkyl, heteroarylalkylcarbonyl, heteroarylcarbonyl, heteroaryloxyalkyl, heteroarylsulfonyl, heterocycle, heterocyclealkyl, heterocyclealkylcarbonyl, heterocyclecarbonyl, heterocyclecarbonyl, heterocycleoxyalkyl, heterocycleoxy, heterocyclesulfonyl and hydroxy, or R 5 and R 6 together with the atom to which they are attached form
- R 7 is selected from the group consisting of hydrogen, alkyl, carboxyalkyl, cycloalkyl, cafboxycycloalkyl, aryl, arylalkyl, aryloxyalkyl, hydroxy, alkoxy, heteroaryl, heteroarylalkyl, heteroaryloxyalkyl, heterocycle, heterocyclealkyl and heterocycleoxyalkyl;
- R 8 and R 9 are each independently selected from the group consisting of hydrogen and alkyl, or R 8 and R 9 taken together with the atom to which they are attached form a ring selected from the group consisting of cycloalkyl and heterocycle;
- R 10 is selected from the group consisting of hydrogen, alkyl, carboxyalkyl, cycloalkyl, carboxycycloalkyl, aryl, arylalkyl, aryloxy, aryloxyalkyl, hydroxy, alkoxy, cycloalkyloxy, heteroaryl, heteroarylalkyl, heteroaryloxy, heteroaryloxyalkyl, heterocycle, heterocyclealkyl, heterocycleoxy, heterocycleoxyalkyl and -N(R 32 R 33 ); R 11 and R 12 are each independently selected from the group consisting of hydrogen and alkyl or R 11 and R 12 taken together with the atom to which they are attached form a ring selected from the group consisting of cycloalkyl and heterocycle;
- R 13 is selected from the group consisting of hydroxy and -N(R 34 R 35 );
- R 14a is selected from the group consisting of carboxyalkyl, cycloalkyl, carboxycycloalkyl, aryl, arylalkyl, aryloxyalkyl, haloalkyl, heteroaryl, heteroarylalkyl, heteroaryloxyalkyl, heterocycle, heterocyclealkyl and heterocycleoxyalkyl;
- R 14b is selected from the group consisting of hydrogen, alkyl, carboxyalkyl, cycloalkyl, carboxycycloalkyl, aryl, arylalkyl, aryloxyalkyl, haloalkyl, heteroaryl, heteroarylalkyl, heteroaryloxyalkyl, heterocycle, heterocyclealkyl and heterocycleoxyalkyl;
- R 15 and R 16 are each independently selected from the group consisting of hydrogen, alkyl, alkylcarbonyl, carboxyalkyl, cycloalkyl, carboxycycloalkyl, aryl, arylalkyl, arylalkylcarbonyl, arylcarbonyl, aryloxyalkyl, heteroaryl, heteroarylalkyl, heteroarylalkylcarbonyl, heteroarylcarbonyl, heteroaryloxyalkyl, heteroarylsulfonyl, heterocycle, heterocyclealkylcarbonyl, heterocyclecarbonyl, heterocyclecarbony
- R 17 is selected from the group consisting of hydrogen, alkyl, carboxyalkyl, cycloalkyl, carboxycycloalkyl, aryl, arylalkyl, aryloxyalkyl, heteroaryl, heteroarylalkyl, heteroaryloxyalkyl, heterocycle, heterocyclealkyl and heterocycleoxyalkyl;
- R 18 and R 19 are each independently selected from the group consisting of hydrogen, alkoxy, alkyl, alkylsufonyl, carboxy, carboxyalkyl, carboxycycloalkyl, cycloalkyl, cycloalkyloxy, cycloalkylsulfonyl, aryl, arylalkyl, aryloxy, aryloxyalkyl, arylsulfonyl, heteroaryl, heteroarylalkyl, heteroaryloxyalkyl, heteroarylsulfonyl, heterocycle, heterocyclealkyl, heterocycleoxy, heterocycleoxy, hetero
- R 20 , R 21 and R 22 are each independently selected from the group consisting of hydrogen, alkyl, aryl, arylalkyl, carboxyalkyl, carboxycycloalkyl, cycloalkyl, haloalkyl, heteroaryl, heteroarylalkyl, heterocycle and heterocyclealkyl;
- R 23 and R 24 are each independently selected from the group consisting of hydrogen, alkyl, alkylcarbonyl, alkylsulfonyl, aryl, arylcarbonyl, arylsulfonyl, carboxyalkyl, carboxycycloalkyl, cycloalkyl, cycloalkylcarbonyl, cycloalkylsulfonyl, heteroaryl, heteroarylcarbonyl, heteroarylsulfonyl, heterocycle, heterocyclecarbonyl and heterocyclesulfonyl;
- R and R are each independently selected from the group consisting of hydrogen, alkoxy, alkyl, alkylcarbonyl, alkylsulfonyl, aryl, arylcarbonyl, aryloxy, arylsulfonyl, carboxyalkyl, carboxycycloalkyl, cycloalkyl, cycloalkylcarbonyl, cycloalkyloxy, cycloalkylsulfonyl, heteroaryl, heteroarylcarbonyl, heteroaryloxy, heteroarylsulfonyl, heterocycle, heterocyclecarbonyl, heterocycleoxy, heterocyclesulfonyl and hydroxy, or R 25 and R 26 together with the nitrogen to which they are attached form a ring selected from the group consisting of heteroaryl and heterocycle;
- R 27 and R 28 are each independently selected from the group consisting of hydrogen, alkyl, aryl, cycloalkyl, heteroaryl and heterocycle or R 27 and R 28 together with the atom to which they are attached form a ring selected from the group consisting of cycloalkyl and heterocycle, or R 27 and R 29 together with the atoms to which they are attached form a ring selected from the group consisting of cycloalkyl and heterocycle, or R and R together with the atoms to which they are attached form a ring selected from the group consisting of cycloalkyl and heterocycle;
- R 29 and R 30 are each independently selected from the group consisting of hydrogen, alkyl, alkoxy, aryl, aryloxy, cycloalkyl, cycloalkyloxy, heteroaryl, heterocycle, and -N(R 36 R 37 ), or R 29 and R 30 together with the atom to which they are attached form a ring selected from the group consisting of cycloalkyl and heterocycle, or R 29 and R 4 together with the atoms to which they are attached form a ring selected from the group consisting of cycloalkyl and heterocycle, or R 29 and E together with the atoms to which they are attached form a ring selected from the group consisting of cycloalkyl and heterocycle;
- R 31 is selected from the group consisting of hydrogen, alkyl, aryl, cycloalkyl, heterocycle and heteroaryl, or R 31 and E together with the atom to which they are attached form a ring selected from the group consisting of heteroaryl and heterocycle, or R
- Another aspect of the present invention is directed toward a compound of formula (I), or a pharmaceutically acceptable salt, prodrug, salt of a prodrug, or a combination thereof, wherein A 2 , A 3 and A 4 are hydrogen;
- R 1 and R 2 are hydrogen; and A 1 , R 3 , R 4 , D and E are as described in the summary of the invention.
- Another aspect of the present invention is directed toward a compound of formula (T), or a pharmaceutically acceptable salt, prodrug, salt of a prodrug, or a combination thereof, wherein
- a 2 , A 3 and A 4 are hydrogen
- R 1 and R 2 are hydrogen
- D is a bond; and A 1 , R 3 , R 4 and E are as described in the summary of the invention.
- Another aspect of the present invention is directed toward a compound of formula (T), or a pharmaceutically acceptable salt, prodrug, salt of a prodrug, or a combination thereof, wherein
- a 2 , A 3 and A 4 are hydrogen; R 1 and R 2 are hydrogen; D is a bond;
- E is selected from the group consisting of alkyl, aryl, and heteroaryl; and A 1 , R 3 , and R 4 are as described in the summary of the invention.
- Another aspect of the present invention is directed toward a compound of formula (I) 5 or a pharmaceutically acceptable salt, prodrug, salt of a prodrug, or a combination thereof, wherein
- a 2 , A 3 and A 4 are hydrogen; R 1 and R 2 are hydrogen; D is a bond;
- E is selected from the group consisting of alkyl, aryl and heteroaryl; R 3 and R 4 are hydrogen; and
- a 1 is selected from the group consisting of heteroaryl, -CO 2 R 17 , -C(O)-N(R 18 R 19 ), alkylsulfonyl and -S(O) 2 -N(R 5 R 6 ); wherein R 5 , R 6 , R 17 , R 18 and R 19 are as described in the summary of the invention.
- Another aspect of the present invention is directed toward a compound of formula (I), or a pharmaceutically acceptable salt, prodrug, salt of a prodrug, or a combination thereof, wherein
- a 2 , A 3 and A 4 are hydrogen; R 1 and R 2 are hydrogen;
- E is selected from the group consisting of alkyl, aryl and heteroaryl;
- R 3 is hydrogen;
- R 4 is alkyl;
- a 1 is selected from the group consisting of heteroaryl, -CO 2 R 17 , -C(O)-N(R 18 R 19 ), alkylsulfonyl and -S(O) 2 -N(R 5 R 6 ); wherein R 5 , R 6 , R 17 , R 18 and R 19 are as described in the summary of the invention.
- Another aspect of the present invention is directed toward a compound of formula (I), or a pharmaceutically acceptable salt, prodrug, salt of a prodrug, or a combination thereof, wherein
- a 2 , A 3 and A 4 are hydrogen; R 1 and R 2 are hydrogen; D is a bond;
- E is selected from the group consisting of alkyl, aryl and heteroaryl
- R 3 and R 4 are alkyl
- a 1 is selected from the group consisting of heteroaryl, -CO 2 R 17 , -C(O)-N(R 18 R 19 ), alkylsulfonyl and -S(O) 2 -N(R 5 R 6 ); wherein R 5 , R 6 , R 17 , R 18 and R 19 are as described in the summary of the invention.
- Another aspect of the present invention is directed toward a compound of formula (T), or a pharmaceutically acceptable salt, prodrug, salt of a prodrug, or a combination thereof, wherein A 2 , A 3 and A 4 are hydrogen;
- R 1 and R 2 are hydrogen
- E is selected from the group consisting of alkyl, aryl and heteroaryl
- R 3 and R 4 together with the atom to which they are attached form a ring selected from the group consisting of cycloalkyl and heterocycle; and A 1 is as described in the summary of the invention.
- Another aspect of the present invention is directed toward a compound of formula (I), or a pharmaceutically acceptable salt, prodrug, salt of a prodrug, or a combination thereof, wherein A 2 , A 3 and A 4 are hydrogen;
- R 1 and R 2 are hydrogen
- E is selected from the group consisting of alkyl, aryl and heteroaryl
- R 3 and R 4 together with the atom to which they are attached form a cycloalkyl ring
- a 1 is selected from the group consisting of heteroaryl, -CO 2 R 17 , -C(O)-N(R 18 R 19 ), alkylsulfonyl and -S(O) 2 -N(R 5 R 6 ); wherein R 5 , R 6 , R 17 , R 18 and R 19 are as described in the summary of the invention.
- Another aspect of the present invention is directed toward a compound of formula (T), or a pharmaceutically acceptable salt, prodrug, salt of a prodrug, or a combination thereof, wherein
- a 2 , A 3 and A 4 are hydrogen; R 1 and R 2 are hydrogen;
- E is selected from the group consisting of alkyl, aryl and heteroaryl
- R 3 and R 4 together with the atom to which they are attached form a heterocycle ring
- a 1 is selected from the group consisting of heteroaryl, -CO 2 R 17 , -C(O)-N(R 18 R 19 ), alkylsulfonyl and -S(O) 2 -N(R 5 R 6 ); wherein R 5 , R 6 , R 17 , R 18 and R 19 are as described in the summary of the invention.
- Another aspect of the present invention is directed toward a compound of formula (I), or a pharmaceutically acceptable salt, prodrug, salt of a prodrug, or a combination thereof, wherein
- a 2 , A 3 and A 4 are hydrogen
- R 1 and R 2 are hydrogen
- D is a bond; R 4 and E together with the atoms to which they are attached form a ring selected from the group consisting of cycloalkyl and heterocycle; and
- a 1 is selected from the group consisting of heteroaryl, -CO 2 R 17 , -C(O)-N(R 18 R 19 ), alkylsulfonyl and -S(O) 2 -N(R 5 R 6 ); wherein R 3 , R 5 , R 6 , R 17 , R 18 and R 19 are as described in the summary of the invention.
- Another aspect of the present invention is directed toward a compound of formula
- a , A and A are hydrogen
- R 1 and R 2 are hydrogen
- R 3 is alkyl
- a 1 is selected from the group consisting of heteroaryl, -CO 2 R 17 , -C(O)-N(R 18 R 19 ), alkylsulfonyl, and -S(O) 2 -N(R 5 R 6 ); wherein R 5 , R 6 , R 17 , R 18 and R 19 are as described in the summary of the invention; and
- R 38 is selected from the group consisting of arylalkyl and heteroarylalkyl wherein the aryl of the arylalkyl and the heteroaryl of the heteroarylalkyl are each independently unsubstituted or substituted with 1, 2 or 3 substituents selected from the group consisting of alkyl, halogen and haloalkyl.
- Another aspect of the present invention is directed toward a compound of formula (III), or a pharmaceutically acceptable salt, prodrug, salt of a prodrug, or a combination thereof,
- a 2 , A 3 and A 4 are hydrogen; R 1 and R 2 are hydrogen;
- R 3 is alkyl
- a 1 is selected from the group consisting of heteroaryl, -CO 2 R 17 , -C(O)-N(R 18 R 19 ), alkylsulfonyl, and -S(O) 2 -N(R 5 R 6 ) ; wherein R 5 , R 6 , R 17 , R 18 and R 19 are as described in the summary of the invention; and R 38 is selected from the group consisting of arylalkyl and heteroarylalkyl wherein the aryl of the arylalkyl and the heteroaryl of the heteroarylalkyl are each independently unsubstituted or substituted with 1, 2 or 3 substituents selected from the group consisting of alkyl, halogen and haloalkyl.
- Another aspect of the present invention is directed toward a compound of formula (I), or a pharmaceutically acceptable salt, prodrug, salt of a prodrug, or a combination thereof, wherein
- a 2 , A 3 and A 4 are hydrogen; R 1 and R 2 are hydrogen;
- D is selected from the group consisting of -C(R 27 R 28 )-X- and -C(R 27 R 28 )-C(R 29 R 30 )- X-; and R 27 , R 28 , R 29 , R 30 , X 5 A 1 , R 3 , R 4 and E are as described in the summary of the invention.
- Another aspect of the present invention is directed toward a compound of formula (I), or a pharmaceutically acceptable salt, prodrug, salt of a prodrug, or a combination thereof, wherein A 2 , A 3 and A 4 are hydrogen;
- R 1 and R 2 are hydrogen
- D is selected from the group consisting of -C(R 27 R 28 )-X- and -C(R 27 R 28 )-C(R 29 R 30 )-
- E is selected from the group consisting of aryl and heteroaryl; and A 1 , R 3 , and R 4 are as described in the summary of the invention.
- Another aspect of the present invention is directed toward a compound of formula (I), or a pharmaceutically acceptable salt, prodrug, salt of a prodrug, or a combination thereof, wherein
- a 2 , A 3 and A 4 are hydrogen; R 1 and R 2 are hydrogen;
- D is selected from the group consisting of -C(R 27 R 28 )-X- and -C(R 27 R 28 )-C(R 29 R 30 )-
- Another aspect of the present invention is directed toward a compound of formula (I), or a pharmaceutically acceptable salt, prodrug, salt of a prodrug, or a combination thereof, wherein
- a 2 , A 3 and A 4 are hydrogen; R 1 and R 2 are hydrogen; D is selected from the group consisting of -C(R 27 R 28 )-X- and -C(R 27 R 28 )-C(R 29 R 30 )-
- R 27 , R 28 , R 29 , and R 30 are each independently selected from the group consisting of hydrogen and alkyl;
- E is selected from the group consisting of aryl and heteroaryl; X is a bond; R 3 and R 4 are hydrogen; and
- a 1 is selected from the group consisting of heteroaryl, -CO 2 R 17 , -C(O)-N(R 18 R 19 ), alkylsulfonyl, and -S(O) 2 -N(R 5 R 6 ); wherein R 5 , R 6 , R 17 , R 18 and R 19 are as described in the summary of the invention.
- Another aspect of the present invention is directed toward a compound of formula (T), or a pharmaceutically acceptable salt, prodrug, salt of a prodrug, or a combination thereof, wherein A 2 , A 3 and A 4 are hydrogen;
- R 1 and R 2 are hydrogen
- D is selected from the group consisting of -C(R 27 R 28 )-X- and -C(R 27 R 28 )-C(R 29 R 30 )- X-; wherein R 27 , R 28 , R 29 , and R 30 are each independently selected from the group consisting of hydrogen and alkyl; E is selected from the group consisting of aryl and heteroaryl;
- X is a bond
- R 3 is hydrogen
- R 4 is alkyl
- a 1 is selected from the group consisting of heteroaryl, -CO 2 R 17 , -C(O)-N(R 18 R 19 ), alkylsulfonyl, and -S(O) 2 -N(R 5 R 6 ); wherein R 5 , R 6 , R 17 , R 18 and R 19 are as described in the summary of the invention.
- Another aspect of the present invention is directed toward a compound of formula (I), or a pharmaceutically acceptable salt, prodrug, salt of a prodrug, or a combination thereof, wherein A 2 , A 3 and A 4 are hydrogen;
- R 1 and R 2 are hydrogen
- D is selected from the group consisting of -C(R 27 R 28 )-X- and -C(R 27 R 28 )-C(R 29 R 30 )- X-; wherein R 5 R 5 R , and R are each independently selected from the group consisting of hydrogen and alkyl; E is selected from the group consisting of aryl and heteroaryl;
- X is a bond
- R 3 and R 4 are alkyl
- a 1 is selected from the group consisting of heteroaryl, -CO 2 R 17 , -C(O)-N(R 18 R 19 ), alkylsulfonyl, and -S(O) 2 -N(R 5 R 6 ); wherein R 5 , R 6 , R 17 , R 18 and R 19 are as described in the summary of the invention.
- Another aspect of the present invention is directed toward a compound of formula (T), or a pharmaceutically acceptable salt, prodrug, salt of a prodrug, or a combination thereof, wherein
- a 2 , A 3 and A 4 are hydrogen
- R 1 and R 2 are hydrogen
- D is selected from the group consisting of -C(R 27 R 28 )-X- and -C(R 27 R 28 )-C(R 29 R 30 )- X-; wherein R 27 , R 28 , R 29 , and R 30 are as described in the summary of the invention;
- E is selected from the group consisting of aryl and heteroaryl
- X is a bond
- R 3 and R 4 together with the atom to which they are attached form a ring selected from the group consisting of cycloalkyl and heterocycle; and A 1 is as described in the summary of the invention.
- Another aspect of the present invention is directed toward a compound of formula (I), or a pharmaceutically acceptable salt, prodrug, salt of a prodrug, or a combination thereof, wherein
- a 2 , A 3 and A 4 are hydrogen; R 1 and R 2 are hydrogen;
- D is selected from the group consisting of -C(R 27 R 28 )-X- and -C(R 27 R 28 )-C(R 29 R 30 )- X-; wherein R , R , R , and R are each independently selected from the group consisting of hydrogen and alkyl;
- E is selected from the group consisting of aryl and heteroaryl; X is a bond;
- R 3 and R 4 together with the atom to which they are attached form a cycloalkyl ring
- a 1 is selected from the group consisting of heteroaryl, -CO 2 R 17 , -C(O)-N(R 18 R 19 ), alkylsulfonyl, and -S(O) 2 -N(R 5 R 6 ); wherein R 5 , R 6 , R 17 , R 18 and R 19 are as described in the summary of the invention.
- Another aspect of the present invention is directed toward a compound of formula (I), or a pharmaceutically acceptable salt, prodrug, salt of a prodrug, or a combination thereof, wherein
- a 2 , A 3 and A 4 are hydrogen; R 1 and R 2 are hydrogen;
- D is selected from the group consisting of -C(R 27 R 28 )-X- and -C(R 27 R 28 )-C(R 29 R 30 )- X-; wherein R 5 R 5 R , and R are each independently selected from the group consisting of hydrogen and alkyl;
- E is selected from the group consisting of aryl and heteroaryl
- X is a bond
- R 3 and R 4 together with the atom to which they are attached form a heterocycle ring
- a 1 is selected from the group consisting of heteroaryl, -CO 2 R 17 , -C(O)-N(R 18 R 19 ), alkylsulfonyl, and -S(O) 2 -N(R 5 R 6 ); wherein R 5 , R 6 , R 17 , R 18 and R 19 are as described in the summary of the invention.
- Another aspect of the present invention is directed toward a compound of formula (I), or a pharmaceutically acceptable salt, prodrug, salt of a prodrug, or a combination thereof, wherein
- a 2 , A 3 and A 4 are hydrogen
- R 1 and R 2 are hydrogen
- D is selected from the group consisting of -C(R 27 R 28 )-X- and -C(R 27 R 28 )-C(R 29 R 30 )- X-; wherein R 27 , R 28 , R 29 , and R 30 are as described in the summary of the invention;
- E is selected from the group consisting of aryl and heteroaryl
- X is selected from the group consisting of -N(R 31 )- and -O-; and A 1 , R 3 , and R 4 are as described in the summary of the invention.
- Another aspect of the present invention is directed toward a compound of formula (I), or a pharmaceutically acceptable salt, prodrug, salt of a prodrug, or a combination thereof, wherein
- a 2 , A 3 and A 4 are hydrogen
- R 1 and R 2 are hydrogen
- D is selected from the group consisting of -C(R 27 R 28 )-X- and -C(R 27 R 28 )-C(R 29 R 30 )- X-; wherein R 5 R 5 R , and R are each independently selected from the group consisting of hydrogen and alkyl;
- E is selected from the group consisting of aryl and heteroaryl;
- X is selected from the group consisting of -N(R 31 )- and -0-;
- R 3 and R 4 are hydrogen;
- a 1 is selected from the group consisting of heteroaryl, -CO 2 R 17 , -C(O)-N(R 18 R 19 ), alkylsulfonyl, and -S(O) 2 -N(R 5 R 6 ); wherein R 5 , R 6 , R 17 , R 18 and R 19 are as described in the summary of the invention.
- Another aspect of the present invention is directed toward a compound of formula (I), or a pharmaceutically acceptable salt, prodrug, salt of a prodrug, or a combination thereof, wherein
- a 2 , A 3 and A 4 are hydrogen; R 1 and R 2 are hydrogen;
- D is selected from the group consisting of -C(R 27 R 28 )-X- and -C(R 27 R 28 )-C(R 29 R 30 )- X-; wherein R 27 , R 28 , R 29 , and R 30 are each independently selected from the group consisting of hydrogen and alkyl;
- E is selected from the group consisting of aryl and heteroaryl
- X is selected from the group consisting of -N(R 31 )- and -O-;
- R 3 is hydrogen
- R 4 is alkyl
- a 1 is selected from the group consisting of heteroaryl, -CO 2 R 17 , -C(O)-N(R 18 R 19 ), alkylsulfonyl, and -S(O) 2 -N(R 5 R 6 ); wherein R 5 , R 6 , R 17 , R 18 and R 19 are as described in the summary of the invention.
- Another aspect of the present invention is directed toward a compound of formula (I), or a pharmaceutically acceptable salt, prodrug, salt of a prodrug, or a combination thereof, wherein
- a 2 , A 3 and A 4 are hydrogen; R 1 and R 2 are hydrogen;
- D is selected from the group consisting of -C(R 27 R 28 )-X- and -C(R 27 R 28 )-C(R 29 R 30 )- X-; wherein R 27 , R 28 , R 29 , and R 30 are each independently selected from the group consisting of hydrogen and alkyl;
- E is selected from the group consisting of aryl and heteroaryl
- X is selected from the group consisting of -N(R 31 )- and -O-;
- R 3 and R 4 are alkyl
- a 1 is selected from the group consisting of heteroaryl, -CO 2 R 17 , -C(O)-N(R 18 R 19 ), alkylsulfonyl, and -S(O) 2 -N(R 5 R 6 ); wherein R 5 , R 6 , R 17 , R 18 and R 19 are as described in the summary of the invention.
- Another aspect of the present invention is directed toward a compound of formula (I), or a pharmaceutically acceptable salt, prodrug, salt of a prodrug, or a combination thereof, wherein A 2 , A J and A 4 are hydrogen;
- R 1 and R 2 are hydrogen
- D is selected from the group consisting of -C(R 27 R 28 )-X- and -C(R 27 R 28 )-C(R 29 R 30 )- X-; wherein R 27 , R 28 , R 29 , and R 30 are each independently selected from the group consisting of hydrogen and alkyl;
- E is selected from the group consisting of aryl and heteroaryl
- X is selected from the group consisting of -N(R 31 )- and -O-;
- R 3 and R 4 together with the atom to which they are attached form a ring selected from the group consisting of cycloalkyl and heterocycle; and
- a 1 is selected from the group consisting of heteroaryl, -CO 2 R 17 , -C(O)-N(R 18 R 19 ), alkylsulfonyl, and -S(O) 2 -N(R 5 R 6 ); wherein R 5 , R 6 , R 17 , R 18 and R 19 are as described in the summary of the invention.
- Another aspect of the present invention is directed toward a compound of formula (I), or a pharmaceutically acceptable salt, prodrug, salt of a prodrug, or a combination thereof, wherein
- a 2 , A 3 and A 4 are hydrogen
- R 1 and R 2 are hydrogen
- D is selected from the group consisting of -C(R 27 R 28 )-X- and -C(R 27 R 28 )-C(R 29 R 30 )-
- R , R , R , and R are each independently selected from the group consisting of hydrogen and alkyl;
- E is selected from the group consisting of aryl and heteroaryl
- X is selected from the group consisting of -N(R 31 )- and -0-
- R 3 and R 4 together with the atom to which they are attached form a cycloalkyl ring
- a 1 is selected from the group consisting of heteroaryl, -CO 2 R 17 , -C(O)-N(R 18 R 19 ), alkylsulfonyl, and -S(O) 2 -N(R 5 R 6 ); wherein R 5 , R 6 , R 17 , R 18 and R 19 are as described in the summary of the invention.
- Another aspect of the present invention is directed toward a compound of formula (I), or a pharmaceutically acceptable salt, prodrug, salt of a prodrug, or a combination thereof, wherein
- a 2 , A 3 and A 4 are hydrogen; R 1 and R 2 are hydrogen; D is selected from the group consisting of -C(R 27 R 28 )-X- and -C(R 27 R 28 )-C(R 29 R 30 )- X-; wherein R 27 , R 28 , R 29 , and R 30 are each independently selected from the group consisting of hydrogen and alkyl;
- E is selected from the group consisting of aryl and heteroaryl
- X is selected from the group consisting of -N(R 31 )- and -O-;
- R 3 and R 4 together with the atom to which they are attached form a heterocycle ring
- a 1 is selected from the group consisting of heteroaryl, -CO 2 R 17 , -C(O)-N(R 18 R 19 ), alkylsulfonyl, and -S(O) 2 -N(R 5 R 6 ); wherein R 5 , R 6 , R 17 , R 18 and R 19 are as described in the summary of the invention.
- Exemplary compounds of the present invention having formula (I) include, but are not limited to,
- Another embodiment of the present invention discloses a method of inhibiting 11- beta-hydroxysteroid dehydrogenase Type I enzyme, comprising administering to a mammal, a therapeutically effective amount of the compound of formula (I).
- Another embodiment of the present invention discloses a method of treating disorders in a mammal by inhibiting 11-beta-hydroxysteroid dehydrogenase Type I enzyme, comprising administering to a mammal, a therapeutically effective amount of the compound of formula (I).
- Another embodiment of the present invention discloses a method of treating non- insulin dependent type 2 diabetes in a mammal by inhibiting 11-beta-hydroxysteroid dehydrogenase Type I enzyme comprising administering to a mammal, a therapeutically effective amount of the compound of formula (I) .
- Another embodiment of the present invention discloses a method of treating insulin resistance in a mammal by inhibiting 11-beta-hydroxysteroid dehydrogenase Type I enzyme comprising administering to a mammal, a therapeutically effective amount of the compound of formula (I).
- Another embodiment of the present invention discloses a method of treating obesity in a mammal by inhibiting 11-beta-hydroxysteroid dehydrogenase Type I enzyme comprising administering to a mammal, a therapeutically effective amount of the compound of formula G ) .
- Another embodiment of the present invention discloses a method of treating lipid disorders in a mammal by inhibiting 11-beta-hydroxysteroid dehydrogenase Type I enzyme comprising administering to a mammal, a therapeutically effective amount of the compound of formula (I).
- Another embodiment of the present invention discloses a method of treating metabolic syndrome in a mammal by inhibiting 11-beta-hydroxysteroid dehydrogenase Type I enzyme comprising administering to a mammal, a therapeutically effective amount of the compound of formula (I).
- Another embodiment of the present invention discloses a method of treating diseases and conditions that are mediated by excessive glucocorticoid action in a mammal by inhibiting 11-beta-hydroxysteroid dehydrogenase Type I enzyme comprising administering to a mammal, a therapeutically effective amount of the compound of formula (I).
- Another embodiment of the present invention discloses a pharmaceutical composition
- a pharmaceutical composition comprising a therapeutically effective amount of the compound of formula (I) in combination with a pharmaceutically suitable carrier.
- alkenyl refers to a straight or branched chain hydrocarbon containing from 2 to 10 carbons and containing at least one carbon-carbon double bond formed by the removal of two hydrogens.
- Representative examples of alkenyl include, but are not limited to, ethenyl, 2-propenyl, 2-methyl-2-propenyl, 3-butenyl, 4-pentenyl, 5- hexenyl, 2-heptenyl, 2-methyl-l-heptenyl, and 3-decenyl.
- alkoxy refers to an alkyl group, as defined herein, appended to the parent molecular moiety through an oxygen atom.
- Representative examples of alkoxy include, but are not limited to, methoxy, ethoxy, propoxy, 2-propoxy, butoxy, tert- butoxy, pentyloxy and hexyloxy.
- alkoxyalkyl refers to an alkoxy group, as defined herein, appended to the parent molecular moiety through an alkyl group, as defined herein.
- Representative examples of alkoxyalkyl include, but are not limited to, tert-butoxymethyl, 2- ethoxyethyl, 2-methoxyethyl and methoxymethyl.
- alkoxycarbonyl refers to an alkoxy group, as defined herein, appended to the parent molecular moiety through a carbonyl group, as defined herein.
- Representative examples of alkoxycarbonyl include, but are not limited to, methoxycarbonyl, ethoxycarbonyl and tert-butoxycarbonyl.
- alkyl refers to a straight or branched chain hydrocarbon containing from 1 to 10 carbon atoms.
- Representative examples of alkyl include, but are not limited to, methyl, ethyl, n-propyl, iso-propyl, n-butyl, sec-butyl, iso-butyl, tert-butyl, n- pentyl, isopentyl, neopentyl, n-hexyl, 3-methylhexyl, 2,2-dimethylpentyl, 2,3-dimethylpentyl, n-heptyl, n-octyl, n-nonyl and n-decyl.
- alkylcarbonyl refers to an alkyl group, as defined herein, appended to the parent molecular moiety through a carbonyl group, as defined herein.
- alkylcarbonyl include, but are not limited to, acetyl, 1-oxopropyl, 2,2-dimethyl-l-oxopropyl, 1-oxobutyl and 1-oxopentyl.
- alkylsulfonyl refers to an alkyl group, as defined herein, appended to the parent molecular moiety through a sulfonyl group, as defined herein.
- Representative examples of alkylsulfonyl include, but are not limited to, methylsulfonyl and ethylsulfonyl.
- alkyl-NH refers to an alkyl group, as defined herein, appended to the parent molecular moiety through a nitrogen atom.
- alkyl-NH-alkyl refers to an alkyl-NH group, as defined herein, appended to the parent molecular moiety through an alkyl group, as defined herein.
- aryl as used herein, means a phenyl group, or a bicyclic or a tricyclic fused ring system.
- Bicyclic fused ring systems are exemplified by a phenyl group appended to the parent molecular moiety and fused to a monocyclic cycloalkyl group, as defined herein, a phenyl group, a monocyclic heteroaryl group, as defined herein, or a monocyclic heterocycle, as defined herein.
- Tricyclic fused ring systems are exemplified by an aryl bicyclic fused ring system, as defined herein and fused to a monocyclic cycloalkyl group, as defined herein, a phenyl group, a monocyclic heteroaryl group, as defined herein, or a monocyclic heterocycle, as defined herein.
- Representative examples of aryl include, but are not limited to, anthracenyl, azulenyl, fluorenyl, indanyl, indenyl, naphthyl, phenyl and tetrahydronaphthyl.
- aryl groups of this invention maybe optionally substituted with 1, 2, 3, 4 or 5 substituents independently selected from alkenyl, alkoxy, alkoxyalkoxy, alkoxyalkyl, alkoxycarbonyl, alkoxycarbonylalkyl, alkyl, alkylcarbonyl, alkynyl, aryl, arylalkenyl, arylalkyl, arylalkoxy, arylcarbonyl, aryloxy, arylsulfonyl, carboxy, carboxyalkyl, cyano, cyanoalkyl, cycloalkylalkoxy, ethylenedioxy, formyl, haloalkoxy, haloalkyl, halogen, heteroaryl, heteroarylalkenyl, heteroarylalkyl, heteroarylalkoxy, heteroarylcarbonyl, heterocycle, heterocyclealkyl, heterocyclealkoxy, heterocyclecarbonyl, heterocycleoxy, hydroxy, hydroxyalkyl
- the substituent aryl, the aryl of arylalkyl, the aryl of arylalkenyl, the aryl of arylalkoxy, the aryl of arylcarbonyl, the aryl of aryloxy, the aryl of arylsulfonyl, the cycloalkyl of cycloalkylalkoxy, the substituent heteroaryl, the heteroaryl of heteroarylalkyl, the heteroaryl of heteroarylalkenyl, the heteroaryl of heteroarylalkoxy, the heteroaryl of heteroarylcarbonyl, the substituent heterocycle, the heterocycle of heterocyclealkyl, the heterocycle of heterocyclealkoxy, the heterocycle of heterocyclecarbonyl, the heterocycle of heterocycleoxy, the heterocycle of heterocyclesulfonyl maybe optionally substituted with 1, 2 or 3 substituents independently selected from the group consisting of alkoxy, alkoxyalkyl, alkoxycarbonyl, alkyl,
- aryl 1 refers to a substituted phenyl group wherein the substituent is a member selected from the group consisting of alkenyl, alkoxy, alkoxyalkoxy, alkoxyalkyl, alkoxycarbonyl, alkoxycarbonylalkyl, alkyl, alkylcarbonyl, alkynyl, aryl, arylcarbonyl, aryloxy, arylsulfonyl, carboxy, carboxyalkyl, cyano, cyanoalkyl, ethylenedioxy, formyl, haloalkoxy, haloalkyl, halogen, heteroaryl, heteroarylalkyl, heteroarylcarbonyl, heterocycle, heterocyclecarbonyl, heterocycleoxy, hydroxy, hydroxyalkyl and nitro, or a bicyclic or a tricyclic fused ring system.
- Bicyclic fused ring systems are exemplified by a phenyl group appended to the parent molecular moiety, which is fused to a cycloalkyl group, as defined herein, a phenyl group, a heteroaryl, as defined herein, or a heterocycle as defined herein.
- Tricyclic fused ring systems are exemplified by an aryl bicyclic fused ring system fused to a cycloalkyl group, as defined herein, a phenyl group, a heteroaryl, as defined herein, or a heterocycle as defined herein.
- Bicyclic and tricyclic fused ring systems of this invention may be optionally substituted with 1, 2, 3, 4 or 5 substituents independently selected from alkenyl, alkoxy, alkoxyalkoxy, alkoxyalkyl, alkoxycarbonyl, alkoxycarbonylalkyl, alkyl, alkylcarbonyl, alkynyl, aryl, arylcarbonyl, aryloxy, arylsulfonyl, carboxy, carboxyalkyl, cyano, cyanoalkyl, ethylenedioxy, formyl, haloalkoxy, haloalkyl, halogen, heteroaryl, heteroarylalkyl, heteroarylcarbonyl, heterocycle, heterocyclecarbonyl, heterocycleoxy, hydroxy, hydroxyalkyl, nitro, R f RgN-, R f R g Nalkyl, R f R g Ncarbonyl and RfR g Nsulfonyl, wherein R
- arylalkenyl refers to an aryl group, as defined herein, appended to the parent molecular moiety through an alkenyl group, as defined herein.
- arylalkyl refers to an aryl group, as defined herein, appended to the parent molecular moiety through an alkyl group, as defined herein.
- arylalkyl include, but are not limited to, benzyl, 2-phenylethyl, 3- phenylpropyl and 2-naphth-2-ylethyl.
- arylalkoxy refers to an aryl group, as defined herein, appended to the parent molecular moiety through an alkoxy group, as defined herein.
- arylcarbonyl refers to an aryl group, as defined herein, appended to the parent molecular moiety through a carbonyl group, as defined herein. Representative examples of arylcarbonyl include, but are not limited to, benzoyl and naphthoyl.
- aryl-NH- refers to an aryl group, as defined herein, appended to the parent molecular moiety through a nitrogen atom.
- aryl-NH-alkyl refers to an aryl-NH- group, as defined herein, appended to the parent molecular moiety through an alkyl group, as defined herein.
- aryloxy refers to an aryl group, as defined herein, appended to the parent molecular moiety through an oxy moiety, as defined herein.
- Representative examples of aryloxy include, but are not limited to phenoxy, naphthyloxy, 3- bromophenoxy, 4-chlorophenoxy, 4-methylphenoxy and 3,5-dimethoxyphenoxy.
- aryloxyalkyl refers to an aryloxy group, as defined herein, appended to the parent molecular moiety through an alkyl group, as defined herein.
- arylsulfonyl refers to an aryl group, as defined herein, appended to the parent molecular moiety through a sulfonyl group, as defined herein.
- Representative examples of arylsulfonyl include, but are not limited to, phenylsulfonyl, A- bromophenylsulfonyl and naphthylsulfonyl.
- carbonyl refers to a -C(O)- group.
- carboxyalkyl refers to a carboxy group as defined herein, appended to the parent molecular moiety through an alkyl group as defined herein.
- carboxycycloalkyl refers to a carboxy group as defined herein, appended to the parent molecular moiety through an cycloalkyl group as defined herein.
- cycloalkyl refers to a monocyclic, bicyclic, or tricyclic ring system.
- Monocyclic ring systems are exemplified by a saturated cyclic hydrocarbon group containing from 3 to 8 carbon atoms. Examples of monocyclic ring systems include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl and cyclooctyl.
- Bicyclic fused ring systems are exemplified by a cycloalkyl group appended to the parent molecular moiety, which is fused to an additional cycloalkyl group, as defined herein, a phenyl group, a heteroaryl, as defined herein, or a heterocycle as defined herein.
- Tricyclic fused ring systems are exemplified by a cycloalkyl bicyclic fused ring system fused to an additional cycloalkyl group, as defined herein, a phenyl group, a heteroaryl, as defined herein, or a heterocycle as defined herein.
- Bicyclic ring systems are also exemplified by a bridged monocyclic ring system in which two non-adjacent carbon atoms of the monocyclic ring are linked by an alkylene bridge of between one and three additional carbon atoms.
- Representative examples of bicyclic ring systems include, but are not limited to, bicyclo[3.1.1]heptane, bicyclo[2.2.1]heptane, bicyclo[2.2.2]octane, bicyclo[3.2.2]nonane, bicyclo[3.3.1]nonane and bicyclo[4.2.1]nonane.
- Tricyclic ring systems are also exemplified by a bicyclic ring system in which two non-adjacent carbon atoms of the bicyclic ring are linked by a bond or an alkylene bridge of between one and three carbon atoms.
- Representative examples of tricyclic-ring systems include, but are not limited to, tricyclo[3.3.1.0 3 ' 7 ]nonane and tricyclo [3.3.1.1 3 ' 7 ] decane (adamantane).
- cycloalkyl groups of this invention may be substituted with 1, 2, 3, 4 or 5 substituents independently selected from alkenyl, alkoxy, alkoxyalkoxy, alkoxyalkyl, alkoxycarbonyl, alkoxycarbonylalkyl, alkyl, alkylcarbonyl, alkynyl, aryl, arylalkyl, arylcarbonyl, aryloxy, arylsulfonyl, carboxy, carboxyalkyl, cyano, cyanoalkyl, cycloalkyl, ethylenedioxy, formyl, haloalkoxy, haloalkyl, halogen, heteroaryl, heteroarylalkyl, heteroarylcarbonyl, heterocycle, heterocyclealkyl, heterocyclecarbonyl, heterocycleoxy, hydroxy, hydroxyalkyl, nitro, RfRgN-, R f RgNalkyl, RfR g Ncarbonyl and R f RgN
- the substituent aryl, the aryl of arylalkyl, the aryl of arylcarbonyl, the aryl of aryloxy, the aryl of arylsulfonyl, the substituent heteroaryl, the heteroaryl of heteroarylalkyl, the heteroaryl of heteroarylcarbonyl, the substituent heterocycle, the heterocycle of heterocyclealkyl, the heterocycle of heterocyclecarbonyl, the heterocycle of heterocycleoxy, the heterocycle of heterocyclesulfonyl may be optionally substituted with 0, 1, 2 or 3 substituents independently selected from the group consisting of alkoxy, alkoxyalkyl, alkoxycarbonyl, alkyl, alkylcarbonyl, alkynyl, carboxy, carboxyalkyl, cyano, haloalkyl, halogen, hydroxy, hydroxyalkyl, nitro, RfRgN-, R f RgNalkyl, RfRgNcarbonyl and R
- cycloalkylalkyl refers to a cycloalkyl group, as defined herein, appended to the parent molecular moiety through an alkyl group, as defined herein.
- Representative examples of cycloalkylalkyl include, but are not limited to, cyclopropylmethyl, 2-cyclobutylethyl, cyclopentylmethyl, cyclohexylmethyl and 4-cycloheptylbutyl.
- cycloalkylalkoxy refers to a cycloalkyl group, as defined herein, appended to the parent molecular moiety through an alkoxy group, as defined herein.
- cycloalkylcarbonyl refers to cycloalkyl group, as defined herein, appended to the parent molecular moiety through a carbonyl group, as defined herein.
- Representative examples of cycloalkylcarbonyl include, but are not limited to, cyclopropylcarbonyl, 2-cyclobutylcarbonyl and cyclohexylcarbonyl.
- cycloalkyloxy refers to cycloalkyl group, as defined herein, appended to the parent molecular moiety through an oxy group, as defined herein.
- cycloalkylsulfonyl refers to cycloalkyl group, as defined herein, appended to the parent molecular moiety through a sulfonyl group, as defined herein.
- Representative examples of cycloalkylsulfonyl include, but are not limited to, cyclohexylsulfonyl and cyclobutylsulfonyl.
- halo refers to -Cl, -Br, -I or -F.
- haloalkyl refers to at least one halogen, as defined herein, appended to the parent molecular moiety through an alkyl group, as defined herein. Representative examples of haloalkyl include, but are not limited to, chloromethyl, 2- fluoroethyl, trifluoromethyl, pentafluoroethyl and 2-chloro-3-fluoropentyl.
- heteroaryl refers to an aromatic monocyclic ring or an aromatic bicyclic ring system.
- the aromatic monocyclic rings are five or six membered rings containing at least one heteroatom independently selected from the group consisting of N, O and S.
- the five membered aromatic monocyclic rings have two double bonds and the six membered aromatic monocyclic rings have three double bonds.
- the bicyclic heteroaryl groups are exemplified by a monocyclic heteroaryl ring appended to the parent molecular moiety and fused to a monocyclic cycloalkyl group, as defined herein, a monocyclic aryl group, as defined herein, a monocyclic heteroaryl group, as defined herein, or a monocyclic heterocycle, as defined herein.
- heteroaryl include, but are not limited to, benzimidazolyl, benzofuranyl, benzothiazolyl, benzothienyl, benzoxazolyl, furyl, imidazolyl, indazolyl, indolyl, indolizinyl, isobenzofuranyl, isoindolyl, isoxazolyl, isoquinolinyl, isothiazolyl, naphthyridinyl, oxadiazolyl, oxazolyl, phthalazinyl, pyridinyl, pyridazinyl, pyrimidinyl, pyrazinyl, pyrazolyl, pyrrolyl, quinolinyl, quinolizinyl, quinoxalinyl, quinazolinyl, tetrazolyl, thiadiazolyl, thiazolyl, thienyl, triazolyl and triazinyl.
- heteroaryls of this invention may be optionally substituted with 1, 2 or 3 substituents independently selected from alkenyl, alkoxy, alkoxyalkoxy, alkoxyalkyl, alkoxycarbonyl, alkoxycarbonylalkyl, alkyl, alkylcarbonyl, alkynyl, aryl, arylalkenyl, arylalkyl, arylcarbonyl, aryloxy, arylsulfonyl, carboxy, carboxyalkyl, cyano, cyanoalkyl, cycloalkyl, ethylenedioxy, formyl, haloalkoxy, haloalkyl, halogen, heteroaryl, heteroarylalkenyl, heteroarylalkyl, heterocycle, heterocyclealkyl, heterocyclecarbonyl, heterocycleoxy, hydroxy, hydroxyalkyl, nitro, R f RgN-, RfR g Nalkyl, R f RgNcarbonyl
- heteroarylalkyl refers to a heteroaryl, as defined herein, appended to the parent molecular moiety through an alkyl group, as defined herein.
- heteroarylalkoxy refers to a heteroaryl, as defined herein, appended to the parent molecular moiety through an alkoxy group, as defined herein.
- heteroaryloxy refers to a heteroaryl, as defined herein, appended to the parent molecular moiety through an oxy group, as defined herein.
- heteroaryloxyalkyl refers to a heteroaryloxy, as defined herein, appended to the parent molecular moiety through an alkyl group, as defined herein.
- heterocycle refers to a non-aromatic monocyclic ring or a non-aromatic bicyclic ring.
- the non-aromatic monocyclic ring is a three, four, five, six, seven, or eight membered ring containing at least one heteroatom, independently selected from the group consisting of N, O and S.
- monocyclic ring systems include, but are not limited to, azetidinyl, aziridinyl, diazepinyl, dithianyl, imidazolinyl, imidazolidinyl, isothiazolinyl, isoxazolinyl, isoxazolidinyl, morpholinyl, oxazolinyl, oxazolidinyl, piperazinyl, piperidinyl, pyranyl, pyrazolinyl, pyrazolidinyl, pyrrolinyl, pyrrolidinyl, tetrahydrofuranyl, tetrahydro-2H-pyranyl, tetrahydro-2H-pyran-2-yl, tetrahydro-2H-pyran-4-yl, tetrahydrothienyl, thiazolinyl, thiazolidinyl, thiomorpholinyl, 1,1- dioxido
- bicyclic heterocycles are exemplified by a monocyclic heterocycle appended to the parent molecular moiety and fused to a monocyclic cycloalkyl group, as defined herein, a monocyclic aryl group, a monocyclic heteroaryl group, as defined herein, or a monocyclic heterocycle, as defined herein.
- Bicyclic ring systems are also exemplified by a bridged monocyclic ring system in which two non-adjacent atoms of the monocyclic ring are linked by a bridge of between one and three atoms selected from the group consisting of carbon, nitrogen and oxygen.
- bicyclic ring systems include but are not limited to, for example, benzopyranyl, benzothiopyranyl, benzodioxinyl, 1,3-benzodioxolyl, cinnolinyl, 1,5- diazocanyl, 3,9-diaza-bicyclo[4.2.1]non-9-yl, 3,7-diazabicyclo[3.3.1]nonane, octahydro- pyrrolo[3,4-c]pyrrole, indolinyl, isoindolinyl, 2,3,4,5-tetrahydro-lH-benzo[c]azepine, 2,3,4,5-tetrahydro-lH-benzo[ ⁇ ]azepine, 2,3,4,5-tetrahydro-lH-benzo[cr
- the heterocycles of this invention may be optionally substituted with 1, 2 or 3 substituents independently selected from oxo, alkenyl, alkoxy, alkoxyalkoxy, alkoxyalkyl, alkoxycarbonyl, alkoxycarbonylalkyl, alkyl, alkylcarbonyl, alkynyl, aryl, arylalkyl, arylcarbonyl, aryloxy, arylsulfonyl, carboxy, carboxyalkyl, cyano, cyanoalkyl, ethylenedioxy, formyl, haloalkoxy, haloalkyl, halogen, heteroaryl, heteroarylalkyl, heterocycle, heterocyclealkyl, heterocyclecarbonyl, heterocycleoxy, hydroxy, hydroxyalkyl, nitro, RfRgN-, RfR g Nalkyl, R f R g Ncarbonyl and R f R g Nsulfonyl, wherein R
- the substituent aryl, the aryl of arylalkyl, the aryl of arylcarbonyl, the aryl of aryloxy, the aryl of arylsulfonyl, the heteroaryl, the heteroaryl of heteroarylalkyl, the substituent heterocycle, the heterocycle of heterocyclealkyl, the heterocycle of heterocyclecarbonyl, the heterocycle of heterocycleoxy, may be optionally substituted with 1, 2 or 3 substituents independently selected from the group consisting of alkoxy, alkoxyalkyl, alkoxycarbonyl, alkyl, alkylcarbonyl, alkynyl, carboxy, carboxyalkyl, cyano, haloalkyl, halogen, hydroxy, hydroxyalkyl, nitro, R f R g N-, R f R g Nalkyl, R f R g Ncarbonyl and R f R g Nsulfonyl.
- heterocyclealkyl refers to a heterocycle, as defined herein, appended to the parent molecular moiety through an alkyl group, as defined herein.
- Representative examples of heterocyclealkyl include, but are not limited to, pyridin-3- ylmethyl and 2-pyrimidin-2-ylpropyl.
- heterocyclealkylcarbonyl refers to a heterocyclealkyl, as defined herein, appended to the parent molecular moiety through a carbonyl group, as defined herein.
- heterocyclealkoxy refers to a heterocycle, as defined herein, appended to the parent molecular moiety through an alkoxy group, as defined herein.
- heterocycleoxy refers to a heterocycle, as defined herein, appended to the parent molecular moiety through an oxy group, as defined herein.
- heterocycleoxyalkyl refers to a heterocycleoxy, as defined herein, appended to the parent molecular moiety through an alkyl group, as defined herein.
- heterocycle-NH- refers to a heterocycle, as defined herein, appended to the parent molecular moiety through a nitrogen atom.
- heterocycle-NH-alkyl refers to a heterocycle-NH-, as defined herein, appended to the parent molecular moiety through an alkyl group, as defined herein.
- heterocyclecarbonyl refers to a heterocycle, as defined herein, appended to the parent molecular moiety through a carbonyl group, as defined herein.
- Representative examples of heterocyclecarbonyl include, but are not limited to, 1- piperidinylcarbonyl, 4-morpholinylcarbonyl, pyridin-3-ylcarbonyl and quinolin-3-ylcarbonyl.
- heterocyclesulfonyl refers to a heterocycle, as defined herein, appended to the parent molecular moiety through a sulfonyl group, as defined herein.
- Representative examples of heterocyclesulfonyl include, but are not limited to, 1- piperidinylsulfonyl, 4-morpholinylsulfonyl, pyridin-3-ylsulfonyl and quinolin-3-ylsulfonyl.
- hydroxy refers to an -OH group.
- hydroxyalkyl refers to a hydroxy group, as defined herein, appended to the parent molecular moiety through an alkyl group, as defined herein.
- hydroxyalkyl include, but are not limited to, hydroxymethyl, 2- hydroxyethyl, 3-hydroxypropyl and 2-ethyl-4-hydroxyheptyl.
- oxy refers to a -O- group.
- sulfonyl refers to a -S(O) 2 - group.
- the present compounds may exist as therapeutically suitable salts.
- the term "therapeutically suitable salt,” refers to salts or zwitterions of the compounds which are water or oil-soluble or dispersible, suitable for treatment of disorders without undue toxicity, irritation and allergic response, commensurate with a reasonable benefit/risk ratio and effective for their intended use.
- the salts may be prepared during the final isolation and purification of the compounds or separately by reacting an amino group of the compounds with a suitable acid.
- a compound may be dissolved in a suitable solvent, such as but not limited to methanol and water and treated with at least one equivalent of an acid, like hydrochloric acid.
- the resulting salt may precipitate out and be isolated by filtration and dried under reduced pressure.
- salts include acetate, adipate, alginate, citrate, aspartate, benzoate, benzenesulfonate, bisulfate, butyrate, camphorate, camphorsulfonate, digluconate, glycerophosphate, hemisulfate, heptanoate, hexanoate, form ate, isethionate, fumarate, lactate, maleate, methanesulfonate, naphthylenesulfonate, nicotinate, oxalate, pamoate, pectinate, persulfate, 3-phenyl ⁇ ro ⁇ ionate, picrate, oxalate, maleate, pivalate, propionate, succinate, tartrate, trichloroacetate, trifluoroacetate, glutamate, para-toluenesulfonate, undecanoate, hydro
- amino groups of the compounds may also be quaternized with alkyl chlorides, bromides and iodides such as methyl, ethyl, propyl, isopropyl, butyl, lauryl, myristyl, stearyl and the like.
- Basic addition salts maybe prepared during the final isolation and purification of the present compounds by reaction of a carboxyl group with a suitable base such as the hydroxide, carbonate, or bicarbonate of a metal cation such as lithium, sodium, potassium, calcium, magnesium, or aluminum, or an organic primary, secondary, or tertiary amine.
- a suitable base such as the hydroxide, carbonate, or bicarbonate of a metal cation such as lithium, sodium, potassium, calcium, magnesium, or aluminum, or an organic primary, secondary, or tertiary amine.
- the present compounds may also exist as therapeutically suitable prodrugs.
- therapeutically suitable prodrug refers to those prodrugs or zwitterions which are suitable for use in contact with the tissues of patients without undue toxicity, irritation and allergic response, are commensurate with a reasonable benefit/risk ratio and are effective for their intended use.
- prodrug refers to compounds that are rapidly transformed in vivo to the parent compounds of formula (I-IXc) for example, by hydrolysis in blood.
- prodrug refers to compounds that contain, but are not limited to, substituents known as “therapeutically suitable esters.”
- therapeuticically suitable ester refers to alkoxycarbonyl groups appended to the parent molecule on an available carbon atom.
- a "therapeutically suitable ester” refers to alkoxycarbonyl groups appended to the parent molecule on one or more available aryl, cycloalkyl and/or heterocycle groups as defined herein.
- Compounds containing therapeutically suitable esters are an example, but are not intended to limit the scope of compounds considered to be prodrugs.
- prodrug ester groups include pivaloyloxymethyl, acetoxymethyl, phthalidyl, indanyl and methoxymethyl, as well as other such groups known in the art.
- Other examples of prodrug ester groups are found in T. Higuchi and V. Stella, Pro-drugs as Novel Delivery Systems, Vol. 14 of the A.C.S. Symposium Series and in Edward B. Roche, ed., Bioreversible Carriers in Drug Design, American Pharmaceutical Association and Pergamon Press, 1987, both of which are incorporated herein by reference.
- Asymmetric centers may exist in the present compounds.
- Individual stereoisomers of the compounds are prepared by synthesis from chiral starting materials or by preparation of racemic mixtures and separation by conversion to a mixture of diastereomers followed by separation or recrystallization, chromatographic techniques, or direct separation of the enantiomers on chiral chromatographic columns.
- Starting materials of particular stereochemistry are either commercially available or are made by the methods described hereinbelow and resolved by techniques well known in the art.
- Geometric isomers may exist in the present compounds.
- the invention contemplates the various geometric isomers and mixtures thereof resulting from the disposal of substituents around a carbon-carbon double bond, a cycloalkyl group, or a heterocycloalkyl group. Substituents around a carbon-carbon double bond are designated as being of Z or E configuration and substituents around a cycloalkyl or heterocycloalkyl are designated as being of cis or trans configuration.
- the invention contemplates the various isomers and mixtures thereof resulting from the disposal of substituents around an adamantane ring system.
- the compounds of this invention can be prepared by a variety of procedures and synthetic routes. Representative procedures and synthetic routes are shown in, but are not limited to, Schemes 1-17. Abbreviations which have been used in the descriptions of the Schemes and the
- Substituted adamantanes of general formula (3) wherein A 5 A 5 A , A , R 5 R , R 5 R 4 , D and E are as defined in formula I, may be prepared as in Scheme 1.
- Substituted adamantamines of general formula (I) 5 purchased, prepared as described herein, or prepared using methodology known to those in the art, may be treated with an acylating agents of general formula (2), wherein Y is chloro, bromo, or fluoro and R 3 , R 4 , D and E are defined as in formula I, in the presence of a base such as diisopropylethylamine to provide amides of general formula (3).
- a 1 , A 2 , A 3 and/or A 4 in amines of formula (1) and D and E in the reagents of formula (2) may exist as or contain a group further substituted with a protecting group such as a carboxylic acid protected as the methyl ester. Examples containing a protected functional group may be required due to the synthetic schemes and the reactivity of said groups and could be later removed to provide the desired compound.
- Such protecting groups can be removed using methodology known to those skilled in the art or as described in T. W. Greene, P. G. M. Wuts "Protective Groups in Organic Synthesis" 3 rd ed. 1999, Wiley & Sons, Inc.
- Substituted adamantane amines of general formula (5) wherein A 1 , A 2 , A 3 , A 4 and R 2 are as defined in formula I, may be prepared as in Scheme 2.
- Substituted adamantane ketones of general formula (4) can be purchased, prepared as described herein, or prepared using methodology known to those skilled in the art.
- Ketones of general formula (4) can be treated with ammonia or primary amines (R 2 NH 2 ) followed by reduction with reagents such as sodium borohydride or H 2 over Pd/C in a solvent like methanol to provide amines of general formula (5).
- a 1 , A 2 , A 3 and/or A 4 in ketones of formula (4) may be a substituent with a functional group containing a protecting group such as a carboxylic acid protected as the methyl ester.
- a protecting group such as a carboxylic acid protected as the methyl ester.
- esters can be hydrolyzed and other protecting groups removed here to provide compounds of general formula (5) or in compounds subsequently prepared from (5) using methodology known to those skilled in the art.
- Substituted adamantanes of general formula (7) wherein A 2 , A 3 and A 4 are as defined in formula I and G is alkyl, cycloalkyl, arylalkyl, or aryl, as defined in the definition of terms, or G is hydrogen or an acid protecting group, may be prepared as in Scheme 3.
- Substituted adamantanes of general formula (6) can be purchased or prepared using methodology known to those in the art.
- Tertiary alcohols of general formula (6) can be treated with oleum and formic acid followed by water or an alcohol GOH to provide polycycles of general formula (7).
- G in formula (7) may be a protecting group such as methyl.
- ester protecting groups can be removed from polycycles of general formula (7) or from compounds subsequently prepared from (7).
- Substituted adamantanes of general formula (10), wherein A 2 , A 3 , A 4 , R 1 , R 2 , R 3 , R 4 , D, E, R 18 and R 19 are as defined in formula I, may be prepared as in Scheme 4.
- Adamantane acids of general formula (8) may be prepared as described herein or using methodology known to those in the art.
- the acids of general formula (8) may be coupled with amines of general formula (9) (wherein R 18 and R 19 are defined as in formula I) with reagents such as O-(benzotrialzol-l-yl)-l,l,3,3-tetramethyluronium tetrafluoroborate (TBTU) to provide amides of general formula (10).
- TBTU O-(benzotrialzol-l-yl)-l,l,3,3-tetramethyluronium tetrafluoroborate
- R 18 and/or R 19 in amides of formula (10) may be a substituent with a functional group containing a protecting group, such as a carboxylic acid protected as the methyl ester.
- a protecting group such as a carboxylic acid protected as the methyl ester.
- esters can be hydrolyzed and other protecting groups removed using methodology known to those skilled in the art.
- Esters of general formula (11) can be mono-alkylated or bis-alkylated to provide esters of general formula (12) wherein R 101 is the acid protecting group, P, as described above.
- the bis-alkylation can be conducted either sequentially or in a one pot reaction.
- Mono or bis-alkylation of esters of general formula (11) can be achieved in the presence of a base such as, but not limited to, sodium hydride, and an alkylating agent such as, but not limited to, alkyl halides (for example, methyl iodide, allyl bromide and the like).
- the reaction is generally performed in a solvent such as, but not limited to, anhydrous N 5 N- dimethylformamide, at a temperature from about 0 0 C to about 23 0 C.
- Removal of the protecting group P can be achieved using methodologies known to those skilled in the art or as described in T. W. Greene, P. G. M.
- esters of formula (13), wherein P is C 1 -C 6 alkyl, unsubstituted or substituted aryl (for example, phenyl) or unsubstituted or substituted arylalkyl (for example, benzyl); and Y is Cl, Br, I, or triflate can be purchased, prepared as described herein, or prepared using methodologies known to those skilled in the art.
- Esters of formula (13) can be converted to boronic esters of formula (14) when treated with a boron source like bis(pinacolato)diboron, a catalyst such as 1,1 '-bis(diphenylphosphino)ferrocenedichloropalladium (II), and a base like potassium acetate.
- a boron source like bis(pinacolato)diboron
- a catalyst such as 1,1 '-bis(diphenylphosphino)ferrocenedichloropalladium (II)
- a base like potassium acetate.
- the conversion is facilitated in a solvent such as, but not limited to, dimethyl sulfoxide, N,N-dimethylforrnamide or toluene, at a temperature of about 80 0 C to about 100 0 C.
- Boronic esters of general formula (14) may be coupled with reagents of formula Z-Y, wherein Z is aryl or heteroaryl and Y is Cl, Br, I, or triflate, a catalyst such as 1,1 '-bis(diphenylphosphino)ferrocenedichloropalladium (II), and a base like sodium carbonate, to provide compounds of formula (15) wherein R 101 is an acid protecting group, P.
- the reaction can be performed in a solvent system like N,N-dimethylformamide and water at a temperature of about 80 0 C to 90 0 C.
- compounds of formula (13) wherein Y is Cl, Br, I, or triflate can be treated with a boronic acid or ester of formula (13 A) or Z-B(OR 102 ) 2 , wherein R 102 is hydrogen or alkyl, in the presence of a catalyst, such as but not limited to, bis(triphenylphospine)palladium (H) chloride or dichlorobis(tri-o-tolylphosphine)palladium (II), and a base such as triethylamine or sodium carbonate, to provide compounds of formula (15) wherein R 101 is an acid protecting group, P.
- the reaction can be effected by heating at a temperature from about 50 0 C to about 180 0 C in solvents such as isopropanol, ethanol, dimethoxyethane, water or dioxane.
- Acids of general formula (19), wherein R 101 is hydrogen, R 3 is as defined in formula (I) and A is a substituent of heterocycle as defined in the definition of terms, can be prepared from malonic acid di-ester of formula (16) wherein P is C 1 -C 6 alkyl or benzyl as shown in Scheme 7.
- Malonic acid di-esters of general formula (16) wherein P is an acid protecting group such as C 1 -C 6 alkyl, unsubstituted or substituted aryl (for example, phenyl) or unsubstituted or substituted arylalkyl (for example, benzyl), can be purchased or prepared using methodologies known to those skilled in the art.
- Malonic acid di-esters of general formula (16) can be treated with one molar equivalent of allyl bromide or 4-bromo-l-butene, using mono alkylation conditions for the conversion of (11) to (12) in Scheme 5, to provide compounds of formula (17).
- Ozonolysis of the terminal olefin of di-ester (17) may be achieved in a solvent system like dichloromethane and methanol at a low temperature of about -78 0 C, by bubbling ozone through the solution, followed by purging the solution with nitrogen gas, and reduction of the intermediate ozonide with dimethyl sulfide to provide aldehyde di-esters of the general formula (18).
- Thiazoles of formula (20) can be purchased or prepared using methodologies known to those skilled in the art.
- Thiazoles of formula (20) may be alkylated by in situ activation with a chloroformate such as, but not limited to, ethyl chloroformate, followed by treatment of a nucleophile such as lithio diethylmalonate (prepared from a malonic acid di-ester in a solution such as tetrahydrofuran with a base such as lithium bis(trimethylsilyl)amide), to afford compounds of formula (21) wherein P 1 and P 2 are C 1 -C 6 alkyl.
- a chloroformate such as, but not limited to, ethyl chloroformate
- a nucleophile such as lithio diethylmalonate (prepared from a malonic acid di-ester in a solution such as tetrahydrofuran with a base such as lithium bis(trimethylsilyl)amide
- the former can be conducted in a solvent such as, but not limited to, tetrahydrofuran, at a temperature around 0 0 C.
- Treatment with the nucleophile can be effected in a solvent such as tetrahydrofuran and at a temperature around 23 0 C.
- the lithio diethylmalonate may be formed in a solvent such as tetrahydrofuran.
- the N-protected malonic acid di-ester adduct of general formula (21) may be oxidized with an agent such as tetrachloro-l,2-benzoquinone in a solvent such as dichloromethane at a temperature around 0 0 C to afford the di-ester of general formula (22).
- Mono-decarboxylation of di-ester (22) may be achieved by heating in a solvent system such as water and dimethyl sulfoxide with a salt such as sodium chloride at a temperature near 180 0 C to provide esters of general formula (
- Acids of formula (27) wherein R 101 is hydrogen, P 3 is -C(O)OCH 2 C 6 H 5 , and R 3 is as defined in formula (I) can be prepared from compounds of formula (24) where P is an acid protecting group such as, C 1 -C 6 alkyl, unsubstituted or substituted aryl (for example, phenyl) or unsubstituted or substituted arylalkyl (for example, benzyl), as shown in Scheme 9.
- P is an acid protecting group such as, C 1 -C 6 alkyl, unsubstituted or substituted aryl (for example, phenyl) or unsubstituted or substituted arylalkyl (for example, benzyl), as shown in Scheme 9.
- Compounds of formula (24) can be purchased or prepared using methodologies known to those skilled in the art.
- Mono alkylation of compounds of formula (25) with halides of formula R 3 -X 3 wherein X 3 is Cl, Br or I, using reaction conditions as described in Scheme 5 provides compounds of formula (26).
- Compounds of formula (28) can be purchased, prepared as described herein, or prepared using methodologies known to those skilled in the art.
- Compounds of formula (28) wherein P is an acid protecting group can be reacted with compounds of formula 7 ⁇ -Y, wherein Y is Cl, Br, I, or triflate such as 6-chloronicotinonitrile, with a base such as sodium hydride, and in an anhydrous solvent system such as tetrahydrofuran and 1,3-dimethyl- 3,4,5,6-tetrahydro-2(lH)-pyrimidinone (DMPU) at a temperature ranging from 0 0 C to 23 0 C to provide esters of general formula (29), wherein R 101 is a protecting group, P.
- G 3 is aryl or heteroaryl
- R 103 is hydrogen, potassium, sodium, lithium, or C 1 -C 6 alkyl, can be prepared as shown in Scheme 11.
- G 3 -Y wherein G 3 is aryl or heteroaryl and Y is Cl, Br, I or triflate can be purchased or prepared using methodologies known to those skilled in the art, as well as, alkyl trimethylsilyl ketene acetals of formula (30) wherein R 103 is C 1 -C 6 alkyl.
- Adamantanes of general formula (34) wherein Y is Cl, Br or I, can be prepared as described herein or prepared using methodologies known to those skilled in the art.
- Olefins of general formula (35) wherein Z 3 is either aryl or heteroaryl can be purchased or prepared using methodologies known to those skilled in the art.
- Adamantanes of general formula (34) can be reacted with olefins of general formula (35), such as, but not limited to, 4- vinylpyridine; a catalyst such as, but not limited to, bis(triphenylphosphine)palladium (II) dichloride; a base such as, but not limited to, triethylamine; and, in a solvent system such as N,N-dimethylformamide at a temperature of near 150 0 C to provide adamantanes of general formula (36).
- olefins of general formula (35) such as, but not limited to, 4- vinylpyridine
- a catalyst such as, but not limited to, bis(triphenylphosphine)palladium (II) dichloride
- a base such as, but not limited to, triethylamine
- a solvent system such as N,N-dimethylformamide at a temperature of near 150 0 C to provide adamantanes of general formula (36).
- Substituted adamantanes of general formula (37) can be prepared as described herein or prepared using methodologies known to those skilled in the art.
- Substituted adamantanes of general formula (37) can be alkylated with alkylating agents Q-Y, wherein Q is alkyl, arylalkyl, heteroarylalkyl, heterocycle alkyl, or cycloalkylalkyl and Y is a leaving group like I, Br, Cl, or triflate, in the presence of a base like potassium carbonate and in a solvent like N,N-dimethylformamide to yield substituted adamantanes of general formula (38).
- Substituted adamantanes of general formula (39), wherein Y is F, Cl, Br, or I, can be prepared as described herein or prepared using methodologies known to those skilled in the art.
- Substituted adamantanes of general formula (39) can be condensed with amines of general formula R 1 ⁇ 111 NH, to provide compounds of formula (40).
- the reaction can be conducted neat in a microwave synthesizer at a temperature near 150 0 C for a period of about 40 minutes.
- R 4 , and D are as defined in formula I; G 8 is aryl or heteroaryl as defined in the definition of terms; Q 1 is C 1 -C 3 alkyl; and, R q and R r are independently hydrogen, alkyl, or heterocyclealkyl, or R q and R r together with the nitrogen to which they are attached form a heterocycle ring, can be prepared as shown in Scheme 16.
- Substituted adamantanes of general formula (41) can be prepared as described herein or prepared using methodologies known to those skilled in the art.
- Substituted adamantanes of general formula (41) can be halogenated with a reagent like N-halosuccinimde (for example, N-chlorosuccinirnide and the like) in the presence of a radical initiator like AIBN and in a solvent like carbon tetrachloride at a temperature near 80 0 C to yield substituted adamantanes of general formula (42), wherein Y is Cl, Br, or I.
- N-halosuccinimde for example, N-chlorosuccinirnide and the like
- Substituted adamantanes of general formula (42) when treated with amines of general formula R q R r NH in a solvent like dichloromethane at a temperature between 23 0 C and 40 0 C provide substituted adamantanes of general formula (43).
- Substituted adamantanes of general formula (6) can be purchased or prepared using methodology known to those in the art. Substituted adamantanes of general formula (6) can be brominated with a reagent like hydrobromic acid in a solvent like water to provide bromides of general formula (44). Adamantanes of general formula (44) when treated with ethylene glycol and a catalytic amount of an acid like p-toluenesulfonic acid in a solvent like benzene provide adamantanes of general formula (45).
- Bromides of general formula (45) can be (a) treated with Rieke zinc in a solvent like tetrahydrofuran; and (b) followed by treatment with reagent (46) (prepared as described in Han, Z.; Krishnamurthy, D.; Grover, P.; Fang, Q. K.; Senanayake, C. H. J. Am. Chem. Soc. 2002, 124, 7880-7881) in a solvent like tetrahydrofuran to provide adamantanes of general formula (47).
- reagent (46) prepared as described in Han, Z.; Krishnamurthy, D.; Grover, P.; Fang, Q. K.; Senanayake, C. H. J. Am. Chem. Soc. 2002, 124, 7880-7881
- Adamantanes of general formula (47) may be treated with lithium amide of formula LiNHR 5 R 6 (prepared in situ by reacting ammonia with lithium or amines of formula R 5 R 6 NH wherein R 5 and R 6 are other than hydrogen, with t-butyl lithium) in a solvent like tetrahydrofuran.
- the resulting sulfanamides can be oxidized with a reagent like osmium tetroxide with a catalyst oxidant like NMO in a solvent like tetrahydrofuran to provide sulfonamides of general formula (48).
- Adamantanes of general formula (48) can be deketalized with reagents like hydrochloric acid in a solvent mixture like water and tetrahydrofuran to provide ketones of formula (49).
- Ketones of formula (49) can be treated with amines of formula R 2 NH 2 followed by reduction with reducing reagents such as, but not limited to, sodium borohydride or hydrogen over Pd/C in a solvent like methanol to provide amines of general formula (50).
- a 2 , A 3 , A 4 , R 2 , R 5 and R 6 in amines of formula (50) may be a substituent with a functional group containing a protecting group such as a carboxylic acid protected as the methyl ester.
- a protecting group such as a carboxylic acid protected as the methyl ester.
- Such esters can be hydrolyzed and other protecting groups removed here or in compounds subsequently prepared from (50) using methodology known to those skilled in the art.
- the reaction solution was cooled to 10 0 C in an ice bath. 20 volumes of 10% NaCl aq (4 L) were cooled to ⁇ 10 0 C, the crude reaction mixture was quenched into the brine solution in batches, maintaining an internal temperature ⁇ 70 0 C. The quenched reaction solution was combined with a second identical reaction mixture for isolation.
- the combined product solutions were extracted 3x5 volumes with CH 2 Cl 2 (3x2.2 L) and the combined CH 2 Cl 2 layers were then washed 1x2 volumes with 10% NaCl (1 L).
- the CH 2 Cl 2 solution was then extracted 3x5 volumes with 10% Na 2 CO 3 (3x2.2L).
- the combined Na 2 CO 3 extracts were washed with 1x2 volumes with CH 2 Cl 2 (1 L).
- the Na 2 CO 3 layer was then adjusted to pH 1-2 with concentrated HCl ( ⁇ 2 volumes, product precipitates out of solution).
- the acidic solution was then extracted 3x5 volumes with CH 2 Cl 2 (3x2.2 L), and the organic layer was washed 1x2 volumes with 10% NaCl.
- the organic solution was then dried over Na 2 SO 4 , filtered, concentrated to -1/4 volume, then chase distilled with 2 volumes EtOAc (1 L). Nucleation occurred during this distillation.
- the suspension was then chase distilled 2x5 volumes (2x2 L) with heptane and cooled to room temperature.
- the suspension was then filtered, and the liquors were recirculated 2x to wash the wet cake.
- the resultant material was dried overnight at 50 0 C, 20 mm Hg to afford the title compound.
- Example 1C E-4-amino-adamantane-l-carboxylic acid methyl ester hydrochloride Methanol (10 volumes, 85 mL) was cooled to 0 0 C. AcCl was added dropwise (5.0 equiv., 15.5 mL), and the solution was warmed to ambient temperature for 15-20 minutes. The product from Example IB (8.53 g, 43.7 mmol, 1.0 equiv.) was added and the reaction solution was heated to 45 0 C for 16 hours (overnight). Consumption of the starting aminoacid was monitored by LC/MS (APCI). The reaction solution was then cooled to room temperature, 10 volumes MeCN (85 mL) was added, distilled to ⁇ 1/4 volume
- the methyl ester of the titled compound obtained from step A (50 mg, 0.12 mmol) was dissolved in 3 N HCl (1 mL), dioxane (0.25 mL), and 4 N HCl (1 mL). The homogenous acid solution was heated to 60 0 C for 24 hours, was cooled to 23 0 C, and was then concentrated under reduced pressure to provide the title compound.
- the methyl ester of the titled compound was prepared according to the method of step
- Example ID A of Example ID substituting 1 -phenyl- 1-cyclopropanecarboxylic acid for X-(A- chlorophenyl)-l-cyclobutanecarboxylic acid, and the crude methyl ester was purified by chromatography on flash silica gel with an eluant gradient of 20-40% ethyl acetate/hexanes.
- the methyl ester of the titled compound was prepared according to the method of step A of Example ID substituting 2-methyl-2-phenyl propionic acid for l-(4-chlorophenyl)-l- cyclobutanecarboxylic acid, and the crude methyl ester was purified by chromatography on flash silica gel with an eluant gradient of 20-40% ethyl acetate/hexanes.
- Step B The methyl ester obtained from step A (49 mg, 0.14 mmol) was dissolved in 3 N HCl (1 mL) and dioxane (0.25 mL), heated to 60 0 C for 24 hours, was cooled to 23 0 C, and was then concentrated under reduced pressure to provide the title compound.
- Example 4 E-A- ⁇ [ 1 -(4-Chloro-phenyiy cyclobutanecarbonyri -amino ⁇ -adamantane- 1 -carboxylic acid amide A solution of the product from step B of Example ID (24 mg, 0.062 mmol) in DCM
- Example 6 i?-4-f 2-Methyl-2-phenyl-propionylamino)-adamantane- 1 -carboxylic acid amide
- the title compound was prepared according to the method of Example 4 substituting the product from step B of Example 3 for the product from step B of Example ID.
- Example 7 iV -2-adamantyl-2-methyl-2-phenylpropanamide
- 2-adamantanamine hydrochloride 38 mg, 0.20 mmol
- 2- phenylisobutyric acid 30 mg, 0.19 mmol
- O-benzotriazol-l-yl- ⁇ -V,N',iV ' '- tetramethyluronium tetrafiuoroborate TBTU
- TBTU O-benzotriazol-l-yl- ⁇ -V,N',iV ' '- tetramethyluronium tetrafiuoroborate
- DMA N,N-dimethylacetamide
- DIEA 80 ⁇ L, 0.46 mmol
- DMSO/MeOH (1:1, 1.5 mL) and purified by preparative HPLC on a Waters Symmetry C8 column (25 mm X 100 mm, 7 urn particle size) using a gradient of 10% to 100% acetonitrile: aqueous ammonium acetate (10 mM) over 8 minutes (10 minute run time) at a flow rate of 40 mL/minute on reverse phase HPLC to afford the title compound upon concentration under reduced pressure.
- step A The methyl ester obtained from step A (47 mg, 0.11 mmol) was dissolved in 5 N aqueous HCl (1 mL) and 4 N HCl in dioxane (2 mL), heated to 60 0 C for 24 hours, was cooled to 23 0 C, and was then concentrated under reduced pressure to provide the title compound.
- the title compound was prepared according to the method as described in Example 4 substituting the product of step B of Example 9A for the product of step B of Example ID, and with the exception that the crude title compound was purified by normal phase flash chromatography with MeOH/DCM (5:95) as eluant.
- step A The methyl ester obtained from step A (51 mg, 0.13 mmol) was dissolved in 5 N aqueous HCl (1 mL) and 4 N HCl in dioxane (2 mL), heated to 60 0 C for 24 hours, was cooled to 23 C, and was then concentrated under reduced pressure to provide the title compound.
- the title compound was prepared according to the method as described in Example 4, substituting the product of step B of Example 1OA for the product of step B of Example ID and with the exception that title compound was purified by normal phase flash chromatography with MeOH/DCM (5:95) as eluant.
- the methyl ester of the title compound was prepared according to the method as described in step A of Example ID, substituting l-(4-chlorophenyl)-l- cyclopentanecarboxylic acid for l-(4-chlorophenyl)-l-cyclobutanecarboxylic acid, and with the exceptions that the methyl ester was purified by reverse phase chromatography.
- the methyl ester obtained from step A (30 mg, 0.072 mmol) was dissolved in 5 N aqueous HCl (0.5 mL) and 4 N HCl in dioxane (1 mL), heated to 60 0 C for 24 hours, was cooled to 23 0 C, and was then concentrated under reduced pressure to provide the title compound.
- the title compound was prepared according to the method as described in Example 4, substituting the product of step B of Example 11 A for the product of step B of Example ID, and with the exception that title compound was purified by normal phase flash chromatography with MeOH/DCM (5:95) as eluant.
- Step B The methyl ester obtained from step A (50 mg, 0.13 mmol) was dissolved in 5 N aqueous HCl (1 mL) and 4 N HCl in dioxane (2 mL), heated to 60 0 C for 24 hours, was cooled to 23 0 C, and was then concentrated under reduced pressure to provide the title compound.
- the methyl ester of the title compound was prepared according to the method as described in step A of Example ID, substituting 1 -phenyl- 1-cyclopentanecarboxylic acid for l-(4-chlorophenyl)-l-cyclobutanecarboxylic acid, and with the exceptions that the methyl ester was purified by reverse phase chromatography.
- the methyl ester obtained from step A (20 mg, 0.052 mmol) was dissolved in 5 N aqueous HCl (0.5 mL) and 4 N HCl in dioxane (1 mL), heated to 60 0 C for 24 hours, was cooled to 23 0 C, and was then concentrated under reduced pressure to provide the title compound.
- Example 13B E-4- [(I -phenylcyclopentvDcarbonyll amino ⁇ adamantane- 1 -carboxamide
- the title compound was prepared according to the method as described in Example 4, substituting the product of step B of Example 13 A for the product of step B of Example ID, and with the exception that title compound was purified by normal phase flash chromatography with MeOH/DCM (5:95) as eluant.
- Step A The methyl ester of the title compound was prepared according to the method as described in step A of Example ID, substituting l-(3-fluorophenyl)-l- cyclopentanecarboxylic acid for l-(4-chlorophenyl)-l-cyclobutanecarboxylic acid, and with the exceptions that the methyl ester was purified by reverse phase chromatography.
- step A The methyl ester obtained from step A (41 mg, 0,10 mmol) was dissolved in 5 N aqueous HCl (0.5 mL) and 4 N HCl in dioxane (1 mL), heated to 60 0 C for 24 hours, was cooled to 23 0 C, and was then concentrated under reduced pressure to provide the title compound.
- Example 15A E-A-( ⁇ [ 1 -(2-chloro-4-fluoro ⁇ henyl)cyclopentyl]carbonvU amino)adamantane- 1 -carboxylic acid Step A
- the methyl ester of the title compound was prepared according to the method as described in step A of Example ID, substituting l-(2-chloro-4-fluorophenyl)-l- cyclopentanecarboxylic acid for l-(4-chlorophenyl)-l-cyclobutanecarboxylic acid, and with the exceptions that the methyl ester was purified by reverse phase chromatography.
- Step B The methyl ester obtained from step A (66 mg, 0.15 mmol) was dissolved in 5 N aqueous HCl (0.5 mL) and 4 N HCl in dioxane (2 mL), heated to 60 0 C for 24 hours, was cooled to 23 0 C, and was then concentrated under reduced pressure to provide the title compound.
- step A The methyl ester obtained from step A (58 mg, 0.15 mmol) was dissolved in 5 N aqueous HCl (0.5 mL) and 4 N HCl in dioxane (2 mL), heated to 60 0 C for 24 hours, was cooled to 23 0 C, and was then concentrated under reduced pressure to provide the title compound.
- the methyl ester of the title compound was prepared according to the method as described in step A of Example ID, substituting l-(2-fluorophenyl)-l- cyclopentanecarboxylic acid for l-(4-chlorophenyl)-l-cyclobutanecarboxylic acid, and with the exceptions that the methyl ester was purified by reverse phase chromatography.
- Step B The methyl ester obtained from step A (56 mg, 0.14 mmol) was dissolved in 5 N aqueous HCl (0.5 mL) and 4 N HCl in dioxane (2 mL), heated to 60 0 C for 24 hours, was cooled to 23 0 C, and was then concentrated under reduced pressure to provide the title compound.
- the methyl ester of the title compound was prepared according to the method as described in step A of Example ID, substituting 1 -methyl- 1-cyclohexanecarboxylic acid for l-(4-chlorophenyl)-l-cyclobutanecarboxylic acid, and with the exceptions that the methyl ester was purified by reverse phase chromatography.
- step A The methyl ester obtained from step A (33 mg, 0.10 mmol) was dissolved in 5 N aqueous HCl (0.5 mL) and 4 N HCl in dioxane (1 mL), heated to 60 0 C for 24 hours, was cooled to 23 0 C, and was then concentrated under reduced pressure to provide the title compound.
- Example 18B E-A- ⁇ ⁇ ( 1 -methylcyclohexyDcarbonyll amino) adamantane- 1 -carboxamide
- the title compound was prepared according to the method as described in Example 4, substituting the product of step B of Example 18A for the product of step B of Example ID.
- the methyl ester of the title compound was prepared according to the method as described in step A of Example ID, substituting l-(2,4-dichlorophenyl)-l- cyclopropanecarboxylic acid for l-(4-chlorophenyl)-l-cyclobutanecarboxylic acid and with the exceptions that the methyl ester was purified by reverse phase chromatography.
- step A The methyl ester obtained from step A (47 mg, 0.11 mmol) was dissolved in 5 N aqueous HCl (0.5 mL) and 4 N HCl in dioxane (2 mL), heated to 60 0 C for 24 hours, was cooled to 23 0 C, and was then concentrated under reduced pressure to provide the title compound.
- the methyl ester of the title compound was prepared according to the method as described in step A of Example ID, substituting l-(4-methoxyphenyl)-l- cyclopropanecarboxylic acid for l-(4-chlorophenyl)-l-cyclobutanecarboxylic acid and with the exceptions that the methyl ester was purified by reverse phase chromatography.
- Step B The methyl ester obtained from step A (43 mg, 0.11 mmol) was dissolved in 5 N aqueous HCl (0.5 mL) and 4 N HCl in dioxane (1 mL), heated to 60 0 C for 24 hours, was cooled to 23 0 C, and was then concentrated under reduced pressure to provide the title compound.
- the methyl ester of the title compound was prepared according to the method as described in step A of Example ID, substituting l-(4-methylphenyl)-l- cyclopropanecarboxylic acid for l-(4-chlorophenyl)-l-cyclobutanecarboxylic acid and with the exceptions that the methyl ester was purified by reverse phase chromatography.
- step A The methyl ester obtained from step A (39 mg, 0.11 mmol) was dissolved in 5 N aqueous HCl (0.5 mL) and 4 N HCl in dioxane (1 mL), heated to 60 0 C for 24 hours, was cooled to 23 0 C, and was then concentrated under reduced pressure to provide the title compound.
- Example 22A To a solution of the product of Example 22A (3.7 g, 13.6 mmoles) in tetrahydrofuran (110.0 ml) and methanol (37.0 ml) was added 2 N sodium hydroxide (19.0 ml) and the solution was stirred at ambient temperature for about 16 hours. The reaction mixture was concentrated in vacuum down to the water layer, was cooled with an ice bath, and was acidified by addition of 2N hydrochloric acid. The precipitate was filtered off and was dried in vacuum to provide the title compound.
- Example 22C To a solution of the product of Example 22C (300 mg, 0.7 mmoles) in 1,2- dimethoxyethane (6.0 ml) was added a solution of pyridine-4-boronic acid (127 mg, 1.0 mmoles) in ethanol (1.0 ml), dichlorobis(tri-o-tolylphosphine)palladium(II) (28 mg, 0.04 mmoles) and a 2M aqueous solution of sodium carbonate (1.7 ml, 3.5 mmoles) and the mixture was stirred under nitrogen in a heavy walled process vial in a microwave synthesizer (Personal Chemistry Smith Synthesizer) at about 140 0 C for about 10 min. The reaction mixture was concentrated in vacuum and the crude product was purified by flash column chromatography on silica gel using hexanes/ethyl acetate (2 : 1) as the mobile phase to provide the title compound.
- pyridine-4-boronic acid 127 mg, 1.0
- Example 23C To a solution of the product of Example 23C (250 mg, 0.69 mmoles) in dioxane (9.0 ml) was added 2N hydrochloric acid (9.0 ml) and the mixture was heated to about 60 °C for about 18 hours. The mixture was cooled, concentrated down to the water layer, the precipitate was filtered off and was dried in vacuum to give the title compound.
- Example 22F The title compound was prepared according to the method of Example 22F, substituting the product of Example 23D for the product of Example 22E.
- Example 24D E-4-(2-Methyl-2-tMophen-3-yl-propionylaminoVadamantane- 1 -carboxylic acid
- the title compound was prepared according to the method of Example 23D, substituting the product of Example 24C for the product of Example 23C.
- Example 22F The title compound was prepared according to the method of Example 22F, substituting the product of Example 24D for the product of Example 22E.
- Example 25B A solution of Example 25B (30 mg, 0.071 mmol), KOTMS (14 mg, 0.11 mmol) in THF (1 mL) was stirred for 12 hours at 23 0 C. The solvent was evaporated in vacuo to collect a solid. To the solid was added TBTU (40 mg, 0.12 mmol), DIEA (22 mg, 0.17 mmol) and DMF (0.5 mL) and stirred for 2 hours at 23 0 C. Ammonium hydroxide-30% by weight (2 mL) was added and stirred at 23 0 C for a further 30 minutes. The reaction was partitioned between EtOAc (8 mL) and water (3mL).
- the organic layer was washed with water (3 mL), dried with MgSO 4 , filtered, and evaporated in vacuo.
- the crude reaction mixture was purified by preparative reverse phase HPLC on a Waters Symmetry C8 column (25 mm X 100 mm, 7 urn particle size) using a gradient of 20% to 100% acetonitrile: water (0.1% TFA) over 18 min at a flow rate of 40 mL/min.to provide the title compound as the trifluoroacetic acid salt.
- Example 22C The title compound was prepared according to the method of Example 22C substituting the product of Example 26C for the product of Example 22B .
- Example 27A E-A- ( I " 1 -(4-Methox v-phenylVcyclopentanecarbonyl] -amino > -adamantane- 1 -carboxylic acid methyl etser A solution of the product of Example 1C (110 mg, 0.45 mmol), l-(4-methoxyphenyi)-
- Example 22F The title compound was prepared according to the method of Example 22F substituting the product of Example 28 A for the product of Example 22E.
- Example 30A was purified by flash chromatography (hexane/EtOAc 100:0 to 85:15) to provide Example 30A as an oil.
- Example 3OA A solution of the product of Example 3OA (1.0 gm, 5 mmol) was dissolved in CH 2 Cl 2 MeOH 10:1 (15 mL) and cooled to -78°C. To the solution was bubbled O 3 over 20 minutes. The reaction solution was purged with N 2 for a further 10 minutes and dimethyl sulfide (DMS) (3.1 gm, 50 mmol) was added and the reaction warmed to ambient temperature and stirred for a further 2 hours. The solvent was evaporated in vacuo and product purified by flash column chromatography (hexane/EtOAc 100:0 to 70:30) to collect Example 30B as an oil.
- DMS dimethyl sulfide
- the crude reaction mixture was purified by preparative reverse phase HPLC on a Waters Symmetry C8 column (25 mm X 100 mm, 7 um particle size) using a gradient of 20% to 100% acetonitrile:water (0.1% TFA) over 18 min at a flow rate of 40 mL/min. to provide the title compound.
- Example 31A A solution of the product of Example 31A (1.1 gm, 5.9 mmol) was dissolved in CH 2 Cl 2 /Me0H 10:1 (15 mL) and cooled to -78 0 C. To the solution was bubbled O 3 over 20 minutes. The reaction solution was purged with N 2 for a further 10 minutes and dimethyl sulfide (DMS) (3.6 gm, 59 mmol) was added and the reaction warmed to ambient temperature and stirred for a further 2 hours. The solvent was evaporated in vacuo and product purified by flash column chromatography (hexane/EtOAc 100:0 to 70:30) to collect Example 3 IB as an oil.
- DMS dimethyl sulfide
- Example 3 ID E-4-raminocarbonyl)-2-adamantyl1-l-benzyl-3-methyl-2-oxopyrrolidine-3-carboxamide
- Example 1C (54 mg, 0.22 mmol), TBTU (92 mg, 0.29 mmol) and DIEA (57 mg, 0.45 mmol) in DMF (1.2 mL) was stirred for 2 hours at 23 0 C.
- the reaction was partitioned between EtOAc (8 ml) and water (4 ml). The organic layer was separated and washed twice with water (4 mL each), dried with MgSO 4 , filtered and evaporated in vacuo.
- the resulting oil was taken in THF (1 mL) and stirred with KOTMS (34 mg, 0.27 mmol) for 12 hours at 23 0 C. The solvent was evaporated in vacuo.
- the crude reaction mixture was purified by preparative reverse phase HPLC on a Waters Symmetry C8 column (25 mm X 100 mm, 7 um particle size) using a gradient of 20% to 100% acetonitrile: water (0.1% TFA) over 18 min at a flow rate of 40 mL/min. to provide the title compound.
- Example 32A 50 mg, 0.17 mmol
- the product of Example 1C 52 mg, 0.21 mmol
- TBTU 87 mg, 0.27 mmol
- DIEA 54 mg, 0.42 mmol
- the reaction was partitioned between EtOAc (8 ml) and water (4 ml). The organic layer was separated and washed twice with water (4 mL each), dried with MgSO 4 , filtered and evaporated in vacuo. The resulting oil was taken in THF (1 mL) and stirred with KOTMS (33 mg, 0.25 mmol) for 12 hours at 23 0 C. The solvent was evaporated in vacuo.
- the crude reaction mixture was purified by preparative reverse phase HPLC on a Waters Symmetry C8 column (25 mm X 100 mm, 7 urn particle size) using a gradient of 20% to 100% acetonitriletwater (0.1% TFA) over 18 min at a flow rate of 40 mL/min. to provide the title compound.
- Example 1C (48 mg, 0.19 mmol), TBTU (82 mg, 0.25 mmol) and DIEA (51 mg, 0.4 mmol) in DMF (1.2 mL) was stirred for 2 hours at 23 0 C.
- the reaction was partitioned between EtOAc (8 ml) and water (4 ml). The organic layer was separated and washed twice with water (4 mL each), dried with MgSO 4 , filtered and evaporated in vacuo.
- the resulting oil was taken in THF (1 mL) and stirred with KOTMS (31 mg, 0.24 mmol) for 12 hours at 23 0 C. The solvent was evaporated in vacuo.
- the crude reaction mixture was purified by preparative reverse phase HPLC on a Waters Symmetry C8 column (25 mm X 100 mm, 7 urn particle size) using a gradient of 20% to 100% acetonitrile:water (0.1% TFA) over 18 min at a flow rate of 40 mL/min. to provide the title compound.
- Example 34A A solution of the product of Example 31B (0.075 gm, 0.4 mmol), 3-chloro- benzylamine (68 mg, 0.47 mmol) and MP-triacetoxy borohydride (431 mg, 1 mmol) in THF (2 mL) was stirred for 12 hours at 23 0 C. The solution was filtered and evaporated in vacuo. The resulting oil was taken up in THF (1.2 mL) and stirred with KOTMS (77 mg, 0.6 mmol) at 23 0 C for 12 hours. The solvent was evaporated in vacuo to provide Example 34A.
- Example 35B E-A- ⁇ 2-Methyl-2-[4-(l -methyl- lH-pyrazol-4-ylVphenyl]-propionylamino ⁇ -adamantane- 1 - carboxylic acid
- the title compound was prepared according to the method of Example 23D, substituting the product of Example 35 A for the product of Example 23C.
- Example 36D E-4-[2-(3-BromophenylV2-methylpropionylamino] -adamantane-1-carboxylic acid methlv ester
- the title compound was prepared according to the method as described in Example
- Example 27C The title compound was prepared according to the method as described in Example 27C, substituting the product of Example 36E for the product of Example 27B.
- Example 22D The title compound was prepared according to the method of Example 22D, substituting 3,5-dimethyl-4-(4,4,5,5-tetramethyl- 1 ,3,2-dioxaborolane-2-yl)isoxazole for pyridine-4-boronic acid.
- Example 37C E-4- ⁇ 2-[4-(3,5-Dimethyl-isoxazol-4-yl)-phenyl1-2-methyl-propionylamino ⁇ -adamantane-l- carboxylic acid amide
- the title compound was prepared according to the method of 35C, substituting the product of Example 37B for the product of Example 35B.
- the crude product was purified by preparative HPLC on a Waters Symmetry C8 column (25 mm x 100 mm, 7 ⁇ m particle size) using a gradient of 10% to 100% acetonitrile : 0.1% aqueous TFA over 8 min (10 min run time) at a flow rate of 40 ml/min.
- Example 38B E-A- [2-Methyl-2-(4- ⁇ yridin-3 -yl-phenyiVpropionylammo] -adamantane- 1 -carboxylic acid
- the trifluoroacetic acid salt of the title compound was prepared according to the method of Example 35C, substituting the product of Example 38B for the product of Example 35B, and with the exception that the crude product was purified by preparative HPLC on a Waters Symmetry C8 column (25 mm x 100 mm, 7 ⁇ m particle size) using a gradient of 10% to 100% acetonitrile : 0.1% aqueous TFA over 8 min (10 min run time) at a flow rate of 40 ml/min.
- Example 39A E-4-( ⁇ [ " 4-(2-Methyl-2-thiophen-2-yl-propionylaminoVadamantane-l-carbonyl1-amino>- methvD-benzoic acid methyl ester
- the title compound was prepared according to the method of Example 22C, substituting the product of Example 23D for Example 22B and substituting methyl A- (aminomethyl)-benzoate hydrochloride for the product of Example 1C.
- Example 35C The title compound was prepared according to the method of Example 35C substituting the product of Example 4OB for the product of Example 35B.
- Example 41 A Potassium: 3 -methyl- 1 -( 1 -methyl- 1 -phenyl-ethyl)-2-oxo-pyrrolidine-3 -carboxylate
- Example 4 IA The solvent was evaporated in vacuo and the residue taken in THF (1.2 mL) and stirred with KOTMS (77 mg, 0.6 mmol) for 12 hours at 23 0 C. The solvent was evaporated in vacuo to provide Example 4 IA.
- the crude reaction mixture was purified by preparative reverse phase HPLC on a Waters Symmetry C8 column (25 mm X 100 mm, 7 um particle size) using a gradient of 20% to 100% acetonitrile ".water (0.1% TFA) over 18 min at a flow rate of 40 mL/min. to provide the title compound.
- Example 42A Potassium; 3 -methyl-2-oxo- 1 -(( 1 R)- 1 -phenylethvDpyrrolidine-3 -carboxylate
- Example 42A A solution of the product of Example 3 IB (0.075 gm, 0.4 mmol), (R)-I- phenylethylamine (58 mg, 0.47 mmol) and MP-triacetoxy borohydride (431 mg, 1 mmol) in THF (2 mL) was stirred for 12 hours at 23 0 C. The solution was filtered and evaporated in vacuo. The resulting oil was taken up in toluene (1.5 mL) and heated at 100 0 C for 5 hours. The solvent was evaporated in vacuo and the residue taken in THF (1.2 mL) and stirred with KOTMS (77 mg, 0.6 mmol) for 12 hours at 23 0 C. The solvent was evaporated in vacuo to provide Example 42A as 1:1 mixture of diastereomers.
- Example 42B E-4-raminocarbonyl)-2-adamantyl]-3-methyl-2-oxo-l-rdRVl-phenylethyl1pyrrolidme-3- carboxamide
- a solution of the product of Example 42 A 50 mg, 0.17 mmol
- the product of Example 1C 52 mg, 0.21 mmol
- TBTU 87 mg, 0.27 mmol
- DIEA 51 mg, 0.4 mmol
- the reaction was partitioned between EtOAc (8 ml) and water (4 ml).
- the organic layer was washed with water (3 mL), dried with MgSO 4 , filtered and evaporated in vacuo.
- the crude reaction mixture was purified by preparative reverse phase HPLC on a Waters Symmetry C8 column (25 mm X 100 mm, 7 ran particle size) using a gradient of 20% to 100% acetonitrile: water (0.1% TFA) over 18 min at a flow rate of 40 mL/min. to provide the title compound as a 1 : 1 mixture of diastereomers.
- Example 43B A solution of the product of Example 31B (0.075 gm, 0.4 mmol), (S)-I- phenylethylamine (58 mg, 0.47 mmol) and MP-triacetoxy borohydride (431 mg, 1 mmol) in THF (2 mL) was stirred for 12 hours at 23 0 C. The solution was filtered and evaporated in vacuo. The resulting oil was taken up in toluene (1.5 mL) and heated at 100 0 C for 5 hours. The solvent was evaporated in vacuo and the residue taken in THF (1.2 mL) and stirred with KOTMS (77 mg, 0.6 mmol) at 23 0 C for 12 hours. The solvent was evaporated in vacuo to provide Example 43 A as a 1:1 mixture of diastereomers.
- Example 43B A solution of the product of Example 31B (0.075 gm, 0.4 mmol), (S)-I- phenylethylamine (58 mg, 0.47
- Example 1C (52 mg, 0.21 mmol), TBTU (87 mg, 0.27 mmol) and DIEA (51 mg, 0.4 mmol) in DMF (1.2 mL) was stirred at 23 0 C for 2 hours.
- the reaction was partitioned between EtOAc (8 ml) and water (4 ml). The organic layer was separated and washed twice with water (4 mL each), dried with MgSO 4 , filtered and evaporated in vacuo.
- the resulting oil was taken in THF (1 mL) and stirred with KOTMS (32 mg, 0.25 mmol) at 23 0 C for 12 hours. The solvent was evaporated in vacuo.
- the crude reaction mixture was purified by preparative reverse phase HPLC on a Waters Symmetry C8 column (25 mm X 100 mm, 7 ran particle size) using a gradient of 20% to 100% acetonitrile: water (0.1% TFA) over 18 min at a flow rate of 40 niL/min. to provide the title compound as a 1:1 mixture of diastereomers.
- Example 44A To a solution of the product of Example 44A (12.9 g, 40.7 mmoles) in dichloromethane (100.0 ml) was added tetrachloro-l,2-benzoquinone (10.0 g, 40.7 mmoles) in portions at about 0 °C, such that the mixture always had time to decolorize to a yellow- orange color. The mixture was then stirred for about 1 hour at 0 0 C and was then washed with saturated aqueous sodium bicarbonate solution (200.0 ml) and brine (100.0 ml).
- Example 45B S-Methyl-piperidine-l ⁇ -dicarboxylic acid 1-benzyl ester To a -78 0 C solution of the product of Example 45A (6.0 g, 20.6 mmoles) in THF (50 mL) was added a solution of lithium bis(trimethylsilyl)amide (1.0 M in THF, 22.7 mmoles). After 35 min, iodomethane (1.4 mL, 22.7 mmoles) was added and the reaction was slowly warmed to room temperature and stirred overnight. The reaction was quenched with aqueous sat. ammonium chloride and extracted with Et 2 O.
- the organic layer was then rinsed with brine, dried over Na 2 SO 4 , filtered, and concentrated in vacuo.
- the crude product was purified by silica gel chromatography employing a solvent gradient (hexane ⁇ 65:35 hexane:EtOAc). The resulting ester was hydrolyzed overnight at room temperature in THF (15 mL), H 2 O (10 mL), and EtOH (15 mL) with NaOH (2.5 g). The solution was concentrated under vacuum; the residue was dissolved in saturated ammonium chloride; and, the solution was extracted with ethyl acetate (3x). The combined ethyl acetate extracts were dried over sodium sulfate, filtered, and concentrated under vacuum to yield the title compound as a white solid.
- Example 45B 2-phenylisobutyric acid
- Example 1C 2-adamantanamine hydrochloride
- Example 45D N-[E-4-(carbomethoxy)-2-adamantyl1-l-3-methylpiperidine-3-carboxamide A solution of the product of Example 45C (0.62 g, 1.32 mmoles) and 10% palladium on carbon (60 mg) in EtOAc (20 mL) was exposed to hydrogen (60 psi) at room temperature for 6 hours. The reaction was incomplete so EtOH was added and the reaction continued for an additional 8 h. The crude product was then filtered away from the catalyst using methanol and isolated after concentration in vacuo to provide the title compound.
- Example 45D To a solution of the product of Example 45D (100 mg, 0.3 mmoles) and 4- chlorobenzaldehyde in dichloroethane (0.75 mL) and acetic acid (0.07 mL, 1,2 mmoles) was added sodium triacetoxyborohydride (127 mg, 0.6 mmoles). The resulting reaction mixture was stirred at room temperature overnight. The reaction was quenched with sat. aqueous NH 4 Cl and extracted with EtOAc. The organic layer was then rinsed with brine, dried over Na 2 SO 4 , filtered, and concentrated in vacuo to provide a crude sample of the title compound.
- Example 45E The crude product from Example 45E was hydrolyzed with an excess of NaOH at room temperature in a solution of water, EtOH, and tetrahydrofuran for 16 hours. The reaction was quenched with sat. aqueous NH 4 Cl and extracted with EtOAc. The organic layer was then rinsed with brine, dried over Na 2 SO 4 , filtered, and concentrated in vacuo. The residue, EDCI (80 mg, 0.42 mmoles), and 1-hydroxybenzotriazole hydrate (56.5 mg, 0.42 mmoles) were dissolved in DMF (0.75 mL) and stirred for 30 min at room temperature. Concentrated NH 4 OH (0.75 mL) was then added and stirring was continued overnight.
- Example 27A The title compound was prepared according to the method as described in Example 27A, substituting the product of Example 46D for l-(4-methoxyphenyl)-l- cyclopentanecarboxylic acid.
- Example 27C The title compound was prepared according to the method as described in Example 27C, substituting the product of Example 46G for the product of Example 27B.
- Example 47 A A solution of the product of Example 30B (0.075 gm, 0.37 mmol), benzylamine (47 mg, 0.44 mmol) and MP-triacetoxy borohydride (420 mg, 0.92 mmol) in THF (2 mL) was stirred for 12 hours at 23 0 C. The solution was filtered and evaporated in vacuo. The resulting oil was taken up in THF (1.2 mL) and stirred with KOTMS (71 mg, 0.55 mmol) for 12 hours at 23 0 C. The solvent was evaporated in vacuo to provide Example 47 A.
- the crude reaction mixture was purified by preparative reverse phase HPLC on a Waters Symmetry C8 column (25 mm X 100 mm, 7 um particle size) using a gradient of 20% to 100% acetonitrile:water (0.1% TFA) over 18 min at a flow rate of 40 mL/min. to provide the title compound.
- Example 35 C The title compound was prepared according to the method of Example 35 C, substituting the product of Example 48E for the product of Example 35B.
- Example 49C 2-rBenzofb]thiophen-3-ylV2-methylpropanoic acid The title compound was prepared according to the method of Example 22B substituting the product of Example 49B for the product of Example 22 A.
- Example 50A The organic layer was dried with MgSO 4 , filtered, and evaporated in vacuo.
- the crude product was purified by flash chromatography (hexane/EtOAc 100:0 to 80:20) to give the methyl ester of Example 50A.
- MTBE Methyl t-butyl ether
- Example 50B E-4- ⁇ [2-(5-fluoropyridin-2-ylV2-methylpropanoyl]amino)adamantane-l-carboxamide A solution of the product of Example 50A (30 mg, 0.15 mmol), the product of
- Example 1C (45 mg, 0.18 mmol), TBTU (77 mg, 0.24 mmol) and DIEA (47 mg, 0.37 mmol) in DMF (1.2 mL) was stirred at 23 0 C for 3 hrs.
- the reaction was diluted with EtOAc (10 mL) and washed twice with water (6 mL) and brine (6 mL).
- the organic layer was dried with MgSO 4 , filtered, and evaporated in vacuo.
- the residue was taken in THF (1 mL) and stirred with KOTMS (29 mg, 0.22 mmol) for 12 hours at 23 0 C. The solvent was evaporated in vacuo.
- the crude reaction mixture was purified by preparative reverse phase HPLC on a Waters Symmetry C8 column (25 mm X 100 mm, 7 urn particle size) using a gradient of 20% to 100% acetonitrile:water (0.1% TFA) over 18 min at a flow rate of 40 mL/min. to provide the title compound as the trifluoroacetic acid salt.
- Example 51A was isolated by filtration.
- the crude reaction mixture was purified by preparative reverse phase HPLC on a Waters Symmetry C8 column (25 mm X 100 mm, 7 urn particle size) using a gradient of 20% to 100% acetonitrile: water (0.1% TFA) over 18 min at a flow rate of 40 niL/min. to provide the title compound as the trifluoroacetic acid salt.
- test compounds to inhibit human 1 l ⁇ -HSD-1 enzymatic activity in vitro was evaluated in a Scintillation Proximity Assay (SPA).
- SPA Scintillation Proximity Assay
- Tritiated-cortisone substrate, NADPH cofactor and titrated compound were incubated with truncated human ll ⁇ -HSD-1 enzyme (24-287AA) at room temperature to allow the conversion to Cortisol to occur.
- the reaction was stopped by adding a non-specific 11 ⁇ -HSD inhibitor, 18 ⁇ -glycyrrhetinic acid.
- the tritiated Cortisol was captured by a mixture of an anti-cortisol monoclonal antibody and SPA beads coated with anti-mouse antibodies.
- the reaction plate was shaken at room temperature and the radioactivity bound to SPA beads was then measured on a ⁇ -scintillation counter.
- the 11- ⁇ HSD-l assay was carried out in 96-well microtiter plates in a total volume of 220 ⁇ l.
- 188 ⁇ l of master mix which contained 17.5 nM 3 H-cortisone, 157.5 nM cortisone and 181 mM NADPH was added to the wells.
- 1 mM G-6-P was also added.
- E.coli lysates overexpressing 11 ⁇ -HSD- 1 enzyme After shaking and incubating plates for 30 minutes at room temperature, reactions were stopped by adding 10 ⁇ l of 1 mM glycyrrhetinic acid. The product, tritiated Cortisol, was captured by adding 10 ⁇ l of 1 ⁇ M monoclonal anti- cortisol antibodies and 100 ⁇ l SPA beads coated with anti-mouse antibodies. After shaking for 30 minutes, plates were read on a liquid scintillation counter Topcount. Percent inhibition was calculated based on the background and the maximal signal. Wells that contained substrate without compound or enzyme were used as the background, while the wells that contained substrate and enzyme without any compound were considered as maximal signal. Percent of inhibition of each compound was calculated relative to the maximal signal and IC 50 curves were generated. This assay was applied to 11 ⁇ -HSD-2 as well, whereby tritiated Cortisol and NAD + were used as substrate and cofactor, respectively.
- Compounds of the present invention are active in the 11- ⁇ HSD-l assay described above and show selectivity for human 11- ⁇ -HSD-l over human 1 l- ⁇ -HSD-2, as indicated in Table 1.
- the data in Table 1 demonstrates that compounds A, B, C and D are active in the human 1 l ⁇ -HSD-1 enzymatic SPA assay described above and the tested compounds show selectivity for l l ⁇ -HSD-1 over ll ⁇ -HSD-2.
- the ll ⁇ -HSD-1 inhibitors of this invention generally have an inhibition constant IC 50 of less than 600 nM and preferably less than 50 nM.
- the compounds preferably are selective, having an inhibition constant IC 50 against ll ⁇ - HSD-2 greater than 1000 nM and preferably greater than 10,000 nM.
- the IC 50 ratio for 11 ⁇ -HSD-2 to 11 ⁇ -HSD- 1 of a compound is at least 10 or greater and preferably 100 or greater..
- Metabolic stability screen each substrate (10 ⁇ M) was incubated with microsomal protein (0.1 - 0.5 mg/ml) in 5OmM potassium phosphate buffer (pH 7.4) in 48-Well plate.
- the enzyme reaction was initiated by the addition of ImM NADPH 5 then incubated at 37°C in a Forma Scientific incubator (Marietta, OH, USA) with gentle shaking.
- the reactions were quenched by the addition of 800 ⁇ l of ACN/MeOH (1:1, v/v), containing 0.5 ⁇ M of internal standard (IS), after 30 min incubation.
- the parent remaining in the incubation mixture was determined by LC/MS.
- the LC/MS system consisted of an Agilent 1100 series (Agilent Technologies, Waldbronn, Germany) and API 2000 (MDS SCIEX, Ontario, Canada).
- a Luna C8(2) 50 x 2.0 mm, particle size 3 ⁇ m, Phenomenex, Torrance, CA, USA was used to quantify each compound at ambient temperature.
- the mobile phase consisted of (A): 10 mM NH 4 AC (pH 3.3) and (B): 100% ACN and was delivered at a flow rate of 0.2 ml/min. Elution was achieved using a linear gradient of 0-100% B over 3 min, then held 100% B for 4 min and returned to 100% A in 1 min. The column was equilibrated for 7 min before the next injection.
- the peak area ratios (each substrate over IS) at each incubation time were expressed as the percentage of the ratios (each substrate over IS) of the control samples (0 min incubation).
- the parent remaining in the incubation mixture was expressed as the percentage of the values at 0 min incubation.
- Compounds A, B, C and D contain a substituted adamantane, whereas the adamantane ring of Compounds E and F is unsubstituted.
- the microsomal, metabolic, stability data in Table 2 demonstrates that substituted adamantane compounds of the present invention may exhibit an increase in metabolic stability compared to unsubstituted adamantane compounds which may lead to longer in vivo half lives and pharmacokinetic advantages over unsubstituted adamantanes.
- Glucocorticoids are steroid hormones that play an important role in regulating multiple physiological processes in a wide range of tissues and organs.
- glucocorticoids are potent regulators of glucose and lipid metabolism. Excess glucocorticoid action may lead to insulin resistance, type 2 diabetes, dyslipidemia, visceral obesity and hypertension.
- Cortisol is the major active and cortisone is the major inactive form of glucocorticoids in humans, while corticosterone and dehydrocorticosterone are the major active and inactive forms in rodents.
- the main determinants of glucocorticoid action were thought to be the circulating hormone concentration and the density of glucocorticoid receptors in the target tissues.
- tissue glucocorticoid levels may also be controlled by 1 l ⁇ -hydroxysteroid dehydrogenases enzymes (1 l ⁇ -HSDs).
- 1 l ⁇ -HSDs There are two 1 l ⁇ - HSD isozymes which have different substrate affinities and cofactors.
- the 1 l ⁇ - hydroxysteroid dehydrogenases type 1 enzyme (1 l ⁇ -HSD-1) is a low affinity enzyme with K m for cortisone in the micromolar range that prefers NADPH/NADP + (nicotinamide adenine dinucleotide) as cofactors.
- 11 ⁇ -HSD-1 is widely expressed and particularly high expression levels are found in liver, brain, lung, adipose tissue and vascular smooth muscle cells.
- the 1 l ⁇ -hydroxysteroid dehydrogenases type 2 enzyme (11 ⁇ -HSD-2) is a NAD + -dependent, high affinity dehydrogenase with a K m for Cortisol in the nanomolar range.
- 11 ⁇ -HSD-2 is found primarily in mineralocorticoid target tissues, such as kidney, colon and placenta.
- Glucocorticoid action is mediated by the binding of glucocorticoids to receptors, such as mineralocorticoid receptors and glucocorticoid receptors. Through binding to its receptor, the main mineralocorticoid aldosterone controls the water and salts balance in the body.
- 1 l ⁇ -HSD-2 converts Cortisol to inactive cortisone, therefore preventing the non-selective mineralocorticoid receptors from being exposed to high levels of Cortisol.
- Mutations in the gene encoding 1 l ⁇ -HSD-2 cause Apparent Mineralocorticoid Excess Syndrome (AME), which is a congenital syndrome resulting in hypokaleamia and severe hypertension.
- AME Patients have elevated Cortisol levels in mineralocorticoid target tissues due to reduced 11 ⁇ -HSD-2 activity.
- the AME symptoms may also be induced by administration of 1 l ⁇ -HSD-2 inhibitor, glycyrrhetinic acid.
- 1 l ⁇ -HSD-2 inhibitor glycyrrhetinic acid.
- the activity of 11 ⁇ -HSD-2 in placenta is probably important for protecting the fetus from excess exposure to maternal glucocorticoids, which may result in hypertension, glucose intolerance and growth retardation.
- the present invention describes selective ll ⁇ -HSD-1 inhibitors.
- Glucocorticoid levels and/or activity may contribute to numerous disorders, including
- Type II diabetes obesity, dyslipidemia, insulin resistance and hypertension.
- Administration of the compounds of the present invention decreases the level of Cortisol and other 1 l ⁇ - hydroxysteroids in target tissues, thereby reducing the effects of glucucocrticoid activity in key target tissues.
- the present invention could be used for the treatment, control, amelioration, prevention, delaying the onset of or reducing the risk of developing the diseases and conditions that are described herein.
- glucocorticoids are potent regulators of glucose and lipid metabolism, glucocorticoid action may contribute or lead to insulin resistance, type 2 diabetes, dyslipidemia, visceral obesity and hypertension.
- Cortisol antagonizes the insulin effect in liver resulting in reduced insulin sensitivity and increased gluconeogenesis. Therefore, patients who already have impaired glucose tolerance have a greater probability of developing type 2 diabetes in the presence of abnormally high levels of Cortisol.
- Previous studies (B. R. Walker et al., J. of Clin. Endocrinology and Met., 80: 3155-3159, 1995) have demonstrated that administration of non-selective ll ⁇ -HSD-1 inhibitor, carbenoxolone, improves insulin sensitivity in humans. Therefore, administration of a therapeutically effective amount of an ll ⁇ -HSD-1 inhibitor may treat, control, ameliorate, delay, or prevent the onset of type 2 diabetes.
- glucocorticoids in vivo has been shown to reduce insulin secretion in rats (B. Billaudel et al., Horm. Metab. Res. 11 : 555-560, 1979). It has also been reported that conversion of dehydrocorticosterone to corticosterone by 11 ⁇ -HSD-1 inhibits insulin secretion from isolated murine pancreatic ⁇ cells. (B. Davani et al., J. Biol. Chem., 275: 34841-34844, 2000), and that incubation of isolated islets with an ll ⁇ -HSD-1 inhibitor improves glucose-stimulated insulin secretion (H Orstater et al., Diabetes Metab. Res. Rev.. 21: 359-366, 2005). Therefore, administration of a therapeutically effective amount of an 1 l ⁇ -HSD-1 inhibitor may treat, control, ameliorate, delay, or prevent the onset of type 2 diabetes by improving glucose-stimulated insulin secretion in the pancreas.
- Abdominal obesity is closely associated with glucose intolerance (C. T. Montaque et al., Diabetes, 49: 883-888, 2000), hyperinsulinemia, hypertriglyceridemia and other factors of metabolic syndrome (also known as syndrome X), such as high blood pressure, elevated VLDL and reduced HDL.
- metabolic syndrome X also known as syndrome X
- Animal data supporting the role of 11 ⁇ -HSD-1 in the pathogenesis of the metabolic syndrome is extensive (Masuzaki, et ah. Science. 294: 2166-2170, 2001; Paterson, J.M., et al; Proc Natl. Acad. Sd. USA. 101: 7088-93, 2004; Montague and O'Rahilly. Diabetes. 49: 883-888, 2000).
- administration of a therapeutically effective amount of an 1 l ⁇ -HSD-1 inhibitor may treat, control, ameliorate, delay, or prevent the onset of obesity.
- Long-term treatment with an 11 ⁇ -HSD-1 inhibitor may also be useful in delaying the onset of obesity, or perhaps preventing it entirely if the patients use an 1 l ⁇ - HSD-I inhibitor in combination with controlled diet, exercise, or in combination or sequence with other pharmacological approaches.
- reducing insulin resistance and/or maintaining serum glucose at normal concentrations and/or reducing obestity compounds of the present invention also have utility in the treatment and prevention of conditions that accompany Type 2 diabetes and insulin resistance, including the metabolic syndrome or syndrome X, obesity, reactive hypoglycemia, and diabetic dyslipidemia.
- 1 l ⁇ -HSD-I is present in multiple tissues, including vascular smooth muscle, where local glucocorticoid levels that are thought to increase insulin resistance, leading to reductions in nitric oxide production, and potentiation of the vasoconstrictive effects of both catecholamines and angiotensin II (M. Pirpiris et al., Hypertension, 19:567-574, 1992, C. Kornel et al., Steroids, 58: 580-587, 1993, B. R. Walker and B. C. Williams, Clin. Sci. 82:597-605, 1992; Hodge, G. et al Exp. Physiol 87: 1-8, 2002).
- Transgenic mice overexpressing 1 l ⁇ -HSD-1 in liver and fat are also hypertensive, a phenotype believed to result from glucocorticoid activation of the renin angiotensin system (Paterson, J.M. et al, PNAS. 101: 7088-93, 2004; Masuzaki, H. et al, J. Clin. Invest. 112: 83-90, 2003). Therefore, administration of a therapeutically effective dose of an 11 ⁇ -HSD-1 inhibitor may treat, control, ameliorate, delay, or prevent the onset of hypertension.
- Cushing's syndrome is a life-threatening metabolic disorder characterized by sustained and elevated glucocorticoid levels caused by the endogenous and excessive production of Cortisol from the adrenal glands.
- Typical Cushingoid characteristics include central obesity, diabetes and/or insulin resistance, moon face, buffalo hump, skin thinning, dyslipidemia, osteoporosis, reduced cognitive capacity, dementia, hypertension, sleep deprivation, and atherosclerosis among others (Principles and Practice of Endocrinology and Metabolism. Edited by Kenneth Becker, Lippincott Williams and WiUrins Pulishers, Philadelphia, 2001; pg 723-8).
- exogenous glucocorticoids such as prednisone or dexamethasone
- Endogenous Cushings typically evolves from pituitary hyperplasia, some other ectopic source of ACTH, or from an adrenal carcinoma or nodular hyperplasia.
- Administration of a therapeutically effective dose of an 1 l ⁇ -HSD-1 inhibitor may reduce local glucocorticoid concentrations and therefore treat, control, ameliorate, delay, or prevent the onset of Gushing' s disease and/or similar symptoms arising from glucocorticoid treatment.
- glucocorticoids include glucocorticoid-induced acute psychosis which is of major concern to physicians when treating patients with these steroidal agents (Wolkowitz et al.; Ann NY Acad Sd. 1032: 191- 4, 2004).
- Conditional mutagenesis studies of the glucocorticoid receptor in mice have also provided genetic evidence that reduced glucocorticoid signaling in the brain results in decreased anxiety (Tranche, F. et al. (1999) Nature Genetics 23: 99-103). Therefore, it is expected that potent, selective 1 l ⁇ -HSD-1 inhibitors would treat, control, ameliorate, delay, or prevent the onset of cognitive decline, dementia, steroid-induced acute psychosis, depression, and/or anxiety.
- mice 1 l ⁇ -HSD-1 knockout mice are resistant to the dyslipidemic effects of a high fat diet and have an improved lipid profile vs wild type controls (Morton N.M. et al, JBC, 276: 41293-41300, 2001), and mice which overexpress 11 ⁇ -HSD-1 in fat exhibit the dyslipidemic phenotype characteristic of metabolic syndrome, including elevated circulating free fatty acids, and triclylgerides (Masuzaki, H., et al Science. 294: 2166-2170, 2001).
- a selective 1 l ⁇ - HSD-I inhibitor has also been shown to reduce elevated plasma triglycerides and free fatty acids in mice on a high fat diet, and significantly reduce aortic content of cholesterol esters, and reduce progression of atherosclerotic plaques in mice (Hermanowski-Vosatka, A. et al. J. Exp. Med. 202: 517-27, 2005).
- the administration of a therapeutically effective amount of an 11 ⁇ -HSD-1 inhibitor would therefore be expected to treat, control, ameliorate, delay, or prevent the onset of dyslipidemia and/or atherosclerosis.
- Glucocorticoids are known to cause a variety of skin related side effects including skin thinning, and impairment of wound healing (Anstead, G. Adv Wound Care. 11 : 277- 85, 1998; Beer, et al; Vitam Horm. 59: 217-39, 2000).
- ll ⁇ -HSD-1 is expressed in human skin fibroblasts, and it has been shown that the topical treatment with the non-selective HSD1/2 inhibitor glycerrhetinic acid increases the potency of topically applied hydrocortisone in a skin vasoconstrictor assay (Hammami, MM, and Siiteri, PK. J. Clin. Endocrinol. Metab. 73: 326-34, 1991).
- Cortisol is an important and well-recognized anti-inflammatory agent (J. Baxer, Pharmac. Ther., 2:605-659, 1976), if present in large amount it also has detrimental effects.
- high glucocorticoid activity shifts the immune response to a humoral response, when in fact a cell based response may be more beneficial to patients.
- Inhibition of 11 ⁇ -HSD-1 activity may reduce glucocorticoid levels, thereby shifting the immuno response to a cell based response.
- the cells that produce the majority of aqueous humor in the eye are the nonpigmented epithelial cells (NPE). These cells have been demonstrated to express 11 ⁇ -HSD- 1 , and consistent with the expression of 11 ⁇ -HSD- 1 , is the finding of elevated ratios of cortisolxortisone in the aqueous humor (Rauz et ah. Invest Ophthalmol Vis Sd. 42: 2037-2042, 2001). Furthermore, it has been shown that patients who have glaucoma, but who are not taking exogenous steroids, have elevated levels of Cortisol vs. cortisone in their aqueous humor (Rauz et al. QJM.
- Glucocorticoids are known to increase bone resorption and reduce bone formation in mammals (Turner et al. Caldf Tissue Int. 54: 311-5, 1995; Lane, NE et al.
- Glucocorticoids such as prednisone and dexamethasone
- Glucocorticoids are also commonly used to treat a variety of inflammatory conditions including arthritis, inflammatory bowl disease, and asthma.
- These steroidal agents have been shown to increase expression of 1 l ⁇ -HSD-1 mRNA and activity in human osteoblasts (Cooper et al; J. Bone Miner Res. 17: 979-986, 2002).
- 11 ⁇ -HSD-I plays a potentially important role in the development of bone-related adverse events as a result of excessive glucocorticoid levels or activity.
- the following diseases, disorders and conditions can be treated, controlled, prevented or delayed, by treatment with the compounds of this invention: (1) hyperglycemia, (2) low glucose tolerance, (3) insulin resistance, (4) lipid disorders, (5) hyperlipidemia, (6) hypertriglyceridemia, (9) hypercholesterolemia, (10) low HDL levels, (11) high LDL levels, (12), atherosclerosis and its sequelae, (13) vascular restensosis, (14) pancreatitis, (15) obdominal obesity, (16) neurodegenerative disease, (17) retinopathy, (18) nephropather, (19), neuropathy, (20) hypertension and other disorders where insulin resistance is a component, and (21) other diseases, disorders, and conditions that can benefit from reduced local glucocorticoid levels.
- compositions of the present compounds comprise an effective amount of the same formulated with one or more therapeutically suitable excipients.
- therapeutically suitable excipient generally refers to pharmaceutically suitable, solid, semi-solid or liquid fillers, diluents, encapsulating material, formulation auxiliary and the like.
- therapeutically suitable excipients include, but are not limited to, sugars, cellulose and derivatives thereof, oils, glycols, solutions, buffers, colorants, releasing agents, coating agents, sweetening agents, flavoring agents, perfuming agents and the like.
- Such therapeutic compositions may be administered parenterally, intracisternally, orally, rectally, intraperitoneally or by other dosage forms known in the art.
- Liquid dosage forms for oral administration include, but are not limited to, emulsions, microemulsions, solutions, suspensions, syrups and elixirs. Liquid dosage forms may also contain diluents, solubilizing agents, emulsifying agents, inert diluents, wetting agents, emulsifiers, sweeteners, flavorants, perfuming agents and the like.
- Injectable preparations include, but are not limited to, sterile, injectable, aqueous, oleaginous solutions, suspensions, emulsions and the like. Such preparations may also be formulated to include, but are not limited to, parenterally suitable diluents, dispersing agents, wetting agents, suspending agents and the like. Such injectable preparations may be sterilized by filtration through a bacterial-retaining filter. Such preparations may also be formulated with sterilizing agents that dissolve or disperse in the injectable media or other methods known in the art.
- the absorption of the compounds of the present invention may be delayed using a liquid suspension of crystalline or amorphous material having poor water solubility.
- the rate of absorption of the compounds generally depends upon the rate of dissolution and crystallinity. Delayed absorption of a parenterally administered compound may also be accomplished by dissolving or suspending the compound in oil.
- Injectable depot dosage forms may also be prepared by microencapsulating the same in biodegradable polymers. The rate of drug release may also be controlled by adjusting the ratio of compound to polymer and the nature of the polymer employed. Depot injectable formulations may also prepared by encapsulating the compounds in liposomes or microemulsions compatible with body tissues.
- Solid dosage forms for oral administration include, but are not limited to, capsules, tablets, gels, pills, powders, granules and the like.
- the drug compound is generally combined with at least one therapeutically suitable excipient, such as carriers, fillers, extenders, disintegrating agents, solution retarding agents, wetting agents, absorbents, lubricants and the like.
- Capsules, tablets and pills may also contain buffering agents.
- Suppositories for rectal administration may be prepared by mixing the compounds with a suitable non-irritating excipient that is solid at ordinary temperature but fluid in the rectum.
- the present drug compounds may also be microencapsulated with one or more excipients.
- Tablets, dragees, capsules, pills and granules may also be prepared using coatings and shells, such as enteric and release or rate controlling polymeric and nonpolymeric materials.
- the compounds may be mixed with one or more inert diluents. Tableting may further include lubricants and other processing aids.
- capsules may contain opacifying agents that delay release of the compounds in the intestinal tract.
- Transdermal patches have the added advantage of providing controlled delivery of the present compounds to the body.
- dosage forms are prepared by dissolving or dispensing the compounds in suitable medium.
- Absorption enhancers may also be used to increase the flux of the compounds across the skin.
- the rate of absorption may be controlled by employing a rate controlling membrane.
- the compounds may also be incorporated into a polymer matrix or gel.
- disorders of the present invention may be treated, prophylatically treated, or have their onset delayed in a patient by administering to the patient a therapeutically effective amount of compound of the present invention in accordance with a suitable dosing regimen.
- a therapeutically effective amount of any one of compounds of formulas (I) is administered to a patient to treat and/or prophylatically treat disorders modulated by the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme.
- the specific therapeutically effective dose level for a given patient population may depend upon a variety of factors including, but not limited to, the specific disorder being treated, the severity of the disorder; the activity of the compound, the specific composition or dosage form, age, body weight, general health, sex, diet of the patient, the time of administration, route of administration, rate of excretion, duration of the treatment, drugs used in combination, coincidental therapy and other factors known in the art.
- the present invention also includes therapeutically suitable metabolites formed by in vivo biotransformation of any of the compounds of formula (I).
- therapeutically suitable metabolite generally refers to a pharmaceutically active compound formed by the in vivo biotransform ation of compounds of formula (I).
- pharmaceutically active metabolites include, but are not limited to, compounds made by adamantane hydroxylation or polyhydroxylation of any of the compounds of formulas(I).
- the total daily dose (single or multiple) of the drug compounds of the present invention necessary to effectively inhibit the action of 11-beta-hydroxysteroid dehydrogenase type 1 enzyme may range from about 0.01 mg/kg/day to about 50 mg/kg/day of body weight and more preferably about 0.1 mg/kg/day to about 25 mg/kg/day of body weight.
- Treatment regimens generally include administering from about 10 mg to about 1000 mg of the compounds per day in single or multiple doses.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Diabetes (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Obesity (AREA)
- Hematology (AREA)
- Endocrinology (AREA)
- Emergency Medicine (AREA)
- Child & Adolescent Psychology (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Pyridine Compounds (AREA)
- Heterocyclic Compounds Containing Sulfur Atoms (AREA)
- Thiazole And Isothizaole Compounds (AREA)
- Hydrogenated Pyridines (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Pyrrole Compounds (AREA)
Abstract
Description








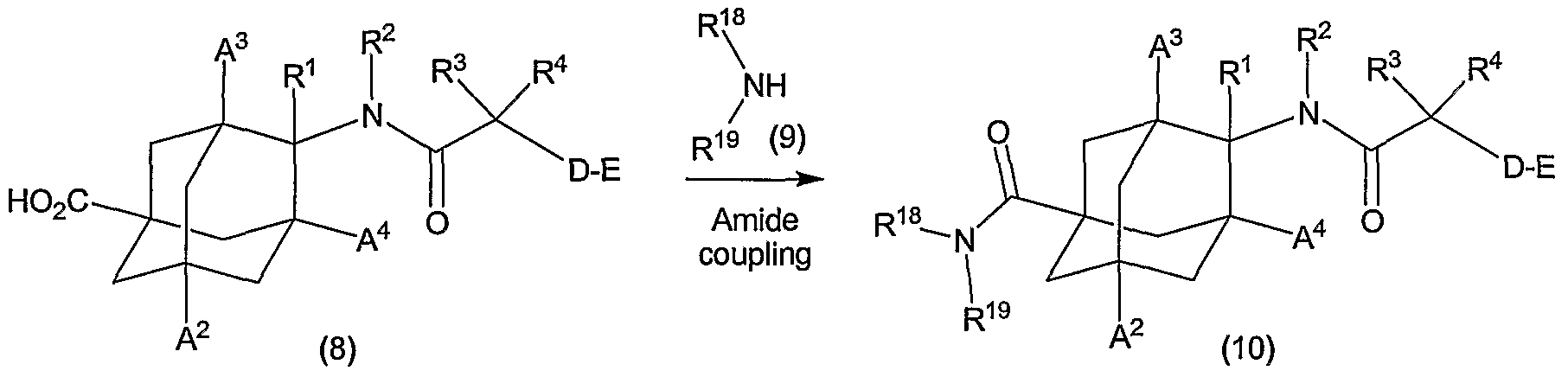

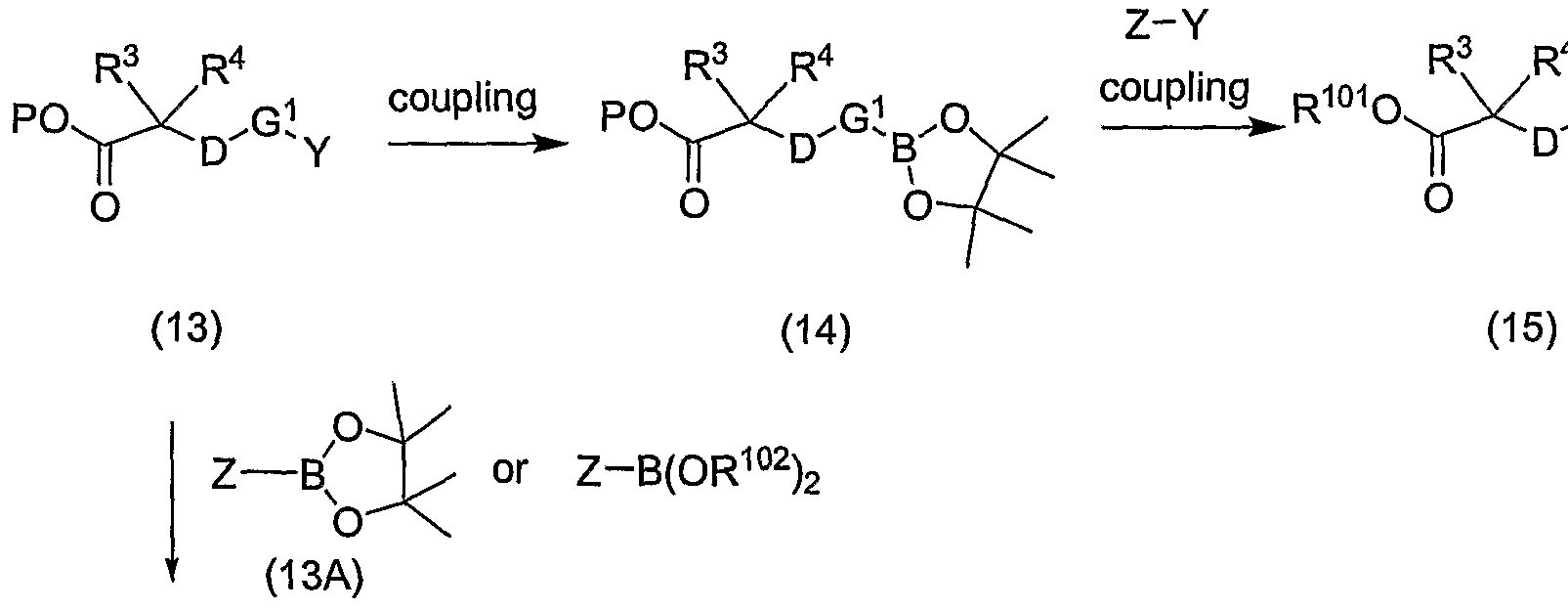












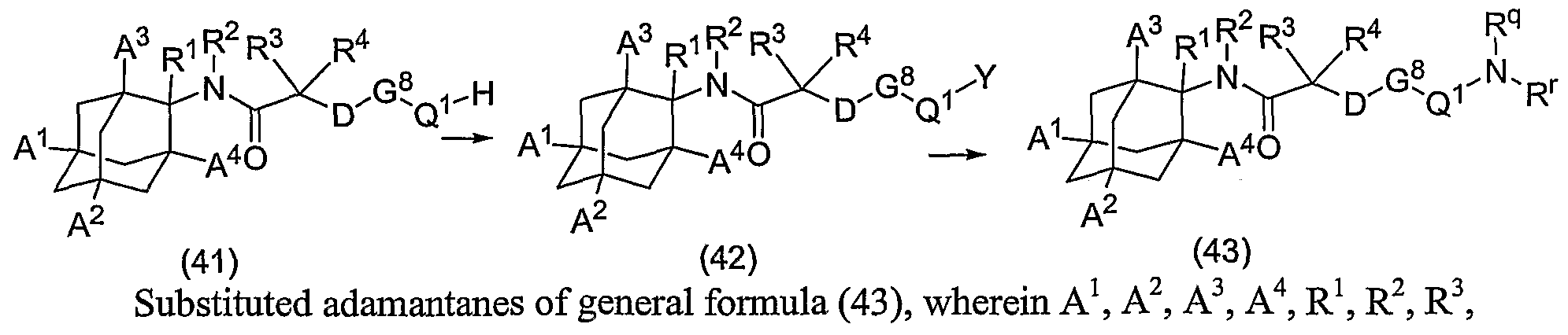


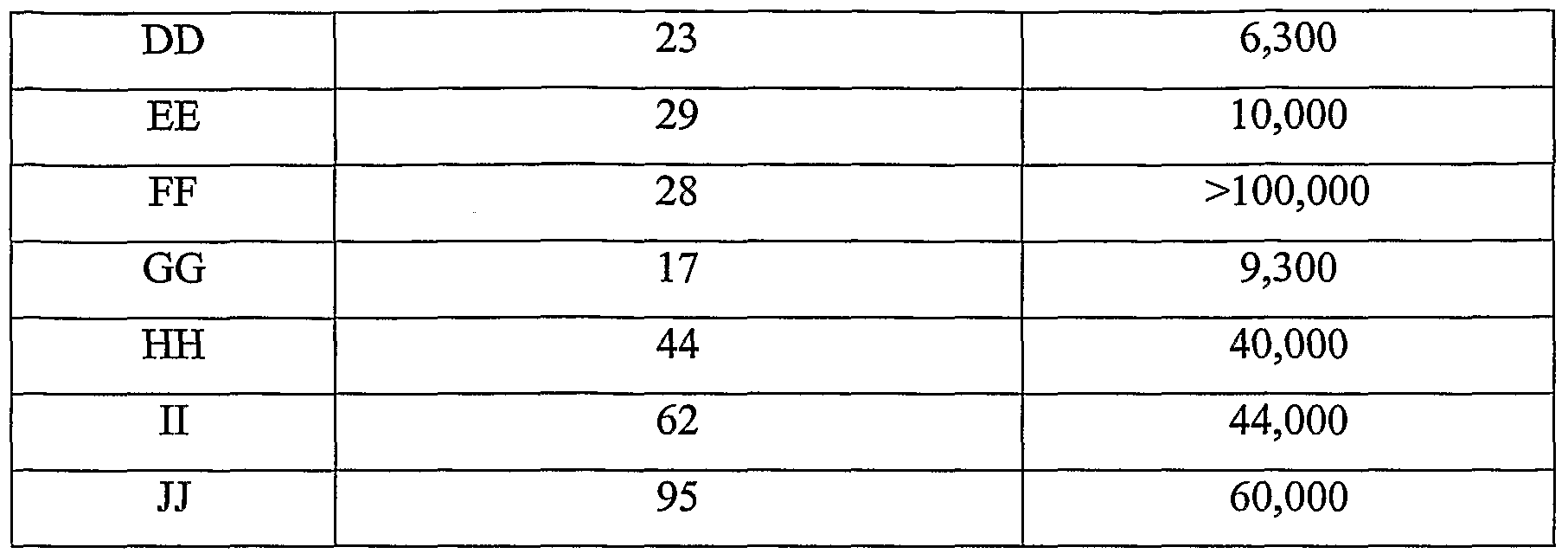

Claims



Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2006203918A AU2006203918B2 (en) | 2005-01-05 | 2006-01-05 | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase Type 1 enzyme |
| KR1020077017918A KR101302627B1 (en) | 2005-01-05 | 2006-01-05 | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| BRPI0606228-8A BRPI0606228A2 (en) | 2005-01-05 | 2006-01-05 | 11-beta-hydroxysteroid dehydrogenase type 1 enzyme inhibitors |
| CA002594116A CA2594116A1 (en) | 2005-01-05 | 2006-01-05 | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| MX2007008239A MX2007008239A (en) | 2005-01-05 | 2006-01-05 | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme. |
| JP2007550479A JP5133702B2 (en) | 2005-01-05 | 2006-01-05 | Inhibitors of 11-β-hydroxysteroid dehydrogenase type 1 enzyme |
| NZ555971A NZ555971A (en) | 2005-01-05 | 2006-01-05 | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| EP06717579A EP1846362A2 (en) | 2005-01-05 | 2006-01-05 | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| IL184329A IL184329A (en) | 2005-01-05 | 2007-07-01 | Amide containing an adamantyl moiety and a carbonyl-bearing substituent on the adamantyl moiety as inhibitor of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| IL213342A IL213342A (en) | 2005-01-05 | 2011-06-02 | Amide containing an adamantyl moiety and a carbonyl- bearing substituent on the adamantyl moiety as inhibitor of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US64149605P | 2005-01-05 | 2005-01-05 | |
| US60/641,496 | 2005-01-05 | ||
| US11/326,277 US7217838B2 (en) | 2005-01-05 | 2006-01-05 | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| US11/326,277 | 2006-01-05 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2006074330A2 true WO2006074330A2 (en) | 2006-07-13 |
| WO2006074330A3 WO2006074330A3 (en) | 2007-01-25 |
Family
ID=36440952
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2006/000402 WO2006074330A2 (en) | 2005-01-05 | 2006-01-05 | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
Country Status (13)
| Country | Link |
|---|---|
| US (3) | US7217838B2 (en) |
| EP (2) | EP1846362A2 (en) |
| JP (1) | JP5133702B2 (en) |
| KR (1) | KR101302627B1 (en) |
| CN (2) | CN102816081A (en) |
| AU (1) | AU2006203918B2 (en) |
| BR (1) | BRPI0606228A2 (en) |
| CA (1) | CA2594116A1 (en) |
| IL (2) | IL184329A (en) |
| MX (1) | MX2007008239A (en) |
| NZ (1) | NZ555971A (en) |
| WO (1) | WO2006074330A2 (en) |
| ZA (1) | ZA200705468B (en) |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007128761A2 (en) | 2006-05-04 | 2007-11-15 | Boehringer Ingelheim International Gmbh | Uses of dpp-iv inhibitors |
| WO2008142986A1 (en) | 2007-05-18 | 2008-11-27 | Shionogi & Co., Ltd. | NITROGEN-CONTAINING HETEROCYCLIC DERIVATIVE HAVING 11 β-HYDROXYSTEROID DEHYDROGENASE TYPE I INHIBITORY ACTIVITY |
| JP2010501589A (en) * | 2006-08-24 | 2010-01-21 | ブリストル−マイヤーズ スクイブ カンパニー | Cyclic 11-β hydroxysteroid dehydrogenase type I inhibitor |
| EP2150109A1 (en) * | 2007-04-24 | 2010-02-10 | High Point Pharmaceuticals, LLC | Pharmaceutical use of substituted amides |
| WO2010044441A1 (en) * | 2008-10-17 | 2010-04-22 | 塩野義製薬株式会社 | Acetic acid amide derivative having inhibitory activity on vascular endothelial lipase |
| US8017638B2 (en) | 2006-03-30 | 2011-09-13 | Shionogi & Co., Ltd. | Isoxazole derivative and isothiazole derivative having inhibitory activity on 11β-hydroxysteroid dehydrogenase type 1 |
| US8324265B2 (en) | 2005-11-21 | 2012-12-04 | Shionogi & Co., Ltd. | Heterocyclic compounds having type I 11β hydroxysteroid dehydrogenase inhibitory activity |
| US8822149B2 (en) | 2007-05-24 | 2014-09-02 | Pacific Edge Limited | Prognosis prediction for melanoma cancer |
| US8952176B2 (en) | 2005-06-07 | 2015-02-10 | Shionogi & Co., Ltd. | Heterocyclic compound having type I 11 β hydroxysteroid dehydrogenase inhibitory activity |
Families Citing this family (59)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20100222316A1 (en) | 2004-04-29 | 2010-09-02 | Abbott Laboratories | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| US8415354B2 (en) * | 2004-04-29 | 2013-04-09 | Abbott Laboratories | Methods of use of inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| US7880001B2 (en) * | 2004-04-29 | 2011-02-01 | Abbott Laboratories | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase Type 1 enzyme |
| TWI350168B (en) | 2004-05-07 | 2011-10-11 | Incyte Corp | Amido compounds and their use as pharmaceuticals |
| EP1758580A4 (en) * | 2004-06-24 | 2008-01-16 | Incyte Corp | N-substituted piperidines and their use as pharmaceuticals |
| US20050288317A1 (en) * | 2004-06-24 | 2005-12-29 | Wenqing Yao | Amido compounds and their use as pharmaceuticals |
| EP1773773A4 (en) * | 2004-06-24 | 2009-07-29 | Incyte Corp | Amido compounds and their use as pharmaceuticals |
| US7687665B2 (en) | 2004-06-24 | 2010-03-30 | Incyte Corporation | 2-methylprop anamides and their use as pharmaceuticals |
| BRPI0514230A (en) * | 2004-08-10 | 2008-06-03 | Incyte Corp | starch compounds and their use as pharmaceuticals |
| JP2008515956A (en) * | 2004-10-12 | 2008-05-15 | ノボ ノルディスク アクティーゼルスカブ | 11.beta.-hydroxysteroid dehydrogenase type 1 active spiro compound |
| US8110581B2 (en) * | 2004-11-10 | 2012-02-07 | Incyte Corporation | Lactam compounds and their use as pharmaceuticals |
| NZ554906A (en) * | 2004-11-10 | 2011-01-28 | Incyte Corp | Lactam compounds and their use as pharmaceuticals |
| EP1824842A4 (en) * | 2004-11-18 | 2009-08-26 | Incyte Corp | Inhibitors of 11- hydroxyl steroid dehydrogenase type 1 and methods of using the same |
| JP5078621B2 (en) * | 2005-01-05 | 2012-11-21 | アボット・ラボラトリーズ | Adamantyl derivatives as inhibitors of 11-β-hydroxysteroid dehydrogenase type 1 enzyme |
| US8198331B2 (en) * | 2005-01-05 | 2012-06-12 | Abbott Laboratories | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| US20090192198A1 (en) | 2005-01-05 | 2009-07-30 | Abbott Laboratories | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| GB0506133D0 (en) * | 2005-03-24 | 2005-05-04 | Sterix Ltd | Compound |
| JP2009508963A (en) * | 2005-09-21 | 2009-03-05 | インサイト・コーポレイション | Amide compounds and their use as pharmaceutical compositions |
| WO2007051811A2 (en) * | 2005-11-01 | 2007-05-10 | Transtech Pharma | Pharmaceutical use of substituted amides |
| US20090124598A1 (en) * | 2005-11-01 | 2009-05-14 | Henrik Sune Andersen | Pharmaceutical use of substituted amides |
| WO2007067504A2 (en) * | 2005-12-05 | 2007-06-14 | Incyte Corporation | Lactam compounds and methods of using the same |
| WO2007084314A2 (en) * | 2006-01-12 | 2007-07-26 | Incyte Corporation | MODULATORS OF 11-ß HYDROXYL STEROID DEHYDROGENASE TYPE 1, PHARMACEUTICAL COMPOSITIONS THEREOF, AND METHODS OF USING THE SAME |
| EP1979318A1 (en) * | 2006-01-31 | 2008-10-15 | Incyte Corporation | Amido compounds and their use as pharmaceuticals |
| TW200808807A (en) * | 2006-03-02 | 2008-02-16 | Incyte Corp | Modulators of 11-β hydroxyl steroid dehydrogenase type 1, pharmaceutical compositions thereof, and methods of using the same |
| US20070208001A1 (en) * | 2006-03-03 | 2007-09-06 | Jincong Zhuo | Modulators of 11- beta hydroxyl steroid dehydrogenase type 1, pharmaceutical compositions thereof, and methods of using the same |
| WO2007107550A1 (en) * | 2006-03-21 | 2007-09-27 | High Point Pharmaceuticals, Llc | Adamantane derivatives for the treatment of the metabolic syndrome |
| JP2009532418A (en) * | 2006-04-07 | 2009-09-10 | ハイ ポイント ファーマシューティカルズ,リミティド ライアビリティ カンパニー | 11β-Hydroxysteroid dehydrogenase type 1 active compound |
| EP2013163A1 (en) * | 2006-05-01 | 2009-01-14 | Incyte Corporation | Tetrasubstituted ureas as modulators of 11-beta hydroxyl steroid dehydrogenase type 1 |
| WO2007137066A2 (en) * | 2006-05-17 | 2007-11-29 | Incyte Corporation | HETEROCYCLIC INHIBITORS OF 11-β HYDROXYL STEROID DEHYDROGENASE TYPE I AND METHODS OF USING THE SAME |
| CA2655282A1 (en) * | 2006-06-16 | 2007-12-21 | High Point Pharmaceuticals, Llc | Pharmaceutical use of substituted piperidine carboxamides |
| CA2657078A1 (en) * | 2006-07-13 | 2008-01-17 | High Point Pharmaceuticals, Llc | 11beta-hydroxysteroid dehydrogenase type 1 active compounds |
| EP1878721A1 (en) * | 2006-07-13 | 2008-01-16 | Novo Nordisk A/S | 4-Piperidylbenzamides as 11-beta-hydroxysteroid dehydrogenase type 1 inhibitors |
| EP1935420A1 (en) * | 2006-12-21 | 2008-06-25 | Merck Sante | 2-Adamantyl-butyramide derivatives as selective 11beta-HSD1 inhibitors |
| JP2010519240A (en) * | 2007-02-23 | 2010-06-03 | ハイ ポイント ファーマシューティカルズ,リミティド ライアビリティ カンパニー | N-adamantylbenzamide as an inhibitor of 11-beta-hydroxysteroid dehydrogenase |
| US8334305B2 (en) * | 2007-02-23 | 2012-12-18 | High Point Pharmaceuticals, Llc | N-adamantyl benzamides as inhibitors of 11-β-hydroxysteroid dehydrogenase |
| US20110003852A1 (en) * | 2007-02-23 | 2011-01-06 | Soren Ebdrup | N-adamantyl benzamides as inhibitors of 11-beta-hydroxysteroid dehydrogenase |
| AU2008219326B2 (en) * | 2007-02-23 | 2012-12-13 | Vtv Therapeutics Llc | N-adamantyl benzamides as inhibitors of 11-beta-hydroxysteroid dehydrogenase |
| US8153798B2 (en) * | 2007-03-09 | 2012-04-10 | High Point Pharmaceuticals, Llc | Indole- and benzimidazole amides as hydroxysteroid dehydrogenase inhibitors |
| WO2008119017A1 (en) * | 2007-03-28 | 2008-10-02 | High Point Pharmaceuticals, Llc | 11beta-hsd1 active compounds |
| CA2683852A1 (en) * | 2007-04-11 | 2008-10-23 | High Point Pharmaceuticals, Llc | Novel compounds |
| CL2008001839A1 (en) | 2007-06-21 | 2009-01-16 | Incyte Holdings Corp | Compounds derived from 2,7-diazaspirocycles, inhibitors of 11-beta hydroxyl steroid dehydrogenase type 1; pharmaceutical composition comprising said compounds; Useful to treat obesity, diabetes, glucose intolerance, type II diabetes, among other diseases. |
| CN101743226B (en) * | 2007-07-17 | 2012-10-10 | 霍夫曼-拉罗奇有限公司 | Inhibitors of 11beta-hydroxysteroid dehydrogenase |
| JP5736098B2 (en) * | 2007-08-21 | 2015-06-17 | アッヴィ・インコーポレイテッド | Pharmaceutical composition for treating central nervous system disorders |
| US8119658B2 (en) | 2007-10-01 | 2012-02-21 | Bristol-Myers Squibb Company | Triazolopyridine 11-beta hydroxysteroid dehydrogenase type I inhibitors |
| EP2322505A4 (en) | 2008-08-29 | 2011-10-19 | Kowa Co | 1-adamantylazetidin-2-one derivative and pharmaceutical preparation comprising same |
| EP2362730A4 (en) | 2008-11-21 | 2012-08-29 | High Point Pharmaceuticals Llc | Adamantyl benzamide compounds |
| EP2243479A3 (en) | 2009-04-20 | 2011-01-19 | Abbott Laboratories | Novel amide and amidine derivates and uses thereof |
| WO2010123838A2 (en) * | 2009-04-20 | 2010-10-28 | Abbott Laboratories | Novel amide and amidine derivatives and uses thereof |
| US8871208B2 (en) * | 2009-12-04 | 2014-10-28 | Abbvie Inc. | 11-β-hydroxysteroid dehydrogenase type 1 (11β-HSD1) inhibitors and uses thereof |
| JP5813106B2 (en) * | 2010-06-25 | 2015-11-17 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Azaspirohexanone as an inhibitor of 11-β-HSD1 for the treatment of metabolic disorders |
| US8513430B2 (en) | 2010-07-27 | 2013-08-20 | High Point Pharmaceuticals, Llc | Substituted thiazol-2-ylamine derivatives, pharmaceutical compositions, and methods of use as 11-beta HSD1 modulators |
| KR101409847B1 (en) * | 2011-08-04 | 2014-06-27 | 현대약품 주식회사 | A COMPOUND FOR INHIBITING 11β-HYDROXY STEROID DEHYDROGENASE 1, AND A PHARMACEUTICAL COMPOSITION COMPRISING THE SAME |
| CA2860192C (en) * | 2011-12-22 | 2020-10-27 | Connexios Life Sciences Pvt. Ltd. | Derivatives of aza adamantane and uses thereof |
| TW201808914A (en) | 2016-05-05 | 2018-03-16 | 嘉來克生命科學有限責任公司 | Modulators of the integrated stress pathway |
| TW201808903A (en) * | 2016-05-05 | 2018-03-16 | 嘉來克生命科學有限責任公司 | Modulators of the integrated stress pathway |
| TW201808888A (en) | 2016-05-05 | 2018-03-16 | 嘉來克生命科學有限責任公司 | Modulators of the integrated stress pathway |
| US11548893B2 (en) | 2017-07-15 | 2023-01-10 | Arisan Therapeutics Inc. | Enantiomerically pure adamantane carboxamides for the treatment of filovirus infection |
| EP3704115A1 (en) | 2017-11-02 | 2020-09-09 | Calico Life Sciences LLC | Modulators of the integrated stress pathway |
| TWI771621B (en) | 2018-10-11 | 2022-07-21 | 美商嘉來克生命科學有限責任公司 | Prodrug modulators of the integrated stress pathway |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004056744A1 (en) * | 2002-12-23 | 2004-07-08 | Janssen Pharmaceutica N.V. | Adamantyl acetamides as hydroxysteroid dehydrogenase inhibitors |
| WO2004089470A2 (en) * | 2003-04-11 | 2004-10-21 | Novo Nordisk A/S | New amide derivatives and pharmaceutical use thereof |
| US20050245534A1 (en) * | 2004-04-29 | 2005-11-03 | Link James T | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase Type 1 enzyme |
| US20050277647A1 (en) * | 2004-04-29 | 2005-12-15 | Link James T | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase Type 1 enzyme |
Family Cites Families (63)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US245534A (en) * | 1881-08-09 | James w | ||
| US3919313A (en) * | 1973-07-23 | 1975-11-11 | Schering Corp | Novel 1-N-({60 -aminoacetyl) aminoadamantanes |
| SU740752A1 (en) | 1977-12-29 | 1980-06-15 | Научно-исследовательский институт фармакологии АМН СССР | Method of preparing 1-hydroxy-4-amino- or 4-n-substituted adamantanes |
| SU803348A1 (en) | 1979-10-12 | 1981-09-30 | Предприятие П/Я В-2343 | Oxyethylammonium derivatives of adamantane possessing antivirus activity |
| US4751292A (en) * | 1985-07-02 | 1988-06-14 | The Plant Cell Research Institute, Inc. | Adamantyl purines |
| EP0336356A3 (en) | 1988-04-05 | 1991-09-25 | Abbott Laboratories | Derivatives of tryptophan as cck antagonists |
| NZ234264A (en) | 1989-06-29 | 1993-05-26 | Warner Lambert Co | N-substituted cycloalkyl and polycycloalkyl alpha-substituted trp-phe- and phenethylamine derivatives, and pharmaceutical compositions |
| US5631281A (en) * | 1989-06-29 | 1997-05-20 | Warner-Lambert Company | N-substituted cycloalkyl and polycycloalkyl α-substituted Trp-Phe- and phenethylamine derivatives |
| EP0668290B1 (en) | 1990-02-28 | 1998-11-11 | Teikoku Seiyaku Kabushiki Kaisha | Physiologically active substance well capable of permeating biomembrane |
| US5244915A (en) * | 1990-08-31 | 1993-09-14 | Warner-Lambert Company | Amico acid derivatives cyclized at the c-terminal |
| JPH06506455A (en) | 1991-02-08 | 1994-07-21 | ケンブリッジ・ニューロサイエンス・インコーポレイテッド | Novel method for identifying guanidine substitutes and their derivatives as neurotransmitter release modulators and neurotransmitter release blockers |
| US5576355A (en) | 1993-06-04 | 1996-11-19 | Mobil Oil Corp. | Diamondoid derivatives for pharmaceutical use |
| US5395846A (en) | 1993-06-25 | 1995-03-07 | Rhone-Poulenc Rorer Pharmaceuticals Inc. | Amino Bi- and tri-carbocyclic aklane bis-aryl squalene synthase inhibitors |
| WO1999002145A1 (en) | 1997-07-07 | 1999-01-21 | Cambridge Neuroscience, Inc. | Combination drug therapies comprising aminoglycoside antibiotics and n,n'-disubstituted guanidines |
| CA2387892A1 (en) | 1999-10-18 | 2001-04-26 | University Of Connecticut | Pyrazole derivatives as cannabinoid receptor antagonists |
| CN1307159C (en) * | 2000-05-31 | 2007-03-28 | 参天制药株式会社 | TNF-alpha prodn. inhibitors |
| AU2002255634A1 (en) | 2001-03-01 | 2002-09-19 | Smithkline Beecham Corporation | Peptide deformylase inhibitors |
| EP1474139B1 (en) | 2002-02-01 | 2007-11-21 | Merck & Co., Inc. | 11-beta-hydroxysteroid dehydrogenase 1 inhibitors useful for the treatment of diabetes, obesity and dyslipidemia |
| WO2003075660A1 (en) | 2002-03-06 | 2003-09-18 | Merck & Co., Inc. | Method of treatment or prevention of obesity |
| US6793240B2 (en) | 2002-07-25 | 2004-09-21 | Autoliv Asp, Inc. | Expandable section for inflatable curtains |
| AU2003269242A1 (en) | 2002-10-11 | 2004-05-04 | Astrazeneca Ab | 1,4-disubstituted piperidine derivatives and their use as 11-betahsd1 inhibitors |
| KR20050059294A (en) | 2002-10-24 | 2005-06-17 | 스테릭스 리미티드 | Inhibitors of 11-beta-hydroxy steroid dehydrogenase type 1 and type 2 |
| US20040122033A1 (en) * | 2002-12-10 | 2004-06-24 | Nargund Ravi P. | Combination therapy for the treatment of obesity |
| US7348456B2 (en) * | 2002-12-19 | 2008-03-25 | Merck & Co., Inc. | Substituted amides |
| JO2397B1 (en) * | 2002-12-20 | 2007-06-17 | ميرك شارب اند دوم كوربوريشن | Triazole Derivatives As Inhibitors Of 11-Beta -Hydroxysteriod Dehydrogenase-1 |
| TW200503994A (en) | 2003-01-24 | 2005-02-01 | Novartis Ag | Organic compounds |
| US20060094699A1 (en) * | 2003-04-11 | 2006-05-04 | Kampen Gita Camilla T | Combination therapy using an 11beta-hydroxysteroid dehydrogenase type 1 inhibitor and a glucocorticoid receptor agonist to minimize the side effects associated with glucocorticoid receptor agonist therapy |
| EP1615697A2 (en) | 2003-04-11 | 2006-01-18 | Novo Nordisk A/S | New pyrazolo[1,5-a] pyrimidine derivatives and pharmaceutical use thereof |
| JP2006522745A (en) | 2003-04-11 | 2006-10-05 | ノボ ノルディスク アクティーゼルスカブ | Pharmaceutical use of substituted 1,2,4-triazoles |
| US7501405B2 (en) * | 2003-04-11 | 2009-03-10 | High Point Pharmaceuticals, Llc | Combination therapy using an 11β-hydroxysteroid dehydrogenase type 1 inhibitor and an antihypertensive agent for the treatment of metabolic syndrome and related diseases and disorders |
| JP2006522747A (en) * | 2003-04-11 | 2006-10-05 | ノボ ノルディスク アクティーゼルスカブ | Pharmaceutical use of condensed 1,2,4-triazole |
| JP2006522750A (en) | 2003-04-11 | 2006-10-05 | ノボ ノルディスク アクティーゼルスカブ | Combination therapy using 11β-hydroxysteroid dehydrogenase type 1 inhibitors and antihypertensive agents to treat metabolic syndrome and related diseases and disorders |
| US7700583B2 (en) * | 2003-04-11 | 2010-04-20 | High Point Pharmaceuticals, Llc | 11β-hydroxysteroid dehydrogenase type 1 active compounds |
| US20060100235A1 (en) * | 2003-04-11 | 2006-05-11 | Novo Nordisk A/S | Pharmaceutical use of substituted 1,2,4-triazoles |
| CA2534221A1 (en) | 2003-08-07 | 2005-02-24 | Merck & Co., Inc. | Pyrazole carboxamides as inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 |
| WO2005042513A1 (en) | 2003-10-23 | 2005-05-12 | Sterix Limited | Phenyl carboxamide and sulfonamide derivatives for use as 11-beta-hydroxysteroid dehydrogenase |
| GB0325745D0 (en) | 2003-11-05 | 2003-12-10 | Astrazeneca Ab | Chemical compounds |
| GB0326029D0 (en) | 2003-11-07 | 2003-12-10 | Astrazeneca Ab | Chemical compounds |
| AU2004305321A1 (en) | 2003-12-19 | 2005-07-07 | Pfizer Inc. | Benzenesulfonylamino-pyridin-2-yl derivatives and related compounds as inhibitors of 11-beta-hydroxysteroid dehydrogenase type 1 (11-beta-hsd-1) for the treatment of diabetes and obesity |
| JP2007261945A (en) | 2004-04-07 | 2007-10-11 | Taisho Pharmaceut Co Ltd | Thiazole derivative |
| GB0408771D0 (en) | 2004-04-20 | 2004-05-26 | Sterix Ltd | Compound |
| US20050261302A1 (en) * | 2004-04-29 | 2005-11-24 | Hoff Ethan D | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase Type 1 enzyme and their therapeutic application |
| EP1745019A1 (en) | 2004-05-06 | 2007-01-24 | Pfizer Inc. | Novel compounds of proline and morpholine derivatives |
| MY148480A (en) | 2004-05-24 | 2013-04-30 | Amgen Inc | Inhibitors of 11-beta-hydroxy steroid dehydrogenase type 1 |
| NZ551602A (en) * | 2004-06-24 | 2010-11-26 | Incyte Corp | Amido compounds and their use as pharmaceuticals |
| US20050288317A1 (en) | 2004-06-24 | 2005-12-29 | Wenqing Yao | Amido compounds and their use as pharmaceuticals |
| US7687665B2 (en) | 2004-06-24 | 2010-03-30 | Incyte Corporation | 2-methylprop anamides and their use as pharmaceuticals |
| EP1758580A4 (en) * | 2004-06-24 | 2008-01-16 | Incyte Corp | N-substituted piperidines and their use as pharmaceuticals |
| WO2006012227A2 (en) | 2004-06-24 | 2006-02-02 | Incyte Corporation | Amido compounds and their use as pharmaceuticals |
| EP1773773A4 (en) | 2004-06-24 | 2009-07-29 | Incyte Corp | Amido compounds and their use as pharmaceuticals |
| PT1773768T (en) | 2004-07-30 | 2018-11-30 | Exelixis Inc | Pyrrole derivatives as pharmaceutical agents |
| US7915252B2 (en) | 2004-08-06 | 2011-03-29 | Merck Sharp & Dohme | Sulfonyl compounds as inhibitors of 11-beta-hydroxysteroid dehydrogenase-1 |
| BRPI0514230A (en) | 2004-08-10 | 2008-06-03 | Incyte Corp | starch compounds and their use as pharmaceuticals |
| EP1786774B1 (en) | 2004-08-30 | 2009-10-14 | Janssen Pharmaceutica N.V. | Tricyclic lactam derivatives as 11-beta hydroxysteroid dehydrogenase inhibitors |
| UA87328C2 (en) | 2004-08-30 | 2009-07-10 | Янссен Фармацевтика Н.В. | N-2 adamantanyl-2-phenoxy-acetamide derivatives as 11-beta hydroxysteroid dehydrogenase inhibitors |
| JP2008515956A (en) | 2004-10-12 | 2008-05-15 | ノボ ノルディスク アクティーゼルスカブ | 11.beta.-hydroxysteroid dehydrogenase type 1 active spiro compound |
| EP1807072B1 (en) | 2004-10-29 | 2009-01-07 | Eli Lilly And Company | Cycloalkyl lactam derivatives as inhibitors of 11-beta-hydroxysteroid dehydrogenase 1 |
| EP1812407A2 (en) | 2004-11-02 | 2007-08-01 | Pfizer, Inc. | Novel compounds of substituted and unsubstituted adamantyl amides |
| EP1659113A1 (en) | 2004-11-08 | 2006-05-24 | Evotec AG | Inhibitors of 11beta-hydroxy steroid dehydrogenase type 1 (11beta-HSD1) |
| EP1666467A1 (en) | 2004-11-08 | 2006-06-07 | Evotec AG | 11Beta-HSD1 Inhibitors |
| EP1655283A1 (en) | 2004-11-08 | 2006-05-10 | Evotec OAI AG | 11beta-HSD1 Inhibitors |
| NZ554906A (en) | 2004-11-10 | 2011-01-28 | Incyte Corp | Lactam compounds and their use as pharmaceuticals |
| EP1888544A2 (en) | 2004-12-17 | 2008-02-20 | Takeda San Diego, Inc. | Hydroxysteroid dehydrogenase inhibitors |
-
2006
- 2006-01-05 BR BRPI0606228-8A patent/BRPI0606228A2/en not_active IP Right Cessation
- 2006-01-05 WO PCT/US2006/000402 patent/WO2006074330A2/en active Application Filing
- 2006-01-05 MX MX2007008239A patent/MX2007008239A/en active IP Right Grant
- 2006-01-05 CN CN2012102653126A patent/CN102816081A/en active Pending
- 2006-01-05 AU AU2006203918A patent/AU2006203918B2/en not_active Ceased
- 2006-01-05 KR KR1020077017918A patent/KR101302627B1/en not_active IP Right Cessation
- 2006-01-05 CN CNA2006800071918A patent/CN101142172A/en active Pending
- 2006-01-05 CA CA002594116A patent/CA2594116A1/en not_active Abandoned
- 2006-01-05 EP EP06717579A patent/EP1846362A2/en not_active Withdrawn
- 2006-01-05 NZ NZ555971A patent/NZ555971A/en not_active IP Right Cessation
- 2006-01-05 US US11/326,277 patent/US7217838B2/en active Active
- 2006-01-05 EP EP14164610.9A patent/EP2835367A1/en not_active Withdrawn
- 2006-01-05 JP JP2007550479A patent/JP5133702B2/en not_active Expired - Fee Related
-
2007
- 2007-04-10 US US11/733,636 patent/US7528282B2/en not_active Expired - Fee Related
- 2007-07-01 IL IL184329A patent/IL184329A/en not_active IP Right Cessation
- 2007-07-04 ZA ZA200705468A patent/ZA200705468B/en unknown
-
2008
- 2008-07-24 US US12/179,405 patent/USRE41135E1/en active Active
-
2011
- 2011-06-02 IL IL213342A patent/IL213342A/en not_active IP Right Cessation
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004056744A1 (en) * | 2002-12-23 | 2004-07-08 | Janssen Pharmaceutica N.V. | Adamantyl acetamides as hydroxysteroid dehydrogenase inhibitors |
| WO2004089470A2 (en) * | 2003-04-11 | 2004-10-21 | Novo Nordisk A/S | New amide derivatives and pharmaceutical use thereof |
| US20050245534A1 (en) * | 2004-04-29 | 2005-11-03 | Link James T | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase Type 1 enzyme |
| US20050277647A1 (en) * | 2004-04-29 | 2005-12-15 | Link James T | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase Type 1 enzyme |
Cited By (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8952176B2 (en) | 2005-06-07 | 2015-02-10 | Shionogi & Co., Ltd. | Heterocyclic compound having type I 11 β hydroxysteroid dehydrogenase inhibitory activity |
| US8324265B2 (en) | 2005-11-21 | 2012-12-04 | Shionogi & Co., Ltd. | Heterocyclic compounds having type I 11β hydroxysteroid dehydrogenase inhibitory activity |
| US8017638B2 (en) | 2006-03-30 | 2011-09-13 | Shionogi & Co., Ltd. | Isoxazole derivative and isothiazole derivative having inhibitory activity on 11β-hydroxysteroid dehydrogenase type 1 |
| EP2351568A2 (en) | 2006-05-04 | 2011-08-03 | Boehringer Ingelheim International GmbH | Uses of dpp-iv inhibitors |
| WO2007128761A2 (en) | 2006-05-04 | 2007-11-15 | Boehringer Ingelheim International Gmbh | Uses of dpp-iv inhibitors |
| JP2010501589A (en) * | 2006-08-24 | 2010-01-21 | ブリストル−マイヤーズ スクイブ カンパニー | Cyclic 11-β hydroxysteroid dehydrogenase type I inhibitor |
| JP2010526777A (en) * | 2007-04-24 | 2010-08-05 | ハイ ポイント ファーマシューティカルズ,リミティド ライアビリティ カンパニー | Pharmaceutical use of substituted amides |
| EP2150109A4 (en) * | 2007-04-24 | 2011-05-04 | High Point Pharmaceuticals Llc | Pharmaceutical use of substituted amides |
| EP2150109A1 (en) * | 2007-04-24 | 2010-02-10 | High Point Pharmaceuticals, LLC | Pharmaceutical use of substituted amides |
| CN101711106B (en) * | 2007-04-24 | 2013-02-06 | 高点制药有限责任公司 | Pharmaceutical use of substituted amides |
| WO2008142986A1 (en) | 2007-05-18 | 2008-11-27 | Shionogi & Co., Ltd. | NITROGEN-CONTAINING HETEROCYCLIC DERIVATIVE HAVING 11 β-HYDROXYSTEROID DEHYDROGENASE TYPE I INHIBITORY ACTIVITY |
| JPWO2008142986A1 (en) * | 2007-05-18 | 2010-08-05 | 塩野義製薬株式会社 | Nitrogen-containing heterocyclic derivatives having type I 11β-hydroxysteroid dehydrogenase inhibitory activity |
| CN103288738B (en) * | 2007-05-18 | 2016-03-16 | 盐野义制药株式会社 | There is the nitogen-contained heterocycle derivant of 11beta-Hydroxysteroid dehydrogenase I type inhibit activities |
| CN101754954B (en) * | 2007-05-18 | 2015-11-25 | 盐野义制药株式会社 | There is the nitogen-contained heterocycle derivant of 11beta-Hydroxysteroid dehydrogenase I type inhibit activities |
| US8383622B2 (en) | 2007-05-18 | 2013-02-26 | Shionogi & Co., Ltd. | Nitrogen-containing heterocyclic derivative having 11β-hydroxysteroid dehydrogenase type I inhibitory activity |
| CN103288738A (en) * | 2007-05-18 | 2013-09-11 | 盐野义制药株式会社 | Nitrogen-containing heterocyclic derivative having 11 beta-hydroxysteroid dehydrogenase type I inhibitory activity |
| JP4671369B2 (en) * | 2007-05-18 | 2011-04-13 | 塩野義製薬株式会社 | Nitrogen-containing heterocyclic derivatives having type I 11β-hydroxysteroid dehydrogenase inhibitory activity |
| US8822149B2 (en) | 2007-05-24 | 2014-09-02 | Pacific Edge Limited | Prognosis prediction for melanoma cancer |
| JP5605844B2 (en) * | 2008-10-17 | 2014-10-15 | 塩野義製薬株式会社 | Acetic acid amide derivatives having vascular endothelial lipase inhibitory activity |
| US8957219B2 (en) | 2008-10-17 | 2015-02-17 | Shionogi & Co., Ltd. | Acetic acid amide derivative having inhibitory activity on endothelial lipase |
| JPWO2010044441A1 (en) * | 2008-10-17 | 2012-03-15 | 塩野義製薬株式会社 | Acetic acid amide derivatives having vascular endothelial lipase inhibitory activity |
| WO2010044441A1 (en) * | 2008-10-17 | 2010-04-22 | 塩野義製薬株式会社 | Acetic acid amide derivative having inhibitory activity on vascular endothelial lipase |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1846362A2 (en) | 2007-10-24 |
| KR20070094954A (en) | 2007-09-27 |
| IL213342A (en) | 2014-09-30 |
| ZA200705468B (en) | 2010-03-31 |
| EP2835367A1 (en) | 2015-02-11 |
| BRPI0606228A2 (en) | 2009-06-09 |
| CN101142172A (en) | 2008-03-12 |
| JP2008526874A (en) | 2008-07-24 |
| US7528282B2 (en) | 2009-05-05 |
| WO2006074330A3 (en) | 2007-01-25 |
| USRE41135E1 (en) | 2010-02-16 |
| NZ555971A (en) | 2011-01-28 |
| AU2006203918B2 (en) | 2011-05-19 |
| IL184329A0 (en) | 2007-10-31 |
| JP5133702B2 (en) | 2013-01-30 |
| KR101302627B1 (en) | 2013-09-10 |
| MX2007008239A (en) | 2007-08-17 |
| US7217838B2 (en) | 2007-05-15 |
| AU2006203918A1 (en) | 2006-07-13 |
| CA2594116A1 (en) | 2006-07-13 |
| IL184329A (en) | 2014-02-27 |
| US20060149070A1 (en) | 2006-07-06 |
| US20070179186A1 (en) | 2007-08-02 |
| CN102816081A (en) | 2012-12-12 |
| IL213342A0 (en) | 2011-07-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2006203918B2 (en) | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase Type 1 enzyme | |
| EP1846363B1 (en) | Adamantyl derivatives as inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme | |
| US20150265613A1 (en) | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme | |
| EP2269977B1 (en) | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase Type 1 enzyme | |
| AU2013206051B8 (en) | Adamantyl derivatives as inhibitors of the 11-beta-hydroxysteroid dehydrogenase Type 1 enzyme | |
| AU2011253958B2 (en) | Adamantyl derivatives as inhibitors of the 11-beta-hydroxysteroid dehydrogenase Type 1 enzyme | |
| AU2013206051B2 (en) | Adamantyl derivatives as inhibitors of the 11-beta-hydroxysteroid dehydrogenase Type 1 enzyme |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200680007191.8 Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 555971 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006203918 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 184329 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 5145/DELNP/2007 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 2594116 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007550479 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2007/008239 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2006203918 Country of ref document: AU Date of ref document: 20060105 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006717579 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020077017918 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: PI0606228 Country of ref document: BR Kind code of ref document: A2 |