WO2006071819A1 - [1h-pyrazolo[3, 4-d]pyrimidin-4-yl]-piperidine or -piperazine compounds as serine-theoronine kinase modulators (p70s6k, atk1 and atk2) for the treatment of immunological, inflammatory and proliferative diseases - Google Patents
[1h-pyrazolo[3, 4-d]pyrimidin-4-yl]-piperidine or -piperazine compounds as serine-theoronine kinase modulators (p70s6k, atk1 and atk2) for the treatment of immunological, inflammatory and proliferative diseases Download PDFInfo
- Publication number
- WO2006071819A1 WO2006071819A1 PCT/US2005/046938 US2005046938W WO2006071819A1 WO 2006071819 A1 WO2006071819 A1 WO 2006071819A1 US 2005046938 W US2005046938 W US 2005046938W WO 2006071819 A1 WO2006071819 A1 WO 2006071819A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- nri
- aryl
- alkyl
- compound
- halo
- Prior art date
Links
- 108091000080 Phosphotransferase Proteins 0.000 title claims abstract description 56
- 102000020233 phosphotransferase Human genes 0.000 title claims abstract description 56
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 title claims abstract description 39
- 201000010099 disease Diseases 0.000 title claims abstract description 36
- 230000002062 proliferating effect Effects 0.000 title claims description 6
- 108010013238 70-kDa Ribosomal Protein S6 Kinases Proteins 0.000 title abstract description 49
- 230000002757 inflammatory effect Effects 0.000 title description 4
- JYIZQPKFJCDDQE-UHFFFAOYSA-N 4-piperidin-1-yl-1h-pyrazolo[3,4-d]pyrimidine Chemical compound C1CCCCN1C1=NC=NC2=C1C=NN2 JYIZQPKFJCDDQE-UHFFFAOYSA-N 0.000 title description 2
- 230000001900 immune effect Effects 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims abstract description 283
- 239000000203 mixture Substances 0.000 claims abstract description 69
- 230000000694 effects Effects 0.000 claims abstract description 66
- 238000000034 method Methods 0.000 claims abstract description 60
- 102000001253 Protein Kinase Human genes 0.000 claims abstract description 23
- 108060006633 protein kinase Proteins 0.000 claims abstract description 20
- 230000001413 cellular effect Effects 0.000 claims abstract description 12
- 101100162366 Caenorhabditis elegans akt-2 gene Proteins 0.000 claims abstract description 11
- 230000005764 inhibitory process Effects 0.000 claims abstract description 11
- 101100322915 Caenorhabditis elegans akt-1 gene Proteins 0.000 claims abstract description 9
- 125000003118 aryl group Chemical group 0.000 claims description 81
- 239000003795 chemical substances by application Substances 0.000 claims description 73
- 125000005843 halogen group Chemical group 0.000 claims description 71
- 125000000217 alkyl group Chemical group 0.000 claims description 69
- 125000003545 alkoxy group Chemical group 0.000 claims description 47
- 125000001424 substituent group Chemical group 0.000 claims description 40
- 125000004122 cyclic group Chemical group 0.000 claims description 30
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 29
- 239000008194 pharmaceutical composition Substances 0.000 claims description 25
- 125000000623 heterocyclic group Chemical group 0.000 claims description 24
- 150000003839 salts Chemical class 0.000 claims description 22
- JCXJVPUVTGWSNB-UHFFFAOYSA-N Nitrogen dioxide Chemical compound O=[N]=O JCXJVPUVTGWSNB-UHFFFAOYSA-N 0.000 claims description 21
- 229910052799 carbon Inorganic materials 0.000 claims description 20
- 229920006395 saturated elastomer Polymers 0.000 claims description 20
- 229910052739 hydrogen Inorganic materials 0.000 claims description 19
- 229910052794 bromium Inorganic materials 0.000 claims description 16
- 125000001072 heteroaryl group Chemical group 0.000 claims description 15
- 229910052760 oxygen Inorganic materials 0.000 claims description 14
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 14
- 125000004429 atom Chemical group 0.000 claims description 13
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 13
- 239000001257 hydrogen Substances 0.000 claims description 13
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 11
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 11
- 239000003937 drug carrier Substances 0.000 claims description 11
- 229910052757 nitrogen Inorganic materials 0.000 claims description 11
- 230000002159 abnormal effect Effects 0.000 claims description 10
- 125000002837 carbocyclic group Chemical group 0.000 claims description 10
- 229910052801 chlorine Inorganic materials 0.000 claims description 9
- 229910052731 fluorine Inorganic materials 0.000 claims description 9
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 9
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims description 8
- 125000002619 bicyclic group Chemical group 0.000 claims description 8
- 125000006413 ring segment Chemical group 0.000 claims description 8
- 238000012216 screening Methods 0.000 claims description 8
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 claims description 7
- 230000002503 metabolic effect Effects 0.000 claims description 7
- 239000002207 metabolite Substances 0.000 claims description 7
- 239000000651 prodrug Substances 0.000 claims description 7
- 229940002612 prodrug Drugs 0.000 claims description 7
- 229910052717 sulfur Inorganic materials 0.000 claims description 7
- 241000124008 Mammalia Species 0.000 claims description 6
- 238000001727 in vivo Methods 0.000 claims description 6
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 claims description 5
- 230000002401 inhibitory effect Effects 0.000 claims description 5
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims description 4
- 125000003342 alkenyl group Chemical group 0.000 claims description 4
- 125000000304 alkynyl group Chemical group 0.000 claims description 4
- 208000035475 disorder Diseases 0.000 claims description 3
- QRKLUICYAXZGJB-UHFFFAOYSA-N 4-[4-(2-fluorophenyl)piperazin-1-yl]-1h-pyrazolo[3,4-d]pyrimidine Chemical compound FC1=CC=CC=C1N1CCN(C=2C=3C=NNC=3N=CN=2)CC1 QRKLUICYAXZGJB-UHFFFAOYSA-N 0.000 claims description 2
- LCGMENDABLGZBC-UHFFFAOYSA-N 4-[4-(3-chlorophenyl)piperazin-1-yl]-1h-pyrazolo[3,4-d]pyrimidine Chemical compound ClC1=CC=CC(N2CCN(CC2)C=2C=3C=NNC=3N=CN=2)=C1 LCGMENDABLGZBC-UHFFFAOYSA-N 0.000 claims description 2
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 claims description 2
- 125000001246 bromo group Chemical group Br* 0.000 claims description 2
- 150000004677 hydrates Chemical class 0.000 claims description 2
- RXDGQPHFVNTZTC-UHFFFAOYSA-N n-(4-phenoxyphenyl)-4-(1h-pyrazolo[3,4-d]pyrimidin-4-yl)piperazine-1-carboxamide Chemical compound C1CN(C=2C=3C=NNC=3N=CN=2)CCN1C(=O)NC(C=C1)=CC=C1OC1=CC=CC=C1 RXDGQPHFVNTZTC-UHFFFAOYSA-N 0.000 claims description 2
- PODFBDRFPZOWFK-UHFFFAOYSA-N tert-butyl 4-(1h-pyrazolo[3,4-d]pyrimidin-4-yl)piperazine-1-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCN1C1=NC=NC2=C1C=NN2 PODFBDRFPZOWFK-UHFFFAOYSA-N 0.000 claims description 2
- KZCSHJAUSSLJGD-UHFFFAOYSA-N ethyl 4-(1h-pyrazolo[3,4-d]pyrimidin-4-yl)piperazine-1-carboxylate Chemical compound C1CN(C(=O)OCC)CCN1C1=NC=NC2=C1C=NN2 KZCSHJAUSSLJGD-UHFFFAOYSA-N 0.000 claims 1
- 230000004060 metabolic process Effects 0.000 abstract description 9
- 230000035755 proliferation Effects 0.000 abstract description 8
- 230000019491 signal transduction Effects 0.000 abstract description 7
- 230000002255 enzymatic effect Effects 0.000 abstract description 6
- 238000013508 migration Methods 0.000 abstract description 6
- 230000001419 dependent effect Effects 0.000 abstract description 5
- 230000005012 migration Effects 0.000 abstract description 5
- 230000004069 differentiation Effects 0.000 abstract description 4
- 238000003782 apoptosis assay Methods 0.000 abstract description 3
- 230000005522 programmed cell death Effects 0.000 abstract description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 96
- -1 CSFIR Proteins 0.000 description 81
- 235000019439 ethyl acetate Nutrition 0.000 description 43
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 42
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 40
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 36
- 230000027455 binding Effects 0.000 description 35
- 239000000243 solution Substances 0.000 description 32
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 31
- 238000006243 chemical reaction Methods 0.000 description 27
- 239000007832 Na2SO4 Substances 0.000 description 26
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 26
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 26
- 239000011541 reaction mixture Substances 0.000 description 26
- 229910052938 sodium sulfate Inorganic materials 0.000 description 26
- 239000012267 brine Substances 0.000 description 25
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 25
- 239000012044 organic layer Substances 0.000 description 22
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 21
- 239000012043 crude product Substances 0.000 description 20
- 102000004169 proteins and genes Human genes 0.000 description 20
- 108090000623 proteins and genes Proteins 0.000 description 20
- 235000018102 proteins Nutrition 0.000 description 19
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 18
- 238000003556 assay Methods 0.000 description 18
- 150000003254 radicals Chemical class 0.000 description 18
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 17
- 206010028980 Neoplasm Diseases 0.000 description 17
- 238000005160 1H NMR spectroscopy Methods 0.000 description 16
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N DMSO Substances CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 15
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 15
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 14
- 210000004027 cell Anatomy 0.000 description 14
- 239000007787 solid Substances 0.000 description 14
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 13
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 13
- 102000004190 Enzymes Human genes 0.000 description 13
- 108090000790 Enzymes Proteins 0.000 description 13
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 13
- 239000003446 ligand Substances 0.000 description 13
- 125000004482 piperidin-4-yl group Chemical group N1CCC(CC1)* 0.000 description 13
- 239000000047 product Substances 0.000 description 13
- 238000003756 stirring Methods 0.000 description 13
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 12
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 12
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 12
- 108020001756 ligand binding domains Proteins 0.000 description 12
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 11
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 11
- 102000005962 receptors Human genes 0.000 description 11
- 108020003175 receptors Proteins 0.000 description 11
- 239000002904 solvent Substances 0.000 description 11
- 102000004022 Protein-Tyrosine Kinases Human genes 0.000 description 10
- 108090000412 Protein-Tyrosine Kinases Proteins 0.000 description 10
- 125000004432 carbon atom Chemical group C* 0.000 description 10
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 10
- 125000004194 piperazin-1-yl group Chemical group [H]N1C([H])([H])C([H])([H])N(*)C([H])([H])C1([H])[H] 0.000 description 10
- 239000000126 substance Substances 0.000 description 10
- 238000012360 testing method Methods 0.000 description 10
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 9
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 9
- 238000005481 NMR spectroscopy Methods 0.000 description 9
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 9
- 201000011510 cancer Diseases 0.000 description 9
- 239000003921 oil Substances 0.000 description 9
- 235000019198 oils Nutrition 0.000 description 9
- 125000004527 pyrimidin-4-yl group Chemical group N1=CN=C(C=C1)* 0.000 description 9
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical group [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 description 8
- 108091008611 Protein Kinase B Proteins 0.000 description 8
- 208000009956 adenocarcinoma Diseases 0.000 description 8
- 125000002947 alkylene group Chemical group 0.000 description 8
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical group BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 8
- 230000004663 cell proliferation Effects 0.000 description 8
- 238000005094 computer simulation Methods 0.000 description 8
- 125000004415 heterocyclylalkyl group Chemical group 0.000 description 8
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 8
- 239000000546 pharmaceutical excipient Substances 0.000 description 8
- 238000002360 preparation method Methods 0.000 description 8
- 239000000725 suspension Substances 0.000 description 8
- DTBKKESEAXYGMM-UHFFFAOYSA-N 3-bromo-2h-pyrazolo[4,3-d]pyrimidine Chemical compound N1=CN=C2C(Br)=NNC2=C1 DTBKKESEAXYGMM-UHFFFAOYSA-N 0.000 description 7
- MDFHFCAMBZNSLV-UHFFFAOYSA-N 3-bromo-4-chloro-2h-pyrazolo[3,4-d]pyrimidine Chemical compound ClC1=NC=NC2=NNC(Br)=C12 MDFHFCAMBZNSLV-UHFFFAOYSA-N 0.000 description 7
- CYCGRDQQIOGCKX-UHFFFAOYSA-N Dehydro-luciferin Natural products OC(=O)C1=CSC(C=2SC3=CC(O)=CC=C3N=2)=N1 CYCGRDQQIOGCKX-UHFFFAOYSA-N 0.000 description 7
- BJGNCJDXODQBOB-UHFFFAOYSA-N Fivefly Luciferin Natural products OC(=O)C1CSC(C=2SC3=CC(O)=CC=C3N=2)=N1 BJGNCJDXODQBOB-UHFFFAOYSA-N 0.000 description 7
- DDWFXDSYGUXRAY-UHFFFAOYSA-N Luciferin Natural products CCc1c(C)c(CC2NC(=O)C(=C2C=C)C)[nH]c1Cc3[nH]c4C(=C5/NC(CC(=O)O)C(C)C5CC(=O)O)CC(=O)c4c3C DDWFXDSYGUXRAY-UHFFFAOYSA-N 0.000 description 7
- 206010025323 Lymphomas Diseases 0.000 description 7
- 102000038030 PI3Ks Human genes 0.000 description 7
- 108091007960 PI3Ks Proteins 0.000 description 7
- 108090000430 Phosphatidylinositol 3-kinases Proteins 0.000 description 7
- 206010039491 Sarcoma Diseases 0.000 description 7
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 7
- 125000001118 alkylidene group Chemical group 0.000 description 7
- 125000003710 aryl alkyl group Chemical group 0.000 description 7
- 230000004071 biological effect Effects 0.000 description 7
- 235000019441 ethanol Nutrition 0.000 description 7
- 150000003840 hydrochlorides Chemical class 0.000 description 7
- IXCSERBJSXMMFS-UHFFFAOYSA-N hydrogen chloride Substances Cl.Cl IXCSERBJSXMMFS-UHFFFAOYSA-N 0.000 description 7
- 229910000041 hydrogen chloride Inorganic materials 0.000 description 7
- 239000003112 inhibitor Substances 0.000 description 7
- 238000000021 kinase assay Methods 0.000 description 7
- 208000030159 metabolic disease Diseases 0.000 description 7
- 239000012074 organic phase Substances 0.000 description 7
- 238000000746 purification Methods 0.000 description 7
- 238000011084 recovery Methods 0.000 description 7
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 7
- 238000006467 substitution reaction Methods 0.000 description 7
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 6
- 102100033810 RAC-alpha serine/threonine-protein kinase Human genes 0.000 description 6
- 229920004890 Triton X-100 Polymers 0.000 description 6
- 239000013504 Triton X-100 Substances 0.000 description 6
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 6
- 150000001732 carboxylic acid derivatives Chemical group 0.000 description 6
- 239000003153 chemical reaction reagent Substances 0.000 description 6
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 6
- 238000004590 computer program Methods 0.000 description 6
- 239000013078 crystal Substances 0.000 description 6
- 239000012153 distilled water Substances 0.000 description 6
- 229910052736 halogen Inorganic materials 0.000 description 6
- 150000002367 halogens Chemical class 0.000 description 6
- 238000011534 incubation Methods 0.000 description 6
- 239000010410 layer Substances 0.000 description 6
- 125000005415 substituted alkoxy group Chemical group 0.000 description 6
- 230000004614 tumor growth Effects 0.000 description 6
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 5
- 201000009030 Carcinoma Diseases 0.000 description 5
- 201000004681 Psoriasis Diseases 0.000 description 5
- 238000004458 analytical method Methods 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 239000000460 chlorine Substances 0.000 description 5
- 239000002552 dosage form Substances 0.000 description 5
- 150000002148 esters Chemical class 0.000 description 5
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 5
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 5
- 125000004043 oxo group Chemical group O=* 0.000 description 5
- 229920001223 polyethylene glycol Polymers 0.000 description 5
- 239000000843 powder Substances 0.000 description 5
- 206010041823 squamous cell carcinoma Diseases 0.000 description 5
- CXWXQJXEFPUFDZ-UHFFFAOYSA-N tetralin Chemical compound C1=CC=C2CCCCC2=C1 CXWXQJXEFPUFDZ-UHFFFAOYSA-N 0.000 description 5
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 4
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 4
- IWSPVFHYFCVQHP-UHFFFAOYSA-N 2-[4-(3-bromo-2h-pyrazolo[3,4-d]pyrimidin-4-yl)piperazin-1-yl]-2-(4-chlorophenyl)-n-[2-(dimethylamino)ethyl]acetamide Chemical compound C1CN(C=2C=3C(Br)=NNC=3N=CN=2)CCN1C(C(=O)NCCN(C)C)C1=CC=C(Cl)C=C1 IWSPVFHYFCVQHP-UHFFFAOYSA-N 0.000 description 4
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 4
- 101150107888 AKT2 gene Proteins 0.000 description 4
- WVDDGKGOMKODPV-UHFFFAOYSA-N Benzyl alcohol Chemical compound OCC1=CC=CC=C1 WVDDGKGOMKODPV-UHFFFAOYSA-N 0.000 description 4
- 229910000906 Bronze Inorganic materials 0.000 description 4
- 0 CC(C1*)*(*IC)C(*)C(*)N1C(OC(C)(C)C)=O Chemical compound CC(C1*)*(*IC)C(*)C(*)N1C(OC(C)(C)C)=O 0.000 description 4
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 4
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 4
- 101000798015 Homo sapiens RAC-beta serine/threonine-protein kinase Proteins 0.000 description 4
- 206010024612 Lipoma Diseases 0.000 description 4
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 4
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 4
- UFWIBTONFRDIAS-UHFFFAOYSA-N Naphthalene Chemical compound C1=CC=CC2=CC=CC=C21 UFWIBTONFRDIAS-UHFFFAOYSA-N 0.000 description 4
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 4
- 102100032315 RAC-beta serine/threonine-protein kinase Human genes 0.000 description 4
- 108010053099 Vascular Endothelial Growth Factor Receptor-2 Proteins 0.000 description 4
- 102100033177 Vascular endothelial growth factor receptor 2 Human genes 0.000 description 4
- 230000004913 activation Effects 0.000 description 4
- 125000002252 acyl group Chemical group 0.000 description 4
- 235000008206 alpha-amino acids Nutrition 0.000 description 4
- 230000006907 apoptotic process Effects 0.000 description 4
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 4
- 230000037396 body weight Effects 0.000 description 4
- 239000010974 bronze Substances 0.000 description 4
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 4
- 239000000969 carrier Substances 0.000 description 4
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 4
- 239000006185 dispersion Substances 0.000 description 4
- 239000003814 drug Substances 0.000 description 4
- 125000001207 fluorophenyl group Chemical group 0.000 description 4
- 235000011187 glycerol Nutrition 0.000 description 4
- 238000004128 high performance liquid chromatography Methods 0.000 description 4
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 4
- KWGKDLIKAYFUFQ-UHFFFAOYSA-M lithium chloride Chemical compound [Li+].[Cl-] KWGKDLIKAYFUFQ-UHFFFAOYSA-M 0.000 description 4
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 4
- 238000002156 mixing Methods 0.000 description 4
- QOHMWDJIBGVPIF-UHFFFAOYSA-N n',n'-diethylpropane-1,3-diamine Chemical compound CCN(CC)CCCN QOHMWDJIBGVPIF-UHFFFAOYSA-N 0.000 description 4
- 235000006408 oxalic acid Nutrition 0.000 description 4
- 230000026731 phosphorylation Effects 0.000 description 4
- 238000006366 phosphorylation reaction Methods 0.000 description 4
- 230000002829 reductive effect Effects 0.000 description 4
- 239000011780 sodium chloride Substances 0.000 description 4
- 239000007858 starting material Substances 0.000 description 4
- 125000000547 substituted alkyl group Chemical group 0.000 description 4
- 125000003107 substituted aryl group Chemical group 0.000 description 4
- GETQZCLCWQTVFV-UHFFFAOYSA-N trimethylamine Chemical compound CN(C)C GETQZCLCWQTVFV-UHFFFAOYSA-N 0.000 description 4
- 239000003656 tris buffered saline Substances 0.000 description 4
- 125000004209 (C1-C8) alkyl group Chemical group 0.000 description 3
- KISWVXRQTGLFGD-UHFFFAOYSA-N 2-[[2-[[6-amino-2-[[2-[[2-[[5-amino-2-[[2-[[1-[2-[[6-amino-2-[(2,5-diamino-5-oxopentanoyl)amino]hexanoyl]amino]-5-(diaminomethylideneamino)pentanoyl]pyrrolidine-2-carbonyl]amino]-3-hydroxypropanoyl]amino]-5-oxopentanoyl]amino]-5-(diaminomethylideneamino)p Chemical compound C1CCN(C(=O)C(CCCN=C(N)N)NC(=O)C(CCCCN)NC(=O)C(N)CCC(N)=O)C1C(=O)NC(CO)C(=O)NC(CCC(N)=O)C(=O)NC(CCCN=C(N)N)C(=O)NC(CO)C(=O)NC(CCCCN)C(=O)NC(C(=O)NC(CC(C)C)C(O)=O)CC1=CC=C(O)C=C1 KISWVXRQTGLFGD-UHFFFAOYSA-N 0.000 description 3
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 3
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 3
- RGJOEKWQDUBAIZ-IBOSZNHHSA-N CoASH Chemical compound O[C@@H]1[C@H](OP(O)(O)=O)[C@@H](COP(O)(=O)OP(O)(=O)OCC(C)(C)[C@@H](O)C(=O)NCCC(=O)NCCS)O[C@H]1N1C2=NC=NC(N)=C2N=C1 RGJOEKWQDUBAIZ-IBOSZNHHSA-N 0.000 description 3
- IGXWBGJHJZYPQS-SSDOTTSWSA-N D-Luciferin Chemical compound OC(=O)[C@H]1CSC(C=2SC3=CC=C(O)C=C3N=2)=N1 IGXWBGJHJZYPQS-SSDOTTSWSA-N 0.000 description 3
- 206010012689 Diabetic retinopathy Diseases 0.000 description 3
- 201000008808 Fibrosarcoma Diseases 0.000 description 3
- 239000007821 HATU Substances 0.000 description 3
- 239000007995 HEPES buffer Substances 0.000 description 3
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 3
- 108060001084 Luciferase Proteins 0.000 description 3
- 239000005089 Luciferase Substances 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- 208000008589 Obesity Diseases 0.000 description 3
- 108700020796 Oncogene Proteins 0.000 description 3
- 102000004160 Phosphoric Monoester Hydrolases Human genes 0.000 description 3
- 108090000608 Phosphoric Monoester Hydrolases Proteins 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Propanedioic acid Natural products OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- 229920002472 Starch Polymers 0.000 description 3
- 102000013530 TOR Serine-Threonine Kinases Human genes 0.000 description 3
- 108010065917 TOR Serine-Threonine Kinases Proteins 0.000 description 3
- 206010043276 Teratoma Diseases 0.000 description 3
- 239000002671 adjuvant Substances 0.000 description 3
- 230000004075 alteration Effects 0.000 description 3
- 150000001408 amides Chemical group 0.000 description 3
- 150000001412 amines Chemical class 0.000 description 3
- 239000012131 assay buffer Substances 0.000 description 3
- 239000011230 binding agent Substances 0.000 description 3
- 230000036983 biotransformation Effects 0.000 description 3
- 239000004202 carbamide Substances 0.000 description 3
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 3
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 3
- 230000019522 cellular metabolic process Effects 0.000 description 3
- 125000000068 chlorophenyl group Chemical group 0.000 description 3
- RGJOEKWQDUBAIZ-UHFFFAOYSA-N coenzime A Natural products OC1C(OP(O)(O)=O)C(COP(O)(=O)OP(O)(=O)OCC(C)(C)C(O)C(=O)NCCC(=O)NCCS)OC1N1C2=NC=NC(N)=C2N=C1 RGJOEKWQDUBAIZ-UHFFFAOYSA-N 0.000 description 3
- 239000005516 coenzyme A Substances 0.000 description 3
- 229940093530 coenzyme a Drugs 0.000 description 3
- 238000004440 column chromatography Methods 0.000 description 3
- KUNSUQLRTQLHQQ-UHFFFAOYSA-N copper tin Chemical compound [Cu].[Sn] KUNSUQLRTQLHQQ-UHFFFAOYSA-N 0.000 description 3
- 239000013058 crude material Substances 0.000 description 3
- KDTSHFARGAKYJN-UHFFFAOYSA-N dephosphocoenzyme A Natural products OC1C(O)C(COP(O)(=O)OP(O)(=O)OCC(C)(C)C(O)C(=O)NCCC(=O)NCCS)OC1N1C2=NC=NC(N)=C2N=C1 KDTSHFARGAKYJN-UHFFFAOYSA-N 0.000 description 3
- 206010012601 diabetes mellitus Diseases 0.000 description 3
- 235000005911 diet Nutrition 0.000 description 3
- 230000037213 diet Effects 0.000 description 3
- 238000006073 displacement reaction Methods 0.000 description 3
- 229940000406 drug candidate Drugs 0.000 description 3
- 238000009510 drug design Methods 0.000 description 3
- 206010016629 fibroma Diseases 0.000 description 3
- 201000011066 hemangioma Diseases 0.000 description 3
- 125000005842 heteroatom Chemical group 0.000 description 3
- YLMAHDNUQAMNNX-UHFFFAOYSA-N imatinib methanesulfonate Chemical compound CS(O)(=O)=O.C1CN(C)CCN1CC1=CC=C(C(=O)NC=2C=C(NC=3N=C(C=CN=3)C=3C=NC=CC=3)C(C)=CC=2)C=C1 YLMAHDNUQAMNNX-UHFFFAOYSA-N 0.000 description 3
- 230000009545 invasion Effects 0.000 description 3
- LWQLLIVTGSMSCT-UHFFFAOYSA-N methyl 2-bromo-2-(4-chlorophenyl)acetate Chemical compound COC(=O)C(Br)C1=CC=C(Cl)C=C1 LWQLLIVTGSMSCT-UHFFFAOYSA-N 0.000 description 3
- 239000002480 mineral oil Substances 0.000 description 3
- 235000010446 mineral oil Nutrition 0.000 description 3
- 235000020824 obesity Nutrition 0.000 description 3
- 230000003647 oxidation Effects 0.000 description 3
- 238000007254 oxidation reaction Methods 0.000 description 3
- 239000006187 pill Substances 0.000 description 3
- 238000002953 preparative HPLC Methods 0.000 description 3
- 108090000765 processed proteins & peptides Proteins 0.000 description 3
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 3
- 125000004076 pyridyl group Chemical group 0.000 description 3
- 230000009467 reduction Effects 0.000 description 3
- 238000010992 reflux Methods 0.000 description 3
- 206010039073 rheumatoid arthritis Diseases 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 239000000377 silicon dioxide Substances 0.000 description 3
- 239000007909 solid dosage form Substances 0.000 description 3
- 230000009870 specific binding Effects 0.000 description 3
- 235000019698 starch Nutrition 0.000 description 3
- 229940124530 sulfonamide Drugs 0.000 description 3
- 150000003456 sulfonamides Chemical class 0.000 description 3
- 239000000829 suppository Substances 0.000 description 3
- 230000004083 survival effect Effects 0.000 description 3
- 239000003826 tablet Substances 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- 230000014616 translation Effects 0.000 description 3
- 229960004441 tyrosine Drugs 0.000 description 3
- 241000701447 unidentified baculovirus Species 0.000 description 3
- 238000005406 washing Methods 0.000 description 3
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 2
- PUPZLCDOIYMWBV-UHFFFAOYSA-N (+/-)-1,3-Butanediol Chemical compound CC(O)CCO PUPZLCDOIYMWBV-UHFFFAOYSA-N 0.000 description 2
- MTCFGRXMJLQNBG-REOHCLBHSA-N (2S)-2-Amino-3-hydroxypropansäure Chemical compound OC[C@H](N)C(O)=O MTCFGRXMJLQNBG-REOHCLBHSA-N 0.000 description 2
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical compound OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 description 2
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 2
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 2
- WORJRXHJTUTINR-UHFFFAOYSA-N 1,4-dioxane;hydron;chloride Chemical compound Cl.C1COCCO1 WORJRXHJTUTINR-UHFFFAOYSA-N 0.000 description 2
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 description 2
- URQJLEHTWPIESE-UHFFFAOYSA-N 3-chloro-2h-pyrazolo[4,3-d]pyrimidine Chemical compound N1=CN=C2C(Cl)=NNC2=C1 URQJLEHTWPIESE-UHFFFAOYSA-N 0.000 description 2
- QSNSCYSYFYORTR-UHFFFAOYSA-N 4-chloroaniline Chemical compound NC1=CC=C(Cl)C=C1 QSNSCYSYFYORTR-UHFFFAOYSA-N 0.000 description 2
- AVPYQKSLYISFPO-UHFFFAOYSA-N 4-chlorobenzaldehyde Chemical compound ClC1=CC=C(C=O)C=C1 AVPYQKSLYISFPO-UHFFFAOYSA-N 0.000 description 2
- 229920001817 Agar Polymers 0.000 description 2
- VVJKKWFAADXIJK-UHFFFAOYSA-N Allylamine Chemical compound NCC=C VVJKKWFAADXIJK-UHFFFAOYSA-N 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- 201000003076 Angiosarcoma Diseases 0.000 description 2
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 2
- 239000004475 Arginine Substances 0.000 description 2
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 2
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 2
- VTYYLEPIZMXCLO-UHFFFAOYSA-L Calcium carbonate Chemical compound [Ca+2].[O-]C([O-])=O VTYYLEPIZMXCLO-UHFFFAOYSA-L 0.000 description 2
- 208000024172 Cardiovascular disease Diseases 0.000 description 2
- 208000010833 Chronic myeloid leukaemia Diseases 0.000 description 2
- OKKJLVBELUTLKV-MZCSYVLQSA-N Deuterated methanol Chemical compound [2H]OC([2H])([2H])[2H] OKKJLVBELUTLKV-MZCSYVLQSA-N 0.000 description 2
- XBPCUCUWBYBCDP-UHFFFAOYSA-N Dicyclohexylamine Chemical compound C1CCCCC1NC1CCCCC1 XBPCUCUWBYBCDP-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- 241000206672 Gelidium Species 0.000 description 2
- 208000032612 Glial tumor Diseases 0.000 description 2
- 206010018338 Glioma Diseases 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 239000004471 Glycine Substances 0.000 description 2
- 108010051975 Glycogen Synthase Kinase 3 beta Proteins 0.000 description 2
- 102100038104 Glycogen synthase kinase-3 beta Human genes 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- 208000001258 Hemangiosarcoma Diseases 0.000 description 2
- WTDHULULXKLSOZ-UHFFFAOYSA-N Hydroxylamine hydrochloride Chemical compound Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- 102000004877 Insulin Human genes 0.000 description 2
- 108090001061 Insulin Proteins 0.000 description 2
- 206010022489 Insulin Resistance Diseases 0.000 description 2
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 2
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 2
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 2
- ODKSFYDXXFIFQN-BYPYZUCNSA-P L-argininium(2+) Chemical compound NC(=[NH2+])NCCC[C@H]([NH3+])C(O)=O ODKSFYDXXFIFQN-BYPYZUCNSA-P 0.000 description 2
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 2
- KDXKERNSBIXSRK-YFKPBYRVSA-N L-lysine Chemical compound NCCCC[C@H](N)C(O)=O KDXKERNSBIXSRK-YFKPBYRVSA-N 0.000 description 2
- COLNVLDHVKWLRT-QMMMGPOBSA-N L-phenylalanine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC=C1 COLNVLDHVKWLRT-QMMMGPOBSA-N 0.000 description 2
- 239000005517 L01XE01 - Imatinib Substances 0.000 description 2
- 208000018142 Leiomyosarcoma Diseases 0.000 description 2
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 2
- 239000004472 Lysine Substances 0.000 description 2
- 102000043136 MAP kinase family Human genes 0.000 description 2
- 108091054455 MAP kinase family Proteins 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 208000034578 Multiple myelomas Diseases 0.000 description 2
- 102000047918 Myelin Basic Human genes 0.000 description 2
- 101710107068 Myelin basic protein Proteins 0.000 description 2
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 2
- 102000008052 Nitric Oxide Synthase Type III Human genes 0.000 description 2
- 108010075520 Nitric Oxide Synthase Type III Proteins 0.000 description 2
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 2
- 206010033128 Ovarian cancer Diseases 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical group [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- 206010035226 Plasma cell myeloma Diseases 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 2
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 2
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical compound [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 102400001320 Transforming growth factor alpha Human genes 0.000 description 2
- 101800004564 Transforming growth factor alpha Proteins 0.000 description 2
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 2
- 108010053096 Vascular Endothelial Growth Factor Receptor-1 Proteins 0.000 description 2
- 102100033178 Vascular endothelial growth factor receptor 1 Human genes 0.000 description 2
- 208000008383 Wilms tumor Diseases 0.000 description 2
- 238000010521 absorption reaction Methods 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 235000010419 agar Nutrition 0.000 description 2
- 235000004279 alanine Nutrition 0.000 description 2
- 150000001299 aldehydes Chemical class 0.000 description 2
- 150000001370 alpha-amino acid derivatives Chemical class 0.000 description 2
- 150000001371 alpha-amino acids Chemical class 0.000 description 2
- 230000033115 angiogenesis Effects 0.000 description 2
- 238000013459 approach Methods 0.000 description 2
- 239000008346 aqueous phase Substances 0.000 description 2
- 239000007864 aqueous solution Substances 0.000 description 2
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 2
- 229960003121 arginine Drugs 0.000 description 2
- 125000004104 aryloxy group Chemical group 0.000 description 2
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical compound [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 2
- 230000001580 bacterial effect Effects 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 239000000440 bentonite Substances 0.000 description 2
- 235000012216 bentonite Nutrition 0.000 description 2
- 229910000278 bentonite Inorganic materials 0.000 description 2
- SVPXDRXYRYOSEX-UHFFFAOYSA-N bentoquatam Chemical compound O.O=[Si]=O.O=[Al]O[Al]=O SVPXDRXYRYOSEX-UHFFFAOYSA-N 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- 229960004217 benzyl alcohol Drugs 0.000 description 2
- SESFRYSPDFLNCH-UHFFFAOYSA-N benzyl benzoate Chemical compound C=1C=CC=CC=1C(=O)OCC1=CC=CC=C1 SESFRYSPDFLNCH-UHFFFAOYSA-N 0.000 description 2
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 229960001948 caffeine Drugs 0.000 description 2
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 2
- 239000011575 calcium Substances 0.000 description 2
- 229910052791 calcium Inorganic materials 0.000 description 2
- 238000004422 calculation algorithm Methods 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 208000002458 carcinoid tumor Diseases 0.000 description 2
- 230000010261 cell growth Effects 0.000 description 2
- 230000012292 cell migration Effects 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 2
- 229960001231 choline Drugs 0.000 description 2
- 238000000576 coating method Methods 0.000 description 2
- 239000012230 colorless oil Substances 0.000 description 2
- 230000000295 complement effect Effects 0.000 description 2
- 238000007796 conventional method Methods 0.000 description 2
- 230000008878 coupling Effects 0.000 description 2
- 238000010168 coupling process Methods 0.000 description 2
- 238000005859 coupling reaction Methods 0.000 description 2
- 108010024505 crosstide peptide Proteins 0.000 description 2
- 238000002425 crystallisation Methods 0.000 description 2
- 230000008025 crystallization Effects 0.000 description 2
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 2
- 230000003436 cytoskeletal effect Effects 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 230000018109 developmental process Effects 0.000 description 2
- 125000004188 dichlorophenyl group Chemical group 0.000 description 2
- 235000014113 dietary fatty acids Nutrition 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 239000003995 emulsifying agent Substances 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 239000002532 enzyme inhibitor Substances 0.000 description 2
- 229940125532 enzyme inhibitor Drugs 0.000 description 2
- 229930195729 fatty acid Natural products 0.000 description 2
- 239000000194 fatty acid Substances 0.000 description 2
- 238000003818 flash chromatography Methods 0.000 description 2
- 238000007667 floating Methods 0.000 description 2
- 125000000524 functional group Chemical group 0.000 description 2
- 230000002496 gastric effect Effects 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 229920000159 gelatin Polymers 0.000 description 2
- 235000019322 gelatine Nutrition 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 230000014509 gene expression Effects 0.000 description 2
- 229940080856 gleevec Drugs 0.000 description 2
- KWIUHFFTVRNATP-UHFFFAOYSA-N glycine betaine Chemical compound C[N+](C)(C)CC([O-])=O KWIUHFFTVRNATP-UHFFFAOYSA-N 0.000 description 2
- 239000003102 growth factor Substances 0.000 description 2
- 230000036541 health Effects 0.000 description 2
- 206010073071 hepatocellular carcinoma Diseases 0.000 description 2
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 2
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 2
- 229960002885 histidine Drugs 0.000 description 2
- 230000007062 hydrolysis Effects 0.000 description 2
- 238000006460 hydrolysis reaction Methods 0.000 description 2
- 208000026278 immune system disease Diseases 0.000 description 2
- 230000006872 improvement Effects 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- PQNFLJBBNBOBRQ-UHFFFAOYSA-N indane Chemical compound C1=CC=C2CCCC2=C1 PQNFLJBBNBOBRQ-UHFFFAOYSA-N 0.000 description 2
- 208000027866 inflammatory disease Diseases 0.000 description 2
- 229940125396 insulin Drugs 0.000 description 2
- 230000003993 interaction Effects 0.000 description 2
- 230000003834 intracellular effect Effects 0.000 description 2
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 2
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 2
- JJWLVOIRVHMVIS-UHFFFAOYSA-N isopropylamine Chemical compound CC(C)N JJWLVOIRVHMVIS-UHFFFAOYSA-N 0.000 description 2
- 201000010260 leiomyoma Diseases 0.000 description 2
- 150000002632 lipids Chemical class 0.000 description 2
- 239000008297 liquid dosage form Substances 0.000 description 2
- 210000004185 liver Anatomy 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 235000018977 lysine Nutrition 0.000 description 2
- 208000002780 macular degeneration Diseases 0.000 description 2
- 229910001629 magnesium chloride Inorganic materials 0.000 description 2
- 235000019359 magnesium stearate Nutrition 0.000 description 2
- 238000004519 manufacturing process Methods 0.000 description 2
- 230000001404 mediated effect Effects 0.000 description 2
- 201000001441 melanoma Diseases 0.000 description 2
- 206010027191 meningioma Diseases 0.000 description 2
- 229930014626 natural product Natural products 0.000 description 2
- 208000015122 neurodegenerative disease Diseases 0.000 description 2
- 125000004433 nitrogen atom Chemical group N* 0.000 description 2
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 2
- 239000004006 olive oil Substances 0.000 description 2
- 235000008390 olive oil Nutrition 0.000 description 2
- 230000000771 oncological effect Effects 0.000 description 2
- 201000008968 osteosarcoma Diseases 0.000 description 2
- 230000002018 overexpression Effects 0.000 description 2
- 150000002923 oximes Chemical class 0.000 description 2
- 239000001301 oxygen Substances 0.000 description 2
- 125000003854 p-chlorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1Cl 0.000 description 2
- 239000002245 particle Substances 0.000 description 2
- 230000001575 pathological effect Effects 0.000 description 2
- 229940124531 pharmaceutical excipient Drugs 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- COLNVLDHVKWLRT-UHFFFAOYSA-N phenylalanine Natural products OC(=O)C(N)CC1=CC=CC=C1 COLNVLDHVKWLRT-UHFFFAOYSA-N 0.000 description 2
- 125000000843 phenylene group Chemical group C1(=C(C=CC=C1)*)* 0.000 description 2
- 239000011574 phosphorus Chemical group 0.000 description 2
- 229910052698 phosphorus Inorganic materials 0.000 description 2
- 125000000587 piperidin-1-yl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 2
- OHEDMAIVEXQCOZ-UHFFFAOYSA-N piperidine;dihydrochloride Chemical compound Cl.Cl.C1CCNCC1 OHEDMAIVEXQCOZ-UHFFFAOYSA-N 0.000 description 2
- 229940068917 polyethylene glycols Drugs 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- WGYKZJWCGVVSQN-UHFFFAOYSA-N propylamine Chemical compound CCCN WGYKZJWCGVVSQN-UHFFFAOYSA-N 0.000 description 2
- 229960004063 propylene glycol Drugs 0.000 description 2
- 235000013772 propylene glycol Nutrition 0.000 description 2
- 150000003212 purines Chemical class 0.000 description 2
- 125000005551 pyridylene group Chemical group 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 238000007423 screening assay Methods 0.000 description 2
- 229940126586 small molecule drug Drugs 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 229940032147 starch Drugs 0.000 description 2
- 238000005556 structure-activity relationship Methods 0.000 description 2
- 239000000758 substrate Substances 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 description 2
- 150000003462 sulfoxides Chemical class 0.000 description 2
- 239000011593 sulfur Substances 0.000 description 2
- 125000004434 sulfur atom Chemical group 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- FCMLWBBLOASUSO-UHFFFAOYSA-N tert-butyl 3-oxopiperazine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCNC(=O)C1 FCMLWBBLOASUSO-UHFFFAOYSA-N 0.000 description 2
- UGKNVKLWKSPWPH-UHFFFAOYSA-N tert-butyl 4-(4-chlorobenzoyl)piperidine-1-carboxylate Chemical compound C1CN(C(=O)OC(C)(C)C)CCC1C(=O)C1=CC=C(Cl)C=C1 UGKNVKLWKSPWPH-UHFFFAOYSA-N 0.000 description 2
- VZGDMQKNWNREIO-UHFFFAOYSA-N tetrachloromethane Chemical compound ClC(Cl)(Cl)Cl VZGDMQKNWNREIO-UHFFFAOYSA-N 0.000 description 2
- YAPQBXQYLJRXSA-UHFFFAOYSA-N theobromine Chemical compound CN1C(=O)NC(=O)C2=C1N=CN2C YAPQBXQYLJRXSA-UHFFFAOYSA-N 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- 125000003396 thiol group Chemical group [H]S* 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 238000013519 translation Methods 0.000 description 2
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 2
- OUYCCCASQSFEME-UHFFFAOYSA-N tyrosine Natural products OC(=O)C(N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-UHFFFAOYSA-N 0.000 description 2
- 229910052720 vanadium Inorganic materials 0.000 description 2
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 2
- 239000001993 wax Substances 0.000 description 2
- 238000009736 wetting Methods 0.000 description 2
- 239000000080 wetting agent Substances 0.000 description 2
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- CCENBZDJMALWCH-UHFFFAOYSA-N (4-chlorophenyl) carbamate Chemical compound NC(=O)OC1=CC=C(Cl)C=C1 CCENBZDJMALWCH-UHFFFAOYSA-N 0.000 description 1
- SRWFWQFRAOFHNK-UHFFFAOYSA-N (4-fluorophenyl)methanone Chemical compound FC1=CC=C([C+]=O)C=C1 SRWFWQFRAOFHNK-UHFFFAOYSA-N 0.000 description 1
- ICLYJLBTOGPLMC-KVVVOXFISA-N (z)-octadec-9-enoate;tris(2-hydroxyethyl)azanium Chemical compound OCCN(CCO)CCO.CCCCCCCC\C=C/CCCCCCCC(O)=O ICLYJLBTOGPLMC-KVVVOXFISA-N 0.000 description 1
- UKAUYVFTDYCKQA-UHFFFAOYSA-N -2-Amino-4-hydroxybutanoic acid Natural products OC(=O)C(N)CCO UKAUYVFTDYCKQA-UHFFFAOYSA-N 0.000 description 1
- WBYWAXJHAXSJNI-VOTSOKGWSA-M .beta-Phenylacrylic acid Natural products [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 1
- 229940058015 1,3-butylene glycol Drugs 0.000 description 1
- CDCMLAUCQJCJOQ-UHFFFAOYSA-N 1-(3-bromo-2h-pyrazolo[3,4-d]pyrimidin-4-yl)-n-(4-chlorophenyl)piperidin-4-amine Chemical compound C1=CC(Cl)=CC=C1NC1CCN(C=2C=3C(Br)=NNC=3N=CN=2)CC1 CDCMLAUCQJCJOQ-UHFFFAOYSA-N 0.000 description 1
- VXNZUUAINFGPBY-UHFFFAOYSA-N 1-Butene Chemical group CCC=C VXNZUUAINFGPBY-UHFFFAOYSA-N 0.000 description 1
- BCMYXYHEMGPZJN-UHFFFAOYSA-N 1-chloro-2-isocyanatoethane Chemical compound ClCCN=C=O BCMYXYHEMGPZJN-UHFFFAOYSA-N 0.000 description 1
- JQZAEUFPPSRDOP-UHFFFAOYSA-N 1-chloro-4-(chloromethyl)benzene Chemical compound ClCC1=CC=C(Cl)C=C1 JQZAEUFPPSRDOP-UHFFFAOYSA-N 0.000 description 1
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 1
- 125000004343 1-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])(*)C([H])([H])[H] 0.000 description 1
- WRMZOPANDOHWJU-UHFFFAOYSA-N 1h-indol-6-ylmethanol Chemical compound OCC1=CC=C2C=CNC2=C1 WRMZOPANDOHWJU-UHFFFAOYSA-N 0.000 description 1
- QUKPALAWEPMWOS-UHFFFAOYSA-N 1h-pyrazolo[3,4-d]pyrimidine Chemical compound C1=NC=C2C=NNC2=N1 QUKPALAWEPMWOS-UHFFFAOYSA-N 0.000 description 1
- UZURTQHPMXADGG-UHFFFAOYSA-N 2,3,3a,4,7,7a-hexahydro-1h-indene Chemical compound C1C=CCC2CCCC21 UZURTQHPMXADGG-UHFFFAOYSA-N 0.000 description 1
- PHXGAJLBHUUAKB-UHFFFAOYSA-N 2,3,3a,5,6,6a-hexahydrofuro[3,2-b]furan Chemical compound O1CCC2OCCC21 PHXGAJLBHUUAKB-UHFFFAOYSA-N 0.000 description 1
- FWUJIDULGWIBPD-UHFFFAOYSA-N 2-(1-phenylhex-5-en-1-yn-3-yl)naphthalene Chemical compound C=1C=C2C=CC=CC2=CC=1C(CC=C)C#CC1=CC=CC=C1 FWUJIDULGWIBPD-UHFFFAOYSA-N 0.000 description 1
- HFNXJOIRWCQTIZ-UHFFFAOYSA-N 2-(4-chlorophenyl)-n-[2-(dimethylamino)ethyl]acetamide Chemical compound CN(C)CCNC(=O)CC1=CC=C(Cl)C=C1 HFNXJOIRWCQTIZ-UHFFFAOYSA-N 0.000 description 1
- XEMIIPOLFGIISJ-UHFFFAOYSA-N 2-(dimethylamino)ethyl n-[1-(3-bromo-2h-pyrazolo[3,4-d]pyrimidin-4-yl)piperidin-4-yl]-n-(4-chlorophenyl)carbamate Chemical compound C1CN(C=2C=3C(Br)=NNC=3N=CN=2)CCC1N(C(=O)OCCN(C)C)C1=CC=C(Cl)C=C1 XEMIIPOLFGIISJ-UHFFFAOYSA-N 0.000 description 1
- MFYSUUPKMDJYPF-UHFFFAOYSA-N 2-[(4-methyl-2-nitrophenyl)diazenyl]-3-oxo-n-phenylbutanamide Chemical compound C=1C=CC=CC=1NC(=O)C(C(=O)C)N=NC1=CC=C(C)C=C1[N+]([O-])=O MFYSUUPKMDJYPF-UHFFFAOYSA-N 0.000 description 1
- GTEXIOINCJRBIO-UHFFFAOYSA-N 2-[2-(dimethylamino)ethoxy]-n,n-dimethylethanamine Chemical compound CN(C)CCOCCN(C)C GTEXIOINCJRBIO-UHFFFAOYSA-N 0.000 description 1
- QUFXMTKDHLGQAE-UHFFFAOYSA-N 2-[2-(dimethylamino)ethoxy]-n,n-dimethylethanamine;dihydrochloride Chemical compound Cl.Cl.CN(C)CCOCCN(C)C QUFXMTKDHLGQAE-UHFFFAOYSA-N 0.000 description 1
- MSWZFWKMSRAUBD-IVMDWMLBSA-N 2-amino-2-deoxy-D-glucopyranose Chemical compound N[C@H]1C(O)O[C@H](CO)[C@@H](O)[C@@H]1O MSWZFWKMSRAUBD-IVMDWMLBSA-N 0.000 description 1
- 125000000022 2-aminoethyl group Chemical group [H]C([*])([H])C([H])([H])N([H])[H] 0.000 description 1
- BFSVOASYOCHEOV-UHFFFAOYSA-N 2-diethylaminoethanol Chemical compound CCN(CC)CCO BFSVOASYOCHEOV-UHFFFAOYSA-N 0.000 description 1
- 229940013085 2-diethylaminoethanol Drugs 0.000 description 1
- LQLJZSJKRYTKTP-UHFFFAOYSA-N 2-dimethylaminoethyl chloride hydrochloride Chemical compound Cl.CN(C)CCCl LQLJZSJKRYTKTP-UHFFFAOYSA-N 0.000 description 1
- WRFPVMFCRNYQNR-UHFFFAOYSA-N 2-hydroxyphenylalanine Chemical compound OC(=O)C(N)CC1=CC=CC=C1O WRFPVMFCRNYQNR-UHFFFAOYSA-N 0.000 description 1
- BKOOMYPCSUNDGP-UHFFFAOYSA-N 2-methylbut-2-ene Chemical group CC=C(C)C BKOOMYPCSUNDGP-UHFFFAOYSA-N 0.000 description 1
- 125000006088 2-oxoazepinyl group Chemical group 0.000 description 1
- 125000004638 2-oxopiperazinyl group Chemical group O=C1N(CCNC1)* 0.000 description 1
- 125000004637 2-oxopiperidinyl group Chemical group O=C1N(CCCC1)* 0.000 description 1
- 125000000094 2-phenylethyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000003903 2-propenyl group Chemical group [H]C([*])([H])C([H])=C([H])[H] 0.000 description 1
- DVGKRPYUFRZAQW-UHFFFAOYSA-N 3 prime Natural products CC(=O)NC1OC(CC(O)C1C(O)C(O)CO)(OC2C(O)C(CO)OC(OC3C(O)C(O)C(O)OC3CO)C2O)C(=O)O DVGKRPYUFRZAQW-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- OHDYDXVLQKCZRK-UHFFFAOYSA-N 3-bromo-2h-pyrazolo[3,4-d]pyrimidine Chemical compound C1=NC=C2C(Br)=NNC2=N1 OHDYDXVLQKCZRK-UHFFFAOYSA-N 0.000 description 1
- FXIUWVNFYZNXCK-UHFFFAOYSA-N 3-bromo-4-[4-(1-phenylethyl)piperazin-1-yl]-2h-pyrazolo[3,4-d]pyrimidine Chemical compound C1CN(C=2C=3C(Br)=NNC=3N=CN=2)CCN1C(C)C1=CC=CC=C1 FXIUWVNFYZNXCK-UHFFFAOYSA-N 0.000 description 1
- WVUULNDRFBHTFG-UHFFFAOYSA-N 3-chloro-n,n-diethylpropan-1-amine Chemical compound CCN(CC)CCCCl WVUULNDRFBHTFG-UHFFFAOYSA-N 0.000 description 1
- JXJSQQWPUMCYGJ-UHFFFAOYSA-N 4-(3-bromo-2h-pyrazolo[3,4-d]pyrimidin-4-yl)-1-[(4-chlorophenyl)methyl]piperazin-2-one Chemical compound C1=CC(Cl)=CC=C1CN1C(=O)CN(C=2C=3C(Br)=NNC=3N=CN=2)CC1 JXJSQQWPUMCYGJ-UHFFFAOYSA-N 0.000 description 1
- 125000001255 4-fluorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1F 0.000 description 1
- 125000004203 4-hydroxyphenyl group Chemical group [H]OC1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- 125000005986 4-piperidonyl group Chemical group 0.000 description 1
- XZIIFPSPUDAGJM-UHFFFAOYSA-N 6-chloro-2-n,2-n-diethylpyrimidine-2,4-diamine Chemical compound CCN(CC)C1=NC(N)=CC(Cl)=N1 XZIIFPSPUDAGJM-UHFFFAOYSA-N 0.000 description 1
- SNZSSCZJMVIOCR-UHFFFAOYSA-N 7-azabicyclo[2.2.1]heptane Chemical compound C1CC2CCC1N2 SNZSSCZJMVIOCR-UHFFFAOYSA-N 0.000 description 1
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 description 1
- 244000215068 Acacia senegal Species 0.000 description 1
- 235000006491 Acacia senegal Nutrition 0.000 description 1
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 1
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 1
- 206010001233 Adenoma benign Diseases 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 102000007299 Amphiregulin Human genes 0.000 description 1
- 108010033760 Amphiregulin Proteins 0.000 description 1
- 101100234530 Arabidopsis thaliana KIN14M gene Proteins 0.000 description 1
- 101100463130 Arabidopsis thaliana PDK gene Proteins 0.000 description 1
- 229930091051 Arenine Natural products 0.000 description 1
- DCXYFEDJOCDNAF-UHFFFAOYSA-N Asparagine Natural products OC(=O)C(N)CC(N)=O DCXYFEDJOCDNAF-UHFFFAOYSA-N 0.000 description 1
- 241000416162 Astragalus gummifer Species 0.000 description 1
- 206010003571 Astrocytoma Diseases 0.000 description 1
- 208000010839 B-cell chronic lymphocytic leukemia Diseases 0.000 description 1
- 210000002237 B-cell of pancreatic islet Anatomy 0.000 description 1
- 206010004146 Basal cell carcinoma Diseases 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 102400001242 Betacellulin Human genes 0.000 description 1
- 101800001382 Betacellulin Proteins 0.000 description 1
- 206010073106 Bone giant cell tumour malignant Diseases 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- 208000026310 Breast neoplasm Diseases 0.000 description 1
- 102000038625 CMGCs Human genes 0.000 description 1
- 108091007913 CMGCs Proteins 0.000 description 1
- AVFPOWPKUSYREA-NSHDSACASA-N C[C@@H](C1)N(Cc(cc2)ccc2Cl)CCN1c1c2c(Br)n[nH]c2ncn1 Chemical compound C[C@@H](C1)N(Cc(cc2)ccc2Cl)CCN1c1c2c(Br)n[nH]c2ncn1 AVFPOWPKUSYREA-NSHDSACASA-N 0.000 description 1
- 102100022789 Calcium/calmodulin-dependent protein kinase type IV Human genes 0.000 description 1
- 208000005623 Carcinogenesis Diseases 0.000 description 1
- 208000009458 Carcinoma in Situ Diseases 0.000 description 1
- 206010008263 Cervical dysplasia Diseases 0.000 description 1
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical group [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 description 1
- 201000005262 Chondroma Diseases 0.000 description 1
- 208000005243 Chondrosarcoma Diseases 0.000 description 1
- 201000009047 Chordoma Diseases 0.000 description 1
- 208000006332 Choriocarcinoma Diseases 0.000 description 1
- WBYWAXJHAXSJNI-SREVYHEPSA-N Cinnamic acid Chemical compound OC(=O)\C=C/C1=CC=CC=C1 WBYWAXJHAXSJNI-SREVYHEPSA-N 0.000 description 1
- YDGDNGZCULEIIU-UHFFFAOYSA-N Clc(cc1)ccc1OC(CC1)CCN1c1c2c(Br)n[nH]c2ncn1 Chemical compound Clc(cc1)ccc1OC(CC1)CCN1c1c2c(Br)n[nH]c2ncn1 YDGDNGZCULEIIU-UHFFFAOYSA-N 0.000 description 1
- 206010048832 Colon adenoma Diseases 0.000 description 1
- 206010010356 Congenital anomaly Diseases 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 229920002785 Croscarmellose sodium Polymers 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 235000019739 Dicalciumphosphate Nutrition 0.000 description 1
- LTMHDMANZUZIPE-AMTYYWEZSA-N Digoxin Natural products O([C@H]1[C@H](C)O[C@H](O[C@@H]2C[C@@H]3[C@@](C)([C@@H]4[C@H]([C@]5(O)[C@](C)([C@H](O)C4)[C@H](C4=CC(=O)OC4)CC5)CC3)CC2)C[C@@H]1O)[C@H]1O[C@H](C)[C@@H](O[C@H]2O[C@@H](C)[C@H](O)[C@@H](O)C2)[C@@H](O)C1 LTMHDMANZUZIPE-AMTYYWEZSA-N 0.000 description 1
- ROSDSFDQCJNGOL-UHFFFAOYSA-N Dimethylamine Chemical compound CNC ROSDSFDQCJNGOL-UHFFFAOYSA-N 0.000 description 1
- 206010061818 Disease progression Diseases 0.000 description 1
- 101100291385 Drosophila melanogaster p38a gene Proteins 0.000 description 1
- 206010013710 Drug interaction Diseases 0.000 description 1
- 208000007033 Dysgerminoma Diseases 0.000 description 1
- 208000000471 Dysplastic Nevus Syndrome Diseases 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 201000009051 Embryonal Carcinoma Diseases 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 206010014733 Endometrial cancer Diseases 0.000 description 1
- 206010014759 Endometrial neoplasm Diseases 0.000 description 1
- 206010014967 Ependymoma Diseases 0.000 description 1
- 102000009024 Epidermal Growth Factor Human genes 0.000 description 1
- VGGSQFUCUMXWEO-UHFFFAOYSA-N Ethene Chemical compound C=C VGGSQFUCUMXWEO-UHFFFAOYSA-N 0.000 description 1
- 239000005977 Ethylene Substances 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- 208000006168 Ewing Sarcoma Diseases 0.000 description 1
- 208000007659 Fibroadenoma Diseases 0.000 description 1
- 206010053717 Fibrous histiocytoma Diseases 0.000 description 1
- 239000004606 Fillers/Extenders Substances 0.000 description 1
- 208000000527 Germinoma Diseases 0.000 description 1
- 208000007569 Giant Cell Tumors Diseases 0.000 description 1
- 201000005409 Gliomatosis cerebri Diseases 0.000 description 1
- 206010018404 Glucagonoma Diseases 0.000 description 1
- WHUUTDBJXJRKMK-UHFFFAOYSA-N Glutamic acid Natural products OC(=O)C(N)CCC(O)=O WHUUTDBJXJRKMK-UHFFFAOYSA-N 0.000 description 1
- 206010018691 Granuloma Diseases 0.000 description 1
- 108010009202 Growth Factor Receptors Proteins 0.000 description 1
- 102000009465 Growth Factor Receptors Human genes 0.000 description 1
- 229920000084 Gum arabic Polymers 0.000 description 1
- 208000002927 Hamartoma Diseases 0.000 description 1
- 101800001649 Heparin-binding EGF-like growth factor Proteins 0.000 description 1
- 102400001369 Heparin-binding EGF-like growth factor Human genes 0.000 description 1
- 206010019629 Hepatic adenoma Diseases 0.000 description 1
- 102100021866 Hepatocyte growth factor Human genes 0.000 description 1
- 208000017604 Hodgkin disease Diseases 0.000 description 1
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101100287682 Homo sapiens CAMK2G gene Proteins 0.000 description 1
- 101100126883 Homo sapiens CAMK4 gene Proteins 0.000 description 1
- 101000898034 Homo sapiens Hepatocyte growth factor Proteins 0.000 description 1
- 101001076408 Homo sapiens Interleukin-6 Proteins 0.000 description 1
- 101000798007 Homo sapiens RAC-gamma serine/threonine-protein kinase Proteins 0.000 description 1
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 1
- 101000868152 Homo sapiens Son of sevenless homolog 1 Proteins 0.000 description 1
- PMMYEEVYMWASQN-DMTCNVIQSA-N Hydroxyproline Chemical compound O[C@H]1CN[C@H](C(O)=O)C1 PMMYEEVYMWASQN-DMTCNVIQSA-N 0.000 description 1
- 208000035150 Hypercholesterolemia Diseases 0.000 description 1
- 208000031226 Hyperlipidaemia Diseases 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 108090000723 Insulin-Like Growth Factor I Proteins 0.000 description 1
- 102000004218 Insulin-Like Growth Factor I Human genes 0.000 description 1
- 208000005045 Interdigitating dendritic cell sarcoma Diseases 0.000 description 1
- 108010044467 Isoenzymes Proteins 0.000 description 1
- 208000002260 Keloid Diseases 0.000 description 1
- SNDPXSYFESPGGJ-BYPYZUCNSA-N L-2-aminopentanoic acid Chemical compound CCC[C@H](N)C(O)=O SNDPXSYFESPGGJ-BYPYZUCNSA-N 0.000 description 1
- XUJNEKJLAYXESH-REOHCLBHSA-N L-Cysteine Chemical compound SC[C@H](N)C(O)=O XUJNEKJLAYXESH-REOHCLBHSA-N 0.000 description 1
- AHLPHDHHMVZTML-BYPYZUCNSA-N L-Ornithine Chemical compound NCCC[C@H](N)C(O)=O AHLPHDHHMVZTML-BYPYZUCNSA-N 0.000 description 1
- ONIBWKKTOPOVIA-BYPYZUCNSA-N L-Proline Chemical compound OC(=O)[C@@H]1CCCN1 ONIBWKKTOPOVIA-BYPYZUCNSA-N 0.000 description 1
- ZGUNAGUHMKGQNY-ZETCQYMHSA-N L-alpha-phenylglycine zwitterion Chemical compound OC(=O)[C@@H](N)C1=CC=CC=C1 ZGUNAGUHMKGQNY-ZETCQYMHSA-N 0.000 description 1
- DCXYFEDJOCDNAF-REOHCLBHSA-N L-asparagine Chemical compound OC(=O)[C@@H](N)CC(N)=O DCXYFEDJOCDNAF-REOHCLBHSA-N 0.000 description 1
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical compound OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- ZDXPYRJPNDTMRX-VKHMYHEASA-N L-glutamine Chemical compound OC(=O)[C@@H](N)CCC(N)=O ZDXPYRJPNDTMRX-VKHMYHEASA-N 0.000 description 1
- FFFHZYDWPBMWHY-VKHMYHEASA-N L-homocysteine Chemical compound OC(=O)[C@@H](N)CCS FFFHZYDWPBMWHY-VKHMYHEASA-N 0.000 description 1
- JTTHKOPSMAVJFE-VIFPVBQESA-N L-homophenylalanine Chemical compound OC(=O)[C@@H](N)CCC1=CC=CC=C1 JTTHKOPSMAVJFE-VIFPVBQESA-N 0.000 description 1
- UKAUYVFTDYCKQA-VKHMYHEASA-N L-homoserine Chemical compound OC(=O)[C@@H](N)CCO UKAUYVFTDYCKQA-VKHMYHEASA-N 0.000 description 1
- AGPKZVBTJJNPAG-WHFBIAKZSA-N L-isoleucine Chemical compound CC[C@H](C)[C@H](N)C(O)=O AGPKZVBTJJNPAG-WHFBIAKZSA-N 0.000 description 1
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 1
- JZKXXXDKRQWDET-QMMMGPOBSA-N L-m-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=CC(O)=C1 JZKXXXDKRQWDET-QMMMGPOBSA-N 0.000 description 1
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 1
- SNDPXSYFESPGGJ-UHFFFAOYSA-N L-norVal-OH Natural products CCCC(N)C(O)=O SNDPXSYFESPGGJ-UHFFFAOYSA-N 0.000 description 1
- LRQKBLKVPFOOQJ-YFKPBYRVSA-N L-norleucine Chemical compound CCCC[C@H]([NH3+])C([O-])=O LRQKBLKVPFOOQJ-YFKPBYRVSA-N 0.000 description 1
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 1
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 1
- OUYCCCASQSFEME-QMMMGPOBSA-N L-tyrosine Chemical compound OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 OUYCCCASQSFEME-QMMMGPOBSA-N 0.000 description 1
- KZSNJWFQEVHDMF-BYPYZUCNSA-N L-valine Chemical compound CC(C)[C@H](N)C(O)=O KZSNJWFQEVHDMF-BYPYZUCNSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 208000002404 Liver Cell Adenoma Diseases 0.000 description 1
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 208000006644 Malignant Fibrous Histiocytoma Diseases 0.000 description 1
- 229910021380 Manganese Chloride Inorganic materials 0.000 description 1
- GLFNIEUTAYBVOC-UHFFFAOYSA-L Manganese chloride Chemical compound Cl[Mn]Cl GLFNIEUTAYBVOC-UHFFFAOYSA-L 0.000 description 1
- 240000003183 Manihot esculenta Species 0.000 description 1
- 235000016735 Manihot esculenta subsp esculenta Nutrition 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- 208000000172 Medulloblastoma Diseases 0.000 description 1
- 206010027406 Mesothelioma Diseases 0.000 description 1
- 208000001145 Metabolic Syndrome Diseases 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 101100268066 Mus musculus Zap70 gene Proteins 0.000 description 1
- 201000003793 Myelodysplastic syndrome Diseases 0.000 description 1
- 208000014767 Myeloproliferative disease Diseases 0.000 description 1
- UEEJHVSXFDXPFK-UHFFFAOYSA-N N-dimethylaminoethanol Chemical compound CN(C)CCO UEEJHVSXFDXPFK-UHFFFAOYSA-N 0.000 description 1
- HTLZVHNRZJPSMI-UHFFFAOYSA-N N-ethylpiperidine Chemical compound CCN1CCCCC1 HTLZVHNRZJPSMI-UHFFFAOYSA-N 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 125000000815 N-oxide group Chemical group 0.000 description 1
- 229910020889 NaBH3 Inorganic materials 0.000 description 1
- 102400000058 Neuregulin-1 Human genes 0.000 description 1
- 108090000556 Neuregulin-1 Proteins 0.000 description 1
- 206010029260 Neuroblastoma Diseases 0.000 description 1
- 201000004404 Neurofibroma Diseases 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 101710163270 Nuclease Proteins 0.000 description 1
- MDDFOASTOUKOHZ-UHFFFAOYSA-N O=C(N(C(CC1)CCN1c1c2c(Br)n[nH]c2ncn1)c(cc1)ccc1Cl)OCCI Chemical compound O=C(N(C(CC1)CCN1c1c2c(Br)n[nH]c2ncn1)c(cc1)ccc1Cl)OCCI MDDFOASTOUKOHZ-UHFFFAOYSA-N 0.000 description 1
- 208000022873 Ocular disease Diseases 0.000 description 1
- 201000010133 Oligodendroglioma Diseases 0.000 description 1
- 108091034117 Oligonucleotide Proteins 0.000 description 1
- 108010038807 Oligopeptides Proteins 0.000 description 1
- 102000015636 Oligopeptides Human genes 0.000 description 1
- AHLPHDHHMVZTML-UHFFFAOYSA-N Orn-delta-NH2 Natural products NCCCC(N)C(O)=O AHLPHDHHMVZTML-UHFFFAOYSA-N 0.000 description 1
- UTJLXEIPEHZYQJ-UHFFFAOYSA-N Ornithine Natural products OC(=O)C(C)CCCN UTJLXEIPEHZYQJ-UHFFFAOYSA-N 0.000 description 1
- 206010031149 Osteitis Diseases 0.000 description 1
- 208000000035 Osteochondroma Diseases 0.000 description 1
- 206010061535 Ovarian neoplasm Diseases 0.000 description 1
- YIKSCQDJHCMVMK-UHFFFAOYSA-N Oxamide Chemical compound NC(=O)C(N)=O YIKSCQDJHCMVMK-UHFFFAOYSA-N 0.000 description 1
- 206010061902 Pancreatic neoplasm Diseases 0.000 description 1
- 208000034038 Pathologic Neovascularization Diseases 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 241001307210 Pene Species 0.000 description 1
- 108010067902 Peptide Library Proteins 0.000 description 1
- 208000007641 Pinealoma Diseases 0.000 description 1
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 1
- 208000006664 Precursor Cell Lymphoblastic Leukemia-Lymphoma Diseases 0.000 description 1
- 101710098940 Pro-epidermal growth factor Proteins 0.000 description 1
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 108010014608 Proto-Oncogene Proteins c-kit Proteins 0.000 description 1
- 102000016971 Proto-Oncogene Proteins c-kit Human genes 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- 102100032314 RAC-gamma serine/threonine-protein kinase Human genes 0.000 description 1
- 108091005682 Receptor kinases Proteins 0.000 description 1
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 1
- 102100029986 Receptor tyrosine-protein kinase erbB-3 Human genes 0.000 description 1
- 101710100969 Receptor tyrosine-protein kinase erbB-3 Proteins 0.000 description 1
- 102100029981 Receptor tyrosine-protein kinase erbB-4 Human genes 0.000 description 1
- 101710100963 Receptor tyrosine-protein kinase erbB-4 Proteins 0.000 description 1
- 206010038389 Renal cancer Diseases 0.000 description 1
- 208000006265 Renal cell carcinoma Diseases 0.000 description 1
- 201000000582 Retinoblastoma Diseases 0.000 description 1
- 208000005678 Rhabdomyoma Diseases 0.000 description 1
- 102000002278 Ribosomal Proteins Human genes 0.000 description 1
- 108010000605 Ribosomal Proteins Proteins 0.000 description 1
- 201000010208 Seminoma Diseases 0.000 description 1
- 208000000097 Sertoli-Leydig cell tumor Diseases 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 235000002595 Solanum tuberosum Nutrition 0.000 description 1
- 244000061456 Solanum tuberosum Species 0.000 description 1
- SSZBUIDZHHWXNJ-UHFFFAOYSA-N Stearinsaeure-hexadecylester Natural products CCCCCCCCCCCCCCCCCC(=O)OCCCCCCCCCCCCCCCC SSZBUIDZHHWXNJ-UHFFFAOYSA-N 0.000 description 1
- 108010090804 Streptavidin Proteins 0.000 description 1
- 208000006011 Stroke Diseases 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- UCKMPCXJQFINFW-UHFFFAOYSA-N Sulphide Chemical compound [S-2] UCKMPCXJQFINFW-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- CBPNZQVSJQDFBE-FUXHJELOSA-N Temsirolimus Chemical compound C1C[C@@H](OC(=O)C(C)(CO)CO)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 CBPNZQVSJQDFBE-FUXHJELOSA-N 0.000 description 1
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 1
- 239000004473 Threonine Substances 0.000 description 1
- 208000024770 Thyroid neoplasm Diseases 0.000 description 1
- 229910010062 TiCl3 Inorganic materials 0.000 description 1
- 229920001615 Tragacanth Polymers 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 1
- 208000015778 Undifferentiated pleomorphic sarcoma Diseases 0.000 description 1
- 208000009311 VIPoma Diseases 0.000 description 1
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Natural products CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 1
- 206010048214 Xanthoma Diseases 0.000 description 1
- 206010048215 Xanthomatosis Diseases 0.000 description 1
- 240000008042 Zea mays Species 0.000 description 1
- 235000005824 Zea mays ssp. parviglumis Nutrition 0.000 description 1
- 235000002017 Zea mays subsp mays Nutrition 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- 239000001089 [(2R)-oxolan-2-yl]methanol Substances 0.000 description 1
- SUMHHCMWZBLBCC-UHFFFAOYSA-N [1-(3-bromo-2h-pyrazolo[3,4-d]pyrimidin-4-yl)piperidin-4-yl]-(4-fluorophenyl)methanol Chemical compound C1CN(C=2C=3C(Br)=NNC=3N=CN=2)CCC1C(O)C1=CC=C(F)C=C1 SUMHHCMWZBLBCC-UHFFFAOYSA-N 0.000 description 1
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 1
- 201000000690 abdominal obesity-metabolic syndrome Diseases 0.000 description 1
- 239000003655 absorption accelerator Substances 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 125000000641 acridinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3C=C12)* 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 239000012042 active reagent Substances 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 125000004442 acylamino group Chemical group 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 125000004423 acyloxy group Chemical group 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- 208000002718 adenomatoid tumor Diseases 0.000 description 1
- 230000001919 adrenal effect Effects 0.000 description 1
- 239000003463 adsorbent Substances 0.000 description 1
- 239000000443 aerosol Substances 0.000 description 1
- 206010064930 age-related macular degeneration Diseases 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 150000001336 alkenes Chemical class 0.000 description 1
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 1
- 125000005907 alkyl ester group Chemical group 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- 125000004644 alkyl sulfinyl group Chemical group 0.000 description 1
- 125000004390 alkyl sulfonyl group Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- BHELZAPQIKSEDF-UHFFFAOYSA-N allyl bromide Chemical compound BrCC=C BHELZAPQIKSEDF-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 229940024606 amino acid Drugs 0.000 description 1
- 235000001014 amino acid Nutrition 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 239000003242 anti bacterial agent Substances 0.000 description 1
- 238000011122 anti-angiogenic therapy Methods 0.000 description 1
- 230000002424 anti-apoptotic effect Effects 0.000 description 1
- 230000009949 anti-apoptotic pathway Effects 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 230000001093 anti-cancer Effects 0.000 description 1
- 230000001028 anti-proliverative effect Effects 0.000 description 1
- 239000003429 antifungal agent Substances 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000004599 antimicrobial Substances 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 238000007080 aromatic substitution reaction Methods 0.000 description 1
- 125000002102 aryl alkyloxo group Chemical group 0.000 description 1
- 150000005840 aryl radicals Chemical class 0.000 description 1
- 125000000732 arylene group Chemical group 0.000 description 1
- 235000009582 asparagine Nutrition 0.000 description 1
- 229960001230 asparagine Drugs 0.000 description 1
- 235000003704 aspartic acid Nutrition 0.000 description 1
- 238000002820 assay format Methods 0.000 description 1
- 238000011914 asymmetric synthesis Methods 0.000 description 1
- 125000002785 azepinyl group Chemical group 0.000 description 1
- 125000002393 azetidinyl group Chemical group 0.000 description 1
- 208000001119 benign fibrous histiocytoma Diseases 0.000 description 1
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 description 1
- 125000002047 benzodioxolyl group Chemical group O1OC(C2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- 235000019445 benzyl alcohol Nutrition 0.000 description 1
- 229960002903 benzyl benzoate Drugs 0.000 description 1
- 125000001584 benzyloxycarbonyl group Chemical group C(=O)(OCC1=CC=CC=C1)* 0.000 description 1
- MSWZFWKMSRAUBD-UHFFFAOYSA-N beta-D-galactosamine Natural products NC1C(O)OC(CO)C(O)C1O MSWZFWKMSRAUBD-UHFFFAOYSA-N 0.000 description 1
- OQFSQFPPLPISGP-UHFFFAOYSA-N beta-carboxyaspartic acid Natural products OC(=O)C(N)C(C(O)=O)C(O)=O OQFSQFPPLPISGP-UHFFFAOYSA-N 0.000 description 1
- 229960003237 betaine Drugs 0.000 description 1
- 239000012867 bioactive agent Substances 0.000 description 1
- 238000010256 biochemical assay Methods 0.000 description 1
- 238000010170 biological method Methods 0.000 description 1
- 229960002685 biotin Drugs 0.000 description 1
- 235000020958 biotin Nutrition 0.000 description 1
- 239000011616 biotin Substances 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 230000036760 body temperature Effects 0.000 description 1
- 210000000988 bone and bone Anatomy 0.000 description 1
- 208000018339 bone inflammation disease Diseases 0.000 description 1
- 201000009480 botryoid rhabdomyosarcoma Diseases 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 210000000481 breast Anatomy 0.000 description 1
- 201000003149 breast fibroadenoma Diseases 0.000 description 1
- 208000003362 bronchogenic carcinoma Diseases 0.000 description 1
- 201000002143 bronchus adenoma Diseases 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- 235000019437 butane-1,3-diol Nutrition 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 229910000019 calcium carbonate Inorganic materials 0.000 description 1
- 235000010216 calcium carbonate Nutrition 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 230000036952 cancer formation Effects 0.000 description 1
- 125000003739 carbamimidoyl group Chemical group C(N)(=N)* 0.000 description 1
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 description 1
- 150000001720 carbohydrates Chemical class 0.000 description 1
- 125000005518 carboxamido group Chemical group 0.000 description 1
- 125000004181 carboxyalkyl group Chemical group 0.000 description 1
- 231100000504 carcinogenesis Toxicity 0.000 description 1
- 230000000747 cardiac effect Effects 0.000 description 1
- 239000004359 castor oil Substances 0.000 description 1
- 235000019438 castor oil Nutrition 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 230000003197 catalytic effect Effects 0.000 description 1
- 230000006369 cell cycle progression Effects 0.000 description 1
- 230000024245 cell differentiation Effects 0.000 description 1
- 230000004709 cell invasion Effects 0.000 description 1
- 230000003833 cell viability Effects 0.000 description 1
- 230000033077 cellular process Effects 0.000 description 1
- 230000006364 cellular survival Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 208000019065 cervical carcinoma Diseases 0.000 description 1
- 210000003679 cervix uteri Anatomy 0.000 description 1
- 229960000541 cetyl alcohol Drugs 0.000 description 1
- 150000005829 chemical entities Chemical class 0.000 description 1
- 125000003636 chemical group Chemical group 0.000 description 1
- 238000007385 chemical modification Methods 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 239000012069 chiral reagent Substances 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- 208000006990 cholangiocarcinoma Diseases 0.000 description 1
- 201000005217 chondroblastoma Diseases 0.000 description 1
- 230000002759 chromosomal effect Effects 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 208000032852 chronic lymphocytic leukemia Diseases 0.000 description 1
- 235000013985 cinnamic acid Nutrition 0.000 description 1
- 229930016911 cinnamic acid Natural products 0.000 description 1
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 description 1
- 229960004106 citric acid Drugs 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 208000009060 clear cell adenocarcinoma Diseases 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 238000012875 competitive assay Methods 0.000 description 1
- 230000009137 competitive binding Effects 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 239000012059 conventional drug carrier Substances 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 235000005822 corn Nutrition 0.000 description 1
- 208000029078 coronary artery disease Diseases 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 229960001681 croscarmellose sodium Drugs 0.000 description 1
- 235000010947 crosslinked sodium carboxy methyl cellulose Nutrition 0.000 description 1
- 201000010305 cutaneous fibrous histiocytoma Diseases 0.000 description 1
- 208000035250 cutaneous malignant susceptibility to 1 melanoma Diseases 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000004210 cyclohexylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 235000018417 cysteine Nutrition 0.000 description 1
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 description 1
- 210000000172 cytosol Anatomy 0.000 description 1
- 125000005507 decahydroisoquinolyl group Chemical group 0.000 description 1
- 230000003111 delayed effect Effects 0.000 description 1
- 230000003831 deregulation Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- 239000003599 detergent Substances 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 229940039227 diagnostic agent Drugs 0.000 description 1
- 239000000032 diagnostic agent Substances 0.000 description 1
- NEFBYIFKOOEVPA-UHFFFAOYSA-K dicalcium phosphate Chemical compound [Ca+2].[Ca+2].[O-]P([O-])([O-])=O NEFBYIFKOOEVPA-UHFFFAOYSA-K 0.000 description 1
- 229940038472 dicalcium phosphate Drugs 0.000 description 1
- 229910000390 dicalcium phosphate Inorganic materials 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- LTMHDMANZUZIPE-PUGKRICDSA-N digoxin Chemical compound C1[C@H](O)[C@H](O)[C@@H](C)O[C@H]1O[C@@H]1[C@@H](C)O[C@@H](O[C@@H]2[C@H](O[C@@H](O[C@@H]3C[C@@H]4[C@]([C@@H]5[C@H]([C@]6(CC[C@@H]([C@@]6(C)[C@H](O)C5)C=5COC(=O)C=5)O)CC4)(C)CC3)C[C@@H]2O)C)C[C@@H]1O LTMHDMANZUZIPE-PUGKRICDSA-N 0.000 description 1
- 229960005156 digoxin Drugs 0.000 description 1
- LTMHDMANZUZIPE-UHFFFAOYSA-N digoxine Natural products C1C(O)C(O)C(C)OC1OC1C(C)OC(OC2C(OC(OC3CC4C(C5C(C6(CCC(C6(C)C(O)C5)C=5COC(=O)C=5)O)CC4)(C)CC3)CC2O)C)CC1O LTMHDMANZUZIPE-UHFFFAOYSA-N 0.000 description 1
- 125000004982 dihaloalkyl group Chemical group 0.000 description 1
- 125000004655 dihydropyridinyl group Chemical group N1(CC=CC=C1)* 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- XNMQEEKYCVKGBD-UHFFFAOYSA-N dimethylacetylene Natural products CC#CC XNMQEEKYCVKGBD-UHFFFAOYSA-N 0.000 description 1
- 125000005879 dioxolanyl group Chemical group 0.000 description 1
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 1
- 208000016097 disease of metabolism Diseases 0.000 description 1
- 230000005750 disease progression Effects 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- PMMYEEVYMWASQN-UHFFFAOYSA-N dl-hydroxyproline Natural products OC1C[NH2+]C(C([O-])=O)C1 PMMYEEVYMWASQN-UHFFFAOYSA-N 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 229940079593 drug Drugs 0.000 description 1
- 239000000890 drug combination Substances 0.000 description 1
- 238000007876 drug discovery Methods 0.000 description 1
- 239000012636 effector Substances 0.000 description 1
- 239000012039 electrophile Substances 0.000 description 1
- 238000002337 electrophoretic mobility shift assay Methods 0.000 description 1
- 201000009409 embryonal rhabdomyosarcoma Diseases 0.000 description 1
- 230000001804 emulsifying effect Effects 0.000 description 1
- 201000003914 endometrial carcinoma Diseases 0.000 description 1
- 239000003623 enhancer Substances 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 239000002702 enteric coating Substances 0.000 description 1
- 238000009505 enteric coating Methods 0.000 description 1
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 1
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 1
- 230000008472 epithelial growth Effects 0.000 description 1
- 210000003238 esophagus Anatomy 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- BEFDCLMNVWHSGT-UHFFFAOYSA-N ethenylcyclopentane Chemical compound C=CC1CCCC1 BEFDCLMNVWHSGT-UHFFFAOYSA-N 0.000 description 1
- 229940093499 ethyl acetate Drugs 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- LYCAIKOWRPUZTN-UHFFFAOYSA-N ethylene glycol Natural products OCCO LYCAIKOWRPUZTN-UHFFFAOYSA-N 0.000 description 1
- 229940012017 ethylenediamine Drugs 0.000 description 1
- 210000003527 eukaryotic cell Anatomy 0.000 description 1
- 230000029142 excretion Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 239000003885 eye ointment Substances 0.000 description 1
- 150000004665 fatty acids Chemical class 0.000 description 1
- 230000001605 fetal effect Effects 0.000 description 1
- 239000000945 filler Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000012467 final product Substances 0.000 description 1
- 108091071773 flk family Proteins 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- RMBPEFMHABBEKP-UHFFFAOYSA-N fluorene Chemical compound C1=CC=C2C3=C[CH]C=CC3=CC2=C1 RMBPEFMHABBEKP-UHFFFAOYSA-N 0.000 description 1
- 125000003983 fluorenyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3CC12)* 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- 238000002825 functional assay Methods 0.000 description 1
- 230000002538 fungal effect Effects 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 108010074605 gamma-Globulins Proteins 0.000 description 1
- 238000001030 gas--liquid chromatography Methods 0.000 description 1
- 208000015419 gastrin-producing neuroendocrine tumor Diseases 0.000 description 1
- 201000000052 gastrinoma Diseases 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000007903 gelatin capsule Substances 0.000 description 1
- 201000003115 germ cell cancer Diseases 0.000 description 1
- 208000005017 glioblastoma Diseases 0.000 description 1
- 229960002442 glucosamine Drugs 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 235000001727 glucose Nutrition 0.000 description 1
- 235000013922 glutamic acid Nutrition 0.000 description 1
- 239000004220 glutamic acid Substances 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- 229960002743 glutamine Drugs 0.000 description 1
- YQEMORVAKMFKLG-UHFFFAOYSA-N glycerine monostearate Natural products CCCCCCCCCCCCCCCCCC(=O)OC(CO)CO YQEMORVAKMFKLG-UHFFFAOYSA-N 0.000 description 1
- SVUQHVRAGMNPLW-UHFFFAOYSA-N glycerol monostearate Natural products CCCCCCCCCCCCCCCCC(=O)OCC(O)CO SVUQHVRAGMNPLW-UHFFFAOYSA-N 0.000 description 1
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 230000012010 growth Effects 0.000 description 1
- 125000001188 haloalkyl group Chemical group 0.000 description 1
- 125000003106 haloaryl group Chemical group 0.000 description 1
- 230000002489 hematologic effect Effects 0.000 description 1
- 208000006359 hepatoblastoma Diseases 0.000 description 1
- 201000002735 hepatocellular adenoma Diseases 0.000 description 1
- 231100000844 hepatocellular carcinoma Toxicity 0.000 description 1
- 125000004446 heteroarylalkyl group Chemical group 0.000 description 1
- 125000005549 heteroarylene group Chemical group 0.000 description 1
- 125000005844 heterocyclyloxy group Chemical group 0.000 description 1
- BXWNKGSJHAJOGX-UHFFFAOYSA-N hexadecan-1-ol Chemical compound CCCCCCCCCCCCCCCCO BXWNKGSJHAJOGX-UHFFFAOYSA-N 0.000 description 1
- 238000013537 high throughput screening Methods 0.000 description 1
- 239000005556 hormone Substances 0.000 description 1
- 229940088597 hormone Drugs 0.000 description 1
- 210000005260 human cell Anatomy 0.000 description 1
- 239000003906 humectant Substances 0.000 description 1
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical compound C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 1
- WGCNASOHLSPBMP-UHFFFAOYSA-N hydroxyacetaldehyde Natural products OCC=O WGCNASOHLSPBMP-UHFFFAOYSA-N 0.000 description 1
- 125000005113 hydroxyalkoxy group Chemical group 0.000 description 1
- 229960002591 hydroxyproline Drugs 0.000 description 1
- 201000001421 hyperglycemia Diseases 0.000 description 1
- 229960003685 imatinib mesylate Drugs 0.000 description 1
- 125000002632 imidazolidinyl group Chemical group 0.000 description 1
- 125000002636 imidazolinyl group Chemical group 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 238000003018 immunoassay Methods 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 201000004933 in situ carcinoma Diseases 0.000 description 1
- 238000012750 in vivo screening Methods 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003406 indolizinyl group Chemical group C=1(C=CN2C=CC=CC12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 150000004001 inositols Chemical class 0.000 description 1
- 206010022498 insulinoma Diseases 0.000 description 1
- 210000002570 interstitial cell Anatomy 0.000 description 1
- 230000031146 intracellular signal transduction Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 201000010985 invasive ductal carcinoma Diseases 0.000 description 1
- 229910052740 iodine Chemical group 0.000 description 1
- 239000011630 iodine Chemical group 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- 229910052742 iron Inorganic materials 0.000 description 1
- 208000028867 ischemia Diseases 0.000 description 1
- 230000000302 ischemic effect Effects 0.000 description 1
- 125000004594 isoindolinyl group Chemical group C1(NCC2=CC=CC=C12)* 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 229960000310 isoleucine Drugs 0.000 description 1
- AGPKZVBTJJNPAG-UHFFFAOYSA-N isoleucine Natural products CCC(C)C(N)C(O)=O AGPKZVBTJJNPAG-UHFFFAOYSA-N 0.000 description 1
- 125000002183 isoquinolinyl group Chemical group C1(=NC=CC2=CC=CC=C12)* 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- 125000004628 isothiazolidinyl group Chemical group S1N(CCC1)* 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 239000007951 isotonicity adjuster Substances 0.000 description 1
- 125000003965 isoxazolidinyl group Chemical group 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 210000001117 keloid Anatomy 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 208000022013 kidney Wilms tumor Diseases 0.000 description 1
- 229940043355 kinase inhibitor Drugs 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 239000000787 lecithin Substances 0.000 description 1
- 235000010445 lecithin Nutrition 0.000 description 1
- 229940067606 lecithin Drugs 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 230000000670 limiting effect Effects 0.000 description 1
- 206010024627 liposarcoma Diseases 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 230000033001 locomotion Effects 0.000 description 1
- 231100000053 low toxicity Toxicity 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 239000008176 lyophilized powder Substances 0.000 description 1
- 229960003646 lysine Drugs 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 159000000003 magnesium salts Chemical class 0.000 description 1
- 239000006249 magnetic particle Substances 0.000 description 1
- 238000012423 maintenance Methods 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 230000003211 malignant effect Effects 0.000 description 1
- 201000004593 malignant giant cell tumor Diseases 0.000 description 1
- 201000000289 malignant teratoma Diseases 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 239000011565 manganese chloride Substances 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000013507 mapping Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 210000002418 meninge Anatomy 0.000 description 1
- JZKXXXDKRQWDET-UHFFFAOYSA-N meta-tyrosine Natural products OC(=O)C(N)CC1=CC=CC(O)=C1 JZKXXXDKRQWDET-UHFFFAOYSA-N 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- GBMDVOWEEQVZKZ-UHFFFAOYSA-N methanol;hydrate Chemical compound O.OC GBMDVOWEEQVZKZ-UHFFFAOYSA-N 0.000 description 1
- 229930182817 methionine Natural products 0.000 description 1
- WWIYGBWRUXQDND-UHFFFAOYSA-N methyl 2-(4-chlorophenyl)acetate Chemical compound COC(=O)CC1=CC=C(Cl)C=C1 WWIYGBWRUXQDND-UHFFFAOYSA-N 0.000 description 1
- 150000004702 methyl esters Chemical class 0.000 description 1
- WBYWAXJHAXSJNI-UHFFFAOYSA-N methyl p-hydroxycinnamate Natural products OC(=O)C=CC1=CC=CC=C1 WBYWAXJHAXSJNI-UHFFFAOYSA-N 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 230000001617 migratory effect Effects 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 230000002297 mitogenic effect Effects 0.000 description 1
- 239000003607 modifier Substances 0.000 description 1
- 125000002950 monocyclic group Chemical group 0.000 description 1
- CQDGTJPVBWZJAZ-UHFFFAOYSA-N monoethyl carbonate Chemical compound CCOC(O)=O CQDGTJPVBWZJAZ-UHFFFAOYSA-N 0.000 description 1
- 125000002757 morpholinyl group Chemical group 0.000 description 1
- 208000010492 mucinous cystadenocarcinoma Diseases 0.000 description 1
- 238000007040 multi-step synthesis reaction Methods 0.000 description 1
- 201000006417 multiple sclerosis Diseases 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- 208000025113 myeloid leukemia Diseases 0.000 description 1
- 208000009091 myxoma Diseases 0.000 description 1
- QUMIGAREEPSVLG-UHFFFAOYSA-N n',n'-diethyl-n-phenylethane-1,2-diamine Chemical compound CCN(CC)CCNC1=CC=CC=C1 QUMIGAREEPSVLG-UHFFFAOYSA-N 0.000 description 1
- BAVMTSYATQUKTC-UHFFFAOYSA-N n',n'-diethyl-n-phenylethane-1,2-diamine;dihydrochloride Chemical compound Cl.Cl.CCN(CC)CCNC1=CC=CC=C1 BAVMTSYATQUKTC-UHFFFAOYSA-N 0.000 description 1
- UDGSVBYJWHOHNN-UHFFFAOYSA-N n',n'-diethylethane-1,2-diamine Chemical compound CCN(CC)CCN UDGSVBYJWHOHNN-UHFFFAOYSA-N 0.000 description 1
- DILRJUIACXKSQE-UHFFFAOYSA-N n',n'-dimethylethane-1,2-diamine Chemical compound CN(C)CCN DILRJUIACXKSQE-UHFFFAOYSA-N 0.000 description 1
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 201000008026 nephroblastoma Diseases 0.000 description 1
- 210000000653 nervous system Anatomy 0.000 description 1
- 230000001537 neural effect Effects 0.000 description 1
- 208000007538 neurilemmoma Diseases 0.000 description 1
- 201000004662 neurofibroma of spinal cord Diseases 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 208000004649 neutrophil actin dysfunction Diseases 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 231100000344 non-irritating Toxicity 0.000 description 1
- 239000012457 nonaqueous media Substances 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 125000002868 norbornyl group Chemical group C12(CCC(CC1)C2)* 0.000 description 1
- 150000007523 nucleic acids Chemical group 0.000 description 1
- 239000012038 nucleophile Substances 0.000 description 1
- NIHNNTQXNPWCJQ-UHFFFAOYSA-N o-biphenylenemethane Natural products C1=CC=C2CC3=CC=CC=C3C2=C1 NIHNNTQXNPWCJQ-UHFFFAOYSA-N 0.000 description 1
- OIPZNTLJVJGRCI-UHFFFAOYSA-M octadecanoyloxyaluminum;dihydrate Chemical compound O.O.CCCCCCCCCCCCCCCCCC(=O)O[Al] OIPZNTLJVJGRCI-UHFFFAOYSA-M 0.000 description 1
- 125000005060 octahydroindolyl group Chemical group N1(CCC2CCCCC12)* 0.000 description 1
- 125000005061 octahydroisoindolyl group Chemical group C1(NCC2CCCCC12)* 0.000 description 1
- 125000002347 octyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 230000003287 optical effect Effects 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 150000002895 organic esters Chemical class 0.000 description 1
- 229960003104 ornithine Drugs 0.000 description 1
- 229910000489 osmium tetroxide Inorganic materials 0.000 description 1
- 208000003388 osteoid osteoma Diseases 0.000 description 1
- 208000008798 osteoma Diseases 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 210000003101 oviduct Anatomy 0.000 description 1
- 125000001715 oxadiazolyl group Chemical group 0.000 description 1
- 125000000160 oxazolidinyl group Chemical group 0.000 description 1
- 125000005968 oxazolinyl group Chemical group 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 125000005476 oxopyrrolidinyl group Chemical group 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 239000006179 pH buffering agent Substances 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- 208000021255 pancreatic insulinoma Diseases 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 239000012188 paraffin wax Substances 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 1
- 239000008177 pharmaceutical agent Substances 0.000 description 1
- 239000002831 pharmacologic agent Substances 0.000 description 1
- 239000012071 phase Substances 0.000 description 1
- 125000001791 phenazinyl group Chemical group C1(=CC=CC2=NC3=CC=CC=C3N=C12)* 0.000 description 1
- 229960003742 phenol Drugs 0.000 description 1
- 125000001484 phenothiazinyl group Chemical group C1(=CC=CC=2SC3=CC=CC=C3NC12)* 0.000 description 1
- 125000001644 phenoxazinyl group Chemical group C1(=CC=CC=2OC3=CC=CC=C3NC12)* 0.000 description 1
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- SONNWYBIRXJNDC-VIFPVBQESA-N phenylephrine Chemical compound CNC[C@H](O)C1=CC=CC(O)=C1 SONNWYBIRXJNDC-VIFPVBQESA-N 0.000 description 1
- UYWQUFXKFGHYNT-UHFFFAOYSA-N phenylmethyl ester of formic acid Natural products O=COCC1=CC=CC=C1 UYWQUFXKFGHYNT-UHFFFAOYSA-N 0.000 description 1
- 150000003906 phosphoinositides Chemical class 0.000 description 1
- 238000003566 phosphorylation assay Methods 0.000 description 1
- 239000003757 phosphotransferase inhibitor Substances 0.000 description 1
- 125000004592 phthalazinyl group Chemical group C1(=NN=CC2=CC=CC=C12)* 0.000 description 1
- 208000024724 pineal body neoplasm Diseases 0.000 description 1
- 201000004123 pineal gland cancer Diseases 0.000 description 1
- IYPZRUYMFDWKSS-UHFFFAOYSA-N piperazin-1-amine Chemical class NN1CCNCC1 IYPZRUYMFDWKSS-UHFFFAOYSA-N 0.000 description 1
- RFIOZSIHFNEKFF-UHFFFAOYSA-M piperazine-1-carboxylate Chemical compound [O-]C(=O)N1CCNCC1 RFIOZSIHFNEKFF-UHFFFAOYSA-M 0.000 description 1
- 125000004193 piperazinyl group Chemical group 0.000 description 1
- FLKRMXAWABTWSH-UHFFFAOYSA-N piperidine-1-sulfonamide Chemical compound NS(=O)(=O)N1CCCCC1 FLKRMXAWABTWSH-UHFFFAOYSA-N 0.000 description 1
- 125000003386 piperidinyl group Chemical group 0.000 description 1
- 229920000768 polyamine Polymers 0.000 description 1
- 229920000642 polymer Polymers 0.000 description 1
- 108091033319 polynucleotide Proteins 0.000 description 1
- 102000040430 polynucleotide Human genes 0.000 description 1
- 239000002157 polynucleotide Substances 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 229920001282 polysaccharide Polymers 0.000 description 1
- 239000005017 polysaccharide Substances 0.000 description 1
- 150000004804 polysaccharides Chemical class 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 239000013641 positive control Substances 0.000 description 1
- 101150063097 ppdK gene Proteins 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 150000003140 primary amides Chemical class 0.000 description 1
- 150000003141 primary amines Chemical class 0.000 description 1
- 230000004647 pro-inflammatory pathway Effects 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 229960002429 proline Drugs 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- IAHIMVFWYADCJJ-UHFFFAOYSA-N prop-1-enylcyclohexane Chemical group CC=CC1CCCCC1 IAHIMVFWYADCJJ-UHFFFAOYSA-N 0.000 description 1
- VVWRJUBEIPHGQF-MDZDMXLPSA-N propan-2-yl (ne)-n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)\N=N\C(=O)OC(C)C VVWRJUBEIPHGQF-MDZDMXLPSA-N 0.000 description 1
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 1
- 229940080818 propionamide Drugs 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 1
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 1
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 125000006239 protecting group Chemical group 0.000 description 1
- 238000000159 protein binding assay Methods 0.000 description 1
- 238000001243 protein synthesis Methods 0.000 description 1
- 125000001042 pteridinyl group Chemical group N1=C(N=CC2=NC=CN=C12)* 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- 125000003072 pyrazolidinyl group Chemical group 0.000 description 1
- 125000003226 pyrazolyl group Chemical group 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 150000003230 pyrimidines Chemical class 0.000 description 1
- 125000000714 pyrimidinyl group Chemical group 0.000 description 1
- 125000000719 pyrrolidinyl group Chemical group 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 229940107700 pyruvic acid Drugs 0.000 description 1
- 125000001453 quaternary ammonium group Chemical group 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 125000002943 quinolinyl group Chemical group N1=C(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 125000004621 quinuclidinyl group Chemical group N12C(CC(CC1)CC2)* 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 1
- 230000035484 reaction time Effects 0.000 description 1
- 230000008707 rearrangement Effects 0.000 description 1
- 230000007115 recruitment Effects 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 238000006722 reduction reaction Methods 0.000 description 1
- 230000021014 regulation of cell growth Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 201000010174 renal carcinoma Diseases 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 239000011347 resin Substances 0.000 description 1
- 229920005989 resin Polymers 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 208000037803 restenosis Diseases 0.000 description 1
- 208000029922 reticulum cell sarcoma Diseases 0.000 description 1
- 201000009410 rhabdomyosarcoma Diseases 0.000 description 1
- 238000002390 rotary evaporation Methods 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 206010039667 schwannoma Diseases 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 208000004548 serous cystadenocarcinoma Diseases 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 150000004760 silicates Chemical class 0.000 description 1
- RMAQACBXLXPBSY-UHFFFAOYSA-N silicic acid Chemical compound O[Si](O)(O)O RMAQACBXLXPBSY-UHFFFAOYSA-N 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 229960002930 sirolimus Drugs 0.000 description 1
- 210000003625 skull Anatomy 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 239000001509 sodium citrate Substances 0.000 description 1
- NLJMYIDDQXHKNR-UHFFFAOYSA-K sodium citrate Chemical compound O.O.[Na+].[Na+].[Na+].[O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O NLJMYIDDQXHKNR-UHFFFAOYSA-K 0.000 description 1
- 235000019333 sodium laurylsulphate Nutrition 0.000 description 1
- 239000004334 sorbic acid Substances 0.000 description 1
- 235000010199 sorbic acid Nutrition 0.000 description 1
- 229940075582 sorbic acid Drugs 0.000 description 1
- 229940035044 sorbitan monolaurate Drugs 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 125000003003 spiro group Chemical group 0.000 description 1
- 239000007921 spray Substances 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 230000000638 stimulation Effects 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 230000009211 stress pathway Effects 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 125000000475 sulfinyl group Chemical group [*:2]S([*:1])=O 0.000 description 1
- 150000003457 sulfones Chemical class 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 230000002195 synergetic effect Effects 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 230000009897 systematic effect Effects 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 229960000235 temsirolimus Drugs 0.000 description 1
- 208000001608 teratocarcinoma Diseases 0.000 description 1
- PWQLFIKTGRINFF-UHFFFAOYSA-N tert-butyl 4-hydroxypiperidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC(O)CC1 PWQLFIKTGRINFF-UHFFFAOYSA-N 0.000 description 1
- SCMFJWWKJBCJDC-UHFFFAOYSA-N tert-butyl n-[4-(4-chlorophenyl)piperidine-4-carbonyl]carbamate Chemical compound C=1C=C(Cl)C=CC=1C1(C(=O)NC(=O)OC(C)(C)C)CCNCC1 SCMFJWWKJBCJDC-UHFFFAOYSA-N 0.000 description 1
- CWXPZXBSDSIRCS-UHFFFAOYSA-N tert-butyl piperazine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCNCC1 CWXPZXBSDSIRCS-UHFFFAOYSA-N 0.000 description 1
- 150000003512 tertiary amines Chemical class 0.000 description 1
- 210000001550 testis Anatomy 0.000 description 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 description 1
- BSYVTEYKTMYBMK-UHFFFAOYSA-N tetrahydrofurfuryl alcohol Chemical compound OCC1CCCO1 BSYVTEYKTMYBMK-UHFFFAOYSA-N 0.000 description 1
- 125000001412 tetrahydropyranyl group Chemical group 0.000 description 1
- 125000004853 tetrahydropyridinyl group Chemical group N1(CCCC=C1)* 0.000 description 1
- 125000005329 tetralinyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 description 1
- 229960004559 theobromine Drugs 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 125000001113 thiadiazolyl group Chemical group 0.000 description 1
- 125000006090 thiamorpholinyl sulfone group Chemical group 0.000 description 1
- 125000001984 thiazolidinyl group Chemical group 0.000 description 1
- 125000002769 thiazolinyl group Chemical group 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- 150000003573 thiols Chemical class 0.000 description 1
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 1
- 125000000341 threoninyl group Chemical group [H]OC([H])(C([H])([H])[H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- 201000002510 thyroid cancer Diseases 0.000 description 1
- YONPGGFAJWQGJC-UHFFFAOYSA-K titanium(iii) chloride Chemical compound Cl[Ti](Cl)Cl YONPGGFAJWQGJC-UHFFFAOYSA-K 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 235000010487 tragacanth Nutrition 0.000 description 1
- 239000000196 tragacanth Substances 0.000 description 1
- 229940116362 tragacanth Drugs 0.000 description 1
- FGMPLJWBKKVCDB-UHFFFAOYSA-N trans-L-hydroxy-proline Natural products ON1CCCC1C(O)=O FGMPLJWBKKVCDB-UHFFFAOYSA-N 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 206010044412 transitional cell carcinoma Diseases 0.000 description 1
- 108091008578 transmembrane receptors Proteins 0.000 description 1
- 102000027257 transmembrane receptors Human genes 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- 229940117013 triethanolamine oleate Drugs 0.000 description 1
- UCPYLLCMEDAXFR-UHFFFAOYSA-N triphosgene Chemical compound ClC(Cl)(Cl)OC(=O)OC(Cl)(Cl)Cl UCPYLLCMEDAXFR-UHFFFAOYSA-N 0.000 description 1
- YFTHZRPMJXBUME-UHFFFAOYSA-N tripropylamine Chemical compound CCCN(CCC)CCC YFTHZRPMJXBUME-UHFFFAOYSA-N 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 229960004799 tryptophan Drugs 0.000 description 1
- 208000022271 tubular adenoma Diseases 0.000 description 1
- 210000004881 tumor cell Anatomy 0.000 description 1
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 1
- 125000001493 tyrosinyl group Chemical group [H]OC1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- 238000011144 upstream manufacturing Methods 0.000 description 1
- 210000003708 urethra Anatomy 0.000 description 1
- 210000004291 uterus Anatomy 0.000 description 1
- 210000001215 vagina Anatomy 0.000 description 1
- 239000004474 valine Substances 0.000 description 1
- 235000015112 vegetable and seed oil Nutrition 0.000 description 1
- 239000008158 vegetable oil Substances 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 208000009540 villous adenoma Diseases 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 210000003905 vulva Anatomy 0.000 description 1
- 230000029663 wound healing Effects 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
Definitions
- This invention relates to compounds for modulating protein kinase enzymatic activity for modulating cellular activities such as proliferation, differentiation, programmed cell death, migration, chemoinvasion and metabolism. Even more specifically, the invention relates to compounds which inhibit, regulate and/or modulate kinase receptor signal transduction pathways related to the changes in cellular activities as mentioned above, compositions which contain these compounds, and methods of using them to treat kinase-dependent diseases and conditions.
- Protein kinases are enzymes that catalyze the phosphorylation of proteins at the hydroxy groups of tyrosine, serine and threonine residues of proteins.
- the kinase complement of the human genome contains 518 putative protein kinase genes [Manning et al, Science, (2002), 298, 1912].
- the consequences of this activity include effects on cell differentiation, proliferation, transcription, translation, metabolism, cell cycle progression, apoptosis, metabolism cytoskeletal rearrangement and movement; i.e., protein kinases mediate the majority of signal transduction in eukaryotic cells.
- Tyrosine kinases can be categorized as receptor type or non-receptor type. Receptor- type tyrosine kinases have an extracellular, a transmembrane, and an intracellular portion, while non-receptor type tyrosine kinases are wholly intracellular.
- Receptor-type tyrosine kinases are comprised of a large number of transmembrane receptors with diverse biological activity. In fact, about 20 different subfamilies of receptor- type tyrosine kinases have been identified.
- Ligands of this subfamily of receptors identified so far include epithelial growth factor, TGF-alpha, amphiregulin, HB-EGF, betacellulin and heregulin.
- the insulin subfamily includes INS-R, IGF-IR, and IR-R.
- the PDGF subfamily includes the PDGF-alpha and -beta receptors, CSFIR, c-kit and FLK-II.
- the FLK family which is comprised of the kinase insert domain receptor (KDR), fetal liver kinase-1 (FLK-I), fetal liver kinase-4 (FLK-4) and the fms-like tyrosine kinase-1 (FIt-I).
- KDR kinase insert domain receptor
- FLK-I fetal liver kinase-1
- FLK-4 fetal liver kinase-4
- FIt-I fms-like tyrosine kinase-1
- the non-receptor type of tyrosine kinases is also comprised of numerous subfamilies, including Src, Frk, Btk, Csk, AbI, Syk/Zap70, Fes/Fps, Fak, Jak, and Ack. Each of these subfamilies is further sub-divided into varying receptors.
- the Src subfamily is one of the largest and includes Src, Yes, Fyn, Lyn, Lck, BIk, Hck, Fgr, and Yrk.
- the Src subfamily of enzymes has been linked to oncogenesis.
- Serine-theoronine kinases play critical roles in intracellular signal transduction and include the multiple families, including STE, CKI, AGC, CAMK, and CMGC. Important subfamilies include, the MAP kinases, p38, JNK and ERK, which modulate signal transduction resulting from such diverse stimuli as mitogenic, stress, proinflammatory and antiapoptotic pathways. Members of the MAP kinase subfamily have been targeted for therapeutic intervention, including p38a, JNK isozymes and Raf.
- kinase modulation relates to oncological indications.
- modulation of protein kinase activity for the treatment of cancer has been demonstrated successfully with the FDA approval of Gleevec® (imatinib mesylate, produced by Novartis Pharmaceutical Corporation of East Hanover, NJ) for the treatment of Chronic Myeloid Leukemia (CML) and gastrointestinal stroma cancers.
- Gleevec is a selective AbI kinase inhibitor.
- the enzyme p70S6 kinase is a serine-theoronine kinase that is a component of the phosphatidylinositol 3 kinase (PDK)/ Akt kinase pathway. Both Akt and p70S6K are downstream of phosphatidylinositol-3 kinase (PI3K), and undergo phosphorylation and activation in response to growth factors such as IGF-I, EGF, TGF- ⁇ and HGF. The enzyme p70S6K modulates protein synthesis by phosphorylation of the S6 ribosomal protein promoting translation.
- Phosphoinositide 3-kinases or PDKs, generate specific inositol lipids implicated in the regulation of cell growth, proliferation, survival, differentiation, metabolism and cytoskeletal changes.
- PI3K lipid products is the serine threonine protein kinase AKT, or protein kinase B (PKB).
- AKT resides in the cytosol in a low-activity conformation.
- AKT Upon cellular stimulation, AKT is activated through recruitment to cellular membranes by PI3K lipid products and by phosphorylation by 3 -prime phosphoinositide-dependent kinase-1 (PDPKl), leading to a cascade of downstream effects through activation of a range of downstream targets including glycogen synthase kinase-3beta (GSK-3beta), mammalian target of rapamycin (mTOR), p70S6 kinase, endothelial nitric oxide synthase (eNOS) and several anti-apoptotic effectors [Hajduch, Eet al (2001) FEBS Lett.
- GSK-3beta glycogen synthase kinase-3beta
- mTOR mammalian target of rapamycin
- eNOS endothelial nitric oxide synthase
- eNOS endothelial nitric oxide syntha
- AKTl plays an important role in neuronal and general cellular survival, and resistance to apoptosis [Dudek H. et al (1997) Science 275: 661-663; Kauffmann-Zeh A, et al (1997) Nature 385: 544-548; Songyang Z. et al (1997) Proc Natl Acad Sci U S A 94: 11345-11350].
- AKTl has been implicated in a wide variety of cancers, including breast and thyroid cancer [Liang J. et al (2002) Nature Med. 8: 1153-1160; Vasco V et al (2004) J. Med. Genet. 41: 161- 170].
- AKTl also play a role in metabolism through control of pancreatic beta-cell growth and survival [Dickson LM, and Rhodes CJ (2004) Am J Physiol Endocrinol Metab. 287:E192-8].
- AKT2 is enriched in insulin-responsive tissues and has been implicated in the metabolic actions of the hormone, such as diabetes [George S et al (2004) Science 304: 1325- 1328].
- AKT2 is amplified and overexpressed in ovarian cancer, and its overexpression contributes to the malignant phenotype of a subset of human ductal pancreatic cancers [Cheng, J. Q. et al (1992) Proc. Nat. Acad. Sci. 89: 9267-9271; Cheng, J. Q. et al (1996) Proc. Nat. Acad. Sci. 93: 3636-3641].
- the identification of small-molecule compounds that specifically inhibit, regulate and/or modulate the signal transduction of kinases, particularly p70S6K, AKTl and AKT2 is desirable as a means to treat or prevent disease states associated with abnormal cell proliferation and metabolism is an object of this invention.
- the present invention provides compounds for modulating the activity of kinases, preferably p70S6, Akt-1 or Akt-2 kinases, and methods of treating diseases mediated by the activity of kinases, prefereably p70S6, Akt-1 or Akt-2 kinases utilizing the compounds and pharmaceutical compositions thereof.
- Diseases mediated by p70S6, Akt-1 and/or Akt-2 kinase activity include, but are not limited to, diseases characterized in part by migration, invasion, proliferation, and other biological activities associated with invasive cell growth, and metabolism.
- the invention provides methods of screening for modulators of kinases activity, preferably p70S6, Akt-1 and/or Akt-2 activity.
- the methods comprise combining a composition of the invention, typically a kinase, preferably p70S6, Akt-1 and/or Akt-2 kinases, and at least one candidate agent and determining the effect of the candidate agent on the kinases activity.
- kits comprising one or more containers filled with one or more of the ingredients of pharmaceutical compounds and/or compositions of the present invention, including, kinase enzyme activity modulators, preferably p70S6, Akt-1 and/or Akt-2 kinase enzyme activity modulators as described herein.
- kinase enzyme activity modulators preferably p70S6, Akt-1 and/or Akt-2 kinase enzyme activity modulators as described herein.
- kits can also include, for example, other compounds and/or compositions (e.g., diluents, permeation enhancers, lubricants, and the like), a device(s) for administering the compounds and/or compositions, and written instructions in a form prescribed by a governmental agency regulating the manufacture, use or sale of pharmaceuticals or biological products, which instructions can also reflects approval by the agency of manufacture, use or sale for human administration.
- other compounds and/or compositions e.g., diluents, permeation enhancers, lubricants, and the like
- a device(s) for administering the compounds and/or compositions e.g., a device(s) for administering the compounds and/or compositions
- written instructions in a form prescribed by a governmental agency regulating the manufacture, use or sale of pharmaceuticals or biological products, which instructions can also reflects approval by the agency of manufacture, use or sale for human administration.
- the invention also provides a diagnostic agent comprising a compound of the invention and, optionally, pharmaceutically acceptable adjuvants and excipients.
- compositions of the invention are used to treat diseases associated with abnormal and or unregulated cellular activities.
- Disease states which can be treated by the methods and compositions provided herein include, but are not limited to, cancer (further discussed below), immunological disorders such as rheumatoid arthritis, graft-host diseases, multiple sclerosis, psoriasis; cardiovascular diseases such as artheroscrosis, myocardioinfarction, ischemia, stroke and restenosis; metabolic disorders and diseases such as diabetes, obesity and hypercholesterolemia; and other inflammatory and degenerative diseases such as interbowel diseases, osteoarthritus, macular degeneration, diabetic retinopathy.
- cancer cancer
- immunological disorders such as rheumatoid arthritis, graft-host diseases, multiple sclerosis, psoriasis
- cardiovascular diseases such as artheroscrosis, myocardioinfarction, ischemia, stroke and restenosis
- metabolic disorders and diseases such as diabetes, obesity and hypercholesterolemia
- the cells may not be in a hyper- or hypo- proliferative and/or migratory state (abnormal state) and still require treatment.
- the cells may be proliferating "normally", but proliferation and migration enhancement may be desired.
- reduction in "normal” cell proliferation and/or migration rate may be desired.
- the present invention comprises a compound for modulating kinase activity, preferably p70S6, AkM and/or Akt-2 kinase activity according to Formula I,
- R 1 is H, halo, cyano, aryl, heteroaryl, Ci -4 alkyl, C 2 -C 6 alkenyl, or C 2 -C 6 alkynyl, wherein the aryl, heteroaryl, alkyl, alkenyl and alkynyl are optionally substituted with one or two groups independently selected from CO 2 RiO, CONR I0 R I i, ORi 0 , and NRi 0 Ri ⁇
- R 2 is H, NH 2 , SH, OH, or Ci-C 2 alkyl
- R 3 , R 4 , R 5 , and R 6 are each independently H, oxo, CO 2 Ri 0 , CONR I0 R I I , Ci -4 alkyl, Ci-C 6 alkoxy, or Ci-C 6 alkoxy-Ci-C 4 alkyl, wherein the Ci-C 4 alkyl, C 1 -C 6 alkoxy and Ci-C 6 alkoxy-Ci-C 4 alkyl in each group are independently optionally substituted with 1 or 2 substituents independently selected from CO 2 Ri 0 , CONRi 0 Rn 5 ORi 0 , andNRioRn, or R 3 and R 5 together with the carbons to which they are attached form a C 3 -C 7 carbocyclic ring, wherein the ring is optionally substituted with H, halo, cyano, nitro, or amino, R 4 and R 6 together with the carbons to which they are attached form a C 3 -C 7 carbocyclic ring, wherein the ring is
- R 3 and R 6 together with the carbons to which they are attached form a bridged C 5 -C 7 carbocyclic ring, wherein the ring is optionally substituted with H, halo, cyano, nitro, or amino, or
- R 4 and R 5 together with the carbons to which they are attached form a bridged C 5 -C 7 carbocyclic ring, wherein the ring is optionally substituted with H, halo, cyano, nitro, or amino;
- L is C 0-4 alkyl, C 2 -C 6 alkenyl, -N(Ri 2 )-, -C(O)N(Ri 2 )-, -N(Ri 2 )C(O)-, -C(O)-, -0-(CH 2 ) n -, or - (CH 2 ) n -0-, wherein n is 1-4;
- Qi is N or CRi 3 , wherein R n is H or C(O)NR 12 (CH 2 ) n NRi 0 Rn;
- V is absent and Ri 3 is not H;
- V is H, OH, NH 2 , Ci-C 6 alkoxy, NRi 0 Ri i, 0(CH 2 ) n NRioRn, O(CH 2 ) n attached to a C or
- N of a 4-7 membered heterocyclyl NRi 2 (CH 2 ) n NRi 0 R ⁇ , NRi 2 C(O)NRi 2 (CH 2 ) n NRi 0 Rii, NRi 2 C(O)(CH 2 ) n NR 10 Rii, (CH 2 ) m O(CH 2 ) n NRioRii, (CH 2 ) m NRi 2 (CH 2 ) n NRi 0 R ⁇ , (CH 2 ) m CHRi 2 (CH 2 ) n NRi 0 Rii, Ci -4 alkyl optionally substituted with OH or NRi 0 Ri l, or
- V is a 4-7 membered unsaturated cyclic containing 1-3 atom of O or N, or
- V is a bicyclic solublizing group
- V is H, (CH 2 ) m O(CH 2 ) n NRioRn, (CH 2 ) m NRi 2 (CH 2 ) n NRi 0 Rn,
- V is a 4-7 membered saturated or unsaturated cyclic or heterocyclic containing 1-3 atoms of O or N, optionally substituted with 1 or 2 Ci-C 3 alkoxy groups or
- V is a "bicyclic solublizing group"
- n 1-3
- n 1-4
- W is Ci-C 6 alkyl, NRi 0 Ri i, or W is aryl, C 3 -C 7 cycloalkyl, heterocyclyl, heteroaryl, or 5-12 membered fused bicylic or tricyclic saturated, partially saturated, or usaturated ring system containing 0-4 ring atoms selected from N, O, and S, wherein each aryl, cycloalkyl, heterocyclyl, heteroaryl, and fused bicyclic or tricyclic ring system is optionally substituted with 1, 2, or 3 substituents independently selected from halo, CN, NO 2 , CF 3 , OH, NRioR ⁇ , Ci-C 6 alkoxy, Ci-C 6 alkyl, NO 2 , C(O)OCi-C 6 alkyl, C(O)NR 12 -C 1 -C 6 alkoxy, C(O)NR 12 -heterocyclyl, aryl, O-aryl, O-CH 2 -
- Rio, Rn and Ri 2 are each independently H or Ci- 6 alkyl which is optionally substituted with aryl or heteroaryl.
- Preferred compounds of Formula I include compounds of Formula II, which are compounds of Formula I wherein Ri is not hydrogen.
- Preferred compounds of Formula II include compounds wherein Ri is halo. [0029] Preferred compounds of Formula II incude compounds wherein R 1 is bromo. [0030] Preferred compounds of Formula II include compounds wherein R 2 is H. [0031] Preferred compounds of Formula II include compounds wherein R 3 , R 4 , R 5 , and R 6 , are each H. [0032] Preferred compounds of Formula I and II include compounds of Formula III, which are compounds of Formula I or Formula II wherein Qi is CH and Q 2 is CH. [0033] Preferred compounds of Formula III include compounds wherein L is a bond. [0034] Preferred compounds of Formula III include compounds wherein W is aryl optionally substituted with 1, 2, or 3 substituents independently selected from halo, CN, NO 2 , CF 3 , OH,
- Preferred compounds of Formula III include compounds wherein W is phenyl optionally substituted with 1, 2, or 3 substituents independently selected from halo, CN, NO 2 ,
- Preferred compounds of Formula III include compounds wherein W is phenyl optionally substituted with 1, or 2 substituents independently selected from halo, CF 3 , Ci- 4 alkyl, and Ci-C 4 alkoxy.
- Preferred compounds of Formula III include compounds wherein W is phenyl optionally substituted with 1, or 2 substituents independently selected from F, Cl and Br.
- Preferred compounds of Formula III include compounds wherein V is H, OH,
- Preferred compounds of Formula III include compounds wherein V is H, OH,
- Preferred compounds of Formula III include compounds wherein Ri is H, halo or Ci-C 2 alkyl and R 2 is H.
- Preferred compounds of Formula III include compounds wherein R 3 , R 4 , R 5 , and R 6 , are each H.
- Preferred compounds of Formula I and II include compounds of Formula IV, which are compounds of Formula I or Formula II wherein Qi is N and Q 2 is CH.
- Preferred compounds of Formula IV include compounds wherein L is a bond.
- Preferred compounds of Formula IV include compounds wherein W is aryl optionally substituted with 1, 2, or 3 substituents independently selected from halo, CN, NO 2 , CF 3 , OH,
- Preferred compounds of Formula IV include compounds wherein W is phenyl optionally substituted with 1, 2, or 3 substituents independently selected from halo, CN, NO 2 ,
- Preferred compounds of Formula IV include compounds wherein W is phenyl optionally substituted with 1, or 2 substituents independently selected from halo, CF 3 , Ci- 4 alkyl, and Ci-C 4 alkoxy.
- Preferred compounds of Formula IV include compounds wherein W is phenyl optionally substituted with 1, or 2 substituents independently selected from F, Cl and Br.
- Preferred compounds of Formula IV include compounds wherein V is H,
- Preferred compounds of Formula IV include compounds wherein V is H,
- Preferred compounds of Formula IV include compounds wherein V is H.
- Preferred compounds of Formula IV include compounds wherein Ri is halo and R 2 is
- Preferred compounds of Formula IV include compounds wherein R 3 , R 4 , R 5 , and R 6 , are each H.
- Preferred compounds of Formula I and II include compounds of Formula V, which are compounds of Formula I or Formula II wherein Qi is CH and Q 2 is N.
- Preferred compounds of Formula V include compounds wherein L is a bond.
- Preferred compounds of Formula V include compounds wherein W is aryl optionally substituted with 1, 2, or 3 substituents independently selected from halo, CN, NO 2 , CF 3 , OH,
- NRi 0 Rn Ci-C 6 alkoxy, Ci-C 6 alkyl, NO 2 , C(O)OCi-C 6 alkyl, C(O)NRi 2 -C 1 -C 6 alkoxy,
- Preferred compounds of Formula V include compounds wherein W is phenyl optionally substituted with 1, 2, or 3 substituents independently selected from halo, CN, NO 2 , CF 3 , OH, NR 10 R 11 , C 1 -C 6 alkoxy, C 1 -C 6 alkyl, NO 2 , C(O)OC 1 -C 6 alkyl, C(O)NR 12 -C 1 -C 6 alkoxy,
- Preferred compounds of Formula V include compounds wherein W is phenyl optionally substituted with 1, or 2 substituents independently selected from halo, CF 3 , C1-4 alkyl, and C 1 -
- Preferred compounds of Formula V include compounds wherein W is phenyl optionally substituted with 1, or 2 substituents independently selected from F, Cl and Br. [0059] Preferred compounds of Formula V include compounds wherein V is H,
- Preferred compounds of Formula V include compounds wherein V is H,
- C(O)C(O)NRi 2 (CH 2 ) n NR 10 Rn, SO 2 (CH 2 ) n NRi 0 Rn, C(O)-C 2 -C 6 alkenyl, or Ci 4 alkyl optionally substituted with NRioRn and Ri 0 and Rn are each CH 3 .
- Preferred compounds of Formula V include compounds wherein Ri is halo and R 2 is H.
- Preferred compounds of Formula V include compounds wherein R 3 , R 4 , R 5 , and R 6 , are each H.
- Preferred compounds of Formula I and II include compounds of Formula VI, which are compounds of Formula I or Formula II wherein Qi is CH or N, and Q 2 and V together form
- Preferred compounds of Formula VI include compounds wherein L is a bond.
- Preferred compounds of Formula VI include compounds wherein W is aryl optionally substituted with 1, 2, or 3 substituents independently selected from halo, CN, NO 2 , CF 3 , OH,
- NRi 0 Rn Ci-C 6 alkoxy, C 1 -C 6 alkyl, NO 2 , C(O)OCi-C 6 alkyl, C(O)NR 12 -C 1 -C 6 alkoxy,
- Preferred compounds of Formula VI include compounds wherein W is phenyl optionally substituted with 1, 2, or 3 substituents independently selected from halo, CN, NO 2 ,
- Preferred compounds of Formula VI include compounds wherein W is phenyl optionally substituted with 1, or 2 substituents independently selected from halo, CF 3 , Cr 4 alkyl, and Ci-C 4 alkoxy.
- Preferred compounds of Formula VI include compounds wherein W is phenyl optionally substituted with 1, or 2 substituents independently selected from F, Cl and Br.
- Preferred compounds of Formula VI include compounds wherein Ri is halo and R 2 is
- Preferred compounds of Formula VI include compounds wherein R 3 , R 4 , R 5 , and R 6 , are each H.
- the compound is according to paragraph [0024], selected from
- Another aspect of the invention is a pharmaceutical composition
- a pharmaceutical composition comprising the compound according to any one of paragraphs [0024]-[0071J and a pharmaceutically acceptable carrier.
- Another aspect of the invention is a metabolite of the compound or the pharmaceutical composition according to any one of paragraphs [0024]-[0072].
- Another aspect of the invention is a method of modulating the in vivo activity of a kinase, the method comprising administering to a subject an effective amount of a composition comprising at least one of: the compound according to any of paragraphs [0024]- [0071], the pharmaceutical composition according to paragraph [0072], a compound explicitly provided against in paragraph [0025], and a pharmaceutical composition comprising a compound, the composition of which was, explicitly provided against in paragraph [0025] and a pharmaceutically acceptable carrier.
- Another aspect of the invention is the method according to paragraph [0074], wherein the kinase is p70S6, Alct-1 and/or Akt-2 kinase.
- Another aspect of the invention is the method according to paragraph [0075], wherein modulating the in vivo activity of a kinase comprises inhibition of the kinase.
- Another aspect of the invention is a method of treating diseases or disorders associated with uncontrolled, abnormal, and/or unwanted cellular activities, the method comprising administering, to a mammal in need thereof, a therapeutically effective amount of a composition comprising at least one of: the compound according to any of paragraphs [0024]- [0071], the pharmaceutical composition according to paragraph [0072], a compound explicitly provided against in paragraph [0025], and a pharmaceutical composition comprising a compound explicitly provided against in paragraph [0025] and a pharmaceutically acceptable carrier.
- Another aspect of the invention is a method of screening for modulator of a kinase, preferably p70S6K, Akt-1 and/or Akt-2 kinase, the method comprising combining at least one of: the compound according to any of paragraphs [0024]-[0071], the pharmaceutical composition according to paragraph [0072], a compound explicitly provided against in paragraph [0025], and a pharmaceutical composition comprising a compound explicitly provided against in paragraph [0025] and a pharmaceutically acceptable carrier, and at least one candidate agent and a kinase and determining the effect of the candidate agent on the activity of said kinase.
- Another aspect of the invention is a method of inhibiting proliferative activity in a cell, the method comprising administering an effective amount of: the compound according to any of paragraphs [0024]-[0071], the pharmaceutical composition according to paragraph [0072], a compound explicitly provided against in paragraph [0025], and a pharmaceutical composition comprising a compound explicitly provided against in paragraph [0022] and a pharmaceutically acceptable carrier.
- Another aspect of the invention is a method of inhibiting abnormal metabolic activity in a cell, the method comprising administering an effective amount of: the compound according to any of paragraphs [0024]-[0071], the pharmaceutical composition according to paragraph [0072], a compound explicitly provided against in paragraph [0025], and a pharmaceutical composition comprising a compound explicitly provided against in paragraph [0025] and a pharmaceutically acceptable carrier.
- the symbol "-" means a single bond
- "s” means a triple bond.
- the symbol refers to a group on a double-bond as occupying either position on the terminus of a double bond to which the symbol is attached; that is, the geometry, E- or Z-, of the double bond is ambiguous.
- the symbol will be used at the end of the bond which was theoretically cleaved in order to separate the group from its parent structural formula.
- a substituent "R” may reside on any atom of the ring system, assuming replacement of a depicted, implied, or expressly defined hydrogen from one of the ring atoms, so long as a stable structure is formed.
- the "R” group may reside on either the 5-membered or the 6- membered ring of the fused ring system.
- the two "R's" may reside on any two atoms of the ring system, again assuming each replaces a depicted, implied, or expressly defined hydrogen on the ring.
- Alkyl is intended to include linear, branched, or cyclic hydrocarbon structures and combinations thereof, inclusively.
- C 8 alkyl may refer to an ra-octyl, wo-octyl, cyclohexylethyl, and the like.
- Lower alkyl refers to alkyl groups of from one to six carbon atoms. Examples of lower alkyl groups include methyl, ethyl, propyl, isopropyl, butyl, s- butyl, /-butyl, isobutyl, pentyl, hexyl and the like.
- Higher alkyl refers to alkyl groups containing more that eight carbon atoms.
- alkyl groups are those of C 20 or below.
- Cycloalkyl is a subset of alkyl and includes cyclic hydrocarbon groups of from three to thirteen carbon atoms. Examples of cycloalkyl groups include c-propyl, c-butyl, c-pentyl, norbornyl, adamantyl and the like, hi this application, alkyl refers to alkanyl, alkenyl, and alkynyl residues (and combinations thereof); it is intended to include cyclohexylmethyl, vinyl, allyl, isoprenyl, and the like.
- alkyl residue having a specific number of carbons when named, all geometric isomers having that number of carbons are intended to be encompassed; thus, for example, either “butyl” or “C 4 alkyl” is meant to include n-butyl, sec- butyl, isobutyl, ⁇ -butyl, isobutenyl and but-2-yne radicals; and for example, "propyl” or “C 3 alkyl” each include 7?-propyl, propenyl, and isopropyl.
- alkenyl and/or alkynyl descriptors are used in a particular definition of a group, for example "C 4 alkyl” along “C 4 alkenyl,” then C 4 alkenyl geometric isomers are not meant to be included in “C 4 alkyl,” but other 4-carbon isomers are, for example C 4 alkynyl.
- C 1-8 alkyl a particular group as “C 1-8 alkyl” while a preferred species may describe the same group as including, “C 1-8 alkyl,” “C 1-6 alkenyl” and "C 1-5 alkynyl.”
- Alkylene refers to straight or branched chain divalent radical consisting solely of carbon and hydrogen atoms, containing no unsaturation and having from one to ten carbon atoms, for example, methylene, ethylene, propylene, n-butylene and the like. Alkylene is a subset of alkyl, referring to the same residues as alkyl, but having two points of attachment and, specifically, fully saturated. Examples of alkylene include ethylene (-CH 2 CH 2 -), propylene (-CH 2 CH 2 CH 2 -), dimethylpropylene (-CH 2 C(CH 3 ) 2 CH 2 -), and cyclohexylpropylene (-CH 2 CH 2 CH(C 6 H 13 )).
- Alkylidene refers to a straight or branched chain unsaturated divalent radical consisting solely of carbon and hydrogen atoms, having from two to ten carbon atoms, for example, ethylidene, propylidene, n-butylidene, and the like. Alkylidene is a subset of alkyl, referring to the same residues as alkyl, but having two points of attachment and, specifically, double bond unsaturation. The unsaturation present includes at least one double bond.
- Alkylidyne refers to a straight or branched chain unsaturated divalent radical consisting solely of carbon and hydrogen atoms having from two to ten carbon atoms, for example, propylid-2-ynyl, ⁇ -butylid-1-ynyl, and the like. Alkylidyne is a subset of alkyl, referring to the same residues as alkyl, but having two points of attachment and, specifically, triple bond unsaturation. The unsaturation present includes at least one triple bond.
- radicals may contain alkyl substitution which itself contains unsaturation.
- 2- (2-phenylethynyl-but-3-enyl)-naphthalene (IUPAC name) contains an n-butylid-3-ynyl radical with a vinyl substituent at the 2-position of said radical.
- Alkoxy or "alkoxyl” refers to the group -O-alkyl, for example including from one to eight carbon atoms of a straight, branched, cyclic configuration, unsaturated chains, and combinations thereof attached to the parent structure through an oxygen atom. Examples include methoxy, ethoxy, propoxy, isopropoxy, cyclopropyloxy, cyclohexyloxy and the like. Lower-alkoxy refers to groups containing one to six carbons.
- Substituted alkoxy refers to the group -O-(substituted alkyl), the substitution on the alkyl group generally containing more than only carbon (as defined by alkoxy).
- One exemplary substituted alkoxy group is "polyalkoxy" or -O-optionally substituted alkylene-optionally substituted alkoxy, and includes groups such as -OCH 2 CH 2 OCH 3 , and glycol ethers such as polyethyleneglycol and -0(CH 2 CH 2 O) x CH 3 , where x is an integer of between about two and about twenty, in another example, between about two and about ten, and in a further example between about two and about five.
- Another exemplary substituted alkoxy group is hydroxyalkoxy or -OCH 2 (CH 2 ) y OH, where y is for example an integer of between about one and about ten, in another example y is an integer of between about one and about four.
- Acyl refers to groups of from one to ten carbon atoms of a straight, branched, cyclic configuration, saturated, unsaturated and aromatic and combinations thereof, attached to the parent structure through a carbonyl functionality.
- One or more carbons in the acyl residue may be replaced by nitrogen, oxygen or sulfur as long as the point of attachment to the parent remains at the carbonyl. Examples include acetyl, benzoyl, propionyl, isobutyryl, t- butoxycarbonyl, benzyloxycarbonyl and the like.
- Lower-acyl refers to groups containing one to six carbons.
- ⁇ -Amino Acids refer to naturally occurring and commercially available amino acids and optical isomers thereof. Typical natural and commercially available ⁇ -amino acids are glycine, alanine, serine, homoserine, threonine, valine, norvaline, leucine, isoleucine, norleucine, aspartic acid, glutamic acid, lysine, ornithine, histidine, arginine, cysteine, homocysteine, methionine, phenylalanine, homophenylalanine, phenylglycine, ortho-tyrosine, meta-tyrosine, para-tyrosine, tryptophan, glutamine, asparagine, proline and hydroxyproline.
- a "side chain of an ⁇ -amino acid” refers to the radical found on the ⁇ -carbon of an ⁇ -amino acid as defined above, for example, hydrogen (for glycine), methyl (for alanine), benzyl (for phenylalanine), and the like.
- Amino refers to the group -NH 2 .
- Substituted amino refers to the gro ⁇ p -N(H)R or -N(R)R where each R is independently selected from the group: optionally substituted alkyl, optionally substituted alkoxy, optionally substituted aryl, optionally substituted heterocyclyl, acyl, carboxy, alkoxycarbonyl, sulfanyl, sulfinyl and sulfonyl, for example, diethylamino, methylsulfonylamino, and furanyl-oxy-sulfonamino.
- Aryl refers to aromatic six- to fourteen-membered carbocyclic ring, for example, benzene, naphthalene, indane, tetralin, fluorene and the like, univalent radicals.
- univalent radicals the aforementioned ring examples are named, phenyl, naphthyl, indanyl, tetralinyl, and fluorenyl.
- Arylene generically refers to any aryl that has at least two groups attached thereto.
- phenylene refers to a divalent phenyl ring radical.
- a phenyl ene thus may have more than two groups attached, but is defined by a minimum of two non-hydrogen groups attached thereto.
- Arylalkyl refers to a residue in which an aryl moiety is attached to a parent structure via one of an alkylene, alkylidene, or alkylidyne radical. Examples include benzyl, phenethyl, phenylvinyl, phenylallyl and the like. Both the aryl, and the corresponding alkylene, alkylidene, or alkylidyne radical portion of an arylalkyl group may be optionally substituted.
- “Lower arylalkyl” refers to an arylalkyl where the "alkyl" portion of the group has one to six carbons; this can also be refered to as C 1-6 arylalkyl.
- Exo-alkenyl refers to a double bond that emanates from an annular carbon, and is not within the ring system, for example the double bond depicted in the formula below.
- two adjacent groups on an aromatic system may be fused together to form a ring structure.
- the fused ring structure may contain heteroatoms and may be optionally substituted with one or more groups.
- saturated carbons of such fused groups i.e. saturated ring structures
- fused-polycyclic or "fused ring system” refers to a polycyclic ring system that contains bridged or fused rings; that is, where two rings have more than one shared atom in their ring structures.
- fused-polycyclics and fused ring systems are not necessarily all aromatic ring systems.
- fused-polycyclics share a vicinal set of atoms, for example naphthalene or 1,2,3,4-tetrahydro-naphthalene.
- a spiro ring system is not a fused-polycyclic by this definition, but fused polycyclic ring systems of the invention may themselves have spiro rings attached thereto via a single ring atom of the fused- polycyclic.
- Halogen refers to fluorine, chlorine, bromine or iodine.
- Haloalkyl and haloaryl refer generically to alkyl and aryl radicals that are substituted with one or more halogens, respectively.
- dihaloaryl refers to aryl and alkyl substituted with a plurality of halogens, but not necessarily a plurality of the same halogen; thus 4-chloro-3 -fluorophenyl is within the scope of dihaloaryl.
- Heteroarylene generically refers to any heteroaryl that has at least two groups attached thereto.
- pyridylene refers to a divalent pyridyl ring radical.
- a pyridylene thus may have more than two groups attached, but is defined by a minimum of two non-hydrogen groups attached thereto.
- Heteroatom refers to O, S, N, or P.
- Heterocyclyl refers to a stable three- to fifteen-membered ring radical that consists of carbon atoms and from one to five heteroatoms selected from the group consisting of nitrogen, phosphorus, oxygen and sulfur.
- the heterocyclyl radical may be a monocyclic, bicyclic or tricyclic ring system, which may include fused or bridged ring systems as well as spirocyclic systems; and the nitrogen, phosphorus, carbon or sulfur atoms in the heterocyclyl radical may be optionally oxidized to various oxidation states.
- the group -S(O) 0-2 - refers to -S- (sulfide), -S(O)- (sulfoxide), and -SO 2 - (sulfone).
- nitrogens particularly but not exclusively, those defined as annular aromatic nitrogens, are meant to include their corresponding N-oxide form, although not explicitly defined as such in a particular example.
- annular nitrogen atoms may be optionally quaternized; and the ring radical may be partially or fully saturated or aromatic.
- heterocyclyl radicals include, but are not limited to, azetidinyl, acridinyl, benzodioxolyl, benzodioxanyl, benzofuranyl, carbazoyl, cinnolinyl, dioxolanyl, indolizinyl, naphthyridinyl, perhydroazepinyl, phenazinyl, phenothiazinyl, phenoxazinyl, phthalazinyl, pteridinyl, purinyl, quinazolinyl, quinoxalinyl, quinolinyl, isoquinolinyl, tetrazoyl, tetrahydroisoquinolyl, piperidinyl, piperazinyl, 2-oxopiperazinyl, 2-oxopiperidinyl, 2-oxopyrrolidinyl, 2-oxoazepinyl, azepiny
- Heteroalicyclic refers specifically to a non-aromatic heterocyclyl radical.
- a heteroalicyclic may contain unsaturation, but is not aromatic.
- Heteroaryl refers specifically to an aromatic heterocyclyl radical.
- Heterocyclylalkyl refers to a residue in which a heterocyclyl is attached to a parent structure via one of an alkylene, alkylidene, or alkylidyne radical. Examples include (4-methylpiperazin-l-yl) methyl, (morpholin-4-yl) methyl, (pyridine-4-yl) methyl, 2-(oxazolin-2-yl) ethyl, 4-(4-methylpiperazin-l-yl)-2-butenyl, and the like. Both the heterocyclyl, and the corresponding alkylene, alkylidene, or alkylidyne radical portion of a heterocyclylalkyl group may be optionally substituted.
- “Lower heterocyclylalkyl” refers to a heterocyclylalkyl where the “alkyl” portion of the group has one to six carbons.
- “Heteroalicyclylalkyl” refers specifically to a heterocyclylalkyl where the heterocyclyl portion of the group is non-aromatic; and “heteroarylalkyl” refers specifically to a heterocyclylalkyl where the heterocyclyl portion of the group is aromatic
- Such terms may be described in more than one way, for example, “lower heterocyclylalkyl” and “heterocyclyl Q ⁇ alkyl” are equivalent terms.
- saturated bridged ring system refers to a bicyclic or polycyclic ring system that is not aromatic. Such a system may contain isolated or conjugated unsaturation, but not aromatic or heteroaromatic rings in its core structure (but may have aromatic substitution thereon). For example, hexahydro-furo[3,2-b]furan, 2,3,3a,4,7,7a-hexahydro-lH-indene, 7-aza- bicyclo[2.2.1]heptane, and l,2,3,4,4a,5,8,8a-octahydro-naphthalene are all included in the class "saturated bridged ring system.
- Spirocyclyl or "spirocyclic ring” refers to a ring originating from a particular annular carbon of another ring.
- a ring atom of a saturated bridged ring system (rings B and B'), but not a bridgehead atom, can be a shared atom between the saturated bridged ring system and a spirocyclyl (ring A) attached thereto.
- a spirocyclyl can be carbocyclic or heteroalicyclic.
- Substituted alkyl, aryl, and heterocyclyl refer respectively to alkyl, aryl, and heterocyclyl, wherein one or more (for example up to about five, in another example, up to about three) hydrogen atoms are replaced by a substituent independently selected from: alkyl (for example, fiuoromethyl, hydroxypropyl, nitromethyl, aminoethyl and the like.), aryl (for example, 4-hydroxyphenyl, 2,3-difluorophenyl, and the like), arylalkyl (for example, 1- phenyl-ethyl, j9 ⁇ ra-methoxyphenyl ethyl and the like), heterocyclylalkyl (for example, 1- pyridin-3-yl-ethyl, N-ethylmorphonlino and the like), heterocyclyl (for example, 5-chloro- pyridin-3-yl, l-methyl-piperidin
- Sulfanyl refers to the groups: -S-(optionally substituted alkyl), -S-(optionally substituted aryl), and -S-(optionally substituted heterocyclyl).
- Sulfmyl refers to the groups: -S(O)-H, -S(O)-(o ⁇ tionally substituted alkyl), -S(O)-optionally substituted aryl), and -S(O)-(optionally substituted heterocyclyl).
- Sulfonyl refers to the groups: -S(O 2 )-H, -S(O 2 )-(optionally substituted alkyl), -S(O 2 )-optionally substituted aryl), -S(O 2 )-(optionally substituted heterocyclyl), -S(O 2 )-(optionally substituted alkoxy), -S(O 2 )-optionally substituted aryloxy), and -S(O 2 )-(optionally substituted heterocyclyloxy).
- Yield for each of the reactions described herein is expressed as a percentage of the theoretical yield.
- Some of the compounds of the invention may have imino, amino, oxo or hydroxy substituents off aromatic heterocyclyl systems.
- imino, amino, oxo or hydroxy substituents may exist in their corresponding tautomeric form, i.e., amino, imino, hydroxy or oxo, respectively.
- the compounds of the invention may have asymmetric carbon atoms, oxidized sulfur atoms or quaternized nitrogen atoms in their structure.
- the compounds of the invention and their pharmaceutically acceptable salts may exist as single stereoisomers, racemates, and as mixtures of enantiomers and diastereomers.
- the compounds may also exist as geometric isomers. All such single stereoisomers, racemates and mixtures thereof, and geometric isomers are intended to.be within the scope of this invention.,
- a particular group with its bonding structure is denoted as being bonded to two partners; that is, a divalent radical, for example, -OCH 2 -, then it is understood that either of the two partners may be bound to the particular group at one end, and the other partner is necessarily bound to the other end of the particular group, unless stated explicitly otherwise.
- divalent radicals are not to be construed as limited to the depicted orientation, for example "-OCH 2 -" is meant to mean not only "-OCH 2 -" as drawn, but also "-CH 2 O-.”
- optically active (R)- and (S)- isomers may be prepared using chiral synthons or chiral reagents, or resolved using conventional techniques.
- Enantiomers may be resolved by methods known to one of ordinary skill in the art, for example by: formation of diastereoisomeric salts or complexes which may be separated, for example, by crystallization; via formation of diastereoisomeric derivatives which may be separated, for example, by crystallization, selective reaction of one enantiomer with an enanliomer-specific reagent, for example enzymatic oxidation or reduction, followed by separation of the modified and unmodified enantiomers; or gas-liquid or liquid chromatography in a chiral environment, for example on a chiral support, such as silica with a bound chiral ligand or in the presence of a chiral solvent.
- enantiomer may be synthesized by asymmetric synthesis using optically active reagents, substrates, catalysts or solvents, or by converting on enantiomer to the other by asymmetric transformation.
- enantiomers enriched in a particular enantiomer, the major component enantiomer may be further enriched (with concomitant loss in yield) by recrystallization.
- Patient for the purposes of the present invention includes humans and other animals, particularly mammals, and other organisms. Thus the methods are applicable to both human therapy and veterinary applications. In a preferred embodiment the patient is a mammal, and in a most preferred embodiment the patient is human.
- Kinase-dependent diseases or conditions refer to pathologic conditions that depend on the activity of one or more protein kinases. Kinases either directly or indirectly participate in the signal transduction pathways of a variety of cellular activities including proliferation, adhesion, migration, differentiation and invasion. Diseases associated with kinase activities include tumor growth, the pathologic neovascularization that supports solid tumor growth, and associated with other diseases where excessive local vascularization is involved such as ocular diseases (diabetic retinopathy, age-related macular degeneration, and the like) and inflammation (psoriasis, rheumatoid arthritis, and the like).
- ocular diseases diabetic retinopathy, age-related macular degeneration, and the like
- inflammation psoriasis, rheumatoid arthritis, and the like.
- phosphatases can also play a role in "kinase-dependent diseases or conditions" as cognates of kinases; that is, kinases phosphorylate and phosphatases dephosphorylate, for example protein substrates. Therefore compounds of the invention, while modulating kinase activity as described herein, may also modulate, either directly or indirectly, phosphatase activity. This additional modulation, if present, may be synergistic (or not) to activity of compounds of the invention toward a related or otherwise interdependent kinase or kinase family, m any case, as stated previously, the compounds of the invention are useful for treating diseases characterized in part by abnormal levels of cell metabolism, proliferation (i.e. tumor growth), programmed cell death (apoptosis), cell migration and invasion and angiogenesis associated with tumor growth.
- proliferation i.e. tumor growth
- apoptosis programmed cell death
- “Therapeutically effective amount” is an amount of a compound of the invention, that when administered to a patient, ameliorates a symptom of the disease.
- the amount of a compound of the invention which constitutes a “therapeutically effective amount” will vary depending on the compound, the disease state and its severity, the age of the patient to be treated, and the like.
- the therapeutically effective amount can be determined routinely by one of ordinary skill in the art having regard to his own knowledge and to this disclosure.
- Cardiac sarcoma (angiosarcoma, fibrosarcoma, rhabdomyosarcoma, liposarcoma), myxoma, rhabdomyoma, fibroma, lipoma and teratoma;
- Lung bronchogenic carcinoma (squamous cell, undifferentiated small cell, undifferentiated large cell, adenocarcinoma), alveolar (bronchiolar) carcinoma, bronchial adenoma, sarcoma, lymphoma, chondromatous hanlartoma, mesothelioma;
- Gastrointestinal esophagus (squamous cell carcinoma, adenocarcinoma, leiomyosarcoma, lymphoma), stomach (carcinoma, lymphoma, leiomyosarcoma), pancreas
- “Pharmaceutically acceptable acid addition salt” refers to those salts that retain the biological effectiveness of the free bases and that are not biologically or otherwise undesirable, formed with inorganic acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, and the like, as well as organic acids such as acetic acid, trifluoroacetic acid, propionic acid, glycolic acid, pyruvic acid, oxalic acid, maleic acid, malonic acid, succinic acid, fumaric acid, tartaric acid, citric acid, benzoic acid, cinnamic acid, mandelic acid, methanesulfonic acid, ethanesulfonic acid, p-toluenesulfonic acid, salicylic acid and the like.
- inorganic acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, and the like
- organic acids such as acetic acid, trifluoro
- “Pharmaceutically acceptable base addition salts” include those derived from inorganic bases such as sodium, potassium, lithium, ammonium, calcium, magnesium, iron, zinc, copper, manganese, aluminum salts and the like. Exemplary salts are the ammonium, potassium, sodium, calcium, and magnesium salts.
- Salts derived from pharmaceutically acceptable organic non-toxic bases include, but are not limited to, salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines and basic ion exchange resins, such as isopropylamine, trimethylamine, diethylamine, triethylamine, tripropylamine, ethanolamine, 2-dimethylaminoethanol, 2-diethylaminoethanol, dicyclohexylamine, lysine, arginine, histidine, caffeine, procaine, hydrabamine, choline, betaine, ethylenediamine, glucosamine, methylglucamine, theobromine, purines, piperazine, piperidine, N-ethylpiperidine, polyamine resins, and the like.
- salts of primary, secondary, and tertiary amines substituted amines including naturally occurring substituted amines, cyclic amines
- Exemplary organic bases are isopropylamine, diethylamine, ethanolamine, trimethylamine, dicyclohexylamine, choline, and caffeine.
- “Prodrug” refers to compounds that are transformed (typically rapidly) in vivo to yield the parent compound of the above formulae, for example, by hydrolysis in blood. Common examples include, but are not limited to, ester and amide forms of a compound having an active form bearing a carboxylic acid moiety.
- esters of the compounds of this invention include, but are not limited to, alkyl esters (for example with between about one and about six carbons) wherein the alkyl group is a straight or branched chain. Acceptable esters also include cycloalkyl esters and arylalkyl esters such as, but not limited to benzyl.
- pharmaceutically acceptable amides of the compounds of this invention include, but are not limited to, primary amides, and secondary and tertiary alkyl amides (for example with between about one and about six carbons). Amides and esters of the compounds of the present invention may be prepared according to conventional methods. A thorough discussion of prodrugs is provided in T. Higuchi and V.
- Methodabolite refers to the break-down or end product of a compound or its salt produced by metabolism or biotransformation in the animal or human body; for example, biotransformation to a more polar molecule such as by oxidation, reduction, or hydrolysis, or to a conjugate (see Goodman and Gilman, "The Pharmacological Basis of Therapeutics” 8. sup. th Ed., Pergamon Press, Gilman et al. (eds), 1990 for a discussion of biotransformation).
- the metabolite of a compound of the invention or its salt may be the biologically active form of the compound in the body.
- a prodrug may be used such that the biologically active form, a metabolite, is released in vivo.
- a biologically active metabolite is discovered serendipitously, that is, no prodrug design per se was undertaken.
- An assay for activity of a metabolite of a compound of the present invention is known to one of skill in the art in light of the present disclosure.
- the compounds of the present invention can exist in unsolvated as well as solvated forms with pharmaceutically acceptable solvents such as water, ethanol, and the like. In general, the solvated forms are considered equivalent to the unsolvated forms for the purposes of the present invention.
- the present invention cover compounds made either using standard organic synthetic techniques, including combinatorial chemistry or by biological methods, such as bacterial digestion, metabolism, enzymatic conversion, and the like.
- Treating covers the treatment of a disease-state in a human, which disease-state is characterized by abnormal cellular proliferation and/or invasion, or metabolism and includes at least one of: (i) preventing the disease-state from occurring in a human, in particular, when such human is predisposed to the disease-state but has not yet been diagnosed as having it; (ii) inhibiting the disease-state, i.e., arresting its development; and (iii) relieving the disease-state, i.e., causing regression of the disease-state.
- Such suitable x-ray quality crystals can be used as part of a method of identifying a candidate agent capable of binding to and modulating the activity of kinases.
- Such methods may be characterized by the following aspects: a) introducing into a suitable computer program, information defining a ligand binding domain of a kinase in a conformation (e.g.
- aspects a-d are not necessarily carried out in the aforementioned order. Such methods may further entail: performing rational drug design with the model of the three- dimensional structure, and selecting a potential candidate agent in conjunction with computer modeling.
- Such methods may further entail: employing a candidate agent, so-determined to fit spatially into the ligand binding domain, in a biological activity assay for kinase modulation, and determining whether said candidate agent modulates kinase activity in the assay. Such methods may also include administering the candidate agent, determined to modulate kinase activity, to a mammal suffering from a condition treatable by kinase modulation, such as those described above.
- compounds of the invention can be used in a method of evaluating the ability of a test agent to associate with a molecule or molecular complex comprising a ligand binding domain of a kinase.
- a method may be characterized by the following aspects: a) creating a computer model of a kinase binding pocket using structure coordinates obtained from suitable x-ray quality crystals of the kinase, b) employing computational algorithms to perform a fitting operation between the test agent and the computer model of the binding pocket, and c) analyzing the results of the fitting operation to quantify the association between the test agent and the computer model of the binding pocket.
- Administration of the compounds of the invention, or their pharmaceutically acceptable salts, in pure form or in an appropriate pharmaceutical composition can be carried out via any of the accepted modes of administration or agents for serving similar utilities.
- administration can be, for example, orally, nasally, parenterally (intravenous, intramuscular, or subcutaneous), topically, transdermally, intravaginally, intravesically, intracistemally, or rectally, in the form of solid, semi-solid, lyophilized powder, or liquid dosage forms, such as for example, tablets, suppositories, pills, soft elastic and hard gelatin capsules, powders, solutions, suspensions, or aerosols, or the like, preferably in unit dosage forms suitable for simple administration of precise dosages.
- compositions will include a conventional pharmaceutical carrier or excipient and a compound of the invention as the/an active agent, and, in addition, may include other medicinal agents, pharmaceutical agents, carriers, adjuvants, etc.
- Compositions of the invention may be used in combination with anticancer or other agents that are generally administered to a patient being treated for cancer.
- Adjuvants include preserving, wetting, suspending, sweetening, flavoring, perfuming, emulsifying, and dispensing agents. Prevention of the action of microorganisms can be ensured by various antibacterial and antifungal agents, for example, parabens, chlorobutanol, phenol, sorbic acid, and the like. It may also be desirable to include isotonic agents, for example sugars, sodium chloride, and the like. Prolonged absorption of the injectable pharmaceutical form can be brought about by the use of agents delaying absorption, for example, aluminum monostearate and gelatin.
- a pharmaceutical composition of the invention may also contain minor amounts of auxiliary substances such as wetting or emulsifying agents, pH buffering agents, antioxidants, and the like, such as, for example, citric acid, sorbitan monolaurate, triethanolamine oleate, butylalted hydroxytoluene, etc.
- auxiliary substances such as wetting or emulsifying agents, pH buffering agents, antioxidants, and the like, such as, for example, citric acid, sorbitan monolaurate, triethanolamine oleate, butylalted hydroxytoluene, etc.
- compositions suitable for parenteral injection may comprise physiologically acceptable sterile aqueous or nonaqueous solutions, dispersions, suspensions or emulsions, and sterile powders for reconstitution into sterile injectable solutions or dispersions.
- suitable aqueous and nonaqueous carriers, diluents, solvents or vehicles include water, eth'anol, polyols (propyleneglycol, polyethyleneglycol, glycerol, and the like), suitable mixtures thereof, vegetable oils (such as olive oil) and injectable organic esters such as ethyl oleate.
- Proper fluidity can be maintained, for example, by the use of a coating such as lecithin, by the maintenance of the required particle size in the case of dispersions and by the use of surfactants.
- One preferable route of administration is oral, using a convenient daily dosage regimen that can be adjusted according to the degree of severity of the disease-state to be treated.
- Solid dosage forms for oral administration include capsules, tablets, pills, powders, and granules.
- the active compound is admixed with at least one inert customary excipient (or carrier) such as sodium citrate or dicalcium phosphate or
- fillers or extenders as for example, starches, lactose, sucrose, glucose, mannitol, and silicic acid
- binders as for example, cellulose derivatives, starch, alignates, gelatin, polyvinylpyrrolidone, sucrose, and gum acacia
- humectants as for example, glycerol
- disintegrating agents as for example, agar-agar, calcium carbonate, potato or tapioca starch, alginic acid, croscarmellose sodium, complex silicates, and sodium carbonate
- solution retarders as for example paraffin
- absorption accelerators as for example,
- Solid dosage forms as described above can be prepared with coatings and shells, such as enteric coatings and others well known in the art. They may contain pacifying agents, and can also be of such composition that they release the active compound or compounds in a certain part of the intestinal tract in a delayed manner. Examples of embedded compositions that can be used are polymeric substances and waxes. The active compounds can also be in microencapsulated form, if appropriate, with one or more of the above-mentioned excipients.
- Liquid dosage forms for oral administration include pharmaceutically acceptable emulsions, solutions, suspensions, syrups, and elixirs. Such dosage forms are prepared, for example, by dissolving, dispersing, etc., a compound(s) of the invention, or a pharmaceutically acceptable salt thereof, and optional pharmaceutical adjuvants in a carrier, such as, for example, water, saline, aqueous dextrose, glycerol, ethanol and the like; solubilizing agents and emulsifiers, as for example, ethyl alcohol, isopropyl alcohol, ethyl carbonate, ethyl acetate, benzyl alcohol, benzyl benzoate, propyleneglycol, 1,3-butyleneglycol, dimethylformamide; oils, in particular, cottonseed oil, groundnut oil, corn germ oil, olive oil, castor oil and sesame oil, glycerol, tetrahydrofurfuryl alcohol
- Suspensions in addition to the active compounds, may contain suspending agents, as for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar and tragacanth, or mixtures of these substances, and the like.
- suspending agents as for example, ethoxylated isostearyl alcohols, polyoxyethylene sorbitol and sorbitan esters, microcrystalline cellulose, aluminum metahydroxide, bentonite, agar-agar and tragacanth, or mixtures of these substances, and the like.
- compositions for rectal administrations are, for example, suppositories that can be prepared by mixing the compounds of the present invention with for example suitable non- irritating excipients or carriers such as cocoa butter, polyethyleneglycol or a suppository wax, which are solid at ordinary temperatures but liquid at body temperature and therefore, melt while in a suitable body cavity and release the active component therein.
- suitable non- irritating excipients or carriers such as cocoa butter, polyethyleneglycol or a suppository wax, which are solid at ordinary temperatures but liquid at body temperature and therefore, melt while in a suitable body cavity and release the active component therein.
- Dosage forms for topical administration of a compound of this invention include ointments, powders, sprays, and inhalants.
- the active component is admixed under sterile conditions with a physiologically acceptable carrier and any preservatives, buffers, or propellants as may be required.
- Ophthalmic formulations, eye ointments, powders, and solutions are also contemplated as being within the scope of this invention.
- the pharmaceutically acceptable compositions will contain about 1% to about 99% by weight of a compound(s) of the invention, or a pharmaceutically acceptable salt thereof, and 99% to 1% by weight of a suitable pharmaceutical excipient.
- the composition will be between about 5% and about 75% by weight of a compound(s) of the invention, or a pharmaceutically acceptable salt thereof, with the rest being suitable pharmaceutical excipients.
- composition to be administered will, in any event, contain a therapeutically effective amount of a compound of the invention, or a pharmaceutically acceptable salt thereof, for treatment of a disease-state in accordance with the teachings of this invention.
- the compounds of the invention are administered in a therapeutically effective amount which will vary depending upon a variety of factors including the activity of the specific compound employed, the metabolic stability and length of action of the compound, the age, body weight, general health, sex, diet, mode and time of administration, rate of excretion, drug combination, the severity of the particular disease-states, and the host undergoing therapy.
- the compounds of the present invention can be administered to a patient at dosage levels in the range of about 0.1 to about 1,000 mg per day. For a normal human adult having a body weight of about 70 kilograms, a dosage in the range of about 0.01 to about 100 mg per kilogram of body weight per day is an example. The specific dosage used, however, can vary.
- the dosage can depend on a number of factors including the requirements of the patient, the severity of the condition being treated, and the pharmacological activity of the compound being used.
- the determination of optimum dosages for a particular patient is well known to one of ordinary skill in the art.
- the protein is bound to a support, and a compound of the invention is added to the assay.
- the compound of the invention is bound to the support and the protein is added.
- Classes of candidate agents among which novel binding agents may be sought include specific antibodies, non-natural binding agents identified in screens of chemical libraries, peptide analogs, etc. Of particular interest are screening assays for candidate agents that have a low toxicity for human cells.
- assays may be used for this purpose, including labeled in vitro protein-protein binding assays, electrophoretic mobility shift assays, immunoassays for protein binding, functional assays (phosphorylation assays, etc.) and the like.
- the determination of the binding of the candidate agent to, for example, p70S6K protein may be done in a number of ways.
- the candidate agent (the compound of the invention) is labeled, for example, with a fluorescent or radioactive moiety and binding determined directly.
- a labeled agent for example a compound of the invention in which at least one atom has been replaced by a detectable isotope
- washing off excess reagent for example a compound of the invention in which at least one atom has been replaced by a detectable isotope
- Various blocking and washing steps may be utilized as is known in the art.
- label herein is meant that the compound is either directly or indirectly labeled with a label which provides a detectable signal, for example, radioisotope, fluorescent tag, enzyme, antibodies, particles such as magnetic particles, chemiluminescent tag, or specific binding molecules, and the like.
- Specific binding molecules include pairs, such as biotin and streptavidin, digoxin and antidigoxin, and the like.
- the complementary member would normally be labeled with a molecule which provides for detection, in accordance with known procedures, as outlined above.
- the label can directly or indirectly provide a detectable signal.
- p70S6K protein may be labeled at tyrosine positions using I, or with fluorophores.
- more than one component may be labeled with different labels; using I for the proteins, for example, and a fluorophor for the candidate agents.
- the compounds of the invention may also be used as competitors to screen for additional drug candidates, "candidate bioactive agent” or “drug candidate” or grammatical equivalents as used herein describe any molecule, e.g., protein, oligopeptide, small organic molecule, polysaccharide, polynucleotide, etc., to be tested for bioactivity. They may be capable of directly or indirectly altering the cellular proliferation or metabolic phenotype or the expression of a cellular proliferation or metabolic sequence, including both nucleic acid sequences and protein sequences. In other cases, alteration of cellular proliferation or metabolic protein binding and/or activity is screened. In the case where protein binding or activity is screened, some embodiments exclude molecules already known to bind to that particular protein. Exemplary embodiments of assays described herein include candidate agents, which do not bind the target protein in its endogenous native state, termed herein as "exogenous" agents. In one example, exogenous agents further exclude antibodies to p70S6K.
- Candidate agents can encompass numerous chemical classes, though typically they are organic molecules having a molecular weight of more than about 100 and less than about 2,500 daltons.
- Candidate agents comprise functional groups necessary for structural interaction with proteins, particularly hydrogen bonding and lipophilic binding, and typically include at least an amine, carbonyl, hydroxyl, ether, or carboxyl group, for example at least two of the functional chemical groups.
- the candidate agents often comprise cyclical carbon or heterocyclyl structures and/or aromatic or polyaromatic structures substituted with one or more of the above functional groups.
- Candidate agents are also found among biomolecules including peptides, saccharides, fatty acids, steroids, purines, pyrimidines, derivatives, structural analogs, or combinations thereof.
- Candidate agents are obtained from a wide variety of sources including libraries of synthetic or natural compounds. For example, numerous means are available for random and directed synthesis of a wide variety of organic compounds and biomolecules, including expression of randomized oligonucleotides. Alternatively, libraries of natural compounds in the form of bacterial, fungal, plant and animal extracts are available or readily produced. Additionally, natural or synthetically produced libraries and compounds are readily modified through conventional chemical, physical and biochemical means. Known pharmacological agents may be subjected to directed or random chemical modifications, such as acylation, alkylation,.esterif ⁇ cation, amidification to produce structural analogs.
- the binding of the candidate agent is determined through the use of competitive binding assays.
- the competitor is a binding moiety known to bind to p70S6K, such as an antibody, peptide, binding partner, ligand, etc. Under certain circumstances, there may be competitive binding as between the candidate agent and the binding moiety, with the binding moiety displacing the candidate agent.
- the candidate agent is labeled. Either the candidate agent, or the competitor, or both, is added first to p70S6K protein for a time sufficient to allow binding, if present. Incubations may be performed at any temperature that facilitates optimal activity, typically between 4°C and 40 0 C.
- Incubation periods are selected for optimum activity, but may also be optimized to facilitate rapid high throughput screening. Typically between 0.1 and 1 hour will be sufficient. Excess reagent is generally removed or washed away. The second component is then added, and the presence or absence of the labeled component is followed, to indicate binding.
- the competitor is added first, followed by the candidate agent.
- Displacement of the competitor is an indication the candidate agent is binding to p70S6K and thus is capable of binding to, and potentially modulating, the activity of the p70S6K.
- either component can be labeled.
- the presence of label in the wash solution indicates displacement by the agent.
- the candidate agent is labeled, the presence of the label on the support indicates displacement.
- the candidate agent is added first, with incubation and washing, followed by the competitor.
- the absence of binding by the competitor may indicate the candidate agent is bound to p70S6K with a higher affinity.
- the candidate agent is labeled, the presence of the label on the support, coupled with a lack of competitor binding, may indicate the candidate agent is capable of binding to p70S6K.
- p70S6K It may be of value to identify the binding site of p70S6K. This can be done in a variety of ways. In one embodiment, once p70S6K has been identified as binding to the candidate agent, the p70S6K is fragmented or modified and the assays repeated to identify the necessary components for binding.
- Modulation is tested by screening for candidate agents capable of modulating the activity of p70S6K comprising the steps of combining a candidate agent with p70S6K, as above, and determining an alteration in the biological activity of the p70S6K.
- the candidate agent should both bind to (although this may not be necessary), and alter its biological or biochemical activity as defined herein.
- the methods include both in vitro screening methods and in vivo screening of cells for alterations in cell viability, morphology, and the like.
- differential screening may be used to identify drug candidates that bind to native p70S6K, but cannot bind to modified p70S6K.
- Positive controls and negative controls can be used in the assays. For example, all control and test samples are performed in at least triplicate to obtain statistically significant results. Incubation of samples is for a time sufficient for the binding of the agent to the protein. Following incubation, samples are washed free of non-specifically bound material and the amount of bound, generally labeled agent determined. For example, where a radiolabel is employed, the samples can be counted in a scintillation counter to determine the amount of bound compound.
- a variety of other reagents can be included in the screening assays. These include reagents like salts, neutral proteins, e.g., albumin, detergents, etc which may be used to facilitate optimal protein-protein binding and/or reduce non-specific or background interactions. Also reagents that otherwise improve the efficiency of the assay, such as protease inhibitors, nuclease inhibitors, anti-microbial agents, etc., may be used. The mixture of components can be added in any order that provides for the requisite binding.
- Such suitable x-ray quality crystals can be used as part of a method of identifying a candidate agent capable of binding to and modulating the activity of p70S6 kinases.
- Such methods may be characterized by the following aspects: a) introducing into a suitable computer program, information defining a ligand binding domain of a p70S6 kinase in a conformation (e.g.
- aspects a-d are not necessarily carried out in the aforementioned order. Such methods may further entail: performing rational drug design with the model of the three-dimensional structure, and selecting a potential candidate agent in conjunction with computer modeling.
- Such methods may further entail: employing a candidate agent, so-determined to fit spatially into the ligand binding domain, in a biological activity assay for p70S6 kinase modulation, and determining whether said candidate agent modulates p70S6 kinase activity in the assay. Such methods may also include administering the candidate agent, determined to modulate p70S6 kinase activity, to a mammal suffering from a condition treatable by p70S6 kinase modulation, such as those described above.
- compounds of the invention can be used in a method of evaluating the ability of a test agent to associate with a molecule or molecular complex comprising a ligand binding domain of a p70S6 kinase.
- a method may be characterized by the following aspects: a) creating a computer model of a p70S6 kinase binding pocket using structure coordinates obtained from suitable x-ray quality crystals of the p70S6 kinase, b) employing computational algorithms to perform a fitting operation between the test agent and the computer model of the binding pocket, and c) analyzing the results of the fitting operation to quantify the association between the test agent and the computer model of the binding pocket.
- Scheme 1 depicts a general synthetic route for exemplary compounds of the invention according to Formula I, and are not intended to be limiting. Specific examples are described subsequently to this general synthetic description. With the description of the general route and the specific examples thereafter, one of ordinary skill in the art would be able to make compounds of the invention as described in the detailed description and claims.
- Scheme 1 shows that in general, compounds according to Formula I can be made, for example, via a linear route, by reacting a compound of Formula X, having a Boc protecting group and an appropriate leaving group from Q 2 , with a nucleophile of group V. The resulting compound is then dissolved in an appropriate solvent such as 1,4-dioxane, and the solution is reacted with Et 3 N and 3-Ri-4-chloro-lR 2 -pyrazolo[3,4-d]pyrimidine to arrive at a compound of Formula I.
- Formula I, Ri-R 6 , Q 1 , Q 2 , L, V and W are as described above.
- 2-Bromo-(p-chlorophenyl)-acetic acid methyl ester was prepared from (4-chlorophenyl)-acetic acid methyl ester (2.5 g, 13.5 mmol) and NBS (2.52 g, 14.1 mmol) in CCl 4 .
- reaction mixture After stirring for several minutes, the reaction mixture became homogeneous. After reaction time of 2.5 h, TLC analysis revealed a lower Rf spot.
- the reaction mixture was diluted with CH 2 Cl 2 (100 mL) and NaHCO 3 (sat'd aq., 100 mL). The organic layer was washed with brine (50 mL). The combined aqueous layers were extracted with CH 2 Cl 2 (2 x 50 mL). The combined organic layers were dried over anhydrous Na 2 SO 4 , filtered, and concentrated to give yellowish oil which solidified overnight to give an off-white solid which was used in subsequent reactions without further purification. (1.4O g, 100%).
- Benzyl alcohol (2) To a round bottom flask, 1.24 g (3.84 mmol) of ketone I was dissolved in 40 rnL of ethanol. To this solution, 145 mg (3.84 mmol) OfNaBH 4 was added and stirred at room temperature for 2 h. The crude reaction was concentrated in vacuum. Ethyl acetate and water were added, and the organic layer was washed with H 2 O (2x) and brine. The organic layer was dried with Na 2 SO 4 , filtered and concentrated to afford 1.25 g (3.84 mmol) of the alcohol 2.
- N-Amino-Piperazine derivatives are prepared according to the general synthetic route above in light of the methods of preparation set forth in Example 1.
- kinase assays were performed by measurement of incorporation of ⁇ - 3 J 3 J ⁇ P ATP into immobilized myelin basic protein (MBP).
- MBP myelin basic protein
- High binding white 384 well plates (Greiner) were coated with MBP (Sigma #M-1891) by incubation of 60 ⁇ l/well of 20 ⁇ g/ml MBP in Tris- buffered saline (TBS; 5OmM Tris pH 8.0, 138mM NaCl, 2.7mM KCl) for 24 hours at 4° C. Plates were washed 3X with lOO ⁇ l TBS.
- kinase reactions were carried out in a total volume of 34 ⁇ l in kinase buffer (5mM Hepes pH 7.6, 15mM NaCl, 0.01% bovine gamma globulin (Sigma #1-5506), 1OmM MgCl 2 , ImM DTT, 0.02% TritonX-100).
- Compound dilutions were performed in DMSO and added to assay wells to a final DMSO concentration of 1%. Each data point was measured in duplicate, and at least two duplicate assays were performed for each individual compound determination. Enzyme was added to final concentrations of 1OnM or 2OnM, for example.
- the above assay procedure can be used to determine the IC 50 for inhibition and/or the inhibition constant, K 1 .
- the IC 50 is defined as the concentration of compound required to reduce the enzyme activity by 50% under the conditions of the assay.
- Exemplary compositions have IC 50 's of, for example, less than about 100 ⁇ M, less than about 10 ⁇ M, less than about 1 ⁇ M, and further for example having IC 50 's of less than about 100 nM, and still further, for example, less than about 10 nM.
- the K, for a compound may be determined from the IC 5O based on three assumptions. First, only one compound molecule binds to the enzyme and there is no cooperativity.
- the concentrations of active enzyme and the compound tested are known (i.e., there are no significant amounts of impurities or inactive forms in the preparations).
- the enzymatic rate of the enzyme-inhibitor complex is zero.
- the rate (i.e., compound concentration) data are fitted to equation (1) below; where V is the observed rate, V max , is the rate of the free enzyme, I 0 is the inhibitor concentration, E 0 is the enzyme concentration, and Ka is the dissociation constant of the enzyme-inhibitor complex.
- ATP concentrations for each assay are selected to be close to the Michaelis-Menten constant (K M ) for each individual kinase.
- K M Michaelis-Menten constant
- Dose-response experiments are performed at 10 different inhibitor concentrations in a 384-well plate format. The data are fitted to four-parameter equation (2) below; where Y is the observed signal, X is the inhibitor concentration, Min is the background signal in the absence of enzyme (0% enzyme activity), Max is the signal in the absence of inhibitor (100% enzyme activity), IC 50 is the inhibitor concentration at 50% enzyme inhibition and H represents the empirical Hill's slope to measure the cooperativity. Typically H is close to unity.
- Biochemical activity of p70S6 kinase was assessed using a Luciferase-Coupled Chemiluminescent Kinase assay (LCCA) format. Kinase activity was measured as the percent ATP remaining following the kinase reaction. Remaining ATP was detected by luciferase- luciferin-coupled chemiluminescence.
- LCCA Luciferase-Coupled Chemiluminescent Kinase assay
- reaction was initiated by mixing test compounds, 5 ⁇ M ATP, 5 ⁇ M RRRLSSLRA peptide and 12nM p70S6K (baculovirus expressed human p70S6 kinase domainresidues 1-421, containing a T412E mutation) in a 2OuL assay buffer (2OmM Tris-HCL pH 7.5, 1OmM MgCl 2 , 0.02% Triton X-100, 10OmM DTT, 2mM MnCl 2 ). The mixture is incubated at ambient temperature for 2hours after which 20 ⁇ L luciferase-luciferin mix is added and the chemiluminescent signal read using a Wallac Victor reader.
- test compounds 5 ⁇ M ATP, 5 ⁇ M RRRLSSLRA peptide and 12nM p70S6K (baculovirus expressed human p70S6 kinase domainresidues 1-421, containing a T412E mutation)
- the luciferase-luciferin mix consists of 50 mM HEPES, pH 7.8, 8.5 ⁇ g/mL oxalic acid (pH 7.8), 5 (or 50) mM DTT, 0.4% Triton X-100, 0.25 mg/mL coenzyme A, 63 uM AMP, 28 ⁇ g/mL luciferin and 40,000 units/mL luciferase.
- Aktl kinase activity was assessed using a Luciferase-Coupled Chemiluminescent Kinase assay (LCCA) format. Kinase activity was measured as the percent ATP remaining following the kinase reaction. Remaining ATP was detected by luciferase- luciferin-coupled chemiluminescence.
- LCCA Luciferase-Coupled Chemiluminescent Kinase assay
- the reaction was initiated by mixing test compounds, l ⁇ M ATP, 5 ⁇ M crosstide peptide and 1.3nM Aktl (baculovirus expressed human Aktl kinase domain residues R144-A480, activated with PDKl) in a 2OuL assay buffer (2OmM Tris-HCL pH 7.5, 1OmM MgC12, 0.01% Triton X-100, ImM DTT, 3mM MnC12). The mixture is incubated at ambient temperature for 3 hours after which 20 ⁇ L luciferase-luciferin mix is added and the chemiluminescent signal read using a Wallac Victor2 reader.
- 2OuL assay buffer 2OmM Tris-HCL pH 7.5, 1OmM MgC12, 0.01% Triton X-100, ImM DTT, 3mM MnC12.
- the luciferase- luciferin mix consists of 50 niM HEPES, pH 7.8, 8.5 ⁇ g/mL oxalic acid (pH 7.8), 5 (or 50) mM DTT, 0.4% Triton X-100, 0.25 mg/mL coenzyme A, 63 uM AMP, 28 ⁇ g/mL luciferin and 40,000 units/mL luciferase.
- Akt2 Kinase Assay 86 Biochemical activity of Akt2 kinase was assessed using a Luciferase-Coupled Chemiluminescent Kinase assay (LCCA) format. Kinase activity was measured as the percent ATP remaining following the kinase reaction. Remaining ATP was detected by luciferase- luciferin-coupled chemiluminescence.
- LCCA Luciferase-Coupled Chemiluminescent Kinase assay
- reaction was initiated by mixing test compounds, 2 ⁇ M ATP, 5 ⁇ M crosstide peptide and 1OnM Akt2 (baculovirus expressed human Akt2 kinase domain residues K146-R480) in a 2OuL assay buffer (2OmM Tris-HCL pH 7.5, 1OmM MgC12, 0.03% Triton X-100, ImM DTT, 3mM MnC12).
- 2OuL assay buffer 2OmM Tris-HCL pH 7.5, 1OmM MgC12, 0.03% Triton X-100, ImM DTT, 3mM MnC12.
- the luciferase-luciferin mix consists of 50 mM HEPES, pH 7.8, 8.5 ⁇ g/mL oxalic acid (pH 7.8), 5 (or 50) mM DTT, 0.4% Triton X-100, 0.25 mg/mL coenzyme A, 63 uM AMP, 28 ⁇ g/mL luciferin and 40,000 units/mL luciferase.
Abstract
Description


























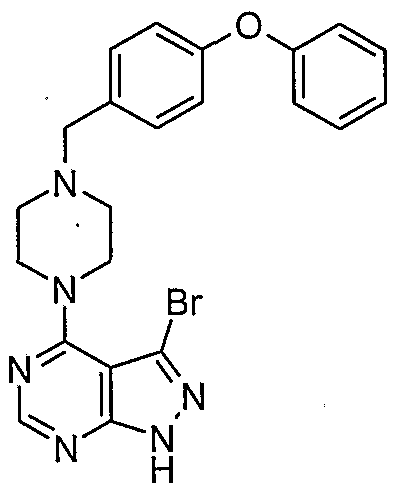






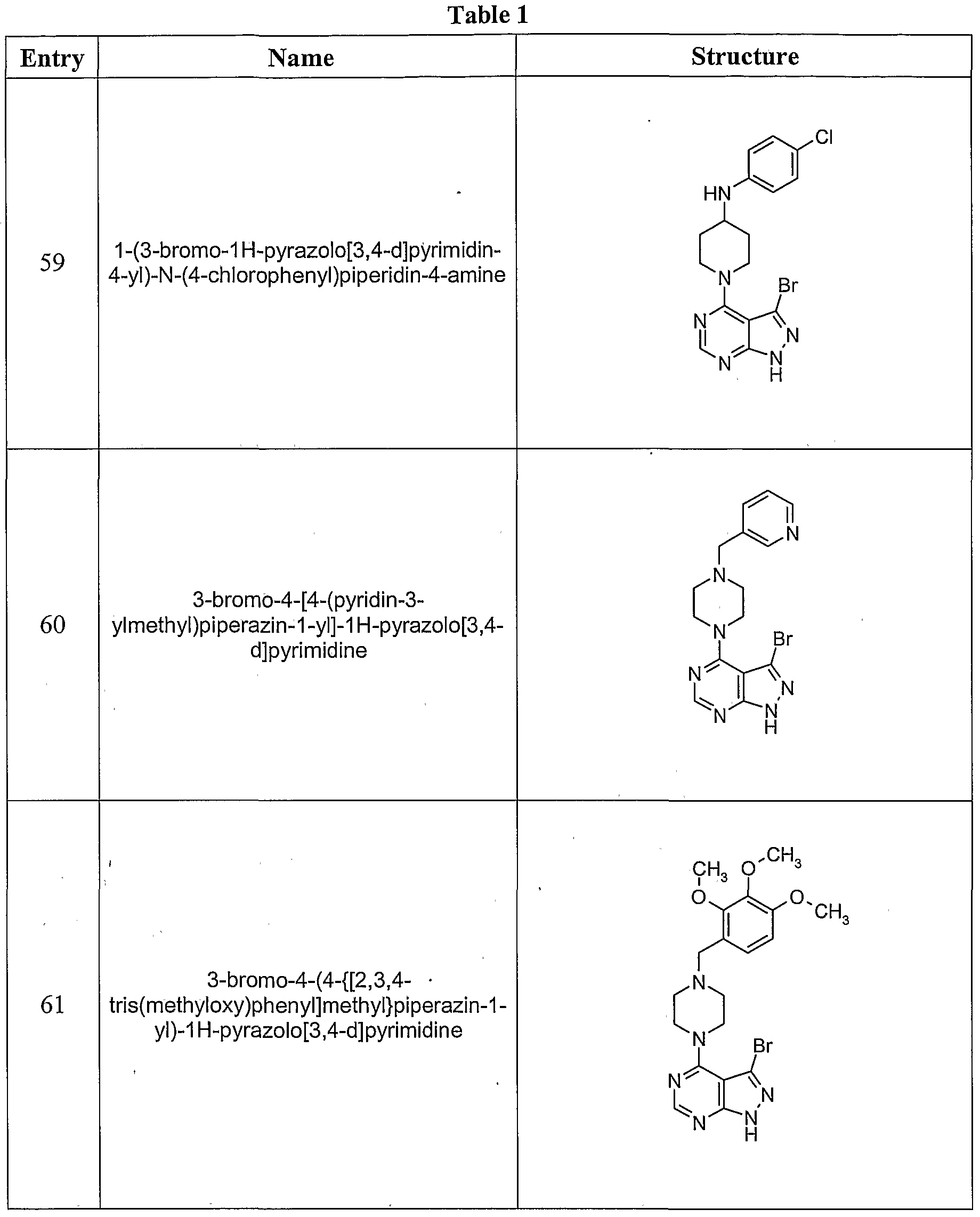


































































Claims





Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2005322085A AU2005322085B2 (en) | 2004-12-28 | 2005-12-27 | [1H-pyrazolo[3, 4-d]pyrimidin-4-yl]-piperidine or -piperazine compounds as serine-threonine kinase modulators (p70S6K, Atk1 and Atk2) for the treatment of immunological, inflammatory and proliferative diseases |
| US11/722,291 US7994172B2 (en) | 2004-12-28 | 2005-12-27 | [1H-pyrazolo[3, 4-D]pyrimidin-4-yl]-piperidine or -piperazine compounds as serine-theoronine kinase modulators (P70s6k, Atk1 and Atk2) for the treatment of immunological, inflammatory and proliferative diseases |
| AT05855490T ATE543821T1 (en) | 2004-12-28 | 2005-12-27 | Ä1H-PYRAZOLOÄ3,4-DÜPYRIMIDINE-4-YLÜ-PIPERIDINE OR PIPERAZINE COMPOUNDS AS SERINE-THREONINE KINASE MODULATORS (P70S6K, ATK1 AND ATK2) FOR THE TREATMENT OF IMMUNOLOGICAL, INFLAMMATORY AND PROLIFERATIVE DISEASES |
| EP05855490A EP1848719B1 (en) | 2004-12-28 | 2005-12-27 | [1h-pyrazolo[3,4-d]pyrimidin-4-yl]-piperidine or -piperazine compounds as serine-threonine kinase modulators (p70s6k, atk1 and atk2) for the treatment of immunological, inflammatory and proliferative diseases |
| JP2007549530A JP5274842B2 (en) | 2004-12-28 | 2005-12-27 | [1H-piperazo [3,4-d] pyrimidin-4-yl] -piperazine as a serine-threonine kinase modulator (p70S6K, Akt-1 and Akt-2) for the treatment of immune, inflammatory and proliferative disorders Or [1H-piperazo [3,4-d] pyrimidin-4-yl] -piperazine compounds |
| CA2590961A CA2590961C (en) | 2004-12-28 | 2005-12-27 | [1h-pyrazolo[3,4-d]pyrimidin-4-yl]-piperidine or -piperazine compounds as serine-threonine kinase modulators (p70s6k, atk1 and atk2) for the treatment of immunological, inflammatory and proliferative diseases |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US64020004P | 2004-12-28 | 2004-12-28 | |
| US60/640,200 | 2004-12-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006071819A1 true WO2006071819A1 (en) | 2006-07-06 |
Family
ID=36121369
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2005/046938 WO2006071819A1 (en) | 2004-12-28 | 2005-12-27 | [1h-pyrazolo[3, 4-d]pyrimidin-4-yl]-piperidine or -piperazine compounds as serine-theoronine kinase modulators (p70s6k, atk1 and atk2) for the treatment of immunological, inflammatory and proliferative diseases |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US7994172B2 (en) |
| EP (1) | EP1848719B1 (en) |
| JP (1) | JP5274842B2 (en) |
| AT (1) | ATE543821T1 (en) |
| AU (1) | AU2005322085B2 (en) |
| CA (1) | CA2590961C (en) |
| WO (1) | WO2006071819A1 (en) |
Cited By (62)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007125321A2 (en) * | 2006-04-25 | 2007-11-08 | Astex Therapeutics Limited | Purine and deazapurine derivatives as pharmaceutical compounds |
| WO2007125325A1 (en) * | 2006-04-25 | 2007-11-08 | Astex Therapeutics Limited | Pharmaceutical compounds |
| WO2008075110A1 (en) * | 2006-12-21 | 2008-06-26 | Astex Therapeutics Limited | Substituted piperidines containing a heteroarylamide or heteroarylphenyl moiety |
| WO2008140947A1 (en) | 2007-05-11 | 2008-11-20 | Eli Lilly And Company | P70 s6 kinase inhibitors |
| WO2009021992A2 (en) | 2007-08-14 | 2009-02-19 | Bayer Schering Pharma Aktiengesellschaft | Fused bicyclic pyrimidines |
| EP2050748A1 (en) * | 2007-10-18 | 2009-04-22 | Bayer Schering Pharma Aktiengesellschaft | Novel fused pyrimidines |
| WO2010056575A1 (en) * | 2008-11-11 | 2010-05-20 | Eli Lilly And Company | P70 s6 kinase inhibitor and egfr inhibitor combination therapy |
| WO2010056574A1 (en) * | 2008-11-11 | 2010-05-20 | Eli Lilly And Company | P70 s6 kinase inhibitor and mtor inhibitor combination therapy |
| JP2010532386A (en) * | 2007-07-05 | 2010-10-07 | アレイ バイオファーマ、インコーポレイテッド | Pyrimidylcyclopentane as an AKT protein kinase inhibitor |
| JP2010532387A (en) * | 2007-07-05 | 2010-10-07 | アレイ バイオファーマ、インコーポレイテッド | Pyrimidylcyclopentane as an AKT protein kinase inhibitor |
| WO2011017009A1 (en) | 2009-08-07 | 2011-02-10 | Merck Patent Gmbh | Novel azaheterocyclic compounds |
| JP2011509309A (en) * | 2008-01-09 | 2011-03-24 | アレイ バイオファーマ、インコーポレイテッド | 5H-cyclopenta [d] pyrimidine as an AKT protein kinase inhibitor |
| US8003651B2 (en) | 2006-07-06 | 2011-08-23 | Array Biopharma Inc. | Pyrimidyl cyclopentanes as AKT protein kinase inhibitors |
| US8044041B2 (en) | 2006-11-15 | 2011-10-25 | Forest Laboratories Holdings Limited | Phthalazine derivatives as inhibitors of protein kinase |
| US8063050B2 (en) | 2006-07-06 | 2011-11-22 | Array Biopharma Inc. | Hydroxylated and methoxylated pyrimidyl cyclopentanes as AKT protein kinase inhibitors |
| US8076338B2 (en) | 2004-04-23 | 2011-12-13 | Exelixis, Inc. | Kinase modulators and methods of use |
| US8101623B2 (en) | 2007-10-11 | 2012-01-24 | Astrazeneca Ab | Substituted pyrrolo[2,3-d]pyrimidine as a protein kinase B inhibitor |
| WO2012016001A1 (en) | 2010-07-29 | 2012-02-02 | Merck Patent Gmbh | Cyclic amine azaheterocyclic carboxamides |
| WO2012013282A1 (en) | 2010-07-29 | 2012-02-02 | Merck Patent Gmbh | Bicyclic azaheterocyclic carboxamides as inhibitors of the kinase p70s6k |
| US8148387B2 (en) | 2008-11-11 | 2012-04-03 | Eli Lilly And Company | AKT and P70 S6 kinase inhibitors |
| WO2012069146A1 (en) | 2010-11-24 | 2012-05-31 | Merck Patent Gmbh | Quinazoline carboxamide azetidines |
| US8247576B2 (en) | 2003-12-23 | 2012-08-21 | Astex Therapeutics Limited | Pyrazole derivatives as protein kinase modulators |
| US8329701B2 (en) | 2006-07-06 | 2012-12-11 | Array Biopharma Inc. | Dihydrofuro pyrimidines as AKT protein kinase inhibitors |
| US8343953B2 (en) | 2005-06-22 | 2013-01-01 | Astex Therapeutics Limited | Pharmaceutical compounds |
| WO2013040044A1 (en) | 2011-09-12 | 2013-03-21 | Merck Patent Gmbh | Aminopyrimidine derivatives for use as modulators of kinase activity |
| WO2013040059A1 (en) | 2011-09-12 | 2013-03-21 | Merck Patent Gmbh | Novel imidazole amines as modulators of kinase activity |
| US8436002B2 (en) | 2009-10-23 | 2013-05-07 | Eli Lilly And Company | AKT inhibitors |
| US8497294B2 (en) | 2007-03-14 | 2013-07-30 | Astex Therapeutics Limited | Compositions comprising (S)-2-amino-1-(4-chlorophenyl)-1-[4-(1H-pyrazol-4-yl)-phenyl]-ethanol as modulator of protein kinases |
| US8541461B2 (en) | 2005-06-23 | 2013-09-24 | Astex Therapeutics Limited | Pharmaceutical combinations comprising pyrazole derivatives as protein kinase modulators |
| US8546407B2 (en) | 2004-10-25 | 2013-10-01 | Astex Therapeutics Limited | Ortho-condensed pyridine and pyrimidine derivatives (e.g., purines) as protein kinases inhibitors |
| US8637532B2 (en) | 2009-02-11 | 2014-01-28 | Merck Patent Gmbh | Amino azaheterocyclic carboxamides |
| WO2014062774A1 (en) * | 2012-10-17 | 2014-04-24 | The University Of North Carolina At Chapel Hill | Pyrazolopyrimidine compounds for the treatment of cancer |
| WO2014078634A1 (en) | 2012-11-16 | 2014-05-22 | Merck Patent Gmbh | Novel imidazol-piperidinyl derivatives as modulators of kinase activity |
| WO2014078637A1 (en) | 2012-11-16 | 2014-05-22 | Merck Patent Gmbh | Novel heterocyclic derivatives as modulators of kinase activity |
| WO2014085528A1 (en) | 2012-11-29 | 2014-06-05 | Merck Patent Gmbh | Azaquinazoline carboxamide derivatives |
| WO2014096423A1 (en) * | 2012-12-20 | 2014-06-26 | Ucb Pharma S.A. | Therapeutically active pyrazolo-pyrimidine derivatives |
| US8785440B2 (en) | 2009-09-04 | 2014-07-22 | Biogen Idec Ma, Inc. | Bruton's tyrosine kinase inhibitors |
| US8835434B2 (en) | 2008-01-09 | 2014-09-16 | Array Biopharma, Inc. | Hydroxylated pyrimidyl cyclopentanes as akt protein kinase inhibitors |
| WO2014143612A1 (en) | 2013-03-11 | 2014-09-18 | Merck Patent Gmbh | 6-[4-(1 h-imidazol-2-yl)piperidin-1 -yl]pyrimidin-4-amine derivatives as modulators of kinase activity |
| US8846683B2 (en) | 2007-07-05 | 2014-09-30 | Array Biopharma, Inc. | Pyrimidyl cyclopentanes as Akt protein kinase inhibitors |
| WO2014039714A3 (en) * | 2012-09-06 | 2014-10-02 | Plexxikon Inc. | Compounds and methods for kinase modulation, and indications therefor |
| US8853216B2 (en) | 2008-01-09 | 2014-10-07 | Array Biopharma, Inc. | Hydroxylated pyrimidyl cyclopentane as AKT protein kinase inhibitor |
| US9150549B2 (en) | 2011-04-01 | 2015-10-06 | Genentech, Inc. | Combinations of AKT inhibitor compounds and erlotinib, and methods of use |
| WO2015149909A1 (en) | 2014-04-03 | 2015-10-08 | Merck Patent Gmbh | Combinations of cancer therapeutics |
| US9303040B2 (en) | 2006-07-06 | 2016-04-05 | Array Biopharma Inc. | Substituted piperazines as AKT inhibitors |
| US9402847B2 (en) | 2011-04-01 | 2016-08-02 | Astrazeneca Ab | Combinations comprising (S)-4-amino-N-(1-(4-chlorophenyl)-3-hydroxypropyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide |
| US9409886B2 (en) | 2007-07-05 | 2016-08-09 | Array Biopharma Inc. | Pyrimidyl cyclopentanes as AKT protein kinase inhibitors |
| US9487525B2 (en) | 2012-04-17 | 2016-11-08 | Astrazeneca Ab | Crystalline forms of (s)-4-amino-n-(1-(4-chlorophenyl)-3-hydroxypropyl)-1-(7h-pyrrolo[2,3-d]pyrimidin-4-yl) piperidine-4-carboxamide |
| US9555030B2 (en) | 2014-04-11 | 2017-01-31 | The University Of North Carolina At Chapel Hill | Therapeutic uses of selected pyrazolopyrimidine compounds with anti-Mer tyrosine kinase activity |
| US9682082B2 (en) | 2011-04-01 | 2017-06-20 | Genentech, Inc. | Combinations of AKT and MEK inhibitor compounds, and methods of use |
| US9737540B2 (en) | 2011-11-30 | 2017-08-22 | Astrazeneca Ab | Combination treatment of cancer |
| US9771330B2 (en) | 2012-11-27 | 2017-09-26 | The University Of North Carolina At Chapel Hill | Pyrimidine compounds for the treatment of cancer |
| US10080753B2 (en) | 2011-12-22 | 2018-09-25 | Merck Patent Gmbh | Heterocyclic carboxamides as modulators of kinase activity |
| US10618887B2 (en) | 2012-06-08 | 2020-04-14 | Sunesis Pharmaceuticals, Inc. | Pyrimidinyl tyrosine kinase inhibitors |
| US10709708B2 (en) | 2016-03-17 | 2020-07-14 | The University Of North Carolina At Chapel Hill | Method of treating cancer with a combination of MER tyrosine kinase inhibitor and an epidermal growth factor receptor (EGFR) inhibitor |
| WO2021123798A1 (en) | 2019-12-19 | 2021-06-24 | Imperial College Innovations Limited | 1h-pyrazolo[3,4-d]pyrimidine compounds useful for the treatment of platinum-resistant cancer |
| USRE48622E1 (en) | 2012-12-20 | 2021-07-06 | UCB Biopharma SRL | Therapeutically active pyrazolo-pyrimidine derivatives |
| WO2021185238A1 (en) | 2020-03-17 | 2021-09-23 | 江苏恒瑞医药股份有限公司 | Fused bicyclic derivative, preparation method therefor, and pharmaceutical use thereof |
| CN113444110A (en) * | 2020-03-25 | 2021-09-28 | 江苏恒瑞医药股份有限公司 | Tetrahydropyrrolopyrazole derivative, preparation method and medical application thereof |
| US11174243B2 (en) | 2016-07-21 | 2021-11-16 | Sunesis Pharmaceuticals, Inc. | Succinate forms and compositions of Bruton's tyrosine kinase inhibitors |
| WO2022101459A1 (en) | 2020-11-16 | 2022-05-19 | Merck Patent Gmbh | Kinase inhibitor combinations for cancer treatment |
| WO2023041061A1 (en) | 2021-09-17 | 2023-03-23 | 江苏恒瑞医药股份有限公司 | Fused bicyclic derivative, pharmaceutically acceptable salt, crystal form thereof and preparation method therefor |
Families Citing this family (31)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8835426B2 (en) * | 2007-02-26 | 2014-09-16 | Vitae Pharmaceuticals, Inc. | Cyclic urea and carbamate inhibitors of 11β-hydroxysteroid dehydrogenase 1 |
| US8575156B2 (en) * | 2007-07-26 | 2013-11-05 | Vitae Pharmaceuticals, Inc. | Cyclic inhibitors of 11β-hydroxysteroid dehydrogenase 1 |
| AR069207A1 (en) * | 2007-11-07 | 2010-01-06 | Vitae Pharmaceuticals Inc | CYCLIC UREAS AS INHIBITORS OF THE 11 BETA - HIDROXI-ESTEROIDE DESHIDROGENASA 1 |
| EP2229368A1 (en) | 2007-12-11 | 2010-09-22 | Vitae Pharmaceuticals, Inc. | Cyclic urea inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| TW200934490A (en) * | 2008-01-07 | 2009-08-16 | Vitae Pharmaceuticals Inc | Lactam inhibitors of 11 &abgr;-hydroxysteroid dehydrogenase 1 |
| CA2711614A1 (en) | 2008-01-08 | 2009-07-16 | Array Biopharma Inc. | Pyrrolopyridines as kinase inhibitors |
| ES2392014T3 (en) | 2008-01-09 | 2012-12-03 | Array Biopharma, Inc. | Pyrazolopyridines as kinase inhibitors |
| WO2009094169A1 (en) | 2008-01-24 | 2009-07-30 | Vitae Pharmaceuticals, Inc. | Cyclic carbazate and semicarbazide inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| WO2009097446A1 (en) * | 2008-01-30 | 2009-08-06 | Genentech, Inc. | Pyrazolopyrimidine pi3k inhibitor compounds and methods of use |
| EP2252598A2 (en) * | 2008-02-11 | 2010-11-24 | Vitae Pharmaceuticals, Inc. | 1,3-oxazepan-2-one and 1,3-diazepan-2-one inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| JP5730021B2 (en) * | 2008-02-15 | 2015-06-03 | ヴァイティー ファーマシューティカルズ,インコーポレイテッド | Cycloalkyllactam derivatives as inhibitors of 11β-hydroxysteroid dehydrogenase 1 |
| JP5538356B2 (en) * | 2008-03-18 | 2014-07-02 | ヴァイティー ファーマシューティカルズ,インコーポレイテッド | Inhibitors of 11β-hydroxysteroid dehydrogenase type 1 |
| CA2722427A1 (en) | 2008-05-01 | 2009-11-05 | Vitae Pharmaceuticals, Inc. | Cyclic inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| US8569292B2 (en) | 2008-05-01 | 2013-10-29 | Vitae Pharmaceuticals, Inc. | Cyclic inhibitors of 11β-hydroxysteroid dehydrogenase 1 |
| CA2723034A1 (en) | 2008-05-01 | 2009-11-05 | Vitae Pharmaceuticals, Inc. | Cyclic inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| US8242111B2 (en) * | 2008-05-01 | 2012-08-14 | Vitae Pharmaceuticals, Inc. | Cyclic inhibitors of 11β-hydroxysteroid dehydrogenase 1 |
| US8114868B2 (en) | 2008-07-25 | 2012-02-14 | Boehringer Ingelheim International Gmbh | Cyclic inhibitors of 11β-hydroxysteroid dehydrogenase 1 |
| TW201016691A (en) | 2008-07-25 | 2010-05-01 | Boehringer Ingelheim Int | Inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| JP5689069B2 (en) | 2008-11-20 | 2015-03-25 | ジェネンテック, インコーポレイテッド | Pyrazolopyridine PI3K inhibitor compounds and methods of use |
| US8637505B2 (en) | 2009-02-04 | 2014-01-28 | Boehringer Ingelheim International Gmbh | Cyclic inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| TW201039034A (en) * | 2009-04-27 | 2010-11-01 | Chunghwa Picture Tubes Ltd | Pixel structure and the method of forming the same |
| UA109255C2 (en) | 2009-04-30 | 2015-08-10 | Берінгер Інгельхайм Інтернешнл Гмбх | Cyclic inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| KR20120061771A (en) * | 2009-04-30 | 2012-06-13 | 비타이 파마슈티컬즈, 인코포레이티드 | Cyclic inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| US8927539B2 (en) | 2009-06-11 | 2015-01-06 | Vitae Pharmaceuticals, Inc. | Cyclic inhibitors of 11β-hydroxysteroid dehydrogenase 1 based on the 1,3-oxazinan-2-one structure |
| US8883778B2 (en) | 2009-07-01 | 2014-11-11 | Vitae Pharmaceuticals, Inc. | Cyclic inhibitors of 11 beta-hydroxysteroid dehydrogenase 1 |
| EP2582698B1 (en) | 2010-06-16 | 2016-09-14 | Vitae Pharmaceuticals, Inc. | Substituted 5-,6- and 7-membered heterocycles, medicaments containing such compounds, and their use |
| EP2585444B1 (en) | 2010-06-25 | 2014-10-22 | Boehringer Ingelheim International GmbH | Azaspirohexanones as inhibitors of 11-beta-hsd1 for the treatment of metabolic disorders |
| KR20130137628A (en) | 2010-11-02 | 2013-12-17 | 베링거 인겔하임 인터내셔날 게엠베하 | Pharmaceutical combinations for the treatment of metabolic disorders |
| EP3762381A4 (en) * | 2018-03-06 | 2022-01-05 | Icahn School of Medicine at Mount Sinai | Serine threonine kinase (akt) degradation / disruption compounds and methods of use |
| CN110396067B (en) * | 2018-04-25 | 2022-07-08 | 复旦大学 | 1, 4-disubstituted-2-piperazinone compound and pharmaceutical application thereof |
| JP2023522965A (en) * | 2020-04-21 | 2023-06-01 | ユニバーシティ オブ マサチューセッツ | Methods and compositions for treating age-related macular degeneration |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6423716B1 (en) | 1998-03-31 | 2002-07-23 | Kyowa Hakko Kogyo Co., Ltd. | Nitrogenous heterocyclic compounds |
| WO2005051304A2 (en) * | 2003-11-21 | 2005-06-09 | Array Biopharma Inc. | Akt protein kinase inhibitors |
| WO2005085249A1 (en) * | 2004-02-27 | 2005-09-15 | F. Hoffmann-La Roche Ag | Fused derivatives of pyrazole |
| WO2005117909A2 (en) * | 2004-04-23 | 2005-12-15 | Exelixis, Inc. | Kinase modulators and methods of use |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003252871A (en) * | 2001-03-29 | 2003-09-10 | Tanabe Seiyaku Co Ltd | Spiroisoquinoline derivative, method of producing the same, and synthetic intermediate therefor |
| JP2004115450A (en) * | 2002-09-27 | 2004-04-15 | Tanabe Seiyaku Co Ltd | Medicinal composition |
| JP4642766B2 (en) * | 2003-05-28 | 2011-03-02 | ユニバーシタ・デグリ・スタディ・ディ・シエナ | 4-Substituted derivatives of pyrazolo [3,4-d] pyrimidine and pyrrolo [2,3-d] pyrimidine and their use |
-
2005
- 2005-12-27 US US11/722,291 patent/US7994172B2/en not_active Expired - Fee Related
- 2005-12-27 AT AT05855490T patent/ATE543821T1/en active
- 2005-12-27 EP EP05855490A patent/EP1848719B1/en active Active
- 2005-12-27 WO PCT/US2005/046938 patent/WO2006071819A1/en active Application Filing
- 2005-12-27 CA CA2590961A patent/CA2590961C/en not_active Expired - Fee Related
- 2005-12-27 AU AU2005322085A patent/AU2005322085B2/en not_active Ceased
- 2005-12-27 JP JP2007549530A patent/JP5274842B2/en not_active Expired - Fee Related
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6423716B1 (en) | 1998-03-31 | 2002-07-23 | Kyowa Hakko Kogyo Co., Ltd. | Nitrogenous heterocyclic compounds |
| WO2005051304A2 (en) * | 2003-11-21 | 2005-06-09 | Array Biopharma Inc. | Akt protein kinase inhibitors |
| WO2005085249A1 (en) * | 2004-02-27 | 2005-09-15 | F. Hoffmann-La Roche Ag | Fused derivatives of pyrazole |
| WO2005117909A2 (en) * | 2004-04-23 | 2005-12-15 | Exelixis, Inc. | Kinase modulators and methods of use |
Non-Patent Citations (9)
| Title |
|---|
| CHENG, J. Q. ET AL., PROC. NAT. ACAD. SCI., vol. 89, 1992, pages 9267 - 9271 |
| CHENG, J. Q. ET AL., PROC. NAT. ACAD. SCI., vol. 93, 1996, pages 3636 - 3641 |
| CHERN, JYH-HAUR ET AL: "Design, synthesis, and structure-activity relationships of pyrazolo[3,4-d]pyrimidines: a novel class of potent enterovirus inhibitors", BIOORGANIC & MEDICINAL CHEMISTRY LETTERS , 14(10), 2519-2525 CODEN: BMCLE8; ISSN: 0960-894X, vol. 14, no. 10, 17 May 2004 (2004-05-17), XP004841231 * |
| MANNING ET AL., SCIENCE, vol. 298, 2002, pages 1912 |
| MATSUNO ET AL., J. MED. CHEM., vol. 46, no. 23, 2003, pages 4910 - 25 |
| MATSUNO, KENJI ET AL: "Potent and Selective Inhibitors of Platelet-Derived Growth Factor Receptor Phosphorylation. 3. Replacement of Quinazoline Moiety and Improvement of Metabolic Polymorphism of 4-[4-(N-Substituted (thio)carbamoyl)-1- piperazinyl]-6,7-dimethoxyquinazoline Derivatives", JOURNAL OF MEDICINAL CHEMISTRY , 46(23), 4910-4925 CODEN: JMCMAR; ISSN: 0022-2623, 2003, XP002362055 * |
| QUINTELA, JOSE M. ET AL: "6-Dimethylamino 1H-Pyrazolo[3,4-d]pyrimidine derivatives as new inhibitors of inflammatory mediators in intact cells", BIOORGANIC & MEDICINAL CHEMISTRY , 11(6), 863-868 CODEN: BMECEP; ISSN: 0968-0896, 2003, XP002376658 * |
| QUINTELA, JOSE M. ET AL: "Pyrazolopyrimidines: synthesis, effect on histamine release from rat peritoneal mast cells and cytotoxic activity", EUROPEAN JOURNAL OF MEDICINAL CHEMISTRY , 36(4), 321-332 CODEN: EJMCA5; ISSN: 0223-5234, 2001, XP004255694 * |
| WU, TOM Y. H. ET AL: "One-Pot Two-Step Microwave-Assisted Reaction in Constructing 4,5-Disubstituted Pyrazolopyrimidines", ORGANIC LETTERS , 3587-3590 CODEN: ORLEF7; ISSN: 1523-7060, vol. 5, no. 20, 2003, XP002376657 * |
Cited By (135)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9283226B2 (en) | 2003-12-23 | 2016-03-15 | Astex Therapeutics Limited | Pyrazole derivatives as protein kinase modulators |
| US8691806B2 (en) | 2003-12-23 | 2014-04-08 | Astex Therapeutics Limited | Pyrazole derivatives as protein kinase modulators |
| US8247576B2 (en) | 2003-12-23 | 2012-08-21 | Astex Therapeutics Limited | Pyrazole derivatives as protein kinase modulators |
| US8076338B2 (en) | 2004-04-23 | 2011-12-13 | Exelixis, Inc. | Kinase modulators and methods of use |
| US8546407B2 (en) | 2004-10-25 | 2013-10-01 | Astex Therapeutics Limited | Ortho-condensed pyridine and pyrimidine derivatives (e.g., purines) as protein kinases inhibitors |
| US8343953B2 (en) | 2005-06-22 | 2013-01-01 | Astex Therapeutics Limited | Pharmaceutical compounds |
| US8541461B2 (en) | 2005-06-23 | 2013-09-24 | Astex Therapeutics Limited | Pharmaceutical combinations comprising pyrazole derivatives as protein kinase modulators |
| WO2007125325A1 (en) * | 2006-04-25 | 2007-11-08 | Astex Therapeutics Limited | Pharmaceutical compounds |
| WO2007125321A3 (en) * | 2006-04-25 | 2007-12-27 | Astex Therapeutics Ltd | Purine and deazapurine derivatives as pharmaceutical compounds |
| US8796293B2 (en) | 2006-04-25 | 2014-08-05 | Astex Therapeutics Limited | Purine and deazapurine derivatives as pharmaceutical compounds |
| WO2007125321A2 (en) * | 2006-04-25 | 2007-11-08 | Astex Therapeutics Limited | Purine and deazapurine derivatives as pharmaceutical compounds |
| US8063050B2 (en) | 2006-07-06 | 2011-11-22 | Array Biopharma Inc. | Hydroxylated and methoxylated pyrimidyl cyclopentanes as AKT protein kinase inhibitors |
| US8846681B2 (en) | 2006-07-06 | 2014-09-30 | Array Biopharma, Inc. | Pyrimidyl cyclopentanes as AKT protein kinase inhibitors |
| US8853199B2 (en) | 2006-07-06 | 2014-10-07 | Array Biopharma, Inc. | Hydroxylated and methoxylated pyrimidyl cyclopentanes as AKT protein kinase inhibitors |
| US9303040B2 (en) | 2006-07-06 | 2016-04-05 | Array Biopharma Inc. | Substituted piperazines as AKT inhibitors |
| US8003651B2 (en) | 2006-07-06 | 2011-08-23 | Array Biopharma Inc. | Pyrimidyl cyclopentanes as AKT protein kinase inhibitors |
| US9359340B2 (en) | 2006-07-06 | 2016-06-07 | Array Biopharma Inc. | Hydroxylated and methoxylated pyrimidyl cyclopentanes as Akt protein kinase inhibitors |
| US8329701B2 (en) | 2006-07-06 | 2012-12-11 | Array Biopharma Inc. | Dihydrofuro pyrimidines as AKT protein kinase inhibitors |
| US8044041B2 (en) | 2006-11-15 | 2011-10-25 | Forest Laboratories Holdings Limited | Phthalazine derivatives as inhibitors of protein kinase |
| WO2008075110A1 (en) * | 2006-12-21 | 2008-06-26 | Astex Therapeutics Limited | Substituted piperidines containing a heteroarylamide or heteroarylphenyl moiety |
| US8497294B2 (en) | 2007-03-14 | 2013-07-30 | Astex Therapeutics Limited | Compositions comprising (S)-2-amino-1-(4-chlorophenyl)-1-[4-(1H-pyrazol-4-yl)-phenyl]-ethanol as modulator of protein kinases |
| US8093383B2 (en) | 2007-05-11 | 2012-01-10 | Eli Lilly And Company | P70 S6 kinase inhibitors |
| WO2008140947A1 (en) | 2007-05-11 | 2008-11-20 | Eli Lilly And Company | P70 s6 kinase inhibitors |
| US8377937B2 (en) | 2007-07-05 | 2013-02-19 | Array Biopharma Inc. | Pyrimidyl cyclopentanes as AKT protein kinase inhibitors |
| US8618097B2 (en) | 2007-07-05 | 2013-12-31 | Array Biopharma, Inc. | Pyrimidyl cyclopentanes as AKT protein kinase inhibitors |
| JP2010532387A (en) * | 2007-07-05 | 2010-10-07 | アレイ バイオファーマ、インコーポレイテッド | Pyrimidylcyclopentane as an AKT protein kinase inhibitor |
| US8846683B2 (en) | 2007-07-05 | 2014-09-30 | Array Biopharma, Inc. | Pyrimidyl cyclopentanes as Akt protein kinase inhibitors |
| JP2010532386A (en) * | 2007-07-05 | 2010-10-07 | アレイ バイオファーマ、インコーポレイテッド | Pyrimidylcyclopentane as an AKT protein kinase inhibitor |
| US9409886B2 (en) | 2007-07-05 | 2016-08-09 | Array Biopharma Inc. | Pyrimidyl cyclopentanes as AKT protein kinase inhibitors |
| WO2009021992A3 (en) * | 2007-08-14 | 2009-04-16 | Bayer Schering Pharma Ag | Fused bicyclic pyrimidines |
| AU2008288392B2 (en) * | 2007-08-14 | 2013-10-24 | Bayer Intellectual Property Gmbh | Fused bicyclic pyrimidines |
| JP2010535848A (en) * | 2007-08-14 | 2010-11-25 | バイエル・シエーリング・ファーマ アクチエンゲゼルシャフト | Fused bicyclic pyrimidine |
| US7776864B2 (en) | 2007-08-14 | 2010-08-17 | Bayer Schering Pharma Ag | Fused bicyclic pyrimidines |
| US8614218B2 (en) | 2007-08-14 | 2013-12-24 | Bayer Intellectual Property Gmbh | Fused bicyclic pyrimidines |
| EA020996B1 (en) * | 2007-08-14 | 2015-03-31 | Байер Интеллекчуал Проперти Гмбх | Fused bicyclic pyrimidines |
| CN101778851B (en) * | 2007-08-14 | 2013-05-08 | 拜耳先灵医药股份有限公司 | Fused bicyclic pyrimidines |
| WO2009021992A2 (en) | 2007-08-14 | 2009-02-19 | Bayer Schering Pharma Aktiengesellschaft | Fused bicyclic pyrimidines |
| US11760760B2 (en) | 2007-10-11 | 2023-09-19 | Astrazeneca Ab | Protein kinase B inhibitors |
| US10059714B2 (en) | 2007-10-11 | 2018-08-28 | Astrazeneca Ab | Protein kinase B inhibitors |
| US8101623B2 (en) | 2007-10-11 | 2012-01-24 | Astrazeneca Ab | Substituted pyrrolo[2,3-d]pyrimidine as a protein kinase B inhibitor |
| US10654855B2 (en) | 2007-10-11 | 2020-05-19 | Astrazeneca Ab | Protein kinase B inhibitors |
| CN101861321B (en) * | 2007-10-11 | 2013-02-06 | 阿斯利康(瑞典)有限公司 | Pyrrolo [2, 3 -D] pyrimidin derivatives as protein kinase B inhibitors |
| US9492453B2 (en) | 2007-10-11 | 2016-11-15 | Astrazeneca Ab | Protein kinase B inhibitors |
| US11236095B2 (en) | 2007-10-11 | 2022-02-01 | Astrazeneca Ab | Protein kinase B inhibitors |
| EP2050748A1 (en) * | 2007-10-18 | 2009-04-22 | Bayer Schering Pharma Aktiengesellschaft | Novel fused pyrimidines |
| US8853216B2 (en) | 2008-01-09 | 2014-10-07 | Array Biopharma, Inc. | Hydroxylated pyrimidyl cyclopentane as AKT protein kinase inhibitor |
| JP2011509309A (en) * | 2008-01-09 | 2011-03-24 | アレイ バイオファーマ、インコーポレイテッド | 5H-cyclopenta [d] pyrimidine as an AKT protein kinase inhibitor |
| US8835434B2 (en) | 2008-01-09 | 2014-09-16 | Array Biopharma, Inc. | Hydroxylated pyrimidyl cyclopentanes as akt protein kinase inhibitors |
| WO2010056574A1 (en) * | 2008-11-11 | 2010-05-20 | Eli Lilly And Company | P70 s6 kinase inhibitor and mtor inhibitor combination therapy |
| EA018824B1 (en) * | 2008-11-11 | 2013-10-30 | Эли Лилли Энд Компани | P70 S6 KINASE INHIBITOR AND mTOR INHIBITOR COMBINATION THERAPY |
| EA018624B1 (en) * | 2008-11-11 | 2013-09-30 | Эли Лилли Энд Компани | P70 s6 kinase inhibitor and egfr inhibitor combination therapy |
| US8148387B2 (en) | 2008-11-11 | 2012-04-03 | Eli Lilly And Company | AKT and P70 S6 kinase inhibitors |
| US8334293B2 (en) | 2008-11-11 | 2012-12-18 | Eli Lilly And Company | P70 S6 kinase inhibitor and EGFR inhibitor combination therapy |
| WO2010056575A1 (en) * | 2008-11-11 | 2010-05-20 | Eli Lilly And Company | P70 s6 kinase inhibitor and egfr inhibitor combination therapy |
| US9040560B2 (en) | 2009-02-11 | 2015-05-26 | Merck Patent Gmbh | Amino azaheterocyclic carboxamides |
| US8637532B2 (en) | 2009-02-11 | 2014-01-28 | Merck Patent Gmbh | Amino azaheterocyclic carboxamides |
| WO2011017009A1 (en) | 2009-08-07 | 2011-02-10 | Merck Patent Gmbh | Novel azaheterocyclic compounds |
| US9023847B2 (en) | 2009-08-07 | 2015-05-05 | Merck Patent Gmbh | Azaheterocyclic compounds |
| US8785440B2 (en) | 2009-09-04 | 2014-07-22 | Biogen Idec Ma, Inc. | Bruton's tyrosine kinase inhibitors |
| US9249146B2 (en) | 2009-09-04 | 2016-02-02 | Biogen Ma Inc. | Bruton'S tyrosine kinase inhibitors |
| US9790229B2 (en) | 2009-09-04 | 2017-10-17 | Sunesis Pharmaceuticals, Inc. | Bruton's tyrosine kinase inhibitors |
| US10577374B2 (en) | 2009-09-04 | 2020-03-03 | Sunesis Pharmaceuticals, Inc. | Bruton's tyrosine kinase inhibitors |
| US8436002B2 (en) | 2009-10-23 | 2013-05-07 | Eli Lilly And Company | AKT inhibitors |
| US10087166B2 (en) | 2010-07-29 | 2018-10-02 | Merck Patent Gmbh | Cyclic amine azaheterocyclic carboxamides |
| US9586938B2 (en) | 2010-07-29 | 2017-03-07 | Merck Patent Gmbh | Cyclic amine azaheterocyclic carboxamides |
| US8710044B2 (en) | 2010-07-29 | 2014-04-29 | Merck Patent Gmbh | Bicyclic azaheterocyclic carboxamides |
| WO2012016001A1 (en) | 2010-07-29 | 2012-02-02 | Merck Patent Gmbh | Cyclic amine azaheterocyclic carboxamides |
| WO2012013282A1 (en) | 2010-07-29 | 2012-02-02 | Merck Patent Gmbh | Bicyclic azaheterocyclic carboxamides as inhibitors of the kinase p70s6k |
| WO2012069146A1 (en) | 2010-11-24 | 2012-05-31 | Merck Patent Gmbh | Quinazoline carboxamide azetidines |
| US9610289B2 (en) | 2011-04-01 | 2017-04-04 | Genentech, Inc. | Combinations of AKT inhibitor compounds and erlotinib, and methods of use |
| US9150548B2 (en) | 2011-04-01 | 2015-10-06 | Genentech, Inc. | Combinations of AKT inhibitor compounds and vemurafenib, and methods of use |
| US9717730B2 (en) | 2011-04-01 | 2017-08-01 | Genentech, Inc. | Combinations of AKT inhibitor compounds and chemotherapeutic agents, and methods of use |
| US9150549B2 (en) | 2011-04-01 | 2015-10-06 | Genentech, Inc. | Combinations of AKT inhibitor compounds and erlotinib, and methods of use |
| US10092567B2 (en) | 2011-04-01 | 2018-10-09 | Genentech, Inc. | Combinations of AKT inhibitor compounds and chemotherapeutic agents, and methods of use |
| US9682082B2 (en) | 2011-04-01 | 2017-06-20 | Genentech, Inc. | Combinations of AKT and MEK inhibitor compounds, and methods of use |
| US9402847B2 (en) | 2011-04-01 | 2016-08-02 | Astrazeneca Ab | Combinations comprising (S)-4-amino-N-(1-(4-chlorophenyl)-3-hydroxypropyl)-1-(7H-pyrrolo[2,3-d]pyrimidin-4-yl)piperidine-4-carboxamide |
| US9346789B2 (en) | 2011-04-01 | 2016-05-24 | Genentech, Inc. | Combinations of AKT inhibitor compounds and abiraterone, and methods of use |
| WO2013040044A1 (en) | 2011-09-12 | 2013-03-21 | Merck Patent Gmbh | Aminopyrimidine derivatives for use as modulators of kinase activity |
| AU2012308681B2 (en) * | 2011-09-12 | 2017-07-13 | Merck Patent Gmbh | Aminopyrimidine derivatives for use as modulators of kinase activity |
| WO2013040059A1 (en) | 2011-09-12 | 2013-03-21 | Merck Patent Gmbh | Novel imidazole amines as modulators of kinase activity |
| US9321760B2 (en) | 2011-09-12 | 2016-04-26 | Merck Patent Gmbh | Aminopyrimidine derivatives for use as modulators of kinase activity |
| CN106946796B (en) * | 2011-09-12 | 2020-05-08 | 默克专利有限公司 | Aminopyrimidine derivatives useful as modulators of kinase activity |
| CN103930407A (en) * | 2011-09-12 | 2014-07-16 | 默克专利有限公司 | Aminopyrimidine derivatives for use as modulators of kinase activity |
| CN106946796A (en) * | 2011-09-12 | 2017-07-14 | 默克专利有限公司 | Aminopyridine derivative as modulators of kinase activity |
| US9662330B2 (en) | 2011-09-12 | 2017-05-30 | Merck Patent Gmbh | Aminopyrimidine derivatives for use as modulators of kinase activity |
| US10080750B2 (en) | 2011-09-12 | 2018-09-25 | Merck Patent Gmbh | Aminopyrimidine derivatives for use as modulators of kinase activity |
| EA028057B1 (en) * | 2011-09-12 | 2017-10-31 | Мерк Патент Гмбх | Aminopyrimidine derivatives for use as modulators of kinase activity |
| US9145392B2 (en) | 2011-09-12 | 2015-09-29 | Merck Patent Gmbh | Imidazole amines as modulators of kinase activity |
| US9737540B2 (en) | 2011-11-30 | 2017-08-22 | Astrazeneca Ab | Combination treatment of cancer |
| US10080753B2 (en) | 2011-12-22 | 2018-09-25 | Merck Patent Gmbh | Heterocyclic carboxamides as modulators of kinase activity |
| US9487525B2 (en) | 2012-04-17 | 2016-11-08 | Astrazeneca Ab | Crystalline forms of (s)-4-amino-n-(1-(4-chlorophenyl)-3-hydroxypropyl)-1-(7h-pyrrolo[2,3-d]pyrimidin-4-yl) piperidine-4-carboxamide |
| US10039766B2 (en) | 2012-04-17 | 2018-08-07 | Astrazeneca Ab | Crystalline forms of (s)-4-amino-n-(1-(4-chlorophenyl)-3-hydroxypropyl)-1-(7h-pyrrolo[2,3-d] pyrimidin-4-y1) piperidine-4-carboxamide |
| US10618887B2 (en) | 2012-06-08 | 2020-04-14 | Sunesis Pharmaceuticals, Inc. | Pyrimidinyl tyrosine kinase inhibitors |
| CN104981247A (en) * | 2012-09-06 | 2015-10-14 | 普莱希科公司 | Compounds and methods for kinase modulation, and indications therefor |
| US10227357B2 (en) | 2012-09-06 | 2019-03-12 | Plexxikon Inc. | Compounds and methods for kinase modulation, and indications therefor |
| WO2014039714A3 (en) * | 2012-09-06 | 2014-10-02 | Plexxikon Inc. | Compounds and methods for kinase modulation, and indications therefor |
| US9562047B2 (en) | 2012-10-17 | 2017-02-07 | The University Of North Carolina At Chapel Hill | Pyrazolopyrimidine compounds for the treatment of cancer |
| WO2014062774A1 (en) * | 2012-10-17 | 2014-04-24 | The University Of North Carolina At Chapel Hill | Pyrazolopyrimidine compounds for the treatment of cancer |
| EP3299362A1 (en) | 2012-11-16 | 2018-03-28 | Merck Patent GmbH | Imidazol-piperidinyl derivatives as modulators of kinase activity |
| US9580443B2 (en) | 2012-11-16 | 2017-02-28 | Merck Patent Gmbh | Heterocyclic derivatives as modulators of kinase activity |
| US9315517B2 (en) | 2012-11-16 | 2016-04-19 | Merck Patent Gmbh | Imidazol-piperidinyl derivatives as modulators of kinase activity |
| WO2014078634A1 (en) | 2012-11-16 | 2014-05-22 | Merck Patent Gmbh | Novel imidazol-piperidinyl derivatives as modulators of kinase activity |
| CN105102435A (en) * | 2012-11-16 | 2015-11-25 | 默克专利有限公司 | Novel imidazol-piperidinyl derivatives as modulators of kinase activity |
| WO2014078637A1 (en) | 2012-11-16 | 2014-05-22 | Merck Patent Gmbh | Novel heterocyclic derivatives as modulators of kinase activity |
| US9850255B2 (en) | 2012-11-16 | 2017-12-26 | Merck Patent Gmbh | Heterocyclic derivatives as modulators of kinase activity |
| CN110078743A (en) * | 2012-11-16 | 2019-08-02 | 默克专利有限公司 | Novel imidazoles-acyclic derivatives as modulators of kinase activity |
| US10287300B2 (en) | 2012-11-16 | 2019-05-14 | Merck Patent Gmbh | Heterocyclic derivatives as modulators of kinase activity |
| US9771330B2 (en) | 2012-11-27 | 2017-09-26 | The University Of North Carolina At Chapel Hill | Pyrimidine compounds for the treatment of cancer |
| US10233160B2 (en) | 2012-11-29 | 2019-03-19 | Merck Patent Gmbh | Substituted pyrido[3,4-d]pyrimidines and pyrido[4,3-d]pyrimidines as p70S6K inhibitors |
| US9440968B2 (en) | 2012-11-29 | 2016-09-13 | Merck Patent Gmbh | Substituted pyrido[3,2-d]pyrimidines for treating cancer |
| US9981925B2 (en) | 2012-11-29 | 2018-05-29 | Merck Patent Gmbh | Substituted benzo[d][1,2,3]triazines as p70S6K inhibitors |
| WO2014085528A1 (en) | 2012-11-29 | 2014-06-05 | Merck Patent Gmbh | Azaquinazoline carboxamide derivatives |
| AU2013366480B2 (en) * | 2012-12-20 | 2017-06-22 | Katholieke Universiteit Leuven, K.U.Leuven R&D | Therapeutically active pyrazolo-pyrimidine derivatives |
| EA027752B1 (en) * | 2012-12-20 | 2017-08-31 | Юсб Биофарма Спрл | Therapeutically active pyrazolo-pyrimidine derivatives |
| USRE48622E1 (en) | 2012-12-20 | 2021-07-06 | UCB Biopharma SRL | Therapeutically active pyrazolo-pyrimidine derivatives |
| WO2014096423A1 (en) * | 2012-12-20 | 2014-06-26 | Ucb Pharma S.A. | Therapeutically active pyrazolo-pyrimidine derivatives |
| CN105008360B (en) * | 2012-12-20 | 2017-09-19 | Ucb生物制药私人有限公司 | The Pyrazolopyrimidine derivative of therapeutic activity |
| US9714248B2 (en) | 2012-12-20 | 2017-07-25 | Ucb Pharma S.A. | Therapeutically active pyrazolo-pyrimidine derivatives |
| WO2014143612A1 (en) | 2013-03-11 | 2014-09-18 | Merck Patent Gmbh | 6-[4-(1 h-imidazol-2-yl)piperidin-1 -yl]pyrimidin-4-amine derivatives as modulators of kinase activity |
| US9458134B2 (en) | 2013-03-11 | 2016-10-04 | Merck Patent Gmbh | Heterocycles as modulators of kinase activity |
| US9867826B2 (en) | 2013-03-11 | 2018-01-16 | Merck Patent Gmbh | Heterocycles as modulators of kinase activity |
| WO2015149909A1 (en) | 2014-04-03 | 2015-10-08 | Merck Patent Gmbh | Combinations of cancer therapeutics |
| US9603850B2 (en) | 2014-04-11 | 2017-03-28 | The University Of North Carolina At Chapel Hill | MerTK-specific pyrazolopyrimidine compounds |
| US9649309B2 (en) | 2014-04-11 | 2017-05-16 | The University Of North Carolina At Chapel Hill | Therapeutic uses of selected pyrimidine compounds with anti-Mer tyrosine kinase activity |
| US10004755B2 (en) | 2014-04-11 | 2018-06-26 | The University Of North Carolina At Chapel Hill | Therapeutic uses of selected pyrrolopyrimidine compounds with anti-mer tyrosine kinase activity |
| US9555030B2 (en) | 2014-04-11 | 2017-01-31 | The University Of North Carolina At Chapel Hill | Therapeutic uses of selected pyrazolopyrimidine compounds with anti-Mer tyrosine kinase activity |
| US9555031B2 (en) | 2014-04-11 | 2017-01-31 | The University Of North Carolina At Chapel Hill | Therapeutic uses of selected pyrrolopyrimidine compounds with anti-mer tyrosine kinase activity |
| US10709708B2 (en) | 2016-03-17 | 2020-07-14 | The University Of North Carolina At Chapel Hill | Method of treating cancer with a combination of MER tyrosine kinase inhibitor and an epidermal growth factor receptor (EGFR) inhibitor |
| US11174243B2 (en) | 2016-07-21 | 2021-11-16 | Sunesis Pharmaceuticals, Inc. | Succinate forms and compositions of Bruton's tyrosine kinase inhibitors |
| WO2021123798A1 (en) | 2019-12-19 | 2021-06-24 | Imperial College Innovations Limited | 1h-pyrazolo[3,4-d]pyrimidine compounds useful for the treatment of platinum-resistant cancer |
| WO2021185238A1 (en) | 2020-03-17 | 2021-09-23 | 江苏恒瑞医药股份有限公司 | Fused bicyclic derivative, preparation method therefor, and pharmaceutical use thereof |
| CN115397815A (en) * | 2020-03-17 | 2022-11-25 | 江苏恒瑞医药股份有限公司 | Condensed bicyclic derivative, preparation method and application thereof in medicine |
| CN113444110A (en) * | 2020-03-25 | 2021-09-28 | 江苏恒瑞医药股份有限公司 | Tetrahydropyrrolopyrazole derivative, preparation method and medical application thereof |
| WO2022101459A1 (en) | 2020-11-16 | 2022-05-19 | Merck Patent Gmbh | Kinase inhibitor combinations for cancer treatment |
| WO2023041061A1 (en) | 2021-09-17 | 2023-03-23 | 江苏恒瑞医药股份有限公司 | Fused bicyclic derivative, pharmaceutically acceptable salt, crystal form thereof and preparation method therefor |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2005322085A1 (en) | 2006-07-06 |
| CA2590961C (en) | 2013-11-26 |
| JP2008525526A (en) | 2008-07-17 |
| US7994172B2 (en) | 2011-08-09 |
| EP1848719B1 (en) | 2012-02-01 |
| ATE543821T1 (en) | 2012-02-15 |
| CA2590961A1 (en) | 2006-07-06 |
| US20080188482A1 (en) | 2008-08-07 |
| JP5274842B2 (en) | 2013-08-28 |
| EP1848719A1 (en) | 2007-10-31 |
| AU2005322085B2 (en) | 2012-07-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1848719B1 (en) | [1h-pyrazolo[3,4-d]pyrimidin-4-yl]-piperidine or -piperazine compounds as serine-threonine kinase modulators (p70s6k, atk1 and atk2) for the treatment of immunological, inflammatory and proliferative diseases | |
| EP1841760B1 (en) | Pyrimidine derivatives as kinase modulators and method of use | |
| US8076338B2 (en) | Kinase modulators and methods of use | |
| CA2491191C (en) | Receptor-type kinase modulators and methods of use | |
| EP1678168B1 (en) | P70s6 kinase modulators and method of use | |
| US7977345B2 (en) | c-MET modulators and method of use | |
| CA2520323C (en) | Tie-2 modulators and methods of use | |
| US8710038B2 (en) | Pyrazole kinase modulators and methods of use | |
| US7626031B2 (en) | Substituted 3-(diarylmethylene)indolin-2-ones and methods of their use |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| ENP | Entry into the national phase |
Ref document number: 2590961 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005322085 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007549530 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005855490 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2005322085 Country of ref document: AU Date of ref document: 20051227 Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11722291 Country of ref document: US |