WO2006071762A2 - Pyrimidine-based inhibitors of dipeptidyl peptidase iv and methods - Google Patents
Pyrimidine-based inhibitors of dipeptidyl peptidase iv and methods Download PDFInfo
- Publication number
- WO2006071762A2 WO2006071762A2 PCT/US2005/046750 US2005046750W WO2006071762A2 WO 2006071762 A2 WO2006071762 A2 WO 2006071762A2 US 2005046750 W US2005046750 W US 2005046750W WO 2006071762 A2 WO2006071762 A2 WO 2006071762A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- agent
- group
- formula
- compound
- inhibitor
- Prior art date
Links
- 0 CN(C)C=C(C(*)=O)C#N Chemical compound CN(C)C=C(C(*)=O)C#N 0.000 description 2
- DMHWDZYUUPEAMT-UHFFFAOYSA-N CCc(c(-c1ccccc1)n1)c(-c(ccc(Cl)c2)c2Cl)c(C(OCC)=O)c1Cl Chemical compound CCc(c(-c1ccccc1)n1)c(-c(ccc(Cl)c2)c2Cl)c(C(OCC)=O)c1Cl DMHWDZYUUPEAMT-UHFFFAOYSA-N 0.000 description 1
- JDHXYBMKGRPHIC-UHFFFAOYSA-N CCc(c(-c1ccccc1)ncc1CN)c1-c(ccc(Cl)c1)c1OC Chemical compound CCc(c(-c1ccccc1)ncc1CN)c1-c(ccc(Cl)c1)c1OC JDHXYBMKGRPHIC-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/26—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/32—One oxygen, sulfur or nitrogen atom
- C07D239/42—One nitrogen atom
Definitions
- the present invention relates to pyrimidine-based inhibitors of dipeptidyl peptidase IV (DPP-4), and to a method for treating multiple diseases or disorders by employing such pyrimidine -based inhibitors alone, or in combination with another type of therapeutic agent.
- DPP-4 dipeptidyl peptidase IV
- Dipeptidyl peptidase IV is a membrane bound non-classical serine aminodipeptidase which is located in a variety of tissues (intestine, liver, lung, kidney) as well as on circulating T-lymphocytes (where the enzyme is known as CD- 26). It is responsible for the metabolic cleavage of certain endogenous peptides (GLP-I (7-36), glucagon) in vivo and has demonstrated proteolytic activity against a variety of other peptides (GHRH, NPY, GLP-2, VIP) in vitro.
- GLP- 1 (7-36) is a 29 amino-acid peptide derived by post-translational processing of proglucagon in the small intestine.
- GLP-l(7-36) has multiple actions in vivo including the stimulation of insulin secretion, inhibition of glucagon secretion, the promotion of satiety, and the slowing of gastric emptying. Based on its physiological profile, the actions of GLP- 1(7-36) are expected to be beneficial in the prevention and treatment of type II diabetes and potentially obesity.
- exogenous administration of GLP- 1(7-36) continuously infusion in diabetic patients has demonstrated efficacy in this patient population.
- GLP- 1(7- 36) is degraded rapidly in vivo and has been shown to have a short half-life in vivo (tl/2 ⁇ l .5 min).
- DPP-4 has been shown to be the primary degrading enzyme of GLP- 1(7-36) in vivo.
- GLP- 1(7-36) is degraded by DPP -4 efficiently to GLP- 1(9-36), which has been speculated to act as a physiological antagonist to GLP-l(7-36).
- inhibition of DPP-4 in vivo should potentiate endogenous levels of GLP-l(7-36) and attenuate formation of its antagonist GLP-I (9- 36) and thus serve to ameliorate the diabetic condition.
- R is a substitutent selected from the group consisting of hydrogen (H), halogen, cyano (CN), CF 3 , alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkylalkyl, bicycloalkyl, bicycloalkylalkyl, alkylthioalkyl, arylalkylthioalkyl, cycloalkenyl, aryl, arylalkyl, heteroaryl, heteroarylalkyl, cycloheteroalkyl and cycloheteroalkylalkyl, wherein any such substituent may optionally be substituted through available carbon atoms with 1, 2, 3, 4 or 5 groups selected from hydrogen, halo, alkyl, polyhaloalkyl, alkoxy, haloalkoxy, polyhaloalkoxy, alkoxycarbonyl, alkenyl, alkynyl, cycloalkyl, cycloalkylalkyl,
- B is selected from the group consisting of a bond, oxygen (O), nitrogen (N) and S(O) 1n ; m is 0, 1 or 2;
- X is a substitutent selected from the group consisting of hydrogen (H), alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkylalkyl, bicycloalkyl, bicycloalkylalkyl, alkylthioalkyl, arylalkylthioalkyl, cycloalkenyl, aryl, arylalkyl, heteroaryl, heteroarylalkyl, cycloheteroalkyl and cycloheteroalkylalkyl, wherein any such substituent may optionally be substituted through available carbon atoms with 1, 2, 3, 4 or 5 groups selected from hydrogen, halo, alkyl, polyhaloalkyl, alkoxy, haloalkoxy, polyhaloalkoxy, alkoxycarbonyl, alkenyl, alkynyl, cycloalkyl, cycloalkylalkyl, polycycloalkyl, heteroarylamino,
- Y is aryl, optionally substituted with 1, 2, 3, 4 or 5 groups selected from hydrogen, halo, alkyl, polyhaloalkyl, alkoxy, haloalkoxy, polyhaloalkoxy, alkoxycarbonyl, alkenyl, alkynyl, cycloalkyl, cycloalkylalkyl, polycycloalkyl, heteroarylamino, arylamino, cycloheteroalkyl, cycloheteroalkylalkyl, hydroxy, hydroxyalkyl, nitro, cyano, amino, substituted amino, alkylamino, dialkylamino, thiol, alkylthio, alkylcarbonyl, acyl, alkoxycarbonyl, aminocarbonyl, alkynylaminocarbonyl, alkylaminocarbonyl, alkenylaminocarbonyl, alkylcarbonyloxy, alkylcarbonylamino, arylcarbonyla
- formula I includes all pharmaceutically acceptable salts, stereoisomers, and prodrug esters of formula I.
- the compounds of formula I possess activity as inhibitors of DPP -4 in vivo and are useful in the treatment of diabetes and the micro- and macrovascular complications of diabetes such as retinopathy, neuropathy, nephropathy, and wound healing. Such diseases and maladies are also sometimes referred to as "diabetic complications”.
- the present invention provides for compounds of formula I, pharmaceutical compositions employing such compounds and for methods of using such compounds, hi particular, the present invention provides a pharmaceutical composition comprising a therapeutically effective amount of a compound of formula I 5 alone or in combination with a pharmaceutically acceptable carrier.
- diabetes especially type II diabetes, including complications of diabetes, including retinopathy, neuropathy, nephropathy and delayed wound healing, and related diseases such as insulin resistance (impaired glucose homeostasis), hyperglycemia, hyperinsulinemia, elevated blood levels of fatty acids or glycerol, obesity, hyperlipidemia including hypertriglyceridemia, Syndrome X, atherosclerosis and hypertension, and for increasing high density lipoprotein levels, wherein a therapeutically effective amount of a compound of formula I is administered to a mammalian, e.g., human, patient in need of treatment.
- a mammalian e.g., human
- the compounds of the invention can be used alone, in combination with other compounds of the present invention, or in combination with one or more other agent(s) active in the therapeutic areas described herein.
- a method is provided for treating diabetes and related diseases as defined above and hereinafter, wherein a therapeutically effective amount of a combination of a compound of formula I and at least one other type of therapeutic agent, such as an antidiabetic agent and/or a hypolipidemic agent, is administered to a human patient in need of treatment.
- Further embodiments of the invention include compounds of formula I wherein n is 1, or compounds of formula I having the structure:
- the compound of formula (I) will be employed in a weight ratio to the antidiabetic agent or other type therapeutic agent (depending upon its mode of operation) within the range from about 0.01:1 to about 500:1, preferably from about 0.1:1 to about 100:1, more preferably from about 0.2:1 to about 10:1.
- Scheme 1 provides a general route to prepare aminomethylpyrimidines of formula (8).
- Acid chlorides of formula (2) may be obtained from commercial sources, or alternatively generated by methods as described herein from the corresponding carboxylic acids of formula (1).
- an acid chloride (2) can be formed by treating a carboxylic acid (1) with (COCl) 2 or SOCl 2 in an inert solvent such as methylene chloride or THF at 0 to 60 °C for 2-48 hours.
- Ketonitriles of formula (4) can be prepared by combining the lithium anion of acetonitrile with an acid chloride of formula (2).
- Acetonitrile (3) can be deprotonated by a strong base such as ⁇ -BuLi in an anhydrous solvent such as THF or diethyl ether at low temperature to give the lithium anion of acetonitrile.
- Acrylnitriles of formula (5) can be prepared by methods known to those skilled in the art such as heating ketonitrile of formula (4) with dimethylformamide dimethylacetal in an inert solvent such as toluene at elevated temperature for 2-48 hours.
- Amidines of formula (6) can either be obtained through commercial sources or conveniently prepared by known methods.
- One example to make the amidines of formula (6) is to start with the corresponding nitrile, treating with HCl followed by NH 3 to provide amidines (6).
- Pyrimidines of formula (7) can be prepared by combining acrylonitriles (5) and amidines (6) by methods known in the art. For example, the process can be performed by heating an acrylonitrile (5) and an amidine (6) with a base such as NaOMe in methanol at room temperature to reflux for 2-48 hours.
- Aminomethylpyrimidines of formula (8) can be prepared from nitriles (7) through a reductive process.
- the reducing agents which may be used for this process include, but are not limited to LAH, CoCl 2 /NaBH 4 , Raney Ni/H 2 , and PdZH 2 .
- Scheme 2 describes an alternative route to prepare aminomethylpyrimidines of formula (8).
- Ketoesters of formula (10) are known in the literature or can be conveniently prepared by known methods known.
- One example to prepare ketoesters of formula (10) is to combine a ketone (9) with a methylcarbonate and a base such as NaH in an inert solvent such as THF at ambient temperature for 2-24 hours.
- Acryloesters of formula (11) can be prepared by the same methods as described in Scheme 1 for acrylonitriles (5).
- Pyrimidine esters of formula (12) can be prepared by combining an acryloester (11) and an amidine (6) using the same methods as described in Scheme 1 for pyrimidines (7).
- Aminomethyl pyrimidines of formula (8) can then be prepared by those skilled in the art through a reduction / oxidation sequence on pyrimidine esters of formula (12) as described in scheme 2.
- the reducing agents that may be used to convert an ester of formula (12) to an alcohol of formula (13) include, but are not limited to DIBAL, LAH, and Red- Al.
- the oxidizing agents that may be used to convert an alcohol of formula (13) to an aldehyde of formula (14) include, but are not limited to Dess-Martin periodinane, Swern, PCC, MnO 2 , and TPAP/NMO.
- formula (15) can be either an oxime or an imine, which can be conveniently prepared by combining an aldehyde of formula (14) with an amine or hydroxylamine.
- the reduction of compounds of formula (15) to aminomethylpyrimidines of formula (8) can be performed by using reducing agents such as Zn/HOAc, PdZH 2 , or Raney Ni/H 2 .
- Scheme 3 provides an alternative route of converting an alcohol of formula (13) to aminomethylpyrimidine of formula (8).
- the chloropyrimidine of formula (16) can be formed from an alcohol of formula (13) by methods known to one skilled in the art.
- One example of such a transformation is to treat an alcohol (13) with SOCl 2 in an inert solvent such as CH 2 Cl 2 at elevated temperature for 2-24 hours.
- the chloropyrimidines of formula (16) can be converted to aminomethylpyrimidines of formula (8) by bubbling NH 3 gas to a solution of chloropyrimidines (16) in a suitable solvent such as methanol.
- Scheme 4 provides an alternative route for converting alcohols of formula (13) to aminomethylpyrimidines of formula (8).
- An alcohol of formula (13) can be converted to a suitable leaving group, such as a mesylate, by treating the alcohol (13) with methanesulfonyl chloride and a base such as triethylamine or pyridine in an inert solvent such as tetrahydrofuran or methylene chloride at 0 to 60 °C for 1 to 24 hours.
- the mesylates of formula (17) can then be converted to azides of formula (18) by known methods.
- One such set of conditions involves treatment of a mesylate (17) with sodium azide in an inert solvent such as DMF at room temperature to 100 °C for 1 to 24 hours.
- the azides of formula (18) can then be reduced to form aminomethylpyrimidines of formula (8).
- Alkylated aminomethylpyrimidines of formula (19) can be prepared from aldehydes of formula (14) as described in scheme 5.
- aldehydes of formula (14) as described in scheme 5.
- One example of such a transformation can be found in: Hart, David J.; Kanai, Kenichi; Thomas, Dudley G.; Yang, Teng Kuei. Journal of Organic Chemistry (1983), 48(3), 289-94.
- Another example of such a transformation is to add a Grignard reagent (R-MgBr) to the aldehyde, followed by oxidation, imine/oxime formation and reduction as described in Scheme 3.
- R-MgBr Grignard reagent
- Scheme 6 describes a route to prepare 6-substituted aminomethylpyrimidines of formula (24).
- Keto esters of formula (20) can either be obtained from commercial sources or conveniently prepared by the methods described in Scheme 2.
- Acryloesters of formula (22) can be prepared by known methods by combining a ketoester of formula (20) and an aldehyde of formula (21).
- One example to prepare an acryloester of formula (22) is through a Knovenagel reaction.
- Pyrimidine esters of formula (23) can be prepared by methods known to those skilled in the art by combining acryloesters of formula (22) and amidines of formula (6) by known methods.
- Pyrimidine esters of formula (25) can either be obtained through commercial sources or conveniently prepared by methods known in the art.
- the ester functionality of (25) can be converted to an alcohol of formula (26) by a reductive process.
- the reducing agents which may be used for this process include, but are not limited to LAH, DIBAL, Red- Al, and NaBH 4 .
- the reaction can be performed by combining an ester (25) and the reducing agent in an inert solvent such as THF or toluene at -78 0 C to elevated temperature for 2-24 hours.
- Pyrimidine aldehydes of formula (27) can be prepared from pyrimidine alcohols of formula (26) by an oxidative process.
- the oxidizing agents which may be used for this process include, but are not limited to PCC, Dess-Martin periodinane, Swern, and TPAP/NMO.
- the reaction can be performed in a solvent such as CH 2 Cl 2 , THF at -30 °C to ambient temperature for 2-24 hours.
- Pyrimidines of formula (29) can be prepared by combining a chloropyrimidine of formula (27) and a boronic acid of formula (28) by a Suzuki coupling process.
- Boronic acids of formula (28) can be obtained from commercial sources or conveniently prepared by methods known in the art.
- a ketoester of formula (22) can be condensed with an alkyl- or arylthioamidine such as (30) to give 2-alkylthiopyrimidines of formula (31) by known methods.
- One such set of conditions is to combine a ketoester of formula (22) with an amidine of formula (30) in a suitable solvent such as DMF at 20 to 100 °C for 1 - 72 hours.
- Molecular sieves can be added to facilitate the reaction.
- the alkylthio compound of formula (31) can be oxidized to an alkylsulfone of formula (32) by known methods.
- the oxidizing agents that may be used for this transformation include, but are not limited to mCPBA, hydrogen peroxide, PCC, and MnO 2 .
- the alkylsulfone of formula (32) can then be displaced by amines to form a 2- aminosubstituted pyrimidine of formula (33).
- An example of one such set of conditions which may be used for this conversion is to combine a sulfone of formula (32) with a primary or secondary amine in a suitable solvent such as methylene chloride, THF or DMF at rt to 100 °C for 1 to 72 hours.
- the ester of formula (33) can be converted to an aminomethylpyrimidine of formula (34) by the same procedures as described in Schemes 2, 3, and 4.
- alkyl or “alk” as used herein alone or as part of another group includes both branched and straight-chain saturated aliphatic hydrocarbon radicals/groups having the specified number of carbon atoms.
- Alkyl refers to a monoradical branched or unbranched saturated hydrocarbon chain, preferably having from 1 to 40 carbon atoms, more preferably 1 to 10 carbon atoms, even more preferably 1 to 6 carbon atoms, such as methyl, ethyl, n- propyl, isopropyl, n-butyl, secondary butyl, tert-butyl, n-hexyl, n-octyl, n-decyl, n- dodecyl, 2-ethyldodecyl, tetradecyl, and the like, unless otherwise indicated.
- alkyl groups can optionally be substituted with one or more substituents selected from a member of the group consisting of such as halo, alkyl, alkoxy, aryl, aryloxy, aryl(aryl) or diaryl, arylalkyl, arylalkyloxy, alkenyl, cycloalkyl, cycloalkylalkyl, cycloalkylalkyloxy, amino, hydroxy, hydroxyalkyl, acyl, heteroaryl, heteroaryloxy, heteroarylalkyl, heteroarylalkoxy, aryloxyalkyl, alkylthio, arylalkylthio, aryloxyaryl, alkylamido, alkanoylamino, arylcarbonylamino, nitro, cyano, thiol, haloalkyl, trihaloalkyl and/or alkylthio.
- Carbocyclic as employed herein alone or as part of another group includes saturated or partially unsaturated (containing 1 or 2 double bonds) cyclic hydrocarbon groups containing 1 to 3 rings, including monocyclic alkyl, bicyclic alkyl (or bicycloalkyl) and tricyclic alkyl, containing a total of 3 to 20 carbons forming the ring, preferably 3 to 10 carbons, forming the ring and which may be fused to 1 or 2 aromatic rings as described for aryl, which includes, for example cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclodecyl and cyclododecyl, cyclohexenyl,
- any of which groups may be optionally substituted with 1 or more substituents such as of the substituents for described herein for alkyl or aryl.
- Aryl or “Ar” as used herein alone or as part of another group refers to an unsaturated aromatic carbocyclic group of from 5 to 20 carbon atoms having a single ring (e.g., phenyl) or multiple condensed (fused) rings (e.g., naphthyl or anthryl).
- Representative examples include, but are not limited to, aromatic radicals such as phenyl, naphthyl, tetrahydronaphthyl, indane and biphenyl.
- such aryl groups can optionally be substituted with one or more substituents selected from a member of the group consisting of hydrogen, halo, haloalkyl, alkyl, haloalkyl, alkoxy, haloalkoxy, alkenyl, trifluoromethyl, trifluoromethoxy, alkynyl, cycloalkyl-alkyl, cycloheteroalkyl, cycloheteroalkylalkyl, aryl, heteroaryl, arylalkyl, aryloxy, aryloxyalkyl, arylalkoxy, arylthio, arylazo, heteroarylalkyl, heteroarylalkenyl, heteroarylheteroaryl, heteroaryloxy, hydroxy, nitro, cyano, amino, any of the alkyl substituents described herein, or substituted amino wherein the amino includes 1 or 2 substituents (which
- cycloheteroalkyl refers to a saturated or unsaturated group having a single ring, multiple condensed rings or multiple covalently joined rings, from 1 to 40 carbon atoms and from 1 to 10 hetero ring atoms, preferably 1 to 4 hetero ring atoms, selected from nitrogen, sulfur, phosphorus, and/or oxygen.
- Heterocycle or “Heterocyclic group” means a stable 5 to 7 membered monocyclic or bicyclic or 7 to 10 membered bicyclic heterocyclic ring that may be saturated, partially unsaturated, or aromatic, and that comprises carbon atoms and from 1 to 4 heteroatoms independently selected from a member of the group consisting of nitrogen, oxygen and sulfur and wherein the nitrogen and sulfur heteroatoms are optionally be oxidized and the nitrogen heteroatom may optionally be quaternized, and including any bicyclic group in which any of the above-defined heterocyclic rings is fused to a benzene ring.
- heterocyclic groups may be substituted on carbon or on a nitrogen, sulfur, phosphorus, and/or oxygen heteroatom, such as, but not limited to, the substituents described for alkyl or aryl herein, so long as the resulting compound is stable.
- substituents described for alkyl or aryl herein such as, but not limited to, the substituents described for alkyl or aryl herein, so long as the resulting compound is stable.
- Heteroaryl as used herein alone or as part of another group embraces unsaturated heterocyclic radicals.
- heteroaryl radicals include unsaturated 3 to 6 membered heteromonocyclic group containing 1 to 4 nitrogen atoms, for example, pyrrolyl, pyrrolinyl, imidazolyl, pyrazolyl, pyridyl, pyrimidyl, pyrazinyl, pyridazinyl, triazolyl (e.g., 4H-l,2,4-triazolyl, lH-l,2,3-triazolyl, 2H-l,2,3-triazolyl, etc.) tetrazolyl (e.g.
- unsaturated condensed heterocyclyl group containing 1 to 5 nitrogen atoms for example, indolyl, isoindolyl, indolizinyl, benzimidazolyl, quinolyl, isoquinolyl, indazolyl, benzotriazolyl, tetrazolopyridazinyl (e.g., tetrazolo[l,5-b]pyridazinyl, etc.), etc.
- unsaturated 3 to 6- membered heteromonocyclic group containing an oxygen atom for example, pyranyl, furyl, etc.
- unsaturated 3 to 6-membered heteromonocyclic group containing a sulfur atom for example, thienyl, etc.
- heteroaryl groups include the following:
- heteroaryl groups can optionally be substituted with one or more substituents, such as those described for alkyl or aryl herein.
- alkenyl refers to straight or branched chain radicals of 2 to 20 carbons, preferably 2 to 12 carbons, and more preferably 1 to 8 carbons in the normal chain, which include one to six double bonds in the normal chain, such as vinyl, 2-propenyl, 3-butenyl, 2-butenyl, 4-pentenyl, 3-pentenyl, 2-hexenyl, 3-hexenyl, 2-heptenyl, 3- heptenyl, 4-heptenyl, 3-octenyl, 3-nonenyl, 4-decenyl, 3-undecenyl, 4-dodecenyl, 4,8,12-tetradecatrienyl, and the like.
- alkenyl group may be substituted with one or substituents, such as those substituents disclosed for alkyl.
- alkynyl refers to straight or branched chain radicals of 2 to 20 carbons, preferably 2 to 12 carbons and more preferably 2 to 8 carbons in the normal chain, which include one triple bond in the normal chain, such as 2-propynyl, 3-butynyl, 2- butynyl, 4-pentynyl, 3-pentynyl, 2-hexynyl, 3-hexynyl, 2-heptynyl, 3-heptynyl, 4- heptynyl, 3-octynyl, 3-nonynyl, 4-decynyl,3-undecynyl, 4-dodecynyl and the like.
- said alkynyl group may be substituted with one or
- cycloalkenyl refers to partially unsaturated cyclic hydrocarbons containing 3 to 12 carbons, preferably 5 to 10 carbons and 1 or 2 double bonds.
- exemplary cycloalkenyl groups include cyclobutenyl, cyclopentenyl, cyclohexenyl, cycloheptenyl, cyclooctenyl, cyclohexadienyl, and cycloheptadienyl.
- said cycloalkenyl group may be substituted with one or substituents, such as those substituents disclosed for alkyl.
- Bicycloalkyl as employed herein alone or as part of another group includes saturated bicyclic ring groups such as, without limitation, [3.3.0]bicyclooctane, [4.3.0]bicyclononane, [4.4.0]bicyclodecane (decalin), [2.2.2]bicyclooctane, and so forth.
- cycloalkenyl as employed herein alone or as part of another group includes partially unsaturated carbocyclic radicals having three to twelve carbon atoms. Examples include, without limitation, cyclobutenyl, cyclopentenyl and cyclohexenyl.
- polycycloalkyl as employed herein alone or as part of another group includes two or more cycloalkyl ring systems, as defined herein, wherein at least one carbon atom is a part of at least two separately identifiable ring systems.
- the polycycloalkyl group may contain bridging between two carbon atoms, for example, bicyclo[1.1.0]butyl, bicyclo[3.2.1]octyl, bicyclo[5.2.0]nonyl, tricycl[2.2.1.0.sup.l
- the polycycloalkyl group may contain one or more fused ring systems, for example, decalinyl (radical from decalin) and perhydroanthracenyl.
- the polycycloalkyl group may contain a spiro union, in which a single atom is the only common member of two rings, for example, spiro[3.4]octyl, spiro[3.3]heptyl and spiro[4.5]decyl.
- halogen or halo as used herein alone or as part of another group refers to chlorine, bromine, fluorine, and iodine as well as CF 3 .
- alkoxy or “alkyloxy” as used herein alone or as part of another group, refers to an alkyl group, as defined herein, appended to a parent molecular moiety through an alkyl group, as defined herein.
- haloalkoxy as used herein alone or as part of another group refers to alkoxy radicals, as defined herein, further substituted with one or more halo atoms, such as fluoro, chloro or bromo, to provide haloalkoxy radicals. Examples include, without limitation, fluoromethoxy, chloromethoxy, trifluoromethoxy, trifluoromethoxy, fluoroethoxy and fluoropropoxy.
- acyl groups include a substituent group attached to a carbonyl, such as alkanoyl, alkenoyl, aroyl, aralkanoyl, heteroaroyl, cycloalkanoyl, cycloheteroalkanoyl and the like.
- bicycloalkylalkyl or “heteroarylalkyl” as used herein alone or as part of another group refers to a cycloalkyl, an aryl, a cyclohetero, a bicycloalkyl or heteroaryl group, as defined herein, appended to a parent molecular moiety through an alkyl group, as defined herein.
- Representative examples of arylalkyl include, but are not limited to, benzyl, 2-phenylethyl, 3-phenylpropyl, and the like.
- cycloheteroalkylalkyl refers to a cycloheteroalkyl group as defined herein, linked through a C atom or heteroatom to a (CH 2 ) r chain, where "r" can be 1 to 10.
- polyhaloalkyl as used herein alone or as part of another group refers to an "alkyl” group as defined above, having 2 to 9, preferably from 2 to 5, halo substiruehts, such as CF 3 CH 2 , CF 3 or CF 3 CF 2 CH 2 .
- polyhaloalkoxy refers to an "alkoxy” or "alkyloxy” group as defined above having 2 to 9, preferably from 2 to 5, halo substituents, such as CF 3 CH 2 O-, CF 3 O- or CF 3 CF 2 CH 2 O-.
- alkylthio or "arylalkylthio” refers to an alkyl group or and arylalkyl group, as defined herein, linked to a parent molecular moiety through a thiol group.
- alkylthioalkyl or “arylalkylthioalkyl” refers to an alkylthio group or and arylalkylthio group, as defined herein, linked to a parent molecular moiety through an alkyl group.
- hydroxy as used herein alone or as part of another group, refers to a -OH group.
- hydroxyalkyl refers to a hydroxyl group, as defined herein, appended to a parent molecular moiety through a alkyl group, as defined herein.
- cyano refers to a -CN group.
- nitro refers to a -NO 2 group.
- alkylsulfinyl denotes respectively divalent radicals -S(O)-.
- alkylsulfinyl as used herein alone or as part of another group, refers to an alkyl group, as defined herein, appended to a parent molecular moiety through a sulfinyl group, as defined herein.
- alkylsulfonyl or “aminosulfonyl”, as used herein, refer to an alkyl or amino group, as defined herein, appended to a parent molecular moiety through a sulfonyl group, as defined herein.
- amino refers to an -NH 3 group or an amine linkage: -NR a - 3 wherein Ra may be as described below in the definition for "substituted amino".
- substituted amino refers to amino substituted with one or two substituents.
- R a and R b may be the same or different and are, for example chosen from hydrogen, alkyl, cycloalkyl, alkenyl, cycloalkenyl, alkynyl, aryl, heteroaryl, heterocyclic, arylalkyl, heteroarylalkyl, cycloheteroalkyl, cycloheteroalkylalkyl, cycloalkylalkyl, haloalklyl, hydrooxyalkyl, alkoxyalkyl or thioalkyl.
- substituents may optionally be further substituted with any of the alkyl substituents as set out above, hi addition, the amino substituents may be taken together with the nitrogen atom to which they are attached to form 1-pyrrolidinyl, 1-piperidinyl, 1- azepinyl, 4-morpholinyl, 4-thiamorpholinyl, 1-piperazinyl, 4-alkyl-lpiperazinyl, 4- arylalkyl-lpiperazinyl, 4-diarylalkyl- 1-piperazinyl, 1-pyrrolindinyl, 1-piperidinyl, or 1-azepinyl, optionally substituted with alkyl, alkoxy, alkylthio, halo, triflouromethyl or hydroxyl.
- dialkylamino refers to a substituted amino group having two alkyl substituents.
- R a and R b are each an alkyl group, as defined herein.
- carbonyl refers to a -C(O)- group.
- aminocarbonyl refers to a -C(O)- group.
- alkenylaminocarbonyl refers to an amino group, alkyl group, alkoxy group, aryl group, alkynylamino group, alkylamino group or an alkenylamino group, as defined herein, appended to a parent molecular moiety through a carbonyl group, as defined herein.
- heteroarylamino refers to a heteroaryl, aryl, alkyl, alkylcarbonyl, arylcarbonyl, alkylsulfonyl, alkylaminocarbonyl or alkoxycarbonyl group as defined herein, appended to a parent molecular moiety through an amino group, as defined herein.
- sulfonamido refers to -S(O) 2 - NR a R b , wherein Ra and Rb are as defined above for "substituted amino".
- alkylcarbonyloxy refers to an "alkyl-CO-O-" group, wherein alkyl is as defined above.
- Optional or “optionally” means that the subsequently described event or circumstance may or may not occur, and that the description includes, without limitation, instances where said event or circumstance occurs and instances in which it does not.
- optionally substituted alkyl means that alkyl may or may not be substituted by those groups enumerated in the definition of substituted alkyl.
- Substituted as used herein, whether express or implied and whether preceded by “optionally” or not, means that any one or more hydrogen on the designated atom (C, N, etc.) is replaced with a selection from the indicated group, provided that the designated atom's normal valency is not exceeded, and that the substitution results in a stable compound.
- prodrug esters as employed herein includes esters and carbonates formed by reacting one or more hydroxyls of compounds of formula I with alkyl, alkoxy, or aryl substituted acylating agents employing procedures known to those skilled in the art to generate acetates, pivalates, methylcarbonates, benzoates and the like.
- Various forms of prodrugs are well known in the art. A comprehensive description of prodrugs and prodrug derivatives are described in: a) The Practice of Medicinal Chemistry, Camille G. Wermuth et al., Ch 31, (Academic Press, 1996); b) Design of Prodrugs, edited by H.
- Bundgaard (Elsevier, 1985); and c) A Textbook of Drug Design and Development, P. Krogsgaard-Larson and H. Bundgaard, eds., Ch. 5, pgs 113-191 (Harwood Academic Publishers, 1991). Said references are incorporated herein by reference.
- diabetes complications include retinopathy, neuropathy and nephropathy, erectile dysfunction, and other known complications of diabetes.
- An administration of a therapeutic agent of the invention includes administration of a therapeutically effective amount of the agent of the invention.
- therapeutically effective amount refers to an amount of a therapeutic agent to treat or prevent a condition treatable by administration of a composition of the invention. That amount is the amount sufficient to exhibit a detectable therapeutic or preventative or ameliorative effect. The effect may include, for example, treatment or prevention of the conditions listed herein.
- the precise effective amount for a subject will depend upon the subject's size and health, the nature and extent of the condition being treated, recommendations of the treating physician, and the therapeutics or combination of therapeutics selected for administration. Thus, it is not useful to specify an exact effective amount in advance.
- other type of therapeutic agents includes, but is not limited to one or more antidiabetic agents (other than DPP-IV inhibitors of formula I), one or more anti-obesity agents, one or more anti-hypertensive agents, one or more anti-platelet agents, one or more anti-atherosclerotic agents and/or one or more lipid-lowering agents (including anti-atherosclerosis agents).
- the compounds of the present invention possess activity as inhibitors of the dipeptidyl peptidase IV which is found in a variety of tissues, such as the intestine, liver, lung and kidney of mammals. Via the inhibition of dipeptidyl peptidase IV in vivo, the compounds of the present invention possess the ability to potentiate endogenous levels of GLP-l(7-36) and attenuate formation of its antagonist GLP-1(9- 36).
- the compounds of the present invention can be administered to mammals, preferably humans, for the treatment of a variety of conditions and disorders, including, but not limited to, treating or delaying the progression or onset of diabetes ⁇ referably Type II, impaired glucose tolerance, insulin resistance, and diabetic complications, such as nephropathy, retinopathy, neuropathy and cataracts), hyperglycemia, hyperinsulinemia, hypercholesterolemia, elevated blood levels of free fatty acids or glycerol, hyperlipidemia, hypertriglyceridemia, obesity, wound healing, tissue ischemia, atherosclerosis and hypertension.
- the compounds of the present invention may also be utilized to increase the blood levels of high density lipoprotein (HDL).
- HDL high density lipoprotein
- the conditions, diseases, and maladies collectively referenced to as "Syndrome X" or Metabolic Syndrome as detailed in Johannsson, J. Clin.
- the present invention includes within its scope pharmaceutical compositions comprising, as an active ingredient, a therapeutically effective amount of at least one of the compounds of formula I, alone or in combination with a pharmaceutical carrier or diluent.
- compounds of the present invention can be used alone, in combination with other compounds of the invention, or in combination with one or more other therapeutic agent(s), e.g., an antidiabetic agent or other pharmaceutically active material.
- therapeutic agent(s) suitable for combination with the compound of the present invention include, but are not limited to, known therapeutic agents useful in the treatment of the aforementioned disorders including: anti-diabetic agents; anti-hyperglycemic agents; hypolipidemic/lipid lowering agents; anti-obesity agents; anti-hypertensive agents, and appetite suppressants.
- Additional therapeutic agents suitable for combination with the compound of the present invention include agents for treating infertility, agents for treating polycystic ovary syndrome, agents for treating a growth disorder and/or frailty, an anti-arthritis agent, agents for preventing inhibiting allograft rejection in transplantation, agents for treating autoimmune disease, an anti-AIDS agent, agents for treating inflammatory bowel disease/syndrome, agents for treating anorexia nervosa and an anti-osteoporosis agent.
- Suitable anti-diabetic agents for use in combination with the compound of the present invention include biguanides (e.g., metformin or phenformin), glucosidase inhibitors (e.g., acarbose or miglitol), insulins (including insulin secretagogues or insulin sensitizers), meglitinides (e.g., repaglinide), sulfonylureas (e.g., glimepiride, glyburide, gliclazide, chlorpropamide and glipizide), biguanide/glyburide combinations (e.g., Glucovance ® ), thiazolidinediones (e.g., troglitazone, rosiglitazone and pioglitazone), PPAR-alpha agonists, PPAR-gamma agonists, PPAR alpha/gamma dual agonists, glycogen phosphorylase inhibitors,
- Suitable thiazolidinediones include Mitsubishi's MCC-555 (disclosed in U.S. Patent No. 5,594,016), Glaxo-Welcome's GL-262570, englitazone (CP-68722, Pfizer) or darglitazone (CP-86325, Pfizer, isaglitazone (MIT/J&J), JTT- 501 (JPNT/P&U), L-895645 (Merck), R-119702 (Sankyo/WL), NN-2344 (Dr. Reddy/NN), or YM-440 (Yamanouchi).
- Examples of PPAR-alpha agonists, PPAR-gamma agonists and PPAR alpha/gamma dual agonists include muraglitizar, peliglitazar, AR-HO39242 (Astra/Zeneca), GW-409544 (Glaxo-Wellcome), GW-501516 (Glaxo-Wellcome), KRP297 (Kyorin Merck) as well as those disclosed by Murakami et al, "A Novel Insulin Sensitizer Acts As a Coligand for Peroxisome Proliferation - Activated Receptor Alpha (PPAR alpha) and PPAR gamma. Effect on PPAR alpha Activation on Abnormal Lipid.
- Suitable aP2 inhibitors include those disclosed in U.S. application Serial No. 09/391,053, filed September 7, 1999, and in U.S. application Serial No. 09/519,079, filed March 6, 2000, employing dosages as set out herein.
- Suitable other DPP4 inhibitors include saxagliptin, those disclosed in WO99/38501, WO99/46272, WO99/67279 (PROBIODRUG), WO99/67278
- glucagon-like peptide- 1 examples include glucagon-like peptide- 1 (GLP-I,) such as GLP-l(l-36) amide, GLP-l(7-36) amide, GLP-l(7-37) (as disclosed in U.S. Patent No. 5,614,492), as well as exenatide (Amylin/Lilly), LY-315902 (Lilly), MK- 0431 (Merck), liraglutide (NovoNordisk), ZP-10 (Zealand Pharmaceuticals AJS), CJC-1131 (Conjuchem hie), and the compounds disclosed in WO 03/033671.
- GLP-I glucagon-like peptide- 1
- hypolipidemic/lipid lowering agents for use in combination with the compound of the present invention include one or more MTP inhibitors, HMG CoA reductase inhibitors, squalene synthetase inhibitors, fibric acid derivatives, ACAT inhibitors, lipoxygenase inhibitors, cholesterol absorption inhibitors, ileal Na /bile acid co-transporter inhibitors, up-regulators of LDL receptor activity, bile acid sequestrants, cholesterol ester transfer protein (e.g., CETP inhibitors, such as CP-529414 (Pfizer) and JTT-705 (Akros Pharma)), PPAR agonists (as described above) and/or nicotinic acid and derivatives thereof.
- MTP inhibitors e.g., HMG CoA reductase inhibitors, squalene synthetase inhibitors, fibric acid derivatives, ACAT inhibitors, lipoxygenase inhibitors, cholesterol absorption inhibitors, ileal Na
- MTP inhibitors which may be employed as described above include those disclosed in U.S. Patent No. 5,595,872, U.S. Patent No. 5,739,135, U.S. Patent No. 5,712,279, U.S. Patent No. 5,760,246, U.S. Patent No. 5,827,875, U.S. Patent No. 5,885,983 and U.S. Patent No. 5,962,440.
- the HMG CoA reductase inhibitors which may be employed in combination with one or more compound of formula I include mevastatin and related compounds, as disclosed in U.S. Patent No. 3,983,140, lovastatin (mevinolin) and related compounds, as disclosed in U.S. Patent No. 4,231,938, pravastatin and related compounds, such as disclosed in U.S. Patent No. 4,346,227, simvastatin and related compounds, as disclosed in U.S. Patent Nos. 4,448,784 and 4,450,171.
- Other HMG CoA reductase inhibitors which may be employed herein include, but are not limited to, fluvastatm, disclosed in U.S. Patent No.
- Preferred hypolipidemic agents are pravastatin, lovastatin, simvastatin, atorvastatin, fluvastatin, cerivastatin, atavastatin and ZD-4522.
- phosphinic acid compounds useful in inhibiting HMG CoA reductase such as those disclosed in GB 2205837, are suitable for use in combination with the compound of the present invention.
- the squalene synthetase inhibitors suitable for use herein include, but are not limited to, ⁇ -phosphono-sulfonates disclosed in U.S. Patent No. 5,712,396, those disclosed by Biller et al., J. Med. Chem., 1988, Vol. 31, No. 10, pp 1869-1871, including isoprenoid ( ⁇ hosphinyl-methyl)phosphonates, as well as other known squalene synthetase inhibitors, for example, as disclosed in U.S. Patent No.
- squalene synthetase inhibitors suitable for use herein include the terpenoid pyrophosphates disclosed by P. Ortiz de Montellano et al, J. Med. Chem., 1977, 20, 243-249, the farnesyl diphosphate analog A and presqualene pyrophosphate (PSQ-PP) analogs as disclosed by Corey and Volante, J. Am. Chem.
- fibric acid derivatives which may be employed in combination the compound of formula I include fenofibrate, gemfibrozil, clofibrate, bezafibrate, ciprofibrate, clinofibrate and the like, probucol, and related compounds, as disclosed in U.S. Patent No.
- bile acid sequestrants such as cholestyramine, colestipol and DEAE-Sephadex (Secholex ® , policexide ® ), as well as lipostabil (Rhone-Poulenc), Eisai E-5050 (an N-substituted ethanolamine derivative), imanixil (HOE-402), tetrahydrolipstatin (THL), istigmastanylphos-phorylcholine (SPC, Roche), aminocyclodextrin (Tanabe Seiyoku), Ajinomoto AJ-814 (azulene derivative), melinamide (Sumitomo), Sandoz 58-035, American Cyanamid CL-277,082 and CL-283,546 (disubstituted urea derivatives), nicotinic acid, acipimox, acifran, neomycin, p-aminosalicylic
- the ACAT inhibitor which may be employed in combination the compound of formula I include those disclosed in Drugs of the Future 24, 9-15 (1999), (Avasimibe); "The ACAT inhibitor, Cl-1011 is effective in the prevention and regression of aortic fatty streak area in hamsters", Nicolosi et al, Atherosclerosis (Shannon, Irel). (1998), 137(1), 77-85; "The pharmacological profile of FCE 27677: a novel ACAT inhibitor with potent hypolipidemic activity mediated by selective suppression of the hepatic secretion of ApoBlOO-containing lipoprotein", Ghiselli, Giancarlo, Cardiovasc. Drug Rev.
- Inhibitors of acyl-Co A cholesterol O-acyl transferase (ACAT) as hypocholesterolemic agents. 6. The first water-soluble ACAT inhibitor with lipid- regulating activity. Inhibitors of acyl-Co A: cholesterol acyltransferase (ACAT). 7. Development of a series of substituted N-phenyl-N'-[(l- phenylcyclopentyl)methyl]ureas with enhanced hypocholesterolemic activity", Stout et al, Chemtracts: Org. Chem. (1995), 8(6), 359-62, or TS-962 (Taisho Pharmaceutical Co. Ltd).
- the hypolipidemic agent may be an up-regulator of LD2 receptor activity, such as MD-700 (Taisho Pharmaceutical Co. Ltd) and LY295427 (Eli Lilly).
- suitable cholesterol absorption inhibitor for use in combination with the compound of the invention include SCH48461 (Schering- Plough), as well as those disclosed in Atherosclerosis 115, 45-63 (1995) and J. Med. Chem. 41, 973 (1998).
- suitable ileal NaVbile acid co-transporter inhibitors for use in combination with the compound of the invention include compounds as disclosed in Drugs of the Future, 24, 425-430 (1999).
- the lipoxygenase inhibitors which may be employed in combination the compound of formula I include 15-lipoxygenase (15-LO) inhibitors, such as benzimidazole derivatives, as disclosed in WO 97/12615, 15-LO inhibitors, as disclosed in WO 97/12613, isothiazolones, as disclosed in WO 96/38144, and 15-LO inhibitors, as disclosed by Sendobry et al "Attenuation of diet-induced atherosclerosis in rabbits with a highly selective 15-lipoxygenase inhibitor lacking significant antioxidant properties", Brit. J. Pharmacology (1997) 120, 1199- 1206, and Cornicelli et al, "15-Lipoxygenase and its Inhibition: A Novel Therapeutic Target for Vascular Disease", Current Pharmaceutical Design, 1999, 5, 11-20.
- 15-LO 15-lipoxygenase
- 15-LO 15-lipoxygenase
- benzimidazole derivatives as disclosed in WO 97/126
- Suitable anti-hypertensive agents for use in combination with the compound of the present invention include beta adrenergic blockers, calcium channel blockers (L-type and T-type; e.g. diltiazem, verapamil, nifedipine, amlodipine and mybefradil), diuretics (e.g., chlorothiazide, hydrochlorothiazide, flumethiazide, hydroflumethiazide, bendroflumethiazide, methylchlorothiazide, trichloromethiazide, polythiazide, benzthiazide, ethacrynic acid tricrynafen, chlorthalidone, furosemide, musolrmine, bumetanide, triamtrenene, amiloride, spironolactone), renin inhibitors, ACE inhibitors (e.g., captopril, zofenopril,
- Dual ET/AII antagonist e.g., compounds disclosed in WO 00/01389
- neutral endopeptidase (NEP) inhibitors neutral endopeptidase (NEP) inhibitors
- vasopepsidase inhibitors dual NEP-ACE inhibitors
- omapatrilat and gemopatrilat e.g., omapatrilat and gemopatrilat
- Suitable anti-obesity agents for use in combination with the compound of the present invention include a beta 3 adrenergic agonist, a lipase inhibitor, a serotonin (and dopamine) reuptake inhibitor, a thyroid receptor beta drug, 5HT2C agonists, (such as Arena APD-356); MCHRl antagonists such as Synaptic
- SNAP-7941 and Takeda T-226926 melanocortin receptor (MC4R) agonists, melanin- concentrating hormone receptor (MCHR) antagonists (such as Synaptic SNAP-7941 and Takeda T-226926), galanin receptor modulators, orexin antagonists, CCK agonists, NPYl or NPY5 antagonsist, NPY2 and NPY4 modulators, corticotropin releasing factor agonists, histamine receptor-3 (H3) modulators, 11-beta-HSD-l inhibitors, adinopectin receptor modulators, monoamine reuptake inhibitors or releasing agents, a ciliary neurotrophic factor (CNTF, such as AXOKINE ® by Regeneron), BDNF (brain-derived neurotrophic factor), leptin and leptin receptor modulators, cannabinoid-1 receptor antagonists (such as SR-141716 (Sanofi) or SLV- 319 (So
- lipase inhibitors which may be optionally employed in combination with compound of the present invention include orlistat or ATL-962 (Alizyme).
- the serotonin (and dopoamine) reuptake inhibitor (or serotonin receptor agonists) which maybe optionally employed in combination with a compound of the present invention maybe BVT-933 (Biovitrum), sibutramine, topiramate (Johnson & Johnson) or axokine (Regeneron).
- the monoamine reuptake inhibitors which may be optionally employed in combination with compound of the present invention include fenfluramine, dexfenfluramine, fluvoxamine, fluoxetine, paroxetine, sertraline, chlorphentermine, cloforex, clortermine, picilorex, sibutramine, dexamphetamine, phentermine, phenylpropanolamine or mazindol.
- the anorectic agent which may be optionally employed in combination with the compound of the present invention include topiramate (Johnson & Johnson), dexamphetamine, phentermine, phenylpropanolamine or mazindol.
- the compound of the invention are utilized in combination with one or more other therapeutic agent(s), either concurrently or sequentially, the following combination ratios and dosage ranges are preferred.
- the other antidiabetic agent is a biguanide
- the compound of formula I will be employed in a weight ratio to biguanide within the range from about
- the compound of formula I will be employed in a weight ratio to the glucosidase inhibitor within the range from about 0.01:1 to about 100:1, preferably from about 0.5 : 1 to about 50:1.
- the compound of formula I will be employed in a weight ratio to the sulfonyl urea in the range from about 0.01:1 to about 100:1, preferably from about
- the compound of formula I will be employed in a weight ratio to the thiazolidinedione in an amount within the range from about 0.01:1 to about 100:1, preferably from about 0.2:1 to about 10:1.
- the thiazolidinedione anti-diabetic agent may be employed in amounts within the range from about 0.01 to about 2000 mg/day which may be administered in single or divided doses one to four times per day.
- the sulfonyl urea and thiazolidinedione may be incorporated in a single tablet with the compound of formula I in amounts of less than about 150 mg.
- metformin or salt thereof may be employed in amounts within the range from about 500 to about 2000 mg per day which may be administered in single or divided doses one to four times daily.
- GLP-I peptides may be administered in oral buccal formulations, by nasal administration or parenterally as described in U.S. Patent Nos. 5,346,701 (TheraTech), 5,614,492 and 5,631,224 which are incorporated herein by reference.
- the compound of formula I will be employed in a weight ratio to the meglitinide, PPAR-gamma agonist, PPAR-alpha/gamma dual agonist, aP2 inhibitor or other DPP4 inhibitor within the range from about 0.01 : 1 to about 100: 1 , preferably from about 0.2:1 to about 10:1.
- the compound of formula I of the invention will be generally be employed in a weight ratio to the hypolipidemic agent (were present), within the range from about 500:1 to about 1:500, preferably from about 100:1 to about 1:100.
- a satisfactory result may be obtained employing the MTP inhibitor in an amount within the range of from about 0.01 mg/kg to about 500 mg and preferably from about 0.1 mg to about 100 mg, one to four times daily.
- a preferred oral dosage form, such as tablets or capsules will contain the MTP inhibitor in an amount of from about 1 to about 500 mg, preferably from about 2 to about 400 mg, and more preferably from about 5 to about 250 mg, one to four times daily.
- an HMG CoA reductase inhibitor in an amount within the range of from about 1 to 2000 mg, and preferably from about 4 to about 200 mg.
- a preferred oral dosage form such as tablets or capsules, will contain the HMG CoA reductase inhibitor in an amount from about 0.1 to about 100 mg, preferably from about 5 to about 80 mg, and more preferably from about 10 to about 40 mg.
- the squalene synthetase inhibitor may be employed in dosages in an amount within the range of from about 10 mg to about 2000 mg and preferably from about 25 mg to about 200 mg.
- a preferred oral dosage form such as tablets or capsules will contain the squalene synthetase inhibitor in an amount of from about 10 to about 500 mg, preferably from about 25 to about 200 mg.
- the compound of the formula I can be administered for any of the uses described herein by any suitable means, for example, orally, such as in the form of tablets, capsules, granules or powders; sublingually; bucally; parenterally, such as by subcutaneous, intravenous, intramuscular, or intrasternal injection or infusion techniques (e.g., as sterile injectable aqueous or non-aqueous solutions or suspensions); nasally, including administration to the nasal membranes, such as by inhalation spray; topically, such as in the form of a cream or ointment; or rectally such as in the form of suppositories; in dosage unit formulations containing non-toxic, pharmaceutically acceptable vehicles or diluents.
- a pharmaceutical composition will be employed containing one or more of the compound of formula I, with or without other antidiabetic agent(s) and/or antihyperlipidemic agent(s) and/or other type therapeutic agents in association with a pharmaceutical vehicle or diluent.
- the pharmaceutical composition can be formulated employing conventional solid or liquid vehicles or diluents and pharmaceutical additives of a type appropriate to the mode of desired administration, such as pharmaceutically acceptable carriers, excipients, binders and the like.
- the compound can be administered to mammalian species including humans, monkeys, dogs, etc.
- a typical injectable preparation may be produced by aseptically placing 250 mg of compound of formula I into a vial, aseptically freeze-drying and sealing. For use, the contents of the vial are mixed with 2 mL of physiological saline, to produce an injectable preparation.
- DPP-4 inhibitory activity of the compounds of the present invention may be determined by use of an in vitro assay system which measures the degree in inhibition of DPP-4-mediated cleavage of an appropriate substrate or pseudo- substrate. Inhibition constants (Ki values) for the DPP-4 inhibitors of the invention may be determined by the method described in the experimental section below.
- PCR Red-tag polymerase, Sigma was performed on Human cDNA from placenta (Clontech) using two primers, ACGCCGACGATGAAGACA and AGGTAAAGAGAAACATTGTT, based on the nucleotide sequence of the human clone (accession number M74777). PCR products were cloned into the pcDN4/HisMax TOPO vector (Invitrogene). For stable transfection of CHO-DG44 cells, DPP4 was rePCRed using primers GGTACCAGCGCAGAGGCTT and CTCGAGCTAAGGTAAAGAGAAACATTG to generate Kpnl and Xhol sites.
- the Kpnl and Xhol sites were used to extract the N- terminal His tagged gene.
- the His tag which could be cleaved and removed by Enterokinase, was included to allow purification using the TALON affinity column.
- the gene was then ligated into the Kpnl and Xhol sites of the pD16 vector for stable transfection.
- Stable cell lines were generated by transfecting the expression vector into Chinese hamster ovary (CHO-DG44) cells using electroporation.
- the CHO-DG44 cell line was grown in PFCHO media supplemented with HT (glycine, hypoxanthine and thymidine, Invitrogene), glutamine and Recombulin (ICN).
- protein was further purified using conventional anion exchange (Sepharose Q) 5 gel filtration (S-200) and high resolution MonoQ columns.
- the final protein yielded a single band on SDS-PAGE gels.
- Amino acid sequence analysis indicated two populations of DPP-4 in the sample. One portion of the protein had 27 amino acids truncated from the N-terminus, while the other was lacking the N- terminal 37 amino acids. This suggests that during isolation the entire transmembrane domain (including His tag) is removed by proteases present in the CHO cells.
- vi and vo are the steady state velocities measured in the presence and absence of inhibitor, E enzyme concentration.
- Ph phenyl
- Cbz carbobenzyloxy or carbobenzoxy or benzyloxycarbonyl
- PtO 2 platinum oxide
- EDCI or EDAC 3-ethyl-3'-(dimemylamino)propyl-carbodiimide hydrochloride (or
- HOBT or HOBT*H 2 O 1 -hydroxybenzotriazole hydrate
- HOAT l-hydroxy-7-azabenzotriazole
- UCT United Chemical Technologies, Inc.; Bristol, PA.
- Example 1 3-(2,4-Dichlorophenyl)-3-oxopropanenitrile.
- Example 1 2-(2,4-Dichlorobenzoyl)-3-(dimethyIamino)acryIonitrile.
- step 1 To a stirred solution of step 1 nitrile (1.74 g, 8.13 mmol) in toluene (50 mL) was added dimethylformamide dimethylacetal (1.35 mL, 10.16 mmol). The resulting brown solution was heated to 50 °C for 1 hr. The solvent was removed under reduced pressure and the residue was diluted with CH 2 Cl 2 (50 mL). The organic layer was washed with saturated NaHCO 3 solution (50 mL) and brine (50 mL), dried (MgSO 4 ), filtered and concentrated under reduced pressure to give the crude product as a brown oil. Purification of the crude product by flash chromatography (silica gel, 40% EtOAc / hexane) afforded 2-(2,4-dichlorobenzoyl)-3-
- Example 1 4-(2,4-Dichlorophenyl)-2-(3,5-dimethoxyphenyl)pyrimidine- 5-carbonitrile.
- Step 2 acrylonitrile (1.5 g, 5.6 mmol) and 3,5- dimethoxybenzamidine hydrochloride (1.2 g, 5.6 mmol) in MeOH (30 mL) was added NaOMe (25% in MeOH, 2.56 mL, 11.2 mmol). The reaction was heated to reflux for 5 hr. Additional NaOMe (25% in MeOH, 2.56 mL, 11.2 mmol) was added and was kept for 16 hr. The reaction was cooled to ambient temperature and quenched by addition OfH 2 O (50 mL).
- TFA salt (4-(2,4-Dimethylphenyl)-2-phenylpyrimidin-5-yl)methanamine, TFA salt was prepared by the methods described in Example 1 using 2,4-dimethylbenzoyl chloride for Step 1 and benzamidine hydrochloride for Step 3.
- Example 8 Methyl 4-(2,4-dichlorophenyl)-6-methyl-2-phenyl-l,4- dihydropyrimidine-5-carboxylate.
- Example 8 4-(2,4-Dichlorophenyl)-6-methyl-2-phenylpyrimidine-5- carbaldehyde oxime.
- Example 8 (4-(2,4-Dichlorophenyl)-6-methyl-2-phenylpyrimidin-5- yl)methanamine.
- Example 10 Diethyl 2-(2,4-dichlorobenzylidene)malonate.
- Example 10 Ethyl 4-(2,4-dichlorophenyl)-6-oxo-2-phenyl-l,6- dihydropyrimidine-5-carboxylate.
- Example 10 Ethyl 4-(2,4-dichlorophenyl)-2,6-diphenylpyrimidine-5- carboxylate.
- the reaction was diluted with EtOAc (15 mL) and the organic layer was washed with IN NaOH (10 mL), saturated Na 2 CO 3 solution (10 mL) and brine (10 mL), dried (MgSO 4 ), filtered and concentrated under reduced pressure.
- the crude reaction product was purified by flash chromatography (silica gel, 20% EtOAc / hexane) to give the alcohol (10 mg, 79%).
- the aqueous layer was extracted with EtOAc (2 x 10 mL) and the combined organic layers were washed with brine, dried (MgSO 4 ), filtered and concentrated under reduced pressure to give the desired azide.
- the azide was dissolved in THF (1 mL) and H 2 O (0.2 mL) and PPh 3 (polymer supported, 3 mmol/g, 16 mg, 0.049 mmol) was added. The reaction was heated to 50 °C for 1 hr and filtered to remove polymer support.
- Example 13 Methyl 2-(benzylthio)-4-(2,4-dichlorophenyl)-6-methyl-l,4- dihydropyrimidine-5-carboxylate.
- the crude reaction product (>98% purity) was moved onto next step without further purification.
Abstract
Compounds are provided having the formula (I) wherein R, B, X and Y are as defined herein.
Description
PYRIMIDINE-BASED INfflBITORS OF DIPEPTIDYL PEPTIDASE IV AND METHODS
CROSS-REFERENCE TO RELATED APPLICATIONS [0001] This application claims the priority benefit of U.S. Provisional Application No. 60/640,110, filed December 29, 2004, the disclosure of which is incorporated herein by reference in its entirety.
FIELD OF THE INVENTION [0002] The present invention relates to pyrimidine-based inhibitors of dipeptidyl peptidase IV (DPP-4), and to a method for treating multiple diseases or disorders by employing such pyrimidine -based inhibitors alone, or in combination with another type of therapeutic agent.
BACKGROUND OF THE INVENTION
[0003] Dipeptidyl peptidase IV (DPP-4) is a membrane bound non-classical serine aminodipeptidase which is located in a variety of tissues (intestine, liver, lung, kidney) as well as on circulating T-lymphocytes (where the enzyme is known as CD- 26). It is responsible for the metabolic cleavage of certain endogenous peptides (GLP-I (7-36), glucagon) in vivo and has demonstrated proteolytic activity against a variety of other peptides (GHRH, NPY, GLP-2, VIP) in vitro. [0004] GLP- 1 (7-36) is a 29 amino-acid peptide derived by post-translational processing of proglucagon in the small intestine. GLP-l(7-36) has multiple actions in vivo including the stimulation of insulin secretion, inhibition of glucagon secretion, the promotion of satiety, and the slowing of gastric emptying. Based on its physiological profile, the actions of GLP- 1(7-36) are expected to be beneficial in the prevention and treatment of type II diabetes and potentially obesity. To support this claim, exogenous administration of GLP- 1(7-36) (continuous infusion) in diabetic patients has demonstrated efficacy in this patient population. Unfortunately GLP- 1(7- 36) is degraded rapidly in vivo and has been shown to have a short half-life in vivo (tl/2~l .5 min). Based on a study of genetically bred DPP-4 KO mice and on in vivo/in vitro studies with selective DPP-4 inhibitors, DPP-4 has been shown to be the
primary degrading enzyme of GLP- 1(7-36) in vivo. GLP- 1(7-36) is degraded by DPP -4 efficiently to GLP- 1(9-36), which has been speculated to act as a physiological antagonist to GLP-l(7-36). Thus, inhibition of DPP-4 in vivo should potentiate endogenous levels of GLP-l(7-36) and attenuate formation of its antagonist GLP-I (9- 36) and thus serve to ameliorate the diabetic condition.
DESCRIPTION OF THE INVENTION
[0005] In accordance with the present invention, compounds of formula (I) are provided

R is a substitutent selected from the group consisting of hydrogen (H), halogen, cyano (CN), CF3, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkylalkyl, bicycloalkyl, bicycloalkylalkyl, alkylthioalkyl, arylalkylthioalkyl, cycloalkenyl, aryl, arylalkyl, heteroaryl, heteroarylalkyl, cycloheteroalkyl and cycloheteroalkylalkyl, wherein any such substituent may optionally be substituted through available carbon atoms with 1, 2, 3, 4 or 5 groups selected from hydrogen, halo, alkyl, polyhaloalkyl, alkoxy, haloalkoxy, polyhaloalkoxy, alkoxycarbonyl, alkenyl, alkynyl, cycloalkyl, cycloalkylalkyl, polycycloalkyl, heteroarylamino, arylamino, cycloheteroalkyl, cycloheteroalkylalkyl, hydroxy, hydroxyalkyl, nitro, cyano, amino, substituted amino, alkylamino, dialkylamino, thiol, alkylthio, alkylcarbonyl, acyl, alkoxycarbonyl, aminocarbonyl, alkynylaminocarbonyl, altylaminocarbonyl, alkenylaminocarbonyl, alkylcarbonyloxy, alkylcarbonylamino, arylcarbonylamino, alkylsulfonylamino, alkylaminocarbonylamino, alkoxycarbonylamino, alkylsulfonyl, aminosulfonyl, alkylsulfinyl, sulfonamido and sulfonyl;
B is selected from the group consisting of a bond, oxygen (O), nitrogen (N) and S(O)1n;
m is 0, 1 or 2;
X is a substitutent selected from the group consisting of hydrogen (H), alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkylalkyl, bicycloalkyl, bicycloalkylalkyl, alkylthioalkyl, arylalkylthioalkyl, cycloalkenyl, aryl, arylalkyl, heteroaryl, heteroarylalkyl, cycloheteroalkyl and cycloheteroalkylalkyl, wherein any such substituent may optionally be substituted through available carbon atoms with 1, 2, 3, 4 or 5 groups selected from hydrogen, halo, alkyl, polyhaloalkyl, alkoxy, haloalkoxy, polyhaloalkoxy, alkoxycarbonyl, alkenyl, alkynyl, cycloalkyl, cycloalkylalkyl, polycycloalkyl, heteroarylamino, arylamino, cycloheteroalkyl, cycloheteroalkylalkyl, hydroxy, hydroxyalkyl, nitro, cyano, amino, substituted amino, alkylamino, dialkylamino, thiol, alkylthio, alkylcarbonyl, acyl, alkoxycarbonyl, aminocarbonyl, alkynylaminocarbonyl, alkylaminocarbonyl, alkenylaminocarbonyl, alkylcarbonyloxy, alkylcarbonylamino, arylcarbonylamino, alkylsulfonylamino, alkylaminocarbonylamino, alkoxycarbonylamino, alkylsulfonyl, aminosulfonyl, alkylsulfinyl, sulfonamide and sulfonyl;
B-X taken together can be a halogen; and
Y is aryl, optionally substituted with 1, 2, 3, 4 or 5 groups selected from hydrogen, halo, alkyl, polyhaloalkyl, alkoxy, haloalkoxy, polyhaloalkoxy, alkoxycarbonyl, alkenyl, alkynyl, cycloalkyl, cycloalkylalkyl, polycycloalkyl, heteroarylamino, arylamino, cycloheteroalkyl, cycloheteroalkylalkyl, hydroxy, hydroxyalkyl, nitro, cyano, amino, substituted amino, alkylamino, dialkylamino, thiol, alkylthio, alkylcarbonyl, acyl, alkoxycarbonyl, aminocarbonyl, alkynylaminocarbonyl, alkylaminocarbonyl, alkenylaminocarbonyl, alkylcarbonyloxy, alkylcarbonylamino, arylcarbonylamino, alkylsulfonylamino, alkylaminocarbonylamino, alkoxycarbonylamino, alkylsulfonyl, aminosulfonyl, alkylsulfinyl, sulfonamido and sulfonyl.
[0006] The definition of formula I above includes all pharmaceutically acceptable salts, stereoisomers, and prodrug esters of formula I. [0007] The compounds of formula I possess activity as inhibitors of DPP -4 in vivo and are useful in the treatment of diabetes and the micro- and macrovascular complications of diabetes such as retinopathy, neuropathy, nephropathy, and wound
healing. Such diseases and maladies are also sometimes referred to as "diabetic complications".
[0008] The present invention provides for compounds of formula I, pharmaceutical compositions employing such compounds and for methods of using such compounds, hi particular, the present invention provides a pharmaceutical composition comprising a therapeutically effective amount of a compound of formula I5 alone or in combination with a pharmaceutically acceptable carrier. [0009] Further provided is a method for treating or delaying the progression or onset of diabetes, especially type II diabetes, including complications of diabetes, including retinopathy, neuropathy, nephropathy and delayed wound healing, and related diseases such as insulin resistance (impaired glucose homeostasis), hyperglycemia, hyperinsulinemia, elevated blood levels of fatty acids or glycerol, obesity, hyperlipidemia including hypertriglyceridemia, Syndrome X, atherosclerosis and hypertension, and for increasing high density lipoprotein levels, wherein a therapeutically effective amount of a compound of formula I is administered to a mammalian, e.g., human, patient in need of treatment.
[0010] The compounds of the invention can be used alone, in combination with other compounds of the present invention, or in combination with one or more other agent(s) active in the therapeutic areas described herein. [0011] In addition, a method is provided for treating diabetes and related diseases as defined above and hereinafter, wherein a therapeutically effective amount of a combination of a compound of formula I and at least one other type of therapeutic agent, such as an antidiabetic agent and/or a hypolipidemic agent, is administered to a human patient in need of treatment. [0012] Further embodiments of the invention include compounds of formula I wherein n is 1, or compounds of formula I having the structure:



[0013] In the above method of the invention, the compound of formula (I) will be employed in a weight ratio to the antidiabetic agent or other type therapeutic agent (depending upon its mode of operation) within the range from about 0.01:1 to about 500:1, preferably from about 0.1:1 to about 100:1, more preferably from about 0.2:1 to about 10:1.
DETAILED DESCRIPTION OF THE INVENTION
[0014] Compounds of formula (I) may be generated by the methods as shown in the following reaction schemes and the descriptions thereof.
SCHEME l
O
Y-COOH - Y-^CI
1 2

base

[0015] Scheme 1 provides a general route to prepare aminomethylpyrimidines of formula (8). Acid chlorides of formula (2) may be obtained from commercial sources, or alternatively generated by methods as described herein from the corresponding carboxylic acids of formula (1). For example, an acid chloride (2) can be formed by treating a carboxylic acid (1) with (COCl)2 or SOCl2 in an inert solvent such as methylene chloride or THF at 0 to 60 °C for 2-48 hours. Ketonitriles of formula (4) can be prepared by combining the lithium anion of acetonitrile with an acid chloride of formula (2). Acetonitrile (3) can be deprotonated by a strong base such as π-BuLi in an anhydrous solvent such as THF or diethyl ether at low temperature to give the lithium anion of acetonitrile. Acrylnitriles of formula (5) can be prepared by methods known to those skilled in the art such as heating ketonitrile of formula (4) with dimethylformamide dimethylacetal in an inert solvent such as toluene at elevated temperature for 2-48 hours. Amidines of formula (6) can either be obtained through commercial sources or conveniently prepared by known methods. One example to make the amidines of formula (6) is to start with the corresponding nitrile, treating with HCl followed by NH3 to provide amidines (6). Pyrimidines of formula (7) can be prepared by combining acrylonitriles (5) and amidines (6) by methods known in the art. For example, the process can be performed by heating an acrylonitrile (5) and an amidine (6) with a base such as NaOMe in methanol at room temperature to reflux for 2-48 hours. Aminomethylpyrimidines of formula (8) can be prepared from nitriles
(7) through a reductive process. The reducing agents which may be used for this process include, but are not limited to LAH, CoCl2/NaBH4, Raney Ni/H2, and PdZH2.
[0016] Scheme 2 describes an alternative route to prepare aminomethylpyrimidines of formula (8).
SCHEME 2 base O O DMF-DMA
A ♦ (MeO)2CO3
Y
9 AA0 /
10
reduction

[0017] Ketoesters of formula (10) are known in the literature or can be conveniently prepared by known methods known. One example to prepare ketoesters of formula (10) is to combine a ketone (9) with a methylcarbonate and a base such as NaH in an inert solvent such as THF at ambient temperature for 2-24 hours. Acryloesters of formula (11) can be prepared by the same methods as described in Scheme 1 for acrylonitriles (5). Pyrimidine esters of formula (12) can be prepared by combining an acryloester (11) and an amidine (6) using the same methods as described in Scheme 1 for pyrimidines (7). Aminomethyl pyrimidines of formula (8) can then be prepared by those skilled in the art through a reduction / oxidation sequence on pyrimidine esters of formula (12) as described in scheme 2. The reducing agents that may be used to convert an ester of formula (12) to an alcohol of formula
(13) include, but are not limited to DIBAL, LAH, and Red- Al. The oxidizing agents that may be used to convert an alcohol of formula (13) to an aldehyde of formula (14) include, but are not limited to Dess-Martin periodinane, Swern, PCC, MnO2, and TPAP/NMO. As understood by those skilled in the art, formula (15) can be either an oxime or an imine, which can be conveniently prepared by combining an aldehyde of formula (14) with an amine or hydroxylamine. The reduction of compounds of formula (15) to aminomethylpyrimidines of formula (8) can be performed by using reducing agents such as Zn/HOAc, PdZH2, or Raney Ni/H2.
[0018] Scheme 3 provides an alternative route of converting an alcohol of formula (13) to aminomethylpyrimidine of formula (8).
SCHEME 3
[0019] The chloropyrimidine of formula (16) can be formed from an alcohol of formula (13) by methods known to one skilled in the art. One example of such a transformation is to treat an alcohol (13) with SOCl2 in an inert solvent such as CH2Cl2 at elevated temperature for 2-24 hours. The chloropyrimidines of formula (16) can be converted to aminomethylpyrimidines of formula (8) by bubbling NH3 gas to a solution of chloropyrimidines (16) in a suitable solvent such as methanol.
[0020] Scheme 4 provides an alternative route for converting alcohols of formula (13) to aminomethylpyrimidines of formula (8).
SCHEME 4


[0021] An alcohol of formula (13) can be converted to a suitable leaving group, such as a mesylate, by treating the alcohol (13) with methanesulfonyl chloride and a base such as triethylamine or pyridine in an inert solvent such as tetrahydrofuran or methylene chloride at 0 to 60 °C for 1 to 24 hours. The mesylates of formula (17) can then be converted to azides of formula (18) by known methods. One such set of conditions involves treatment of a mesylate (17) with sodium azide in an inert solvent such as DMF at room temperature to 100 °C for 1 to 24 hours. The azides of formula (18) can then be reduced to form aminomethylpyrimidines of formula (8). The reducing agents that may be used for this transformation include, but are not limited to triphenylphosphine, trialkylphosphine (including polymer supported phosphines), lithium aluminum hydride, hydrogen with palladium, and platinum containing catalysts. [0022] Alkylated aminomethylpyrimidines of formula (19) can be prepared from aldehydes of formula (14) as described in scheme 5. One example of such a transformation can be found in: Hart, David J.; Kanai, Kenichi; Thomas, Dudley G.; Yang, Teng Kuei. Journal of Organic Chemistry (1983), 48(3), 289-94. Another example of such a transformation is to add a Grignard reagent (R-MgBr) to the aldehyde, followed by oxidation, imine/oxime formation and reduction as described in Scheme 3.
SCHEME 5
[0023] Scheme 6 describes a route to prepare 6-substituted aminomethylpyrimidines of formula (24).
SCHEME 6

23 24
[0024] Keto esters of formula (20) can either be obtained from commercial sources or conveniently prepared by the methods described in Scheme 2. Acryloesters of formula (22) can be prepared by known methods by combining a ketoester of formula (20) and an aldehyde of formula (21). One example to prepare an acryloester of formula (22) is through a Knovenagel reaction. Pyrimidine esters of formula (23) can be prepared by methods known to those skilled in the art by combining acryloesters of formula (22) and amidines of formula (6) by known methods. For example, combining an acryloester of formula (22) and an amidine of formula (6) in the presence of a suitable base such as triethylamine, pyridine, NaOMe or KOAc in an inert solvent such as toluene, chloroform, benzene or DMF at elevated temperature gives pyrimidine esters of formula (23). The conversion of pyrimidine esters of formula (23) to aminomethylpyrimidines of formula (24) follows the same procedures as described in Schemes 2, 3 and 4.
[0025] Scheme 7 describes an alternative route to prepare aminomethylpyrimidines of formula (8).
SCHEME 7 oxidation



[0026] Pyrimidine esters of formula (25) can either be obtained through commercial sources or conveniently prepared by methods known in the art. The ester functionality of (25) can be converted to an alcohol of formula (26) by a reductive process. The reducing agents which may be used for this process include, but are not limited to LAH, DIBAL, Red- Al, and NaBH4. The reaction can be performed by combining an ester (25) and the reducing agent in an inert solvent such as THF or toluene at -78 0C to elevated temperature for 2-24 hours. Pyrimidine aldehydes of formula (27) can be prepared from pyrimidine alcohols of formula (26) by an oxidative process. The oxidizing agents which may be used for this process include, but are not limited to PCC, Dess-Martin periodinane, Swern, and TPAP/NMO. The reaction can be performed in a solvent such as CH2Cl2, THF at -30 °C to ambient temperature for 2-24 hours. Pyrimidines of formula (29) can be prepared by combining a chloropyrimidine of formula (27) and a boronic acid of formula (28) by a Suzuki coupling process. Boronic acids of formula (28) can be obtained from commercial sources or conveniently prepared by methods known in the art. Examples of suitable palladium-catalyzed Suzuki coupling process can be found in: Palladium reagents and catalysts: innovations in organic synthesis, by Tsuji, Jiro; Palladium reagents in organic syntheses by Richard F. Heck. The aminomethylpyrimidines of formula (8) can be synthesized from compounds of formula (29) according the chemistry described in Schemes 2, 3 and 4.
[0027] 2-Amino-substituted pyrimidines of formula (x) can be prepared by methods described in Scheme 8.
SCHEME 8
oxidatlon ,


[0028] A ketoester of formula (22) can be condensed with an alkyl- or arylthioamidine such as (30) to give 2-alkylthiopyrimidines of formula (31) by known methods. One such set of conditions is to combine a ketoester of formula (22) with an amidine of formula (30) in a suitable solvent such as DMF at 20 to 100 °C for 1 - 72 hours. Molecular sieves can be added to facilitate the reaction. The alkylthio compound of formula (31) can be oxidized to an alkylsulfone of formula (32) by known methods. The oxidizing agents that may be used for this transformation include, but are not limited to mCPBA, hydrogen peroxide, PCC, and MnO2. The alkylsulfone of formula (32) can then be displaced by amines to form a 2- aminosubstituted pyrimidine of formula (33). An example of one such set of conditions which may be used for this conversion is to combine a sulfone of formula (32) with a primary or secondary amine in a suitable solvent such as methylene chloride, THF or DMF at rt to 100 °C for 1 to 72 hours. The ester of formula (33) can be converted to an aminomethylpyrimidine of formula (34) by the same procedures as described in Schemes 2, 3, and 4.
DEFINITIONS
[0029] The following definitions apply to the terms as used throughout this specification, unless otherwise limited in specific instances. [0030] Unless otherwise indicated, the term "alkyl" or "alk" as used herein alone or as part of another group includes both branched and straight-chain saturated
aliphatic hydrocarbon radicals/groups having the specified number of carbon atoms. In particular, "Alkyl" refers to a monoradical branched or unbranched saturated hydrocarbon chain, preferably having from 1 to 40 carbon atoms, more preferably 1 to 10 carbon atoms, even more preferably 1 to 6 carbon atoms, such as methyl, ethyl, n- propyl, isopropyl, n-butyl, secondary butyl, tert-butyl, n-hexyl, n-octyl, n-decyl, n- dodecyl, 2-ethyldodecyl, tetradecyl, and the like, unless otherwise indicated. Unless otherwise constrained by the definition for the alkyl substituent, such alkyl groups can optionally be substituted with one or more substituents selected from a member of the group consisting of such as halo, alkyl, alkoxy, aryl, aryloxy, aryl(aryl) or diaryl, arylalkyl, arylalkyloxy, alkenyl, cycloalkyl, cycloalkylalkyl, cycloalkylalkyloxy, amino, hydroxy, hydroxyalkyl, acyl, heteroaryl, heteroaryloxy, heteroarylalkyl, heteroarylalkoxy, aryloxyalkyl, alkylthio, arylalkylthio, aryloxyaryl, alkylamido, alkanoylamino, arylcarbonylamino, nitro, cyano, thiol, haloalkyl, trihaloalkyl and/or alkylthio. [0031] Unless otherwise indicated, the term "cycloalkyl", "carbocycle" or
"carbocyclic" as employed herein alone or as part of another group includes saturated or partially unsaturated (containing 1 or 2 double bonds) cyclic hydrocarbon groups containing 1 to 3 rings, including monocyclic alkyl, bicyclic alkyl (or bicycloalkyl) and tricyclic alkyl, containing a total of 3 to 20 carbons forming the ring, preferably 3 to 10 carbons, forming the ring and which may be fused to 1 or 2 aromatic rings as described for aryl, which includes, for example cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, cyclooctyl, cyclodecyl and cyclododecyl, cyclohexenyl,


[0032] The term "Aryl" or "Ar" as used herein alone or as part of another group refers to an unsaturated aromatic carbocyclic group of from 5 to 20 carbon atoms having a single ring (e.g., phenyl) or multiple condensed (fused) rings (e.g., naphthyl or anthryl). Representative examples include, but are not limited to, aromatic radicals such as phenyl, naphthyl, tetrahydronaphthyl, indane and biphenyl. Unless otherwise constrained by the definition for the aryl substituent, such aryl groups can optionally be substituted with one or more substituents selected from a member of the group consisting of hydrogen, halo, haloalkyl, alkyl, haloalkyl, alkoxy, haloalkoxy, alkenyl, trifluoromethyl, trifluoromethoxy, alkynyl, cycloalkyl-alkyl, cycloheteroalkyl, cycloheteroalkylalkyl, aryl, heteroaryl, arylalkyl, aryloxy, aryloxyalkyl, arylalkoxy, arylthio, arylazo, heteroarylalkyl, heteroarylalkenyl, heteroarylheteroaryl, heteroaryloxy, hydroxy, nitro, cyano, amino, any of the alkyl substituents described herein, or substituted amino wherein the amino includes 1 or 2 substituents (which are alkyl, aryl or any of the other aryl compounds mentioned in the definitions), thiol, alkylthio, arylthio, heteroarylthio, arylthioalkyl, alkoxyarylthio, alkylcarbonyl, arylcarbonyl, alkyl-aminocarbonyl, arylaminocarbonyl, alkoxycarbonyl, aminocarbonyl, alkylcarbonyloxy, arylcarbonyloxy, alkylcarbonylamino, arylcarbonylamino, arylsulfinyl, arylsulfinylalkyl, arylsulfonylamino or arylsulfon- aminocarbonyl and/or any of the alkyl substituents set out herein. [0033] Unless otherwise indicated, the term "cycloheteroalkyl", "heterocyclo", "heterocyclic group" or "heterocyclyl" as used herein alone or as part of another group refers to a saturated or unsaturated group having a single ring, multiple condensed rings or multiple covalently joined rings, from 1 to 40 carbon atoms and from 1 to 10 hetero ring atoms, preferably 1 to 4 hetero ring atoms, selected from nitrogen, sulfur, phosphorus, and/or oxygen. Preferably, "Heterocycle" or "Heterocyclic group" means a stable 5 to 7 membered monocyclic or bicyclic or 7 to 10 membered bicyclic heterocyclic ring that may be saturated, partially unsaturated, or aromatic, and that comprises carbon atoms and from 1 to 4 heteroatoms independently selected from a member of the group consisting of nitrogen, oxygen and sulfur and wherein the
nitrogen and sulfur heteroatoms are optionally be oxidized and the nitrogen heteroatom may optionally be quaternized, and including any bicyclic group in which any of the above-defined heterocyclic rings is fused to a benzene ring. The heterocyclic groups may be substituted on carbon or on a nitrogen, sulfur, phosphorus, and/or oxygen heteroatom, such as, but not limited to, the substituents described for alkyl or aryl herein, so long as the resulting compound is stable. For example:



[0034] "Heteroaryl" as used herein alone or as part of another group embraces unsaturated heterocyclic radicals. Examples of heteroaryl radicals include unsaturated 3 to 6 membered heteromonocyclic group containing 1 to 4 nitrogen atoms, for example, pyrrolyl, pyrrolinyl, imidazolyl, pyrazolyl, pyridyl, pyrimidyl, pyrazinyl, pyridazinyl, triazolyl (e.g., 4H-l,2,4-triazolyl, lH-l,2,3-triazolyl, 2H-l,2,3-triazolyl, etc.) tetrazolyl (e.g. lH-tetrazolyl, 2H-tetrazolyl, etc.), etc.; unsaturated condensed heterocyclyl group containing 1 to 5 nitrogen atoms, for example, indolyl, isoindolyl,
indolizinyl, benzimidazolyl, quinolyl, isoquinolyl, indazolyl, benzotriazolyl, tetrazolopyridazinyl (e.g., tetrazolo[l,5-b]pyridazinyl, etc.), etc.; unsaturated 3 to 6- membered heteromonocyclic group containing an oxygen atom, for example, pyranyl, furyl, etc.; unsaturated 3 to 6-membered heteromonocyclic group containing a sulfur atom, for example, thienyl, etc.; unsaturated 3- to 6-membered heteromonocyclic group containing 1 to 2 oxygen atoms and 1 to 3 nitrogen atoms, for example, oxazolyl, isoxazolyl, oxadiazolyl (e.g., 1,2,4-oxadiazolyl, 1,3,4-oxadiazolyl, 1,2,5- oxadiazolyl, etc.) etc.; unsaturated condensed heterocyclyl group containing 1 to 2 oxygen atoms and 1 to 3 nitrogen atoms (e.g. benzoxazolyl, benzoxadiazolyl, etc.); unsaturated 3 to 6-membered heteromonocyclic group containing 1 to 2 sulfur atoms and 1 to 3 nitrogen atoms, for example, thiazolyl, tbiadiazolyl (e.g., 1,2,4-thiadiazolyl, 1,3,4-thiadiazolyl, 1,2,5-thiadiazolyl, etc.) etc.; unsaturated condensed heterocyclyl group containing 1 to 2 sulfur atoms and 1 to 3 nitrogen atoms (e.g., benzothiazolyl, benzothiadiazolyl, etc.) and the like. Further, examples of heteroaryl groups include the following:




[0035] Unless otherwise indicated, the term "alkenyl" as used herein alone or as part of another group refers to straight or branched chain radicals of 2 to 20 carbons, preferably 2 to 12 carbons, and more preferably 1 to 8 carbons in the normal chain, which include one to six double bonds in the normal chain, such as vinyl, 2-propenyl, 3-butenyl, 2-butenyl, 4-pentenyl, 3-pentenyl, 2-hexenyl, 3-hexenyl, 2-heptenyl, 3- heptenyl, 4-heptenyl, 3-octenyl, 3-nonenyl, 4-decenyl, 3-undecenyl, 4-dodecenyl, 4,8,12-tetradecatrienyl, and the like. Optionally, said alkenyl group may be substituted with one or substituents, such as those substituents disclosed for alkyl. [0036] Unless otherwise indicated, the term "alkynyl" as used herein alone or as part of another group refers to straight or branched chain radicals of 2 to 20 carbons, preferably 2 to 12 carbons and more preferably 2 to 8 carbons in the normal chain, which include one triple bond in the normal chain, such as 2-propynyl, 3-butynyl, 2- butynyl, 4-pentynyl, 3-pentynyl, 2-hexynyl, 3-hexynyl, 2-heptynyl, 3-heptynyl, 4- heptynyl, 3-octynyl, 3-nonynyl, 4-decynyl,3-undecynyl, 4-dodecynyl and the like. Optionally, said alkynyl group may be substituted with one or substituents, such as those substituents disclosed for alkyl.
[0037] The term "cycloalkenyl" as employed herein alone or as part of another group refers to partially unsaturated cyclic hydrocarbons containing 3 to 12 carbons, preferably 5 to 10 carbons and 1 or 2 double bonds. Exemplary cycloalkenyl groups include cyclobutenyl, cyclopentenyl, cyclohexenyl, cycloheptenyl, cyclooctenyl, cyclohexadienyl, and cycloheptadienyl. Optionally, said cycloalkenyl group may be substituted with one or substituents, such as those substituents disclosed for alkyl. [0038] The term "Bicycloalkyl" as employed herein alone or as part of another group includes saturated bicyclic ring groups such as, without limitation, [3.3.0]bicyclooctane, [4.3.0]bicyclononane, [4.4.0]bicyclodecane (decalin), [2.2.2]bicyclooctane, and so forth.
[0039] The term "cycloalkenyl" as employed herein alone or as part of another group includes partially unsaturated carbocyclic radicals having three to twelve carbon
atoms. Examples include, without limitation, cyclobutenyl, cyclopentenyl and cyclohexenyl.
[0040] The term "polycycloalkyl" as employed herein alone or as part of another group includes two or more cycloalkyl ring systems, as defined herein, wherein at least one carbon atom is a part of at least two separately identifiable ring systems. The polycycloalkyl group may contain bridging between two carbon atoms, for example, bicyclo[1.1.0]butyl, bicyclo[3.2.1]octyl, bicyclo[5.2.0]nonyl, tricycl[2.2.1.0.sup.l
]heptyl, norbornyl and pinanyl. The polycycloalkyl group may contain one or more fused ring systems, for example, decalinyl (radical from decalin) and perhydroanthracenyl. The polycycloalkyl group may contain a spiro union, in which a single atom is the only common member of two rings, for example, spiro[3.4]octyl, spiro[3.3]heptyl and spiro[4.5]decyl.
[0041] The term "halogen" or "halo" as used herein alone or as part of another group refers to chlorine, bromine, fluorine, and iodine as well as CF3. [0042] The term "alkoxy" or "alkyloxy" as used herein alone or as part of another group, refers to an alkyl group, as defined herein, appended to a parent molecular moiety through an alkyl group, as defined herein.
[0043] The term " haloalkoxy " as used herein alone or as part of another group refers to alkoxy radicals, as defined herein, further substituted with one or more halo atoms, such as fluoro, chloro or bromo, to provide haloalkoxy radicals. Examples include, without limitation, fluoromethoxy, chloromethoxy, trifluoromethoxy, trifluoromethoxy, fluoroethoxy and fluoropropoxy.
[0044] The term "acyl" as employed herein by itself or part of another group, as
defined herein, refers to an organic radical linked to a carbonyl v c ' group; examples of acyl groups include a substituent group attached to a carbonyl, such as alkanoyl, alkenoyl, aroyl, aralkanoyl, heteroaroyl, cycloalkanoyl, cycloheteroalkanoyl and the like.
[0045] The term "cycloalkylalkyl", "arylalkyl", "cycloheteroalkyl",
"bicycloalkylalkyl" or "heteroarylalkyl" as used herein alone or as part of another group, refers to a cycloalkyl, an aryl, a cyclohetero, a bicycloalkyl or heteroaryl group, as defined herein, appended to a parent molecular moiety through an alkyl group, as
defined herein. Representative examples of arylalkyl include, but are not limited to, benzyl, 2-phenylethyl, 3-phenylpropyl, and the like.
[0046] The term "cycloheteroalkylalkyl" as used herein alone or as part of another group refers to a cycloheteroalkyl group as defined herein, linked through a C atom or heteroatom to a (CH2)r chain, where "r" can be 1 to 10.
[0047] The term "polyhaloalkyl" as used herein alone or as part of another group refers to an "alkyl" group as defined above, having 2 to 9, preferably from 2 to 5, halo substiruehts, such as CF3CH2, CF3 or CF3CF2CH2.
[0048] The term "polyhaloalkoxy" as used herein refers to an "alkoxy" or "alkyloxy" group as defined above having 2 to 9, preferably from 2 to 5, halo substituents, such as CF3CH2O-, CF3O- or CF3CF2CH2O-.
[0049] The term "thiol" or "thio" as used herein alone or as part of another group, refers to (-S) or (-S-).
[0050] The term "alkylthio" or "arylalkylthio" refers to an alkyl group or and arylalkyl group, as defined herein, linked to a parent molecular moiety through a thiol group.
[0051] The term "alkylthioalkyl" or "arylalkylthioalkyl" refers to an alkylthio group or and arylalkylthio group, as defined herein, linked to a parent molecular moiety through an alkyl group. [0052] The term "hydroxy" as used herein alone or as part of another group, refers to a -OH group.
[0053] The term "hydroxyalkyl" as used herein alone or as part of another group, refers to a hydroxyl group, as defined herein, appended to a parent molecular moiety through a alkyl group, as defined herein. [0054] The term "cyano" as used herein alone or as part of another group, refers to a -CN group.
[0055] The term "nitro" as used herein, refers to a -NO2 group.
[0056] The term "sulfinyl", whether used alone or linked to other terms such as alkylsulfinyl, denotes respectively divalent radicals -S(O)-.
[0057] The term " alkylsulfinyl " as used herein alone or as part of another group, refers to an alkyl group, as defined herein, appended to a parent molecular moiety through a sulfinyl group, as defined herein.
[0058] The term "sulfonyl" as used herein alone or as part of another group, refers to an SO2 group.
[0059] The term "alkylsulfonyl" or "aminosulfonyl", as used herein, refer to an alkyl or amino group, as defined herein, appended to a parent molecular moiety through a sulfonyl group, as defined herein. [0060] The term "amino" as employed herein, refers to an -NH3 group or an amine linkage: -NRa-3 wherein Ra may be as described below in the definition for "substituted amino".
[0061] The term "substituted amino" as employed herein alone or as part of another group refers to amino substituted with one or two substituents. For example, NRa Rb, wherein Ra and Rb may be the same or different and are, for example chosen from hydrogen, alkyl, cycloalkyl, alkenyl, cycloalkenyl, alkynyl, aryl, heteroaryl, heterocyclic, arylalkyl, heteroarylalkyl, cycloheteroalkyl, cycloheteroalkylalkyl, cycloalkylalkyl, haloalklyl, hydrooxyalkyl, alkoxyalkyl or thioalkyl. These substituents may optionally be further substituted with any of the alkyl substituents as set out above, hi addition, the amino substituents may be taken together with the nitrogen atom to which they are attached to form 1-pyrrolidinyl, 1-piperidinyl, 1- azepinyl, 4-morpholinyl, 4-thiamorpholinyl, 1-piperazinyl, 4-alkyl-lpiperazinyl, 4- arylalkyl-lpiperazinyl, 4-diarylalkyl- 1-piperazinyl, 1-pyrrolindinyl, 1-piperidinyl, or 1-azepinyl, optionally substituted with alkyl, alkoxy, alkylthio, halo, triflouromethyl or hydroxyl. [0062] The term "dialkylamino" as employed herein alone, or as part of another group, refers to a substituted amino group having two alkyl substituents. For example, NRa Rb, wherein Ra and Rb are each an alkyl group, as defined herein. [0063] The term "carbonyl" as used herein, refers to a -C(O)- group. [0064] The term "aminocarbonyl", "alkylcarbonyl", "alkoxycarbonyl", "arylcarbonyl", "alkynylamήiocarbonyl", "alkylaminocarbonyl" and
"alkenylaminocarbonyl" as used herein, refer to an amino group, alkyl group, alkoxy group, aryl group, alkynylamino group, alkylamino group or an alkenylamino group,
as defined herein, appended to a parent molecular moiety through a carbonyl group, as defined herein.
[0065] The term "heteroarylamino", "arylamino", "alkylamino", "alkylcarbonylamino", "arylcarbonylamino", "alkylsulfonylamino", "alkylaminocarbonylamino" or "alkoxycarbonylamino" as used herein, refers to a heteroaryl, aryl, alkyl, alkylcarbonyl, arylcarbonyl, alkylsulfonyl, alkylaminocarbonyl or alkoxycarbonyl group as defined herein, appended to a parent molecular moiety through an amino group, as defined herein. [0066] The term "sulfonamido" refers to -S(O)2- NRa Rb, wherein Ra and Rb are as defined above for "substituted amino".
[0067] The term "alkylcarbonyloxy" as used herein, refers to an "alkyl-CO-O-" group, wherein alkyl is as defined above.
[0068] "Optional" or "optionally" means that the subsequently described event or circumstance may or may not occur, and that the description includes, without limitation, instances where said event or circumstance occurs and instances in which it does not. For example, optionally substituted alkyl means that alkyl may or may not be substituted by those groups enumerated in the definition of substituted alkyl. [0069] "Substituted," as used herein, whether express or implied and whether preceded by "optionally" or not, means that any one or more hydrogen on the designated atom (C, N, etc.) is replaced with a selection from the indicated group, provided that the designated atom's normal valency is not exceeded, and that the substitution results in a stable compound. For instance, when a CH2 is substituted by a keto substituent (=0), then 2 hydrogens on the atom are replaced. Combinations of substituents and/or variables are permissible only if such combinations result in stable compounds. Further, when more than one position in a given structure may be substituted with a substituent selected from a specified group, the substituents may be either the same or different at every position.
[0070] The term "prodrug esters" as employed herein includes esters and carbonates formed by reacting one or more hydroxyls of compounds of formula I with alkyl, alkoxy, or aryl substituted acylating agents employing procedures known to those skilled in the art to generate acetates, pivalates, methylcarbonates, benzoates and the like.
[0071] Various forms of prodrugs are well known in the art. A comprehensive description of prodrugs and prodrug derivatives are described in: a) The Practice of Medicinal Chemistry, Camille G. Wermuth et al., Ch 31, (Academic Press, 1996); b) Design of Prodrugs, edited by H. Bundgaard, (Elsevier, 1985); and c) A Textbook of Drug Design and Development, P. Krogsgaard-Larson and H. Bundgaard, eds., Ch. 5, pgs 113-191 (Harwood Academic Publishers, 1991). Said references are incorporated herein by reference.
[0072] The conditions, diseases and maladies collectively referred to as "diabetic complications" include retinopathy, neuropathy and nephropathy, erectile dysfunction, and other known complications of diabetes.
[0073] An administration of a therapeutic agent of the invention includes administration of a therapeutically effective amount of the agent of the invention. The term "therapeutically effective amount" as used herein refers to an amount of a therapeutic agent to treat or prevent a condition treatable by administration of a composition of the invention. That amount is the amount sufficient to exhibit a detectable therapeutic or preventative or ameliorative effect. The effect may include, for example, treatment or prevention of the conditions listed herein. The precise effective amount for a subject will depend upon the subject's size and health, the nature and extent of the condition being treated, recommendations of the treating physician, and the therapeutics or combination of therapeutics selected for administration. Thus, it is not useful to specify an exact effective amount in advance. [0074] The term "other type of therapeutic agents" as employed herein includes, but is not limited to one or more antidiabetic agents (other than DPP-IV inhibitors of formula I), one or more anti-obesity agents, one or more anti-hypertensive agents, one or more anti-platelet agents, one or more anti-atherosclerotic agents and/or one or more lipid-lowering agents (including anti-atherosclerosis agents).
UTILITIES AND COMBINATIONS
A. Utilities
[0075] The compounds of the present invention possess activity as inhibitors of the dipeptidyl peptidase IV which is found in a variety of tissues, such as the intestine, liver, lung and kidney of mammals. Via the inhibition of dipeptidyl peptidase IV in vivo, the compounds of the present invention possess the ability to potentiate endogenous levels of GLP-l(7-36) and attenuate formation of its antagonist GLP-1(9- 36). [0076] Accordingly, the compounds of the present invention can be administered to mammals, preferably humans, for the treatment of a variety of conditions and disorders, including, but not limited to, treating or delaying the progression or onset of diabetesφreferably Type II, impaired glucose tolerance, insulin resistance, and diabetic complications, such as nephropathy, retinopathy, neuropathy and cataracts), hyperglycemia, hyperinsulinemia, hypercholesterolemia, elevated blood levels of free fatty acids or glycerol, hyperlipidemia, hypertriglyceridemia, obesity, wound healing, tissue ischemia, atherosclerosis and hypertension. The compounds of the present invention may also be utilized to increase the blood levels of high density lipoprotein (HDL). [0077] In addition, the conditions, diseases, and maladies collectively referenced to as "Syndrome X" or Metabolic Syndrome as detailed in Johannsson, J. Clin.
Endocrinol. Metab., 82, 727-34 (1997), maybe treated employing the compounds of the invention.
B. Combinations [0078] The present invention includes within its scope pharmaceutical compositions comprising, as an active ingredient, a therapeutically effective amount of at least one of the compounds of formula I, alone or in combination with a pharmaceutical carrier or diluent. Optionally, compounds of the present invention can be used alone, in combination with other compounds of the invention, or in combination with one or more other therapeutic agent(s), e.g., an antidiabetic agent or other pharmaceutically active material.
[0079] Other "therapeutic agent(s)" suitable for combination with the compound of the present invention include, but are not limited to, known therapeutic agents useful in the treatment of the aforementioned disorders including: anti-diabetic agents; anti-hyperglycemic agents; hypolipidemic/lipid lowering agents; anti-obesity agents; anti-hypertensive agents, and appetite suppressants. Additional therapeutic agents suitable for combination with the compound of the present invention include agents for treating infertility, agents for treating polycystic ovary syndrome, agents for treating a growth disorder and/or frailty, an anti-arthritis agent, agents for preventing inhibiting allograft rejection in transplantation, agents for treating autoimmune disease, an anti-AIDS agent, agents for treating inflammatory bowel disease/syndrome, agents for treating anorexia nervosa and an anti-osteoporosis agent. [0080] Examples of suitable anti-diabetic agents for use in combination with the compound of the present invention include biguanides (e.g., metformin or phenformin), glucosidase inhibitors (e.g., acarbose or miglitol), insulins (including insulin secretagogues or insulin sensitizers), meglitinides (e.g., repaglinide), sulfonylureas (e.g., glimepiride, glyburide, gliclazide, chlorpropamide and glipizide), biguanide/glyburide combinations (e.g., Glucovance®), thiazolidinediones (e.g., troglitazone, rosiglitazone and pioglitazone), PPAR-alpha agonists, PPAR-gamma agonists, PPAR alpha/gamma dual agonists, glycogen phosphorylase inhibitors, inhibitors of fatty acid binding protein (aP2), glucagon-like peptide- 1 (GLP-I) or other agonists of the GLP-I receptor, STLT2 inhibitors and other dipeptidyl peptidase IV (DPP4) inhibitors.
[0081] Other suitable thiazolidinediones include Mitsubishi's MCC-555 (disclosed in U.S. Patent No. 5,594,016), Glaxo-Welcome's GL-262570, englitazone (CP-68722, Pfizer) or darglitazone (CP-86325, Pfizer, isaglitazone (MIT/J&J), JTT- 501 (JPNT/P&U), L-895645 (Merck), R-119702 (Sankyo/WL), NN-2344 (Dr. Reddy/NN), or YM-440 (Yamanouchi).
[0082] Examples of PPAR-alpha agonists, PPAR-gamma agonists and PPAR alpha/gamma dual agonists include muraglitizar, peliglitazar, AR-HO39242 (Astra/Zeneca), GW-409544 (Glaxo-Wellcome), GW-501516 (Glaxo-Wellcome), KRP297 (Kyorin Merck) as well as those disclosed by Murakami et al, "A Novel Insulin Sensitizer Acts As a Coligand for Peroxisome Proliferation - Activated
Receptor Alpha (PPAR alpha) and PPAR gamma. Effect on PPAR alpha Activation on Abnormal Lipid. Metabolism in Liver of Zucker Fatty Rats", Diabetes 47, 1841- 1847 (1998), WO 01/21602 and in U.S patent 6,653,314, the disclosure of which is incorporated herein by reference, employing dosages as set out therein, which compounds designated as preferred are preferred for use herein.
[0083] Suitable aP2 inhibitors include those disclosed in U.S. application Serial No. 09/391,053, filed September 7, 1999, and in U.S. application Serial No. 09/519,079, filed March 6, 2000, employing dosages as set out herein. [0084] Suitable other DPP4 inhibitors include saxagliptin, those disclosed in WO99/38501, WO99/46272, WO99/67279 (PROBIODRUG), WO99/67278
(PROBIODRUG), WO99/61431 (PROBIODRUG), NVP-DPP728A (l-[[[2-[(5- cyanopyridin-2-yl)amino] ethyl] amino] acetyl] -2-cyano-(S)-pyrrolidine) (Novartis) as disclosed by Hughes et al, Biochemistry, 38(36), 11597-11603, 1999, TSL-225 (tryptophyl-l,2,3,4-tetrahydroisoquinoline-3-carboxylic acid (disclosed by Yamada et al, Bioorg. & Med. Chem. Lett. 8 (1998) 1537-1540), 2-cyanopyrrolidides and 4- cyanopyrrolidides, as disclosed by Ashworth et al, Bioorg. & Med. Chem. Lett., Vol. 6, No. 22, pp 1163-1166 and 2745-2748 (1996), the compounds disclosed in U.S. application Serial No. 10/899641, WO 01/868603 and U.S. patent 6,395,767, employing dosages as set out in the above references. [0085] Other suitable meglitinides include nateglinide (Novartis) or KAD 1229 (PF/Kissei).
[0086] Examples of suitable anti-hyperglycemic agents for use in combination with the compound of the present invention include glucagon-like peptide- 1 (GLP-I,) such as GLP-l(l-36) amide, GLP-l(7-36) amide, GLP-l(7-37) (as disclosed in U.S. Patent No. 5,614,492), as well as exenatide (Amylin/Lilly), LY-315902 (Lilly), MK- 0431 (Merck), liraglutide (NovoNordisk), ZP-10 (Zealand Pharmaceuticals AJS), CJC-1131 (Conjuchem hie), and the compounds disclosed in WO 03/033671. [0087] Examples of suitable hypolipidemic/lipid lowering agents for use in combination with the compound of the present invention include one or more MTP inhibitors, HMG CoA reductase inhibitors, squalene synthetase inhibitors, fibric acid derivatives, ACAT inhibitors, lipoxygenase inhibitors, cholesterol absorption inhibitors, ileal Na /bile acid co-transporter inhibitors, up-regulators of LDL receptor
activity, bile acid sequestrants, cholesterol ester transfer protein (e.g., CETP inhibitors, such as CP-529414 (Pfizer) and JTT-705 (Akros Pharma)), PPAR agonists (as described above) and/or nicotinic acid and derivatives thereof. [0088] MTP inhibitors which may be employed as described above include those disclosed in U.S. Patent No. 5,595,872, U.S. Patent No. 5,739,135, U.S. Patent No. 5,712,279, U.S. Patent No. 5,760,246, U.S. Patent No. 5,827,875, U.S. Patent No. 5,885,983 and U.S. Patent No. 5,962,440.
[0089] The HMG CoA reductase inhibitors which may be employed in combination with one or more compound of formula I include mevastatin and related compounds, as disclosed in U.S. Patent No. 3,983,140, lovastatin (mevinolin) and related compounds, as disclosed in U.S. Patent No. 4,231,938, pravastatin and related compounds, such as disclosed in U.S. Patent No. 4,346,227, simvastatin and related compounds, as disclosed in U.S. Patent Nos. 4,448,784 and 4,450,171. Other HMG CoA reductase inhibitors which may be employed herein include, but are not limited to, fluvastatm, disclosed in U.S. Patent No. 5,354,772, cerivastatin, as disclosed in U.S. Patent Nos. 5,006,530 and 5,177,080, atorvastatin, as disclosed in U.S. Patent Nos. 4,681,893, 5,273,995, 5,385,929 and 5,686,104, atavastatin (Nissan/Sankyo's nisvastatin (NK-104)), as disclosed in U.S. Patent No. 5,011,930, visastatin (Shionogi-Astra/Zeneca (ZD-4522)), as disclosed in U.S. Patent No. 5,260,440, and related statin compounds disclosed in U.S. Patent No. 5,753,675, pyrazole analogs of mevalonolactone derivatives, as disclosed in U.S. Patent No. 4,613,610, indene analogs of mevalonolactone derivatives, as disclosed in PCT application WO 86/03488, 6-[2-(substituted-pyrrol-l-yl)-aucyl)pyran-2-ones and derivatives thereof, as disclosed in U.S. Patent No. 4,647,576, Searle's SC-45355 (a 3-substituted pentanedioic acid derivative) dichloroacetate, imidazole analogs of mevalonolactone, as disclosed in PCT application WO 86/07054, 3-carboxy-2-hydroxy-propane- phosphonic acid derivatives, as disclosed in French Patent No. 2,596,393, 2,3- disubstituted pyrrole, furan and thiophene derivatives, as disclosed in European Patent Application No. 0221025, naphthyl analogs of mevalonolactone, as disclosed in U.S. Patent No. 4,686,237, octahydronaphthalenes, such as disclosed in U.S. Patent No. 4,499,289, keto analogs of mevinolin (lovastatin), as disclosed in European Patent
Application No.0142146 A2, and quinoline and pyridine derivatives, as disclosed in U.S. Patent No. 5,506,219 and 5,691,322.
[0090] Preferred hypolipidemic agents are pravastatin, lovastatin, simvastatin, atorvastatin, fluvastatin, cerivastatin, atavastatin and ZD-4522. [0091] In addition, phosphinic acid compounds useful in inhibiting HMG CoA reductase, such as those disclosed in GB 2205837, are suitable for use in combination with the compound of the present invention.
[0092] The squalene synthetase inhibitors suitable for use herein include, but are not limited to, α-phosphono-sulfonates disclosed in U.S. Patent No. 5,712,396, those disclosed by Biller et al., J. Med. Chem., 1988, Vol. 31, No. 10, pp 1869-1871, including isoprenoid (ρhosphinyl-methyl)phosphonates, as well as other known squalene synthetase inhibitors, for example, as disclosed in U.S. Patent No. 4,871,721 and 4,924,024 and in Biller, S.A., Neuenschwander, K., Ponpipom, M.M., and Poulter, CD., Current Pharmaceutical Design, 2, 1-40 (1996). [0093] In addition, other squalene synthetase inhibitors suitable for use herein include the terpenoid pyrophosphates disclosed by P. Ortiz de Montellano et al, J. Med. Chem., 1977, 20, 243-249, the farnesyl diphosphate analog A and presqualene pyrophosphate (PSQ-PP) analogs as disclosed by Corey and Volante, J. Am. Chem. Soc, 1976, 98, 1291-1293, phosphinylphosphonates reported by McClard, R.W. et al, J.A.C.S., 1987, 109, 5544 and cyclopropanes reported by Capson, T.L., PhD dissertation, June, 1987, Dept. Med. Chem. U of Utah, Abstract, Table of Contents, pp 16, 17, 40-43, 48-51, Summary.
[0094] The fibric acid derivatives which may be employed in combination the compound of formula I include fenofibrate, gemfibrozil, clofibrate, bezafibrate, ciprofibrate, clinofibrate and the like, probucol, and related compounds, as disclosed in U.S. Patent No. 3,674,836, probucol and gemfibrozil being preferred, bile acid sequestrants, such as cholestyramine, colestipol and DEAE-Sephadex (Secholex®, Policexide®), as well as lipostabil (Rhone-Poulenc), Eisai E-5050 (an N-substituted ethanolamine derivative), imanixil (HOE-402), tetrahydrolipstatin (THL), istigmastanylphos-phorylcholine (SPC, Roche), aminocyclodextrin (Tanabe Seiyoku), Ajinomoto AJ-814 (azulene derivative), melinamide (Sumitomo), Sandoz 58-035, American Cyanamid CL-277,082 and CL-283,546 (disubstituted urea derivatives),
nicotinic acid, acipimox, acifran, neomycin, p-aminosalicylic acid, aspirin, poly(diallylmethylamine) derivatives, such as disclosed in U.S. Patent No. 4,759,923, quaternary amine poly(diallyldimethylammonium chloride) and ionenes, such as disclosed in U.S. Patent No. 4,027,009, and other known serum cholesterol lowering agents.
[0095] The ACAT inhibitor which may be employed in combination the compound of formula I include those disclosed in Drugs of the Future 24, 9-15 (1999), (Avasimibe); "The ACAT inhibitor, Cl-1011 is effective in the prevention and regression of aortic fatty streak area in hamsters", Nicolosi et al, Atherosclerosis (Shannon, Irel). (1998), 137(1), 77-85; "The pharmacological profile of FCE 27677: a novel ACAT inhibitor with potent hypolipidemic activity mediated by selective suppression of the hepatic secretion of ApoBlOO-containing lipoprotein", Ghiselli, Giancarlo, Cardiovasc. Drug Rev. (1998), 16(1), 16-30; "RP 73163: a bioavailable alkylsulfinyl-diphenylimidazole ACAT inhibitor", Smith, C, et al, Bioorg. Med. Chem. Lett. (1996), 6(1), 47-50; "ACAT inhibitors: physiologic mechanisms for hypolipidemic and anti-atherosclerotic activities in experimental animals", Krause et al, Editor(s): Ruffolo, Robert R., Jr.; Hollinger, Mannfred A., Inflammation: Mediators Pathways (1995), 173-98, Publisher: CRC, Boca Raton, FIa.; "ACAT inhibitors: potential anti-atherosclerotic agents", Sliskovic et al, Curr. Med. Chem. (1994), 1(3), 204-25; "Inhibitors of acyl-Co A: cholesterol O-acyl transferase (ACAT) as hypocholesterolemic agents. 6. The first water-soluble ACAT inhibitor with lipid- regulating activity. Inhibitors of acyl-Co A: cholesterol acyltransferase (ACAT). 7. Development of a series of substituted N-phenyl-N'-[(l- phenylcyclopentyl)methyl]ureas with enhanced hypocholesterolemic activity", Stout et al, Chemtracts: Org. Chem. (1995), 8(6), 359-62, or TS-962 (Taisho Pharmaceutical Co. Ltd).
[0096] The hypolipidemic agent may be an up-regulator of LD2 receptor activity, such as MD-700 (Taisho Pharmaceutical Co. Ltd) and LY295427 (Eli Lilly). [0097] Examples of suitable cholesterol absorption inhibitor for use in combination with the compound of the invention include SCH48461 (Schering- Plough), as well as those disclosed in Atherosclerosis 115, 45-63 (1995) and J. Med. Chem. 41, 973 (1998).
[0098] Examples of suitable ileal NaVbile acid co-transporter inhibitors for use in combination with the compound of the invention include compounds as disclosed in Drugs of the Future, 24, 425-430 (1999).
[0099] The lipoxygenase inhibitors which may be employed in combination the compound of formula I include 15-lipoxygenase (15-LO) inhibitors, such as benzimidazole derivatives, as disclosed in WO 97/12615, 15-LO inhibitors, as disclosed in WO 97/12613, isothiazolones, as disclosed in WO 96/38144, and 15-LO inhibitors, as disclosed by Sendobry et al "Attenuation of diet-induced atherosclerosis in rabbits with a highly selective 15-lipoxygenase inhibitor lacking significant antioxidant properties", Brit. J. Pharmacology (1997) 120, 1199- 1206, and Cornicelli et al, "15-Lipoxygenase and its Inhibition: A Novel Therapeutic Target for Vascular Disease", Current Pharmaceutical Design, 1999, 5, 11-20.
[00100] Examples of suitable anti-hypertensive agents for use in combination with the compound of the present invention include beta adrenergic blockers, calcium channel blockers (L-type and T-type; e.g. diltiazem, verapamil, nifedipine, amlodipine and mybefradil), diuretics (e.g., chlorothiazide, hydrochlorothiazide, flumethiazide, hydroflumethiazide, bendroflumethiazide, methylchlorothiazide, trichloromethiazide, polythiazide, benzthiazide, ethacrynic acid tricrynafen, chlorthalidone, furosemide, musolrmine, bumetanide, triamtrenene, amiloride, spironolactone), renin inhibitors, ACE inhibitors (e.g., captopril, zofenopril, fosinopril, enalapril, ceranopril, cilazopril, delapril, pentopril, quinapril, ramipril, lisinopril), AT-I receptor antagonists (e.g., losartan, irbesartan, valsartan), ET receptor antagonists (e.g., sitaxsentan, atrsentan and compounds disclosed in U.S. Patent Nos. 5,612,359 and 6,043,265), Dual ET/AII antagonist (e.g., compounds disclosed in WO 00/01389), neutral endopeptidase (NEP) inhibitors, vasopepsidase inhibitors (dual NEP-ACE inhibitors) (e.g., omapatrilat and gemopatrilat), and nitrates.
[00101] Examples of suitable anti-obesity agents for use in combination with the compound of the present invention include a beta 3 adrenergic agonist, a lipase inhibitor, a serotonin (and dopamine) reuptake inhibitor, a thyroid receptor beta drug, 5HT2C agonists, (such as Arena APD-356); MCHRl antagonists such as Synaptic
SNAP-7941 and Takeda T-226926, melanocortin receptor (MC4R) agonists, melanin- concentrating hormone receptor (MCHR) antagonists (such as Synaptic SNAP-7941
and Takeda T-226926), galanin receptor modulators, orexin antagonists, CCK agonists, NPYl or NPY5 antagonsist, NPY2 and NPY4 modulators, corticotropin releasing factor agonists, histamine receptor-3 (H3) modulators, 11-beta-HSD-l inhibitors, adinopectin receptor modulators, monoamine reuptake inhibitors or releasing agents, a ciliary neurotrophic factor (CNTF, such as AXOKINE® by Regeneron), BDNF (brain-derived neurotrophic factor), leptin and leptin receptor modulators, cannabinoid-1 receptor antagonists (such as SR-141716 (Sanofi) or SLV- 319 (Solvay)), and/or an anorectic agent. [00102] The beta 3 adrenergic agonists which may be optionally employed in combination with compound of the present invention include AJ9677
(Takeda/Dainippon), L750355 (Merck), or CP331648 (Pfizer,) or other known beta 3 agonists, as disclosed in U.S. Patent Nos. 5,541,204, 5,770,615, 5,491,134, 5,776,983 and 5,488,064. [00103] Examples of lipase inhibitors which may be optionally employed in combination with compound of the present invention include orlistat or ATL-962 (Alizyme).
[00104] The serotonin (and dopoamine) reuptake inhibitor (or serotonin receptor agonists) which maybe optionally employed in combination with a compound of the present invention maybe BVT-933 (Biovitrum), sibutramine, topiramate (Johnson & Johnson) or axokine (Regeneron).
[00105] Examples of thyroid receptor beta compounds which may be optionally employed in combination with the compound of the present invention include thyroid receptor ligands, such as those disclosed in WO97/21993 (U. CaI SF), WO99/00353 (KaroBio) and GB98/284425 (KaroBio). [00106] The monoamine reuptake inhibitors which may be optionally employed in combination with compound of the present invention include fenfluramine, dexfenfluramine, fluvoxamine, fluoxetine, paroxetine, sertraline, chlorphentermine, cloforex, clortermine, picilorex, sibutramine, dexamphetamine, phentermine, phenylpropanolamine or mazindol. [00107] The anorectic agent which may be optionally employed in combination with the compound of the present invention include topiramate (Johnson & Johnson), dexamphetamine, phentermine, phenylpropanolamine or mazindol.
[00108] The aforementioned patents and patent applications are incorporated herein by reference.
[00109] The above other therapeutic agents, when employed in combination with the compound of the present invention maybe used, for example, in those amounts indicated in the Physician's Desk Reference, as in the patents set out above or as otherwise determined by one of ordinary skill in the art.
[00110] Where the compound of the invention are utilized in combination with one or more other therapeutic agent(s), either concurrently or sequentially, the following combination ratios and dosage ranges are preferred. [00111] Where the other antidiabetic agent is a biguanide, the compound of formula I will be employed in a weight ratio to biguanide within the range from about
0.01:1 to about 100:1, preferably from about 0.1:1 to about 5:1.
[00112] The compound of formula I will be employed in a weight ratio to the glucosidase inhibitor within the range from about 0.01:1 to about 100:1, preferably from about 0.5 : 1 to about 50:1.
[00113] The compound of formula I will be employed in a weight ratio to the sulfonyl urea in the range from about 0.01:1 to about 100:1, preferably from about
0.2:1 to about 10:1.
[00114] The compound of formula I will be employed in a weight ratio to the thiazolidinedione in an amount within the range from about 0.01:1 to about 100:1, preferably from about 0.2:1 to about 10:1.
[00115] Where present, the thiazolidinedione anti-diabetic agent may be employed in amounts within the range from about 0.01 to about 2000 mg/day which may be administered in single or divided doses one to four times per day. [00116] Optionally, the sulfonyl urea and thiazolidinedione may be incorporated in a single tablet with the compound of formula I in amounts of less than about 150 mg.
[00117] Where present, metformin or salt thereof may be employed in amounts within the range from about 500 to about 2000 mg per day which may be administered in single or divided doses one to four times daily. [00118] Where present GLP-I peptides may be administered in oral buccal formulations, by nasal administration or parenterally as described in U.S. Patent Nos.
5,346,701 (TheraTech), 5,614,492 and 5,631,224 which are incorporated herein by reference.
[00119] The compound of formula I will be employed in a weight ratio to the meglitinide, PPAR-gamma agonist, PPAR-alpha/gamma dual agonist, aP2 inhibitor or other DPP4 inhibitor within the range from about 0.01 : 1 to about 100: 1 , preferably from about 0.2:1 to about 10:1.
[00120] The compound of formula I of the invention will be generally be employed in a weight ratio to the hypolipidemic agent (were present), within the range from about 500:1 to about 1:500, preferably from about 100:1 to about 1:100. [00121] For oral administration, a satisfactory result may be obtained employing the MTP inhibitor in an amount within the range of from about 0.01 mg/kg to about 500 mg and preferably from about 0.1 mg to about 100 mg, one to four times daily. [00122] A preferred oral dosage form, such as tablets or capsules, will contain the MTP inhibitor in an amount of from about 1 to about 500 mg, preferably from about 2 to about 400 mg, and more preferably from about 5 to about 250 mg, one to four times daily.
[00123] For oral administration, a satisfactory result may be obtained employing an HMG CoA reductase inhibitor in an amount within the range of from about 1 to 2000 mg, and preferably from about 4 to about 200 mg. [00124] A preferred oral dosage form, such as tablets or capsules, will contain the HMG CoA reductase inhibitor in an amount from about 0.1 to about 100 mg, preferably from about 5 to about 80 mg, and more preferably from about 10 to about 40 mg. [00125] The squalene synthetase inhibitor may be employed in dosages in an amount within the range of from about 10 mg to about 2000 mg and preferably from about 25 mg to about 200 mg.
[00126] A preferred oral dosage form, such as tablets or capsules will contain the squalene synthetase inhibitor in an amount of from about 10 to about 500 mg, preferably from about 25 to about 200 mg. [00127] The compound of the formula I can be administered for any of the uses described herein by any suitable means, for example, orally, such as in the form of tablets, capsules, granules or powders; sublingually; bucally; parenterally, such as by
subcutaneous, intravenous, intramuscular, or intrasternal injection or infusion techniques (e.g., as sterile injectable aqueous or non-aqueous solutions or suspensions); nasally, including administration to the nasal membranes, such as by inhalation spray; topically, such as in the form of a cream or ointment; or rectally such as in the form of suppositories; in dosage unit formulations containing non-toxic, pharmaceutically acceptable vehicles or diluents.
[00128] hi carrying out a preferred method of the invention for treating any of the diseases disclosed herein, such as diabetes and related diseases, a pharmaceutical composition will be employed containing one or more of the compound of formula I, with or without other antidiabetic agent(s) and/or antihyperlipidemic agent(s) and/or other type therapeutic agents in association with a pharmaceutical vehicle or diluent. The pharmaceutical composition can be formulated employing conventional solid or liquid vehicles or diluents and pharmaceutical additives of a type appropriate to the mode of desired administration, such as pharmaceutically acceptable carriers, excipients, binders and the like. The compound can be administered to mammalian species including humans, monkeys, dogs, etc. by an oral route, for example, in the form of tablets, capsules, beads, granules or powders, or they can be administered by a parenteral route in the form of injectable preparations, or they can be administered intranasally or in transdermal patches. Typical solid formulations will contain from about 10 to about 500 mg of a compound of formula I. The dose for adults is preferably between 10 and 2,000 mg per day, which can be administered in a single dose or in the form of individual doses from 1-4 times per day. [00129] A typical injectable preparation may be produced by aseptically placing 250 mg of compound of formula I into a vial, aseptically freeze-drying and sealing. For use, the contents of the vial are mixed with 2 mL of physiological saline, to produce an injectable preparation.
[00130] It will be understood that the specific dose level and frequency of dosage for any particular subject can be varied and will depend upon a variety of factors including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the species, age, body weight, general health, sex and diet of the subject, the mode and time of administration, rate of excretion, drug combination, and severity of the particular condition.
[00131] DPP-4 inhibitory activity of the compounds of the present invention may be determined by use of an in vitro assay system which measures the degree in inhibition of DPP-4-mediated cleavage of an appropriate substrate or pseudo- substrate. Inhibition constants (Ki values) for the DPP-4 inhibitors of the invention may be determined by the method described in the experimental section below.
Cloning, Expression and Purification of human DPP-4
[00132] To generate human DPP-4, PCR (Red-tag polymerase, Sigma) was performed on Human cDNA from placenta (Clontech) using two primers, ACGCCGACGATGAAGACA and AGGTAAAGAGAAACATTGTT, based on the nucleotide sequence of the human clone (accession number M74777). PCR products were cloned into the pcDN4/HisMax TOPO vector (Invitrogene). For stable transfection of CHO-DG44 cells, DPP4 was rePCRed using primers GGTACCAGCGCAGAGGCTT and CTCGAGCTAAGGTAAAGAGAAACATTG to generate Kpnl and Xhol sites. The Kpnl and Xhol sites were used to extract the N- terminal His tagged gene. The His tag, which could be cleaved and removed by Enterokinase, was included to allow purification using the TALON affinity column. The gene was then ligated into the Kpnl and Xhol sites of the pD16 vector for stable transfection. Stable cell lines were generated by transfecting the expression vector into Chinese hamster ovary (CHO-DG44) cells using electroporation. The CHO-DG44 cell line was grown in PFCHO media supplemented with HT (glycine, hypoxanthine and thymidine, Invitrogene), glutamine and Recombulin (ICN). Then 1x107 cells/ml were collected, transfected with 60 μg of DNA using electroporation at 300V, and then transferred to a T75 flask. On the third day following transfection, the HT supplement was removed and selection was initiated with methotrexate (MTX, 10 nM, ICN). After a further 10 days the cells were plated into individual wells of 96 well plates. Every 10 days the concentration of MTX was increased two to three fold, up to a maximum of 400 nM. Final stable cell line selection was based on yield and activity of the expressed protein. [00133] An attempt to purify recombinant DPP-4 using Talon resin was not efficient, resulting in small yields, with most of the DPP activity passing through the column. Therefore, protein was further purified using conventional anion exchange
(Sepharose Q)5 gel filtration (S-200) and high resolution MonoQ columns. The final protein yielded a single band on SDS-PAGE gels. Amino acid sequence analysis indicated two populations of DPP-4 in the sample. One portion of the protein had 27 amino acids truncated from the N-terminus, while the other was lacking the N- terminal 37 amino acids. This suggests that during isolation the entire transmembrane domain (including His tag) is removed by proteases present in the CHO cells. Total protein concentration was measured using the Bradford dye method and the amount of the active DPP-4 was determined by titrating the enzyme with a previously characterized inhibitor (Ki = 0.4 nM). No biphasic behavior was observed during inhibition or catalysis, suggesting that both protein populations are functionally identical.
DPP-4 inhibition assays.
[00134] Inhibition of human DPP-4 activity was measured under steady-state conditions by following the absorbance increase at 405 nm upon the cleavage of the pseudosubstrate, Gly-Pro-pNA. Assays were performed in 96-well plates using a Thermomax plate reader. Typically reactions contained 100 μl of ATE buffer (100 mM Aces, 52 mM Tris, 52 mM ethanolamine, pH 7.4), 0.45 nM enzyme, either 120 or 1000 μM of substrate (S<Km and S>Km, Km = 180 μM) and variable concentration of the inhibitor. To ensure steady-state conditions for slow-binding inhibitors, enzyme was preincubated with the compound for 40 minutes prior to substrate addition, to initiate the reaction. AU serial inhibitor dilutions were in DMSO and final solvent concentration did not exceed 1%.
[00135] Inhibitor potency was evaluated by fitting inhibition data to the binding isotherm:
Error! Objects cannot be created from editing field codes. (1)
where vi is the initial reaction velocity at different concentrations of inhibitor I; v is the control velocity in the absence of inhibitor, range is the difference between the uninhibited velocity and background; background is the rate of spontaneous substrate hydrolysis in the absent of enzyme, n is the Hill coefficient.
[00136] Calculated IC50 at each substrate concentration were converted to Eli assuming competitive inhibition according to:
Error! Objects cannot be created from editing field codes. (2)
[00137] AU inhibitors were competitive as judged by a very good agreement of Ki values obtained from the assays at high and low substrate concentrations. In cases where IC50 at the low substrate concentration was close to the enzyme concentration used in the assay, the data were fit to the Morrison equation1, to account for the depletion of the free inhibitor:
Error! Objects cannot be created from editing field codes. (3)
where vi and vo are the steady state velocities measured in the presence and absence of inhibitor, E enzyme concentration.
[00138] Each IC50 was further refined to Ki, to account for the substrate concentration in the assay using equation (2).
ABBREVIATIONS [00139] The following abbreviations are employed in the Examples and elsewhere herein:
Ph = phenyl
Bn = benzyl z-Bu = iso-butyl Me = methyl
Et = ethyl
Pr = propyl
Bu = butyl
TMS = trimethylsilyl FMOC = fluorenylmethoxycarbonyl
1 Morrison, JP, Walsh, CT. Advances in Enzymology. 61 (1988), 201-206.
Boc or BOC = fert-butoxycarbonyl
Cbz = carbobenzyloxy or carbobenzoxy or benzyloxycarbonyl
HOAc or AcOH = acetic acid
DMF = N,N-dimethylformamide DMSO = dimethylsulfoxide
EtOAc = ethyl acetate
THF = tetrahydrofuran
TFA = trifluoroacetic acid
Et2NH = diethylamine NMM = N-methyl moφholine rø-BuLi = n-butyllithium
Pd/C = palladium on carbon
PtO2 = platinum oxide
TEA = triethylamine min = minute(s) h or hr = hour(s)
L = liter mL = milliliter μL = microliter g = gram(s) mg = milligram(s) mol = mole(s) mmol = millimole(s) meq = milliequivalent rt = room temperature sat or sat'd = saturated aq. = aqueous
TLC = thin layer chromatography tR = retention time mp = melting point
HPLC = high performance liquid chromatography
LC/MS = high performance liquid chromatography/mass spectrometry
MS or Mass Spec = mass spectrometry
NMR = nuclear magnetic resonance
EDCI or EDAC = 3-ethyl-3'-(dimemylamino)propyl-carbodiimide hydrochloride (or
1 -[(3-(dimethyl)amino)propyl])-3-ethylcarbodiimide hydrochloride) HOBT or HOBT*H2O = 1 -hydroxybenzotriazole hydrate HOAT = l-hydroxy-7-azabenzotriazole PyBOP reagent = benzotriazol-1-yloxy-tripyrrolidino phosphonium hexafluorophosphate equiv = equivalent(s) UCT = United Chemical Technologies, Inc.; Bristol, PA.
EXAMPLES
[00140] The following examples are provided to describe the invention in further detail. These examples, which set forth the best mode presently contemplated for carrying out the invention, are intended to illustrate and not to limit the invention. [00141] In general, preferred compounds of the present invention, such as the compounds disclosed in the following examples, have been identified to inhibit the catalytic activity of dipeptidyl peptidase IV at concentrations equivalent to, or more potently than, 10 μM, preferably 5 μM, more preferably 3 μM, thereby corroborating the utility of the compounds of the present invention as effective inhibitors dipeptidyl peptidase IV. Potencies can be calculated and. expressed as either inhibition constants (Ki values) or as IC50 (inhibitory concentration 50%) values, and refer to activity measured employing the in vitro assay system described herein.
EXAMPLE 1


[00142] To a stirred solution of acetonitrile (2.0 mL, 38.2 mmol) in THF (50 mL) at -78 °C was added nBuLi (1.81 M in hexane, 16 mL, 28.7 mmol). The resulting slurry was kept at -78 °C for 15 min and 2,4-dichlorobenzoyl chloride (2.0 g, 9.55 mmol) was added dropwise to the acetonitrile anion. After 40 min, the reaction was quenched by addition of saturated NH4Cl (30 mL). THF was removed under reduced pressure and the suspension was filtered. The solid was washed with H2O (100 mL) and dried to give 3-(2,4-dichlorophenyl)-3-oxopropanenitrile (2.0 g, 98%, >95% purity) as a light yellow solid.
[00143] 1H NMR (400 MHz, CDCl3) 7.64 (d, J = 8.32 Hz, IH), 7.52 (d, J = 1.76
Hz, IH), 7.41 (dd, J = 1.76, 8.32 Hz, IH), 4.13 (s, 2H).
[00144] HPLC Phenomenex LUNA C-18 4.6 X 50 mm, 0 to 100% B over 4 minutes, 1 minutes hold time, A = 90% water, 10% methanol, 0.2% phosphoric acid,
B = 10% water, 90% methanol, 0.2% phosphoric acid, RT = 2.73 min, 95.5% homogeneity index.
[00145] LCMS: Anal. Calcd. for C9H5Cl2NO 212.97 found: 211.89 (M-H)".
Example 1, Step 2. 2-(2,4-Dichlorobenzoyl)-3-(dimethyIamino)acryIonitrile.

[00146] To a stirred solution of step 1 nitrile (1.74 g, 8.13 mmol) in toluene (50 mL) was added dimethylformamide dimethylacetal (1.35 mL, 10.16 mmol). The resulting brown solution was heated to 50 °C for 1 hr. The solvent was removed under reduced pressure and the residue was diluted with CH2Cl2 (50 mL). The organic layer was washed with saturated NaHCO3 solution (50 mL) and brine (50 mL), dried (MgSO4), filtered and concentrated under reduced pressure to give the crude product as a brown oil. Purification of the crude product by flash chromatography (silica gel, 40% EtOAc / hexane) afforded 2-(2,4-dichlorobenzoyl)-3-
(dimethylamino)acrylonitrile (1.5 g, 69%) as a light brown oil.
[00147] 1H NMR (400 MHz, CDCl3) 7.43 (s, IH), 7.30 (s, 2H), 3.48 (s, 3H), 3.32
(s, 3H).
Example 1, Step 3. 4-(2,4-Dichlorophenyl)-2-(3,5-dimethoxyphenyl)pyrimidine- 5-carbonitrile.

[00148] To a stirred solution of Step 2 acrylonitrile (1.5 g, 5.6 mmol) and 3,5- dimethoxybenzamidine hydrochloride (1.2 g, 5.6 mmol) in MeOH (30 mL) was added NaOMe (25% in MeOH, 2.56 mL, 11.2 mmol). The reaction was heated to reflux for 5 hr. Additional NaOMe (25% in MeOH, 2.56 mL, 11.2 mmol) was added and was kept for 16 hr. The reaction was cooled to ambient temperature and quenched by addition OfH2O (50 mL). The reaction was filtered and the solid was washed with
MeOH (40 mL) to give 4-(2,4-dichlorophenyl)-2-(3,5-dimethoxyphenyl)pyrimidine-5- carbonitrile (960 mg, 38.4%) as a white solid.
[00149] 1H NMR (400 MHz, CDCl3) 9.10 (s, IH), 7.71 (d, J = 2.2 Hz, 2H), 7.62 (d, J = 1.75 Hz, IH), 7.51 (d, J = 7.78 Hz, IH), 7.47 (dd, J = 1.75, 7.76 Hz, IH), 6.68 (t, J = 2.1 Hz, IH), 3.88 (s, 6H).
[00150] 13HNMR (400 MHz, CDCl3) 166.44, 165.67, 161.25, 160.79, 137.63, 137.59, 133.62, 133.31, 131.66, 130.49, 127.71, 114.93, 107.07, 106.89, 105.64, 55.65.
[00151] HPLC Phenomenex LUNA C-18 4.6 x 50 mm, 0 to 100% B over 4 minutes, 1 minutes hold time, A = 90% water, 10% methanol, 0.2% phosphoric acid, B = 10% water, 90% methanol, 0.2% phosphoric acid, RT = 4.14 min. [00152] LCMS: Anal. Calcd. for Ci9Hi3Cl2N3O2 385.04 found: 386.15 (M+H)+.
Example 1, Step 4. (4-(2,4-Dichlorophenyl)-2-(3,5-dimethoxyphenyl)pyrimidin- 5-yl)methanamine.

[00153] To a stirred solution of Step 3 carbonitrile (35 mg, 0.09 mmol) in THF (2 mL) and H2O (1 mL) was added CoCl26H2O (20 mg, 0.09 mmol) followed by NaBH4 (17 mg, 0.45 mmol) in H2O (0.5 mL). A black precipitate formed immediately with gas evolution. After 30 min, the reaction was filtered and diluted with CH2Cl2 (6 mL). The organic layer was washed with saturated NaHCO3 solution (5 mL) and brine (5 mL), dried (MgSO4), filtered and concentrated under reduced pressure to give the crude product as a yellow oil. Purification of the crude product by reverse-phase preparative HPLC provided (4-(2,4-dichlorophenyl)-2-(3,5- dimethoxyphenyl)pyrimidin-5-yl)methanamine, TFA salt (20 mg, 44%) as a light yellow solid.
[00154] 1H NMR (400 MHz, CD3OD) 9.06 (s, IH), 7.41 (s, IH), 7.64 (d, J = 2.64 Hz3 2H), 7.61 (dd, J = 1.76, 7.75 Hz, IH), 7.55 (d, J = 7.76 Hz, IH), 6.66 (d, J = 1.80 Hz, IH), 4.02 (br s, 2H), 3.84 (s, 6H).
[00155] HPLC Phenomenex LUNA C-18 4.6 x 50 mm, 0 to 100% B over 4 minutes, 1 minutes hold time, A = 90% water, 10% methanol, 0.1% trifluoroacetic acid, B = 10% water, 90% methanol, 0.1% trifluoroacetic acid, RT = 3.19 min, 97% homogeneity index.
[00156] HRMS: Anal. Calcd. for C19H17Cl2N3O2 390.0776 found: 390.0778 (M+H)+.
EXAMPLE 2

[00157] (4-(2,4-Dichlorophenyl)-2-phenylpyrimidin-5-yl)methanamine, TFA salt was prepared by the methods described in Example 1, Step 3 and Step 4 using Example 1, Step 2 acrylonitrile and benzamidine hydrochloride.
[00158] 1H NMR (400 MHz, CD3OD) 9.07 (s, IH), 8.46 (dd, J = 1.76, 8.36 Hz, 2H), 7.76 (d, J = 1.44 Hz, IH), 7.47-7.63 (m, 5H), 4.16 (br s, 2H). [00159] HPLC Phenomenex LUNA C-18 4.6 x 50 mm, 0 to 100% B over 4 minutes, 1 minutes hold time, A = 90% water, 10% methanol, 0.2% phosphoric acid, B = 10% water, 90% methanol, 0.2% phosphoric acid, RT = 2.82 min, 98% homogeneity index. [00160] LCMS: Anal. Calcd. for C17H13Cl2N3 329.05 found: 330.14 (M+H)+.

[00161] (4-(2,4-Dimethylphenyl)-2-methylρyrimidin-5-yl)methanamine, TFA salt was prepared by the methods described in Example 1, using 2,4- dimethylbenzaldehyde for Step 1 and acetamidine for Step 3.
[00162] 1H NMR (400 MHz, CD3OD) 8.79 (s, IH), 7.13 (s, IH), 7.06 (d, J = 8.8 Hz, IH), 7.03 (d, J = 8.8 Hz, IH), 3.91 (s, 2H), 2.63 (s, 3H), 2.27 (s, 3H), 2.00 (s, 3H). [00163] HPLC Phenomenex LUNA C-18 4.6 X 50 mm, 0 to 100% B over 4 minutes, 1 minutes hold time, A = 90% water, 10% methanol, 0.2% phosphoric acid, B = 10% water, 90% methanol, 0.2% phosphoric acid, RT = 2.64 min, 95% homogeneity index. [00164] HRMS: Anal. Calcd. for C14H17N3 228.1501 found: 228.1491 (M+H)+.
EXAMPLE 4

[00165] (4-(4-Chlorophenyl)-2-phenylpyrimidin-5-yl)methanamine, TFA salt was prepared by the methods described in Example 1 using 4-chlorobenzoyl chloride for
Step 1 and benzamidine hydrochloride for Step 3.
[00166] 1H NMR (400 MHz, CD3OD) 8.93 (s, IH), 8.40 (dd, J = 1.76, 7.88 Hz, 2H), 7.63 (d, J = 8.32 Hz, 2H), 7.52 (d, J = 8.80 Hz, 2H), 7.42 (m, IH), 7.41 (d, J =
7.04 Hz, 2H), 4.26 (s, 2H).
[00167] HPLC Phenomenex LUNA C-18 4.6 x 75 mm, 0 to 100% B over 8 minutes, 2 minutes hold time, A = 90% water, 10% methanol, 0.2% phosphoric acid,
B = 10% water, 90% methanol, 0.2% phosphoric acid, RT = 5.05 min, 96% homogeneity index.
[00168] HRMS: Anal. Calcd. for C17H15ClN3 296.0955 found: 296.0947 (M+H)+.
EXAMPLE 5

[00169] (4-(2-Chlorophenyl)-2-phenylpyrimidin-5-yl)methanamine, TFA salt was prepared by the methods described in Example 1 using 2-chlorobenzoyl chloride for Step 1 and benκamidine hydrochloride for Step 3. [00170] 1H NMR (400 MHz, CD3OD) 9.08 (s, IH), 8.47 (dd, J = 1.32, 7.48 Hz, 2H), 7.47-7.67 (m, 7H), 4.01 (br s, 2H).
[00171] HPLC Phenomenex LUNA C-18 4.6 X 75 mm, 0 to 100% B over 8 minutes, 2 minutes hold time, A = 90% water, 10% methanol, 0.2% phosphoric acid, B = 10% water, 90% methanol, 0.2% phosphoric acid, RT = 4.92 min, 95% homogeneity index.
[00172] HRMS: Anal. Calcd. for C17H15ClN3 296.0955 found: 296.0945 (M+H)+.
EXAMPLE 6

[00175] HPLC Phenomenex LIMA C-18 4.6 X 75 mm, 0 to 100% B over 8 minutes, 2 minutes hold time, A = 90% water, 10% methanol, 0.2% phosphoric acid, B = 10% water, 90% methanol, 0.2% phosphoric acid, RT = 5.49 min, 98% homogeneity index.
[00176] HRMS: Anal. Calcd. for Ci8Hi7ClN3O 326.1060 found: 326.1048 (M+H)+.
EXAMPLE 7

[00177] (4-(2,4-Dimethylphenyl)-2-phenylpyrimidin-5-yl)methanamine, TFA salt was prepared by the methods described in Example 1 using 2,4-dimethylbenzoyl chloride for Step 1 and benzamidine hydrochloride for Step 3.
[00178] 1R NMR (400 MHz, CD3OD) 9.14 (s, IH), 8.43 (dd, J = 1.75, 7.60 Hz, 2H), 7.53 (m, 3H), 7.26 (m, 3H), 4.13 (s, 2H), 2.42 (s, 3H), 2.14 (s, 3H). [00179] HPLC Phenomenex LUNA C-18 4.6 X 75 mm, 0 to 100% B over 8 minutes, 2 minutes hold time, A = 90% water, 10% methanol, 0.2% phosphoric acid, B = 10% water, 90% methanol, 0.2% phosphoric acid, RT = 5.39 min, 99% homogeneity index. [00180] HRMS: Anal. Calcd. for Ci9H20N3 290.1657 found: 290.1643 (M+H)+.
EXAMPLE 8

Example 8, Step 1. Methyl 2-(2,4-dichlorobenzylidene)-3-oxobutanoate.

[00181] To a stirred solution of methyl acetoacetate (345 mg, 2.97 mmol) and 2,4- dichlorobenzaldehyde (500 mg, 2.86 mmol) in 2-propanol (5 mL) was added AcOH (7 mg, 0.11 mmol) and dimethylamine (0.06 mL, 2M in THF, 0.11 mmol). The reaction was heated to 40 °C for 4 hrs followed by cooling to ambient temperature for 15 hrs. The reaction was concentrated and purified by flash chromatography (silica gel, 30% EtOAc / hexane) to give methyl 2-(2,4-dichlorobenzylidene)-3-oxobutanoate as a mixture of two isomers (colorless oil, 610 mg, 78%).
[00182] 1H NMR (400 MHz, CDCl3) Fast eluting isomer: 7.86 (s, IH), 7.45 (d, J = 2.2 Hz, IH), 7.25 (d, J = 6.8 Hz, IH), 7.21 (dd, J = 2.2, 7.0 Hz, IH), 3.85 (s, 3H), 2.24 (s, 3H). Slow eluting isomer: 7.78 (s, IH), 7.45 (d, J = 2.2 Hz, IH), 7.34 (d, J = 7.0 Hz, IH), 7.25 (dd, J = 2.2, 7.0 Hz, IH), 3.73 (s, 3H), 2.44 (s, 3H).
Example 8, Step 2. Methyl 4-(2,4-dichlorophenyl)-6-methyl-2-phenyl-l,4- dihydropyrimidine-5-carboxylate.

[00183] To a stirred solution of methyl 2-(2,4-dichlorobenzylidene) -3- oxobutanoate (480 mg, 1.8 mmol) and benzamidine HCl salt (275 mg, 1.8 mmol) in DMF (6 mL) was added NaOAc (144 mg, 1.8 mmol). The reaction was heated to 60°C for 3 days and was quenched by IN HCl (10 mL). The reaction was diluted with EtOAc (10 mL) and the organic layer was washed with IN HCl, saturated NH4Cl solution (10 mL) and brine (10 mL), dried (MgSO4), filtered and concentrated under reduced pressure to give the crude product as a yellow oil (600 mg). The crude reaction product was moved onto next step without further purification. [00184] LCMS: Anal. Calcd. for C19H16Cl2N2O2 374.06 found: 375.00 (M+H)+.
Example 8, Step 3. Methyl 4-(2,4-dichlorophenyl)-6-methyl-2-phenylpyrimidine- 5-carboxylate.

[00185] To a stirred solution of methyl 4-(2,4-dichlorophenyl)-6-methyl-2-phenyl- l,4-dihydropyrimidine-5-carboxylate (600 mg, 1.76 mmol) in PhCH3 (6 mL) was added MnO2 (227 mg, 2.64 mmol) and the reaction was heated to 95 0C for 14 hrs. The reaction was filtered through a pad of celite, concentrated under reduced pressure, and purified by flash chromatography (silica gel, 30% EtOAc / hexane) to give methyl 4-(2,4-dichlorophenyl)-6-methyl-2-phenylpvrimidine-5-carboxylate (150 mg, 23% for
2 steps) and 4-(2,4-dichlorophenyl)-6-methyl-2-phenylpyrimidine-5-carboxylic acid (100 mg, 15% for 2 steps).
[00186] 1R NMR (400 MHz, CDCl3) 8.44 (dd, J = 1.3, 7.6 Hz, 2H), 7.38-7.47 (m, 4H), 7.30 (d, J = 1.3 Hz, 2H), 3.62 (s, 3H), 2.71 (s, 3H). [00187] For acid 1H NMR (400 MHz, CDC13/CD3OD) 8.04 (dd, J = 1.3, 7.9 Hz, 2H), 7.41-7.57 (m, 3H), 7.37 (s, IH), 7.26 (d, J = 7.9 Hz, 2H), 2.48 (s, 3H). [00188] HPLC Phenomenex LUNA C-18 4.6 X 50 mm, 0 to 100% B over 4 minutes, 1 minutes hold time, A = 90% water, 10% methanol, 0.2% phosphoric acid, B = 10% water, 90% methanol, 0.2% phosphoric acid, RT = 3.63 min, 90% homogeneity index.
Example 8, Step 4. (4-(2,4-Dichlorophenyl)-6-methyl-2-phenylpyrimidin-5- yl)methanol.

(MgSO4), filtered and concentrated under reduced pressure to give the crude product as a white solid (75 mg). The crude reaction product was moved onto next step without further purification. [00190] 1H NMR (400 MHz, CDCl3) 8.36 (dd, J = 1.2, 7.5 Hz, 2H), 7.47 (s, IH), 7.39 (m, 3H), 7.30 (dd, J = 2.6, 8.0 Hz, 2H), 4.52 (hr s, IH), 4.42 (br s, IH), 2.72 (s, 3H).
Example 8, Step 5. 4-(2,4-Dichlorophenyl)-6-methyl-2-phenyIpyrimidine-5- carb aldehyde.

[00191] To a stirred solution of (4-(2,4-dichlorophenyl)-6-methyl-2- phenylpyrimidin-5-yl)methanol (75 mg, 0.2 mmol) in CH2Cl2 (6 mL) was added
Dess-Martin periodinane (102 mg, 0.24 mmol). The reaction was kept for 2 hrs and was diluted with EtOAc (10 mL).The organic layer was washed with saturated
NaHCO3 solution (10 mL), and brine (10 mL), dried (MgSO4), filtered and concentrated under reduced pressure to give the crude product as a white solid (75 mg). The crude product was purified by flash chromatography (silica gel, 30% EtOAc
/ hexane) to give 4-(2,4-dichlorophenyl)-6-methyl-2-phenylpyrimidine-5- carbaldehyde (65 mg, 91% for 2 steps) as a white solid.
[00192] 1H NMR (400 MHz, CDCl3) 9.90 (s, IH), 8.47 (dd, J = 1.8, 8.4 Hz, 2H),
7.37-7.49 (m, 6H), 2.88 (s, 3H).
Example 8, Step 5. 4-(2,4-Dichlorophenyl)-6-methyl-2-phenylpyrimidine-5- carbaldehyde oxime.

[00193] To a stirred solution of 4-(2,4-dichlorophenyl)~6-methyl-2- phenylpyrimidine-5-carbaldehyde (24 mg, 0.07 mmol) in EtOH (4 mL) was added NH2OHHCl (10 mg, 0.14 mmol) and Et3N (50 μL). The reaction was heated to 70 0C for 3 hrs and was concentrated under reduced pressure. The residue was dissolved in
EtOAc (10 mL) and the organic layer was washed by saturated NH4Cl solution (10 HiL) and brine, dried (MgSO4), filtered and concentrated under reduced pressure to give the crude product as a white solid (24 mg). The crude reaction product was moved onto next step without further purification. [00194] HPLC Phenomenex LUNA C-18 4.6 X 50 mm, 0 to 100% B over 4 minutes, 1 minutes hold time, A = 90% water, 10% methanol, 0.1% trifluoroacetic acid, B = 10% water, 90% methanol, 0.1% trifluoroacetic acid, RT = 4.09 min, 95% homogeneity index.
[00195] LCMS: Anal. Calcd. for C18H13Cl2N3O 357.04 found: 357.97 (M+H)+.
Example 8, Step 5. (4-(2,4-Dichlorophenyl)-6-methyl-2-phenylpyrimidin-5- yl)methanamine.

[00196] To a stirred solution 4-(2,4-dichlorophenyl)-6-methyl-2-phenylpyrimidiiie- 5-carbaldehyde oxime (24 mg, 0.07 mmol) in EtOH (4 mL) was added Zn (14 mg, 0.21 mmol), NH4OAc (16 mg, 0.21 mmol) and NH4OH (30 μL, 28% in H2O, 0.21 mmol). The reaction was heated to 78 °C for 15 hrs. Additional 3 eqs of Zn, NH4OAc, NH4OH were added and after 3 hrs, the reaction was concentrated under reduced pressure and diluted with EtOAc (10 mL). The organic layer was washed by saturated NaHCO3 solution (10 mL) and brine, dried (MgSO4), filtered and concentrated under reduced pressure to give the crude product. The crude product was purified by reverse-phase preparative HPLC to provide (4-(2,4-dichlorophenyl)-6-methyl-2- phenylpyrimidin-5-yl)methanamine, TFA salt (20 mg, 63% for 2 steps) as a light yellow solid. [00197] 1H NMR (400 MHz, CD3OD) 8.44 (dd, J = 1.6, 8.4 Hz, 2H), 7.72 (d, J = 1.3 Hz, IH), 7.46-7.62 (m, 5H), 4.27 (d, J = 14.5 Hz, IH), 4.00 (d, J = 14.5 Hz, IH), 2.82 (s, 3H).
[00198] HPLC Phenomenex LUNA C-18 4.6 X 50 mm, 0 to 100% B over 4 minutes, 1 minutes hold time, A = 90% water, 10% methanol, 0.2% phosphoric acid, B = 10% water, 90% methanol, 0.2% phosphoric acid, RT = 2.83 min, 99% homogeneity index.
[00199] LCMS: Anal. Calcd. for Ci8H15Cl2N3 343.06 found: 343.99 (M+H)+. [00200] HRMS: Anal. Calcd. for C18H16Cl2N3 344.0721 found: 344.0728 (M+H)+.
EXAMPLE 9

Example 9, Step 1. (4-Chloro-2-phenylpyrimidin-5-yl)methanol.

[00201] To a stirred solution of methyl 4-chloro-2-phenylpyrimidine -5-carboxylate (300 mg, 1.14 mmol) in CH2Cl2 (15 mL) at -78 0C was added DIBAL (1.5 M in PhCH3, 1.5 mL, 2.28 mmol). The reaction was kept for 2 hrs and was quenched by saturated aqueous potassium sodium tartrate solution (10 mL). The reaction was diluted with EtOAc (15 mL) and the organic layer was washed with IN NaOH (10 mL), saturated Na2CO3 solution (10 mL) and brine (10 mL), dried (MgSO4), filtered and concentrated under reduced pressure to give the crude product as a yellow solid (242 mg). The crude reaction product was moved onto next step without further purification.
[00202] 1H NMR (400 MHz, CD3OD) 8.73 (s, IH), 8.30 (d, J = 1.3, 6.2 Hz, 2H), 7.38-7.41 (m, 3H), 4.64 (s, 2H).
[00203] HPLC Phenomenex LUNA C-18 4.6 X 75 mm, 0 to 100% B over 8 minutes, 2 minutes hold time, A = 90% water, 10% methanol, 0.2% phosphoric acid, B = 10% water, 90% methanol, 0.2% phosphoric acid, RT = 6.17 min, 95% homogeneity index. [00204] LCMS: Anal. Calcd. for C11H9ClN2O 220.04 found: 221.04 (M+H)+.
Example 9, Step 2. 4-Chloro-2-phenylpyrimidine-5-carb aldehyde.
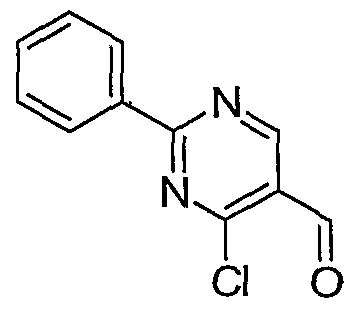
[00205] To a stirred (4-chloro-2-phenylpyrimidin-5-yl)methanol (242 mg, 1.10 mmol) in CH2Cl2 (11 mL) was added Dess-Martin periodinane (553 mg, 1.43 mmol).
The reaction was kept for 2 hrs and was diluted with EtOAc (10 mL).The organic layer was washed with saturated NaHCO3 solution (10 mL), and brine (10 mL), dried
(MgSO4), filtered and concentrated under reduced pressure to give the crude product as a white solid (250 mg). The crude product was purified by flash chromatography (silica gel, 20% EtOAc / hexane) to give 4-chloro-2-phenylpyrimidine-5-carbaldehyde
(205 mg, 82% for 2 steps) as a white solid.
[00206] 1H NMR (400 MHz, CDCl3) 10.43 (s, IH), 9.12 (s, IH), 8.51 (dd, J = 1.4,
6.2 Hz, 2H), 7.48-7.59 (m, 3H).
[00207] HPLC Phenomenex LUNA C- 18 4.6 x 75 mm, 0 to 100% B over 8 minutes, 2 minutes hold time, A = 90% water, 10% methanol, 0.2% phosphoric acid,
B = 10% water, 90% methanol, 0.2% phosphoric acid, RT = 6.60 min, 99% homogeneity index.
[00208] LCMS: Anal. Calcd. for C11H7ClN2O 218.02 found: 219.06 (M+ H)+.
Example 9, Step 3. 4-(4-ChIoro-2-methylphenyl)-2-phenylpyrimidine-5- carbaldehyde.

[00209] To a stirred solution of 4-chloro-2-phenylpyrimidine-5-carbaldehyde (50 mg, 0.23 mmol) and 4-chloro-o-toluene boronic acid (48 mg, 0.29 mmol) in dioxane (1 mL) and H2O (0.5 niL) was added Pd(PPh3)4 (26.5 mg, 0.02 mmol), K2CO3 (126.5 mg, 0.92 mmol). The reaction was heated to 85 °C for 6 hrs. After concentration under reduced pressure, the residue was diluted with EtOAc (10 mL) and the organic layer was washed with saturated NH4Cl solution (10 mL), and brine (10 mL), dried (MgSO4), filtered and concentrated under reduced pressure to give the crude product (70 mg). The crude product was purified by flash chromatography (silica gel, 20% EtOAc / hexane) to give 4-(4-chloro-2-methylphenyl)-2-phenylpyrimidine-5- carbaldehyde (63 mg, 75%) as a white solid. [00210] 1H NMR (400 MHz, CDCl3) 9.82 (s, IH), 9.26 (s, IH), 8.48 (dd, J = 1.8, 8.4 Hz, 2H), 7.41-7.52 (m, 3H), 7.12-7.37 (m, 3H), 2.26 (s, 3H).
[00211] HPLC Phenomenex LUNA C-18 4.6 x 75 mm, 0 to 100% B over 8 minutes, 2 minutes hold time, A = 90% water, 10% methanol, 0.2% phosphoric acid, B = 10% water, 90% methanol, 0.2% phosphoric acid, RT = 7.84 min, 90% homogeneity index. [00212] LCMS: Anal. Calcd. for C18H13ClN2O 308.07 found: 309.06 (M+H)+.
Example 9, Step 4. 4-(4-Chloro-2-methylphenyl)-2-phenyIpyrimidine-5- carb aldehyde oxime.
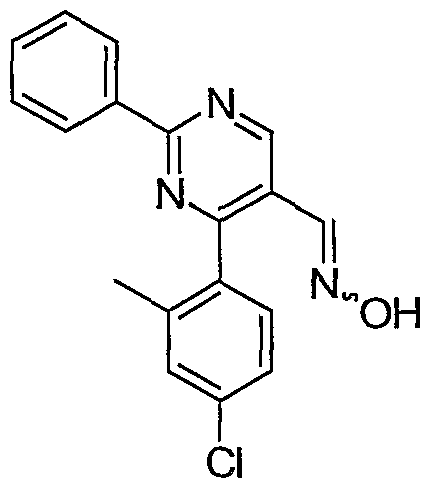
[00213] To a stirred solution of 4-(4-ctøoiO-2-methylphenyl)-2-phenylpyrimidine- 5-carbaldehyde (63 mg, 0.20 mmol) in EtOH (4 mL) was added NH2OHHCl (28.3 mg, 0.41 mmol) and pyridine (430 μL). The reaction was heated to 70 °C for 3 hrs and was concentrated under reduced pressure. The residue was dissolved in EtOAc (10 mL) and the organic layer was washed by saturated NH4Cl solution (10 mL) and brine, dried (MgSO4), filtered and concentrated under reduced pressure to give the crude product as a light yellow foam (69 mg). The crude reaction product was moved onto next step without further purification.
[00214] HPLC Phenomenex LUNA C-18 4.6 x 75 mm, 0 to 100% B over 8 minutes, 2 minutes hold time, A = 90% water, 10% methanol, 0.2% phosphoric acid, B = 10% water, 90% methanol, 0.2% phosphoric acid, RT = 8.26 min, 95% homogeneity index.
[00215] LCMS: Anal. Calcd. for C18H14ClN3O 323.08 found: 324.06 (M+H)+.
Example 9, Step 5. (4-(4-Chloro-2-methylphenyl)-2-phenylpyrimidin-5- yl)methanamine.

[00216] To a stirred solution (4-(4-chloro-2-methylphenyl)-2-phenylpyrimidin-5- yl)methanamine (69 mg, 0.21 mmol) in EtOH (5 mL) was added Zn (28 mg, 0.43
mmol), NH4OAc (33 mg, 0.43 mmol) and NH4OH (58 μL, 28% in H2O, 0.43 mmol). The reaction was heated to 78 °C for 10 hrs. Additional 2 eqs of Zn, NH4OAc, NH4OH were added and after 3 hrs, the reaction was concentrated under reduced pressure and diluted with EtOAc (10 mL). The organic layer was washed by saturated NaHCO3 solution solution (10 mL) and brine, dried (MgSO4), filtered and concentrated under reduced pressure to give the crude product. The crude product was purified by reverse-phase preparative HPLC to provide (4-(4-chloro-2-methylphenyl)- 2-phenylpyrimidin-5-yl)methanamine, TFA salt (27 mg, 40% for 2 steps) as a white solid. [00217] 1H NMR (400 MHz, CD3OD) 9.04 (s, IH), 8.44 (dd, J = 1.3, 8.3 Hz, 2H), 7.44.7.54 (m5 4H), 7.38 (dd, J = 1.8, 6.2 Hz, IH), 7.9 (d, J = 7.9 Hz, IH), 4.07 (s, 2H), 2.21 (s, 3H).
[00218] HPLC Phenomenex LUNA C-18 4.6 X 75 mm, 0 to 100% B over 8 minutes, 2 minutes hold time, A = 90% water, 10% methanol, 0.2% phosphoric acid, B = 10% water, 90% methanol, 0.2% phosphoric acid, RT = 5.52 min, 99% homogeneity index.
[00219] LCMS: Anal. Calcd. for C18H16ClN3 309.10 found: 310.07 (M+H)+. [00220] HRMS: Anal. Calcd. for C18H17ClN3 310.1111 found: 310.1101 (M+H)+.
EXAMPLE 10

Example 10, Step 1. Diethyl 2-(2,4-dichlorobenzylidene)malonate.

Example 10, Step 2. Ethyl 4-(2,4-dichlorophenyl)-6-oxo-2-phenyl-l,6- dihydropyrimidine-5-carboxylate.


[00225] To a stirred solution of ethyl 4-(2,4-dichlorophenyl)-6-oxo-2-phenyl- 1 ,6- dihydropyrimidine-5-carboxylate (372 mg, 0.91 mmol) in doixane (6 mL) was added POCl3 (0.4 mL, 4.55 mmol) and N,N-dimethylaniline (11.5 μL, 0.09 mmol). After 4 hrs at 70 0C5 the reaction was concentrated under reduced pressure and diluted with EtOAc (100 mL). The organic layer was extracted with saturated NaHCO3 solution (2 x 60 mL), brine (50 mL), dried (MgSO4), filtered and concentrated under reduced pressure. The crude reaction product was purified by flash chromatography (silica gel, 30% EtOAc / hexane) to give ethyl 4-chloro-6-(2,4-dichlorophenyl)-2- phenylpyrimidine-5-carboxylate (360 mg, 97% for 3 steps) as a clear oil. [00226] 1H NMR (400 MHz, CDCl3) 8.48 (d, J = 6.6 Hz, 2H), 7.47-7.58 (m, 6H)5 4.21 (q, J = 7.0 Hz, 2H), 1.12 (t, J = 7.0 Hz, 3H).
Example 10, Step 4. Ethyl 4-(2,4-dichlorophenyl)-2,6-diphenylpyrimidine-5- carboxylate.

[00227] A solution of ethyl 4-chloro-6-(2,4-dichlorophenyl)-2-phenylpyrimidine-5- carboxylate (30 mg, 0.07 mmol) and phenyl boronic acid (14 mg, 0.11 mmol) in toluene (3 mL) was degassed with argon for 15 minutes. To this solution was added Pd(PPh3)4 (9 mg, 0.007 mmol), Na2CO3 (23 mg, 0.22 mmol) and the reaction was
heated to reflux for 3 days. The reaction was diluted with EtOAc (20 mL) and extracted with saturated NH4Cl solution (2 χ 20 mL), brine (20 mL). The organic layer was dried (MgSO4), filtered and concentrated under reduced pressure. The crude reaction product was purified by flash chromatography (silica gel, 20% EtOAc / hexane) to give ethyl 4-(2,4-dichlorophenyl)-2,6-diphenylpyrimidine-5-carboxylate (14 mg, 42%).
[00228] 1H NMR (400 MHz, CDCl3) 8.58 (dd, J = 1.3, 8.4 Hz, 2H), 7.80 (d, J = 8.0 Hz, 2H)3 7.46-7.57 (m, 7H), 7.36-7.43 (m, 2H), 4.00 (q, J = 7.0 Hz, 2H), 0.91 (t, J = 7.0 Hz, 3H).
Example 10, Steps 5-8. (4-(2,4-Dichlorophenyl)-2,6-diphenylpyrimidin-5- yl)methanamine.

[00230] To a stirred solution of alcohol (10 mg, 0.025 mmol) in CH2Cl2 (2 mL) was added MsCl (4 μL, 0.049 mmol) and Et3N (17 μL, 0.12 mmol). The reaction was kept at ambient temperature for 16 hrs and was quenched by addition of H2O (5 mL). The organic layer was extracted with H2O and brine, dried (MgSO4), filtered and concentrated under reduced pressure to afford the desired mesylate. The crude
reaction product was dissolve in DMF (2 mL) and NaN3 (2.5 mg, 0.037 mmol) was added. The reaction was heated to 50 °C for 1 hr and was quenched by H2O (5 mL). The aqueous layer was extracted with EtOAc (2 x 10 mL) and the combined organic layers were washed with brine, dried (MgSO4), filtered and concentrated under reduced pressure to give the desired azide. The azide was dissolved in THF (1 mL) and H2O (0.2 mL) and PPh3 (polymer supported, 3 mmol/g, 16 mg, 0.049 mmol) was added. The reaction was heated to 50 °C for 1 hr and filtered to remove polymer support. The filtrated was concentrated under reduced pressure and purified by reverse-phase preparative HPLC to provide (4-(2,4-dichlorophenyl)-2,6- diphenylpyrimidin-5-yl)methanamine, TFA salt (5 mg, 50% for 3 steps) as a white solid.
[00231] 1H NMR (400 MHz, CD3OD) 8.49 (dd, J = 1.8, 8.4 Hz, 2H), 7.74-7.82 (m, 3H), 7.60-7.70 (m, 5H), 7.47-7.59 (m, 3H), 4.37 (d, J = 14.5 Hz, IH), 4.13 (d, J = 14.5 Hz, IH). [00232] HPLC Phenomenex LUNA C-18 4.6 X 50 mm, 0 to 100% B over 4 minutes, 1 minutes hold time, A = 90% water, 10% methanol, 0.1% TFA, B = 10% water, 90% methanol, 0.1% TFA, RT = 3.52 min, 99% homogeneity index. [00233] LCMS: Anal. Calcd. for C23H17Cl2N3 405.08 found: 406.19 (M+H)+. [00234] HRMS: Anal. Calcd. for C23H18Cl2N3 406.0878 found: 406.0895 (M+H)+.
EXAMPLE 11
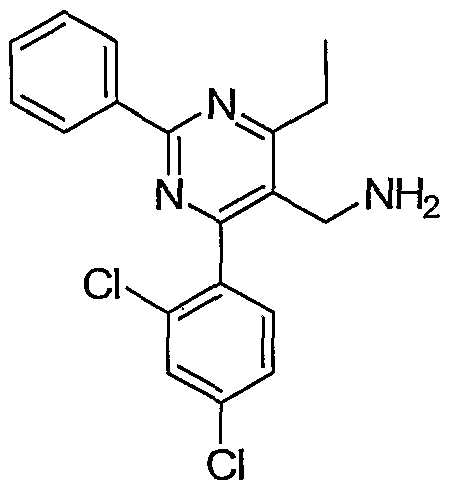
[00235] (4-(2,4-Dichlorophenyl)-6-ethyl-2-phenylpyrimidin-5-yl)methanamine, TFA salt was prepared by the methods described in Example 8, using methyl 3- oxopentanoate for Step 1.
[00236] 1H NMR (400 MHz, CD3OD) 8.38 (dd, J = 1.76, 6.16 Hz5 IH), 7.64 (d, J =
1.76 Hz, IH), 7.37-7.53 (m, 6H), 4.17 (d, J = 14.5 Hz, IH), 3.89 (d, J = 15.5 Hz, IH),
2.99 (m, 2H), 1.43 (t, J = 7.48 Hz, 3H).
[00237] HPLC Phenomenex LUNA C-18 4.6 X 50 mm, 0 to 100% B over 8 minutes, 2 minutes hold time, A = 90% water, 10% methanol, 0.2% phosphoric acid,
B = 10% water, 90% methanol, 0.2% phosphoric acid, RT = 5.84 min, 100% homogeneity index.
[00238] LCMS: Anal. Calcd. for C19H17Cl2N3 357.08 found: 358.06 (M+H)+.
[00239] LCMS: Anal. Calcd. for C19H18Cl2N3 358.0878 found: 358.00884
(M+H)+.

[00240] (4-(2,4-Dichlorophenyl)-2,6-dimethyl-pyrimidin-5-yl)methanamine, TFA salt was prepared by the methods described in Example 8, using acetamidine, hydrochloride for Step 2.
[00241] 1H NMR (400 MHz, CD3OD) 7.60 (d, J = 1.96 Hz, IH), 7.45 (dd5 J = 1.96,
8.32 Hz, IH), 7.36 (d, J = 8.32 Hz5 IH), 4.11 (d, J = 14.96 Hz, IH)5 3.83 (d, J = 14.96
Hz5 IH)5 2.61 (s, 3H)5 2.60 (s, 3H). [00242] HPLC Phenomenex LUNA C-18 4.6 X 50 mm, 0 to 100% B over 8 minutes, 2 minutes hold time, A = 90% water, 10% methanol, 0.2% phosphoric acid,
B = 10% water, 90% methanol, 0.2% phosphoric acid, RT = 3.10 mill, 100% homogeneity index.
[00243] LCMS: Anal. Calcd. for C13H13Cl2N3 281.05 found: 282.19 (M+H)+. [00244] HRMS: Anal. Calcd. for C13H14Cl2N3 282.0565 found: 282.0569 (M+H)+.
EXAMPLE 13

Example 13, Step 1. Methyl 2-(benzylthio)-4-(2,4-dichlorophenyl)-6-methyl-l,4- dihydropyrimidine-5-carboxylate.

[00245] To a stirred solution of methyl 2-(2,4-dichlorobenzylidene)-3- oxobutanoate from Example 8, Step 1 (60 mg, 0.22 mmol) and benzylthiourea HCl salt (44 mg, 0.22 mmol) in DMF (2 mL) was added molecular sieves (3 A). The reaction was heated to 90 °C for 16 hrs and was diluted with EtOAc (15 mL) and filtered to remove molecular sieves. The organic layer was extracted with H2O (2 x 10 mL), brine, dried (MgSO4), filtered and concentrated under reduced pressure to afford the desired methyl 2-(benzylthio)-4-(2,4-dichlorophenyl)-6-methyl-l,4- dihydropyrimidine-5-carboxylate. The crude reaction product (>95% purity) was moved onto next step without further purification.
Example 13, Step 2. Methyl 2-(benzylthio)-4-(2,4-dichIorophenyl)-6- methylpyrimidine-5-carboxylate.

[00246] The crude methyl 2-(benzylthio)-4-(2,4-dichloroρhenyl)-6-methyl- 1 ,4- dihydropyrrmidine-5-carboxylate (0.22 mmol) from Step 1 was dissolved in CH2Cl2 (2 mL) and DDQ (50 mg, 0.22 mmol) was added. After 1 hr, The reaction was diluted with cyclohexane/EtOAc solution (4:1, 10 mL) and the organic layer was extracted with saturated NaHCO3 solution (2 x 6 mL), brine (5 mL). The organic layer dried (MgSO4), filtered and concentrated under reduced pressure to give methyl 2- (benzylthio)-4-(2,4-dichlorophenyl)-6-methylρyrimidine-5-carboxylate. The crude reaction product (>95% purity) was moved onto next step without further purification. [00247] 1H NMR (400 MHz, CDCl3) 7.45 (d, J = 1.3 Hz, IH), 7.41 (d, J = 6.2 Hz, IH), 7.20-7.37 (m, 6H), 4.41 (s, 2H), 3.63 (s, 3H), 2.66 (s, 3H).
Example 13, Step 3. Methyl 2-(benzylsulfonyl)-4-(2,4-dichlorophenyl)-6- methylpyrimidine-5-carboxylate.

[00248] To crude methyl 2-(benzylthio)-4-(2,4-dichlorophenyl)-6- methylpyrirnidine-5-carboxylate (0.22 mmol) from Step 2 in CH2Cl2 (3 mL) was added mCPBA (105 mg, 0.55 mmol). After 16 hrs, the reaction was concentrated under reduced pressure and diluted with EtOAc (10 mL). The organic layer was extracted with saturated NaHCO3 solution (2 x 6 mL), brine (5 mL), dried (MgSO4),
filtered and concentrated under reduced pressure. The crude reaction product was purified by flash chromatography (silica gel, 20% EtOAc / hexane) to give methyl 2- (benzylsulfonyl)-4-(2,4-dichlorophenyl)-6-methylpyrimidine-5-carboxylate (80 mg, 80% for 3 steps) as a clear oil. [00249] 1H NMR (400 MHz, CDCl3) 7.51 (s, IH), 7.26-7.42 (m, 7H), 4.80 (s, 2H), 3.72 (s, 3H), 2.79 (s, 3H).
[00250] HPLC Phenomenex LUNA C-18 4.6 x 50 mm, 0 to 100% B over 4 minutes, 1 minutes hold time, A = 90% water, 10% methanol, 0.1% TFA, B = 10% water, 90% methanol, 0.1% TFA, RT = 3.48 min. [00251] LCMS: Anal. Calcd. for C20H16Cl2N2O4S 450.02 found: 451.15 (M+H)+.
Example 13, Step 4. Methyl 4-(2,4-dichlorophenyl)-6-methyl-2- thiomorpholmopyrimidine-5-carboxylate.

The crude reaction product (>98% purity) was moved onto next step without further purification.
[00253] 1H NMR (400 MHz, CDCl3) 7.43 (d, J = 1.8 hz, IH), 7.31 (dd, J = 1.8, 8.4 Hz, IH), 7.22 (d, J = 8.4 Hz, IH), 4.21 (m, 4H), 3.55 (s, 3H), 2.66 (m5 4H), 2.58 (s, 3H).
Example 13, Step 5-8. (4-(2,4-Dichlorophenyl)-6-methyl-2- thiomorphoIinopyrimidin-5-yl)methanamine, TFA salt.

[00254] (4-(2,4-Dichlorophenyl)-6-methyl-2-thiomorpholinopyrimidin-5- yl)methanamine, TFA salt was prepared by the methods described in Example 10, Step 5-8.
[00255] 1H NMR (400 MHz, CD3OD) 7.67 (d, J = 1.2 Hz5 IH), 7.52 (dd, J = 1.2, 7.6 Hz, IH), 7.40 (d, J = 7.6 Hz5 IH)5 4.17 (m, 4H)5 4.06 (d, J = 15.2 Hz5 IH)5 3.78 (d, J = 15.2 Hz5 IH)5 2.63 (m, 4H)5 2.53 (s, 3H). [00256] HPLC Phenomenex LUNA C-18 4.6 x 50 mm, 0 to 100% B over 4 minutes, 1 minutes hold time, A = 90% water, 10% methanol, 0.1% TFA5 B = 10% water, 90% methanol, 0.1% TFA5 RT = 3.37 min, 95% homogeneity index. [00257] LCMS: Anal. Calcd. for C16H18Cl2N4S 368.06 found: 369.18 (M+H)+.
Claims
1. A compound of formula (I)

(I) wherein: n = 1 or 2;
R is a substitutent selected from the group consisting of hydrogen (H), halogen, cyano (CN), CF3, alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkylalkyl, bicycloalkyl, bicycloalkylalkyl, alkylthioalkyl, arylalkylthioalkyl, cycloalkenyl, aryl, arylalkyl, heteroaryl, heteroarylalkyl, cycloheteroalkyl and cycloheteroalkylalkyl, wherein any such substituent may optionally be substituted through available carbon atoms with 1, 2, 3, 4 or 5 groups selected from hydrogen, halo, alkyl, polyhaloalkyl, alkoxy, haloalkoxy, polyhaloalkoxy, alkoxycarbonyl, alkenyl, alkynyl, cycloalkyl, cycloalkylalkyl, polycycloalkyl, heteroarylamino, arylamino, cycloheteroalkyl, cycloheteroalkylalkyl, hydroxy, hydroxyalkyl, nitro, cyano, amino, substituted amino, alkylamino, dialkylamino, thiol, alkylthio, alkylcarbonyl, acyl, alkoxycarbonyl, aminocarbonyl, alkynylaminocarbonyl, alkylaminocarbonyl, alkenylaminocarbonyl, alkylcarbonyloxy, alkylcarbonylamino, arylcarbonylamino, alkylsulfonylamino, alkylaminocarbonylamino, alkoxycarbonylamino, alkylsulfonyl, aminosulfonyl, alkylsulfinyl, sulfonamido and sulfonyl;
B is selected from the group consisting of a bond, oxygen (O), nitrogen (N) and S(O)m; m is 0, 1 or 2;
X is a substitutent selected from the group consisting of hydrogen (H), alkyl, alkenyl, alkynyl, cycloalkyl, cycloalkylalkyl, bicycloalkyl, bicycloalkylalkyl, alkylthioalkyl, arylalkylthioalkyl, cycloalkenyl, aryl, arylalkyl, heteroaryl, heteroarylalkyl, cycloheteroalkyl and cycloheteroalkylalkyl, wherein any such substituent may optionally be substituted through available carbon atoms with 1, 2, 3, 4 or 5 groups selected from hydrogen, halo, alkyl, polyhaloalkyl, alkoxy, haloalkoxy, polyhaloalkoxy, alkoxycarbonyl, alkenyl, alkynyl, cycloalkyl, cycloalkylalkyl, polycycloalkyl, heteroarylamino, arylamino, cycloheteroalkyl, cycloheteroalkylalkyl, hydroxy, hydroxyalkyl, nitro, cyano, amino, substituted amino, alkylamino, dialkylamino, thiol, alkylthio, alkylcarbonyl, acyl, alkoxycarbonyl, aminocarbonyl, alkynylamitiocarbonyl, alkylaminocarbonyl, alkenylaminocarbonyl, alkylcarbonyloxy, alkylcarbonylamino, arylcarbonylamino, alkylsulfonylamino, alkylaminocarbonylamino, alkoxycarbonylamino, alkylsulfonyl, aminosulfonyl, alkylsulfinyl, sulfonamido and sulfonyl; B-X taken together can be a halogen; and
Y is aryl, optionally substituted with 1, 2, 3, 4 or 5 groups selected from hydrogen, halo, alkyl, polyhaloalkyl, alkoxy, haloalkoxy, polyhaloalkoxy, alkoxycarbonyl, alkenyl, alkynyl, cycloalkyl, cycloalkylalkyl, polycycloalkyl, heteroarylamino, arylamino, cycloheteroalkyl, cycloheteroalkylalkyl, hydroxy, hydroxyalkyl, nitro, cyano, amino, substituted amino, alkylamino, dialkylamino, thiol, alkylthio, alkylcarbonyl, acyl, alkoxycarbonyl, aminocarbonyl, alkynylaminocarbonyl, alkylaminocarbonyl, alkenylaminocarbonyl, alkylcarbonyloxy, alkylcarbonylamino, arylcarbonylamino, alkylsulfonylamino, alkylaminocarbonylamino, alkoxycarbonylamino, alkylsulfonyl, aminosulfonyl, alkylsulfinyl, sulfonamido and sulfonyl; including pharmaceutically acceptable salts thereof, and prodrug esters thereof, and all stereoisomers thereof.
2. The compound as defined in Claim 1 wherein n is 1.
3. The compound as defined in Claim 1 selected from 


4. A pharmaceutical composition comprising a compound as defined in Claim 1 and a pharmaceutically acceptable carrier therefore.
5. A pharmaceutical combination comprising a compound of formula I as defined in Claim 1 and at least one therapeutic agent selected from the group consisting of an antidiabetic agent, an anti-obesity agent, a anti-hypertensive agent, an anti-atherosclerotic agent and a lipid-lowering agent.
6. The pharmaceutical combination as defined in Claim 5 wherein the therapeutic agent is an antidiabetic agent.
7. The combination as defined in Claim 6 wherein the antidiabetic agent is at least one agent selected from the group consisting of a biguanide, a sulfonyl urea, a glucosidase inhibitor, a PPAR gamma agonist, a PPAR alpha/gamma dual agonist, an aP2 inhibitor, a SGLT2 inhibitor, an insulin sensitizer, a glucagon-like peptide-1 (GLP-I), insulin and a meglitinide.
8. The combination as defined in Claim 7 wherein the antidiabetic agent is at least one agent selected from the group consisting of metformin, glyburide, glimepiride, glipyride, glipizide, chlorpropamide, gliclazide, acarbose, miglitol, pioglitazone, troglitazone, rosiglitazone, insulin, isaglitazone, repaglinide and nateglinide.
9. The combination as defined in Claim 6 wherein the compound of formula I is present in a weight ratio to the antidiabetic agent in the range of about 0.01 to about 300:1.
10. The combination as defined in Claim 5 wherein the anti-obesity agent is at least one agent selected from the group consisting of a beta 3 adrenergic agonist, a lipase inhibitor, a serotonin (and dopamine) reuptake inhibitor, a thyroid receptor beta compound and an anorectic agent.
11. The combination as defined in Claim 10 wherein the anti-obesity agent is at least one agent selected from the group consisting of orlistat, sibutramine, topiramate, axokine, dexamphetamine, phentermine, phenylpropanolamine and mazindol.
12. The combination as defined in Claim 5 wherein the lipid lowering agent is at least one agent selected from the group consisting of an MTP inhibitor, cholesterol ester transfer protein, an HMG CoA reductase inhibitor, a squalene synthetase inhibitor, a fibric acid derivative, an upregulator of LDL receptor activity, a lipoxygenase inhibitor, or an ACAT inhibitor.
13. The combination as defined in Claim 12 wherein the lipid lowering agent is at least one agent selected from the group consisting of pravastatin, lovastatin, simvastatin, atorvastatin, cerivastatin, fluvastatin, nisvastatin, visastatin, fenofibrate, gemfibrozil, clofϊbrate and avasimibe.
14. The combination as defined in Claim 5 wherein the compound of formula I is present in a weight ratio to the lipid-lowering agent in the range of about 0.01 to about 100:1.
15. A method for treating or delaying the progression or onset of diabetes, diabetic retinopathy, diabetic neuropathy, diabetic nephropathy, wound healing, insulin resistance, hyperglycemia, hyperinsulinemia, Syndrome X, diabetic complications, elevated blood levels of free fatty acids or glycerol, hyperlipidemia, obesity, hypertriglyceridemia, atherosclerosis or hypertension, which comprises administering to a mammalian species in need of treatment a therapeutically effective amount of a compound as defined in Claim 1.
16. A method according to claim 15 further comprising administering, concurrently or sequentially, a therapeutically effective amount of at least one additional therapeutic agent selected from the group consisting of an antidiabetic agent, an anti-obesity agent, a anti-hypertensive agent, an anti-atherosclerotic agent, an agent for inhibiting allograft rejection in transplantation and a lipid-lowering agent.
17. A pharmaceutical composition that inhibits DPP-IV containing a compound as defined in Claim 1.
18. A method of inhibiting DPP-IV comprising administering a pharmaceutical composition comprising a compound as defined in Claim 1.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP05855333.0A EP1831180B1 (en) | 2004-12-29 | 2005-12-23 | Pyrimidine-based inhibitors of dipeptidyl peptidase iv and methods |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US64011004P | 2004-12-29 | 2004-12-29 | |
| US60/640,110 | 2004-12-29 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2006071762A2 true WO2006071762A2 (en) | 2006-07-06 |
| WO2006071762A3 WO2006071762A3 (en) | 2006-11-23 |
Family
ID=36615438
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2005/046750 WO2006071762A2 (en) | 2004-12-29 | 2005-12-23 | Pyrimidine-based inhibitors of dipeptidyl peptidase iv and methods |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US7589088B2 (en) |
| EP (1) | EP1831180B1 (en) |
| WO (1) | WO2006071762A2 (en) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006119451A1 (en) * | 2005-05-04 | 2006-11-09 | Vertex Pharmaceuticals Incorporated | Pyrimidines and pyrazines useful as modulators of ion channels |
| WO2007120702A2 (en) | 2006-04-11 | 2007-10-25 | Arena Pharmaceuticals, Inc. | Use of gpr119 receptor agonists for increasing bone mass and for treating osteoporosis, and combination therapy relating thereto |
| US7880008B2 (en) | 2005-05-31 | 2011-02-01 | Vertex Pharmaceuticals Incorporated | Heterocycles useful as modulators of ion channels |
| US10064850B2 (en) | 2007-04-11 | 2018-09-04 | Omeros Corporation | Compositions and methods for prophylaxis and treatment of addictions |
| US11241420B2 (en) | 2007-04-11 | 2022-02-08 | Omeros Corporation | Compositions and methods for prophylaxis and treatment of addictions |
Families Citing this family (82)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8645137B2 (en) | 2000-03-16 | 2014-02-04 | Apple Inc. | Fast, language-independent method for user authentication by voice |
| DOP2006000008A (en) | 2005-01-10 | 2006-08-31 | Arena Pharm Inc | COMBINED THERAPY FOR THE TREATMENT OF DIABETES AND RELATED AFFECTIONS AND FOR THE TREATMENT OF AFFECTIONS THAT IMPROVE THROUGH AN INCREASE IN THE BLOOD CONCENTRATION OF GLP-1 |
| US8677377B2 (en) | 2005-09-08 | 2014-03-18 | Apple Inc. | Method and apparatus for building an intelligent automated assistant |
| DK1971862T3 (en) | 2006-04-11 | 2011-02-14 | Arena Pharm Inc | Methods of Using GPR119 Receptor to Identify Compounds Useful for Increasing Bone Mass in a Person |
| US9318108B2 (en) | 2010-01-18 | 2016-04-19 | Apple Inc. | Intelligent automated assistant |
| US8996376B2 (en) | 2008-04-05 | 2015-03-31 | Apple Inc. | Intelligent text-to-speech conversion |
| EP2108960A1 (en) | 2008-04-07 | 2009-10-14 | Arena Pharmaceuticals, Inc. | Methods of using A G protein-coupled receptor to identify peptide YY (PYY) secretagogues and compounds useful in the treatment of conditons modulated by PYY |
| US8676904B2 (en) | 2008-10-02 | 2014-03-18 | Apple Inc. | Electronic devices with voice command and contextual data processing capabilities |
| US10241752B2 (en) | 2011-09-30 | 2019-03-26 | Apple Inc. | Interface for a virtual digital assistant |
| US10241644B2 (en) | 2011-06-03 | 2019-03-26 | Apple Inc. | Actionable reminder entries |
| US9431006B2 (en) | 2009-07-02 | 2016-08-30 | Apple Inc. | Methods and apparatuses for automatic speech recognition |
| US8682667B2 (en) | 2010-02-25 | 2014-03-25 | Apple Inc. | User profiling for selecting user specific voice input processing information |
| CZ305457B6 (en) | 2011-02-28 | 2015-09-30 | Ústav organické chemie a biochemie, Akademie věd ČR v. v. i. | Pyrimidine compounds inhibiting formation of nitrogen monoxide and prostaglandin E2, process for their preparation and use |
| US9262612B2 (en) | 2011-03-21 | 2016-02-16 | Apple Inc. | Device access using voice authentication |
| US8994660B2 (en) | 2011-08-29 | 2015-03-31 | Apple Inc. | Text correction processing |
| US9280610B2 (en) | 2012-05-14 | 2016-03-08 | Apple Inc. | Crowd sourcing information to fulfill user requests |
| US9721563B2 (en) | 2012-06-08 | 2017-08-01 | Apple Inc. | Name recognition system |
| US9547647B2 (en) | 2012-09-19 | 2017-01-17 | Apple Inc. | Voice-based media searching |
| US8652527B1 (en) | 2013-03-13 | 2014-02-18 | Upsher-Smith Laboratories, Inc | Extended-release topiramate capsules |
| US9101545B2 (en) | 2013-03-15 | 2015-08-11 | Upsher-Smith Laboratories, Inc. | Extended-release topiramate capsules |
| WO2014197336A1 (en) | 2013-06-07 | 2014-12-11 | Apple Inc. | System and method for detecting errors in interactions with a voice-based digital assistant |
| US9582608B2 (en) | 2013-06-07 | 2017-02-28 | Apple Inc. | Unified ranking with entropy-weighted information for phrase-based semantic auto-completion |
| WO2014197334A2 (en) | 2013-06-07 | 2014-12-11 | Apple Inc. | System and method for user-specified pronunciation of words for speech synthesis and recognition |
| WO2014197335A1 (en) | 2013-06-08 | 2014-12-11 | Apple Inc. | Interpreting and acting upon commands that involve sharing information with remote devices |
| US10176167B2 (en) | 2013-06-09 | 2019-01-08 | Apple Inc. | System and method for inferring user intent from speech inputs |
| DE112014002747T5 (en) | 2013-06-09 | 2016-03-03 | Apple Inc. | Apparatus, method and graphical user interface for enabling conversation persistence over two or more instances of a digital assistant |
| US9842101B2 (en) | 2014-05-30 | 2017-12-12 | Apple Inc. | Predictive conversion of language input |
| US9430463B2 (en) | 2014-05-30 | 2016-08-30 | Apple Inc. | Exemplar-based natural language processing |
| US9338493B2 (en) | 2014-06-30 | 2016-05-10 | Apple Inc. | Intelligent automated assistant for TV user interactions |
| US9818400B2 (en) | 2014-09-11 | 2017-11-14 | Apple Inc. | Method and apparatus for discovering trending terms in speech requests |
| US10789041B2 (en) | 2014-09-12 | 2020-09-29 | Apple Inc. | Dynamic thresholds for always listening speech trigger |
| US9646609B2 (en) | 2014-09-30 | 2017-05-09 | Apple Inc. | Caching apparatus for serving phonetic pronunciations |
| US9886432B2 (en) | 2014-09-30 | 2018-02-06 | Apple Inc. | Parsimonious handling of word inflection via categorical stem + suffix N-gram language models |
| US9668121B2 (en) | 2014-09-30 | 2017-05-30 | Apple Inc. | Social reminders |
| US10074360B2 (en) | 2014-09-30 | 2018-09-11 | Apple Inc. | Providing an indication of the suitability of speech recognition |
| US10127911B2 (en) | 2014-09-30 | 2018-11-13 | Apple Inc. | Speaker identification and unsupervised speaker adaptation techniques |
| US9865280B2 (en) | 2015-03-06 | 2018-01-09 | Apple Inc. | Structured dictation using intelligent automated assistants |
| US9886953B2 (en) | 2015-03-08 | 2018-02-06 | Apple Inc. | Virtual assistant activation |
| US9721566B2 (en) | 2015-03-08 | 2017-08-01 | Apple Inc. | Competing devices responding to voice triggers |
| US10567477B2 (en) | 2015-03-08 | 2020-02-18 | Apple Inc. | Virtual assistant continuity |
| US9899019B2 (en) | 2015-03-18 | 2018-02-20 | Apple Inc. | Systems and methods for structured stem and suffix language models |
| US9842105B2 (en) | 2015-04-16 | 2017-12-12 | Apple Inc. | Parsimonious continuous-space phrase representations for natural language processing |
| US10083688B2 (en) | 2015-05-27 | 2018-09-25 | Apple Inc. | Device voice control for selecting a displayed affordance |
| US10127220B2 (en) | 2015-06-04 | 2018-11-13 | Apple Inc. | Language identification from short strings |
| US9578173B2 (en) | 2015-06-05 | 2017-02-21 | Apple Inc. | Virtual assistant aided communication with 3rd party service in a communication session |
| US10101822B2 (en) | 2015-06-05 | 2018-10-16 | Apple Inc. | Language input correction |
| US10255907B2 (en) | 2015-06-07 | 2019-04-09 | Apple Inc. | Automatic accent detection using acoustic models |
| US10186254B2 (en) | 2015-06-07 | 2019-01-22 | Apple Inc. | Context-based endpoint detection |
| US11025565B2 (en) | 2015-06-07 | 2021-06-01 | Apple Inc. | Personalized prediction of responses for instant messaging |
| US10671428B2 (en) | 2015-09-08 | 2020-06-02 | Apple Inc. | Distributed personal assistant |
| US10747498B2 (en) | 2015-09-08 | 2020-08-18 | Apple Inc. | Zero latency digital assistant |
| US9697820B2 (en) | 2015-09-24 | 2017-07-04 | Apple Inc. | Unit-selection text-to-speech synthesis using concatenation-sensitive neural networks |
| US11010550B2 (en) | 2015-09-29 | 2021-05-18 | Apple Inc. | Unified language modeling framework for word prediction, auto-completion and auto-correction |
| US10366158B2 (en) | 2015-09-29 | 2019-07-30 | Apple Inc. | Efficient word encoding for recurrent neural network language models |
| US11587559B2 (en) | 2015-09-30 | 2023-02-21 | Apple Inc. | Intelligent device identification |
| US10691473B2 (en) | 2015-11-06 | 2020-06-23 | Apple Inc. | Intelligent automated assistant in a messaging environment |
| US10049668B2 (en) | 2015-12-02 | 2018-08-14 | Apple Inc. | Applying neural network language models to weighted finite state transducers for automatic speech recognition |
| US10223066B2 (en) | 2015-12-23 | 2019-03-05 | Apple Inc. | Proactive assistance based on dialog communication between devices |
| US10446143B2 (en) | 2016-03-14 | 2019-10-15 | Apple Inc. | Identification of voice inputs providing credentials |
| US9934775B2 (en) | 2016-05-26 | 2018-04-03 | Apple Inc. | Unit-selection text-to-speech synthesis based on predicted concatenation parameters |
| US9972304B2 (en) | 2016-06-03 | 2018-05-15 | Apple Inc. | Privacy preserving distributed evaluation framework for embedded personalized systems |
| US10249300B2 (en) | 2016-06-06 | 2019-04-02 | Apple Inc. | Intelligent list reading |
| US10049663B2 (en) | 2016-06-08 | 2018-08-14 | Apple, Inc. | Intelligent automated assistant for media exploration |
| DK179309B1 (en) | 2016-06-09 | 2018-04-23 | Apple Inc | Intelligent automated assistant in a home environment |
| US10067938B2 (en) | 2016-06-10 | 2018-09-04 | Apple Inc. | Multilingual word prediction |
| US10509862B2 (en) | 2016-06-10 | 2019-12-17 | Apple Inc. | Dynamic phrase expansion of language input |
| US10490187B2 (en) | 2016-06-10 | 2019-11-26 | Apple Inc. | Digital assistant providing automated status report |
| US10586535B2 (en) | 2016-06-10 | 2020-03-10 | Apple Inc. | Intelligent digital assistant in a multi-tasking environment |
| US10192552B2 (en) | 2016-06-10 | 2019-01-29 | Apple Inc. | Digital assistant providing whispered speech |
| DK179343B1 (en) | 2016-06-11 | 2018-05-14 | Apple Inc | Intelligent task discovery |
| DK201670540A1 (en) | 2016-06-11 | 2018-01-08 | Apple Inc | Application integration with a digital assistant |
| DK179415B1 (en) | 2016-06-11 | 2018-06-14 | Apple Inc | Intelligent device arbitration and control |
| DK179049B1 (en) | 2016-06-11 | 2017-09-18 | Apple Inc | Data driven natural language event detection and classification |
| US10043516B2 (en) | 2016-09-23 | 2018-08-07 | Apple Inc. | Intelligent automated assistant |
| US10593346B2 (en) | 2016-12-22 | 2020-03-17 | Apple Inc. | Rank-reduced token representation for automatic speech recognition |
| DK201770439A1 (en) | 2017-05-11 | 2018-12-13 | Apple Inc. | Offline personal assistant |
| DK179496B1 (en) | 2017-05-12 | 2019-01-15 | Apple Inc. | USER-SPECIFIC Acoustic Models |
| DK179745B1 (en) | 2017-05-12 | 2019-05-01 | Apple Inc. | SYNCHRONIZATION AND TASK DELEGATION OF A DIGITAL ASSISTANT |
| DK201770431A1 (en) | 2017-05-15 | 2018-12-20 | Apple Inc. | Optimizing dialogue policy decisions for digital assistants using implicit feedback |
| DK201770432A1 (en) | 2017-05-15 | 2018-12-21 | Apple Inc. | Hierarchical belief states for digital assistants |
| DK179549B1 (en) | 2017-05-16 | 2019-02-12 | Apple Inc. | Far-field extension for digital assistant services |
| CN111423362A (en) * | 2020-03-11 | 2020-07-17 | 山东省药学科学院 | Preparation method of two high-purity clevidipine butyrate impurities |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003068757A1 (en) * | 2002-02-13 | 2003-08-21 | F. Hoffmann-La Roche Ag | Novel pyridin- and pyrimidin-derivatives |
Family Cites Families (66)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3674836A (en) | 1968-05-21 | 1972-07-04 | Parke Davis & Co | 2,2-dimethyl-{11 -aryloxy-alkanoic acids and salts and esters thereof |
| US4027009A (en) | 1973-06-11 | 1977-05-31 | Merck & Co., Inc. | Compositions and methods for depressing blood serum cholesterol |
| JPS5612114B2 (en) | 1974-06-07 | 1981-03-18 | ||
| US4231938A (en) | 1979-06-15 | 1980-11-04 | Merck & Co., Inc. | Hypocholesteremic fermentation products and process of preparation |
| MX7065E (en) | 1980-06-06 | 1987-04-10 | Sankyo Co | A MICROBIOLOGICAL PROCEDURE FOR PREPARING DERIVATIVES OF ML-236B |
| US4450171A (en) | 1980-08-05 | 1984-05-22 | Merck & Co., Inc. | Antihypercholesterolemic compounds |
| US4448784A (en) | 1982-04-12 | 1984-05-15 | Hoechst-Roussel Pharmaceuticals, Inc. | 1-(Aminoalkylphenyl and aminoalkylbenzyl)-indoles and indolines and analgesic method of use thereof |
| US5354772A (en) | 1982-11-22 | 1994-10-11 | Sandoz Pharm. Corp. | Indole analogs of mevalonolactone and derivatives thereof |
| US4499289A (en) | 1982-12-03 | 1985-02-12 | G. D. Searle & Co. | Octahydronapthalenes |
| CA1327360C (en) | 1983-11-14 | 1994-03-01 | William F. Hoffman | Oxo-analogs of mevinolin-like antihypercholesterolemic agents |
| US4613610A (en) | 1984-06-22 | 1986-09-23 | Sandoz Pharmaceuticals Corp. | Cholesterol biosynthesis inhibiting pyrazole analogs of mevalonolactone and its derivatives |
| US4686237A (en) | 1984-07-24 | 1987-08-11 | Sandoz Pharmaceuticals Corp. | Erythro-(E)-7-[3'-C1-3 alkyl-1'-(3",5"-dimethylphenyl)naphth-2'-yl]-3,5-dihydroxyhept-6-enoic acids and derivatives thereof |
| US4647576A (en) | 1984-09-24 | 1987-03-03 | Warner-Lambert Company | Trans-6-[2-(substitutedpyrrol-1-yl)alkyl]-pyran-2-one inhibitors of cholesterol synthesis |
| JPS62501009A (en) | 1984-12-04 | 1987-04-23 | サンド・アクチエンゲゼルシヤフト | Indene congeners of mevalonolactone and derivatives thereof |
| US4668794A (en) | 1985-05-22 | 1987-05-26 | Sandoz Pharm. Corp. | Intermediate imidazole acrolein analogs |
| AU598775B2 (en) | 1985-10-25 | 1990-07-05 | Sandoz Ag | Heterocyclic analogs of mevalonolactone |
| FR2596393B1 (en) | 1986-04-01 | 1988-06-03 | Sanofi Sa | HYDROXY-3 DIHYDROXYOXOPHOSPHORIO-4 BUTANOIC ACID DERIVATIVES, THEIR PREPARATION PROCESS, THEIR USE AS MEDICAMENTS AND THE COMPOSITIONS CONTAINING THEM |
| US5614492A (en) | 1986-05-05 | 1997-03-25 | The General Hospital Corporation | Insulinotropic hormone GLP-1 (7-36) and uses thereof |
| US4681893A (en) | 1986-05-30 | 1987-07-21 | Warner-Lambert Company | Trans-6-[2-(3- or 4-carboxamido-substituted pyrrol-1-yl)alkyl]-4-hydroxypyran-2-one inhibitors of cholesterol synthesis |
| GB2205837B (en) | 1987-05-22 | 1991-11-20 | Squibb & Sons Inc | Phosphorus-containing hmg-coa reductase inhibitors |
| US4759923A (en) | 1987-06-25 | 1988-07-26 | Hercules Incorporated | Process for lowering serum cholesterol using poly(diallylmethylamine) derivatives |
| JP2569746B2 (en) | 1987-08-20 | 1997-01-08 | 日産化学工業株式会社 | Quinoline mevalonolactones |
| US4924024A (en) | 1988-01-11 | 1990-05-08 | E. R. Squibb & Sons, Inc. | Phosphorus-containing squalene synthetase inhibitors, new intermediates and method |
| US4871721A (en) | 1988-01-11 | 1989-10-03 | E. R. Squibb & Sons, Inc. | Phosphorus-containing squalene synthetase inhibitors |
| NO177005C (en) | 1988-01-20 | 1995-07-05 | Bayer Ag | Analogous process for the preparation of substituted pyridines, as well as intermediates for use in the preparation |
| US5506219A (en) | 1988-08-29 | 1996-04-09 | E. R. Squibb & Sons, Inc. | Pyridine anchors for HMG-CoA reductase inhibitors |
| US5753675A (en) | 1989-03-03 | 1998-05-19 | Novartis Pharmaceuticals Corporation | Quinoline analogs of mevalonolactone and derivatives thereof |
| FI94339C (en) | 1989-07-21 | 1995-08-25 | Warner Lambert Co | Process for the preparation of pharmaceutically acceptable [R- (R *, R *)] - 2- (4-fluorophenyl) -, - dihydroxy-5- (1-methylethyl) -3-phenyl-4 - [(phenylamino) carbonyl] -1H- for the preparation of pyrrole-1-heptanoic acid and its pharmaceutically acceptable salts |
| US5177080A (en) | 1990-12-14 | 1993-01-05 | Bayer Aktiengesellschaft | Substituted pyridyl-dihydroxy-heptenoic acid and its salts |
| JP2648897B2 (en) | 1991-07-01 | 1997-09-03 | 塩野義製薬株式会社 | Pyrimidine derivatives |
| US5595872A (en) | 1992-03-06 | 1997-01-21 | Bristol-Myers Squibb Company | Nucleic acids encoding microsomal trigyceride transfer protein |
| DK36392D0 (en) | 1992-03-19 | 1992-03-19 | Novo Nordisk As | USE OF CHEMICAL COMPOUND |
| US5470845A (en) | 1992-10-28 | 1995-11-28 | Bristol-Myers Squibb Company | Methods of using α-phosphonosulfonate squalene synthetase inhibitors including the treatment of atherosclerosis and hypercholesterolemia |
| US5594016A (en) | 1992-12-28 | 1997-01-14 | Mitsubishi Chemical Corporation | Naphthalene derivatives |
| ES2133158T3 (en) | 1993-01-19 | 1999-09-01 | Warner Lambert Co | FORMULATION CI-981 ORAL, STABLE AND PREPARATION PROCESS OF THE SAME. |
| US5346701A (en) | 1993-02-22 | 1994-09-13 | Theratech, Inc. | Transmucosal delivery of macromolecular drugs |
| US5739135A (en) | 1993-09-03 | 1998-04-14 | Bristol-Myers Squibb Company | Inhibitors of microsomal triglyceride transfer protein and method |
| US5776983A (en) | 1993-12-21 | 1998-07-07 | Bristol-Myers Squibb Company | Catecholamine surrogates useful as β3 agonists |
| US5488064A (en) | 1994-05-02 | 1996-01-30 | Bristol-Myers Squibb Company | Benzo 1,3 dioxole derivatives |
| US5385929A (en) | 1994-05-04 | 1995-01-31 | Warner-Lambert Company | [(Hydroxyphenylamino) carbonyl] pyrroles |
| US5612359A (en) | 1994-08-26 | 1997-03-18 | Bristol-Myers Squibb Company | Substituted biphenyl isoxazole sulfonamides |
| US5491134A (en) | 1994-09-16 | 1996-02-13 | Bristol-Myers Squibb Company | Sulfonic, phosphonic or phosphiniic acid β3 agonist derivatives |
| US5541204A (en) | 1994-12-02 | 1996-07-30 | Bristol-Myers Squibb Company | Aryloxypropanolamine β 3 adrenergic agonists |
| US5620997A (en) | 1995-05-31 | 1997-04-15 | Warner-Lambert Company | Isothiazolones |
| WO1997012613A1 (en) | 1995-10-05 | 1997-04-10 | Warner-Lambert Company | Method for treating and preventing inflammation and atherosclerosis |
| ATE344279T1 (en) | 1995-12-13 | 2006-11-15 | Univ California | CRYSTALS OF THE LIGAND-BINDING DOMAIN OF THE THYROID HORMONE RECEPTOR COMPLEXED WITH A LIGAND |
| US5770615A (en) | 1996-04-04 | 1998-06-23 | Bristol-Myers Squibb Company | Catecholamine surrogates useful as β3 agonists |
| US5962440A (en) | 1996-05-09 | 1999-10-05 | Bristol-Myers Squibb Company | Cyclic phosphonate ester inhibitors of microsomal triglyceride transfer protein and method |
| US5827875A (en) | 1996-05-10 | 1998-10-27 | Bristol-Myers Squibb Company | Inhibitors of microsomal triglyceride transfer protein and method |
| US5885983A (en) | 1996-05-10 | 1999-03-23 | Bristol-Myers Squibb Company | Inhibitors of microsomal triglyceride transfer protein and method |
| US5760246A (en) | 1996-12-17 | 1998-06-02 | Biller; Scott A. | Conformationally restricted aromatic inhibitors of microsomal triglyceride transfer protein and method |
| TW536540B (en) | 1997-01-30 | 2003-06-11 | Bristol Myers Squibb Co | Endothelin antagonists: N-[[2'-[[(4,5-dimethyl-3-isoxazolyl)amino]sulfonyl]-4-(2-oxazolyl)[1,1'-biphenyl]-2-yl]methyl]-N,3,3-trimethylbutanamide and N-(4,5-dimethyl-3-isoxazolyl)-2'-[(3,3-dimethyl-2-oxo-1-pyrrolidinyl)methyl]-4'-(2-oxazolyl)[1,1'-biphe |
| GB9713739D0 (en) | 1997-06-27 | 1997-09-03 | Karobio Ab | Thyroid receptor ligands |
| WO1999038501A2 (en) | 1998-02-02 | 1999-08-05 | Trustees Of Tufts College | Method of regulating glucose metabolism, and reagents related thereto |
| JP2002506075A (en) | 1998-03-09 | 2002-02-26 | フォンダテッヒ・ベネルクス・ナムローゼ・フェンノートシャップ | Serine peptidase modulator |
| DE19823831A1 (en) | 1998-05-28 | 1999-12-02 | Probiodrug Ges Fuer Arzneim | New pharmaceutical use of isoleucyl thiazolidide and its salts |
| DE19828113A1 (en) | 1998-06-24 | 2000-01-05 | Probiodrug Ges Fuer Arzneim | Prodrugs of Dipeptidyl Peptidase IV Inhibitors |
| DE19828114A1 (en) | 1998-06-24 | 2000-01-27 | Probiodrug Ges Fuer Arzneim | Produgs of unstable inhibitors of dipeptidyl peptidase IV |
| DE69940063D1 (en) | 1998-07-06 | 2009-01-22 | Bristol Myers Squibb Co | BIPHENYLSULFONAMIDE AS A DOUBLE-ACTIVE RECEPTOR ANTAGONIST OF ANGIOTENSIN AND ENDOTHELIN |
| US6548529B1 (en) | 1999-04-05 | 2003-04-15 | Bristol-Myers Squibb Company | Heterocyclic containing biphenyl aP2 inhibitors and method |
| TW200514783A (en) | 1999-09-22 | 2005-05-01 | Bristol Myers Squibb Co | Substituted acid derivatives useful as antiodiabetic and antiobesity agents and method |
| US6414002B1 (en) | 1999-09-22 | 2002-07-02 | Bristol-Myers Squibb Company | Substituted acid derivatives useful as antidiabetic and antiobesity agents and method |
| US6395767B2 (en) | 2000-03-10 | 2002-05-28 | Bristol-Myers Squibb Company | Cyclopropyl-fused pyrrolidine-based inhibitors of dipeptidyl peptidase IV and method |
| CN1630709A (en) | 2001-10-18 | 2005-06-22 | 百时美施贵宝公司 | Human glucagon-like-peptide-1 mimics and their use in the treatment of diabetes and related conditions |
| WO2004087679A1 (en) * | 2003-04-01 | 2004-10-14 | Aponetics Ag | 2, 4, 6-trisubstituted pyrimidine derivatives useful for the treatment of neoplastic and autoimmune diseases |
| US6995183B2 (en) | 2003-08-01 | 2006-02-07 | Bristol Myers Squibb Company | Adamantylglycine-based inhibitors of dipeptidyl peptidase IV and methods |
-
2005
- 2005-12-21 US US11/314,795 patent/US7589088B2/en active Active
- 2005-12-23 WO PCT/US2005/046750 patent/WO2006071762A2/en active Application Filing
- 2005-12-23 EP EP05855333.0A patent/EP1831180B1/en not_active Not-in-force
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003068757A1 (en) * | 2002-02-13 | 2003-08-21 | F. Hoffmann-La Roche Ag | Novel pyridin- and pyrimidin-derivatives |
Non-Patent Citations (2)
| Title |
|---|
| PETERS J.U. ET AL: "An aminomethylpyrimidine DPP-IV onhibitor with improved properties" BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, vol. 14, 5 July 2004 (2004-07-05), pages 3575-3578, XP002397580 * |
| PETERS J.U. ET AL: "Aminomethylpyrimidines as novel DPP-IV inhibitors" BIOORGANIC & MEDICINAL CHEMISTRY LETTERS, vol. 14, 22 March 2004 (2004-03-22), pages 1491-1493, XP002397579 * |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006119451A1 (en) * | 2005-05-04 | 2006-11-09 | Vertex Pharmaceuticals Incorporated | Pyrimidines and pyrazines useful as modulators of ion channels |
| US7858786B2 (en) | 2005-05-04 | 2010-12-28 | Vertex Pharmaceuticals Incoropated | Pyrimidines and pyrazines useful as modulators of ion channels |
| US7880008B2 (en) | 2005-05-31 | 2011-02-01 | Vertex Pharmaceuticals Incorporated | Heterocycles useful as modulators of ion channels |
| US8329702B2 (en) | 2005-05-31 | 2012-12-11 | Vertex Pharmaceuticals Incorporated | Heterocycles useful as modulators of ion channels |
| WO2007120702A2 (en) | 2006-04-11 | 2007-10-25 | Arena Pharmaceuticals, Inc. | Use of gpr119 receptor agonists for increasing bone mass and for treating osteoporosis, and combination therapy relating thereto |
| EP2253311A2 (en) | 2006-04-11 | 2010-11-24 | Arena Pharmaceuticals, Inc. | Use of GPR119 receptor agonists for increasing bone mass and for treating osteoporosis, as well as combination therapy relating thereto |
| US10064850B2 (en) | 2007-04-11 | 2018-09-04 | Omeros Corporation | Compositions and methods for prophylaxis and treatment of addictions |
| US11241420B2 (en) | 2007-04-11 | 2022-02-08 | Omeros Corporation | Compositions and methods for prophylaxis and treatment of addictions |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1831180A2 (en) | 2007-09-12 |
| EP1831180B1 (en) | 2015-07-29 |
| WO2006071762A3 (en) | 2006-11-23 |
| US20060142576A1 (en) | 2006-06-29 |
| US7589088B2 (en) | 2009-09-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7589088B2 (en) | Pyrimidine-based inhibitors of dipeptidyl peptidase IV and methods | |
| EP1836206B1 (en) | Azolopyrimidine-based inhibitors of dipeptidyl peptidase iv and methods | |
| EP2086975B1 (en) | 7,8-dihydro-1,6-naphthyridin-5(6h)-ones and related bicyclic compounds as inhibitors of dipeptidyl peptidase iv and methods | |
| US7521557B2 (en) | Pyrrolopyridine-based inhibitors of dipeptidyl peptidase IV and methods | |
| EP1553937B1 (en) | Glycinenitrile-based inhibitors of dipeptidyl peptidase iv | |
| US6670380B2 (en) | Pyridone inhibitors of fatty acid binding protein and method | |
| US6995183B2 (en) | Adamantylglycine-based inhibitors of dipeptidyl peptidase IV and methods | |
| US7488725B2 (en) | Pyrrolidinyl beta-amino amide-based inhibitors of dipeptidyl peptidase IV and methods | |
| US20030087843A1 (en) | O-pyrazole glucoside SGLT2 inhibitors and method of use |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005855333 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |