WO2006065480A2 - Tetrahydrocarbazoles as active agents for inhibiting vegf production by translational control - Google Patents
Tetrahydrocarbazoles as active agents for inhibiting vegf production by translational control Download PDFInfo
- Publication number
- WO2006065480A2 WO2006065480A2 PCT/US2005/042483 US2005042483W WO2006065480A2 WO 2006065480 A2 WO2006065480 A2 WO 2006065480A2 US 2005042483 W US2005042483 W US 2005042483W WO 2006065480 A2 WO2006065480 A2 WO 2006065480A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituted
- unsubstituted
- compound
- alkyl
- heterocyclic ring
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *C(*C(*)c1c(C2*)[n](*)c3c1c(*)c(*)c(*)c3*)C2*=C Chemical compound *C(*C(*)c1c(C2*)[n](*)c3c1c(*)c(*)c(*)c3*)C2*=C 0.000 description 6
- UMKJRDACNBGELF-UHFFFAOYSA-N BC(CCCC1)C1=O Chemical compound BC(CCCC1)C1=O UMKJRDACNBGELF-UHFFFAOYSA-N 0.000 description 1
- YJGKFYMZHYSDSA-UHFFFAOYSA-N Brc1ccc2[nH]c(C(CCC3)N(C4)Cc5c4cccc5)c3c2c1 Chemical compound Brc1ccc2[nH]c(C(CCC3)N(C4)Cc5c4cccc5)c3c2c1 YJGKFYMZHYSDSA-UHFFFAOYSA-N 0.000 description 1
- GFISDBXSWQMOND-UHFFFAOYSA-N COC(CC1)OC1OC Chemical compound COC(CC1)OC1OC GFISDBXSWQMOND-UHFFFAOYSA-N 0.000 description 1
- PQOXQVYPYLASPX-CZIZESTLSA-N Cc(cc1)cc2c1[nH]c1c2CCC/C1=N\CCN1CCOCC1 Chemical compound Cc(cc1)cc2c1[nH]c1c2CCC/C1=N\CCN1CCOCC1 PQOXQVYPYLASPX-CZIZESTLSA-N 0.000 description 1
- MARJSQOYNSPNPV-UHFFFAOYSA-N Fc(cc1)ccc1OC(CCC1)c2c1c1cc(Br)ccc1[nH]2 Chemical compound Fc(cc1)ccc1OC(CCC1)c2c1c1cc(Br)ccc1[nH]2 MARJSQOYNSPNPV-UHFFFAOYSA-N 0.000 description 1
- FLPWBPSIEMPYOY-UHFFFAOYSA-N NC(CCC1)c2c1c1cc(Br)ccc1[nH]2 Chemical compound NC(CCC1)c2c1c1cc(Br)ccc1[nH]2 FLPWBPSIEMPYOY-UHFFFAOYSA-N 0.000 description 1
- BWLLTVHXDXVUIQ-UHFFFAOYSA-N NC(CCC1)c2c1c1cc(Cl)ccc1[nH]2 Chemical compound NC(CCC1)c2c1c1cc(Cl)ccc1[nH]2 BWLLTVHXDXVUIQ-UHFFFAOYSA-N 0.000 description 1
- UZMYPCOHBHVFJE-UHFFFAOYSA-N O=C(CCC1)c2c1c1cc(Br)ccc1[nH]2 Chemical compound O=C(CCC1)c2c1c1cc(Br)ccc1[nH]2 UZMYPCOHBHVFJE-UHFFFAOYSA-N 0.000 description 1
- RLUDCSPDYQFHLI-UHFFFAOYSA-N O=C(c1c(C2)cccc1)N2C(CCC1)c2c1c(cc(cc1)Br)c1[nH]2 Chemical compound O=C(c1c(C2)cccc1)N2C(CCC1)c2c1c(cc(cc1)Br)c1[nH]2 RLUDCSPDYQFHLI-UHFFFAOYSA-N 0.000 description 1
- WSEJZRIZDQWMKQ-UHFFFAOYSA-N O=Cc1c(C=O)[s]cc1 Chemical compound O=Cc1c(C=O)[s]cc1 WSEJZRIZDQWMKQ-UHFFFAOYSA-N 0.000 description 1
- ODJMJEOJZGQCCD-UHFFFAOYSA-N OC(CCC1)c2c1c1cc(Br)ccc1[nH]2 Chemical compound OC(CCC1)c2c1c1cc(Br)ccc1[nH]2 ODJMJEOJZGQCCD-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/34—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having five-membered rings with one oxygen as the only ring hetero atom, e.g. isosorbide
- A61K31/343—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having five-membered rings with one oxygen as the only ring hetero atom, e.g. isosorbide condensed with a carbocyclic ring, e.g. coumaran, bufuralol, befunolol, clobenfurol, amiodarone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/4035—Isoindoles, e.g. phthalimide
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/407—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with other heterocyclic ring systems, e.g. ketorolac, physostigmine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/415—1,2-Diazoles
- A61K31/4162—1,2-Diazoles condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4184—1,3-Diazoles condensed with carbocyclic rings, e.g. benzimidazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4164—1,3-Diazoles
- A61K31/4188—1,3-Diazoles condensed with other heterocyclic ring systems, e.g. biotin, sorbinil
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/42—Oxazoles
- A61K31/424—Oxazoles condensed with heterocyclic ring systems, e.g. clavulanic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/4245—Oxadiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/08—Indoles; Hydrogenated indoles with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, directly attached to carbon atoms of the hetero ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/56—Ring systems containing three or more rings
- C07D209/80—[b, c]- or [b, d]-condensed
- C07D209/90—Benzo [c, d] indoles; Hydrogenated benzo [c, d] indoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
Definitions
- the present invention relates to methods, compounds, and compositions for inhibiting angiogenesis. More particularly, the present invention relates to methods, compounds, and compositions for inhibiting VEGF production.
- VEGF Vascular Endothelial Growth Factor
- VEGF also known as VEGF-A
- VEGF-A was initially identified for its ability to induce vascular permeability and to promote vascular endothelial cell proliferation (8-10).
- VEGF is encoded by a single gene that gives rise to four isoforms by alternative splicing (11). All four isoforms share the same unusually long and GC rich 5'-UTR, as well as a 3'-UTR that includes multiple RNA stability determinants.
- the receptors VEGFR-2 also known as KDR or FIk-I
- VEGFR-I previously known as Fltl
- the highly specific VEGFR-2 receptor is expressed on endothelial cells.
- VEGFR-I inhibits growth either by acting as a decoy or by suppressing signaling pathways through VEGFR-2 (15).
- angiogenesis regulators including VEGF, the FGFs, PDGF, TGF, EGF, IL-8, IL-6, and the angiopoietins, etc.
- VEGF and its receptor have been demonstrated to have a central role in tumor angiogenesis, especially in the early stages of tumor growth (19). Indeed, increased levels of VEGF expression have been correlated with microvessel density in primary tumor tissues (20). Moreover, increased levels of the VEGF transcript are found in virtually all of the common solid tumors (21).
- VEGF null embryonic stem cells showed a dramatically reduced ability to form tumors in nude mice (23).
- Direct evidence for the involvement of VEGF in tumorigenesis was demonstrated by using specific antibodies against VEGF in human xenografts implanted in nude mice (24, 25). In these studies, the inhibition of tumor growth correlated positively with decreased vessel formation in the antibody-treated tumors.
- VEGF vascular endothelial growth factor
- CNV retinal/choroidal neovascularization
- studies using transgenic mice demonstrated that overexpression of VEGF in retinal pigment epithelial cells or photoreceptor cells results in choroidal or retinal neovascularization (31, 32).
- neutralizing antibodies, soluble receptor, receptor antagonists, or siRNA have proven efficacious in reducing VEGF- mediated blood vessel formation in animal models and in the clinic (33, 34-37).
- VEGF expression is regulated by a number of factors and agents including cytokines, growth factors, steroid hormones and chemicals, and mutations that modulate the activity of oncogenes such as ras or the tumor suppressor gene VHL (38, 39). Nevertheless, hypoxia is the most significant physiologicsignal for regulating VEGF expression. Hypoxia results in enhanced VEGF expression by increasing both the transcription rate and stability of the VEGF transcript (40-42). Hypoxia-inducible factor l ⁇ (HIF- lot) is a transcription factor that increases VEGF gene expression in cells undergoing hypoxia by binding to the hypoxia response element (HRE) located in the VEGF promoter (43, 44).
- HRE hypoxia response element
- VEGF mRNA stability is also greatly enhanced as a consequence of the binding of factors to elements in the 3'-UTR (45).
- translation initiation of the VEGF transcript is uniquely regulated. Under hypoxic conditions, translation of most cellular transcripts mediated by cap-dependent translation initiation process is greatly impaired (46). Initiation of translation of the VEGF mRNA, however, is unique under hypoxic conditions in that it is mediated via an internal ribosome entry site (IRES) within the VEGF 5'UTR (41, 42, 47, 48).
- IRS internal ribosome entry site
- tumor growth can be inhibited by the prevention of neovascularization (26, 49).
- Tumor vessels are generally immature and constantly undergo remodeling (1, 50).
- Active and aberrant angiogenesis is the result of a disruption in the normal balance of proangiogenic and anti-angiogenic factors, including various cytokines, growth factors and steroid hormones.
- proangiogenic and anti-angiogenic factors including various cytokines, growth factors and steroid hormones.
- VEGF and its receptor are most attractive (1, 12).
- VEGF is a valid target for the development of tumor therapy. Indeed, a number of clinical trials are underway using VEGF inhibitors (17, 25).
- VEGF vascular endothelial growth factor
- siRNA small interfering RNAs directed against murine VEGF significantly inhibited ocular neovascularization after laser photocoagulation in a mouse model (58).
- VEGF activity has been used to inhibit VEGF activity, including (1) neutralization of VEGF activity by using a specific antibody, soluble VEGF receptor or aptamer oligos against the VEGF /VEGFR interaction (24, 26, 27, 49, 51, 59, 60); (2) inhibition of VEGFR mediated signal transduction by specific small molecule tyrosine kinase inhibitors (52, 61, 62); and (3) inhibition of VEGF/VEGFR expression by using antisense, siRNA or ribozyme (58, 63-65). Although all of these approaches show significant inhibition of angiogenesis in vivo, they all possess significant limitations.
- VEGF vascular endothelial growth factor
- compounds of Formulas (I) to (VIII) are provided which are useful in the inhibition of VEGF production and/or in the inhibition of angiogenesis, and/or in the treatment of cancer, diabetic retinopathy, rheumatoid arthritis, psoriasis, atherosclerosis, obesity, chronic inflammation or exudative macular degeneration.
- methods are provided for the inhibition of VEGF production and/or the inhibition of angi ⁇ genesis, and/or the treatment of cancer, diabetic retinopathy, rheumatoid arthritis, psoriasis, atherosclerosis, obesity, chronic inflammation or exudative macular degeneration using the compounds described herein.
- the invention is directed to methods for inhibiting VEGF production comprising administering a VEGF-inhibiting amount of at least one compound of the invention to a subject in need thereof.
- methods for inhibiting angiogenesis comprising administering an anti-angiogenic amount of at least one compound of the invention to a subject in need thereof.
- methods for treating cancer, diabetic retinopathy, rheumatoid arthritis, psoriasis, atherosclerosis, obesity, chronic inflammation or exudative macular degeneration comprising administering a therapeutically effective amount of at least one compound of the invention to a subject in need thereof.
- Embodiment 1 A method for inhibiting VEGF production in a subject, comprising administering a VEGF-inhibiting amount of a compound selected from the group consisting of the compounds of Formula (I) to Formula (VIII), or an enantiomer, a diastereomer, a pharmaceutically acceptable salt, a prodrug, a solvate or a mixture thereof to a subject in need thereof.
- Embodiment 2 A method for inhibiting angiogenesis in a subject, comprising administering an anti-angiogenic amount of a compound selected from the group consisting of the compounds of Formula (I) to Formula (VIII), or an enantiomer, a diastereomer, a pharmaceutically acceptable salt, a prodrug, a solvate or a mixture thereof to a subject in need thereof.
- Embodiment 3 A method for treating cancer in a subject, comprising administering a therapeutically effective amount of a compound selected from the group consisting of the compounds of Formula (I) to Formula (VIII), or an enantiomer, a diastereomer, a pharmaceutically acceptable salt, a prodrug, a solvate or a mixture thereof to a subject in need thereof.
- a compound selected from the group consisting of the compounds of Formula (I) to Formula (VIII), or an enantiomer, a diastereomer, a pharmaceutically acceptable salt, a prodrug, a solvate or a mixture thereof to a subject in need thereof.
- a method for treating diabetic retinopathy in a subject comprising administering a therapeutically effective amount of a compound selected from the group consisting of the compounds of Formula (I) to Formula (VIII), or an enantiomer, a diastereomer, a pharmaceutically acceptable salt, a prodrug, a solvate or a mixture thereof to a subject in need thereof.
- Embodiment 5 A method for treating exudative macular degeneration in a subject, comprising administering a therapeutically effective amount of a compound selected from the group consisting of the compounds of Formula (I) to Formula (VIII), or an enantiomer, a diastereomer, a pharmaceutically acceptable salt, a prodrug, a solvate or a mixture thereof to a subject in need thereof.
- Embodiment 6 A method for treating rheumatoid arthritis in a subject, comprising administering a therapeutically effective amount of a compound selected from the group consisting of the compounds of Formula (I) to Formula (VIII), or an enantiomer, a diastereomer, a pharmaceutically acceptable salt, a prodrug, a solvate or a mixture thereof to a subject in need thereof.
- Embodiment 7 A method for treating psoriasis in a subject, comprising administering a therapeutically effective amount of a compound selected from the group consisting of the compounds of Formula (I) to Formula (VIII), or an enantiomer, a diastereomer, a pharmaceutically acceptable salt, a prodrug, a solvate or a mixture thereof to a subject in need thereof.
- Embodiment 8 A method for atherosclerosis in a subject, comprising administering a therapeutically effective amount of a compound selected from the group consisting of the compounds of Formula (I) to Formula (VIII), or an enantiomer, a diastereomer, a pharmaceutically acceptable salt, a prodrug, a solvate or a mixture thereof to a subject in need thereof.
- Embodiment 9 A method for treating obesity in a subject, comprising administering a therapeutically effective amount of a compound selected from the group consisting of the compounds of Formula (I) to Formula (VIII), or an enantiomer, a diastereomer, a pharmaceutically acceptable salt, a prodrug, a solvate or a mixture thereof to a subject in need thereof.
- Embodiment 10 A method for treating chronic inflammation in a subject, comprising administering a therapeutically effective amount of a compound selected from the group consisting of the compounds of Formula (I) to Formula (VIII), or an enantiomer, a diastereomer, a pharmaceutically acceptable salt, a prodrug, a solvate or a mixture thereof to a subject in need thereof.
- Embodiment 11 A method of selectively inhibiting VEGF in cells comprising exposing the cells to an effective amount of at least one compound of Formula (I) to Formula (VIII), or an enantiomer, a diastereomer, a pharmaceutically acceptable salt, a prodrug, a solvate or a mixture thereof, under conditions and for a time sufficient to selectively inhibit VEGF therein.
- Embodiment 12 A method of selectively inhibiting VEGF in cells which comprises exposing the cells to an effective amount of a composition including a pharmaceutically acceptable excipient and at least one compound of Formula (I) to Formula (VIII), or an enantiomer, a diastereomer, a pharmaceutically acceptable salt, a prodrug, a solvate or a mixture thereof, under conditions and for a time sufficient to selectively inhibit VEGF therein.
- Embodiment 13 A method for treating or preventing a disease whose onset or progress is aided by abberant VEGF production, which comprises administering to a subject in need thereof a therapeutically effective amount of at least one compound of Formula (I) to Formula (VIII), or an enantiomer, a diastereomer, a pharmaceutically acceptable salt, a prodrug, a solvate or a mixture thereof, under conditions and for a time sufficient to selectively inhibit VEGF therein.
- Embodiment 14 A method for inhibiting abberant angiogenesis, which comprises administering to a subject in need thereof a therapeutically effective amount of at least one compound of Formula (I) to Formula (VIII), or an enantiomer, a diastereomer, a pharmaceutically acceptable salt, a prodrug, a solvate or a mixture thereof, under conditions and for a time sufficient to selectively inhibit VEGF therein.
- Embodiment 15 A pharmaceutical composition comprising a compound selected from the group consisting of the compounds of Formula (I) to Formula (VIII), or enantiomer, a diastereomer, a pharmaceutically acceptable salt, a prodrug, a solvate or a mixture thereof and a pharmaceutically acceptable excipient.
- a VEGF-inhibiting composition comprising at least one compound of Formula (I) to Formula (VIII), or an enantiomer, a diastereomer, a pharmaceutically acceptable salt, a prodrug, a solvate or a mixture thereof.
- the VEGF-inhibiting composition can include a pharmaceutically acceptable excipient.
- Embodiment 17 The use of a compound of Formula (I) through Formula (VIII) for the preparation of a pharmaceutical composition.
- Embodiment 18 The use of a compound selected from Compounds 191 through 239.
- VEGF post-transcriptionally have been identified, and methods for their use provided.
- Compounds of the present invention may be useful in the inhibition of VEGF production and/or in the inhibition of angiogenesis and/or in the treatment or prevention of diseases whose onset or progress is aided by abberant VEGF production or abberant angiogenesis.
- certain compounds of the invention may be include a chiral center, and as such may exist as racemic mixtures or as enantiomerically pure compositions.
- the compounds may exist as R or S isomers in enantiomerically pure compositions.
- Figure 1 is a graph showing a dose-response ELISA assay and, in parallel, a dose- response cytotoxicity assay for a typical compound of the present invention. Dose-response curves were plotted using percentage inhibition of VEGF post-transcriptional expression versus concentration of a compound of the present invention.
- VEGF Vascular Endothelial Growth Factor
- angiogenesis vascular Endothelial Growth Factor
- compounds that inhibit the expression of VEGF post-transcriptionally have been identified and methods for their use provided.
- the compounds of the invention have low nonomolar activity for the inhibition of VEGF expression.
- compounds are provided which are useful in the inhibition of VEGF production and/or in the inhibition of angiogenesis, and/or in the treatment of cancer, diabetic retinopathy, rheumatoid arthritis, psoriasis, atherosclerosis, obesity, chronic inflammation or exudative macular degeneration.
- the compounds of the invention specifically inhibit VEGF production.
- the compounds of the invention inhibit VEGF production as well as that of other angiogenesis factors such as FGF -2.
- pan-angiogenic inhibitors may be preferred for the treatment of ocular neovascular disorders.
- certain compounds of the invention may be include a chiral center, and as such may exist as racemice mixtures or as enantiomerically pur compositions.
- the compounds may exist as R or S isomers in enantiomerically pure compositions.
- enantiomerically pure refers to compositions consisting substantially of a single isomer, preferably consisting of 75%, 80%, 85%, 90%, 92%, 95%, 98%, 99%, or 100% of a single isomer.
- Preferred compounds of the present invention include those of Formula (I) as shown below:
- X is -NR 9 R 10 , -N(alkyl)-C(O)-aryl, -N(alkyl)-C(O)-halogen substituted aryl, oxo, OR 9 , H, substituted or unsubstituted phenylaminocarbonyl, substituted or unsubstituted phenyl, oxime, substituted or unsubstituted alkoxycarbonyl, hydroxycarbonyl (i.e., -COOH), or a substituted or unsubstituted heterocyclic ring;
- R 9 and R 10 are each independently H, substituted or unsubstituted C 1-6 alkyl, substituted or unsubstituted C 1 -C 6 alcohol, substituted or unsubstituted carbonyl (i.e., -C(O)H), substituted or unsubstituted mono- or bi- cyclic cycloalkyl, substituted or unsubstituted mono- or bi- cyclic heterocyclic ring, substituted or unsubstituted aryl, substituted or unsubstituted alkenyl, substituted or unsubstituted sulfonyl, substituted or unsubstituted arylalkyl, substituted or unsubstituted heterocycloalkyl, substituted or unsubstituted aminothiocarbonyl wherein at least one of R 9 and R 10 is H, or R 9 and R 10 together with the atom to which they are attached form a mono- or bi-cyclic heterocyclic ring, where
- n is 0, 1 or 2, wherein when n is 0 then R 2 is absent;
- R 4 , R 5 , R 6 and R 7 are each independently -H, -OH, substituted or unsubstituted C 1-6 alkyl, substituted or unsubstituted carbonyl (i.e., -C(O)H), substituted or unsubstituted alkoxy, halo, haloalkyl, haloalkoxy, nitro, cyano, substituted or unsubstituted heterocyclic ring, substituted or unsubstituted amino, substituted or unsubstituted phenyl, substituted or unsubstituted phenoxy, substituted or unsubstituted cycloalkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, or substituted or unsubstituted alkoxycarbonyl, or hydroxycarbonyl;
- W is N, O, or S
- R 8 is H, C 1-3 alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted carbonyl (i.e. -C(O)H), or together with X forms a substituted or unsubstituted 5- 11 membered mono- or bi- cyclic heterocyclic ring, with the proviso that when W is O or S, R 8 is absent; and
- alkyl denotes an optionally substituted, branched or straight- chained saturated hydrocarbon radical.
- alkyl include a C 1 to C 4 alkyl, a C 1 to C 8 alkyl, or C 1 to C 12 alkyl.
- alkenyl denotes an optionally substituted, branched or straight-chained unsaturated hydrocarbon radical having at least one carbon - carbon double bond.
- alkynyl denotes an optionally substituted, branched or straight-chained aliphatic hydrocarbon radical having at least one carbon - carbon triple bond.
- aromatic ring denotes an optionally substituted, monocyclic aromatic hydrocarbon ring.
- the aromatic ring may be a part of an aromatic bicyclic ring system, such as napthyl.
- the ring to which the aromatic ring is attached in the bicyclic ring system may be an aliphatic ring.
- aryl denotes an optionally substituted, stable 5 to 7 membered monocyclic hydrocarbon radical or a stable 8 to 11 membered bicyclic aromatic hydrocarbon radical.
- cycloalkyl denotes the radical of an optionally substituted, aliphatic hydrocarbon ring having three to ten carbon atoms.
- cycloalkylalkyl denotes an optionally substituted alkyl radical having a cycloalkyl substituent.
- heteroatom denotes an atom that is any element other than carbon or hydrogen.
- heterocycle and “heterocyclic ring” denote an optionally substituted stable 5 to 7 membered monocyclic hydrocarbon ring or an optionally substituted stable 8 to 11 membered bicyclic hydrocarbon ring, in which one to four carbon atoms have been replaced with a heteroatom selected from the group consisting of N, O, and S.
- substitution may take place on either ring.
- the heterocycle may be saturated or unsaturated, and aliphatic or aromatic.
- R 26 is H.
- the phrase "pharmaceutically acceptable salts” refers to those salts derived from organic and inorganic acids such as: acetic, lactic, citric, cinnamic, tartaric, succinic, fumaric, maleic, malonic, mandelic, malic, oxalic, propionic, hydrochloric, hydrobromic, phosphoric, nitric, sulfuric, glycolic, pyruvic, methanesulfonic, ethanesulfonic, toluenesulfonic, salicylic, benzoic, and similarly acceptable acids.
- organic and inorganic acids such as: acetic, lactic, citric, cinnamic, tartaric, succinic, fumaric, maleic, malonic, mandelic, malic, oxalic, propionic, hydrochloric, hydrobromic, phosphoric, nitric, sulfuric, glycolic, pyruvic, methanesulfonic, ethane
- aminothiocarbonyl denotes a radical in which an amino group is bonded to the carbon of a thiocarbonyl group.
- a thiocarbonyl group is one in which a carbon atom is connected to a sulfur atom through a double bond. The point of attachment of the aminothiocarbonyl radical to the indicated atom is the carbon atom of the thiocarbonyl moiety.
- substituted denotes that a moiety has one or more of its hydrogen atoms replaced by one or more substituents.
- substituents include, but are not limited to:
- R 30 and R 31 are each independently H or alkyl
- R 30 and R 31 together with the nitrogen to which they are bound may form a 5-7 membered nitrogen containing heterocyclic ring.
- X is phenyl. In another embodiment of Formula (I), X is substituted phenyl. In a preferred embodiment of compounds of Formula (I), where X is substituted phenyl, the substituted phenyl may also bear one or more R a groups alone or in combination with those moieties recited for the term "substituted" above.
- R 3 is independently selected from halogen; -OR d group; a 5 to 6 membered heterocycle; a 5 to 6 membered heteroaryl; or a C 1 to C 6 alkyl group, wherein the alkyl group is optionally substituted with one or more independently selected halogen groups;
- R b is hydroxyl; an amino; an alkylamino, wherein the alkylamino is optionally substituted with a hydroxyl, an amino, an alkylamino, a C 1 to C 4 alkoxy, a 3 to 12 membered heterocycle optionally substituted with at least one independently selected C 1 to C 6 alkyl, oxo, - C(O)O-R cC , or a 5 to 12 membered heteroaryl optionally substituted with a C 1 to C 4 alkyl; a C 1 to C 4 alkoxy; a C 2 to C 8 alkenyl; a C 2 to Cg alkynyl; a C 6 to
- the present invention provides compounds, including a preferred class of compounds within Formula (I), including those compounds of Formula (Ia) shown below:
- Formula (Ia) an enantiomer, a diastereomer, a pharmaceutically acceptable salt, a prodrug, a solvate or a mixture thereof, wherein:
- X is NR 9 R 10 , -N(alkyl)-C(O)-aryl, -N(alkyl)-C(O)-halogen substituted aryl, oxo, OR 9 , H, substituted or unsubstituted phenylaminocarbonyl, substituted or unsubstituted phenyl, oxime, substituted or unsubstituted alkoxycarbonyl, hydroxycarbonyl or a substituted or unsubstituted heterocyclic ring;
- R 9 and R 10 are each independently H, substituted or unsubstituted C 1-6 alkyl, substituted or unsubstituted carbonyl (i.e., -C(O)H), substituted or unsubstituted mono- or bi- cyclic cycloalkyl, substituted or unsubstituted mono- or bi- cyclic heterocyclic ring, substituted or unsubstituted aryl, substituted or unsubstituted sulfonyl, substituted or unsubstituted arylalkyl, substituted or unsubstituted heterocycloalkyl, wherein at least one of R 9 and R 10 is H, or R 9 and R 10 together with the atom to which they are attached form a mono- or bi-cyclic heterocyclic ring, wherein at least one ring contains one or two heteroatoms;
- R 1 , R 2 and R 3 are each independently H or alkyl, wherein R 1 may optionally form a substituted or unsubstituted 5-1 1 membered mono- or bi- heterocyclic ring with X;
- n is 0, 1 or 2, wherein when n is 0 then R 2 is absent;
- R 4 R 6 and R 7 are each independently H, OH, C 1-6 alkyl, substituted or unsubstituted carbonyl (i.e. -C(O)H), halo, haloalkyl, haloalkoxy, cyano, substituted or unsubstituted mono-cyclic heterocyclic ring, substituted or unsubstituted amino, substituted or unsubstituted phenyl, substituted or unsubstituted phenoxy, substituted or unsubstituted alkenyl, or substituted or unsubstituted alkynyl;
- R 5 are each independently H, OH, substituted or unsubstituted C 2-6 alkyl, substituted or unsubstituted alkoxy, substituted or unsubstituted carbonyl (i.e. -C(O)H), halo, haloalkyl, haloalkoxy, cyano, substituted or unsubstituted heterocyclic ring, substituted or unsubstituted amino, substituted or unsubstituted phenyl, substituted or unsubstituted phenoxy, substituted or unsubstituted cycloalkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, or substituted or unsubstituted alkoxycarbonyl, or hydroxycarbonyl;
- R 8 is H, C i- 3 alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted carbonyl (i.e. -C(O)H), or R together with X forms a substituted or unsubstituted 5-11 membered mono- or bi- cyclic heterocyclic ring, with the proviso that when X, R 9 and R 10 form an unsubstituted pyrrole, then R 5 is not bromine when R is H, and R may together with X form a substituted or unsubstituted heterocyclic ring; and
- W is N, O, or S, with the proviso that when W is O or S, R 8 is absent.
- the present invention provides VEGF-inhibiting compounds, including a compound of Formula (Ib):
- Formula (Ib) an enantiomer, a diastereomer, a pharmaceutically acceptable salt, a prodrug, a solvate or a mixture thereof, wherein:
- X is NR 9 R 10 , -N(alkyl)-C(O)-aryl, -N(alkyl)-C(O)-halogen substituted aryl, oxo, OR 9 , H, substituted or unsubstituted phenylaminocarbonyl, substituted or unsubstituted phenyl, oxime, substituted or unsubstituted alkoxycarbonyl, hydroxycarbonyl, or a substituted or unsubstituted heterocyclic ring;
- R 9 and R 10 are each independently H, substituted or unsubstituted C 1-6 alkyl, substituted or unsubstituted carbonyl (i.e., -C(O)H), substituted or unsubstituted mono- or bi- cyclic cycloalkyl, substituted or unsubstituted mono- or bi- cyclic heterocyclic ring, substituted or unsubstituted aryl, substituted or
- R 9 and R 1 together with the atom to which they are attached form a mono- or bi-cyclic heterocyclic ring, wherein at least one ring contains one or two heteroatoms;
- R 1 , R 2 and R 3 are each independently H or alkyl, wherein R 1 may optionally form a substituted or unsubstituted 5-11 membered mono- or bi- heterocyclic ring with X;
- n 0, 1 or 2, wherein when n is 0 then R 2 is absent;
- R 4 5 R 5 , R 6 and R 7 are each independently H, OH, substituted or unsubstituted C 1-
- R 8 is H, C 1-3 alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted carbonyl (i.e., -C(O)H), with the proviso that when X, R 9 and R 10 form an unsubstituted pyrrole, then R 5 is not bromine when R 8 is H;
- W is N, O or S, with the proviso that when W is O or S, R 8 is absent.
- X is selected from the following:
- X is NR 9 R 10 .
- Preferable substituents for NR 9 R 10 include the following:
- X is -OR 9 , wherein R 9 is phenol.
- R and R ° may be substituted with one or more of the same or different: halogen, C 1-6 alkyl, C 1-6 alcohol, hydroxyl, cyano, oxo, alkoxy, carbonyl (i.e., -C(O)H) , substituted or unsubstituted alkoxycarbonyl, haloalkyl, haloalkoxy, substituted or unsubstituted heterocyclic ring, substituted or unsubstituted cycloalkyl, substituted or unsubstituted amino, or substituted or unsubstituted phenyl.
- halogen C 1-6 alkyl, C 1-6 alcohol, hydroxyl, cyano, oxo, alkoxy, carbonyl (i.e., -C(O)H) , substituted or unsubstituted alkoxycarbonyl, haloalkyl, haloalkoxy, substituted or unsubstituted heterocyclic
- R and R 10 may be substituted with one or more of the same or different halogen, methyl, isopropyl, ethyl, -(CH 2 ) 3 CH 3 or -C(CH 3 ) 3> .
- R 1 is H or methyl.
- R 2 and R 3 are H.
- R 4 is H, Br, or Cl.
- R 5 is Br, Cl, methyl, trifluoromethyl, trifluoromethoxy, methoxy, methoxycarbonyl, hydroxycarbonyl, morpholino, pyrrolidino, or -C(O)NH 2 .
- R is H or Br.
- R 7 is H or Br.
- R 8 is H, methyl, acyl, or t-butoxycarbonyl.
- R 5 is halogen, and R 1 , R 2 , and R 3 are H or -OH.
- R 5 is Br, R 1 is H or -OH, and R 2 , R 3 , R 4 , R 6 , R 7 and R 8 are H.
- R 5 is halogen
- n is 1 or 2
- R 1 , R 2 , and R 3 are H or -OH.
- R 5 is -CF 3 , Br or Cl
- R 1 is H or -OH
- n is 1 or 2
- R 2 , R 3 , R 4 , R 6 , R 7 and R 8 are H.
- X is phenyl substituted with a substituent selected from the group consisting of:
- X is -NR 9 R 10 ;
- R 9 and R 1 are each independently H, substituted carbonyl, substituted or unsubstituted mono- or bi- cyclic cycloalkyl, substituted or unsubstituted mono- or bi- cyclic heterocyclic ring, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted heterocycloalkyl, wherein at least one of R 9 and R 10 is H, or R 9 and R 10 together with the atom to which they are attached form a mono- or bi-cyclic heterocyclic ring, wherein at least one ring contains one or two heteroatoms.
- R 9 is H
- R 10 is a substituted or unsubstituted aryl.
- compounds of Formulas (I), (Ia), and (Ib) are provided, wherein X is -N(alkyl)-C(O)-aryl or -N(alkyl)-C(O)-halogen substituted aryl.
- -N(alkyl)-C(O)-aryl is -N(C 1 to C 6 alkyl)-C(O)-(C 6 to C 8 aryl).
- -N(alkyl)-C(O)- halogen substituted aryl is -N(C 1 to C 6 alkyl)-C(O)-(C 6 to Cg halogen substituted aryl).
- compounds of Formulas (I), (Ia), and (Ib) are provided, wherein X is not -N(alkyl)-C(O)-aryl.
- a more preferred embodiment of the invention provides for compounds of Formula (I) where X is substituted or unsubstituted phenyl.
- X is substituted or unsubstituted alkoxycarbonyl, or a hydroxycarbonyl.
- X is a substituted or unsubstituted heterocyclic ring.
- Other preferred embodiments of compounds of Formula (I) include compounds of Formula (Ia) and compounds of Formula (Ib).
- R 5 is Br or Cl.
- Another preferred class of compounds include those compounds of Formula (II) as shown below:
- R 5 is halo, C1-C 3 haloalkyl, or substituted or unsubstituted C 1 -C 6 alkyl;
- R 11 and R 12 are each, independently, H, halo, substituted or unsubstituted C 1 -C 6 alkyl, substituted or unsubstituted C 1 -C 6 alkoxy, phenoxy, and substituted or unsubstituted phenyl; or
- R 11 and R 12 taken together with the atom or atoms to which they are attached, may optionally form a five or six membered carbocyclic or heterocyclic ring, containing, including the atoms to which R 11 and R 12 are attached, one to three heteroatoms selected from the group consisting of N, O, and S.
- Another preferred class of compounds includes those of Formula (III):
- R 5 is halo and substituted or unsubstituted C 1 -C 6 alkyl; and (b) R 11 is H, halo, substituted or unsubstituted C 1 -C 6 alkyl, substituted or unsubstituted phenoxy, substituted or unsubstituted C 1 -C 6 alkoxy, and substituted or unsubstituted phenyl.
- R 11 is H, halo, substituted or unsubstituted C 1 -C 6 alkoxy, or substituted or unsubstituted C 1 -C 6 alkyl.
- preferred compounds include those compounds of Formula
- R 5 is halo or substituted or unsubstituted C 1 -C 6 alkyl
- R 14 and R 15 are each, independently, H, halo, substituted or unsubstituted phenoxy, cyano, substituted or unsubstituted C 1 -C 6 alkyl; or
- R 14 and R 15 taken together with the atom or atoms to which they are attached, may optionally form a five or six membered heterocyclic ring, containing, including the atoms to which R 14 and R 15 are attached, one to three heteroatoms selected from the group consisting of N, O, and S.
- preferred compounds include those of Formula (VI):
- R 5 is halo or substituted or unsubstituted CpC 6 alkyl
- Q is N, O, or S, with the proviso that when Q is O or S, R 17 is absent;
- R 17 is H or alkyl
- R 18 and R 19 are each, independently, selected from the group consisting of H, halo, substituted or unsubstituted C 1 -C 6 alkyl, substituted or unsubstituted phenyl, and nitro; or
- R l8 and R 19 taken together with the atoms to which they are attached, may optionally form a carbocyclic aromatic ring.
- Still other preferred compounds include those of Formula (VII):
- R 5 is halo or C 1 -C 6 alkyl
- R 20 is H or oxo
- R 21 and R 22 are H;
- R 21 and R 22 taken together with the atoms to which they are attached, may optionally form a carbocyclic aromatic ring, or a five or six membered heterocyclic ring, the heterocyclic ring containing, including the atoms to which R 21 and R 22 are attached, one to three heteroatoms selected from the group consisting of N, O, and S; and
- R 23 is H or oxo.
- compounds of the present invention include those of
- R 5 is halo or C i -C 6 alkyl
- R 23 and R 24 are selected from the group consisting of H, substituted of unsubstituted alkoxycarbonyl, substituted or unsubstituted alkylcarbonyl, formyl, cyano, and substituted or unsubstituted phenylaminoalkyl; or R 23 and R 24 , taken together with the atoms to which they are attached, may optionally form a carbocyclic aromatic ring, or a five or six membered heterocyclic ring, the heterocyclic ring containing, including the atoms to which R 23 and R 24 are attached, one to three heteroatoms selected from the group consisting of N, O, and S; and
- R is substituted or unsubstituted carbonyl.
- Formula (VIII) are provided with the proviso that the compounds are not any of Compounds 156 through 188.
- a compound is selected from Compounds 191 through 239, or an enantiomer, a diastereomer, a pharmaceutically acceptable salt, a prodrug, a solvate or a mixture thereof.
- methods are provided for the inhibition of VEGF production and/or the inhibition of angiogenesis, and/or the treatment of cancer, diabetic retinopathy, rheumatoid arthritis, psoriasis, atherosclerosis, obesity, chronic inflammation or exudative macular degeneration using one or more compounds of the present invention.
- the present invention relates to a method for treating aberrant angiogenesis, including administering to a mammal in need thereof a compound of the invention.
- the present invention relates to a method for treating overexpression of vascular endothelial growth factor, including administering to a mammal in need thereof a compound of the invention.
- the present invention relates to a method for treating cancer, including administering to a mammal suffering from such a condition a compound of the invention.
- the present invention relates to a method for treating ocular neovascular disorders, including administering to a mammal suffering from such a condition a compound of the invention.
- the invention is directed to methods for inhibiting VEGF in cells, which methods include exposing the cells to an effective amount of at least one compound of the invention.
- a compound of the present invention may be administered to a subject in need of inhibition of VEGF production.
- Compounds of the present invention can be administered neat or can be formulated with a pharmaceutically acceptable excipient.
- VEGF vascular endothelial growth factor
- inhibiting VEGF By the terms “inhibiting VEGF”, “inhibition of VEGF”, and the like, it is meant that the post-transcriptional expression or production of VEGF in cells treated with a compound of the present invention for a sufficient period of time is lower in relation to untreated cells. As such, VEGF activity (e.g., its pro-angiogenic activity) would also be reduced.
- compounds of the present invention inhibit VEGF expression in cells during culture by an amount at least 10% relative to untreated cells. In one embodiment, compounds of the present invention inhibit VEGF expression in cells by an amount at least about 25% relative to untreated cells. In another embodiment, the compounds inhibit VEGF expression in cells by an amount at least about 50% relative to untreated cells.
- the compounds inhibit VEGF expression in cells by an amount of at least about 75% relative to untreated cells.
- methods for inhibiting abberant angiogenesis include administering to a subject in need thereof a therapeutically effective amount of at least one compound of the invention.
- methods for treating or preventing a disease whose onset or progress is aided by abberant VEGF production include administering to a subject in need thereof a therapeutically effective amount of at least one compound of the invention.
- the disease is selected from cancer, diabetic retinopathy, rheumatoid arthritis, psoriasis, atherosclerosis, obesity, chronic inflammation and exudative macular degeneration. Without intending to be limited by theory, it is believed that the methods of the present invention act through a combination of mechanisms that modulate the activity of VEGF.
- one or more compounds may be administered to the subject via any drug delivery route known in the art.
- Specific exemplary administration routes include oral, ocular, rectal, buccal, topical, nasal, opthamalic, subcutaneous, intramuscular, intravenous (bolus and infusion), intracerebral, transdermal and pulmonary.
- therapeutically effective amount refers to an amount of a pharmaceutical agent to treat, meliorate or prevent the identified disease or condition, or to exhibit a detectable therapeutic or inhibitory effect.
- the effect can be detected by, for example, assays disclosed in the following examples.
- the precise effective amount for a subject will depend upon the subject's body weight, size, and health; the nature and extent of the condition; and the therapeutic or combination of therapeutics selected for administration.
- Therapeutically effective amounts for a given situation can be determined by routine experimentation that is within the skill and judgment of the clinician.
- the therapeutically effective amount can be estimated initially either in cell culture assays, e.g., of neoplastic cells, or in animal models, usually rats, mice, rabbits, dogs, or pigs.
- the animal model may also be used to determine the appropriate concentration range and route of administration. Such information can then be used to determine useful doses and routes for administration in humans.
- Therapeutic/prophylactic efficacy and toxicity may be determined by standard pharmaceutical procedures in cell cultures or experimental animals, e.g., ED 50 (the dose therapeutically effective in 50% of the population) and LD 50 (the dose lethal to 50% of the population).
- the dose ratio between therapeutic and toxic effects is the therapeutic index, and it can be expressed as the ratio, ED 50 ZLDs O .
- compositions that exhibit large therapeutic indices are preferred.
- the data obtained from cell culture assays and animal studies may be used in formulating a range of dosage for human use.
- the dosage contained in such compositions is preferably within a range of circulating concentrations that include an ED 50 with little or no toxicity.
- the dosage may vary within this range depending upon the dosage form employed, sensitivity of the patient, and the route of administration.
- the concentration-biological effect relationships observed with regard to the compound(s) of the present invention indicate an initial target plasma concentration ranging from approximately 5 ⁇ g/mL to approximately 100 ⁇ g/mL, preferably from approximately 10 ⁇ g/mL to approximately 50 ⁇ g/mL, more preferably from approximately 10 ⁇ g/niL to approximately 25 ⁇ g/niL.
- the compounds of the invention may be administered at doses that vary from 0.1 ⁇ g to 100,000 mg, depending upon the route of administration.
- Guidance as to particular dosages and methods of delivery is provided in the literature and is generally available to practitioners in the art.
- the dose will be in the range of about lmg/day to about 10g/day, or about O.lg to about 3g/day, or about 0.3g to about 3g/day, or about 0.5g to about 2g/day, in single, divided, or continuous doses for a patient weighing between about 40 to about 100 kg (which dose may be adjusted for patients above or below this weight range, particularly children below 40 kg).
- the exact dosage will be determined by the practitioner, in light of factors related to the subject that requires treatment. Dosage and administration are adjusted to provide sufficient levels of the active agent(s) or to maintain the desired effect. Factors which may be taken into account include the severity of the disease state, general health of the subject, age, weight, and gender of the subject, diet, time and frequency of administration, drug combination(s), reaction sensitivities, and tolerance/response to therapy. Long-acting pharmaceutical compositions may be administered every 3 to 4 days, every week, or once every two weeks depending on half-life and clearance rate of the particular formulation.
- a method of inhibiting VEGF production comprising administering to a patient in need thereof a pharmaceutical composition comprising a compound selected from any of compounds 156 through 188, a compound of Formula I,
- Formula (I) an enantiomer, a diastereomer, a pharmaceutically acceptable salt, a prodrug, a solvate or a mixture thereof, wherein (a) X is -NR 9 R 10 , -N(alkyl)-C(O)-aryl, -N(alkyl)-C(O)-halogen substituted aryl, oxo, OR 9 , H, substituted or unsubstituted phenylaminocarbonyl, substituted or unsubstituted phenyl, oxime, substituted or unsubstituted alkoxycarbonyl, hydroxycarbonyl (i.e., -COOH), or a substituted or unsubstituted heterocyclic ring; where R 9 and R 10 are each independently H, substituted or unsubstituted C -6 alkyl, substituted or unsubstituted C 1 -C 6 alcohol, substituted or unsubstituted carbonyl (i
- n 0, 1 or 2, wherein when n is 0 then R is absent;
- R 4 , R 5 , R 6 and R 7 are each independently -H, -OH, substituted or unsubstituted C 1-6 alkyl, substituted or unsubstituted carbonyl (i.e., -C(O)H), substituted or unsubstituted alkoxy, halo, haloalkyl, haloalkoxy, nitro, cyano, substituted or unsubstituted heterocyclic ring, substituted or unsubstituted amino, substituted or unsubstituted phenyl, substituted or unsubstituted phenoxy, substituted or unsubstituted cycloalkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, or substituted or unsubstituted alkoxycarbonyl, or hydroxycarbonyl;
- W is N, O, or S
- R is H, C 1-3 alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted carbonyl (i.e., -C(O)H), or together with X forms a substituted or unsubstituted 5- 11 membered mono- or bi- cyclic heterocyclic ring, with the proviso that when W is O or S, R 8 is absent.
- the present invention provides a method of inhibiting VEGF production comprising administering to a patient in need thereof a pharmaceutical composition comprising a compound selected from any of compounds 156 through 188.
- a method of inhibiting VEGF production comprising administering to a patient in need thereof a pharmaceutical composition comprising a compound of Formula (I) with the proviso that the compound is not selected from any of compounds 156 through 188.
- a method of inhibitng angiogenesis comprising administering to a patient in need thereof a pharmaceutical composition comprising a compound selected from any of compounds 156 through 188, a compound of Formula I,
- X is -NR 9 R 10 , -N(alkyl)-C(O)-aryl, -N(alkyl)-C(O)-halogen substituted aryl, oxo, OR 9 , H, substituted or unsubstituted phenylaminocarbonyl, substituted or unsubstituted phenyl, oxime, substituted or unsubstituted alkoxycarbonyl, hydroxycarbonyl (i.e., -COOH), or a substituted or unsubstituted heterocyclic ring;
- R 9 and R 10 are each independently H, substituted or unsubstituted C 1-6 alkyl, substituted or unsubstituted C 1 -C 6 alcohol, substituted or unsubstituted carbonyl (i.e., -C(O)H), substituted or unsubstituted mono- or bi- cyclic cycloalkyl, substituted or unsubstituted mono- or bi- cyclic heterocyclic ring, substituted or unsubstituted aryl, substituted or unsubstituted alkenyl, substituted or unsubstituted sulfonyl, substituted or unsubstituted arylalkyl, substituted or unsubstituted heterocycloalkyl, substituted or unsubstituted aminothiocarbonyl wherein at least one of R 9 and R 10 is H, or R 9 and R 10 together with the atom to which they are attached form a mono- or bi-cyclic heterocyclic ring, where
- R 1 , R 2 and R 3 are each independently -H, -OH, or alkyl, wherein R 1 may optionally form a substituted or unsubstituted 5-11 membered mono- or bi- heterocyclic ring with X;
- n is O, 1 or 2, wherein when n is O then R 2 is absent;
- R 4 ; R 5 , R 6 and R 7 are each independently -H, -OH, substituted or unsubstituted C 1-6 alkyl, substituted or unsubstituted carbonyl (i.e., -C(O)H), substituted or unsubstituted alkoxy, halo, haloalkyl, haloalkoxy, nitro, cyano, substituted or unsubstituted heterocyclic ring, substituted or unsubstituted amino, substituted or unsubstituted phenyl, substituted or unsubstituted phenoxy, substituted or unsubstituted cycloalkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, or substituted or unsubstituted alkoxycarbonyl, or hydroxycarbonyl;
- W is N, O, or S; and (f) R 8 is H, C 1 -3 alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted carbonyl (i.e., -C(O)H), or together with X forms a substituted or unsubstituted 5- 11 membered mono- or bi- cyclic heterocyclic ring, with the proviso that when W is O or S, R 8 is absent.
- the present invention provides a method of inhibiting angiogenesis comprising administering to a patient in need thereof a pharmaceutical composition comprising a compound selected from any of compounds 156 through 188.
- a method of inhibiting angiogenesis comprising administering to a patient in need thereof a pharmaceutical composition comprising a compound of Formula (I) with the proviso that the compound is not selected from any of compounds 156 through 188.
- a method of treating cancer, diabetic retinopathy, rheumatoid arthritis, psoriasis, atherosclerosis, obesity, chronic inflammation or exudative macular degeneration comprising administering to a patient in need thereof a pharmaceutical composition comprising a compound selected from any of compounds 156 through 188, a compound of Formula I,
- X is -NR 9 R 10 , -N(alkyl)-C(O)-aryl, -N(alkyl)-C(O)-halogen substituted aryl, oxo, OR 9 , H, substituted or unsubstituted phenylaminocarbonyl, substituted or unsubstituted phenyl, oxime, substituted or unsubstituted alkoxycarbonyl, hydroxycarbonyl (i.e., -COOH), or a substituted or unsubstituted heterocyclic ring;
- R 9 and R 10 are each independently H, substituted or unsubstituted C 1-6 alkyl, substituted or unsubstituted C 1 -C 6 alcohol, substituted or unsubstituted carbonyl (i.e., -C(O)H), substituted or unsubstituted mono- or bi- cyclic cycloalkyl, substituted or unsubstituted mono- or bi- cyclic heterocyclic ring, substituted or unsubstituted aryl, substituted or unsubstituted alkenyl, substituted or unsubstituted sulfonyl, substituted or unsubstituted arylalkyl, substituted or unsubstituted heterocycloalkyl, substituted or unsubstituted aminothiocarbonyl wherein at least one of R 9 and R 10 is H, or R 9 and R 10 together with the atom to which they are attached form a mono- or bi-cyclic heterocyclic ring, where
- R 1 , R 2 and R 3 are each independently -H, -OH, or alkyl, wherein R 1 may optionally form a substituted or unsubstituted 5-11 membered mono- or bi- heterocyclic ring with X;
- n 0, 1 or 2, wherein when n is 0 then R 2 is absent;
- R 4 ; R 5 , R 6 and R 7 are each independently -H, -OH, substituted or unsubstituted C 1-6 alkyl, substituted or unsubstituted carbonyl (i.e., -C(O)H), substituted or unsubstituted alkoxy, halo, haloalkyl, haloalkoxy, nitro, cyano, substituted or unsubstituted heterocyclic ring, substituted or unsubstituted amino, substituted or unsubstituted phenyl, substituted or unsubstituted phenoxy, substituted or unsubstituted cycloalkyl, substituted or unsubstituted alkenyl, substituted or unsubstituted alkynyl, or substituted or unsubstituted alkoxycarbonyl, or hydroxycarbonyl ;
- W is N, O, or S; and (f) R 8 is H, C 1-3 alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted carbonyl (i.e., -C(O)H), or together with X forms a substituted or unsubstituted 5- 11 membered mono- or bi- cyclic heterocyclic ring, with the proviso that when W is O or S, R 8 is absent.
- the present invention provides a method of treating cancer, diabetic retinopathy, rheumatoid arthritis, psoriasis, atherosclerosis, obesity, chronic inflammation or exudative macular degeneration comprising administering to a patient in need thereof a pharmaceutical composition comprising a compound selected from any of compounds 156 through 188.
- a method of treating cancer, diabetic retinopathy, rheumatoid arthritis, psoriasis, atherosclerosis, obesity, chronic inflammation or exudative macular degeneration comprising administering to a patient in need thereof a pharmaceutical composition comprising a compound of Formula (I) with the proviso that the compound is not selected from any of compounds 156 through 188.
- a method of inhibiting VEGF production comprising administering to a patient in need thereof a pharmaceutical composition comprising a compound selected from any of compounds 156 through 188, a compound of Formula (II) through Formula (VIII).
- a method of inhibiting VEGF production comprising administering to a patient in need thereof a pharmaceutical composition comprising a compound of Formula (II) through Formula (VIII) with the proviso that the compound is not selected from any of compounds 156 through 188.
- a method of inhibiting angiogenesis comprising administering to a patient in need thereof a pharmaceutical composition comprising a compound selected from any of compounds 156 through 188, a compound of Formula (II) through Formula (VIII).
- a method of inhibiting angiogenesis comprising administering to a patient in need thereof a pharmaceutical composition comprising a compound of Formula (II) through Formula (VIII) with the proviso that the compound is not selected from any of compounds 156 through 188.
- a method of treating cancer, diabetic retinopathy, rheumatoid arthritis, psoriasis, atherosclerosis, obesity, chronic inflammation or exudative macular degeneration comprising administering to a patient in need thereof a pharmaceutical composition comprising a compound selected from any of compounds 156 through 188, a compound of Formula (II) through Formula (VIII).
- a method of treating cancer, diabetic retinopathy, rheumatoid arthritis, psoriasis, atherosclerosis, obesity, chronic inflammation or exudative macular degeneration comprising administering to a patient in need thereof a pharmaceutical composition comprising a compound of Formula (II) through Formula (VIII) with the proviso that the compound is not selected from any of compounds 156 through 188.
- the present invention provides a method for selectively inhibiting VEGF in cells, by exposing the cells to an effective amount of at least one compound selected from the following:
- Thiophene-2-carboxylic acid (6-bromo-2,3,4,9-tetrahydro- 1 H-carbazol- 1 -yl)-amide; l-Benzooxazol-2-yl-6-bromo-2,3,4,9-tetrahydro-l H-carbazole;
- the compound of Formula (I) is selected from the following: 6-Bromo-l-thieno[2,3-c]pyrrol-5-yl-2,3,4,9-tetrahydro-1H-carbazole;
- Furan-2-carboxylic acid (6-bromo-2,3,4,9-tetrahydro-1H-carbazol-l-yl)-amide
- Thiophene-2-carboxylic acid (6-bromo-2, 3, 4,9-tetrahydro-l H-carbazol- 1 -yl)-amide; l-Benzooxazol-2-yl-6-bromo-2,3,4,9-tetrahydro-1H-carbazole; N-(6-Bromo-2,3,4,9-tetrahydro-1H-carbazol-l-yl)-4-methoxy-benzamide;
- the invention includes compounds produced by a process comprising contacting a compound of this invention with a mammalian tissue or a mammal for a period of time sufficient to yield a metabolic product thereof.
- Such products typically are identified by preparing a radio-labeled ⁇ e.g.
- C ⁇ or H ⁇ ) compound of the invention administering it in a detectable dose ⁇ e.g., greater than about 0.5 mg/kg) to a mammal such as rat, mouse, guinea pig, monkey, or to man, allowing sufficient time for metabolism to occur (typically about 30 seconds to 30 hours), and isolating its conversion products from urine, blood or other biological samples.
- a detectable dose ⁇ e.g., greater than about 0.5 mg/kg
- a mammal such as rat, mouse, guinea pig, monkey, or to man
- sufficient time for metabolism to occur typically about 30 seconds to 30 hours
- the metabolite structures are determined in conventional fashion, e.g. , by MS or NMR analysis. In general, analysis of metabolites may be done in the same way as conventional drug metabolism studies well-known to those skilled in the art.
- the conversion products so long as they are not otherwise found in vivo, are useful in diagnostic assays for therapeutic dosing of the compounds of the invention even
- compositions useful in the methods of the invention are provided.
- the pharmaceutical compositions of the invention may be formulated with pharmaceutically acceptable excipients such as carriers, solvents, stabilizers, adjuvants, diluents, etc , depending upon the particular mode of administration and dosage form.
- the pharmaceutical compositions should generally be formulated to achieve a physiologically compatible pH, and may range from a pH of about 3 to a pH of about 11, preferably about pH 3 to about pH 7, depending on the formulation and route of administration. In alternative embodiments, it may be preferred that the pH is adjusted to a range from about pH 5 to about pH 8.
- the pH is adjusted to a range from about pH 4 to about pH 7.
- the pharmaceutical compositions of the invention comprise a therapeutically or prophylactically effective amount of one or more, two or more, or three or more compounds of the present invention, together with one or more pharmaceutically acceptable excipients.
- pharmaceutical compositions of the invention may comprise one or more compounds of Formula (I) through Formula (VIII) and one or more pharmaceutically acceptable excipients.
- pharmaceutical compositions of the invention may comprise one or more of Compounds 159 through 188 and one or more pharmaceutically acceptable excipients.
- compositions of the invention may comprise one or more compounds of Formula (I) through Formula (VIII) and one or more of Compounds 159 through 188 together with one or more pharmaceutically acceptable excipients.
- a pharmaceutical composition comprises one or more compounds of the invention and one ore more pharmaceutical excipients, with the proviso that the pharmaceutical composition does not comprise Compounds 159 through 188.
- a pharmaceutical composition comprises one or more, two or more, or three or more compounds selected from the group consisting of Compound No. 191 to 239, or a hydrate, enantiomer, a diastereomer, a pharmaceutically accpetable salt, prodrug, solvate or a mixture of , said one or more, two or more, or three or more compounds, and one or more pharmaceutically acceptable excipients.
- compositions of the invention may comprise a combination of compounds of the present invention, or may include a second active ingredient useful in the treatment of cancer, diabetic retinopathy, rheumatoid arthritis, psoriasis, atherosclerosis, obesity, chronic inflammation, or exudative macular degeneration.
- Formulations of the present invention are most typically solids, liquid solutions, emulsions or suspensions, while inhaleable formulations for pulmonary administration are generally liquids or powders, with powder formulations being generally preferred.
- a preferred pharmaceutical composition of the invention may also be formulated as a lyophilized solid that is reconstituted with a physiologically compatible solvent prior to administration.
- Alternative pharmaceutical compositions of the invention may be formulated as syrups, creams, ointments, tablets, and the like.
- pharmaceutically acceptable excipient refers to an excipient for administration of a pharmaceutical agent, such as the compounds of the present invention.
- the term refers to any pharmaceutical excipient that may be administered without undue toxicity.
- Pharmaceutically acceptable excipients are determined in part by the particular composition being administered, as well as by the particular method used to administer the composition. Accordingly, there exists a wide variety of suitable formulations of pharmaceutical compositions of the present invention ⁇ see, e.g., Remington's Pharmaceutical Sciences).
- Suitable excipients may be carrier molecules that include large, slowly metabolized macromolecules such as proteins, polysaccharides, polylactic acids, polyglycolic acids, polymeric amino acids, amino acid copolymers, and inactive virus particles.
- Other exemplary excipients include antioxidants such as ascorbic acid; chelating agents such as EDTA; carbohydrates such as dextrin, hydroxyalkylcellulose, hydroxyalkylmethylcellulose, stearic acid; liquids such as oils, water, saline, glycerol and ethanol; wetting or emulsifying agents; pH buffering substances; and the like. Liposomes are also included within the definition of pharmaceutically acceptable excipients.
- compositions of the invention may be formulated in any form suitable for the intended method of administration.
- tablets, troches, lozenges, aqueous or oil suspensions, non-aqueous solutions, dispersible powders or granules (including micronized particles or nanoparticles), emulsions, hard or soft capsules, syrups or elixirs may be prepared.
- Compositions intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions, and such compositions may contain one or more agents including sweetening agents, flavoring agents, coloring agents and preserving agents, in order to provide a palatable preparation.
- compositions particularly suitable for use in conjunction with tablets include, for example, inert diluents, such as celluloses, calcium or sodium carbonate, lactose, calcium or sodium phosphate; disintegrating agents, such as croscarmellose sodium, cross-linked povidone, maize starch, or alginic acid; binding agents, such as povidone, starch, gelatin or acacia; and lubricating agents, such as magnesium stearate, stearic acid or talc. Tablets may be uncoated or may be coated by known techniques including microencapsulation to delay disintegration and adsorption in the gastrointestinal tract and thereby provide a sustained action over a longer period. For example, a time delay material such as glyceryl monostearate or glyceryl distearate alone or with a wax may be employed.
- inert diluents such as celluloses, calcium or sodium carbonate, lactose, calcium or sodium phosphate
- disintegrating agents such
- Formulations for oral use may be also presented as hard gelatin capsules where the active ingredient is mixed with an inert solid diluent, for example celluloses, lactose, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with nonaqueous or oil medium, such as glycerin, propylene glycol, polyethylene glycol, peanut oil, liquid paraffin or olive oil.
- an inert solid diluent for example celluloses, lactose, calcium phosphate or kaolin
- nonaqueous or oil medium such as glycerin, propylene glycol, polyethylene glycol, peanut oil, liquid paraffin or olive oil.
- compositions of the invention may be formulated as suspensions comprising a compound of the present invention in admixture with at least one pharmaceutically acceptable excipient suitable for the manufacture of a suspension.
- pharmaceutical compositions of the invention may be formulated as dispersible powders and granules suitable for preparation of a suspension by the addition of suitable excipients.
- Excipients suitable for use in connection with suspensions include suspending agents, such as sodium carboxymethylcellulose, methylcellulose, hydroxypropyl methylcelluose, sodium alginate, polyvinylpyrrolidone, gum tragacanth, gum acacia, dispersing or wetting agents such as a naturally occurring phosphatide (e.g., lecithin), a condensation product of an alkylene oxide with a fatty acid (e.g., polyoxyethylene stearate), a condensation product of ethylene oxide with a long chain aliphatic alcohol (e.g., heptadecaethyleneoxycethanol), a condensation product of ethylene oxide with a partial ester derived from a fatty acid and a hexitol anhydride (e.g., polyoxyethylene sorbitan monooleate); and thickening agents, such as carbomer, beeswax, hard paraffin or cetyl alcohol.
- suspending agents such as sodium carboxymethylcellulose
- the suspensions may also contain one or more preservatives such as acetic acid, methyl and/or n-propyl p-hydroxy-benzoate; one or more coloring agents; one or more flavoring agents; and one or more sweetening agents such as sucrose or saccharin.
- preservatives such as acetic acid, methyl and/or n-propyl p-hydroxy-benzoate
- coloring agents such as acetic acid, methyl and/or n-propyl p-hydroxy-benzoate
- flavoring agents such as sucrose or saccharin.
- sweetening agents such as sucrose or saccharin.
- the pharmaceutical compositions of the invention may also be in the form of oil-in- water emulsions.
- the oily phase may be a vegetable oil, such as olive oil or arachis oil, a mineral oil, such as liquid paraffin, or a mixture of these.
- Suitable emulsifying agents include naturally-occurring gums, such as gum acacia and gum tragacanth; naturally occurring phosphatides, such as soybean lecithin, esters or partial esters derived from fatty acids; hexitol anhydrides, such as sorbitan monooleate; and condensation products of these partial esters with ethylene oxide, such as polyoxyethylene sorbitan monooleate.
- the emulsion may also contain sweetening and flavoring agents.
- Syrups and elixirs may be formulated with sweetening agents, such as glycerol, sorbitol or sucrose. Such formulations may also contain a demulcent, a preservative, a flavoring or a coloring agent.
- sweetening agents such as glycerol, sorbitol or sucrose.
- Such formulations may also contain a demulcent, a preservative, a flavoring or a coloring agent.
- compositions of the invention may be in the form of a sterile injectable preparation, such as a sterile injectable aqueous emulsion or oleaginous suspension.
- a sterile injectable preparation such as a sterile injectable aqueous emulsion or oleaginous suspension.
- This emulsion or suspension may be formulated according to the known art using those suitable dispersing or wetting agents and suspending agents which have been mentioned above.
- the sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parenterally acceptable diluent or solvent, such as a solution in 1,2-propane-diol.
- the sterile injectable preparation may also be prepared as a lyophilized powder.
- arid solvents that may be employed are water, Ringer's solution, and isotonic sodium chloride solution.
- sterile fixed oils may be employed as a solvent or suspending medium.
- any bland fixed oil may be employed including synthetic mono- or diglycerides.
- fatty acids such as oleic acid may likewise be used in the preparation of injectables.
- the compounds of the present invention useful in the methods of the present invention are substantially insoluble in water and are sparingly soluble in most pharmaceutically acceptable protic solvents and in vegetable oils.
- the compounds are generally soluble in medium chain fatty acids (e.g., caprylic and capric acids) or triglycerides and have high solubility in propylene glycol esters of medium chain fatty acids.
- medium chain fatty acids e.g., caprylic and capric acids
- triglycerides e.g., triglycerides
- compounds which have been modified by substitutions or additions of chemical or biochemical moieties which make them more suitable for delivery e.g., increase solubility, bioactivity, palatability, decrease adverse reactions, etc.
- esterification glycosylation, PEGylation, etc.
- the compounds of the present invention may be formulated for oral administration in a lipid-based formulation suitable for low solubility compounds. Lipid-based formulations can generally enhance the oral bioavailability of such compounds.
- a preferred pharmaceutical composition of the invention comprises a therapeutically or prophylactically effective amount of a compound of the present invention, together with at least one pharmaceutically acceptable excipient selected from the group consisting of: medium chain fatty acids or propylene glycol esters thereof (e.g., propylene glycol esters of edible fatty acids such as caprylic and capric fatty acids) and pharmaceutically acceptable surfactants such as polyoxyl 40 hydrogenated castor oil.
- cyclodextrins may be added as aqueous solubility enhancers.
- Preferred cyclodextrins include hydroxypropyl, hydroxyethyl, glucosyl, maltosyl and maltotriosyl derivatives of ⁇ -, ⁇ -, and ⁇ -cyclodextrin.
- a particularly preferred cyclodextrin solubility enhancer is hydroxypropyl- ⁇ -cyclodextrin (HPBC), which may be added to any of the above-described compositions to further improve the aqueous solubility characteristics of the compounds of the present invention.
- the composition comprises 0.1% to 20% hydroxypropyl- ⁇ -cyclodextrin, more preferably 1% to 15% hydroxypropyl- ⁇ -cyclodextrin, and even more preferably from 2.5% to 10% hydroxypropyl- ⁇ - cyclodextrin.
- solubility enhancer employed will depend on the amount of the compound of the present invention in the composition.
- any compound of the present invention with one or more other active ingredients useful in the treatment of cancer, exudative macular degeneration, rheumatoid arthritis, psoriasis, atherosclerosis, obesity, chronic inflammation or diabetic reinopathy, including compounds, in a unitary dosage form, or in separate dosage forms intended for simultaneous or sequential administration to a patient in need of treatment.
- the combination may be administered in two or more administrations.
- the combination of active ingredients may be: (1) co-formulated and administered or delivered simultaneously in a combined formulation; (2) delivered by alternation or in parallel as separate formulations; or (3) by any other combination therapy regimen known in the art.
- the methods of the invention may comprise administering or delivering the active ingredients sequentially, e.g., in separate solution, emulsion, suspension, tablets, pills or capsules, or by different injections in separate syringes.
- an effective dosage of each active ingredient is administered sequentially, i.e., serially, whereas in simultaneous therapy, effective dosages of two or more active ingredients are administered together.
- Various sequences of intermittent combination therapy may also be used.
- Z includes, but is not limited to, substituted or unsubstituted carbonyl (i.e., unsubstituted carbonyl is -C(O)H), C 1 -C 6 alkoxycarbonyl, substituted or unsubstituted aminocarbonyl, or sulfonyl.
- Scheme A depicts two pathways by which compounds encompassed by Formulas (I), (Ia), and I(b) are obtained. Also shown is a standard procedure for converting carbonyl compounds of the invention to amino compounds of the invention.
- Scheme B depicts a carbonyl compound encompassed by formulas (I), I(a), and I(b), which, by means of a typical synthetic strategy, may be oxidized to an alcohol compound of the invention. Further compounds of the invention may then be obtained from the alcohol. For example, in Scheme B, the alcohol compound of the invention is converted to a variety of other compounds, including, among other compounds, ethers and esters.
- Scheme C depicts a carbonyl compound encompassed by formulas (I), I(a), and I(b), which, by means of a typical synthetic strategy, may be oxidized to an alcohol compound of the invention. Further compounds of the invention may then be obtained from the alcohol. For example, in Scheme B, the alcohol compound of the invention is converted to a variety of other compounds, including, among other compounds, ethers and esters.
- Scheme C depicts a typical synthetic sequence used in the preparation of an amino substituted compound of the invention.
- the amino substituted compound of the invention may itself serve as an intermediate for a large variety of compounds encompassed by Formulas (Ia), (Ib), and (Ic).
- Scheme D depicts a typical route by which compounds of Formula (VIII) are obtained from compounds encompassed by Formulas (Ia), (Ib), and (Ic).
- Scheme E depicts typical syntheses of compounds of the invention wherein the amino substituent is incorporated into various bicyclic ring systems.
- the Br substituent may be replaced with a variety of substituents, including, for example, cyano, amino, and carbonyl.
- R 51 and R 52 are selected from the group consisting of H, substituted and unsubstituted C 1 -C 6 alkyl, and substituted and unsubstituted C 1 -C 6 alkylcarbonyl.
- Scheme G depicts the synthesis of a typical intermediate, and compounds of the invention obtained from the intermediate.
- the intermediate is itself a compound of the invention.
- Scheme H depicts a typical synthetic pathway in which the carbonyl moiety of the cyclohexenone ring portion of the starting compound provides a basis for the incorporation of a fourth ring into tricyclic ring system, providing a compound of the invention.
- Scheme I depicts a typical means of introducing a heterocyclic ring into a compound of the invention, thereby providing a new compound of the invention.
- Scheme J depicts a typical means of introducing a heterocyclic ring into a compound of the invention, thereby providing a new compound of the invention.
- Scheme J depicts typical syntheses of compounds having a phenol or phenyl ether group.
- the synthesis methods described herein may employ a variety of commercially available starting materials, starting materials known in the literature, and readily-prepared starting materials prepared by employing standard synthetic methods and procedures.
- Standard synthetic methods and procedures for the preparation of organic molecules and functional group transformations and manipulations can be obtained from the relevant scientific literature or from standard reference textbooks in the field.
- recognized reference textbooks of organic synthesis include for example: Smith, M. B.; March, J. March's Advanced Organic Chemistry: Reactions, Mechanisms, and Structure, 5 th ed.; John Wiley & Sons: New York, 2001; and Greene, T. W.; Wuts, P.G. M. Protective Groups in Organic Synthesis, 3 rd ; John Wiley & Sons: New York, 1999.
- the foregoing descriptions of synthetic methods are designed to illustrate, but not limit, general procedures for the preparation of compounds of the invention.
- Compound 5 structure assignment is based on the following references: JOC, 1997, 5392 and JCS, CC, 1985, 1183.
- Compound 1 is prepared by the same procedure.
- Compound 38 is prepared in the same manner as that of compound 37, Procedure II.
- Compound 49 is prepared in the same manner as that of compound 17, Procedure VI.
- Compound 16 is made in the same manner.
- Compound 107 is prepared from 105 in the same manner of that of compound 17, procedure Via.
- a mixture of the 4-bromo-aniline (solid) and 3-bromo-2-oxo-cyclohexanecarboxylic acid ethyl ester (oil) is heated at 150 °C under high vacuum.
- the solid is dissolved, whereupon the whole mass becomes solid. After about 1 hour, the solid melts into dark brown oil.
- the reaction mixture solidifies.
- the solids are treated with DCM and partially dissolved.
- the mixture is filtered and washed with DCM.
- the filtrate is concentrated under vacuum and chromatographed to give 90 as a yellow solid. Yield: 1.4 g, 36%.
- ester 90 (0.85 g, 2.64 mmol) in THF (10 mL) is added 5 N NaOH (2.6 mL, 13 mmol). After heating at 90 °C for 2 hours, the mixture is concentrated under vacuum to remove THF, diluted with water and washed with EtOAc. The aqueous layer is acidified to pH 3 with 6N HCl and extracted with DCM (3x). The combined organics are concentrated under vacuum to give 85 as a brownish solid. Yield: 0.71 g, 92%.
- EXAMPLE 2 Assay to Evaluate Effect on Hypoxia-Inducible Endogenous VEGF Expression
- VEGF protein levels are monitored by an ELISA assay (R&D Systems). Briefly, HeLa cells are cultured for 24-48 hours under hypoxic conditions (1% O 2 , 5% CO 2 , balanced with nitrogen) in the presence or absence of a compound of the invention. The conditioned media is then assayed by ELISA, and the concentration of VEGF is calculated from the standard ELISA curve of each assay. A dose-response analysis is performed using the ELISA assay and conditions described above. A series of different concentrations (e.g., seven) are analyzed.
- a dose- response cytotoxicity assay is performed using Cell Titer GIo (Promega) under the same conditions as the ELISA to ensure that the inhibition of VEGF expression is not due to cytotoxicity.
- Dose-response curves are plotted using percentage inhibition versus concentration of the compound, and EC 5O and CC 50 values are generated for each compound with the maximal inhibition set as 100% and the minimal inhibition as 0%.
- Figure 1 and Table 1 below show the ability of a typical compound of the invention to inhibit endogenous VEGF production in tumor cells under hypoxic conditions.
- the ELISA EC 50 is 0.0098 ⁇ m, while its CC 50 (50% cytotoxicity) is greater than 1.68 ⁇ m.
- the EC 50 values for a series of compounds, which can be employed in the compositions and methods of the invention, are provided in Table 1 below.
- Compounds 156-188 in Table 1 are commercially available.
- each of the represented compounds is followed by one to five stars. The number of stars next to a particular compound indicates that compound's EC 50 (the effective concentration required to lower the amount of VEGF translation by 50%), according to the following scale:
- EXAMPLE 3 Compounds of the Invention Inhibit VEGF Expression and Tumor Growth in an In Vivo Tumor Growth PD Model
- HTl 080 cells a human fibrosarcoma cell line
- mice were implanted subcutaneously in nude mice. After seven days, mice were administrated compounds orally at a desired dosage range, e.g., 200mg/kg/day, for seven days.
- the tumors were then excised from mice and homogenized in Tris-HCl buffer containing proteinase inhibitors.
- Intratumor VEGF levels were subsequently measured using a human VEGF ELISA kit (R&D System). Protein concentrations of the homogenates were measured with a Bio-Rad Protein assay kit and intratumor VEGF levels were normalized to the protein concentrations.
- Preferred compounds of the invention when used for one week on a 100 mm 3 tumor, will generally inhibit tumor growth by at least 50%, as compared to the vehicle-treated control groups (data not shown).
- the compounds shown below in Table 2 numbered 156-188 are commercially available. These compounds are generally known as drug-like compounds, and were purchased for the purpose of determining new uses of the compounds. Their commercial information is as follows:
- Vascular endothelial growth factor, interleukin 8, platelet- derived endothelial cell growth factor, and basic fibroblast growth factor promote angiogenesis and metastasis in human melanoma xenografts. Cancer Res. 60(17):4932-8, 2000.
- Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 246:1306-1309, 1989.
- VEGF-TRAP(R 1R2) suppresses choroidal neovascularization and VEGF-induced breakdown of the blood-retinal barrier. J Cell Physiol. 195(2):241-8, 2003.
- tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399:271-275, 1999.
- Levy AP Post-transcriptional regulation of vascular endothelial growth factor by hypoxia. J. Biol. Chem. 271 : 2746-2753, 1996.
- Semenza GL Regulation of mammalian 02 homeostasis by hypoxia-inducible factor 1. Annu. Rev. Cell. Dev. Biol, 5:551-578, 1999.
- Goldberg I Furneaux H and Levy AP. A 40 bp element that mediates stabilization of VEGF mRNA by HuR. J. Biol. Cell. J Biol Chem. 2002 Apr 19;277(16):13635- 40, 2002.
- VEGF vascular endothelial growth factor
- Fong TA, et al. SU5416 is a potent and selective inhibitor of the vascular endothelial growth factor receptor (FIk-I /KDR) that inhibits tyrosine kinase catalysis, tumor vascularizatiion, and growth of multiple tumor types.
- FIk-I /KDR vascular endothelial growth factor receptor
- Tolentino MJ Small interfering RNA (siRNA) targeting VEGF effectively inhibits ocular neovascularization in a mouse model. MoI Vis. 30;9:210-6, 2003. 59. Asano M, Yukita A, Suzuki H. Wide spectrum of antitumor activity of a neutralizing monoclonal antibody to human vascular endothelial growth factor. Jpn J Cancer Res. 90(l):93-100,1999.
- siRNA Small interfering RNA
- VEGF vascular endothelial growth factor
- Laird AD. et al. SU6668 is a potent antiangiogenic and antitumor agent that induces regression of established tumors. Cancer Res. 60(15):4152-60, 2000. 62. Wedge SR, Ogilvie DJ, Dukes M, Kendrew J, Curwen JO, Hennequin LF,
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Organic Chemistry (AREA)
- Epidemiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Cardiology (AREA)
- Immunology (AREA)
- Obesity (AREA)
- Rheumatology (AREA)
- Endocrinology (AREA)
- Heart & Thoracic Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Ophthalmology & Optometry (AREA)
- Pain & Pain Management (AREA)
- Vascular Medicine (AREA)
- Child & Adolescent Psychology (AREA)
- Emergency Medicine (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Urology & Nephrology (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Indole Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Furan Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Other In-Based Heterocyclic Compounds (AREA)
Abstract
Description

















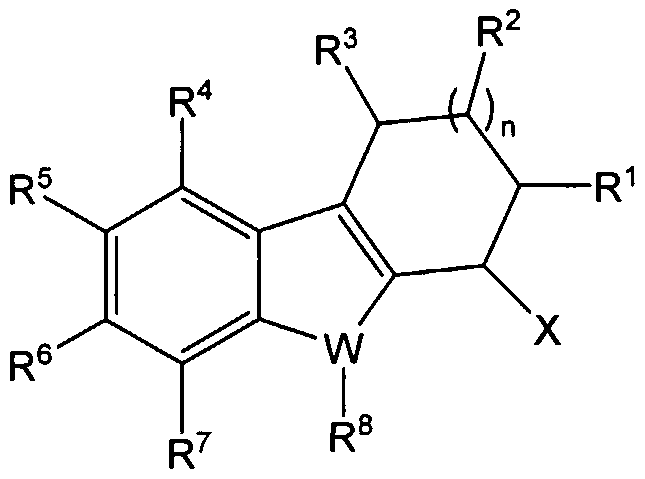


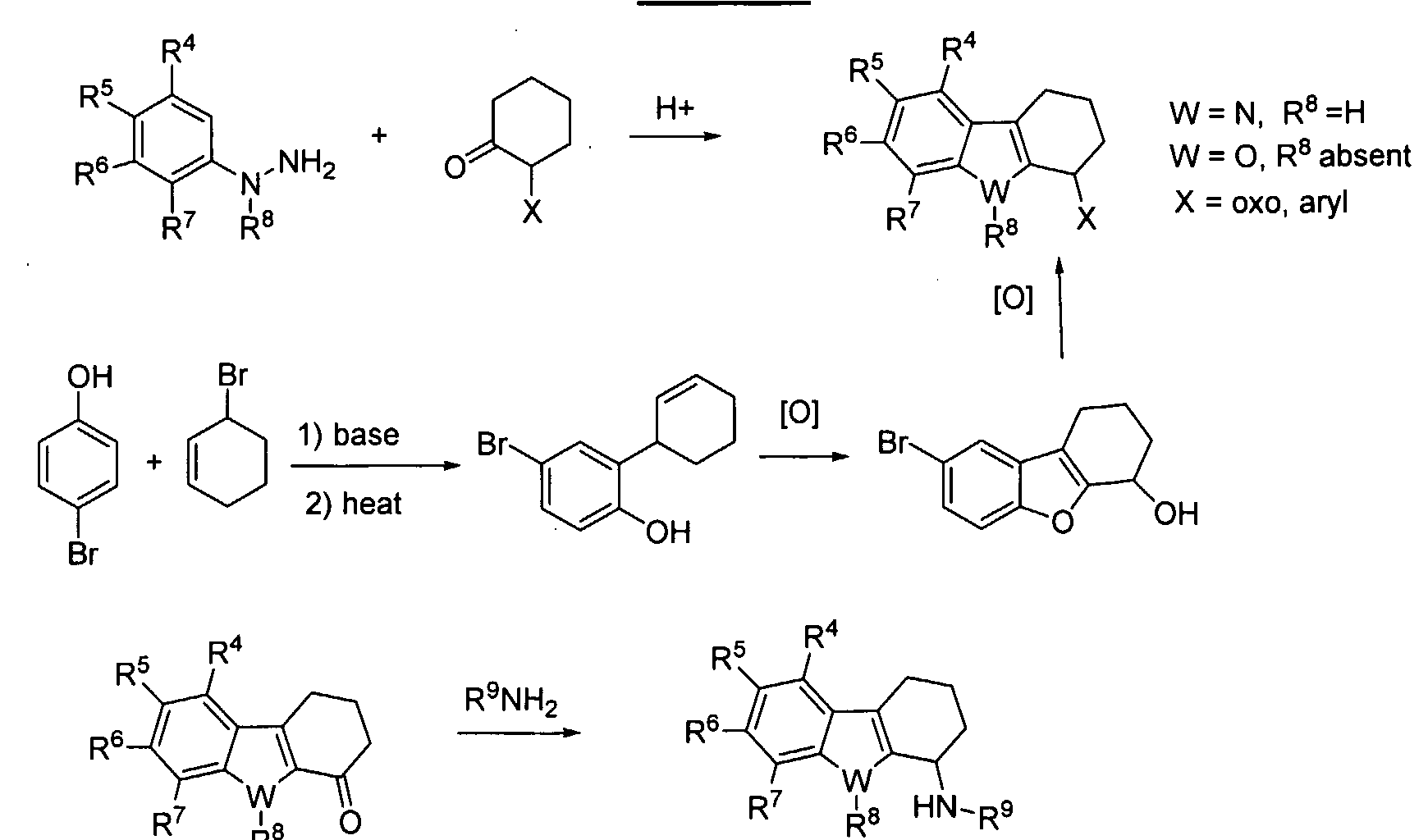













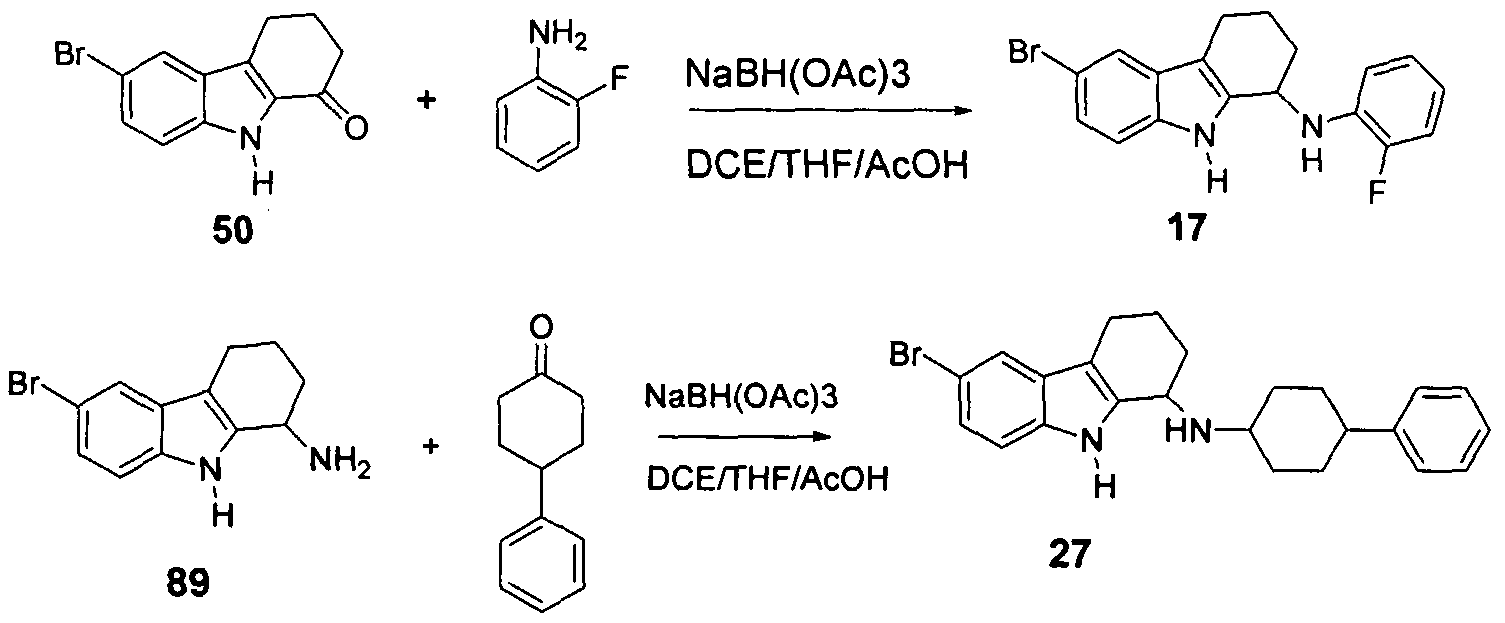


















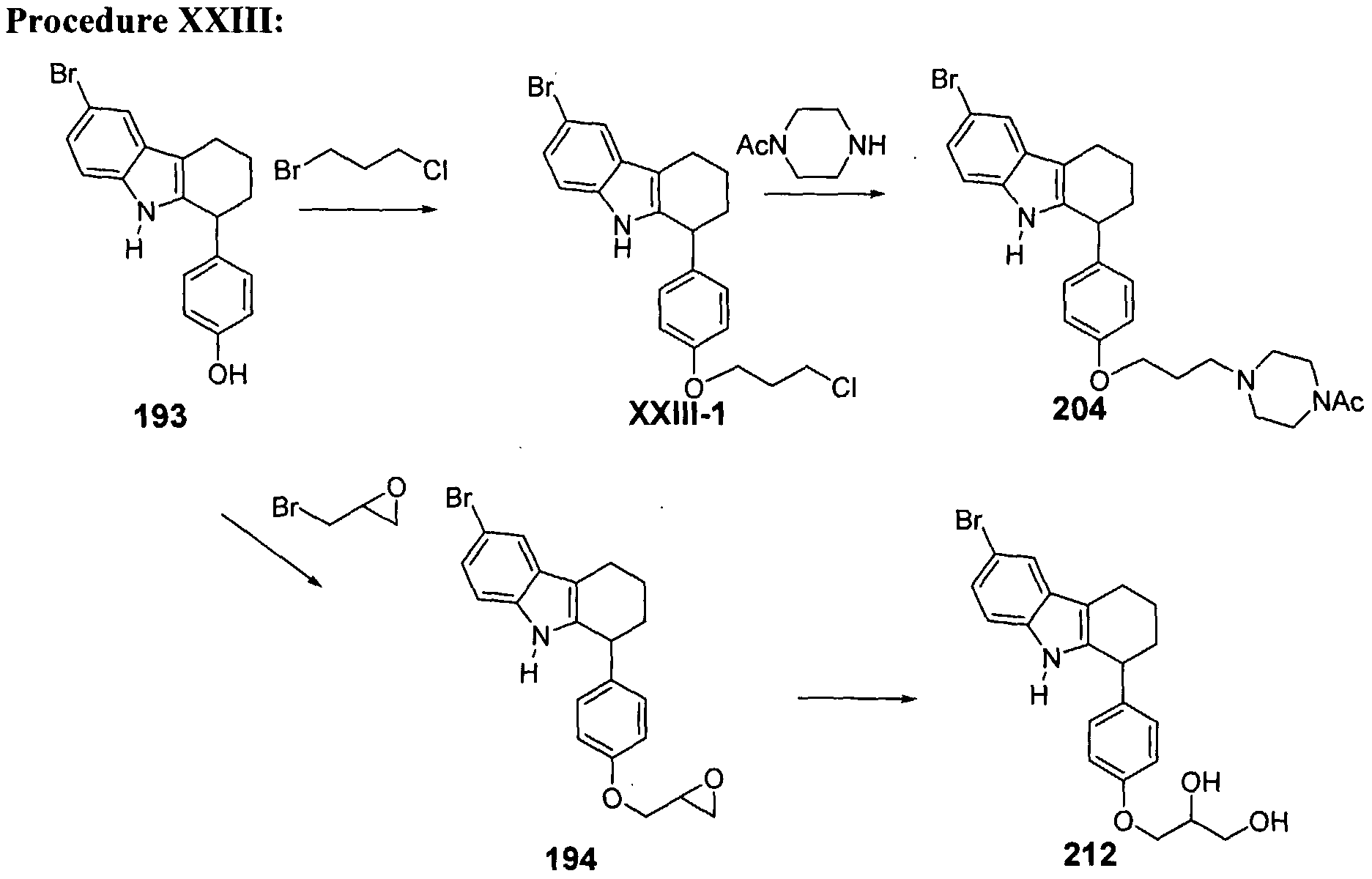
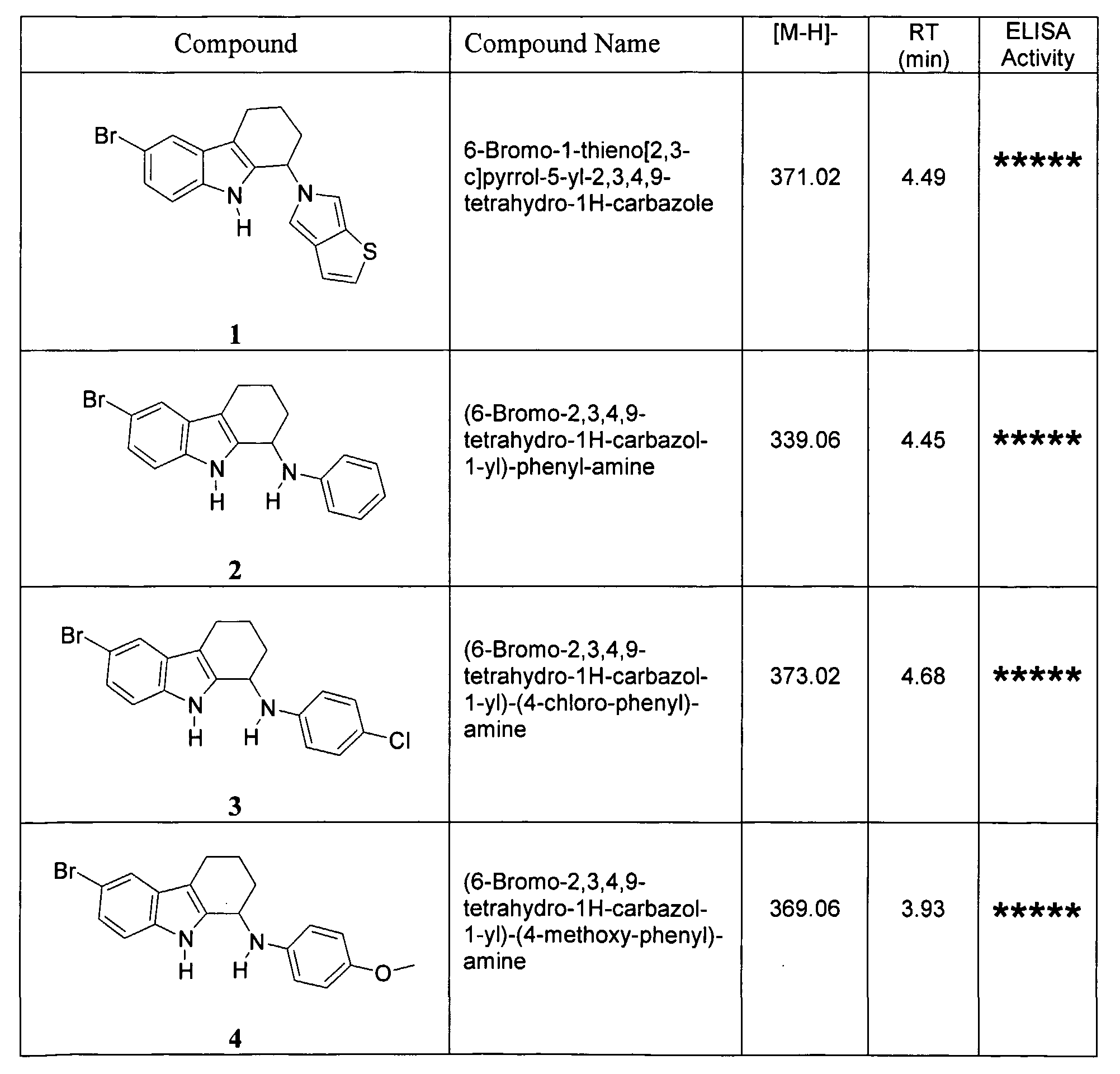
































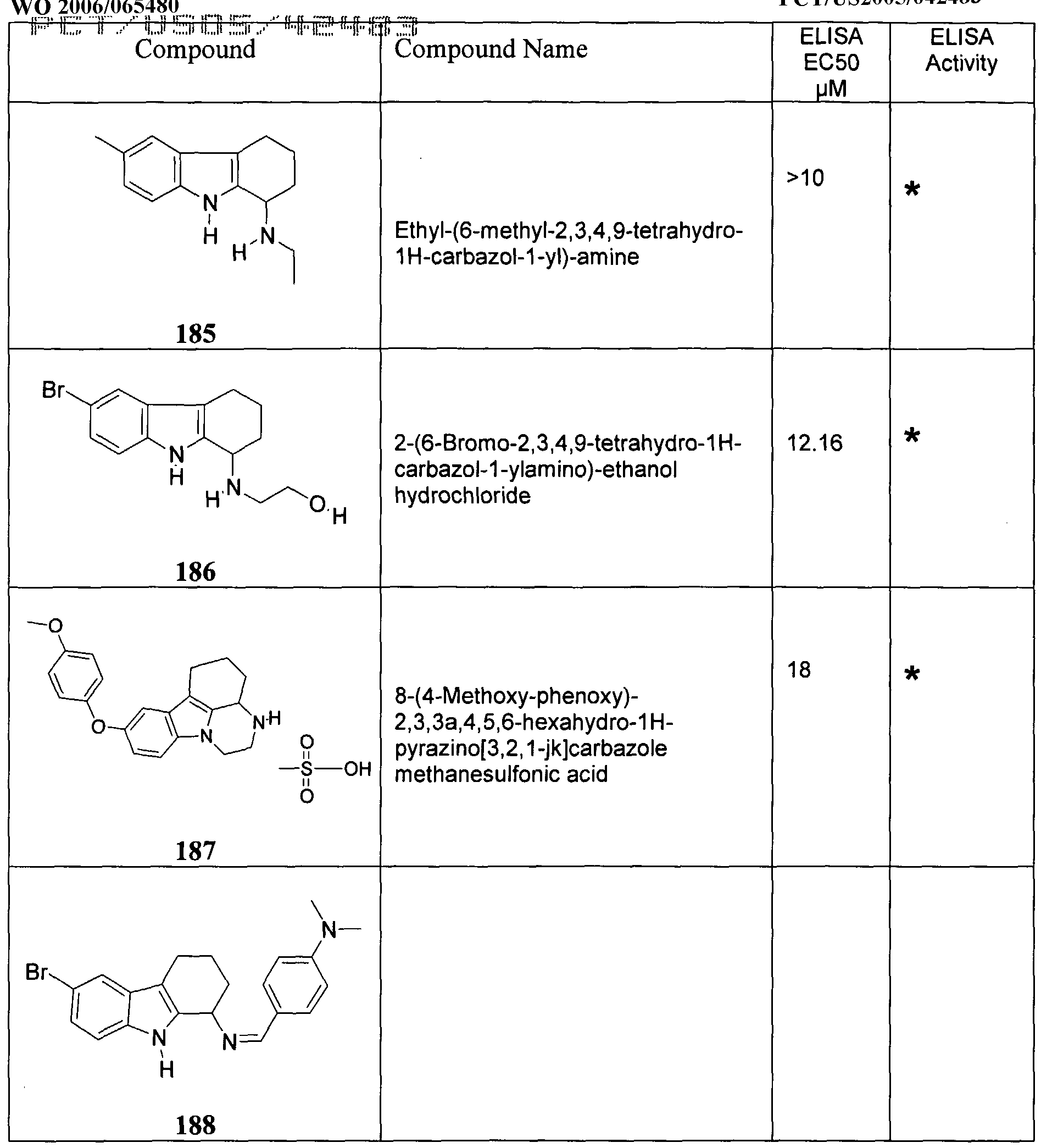




















Claims










Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP05852075A EP1817025A2 (en) | 2004-11-23 | 2005-11-23 | Tetrahydrocarbazoles as active agents for inhibiting vegf production by translational control |
| US11/720,055 US8946444B2 (en) | 2004-11-23 | 2005-11-23 | Tetrahydrocarbazoles as active agents for inhibiting VEGF production by translational control |
| CA002588384A CA2588384A1 (en) | 2004-11-23 | 2005-11-23 | Tetrahydrocarbazoles as active agents for inhibiting vegf production by translational control |
| MX2007006179A MX2007006179A (en) | 2004-11-23 | 2005-11-23 | Tetrahydrocarbazoles as active agents for inhibiting vegf production by translational control. |
| JP2007543449A JP2008520741A (en) | 2004-11-23 | 2005-11-23 | Tetrahydrocarbazole as an activator to inhibit translation-controlled VEGF production |
| US14/605,721 US9271960B2 (en) | 2004-11-23 | 2015-01-26 | Tetrahydrocarbazoles as active agents for inhibiting VEGF production by translational control |
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US62988904P | 2004-11-23 | 2004-11-23 | |
| US60/629,889 | 2004-11-23 | ||
| US63373804P | 2004-12-06 | 2004-12-06 | |
| US60/633,738 | 2004-12-06 | ||
| US63928304P | 2004-12-27 | 2004-12-27 | |
| US60/639,283 | 2004-12-27 |
Related Child Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US11/720,055 A-371-Of-International US8946444B2 (en) | 2004-11-23 | 2005-11-23 | Tetrahydrocarbazoles as active agents for inhibiting VEGF production by translational control |
| US14/605,721 Continuation US9271960B2 (en) | 2004-11-23 | 2015-01-26 | Tetrahydrocarbazoles as active agents for inhibiting VEGF production by translational control |
| US14/605,721 Division US9271960B2 (en) | 2004-11-23 | 2015-01-26 | Tetrahydrocarbazoles as active agents for inhibiting VEGF production by translational control |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2006065480A2 true WO2006065480A2 (en) | 2006-06-22 |
| WO2006065480A3 WO2006065480A3 (en) | 2006-08-03 |
Family
ID=36143248
Family Applications (3)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2005/042483 Ceased WO2006065480A2 (en) | 2004-11-23 | 2005-11-23 | Tetrahydrocarbazoles as active agents for inhibiting vegf production by translational control |
| PCT/US2005/042484 Ceased WO2006058088A2 (en) | 2004-11-23 | 2005-11-23 | Carbazole, carboline and indole derivatives useful in the inhibition of vegf production |
| PCT/US2005/042482 Ceased WO2006065479A2 (en) | 2004-11-23 | 2005-11-23 | Substituted phenols as active agents inhibiting vegf production |
Family Applications After (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2005/042484 Ceased WO2006058088A2 (en) | 2004-11-23 | 2005-11-23 | Carbazole, carboline and indole derivatives useful in the inhibition of vegf production |
| PCT/US2005/042482 Ceased WO2006065479A2 (en) | 2004-11-23 | 2005-11-23 | Substituted phenols as active agents inhibiting vegf production |
Country Status (6)
| Country | Link |
|---|---|
| US (3) | US8143257B2 (en) |
| EP (3) | EP1817025A2 (en) |
| JP (3) | JP2008520741A (en) |
| CA (3) | CA2588384A1 (en) |
| MX (3) | MX2007006179A (en) |
| WO (3) | WO2006065480A2 (en) |
Cited By (30)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20100267712A1 (en) * | 2007-09-27 | 2010-10-21 | The United States of America, as represented by the Secretary, Department of Health and | Isoindoline compounds for the treatment of spinal muscular atrophy and other uses |
| US7935722B2 (en) | 2005-06-24 | 2011-05-03 | Eli Lilly And Company | Tetrahydrocarbazole derivatives useful as androgen receptor modulators |
| US7968587B2 (en) | 2006-11-20 | 2011-06-28 | Eli Lilly And Company | Tetrahydrocyclopenta[b]indole compounds as androgen receptor modulators |
| US8486943B2 (en) | 2008-05-16 | 2013-07-16 | Eli Lilly And Company | Tetrahydrocyclopenta[b]indole androgen receptor modulators |
| WO2013151668A2 (en) | 2012-04-02 | 2013-10-10 | modeRNA Therapeutics | Modified polynucleotides for the production of secreted proteins |
| WO2013151666A2 (en) | 2012-04-02 | 2013-10-10 | modeRNA Therapeutics | Modified polynucleotides for the production of biologics and proteins associated with human disease |
| EP2690091A1 (en) | 2012-07-25 | 2014-01-29 | Studiengesellschaft Kohle mbH | Process for preparing substituted indole derivatives |
| WO2014041125A1 (en) * | 2012-09-13 | 2014-03-20 | Baden-Württemberg Stiftung Gmbh | Specific inhibitors of protein p21 as therapeutic agents |
| EP2759540A3 (en) * | 2006-08-04 | 2014-08-20 | Universita' Degli Studi di Bari | Beta-3 receptor ligands and their use in therapy |
| US20140303382A1 (en) * | 2011-10-20 | 2014-10-09 | Siena Biotech S.P.A. | Process for the preparation of 6-chloro-2,3,4,9-tetrahydro-1h-carbazole-1-carboxamide and intermediates thereof |
| US8980864B2 (en) | 2013-03-15 | 2015-03-17 | Moderna Therapeutics, Inc. | Compositions and methods of altering cholesterol levels |
| WO2015051214A1 (en) | 2013-10-03 | 2015-04-09 | Moderna Therapeutics, Inc. | Polynucleotides encoding low density lipoprotein receptor |
| US9181319B2 (en) | 2010-08-06 | 2015-11-10 | Moderna Therapeutics, Inc. | Engineered nucleic acids and methods of use thereof |
| WO2016014846A1 (en) | 2014-07-23 | 2016-01-28 | Moderna Therapeutics, Inc. | Modified polynucleotides for the production of intrabodies |
| US9255129B2 (en) | 2012-04-02 | 2016-02-09 | Moderna Therapeutics, Inc. | Modified polynucleotides encoding SIAH E3 ubiquitin protein ligase 1 |
| US9266832B2 (en) | 2010-12-13 | 2016-02-23 | Katholieke Universiteit Levun | Compounds for the treatment of neurodegenerative diseases |
| US9403767B2 (en) | 2008-03-27 | 2016-08-02 | Gruenenthal Gmbh | Substituted 4-aminocyclohexane derivatives |
| US9572897B2 (en) | 2012-04-02 | 2017-02-21 | Modernatx, Inc. | Modified polynucleotides for the production of cytoplasmic and cytoskeletal proteins |
| US9587003B2 (en) | 2012-04-02 | 2017-03-07 | Modernatx, Inc. | Modified polynucleotides for the production of oncology-related proteins and peptides |
| US9657295B2 (en) | 2010-10-01 | 2017-05-23 | Modernatx, Inc. | Modified nucleosides, nucleotides, and nucleic acids, and uses thereof |
| US9950068B2 (en) | 2011-03-31 | 2018-04-24 | Modernatx, Inc. | Delivery and formulation of engineered nucleic acids |
| US10022425B2 (en) | 2011-09-12 | 2018-07-17 | Modernatx, Inc. | Engineered nucleic acids and methods of use thereof |
| US10040804B2 (en) | 2016-12-21 | 2018-08-07 | Biotheryx, Inc. | Compounds targeting proteins, compositions, methods, and uses thereof |
| US11458126B2 (en) | 2017-08-01 | 2022-10-04 | Ptc Therapeutics, Inc. | DHODH inhibitor for use in treating hematologic cancers |
| US11613538B2 (en) | 2009-05-27 | 2023-03-28 | Ptc Therapeutics, Inc. | Method of inhibiting or reducing a viral infection |
| US11744801B2 (en) | 2017-08-31 | 2023-09-05 | Modernatx, Inc. | Methods of making lipid nanoparticles |
| US11786607B2 (en) | 2017-06-15 | 2023-10-17 | Modernatx, Inc. | RNA formulations |
| US12090235B2 (en) | 2018-09-20 | 2024-09-17 | Modernatx, Inc. | Preparation of lipid nanoparticles and methods of administration thereof |
| WO2024230729A1 (en) | 2023-05-10 | 2024-11-14 | 杭州拿因生物科技有限责任公司 | Tetrahydrocarbazole derivative, pharmaceutical composition, and use thereof |
| EP4725481A1 (en) * | 2024-10-10 | 2026-04-15 | Leibniz-Institut für Virologie | 1,2,3,4-tetrahydrocarbazole derivatives for use in treating an adenoviral infection |
Families Citing this family (102)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE10252667A1 (en) * | 2002-11-11 | 2004-05-27 | Grünenthal GmbH | New spiro-((cyclohexane)-tetrahydropyrano-(3,4-b)-indole) derivatives, are ORL1 receptor ligands useful e.g. for treating anxiety, depression, epilepsy, senile dementia, withdrawal symptoms or especially pain |
| US7767689B2 (en) | 2004-03-15 | 2010-08-03 | Ptc Therapeutics, Inc. | Carboline derivatives useful in the treatment of cancer |
| US8076352B2 (en) | 2004-03-15 | 2011-12-13 | Ptc Therapeutics, Inc. | Administration of carboline derivatives useful in the treatment of cancer and other diseases |
| MXPA06010543A (en) | 2004-03-15 | 2007-03-26 | Ptc Therapeutics Inc | Carboline derivatives useful in the inhibition of angiogenesis. |
| US8076353B2 (en) | 2004-03-15 | 2011-12-13 | Ptc Therapeutics, Inc. | Inhibition of VEGF translation |
| US9512125B2 (en) | 2004-11-19 | 2016-12-06 | The Regents Of The University Of California | Substituted pyrazolo[3.4-D] pyrimidines as anti-inflammatory agents |
| DE102005016460A1 (en) * | 2005-04-11 | 2006-10-19 | Grünenthal GmbH | Spriocyclic cyclohexane derivatives for the treatment of substance dependence |
| WO2007062175A2 (en) * | 2005-11-21 | 2007-05-31 | Amgen Inc. | Spiro-substituted tricyclic heterocycles cxcr3 antagonists |
| EP3219705B1 (en) | 2005-12-28 | 2020-03-11 | Vertex Pharmaceuticals Incorporated | Pharmaceutical compositions of the amorphous form of n-[2,4-bis(1,1-dimethylethyl)-5-hydroxyphenyl]-1,4-dihydro-4-oxoquinoline-3-carboxamide |
| WO2007114926A2 (en) | 2006-04-04 | 2007-10-11 | The Regents Of The University Of California | Kinase antagonists |
| ZA200810231B (en) | 2006-07-19 | 2010-07-28 | Du Pont | Process for making 3-substituted 2-amino-5-halobenzamides |
| DE102007009235A1 (en) * | 2007-02-22 | 2008-09-18 | Grünenthal GmbH | Spirocyclic cyclohexane derivatives |
| GB2467670B (en) | 2007-10-04 | 2012-08-01 | Intellikine Inc | Chemical entities and therapeutic uses thereof |
| JP5366211B2 (en) * | 2007-12-18 | 2013-12-11 | 国立大学法人富山大学 | Condensed tricyclic compounds having aldose reductase inhibitory activity |
| EP3613743B1 (en) | 2008-01-04 | 2022-03-16 | Intellikine, LLC | Processes for the preparation of 1h-pyrazolo[3,4-d]pyrimidin-4-amine derivatives |
| US8193182B2 (en) | 2008-01-04 | 2012-06-05 | Intellikine, Inc. | Substituted isoquinolin-1(2H)-ones, and methods of use thereof |
| US8993580B2 (en) | 2008-03-14 | 2015-03-31 | Intellikine Llc | Benzothiazole kinase inhibitors and methods of use |
| US8637542B2 (en) | 2008-03-14 | 2014-01-28 | Intellikine, Inc. | Kinase inhibitors and methods of use |
| AU2009228647B2 (en) | 2008-03-27 | 2013-07-04 | Grunenthal Gmbh | Substituted spirocyclic cyclohexane derivatives |
| ES2375543T3 (en) * | 2008-03-27 | 2012-03-01 | Grünenthal GmbH | DERIVATIVES OF ESPIRO (5.5) UNDECANO. |
| CN102046591B (en) * | 2008-03-27 | 2014-12-03 | 格吕伦塔尔有限公司 | Hydroxymethylcyclohexylamine |
| JP5529843B2 (en) | 2008-03-27 | 2014-06-25 | グリュネンタール・ゲゼルシャフト・ミト・ベシュレンクテル・ハフツング | Substituted cyclohexyldiamine |
| JP5788316B2 (en) | 2008-07-08 | 2015-09-30 | インテリカイン, エルエルシー | Kinase inhibitors and methods of use |
| US20110224223A1 (en) | 2008-07-08 | 2011-09-15 | The Regents Of The University Of California, A California Corporation | MTOR Modulators and Uses Thereof |
| US8703778B2 (en) | 2008-09-26 | 2014-04-22 | Intellikine Llc | Heterocyclic kinase inhibitors |
| WO2010045542A2 (en) | 2008-10-16 | 2010-04-22 | The Regents Of The University Of California | Fused ring heteroaryl kinase inhibitors |
| US8476431B2 (en) | 2008-11-03 | 2013-07-02 | Itellikine LLC | Benzoxazole kinase inhibitors and methods of use |
| WO2010056309A2 (en) * | 2008-11-14 | 2010-05-20 | The Scripps Research Institute | Methods and compositions related to targeting monoacylglycerol lipase |
| EP2427195B1 (en) | 2009-05-07 | 2019-05-01 | Intellikine, LLC | Heterocyclic compounds and uses thereof |
| MX2011012517A (en) | 2009-05-27 | 2012-01-27 | Ptc Therapeutics Inc | Processes for the preparation of substituted tetrahydro beta-carbolines. |
| WO2010138685A1 (en) | 2009-05-27 | 2010-12-02 | Ptc Therapeutics, Inc. | Methods for treating prostate conditions |
| US8697662B2 (en) | 2009-05-27 | 2014-04-15 | Ptc Therapeutics, Inc. | Methods for treating Kaposi sarcoma |
| IN2012DN00352A (en) | 2009-06-16 | 2015-08-21 | Bikam Pharmaceuticals Inc | |
| CN102574787B (en) * | 2009-07-30 | 2014-12-31 | 新加坡国立大学 | Small molecule inhibitors of isoprenylcysteine carboxyl methyltransferase with potential anticancer activity |
| US8980899B2 (en) | 2009-10-16 | 2015-03-17 | The Regents Of The University Of California | Methods of inhibiting Ire1 |
| US9556201B2 (en) * | 2009-10-29 | 2017-01-31 | Glaxosmithkline Llc | Bicyclic pyridines and analogs as sirtuin modulators |
| MX338831B (en) | 2010-02-04 | 2016-05-03 | Radius Health Inc | Selective androgen receptor modulators. |
| EP2568806B1 (en) | 2010-05-12 | 2016-05-11 | Radius Health, Inc. | Therapeutic regimens |
| CA2799579A1 (en) | 2010-05-21 | 2011-11-24 | Intellikine, Inc. | Chemical compounds, compositions and methods for kinase modulation |
| WO2011155983A1 (en) * | 2010-06-07 | 2011-12-15 | Bikam Pharmaceuticals Inc. | Opsin-binding ligands, compositions and methods of use |
| US9133182B2 (en) | 2010-09-28 | 2015-09-15 | Radius Health, Inc. | Selective androgen receptor modulators |
| JP2013545749A (en) | 2010-11-10 | 2013-12-26 | インフィニティー ファーマシューティカルズ, インコーポレイテッド | Heterocyclic compounds and uses thereof |
| WO2012071369A2 (en) | 2010-11-24 | 2012-05-31 | The Trustees Of Columbia University In The City Of New York | A non-retinoid rbp4 antagonist for treatment of age-related macular degeneration and stargardt disease |
| TWI674262B (en) | 2011-01-10 | 2019-10-11 | 美商英菲尼提製藥股份有限公司 | Processes for preparing isoquinolinones and solid forms of isoquinolinones |
| WO2012105610A1 (en) * | 2011-02-02 | 2012-08-09 | 公立大学法人名古屋市立大学 | Medicinal agent for prevention or treatment of diseases associated with intraocular neovascularization and/or intraocular vascular hyperpermeability |
| US9295673B2 (en) | 2011-02-23 | 2016-03-29 | Intellikine Llc | Combination of mTOR inhibitors and P13-kinase inhibitors, and uses thereof |
| ES2587856T3 (en) | 2011-03-18 | 2016-10-27 | Ono Pharmaceutical Co., Ltd. | Tetrahydrocarboline derivative |
| WO2013012915A1 (en) | 2011-07-19 | 2013-01-24 | Infinity Pharmaceuticals Inc. | Heterocyclic compounds and uses thereof |
| MX2014000648A (en) | 2011-07-19 | 2014-09-25 | Infinity Pharmaceuticals Inc | Heterocyclic compounds and uses thereof. |
| KR20140075693A (en) | 2011-08-29 | 2014-06-19 | 인피니티 파마슈티칼스, 인코포레이티드 | Heterocyclic compounds and uses thereof |
| MX370814B (en) | 2011-09-02 | 2020-01-08 | Univ California | Substituted pyrazolo[3,4-d]pyrimidines and uses thereof. |
| CN103030643B (en) * | 2011-09-29 | 2015-04-29 | 中国科学院化学研究所 | Six-ring indole alkaloid and preparation method thereof |
| RU2648950C2 (en) | 2011-10-03 | 2018-04-02 | Модерна Терапьютикс, Инк. | Modified nucleosides, nucleotides and nucleic acids and their application |
| CN104114572A (en) | 2011-12-16 | 2014-10-22 | 现代治疗公司 | Modified nucleosides, nucleotides and nucleic acid compositions |
| EP2606726A1 (en) | 2011-12-21 | 2013-06-26 | Bayer CropScience AG | N-Arylamidine-substituted trifluoroethylsulfide derivatives as acaricides and insecticides |
| US8940742B2 (en) | 2012-04-10 | 2015-01-27 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| WO2013166037A1 (en) | 2012-05-01 | 2013-11-07 | The Trustees Of Columbia University In The City Of New York | Non-retinoid antagonists for treatment of eye disorders |
| US8828998B2 (en) | 2012-06-25 | 2014-09-09 | Infinity Pharmaceuticals, Inc. | Treatment of lupus, fibrotic conditions, and inflammatory myopathies and other disorders using PI3 kinase inhibitors |
| MX2015003874A (en) | 2012-09-26 | 2015-12-16 | Univ California | Modulation of ire1. |
| EP2914296B2 (en) | 2012-11-01 | 2021-09-29 | Infinity Pharmaceuticals, Inc. | Treatment of cancers using pi3 kinase isoform modulators |
| WO2014081507A1 (en) | 2012-11-26 | 2014-05-30 | Moderna Therapeutics, Inc. | Terminally modified rna |
| US9637450B2 (en) | 2013-03-14 | 2017-05-02 | The Trustees Of Columbia University In The City Of New York | Octahydrocyclopentapyrroles, their preparation and use |
| US9938291B2 (en) | 2013-03-14 | 2018-04-10 | The Trustess Of Columbia University In The City Of New York | N-alkyl-2-phenoxyethanamines, their preparation and use |
| EP2968304B1 (en) | 2013-03-14 | 2018-10-10 | The Trustees of Columbia University in the City of New York | 4-phenylpiperidines, their preparation and use |
| US9944644B2 (en) | 2013-03-14 | 2018-04-17 | The Trustees Of Columbia University In The City Of New York | Octahydropyrrolopyrroles their preparation and use |
| US9481667B2 (en) | 2013-03-15 | 2016-11-01 | Infinity Pharmaceuticals, Inc. | Salts and solid forms of isoquinolinones and composition comprising and methods of using the same |
| MX2015014666A (en) | 2013-04-17 | 2016-03-01 | Pfizer | N-piperidin-3-ylbenzamide derivatives for treating cardiovascular diseases. |
| US10561707B2 (en) * | 2013-09-08 | 2020-02-18 | Technion Research And Development Foundation Ltd. | Semaphorin 3C variants, compositions comprising said variants and methods of use thereof in treating eye diseases |
| US10023626B2 (en) | 2013-09-30 | 2018-07-17 | Modernatx, Inc. | Polynucleotides encoding immune modulating polypeptides |
| MX2021012208A (en) | 2013-10-04 | 2023-01-19 | Infinity Pharmaceuticals Inc | Heterocyclic compounds and uses thereof. |
| WO2015051241A1 (en) | 2013-10-04 | 2015-04-09 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| NZ724368A (en) | 2014-03-19 | 2023-07-28 | Infinity Pharmaceuticals Inc | Heterocyclic compounds for use in the treatment of pi3k-gamma mediated disorders |
| KR102359214B1 (en) | 2014-04-04 | 2022-02-07 | 델 마 파마슈티컬스 | Use of dianhydrogalactitol and analogs or derivatives thereof to treat non-small-cell carcinoma of the lung and ovarian cancer |
| WO2015160975A2 (en) | 2014-04-16 | 2015-10-22 | Infinity Pharmaceuticals, Inc. | Combination therapies |
| EP4036094B1 (en) | 2014-04-30 | 2025-12-24 | The Trustees of Columbia University in the City of New York | Substituted 4-phenylpiperidines, their preparation and use |
| CN111978319B (en) | 2014-06-27 | 2023-08-11 | 诺格拉制药有限公司 | Aryl receptor modulators and methods of making and using the same |
| US10570146B2 (en) * | 2014-07-25 | 2020-02-25 | Northeastern University | Urea/carbamates FAAH MAGL or dual FAAH/MAGL inhibitors and uses thereof |
| WO2016040505A1 (en) * | 2014-09-10 | 2016-03-17 | Epizyme, Inc. | Smyd inhibitors |
| WO2016054491A1 (en) | 2014-10-03 | 2016-04-07 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| CN107250113B (en) | 2014-10-07 | 2019-03-29 | 弗特克斯药品有限公司 | Co-crystals of modulators of cystic fibrosis transmembrane conductance regulator proteins |
| LT3319959T (en) | 2015-07-06 | 2021-12-27 | Alkermes, Inc. | HISTON DEACETYLASE HETERO-HALOGEN INHIBITORS |
| EP3319968A1 (en) | 2015-07-06 | 2018-05-16 | Rodin Therapeutics, Inc. | Heterobicyclic n-aminophenyl-amides as inhibitors of histone deacetylase |
| EP4585268A3 (en) | 2015-09-14 | 2025-10-15 | Twelve Therapeutics, Inc. | Solid forms of isoquinolinone derivatives, process of making, compositions comprising, and methods of using the same |
| US10759806B2 (en) | 2016-03-17 | 2020-09-01 | Infinity Pharmaceuticals, Inc. | Isotopologues of isoquinolinone and quinazolinone compounds and uses thereof as PI3K kinase inhibitors |
| WO2017214269A1 (en) | 2016-06-08 | 2017-12-14 | Infinity Pharmaceuticals, Inc. | Heterocyclic compounds and uses thereof |
| LT3474841T (en) | 2016-06-22 | 2022-06-10 | Ellipses Pharma Ltd | Ar+ breast cancer treatment methods |
| RU2754507C2 (en) | 2016-06-24 | 2021-09-02 | Инфинити Фармасьютикалз, Инк. | Combination therapy |
| US11168068B2 (en) | 2016-07-18 | 2021-11-09 | Janssen Pharmaceutica Nv | Tau PET imaging ligands |
| US10624343B2 (en) | 2016-08-26 | 2020-04-21 | Sumitomo Chemical Company, Limited | Phenyl urea compound, and use thereof |
| US10893676B2 (en) | 2016-08-26 | 2021-01-19 | Sumitomo Chemical Company, Limited | 1-acetyl-3-phenyl urea compound, and use thereof |
| RS62959B1 (en) | 2017-01-11 | 2022-03-31 | Alkermes Inc | Bicyclic inhibitors of histone deacetylase |
| WO2018165612A1 (en) | 2017-03-10 | 2018-09-13 | Rutgers, The State University Of New Jersey | Bacterial efflux pump inhibitors |
| CN110770224B (en) | 2017-03-10 | 2022-11-18 | 罗格斯新泽西州立大学 | Indole derivatives as efflux pump inhibitors |
| US11938114B2 (en) | 2017-03-10 | 2024-03-26 | Rutgers, The State University Of New Jersey | Bacterial efflux pump inhibitors |
| EP3630109A4 (en) | 2017-05-26 | 2021-03-17 | Rutgers, the State University of New Jersey | Bacterial efflux pump inhibitors |
| WO2019005841A1 (en) * | 2017-06-26 | 2019-01-03 | Rutgers, The State University Of New Jersey | Therapeutic compounds and methods to treat infection |
| US11225475B2 (en) | 2017-08-07 | 2022-01-18 | Alkermes, Inc. | Substituted pyridines as inhibitors of histone deacetylase |
| US10882821B1 (en) | 2017-09-26 | 2021-01-05 | The Board Of Trustees Of The Leland Stanford Junior University | Enantiomeric compound for the reduction of the deleterious activity of extended nucleotide repeat containing genes |
| CN111787916B (en) | 2018-01-11 | 2023-09-05 | 森陶鲁斯治疗公司 | Dihydroceramide desaturase inhibitors for use in the treatment of diseases |
| EP4267578A4 (en) | 2020-12-23 | 2024-11-20 | Recurium IP Holdings, LLC | ESTROGEN RECEPTOR MODULATORS |
| EP4434968A4 (en) * | 2021-11-19 | 2025-10-22 | Nain Biotech Hangzhou Co Ltd | Tetrahydrocarbazole compound and pharmaceutical composition and use thereof |
| KR20260040871A (en) * | 2024-09-19 | 2026-03-26 | 이화여자대학교 산학협력단 | Pyrazinocarbazole derivatives or pharmaceutically acceptable salts thereof and uses thereof |
Family Cites Families (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE1003577B (en) * | 1955-02-11 | 1957-02-28 | Agfa Ag | Process for producing correct-sided positive images by heat development |
| BE793493A (en) * | 1971-12-30 | 1973-06-29 | Hoffmann La Roche | TRICYCLIC COMPOUNDS |
| BG25793A3 (en) | 1973-07-18 | 1978-12-12 | Schering Aktiengesellschaft | METHOD FOR OBTAINING CARBAZOLE DERIVATIVES |
| US4014890A (en) * | 1976-03-23 | 1977-03-29 | Pfizer Inc. | Process for preparing indole derivatives |
| DE3217563A1 (en) * | 1982-05-11 | 1983-11-17 | Cassella Ag, 6000 Frankfurt | METHOD FOR THE PRODUCTION OF SUBSTITUTED 1,2,3,4-TETRAHYDRO-9-CYANMETHYL-CARBAZOL-1-ONE, IF SUBSTITUTED |
| US4880817A (en) * | 1987-06-09 | 1989-11-14 | Ortho Pharmaceutical Corporation | O-functionalized derivatives of substituted isoquinolin-3-ols having cardiotonic and/or phosphodiesterase fraction III inhibiting properties and/or renal vasodilating properties |
| US5166204A (en) * | 1989-11-01 | 1992-11-24 | Toyama Chemical Co., Ltd. | Isoindole derivatives and salts thereof and antitumor agent comprising the same |
| US5100891A (en) * | 1991-01-18 | 1992-03-31 | Hoechst-Roussel Pharmaceuticals Inc. | Substituted 1,2,3,4-tetrahydrocyclopent[b]indoles, 1,2,3,3a,4,8a-hexahydrocyclopent[B]indoles and related compounds |
| US5192789A (en) * | 1991-01-18 | 1993-03-09 | Hoechst-Roussel Pharmaceuticals Incorporated | Substituted 1,2,3,4-tetrahydrocyclopent[b]indoles, 1,2,3,3a,4,8a-hexahydrocyclopent[b]indoles and related compounds |
| US5298626A (en) * | 1991-01-18 | 1994-03-29 | Hoechst-Roussel Pharmaceuticals Incorporated | Select cyclopent[b]indoles |
| US5451600A (en) * | 1994-04-19 | 1995-09-19 | Hoffmann-La Roche Inc. | Substituted tetrahydrobenzopyrrolylfuranoic acid derivatives as phospholipase A2 inhibitors |
| US5892041A (en) * | 1996-08-12 | 1999-04-06 | Neurogen Corporation | Fused indolecarboxamides: dopamine receptor subtype specific ligands |
| FR2763589B1 (en) * | 1997-05-22 | 2001-01-05 | Centre Nat Rech Scient | COMPOUNDS FOR THE PREPARATION OF MEDICINES FOR THE TREATMENT OF CONDITIONS INVOLVING EXTRACELLULAR GLUTAMATE, AND PROCESSES FOR OBTAINING THEM |
| AR016133A1 (en) * | 1997-07-31 | 2001-06-20 | Wyeth Corp | CARBAMILOXI COMPOUND INHIBITING THE ADHESION OF LEUKOCYTES THROUGH VLA-4, COMPOUNDS THAT ARE DRUGS OF THESE COMPOUNDS, PHARMACEUTICAL COMPOSITION, METHOD FOR SETTING VLA-4 TO A BIOLOGICAL SAMPLE, METHOD FOR THE TREATMENT OF A TREATMENT |
| US6514981B1 (en) * | 1998-04-02 | 2003-02-04 | Sugen, Inc. | Methods of modulating tyrosine protein kinase function with indolinone compounds |
| GB9918962D0 (en) * | 1999-08-11 | 1999-10-13 | Cerebrus Ltd | Chemical compounds xxii |
| CA2385882C (en) * | 1999-09-24 | 2009-11-24 | Genentech, Inc. | Tyrosine derivatives |
| DE19962936A1 (en) | 1999-12-24 | 2001-06-28 | Bayer Ag | New beta-aminoacid amide derivatives, are integrin antagonists useful for treating inflammatory, autoimmune or immunological disorders e.g. asthma, diabetes or rheumatoid arthritis |
| KR20040068240A (en) * | 2001-12-14 | 2004-07-30 | 노보 노르디스크 에이/에스 | Compounds and uses thereof for decreasing activity of hormone-sensitive lipase |
| US20040023961A1 (en) * | 2002-02-11 | 2004-02-05 | Bayer Corporation | Aryl ureas with raf kinase and angiogenisis inhibiting activity |
| JP4743382B2 (en) * | 2002-06-06 | 2011-08-10 | 株式会社医薬分子設計研究所 | O-substituted hydroxyaryl derivatives |
| WO2004020434A1 (en) * | 2002-08-30 | 2004-03-11 | Eisai Co., Ltd. | Azaarene derivatives |
| EP1558591B1 (en) * | 2002-10-07 | 2014-05-07 | The Regents of The University of California | Modulation of anxiety through blockade of anandamide hydrolysis |
| EP1581217A4 (en) * | 2002-11-01 | 2007-07-11 | Merck & Co Inc | CARBONYLAMINO-BENZIMIDAZOLE DERIVATIVES AS MODULATORS OF THE ANDROGEN RECEPTOR |
| WO2004069831A1 (en) * | 2003-02-10 | 2004-08-19 | Glenmark Pharmaceuticals Ltd. | Tricyclic compounds useful for the treatment of inflammatory and allergic disorders: process for their preparation |
| BRPI0407544A (en) * | 2003-02-17 | 2006-02-14 | Pharmacia Italia Spa | tetracyclic pyrazole derivatives as kinase inhibitors, process for their preparation and pharmaceutical compositions comprising them |
| AU2003902023A0 (en) * | 2003-04-29 | 2003-05-15 | The Australian National University | A method of indole synthesis |
| KR20060017545A (en) * | 2003-06-10 | 2006-02-23 | 스미스클라인 비참 코포레이션 | Tetrahydrocarbazole Derivatives and Their Pharmaceutical Uses |
| DE602004022169D1 (en) * | 2003-06-12 | 2009-09-03 | Smithkline Beecham Corp | TETRAHYDROCARBAZOL DERIVATIVES AND THEIR PHARMACEUTICAL USE |
| AR044927A1 (en) * | 2003-06-25 | 2005-10-12 | Elan Pharm Inc | METHODS AND COMPOSITIONS TO TREAT REUMATOID ARTHRITIS |
| CA2538759C (en) * | 2003-09-12 | 2015-11-03 | Elixir Pharmaceuticals, Inc. | Substituted heterocyclic compounds as sirtuin inhitibitors |
| JP2007509057A (en) * | 2003-10-15 | 2007-04-12 | カイロン コーポレイション | Compositions and methods for virus inhibition |
| WO2005097162A2 (en) * | 2004-04-01 | 2005-10-20 | Elan Pharmaceuticals, Inc. | Steroid sparing agents and their use |
| BRPI0508762A (en) * | 2004-04-16 | 2007-08-14 | Genentech Inc | b-cell depletion method in mammals, b-cell depletion efficacy method, b-cell malignancy or neoplasm treatment method, b-cell autoimmune dysfunction relief method, b-cell depletion method composition |
-
2005
- 2005-11-23 US US11/720,061 patent/US8143257B2/en not_active Expired - Fee Related
- 2005-11-23 JP JP2007543449A patent/JP2008520741A/en active Pending
- 2005-11-23 EP EP05852075A patent/EP1817025A2/en not_active Withdrawn
- 2005-11-23 JP JP2007543450A patent/JP2008520742A/en active Pending
- 2005-11-23 MX MX2007006179A patent/MX2007006179A/en not_active Application Discontinuation
- 2005-11-23 MX MX2007006178A patent/MX2007006178A/en not_active Application Discontinuation
- 2005-11-23 US US11/720,055 patent/US8946444B2/en not_active Expired - Fee Related
- 2005-11-23 CA CA002588384A patent/CA2588384A1/en not_active Abandoned
- 2005-11-23 MX MX2007006180A patent/MX2007006180A/en not_active Application Discontinuation
- 2005-11-23 WO PCT/US2005/042483 patent/WO2006065480A2/en not_active Ceased
- 2005-11-23 WO PCT/US2005/042484 patent/WO2006058088A2/en not_active Ceased
- 2005-11-23 WO PCT/US2005/042482 patent/WO2006065479A2/en not_active Ceased
- 2005-11-23 EP EP05852076A patent/EP1824821A2/en not_active Withdrawn
- 2005-11-23 CA CA002588607A patent/CA2588607A1/en not_active Abandoned
- 2005-11-23 JP JP2007543448A patent/JP2008520740A/en active Pending
- 2005-11-23 CA CA002588389A patent/CA2588389A1/en not_active Abandoned
- 2005-11-23 EP EP05852074A patent/EP1828195A2/en not_active Withdrawn
-
2015
- 2015-01-26 US US14/605,721 patent/US9271960B2/en not_active Expired - Lifetime
Cited By (48)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7935722B2 (en) | 2005-06-24 | 2011-05-03 | Eli Lilly And Company | Tetrahydrocarbazole derivatives useful as androgen receptor modulators |
| EP2759540A3 (en) * | 2006-08-04 | 2014-08-20 | Universita' Degli Studi di Bari | Beta-3 receptor ligands and their use in therapy |
| US7968587B2 (en) | 2006-11-20 | 2011-06-28 | Eli Lilly And Company | Tetrahydrocyclopenta[b]indole compounds as androgen receptor modulators |
| US20100267712A1 (en) * | 2007-09-27 | 2010-10-21 | The United States of America, as represented by the Secretary, Department of Health and | Isoindoline compounds for the treatment of spinal muscular atrophy and other uses |
| US9403767B2 (en) | 2008-03-27 | 2016-08-02 | Gruenenthal Gmbh | Substituted 4-aminocyclohexane derivatives |
| US8486943B2 (en) | 2008-05-16 | 2013-07-16 | Eli Lilly And Company | Tetrahydrocyclopenta[b]indole androgen receptor modulators |
| US11613538B2 (en) | 2009-05-27 | 2023-03-28 | Ptc Therapeutics, Inc. | Method of inhibiting or reducing a viral infection |
| US9181319B2 (en) | 2010-08-06 | 2015-11-10 | Moderna Therapeutics, Inc. | Engineered nucleic acids and methods of use thereof |
| US9937233B2 (en) | 2010-08-06 | 2018-04-10 | Modernatx, Inc. | Engineered nucleic acids and methods of use thereof |
| US10064959B2 (en) | 2010-10-01 | 2018-09-04 | Modernatx, Inc. | Modified nucleosides, nucleotides, and nucleic acids, and uses thereof |
| US9657295B2 (en) | 2010-10-01 | 2017-05-23 | Modernatx, Inc. | Modified nucleosides, nucleotides, and nucleic acids, and uses thereof |
| US9266832B2 (en) | 2010-12-13 | 2016-02-23 | Katholieke Universiteit Levun | Compounds for the treatment of neurodegenerative diseases |
| US9950068B2 (en) | 2011-03-31 | 2018-04-24 | Modernatx, Inc. | Delivery and formulation of engineered nucleic acids |
| US10022425B2 (en) | 2011-09-12 | 2018-07-17 | Modernatx, Inc. | Engineered nucleic acids and methods of use thereof |
| US10751386B2 (en) | 2011-09-12 | 2020-08-25 | Modernatx, Inc. | Engineered nucleic acids and methods of use thereof |
| US20140303382A1 (en) * | 2011-10-20 | 2014-10-09 | Siena Biotech S.P.A. | Process for the preparation of 6-chloro-2,3,4,9-tetrahydro-1h-carbazole-1-carboxamide and intermediates thereof |
| US10329254B2 (en) | 2011-10-20 | 2019-06-25 | Aop Orphan Pharmaceuticals Ag | Process for the preparation of 6-chloro-2,3,4,9-tetrahydro-1H-carbazole-1-carboxamide and intermediates thereof |
| US10501512B2 (en) | 2012-04-02 | 2019-12-10 | Modernatx, Inc. | Modified polynucleotides |
| WO2013151736A2 (en) | 2012-04-02 | 2013-10-10 | modeRNA Therapeutics | In vivo production of proteins |
| US9255129B2 (en) | 2012-04-02 | 2016-02-09 | Moderna Therapeutics, Inc. | Modified polynucleotides encoding SIAH E3 ubiquitin protein ligase 1 |
| US9572897B2 (en) | 2012-04-02 | 2017-02-21 | Modernatx, Inc. | Modified polynucleotides for the production of cytoplasmic and cytoskeletal proteins |
| US9587003B2 (en) | 2012-04-02 | 2017-03-07 | Modernatx, Inc. | Modified polynucleotides for the production of oncology-related proteins and peptides |
| WO2013151668A2 (en) | 2012-04-02 | 2013-10-10 | modeRNA Therapeutics | Modified polynucleotides for the production of secreted proteins |
| US9782462B2 (en) | 2012-04-02 | 2017-10-10 | Modernatx, Inc. | Modified polynucleotides for the production of proteins associated with human disease |
| US9814760B2 (en) | 2012-04-02 | 2017-11-14 | Modernatx, Inc. | Modified polynucleotides for the production of biologics and proteins associated with human disease |
| US9828416B2 (en) | 2012-04-02 | 2017-11-28 | Modernatx, Inc. | Modified polynucleotides for the production of secreted proteins |
| US9827332B2 (en) | 2012-04-02 | 2017-11-28 | Modernatx, Inc. | Modified polynucleotides for the production of proteins |
| US9878056B2 (en) | 2012-04-02 | 2018-01-30 | Modernatx, Inc. | Modified polynucleotides for the production of cosmetic proteins and peptides |
| WO2013151666A2 (en) | 2012-04-02 | 2013-10-10 | modeRNA Therapeutics | Modified polynucleotides for the production of biologics and proteins associated with human disease |
| WO2014016296A1 (en) | 2012-07-25 | 2014-01-30 | Studiengesellschaft Kohle Mbh | Process for preparing substituted indole derivatives |
| US9399621B2 (en) | 2012-07-25 | 2016-07-26 | Studiengesellschaft Kohle Mbh | Process for preparing substituted indole derivatives |
| EP2690091A1 (en) | 2012-07-25 | 2014-01-29 | Studiengesellschaft Kohle mbH | Process for preparing substituted indole derivatives |
| WO2014041125A1 (en) * | 2012-09-13 | 2014-03-20 | Baden-Württemberg Stiftung Gmbh | Specific inhibitors of protein p21 as therapeutic agents |
| US8980864B2 (en) | 2013-03-15 | 2015-03-17 | Moderna Therapeutics, Inc. | Compositions and methods of altering cholesterol levels |
| WO2015051214A1 (en) | 2013-10-03 | 2015-04-09 | Moderna Therapeutics, Inc. | Polynucleotides encoding low density lipoprotein receptor |
| US10323076B2 (en) | 2013-10-03 | 2019-06-18 | Modernatx, Inc. | Polynucleotides encoding low density lipoprotein receptor |
| WO2016014846A1 (en) | 2014-07-23 | 2016-01-28 | Moderna Therapeutics, Inc. | Modified polynucleotides for the production of intrabodies |
| US10889593B2 (en) | 2016-12-21 | 2021-01-12 | Biotheryx, Inc. | Compounds targeting proteins, compositions, methods, and uses thereof |
| US10336771B2 (en) | 2016-12-21 | 2019-07-02 | Biotheryx, Inc. | Compounds targeting proteins, compositions, methods, and uses thereof |
| US11345714B2 (en) | 2016-12-21 | 2022-05-31 | Biotheryx, Inc. | Compounds targeting proteins, compositions, methods, and uses thereof |
| US10040804B2 (en) | 2016-12-21 | 2018-08-07 | Biotheryx, Inc. | Compounds targeting proteins, compositions, methods, and uses thereof |
| US11786607B2 (en) | 2017-06-15 | 2023-10-17 | Modernatx, Inc. | RNA formulations |
| US11458126B2 (en) | 2017-08-01 | 2022-10-04 | Ptc Therapeutics, Inc. | DHODH inhibitor for use in treating hematologic cancers |
| US11744801B2 (en) | 2017-08-31 | 2023-09-05 | Modernatx, Inc. | Methods of making lipid nanoparticles |
| US12357575B2 (en) | 2017-08-31 | 2025-07-15 | Modernatx, Inc. | Methods of making lipid nanoparticles |
| US12090235B2 (en) | 2018-09-20 | 2024-09-17 | Modernatx, Inc. | Preparation of lipid nanoparticles and methods of administration thereof |
| WO2024230729A1 (en) | 2023-05-10 | 2024-11-14 | 杭州拿因生物科技有限责任公司 | Tetrahydrocarbazole derivative, pharmaceutical composition, and use thereof |
| EP4725481A1 (en) * | 2024-10-10 | 2026-04-15 | Leibniz-Institut für Virologie | 1,2,3,4-tetrahydrocarbazole derivatives for use in treating an adenoviral infection |
Also Published As
| Publication number | Publication date |
|---|---|
| US20090042866A1 (en) | 2009-02-12 |
| EP1824821A2 (en) | 2007-08-29 |
| CA2588607A1 (en) | 2006-06-01 |
| US20080261956A1 (en) | 2008-10-23 |
| JP2008520740A (en) | 2008-06-19 |
| MX2007006179A (en) | 2007-06-20 |
| US9271960B2 (en) | 2016-03-01 |
| US20150141418A1 (en) | 2015-05-21 |
| CA2588389A1 (en) | 2006-06-22 |
| WO2006065480A3 (en) | 2006-08-03 |
| US8946444B2 (en) | 2015-02-03 |
| JP2008520741A (en) | 2008-06-19 |
| MX2007006180A (en) | 2007-06-20 |
| EP1817025A2 (en) | 2007-08-15 |
| JP2008520742A (en) | 2008-06-19 |
| WO2006065479A2 (en) | 2006-06-22 |
| US8143257B2 (en) | 2012-03-27 |
| WO2006058088A2 (en) | 2006-06-01 |
| WO2006058088A3 (en) | 2006-12-07 |
| CA2588384A1 (en) | 2006-06-22 |
| WO2006065479A3 (en) | 2006-08-03 |
| MX2007006178A (en) | 2007-06-20 |
| EP1828195A2 (en) | 2007-09-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US9271960B2 (en) | Tetrahydrocarbazoles as active agents for inhibiting VEGF production by translational control | |
| KR101188778B1 (en) | Carboline derivatives useful in the inhibition of angiogenesis | |
| CN101594862B (en) | Substituted indazole derivatives as kinase inhibitors | |
| US5618833A (en) | 1-benzyl-1,3-dihydroindol-2-one derivatives, their preparation and the pharmaceutical compositions in which they are present | |
| KR20210102328A (en) | THRβ receptor agonist compounds and methods for their preparation and uses | |
| EP1732543B1 (en) | Tetracyclic carboline deratives for inhibiting angiogenesis | |
| US8569355B2 (en) | Pyrone-indole derivatives and process for their preparation | |
| US7141581B2 (en) | Indazole compounds and pharmaceutical compositions for inhibiting protein kinases, and methods for their use | |
| AU2012388221A1 (en) | Pro-neurogenic compounds | |
| JP2002535331A (en) | Novel angiogenesis inhibitors | |
| CA2491477A1 (en) | Indolin phenylsulfonamide derivatives | |
| BRPI0012352B1 (en) | pharmaceutically acceptable salt or indazole compounds and pharmaceutical composition | |
| US20030008909A1 (en) | 3,3-substituted indoline derivatives | |
| JP2008536876A (en) | Carboline derivatives useful for cancer treatment | |
| JPWO2002094809A1 (en) | 3-quinolin-2 (1H) -ylideneindoline-2-one derivatives | |
| JPH09502177A (en) | Indole and indoline derivatives for 5HT1D receptor antagonists | |
| CN101103000B (en) | Carbazole, Carboline and Indole Derivatives for Inhibition of VEGF Production | |
| AU2017300841A1 (en) | Imidazolyl-substituted indole derivatives binding 5-HT7 serotonin receptor and pharmaceutical compositions thereof | |
| CN120058702A (en) | Heterocyclic TEAD inhibitors | |
| US6107292A (en) | Indenoindoles and benzocarbazoles as estrogenic agents |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KM KN KP KR KZ LC LK LR LS LT LU LV LY MA MD MG MK MN MW MX MZ NA NG NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SM SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU LV MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2588384 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2007/006179 Country of ref document: MX Ref document number: 2007543449 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2005852075 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005852075 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200580046673.X Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005852075 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11720055 Country of ref document: US |