WO2006055434A2 - Macrocyclic tertiary amine beta-secretase inhibitors for the treatment of alzheimer's disease - Google Patents
Macrocyclic tertiary amine beta-secretase inhibitors for the treatment of alzheimer's disease Download PDFInfo
- Publication number
- WO2006055434A2 WO2006055434A2 PCT/US2005/040984 US2005040984W WO2006055434A2 WO 2006055434 A2 WO2006055434 A2 WO 2006055434A2 US 2005040984 W US2005040984 W US 2005040984W WO 2006055434 A2 WO2006055434 A2 WO 2006055434A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- group
- aryl
- cycloalkyl
- mmol
- Prior art date
Links
- 0 *C(Cc1cccc(*2)c1)(*c1cc2c(*)c(*)c1*)N Chemical compound *C(Cc1cccc(*2)c1)(*c1cc2c(*)c(*)c1*)N 0.000 description 5
- MFOIMMLUCRRNIC-UHFFFAOYSA-N CC1=[O](C2)C2C=CO1 Chemical compound CC1=[O](C2)C2C=CO1 MFOIMMLUCRRNIC-UHFFFAOYSA-N 0.000 description 1
- GRAYGCZWJPKJIG-UHFFFAOYSA-N CCCN(c1cc(C(O)=O)cc(C(OC)=O)c1)S(C)(=O)=O Chemical compound CCCN(c1cc(C(O)=O)cc(C(OC)=O)c1)S(C)(=O)=O GRAYGCZWJPKJIG-UHFFFAOYSA-N 0.000 description 1
- DUZLZTAQNSURNV-UHFFFAOYSA-N CCCN(c1cc(CBr)cc(/C=[O]\C)c1)[S](C)(C)(=O)=O Chemical compound CCCN(c1cc(CBr)cc(/C=[O]\C)c1)[S](C)(C)(=O)=O DUZLZTAQNSURNV-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D273/00—Heterocyclic compounds containing rings having nitrogen and oxygen atoms as the only ring hetero atoms, not provided for by groups C07D261/00 - C07D271/00
- C07D273/01—Heterocyclic compounds containing rings having nitrogen and oxygen atoms as the only ring hetero atoms, not provided for by groups C07D261/00 - C07D271/00 having one nitrogen atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D267/00—Heterocyclic compounds containing rings of more than six members having one nitrogen atom and one oxygen atom as the only ring hetero atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D313/00—Heterocyclic compounds containing rings of more than six members having one oxygen atom as the only ring hetero atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D321/00—Heterocyclic compounds containing rings having two oxygen atoms as the only ring hetero atoms, not provided for by groups C07D317/00 - C07D319/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D323/00—Heterocyclic compounds containing more than two oxygen atoms as the only ring hetero atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D498/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D498/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and oxygen atoms as the only ring hetero atoms in which the condensed system contains two hetero rings
- C07D498/08—Bridged systems
Definitions
- the invention is directed to the field of compounds which are inhibitors of the the activity of the ⁇ -secretase enzyme, and to the use of the compounds for the treatment of diseases in which the ⁇ - secretase enzyme is involved, such as Alzheimer's disease.
- Alzheimer's disease is characterized by the abnormal deposition of amyloid in the brain in the form of extra-cellular plaques and intra-cellular neurofibrillary tangles.
- the rate of amyloid accumulation is a combination of the rates of formation, aggregation and egress from the brain. It is generally accepted that the main constituent of amyloid plaques is the 4kD amyloid protein ( ⁇ A4, also referred to as A ⁇ , ⁇ -protein and ⁇ AP) which is a proteolytic product of a precursor protein of much larger size.
- the amyloid precursor protein (APP or A ⁇ PP) has a receptor-like structure with a large ectodomain, a membrane spanning region and a short cytoplasmic tail.
- the A ⁇ domain encompasses parts of both extra-cellular and transmembrane domains of APP, thus its release implies the existence of two distinct proteolytic events to generate its NH 2 - and COOH-termini. At least two secretory mechanisms exist which release APP from the membrane and generate soluble, COOH-truncated forms of APP (APP s ). Proteases that release APP and its fragments from the membrane are termed "secretases.” Most APP s is released by a putative ⁇ -secretase which cleaves within the A ⁇ protein to release ⁇ -APP s and precludes the release of intact A ⁇ .
- ⁇ -secretase a ⁇ - secretase
- CTFs COOH-terminal fragments
- BACE amyloid precursor protein-cleaving enzyme
- therapeutic agents that can inhibit ⁇ -secretase or BACE may be useful for the treatment of Alzheimer's disease.
- the compounds of the present invention are useful for treating Alzheimer's disease by inhibiting the activity of ⁇ -secretase or BACE, thus preventing the formation of insoluble A ⁇ and arresting the production of A ⁇ .
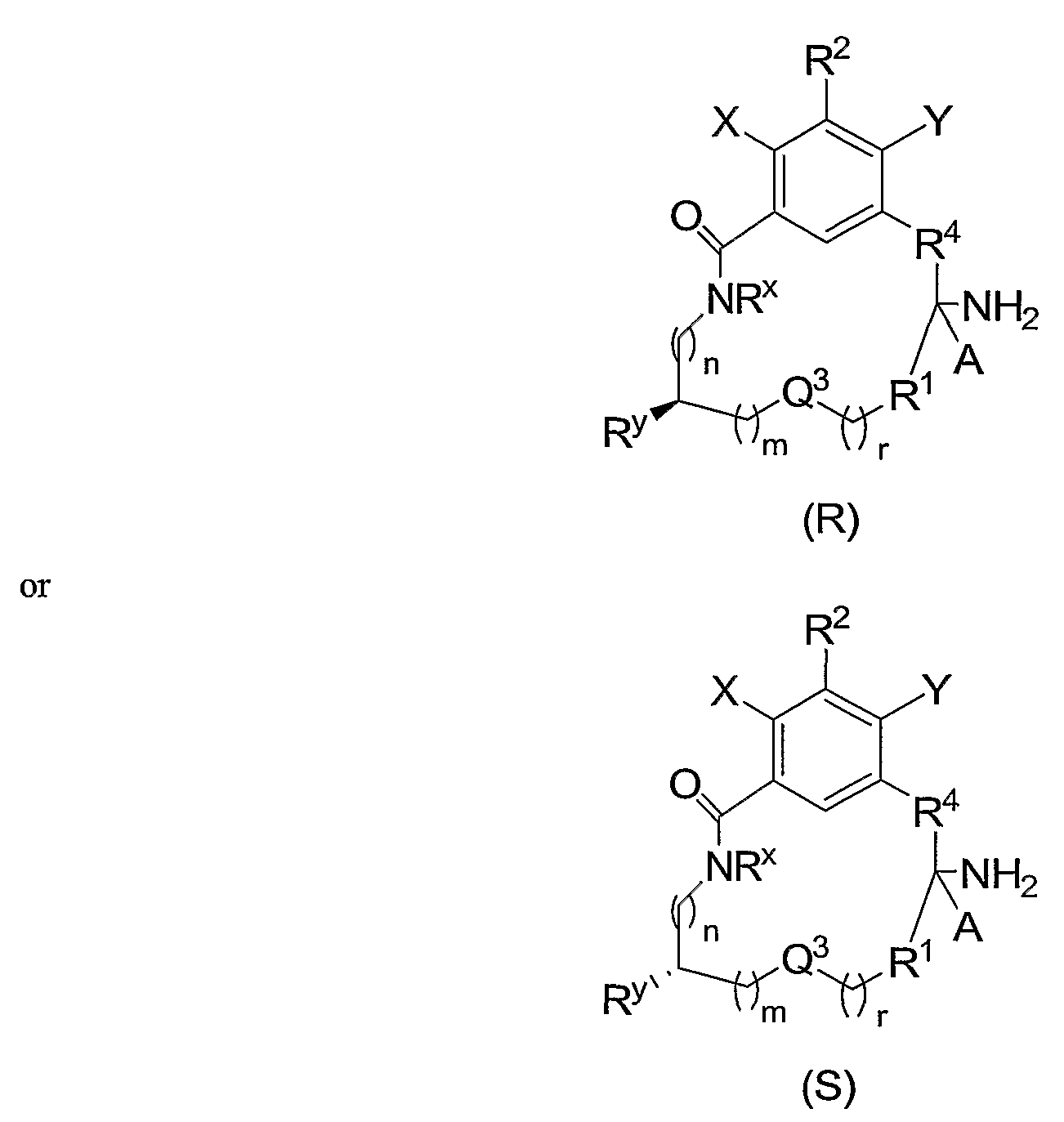
- the present invention is directed to novel macrocyclic tertiary amine compounds represented by general formula (I)
- the invention is also directed to pharmaceutical compositions which include an effective amount of a compound of formula (I), or pharmaceutically acceptable salts thereof, and individual enantiomers and diastereomers thereof, and a pharmaceutically acceptable carrier.
- the invention is also directed to methods of treating mammals for diseases in which the ⁇ -secretase enzyme is involved, such as Alzheimer's disease, and the use of the compounds and pharmaceutical compositions of the invention in the treatment of such diseases.
- the present invention is directed to compounds of formula (I):
- X and Y are selected from the group consisting of (1) hydrogen, (2) -C1-3 alkyl,
- A is selected from the group consisting of
- heteroaryl selected from the group consisting of pyrazinyl, pyrazolyl, pyridazinyl, pyridyl, pyrimidinyl, pyrrolyl, tetrazolyl, furanyl, imidazolyl, triazinyl, pyranyl, thiazolyl, thienyl, thiophenyl, triazolyl, oxazolyl, isoxazolyl, thiazolyl, oxadiazolyl, indolyl, quinolinyl, isoquinolinyl, benzimidazolyl and benzoxazolyl,
- aryl and heteroaryl groups are unsubstituted or substituted with one or more (i) halo
- R1 is selected from the group consisting of (1) -C6-10 arylene, or
- heteroarylene selected from the group consisting of divalent pyrazinyl, pyrazolyl, pyridazinyl, pyridyl, pyrimidinyl, pyrrolyl, tetrazolyl, furanyl, imidazolyl, triazinyl, pyranyl, thiazolyl, thienyl, thiophenyl, triazolyl, oxazolyl, isoxazolyl, thiazolyl, oxadiazolyl, indolyl, quinolinyl, isoquinolinyl, benzimidazolyl and benzoxazolyl,
- arylene or heteroarylene is unsubstituted or substituted with one or more
- R2 is selected from the group consisting of: (1) (R5-SO2)N(R6)-, wherein R5 is selected from the group consisting of: (1) (R5-SO2)N(R6)-, wherein R5 is selected from the group consisting of: (1) (R5-SO2)N(R6)-, wherein R5 is selected from the group consisting of: (1) (R5-SO2)N(R6)-, wherein R5 is selected from the group consisting of: (1) (R5-SO2)N(R6)-, wherein R5 is
- heteroaryl selected from the group consisting of pyrazinyl, pyrazolyl, pyridazinyl, pyridyl, pyrimidinyl, pyrrolyl, tetrazolyl, furanyl, imidazolyl, triazinyl, pyranyl, thiazolyl, thienyl, thiophenyl, triazolyl, oxazolyl, isoxazolyl, thiazolyl, oxadiazolyl, indolyl, quinolinyl, isoquinolinyl, benzimidazolyl and benzoxazolyl,
- alkyl, alkenyl, alkynyl, cycloalkyl, aryl and heteroaryl is unsubstituted or substituted with one or more (i) halo
- heteroaryl selected from the group consisting of pyrazinyl, pyrazolyl, pyridazinyl, pyridyl, pyrimidinyl, pyrrolyl, tetrazolyl, furanyl, imidazolyl, triazinyl, pyranyl, thiazolyl, thienyl, thiophenyl, triazolyl, oxazolyl, isoxazolyl, thiazolyl, oxadiazolyl, indolyl, quinolinyl, isoquinolinyl, benzimidazolyl and benzoxazolyl, and said aryl and heteroaryl is unsubstituted or substituted with one or more
- R6 is selected from the group consisting of
- heteroaryl selected from the group consisting of pyrazinyl, pyrazolyl, pyridazinyl, pyridyl, pyrimidinyl, pyrrolyl, tetrazolyl, furanyl, imidazolyl, triazinyl, pyranyl, thiazolyl, thienyl, thiophenyl, triazolyl, oxazolyl, isoxazolyl, thiazolyl, oxadiazolyl, indolyl, quinolinyl, isoquinolinyl, benzimidazolyl and benzoxazolyl,
- alkyl, alkenyl, alkynyl, aryl or heteroaryl is unsubstituted or substituted with one or more
- heteroaryl selected from the group consisting of pyrazinyl, pyrazolyl, pyridazinyl, pyridyl, pyrimidinyl, pyrrolyl, tetrazolyl, furanyl, imidazolyl, triazinyl, pyranyl, thiazolyl, thienyl, thiophenyl, triazolyl, oxazolyl, isoxazolyl, thiazolyl, oxadiazolyl, indolyl, quinolinyl, isoquinolinyl, benzimidazolyl and benzoxazolyl;
- cycloalkyl, aryl or heteroaryl is unsubstituted or substituted with one or more
- R5 and R6 may be linked to form a group -CH2(CH2)pCH2-;
- heteroaryl selected from the group consisting of pyrazinyl, pyrazolyl,pyridazinyl, pyridyl, pyrimidinyl, pyrrolyl, tetrazolyl, furanyl, imidazolyl, triazinyl, pyranyl, thiazolyl, thienyl, thiophenyl, triazolyl, oxazolyl, isoxazolyl, thiazolyl, oxadiazolyl, indolyl, quinolinyl, isoquinolinyl, benzimidazolyl and benzoxazolyl, wherein said heteroaryl is unsubstituted or substituted with one or more (i) halo, ( ⁇ ) -OH, (iii) -CN,
- R3 is selected from the group consisting of
- R x is selected from the group consisting of
- RY is selected from the group consisting of (a) hydrogen, (b) -C1-10 alkyl, (c)-C2-10 alkenyl, (d) -C2-10 alkynyl,
- heteroaryl is selected from the group consisting of pyrazinyl, pyrazolyl, pyridazinyl, pyridyl, pyrimidinyl, pyrrolyl, tetrazolyl, furanyl, imidazolyl, triazinyl, pyranyl, thiazolyl, thienyl, thiophenyl, triazolyl, oxazolyl, isoxazolyl, thiazolyl, oxadiazolyl, indolyl, quinolinyl, isoquinolinyl, benzimidazolyl and benzoxazolyl,
- RY alkyl, alkylene, alkenyl,alkynyl, cycloalkyl and heteroaryl groups are unsubstituted with one or more (i) halo,
- (c) CH 2 wherein said alkyl, alkylene, cycloalkyl, aryl or heteroaryl Ry groups are unsubstituted or substituted with one or more (i) halo, (ii) -C1-10 alkyl, (iii) -OH, (iv) -CN, or (v) -O-C1-10 alkyl, or (vi) -C3-8 cycloalkyl;
- Q3, Q4 and Q5 are selected from the group consisting of (a) -CH 2 - (b) -O-, and (c) -NH-;
- R7 and R8 are selected from the group consisting of (I) -C1-10 alkyl, and
- R9 is selected from the group consisting of (1) -C1-10 alkyl
- R9 is NR7R8
- n 0, 1 or 2
- p is 1, 2, 3, 4 or 5
- q is 2, 3, 4 or 5
- r is 0, 1 or 2.
- X and Y are both hydrogen.
- R1 is unsubstituted or substituted -C6-10 arylene, preferably unsubstituted phenylene.
- R4 is -(CH2)n-Q2 -(CH2)m, wherein Q2 is selected from the group consisting of
- n and m are preferably each 1.
- R3 is as depicted in paragraph (1) below:
- R ⁇ is preferably hydrogen.
- Ry is preferably selected from the group consisting of
- heteroaryl is selected from the group consisting of pyrazinyl, pyrazolyl, pyridazinyl, pyridyl, pyrimidinyl, pyrrolyl, tetrazolyl, furanyl, imidazolyl, triazinyl, pyranyl, thiazolyl, thienyl, thiophenyl, triazolyl, oxazolyl, isoxazolyl, thiazolyl, oxadiazolyl, indolyl, quinolinyl, isoquinolinyl, benzimidazolyl and benzoxazolyl, wherein said alkyl, alkylene, alkenyl, alkynyl, cycloalkyl, aryl or heteroaryl are unsubstituted or substituted with one or more (i) halo,
- Q3 is preferably -O- or -CH2
- m is preferably 1
- n and r are each preferably O.
- R x and Ry are not both hydrogen.
- R3 is as depicted in paragraph (2) below:
- RY is preferably selected from the group consisting of (a) -C1-10 alkyl,
- heteroaryl is selected from the group consisting of pyrazinyl, pyrazolyl, pyridazinyl, pyridyl, pyrimidinyl, pyrrolyl, tetrazolyl, furanyl, imidazolyl, triazinyl, pyranyl, thiazolyl, thienyl, thiophenyl, triazolyl, oxazolyl, isoxazolyl, thiazolyl, oxadiazolyl, indolyl, quinolinyl, isoquinolinyl, benzimidazolyl and benzoxazolyl, wherein said alkyl, alkenyl, alkynyl, cycloalkyl, aryl or heteroaryl are unsubstituted or substituted with one or
- (b) CH-C0-6 alkylene-C6-10 aryl, wherein said alkyl, alkylene, aryl or heteroaryl groups are unsubstituted or substituted with one or more
- Q4 is — O— or -CH2— and Q5 is — O— or -CH2-.
- n and m are each 1 , and r is preferably 0.
- A is selected from the group consisting of
- heteroaryl selected from the group consisting of pyrazinyl, pyrazolyl, pyridazinyl, pyridyl, pyrimidinyl, pyrrolyl, tetrazolyl, furanyl, imidazolyl, triazinyl, pyranyl, thiazolyl, thienyl, thiophenyl, triazolyl, oxazolyl, isoxazolyl, thiazolyl, oxadiazolyl, indolyl, quinolinyl, isoquinolinyl, benzimidazolyl and benzoxazolyl.
- A is -C1 -10 alkyl
- alkyl (preferably methyl), wherein said alkyl is unsubstituted or substituted with one or more halo (preferably fiuoro).
- R2 is selected from the group consisting of (R5-SO2)N(R6)-, wherein R5 is -C1-6 alkyl, wherein said alkyl is unsubstituted or substituted with one or more
- R6 is selected from the group consisting of
- R5 and R6 are linked to form a group -CH2(CH2)pCH2-.
- Another preferred R2 group is-C6-10 aryl, unsubstituted or substituted as described above.
- Preferred aryl groups are phenyl groups, unsubstituted or substituted with cyano.
- a preferred R2 substituent is shown below:
- R2 substituent is heteroaryl, either unsubstituted or substituted as described above.
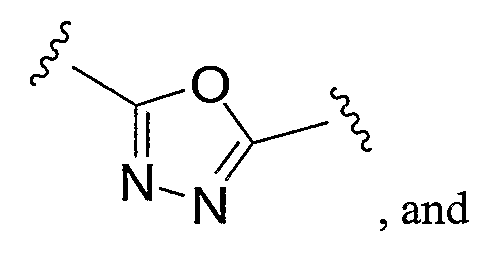
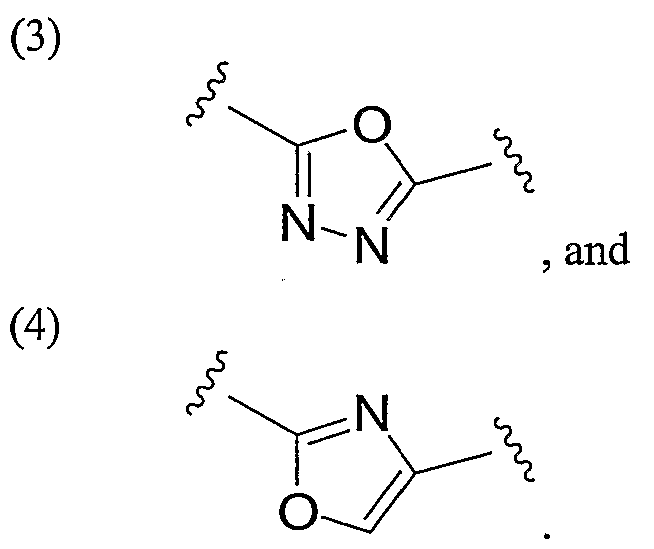
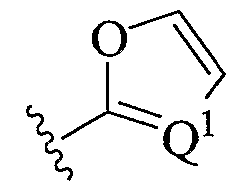
- a preferred heteroaryl group is furanyl or oxazolyl, either unsubstituted or substituted as described above.
- a preferred furanyl or oxazolyl substituent is depicted below:
- Q1 is selected from the group consisting of
- Another embodiment of the present invention is directed to compounds of formula (H):
- X and Y are both hydrogen.
- R4 is -(CH2)n-Q2 - (CH2)m- > wherein Q2 is selected from the group consisting of
- n and m are preferably each 1.
- R3 is as depicted in paragraph (1) below:
- R ⁇ is preferably hydrogen.
- RY is preferably selected from the group consisting of (a) -C1-10 alkyl, (b)-C2-10 alkenyl,
- heteroaryl is selected from the group consisting of pyrazinyl, pyrazolyl, pyridazinyl, pyridyl, pyrimidinyl, pyrrolyl, tetrazolyl, furanyl, imidazolyl, triazinyl, pyranyl, thiazolyl, thienyl, thiophenyl, triazolyl, oxazolyl, isoxazolyl, thiazolyl, oxadiazolyl, indolyl, quinolinyl, isoquinolinyl, benzimidazolyl and benzoxazolyl, wherein said alkyl, alkylene, alkenyl, alkynyl, cycloalkyl, aryl or heteroaryl are unsubstituted or substituted with one or more (i) halo,
- RY is preferably selected from the group consisting of
- R3 is as depicted in paragraph (2) below:
- Ry is preferably selected from the group consisting of (a) -C1-10 alkyl,
- heteroaryl is selected from the group consisting of pyrazinyl, pyrazolyl, pyridazinyl, pyridyl, pyrimidinyl, pyrrolyl, tetrazolyl, furanyl, imidazolyl, triazinyl, pyranyl, thiazolyl, thienyl, thiophenyl, triazolyl, oxazolyl, isoxazolyl, thiazolyl, oxadiazolyl, indolyl, quinolinyl, isoquinolinyl, benzimidazolyl and benzoxazolyl, wherein said alkyl, alkylene, alkenyl, alkynyl, cycloalkyl, aryl or heteroaryl are unsubstituted
- (b) CH-C0-6 alkylene-C6-10 aryl, wherein said alkyl, alkylene, aryl or heteroaryl groups are unsubstituted or substituted with one or more
- Q4 is -O- or -CH2- and Q5 is -O- or — CH2 -.
- n and m are each 1 and r is preferably 0.
- A is selected from the group consisting of
- heteroaryl selected from the group consisting of pyrazinyl, pyrazolyl, pyridazinyl, pyridyl, pyrimidinyl, pyrrolyl, tetrazolyl, furanyl, imidazolyl, triazinyl, pyranyl, thiazolyl, thienyl, thiophenyl, triazolyl, oxazolyl, isoxazolyl, thiazolyl, oxadiazolyl, indolyl, quinolinyl, isoquinolinyl, benzimidazolyl and benzoxazolyl.
- A is -C1-10 alkyl
- alkyl (preferably methyl), wherein said alkyl is unsubstituted or substituted with one or more halo (preferably fluoro).
- R2 is selected from the group consisting of (R5-SO2)N(R6)-, wherein R5 is -C1-6 alkyl, wherein said alkyl is unsubstituted or substituted with one or more
- R6 is selected from the group consisting of
- R5 and R6 may be linked to form a group -CH2(CH2) p CH2-.
- Another preferred R2 group is— C6-10 aryl, unsubstituted or substituted as described above.
- Preferred aryl groups are phenyl groups, unsubstituted or substituted with cyano.
- a preferred R2 substituent is shown below:
- R2 substituent is heteroaryl, either unsubstituted or substituted as described above.
- Preferred heteroaryl groups include furanyl or oxazolyl, either unsubstituted or substituted as described above.
- a preferred furanyl or oxazolyl substituent is depicted below:
- Ql is selected from the group consisting of
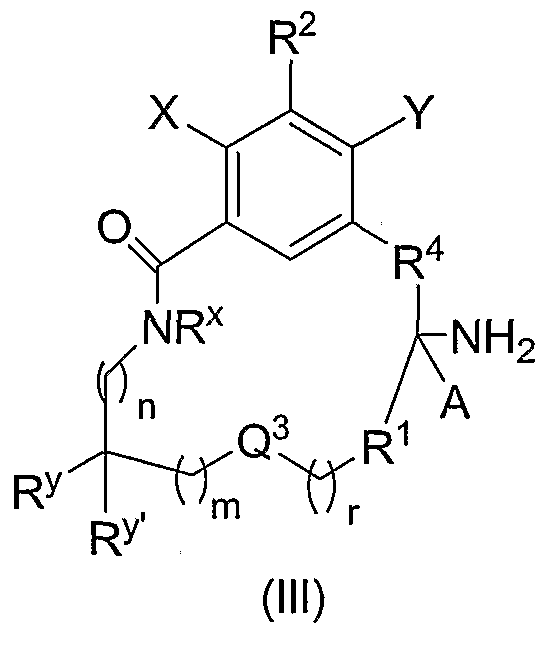
- Another embodiment of the present invention is directed to compounds of the formula (III)
- A, X, Y, R1, R2, R4, R x , R y , R y' Q3, m, n and r are as defined above, and pharmaceutically acceptable salts thereof, and individual enantiomers and diastereomers thereof.
- X and Y are both hydrogen.
- R4 is -(CH2)n-Q2 -(CH2)m, wherein Q2 is selected from the group consisting of (I)-O-,
- n and m are preferably each 1.
- A is selected from the group consisting of
- heteroaryl selected from the group consisting of pyrazinyl, pyrazolyl, pyridazinyl, pyridyl, pyrimidinyl, pyrrolyl, tetrazolyl, furanyl, imidazolyl, triazinyl, pyranyl, thiazolyl, thienyl, thiophenyl, triazolyl, oxazolyl, isoxazolyl, thiazolyl, oxadiazolyl, indolyl, quinolinyl, isoquinolinyl, benzimidazolyl and benzoxazolyl.
- A is -Ci_io alkyl
- alkyl (preferably methyl), wherein said alkyl is unsubstituted or substituted with one or more halo (preferably fluoro).
- R2 is selected from the group consisting of (R5-SO2)N(R6)-, wherein R5 is -C 1-6 alkyl, wherein said alkyl is unsubstituted or substituted with one or more
- R6 is selected from the group consisting of (a) hydrogen,
- R5 and R6 may be linked to form a group -CH2(CH2)pCH2-.
- Another preferred R2 group is-C6-10 aryl, unsubstituted or substituted as described above.
- Preferred aryl groups are phenyl groups, unsubstituted or substituted with cyano.
- a preferred R2 substituent is shown below:
- R2 substituent is ; wherein p is 1, 2 or 3.
- R2 substituent is heteroaryl, either unsubstituted or substituted as described above.
- Preferred heteroaryl groups include furanyl and oxazolyl, either unsubstituted or substituted as described above.
- a preferred furanyl or oxazolyl substiturent is depicted below:
- Q1 is selected from the group consisting of
- Another embodiment of the present invention is directed to compounds of the formula (IV):
- A, X,Y, R1, R 2 , R4, Ry, Ry' Q4, Q5, m, n and r are as defined above, and pharmaceutically acceptable salts thereof, and individual enantiomers and diastereomers thereof.
- X and Y are both hydrogen.
- R4 is -(CH2) n Q2 - (CH2)m, wherein Q2 is selected from the group consisting of (I)-O-,
- n and m are preferably each 1.
- A is selected from the group consisting of
- A is — C1-10 alkyl (preferably methyl), wherein said alkyl is unsubstituted or substituted with one or more halo (preferably fluoro).
- R2 is selected from the group consisting of (R5-SO2)N(R6)-, wherein R5 is -C1-6 alkyl, wherein said alkyl is unsubstituted or substituted with one or more (i) halo,
- R6 is selected from the group consisting of (a) hydrogen, (b) -C1-6 alkyl, or
- R 5 and R6 may be linked to form a group -CH2(CH2)pCH2 ⁇ .
- R2 group is-C6-10 aryl, unsubstituted or substituted as described above.
- Preferred aryl groups are phenyl groups, unsubstituted or substituted with cyano.
- a preferred R2 substituent is shown below:
- R2 substituent is heteroaryl, either unsubstituted or substituted as described above.
- Preferred heteroaryl groups include furanyl or oxazolyl, either unsubstituted or substituted as described above.
- a preferred furanyl or oxazolyl substituent is depicted below:
- Ql is selected from the group consisting of (a) N, and
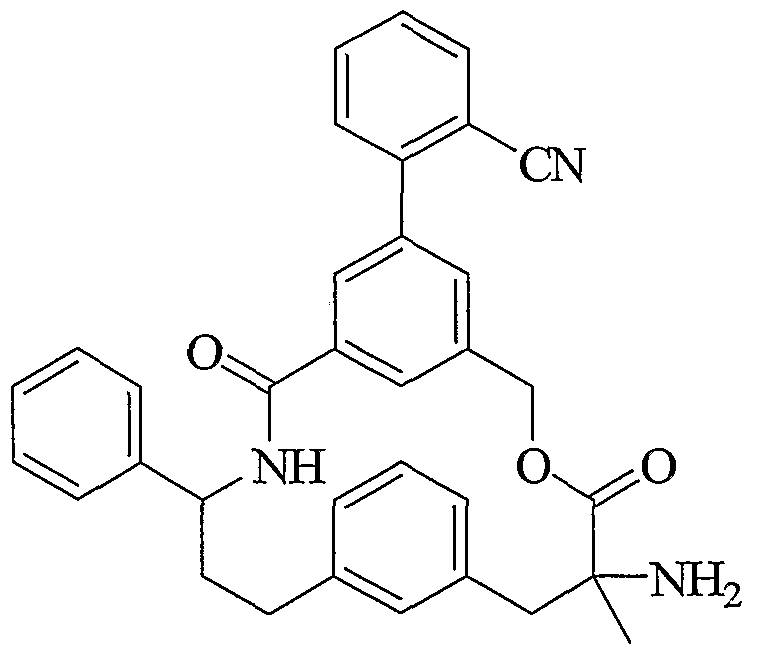
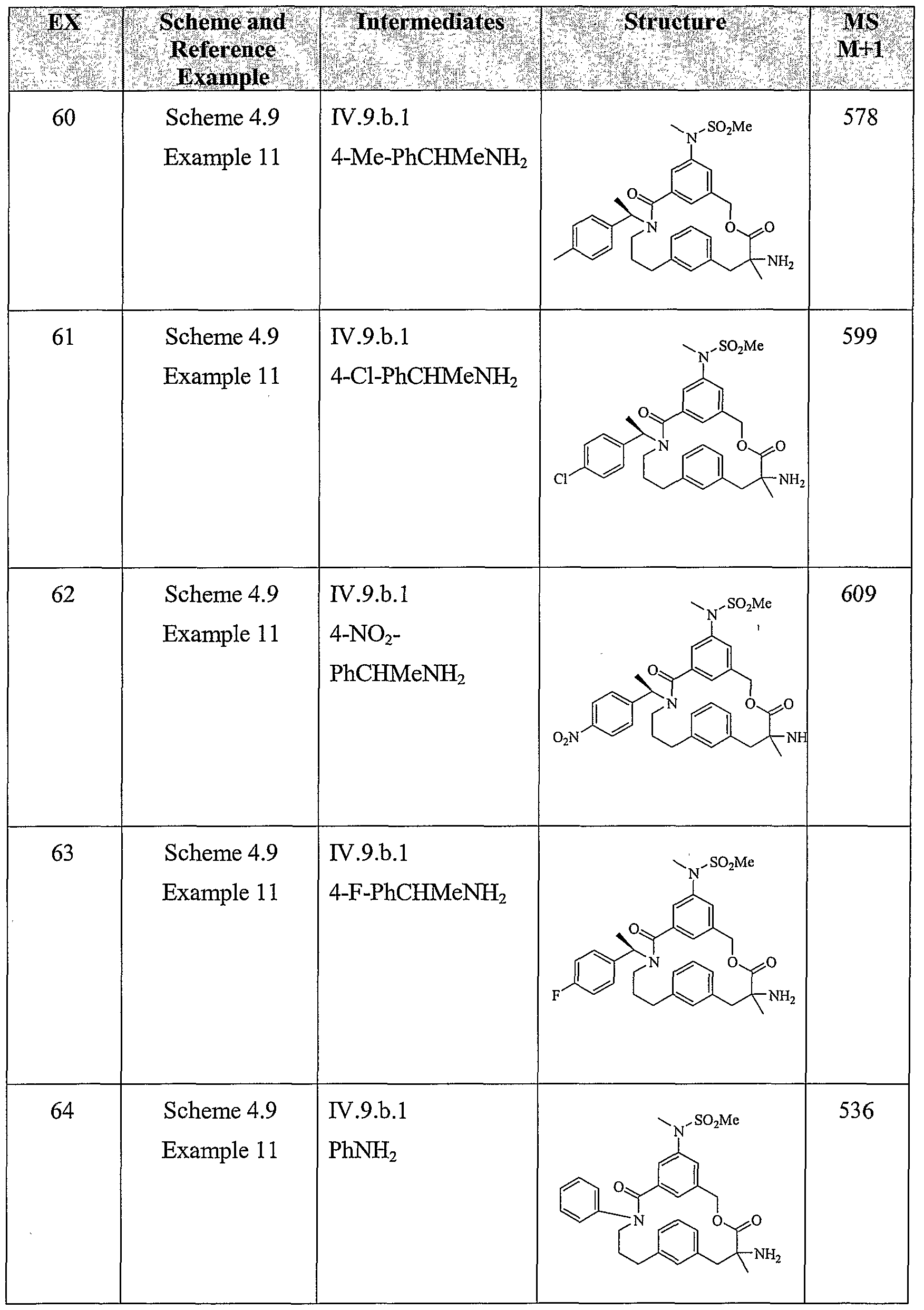
- Another embodiment of the present invention includes a compound which is selected from the title compounds of the following Examples and pharmaceutically acceptable salts thereof.
- alkyl by itself or as part of another substituent, means a saturated straight or branched chain hydrocarbon radical having the number of carbon atoms designated (e.g., C ⁇ . io alkyl means an alkyl group having from one to ten carbon atoms).
- Preferred alkyl groups for use in the invention are C1-6 alkyl groups, having from one to six carbon atoms.
- Exemplary alkyl groups include methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, tert-butyl, pentyl, hexyl, and the like.
- alkylene by itself or as part of another substituent, means a saturated straight or branched chain divalent hydrocarbon radical having the number of carbon atoms designated.
- Co alkylene means a bond.
- cycloalkyl by itself or as part of another substituent, means a saturated cyclic hydrocarbon radical having the number of carbon atoms designated (e.g., C3.8 cycloalkyl means a cycloalkyl group having from three to eight carbon atoms).
- exemplary cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl and the like.
- alkenyl by itself of as part of another substituent, means a straight or branched chain hydrocarbon radical having a single carbon-carbon double bond and having the number of carbon atoms designated (e.g., C2-10 alkenyl means an alkenyl group having from one to ten carbon atoms).
- Preferred alkenyl groups for use in the invention are C2-6 alkenyl groups, having from two to six carbon atoms.
- Exemplary alkenyl groups include ethenyl, n-propenyl, isopropenyl, butenyl, and the like.
- alkynyl by itself or as part of another substituent, means a saturated straight or branched chain hydrocarbon radical having the number of carbon atoms designated (e.g., C2-10 alkynyl means an alkynyl group having from two to ten carbon atoms).
- Preferred alkynyl groups for use in the invention are C2-6 alkynyl groups, having from two to six carbon atoms.
- Exemplary alkynyl groups include ethynyl and propynyl.
- aryl by itself or as part of another substituent, means an aromatic or cyclic radical having the number of carbon atoms designated (e.g., C6-10 aryl means an aryl group having from six to ten carbons atoms).
- aryl includes multiple ring systems as well as single ring systems.
- Preferred aryl groups for use in the invention include phenyl and naphthyl.
- arylene by itself or as part of another substituent, means a divalent aromatic or cyclic radical, having the number of carbon atoms designated (e.g., C6-10 arylene means an arylene group having from six to ten carbons atoms).
- arylene includes multiple ring systems as well as single ring systems.
- Preferred arylene groups for use in the invention include phenylene and naphthylene.
- heteroaryl by itself or as part of another substituent, means an aromatic cyclic radical having at least one ring heteroatom (O, N or S).
- heteroaryl includes multiple ring systems as well as single ring systems.
- heteroaryl groups for use in the invention include furyl, pyranyl, benzofuranyl, isobenzofuranyl, chromenyl, thienyl, benzothiophenyl, pyrrolyl, pyrazolyl, imidazolyl, pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, indolyl, benzimidazolyl, quinolinyl, isoquinolinyl, tetrazolyl, indazolyl, napthyridinyl, triazolyl, oxazolyl, oxadiazolyl, thiazolyl, thiadiazolyl, isoxazolyl, tetrahydroquinolinyl, tetrahydroisoquinolinyl and dihydroindolyl.
- the substituent When a heteroaryl group as defined herein is substituted, the substituent may be bonded to a ring carbon atom of the heteroaryl group, or on a ring heteroatom ⁇ i.e., a nitrogen, oxygen or sulfur), which has a valence which permits substitution. Preferably, the substituent is bonded to a ring carbon atom.
- the point of attachment may be at a ring carbon atom of the heteroaryl group, or on a ring heteroatom ⁇ i.e., a nitrogen, oxygen or sulfur), which has a valence which permits attachment.
- the attachment is at a ring carbon atom.
- heteroarylene by itself or as part of another substituent, means an aromatic cyclic divalent radical having at least one ring heteroatom (O, N or S).
- halo or “halogen” includes fluoro, chloro, bromo and iodo.
- the compounds of the instant invention have at least one asymmetric center. Additional asymmetric centers may be present depending upon the nature of the various substituents on the molecule.
- racemic mixtures of the compounds may be separated so that the individual enantiomers are isolated.
- the separation can be carried out by methods well known in the art, such as the coupling of a racemic mixture of compounds to an enantiomerically pure compound to form a diastereomeric mixture, followed by separation of the individual diastereomers by standard methods, such as fractional crystallization or chromatography.
- the coupling reaction is often the formation of salts using an enantiomerically pure acid or base.
- the diastereomeric derivatives may then be converted to the pure enantiomers by cleavage of the added chiral residue.
- the racemic mixture of the compounds can also be separated directly by chromatographic methods utilizing chiral stationary phases, which methods are well known in the art.
- any enantiomer of a compound may be obtained by stereoselective synthesis using optically pure starting materials or reagents of known configuration by methods well known in the art.
- the carbon to which A and R4 are bonded is a chiral carbon.
- the compounds of formulas (I)-(IV) may be present as racemates, or in the stereochemically pure (R) or (S) forms.
- the present invention encompasses all such isomeric forms.
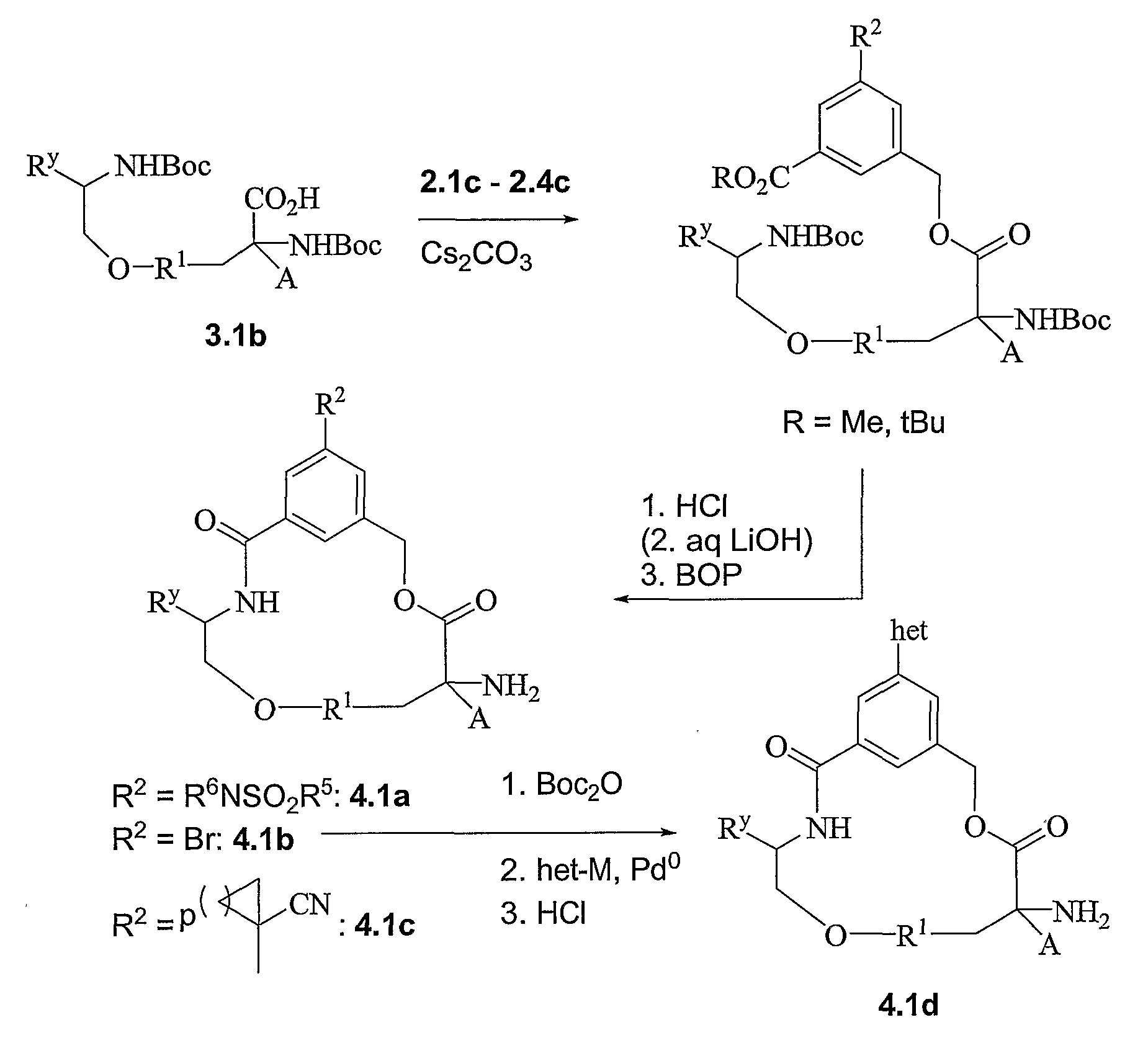
- Scheme 1.1 describes the preparation of hydroxyl derivatives of type 1.1a, their triflate analogs 1.1b and 1.1c. Starting from glycine Schiff base, more elaborated bromides of type l.ld and l.le can be prepared.
- Scheme 2.1 describes a sulfonylation, alkylation, monohydrolysis sequence leading to monoacids of type 2.1a. Reduction to hydroxymethyl derivatives 2.1b, bromination to bromomethyl derivatives 2.1c or protection with TBS (2.Id) is described as well. Acylhydrazide derivatives of type 2.1e are obtained from the corresponding acids.
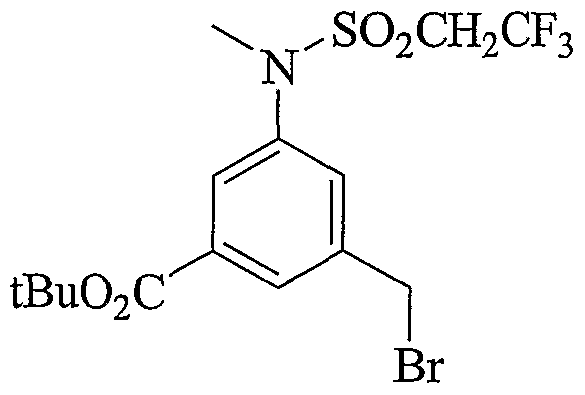
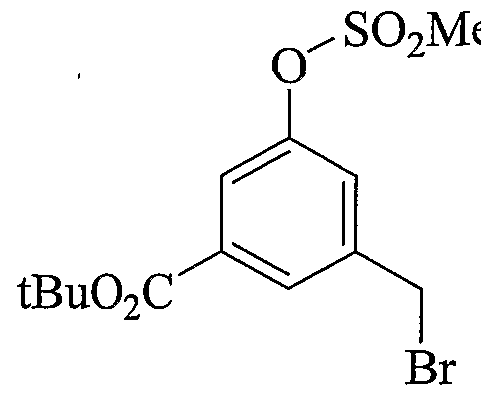
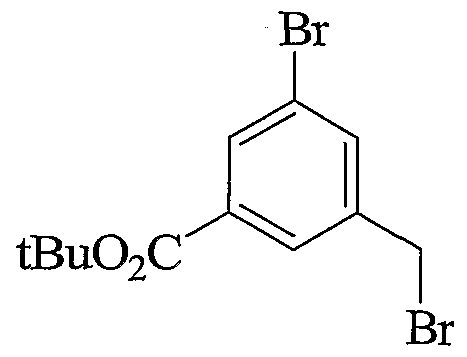
- Scheme 2.2 describes very similar preparation as in scheme 2.1 with the incorporation of a tert- butyl ester that can be removed under non-hydrolytic conditions. Alternate mode of alkylation/sulfonylation is also represented.
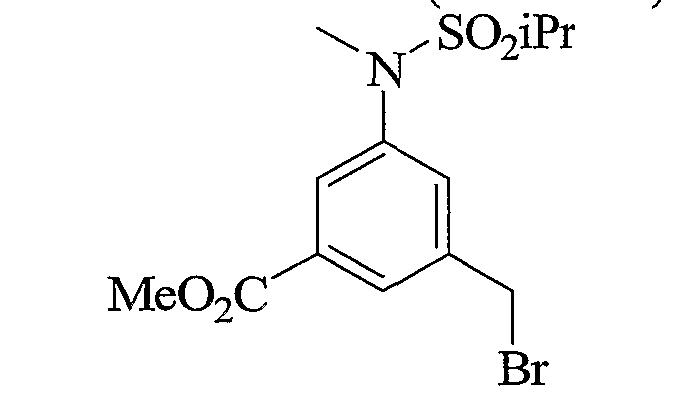
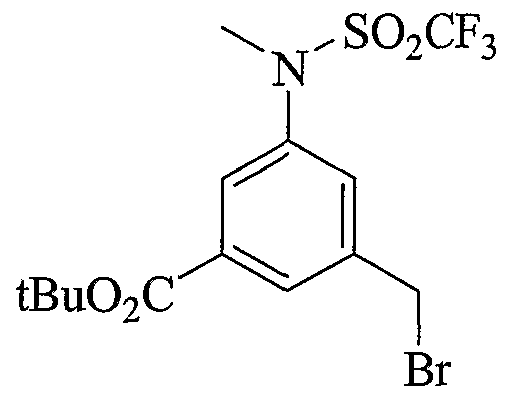
- Scheme 2.3 is similar to schemes 2.1 and 2.2, with the incorporation of an aryl bromide useful to introduce various aryl groups, sulfonamides and heterocycles later in the syntheses or early on as described in the 2 nd part of the scheme.
- Scheme 2.3 is similar to schemes 2.1 and 2.2, with the incorporation of an aryl bromide useful to introduce various aryl groups, sulfonamides and heterocycles later in the syntheses or early on as described in the 2 nd part of the scheme.
- Scheme 2.4 describes the preparation of similar intermediates that display cyano-spirocyclic groups to replace the alkyl-sulfonamides described in schemes 2.1 and 2.2.
- Scheme 2.5 describes the preparation of phenols of type 2.5b and 2.5d, along with their triflate derivatives of type 2.5c and 2.5e.
- Scheme 3.1 and 3.2 illustrate the preparation of carboxylic acids of type 3.1-2a and alcohols of type 3.1- 2c.
- Schemes 4.1-10 illustrate the assembly of various intermediates and their final elaboration to macrocycles.
- substantially pure means that the isolated material is at least 90% pure, and preferably 95% pure, and even more preferably 99% pure as assayed by analytical techniques known in the art.
- salts refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic or organic bases and inorganic or organic acids.
- Salts derived from inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic salts, manganous, potassium, sodium, zinc, and the like. Particularly preferred are the ammonium, calcium, magnesium, potassium, and sodium salts. Salts in the solid form may exist in more than one crystal structure, and may also be in the form of hydrates.
- Salts derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, and basic ion exchange resins, such as arginine, betaine, caffeine, choline, N,N'-dibenzylethylene-diamine, diethylamine, 2-diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethyl- morpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine, and the like.
- basic ion exchange resins
- salts may be prepared from pharmaceutically acceptable non-toxic acids, including inorganic and organic acids.
- acids include acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethanesulfonic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, nitric, pamoic, pantothenic, phosphoric, succinic, sulfuric, tartaric, p-toluenesulfonic, triflouoroacetic acid and the like.
- Particularly preferred are citric, hydrobromic, hydrochloric, maleic, phosphoric, sulfuric, fumaric, tartaric and trifluoroacetic acids.
- the present invention is directed to the use of the compounds disclosed herein as inhibitors of ⁇ - secretase enzyme activity or ⁇ -site amyloid precursor protein-cleaving enzyme ("BACE") activity, in a patient or subject such as a mammal in need of such inhibition, comprising the administration of an effective amount of the compound.
- BACE ⁇ -secretase enzyme
- ⁇ -site amyloid precursor protein- cleaving enzyme and “BACE” are used interchangeably in this specification.
- ⁇ -secretase enzyme ⁇ -site amyloid precursor protein- cleaving enzyme
- BACE ⁇ -site amyloid precursor protein-cleaving enzyme
- the present invention is further directed to a method for the manufacture of a medicament or a composition for inhibiting ⁇ -secretase enzyme activity in humans and animals comprising combining a compound of the present invention with a pharmaceutical carrier or diluent.
- the compounds of the present invention have utility in treating Alzheimer's disease.
- the compounds may be useful for the prevention of dementia of the Alzheimer's type, as well as for the treatment of early stage, intermediate stage or late stage dementia of the Alzheimer's type.
- the compounds may also be useful in treating diseases mediated by abnormal cleavage of amyloid precursor protein (also referred to as APP), and other conditions that may be treated or prevented by inhibition of ⁇ -secretase.
- APP amyloid precursor protein
- Such conditions include mild cognitive impairment, Trisomy 21 (Down Syndrome), cerebral amyloid angiopathy, degenerative dementia, Hereditary Cerebral Hemorrhage with Amyloidosis of the Dutch-Type (HCHWA-D), Creutzfeld- Jakob disease, prion disorders, amyotrophic lateral sclerosis, progressive supranuclear palsy, head trauma, stroke, Down syndrome, pancreatitis, inclusion body myositis, other peripheral amyloidoses, diabetes and atherosclerosis.
- the subject or patient to whom the compounds of the present invention is administered is generally a human being, male or female, in whom inhibition of ⁇ -secretase enzyme activity is desired, but may also encompass other mammals, such as dogs, cats, mice, rats, cattle, horses, sheep, rabbits, monkeys, chimpanzees or other apes or primates, for which inhibition of ⁇ -secretase enzyme activity or treatment of the above noted disorders is desired.
- the compounds of the present invention may be used in combination with one or more other drugs in the treatment of diseases or conditions for which the compounds of the present invention have utility, where the combination of the drugs together are safer or more effective than either drug alone. Additionally, the compounds of the present invention may be used in combination with one or more other drugs that treat, prevent, control, ameliorate, or reduce the risk of side effects or toxicity of the compounds of the present invention. Such other drugs may be administered, by a route and in an amount commonly used therefor, contemporaneously or sequentially with the compounds of the present invention. Accordingly, the pharmaceutical compositions of the present invention include those that contain one or more other active ingredients, in addition to the compounds of the present invention. The combinations may be administered as part of a unit dosage form combination product, or as a kit or treatment protocol wherein one or more additional drugs are administered in separate dosage forms as part of a treatment regimen.
- combinations of the compounds of the present invention with other drugs in either unit dose or kit form include combinations with anti-Alzheimer's agents, for example other beta- secretase inhibitors or gamma-secretase inhibitors; tau phosphorylation inhibitors; Ml receptor positive allosteric modulators; blockers of A/3 oligomer formation; 5-HT modulators, such as PRX-03140, GSK 742467, SGS-518, FK-962, SL-65.0155, SRA-333 and xaliproden; p25/CDK5 inhibitors; NK1/NK3 receptor antagonists; COX-2 inhibitors; HMG-CoA reductase inhibitors; NSAIDs including ibuprofen; vitamin E; anti-amyloid antibodies, including anti-amyloid humanized monoclonal antibodies; anti ⁇ inflammatory compounds such as (R)-flurbiprofen, nitroflurbiprofen, rosiglitazone, ND- 1251, VP-
- MARK MARK
- P-450 inhibitors such as ritonavir
- P-450 inhibitors such as ritonavir
- other drugs that affect receptors or enzymes that either increase the efficacy, safety, convenience, or reduce unwanted side effects or toxicity of the compounds of the present invention.
- composition as used herein is intended to encompass a product comprising specified ingredients in predetermined amounts or proportions, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts.
- This term in relation to pharmaceutical compositions is intended to encompass a product comprising one or more active ingredients, and an optional carrier comprising inert ingredients, as well as any product which results, directly or indirectly, from combination, complexation or aggregation of any two or more of the ingredients, or from dissociation of one or more of the ingredients, or from other types of reactions or interactions of one or more of the ingredients.
- compositions are prepared by uniformly and intimately bringing the active ingredient into association with a liquid carrier or a finely divided solid carrier or both, and then, if necessary, shaping the product into the desired formulation.
- the active object compound is included in an amount sufficient to produce the desired effect upon the process or condition of diseases.
- the pharmaceutical compositions of the present invention encompass any composition made by admixing a compound of the present invention and a pharmaceutically acceptable carrier.
- compositions intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions and such compositions may contain one or more agents selected from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents in order to provide pharmaceutically elegant and palatable preparations.
- Tablets may contain the active ingredient in admixture with non-toxic pharmaceutically acceptable excipients which are suitable for the manufacture of tablets.
- excipients may be, for example, inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, for example, corn starch, or alginic acid; binding agents, for example starch, gelatin or acacia, and lubricating agents, for example magnesium stearate, stearic acid or talc.
- the tablets may be uncoated or they may be coated by known techniques to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period.
- compositions for oral use may also be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, for example peanut oil, liquid paraffin, or olive oil.
- an inert solid diluent for example, calcium carbonate, calcium phosphate or kaolin
- water or an oil medium for example peanut oil, liquid paraffin, or olive oil.
- compositions include aqueous suspensions, which contain the active materials in admixture with excipients suitable for the manufacture of aqueous suspensions.
- oily suspensions may be formulated by suspending the active ingredient in a vegetable oil, for example arachis oil, olive oil, sesame oil or coconut oil, or in a mineral oil such as liquid paraffin. Oily suspensions may also contain various excipients.
- the pharmaceutical compositions of the invention may also be in the form of oil-in-water emulsions, which may also contain excipients such as sweetening and flavoring agents.
- the pharmaceutical compositions may be in the form of a sterile injectable aqueous or oleaginous suspension, which may be formulated according to the known art, or may be administered in the form of suppositories for rectal administration of the drug.
- the compounds of the present invention may also be administered by inhalation, by way of inhalation devices known to those skilled in the art, or by a transdermal patch.
- pharmaceutically acceptable it is meant the carrier, diluent or excipient must be compatible with the other ingredients of the formulation and not deleterious to the recipient thereof.
- administering a should be understood to mean providing a compound of the invention to the individual in need of treatment in a form that can be introduced into that individual's body in a therapeutically useful form and therapeutically useful amount, including, but not limited to: oral dosage forms, such as tablets, capsules, syrups, suspensions, and the like; injectable dosage forms, such as IV, IM, or IP, and the like; transdermal dosage forms, including creams, jellies, powders, or patches; buccal dosage forms; inhalation powders, sprays, suspensions, and the like; and rectal suppositories.
- oral dosage forms such as tablets, capsules, syrups, suspensions, and the like
- injectable dosage forms such as IV, IM, or IP, and the like
- transdermal dosage forms including creams, jellies, powders, or patches
- buccal dosage forms inhalation powders, sprays, suspensions, and the like
- rectal suppositories rectal suppositories.
- an effective amount or “therapeutically effective amount” means the amount of the subject compound that will elicit the biological or medical response of a tissue, system, animal or human that is being sought by the researcher, veterinarian, medical doctor or other clinician.
- treatment refers to the treatment of the mentioned conditions, particularly in a patient who demonstrates symptoms of the disease or disorder.
- compositions containing compounds of the present invention may conveniently be presented in unit dosage form and may be prepared by any of the methods well known in the art of pharmacy.
- unit dosage form is taken to mean a single dose wherein all active and inactive ingredients are combined in a suitable system, such that the patient or person adminstering the drug to the patient can open a single container or package with the entire dose contained therein, and does not have to mix any components together from two or more containers or packages.
- Typical examples of unit dosage forms are tablets or capsules for oral administration, single dose vials for injection, or suppositories for rectal administration. This list of unit dosage forms is not intended to be limiting in any way, but merely to represent typical examples of unit dosage forms.
- compositions containing compounds of the present invention may conveniently be presented as a kit, whereby two or more components, which may be active or inactive ingredients, carriers, diluents, and the like, are provided with instructions for preparation of the actual dosage form by the patient or person adminstering the drug to the patient.
- kits may be provided with all necessary materials and ingredients contained therein, or they may contain instructions for using or making materials or components that must be obtained independently by the patient or person administering the drug to the patient.
- the compounds of the present invention are administered at a daily dosage of from about 0.1 mg to about 100 mg per kilogram of animal body weight, preferably given as a single daily dose or in divided doses two to six times a day, or in sustained release form.
- the total daily dosage is from about 1.0 mg to about 2000 mg, preferably from about 0.1 mg to about 20 mg per kg of body weight. In the case of a 70 kg adult human, the total daily dose will generally be from about 7 mg to about 1 ,400 mg. This dosage regimen may be adjusted to provide the optimal therapeutic response.
- the compounds may be administered on a regimen of 1 to 4 times per day, preferably once or twice per day.
- Specific dosages of the compounds of the present invention, or pharmaceutically acceptable salts thereof, for administration include 1 mg, 5 mg, 10 mg, 30 mg, 80 mg, 100 mg, 150 mg, 300 mg and 500 mg.
- Pharmaceutical compositions of the present invention may be provided in a formulation comprising about 0.5 mg to 1000 mg active ingredient; more preferably comprising about 0.5 mg to 500 mg active ingredient; or 0.5 mg to 250 mg active ingredient; or 1 mg to 100 mg active ingredient.
- Specific pharmaceutical compositions useful for treatment may comprise about 1 mg, 5 mg, 10 mg, 30 mg, 80 mg, 100 mg, 150 mg, 300 mg and 500 mg of active ingredient.
- FRET fluorescence resonance energy transfer
- a homogeneous end point fluorescence resonance energy transfer (FRET) assay is employed with the substrate ([TAMRA-S-CO-EEISEVNLDAEF-NHQSY] QFRET), which is cleaved by BACE 1 to release the fluorescence from TAMRA.
- the Km of the substrate is not determined due to the limit of solubility of the substrate.
- a typical reaction contains approximately 30 nM enzyme, 1.25 ⁇ M of the substrate, and buffer (50 mM NaOAc, pH 4.5, 0.1 mg/ml BSA, 0.2% CHAPS, 15 mM EDTA and 1 mM deferoxamine) in a total reaction volume of 100 ⁇ l.
- the reaction is proceeded for 30 min and the liberation of TAMRA fragment is measured in a 96-well plate LJL Analyst AD using an excitation wavelength of 530 nm and an emission wavelength of 580 nm. Under these conditions, less than 10% of substrate is processed by BACE 1.
- the enzyme used in these studies is soluble (transmembrane domain and cytoplasmic extension excluded) human protein produced in a baculovirus expression system.
- solutions of inhibitor in DMSO four concentrations of the inhibitors are prepared: ImM, 100 ⁇ M, 10 ⁇ M, 1 ⁇ M) are included in the reactions mixture (final DMSO concentration is 0.8%). All experiments are conducted at rt using the standard reaction conditions described above.
- HPLC assay A homogeneous end point HPLC assay is used with the substrate (coumarin-CO-REVNFEVEFR), which is cleaved by BACE 1 to release the N-terminal fragment attached with coumarin.
- the Km of the substrate is greater than 100 ⁇ M and can not be determined due to the limit of solubility of the substrate.
- a typical reaction contains approximately 2 nM enzyme, 1.0 ⁇ M of the substrate, and buffer (50 mM NaOAc, pH 4.5, 0.1 mg/ml BSA, 0.2% CHAPS, 15 mM EDTA and 1 mM deferoxamine) in a total reaction volume of 100 ⁇ l.
- the reaction is proceeded for 30 min and is stopped by the addition of 25 ⁇ L of 1 M Tris-HCl, pH 8.0.
- the resulting reaction mixture is loaded on the HPLC and the product is separated from substrate with 5 min linear gradient. Under these conditions, less than 10% of substrate is processed by BACE 1.
- the enzyme used in these studies is soluble (transmembrane domain and cytoplasmic extension excluded) human protein produced in a baculovirus expression system.
- solutions of inhibitor in DMSO (12 concentrations of the inhibitors are prepared and the concentration rage is dependent on the potency predicted by FRET) are included in the reaction mixture (final DMSO concentration is 10 %). All experiments are conducted at rt using the standard reaction conditions described above.
- To determine the IC50 of the compound four parameters equation is used for curve fitting. The errors in reproducing the dissociation constants are typically less than two-fold.
- the compounds of the following examples had activity in inhibiting the beta- secretase enzyme in one or both of the aforementioned assays, generally with an IC50 from about 1 nM to 100 ⁇ M. Such a result is indicative of the intrinsic activity of the compounds in use as inhibitors of the beta-secretase enzyme activity.
- HMDS hexamethyldisilazane
- TMAD N,N,N',N'-Tetramethylazocarboxamide
- DIAD Diisopropylazodicarboxylate
- HOAt l-hydroxy-7-azabenzotriazole
- EDC 1 -Ethyl-3 -(3 -dimemylaminopropyl)-carbodiimide
- DPPA diphenylphosphorylazide
- TPAP tetrapropylammonium perruthenate
- BSA bovine serum albumin
- CHAPS 3-[(3-cholamidopropyl)dimethylammonio]-2-hydroxy-l-propanesulfonate
- rt room temperature
- HPLC high performance liquid chromatography
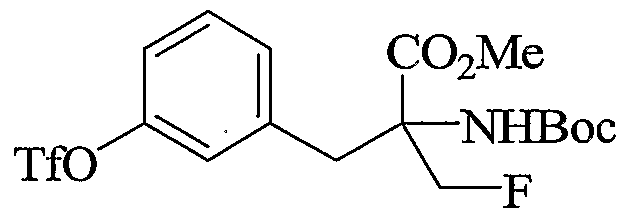
- Step A l-((3-(Bromomethyl)phenoxy)methyl)benzene
- Step B 2-Amino-3-(3-benzyloxy)phenyl)-2-(fluoromethyl)propanenitrile
- Step C 2-Amino-2-(fluoromethyl)-3-(3-hydroxyphenyl)propanoic acid
- aqueous HCl 6N, 60 mL
- the reaction mixture was diluted with H 2 O and extracted with ether.
- the pH of the aqueous phase was brought to 5.5 and solid impurities were filtered.
- Step E Methyl 2-tert-butoxycarbonylamino-2-(fluoromethyl)-3-(3-hydroxyphenyl)propanoate
- a suspension of methyl 2-amino-2-(fluoromethyl)-3-(3-hydroxyphenyl)propanoate (1 g, 3.8 mmol) in DMF/tert-butanol (1:1, 2.6 mL) was added a solution of di-tert-buty ⁇ dicarbonate (1.6 g, 7.6 mmol) in DMF/tert-butanol (0.9 mL) followed by sodium bicarbonate (1.1 g, 13.3 mmol).
- Step F 3-(2-(Methoxycarbonyl)-2-tert-butoxycarbonylamino-3-fluoropropyl)phenyl trifluoromethanesulfonate
- reaction mixture was allowed to warm to rt over 16h, quenched with aq NH 4 Cl and water, extracted with EtOAc, washed with aq LiCl (x3), dried over Na 2 SO 4 , concentrated in vacuo, and purified by flash chromatography (12Og silica, 0-15% EtOAc in hexanes) to provide methyl 3 -bromo-N-(diphenylmethylene)- ⁇ -methylphenylalaninate .
- Step B Bromination To a solution of alcohol (0.710 g, 2.60 mmol) from Step A and carbon tetrabromide (1.12 g, 3.38 mmol) in 25 mL anhydrous CH 2 Cl 2 under an atmosphere of argon was added a solution of triphenylphosphine (0.818 g, 3.12 mmol) in 5 mL anhydrous CH 2 Cl 2 slowly via syringe. After 2 hr, additional carbon tetrabromide (0.224 g, 0.675 mmol) and triphenylphosphine (0.164 g, 0.623 mmol) were added. After an additional 1 hr, it was concentrated in vacuo.
- Step A Coupling To a solution of intermediate H.l.a.l (0.520 g, 1.810 mmol) and Boc-hydrazine (0.359 g, 2.715 mmol) in 8 mL CH 2 Cl 2 was added Hunig's base (0.950 mL, 5.43 mmol) and BOP-reagent (0.881 g, 1.991 mmol). After 30 min, the reaction was poured onto a silica gel column and purified by normal phase chromatography (5->75% EtOAc/hexanes) to afford the desired product as a white foam.
- Step A tBu ester Installment
- StepD Nitro reduction A solution of methyl 2-chloro-3-nitro-5-vinylbenzoate (1.75 g, 7.2 mmol) and SnCl 2 (4.1 g, 18.1 mmol) in EtOH (50 mL) was stirred at 75 °C for 16 h. The reaction mixture was cooled to RT, diluted with water and EtOAc, stirred at RT for 10 min, and filtered on cellite.
- StepE Mesylation As described in the preparation of intermediate K.l.a.1, step A.
- step B As described in the preparation of intermediate H.l.a.l, step B.
- step B As described in the preparation of intermediate II.2.C.2, step B.
- step A to give ter/-butyl 2-chloro-3- [methyl(methylsulfonyl)amino]-5-vinylbenzoate.
- Stepl Reductive ozonolysis Through a solution of tert-butyl 2-chloro-3-[methyl(methylsulfonyl)amino]-5-vinylbenzoate (700 mg, 2 mmol) in DCM (7 mL) and MeOH (3 mL) cooled to -78 °C was bubbled ozone until the solution remained blue. After 5 min stirring at -78 °C, MeOH (4 mL) and NaBH 4 (115 mg, 3 mmol) were added and the reaction mixture was allowed to warm to RT.
- reaction mixture was diluted with EtOAc, washed with 10% KHSO 4 , brine, dried over sodium sulfate and concentrated in vacuo to provide tert- butyl 2-chloro-5-(hydroxymethyl)-3-[methyl(methylsulfonyl)amino]benzoate, used crude in the bromination step.
- step B As described in the preparation of intermediate II.l.cl, step B, to provide tert-buty ⁇ 5-(bromomethyl)-2- chloro-3-[methyl(methylsulfonyl)amino]benzoate.
- Step D Lithium borohydride reduction To a 0°C solution of tert-butyl 3-(hydroxymethyl)-5-[(methylsulfonyl)oxy]benzoate (0.330 g, 0.99 mmol) in THF (15 mL) was added 2 M lithium borohydride solution (0.524 mL, 1.05 mmol). After 1 hour, added 2 more equivalents LiBH 4 solution and warmed reaction to room temp over 18 h. Quenched reaction dropwise with MeOH and then concentrated reaction mixture in vacuo. The crude material was diluted with EtOAc and washed with sat.
- Step A Bis Hydrolysis To a solution of dimethyl 5-bromoiso ⁇ hthalate (10 g, 36.6 mmol) in MeOH (200 mL) and THF (200 mL) was added IN NaOH (91.5 mL, 91.5 mmol) and the reaction mixture was stirred at rt for 5 h, quenched with IN HCl (92 mL), concentrated in vacuo to ca. 250 mL. The white solid was filtered, washed with water and dried over P 2 O 5 , under high vacuum, at 50 °C.
- Step B Mono tBu Esterif ⁇ cation
- reaction mixture was diluted with 10% KHSO 4 , filtered on celite, extracted with EtOAc, washed with aq LiCl (x3), dried over Na 2 SO 4 , and concentrated in vacuo to provide the corresponding mono tBu ester.
- StepA Pd 0 coupling
- StepB-F hydrolysis, tBu ester installation, Me ester hydrolysis, borane reduction, bromination, as described above.
- StepA Pd coupling of aniline to fert-butyl methyl 5-hromoisophthalate
- Step E Acid Reduction and Bromination
- IM in THF 5.6 mL, 5.6 mmol
- IM in THF 5.6 mL
- 5.6 mmol borane-tetrahydrofuran complex
- reaction mixture was diluted with water, the pH was adjusted to pH 7-8 with IN HCl, the resulting mixture was extracted with EtOAc, washed with aq LiCl (x3), dried over Na 2 S ⁇ 4 , and concentrated in vacuo to provide the corresponding benzyl ether
- Step B Monohydrolysis Monohydrolysis of the previous diester with IN NaOH in MeOH/THF, according to preparation of intermediate II.l.a.l, step C, followed by purification by flash chromatography (30Og silica, 0-50% (0.5% HOAc in EtOAc) in hexanes) provided the corresponding monoacid.
- Step C Curtius Rearrangement
- the previous monoacid (5.98 g, 20.9 mmol), triethyl amine (16.1 mL, 31.3 mmol), and diphenyphosphoryl azide (8.62 g, 31.3 mmol) were dissolved in anhydrous tert-butyl alcohol (200 mL) and allowed to stir under reflux, 110°C, for 16 hours.
- the crude reaction mixture is then concentrated in vacuo, then diluted with EtOAc and washed with deionized water (x3), brine (x3), dried over sodium sulfate, and concentrated in vacuo.
- the crude mixture was then purified using flash chromatography (145g silica, 0-30% EtOAc in hexanes) to afford the corresponding ester carbamate.
- Step D Alkylation and Debocing
- the previous ester carbamate (7.3 g, 20.5 rnmol) was dissolved in DMF (40 mL) and cooled to 0°C, the 1.0 M solution of NaHMDS (22.5 mL, 22.5 rnmol) was then added dropwise via syringe. After stirring 0.5 h at 0°C, the MeI (1.53 mL, 24.5 rnmol) was added dropwise via syringe and the reaction was allowed to warm slowly to rt and stir for an additional 16 h. The crude reaction mixture was quenched with deionized water and diluted with DCM.
- the biphasic system was washed with DI water (x3), brine (x3), dried over sodium sulfate, and concentrated in vacuo.
- the crude mixture was then purified using flash chromatography (145g silica, 0-25% EtOAc in hexanes) to afford the corresponding N-methyl carbamate.
- the N-methyl carbamate (6.5 g, 17.5 mmol) was then dissolved in a 4.0 M of HCl in 1,4- dioxane (43.8 mL, 175 mmol) and allowed to stir at rt for 16 h, the reaction was then concentrated in vacuo to afford to corresponding N-methyl amino ester.
- Step G Hydrogenolysis of Bn Ether
- the previous silyl ether (3.14 g, 7.2 mmol) was dissolved in 120 mL of degassed EtOAc and placed under argon and Pd/C (0.08 g, 0.73 mmol) was added in one portion.
- Hydrogen 144 mmol was added via a three way adaptor and the system was purged under reduced pressure, then exposed to hydrogen. This process of purging and exposure to hydrogen was repeated three times. The reaction was allowed to stir at rt for 16 h. The crude reaction mixture was filtered over celite and washed with EtOAc, dried over sodium sulfate, and concentrated in vacuo.
- Step A Alkylation To a solution of intermediate I.l.a.l (0.050 g, 0.162 mmol) and 2-bromoacetophenone (0.032 g, 0.162 mmol) in 1 mL anhydrous DMF under an atmosphere of argon was added Cs 2 CO 3 (0.029 g, 0.089 mmol). After 24 hr, the crude reaction mixture was purified by reverse phase preparative HPLC (5 -> 95% CH3CN/H2O, 0.1% added TFA, Cl 8 PRO YMC 20x150 mm) to afford methyl N-(fert-butoxycarbonyl)- alpha-methyl-3-(2-oxo-2-phenylethoxy)phenylalaninate as a pale yellow oil.
- Step B Reductive Animation To a solution of methyl N-(f ⁇ rt-butoxycarbonyl)-alpha-methyl-3-(2-oxo-2-phenylethoxy)phenylalaninate (0.600 g, 1.40 mmol), 4A sieves (spatula tip), acetic acid (0.089 mL, 1.54 mmol), and benzylamine (0.184 mL, 1.68 mmol) in 10.0 mL dichloroethane was added sodium triacetoxyborohydride (0.357 g, 1.68 mmol).
- Step C Hydrogenolysis To a degassed solution of methyl 3-[2-(benzylamino)-2-phenylethoxy]-N-(tert-butoxycarbonyl)-alpha- methylphenylalaninate (0.457 g, 0.881 mmol) in 10 mL EtOAc was added palladium hydroxide (0.198 g, 1.41 mmol). The resulting mixture was hydrogenated under 1 atm at rt. After 60 hr., the reaction mixture was filtered over celite and concentrated in vacuo to give the corresponding amine as a yellow foam.
- Step A-D conversion of benzaldehyde to tert -butyl (l-phenylprop-2-en-l-yl)carbamate was performed using vinyl Grignard as described in D.A. Cogan et al. Tetrahedron 55 (1999) 8883-8904, followed by standard Boc installation.
- Step E Hydroboration and Pd 0 Coupling
- Solid tert-butyl (l-phenylprop-2-en-l-yl)carbamate (0.436 g, 1.87 mmol) was placed in an oven-dried flask under an atmosphere of argon and dissolved in 9-borabicyclo[3.3.1]nonane (3.91 mL, 1.95 mmol, 0.5M solution in THF) and heated to 70°C.
- StepA Stille coupling to intermediate I.l.c.1
- the organic layer was added water (100 ml) and KF (5g) and stirred for 1 hour.
- the organic layer was dried, concentrated and purified by flash column chromatography (30% EtO Ac/Hex) affording 1.08 g desired product.
- StepA Coupling of intermediates IL2.C.1 and i ⁇ .2.e.l
- intermediate II.2.C.1 (0.100 g, 0.264 mmol) and intermediate IH.2.e.l (0.084 g, 0.264 mmol) in 1 mL DMF was added cesium carbonate (0.095 g, 0.291 mmol).
- cesium carbonate 0.095 g, 0.291 mmol.
- the reaction was diluted with LiCl (aq) (25 mL) and extracted with EtOAc (2 x 25 mL). The organic layers were combined, washed with LiCl (aq) and brine, dried over sodium sulfate, and concentrated in vacuo.
- Ether formation with intermediates H.l.c.2 and III.l.c.1 was performed using a similar procedure as described in the preparation of intermediate IV.4.e.2 to give methyl 3- ⁇ [2-[(fert-butoxycarbonyl)amino]- 3-(3- ⁇ 2-[(ter/-butoxycarbonyl)amino]-2-phenylethoxy ⁇ phenyl)-2-methylpropoxy]methyl ⁇ -5- [(methylsulfonyl)(propyl)amino]benzoate as a colorless oil.
- Step B Boc Removal, Hydrolysis.
- the resulting deprotected material was taken up in 1.5 mL tetrahydrofuran, and IN LiOH (0.350 mL, 0.350 mmol) was added. After 6 hr., it was acidified to pH 4 with IN HCl (0.380 mL, 0.380 mmol) and concentrated under reduced pressure to give the resulting acid.
- Step C BOP Cyclization To a solution of acid from step B (0.01Og, 0.018 mmol) in 5 mL DMF was added diisopropylethylamine (0.005 mL, 0.026 mmol) and benzotriazol-l-yloxytris(dimethylamino)-phosphonium hexafluorophosphate (0.009 g, 0.021 mmol). After 1 hr, the crude reaction mixture was purified by preparative HPLC (5 -> 95% CH3CN/H2O, 0.1% added TFA, C18 PRO YMC 20x150 mm) to afford
- Example 1 as a white solid.
- Step B Boc and tBu ester Removal
- Step A Ester formation (intermediates II.3.C and IH.2.b.l.l), using a similar procedure as described in the preparation of Example 2.
- Step B Boc and tfiu ester removal, macrolactamization, using a similar procedure as described in the preparation of Example 2.
- Step D Pd 0 coupling of MeNMs
- the aryl sulfonamide from Step D was treated with 4N HCl in dioxane (5 mL) for 1 h 45, concentrated in vacuo and purified by ion exchange chromatography (2 g SCX, MeOH then 2M NH 3 in MeOH) to provide Example 3 as a white solid.
- Step D Pd 0 coupling of 2-CN-Ph-ZnI
- 2-cyanophenylzinc bromide solution 0.5 M in THF, 2.50 mL, 1.251 mmol.
- Pd(PPh 3 ) 4 0.072 g, 0.063 mmol was added.
- the reaction mixture was purged with argon, it was microwaved at 75 °C for 50 min.
- the reaction was diluted with EtOAc and water. The layers were separated, and the aqueous layer was extracted with EtOAc (2x).
- Step A 9-BBN, Pd 0 coupling of intermediate IV.4.e.2 and tert ⁇ buty ⁇ l-ethylprop-2-enylcarbamate Solid tert-butyl l-ethylprop-2-enylcarbamate (0.028 g, 0.135 mmol, prepared from propionaldehyde and vinyl Grignard according to D.A. Cogan et al.
- Tetrahedron 55 (1999) 8883-8904, followed by standard Boc installation) was placed in an oven-dried flask under an atmosphere of argon and dissolved in 9- borabicyclo[3.3.1]nonane (0.532 mL, 0.176 mmol, 0.5M solution in THF) and heated to 70°C.
- Step B Boc and ⁇ Bu Ester Removal Deprotection of 3 tot-butyl 3- ⁇ [2-[(tert-butoxycarbonyl)amino]-3-(3- ⁇ 3-[(tert- butoxycarbonyl)ammo]pentyl ⁇ phenyl)-2-methylpropoxy]methyl ⁇ -5-
- Step A Mitsunobu etherif ⁇ cation (intermediates II.5.d.l and DI.3.a.l) Intermediate m.3.a.l (0.22 g, 0.574 mmol), intermediate ⁇ .5.d.l (0.21 g, 0.603 mmol), and tri-n-butyl phosphine (0.22 mL, 0.862 mmol) were dissolved in 10 mL of anhydrous toluene and placed under argon atmosphere. TMAD (0.148 g, 0.862 mmol) was added in one portion and the reaction was allowed to stir at rt for 16 h. The reaction was then concentrated and purified using flash chromatography (4Og silica, 10-40% EtOAc in hexanes) to afford the corresponding phenolic ether.
- Step B TBS removal and hydrolysis
- TBAF 0.52 mL, 0.524 mmol
- the reaction was allowed to stir at RT for 16h.
- the reaction was then concentrated and purified using flash chromatography (40g silica, 10- 70% EtOAc in hexanes) to afford the corresponding benzylic alcohol.
- the previous benzylic alcohol (0.193 g, 0.326 mmol) was dissolved in 5 mL of THF.
- a 1.0 M solution of LiOH (3.26 mL, 3.26 mmol) was added in one portion and the reaction was allowed to stir at 50°C for 16 h.
- Step A hydroboration, Pd 0 coupling (intermediates II.5.e.l and III.4.a.l.l) Intermediate IIL4.a.l.l (92 mg, 0.25 mmol) was placed in an oven dried round bottom flask and dissolved in 0.5 M solution of 9-BBN (0.59 mL, 0.29 mmol) and the reaction was allowed to stir at 75°C for 45 min.
- Step B TBS removal and hydrolysis, as described in the preparation of Example 6.
- Step C Macrolactonization, as described in the preparation of Example 6.
- Step D Boc removal, as described in the preparation of Example 6 to provide Example 7.
- Step A Phenol alkylation (intermediates I.l.a.l and IQ.5.a.l)
- Step B TBS removal and hydrolysis, as described in the preparation of Example 6.
- Step C Macrolactonization, as described in the preparation of example Example 6.
- Step D Boc removal, as described in the preparation of Example 6 to provide Example 8.
- Example 9 1 H NMR (two diastereomers)(400 MHz, CD 3 OD) ⁇ 7.21-1.17 (m, 2H), 7.06-6.98 (m, 4H), 6.86-6.79 (m, 4H), 6.73-6.69 (m, 2H), 6.44-6.29 (m, 2H), 5.36-5.21 (m, 2H), 5.19-5.05 (m, 2H), 4.58-4.24 (m, 3H), 4.25-4.15 (m, 3H), 4.10-3.85 (m, 3H), 3.26 (s, 3H), 3.24 (s, 3H), 3.03-2.97 (m, 2H), 2.82 (s, 3H), 2.80 (s, 3H), 2.75-2.70 (m, 2H), 2.51-2.49 (m, 2H), 2.42 (br s, 2H), 1.65 (s, 3H), 1.64 (s, 3H).
- Step A Coupling of acylhydrazide Il.l.e.l and acid m.2.b.l.l (EDC, HOAt), followed by cyclodehydration (Burgess reagent, heat).
- Step B Boc and Me ester Removal, as described for the preparation of Example 1.
- Step C BOP Cyclization, as described for the preparation of Example 1. HRMS calculated for C 29 H 31 N 5 O 4 S: 546.2170, found: 546.2160.
- StepA Reductive animation
- StepB Boc and tBu ester removal, as described in example 2, stepB.
- StepC BOP cychzation, as described m example 2, stepC.
- MS M+l 550
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Hospice & Palliative Care (AREA)
- Psychiatry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Heterocyclic Compounds That Contain Two Or More Ring Oxygen Atoms (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Pyrane Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
Description






























































































































Claims









Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP05825640.5A EP1814537B1 (en) | 2004-11-17 | 2005-11-14 | Macrocyclic tertiary amine beta-secretase inhibitors for the treatment of alzheimer's disease |
| US11/667,913 US7678783B2 (en) | 2004-11-17 | 2005-11-14 | Macrocyclic tertiary amine beta-secretase inhibitors for the treatment of alzheimer's disease |
| JP2007543137A JP2008520670A (en) | 2004-11-17 | 2005-11-14 | Macrocyclic tertiary amine beta-secretase inhibitors for the treatment of Alzheimer's disease |
| CA002587083A CA2587083A1 (en) | 2004-11-17 | 2005-11-14 | Macrocyclic tertiary amine beta-secretase inhibitors for the treatment of alzheimer's disease |
| AU2005306701A AU2005306701A1 (en) | 2004-11-17 | 2005-11-14 | Macrocyclic tertiary amine beta-secretase inhibitors for the treatment of Alzheimer's disease |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US62883004P | 2004-11-17 | 2004-11-17 | |
| US60/628,830 | 2004-11-17 | ||
| US65303605P | 2005-02-15 | 2005-02-15 | |
| US60/653,036 | 2005-02-15 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2006055434A2 true WO2006055434A2 (en) | 2006-05-26 |
| WO2006055434A3 WO2006055434A3 (en) | 2007-07-05 |
Family
ID=36407657
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2005/040984 WO2006055434A2 (en) | 2004-11-17 | 2005-11-14 | Macrocyclic tertiary amine beta-secretase inhibitors for the treatment of alzheimer's disease |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US7678783B2 (en) |
| EP (1) | EP1814537B1 (en) |
| JP (1) | JP2008520670A (en) |
| AU (1) | AU2005306701A1 (en) |
| CA (1) | CA2587083A1 (en) |
| WO (1) | WO2006055434A2 (en) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008055945A1 (en) | 2006-11-09 | 2008-05-15 | Probiodrug Ag | 3-hydr0xy-1,5-dihydr0-pyrr0l-2-one derivatives as inhibitors of glutaminyl cyclase for the treatment of ulcer, cancer and other diseases |
| WO2008065141A1 (en) | 2006-11-30 | 2008-06-05 | Probiodrug Ag | Novel inhibitors of glutaminyl cyclase |
| WO2008104580A1 (en) | 2007-03-01 | 2008-09-04 | Probiodrug Ag | New use of glutaminyl cyclase inhibitors |
| WO2011029920A1 (en) | 2009-09-11 | 2011-03-17 | Probiodrug Ag | Heterocylcic derivatives as inhibitors of glutaminyl cyclase |
| US7951949B2 (en) | 2004-11-23 | 2011-05-31 | Merck, Sharp & Dohme, Corp. | Macrocyclic aminopyridyl beta-secretase inhibitors for the treatment of Alzheimer's disease |
| WO2011107530A2 (en) | 2010-03-03 | 2011-09-09 | Probiodrug Ag | Novel inhibitors |
| WO2011110613A1 (en) | 2010-03-10 | 2011-09-15 | Probiodrug Ag | Heterocyclic inhibitors of glutaminyl cyclase (qc, ec 2.3.2.5) |
| WO2011131748A2 (en) | 2010-04-21 | 2011-10-27 | Probiodrug Ag | Novel inhibitors |
| WO2012123563A1 (en) | 2011-03-16 | 2012-09-20 | Probiodrug Ag | Benz imidazole derivatives as inhibitors of glutaminyl cyclase |
| EP2865670A1 (en) | 2007-04-18 | 2015-04-29 | Probiodrug AG | Thiourea derivatives as glutaminyl cyclase inhibitors |
| EP3461819A1 (en) | 2017-09-29 | 2019-04-03 | Probiodrug AG | Inhibitors of glutaminyl cyclase |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20110107664A1 (en) * | 2009-11-10 | 2011-05-12 | Biovantage Resources, Inc. | Nutrient System and Methods |
| SG11201704628VA (en) * | 2015-02-05 | 2017-07-28 | Merck Patent Gmbh | Macrocyclic compounds as irak1/4 inhibitors and uses thereof |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU2001229320A1 (en) | 2000-01-12 | 2001-07-24 | Merck And Co., Inc. | Inhibitors of prenyl-protein transferase |
| US6333410B1 (en) | 2000-08-18 | 2001-12-25 | Immunogen, Inc. | Process for the preparation and purification of thiol-containing maytansinoids |
| CA2450167A1 (en) * | 2001-06-12 | 2002-12-19 | Elan Pharmaceuticals, Inc. | Macrocycles useful in the treatment of alzheimer's disease |
| WO2003106405A1 (en) | 2002-06-01 | 2003-12-24 | Sunesis Pharmaceuticals, Inc. | Aspartyl protease inhibitors |
| WO2004043916A1 (en) | 2002-11-12 | 2004-05-27 | Merck & Co., Inc. | Phenylcarboxamide beta-secretase inhibitors for the treatment of alzheimer's disease |
| US7371853B2 (en) | 2003-01-07 | 2008-05-13 | Merck & Co., Inc. | Macrocyclic β-secretase inhibitors for the treatment of Alzheimer's disease |
| EP1644322A1 (en) | 2003-06-16 | 2006-04-12 | Sunesis Pharmaceuticals, Inc. | Aspartyl protease inhibitors |
| AU2004255183A1 (en) | 2003-06-30 | 2005-01-20 | Merck & Co., Inc. | N-alkyl phenylcarboxamide beta-secretase inhibitors for the treatment of Alzheimer's disease |
| WO2005004803A2 (en) | 2003-07-01 | 2005-01-20 | Merck & Co., Inc. | Phenylcarboxylate beta-secretase inhibitors for the treatment of alzheimer's disease |
| CN1835936A (en) | 2003-08-14 | 2006-09-20 | 默克公司 | Macrocyclic beta-secretase inhibitors for the treatment of alzheimer's disease |
| DE602004014170D1 (en) | 2003-10-03 | 2008-07-10 | Merck & Co Inc | BENZYL ETHER AND BENZYLAMINO BETA SECRETASE INHIBITORS FOR THE TREATMENT OF ALZHEIMER DISEASE |
| ATE517861T1 (en) | 2003-11-24 | 2011-08-15 | Merck Sharp & Dohme | BENZYL ETHER AND BENZYLAMIN COMPOUNDS AS INHIBITORS OF BETA-SECRETASE FOR THE TREATMENT OF ALZHEIMER'S DISEASE |
| EP1697308B1 (en) | 2003-12-19 | 2014-03-19 | Merck Sharp & Dohme Corp. | Phenylamide and pyridylamide beta-secretase inhibitors for the treatment of alzheimer's disease |
| EP1740559B1 (en) | 2004-04-20 | 2014-10-15 | Merck Sharp & Dohme Corp. | 1,3,5-substituted phenyl derivative compounds useful as beta-secretase inhibitors for the treatment of alzheimer's disease |
| JP4764418B2 (en) | 2004-04-20 | 2011-09-07 | メルク・シャープ・エンド・ドーム・コーポレイション | 2,4,6-substituted pyridyl derivative compounds useful as β-secretase inhibitors for the treatment of Alzheimer's disease |
| US20070298029A1 (en) | 2004-08-09 | 2007-12-27 | Carsten Hopf | Treatment of Neurodegenerative Diseases by the Use of Degs Inhibitors |
| US7951949B2 (en) * | 2004-11-23 | 2011-05-31 | Merck, Sharp & Dohme, Corp. | Macrocyclic aminopyridyl beta-secretase inhibitors for the treatment of Alzheimer's disease |
-
2005
- 2005-11-14 US US11/667,913 patent/US7678783B2/en active Active
- 2005-11-14 EP EP05825640.5A patent/EP1814537B1/en not_active Not-in-force
- 2005-11-14 WO PCT/US2005/040984 patent/WO2006055434A2/en active Application Filing
- 2005-11-14 AU AU2005306701A patent/AU2005306701A1/en not_active Abandoned
- 2005-11-14 CA CA002587083A patent/CA2587083A1/en not_active Abandoned
- 2005-11-14 JP JP2007543137A patent/JP2008520670A/en active Pending
Non-Patent Citations (5)
| Title |
|---|
| H. FUKUMOTO ET AL., ARCH. NEUROL., vol. 59, September 2002 (2002-09-01), pages 1381 - 1389 |
| J.T. HUSE ET AL., J. BIOL. CHEM., vol. 277, no. 18, 3 May 2002 (2002-05-03), pages 16278 - 16284 |
| K.C. CHEN; W.J. HOWE, BIOCHEM. BIOPHYS. RES. COMM, vol. 292, 2002, pages 702 - 708 |
| R. N. ROSENBERG, ARCH. NEUROL., vol. 59, September 2002 (2002-09-01), pages 1367 - 1368 |
| See also references of EP1814537A4 |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7951949B2 (en) | 2004-11-23 | 2011-05-31 | Merck, Sharp & Dohme, Corp. | Macrocyclic aminopyridyl beta-secretase inhibitors for the treatment of Alzheimer's disease |
| WO2008055945A1 (en) | 2006-11-09 | 2008-05-15 | Probiodrug Ag | 3-hydr0xy-1,5-dihydr0-pyrr0l-2-one derivatives as inhibitors of glutaminyl cyclase for the treatment of ulcer, cancer and other diseases |
| WO2008065141A1 (en) | 2006-11-30 | 2008-06-05 | Probiodrug Ag | Novel inhibitors of glutaminyl cyclase |
| WO2008104580A1 (en) | 2007-03-01 | 2008-09-04 | Probiodrug Ag | New use of glutaminyl cyclase inhibitors |
| EP2481408A2 (en) | 2007-03-01 | 2012-08-01 | Probiodrug AG | New use of glutaminyl cyclase inhibitors |
| EP2865670A1 (en) | 2007-04-18 | 2015-04-29 | Probiodrug AG | Thiourea derivatives as glutaminyl cyclase inhibitors |
| WO2011029920A1 (en) | 2009-09-11 | 2011-03-17 | Probiodrug Ag | Heterocylcic derivatives as inhibitors of glutaminyl cyclase |
| WO2011107530A2 (en) | 2010-03-03 | 2011-09-09 | Probiodrug Ag | Novel inhibitors |
| WO2011110613A1 (en) | 2010-03-10 | 2011-09-15 | Probiodrug Ag | Heterocyclic inhibitors of glutaminyl cyclase (qc, ec 2.3.2.5) |
| WO2011131748A2 (en) | 2010-04-21 | 2011-10-27 | Probiodrug Ag | Novel inhibitors |
| WO2012123563A1 (en) | 2011-03-16 | 2012-09-20 | Probiodrug Ag | Benz imidazole derivatives as inhibitors of glutaminyl cyclase |
| EP3461819A1 (en) | 2017-09-29 | 2019-04-03 | Probiodrug AG | Inhibitors of glutaminyl cyclase |
Also Published As
| Publication number | Publication date |
|---|---|
| US7678783B2 (en) | 2010-03-16 |
| WO2006055434A3 (en) | 2007-07-05 |
| CA2587083A1 (en) | 2006-05-26 |
| EP1814537A2 (en) | 2007-08-08 |
| US20080269302A1 (en) | 2008-10-30 |
| EP1814537B1 (en) | 2014-11-12 |
| EP1814537A4 (en) | 2009-09-16 |
| AU2005306701A1 (en) | 2006-05-26 |
| JP2008520670A (en) | 2008-06-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1814537B1 (en) | Macrocyclic tertiary amine beta-secretase inhibitors for the treatment of alzheimer's disease | |
| EP1817312B1 (en) | Macrocyclic aminopyridyl beta-secretase inhibitors for the treatment of alzheimer's disease | |
| EP1855679B1 (en) | Aminomethyl beta-secretase inhibitors for the treatment of alzheimer's disease | |
| EP1673078B1 (en) | Benzylether and benzylamino beta-secretase inhibitors for the treatment of alzheimer's disease | |
| JP5676379B2 (en) | Fused ring compounds and uses thereof | |
| JP2010510962A (en) | Heteromonocyclic compounds and uses thereof | |
| WO2006078577A1 (en) | Tertiary carbinamines having substituted heterocycles, which are active as inhibitors of beta-secretase, for the treatment of alzheimer's disease | |
| AU2005257904A1 (en) | Pyrrolidin-3-yl compounds useful as beta-secretase inhibitors for the treatment of Alzheimer's disease | |
| US20080132477A1 (en) | Macrocyclic Compounds Useful as Bace Inhibitors | |
| JP2024123202A (en) | Macrocyclic compounds and uses thereof | |
| WO2007019078A2 (en) | Tricyclic beta-secretase inhibitors for the treatment of alzheimer's disease | |
| EP4364737A2 (en) | Triazolopyridinyl compounds as kinase inhibitors | |
| WO2007019080A2 (en) | Tricyclic beta-secretase inhibitors for the treatment of alzheimer's disease | |
| WO2007019111A2 (en) | Cyclic ketal beta-secretase inhibitors for the treatment of alzheimer's disease |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200580039377.7 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KM KN KP KR KZ LC LK LR LS LT LU LV LY MA MD MG MK MN MW MX MZ NA NG NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SM SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU LV MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1654/CHENP/2007 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005306701 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2587083 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005825640 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007543137 Country of ref document: JP Ref document number: 11667913 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2005306701 Country of ref document: AU Date of ref document: 20051114 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005825640 Country of ref document: EP |