WO2006049968A1 - Pyrimidine and quinoline potentiators of metabotropic glutamate receptors - Google Patents
Pyrimidine and quinoline potentiators of metabotropic glutamate receptors Download PDFInfo
- Publication number
- WO2006049968A1 WO2006049968A1 PCT/US2005/038435 US2005038435W WO2006049968A1 WO 2006049968 A1 WO2006049968 A1 WO 2006049968A1 US 2005038435 W US2005038435 W US 2005038435W WO 2006049968 A1 WO2006049968 A1 WO 2006049968A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phenyl
- ylmethyl
- pyrimidin
- trifluoro
- compound
- Prior art date
Links
- 0 *N(*C1=C*=C=CN=C1)c1ccccc1 Chemical compound *N(*C1=C*=C=CN=C1)c1ccccc1 0.000 description 1
- KTGOFUPLLGQSBB-UHFFFAOYSA-N O=S(CC(F)(F)F)(N(Cc1cncnc1)c1cccc(OC2CCCC2)c1)=O Chemical compound O=S(CC(F)(F)F)(N(Cc1cncnc1)c1cccc(OC2CCCC2)c1)=O KTGOFUPLLGQSBB-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/26—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/06—Antimigraine agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/04—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, directly attached to the ring carbon atoms
- C07D215/06—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, directly attached to the ring carbon atoms having only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, attached to the ring nitrogen atom
Definitions
- the excitatory amino acid L-glutamate (sometimes referred to herein simply as glutamate) through its many receptors mediates most of the excitatory neurotransmission within the mammalian central nervous system (CNS).
- the excitatory amino acids, including glutamate, are of great physiological importance, playing a role in a variety of physiological processes, such as long-term potentiation (learning and memory), the development of synaptic plasticity, motor control, respiration, cardiovascular regulation, and sensory perception.
- Glutamate acts via at least two distinct classes of receptors.
- One class is composed of the ionotropic glutamate (iGlu) receptors that act as ligand-gated ionic channels. Via activation of the iGlu receptors, glutamate is thought to regulate fast neuronal transmission within the synapse of two connecting neurons in the CNS.
- the second general type of receptor is the G-protein or second messenger-linked "metabotropic" glutamate (mGluR) receptor. Both types of receptors appear not only to mediate normal synaptic transmission along excitatory pathways, but also participate in the modification of synaptic connections during development and throughout life. Schoepp, Bockaert, and Sladeczek, Trends in Pharmacol. Sci., 11, 508 (1990); McDonald and Johnson, Brain Research Reviews, 15, 41 (1990).
- the present invention relates to potentiators of mGlu receptors, in particular mGluR2 receptors.
- the mGluR receptors belong to the Type III G- protein coupled receptor (GPCR) superfamily. This superfamily of GPCR'sf including the calcium-sensing receptors, GABAB receptors and pheromone receptors, which are unique in that they are activated by binding of effectors to the amino-terminus portion of the receptor protein.
- GPCR G- protein coupled receptor
- the mGlu receptors are thought to mediate glutamate's demonstrated ability to modulate intracellular signal transduction pathways. Ozawa, Kamiya and Tsuzuski, Prog. Neurobio., 54, 581 (1998).
- the Group I mGluR receptors which include the mGlulR and mGlu5R, are known to activate phospholipase C (PLC) via Gaq-proteins thereby resulting in the increased hydrolysis of phosphoinositides and intracellular calcium mobilization.
- PLC phospholipase C
- the Group II mGlu receptors consist of the two distinct receptors, mGluR2 and mGluR3 receptors. Both have been found to be negatively coupled to adenylate cyclase via activation of Gai- protein. These receptors can be activated by a selective compound such as lS,2S,SR,6S-2 aminobicyclo[3.1.0]hexane-2,6-dicarboxylate. Monn, et al., J. Med. Chem., 40, 528 (1997); Schoepp, et al., Neuropharmacol., 36, 1 (1997).
- the Group HI mGlu receptors including mGluR4, mGluR6, mGluR7 and mGluR8, are negatively coupled to adenylate cyclase via Gai and are potently activated by L-AP4 (L- (+) -2-amino-4- phosphonobutyric acid). Schoepp, Neurochem. Int., 24, 439 (1994).
- the present invention is directed to compounds which are potentiators of metabotropic glutamate receptors, including the mGluR2 receptor, and which are useful in the treatment or prevention of neurological and psychiatric disorders associated with glutamate dysfunction and diseases in which metabotropic glutamate receptors are involved.
- the invention is also directed to pharmaceutical compositions comprising these compounds and the use of these compounds and compositions in the prevention or treatment of such diseases in which metabotropic glutamate receptors are involved.
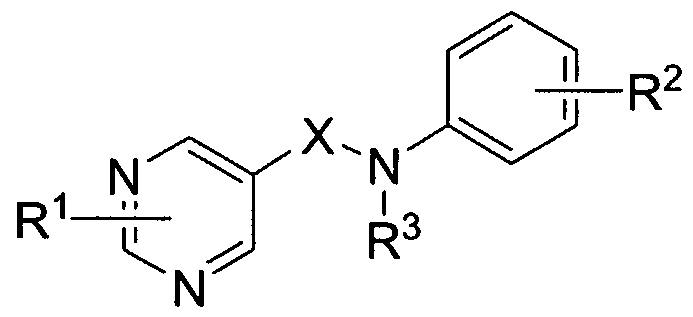
- the present invention is directed to compounds of the formula I:
- X is selected from the group consisting of:
- Y is C wheren Rl at the adjacent position is joined to Y as a phenyl group to form a quinolinyl ring;
- Rl and R2 may include multiple substituents and are independently selected from the group consisting of:
- phenyl which is unsubstituted or substituted with fluoro, bromo, -Ci-6alkyl, -CF3 or -O-Ci-6alkyl, (4) -O-Ci-galkyl, unsubstituted or substituted with halogen,
- R3 is selected from the group consisting of:
- R ⁇ is independently selected from:
- RIO is independently selected from:
- An embodiment of the present invention includes compounds of the formula Ia:
- Rl, R2, Ry, A and X are defined herein; or a pharmaceutically acceptable salt thereof or an individual enantiomer or diastereomer thereof.
- An embodiment of the present invention includes compounds of the formula Ib:
- An embodiment of the present invention includes compounds wherein X is -CH2-.
- An embodiment of the present invention includes compounds wherein X is -CH2CH2-.
- An embodiment of the present invention includes compounds wherein
- Rl is hydrogen
- R2 is selected from the group consisting of:
- Ci- ⁇ alkyl unsubstituted or substituted with halogen or phenyl, which is unsubstituted or substituted with fluoro, bromo, -Ci_6alkyl, -CF3 or
- R.2 is selected from the group consisting of:
- Ci_6alkyl unsubstituted or substituted with halogen or phenyl, which is unsubstituted or substituted with fluoro, bromo, -CH3, -CF3 or -O-CH3,
- An embodiment of the present invention includes compounds wherein R.2 is selected from the group consisting of: (1) hydrogen,
- An embodiment of the present invention includes compounds wherein R3 is hydrogen.
- An embodiment of the present invention includes compounds wherein R3 is -S ⁇ 2-Ci_6alkyl, which is unsubstituted or substituted with fluoro.
- An embodiment of the present invention includes compounds wherein R3 is -SO2-CH2CF3.
- Specific embodiments of the present invention include a compound which selected from the group consisting of: N -[3-(cyclopentyloxy)phenyl]-2,2,2-trifluoro-N-(pyrimidin-5-ylmethyl)ethanesulfonamide; 2,2,2-trifluoro-N-(pyrimidin-5-ylmethyl)-N- ⁇ 3-[2-(trifluoromethyl)benzyl]phenyl ⁇ ethanesulfonamide;
- the compounds of the present invention are potentiators of metabotropic glutamate (mGluR) receptor function, in particular they are potentiators of mGluR2 receptors. That is, the compounds of the present invention do not appear to bind at the glutamate recognition site on the mGluR receptor, but in the presence of glutamate or a glutamate agonist, the compounds of the present invention increase mGluR receptor response.
- the present potentiators are expected to have their effect at mGluR receptors by virtue of their ability to increase the response of such receptors to glutamate or glutamate agonists, enhancing the function of the receptors.
- the compounds of the present invention would be expected to increase the effectiveness of glutamate and glutamate agonists of the mGluR2 receptor.
- the potentiators of the present invention are expected to be useful in the treatment of various neurological and psychiatric disorders associated with glutamate dysfunction described to be treated herein and others that can be treated by such potentiators as are appreciated by those skilled in the art.
- the compounds of the present invention may contain one or more asymmetric centers and can thus occur as racemates and racemic mixtures, single enantiomers, diastereomeric mixtures and individual diastereomers. Additional asymmetric centers may be present depending upon the nature of the various substituents on the molecule. Each such asymmetric center will independently produce two optical isomers and it is intended that all of the possible optical isomers and diastereomers in mixtures and as pure or partially purified compounds are included within the ambit of this invention. The present invention is meant to comprehend all such isomeric forms of these compounds.
- Formula I shows the structure of the class of compounds without preferred stereochemistry.
- racemic mixtures of the compounds may be separated so that the individual enantiomers are isolated.
- the separation can be carried out by methods well known in the art, such as the coupling of a racemic mixture of compounds to an enantiomerically pure compound to form a diastereomeric mixture, followed by separation of the individual diastereomers by standard methods, such as fractional crystallization or chromatography.
- the coupling reaction is often the formation of salts using an enantiomerically pure acid or base.
- the diasteromeric derivatives may then be converted to the pure enantiomers by cleavage of the added chiral residue.
- the racemic mixture of the compounds can also be separated directly by chromatographic methods utilizing chiral stationary phases, which methods are well known in the art.
- any enantiomer of a compound may be obtained by stereoselective synthesis using optically pure starting materials or reagents of known configuration by methods well known in the art.
- Ci- ⁇ alkyl is defined to identify the group as having 1, 2, 3, 4, 5 or 6 carbons in a linear or branched arrangement, such that Ci-8alkyl specifically includes methyl, ethyl, n-propyl, iso-propyl, n-butyl, iso-butyl, tert-butyl, pentyl, and hexyl.
- a group which is designated as being independently substituted with substituents may be independently substituted with multiple numbers of such substituents.
- salts refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic or organic bases and inorganic or organic acids.
- Salts derived from inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic salts, manganous, potassium, sodium, zinc, and the like. Particularly preferred are the ammonium, calcium, magnesium, potassium, and sodium salts. Salts in the solid form may exist in more than one crystal structure, and may also be in the form of hydrates.
- Salts derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, and basic ion exchange resins, such as arginine, betaine, caffeine, choline, N,N'-dibenzylethylene- diamine, diethylamine, 2-diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethyl-morpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine, and the like.
- basic ion exchange resins such as
- salts may be prepared from pharmaceutically acceptable non-toxic acids, including inorganic and organic acids.
- acids include acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethanesulfonic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, nitric, pamoic, pantothenic, phosphoric, succinic, sulfuric, tartaric, p-toluenesulfonic acid, and the like.
- Exemplifying the invention is the use of the compounds disclosed in the Examples and herein.
- Specific compounds within the present invention include a compound which selected from the group consisting of the compounds disclosed in the following Examples and pharmaceutically acceptable salts thereof and individual diastereomers thereof.
- the subject compounds are useful in a method of potentiating metabotorpic glutamate receptor activity in a patient such as a mammal in need of such inhibition comprising the administration of an effective amount of the compound.
- the present invention is directed to the use of the compounds disclosed herein as potentiators of metabotorpic glutamate receptor activity. In addition to primates, especially humans, a variety of other mammals can be treated according to the method of the present invention.
- the present invention is further directed to a method for the manufacture of a medicament for potentiating metabotorpic glutamate receptor activity in humans and animals comprising combining a compound of the present invention with a pharmaceutical carrier or diluent.
- the subject treated in the present methods is generally a mammal, preferably a human being, male or female, in whom potentiation of metabotorpic glutamate receptor activity is desired.
- the term "therapeutically effective amount” means the amount of the subject compound that will elicit the biological or medical response of a tissue, system, animal or human that is being sought by the researcher, veterinarian, medical doctor or other clinician. It is recognized that one skilled in the art may affect the neurological and psychiatric disorders by treating a patient presently afflicted with the disorders or by prophylactically treating a patient afflicted with the disorders with an effective amount of the compound of the present invention.
- treatment and “treating” refer to all processes wherein there may be a slowing, interrupting, arresting, controlling, or stopping of the progression of the neurological and psychiatric disorders described herein, but does not necessarily indicate a total elimination of all disorder symptoms, as well as the prophylactic therapy of the mentioned conditions, particularly in a patient who is predisposed to such disease or disorder.
- composition as used herein is intended to encompass a product comprising the specified ingredients in the specified amounts, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts.
- compositions of the present invention encompass any composition made by admixing a compound of the present invention and a pharmaceutically acceptable carrier.
- pharmaceutically acceptable it is meant the carrier, diluent or excipient must be compatible with the other ingredients of the formulation and not deleterious to the recipient thereof.
- administering a should be understood to mean providing a compound of the invention or a prodrug of a compound of the invention to the individual in need of treatment.
- the utility of the compounds in accordance with the present invention as inhibitors of metabotropic glutamate receptor activity, in particular mGluR2 activity, may be demonstrated by methodology known in the art. Inhibition constants are determined as follows. The compounds of the present invention were tested in a [ 35 S]-GTPyS assay. The stimulation of [ 35 S]-GTPyS binding is a common functional assay to monitor God-coupled receptor in native and recombinant receptor membrane preparation.
- Membrane from cells stably expressing hmGlu2 CHO-Kl were incubated in a 96 well plate for 1 hour in the presence of GTPyS 35 (0.05nM), GDP (5 ⁇ M) and compounds.
- the reaction was stopped by rapid filtration over Unifilter GF/B plate (Packard, Bioscience, Meriden CT) using a 96- well cell harvester (Brandel Gaithersburg, MD). The filter plates were counted using Topcount counter (Packard, Bioscience, Meriden CT, USA).
- the activation (agonist) or the potentiation of glutamate (potentiator) curves were fitted with a four parameters logistic equation giving EC 50 and Hill coefficient using the iterative non linear curve fitting software GraphPad (San Diego CA, USA).
- the compounds of the following examples had activity in potentiating the mGluR2 receptor in the aforementioned assays, generally with an EC50 of less than about 10 ⁇ M.
- Preferred compounds within the present invention had activity in potentiating the mGluR2 receptor in the aforementioned assays with an EC50 of less than about 1 ⁇ M. Such a result is indicative of the intrinsic activity of the compounds in use as potentiators of mGluR2 receptor activity.
- Metabotropic glutamate receptors including the mGluR2 receptor have been implicated in a wide range of biological functions. This has suggested a potential role for these receptors in a variety of disease processes in humans or other species.
- the compounds of the present invention have utility in treating, preventing, ameliorating, controlling or reducing the risk of a variety of neurological and psychiatric disorders associated with glutamate dysfunction, including one or more of the following conditions or diseases: acute neurological and psychiatric disorders such as cerebral deficits subsequent to cardiac bypass surgery and grafting, stroke, cerebral ischemia, spinal cord trauma, head trauma, perinatal hypoxia, cardiac arrest, hypoglycemic neuronal damage, dementia (including AIDS-induced dementia), Alzheimer's disease, Huntington's Chorea, amyotrophic lateral sclerosis, ocular damage, retinopathy, cognitive disorders, idiopathic and drug-induced Parkinson's disease, muscular spasms and disorders associated with muscular spasticity including tremors, epilepsy, convulsions, migraine (including migraine headache),.
- acute neurological and psychiatric disorders such as cerebral deficits subsequent to cardiac bypass surgery and grafting, stroke, cerebral ischemia, spinal cord trauma, head trauma, perinatal hypoxia, cardiac arrest,
- urinary incontinence substance tolerance
- substance withdrawal including, substances such as opiates, nicotine, tobacco products, alcohol, benzodiazepines, cocaine, sedatives, hypnotics, etc.
- psychosis schizophrenia, anxiety (including generalized anxiety disorder, panic disorder, and obsessive compulsive disorder), mood disorders (including depression, mania, bipolar disorders), trigeminal neuralgia, hearing loss, tinnitus, macular degeneration of the eye, emesis, brain edema, pain (including acute and chronic pain states, severe pain, intractable pain, neuropathic pain, and post-traumatic pain), tardive dyskinesia, sleep disorders (including narcolepsy), attention deficit/hyperactivity disorder, and conduct disorder.
- anxiety including generalized anxiety disorder, panic disorder, and obsessive compulsive disorder
- mood disorders including depression, mania, bipolar disorders
- trigeminal neuralgia including depression, mania, bipolar disorders
- hearing loss including depression,
- the present invention provides a method for treating migraine, comprising: administering to a patient in need thereof an effective amount of a compound of formula I.
- the present invention provides a method for preventing or treating anxiety, comprising: administering to a patient in need thereof an effective amount of a compound of formula I.
- Particularly preferred anxiety disorders are generalized anxiety disorder, panic disorder, and obsessive compulsive disorder.
- the present invention provides a method for treating schizophrenia, comprising: administering to a patient in need thereof an effective amount of a compound of formula I.
- the present invention provides a method for treating epilepsy, comprising: administering to a patient in need thereof an effective amount of a compound of formula I.
- migraine migraine
- anxiety disorders are particularly preferred.
- anxiety disorders are generalized anxiety disorder, panic disorder, and obsessive compulsive disorder.
- the present invention provides a method for treating migraine, comprising: administering to a patient in need thereof an effective amount of a compound of formula I or a pharmaceutical composition thereof.
- migraine is defined as a symptom complex of periodic headaches, usually temporal and unilateral, often with irritability, nausea, vomiting, constipation or diarrhea, and photophobia.
- migraine includes these periodic headaches, both temporal and unilateral, the associated irritability, nausea, vomiting, constipation or diarrhea, photophobia, and other associated symptoms.
- migraine includes these periodic headaches, both temporal and unilateral, the associated irritability, nausea, vomiting, constipation or diarrhea, photophobia, and other associated symptoms.
- the present invention provides a method for treating anxiety, comprising: administering to a patient in need thereof an effective amount of a compound of formula I or a pharmaceutical composition thereof.
- anxiety includes treatment of those anxiety disorders and related disorder as described in the DSM-IV.
- the skilled artisan will recognize that there are alternative nomenclatures, nosologies, and classification systems for neurological and psychiatric disorders, and particular anxiety, and that these systems evolve with medical scientific progress.
- the term “anxiety” is intended to include like disorders that are described in other diagnostic sources.
- the present invention provides a method for treating depression, comprising: administering to a patient in need thereof an effective amount of a compound of formula I or a pharmaceutical composition thereof.
- DSM-IV Diagnostic and Statistical Manual of Mental Disorders (1994, American Psychiatric Association, Washington, D.C.) (1994, American Psychiatric Association, Washington, D.C.), provides a diagnostic tool including depression and related disorders.
- Depressive disorders include, for example, single episodic or recurrent major depressive disorders, and dysthymic disorders, depressive neurosis, and neurotic depression; melancholic depression including anorexia, weight loss, insomnia and early morning waking, and psychomotor retardation; atypical depression (or reactive depression) including increased appetite, hypersomnia, psychomotor agitation or irritability, anxiety and phobias; seasonal affective disorder; or bipolar disorders or manic depression, for example, bipolar I disorder, bipolar II disorder and cyclothymic disorder.
- depression includes treatment of those depression disorders and related disorder as described in the DSM-IV.
- the present invention provides a method for treating epilepsy, comprising: administering to a patient in need thereof an effective amount of a compound of formula I or a pharmaceutical composition thereof.
- epilepsy there are several types and subtypes of seizures associated with epilepsy, including idiopathic, symptomatic, and cryptogenic. These epileptic seizures can be focal (partial) or generalized. They can also be simple or complex.
- Epilepsy is described in the art, such as Epilepsy: A comprehensive textbook. Ed. by Jerome Engel, Jr. and Timothy A. Pedley. (Lippincott-Raven, Philadelphia, 1997).
- the International Classification of Diseases, Ninth Revision, (ICD-9) provides a diagnostic tool including epilepsy and related disorders.
- epilepsy includes these all types and subtypes.
- the skilled artisan will recognize that there are alternative nomenclatures, nosologies, and classification systems for neurological and psychiatric disorders, including epilepsy, and that these systems evolve with medical scientific progress.
- the subject compounds are further useful in a method for the prevention, treatment, control, amelioration, or reducation of risk of the diseases, disorders and conditions noted herein.
- the subject compounds are further useful in a method for the prevention, treatment, control, amelioration, or reduction of risk of the aforementioned diseases, disorders and conditions in combination with other agents, including an mGluR agonist.
- the term "potentiated amount" refers to an amount of an mGluR agonist, that is, the dosage of agonist which is effective in treating the neurological and psychiatric disorders described herein when administered in combination with an effective amount of a compound of the present invention.
- a potentiated amount is expected to be less than the amount that is required to provided the same effect when the mGluR agonist is administered without an effective amount of a compound of the present invention.
- a potentiated amount can be readily determined by the attending diagnostician, as one skilled in the art, by the use of conventional techniques and by observing results obtained under analogous circumstances.
- the dose of an mGluR agonist to be administered in combination with a compound of formula I a number of factors are considered by the attending diagnostician, including, but not limited to: the mGluR agonist selected to be administered, including its potency and selectivity; the compound of formula I to be coadministered; the species of mammal; its size, age, and general health; the specific disorder involved; the degree of involvement or the severity of the disorder; the response of the individual patient; the modes of administration; the bioavailability characteristics of the preparations administered; the dose regimens selected; the use of other concomitant medication; and other relevant circumstances.
- a potentiated amount of an mGluR agonist to be administered in combination with an effective amount of a compound of formula I is expected to vary from about 0.1 milligram per kilogram of body weight per day (mg/kg/day) to about 100 mg/kg/day and is expected to be less than the amount that is required to provided the same effect when administered without an effective amount of a compound of formula I.
- Preferred amounts of a co-administered mGlu agonist are able to be determined by one skilled in the art.
- the compounds of the present invention may be used in combination with one or more other drugs in the treatment, prevention, control, amelioration, or reduction of risk of diseases or conditions for which compounds of Formula I or the other drugs may have utility, where the combination of the drugs together are safer or more effective than either drug alone.
- Such other drug(s) may be administered, by a route and in an amount commonly used therefor, contemporaneously or sequentially with a compound of Formula I.
- a pharmaceutical composition in unit dosage form containing such other drugs and the compound of Formula I is preferred.
- the combination therapy may also includes therapies in which the compound of Formula I and one or more other drugs are administered on different overlapping schedules.
- the compounds of the present invention and the other active ingredients may be used in lower doses than when each is used singly.
- the pharmaceutical compositions of the present invention include those that contain one or more other active ingredients, in addition to a compound of Formula I.
- the above combinations include combinations of a compound of the present invention not only with one other active compound, but also with two or more other active compounds.
- compounds of the present invention may be used in combination with other drugs that are used in the prevention, treatment, control, amelioration, or reduction of risk of the diseases or conditions for which compounds of the present invention are useful.
- Such other drugs may be administered, by a route and in an amount commonly used therefor, contemporaneously or sequentially with a compound of the present invention.
- a pharmaceutical composition containing such other drugs in addition to the compound of the present invention is preferred.
- the pharmaceutical compositions of the present invention include those that also contain one or more other active ingredients, in addition to a compound of the present invention.
- the weight ratio of the compound of the compound of the present invention to the second active ingredient may be varied and will depend upon the effective dose of each ingredient. Generally, an effective dose of each will be used. Thus, for example, when a compound of the present invention is combined with another agent, the weight ratio of the compound of the present invention to the other agent will generally range from about 1000:1 to about 1:1000, preferably about 200:1 to about 1:200. Combinations of a compound of the present invention and other active ingredients will generally also be within the aforementioned range, but in each case, an effective dose of each active ingredient should be used. In such combinations the compound of the present invention and other active agents may be administered separately or in conjunction. In addition, the administration of one element may be prior to, concurrent to, or subsequent to the administration of other agent(s).
- the compounds of the present invention may be administered by oral, parenteral (e.g., intramuscular, intraperitoneal, intravenous, ICV, intracisternal injection or infusion, subcutaneous injection, or implant), by inhalation spray, nasal, vaginal, rectal, sublingual, or topical routes of administration and may be formulated, alone or together, in suitable dosage unit formulations containing conventional non-toxic pharmaceutically acceptable carriers, adjuvants and vehicles appropriate for each route of administration.
- parenteral e.g., intramuscular, intraperitoneal, intravenous, ICV, intracisternal injection or infusion, subcutaneous injection, or implant
- inhalation spray nasal, vaginal, rectal, sublingual, or topical routes of administration
- nasal, vaginal, rectal, sublingual, or topical routes of administration may be formulated, alone or together, in suitable dosage unit formulations containing conventional non-toxic pharmaceutically acceptable carriers, adjuvants and vehicles appropriate for each route of administration.
- the compounds of the invention are effective for
- compositions for the administration of the compounds of this invention may conveniently be presented in dosage unit form and may be prepared by any of the methods well known in the art of pharmacy. All methods include the step of bringing the active ingredient into association with the carrier which constitutes one or more accessory ingredients.
- the pharmaceutical compositions are prepared by uniformly and intimately bringing the active ingredient into association with a liquid carrier or a finely divided solid carrier or both, and then, if necessary, shaping the product into the desired formulation.
- the active object compound is included in an amount sufficient to produce the desired effect upon the process or condition of diseases.
- composition is intended to encompass a product comprising the specified ingredients in the specified amounts, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts.
- compositions intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions and such compositions may contain one or more agents selected from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents in order to provide pharmaceutically elegant and palatable preparations.
- Tablets contain the active ingredient m admixture with non-toxic pharmaceutically acceptable excipients which are suitable for the manufacture of tablets.
- excipients may be for example, inert diluents, such as calcium carbonate, sodium carbonate, lactose, calcium phosphate or sodium phosphate; granulating and disintegrating agents, for example, corn starch, or algmic acid; binding agents, for example starch, gelatin or acacia, and lubricating agents, for example magnesium stearate, stearic acid or talc.
- the tablets may be uncoated or they may be coated by known techniques to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer pe ⁇ od.
- compositions for oral use may also be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolm, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, for example peanut oil, liquid paraffin, or olive oil.
- an inert solid diluent for example, calcium carbonate, calcium phosphate or kaolm
- water or an oil medium for example peanut oil, liquid paraffin, or olive oil.
- Aqueous suspensions contain the active materials in admixture with excipients suitable for the manufacture of aqueous suspensions.
- Oily suspensions may be formulated by suspending the active ingredient in a suitable oil.
- Oil-m-water emulsions may also be employed.
- Dispersible powders and granules suitable for preparation of an aqueous suspension by the addition of water provide the active ingredient in admixture with a dispersing or wetting agent, suspending agent and one or more preservatives.
- Pharmaceutical compositions of the present compounds may be in the form of a ste ⁇ le injectable aqueous or oleagenous suspension.
- the compounds of the present invention may also be administered in the form of suppositories for rectal administration.
- creams, ointments, jellies, solutions or suspensions, etc., containing the compounds of the present invention may be employed.
- the compounds of the present invention may also be formulated for administered by inhalation.
- the compounds of the present invention may also be administered by a transdermal patch by methods known in the art.
- the pharmaceutical composition and method of the present invention may further comprise other therapeutically active compounds as noted herein which are usually applied in the treatment of the above mentioned pathological conditions.
- an approp ⁇ ate dosage level will generally be about 0.01 to 500 mg per kg patient body weight per day which can be administered in single or multiple doses.
- the dosage level will be about 0.1 to about 250 mg/kg per day; more preferably about 0.5 to about 100 mg/kg per day.
- a suitable dosage level may be about 0.01 to 250 mg/kg per day, about 0.05 to 100 mg/kg per day, or about 0.1 to 50 mg/kg per day.
- the dosage may be 0.05 to 0.5, 0.5 to 5 or 5 to 50 mg/kg per day.
- the compositions are preferably provided in the form of tablets containing 1.0 to 1000 milligrams of the active ingredient, particularly 1.0, 5.0, 10.0, 15.0. 20.0, 25.0, 50.0, 75.0, 100.0, 150.0, 200.0, 250.0, 300.0, 400.0, 500.0, 600.0, 750.0, 800.0, 900.0, and 1000.0 milligrams of the active ingredient for the symptomatic adjustment of the dosage to the patient to be treated.
- the compounds may be administered on a regimen of 1 to 4 times per day, preferably once or twice per day.
- the compounds of the present invention are administered at a daily dosage of from about 0.1 milligram to about 100 milligram per kilogram of animal body weight, preferably given as a single daily dose or in divided doses two to six times a day, or in sustained release form.
- the total daily dosage is from about 1.0 milligrams to about 1000 milligrams, preferably from about 1 milligrams to about 50 milligrams. In the case of a 70 kg adult human, the total daily dose will generally be from about 7 milligrams to about 350 milligrams.
- This dosage regimen may be adjusted to provide the optimal therapeutic response. It will be understood, however, that the specific dose level and frequency of dosage for any particular patient may be varied and will depend upon a variety of factors including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the age, body weight, general health, sex, diet, mode and time of administration, rate of excretion, drug combination, the severity of the particular condition, and the host undergoing therapy.
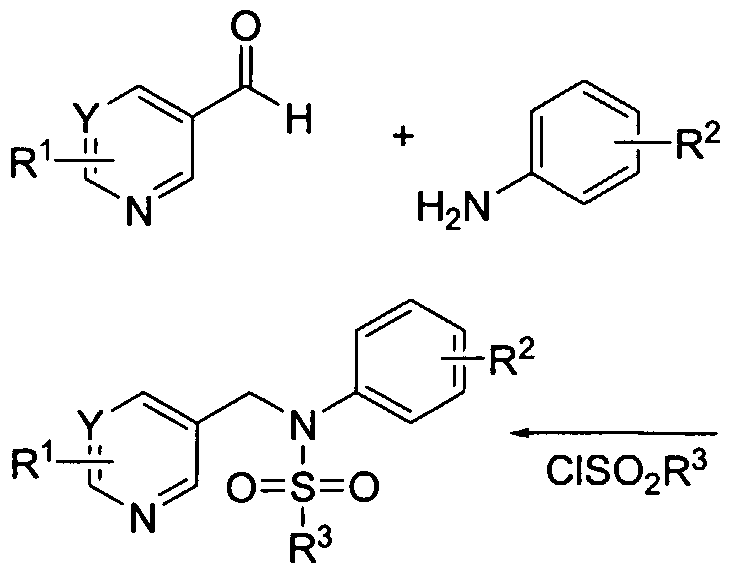
- Several methods for preparing the compounds of this invention are illustrated in the following Schemes and Examples. Starting materials are made according to procedures known in the art or as illustrated herein. The compounds of the present invention can be prepared in a variety of fashions.
- An appropriately substituted pyrimidine (or quinoline) can be prepared via reductive amination followed by sulfonylation as illustrated in Scheme 1.
- a substituted pyrimidine aldehyde (either purchased commercially or prepared using techniques well known in the art) is reacted with a substituted aniline in the presence of a reducing agent, such as sodium borohydride or sodium triacetoxyborohydride, can be used in solvents such as methanol or dichloroethane.
- the reaction generally proceeds either by refluxing the reagents in methanol then adding sodium borohydride at 0 0 C and allowing the reaction to warm to ambient temperature over a period of several hours, or by adding sodium triacetoxyboro-hydride to the reagents at ambient temperature and then maintaining ambient temperature for several more hours.
- the product from the reaction can be isolated and purified employing standard techniques such as solvent extraction, chromatography, crystallization, distillation and the like.
- the product obtained is then sulfonylated with variously substituted sulfonyl chloride compounds. These sulfonyl chloride compounds are reacted in the presence of a base (potassium carbonate, pyridine, and the like) in a suitable solvent (dichloroethane, pyridine, etc). The reaction is generally run at ambient temperature for several hours.
- the product from the reaction can be isolated and purified employing standard techniques such as solvent extraction, chromatography, crystallization, distillation and the like.
- the final product may be further modified, for example, by manipulation of substituents.
- manipulations may include, but are not limited to, reduction, oxidation, alkylation, acylation, and hydrolysis reactions which are commonly known to those skilled in the art.
- Diisopropyl azodicarboxylate (0.45 mL, 2.3 mmol) was added dropwise to a stirred solution of 3-aminophenol (250 mg, 2.3 mmol), cyclopentanol (0.25 mL, 2.8 mmol), and triphenylphosphine (600 mg, 2.3 mmol) in tetrahydrofuran (1.4 mL) at 0 0 C. After stirring for an additional 12hr at room temperature, the reaction mixture was rotovapped in vacuo to remove volatile solvents.
- reaction mixture was then rotovapped in vacuo to remove volatile solvents.
- residue was directly purified via column chromatography on silica gel (eluting 15-100% ethyl acetate/hexanes) to give 200 mg (83%) of [3-(cyclopentyloxy)phenyl]- (pyrimidin-5-ylmethyl)amine as brown oil.
- Acetic acid 131 mg, 2 .19 mmol
- tetrabutylammonium fluoride 1.0 M in tetrahydrofuran, 2.19 mL, 2.19 mmol
- N -[3-( ⁇ [ tert - butyl(dimethyl)silyl]oxy ⁇ methyl)phenyl]-2,2,2-trifluoro-N -(pyrimidin-5-ylmethyl) ethanesulfonamide 520 mg, 1.09 mmol
- tetrahydrofuran 10 mL
Landscapes
- Health & Medical Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Public Health (AREA)
- Medicinal Chemistry (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Pain & Pain Management (AREA)
- Psychiatry (AREA)
- Hospice & Palliative Care (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
- Quinoline Compounds (AREA)
Abstract
Description










Claims


Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2005302608A AU2005302608A1 (en) | 2004-10-28 | 2005-10-24 | Pyrimidine and quinoline potentiators of metabotropic glutamate receptors |
| JP2007539056A JP2008518913A (en) | 2004-10-28 | 2005-10-24 | Pyrimidine and quinoline potentiators of metabotropic glutamate receptors |
| CA002585210A CA2585210A1 (en) | 2004-10-28 | 2005-10-24 | Pyrimidine and quinoline potentiators of metabotropic glutamate receptors |
| US11/665,422 US20070287716A1 (en) | 2004-10-28 | 2005-10-24 | Pyrimidine and Quinoline Potentiators of Metabotropic Glutamate Receptors |
| EP05812616A EP1809608A4 (en) | 2004-10-28 | 2005-10-24 | Pyrimidine and quinoline potentiators of metabotropic glutamate receptors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US62286804P | 2004-10-28 | 2004-10-28 | |
| US60/622,868 | 2004-10-28 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2006049968A1 true WO2006049968A1 (en) | 2006-05-11 |
Family
ID=36319508
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2005/038435 WO2006049968A1 (en) | 2004-10-28 | 2005-10-24 | Pyrimidine and quinoline potentiators of metabotropic glutamate receptors |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20070287716A1 (en) |
| EP (1) | EP1809608A4 (en) |
| JP (1) | JP2008518913A (en) |
| CN (1) | CN101048384A (en) |
| AU (1) | AU2005302608A1 (en) |
| CA (1) | CA2585210A1 (en) |
| WO (1) | WO2006049968A1 (en) |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7598423B2 (en) | 2004-11-22 | 2009-10-06 | Eli Lilly And Company | Potentiators of glutamate receptors |
| WO2010114726A1 (en) * | 2009-03-31 | 2010-10-07 | Merck Sharp & Dohme Corp. | Aminobenzotriazole derivatives |
| US7960417B2 (en) | 2005-02-24 | 2011-06-14 | Merck Sharp & Dohme Corp. | Benzazole potentiators of metabotropic glutamate receptors |
| WO2014100425A1 (en) * | 2012-12-20 | 2014-06-26 | Aldexa Therapeutics, Inc. | Peri-carbinols |
| US9278960B2 (en) | 2011-11-03 | 2016-03-08 | Merck Sharp & Dohme Corp. | Quinoline carboxamide and quinoline carbonitrile derivatives as mGluR2-negative allosteric modulators, compositions, and their use |
| US9364471B2 (en) | 2005-05-26 | 2016-06-14 | Aldeyra Therapeutics, Inc. | Compositions and methods of treating retinal disease |
| US9687481B2 (en) | 2013-01-23 | 2017-06-27 | Aldeyra Therapeutics, Inc. | Toxic aldehyde related diseases and treatment |
| US9814701B2 (en) | 2009-12-11 | 2017-11-14 | Aldeyra Therapeutics, Inc. | Compositions and methods for the treatment of macular degeneration |
| US10111862B2 (en) | 2013-01-25 | 2018-10-30 | Aldeyra Therapeutics, Inc. | Traps in the treatment of macular degeneration |
| US10414732B2 (en) | 2017-03-16 | 2019-09-17 | Aldeyra Therapeutics, Inc. | Polymorphic compounds and uses thereof |
| US10550085B2 (en) | 2015-08-21 | 2020-02-04 | Aldeyra Therapeutics, Inc. | Deuterated compounds and uses thereof |
| US11040039B2 (en) | 2017-10-10 | 2021-06-22 | Aldeyra Therapeutics, Inc. | Treatment of inflammatory disorders |
| US11129823B2 (en) | 2016-05-09 | 2021-09-28 | Aldeyra Therapeutics, Inc. | Combination treatment of ocular inflammatory disorders and diseases |
| US12006298B2 (en) | 2018-08-06 | 2024-06-11 | Aldeyra Therapeutics, Inc. | Polymorphic compounds and uses thereof |
| US12029735B2 (en) | 2019-05-02 | 2024-07-09 | Aldeyra Therapeutics, Inc. | Polymorphic compounds and uses thereof |
| US12064516B2 (en) | 2020-05-13 | 2024-08-20 | Aldeyra Therapeutics, Inc. | Pharmaceutical formulations and uses thereof |
| US12098132B2 (en) | 2019-05-02 | 2024-09-24 | Aldeyra Therapeutics, Inc. | Process for preparation of aldehyde scavenger and intermediates |
| US12128013B2 (en) | 2023-08-22 | 2024-10-29 | Aldeyra Therapeutics, Inc. | Toxic aldehyde related diseases and treatment |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| BR112014010042B1 (en) | 2011-10-26 | 2022-03-03 | Allergan, Inc | Amide derivatives of substituted n-urea amino acids as formyl peptide receptor-like 1 (fprl-1) receptor modulators |
| CN106905388B (en) * | 2017-02-16 | 2021-01-01 | 重庆西南制药二厂有限责任公司 | Method for synthesizing gastrodin |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4874775A (en) * | 1985-06-11 | 1989-10-17 | Eli Lilly And Company | Agriculturally useful sulfonamides |
| US6482834B2 (en) * | 1997-05-28 | 2002-11-19 | Aventis Pharmaceuticals Inc. | Quinoline and quinoxaline compounds which inhibit platelet-derived growth factor and/or p56lck tyrosine kinases |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE1802922A1 (en) * | 1967-10-18 | 1969-05-14 | Yamanouchi Pharma Co Ltd | New 5-aminomethyl-2,4-dihydroxy-6-methylpyrimidine derivatives |
| US4182764A (en) * | 1973-10-11 | 1980-01-08 | Merck & Co., Inc. | Tetrazole derivatives of [1-oxo-2-aryl or thienyl-2-substituted-5-indanyloxy(or thio)]alkanoic acids |
| US4177285A (en) * | 1973-10-11 | 1979-12-04 | Merck & Co., Inc. | [1-Oxo-2-thienyl-2-substituted-5-indanyloxy (or thio)]alkanoic acids and derivatives thereof |
| PL98342B1 (en) * | 1974-07-30 | 1978-04-29 | METHOD FOR THE PRODUCTION OF 1-KETO-2-ARYL- / OR THENYL / -2-SUBSTITUTED-INDANOXY- / OR THIO / -5-ALCOXYLIC ACID | |
| US6800651B2 (en) * | 2000-02-03 | 2004-10-05 | Eli Lilly And Company | Potentiators of glutamate receptors |
| CZ20031986A3 (en) * | 2001-01-22 | 2003-12-17 | Memory Pharmaceuticals Corporation | N-substituted anilines and diphenylamines inhibiting PDE4 and pharmaceutical composition in which they are comprised |
| CA2521124A1 (en) * | 2003-04-10 | 2004-10-21 | F. Hoffmann-La Roche Ag | Pyrimido compounds |
-
2005
- 2005-10-24 US US11/665,422 patent/US20070287716A1/en not_active Abandoned
- 2005-10-24 EP EP05812616A patent/EP1809608A4/en not_active Withdrawn
- 2005-10-24 CA CA002585210A patent/CA2585210A1/en not_active Abandoned
- 2005-10-24 JP JP2007539056A patent/JP2008518913A/en not_active Withdrawn
- 2005-10-24 AU AU2005302608A patent/AU2005302608A1/en not_active Abandoned
- 2005-10-24 CN CNA2005800370633A patent/CN101048384A/en active Pending
- 2005-10-24 WO PCT/US2005/038435 patent/WO2006049968A1/en active Application Filing
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4874775A (en) * | 1985-06-11 | 1989-10-17 | Eli Lilly And Company | Agriculturally useful sulfonamides |
| US6482834B2 (en) * | 1997-05-28 | 2002-11-19 | Aventis Pharmaceuticals Inc. | Quinoline and quinoxaline compounds which inhibit platelet-derived growth factor and/or p56lck tyrosine kinases |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP1809608A4 * |
Cited By (34)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7678794B2 (en) | 2004-11-22 | 2010-03-16 | Eli Lilly And Company | Potentiators of glutamate receptors |
| US7803938B2 (en) | 2004-11-22 | 2010-09-28 | Eli Lilly And Company | Potentiators of glutamate receptors |
| US7816523B2 (en) | 2004-11-22 | 2010-10-19 | Eli Lilly And Company | Potentiators of glutamate receptors |
| US7858646B2 (en) | 2004-11-22 | 2010-12-28 | Eli Lilly And Company | Potentiators of glutamate receptors |
| US7598423B2 (en) | 2004-11-22 | 2009-10-06 | Eli Lilly And Company | Potentiators of glutamate receptors |
| US7960417B2 (en) | 2005-02-24 | 2011-06-14 | Merck Sharp & Dohme Corp. | Benzazole potentiators of metabotropic glutamate receptors |
| US10913722B2 (en) | 2005-05-26 | 2021-02-09 | Aldeyra Therapeutics, Inc. | Compositions and methods of treating retinal disease |
| US9364471B2 (en) | 2005-05-26 | 2016-06-14 | Aldeyra Therapeutics, Inc. | Compositions and methods of treating retinal disease |
| US11724987B2 (en) | 2005-05-26 | 2023-08-15 | Aldeyra Therapeutics, Inc. | Compositions and methods of treating retinal disease |
| WO2010114726A1 (en) * | 2009-03-31 | 2010-10-07 | Merck Sharp & Dohme Corp. | Aminobenzotriazole derivatives |
| US12097188B2 (en) | 2009-12-11 | 2024-09-24 | Aldeyra Therapeutics, Inc. | Compositions and methods for the treatment of macular degeneration |
| US9814701B2 (en) | 2009-12-11 | 2017-11-14 | Aldeyra Therapeutics, Inc. | Compositions and methods for the treatment of macular degeneration |
| US9278960B2 (en) | 2011-11-03 | 2016-03-08 | Merck Sharp & Dohme Corp. | Quinoline carboxamide and quinoline carbonitrile derivatives as mGluR2-negative allosteric modulators, compositions, and their use |
| US9663506B2 (en) | 2011-11-03 | 2017-05-30 | Merck Sharp & Dohme Corp. | Quinoline carboxamide and quinoline carbonitrile derivatives as mGluR2-negative allosteric modulators, compositions, and their use |
| US9636337B2 (en) | 2011-11-03 | 2017-05-02 | Merck Sharp & Dohme Corp. | Quinoline carboxamide and quinoline carbonitrile derivatives as mGluR2-negative allosteric modulators, compositions, and their use |
| US9604997B2 (en) | 2012-12-20 | 2017-03-28 | Aldeyra Therapeutics, Inc. | Peri-carbinols |
| WO2014100425A1 (en) * | 2012-12-20 | 2014-06-26 | Aldexa Therapeutics, Inc. | Peri-carbinols |
| US10213395B2 (en) | 2013-01-23 | 2019-02-26 | Aldeyra Therapeutics, Inc. | Toxic aldehyde related diseases and treatment |
| US10588874B2 (en) | 2013-01-23 | 2020-03-17 | Aldeyra Therapeutics, Inc. | Toxic aldehyde related diseases and treatment |
| US11007157B2 (en) | 2013-01-23 | 2021-05-18 | Aldeyra Therapeutics, Inc. | Toxic aldehyde related diseases and treatment |
| US9687481B2 (en) | 2013-01-23 | 2017-06-27 | Aldeyra Therapeutics, Inc. | Toxic aldehyde related diseases and treatment |
| US10543181B2 (en) | 2013-01-23 | 2020-01-28 | Aldeyra Therapeutics, Inc. | Toxic aldehyde related diseases and treatment |
| US10111862B2 (en) | 2013-01-25 | 2018-10-30 | Aldeyra Therapeutics, Inc. | Traps in the treatment of macular degeneration |
| US10550085B2 (en) | 2015-08-21 | 2020-02-04 | Aldeyra Therapeutics, Inc. | Deuterated compounds and uses thereof |
| US11046650B2 (en) | 2015-08-21 | 2021-06-29 | Aldeyra Therapeutics, Inc. | Deuterated compounds and uses thereof |
| US11129823B2 (en) | 2016-05-09 | 2021-09-28 | Aldeyra Therapeutics, Inc. | Combination treatment of ocular inflammatory disorders and diseases |
| US10414732B2 (en) | 2017-03-16 | 2019-09-17 | Aldeyra Therapeutics, Inc. | Polymorphic compounds and uses thereof |
| US11583529B2 (en) | 2017-10-10 | 2023-02-21 | Aldeyra Therapeutics, Inc. | Treatment of inflammatory disorders |
| US11040039B2 (en) | 2017-10-10 | 2021-06-22 | Aldeyra Therapeutics, Inc. | Treatment of inflammatory disorders |
| US12006298B2 (en) | 2018-08-06 | 2024-06-11 | Aldeyra Therapeutics, Inc. | Polymorphic compounds and uses thereof |
| US12029735B2 (en) | 2019-05-02 | 2024-07-09 | Aldeyra Therapeutics, Inc. | Polymorphic compounds and uses thereof |
| US12098132B2 (en) | 2019-05-02 | 2024-09-24 | Aldeyra Therapeutics, Inc. | Process for preparation of aldehyde scavenger and intermediates |
| US12064516B2 (en) | 2020-05-13 | 2024-08-20 | Aldeyra Therapeutics, Inc. | Pharmaceutical formulations and uses thereof |
| US12128013B2 (en) | 2023-08-22 | 2024-10-29 | Aldeyra Therapeutics, Inc. | Toxic aldehyde related diseases and treatment |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1809608A4 (en) | 2009-11-04 |
| AU2005302608A1 (en) | 2006-05-11 |
| EP1809608A1 (en) | 2007-07-25 |
| CA2585210A1 (en) | 2006-05-11 |
| US20070287716A1 (en) | 2007-12-13 |
| CN101048384A (en) | 2007-10-03 |
| JP2008518913A (en) | 2008-06-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1809608A1 (en) | Pyrimidine and quinoline potentiators of metabotropic glutamate receptors | |
| EP1807073A2 (en) | Heterocyclic indanone potentiators of metabotropic glutamate receptors | |
| AU2003262805A1 (en) | Acetophenone potentiators of metabotropic glutamate receptors | |
| US7960417B2 (en) | Benzazole potentiators of metabotropic glutamate receptors | |
| US20080293684A1 (en) | Heterocyclic Acetophenone Potentiators of Metabotropic Glutamate Receptors | |
| US20080312286A1 (en) | Indanone Potentiators of Metabotropic Glutamate Receptors | |
| US20110065669A1 (en) | Oxazolobenzimidazole derivatives | |
| US7507836B2 (en) | Benzamide modulators of metabotropic glutamate receptors | |
| US20050222200A1 (en) | Amide derivatives as ion channel ligands and pharmaceutical compositions and methods of using the same | |
| EP2705025A1 (en) | Hydroxymethyl biaryl benzotriazole derivatives |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KM KP KR KZ LC LK LR LS LT LU LV LY MA MD MG MK MN MW MX MZ NA NG NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SM SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU LV MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1331/CHENP/2007 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005302608 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11665422 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007539056 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2585210 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005812616 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2005302608 Country of ref document: AU Date of ref document: 20051024 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005302608 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200580037063.3 Country of ref document: CN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005812616 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 11665422 Country of ref document: US |