WO2006046030A2 - Tetracyclic indole derivatives as antiviral agents - Google Patents
Tetracyclic indole derivatives as antiviral agents Download PDFInfo
- Publication number
- WO2006046030A2 WO2006046030A2 PCT/GB2005/004127 GB2005004127W WO2006046030A2 WO 2006046030 A2 WO2006046030 A2 WO 2006046030A2 GB 2005004127 W GB2005004127 W GB 2005004127W WO 2006046030 A2 WO2006046030 A2 WO 2006046030A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- cyclohexyl
- ring
- compound
- methyl
- Prior art date
Links
- 0 *C(CC[n]1c2c3)C(*)c(cccc4)c4-c1c(C1CCCCC1)c2ccc3C(O)=O Chemical compound *C(CC[n]1c2c3)C(*)c(cccc4)c4-c1c(C1CCCCC1)c2ccc3C(O)=O 0.000 description 3
- SORARJZLMNRBAQ-UHFFFAOYSA-N CNCCCN(C)C Chemical compound CNCCCN(C)C SORARJZLMNRBAQ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D245/00—Heterocyclic compounds containing rings of more than seven members having two nitrogen atoms as the only ring hetero atoms
- C07D245/04—Heterocyclic compounds containing rings of more than seven members having two nitrogen atoms as the only ring hetero atoms condensed with carbocyclic rings or ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/407—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with other heterocyclic ring systems, e.g. ketorolac, physostigmine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D267/00—Heterocyclic compounds containing rings of more than six members having one nitrogen atom and one oxygen atom as the only ring hetero atoms
- C07D267/22—Eight-membered rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/12—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains three hetero rings
- C07D491/14—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D495/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms
- C07D495/12—Heterocyclic compounds containing in the condensed system at least one hetero ring having sulfur atoms as the only ring hetero atoms in which the condensed system contains three hetero rings
- C07D495/14—Ortho-condensed systems
Definitions
- the present invention relates to tetracyclic indole compounds, to pharmaceutical compositions containing them, to their use in the prevention and treatment of hepatitis C infections and to methods of preparation of such compounds and compositions.
- HCV Hepatitis C
- A, Z, R 1 , R 2 , R 3 , R 4 and n are defined therein, as useful in compositions and methods for treating psychiatric and neurological disorders.
- this document does not disclose the use of tetracyclic indole derivatives in treating or preventing viral infections.
- A, X, Cy, G 1 , G 2 , G 3 , G 4 , G 5 , G 6 , R 1 , R 2 , R 3 , R 4 , R 5 , R 6 and a are defined therein, and their use as HCV polymerase inhibitors.
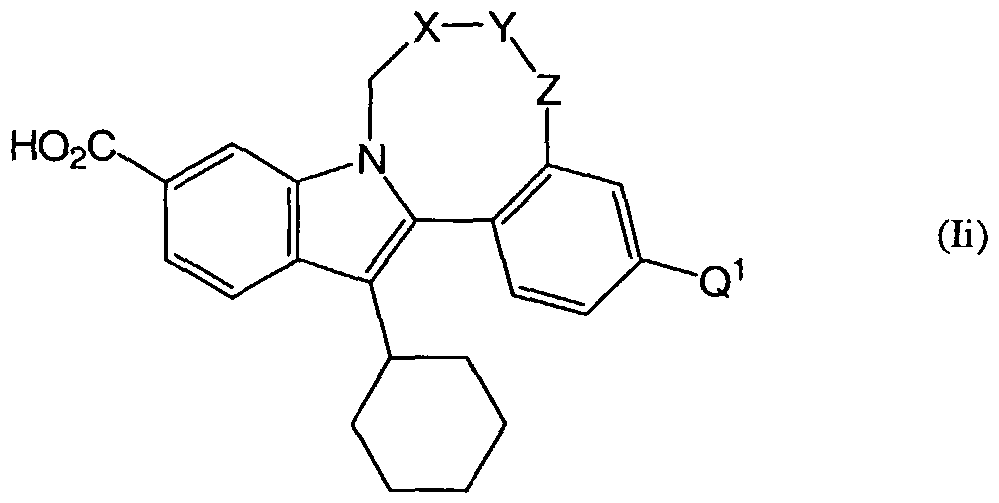
- the present invention provides the compound of the formula (I): wherein
- A is C 3 .gcycloalkyl, optionally substituted by halogen, hydroxy, C ⁇ alkyl or C ⁇ alkoxy;
- Ar is a moiety containing at least one aromatic ring and possesses 5, 6, 9 or 10 ring atoms, optionally containing 1, 2 or 3 heteroatoms independently selected from N, O and S, such as phenyl, pyridyl, pyridazinyl, pyrimidinyl, pyrazinyl, thienyl, furanyl, pyrazolyl and imidazolyl, which ring is optionally substituted by groups Q 1 and Q 2 ;
- Q 1 is halogen, hydroxy, C 1-6 alkyl, Ci -6 alkoxy, (CH 2 )o- 3 aryl, heteroaryl, CONR c R d , (CH 2 )o- 3 NR c R d , 0(CH 2 )o- 3 C 3 . 8 cycloalkyl ) O(CH 2 )i- 3 NR c R d , O(CH 2 ) 0 - 3 CONR c R d , 0(CH 2 )o- 3 C0 2 H, O(CH 2 ) 0 . 3 aryl, 0(CH 2 )o. 3 heteroaryl, OCHR e R f or 0(CH 2 )o- 3 S(0) 2 (CH 2 )Q. 3 NR c R d ;
- R c and R d are independently selected from hydrogen, Ci. 6 alkyl and C(O)Ci_ 6 alkyl; or R c and R d , together with the nitrogen atom to which they are attached, form a heteroaliphatic ring of 4 to 7 ring atoms, optionally containing 1 or 2 more heteroatoms independently selected from O and S and/or 1 or 2 groups independently selected from NH and NC ⁇ alkyl, where said ring is optionally substituted by halogen, hydroxy, C ⁇ alkyl or Ci_ 4 alkoxy;
- R e and R f are independently selected from hydrogen, Ci_ 4 alkyl and Q ⁇ alkoxy; or R e and R f are linked by a heteroatom selected from N, O and S to form a heteroaliphatic ring of 4 to 7 ring atoms, where said ring is optionally substituted by halogen, hydroxy, Ci_ 4 alkyl or and where said Ci. 4 alkyl, Ci. 4 alkoxy and aryl groups are optionally substituted by halogen or hydroxy;
- Q 2 is halogen, hydroxy, Ci. 4 alkyl or Q ⁇ alkoxy, where said Ci. 4 alkyl and Q. 4 alkoxy groups are optionally substituted by halogen or hydroxy; or Q 1 and Q 2 may be linked to form a ring of 4 to 7 atoms, where said ring optionally contains 1 or 2 heteroatoms independently selected from N, O and S, and is optionally substituted by halogen, hydroxy, Ci_ 4 alkyl or Ci. 4 alkoxy; one of R 1 and R 2 is CO 2 H, C(O)NHS(O) 2 NR 3 R", C(O)NHS(O) 2 Ci. 6 alkyl, C(0)NHS(0) 2 (CH 2 )o. 3 C0 2 R c or C(O)NHS(O) 2 (CH 2 ) 0 - 3 aryl, and the other of R 1 and R 2 is hydrogen;
- Y is -CR 14a R 15a - or NR 14a ;
- Z is O, -CHR 10 - or -CHR 10 CH 2 -;
- R 10 is hydrogen, hydroxy, C ⁇ alkyl, C 2 . 6 alkenyl, oxo, 0(CH 2 ) L3 NR 0 R 11 or N(CH 2 ) L3 NR 0 R";
- R 14 , R 14a , R 15 and R 15a are each independently selected from hydrogen, hydroxy, C,. 6 alkyl,
- R 16 and R 17 are independently selected from hydrogen, Ci. 6 alkyl, (CH 2 ) 0 . 4 NR 18 R 19 , (CH 2 ) 0 . 3 Het, (CH 2 )o_ 3 heteroaryl, (CH 2 ) 0 - 3 C(O)(CH 2 ) 0 - 3 NR 18 R 19 or (CH 2 ) 0 - 3 C 3 . 8 cycloalkyl, optionally substituted by Ci. 6 alkyl, (CH 2 ) 0 . 3 OH or (CH 2 ) 0 . 3 Ci.
- R 16 and R 17 together with the nitrogen atom to which they are attached, form a heteroaliphatic ring of 4 to 7 ring atoms, which ring may optionally contain 1 or 2 more heteroatoms selected from O and S and/or 1 or 2 groups independently selected from S(O), S(O) 2 , NH, NCi. 4 alkyl and N(CH 2 ) 0 . 3 Ci. 4 alkoxy, and which ring is optionally substituted by halogen, hydroxy, C 1-4 alkyl or Ci. 4 alkoxy;
- R 18 and R 19 are independently selected from hydrogen, Ci. 6 alkyl and heteroaryl; or R 18 and R 19 , together with the nitrogen atom to which they are attached, form a heteroaliphatic ring of 4 to 7 ring atoms, which ring may optionally contain 1 or 2 more heteroatoms selected from O and S and/or 1 or 2 groups selected from S(O), S(O) 2 , NH and NCi. 4 alkyl, and which ring is optionally substituted by halogen, hydroxy, Ci.
- Y is -CR 14a R 15a - and X is NR 14 .
- X is -CHR 14 -;
- Y is -CH 2 - or NR 14a ;
- Z is O or -CH 2 -;
- Q 1 , R 14 and R l4a are as defined in relation to formula (I); with the proviso that when Y is NR 14a , Z is not O.
- Y is -CH 2 - when Z is O.
- Y is NR 14a when Z is -CH 2 -.
- Y is NR 14a when X is -CH 2 -.
- Y is -CH 2 - when R 14 is other than hydrogen.
- R 14 is (CH 2 ) O4 OR 16 or (CH 2 ) 0 .iNR 16 R 17 where R 16 and R 17 are as defined in relation to formula (I).
- R 14 is OR 16 or NR 16 R 17 .
- R 16 is hydrogen, (CH 2 ) L3 (C LS aIkOXy) or (CH 2 ) L3 NR 18 R 19 where R 18 and R 19 are as defined in relation to formula (I). More preferably, R 16 is hydrogen, (CH 2 ) 2 . 3 (Ci. 4 alkoxy), (CH 2 ) 2 . 3 N(C,. 4 alkyl) 2 or (CH 2 ) 2 .
- R 18 and R 19 together with the nitrogen atom to which they are attached, form a 5- or 6- membered heteroaliphatic ring, which ring may optionally contain 1 or 2 more heteroatoms selected from O and S and/or 1 or 2 groups selected from S(O), S(O) 2 , NH and Examples of suitable R 16 groups include hydrogen,
- R 14 is NR 16 R 17
- R 16 is hydrogen, C M alkyl, C 3 . 8 cycloalkyl, (CH 2 )L 3 NR 18 R 19 , (CH 2 )o. 3 Ci. 4 alkoxy, (CH 2 ) 0 . 2 C(0)(CH 2 )o. 2 N(Ci. 4 alkyl) 2 or Het, optionally substituted by C M alkyl, where R 18 and R 19 are independently selected from hydrogen, Ci.
- R 18 and R 19 together with the nitrogen atom to which they are attached, form a 5- or 6-membered heteroaliphatic ring, which ring may optionally contain 1 or 2 more heteroatoms selected from O and S and/or 1 or 2 groups selected from S(O), S(O) 2 , NH and NC M alkyl.
- R 17 is hydrogen or C h alky!. More preferably, R 17 is hydrogen, methyl, ethyl or i-propyl.
- R 14 is NR 16 R 17
- R 16 and R 17 together with the nitrogen atom to which they are attached, form a 5- or 6-membered heteroaliphatic ring, which ring may optionally contain 1 or 2 more O atoms and/or 1 or 2 groups selected from NH, NCi. 4 alkyl and N(CH 2 )o- 3 Ci. 4 alkoxy.
- suitable NR 16 R 17 groups include:
- R 14a is hydrogen, Ci -6 alkyl, C ⁇ alkenyl, Het, (CH 2 )o. 3 NR 16 R 17 or C(O)(CH 2 V 3 NR 16 R 17 , where R 16 and R 17 are as defined in relation to formula (I).
- R 14a is hydrogen, C ⁇ alkyl, Cwalkenyl, Het, (CH 2 ) 2 NR 16 R 17 or C(O)(CH 2 ) L2 NR 16 R 17 , where R 16 and R 17 are independently Q ⁇ alkyl, or R 16 and R 17 , together with the nitrogen atom to which they are attached, form a 5- or 6-membered heteroaliphatic ring, which ring may optionally contain 1 or 2 more O atoms and/or 1 or 2 groups selected from NH and NCi. 4 alkyl.
- suitable R 14a groups include hydrogen, methyl, ethyl, propyl, butyl, pentyl, allyl,
- Q 1 is hydrogen, halogen, hydroxy, Ci_ 6 alkoxy,
- Q 1 is hydrogen, fluorine, chlorine, hydroxy, Ci_ 4 alkoxy, O(CH 2 ) 0 . 3 C(O)N(Ci. 4 alkyl) 2 , 0(CH 2 )o iaryl or O(CH 2 ) 0 .iheteroaryl. More preferably, Q 1 is hydrogen, fluorine, chlorine, hydroxy, Ci. 3 alkoxy, O(CH 2 ),. 2 C(O)N(C,.
- Q 1 is hydrogen, fluorine, chlorine, hydroxy, methoxy, ethoxy, i-propoxy, OCH 2 C(O)N(CH 3 ) 2 , benzyloxy, O-pyridinyl, OCH 2 pyridinyl, OCH 2 pyridazinyl, OCH 2 pyrimidinyl or O-pyrazinyl.
- A is cyclopentyl or cyclohexyl, optionally substituted by halogen, hydroxy, d. 4 alkyl or C M alkoxy;
- Q 1 is halogen, hydroxy, C ⁇ alkyl, C,. 6 alkoxy, aryl, heteroaryl, CONR c R d , (CH 2 )o. 3 NR c R d , O(CH 2 )i. 3 NR c R d , O(CH 2 ) 0 . 3 CONR c R d , O(CH 2 ) 0 . 3 aryl, O(CH 2 ) 0 .
- R c and R d are each independently selected from hydrogen, C M alkyl and C(O)Ci- 4 alkyl; or R c , R d and the nitrogen atom to which they are attached form a heteroaliphatic ring of 4 to 7 ring atoms, where said ring is optionally substituted by halogen, hydroxy, Ci_ 4 alkyl or Ci. 4 alkoxy;
- R e and R f are each independently selected from hydrogen and Ci. 4 alkoxy; or R e and R f are linked by a heteroatom selected from N, O and S to form a heteroaliphatic ring of 4 to 7 ring atoms, where said ring is optionally substituted by halogen, hydroxy, C M alkyl or Ci_ 4 alkoxy; and wherein said C].
- 4 alkyl, Ci_ 4 alkoxy and aryl groups are optionally substituted by halogen or hydroxy;
- Q 2 is halogen, hydroxy, C M alkyl or Q. 4 alkoxy, where said Ci_ 4 alkyl and Ci_ 4 alkoxy groups are optionally substituted by halogen or hydroxy; or Q 1 and Q 2 may be linked by a bond or a heteroatom selected from N, O and S to form a ring of
- Y is -CR 14a R I5a - or NR 14a ;
- Z is O, -CH 2 - or -CH 2 CH 2 -;
- R 14 , R 14a , R 1S and R I5a are each independently selected from hydrogen, hydroxy, Ci. 6 alkyl,
- R 16 and R 17 are independently selected from hydrogen, C ⁇ alkyl and (CH 2 V 4 NR 18 R 19 ; or R 16 , R 17 and the nitrogen atom to which they are attached form a heteroaliphatic ring of 4 to 7 ring atoms, which ring may optionally contain 1 or 2 more heteroatoms selected from O or S or a group S(O), S(O) 2 , NH or NQ. 4 alkyl, and which ring is optionally substituted by halogen, hydroxy, C M alkyl or Ci. 4 alkoxy;
- R 18 and R 19 are independently selected from hydrogen and d. 6 alkyl; or R 18 , R 19 and the nitrogen atom to which they are attached form a heteroaliphatic ring of 4 to 7 ring atoms, which ring may optionally contain 1 or 2 more heteroatoms selected from O or S or a group S(O), S(O) 2 , NH or NCi. 4 alkyl, and which ring is optionally substituted by halogen, hydroxy, Ci_ 4 alkyl or Q. 4 alkoxy; and pharmaceutically acceptable salts thereof; with the proviso that: when Z is O, then Q 1 does not contain a ring that is either a C 6 -i 4 aryl group, a C 3 .
- A is cyclohexyl, optionally substituted by halogen, hydroxy, Q ⁇ alkyl or Ci_ 4 alkoxy.
- A is unsubstituted or substituted by fluorine, chlorine, methyl or methoxy. More preferably, A is unsubstituted.
- Q 1 is halogen, hydroxy, Ci_ 4 alkyl or Ci. 4 alkoxy. More preferably, Q 1 is fluorine, chlorine, methyl or methoxy.
- Q 2 is absent.
- Another favoured group of compounds of the present invention is of formula (Iaa) and pharmaceutically acceptable salts thereof:
- Q 1 is halogen or absent.
- Q 1 is fluorine, chlorine or absent.
- R 14a is (CH 2 )o- 3 NR I6 R 17 or C(O)(CH 2 V 3 NR 16 R 17 where R 16 and R 17 are as defined in relation to formula (Ia).
- R 16 and R 17 are independently selected from hydrogen and Ci. 6 alkyl. More preferably, R 16 and R 17 are independently selected from hydrogen, methyl and ethyl.
- R 16 and R 17 are both methyl.
- suitable R 14 groups include:
- Q 1 is absent, fluorine or chlorine.
- Q 1 is absent.
- Q 2 is absent or fluorine.
- Q 2 is absent.
- R 14 is (CH 2 ) 0 . t OR 16 or (CH 2 ) 0 .iNR 16 R 17 , where R 16 and R 17 are as defined in relation to formula (I).
- R 14 is OR 16 or NR 16 R 17 .
- R 16 is hydrogen, (CH 2 )i. 3 (C I . 6 alkoxy) or (CH 2 )i. 3 NR 18 R 19 where R 18 and R 19 are as defined in relation to formula (I). More preferably, R 16 is hydrogen, (CH 2 ) 2 . 3 (Ci- 4 alkoxy), (CH 2 ) 2 . 3 N(Ci. 4 alkyl) 2 or (CH 2 ) 2 .
- R 18 and R 19 together with the nitrogen atom to which they are attached, form a 5- or 6- membered heteroaliphatic ring, which ring may optionally contain 1 or 2 more heteroatoms selected from O and S and/or 1 or 2 groups selected from S(O), S(O) 2 , NH and NCi. 4 alkyl.
- suitable R 16 groups include hydrogen,
- R 14 is NR 16 R 17 , preferably R 16 is hydrogen, C M alkyl, C 3 . 8 cycloalkyl, (CH 2 ),. 3 NR 18 R 19 ,
- R 18 and R 19 are independently selected from hydrogen, Ci_ 4 alkyl and heteroaryl, or R 18 and R 19 , together with the nitrogen atom to which they are attached, form a 5- or 6-membered heteroaliphatic ring, which ring may optionally contain 1 or 2 more heteroatoms selected from O and S and/or 1 or 2 groups selected from S(O), S(O) 2 , NH and NC,. 4 alkyl.
- R 14 is NR 16 R 17
- R 17 is hydrogen or C ⁇ alkyl. More preferably, R 17 is hydrogen, methyl, ethyl or i-propyl.
- R 14 is NR 16 R 17
- R 16 and R 17 together with the nitrogen atom to which they are attached, form a 5- or 6-membered heteroaliphatic ring, which ring may optionally contain 1 or 2 more O atoms and/or 1 or 2 groups selected from NH, NC M alkyl and N(CH 2 )o- 3 Ci- 4 alkoxy.
- suitable NR 16 R 17 groups include:
- R 15 is hydrogen
- R 10 is hydrogen, hydroxy, oxo, OCH 2 CH 2 NR c R d or NHCH 2 CH 2 NR c R d where R c and R d are as defined in relation to formula (I).
- suitable NR c R d groups are NH 2 , NH(CH 3 ), N(CH 3 ) 2 and pyrrolidinyl.
- R 14 is hydrogen, (CH 2 V 3 OR 16 or (CH 2 ) 0 . 3 NR 16 R 17 where R 16 and R 17 are as defined in relation to formula (I).
- R 14 is hydrogen, O(CH 2 )i. 3 NR 18 R 19 or NH(CH 2 ),. 3 NR 18 R 19 where R 18 and R 19 are as defined in relation to formula (I). More preferably, R 14 is hydrogen or
- alkyl or "alkoxy" as a group or part of a group means that the group is straight or branched.
- suitable alkyl groups include methyl, ethyl, n-propyl, i-propyl, n-butyl, s-butyl and t-butyl.
- suitable alkoxy groups include methoxy, ethoxy, n-propoxy, i-propoxy, n-butoxy, s-butoxy and t-butoxy.
- cycloalkyl groups referred to herein may represent, for example, cyclopropyl, cyclobutyl, cyclopentyl or cyclohexyl.
- a suitable cycloalkylalkyl group may be, for example, cyclopropylmethyl.
- alkenyl as a group or part of a group means that the group is straight or branched.
- suitable alkenyl groups include vinyl and allyl.
- halogen means fluorine, chlorine, bromine and iodine.
- aryl as a group or part of a group means a carbocyclic aromatic ring. Examples of suitable aryl groups include phenyl and naphthyl.
- heteroaryl as a group or part of a group means a 5- to 10-membered heteroaromatic ring system containing 1 to 4 heteroatoms selected from N, O and S.
- Such groups include pyrrolyl, furanyl, thienyl, pyridyl, pyrazolyl, imidazolyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, pyrazinyl, pyrimidinyl, pyridazinyl, triazolyl, oxadiazolyl, thiadiazolyl, triazinyl, tetrazolyl, indolyl, benzothienyl, benzimidazolyl and quinolinyl.
- Het as a group or part of a group means a heteroaliphatic ring of 4 to 7 atoms, which ring may contain 1, 2 or 3 heteroatoms selected from N, O and S or a group S(O), S(O) 2 , NH or NC,. 4 alkyl.
- substituents may be present.
- Optional substituents may be attached to the compounds or groups which they substitute in a variety of ways, either directly or through a connecting group of which the following are examples: amine, amide, ester, ether, thioether, sulfonamide, sulfamide, sulfoxide, urea, thiourea and urethane.
- an optional substituent may itself be substituted by another substituent, the latter being connected directly to the former or through a connecting group such as those exemplified above.
- Specific compounds within the scope of this invention include those named in the Examples and
- the salts of the compounds of formula (I) will be non-toxic pharmaceutically acceptable salts.
- Other salts may, however, be useful in the preparation of the compounds according to the invention or of their non-toxic pharmaceutically acceptable salts.
- Suitable pharmaceutically acceptable salts of the compounds of this invention include acid addition salts which may, for example, be formed by mixing a solution of the compound according to the invention with a solution of a pharmaceutically acceptable acid such as hydrochloric acid, fumaric acid, p-toluenesulfonic acid, maleic acid, succinic acid, acetic acid, citric acid, tartaric acid, carbonic acid, phosphoric acid or sulfuric acid.
- a pharmaceutically acceptable acid such as hydrochloric acid, fumaric acid, p-toluenesulfonic acid, maleic acid, succinic acid, acetic acid, citric acid, tartaric acid, carbonic acid, phosphoric acid or sulfuric acid.
- Salts of amine groups may also comprise quaternary ammonium salts in which the amino nitrogen atom carries a suitable organic group such as an alkyl, alkenyl, alkynyl or aralkyl moiety.
- suitable pharmaceutically acceptable salts thereof may include metal salts such as alkali metal salts, e.g. sodium or potassium salts; and alkaline earth metal salts, e.g. calcium or magnesium salts.
- the salts may be formed by conventional means, such as by reacting the free base form of the product with one or more equivalents of the appropriate acid in a solvent or medium in which the salt is insoluble, or in a solvent such as water which is removed in vacuo or by freeze drying or by exchanging the anions of an existing salt for another anion on a suitable ion exchange resin.
- prodrugs of the compounds of formula (I) above include within its scope prodrugs of the compounds of formula (I) above.
- prodrugs will be functional derivatives of the compounds of formula (I) which are readily convertible in vivo into the required compound of formula (I).
- Conventional procedures for the selection and preparation of suitable prodrug derivatives are described, for example, in "Design of Prodrugs", ed. H. Bundgaard, Elsevier, 1985.
- a prodrug may be a pharmacologically inactive derivative of a biologically active substance (the "parent drug” or "parent molecule") that requires transformation within the body in order to release the active drug, and that has improved delivery properties over the parent drug molecule.
- the transformation in vivo may be, for example, as the result of some metabolic process, such as chemical or enzymatic hydrolysis of a carboxylic, phosphoric or sulfate ester, or reduction or oxidation of a susceptible functionality.
- the present invention includes within its scope solvates of the compounds of formula (I) and salts thereof, for example, hydrates.
- the present invention also includes within its scope N-oxides of the compounds of formula (I).
- the present invention also includes within its scope any enantiomers, diastereomers, geometric isomers and tautomers of the compounds of formula (I). It is to be understood that all such isomers and mixtures thereof are encompassed within the scope of the invention.
- the present invention further provides a compound of formula (I) or a pharmaceutically acceptable salt thereof for use in therapy.
- the invention provides the use of a compound of formula (I) as defined above, or a pharmaceutically acceptable salt thereof, for the manufacture of a medicament for treatment or prevention of infection by hepatitis C virus in a human or animal.
- a further aspect of the invention provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of formula (I) as defined above, or a pharmaceutically acceptable salt thereof, in association with a pharmaceutically acceptable carrier.
- the composition may be in any suitable form, depending on the intended method of administration. It may for example be in the form of a tablet, capsule or liquid for oral administration, or of a solution or suspension for administration parenterally.
- compositions optionally also include one or more other agents for the treatment of viral infections such as an antiviral agent, or an immunomodulatory agent such as ⁇ -, ⁇ - or ⁇ - interferon.
- agents for the treatment of viral infections such as an antiviral agent, or an immunomodulatory agent such as ⁇ -, ⁇ - or ⁇ - interferon.
- the invention provides a method of inhibiting hepatitis C virus polymerase and/or of treating or preventing an illness due to hepatitis C virus, the method involving administering to a human or animal (preferably mammalian) subject suffering from the condition a therapeutically or prophylactically effective amount of the pharmaceutical composition described above or of a compound of formula (I) as defined above, or a pharmaceutically acceptable salt thereof.
- Effective amount means an amount sufficient to cause a benefit to the subject or at least to cause a change in the subject's condition.
- the dosage rate at which the compound is administered will depend on a variety of factors including the activity of the specific compound employed, the metabolic stability and length of action of that compound, the age of the patient, body weight, general health, sex, diet, mode and time of administration, rate of excretion, drug combination, the severity of the particular condition and the host undergoing therapy. Suitable dosage levels may be of the order of 0.02 to 5 or 10 g per day, with oral dosages two to five times higher. For instance, administration of from 10 to 50 mg of the compound per kg of body weight from one to three times per day may be in order. Appropriate values are selectable by routine testing. The compound may be administered alone or in combination with other treatments, either simultaneously or sequentially.
- it may be administered in combination with effective amounts of antiviral agents, immunomodulators, anti-infectives or vaccines known to those of ordinary skill in the art. It may be administered by any suitable route, including orally, intravenously, cutaneously and subcutaneously. It may be administered directly to a suitable site or in a manner in which it targets a particular site, such as a certain type of cell. Suitable targeting methods are already known.
- An additional aspect of the invention provides a method of preparation of a pharmaceutical composition, involving admixing at least one compound of formula (I) as defined above, or a pharmaceutically acceptable salt thereof, with one or more pharmaceutically acceptable adjuvants, diluents or carriers and/or with one or more other therapeutically or prophylactically active agents.
- the present invention also provides a process for the preparation of compounds of formula (I).
- compounds of formula (I) where Y is NR 14 may be prepared by internal ring closure of a compound of formula (II):
- R 1 , R 2 , A, Ar, W, X and Z are as defined in relation to formula (I).
- the reaction is conveniently performed in the presence of a coupling reagent, such as HATU or TBTU, and a base, such as diisopropylethylamine or triethylamine, in a solvent.
- a coupling reagent such as HATU or TBTU
- a base such as diisopropylethylamine or triethylamine
- compounds of formula (I) may be prepared by internal ring closure of a compound of formula (IH):
- R 1 , R 2 , A, Ar, W, X, Y and Z are as defined in relation to formula (I) and E is hydrogen or bromine.
- the reaction is conveniently performed in the presence of a Pd(O) catalyst, such as PdCl 2 (dppf), a dioxoborolane, such as 4,4,4',4',5,5,5',5'-octamethyl-2,2'-bi-l,3,2-dioxaborolane, and a base, such as potassium acetate, in a suitable solvent, such as DMF, under a nitrogen atmosphere.
- Pd(O) catalyst such as PdCl 2 (dppf)
- a dioxoborolane such as 4,4,4',4',5,5,5',5'-octamethyl-2,2'-bi-l,3,2-dioxaborolane
- a base such as potassium acetate
- compounds of formula (I) may be prepared by internal ring closure of a compound of formula (IV):
- R 1 , R 2 , A, Ar, Y and Z are as defined in relation to formula (I) and X' is X as defined in relation to formula (I) or is converted to X during or after the cyclisation reaction, and W" is W as defined in relation to formula (I) or converted to W during or after the cyclisation reaction.
- W and X' may be suitable activated precursors of groups W and X respectively which can be converted into W and X during the ring closure or after it using methods described in the accompanying Schemes and Examples or known to the person skilled in the art.
- W may be CH 2 -halogen or W and X' together may be an epoxide or aziridine group.
- W' is CH 2 -halogen, such as CH 2 -Br
- the reaction is conveniently performed in the presence of a base, such as sodium hydroxide, in a suitable solvent, such as DMF.
- a base such as sodium hydroxide
- a suitable solvent such as DMF.
- the compound of formula (I) where R 1 is CO 2 CH 3 may be converted into the compound of formula (I) where R 1 is CO 2 H by conversion of the ester to the carboxylic acid, for example, by treatment with BBr 3 in a suitable solvent, such as dichloromethane, or with NaOH in a suitable solvent, such as dioxane, THF and/or methanol.
- a suitable solvent such as dichloromethane
- NaOH in a suitable solvent, such as dioxane, THF and/or methanol.
- a borane reagent such as BH 3 »Me 2 S
- 2-bromoindole intermediate (prepared as described in published International patent application WO2004/087714) was functionalized on the indole nitrogen to introduce precursor functionality WVX' to either or both of the elements -CHz-IX of the tether.
- Pd-mediated cross-coupling methodology eg, Suzuki, Stille etc
- ZVY' the C2 aromatic bearing pre-cursor functionality
- Functional group manipulation followed by ring closure afforded the tetracyclic system. Ester deprotection then yielded the target indole carboxylic acids, with the C2 aromatic tethered to the indole nitrogen.
- the C2 aromatic was introduced at the outset via Pd-mediated cross-coupling methodology (Suzuki, Stille etc).
- the tether was then built up, with cyclisation onto the indole nitrogen finally closing the ring. Ester deprotection then yielded the target indole carboxylic acids, with the C2 aromatic tethered to the indole nitrogen.
- C2-tethered indole carboxylic acids arising from Methods A-D were further derivatised through manipulation of the carboxylate functionality to give compounds bearing a carboxylate replacement or carboxamide.
- the protecting groups may be removed at a convenient subsequent stage using methods known from the art.
- the compounds of the invention were tested for inhibitory activity against the HCV RNA dependent RNA polymerase (NS5B) in an enzyme inhibition assay (example i)) and in a cell based sub-genomic replication assay (example ii)).
- the compounds have IC50's below 5 ⁇ M in the enzyme assay and several examples have EC50' s below 2 ⁇ M in the cell based assay.
- Compound names in the examples were generated using software from ACDLabs (version 6.0).
- WO 96/37619 describes the production of recombinant HCV RdRp from insect cells infected with recombinant baculovirus encoding the enzyme.
- the purified enzyme was shown to possess in vitro RNA polymerase activity using RNA as template.
- the reference describes a polymerisation assay using poly(A) and oligo(U) as a primer or an heteropolymeric template. Incorporation of tritiated UTP or NTPs is quantified by measuring acid-insoluble radioactivity. This assay has been employed to screen the various compounds described above as inhibitors of HCV RdRp.
- Cells were seeded into 96 well plates at a density of 10 4 cells per well in a final volume of 0.1 ml of DMEM/10% FCS. Two hours after plating, 50 ⁇ l of DMEM/10% FCS containing a 3x concentration of inhibitor were added, cells were incubated for 96 hours and then fixed for 10' with ice-cold isopropanol. Each condition was tested in duplicate and average absorbance values were used for calculations. The cells were washed twice with PBS, blocked with 5% non-fat dry milk in PBS + 0.1% Triton XlOO + 0.02% SDS (PBSTS) and then incubated o/n at 4° C with the 10E5/24 mab diluted in Milk/PBSTS.
- PBSTS Triton XlOO + 0.02% SDS
- Ai absorbance value of HBIlO cells supplemented with the indicated inhibitor concentration.
- a 0 absorbance value of HBIlO cells incubated without inhibitor.
- Reagents were usually obtained directly from commercial suppliers (and used as supplied) but a limited number of compounds from in-house corporate collections were utilised. In the latter case the reagents are readily accessible using routine synthetic steps that are either reported in the scientific literature or are known to those skilled in the art.
- 1 H NMR spectra were recorded on Bruker AM series spectrometers operating at (reported) frequencies between 300 and 600 MHz. Chemical shifts ( ⁇ ) for signals corresponding to non- exchangeable protons (and exchangeable protons where visible) are recorded in parts per million (ppm) relative to tetramethylsilane and are measured using the residual solvent peak as reference.
- Zhan catalyst I [l,3-bis(2,4,6-trimethylphenyl)-4,5-dihydro-imidazol-2-ylidene]-[4-chloro-l- isopropxy-benzylidine]ruthenium-dichloride: commercially available from ZannanPharma Ltd. (www.zannanpharma.com); methyl (aminosulfonyl)acetate was prepared in analogous fashion to related esters of aminosulfonyl acetic acid: eg. Tetrahedron Lett. 1989, 30 (22), 2869; Bull. Soc. Chim. France 1975, 3, 807.
- Example 1 14-cyclohexyl-6-[2-(dimethylamino)ethyl]-7-oxo-5,6,7,8 tetrahydroindolo[2,l- a][2,5]benzodiazocine-ll-carboxylic acid
- Step 1 methyl 2-bromo-l-(2-tert-butoxy-2-oxoethyl) ⁇ 3-cvclohe ⁇ yl-lH-indole-6-carboxylate NaH (1.4 eq, 60 % dispersion in mineral oil) was added to a solution of methyl 2-bromo-3-cyclohexyl- lH-indole-6-carboxylate (prepared as described in published International Patent application WO 2004/065367, from commercially available methyl indole-6-carboxylate) in DMF (0.2 M) and the solution stirred at RT for Ih.
- Step 2 methyl l-(2-tert-butoxy-2-oxoethyl)-3-cyclohexyl-2-(2-formylphenyl)-lH-indole-6-carboxylate
- methyl 2-bromo-l-(2-te/?-butoxy-2-oxoethyl)-3-cyclohexyl-lH-indole-6-carboxylate in 1,4-dioxane (0.15 M) was added Na 2 CO 3 (6 eq, 2 M solution), (2-formylphenyl)boronic acid (1.5 eq) and bis(triphenylphosphine)palladium(II) dichl ⁇ ride (0.2 eq) and the mixture heated at reflux for 45 min.
- Step 3 methyl l-(2-tert-butoxy-2-oxoethyl)-3-cyclohexyl-2-f2-( ⁇ [2- (dimethylamino ⁇ ethyl 1 amino lmethyl )phenyl]-lH-indole-6-carboxylate
- N,N-dimethylethane-l,2-diamine (10 eq) was added and the pH adjusted to pH 6 with acetic acid.
- Step 4 [3-cyclohexyl-2-[2-(lf2-(dimethylamino)ethyllamino)methyl)vhenyll-6-(methoxycarbonyl)-lH- indol-1-yll acetic acid
- methyl l-(2-terf-butoxy-2-oxoethyl)-3-cyclohexyl-2-[2-( ⁇ [2-(dimethylamino)ethyl] amino ⁇ methyl)phenyl]-lH-indole-6-carboxylate DCM/H 2 0 (2:1; 0.15 M
- Step 5 methyl 14-cyclohexyl-6-(2-(dimethylamino)ethyl]-7-oxo-5.6.7.8-tetrahvdroindolo[2.1 - a! [2.51benzodiazocine- 11 -carboxylate
- Step 6 14-cyclohexyl-6-I2-(dimethylamino)ethyll-7-oxo-5.6.7.8-tetrahydroindolof2,l- a] [2,5 lbenzodiazocine-11 -carboxylic acid
- Step 1 methyl l-(2-tert-butoxy-2-oxoethyl)-3-cyclohexyl-2-(2- ⁇ [(2-morpholin-4- ylethyl)aminolmethyl)phenyl)-lH-indole-6-carboxylate
- Step 3 methyl 14-cvclohexyl-6-(2-morpholin-4-ylethyl)-7-oxo-5.6,7,8-tetrahvdroindolo[2.1- a 1 [2.5]benzodiazocine-l 1 -carboxylate
- the title compound was prepared using the same procedure described for methyl 14-cyclohexyl-6-[2- (dimethylamino)ethyl]-7-oxo-5,6,7,8-tetrahydroindolo[2,l-a][2,5]benzodiazocine-ll-carboxylate in Example 1, Step 5.
- the crude was used in the next step without further purification; MS (ES + ) m/z 516 (M+H) +
- Step 4 methyl 14-cyclohexyl-6-(2-morpholin-4-ylethyl)-5,6.7.8-tetrahydroindolof2.1- a][2,5 lbenz.odiazocine-11 -carboxylate
- Step 5 14-cyclohexyl-6-(2-morpholin-4-ylethyl)-5,6.7,8-tetrahydroindolo[2.1-alf2.51benzodiazocine-ll- carboxylic acid
- the crude methyl 14-cyclohexyl-6-(2-mo ⁇ holin-4-ylethyl)-5,6,7,8-tetrahydroindolo[2,l- a][2,5]benzodiazocine-l l-carboxylate was dissolved in THF/MeOH (1:1) and to that solution an excess of NaOH (IN) was added. The solution was stirred at RT overnight. The solvent was evaporated in vacuo.
- Step 1 methyl 14-cyclohex ⁇ l-6-[2-(dimethylamino)ethyll-3-methoxy-5.6,7,8-tetrahydroindolo[2.1- a] [2.51 benzodiazocine- 11 -carboxylate
- Step 2 14-cyclohexyl-6-[2-(dimethylamino )ethylJ-3-methoxy-5.6.7, 8-tetrahydroindolo[2.1 - a] [2.5 ] benzodiazocine- 11 -carboxylic acid
- the crude methyl 14-cyclohexyl-6-[2-(dimethylamino)ethyl]-3-methoxy-5,6,7,8-tetrahydroindolo[2,l- a][2,5]benzodiazocine-l 1-carboxylate was dissolved in dioxane (0.06 M) and to that solution 10 eq of an aqueous solution of NaOH (2N) were added.
- Step 1 methyl 2-bromo-3-cyclohexyl-l -( 1.3-dioxolan-2-ylmethyl)-l H-indole-6-carboxylate NaH (1.5 eq, 60 % dispersion in mineral oil) was added to a solution of methyl 2-bromo-3-cyclohexyl- lH-indole-6-carboxylate (prepared as described in published International Patent application WO 2004/065367, from commercially available methyl indole-6-carboxylate) in DMF (0.1 M) and once effervescence had subsided the solution was stirred at RT for a further 30 min.
- DMF 0.1 M
- Step 2 methyl 3-c ⁇ clohexyl-l -( 1.3-dioxolan-2-ylmethyl)-2-(2-formyl-4-methoxyphenyl)-l H-indole-6- carboxylate
- Step 3 methyl 3-cvclohexyl-l -( 1.3-dioxolan-2-ylmethyl)-2-(4-methoxy-2-[(methylamino)methyl]phenyll- lH-indole-6-carboxylate
- Step 4 methyl 3-cvclohexyl-2-f4-methoxy-2-f(methylamino)methyllphenyl ⁇ -l-(2-oxoethyl)-lH-indole-6- carboxylate
- Step 5 14-cyclohex ⁇ l-3-methoxy-6-methyl-5.6.7 '.8-tetrahydwindolof2.1 -a] f2,51benzodiazocine-l 1 - carboxylic acid
- Acetic acid was added dropwise to a stirred solution of methyl 3-cyclohexyl-2- ⁇ 4-methoxy-2- [(methylamino)methyl]phenyl ⁇ -l-(2-oxoethyl)-lH-indole-6-carboxylate in MeOH (0.005 M) at RT, to adjust the pH to pH 6.
- the mixture was stirred for 10 min prior to introducing 3.2 eq Of NaCNBH 3 .
- RP- HPLC analysis of the reaction mixture after 1 h confirmed the complete conversion of the aminoaldehyde to the desired cyclic amine.
- the reaction was diluted with an equal volume of THF and 100 eq of NaOH (2 M aqueous solution) introduced.
- Example 5 methyl ( ⁇ [(14-cyclohexyl-3-methoxy-6-methyI-5,6,7,8-tetrahydroindolo[2,l- al ⁇ Slbenzodiazocin-ll-ytycarbonyllaminolsulfonyOacetate
- the resultant aqueous slurry was diluted with MeCN and water and freeze dried to leave a white powder.
- the crude was then purified by automated RP-MS-HPLC (stationary phase: column Waters XTERRA prep. C18, 5 um, 19 x 100 mm. Mobile phase: MeCN/H 2 0 buffered with 0.1 % TFA). Fractions containing the pure compound were combined and freeze dried to afford the title compound as a white powder (72 %).
- Example 7 14-cyclohexyI-N-[(dimethylamino)suIfonyl]-3-methoxy-6-methyl-5,6,7,8- tetrahydroindolo[2,l- ⁇ ][2,5]benzodiazocine-ll-carboxamide 1.5 eq of DMAP was added to the trifluoroacetate salt of M-cyclohexyl-S-methoxy- ⁇ -methyl-S. ⁇ J. ⁇ - tetrahydroindoloP.l-aJP.SJbenzodiazocine-l l-carboxylic acid (from Example 4) in dry DCM (0.1 M).
- Step 1 l-bromo-2-(bromomethyl)-4-chlorobenzene
- l-bromo-2-(bromomethyl)-4-chlorobenzene was prepared according to literature precedent (J. Am. Chem. Soc. 2002, 124 (7), 1354): a suspension of l-bromo-2-methyl-4-chlorobenzene (1 eq), NBS (1 eq) and benzoyl peroxide (0.004 eq) in CCl 4 (0.7 M) was heated at reflux for 4 h. The reaction was then filtered whilst hot and the volatiles reduced in vacuo. PE was added, and the resultant precipitate filtered off and dried in vacuo to afford the title compound (45 %).
- 2-bromo-5-chlorobenzaldehyde was prepared according to literature precedent (/. Am. Chem. Soc. 2002, 124 (7), 1354): a mixture of activated powdered 4A molecular sieves (800 mg/mmol substrate), N- methylmorpholine-N-oxide (2 eq) and 2-bromomethyl-5-chlorobenzaldehyde (1 eq) in MeCN (0.16 M) was stirred at 0 0 C for 2 h. The reaction was then filtered through a pad of celite and concentrated in vacuo to afford the title compound (92 %)
- Step 4 tert-butyl S-cvclohexyl-lH-indole- ⁇ -carboxylate
- Step 6 tert-butyl 2-bromo-3-cyclohexyl-l-(2-methoxy-2-oxoethyl)-lH-indole-6-carboxylate
- Step 7 f2-bromo-6-(tert-butoxycarbonyl)-3-cvclohexyl-lH-indol-l-yllacetic acid
- LiOH monohydrate (4 eq) in H 2 O (0.05 M)
- ferf-butyl 2-bromo-3- cyclohexyl-l-(2-methoxy-2-oxoethyl)-lH-indole-6-carboxylate (1 eq) in a mixture THF : CH 3 OH (1 : 1) (from Step 6, 0.05 M).
- the reaction was heated at 50 0 C for 2 h, before being allowed to cool to RT and reducing the volatiles in vacuo.
- Step 8 tert-butyl-2-bromo-l-(2- ⁇ (2-bromo-5-chlorobenzyl)[2-(dimethylamino)ethyllamino ⁇ -2-oxoethyl)-
- Step 10 3-chloro-14-cyclohexyl-6-[2-(dimethylamino)ethyll-7-oxo-5,6.7.8-tetrahydroindolo[2,l- a ][2.5]benzodiazocine-l 1 -carboxylic acid
- Step 1 methyl 3-cyclohexyl-2-(2-hydroxyphenyl)-lH-indole-6-carboxylate
- Step 2 methyl 3-cvclohexyl-2-(2-[(2S)-oxiran-2-ylmetho ⁇ ylphenvU-lH-indole-6-carboxylate
- cesium fluoride 3 eq
- (5)-glycidyl 3-nitrobenzenesulfonate 1.1 eq
- Step 3 methyl (7S)-14-cyclohexyl-7-hydroxy-7 ⁇ 8-dihydro-6H-indoloj 1 ,2-e] 71.51benzoxazocine-l 1- carboxylate
- Step 4 methyl 14-cyclohexyl-7-oxo-7.8-dihydro-6H-indolofl.2-e1fl.5]benzoxazocine-ll-carboxylate
- Step 5 methyl 14-cyclohexyl-7-ff2-(dimethylamino)ethyllaminof-7,8-dihydro-6H-indolofl.2- e ][ 1.5]benzoxazocine-l 1 -carboxylate
- Step 6 N'-(l 1 -carboxy-14-cyclohexyl-l '.8-dihydro-6H-indolol 1,2-eJI r l,5]benzoxazocin-'/ 7 -yl)-N '.N- dimethylethane-1.2-diaminium bis(trifluoroacetate)
- THWMeOH 0.05 M, 1 : 1, Wv
- reaction mixture was brought to pH 2 by the dropwise addition of HCl (1 N), then diluted with MeCN and purified by RP-HPLC (stationary phase: column Waters XTERRA prep. C18, 5 um, 19 x 100 mm. Mobile phase: MeCN/H 2 O buffered with 0.1 % TFA). Fractions containing the pure compound were combined and freeze dried to afford the title compound as a white powder (60 % over two steps).
- Example 11 iV-(ll-carboxy-14-cyclohexyl-7,8-dihydro-6H-indolo[l,2-e][l,5]benzoxazocin-7-yl)- yV ⁇ V' ⁇ V'-trimethylethane-l,2-diaminium bis(trifluoroacetate)
- Step 1 methyl 14-cyclohexyl-7-[[2-(dimethylamino)ethyll(methyl)aminol-7.8-dihydro-6H-indolo[1.2- e 1 ( 1 ,51benzoxazocine-l 1 -carboxylate
- Step 2 N-(ll-carboxy-14-cyclohexyl-7,8-dihydro-6H-indolofl,2-e]fl.51benzoxazocin-7-yl)-N.N'.N'- trimethylethane-1.2-diaminium bis(trifluoroacetate)
- Example 12 iV'-[(7/ ⁇ )-ll-carboxy-14-cyclohexyl-7,8-dihydro-6H-indolo[l,2-e][l,5]benzoxazocin-7- yl]- ⁇ '-trimethylethane-l,2-diaminium bis(trifluoroacetate)
- Step 2 methyl ⁇ 7R )- 7 -azido-H-cyclohexyl- 7.8-dihydro-6H-indolol 1.2-el f 1.51benzoxazocine-l 1 - carboxylate
- azidotrimethylsilane 3.5 eq.
- tetrabutylammonium triphenyldifluorosilicate 3.5 eq.
- Step 3 methyl (7R)-7-amino-14-cyclohexyl-7 ,8-dihydro-6H-indolof 1 ,2-elf 1 ,51benzoxaz.ocine-l 1 - carboxylate
- MeOH 0.1 M
- Step 5 methyl (7R)-13-cyclohexyl-7-[f2-(dimethylamino)ethyll(methyl)aminol-6,7,8.8a- tetrahydrobenzofblindenof2.1-d]oxocine-10-carboxylate
- Step 6 N' -f (7 R)- 11 -carboxy- 14-cyclohexyl- 7.8-dihydro-6H-indolol 1 ,2 -elf 1.5 lbenzoxaz.ocin-7-yll-
- Example 13 N'-[(7S)-ll-carboxy-14-cyclohexyl-7,8-dihydro-6H-indolo[l,2-e][l,5]benzoxazocin-7- yl]-N,N,N'-trimethylethane-l,2-diaminium bis(trifluoroacetate)
- Step 1 methyl 14-cyclohexyl-7-(4-methylpiperazin-l-yl)-7,8-dihydro-6H-indolo[l,2- elfl.5 ]benzoxazocine-l 1 -carboxylate
- Step 2 1-(11 -carboxy-14-cyclohexyl- 7, 8-dihydro-6H-indolol 1.2 -elf 1,5 Ibenzoxazocin- 7-yl)-4- methylpiperazinediium bisftrifluoroacetate )
- Example 15 (+)l-(ll-carboxy-14-cyclohexyl-7,8-dihydro-6H-indolo[l,2-e][l,5]benzoxazocin-7-yl)- 4-methylpiperazinediium bis(trifluoroacetate) and (-)l-(ll-carboxy-14-cycIohexyl-7,8-dihydro-6H-indolo[l,2-e][l,5]benzoxazocin-7-yl)-4- methylpiperazinediium bis(trifluoroacetate)
- the title compounds were obtained by resolution of the racemate (synthesized as described in example 14) with chiral HPLC (stationary phase: column, chiralpak AD, amilose carbamate, 10 ⁇ m, 20 x 250 mm. Mobile phase: n-hexane / 97%EtOH, 3%MeOH buffered with 0.2 % TFA).
- Step 1 methyl 14-cyclohexyl-7-[(2-pyrrolidin-l-ylethyl)aminol-7,8-dihydro-6H-indolo(1.2- ejfl.5 Ibenzoxazocine- 11 -carboxylate
- Step 2 methyl 14-cyclohexyl-7-fmethyl(2-pyrrolidin-l-ylethyl)amino1-7.8-dihydro-6H-indolofl.2- elf 1.5 Ibenzoxazocine- 11 -carboxylate
- MeOH MeOH
- reaction mixture was concentrated in vacuo and sat. aq. NaHCO 3 and CH 2 Cl 2 were added after 1.5 h.
- Step 3 ( ⁇ )-l-f2-f(ll-carboxy-14-cyclohexyl-7.8-dihydro-6H-indolofl,2-e][l,51benzoxazocin-7- yl )( methyl )ammonio 1 ethyl ipyrrolidinium bisftrifluoroacetate )
- Example 17 (+)- and (-)-l- ⁇ 2-[(ll-carboxy-14-cyclohexyl-7,8-dihydro-6H-indolo[l,2- e][l,5]benzoxazocin-7-yl)(methyl)ammonio]ethyl ⁇ pyrrolidinium bis(trifluoroacetate)
- Example 18 14-cyclohexyl-6-(N,N-dimethylglycyI)-3-fluoro-5,6,7,8-tetrahydroindolo[2,l- a][2,5]benzodiazocinc-ll-carboxylic acid
- Step 1 methyl l-fl-ffftert-butoxycarbonyttaminolmethyli ⁇ -fluorophenvtt-S-cvclohexyl-lH-indole- ⁇ - carboxylate
- Step 2 methyl 2-(2-tert-butoxycarbonyl)aminolmethyl)-4-fluorophenyl ) -3-cyclohexyl-l-(2-metho ⁇ y-2- oxoethyl)-lH-indole-6-carboxylate
- Methyl bromoacetate (4 eq) was added to a mixture of methyl 2-(2- ⁇ [(tert- butoxycarbonyl)amino]methyl ⁇ -4-fluorophenyl)-3-cyclohexyl-lH-indole-6-carboxylate (1 eq., from Step 1) and K 2 CO 3 (6 eq) in dry DMSO (0.2 M). The mixture was stirred at 60 0 C for 48 h. At this time the reaction was allowed to cool to RT, diluted with EtOAc. The organic phase was washed with H 2 O
- Step 3 methyl 2-[2-(aminomethyl)-4-fluorophenyll-3 ⁇ cvclohexyl-l-(2-methoxy-2-oxoethyl)-lH-indole-6- carboxylate
- Step 4 methyl M-cyclohexyl-S-fluoro-J-oxoS.6.7.8-tetrahvdroindolo[2.1 -a If 2, 5 lbenzodiaz.ocine-11 - carboxylate
- Step 5 methyl 14-cyclohexyl-3-fluoro-5,6,7.8-tetrahvdroindolof2.1-a][2.51benzodiazocine-ll- carboxylate
- Step 6 methyl 14-cyclohexyl-6-(N.N-dimethylelycyl)-3-fluoro-5,6,7.8-tetrahydroindolo[2.1- a If 2, 5 lbenz.odiaz.ocine-11 -carboxylate
- Step 7 14-cvclohexyl-6-(N.N-dimethylglvcyl )-3-fluoro-5, 6, 7.8-tetrahvdroindolo[2, 1 - a] [2,51benz.odiaz.ocine-l 1-carboxylic acid
- Example 19 14-cyclohexyl-6-[2-(dimethylamino)ethyI]-3-fluoro-5,6,7,8-tetrahydroindolo[2,l- ⁇ ][2,5]benzodiazocine-ll-carboxylic acid
- Step 1 methyl 14-cyclohex ⁇ l-6-[2-(dimethylamino)ethyll-3-fluoro-5.6.7.8-tetrahydroindolof2.1- a ][2.5]benzodiazocine-l 1 -carboxylate
- Step 2 14-cyclohexyl-6-[2-(dimethylamino)ethyl]-3-fluoro-5.6.7.8-tetrahydroindolo[2.1- all 2, 5 Jbenzodiazocine- 11 -carboxylic acid
- Example 20 14-cyclohexyl-6-[2-(dimethylamino)ethyI]-5,6,7,8-tetrahydroindolo[2,l- a][2,5]benzodiazocine-ll-carboxylic acid
- Step 1 Methyl 2-bromo-3-cyclohexyl-l-(2.2-dimethoxyethyl)-lH-indole-6-carboxylate
- Step 2 Methyl 3-cyclohexyl-l-(2,2-dimethoxyethyl)-2-(2-formylphenyl)-lH-indole-6-carboxylate
- Step 4 Methyl 14-cyclohexyl-6-[2-(dimethylamino)ethyl1-5.6,7,8-tetrahydroindolof2.1- aU2.51benzodiazocine-ll-carboxylate To a stirred solution of methyl 3-cyclohexyl- 1 -(2,2-dimethoxyethyl)-2-[2-( ⁇ [2-
- Step 5 14-cyclohex ⁇ l-6-f2-(dimethylamino )ethyl /-5, 6.7.8-tetrahydroindolof2.1 -a ][2.5]benzo diazocine- 11 -carboxylic acid
- Step 1 methyl 2-f4-chloro-2(ethoxycarbonyl)phenyl-3-cyclohexyl-lH-indole-6-carboxylate
- Step 2 methyl 2-[4-chloro-2(hydroxymethyl)phenyl-3-cyclohexyl-lH-indole-6-carboxylate
- THF methyl 2-[4-chloro-2(ethoxycarbony l)pheny 1-3 -cyclohexyl-lH-indole-6-carboxy late
- BH 3 .THF 1 M solution in THF, 2 eq
- the solution was diluted with EtOAc.
- the organic phase was washed with saturated aqueous NaHCO 3 and brine.
- Step 3 methyl 2-(4-chloro-2-(hvdroxymethyl)phenyl-3-cyclohexyl-l-(2-methoxy-2-oxoethyl)-lH-indole-6- carboxylate
- Step 4 methyl 2-(4-chloro-2-formylphenyl)'3-cyclohexyl-l-(2-methoxy-2-oxoethyl)-lH-indole-6- carboxylate
- Step 5 methyl 2[4-chloro-2-( ⁇ [2-(dimethylamino)ethyllamino ⁇ methyl)phenyl)-3-cyclohex ⁇ l-l-(2- methoxy-2-oxoethyl)-lH-indole-6-carboxylate
- Step 6 methyl 3-chloro-14-cyclohexyl-6-f2-(dimethylamino)ethyll-7-oxo-5.6.7.8-tetrahydroindolof2,l- al 12.51benzodiazocine-l 1 -carboxylate
- Step 8 3-chloro-14-cyclohexyl-6-[2-(dimethylamino)ethyl] -5.6,7 ' ,8-tetrahydroindolo[2,l ' - al[2.5 lbenz.odiazocine-11 -carboxylic acid
- Methyl 3-chloro-14-cyclohexyl-6-[2-(dimethylamino)ethyl]-5,6,7, 8-tetrahydroindolo[2,l- a][2,5]benzodiazocine-l 1-carboxylate was dissolved in a solution THF: MeOH (1 : 1) (0.02 M) and to that solution 7 eq of an aqueous solution of NaOH (IN) were added.
- Step 1 2- ⁇ [(7S)-11 -carboxy-M-cyclohexyl ⁇ .S-dihydro- ⁇ H-indolol 1 ,2-elf 1.51benz.oxaz.ocin-7-yllo ⁇ y ⁇ - N, N-dimethylethanaminium trifluoroacetate
- Example 25 l-(2- ⁇ [(7S)-ll-carboxy-14-cyclohexyl-7,8-dihydro-6H-indolo[l,2-e][l,5]benzoxazocin- 7-yl]oxy ⁇ ethyl)pyrroIidinium trifluoroacetate
- reaction mixture was concentration in vacuo, redissolved with DMSO and purified by RP-HPLC (stationary phase: column Waters XTERRA prep. C18, 5 um, 19 x 100 mm. Mobile phase: MeCN/H 2 0 buffered with 0.1 % TFA). Fractions containing the pure compound were combined and freeze dried to afford the title compound as a white powder (65 %).
- Example 26 (2Z)-(ll-carboxy-14-cyclohexyl-6H-indolo[l,2-e][l,5]benzoxazocin-7(8H)-ylidene)- N,N-dimethyIethanaminium trifluoroacetate and (2Z?)-(ll-carboxy-14-cyclohexyl-6H-indolo[l,2- e][l,5]benzoxazocin-7(8H)-yIidene)- N,N-dimethylethanaminium trifluoroacetate
- Step 1 (2Z)-2-[14-cyclohexyl-ll-(methoxycarbonyl)-6H-indolo[l,2-elfl,51benz.oxazocin-7(8H)-ylidenel- N.N-dimethylethanaminium trifluoroacetate and (2E)-2-[ 14-cyclohexyl-l 1 -(methoxycarbonyl)-6H- indolofl ,2 -elf 1.5 Ibenzoxazocin- 7(8H)-ylidene]-N, N-dimethylethanaminium trifluoroacetate To a suspension of 1.5 eq of [2-(dimethylamino)ethyl](triphenyl)phosphonium bromide in THF (0.12 M) cooled at -78 0 C, was added n-BuLi (1.6 eq, 1.6 M in hexanes).
- Step 2 (2Z)-(ll-carboxy-14-cyclohexyl-6H-indolo[L2-ellL51benz.oxaz.ocin-7(8H)-ylidene)- NN- dimethylethanaminium trifluoroacetate and (2E)-( 11 -carboxy-M-cyclohexyl- ⁇ H-indolof 1 ,2- elfl,5]benzoxazocin-7(8H)-ylidene)- N, N-dimethylethanaminium trifluoroacetate
- KOH 5 eq, IN
- Example 28 N-(ll-carboxy-14-cyclohexyl-3-fluoro-7,8-dihydro-6H-indolo[l,2-e][l,5]benzoxazocin- 7-yl)- ⁇ yV'//'-trimethyIethane-l,2-diaminium bis(trifluoroacetate)
- Step 1 methyl 3-cvclohe ⁇ yl'2-(4-fluoro-2-hydroxyphenyl)-lH-indole-6-carboxylate
- methyl 2-bromo-3-cyclohexyl-l//-indole-6-carboxylate prepared as described in WO2004087714 from commercially available methyl indole-6-carboxylate
- EtOH 5:2, v/v, 0.2 M
- Step 2 methyl 3-cyclohe ⁇ yl-2-(4-fluoro-2-[(2S)-oxiran-2-ylmetho ⁇ ylphenyl ⁇ -lH-indole-6-carboxylate
- cesium fluoride 3 eq.
- (5)-glycidyl 3-nitrobenzenesulfonate 1.1 eq.
- the resulting mixture was stirred at RT overnight then diluted with EtOAc and washed with water and brine. Drying over Na 2 SO 4 , filtration and concentration i. vac. gave the crude product, which was purified by flash chromatography (1:5 EtOAc/PE) to afford the title compound as colourless foam (70%); MS (ES + ) m/z 424 (M+H) + .
- Step 3 methyl (7S)-14-cvclohexyl-3-fluoro-7-hydroxy-7,8-dihydro-6H-indolof 1.2-elH, 51benzoxazocine- 11 -carboxylate
- a solution of the foregoing product (from Step 2) in dry DMF (0.05 M) was cooled to 0 0 C and a IM solution of sodium bis(trimethylsilyl)amide in THF (1.1 eq.) was added dropwise. The reaction was allowed to reach RT and stirred for 3 h.
- the reaction mixture was diluted with EtOAc and washed with hydrochloric acid (1 N), water and brine. Drying over Na 2 SO 4 , filtration and concentration i. vac.
- Step 5 methyl 14-cvclohexyl-7- ⁇ [2-(dimethylamino)ethyllamino ⁇ -3-fluoro-7.8-dihydro-6H-indolo[1.2- e ]fl.5]benzoxazocine-ll -carboxylate
- Step 6 methyl 14-cyclohexyl-7-[!2-(dimethylamino)ethyll(methyl)aminol-3-fluoro-7.8-dihydro-6H- indolof 1 ,2-elf 1.5]benzoxazocine-l 1 -carboxylate
- Step 7 N-(ll-carbo ⁇ y-14-cyclohexyl-7.8-dihydro-6H-indolofl.2-eiri.51benzoxazocin-7-yl)-N.N'.N'- trimethylethane-1 , 2 -diaminium bis(trifluoroacetate )
- Example 29 (7/? / )-14-cyclohexyl-7-[[2-(dimethylamino)ethyl](methyl)amino]-3-fluoro-7,8-dihydro- ⁇ H-indolotl j Z-eJtl j Slbenzoxazocine-ll-carboxylic acid
- NMR and MS as reported for the racemic mixture in example 1.
- the second peak was (7S)-14- cyclohexyl-7-[[2-(dimethylamino)ethyl](methyl)amino]-3-fluoro-7,8-dihydro-6H-indolo[l,2- e][l,5]benzoxazocine-ll-carboxylic acid (97% ee).
- Example 30 l-(2- ⁇ [(75)-ll-carboxy-14-cyclohexyl-3-fluoro-7,8-dihydro-6 ⁇ -indolo[l,2- e][l,5]benzoxazocin-7-yl]oxy ⁇ ethyl)pyrrolidinium trifluoroacetate
- methyl (75)-14-cyclohexyl-3-fluoro-7-hydroxy-7,8-dihydro-6H-indolo[l,2- e][l,5]benzoxazocine-l l-carboxylate prepared as described in Example 28, Steps 1-3
- toluene 0.05 M
- Step 1 methyl 14-cyclohex ⁇ l-7-meiks ⁇ lene-7.&-dihydro-6H-indolo ⁇ l.2-e]fl.5]benzoxazocine-ll- carboxylate
- methyl 3-cyclohexyl-2-(2-hydroxyphenyl)-lH-indole-6-carboxylate prepared as described in example 9, step 1) in dry DMF (0.06 M) was added NaH (2.5 eq., 60% suspension in mineral oil). After 30 min, 3-chloro-2-(chloromethyl)prop-l-ene (1.2 eq.) was added dropwise via syringe and the solution stirred at RT for 60 min.
- Step 2 methyl l4-cyclohexyl-7-(hydroxymethyl)-7, 8-dihydro-6H-indolo[ 1 ,2-e J ! 1.5]benzoxazocine-l 1 - carboxylate
- Step3 methyl 14-cyclohexyl-7 -formyl-7 ⁇ 8-dihydro-6H-indolol 1 ,2-e] '/ 1 ,51benzoxazocine-l 1 -carboxylate
- reaction mixture was diluted with EtOAc and washed with a 1 : 1 (v/v) mixture of sodium thiosulfate and NaHC ⁇ 3 (both aqueous saturated solutions), then with brine. Drying over Na 2 SO 4 , filtration and concentration i. vac. gave the crude methyl 14- cyclohexyl-T-formyl ⁇ . ⁇ -dihydro- ⁇ H-indolof l ⁇ -eKl.SJbenzoxazocine-l 1-carboxylate, which was used without further purification.
- Step 4 14-cyclohexyl-7-f(dimethylamino)methyl]-7.8-dihydro-6H-indolofl.2-e]fl.5]benzoxazocine-ll- carboxylic acid
- the foregoing aldehyde (from Step 3) was reductively aminated using dimethylamine and sodium triacetoxy sodiumborohydride and the ester from the resulting product hydrolyzed as described in Example 9, Step 6.
- Purification of the crude product by RP-HPLC stationary phase: column Waters Symmetry prep. C18, 7 urn, 19 x 150 mm.
- Example 32 Preparation of 13-cyclohexyl-5-[2-(dimethylamino)ethyl]-4,5,6,7- tetrahydrothienopSS'r ⁇ HMJdiazocinofl ⁇ - ⁇ Jindole-lO-carboxylic acid
- Step 1 Methyl l-(2'tert-butoxy-2-oxoethyl)-3-cyclohexyl-2-(2-formyl-3-thienyl)-lH-indole-6-carboxylate Methyl 2-bromo-l-(2-tert-butoxy-2-oxoethyl)-3-cyclohexyl-lH-indole-6-carboxylate (prepared as described in Example 1, Step 1) and (2-formyl-3-thienyl) boronic acid (1.5 eq) were dissolved in dioxane (0.07 M) and 2M aqueous Na 2 CO 3 (6 eq) was added.
- Step 2 13-cyclohexyl-5-[2-(dimethylamino)ethyl]-4,5.6,7-tetrahydrothieno[2 ⁇ 3':6.7Hl,41diazocinofl,8- a] indole- 10-carboxylic acid
- Example 33 Preparation of 13-cyclohexyI-5-[2-(dimethylamino)ethyl]-4,5,6,7- tetrahydrothieno[3 ⁇ 2':6/7][l,4]diazocino[l,8- ⁇ ]indole-10-carboxylic acid
- Step 2 13-cyclohexyl-5-[2-(dimethylamino)ethyl]-4,5,6,7-tetrahydrothienof 3'.4':6.7] f 1 ,41diazocinof 1.8- a] indole- 10-carboxylic acid
- Example 32 Using the foregoing (4-formyl-3-thienyl)boronic acid the title compound was prepared following the procedures in Example 32. 56% yield after RP-HPLC purification and lyophilisation (Conditions: Column: Waters X-TERRA MS C18, 10 micron, 19 x 150 mm; Gradient: A: H 2 O + 0.1% TFA; B: MeCN + 0.1% TFA; 75% A isocratic for 3 min, linear to 20% A in 12 min).
- Step 1 Methyl 3-cyclohexyl-2-(2-vinylphenyl)-lH-indole-6-carboxylate Methyl 2-bromo-3-cyclohexyl-lH-indole-6-carboxylate (prepared as described in WO 2004065367) and (2-vinylphenyl)boronic acid (1.5 eq) were dissolved in dioxane (0.07 M) and 2M aqueous Na 2 CO 3 (6 eq) was added.
- Step 2 Methyl l-but-3-en-l-yl-3-cyclohexyl-2-(2-vinylphenyl)-lH-indole-6-carbox ⁇ late
- 60% NaH (1.5 eq) in mineral oil was added at 0 0 C; after stirring for 45 min at RT, 4-bromobut-l-ene (1.5 eq) was added and the suspension was stirred for 5 h at 40 0 C and then 1 day at RT (NaH and 4-bromobut-l- ene were added several times).
- Step 3 Methyl 14-cyclohexyl-7.8-dihydroindolo[2,l-a][2]benzazocine-l]-carboxylate
- Step 4 14- Cvclohexyl-5-(2 -pyrrolidin- 1 -ylethoxy )-5.6.7.8-tetrahydroindolo[2, l-aH2 Ibenzazocine- 11- carboxylic acid
- Step 1 Methyl 14-cvclohexyl-5,6-dihydroxy-5.6,7.8-tetrahydroindolo[2,l-a][2]benz.az.ocine-ll- carboxylate
- Step 2 Methyl 15-cyclohexyl-6-oxo-4b,7a,8,9-tetrahydro[l,3]dioxolo[4.5-elindolo[2,l- a ] ⁇ 2 ]benzazocine-12-carboxylate
- Step 4 14-cyclohexyl-6-(2-pyrrolidin-lylethoxy)-5,6, 7,8-tetrahvdroindolo[2, l-a][21benzazocine-ll - carboxylate
- Example 44 14-cyclohexyl-3-fluoro-6-[(4-methylpiperazin-l-yl)acetyl]-5,6,7,8- tetrahydroindolo[2,l -a] [2,5] benzodiazocine- 11-car boxy lie acid :
- Step 1 14-cyclohexyl-3-fluoro-5.6, 7,8-tetrahydroindolo[2.1-aH2,51benz.odiaz.ocine-ll-carbox ⁇ lic acid
- Step 2 14-cyclohexyl-3-fluoro-6-[(4-methylpiperaz.in-l-yl)acet ⁇ ll-5.6.7.8-tetrahydroindolo[2.1- al[2,5]benzodiazocine-ll-carboxylic acid
- DCM 0.2 M
- 4 eq of DIPEA and 2 eq of HATU were added and the mixture stirred at RT for 1 h.
- Example 45 14-cyclohexyl-6- ⁇ [2-(dimethylamino)ethyl]suIfonyl ⁇ -3-fluoro-5,6,7,8- tetrahydroindolo[2,l-a][2,5]benzodiazocine-ll-carboxylic acid
- Example 46 14-cyclohexyl-3-fluoro-6- ⁇ [(2R)-l-methylpyrroIidin-2-yl]methyl ⁇ -5,6,7,8- tetrahydroindolo[2,l -a] [2,5] bcnzodiazoci ne- 11 -carboxylic acid
- Step 1 methyl 14-cvclohexyl-3-fluoro-6-( 1 -methyl-D-prolyl)-5.6.7.8-tetrahydroindolo[2.1 -a J [2. 5 lbenzodiaz.ocine-11 -carboxylate
- Step 2 methyl 14-cyclohexyl-3-fluoro-6- ⁇ f(2R)-l-methylpyrrolidin-2-yllmethyl ⁇ -5.6.7.8- tetrahydroindolo[2.1 -a If 2, 5 Wenzodiazocine-11 -carboxylate
- Step 3 14-cyclohexyl-3-fluoro-6-ff(2R)-l-methylpyrrolidin-2-yl]methylf-5.6.7,8-tetrahydroindolof2,l- ajf2.5]benzodiazocine-l 1 -carboxylic acid
- Example 47 14-cyclohexyl-3-fluoro-6-(lH-imidazol-l-ylacetyI)-5,6,7,8-tetrahydroindolo[2,l- a][2,5]benzodiazocine-ll-carboxylic acid
- Step 1 methyl 14-cyclohex ⁇ l-3-fluoro-6-( 1 H-imidazol-1 -ylacetyl)-5.6,7,8-tetrahydroindolo[2.] -a] (2,
- Step 2 14-cyclohexyl-3-fluoro-6-(lH-imidazol-l-ylacetyl)-5.6.7,8-tetrahydroindolof2.1- alf2.5 Ibenzodiazocine- 11 -carboxylic acid
- Example 48 14-cyclohexyl-6-(iV ⁇ V-dimethylglycyl)-5,6,7,8-tetrahydroindolo[2,l- ⁇ ][2,5]benzodiazocine-ll-carboxylic acid
- Step 1 methyl 3-cvclohexyl- 1 -(1.3-dioxolan-2 -ylmethyl )-2-f2 - ⁇ (hydroxyimino )methyl 1 phenyl ⁇ -l H-indole- 6-carboxylate
- Step 2 methyl 2-[2-(aminomethyl)phenyll-3-cyclohexyl-l-(].3-dioxolan-2-ylmethyl)-lH-indole-6- carboxylate
- Step 3 methyl 14-cyclohexyl-5,6.7.8-tetrahydroindolo[2,l-alf2.5]benz.odiazocine-ll-carboxylate 3M aqueous HCl (16 eq) was added to a solution of methyl 2-[2-(aminomethyl)phenyl]-3-cyclohexyl-l- (l,3-dioxolan-2-ylmethyl)-lH-indole-6-carboxylate in THF (0.03 M). The reaction was heated with stirring at reflux for 24 h, before being allowed to cool to RT, basified with aqueous NaOH (2N) and extracted into EtOAc (3 times).
- Step 4 methyl 14-cyclohexyl-6-(N. N-dimethyl ⁇ lvcyl)-5,6.7.8-tetrahydroindolo[2J- aj[2,5 Jbenzodiazocine- 11 -carboxylate
- Step 5 14-cyclohexyl-6-(N. N-dimethylslycyl)-5,6, 7,8-tetrahydroindolo[2.1-aU2.51benzodiaz.ocine-l 1 - carboxylic acid
- Example 49 14-cyclohexyl-6-[(l-methyl-lH-pyrazol-4-yl)methyl]-5,6,7,8-tetrahydroindolo[2,l- a][2,5]benzodiazocine-ll-carboxylic acid
- Step 1 methyl 14-cvclohexyl-6-f(l -methyl- 1 H-pyrazol-4-yl )methyl]-5.6.7, 8-tetrahvdroindolo[2, 1 - a] [2,5 Jbenzodiazocine-] 1 -carboxylate
- Step 2 14-cyclohexyl-6-[(l-methyl-lH-pyrazol-4-yl)methyll-5.6.7,8-tetrahydroindolo[2,l- a] [2,5 Jbenzodiazocine- 11 -carboxylic acid
- BBr 3 (5 eq, 1 M sol. in CH 2 Cl 2 ) was added to a solution of methyl 14-cyclohexyl-6-[(l-methyl-lH- pyrazol-4-yl)methyl]-5,6,7,8-tetrahydroindolo[2,l-a][2,5]benzodiazocine-l 1-carboxylate in CH 2 Cl 2 (0.06 M), and the mixture stirred at RT for 40 mins. The volatiles were removed in vacuo. The crude was then purified by prep RP-HPLC (stationary phase: column Waters XTERRA prep. C18, 5 um, 19 xl50 mm. Mobile phase: MeCN/H 2 O buffered with 0.1 % TFA).
- Example 50 14-cyclohexyl-6- ⁇ 2-[methyl(pyridin-3-ylmethyl)amino]ethyl ⁇ -5,6,7,8- tetrahydroindolo ⁇ l-alP ⁇ Jbenzodiazocine-ll-carboxylic acid
- Step 1 Tert-butyl N-/2-[3-cvclohexyl-l-(2.2-dimethoxyethyl)-6-(methoxycarbonyl)-lH-indol-2- y/ J benzyl fslycinate
- Step 3 fl4-cyclohexyl-ll-(methoxycarbonyl)-7.8-dihydroindolof2.1-a][2.5]benzodiazocin-6(5H)- yl] acetic acid
- Step 4 methyl 14-cyclohexyl-6-f2-fmethyl(pyridin-3-ylmethyl)amino]-2-oxoethyl!-5.6.7.8- tetrahydroindolo[2, 1 -all 2, 5 Ibenzodiazocine- 11 -carboxylate
- Step 5 methyl 14-cyclohexyl-6-f2-[methyl(pyridin-3-ylmethyl)amino]ethyl ⁇ -5.6.7,8-tetrahydroindolo[2.1- a] [2,5 Ibenzodiazocine- 11 -carboxylate
- BH 3 -Me 2 S (20 eq, 2 M solution in THF) was added.
- Step 6 14-cyclohexyl-6- ⁇ 2-fmethyl(pyridin-3-ylmethyl)aminolethyl ⁇ -5,6,7.8-tetrahydroindolo[2,l- all 2.5 Ibenzodiazocine- 11 -carboxylic acid
- Example 51 13-cyclohexyl-5-[2-(dimethylamino)ethyl]-4,5,6,7- tetrahydrofuro[3',2':6,7][l,4]diazocino[l,8- ⁇ ]indole-10-carboxylic acid
- Step 1 methyl l-(2-tert-butoxy-2-oxoethyl)-3-cyclohexyl-2-(3-formyl-2-furyl)-lH-indole-6-carboxylate
- methyl 2-bromo-l-(2-te/t-butoxy-2-oxoethyl)-3-cyclohexyl-lH-indole-6-carboxylate prepared as described in Example 1, Step 1) in dioxane (0.04 M) was added Na 2 COs (5 eq, 2 M aqueous solution), 3-formylfuran-2-boronic acid (1.4 eq) and bis(triphenylphosphine)palladium(II) dichloride (0.2 eq).
- Step 2 methyl l-(2-tert-butoxy-2-oxoethyl)-3-cyclohexyl-2-[3-( ⁇ [2-(dimethylamino)ethyll amino imethyl)- 2-furyll-l H-indole-6-carbo ⁇ ylate
- Step 4 methyl 13-cvclohexyl-5-[2-(dimethylamino)ethyll-4.5.6.7-tetrahydrofurot3'.2' :6,7) 11 Aldiazocinol 1 ,8-al indole- 10-carboxylate
- Step 5 13-cyclohexyl-5-[2-(dimethylamino)ethyll-4,5,6, 7-tetrahydrofuro[3',2':6.71fl.41dia7.ocinofl.8- a] indole- 10-carboxylic acid
- Example 52 15-cyclohexyI-6-[2-(dimethylamino)ethyl]-7-oxo-6,7,8,9-tetrahydro-5H-indolo[2,l- a][2,6]benzodiazonine-12-carboxylic acid
- Step 1 3-[2-bromo-3-cyclohexyl-6-(methoxycarbonyl)-lH-indol-l-yl]propanoic acid NaH (3.5 eq, 60 % dispersion in mineral oil) was added to a solution of methyl 2-bromo-3-cyclohexyl- lH-indole-6-carboxylate (prepared as described in WO 2004065367, from commercially available methyl indole-6-carboxylate) in DMF (0.2 M) and the solution allowed to stir at RT for 1 h.
- Step 2 methyl 2-bromo-3-cyclohexyl-l-(3-methoxy-3-oxopropyl)-lH-indole-6-carboxylate
- Step 3 methyl 3-cyclohe ⁇ yl-2-(2-formylphenyl)-l-(3-methoxy-3-oxopropyl)-lH-indole-6-carboxylate
- methyl 2-bromo-3-cyclohexyl-l-(3-methoxy-3-oxopropyl)-lH-indole-6-carboxylate in dioxane (0.15 M) were added Na 2 C ⁇ 3 (6 eq, 2 M aqueous solution), 1.6 eq of (2-formylphenyl)boronic acid and 0.2 eq of bis(triphenylphosphine)-palladium(IT) dichloride.
- Step 4 methyl 3-cyclohexyl-2-[2-(lf2-(dimethylamino)ethyllamino ⁇ methyl)phenyll-l-(3-methoxy-3- oxopropyl)-lH-indole-6-carboxylate
- Lithium hydroxide monohydrate (1 eq) was added to a solution of methyl 3-cyclohexyl-2-[2-( ⁇ [2- (dimethylamino)ethyl]amino ⁇ methyl)phenyl]-l-(3-methoxy-3-oxopropyl)-lH-indole-6-carboxylate in a mixture THF:H 2 O (4: 1, 0.1 M). The mixture stirred at RT for 1 h. The reaction was quenched with 1 N HCl and the solvent evaporated in vacuo. The residue was washed with the minimum amount of Et 2 O and the solid residue filtered to obtain the title compound (82 %); MS (ES + ) m/z 506 (M+H) + .
- Step 6 methyl 15-cyclohexyl-6-[2-(dimethylamino)ethyl]-7-oxo-6.7.8,9-tetrahydro-5H-indolof2.1- a][2,6]benzodiazonine-12-carboxylate
- Step 7 15-cyclohexyl-6-[2-(dimethylamino)ethyl]-7-oxo-6.7.8.9-tetrahvdro-5H-indolo[2.1- a]f2.61benzodiazonine-12-carboxylic acid
- DCM 0.04 M
- 7 eq BBr 3 (1 M solution in DCM) were added. The solution stirred at RT for 20 mins.
- Step 2 14-cyclohexyl-6f2-(dimethylamino)ethyll-N-(morpholin-4-ylsulfonyl)-5.6.7.8- tetrahy ⁇ -pindolol r 2, 1 -a] [2,51benzodiazocine-l 1 -carboxamide
Abstract
Description





































Claims








Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BRPI0516972-0A BRPI0516972A (en) | 2004-10-26 | 2005-10-25 | compound, use thereof, pharmaceutical composition, method for inhibiting hepatitis c virus polymerase and / or treating or preventing a disease due to hepatitis c virus, and, process for preparing a compound |
| RU2007119562/04A RU2007119562A (en) | 2004-10-26 | 2005-10-25 | TETRACYCLIC DERIVATIVES OF INDOLES AS ANTIVIRAL AGENTS |
| CA002585084A CA2585084A1 (en) | 2004-10-26 | 2005-10-25 | Tetracyclic indole derivatives as antiviral agents |
| JP2007538502A JP2008517986A (en) | 2004-10-26 | 2005-10-25 | Tetracyclic indole derivatives as antiviral agents |
| EP05798379A EP1807403A2 (en) | 2004-10-26 | 2005-10-25 | Tetracyclic indole derivatives as antiviral agents |
| MX2007004979A MX2007004979A (en) | 2004-10-26 | 2005-10-25 | Tetracyclic indole derivatives as antiviral agents. |
| AU2005298403A AU2005298403A1 (en) | 2004-10-26 | 2005-10-25 | Tetracyclic indole derivatives as antiviral agents |
| IL182610A IL182610A0 (en) | 2004-10-26 | 2007-04-17 | Tetracyclic indole derivatives as antiviral agents |
| NO20072689A NO20072689L (en) | 2004-10-26 | 2007-05-25 | Tetracyclic indole derivatives as antivirals |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0423767.3 | 2004-10-26 | ||
| GB0423767A GB0423767D0 (en) | 2004-10-26 | 2004-10-26 | Therapeutic compounds |
| GB0512519.0 | 2005-06-21 | ||
| GB0512519A GB0512519D0 (en) | 2005-06-21 | 2005-06-21 | Therapeutic compounds |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2006046030A2 true WO2006046030A2 (en) | 2006-05-04 |
| WO2006046030A3 WO2006046030A3 (en) | 2006-07-06 |
Family
ID=36101507
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2005/004127 WO2006046030A2 (en) | 2004-10-26 | 2005-10-25 | Tetracyclic indole derivatives as antiviral agents |
| PCT/GB2005/004144 WO2006046039A2 (en) | 2004-10-26 | 2005-10-25 | Tetracyclic indole derivatives as antiviral agents |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2005/004144 WO2006046039A2 (en) | 2004-10-26 | 2005-10-25 | Tetracyclic indole derivatives as antiviral agents |
Country Status (19)
| Country | Link |
|---|---|
| US (1) | US7662809B2 (en) |
| EP (2) | EP1807403A2 (en) |
| JP (2) | JP2008517987A (en) |
| KR (1) | KR20070068427A (en) |
| AR (1) | AR051469A1 (en) |
| AU (2) | AU2005298403A1 (en) |
| BR (1) | BRPI0516972A (en) |
| CA (2) | CA2585084A1 (en) |
| CR (1) | CR9069A (en) |
| EC (1) | ECSP077412A (en) |
| IL (1) | IL182610A0 (en) |
| MA (1) | MA29037B1 (en) |
| MX (1) | MX2007004979A (en) |
| NI (1) | NI200700102A (en) |
| NO (1) | NO20072689L (en) |
| PE (1) | PE20060607A1 (en) |
| RU (1) | RU2007119562A (en) |
| TW (1) | TW200630344A (en) |
| WO (2) | WO2006046030A2 (en) |
Cited By (76)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007033175A1 (en) * | 2005-09-13 | 2007-03-22 | Bristol-Myers Squibb Company | Indolobenzazepine hcv ns5b inhibitors |
| WO2007054741A1 (en) * | 2005-11-10 | 2007-05-18 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Tetracyclic indole derivatives as antiviral agents |
| WO2007092000A1 (en) * | 2006-02-06 | 2007-08-16 | Bristol-Myers Squibb Company | Inhibitors of hcv replication |
| WO2007129119A1 (en) * | 2006-05-08 | 2007-11-15 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Pentacyclic indole derivatives as antiviral agents |
| WO2007136982A1 (en) * | 2006-05-17 | 2007-11-29 | Bristol-Myers Squibb Company | Cyclopropyl fused indolobenzazepine hcv ns5b inhibitors |
| WO2007140109A1 (en) * | 2006-05-22 | 2007-12-06 | Bristol-Myers Squibb Company | Cyclopropyl fused indolobenzazepine hcv ns5b inhibitors |
| WO2007140254A2 (en) * | 2006-05-25 | 2007-12-06 | Bristol-Myers Squibb Company | Cyclopropyl fused indolobenzazepine hcv ns5b inhibitors |
| WO2008011521A2 (en) * | 2006-07-20 | 2008-01-24 | Genelabs Technologies, Inc. | Polycyclic viral inhibitors |
| US7348425B2 (en) | 2004-08-09 | 2008-03-25 | Bristol-Myers Squibb Company | Inhibitors of HCV replication |
| WO2008075103A1 (en) * | 2006-12-20 | 2008-06-26 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Antiviral indoles |
| US7399758B2 (en) | 2005-09-12 | 2008-07-15 | Meanwell Nicholas A | Cyclopropyl fused indolobenzazepine HCV NS5B inhibitors |
| WO2008099020A1 (en) * | 2007-02-16 | 2008-08-21 | Tibotec Pharmaceuticals Ltd. | 1,1-dioxo-1-thia-5,10-diazadibenzocycloheptenes useful as hepatitis c virus inhibitors |
| WO2008099019A1 (en) * | 2007-02-16 | 2008-08-21 | Tibotec Pharmaceuticals Ltd. | 6-hydroxy-dibenzodiazepinones useful as hepatitis c virus inhibitors |
| WO2008112473A1 (en) * | 2007-03-14 | 2008-09-18 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis c |
| WO2008111978A1 (en) | 2007-03-13 | 2008-09-18 | Bristol-Myers Squibb Company | Cyclopropyl fused indolobenzazepine hcv ns5b inhibitors |
| WO2008112848A1 (en) | 2007-03-14 | 2008-09-18 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis c |
| WO2008112841A1 (en) * | 2007-03-14 | 2008-09-18 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis c |
| WO2008136815A2 (en) * | 2006-12-22 | 2008-11-13 | Schering Corporation | 5, 6-ring annulated indole derivatives and use thereof |
| US7452876B2 (en) | 2006-06-08 | 2008-11-18 | Bristol-Myers Squibb Company | Cyclopropyl fused indolobenzazepine HCV NS5B inhibitors |
| US7456165B2 (en) | 2006-02-08 | 2008-11-25 | Bristol-Myers Squibb Company | HCV NS5B inhibitors |
| GB2451184A (en) * | 2007-07-17 | 2009-01-21 | Angeletti P Ist Richerche Bio | Macrocyclic hepatitis C virus NS3 protease inhibitors having a 6-(N-sulphamoyl(carbamoyl))-[7-aza-]indole moiety within the macrocyclic ring path |
| WO2009023487A1 (en) | 2007-08-09 | 2009-02-19 | Bristol-Myers Squibb Company | Tetracyclic compounds for the treatment of hepatitis c |
| US7517872B2 (en) | 2007-02-22 | 2009-04-14 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis C |
| US7521442B2 (en) | 2006-05-25 | 2009-04-21 | Bristol-Myers Squibb Company | Cyclopropyl fused indolobenzazepine HCV NS5B inhibitors |
| US7521443B2 (en) | 2006-05-17 | 2009-04-21 | Bristol-Myers Squibb Company | Cyclopropyl fused indolobenzazepine HCV NS5B inhibitors |
| US7538103B2 (en) | 2007-03-15 | 2009-05-26 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis C |
| US7538102B2 (en) | 2007-03-14 | 2009-05-26 | Bristol-Myers Squibb Company | Compounds for the treatment of Hepatitis C |
| WO2009067481A1 (en) * | 2007-11-21 | 2009-05-28 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis c |
| US7541351B2 (en) | 2007-01-11 | 2009-06-02 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis C |
| US7541352B2 (en) | 2007-02-02 | 2009-06-02 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis C |
| WO2009076747A1 (en) | 2007-12-19 | 2009-06-25 | Boehringer Ingelheim International Gmbh | Viral polymerase inhibitors |
| WO2009086139A1 (en) * | 2007-12-21 | 2009-07-09 | Genelabs Technologies, Inc. | Condensed pentacyclic derivatives for use in the treatment of flaviviridae infections |
| US7652004B2 (en) | 2007-08-09 | 2010-01-26 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis C |
| US7662809B2 (en) | 2004-10-26 | 2010-02-16 | Istituto Di Richerche Di Biologia Molecolare P Angeletti Spa | Tetracyclic indole derivatives as antiviral agents |
| WO2010080874A1 (en) | 2009-01-07 | 2010-07-15 | Scynexis, Inc. | Cyclosporine derivative for use in the treatment of hcv and hiv infection |
| WO2010082050A1 (en) | 2009-01-16 | 2010-07-22 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti S.P.A. | Macrocyclic and 7-aminoalkyl-substituted benzoxazocines for treatment of hepatitis c infections |
| US7767660B2 (en) | 2006-12-20 | 2010-08-03 | Istituto Di Richerche Di Biologia Molecolare P. Angeletti Spa | Antiviral indoles |
| US7781422B2 (en) | 2006-12-20 | 2010-08-24 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Antiviral indoles |
| US7789125B2 (en) | 2004-01-09 | 2010-09-07 | United Technologies Corporation | Extended impingement cooling device and method |
| US7795247B2 (en) | 2004-10-26 | 2010-09-14 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Tetracyclic indole derivatives as antiviral agents |
| US7973040B2 (en) | 2008-07-22 | 2011-07-05 | Merck Sharp & Dohme Corp. | Macrocyclic quinoxaline compounds as HCV NS3 protease inhibitors |
| US7977331B1 (en) | 2004-02-24 | 2011-07-12 | Japan Tobacco Inc. | Tetracyclic fused heterocyclic compound and use thereof as HCV polymerase inhibitor |
| US7998951B2 (en) | 2007-03-05 | 2011-08-16 | Bristol-Myers Squibb Company | HCV NS5B inhibitors |
| US8119628B2 (en) | 2008-03-27 | 2012-02-21 | Bristol-Myers Squibb Company | Pyrrolidine fused indolobenzadiazepine HCV NS5B inhibitors |
| US8124601B2 (en) | 2007-11-21 | 2012-02-28 | Bristol-Myers Squibb Company | Compounds for the treatment of Hepatitis C |
| US8133884B2 (en) | 2008-05-06 | 2012-03-13 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis C |
| US8138164B2 (en) | 2006-10-24 | 2012-03-20 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| US8138171B2 (en) | 2008-03-27 | 2012-03-20 | Bristol-Myers Squibb Company | Dioxolane and dioxolanone fused indolobenzadiazepine HCV NS5B inhibitors |
| US8143305B2 (en) | 2007-08-29 | 2012-03-27 | Schering Corporation | 2,3-substituted indole derivatives for treating viral infections |
| US8143244B2 (en) | 2009-02-26 | 2012-03-27 | Bristol-Myers Squibb Company | Cyclopropyl fused indolobenzazepine HCV NS5B inhibitors |
| US8143243B2 (en) | 2007-08-09 | 2012-03-27 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis C |
| WO2012041227A1 (en) * | 2010-09-29 | 2012-04-05 | Merck Sharp & Dohme Corp. | Tetracyclic heterocycle compounds for treating hepatitis c viral infection |
| US8178520B2 (en) | 2006-05-15 | 2012-05-15 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Macrocyclic compounds as antiviral agents |
| US8178523B2 (en) | 2008-03-27 | 2012-05-15 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis C |
| CN101443336B (en) * | 2006-05-17 | 2012-07-04 | 百时美施贵宝公司 | Cyclopropyl fused indolobenzazepine HVC ns5b inhibitors |
| WO2012092484A2 (en) | 2010-12-29 | 2012-07-05 | Inhibitex, Inc. | Substituted purine nucleosides, phosphoroamidate and phosphorodiamidate derivatives for treatment of viral infections |
| EP2481807A2 (en) | 2007-03-09 | 2012-08-01 | Merck Sharp & Dohme Corporation | In vivo HCV resistance to anti-viral inhibitors |
| EP2494991A1 (en) | 2007-05-04 | 2012-09-05 | Vertex Pharmaceuticals Incorporated | Combination therapy for the treatment of HCV infection |
| US8278322B2 (en) | 2005-08-01 | 2012-10-02 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| US8309540B2 (en) | 2006-10-24 | 2012-11-13 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| US8314062B2 (en) | 2006-06-23 | 2012-11-20 | Instituto Di Ricerche Di Biologia Molecolare P. Angeletti S.P.A. | Macrocyclic compounds as antiviral agents |
| US8377873B2 (en) | 2006-10-24 | 2013-02-19 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| US8377928B2 (en) | 2007-11-16 | 2013-02-19 | Merck Sharp & Dohme Corp. | 3-aminosulfonyl substituted indole derivatives and methods of use thereof |
| US8377874B2 (en) | 2006-10-27 | 2013-02-19 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| US8404845B2 (en) | 2007-08-29 | 2013-03-26 | Merck Sharp & Dohme Corp. | 2,3-substituted azaindole derivatives for treating viral infections |
| US8431568B2 (en) | 2008-03-27 | 2013-04-30 | Bristol-Myers Squibb Company | Aromatic heterocyclic fused indolobenzadiazepine HCV NS5B inhibitors |
| US8461107B2 (en) | 2008-04-28 | 2013-06-11 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| US8546420B2 (en) | 2006-12-22 | 2013-10-01 | Merck Sharp & Dohme Corp. | 4, 5-ring annulated indole derivatives for treating or preventing of HCV and related viral infections |
| US8557848B2 (en) | 2006-12-22 | 2013-10-15 | Merck Sharp & Dohme Corp. | 4,5-ring annulated indole derivatives for treating or preventing of HCV and related viral infections |
| US8614229B2 (en) | 2007-08-29 | 2013-12-24 | Merck Sharp & Dohme Corp. | Substituted indole derivatives and methods of use thereof |
| US8765757B2 (en) | 2007-11-16 | 2014-07-01 | Merck Sharp & Dohme Corp. | 3-heterocyclic substituted indole derivatives and methods of use thereof |
| US8901139B2 (en) | 2008-06-13 | 2014-12-02 | Merck Sharp & Dohme Corp. | Tricyclic indole derivatives and methods of use thereof |
| US8927569B2 (en) | 2007-07-19 | 2015-01-06 | Merck Sharp & Dohme Corp. | Macrocyclic compounds as antiviral agents |
| US9090671B2 (en) | 2008-06-06 | 2015-07-28 | Scynexis, Inc. | Macrocyclic peptides |
| US9738661B2 (en) | 2006-10-27 | 2017-08-22 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| US11234977B2 (en) | 2017-12-20 | 2022-02-01 | Novartis Ag | Fused tricyclic pyrazolo-dihydropyrazinyl-pyridone compounds as antivirals |
Families Citing this family (19)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7659263B2 (en) | 2004-11-12 | 2010-02-09 | Japan Tobacco Inc. | Thienopyrrole compound and use thereof as HCV polymerase inhibitor |
| WO2006119061A2 (en) | 2005-05-02 | 2006-11-09 | Merck & Co., Inc. | Hcv ns3 protease inhibitors |
| EP2385951A4 (en) | 2009-01-09 | 2013-05-29 | Univ Cardiff | Phosphoramidate derivatives of guanosine nucleoside compounds for treatment of viral infections |
| UA108351C2 (en) | 2009-03-27 | 2015-04-27 | Hepatitis C Virus Replication Inhibitors | |
| US20190127365A1 (en) | 2017-11-01 | 2019-05-02 | Merck Sharp & Dohme Corp. | Inhibitors of hepatitis c virus replication |
| US9260192B2 (en) | 2009-07-27 | 2016-02-16 | Textron Innovations Inc. | Active vent and re-inflation system for a crash attentuation airbag |
| WO2011014487A1 (en) | 2009-07-30 | 2011-02-03 | Merck Sharp & Dohme Corp. | Hepatitis c virus ns3 protease inhibitors |
| WO2011103433A1 (en) | 2010-02-18 | 2011-08-25 | Medivation Technologies, Inc. | Pyrido[4,3-b]indole and pyrido[3,4-b]indole derivatives and methods of use |
| US9193728B2 (en) | 2010-02-18 | 2015-11-24 | Medivation Technologies, Inc. | Fused tetracyclic pyrido [4,3-B] indole and pyrido [3,4-B] indole derivatives and methods of use |
| US9040519B2 (en) | 2010-02-18 | 2015-05-26 | Medivation Technologies, Inc. | Fused tetracyclic pyrido [4,3-B] indole and pyrido [3,4-B] indole derivatives and methods of use |
| US9254292B2 (en) | 2010-09-29 | 2016-02-09 | Merck Sharp & Dohme Corp. | Fused tetracycle derivatives and methods of use thereof for the treatment of viral diseases |
| WO2012112961A1 (en) | 2011-02-18 | 2012-08-23 | Medivation Technologies, Inc. | Compounds and methods of treating hypertension |
| WO2012122716A1 (en) | 2011-03-17 | 2012-09-20 | Merck Sharp & Dohme Corp. | Tetracyclic xanthene derivatives and methods of use thereof for treatment of viral diseases |
| TW201600087A (en) | 2011-10-21 | 2016-01-01 | 艾伯維有限公司 | Methods for treating HCV |
| US8492386B2 (en) | 2011-10-21 | 2013-07-23 | Abbvie Inc. | Methods for treating HCV |
| US8466159B2 (en) | 2011-10-21 | 2013-06-18 | Abbvie Inc. | Methods for treating HCV |
| DK2583677T1 (en) | 2011-10-21 | 2015-01-19 | Abbvie Inc | Methods for treatment of HCV comprising at least two direct-acting antiviral agents ribavirin, interferon but not |
| WO2014110687A1 (en) | 2013-01-16 | 2014-07-24 | Merck Sharp & Dohme Corp. | Thiazolyl-substitued tetracyclic compounds and methods of use thereof for treatment of viral diseases |
| JP7129703B2 (en) | 2016-04-28 | 2022-09-02 | エモリー ユニバーシティー | Alkyne-Containing Nucleotide and Nucleoside Therapeutic Compositions and Uses Associated Therewith |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1993000334A1 (en) * | 1991-06-27 | 1993-01-07 | Fidia - Georgetown Institute For The Neurosciences | Indole derivatives, pharmaceutical compositions and methods of treating neurological and psychiatric disorders |
| WO2003099824A1 (en) * | 2002-05-21 | 2003-12-04 | Wyeth | R-enantiomers of pyranoindole derivatives against hepatitis c |
| WO2005080399A1 (en) * | 2004-02-24 | 2005-09-01 | Japan Tobacco Inc. | Fused heterotetracyclic compounds and use tehreof as hcv polymerase inhibitor |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| IT1278077B1 (en) | 1995-05-25 | 1997-11-17 | Angeletti P Ist Richerche Bio | METHODOLOGY TO REPRODUCE IN VITRO THE ACTIVITIES OF RNA-DEPENDENT RNA POLYMERASE AND OF TERMINAL NUCLEOTIDYLTRANSPHERASE CODED BY THE |
| JP4299540B2 (en) | 2001-01-23 | 2009-07-22 | イステイチユート・デイ・リチエルケ・デイ・ビオロジア・モレコラーレ・ピ・アンジエレツテイ・エツセ・ピー・アー | Hepatitis C virus replicon and replicon enhanced cells |
| AU2002368247A1 (en) * | 2002-09-27 | 2004-04-19 | Mitsubishi Paper Mills Limited | Electrophotographic transfer sheet |
| GB0307891D0 (en) | 2003-04-04 | 2003-05-14 | Angeletti P Ist Richerche Bio | Chemical compounds,compositions and uses |
| US7153848B2 (en) * | 2004-08-09 | 2006-12-26 | Bristol-Myers Squibb Company | Inhibitors of HCV replication |
| US7795247B2 (en) | 2004-10-26 | 2010-09-14 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Tetracyclic indole derivatives as antiviral agents |
| MX2007004979A (en) | 2004-10-26 | 2007-06-14 | Angeletti P Ist Richerche Bio | Tetracyclic indole derivatives as antiviral agents. |
-
2005
- 2005-10-25 MX MX2007004979A patent/MX2007004979A/en active IP Right Grant
- 2005-10-25 EP EP05798379A patent/EP1807403A2/en not_active Withdrawn
- 2005-10-25 CA CA002585084A patent/CA2585084A1/en not_active Abandoned
- 2005-10-25 EP EP05798382A patent/EP1807397A2/en not_active Withdrawn
- 2005-10-25 RU RU2007119562/04A patent/RU2007119562A/en not_active Application Discontinuation
- 2005-10-25 BR BRPI0516972-0A patent/BRPI0516972A/en not_active IP Right Cessation
- 2005-10-25 WO PCT/GB2005/004127 patent/WO2006046030A2/en active Application Filing
- 2005-10-25 JP JP2007538504A patent/JP2008517987A/en active Pending
- 2005-10-25 AU AU2005298403A patent/AU2005298403A1/en not_active Abandoned
- 2005-10-25 CA CA002585113A patent/CA2585113A1/en not_active Abandoned
- 2005-10-25 JP JP2007538502A patent/JP2008517986A/en active Pending
- 2005-10-25 KR KR1020077009541A patent/KR20070068427A/en not_active Application Discontinuation
- 2005-10-25 US US11/666,583 patent/US7662809B2/en not_active Expired - Fee Related
- 2005-10-25 WO PCT/GB2005/004144 patent/WO2006046039A2/en active Application Filing
- 2005-10-25 AU AU2005298412A patent/AU2005298412B2/en not_active Ceased
- 2005-10-25 PE PE2005001247A patent/PE20060607A1/en not_active Application Discontinuation
- 2005-10-26 AR ARP050104493A patent/AR051469A1/en unknown
- 2005-10-26 TW TW094137552A patent/TW200630344A/en unknown
-
2007
- 2007-04-16 NI NI200700102A patent/NI200700102A/en unknown
- 2007-04-17 IL IL182610A patent/IL182610A0/en unknown
- 2007-04-19 CR CR9069A patent/CR9069A/en not_active Application Discontinuation
- 2007-04-23 EC EC2007007412A patent/ECSP077412A/en unknown
- 2007-05-23 MA MA29932A patent/MA29037B1/en unknown
- 2007-05-25 NO NO20072689A patent/NO20072689L/en not_active Application Discontinuation
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1993000334A1 (en) * | 1991-06-27 | 1993-01-07 | Fidia - Georgetown Institute For The Neurosciences | Indole derivatives, pharmaceutical compositions and methods of treating neurological and psychiatric disorders |
| WO2003099824A1 (en) * | 2002-05-21 | 2003-12-04 | Wyeth | R-enantiomers of pyranoindole derivatives against hepatitis c |
| WO2005080399A1 (en) * | 2004-02-24 | 2005-09-01 | Japan Tobacco Inc. | Fused heterotetracyclic compounds and use tehreof as hcv polymerase inhibitor |
Cited By (116)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7789125B2 (en) | 2004-01-09 | 2010-09-07 | United Technologies Corporation | Extended impingement cooling device and method |
| US7977331B1 (en) | 2004-02-24 | 2011-07-12 | Japan Tobacco Inc. | Tetracyclic fused heterocyclic compound and use thereof as HCV polymerase inhibitor |
| US7348425B2 (en) | 2004-08-09 | 2008-03-25 | Bristol-Myers Squibb Company | Inhibitors of HCV replication |
| US7795247B2 (en) | 2004-10-26 | 2010-09-14 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Tetracyclic indole derivatives as antiviral agents |
| US7662809B2 (en) | 2004-10-26 | 2010-02-16 | Istituto Di Richerche Di Biologia Molecolare P Angeletti Spa | Tetracyclic indole derivatives as antiviral agents |
| US8278322B2 (en) | 2005-08-01 | 2012-10-02 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| US7399758B2 (en) | 2005-09-12 | 2008-07-15 | Meanwell Nicholas A | Cyclopropyl fused indolobenzazepine HCV NS5B inhibitors |
| US7485633B2 (en) | 2005-09-12 | 2009-02-03 | Bristol-Myers Squibb Company | Cyclopropyl fused indolobenzazepine HCV NS5B inhibitors |
| US7473688B2 (en) | 2005-09-13 | 2009-01-06 | Bristol-Myers Squibb Company | Indolobenzazepine HCV NS5B inhibitors |
| WO2007033175A1 (en) * | 2005-09-13 | 2007-03-22 | Bristol-Myers Squibb Company | Indolobenzazepine hcv ns5b inhibitors |
| WO2007054741A1 (en) * | 2005-11-10 | 2007-05-18 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Tetracyclic indole derivatives as antiviral agents |
| WO2007092000A1 (en) * | 2006-02-06 | 2007-08-16 | Bristol-Myers Squibb Company | Inhibitors of hcv replication |
| US7456165B2 (en) | 2006-02-08 | 2008-11-25 | Bristol-Myers Squibb Company | HCV NS5B inhibitors |
| JP2009536189A (en) * | 2006-05-08 | 2009-10-08 | イステイチユート・デイ・リチエルケ・デイ・ビオロジア・モレコラーレ・ピ・アンジエレツテイ・エツセ・ピー・アー | Pentacyclic indole derivatives as antiviral agents |
| AU2007246850B2 (en) * | 2006-05-08 | 2012-07-05 | Istituto Di Richerche Di Biologia Molecolare P. Angeletti Spa | Pentacyclic indole derivatives as antiviral agents |
| WO2007129119A1 (en) * | 2006-05-08 | 2007-11-15 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Pentacyclic indole derivatives as antiviral agents |
| US8232390B2 (en) | 2006-05-08 | 2012-07-31 | Istituto Di Richerche Di Biologia Molecolare P. Angeletti Spa | Pentacyclic indole derivatives as antiviral agents |
| US8178520B2 (en) | 2006-05-15 | 2012-05-15 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Macrocyclic compounds as antiviral agents |
| AU2007253998B2 (en) * | 2006-05-17 | 2012-03-15 | Bristol-Myers Squibb Holdings Ireland Unlimited Company | Cyclopropyl fused indolobenzazepine HCV NS5B inhibitors |
| JP2009537557A (en) * | 2006-05-17 | 2009-10-29 | ブリストル−マイヤーズ スクイブ カンパニー | Cyclopropyl condensed indolobenzazepine HCVNS5B inhibitor |
| KR101417145B1 (en) * | 2006-05-17 | 2014-07-08 | 브리스톨-마이어스 스큅 컴퍼니 | Cyclopropyl fused indolobenzazepine hcv ns5b inhibitors |
| CN101443336B (en) * | 2006-05-17 | 2012-07-04 | 百时美施贵宝公司 | Cyclopropyl fused indolobenzazepine HVC ns5b inhibitors |
| US7521443B2 (en) | 2006-05-17 | 2009-04-21 | Bristol-Myers Squibb Company | Cyclopropyl fused indolobenzazepine HCV NS5B inhibitors |
| US7456166B2 (en) | 2006-05-17 | 2008-11-25 | Bristol-Myers Squibb Company | Cyclopropyl fused indolobenzazepine HCV NS5B inhibitors |
| EA015286B1 (en) * | 2006-05-17 | 2011-06-30 | Бристол-Маерс Сквибб Компани | Cyclopropyl fused indolobenzazepine hcv ns5b inhibitors |
| WO2007136982A1 (en) * | 2006-05-17 | 2007-11-29 | Bristol-Myers Squibb Company | Cyclopropyl fused indolobenzazepine hcv ns5b inhibitors |
| NO341663B1 (en) * | 2006-05-17 | 2017-12-18 | Bristol Myers Squibb Holdings Ireland | Cyclopropyl-condensed indolobenzzaepine HCV NS5B inhibitors |
| TWI401244B (en) * | 2006-05-17 | 2013-07-11 | 必治妥美雅史谷比公司 | Cyclopropyl fused indolobenzazepine hcv ns5b inhibitors |
| US7521441B2 (en) | 2006-05-22 | 2009-04-21 | Bristol-Myers Squibb Company | Cyclopropyl fused indolobenzazepine HCV NS5B inhibitors |
| WO2007140109A1 (en) * | 2006-05-22 | 2007-12-06 | Bristol-Myers Squibb Company | Cyclopropyl fused indolobenzazepine hcv ns5b inhibitors |
| WO2007140254A2 (en) * | 2006-05-25 | 2007-12-06 | Bristol-Myers Squibb Company | Cyclopropyl fused indolobenzazepine hcv ns5b inhibitors |
| US7456167B2 (en) | 2006-05-25 | 2008-11-25 | Bristol-Myers Squibb Company | Cyclopropyl fused indolobenzazepine HCV NS5B inhibitors |
| WO2007140254A3 (en) * | 2006-05-25 | 2008-01-24 | Bristol Myers Squibb Co | Cyclopropyl fused indolobenzazepine hcv ns5b inhibitors |
| US7521442B2 (en) | 2006-05-25 | 2009-04-21 | Bristol-Myers Squibb Company | Cyclopropyl fused indolobenzazepine HCV NS5B inhibitors |
| CN101490054B (en) * | 2006-05-25 | 2012-05-16 | 百时美施贵宝公司 | Cyclopropyl fused indolobenzazepine hcv ns5b inhibitors |
| JP2009538347A (en) * | 2006-05-25 | 2009-11-05 | ブリストル−マイヤーズ スクイブ カンパニー | Cyclopropyl condensed indolobenzazepine HCVNS5B inhibitor |
| US7452876B2 (en) | 2006-06-08 | 2008-11-18 | Bristol-Myers Squibb Company | Cyclopropyl fused indolobenzazepine HCV NS5B inhibitors |
| US8314062B2 (en) | 2006-06-23 | 2012-11-20 | Instituto Di Ricerche Di Biologia Molecolare P. Angeletti S.P.A. | Macrocyclic compounds as antiviral agents |
| WO2008011521A2 (en) * | 2006-07-20 | 2008-01-24 | Genelabs Technologies, Inc. | Polycyclic viral inhibitors |
| WO2008011521A3 (en) * | 2006-07-20 | 2008-06-26 | Genelabs Tech Inc | Polycyclic viral inhibitors |
| US8138164B2 (en) | 2006-10-24 | 2012-03-20 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| US8309540B2 (en) | 2006-10-24 | 2012-11-13 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| US8377873B2 (en) | 2006-10-24 | 2013-02-19 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| US9738661B2 (en) | 2006-10-27 | 2017-08-22 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| US8377874B2 (en) | 2006-10-27 | 2013-02-19 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| WO2008075103A1 (en) * | 2006-12-20 | 2008-06-26 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Antiviral indoles |
| US8101595B2 (en) | 2006-12-20 | 2012-01-24 | Istituto di Ricerche di Biologia Molecolare P. Angletti SpA | Antiviral indoles |
| JP2010513450A (en) * | 2006-12-20 | 2010-04-30 | イステイチユート・デイ・リチエルケ・デイ・ビオロジア・モレコラーレ・ピ・アンジエレツテイ・エツセ・ピー・アー | Antiviral indole |
| AU2007335962B2 (en) * | 2006-12-20 | 2012-09-06 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Antiviral indoles |
| CN103224506A (en) * | 2006-12-20 | 2013-07-31 | P.安杰莱蒂分子生物学研究所 | Antiviral indoles |
| US7767660B2 (en) | 2006-12-20 | 2010-08-03 | Istituto Di Richerche Di Biologia Molecolare P. Angeletti Spa | Antiviral indoles |
| US7781422B2 (en) | 2006-12-20 | 2010-08-24 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Antiviral indoles |
| US8268803B2 (en) | 2006-12-22 | 2012-09-18 | Merck Sharp & Dohme Corp. | 5, 6-ring annulated indole derivatives and use thereof |
| US8546420B2 (en) | 2006-12-22 | 2013-10-01 | Merck Sharp & Dohme Corp. | 4, 5-ring annulated indole derivatives for treating or preventing of HCV and related viral infections |
| WO2008136815A3 (en) * | 2006-12-22 | 2009-05-22 | Schering Corp | 5, 6-ring annulated indole derivatives and use thereof |
| WO2008136815A2 (en) * | 2006-12-22 | 2008-11-13 | Schering Corporation | 5, 6-ring annulated indole derivatives and use thereof |
| US8557848B2 (en) | 2006-12-22 | 2013-10-15 | Merck Sharp & Dohme Corp. | 4,5-ring annulated indole derivatives for treating or preventing of HCV and related viral infections |
| US7541351B2 (en) | 2007-01-11 | 2009-06-02 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis C |
| US7541352B2 (en) | 2007-02-02 | 2009-06-02 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis C |
| WO2008099020A1 (en) * | 2007-02-16 | 2008-08-21 | Tibotec Pharmaceuticals Ltd. | 1,1-dioxo-1-thia-5,10-diazadibenzocycloheptenes useful as hepatitis c virus inhibitors |
| WO2008099019A1 (en) * | 2007-02-16 | 2008-08-21 | Tibotec Pharmaceuticals Ltd. | 6-hydroxy-dibenzodiazepinones useful as hepatitis c virus inhibitors |
| US7517872B2 (en) | 2007-02-22 | 2009-04-14 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis C |
| US7998951B2 (en) | 2007-03-05 | 2011-08-16 | Bristol-Myers Squibb Company | HCV NS5B inhibitors |
| EP2481807A2 (en) | 2007-03-09 | 2012-08-01 | Merck Sharp & Dohme Corporation | In vivo HCV resistance to anti-viral inhibitors |
| WO2008111978A1 (en) | 2007-03-13 | 2008-09-18 | Bristol-Myers Squibb Company | Cyclopropyl fused indolobenzazepine hcv ns5b inhibitors |
| JP2010521454A (en) * | 2007-03-13 | 2010-06-24 | ブリストル−マイヤーズ スクイブ カンパニー | Cyclopropyl condensed indolobenzazepine HCVNS5B inhibitor |
| CN101679437B (en) * | 2007-03-13 | 2013-04-17 | 百时美施贵宝公司 | Cyclopropyl fused indolobenzazepine hcv ns5b inhibitors |
| CN101679442B (en) * | 2007-03-14 | 2013-02-20 | 百时美施贵宝公司 | Compounds for the treatment of hepatitis c |
| WO2008112841A1 (en) * | 2007-03-14 | 2008-09-18 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis c |
| EA016614B1 (en) * | 2007-03-14 | 2012-06-29 | Бристол-Маерс Сквибб Компани | Compounds for the treatment of hepatitis c |
| US7538102B2 (en) | 2007-03-14 | 2009-05-26 | Bristol-Myers Squibb Company | Compounds for the treatment of Hepatitis C |
| JP2010521482A (en) * | 2007-03-14 | 2010-06-24 | ブリストル−マイヤーズ スクイブ カンパニー | Compounds for the treatment of hepatitis C |
| WO2008112848A1 (en) | 2007-03-14 | 2008-09-18 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis c |
| US7541353B2 (en) | 2007-03-14 | 2009-06-02 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis C |
| US7547690B2 (en) | 2007-03-14 | 2009-06-16 | Bristol-Myers Squibb Company | Compounds for the treatment of Hepatitis C |
| JP2010521476A (en) * | 2007-03-14 | 2010-06-24 | ブリストル−マイヤーズ スクイブ カンパニー | Compounds for the treatment of hepatitis C |
| US7521444B2 (en) | 2007-03-14 | 2009-04-21 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis C |
| WO2008112473A1 (en) * | 2007-03-14 | 2008-09-18 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis c |
| US7538103B2 (en) | 2007-03-15 | 2009-05-26 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis C |
| EP2494991A1 (en) | 2007-05-04 | 2012-09-05 | Vertex Pharmaceuticals Incorporated | Combination therapy for the treatment of HCV infection |
| GB2451184A (en) * | 2007-07-17 | 2009-01-21 | Angeletti P Ist Richerche Bio | Macrocyclic hepatitis C virus NS3 protease inhibitors having a 6-(N-sulphamoyl(carbamoyl))-[7-aza-]indole moiety within the macrocyclic ring path |
| GB2451184B (en) * | 2007-07-17 | 2011-09-21 | Angeletti P Ist Richerche Bio | Novel anti-viral macrocyclic indole compounds |
| US7989438B2 (en) | 2007-07-17 | 2011-08-02 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti Spa | Therapeutic compounds |
| US8927569B2 (en) | 2007-07-19 | 2015-01-06 | Merck Sharp & Dohme Corp. | Macrocyclic compounds as antiviral agents |
| WO2009023487A1 (en) | 2007-08-09 | 2009-02-19 | Bristol-Myers Squibb Company | Tetracyclic compounds for the treatment of hepatitis c |
| US7652004B2 (en) | 2007-08-09 | 2010-01-26 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis C |
| US7642251B2 (en) | 2007-08-09 | 2010-01-05 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis C |
| US8143243B2 (en) | 2007-08-09 | 2012-03-27 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis C |
| JP2010535799A (en) * | 2007-08-09 | 2010-11-25 | ブリストル−マイヤーズ スクイブ カンパニー | Tetracyclic compounds for treating hepatitis C |
| US8614229B2 (en) | 2007-08-29 | 2013-12-24 | Merck Sharp & Dohme Corp. | Substituted indole derivatives and methods of use thereof |
| US8143305B2 (en) | 2007-08-29 | 2012-03-27 | Schering Corporation | 2,3-substituted indole derivatives for treating viral infections |
| US8404845B2 (en) | 2007-08-29 | 2013-03-26 | Merck Sharp & Dohme Corp. | 2,3-substituted azaindole derivatives for treating viral infections |
| US8765757B2 (en) | 2007-11-16 | 2014-07-01 | Merck Sharp & Dohme Corp. | 3-heterocyclic substituted indole derivatives and methods of use thereof |
| US8377928B2 (en) | 2007-11-16 | 2013-02-19 | Merck Sharp & Dohme Corp. | 3-aminosulfonyl substituted indole derivatives and methods of use thereof |
| EP2518073A1 (en) * | 2007-11-21 | 2012-10-31 | Bristol-Myers Squibb Company | Compounds for the treatment of Hepatitis C |
| WO2009067481A1 (en) * | 2007-11-21 | 2009-05-28 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis c |
| US8129367B2 (en) | 2007-11-21 | 2012-03-06 | Bristol-Myers Squibb Company | Compounds for the treatment of Hepatitis C |
| US8124601B2 (en) | 2007-11-21 | 2012-02-28 | Bristol-Myers Squibb Company | Compounds for the treatment of Hepatitis C |
| WO2009076747A1 (en) | 2007-12-19 | 2009-06-25 | Boehringer Ingelheim International Gmbh | Viral polymerase inhibitors |
| WO2009086139A1 (en) * | 2007-12-21 | 2009-07-09 | Genelabs Technologies, Inc. | Condensed pentacyclic derivatives for use in the treatment of flaviviridae infections |
| US8138171B2 (en) | 2008-03-27 | 2012-03-20 | Bristol-Myers Squibb Company | Dioxolane and dioxolanone fused indolobenzadiazepine HCV NS5B inhibitors |
| US8431568B2 (en) | 2008-03-27 | 2013-04-30 | Bristol-Myers Squibb Company | Aromatic heterocyclic fused indolobenzadiazepine HCV NS5B inhibitors |
| US8119628B2 (en) | 2008-03-27 | 2012-02-21 | Bristol-Myers Squibb Company | Pyrrolidine fused indolobenzadiazepine HCV NS5B inhibitors |
| US8178523B2 (en) | 2008-03-27 | 2012-05-15 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis C |
| US8461107B2 (en) | 2008-04-28 | 2013-06-11 | Merck Sharp & Dohme Corp. | HCV NS3 protease inhibitors |
| US8133884B2 (en) | 2008-05-06 | 2012-03-13 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis C |
| US9090671B2 (en) | 2008-06-06 | 2015-07-28 | Scynexis, Inc. | Macrocyclic peptides |
| US8901139B2 (en) | 2008-06-13 | 2014-12-02 | Merck Sharp & Dohme Corp. | Tricyclic indole derivatives and methods of use thereof |
| US7973040B2 (en) | 2008-07-22 | 2011-07-05 | Merck Sharp & Dohme Corp. | Macrocyclic quinoxaline compounds as HCV NS3 protease inhibitors |
| US8080654B2 (en) | 2008-07-22 | 2011-12-20 | Insituto di Ricerche di Biologia Molecolare P. Angeletti SpA | Macrocyclic quinoxaline compounds as HCV NS3 protease inhibitors |
| WO2010080874A1 (en) | 2009-01-07 | 2010-07-15 | Scynexis, Inc. | Cyclosporine derivative for use in the treatment of hcv and hiv infection |
| WO2010082050A1 (en) | 2009-01-16 | 2010-07-22 | Istituto Di Ricerche Di Biologia Molecolare P. Angeletti S.P.A. | Macrocyclic and 7-aminoalkyl-substituted benzoxazocines for treatment of hepatitis c infections |
| US8143244B2 (en) | 2009-02-26 | 2012-03-27 | Bristol-Myers Squibb Company | Cyclopropyl fused indolobenzazepine HCV NS5B inhibitors |
| WO2012041227A1 (en) * | 2010-09-29 | 2012-04-05 | Merck Sharp & Dohme Corp. | Tetracyclic heterocycle compounds for treating hepatitis c viral infection |
| WO2012092484A2 (en) | 2010-12-29 | 2012-07-05 | Inhibitex, Inc. | Substituted purine nucleosides, phosphoroamidate and phosphorodiamidate derivatives for treatment of viral infections |
| US11234977B2 (en) | 2017-12-20 | 2022-02-01 | Novartis Ag | Fused tricyclic pyrazolo-dihydropyrazinyl-pyridone compounds as antivirals |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2006046030A3 (en) | 2006-07-06 |
| AU2005298403A1 (en) | 2006-05-04 |
| RU2007119562A (en) | 2008-12-10 |
| CA2585113A1 (en) | 2006-05-04 |
| AU2005298412B2 (en) | 2011-06-09 |
| JP2008517987A (en) | 2008-05-29 |
| WO2006046039A3 (en) | 2006-07-06 |
| BRPI0516972A (en) | 2008-09-30 |
| IL182610A0 (en) | 2007-07-24 |
| TW200630344A (en) | 2006-09-01 |
| NI200700102A (en) | 2007-10-18 |
| AR051469A1 (en) | 2007-01-17 |
| EP1807397A2 (en) | 2007-07-18 |
| MA29037B1 (en) | 2007-11-01 |
| KR20070068427A (en) | 2007-06-29 |
| US7662809B2 (en) | 2010-02-16 |
| CA2585084A1 (en) | 2006-05-04 |
| EP1807403A2 (en) | 2007-07-18 |
| CR9069A (en) | 2007-10-01 |
| ECSP077412A (en) | 2007-05-30 |
| AU2005298412A1 (en) | 2006-05-04 |
| WO2006046039A2 (en) | 2006-05-04 |
| JP2008517986A (en) | 2008-05-29 |
| US20080261938A1 (en) | 2008-10-23 |
| MX2007004979A (en) | 2007-06-14 |
| PE20060607A1 (en) | 2006-07-14 |
| NO20072689L (en) | 2007-07-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1807403A2 (en) | Tetracyclic indole derivatives as antiviral agents | |
| US7795247B2 (en) | Tetracyclic indole derivatives as antiviral agents | |
| US8101595B2 (en) | Antiviral indoles | |
| JP5276582B2 (en) | Pentacyclic indole derivatives as antiviral agents | |
| US7973026B2 (en) | Thienopyrroles as antiviral agents | |
| WO2007029029A2 (en) | Tetracyclic indole derivatives as antiviral agents | |
| US20090136449A1 (en) | Tetracyclic Indole Derivatives as Antiviral Agents | |
| US20070167447A1 (en) | Indole acetamides as inhibitors of the hepatitis c virus ns5b polymerase | |
| US7767660B2 (en) | Antiviral indoles | |
| US20080153895A1 (en) | Antiviral indoles |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BW BY BZ CA CH CN CO CR CU CZ DK DM DZ EC EE EG ES FI GB GD GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV LY MD MG MK MN MW MX MZ NA NG NO NZ OM PG PH PL PT RO RU SC SD SG SK SL SM SY TJ TM TN TR TT TZ UG US UZ VC VN YU ZA ZM |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GM KE LS MW MZ NA SD SZ TZ UG ZM ZW AM AZ BY KG MD RU TJ TM AT BE BG CH CY DE DK EE ES FI FR GB GR HU IE IS IT LU LV MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 554162 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005298403 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1200700727 Country of ref document: VN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 182610 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: CR2007-009069 Country of ref document: CR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2585084 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005798379 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/a/2007/004979 Country of ref document: MX Ref document number: 2007538502 Country of ref document: JP Ref document number: 200580036540.4 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12007500905 Country of ref document: PH Ref document number: 1020077009541 Country of ref document: KR |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 3791/DELNP/2007 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: DZP2007000315 Country of ref document: DZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007119562 Country of ref document: RU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005798379 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: PI0516972 Country of ref document: BR |