WO2006044174A2 - Aryl substituted 8-azabicyclo[3.2.1]octane compounds as ligands of the melanin concentrating hormone receptor - Google Patents
Aryl substituted 8-azabicyclo[3.2.1]octane compounds as ligands of the melanin concentrating hormone receptor Download PDFInfo
- Publication number
- WO2006044174A2 WO2006044174A2 PCT/US2005/035457 US2005035457W WO2006044174A2 WO 2006044174 A2 WO2006044174 A2 WO 2006044174A2 US 2005035457 W US2005035457 W US 2005035457W WO 2006044174 A2 WO2006044174 A2 WO 2006044174A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- compound
- alkoxy
- salt
- hydroxy
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC(C(*)(*)*(C(CC1)C2)C1CC2(*)C(*)=NC)=C*=*C=I Chemical compound CC(C(*)(*)*(C(CC1)C2)C1CC2(*)C(*)=NC)=C*=*C=I 0.000 description 3
- JJHRUGVMUFXOHS-UHFFFAOYSA-M CCN(C(CC1)C2)C1CC2(O)[AlH]I Chemical compound CCN(C(CC1)C2)C1CC2(O)[AlH]I JJHRUGVMUFXOHS-UHFFFAOYSA-M 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D451/00—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof
- C07D451/02—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof containing not further condensed 8-azabicyclo [3.2.1] octane or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane; Cyclic acetals thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/22—Anxiolytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D451/00—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof
- C07D451/02—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof containing not further condensed 8-azabicyclo [3.2.1] octane or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane; Cyclic acetals thereof
- C07D451/04—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof containing not further condensed 8-azabicyclo [3.2.1] octane or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane; Cyclic acetals thereof with hetero atoms directly attached in position 3 of the 8-azabicyclo [3.2.1] octane or in position 7 of the 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D451/00—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof
- C07D451/02—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof containing not further condensed 8-azabicyclo [3.2.1] octane or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane; Cyclic acetals thereof
- C07D451/04—Heterocyclic compounds containing 8-azabicyclo [3.2.1] octane, 9-azabicyclo [3.3.1] nonane, or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane or granatane alkaloids, scopolamine; Cyclic acetals thereof containing not further condensed 8-azabicyclo [3.2.1] octane or 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring systems, e.g. tropane; Cyclic acetals thereof with hetero atoms directly attached in position 3 of the 8-azabicyclo [3.2.1] octane or in position 7 of the 3-oxa-9-azatricyclo [3.3.1.0<2,4>] nonane ring system
- C07D451/06—Oxygen atoms
Definitions
- Aryl substituted 8-azabicyclo[3.2.1]octane compounds and analogues thereof that are melanin concentrating hormone receptor modulators are provided herein. Methods for using such compounds for treating a variety of metabolic, eating and sexual disorders, and as probes for the detection and localization of MCH receptors are also described.
- MCH Melanin concentrating hormone
- mice having the ob/ob genotype exhibit a 50-80% increase in MCH mRNA expression as compared to leaner ob/+ genotype mice, and prepro-MCH knockout mice, as well as MCH receptor knockout mice, are leaner than normal mice, due to hypophagia and an increased metabolic rate.
- MCH activity is mediated via binding to specific receptors.
- G protein-coupled receptors e.g., neuropeptide Y (NPY) and beta-adrenergic receptors
- MCH receptors are membrane- spanning proteins, generally found on cell surfaces, that consist of a single contiguous amino acid chain comprising an extracellular N-terminal domain, seven membrane-spanning alpha helical domains (connected by three intracellular loop domains alternating with three extracellular loop domains), and an intracellular C-terminal domain.
- Signal transduction is typically initiated by the binding of extracellular MCH to the receptor. This elicits conformational changes in the extracellular domains.
- MCHlR Human Melanin Concentrating Hormone Receptor- 1
- MCHlR expression is found in olfactory tubercle, cerebral cortex, substantia nigra, basal forebrain CAl, CA2, and CA3 fields of the hippocampus, amygdala, and in nuclei of the hypothalamus, thalamus, midbrain and hindbrain. Strong signals are observed in the ventromedial and dorsomedial nuclei of the hypothalamus, two areas of the brain involved in feeding behavior. Upon binding MCH, MCHlR recombinantly expressed in HEK 293 cells mediates a dose dependent release of intracellular calcium.
- MCHlR Cells expressing MCHlR also exhibit a pertussis toxin sensitive dose-dependent inhibition of forskolin-elevated cyclic AMP, indicating that the receptor couples to a Gj/ o G-protein alpha subunit.
- MCH2R A second MCH receptor (designated MCH2R) has also been identified.
- MCH2R has an overall amino acid identity of more than 30% with MCHlR, and is detected specifically in the same regions of the brain as MCHlR.
- Monkey and canine MCH2R sequences, as well as various chimeric MCH2R proteins, have been disclosed in U.S. Patent Application Serial Number 10/291,990 (published as 2003/0148457 on August 7, 2003).
- Agents capable of modulating MCH receptor activity are highly desirable for the treatment of a variety of diseases and disorders, including obesity, eating disorders (e.g., bulimia and anorexia), sexual disorders (e.g., anorgasmic or psychogenic impotence) and metabolic disorders, such as diabetes.
- eating disorders e.g., bulimia and anorexia
- sexual disorders e.g., anorgasmic or psychogenic impotence
- metabolic disorders such as diabetes.
- Small molecule, non-peptide antagonists of MCH receptors would be of particular value for such therapies.
- the present invention fulfills this need, and provides further related advantages.
- the present invention provides aryl substituted 8-azabicyclo[3.2.1]octane compounds and analogues thereof of Formula I and pharmaceutically acceptable salts thereof.
- W is absent or CR 5 R 6 , wherein R 5 and R 5 are independently hydrogen, halogen, hydroxy, Q- C 4 alkyl, Ci-C 4 alkoxy, C 2 -C 4 alkenyl, or haloCi-C 2 alkyl, or R 5 and R 6 are taken together to form an oxo group; Yi, Y 2 , Y 3 , Y 4 and Y 5 are each nitrogen or CRi, wherein no more than 3 of Yi, Y 2 , Y 3 , Y 4 , and Y 5 are nitrogen.
- Each Ri is independently:
- C 8 alkylether ammoCi-C 6 alkyl, mono- or di-(Ci-C 6 alkyl)ammoC 0 -C 6 alkyl, mono- or di-Q-
- any two adjacent Ri may be joined to form a fused 5- or 6-membered carbocycle or heterocycle, each of which is substituted with 0 to 3 substituents independently chosen from halogen, hydroxy, nitro, cyano, amino, Ci-C 4 alkyl, Ci-C 4 alkoxy, haloCi-C 4 alkyl, and haloCi ⁇ C 4 alkoxy.
- substituents independently chosen from halogen, hydroxy, nitro, cyano, amino, Ci-C 4 alkyl, Ci-C 4 alkoxy, haloCi-C 4 alkyl, and haloCi ⁇ C 4 alkoxy.
- R 2 and R 3 are independently hydrogen, halogen, hydroxy, Ci-C 4 alkyl, C ! -C 4 alkoxy, C 2 - C 4 alkenyl, or haloC r C 2 alkyl; or R 2 and R 3 are taken together to form an oxo group; or R 3 is taken together with R 9 to form a fused 5- to 10-membered carbocycle or heterocycle.
- R 4 is hydrogen, halogen, hydroxy, amino, Ci-C 4 alkyl, Ci-C 4 alkoxy, mono- or di-Q- C 4 alkylamino, -NHCHO, C 2 -C 4 alkanoylamino, haloCi-C 2 alkyl, or haloQ-Qalkoxy.
- P is nitrogen or CR 7 .
- Q is nitrogen or CRg.
- U is nitrogen or CR 9 .
- T is nitrogen or CRi 0 .
- X is nitrogen or CRn.
- R 7 is: (i) hydrogen, halogen, hydroxy, nitro, cyano, -COOH, or a group of the formula -L-M; or (ii) taken together with R 8 to form a fused 5- or 6-membered carbocycle or heterocycle.
- R 8 is:
- R 9 is: (i) hydrogen, halogen, hydroxy, nitro, cyano, -COOH, or a group of the formula -L-M; or (ii) taken together with Ri 0 to form a fused 5- to 10-membered carbocycle or heterocycle.
- R 11 is:
- G is C r C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, haloQ-Qalkyl, aminoC r C 6 alkyl, or a 5- to 10- membered cycloalkyl or heterocycloalkyl; each of which is substituted with 0 to 3 substituents independently chosen from halogen, amino, and haloC 1 -C 2 alkoxy, and
- G is further substituted with 0 to 1 substituent chosen from
- each R 13 is independently hydrogen, CrC ⁇ alkyl, C 2 - C 6 alkenyl, C 2 -C 6 alkynyl, or haloCi-C 6 alkyl.
- Each M is independently hydrogen, CrC 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, haloQ-C ⁇ alkyl, aminoCrC ⁇ allcyl, or a 5- to 10-membered cycloalkyl or heterocycloalkyl.
- compounds as described herein are MCH receptor modulators and exhibit a K; of no greater than 1 micromolar, 500 nanomolar, 100 nanomolar, or 10 nanomolar in a
- MCH receptor binding assay and/or have an EC 50 or IC 50 value of no greater than 1 micromolar, 500 nanomolar, 100 nanomolar, or 10 nanomolar in an assay for determining MCH receptor agonist or antagonist activity.
- compounds or salts as described herein are labeled with a detectable marker (e.g., radiolabeled or fluorescein conjugated).
- a detectable marker e.g., radiolabeled or fluorescein conjugated
- compositions comprising at least one compound or salts as described herein (i.e., a compound Formula I or a pharmaceutically acceptable salt thereof) in combination with a physiologically acceptable carrier or excipient.
- a pharmaceutical composition provided herein may further comprise one or more additional active agents (i.e., drugs).
- Pharmaceutical compositions provided herein may be formulated, for example, as an injectable fluid, an aerosol, a cream, a gel, a pill, a capsule, a syrup, or a transdermal patch.
- the present invention further provides, within other aspects, methods for treating a disease or disorder associated with MCH receptor activation, comprising administering to a patient in need of such treatment a therapeutically effective amount of a MCH receptor modulator as described above.
- diseases and disorders include, for example, eating disorders (e.g., obesity and bulimia nervosa), sexual disorders, diabetes, heart disease, and stroke.
- the MCH receptor modulator may be administered orally, or via another means such as intranasally, intravenously, or topically.
- the patient is a human, companion animal, or livestock animal.
- Methods are provided, within other aspects, for determining the presence or absence of MCH receptor in a sample, comprising: contacting a sample with a compound as described above under conditions that permit binding of the compound to MCH receptor; and detecting a level of the compound bound to MCH receptor.
- the compound is radiolabeled
- the step of detection comprises: separating unbound compound from bound compound; and determining an amount of bound compound in the sample. Detection may be achieved, for example, using autoradiography.
- the present invention further provides, within other aspects, methods for modulating binding of ligand to MCH receptor.
- Certain such methods are performed in vitro, and comprise contacting MCH receptor with MCH receptor modulator, as described above under conditions and in an amount sufficient to detectably modulate MCH binding to MCH receptor.
- Other such methods may be performed in vivo, and comprise contacting cells expressing MCH receptor with a compound or modulator as described above in an amount sufficient to detectably modulate MCH binding to cells expressing a cloned MCH receptor in vitro.
- Methods are further provided for modulating binding of MCH to MCH receptor in a patient, comprising administering to a patient (i.e., a human or non-human animal) a compound or modulator as described above.
- Patients include, for example, companion animals such as dogs.
- the present invention provides methods for modulating the signal- transducing activity of MCH receptor, comprising contacting an MCH receptor, either in vivo or in vitro, with an amount of an MCH receptor modulator sufficient to detectably alter MCH receptor activity, under conditions suitable for binding of MCH to MCH receptor.
- the MCH receptor is a MCHlR.
- Packaged pharmaceutical preparations comprising: (a) a pharmaceutical composition as described above in a container; and (b) instructions for using the composition to treat a patient suffering from a disease or disorder associated with MCH receptor activation.
- diseases include, for example eating disorders (e.g., obesity and bulimia nervosa), sexual disorders, diabetes, heart disease, and stroke, are also provided herein.
- the present invention provides aryl substituted 8-azabicyclo[3.2.1]octane compounds and analogues thereof of Formula I.
- Certain preferred compounds are MCH receptor modulators that may be used in vitro or in vivo, to inhibit MCH binding to MCH receptors, activate
- MCH receptors or to otherwise modulate MCH receptor activity in a variety of contexts, as discussed in further detail below.
- TERMINOLOGY Compounds are generally described herein using standard nomenclature. For compounds having asymmetric centers, it should be understood that (unless otherwise specified) all of the optical isomers and mixtures thereof are encompassed. In addition, compounds with carbon-carbon double bonds may occur in Z- and E- forms, with all isomeric forms of the compounds being included unless otherwise specified. Where a compound exists in various tautomeric forms, a recited compound is not limited to any one specific tautomer, but rather is intended to encompass all tautomeric forms. Compound descriptions are intended to encompass compounds with all possible isotopes of atoms occurring in the compounds. Isotopes are those atoms having the same atomic number but different mass numbers.
- isotopes of hydrogen include tritium and deuterium and isotopes of carbon include 11 C, 13 C, and 14 C.
- Certain compounds are described herein using a general formula that includes variables (e.g., R b R 2 , R 3 ). Unless otherwise specified, each variable within such a formula is defined independently of any other variable, and any variable that occurs more than one time in a formula is defined independently at each occurrence.
- the variables e.g. Ri, R 2 , R 3
- aryl substituted 8-azabicyclo[3.2.1]octane compounds and analogues thereof as used herein encompass all compounds that satisfy Formula I, including any enantiomers, racemates, and stereoisomers, as well as all pharmaceutically acceptable salts of such compounds.
- a “pharmaceutically acceptable salt” of a compound recited herein is an acid or base salt that is suitable for use in contact with the tissues of human beings or animals without excessive toxicity carcinogenicity, and preferably without irritation, allergic response, or other problem or complication.
- Such salts include mineral and organic acid salts of basic residues such as amines, as well as alkali or organic salts of acidic residues such as carboxylic acids.
- Specific pharmaceutical salts include, but are not limited to, salts of acids such as hydrochloric, phosphoric, hydrobromic, malic, glycolic, fumaric, sulfuric, sulfamic, sulfanilic, formic, toluenesulfonic, methanesulfonic, benzene sulfonic, ethane disulfonic, 2-hydroxyethylsulfonic, nitric, benzoic, 2-acetoxybenzoic, citric, tartaric, lactic, stearic, salicylic, glutamic, ascorbic, pamoic, succinic, fumaric, maleic, propionic, hydroxymaleic, hydroiodic, phenylacetic, alkanoic such as acetic, HOOC-(CH 2 ) n -COOH where n is 0-4, and the like.
- acids such as hydrochloric, phosphoric, hydrobromic, malic, glycolic, fumaric, sulfuric
- pharmaceutically acceptable cations include, but are not limited to sodium, potassium, calcium, aluminum, lithium, and ammonium.
- pharmaceutically acceptable salts for the compounds provided herein, including those listed by Remington's Pharmaceutical Sciences, 17th ed., Mack Publishing Company, Easton, PA, p. 1418 (1985).
- a pharmaceutically acceptable acid or base salt can be synthesized from a parent compound that contains a basic or acidic moiety by any conventional chemical method.
- such salts can be prepared by reacting the free acid or base forms of these compounds with a stoichiometric amount of the appropriate base or acid in water or in an organic solvent, or in a mixture of the two; generally, the use of nonaqueous media, such as ether, ethyl acetate, ethanol, isopropanol, or acetonitrile, is preferred.
- Pharmaceutically acceptable salts of the aryl substituted 8- azabicyclo[3.2.1]octane compounds and analogues thereof disclosed herein are a preferred compound form.
- each compound of Formula I may, but need not, be formulated as a hydrate, solvate or non-covalent complex.
- the various crystal forms and polymorphs are within the scope of the present invention.
- prodrugs of the compounds of Formula I are also provided herein.
- a "prodrug” is a compound that may not fully satisfy the structural requirements of the compounds provided herein, but is modified in vivo, following administration to a patient, to produce a compound of Formula I.
- a prodrug may be an acylated derivative of a compound as provided herein.
- Prodrugs include compounds wherein hydroxy, amine or sulfhydryl groups are bonded to any group that, when administered to a mammalian subject, cleaves to form a free hydroxyl, amino or sulfhydryl group, respectively.
- Examples of prodrugs include, but are not limited to, acetate, formate, and benzoate derivatives of alcohol and amine functional groups within the compounds provided herein.
- Prodrugs of the compounds provided herein may be prepared by modifying functional groups present in the compounds in such a way that the modifications are cleaved in vivo to yield the parent compounds.
- alkyl refers to a straight chain or branched chain saturated aliphatic hydrocarbon.
- An alkyl group may be bonded to an atom within a molecule of interest via any chemically suitable portion.
- Alkyl groups include groups having from 1 to 8 carbon atoms (C]- C 8 alkyl), from 1 to 6 carbon atoms (Ci-C 6 alkyl), and from 1 to 4 carbon atoms (Ci-C 4 alkyl), such as methyl, ethyl, propyl, isopropyl, n-butyl, sec-butyl, tert-butyl, pentyl, 2-pentyl, isopentyl, neopentyl, hexyl, 2-hexyl, 3-hexyl, and 3-methylpentyl.
- C 0 -C n alkyl refers to a single covalent bond (C 0 ) or an alkyl group having from 1 to n carbon atoms.
- Co-C 6 alkyl refers to a single covalent bond or a Ci-C ⁇ alkyl group.
- alkenyl refers to straight or branched chain alkene groups, in which at least one unsaturated carbon-carbon double bond is present.
- Alkenyl groups include C 2 -C 8 alkenyl, C2- C ⁇ alkenyl, and C2-C4alkenyl groups, which have from 2 to 8, 2 to 6, or 2 to 4 carbon atoms, respectively, such as ethenyl, allyl, or isopropenyl.
- Alkynyl refers to straight or branched chain alkyne groups, which have one or more unsaturated carbon-carbon bonds, at least one of which is a triple bond.
- Alkynyl groups include C 2 -C 3 alkynyl, C 2 -C 6 alkynyl, and C 2 -C 4 alkynyl groups, which have from 2 to 8, 2 to 6, or 2 to 4 carbon atoms, respectively.
- Alkenyl and alkynyl groups may be straight or branched chain.
- alkoxy as used herein, is meant an alkyl group as described above attached via an oxygen bridge.
- Alkoxy groups include Ci-C 8 alkoxy, Ci-C 6 alkoxy, and Ci-C 4 alkoxy groups, which have from 1 to 8, 1 to 6, or 1 to 4 carbon atoms, respectively.
- Alkoxy groups include, for example, methoxy, ethoxy, propoxy, isopropoxy, n-butoxy, sec-butoxy, tert-butoxy, n-pentoxy, 2-pentoxy, 3- pentoxy, isopentoxy, neopentoxy, hexoxy, 2-hexoxy, 3-hexoxy, and 3-methylpentoxy.
- alkylthio refers to an alkyl group as described above attached via a sulfur bridge
- alkylsufonyl refers to an alkyl groups as described above attached via an -(SO 2 )- bridge.
- alkoxyalkyl is an alkoxy group as described above attached via an oxygen bridge to an alkyl group, as described above, attached via a single covalent bond of the alkyl carbon.
- the alkoxy moiety of the alkoxycarbonyl group has the indicated number of carbon atoms; the carbon of the keto bridge is not included in this number.
- Alkylamino refers to a secondary or tertiary amine having the general structure -NH(alkyl) or -N(alkyl)(alkyl), wherein each alkyl may be the same or different.
- groups include, for example, mono- and di-(C 1 -C 8 alkyl)amino groups, in which each alkyl is straight or branched and may be the same or different and contains the indicated number of carbon atoms, for example from 1 to 8 carbon atoms, from 1 to 6 carbon atoms, or from 1 to 4 carbon atoms, from as well as mono- and di-(Ci-C 6 alkyl)amino groups and mono- and di-(C 1 -C 4 alkyl)amino groups.
- Alkylaminoalkyl refers to an alkylamino group linked via an alkyl group (i.e., a group having the general structure -alkyl- NH-alkyl or -alkyl-N(alkyl)(alkyl)) in which each alkyl is selected independently.
- alkyl group i.e., a group having the general structure -alkyl- NH-alkyl or -alkyl-N(alkyl)(alkyl)
- alkyl is selected independently.
- Such groups include, for example, mono- and di-(Ci-C 6 alkyl)aminoCi- C 6 alkyl and mono- and di-(Ci-C 4 alkyl)aminoCi-C 4 alkyl, in which each alkyl may be the same or different, and is straight or branched.
- “Mono- or di-(C 1 -C 6 alkyl)aminoCo-C6alkyl” refers to a mono- or di-(C r C 6 alkyl)amino group linked via a single covalent bond or a Q-C ⁇ alkyl group.
- Alkyl ether indicates an alkyl group as described herein attached via an oxygen linker to another alkyl group.
- an aromatic moiety is substituted with an oxo group, the aromatic ring is replaced with the corresponding partially unsaturated ring.
- a pyridyl group substituted with oxo is a pyridone.
- the term "mono- and/ or di-alkylsulfonamide" refers to groups of formula
- alkylsulfonyl refers to a substituent of the formula (alkyl)-SO 2 - in which the alkyl group has the indicated number of carbon atoms, and the point of attachment is on the sulfur atom.
- aminoalkyl is an alkyl group as defined above substituted with at least one amino substituent. When indicated an aminoalkyl group may be further substituted with additional amino or non-amino substituent.
- aryl indicates aromatic groups containing only carbon in the aromatic ring or rings. Such aromatic groups may be further substituted with carbon or non-carbon atoms or groups. Typical aryl groups contain 1 or 2 separate, fused, or pendant rings and from 6 to about 12 ring atoms, without heteroatoms as ring members. Where indicated aryl groups may be substituted. Such substitution may include fusion to a 5 to 7-membered saturated cyclic group that optionally contains 1 or 2 heteroatoms independently chosen from N, O, and S, to form, for example, a 3,4-methylenedioxy-phenyl group.
- Aryl groups include, for example, phenyl, naphthyl, including 1- naphthyl and 2-naphthyl, and bi-phenyl.
- (aryl)alkyl aryl and alkyl are as defined above, and the point of attachment is on the alkyl group.
- (phenyl)C 0 -C 2 alkyl indicates a phenyl group indicates a phenyl group that is directly attached via a single covalent bond ((phenyl)C o alkyl) or attached through an alkyl group having from 1 to about 2 carbon atoms.
- an aryl group may be attached through other linker groups, for example included herein are arylCi-C 6 alkanoylamino and (aryl)alkoxy groups, in which aryl carries the definition set forth above, and is attached via the indicated linker group.
- a “carbocycle” has from 1 to 3 fused, pendant, or spiro rings, containing only carbon ring members.
- a heterocyclic ring comprises contains from 3 to 8 ring members (rings having from 4 or 5 to 7 ring members are recited in certain embodiments) and carbocycles comprising fused, pendant, or spiro rings typically contain from 9 to 14 ring members.
- Carbocycles may be optionally substituted with a variety of substituents, as indicated. Unless otherwise specified, a carbocycle may be a cycloalkyl group ⁇ i.e., each ring is saturated), a partially saturated group, or an aryl group ⁇ i.e., at least one ring within the group is aromatic).
- a carbocyclic group may generally be linked via any ring or substituent atom, provided that a stable compound results.
- Certain carbocyclic groups are 4- to 7-membered or 5- to 7-membered groups that are optionally substituted.
- Representative aromatic carbocycles are phenyl, naphthyl and biphenyl.
- preferred carbocycles are carbocycles having a single ring, such as phenyl and 3- to 7- membered cycloalkyl groups.
- a carbocycle may be directly attached or attached via an indicated linker group.
- (carbocycle)alkyl, (carbocycle)alkoxy, and (carbocycle)alkylamino substituents are present in some embodiments described herein.
- “carbocycle” carries the definition set forth above and is covalently bound to the indicated linker group, which carries the definition set forth above.
- a “cycloalkyl” group is a carbocycle as described above, which is fully saturated.
- preferred cycloalkyl groups are 3- to 7-membered cycloalkyl groups having a single saturated ring, e.g. cyclopropyl, cyclopentyl, and cyclohexyl.
- a "cycloalkylC 0 -C n alkyl” is a cycloalkyl group linked via a single covalent bond or a Ci-C n alkyl group, e.g. a Ci-C 4 alkyl group.
- a "cycloalkenyl” group is a 3- to 7- membered carbocycle having at least one carbon-carbon double bond, but which is not fully aromatic, e.g. a cycloalkenyl group.
- halogen refers to fluorine, chlorine, bromine and iodine.
- a “haloalkyl” is a branched or straight-chain alkyl group, substituted with 1 or more halogen atoms ⁇ e.g., "haloQ-C ⁇ alkyl” groups have from 1 to 6 carbon atoms; "haloC 1 -C 4 alkyl” groups have from 1 to 4 carbon atoms).
- haloalkyl groups include, but are not limited to, mono-, di-, or trifluoromethyl; mono-, di-, or trichloromethyl; mono-, di-, tri-, tetra-, or pentafluoroethyl; mono-, di-, tri-, tetra- or pentachloroethyl; and 1, 2,2,2 -tetrafluoro-l-trifluoromethyl-ethyl.
- Typical haloalkyl groups are trifluoromethyl and difluoromethyl.
- Haloalkoxy indicates a haloalkyl group as defined above attached through an oxygen bridge.
- haloCi-C 6 alkoxy groups have from 1 to 6 carbon atoms.
- hydroxyalkyl is an alkyl group as defined herein, having the indicated number of carbon atoms, and substituted with at least one hydroxyl substituent (-OH). When indicated, hydroxyalkyl groups, like other groups described herein, may be additionally substituted.
- a dash (“-") that is not between two letters or symbols is used to indicate a point of attachment for a substituent.
- -CONH 2 is attached through the carbon atom.
- heteroatom is oxygen, sulfur, or nitrogen.
- a “heterocycle” has from 1 to 3 fused, pendant, or spiro rings, at least one of which is a heterocyclic ring ⁇ i.e., one or more ring atoms is a heteroatom, with the remaining ring atoms being carbon).
- a heterocyclic ring comprises 1, 2, 3, or 4 heteroatoms; within certain embodiments each heterocyclic ring has 1 or 2 heteroatoms per ring.
- Each heterocyclic ring generally contains from 3 to 8 ring members (rings having from 4 or 5 to 7 ring members are recited in certain embodiments) and heterocycles comprising fused, pendant, or spiro rings typically contain from 9 to 14 ring members.
- heterocycles comprise a sulfur atom as a ring member; in certain embodiments the sulfur atom is oxidized to SO or SO 2 .
- Heterocycles may be optionally substituted with a variety of substituents, as indicated.
- a heterocycle may be a heterocycloalkyl group (i.e., each ring is saturated), a partially saturated group, or a heteroaryl group (i.e., at least one ring within the group is aromatic).
- a heterocyclic group may generally be linked via any ring or substituent atom, provided that a stable compound results.
- N-linked heterocyclic groups are linked via a component nitrogen atom.
- Certain heterocyclic groups are 4- to 7-membered or 5- to 7-membered groups that are optionally substituted.
- 4-to 7-membered heterocycloalkyl groups include, for example, piperidinyl, piperazinyl, pyrrolidinyl, azepanyl, morpholino, thiomorpholino, and l,l-dioxo-thiomorpholin-4-yl. Such groups may be substituted as indicated.
- Representative aromatic heterocycles are azocinyl, pyridyl, pyrimidyl, imidazolyl, and tetrazolyl.
- preferred heterocycles are 5- to 7-membered heterocycle having a single saturated, partially unsaturated or aromatic heterocyclic ring with 5 to 7 ring members, 1 or 2 ring members independently chosen from N, O, and S, with remaining ring members being carbon.
- a heterocycle may be directly attached or attached via an indicated linker group.
- (heterocycle)alkyl, (heterocycle)alkoxy, and (heterocycle)alkylamino substituents are present in some embodiments described herein.
- “heterocycle” carries the definition set forth above and is covalently bound to the indicated linker group, which carries the definition set forth above.
- heteroaryl indicates a stable 5- to 7-membered monocyclic aromatic ring which contains from 1 to 3, or preferably from 1 to 2, heteroatoms chosen from N, O, and S, with remaining ring atoms being carbon or a stable bicyclic or tricyclic system containing at least one 5- to 7-membered aromatic ring which contains from 1 to 3, or preferably from 1 to 2, heteroatoms chosen from N, O, and S, with remaining ring atoms being carbon.
- the total number of S and O atoms in the heteroaryl group exceeds 1, these heteroatoms are not adjacent to one another. It is preferred that the total number of S and O atoms in the heteroaryl group is not more than 2.
- heteroaryl groups include, but are not limited to, oxazolyl, pyranyl, pyrazinyl, pyrazolopyrimidinyl, pyrazolyl, pyridizinyl, pyridyl, pyrimidinyl, pyrrolyl, quinolinyl, tetrazolyl, thiazolyl, thienylpyrazolyl, thiophenyl, triazolyl, benzo[d]oxazolyl, benzofuranyl, benzothiazolyl, benzothiophenyl, benzoxadiazolyl, dihydrobenzodioxynyl, furanyl, imidazolyl, indolyl, and isoxazolyl.
- a “heterocyclolalkyl” group is a heterocycle as described above, which is fully saturated.
- preferred heterocycloalkyl groups have a single saturated ring with 5 to 7 ring members, 1 or 2 ring members independently chosen from N, O, and S, with remaining ring members being carbon.
- a “heterocycloalkylC 0 -C n alkyl” is a heterocycloalkyl group linked via a single covalent bond or C r C n alkyl group, e.g., a Ci-C 4 alkyl group.
- heterocycloalkenyl is a 5- to 7-membered heterocycle having a single ring that contains at least one carbon-carbon, carbon- nitrogen, or nitrogen-nitrogen double bond, but which is not fully aromatic, with 1 or 2 ring members independently chosen from N, O, and S, with remaining ring members being carbon.
- a "substituent,” as used herein, refers to a molecular moiety that is covalently bonded to an atom within a molecule of interest.
- a ring substituent may be a moiety such as a halogen, alkyl group, haloalkyl group or other group discussed herein that is covalently bonded to an atom (preferably a carbon or nitrogen atom) that is a ring member.
- Substituents or aromatic groups are generally covalently bonded to a ring carbon atom.
- substitution refers to replacing a hydrogen atom in a molecular structure with a substituent, such that the valence on the designated atom is not exceeded, and such that a chemically stable compound (i.e., a compound that can be isolated, characterized and tested for biological activity) results from the substitution.
- Groups that are "optionally substituted” are unsubstituted or are substituted by other than hydrogen at one or more available positions, typically 1, 2, 3, 4 or 5 positions, by one or more suitable groups (which may be the same or different).
- Such optional substituents include, for example, hydroxy, halogen, cyano, nitro, Ci-C 8 alkyl, C 2 -C 8 alkenyl, C 2 -C 8 alkynyl, CVCgalkoxy, C 2 -C 8 alkyl ether, C 3 -C 8 alkanone, Ci-C 8 alkylthio, amino, mono- or di ⁇ Q-Csalkytyamino, Ci-C 8 haloalkyl, Q- C 8 haloalkoxy, Cx-Qalkanoyl, C 2 -C 8 alkanoyloxy, Q-Csalkoxycarbonyl, -COOH, -CONH 2 , mono- or di-(Ci-C8alkyl)a
- Optional substitution is also indicated by the phrase "substituted with 0 to X substituents," where X is the maximum number of possible substituents.
- Certain optionally substituted groups are substituted with from 0 to 2, 3 or 4 independently selected substituents ⁇ i.e., are unsubstituted or substituted with up to the recited maximum number of substitutents).
- MCH receptor refers to any naturally-occurring mammalian (especially human, monkey, or canine) MCH type 1 or type 2 receptor, as well as chimeric receptors in which one or more domains of a naturally-occurring MCHlR or MCH2R are replaced with a corresponding domain of a different G protein-coupled receptor, such that the ability of the chimeric receptor to bind MCH and mediate a dose-dependent release of intracellular calcium is not diminished.
- MCH receptors for use within the various assays and other methods described herein include, for example, recombinantly expressed human MCH receptor (e.g., Genbank Accession No. Z86090; SEQ ID NO:29 of U.S.
- MCH receptor modulator also referred to herein as a “modulator,” is a compound that alters (increases or decreases) MCH receptor activation and/or MCH receptor-mediated signal transduction.
- MCH receptor modulators specifically provided herein are aryl-substituted piperazine derivatives.
- a modulator may be a MCH receptor agonist or antagonist.
- a modulator may exhibit an EC 50 or IC 50 at MCH receptor that is less than 1 micromolar, 500 nM, 200 nM, 100 nM, 50 nM, 25 nM or 10 nM in a standard calcium mobilization assay (as described in Example 13, herein) and/or an agonist-stimulated GTP gamma 35 S binding assay (as described in Example 11, herein).
- a modulator may be a MCH receptor agonist or antagonist, although, for certain purposes described herein, a modulator preferably inhibits MCH receptor activation resulting from binding of MCH ⁇ i.e., the modulator is an antagonist).
- a MCH receptor modulator binds with "high affinity” if the K; at a MCH receptor is less than 1 micromolar, preferably less than 500 nanomolar, 100 nanomolar, or 10 nanomolar.
- a modulator binds "specifically" to MCH receptor if it binds to a MCH receptor (total binding minus nonspecific binding) with a K; that is 10-fold, preferably 100-fold, and more preferably 1000-fold, less than the K; measured for modulator binding to other G protein-coupled receptors.
- a modulator may have a IQ of 500 nanomolar or less in an MCH receptor ligand binding assay and a K; of at least 1 micromolar in a dopamine receptor ligand binding assay, such as the assay described in Example 7 (pages 111-112) of PCT International Publication Number WO 02/094799, which is hereby incorporated by reference.
- Representative assays for determining Kj at MCH receptor are provided in Examples 9 and 12, herein.
- a modulator is considered an "antagonist" if it detectably inhibits MCH binding to MCH receptor and/or MCH-mediated signal transduction (using, for example, the representative assay provided in Example 9 or Example 12).
- an antagonist has an IC 50 value of less than 1 micromolar, preferably less than 100 nanomolar, and more preferably less than 10 nanomolar within the assay provided in Example 9 and/ or the assay provided in Example 12.
- MCH receptor antagonists include neutral antagonists and inverse agonists.
- An "inverse agonist” is a compound that reduces the activity of MCH receptor below its basal activity level in the absence of added ligand. Inverse agonists may also inhibit the activity of MCH at MCH receptor, and/or may also inhibit binding of MCH to MCH receptor.
- the ability of a compound to inhibit the binding of MCH to MCH receptor may be measured by a binding assay, such as the binding assay given in Examples 9 or 12.
- the basal activity of MCH receptor, as well as the reduction in MCH receptor activity due to the presence of antagonist may be determined from a calcium mobilization assay, such as the assay of Example 13, or an agonist-stimulated GTP gamma 35 S binding assay, such as the assay described in Example 11.
- a "neutral antagonist" of MCH receptor is a compound that inhibits the activity of MCH at MCH receptor, but does not significantly change the basal activity of the receptor (e.g., within an assay as described in Example 11 or Example 13 performed in the absence of ligand, MCH receptor activity is reduced by no more than 10%, more preferably by no more than 5%, and even more preferably by no more than 2%; most preferably, there is no detectable reduction in activity).
- Neutral antagonists may also inhibit ligand binding to MCH receptor.
- MCH receptor agonist is a compound that elevates the activity of the receptor above the basal activity level of the receptor (i.e., enhances MCH receptor activation and/or MCH receptor-mediated signal transduction).
- MCH receptor agonist activity may be identified using the representative assays provided in Examples 11 and 13. In general, such an agonist has an EC 50 value of less than 1 micromolar, preferably less than 100 nanomolar, and more preferably less than 10 nanomolar within one or both of the assays provided in Examples 11 and 13.
- a “therapeutically effective amount” (or dose) is an amount that, upon administration, is sufficient to provide a discernible patient benefit. For example, a therapeutically effective amount may reduce symptom severity or frequency, or to result in detectable weight loss.
- a therapeutically effective amount may improve patient status or outcome and/or prevent or delay disease or symptom onset.
- a therapeutically effective amount or dose generally results in a concentration of compound in a body fluid (such as blood, plasma, serum, CSF, synovial fluid, lymph, cellular interstitial fluid, tears or urine) that is sufficient to alter the binding of ligand to MCH receptor in vitro (using the assay provided in Example 9 or Example 12) and/or MCH-mediated signal transduction (using an assay provided in Example 11 or Example 13).
- a body fluid such as blood, plasma, serum, CSF, synovial fluid, lymph, cellular interstitial fluid, tears or urine
- a “disease or disorder associated with MCH receptor activation,” as used herein is any condition that is characterized by inappropriate stimulation of MCH receptor, regardless of the amount of MCH present locally, and/or that is responsive to modulation of MCH receptor activity (i.e., the condition or a symptom thereof is alleviated by such modulation).
- Such conditions include, for example, metabolic disorders (such as diabetes), heart disease, stroke, eating disorders (such as obesity and bulimia nervosa) and sexual disorders such as anorgasmic and psychogenic impotence, as well as other diseases and disorders recited herein.
- a "patient” is any individual treated with a MCH modulator as provided herein. Patients include humans, as well as other animals such as companion animals (e.g., dogs and cats) and livestock. Patients may be experiencing one or more symptoms of a condition responsive to MCH receptor modulation, or may be free of such symptom(s) (i.e., treatment may be prophylactic).
- the present invention provides and comprises aryl substituted 8- azabicyclo[3.2.1]octane compounds and analogues thereof of Formula I described above, as well as pharmaceutically acceptable salts thereof,
- W is CR 5 Rg; e.g. compounds and salts of Formula III are provided herein:
- R 5 and R 6 are independently hydrogen or methyl. iv. R 5 and R 6 are taken together to form an oxo group, e.g compounds and salts of Formula IV are provided herein:
- Y 1 , Y 2 , Y 3 , Y 4 and Y 5 are nitrogen.
- One of Y 1 , Y 4 , and Y 5 is nitrogen.
- Y 1 , Y 2 , Y 3 , Y 4 , and Y 5 are all CRi, e.g., compounds and salts of Formula V and Formula VI are provided herein:
- Each R 1 is independently hydrogen, halogen, hydroxy, nitro, cyano, amino, Q- C 4 alkyl, C 2 -C 4 alkenyl, C 1 -C 4 alkoxy, haloC r C 2 alkyl, haloCrQalkoxy, hydroxyQ-Qalkyl, C 1 - C 4 alkylthio, aminoC]-C 6 alkyl, mono- or di-(C 1 -C 4 alkyl)amino, or (C 3 -C 7 cycloalkyl)Co-C 2 alkyl. ix.
- Each R 1 is independently hydrogen, halogen, C r C 2 alkyl, C 2 -C 4 alkenyl, Ci-C 2 alkoxy, haloQ-Qalkyl, or haloC 1 -C 2 alkoxy.
- x. Y 1 , Y 2 , Y 3 , Y 4 , and Y 5 are all CR 1 ; and the R 1 Of Y 1 , Y 4 , and Y 5 are all hydrogen, e.g. compounds and salts of Formula VII and Formula VlH are provided herein:
- R 1 of Y 2 is halogen, methyl, methoxy, or trifluoromethyl
- Ri of Y 3 is hydrogen, halogen, methyl, methoxy, or trifluoromethyl
- xii. R 1 of Y 2 is hydrogen, halogen, methyl, methoxy, or trifluoromethyl
- R 1 of Y 3 is halogen, methyl, methoxy, or trifluoromethyl.
- R 1 OfY 2 is methoxy or trifluoromethyl, and R 1 of Y 3 is chloro.
- xiv. R 2 and R 3 are independently hydrogen or methyl.
- xv. R 2 and R 3 are both hydrogen.
- xvi. R 2 is joined with R 3 to form an oxo group.
- R 4 is hydrogen, hydroxy, amino, -NHCHO, or C 2 alkanoylamino.
- P is nitrogen or CR 7 ;
- Q is CR 8 ;
- U is CR 9 ;
- T is CRi 0 ; and
- X is CRn, e.g. compounds and salts of Fo ⁇ nula DC are provided herein
- P is CR 7 ; Q is nitrogen; U is CR 9 ; T is CR 10 ; and X is CRn, e.g. compounds and salts of Fo ⁇ nula X are provided herein:
- R 7 , R 8 , R 9 , and R 10 are independently hydrogen, halogen, hydroxy, Q-C 6 alkyl, Q- C 6 alkoxy, mono- or di-Q-C 6 alkylamino, haloQ-C 2 alkyl, or haloC r C 2 alkoxy.
- R 7 and R 8 are independently hydrogen, Ci-C 2 alkyl, or Ci-C 2 alkoxy; and R 9 and R 10 are hydrogen.
- R 7 and R 8 are both methyl.
- Rn is a group of the formula -L-G. xxviii.
- G is Ci-C 6 alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, haloQ-C ⁇ alkyl, aminoCi-C 6 alkyl, or a 5- to 10-membered cycloalkyl or heterocycloalkyl; each of which is substituted with 0 to 3 substituents independently chosen from halogen, amino, and haloCi-C 2 alkoxy.
- heterocycle C 0 -C 6 alkoxy, (carbocycle)C 0 -C 6 alkylamino, and (heterocycle)C 0 -C 6 alkylamino, wherein the carbocycle is phenyl, naphthyl, Q-C 7 cycloalkyl, or C 3 -C 7 cycloalkenyl, and the heterocycle is pyrrolidinyl, tefrahydrofuranyl, dioxolanyl, tetrahydropyranyl, isothiazolidinyl, piperidinyl, piperazinyl, morpholinyl, thiomorpholinyl, py ⁇ -olyl, dihydropyrrolyl, pyrazolyl, furanyl, thienyl, pyrazolyl, oxazolyl, thiazolyl, thiadiazolyl, isoxazolyl, imidiazolyl, triazolyl
- G is Q-C 6 alkyl substituted with at least one substituent independently chosen from (carbocycle)C 0 -C 6 alkyl and (heterocycle)Co-C 6 alkyl, wherein the carbocycle is phenyl or C 3 - C 7 cycloalkyl, and the heterocycle is pyrrolidinyl, tetrahydrofuranyl, piperidinyl, piperazinyl, morpholinyl, thiomorpholinyl, pyrrolyl, pyrazolyl, furanyl, thienyl, imidiazolyl, pyridinyl, pyrimidinyl, or pyrazinyl, each of which (carbocycle)C 0 -C 6 alkyl and (heterocycle)C 0 -C 6 alkyl is substituted with from O to 3 substituents independently chosen from halogen, hydroxy, oxo, Q- C 2 alkoxy, mono- and di-Ci-
- xxxiv. Rn is taken together with R 8 to form a fused carbocycle or heterocycle; which is substituted with O to 3 substituents independently chosen from halogen, amino, cyano, hydroxy, oxo, Q-C 6 alkyl, (Ci-C 6 alkoxy)C 0 -C 6 alkoxy, mono- and di-(Q-C 6 alkyl)aminoCo-C 6 alkyl, C 2 -C 4 alkanoyl, C 3 -Qcycloalkyl, Q-C 4 alkoxycarbonyl, haloQ-C 2 alkyl, and haloQ-C 2 alkoxy.
- substituents independently chosen from halogen, amino, cyano, hydroxy, oxo, Q-C 6 alkyl, (Ci-C 6 alkoxy)C 0 -C 6 alkoxy, mono- and di-(Q-C 6 alkyl)aminoCo-C 6 alkyl, C 2 -
- xxxv Ri i is taken together with R 8 to form a fused 5 or 6 membered heterocycloalkyl ⁇ ng having 1 or 2 oxygen atoms; which is substituted with 0 to 2 substituents independently chosen from halogen, hydroxy, methyl, and methoxy xxxvi.
- Rn is taken together with R 8 to form a fused 5 or 6 membered heterocycloalkyl ⁇ ng having 1 or 2 oxygen atoms; which is substituted with 0 to 2 substituents independently chosen from halogen, hydroxy, methyl, and methoxy and R 7 , R 9 , and R] 0 are independently hydrogen or methyl
- Certain aryl substituted 8-azabicyclo[3 2 l]octane compounds and analogues thereof provided herein detectably alter (modulate) MCH binding to MCHRl and/or MCHR2 receptor, as determined using a standard in vitro MCH receptor binding assay and/or calcium mobilization assay.
- MCH receptor ligand binding assay refer to either of the standard in vitro receptor binding assay provided in Examples 9 and 12. Withm such assays, the receptor is incubated with labeled MCH (or other suitable ligand) and a test compound.
- a test compound that detectably modulates binding of ligand to MCH receptor will result in a decrease or increase in the amount of label bound to the MCH receptor preparation, relative to the amount of label bound in the absence of the compound
- such a compound will exhibit a K 1 at an MCH receptor that is less than 1 micromolar, more preferably less than 500 nM, 100 nM, 20 nM or 10 nM, within an assay performed as desc ⁇ bed in Example 9 and/or within an assay performed as desc ⁇ bed in Example 12.
- Certain preferred compounds are MCH receptor antagonists, and exhibit IC 50 values of about 4 micromolar or less, more preferably 1 micromolar or less, still more preferably about 100 nanomolar or less, or 10 nanomolar or less within a standard in vitro MCH receptor mediated calcium mobilization assay, as provided in Example 13 and/or an agonist-stimulated GTP gamma 35 S binding assay, as described in Example 11.
- MCH receptor modulators provided herein may be evaluated for certain pharmacological properties including, but not limited to, oral bioavailability (preferred compounds are orally bioavailable to an extent allowing for therapeutically effective concentrations of the compound to be achieved at oral doses of less than 140 mg/kg, preferably less than 50 mg/kg, more preferably less than 30 mg/kg, even more preferably less than 10 mg/kg, still more preferably less than 1 mg/kg), toxicity (a preferred MCH receptor modulator is nontoxic when a therapeutically effective amount is administered to a subject), side effects (a preferred MCH receptor modulator produces side effects comparable to placebo when a therapeutically effective amount of the compound is administered to a subject), serum protein binding and in vitro and in vivo half-life (a preferred MCH receptor modulator exhibits an in vitro half-life that is equal to an in vivo half-life allowing for Q.I.D.
- oral bioavailability preferred compounds are orally bioavailable to an extent allowing for therapeutically effective concentrations of the compound to be achieved at
- T.I.D. dosing preferably T.I.D. dosing, more preferably B.I.D. dosing, and most preferably once-a-day dosing).
- differential penetration of the blood brain barrier may be desirable for MCH receptor modulators used to treat CNS disorders, while low brain levels of MCH receptor modulators used to treat peripheral disorders are preferred.
- Routine assays that are well known in the art may be used to assess these properties and identify superior compounds for a particular use. For example, assays used to predict bioavailability include transport across human intestinal cell monolayers, including Caco-2 cell monolayers. Penetration of the blood brain barrier of a compound in humans may be predicted from the brain levels of the compound in laboratory animals given the compound (e.g., intravenously).
- Serum protein binding may be predicted from albumin binding assays.
- Compound half-life is inversely proportional to the frequency of dosage of a compound.
- In vitro half-lives of compounds may be predicted from assays of microsomal half-life as described within Example 15, herein.
- preferred MCH receptor modulators provided herein are nontoxic.
- the term "nontoxic” as used herein shall be understood in a relative sense and is intended to refer to any substance that has been approved by the United States Food and Drug Administration (“FDA”) for administration to mammals (preferably humans) or, in keeping with established criteria, is susceptible to approval by the FDA for administration to mammals (preferably humans).
- FDA United States Food and Drug Administration
- a highly preferred nontoxic compound generally satisfies one or more of the following criteria when administered in minimum therapeutically effective amounts, or when contacted with cells at a concentration that is sufficient to inhibit the binding of MCH receptor ligand to MCH receptor in vitro : (1) does not substantially inhibit cellular ATP production; (2) does not significantly prolong heart QT intervals; (3) does not cause substantial liver enlargement and (4) does not cause substantial release of liver enzymes.
- a compound that does not substantially inhibit cellular ATP production is a compound that satisfies the criteria set forth in Example 14, herein.
- cells treated as described in Example 14 with 100 ⁇ M of such a compound exhibit ATP levels that are at least 50% of the ATP levels detected in untreated cells.
- such cells exhibit ATP levels that are at least 80% of the ATP levels detected in untreated cells.
- the concentration of compound used in such assays is generally at least 10-fold, 100-fold or 1000-fold greater than the EC 50 or IC 50 for the modulator in the assay of Example 11 or 13.
- a compound that does not significantly prolong heart QT intervals is a compound that does not result in a statistically significant prolongation of heart QT intervals (as determined by electrocardiography) in guinea pigs, minipigs or dogs upon administration of a dose that yields a serum concentration equal to the EC 50 or IC 50 for the compound.
- a dose of 0.01, 0.05, 0.1, 0.5, 1, 5, 10, 40 or 50 mg/kg administered parenterally or orally does not result in a statistically significant prolongation of heart QT intervals.
- statically significant is meant results varying from control at the p ⁇ 0.1 level or more preferably at the p ⁇ 0.05 level of significance as measured using a standard parametric assay of statistical significance such as a student's T test.
- a compound does not cause substantial liver enlargement if daily treatment of laboratory rodents (e.g., mice or rats) for 5-10 days with a dose that yields a serum concentration equal to the EC 50 or ICso for the compound results in an increase in liver to body weight ratio that is no more than 100% over matched controls. In more highly preferred embodiments, such doses do not cause liver enlargement of more than 75% or 50% over matched controls. If non-rodent mammals (e.g., dogs) are used, such doses should not result in an increase of liver to body weight ratio of more than 50%, preferably not more than 25%, and more preferably not more than 10% over matched untreated controls. Preferred doses within such assays include 0.01, 0.05.
- a compound does not promote substantial release of liver enzymes if administration of twice the minimum dose that yields a serum concentration equal to the EC50 or IC 5 0 for the compound does not elevate serum levels of ALT, LDH or AST in laboratory rodents by more than 100% over matched mock-treated controls. In more preferred embodiments, such doses do not elevate such serum levels by more than 75% or 50% over matched controls.
- compound does not promote substantial release of liver enzymes if, in an in vitro hepatocyte assay, concentrations (in culture media or other such solutions that are contacted and incubated with hepatocytes in vitro) that are equal to the EC 50 or IC 50 for the compound do not cause detectable release of any of such liver enzymes into culture medium above baseline levels seen in media from matched mock-treated control cells. In more highly preferred embodiments, there is no detectable release of any of such liver enzymes into culture medium above baseline levels when such compound concentrations are five-fold, and preferably ten-fold the EC 50 or IC 50 for the compound.
- certain preferred compounds do not inhibit or induce microsomal cytochrome P450 enzyme activities, such as CYP 1A2 activity, CYP2A6 activity, CYP2C9 activity, CYP2C19 activity, CYP2D6 activity, CYP2E1 activity or CYP3A4 activity at a concentration equal to the EC 50 or IC 50 for the compound.
- microsomal cytochrome P450 enzyme activities such as CYP 1A2 activity, CYP2A6 activity, CYP2C9 activity, CYP2C19 activity, CYP2D6 activity, CYP2E1 activity or CYP3A4 activity at a concentration equal to the EC 50 or IC 50 for the compound.
- Certain preferred compounds are not clastogenic (e.g., as determined using a mouse erythrocyte precursor cell micronucleus assay, an Ames micronucleus assay, a spiral micronucleus assay or the like) at a concentration equal the EC 50 or IC 50 for the compound.
- certain preferred MCH receptor modulators do not induce sister chromatid exchange (e.g., in Chinese hamster ovary cells) at such concentrations.
- MCH receptor modulators provided herein may be isotopically-labeled or radiolabeled.
- compounds of Formula I may have one or more atoms replaced by an atom of the same element having an atomic mass or mass number different from the atomic mass or mass number usually found in nature.
- isotopes that can be present in the compounds provided herein include isotopes of hydrogen, carbon, nitrogen, oxygen, phosphorous, fluorine and chlorine, such as 2 H, 3 H, 11 C, 13 C, 14 C, 15 N, 18 O, 17 0, 31 P, 32 P, 35 S, 18 F and 36 Cl.
- substitution with heavy isotopes such as deuterium (i.e., 2 H) can afford certain therapeutic advantages resulting from greater metabolic stability, for example increased in vivo half-life or reduced dosage requirements and, hence, may be preferred in some circumstances.
- Aryl-substituted piperazine derivatives can be administered as the neat chemical, but are preferably administered as a pharmaceutical composition comprising such a compound, together with at least one physiologically acceptable carrier or excipient.
- Representative carriers include, for example, water, buffers (e.g., neutral buffered saline or phosphate buffered saline), ethanol, mineral oil, vegetable oil, dimethylsulfoxide, carbohydrates (e.g., glucose, mannose, sucrose or dextrans), mannitol and proteins. Additional optional components include, adjuvants, diluents, polypeptides or amino acids such as glycine, antioxidants, chelating agents such as EDTA or glutathione and/or preservatives.
- Preferred pharmaceutical compositions are formulated for oral delivery to humans or other animals (e.g., companion animals such as dogs).
- Pharmaceutical carriers must be of sufficiently high purity and sufficiently low toxicity to render them suitable for administration to the animal being treated.
- the carrier can be inert or it can possess pharmaceutical benefits.
- the amount of carrier employed in conjunction with the compound is sufficient to provide a practical quantity of material for administration per unit dose of the compound.
- Representative pharmaceutically acceptable carriers or components thereof are sugars, such as lactose, glucose and sucrose; starches, such as corn starch and potato starch; cellulose and its derivatives, such as sodium carboxymethyl cellulose, ethyl cellulose and methyl cellulose; powdered tragacanth; malt; gelatin; talc; solid lubricants, such as stearic acid and magnesium stearate; calcium sulfate; synthetic oils; vegetable oils, such as peanut oil, cottonseed oil, sesame oil, olive oil and corn oil; polyols such as propylene glycol, glycerine, sorbitol, mannitol and polyethylene glycol; alginic acid; phosphate buffer solutions; emulsifiers, such as the TWEENS; wetting agents, such as sodium lauryl sulfate; coloring agents; flavoring agents; tableting agents; stabilizers; antioxidants; preservatives; pyrogen-free water; isotonic saline
- a suitable pharmaceutical carriers or excipients suitable pharmaceutical carriers or excipients.
- methods for solubilizing compounds include, but are not limited to, using cosolvents such as dimethylsulfoxide (DMSO), using surfactant, such as TWEEN, or dissolution in aqueous sodium bicarbonate.
- DMSO dimethylsulfoxide
- surfactant such as TWEEN
- dissolution in aqueous sodium bicarbonate Upon mixing or addition of the compound(s), the resulting mixture may be a solution, suspension, emulsion or the like. The form of the resulting mixture depends upon a number of factors, including the intended mode of administration and the solubility of the compound in the chosen carrier.
- compositions may be formulated for administration by any suitable route, including orally, topically, parenterally, by inhalation or spray, sublingually, transdermally, via buccal administration, rectally, as an ophthalmic solution or by other means, and may be prepared in dosage unit formulations.
- Dosage formulations suitable for oral use include, for example, tablets, troches, lozenges, liquid solutions, aqueous or oily suspensions, dispersible powders or granules, emulsions, hard or soft capsules, tinctures, syrups or elixirs.
- compositions intended for oral use may further contain one or more optional agents, such as sweetening agents (e.g., glycerol, propylene glycol, sorbitol or sucrose), flavoring agents, coloring agents and preserving agents, in order to provide pharmaceutically appealing and palatable preparations.
- sweetening agents e.g., glycerol, propylene glycol, sorbitol or sucrose
- flavoring agents e.g., glycerol, propylene glycol, sorbitol or sucrose
- coloring agents e.g., sorbitol or sucrose
- preserving agents e.glycerol, sorbitol or sucrose
- Such formulations may also contain a demulcent.
- Typical components of carriers for syrups, elixirs, emulsions and suspensions include ethanol, glycerol, propylene glycol, polyethylene glycol, liquid sucrose, sorbitol and water.
- Orally Administered Liquid Formulations Compounds provided herein can be incorporated into oral liquid preparations such as, for example, aqueous or oily suspensions, solutions, emulsions, syrups or elixirs. Moreover, formulations containing these compounds can be presented as a dry product for constitution with water or other suitable vehicle before use.
- Such liquid preparations may further contain one or more conventional additives, such as suspending agents (e.g., sorbitol syrup, methyl cellulose, glucose/sugar, syrup, gelatin, hydroxyethyl cellulose, carboxymethyl cellulose, aluminum stearate gel and hydrogenated edible fats); emulsifying agents (e.g., lecithin, sorbitan monsoleate or acacia); and/or non-aqueous vehicles such as edible oils (e.g., almond oil, fractionated coconut oil, silyl esters, propylene glycol and ethyl alcohol) and preservatives (e.g., methyl or propyl p-hydroxybenzoate and sorbic acid).
- suspending agents e.g., sorbitol syrup, methyl cellulose, glucose/sugar, syrup, gelatin, hydroxyethyl cellulose, carboxymethyl cellulose, aluminum stearate gel and hydrogenated edible fats
- emulsifying agents e.
- Suspensions contain the active material(s) in admixture with excipients (e.g., suspending agents, wetting agents and/or preservatives) suitable for the manufacture of aqueous suspensions.
- Suspending agents include, for example, sodium carboxymethylcellulose, methylcellulose, hydropropylmethylcellulose, AVICEL RC-591, sodium alginate, polyvinylpyrrolidone, gum tragacanth and gum acacia.
- Dispersing or wetting agents include, for example, lecithin, polysorbate 80, naturally-occurring phosphatides such as lecithin, condensation products of an alkylene oxide with fatty acids (e.g., polyoxyethylene stearate), condensation products of ethylene oxide with long chain aliphatic alcohols (e.g., heptadecaethyleneoxycetanol), condensation products of ethylene oxide with partial esters derived from fatty acids and a hexitol (e.g., polyoxyethylene sorbitol substitute), or condensation products of ethylene oxide with partial esters derived from fatty acids and hexitol anhydrides (e.g., polyethylene sorbitan substitute).
- Representative preservatives include, for example, ethyl- or n-propyl- p-hydroxybenzoate, sodium benzoate and methyl paraben.
- Oily suspensions may be formulated by suspending the active ingredients in a vegetable oil (e.g., peanut oil, olive oil, sesame oil or coconut oil), a mineral oil (such as liquid paraffin) or a mixture of such oils.
- the oily suspensions may further contain a thickening agent, such as beeswax, hard paraffin or cetyl alcohol. Sweetening agents, such as those set forth above, and flavoring agents may be added to improve palatability. If desired, these compositions may be preserved by the addition of an anti-oxidant such as ascorbic acid.
- Emulsions Pharmaceutical compositions provided herein may also be in the form of oil-in-water emulsions.
- the oily phase may be a vegetable oil, mineral oil, or mixture thereof as described above.
- Suitable emulsifying agents include naturally-occurring gums (e.g., gum acacia or gum tragacanth), naturally-occurring phosphatides (e.g., soy bean phosphatide, lecithin and esters or partial esters derived from fatty acids and hexitol), and anhydrides (e.g., sorbitan monoleate and condensation products of the above partial esters with ethylene oxide, such as polyoxyethylene sorbitan monoleate).
- Naturally-occurring gums e.g., gum acacia or gum tragacanth
- naturally-occurring phosphatides e.g., soy bean phosphatide, lecithin and esters or partial esters derived from fatty acids and hexitol
- anhydrides e.g., sorbitan monoleate and condensation products of the above partial esters with ethylene oxide, such as polyoxyethylene sorbitan monoleate.
- Dispersible powders and granules suitable for preparation of an aqueous suspension by the addition of water provide the active ingredient in admixture with a dispersing or wetting agent, suspending agent and one or more preservatives.
- a dispersing or wetting agent e.g., kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, kaolin, ka
- Tablets typically comprise conventional pharmaceutically compatible inert diluents, such as calcium carbonate, sodium carbonate, mannitol, lactose and cellulose; binders such as starch, gelatin and sucrose; disintegrants such as starch, alginic acid and croscarmelose; and/or lubricants such as magnesium stearate, stearic acid and talc. Glidants such as silicon dioxide can be used to improve flow characteristics of the powder mixture. Coloring agents, such as the FD&C dyes, can be added for appearance. Sweeteners and flavoring agents, such as aspartame, saccharin, menthol, peppermint and fruit flavors, are useful adjuvants for chewable tablets. Capsules (including time release and sustained release formulations) typically comprise one or more solid diluents disclosed above. The selection of carrier components often depends on secondary considerations such as taste, cost and shelf stability.
- compositions may also be coated by conventional methods, typically with pH-dependent or time-dependent coatings, such that the subject compound is released in the gastrointestinal tract in the vicinity of the desired topical application, or at various times to extend the desired action.
- coatings typically include, but are not limited to, one or more of cellulose acetate phthalate, polyvinylacetate phthalate, hydroxypropyl methylcellulose phthalate, ethyl cellulose, Eudragit coatings, waxes and shellac.
- Formulations for oral use may also be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, such as calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, such as peanut oil, liquid paraffin or olive oil.
- an inert solid diluent such as calcium carbonate, calcium phosphate or kaolin
- an oil medium such as peanut oil, liquid paraffin or olive oil.
- compositions may be in the form of a sterile injectable aqueous or oleaginous suspension.
- a suspension may be formulated according to the known art using dispersing or wetting agents and suspending agents as described above.
- the sterile injectable preparation may also be a sterile injectable solution or suspension in a non-toxic parentally acceptable diluent or solvent (e.g., as a solution in 1,3-butanediol).
- a non-toxic parentally acceptable diluent or solvent e.g., as a solution in 1,3-butanediol.
- the acceptable vehicles and solvents that may be employed are water, Ringer's solution and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- any bland fixed oil synthetic ⁇ e.g., synthetic mono- or diglycerides
- fatty acids such as oleic acid are useful in the preparation of injectable formulations.
- compositions may be administered parenterally in a sterile medium.
- Parenteral administration includes subcutaneous injections, intravenous, intramuscular, intrathecal injection or infusion techniques.
- the active agent(s) depending on the vehicle and concentration used, can either be suspended or dissolved in the vehicle.
- Adjuvants such as local anesthetics, preservatives and buffering agents can also be dissolved in the vehicle.
- at least about 90% by weight of the total composition is carrier.
- Preferred carriers for parenteral administration include propylene glycol, ethyl oleate, pyrrolidone, ethanol and sesame oil.
- compositions may also be administered rectally, in the form of suppositories.
- Such compositions can be prepared by mixing the active ingredient(s) with a suitable non-irritating excipient that is solid at ordinary temperatures but liquid at rectal temperature and will therefore melt in the rectum to release the drug.
- suitable non-irritating excipient include cocoa butter and polyethylene glycols.
- compositions may be formulated for local or topical application, such as for topical application to the skin or mucous membranes.
- Topical compositions may be in any suitable form including, for example, solutions, creams, ointments, gels, lotions, milks, cleansers, moisturizers, sprays, skin patches and the like.
- solutions may, for example, be formulated as 0.01%-10% isotonic solutions, pH about 5-7, with appropriate salts.
- Pharmaceutical compositions may also be formulated for transdermal administration as a transdermal patch.
- Topical compositions containing the active compound can be admixed with a variety of carrier materials well known in the art, such as, for example, water, alcohols, aloe vera gel, allantoin, glycerine, vitamin A and E oils, mineral oil, propylene glycol, PPG-2 myristyl propionate and the like.
- carrier materials such as, for example, water, alcohols, aloe vera gel, allantoin, glycerine, vitamin A and E oils, mineral oil, propylene glycol, PPG-2 myristyl propionate and the like.
- Other materials suitable for use in topical earners include, for example, emollients, solvents, humectants, thickeners and powders.
- emollients such as stearyl alcohol, glyceryl monoricinoleate, glyceryl monostearate, propane- 1,2-diol, butane- 1, 3 -diol, mink oil, cetyl alcohol, iso-propyl isostearate, stearic acid, iso-butyl palmitate, isocetyl stearate, oleyl alcohol, isopropyl laurate, hexyl laurate, decyl oleate, octadecan-2-ol, isocetyl alcohol, cetyl palmitate, dimethylpolysiloxane, di-n-butyl sebacate, iso-propyl myristate, iso-propyl palmitate, iso-propyl stearate, butyl stearate, polyethylene glycol,
- emollients such as stearyl alcohol, glyceryl monoricinoleate, g
- compositions may also be topically administered in the form of liposome delivery systems, such as small unilamellar vesicles, large unilamellar vesicles and multilamellar vesicles.
- Liposomes can be formed from a variety of phospholipids, such as cholesterol, stearylamine or phosphatidylcholines. Other Formiilatiorts and Additional Components
- compositions useful for attaining systemic delivery of the subject compounds include sublingual, buccal and nasal dosage forms.
- Such compositions typically comprise one or more soluble filler substances such as sucrose, sorbitol and mannitol, and/or binders such as acacia, microcrystalline cellulose, carboxymethyl cellulose and hydroxypropyl methylcellulose. Glidants, lubricants, sweeteners, colorants, antioxidants and flavoring agents disclosed above may also be included.
- compositions for inhalation are typically provided in the form of a solution, suspension or emulsion that can be administered as a dry powder or in the form of an aerosol using a conventional propellant ⁇ e.g., dichlorodifluoromethane or trichlorofluoromethane).
- a conventional propellant e.g., dichlorodifluoromethane or trichlorofluoromethane
- a pharmaceutical composition may be conveniently added to food or drinking water ⁇ e.g., for administration to non- human animals including companion animals, such as dogs and cats and livestock).
- Animal feed and drinking water compositions may be formulated so that the animal takes in an appropriate quantity of the composition along with its diet. It may also be convenient to present the composition as a premix for addition to feed or drinking water.
- compositions may also optionally comprise an activity enhancer.
- the activity enhancer can be chosen from a wide variety of molecules that function in different ways to enhance MCH receptor modulator effect. Particular classes of activity enhancers include skin penetration enhancers and absorption enhancers.
- compositions provided herein may also contain additional active agents, which can be chosen from a wide variety of molecules and can function in different ways to enhance the therapeutic effects of a MCH receptor modulator, or to provide a separate therapeutic effect that does not substantially interfere with the activity of the MCH receptor modulator.
- additional active agents when present, are typically employed in the compositions described herein at a level ranging from about 0.01% to about 50% by weight of the composition, preferably 0.1% to 25%, 0.2% to 15, 0.5% to 10% or 0.5% to 5% by weight of the composition.
- compositions intended for the treatment of obesity and/or eating disorders may further comprise leptin, a leptin receptor agonist, a melanocortin receptor 4 (MC4) agonist, sibutramine, dexfenfluramine, a growth hormone secretagogue, a beta-3 agonist, a 5HT-2 agonist, an orexin antagonist, a neuropeptide Yi or Y 5 antagonist, a galanin antagonist, a CCK agonist, a GLP-I agonist, a cannabinoid receptor antagonist ⁇ e.g., a CBl antagonist) and/or a corticotropin-releasing hormone agonist.
- leptin a leptin receptor agonist
- a melanocortin receptor 4 (MC4) agonist sibutramine
- dexfenfluramine a growth hormone secretagogue
- beta-3 agonist a beta-3 agonist
- 5HT-2 agonist an orexin antagonist
- a neuropeptide Yi or Y 5 antagonist
- an additional active agent is a CBl antagonist.
- Representative CBl antagonists include, for example, certain pyrimidines ⁇ e.g., PCT International Application Publication No. WO 04/029,204), pyrazines (e.g., PCT International Application Publication Nos. WO 01/111,038; WO 04/111,034 and WO 04/111,033), azetidine derivatives (e.g., US Patent Nos.
- WO 03/063781 and WO 03/040107 substituted furo[2,3-b]pyridine derivatives (e.g., PCT International Application Publication No. WO 04/012671); substituted aryl amides (e.g., PCT International Application Publication Nos. WO 03/087037 and WO 03/077847); substituted bicyclic or spirocyclic amides (e.g., PCT International Application Publication Nos. WO 03/086288 and WO 03/082190); and substituted 2,3-diphenyl pyridines (e.g., PCT International Application Publication No. WO 03/082191).
- Other CBl antagonists are cannabidiol and its derivatives.
- Preferred CBl antagonists include, for example, aryl substituted pyrazole carboxamides such as SR-141716A (N- ⁇ iperidin-l-yl)-5-(4-chlorophenyl)-l- (2,4-dichlorophenyl)-4-methyl-l-H-pyrazole-3-carboxamide, also known as RHVIONABANTTM or ACOMPLIATM) as well analogues thereof such as AM251 (N-piperidin-l-yl)-5-(4-iodophenyl)-l- (2,4-dichlorophenyl)-4-methyl-l-H-pyrazole-3-carboxamide) and AM281 (N-(morpholin-4-yl)-l- (2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-l-H-pyrazole-3-carboxamide); various azetidine compounds (e.g., US Patent Nos
- compositions may be packaged for treating or preventing a disease or disorder that is associated with MC ⁇ receptor activation (e.g., treatment of metabolic disorders such as diabetes, heart disease, metabolic syndrome, stroke, obesity and eating disorders such as bulimia, fluid balance disorders, skin disorders such as pigmentation disorders and vitiligo, neuropsychiatric disorders such as anxiety, depression, reward system disorders and cognitive deficits, alterations in NMDA receptor function, reproductive function disorders or sexual disorders such as anorgasmic or psychogenic impotence), or for promoting weight loss.
- Pharmaceutical compositions may also be packaged for modulating bone mass (i.e., inhibiting loss of bone mass and/or stimulating an increase in bone mass).
- Packaged pharmaceutical preparations comprise a container holding a therapeutically effective amount of MCH receptor modulator as described herein and instructions (e.g., labeling) indicating that the contained composition is to be used for promoting weight loss, modulating bone mass or for treating or preventing a disease or disorder that is associated with MCH receptor activation in the patient.
- Prescribing information may be provided separately to a patient or health care provider, or may be provided as a label or package insert. Prescribing information may include, for example, efficacy, dosage and administration, contraindication and adverse reaction information pertaining to the pharmaceutical formulation.
- Certain packaged pharmaceutical preparations further include a second therapeutic agent as discussed above. Dosages
- Aryl-substituted piperazine derivatives are generally present within a pharmaceutical composition in a therapeutically effective amount.
- Compositions providing dosage levels ranging from about 0.1 mg to about 140 mg per kilogram of body weight per day are preferred (about 0.5 mg to about 7 g per human patient per day), with dosages ranging from 0.1 mg to 50 mg, 30 mg or 10 mg particularly preferred.
- the amount of active ingredient that may be combined with the carrier to produce a single dosage form will vary depending upon the patient to be treated and the particular mode of administration. Dosage unit forms generally contain from about 1 mg to about 500 mg of an active ingredient.
- Dosage units generally contain from about 10 ⁇ g to about 500 mg of each active ingredient. Optimal dosages may be established using routine testing and procedures that are well known in the art.
- the present invention provides methods for inhibiting the development or progression of a disease or disorder responsive to MCH receptor modulation.
- therapeutic methods provided herein may be used to treat a patient already afflicted with such a disease or disorder, or may be used to prevent or delay the onset of such a disease or disorder in a patient who is free of detectable disease or disorder that is associated with MCH receptor activation.
- a disease or disorder is "associated with MCH receptor activation" if it is characterized by inappropriate stimulation of MCH receptor, regardless of the amount of MCH present locally, and/or is responsive to modulation of MCH receptor activity.
- Such conditions include, for example, metabolic disorders (such as diabetes), heart disease, metabolic syndrome, stroke, obesity and eating disorders (such as bulimia nervosa), fluid balance disorders, skin disorders such as pigmentation disorders and vitiligo, alterations in NMDA receptor function, and sexual disorders such as anorgasmic or psychogenic impotence. These conditions may be diagnosed and monitored using criteria that have been established in the art.
- MCH antagonists provided herein may be used to promote weight loss in patients, and MCH agonists provided herein may be used to promote weight gain in patients.
- MCH antagonists may also be used to modulate a patient's bone mass (i.e., to inhibit loss of bone mass and/or stimulate an increase in bone mass).
- Patients may include humans, domesticated companion animals (pets, such as dogs and cats) and livestock animals, with dosages and treatment regimes as described above. Additional conditions that are associated with MCH receptor activation include:
- Cognitive impairment and memory disorders such as Alzheimer's disease, Parkinson's disease, mild cognitive impairment (MCI), age-related cognitive decline (ARCD), stroke, traumatic brain injury, ADDS associated dementia, and dementia associated with depression, anxiety and psychosis (including schizophrenia and hallucinatory disorders);
- Anxiety, depression and other mood disorders including general anxiety disorder (GAD), agoraphobia, panic disorder with and without agoraphobia, social phobia, specific phobia, post traumatic stress disorder, obsessive compulsive disorder (OCD), dysthymia, adjustment disorders with disturbance of mood and anxiety, separation anxiety disorder, anticipatory anxiety acute stress disorder, adjustment disorders and cyclothymia;
- GAD general anxiety disorder
- OCD obsessive compulsive disorder
- Reward system disorders such as addiction (e.g., opioid, nicotine or alcohol);
- Pain such as migraine, peripheral inflammatory pain, neuropathic pain and sympathetic nervous system associated pain; and Peripheral indications such as respiratory disorders (e.g., asthma), urinary disorders (e.g., urinary incontinence), gastrointestinal disorders, reproductive function disorders and cardiovascular disorders (e.g., arteriosclerosis and hypertension).
- respiratory disorders e.g., asthma
- urinary disorders e.g., urinary incontinence
- gastrointestinal disorders e.g., urinary incontinence
- reproductive function disorders e.g., arteriosclerosis and hypertension
- Frequency of dosage may vary depending on the compound used and the particular disease to be treated or prevented. In general, for treatment of most disorders, a dosage regimen of 4 times daily or less is preferred. For the treatment of eating disorders and obesity, a dosage regimen of 1 or 2 times daily is particularly preferred. For the treatment of impotence a single dose that rapidly reaches effective concentrations is desirable. It will be understood, however, that the specific dose level for any particular patient will depend upon a variety of factors including the activity of the specific compound employed, the patient's age, body weight, general health, sex and diet, the time and route of administration, the rate of excretion, any coadministered drugs and the severity of the particular disease. In certain embodiments, administration at meal times is preferred.
- patients may generally be monitored for therapeutic effectiveness using assays suitable for the condition being treated or prevented, which will be familiar to those of ordinary skill in the art.
- methods for treating a patient comprising diagnosing the patient as having a disease or disorder associated with MCH receptor activation, correlating the diagnosis of the disease or disorder with the need for MCH modulator administration, and administering an a effective amount of an aryl-substituted piperazine derivative provided herein.
- a method for treating a patient comprising administering an effective amount of an aryl-substituted piperazine derivative of Formula I to a patient having a disease or disorder associated with MCH receptor activation is also provided herein.
- the disease or disorder associated with MCH receptor activation is obesity, an eating disorder, a sexual disorder, diabetes, heart disease, metabolic syndrome, stroke, anxiety, depression, a skin pigmentation disorder, a reward system disorder, a cognitive disorder, or a fluid balance disorder.
- the MCH receptor modulator is used to modulate bone mass.
- the aryl-substituted piperazine derivative of Formula I is administered orally, intranasally, intravenously or topically.
- MCH receptor modulators provided herein may be used within combination therapy for the treatment of conditions associated with MCH receptor modulation.
- a MCH receptor modulator is administered to a patient along with a second therapeutic agent that is not primarily a MCH receptor modulator, but that is appropriate for treatment of the condition(s) of interest.
- the MCH receptor modulator and second therapeutic agent(s) may be present in the same pharmaceutical composition, or may be administered separately in either order. Suitable second therapeutic agents include those listed above.
- Suitable dosages for MCH receptor modulator(s) within such combination therapy are generally as described herein. Dosages and methods of administration of other therapeutic agents can be found, for example, in the manufacturer's instructions in the Physician's Desk Reference.
- the combination administration results in a reduction of the dosage of the second therapeutic agent required to produce a therapeutic effect ⁇ i.e., a decrease in the minimum therapeutically effective amount).
- the dosage of second therapeutic agent in a combination or combination treatment method of the invention is less than the maximum dose advised by the manufacturer for administration of the second therapeutic agent without combination administration of a MCH receptor modulator.
- this dosage is less than 3 A, even more preferably less than Vi, and highly preferably, less than 1 A of the maximum dose, while most preferably the dose is less than 10% of the maximum dose advised by the manufacturer for administration of the second therapeutic agent(s) when administered without combination administration of a MCH receptor modulator. It will be apparent that the dosage amount of MCH receptor modulator component of the combination needed to achieve the desired effect may similarly be affected by the dosage amount and potency of the second therapeutic agent component of the combination.
- the combination administration of a MCH receptor modulator with a second therapeutic agent is accomplished by packaging one or more MCH receptor modulators and one or more second therapeutic agents in the same package, either in separate containers within the package or in the same container as a mixture of one or more MCH receptor modulators and one or more second therapeutic agents.
- Preferred mixtures are formulated for oral administration (e.g., as pills, capsules, tablets or the like).
- the package comprises a label or package insert indicating that the one or more MCH receptor modulators and one or more second therapeutic agents are to be taken together for the treatment of a condition that is associated with MCH receptor activation, such as obesity.
- one or more MCH receptor modulators provided herein are used along with one or more CBl antagonists within a combination therapy. Such combinations are of particular use for weight management, to reduce appetite and/or food intake or to prevent or treat obesity (e.g., promote weight loss).
- Patients may include humans, domesticated companion animals and livestock animals, with dosages and treatment regimes as described above.
- the MCH receptor modulator(s) may be administered to the patient at the same time as the CBl antagonist(s) (e.g., as a single dosage unit), or may be administered separately (before or after CBl antagonist).
- the MCH receptor modulator(s) and CBl antagonist(s) are ultimately simultaneously present at effective concentrations in a body fluid (e.g., blood) of the patient.
- An effective concentration of MCH receptor modulator or CBl antagonist is a concentration that is sufficient to reduce one or more of food consumption, appetite and/or body mass index in the patient when repeatedly coadministered as described herein.
- the present invention provides a variety of in vitro uses for the compounds provided herein.
- such compounds may be used as probes for the detection and localization of MCH receptors, in samples such as tissue sections, as positive controls in assays for receptor activity, as standards and reagents for determining the ability of a candidate agent to bind to MCH receptor, or as radiotracers for positron emission tomography (PET) imaging or for single photon emission computerized tomography (SPECT).
- PET positron emission tomography
- SPECT single photon emission computerized tomography
- Such assays can be used to characterize MCH receptors in living subjects.
- Compounds provided herein are also useful as standards and reagents in determining the ability of a test compound to bind to MCH receptor.
- a sample may be incubated with a compound as provided herein under conditions that permit binding of the compound to MCH receptor.
- the amount of compound bound to MCH receptor in the sample is then detected.
- a compound may be labeled using any of a variety of well-known techniques (e.g., radiolabeled with a radionucleide such as tritium, as described herein), and incubated with the sample (which may be, for example, a preparation of cultured cells, a tissue preparation or a fraction thereof).
- a suitable incubation time may generally be determined by assaying the level of binding that occurs over a period of time.
- unbound compound is removed, and bound compound detected using any method for the label employed (e.g., autoradiography or scintillation counting for radiolabeled compounds; spectroscopic methods may be used to detect luminescent groups and fluorescent groups).
- a matched sample may be simultaneously contacted with radiolabeled compound and a greater amount of unlabeled compound. Unbound labeled and unlabeled compound is then removed in the same fashion, and bound label is detected. A greater amount of detectable label in the test sample than in the control indicates the presence of MCH receptor in the sample.
- Detection assays including receptor autoradiography (receptor mapping) of MCH receptors in cultured cells or tissue samples may be performed as described by Kuhar in sections 8.1.1 to 8.1.9 of Current Protocols in Pharmacology (1998) John Wiley & Sons, New York.
- Compounds provided herein may also be used within a variety of well-known cell culture and cell separation methods.
- compounds may be linked to the interior surface of a tissue culture plate or other cell culture support, for use in immobilizing MCH receptor-expressing cells for screens, assays and growth in culture.
- Compounds may also be used to facilitate cell identification and sorting in vitro, permitting the selection of cells expressing a MCH receptor.
- the compound(s) for use in such methods are labeled as described herein.
- a compound linked to a fluorescent marker such as fluorescein, is contacted with the cells, which are then analyzed by fluorescence activated cell sorting (FACS).
- FACS fluorescence activated cell sorting
- methods for modulating binding of MCH to an MCH receptor in vitro or in vivo, comprising contacting a MCH receptor with a sufficient amount of a modulator provided herein, under conditions suitable for binding of MCH to the receptor.
- MCH binding to receptor is inhibited by the modulator.
- the MCH receptor may be present in solution, in a cultured or isolated cell preparation or within a patient.
- the MCH receptor is a MCHlR receptor present in the hypothalamus.
- the amount of compound contacted with the receptor should be sufficient to modulate MCH binding to MCH receptor in vitro within, for example, a binding assay as described in Example 9 and/or Example 12.
- MCH receptor preparations used to determine in vitro binding may be obtained from a variety of sources, such as from HEK 293 cells or Chinese Hamster Ovary (CHO) cells transfected with a MCH receptor expression vector, as described herein.
- the MCH receptor may be present in solution, in a cultured or isolated cell preparation or within a patient.
- the amount of modulator contacted with the receptor should be sufficient to modulate MCH receptor signal transducing activity in vitro within, for example, a calcium mobilization assay as described in Example 13 and/or an agonist-stimulated GTP gamma 35 S binding assay as described in Example 11.
- An effect on signal-transducing activity may be assessed as an alteration in the electrophysiology of the cells, using standard techniques, such as intracellular patch clamp recording or patch clamp recording. If the receptor is present in an animal, an alteration in the electrophysiology of the cell may be detected as a change in the animal's feeding behavior.
- a MCH receptor modulator may contain one or more asymmetric carbon atoms, so that the compound can exist in different stereoisomeric forms.
- Such forms can be, for example, racemates or optically active forms.
- All stereoisomers are encompassed by the present invention. Nonetheless, it may be desirable in certain instances to obtain single enantiomers ⁇ i.e., optically active forms).
- Standard methods for preparing single enantiomers include asymmetric synthesis and resolution of the racemates. Resolution of the racemates can be accomplished, for example, by conventional methods such as crystallization in the presence of a resolving agent, or chromatography using, for example a chiral HPLC column.
- Compounds may be radiolabeled by carrying out their synthesis using precursors comprising at least one atom that is a radioisotope.
- Each radioisotope is preferably carbon (e.g., 14 C), hydrogen ⁇ e.g., 3 H or 2 H), fluorine ⁇ e.g., 18 F or 19 F), sulfur ⁇ e.g., 35 S) or iodine ⁇ e.g., 125 I).
- Tritium labeled compounds may also be prepared catalytically via platinum-catalyzed exchange in tritiated acetic acid, acid-catalyzed exchange in tritiated trifluoroacetic acid, or heterogeneous-catalyzed exchange with tritium gas using the compound as substrate.
- certain precursors may be subjected to tritium-halogen exchange with tritium gas, tritium gas reduction of unsaturated bonds, or reduction using sodium borotritide, as appropriate.
- Preparation of radiolabeled compounds may be conveniently performed by a radioisotope supplier specializing in custom synthesis of radiolabeled probe compounds.
- a commercially available N-protected tropinone derivative such as but not limited to N-carbethoxy-4-tropinone, tropinone, BOC-nortropinone is reacted with an arylorganometallic reagent such as but not limited to arylmagnesium halides (Grignard reagents), aryllithium, arylsodium, arylzinc halides, aryl copper halides, or arylcerium halides in an inert reaction solvent such as but not limited to ethyl ether, tetrahydrofuran, diisopropyl ether, methyl-tert-butyl ether, benzene, toluene and the like at a reaction temperature between -78 0 C and 120 0 C, more preferably between O 0 C and 60 0 C to furnish the ⁇ -protected 3-aryl-3-hydroxy-8-azabicyclo[3.2.1]octane.
- the addition of the organometallic reagent takes place highly preferably from the less-hindered side (see for example Langbein, A. et al. 8-Arylalkyl-3-phenyl-3-nortropanols, their acid addition salts and their pharmaceutical compositions. US 4,393,069, 7/12/83).
- the protecting group on the nitrogen atom can then be removed using a number of methods familiar to those skilled in the art (see for example Greene, T.W. and Wuts, P.G.M., Protective Groups in Organic Synthesis, John Wiley and Sons, Inc., New York, 1999).
- the last step can be carried out by reacting the 3-aryl-8- azabicyclo[3.2.1]octan-3-ol with an appropriately substituted benzylic chloride, bromide, iodide, tosylate, mesylate, benzenesulfonate and the like in a solvent such as EtOH, DMF, DMSO, acetonitrile, propionitrile, acetone and the like in the presence of a base such as but not limited to K 2 CO 3 , Na 2 CO 3 , NaHCO 3 , Cs 2 CO 3 and the like at temperatures between room temperature and 140 0 C.
- EXAMPLE 1 SYNTHESIS OF 8-(4-METHOXY-2,3-DIMETP ⁇ YLBENZYL)-3-(4-CHLORO-3-
- step 1 The ethyl 3-(4-chloro-3-methoxyphenyl)-3-hydroxy-8-azabicyclo[3.2. l]octane-8-carboxylate obtained in step 1 (11.1 g) is treated with KOH (48 g, 0.86 mol), and hydrazine monohydrate (8 mL) in HOCH 2 CH 2 OH (240 mL) at reflux for 4h. The reaction mixture is then cooled to room temperature, poured into 200 mL of water and extracted with CHCl 3 (3x).
- the mixture is stirred at room temperature for 2h.
- the solvent is removed under reduced pressure.
- the residue is dried on high vacuum pump overnight, and used in the next step without further purification.
- Step 5 8-(4-(3-(Dimethylamino)propoxy)-2, 3-dimethylbenzyl)-3-(4-chIoro-3-methoxyphenyl)-8-aza- bicyclo[3.2.1] octa ⁇ -3-ol
- 3-(4-chloro-3-methoxyphenyl)-8-(4-hydroxy-2,3-dimethylbenzyl)-8- azabicyclo[3.2.1]octan-3-ol obtained in step 4 (20 mg, 0.05 mmol) in anhydrous DMF (1 mL) is added Cs 2 CO 3 (24.2 mg, 0.075 mmol), followed by Me 2 NCH 2 CH 2 CH 2 ClHCl (0.06 mmol) and catalytic amounts of NaI.
- Example 2 To a solution of 3-(4-chloro-3-memoxyphenyl)-8-(4-hydroxy-2,3-dimethylbenzyl)-8- azabicyclo[3.2.1]octan-3-ol obtained in step 4, Example 2, (20 mg, 0.05 mmol) in anhydrous DMF (1 mL) is added Cs 2 CO 3 (24.2 mg, 0.075 mmol), followed by 2-chloro-N,N-dimethylacetamide (0.06 mmol) and catalytic amounts of NaI. The mixture is heated at 6O 0 C overnight. The reaction is then cooled to room temperature, diluted with ethyl acetate, washed with water, brine, dried over Na 2 SO 4 and concentrated under reduced pressure.
- the MCHlR receptor source is a rat striatum homogenate.
- the rats are na ⁇ ve Sprague Dawley or Wistar rats which are not food deprived overnight, and weigh roughly 250 ⁇ 25 grams.
- the stratum is rapidly/carefully dissected away from the cortex, mid-bram and hippocampus.
- the stratum is weighed, and homogenized in Prep buffer (50 mM T ⁇ s, pH 7.4, 10 mM MgCl 2 , 2 mM EGTA- 23 mL per gram of striatum, typically 150 mg of tissue plus 3.5 mL of prep buffer), homogenizing for 30 seconds using a BRINKMAN POLYTRON at setting 5.
- Prep buffer 50 mM T ⁇ s, pH 7.4, 10 mM MgCl 2 , 2 mM EGTA- 23 mL per gram of striatum, typically 150 mg of tissue plus 3.5 mL of prep buffer
- rat striatal membranes The protein concentration of the resulting membrane preparation (hereinafter "rat striatal membranes") is conveniently measured using a Bradford protein assay (Bio-Rad Laboratories, Hercules, CA).
- EXAMPLE 9 RADIOLIGAND BINDING ASSAYS This Example illustrates a standard assay of Melanin Concentrating Hormone receptor binding that may be used to determine the binding affinity of compounds for the MCH receptor.
- 125 I- labeled S36057 New England Nuclear Corp., Boston, MA
- a stable analogue of MCH is used as the radioligand.
- Purified rat striatal membranes prepared by the method given in Example 8 above, are resuspended by Dounce homogenization (tight pestle) in binding buffer (50 mM T ⁇ s pH. 7.4, 1.0 mM Mg Cl 2 , 5 mM KCl 3 1 mM CaCl 2 , 120 mM NaCl, 1 mM bacitracin, 0.02 mg/mL Aprotmm & 0.1% BSA).
- the optimal rat striatal homogenate input has been determined, via a protein linearity expe ⁇ ment, to be 275 ⁇ g / data point / 250 ⁇ L.
- this amount of protein binds 10-15% of the input radioligand.
- the specific binding signal is routinely 50%.
- Non specific binding is defined with l ⁇ M MCH.
- Displacement binding studies, designed to determine the IC 5 o/K, of exogenously added compounds, are run at 30 pM [ I25 I]-S36057.
- rat st ⁇ atal membranes (275 ⁇ g) are added to polypropylene tubes containing 25 pM - 0.5 nM [ 125 I]S36057.
- Nonspecific binding is determined in the presence of 10 ⁇ M MCH (Toc ⁇ s Cookson Inc., Ellisville, MO, USA) and accounts for less than 10 % of total binding.
- GTP7S is added to duplicate tubes at the final concentration of 50 ⁇ M.
- membranes 275 ⁇ .g are added to polypropylene tubes containing 0.03 nM [ 125 I]S36057.
- Non-radiolabeled displacers are added to separate assays at concentrations ranging from 10 "10 M to 10 "5 M to yield a final volume of 0.250 mL.
- Nonspecific binding is determined in the presence of 10 ⁇ M MCH and accounts for less than 30% of total binding.
- WO 03/059289 Chinese hamster ovary (CHO) cells (American Type Culture Collection, Manassas, VA) via calcium precipitation.
- CHO Chinese hamster ovary
- CHO mMCHlR cell pellets are resuspended in homogenization buffer (10 mM HEPES, 250 mM sucrose, 0.5 ⁇ g/mL leupeptin, 2 ⁇ g/mL Aprotinin, 200 ⁇ M PMSF, and 2.5 mM EDTA, pH 7.4) and homogenized using a BRINKMAN POLYTRON homogenizer (setting 5 for 30 seconds).
- the homogenate is centrifuged (536 x g/ 10 min/ 4 0 C) to pellet the nuclei.
- the supernatant containing isolated membranes is decanted to a clean centrifuge tube, centrifuged (48,000 X g/ 30 min, 4 0 C) and the resulting pellet resuspended in 30 mL homogenization buffer. This centrifugation and resuspension step is repeated twice. The final pellet is resuspended in ice cold Dulbecco's PBS containing 5 mM EDTA and stored in frozen aliquots at -8O 0 C until needed.
- the protein concentration of the resulting membrane preparation (hereinafter "P2 membranes") is conveniently measured using a Bradford protein assay (Bio-Rad Laboratories, Hercules, CA).
- GTP binding activity can be used to identify agonist and antagonist compounds and to differentiate neutral antagonist compounds from those that possess inverse agonist activity. This activity can also be used to detect partial agonism mediated by antagonist compounds. A compound being analyzed in this assay is referred to herein as a "test compound.”
- Agonist-stimulated GTP binding on purified P2 membranes is assessed using MCH as agonist in order to ascertain the level of signal, and EC 50 value of MCH as measured by GTP binding.
- P2 membranes from the CHO cells are resuspended by Dounce homogenization (tight pestle) in GTP binding assay buffer (50 niM Tris pH 7.4, 120 mM NaCl, 5 mM MgC12, 2 mM EGTA, 0.1% BSA, 0.1 mM bacitracin, 100 KIU/mL aprotinin, 5 ⁇ M GDP, 10 ⁇ g/mL saponin) and added to reaction tubes at a concentration of 50 ⁇ g protein/reaction tube. After adding increasing doses of the agonist MCH at concentrations ranging from 10 "12 M to 10 "6 M, reactions are initiated by the addition of 100 pM GTP gamma 35 S.
- GTP binding assay buffer 50 niM Tris pH 7.4, 120 mM NaCl, 5 mM MgC12, 2 mM EGTA, 0.1% BSA, 0.1 mM bacitracin, 100 KIU/mL aprotin
- non-radiolabeled test compounds e.g., compounds provided herein
- Neutral antagonists are those test compounds that reduce the MCH stimulated GTP binding activity towards, but not below, baseline (the level of GTP bound by membranes in this assay in the absence of added MCH or other agonist and in the further absence of any test compound).
- An antagonist test compound that elevates GTP binding activity above baseline in the absence of added MCH in this GTP binding assay is characterized as having partial agonist activity.
- Preferred antagonist compounds described herein do not elevate GTP binding activity under such conditions more than 10% above baseline, preferably not more than 5% above baseline, and most preferably not more than 2% above baseline.
- G-alpha-bound (and thereby membrane-bound) GTP gamma 35 S is determined by measuring the bound radioactivity, preferably by liquid scintillation spectrometry of the washed filters.
- Non-specific binding is determined using 10 mM GTP gamma 35 S and typically represents less than 10% of total binding. Data is expressed as percent above basal (baseline).
- SIGMAPLOT software is then used to generate K; as described by Cheng and Prusoff (1973) Biochem Pharmacol. 22(23):3099-10&.
- Preferred compounds are MCHl receptor antagonists that do not possess significant (e.g., greater than 5%) agonist activity in any of the MCH mediated functional assays discussed herein. Specifically, this undesired agonist activity can be evaluated, for example, in the GTP binding assay described above, by measuring small molecule mediated GTP binding in the absence of the agonist, MCH.
- the preferred extent of MCHlR agonist activity exhibited by compounds of the invention is less than 10%, more preferably less than 5% and most preferably less than 2% of the response elicited by the agonist, MCH.
- This Example illustrates a standard assay of melanin concentrating hormone receptor binding that may be used to determine the binding affinity of compounds for the MCH receptor.
- Cynomolgus macaque hypothalamus MCHl cDNA is prepared and cloned into PCDNA3.1 (INVITROGEN Corp., Carlsbad, CA), and HEK293 cells (American Type Culture Collection, Manassas, VA) are stably transfected with the MCHl expression vector as described in PCT International Application publication number WO 03/059289, which published on July 24, 2003.
- the disclosure of WO 03/059289 at page 52 directed to the preparation and storage of the transfected HEK293 cells is hereby incorporated by reference.
- pellets are thawed by addition of wash buffer (25 mM Hepes with 1.0 mM CaCl 2 , 5.0 mM MgCl 2 , 120 mM NaCl, pH 7.4) and homogenized for 30 seconds using a BRINKMAN POLYTRON, setting 5.
- wash buffer 25 mM Hepes with 1.0 mM CaCl 2 , 5.0 mM MgCl 2 , 120 mM NaCl, pH 7.4
- BRINKMAN POLYTRON setting 5.
- Cells are centrifuged for 10 minutes at 48,000 x g. The supernatant is discarded and the pellet is resuspended in fresh wash buffer, and homogenized again. An aliquot of this membrane homogenate is used to determine protein concentration via the Bradford method (BIO-RAD Protein Assay Kit, #500-0001, BIO-RAD, Hercules, CA).
- a 1- liter culture of cells typically yields 50-75 mg of total membrane protein.
- the homogenate is centrifuged as before and resuspended to a protein concentration of 333 ⁇ g/mL in binding buffer (Wash buffer + 0.1% BSA and 1.0 ⁇ M final phosphoramidon) for an assay volume of 50 ⁇ g membrane protein/150 ⁇ L binding buffer.
- Phosphoramidon was from SIGMA BIOCHEMICALS, St. Louis, MO (cat# R-7385). Competition binding assays are performed at room temperature in Falcon 96 well round bottom polypropylene plates.
- Each assay well contains 150 ⁇ L of MCH receptor-containing membranes prepared as described above, 50 ⁇ L 125 I-Tyr MCH, 50 ⁇ L binding buffer, and 2 ⁇ L test compound in DMSO.
- Non-specific binding is defined as the binding measured in the presence of 1 ⁇ M unlabeled MCH.
- MCH is purchased from BACHEM U.S.A., King of Prussia, PA (cat # H-1482).
- Assay wells used to determine MCH binding contain 150 ⁇ L of MCH receptor containing membranes, 50 ⁇ L 125 I- Tyr MCH, 25 ⁇ L binding buffer and 25 ⁇ L binding buffer. Assay plates are incubated for 1 hour at room temperature. Membranes are harvested onto
- PEI polyethyleneimine
- the concentration of I25 I-Tyr MCH is varied from 7 to 1,000 pM.
- K values are below 1 micromolar, preferably below 500 nanomolar, more preferably below 100 nanomolar.
- This Example illustrates a representative functional assay for monitoring the response of cells expressing melanin concentrating hormone receptors to melanin concentrating hormone. This assay can also be used to determine if test compounds act as agonists or antagonists of melanin concentrating hormone receptors.
- Chinese Hamster Ovary (CHO) cells (American Type Culture Collection; Manassas, VA) are stably transfected with the MCH expression vector via calcium phosphate precipitation, and are grown to a density of 15,000 cells/well in FALCONTM black-walled, clear-bottomed 96-well plates (#3904, BECTON-DICKINSON, Franklin Lakes, NJ) in Ham's F12 culture medium (MEDIATECH, Herndon, VA) supplemented with 10% fetal bovine serum, 25 mM HEPES and 500 ⁇ g/mL (active) G418. Prior to running the assay, the culture medium is emptied from the 96 well plates.
- Fluo-3 calcium sensitive dye (Molecular Probes, Eugene, OR) is added to each well (dye solution: 1 mg FLUO-3 AM, 440 ⁇ L DMSO and 440 ⁇ L 20% pluronic acid in DMSO, diluted 1:4, 50 ⁇ L diluted solution per well). Plates are covered with aluminum foil and incubated at 37 0 C for 1-2 hours.
- KRH buffer 0.05 mM KCl, 0.115 M NaCl, 9.6 mM NaH 2 PO 4 , 0.01 mM MgSO 4 , 25 mM HEPES, pH 7.4
- Fluorescence response is monitored upon the addition of either human MCH receptor or test compound by a FLDPRTM plate reader (Molecular Devices, Sunnyvale, CA) by excitation at 480 nm and emission at 530 nm.
- the EC 50 of MCH is first determined. An additional 20 ⁇ L of KRH buffer and 1 ⁇ L DMSO are added to each well of cells, prepared as described above. 100 ⁇ L human MCH in KRH buffer is automatically transferred by the FLEPR instrument to each well. An 8- point concentration response curve, with final MCH concentrations of 1 nM to 3 ⁇ M, is used to determine MCH EC 50 .
- Test compounds are dissolved in DMSO, diluted in 20 ⁇ L KRH buffer, and added to cells prepared as described above.
- the 96 well plates containing prepared cells and test compounds are incubated in the dark, at room temperature for 0.5-6 hours. It is important that the incubation not continue beyond 6 hours.
- 100 ⁇ L human MCH diluted in KRH buffer to 2 x EC 50 is automatically added by the FLLPR instrument to each well of the 96 well plate for a final sample volume of 200 ⁇ L and a final MCH concentration Of EC 50 .
- the final concentration of test compounds in the assay wells is between 1 nM and 5 ⁇ M.
- cells exposed to one EC 50 of MCH exhibit a fluorescence response of about 10,000 Relative Fluorescence Units.
- Cells incubated with antagonists of the MCH receptor exhibit a response that is significantly less than that of the control cells to the p ⁇ 0.05 level, as measured using a parametric test of statistical significance.
- antagonists of the MCH receptor decrease the fluorescence response by about 20%, preferably by about 50%, and most preferably by at least 80% as compared to matched controls.
- the ability of a compound to act as an agonist of the MCH receptor is determined by measuring the fluorescence response of cells expressing MCH receptors, using the methods described above, in the absence of MCH. Compounds that cause cells to exhibit fluorescence above background are MCH receptor agonists.
- This Example illustrates the evaluation of compound toxicity using a Madin Darby canine kidney (MDCK) cell cytotoxicity assay.
- test compound 1 ⁇ L is added to each well of a clear bottom 96-well plate (PACKARD, Meriden, CT) to give final concentration of compound in the assay of 10 ⁇ M, 100 ⁇ M or 200 ⁇ M. Solvent without test compound is added to control wells.
- MDCK cells ATCC no. CCL-34 (American Type Culture Collection, Manassas, VA), are maintained in sterile conditions following the instructions in the ATCC production information sheet.
- Confluent MDCK cells are trypsinized, harvested, and diluted to a concentration of 0.1 x 10 6 cells/mL with warm (37 0 C) medium (VITACELL Minimum Essential Medium Eagle, ATCC catalog # 30- 2003). 100 ⁇ L of diluted cells is added to each well, except for five standard curve control wells that contain 100 ⁇ L of warm medium without cells. The plate is then incubated at 37°C under 95% O 2 , 5% CO 2 for 2 hours with constant shaking.
- mammalian cell lysis solution from the PACKARD (Meriden, CT) ATP-LITE-M Luminescent ATP detection kit
- PACKARD TOPSEAL stickers from the PACKARD (Meriden, CT) ATP-LITE-M Luminescent ATP detection kit
- the ATP-LITE-M Luminescent ATP detection kit is generally used according to the manufacturer's instructions to measure ATP production in treated and untreated MDCK cells. PACKARD ATP LITE-M reagents are allowed to equilibrate to room temperature. Once equilibrated, the lyophilized substrate solution is reconstituted in 5.5 mL of substrate buffer solution (from kit). Lyophilized ATP standard solution is reconstituted in deionized water to give a 10 mM stock.
- PACKARD substrate solution 50 ⁇ L is added to all wells, which are then covered, and the plates are shaken at approximately 700 rpm on a suitable shaker for 2 minutes.
- a white PACKARD sticker is attached to the bottom of each plate and samples are dark adapted by wrapping plates in foil and placing in the dark for 10 minutes.
- Luminescence is then measured at 22°C using a luminescence counter (e.g., PACKARD TOPCOUNT Microplate Scintillation and Luminescence Counter or TECAN SPECTRAFLUOR PLUS), and ATP levels calculated from the standard curve.
- ATP levels in cells treated with test compound(s) are compared to the levels determined for untreated cells.
- Cells treated with 10 ⁇ M of a preferred test compound exhibit ATP levels that are at least 80%, preferably at least 90%, of the untreated cells.
- a 100 ⁇ M concentration of the test compound is used, cells treated with preferred test compounds exhibit ATP levels that are at least 50%, preferably at least 80%, of the ATP levels detected in untreated cells.
- This Example illustrates the evaluation of compound half-life values (ti /2 values) using a representative liver microsomal half-life assay.
- liver microsomes are obtained from XenoTech LLC (Kansas City, KS). Such liver microsomes may also be obtained from Ih Vitro Technologies (Baltimore, MD) or Tissue Transformation Technologies (Edison, NJ). Six test reactions are prepared, each containing 25 ⁇ L microsomes, 5 ⁇ L of a 100 ⁇ M solution of test compound, and 399 ⁇ L 0.1 M phosphate buffer (19 mL 0.1 M NaH 2 PO 4 , 81 mL 0.1 M Na 2 HPO 4 , adjusted to pH 7.4 with H 3 PO 4 ).
- a seventh reaction is prepared as a positive control containing 25 ⁇ L microsomes, 399 ⁇ L 0.1 M phosphate buffer, and 5 ⁇ L of a 100 ⁇ M solution of a compound with known metabolic properties (e.g., DIAZEPAM or CLOZAPINE). Reactions are preincubated at 39 0 C for 10 minutes.
- a compound with known metabolic properties e.g., DIAZEPAM or CLOZAPINE
- Cofactor mixture is prepared by diluting 16.2 mg NADP and 45.4 mg glucose-6-phosphate in 4 mL 100 mM MgCl 2 .
- Glucose-6-phosphate dehydrogenase solution is prepared by diluting 214.3 ⁇ L glucose-6-phosphate dehydrogenase suspension (Roche Molecular Biochemicals; Indianapolis, IN) into 1285.7 ⁇ L distilled water.
- 71 ⁇ L of starting reaction mixture (3 mL cofactor mixture; 1.2 mL glucose-6-phosphate dehydrogenase solution) is added to 5 of the 6 test reactions and to the positive control.
- 71 ⁇ L 100 mM MgCl 2 is added to the sixth test reaction, which is used as a negative control.
- 75 ⁇ L of each reaction mix is pipetted into a well of a 96-well deep-well plate containing 75 ⁇ L ice-cold acetonitrile.
- Samples are vortexed and centrifuged 10 minutes at 3500 rpm (Sorval T 6000D centrifuge, HlOOOB rotor).
- 75 ⁇ L of supernatant from each reaction is transferred to a well of a 96-well plate containing 150 ⁇ L of a 0.5 ⁇ M solution of a compound with a known LCMS profile (internal standard) per well.
- LCMS analysis of each sample is carried out and the amount of unmetabolized test compound is measured as AUC, compound concentration vs. time is plotted, and the t 1/2 value of the test compound is extrapolated.
- Preferred compounds provided herein exhibit in vitro t 1/2 values of greater than 10 minutes and less than 4 hours, preferably between 30 minutes and 1 hour, in human liver microsomes.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Hematology (AREA)
- Diabetes (AREA)
- Physical Education & Sports Medicine (AREA)
- Psychiatry (AREA)
- Rheumatology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Obesity (AREA)
- Heart & Thoracic Surgery (AREA)
- Child & Adolescent Psychology (AREA)
- Endocrinology (AREA)
- Reproductive Health (AREA)
- Dermatology (AREA)
- Pain & Pain Management (AREA)
- Hospice & Palliative Care (AREA)
- Cardiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicinal Preparation (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description


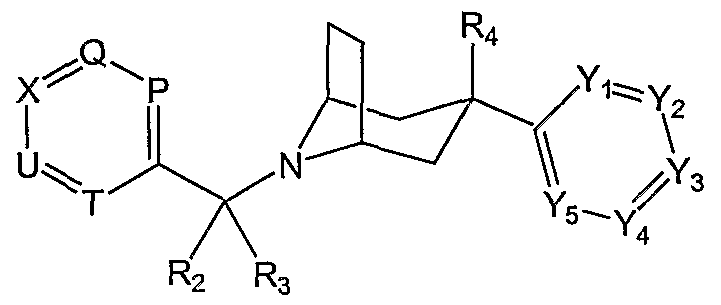


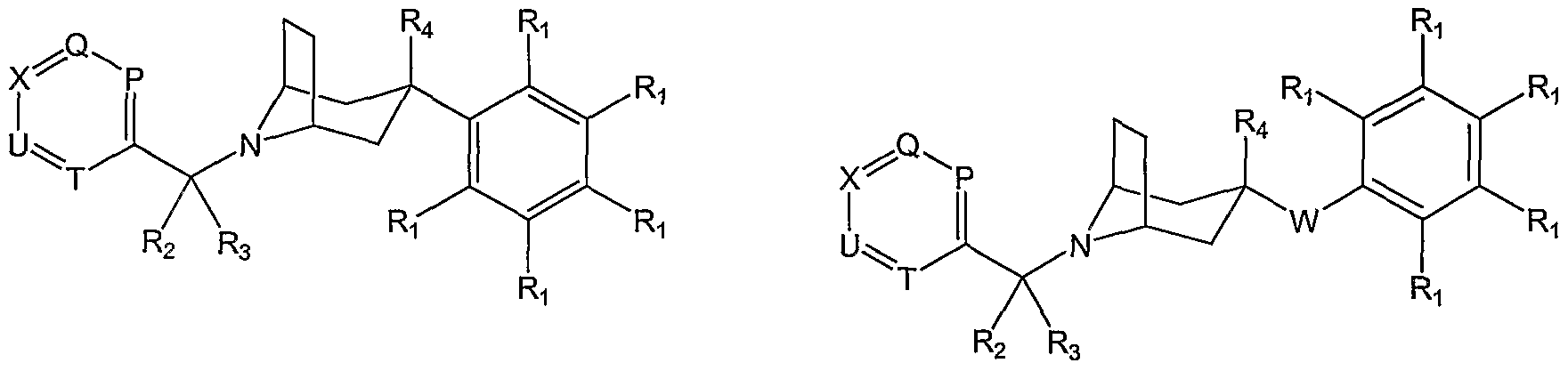




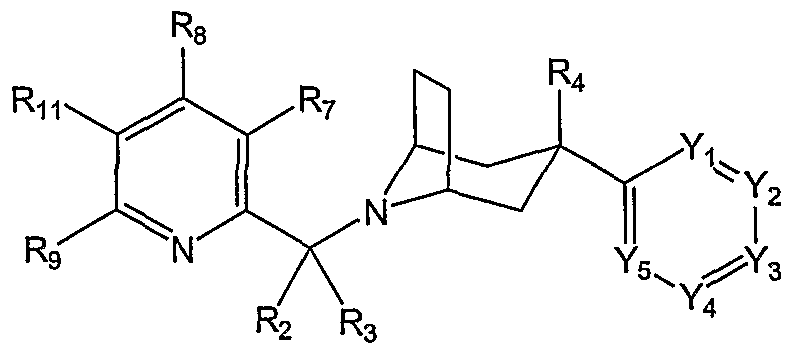














Claims

Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA002581745A CA2581745A1 (en) | 2004-10-13 | 2005-10-04 | Aryl substituted 8-azabicyclo[3.2.1]octane compounds as ligands of the melanin concentrating hormone receptor |
| EP05812516A EP1809626A2 (en) | 2004-10-13 | 2005-10-04 | Aryl substituted 8-azabicyclo[3.2.1]octane compounds as ligands of the melanin concentrating hormone receptor |
| AU2005296124A AU2005296124A1 (en) | 2004-10-13 | 2005-10-04 | Aryl substituted 8-azabicyclo[3.2.1]octane compounds as ligands of the melanin concentrating hormone receptor |
| JP2007536725A JP2008515972A (en) | 2004-10-13 | 2005-10-04 | Aryl-substituted 8-azabicyclo [3.2.1] octane compounds as ligands for melanin-concentrating hormone receptors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US61829204P | 2004-10-13 | 2004-10-13 | |
| US60/618,292 | 2004-10-13 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| WO2006044174A2 true WO2006044174A2 (en) | 2006-04-27 |
| WO2006044174A3 WO2006044174A3 (en) | 2006-06-08 |
| WO2006044174A9 WO2006044174A9 (en) | 2006-07-20 |
Family
ID=36088256
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2005/035457 Ceased WO2006044174A2 (en) | 2004-10-13 | 2005-10-04 | Aryl substituted 8-azabicyclo[3.2.1]octane compounds as ligands of the melanin concentrating hormone receptor |
Country Status (5)
| Country | Link |
|---|---|
| EP (1) | EP1809626A2 (en) |
| JP (1) | JP2008515972A (en) |
| AU (1) | AU2005296124A1 (en) |
| CA (1) | CA2581745A1 (en) |
| WO (1) | WO2006044174A2 (en) |
Cited By (39)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| FR2911874A1 (en) * | 2007-01-25 | 2008-08-01 | Sanofi Aventis Sa | New urea derivatives of tropane are 11-beta-hydroxy steroid dehydrogenase type 1 modulators, useful to treat e.g. obesity, diabetes, insulin resistance, metabolic syndrome, Cushing's syndrome, hypertension and atherosclerosis |
| WO2009137270A3 (en) * | 2008-05-09 | 2009-12-30 | H. Lundbeck A/S | Azetidine derivatives |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| WO2010009195A1 (en) * | 2008-07-16 | 2010-01-21 | Schering Corporation | Bicyclic heterocycle derivatives and use thereof as gpr119 modulators |
| WO2010023161A1 (en) * | 2008-08-25 | 2010-03-04 | Boehringer Ingelheim International Gmbh | Aryl- and heteroarylcarbonyl derivatives of substituted nortropanes, medicaments containing such compounds and their use |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120050A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| WO2012120051A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Benzyl-oxathiazine derivates substituted with adamantane or noradamantane, medicaments containing said compounds and use thereof |
| WO2012120058A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives which are substituted with benzyl or heteromethylene groups, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120057A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| US8497281B2 (en) | 2009-11-06 | 2013-07-30 | Vitae Pharmaceuticals, Inc. | Aryl- and heteroarylcarbonyl derivatives of hexahydroindenopyridine and octahydrobenzoquinoline |
| US8637505B2 (en) | 2009-02-04 | 2014-01-28 | Boehringer Ingelheim International Gmbh | Cyclic inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| US8673899B2 (en) | 2008-05-01 | 2014-03-18 | Vitae Pharmaceuticals, Inc. | Cyclic inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| US8680093B2 (en) | 2009-04-30 | 2014-03-25 | Vitae Pharmaceuticals, Inc. | Cyclic inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| US8686149B2 (en) | 2010-11-05 | 2014-04-01 | Boehringer-Ingelheim International Gmbh | Aryl- and heteroarylcarbonyl derivatives of hexahydroindenopyridine and octahydrobenzoquinoline |
| US8703765B2 (en) | 2009-06-02 | 2014-04-22 | Boehringer Ingelheim International Gmbh | Cyclic inhibitors of 11β-hydroxysteroid dehydrogenase 1 |
| US8735585B2 (en) | 2011-08-17 | 2014-05-27 | Boehringer Ingelheim International Gmbh | Indenopyridine derivatives |
| US8748444B2 (en) | 2007-12-11 | 2014-06-10 | Vitae Pharmaceuticals, Inc. | Cyclic urea inhibitors of 11β-hydroxysteroid dehydrogenase 1 |
| US8754076B2 (en) | 2008-07-25 | 2014-06-17 | Vitae Pharmaceuticals, Inc./Boehringer-Ingelheim | Cyclic inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| US8765780B2 (en) | 2008-05-13 | 2014-07-01 | Boehringer Ingelheim International Gmbh | Alicyclic carboxylic acid derivatives of benzomorphans and related scaffolds, medicaments containing such compounds and their use |
| US8765744B2 (en) | 2010-06-25 | 2014-07-01 | Boehringer Ingelheim International Gmbh | Azaspirohexanones |
| US8829027B2 (en) | 2008-10-23 | 2014-09-09 | Boehringer Ingelheim International Gmbh | Urea derivatives of substituted nortropanes, medicaments containing such compounds and their use |
| US8846668B2 (en) | 2008-07-25 | 2014-09-30 | Vitae Pharmaceuticals, Inc. | Inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| US8846613B2 (en) | 2010-11-02 | 2014-09-30 | Boehringer Ingelheim International Gmbh | Pharmaceutical combinations for the treatment of metabolic disorders |
| US8859580B2 (en) | 2007-11-16 | 2014-10-14 | Boehringer Ingelheim International Gmbh | Aryl- and heteroarylcarbonyl derivatives of benzomorphanes and related scaffolds, medicaments containing such compounds and their use |
| US8883778B2 (en) | 2009-07-01 | 2014-11-11 | Vitae Pharmaceuticals, Inc. | Cyclic inhibitors of 11 beta-hydroxysteroid dehydrogenase 1 |
| US8927539B2 (en) | 2009-06-11 | 2015-01-06 | Vitae Pharmaceuticals, Inc. | Cyclic inhibitors of 11β-hydroxysteroid dehydrogenase 1 based on the 1,3-oxazinan-2-one structure |
| US8933072B2 (en) | 2010-06-16 | 2015-01-13 | Vitae Pharmaceuticals, Inc. | Substituted 5-,6- and 7-membered heterocycles, medicaments containing such compounds, and their use |
| US10800757B2 (en) | 2017-10-27 | 2020-10-13 | Boehringer Ingelheim International Gmbh | Inhibitors of TRPC6 |
| US10906888B2 (en) | 2016-07-14 | 2021-02-02 | Pfizer Inc. | Pyrimidine carboxamides as inhibitors of Vanin-1 enzyme |
| US12059408B2 (en) | 2020-08-13 | 2024-08-13 | Boehringer Ingelheim International Gmbh | Treatment of cognitive impairment associated with schizophrenia |
| US12275722B2 (en) | 2020-10-13 | 2025-04-15 | Boehringer Ingelheim International Gmbh | Process of reworking a crystalline form of a glycine transport inhibitor |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2678577A1 (en) | 2007-02-26 | 2008-09-04 | Vitae Pharmaceuticals, Inc. | Cyclic urea and carbamate inhibitors of 11.beta.-hydroxysteroid dehydrogenase 1 |
| AR069207A1 (en) | 2007-11-07 | 2010-01-06 | Vitae Pharmaceuticals Inc | CYCLIC UREAS AS INHIBITORS OF THE 11 BETA - HIDROXI-ESTEROIDE DESHIDROGENASA 1 |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MXPA03000174A (en) * | 2000-07-06 | 2003-09-22 | Neurogen Corp | Melanin concentrating hormone receptor ligands. |
| JP2005515961A (en) * | 2001-05-22 | 2005-06-02 | ニューロジェン コーポレイション | Melanin-concentrating hormone receptor ligand: substituted 1-benzyl-4-arylpiperazine analogues |
-
2005
- 2005-10-04 EP EP05812516A patent/EP1809626A2/en not_active Withdrawn
- 2005-10-04 JP JP2007536725A patent/JP2008515972A/en active Pending
- 2005-10-04 AU AU2005296124A patent/AU2005296124A1/en not_active Abandoned
- 2005-10-04 WO PCT/US2005/035457 patent/WO2006044174A2/en not_active Ceased
- 2005-10-04 CA CA002581745A patent/CA2581745A1/en not_active Abandoned
Cited By (51)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008017381A1 (en) | 2006-08-08 | 2008-02-14 | Sanofi-Aventis | Arylaminoaryl-alkyl-substituted imidazolidine-2,4-diones, processes for preparing them, medicaments comprising these compounds, and their use |
| FR2911874A1 (en) * | 2007-01-25 | 2008-08-01 | Sanofi Aventis Sa | New urea derivatives of tropane are 11-beta-hydroxy steroid dehydrogenase type 1 modulators, useful to treat e.g. obesity, diabetes, insulin resistance, metabolic syndrome, Cushing's syndrome, hypertension and atherosclerosis |
| US8859580B2 (en) | 2007-11-16 | 2014-10-14 | Boehringer Ingelheim International Gmbh | Aryl- and heteroarylcarbonyl derivatives of benzomorphanes and related scaffolds, medicaments containing such compounds and their use |
| US8748444B2 (en) | 2007-12-11 | 2014-06-10 | Vitae Pharmaceuticals, Inc. | Cyclic urea inhibitors of 11β-hydroxysteroid dehydrogenase 1 |
| US8673899B2 (en) | 2008-05-01 | 2014-03-18 | Vitae Pharmaceuticals, Inc. | Cyclic inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| WO2009137270A3 (en) * | 2008-05-09 | 2009-12-30 | H. Lundbeck A/S | Azetidine derivatives |
| US8765780B2 (en) | 2008-05-13 | 2014-07-01 | Boehringer Ingelheim International Gmbh | Alicyclic carboxylic acid derivatives of benzomorphans and related scaffolds, medicaments containing such compounds and their use |
| US9073870B2 (en) | 2008-05-13 | 2015-07-07 | Boehringer Ingelheim International Gmbh | Alicyclic carboxylic acid derivatives of benzomorphans and related scaffolds, medicaments containing such compounds and their use |
| WO2010003624A2 (en) | 2008-07-09 | 2010-01-14 | Sanofi-Aventis | Heterocyclic compounds, processes for their preparation, medicaments comprising these compounds, and the use thereof |
| CN102159572A (en) * | 2008-07-16 | 2011-08-17 | 先灵公司 | Bicyclic heterocycle derivatives and use thereof as gpr119 modulators |
| US8822480B2 (en) | 2008-07-16 | 2014-09-02 | Merck Sharp & Dohme Corp. | Bicyclic heterocycle derivatives and use thereof as GPR119 modulators |
| WO2010009195A1 (en) * | 2008-07-16 | 2010-01-21 | Schering Corporation | Bicyclic heterocycle derivatives and use thereof as gpr119 modulators |
| US8846668B2 (en) | 2008-07-25 | 2014-09-30 | Vitae Pharmaceuticals, Inc. | Inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| US8754076B2 (en) | 2008-07-25 | 2014-06-17 | Vitae Pharmaceuticals, Inc./Boehringer-Ingelheim | Cyclic inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| JP2012500821A (en) * | 2008-08-25 | 2012-01-12 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Aryl and heteroarylcarbonyl derivatives of substituted nortropanes, medicaments containing said compounds and uses thereof |
| US8609690B2 (en) | 2008-08-25 | 2013-12-17 | Boehringer Ingelheim International Gmbh | Aryl- and heteroarylcarbonyl derivatives of substituted nortropanes, medicaments containing such compounds and their use |
| WO2010023161A1 (en) * | 2008-08-25 | 2010-03-04 | Boehringer Ingelheim International Gmbh | Aryl- and heteroarylcarbonyl derivatives of substituted nortropanes, medicaments containing such compounds and their use |
| US8829027B2 (en) | 2008-10-23 | 2014-09-09 | Boehringer Ingelheim International Gmbh | Urea derivatives of substituted nortropanes, medicaments containing such compounds and their use |
| WO2010068601A1 (en) | 2008-12-08 | 2010-06-17 | Sanofi-Aventis | A crystalline heteroaromatic fluoroglycoside hydrate, processes for making, methods of use and pharmaceutical compositions thereof |
| US8637505B2 (en) | 2009-02-04 | 2014-01-28 | Boehringer Ingelheim International Gmbh | Cyclic inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| US8680093B2 (en) | 2009-04-30 | 2014-03-25 | Vitae Pharmaceuticals, Inc. | Cyclic inhibitors of 11beta-hydroxysteroid dehydrogenase 1 |
| US8703765B2 (en) | 2009-06-02 | 2014-04-22 | Boehringer Ingelheim International Gmbh | Cyclic inhibitors of 11β-hydroxysteroid dehydrogenase 1 |
| US8927539B2 (en) | 2009-06-11 | 2015-01-06 | Vitae Pharmaceuticals, Inc. | Cyclic inhibitors of 11β-hydroxysteroid dehydrogenase 1 based on the 1,3-oxazinan-2-one structure |
| US8883778B2 (en) | 2009-07-01 | 2014-11-11 | Vitae Pharmaceuticals, Inc. | Cyclic inhibitors of 11 beta-hydroxysteroid dehydrogenase 1 |
| WO2011023754A1 (en) | 2009-08-26 | 2011-03-03 | Sanofi-Aventis | Novel crystalline heteroaromatic fluoroglycoside hydrates, pharmaceuticals comprising these compounds and their use |
| US8497281B2 (en) | 2009-11-06 | 2013-07-30 | Vitae Pharmaceuticals, Inc. | Aryl- and heteroarylcarbonyl derivatives of hexahydroindenopyridine and octahydrobenzoquinoline |
| US9663470B2 (en) | 2009-11-06 | 2017-05-30 | Vitae Pharmaceuticals, Inc. | Aryl- and heteroarylcarbonyl derivatives of hexahydroindenopyridine and octahydrobenzoquinoline |
| US9328072B2 (en) | 2009-11-06 | 2016-05-03 | Vitae Pharmaceuticals, Inc. | Aryl-and heteroarylcarbonyl derivatives of hexahydroindenopyridine and octahydrobenzoquinoline |
| US9090605B2 (en) | 2010-06-16 | 2015-07-28 | Vitae Pharmaceuticals, Inc. | Substituted 5-,6- and 7-membered heterocycles, medicaments containing such compounds, and their use |
| US8933072B2 (en) | 2010-06-16 | 2015-01-13 | Vitae Pharmaceuticals, Inc. | Substituted 5-,6- and 7-membered heterocycles, medicaments containing such compounds, and their use |
| US8765744B2 (en) | 2010-06-25 | 2014-07-01 | Boehringer Ingelheim International Gmbh | Azaspirohexanones |
| US8846613B2 (en) | 2010-11-02 | 2014-09-30 | Boehringer Ingelheim International Gmbh | Pharmaceutical combinations for the treatment of metabolic disorders |
| US8686149B2 (en) | 2010-11-05 | 2014-04-01 | Boehringer-Ingelheim International Gmbh | Aryl- and heteroarylcarbonyl derivatives of hexahydroindenopyridine and octahydrobenzoquinoline |
| US9120769B2 (en) | 2010-11-05 | 2015-09-01 | Boehringer-Ingelheim International Gmbh | Aryl-and heteroarylcarbonyl derivatives of hexahydroindenopyridine and octahydrobenzoquinoline |
| WO2012120052A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives substituted with carbocycles or heterocycles, method for producing same, drugs containing said compounds, and use thereof |
| WO2012120053A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Branched oxathiazine derivatives, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120056A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Tetrasubstituted oxathiazine derivatives, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120054A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120051A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Benzyl-oxathiazine derivates substituted with adamantane or noradamantane, medicaments containing said compounds and use thereof |
| WO2012120055A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Di- and tri-substituted oxathiazine derivates, method for the production thereof, use thereof as medicine and drug containing said derivatives and use thereof |
| WO2012120058A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Oxathiazine derivatives which are substituted with benzyl or heteromethylene groups, method for producing them, their use as medicine and drug containing said derivatives and the use thereof |
| WO2012120057A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| WO2012120050A1 (en) | 2011-03-08 | 2012-09-13 | Sanofi | Novel substituted phenyl-oxathiazine derivatives, method for producing them, drugs containing said compounds and the use thereof |
| US8735585B2 (en) | 2011-08-17 | 2014-05-27 | Boehringer Ingelheim International Gmbh | Indenopyridine derivatives |
| US8975405B2 (en) | 2011-08-17 | 2015-03-10 | Boehringer Ingelheim International Gmbh | Indenopyridine derivatives |
| US10906888B2 (en) | 2016-07-14 | 2021-02-02 | Pfizer Inc. | Pyrimidine carboxamides as inhibitors of Vanin-1 enzyme |
| US10800757B2 (en) | 2017-10-27 | 2020-10-13 | Boehringer Ingelheim International Gmbh | Inhibitors of TRPC6 |
| US10889568B2 (en) | 2017-10-27 | 2021-01-12 | Boehringer Ingelheim International Gmbh | Inhibitors of TRPC6 |
| USRE49699E1 (en) | 2017-10-27 | 2023-10-17 | Boehringer Ingelheim International Gmbh | Inhibitors of TRPC6 |
| US12059408B2 (en) | 2020-08-13 | 2024-08-13 | Boehringer Ingelheim International Gmbh | Treatment of cognitive impairment associated with schizophrenia |
| US12275722B2 (en) | 2020-10-13 | 2025-04-15 | Boehringer Ingelheim International Gmbh | Process of reworking a crystalline form of a glycine transport inhibitor |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2005296124A1 (en) | 2006-04-27 |
| CA2581745A1 (en) | 2006-04-27 |
| WO2006044174A3 (en) | 2006-06-08 |
| WO2006044174A9 (en) | 2006-07-20 |
| EP1809626A2 (en) | 2007-07-25 |
| JP2008515972A (en) | 2008-05-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2006044174A2 (en) | Aryl substituted 8-azabicyclo[3.2.1]octane compounds as ligands of the melanin concentrating hormone receptor | |
| US20070249616A1 (en) | Substituted 1-Benzyl-4-Substituted Piperazine Analogues | |
| AU774867B2 (en) | Heteroaryl diazabicycloalkanes, their preparation and use | |
| US6815438B2 (en) | Heteroaryl-diazabicycloalkanes | |
| US20060009456A1 (en) | Aryl-substituted piperazine derivatives | |
| US7160879B2 (en) | Melanin concentrating hormone receptor ligands: substituted 2-(4-benzyl-piperazin-1-ylmethyl)- and 2-(4-benzyl-diazepan-1-ylmethyl)-1H-benzoimidazole analogues | |
| JP2007077144A (en) | Amino-substituted pyrazolo [1,5-a] -1,5-pyrimidines and pyrazolo [1,5-a] -1,3,5-triazines | |
| US6680328B2 (en) | 8-azabicyclo[3.2.1]oct-2-ene and -octane derivatives | |
| US6906075B2 (en) | Melanin concentrating hormone receptor ligands: substituted benzoimidazole analogues | |
| US7064118B2 (en) | Heteroaryl diazacycloalkanes, their preparation and use | |
| WO2006015279A1 (en) | Heterocyclic diamine compounds as ligands of the melanin concentrating hormone receptor useful for the treatment of obesity, diabetes, eating and sexual disorders | |
| CA2641767A1 (en) | Novel diazabicycloalkane derivatives and their medical use | |
| JP2003137872A (en) | Substituted 2-cyclohexyl-4-phenyl-1h-imidazole derivative | |
| US20050239791A1 (en) | Substituted 1-heteroaryl-4-substituted piperazine and piperidine analogues | |
| US20080161344A1 (en) | Melanin Concentrating Hormone Receptor Ligands: Substituted Tetrahydroisoquinoline Analogues |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KM KP KR KZ LC LK LR LS LT LU LV LY MA MD MG MK MN MW MX MZ NA NG NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SM SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU LV MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2005296124 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005812516 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2581745 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2005296124 Country of ref document: AU Date of ref document: 20051004 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005296124 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007536725 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005812516 Country of ref document: EP |