WO2006042027A2 - Immunogenic and therapeutic compositions for streptococcus pyogenes - Google Patents
Immunogenic and therapeutic compositions for streptococcus pyogenes Download PDFInfo
- Publication number
- WO2006042027A2 WO2006042027A2 PCT/US2005/036009 US2005036009W WO2006042027A2 WO 2006042027 A2 WO2006042027 A2 WO 2006042027A2 US 2005036009 W US2005036009 W US 2005036009W WO 2006042027 A2 WO2006042027 A2 WO 2006042027A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- antigen
- gas
- antigens
- protein
- proteins listed
- Prior art date
Links
- 239000000203 mixture Substances 0.000 title claims description 173
- 241000193996 Streptococcus pyogenes Species 0.000 title claims description 39
- 230000002163 immunogen Effects 0.000 title description 35
- 230000001225 therapeutic effect Effects 0.000 title description 10
- 108091007433 antigens Proteins 0.000 claims abstract description 671
- 102000036639 antigens Human genes 0.000 claims abstract description 671
- 239000000427 antigen Substances 0.000 claims abstract description 669
- 208000015181 infectious disease Diseases 0.000 claims abstract description 41
- 230000036039 immunity Effects 0.000 claims abstract description 13
- 108090000623 proteins and genes Proteins 0.000 claims description 342
- 102000004169 proteins and genes Human genes 0.000 claims description 328
- 108090000765 processed proteins & peptides Proteins 0.000 claims description 114
- 102000004196 processed proteins & peptides Human genes 0.000 claims description 98
- 238000000034 method Methods 0.000 claims description 80
- 241000894006 Bacteria Species 0.000 claims description 72
- 239000002671 adjuvant Substances 0.000 claims description 69
- 229960005486 vaccine Drugs 0.000 claims description 64
- 230000001580 bacterial effect Effects 0.000 claims description 56
- 150000001413 amino acids Chemical class 0.000 claims description 54
- 108020004707 nucleic acids Proteins 0.000 claims description 39
- 102000039446 nucleic acids Human genes 0.000 claims description 39
- 150000007523 nucleic acids Chemical class 0.000 claims description 39
- 229920001184 polypeptide Polymers 0.000 claims description 39
- 238000004519 manufacturing process Methods 0.000 claims description 31
- 125000003275 alpha amino acid group Chemical group 0.000 claims description 21
- 239000013543 active substance Substances 0.000 claims description 15
- 101710085938 Matrix protein Proteins 0.000 claims description 13
- 101710127721 Membrane protein Proteins 0.000 claims description 13
- 239000003814 drug Substances 0.000 claims description 13
- 102000014914 Carrier Proteins Human genes 0.000 claims description 12
- 101710116435 Outer membrane protein Proteins 0.000 claims description 10
- 239000013604 expression vector Substances 0.000 claims description 10
- 101150077246 gas5 gene Proteins 0.000 claims description 10
- 230000001939 inductive effect Effects 0.000 claims description 10
- 108010078791 Carrier Proteins Proteins 0.000 claims description 9
- 102100031487 Growth arrest-specific protein 6 Human genes 0.000 claims description 9
- 101000923005 Homo sapiens Growth arrest-specific protein 6 Proteins 0.000 claims description 9
- 230000001086 cytosolic effect Effects 0.000 claims description 8
- 102000004127 Cytokines Human genes 0.000 claims description 7
- 108090000695 Cytokines Proteins 0.000 claims description 7
- 108020001507 fusion proteins Proteins 0.000 claims description 7
- 102000037865 fusion proteins Human genes 0.000 claims description 7
- 241000606768 Haemophilus influenzae Species 0.000 claims description 5
- 206010061598 Immunodeficiency Diseases 0.000 claims description 5
- 101150101889 GAS4 gene Proteins 0.000 claims description 4
- 201000005702 Pertussis Diseases 0.000 claims description 4
- 231100000699 Bacterial toxin Toxicity 0.000 claims description 3
- 241000193163 Clostridioides difficile Species 0.000 claims description 3
- 239000000688 bacterial toxin Substances 0.000 claims description 3
- 239000003937 drug carrier Substances 0.000 claims description 3
- 102100037840 Dehydrogenase/reductase SDR family member 2, mitochondrial Human genes 0.000 claims description 2
- 102000002812 Heat-Shock Proteins Human genes 0.000 claims description 2
- 108010004889 Heat-Shock Proteins Proteins 0.000 claims description 2
- 241000588650 Neisseria meningitidis Species 0.000 claims description 2
- 101710188053 Protein D Proteins 0.000 claims description 2
- 101710132893 Resolvase Proteins 0.000 claims description 2
- 101710084578 Short neurotoxin 1 Proteins 0.000 claims description 2
- 101710182223 Toxin B Proteins 0.000 claims description 2
- 101710182532 Toxin a Proteins 0.000 claims description 2
- 239000003102 growth factor Substances 0.000 claims description 2
- 229940088597 hormone Drugs 0.000 claims description 2
- 239000005556 hormone Substances 0.000 claims description 2
- 238000012258 culturing Methods 0.000 claims 1
- 235000018102 proteins Nutrition 0.000 description 266
- 239000007789 gas Substances 0.000 description 251
- 210000004027 cell Anatomy 0.000 description 68
- 235000001014 amino acid Nutrition 0.000 description 55
- 230000003612 virological effect Effects 0.000 description 35
- 241000700605 Viruses Species 0.000 description 30
- 241000699670 Mus sp. Species 0.000 description 29
- -1 Linker amino acid Chemical group 0.000 description 28
- 230000029087 digestion Effects 0.000 description 25
- 230000028993 immune response Effects 0.000 description 25
- 241000588724 Escherichia coli Species 0.000 description 24
- 108010052285 Membrane Proteins Proteins 0.000 description 24
- 241000282414 Homo sapiens Species 0.000 description 22
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 22
- 239000002953 phosphate buffered saline Substances 0.000 description 22
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 21
- 238000009472 formulation Methods 0.000 description 20
- 239000012634 fragment Substances 0.000 description 20
- 239000002158 endotoxin Substances 0.000 description 19
- 210000002966 serum Anatomy 0.000 description 19
- 238000012413 Fluorescence activated cell sorting analysis Methods 0.000 description 18
- 238000001943 fluorescence-activated cell sorting Methods 0.000 description 18
- 230000003053 immunization Effects 0.000 description 18
- 238000002649 immunization Methods 0.000 description 18
- 229920006008 lipopolysaccharide Polymers 0.000 description 18
- 239000012528 membrane Substances 0.000 description 18
- 239000002773 nucleotide Substances 0.000 description 18
- 125000003729 nucleotide group Chemical group 0.000 description 18
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 18
- 238000004458 analytical method Methods 0.000 description 17
- 210000002421 cell wall Anatomy 0.000 description 17
- 150000001720 carbohydrates Chemical class 0.000 description 16
- 229930182490 saponin Natural products 0.000 description 16
- 150000007949 saponins Chemical class 0.000 description 16
- 235000017709 saponins Nutrition 0.000 description 16
- 238000004885 tandem mass spectrometry Methods 0.000 description 16
- 102000004142 Trypsin Human genes 0.000 description 15
- 108090000631 Trypsin Proteins 0.000 description 15
- 239000012588 trypsin Substances 0.000 description 15
- 102000018697 Membrane Proteins Human genes 0.000 description 14
- 239000003153 chemical reaction reagent Substances 0.000 description 14
- 239000000839 emulsion Substances 0.000 description 14
- 230000004044 response Effects 0.000 description 14
- 239000000243 solution Substances 0.000 description 14
- 102000035195 Peptidases Human genes 0.000 description 13
- 108091005804 Peptidases Proteins 0.000 description 13
- 108010076504 Protein Sorting Signals Proteins 0.000 description 13
- 241000193990 Streptococcus sp. 'group B' Species 0.000 description 13
- 239000002775 capsule Substances 0.000 description 13
- 239000002245 particle Substances 0.000 description 13
- 238000002360 preparation method Methods 0.000 description 13
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 12
- 238000007912 intraperitoneal administration Methods 0.000 description 12
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 12
- 239000000047 product Substances 0.000 description 12
- 239000001397 quillaja saponaria molina bark Substances 0.000 description 12
- 108010067770 Endopeptidase K Proteins 0.000 description 11
- 102000005720 Glutathione transferase Human genes 0.000 description 11
- 108010070675 Glutathione transferase Proteins 0.000 description 11
- 108090001030 Lipoproteins Proteins 0.000 description 11
- 102000004895 Lipoproteins Human genes 0.000 description 11
- 241000699666 Mus <mouse, genus> Species 0.000 description 11
- 108091034117 Oligonucleotide Proteins 0.000 description 11
- 239000004365 Protease Substances 0.000 description 11
- 241000725643 Respiratory syncytial virus Species 0.000 description 11
- 150000004676 glycans Chemical class 0.000 description 11
- 229920001282 polysaccharide Polymers 0.000 description 11
- 239000005017 polysaccharide Substances 0.000 description 11
- 238000011282 treatment Methods 0.000 description 11
- 102100038385 Coiled-coil domain-containing protein R3HCC1L Human genes 0.000 description 10
- 241000711902 Pneumovirus Species 0.000 description 10
- WNROFYMDJYEPJX-UHFFFAOYSA-K aluminium hydroxide Chemical compound [OH-].[OH-].[OH-].[Al+3] WNROFYMDJYEPJX-UHFFFAOYSA-K 0.000 description 10
- 238000010367 cloning Methods 0.000 description 10
- 206010022000 influenza Diseases 0.000 description 10
- 229910052500 inorganic mineral Inorganic materials 0.000 description 10
- JMUHBNWAORSSBD-WKYWBUFDSA-N mifamurtide Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@@H](OC(=O)CCCCCCCCCCCCCCC)COP(O)(=O)OCCNC(=O)[C@H](C)NC(=O)CC[C@H](C(N)=O)NC(=O)[C@H](C)NC(=O)[C@@H](C)O[C@H]1[C@H](O)[C@@H](CO)OC(O)[C@@H]1NC(C)=O JMUHBNWAORSSBD-WKYWBUFDSA-N 0.000 description 10
- 229960005225 mifamurtide Drugs 0.000 description 10
- 239000011707 mineral Substances 0.000 description 10
- 238000000746 purification Methods 0.000 description 10
- 150000003839 salts Chemical class 0.000 description 10
- 239000003053 toxin Substances 0.000 description 10
- 231100000765 toxin Toxicity 0.000 description 10
- 108700012359 toxins Proteins 0.000 description 10
- KIUKXJAPPMFGSW-DNGZLQJQSA-N (2S,3S,4S,5R,6R)-6-[(2S,3R,4R,5S,6R)-3-Acetamido-2-[(2S,3S,4R,5R,6R)-6-[(2R,3R,4R,5S,6R)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-2-carboxy-4,5-dihydroxyoxan-3-yl]oxy-5-hydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-3,4,5-trihydroxyoxane-2-carboxylic acid Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@H](O3)C(O)=O)O)[C@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](C(O)=O)O1 KIUKXJAPPMFGSW-DNGZLQJQSA-N 0.000 description 9
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 9
- 241000709661 Enterovirus Species 0.000 description 9
- 101000743767 Homo sapiens Coiled-coil domain-containing protein R3HCC1L Proteins 0.000 description 9
- 241000701085 Human alphaherpesvirus 3 Species 0.000 description 9
- 208000019802 Sexually transmitted disease Diseases 0.000 description 9
- 230000003115 biocidal effect Effects 0.000 description 9
- 239000000872 buffer Substances 0.000 description 9
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 9
- PRAKJMSDJKAYCZ-UHFFFAOYSA-N dodecahydrosqualene Natural products CC(C)CCCC(C)CCCC(C)CCCCC(C)CCCC(C)CCCC(C)C PRAKJMSDJKAYCZ-UHFFFAOYSA-N 0.000 description 9
- 229920002674 hyaluronan Polymers 0.000 description 9
- 229960003160 hyaluronic acid Drugs 0.000 description 9
- 238000011534 incubation Methods 0.000 description 9
- 244000052769 pathogen Species 0.000 description 9
- 238000003752 polymerase chain reaction Methods 0.000 description 9
- 108091026890 Coding region Proteins 0.000 description 8
- 241000711573 Coronaviridae Species 0.000 description 8
- 108020004414 DNA Proteins 0.000 description 8
- 241000701044 Human gammaherpesvirus 4 Species 0.000 description 8
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 8
- 238000003556 assay Methods 0.000 description 8
- 238000006243 chemical reaction Methods 0.000 description 8
- 239000003795 chemical substances by application Substances 0.000 description 8
- 230000002538 fungal effect Effects 0.000 description 8
- 230000004927 fusion Effects 0.000 description 8
- 239000011859 microparticle Substances 0.000 description 8
- 229940035032 monophosphoryl lipid a Drugs 0.000 description 8
- 230000001681 protective effect Effects 0.000 description 8
- 230000004083 survival effect Effects 0.000 description 8
- 239000000725 suspension Substances 0.000 description 8
- 239000013598 vector Substances 0.000 description 8
- YYGNTYWPHWGJRM-UHFFFAOYSA-N (6E,10E,14E,18E)-2,6,10,15,19,23-hexamethyltetracosa-2,6,10,14,18,22-hexaene Chemical compound CC(C)=CCCC(C)=CCCC(C)=CCCC=C(C)CCC=C(C)CCC=C(C)C YYGNTYWPHWGJRM-UHFFFAOYSA-N 0.000 description 7
- 108020004705 Codon Proteins 0.000 description 7
- 101710154606 Hemagglutinin Proteins 0.000 description 7
- 241000701806 Human papillomavirus Species 0.000 description 7
- 241000712045 Morbillivirus Species 0.000 description 7
- 102000011931 Nucleoproteins Human genes 0.000 description 7
- 108010061100 Nucleoproteins Proteins 0.000 description 7
- 101710093908 Outer capsid protein VP4 Proteins 0.000 description 7
- 101710135467 Outer capsid protein sigma-1 Proteins 0.000 description 7
- 229910019142 PO4 Inorganic materials 0.000 description 7
- 101710176177 Protein A56 Proteins 0.000 description 7
- 241000193998 Streptococcus pneumoniae Species 0.000 description 7
- BHEOSNUKNHRBNM-UHFFFAOYSA-N Tetramethylsqualene Natural products CC(=C)C(C)CCC(=C)C(C)CCC(C)=CCCC=C(C)CCC(C)C(=C)CCC(C)C(C)=C BHEOSNUKNHRBNM-UHFFFAOYSA-N 0.000 description 7
- 239000002253 acid Substances 0.000 description 7
- ILRRQNADMUWWFW-UHFFFAOYSA-K aluminium phosphate Chemical compound O1[Al]2OP1(=O)O2 ILRRQNADMUWWFW-UHFFFAOYSA-K 0.000 description 7
- 239000003242 anti bacterial agent Substances 0.000 description 7
- 238000013459 approach Methods 0.000 description 7
- 238000005119 centrifugation Methods 0.000 description 7
- 229940079593 drug Drugs 0.000 description 7
- 239000000499 gel Substances 0.000 description 7
- 239000000185 hemagglutinin Substances 0.000 description 7
- 239000008188 pellet Substances 0.000 description 7
- 235000021317 phosphate Nutrition 0.000 description 7
- 239000000523 sample Substances 0.000 description 7
- 229940031439 squalene Drugs 0.000 description 7
- TUHBEKDERLKLEC-UHFFFAOYSA-N squalene Natural products CC(=CCCC(=CCCC(=CCCC=C(/C)CCC=C(/C)CC=C(C)C)C)C)C TUHBEKDERLKLEC-UHFFFAOYSA-N 0.000 description 7
- 229940031000 streptococcus pneumoniae Drugs 0.000 description 7
- 239000006228 supernatant Substances 0.000 description 7
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 6
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 6
- 241000711549 Hepacivirus C Species 0.000 description 6
- 201000005505 Measles Diseases 0.000 description 6
- 201000009906 Meningitis Diseases 0.000 description 6
- 208000005647 Mumps Diseases 0.000 description 6
- 108091028043 Nucleic acid sequence Proteins 0.000 description 6
- 238000004873 anchoring Methods 0.000 description 6
- 230000027455 binding Effects 0.000 description 6
- 230000015572 biosynthetic process Effects 0.000 description 6
- 206010013023 diphtheria Diseases 0.000 description 6
- 235000019253 formic acid Nutrition 0.000 description 6
- 238000006062 fragmentation reaction Methods 0.000 description 6
- 238000009396 hybridization Methods 0.000 description 6
- 238000001727 in vivo Methods 0.000 description 6
- 238000004949 mass spectrometry Methods 0.000 description 6
- 238000012544 monitoring process Methods 0.000 description 6
- 238000010172 mouse model Methods 0.000 description 6
- 208000010805 mumps infectious disease Diseases 0.000 description 6
- 239000013642 negative control Substances 0.000 description 6
- 231100000252 nontoxic Toxicity 0.000 description 6
- 230000003000 nontoxic effect Effects 0.000 description 6
- 239000007764 o/w emulsion Substances 0.000 description 6
- 230000001717 pathogenic effect Effects 0.000 description 6
- 239000010452 phosphate Substances 0.000 description 6
- 235000019419 proteases Nutrition 0.000 description 6
- 239000011780 sodium chloride Substances 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- 230000009885 systemic effect Effects 0.000 description 6
- 241000588832 Bordetella pertussis Species 0.000 description 5
- 241000701022 Cytomegalovirus Species 0.000 description 5
- 102000003886 Glycoproteins Human genes 0.000 description 5
- 108090000288 Glycoproteins Proteins 0.000 description 5
- 241001465754 Metazoa Species 0.000 description 5
- 229930182555 Penicillin Natural products 0.000 description 5
- 241000702670 Rotavirus Species 0.000 description 5
- 241001505901 Streptococcus sp. 'group A' Species 0.000 description 5
- 206010043376 Tetanus Diseases 0.000 description 5
- 208000004006 Tick-borne encephalitis Diseases 0.000 description 5
- 108010067390 Viral Proteins Proteins 0.000 description 5
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 5
- 229910052782 aluminium Inorganic materials 0.000 description 5
- 210000004369 blood Anatomy 0.000 description 5
- 239000008280 blood Substances 0.000 description 5
- 210000004899 c-terminal region Anatomy 0.000 description 5
- 238000003776 cleavage reaction Methods 0.000 description 5
- 230000021615 conjugation Effects 0.000 description 5
- 238000010790 dilution Methods 0.000 description 5
- 239000012895 dilution Substances 0.000 description 5
- 201000010099 disease Diseases 0.000 description 5
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 5
- 238000002474 experimental method Methods 0.000 description 5
- 239000000284 extract Substances 0.000 description 5
- 238000013467 fragmentation Methods 0.000 description 5
- 230000004957 immunoregulator effect Effects 0.000 description 5
- 230000003308 immunostimulating effect Effects 0.000 description 5
- 239000007924 injection Substances 0.000 description 5
- 238000002347 injection Methods 0.000 description 5
- 239000000463 material Substances 0.000 description 5
- 239000011159 matrix material Substances 0.000 description 5
- 230000004048 modification Effects 0.000 description 5
- 238000012986 modification Methods 0.000 description 5
- 239000013641 positive control Substances 0.000 description 5
- 230000008569 process Effects 0.000 description 5
- 238000012545 processing Methods 0.000 description 5
- 230000000241 respiratory effect Effects 0.000 description 5
- 230000007017 scission Effects 0.000 description 5
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 5
- 238000001228 spectrum Methods 0.000 description 5
- 238000007920 subcutaneous administration Methods 0.000 description 5
- 238000003786 synthesis reaction Methods 0.000 description 5
- 238000012360 testing method Methods 0.000 description 5
- 230000029069 type 2 immune response Effects 0.000 description 5
- 241001430294 unidentified retrovirus Species 0.000 description 5
- 239000000277 virosome Substances 0.000 description 5
- 241000193738 Bacillus anthracis Species 0.000 description 4
- 125000001433 C-terminal amino-acid group Chemical group 0.000 description 4
- 108090000565 Capsid Proteins Proteins 0.000 description 4
- 102100023321 Ceruloplasmin Human genes 0.000 description 4
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 4
- 241000710831 Flavivirus Species 0.000 description 4
- ZHNUHDYFZUAESO-UHFFFAOYSA-N Formamide Chemical compound NC=O ZHNUHDYFZUAESO-UHFFFAOYSA-N 0.000 description 4
- NYHBQMYGNKIUIF-UUOKFMHZSA-N Guanosine Chemical compound C1=NC=2C(=O)NC(N)=NC=2N1[C@@H]1O[C@H](CO)[C@@H](O)[C@H]1O NYHBQMYGNKIUIF-UUOKFMHZSA-N 0.000 description 4
- 241000724675 Hepatitis E virus Species 0.000 description 4
- 241000709721 Hepatovirus A Species 0.000 description 4
- 241000725303 Human immunodeficiency virus Species 0.000 description 4
- 108060003951 Immunoglobulin Proteins 0.000 description 4
- FFEARJCKVFRZRR-BYPYZUCNSA-N L-methionine Chemical compound CSCC[C@H](N)C(O)=O FFEARJCKVFRZRR-BYPYZUCNSA-N 0.000 description 4
- 241000588655 Moraxella catarrhalis Species 0.000 description 4
- 125000001429 N-terminal alpha-amino-acid group Chemical group 0.000 description 4
- 108010006232 Neuraminidase Proteins 0.000 description 4
- 102000005348 Neuraminidase Human genes 0.000 description 4
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 4
- 241000125945 Protoparvovirus Species 0.000 description 4
- 241000589517 Pseudomonas aeruginosa Species 0.000 description 4
- 101500027983 Rattus norvegicus Octadecaneuropeptide Proteins 0.000 description 4
- 108010008281 Recombinant Fusion Proteins Proteins 0.000 description 4
- 102000007056 Recombinant Fusion Proteins Human genes 0.000 description 4
- 241000702263 Reovirus sp. Species 0.000 description 4
- 241000191967 Staphylococcus aureus Species 0.000 description 4
- 241000193985 Streptococcus agalactiae Species 0.000 description 4
- 101000815632 Streptococcus suis (strain 05ZYH33) Rqc2 homolog RqcH Proteins 0.000 description 4
- 241000223238 Trichophyton Species 0.000 description 4
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 4
- 241000607626 Vibrio cholerae Species 0.000 description 4
- 241000607479 Yersinia pestis Species 0.000 description 4
- 238000001042 affinity chromatography Methods 0.000 description 4
- AZDRQVAHHNSJOQ-UHFFFAOYSA-N alumane Chemical class [AlH3] AZDRQVAHHNSJOQ-UHFFFAOYSA-N 0.000 description 4
- 229940088710 antibiotic agent Drugs 0.000 description 4
- 210000003719 b-lymphocyte Anatomy 0.000 description 4
- 239000000227 bioadhesive Substances 0.000 description 4
- 239000004202 carbamide Substances 0.000 description 4
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 4
- 239000002299 complementary DNA Substances 0.000 description 4
- 150000001875 compounds Chemical class 0.000 description 4
- 229920001577 copolymer Polymers 0.000 description 4
- 239000003599 detergent Substances 0.000 description 4
- 102000036072 fibronectin binding proteins Human genes 0.000 description 4
- 208000029570 hepatitis D virus infection Diseases 0.000 description 4
- 210000004408 hybridoma Anatomy 0.000 description 4
- 230000005847 immunogenicity Effects 0.000 description 4
- 102000018358 immunoglobulin Human genes 0.000 description 4
- 230000001965 increasing effect Effects 0.000 description 4
- 239000007928 intraperitoneal injection Substances 0.000 description 4
- 238000001990 intravenous administration Methods 0.000 description 4
- GZQKNULLWNGMCW-PWQABINMSA-N lipid A (E. coli) Chemical class O1[C@H](CO)[C@@H](OP(O)(O)=O)[C@H](OC(=O)C[C@@H](CCCCCCCCCCC)OC(=O)CCCCCCCCCCCCC)[C@@H](NC(=O)C[C@@H](CCCCCCCCCCC)OC(=O)CCCCCCCCCCC)[C@@H]1OC[C@@H]1[C@@H](O)[C@H](OC(=O)C[C@H](O)CCCCCCCCCCC)[C@@H](NC(=O)C[C@H](O)CCCCCCCCCCC)[C@@H](OP(O)(O)=O)O1 GZQKNULLWNGMCW-PWQABINMSA-N 0.000 description 4
- 239000002502 liposome Substances 0.000 description 4
- 238000004811 liquid chromatography Methods 0.000 description 4
- 238000000816 matrix-assisted laser desorption--ionisation Methods 0.000 description 4
- 229930182817 methionine Natural products 0.000 description 4
- 229940046166 oligodeoxynucleotide Drugs 0.000 description 4
- 239000003960 organic solvent Substances 0.000 description 4
- 238000012261 overproduction Methods 0.000 description 4
- 229940049954 penicillin Drugs 0.000 description 4
- 239000012071 phase Substances 0.000 description 4
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 4
- 229920002704 polyhistidine Polymers 0.000 description 4
- 229920000642 polymer Polymers 0.000 description 4
- 230000000069 prophylactic effect Effects 0.000 description 4
- 238000012552 review Methods 0.000 description 4
- 201000005404 rubella Diseases 0.000 description 4
- 241000894007 species Species 0.000 description 4
- 108091005703 transmembrane proteins Proteins 0.000 description 4
- 102000035160 transmembrane proteins Human genes 0.000 description 4
- 239000003643 water by type Substances 0.000 description 4
- ATRRKUHOCOJYRX-UHFFFAOYSA-N Ammonium bicarbonate Chemical compound [NH4+].OC([O-])=O ATRRKUHOCOJYRX-UHFFFAOYSA-N 0.000 description 3
- 229910000013 Ammonium bicarbonate Inorganic materials 0.000 description 3
- 241000228212 Aspergillus Species 0.000 description 3
- 108010059574 C5a peptidase Proteins 0.000 description 3
- 241000222120 Candida <Saccharomycetales> Species 0.000 description 3
- 101710132601 Capsid protein Proteins 0.000 description 3
- 108010039939 Cell Wall Skeleton Proteins 0.000 description 3
- 241000606153 Chlamydia trachomatis Species 0.000 description 3
- 241000193449 Clostridium tetani Species 0.000 description 3
- 241000991587 Enterovirus C Species 0.000 description 3
- 101710181478 Envelope glycoprotein GP350 Proteins 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 241000531123 GB virus C Species 0.000 description 3
- 108010015899 Glycopeptides Proteins 0.000 description 3
- 102000002068 Glycopeptides Human genes 0.000 description 3
- 108060003393 Granulin Proteins 0.000 description 3
- 101710133291 Hemagglutinin-neuraminidase Proteins 0.000 description 3
- 241000282412 Homo Species 0.000 description 3
- 108090000978 Interleukin-4 Proteins 0.000 description 3
- 108010002616 Interleukin-5 Proteins 0.000 description 3
- 108090001005 Interleukin-6 Proteins 0.000 description 3
- 241000589242 Legionella pneumophila Species 0.000 description 3
- 241000187479 Mycobacterium tuberculosis Species 0.000 description 3
- 108700020354 N-acetylmuramyl-threonyl-isoglutamine Proteins 0.000 description 3
- 241000588653 Neisseria Species 0.000 description 3
- 206010028980 Neoplasm Diseases 0.000 description 3
- 108090001074 Nucleocapsid Proteins Proteins 0.000 description 3
- 241000283973 Oryctolagus cuniculus Species 0.000 description 3
- 101710160102 Outer membrane protein B Proteins 0.000 description 3
- 201000007100 Pharyngitis Diseases 0.000 description 3
- 229920003171 Poly (ethylene oxide) Polymers 0.000 description 3
- 229920002732 Polyanhydride Polymers 0.000 description 3
- 229920001710 Polyorthoester Polymers 0.000 description 3
- 101710194807 Protective antigen Proteins 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 229930182558 Sterol Natural products 0.000 description 3
- 231100000650 Toxic shock syndrome Toxicity 0.000 description 3
- VQQVWGVXDIPORV-UHFFFAOYSA-N Tryptanthrine Chemical class C1=CC=C2C(=O)N3C4=CC=CC=C4C(=O)C3=NC2=C1 VQQVWGVXDIPORV-UHFFFAOYSA-N 0.000 description 3
- 108010059993 Vancomycin Proteins 0.000 description 3
- 230000004075 alteration Effects 0.000 description 3
- 229940037003 alum Drugs 0.000 description 3
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 3
- 125000000539 amino acid group Chemical group 0.000 description 3
- 235000012538 ammonium bicarbonate Nutrition 0.000 description 3
- 239000001099 ammonium carbonate Substances 0.000 description 3
- 230000003698 anagen phase Effects 0.000 description 3
- 238000010171 animal model Methods 0.000 description 3
- 230000005875 antibody response Effects 0.000 description 3
- 230000002238 attenuated effect Effects 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 108091008324 binding proteins Proteins 0.000 description 3
- 230000000903 blocking effect Effects 0.000 description 3
- 238000004113 cell culture Methods 0.000 description 3
- 210000004520 cell wall skeleton Anatomy 0.000 description 3
- 230000036755 cellular response Effects 0.000 description 3
- 229940038705 chlamydia trachomatis Drugs 0.000 description 3
- 235000012000 cholesterol Nutrition 0.000 description 3
- 238000001514 detection method Methods 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 238000001962 electrophoresis Methods 0.000 description 3
- 238000005516 engineering process Methods 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- 238000005194 fractionation Methods 0.000 description 3
- 230000006870 function Effects 0.000 description 3
- 230000009036 growth inhibition Effects 0.000 description 3
- ZRALSGWEFCBTJO-UHFFFAOYSA-O guanidinium Chemical compound NC(N)=[NH2+] ZRALSGWEFCBTJO-UHFFFAOYSA-O 0.000 description 3
- 229940047650 haemophilus influenzae Drugs 0.000 description 3
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 3
- 210000004201 immune sera Anatomy 0.000 description 3
- 229940042743 immune sera Drugs 0.000 description 3
- 230000000984 immunochemical effect Effects 0.000 description 3
- 238000012405 in silico analysis Methods 0.000 description 3
- 229940115932 legionella pneumophila Drugs 0.000 description 3
- 230000021633 leukocyte mediated immunity Effects 0.000 description 3
- 150000002632 lipids Chemical class 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 238000001840 matrix-assisted laser desorption--ionisation time-of-flight mass spectrometry Methods 0.000 description 3
- 229940041323 measles vaccine Drugs 0.000 description 3
- 230000001404 mediated effect Effects 0.000 description 3
- 239000002609 medium Substances 0.000 description 3
- 239000000178 monomer Substances 0.000 description 3
- 230000003232 mucoadhesive effect Effects 0.000 description 3
- 230000016379 mucosal immune response Effects 0.000 description 3
- 125000001446 muramyl group Chemical group N[C@@H](C=O)[C@@H](O[C@@H](C(=O)*)C)[C@H](O)[C@H](O)CO 0.000 description 3
- 230000003472 neutralizing effect Effects 0.000 description 3
- 229920001542 oligosaccharide Polymers 0.000 description 3
- 150000002482 oligosaccharides Chemical class 0.000 description 3
- 238000005457 optimization Methods 0.000 description 3
- 238000007747 plating Methods 0.000 description 3
- 229920000747 poly(lactic acid) Polymers 0.000 description 3
- 239000002745 poly(ortho ester) Substances 0.000 description 3
- 229920001610 polycaprolactone Polymers 0.000 description 3
- 239000004632 polycaprolactone Substances 0.000 description 3
- 229920000056 polyoxyethylene ether Polymers 0.000 description 3
- 238000011321 prophylaxis Methods 0.000 description 3
- 235000019833 protease Nutrition 0.000 description 3
- 108020001580 protein domains Proteins 0.000 description 3
- 238000011084 recovery Methods 0.000 description 3
- 238000012216 screening Methods 0.000 description 3
- 238000000926 separation method Methods 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 239000007921 spray Substances 0.000 description 3
- 235000003702 sterols Nutrition 0.000 description 3
- 238000006467 substitution reaction Methods 0.000 description 3
- 208000006379 syphilis Diseases 0.000 description 3
- 229960000814 tetanus toxoid Drugs 0.000 description 3
- 238000002560 therapeutic procedure Methods 0.000 description 3
- 150000003584 thiosemicarbazones Chemical class 0.000 description 3
- 231100000331 toxic Toxicity 0.000 description 3
- 230000002588 toxic effect Effects 0.000 description 3
- AVWQQPYHYQKEIZ-UHFFFAOYSA-K trisodium;2-dodecylbenzenesulfonate;3-dodecylbenzenesulfonate;4-dodecylbenzenesulfonate Chemical compound [Na+].[Na+].[Na+].CCCCCCCCCCCCC1=CC=C(S([O-])(=O)=O)C=C1.CCCCCCCCCCCCC1=CC=CC(S([O-])(=O)=O)=C1.CCCCCCCCCCCCC1=CC=CC=C1S([O-])(=O)=O AVWQQPYHYQKEIZ-UHFFFAOYSA-K 0.000 description 3
- 241001529453 unidentified herpesvirus Species 0.000 description 3
- 229940125575 vaccine candidate Drugs 0.000 description 3
- MYPYJXKWCTUITO-LYRMYLQWSA-N vancomycin Chemical compound O([C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1OC1=C2C=C3C=C1OC1=CC=C(C=C1Cl)[C@@H](O)[C@H](C(N[C@@H](CC(N)=O)C(=O)N[C@H]3C(=O)N[C@H]1C(=O)N[C@H](C(N[C@@H](C3=CC(O)=CC(O)=C3C=3C(O)=CC=C1C=3)C(O)=O)=O)[C@H](O)C1=CC=C(C(=C1)Cl)O2)=O)NC(=O)[C@@H](CC(C)C)NC)[C@H]1C[C@](C)(N)[C@H](O)[C@H](C)O1 MYPYJXKWCTUITO-LYRMYLQWSA-N 0.000 description 3
- 229960003165 vancomycin Drugs 0.000 description 3
- MYPYJXKWCTUITO-UHFFFAOYSA-N vancomycin Natural products O1C(C(=C2)Cl)=CC=C2C(O)C(C(NC(C2=CC(O)=CC(O)=C2C=2C(O)=CC=C3C=2)C(O)=O)=O)NC(=O)C3NC(=O)C2NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(CC(C)C)NC)C(O)C(C=C3Cl)=CC=C3OC3=CC2=CC1=C3OC1OC(CO)C(O)C(O)C1OC1CC(C)(N)C(O)C(C)O1 MYPYJXKWCTUITO-UHFFFAOYSA-N 0.000 description 3
- 229940118696 vibrio cholerae Drugs 0.000 description 3
- 230000001018 virulence Effects 0.000 description 3
- 238000001262 western blot Methods 0.000 description 3
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 2
- HNSDLXPSAYFUHK-UHFFFAOYSA-N 1,4-bis(2-ethylhexyl) sulfosuccinate Chemical compound CCCCC(CC)COC(=O)CC(S(O)(=O)=O)C(=O)OCC(CC)CCCC HNSDLXPSAYFUHK-UHFFFAOYSA-N 0.000 description 2
- RKDVKSZUMVYZHH-UHFFFAOYSA-N 1,4-dioxane-2,5-dione Chemical compound O=C1COC(=O)CO1 RKDVKSZUMVYZHH-UHFFFAOYSA-N 0.000 description 2
- WXTMDXOMEHJXQO-UHFFFAOYSA-N 2,5-dihydroxybenzoic acid Chemical compound OC(=O)C1=CC(O)=CC=C1O WXTMDXOMEHJXQO-UHFFFAOYSA-N 0.000 description 2
- QKNYBSVHEMOAJP-UHFFFAOYSA-N 2-amino-2-(hydroxymethyl)propane-1,3-diol;hydron;chloride Chemical compound Cl.OCC(N)(CO)CO QKNYBSVHEMOAJP-UHFFFAOYSA-N 0.000 description 2
- RHKWIGHJGOEUSM-UHFFFAOYSA-N 3h-imidazo[4,5-h]quinoline Chemical class C1=CN=C2C(N=CN3)=C3C=CC2=C1 RHKWIGHJGOEUSM-UHFFFAOYSA-N 0.000 description 2
- JJTUDXZGHPGLLC-IMJSIDKUSA-N 4511-42-6 Chemical compound C[C@@H]1OC(=O)[C@H](C)OC1=O JJTUDXZGHPGLLC-IMJSIDKUSA-N 0.000 description 2
- GOZMBJCYMQQACI-UHFFFAOYSA-N 6,7-dimethyl-3-[[methyl-[2-[methyl-[[1-[3-(trifluoromethyl)phenyl]indol-3-yl]methyl]amino]ethyl]amino]methyl]chromen-4-one;dihydrochloride Chemical compound Cl.Cl.C=1OC2=CC(C)=C(C)C=C2C(=O)C=1CN(C)CCN(C)CC(C1=CC=CC=C11)=CN1C1=CC=CC(C(F)(F)F)=C1 GOZMBJCYMQQACI-UHFFFAOYSA-N 0.000 description 2
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 2
- 108010088751 Albumins Proteins 0.000 description 2
- 102000009027 Albumins Human genes 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- 241000271566 Aves Species 0.000 description 2
- 235000014469 Bacillus subtilis Nutrition 0.000 description 2
- FERIUCNNQQJTOY-UHFFFAOYSA-N Butyric acid Chemical compound CCCC(O)=O FERIUCNNQQJTOY-UHFFFAOYSA-N 0.000 description 2
- 241000222122 Candida albicans Species 0.000 description 2
- 241000283707 Capra Species 0.000 description 2
- 229930186147 Cephalosporin Natural products 0.000 description 2
- 241000606161 Chlamydia Species 0.000 description 2
- 241001647372 Chlamydia pneumoniae Species 0.000 description 2
- 208000001726 Classical Swine Fever Diseases 0.000 description 2
- 208000035473 Communicable disease Diseases 0.000 description 2
- 108010060123 Conjugate Vaccines Proteins 0.000 description 2
- MIKUYHXYGGJMLM-GIMIYPNGSA-N Crotonoside Natural products C1=NC2=C(N)NC(=O)N=C2N1[C@H]1O[C@@H](CO)[C@H](O)[C@@H]1O MIKUYHXYGGJMLM-GIMIYPNGSA-N 0.000 description 2
- 102100038284 Cytospin-B Human genes 0.000 description 2
- DYDCUQKUCUHJBH-UWTATZPHSA-N D-Cycloserine Chemical compound N[C@@H]1CONC1=O DYDCUQKUCUHJBH-UWTATZPHSA-N 0.000 description 2
- DYDCUQKUCUHJBH-UHFFFAOYSA-N D-Cycloserine Natural products NC1CONC1=O DYDCUQKUCUHJBH-UHFFFAOYSA-N 0.000 description 2
- NYHBQMYGNKIUIF-UHFFFAOYSA-N D-guanosine Natural products C1=2NC(N)=NC(=O)C=2N=CN1C1OC(CO)C(O)C1O NYHBQMYGNKIUIF-UHFFFAOYSA-N 0.000 description 2
- 102000053602 DNA Human genes 0.000 description 2
- 241001466953 Echovirus Species 0.000 description 2
- 102100038132 Endogenous retrovirus group K member 6 Pro protein Human genes 0.000 description 2
- 241000194032 Enterococcus faecalis Species 0.000 description 2
- 102000004190 Enzymes Human genes 0.000 description 2
- 108090000790 Enzymes Proteins 0.000 description 2
- 201000000297 Erysipelas Diseases 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- 241000590002 Helicobacter pylori Species 0.000 description 2
- 241000700721 Hepatitis B virus Species 0.000 description 2
- 101000585548 Homo sapiens PAXIP1-associated glutamate-rich protein 1 Proteins 0.000 description 2
- 241000701074 Human alphaherpesvirus 2 Species 0.000 description 2
- 241000701041 Human betaherpesvirus 7 Species 0.000 description 2
- 241001502974 Human gammaherpesvirus 8 Species 0.000 description 2
- 241000701027 Human herpesvirus 6 Species 0.000 description 2
- 206010021531 Impetigo Diseases 0.000 description 2
- 108010065805 Interleukin-12 Proteins 0.000 description 2
- 108010002350 Interleukin-2 Proteins 0.000 description 2
- 241000713666 Lentivirus Species 0.000 description 2
- 108010014603 Leukocidins Proteins 0.000 description 2
- 241000186779 Listeria monocytogenes Species 0.000 description 2
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- 241000712079 Measles morbillivirus Species 0.000 description 2
- 241001480037 Microsporum Species 0.000 description 2
- WPNJAUFVNXKLIM-UHFFFAOYSA-N Moxonidine Chemical compound COC1=NC(C)=NC(Cl)=C1NC1=NCCN1 WPNJAUFVNXKLIM-UHFFFAOYSA-N 0.000 description 2
- 102000016943 Muramidase Human genes 0.000 description 2
- 108010014251 Muramidase Proteins 0.000 description 2
- 241001529936 Murinae Species 0.000 description 2
- 241000711466 Murine hepatitis virus Species 0.000 description 2
- 108010062010 N-Acetylmuramoyl-L-alanine Amidase Proteins 0.000 description 2
- 206010028885 Necrotising fasciitis Diseases 0.000 description 2
- PXHVJJICTQNCMI-UHFFFAOYSA-N Nickel Chemical compound [Ni] PXHVJJICTQNCMI-UHFFFAOYSA-N 0.000 description 2
- 101710144128 Non-structural protein 2 Proteins 0.000 description 2
- 101710144111 Non-structural protein 3 Proteins 0.000 description 2
- 101710144121 Non-structural protein 5 Proteins 0.000 description 2
- 241000714209 Norwalk virus Species 0.000 description 2
- 241001631646 Papillomaviridae Species 0.000 description 2
- 241000711504 Paramyxoviridae Species 0.000 description 2
- 102000008153 Peptide Elongation Factor Tu Human genes 0.000 description 2
- 108010049977 Peptide Elongation Factor Tu Proteins 0.000 description 2
- 241000710778 Pestivirus Species 0.000 description 2
- 108010089430 Phosphoproteins Proteins 0.000 description 2
- 102000007982 Phosphoproteins Human genes 0.000 description 2
- 241000709664 Picornaviridae Species 0.000 description 2
- 206010035148 Plague Diseases 0.000 description 2
- 208000000474 Poliomyelitis Diseases 0.000 description 2
- 241001505332 Polyomavirus sp. Species 0.000 description 2
- 108010076039 Polyproteins Proteins 0.000 description 2
- 241000605862 Porphyromonas gingivalis Species 0.000 description 2
- 101710186352 Probable membrane antigen 3 Proteins 0.000 description 2
- 101710181078 Probable membrane antigen 75 Proteins 0.000 description 2
- 108020004511 Recombinant DNA Proteins 0.000 description 2
- 241000710801 Rubivirus Species 0.000 description 2
- 241000293871 Salmonella enterica subsp. enterica serovar Typhi Species 0.000 description 2
- 241000219287 Saponaria Species 0.000 description 2
- 206010039587 Scarlet Fever Diseases 0.000 description 2
- 241000700584 Simplexvirus Species 0.000 description 2
- 101710200413 Small hydrophobic protein Proteins 0.000 description 2
- PRXRUNOAOLTIEF-ADSICKODSA-N Sorbitan trioleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@@H](OC(=O)CCCCCCC\C=C/CCCCCCCC)[C@H]1OC[C@H](O)[C@H]1OC(=O)CCCCCCC\C=C/CCCCCCCC PRXRUNOAOLTIEF-ADSICKODSA-N 0.000 description 2
- 241000191940 Staphylococcus Species 0.000 description 2
- 241001147691 Staphylococcus saprophyticus Species 0.000 description 2
- 208000017757 Streptococcal toxic-shock syndrome Diseases 0.000 description 2
- 229930006000 Sucrose Natural products 0.000 description 2
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 2
- 230000024932 T cell mediated immunity Effects 0.000 description 2
- 101710178472 Tegument protein Proteins 0.000 description 2
- 206010044251 Toxic shock syndrome streptococcal Diseases 0.000 description 2
- 241000711484 Transmissible gastroenteritis virus Species 0.000 description 2
- 241000589884 Treponema pallidum Species 0.000 description 2
- 241000893966 Trichophyton verrucosum Species 0.000 description 2
- 241000711970 Vesiculovirus Species 0.000 description 2
- 241000606834 [Haemophilus] ducreyi Species 0.000 description 2
- 238000002835 absorbance Methods 0.000 description 2
- 150000007513 acids Chemical class 0.000 description 2
- 238000012382 advanced drug delivery Methods 0.000 description 2
- 159000000013 aluminium salts Chemical class 0.000 description 2
- 229910000329 aluminium sulfate Inorganic materials 0.000 description 2
- 230000003321 amplification Effects 0.000 description 2
- 230000000844 anti-bacterial effect Effects 0.000 description 2
- 230000000890 antigenic effect Effects 0.000 description 2
- 229940065181 bacillus anthracis Drugs 0.000 description 2
- 229920002988 biodegradable polymer Polymers 0.000 description 2
- 239000004621 biodegradable polymer Substances 0.000 description 2
- 239000012472 biological sample Substances 0.000 description 2
- 239000006161 blood agar Substances 0.000 description 2
- 201000011510 cancer Diseases 0.000 description 2
- 235000014633 carbohydrates Nutrition 0.000 description 2
- 229910052799 carbon Inorganic materials 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 230000032823 cell division Effects 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 229940124587 cephalosporin Drugs 0.000 description 2
- 150000001780 cephalosporins Chemical class 0.000 description 2
- 201000004308 chancroid Diseases 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 238000010961 commercial manufacture process Methods 0.000 description 2
- 229940031670 conjugate vaccine Drugs 0.000 description 2
- 238000010276 construction Methods 0.000 description 2
- 229960003077 cycloserine Drugs 0.000 description 2
- 230000001419 dependent effect Effects 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 230000018109 developmental process Effects 0.000 description 2
- 238000010586 diagram Methods 0.000 description 2
- 239000003085 diluting agent Substances 0.000 description 2
- 238000009826 distribution Methods 0.000 description 2
- 238000000635 electron micrograph Methods 0.000 description 2
- 229940032049 enterococcus faecalis Drugs 0.000 description 2
- 229940088598 enzyme Drugs 0.000 description 2
- 210000002615 epidermis Anatomy 0.000 description 2
- 210000003527 eukaryotic cell Anatomy 0.000 description 2
- 229930182470 glycoside Natural products 0.000 description 2
- 230000013595 glycosylation Effects 0.000 description 2
- 238000006206 glycosylation reaction Methods 0.000 description 2
- 239000001963 growth medium Substances 0.000 description 2
- 229940029575 guanosine Drugs 0.000 description 2
- 150000003278 haem Chemical class 0.000 description 2
- 229940037467 helicobacter pylori Drugs 0.000 description 2
- 238000004128 high performance liquid chromatography Methods 0.000 description 2
- 125000000487 histidyl group Chemical group [H]N([H])C(C(=O)O*)C([H])([H])C1=C([H])N([H])C([H])=N1 0.000 description 2
- 102000053056 human PAGR1 Human genes 0.000 description 2
- 230000002209 hydrophobic effect Effects 0.000 description 2
- 210000000987 immune system Anatomy 0.000 description 2
- 238000003018 immunoassay Methods 0.000 description 2
- 239000002955 immunomodulating agent Substances 0.000 description 2
- 229940121354 immunomodulator Drugs 0.000 description 2
- 238000000126 in silico method Methods 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 229940029583 inactivated polio vaccine Drugs 0.000 description 2
- 238000003780 insertion Methods 0.000 description 2
- 230000037431 insertion Effects 0.000 description 2
- 238000001361 intraarterial administration Methods 0.000 description 2
- 230000003834 intracellular effect Effects 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- 238000002955 isolation Methods 0.000 description 2
- BPHPUYQFMNQIOC-NXRLNHOXSA-N isopropyl beta-D-thiogalactopyranoside Chemical compound CC(C)S[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O BPHPUYQFMNQIOC-NXRLNHOXSA-N 0.000 description 2
- 101150000417 izt gene Proteins 0.000 description 2
- 108010045069 keyhole-limpet hemocyanin Proteins 0.000 description 2
- 239000006166 lysate Substances 0.000 description 2
- 239000004325 lysozyme Substances 0.000 description 2
- 229960000274 lysozyme Drugs 0.000 description 2
- 235000010335 lysozyme Nutrition 0.000 description 2
- 229920002521 macromolecule Polymers 0.000 description 2
- 238000001906 matrix-assisted laser desorption--ionisation mass spectrometry Methods 0.000 description 2
- 238000000074 matrix-assisted laser desorption--ionisation tandem time-of-flight detection Methods 0.000 description 2
- 210000001806 memory b lymphocyte Anatomy 0.000 description 2
- 108020004999 messenger RNA Proteins 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 230000000813 microbial effect Effects 0.000 description 2
- 239000004005 microsphere Substances 0.000 description 2
- 238000001823 molecular biology technique Methods 0.000 description 2
- 229940095293 mumps vaccine Drugs 0.000 description 2
- 108010009719 mutanolysin Proteins 0.000 description 2
- 238000002170 nanoflow liquid chromatography-tandem mass spectrometry Methods 0.000 description 2
- 201000007970 necrotizing fasciitis Diseases 0.000 description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 2
- 238000003199 nucleic acid amplification method Methods 0.000 description 2
- 229940066429 octoxynol Drugs 0.000 description 2
- 229920002113 octoxynol Polymers 0.000 description 2
- 230000008520 organization Effects 0.000 description 2
- UNEIHNMKASENIG-UHFFFAOYSA-N para-chlorophenylpiperazine Chemical compound C1=CC(Cl)=CC=C1N1CCNCC1 UNEIHNMKASENIG-UHFFFAOYSA-N 0.000 description 2
- 238000007911 parenteral administration Methods 0.000 description 2
- 210000003819 peripheral blood mononuclear cell Anatomy 0.000 description 2
- 150000003904 phospholipids Chemical class 0.000 description 2
- 235000013550 pizza Nutrition 0.000 description 2
- 239000013612 plasmid Substances 0.000 description 2
- 229920001606 poly(lactic acid-co-glycolic acid) Polymers 0.000 description 2
- 229920002627 poly(phosphazenes) Polymers 0.000 description 2
- 229920002721 polycyanoacrylate Polymers 0.000 description 2
- 108010094020 polyglycine Proteins 0.000 description 2
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 description 2
- 229920000053 polysorbate 80 Polymers 0.000 description 2
- 230000002516 postimmunization Effects 0.000 description 2
- 230000001323 posttranslational effect Effects 0.000 description 2
- 239000002243 precursor Substances 0.000 description 2
- 238000007639 printing Methods 0.000 description 2
- 230000007026 protein scission Effects 0.000 description 2
- 230000002797 proteolythic effect Effects 0.000 description 2
- 238000000575 proteomic method Methods 0.000 description 2
- 230000002685 pulmonary effect Effects 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 208000023504 respiratory system disease Diseases 0.000 description 2
- 238000004007 reversed phase HPLC Methods 0.000 description 2
- 229960003131 rubella vaccine Drugs 0.000 description 2
- 239000012047 saturated solution Substances 0.000 description 2
- 238000010845 search algorithm Methods 0.000 description 2
- 238000012163 sequencing technique Methods 0.000 description 2
- 239000013605 shuttle vector Substances 0.000 description 2
- 238000002741 site-directed mutagenesis Methods 0.000 description 2
- 150000003432 sterols Chemical class 0.000 description 2
- 230000000638 stimulation Effects 0.000 description 2
- 108010075242 streptolysin S Proteins 0.000 description 2
- 238000012799 strong cation exchange Methods 0.000 description 2
- 229940031626 subunit vaccine Drugs 0.000 description 2
- 239000005720 sucrose Substances 0.000 description 2
- 239000004094 surface-active agent Substances 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 206010044325 trachoma Diseases 0.000 description 2
- 238000001890 transfection Methods 0.000 description 2
- 230000014616 translation Effects 0.000 description 2
- 230000032258 transport Effects 0.000 description 2
- XETCRXVKJHBPMK-MJSODCSWSA-N trehalose 6,6'-dimycolate Chemical compound C([C@@H]1[C@H]([C@H](O)[C@@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](COC(=O)C(CCCCCCCCCCC3C(C3)CCCCCCCCCCCCCCCCCC)C(O)CCCCCCCCCCCCCCCCCCCCCCCCC)O2)O)O1)O)OC(=O)C(C(O)CCCCCCCCCCCCCCCCCCCCCCCCC)CCCCCCCCCCC1CC1CCCCCCCCCCCCCCCCCC XETCRXVKJHBPMK-MJSODCSWSA-N 0.000 description 2
- 201000008827 tuberculosis Diseases 0.000 description 2
- 241000712461 unidentified influenza virus Species 0.000 description 2
- 238000010200 validation analysis Methods 0.000 description 2
- 238000005406 washing Methods 0.000 description 2
- DQJCDTNMLBYVAY-ZXXIYAEKSA-N (2S,5R,10R,13R)-16-{[(2R,3S,4R,5R)-3-{[(2S,3R,4R,5S,6R)-3-acetamido-4,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy}-5-(ethylamino)-6-hydroxy-2-(hydroxymethyl)oxan-4-yl]oxy}-5-(4-aminobutyl)-10-carbamoyl-2,13-dimethyl-4,7,12,15-tetraoxo-3,6,11,14-tetraazaheptadecan-1-oic acid Chemical compound NCCCC[C@H](C(=O)N[C@@H](C)C(O)=O)NC(=O)CC[C@H](C(N)=O)NC(=O)[C@@H](C)NC(=O)C(C)O[C@@H]1[C@@H](NCC)C(O)O[C@H](CO)[C@H]1O[C@H]1[C@H](NC(C)=O)[C@@H](O)[C@H](O)[C@@H](CO)O1 DQJCDTNMLBYVAY-ZXXIYAEKSA-N 0.000 description 1
- ASWBNKHCZGQVJV-UHFFFAOYSA-N (3-hexadecanoyloxy-2-hydroxypropyl) 2-(trimethylazaniumyl)ethyl phosphate Chemical compound CCCCCCCCCCCCCCCC(=O)OCC(O)COP([O-])(=O)OCC[N+](C)(C)C ASWBNKHCZGQVJV-UHFFFAOYSA-N 0.000 description 1
- YHQZWWDVLJPRIF-JLHRHDQISA-N (4R)-4-[[(2S,3R)-2-[acetyl-[(3R,4R,5S,6R)-3-amino-4-[(1R)-1-carboxyethoxy]-5-hydroxy-6-(hydroxymethyl)oxan-2-yl]amino]-3-hydroxybutanoyl]amino]-5-amino-5-oxopentanoic acid Chemical compound C(C)(=O)N([C@@H]([C@H](O)C)C(=O)N[C@H](CCC(=O)O)C(N)=O)C1[C@H](N)[C@@H](O[C@@H](C(=O)O)C)[C@H](O)[C@H](O1)CO YHQZWWDVLJPRIF-JLHRHDQISA-N 0.000 description 1
- HLHSUNWAPXINQU-GQCTYLIASA-N (E)-3-(3,4-dihydroxyphenyl)-N-prop-2-ynylprop-2-enamide Chemical compound OC=1C=C(C=CC=1O)/C=C/C(=O)NCC#C HLHSUNWAPXINQU-GQCTYLIASA-N 0.000 description 1
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 1
- PXFBZOLANLWPMH-UHFFFAOYSA-N 16-Epiaffinine Natural products C1C(C2=CC=CC=C2N2)=C2C(=O)CC2C(=CC)CN(C)C1C2CO PXFBZOLANLWPMH-UHFFFAOYSA-N 0.000 description 1
- UFBJCMHMOXMLKC-UHFFFAOYSA-N 2,4-dinitrophenol Chemical compound OC1=CC=C([N+]([O-])=O)C=C1[N+]([O-])=O UFBJCMHMOXMLKC-UHFFFAOYSA-N 0.000 description 1
- LMSDCGXQALIMLM-UHFFFAOYSA-N 2-[2-[bis(carboxymethyl)amino]ethyl-(carboxymethyl)amino]acetic acid;iron Chemical compound [Fe].OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O LMSDCGXQALIMLM-UHFFFAOYSA-N 0.000 description 1
- PFCLMNDDPTZJHQ-XLPZGREQSA-N 2-amino-7-[(2r,4s,5r)-4-hydroxy-5-(hydroxymethyl)oxolan-2-yl]-1h-pyrrolo[2,3-d]pyrimidin-4-one Chemical compound C1=CC=2C(=O)NC(N)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 PFCLMNDDPTZJHQ-XLPZGREQSA-N 0.000 description 1
- LOVXREQUMZKFCM-UHFFFAOYSA-N 4-[2-[1-(1,3-benzodioxol-5-yl)propan-2-ylamino]-1-hydroxyethyl]benzene-1,2-diol;hydron;chloride Chemical compound Cl.C=1C=C2OCOC2=CC=1CC(C)NCC(O)C1=CC=C(O)C(O)=C1 LOVXREQUMZKFCM-UHFFFAOYSA-N 0.000 description 1
- UHPMCKVQTMMPCG-UHFFFAOYSA-N 5,8-dihydroxy-2-methoxy-6-methyl-7-(2-oxopropyl)naphthalene-1,4-dione Chemical compound CC1=C(CC(C)=O)C(O)=C2C(=O)C(OC)=CC(=O)C2=C1O UHPMCKVQTMMPCG-UHFFFAOYSA-N 0.000 description 1
- 101710166488 6 kDa early secretory antigenic target Proteins 0.000 description 1
- 230000005730 ADP ribosylation Effects 0.000 description 1
- 208000030507 AIDS Diseases 0.000 description 1
- 241000235389 Absidia Species 0.000 description 1
- 102000007698 Alcohol dehydrogenase Human genes 0.000 description 1
- 108010021809 Alcohol dehydrogenase Proteins 0.000 description 1
- 241000710929 Alphavirus Species 0.000 description 1
- 241000223600 Alternaria Species 0.000 description 1
- 241000429837 Alternaria caespitosa Species 0.000 description 1
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 1
- 206010059313 Anogenital warts Diseases 0.000 description 1
- 101100420868 Anuroctonus phaiodactylus phtx gene Proteins 0.000 description 1
- 241000710189 Aphthovirus Species 0.000 description 1
- 241000228197 Aspergillus flavus Species 0.000 description 1
- 241001225321 Aspergillus fumigatus Species 0.000 description 1
- 241000132177 Aspergillus glaucus Species 0.000 description 1
- 241000351920 Aspergillus nidulans Species 0.000 description 1
- 241000228245 Aspergillus niger Species 0.000 description 1
- 241001465318 Aspergillus terreus Species 0.000 description 1
- 241000711404 Avian avulavirus 1 Species 0.000 description 1
- BXTVQNYQYUTQAZ-UHFFFAOYSA-N BNPS-skatole Chemical compound N=1C2=CC=CC=C2C(C)(Br)C=1SC1=CC=CC=C1[N+]([O-])=O BXTVQNYQYUTQAZ-UHFFFAOYSA-N 0.000 description 1
- 244000063299 Bacillus subtilis Species 0.000 description 1
- 101100007857 Bacillus subtilis (strain 168) cspB gene Proteins 0.000 description 1
- 101000996791 Banna virus (strain Indonesia/JKT-6423/1980) Non-structural protein 2 Proteins 0.000 description 1
- 241000606685 Bartonella bacilliformis Species 0.000 description 1
- 241000235579 Basidiobolus Species 0.000 description 1
- 241000335423 Blastomyces Species 0.000 description 1
- 201000004569 Blindness Diseases 0.000 description 1
- 102000015081 Blood Coagulation Factors Human genes 0.000 description 1
- 108010039209 Blood Coagulation Factors Proteins 0.000 description 1
- 241000589969 Borreliella burgdorferi Species 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 101000800130 Bos taurus Thyroglobulin Proteins 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- 241000711895 Bovine orthopneumovirus Species 0.000 description 1
- 241000712005 Bovine respirovirus 3 Species 0.000 description 1
- 108010071134 CRM197 (non-toxic variant of diphtheria toxin) Proteins 0.000 description 1
- 101100039010 Caenorhabditis elegans dis-3 gene Proteins 0.000 description 1
- 241000714198 Caliciviridae Species 0.000 description 1
- 241000222173 Candida parapsilosis Species 0.000 description 1
- 241000222178 Candida tropicalis Species 0.000 description 1
- 241000282461 Canis lupus Species 0.000 description 1
- 101710197665 Capsid protein VP2 Proteins 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 241000710190 Cardiovirus Species 0.000 description 1
- 102100035882 Catalase Human genes 0.000 description 1
- 108010053835 Catalase Proteins 0.000 description 1
- 241000700198 Cavia Species 0.000 description 1
- 241000700199 Cavia porcellus Species 0.000 description 1
- 206010007882 Cellulitis Diseases 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- LZZYPRNAOMGNLH-UHFFFAOYSA-M Cetrimonium bromide Chemical compound [Br-].CCCCCCCCCCCCCCCC[N+](C)(C)C LZZYPRNAOMGNLH-UHFFFAOYSA-M 0.000 description 1
- 229920001661 Chitosan Polymers 0.000 description 1
- 206010008631 Cholera Diseases 0.000 description 1
- 101710164918 Choline-binding protein Proteins 0.000 description 1
- 241001668502 Cladophialophora carrionii Species 0.000 description 1
- 241000222290 Cladosporium Species 0.000 description 1
- 241001508813 Clavispora lusitaniae Species 0.000 description 1
- 108010065152 Coagulase Proteins 0.000 description 1
- 101710094648 Coat protein Proteins 0.000 description 1
- 241000223205 Coccidioides immitis Species 0.000 description 1
- 108700010070 Codon Usage Proteins 0.000 description 1
- 241000702669 Coltivirus Species 0.000 description 1
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 1
- 208000000907 Condylomata Acuminata Diseases 0.000 description 1
- 241001480517 Conidiobolus Species 0.000 description 1
- 102100031673 Corneodesmosin Human genes 0.000 description 1
- FBUKMFOXMZRGRB-UHFFFAOYSA-N Coronaric acid Natural products CCCCCC=CCC1OC1CCCCCCCC(O)=O FBUKMFOXMZRGRB-UHFFFAOYSA-N 0.000 description 1
- 241000186216 Corynebacterium Species 0.000 description 1
- 201000007336 Cryptococcosis Diseases 0.000 description 1
- 241000221204 Cryptococcus neoformans Species 0.000 description 1
- 241000235555 Cunninghamella Species 0.000 description 1
- 241000223208 Curvularia Species 0.000 description 1
- 241000371644 Curvularia ravenelii Species 0.000 description 1
- 108010041986 DNA Vaccines Proteins 0.000 description 1
- 230000004544 DNA amplification Effects 0.000 description 1
- 238000001712 DNA sequencing Methods 0.000 description 1
- 229940021995 DNA vaccine Drugs 0.000 description 1
- 102000004163 DNA-directed RNA polymerases Human genes 0.000 description 1
- 108090000626 DNA-directed RNA polymerases Proteins 0.000 description 1
- 102100035784 Decorin Human genes 0.000 description 1
- 108090000738 Decorin Proteins 0.000 description 1
- 208000001490 Dengue Diseases 0.000 description 1
- 206010012310 Dengue fever Diseases 0.000 description 1
- 229920002307 Dextran Polymers 0.000 description 1
- 108010016626 Dipeptides Proteins 0.000 description 1
- 108010053187 Diphtheria Toxin Proteins 0.000 description 1
- 102000016607 Diphtheria Toxin Human genes 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 206010014596 Encephalitis Japanese B Diseases 0.000 description 1
- 241000305071 Enterobacterales Species 0.000 description 1
- 101000742439 Enterobacteria phage T4 Head formation protein Proteins 0.000 description 1
- 241000194033 Enterococcus Species 0.000 description 1
- 241000194031 Enterococcus faecium Species 0.000 description 1
- 101710146739 Enterotoxin Proteins 0.000 description 1
- 241000146324 Enterovirus D68 Species 0.000 description 1
- 101710121417 Envelope glycoprotein Proteins 0.000 description 1
- 101710091045 Envelope protein Proteins 0.000 description 1
- 241001480035 Epidermophyton Species 0.000 description 1
- 101001069760 Eumenes pomiformis Venom peptide 6 Proteins 0.000 description 1
- 241000223682 Exophiala Species 0.000 description 1
- 241000724791 Filamentous phage Species 0.000 description 1
- 241000192125 Firmicutes Species 0.000 description 1
- 241000122862 Fonsecaea Species 0.000 description 1
- 241000710198 Foot-and-mouth disease virus Species 0.000 description 1
- 241000233866 Fungi Species 0.000 description 1
- 241000223218 Fusarium Species 0.000 description 1
- 241000178292 Geotrichum clavatum Species 0.000 description 1
- 206010018366 Glomerulonephritis acute Diseases 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Chemical compound OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- BCCRXDTUTZHDEU-VKHMYHEASA-N Gly-Ser Chemical compound NCC(=O)N[C@@H](CO)C(O)=O BCCRXDTUTZHDEU-VKHMYHEASA-N 0.000 description 1
- JZNWSCPGTDBMEW-UHFFFAOYSA-N Glycerophosphorylethanolamin Natural products NCCOP(O)(=O)OCC(O)CO JZNWSCPGTDBMEW-UHFFFAOYSA-N 0.000 description 1
- 102100021181 Golgi phosphoprotein 3 Human genes 0.000 description 1
- 206010018612 Gonorrhoea Diseases 0.000 description 1
- 208000031886 HIV Infections Diseases 0.000 description 1
- 101150029115 HOPX gene Proteins 0.000 description 1
- 241001466963 Hawaii calicivirus Species 0.000 description 1
- 101710178376 Heat shock 70 kDa protein Proteins 0.000 description 1
- 108010006464 Hemolysin Proteins Proteins 0.000 description 1
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical compound OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 1
- 208000005331 Hepatitis D Diseases 0.000 description 1
- 208000001688 Herpes Genitalis Diseases 0.000 description 1
- 108091006054 His-tagged proteins Proteins 0.000 description 1
- 241000228404 Histoplasma capsulatum Species 0.000 description 1
- 108090000144 Human Proteins Proteins 0.000 description 1
- 102000003839 Human Proteins Human genes 0.000 description 1
- 206010020460 Human T-cell lymphotropic virus type I infection Diseases 0.000 description 1
- 241000714260 Human T-lymphotropic virus 1 Species 0.000 description 1
- 241000714259 Human T-lymphotropic virus 2 Species 0.000 description 1
- 241000713340 Human immunodeficiency virus 2 Species 0.000 description 1
- 241000702617 Human parvovirus B19 Species 0.000 description 1
- 241000829111 Human polyomavirus 1 Species 0.000 description 1
- 108010003272 Hyaluronate lyase Proteins 0.000 description 1
- 102000001974 Hyaluronidases Human genes 0.000 description 1
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 1
- 108010007403 Immediate-Early Proteins Proteins 0.000 description 1
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 1
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 1
- 102000008070 Interferon-gamma Human genes 0.000 description 1
- 108010074328 Interferon-gamma Proteins 0.000 description 1
- 102000014150 Interferons Human genes 0.000 description 1
- 108010050904 Interferons Proteins 0.000 description 1
- 108010002586 Interleukin-7 Proteins 0.000 description 1
- 108010063738 Interleukins Proteins 0.000 description 1
- 102000015696 Interleukins Human genes 0.000 description 1
- 102000012011 Isocitrate Dehydrogenase Human genes 0.000 description 1
- 108010075869 Isocitrate Dehydrogenase Proteins 0.000 description 1
- 201000005807 Japanese encephalitis Diseases 0.000 description 1
- 241000710842 Japanese encephalitis virus Species 0.000 description 1
- 239000007836 KH2PO4 Substances 0.000 description 1
- 241000588748 Klebsiella Species 0.000 description 1
- 241000588747 Klebsiella pneumoniae Species 0.000 description 1
- 244000285963 Kluyveromyces fragilis Species 0.000 description 1
- 235000014663 Kluyveromyces fragilis Nutrition 0.000 description 1
- ROHFNLRQFUQHCH-YFKPBYRVSA-N L-leucine Chemical compound CC(C)C[C@H](N)C(O)=O ROHFNLRQFUQHCH-YFKPBYRVSA-N 0.000 description 1
- 241000194034 Lactococcus lactis subsp. cremoris Species 0.000 description 1
- 229920002884 Laureth 4 Polymers 0.000 description 1
- 241001071861 Lethrinus genivittatus Species 0.000 description 1
- ROHFNLRQFUQHCH-UHFFFAOYSA-N Leucine Natural products CC(C)CC(N)C(O)=O ROHFNLRQFUQHCH-UHFFFAOYSA-N 0.000 description 1
- 101710170970 Leukotoxin Proteins 0.000 description 1
- 208000016604 Lyme disease Diseases 0.000 description 1
- 108010074338 Lymphokines Proteins 0.000 description 1
- 102000008072 Lymphokines Human genes 0.000 description 1
- 241000711828 Lyssavirus Species 0.000 description 1
- 241000701076 Macacine alphaherpesvirus 1 Species 0.000 description 1
- 102000007651 Macrophage Colony-Stimulating Factor Human genes 0.000 description 1
- 108010046938 Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 241001539803 Magnusiomyces capitatus Species 0.000 description 1
- 101710125418 Major capsid protein Proteins 0.000 description 1
- 241000555676 Malassezia Species 0.000 description 1
- 241000555688 Malassezia furfur Species 0.000 description 1
- 241001559185 Mammalian rubulavirus 5 Species 0.000 description 1
- 102000003939 Membrane transport proteins Human genes 0.000 description 1
- 108090000301 Membrane transport proteins Proteins 0.000 description 1
- 101000773513 Methanopyrus kandleri (strain AV19 / DSM 6324 / JCM 9639 / NBRC 100938) Uncharacterized protein MK0525 Proteins 0.000 description 1
- 241000235048 Meyerozyma guilliermondii Species 0.000 description 1
- 241000893980 Microsporum canis Species 0.000 description 1
- 241001460074 Microsporum distortum Species 0.000 description 1
- 241001260008 Microsporum equinum Species 0.000 description 1
- 241001092142 Molina Species 0.000 description 1
- 241000588621 Moraxella Species 0.000 description 1
- 241000235575 Mortierella Species 0.000 description 1
- 241000235395 Mucor Species 0.000 description 1
- 241000711386 Mumps virus Species 0.000 description 1
- MSFSPUZXLOGKHJ-UHFFFAOYSA-N Muraminsaeure Natural products OC(=O)C(C)OC1C(N)C(O)OC(CO)C1O MSFSPUZXLOGKHJ-UHFFFAOYSA-N 0.000 description 1
- 241000711941 Murine orthopneumovirus Species 0.000 description 1
- 241000711408 Murine respirovirus Species 0.000 description 1
- 241000202934 Mycoplasma pneumoniae Species 0.000 description 1
- 201000002481 Myositis Diseases 0.000 description 1
- 125000000729 N-terminal amino-acid group Chemical group 0.000 description 1
- 241000893976 Nannizzia gypsea Species 0.000 description 1
- 241000264375 Nannizzia nana Species 0.000 description 1
- 241000588654 Neisseria cinerea Species 0.000 description 1
- 241000588649 Neisseria lactamica Species 0.000 description 1
- 239000000020 Nitrocellulose Substances 0.000 description 1
- 208000035823 Non-specific autoimmune cerebellar ataxia without characteristic antibodies Diseases 0.000 description 1
- 101710138767 Non-structural glycoprotein 4 Proteins 0.000 description 1
- 241001263478 Norovirus Species 0.000 description 1
- 108020004711 Nucleic Acid Probes Proteins 0.000 description 1
- 101710141454 Nucleoprotein Proteins 0.000 description 1
- GOWLTLODGKPXMN-MEKRSRHXSA-N OM-174 Chemical compound O1[C@H](OP(O)(O)=O)[C@H](NC(=O)C[C@H](O)CCCCCCCCCCC)[C@@H](O)[C@H](O)[C@H]1CO[C@H]1[C@H](NC(=O)C[C@H](CCCCCCCCCCC)OC(=O)CCCCCCCCCCC)[C@@H](O)[C@H](OP(O)(O)=O)[C@@H](CO)O1 GOWLTLODGKPXMN-MEKRSRHXSA-N 0.000 description 1
- 108010079246 OMPA outer membrane proteins Proteins 0.000 description 1
- 206010030113 Oedema Diseases 0.000 description 1
- 108010038807 Oligopeptides Proteins 0.000 description 1
- 102000015636 Oligopeptides Human genes 0.000 description 1
- 241000702259 Orbivirus Species 0.000 description 1
- 241000702244 Orthoreovirus Species 0.000 description 1
- 240000007594 Oryza sativa Species 0.000 description 1
- 235000007164 Oryza sativa Nutrition 0.000 description 1
- 108700006640 OspA Proteins 0.000 description 1
- 101710203389 Outer membrane porin F Proteins 0.000 description 1
- 101710160107 Outer membrane protein A Proteins 0.000 description 1
- 101710098399 Outer membrane protein YopN Proteins 0.000 description 1
- 238000012408 PCR amplification Methods 0.000 description 1
- 241001236817 Paecilomyces <Clavicipitaceae> Species 0.000 description 1
- 108090000526 Papain Proteins 0.000 description 1
- 241000526686 Paracoccidioides brasiliensis Species 0.000 description 1
- 208000002606 Paramyxoviridae Infections Diseases 0.000 description 1
- 241000228143 Penicillium Species 0.000 description 1
- 108010013639 Peptidoglycan Proteins 0.000 description 1
- 108010081690 Pertussis Toxin Proteins 0.000 description 1
- 206010057249 Phagocytosis Diseases 0.000 description 1
- 206010034839 Pharyngitis streptococcal Diseases 0.000 description 1
- 102000012288 Phosphopyruvate Hydratase Human genes 0.000 description 1
- 108010022181 Phosphopyruvate Hydratase Proteins 0.000 description 1
- 101710099976 Photosystem I P700 chlorophyll a apoprotein A1 Proteins 0.000 description 1
- 108010004729 Phycoerythrin Proteins 0.000 description 1
- 241000235645 Pichia kudriavzevii Species 0.000 description 1
- 235000008331 Pinus X rigitaeda Nutrition 0.000 description 1
- 235000011613 Pinus brutia Nutrition 0.000 description 1
- 241000018646 Pinus brutia Species 0.000 description 1
- 241000305299 Pithomyces Species 0.000 description 1
- 241000233872 Pneumocystis carinii Species 0.000 description 1
- 101710183389 Pneumolysin Proteins 0.000 description 1
- 229940124867 Poliovirus vaccine Drugs 0.000 description 1
- 241000276498 Pollachius virens Species 0.000 description 1
- 229920000954 Polyglycolide Polymers 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- 229920001213 Polysorbate 20 Polymers 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- 102000017033 Porins Human genes 0.000 description 1
- 108010013381 Porins Proteins 0.000 description 1
- 201000009754 Powassan encephalitis Diseases 0.000 description 1
- 241000288906 Primates Species 0.000 description 1
- 101710083689 Probable capsid protein Proteins 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 102000001253 Protein Kinase Human genes 0.000 description 1
- 101710188315 Protein X Proteins 0.000 description 1
- 241000589516 Pseudomonas Species 0.000 description 1
- 101900161471 Pseudomonas aeruginosa Exotoxin A Proteins 0.000 description 1
- 206010037294 Puerperal pyrexia Diseases 0.000 description 1
- 101710178842 Putative penicillin-binding protein Proteins 0.000 description 1
- 241000711798 Rabies lyssavirus Species 0.000 description 1
- 229940124861 Rabies virus vaccine Drugs 0.000 description 1
- 241000700159 Rattus Species 0.000 description 1
- 101100431670 Rattus norvegicus Ybx3 gene Proteins 0.000 description 1
- 102100022648 Reticulon-2 Human genes 0.000 description 1
- 241001361634 Rhizoctonia Species 0.000 description 1
- 241000235527 Rhizopus Species 0.000 description 1
- 102000002278 Ribosomal Proteins Human genes 0.000 description 1
- 108010000605 Ribosomal Proteins Proteins 0.000 description 1
- 241000606701 Rickettsia Species 0.000 description 1
- 241000283984 Rodentia Species 0.000 description 1
- 241000710799 Rubella virus Species 0.000 description 1
- 208000032108 Russian spring-summer encephalitis Diseases 0.000 description 1
- 241000315672 SARS coronavirus Species 0.000 description 1
- 102100021798 SH2 domain-containing protein 3C Human genes 0.000 description 1
- 241000235070 Saccharomyces Species 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- 241000293026 Saksenaea Species 0.000 description 1
- 241000293869 Salmonella enterica subsp. enterica serovar Typhimurium Species 0.000 description 1
- 241000132889 Scedosporium Species 0.000 description 1
- 206010040047 Sepsis Diseases 0.000 description 1
- 206010040070 Septic Shock Diseases 0.000 description 1
- 102000012479 Serine Proteases Human genes 0.000 description 1
- 108010022999 Serine Proteases Proteins 0.000 description 1
- 102100031056 Serine protease 57 Human genes 0.000 description 1
- 101710197596 Serine protease 57 Proteins 0.000 description 1
- 241000710960 Sindbis virus Species 0.000 description 1
- 240000002493 Smilax officinalis Species 0.000 description 1
- 235000008981 Smilax officinalis Nutrition 0.000 description 1
- 241000509413 Snow Mountain virus Species 0.000 description 1
- DBMJMQXJHONAFJ-UHFFFAOYSA-M Sodium laurylsulphate Chemical compound [Na+].CCCCCCCCCCCCOS([O-])(=O)=O DBMJMQXJHONAFJ-UHFFFAOYSA-M 0.000 description 1
- 229920002125 Sokalan® Polymers 0.000 description 1
- 239000004147 Sorbitan trioleate Substances 0.000 description 1
- 241001149962 Sporothrix Species 0.000 description 1
- 241001149963 Sporothrix schenckii Species 0.000 description 1
- 241000713675 Spumavirus Species 0.000 description 1
- 206010041896 St. Louis Encephalitis Diseases 0.000 description 1
- 241000191963 Staphylococcus epidermidis Species 0.000 description 1
- 235000014962 Streptococcus cremoris Nutrition 0.000 description 1
- 244000057717 Streptococcus lactis Species 0.000 description 1
- 235000014897 Streptococcus lactis Nutrition 0.000 description 1
- 241000320123 Streptococcus pyogenes M1 GAS Species 0.000 description 1
- 108700037929 Streptococcus pyogenes SpeA Proteins 0.000 description 1
- 101710172711 Structural protein Proteins 0.000 description 1
- 101710137302 Surface antigen S Proteins 0.000 description 1
- 210000001744 T-lymphocyte Anatomy 0.000 description 1
- 241001523006 Talaromyces marneffei Species 0.000 description 1
- 102000009843 Thyroglobulin Human genes 0.000 description 1
- 102000008235 Toll-Like Receptor 9 Human genes 0.000 description 1
- 108010060818 Toll-Like Receptor 9 Proteins 0.000 description 1
- 206010044248 Toxic shock syndrome Diseases 0.000 description 1
- 241000223997 Toxoplasma gondii Species 0.000 description 1
- 241000390203 Trachoma Species 0.000 description 1
- 108010031133 Transferrin-Binding Protein A Proteins 0.000 description 1
- 108010031127 Transferrin-Binding Protein B Proteins 0.000 description 1
- 101800001690 Transmembrane protein gp41 Proteins 0.000 description 1
- 241000893963 Trichophyton concentricum Species 0.000 description 1
- 241000893962 Trichophyton equinum Species 0.000 description 1
- 241001045770 Trichophyton mentagrophytes Species 0.000 description 1
- 241001609979 Trichophyton quinckeanum Species 0.000 description 1
- 241000223229 Trichophyton rubrum Species 0.000 description 1
- 241001480048 Trichophyton tonsurans Species 0.000 description 1
- 241001480050 Trichophyton violaceum Species 0.000 description 1
- 241000223231 Trichosporon beigelii Species 0.000 description 1
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 1
- 241000711955 Turkey rhinotracheitis virus Species 0.000 description 1
- 108010073429 Type V Secretion Systems Proteins 0.000 description 1
- 208000037386 Typhoid Diseases 0.000 description 1
- 101710100170 Unknown protein Proteins 0.000 description 1
- 108010046334 Urease Proteins 0.000 description 1
- 206010051511 Viral diarrhoea Diseases 0.000 description 1
- 208000036142 Viral infection Diseases 0.000 description 1
- 201000006449 West Nile encephalitis Diseases 0.000 description 1
- 208000003152 Yellow Fever Diseases 0.000 description 1
- 241000607447 Yersinia enterocolitica Species 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- UZQJVUCHXGYFLQ-AYDHOLPZSA-N [(2s,3r,4s,5r,6r)-4-[(2s,3r,4s,5r,6r)-4-[(2r,3r,4s,5r,6r)-4-[(2s,3r,4s,5r,6r)-3,5-dihydroxy-6-(hydroxymethyl)-4-[(2s,3r,4s,5s,6r)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxyoxan-2-yl]oxy-3,5-dihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy-3,5-dihydroxy-6-(hy Chemical compound O([C@H]1[C@H](O)[C@@H](CO)O[C@H]([C@@H]1O)O[C@H]1[C@H](O)[C@@H](CO)O[C@H]([C@@H]1O)O[C@H]1CC[C@]2(C)[C@H]3CC=C4[C@@]([C@@]3(CC[C@H]2[C@@]1(C=O)C)C)(C)CC(O)[C@]1(CCC(CC14)(C)C)C(=O)O[C@H]1[C@@H]([C@@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O[C@H]4[C@@H]([C@@H](O[C@H]5[C@@H]([C@@H](O)[C@H](O)[C@@H](CO)O5)O)[C@H](O)[C@@H](CO)O4)O)[C@H](O)[C@@H](CO)O3)O)[C@H](O)[C@@H](CO)O2)O)[C@H](O)[C@@H](CO)O1)O)[C@@H]1O[C@H](CO)[C@@H](O)[C@H](O)[C@H]1O UZQJVUCHXGYFLQ-AYDHOLPZSA-N 0.000 description 1
- 241000222126 [Candida] glabrata Species 0.000 description 1
- 229960000583 acetic acid Drugs 0.000 description 1
- 230000021736 acetylation Effects 0.000 description 1
- 238000006640 acetylation reaction Methods 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 230000001154 acute effect Effects 0.000 description 1
- 231100000851 acute glomerulonephritis Toxicity 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 238000011374 additional therapy Methods 0.000 description 1
- 230000000240 adjuvant effect Effects 0.000 description 1
- 241001148470 aerobic bacillus Species 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 150000005215 alkyl ethers Chemical class 0.000 description 1
- 229910021502 aluminium hydroxide Inorganic materials 0.000 description 1
- 229940047712 aluminum hydroxyphosphate Drugs 0.000 description 1
- 229940103272 aluminum potassium sulfate Drugs 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 239000000908 ammonium hydroxide Substances 0.000 description 1
- BFNBIHQBYMNNAN-UHFFFAOYSA-N ammonium sulfate Chemical compound N.N.OS(O)(=O)=O BFNBIHQBYMNNAN-UHFFFAOYSA-N 0.000 description 1
- 229910052921 ammonium sulfate Inorganic materials 0.000 description 1
- 235000011130 ammonium sulphate Nutrition 0.000 description 1
- 208000025009 anogenital human papillomavirus infection Diseases 0.000 description 1
- 201000004201 anogenital venereal wart Diseases 0.000 description 1
- 238000010913 antigen-directed enzyme pro-drug therapy Methods 0.000 description 1
- 230000027645 antigenic variation Effects 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 238000013528 artificial neural network Methods 0.000 description 1
- 229940091771 aspergillus fumigatus Drugs 0.000 description 1
- 244000052616 bacterial pathogen Species 0.000 description 1
- 230000007924 bacterial virulence factor Effects 0.000 description 1
- 229940092528 bartonella bacilliformis Drugs 0.000 description 1
- 239000011324 bead Substances 0.000 description 1
- 229960003237 betaine Drugs 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 239000012620 biological material Substances 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- 230000006287 biotinylation Effects 0.000 description 1
- 238000007413 biotinylation Methods 0.000 description 1
- 239000003114 blood coagulation factor Substances 0.000 description 1
- 208000008921 border disease Diseases 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 206010006451 bronchitis Diseases 0.000 description 1
- 230000009172 bursting Effects 0.000 description 1
- SNCZNSNPXMPCGN-UHFFFAOYSA-N butanediamide Chemical compound NC(=O)CCC(N)=O SNCZNSNPXMPCGN-UHFFFAOYSA-N 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 239000001506 calcium phosphate Substances 0.000 description 1
- 229910000389 calcium phosphate Inorganic materials 0.000 description 1
- 235000011010 calcium phosphates Nutrition 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 244000309466 calf Species 0.000 description 1
- 229940095731 candida albicans Drugs 0.000 description 1
- 208000032343 candida glabrata infection Diseases 0.000 description 1
- 229940055022 candida parapsilosis Drugs 0.000 description 1
- 210000000234 capsid Anatomy 0.000 description 1
- MPBRYMWMMKKRGC-UHFFFAOYSA-M carbocyanin DBTC Chemical compound [Br-].C1=CC=CC2=C([N+](=C(C=C(C)C=C3N(C4=C5C=CC=CC5=CC=C4S3)CC)S3)CC)C3=CC=C21 MPBRYMWMMKKRGC-UHFFFAOYSA-M 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 230000021523 carboxylation Effects 0.000 description 1
- 238000006473 carboxylation reaction Methods 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 235000021466 carotenoid Nutrition 0.000 description 1
- 150000001747 carotenoids Chemical class 0.000 description 1
- 125000002091 cationic group Chemical group 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 230000030570 cellular localization Effects 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 239000013522 chelant Substances 0.000 description 1
- 150000001841 cholesterols Chemical class 0.000 description 1
- 238000013375 chromatographic separation Methods 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 239000007979 citrate buffer Substances 0.000 description 1
- 229960002227 clindamycin Drugs 0.000 description 1
- KDLRVYVGXIQJDK-AWPVFWJPSA-N clindamycin Chemical compound CN1C[C@H](CCC)C[C@H]1C(=O)N[C@H]([C@H](C)Cl)[C@@H]1[C@H](O)[C@H](O)[C@@H](O)[C@@H](SC)O1 KDLRVYVGXIQJDK-AWPVFWJPSA-N 0.000 description 1
- 238000011260 co-administration Methods 0.000 description 1
- 238000005354 coacervation Methods 0.000 description 1
- 230000001332 colony forming effect Effects 0.000 description 1
- 230000009137 competitive binding Effects 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 230000009918 complex formation Effects 0.000 description 1
- 230000001010 compromised effect Effects 0.000 description 1
- 238000004132 cross linking Methods 0.000 description 1
- 101150110403 cspA gene Proteins 0.000 description 1
- 101150068339 cspLA gene Proteins 0.000 description 1
- ATDGTVJJHBUTRL-UHFFFAOYSA-N cyanogen bromide Chemical compound BrC#N ATDGTVJJHBUTRL-UHFFFAOYSA-N 0.000 description 1
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical class NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 description 1
- 230000007123 defense Effects 0.000 description 1
- 239000008367 deionised water Substances 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- 238000002716 delivery method Methods 0.000 description 1
- 208000025729 dengue disease Diseases 0.000 description 1
- 238000001784 detoxification Methods 0.000 description 1
- 238000002405 diagnostic procedure Methods 0.000 description 1
- 239000000539 dimer Substances 0.000 description 1
- 229960003983 diphtheria toxoid Drugs 0.000 description 1
- 210000001840 diploid cell Anatomy 0.000 description 1
- 229940042396 direct acting antivirals thiosemicarbazones Drugs 0.000 description 1
- BNIILDVGGAEEIG-UHFFFAOYSA-L disodium hydrogen phosphate Chemical compound [Na+].[Na+].OP([O-])([O-])=O BNIILDVGGAEEIG-UHFFFAOYSA-L 0.000 description 1
- 229910000397 disodium phosphate Inorganic materials 0.000 description 1
- 235000019800 disodium phosphate Nutrition 0.000 description 1
- 239000012153 distilled water Substances 0.000 description 1
- 238000001493 electron microscopy Methods 0.000 description 1
- 238000004520 electroporation Methods 0.000 description 1
- 238000002101 electrospray ionisation tandem mass spectrometry Methods 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 230000000369 enteropathogenic effect Effects 0.000 description 1
- 230000000688 enterotoxigenic effect Effects 0.000 description 1
- 239000000147 enterotoxin Substances 0.000 description 1
- 231100000655 enterotoxin Toxicity 0.000 description 1
- 210000003743 erythrocyte Anatomy 0.000 description 1
- 230000017188 evasion or tolerance of host immune response Effects 0.000 description 1
- XUFQPHANEAPEMJ-UHFFFAOYSA-N famotidine Chemical compound NC(N)=NC1=NC(CSCCC(N)=NS(N)(=O)=O)=CS1 XUFQPHANEAPEMJ-UHFFFAOYSA-N 0.000 description 1
- 230000001605 fetal effect Effects 0.000 description 1
- 244000053095 fungal pathogen Species 0.000 description 1
- 238000001502 gel electrophoresis Methods 0.000 description 1
- 102000034356 gene-regulatory proteins Human genes 0.000 description 1
- 108091006104 gene-regulatory proteins Proteins 0.000 description 1
- 230000002068 genetic effect Effects 0.000 description 1
- 201000004946 genital herpes Diseases 0.000 description 1
- 229910001679 gibbsite Inorganic materials 0.000 description 1
- 239000012362 glacial acetic acid Substances 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- KWIUHFFTVRNATP-UHFFFAOYSA-N glycine betaine Chemical compound C[N+](C)(C)CC([O-])=O KWIUHFFTVRNATP-UHFFFAOYSA-N 0.000 description 1
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 1
- 239000010931 gold Substances 0.000 description 1
- 229910052737 gold Inorganic materials 0.000 description 1
- 239000010440 gypsum Substances 0.000 description 1
- 229910052602 gypsum Inorganic materials 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 108010028403 hemagglutinin esterase Proteins 0.000 description 1
- 239000003228 hemolysin Substances 0.000 description 1
- 229920000669 heparin Polymers 0.000 description 1
- 229960002897 heparin Drugs 0.000 description 1
- 208000006454 hepatitis Diseases 0.000 description 1
- 231100000283 hepatitis Toxicity 0.000 description 1
- 108700039582 histidine triad Proteins 0.000 description 1
- 229930186900 holotoxin Natural products 0.000 description 1
- 230000028996 humoral immune response Effects 0.000 description 1
- 229960002773 hyaluronidase Drugs 0.000 description 1
- 239000000017 hydrogel Substances 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- 150000004679 hydroxides Chemical class 0.000 description 1
- FAHBNUUHRFUEAI-UHFFFAOYSA-M hydroxidooxidoaluminium Chemical compound O[Al]=O FAHBNUUHRFUEAI-UHFFFAOYSA-M 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 229960002751 imiquimod Drugs 0.000 description 1
- DOUYETYNHWVLEO-UHFFFAOYSA-N imiquimod Chemical compound C1=CC=CC2=C3N(CC(C)C)C=NC3=C(N)N=C21 DOUYETYNHWVLEO-UHFFFAOYSA-N 0.000 description 1
- 238000003119 immunoblot Methods 0.000 description 1
- 230000002998 immunogenetic effect Effects 0.000 description 1
- 230000016784 immunoglobulin production Effects 0.000 description 1
- 229940072221 immunoglobulins Drugs 0.000 description 1
- 238000003126 immunogold labeling Methods 0.000 description 1
- 230000002055 immunohistochemical effect Effects 0.000 description 1
- 230000002434 immunopotentiative effect Effects 0.000 description 1
- 238000001114 immunoprecipitation Methods 0.000 description 1
- 239000003022 immunostimulating agent Substances 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 201000001371 inclusion conjunctivitis Diseases 0.000 description 1
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 230000002458 infectious effect Effects 0.000 description 1
- 208000037797 influenza A Diseases 0.000 description 1
- 208000037798 influenza B Diseases 0.000 description 1
- 208000037799 influenza C Diseases 0.000 description 1
- 229960003971 influenza vaccine Drugs 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 239000002054 inoculum Substances 0.000 description 1
- 229960003130 interferon gamma Drugs 0.000 description 1
- 229940047124 interferons Drugs 0.000 description 1
- 229940047122 interleukins Drugs 0.000 description 1
- 230000000968 intestinal effect Effects 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 238000004255 ion exchange chromatography Methods 0.000 description 1
- FZWBNHMXJMCXLU-BLAUPYHCSA-N isomaltotriose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1OC[C@@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@@H](OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O)O1 FZWBNHMXJMCXLU-BLAUPYHCSA-N 0.000 description 1
- 238000005304 joining Methods 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 230000002147 killing effect Effects 0.000 description 1
- 238000002372 labelling Methods 0.000 description 1
- JJTUDXZGHPGLLC-UHFFFAOYSA-N lactide Chemical compound CC1OC(=O)C(C)OC1=O JJTUDXZGHPGLLC-UHFFFAOYSA-N 0.000 description 1
- 239000004816 latex Substances 0.000 description 1
- 229920000126 latex Polymers 0.000 description 1
- 229940062711 laureth-9 Drugs 0.000 description 1
- 231100000518 lethal Toxicity 0.000 description 1
- 230000001665 lethal effect Effects 0.000 description 1
- 210000000265 leukocyte Anatomy 0.000 description 1
- 230000029226 lipidation Effects 0.000 description 1
- 238000004895 liquid chromatography mass spectrometry Methods 0.000 description 1
- 238000001294 liquid chromatography-tandem mass spectrometry Methods 0.000 description 1
- 239000006193 liquid solution Substances 0.000 description 1
- 229960003213 live attenuated measles Drugs 0.000 description 1
- 229960003897 live attenuated mumps Drugs 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 238000011068 loading method Methods 0.000 description 1
- 230000004807 localization Effects 0.000 description 1
- 230000005923 long-lasting effect Effects 0.000 description 1
- 210000005265 lung cell Anatomy 0.000 description 1
- 210000004698 lymphocyte Anatomy 0.000 description 1
- 208000001581 lymphogranuloma venereum Diseases 0.000 description 1
- 235000018977 lysine Nutrition 0.000 description 1
- 125000003588 lysine group Chemical class [H]N([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])(N([H])[H])C(*)=O 0.000 description 1
- 230000002934 lysing effect Effects 0.000 description 1
- 239000012139 lysis buffer Substances 0.000 description 1
- 210000002540 macrophage Anatomy 0.000 description 1
- 229910001629 magnesium chloride Inorganic materials 0.000 description 1
- 230000000873 masking effect Effects 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000010534 mechanism of action Effects 0.000 description 1
- MYWUZJCMWCOHBA-VIFPVBQESA-N methamphetamine Chemical compound CN[C@@H](C)CC1=CC=CC=C1 MYWUZJCMWCOHBA-VIFPVBQESA-N 0.000 description 1
- 125000001360 methionine group Chemical group N[C@@H](CCSC)C(=O)* 0.000 description 1
- 238000002493 microarray Methods 0.000 description 1
- 244000005700 microbiome Species 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 238000010369 molecular cloning Methods 0.000 description 1
- 210000005087 mononuclear cell Anatomy 0.000 description 1
- 229910000402 monopotassium phosphate Inorganic materials 0.000 description 1
- 235000019796 monopotassium phosphate Nutrition 0.000 description 1
- 230000000877 morphologic effect Effects 0.000 description 1
- 230000001459 mortal effect Effects 0.000 description 1
- 101150047914 mpt51 gene Proteins 0.000 description 1
- 210000003097 mucus Anatomy 0.000 description 1
- 230000035772 mutation Effects 0.000 description 1
- JXTPJDDICSTXJX-UHFFFAOYSA-N n-Triacontane Natural products CCCCCCCCCCCCCCCCCCCCCCCCCCCCCC JXTPJDDICSTXJX-UHFFFAOYSA-N 0.000 description 1
- 210000004898 n-terminal fragment Anatomy 0.000 description 1
- 230000007935 neutral effect Effects 0.000 description 1
- 229910052759 nickel Inorganic materials 0.000 description 1
- 229920001220 nitrocellulos Polymers 0.000 description 1
- 239000002736 nonionic surfactant Substances 0.000 description 1
- 239000002853 nucleic acid probe Substances 0.000 description 1
- 238000001821 nucleic acid purification Methods 0.000 description 1
- 238000010397 one-hybrid screening Methods 0.000 description 1
- 230000014207 opsonization Effects 0.000 description 1
- 230000000625 opsonophagocytic effect Effects 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 239000006179 pH buffering agent Substances 0.000 description 1
- 238000012856 packing Methods 0.000 description 1
- 230000000242 pagocytic effect Effects 0.000 description 1
- 229940055729 papain Drugs 0.000 description 1
- 235000019834 papain Nutrition 0.000 description 1
- 108700010867 paramyxovirus proteins Proteins 0.000 description 1
- 150000002960 penicillins Chemical class 0.000 description 1
- 230000007030 peptide scission Effects 0.000 description 1
- 230000002093 peripheral effect Effects 0.000 description 1
- 108010021711 pertactin Proteins 0.000 description 1
- 238000002823 phage display Methods 0.000 description 1
- 230000008782 phagocytosis Effects 0.000 description 1
- 239000008363 phosphate buffer Substances 0.000 description 1
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 1
- WTJKGGKOPKCXLL-RRHRGVEJSA-N phosphatidylcholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCC=CCCCCCCCC WTJKGGKOPKCXLL-RRHRGVEJSA-N 0.000 description 1
- 150000008104 phosphatidylethanolamines Chemical class 0.000 description 1
- 150000003013 phosphoric acid derivatives Chemical class 0.000 description 1
- 230000026731 phosphorylation Effects 0.000 description 1
- 238000006366 phosphorylation reaction Methods 0.000 description 1
- 230000004962 physiological condition Effects 0.000 description 1
- 230000004260 plant-type cell wall biogenesis Effects 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- ONJQDTZCDSESIW-UHFFFAOYSA-N polidocanol Chemical compound CCCCCCCCCCCCOCCOCCOCCOCCOCCOCCOCCOCCOCCO ONJQDTZCDSESIW-UHFFFAOYSA-N 0.000 description 1
- 229960001539 poliomyelitis vaccine Drugs 0.000 description 1
- 229920001983 poloxamer Polymers 0.000 description 1
- 229920000771 poly (alkylcyanoacrylate) Polymers 0.000 description 1
- 229940065514 poly(lactide) Drugs 0.000 description 1
- 229920002401 polyacrylamide Polymers 0.000 description 1
- 239000004584 polyacrylic acid Substances 0.000 description 1
- 229920000447 polyanionic polymer Polymers 0.000 description 1
- 108091033319 polynucleotide Proteins 0.000 description 1
- 102000040430 polynucleotide Human genes 0.000 description 1
- 239000002157 polynucleotide Substances 0.000 description 1
- 229920005862 polyol Polymers 0.000 description 1
- 150000003077 polyols Chemical class 0.000 description 1
- 229940051841 polyoxyethylene ether Drugs 0.000 description 1
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 1
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 229920000136 polysorbate Polymers 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 230000004481 post-translational protein modification Effects 0.000 description 1
- GRLPQNLYRHEGIJ-UHFFFAOYSA-J potassium aluminium sulfate Chemical compound [Al+3].[K+].[O-]S([O-])(=O)=O.[O-]S([O-])(=O)=O GRLPQNLYRHEGIJ-UHFFFAOYSA-J 0.000 description 1
- GNSKLFRGEWLPPA-UHFFFAOYSA-M potassium dihydrogen phosphate Chemical compound [K+].OP(O)([O-])=O GNSKLFRGEWLPPA-UHFFFAOYSA-M 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 238000011809 primate model Methods 0.000 description 1
- 210000001236 prokaryotic cell Anatomy 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 230000000644 propagated effect Effects 0.000 description 1
- 229940021993 prophylactic vaccine Drugs 0.000 description 1
- 230000013777 protein digestion Effects 0.000 description 1
- 108060006633 protein kinase Proteins 0.000 description 1
- 239000012460 protein solution Substances 0.000 description 1
- 238000001243 protein synthesis Methods 0.000 description 1
- 230000006337 proteolytic cleavage Effects 0.000 description 1
- 210000001938 protoplast Anatomy 0.000 description 1
- 239000002510 pyrogen Substances 0.000 description 1
- HNJBEVLQSNELDL-UHFFFAOYSA-N pyrrolidin-2-one Chemical compound O=C1CCCN1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 description 1
- 238000011555 rabbit model Methods 0.000 description 1
- 238000003127 radioimmunoassay Methods 0.000 description 1
- 230000009257 reactivity Effects 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 238000003259 recombinant expression Methods 0.000 description 1
- 238000010188 recombinant method Methods 0.000 description 1
- 230000006798 recombination Effects 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000003362 replicative effect Effects 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 201000003068 rheumatic fever Diseases 0.000 description 1
- 235000009566 rice Nutrition 0.000 description 1
- 229960002181 saccharomyces boulardii Drugs 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 208000013223 septicemia Diseases 0.000 description 1
- 230000001568 sexual effect Effects 0.000 description 1
- 238000004904 shortening Methods 0.000 description 1
- 238000001542 size-exclusion chromatography Methods 0.000 description 1
- 239000000344 soap Substances 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 238000000527 sonication Methods 0.000 description 1
- 235000019337 sorbitan trioleate Nutrition 0.000 description 1
- 229960000391 sorbitan trioleate Drugs 0.000 description 1
- 238000001179 sorption measurement Methods 0.000 description 1
- 230000009870 specific binding Effects 0.000 description 1
- 230000007480 spreading Effects 0.000 description 1
- 238000003892 spreading Methods 0.000 description 1
- 229940032094 squalane Drugs 0.000 description 1
- 239000007858 starting material Substances 0.000 description 1
- 238000007619 statistical method Methods 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 208000016765 streptococcal sore throat Diseases 0.000 description 1
- 239000000758 substrate Substances 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- 239000013589 supplement Substances 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 238000007910 systemic administration Methods 0.000 description 1
- 229940021747 therapeutic vaccine Drugs 0.000 description 1
- 238000004809 thin layer chromatography Methods 0.000 description 1
- RYYWUUFWQRZTIU-UHFFFAOYSA-K thiophosphate Chemical compound [O-]P([O-])([O-])=S RYYWUUFWQRZTIU-UHFFFAOYSA-K 0.000 description 1
- 229960002175 thyroglobulin Drugs 0.000 description 1
- 210000001578 tight junction Anatomy 0.000 description 1
- 238000004448 titration Methods 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 238000013518 transcription Methods 0.000 description 1
- 230000035897 transcription Effects 0.000 description 1
- 230000002103 transcriptional effect Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 238000013519 translation Methods 0.000 description 1
- STCOOQWBFONSKY-UHFFFAOYSA-N tributyl phosphate Chemical compound CCCCOP(=O)(OCCCC)OCCCC STCOOQWBFONSKY-UHFFFAOYSA-N 0.000 description 1
- QORWJWZARLRLPR-UHFFFAOYSA-H tricalcium bis(phosphate) Chemical compound [Ca+2].[Ca+2].[Ca+2].[O-]P([O-])([O-])=O.[O-]P([O-])([O-])=O QORWJWZARLRLPR-UHFFFAOYSA-H 0.000 description 1
- 229940055035 trichophyton verrucosum Drugs 0.000 description 1
- 239000013638 trimer Substances 0.000 description 1
- 150000004043 trisaccharides Chemical group 0.000 description 1
- 102000003390 tumor necrosis factor Human genes 0.000 description 1
- 201000008297 typhoid fever Diseases 0.000 description 1
- 238000005199 ultracentrifugation Methods 0.000 description 1
- 241001148471 unidentified anaerobic bacterium Species 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 229920002554 vinyl polymer Polymers 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
- 238000009736 wetting Methods 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- 229940098232 yersinia enterocolitica Drugs 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
- PAPBSGBWRJIAAV-UHFFFAOYSA-N ε-Caprolactone Chemical compound O=C1CCCCCO1 PAPBSGBWRJIAAV-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/195—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from bacteria
- C07K14/315—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from bacteria from Streptococcus (G), e.g. Enterococci
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/505—Medicinal preparations containing antigens or antibodies comprising antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/51—Medicinal preparations containing antigens or antibodies comprising whole cells, viruses or DNA/RNA
- A61K2039/52—Bacterial cells; Fungal cells; Protozoal cells
- A61K2039/523—Bacterial cells; Fungal cells; Protozoal cells expressing foreign proteins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/51—Medicinal preparations containing antigens or antibodies comprising whole cells, viruses or DNA/RNA
- A61K2039/53—DNA (RNA) vaccination
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
Definitions
- This invention is in the fields of immunology and vaccinology.
- it relates to antigens derived from Streptococcus pyogenes and their use in immunization.
- Group A streptococcus (“GAS,” S. pyogenes) is a frequent human pathogen, estimated to be present in between 5-15% of normal individuals without signs of disease.
- GAS Group A streptococcus
- An acute infection occurs, however, when host defenses are compromised, when the organism is able to exert its virulence, or when the organism is introduced to vulnerable tissues or hosts.
- Related diseases include puerperal fever, scarlet fever, erysipelas, pharyngitis, impetigo, necrotizing fasciitis, myositis, and streptococcal toxic shock syndrome.
- GAS bacteria are gram positive, non-spore forming coccus-shaped bacteria which typically exist in chains or in pairs of cells. GAS bacteria are subdivided according to serotyping based on a large, highly variable cell surface antigen call the M protein. Lancefield, J. Exp. Med. 47, 9-10, 1928; Lancefield, J. Immunol. 89, 307-13, 1962. DNA sequencing of genes encoding M proteins has become the most common method of determining GASM types (emm sequence types). To date 124 different M types have been identified; 22 of these types were identified between 1995 and 1998 (Facklam et al, Clin. Infect. Dis. 34, 28-38, 2002).
- Ml, M28, M12, M3, Mi l, and M6 are among the most prevalent GAS types worldwide. Li et al, Infect. Dis. 188, 1587-92, 2003; O'Brien et al, Clin. Infect. Dis. 35, 268-76, 2002.
- S. pyogenes infections can be treated using antibiotics, there is a need in the art for prophylactic vaccines to prevent the onset of disease, as well as for additional therapies for treating S. pyogenes infections.
- FIG. 1 Alignment of the amino acid sequences of GAS40 proteins from GASM strains SF370, 2580, 3280, 3348, 3789, and 2913 (SEQ ID NO:17) 2634 (SEQ ID NO:18), 2726 (SEQ ID NO: 19), 2721 (SEQ ID NO:20), 3040 and 3135 (SEQ ID NO:21), 2722 (SEQ ID NO:22), 2728 (SEQ ID NO:23), 4883 (SEQ ID NO:24), 2724 (SEQ ID NO:25), 2894, 3650, 5529, and 3776 (SEQ ID NO:26), 2720 (SEQ ID NO:27), 2725 (SEQ ID NO:28), 4538 (SEQ ID NO:29), 5531 (SEQ ID NO:30), 5481 (SEQ ID NO:31), 4959 (SEQ ID NO:32), D2071 (SEQ ID NO:33), 4436 (SEQ ID NO:34), 2727 (SEQ ID NO:35), 2719 (SEQ ID NO:36
- FIG. IA amino acids 1-50; FIG. IB, amino acids 51-100; FIG. 1C, amino acids 101-150; FIG. ID, amino acids 151-200; FIG. IE, amino acids 201-250; FIG. IF, amino acids 251-300; FIG. IG, amino acids 301-350; FIG. IH, amino acids 351-400; FIG. II, amino acids 401-450; FIG. IJ, amino acids 451-500; FIG. IK, amino acids 501-550; FIG. IL, amino acids 551-600; FIG. IM, amino acids 601-650; FIG. IN, amino acids 651-700; FIG. 10, amino acids 701-750; FIG. IP, amino acids 751-800; FIG. IQ, amino acids 801-850; FIG. IR, amino acids 851-873.
- FIG. 2. Results of FACS analysis demonstrating that GAS40 proteins are exposed on the cell surface of strains of different M types.
- FIG. 3. Results of FACS analysis demonstrating that antisera directed against native GAS40 protein detects GAS40 protein on the cell surface of strains of different M types (strains DSM2071, 2634, a hypocapsulated mutant of DSM2071, 2727, SF370, 2720, 3789, 2725, 2580, 2894, 2728, 2913, 2726, 3348, and 3280).
- FIG. 4A-B Results of FACS analysis demonstrating that antisera directed against a "GST-GAS40" antigen detects GAS40 protein on the cell surface of strains of different M types.
- FIG. 4A strains 3789, 4883, a hypocapsulated mutant of DSM2071, 5476, SF370, DSM2071, 2720, 2723, 2728, 2724, 2580, 2725, 2719, 2726, 3776.
- FIG. 4B strains 4436, 2721, 4959, 2727, 5468, 3650, 2634, 4088, 4538, 2722.
- FIG. 5A-B Results of FACS analysis demonstrating that antisera directed against a GAS40a antigen detects GAS40 protein on the cell surface of strains of different M types.
- FIG. 5A DSM2071, SF370, 2721, 3280, 2728, 3789, a hypocapsulated mutant of DSM2071, 4883, 5476, 2725, 2720, 2726, 2723, 2728, 2724, and 2580;
- FIG. 5B 2719, 5468, 3776, 2634, 4436, 2721, 4959, 2727, 3650, 4088, 4538, and 2722.
- FIG. 6A-B Results of FACS analysis demonstrating that antisera directed against a GAS40aCH antigen detects GAS40 protein on the cell surface of strains of different M types.
- FIG. 6A strains DSM2071, 3280, 2721, 3789, 2728, 4883, a hypocapsulated mutant of DSM2071, 5476, SF370, 2720, 2723, 2580, 2724, 2719, 2725, 3776, 2726, 4436, 2728, and 4959;
- FIG. 6B strains 5468, 4088, 2634, 4538, 2721, 2722, 2727, and 3650.
- FIG. 7 Results of FACS analysis demonstrating that antisera directed against a GAS40/GAS117 hybrid antigen detects GAS40 protein on the cell surface of strains of different M types (strains 2720, 2726, 2725, 3280, 2580, 2728).
- FIG. 8 Results of FACS analysis demonstrating that antisera directed against a GAS117/GAS40 hybrid antigen detects GAS40 protein on the cell surface of strains of different M types (strains DSM2071, 2634, a hypocapsulated mutant of DSM2071, 2727, 3789, 2720, SF370, 2725, 258O 5 2894, 2728, 2913, 2726, 3348, 3280).
- FIG. 9A-B Results of FACS analysis demonstrating that antisera directed against a GAS40aRR antigen detects GAS40 protein on the cell surface of strains of different M types.
- FIG. 9A strains DSM2071, 3280, 2721, 4789, 2728, 4883, a hypocapsulated mutant of DSM2071, 5476, SF370, 2720, 2723, 2580, 2724, 2719, 2725, 3776, 2726, 4436, 2728, 4959;
- FIG. 9B strains 5468, 4088, 2634, 4538, 2721, 2722, 2727, 3650.
- FIG. 10A-B Results of FACS analysis demonstrating that antisera directed against a GAS40aNH antigen detects GAS40 protein on the cell surface of. strains of different M types.
- FIG. 1OA strains DSM2071, 3280, 2721, 3789, 2728, 4883 a hypocapsulated mutant of DSM2071, 5476, SF370, 2720, 2723, 2580, 2724, 2719, 2725, 3776, 2726, 4436, 2728, 4959;
- FIG. 1OB strains 5468, 4088, 2634, 4538, 2721, 2722, 2727, 3650.
- FIG. HA-B Results of FACS analysis demonstrating that antisera directed against a GAS40aRRNH antigen detects GAS40 protein on the cell surface of strains of different M types.
- FIG. 1 IA strains DSM2071, 3280, 2721, 3789, 2728, 4883, a hypocapsulated mutant of DSM2071, 5476, SF370, 2720, 2723, 2580, 2724, 2719, 2725, 3776, 2726, 4436, 2728, 4959;
- FIGS. HB strains 5468, 4088, 2634, 4538, 2721, 2722, 2727, 3650.
- FIG. 12 A-B Diagram of expression vectors and recombinant GAS antigens.
- FIG. 12A expression vectors pET-21+ and pGEX;
- FIG. 12B encoded recombinant proteins.
- FIG. 13 Schematic view of mouse model.
- FIG. 14 Mouse model results.
- FIG. 15 Schematic view of GAS40 structure.
- FIG. 16 Western blots showing expression of GAS40 in different GAS serotypes.
- FIG. 17 FACS pictograms showing surface expression of GAS40.
- FIG. 18 Photomicrographs showing distribution of GAS40 on the bacterial cell surface.
- FIG. 19 Graph illustrating bactericidal properties of anti-GAS40 antibodies.
- FIG. 20 Graph illustrating opsonization properties of anti-GAS40 antibodies.
- FIG. 21 Schematic view of GAS40 domains.
- FIG. 22 Graph illustrating time course survival results for mice immunized with GAS40N (SEQ ID NO:930).
- FIG. 23 FACS data demonstrating that GAS40 is surface exposed across different M strains.
- FIG. 24 Western blots and FACS graphs demonstrating that the four monoclonal antibodies tested do not bind to a GAS40N epitope.
- FIG. 25A-B Peptides derived from proteinase K digestion of GAS190 aligned with the full-length amino acid sequence of GAS190 (SEQ ID NO:117).
- FIG. 25A individual peptides (SEQ ID NOS:932-949);
- FIG. 25B schematic.
- FIG. 26A-B Peptides derived from trypsin digestion of GAS 190 aligned with the full- length amino acid sequence of GAS 190 (SEQ ID NO: 117).
- FIG. 26A individual peptides (SEQ ID NOS:950-961);
- FIG. 26B schematic.
- FIG. 27 Summary of predicted LPXTG (SEQ ID NO:931) proteins.
- FIG. 28-104 Topological representations of identified membrane-associated proteins. The protease cleavage sites are in red. LPXTG, SEQ ID NO:931.
- FIG. 105 A-B Bioinformatics-based topology predictions of all the predicted membrane proteins identified and their matching with identified peptides. Proteins are ordered by the number of predicted transmembrane domains (TMD) and, within, by their TIGR accession number.
- FIG. 105 A shows the proteins whose peptides identified by proteomics matched extracellular domains predicted by PSORT.
- FIG. 105B shows those membrane proteins whose peptides identified by proteomics matched cytoplasmic domains predicted by PSORT.
- FIG. 106 Comparison between found and predicted proteins for each of the four types of surface-associated proteins in Streptococcus pyogenes and FACS responses of those identified.
- LPXTG proteins 17 proteins containing the LPXTG (SEQ ID NO:931)- anchoring motif to the cell wall were predicted to be present in the genome; 12 (71%) were found and 5 (29%) were not. Of those identified, 11 were tested and all of them were positive.
- Membrane proteins 489 membrane proteins were predicted by in silico analysis; 452 (92%) were not found, whereas the number of found proteins was 37 (8%); 15 were not tested by FACS. Of those tested, 17 (77%) exhibited a positive response; 5 (23%) were negative.
- Lipoproteins 11 lipoproteins out of 28 predicted by in silico analysis (39%) were found; 17 (61%) were not found. AU of those identified were FACS- tested, and 9 (81%) were positive; 2 lipoproteins (19%) exhibited a negative response. Secreted proteins: 67 secreted proteins were predicted; 59 (88%) were not found, and 8 (12%) were found. Of these, one was not tested by FACS. Out of those tested, 6 (86%) were positive, and only one (14%) was negative.
- FIG. 107 Electron micrograph of membrane-delimited structures produced upon penicillin treatment of GAS bacteria.
- FIG. 108 Graph showing hyaluronic acid content of Ml, M3, M6, and M23 GAS bacteria (fg/CFU).
- FIG. 109A-C FACS pictograms of surface-exposed GAS antigens.
- FIG. 11 OA-C. FACS pictograms of surface-exposed GAS antigens.
- FIG. 11 IA-C. FACS pictograms of surface-exposed GAS antigens.
- FIG. 112. Graph showing prevalent immunoreactive antigens identified from serum samples of 6 healthy donors.
- compositions for preventing and/or treating S. pyogenes infection comprise one or more active agents, which can be GAS antigens expressed on the surface of GAS bacteria, nucleic acid molecules encoding the GAS antigens, and/or antibodies which selectively bind to the GAS antigens.
- GAS antigens include (1) naturally occurring immunogenic proteins of a GAS bacterium, (2) immunogenic portions of such proteins, and (3) engineered proteins or portions of proteins with amino acid sequences which retain immunogenicity and which are at least 50% identical to the amino acid sequence of a naturally occurring GAS immunogenic protein or portion thereof, such as homologs, orthologs, allelic variants, and mutants.
- the degree of sequence identity is preferably greater than 50% (e.g., 60%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or more).
- a GAS antigen is shorter than a GAS protein by at least one amino acid (e.g., 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, 50, 55, 60, 70, 76, 80, 85, 90, 95, 100, or more amino acids). More preferably a GAS antigen lacks a transmembrane domain. Even more preferably, a GAS antigen comprises a surface-exposed domain.
- the invention also includes various polypeptide fragments (including immunogenic portions) of the identified GAS proteins.
- the length of a fragment may vary depending on the amino acid sequence of the particular GAS antigen.
- fragments of GAS proteins comprise at least 7 contiguous amino acids (e.g., 8, 10, 12, 14, 16, 18, 20, 25, 29, 30, 35, 40, 50, 60, 70, 80, 9O 5 100, 150, 178, 200, 203, 250 or more contiguous amino acids).
- the fragment comprises one or more epitopes.
- the fragment may comprise at least one T-cell or, preferably, a B-cell epitope of the sequence.
- T- and B-cell epitopes can be identified empirically (e.g., using PEPSCAN (Geysen et al (1984) PNAS USA 81:3998-4002; Carter (1994) Methods MoI. Biol. 36:207-223, or similar methods), or they can be predicted (e.g., using the Jameson- Wolf antigenic index (Jameson, BA et al 1988, CABIOS 4(1): 1818-186), matrix-based approaches (Raddrizzani and Hammer (2000) Brief Bioinform.
- Other preferred fragments include (1) the N-terminal signal peptides of each identified GAS protein, (2) the identified GAS protein without its N-terminal signal peptide, (3) each identified GAS protein wherein up to 10 amino acid residues (e.g., 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25 or more) are deleted from the N-terminus and/or the C-terminus, and (4) GAS polypeptides without their N-terminal amino acid residue. Some fragments omit one or more domains of the protein (e.g., omission of a signal peptide, a cytoplasmic domain, a transmembrane domain, and/or an extracellular domain).
- GAS antigens consist of immunogenic portions of GAS proteins, which can be surface exposed domains as disclosed herein.
- Other GAS antigens are "hybrid GAS antigens," which comprise one or more immunogenic portions of a full-length GAS protein.
- Hybrid GAS antigens which also can include full-length GAS antigens, are described in detail below.
- Other fusion proteins can comprise, for example, one or more additional antigens and/or a tag protein, such as polyhistidine (HIS) or glutathione-S- transferase (GST).
- HIS polyhistidine
- GST glutathione-S- transferase
- a GAS antigen is expressed on the surface of a GAS bacterium, most preferably on the surface of more than one M type (e.g., 2, 3, 4, 5, 6, 7, 8, or 9 M types), particularly Ml, M3, M6, Mi l, M 12, and/or M23 GAS types.
- M type e.g., 2, 3, 4, 5, 6, 7, 8, or 9 M types
- Ml e.g., M3, M6, Mi l, M 12, and/or M23 GAS types.
- GAS antigens also preferably are found on the surface of at least two different strains (e.g., 2, 3, 4, 5, 6, 7, 8, . 9, 10, 11, 12, 13, or 14 or more strains).
- Preferred GAS antigens are highly conserved among multiple M types and/or multiple strains within an M type. See Table 2, which lists full-length GAS proteins and M types on which the proteins are expressed.
- compositions of the invention comprise one or more GAS antigens which are expressed on the surface of an Ml, M3, M6, Mi l, M 12, and/or M23 type.
- GAS antigens of this type GAS 99, 166, 96, 103, 188, 76, 108, 142, 190, 22, 56, 77, 67, 75, 93, 18, 23, 69, 206, 249, 123, 143, 68, 25, 30, 97, 105, 187, 195, 242, 81, 101, 6, 62, 49, 63, 85, 89, 100, 179, 205, 291, 98, 104, 36, 92, 158, 178, 218, 175, 78, 131, 29, 82, 91, 165, 327, 219, 60, 86, 380, 207, 271, 74, and 685 antigens.
- a GAS antigen is exposed on at least 10 M types (e.g., GAS 5, 22, 40, 56, 67, 76, 77, 96, 99, 103, 108, 142, 166, 188, and 190).
- GAS antigens of the invention also include surface-exposed domains of GAS proteins 4, 5, 15, 16, 23, 24, 25, 40, 49, 54, 57, 63, 64, 68, 72, 84, 86, 87, 89, 98, 102, 103, 108, 143, 149, 152, 157, 158, 163, 166, 168, 171, 177, 188, 190, 191, 192, 193, 194, 195, 198, 201, 224, 251, 259, 262, 264, 268, 277, 282, 299, 382, 405, 406, 425, 433, 460, 469, 493, 500, 545, 558, 587, 645, 650, 685, 362-1, spy0080a, spyO272, spyO461, spyO611, spyO717, spyO792, spylO29, s ⁇ ylO73, spyl260, spyl613, spyl835, spy2005, spy2093, spy2178, NT
- GAS antigens include surface-exposed domains of GAS proteins 5, 10, 23, 24, 49, 56, 63, 67, 72, 78, 81, 83, 84, 86, 89, 98, 100, 103, 157, 160, 177, 192, 194, 201, 205, 284, 286, 292, 382, 396, 405, 406, 500, spy0047 , spyOO53, spy0056, spy0063, spy0069, spy0098, spyO127, spyO274, spyO611, spyO666, s ⁇ yO686, s ⁇ y0688, spyO731, spyO913, spyl200, spyl281, spyl721, spyl750, spyl805, spy2070, spy2092, spy2178, and gi- 21909751 (see Table 8).
- Still others include surface-exposed domains of GAS proteins 16, 57, 68, 143, 158, 166, 171, 188, 190, 191, 192, 23, NT01SP0246, 49, 685, 63, 108, 84, 86, 89, 98, 103, 4, 149, 152, 157, 72, 405, 406, 299, 168, 251, 259, 262, 177, 264, 268, 277, 193, 194, 282, 195, 201, 40, 224, 163, 500, 198, 433, 54, 545, 469, 587, 645, 425, 493, 460, 558, 650, 5, 24, 25, 64, 87, 362-1, 382, 102, NT01SP0485, NT01SP0572, NT01SP0634, and NT01SP0877 (see Table 9).
- SEQ ID NOS:591-649 Some surface-exposed domains are shown in SEQ ID NOS:591-649.
- Other surface- exposed GAS antigens comprise at least 7 contiguous amino acids selected from the group consisting of SEQ ID NOS:1-281 (i.e., 7, 8, 9, 10, 11, 12, 13, 14, 15, 20, 25, 50, 75, or 100 or more).
- GAS antigens also include the surface-exposed domains of GAS proteins 35, 54, 70, 414, 421, 425, 426, 428, 433, 434, 437, 438, 439, 457, 461, 465, 469, 472, 473, 474, 475, 477, 478, 486, 492, 494, 495, 535, 538, 540, 543, 553, 560, 561, 564, 565, 574, 576, 577, 579, 586, 587, 591, 592, 607, 609, 625, 626, 636, 640, 643, 649, 653, 657, and 663.
- More preferred GAS antigens comprise a surface-exposed domain of 35, 414, 437, 438, 461, 465-2, 469, 472, 473, 475, 478, 495, 538, 553, 561, 577-2, 591, 593, 636, 643, 649, or 663.
- Even more preferred surface-exposed GAS antigens comprise a surface-exposed domain of GAS472, GAS473, or GAS553.
- GAS antigens include GASl 17, GAS130, GAS277, GAS236, GAS389, GAS504, GAS509, GAS366, GAS159, GAS217, GAS309, GAS372, GAS039, GAS042, GAS058, GAS290, GAS511, GAS533, GAS527, GAS294, GAS253, GAS529, GAS045, GAS095, GAS193, GAS137, GAS084, GAS384, GAS202, and GAS057 antigens, as well as M protein, GAS fibronectin-binding protein, GAS streptococcal heme-associated protein, and streptolysin S antigens.
- Preferred groups of GAS antigens for use in vaccines of the present invention include:
- GAS 680 is annotated as a predicted CoA-binding protein and corresponds to Ml GenBank accession numbers GL13621481 and GL71909974, to M49 GenBank accession number GL56808534, to Ml 8 GenBank accession number GI: 19747454, to M3 GenBank accession number GI: 28895062, and is also referred to as 'SpyO186' or 'M5005_Spy_0160' (Ml), 'SpyoMO 1000450' (M49), 'spyM18_0185' (M18) and 'SPs0150' (M3).
- GAS vaccines of the invention preferably include all or a surface portion of GAS57 and/or GAS40.
- GAS40 antigens are particularly useful in compositions of the invention because GAS40 proteins are highly conserved both in many M types and in multiple strains of these M types (see FIG. 1). GAS40 proteins are described in detail in WO 05/032582. See also FIG. 15. GAS40 consistently provides protection in the animal model of systemic immunization and challenge and induction of bactericidal antibodies (see the specific Examples, below). GAS40 is an extremely highly conserved protein and appears to be exposed on the surface of most M serotypes (the only exception observed thus far is the M3 serotype).
- GAS40 proteins Amino acid sequences of a number of GAS40 proteins from various M strains are provided in SEQ ID NOS: 17-43.
- the amino acid sequences of several GAS40 proteins also are contained in GenBank and have accession numbers GI: 13621545 and GL15674449 (Ml); accession number GI: 21909733 (M3), and accession number GI: 19745402 (Ml 8).
- GAS40 proteins also are known as "SpyO269” (Ml), “SpyM3_0197” (M3), "SpyM18_0256” (Ml 8) and "prgA.”
- a GAS40 protein typically contains a leader peptide sequence (e.g., amino acids 1 - 26 of SEQ ID NO:17), a first coiled-coil region (e.g., amino acids 58 - 261 of SEQ ID NO: 17), a second coiled coil region (e.g., amino acids 556 - 733 of SEQ ID NO:17), a leucine zipper region (e.g., amino acids 673 - 701 of SEQ ID NO:17) and a transmembrane region (e.g., amino acids 855 - 866 of SEQ ID NO: 17).
- a leader peptide sequence e.g., amino acids 1 - 26 of SEQ ID NO:17
- a first coiled-coil region e.g., amino acids 58 - 261 of SEQ ID NO: 17
- a second coiled coil region e.g., amino acids 556 - 733 of SEQ ID NO:17
- Preferred fragments of a GAS40 protein lack one or more amino acids (e.g.,1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25 or more) from the C-terminus and/or one or more amino acids (e.g.,1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25 or more) from the N-terminus of the GAS40 protein.
- the leader sequence is removed.
- the transmembrane region is removed.
- Other fragments may omit one or more other domains of the GAS40 protein.
- the coiled-coil regions of GAS40 are likely involved in the formation of oligomers such as dimers or trimers. Such oligomers could be homomers (containing two or more GAS40 proteins oligomerized together) or heteromers (containing one or more additional GAS proteins oligomerized with GAS40). Alternatively, two coiled-coil regions may interact together within the GAS40 protein to form oligomeric reactions between the first and second coiled-coil regions.
- the GAS40 antigen is in the form of an oligomer. Some oligomers comprise two more GAS40 antigens. Other oligomers comprise a GAS40 antigen oligomerized to a second GAS antigen.
- GAS antigens include fusion proteins comprising GAS40 and GASl 17.
- GAS40 hybrid antigen in which the GAS 40 protein is placed to the N- terminus of the GASl 17 protein and a HIS tag is added to the C terminus of the GASl 17 protein (SEQ ID NO:234).
- 117/40 is a GAS40 hybrid antigen in which GASl 17 is fused to GAS40 by the linker sequence YASGGGS (SEQ ID NO:278). Its amino acid sequence is shown in SEQ ID NO:233.
- GAS40a-HIS is a GAS40 antigen with a HIS tag but without the leader and hydrophobic sequences (SEQ ID NO:235).
- a nucleotide sequence encoding GAS40a is shown in SEQ ID NO:892 (codon 824, AGA in the wild-type sequence, was mutagenized to CGT).
- GAS40aRR is similar to GAS40a except that two additional AGA codons (334 and 335) in the coding sequence were mutated to CGT.
- GAS antigens can be present in compositions of the invention as individual separate polypeptides. Alternatively, at least two (e.g., 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20) of any of the GAS antigens described above can be expressed as a single polypeptide chain, i.e., a "hybrid GAS antigen.”
- Hybrid GAS antigens offer two principal advantages. First, a polypeptide which may be unstable or poorly expressed on its own can be assisted by adding a suitable hybrid partner which overcomes the problem. Second, commercial manufacture is simplified because only one expression and purification produces two polypeptides, both of which are antigenically useful.
- a hybrid GAS antigen can comprise two or more amino acid sequences for GAS40 antigens and/or one or more other GAS antigens of the invention.
- Hybrids can comprise amino acid sequences from two, three, four, five, six, seven, eight, nine, or ten or more GAS antigens.
- a GAS antigen can be present in more than one hybrid GAS antigen and/or as a non hybrid GAS antigen.
- a hybrid GAS antigen comprises at least two GAS antigens (e.g., I 3 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19 or 20) expressed as a single polypeptide chain.
- Preferred hybrid GAS antigens comprise at least one surface-exposed and/or surface- associated GAS antigen.
- Hybrid GAS antigens offer two principal advantages. First, a polypeptide which may be unstable or poorly expressed on its own can be assisted by adding a suitable hybrid partner which overcomes the problem. Second, commercial manufacture is simplified because only one expression and purification produces two polypeptides, both of which are antigenically useful.
- Hybrid GAS antigens can be represented by the formula:
- X is an amino acid sequence of a surface-exposed and/or surface-associated or secondary GAS antigen
- L is an optional linker amino acid sequence
- A is an optional N-terminal amino acid sequence
- B is an optional C-terminal amino acid sequence
- n is 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14 or 15.
- an -X- moiety has a leader peptide sequence in its wild-type form, this may be included or omitted in the hybrid antigen.
- the leader peptides will be deleted except for that of the -X- moiety located at the N-terminus of the hybrid protein i.e. the leader peptide Of X 1 will be retained, but the leader peptides of X 2 ... X n will be omitted. This is equivalent to deleting all leader peptides and using the leader peptide of X 1 as moiety -A-.
- linker amino acid sequence -L- may be present or absent.
- the hybrid may be NH 2 -X 1 -L 1 -X 2 -L 2 -COOH, NH 2 -X 1 -X 2 -COOH, NH 2 -X 1 -L 1 -X 2 -COOH, NH 2 -X 1 -X 2 -L 2 -COOH, etc.
- Linker amino acid sequence(s) -L- will typically be short, e.g.,20 or fewer amino acids (i.e., 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, 3, 2, 1).
- Other suitable linker amino acid sequences will be apparent to those skilled in the art.
- a useful linker is GSGGGG (SEQ ID NO:280), with the Gly-Ser dipeptide being formed from a Bam ⁇ I restriction site, which aids cloning and manipulation, and the (GIy) 4 tetrapeptide being a typical poly-glycine linker.
- oligopeptide e.g., with 1, 2, 3, 4, 5, 6, 7 or 8 amino acids
- [78] -B- is an optional C-terminal amino acid sequence.
- This will typically be short, e.g., 40 or fewer amino acids (i.e., 40, 39, 38, 37, 36, 35, 34, 33, 32, 31, 30, 29, 28, 27, 26, 25, 24, 23, 22, 21, 20, 19, 18, 17, 16, 15, 14, 13, 12, 11, 10, 9, 8, 7, 6, 5, 4, 3, 2, 1).
- Other suitable C-terminal amino acid sequences will be apparent to those skilled in the art.
- GAS antigens can be identified using any one or combination of several proteomics approaches as outlined below. These proteomics strategies have great potential for shortening the time needed for vaccine discovery when compared with other strategies, such as reverse vaccinology. Surface-exposed and/or surface-associated GAS antigens identified by these methods can be used as active agents in compositions for preventing and for treating S. pyogenes infections.
- Example 13 One embodiment is described in Example 13. Briefly, the surface of whole GAS bacterial cells is digested in vivo under physiological conditions using reagents which cleave proteins. Typically the reagents are proteases (e.g., trypsin, protease K, papain), although any protein cleavage reagent can be used.
- proteases e.g., trypsin, protease K, papain
- reagents include, for example, formic acid, hydroxylamine, BNPS-skatole (3-bromo-3-methyl-2-(o-nitrophenyl- sulfenyl)- indolenine), which cleaves at Trp residues), cyanogen bromide (which cleaves polypeptides on the carboxyl side of methionine residues), metal chelate reagents such as Fe-EDTA, and the like.
- Proteases can be either free or anchored, this latter condition favouring the identification of surface extruding regions. Combinations of more than one protein cleavage reagent can be used.
- the recovered peptides are then separated by liquid chromatography and identified by tandem mass spectrometry. The actual accessibility of identified proteins to the immune system can be assessed by fluorescence-activated cell sorting (FACS) analysis. This proteomic approach permits validation of software-based topology predictions and vice versa.
- FACS fluorescence-activated cell sorting
- Example 14 Another embodiment is described in Example 14. This approach involves overproduction of membrane-delimited from GAS bacteria after antibiotic treatment. See Hakenbeck et al, J. Bacteriol. 155, 1372-81, 1983, which is incorporated herein by reference. Either wild-type GAS bacteria or mutant GAS bacteria, for example those with "leaky” or destabilized peptidoglycan cell walls, can be used in this method. GAS bacteria naturally produce membrane-delimited structures which are released into the growth medium. When the bacteria are treated with an antibiotic that interferes with the synthesis of the cell-wall such as penicillins, cephalosporins, glycopeptides and cycloserine, production of these membrane-delimited structures increases.
- Vancomycin a glycopeptide which inhibits both cell wall synthesis and the sortase, interferes with surface protein anchoring which is catalyzed by sortases and can be used to further increase the overproduction of membrane-delimited structures.
- the membrane-delimited structures contain GAS proteins which are potential vaccine candidates.
- the GAS proteins can be separated by electrophoresis and identified using mass spectrometry (e.g., MALDI-TOF). Alternatively, the GAS proteins can be digested with proteases, and the resulting fragments can be separated by liquid chromatography and identified using tandem mass spectrometry.
- Example 15 A third embodiment is described in Example 15.
- cell wall and/or membrane fractions are generated by chemical cell fractionation of bacterial cells using, for example, 6 M guanidinium, urea, or SDS.
- the cell wall is insoluble in these reagents. This property allows the isolation of the cell wall and identification of anchored cell wall proteins. GAS proteins in these fractions can be separated and identified as described above.
- a fourth embodiment involves labeling cell surface GAS proteins (e.g., by biotinylation), lysing the cells, and isolating labeled proteins using affinity chromatography.
- the isolated proteins can be separated by electrophoresis and identified using mass spectrometry.
- the isolated proteins can be digested in solution, followed by isolation of labeled peptides by affinity chromatography, separation of the labeled peptides by liquid chromatography, and identification of the labeled peptides using tandem mass spectrometry.
- affinity chromatography separation of the labeled peptides by liquid chromatography
- identification of the labeled peptides using tandem mass spectrometry.
- a mutant can be used which harbors a deleted gene for one of the more abundant known surface-exposed antigens, such as M protein and C5a peptidase. These mutants will increase the probability of spotting previously unidentified, less abundant surface proteins.
- SpyO416 is a 1647 amino acid protein, carrying a C-terminal LPXTG-like motif, which shares 48% similarity with the C5a peptidase precursor. See SEQ ID NO: 118.
- the protein has a Ca-dependent serine protease activity (Fernandez-Espla, App. Env. Microbiol., 2000) which maps within the first 600 amino acids of the protein.
- SpyO416 has a homolog in Group B Streptococcus (GBS) (cspA) which was proposed to be involved in GBS virulence by potentially protecting the bacterium from opsonophagocytic killing (Harris et at , J. Clin. Invest. Ill, 61-70, 2003). Lei and co-workers recently found that a 31 kDa N-terminal fragment of SpyO416 is released in the supernatant of GAS cultures and that the protein is well recognized by sera from GAS-infected patients (Lei et at, Inf. Immunol. 68, 6807-18, 2000).
- GAS Group B Streptococcus
- the protein appears to be highly conserved (over 98%) and preliminary data on surface expression on a panel of 20 different GAS strains indicates that SpyO416 is a major component of over 70% of the circulating strains. It is, therefore, a preferred antigen for use in immunogenic compositions, either alone or in combination with one or more other GAS antigens.
- sequence listing provides coding sequences for the surface-exposed and/or surface- associated domains disclosed herein and their full-length proteins, as well as for the additional disclosed secondary GAS antigens.
- Any nucleotide sequence which encodes a particular antigen can be used in a compositions of the invention, for example as a DNA vaccine, or to produce a GAS antigen recombinantly, as described below.
- the full genomic sequences of at least three GAS strains are publicly available and can be used to obtain coding sequences for GAS antigens.
- the genomic sequence of an Ml GAS strain is reported in Ferretti et al, Proc. Natl. Acad. Sci. U.S.A. 98, 4658-63, 2002.
- the genomic sequence of an M3 GAS strain is reported in Beres et al , Proc. Natl. Acad. Sci. U.S.A. 99, 10078 - 83, 2002.
- the genomic sequence of an Ml 8 GAS strain is reported in Smooet et al, Proc. Natl. Acad. Sci. U.S.A. 99, 4668 - 73, 2002.
- the invention includes nucleic acid molecules which encode the identified GAS proteins and protein fragments.
- the invention also includes nucleic acid molecules comprising nucleotide sequences having at least 50% sequence identity to such molecules. Depending on the particular sequence, the degree of sequence identity is preferably greater than 50% (e.g., 60%, 70%, 75%, 80%, 85%, 90%, 91%, 92%, 93%, 94%, 95%, 96%, 97%, 98%, 99% or more).
- the invention also provides nucleic acid molecules which can hybridize to these molecules. Hybridization reactions can be performed under conditions of different "stringency.” Conditions which increase stringency of a hybridization reaction are widely known and published in the art. See, e.g., page 7.52 of Sambrook et al, Molecular Cloning: A Laboratory Manual, 1989.
- Examples of relevant conditions include (in order of increasing stringency): incubation temperatures of 25oC, 37 oC, 50 oC, 55 oC, and 68 oC; buffer concentrations of 1OX SSC, 6X SSC, IX SSC, and 0.1X SSC (where SSC is 0.15 M NaCl and 15 mM citrate buffer) and their equivalents using other buffer systems; formamide concentrations of 0%, 25%, 50%, and 75%; incubation times from 5 minutes to 24 hours; 1, 2, or more washing steps; wash incubation times of 1, 2, or 15 minutes; and wash solutions of 6X SSC, IX SSC, 0.1X SSC, or de-ionized water.
- Hybridization techniques and their optimization are well known in the art.
- nucleic acid molecules of the invention hybridize to a target under low stringency conditions; in other embodiments, nucleic acid molecules of the invention hybridize under intermediate stringency conditions; in preferred embodiments, nucleic acid molecules of the invention hybridize under high stringency conditions.
- An example of a low stringency hybridization condition is 50oC and 1OX SSC.
- An example of an intermediate stringency hybridization condition is 55oC and IX SSC.
- An example of a high stringency hybridization condition is 68oC and 0.1X SSC.
- Nucleic acid molecules comprising fragments of these sequences are also included in the invention. These comprise at least n consecutive nucleotides of these sequences and, depending on the particular sequence, n is 10 or more (e.g., 12, 14, 15, 18, 20, 25, 30, 35, 40, 50, 60, 70, 80, 90, 100, 150, 200, or more).
- Nucleic acids (and polypeptides) of the invention may include sequences which:
- (c) have 1, 2, 3, 4, 5, 6, 7, 8, 9, or 10 single nucleotide or amino acid alterations (deletions, insertions, substitutions), which may be at separate locations or may be contiguous, as compared to the sequences of (a) or (b); and,
- a moving window of x monomers moving from start (N-terminus or 5') to end (C-terminus or 3'), such that for an alignment that extends to p monomers (where p>x) there are p-x+1 such windows, each window has at least x-y identical aligned monomers, where: x is selected from 20, 25, 30, 35, 40, 45, 50, 60, 70, 80, 90, 100, 150, 200; y is selected from 0.50, 0.60, 0.70, 0.75, 0.80, 0.85, 0.90, 0.91, 0.92, 0.93, 0.94, 0.95, 0.96, 0.97, 0.98, 0.99; and if x-y is not an integer then it is rounded up to the nearest integer.
- This algorithm is conveniently implemented in the needle tool in the EMBOSS package [Rice et a ⁇ . (2000) Trends Genet. 16:276-277].
- nucleic acids and polypeptides of the invention may additionally have further sequences to the N-terminus/5' and/or C-terminus/3' of these sequences (a) to (d).
- Antibodies can be generated to bind specifically to a surface-exposed and/or surface- associated GAS antigen or to a secondary GAS or non-GAS polypeptide antigen disclosed herein.
- the term "antibody” includes intact immunoglobulin molecules, as well as fragments thereof which are capable of binding an antigen. These include hybrid (chimeric) antibody molecules (e.g., Winter et al, Nature 349, 293-99, 1991; U.S. Patent 4,816,567); F(ab')2 and F(ab) fragments and Fv molecules; non-covalent heterodimers (e.g., Inbar et al, Proc. Natl. Acad. Sci. U.S.A.
- sFv single-chain Fv molecules
- minibodies e.g., Pack et al, Biochem 31, 1579-84, 1992; Cumber et al, J. Immunology 149B, 120-26, 1992
- humanized antibody molecules e.g., Riechmann et al, Nature 332, 323-27, 1988; Verhoeyan et al, Science 239, 1534-36, 1988; and U.K.
- the antibodies are monoclonal antibodies. Methods of obtaining monoclonal antibodies are well known in the art.
- epitopes typically require more, e.g., at least 15, 25, or 50 amino acids.
- Various immunoassays e.g., Western blots, ELISAs, radioimmunoassays, immunohistochemical assays, immunoprecipitations, or other immunochemical assays known in the art
- Western blots e.g., Western blots, ELISAs, radioimmunoassays, immunohistochemical assays, immunoprecipitations, or other immunochemical assays known in the art
- Numerous protocols for competitive binding or immunoradiometric assays are well known in the art.
- Such immunoassays typically involve the measurement of complex formation between an immunogen and an antibody which specifically binds to the immunogen.
- a preparation of antibodies which specifically bind to a particular antigen typically provides a detection signal at least 5-, 10-, or 20-fold higher than a detection signal provided with other proteins when used in an immunochemical assay.
- the antibodies do not detect other proteins in immunochemical assays and can imniunoprecipitate the particular antigen from solution.
- GAS antigens or non-GAS polypeptide antigens can be used to immunize a mammal, such as a mouse, rat, rabbit, guinea pig, monkey, or human, to produce polyclonal antibodies.
- an antigen can be conjugated to a carrier protein, such as bovine serum albumin, thyroglobulin, and keyhole limpet hemocyanin.
- a carrier protein such as bovine serum albumin, thyroglobulin, and keyhole limpet hemocyanin.
- various adjuvants can be used to increase the immunological response.
- adjuvants include, but are not limited to, Freund's adjuvant, mineral gels (e.g., aluminum hydroxide), and surface active substances (e.g.
- Monoclonal antibodies which specifically bind to an antigen can be prepared using any technique which provides for the production of antibody molecules by continuous cell lines in culture. These techniques include, but are not limited to, the hybridoma technique, the human B cell hybridoma technique, and the EBV hybridoma technique (Kohler et al, Nature 256, 495 497, 1985; Kozbor et al, J. Immunol. Methods 81, 31 42, 1985; Cote et al, Proc. Natl. Acad. Sci. 80, 2026 2030, 1983; Cole et al, MoL Cell Biol. 62, 109 120, 1984).
- chimeric antibodies the splicing of mouse antibody genes to human antibody genes to obtain a molecule with appropriate antigen specificity and biological activity, can be used (Morrison et al , Proc. Natl. Acad. Sci. 81, 6851 6855, 1984; Neuberger et al, Nature 312, 604 608, 1984; Takeda et al, Nature 314, 452 454, 1985).
- Monoclonal and other antibodies also can be "humanized” to prevent a patient from mounting an immune response against the antibody when it is used therapeutically.
- Such antibodies may be sufficiently similar in sequence to human antibodies to be used directly in therapy or may require alteration of a few key residues. Sequence differences between rodent antibodies and human sequences can be minimized by replacing residues which differ from those in the human sequences by site directed mutagenesis of individual residues or by grating of entire complementarity determining regions.
- humanized antibodies can be produced using recombinant methods, as described below.
- Antibodies which specifically bind to a particular antigen can contain antigen binding sites which are either partially or fully humanized, as disclosed in U.S. 5,565,332.
- Single-chain antibodies also can be constructed using a DNA amplification method, such as PCR, using hybridoma cDNA as a template (Thirion et al, 1996, Eur. J. Cancer Prev. 5, 507-11).
- Single-chain antibodies can be mono- or bispecific, and can be bivalent or tetravalent. Construction of tetravalent, bispecific single-chain antibodies is taught, for example, in Coloma & Morrison, 1997, Nat. Biotechnol. 15, 159-63. Construction of bivalent, bispecific single-chain antibodies is taught in Mallender & Voss, 1994, J. Biol. Chem. 269, 199-206.
- a nucleotide sequence encoding a single-chain antibody can be constructed using manual or automated nucleotide synthesis, cloned into an expression construct using standard recombinant DNA methods, and introduced into a cell to express the coding sequence, as described below.
- single-chain antibodies can be produced directly using, for example, filamentous phage technology (Verhaar et al, 1995, Int. J. Cancer 61, 497- 501; Nicholls et al, 1993, J. Immunol. Meth. 165, 81-91).
- Antibodies which specifically bind to a particular antigen also can be produced by inducing in vivo production in the lymphocyte population or by screening immunoglobulin libraries or panels of highly specific binding reagents as disclosed in the literature (Orlandi et al, Proc. Natl. Acad. Sci. 86, 3833 3837, 1989; Winter et al, Nature 349, 293 299, 1991).
- Chimeric antibodies can be constructed as disclosed in WO 93/03151. Binding proteins which are derived from immunoglobulins and which are multivalent and multispecific, such as the "diabodies" described in WO 94/13804, also can be prepared.
- Antibodies can be purified by methods well known in the art. For example, antibodies can be affinity purified by passage over a column to which the relevant antigen is bound. The bound antibodies can then be eluted from the column using a buffer with a high salt concentration.
- Any nucleotide sequence which encodes a particular antigen can be used to produce that antigen recombinantly. If desired, an antibody can be produced recombinantly once its amino acid sequence is known.
- nucleic acid molecules encoding surface-exposed and/or surface- associated or secondary GAS antigens can be isolated from the appropriate S. pyogenes bacterium using standard nucleic acid purification techniques or can be synthesized using an amplification technique, such as the polymerase chain reaction (PCR), or by using an automatic synthesizer. Methods for isolating nucleic acids are routine and are known in the art. Any such technique for obtaining nucleic acid molecules can be used to obtain a nucleic acid molecule which encodes a particular antigen.
- PCR polymerase chain reaction
- Sequences encoding a particular antigen or antibody can be synthesized, in whole or in part, using chemical methods well known in the art (see Caruthers et ah, Nucl. Acids Res. Symp. Ser. 215 223, 1980; Horn et al. Nucl. Acids Res. Symp. Ser. 225 232, 1980).
- cDNA molecules can be made with standard molecular biology techniques, using mRNA as a template. cDNA molecules can thereafter be replicated using molecular biology techniques well known in the art. An amplification technique, such as PCR, can be used to obtain additional copies of polynucleotides of the invention, using either genomic DNA or cDNA as a template.
- nucleotide sequences can be engineered using methods generally known in the art to alter antigen-encoding sequences for a variety of reasons, including but not limited to, alterations which modify the cloning, processing, and/or expression of the polypeptide or mRNA product.
- DNA shuffling by random fragmentation and PCR reassembly of gene fragments and synthetic oligonucleotides can be used to engineer the nucleotide sequences.
- site directed mutagenesis can be used to insert new restriction sites, alter glycosylation patterns, change codon preference, produce splice variants, introduce mutations, and so forth.
- Sequence modifications such as the addition of a purification tag sequence or codon optimization, can be used to facilitate expression.
- the N-terminal leader sequence may be replaced with a sequence encoding for a tag protein such as polyhistidine ("HIS") or glutathione S-transferase ("GST").
- tag proteins may be used to facilitate purification, detection, and stability of the expressed protein.
- Codons preferred by a particular prokaryotic or eukaryotic host can be selected to increase the rate of protein expression or to produce an RNA transcript having desirable properties, such as a half life which is longer than that of a transcript generated from the naturally occurring sequence.
- a nucleic acid molecule which encodes an antigen or antibody can be inserted into an expression vector which contains the necessary elements for the transcription and translation of the inserted coding sequence.
- Methods which are well known to those skilled in the art can be used to construct expression vectors containing coding sequences and appropriate transcriptional and translational control elements. These methods include in vitro recombinant DNA techniques, synthetic techniques, and in vivo genetic recombination.
- the heterologous host can be prokaryotic or eukaryotic.
- E. coli is a preferred host cell, but other suitable hosts include Lactococcus lactis, Lactococcus cremoris, Bacillus subtilis, Vibrio cholerae, Salmonella typhi, Salmonella typhimurium, Neisseria lactamica, Neisseria cinerea, Mycobacteria ⁇ e.g., M. tuberculosis), yeasts, etc.
- a host cell strain can be chosen for its ability to modulate the expression of the inserted sequences or to process the expressed polypeptide in the desired fashion. Such modifications of the polypeptide include, but are not limited to, acetylation, carboxylation, glycosylation, phosphorylation, lipidation, and acylation. Post translational processing which cleaves a "prepro" form of the polypeptide also can be used to facilitate correct insertion, folding and/or function. Different host cells which have specific cellular machinery and characteristic mechanisms for post translational activities are available from the American Type Culture Collection (ATCC; 10801 University Boulevard, Manassas, VA 20110-2209) and can be chosen to ensure the correct modification and processing of a foreign protein. See WO 01/98340.
- ATCC American Type Culture Collection
- Expression constructs can be introduced into host cells using well-established techniques which include, but are not limited to, transferrin-polycation-mediated DNA transfer, transfection with naked or encapsulated nucleic acids, liposome-mediated cellular fusion, intracellular transportation of DNA-coated latex beads, protoplast fusion, viral infection, electroporation, "gene gun” methods, and DEAE- or calcium phosphate-mediated transfection.
- Host cells transformed with expression vectors can be cultured under conditions suitable for the expression and recovery of the protein from cell culture.
- the protein produced by a transformed cell can be secreted or contained intracellularly depending on the nucleotide sequence and/or the expression vector used.
- expression vectors can be designed to contain signal sequences which direct secretion of soluble antigens through a prokaryotic or eukaryotic cell membrane.
- Antigens used in the invention can be isolated from the appropriate Streptococcus pyogenes bacterium or from a host cell engineered to produce GAS or non-GAS antigens.
- a purified polypeptide antigen is separated from other components in the cell, such as proteins, carbohydrates, or lipids, using methods well-known in the art. Such methods include, but are not limited to, size exclusion chromatography, ammonium sulfate fractionation, ion exchange chromatography, affinity chromatography, and preparative gel electrophoresis.
- a preparation of purified polypeptide antigens is at least 80% pure; preferably, the preparations are 90%, 95%, or 99% pure. Purity of the preparations can be assessed by any means known in the art, such as SDS-polyacrylamide gel electrophoresis. Where appropriate, polypeptide antigens can be solubilized, for example, with urea.
- GAS antigens as well as other antigens used in compositions of the invention, can be synthesized, for example, using solid phase techniques. See, e.g., Merrifield, J. Am. Chem. Soc. 85, 2149 54, 1963; Roberge et al, Science 269, 202 04, 1995. Protein synthesis can be performed using manual techniques or by automation. Automated synthesis can be achieved, for example, using Applied Biosystems 43 IA Peptide Synthesizer (Perkin Elmer). Optionally, fragments of a surface-exposed and/or surface- associated GAS antigen can be separately synthesized and combined using chemical methods to produce a full-length molecule.
- Nucleic acid molecules which encode antibodies or polypeptide antigens can be synthesized by conventional methodology, such as the phosphate triester method (Hunkapiller, M. et al. (1984), Nature 310: 105-111) or by the chemical synthesis of nucleic acids (Grantham, R. et al. (1981), Nucleic Acids Res. 9: r43-r74). Immunogenic, Diagnostic, and Therapeutic Compositions
- the invention also provides compositions for use as medicaments (e.g., as immunogenic compositions or vaccines) or as diagnostic reagents for detecting a GAS infection in a host subject. It also provides the use of the compositions in the manufacture of: (i) a medicament for treating or preventing infection due to GAS bacteria; (ii) a diagnostic reagent for detecting the presence of GAS bacteria or of antibodies raised against GAS bacteria; and/or (iii) a reagent which can raise antibodies against GAS bacteria.
- GAS antigens or nucleic acids encoding the antigens can be used in the manufacture of a diagnostic reagent for detecting the presence of a GAS infection or for detecting antibodies raised against GAS bacteria, or in the manufacture of a reagent which can raise antibodies against GAS bacteria.
- Nucleic acids encoding GAS antigens can be detected by contacting a nucleic acid probe with a biological sample under hybridizing conditions to form duplexes and detecting the duplexes as is known in the art.
- a GAS antigen can be detected using antibodies which specifically bind to the GAS antigen.
- antibodies to GAS antigens can be used to detect GAS antigens by contacting a biological sample under conditions suitable for the formation of antibody- antigen complexes and detecting any complexes formed.
- the invention also provides kits comprising reagents suitable for use these methods.
- compositions of the invention are useful for preventing and/or treating S. pyogenes infection.
- Compositions containing GAS antigens are preferably immunogenic compositions, and are more preferably vaccine compositions.
- the pH of such compositions preferably is between 6 and 8, preferably about 7.
- the pH can be maintained by the use of a buffer.
- the composition can be sterile and/or pyrogen free.
- the composition can be isotonic with respect to humans.
- Vaccines according to the invention may be used either prophylactically or therapeutically, but will typically be prophylactic.
- the invention includes a method for the therapeutic or prophylactic treatment of a Streptococcus pyogenes infection.
- the animal is preferably a mammal, most preferably a human.
- the methods involve administering to the animal a therapeutic or prophylactic amount of the immunogenic compositions of the invention.
- compositions of the invention comprise at least two surface-exposed GAS antigens as described above.
- Other compositions of the invention comprise at least one nucleic acid molecule which encodes two surface-exposed GAS antigens.
- Still other compositions of the invention comprise at least two antibodies, each of which specifically binds to one of two surface-exposed GAS antigens.
- Preferred compositions of the invention comprise at least one of the surface-exposed GAS antigens is a GAS40 antigen and the other antigen is any other GAS antigen; at least one nucleic acid molecule encoding the two antigens, or at least two antibodies which specifically bind to the two antigens.
- Some compositions comprise one or more additional GAS antigens, a nucleic acid molecule encoding the additional antigen(s), or an antibody which specifically binds to the additional antigen(s); of these antigens, GASl 17 is preferred.
- compositions of the invention comprise a nucleic acid molecule which encodes the at least two GAS antigens and, optionally, other antigens which can be included in the composition (see below).
- a nucleic acid molecule which encodes the at least two GAS antigens and, optionally, other antigens which can be included in the composition (see below).
- Donnelly et al (1997) Ann. Rev Immunol 15:617-648; Scott-Taylor & Dalgleish (2000) Expert Opin Investig Drugs 9:471-480; tendopoulos & Plebanski (2000) Curr Opin MoI Ther 2:441-447; Ilan (1999) Curr Opin MoI Ther 1:116-120; Dubensky et al.
- compositions for treating S. pyogenes infections comprise at least one antibody which specifically binds to a GAS antigen and, optionally, an antibody which specifically binds to a non-GAS antigen.
- compositions of the invention are immunogenic and comprise one or more polypeptide antigens, while other immunogenic compositions comprise nucleic acid molecules which encode a surface-exposed and/or surface- associated GAS antigen and, optionally, a secondary GAS antigen or a non-GAS antigen.
- nucleic acid molecules which encode a surface-exposed and/or surface- associated GAS antigen and, optionally, a secondary GAS antigen or a non-GAS antigen.
- the nucleic acid molecule is a DNA molecule, e.g., in the form of a plasmid.
- compositions of the invention comprise at least one active agent.
- compositions for preventing S. pyogenes infection can comprise as an active agent either a polypeptide comprising a GAS antigen of the invention or a nucleic acid molecule which encodes the polypeptide.
- compositions of the invention can include one or more additional active agents.
- additional active agents include, but are not limited to, (a) another GAS antigen of the invention, preferably a surface-exposed antigen, (b) a polypeptide antigen which is useful in a pediatric vaccine, (c) a polypeptide antigen which is useful in a vaccine for elderly or immunocompromised individuals, (d) a nucleic acid molecule encoding (a)-(c), and an antibody which specifically binds to (a)-(c).
- compositions of the invention may be administered in conjunction with one or more antigens for use in therapeutic, prophylactic, or diagnostic methods of the present invention.
- Preferred antigens include those listed below. Additionally, the compositions of the present invention may be used to treat or prevent infections caused by any of the below-listed pathogens. In addition to combination with the antigens described below, the compositions of the invention may also be combined with an adjuvant as described herein.
- Antigens for use with the invention include, but are not limited to, one or more of the following antigens set forth below, or antigens derived from one or more of the pathogens set forth below:
- Bacterial antigens suitable for use in the invention include proteins, polysaccharides, lipopolysaccharides, and outer membrane vesicles which may be isolated, purified or derived from a bacteria.
- bacterial antigens may include bacterial lysates and inactivated bacteria formulations.
- Bacteria antigens may be produced by recombinant expression.
- Bacterial antigens preferably include epitopes which are exposed on the surface of the bacteria during at least one stage of its life cycle. Bacterial antigens are preferably conserved across multiple serotypes.
- Bacterial antigens include antigens derived from one or more of the bacteria set forth below as well as the specific antigens examples identified below.
- Meningitides antigens may include proteins (such as those identified in References 1 - 7), saccharides (including a polysaccharide, oligosaccharide or lipopolysaccharide), or outer-membrane vesicles (References 8, 9, 10, 11) purified or derived from N. meningitides serogroup such as A, C, Wl 35, Y, and/or B. Meningitides protein antigens may be selected from adhesions, autotransporters, toxins, Fe acquisition proteins, and membrane associated proteins (preferably integral outer membrane protein).
- Streptococcus pneumoniae antigens may include a saccharide (including a polysaccharide or an oligosaccharide) and/or protein from Streptococcus pneumoniae. Saccharide antigens may be selected from serotypes 1, 2, 3, 4, 5, 6B, 7F, 8, 9N, 9V, 1OA, HA, 12F, 14, 15B, 17F, 18C, 19A, 19F, 20, 22F, 23F, and 33F. Protein antigens may be selected from a protein identified in WO 98/18931, WO 98/18930, US Patent No. 6,699,703, US Patent No.
- Streptococcus pneumoniae proteins may be selected from the Poly Histidine Triad family (PhtX), the Choline Binding Protein family (CbpX), CbpX truncates, LytX family, LytX truncates, CbpX truncate-LytX truncate chimeric proteins, pneumolysin (Ply), PspA, PsaA, Spl28, SpIOl, Spl30, Spl25 or Spl33.
- PhtX Poly Histidine Triad family
- CbpX Choline Binding Protein family
- CbpX truncates CbpX truncates
- LytX family LytX truncates
- pneumolysin (Ply) PspA, PsaA, Spl28, SpIOl, Spl30, Spl25 or Spl33.
- Streptococcus pyogenes Group A Streptococcus antigens may include a protein identified in WO 02/34771 or WO 2005/032582 (including GAS 40), fusions of fragments of GAS M proteins (including those described in WO 02/094851, and Dale, Vaccine (1999) 17:193-200, and Dale, Vaccine 14(10): 944-948), fibronectin binding protein (Sfbl), Streptococcal heme-associated protein (Shp), and Streptolysin S (SagA).
- Moraxella antigens include antigens identified in WO 02/18595 and WO 99/58562, outer membrane protein antigens (HMW-OMP), C-antigen, and/or LPS.
- HMW-OMP outer membrane protein antigens
- C-antigen C-antigen
- LPS LPS
- Pertussis antigens include petussis holotoxin (PT) and filamentous haemagglutinin (FHA) from B. pertussis, optionally also combination with pertactin and/or agglutinogens 2 and 3 antigen.
- PT petussis holotoxin
- FHA filamentous haemagglutinin
- Staphylococcus aureus antigens include S. aureus type 5 and 8 capsular polysaccharides optionally conjugated to nontoxic recombinant Pseudomonas aeruginosa exotoxin A, such as Staph V AXTM, or antigens derived from surface proteins, invasins (leukocidin, kinases, hyaluronidase), surface factors that inhibit phagocytic engulfment (capsule, Protein A), carotenoids, catalase production, Protein A, coagulase, clotting factor, and/or membrane-damaging toxins (optionally detoxified) that lyse eukaryotic cell membranes (hemolysins, leukotoxin, leukocidin).
- Staphylococcus epidermis S. epidermidis antigens include slime-associated antigen (SAA).
- Tetanus antigens include tetanus toxoid (TT), preferably used as a carrier protein in conjunction/conjugated with the compositions of the present invention.
- Diphtheria antigens include diphtheria toxin, preferably detoxified, such as CRMl 97. Additionally antigens capable of modulating, inhibiting or associated with ADP ribosylation are contemplated for combination/co- administration/conjugation with the compositions of the present invention.
- the diphtheria toxoids may be used as carrier proteins.
- Hib antigens include a Hib saccharide antigen.
- Pseudomonas aeruginosa Pseudomonas antigens include endotoxin A, Wzz protein, P. aeruginosa LPS, more particularly LPS isolated from PAOl (05 serotype), and/or Outer Membrane Proteins, including Outer Membrane Proteins F (OprF) (Infect Immun. 2001 May; 69(5): 3510-3515).
- Legionella pneumophila Bacterial antigens may be derived from Legionella pneumophila.
- Streptococcus agalactiae Group B Streptococcus antigens include a protein or saccharide antigen identified in WO 02/34771, WO 03/093306, WO 04/041157, or WO 2005/002619 (including proteins GBS 80, GBS 104, GBS 276 and GBS 322, and including saccharide antigens derived from serotypes Ia, Ib, Ia/c, II, III, IV, V, VI, VII and VIII).
- Neiserria gonorrhoeae antigens include Por (or porin) protein, such as PorB (see Zhu et al., Vaccine (2004) 22:660 - 669), a transferring binding protein, such as TbpA and TbpB (See Price et al., Infection and Immunity (2004) 71(1):277 - 283), a opacity protein (such as Opa), a reduction-modifiable protein (Rmp), and outer membrane vesicle (OMV) preparations (see Plante et al., J Infectious Disease (2000) 182:848 - 855), also see e.g. WO99/24578, WO99/36544, WO99/57280, WO02/079243).
- PorB see Zhu et al., Vaccine (2004) 22:660 - 669
- TbpA and TbpB See Price et al., Infection and Immunity (2004) 71(1):2
- Chlamydia trachomatis antigens include antigens derived from serotypes A, B, Ba and C (agents of trachoma, a cause of blindness), serotypes Ll, L2 & L3 (associated with Lymphogranuloma venereum), and serotypes, D-K.
- Chlamydia trachomas antigens may also include an antigen identified in WO 00/37494, WO 03/049762, WO 03/068811, or WO 05/002619, including PepA (CT045), LcrE (CT089), ArtJ (CT381), DnaK (CT396), CT398, OmpH-like (CT242), L7/L12 (CT316), OmcA (CT444), AtosS (CT467), CT547, Eno (CT587), HrtA (CT823), and MurG (CT761).
- Treponema pallidum Syphilis antigens include TmpA antigen.
- Ducreyi antigens include outer membrane protein (DsrA).
- Antigens include a trisaccharide repeat or other Enterococcus derived antigens provided in US Patent No. 6,756,361.
- H. pylori antigens include Cag, Vac, Nap, HopX, HopY and/or urease antigen.
- Staphylococcus saprophyticus Antigens include the 160 kDa hemagglutinin of S. saprophyticus antigen.
- Yersinia enterocolitica antigens include LPS (Infect Immun. 2002 August; 70(8): 4414).
- E. coli antigens may be derived from enterotoxigenic E. coli (ETEC), enteroaggregative E. coli (EAggEC), diffusely adhering E. coli (DAEC), enteropathogenic E. coli (EPEC) 5 and/or enterohemorrhagic E. coli (EHEC).
- ETEC enterotoxigenic E. coli
- EAggEC enteroaggregative E. coli
- DAEC diffusely adhering E. coli
- EPEC enteropathogenic E. coli
- EHEC enterohemorrhagic E. coli
- LF lethal factor
- EF edema factor
- Plague antigens include Fl capsular antigen (Infect Immun. 2003 Jan; 71(1)): 374-383, LPS (Infect Immun. 1999 Oct; 67(10): 5395), Yersinia pestis V antigen (Infect Immun. 1997 Nov; 65(11): 4476-4482).
- Tuberculosis antigens include lipoproteins, LPS, BCG antigens, a fusion protein of antigen 85B (Ag85B) and/or ESAT-6 optionally formulated in cationic lipid vesicles (Infect Immun. 2004 October; 72(10): 6148), Mycobacterium tuberculosis (Mtb) isocitrate dehydrogenase associated antigens (Proc Natl Acad Sci U S A. 2004 Aug 24; 101(34): 12652), and/or MPT51 antigens (Infect Immun. 2004 July; 72(7): 3829).
- Antigens include outer membrane proteins, including the outer membrane protein A and/or B (OmpB) (Biochim Biophys Acta. 2004 Nov l;1702(2):145), LPS, and surface protein antigen (SPA) (J Autoimmun. 1989 Jun;2 Suppl:81).
- OmpB outer membrane protein A and/or B
- SPA surface protein antigen
- Bacterial antigens may be derived from Listeria monocytogenes.
- Chlamydia pneumoniae Antigens include those identified in WO 02/02606.
- Vibrio cholerae Antigens include proteinase antigens,- LPS, particularly lipopolysaccharides of Vibrio cholerae II, Ol Inaba O-specific polysaccharides, V. cholera 0139, antigens of IEM108 vaccine (Infect Immun. 2003 Oct;71(10):5498-504), and/or Zonula occludens toxin (Zot).
- proteinase antigens include proteinase antigens,- LPS, particularly lipopolysaccharides of Vibrio cholerae II, Ol Inaba O-specific polysaccharides, V. cholera 0139, antigens of IEM108 vaccine (Infect Immun. 2003 Oct;71(10):5498-504), and/or Zonula occludens toxin (Zot).
- Salmonella typhi typhoid fever
- Antigens include capsular polysaccharides preferably conjugates (Vi, i.e. vax-TyVi).
- Borrelia burgdorferi Lyme disease
- Antigens include lipoproteins (such as OspA, OspB, Osp C and Osp D), other surface proteins such as OspE-related proteins (Erps), decorin-binding proteins (such as DbpA), and antigenically variable VI proteins. , such as antigens associated with P39 and Pl 3 (an integral membrane protein, Infect Immun. 2001 May; 69(5): 3323-3334), VIsE Antigenic Variation Protein (J Clin Microbiol. 1999 Dec; 37(12): 3997).
- Antigens include P. gingivalis outer membrane protein (OMP).
- Klebsiella Antigens include an OMP, including OMP A, or a polysaccharide optionally conjugated to tetanus toxoid.
- Further bacterial antigens of the invention may be capsular antigens, polysaccharide antigens or protein antigens of any of the above. Further bacterial antigens may also include an outer membrane vesicle (OMV) preparation. Additionally, antigens include live, attenuated, and/or purified versions of any of the aforementioned bacteria.
- the antigens of the present invention may be derived from gram-negative or gram-positive bacteria.
- the antigens of the present invention may be derived from aerobic or anaerobic bacteria.
- any of the above bacterial-derived saccharides can be conjugated to another agent or antigen, such as a carrier protein (for example CRMl 97 ).
- a carrier protein for example CRMl 97
- Such conjugation may be direct conjugation effected by reductive animation of carbonyl moieties on the saccharide to amino groups on the protein, as provided in US Patent No. 5,360,897 and Can J Biochem Cell Biol. 1984 May;62(5):270-5.
- the saccharides can be conjugated through a linker, such as, with succinamide or other linkages provided in Bioconjugate Techniques, 1996 and CRC, Chemistry of Protein Conjugation and Cross-Linking, 1993.
- a linker such as, with succinamide or other linkages provided in Bioconjugate Techniques, 1996 and CRC, Chemistry of Protein Conjugation and Cross-Linking, 1993.
- Viral antigens suitable for use in the invention include inactivated (or killed) virus, attenuated virus, split virus formulations, purified subunit formulations, viral proteins which may be isolated, purified or derived from a virus, and Virus Like Particles (VLPs).
- Viral antigens may be derived from viruses propagated on cell culture or other substrate. Alternatively, viral antigens may be expressed recombinantly.
- Viral antigens preferably include epitopes which are exposed on the surface of the virus during at least one stage of its life cycle. Viral antigens are preferably conserved across multiple serotypes or isolates. Viral antigens include antigens derived from one or more of the viruses set forth below as well as the specific antigens examples identified below.
- Orthomyxovirus Viral antigens may be derived from an Orthomyxovirus, such as Influenza A, B and C. Orthomyxovirus antigens may be selected from one or more of the viral proteins, including hemagglutinin (HA), neuraminidase (NA), nucleoprotein (NP), matrix protein (Ml), membrane protein (M2), one or more of the transcriptase components (PBl, PB2 and PA). Preferred antigens include HA and NA.
- HA hemagglutinin
- NA neuraminidase
- NP nucleoprotein
- Ml matrix protein
- M2 membrane protein
- Preferred antigens include HA and NA.
- Influenza antigens may be derived from interpandemic (annual) flu strains.
- influenza antigens may be derived from strains with the potential to cause pandemic a pandemic outbreak (i.e., influenza strains with new haemagglutinin compared to the haemagglutinin in currently circulating strains, or influenza strains which are pathogenic in avian subjects and have the potential to be transmitted horizontally in the human population, or influenza strains which are pathogenic to humans).
- Viral antigens may be derived from Paramyxoviridae viruses, such as Pneumoviruses (RSV), Paramyxoviruses (PIV) and Morbilliviruses (Measles).
- RSV Pneumoviruses
- PMV Paramyxoviruses
- Measles Morbilliviruses
- Pneumovirus Viral antigens may be derived from a Pneumovirus, such as Respiratory syncytial virus (RSV), Bovine respiratory syncytial virus, Pneumonia virus of mice, and Turkey rhinotracheitis virus.
- the Pneumovirus is ' RSV.
- Pneumovirus antigens may be selected from one or more of the following proteins, including surface proteins Fusion (F), Glycoprotein (G) and Small Hydrophobic protein (SH), matrix proteins M and M2, nucleocapsid proteins N, P and L and nonstructural proteins NSl and NS2.
- Preferred Pneumovirus antigens include F, G and M. See e.g., J Gen Virol. 2004 Nov; 85(Pt 11):3229).
- Pneumovirus antigens may also be formulated in or derived from chimeric viruses.
- chimeric RSV/PIV viruses may comprise components of both RSV and PIV.
- Paramyxovirus Viral antigens may be derived from a Paramyxovirus, such as Parainfluenza virus types 1 - 4 (PIV), Mumps, Sendai viruses, Simian virus 5, Bovine parainfluenza virus and Newcastle disease virus.
- the Paramyxovirus is PIV or Mumps.
- Paramyxovirus antigens may be selected from one or more of the following proteins: Hemagglutinin -Neuraminidase (HN), Fusion proteins Fl and F2, Nucleoprotein (NP), Phosphoprotein (P), Large protein (L), and Matrix protein (M).
- HN Hemagglutinin -Neuraminidase
- NP Nucleoprotein
- Phosphoprotein P
- Large protein L
- Matrix protein M
- Preferred Paramyxovirus proteins include HN, Fl and F2.
- Paramyxovirus antigens may also be formulated in or derived from chimeric viruses.
- chimeric RSV/PIV viruses may comprise components of both RSV and PIV.
- Commercially available mumps vaccines include live attenuated mumps virus, in either a monovalent form or in combination with measles and rubella vaccines (MMR).
- Morbillivirus Viral antigens may be derived from a Morbillivirus, such as Measles. Morbilli virus antigens may be selected from one or more of the following proteins: hemagglutinin (H), Glycoprotein (G), Fusion factor (F), Large protein (L), Nucleoprotein (NP), Polymerase phosphoprotein (P), and Matrix (M). Commercially available measles vaccines include live attenuated measles virus, typically in combination with mumps and rubella (MMR).
- MMR Matrix
- Picornavirus Viral antigens may be derived from Picornaviruses, such as Enteroviruses, Rhinoviruses, Heparnavirus, Cardioviruses and Aphthoviruses. Antigens derived from Enteroviruses, such as Poliovirus are preferred.
- Enterovirus Viral antigens may be derived from an Enterovirus, such as Poliovirus types 1, 2 or 3, Coxsackie A virus types 1 to 22 and 24, Coxsackie B virus types 1 to 6, Echovirus (ECHO) virus) types 1 to 9, 11 to 27 and 29 to 34 and Enterovirus 68 to 71.
- the Enterovirus is poliovirus.
- Enterovirus antigens are preferably selected from one or more of the following Capsid proteins VPl, VP2, VP3 and VP4.
- Commercially available polio vaccines include Inactivated Polio Vaccine (IPV) and Oral poliovirus vaccine (OPV).
- Heparnavirus Viral antigens may be derived from an Heparnavirus, such as Hepatitis A virus (HAV).
- HAV Hepatitis A virus
- Commercially available HAV vaccines include inactivated HAV vaccine.
- Togavirus Viral antigens may be derived from a Togavirus, such as a Rubivirus, an Alphavirus, or an Arterivirus. Antigens derived from Rubivirus, such as Rubella virus, are preferred. Togavirus antigens may be selected from El, E2, E3, C, NSP-I, NSPO-2, NSP-3 or NSP-4. Togavirus antigens are preferably selected from El, E2 or E3.
- Commercially available Rubella vaccines include a live cold-adapted virus, typically in combination with mumps and measles vaccines (MMR).
- Flavivirus Viral antigens may be derived from a Flavivirus, such as Tick-borne encephalitis (TBE), Dengue (types 1, 2, 3 or 4), Yellow Fever, Japanese encephalitis, West Nile encephalitis, St. Louis encephalitis, Russian spring-summer encephalitis, Powassan encephalitis. Flavivirus antigens may be selected from PrM, M, C, E, NS-I, NS-2a, NS2b, NS3, NS4a, NS4b, and NS5. Flavivirus antigens are preferably selected from PrM, M and E. Commercially available TBE vaccine include inactivated virus vaccines.
- TBE vaccine include inactivated virus vaccines.
- Pestivirus Viral antigens may be derived from a Pestivirus, such as Bovine viral diarrhea (BVDV), Classical swine fever (CSFV) or Border disease (BDV).
- BVDV Bovine viral diarrhea
- CSFV Classical swine fever
- BDV Border disease
- Hepadnavirus Viral antigens may be derived from a Hepadnavirus, such as Hepatitis B virus. Hepadnavirus antigens may be selected from surface antigens (L, M and S), core antigens (HBc, HBe). Commercially available HBV vaccines include subunit vaccines comprising the surface antigen S protein.
- Hepatitis C virus Viral antigens may be derived from a Hepatitis C virus (HCV). HCV antigens may be selected from one or more of El, E2, E1/E2, NS345 polyprotein, NS 345-core polyprotein, core, and/or peptides from the nonstructural regions (Houghton et al., Hepatology (1991) 14:381).
- Rhabdovirus Viral antigens may be derived from a Rhabdovirus, such as a Lyssavirus (Rabies virus) and Vesiculovirus (VSV). Rhabdovirus antigens may be selected from glycoprotein (G), nucleoprotein (N), large protein (L), nonstructural proteins (NS).
- Rhabdovirus antigens may be selected from glycoprotein (G), nucleoprotein (N), large protein (L), nonstructural proteins (NS).
- G glycoprotein
- N nucleoprotein
- L large protein
- NS nonstructural proteins
- Rabies virus vaccine comprise killed virus grown on human diploid cells or fetal rhesus lung cells.
- Viral antigens may be derived from Calciviridae, such as Norwalk virus, and Norwalk-like Viruses, such as Hawaii Virus and Snow Mountain Virus.
- Coronavirus Viral antigens may be derived from a Coronavirus, SARS, Human respiratory coronavirus, Avian infectious bronchitis (IBV), Mouse hepatitis virus (MHV), and Porcine transmissible gastroenteritis virus (TGEV). Coronavirus antigens may be selected from spike (S), envelope (E), matrix (M), nucleocapsid (N), and Hemagglutinin- esterase glycoprotein (HE). Preferably, the Coronavirus antigen is derived from a SARS virus. SARS viral antigens are described in WO 04/92360;
- Retrovirus Viral antigens may be derived from a Retrovirus, such as an Oncovirus, a Lentivirus or a Spumavirus.
- Oncovirus antigens may be derived from HTLV-I, HTLV-2 or HTL V-5.
- Lentivirus antigens may be derived from HIV-I or HIV-2.
- Retrovirus antigens may be selected from gag, pol, env, tax, tat, rex, rev, nef, vif, vpu, and vpr.
- HIV antigens may be selected from gag (p24gag and p55gag), env (gpl60 and gp41), pol, tat, nef, rev vpu, miniproteins, (preferably p55 gag and gpl40v delete). HIV antigens may be derived from one or more of the following strains: HIVIIIb, HIVSF2, HIVLAV, HIVLAI, HIVMN, HIV-1CM235, HIV-1US4.
- Reovirus Viral antigens may be derived from a Reovirus, such as an Orthoreovirus, a Rotavirus, an Orbivirus, or a Colti virus.
- Reovirus antigens may be selected from structural proteins ⁇ l, ⁇ 2, ⁇ 3, ⁇ l, ⁇ 2, ⁇ l, ⁇ 2, or ⁇ 3, or nonstructural proteins ⁇ NS, ⁇ NS, or ⁇ ls.
- Preferred Reovirus antigens may be derived from a Rotavirus.
- Rotavirus antigens may be selected from VPl, VP2, VP3, VP4 (or the cleaved product VP5 and VP8), NSP 1, VP6, NSP3, NSP2, VP7, NSP4, or NSP5.
- Preferred Rotavirus antigens include VP4 (or the cleaved product VP5 and VP8), and VP7.
- Parvovirus Viral antigens may be derived from a Parvovirus, such as Parvovirus B19. Parvovirus antigens may be selected from VP-I, VP-2, VP-3, NS-I and NS-2. Preferably, the Parvovirus antigen is capsid protein VP-2.
- HDV Delta hepatitis virus
- Viral antigens may be derived HDV, particularly ⁇ -antigen from HDV (see, e.g., U.S. Patent No. 5,378,814).
- HEV Hepatitis E virus
- Hepatitis G virus Viral antigens may be derived from HGV.
- Human Herpesvirus Viral antigens may be derived from a Human Herpesvirus, such as Herpes Simplex Viruses (HSV) 5 Varicella-zoster virus (VZV), Epstein-Barr virus (EBV), Cytomegalovirus (CMV), Human Herpesvirus 6 (HHV6), Human Herpesvirus 7 (HHV7), and Human Herpesvirus 8 (HHV8).
- Human Herpesvirus antigens may be selected from immediate early proteins ( ⁇ ), early proteins ( ⁇ ), and late proteins ( ⁇ ).
- HSV antigens may be derived from HSV-I or HSV-2 strains.
- HSV antigens may be selected from glycoproteins gB, gC, gD and gH, fusion protein (gB), or immune escape proteins (gC, gE, or gl).
- VZV antigens may be selected from core, nucleocapsid, tegument, or envelope proteins.
- a live attenuated VZV vaccine is commercially available.
- EBV antigens may be selected from early antigen (EA) proteins, viral capsid antigen (VCA), and glycoproteins of the membrane antigen (MA).
- CMV antigens may be selected from capsid proteins, envelope glycoproteins (such as gB and gH), and tegument proteins
- Papovaviruses Antigens may be derived from Papovaviruses, such as Papillomaviruses and Polyomaviruses.
- Papillomaviruses include HPV serotypes 1, 2, 4, 5, 6, 8, 11, 13, 16, 18, 31, 33, 35, 39, 41, 42, 47, 51, 57, 58, 63 and 65.
- HPV antigens are derived from serotypes 6, 11, 16 or 18.
- HPV antigens may be selected from capsid proteins (Ll) and (L2), or El - E7, or fusions thereof.
- HPV antigens are preferably formulated into virus-like particles (VLPs).
- Polyomyavirus viruses include BK virus and JK virus.
- Polyomavirus antigens may be selected from VPl, VP2 or VP3.
- Fungal antigens for use in the invention may be derived from one or more of the fungi set forth below.
- Fungal antigens may be derived from Dermatophytres, including: Epidermophyton floccusum, Microsporum audouini, Microsporum canis, Microsporum distortum, Microsporum equinum, Microsporum gypsum, Microsporum nanum, Trichophyton concentricum, Trichophyton equinum, Trichophyton gallinae, Trichophyton gypseum, Trichophyton megnini, Trichophyton mentagrophytes, Trichophyton quinckeanum, Trichophyton rubrum, Trichophyton schoenleini, Trichophyton tonsurans, Trichophyton verrucosum, T. verrucosum var.
- Fungal pathogens may be derived from Aspergillus fumigatus, Aspergillus flavus, Aspergillus niger, Aspergillus nidulans, Aspergillus terreus, Aspergillus sydowi, Aspergillus flavatus, Aspergillus glaucus, Blastoschizomyces capitatus, Candida albicans, Candida enolase, Candida tropicalis, Candida glabrata, Candida krusei, Candida parapsilosis, Candida stellatoidea, Candida kusei, Candida parakwsei, Candida lusitaniae, Candida pseudotropicalis, Candida guilliermondi, Cladosporium carrionii, Coccidioides immitis, Blastomyces dermatidis, Cryptococc
- a solubilized fraction extracted and separated from an insoluble fraction obtainable from fungal cells of which cell wall has been substantially removed or at least partially removed characterized in that the process comprises the steps of: obtaining living fungal cells; obtaining fungal cells of which cell wall has been substantially removed or at least partially removed; bursting the fungal cells of which cell wall has been substantially removed or at least partially removed; obtaining an insoluble fraction; and extracting and separating a solubilized fraction from the insoluble fraction.
- compositions of the invention may include one or more antigens derived from a sexually transmitted disease (STD).
- STD sexually transmitted disease
- Such antigens may provide for prophylactis or therapy for STD 's such as chlamydia, genital herpes, hepatits (such as HCV), genital warts, gonorrhoea, syphilis and/or chancroid (See, WO00/15255).
- Antigens may be derived from one or more viral or bacterial STD 's.
- Viral STD antigens for use in the invention may be derived from, for example, HIV, herpes simplex virus (HSV-I and HSV-2), human papillomavirus (HPV), and hepatitis (HCV).
- Bacterial STD antigens for use in the invention may be derived from, for example, Neiserria gonorrhoeae, Chlamydia trachomatis, Treponema pallidum, Haemophilus ducreyi, E. coli, and Streptococcus agalactiae. Examples of specific antigens derived from these pathogens are described above.
- compositions of the invention may include one or more antigens derived from a pathogen which causes respiratory disease.
- respiratory antigens may be derived from a respiratory virus such as Orthomyxoviruses (influenza), Pneumovirus (RSV), Paramyxovirus (PIV), Morbillivirus (measles), Togavirus (Rubella), VZV, and Coronavirus (SARS).
- Respiratory antigens may be derived from a bacteria which causes respiratory disease, such as Streptococcus pneumoniae, Pseudomonas aeruginosa, Bordetella pertussis, Mycobacterium tuberculosis, Mycoplasma pneumoniae, Chlamydia pneumoniae, Bacillus anthracis, and Moraxella catarrhalis. Examples of specific antigens derived from these pathogens are described above.
- compositions of the invention may include one or more antigens suitable for use in pediatric subjects.
- Pediatric subjects are typically less than about 3- years old, or less than about 2 years old, or less than about 1 years old.
- Pediatric antigens may be administered multiple times over the course of 6 months, 1, 2 or 3 years.
- Pediatric antigens may be derived from a virus which may target pediatric populations and/or a virus from which pediatric populations are susceptible to infection.
- Pediatric viral antigens include antigens derived from one or more of Orthomyxovirus (influenza), Pneumovirus (RSV), Paramyxovirus (PIV and Mumps), Morbillivirus (measles), Togavirus (Rubella), Enterovirus (polio), HBV, Coronavirus (SARS), and Varicella-zoster virus (VZV), Epstein Barr virus (EBV).
- Orthomyxovirus influenza
- RSV Pneumovirus
- PIV and Mumps Paramyxovirus
- Morbillivirus measles
- Togavirus Rubella
- Enterovirus polio
- HBV HBV
- Coronavirus Coronavirus
- VZV Varicella-zoster virus
- EBV Epstein Barr virus
- Pediatric bacterial antigens include antigens derived from one or more of Streptococcus pneumoniae, Neisseria meningitides, Streptococcus pyogenes (Group A Streptococcus), Moraxella catarrhalis, Bordetella pertussis, Staphylococcus aureus, Clostridium tetani (Tetanus), Comynebacterium diphtheriae (Diphtheria), Haemophilus influenzae B (Hib), Pseudomonas aeruginosa, Streptococcus agalactiae (Group B Streptococcus), and E. coli. Examples of specific antigens derived from these pathogens are described above.
- compositions of the invention may include one or more antigens suitable for use in elderly or immunocompromised individuals. Such individuals may need to be vaccinated more frequently, with higher doses or with adjuvanted formulations to improve their immune response to the targeted antigens.
- Antigens which may be targeted for use in Elderly or Immunocompromised individuals include antigens derived from one or more of the following pathogens: Neisseria meningitides, Streptococcus pneumoniae, Streptococcus pyogenes (Group A Streptococcus), Moraxella catarrhalis, Bordetella pertussis, Staphylococcus aureus, Staphylococcus epidermis, Clostridium tetani (Tetanus), Comynebacterium diphtheriae (Diphtheria), Haemophilus influenzae B (Hib), Pseudomonas aeruginosa, Legionella pneumophila, Streptococcus agalactiae (Group B Streptococcus), Enterococcus faecalis, Helicobacter pylori, Clamydia pneumoniae, Orthomyxovirus (influenza), Pneumovirus (RS
- compositions of the invention may include one or more antigens suitable for use in adolescent subjects.
- Adolescents may be in need of a boost of a previously administered pediatric antigen.
- Pediatric antigens which may be suitable for use in adolescents are described above.
- adolescents may be targeted to receive antigens derived from an STD pathogen in order to ensure protective or therapeutic immunity before the beginning of sexual activity.
- STD antigens which may be suitable for use in adolescents are described above.
- methods of producing microparticles having adsorbed antigens comprise: (a) providing an emulsion by dispersing a mixture comprising (i) water, (ii) a detergent, (iii) an organic solvent, and (iv) a biodegradable polymer selected from the group consisting of a poly( ⁇ -hydroxy acid), a polyhydroxy butyric acid, a polycaprolactone, a polyorthoester, a polyanhydride, and a polycyanoacrylate.
- the polymer is typically present in the mixture at a concentration of about 1% to about 30% relative to the organic solvent, while the detergent is typically present in the mixture at a weight-to-weight detergent-to-polymer ratio of from about 0.00001:1 to about 0.1:1 (more typically about 0.0001:1 to about 0.1:1, about 0.001:1 to about 0.1:1, or about 0.005:1 to about 0.1:1); (b) removing the organic solvent from the emulsion; and (c) adsorbing an antigen on the surface of the microparticles.
- the biodegradable polymer is present at a concentration of about 3% to about 10% relative to the organic solvent.
- Microparticles for use herein will be formed from materials that are sterilizable, non ⁇ toxic and biodegradable. Such materials include, without limitation, poly( ⁇ -hydroxy acid), polyhydroxybutyric acid, polycaprolactone, polyorthoester, polyanhydride, PACA, and polycyanoacrylate.
- microparticles for use with the present invention are derived from a poly( ⁇ -hydroxy acid), in particular, from a poly(lactide) ("PLA”) or a copolymer of D,L-lactide and glycolide or glycolic acid, such as a poly(D,L-lactide-co- glycolide) (“PLG” or "PLGA”), or a copolymer of D,L-lactide and caprolactone.
- PLA poly(lactide)
- PLA poly(lactide)
- PLA poly(D,L-lactide-co- glycolide)
- caprolactone a copolymer of D,L-lactide and caprolactone
- microparticles may be derived from any of various polymeric starting materials which have a variety of molecular weights and, in the case of the copolymers such as PLG, a variety of lactide: glycolide ratios, the selection of which will be largely a matter of choice, depending in part on the coadministered macromolecule. These parameters are discussed more fully below.
- Further antigens may also include an outer membrane vesicle (OMV) preparation.
- OMV outer membrane vesicle
- a saccharide or carbohydrate antigen is used, it is preferably conjugated to a carrier protein in order to enhance immunogenicity.
- a carrier protein in order to enhance immunogenicity. See Ramsay et al. (2001) Lancet 357(9251):195-196; Lindberg (1999) Vaccine 17 Suppl 2:S28-36; Buttery & Moxon (2000) J R Coll Physicians Lond 34:163-168; Ahmad & Chapnick (1999) Infect Dis Clin North Am 13:113-133, vii; Goldblatt (1998) J. Med. Microbiol. 47:563-567; European patent 0 477 508; US Patent No. 5,306,492; WO98/42721; Conjugate Vaccines (eds.
- Preferred carrier proteins are bacterial toxins or toxoids, such as diphtheria or tetanus toxoids.
- the CRM 197 diphtheria toxoid is particularly preferred.
- carrier polypeptides include the N. meningitidis outer membrane protein (EP-A- 0372501), synthetic peptides (EP-A-0378881 and EP-A 0427347), heat shock proteins (WO 93/17712 and WO 94/03208), pertussis proteins (WO 98/58668 and EP A 0471177), protein D from H. influenzae (WO 00/56360), cytokines (WO 91/01146), lymphokines, hormones, growth factors, toxin A or B from C. difficile (WO 00/61761), iron-uptake proteins ( WO 01/72337), etc.
- a mixture comprises capsular saccharide from both serigraphs A and C
- the ratio (w/w) of MenA saccharide:MenC saccharide is greater than 1 (e.g., 2:1, 3:1, 4:1, 5:1, 10:1 or higher).
- Different saccharides can be conjugated to the same or different type of carrier protein. Any suitable conjugation reaction can be used, with any suitable linker where necessary.
- Toxic protein antigens may be detoxified where necessary e.g., detoxification of pertussis toxin by chemical and/or genetic means.
- Pharmaceutically acceptable carriers may be used.
- compositions of the invention will typically, in addition to the components mentioned above, comprise one or more "pharmaceutically acceptable carriers.” These include any carrier which does not itself induce the production of antibodies harmful to the individual receiving the composition. Suitable carriers typically are large, slowly metabolized macromolecules such as proteins, polysaccharides, polylactic acids, polyglycolic acids, polymeric amino acids, amino acid copolymers, and lipid aggregates (such as oil droplets or liposomes). Such carriers are well known to those of ordinary skill in the art. A composition may also contain a diluent, such as water, saline, glycerol, etc.
- a diluent such as water, saline, glycerol, etc.
- auxiliary substance such as a wetting or emulsifying agent, pH buffering substance, and the like, may be present.
- auxiliary substance such as a wetting or emulsifying agent, pH buffering substance, and the like.
- Vaccines of the invention may be administered in conjunction with other immunoregulatory agents.
- compositions will usually include an adjuvant.
- adjuvants for use with the invention include, but are not limited to, one or more of the following set forth below:
- Mineral containing compositions suitable for use as adjuvants in the invention include mineral salts, such as aluminum salts and calcium salts.
- the invention includes mineral salts such as hydroxides (e.g. oxyhydroxides), phosphates (e.g. hydroxyphosphates, orthophosphates), sulfates, etc. (e.g. see chapters 8 & 9 of Vaccine Design... (1995) eds. Powell & Newman. ISBN: 030644867X. Plenum.), or mixtures of different mineral compounds (e.g. a mixture of a phosphate and a hydroxide adjuvant, optionally with an excess of the phosphate), with the compounds taking any suitable form (e.g. gel, crystalline, amorphous, etc.), and with adsorption to the salt(s) being preferred.
- the mineral containing compositions may also be formulated as a particle of metal salt (WO00/23105).
- Aluminum salts may be included in vaccines of the invention such that the dose of Al 3+ is between 0.2 and 1.0 mg per dose.
- the aluminum based adjuvant for use in the present invention is alum (aluminum potassium sulfate (A1K(SO 4 ) 2 )), or an alum derivative, such as that formed in- situ by mixing an antigen in phosphate buffer with alum, followed by titration and precipitation with a base such as ammonium hydroxide or sodium hydroxide.
- alum aluminum potassium sulfate (A1K(SO 4 ) 2 )
- A1K(SO 4 ) 2 aluminum potassium sulfate
- an alum derivative such as that formed in- situ by mixing an antigen in phosphate buffer with alum, followed by titration and precipitation with a base such as ammonium hydroxide or sodium hydroxide.
- Aluminum-based adjuvant for use in vaccine formulations of the present invention is aluminum hydroxide adjuvant (Al(OH) 3 ) or crystalline aluminum oxyhydroxide (AlOOH), which is an excellent adsorbant, having a surface area of approximately 500m 2 /g.
- Al(OH) 3 aluminum hydroxide adjuvant
- AlOOH crystalline aluminum oxyhydroxide
- AlPO 4 aluminum phosphate adjuvant
- AlPO 4 aluminum hydroxyphosphate, which contains phosphate groups in place of some or all of the hydroxyl groups of aluminum hydroxide adjuvant is provided.
- Preferred aluminum phosphate adjuvants provided herein are amorphous and soluble in acidic, basic and neutral media.
- the adjuvant of the invention comprises both aluminum phosphate and aluminum hydroxide, hi a more particular embodiment thereof, the adjuvant has a greater amount of aluminum phosphate than aluminum hydroxide, such as a ratio of 2:1, 3:1, 4:1, 5:1, 6:1, 7:1, 8:1, 9:1 or greater than 9:1, by weight aluminum phosphate to aluminum hydroxide. More particular still, aluminum salts in the vaccine are present at 0.4 to 1.0 mg per vaccine dose, or 0.4 to 0.8 mg per vaccine dose, or 0.5 to 0.7 mg per vaccine dose, or about 0.6 mg per vaccine dose.
- the preferred aluminum-based adjuvant(s), or ratio of multiple aluminum- based adjuvants, such as aluminum phosphate to aluminum hydroxide is selected by optimization of electrostatic attraction between molecules such that the antigen carries an opposite charge as the adjuvant at the desired pH.
- pretreatment of aluminum hydroxide with phosphate lowers its isoelectric point, making it a preferred adjuvant for more basic antigens.
- Oil-emulsion compositions suitable for use as adjuvants in the invention include squalene-water emulsions, such as MF59 (5% Squalene, 0.5% TWEENTM 80, and 0.5% Span 85, formulated into submicron particles using a microfluidizer). See WO90/14837. See also, Podda, Vaccine (2001) 19: 2673-2680; Frey et al, Vaccine (2003) 21:4234- 4237. MF59 is used as the adjuvant in the FLUADTM influenza virus trivalent subunit vaccine.
- MF59 5% Squalene, 0.5% TWEENTM 80, and 0.5% Span 85
- Particularly preferred adjuvants for use in the compositions are submicron oil-in-water emulsions.
- Preferred submicron oil-in-water emulsions for use herein are squalene/water emulsions optionally containing varying amounts of MTP-PE, such as a submicron oil-in- water emulsion containing 4-5% w/v squalene, 0.25-1.0% w/v TWEENTM 8OD (polyoxyelthylenesorbitan monooleate), and/or 0.25-1.0% SPAN 85TM (sorbitan trioleate), and, optionally, N-acetylmuramyl-L-alanyl-D-isogluatminyl-L-alanine-2-(r-2'- dipalmitoyl-sn-glycero-3 -huydroxyphosphophoryloxy)-ethylamine (MTP-PE), for example, the submicron oil-in-water
- MF59 contains 4-5% w/v Squalene (e.g. 4.3%), 0.25-0.5% w/v TWEENTM 80, and 0.5% w/v SPAN 85TM and optionally contains various amounts of MTP-PE, formulated into submicron particles using a microfluidizer such as Model HOY microfluidizer (Microfluidics, Newton, MA).
- a microfluidizer such as Model HOY microfluidizer (Microfluidics, Newton, MA).
- MTP-PE may be present in an amount of about 0-500 ⁇ g/dose, more preferably 0-250 ⁇ g/dose and most preferably, 0-100 ⁇ g/dose.
- MF59-0 refers to the above submicron oil-in-water emulsion lacking MTP-PE
- MF59-MTP denotes a formulation that contains MTP-PE.
- MF59-100 contains 100 ⁇ g MTP-PE per dose, and so on.
- MF69 another submicron oil-in-water emulsion for use herein, contains 4.3% w/v squalene, 0.25% w/v TWEENTM 80, and 0.75% w/v SPAN 85TM and optionally MTP-PE.
- MF75 also known as SAF, containing 10% squalene, 0.4% TWEENTM 80, 5% pluronic-blocked polymer L 121, and thr-MDP, also microfluidized into a submicron emulsion.
- MF75-MTP denotes an MF75 formulation that includes MTP, such as from 100-400 ⁇ g MTP-PE per dose.
- CFA Complete Freund's adjuvant
- IFA incomplete Freund's adjuvant
- Saponin formulations may also be used as adjuvants in the invention.
- Saponins are a heterologous group of sterol glycosides and triterpenoid glycosides that are found in the bark, leaves, stems, roots and even flowers of a wide range of plant species. Saponins isolated from the bark of the Quillaia saponaria Molina tree have been widely studied as adjuvants. Saponins can also be commercially obtained from Smilax ornata (sarsaprilla), Gypsophilla paniculata (brides veil), and Saponaria officianalis (soap root).
- Saponin adjuvant formulations include purified formulations, such as QS21, as well as lipid formulations, such as ISCOMs.
- Saponin compositions have been purified using High Performance Thin Layer Chromatography (HP-TLC) and Reversed Phase High Performance Liquid Chromatography (RP-HPLC). Specific purified fractions using these techniques have been identified, including QS7, QS 17, QS 18, QS21, QH-A, QH-B and QH-C.
- the saponin is QS21.
- a method of production of QS21 is disclosed in U.S. Patent 5,057,540.
- Saponin formulations may also comprise a sterol, such as cholesterol (see WO96/33739).
- ISCOMs Immunostimulating Complexes
- phospholipid such as phosphatidylethanolamine or phosphatidylcholine.
- Any known saponin can be used in ISCOMs.
- the ISCOM includes one or more of Quil A, QHA and QHC.
- ISCOMS may be devoid of (an) additional detergent(s). See WOOO/07621.
- VLPs Virosomes and Virus Like Particles
- Virosomes and Virus Like Particles can also be used as adjuvants in the invention.
- These structures generally contain one or more proteins from a virus optionally combined or formulated with a phospholipid. They are generally non-pathogenic, non- replicating and generally do not contain any of the native viral genome.
- the viral proteins may be recombinantly produced or isolated from whole viruses.
- viral proteins suitable for use in virosomes or VLPs include proteins derived from influenza virus (such as HA or NA), Hepatitis B virus (such as core or capsid proteins), Hepatitis E virus, measles virus, Sindbis virus, Rotavirus, Foot-and-Mouth Disease virus, Retrovirus, Norwalk virus, human Papilloma virus, HIV, RNA-phages, Q ⁇ -phage (such as coat proteins), GA-phage, fr-phage, AP205 phage, and Ty (such as retrotransposon Ty protein pi).
- influenza virus such as HA or NA
- Hepatitis B virus such as core or capsid proteins
- Hepatitis E virus measles virus
- Sindbis virus Rotavirus
- Foot-and-Mouth Disease virus Retrovirus
- Norwalk virus Norwalk virus
- human Papilloma virus HIV
- RNA-phages Q ⁇ -phage (such as coat proteins)
- GA-phage such as fr-phage,
- VLPs are discussed further in WO03/024480, WO03/024481, and Niikura et al, Virology (2002) 293:273-280; Lenz et al, Journal of Immunology (2001) 5246-5355; Pinto, et al, Journal of Infectious Diseases (2003) 188:327-338; and Gerber et al, Journal of Virology (2001) 75(10):4752-4760. Virosomes are discussed further in, for example, Gluck et al, Vaccine (2002) 20:B10 -B 16.
- Immunopotentiating reconstituted influenza virosomes are used as the subunit antigen delivery system in the intranasal trivalent INFLEXALTM product ⁇ Mischler & Metcalfe (2002) Vaccine 20 Suppl 5 :B 17-23 ⁇ and the INFLUVAC PLUSTM product.
- Adjuvants suitable for use in the invention include bacterial or microbial derivatives such as:
- Such derivatives include Monophosphoryl lipid A (MPL) and 3-O-deacylated MPL (3dMPL).
- 3dMPL is a mixture of 3 De-O-acylated monophosphoryl lipid A with 4, 5 or 6 acylated chains.
- a preferred "small particle" form of 3 De-O-acylated monophosphoryl lipid A is disclosed in EP 0 689 454.
- Such "small particles" of 3dMPL are small enough to be sterile filtered through a 0.22 micron membrane (see EP 0 689 454).
- Other non ⁇ toxic LPS derivatives include monophosphoryl lipid A mimics, such as aminoalkyl glucosaminide phosphate derivatives e.g. RC 529. See Johnson et al (1999) Bioorg Med Chem Lett 9:2273-2278.
- Lipid A derivatives include derivatives of lipid A from Escherichia coli such as OM- 174.
- OM-174 is described for example in Meraldi et al, Vaccine (2003) 21:2485-2491; and Pajak, et al, Vaccine (2003) 21:836-842.
- Immunostimulatory oligonucleotides suitable for use as adjuvants in the invention include nucleotide sequences containing a CpG motif (a sequence containing an unmethylated cytosine followed by guanosine and linked by a phosphate bond). Bacterial double stranded RNA or oligonucleotides containing palindromic or poly(dG) sequences have also been shown to be immunostimulatory.
- the CpG' s can include nucleotide modifications/analogs such as phosphorothioate modifications and can be double-stranded or single-stranded.
- the guanosine may be replaced with an analog such as 2'-deoxy-7-deazaguanosine. See Kandimalla, et al, Nucleic Acids Research (2003) 31(9): 2393-2400; WO02/26757 and WO99/62923 for examples of possible analog substitutions.
- the CpG sequence may be directed to TLR9, such as the motif GTCGTT or TTCGTT. See Kandimalla, et al, Biochemical Society Transactions (2003) 31 (part 3): 654-658.
- the CpG sequence may be specific for inducing a ThI immune response, such as a CpG- A ODN, or it may be more specific for inducing a B cell response, such a CpG-B ODN.
- CpG-A and CpG-B ODNs are discussed in Blackwell, et al, J. Immunol. (2003) 170(8):4061-4068; Krieg, TRENDS in Immunology (2002) 23(2): 64-65 and WO01/95935.
- the CpG is a CpG-A ODN.
- the CpG oligonucleotide is constructed so that the 5' end is accessible for receptor recognition.
- two CpG oligonucleotide sequences may be attached at their 3' ends to form "immunomers". See, for example, Kandimalla, et al, BBRC (2003) 306:948-953; Kandimalla, et al, Biochemical Society Transactions (2003) 31(part 3):664-658; Bhagat et al, BBRC (2003) 300:853-861 and WO03/035836. (4) ADP-ribosylating toxins and detoxified derivatives thereof.
- Bacterial ADP-ribosylating toxins and detoxified derivatives thereof may be used as adjuvants in the invention.
- the protein is derived from E. coli (i.e., E. coli heat labile enterotoxin "LT), cholera ("CT"), or pertussis ("PT").
- LT E. coli heat labile enterotoxin
- CT cholera
- PT pertussis
- the use of detoxified ADP- ribosylating toxins as mucosal adjuvants is described in WO95/17211 and as parenteral adjuvants in WO98/42375.
- the adjuvant is a detoxified LT mutant such as LT-K63, LT-R72, and LTRl 92G.
- ADP-ribosylating toxins and detoxified derivatives thereof, particularly LT-K63 and LT-R72, as adjuvants can be found in the following references: Beignon, et al, Infection and Immunity (2002) 70(6):3012-3019; Pizza, et al, Vaccine (2001) 19:2534-2541; Pizza, et al, Int. J. Med. Microbiol (2000) 290(4-5):455-461; Scharton-Kersten et al, Infection and Immunity (2000) 68(9):5306- 5313; Ryan et al, Infection and Immunity (1999) 67(12):6270-6280; Partidos et al, Immunol. Lett.
- Numerical reference for amino acid substitutions is preferably based on the alignments of the A and B subunits of ADP-ribosylating toxins set forth in Domenighini et al, MoI. Microbiol (1995) 15(6):1165-1167.
- Bioadhesives and mucoadhesives may also be used as adjuvants in the invention.
- Suitable bioadhesives include esterified hyaluronic acid microspheres (Singh et al (2001) J. Cont. ReIe. 70:267-276) or mucoadhesives such as cross-linked derivatives of polyacrylic acid, polyvinyl alcohol, polyvinyl pyrollidone, polysaccharides and carboxymethylcellulose. Chitosan and derivatives thereof may also be used as adjuvants in the invention. See WO99/27960.
- Microparticles may also be used as adjuvants in the invention.
- Microparticles i.e. a particle of ⁇ 100nm to ⁇ 150 ⁇ m in diameter, more preferably ⁇ 200nm to ⁇ 30 ⁇ m in diameter, and most preferably ⁇ 500nm to ⁇ 10 ⁇ m in diameter
- materials that are biodegradable and non toxic e.g. a poly( ⁇ -hydroxy acid), a polyhydroxybutyric acid, a polyorthoester, a polyanhydride, a polycaprolactone, etc.
- a negatively-charged surface e.g. with SDS
- a positively-charged surface e.g. with a cationic detergent, such as CTAB
- liposome formulations suitable for use as adjuvants are described in US Patent No. 6,090,406, US Patent No. 5,916,588, and EP 0 626 169.
- Adjuvants suitable for use in the invention include polyoxyethylene ethers and polyoxyethylene esters. WO99/52549. Such formulations further include polyoxyethylene sorbitan ester surfactants in combination with an octoxynol (WOO 1/21207) as well as polyoxyethylene alkyl ethers or ester surfactants in combination with at least one additional non-ionic surfactant such as an octoxynol (WO01/21152).
- Preferred polyoxyethylene ethers are selected from the following group: polyoxyethylene-9-lauryl ether (laureth 9), polyoxyethylene-9-steoryl ether, polyoxytheylene-8-steoryl ether, polyoxyethylene-4-lauryl ether, polyoxyethylene-35- lauryl ether, and polyoxyethylene-23-lauryl ether.
- PCPP J. Polyphosphazene
- PCPP formulations are described, for example, in Andrianov et ah, "Preparation of hydrogel microspheres by coacervation of aqueous polyphophazene solutions", Biomaterials (1998) 19(l-3):109-115 and Payne et al, "Protein Release from Polyphosphazene Matrices", Adv. Drug. Delivery Review (1998) 31(3):185-196.
- muramyl peptides suitable for use as adjuvants in the invention include N- acetyl-muramyl-L-threonyl-D-isoglutamine (thr-MDP), N-acetyl-normuramyl-1-alanyl-d- isoglutamine (nor-MDP), and N acetylmuramyl-l-alanyl-d-isoglutaminyl-l-alanine-2-(r- 2'-dipalmitoyl-sn-glycero-3-hydroxyphosphoryloxy)-ethylamine MTP-PE).
- imidazoquinoline compounds suitable for use adjuvants in the invention include Imiquimod and its analogues, described further in Stanley, Clin Exp Dermatol (2002) 27(7):571-577; Jones, Curr Opin Investig Drugs (2003) 4(2):214-218; and U.S. Patents 4,689,338, 5,389,640, 5,268,376, 4,929,624, 5,266,575, 5,352,784, 5,494,916, 5,482,936, 5,346,905, 5,395,937, 5,238,944, and 5,525,612.
- thiosemicarbazone compounds as well as methods of formulating, manufacturing, and screening for compounds all suitable for use as adjuvants in the invention include those described in WO04/60308.
- the thiosemicarbazones are particularly effective in the stimulation of human peripheral blood mononuclear cells for the production of cytokines, such as TNF- ⁇ .
- tryptanthrin compounds as well as methods of formulating, manufacturing, and screening for compounds all suitable for use as adjuvants in the invention include those described in WO04/64759.
- the tryptanthrin compounds are particularly effective in the stimulation of human peripheral blood mononuclear cells for the production of cytokines, such as TNF- ⁇ .
- the invention may also comprise combinations of aspects of one or more of the adjuvants identified above.
- the following adjuvant compositions may be used in the invention:
- RibiTM adjuvant system (RAS), (Ribi Immunochem) containing 2% Squalene, 0.2% Tween 80, and one or more bacterial cell wall components from the group consisting of monophosphorylipid A (MPL), trehalose dimycolate (TDM), and cell wall skeleton (CWS), preferably MPL + CWS (DetoxTM); and
- one or more mineral salts such as an aluminum salt
- a non-toxic derivative of LPS such as 3dPML
- Human immunomodulators suitable for use as adjuvants in the invention include cytokines, such as interleukins (e.g. IL-I, IL-2, IL-4, IL-5, IL-6, IL-7, IL-12, etc.), interferons (e.g. interferon- ⁇ ), macrophage colony stimulating factor, and tumor necrosis factor.
- cytokines such as interleukins (e.g. IL-I, IL-2, IL-4, IL-5, IL-6, IL-7, IL-12, etc.), interferons (e.g. interferon- ⁇ ), macrophage colony stimulating factor, and tumor necrosis factor.
- Aluminum salts and MF59 are preferred adjuvants for use with injectable influenza vaccines.
- Bacterial toxins and bioadhesives are preferred adjuvants for use with mucosally-delivered vaccines, such as nasal vaccines.
- the invention provides methods for inducing or increasing an immune response to a GAS antigen using the compositions described above.
- the immune response is preferably protective and can include antibodies and/or cell-mediated immunity (including systemic and mucosal immunity). Immune responses include booster responses.
- Compositions comprising antibodies can be used to treat S. pyogenes infections.
- a vaccine intended for children may also be administered to adults e.g., to assess safety, dosage, immunogenicity, etc.
- Streptococcus pyogenes which can be prevented or treated according to the invention include, but are not limited to, pharyngitis (such as streptococcal sore throat), scarlet fever, impetigo, erysipelas, cellulitis, septicemia, toxic shock syndrome, necrotizing fasciitis, and sequelae such as rheumatic fever and acute glomerulonephritis.
- the compositions may also be effective against other streptococcal bacteria, e.g., GBS.
- One way of assessing efficacy of therapeutic treatment involves monitoring GAS infection after administration of the composition of the invention.
- One way of assessing efficacy of prophylactic treatment involves monitoring immune responses against the GAS antigens in the compositions of the invention after administration of the composition.
- Another way of assessing the immunogenicity of the component proteins of the immunogenic compositions of the present invention is to express the proteins recombinantly and to screen patient sera or mucosal secretions by immunoblot. A positive reaction between the protein and the patient serum indicates that the patient has previously mounted an immune response to the protein in question; i.e., the protein is an immunogen. This method may also be used to identify immunodominant proteins and/or epitopes.
- Another way of checking efficacy of therapeutic treatment involves monitoring GAS infection after administration of the compositions of the invention.
- One way of checking efficacy of prophylactic treatment involves monitoring immune responses both systemically (such as monitoring the level of IgGl and IgG2a production) and mucosally (such as monitoring the level of IgA production) against the GAS antigens in the compositions of the invention after administration of the composition.
- GAS serum specific antibody responses are determined post-immunization but pre-challenge whereas mucosal GAS-specific antibody body responses are determined post- immunization and post-challenge.
- the vaccine compositions of the present invention can be evaluated in in vitro and in vivo animal models prior to host, e.g., human, administration.
- Particularly useful mouse models include those in which intraperitoneal immunization is followed by either intraperitoneal challenge or intranasal challenge.
- a model in which intraperitoneal immunization is followed by intraperitoneal challenge is illustrated in FIG. 13.
- the efficacy of immunogenic compositions of the invention can also be determined in vivo by challenging animal models of GAS infection, e.g., guinea pigs or mice, with the immunogenic compositions.
- the immunogenic compositions may or may not be derived from the same serotypes as the challenge serotypes.
- the immunnogenic compositions are derivable from the same serotypes as the challenge serotypes.
- the immunogenic composition and/or the challenge serotype are derivable from the group of GAS serotypes consisting of Ml, M3, M23 and/or combinations thereof.
- In vivo efficacy models include but are not limited to: (i) a murine infection model using human GAS serotypes; (ii) a murine disease model which is a murine model using a mouse-adapted GAS strain, such as the M23 strain which is particularly virulent in mice, and (iii) a primate model using human GAS isolates.
- the immune response may be one or both of a THl immune response and a TH2 response.
- the immune response may be an improved or an enhanced or an altered immune response.
- the immune response may be one or both of a systemic and a mucosal immune response.
- Preferably the immune response is an enhanced system and/or mucosal response.
- an enhanced systemic and/or mucosal immunity is reflected in an enhanced THl and/or TH2 immune response.
- the enhanced immune response includes an increase in the production of IgGl and/or IgG2a and/or IgA.
- the mucosal immune response is a TH2 immune response.
- the mucosal immune response includes an increase in the production of IgA.
- Activated TH2 cells enhance antibody production and are therefore of value in responding to extracellular infections.
- Activated TH2 cells may secrete one or more of IL-4, IL-5, IL-6, and IL-IO.
- a TH2 immune response may result in the production of IgGl, IgE, IgA and memory B cells for future protection.
- a TH2 immune response may include one or more of an increase in one or more of the cytokines associated with a TH2 immune response (such as IL-4, IL-5, IL-6 and IL-IO), or an increase in the production of IgGl, IgE, IgA and memory B cells.
- the enhanced TH2 immune resonse will include an increase in IgGl production.
- a THl immune response may include one or more of an increase in CTLs, an increase in one or more of the cytokines associated with a THl immune response (such as IL-2, IFN ⁇ , and TNF ⁇ ), an increase in activated macrophages, an increase in NK activity, or an increase in the production of IgG2a.
- the enhanced THl immune response will include an increase in IgG2a production.
- Immunogenic compositions of the invention in particular, immunogenic composition comprising one or more GAS antigens of the present invention may be used either alone or in combination with other GAS antigens optionally with an immunoregulatory agent capable of eliciting a ThI and/or Th2 response.
- the invention also comprises an immunogenic composition
- an immunogenic composition comprising one or more immunoregulatory agent, such as a mineral salt, such as an aluminium salt and an oligonucleotide containing a CpG motif.
- the immunogenic composition includes both an aluminium salt and an oligonucleotide containing a CpG motif.
- the immunogenic composition includes an ADP ribosylating toxin, such as a detoxified ADP ribosylating toxin and an oligonucleotide containing a CpG motif.
- one or more of the immunoregulatory agents include an adjuvant.
- the adjuvant may be selected from one or more of the group consisting of a THl adjuvant and TH2 adjuvant, further discussed below.
- compositions of the invention will preferably elicit both a cell mediated immune response as well as a humoral immune response in order to effectively address a GAS infection.
- This immune response will preferably induce long lasting (e.g., neutralizing) antibodies and a cell mediated immunity that can quickly respond upon exposure to one or more GAS antigens.
- the immunogenic composition comprises one or more GAS antigen(s) which elicits a neutralizing antibody response and one or more GAS antigen(s) which elicit a cell mediated immune response.
- the neutralizing antibody response prevents or inhibits an initial GAS infection while the cell-mediated immune response capable of eliciting an enhanced ThI cellular response prevents further spreading of the GAS infection.
- the immunogenic composition comprises one or more GAS surface antigens and one or more GAS cytoplasmic antigens.
- the immunogenic composition comprises one or more GAS40 surface antigens or the like and one or other antigens, such as a cytoplasmic antigen capable of eliciting a ThI cellular response.
- compositions of the invention will generally be administered directly to a patient.
- the compositions of the present invention may be administered, either alone or as part of a composition, via a variety of different routes. Certain routes may be favored for certain compositions, as resulting in the generation of a more effective immune response, preferably a CMI response, or as being less likely to induce side effects, or as being easier for administration.
- Delivery methods include parenteral injection (e.g., subcutaneous, intraperitoneal, intravenous, intramuscular, or interstitial injection) and rectal, oral (e.g., tablet, spray), vaginal, topical, transdermal (e.g., see WO 99/27961), transcutaneous (e.g., see WO02/074244 and WO02/064162), intranasal (e.g., see WO03/028760), ocular, aural, and pulmonary or other mucosal administration.
- parenteral injection e.g., subcutaneous, intraperitoneal, intravenous, intramuscular, or interstitial injection
- rectal e.g., oral
- vaginal topical
- transdermal e.g., see WO 99/27961
- transcutaneous e.g., see WO02/074244 and WO02/064162
- intranasal e.g., see WO03/028760
- compositions of the present invention may be administered via a systemic route or a mucosal route or a transdermal route or it may be administered directly into a specific tissue.
- systemic administration includes but is not limited to any parenteral routes of administration.
- parenteral administration includes but is not limited to subcutaneous, intraperitoneal, intravenous, intraarterial, intramuscular, or intrasternal injection, intravenous, intraarterial, or kidney dialytic infusion techniques.
- the systemic, parenteral administration is intramuscular injection.
- the term “mucosal administration” includes but is not limited to oral, intranasal, intravaginal, intrarectal, intratracheal, intestinal and ophthalmic administration.
- Dosage treatment can be a single dose schedule or a multiple dose schedule. Multiple doses may be used in a primary immunization schedule and/or in a booster immunization schedule. In a multiple dose schedule the various doses may be given by the same or different routes e.g., a parenteral prime and mucosal boost, a mucosal prime and parenteral boost, etc.
- compositions of the invention may be prepared in various forms.
- a composition can be prepared as an injectable, either as a liquid solution or a suspension.
- Solid forms suitable for solution in, or suspension in, liquid vehicles prior to injection can also be prepared (e.g., a lyophilized composition).
- a composition can be prepared for oral administration, such as a tablet or capsule, as a spray, or as a syrup (optionally flavored).
- a composition can be prepared for pulmonary administration, e.g., as an inhaler, using a fine powder or a spray.
- a composition can be prepared as a suppository or pessary.
- a composition can be prepared for nasal, aural or ocular administration e.g., as drops.
- a composition can be in kit form, designed such that a combined composition is reconstituted just prior to administration to a patient. Such kits may comprise one or more GAS or other antigens in liquid form and one or more lyophilized antigens.
- Immunogenic compositions used as vaccines comprise an immunologically effective amount of GAS or other antigens (or nucleic acid molecules encoding the antigens) or antibodies, as well as any other components, as needed, such as antibiotics.
- An "immunologically effective amount” is an amount which, when administered to an individual, either in a single dose or as part of a series, increases a measurable immune response or prevents or reduces a clinical symptom.
- the immunogenic compositions of the present invention may be administered in combination with an antibiotic treatment regime.
- the antibiotic is administered prior to administration of the antigen of the invention or the composition comprising the one or more GAS antigens of the invention.
- the antibiotic is administered subsequent to the administration of the one or more surface-exposed and/or surface-associated GAS antigens of the invention or the composition comprising the one or more surface-exposed and/or surface-associated GAS antigens of the invention.
- antibiotics suitable for use in the treatment of a GAS infection include but are not limited to penicillin or a derivative thereof or clindamycin, cephalosporins, glycopeptides (e.g., vancomycin), and cycloserine.
- the amount of active agent in a composition varies, however, depending upon the health and physical condition of the individual to be treated, age, the taxonomic group of individual to be treated (e.g., non-human primate, primate, etc.), the capacity of the individual's immune system to synthesize antibodies, the degree of protection desired, the formulation of the vaccine, the treating doctor's assessment of the medical situation, and other relevant factors.
- the amount will fall in a relatively broad range which can be determined through routine trials.
- Kits [278] The invention also provides kits comprising one or more containers of compositions of the invention.
- Compositions can be in liquid form or can be lyophilized, as can individual antigens.
- Suitable containers for the compositions include, for example, bottles, vials, syringes, and test tubes.
- Containers can be formed from a variety of materials, including glass or plastic.
- a container may have a sterile access port (for example, the container may be an intravenous solution bag or a vial having a stopper pierceable by a hypodermic injection needle).
- the kit can further comprise a second container comprising a pharmaceutically- acceptable buffer, such as phosphate-buffered saline, Ringer's solution, or dextrose solution. It can also contain other materials useful to the end-user, including other buffers, diluents, filters, needles, and syringes.
- a pharmaceutically- acceptable buffer such as phosphate-buffered saline, Ringer's solution, or dextrose solution. It can also contain other materials useful to the end-user, including other buffers, diluents, filters, needles, and syringes.
- the kit can also comprise a second or third container with another active agent, for example an antibiotic '
- the kit can also comprise a package insert containing written instructions for methods of inducing immunity against S. pyogenes or for treating S. pyogenes infections.
- the package insert can be an unapproved draft package insert or can be a package insert approved by the Food and Drug Administration (FDA) or other regulatory body.
- FDA Food and Drug Administration
- E. coli expression vectors pET and pGex were used to express GAS antigens either as HIS-tagged proteins or as HIS-GST fusions, i.e., an amino- terminal histidine tag and a carboxy terminal GST (wherever "GST" is not specified, only the HIS-tagged antigen was expressed. See Figures 4 A and 4B. In some cases, urea was used to solubilize the antigen. Briefly, PCR reactions were performed to amplify GAS antigen coding sequences, then the amplified products were digested overnight. The digested PCR products were then purified and ligated overnight with either pET or pGEX vectors. E.
- coli strains BL21(DE3) and BL21 were transformed with the pGEX and pET ligation products, respectively, plated, and incubated at 37 °C. Two PCR-positive colonies from each transformation were inoculated and grown overnight. Protein expression in these clones was induced with IPTG and assessed by SDS-PAGE analysis of E. coli extracts. Glycerol batches of the GAS antigen-expressing clones were then prepared.
- Genomic DNAs were prepared from GAS strains of different M types, and the complete protein sequence of the full-length GAS40 antigen was obtained. The results are shown in FIG. 1 and in Table 5.
- mice were immunized with various GAS40 antigens. Sera of these mice were used for FACS analysis on native bacterial cells of various S. pyogenes strains. The ability of these antisera to detect GAS40 protein on the bacterial cell surface was assessed by calculating the difference between the FACS value obtained with the immune sera and that obtained with the preimmune sera. The results are shown in FIGS. 3-11.
- FIG. 3 is the GAS40 protein having the amino acid sequence shown in SEQ ID NO: 17 and is encoded by a nucleotide sequence derived from the genomic sequence of the SF 370 strain.
- GST40 (FIGS. 4A-4B) is a hybrid GAS40 antigen with glutathione-S-transferase in place of the leader sequence at its N terminus.
- "40a” (FIGS. 5A-5B) is a GAS40 antigen with a HIS tag but without the leader and hydrophobic sequences (SEQ ID NO:235).
- nucleotide sequence shown in SEQ ID NO:892 was cloned into vector pET21b+ (Novagen) using the Ndel and Notl restriction sites. The carboxyl terminal 12 amino acids were introduced with the vector. Codon 824 (AGA in the wild-type sequence) was mutagenized to CGT.
- "4OaCH” (FIGS. 6A-6B) is a GAS40 antigen with a HIS tag at its carboxyl terminus and two additional amino acids at its N terminus. Three nucleotide changes introduced with the cloning of its coding sequence into the pSM214gCH shuttle vector (at nucleotides 198, 222, and 1115). One amino acid (amino acid 372) was changed from Phe to Ser.
- "40/117" (FIG. 7) is a GAS40 hybrid antigen in which the GAS40 protein is placed to the N-terminus of the GASl 17 protein and a HIS tag is added to the C terminus of the GASl 17 protein (SEQ ID NO:234).
- SEQ ID NO:891 is a nucleotide sequence which codes for this antigen.
- 117/40 is a GAS40 hybrid antigen in which GASl 17 to GAS40 by the linker sequence YASGGGS (SEQ ID NO:278). Its amino acid sequence is shown in SEQ ID NO:233; a coding sequence is shown in SEQ ID NO:890.
- "4OaNH” (FIGS. 10A- 10B) is a GAS40 antigen with the HIS tag at its N terminus. Its coding sequence was cloned into the E. coli/B. subtilis expression shuttle vector pSM214gNH, which uses a constitutive promoter instead of an IPTG inducible promoter. The amino terminal nine amino acids are introduced with the cloning. It contains two nucleotide changes which most likely occurred during PCR amplification and do not result in amino acid changes (nucleotides 356 and 1547).
- Groups of 10 CDl female mice aged between 6 and 7 weeks are immunized with two or more GAS antigens of the invention, (20 ⁇ g of each recombinant GAS antigen), suspended in 100 ⁇ l of suitable solution. Each group received 3 doses at days 0, 21, and 45. Immunization was performed through intra-peritoneal injection of the protein with an equal volume of Complete Freund's Adjuvant (CFA) for the first dose and Incomplete Freund's Adjuvant (IFA) for the following two doses. In each immunization scheme negative and positive control groups were used. See FIG. 13.
- CFA Complete Freund's Adjuvant
- IFA Incomplete Freund's Adjuvant
- mice were immunized with E. coli proteins eluted from the purification columns following processing of total bacterial extract from an E. coli strain containing either the pET21b or the pGEX-NNH vector (thus expressing GST only) without any cloned GAS ORF (groups can be indicated as HisStop or GSTStop respectively).
- mice were immunized with purified GAS M cloned from either GAS SF370 or GAS DSM 2071 strains (groups indicated as 192SF and 192DSM respectively).
- DSM 2071 was grown at 37° C in THY broth until the OD 600 was 0.4. Bacteria were pelletted by centrifugation, washed once with PBS, suspended, and diluted with PBS to obtain the appropriate concentration of bacteria/ml and administered to mice by intraperitoneal injection. Between 50 and 100 bacteria were given to each mouse, as determined by plating aliquots of the bacterial suspension on 5 THY plates. Animals were observed daily and checked for survival. The results obtained after intraperitoneal challenge are shown in FIG. 22 and Table 6A. The results obtained after intranasal challenge are shown in Table 6B (note the increased survival rate).
- GAS40 appeared to be particularly promising, giving protection efficacy above 50% against mouse challenge with a heterologous GAS strain.
- GAS were grown in THYE medium to mid-log phase, washed and resuspended in PBS.
- Formvar-carbon-coated nickel grids were floated on drops of GAS suspensions for 5 min, fixed in 2% PFA for 5 min, and placed in blocking solution (PBS containing 1% normal rabbit serum and 1% BSA) for 30 min.
- the grids were then floated on drops of primary antiserum diluted 1 :20 in blocking solution for 30min at RT, washed, and floated on secondary antibody conjugated to 10 nm gold particles diluted 1:10 in 1% BSA for 30 min.
- Bacterial cells were grown in THY medium until they reached the middle exponential phase (OD600 0.4) at 37°C. Bacteria were washed twice in chilled saline solution and suspended in MEM medium with the volume being adjusted for each strain depending on the amount of bacteria used. Bacterial cells were kept in ice until used in the assay.
- PMN were prepared from buffy coats of heparinized blood from healthy volunteers. The buffy coat was incubated for 30 minutes in a solution containing dextran, NaCl and Heparin (rate 1:1). After incubation the supernatant, enriched in leukocytes, was removed, transferred to a clean tube, and centrifuged at 700xg for 20 minutes. A short wash in water was performed to break red blood cells, and then a solution of NaCl was added to restore the appropriate salt concentration. After this step cells were centrifuged, washed, and suspended in MEM at a suitable concentration.
- Cell extracts were obtained by growing the cells at 37°C to OD 600 0.32 in 10 ml of THY. The cell pellet was washed in PBS, resuspended in lysis buffer (40% sucrose, 0.1 M KPO 4 pH 6.2, MgCl 2 10 mM, and Roche's COMPLETETM EDTA-free), and digested for 3 hours with 400U of mutanolysin and 2 mg/ml lysozyme. The insoluble fraction was separated by centrifugation and the supernatant was analyzed.
- lysis buffer 50% sucrose, 0.1 M KPO 4 pH 6.2, MgCl 2 10 mM, and Roche's COMPLETETM EDTA-free
- GAS40 is surface-exposed across different M strains
- FIG. 23 demonstrates that GAS40 is surface exposed across different M strains. The data were obtained using convalescent sera from patients with or recovered from a GAS infection.
- GAS40N does not react with four anti-GAS40 monoclonal antibodies
- FIG. 24 demonstrates that four different monoclonal antibodies against GAS40 (28A8, 29G2, 2E4 and 4E6) do not react with the GAS40N part of the molecule.
- the FACS graphs shown in FIG. 24 demonstrate that the four GAS40 monoclonal antibodies do not appear to bind to any surface exposed molecules on the M23 strain (no shift in graph peaks), whereas at least three of the four GAS40 monoclonal antibodies appear to bind to surface exposed molecules on the M4 strain. This result may be explained by the fact that the M23 is a capsulated strain and so the capsule is blocking the surface exposed GAS40 molecules.
- peptides were separated by two-dimensional nano-liquid chromatography (2-D LC), spotted directly onto a MALDI target, and analyzed by MALDI TOF-TOF (off-line coupled 2-D LC/MALDI MS/MS).
- the chromatographic system (Dionex, Amsterdam, The Netherlands) consisted of a FAMOS autosampler, an UltiMate micropump with UV detector and a Switchos column-switching device, as described in (Mitulovic et al, Proteomics. 2004 Sep;4(9):2545-57).
- the UltiMate pump was set to operate at a flow rate 300 nL/min.
- the flow of the Switchos loading pump which was used to carry the sample from the sample loop to the first column, was set to operate at 30 ⁇ L/min.
- peptide separation was performed as follows. In the first dimension, peptides were loaded on a strong cation exchange (SCX) column (10 cm x 320 ⁇ m i. d.) and eluted isocratically by applying 5 increasing NaCl concentrations (0.01, 0.05, 0.1, 0.5 and 1 M). In the second dimension, peptides were separated by a reversed phase Cl 8 analytical column (15 cm x 75 ⁇ m i. d., Cl 8 PEPMAP 100TM, 3 ⁇ m, 100 A) through a C18 trap column (PEPMAPTM C18 ⁇ -precolumn, 300 ⁇ m i.d. x 1 mm, Dionex). Peptides were eluted with a 45-min gradient from 5 to 50% of 80% ACN in 0.1% formic acid at a flow rate of 300 nl/min.
- SCX strong cation exchange
- Mass spectrometry analysis was performed automatically with an Ultraflex MALDI TOF-TOF instrument, under the control of the WARP LC software (Bruker Daltoniks). First, MS spectra of all the spotted fractions were acquired for peak selection and further MS/MS spectra acquisition of selected peaks. Searching and identification of peptides were performed in batch mode with a licensed version of MASCOT in a local database.
- the MASCOT search parameters were: (1) species: & pyogenes strain SF370; (2) allowed number of missed cleavages (only for trypsin digestion): 6; (3) variable post- translational modifications: methionine oxidation; (4) peptide tolerance: ⁇ 300 ppm; (5) MS/MS tolerance: ⁇ 1.5 Da and (6): peptide charge: +1.
- peptides were separated by nanoLC-MS/MS on a CapLC HPLC system (Waters, Milford, MA, USA) connected to a Q-ToF Micro ESI mass spectrometer equipped with a nanospray source (Waters, Milford, MA, USA). Samples were resuspended in 5% (v/v) ACN, 0.1% (v/v) formic acid (Solvent A) and loaded on a C18 trap column (300 ⁇ m i.d. x 5 mm, LC Packings, Amsterdam, The Netherlands). After 3 min, the flow was switched to an Atlantis Cl 8 NanoEase column (lOO ⁇ m i.d. x 100mm, Waters, Milford, MA, USA) and a solvent gradient was started. The applied flow rate was of 4 ⁇ L/min and a flow splitter was set up to direct a nanoflow of 400 nL/min through the analytical column.
- Bacteria were washed once by adding 100 ⁇ l of PBS-0.1% BSA, centrifuged for 10 minutes at 3,500 x g, at 4°C, resuspended in 200 ⁇ l of PBS-0.1% BSA, and centrifuged again. The bacteria were resuspended in 10 ⁇ l of phycoerythrin-conjugated goat anti- mouse IgG F(ab') 2 fragment (Jackson Immunoresearch Laboratories Inc.) in PBS-0.1% BSA to a final dilution of 1:100, and incubated on ice for 30 minutes in the dark. Bacteria were then washed by adding 180 ⁇ l of PBS-0.1% BSA, and centrifuged for 10 minutes at 3,500 x g, at 4°C, and resuspended in 200 ⁇ l of PBS.
- the bacterial resuspension was then analyzed using a FACS Calibur instrument (Becton Dickinson, Mountain View, CA USA) 5 and 10,000 events were acquired. Data were analyzed using Cell Quest Software (Becton Dickinson) by drawing a morphological dot plot (using forward and side scatter parameters) on bacterial signals. A histogram plot was then created on FL2 intensity of fluorescence log scale.
- the 72 proteins could be grouped into 4 major families: the cell wall-anchored protein family containing LPXTG (SEQ ID NO:931)-like motifs (12 proteins), the lipoprotein family (11 proteins), the transmembrane protein family carrying one or more transmembrane spanning regions (37 proteins) and the family of secreted proteins (8 proteins).
- the total number of GAS SF370 proteins which could be attributed to each of these protein families are 17, 28, 489 and 67, respectively. Therefore, while a large proportion of all predicted cell-wall anchored proteins and lipoproteins were identified, less than 7% of secreted proteins and membrane spanning proteins were found exposed on the cell surface.
- Proteinase K released peptides corresponding to 30 proteins; 19 proteins were identified by both enzymes. Proteinase K was especially useful for the recovery of peptides corresponding to membrane proteins with a high number of transmembrane domains (TMD): e.g., proteins NT01SP0454, NT01SP0906 and NT01SP1664, with 7, 10 and 4 TMD, respectively, which were not identified from trypsin digestion.
- TMD transmembrane domains
- FIGS. 25 A-B and 26A-B show the high coverage obtained after digestion with both proteases of the cell-wall protein NTO ISP 1652 (GAS 190; SEQ ID NO: 117).
- GAS 190 contains the anchoring sequence LPXTG (SEQ ID NO:931). It has been previously described as "OrfX" and belonging to the vir regulon, which organizes the expression of several bacterial virulence factors under the control of the Mga regulator.
- the function of GAS 190 is, so far, unknown, although it may be a fibronectin-binding protein.
- the wall- associated region can cover from about 50 to as many as 125 amino acid residues.
- FIGS. 25A SEQ ID NOS:932-949) and 25B show the coverage of the GAS190 protein sequence (50.6%) by the 35 proteinase K-generated peptides.
- the zone lacking coverage by tryptic peptides shown in FIG. 25B between arrows) was widely represented under proteinase K digestion, although a high degree of redundant information was obtained.
- Most of the peptides identified corresponded to the most exposed region of the protein, i.e., the first half of the sequence from the N-terminus.
- VDGIPPISLTQK SEQ ID NO: 969
- Peptides having more than one trypsin-missed cleavage site (6 out of 11) were relatively abundant.
- the peptide IKTAPDKDKLLFTYHSEYMTAVK SEQ ID NO:970
- Proteins containing cell wall-anchoring motifs showed the highest degree of coverage. Proteins with at least one predicted TMD (according to PSORT prediction) were identified, in general, from a low number of peptides (Table 9).
- FIG. 107 is an electron micrograph showing membrane- delimited structures produced upon penicillin treatment.
- Proteins identified by this method are shown in Table 8. The majority of the identified proteins are surface-exposed proteins (secreted, membrane-bound, or lipoproteins). No cell wall proteins were observed either in this fraction or in the membrane-delimited structures. Thus, most of the protein content of the membrane-delimited structures is of potential interest for vaccine development.
- PBS phosphate-buffered saline
- Bacteria were re-suspended in 20 ml of 6M guanidinium (urea or SDS also could be substituted for guanidinium), 200 mM Tris HCl and disrupted with 3 cycles at 1.7 kBar in a Basic Z 0.75V Model Cell Disrupter equipped with an "one shot head" (Constant System Ltd, Northants, England). The lysate was then centrifuged at 20,000 x g for 20 minutes at 4°C. The resulting supernatant was filtered through a 0.22 ⁇ m membrane and spun in an ultracentrifuge at 200,000 x g for 2 hours at 4°C. The pellet was washed with PBS (200,000 x g for 30 minutes at 4°C).
- GAS is surrounded by a hyaluronic acid-based capsule whose thickness can vary from strain to strain.
- Capsule plays an important role in bacterial virulence, and in general the highly capsulated strains are the most virulent strains. It is expected that the number of proteins with accessible external domains will depend on the thickness of the surrounding capsule. To verify that, we characterized the surfome of the highly capsulated strain M3.
- NTOl SP 1255 a second putative cell division protein homologous to FtsZ
- the four peptides of NTOl SP 1255 are located within the C-terminal part of the molecule, a region that in Bartonella bacilliformis was found immunogenic and surface-exposed (Padmalayam et al, J. Bacteriol. 179, 4545-52, 1997).
- TMR transmembrane regions
- NT01SP0947 has two TMRs the second of which with a very poor PSORT score (-0.32 as opposed to -8.12 of the first TMR). If the second TMR were in fact not real, the C- terminal part of the molecule, which carries a typical sortase domain, would be exposed on the surface, which would be consistent with the sortases mechanism of action (Paterson & Mitchell, Trends Microbiol. 12, 89-95, 2004).
- NTO ISPO 154 a putative glycine-betaine binding permease protein, is predicted to have 6 TMRs, one of which with a poor score. Again, if the weak TMR is neglected, the topological organization would change and the C-terminal region, where the two MS/MS identified peptides fall, would become surface-exposed. Indeed, polyclonal antibodies against the C-terminal domain of the protein efficiently bound GAS SF370 whole cells when tested by FACS.
- CFUs colony forming units
- NT01SP0336 which corresponds to GAS 57 in the SF370 serotype, is a putative cell envelope proteinase carrying a typical cell anchoring LPXTG (SEQ ID NO:931) motif.
- GAS57 provided little or no protection in a mouse model in which intraperitoneal immunization was followed by intraperitoneal challenge.
- GAS40 was protective in both models, even though the survival rate in the intranasal challenge model was higher.
- mice in the negative control group were immunized with E. coli proteins eluted from the purification columns following processing of total bacterial extract from an E. coli strain containing either the pET21b or the pGEX-NNH vector (thus expressing GST only) without any cloned GAS ORF (indicated as HisStop or GSTStop, respectively).
- mice in the positive control groups were immunized with purified GAS M cloned from either GAS SF370 or GAS DSM 2071 strains (groups indicated as 192SF and 192DSM respectively).
- mice Serum from each mouse was collected before the first immunization and two weeks after the last immunization. The sera of mice in each group were pooled. Mice were infected with one of GAS strains 2071 (M23), 3348 (Ml), or 2728 (M12) about a week after the last immunization. For infection, GAS strains were grown at 37° C in THY broth until OD600 0.4. Bacteria were collected by centrifugation, washed once with PBS, suspended, and diluted with PBS to obtain the appropriate concentration of bacteria/ml and administered to mice by intraperitoneal injection. Between 50 and 100 bacteria were given to each mouse, as determined by plating aliquots of the bacterial suspension on 5 THY plates. Animals were observed daily and checked for survival.
- Domains GAS35, GAS414, GAS437, GAS438, GAS461, GAS465-2, GAS469, GAS472, GAS473, GAS475, GAS478, GAS495, GAS538, GAS553, GAS561, GAS577-2, GAS591, GAS593, GAS636, GAS643, GAS649, and GAS663 are surface-exposed on the surface of at least two of the three GAS strains tested. Domains GAS472, GAS473, and GAS553 are surface-exposed on all three of the tested GAS strains.
- FIG. 112 shows a diagram where the prevalent immunoreactive antigens are mapped and clustered according to signal intensity and response frequency for each of the 6 donors.
- Each chip contains at least one immunoglobulin and two BSA (Cy3 and Cy5-labelled, Amersham Bioscences) standard curves. After each printing process, each slide was scanned to check the signals of the Cy3 and Cy5 -labeled BSA curves.
- Fluorescence signals were detected with a ScanArray 5000 Unit (Packard, Billerica, MA, USA) at high resolution (10 ⁇ m pixel size) and quantified with the ImaGene 6.0 software (Biodiscovery Inc, CA, USA) Elaboration of the collected data was performed using software which normalizes the data by interpolating the least-mean squares of the Ig controls to a sigmoid curve. The titer value corresponding to each experimental intensity signal is then referred to such sigmoid and normalized to a theoretic sigmoid curve extending over the whole dynamic range of the scanner.
- GAS proteins identified after protease digestion of the bacterial cell surface (LPXTG, SEQ ID NO:931; RGD LPXTG, SEQ ID NO:932)
Abstract
Description








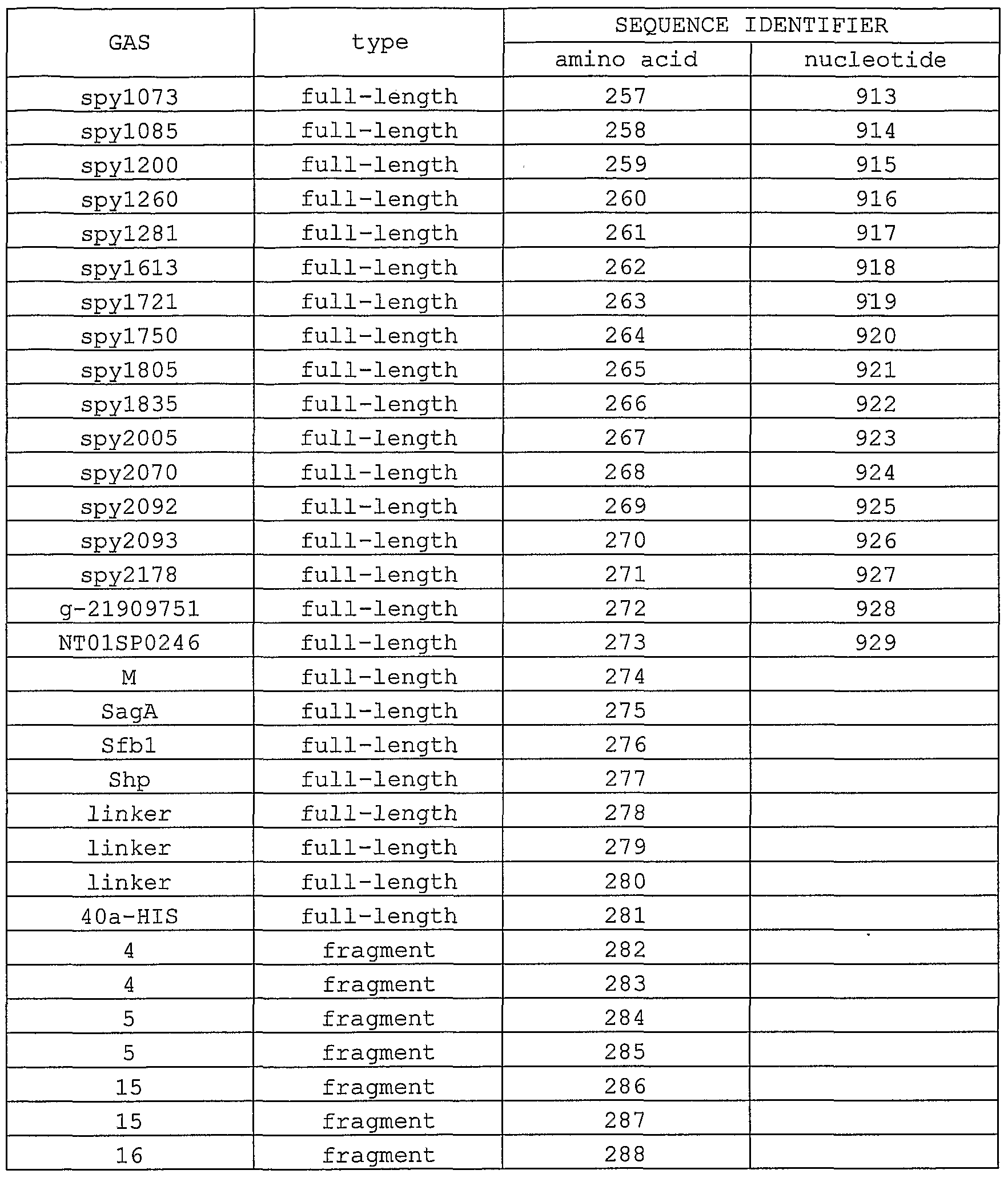






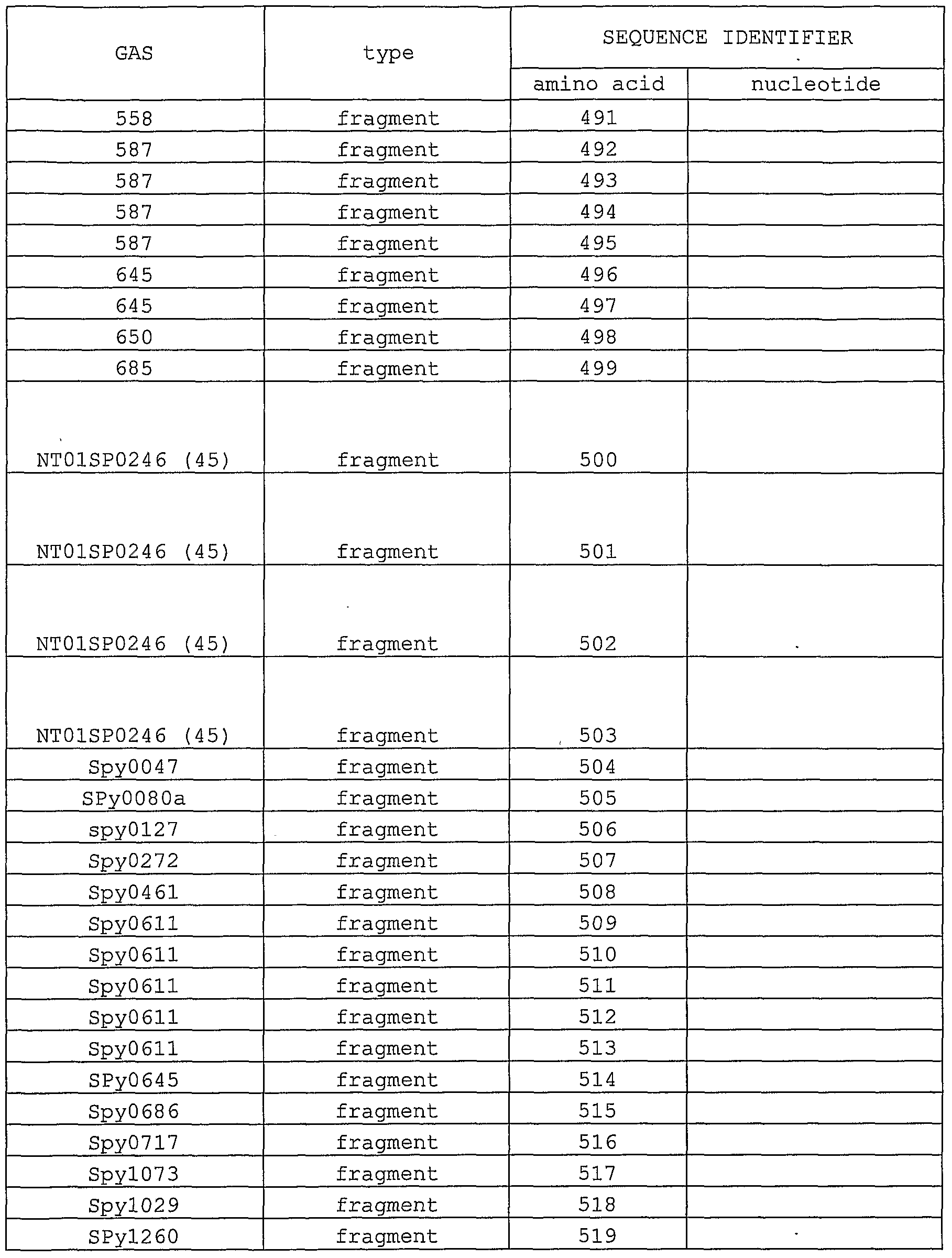


























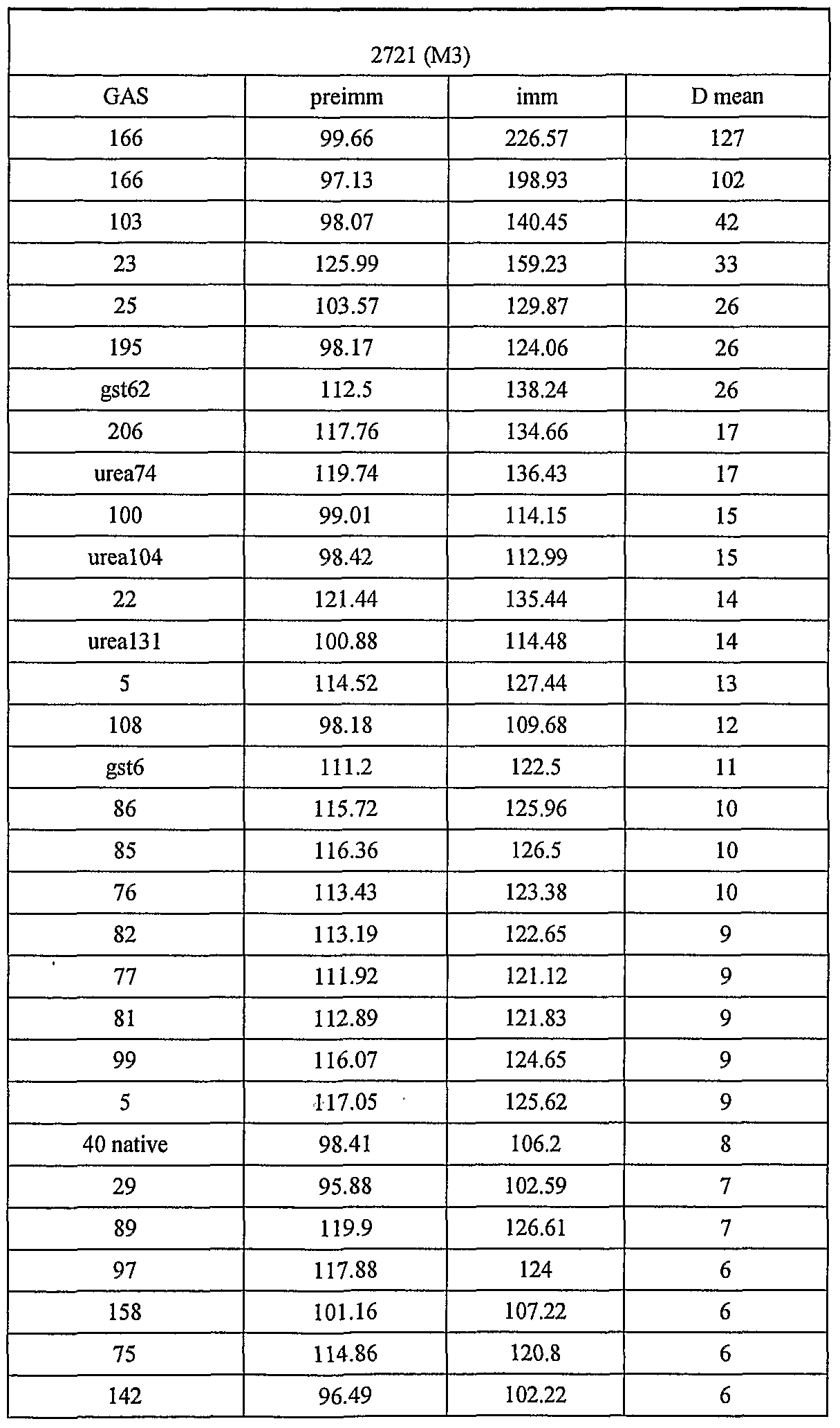


















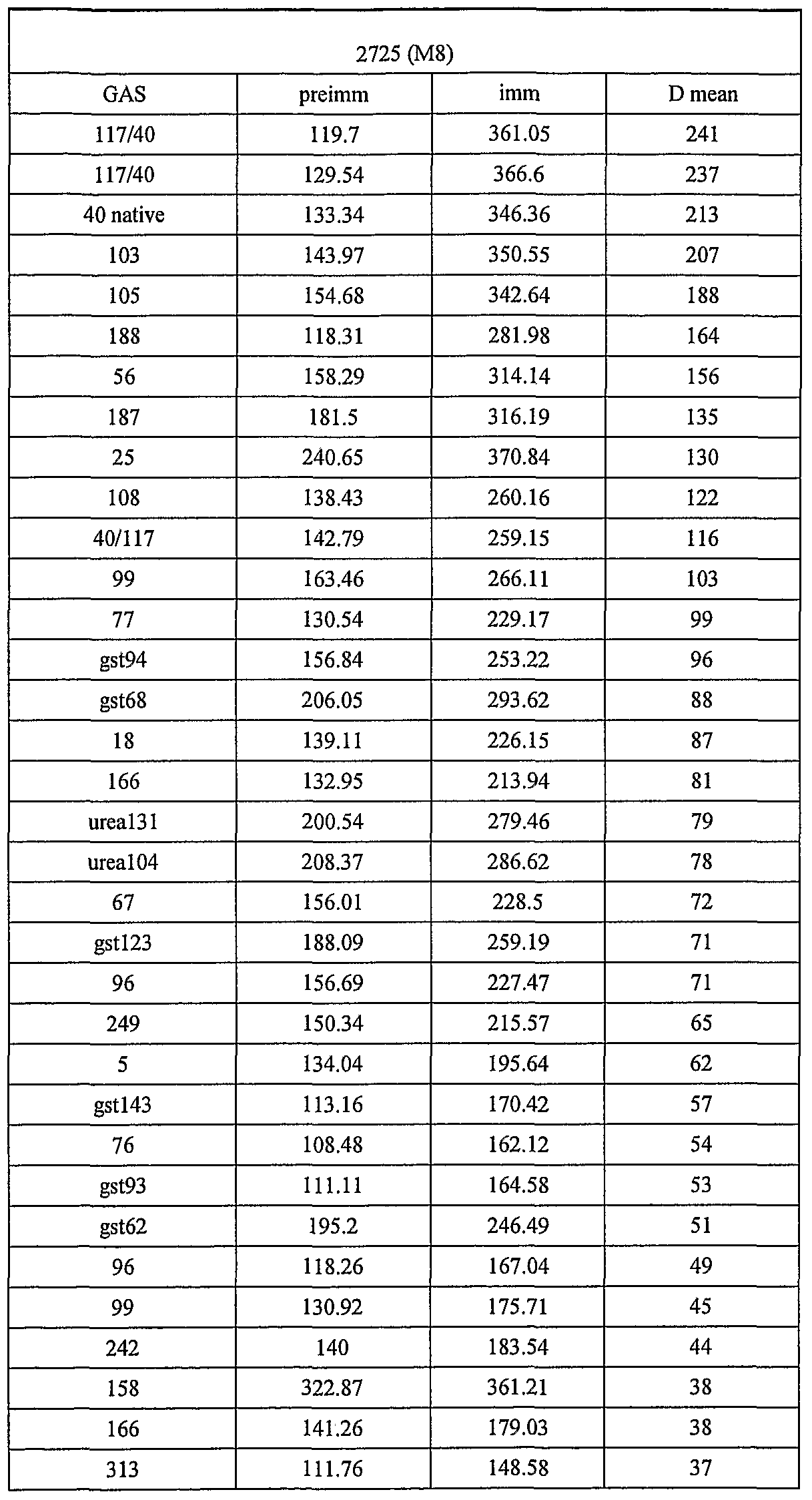



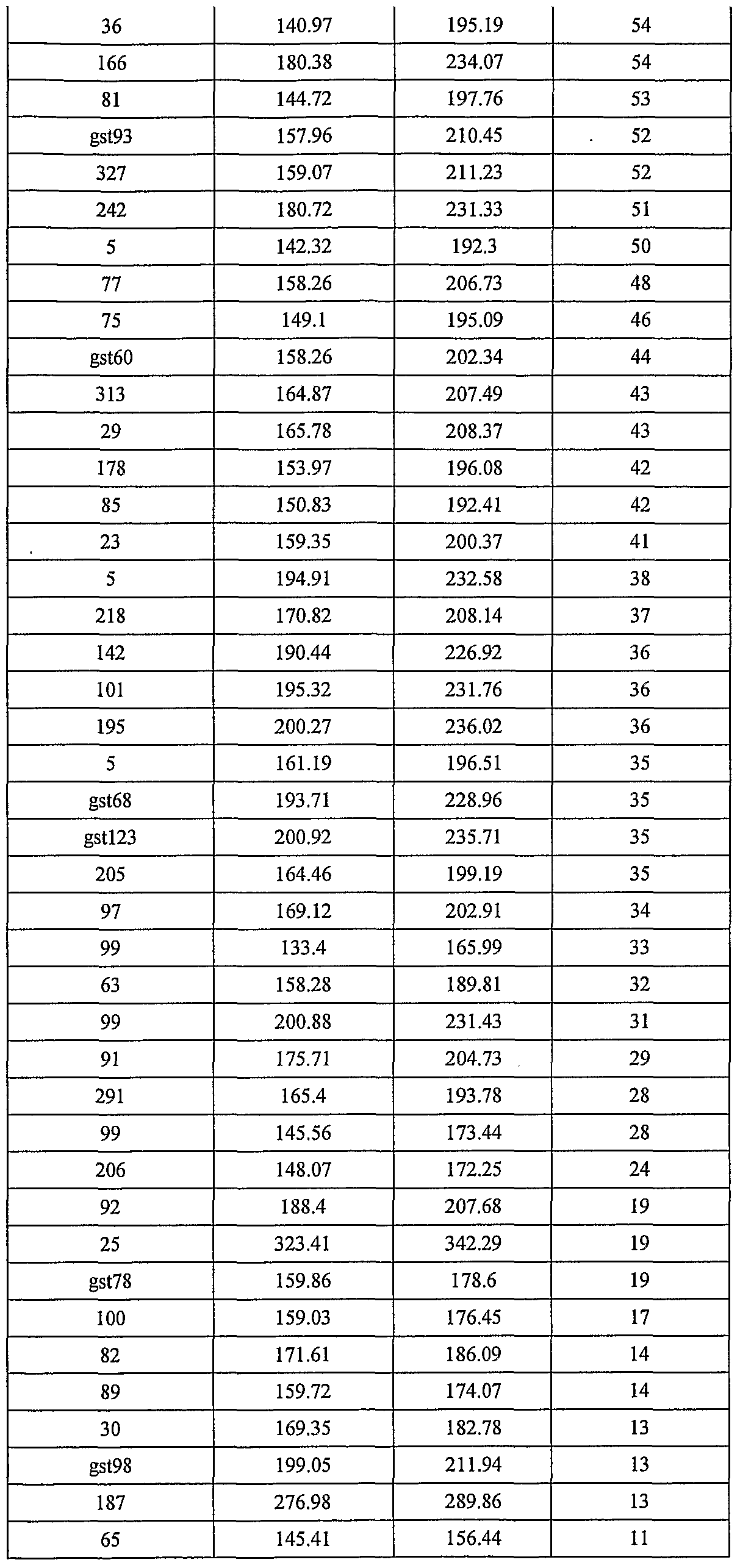
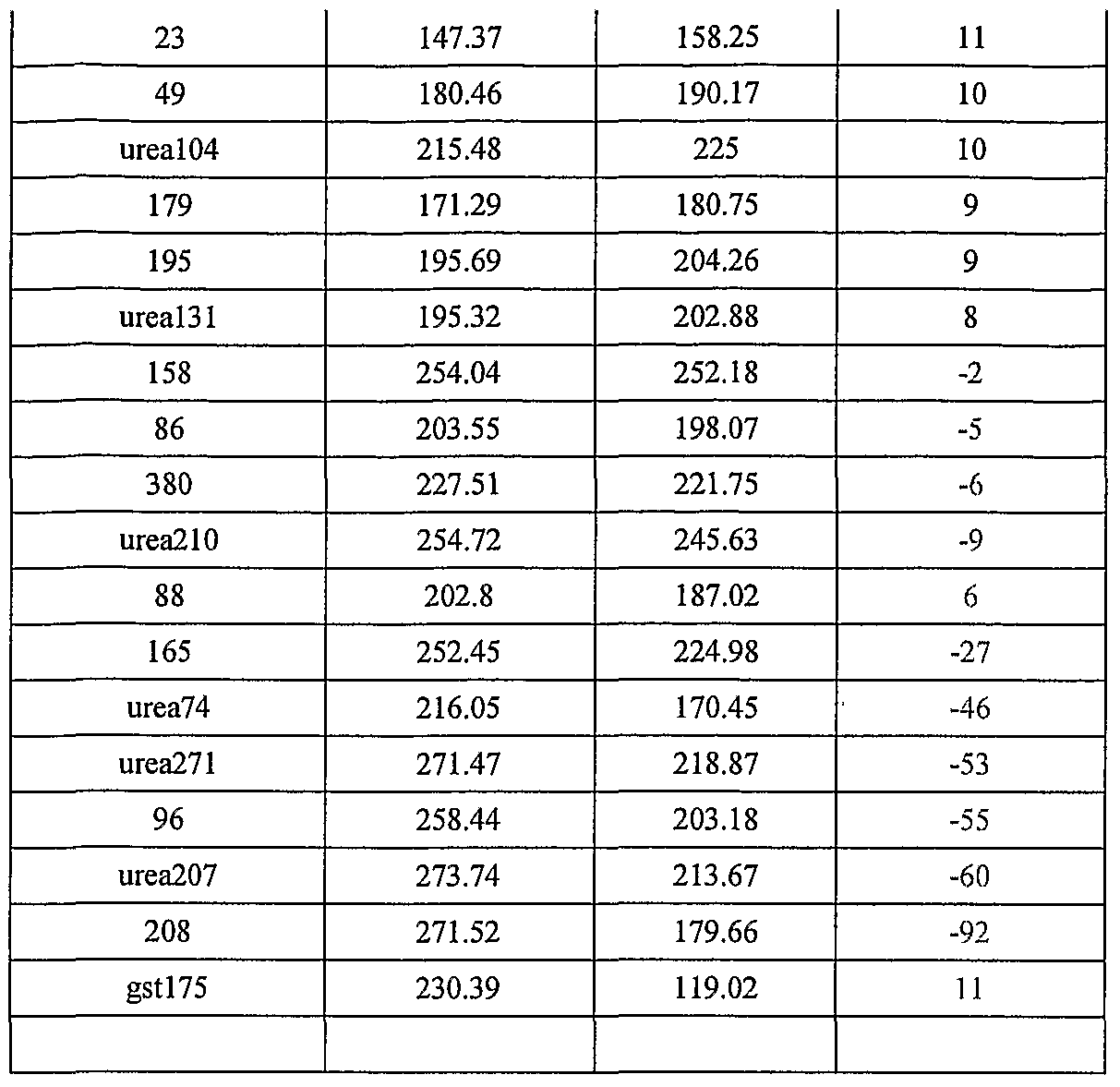
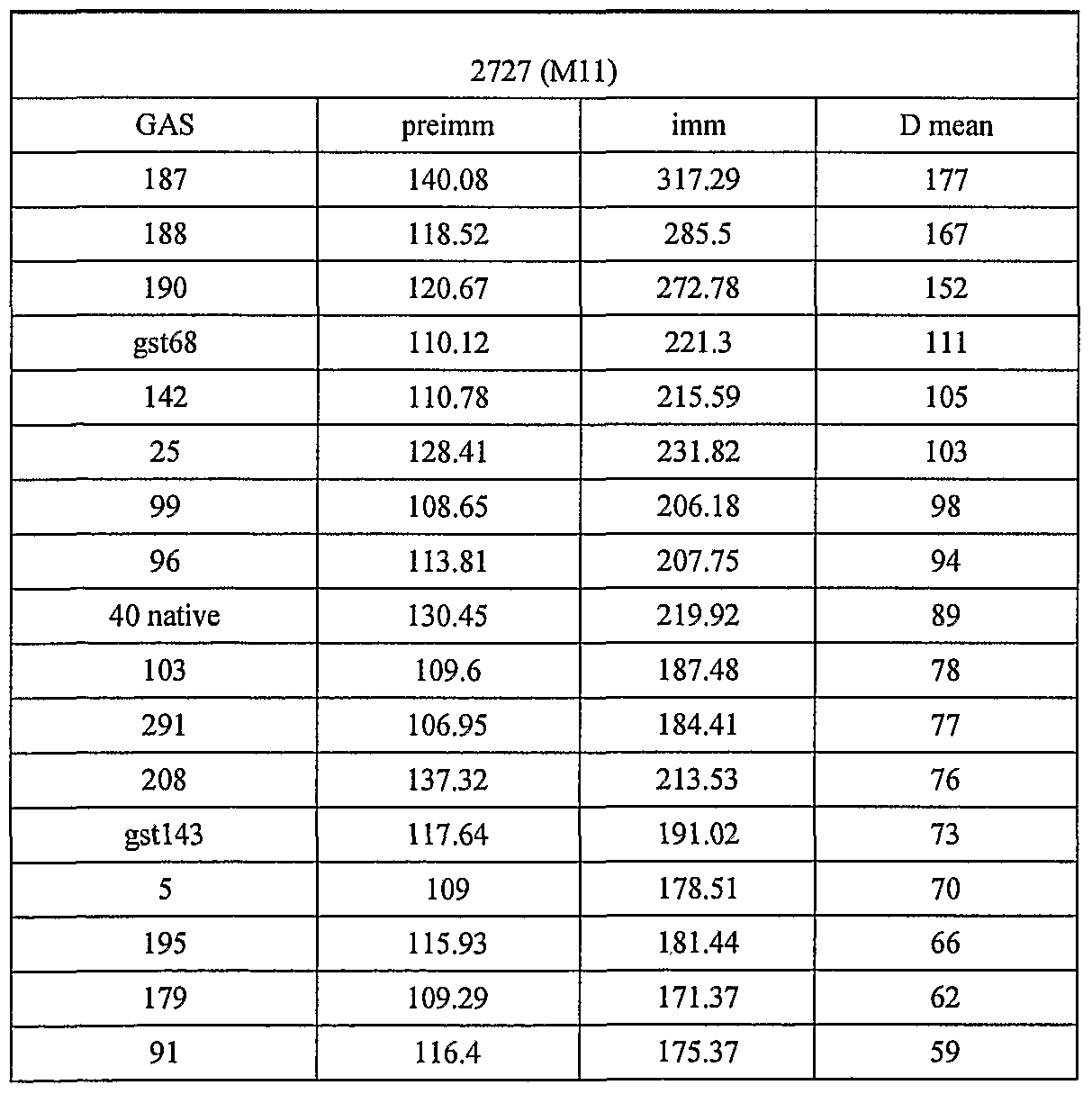




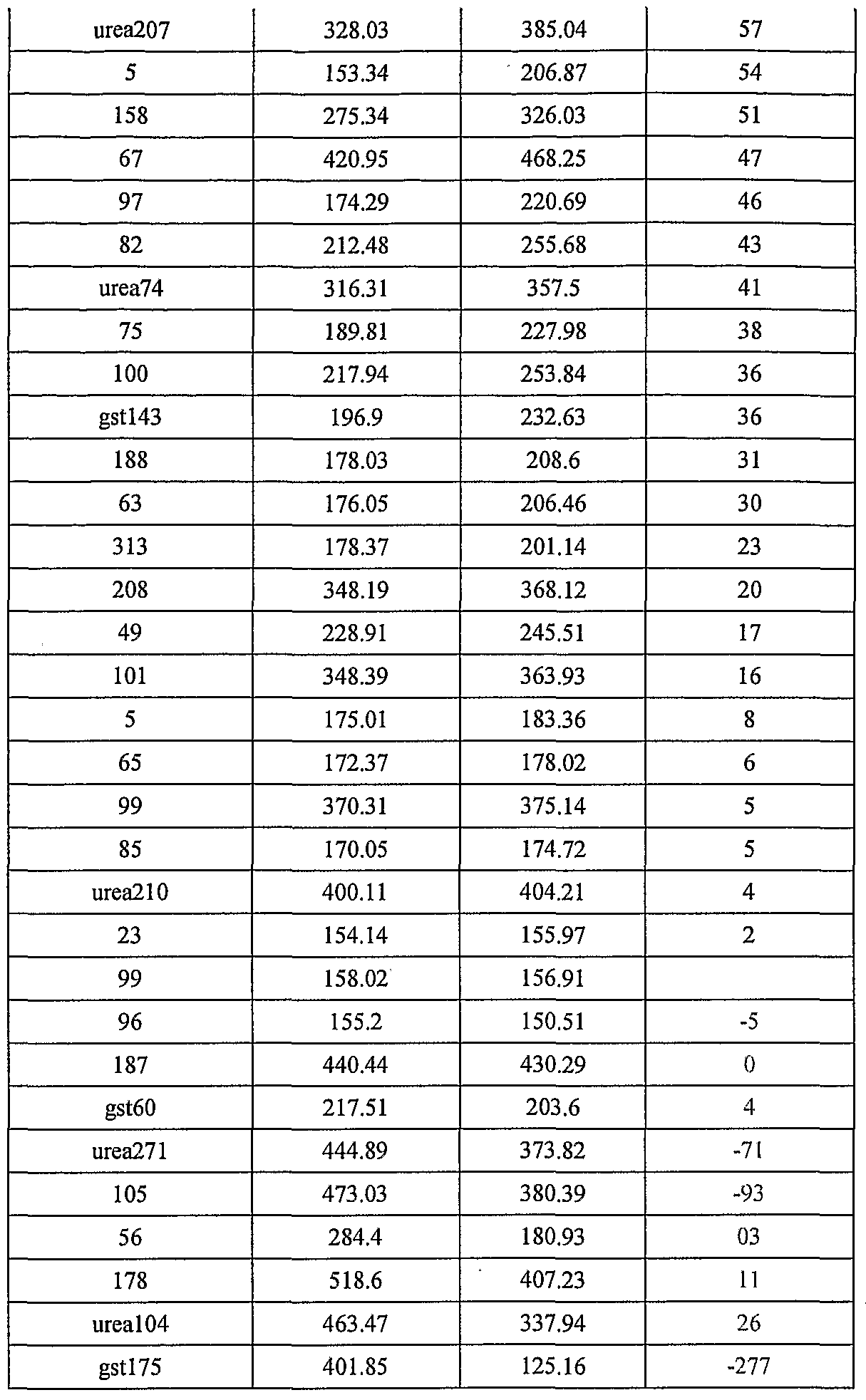














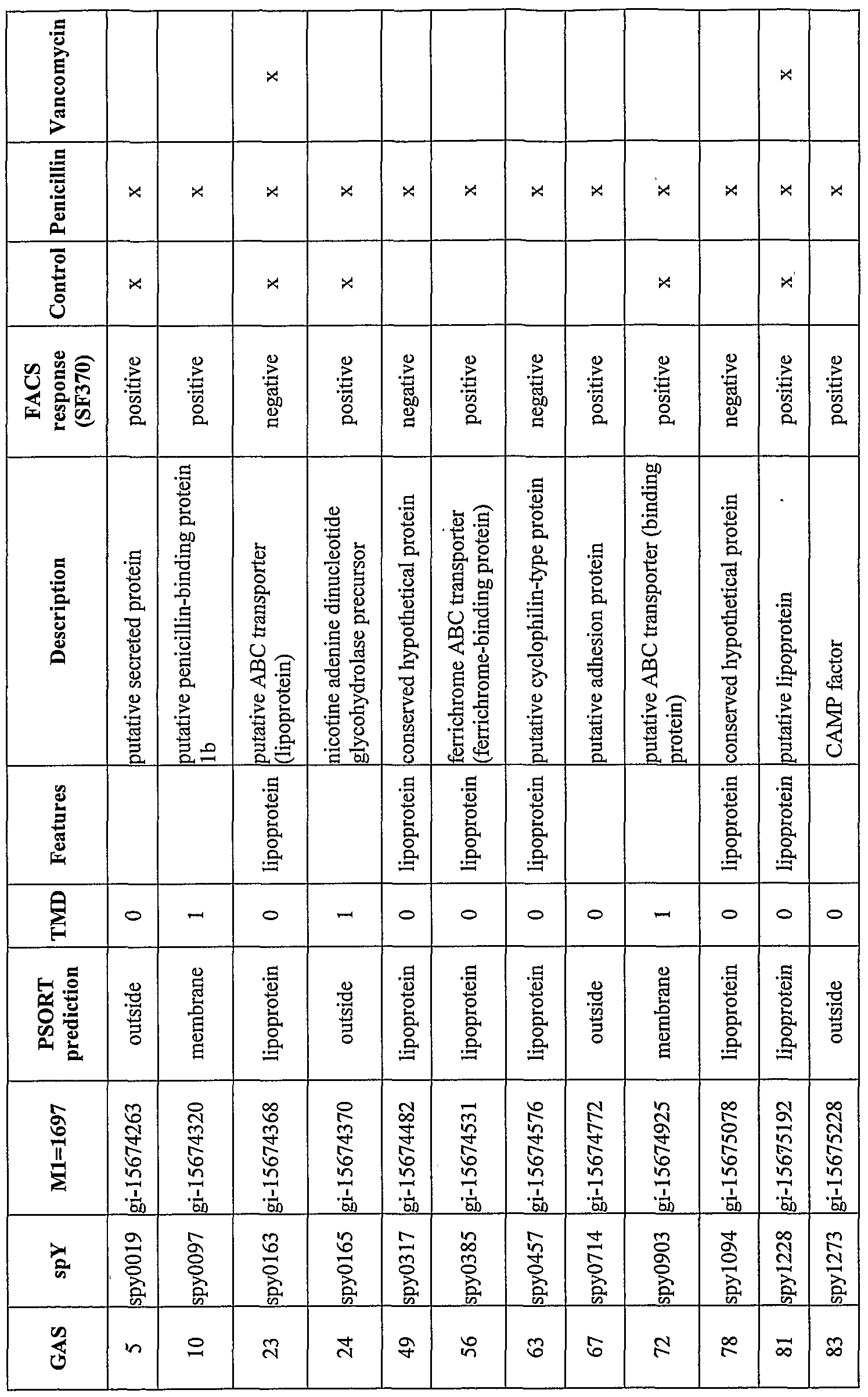





















Claims
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA2583803A CA2583803C (en) | 2004-10-08 | 2005-10-11 | Immunogenic and therapeutic compositions for streptococcus pyogenes |
| JP2007535816A JP2008544949A (en) | 2004-10-08 | 2005-10-11 | Immunostimulatory and therapeutic compositions for pyogenic streptococci |
| AU2005294275A AU2005294275B2 (en) | 2004-10-08 | 2005-10-11 | Immunogenic and therapeutic compositions for Streptococcus pyogenes |
| US11/792,038 US7838010B2 (en) | 2004-10-08 | 2005-10-11 | Immunogenic and therapeutic compositions for Streptococcus pyogenes |
| EP05817357A EP1807446A2 (en) | 2004-10-08 | 2005-10-11 | Immunogenic and therapeutic compositions for streptococcus pyogenes |
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US61685404P | 2004-10-08 | 2004-10-08 | |
| US60/616,854 | 2004-10-08 | ||
| US65273605P | 2005-02-15 | 2005-02-15 | |
| US60/652,736 | 2005-02-15 | ||
| US70112105P | 2005-07-21 | 2005-07-21 | |
| US60/701,121 | 2005-07-21 | ||
| US70520905P | 2005-08-04 | 2005-08-04 | |
| US60/705,209 | 2005-08-04 |
Publications (3)
| Publication Number | Publication Date |
|---|---|
| WO2006042027A2 true WO2006042027A2 (en) | 2006-04-20 |
| WO2006042027A3 WO2006042027A3 (en) | 2006-08-24 |
| WO2006042027A8 WO2006042027A8 (en) | 2006-11-02 |
Family
ID=35720938
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2005/036009 WO2006042027A2 (en) | 2004-10-08 | 2005-10-11 | Immunogenic and therapeutic compositions for streptococcus pyogenes |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US7838010B2 (en) |
| EP (1) | EP1807446A2 (en) |
| JP (2) | JP2008544949A (en) |
| AU (1) | AU2005294275B2 (en) |
| CA (1) | CA2583803C (en) |
| WO (1) | WO2006042027A2 (en) |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008108830A2 (en) * | 2006-10-30 | 2008-09-12 | Novartis Ag | Immunogenic and therapeutic compositions for streptococcus pyogenes |
| WO2009060217A3 (en) * | 2007-11-06 | 2009-07-30 | Novartis Ag | Streptococcus pyogenes classification |
| WO2009155484A3 (en) * | 2008-06-20 | 2010-03-11 | Wyeth Llc | Compositions and methods of use of orf1358 from beta-hemolytic streptococcal strains |
| JP2010512361A (en) * | 2006-12-13 | 2010-04-22 | ハンサ メディカル アクチボラゲット | Use of endoglycosidase EndoS to treat immunoglobulin G-mediated diseases |
| WO2010053986A1 (en) * | 2008-11-05 | 2010-05-14 | Wyeth | Multicomponent immunogenic composition for the prevention of beta-hemolytic streptococcal (bhs) disease |
| WO2010076618A1 (en) * | 2008-09-17 | 2010-07-08 | Novartis Ag | Combination gas vaccines and therapeutics |
| WO2010079464A1 (en) | 2009-01-12 | 2010-07-15 | Novartis Ag | Cna_b domain antigens in vaccines against gram positive bacteria |
| WO2010100627A1 (en) * | 2009-03-05 | 2010-09-10 | Novartis Ag | Diagnosis of neuropsychiatric and behavioural disorders |
| US20100285047A1 (en) * | 2007-07-03 | 2010-11-11 | Kowthar Salim | Therapeutics and diagnostics for group a streptococci |
| WO2010136897A2 (en) | 2009-05-28 | 2010-12-02 | Novartis Ag | Expression of recombinant proteins |
| EP2287188A1 (en) * | 2006-07-07 | 2011-02-23 | Intercell AG | Small Streptococcus pyogenes antigens and their use |
| WO2011098772A1 (en) * | 2010-02-11 | 2011-08-18 | Isis Innovation Limited | Peptide tag systems that spontaneously form an irreversible link to protein partners via isopeptide bonds |
| US8137673B2 (en) | 2000-10-27 | 2012-03-20 | Novartis Vaccines And Diagnostics, Inc. | Nucleic acids and proteins from Streptococcus groups A & B |
| RU2471497C2 (en) * | 2007-09-12 | 2013-01-10 | Новартис Аг | Mutant antigens gas57 and gas57 antibodies |
| US9056912B2 (en) | 2003-07-31 | 2015-06-16 | Novartis Vaccines And Diagnostics, Srl | Immunogenic compositions for Streptococcus pyogenes |
| WO2015169773A3 (en) * | 2014-05-07 | 2016-01-21 | Glaxosmithkline Biologicals S.A. | Mutants of spy0269 |
| WO2018199775A1 (en) * | 2017-04-26 | 2018-11-01 | Auckland Uniservices | Analytical and therapeutic methods and compositions, and uses thereof |
| WO2024083873A1 (en) | 2022-10-18 | 2024-04-25 | Glaxosmithkline Biologicals Sa | Vaccine |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU1686302A (en) * | 2000-12-21 | 2002-07-01 | Shire Biochem Inc | Streptococcus pyogenes antigens and corresponding dna fragments |
| US20100255002A1 (en) * | 2003-06-26 | 2010-10-07 | Chiron Corporation | Immunogenic compositions for chlamydia trachomatis |
| US8945589B2 (en) | 2003-09-15 | 2015-02-03 | Novartis Vaccines And Diagnostics, Srl | Immunogenic compositions for Streptococcus agalactiae |
| EP1807446A2 (en) | 2004-10-08 | 2007-07-18 | Novartis Vaccines and Diagnostics, Inc. | Immunogenic and therapeutic compositions for streptococcus pyogenes |
| WO2009022236A2 (en) | 2007-08-16 | 2009-02-19 | Tripep Ab | Immunogen platform |
| BRPI1100857A2 (en) * | 2011-03-18 | 2013-05-21 | Alexandre Eduardo Nowill | immunomodulatory agent and combinations thereof, their use and immunotherapeutic method for real time recontextualization, reprogramming and rebuilding of the immune system |
| WO2012174455A2 (en) * | 2011-06-17 | 2012-12-20 | University Of Tennessee Research Foundation | Group a streptococcus multivalent vaccine |
| CN106794236B (en) * | 2014-04-15 | 2021-09-03 | 格里菲斯大学 | Group A streptococcus vaccine |
| US10738338B2 (en) | 2016-10-18 | 2020-08-11 | The Research Foundation for the State University | Method and composition for biocatalytic protein-oligonucleotide conjugation and protein-oligonucleotide conjugate |
| TWI598360B (en) * | 2016-12-19 | 2017-09-11 | 義守大學 | Fsbm recombinant protein and use thereof |
| CN108379572A (en) * | 2018-01-25 | 2018-08-10 | 暨南大学 | A kind of streptococcus pyogenes HtsA protein vaccines and preparation method and application |
| CN110922456B (en) * | 2019-12-28 | 2023-02-24 | 重庆艾力彼生物科技有限公司 | Pseudomonas aeruginosa vaccine recombinant protein reaSBP-ExoU, and preparation method and application thereof |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004078907A2 (en) * | 2003-03-04 | 2004-09-16 | Intercell Ag | Streptococcus pyogenes antigens |
Family Cites Families (90)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4281061A (en) * | 1979-07-27 | 1981-07-28 | Syva Company | Double antibody for enhanced sensitivity in immunoassay |
| US4454121A (en) | 1982-07-27 | 1984-06-12 | The University Of Tennessee Research Corporation | Synthetic peptides corresponding to antigenic determinants of the M protein of Streptococcus pyogenes |
| US5098827A (en) | 1988-02-26 | 1992-03-24 | The University Of Florida | Novel bacterial markers for pathogenic group B streptococci |
| GB8827038D0 (en) | 1988-11-18 | 1988-12-21 | Kehoe M | Streptolysin o antigens & uses |
| US5354846A (en) | 1988-11-18 | 1994-10-11 | Michael Kehoe | Streptolysin O antigen derivatives, its production and uses |
| WO1990006951A1 (en) | 1988-12-16 | 1990-06-28 | De Staat Der Nederlanden Vertegenwoordigd Door De Minister Van Welzijn, Volksgezondheid En Cultuur | Pneumolysin mutants and pneumococcal vaccines made therefrom |
| GB2233977B (en) | 1989-01-04 | 1993-03-31 | Michael Kehoe | Cytolytic streptolysin o mutants and uses |
| US5821088A (en) | 1990-05-11 | 1998-10-13 | Siga Pharmaceuticals, Inc. | Use of gram-positive bacteria to express recombinant proteins |
| US6737521B1 (en) | 1990-05-11 | 2004-05-18 | The Rockefeller University | Delivery and expression of a hybrid surface protein on the surface of gram positive bacteria |
| US5391712A (en) | 1991-08-30 | 1995-02-21 | Beckman Instruments, Inc. | Non-hemolytic streptolysin O variants |
| US5378620A (en) | 1991-08-30 | 1995-01-03 | Beckman Instruments, Inc. | Streptolysin O derivatives |
| DE4240056A1 (en) | 1992-11-28 | 1994-06-01 | Boehringer Mannheim Gmbh | Streptolysin O peptide antigens and method for the determination of streptolysin antibodies |
| ES2217258T3 (en) | 1993-02-01 | 2004-11-01 | Beckman Coulter Inc | MITOGEN FACTOR, GEN OF THE SAME AND PROCEDURE FOR YOUR MICRODETECTION. |
| EP1380648A3 (en) | 1993-06-23 | 2004-04-14 | Beckman Coulter, Inc. | Recombinant DNAase B derived from streptococcus pyogenes |
| US5585098A (en) | 1993-11-23 | 1996-12-17 | Ovimmune, Inc. | Oral administration of chicken yolk immunoglobulins to lower somatic cell count in the milk of lactating ruminants |
| EP0804559B1 (en) | 1994-10-07 | 2005-06-01 | Rockefeller University | Enzyme for cleavage of the anchor region of surface proteins from gram positive bacteria |
| AUPM885194A0 (en) | 1994-10-14 | 1994-11-10 | Council Of The Queensland Institute Of Medical Research, The | Synthetic peptides and vaccines comprising same |
| US5799981A (en) * | 1995-05-12 | 1998-09-01 | Global Healthcomm, Inc. | Pharmaceutical marketing device and system |
| EP0743617B1 (en) * | 1995-05-18 | 2002-03-06 | Canon Kabushiki Kaisha | Image processing method and apparatus thereof |
| US6284884B1 (en) | 1995-06-07 | 2001-09-04 | North American Vaccine, Inc. | Antigenic group B streptococcus type II and type III polysaccharide fragments having a 2,5-anhydro-D-mannose terminal structure and conjugate vaccine thereof |
| US6936259B2 (en) | 1995-06-08 | 2005-08-30 | University Of Saskatchewan | CAMP factor of Streptococcus uberis |
| US5819228A (en) * | 1995-10-31 | 1998-10-06 | Utilimed, Inc. | Health care payment system utilizing an intensity adjustment factor applied to provider episodes of care |
| US6014634A (en) * | 1995-12-26 | 2000-01-11 | Supermarkets Online, Inc. | System and method for providing shopping aids and incentives to customers through a computer network |
| US5846547A (en) | 1996-01-22 | 1998-12-08 | Regents Of The University Of Minnesota | Streptococcal C5a peptidase vaccine |
| US6346392B1 (en) | 1996-11-27 | 2002-02-12 | Smithkline Beecham Corporation | Polynucleotides encoding a novel glutamine transport ATP-binding protein |
| US6272481B1 (en) * | 1996-05-31 | 2001-08-07 | Lucent Technologies Inc. | Hospital-based integrated medical computer system for processing medical and patient information using specialized functional modules |
| IES960488A2 (en) | 1996-07-03 | 1997-11-19 | Trinity College Dublin | Subunit Vaccine for Streptococcus Equi |
| EP0821070A1 (en) | 1996-07-22 | 1998-01-28 | Carelli, Claude Marcel Henri | Pit-1 gene polymorphism and trait selection in animals |
| DE69737125T3 (en) | 1996-10-31 | 2015-02-26 | Human Genome Sciences, Inc. | Streptococcus pneumoniae antigens and vaccines |
| EP1007069A1 (en) | 1996-11-01 | 2000-06-14 | Smithkline Beecham Corporation | Novel coding sequences |
| US7033765B1 (en) | 1997-02-20 | 2006-04-25 | Toronto Research Chemicals, Inc. | Site-specific drug delivery |
| US6426074B1 (en) | 1997-03-19 | 2002-07-30 | The Brigham And Women's Hospital Inc. | Group B Streptococcus vaccine |
| US6256614B1 (en) * | 1997-04-17 | 2001-07-03 | Jeff H. Wecker | Internet system for producing electronic reward cards |
| JP2002516571A (en) | 1997-05-06 | 2002-06-04 | ヒューマン ジノーム サイエンシーズ,インコーポレイテッド | Enterococcus faecalis polynucleotides and polypeptides |
| US6635623B1 (en) | 1997-06-13 | 2003-10-21 | Baylor College Of Medicine | Lipoproteins as nucleic acid vectors |
| CA2303478C (en) | 1997-09-12 | 2008-09-02 | James B. Dale | Group a streptococcal vaccines |
| EP1037997A1 (en) | 1997-09-26 | 2000-09-27 | Medimmune, Inc. | Lmb gene of streptococcus agalactiae |
| US6406883B1 (en) | 1997-09-26 | 2002-06-18 | Luetticken Rudolf | Lmb gene of Streptococcus agalactiae |
| ATE338812T1 (en) | 1997-10-17 | 2006-09-15 | Nestle Sa | NEW LACTOBACTERIA STRAIN |
| US6035276A (en) * | 1997-10-17 | 2000-03-07 | Veritas Medical Services, Inc. | Medical practitioner credentialing system |
| WO1999026969A1 (en) | 1997-11-21 | 1999-06-03 | University Of Otago | Zoocin a immunity factor |
| JP2002508156A (en) | 1997-12-31 | 2002-03-19 | ストレスジェン バイオテクノロジーズ コーポレイション | Streptococcus heat shock proteins of the Hsp60 family |
| US7041814B1 (en) | 1998-02-18 | 2006-05-09 | Genome Therapeutics Corporation | Nucleic acid and amino acid sequences relating to Enterobacter cloacae for diagnostics and therapeutics |
| TR200002437T2 (en) | 1998-02-20 | 2000-11-21 | Biochem Pharma Inc. | Group B streptococcus antigens. |
| GB9808327D0 (en) | 1998-04-20 | 1998-06-17 | Chiron Spa | Antidiotypic compounds |
| US6660520B2 (en) | 1998-06-05 | 2003-12-09 | Smithkline Beecham Corporation | Nrde |
| US6936252B2 (en) | 1998-07-27 | 2005-08-30 | Microbial Technics Limited | Streptococcus pneumoniae proteins and nucleic acid molecules |
| US7098182B2 (en) | 1998-07-27 | 2006-08-29 | Microbial Technics Limited | Nucleic acids and proteins from group B streptococcus |
| US7128918B1 (en) | 1998-12-23 | 2006-10-31 | Id Biomedical Corporation | Streptococcus antigens |
| US6076525A (en) * | 1999-01-28 | 2000-06-20 | Hoffman; Michael D. | Frame for prone surgical positioning |
| US7101692B2 (en) | 1999-04-15 | 2006-09-05 | The Regents Of The University Of California | Identification of sortase gene |
| US6833356B1 (en) | 1999-08-25 | 2004-12-21 | Medimmune, Inc. | Pneumococcal protein homologs and fragments for vaccines |
| WO2001022920A2 (en) | 1999-09-29 | 2001-04-05 | Human Genome Sciences, Inc. | Colon and colon cancer associated polynucleotides and polypeptides |
| US6777547B1 (en) | 2000-01-31 | 2004-08-17 | Andreas Podbielski | Collagen-binding proteins from streptococcus pyogenes |
| EP1268774A2 (en) | 2000-03-21 | 2003-01-02 | Elitra Pharmaceuticals, Inc. | Identification of essential genes in prokaryotes |
| AUPQ801700A0 (en) | 2000-06-07 | 2000-06-29 | Peplin Research Pty Ltd | Enzyme and viral activation |
| ES2316458T3 (en) | 2000-07-06 | 2009-04-16 | Id Biomedical Corporation | STREPTOCOCCUS PYOGENES ANTIGEN. |
| ES2286133T3 (en) | 2000-08-08 | 2007-12-01 | St. Jude Children's Research Hospital | NUCLEIC ACIDS OF GROUP B STREPTOCOCO POLYPEPTIDES AND THERAPEUTIC AND VACCINE COMPOSITIONS OF THE SAME. |
| US7160547B2 (en) | 2000-10-10 | 2007-01-09 | University Of Tennessee Research Corporation | Streptococcal streptolysin S vaccines |
| US20020086023A1 (en) | 2000-10-10 | 2002-07-04 | University Of Tennessee Research Corporation | Streptococcal streptolysin S vaccines |
| MXPA03003690A (en) | 2000-10-27 | 2004-05-05 | Chiron Spa | Nucleic acids and proteins from streptococcus groups a b. |
| GB0107658D0 (en) | 2001-03-27 | 2001-05-16 | Chiron Spa | Streptococcus pneumoniae |
| JP2004533236A (en) | 2001-04-13 | 2004-11-04 | ワイエス | Surface protein of Streptococcus pyogenes |
| US20070128229A1 (en) | 2002-04-12 | 2007-06-07 | Wyeth | Surface proteins of Streptococcus pyogenes |
| WO2002094851A2 (en) | 2001-05-18 | 2002-11-28 | The Government Of The United States Of America, Asrepresented By The Secretary, Department Of Healthand Human Services, Centers For Disease Control And Prevention, Technology Transfer Office | Peptide vaccines against group a streptococci |
| GB0118249D0 (en) | 2001-07-26 | 2001-09-19 | Chiron Spa | Histidine vaccines |
| US20060073530A1 (en) | 2001-08-15 | 2006-04-06 | Olaf Schneewind | Methods and compositions involving sortase B |
| US20040029129A1 (en) | 2001-10-25 | 2004-02-12 | Liangsu Wang | Identification of essential genes in microorganisms |
| GB2385274B (en) | 2002-02-13 | 2004-04-14 | Ming-Jeng Shue | Vaginal suppository delivery device |
| US20050181388A1 (en) | 2002-04-02 | 2005-08-18 | Affinium Pharmaceuticals, Inc. | Novel purified polypeptides from bacteria |
| AU2003213949A1 (en) | 2002-04-08 | 2003-10-27 | Affinium Pharmaceuticals, Inc. | Purified polypeptides involved in membrane biogenesis |
| GB0210128D0 (en) | 2002-05-02 | 2002-06-12 | Chiron Spa | Nucleic acids and proteins from streptococcus groups A & B |
| US20070053924A1 (en) | 2002-08-26 | 2007-03-08 | Herve Tettelin | Conserved and specific streptococcal genomes |
| WO2004041157A2 (en) | 2002-09-13 | 2004-05-21 | Chiron Corporation | Group b streptococcus vaccine |
| TW566366U (en) | 2002-09-27 | 2003-12-11 | Wus Tech Co Ltd | Labor-saving portable battery equipment for power-driven walking assisted scooter |
| AU2003274011B2 (en) | 2002-10-15 | 2010-03-04 | Intercell Ag | Nucleic acids coding for adhesion factor of group B streptococcus, adhesion factors of group B streptococcus and further uses thereof |
| AU2004235952A1 (en) | 2003-05-07 | 2004-11-18 | Intercell Ag | Streptococcus agalactiae antigens I + II |
| EP1648500B1 (en) * | 2003-07-31 | 2014-07-09 | Novartis Vaccines and Diagnostics, Inc. | Immunogenic compositions for streptococcus pyogenes |
| AU2003904237A0 (en) | 2003-08-08 | 2003-08-21 | Garvan Institute Of Medical Research | Novel translocation assay |
| US8945589B2 (en) | 2003-09-15 | 2015-02-03 | Novartis Vaccines And Diagnostics, Srl | Immunogenic compositions for Streptococcus agalactiae |
| JP2007520718A (en) | 2004-02-06 | 2007-07-26 | カウンシル オブ サイエンティフィック アンド インダストリアル リサーチ | A computer-based method for identifying therapeutic adhesins and adhesin-like proteins |
| US20060041961A1 (en) | 2004-03-25 | 2006-02-23 | Abad Mark S | Genes and uses for pant improvement |
| WO2005108419A1 (en) | 2004-05-07 | 2005-11-17 | Lea-Ann Kirkham | Mutant cholesterol-binding cytolysin proteins |
| EP1784211A4 (en) | 2004-07-29 | 2010-06-30 | Novartis Vaccines & Diagnostic | Immunogenic compositions for gram positive bacteria such as streptococcus agalactiae |
| EP1817432A2 (en) | 2004-10-05 | 2007-08-15 | Wyeth a Corporation of the State of Delaware | Probe arrays for detecting multiple strains of different species |
| EP1807446A2 (en) | 2004-10-08 | 2007-07-18 | Novartis Vaccines and Diagnostics, Inc. | Immunogenic and therapeutic compositions for streptococcus pyogenes |
| CA2591510A1 (en) | 2004-12-22 | 2006-06-29 | Novartis Vaccines And Diagnostics, Inc. | Group b streptococcus |
| EP1770171A1 (en) | 2005-09-29 | 2007-04-04 | Universität Zu Köln | DNA microarray for rapid identification of Candida albicans in blood cultures. |
| JP2009542196A (en) | 2006-07-07 | 2009-12-03 | インターセル アーゲー | Small Streptococcus pyogenes antigens and their use |
| WO2009081274A2 (en) | 2007-12-21 | 2009-07-02 | Novartis Ag | Mutant forms of streptolysin o |
-
2005
- 2005-10-11 EP EP05817357A patent/EP1807446A2/en not_active Withdrawn
- 2005-10-11 CA CA2583803A patent/CA2583803C/en active Active
- 2005-10-11 US US11/792,038 patent/US7838010B2/en active Active
- 2005-10-11 WO PCT/US2005/036009 patent/WO2006042027A2/en active Application Filing
- 2005-10-11 JP JP2007535816A patent/JP2008544949A/en not_active Withdrawn
- 2005-10-11 AU AU2005294275A patent/AU2005294275B2/en active Active
-
2012
- 2012-03-27 JP JP2012071927A patent/JP2012126742A/en active Pending
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004078907A2 (en) * | 2003-03-04 | 2004-09-16 | Intercell Ag | Streptococcus pyogenes antigens |
Non-Patent Citations (5)
| Title |
|---|
| DATABASE UniProt [Online] 1 October 2002 (2002-10-01), XP002367841 retrieved from EBI Database accession no. Q8P318 -& NAKAGAWA ICHIRO ET AL: "Genome sequence of an M3 strain of Streptococcus pyogenes reveals a large-scale genomic rearrangement in invasive strains and new insights into phage evolution." GENOME RESEARCH, vol. 13, no. 6a, June 2003 (2003-06), pages 1042-1055, XP002367632 ISSN: 1088-9051 * |
| DATABASE UniProt [Online] 23 November 2004 (2004-11-23), XP002367843 retrieved from EBI Database accession no. Q5XEL1 -& BANKS D J ET AL: "PROGRESS TOWARD CHARACTERIZATION OF THE GROUP A STREPTOCOCCUS METAGENOME: COMPLETE GENOME SEQUENCE OF A MACROLIDE-RESISTANT SEROTYPE M6 STRAIN" JOURNAL OF INFECTIOUS DISEASES, CHICAGO, IL, US, vol. 190, no. 4, 15 August 2004 (2004-08-15), pages 727-738, XP008047099 ISSN: 0022-1899 * |
| DATABASE UniProt [Online] 5 July 2004 (2004-07-05), XP002367842 retrieved from EBI Database accession no. Q7CNQ7 -& SMOOT J C ET AL: "Genome sequence and comparative microarray analysis of serotype M18 group A Streptococcus strains associated with acute rheumatic fever outbreaks" PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES OF USA, NATIONAL ACADEMY OF SCIENCE, WASHINGTON, DC, US, vol. 99, no. 7, 2 April 2002 (2002-04-02), pages 4668-4673, XP002267116 ISSN: 0027-8424 * |
| FERRETTI J J ET AL: "Complete genome sequence of an M1 strain of Streptococcus pyogenes" PROCEEDINGS OF THE NATIONAL ACADEMY OF SCIENCES OF USA, NATIONAL ACADEMY OF SCIENCE, WASHINGTON, DC, US, vol. 98, no. 8, 10 April 2001 (2001-04-10), pages 4658-4663, XP002168716 ISSN: 0027-8424 * |
| OLIVE C ET AL: "Protection of mice from group A streptococcal infection by intranasal immunisation with a peptide vaccine that contains a conserved M protein B cell epitope and lacks a T cell autoepitope" VACCINE, BUTTERWORTH SCIENTIFIC. GUILDFORD, GB, vol. 20, no. 21-22, 21 June 2002 (2002-06-21), pages 2816-2825, XP004357806 ISSN: 0264-410X * |
Cited By (41)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9738693B2 (en) | 2000-10-27 | 2017-08-22 | Novartis Ag | Nucleic acids and proteins from streptococcus groups A and B |
| US10428121B2 (en) | 2000-10-27 | 2019-10-01 | Novartis Ag | Nucleic acids and proteins from streptococcus groups A and B |
| US9840538B2 (en) | 2000-10-27 | 2017-12-12 | Novartis Ag | Nucleic acids and proteins from Streptococcus groups A and B |
| US8137673B2 (en) | 2000-10-27 | 2012-03-20 | Novartis Vaccines And Diagnostics, Inc. | Nucleic acids and proteins from Streptococcus groups A & B |
| US9056912B2 (en) | 2003-07-31 | 2015-06-16 | Novartis Vaccines And Diagnostics, Srl | Immunogenic compositions for Streptococcus pyogenes |
| US8529911B2 (en) | 2006-07-07 | 2013-09-10 | Intercell Austria Ag | Small Streptococcus pyogenes antigens and their use |
| AU2007271350B2 (en) * | 2006-07-07 | 2013-03-21 | Intercell Ag | Small Streptococcus pyogenes antigens and their use |
| EP2287188A1 (en) * | 2006-07-07 | 2011-02-23 | Intercell AG | Small Streptococcus pyogenes antigens and their use |
| WO2008108830A3 (en) * | 2006-10-30 | 2008-10-30 | Novartis Ag | Immunogenic and therapeutic compositions for streptococcus pyogenes |
| JP2010508276A (en) * | 2006-10-30 | 2010-03-18 | ノバルティス アーゲー | Immunogenic and therapeutic compositions for Streptococcus pyogenes |
| WO2008108830A2 (en) * | 2006-10-30 | 2008-09-12 | Novartis Ag | Immunogenic and therapeutic compositions for streptococcus pyogenes |
| JP2010512361A (en) * | 2006-12-13 | 2010-04-22 | ハンサ メディカル アクチボラゲット | Use of endoglycosidase EndoS to treat immunoglobulin G-mediated diseases |
| US8889128B2 (en) | 2006-12-13 | 2014-11-18 | Hansa Medical Ab | Use of the endoglycosidase endos for treating immunoglobulin G mediated diseases |
| US20100285047A1 (en) * | 2007-07-03 | 2010-11-11 | Kowthar Salim | Therapeutics and diagnostics for group a streptococci |
| RU2471497C2 (en) * | 2007-09-12 | 2013-01-10 | Новартис Аг | Mutant antigens gas57 and gas57 antibodies |
| US9102741B2 (en) | 2007-09-12 | 2015-08-11 | Novartis Ag | GAS57 mutant antigens and GAS57 antibodies |
| WO2009060217A3 (en) * | 2007-11-06 | 2009-07-30 | Novartis Ag | Streptococcus pyogenes classification |
| JP2011525113A (en) * | 2008-06-20 | 2011-09-15 | ワイス・エルエルシー | Composition of ORF1358 from β-hemolytic Streptococcus strain and method of use |
| US8071112B2 (en) | 2008-06-20 | 2011-12-06 | Wyeth Llc | Compositions and methods of use of ORF-1358 from beta-hemolytic streptococcal strains |
| WO2009155484A3 (en) * | 2008-06-20 | 2010-03-11 | Wyeth Llc | Compositions and methods of use of orf1358 from beta-hemolytic streptococcal strains |
| US7914798B2 (en) | 2008-06-20 | 2011-03-29 | Wyeth Llc | Compositions and methods of use of ORF1358 from beta-hemolytic streptococcal strains |
| AU2009334428B2 (en) * | 2008-09-17 | 2014-02-20 | Glaxosmithkline Biologicals S.A. | Combination gas vaccines and therapeutics |
| JP2012502969A (en) * | 2008-09-17 | 2012-02-02 | ノバルティス アーゲー | Combined GAS vaccine and therapy |
| WO2010076618A1 (en) * | 2008-09-17 | 2010-07-08 | Novartis Ag | Combination gas vaccines and therapeutics |
| US8563001B2 (en) * | 2008-11-05 | 2013-10-22 | Regents Of The University Of Minnesota | Multicomponent immunogenic composition for the prevention of beta-hemolytic streptococcal (BHS) disease |
| CN107050442A (en) * | 2008-11-05 | 2017-08-18 | 惠氏有限责任公司 | Multicomponent immunogenic composition for preventing β Streptococcus hemolyticus (BHS) disease |
| CN102203122A (en) * | 2008-11-05 | 2011-09-28 | 惠氏有限责任公司 | Multicomponent immunogenic composition for the prevention of beta-hemolytic streptococcal (bhs) disease |
| WO2010053986A1 (en) * | 2008-11-05 | 2010-05-14 | Wyeth | Multicomponent immunogenic composition for the prevention of beta-hemolytic streptococcal (bhs) disease |
| RU2478396C2 (en) * | 2008-11-05 | 2013-04-10 | ВАЙЕТ ЭлЭлСи | MULTI-COMPONENT IMMUNOGENIC COMPOSITION FOR PREVENTING DOSEASE, CAUSED BY β-HEMOLYTIC STREPTOCCOCI (BHS) |
| WO2010079464A1 (en) | 2009-01-12 | 2010-07-15 | Novartis Ag | Cna_b domain antigens in vaccines against gram positive bacteria |
| US9102740B2 (en) | 2009-01-12 | 2015-08-11 | Novartis Ag | Cna-B domain antigens in vaccines against gram positive bacteria |
| US8465751B2 (en) * | 2009-01-12 | 2013-06-18 | Novartis Ag | Cna—B domain antigens in vaccines against gram positive bacteria |
| US20120034230A1 (en) * | 2009-01-12 | 2012-02-09 | Manetti Andrea Guido Oreste | Cna-B DOMAIN ANTIGENS IN VACCINES AGAINST GRAM POSITIVE BACTERIA |
| WO2010100627A1 (en) * | 2009-03-05 | 2010-09-10 | Novartis Ag | Diagnosis of neuropsychiatric and behavioural disorders |
| WO2010136897A2 (en) | 2009-05-28 | 2010-12-02 | Novartis Ag | Expression of recombinant proteins |
| WO2011098772A1 (en) * | 2010-02-11 | 2011-08-18 | Isis Innovation Limited | Peptide tag systems that spontaneously form an irreversible link to protein partners via isopeptide bonds |
| US10527609B2 (en) | 2010-02-11 | 2020-01-07 | Oxford University Innovation Limited | Peptide tag systems that spontaneously form an irreversible link to protein partners via isopeptide bonds |
| WO2015169773A3 (en) * | 2014-05-07 | 2016-01-21 | Glaxosmithkline Biologicals S.A. | Mutants of spy0269 |
| WO2018199775A1 (en) * | 2017-04-26 | 2018-11-01 | Auckland Uniservices | Analytical and therapeutic methods and compositions, and uses thereof |
| CN110785662A (en) * | 2017-04-26 | 2020-02-11 | 奥克兰联合服务有限公司 | Analytical and therapeutic methods and compositions and uses thereof |
| WO2024083873A1 (en) | 2022-10-18 | 2024-04-25 | Glaxosmithkline Biologicals Sa | Vaccine |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2006042027A8 (en) | 2006-11-02 |
| EP1807446A2 (en) | 2007-07-18 |
| JP2008544949A (en) | 2008-12-11 |
| US20090117113A1 (en) | 2009-05-07 |
| AU2005294275B2 (en) | 2012-09-13 |
| US7838010B2 (en) | 2010-11-23 |
| WO2006042027A3 (en) | 2006-08-24 |
| CA2583803A1 (en) | 2006-04-20 |
| JP2012126742A (en) | 2012-07-05 |
| CA2583803C (en) | 2014-11-25 |
| AU2005294275A1 (en) | 2006-04-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7838010B2 (en) | Immunogenic and therapeutic compositions for Streptococcus pyogenes | |
| US8377446B2 (en) | Serum resistance factors of gram positive bacteria | |
| US8409589B2 (en) | Mutant forms of streptolysin O | |
| AU2009334428B2 (en) | Combination gas vaccines and therapeutics | |
| US20110038879A1 (en) | Immunogenic and therapeutic compositions for streptococcus pyogenes | |
| US9102741B2 (en) | GAS57 mutant antigens and GAS57 antibodies | |
| AU2013203022B2 (en) | GAS57 mutant antigens and GAS57 antibodies | |
| AU2013203063A1 (en) | Combination gas vaccines and therapeutics |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KM KP KR KZ LC LK LR LS LT LU LV LY MA MD MG MK MN MW MX MZ NA NG NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SM SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU LV MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2007535816 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2583803 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005294275 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005817357 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2005294275 Country of ref document: AU Date of ref document: 20051011 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005817357 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11792038 Country of ref document: US |