WO2006028962A2 - 1-alkoxy 1h-imidazo ring systems and methods - Google Patents
1-alkoxy 1h-imidazo ring systems and methods Download PDFInfo
- Publication number
- WO2006028962A2 WO2006028962A2 PCT/US2005/031310 US2005031310W WO2006028962A2 WO 2006028962 A2 WO2006028962 A2 WO 2006028962A2 US 2005031310 W US2005031310 W US 2005031310W WO 2006028962 A2 WO2006028962 A2 WO 2006028962A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- alkyl
- aryl
- alkenyl
- hydrogen
- Prior art date
Links
- 0 C[C@@](*)(C=CC)C=Nc(c1c(c(N)n2)nc(*)[n]1O*)c2[Pm]C Chemical compound C[C@@](*)(C=CC)C=Nc(c1c(c(N)n2)nc(*)[n]1O*)c2[Pm]C 0.000 description 8
- SSJFZCLLSUOMFA-UHFFFAOYSA-N CC(C)(C)c1nc(c(N)nc2c3cccc2)c3[n]1OC Chemical compound CC(C)(C)c1nc(c(N)nc2c3cccc2)c3[n]1OC SSJFZCLLSUOMFA-UHFFFAOYSA-N 0.000 description 1
- XRPIRUHGBDPVCF-UHFFFAOYSA-N CC(C)O[n]1c2c(cccc3)c3nc(N)c2nc1C Chemical compound CC(C)O[n]1c2c(cccc3)c3nc(N)c2nc1C XRPIRUHGBDPVCF-UHFFFAOYSA-N 0.000 description 1
- HPDMINAXSSPBPB-UHFFFAOYSA-N CCO[n]1c2c(cccc3)c3nc(N)c2nc1C Chemical compound CCO[n]1c2c(cccc3)c3nc(N)c2nc1C HPDMINAXSSPBPB-UHFFFAOYSA-N 0.000 description 1
- HYHAPQHWQIPEFN-UHFFFAOYSA-N Cc1nc2c(N)nc(cccc3)c3c2[n]1OCc(cccc1)c1[N+]([O-])=O Chemical compound Cc1nc2c(N)nc(cccc3)c3c2[n]1OCc(cccc1)c1[N+]([O-])=O HYHAPQHWQIPEFN-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains three hetero rings
- C07D471/14—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/02—Nasal agents, e.g. decongestants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/14—Drugs for dermatological disorders for baldness or alopecia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/10—Antimycotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/16—Antivirals for RNA viruses for influenza or rhinoviruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
- A61P31/22—Antivirals for DNA viruses for herpes viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/02—Antiprotozoals, e.g. for leishmaniasis, trichomoniasis, toxoplasmosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/04—Immunostimulants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02A—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE
- Y02A50/00—TECHNOLOGIES FOR ADAPTATION TO CLIMATE CHANGE in human health protection, e.g. against extreme weather
- Y02A50/30—Against vector-borne diseases, e.g. mosquito-borne, fly-borne, tick-borne or waterborne diseases whose impact is exacerbated by climate change
Definitions
- Certain lH-imidazo[4,5-c]quinolin-4-amines and 1- and 2-substituted derivatives thereof were later found to be useful as antiviral agents, bronchodilators and immunomodulators. Subsequently, certain substituted lH-imidazo[4,5-c] pyridine- amine, quinolin-4-amine, tetrahydroquinolin-4-amine, naphthyridin-4-amine, and tetrahydronaphthyridin-4-amine compounds as well as certain analogous thiazolo and oxazolo compounds were synthesized and found to be useful as immune response modifiers (IRMs), rendering them useful in the treatment of a variety of disorders.
- IRMs immune response modifiers
- the present invention provides compounds of the Formula I:
- R 1 , R 2; R 3 , R", R, R A , R B , RA I , R B I, m 5 n, P 5 and G are as defined below; and pharmaceutically acceptable salts thereof.
- the compounds of Formulas I, II, III, IV, V, VI, VII, VIII, IX, X, XI, XII, XIII, and XIV are useful as immune response modifiers (IRMs) due to their ability to modulate cytokine biosynthesis (e.g., induce or inhibit the biosynthesis or production of one or more cytokines) and otherwise modulate the immune response when administered to animals.
- IRMs immune response modifiers
- Compounds can be tested for induction of cytokine biosynthesis by incubating human peripheral blood mononuclear cells (PBMC) in a culture with the compound(s) at a concentration range of 30 to 0.014 ⁇ M and analyzing for interferon ( ⁇ ) or tumor necrosis factor ( ⁇ ) in the culture supernatant.
- Compounds can be tested for inhibition of cytokine biosynthesis by incubating mouse macrophage cell line Raw 264.7 in a culture with the compound(s) at a single concentration of, for example, 5 ⁇ M and analyzing for tumor necrosis factor ( ⁇ ) in the culture supernatant.
- cytokine biosynthesis for example, induce the biosynthesis of one or more cytokines, makes the compounds useful in the treatment of a variety of conditions such as viral diseases and neoplastic diseases, that are responsive to such changes in the immune response.
- the present invention provides pharmaceutical compositions containing the immune response modifier compounds, and methods of inducing cytokine biosynthesis in animal cells, treating a viral disease in an animal, and/or treating a neoplastic disease in an animal by administering to the animal one or more compounds of the Formulas I, II, III, IV, V, VI, VII, VIII, IX, X, XI, XII, XIII, and/or XIV, and/or pharmaceutically acceptable salts thereof.
- the invention provides methods of synthesizing the compounds of Formulas I, II, III, IV, V, VI, VII, VIII, IX, X, XI, XII, XIII, and XIV and intermediates useful in the synthesis of these compounds.
- R 1 , R 2 , R 3 , R", R, R A , R B , R AI , R BI , m, n, p, and G are as defined below.
- the present invention provides a compound of Formula I:
- R 1 is selected from the group consisting of: -R 4 ,
- R A and R B are each independently selected from the group consisting of: hydrogen, halogen, alkyl, alkenyl, alkoxy, alkylthio, and
- R A and R B form a fused benzene or pyridine ring which is unsubstituted or substituted by one or more R" groups; or when taken together, R A and R B form a fused cyclohexene or tetrahydropyridine ring which is unsubstituted or substituted by one or more R groups; R is selected from the group consisting of: halogen, hydroxyl, alkyl, alkenyl, haloalkyl, alkoxy, alkylthio, and
- R 4 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, heteroaryl, and heterocyclyl wherein the alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, heteroaryl, and heterocyclyl groups can be unsubstituted or substituted by one or more substituents independently selected from the group consisting of alkyl, alkoxy, haloalkyl, haloalkoxy, halogen, nitro, hydroxy, mercapto, cyano, carboxy, formyl, aryl, aryloxy, arylalkoxy, heteroaryl, heteroaryloxy, heteroarylalkoxy, heterocyclyl, heterocyclylalkylenyl, amino, alkylamino, (arylalkylenyl)amino, dialkylamino, and in the
- R 6 is selected from the group consisting of hydrogen, alkyl, and arylalkylenyl
- R 8 is C 2-7 alkylene
- R 9 is selected from the group consisting of hydrogen, alkyl, and arylalkylenyl; or R 9 and R 4 together with the nitrogen atom to which R 9 is bonded can join to form the group
- R 12 is selected from the group consisting of hydrogen and alkyl
- A is selected from the group consisting Of -CH(R 6 )-, -O-, -N(R 6 )-, -N(Y-R 4 )-, and -N(X-N(Re)-Y-R 4 )-;
- X is C 2-20 alkylene; X" is C 1-20 alkylene; Y is selected from the group consisting of -C(R 7 )-, -C(R 7 )-O-, -S(O) 2 -,
- a and b are independently integers from 1 to 4 with the proviso that when A is -O-, -N(R 6 )-, -N(Y-R 4 )-, or -N(X-N(Re)-Y-R 4 )- then a and b are independently integers from 2 to 4;
- R" hydrogen or a non-interfering substituent;
- R'" is a non-interfering substituent; or a pharmaceutically acceptable salt thereof.
- the present invention provides a compound of Formula II:
- R 1 is selected from the group consisting of:
- R 2 is selected from the group consisting of: hydrogen, alkyl, alkenyl, aryl, heteroaryl, heterocyclyl, alkyl-Z-alkylenyl, aryl-Z-alkylenyl, alkenyl-Z-alkylenyl, and alkyl or alkenyl substituted by one or more substituents selected from the group consisting of: hydroxy, halogen, -N(R 6 ) 2 , -C(R 7 )-N(R 6 ) 2 , -S(O) 2 -N(Re) 2 ,
- R A and R B are each independently selected from the group consisting of: hydrogen, halogen, alkyl, alkenyl, alkoxy, alkylthio, and
- R A and R B form a fused benzene or pyridine ring which is unsubstituted or substituted by one or more R groups, or substituted by one R 3 group, or substituted by one R 3 group and one R group, or substituted by one R 3 group and two R groups; or when taken together, RA and R B form a fused cyclohexene or tetrahydropyridine ring which is unsubstituted or substituted by one or more R groups;
- R is selected from the group consisting of: halogen, hydroxy, alkyl, alkenyl, haloalkyl, alkoxy, alkylthio, and
- R 3 is selected from the group consisting of:
- R 4 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, heteroaryl, and heterocyclyl wherein the alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, heteroaryl, and heterocyclyl groups can be unsubstituted or substituted by one or more substituents independently selected from the group consisting of alkyl, alkoxy, haloalkyl, haloalkoxy, halogen, nitro, hydroxy, mercapto, cyano, carboxy, formyl, aryl, aryloxy, arylalkoxy, heteroaryl, heteroaryloxy, heteroarylalkoxy, heterocyclyl, heterocyclylalkylenyl, amino, alkylamino, (arylalkylalkylenyl, amino, alkylamino, (arylalkylalkyleny
- R 5 is selected from the group consisting of:
- X is C 2-2O alkylene; X" is C 1-20 alkylene;
- Y is selected from the group consisting Of -C(R 7 )-, -C(R 7 )-O-, -S(O) 2 -, -S(O) 2 -N(R 6 )-, and -C(R 7 )-N(R 9 )-;
- Z is selected from the group consisting of -O- and -S(O) 0-2 -;
- A is selected from the group consisting Of-CH(R 6 )-, -0-, -N(R 6 )-, -N(Y-R 4 )-, and -N(X-N(Re)-Y-R 4 )-;
- a and b are independently integers from 1 to 4 with the proviso that when A is -0-, -N(R 6 )-, -N(Y-R 4 )-, or -N(X-N(Re)-Y-R 4 )- then a and b are independently integers from 2 to 4;
- R 4 ' is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl wherein the alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl groups can be unsubstituted or substituted by one or more substituents independently selected from the group consisting of alkyl, alkoxy, hydroxyalkyl, haloalkyl, haloalkoxy, hal
- R 5 ' is selected from the group consisting of:
- X' is selected from the group consisting of alkylene, alkenylene, alkynylene, arylene, heteroarylene, and heterocyclylene wherein the alkylene, alkenylene, and alkynylene groups can be optionally interrupted or terminated by arylene, heteroarylene, or heterocyclylene and optionally interrupted by one or more -O- groups;
- Y' is selected from the group consisting of: -S(O) 0-2 -, -S(O) 2 -N(R 11 )-, -C(R 7 )-, -C(Ry)-O-,
- Z' is a bond or -O-
- A' is selected from the group consisting Of-CH 2 -, -O-, -C(O)-, -S(O) 0-2 -, and -N(R 4 ')-;
- Q is selected from the group consisting of a bond, -C(R 7 )-, -C(R 7 )-C(R 7 )-, -S(O) 2 -, -C(R 7 VNCRn)-W-, -S(O) 2 -N(R 11 )-, -C(R 7 )-0-, and -C(R 7 )-N(OR 12 )-;
- V is selected from the group consisting Of -C(R 7 )-, -0-C(R 7 )-, -N(Rn)-C(R 7 )-, and -S(O) 2 -;
- W is selected from the group consisting of a bond, -C(O)-, and -S(O) 2 -;
- c and d are independently integers from 1 to 6 with the proviso that c + d is ⁇ 7, and when A' is -O- or -N(R 4 ')- then c and d are independently integers from 2 to 4;
- R 6 is selected from the group consisting of hydrogen, alkyl, and arylalkylenyl;
- R 8 is C 2-7 alkylene;
- R 9 is selected from the group consisting of hydrogen, alkyl, and arylalkylenyl; or R 9 and R 4 together with the nitrogen atom to which R 9 is bonded can join to form the group
- R 10 is C 3-8 alkylene
- R 11 is selected from the group consisting of hydrogen, C 1-1O alkyl, C 2-10 alkenyl, C 1-10 alkoxyC 2-10 alkylenyl, and arylC ⁇ o alkylenyl;
- R 12 is selected from the group consisting of hydrogen and alkyl; or a pharmaceutically acceptable salt thereof.
- the present invention provides a compound of Formula III:
- R 1 is selected from the group consisting of:
- R 2 is selected from the group consisting of: hydrogen, alkyl, alkenyl, aryl, heteroaryl, heterocyclyl, alkyl-Z-alkylenyl, aryl-Z-alkylenyl, alkenyl-Z-alkylenyl, and alkyl or alkenyl substituted by one or more substituents selected from the group consisting of: hydroxy, halogen, -N(R 6 ) 2 , -C(R 7 )-N(R 6 ) 2 , -S(O) 2 -N(Re) 2 , -N(R6)-C(R 7 )-Ci-io alkyl, -N(R O )-S(O) 2 -C 1-10 alkyl, -C(O)-C 1-I0 alkyl, -C(O)-O-C
- RAI and R B1 are each independently selected from the group consisting of: hydrogen, halogen, alkyl, alkenyl, alkoxy, alkylthio, and
- R 4 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, heteroaryl, and heterocyclyl wherein the alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, heteroaryl, and heterocyclyl groups can be unsubstituted or substituted by one or more substituents independently selected from the group consisting of alkyl, alkoxy, haloalkyl, haloalkoxy, halogen, nitro, hydroxy, mercapto, cyano, carboxy, formyl, aryl, aryloxy, arylalkoxy, heteroaryl, heteroaryloxy, heteroarylalkoxy, heterocyclyl, heterocyclylalkylenyl, amino, alkylamino, (arylalkylenyl)amino, dialkylamino, and in the case
- R 5 is selected from the group consisting of:
- R 6 is selected from the group consisting of hydrogen, alkyl, and arylalkylenyl;
- R 8 is C 2-7 alkylene;
- R 9 is selected from the group consisting of hydrogen, alkyl, and arylalkylenyl; or R 9 and R 4 together with the nitrogen atom to which R 9 is bonded can join to form the group
- R 12 is selected from the group consisting of hydrogen and alkyl;
- A is selected from the group consisting Of -CH(R 6 )-, -O-, -N(R 6 )-, -N(Y-R 4 )-, and -N(X-N(Re)-Y-R 4 )-;
- X is C 2-2O alkylene; X" is C 1-20 alkylene;
- Y is selected from the group consisting of -C(R 7 )-, -C(R 7 )-O-, -S(O) 2 -, -S(O) 2 -N(R 6 )-, and -C(R 7 )-N(R 9 )-;
- Z is selected from the group consisting of -O- and -S(O) 0-2 -; and a and b are independently integers from 1 to 4 with the proviso that when A is -0-, -N(R 6 )-, -N(Y-R 4 )-, or -N(X-N(Re)-Y-R 4 )- then a and b are independently integers from 2 to 4; or a pharmaceutically acceptable salt thereof.
- the present invention provides a compound of Formula IV:
- R 1 is selected from the group consisting of: -R 4 ,
- R 2 is selected from the group consisting of: hydrogen, alkyl, alkenyl, aryl, heteroaryl, heterocyclyl, alkyl-Z-alkylenyl, aryl-Z-alkylenyl, alkenyl-Z-alkylenyl, and alkyl or alkenyl substituted by one or more substituents selected from the group consisting of: hydroxy, halogen,
- R is selected from the group consisting of: halogen, hydroxyl, alkyl, alkenyl, haloalkyl, alkoxy, alkylthio, and -N(R 12 ) 2 ;
- R 3 is selected from the group consisting of:
- n is an integer from 0 to 4; m is 0 or 1, with the proviso that when m is 1, n is 0, 1, or 2; R 4 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, heteroaryl, and heterocyclyl wherein the alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, heteroaryl, and heterocyclyl groups can be unsubstituted or substituted by one or more substituents independently selected from the group consisting of alkyl, alkoxy, haloalkyl, haloalkoxy, halogen, nitro, hydroxy, mercapto, cyano, carboxy, formyl, aryl, ary
- R 5 is selected from the group consisting of:
- X is C 2-2O alkylene; X" is C 1-20 alkylene;
- Y is selected from the group consisting Of -C(R 7 )-, -C(R 7 )O-, -S(O) 2 -, -S(O) 2 -N(R 6 )-, and -C(R 7 )-N(R 9 )-;
- Z is selected from the group consisting of -O- and -S(O) 0-2 -;
- A is selected from the group consisting Of -CH(R 6 )-, -0-, -N(R 6 )-, -N(Y-R 4 )-, and -N(X-N(Re)-Y-R 4 )-;
- a and b are independently integers from 1 to 4 with the proviso that when A is -0-, -N(R 6 )-, -N(Y-R 4 )-, or -N(X-N(Re)-Y-R 4 )- then a and b are independently integers from 2 to 4;
- R 4 ' is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl wherein the alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl groups can be unsubstituted or substituted by one or more substituents independently selected from the group consisting of alkyl, alkoxy, hydroxyalkyl, haloalkyl, haloalkoxy, hal
- R 5 ' is selected from the group consisting of:
- X' is selected from the group consisting of alkylene, alkenylene, alkynylene, arylene, heteroarylene, and heterocyclylene wherein the alkylene, alkenylene, and alkynylene groups can be optionally interrupted or terminated by arylene, heteroarylene, or heterocyclylene and optionally interrupted by one or more -O- groups;
- Y' is selected from the group consisting of: -S(O) 0-2 -, -S(O) 2 -N(R 11 )-, -C(R 7 )-, -C(Rv)-O-,
- Z' is a bond or -O-
- a 1 is selected from the group consisting Of-CH 2 -, -O-, -C(O)-, -S(O) 0-2 -, and -N(R 4 ')-;
- Q is selected from the group consisting of a bond, -C(R 7 )-, -C(R 7 )-C(R 7 )-, -S(O) 2 -, -C(Rv)-N(Rn)-W-, -S(O) 2 -N(R 11 )-, -C(Rv)-O-, and -C(Rv)-N(ORi 2 )-;
- V is selected from the group consisting of -C(R 7 )-, -0-C(R 7 )-, -N(Rn)-C(R 7 )-, and -S(O) 2 -;
- W is selected from the group consisting of a bond, -C(O)-, and -S(O) 2 -;
- c and d are independently integers from 1 to 6 with the proviso that c + d is ⁇ 7, and when A' is -O- or -N(R 4 ')- then c and d are independently integers from 2 to 4;
- R 6 is selected from the group consisting of hydrogen, alkyl, and arylalkylenyl;
- R 8 is C 2-7 alkylene;
- R 9 is selected from the group consisting of hydrogen, alkyl, and arylalkylenyl; or R 9 and R 4 together with the nitrogen atom to which R 9 is bonded can join to form the group
- R 10 is C 3-8 alkylene
- Rn is selected from the group consisting of hydrogen, C 1-10 alkyl, C 2- I 0 alkenyl, C 1-10 alkoxyC 2-10 alkylenyl, and arylC 1-10 alkylenyl;
- R 12 is selected from the group consisting of hydrogen and alkyl; or a pharmaceutically acceptable salt thereof.
- the present invention provides a compound of Formula V:
- Ri is selected from the group consisting of: -R 4 ,
- R 2 is selected from the group consisting of: hydrogen, alkyl, alkenyl, aryl, heteroaryl, heterocyclyl, alkyl-Z-alkylenyl, aryl-Z-alkylenyl, alkenyl-Z-alkylenyl, and alkyl or alkenyl substituted by one or more substituents selected from the group consisting of: hydroxy, halogen,
- R is selected, from the group consisting of: halogen, hydroxy, alkyl, alkenyl, haloalkyl, alkoxy, alkylthio, and -N(R 12 ) 2 ; n is an integer from 0 to 4;
- R 4 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, heteroaryl, and heterocyclyl wherein the alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, heteroaryl, and heterocyclyl groups can be unsubstituted or substituted by one or more substituents independently selected from the group consisting of alkyl, alkoxy, haloalkyl, haloalkoxy, halogen, nitro, hydroxy, mercapto, cyano, carboxy, formyl, aryl, aryloxy, arylalkoxy, heteroaryl, heteroaryloxy, heteroarylalkoxy, heterocyclyl, heterocyclylalkylenyl, amino, alkylamino, (arylalkylenyl)amino, dialkylamino, and in the case of alkyl, alkeny
- R 6 is selected from the group consisting of hydrogen, alkyl, and arylalkylenyl
- Rg is C 2-7 alkylene; Rg is selected from the group consisting of hydrogen, alkyl, and arylalkylenyl; or
- R 9 and R 4 together with the nitrogen atom to which R 9 is bonded can join to form the group
- R 12 is selected from the group consisting of hydrogen and alkyl;
- A is selected from the group consisting Of -CH(R 6 )-, -0-, -N(R 6 )-, -N(Y-R 4 )-, and
- X is C 2-20 alkylene; X" is C 1-2 O alkylene;
- Y is selected from the group consisting of -C(R 7 )-, -C(R 7 )-O-, -S(O) 2 -, -S(O) 2 -N(R 6 )-, and -C(R 7 )-N(R 9 )-;
- Z is selected from the group consisting of -O- and -S(O) 0-2 -; and a and b are independently integers from 1 to 4 with the proviso that when A is -0-, -N(R 6 )-, -N(Y-R 4 )-, or -N(X-N(Re)-Y-R 4 )- then a and b are independently integers from 2 to 4; or a pharmaceutically acceptable salt thereof.
- the present invention provides a compound selected from the group consisting of the following Formulas VI, VII, VIII, and IX (preferably, a compound of Formula VI):
- R 1 is selected from the group consisting of:
- R 2 is selected from the group consisting of: hydrogen, alkyl, alkenyl, aryl, heteroaryl, heterocyclyl, alkyl-Z-alkylenyl, aryl-Z-alkylenyl, alkenyl-Z-alkylenyl, and alkyl or alkenyl substituted by one or more substituents selected from the group consisting of: hydroxy, halogen,
- R is selected from the group consisting of: halogen, hydroxyl, alkyl, alkenyl, haloalkyl, alkoxy, alkylthio, and R 3 is selected from the group consisting of:
- R 4 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, heteroaryl, and heterocyclyl wherein the alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, heteroaryl, and heterocyclyl groups can be unsubstituted or substituted by one or more substituents independently selected from the group consisting of alkyl, alkoxy, haloalkyl, haloalkoxy, halogen, nitro, hydroxy, mercapto, cyano, carboxy, formyl, aryl, aryloxy, arylalkoxy, heteroaryl, heteroaryloxy, heteroarylalkoxy, heterocyclyl, heterocyclylalkoxy, heterocyclyl, heterocyclylalkoxy, heterocyclyl, heterocyclylal
- X is C 2-20 alkylene; X" is C 1-20 alkylene;
- Y is selected from the group consisting Of -C(R 7 )-, -C(R 7 )-O-, -S(O) 2 -, -S(O) 2 -N(R 6 )-, and -C(R 7 )-N(R 9 )-;
- Z is selected from the group consisting of -O- and -S(0)o -2 -;
- A is selected from the group consisting of -CH(R 6 )-, -O-, -N(R 6 )-, -N(Y-R 4 )-, and -N(X-N(Re)-Y-R 4 )-;
- a and b are independently integers from 1 to 4 with the proviso that when A is -O-, -N(R 6 )-, -N(Y-R 4 )-, or -N(X-N(Re)-Y-R 4 )- then a and b are independently integers from 2 to 4;
- R 4 1 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl wherein the alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl groups can be unsubstituted or substituted by one or more substituents independently selected from the group consisting of alkyl, alkoxy, hydroxyalkyl, haloalkyl, haloalkoxy, halogen
- R 5 1 is selected from the group consisting of:
- X' is selected from the group consisting of alkylene, alkenylene, alkynylene, arylene, heteroarylene, and heterocyclylene wherein the alkylene, alkenylene, and alkynylene groups can be optionally interrupted or terminated by arylene, heteroarylene, or heterocyclylene and optionally interrupted by one or more -O- groups;
- Y' is selected from the group consisting of:
- Z 1 is a bond or -O-;
- A' is selected from the group consisting Of-CH 2 -, -O-, -C(O)-, -S(O) 0-2 -, and -N(R 4 1 )-;
- Q is selected from the group consisting of a bond, -C(R 7 )-, -C(R 7 )-C(R 7 )-, -S(O) 2 -, -C(Ry)-N(Rn)-W-, -S(O) 2 -N(R 11 )-, -C(Ry)-O-, and -C(Ry)-N(ORi 2 )-;
- V is selected from the group consisting Of -C(R 7 )-, -0-C(R 7 )-, -N(Rn)-C(R 7 )-, and -S(O) 2 -;
- W is selected from the group consisting of a bond, -C(O)-, and -S(O) 2 -; c and d are independently integers from 1 to 6 with the proviso that c + d is ⁇ 7, and when A' is -O- or -N(R 4 ')- then c and d are independently integers from 2 to 4;
- R 6 is selected from the group consisting of hydrogen, alkyl, and arylalkylenyl
- R 8 is C 2-7 alkylene
- Rg is selected from the group consisting of hydrogen, alkyl, and arylalkylenyl; or Rg and R 4 together with the nitrogen atom to which Rg is bonded can join to form the group
- R 1O is C 3-8 alkylene
- R 11 is selected from the group consisting of hydrogen, C 1-10 alkyl, C 2-10 alkenyl, C 1-10 alkoxyC 2-10 alkylenyl, and arylQ-io alkylenyl;
- R 12 is selected from the group consisting of hydrogen and alkyl; or a pharmaceutically acceptable salt thereof.
- the present invention provides a compound selected from the group consisting of the following Formulas X, XI, XII, and XIII:
- R 1 is selected from the group consisting of: -R 4 , -X-R 5 ,
- R 2 is selected from the group consisting of: hydrogen, alkyl, alkenyl, aryl, heteroaryl, heterocyclyl, alkyl-Z-alkylenyl, aryl-Z-alkylenyl, alkenyl-Z-alkylenyl, and alkyl or alkenyl substituted by one or more substituents selected from the group consisting of: hydroxy, halogen,
- R is selected from the group consisting of: halogen, hydroxy, alkyl, alkenyl, haloalkyl, alkoxy, alkylthio, and
- p is an integer from 0 to 3;
- R 4 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, lieteroaryl, and heterocyclyl wherein the alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, heteroaryl, and heterocyclyl groups can be unsubstituted or substituted by one or more substituents independently selected from the group consisting of alkyl, alkoxy, haloalkyl, haloalkoxy, halogen, nitro, hydroxy, mercapto, cyano, carboxy, formyl, aryl, aryloxy, arylalkoxy, heteroaryl, heteroaryloxy, heteroarylalkoxy, heterocyclyl, heterocyclylalkylenyl, amino, alkylamino, (arylalkylenyl)amino, dialkylamino, and in the case of alkyl, al
- R 5 is selected from the group consisting of:
- R 6 is selected from the group consisting of hydrogen, alkyl, and arylalkylenyl;
- R 8 is C 2-7 alkylene;
- R 9 is selected from the group consisting of hydrogen, alkyl, and arylalkylenyl; or R 9 and R 4 together with the nitrogen atom to which R 9 is bonded can join to form the group
- R 12 is selected from the group consisting of hydrogen and alkyl;
- A is selected from the group consisting of -CH(R 6 )-, -O-, -N(R 6 )-, -N(Y-R 4 )-, and -N(X-N(Re)-Y-R 4 )-;
- X is C 2-20 alkylene
- X" is C 1-20 alkylene; Y is selected from the group consisting of -C(R 7 )-, -C(R 7 )-O-, -S(O) 2 -,
- Z is selected from the group consisting of -O- and -S(0)o -2 -; and a and b are independently integers from 1 to 4 with the proviso that when A is -O-, -N(R 6 )-, -N(Y-R 4 )-, or -N(X-N(R ⁇ -Y-R 4 )- then a and b are independently integers from 2 to 4; or a pharmaceutically acceptable salt thereof.
- the present invention provides a compound of Formula XIV:
- G is selected from the group consisting of:
- R' and R"" are each independently C 1-10 alkyl, C 3-7 cycloalkyl, phenyl, or benzyl, each of which may be unsubstituted or substituted by one or more substitutents selected from the group consisting of halogen, hydroxy, nitro, cyano, carboxy, C 1-6 alkyl, C 1-4 alkoxy, aryl, heteroaryl, arylC 1-4 alkylenyl, heteroarylC 1-4 alkylenyl, haloC 1-4 alkyl, haloC 1-4 alkoxy, -0-C(O)-CH 3 , -C(O)-O-CH 3 , -C(O)-NH 2 , -0-CH 2 -C(O)-NH 2 , -NH 2 , and -S(O) 2 -NH 2 ;
- ⁇ -aminoacyl is an acyl group derived from an amino acid selected from the group consisting of racemic
- Y 0 is selected from the group consisting of C 1-6 alkyl, carboxyC 1-6 alkyl, aminoC 1-4 alkyl, mono-iV-C 1-6 alkylaminoC 1-4 alkyl, and di-N,N-C ⁇ -6 alkylaminoC 1-4 alkyl;
- Y 1 is selected from the group consisting of mono-N-C 1-6 alkylamino, di--V,_V-C 1 - 6 alkylamino, morpholin-4-yl, piperidin-1-yl, pyrrolidin-1-yl, and 4-C 1-4 alkylpiperazin-l-yl.
- R 1 is selected from the group consisting of: -R 4 , -X-R 5 ,
- R A and R B are each independently selected from the group consisting of: hydrogen, halogen, alkyl, alkenyl, alkoxy, alkylthio, and
- R A and R B form a fused benzene or pyridine ring which is unsubstituted or substituted by one or more R'" groups; or when taken together, RA and R B form a fused cyclohexene or tetrahydropyridine ring which is unsubstituted or substituted by one or more R groups;
- R is selected from the group consisting of: halogen, hydroxyl, alkyl, alkenyl, haloalkyl, alkoxy, alkylthio, and
- R 4 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, heteroaryl, and heterocyclyl wherein the alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, heteroaryl, and heterocyclyl groups can be unsubstituted or substituted by one or more substituents independently selected from the group consisting of alkyl, alkoxy, haloalkyl, haloalkoxy, halogen, nitro, hydroxy, mercapto, cyano, carboxy, formyl, aryl, aryloxy, arylalkoxy, heteroaryl, heteroaryloxy, heteroarylalkoxy, heterocyclyl, heterocyclylalkylenyl, amino, alkylamino, (arylalkylenyl)amino, dialkylamino, and in
- R 6 is selected from the group consisting of hydrogen, alkyl, and arylalkylenyl
- R 8 is C 2-7 alkylene
- R 9 is selected from the group consisting of hydrogen, alkyl, and arylalkylenyl; or R 9 and R 4 together with the nitrogen atom to which R 9 is bonded can join to form the group
- R 12 is selected from the group consisting of hydrogen and alkyl
- A is selected from the group consisting of -CH(R 6 )-, -O-, -N(R 6 )-, -N(Y-R 4 )-, and -N(X-N(Re)-Y-R 4 )-;
- X is C 2-20 alkylene; X" is C 1-20 alkylene; Y is selected from the group consisting Of -C(R 7 )-, -C(R 7 )-O-, -S(O) 2 -,
- a and b are independently integers from 1 to 4 with the proviso that when A is -O-, -N(R 6 )-, -N(Y-R 4 )-, or -N(X-N(Re)-Y-R 4 )- then a and b are independently integers from 2 to 4;
- R" hydrogen or a non-interfering substituent;
- R'" is a non-interfering substituent;or a pharmaceutically acceptable salt thereof.
- each one of the following variables e.g., R, R", R'", R 1 , R 2 , R 3 , R 4 ', R A , R B , m, n, p, A, G, and so on
- each one of the following variables e.g., R, R", R'", R 1 , R 2 , R 3 , R 4 ', R A , R B , m, n, p, A, G, and so on
- each of the resulting combinations of variables is an embodiment of the present invention.
- each of R" and R'" is independently a non-interfering substituent.
- each R" is independently selected from the group consisting of hydrogen and non-interfering substituents.
- non-interfering means that the immunomodulator activity (for example, the ability to induce the biosynthesis of one or more cytokines or the ability to inhibit the biosynthesis of one or more cytokines) of the compound, which includes a non-interfering substituent, is not destroyed.
- Illustrative R" groups include those described herein for R 2 .
- Illustrative R'" groups include those described herein for R and R 3 .
- RA and RB are each independently selected from the group consisting of: hydrogen, halogen, alkyl, alkenyl, alkoxy, alkylthio, and -N(R 12 ) 2 ; or when taken together, RA and R B form a fused benzene or pyridine ring which is unsubstituted or substituted by one or more R'" groups; or when taken together, R A and R B form a fused cyclohexene or tetrahydropyridine ring which is unsubstituted or substituted by one or more R groups.
- R A and R B are each independently selected from the group consisting of: hydrogen, halogen, alkyl, alkenyl, alkoxy, alkylthio, and -N(R 12 ) 2 ; or when taken together, R A and R B form a fused benzene or pyridine ring which is unsubstituted or substituted by one or more R groups, or substituted by one R 3 group, or substituted by one R 3 group and one R group, or substituted by one R 3 group and two R groups; or when taken together, R A and R B form a fused cyclohexene or tetrahydropyridine ring which is unsubstituted or substituted by one or more R groups.
- R A and R B are each independently selected from the group consisting of: hydrogen, halogen, alkyl, alkenyl, alkoxy, alkylthio, and -N(Rj 2 ) 2 .
- R A and R B are each independently selected from hydrogen and alkyl.
- R A and R B are each methyl.
- R A and R B form a fused benzene or pyridine ring which is unsubstituted or substituted by one or more R'" groups.
- R A and R B form a fused benzene or pyridine ring which is unsubstituted or substituted by one or more R groups, or substituted by one R 3 group, or substituted by one R 3 group and one R group, or substituted by one R 3 group and two R groups.
- RA and R B form a fused benzene ring.
- the fused benzene ring is unsubstituted.
- R A and R B form a fused pyridine ring.
- the fused pyridine ring is
- the highlighted bond indicates the position where the ring is fused.
- the fused pyridine ring is unsubstituted.
- RA and R B form a fused cyclohexene or tetrahydropyridine ring which is unsubstituted or substituted by one or more R groups.
- R A and R 5 form a fused cyclohexene ring.
- the fused cyclohexene ring is unsubstituted.
- R A and R B form a fused tetrahydropyridine ring.
- the fused tetrahydropyridine ring is wherein the highlighted bond indicates the position where the ring is fused.
- the fused tetrahydropyridine ring is unsubstituted.
- fused cyclohexene ring and fused tetrahydropyridine ring are each fused to the ring system such that the unsaturated carbon atoms of each of these rings are in common with the pyridine ring.
- a fused cyclohexene ring is illustrated within the following formulas:
- R A1 and R BI are each independently selected from the group consisting of: hydrogen, halogen, alkyl, alkenyl, alkoxy, alkylthio, and -N(R 12 ) 2 .
- R AI and R BI are each independently selected from hydrogen and alkyl.
- R A1 and R BI are each methyl.
- R is selected from the group consisting of: halogen, hydroxyl, alkyl, alkenyl, haloalkyl, alkoxy, alkylthio, and -N(R 12 ) 2 .
- R 1 is selected from the group consisting of: -R 4 , -X-R 5 , -X-N(Rg)-Y-R 4 , -X"-C(R 7 )-N(R 9 )-R 4 , -X"-C(R 7 )-O-N(R 6 )-R 4 , -X-S(O) 2 -N(R( J )-R 4 , and -X-O-R 4 .
- R 1 is -R 4 , -X-N(Rg)-Y-R 4 , or -X"-C(R 7 )-N(R 9 )-R4.
- R 1 is -R 4 .
- -R 4 is alkyl, aryl, or arylalkylenyl.
- R 1 is selected from the group consisting of hydrogen, methyl, ethyl, ?z-propyl, isopropyl, 7j-butyl, tert-butyl, isobutyl, cyclohexyl, benzyl, phenyl, 3-phenylpropyl, 2-nitrobenzyl, 2-phenoxyethyl, and (pyridin-3-yl)methyl.
- R 1 is (and, hence, R 4 is) methyl, ethyl, n-propyl, n-butyl, isobutyl, cyclohexyl, phenyl, 3-phenylpropyl, or (pyridin-3-yl)methyl.
- R 1 is selected from the group consisting of hydrogen, methyl, ethyl, isopropyl, tert-butyl, isobutyl, benzyl, 2-nitrobenzyl, and 2-phenoxyethyl.
- R 1 is -X-N(Re)-Y-R 4 or -X"-C(R 7 )-N(R 9 )-R 4 .
- R 1 is 3-[(methanesulfonyl)amino]propyl
- R 1 is 3-(acetylamino)propyl, 3-[(isopropylcarbonyl)amino]propyl, 3-[(cyclohexylcarbonyl)amino]propyl, 3-[(morpholin-4-ylcarbonyl)amino]propyl, 3 - ⁇ [(isopropylamino)carbonyl] amino ⁇ propyl, 2-(morpholin-4-yl)-2-oxoethyl, or carbamoylmethyl.
- R 1 is 3-[(methanesulfonyl)amino]propyl
- R 1 is methyl, ethyl, n-propyl, isopropyl, ra-butyl, isobutyl, cyclohexyl, benzyl, phenyl, 3-phenylpropyl, (pyridin-3-yl)methyl, 3 - [(methanesulfonyl)amino]propyl, 3 -(acetylamino)propyl, 3-[(isopropylcarbonyl)amino]propyl, 3-[(cyclohexylcarbonyl)amino]propyl, 3-[(morpholin-4-ylcarbonyl)amino]propyl, 3- ⁇ [(isopropylamino)carbonyl]amino ⁇ propyl, or 2-(mo ⁇ holin-4-yl)-2-oxoethyl.
- R 2 is selected from the group consisting of: hydrogen, alkyl, alkenyl, aryl, heteroaryl, heterocyclyl, alkyl-Z-alkylenyl, aryl-Z-alkylenyl, alkenyl-Z-alkylenyl, and alkyl or alkenyl substituted by one or more substituents selected from the group consisting of: hydroxy, halogen, -N(R 6 ) 2 , -C(R 7 )-N(R 6 ) 2 , -S(O) 2 -N(R 6 ) 2 , -N(R 6 )-C(R 7 )-C 1-10 alkyl, -N(R 6 )-S(O) 2 -C M0 alkyl, -C(O)-C 1-10 alkyl, -C(O)-O-Ci -I0 alkyl, -N 3 , aryl, heteroaryl, heterocyclyl, al
- R 2 is selected from the group consisting of hydrogen, alkyl, hydroxyalkyl, alkoxyalkylenyl, heterocyclyl arylalkylenyl, and aryl.
- R 2 is selected from the group consisting of hydrogen, alkyl, hydroxyalkyl, and alkoxyalkylenyl.
- R 2 is selected from the group consisting of hydrogen, methyl, ethyl, r ⁇ -propyl, isopropyl, isopropenyl, r ⁇ -butyl, sec-butyl, tert-butyl, methoxymethyl, ethoxymethyl, 2-methoxyethyl, hydroxymethyl, 1-hydroxyethyl, 2-hydroxyethyl, 2-hydroxy-l-methylethyl, 3-hydroxypropyl, cyclohexyl, cyclopentyl, tetrahydropyran-4-yl, tetrahydrofuran-3-yl, and phenyl.
- R 2 is selected from the group consisting of hydrogen, methyl, ethyl, » -propyl, n -butyl, methoxymethyl, ethoxymethyl, 2-methoxyethyl, hydroxymethyl, 2-hydroxyethyl, and 3- hydroxypropyl.
- R 2 is selected from the group consisting of hydrogen, methyl, ethyl, n-propyl, isopropyl, ra-butyl, ethoxymethyl, hydroxymethyl, and phenyl.
- R 2 is hydrogen, methyl, ethyl, ⁇ -propyl, ⁇ -butyl, methoxymethyl, ethoxymethyl, 2-methoxyethyl, hydroxymethyl, 2-hydroxyethyl, 3-hydroxypropyl, isopropyl, sec-butyl, tert-butyl, isopropenyl, cyclopentyl, cyclohexyl, 1-hydroxyethyl, 2-hydroxy-l-methylethyl, tetrahydrofuran-3-yl, or tetrahydropyran-4-yl.
- R 3 is selected from the group consisting of: -Z-R 4 ,
- R 3 is -Z-R 4 ', or -Z-X'-Y'-Rj.
- R 3 is -Z-R 4 .
- R 3 is phenyl or pyridin-3-yl.
- R 4 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, heteroaryl, and heterocyclyl wherein the alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, heteroaryl, and heterocyclyl groups can be unsubstituted or substituted by one or more substituents independently selected from the group consisting of alkyl, alkoxy, haloalkyl, haloalkoxy, halogen, nitro, hydroxy, mercapto, cyano, carboxy, formyl, aryl, aryloxy, arylalkoxy, heteroaryl, heteroaryloxy, heteroarylalkoxy, heterocyclyl, heterocyclylalkylenyl, amino, alkylamino, (arylalkylenyl)amino, dialkylamino, and in the case of alkyl
- R 4 is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl, heteroaryl, and heterocyclyl wherein the alkyl, alkenyl, alkynyl, aryl, heteroaryl, and heterocyclyl groups can be unsubstituted or substituted by one or more substituents independently selected from the group consisting of alkyl, alkoxy, haloalkyl, haloalkoxy, halogen, nitro, hydroxy, mercapto, cyano, carboxy, formyl, aryl, aryloxy, arylalkoxy, heteroaryl, heteroaryloxy, heteroarylalkoxy, heterocyclyl, heterocyclylalkylenyl, amino, alkylamino, (arylalkylenyl)amino, dialkylamino, and in the case of alkyl, alkenyl, alkynyl, and heterocyclyl, o
- R 4 ' is selected from the group consisting of hydrogen, alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl wherein the alkyl, alkenyl, alkynyl, aryl, arylalkylenyl, aryloxyalkylenyl, alkylarylenyl, heteroaryl, heteroarylalkylenyl, heteroaryloxyalkylenyl, alkylheteroarylenyl, and heterocyclyl groups can be unsubstituted or substituted by one or more substituents independently selected from the group consisting of alkyl, alkoxy, hydroxyalkyl, haloalkyl, haloalk
- R 5 1 is selected from the group consisting of:
- R 6 is selected from the group consisting of hydrogen, alkyl, and arylalkylenyl.
- R 6 is hydrogen.
- R 8 is C 2-7 alkylene.
- R 9 is selected from the group consisting of hydrogen, alkyl, and arylalkylenyl.
- R 9 is hydrogen.
- R 9 and R 4 together with the nitrogen atom to which R 9 is
- R 10 is C 3-8 alkylene.
- R 11 is selected from the group consisting of hydrogen, CM O alkyl, C 2-10 alkenyl, Ci -10 alkoxyC 2-10 alkylenyl, and arylCi -10 alkylenyl.
- Ri 2 is selected from the group consisting of hydrogen and alkyl.
- A is selected from the group consisting of: -CH(R 6 )-,
- A is -0-.
- A is -0-, -N(R 6 )-, -N(Y-R 4 )-, or -N(X-N(Re)-Y-R 4 )-.
- A' is selected from the group consisting Of-CH 2 -, -0-, -C(O)-, -S(O) 0-2 -, and -N(R 4 ')-.
- a 1 is -O- or -N(R 4 1 )-.
- Q is selected from the group consisting of a bond,
- V is selected from the group consisting Of -C(R 7 )-, -0-C(R 7 )-, -N(Rn)-C(R 7 )-, and -S(O) 2 -.
- W is selected from the group consisting of a bond, -C(O)-, and -S(O) 2 -.
- X is C 2-20 alkylene.
- X is C 2-4 alkylene.
- X' is selected from the group consisting of alkylene, alkenylene, alkynylene, arylene, heteroarylene, and heterocyclylene wherein the alkylene, alkenylene, and alkynylene groups can be optionally interrupted or terminated by arylene, heteroarylene, or heterocyclylene and optionally interrupted by one or more -O- groups.
- X" is C 1-20 alkylene.
- X" is C 1-4 alkylene.
- Y is selected from the group consisting of -C(R 7 )-, -C(R 7 )O-, -S(O) 2 -, -S(O) 2 -N(R 6 )-, and -C(R 7 )-N(R 9 )-.
- Y is -C(R 7 )-, -S(O) 2 -, or -C(R 7 )-N(R 9 )-.
- R 7 0.
- R 9 is hydrogen.
- Y' is selected from the group consisting of: -S(O) 0-2 -, -S(O) 2 -N(R 11 )-, -C(R 7 )-, -C(Ry)-O-, -0-C(R 7 )-, -0-C(O)-O-, -N(Rn)-Q-, -C(R 7 )-N(R n )-,
- Z is selected from the group consisting of -O- and -S(O) 0-2 -.
- Z' is a bond or -0-.
- Z' is a bond.
- a and b are independently integers from 1 to 4.
- a and b are independently integers from 2 to 4.
- c and d are independently integers from 1 to 6.
- c + d is ⁇ 7.
- A' is -O- and c and d are independently integers from 2 to 4.
- a 1 is -N(R 4 1 )- and c and d are independently integers from 2 to 4.
- m is 0 or 1.
- m is 0.
- m is 1.
- when m is 1, n is 0, 1, or 2.
- n is 0.
- n is 1.
- n is 2.
- m is 1 and p is 0.
- m is 1 and p is 1.
- m is 1 and p is 2.
- n is an integer from 0 to 4. For certain embodiments, n is 0. For certain embodiments, n is 0, 1, or 2.
- p is an integer from 0 to 3. For certain embodiments, p is 0. For certain embodiments, p is 0, 1, or 2.
- n is 0.
- m is 0 and p is 0.
- R' and R"" are each independently C 1-10 alkyl, C 3-7 cycloalkyl, phenyl, or benzyl, each of which may be unsubstituted or substituted by one or more substitutents selected from the group consisting of halogen, hydroxy, nitro, cyano, carboxy, C 1-6 alkyl, C 1-4 alkoxy, aryl, heteroaryl, arylC 1-4 alkylenyl, heteroarylC 1-4 alkylenyl, haloC 1-4 alkyl, haloCi.4 alkoxy, -0-C(O)-CH 3 , -C(O)-O-CH 3 , -C(O)-NH 2 , -0-CH 2 -C(O)-NH 2 , -NH 2 , and -S(O) 2 -NH 2 .
- ⁇ -aminoacyl is an acyl group derived from an amino acid selected from the group consisting of racemic, D-, and L-amino acids.
- Y 2 is selected from the group consisting of hydrogen, C 1-6 alkyl, and benzyl.
- Y 0 is selected from the group consisting of C 1-6 alkyl, carboxyC 1-6 alkyl, aminoC 1-4 alkyl, mono-iV-C 1-6 alkylaminoC 1-4 alkyl, and di-N,JV-C 1-6 alkylaminoC 1-4 alkyl.
- Y 1 is selected from the group consisting of mono-N-C 1-6 alkylamino, di-iV,N-C 1-6 alkylamino, morpholin-4-yl, piperidin-1-yl, pyrrolidin-1-yl, and 4-C 1-4 alkylpiperazin-1-yl.
- alkyl As used herein, the terms “alkyl”, “alkenyl”, “alkynyl” and the prefix “alk ⁇ ” are inclusive of both straight chain and branched chain groups and of cyclic groups, e.g., cycloalkyl and cycloalkenyl. Unless otherwise specified, these groups contain from 1 to 20 carbon atoms, with alkenyl groups containing from 2 to 20 carbon atoms, and alkynyl groups containing from 2 to 20 carbon atoms. In some embodiments, these groups have a total of up to 10 carbon atoms, up to 8 carbon atoms, up to 6 carbon atoms, or up to 4 carbon atoms.
- Cyclic groups can be monocyclic or polycyclic and preferably have from 3 to 10 ring carbon atoms.
- Exemplary cyclic groups include cyclopropyl, cyclopropylmethyl, cyclopentyl, cyclohexyl, adamantyl, and substituted and unsubstituted bornyl, norbornyl, and norbornenyl.
- alkylene "-alkylene-”, “alkenylene”, “-alkenylene-”, “alkynylene”, and “-alkynylene-” are the divalent forms of the “alkyl”, “alkenyl”, and “alkynyl” groups defined above.
- alkylenyl “alkenylenyl”, and “alkynylenyl” are used when “alkylene”, “alkenylene”, and “alkynylene", respectively, are substituted.
- an arylalkylenyl group comprises an "alkylene” moiety to which an aryl group is attached.
- haloalkyl is inclusive of alkyl groups that are substituted by one or more halogen atoms, including perfluorinated groups. This is also true of other groups that include the prefix "halo-". Examples of suitable haloalkyl groups are chloromethyl, trifluoromethyl, and the like.
- aryl as used herein includes carbocyclic aromatic rings or ring systems. Examples of aryl groups include phenyl, naphthyl, biphenyl, fluorenyl and indenyl.
- heteroatom refers to the atoms O, S, or N.
- heteroaryl includes aromatic rings or ring systems that contain at least one ring heteroatom (e.g., O, S, N).
- heteroaryl includes a ring or ring system that contains 2 to 12 carbon atoms, 1 to 3 rings, 1 to 4 heteroatoms, and O, S, and/or N as the heteroatoms.
- Suitable heteroaryl groups include furyl, thienyl, pyridyl, quinolinyl, isoquinolinyl, indolyl, isoindolyl, triazolyl, pyrrolyl, tetrazolyl, imidazolyl, pyrazolyl, oxazolyl, thiazolyl, benzofuranyl, benzothiophenyl, carbazolyl, benzoxazolyl, pyrimidinyl, benzimidazolyl, quinoxalinyl, benzothiazolyl, naphthyridinyl, isoxazolyl, isothiazolyl, purinyl, quinazolinyl, pyrazinyl, 1-oxidopyridyl, pyridazinyl, triazinyl, tetrazinyl, oxadiazolyl, thiadiazolyl, and so on.
- heterocyclyl includes non-aromatic rings or ring systems that contain at least one ring heteroatom (e.g., O, S, N) and includes all of the fully saturated and partially unsaturated derivatives of the above mentioned heteroaryl groups.
- heterocyclyl includes a ring or ring system that contains 2 to 12 carbon atoms, 1 to 3 rings, 1 to 4 heteroatoms, and O, S, and N as the heteroatoms.
- heterocyclic groups include pyrrolidinyl, tetrahydrofuranyl, morpholinyl, thiomorpholinyl, 1,1-dioxothiomorpholinyl, piperidinyl, piperazinyl, thiazolidinyl, imidazolidinyl, isothiazolidinyl, tetrahydropyranyl, quinuclidinyl, homopiperidinyl (azepanyl), 1,4-oxazepanyl, homopiperazinyl (diazepanyl), 1,3-dioxolanyl, aziridinyl, azetidinyl, dihydroisoquinolin-(lH)-yl, octahydroisoquinolin-(lH)-yl, dihydroquinolin-(2H)-yl, octahydroquinolin-(2H)-yl, dihydro-lH-imi
- heterocyclyl includes bicylic and tricyclic heterocyclic ring systems. Such ring systems include fused and/or bridged rings and spiro rings. Fused rings can include, in addition to a saturated or partially saturated ring, an aromatic ring, for example, a benzene ring. Spiro rings include two rings joined by one spiro atom and three rings joined by two spiro atoms.
- heterocyclyl contains a nitrogen atom
- the point of attachment of the heterocyclyl group may be the nitrogen atom
- arylene is the divalent forms of the "aryl”, “heteroaryl”, and “heterocyclyl” groups defined above.
- arylenyl is used when “arylene”, “heteroarylene”, and “heterocyclylene”, respectively, are substituted.
- an alkylarylenyl group comprises an arylene moiety to which an alkyl group is attached.
- each group is independently selected, whether explicitly stated or not.
- each R 12 group is independently selected for the formula -N(R 12 ) 2 .
- each R 6 group is independently selected when an R 1 and an R 2 group both contain an R 6 group.
- each R 6 group is independently selected when more than one
- R / 8 group is present (i.e., R 5 and R 5 ' both contain a 8 group) each R 8 group is independently selected and each R 7 group is independently selected.
- the invention is inclusive of the compounds described herein (including intermediates) in any of their pharmaceutically acceptable forms, including isomers (e.g., diastereomers and enantiomers), salts, solvates, polymorphs, prodrugs, and the like.
- isomers e.g., diastereomers and enantiomers
- salts e.g., sodium bicarbonate
- solvates e.g., sodium bicarbonate
- polymorphs e.g., sodium bicarbonate
- prodrugs e.g., sodium bicarbonate
- the term “compound” includes any or all of such forms, whether explicitly stated or not (although at times, “salts" are explicitly stated).
- prodrug means a compound that can be transformed in vivo to yield an immune response modifying compound in any of the salt, solvated, polymorphic, or isomeric forms described above.
- the prodrug itself, may be an immune response modifying compound in any of the salt, solvated, polymorphic, or isomeric forms described above.
- the transformation may occur by vaious mechanisms, such as through a chemical (e.g., solvolysis or hydrolysis, for example, in the blood) or enzymatic biotransformation.
- a discussion of the use of prodrugs is provided by T. Higuchi and W. Stella, "Pro-drugs as Novel Delivery Systems," Vol. 14 of the A. C. S. Symposium Series, and in Bioreversible Carriers in Drug Design, ed. Edward B. Roche, American Pharmaceutical Association and Pergamon Press, 1987.
- Compounds of the invention may be synthesized by synthetic routes that include processes analogous to those well known in the chemical arts, particularly in light of the description contained herein.
- the starting materials are generally available from commercial sources such as Aldrich Chemicals (Milwaukee, Wisconsin, USA) or are readily prepared using methods well known to those skilled in the art (e.g., prepared by methods generally described in Louis F. Fieser and Mary Fieser, Reagents for Organic Synthesis, v. 1-19, Wiley, New York, (1967-1999 ed.); Alan R. Katritsky, Otto Meth- Cohn, Charles W. Rees, Comprehensive Organic Functional Group Transformations, v 1- 6, Pergamon Press, Oxford, England, (1995); Barry M.
- reaction schemes depicted below provide potential routes for synthesizing the compounds of the present invention as well as key intermediates.
- EXAMPLES section below For more detailed description of the individual reaction steps, see the EXAMPLES section below.
- Other synthetic routes may be used to synthesize the compounds of the invention.
- specific starting materials and reagents are depicted in the reaction schemes and discussed below, other starting materials and reagents can be easily substituted to provide a variety of derivatives and/or reaction conditions.
- many of the compounds prepared by the methods described below can be further modified in light of this disclosure using conventional methods well known to those skilled in the art.
- a 4-chloro-3-nitroquinoline of Formula XX is reduced to provide a 3-amino-4-chloroquinoline of Formula XXI.
- the reduction can be carried out using a conventional heterogeneous hydrogenation catalyst such as platinum on carbon or palladium on carbon.
- a platinum catalyst is preferred.
- the reaction can be conveniently carried out on a Parr apparatus in a suitable solvent such as toluene and/or isopropanol.
- Many compounds of Formula XX are known or can be prepared using known synthetic methods, see for example, U.S. Patent Nos. 4,689,338; 5,175,296; 5,367,076; and 5,389,640; and the documents cited therein.
- XXI is known.
- 3-amino-4-chloroquinoline, 3-amino-4,5- dichloroquinoline, and 3-amino-4,7-dichloroquinoline have been prepared by Surrey et al., Journal of the American Chemical Society, 73, pp. 2413-2416 (1951).
- step (1) an aqueous solution of sodium dithionite can be added to a solution or suspension of the compound of Formula XX in a suitable solvent such as ethanol or isopropanol.
- a suitable solvent such as ethanol or isopropanol.
- the reaction can be carried out at an elevated temperature, for example, at reflux, or at ambient temperature.
- step (2) of Reaction Scheme I a 3-amino-4-chloroquinoline of Formula XXI is reacted with an acyl halide of Formula R 2 C(O)Cl or R 2 C(O)Br to provide an N-(4- chloroquinolin-3-yl) amide of Formula XXII.
- the acyl halide is added to a solution of a compound of Formula XXI in a suitable solvent such as anhydrous dichloromethane optionally in the presence of a base such as triethylamine.
- a suitable solvent such as anhydrous dichloromethane optionally in the presence of a base such as triethylamine.
- the reaction can be run at a reduced temperature, for example, 0° C, or at ambient temperature.
- the compound of Formula XXI can be reacted with a formylating agent such as, for example, diethoxymethyl acetate.
- compounds wherein R 2 is hydrogen can be prepared utilizing amidines such as iV-(4-chloroquinolin-3-yl)-N,N- dimethylimidoformamide.
- XXII is reacted with a hydroxylamine hydrochloride of Formula R 1 ONH 2 ⁇ HCl and cyclized to provide a lH-imidazo[4,5-c]quinoline of Formula XXIII.
- the hydroxylamine hydrochloride is added to a solution of a compound of Formula XXII in an alcoholic solvent.
- the reaction can be carried out at an elevated temperature, for example, at reflux.
- the compound of Formula XXII and the hydroxylamine are refluxed in ethanol.
- Some hydroxylamine hydrochlorides of Formula R 1 ONH 2 ⁇ HCl are commercially available, others can be prepared using known synthetic methods.
- a lH-imidazo[4,5-c]quinoline of Formula XXIII is oxidized to provide an N-oxide of Formula XXIV using a conventional oxidizing agent that is capable of forming iV-oxides.
- the reaction is carried out by treating a solution of a compound of Formula XXIII in a suitable solvent such as chloroform or dichloromethane with 3-chloroperoxybenzoic acid at ambient temperature.
- an N-oxide of Formula XXIV is animated to provide a lH-imidazo[4,5-c]quinolin-4-amine of the Formula XXV, which is a subgenus 5 of compounds of Formulas I, II, and IV.
- Step (5) can be carried out by the activation of an iV-oxide of Formula XXIV by conversion to an ester and then reacting the ester with an aminating agent.
- Suitable activating agents include alkyl- or arylsulfonyl chorides (e.g., benzenesulfonyl choride, methanesulfonyl choride, andp-toluenesulfonyl chloride).
- Suitable aminating agents include ammonia (e.g. in the form of ammonium hydroxide)
- ammonium salts e.g., ammonium carbonate, ammonium bicarbonate, ammonium phosphate.
- the reaction can be carried out by dissolving a compound of Formula XXIV in a suitable solvent such as chloroform, adding ammonium hydroxide to the solution, and then adding benzenesulfonyl chloride.
- a suitable solvent such as chloroform
- step (4) the oxidation of step (4) and the animation of step (5) can be carried out sequentially without isolating the product of the oxidation to provide a IH- imidazo[4,5-c]quinolin-4-amine of the Formula XXV.
- step (4) after the IH- imidazo[4,5-c]quinoline of Formula XXIII is consumed by reaction with 3- chloroperoxybenzoic acid as described in step (4), the aminating and acylating agents are 0 added to the reaction mixture as in step (5).
- the product or a pharmaceutically acceptable salt thereof can be isolated using conventional methods.
- step (5) can be carried out by the reaction of an iV-oxide of Formula XXrV with trichloroacetyl isocyanate followed by hydrolysis of the resulting intennediate to provide a lH-imidazo[4,5-c]quinolin-4-amine of the Formula XXV.
- the reaction is
- step 25 conveniently carried out in two steps by (i) adding trichloroacetyl isocyanate to a solution of the iV-oxide of Formula XXIV in a solvent such as dichloromethane and stirring at ambient temperature to provide an isolable amide intermediate.
- step (ii) a solution of the intermediate in methanol is treated with a base such as sodium methoxide at ambient temperature.
- a base such as sodium methoxide
- R, R 1 , R 2 , and m are as defined above;
- E is carbon (imidazoquinoline ring) or nitrogen (imidazonaphthyridine ring); n is an integer from 0 to 4 (imidazoquinoline ring) or 0 to 3 (imidazonaphthyridine ring) with the proviso that when m is 1, n is 0 or 1; and D is -Br, -I, or -OCH 2 Ph; wherein Ph is phenyl.
- an aniline or aminopyridine of Formula XXVI is treated with the condensation product generated from 2,2-dimethyl-l,3-dioxane-4,6-dione (Meldrum's acid) and triethyl orthoformate to provide an imine of Formula XXVII.
- the reaction is conveniently carried out by adding a solution of an aniline or aminopyridine of Formula XXVI to a heated mixture of Meldrum's acid and triethyl orthoformate and heating the reaction at an elevated temperature.
- Many anilines and aminopyridines of Formula XXVI are commercially available; others can be prepared by known synthetic methods.
- benzyloxypyridines of Formula XXVI can be prepared using the method of Holladay et al., Biorg. Med. Chem. Lett, 8, pp. 2797-2802, (1998).
- step (2) of Reaction Scheme II an imine of Formula XXVII undergoes thermolysis and cyclization to provide a [l,5]naphthyridin-4-ol or a quinolin-4-ol of Formula XXVIII.
- the reaction is conveniently carried out in a medium such as DOWTHERM A heat transfer fluid at a temperature between 200 and 250 0 C.
- step (3) of Reaction Scheme II a [l,5]naphthyridin-4-ol or a quinolin-4-ol of Formula XXVIII is nitrated under conventional nitration conditions to provide a 3- mtro[l,5]naphthyridin-4-ol or 3-nitroquinolin-4-ol of Formula XXIX.
- the reaction is conveniently carried out by adding nitric acid to a compound of Formula XXVIII in a suitable solvent such as propionic acid and heating the mixture at an elevated temperature.
- step (4) of Reaction Scheme II a 3-nitro[l,5]naphthyridin-4-ol or 3- nitroquinolin-4-ol of Formula XXIX is reduced to provide a 3-amino[l,5]naphthyridin-4- ol or 3-aminoquinolin-4-ol of Formula XXX.
- the reduction can be carried out using the methods described in step (1) of Reaction Scheme I.
- a 3-amino[l,5]naphthyridin-4-ol or 3- aminoquinolin-4-ol of Formula XXX is chlorinated using conventional chlorination chemistry to provide a 3-amino-4-chloro[l,5]naphthyridine or 3-amino-4-chloroquinoline of Formula XXXI.
- the reaction is conveniently carried out by treating a compound of Formula XXX with phosphorous oxychloride in a suitable solvent such as N 1 N- dimethylformamide (DMF).
- DMF N 1 N- dimethylformamide
- the reaction can be carried out at ambient temperature or at an elevated temperature such as 100 °C.
- step (6) of Reaction Scheme II a 3-ammo-4-chloro[l,5]naphthyridine or 3- amino-4-chloroquinoline of Formula XXXI is reacted with an acyl halide of Formula R 2 C(O)Cl or R 2 C(O)Br to provide an N-(4-chloro[l,5]naphthyridm-3-yl) amide or /V-(4- chloroquinolin-3-yl) amide of Formula XXXII.
- the reaction can be carried out using the method described in step (2) of Reaction Scheme I.
- step (7) of Reaction Scheme II aniV-(4-chloro[l,5]naphthyridin-3-yl) amide or /V-(4-chloroqumolin-3-yl) amide of Formula XXXII is reacted with a hydroxylamine hydrochloride of Formula R 1 ONH 2 ⁇ HCl and cyclized to provide a lH-imidazo[4,5- c][l,5]naphthyridine or a lH-imidazo[4,5-c]quinoline of Formula XXXIII.
- the reaction can be carried out using the method described in step (3) of Reaction Scheme I.
- a lH-imidazo[4,5-c][l,5]naphthyridine or a lH-imidazo[4,5-c]quinoline of Formula XXXIII is oxidized to provide an TV-oxide of Formula XXXIV and then animated to provide a lH-imidazo[4,5-c][l,5]naphthyridin-4- amine or a lH-imidazo[4,5-c]quinolin-4-amine of the Formula XXXV, which is a subgenus of compounds of Formulas I and II.
- the reactions can be carried out using the methods described in steps (4) and (5) of Reaction Scheme I, and the product or a pharmaceutically acceptable salt thereof can be isolated using conventional methods.
- compounds in Reaction Scheme II can be further elaborated using known synthetic methods.
- the acid chloride used in step (6) of Reaction Scheme II may contain a protected hydroxy or amino group.
- Some acid chlorides of this type, for example acetoxyacetyl chloride, are commercially available. Others can be prepared by known synthetic methods.
- the protected hydroxy or amino group may be deprotected and further functionalized before step (8) of Reaction Scheme II.
- this type of functionalization of an R 2 group see U.S. Patent No. 5,389,640 (Gerster et al.).
- Compounds of the invention can be prepared according to Reaction Scheme III, where R, R 1 , R 2 , and n are as defined above, Hal is Br or I, and R 3a is as defined below.
- Formula XXXVa is a subset of Formula XXXV where D is Br or I.
- Compounds of Formula XXXVa can be prepared according to the method of Reaction Scheme II.
- Reaction Scheme III can be carried out using known palladium-catalyzed coupling reactions such as Suzuki coupling, Stille coupling, Sonogashira coupling, and the Heck reaction.
- a lH-imidazo[4,5-c][l,5]naphthyridin-4-amine or a lH-imidazo[4,5-c]qumolin-4-amine of Formula XXXVa undergoes Suzuki coupling with a boronic acid of Formula R 3a -B(OH) 2 , an anhydride thereof, or a boronic acid ester of Formula R 3a -B(O-alkyl) 2 to provide a lH-imidazo[4,5-c][l,5]naphthyridin-4-amine or a lH-imidazo[4,5-c]quinolin-4-amine of Formula XXXVI, a subgenus of Formula II, wherein R 3a is -Z-R 4 ', -Z'-X'-RV, -Z'-X'-Y'-RV, or -Z'-X'-R 5 '; -Z' is a bond; -Z'
- the coupling is carried out by combining a compound of Formula XXXVa with a boronic acid or an ester or anhydride thereof in the presence of palladium (II) acetate, triphenylphosphine, and a base such as sodium carbonate in a suitable solvent such as n-propanol.
- the reaction can be carried out at an elevated temperature (e.g., 80-100 0 C).
- R 33 -B(OH) 2 , anhydrides thereof, and boronic acid esters of Formula R 3a -B(O-alkyl) 2 are commercially available; others can be readily prepared using known synthetic methods. See, for example, Li, W. et al, J. Org. Chem., 67, 5394-5397 (2002).
- the product of Formula XXXVI or a pharmaceutically acceptable salt thereof can be isolated by conventional methods.
- the Heck reaction can also be used in Reaction Scheme III to provide compounds of Formula XXXVI, wherein R 3a is -Z-X-R 4 ' or -Z'-X'-Y'-RV; -Z' is a bond; -X'- is alkenylene optionally terminated by arylene or heteroarylene; and R 4 ' and Y' are as defined above.
- the Heck reaction is carried out by coupling a lH-imidazo[4,5-c][l,5]naphthyridine-4-amine or a lH-imidazo[4,5-c]quinolin-4-amine of Formula XXXVa with a vinyl-substituted arylene or heteroarylene compound.
- arylene or heteroarylene compounds such as 2-vinylpyridine, 3-vinylpyridine, and 4-vinylpyridine
- the reaction is conveniently carried out by combining the lH-imidazo[4,5-c][l,5]naphthyridine-4-amine or lH-imidazo[4,5-c]quinolin-4-amine of Formula XXXVa and the vinyl-substituted compound in the presence of palladium (II) acetate, triphenylphosphine or tri-ort/zo-tolylphosphine, and a base such as triethylamine in a suitable solvent such as acetonitrile or toluene.
- the reaction can be carried out at an elevated temperature such as 100-120 °C under an inert atmosphere.
- the product or pharmaceutically acceptable salt thereof can be isolated using conventional methods.
- Step (1) of Reaction Scheme IV a 2,4-dichloro-3-nitropyridine of Formula XXXVII is reacted with a hydroxylamine hydrochloride of Formula R 1 ONH 2 ⁇ HCl to provide an N-(2-chloro-3-nitropyridin-4-yl)hydroxylamine of Formula XXXVIII.
- the reaction can be carried out by combining the hydroxylamine with a compound of Formula XXXVII in the presence of a base such as triethylamine in an inert solvent such as DMF.
- a base such as triethylamine
- an inert solvent such as DMF.
- the reaction can be carried out at an elevated temperature (about 80 0 C).
- 2,4-dichloro-3-nitropyridines of the Formula XXXVII are known and can be readily prepared using known synthetic methods. (See, for example, Dellaria et al, U.S. Pat. No. 6,525,064 and the references cited therein.)
- step (2) of Reaction Scheme IV an N-(2-chloro-3-nitropyridin-4- yl)hydroxylamine of Formula XXXVIII is reacted with t ⁇ -(4-methoxybenzyl)amine to provide an N- ⁇ 2-[ ⁇ zi?-(4-methoxybenzyl)ammo]-3-nitropyridm-4-yl ⁇ hydroxylamine of Formula XXXIX.
- the reaction can be carried out by adding the bfs-(4-methoxybenzyl)amine to a solution of a compound of Formula XXXVIII in a suitable solvent such as toluene in the presence of a base such as triethylamine.
- the reaction can be carried out at an elevated temperature (about 90 0 C).
- step (3) of Reaction Scheme IV an N- ⁇ 2-[ ⁇ w-(4-methoxybenzyl)amino]-3- nitropyridin-4-yl ⁇ hydroxylamine of Formula XXXIX is reduced to provide an N- ⁇ 3-ammo-2-[&z5-(4-methoxybenzyl)amino]pyridin-4-yl ⁇ hydroxylamine of Formula XL.
- the reduction can be carried out by treating a compound of Formula XXXIX with NiBH 4 .
- NiBH 4 is generated in situ by adding sodium borohydride to a mixture of nickel (II) chloride heptahydrate and methanol. A solution of a compound of Formula XXXIX in a suitable solvent such as 9:2 methanol:dichloromethane is then added to the catalyst. The reaction can be run at ambient temperature.
- step (4) of Reaction Scheme IV an N- ⁇ 3-ammo-2-[bis-(4- methoxybenzyl)amino]pyridin-4-yl ⁇ hydroxylamine of Formula XL is reacted with an acyl halide of Formula R 2 C(O)Cl or R 2 C(O)Br to provide an iV-(pyridin-3-yi)amide of Formula XLI.
- the reaction can be carried out using the method described in step (2) of Reaction
- step (5) of Reaction Scheme IV an 7V-(pyridin-3-yl)amide of Formula XLI is cyclized to provide a lH-imidazo[4,5-c]pyridine of Formula XLII.
- the reaction can be carried out by heating an amide of Formula XLI in toluene in the presence of pyridine hydrochloride.
- the reaction can be carried out at an elevated temperature, for example, at reflux.
- step (6) of Reaction Scheme IV the 4-methoxybenzyl groups on a lH-imidazo[4,5-c]pyridine of Formula XLII are removed by acid hydrolysis to provide a lH-imidazo[4,5-c]pyridin-4-amine of Formula III.
- the reaction can be carried out by treating a compound of Formula XLII with trifluoroacetic acid.
- the reaction can be carried out at ambient temperature.
- the product or a pharmaceutically acceptable salt thereof can be isolated using conventional methods.
- Step (1) the amino group of a compound of Formula XLIII can be acylated to provide a compound of Formula XLIV.
- the reaction can be conveniently carried out by reacting a compound of Formula XLIII with an alkyl malonyl chloride in the presence of a base such as triethylamine in a suitable solvent such as dichloromethane.
- a compound of Formula XLW can be cyclized to provide a compound of Formula XLV.
- the reaction can be conveniently carried out by adding a solution of a compound of Formula XLIV in a suitable solvent such as tetrahydrofuran (THF) to a suspension of sodium hydride (or other base capable of removing a malonyl methylene proton) in a suitable solvent such as THF.
- THF tetrahydrofuran
- the reaction can be run at an elevated temperature, for example, the reflux temperature.
- a compound of Formula XLV can be hydrolyzed and decarboxylated to provide a compound of Formula XLVI.
- the reaction can be carried out by conventional methods, for example, by combining a compound of Formula XLV with an acid, such as hydrochloric acid, with heating.
- a compound of Formula XLVI can be nitrated to provide a compound of Formula XLVII.
- the reaction can be carried out under conventional nitration conditions, such as by heating a compound of Formula XLVI in the presence of nitric acid, preferably in a solvent such acetic acid.
- a compound of Formula XLVII can be chlorinated to provide a 2,4-dichloro-3-nitro-5,6,7,8-tetrahydroquinoline of Formula XLVIII.
- the reaction can be carried out by combining a compound of Formula XLVII with a conventional chlorinating agent (e.g., phosphorus oxychloride, thionyl chloride, phosgene, oxalyl chloride, or phosphorus pentachloride), optionally in a solvent such as N,iV-dimethylformamide (DMF) or dichloromethane, with heating (e.g., at the reflux temperature).
- a conventional chlorinating agent e.g., phosphorus oxychloride, thionyl chloride, phosgene, oxalyl chloride, or phosphorus pentachloride
- a solvent such as N,iV-dimethylformamide (DMF) or dichloromethane
- step (6) of Reaction Scheme V 2,4-dichloro-3-nitro-5,6,7,8-tetrahydroquinoline of Formula XLVIII is reduced to provide a 3-amino-2,4-dichloro-5,6,7,8- tetrahydroquinoline of Formula XLIX.
- the reduction can be carried out using the methods described in step (1) of Reaction Scheme I.
- step (7) of Reaction Scheme V a 3-amino-2,4-dichloro-5,6,7,8- tetrahydroquinoline of Formula XLIX is reacted with an acyl halide of Formula R 2 C(O)Cl or R 2 C(O)Br to provide anN-(4-chloro-5,6,7,8-tetrahydroquinolin-3-yl) amide of Formula L.
- the reaction can be carried out using the method described in step (2) of Reaction Scheme I.
- step (8) of Reaction Scheme V anN-(4-chloro-5,6,7,8-tetrahydroquinolin-3-yl) amide of Formula L is reacted with a hydroxylamine hydrochloride of Formula R 1 ONH 2 ⁇ HCl and cyclized to provide a 4-chloro-6,7,8,9-tetrahydro-l#-imidazo[4,5- c]quinoline of Formula LI.
- the reaction can be carried out using the method described in step (3) of Reaction Scheme I.
- step (1) of Reaction Scheme VI a 3-amino[l,5]naphthyridin-4-ol or 3- aminoquinolin-4-ol of Formula XXX is reacted with an acyl halide of Formula R 2 C(O)Cl or R 2 C(O)Br to provide an N-(4-hydroxy[l,5]naphthyridin-3-yl) amide or JV-(4- hydroxyquinolin-3-yl) amide of Formula LVIII.
- the reaction can be carried out using the method described in step (2) of Reaction Scheme I.
- an7V-(4-hydroxy[l,5]naphthyridin-3-yl) amide or N-(4 ⁇ hydroxyquinolin-3-yl) amide of Formula LVIII is reacted with a triflating reagent such as iV-phenyl-bis(trifluoromethanesulfonamide) to provide a [l,5]naphthyridin-3-yl trifluoromethanesulfonate or quinolin-3-yl trifluoromethanesulfonate of Formula LIX.
- step (3) of Reaction Scheme VI a [l,5]naphthyridin-3-yl trifluoromethanesulfonate or quinolin-3-yl trifluoromethanesulfonate of Formula LIX is reacted with a hydroxylamine hydrochloride of Formula R 1 ONH 2 ⁇ HCl and cyclized to provide a lH-imidazo[4,5-c][l,5]naphthyridine or a l ⁇ f-imidazo[4,5-c]quinoline of Formula XXXIII.
- the reaction can be carried out using the method described in step (3) of Reaction Scheme I.
- a lH " -imidazo[4,5-c][l,5]naphthyridine or a lH-imidazo[4,5-c]quinoline of Formula XXXIII is oxidized to provide an N-oxide of Formula XXXIV and then aminated to provide a lH-imidazo[4,5-c][l,5]naphthyridin-4- amine or a lH " -imidazo[4,5-c]quinolin-4-amine of the Formula XXXV, which is a subgenus of compounds of Formulas I and II.
- the reactions can be carried out using the methods described in steps (4) and (5) of Reaction Scheme I, and the product or a pharmaceutically acceptable salt thereof can be isolated using conventional methods.
- Step (1) of Reaction Scheme VII an N-(4-chloro[l,5]naphthyridin-3-yl) amide or N-(4-chloroquinolin-3-yl) amide of Formula XXXII is reacted with a hydroxylamine of the Formula BoC-CH 2 -X-CH 2 -ONH 2 and cyclized to provide a lH-imidazo[4,5- c][l,5]naphthyridine or a lH-imidazo[4,5-c]quinoline of Formula LX.
- the reaction can be carried out using the method described in step (3) of Reaction Scheme I. ⁇ ydroxylamines of the Formula Boc-C ⁇ 2 -X-C ⁇ 2 -
- step (2) of Reaction Scheme VII the tert-butoxycarbonyl protecting group is removed under acidic conditions to provide an amino substituted lH-imidazo[4,5- c][l,5]naphthyridine or lH-imidazo[4,5-c]quinoline of Formula LXI.
- a compound of Formula LX can be combined with a solution of hydrogen chloride in ethanol and heated.
- step (3) of Reaction Scheme VII an amino substituted l/J-imidazo[4,5- c][l,5]naphthyridine or lH-imidazo[4,5-cJquinoline of Formula LXI is converted to a substituted lH-imidazo[4,5-c][l,5]naphthyridine or lH-imidazo[4,5-c]qumoline of Formula LXII using conventional methods.
- a lH-imidazo[4,5- c][l,5]naphthyridine or a lH-imidazo[4,5-c]quinoline of the Formula LXI can react with an acid chloride of Formula R 4 C(O)Cl to provide a compound of Formula LXII in which Y is -C(O)-.
- a compound of the Formula LXI can react with sulfonyl chloride OfFOmIuIaR 4 S(O) 2 Cl or a sulfonic anhydride of Formula (R 4 S(O) 2 ⁇ O to provide a compound of Formula LXII in which Y is -S(O) 2 -.
- a compound of the Formula LXI can also react with a chloroformate of Formula R 4 CO(O)Cl to provide a compound of Formula LXII in which Y is -C(O)-O-.
- Numerous acid chlorides of Formula R 4 C(O)Cl, sulfonyl chlorides of Formula R 4 S(O) 2 Cl, sulfonic anhydrides of Formula (R 4 S(O) ⁇ 2 O, and chloroformates of Formula R 4 CO(O)Cl are commercially available; others can be readily prepared using known synthetic methods.
- the reaction can be conveniently carried out by adding the acid chloride of Formula R 4 C(O)Cl, chloroformate of Formula R 4 CO(O)Cl, sulfonyl chloride of Formula R 4 S(O) 2 Cl, or sulfonic anhydride of Formula (R 4 S(O) ⁇ 2 O to a solution of a compound of Formula LXI and a base such as triethylamine in a suitable solvent such as chloroform, dichloromethane, or acetonitrile.
- the reaction can be carried out at ambient temperature or at a sub-ambient temperature such as 0 0 C.
- reaction can be carried out at ambient temperature or at a sub-ambient temperature such as O °C.
- a compound of Formula LXI can be treated with carbamoyl chlorides of Formula R 4 N-(Rg)-C(O)Cl or Formula
- Sulfamides of Formula LXII where Y is -S(O) 2 -N(R 6 )- wherein R 6 is as defined above, can be prepared by reacting a compound of Formula LXI with sulfuryl chloride to generate a sulfamoyl chloride in situ, and then reacting the sulfamoyl chloride with an amine of formula HN(R 6 )R 4 .
- sulfamides of Formula LXII can be prepared by reacting a compound of Formula LXI with a sulfamoyl chloride of Formula R 4 (R 6 )N-S(O) 2 Cl under the reaction conditions described above for reaction of compounds of Formula LXI with sulfonyl chlorides.
- Many amines of Formula HN(R 6 )R 4 , and some sulfamoyl chlorides of Formula R 4 (R 6 )N-S(O) 2 Cl are commercially available; others can be prepared using known synthetic methods.
- a lH-imidazo[4,5- c][l,5]naphthyridine or a lH " -imidazo[4,5-c]quinoline of Formula LXII is oxidized to provide an N-oxide of Formula LXIII and then aminated to provide a lH-imidazo[4,5- c][l,5]naphthyridin-4-amine or a l/f-imidazo[4,5-c]quinolin-4-amine of the Formula LXIV.
- the reactions can be carried out using the methods described in steps (4) and (5) of Reaction Scheme I, and the product or a pharmaceutically acceptable salt thereof can be isolated using conventional methods.
- Prodrugs can be prepared in a variety of ways.
- a compound wherein R 2 is hydroxyalkyl can be converted into a prodrug wherein R 2 is, for example, -alkylenyl-O-C(R 7 )-R4, -alkylenyl-O-C(R 7 )-O-R 4 , or -alkylenyl-O-C(R 7 )-N(R6)-R4, wherein R 4 , R 6 , and R 7 are as defined above, using methods known to one skilled in the art.
- a compound wherein R is hydroxy may also be converted to an ester, an ether, a carbonate, or a carbamate.
- a prodrug can be formed by the replacement of the hydrogen atom of the alcohol group with a group such as C 1-6 alkanoyloxymethyl, 1-(C 1-6 alkanoyloxy)ethyl, 1-methyl-l -(C 1-6 alkanoyloxy)ethyl, C 1-6 alkoxycarbonyloxymethyl, N-(Ci - 6 alkoxycarbonyl)aminomethyl, succinoyl, C 1-6 alkanoyl, ⁇ -ammoC 1-4 alkanoyl, arylacyl, -P(O)(OH) 2 , -P(O)(O-C 1-6 alkyl) 2 , C 1-6 alkoxycarbonyl, C 1-6 alkylcarbamoyl, and ⁇ -aminoacyl or ⁇ -aminoacyl- ⁇ -aminoacyl, where each ⁇ -aminoacyl group is independently selected from the naturally occurring race
- prodrugs are esters made from carboxylic acids containing one to six carbon atoms, unsubstituted or substituted benzoic acid esters, or esters made from naturally occurring L-amino acids.
- Prodrugs can also be made from a compound containing an amino group by conversion of the amino group to a functional group such as an amide, carbamate, urea, amidine, or another hydroylizable group using conventional methods.
- R 1 and R"" are each independently C 1-10 alkyl, C 3-7 cycloalkyl, phenyl, or benzyl, each of which may be unsubstituted or substituted by one or more substitutents selected from the group consisting of halogen, hydroxy, nitro, cyano, carboxy, C 1-6 alkyl, C 1-4 alkoxy, aryl, heteroaryl, arylC 1-4 alkylenyl, heteroarylC 1-4 alkylenyl, haloC 1-4 alkyl, haloC 1-4 alkoxy, -0-C(O)-CH 3 , -C(O)-O-CH 3 , -C(O)-NH 2 , -0-CH 2 -C(O)
- compositions and Biological Activity contain a therapeutically effective amount of a compound or salt of the invention as described above in combination with a pharmaceutically acceptable carrier.
- a therapeutically effective amount and “effective amount” mean an amount of the compound or salt sufficient to induce a therapeutic or prophylactic effect, such as cytokine induction, immunomodulation, antitumor activity, and/or antiviral activity.
- a therapeutic or prophylactic effect such as cytokine induction, immunomodulation, antitumor activity, and/or antiviral activity.
- the exact amount of active compound or salt used in a pharmaceutical composition of the invention will vary according to factors known to those of skill in the art, such as the physical and chemical nature of the compound or salt, the nature of the carrier, and the intended dosing regimen, it is anticipated that the compositions of the invention will contain sufficient active ingredient to provide a dose of about 100 nanograms per kilogram (ng/kg) to about 50 milligrams per kilogram (mg/kg), preferably about 10 micrograms per kilogram ( ⁇ g/kg) to about 5 mg/kg, of the compound or salt to the subject.
- a variety of dosage forms maybe used, such as tablets, lozenges, capsules
- the compounds or salts of the invention can be administered as the single therapeutic agent in the treatment regimen, or the compounds or salts of the invention may be administered in combination with one another or with other active agents, including additional immune response modifiers, antivirals, antibiotics, antibodies, proteins, peptides, oligonucleotides, etc.
- Cytokines whose production may be induced by the administration of compounds or salts of the invention generally include interferon- ⁇ (IFN- ⁇ ) and/or tumor necrosis factor- ⁇ (TNF- ⁇ ) as well as certain interleukins (IL).
- IFN- ⁇ interferon- ⁇
- TNF- ⁇ tumor necrosis factor- ⁇
- IL interleukins
- Cytokines whose biosynthesis may be induced by compounds or salts of the invention include IFN- ⁇ , TNF- ⁇ , IL-I, IL-6, IL- 10 and IL- 12, and a variety of other cytokines. Among other effects, these and other cytokines can inhibit virus production and tumor cell growth, making the compounds or salts useful in the treatment of viral diseases and neoplastic diseases. Accordingly, the invention provides a method of inducing cytokine biosynthesis in an animal comprising administering an effective amount of a compound or salt or composition of the invention to the animal.
- the animal to which the compound or salt or composition is administered for induction of cytokine biosynthesis may have a disease as described infra, for example a viral disease or a neoplastic disease, and admim ' stration of the compound or salt may provide therapeutic treatment.
- the compound or salt may be administered to the animal prior to the animal acquiring the disease so that administration of the compound or salt may provide a prophylactic treatment.
- compounds or salts of the invention can affect other aspects of the innate immune response. For example, natural killer cell activity may be stimulated, an effect that may be due to cytokine induction.
- the compounds or salts may also activate macrophages, which in turn stimulate secretion of nitric oxide and the production of additional cytokines. Further, the compounds or salts may cause proliferation and differentiation of B-lymphocytes.
- Compounds or salts of the invention can also have an effect on the acquired immune response.
- T H I T helper type 1
- T H 2 T helper type 2
- TNF- ⁇ tumor necrosis factor- ⁇
- the invention provides a method of inhibiting TNF- ⁇ biosynthesis in an animal comprising administering an effective amount of a compound or salt or composition of the invention to the animal.
- the animal to which the compound or salt or composition is administered for inhibition of TNF- ⁇ biosynthesis may have a disease as described infra, for example an autoimmune disease, and administration of the compound or salt may provide therapeutic treatment.
- the compound or salt may be administered to the animal prior to the animal acquiring the disease so that administration of the compound or salt may provide a prophylactic treatment.
- the compound or salt or composition may be administered alone or in combination with one or more active components as in, for example, a vaccine adjuvant.
- the compound or salt and other component or components may be administered separately; together but independently such as in a solution; or together and associated with one another such as (a) covalently linked or (b) non-covalently associated, e.g., in a colloidal suspension.
- Conditions for which compounds or salts identified herein may be used as treatments include, but are not limited to:
- viral diseases such as, for example, diseases resulting from infection by an adenovirus, a herpesvirus (e.g., HSV-I, HSV-II, CMV, or VZV), a poxvirus (e.g., an orthopoxvirus such as variola or vaccinia, or molluscum contagiosum), a picornavirus
- a herpesvirus e.g., HSV-I, HSV-II, CMV, or VZV
- a poxvirus e.g., an orthopoxvirus such as variola or vaccinia, or molluscum contagiosum
- a picornavirus e.g., an orthopoxvirus such as variola or vaccinia, or molluscum contagiosum
- a coronavirus e.g., SARS
- a papovavirus e.g., papillomaviruses, such as those that cause genital warts, common warts, or plantar warts
- a hepadnavirus e.g., hepatitis B virus
- a fiavivirus e.g., hepatitis C virus or Dengue virus
- a retrovirus e.g., a lentivirus such as HIV
- bacterial diseases such as, for example, diseases resulting from infection by bacteria of, for example, the genus Escherichia, Enterobacter, Salmonella, Staphylococcus, Shigella, Listeria, Aerobacter, Helicobacter, Klebsiella, Proteus, Pseudomonas, Streptococcus, Chlamydia, Mycoplasma, Pneumococcus, Neisseria, Clostridium, Bacillus, Corynebacterium, Mycobacterium, Campylobacter, Vibrio, Serratia, Providencia, Chromobacterium, Brucella, Yersinia, Haemophilus, or Bordetella;
- infectious diseases such as chlamydia, fungal diseases including but not limited to candidiasis, aspergillosis, histoplasmosis, cryptococcal meningitis, or parasitic diseases including but not limited to malaria, Pneumocystis carnii pneumonia, leishmaniasis, cryptosporidiosis, toxoplasmosis, and trypanosome infection;
- neoplastic diseases such as intraepithelial neoplasias, cervical dysplasia, actinic keratosis, basal cell carcinoma, squamous cell carcinoma, renal cell carcinoma, Kaposi's sarcoma, melanoma, leukemias including but not limited to myelogeous leukemia, chronic lymphocytic leukemia, multiple myeloma, non-Hodgkin's lymphoma, cutaneous T-cell lymphoma, B-cell lymphoma, and hairy cell leukemia, and other cancers;
- neoplastic diseases such as intraepithelial neoplasias, cervical dysplasia, actinic keratosis, basal cell carcinoma, squamous cell carcinoma, renal cell carcinoma, Kaposi's sarcoma, melanoma
- leukemias including but not limited to myelogeous leukemia, chronic lymphocytic leukemia, multiple
- atopic diseases such as atopic dermatitis or eczema, eosinophilia, asthma, allergy, allergic rhinitis, and Ommen's syndrome;
- autoimmune diseases such as systemic lupus erythematosus, essential thrombocythaemia, multiple sclerosis, discoid lupus, alopecia areata; and (g) diseases associated with wound repair such as, for example, inhibition of keloid formation and other types of scarring (e.g., enhancing wound healing, including chronic wounds).
- a compound or salt of the present invention may be useful as a vaccine adjuvant for use in conjunction with any material that raises either humoral and/or cell mediated immune response, such as, for example, live viral, bacterial, or parasitic immunogens; inactivated viral, tumor-derived, protozoal, organism-derived, fungal, or bacterial immunogens, toxoids; toxins; self-antigens; polysaccharides; proteins; glycoproteins; peptides; cellular vaccines; DNA vaccines; autologous vaccines; recombinant proteins; and the like, for use in connection with, for example, BCG, cholera, plague, typhoid, hepatitis A, hepatitis B, hepatitis C, influenza A, influenza B, parainfluenza, polio, rabies, measles, mumps, rubella, yellow fever, tetanus, diphtheria, hemophilus influenza b, tuberculosis, mening
- Compounds or salts of the present invention may be particularly helpful in individuals having compromised immune function.
- compounds or salts may be used for treating the opportunistic infections and tumors that occur after suppression of cell mediated immunity in, for example, transplant patients, cancer patients and HIV patients.
- one or more of the above diseases or types of diseases for example, a viral disease or a neoplastic disease may be treated in an animal in need thereof (having the disease) by administering a therapeutically effective amount of a compound or salt of the invention to the animal.
- An amount of a compound or salt effective to induce or inhibit cytokine biosynthesis is an amount sufficient to cause one or more cell types, such as monocytes, macrophages, dendritic cells and B-cells to produce an amount of one or more cytokines such as, for example, IFN- ⁇ , TNF- ⁇ , IL-I, IL-6, IL-10 and IL- 12 that is increased (induced) or decreased (inhibited) over a background level of such cytokines.
- the precise amount will vary according to factors known in the art but is expected to be a dose of about 100 ng/kg to about 50 mg/kg, preferably about 10 ⁇ g/kg to about 5 mg/kg.
- the invention also provides a method of treating a viral infection in an animal and a method of treating a neoplastic disease in an animal comprising administering an effective amount of a compound or salt or composition of the invention to the animal.
- An amount effective to treat or inhibit a viral infection is an amount that will cause a reduction in one or more of the manifestations of viral infection, such as viral lesions, viral load, rate of virus production, and mortality as compared to untreated control animals.
- the precise amount that is effective for such treatment will vary according to factors known in the art but is expected to be a dose of about 100 ng/kg to about 50 mg/kg, preferably about 10 ⁇ g/kg to about 5 mg/kg.
- An amount of a compound or salt effective to treat a neoplastic condition is an amount that will cause a reduction in tumor size or in the number of tumor foci.
- the precise amount will vary according to factors known in the art but is expected to be a dose of about 100 ng/kg to about 50 mg/kg, preferably about 10 ⁇ g/kg to about 5 mg/kg.
- Butyryl chloride (0.72 mL, 6.94 mmol, 1.2 eq.) and triethylamine (1.13 niL, 8.09 mmol, 1.4 eq) were added to a solution of 3-amino-4-chloroquinoline (1.03 g, 5.78 mmol, 1.0 eq) in anhydrous dichloromethane (25 mL).
- the reaction mixture was stirred at ambient temperature for 3 hours and then washed sequentially with saturated aqueous sodium bicarbonate and brine, dried over magnesium sulfate, and concentrated under reduced pressure.
- the crude product was purified by column chromatography (silica gel eluting with 3% methanol in dichloromethane) to provide 0.70 g of iV-(4-chloroquinolin-3- yl)butyramide.
- Part B A solution of the material from Part A (0.70 g, 2.9 mmol, 1.0 eq) and O- methylhydroxylamine hydrochloride (0.336 g, 4.0 mmol, 1.4 eq) in ethanol (50 mL) was heated at reflux for 2 hours and then concentrated under reduced pressure. Analysis of the residue by NMR and mass spectroscopy indicated that the reaction was about 30-35 % complete. The residue was dissolved in ethanol (35 mL).
- Triethylamine (0.47 mL) and O-methylhydroxylamine hydrochloride (0.24 g) were added.
- the reaction mixture was heated at reflux for 2 hours at which time analysis by high performance liquid chromatography (HPLC) indicated that the reaction had not progressed.
- Additional O- methylhydroxylamine hydrochloride (0.500 g) was added and the reaction was heated at reflux for 1 hour at which time analysis by HPLC indicated that the reaction was complete.
- the reaction mixture was concentrated under reduced pressure.
- the reaction mixture was diluted with 10% sodium hydroxide and the layers were separated. The organic layer was washed sequentially with saturated aqueous sodium bicarbonate and brine, dried over magnesium sulfate, and concentrated under reduced pressure. The residue was purified by column chromatography (silica gel eluting with 4% methanol in dichloromethane) followed by recrystallization from 1% dichloromethane and methanol/water to provide 0.151 g of l-methoxy-2-propyl-lH- imidazo[4,5-c]quinolin-4-amine as off-white needles, mp 210-211 °C.
- Example 1 Part C The crude product was purified by column chromatography (silica gel eluting with 4% methanol in chloroform) followed by recrystallization from a mixture of water and 2% dichloromethane in methanol to provide 0.185 g of 1 -ethoxy-2-propyl- IH- imidazo[4,5-c]quinolin-4-amine as brownish needles, mp 198-199 °C.
- Example 1 Part C The crude product was recrystallized 3 times from a mixture of methanol and water to provide 0.244 g of 2-ethyl-l-isopropoxy-lH-imidazo[4,5- c]quinolin-4-amine as a tan powder, mp 242-243 0 C. MS (APCI) m/z 271.40 (M + H + );
- Acetyl chloride (3.41 mL, 1.25 eq) and triethylamine (6.79 mL, 1.4 eq) were added sequentially to a solution of 3-amino-4-chloroquinoline (6.22 g, 1.0 eq) in dichloromethane (100 mL).
- the reaction mixture was stirred overnight and then washed sequentially with aqueous saturated sodium bicarbonate and brine, dried over magnesium sulfate, and then concentrated under reduced pressure to provide 6.68 g of N-(4- chloroquinolin-3-yl)acetamide.
- Example 1 Part C The crude product was sonicated with 25% sodium hydroxide. The resulting powder was isolated by filtration and then washed with water to provide 151 mg of l-isopropoxy-2-methyl-lH " -imidazo[4,5-c]quinolm-4-amme as a brown powder, mp
- l-Methoxy-2-methyl-l/f-imidazo[4,5-c]qumolin-4-amine was prepared according to the method of Example 6 using O-methylhydroxylamine hydrochloride in lieu of O- isopropylhydroxylamine hydrochloride in Part B.
- the crude product was purified by column chromatography (silica gel eluting with 3% methanol in chloroform) followed by recrystallization from a mixture of water and 10% dichloromethane in methanol to provide 299 mg of l-methoxy-2-methyl-lH-imidazo[4,5-c]quinolin-4-amine as a brown powder, mp 237-238 0 C.
- l-Isobutoxy-2-methyl-lH-imidazo[4,5-c]quinolin-4-amine was prepared according to the method of Example 6 using O-isobutyllhydroxylamine hydrochloride in lieu of O- isopropylhydroxylamine hydrochloride in Part B.
- the crude product was slurried 4 times with 25% sodium hydroxide and then recrystallized from a mixture of water and 5% dichloromethane in methanol to provide 370 mg of l-isobutoxy-2-methyl-lH-imidazo[4,5- c]quinolin-4-amine as brown crystals, mp 215-216 °C.
- l-Ethoxy-2-memyl-lH-imidazo[4,5-c]quinolin-4-amine was prepared according to the method of Example 6 using ethoxyamine hydrochloride in lieu of O- isopropylhydroxylamine hydrochloride in Part B.
- the crude product was recrystallized from a mixture of water and 2% dichloromethane in methanol and then from a mixture of water and methanol to provide l-ethoxy-2-methyl-li7-imidazo[4 3 5-c]qumolin-4-amine as brown crystals, mp 219-221 0 C.
- the crude product was purified by column chromatography (230 g of silica gel eluted initially with a gradient of 20-50% ethyl acetate in hexanes and then with 4/3/3 dichloromethane/ethyl acetate/hexanes) to provide 1.01 g of N-(4-chloroquinolin-3-yl)benzamide as a white solid.
- Part B A mixture ofN-(4-chloroquinolin-3-yl)benzamide (0.020 g, 1 eq), O- methylhydroxylamine hydrochloride (3 eq), and isopropanol was heated at 80 0 C for 17 hours. The reaction was also run using ethanol in lieu of isopropanol.
- the heterogeneous mixture was filtered and then triturated with hexanes to provide 2.82 g of l-methoxy-2-phenyl-lH- imidazo[4,5-c]quinoline 5iV-oxide as a light tan solid.
- Part D Ammonium hydroxide (48 mL of 15M) was added to a solution of the material from Part C (1 eq) in dichloromethane (100 mL) and stirred vigorously at 0 0 C. Solid tosyl chloride (2.7 g, 1.5 eq) was added over a period of 1 minute. After 15 minutes the ice bath was removed. After 30 minutes the reaction mixture was diluted with dichloromethane and washed with saturated aqueous potassium carbonate.
- Ammonium hydroxide 49 mL of 15M was added to a solution of material from whom Part B (3.126 g, 1 eq) in dichloromethane (100 mL) and stirred vigorously at 0 0 C. Solid tosyl chloride (2.8 g, 1.5 eq) was added over a period of 1 minute. After 15 minutes the ice bath was removed. After 30 minutes the reaction mixture was diluted with dichloromethane, washed sequentially with saturated aqueous potassium carbonate, water, and brine, dried over sodium sulfate, filtered, and then concentrated under reduced pressure. The residue was triturated with hexanes/ether to provide 2.3 g of a tan solid.
- This material was purified by column chromatography (200 g of silica gel eluted with a gradient of 5 - 10% CMA in chloroform; CMA is a 80/12/2 v/v/v chloroform/methanol/concentrated ammonium hydroxide) to provide 1.15 g of a tan solid.
- This material was dissolved in hot acetonitrile (20 mL). After 17 hours a solid was isolated by filtration and then dried at 100 °C under high vacuum for 17 hours to provide 0.88 g of l-isopropoxy-2-phenyl-lH-imidazo[4,5-c]quinolin-4-amine as a tan powder, mp 188.0-191 °C.
- Ammonium hydroxide 50 mL of 15M was added to a solution of material from Part C (3.371 g, 1 eq) in dichloromethane (200 mL) and stirred vigorously at 0 °C. Solid tosyl chloride (2.9 g, 1.5 eq) was added over a period of 1 minute. After 15 minutes the ice bath was removed. After 30 minutes the reaction mixture was diluted with dichloromethane, washed sequentially with saturated aqueous potassium carbonate, water, and brine, dried over sodium sulfate, filtered, and then concentrated under reduced pressure. The residue was triturated with hexanes/ether to provide a solid.
- Ammonium hydroxide (23 mL of 15M) was added to a solution of material from Part B (1.37 g,l eq) in dichloromethane (50 mL) and stirred vigorously at 0 0 C. Solid tosyl chloride (1.0 g, 1.2 eq) was added over a period of 1 minute. After 15 minutes the ice bath was removed. After 1 hour the reaction mixture was diluted with dichloromethane, washed sequentially with saturated aqueous potassium carbonate, water, and brine, dried over sodium sulfate, filtered, and then concentrated under reduced pressure. The residue was triturated with hexanes to provide a solid.
- the filtrate was concentrated under reduced pressure to provide 5 g of crude iV-(4-chloroquinolin-3-yl)trimethylacetamide.
- Part B Under a nitrogen atmosphere, a mixture of N-(4-chloroquinolin-3- yl)trimethylacetamide (6.00 g, 1 eq), 0-isopropylhydroxylamine hydrochloride (7.6 g, 3 eq), and anhydrous isopropanol (80 mL) was heated at 80 °C for 19 hours. The reaction mixture was concentrated under reduced pressure.
- Acetic anhydride (16 mL, 170 mmol) was cooled to 0 °C, and formic acid (7.2 mL of 97% pure material, 190 mmol) was added over a period often mintues. The solution was stirred for 2.5 hours at room temperature and then added to a stirred solution of 3- amino-4-chloroquinoline (10.0 g, 56.0 mmol) in tetrahydroforan (THF) (100 mL). The reaction was stirred for one hour at room temperature and then concentrated under reduced pressure. Methanol (50 mL) was added to the residue, stirred for 30 minutes, and removed under reduced pressure.
- the crude animation product (2.8 g) was purified by column chromatography on silica gel (eluting with 5% methanol in dichloromethane containing 3 mL of aqueous ammonium hydroxide per liter of eluent) followed by recrystallization from methanol/water and then from isopropanol/water.
- the crystals were washed with 25% w/w aqueous sodium hydroxide (5 x 25 ml) and water (2 x 25 mL) and dried overnight in a vacuum oven at 70 0 C to provide 0.338 g of l-methoxy-l/J-imidazo[4,5- c]quinolin-4-amine as a brown solid, mp 238-240 0 C.
- the method of Part D of Example 20 was used to aminate the material from Part D using the following modifications.
- the reaction with trichloroacetyl isocyante was stirred for four days. After the reaction with sodium methoxide, the solution was concentrated under reduced pressure, the residue was stirred with methanol (15 mL), collected by filtration, and recrystallized from methanol/water (75 mL). A second recrystallization was carried out after hot filtration of the product in methanol (150 mL). The filtrate was concentrated to 50 mL, and water (5 mL) was added.
- Trichloroacetyl isocyanate (1.2 niL, 0.010 mol) was added to a solution of the material from Part B in dichloromethane (75 mL), and the reaction was stirred 45 minutes at room temperature. An analysis by LC/MS indicated the reaction was incomplete, and addition trichloroacetyl isocyanate (0.50 mL) was added. The reaction was stirred for one hour, and methanol (20 mL) was added. The mixture was stirred for 15 minutes and concentrated under reduced pressure. The residue was dissolved in methanol (25 mL), and sodium methoxide (0.5 mL of a 4.5 N solution in methanol) was added. The mixture was stirred at room temperature overnight, and a solid was present.
- Part C of Example 20 The method of Part C of Example 20 was used to oxidize 2-methyl-l-(2- phenoxyethoxy)-lH-imidazo[4,5-c]quinoline (2.7 g, 8.5 mmol) with the modification that aqueous sodium hydroxide (35 mL of 15% w/w) was used in lieu of saturated aqueous sodium bicarbonate during the work-up procedure.
- Trichloroacetyl isocyanate (0.21 mL, 1.8 mmol) was added to a chilled (ice bath) solution of the N-oxide from Part E in dichloromethane (20 mL). After 15 minutes the ice bath was removed and the reaction mixture was stirred at ambient temperature. After 1 hour methanol (5 mL) was added. The reaction mixture was stirred for 5 minutes and then concentrated under reduced pressure. The residue was combined with methanol (10 mL) and sodium methoxide (0.5 mL of 25% in methanol).
- Certain exemplary compounds including some of those described above in the Examples, have the following Formulas (LII, LIII, LIV, LV, LVI, or LVII) and the following R 1 and R 2 substituents, wherein each line of the table is matched with Formula LII, LIII, LIV, LV, LVI, or LVII to represent a specific compound.
- Compounds of the invention have been found to modulate cytokine biosynthesis by inducing the production of interferon ⁇ and/or tumor necrosis factor ⁇ in human cells when tested using the method described below.
- cytokine induction An in vitro human blood cell system is used to assess cytokine induction. Activity is based on the measurement of interferon ( ⁇ ) and tumor necrosis factor ( ⁇ ) (IFN- ⁇ and TNF- ⁇ , respectively) secreted into culture media as described by Testerman et al. in “Cytokine Induction by the Immunomodulators Imiquimod and S-27609,” Journal of Leukocyte Biology, 58, 365-372 (September, 1995).
- ⁇ interferon
- ⁇ tumor necrosis factor
- PBMC Peripheral blood mononuclear cells
- HISTOPAQUE- 1077 Sigma, St. Louis, MO
- Ficoll-Paque Plus Amersham Biosciences Piscataway, NJ
- Blood is diluted 1:1 with Dulbecco's Phosphate Buffered Saline (DPBS) or Hank's Balanced Salts Solution (HBSS).
- DPBS Dulbecco's Phosphate Buffered Saline
- HBSS Hank's Balanced Salts Solution
- PBMC whole blood is placed in Accuspin (Sigma) or LeucoSep (Greiner Bio-One, Inc., Longwood, FL) centrifuge frit tubes containing density gradient medium.
- the PBMC layer is collected and washed twice with DPBS or HBSS and re-suspended at 4 x 10 6 cells/mL in RPMI complete.
- the PBMC suspension is added to 96 well flat bottom sterile tissue culture plates containing an equal volume of RPMI complete media containing test compound.
- the compounds are solubilized in dimethyl sulfoxide (DMSO).
- DMSO concentration should not exceed a final concentration of 1% for addition to the culture wells.
- the compounds are generally tested at concentrations ranging from 30-0.014 ⁇ M.
- Controls include cell samples with media only, cell samples with DMSO only (no compound), and cell samples with reference compound. Incubation
- test compound is added at 60 ⁇ M to the first well containing RPMI complete and serial 3 fold dilutions are made in the wells.
- the PBMC suspension is then added to the wells in an equal volume, bringing the test compound concentrations to the desired range (usually 30-0.014 ⁇ M).
- the final concentration of PBMC suspension is 2 x 10 6 cells/mL.
- the plates are covered with sterile plastic lids, mixed gently and then incubated for 18 hours to 24 hours at 37°C in a 5% carbon dioxide atmosphere.
- IFN- ⁇ concentration is determined with a human multi-subtype colorimetric sandwich ELISA (Catalog Number 41105) from PBL Biomedical Laboratories,
- TNF- ⁇ concentration is determined by ORIGEN M-Series Immunoassay and read on an IGEN M- 8 analyzer from BioVeris Corporation, formerly known as IGEN
- the immunoassay uses a human TNF- ⁇ capture and detection antibody pair (Catalog Numbers AHC3419 and AHC3712) from Biosource
- the data output of the assay consists of concentration values of TNF- ⁇ and
- IFN- ⁇ (y-axis) as a function of compound concentration (x-axis).
- the reference compound used is 2-[4-amino-2-ethoxymethyl ⁇ 6,7,8,9-tetrahydro- ⁇ , ⁇ - dimethyl-lH-imidazo[4,5-c]quinolin-l-yl]ethanol hydrate (U.S. Patent No. 5,352,784; Example 91) and the expected area is the sum of the median dose values from the past 61 experiments.
- the minimum effective concentration is calculated based on the background- subtracted, reference-adjusted results for a given experiment and compound.
- the minimum effective concentration ( ⁇ molar) is the lowest of the tested compound concentrations that induces a response over a fixed cytokine concentration for the tested cytokine (usually 20 pg/mL for IFN- ⁇ and 40 pg/mL for TNF- ⁇ ).
- the maximal response is the maximal amount of cytokine (pg/ml) produced in the dose-response.
- PBMC Peripheral blood mononuclear cells
- HISTOPAQUE- 1077 Sigma, St. Louis, MO
- Ficoll-Paque Plus Amersham Biosciences Piscataway, NJ
- Whole blood is placed in Accuspin (Sigma) or LeucoSep (Greiner Bio-One, Inc., Longwood, FL) centrifuge frit tubes containing density gradient medium.
- the PBMC layer is collected and washed twice with DPBS or HBSS and re- suspended at 4 x 10 6 cells/mL in RPMI complete (2-fold the final cell density).
- the PBMC suspension is added to 96-well flat bottom sterile tissue culture plates.
- the compounds are solubilized in dimethyl sulfoxide (DMSO).
- DMSO dimethyl sulfoxide
- Controls include cell samples with media only, cell samples with DMSO only (no compound), and cell samples with a reference compound 2-[4-amino-2-ethoxymemyl-6,7,8,9-tetrahydro- ⁇ , ⁇ -dimethyl- lH-imidazo[4,5-c]quinolin-l-yl]ethanol hydrate (U.S. Patent No. 5,352,784; Example 91) on each plate.
- test compound is added at 7.5 mM to the first well of a dosing plate and serial 3 fold dilutions are made for the 7 subsequent concentrations in DMSO.
- RPMI Complete media is then added to the test compound dilutions in order to reach a final compound concentration of 2-fold higher (60 - 0.028 ⁇ M) than the final tested concentration range.
- Incubation Compound solution is then added to the wells containing the PBMC suspension bringing the test compound concentrations to the desired range (usually 30 ⁇ M - 0.014 ⁇ M) and the DMSO concentration to 0.4%.
- the final concentration of PBMC suspension is 2x10 6 cells/mL.
- the plates are covered with sterile plastic lids, mixed gently and then incubated for 18 to 24 hours at 37°C in a 5% carbon dioxide atmosphere.
- MSD MULTI-SPOT plates contain within each well capture antibodies for human
- TNF- ⁇ and human IFN- ⁇ that have been pre-coated on specific spots.
- Each well contains four spots: one human TNF- ⁇ capture antibody (MSD) spot, one human IFN- ⁇ capture antibody (PBL Biomedical Laboratories, Piscataway, NJ) spot, and two inactive bovine serum albumin spots.
- the human TNF- ⁇ capture and detection antibody pair is from MesoScale Discovery.
- the human IFN- ⁇ multi-subtype antibody (PBL Biomedical Laboratories) captures all IFN- ⁇ subtypes except IFN- ⁇ F (IFNA21). Standards consist of recombinant human TNF- ⁇ (R&D Systems, Minneapolis, MN) and IFN- ⁇ (PBL Biomedical Laboratories).
- Samples and separate standards are added at the time of analysis to each MSD plate.
- Two human IFN- ⁇ detection antibodies Cat. Nos. 21112 & 21100, PBL
- the cytokine-specific detection antibodies are labeled with the SULFO-TAG reagent (MSD).
- MSD SULFO-TAG reagent
- each well's electrochemoluminescent levels are read using MSD's SECTOR HTS READER. Results are expressed in pg/mL upon calculation with known cytokine standards.
- the data output of the assay consists of concentration values of TNF- ⁇ or IFN- ⁇ (y-axis) as a function of compound concentration (x-axis).
- a plate-wise scaling is performed within a given experiment aimed at reducing plate-to-plate variability associated within the same experiment.
- the greater of the median DMSO (DMSO control wells) or the experimental background (usually 20 pg/mL for IFN- ⁇ and 40 pg/mL for TNF- ⁇ ) is subtracted from each reading. Negative values that may result from background subtraction are set to zero.
- Each plate within a given experiment has a reference compound that serves as a control. This control is used to calculate a median expected area under the curve across all plates in the assay.
- a plate- wise scaling factor is calculated for each plate as a ratio of the area of the reference compound on the particular plate to the median expected area for the entire experiment.
- the data from each plate are then multiplied by the plate-wise scaling factor for all plates. Only data from plates bearing a scaling factor of between 0.5 and 2.0 (for both cytokines IFN- ⁇ , TNF- ⁇ ) are reported. Data from plates with scaling factors outside the above- mentioned interval are retested until they bear scaling factors inside the above mentioned interval. The above method produces a scaling of the y- values without altering the shape of the curve.
- the reference compound used is 2-[4-amino-2-ethoxymethyl-6,7,8,9- tetrahydro- ⁇ , ⁇ -dimethyl-l/i ' -imidazo[4,5-c]quinolm-l-yl]ethanol hydrate (U.S. Patent No. 5,352,784; Example 91).
- the median expected area is the median area across all plates that are part of a given experiment.
- a second scaling may also be performed to reduce inter-experiment variability (across multiple experiments). All background-subtracted values are multiplied by a single adjustment ratio to decrease experiment-to-experiment variability.
- the adjustment ratio is the area of the reference compound in the new experiment divided by the expected area of the reference compound based on an average of previous experiments (unadjusted readings). This results in the scaling of the reading (y-axis) for the new data without changing the shape of the dose-response curve.
- the reference compound used is 2-[4- amino-2-ethoxymethyl-6,7,8,9-tetrahydro- ⁇ , ⁇ -dimethyl-lH-imidazo[4,5-c]quinolin-l- yl]ethanol hydrate (U.S. Patent No. 5,352,784; Example 91) and the expected area is the sum of the median dose values from an average of previous experiments.
- the minimum effective concentration is calculated based on the background- subtracted, reference-adjusted results for a given experiment and compound.
- the minimum effective concentration ( ⁇ molar) is the lowest of the tested compound concentrations that induces a response over a fixed cytokine concentration for the tested cytokine (usually 20 pg/mL for IFN- ⁇ and 40 pg/mL for TNF- ⁇ ).
- the maximal response is the maximal amount of cytokine (pg/ml) produced in the dose-response.
- Certain compounds of the invention may modulate cytokine biosynthesis by inhibiting production of tumor necrosis factor ⁇ (TNF- ⁇ ) when tested using the method described below.
- TNF- ⁇ tumor necrosis factor ⁇
- the mouse macrophage cell line Raw 264.7 is used to assess the ability of compounds to inhibit tumor necrosis factor- ⁇ (TNF- ⁇ ) production upon stimulation by lipopolysaccharide (LPS).
- TNF- ⁇ tumor necrosis factor- ⁇
- LPS lipopolysaccharide
- APC Blood Cell Preparation for Culture Raw cells
- the cell suspension is brought to 3 x 10 5 cells/mL in RPMI with 10 % fetal bovine serum (FBS).
- FBS fetal bovine serum
- Cell suspension 100 ⁇ L is added to 96-well flat bottom sterile tissues culture plates (Becton Dickinson Labware, Lincoln Park, NJ). The final concentration of cells is 3 x 10 4 cells/well. The plates are incubated for 3 hours. Prior to the addition of test compound the medium is replaced with colorless RPMI medium with 3 % FBS.
- the compounds are solubilized in dimethyl sulfoxide (DMSO).
- DMSO concentration should not exceed a final concentration of 1% for addition to the culture wells.
- Compounds are tested at 5 ⁇ M.
- LPS Lipopolysaccaride from Salmonella typhimurimn, Sigma- Aldrich
- EC 70 concentration as measured by a dose response assay.
- test compound (l ⁇ l) is added to each well.
- the plates are mixed on a microtiter plate shaker for 1 minute and then placed in an incubator. Twenty minutes later the solution of LPS (1 ⁇ L, EC 70 concentration - 10 ng/ml) is added and the plates are mixed for 1 minute on a shaker. The plates are incubated for 18 to 24 hours at 37 0 C in a 5 % carbon dioxide atmosphere.
- TNF- ⁇ concentration is determined by ELISA using a mouse TNF- ⁇ kit (from Bio source International, Camarillo, CA). Results are expressed in pg/mL. TNF- ⁇ expression upon LPS stimulation alone is considered a 100% response.
- Dose Response Assay Blood Cell Preparation for Culture Raw cells (ATCC) are harvested by gentle scraping and then counted. The cell suspension is brought to 4 x 10 5 cells/mL in RPMI with 10 % FBS. Cell suspension (250 ⁇ L) is added to 48-well flat bottom sterile tissues culture plates (Costar, Cambridge, MA). The final concentration of cells is 1 x 10 5 cells/well. The plates are incubated for 3 hours. Prior to the addition of test compound the medium is replaced with colorless RPMI medium with 3 % FBS.
- the compounds are solubilized in dimethyl sulfoxide (DMSO).
- DMSO concentration hould not exceed a final concentration of 1% for addition to the culture wells.
- Compounds are tested at 0.03, 0.1, 0.3, 1, 3, 5 and 10 ⁇ M.
- LPS Lipopolysaccaride from Salmonella typhimurium, Sigma- Aldrich
- EC 70 concentration as measured by dose response assay.
- test compound 200 ⁇ l
- the plates are mixed on a microtiter plate shaker for 1 minute and then placed in an incubator. Twenty minutes later the solution of LPS (200 ⁇ L, EC 7O concentration approximately 10 ng/ml) is added and the plates are mixed for 1 minute on a shaker. The plates are incubated for 18 to 24 hours at 37 0 C in a 5 % carbon dioxide atmosphere.
- TNF- ⁇ Analysis Following the incubation the supernatant is removed with a pipet. TNF- ⁇ concentration is determined by ELISA using a mouse TNF- ⁇ kit (from Biosource International, Camarillo, CA). Results are expressed in pg/mL. TNF- ⁇ expression upon LPS stimulation alone is considered a 100% response.
Abstract
Description













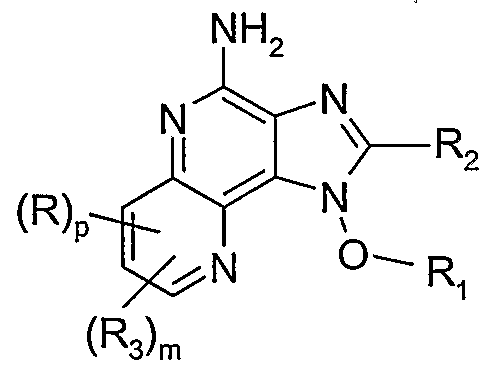
























































































Claims













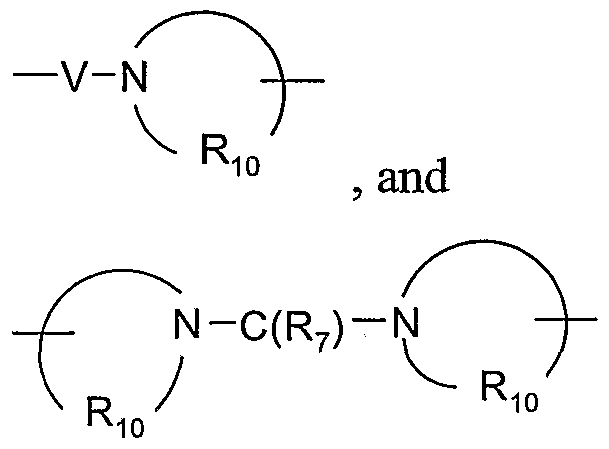






Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2007530396A JP5209312B2 (en) | 2004-09-02 | 2005-09-01 | 1-alkoxy 1H-imidazo ring systems and methods |
| AT05794068T ATE555786T1 (en) | 2004-09-02 | 2005-09-01 | 1-ALKOXY 1H-IMIDAZO RING SYSTEMS AND METHODS |
| US11/574,463 US7579359B2 (en) | 2004-09-02 | 2005-09-01 | 1-alkoxy 1H-imidazo ring systems and methods |
| EP05794068A EP1789042B1 (en) | 2004-09-02 | 2005-09-01 | 1-alkoxy 1h-imidazo ring systems and methods |
| AU2005282726A AU2005282726B2 (en) | 2004-09-02 | 2005-09-01 | 1-alkoxy 1H-imidazo ring systems and methods |
| CA2578741A CA2578741C (en) | 2004-09-02 | 2005-09-01 | 1-alkoxy 1h-imidazo ring systems and methods |
| PL05794068T PL1789042T3 (en) | 2004-09-02 | 2005-09-01 | 1-alkoxy 1h-imidazo ring systems and methods |
| ES05794068T ES2384390T3 (en) | 2004-09-02 | 2005-09-01 | 1-alkoxy-1H-imidazo cyclic systems and associated methods |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US60662904P | 2004-09-02 | 2004-09-02 | |
| US60/606,629 | 2004-09-02 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2006028962A2 true WO2006028962A2 (en) | 2006-03-16 |
| WO2006028962A3 WO2006028962A3 (en) | 2006-10-26 |
Family
ID=36036867
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2005/031310 WO2006028962A2 (en) | 2004-09-02 | 2005-09-01 | 1-alkoxy 1h-imidazo ring systems and methods |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US7579359B2 (en) |
| EP (1) | EP1789042B1 (en) |
| JP (1) | JP5209312B2 (en) |
| AT (1) | ATE555786T1 (en) |
| AU (1) | AU2005282726B2 (en) |
| CA (1) | CA2578741C (en) |
| ES (1) | ES2384390T3 (en) |
| PL (1) | PL1789042T3 (en) |
| WO (1) | WO2006028962A2 (en) |
Cited By (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1937683A1 (en) * | 2005-09-23 | 2008-07-02 | Coley Pharmaceutical Group, Inc. | METHOD FOR 1H-IMIDAZOÝ4,5-c¨PYRIDINES AND ANALOGS THEREOF |
| US7879849B2 (en) | 2003-10-03 | 2011-02-01 | 3M Innovative Properties Company | Pyrazolopyridines and analogs thereof |
| US7906506B2 (en) | 2006-07-12 | 2011-03-15 | 3M Innovative Properties Company | Substituted chiral fused [1,2] imidazo [4,5-c] ring compounds and methods |
| US7915281B2 (en) | 2004-06-18 | 2011-03-29 | 3M Innovative Properties Company | Isoxazole, dihydroisoxazole, and oxadiazole substituted imidazo ring compounds and method |
| US7923429B2 (en) | 2003-09-05 | 2011-04-12 | 3M Innovative Properties Company | Treatment for CD5+ B cell lymphoma |
| US7943610B2 (en) | 2005-04-01 | 2011-05-17 | 3M Innovative Properties Company | Pyrazolopyridine-1,4-diamines and analogs thereof |
| US7943636B2 (en) | 2005-04-01 | 2011-05-17 | 3M Innovative Properties Company | 1-substituted pyrazolo (3,4-C) ring compounds as modulators of cytokine biosynthesis for the treatment of viral infections and neoplastic diseases |
| US7943609B2 (en) | 2004-12-30 | 2011-05-17 | 3M Innovative Proprerties Company | Chiral fused [1,2]imidazo[4,5-C] ring compounds |
| US7968563B2 (en) | 2005-02-11 | 2011-06-28 | 3M Innovative Properties Company | Oxime and hydroxylamine substituted imidazo[4,5-c] ring compounds and methods |
| US8034938B2 (en) | 2004-12-30 | 2011-10-11 | 3M Innovative Properties Company | Substituted chiral fused [1,2]imidazo[4,5-c] ring compounds |
| CN103097386A (en) * | 2010-08-17 | 2013-05-08 | 3M创新有限公司 | Lipidated immune response modifier compound compositions, formulations, and methods |
| WO2014107663A2 (en) | 2013-01-07 | 2014-07-10 | The Trustees Of The University Of Pennsylvania | Compositions and methods for treating cutaneous t cell lymphoma |
| US8871782B2 (en) | 2003-10-03 | 2014-10-28 | 3M Innovative Properties Company | Alkoxy substituted imidazoquinolines |
| US9248127B2 (en) | 2005-02-04 | 2016-02-02 | 3M Innovative Properties Company | Aqueous gel formulations containing immune response modifiers |
| EP3085373A1 (en) | 2006-02-22 | 2016-10-26 | 3M Innovative Properties Company | Immune response modifier conjugates |
| EP3153180A1 (en) | 2011-06-03 | 2017-04-12 | 3M Innovative Properties Company | Heterobifunctional linkers with polyethylene glycol segments and immune response modifier conjugates made therefrom |
| US10005772B2 (en) | 2006-12-22 | 2018-06-26 | 3M Innovative Properties Company | Immune response modifier compositions and methods |
| WO2019166946A1 (en) | 2018-02-28 | 2019-09-06 | Pfizer Inc. | Il-15 variants and uses thereof |
| WO2019224716A2 (en) | 2018-05-23 | 2019-11-28 | Pfizer Inc. | Antibodies specific for gucy2c and uses thereof |
| WO2019224715A1 (en) | 2018-05-23 | 2019-11-28 | Pfizer Inc. | Antibodies specific for cd3 and uses thereof |
| US10526309B2 (en) | 2015-10-02 | 2020-01-07 | The University Of North Carolina At Chapel Hill | Pan-TAM inhibitors and Mer/Axl dual inhibitors |
| WO2020128893A1 (en) | 2018-12-21 | 2020-06-25 | Pfizer Inc. | Combination treatments of cancer comprising a tlr agonist |
| WO2020160054A1 (en) * | 2019-02-01 | 2020-08-06 | Canwell Biotech Limited | Imidazoquinoline amine derivatives, pharmaceutical composition, use thereof |
| WO2021124073A1 (en) | 2019-12-17 | 2021-06-24 | Pfizer Inc. | Antibodies specific for cd47, pd-l1, and uses thereof |
| WO2022013775A1 (en) | 2020-07-17 | 2022-01-20 | Pfizer Inc. | Therapeutic antibodies and their uses |
| US11306083B2 (en) | 2017-12-20 | 2022-04-19 | 3M Innovative Properties Company | Amide substituted imidazo[4,5-C]quinoline compounds with a branched chain linking group for use as an immune response modifier |
| US20230270819A1 (en) * | 2019-12-20 | 2023-08-31 | Nammi Therapeutics, Inc. | Formulated and/or Co-Formulated Liposome Compositions Containing Toll--Like Receptor ("TLR") Agonist Prodrugs Useful In The Treatment of Cancer and Methods Thereof |
| EP4051270A4 (en) * | 2019-10-29 | 2024-02-28 | Prime Reach Trading Ltd | 4-amino-imidazoquinoline compounds and use thereof |
Families Citing this family (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20040265351A1 (en) | 2003-04-10 | 2004-12-30 | Miller Richard L. | Methods and compositions for enhancing immune response |
| US7897597B2 (en) | 2003-08-27 | 2011-03-01 | 3M Innovative Properties Company | Aryloxy and arylalkyleneoxy substituted imidazoquinolines |
| KR20060120069A (en) | 2003-10-03 | 2006-11-24 | 쓰리엠 이노베이티브 프로퍼티즈 컴파니 | Pyrazolopyridines and analogs thereof |
| BRPI0416936A (en) | 2003-11-25 | 2007-01-16 | 3M Innovative Properties Co | substituted imidazo ring systems and methods |
| US8541438B2 (en) | 2004-06-18 | 2013-09-24 | 3M Innovative Properties Company | Substituted imidazoquinolines, imidazopyridines, and imidazonaphthyridines |
| AU2006212765B2 (en) | 2005-02-09 | 2012-02-02 | 3M Innovative Properties Company | Alkyloxy substituted thiazoloquinolines and thiazolonaphthyridines |
| EP1877056A2 (en) | 2005-02-09 | 2008-01-16 | Coley Pharmaceutical Group, Inc. | Oxime and hydroxylamine substituted thiazoloý4,5-c¨ring compounds and methods |
| US8658666B2 (en) * | 2005-02-11 | 2014-02-25 | 3M Innovative Properties Company | Substituted imidazoquinolines and imidazonaphthyridines |
| JP2008543725A (en) | 2005-02-23 | 2008-12-04 | コーリー ファーマシューティカル グループ,インコーポレイテッド | Hydroxyalkyl substituted imidazoquinolines |
| US8343993B2 (en) | 2005-02-23 | 2013-01-01 | 3M Innovative Properties Company | Hydroxyalkyl substituted imidazonaphthyridines |
| US8158794B2 (en) | 2005-02-23 | 2012-04-17 | 3M Innovative Properties Company | Hydroxyalkyl substituted imidazoquinoline compounds and methods |
| CA2598437A1 (en) | 2005-02-23 | 2006-08-31 | Coley Pharmaceutical Group, Inc. | Method of preferentially inducing the biosynthesis of interferon |
| ZA200803029B (en) * | 2005-09-09 | 2009-02-25 | Coley Pharm Group Inc | Amide and carbamate derivatives of alkyl substituted /V-[4-(4-amino-1H-imidazo[4,5-c] quinolin-1-yl)butyl] methane-sulfonamides and methods |
| CA2621831A1 (en) | 2005-09-09 | 2007-03-15 | Coley Pharmaceutical Group, Inc. | Amide and carbamate derivatives of n-{2-[4-amino-2- (ethoxymethyl)-1h-imidazo[4,5-c]quinolin-1-yl]-1,1-dimethylethyl}methanesulfonamide and methods |
| ES2429170T3 (en) | 2005-11-04 | 2013-11-13 | 3M Innovative Properties Company | 1H-Imidazoquinolines substituted with hydroxyl and alkoxy and methods |
| WO2007106854A2 (en) | 2006-03-15 | 2007-09-20 | Coley Pharmaceutical Group, Inc. | Hydroxy and alkoxy substituted 1h-imidazonaphthyridines and methods |
| WO2008030511A2 (en) | 2006-09-06 | 2008-03-13 | Coley Pharmaceuticial Group, Inc. | Substituted 3,4,6,7-tetrahydro-5h, 1,2a,4a,8-tetraazacyclopenta[cd]phenalenes |
| EP2717919B1 (en) | 2011-06-03 | 2016-08-03 | 3M Innovative Properties Company | Heterobifunctional linkers with polyethylene glycol segments and immune response modifier conjugates made therefrom |
| WO2017040233A1 (en) | 2015-08-31 | 2017-03-09 | 3M Innovative Properties Company | GUANIDINE SUBSTITUTED IMIDAZO[4,5-c] RING COMPOUNDS |
| WO2017040234A1 (en) * | 2015-08-31 | 2017-03-09 | 3M Innovative Properties Company | IMIDAZO[4,5-c] RING COMPOUNDS CONTAINING SUBSTITUTED GUANIDINE GROUPS |
| AR109629A1 (en) * | 2016-09-08 | 2019-01-09 | Medimmune Llc | COMPOSITIONS FOR THE LOCAL ADMINISTRATION OF TOLL TYPE RECEIVER AGONISTS |
| WO2018160552A1 (en) | 2017-03-01 | 2018-09-07 | 3M Innovative Properties Company | IMIDAZO[4,5-c] RING COMPOUNDS CONTAINING GUANIDINE SUBSTITUTED BENZAMIDE GROUPS |
Family Cites Families (93)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3314941A (en) * | 1964-06-23 | 1967-04-18 | American Cyanamid Co | Novel substituted pyridodiazepins |
| ZA848968B (en) * | 1983-11-18 | 1986-06-25 | Riker Laboratories Inc | 1h-imidazo(4,5-c)quinolines and 1h-imidazo(4,5-c)quinolin-4-amines |
| IL73534A (en) * | 1983-11-18 | 1990-12-23 | Riker Laboratories Inc | 1h-imidazo(4,5-c)quinoline-4-amines,their preparation and pharmaceutical compositions containing certain such compounds |
| US5238944A (en) * | 1988-12-15 | 1993-08-24 | Riker Laboratories, Inc. | Topical formulations and transdermal delivery systems containing 1-isobutyl-1H-imidazo[4,5-c]quinolin-4-amine |
| US5756747A (en) * | 1989-02-27 | 1998-05-26 | Riker Laboratories, Inc. | 1H-imidazo 4,5-c!quinolin-4-amines |
| US5037986A (en) * | 1989-03-23 | 1991-08-06 | Minnesota Mining And Manufacturing Company | Olefinic 1H-imidazo[4,5-c]quinolin-4-amines |
| US4929624A (en) * | 1989-03-23 | 1990-05-29 | Minnesota Mining And Manufacturing Company | Olefinic 1H-imidazo(4,5-c)quinolin-4-amines |
| NZ232740A (en) | 1989-04-20 | 1992-06-25 | Riker Laboratories Inc | Solution for parenteral administration comprising a 1h-imidazo(4,5-c) quinolin-4-amine derivative, an acid and a tonicity adjuster |
| US4988815A (en) * | 1989-10-26 | 1991-01-29 | Riker Laboratories, Inc. | 3-Amino or 3-nitro quinoline compounds which are intermediates in preparing 1H-imidazo[4,5-c]quinolines |
| ATE121088T1 (en) * | 1990-10-05 | 1995-04-15 | Minnesota Mining & Mfg | METHOD FOR PRODUCING IMIDAZO(4,5- C>QUINOLINE-4-AMINES. |
| US5175296A (en) * | 1991-03-01 | 1992-12-29 | Minnesota Mining And Manufacturing Company | Imidazo[4,5-c]quinolin-4-amines and processes for their preparation |
| US5389640A (en) * | 1991-03-01 | 1995-02-14 | Minnesota Mining And Manufacturing Company | 1-substituted, 2-substituted 1H-imidazo[4,5-c]quinolin-4-amines |
| US5268376A (en) * | 1991-09-04 | 1993-12-07 | Minnesota Mining And Manufacturing Company | 1-substituted 1H-imidazo[4,5-c]quinolin-4-amines |
| US5266575A (en) * | 1991-11-06 | 1993-11-30 | Minnesota Mining And Manufacturing Company | 2-ethyl 1H-imidazo[4,5-ciquinolin-4-amines |
| IL105325A (en) * | 1992-04-16 | 1996-11-14 | Minnesota Mining & Mfg | Immunogen/vaccine adjuvant composition |
| US5395937A (en) * | 1993-01-29 | 1995-03-07 | Minnesota Mining And Manufacturing Company | Process for preparing quinoline amines |
| US5352784A (en) * | 1993-07-15 | 1994-10-04 | Minnesota Mining And Manufacturing Company | Fused cycloalkylimidazopyridines |
| JPH09500128A (en) * | 1993-07-15 | 1997-01-07 | ミネソタ マイニング アンド マニュファクチャリング カンパニー | Imidazo [4,5-c] pyridin-4-amine |
| US5482936A (en) * | 1995-01-12 | 1996-01-09 | Minnesota Mining And Manufacturing Company | Imidazo[4,5-C]quinoline amines |
| JPH09208584A (en) | 1996-01-29 | 1997-08-12 | Terumo Corp | Amide derivative, pharmaceutical preparation containing the same, and intermediate for synthesizing the same |
| US5741908A (en) * | 1996-06-21 | 1998-04-21 | Minnesota Mining And Manufacturing Company | Process for reparing imidazoquinolinamines |
| US5693811A (en) * | 1996-06-21 | 1997-12-02 | Minnesota Mining And Manufacturing Company | Process for preparing tetrahdroimidazoquinolinamines |
| JP4391592B2 (en) * | 1996-10-25 | 2009-12-24 | スリーエム カンパニー | Immune response modifying compounds for the treatment of TH2-mediated and related diseases |
| US5939090A (en) * | 1996-12-03 | 1999-08-17 | 3M Innovative Properties Company | Gel formulations for topical drug delivery |
| US6069149A (en) * | 1997-01-09 | 2000-05-30 | Terumo Kabushiki Kaisha | Amide derivatives and intermediates for the synthesis thereof |
| JPH1180156A (en) | 1997-09-04 | 1999-03-26 | Hokuriku Seiyaku Co Ltd | 1-(substitutedaryl)alkyl-1h-imidazopyridin-4-amine derivative |
| UA67760C2 (en) * | 1997-12-11 | 2004-07-15 | Міннесота Майнінг Енд Мануфакчурінг Компані | Imidazonaphthyridines and use thereof to induce the biosynthesis of cytokines |
| JPH11222432A (en) | 1998-02-03 | 1999-08-17 | Terumo Corp | Preparation for external use containing amide derivative inducing interferon |
| US6110929A (en) * | 1998-07-28 | 2000-08-29 | 3M Innovative Properties Company | Oxazolo, thiazolo and selenazolo [4,5-c]-quinolin-4-amines and analogs thereof |
| JP2000119271A (en) | 1998-08-12 | 2000-04-25 | Hokuriku Seiyaku Co Ltd | 1h-imidazopyridine derivative |
| CA2348741C (en) * | 1998-10-30 | 2010-04-20 | Nitromed Inc. | Nitrosasted and nitrosylated nonsteroidal antiinflammatory compounds, comositions and methods of use |
| US20020058674A1 (en) * | 1999-01-08 | 2002-05-16 | Hedenstrom John C. | Systems and methods for treating a mucosal surface |
| CA2361936C (en) * | 1999-01-08 | 2009-06-16 | 3M Innovative Properties Company | Formulations comprising imiquimod or other immune response modifiers for treating mucosal conditions |
| US6558951B1 (en) * | 1999-02-11 | 2003-05-06 | 3M Innovative Properties Company | Maturation of dendritic cells with immune response modifying compounds |
| JP2000247884A (en) | 1999-03-01 | 2000-09-12 | Sumitomo Pharmaceut Co Ltd | Arachidonic acid-induced skin disease-treating agent |
| US6756382B2 (en) * | 1999-06-10 | 2004-06-29 | 3M Innovative Properties Company | Amide substituted imidazoquinolines |
| US6573273B1 (en) * | 1999-06-10 | 2003-06-03 | 3M Innovative Properties Company | Urea substituted imidazoquinolines |
| US6451810B1 (en) * | 1999-06-10 | 2002-09-17 | 3M Innovative Properties Company | Amide substituted imidazoquinolines |
| US6541485B1 (en) * | 1999-06-10 | 2003-04-01 | 3M Innovative Properties Company | Urea substituted imidazoquinolines |
| US6331539B1 (en) * | 1999-06-10 | 2001-12-18 | 3M Innovative Properties Company | Sulfonamide and sulfamide substituted imidazoquinolines |
| US6376669B1 (en) * | 1999-11-05 | 2002-04-23 | 3M Innovative Properties Company | Dye labeled imidazoquinoline compounds |
| US6894060B2 (en) | 2000-03-30 | 2005-05-17 | 3M Innovative Properties Company | Method for the treatment of dermal lesions caused by envenomation |
| US20020055517A1 (en) * | 2000-09-15 | 2002-05-09 | 3M Innovative Properties Company | Methods for delaying recurrence of herpes virus symptoms |
| JP2002145777A (en) | 2000-11-06 | 2002-05-22 | Sumitomo Pharmaceut Co Ltd | Therapeutic agent for arachidonic acid-induced dermatosis |
| US6664260B2 (en) * | 2000-12-08 | 2003-12-16 | 3M Innovative Properties Company | Heterocyclic ether substituted imidazoquinolines |
| US6664265B2 (en) | 2000-12-08 | 2003-12-16 | 3M Innovative Properties Company | Amido ether substituted imidazoquinolines |
| US6664264B2 (en) * | 2000-12-08 | 2003-12-16 | 3M Innovative Properties Company | Thioether substituted imidazoquinolines |
| UA75622C2 (en) * | 2000-12-08 | 2006-05-15 | 3M Innovative Properties Co | Aryl ether substituted imidazoquinolines, pharmaceutical composition based thereon |
| US6677348B2 (en) * | 2000-12-08 | 2004-01-13 | 3M Innovative Properties Company | Aryl ether substituted imidazoquinolines |
| US6545017B1 (en) * | 2000-12-08 | 2003-04-08 | 3M Innovative Properties Company | Urea substituted imidazopyridines |
| CA2430206A1 (en) * | 2000-12-08 | 2002-06-13 | 3M Innovative Properties Company | Screening method for identifying compounds that selectively induce interferon alpha |
| US6545016B1 (en) * | 2000-12-08 | 2003-04-08 | 3M Innovative Properties Company | Amide substituted imidazopyridines |
| US20020107262A1 (en) * | 2000-12-08 | 2002-08-08 | 3M Innovative Properties Company | Substituted imidazopyridines |
| US6525064B1 (en) * | 2000-12-08 | 2003-02-25 | 3M Innovative Properties Company | Sulfonamido substituted imidazopyridines |
| US6677347B2 (en) * | 2000-12-08 | 2004-01-13 | 3M Innovative Properties Company | Sulfonamido ether substituted imidazoquinolines |
| US6667312B2 (en) * | 2000-12-08 | 2003-12-23 | 3M Innovative Properties Company | Thioether substituted imidazoquinolines |
| US6660735B2 (en) | 2000-12-08 | 2003-12-09 | 3M Innovative Properties Company | Urea substituted imidazoquinoline ethers |
| US6660747B2 (en) * | 2000-12-08 | 2003-12-09 | 3M Innovative Properties Company | Amido ether substituted imidazoquinolines |
| US20030133913A1 (en) * | 2001-08-30 | 2003-07-17 | 3M Innovative Properties Company | Methods of maturing plasmacytoid dendritic cells using immune response modifier molecules |
| JP4445262B2 (en) * | 2001-10-09 | 2010-04-07 | アムジェン インコーポレイテッド | Imidazole derivatives as anti-inflammatory agents |
| JP2005519990A (en) * | 2001-10-12 | 2005-07-07 | ユニバーシティ オブ アイオワ リサーチ ファウンデーション | Methods and products for enhancing immune responses using imidazoquinoline compounds |
| ES2318615T3 (en) * | 2001-11-16 | 2009-05-01 | Coley Pharmaceutical Group, Inc. | N- (4- (4-AMINO-2-ETIL-1H-IMIDAZO (4,5-C) QUINOLIN-1-IL) BUTIL) METHANOSULPHONAMIDE, A PHARMACEUTICAL COMPOSITION THAT UNDERSTANDS AND USES ITSELF. |
| WO2004080430A2 (en) * | 2003-03-13 | 2004-09-23 | 3M Innovative Properties Company | Methods of improving skin quality |
| DK1450804T3 (en) * | 2001-11-29 | 2009-01-05 | 3M Innovative Properties Co | Pharmaceutical formula rings comprising an immune response modifier |
| US6677349B1 (en) * | 2001-12-21 | 2004-01-13 | 3M Innovative Properties Company | Sulfonamide and sulfamide substituted imidazoquinolines |
| SI1478327T1 (en) * | 2002-02-22 | 2015-08-31 | Meda Ab | Method of reducing and treating uvb-induced immunosuppression |
| US20030185835A1 (en) * | 2002-03-19 | 2003-10-02 | Braun Ralph P. | Adjuvant for vaccines |
| GB0211649D0 (en) * | 2002-05-21 | 2002-07-03 | Novartis Ag | Organic compounds |
| EP1511746A2 (en) * | 2002-05-29 | 2005-03-09 | 3M Innovative Properties Company | Process for imidazo[4,5-c]pyridin-4-amines |
| EP1513524A4 (en) * | 2002-06-07 | 2008-09-03 | 3M Innovative Properties Co | Ether substituted imidazopyridines |
| CA2493227A1 (en) * | 2002-07-23 | 2004-01-29 | Biogal Gyogyszergyar Rt. | Preparation of 1h-imidazo[4,5-c]quinolin-4-amines via1h-imidazo [4,5-c]quinolin-4-phthalimide intermediates |
| AU2003254221A1 (en) * | 2002-07-26 | 2004-02-16 | Biogal Gyogyszergyar Rt. | Preparation of 1h-imidazo (4,5-c) quinolin-4-amines via novel 1h-imidazo (4,5-c) quinolin-4-cyano and 1h-imidazo (4,5-c) quinolin-4-carboxamide intermediates |
| DE60335010D1 (en) * | 2002-08-15 | 2010-12-30 | 3M Innovative Properties Co | IMMUNOSTIMULATORY COMPOSITIONS AND METHOD FOR STIMULATING AN IMMUNE RESPONSE |
| US6818650B2 (en) * | 2002-09-26 | 2004-11-16 | 3M Innovative Properties Company | 1H-imidazo dimers |
| WO2004053452A2 (en) * | 2002-12-11 | 2004-06-24 | 3M Innovative Properties Company | Assays relating to toll-like receptor activity |
| WO2004053057A2 (en) * | 2002-12-11 | 2004-06-24 | 3M Innovative Properties Company | Gene expression systems and recombinant cell lines |
| AU2003301052A1 (en) * | 2002-12-20 | 2004-07-22 | 3M Innovative Properties Company | Aryl / hetaryl substituted imidazoquinolines |
| WO2004060319A2 (en) * | 2002-12-30 | 2004-07-22 | 3M Innovative Properties Company | Immunostimulatory combinations |
| WO2004071459A2 (en) * | 2003-02-13 | 2004-08-26 | 3M Innovative Properties Company | Methods and compositions related to irm compounds and toll-like receptor 8 |
| EP1599726A4 (en) * | 2003-02-27 | 2009-07-22 | 3M Innovative Properties Co | Selective modulation of tlr-mediated biological activity |
| US8110582B2 (en) * | 2003-03-04 | 2012-02-07 | 3M Innovative Properties Company | Prophylactic treatment of UV-induced epidermal neoplasia |
| TW200505458A (en) * | 2003-03-07 | 2005-02-16 | 3M Innovative Properties Co | 1-amino 1h-imidazoquinolines |
| US7163947B2 (en) * | 2003-03-07 | 2007-01-16 | 3M Innovative Properties Company | 1-Amino 1H-imidazoquinolines |
| AU2004220465A1 (en) * | 2003-03-13 | 2004-09-23 | 3M Innovative Properties Company | Method of tattoo removal |
| WO2004080293A2 (en) * | 2003-03-13 | 2004-09-23 | 3M Innovative Properties Company | Methods for diagnosing skin lesions |
| US20040192585A1 (en) * | 2003-03-25 | 2004-09-30 | 3M Innovative Properties Company | Treatment for basal cell carcinoma |
| US20040191833A1 (en) | 2003-03-25 | 2004-09-30 | 3M Innovative Properties Company | Selective activation of cellular activities mediated through a common toll-like receptor |
| EP1617871A4 (en) * | 2003-04-10 | 2010-10-06 | 3M Innovative Properties Co | Delivery of immune response modifier compounds using metal-containing particulate support materials |
| WO2004096144A2 (en) * | 2003-04-28 | 2004-11-11 | 3M Innovative Properties Company | Compositions and methods for induction of opioid receptors |
| EP1653914A4 (en) | 2003-08-12 | 2008-10-29 | 3M Innovative Properties Co | Oxime substituted imidazo-containing compounds |
| EP1667694B1 (en) * | 2003-09-05 | 2010-04-28 | Anadys Pharmaceuticals, Inc. | Tlr7 ligands for the treatment of hepatitis c |
| CA2536530A1 (en) * | 2003-10-01 | 2005-04-14 | Taro Pharmaceuticals U.S.A., Inc. | Method of preparing 4-amino-1h-imidazo(4,5-c)quinolines and acid addition salts thereof |
| ITMI20032121A1 (en) * | 2003-11-04 | 2005-05-05 | Dinamite Dipharma Spa In Forma Abbr Eviata Dipharm | PROCEDURE FOR THE PREPARATION OF IMIQUIMOD AND ITS INTERMEDIATES |
-
2005
- 2005-09-01 JP JP2007530396A patent/JP5209312B2/en not_active Expired - Fee Related
- 2005-09-01 AT AT05794068T patent/ATE555786T1/en active
- 2005-09-01 ES ES05794068T patent/ES2384390T3/en active Active
- 2005-09-01 EP EP05794068A patent/EP1789042B1/en active Active
- 2005-09-01 AU AU2005282726A patent/AU2005282726B2/en not_active Ceased
- 2005-09-01 CA CA2578741A patent/CA2578741C/en active Active
- 2005-09-01 PL PL05794068T patent/PL1789042T3/en unknown
- 2005-09-01 WO PCT/US2005/031310 patent/WO2006028962A2/en active Application Filing
- 2005-09-01 US US11/574,463 patent/US7579359B2/en active Active
Non-Patent Citations (2)
| Title |
|---|
| None |
| See also references of EP1789042A4 |
Cited By (47)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7923429B2 (en) | 2003-09-05 | 2011-04-12 | 3M Innovative Properties Company | Treatment for CD5+ B cell lymphoma |
| US8871782B2 (en) | 2003-10-03 | 2014-10-28 | 3M Innovative Properties Company | Alkoxy substituted imidazoquinolines |
| US7879849B2 (en) | 2003-10-03 | 2011-02-01 | 3M Innovative Properties Company | Pyrazolopyridines and analogs thereof |
| US7915281B2 (en) | 2004-06-18 | 2011-03-29 | 3M Innovative Properties Company | Isoxazole, dihydroisoxazole, and oxadiazole substituted imidazo ring compounds and method |
| US7943609B2 (en) | 2004-12-30 | 2011-05-17 | 3M Innovative Proprerties Company | Chiral fused [1,2]imidazo[4,5-C] ring compounds |
| US8034938B2 (en) | 2004-12-30 | 2011-10-11 | 3M Innovative Properties Company | Substituted chiral fused [1,2]imidazo[4,5-c] ring compounds |
| US10071156B2 (en) | 2005-02-04 | 2018-09-11 | 3M Innovative Properties Company | Aqueous gel formulations containing immune response modifiers |
| US9248127B2 (en) | 2005-02-04 | 2016-02-02 | 3M Innovative Properties Company | Aqueous gel formulations containing immune response modifiers |
| US7968563B2 (en) | 2005-02-11 | 2011-06-28 | 3M Innovative Properties Company | Oxime and hydroxylamine substituted imidazo[4,5-c] ring compounds and methods |
| US7943610B2 (en) | 2005-04-01 | 2011-05-17 | 3M Innovative Properties Company | Pyrazolopyridine-1,4-diamines and analogs thereof |
| US7943636B2 (en) | 2005-04-01 | 2011-05-17 | 3M Innovative Properties Company | 1-substituted pyrazolo (3,4-C) ring compounds as modulators of cytokine biosynthesis for the treatment of viral infections and neoplastic diseases |
| EP1937683A1 (en) * | 2005-09-23 | 2008-07-02 | Coley Pharmaceutical Group, Inc. | METHOD FOR 1H-IMIDAZOÝ4,5-c¨PYRIDINES AND ANALOGS THEREOF |
| EP1937683A4 (en) * | 2005-09-23 | 2010-08-25 | Coley Pharm Group Inc | METHOD FOR 1H-IMIDAZOÝ4,5-c¨PYRIDINES AND ANALOGS THEREOF |
| EP3085373A1 (en) | 2006-02-22 | 2016-10-26 | 3M Innovative Properties Company | Immune response modifier conjugates |
| US7906506B2 (en) | 2006-07-12 | 2011-03-15 | 3M Innovative Properties Company | Substituted chiral fused [1,2] imidazo [4,5-c] ring compounds and methods |
| US10144735B2 (en) | 2006-12-22 | 2018-12-04 | 3M Innovative Properties Company | Immune response modifier compositions and methods |
| US10005772B2 (en) | 2006-12-22 | 2018-06-26 | 3M Innovative Properties Company | Immune response modifier compositions and methods |
| CN105294684B (en) * | 2010-08-17 | 2018-04-06 | 3M创新有限公司 | Composition, preparation and the method for lipidization immune response modifier compound |
| US10052380B2 (en) | 2010-08-17 | 2018-08-21 | 3M Innovative Properties Company | Lipidated immune response modifier compound compositions, formulations, and methods |
| CN105294684A (en) * | 2010-08-17 | 2016-02-03 | 3M创新有限公司 | Lipidated immune response modifier compound compositions, formulations, and methods |
| EP3222621A1 (en) * | 2010-08-17 | 2017-09-27 | 3M Innovative Properties Company | Lipidated immune response modifier compound and its medical use |
| US9795669B2 (en) | 2010-08-17 | 2017-10-24 | 3M Innovative Properties Company | Lipidated immune response modifier compound compositions, formulations, and methods |
| US11524071B2 (en) | 2010-08-17 | 2022-12-13 | 3M Innovative Properties Company | Lipidated immune response modifier compound compositions, formulations, and methods |
| EP2606047A4 (en) * | 2010-08-17 | 2014-07-02 | 3M Innovative Properties Co | Lipidated immune response modifier compound compositions, formulations, and methods |
| US10821176B2 (en) | 2010-08-17 | 2020-11-03 | 3M Innovative Properties Company | Lipidated immune response modifier compound compositions, formulations, and methods |
| EP2606047A1 (en) * | 2010-08-17 | 2013-06-26 | 3M Innovative Properties Company | Lipidated immune response modifier compound compositions, formulations, and methods |
| CN103097386A (en) * | 2010-08-17 | 2013-05-08 | 3M创新有限公司 | Lipidated immune response modifier compound compositions, formulations, and methods |
| US10383938B2 (en) | 2010-08-17 | 2019-08-20 | 3M Innovative Properties Company | Lipidated immune response modifier compound compositions, formulations, and methods |
| EP3153180A1 (en) | 2011-06-03 | 2017-04-12 | 3M Innovative Properties Company | Heterobifunctional linkers with polyethylene glycol segments and immune response modifier conjugates made therefrom |
| WO2014107663A2 (en) | 2013-01-07 | 2014-07-10 | The Trustees Of The University Of Pennsylvania | Compositions and methods for treating cutaneous t cell lymphoma |
| EP3756669A1 (en) | 2013-01-07 | 2020-12-30 | The Trustees of the University of Pennsylvania | Compositions for use for treating cutaneous t cell lymphoma |
| US10526309B2 (en) | 2015-10-02 | 2020-01-07 | The University Of North Carolina At Chapel Hill | Pan-TAM inhibitors and Mer/Axl dual inhibitors |
| US11306083B2 (en) | 2017-12-20 | 2022-04-19 | 3M Innovative Properties Company | Amide substituted imidazo[4,5-C]quinoline compounds with a branched chain linking group for use as an immune response modifier |
| WO2019166946A1 (en) | 2018-02-28 | 2019-09-06 | Pfizer Inc. | Il-15 variants and uses thereof |
| WO2019224716A2 (en) | 2018-05-23 | 2019-11-28 | Pfizer Inc. | Antibodies specific for gucy2c and uses thereof |
| WO2019224715A1 (en) | 2018-05-23 | 2019-11-28 | Pfizer Inc. | Antibodies specific for cd3 and uses thereof |
| US11434292B2 (en) | 2018-05-23 | 2022-09-06 | Pfizer Inc. | Antibodies specific for CD3 and uses thereof |
| WO2020128893A1 (en) | 2018-12-21 | 2020-06-25 | Pfizer Inc. | Combination treatments of cancer comprising a tlr agonist |
| US10954239B2 (en) | 2019-02-01 | 2021-03-23 | Canwell Biotech Limited | Imidazoquinoline amine derivatives, pharmaceutical compositions and therapeutic methods thereof |
| WO2020160054A1 (en) * | 2019-02-01 | 2020-08-06 | Canwell Biotech Limited | Imidazoquinoline amine derivatives, pharmaceutical composition, use thereof |
| CN114585621A (en) * | 2019-02-01 | 2022-06-03 | 康威(广州)生物科技有限公司 | Imidazoquinoline amine derivative, and pharmaceutical composition and application thereof |
| CN114585621B (en) * | 2019-02-01 | 2023-11-14 | 康威(广州)生物科技有限公司 | Imidazoquinoline amine derivative, pharmaceutical composition and application thereof |
| EP4051270A4 (en) * | 2019-10-29 | 2024-02-28 | Prime Reach Trading Ltd | 4-amino-imidazoquinoline compounds and use thereof |
| WO2021124073A1 (en) | 2019-12-17 | 2021-06-24 | Pfizer Inc. | Antibodies specific for cd47, pd-l1, and uses thereof |
| US20230270819A1 (en) * | 2019-12-20 | 2023-08-31 | Nammi Therapeutics, Inc. | Formulated and/or Co-Formulated Liposome Compositions Containing Toll--Like Receptor ("TLR") Agonist Prodrugs Useful In The Treatment of Cancer and Methods Thereof |
| US11896646B2 (en) * | 2019-12-20 | 2024-02-13 | Nammi Therapeutics, Inc. | Formulated and/or co-formulated liposome compositions containing toll-like receptor (“TLR”) agonist prodrugs useful in the treatment of cancer and methods thereof |
| WO2022013775A1 (en) | 2020-07-17 | 2022-01-20 | Pfizer Inc. | Therapeutic antibodies and their uses |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2008511677A (en) | 2008-04-17 |
| ES2384390T3 (en) | 2012-07-04 |
| AU2005282726A1 (en) | 2006-03-16 |
| US20090005376A1 (en) | 2009-01-01 |
| CA2578741C (en) | 2014-01-14 |
| AU2005282726B2 (en) | 2011-06-02 |
| ATE555786T1 (en) | 2012-05-15 |
| JP5209312B2 (en) | 2013-06-12 |
| PL1789042T3 (en) | 2012-09-28 |
| CA2578741A1 (en) | 2006-03-16 |
| EP1789042B1 (en) | 2012-05-02 |
| EP1789042A2 (en) | 2007-05-30 |
| WO2006028962A3 (en) | 2006-10-26 |
| US7579359B2 (en) | 2009-08-25 |
| EP1789042A4 (en) | 2009-07-22 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1789042B1 (en) | 1-alkoxy 1h-imidazo ring systems and methods | |
| US8143270B2 (en) | 2-amino 1H-in-imidazo ring systems and methods | |
| EP1831221B1 (en) | Substituted chiral fused 1,2 imidazo 4,5-c ring compounds | |
| US7968563B2 (en) | Oxime and hydroxylamine substituted imidazo[4,5-c] ring compounds and methods | |
| US20040176367A1 (en) | 1-Amino 1H-imidazoquinolines | |
| US20070287725A1 (en) | Isoxazole, Dihydroisoxazole, And Oxadiazole Substituted Imidazo Ring Compounds And Method | |
| WO2005123079A2 (en) | Urea substituted imidazopyridines, imidazoquinolines, and imidazonaphthyridines | |
| US20090030030A1 (en) | Arylalkenyl and arylalkynyl substituted imidazoquinolines | |
| WO2005094531A2 (en) | Amide substituted imidazopyridines, imidazoquinolines, and imidazonaphthyridines | |
| WO2006093514A2 (en) | Aryl and arylalkylenyl substituted thiazoloquinolines and thiazolonaphthyridines | |
| WO2007120121A2 (en) | Oxime and hydroxylamine substituted thiazolo[4,5-c] ring compounds and methods | |
| WO2006091567A2 (en) | Hydroxyalkyl substituted imidazoquinoline compounds and methods | |
| WO2006098852A2 (en) | Hydroxyalkyl substituted imidazoquinolines | |
| AU2006216997A1 (en) | Substituted imidazoquinolines and imidazonaphthyridines | |
| WO2008008432A2 (en) | Substituted chiral fused( 1,2) imidazo (4,5-c) ring compounds and methods | |
| EP1686992A2 (en) | Hydroxylamine and oxime substituted imidazoquinolines, imidazopyridines, and imidazonaphthyridines |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KM KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NG NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SM SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU LV MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 11574463 Country of ref document: US Ref document number: 2578741 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007530396 Country of ref document: JP Ref document number: 2005282726 Country of ref document: AU |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005794068 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: 2005282726 Country of ref document: AU Date of ref document: 20050901 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005282726 Country of ref document: AU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005794068 Country of ref document: EP |