PYRIMIDINE SULPHONAMIDE DERIVATIVES AS CHEMOKINE RECEPTOR MODULATORS
The present invention relates to certain heterocyclic compounds, processes and intermediates used in their preparation, pharmaceutical compositions containing them and their use in therapy.
Chemokines play an important role in immune and inflammatory responses in various diseases and disorders, including asthma and allergic diseases, as well as autoimmune pathologies such as rheumatoid arthritis and atherosclerosis. These small secreted molecules are a growing superfamily of 8-14 kDa proteins characterised by a conserved cysteine motif. At the present time, the chemokine superfamily comprises three groups exhibiting characteristic structural motifs, the C-X-C, C-C and C-X3-C families. The C-X-C and C-C families have sequence similarity and are distinguished from one another on the basis of a single amino acid insertion between the NH-proximal pair of cysteine residues. The C-X3-C family is distinguished from the other two families on the basis of having a triple amino acid insertion between the NH-proximal pair of cysteine residues.
The C-X-C chemokines include several potent chemoattractants and activators of neutrophils such as interleukin-8 (EL-8) and neutrophil- activating peptide 2 (NAP-2).
The C-C chemokines include potent chemoattractants of monocytes and lymphocytes but not neutrophils. Examples include human monocyte chemotactic proteins 1- 3 (MCP-I, MCP-2 and MCP-3), RANTES (Regulated on Activation, Normal T Expressed and Secreted), eotaxin and the macrophage inflammatory proteins lα and lβ (MIP- lα and MIP- lβ).
The C-X3-C chemokine (also known as fractalkine) is a potent chemoattractant and activator of microglia in the central nervous system (CNS) as well as of monocytes, T cells, NK cells and mast cells.
Studies have demonstrated that the actions of the chemokines are mediated by subfamilies of G protein-coupled receptors, among which are the receptors designated CCRl, CCR2, CCR2A, CCR2B, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR9, CCRlO and CCRl 1 (for the C-C family); CXCRl, CXCR2, CXCR3, CXCR4 and CXCR5 (for the C-X-C family) and CX3CRl for the C-X3-C family. These receptors represent good targets for drug development since agents which modulate these receptors would be useful in the treatment of disorders and diseases such as those mentioned above.
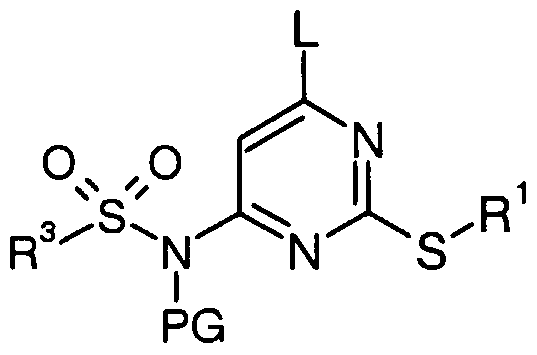
In our PCT patent application WO 2004/011443 we disclosed amino-substituted pyrimidine sulfonamides for use as modulators of chemokine receptors.
The present invention now provides a compound of formula (1), or a pharmaceutically acceptable salt, solvate or in vivo hydro lysable ester thereof:
(1)
wherein R1 is a group selected from C3-7carbocyclyl, Ci.salkyl, C2_6alkenyl and C2-6alkynyl; wherein the group is optionally substituted by 1 , 2 or 3 substituents independently selected from fluoro, nitrite, -OR4, -NR5R6, -CONR5R6, -COOR7, -NR8COR9, -SR10, -SO2R10, -SO2NR5R6, -NR8SO2R9, phenyl or heteroaryl; wherein phenyl and heteroaryl are optionally substituted by 1, 2 or 3 substituents independently selected from halo, cyano, nitro, -OR4, - NR5R6, -CONR5R6, -COOR7, -NR8COR9, -SR10, -SO2R10, -SO2NR5R6, -NR8SO2R9, C1-6alkyl and trifluoromethyl;
X is -CH2-, a bond, oxygen, sulphur, sulphoxide, or sulphone;
R
2 is C
3-7carbocyclyl, optionally substituted by 1, 2 or 3 substituents independently selected from: fluoro, -OR
4, -NR
5R
6 -CONR
5R
6, -COOR
7, -NR
8COR
9, -SR
10, -SO
2R
10, -SO
2NR
5R
6, -NR
8SO
2R
9; or R
2 is a 3-8 membered ring optionally containing 1, 2 or 3 atoms selected from O, S, -NR
8 and whereby the ring is optionally substituted by 1 ,2 or 3 substituents indepedently selected from Ci ^alkyl, fluoro, -OR
4, -NR
5R
6 -CONR
5R
6, -COOR
7, -NR
8COR
9, -SR
10, -SO
2R
10, -SO
2NR
5R
6, -NR
8SO
2R
9;
or R
2 is phenyl or heteroaryl, each of which is optionally substituted by 1, 2 or 3 substituents independently selected from halo, cyano, nitro, -OR
4, -NR
5R
6, -CONR
5R
6, -NR
8COR
9, - SO
2NR
5R
6, -NR
8SO
2R
9, C
1-6alkyl and trifluoromethyl; or R
2 is a group selected from C
t-salkyl, C
2-6alkenyl or C
2-
6alkynyl wherein the group is substituted by 1, 2 or 3 substituents independently selected from hydroxy, amino, Ci
-6alkoxy, Ci-6alkylamino,
N-(Ci-
6alkyl)-Λf -(phenyl)amino, iV-Cuealkylcarbamoyl, iy,N-di(Ci.6alkyl)carbamoyl, N-(C
1.
6alkyl)-N-(phenyi)carbamoyl, carboxy, phenoxycarbonyl, -NR
8COR
9, -SO
2R
10, -SO
2NR
5R
6 ,-NR
8SO
2R
9 and -CONR
5R
6;
R3 is trifluoromethyl or a group-NR5R6,
or R is phenyl, napthyl, monocyclic or bicyclic heteroaryl wherein a heteroring may be partially or fully saturated and one or more ring carbon atoms may form a carbonyl group, and wherein each phenyl or heteroaryl group is optionally substituted by 1, 2 or 3 substituents independently selected from halo, cyano, nitro, phenyl, heteroaryl, -OR4, -NR5R6, -CONR5R6, -COR7 , , -COR20,--COOR7, -NR8COR9, -SR10, -SO2R10, -SO2NR5R6, -NR8SO2R9, trifluoromethyl or Q-όalkyl [optionally further substituted by 1, 2 or 3 substituents independently selected from halo, cyano, nitro, -OR20 , -COOR20, -COR20, -NR18R19, -CONR18R19, -NR18COR19, -SO2R20, -SO2NR18R19, NR18SO2R19, phenyl or monocyclic or bicyclic heteroaryl, wherein a heteroring may be partially or fully saturated; and wherein each phenyl or heteroaryl group is optionally substituted by 1, 2 or 3 substituents independently selected from halo, cyano, nitro, -OR20, -NR5R6, -CONR5R6, -COR7' -COOR7, -NR8COR9, -SR10, -SO2R10, -SO2NR5R6, -NR8SO2R9, heteroaryl, Ci-6alkyl (optionally further substituted by 1, 2 or 3 substituents independently selected from halo, cyano, nitro, -OR , -COOR20, -COR20, -NR18R19, -CONR18R19, -NR18COR19, -SO2R20, -SO2NR18R19, NR18SO2R19.
or R3 is a group selected from C3-7carbocyclyl, C^aUcyl, C2-6alkenyl and C2.6alkynyl whereby the group is optionally substituted by 1, 2 or 3 substituents independently selected from halo, -OR4, -NR5R6, -CONR5R6, -COR7,-COOR7, -NR8COR9, -SR10, -SO2R10, -SO2NR5R6, -NR8SO2R9, phenyl or monocyclic or bicyclic heteroaryl, wherein a heteroring may be partially or fully saturated; and wherein each phenyl or monocyclic or bicyclic heteroaryl
group is optionally substituted by 1, 2 or 3 substituents independently selected from halo, cyano, nitro, -OR4, -NR5R6, -CONR5R6, -COR7' -COOR7, -NR8COR9, -SR10, -SO2R10, -SO2NR5R6, -NR8SO2R9, Ci-6alkyl, or trifluoromethyl;
R4 is hydrogen or a group selected from C^aUcyl and phenyl, wherein the group is optionally substituted by 1 or 2 substituents independently selected from halo, phenyl, -OR11 and - NR12R13;
R5 and R6 are independently hydrogen or a group selected from C1-6alk;yl and phenyl and monocyclic or bicyclic heteroaryl, wherein a heteroring may be partially or fully saturated; wherein the group is optionally substituted by 1, 2 or 3 substituents independently selected from halo, phenyl, -OR14,-NR15R16, -COOR14, -CONR15R16, -NR15COR16, -SO2R10, - SO2NR15R16 and NR15SO2R16; or R5 and R6 together with the nitrogen atom to which they are attached form a 4- to
7-membered saturated heterocyclic ring system optionally containing a further heteroatom selected from oxygen , -S0(n)- (where n = O, 1 or 2) and nitrogen atoms, in which the ring is optionally substituted by 1, 2 or 3 substituents independently selected from phenyl, heteroaryl, -OR14, -COR20, -COOR14, -NR15R16, -CONR15R16, -NR15COR16, -SO2R10, -SO2NR15R16, NR15SO2R16 or Q-ealkyl (optionally further substituted by 1 or 2 or 3 substituents independently selected from halo, -NR15R16 and -OR17 or cyano, nitro, -OR20 , -COOR20, -COR20, -NR18R19, -CONR18R19, -NR18COR19, -SO2R20, -SO2NR18R19, and NR18SO2R19 groups).
R10 is hydrogen or a group selected from C^aUcyl or phenyl, wherein the group is optionally substituted by 1, 2 or 3 substituents independently selected from halo, phenyl, -OR17 and - NR15R16; and each of R7, R8, R9, R11, R12, R13, R14 R15, R16, R17 is independently hydrogen, Ci-6alkyl or phenyl.
R18, R19, and R20 are hydrogen or a group selected from C^aUcyl or heteroaryl (wherein a heteroring may be partially or fully saturated) or phenyl, wherein the group is optionally substituted by 1, 2 or 3 substituents independently selected from halo, nitro, -CN, -OR4, -
NR8R9, -CONR8R9, -COR7' -COOR7, -NR8COR9, -SR10, -SO2R10, -SO2NR8R9, -NR8SO2R9, C1-6alkyl or heteroaryl.
Certain compounds of formula (1) are capable of existing in stereoisomeric forms. It will be understood that the invention encompasses all geometric and optical isomers of the compounds of formula (1) and mixtures thereof including racemates.
The synthesis of optically active forms may be carried out by standard techniques of organic chemistry well known in the art, for example by synthesis from optically active starting materials or by resolution of a racemic form. Similarly, the above-mentioned activity may be evaluated using the standard laboratory techniques referred to hereinafter. Within the present invention it is to be understood that a compound of formula (1) or a salt, solvate or in vivo hydrolysable ester thereof may exhibit the phenomenon of tautomerism and that the formulae drawings within this specification can represent only one of the possible tautomeric forms. It is to be understood that the invention encompasses any tautomeric form and mixtures thereof and is not to be limited merely to any one tautomeric form utilised within the formulae drawings. The formulae drawings within this specification can represent only one of the possible tautomeric forms and it is to be understood that the specification encompasses all possible tautomeric forms of the compounds drawn not just those forms which it has been possible to show graphically herein.
It is also to be understood that certain compounds of formula (1) and salts thereof can exist in solvated as well as unsolvated forms such as, for example, hydrated forms. It is to be understood that the invention encompasses all such solvated forms.
The present invention relates to the compounds of formula (1) as hereinbefore defined as well as to the salts thereof. Salts for use in pharmaceutical compositions will be pharmaceutically acceptable salts, but other salts may be useful in the production of the compounds of formula (1) and their pharmaceutically acceptable salts. Pharmaceutically acceptable salts of the invention may, for example, include acid addition salts of the compounds of formula (1) as hereinbefore defined which are sufficiently basic to form such salts. Such acid addition salts include for example salts with inorganic or organic acids affording pharmaceutically acceptable anions such as with hydrogen halides (especially hydrochloric or hydrobromic acid of which hydrochloric acid is particularly preferred) or with sulphuric or phosphoric acid, or with trifluoro acetic, citric or maleic acid. Suitable salts include hydrochlorides, hydrobromides, phosphates, sulphates, hydrogen sulphates,
alkylsulphonates, arylsulphonates, acetates, benzoates, citrates, maleates, fumarates, succinates, lactates, tartrates, oxalates, methanesulphonates or/?-toluenesulphonates. Pharmaceutically acceptable salts of the invention may also include basic addition salts of the compounds of formula (1) as hereinbefore defined which are sufficiently acidic to form such salts. Such salts may be formed with an inorganic or organic base which affords a pharmaceutically acceptable cation. Such salts with inorganic or organic bases include for example an alkali metal salt, such as a lithium, sodium or potassium salt, an alkaline earth metal salt such as a calcium or magnesium salt, an ammonium salt or an organic amine salt, for example a salt with methylamine, dimethylamine, trimethylamine, triethylamine, piperidine, morpholine or tris-(2-hydroxyethyl)amine. Other basic addition salts include aluminium, zinc, benzathine, chloroprocaine, choline, diethanolamine, ethanolamine, ethyldiamine, meglumine, tromethamine or procaine.
The present invention further relates to an in vivo hydro lysable ester of a compound of formula (1). An in vivo hydrolysable ester of a compound of formula (1) which contains carboxy or hydroxy group is, for example a pharmaceutically acceptable ester which is cleaved in the human or animal body to produce the parent acid or alcohol. Such esters can be identified by administering, for example, intravenously to a test animal, the compound under test and subsequently examining the test animal's body fluid.
Suitable pharmaceutically acceptable esters for carboxy include Q-βalkoxymethyl esters for example methoxymethyl, Ci-βalkanoyloxymethyl esters for example pivaloyloxymethyl, phthalidyl esters, C3.8cycloalkoxycarbonyloxyC1-6alkyl esters for example 1-cyclohexylcarbonyloxyethyl; l,3-dioxolen-2-onylmethyl esters for example 5-methyl-l,3-dioxolen-2-onylmethyl; and C^alko xycarbony Io xy ethyl esters.for example 1-methoxycarbonyloxyethyl and may be formed at any carboxy group in the compounds of this invention.
Suitable pharmaceutically-acceptable esters for hydroxy include inorganic esters such as phosphate esters (including phosphoramidic cyclic esters) and α-acyloxyalkyl ethers and related compounds which as a result of the in vivo hydrolysis of the ester breakdown to give the parent hydroxy group/s. Examples of α-acyloxyalkyl ethers include acetoxymethoxy and 2,2-dimethylpropionyloxymethoxy. A selection of in-vivo hydrolysable ester forming groups for hydroxy include Ci-ioalkanoyl, for example acetyl; benzoyl; phenylacetyl; substituted benzoyl and phenylacetyl, Q-ioalkoxycarbonyl (to give alkyl carbonate esters), for
example ethoxycarbonyl; di-(Cr4)alkylcarbamoyl and N-(di-(Cr4)alkylaminoethyl)-//- (Cr4)HUCyICaTbBmOyI (to give carbamates); di-(Ci-4)alkylaminoacetyl and carboxyacetyl. Examples of ring substituents on phenylacetyl and benzoyl include aminomethyl, (Ci. 4)alkylaminomethyl and di-((Ci-4)alkyl)aminomethyl, and morpholino or piperazino linked from a ring nitrogen atom via a methylene linking group to the 3- or 4- position of the benzoyl ring. Other interesting in- vivo hydrolysable esters include, for example, RΛC(O)O(Ci-6)alkyl- CO-, wherein RΛ is for example, benzyloxy-(Ci-4)alkyl, or phenyl). Suitable substituents on a phenyl group in such esters include, for example, 4-(Ci-4)piperazino-(Ci-4)alkyl, piperazino- (Ci-4)alkyl and morpho lino -(Cr4) alky 1. In this specification the term "alkyl" includes both straight-chain and branched- chain alkyl groups. However references to individual alkyl groups such as "propyl" are specific for the straight chain version only and references to individual branched-chain alkyl groups such as f-butyl are specific for the branched chain version only. For example, "Ci.3alkyl" includes methyl, ethyl, propyl and isopropyl and examples of "Ci-6alkyl" include the examples of "Ci-3alkyl"and additionally t-butyl, pentyl, 2,3-dimethylpropyl, 3- methylbutyl and hexyl. Examples of "Q-salkyl" include the examples of "Ci-όalkyl" and additionally heptyl, 2,3-dimethylpentyl, 1-propylbutyl and octyl. An analogous convention applies to other terms, for example "C2.6alkenyl" includes vinyl, allyl, 1-propenyl , 2-butenyl, 3-butenyl, 3-methylbut-l-enyl, 1-pentenyl and 4-hexenyl and examples of "C2-6alkynyl" includes ethynyl, 1-propynyl, 3-butynyl, 2-pentynyl and l-methylpent-2-ynyl.
"C3.7carbocyclyl" is a saturated, partially saturated or unsaturated, monocyclic ring containing 3 to 7 carbon ring atoms wherein a -CH2- group can optionally be replaced by a -C(O)-. Suitable examples of "carbocyclyl" are cyclopropyl, cyclopentyl, cyclobutyl, cyclohexyl, cyclohexenyl, 4-oxocyclohex-l-yl and 3-oxocyclohept-5-en-l-yl. The term "halo" refers to fluoro, chloro, bromo and iodo.
Examples of "Ci-βalkoxy" include methoxy, ethoxy, propoxy, isopropoxy, butyloxy, pentyloxy, 1-ethylpropoxy and hexyloxy. Examples of "Ci-6alkylamino" include methylamino, ethylamino, propylamino, butylamino and 2-methylpropyhnino. Examples of "di(Ci.6alkyl)amino" include dimethylamino, N-methyl-N-ethylamino, diethylamino, N- propyl-N-3-methylbutylamino. Examples of 'W-(Ci.6alkyl)-N-(phenyl)amino" include N- methyl-N-phenylamino, N-propyl-N-phenylamino and N-(2-methylbutyl)-iV-phenylamino. Examples of 'W-(Ci-6alkyi)carbamoyr are iV-methylcarbamoyl, N-ethylcarbamoyl and N-(2-
ethylbutylcarbamoyl. Examples of 'W-(C1-6alkyl)-N-(phenyl)carbamoyl" include N-methy\-N- phenylcarbamoyl, JV-butyl-N-phenylcarbamoyl and Λ^(3-methylpentyl)-N-(phenyl)carbamoyl. Examples of 'W.N-d^C^ealky^carbamoyl" include ΛζN-dimethylcarbamoyl, N-methyl-iV- ethylcarbamoyl and iV-propyl-N-(2-methylbutyl)carbamoyl. Examples of "Ci-βalkylthio" include methylthio, ethylthio, propylthio, butylthio and 2-methylbutylthio.
'Ηeteroaryl" is a monocyclic or bicyclic aryl ring, containing 5 to 10 ring atoms of which 1, 2, 3 or 4 ring atoms are chosen from nitrogen, sulphur or oxygen. Examples of heteroaryl include pyrrolyl, furanyl, thienyl, thiazolyl, isothiazolyl, oxazolyl, isoxazolyl, imidazolyl, pyrazolyl, triazolyl, tetrazolyl, isoxadiazolyl, oxadiazolyl, isothiadiazolyl, thiadiazolyl, pyridyl, pyrimidinyl, pyrazinyl, pyridazinyl, triazinyl, pyridinonyl, pyrimidindionyl, benzfuranyl, benzthieno, indolyl, benzimidazolyl, benzoxazolyl, benzthiazolyl, indazolyl, benzisoxazolyl, benzisothiazolyl, benztriazolyl, quinolinyl, isoquinolinyl, 4H-chromen-4-onyl, tetrahydroquinolinyl, tetrahydroisoquinolinyl, and naphthiridinyl. Conveniently heteroaryl is selected from imidazolyl, pyrazolyl, thiazolyl, isoxazolyl, furanyl, thienyl, isoxazolyl, or indazolyl. Fully saturated heterocyclic rings include examples such as oxetanyl, azetidinyl, pyrrolidinyl, tetrahydrofuranyl, isoxazolidinyl, tetrahydropyranyl, piperidinyl, piperazinyl, piperazinonyl, morpholinyl, thiomorpholinyl, thiomoφholinyl-1 -oxide, thiomorpholiny 1-1,1 -dioxide, oxazinanonyl, quinuclidinyl, homopiperidinyl and homopiperazinyl, 9-methyl-3,9-diazabicyclo[4.2.1]nonanyl and tetrahydropyridinyl.
Examples of "a 3-8 membered ring optionally containing 1, 2 or 3 atoms selected from O, S and NR8" include oxetanyl, azetidinyl, benzodiazolyl, pyrrolidinyl, tetrahydrofuranyl, isoxazolidinyl, tetrahydrothiophenyl, tetrahydropyranyl, piperidinyl, piperazinyl, piperazinonyl, morpholinyl, thiomorpholinyl, thiomoφho liny 1-1 -oxide, thiomorpholiny 1-1,1 -dioxide, oxazinanonyl, quinuclidinyl, homopiperidinyl and homopiperazinyl tetrahydrodioxanyl. Examples of "a 4- to 7-membered saturated heterocyclic ring system" include azetidinyl, pyrrolidinyl, isoxazolidinyl, piperidinyl, piperazinyl, piperazinonyl, homopiperazinyl, thiomorpholinyl, thiomorpholinyl-l -oxide, thiomorpholiny 1-1,1 -dioxide, oxazinanonyl, quinuclidinyl and morpholinyl, Where optional substituents are chosen from "1, 2 or 3" groups it is to be understood that this definition includes all substituents being chosen from one of the specified
groups or the substituents being chosen from two or more of the specified groups. An analogous convention applies to substituents chosen from "l or 2" groups. Convenient values of R1, R2, R3, and X are as follows:
R1 is Ci-βalkyl, wherein the group is substituted by phenyl optionally substituted by 1, 2 or 3 substituents independently selected from fluoro, chloro, bromo, methoxy, methyl and trifluoromethyl.
X is -CH2-, a bond, oxygen , sulphur, sulphoxide, or sulphone;
R is C1-8alkyl wherein the group is optionally substituted by 1, 2 or 3 substituents independantly selected from d-βalkoxy, hydroxy and fluoro; or R is a 5-6 membered ring optionally containing 1,2 or 3 heteroatoms selected from O, S, -
NR8 and wherby the ring is optionally substituted by -OR4.
R3 is C3-7carbocyclyl, d-salkyl, -NR5R6, phenyl, monocyclic or bicyclic heteroaryl wherein a heteroring may be partially or fully saturated and one or more ring carbon atoms may form a carbonyl group, and wherein each phenyl or heteroaryl group is optionally substituted by 1, 2 or 3 substituents independently selected from cyano, heteroaryl, -OR4, -NR5R6, -CONR5R6,
-COR7", , -COR20, -NR8COR9, -SO2R10, -SO2NR5R6, Ci-6alkyl [optionally further substituted by 1, 2 or 3 substituents independently selected from -OR20 , -COR20, -NR18R19, -CONR18R19, phenyl or monocyclic or bicyclic heteroaryl, wherein a heteroring may be partially or fully saturated; and wherein each phenyl or heteroaryl group is optionally substituted by 1, 2 or 3 substituents independently selected fromnitro, -OR20, -NR5R6, -NR8COR9, heteroaryl, d-όalkyl (optionally further substituted by 1, 2 or 3 substituents independently selected from cyano, -OR20).
Convenient values of R4 -R17 are as follows:
R4 is hydrogen or Ci-6alkyl; R5 and R6 are a group selected from d-βalkyl or R5 and R6 together with the nitrogen atom to which they are attached form a 4- to 7-membered saturated heterocyclic ring optionally containing a further heteroatom selected from oxygen and nitrogen atoms.
R7, R8, R9, R11, R12, R13, R14 R15, R16, R17 are independently hydrogen, Ci-6alkyl or phenyl.
Convenient values of R18-R20 are as follows: R18, R19 and R20 are hydrogen, phenyl, heteroaryl, or d-βalkyl (optionally further substituted by NR8R9).
Preferred values of R1, R2, R3, and X are as follows:
R1 is Ci-3alkyl (such as -CH2-, -(CH2)2-, -(CH2)3-, -CH2(CH3)- or -CH2(CH3)CH2-) wherein the group is substituted by phenyl optionally substituted by 1, 2 or 3 substituents independently selected from fluoro and chloro. Benzyl is particularly preferred. X is -CH2-, a bond, oxygen, or sulphur. Oxygen is particularly preferred.
R2 is C1-8 alkyl, such as C1-4 alkyl, wherein the group is optionally substituted by 1 or 2 substituents independently selected from Ci.3alkoxy (such as methoxy, ethoxy, cyclopropyloxy or isopropyloxy), hydroxy and fluoro, hydroxy is particularly preferred; or R2 is a 5-membered ring optionally containing a heteroatom selected from O or -NR8 and wherby the ring is optionially substituted by -OR4.
R3 is C1-3alkyl (such as methyl, ethyl, isopropyl or cyclopropyl) or -NR5R6 (such as azetidinyl, pyrolidinyl, morphotinyl, piperidinyl, piperazinyl) or phenyl or a monocyclic or bicyclic heteroaryl group (such as 1-methylimidazolyl or 1,2-dimethylimidazolyl). Preferred values of R4-R17 are as follows: R4 is hydrogen, or C1-3alkyl (such as methyl, ethyl, cyclopropyl or isopropyl)
R5 and R6 are a group selected from C1-2alkyl (such as methyl and ethyl) or R5 and R6 together with the nitrogen atom to which they are attached form a 4- to 6-membered saturated heterocyclic ring (such as azetidinyl, pyrolidinyl, piperidinyl) or optionally containing a further heteroatom selected from oxygen (such as morpholinyl) or nitrogen (such as . piperazinyl).
R7, R8, R9, R11, R12, R13, R14 R15, R16, R17 are independently hydrogen, or C1-2alkyl (such as methyl or ethyl).
Preferred values of R18-R20 are as follows: R18, R19 and R20 are hydrogen or C1-6alkyl (optionally further substituted by NR8R9). Such values may be used where appropriate with any of the definitions, claims or embodiments defined hereinbefore or hereinafter.
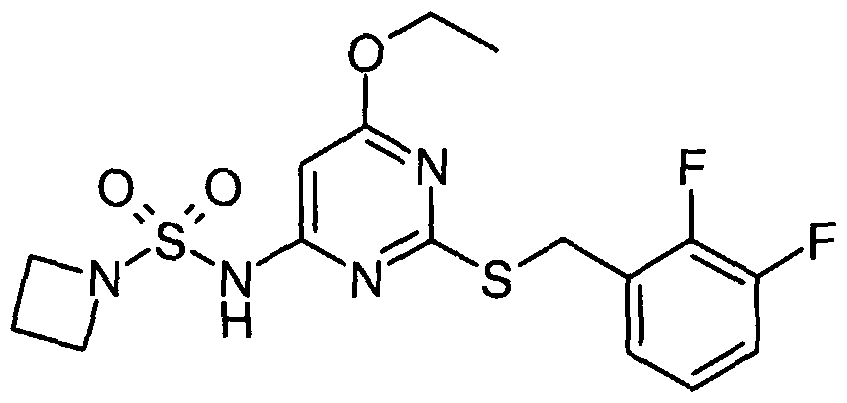
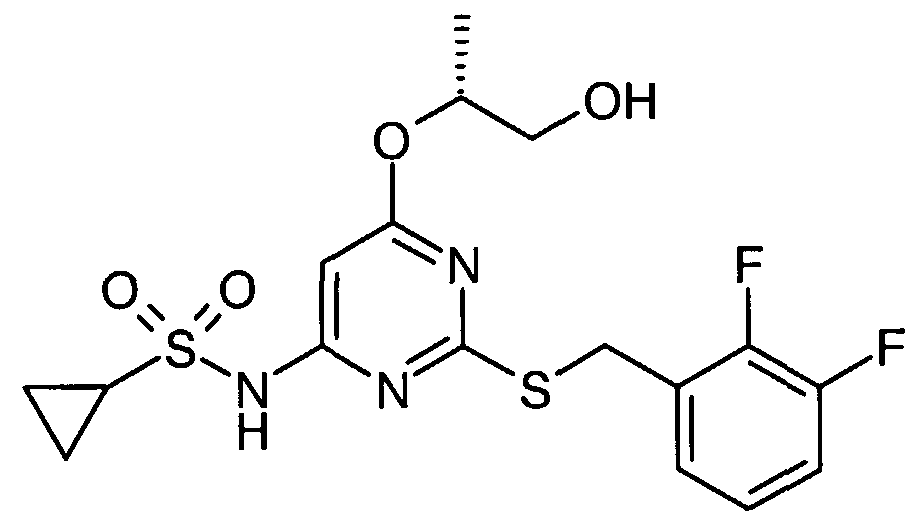
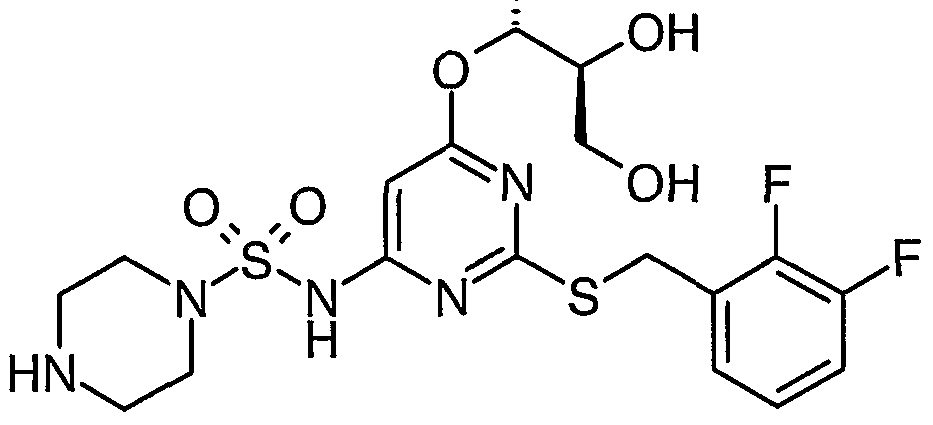
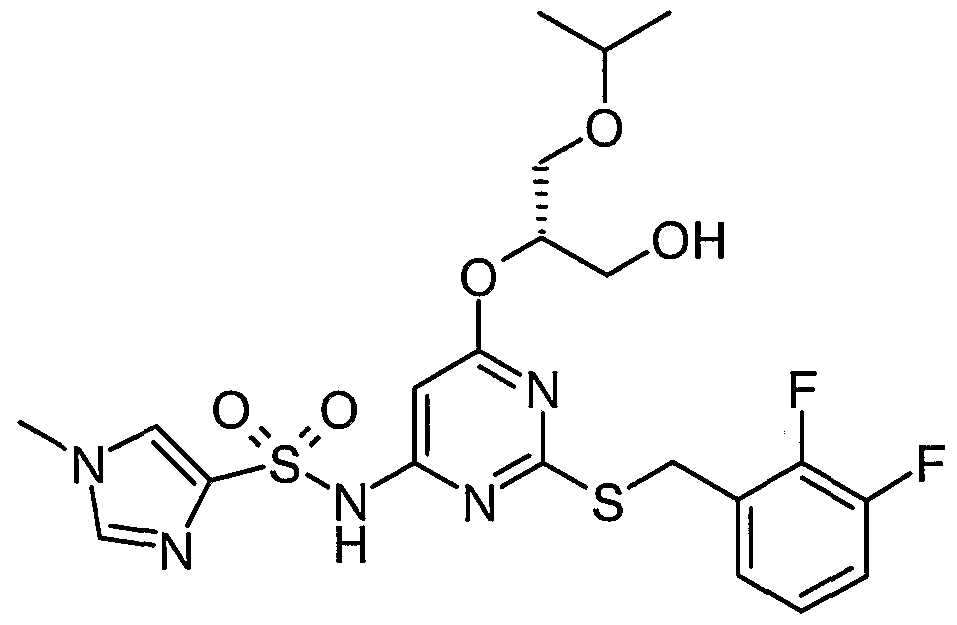
Particular compounds of the invention include:
N- [2- [[(2,3-difluorophenyl)methyl] thio] -6- [2-hydroxy- 1 -(hydroxymethyl)ethoxy] -A- pyrimidinyl]-l-azetidinesulfonamide R, S) N-[2-[[(2,3-dMuorophenyl)methyl]tMo]-6-[3,4-dihydroxybutyl]pyrimidin-4-yl]azetidine- 1-sulphonamide; and
N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[3-hydroxy-2-(hydroxymethyl)propyl] pyrimidin-
4- yl] azetidine- 1 - sulphonamide
N-(2-[(2,3-difluorobenzyl)thio]-6-{[(lR,2R)-2-hydroxy-l-methylpropyl]oxy}pyriinidin-4- y 1) azetidine- 1 - sulfonamide : and N-(2-[(2,3-difluorobenzyl)thio]-6-{[(15,2S)-2-hydroxy-l-methylρropyl]oxy}pyrimidin-4- yl)azetidine- 1- sulfonamide
N-[2-[[(2,3-difluoroρhenyl)methyl]thio]-6-[[(25)-2,3-dihydroxyρropyl]oxy]-4-pyrimidinyl]-l- azetidinesulfonamide
N- [2- [[(2,3-difluorophenyl)methyl] thio] -6- [2-hydroxy- 1 -(hydroxymethyl)- 1 -methylethoxy] -4- pyrimidinyl] - 1 -azetidinesulfonamide
N- [2- [[(2,3-difluorophenyl)methyl] thio] -6- [( l/?)-2-hydroxy- 1 -methylethoxy] -4-pyrimidinyl] -
2-thiazolesulfonamide
N- [2- [ [(2,3-difluorophenyl)methyl] thio] -6- [( l/?)-2-hydroxy- 1 -methylethoxy] -4-pyrimidinyl] -
4-pyridinesuhconamide N- [2- [[(2 ,3-difluorophenyl)methyl] thio] -6-[( l/?)-2-hydroxy- 1 -methylethoxy] -4-pyrimidinyl] - l-piperazinesulfonamide iV-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(l/?)-2-hydroxy-l- methylethoxy] -4-pyrimidinyl] - 1 ,6-dihydro- 1 -methyl-6-oxo-3-pyridinesulfonamide
N- [2- [ [(2,3-difluorophenyl)methy 1] thio] -6- [( l/?)-2-hydroxy- 1 -methylethoxy] -4-pyrimidinyl] - l-azetidinesulfonamide N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(li?)-2-hydroxy-l- methylethoxy] -4-pyrimidinyl] -methanesulfonamide
N- [2- [[(2,3-difluorophenyl)methyl] thio] -6-[( l/?)-2-hydroxy- 1 -methylethoxy] -4-pyrimidinyl] -
4-moφholinesulfonamide .
N- [2- [[(2,3-difluorophenyl)methyl] thio] -6- [( lJR)-2-hydroxy- 1 -methylethoxy] -4-pyrimidinyl] -
1 -pyrrolidinesulfonamide N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[( li?)-2-hydroxy- 1 -methylethoxy] -4-pyrimidinyl] - cyclopropanesulfonamide
N- [2- [ [(2,3-difluorophenyl)methyl] thio] -6-[( l/?)-2-hydroxy- 1 -methylethoxy] -4-pyrimidinyl] -
1 -methyl- lH-irmdazole-4-sulfonamide
N-[2-[[(2,3-Difluorophenyl)methyl]tWo]-6-methoxypyrimidin-4-yl]azetidine-l-sulfonamide N-[2-[[(2,3-Difluorophenyl)methyl]tMo]-6-methoxypyriπύdin-4-yl]piperazine-l-suhconamide
N-[2-[[(2,3-Difluorophenyl)methyl]tWo]-6-methoxypyrirnidin-4-yl]-l-methyl -lΗ-imidazole-
4-sunOnamide
N-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-{[(li?,2/?)-2,3-dihydroxy-l-methylproρyl]oxy}-4- pyrimidinyl]-l-azetidinesulfonamide
N-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-[[(l/?,2/?)-2,3-dihydroxy-l-methylpropyl]oxy]-4- pyrimidinyl]-methanesulfonamide N- [2- [ [(2,3-Difluoroρhenyl)methyl] thio] -6- { [( lΛ,2S)-2,3-dihydroxy- 1 -methylpropyl] oxy } -A- pyrimidiny 1] - 1 - azetidinesulfonamide
N-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-{[(l/?,2>S)-2,3-dihydroxy-l-methylpropyl]oxy}-4- pyrimidinyl] - 1 -piperazinesulfonamide
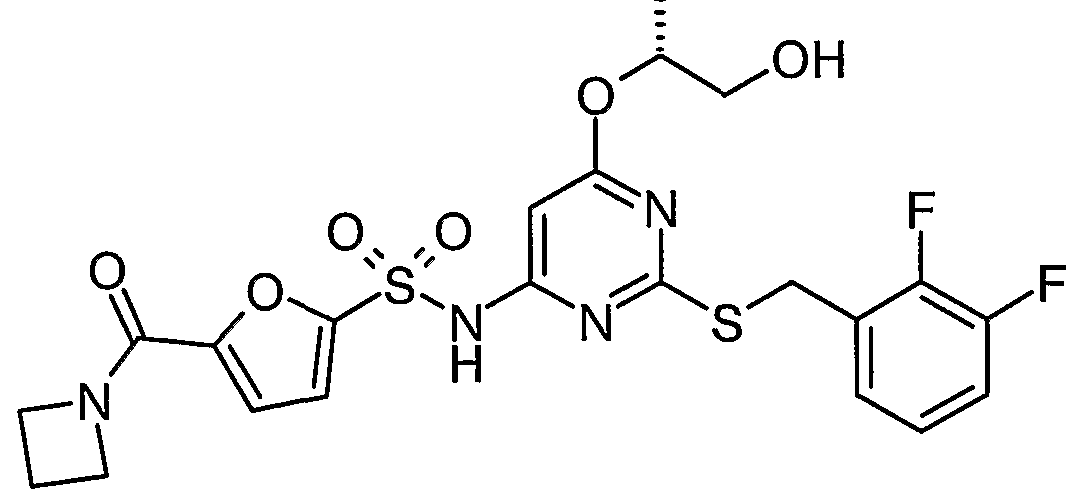
5-(azetidin-l-ylcarbonyl)-N-{2-[(2,3-difluorobenzyl)thio]-6-[(l/?)-2-hydroxy-l- methylethoxy]pyrirnidin-4-yl}furan-2-sulfonamide
Each of the above mentioned compounds and the pharmaceutically acceptable salts, solvates or in vivo hydrolysable esters thereof, taken individually is a particular aspect of the invention.
The present invention further provides a process for the preparation of compounds of formula (1) as defined above which comprises:
(a) treating a compound of formula (2a):
wherein R1, R2 and X are as defined in formula (1) and L is a leaving group such as halogen with sulfonamides (R3SO2NH2) where R3 is as defined in formula (1). and optionally thereafter (i), (ϋ), (in), (iv), or (v) in any order: i) removing any protecting groups; ii) converting the compound of formula (1) into a further compound of formula (1) iii) forming a salt iv) forming a prodrug v) forming an in vivo hydrolysable ester.
Reaction of compounds of formula (2a) wherein R1, R2 and X are as defined in formula (1) with sulfonamides (R3SO2NH2), where R3 is as defined in formula (1), can be
carried out in the presence of a suitable base, solvent and catalyst heated thermally or by microwaves. Examples of suitable bases include metal carbonates such as those from cesium, potassium, lithium or sodium. Most preferably Cesium carbonate is used. Suitable solvents include toluene and ethers such as anisole, tetrahydrofuran, 1,4-dioxane, glyme and diglyme. Preferably 1,4-dioxane is used. The temperature of the reaction can be performed between 1O0C and 12O0C, preferably at 1000C. Examples of suitable catalysts include a suitable palladium(O) source such as palladium tris(dibenzylideneacetone)dipalladium(0) (Pd2(dba)3 ), or tetrakistriphenylphosphinepalladium (Pd(PhJ)4) (either in 0.01-0.5 mol equivalents) in the presence of a suitable ligand such as (9,9-dimethyl-9H-xanthene-4,5-diyl)bis[diphenyl- phosphine (Xantphos), or 2-dicyclohexyl-phosphino-2'-(N,N-dimethylamino)biphenyl or 2- dicyclohexyl-phosphino-2',4',6'-tri-isopropyl,l,r-biphenyl (XPΗOS) (either in 0.01-0.5 mol equivalents). Preferably the catalyst combination is tris(dibenzylideneacetone)dipalladium(0) (Pd2(dba)3 ) with 2-dicyclohexyl-phosphino-2',4',6'-tri-isopropyl,l,l'-biphenyl (Xphos) in 0.01-0.5 mol equivalents in 1,4-dioxane at 1000C with cesium carbonate as the base; or (b) treating a compound of formula (2b):
(2b) wherein R1 and R3 are as defined in formula (1), L is a leaving group such as halogen, PG is a convenient protecting group or hydrogen and where X is oxygen or sulphur, with alcohols ΗOR2 or thiols ΗSR2 respectively wherein R2 is as defined in formula (1) in the presence of a suitable base and solvent, and optionally thereafter (i), (ii), (in), (iv), or (v) in any order: i) removing any protecting groups; ii) converting the compound of formula (1) into a further compound of formula (1) iii) forming a salt iv) forming a prodrug v) forming an in vivo hydrolysable ester.
Examples of suitable bases include the alkali metal hydrides such as Na or K, or metal alkoxides such as Li, Na or K-tert-butoxide, alkali metal hexamethyldisilazides such as Li, Na
or K-hexamethyldisilazide, or metal carbonates such as Na, K, Cs. Suitable solvents include iV.iV-dimethylamides, l-methyl-2-pyrolidinone, toluene and ethers such as anisole, tetrahydrofuran, 1,4-dioxane, glyme and diglyme.
Also, compounds of formula (1) wherein R'and R3 are as defined in formula (1), L is a leaving group such as halogen, PG is a convenient protecting group or hydrogen and X is - CH2- or a bond, can be prepared from compounds of formula (2b) wherein R2 is as defined in formula (1) by 'treatment with a suitably protected alkene under "Heck coupling" type reaction conditions (Synlett, 2003, no 8 ppl 133- 1136) or with a suitably protected boronic acid or ester under "Suzuki coupling" type reaction conditions (JACS, 1999, no 121, pp9550-9561, JACS 2001, no 123, ppl0099- 10100) in the presence of a suitable palladium catalyst, ligand, salt, base and solvent with thermal or microwave heating.
For 'Ηeck" type couplings, examples of suitable palladium catalysts, salts, bases and solvents include tris(dibenzylideneacetone)dipalladium(0) (Pd2(dba)3 ), or palladium di- acatate (Pd(OAc)2); added salts include potassium chloride, tetra-n-butylammonium chloride; and bases include tri-n-butylamine or di-isopropylethylamine; and solvents include N,N- dimethylformamide or N-methyl-pyrrolidin-2-one.
For "Suzuki" type couplings, examples of suitable palladium catalysts, ligands, salts, bases and solvents include palladium di-acetate; with ligands tri-cyclohexylphosphine, or 2,2'bis-dicyclohexyl-phosphino- 1 , 1 '-biphenyl or di-t-butyl-phosphino- 1 , 1 '-biphenyl or tri-t- butylphosphine; with salts potassium phosphate (K3PO4) or potassium fluoride in solvents tetrahydrofuran or 1,4-dioxane.
Compounds of formula (2a) wherein R1, and R2 are as defined in formula (1), and X is oxygen or sulphur can be prepared from compounds of formula (3) wherein R1 is as defined in formula (1) and L is a leaving group such as halogen by treatment with alcohols HOR2 or thiols HSR2 wherein R2 is as defined in formula (1) in the presence of a suitable base and solvent.
(3)
Examples of suitable bases include the alkali metal hydrides such as Na or K, or metal alkoxides such as Li, Na or K-tert-butoxide, alkali metal hexamethyldisilazides such as Li, Na or K-hexamethyldisilazide, or metal carbonates such as Na, K, Cs. Suitable solvents include ΛζiV-dimethylamides, l-methyl-2-pyrolidinone, ethers such as tetrahydrofuran, 1,4-dioxane, glyme and diglyme. Preferably sodium hydride in tetrahydrofuran at ambient to reflux temperature is employed.
Also, compounds of formula (2a) wherein R*and R2 are as defined in formula (1), and X is -CH2- or a bond can be prepared from compounds of formula (3) wherein R1 is as defined in formula (1) and L is a leaving group such as halogen, by treatment with a suitably protected alkene under "Heck coupling" type reaction conditions (Synlett, 2003, no 8 ppl 133- 1136) or with a suitably protected boronic acid or ester under "Suzuki coupling" type reaction conditions (JACS, 1999, no 121, pp9550-9561, JACS 2001, no 123, ppl0099- 10100) in the presence of a suitable palladium catalyst, ligand, salt, base and solvent with thermal or microwave heating. For 'Ηeck" type couplings, examples of suitable palladium catalysts, salts, bases and solvents include tris(dibenzylideneacetone)dipalladium(0) (Pd2(dba)3 ), or palladium di- acatate (Pd(OAc)2); added salts include potassium chloride, tetra-n-butylammonium chloride; and bases include tri-n-butylamine or di-isopropylethylamine; and solvents include N,N- dimethylformamide or N-methyl-pyrrolidin-2-one. Preferably palladium di-acetate, with salt tetra-n-butylammonium chloride, with base tri-n-butylamine in solvent NN,- dimemylformamide at 95°C is employed.
For "Suzuki" type couplings, Examples of suitable palladium catalysts, ligands, salts, bases and solvents include palladium di-acetate; with ligands tri-cyclohexylphosphine, or 2,2'bis-dicyclohexyl-phosphino-l,l'-biρhenyl or di-t-butyl-phosphino-l,l'-biphenyl or tri-t- butylphosphine; with salts potassium phosphate (K3PO4) or potassium fluoride in solvents tetrahydrofuran or 1,4-dioxane. Preferably palladium di-acetate with ligand 2,2'bis- dicyclohexyl-phosphino-l,l'-biρhenyl with salt potassium phosphate (K3PO4) in solvent tetrahydrofuran at reflux temperature is employed.
Compounds of formula (2b) wherein R1 and R3 are as defined in formula (1), L is a leaving group such as halogen and PG is a suitable protecting group or halogen may be prepared by reaction of compounds of formula (3), wherein R1 is as defined in formula (1) and L is a leaving group such as halogen with sulfonamides (R3SO2NHPG) where R3 is as
defined in formula (1) and PG is a suitable protecting group or hydrogen, in the presence of a suitable base, solvent and catalyst heated thermally or by microwaves, and optionally thereafter (i) or (ii) in any order: i) adding any protecting groups; ii) converting the compound of formula (2b) into a further compound of formula (2b)
Examples of suitable bases include the alkali metal hydrides such as Na or K, or metal alkoxides such as Li, Na or K-tert-butoxide, alkali metal hexamethyldisilazides such as Li, Na or K-hexamethyldisilazide, or metal carbonates such as Na, K, Cs. Suitable solvents include acetonitrile, tetrahydrofuran, 1,4-dioxane, glyme and diglyme. The temperature of the reaction can be performed between 10°C and 12O0C. Examples of suitable catalysts include a suitable palladium(0) source such as tetrakistriphenylphosphinepalladium (Pd(Pl^)4) or tris(dibenzylideneacetone)dipalladium(0) (Pd2(dba)3 ) in the presence of a suitable ligand such as (9,9-dimethyl-9H-xanthene-4,5-diyl)bis[diphenyl-phosphine (Xantphos), or 2- dicyclohexyl-phosphino-2'-(N,N-dimethylamino)biphenyl or 2-dicyclohexyl-phosphino- 2\4',6'-tri-isopropyU,l'-biphenyl (XPΗOS).
Compounds of formula (3) wherein R1 is as defined in formula (1) and L is halogen may be prepared from compounds of formula (3) wherein R1 is as defined in formula (1) and L is OH by reaction with a halogenating agent such as phosphorous oxychloride. The reaction may be carried out in the presence of N,N-dimethylaniline at reflux. Compounds of formula (3) wherein R1 is as defined in formula (1) and L is OH;
(4) may be prepared from compounds of formula (4) wherein L is OH by reaction with alkylhalides (R1A) where R1 is as defined in formula (1) and A is halogen in the presence of a suitable base and solvent.
Examples of suitable bases include the alkali metal hydroxides such as Li, Na, or K, or metal carbonates such as Li, Na, K or Cs, or metal acetates such as Li, Na, K or Cs, or metal alkoxides such as Li, Na, K tert-butoxide. Suitable solvents include water, N,N- dirnethylamides, l-methyl-2-pyrolidinone, ethers such as tetrahydrofuran, 1,4-dioxane, glyme
-23-
The present invention also provides a pharmaceutical composition comprising a compound of formula (1), or a pharmaceutically acceptable salt, solvate or in vivo hydro lysable ester thereof, as hereinbefore defined, in association with a pharmaceutically acceptable adjuvant, diluent or carrier. The invention further provides a process for the preparation of a pharmaceutical composition of the invention which comprises mixing a compound of formula (1), or a pharmaceutically acceptable salt, solvate or in vivo hydrolysable ester thereof, as hereinbefore defined, with a pharmaceutically acceptable adjuvant, diluent or carrier. The pharmaceutical compositions may be administered topically (e.g. to the lung and/or airways or to the skin) in the form of solutions, suspensions, heptafluoroalkane aerosols and dry powder formulations; or systemically, e.g. by oral administration in the form of tablets, capsules, syrups, powders or granules, or by parenteral administration in the form of solutions or suspensions, or by subcutaneous administration or by rectal administration in the form of suppositories or transdermally. Preferably the compounds of the invention are administered orally. In addition to their use as therapeutic medicines, the compounds of formula (1) and their pharmaceutically acceptable salts, solvate or in vivo hydrolysable esters are also useful as pharmacological tools in the development and standardisation of in vitro and in vivo test systems for the evaluation of the effect of chemokine modulation activity in labatory animals such as cats, dogs, rabbits, monkeys, rats and mice, as part of the search for new therapeutic agents.
The invention further relates to combination therapies wherein a compound of formula (1) or a pharmaceutically acceptable salts, solvate or in vivo hydrolysable ester thereof, or a pharmaceutical composition or formulation comprising a compound of formula (1) is administered concurrently or sequentially with therapy and/or an agent for the treatment of any one of asthma, allergic rhinitis, cancer, COPD, rheumatoid arthritis, psoriasis, inflammatory bowel disease, irritable bowel syndrome, osteoarthritis or osteoporosis. hi particular, for the treatment of the inflammatory diseases rheumatoid arthritis, psoriasis, inflammatory bowel disease, irritable bowel syndrome, COPD, asthma and allergic rhinitis the compounds of the invention may be combined with agents such as TNF-α inhibitors such as anti-TNF monoclonal antibodies (such as Remicade, CDP-870 and D.sub2.E.sub7.) and TNF receptor immunoglobulin molecules (such as EnbreLreg.), non¬ selective COX-I / COX-2 inhibitors (such as piroxicam, diclofenac, propionic acids such as
-24- naproxen, flubiprofen, fenoprofen, ketoprofen and ibuprofen, fenamates such as mefenamic acid, indomethacin, sulindac, apazone, pyrazolones such as phenylbutazone, salicylates such as aspirin), COX-2 inhibitors (such as meloxicam, celecoxib, rofecoxib, valdecoxib and etoricoxib) low dose methotrexate, lefunomide; ciclesonide; hydroxychloroquine, d- 5 penicillamine, auranofin or parenteral or oral gold. For inflammatory bowel disease and irritable bowel disorder further convenient agents include sulphasalazine and 5-ASAs, topical and systemic steroids, immunomodulators and immunosuppressants, antibiotics, probiotics and anti-integrins.
The present invention still further relates to the combination of a compound of the 10 invention together with a leukotriene biosynthesis inhibitor, 5-lipoxygenase (5-LO) inhibitor or 5-lipoxygenase activating protein (FLAP) antagonist such as zileuton; ABT-761; fenleuton; tepoxalin; Abbott-79175; Abbott-85761; N-(5-substituted)-thiophene-2-alkylsulfonamides; 2,6-di-tert-butylphenol hydrazones; methoxytetrahydropyrans such as Zeneca ZD-2138; the compound SB-210661; pyridinyl-substituted 2-cyanonaphthalene compounds such as L- 15 739,010; 2-cyanoquinoline compounds such as L-746,530; indole and quinoline compounds such as MK-591, MK-886, and BAY x 1005.
The present invention still further relates to the combination of a compound of the invention together with a receptor antagonist for leukotrienes LTB.sub4., LTC.suM., LTD.suM., and LTE.suM. selected from the group consisting of the phenothiazin-3-ones 20 such as L-651,392; amidino compounds such as CGS-25019c; benzoxalamines such as ontazolast; benzenecarboximidamides such as BIIL 284/260; and compounds such as zafirlukast, ablukast, montelukast, pranlukast, verlukast (MK-679), RG- 12525, Ro-245913, iralukast (CGP 45715A), and BAY x 7195.
The present invention still further relates to the combination of a compound of the 25 invention together with a PDE4 inhibitor including inhibitors of the isoform PDE4D.
The present invention still further relates to the combination of a compound of the invention together with a antihistaminic H.subl. receptor antagonists such as cetirizine, loratadine, desloratadine, fexofenadine, astemizole, azelastine, and chlorpheniramine.
The present invention still further relates to the combination of a compound of the 30 invention together with a gastroprotective H.sub2. receptor antagonist.
The present invention still further relates to the combination of a compound of the invention together with an α.subl.- and α.sub2. -adrenoceptor agonist vasoconstrictor
-25- sympathomimetic agent, such as propylhexedrine, phenylephrine, phenylpropanolamine, pseudoephedrine, naphazoline hydrochloride, oxymetazoline hydrochloride, tetrahydrozoline hydrochloride, xylometazoline hydrochloride, and ethylnorepinephrine hydrochloride.
The present invention still further relates to the combination of a compound of the invention together with anticholinergic agents such as ipratropium bromide; tiotropium bromide; oxitropium bromide; pirenzepine; and telenzepine.
The present invention still further relates to the combination of a compound of the invention together with a β.subl.- to β.sub4. -adrenoceptor agonists such as metaproterenol, isoproterenol, isoprenaline, albuterol, salbutamol, formoterol, salmeterol, terbutaline, orciprenaline, bitolterol mesylate, and pirbuterol; or methylxanthanines including theophylline and aminophylline; sodium cromoglycate; or muscarinic receptor (Ml, M2, and M3) antagonist.
The present invention still further relates to the combination of a compound of the invention together with an insulin- like growth factor type I (IGF-I) mimetic. The present invention still further relates to the combination of a compound of the invention together with an inhaled glucocorticoid with reduced systemic side effects, such as prednisone, prednisolone, flunisolide, triamcinolone acetonide, beclomethasone dipropionate, budesonide, fluticasone propionate, and mometasone furoate.
The present invention still further relates to the combination of a compound of the invention together with an inhibitor of matrix metalloproteases (MMPs), Le., the stromelysins, the collagenases, and the gelatinases, as well as aggrecanase; especially collagenase- 1 (MMP- 1), collagenase-2 (MMP-8), collagenase-3 (MMP-13), stromelysin-1 (MMP-3), stromelysin-2 (MMP-10), and stromelysin-3 (MMP-11) and MMP- 12.
The present invention still further relates to the combination of a compound of the invention together with other modulators of chemokine receptor function such as CCRl,
CCR2, CCR2A, CCR2B, CCR3, CCR4, CCR5, CCR6, CCR7, CCR8, CCR9, CCRlO and
CCRIl (for the C-C family); CXCRl, CXCR3, CXCR4 and CXCR5 (for the C-X-C family) and CX3CRl for the C-X3-C family.
The present invention still further relates to the combination of a compound of the invention together with antiviral agents such as Viracept, AZT, aciclovir and famciclovir, and antisepsis compounds such as Valant.
-26-
The present invention still further relates to the combination of a compound of the invention together with cardiovascular agents such as calcium channel blockers, lipid lowering agents such as statins, fibrates, beta-blockers, Ace inhibitors, Angiotensin-2 receptor antagonists and platelet aggregation inhibitors. The present invention still further relates to the combination of a compound of the invention together with CNS agents such as antidepressants (such as sertraline), anti¬ parkinsonian drugs (such as deprenyl, L-dopa, Requip, Mirapex, MAOB inhibitors such as selegine and rasagiline, comP inhibitors such as Tasmar, A-2 inhibitors, dopamine reuptake inhibitors, NMDA antagonists, Nicotine agonists, Dopamine agonists and inhibitors of neuronal nitric oxide synthase), and anti- Alzheimer's drugs such as donepezil, tacrine, COX-2 inhibitors, propentofylline or metryfonate.
The present invention still further relates to the combination of a compound of the invention together with (i) tryptase inhibitors; (ii) platelet activating factor (PAF) antagonists; (iii) interleukin converting enzyme (ICE) inhibitors; (iv) IMPDH inhibitors; (v) adhesion molecule inhibitors including VLA-4 antagonists; (vi) cathepsins; (vii) MAP kinase inhibitors; (vϋi) glucose-6 phosphate dehydrogenase inhibitors; (ix) kinin-B.subl. - and B.sub2. -receptor antagonists; (x) anti-gout agents, e.g., colchicine; (xi) xanthine oxidase inhibitors, e.g., allopurinol; (xii) uricosuric agents, e.g., probenecid, sulfinpyrazone, and benzbromarone; (xiii) growth hormone secretagogues; (xiv) transforming growth factor (TGFβ); (xv) platelet-derived growth factor (PDGF); (xvi) fibroblast growth factor, e.g., basic fibroblast growth factor (bFGF); (xvii) granulocyte macrophage colony stimulating factor (GM-CSF); (xviii) capsaicin cream; (xix) Tachykinin NK.subl. and NK.sub3. receptor antagonists selected from the group consisting of NKP-608C; SB-233412 (talnetant); and D- 4418; (xx) elastase inhibitors selected from the group consisting of UT-77 and ZD-0892; (xxi) TNFD converting enzyme inhibitors (TACE); (xxii) induced nitric oxide synthase inhibitors (iNOS) or (xxiii) chemoattractant receptor-homologous molecule expressed on TH2 cells, (CRTH2 antagonists).
The compounds of the present invention may also be used in combination with osteoporosis agents such as raloxifene, droloxifene, lasofoxifene or fosomax and immunosuppressant agents such as FK-506, rapamycin, cyclosporine, azathioprine, and methotrexate;.
-27-
The compounds of the invention may also be used in combination with existing therapeutic agents for the treatment of osteoarthritis. Suitable agents to be used in combination include standard non-steroidal anti- inflammatory agents (hereinafter NSAID's) such as piroxicam, diclofenac, propionic acids such as naproxen, flubiprofen, fenoprofen, ketoprofen and ibuprofen, fenamates such as mefenamic acid, indomethacin, sulindac, apazone, pyrazolones such as phenylbutazone, salicylates such as aspirin, COX-2 inhibitors such as celecoxib, valdecoxib, rofecoxib and etoricoxib, analgesics and intraarticular therapies such as corticosteroids and hyaluronic acids such as hyalgan and synvisc and P2X7 receptor antagonists. The compounds of the invention can also be used in combination with existing therapeutic agents for the treatment of cancer. Suitable agents to be used in combination include:
(i) antiproliferative/antineoplastic drugs and combinations thereof, as used in medical oncology, such as alkylating agents (for example cis-platin, carboplatin, cyclophosphamide, nitrogen mustard, melphalan, chlorambucil, busulphan and nitrosoureas); antimetabolites (for example antifolates such as fluoropyrimidines like 5-fluorouracil and tegafur, raltitrexed, methotrexate, cytosine arabinoside, hydroxyurea, gemcitabine and paclitaxel (Taxol®); antitumour antibiotics (for example anthracyclines like adriamycin, bleomycin, doxorubicin, daunomycin, epirubicin, idarubicin, mitomycin-C, dactinomycin and mithramycin); antimitotic agents (for example vinca alkaloids like vincristine, vinblastine, vindesine and vinorelbine and taxoids like taxol and taxotere); and topoisomerase inhibitors (for example epipodophyllo toxins like etoposide and teniposide, amsacrine, topotecan and camptothecin); (ii) cytostatic agents such as antioestrogens (for example tamoxifen, toremifene, raloxifene, droloxifene and iodoxyfene), oestrogen receptor down regulators (for example fulvestrant), antiandrogens (for example bicalutamide, flutamide, nilutamide and cyproterone acetate), LHRH antagonists or LHRH agonists (for example goserelin, leuprorelin and busereUn), progestogens (for example megestrol acetate), aromatase inhibitors (for example as anastrozole, letrozole, vorazole and exemestane) and inhibitors of 5α-reductase such as finasteride; (iϋ) Agents which inhibit cancer cell invasion (for example metalloproteinase inhibitors like marimastat and inhibitors of urokinase plasminogen activator receptor function);
-28-
(iv) inhibitors of growth factor function, for example such inhibitors include growth factor antibodies, growth factor receptor antibodies (for example the anti-erbb2 antibody trastuzumab [Herceptin™] and the anti-erbbl antibody cetuximab [C225]) , farnesyl transferase inhibitors, tyrosine kinase inhibitors and serine/threonine kinase inhibitors, for example inhibitors of the epidermal growth factor family (for example EGFR family tyrosine kinase inhibitors such as N-(3-chloro-4-fluorophenyl)-7-methoxy-6-(3- morphohnopropoxy)quinazolin-4-amine (gefitinib, AZD 1839), N-(3-ethynylphenyl)-6,7- bis(2-methoxyethoxy)quinazolin-4-amine (erlotinib, OSI-774) and 6-acrylamido-N-(3-chloro- 4-fluorophenyl)-7-(3-morphotoopropoxy)quinazolin-4-amine (CI 1033)), for example inhibitors of the platelet-derived growth factor family and for example inhibitors of the hepatocyte growth factor family;
(v) antiangiogenic agents such as those which inhibit the effects of vascular endothelial growth factor, (for example the anti- vascular endothelial cell growth factor antibody bevacizumab [Avastin™], compounds such as those disclosed in International Patent Applications WO 97/22596, WO 97/30035, WO 97/32856 and WO 98/13354) and compounds that work by other mechanisms (for example linomide, inhibitors of integrin αvβ3 function and angio statin);
(vi) vascular damaging agents such as Combretastatin A4 and compounds disclosed in International Patent Applications WO 99/02166, WO00/40529, WO 00/41669, WO01/92224, WO02/04434 and WO02/08213;
(vii) antisense therapies, for example those which are directed to the targets listed above, such as ISIS 2503, an anti-ras antisense;
(viii) gene therapy approaches, including for example approaches to replace aberrant genes such as aberrant ρ53 or aberrant BRCAl or BRCA2, GDEPT (gene-directed enzyme pro-drug therapy) approaches such as those using cytosine deaminase, thymidine kinase or a bacterial nitroreductase enzyme and approaches to increase patient tolerance to chemotherapy or radiotherapy such as multi-drug resistance gene therapy; and
(ix) immunotherapy approaches, including for example ex-vivo and in- vivo approaches to increase the immunogenicity of patient tumour cells, such as transfection with cytokines such as interleukin 2, interleukin 4 or granulocyte-macrophage colony stimulating factor, approaches to decrease T-cell anergy, approaches using transfected immune cells such as
-29- cytokine-transfected dendritic cells, approaches using cytokine-transfected tumour cell lines and approaches using anti-idiotypic antibodies.
-30- Pharmacological Data
Lijgand Binding Assay
[ I] EL- 8 (human, recombinant) was purchased from Amersham, U.K. with a specific activity of 2,000Ci/mmol. All other chemicals were of analytical grade. High levels of hrCXCR2 were expressed in HEK 293 cells (human embryo kidney 293 cells ECACC No. 85120602) (Lee et al. [YWl) J. Biol. Chem. 267, ppl 6283- 16291). hrCXCR2 cDNA was amplified and cloned from human neutrophil mRNA. The DNA was cloned into PCRScript (Stratagene) and clones were identified using DNA. The coding sequence was sub-cloned into the eukaryotic expression vector RcCMV (Invitrogen). Plasmid DNA was prepared using Quiagen Megaprep 2500 and transfected into HEK 293 cells using Lipofectamine reagent (Gibco BRL). Cells of the highest expressing clone were harvested in phosphate-buffered saline containing 0.2%(w/v) ethylenediaminetetraacetic acid (EDTA) and centrifuged (20Og, 5min.). The cell pellet was resuspended in ice cold homogenisation buffer [1OmM HEPES (pH 7.4), ImM dithiothreitol, ImM EDTA and a panel of protease inhibitors (ImM phenyl methyl sulphonyl fluoride, 2μg/ml soybean trypsin inhibitor, 3mM benzamidine, 0.5μg/ml leupeptin and lOOμg/ml bacitracin)] and the cells left to swell for 10 minutes. The cell preparation was disrupted using a hand held glass mortar/PTFE pestle homogeniser and cell membranes harvested by centrifugation (45 minutes, 100,000g, 4°C). The membrane preparation was stored at -7O0C in homogenisation buffer supplemented with Tyrode's salt solution (137mM NaCl, 2.7mM KCl, 0.4mM NaH2PO4), 0.1%(w/v) gelatin and 10%(v/v) glycerol.
All assays were performed in a 96- well MultiScreen 0.45μm filtration plates (Millipore, U.K.). Each assay contained -5OpM [125I]DL-8 and membranes (equivalent to -200,000 cells) in assay buffer [Tyrode's salt solution supplemented with 1OmM HEPES (pH 7.4), 1.8mM CaCl2, ImM MgCl2, 0.125mg/ml bacitracin and 0.1%(w/v) gelatin]. In addition, a compound of formula (I) according to the Examples was pre-dissolved in DMSO and added to reach a final concentration of l%(v/v) DMSO. The assay was initiated with the addition of membranes and after 1.5 hours at room temperature the membranes were harvested by filtration using a Millipore MultiScreen vacuum manifold and washed twice with assay buffer (without bacitracin). The backing plate was removed from the MultiScreen plate assembly, the filters dried at room temperature, punched out and then counted on a Cobra γ-counter.
The compounds of formula (I) according to the Examples 1 - 156 were found to have ρIC50 values of greater than (>) 5.0.
-31-
Intracellular Calcium Mobilisation Assay
Human neutrophils were prepared from EDTA-treated peripheral blood, as previously described (BaIy et al. (1997) Methods in Enzymology 287 pp70-72), in storage buffer [Tyrode's salt solution (137mM NaCl, 2.7mM KCl, 0.4mM NaH2PO4) supplemented with 5.7mM glucose and 1OmM HEPES (pH 7.4)].
The chemokine GROα (human, recombinant) was purchased from R&D Systems (Abingdon, U.K.). All other chemicals were of analytical grade. Changes in intracellular free calcium were measured fluorometrically by loading neutrophils with the calcium sensitive fluorescent dye, fluo-3, as described previously (Merritt et al. (1990) Biochem. J. 269, pp513- 519). Cells were loaded for 1 hour at 37°C in loading buffer (storage buffer with 0. l%(w/v) gelatin) containing 5μM fluo-3 AM ester, washed with loading buffer and then resuspended in Tyrode's salt solution supplemented with 5.7mM glucose, 0. l%(w/v) bovine serum albumin (BSA), 1.8mM CaCl2 and ImM MgCl2. The cells were pipetted into black walled, clear bottom, 96 well micro plates (Costar, Boston, U.S.A.) and centrifuged (20Og, 5 minutes, room temperature).
A compound of formula (I) according to the Examples was pre-dissolved in DMSO and added to a final concentration of 0. l%(v/v) DMSO. Assays were initiated by the addition of an A50 concentration of GROD and the transient increase in fluo-3 fluorescence (ΛEX =490nm and λEm = 520nm) monitored using a FLIPR (Fluorometric Imaging Plate Reader, Molecular Devices, Sunnyvale, U.S.A.).
The compounds of formula (I) according to the Examples were tested and found to be antagonists of the CXCR2 receptor in human neutrophils.
The invention will now be illustrated by the following non-limiting Examples in which, unless stated otherwise:
(i) when given Nuclear Magnetic Resonance (NMR) spectra were measured on a Varian Unity Inova 300 or 400 MHz spectrometer. 1H NMR data is quoted in the form of delta values for major diagnostic protons, given in parts per million (ppm) relative to tetramethylsilane (TMS) as an internal standard. (ϋ) Mass Spectrometry (MS) spectra were measured on a Finnigan Mat SSQ7000 or Micromass Platform spectrometer.
-32-
(iii) the title and sub-titled compounds of the Examples and methods were named using the ACD/Name program (version 4.55) from Advanced Chemical Development
Inc, Canada.
(iv) Normal phase column chromatography and normal phase HPLC was conducted using a silica column. Reverse phase High Pressure Liquid Chromatography
(HPLC) purification was performed using either a Waters Micromass LCZ with a
Waters 600 pump controller, Waters 2487 detector and Gilson FC024 fraction collector or a Waters Delta Prep 4000 or a Gilson Auto Purification System, using a Symmetry, NovaPak or Ex- Terra reverse phase silica column. (v) The following abbreviations are used:
AcOH acetic acid
CHCl3 chloroform
DCM dichloromethane
DMF N,ΛT-dimethylformarnide DMSO dimethy Sulfoxide
Et2O diethyl ether
EtOAc ethyl acetate
MgSO4 magnesium sulfate
NMP l-methylpyrrolidin-2-one THF tetrahydrofuran
H2O water
NH3 ammonia
-33-
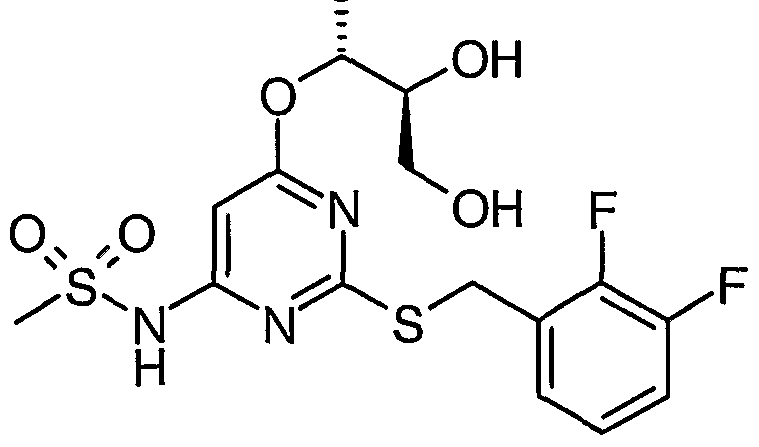
Example 1 yV-[2-[[(2,3-difluorophenyI)methyI]thio]-6-[2-hydroxy-l-(hydroxymethyl)ethoxy]-4- pyrimidinyl]-l-azetidinesulfonamide
To a suspension of N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(2-phenyl-l,3-dioxan-5- yl)oxy]-4-pyrimidinyl]-l-azetidinesulfonamide (the product of step iv) (220 mg) in methanol (5 ml)/ water (0.1 ml) was added pyridinium p-toluenesulfonate (20 mg) and the mixture was stirred at ambient temperature for 1.5 hour, then at reflux for 20 hour. The reaction mixture was evaporated, suspended in water and extracted with ethyl acetate (x2). The combined organic layers were dried with magnesium sulfate, filtered and evaporated. The residue was purified by column chromatography on silica gel using a 98:2 mixture of methylene chloride and methanol as eluent to give the title compound as a white solid. Yield: 120mg MS: APCI(+ve) 463, [M+H+] 1U NMR: (DMSO) δ 2.13 (quintet, 2H), 3.57 (m, 4H), 3.89 (t, 4), 4.44 (s, 2H), 4.78 (t, 2H), 5.13 (quintet, IH), 6.15 (s, IH), 7.17 (dq, IH), 7.36 (dq, IH), 7.45 (dt, IH), 11.11 (bs, IH);
The intermediates for this compound were prepared as follows: i) 2-[(2,3-Difluorobenzyl)thio]pyrimidine-4,6-diol To a slurry of 2-mercaptopyrimidine-4,6-diol (55.6g) in water (735ml) was added sodium acetate (47.4g) with stirring forming a complete solution over 20 minutes. A solution of 2,3- difluorobenzyl bromide (80g) in acetonitrile (73.5ml) was then added dropwise over 15 minutes and the resulting mixture heated at 4O0C with stirring for 18h. After cooling to ambient temperature the resulting precipitate was then filtered and washed with H2O (IL) before drying in vacuo at 1000C to afford the subtitle compound as a cream solid. Yield: 101.5g.
-34-
1H NMR: δ (DMSO) 7.74 (IH, s), 7.39 - 7.32 (2H, m), 7.21 - 7.15 (IH, m), 4.48 (2H, s).
ii) 4,6-Dichloro-2-[(2,3-difluorobenzyl)thio]pyrimidine
To a mixture of the the subtitle product of step i) (101.5g) with benzyltriethylammonium chloride (8.6g) in 1,2-dimethoxyethane (550ml) was added phosphorus oxychloride (70ml) and the mixture heated at 850C for 5h. The reaction was allowed to cool and solvents and excess phosphorus oxychloride were removed in vacuo before partitioning between ethyl acetate and ice water. The layers were separated and the dried (MgSO4) organics concentrated in vacuo to afford the crude product as a pale brown oil which solidified on standing. The crude product was purified by column chromatography (4% EtOAc / iso- hexane) to yield the subtitle compound as a white solid. Yield: 9Og. 1H NMR: 5 (DMSO) 7.74 (IH, s), 7.39-7.32 (2H, m), 7.21-7.15 (IH, m) 4.48 (2H, s)
iii) 4-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-[(2-phenyl-l,3-dioxan-5-yl)oxy]- pyrimidine
To a solution of 2-phenyl-l,3-dioxan-5-ol (484mg) in anhydrous tetrahydrofuran (10ml) at O0C was added 60% sodium hydride (1 lOmg) and the mixture was heated to reflux for 25 minutes. On allowing to cool to ambient temperature 4,6-Dichloro-2-[(2,3- difluorobenzyl)thio]pyrimidine (the product of step (ii) (75 mg) was added and the reaction was heated to reflux for a further 90 minutes. The reaction mixture was allowed to cool, diluted with water and extracted with ethyl acetate (x3). The combined organic layers were dried with magnesium sulfate, filtered and evaporated. The residue was purified by column chromatography on silica using a 95:5 to 90: 10 mixture of iso-hexane and ethyl acetate as eluent to give the sub-title compound as a white solid. Yield: 350mg MS: APCI(+ve) 451 [M+H+]
iv) Λr-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(2-phenyl-l,3-dioxan-5-yl)oxy]-4- py rimidinyl] - 1 -azetidinesulfonamide
A mixture of azetidine-1-sulphonamide (420mg), tris(dibenzylideneacetone)dipalladium (0) (71 mg), 2-dicyclohexylphosphino-2',4',6'-tri- isopropyl-l,l'-biphenyl (XPHOS) (37mg), cesium carbonate (380mg) and 4-chloro-2-[[(2,3- difluorophenyl)methyl]thio]-6-[(2-phenyl- 1 ,3-dioxan-5-yl)oxy]-ρyrimidine (350mg) in
-35- anhydrous dioxane (8ml) was heated to reflux in a microwave at 1000C, 300W, open vessel with cooling for 10 minutes. The reaction mixture was diluted with methylene chloride, filtered through arbocel and the filtrate evaporated. The residue was purified by column chromatography on silica using a 80:20 to 70:30 mixture of iso-hexane and ethyl acetate as eluent to give the sub-title compound as a white solid. Yield: 220mg. MS: APCI(+ve) 551 [M+H+]
Example 2 ^,5> N-[2-[[(2,3-di£luorophenyl)methyl]thio]-6-[3,4-dihydroxybutyl]pyrlmidin-4- yl]azetidine- 1 -sulphonamide
A solution of N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[2-(2,2-dimethyl[l,3]dioxolan-4yl)- ethyl]-pyrimidin-4-yl]azetidine-l-sulphonamide (the product of step iii) (43mg) and pyridinium/jαra-toluenesulphonate (43mg) in methanol (ImI) and one drop of water was heated at 60°C for 1.5h. The solution was cooled and the solvent evaporated under reduced pressure. The residue was dissolved in dichloromethane and washed with water, dried (MgSO4) and the solvent evaporated under reduced pressure. The residual yellow solid was purified by preparative plate chromatography eluting with ethyl acetate. The isolated product was dissolved in dichloromethane and the solvent evaporated at room temperature under reduced pressure to give the title product as a white solid. Yield 20mg. MS: APCI(-ve) 459 [M-I] 1H NMR: 5 (DMSO) 11.18 (s,lH), 7.44 (t,lH), 7.33 (q,lH), 7.14 (m,lH), 6.66 (s,lH),
4.57(d,lH), 4.51 (t,lH), 4.45 (s,2H), 3.93 (t,4H), 3.41 (m,lH), 3.26 (m,lH), 2.71 (m,lH), 2.65 (m,lH), 2.12 (p,2H), 1.82 (m,lH), 1.53 (mJH).
-36-
The intermediates for this compound were prepared as follows:
i) (ds/trans) 4-Chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-[2-(2,2- dimethyl[l,3]dioxolan-4-yl)- vinyl] -pyrimidine A mixture of 4,6-dichloro-2-[(2,3-difluorobenzyl)thio]pyrimidine (product of example 1 step ii) (0.5g), tris(dibenzylideneacetone)dipalladium(0) (45mg), 2,2-dimethy 1-4- vinyl- 1,3- dioxolane(630mg), tri-n-butylamine (610mg) and tetra-n-butylammonium chloride (460mg) in anhydrous N,N-dimethylformamide (6.5ml) were heated at 900C for 3h. then stirred at room temperature overnight. The reaction mixture was partitioned between ethyl acetate and water. The organic phase was washed with water and brine, dried (MgSO4) and the solvent evaporated under reduced pressure. The residue was purified by flash silica-gel chromatography eluting with 10% diethyl ether in iso-hexane to give the sub-title compound as a yellow viscous oil. Yield: 98mg. MS: APCI (+ve) 399 [M+ 1]
ii) fR,S/) 4-Chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-[2-(2,2-dimethyl[l,3]dioxolaii- 4-yl)ethyl]- pyrimidine
A solution of the product of step i) (96.8mg) in ethanol (10ml) was hydrogenated over platinum oxide (5mg) at 3 atmospheres over 2 days. Further platinum oxide (20mg) was added and the mixture was hydrogenated for further 3 days at 5 atmospheres. The catalyst was filtered (Celite) and the filtrate evaporated under reduced pressure. The residue was purified by flash silica-gel chromatography eluting with 10% diethyl ether in /sO-hexane to give the sub-title compound as a viscous oil. Yield: 33mg. MS: APCI (+ve) 401 [M+l]
m) (R,S) N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[2-(2,2-dimethyl[l,3]dioxolan-4yl)- ethyl]-pyrimidin-4-yl]azetidine-l-sulphonamide
A solution of the product of step ii) (47mg), tris(dibenzylideneacetone)dipalladium(0) (6mg), azetidine-1-sulphonamide (62mg), 2-dicyclohexyl-phosphino-2',4',6'-tri-isopropyl,l,r- biphenyl (XPHOS) (6mg) and cesium carbonate (52mg) in anhydrous dioxane (ImI) was heated at 1000C for 45min. The reaction mixture was partitioned between ethyl acetate and
-37- water. Acetic acid (0.2ml) was added and the separated organic phase was washed with water and brine, dried (MgSO4) and the solvent evaporated under reduced pressure. The residue was purified by flash silica-gel chromatography eluting with 40% ethyl acetate in «σ-hexane to give the sub-title compound as a yellow viscous oil. Yield: 46mg. MS: APCI (+ve) 501 [M+l]
Example 3
N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[3-hydroxy-2-(hydroxymethyl)propyl] pyriniidin-4-yl]azetidiπe-l-suIphonamide
A solution of N-[2-[[(2,3-difluoroρhenyl)methyl]thio]-6-[(2,2-dimethyl-l,3-dioxan-5- yl)methyl]-4-pyrirnidπiyl]azetidine-l-sulphonarnide (the product of step ii) (78mg) and pyridirmim/?αrα-toluenesulphonate (79mg) in methanol (1.8ml) and one drop of water was heated at 6O0C for 15min. The solution was cooled and the solvent evaporated under reduced pressure. The residue was dissolved in dichloromethane and washed with 2N hydrochloric acid and water, dried (MgSO4) and the solvent evaporated under reduced pressure to give a viscous yellow oil (17mg). The aqueous washings were combined, the pH adjusted to 5 with aqueous sodium bicarbonate and then extracted with ethyl acetate. The organic solution was dried (MgSO4) and the solvent evaporated under reduced pressure. The residual viscous oil was dissolved in dichloromethane and the solvent evaporated at room temperature under reduced pressure to give the title product as a white solid. Yield 62mg. MS: APCI (-ve) 459 [M-I] 1H NMR: δ (DMSO) 11.17 (s,lH), 7.44 (t,lH), 7.33 (m,lH), 7.14 (m,lH), 6.65 (s,lH), 4.45 (s,4H), 3.92 (t,4H), 3.38 (m,4H), 2.57 (d,2H), 2.12 (p,2H), 1.98 (m,lH)
-38-
The intermediates for this compound were prepared as follows:
i) 4-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-[(2,2-dimethyl-l,3-dioxan-5- yl)methyl]pyrimidine
A solution of 0.5M 9-borabicyclo[3.3.1]nonane (9-BBN) in tetrahydrofuran (17.12ml) and 2,2-dimethyl-5-methylene-l,3-dioxane (Tet. Lett. (1988) 29 (45) 5703 - 5706) (1.3g) was heated at 45°C for 18h. The solution was cooled and added to mixture of palladium(II) acetate, potassium phosphate (1.16g), (biphenyl-2-yl)dicyclohexyl- phosphine (0.14g) and 4,6-dichloro-2-[(2,3-difluorobenzyl)thio]pyrimidine (1.5g) stirred under nitrogen. The mixture was heated in a microwave at 70°C, 250W for a total of 1.5h, then 7O0C on a hot-plate for 2 days. The reaction mixture was adsorbed onto silica-gel, the solvent evaporated under reduced pressure and the residue purified by flash silica-gel chromatography eluting with 20% ethyl acetate in iso-htxane to give a yellow oil. The oil was further purified by flash silica-gel chromatography eluting with dichloromethane to give the sub- title product as a viscous oil. Yield: llOmg. MS: APCI (-ve) 399 [M-I]
ii) iV-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(2,2-dimethyl-l,3-dioxan-5-yl)methyl]-4- pyrimidinyl]azetidine-l-sulphonamide
A solution of the product of step i) (109mg), tris(dibenzylideneacetone)dipalladium(0) (14mg), azetidine-1-sulphonamide (145mg), 2-dicyclohexyl-phosphino-2',4',6'-tri- isopropyl,l,r-biphenyl (XPHOS)(14mg) and cesium carbonate (120mg) in anhydrous dioxane (2.3ml) was heated at 1000C for 45min. The reaction mixture was partitioned between ethyl acetate and water. Acetic acid (0.2ml) was added and the separated organic phase was washed with water and brine, dried (MgSO4) and the solvent evaporated under reduced pressure. The residue was purified by flash silica-gel chromatography eluting with 40% ethyl acetate in isø-hexane to give the sub-title compound as a yellow viscous oil. Yield: 78mg. MS: APCI (-ve) 499 [M-I]
-39-
Example 4
N-(2-[(2,3-difluorobenzyl)thio]-6-{[(lR,2R)-2-hydroxy-l-methylpropyl]oxy}pyriiiiidin-4- y 1) azeti dine- 1 -sulfonamide
The title compound was prepared according to the procedure outlined in example 1 step (iv) using a mixture of azetidine-1 -sulfonamide (150mg), tris(dibenzylideneacetone)dipalladium
(0) (25 mg), 2-dicyclohexylρhosphino-2',4',6'-tri-isoρroρyl-l,l'-biρhenyl (XPHOS) (25mg), cesium carbonate (244mg) and (2i?,3i?)-3-({6-chloro-2-[(2,3-difluorobenzyl)thio]pyrimidin-4- yl}oxy)butan-2-ol (200mg) in anhydrous dioxane (10ml). Purification was by reverse phase
HPLC eluting with acetonitrile / aq. 0.1% ammonium acetate mixtures to give title compound as a white solid. Yield: 79mg
MS: APCI (+ve) 461 [M+l]
1H NMR: δ (CDCl3) 7.26-7.22 (IH, m), 7.10-6.99 (2H, m), 6.33 (IH, s), 5.07 -5.00 (IH, m), 4.37 (2H, s), 4.02 (4H, t), 3.89-3.82 (IH, m), 2.25 (2H, quintet), 1.26-1.21(6H, m)
The intermediates for this compound were prepared as follows:
i) (2R,3R)-3-({6-chloro-2-[(2,3-difluorobenzyl)thio]pyrimidin-4-yl}oxy)butan-2-ol To a solution of (2/?,3i?)-butane-2,3-diol (250mg) and 4,6-Dichloro-2-[(2,3- difluorobenzyl)thio]pyrimidine (the product of example 1 step (ii)) (427mg) in anhydrous tetrahydrofuran (20ml) at ambient temperature was added 60% sodium hydride (33.4mg). After stirring for 15 minutes the reaction mixture was partitioned between aq. ammonium chloride solution and ethyl acetate. The organics collected, dried (MgSO4) and solvents removed under vacuo to give the subtitle compound as colourless gum. Yield: 525mg. MS: APCI(+ve) 361 [M+H+]
-40-
Example 5
N-(2-[(2,3-difluorobenzyl)thio]-6-{[(15,25)-2-hydroxy-l-methylpropyl]oxy}pyrimidin-4- yl) azetidine- 1 -sulfonamide
The title compound was prepared according to the procedure outlined in example 4 using a mixture of azetidine- 1-sulfonamide (150mg), tris(dibenzylideneacetone)dipalladium (0) (25 mg), 2-dicyclohexylphosρnino-2',4',6'-tri-isopropyl-l,l'-biphenyl (XPHOS) (25mg), cesium carbonate (244mg) and (25,3S)-3-({6-chloro-2-[(2,3-difluorobenzyl)thio]ρyrimidin-4- yl}oxy)butan-2-ol (200mg) in anhydrous dioxane (10ml). Purification was by reverse phase HPLC eluting with acetonitrile / aq. 0.1% ammonium acetate mixtures to give title compound as a white solid. Yield: 60mg MS: APCI (+ve) 461 [M+ 1]
1H NMR: 5 (CDCl3) 7.25-7.21 (IH, m), 7.10-6.99 (2H, m), 6.32 (IH, s), 5.07 -5.00 (IH, m), 4.37 (2H, s), 4.02 (4H, t), 3.88-3.81 (IH, m), 2.26 (2H, quintet), 1.26-1.21(6H, m)
The intermediates for this compound were prepared as follows:
i) (25,35)-3-({6-chloro-2-[(2,3-difluorobenzyl)thio]pyrimidin-4-yl}oxy)butan-2-ol
The subtitle compound was prepared according to the procedure outlined in example 4 step (i) using (25,35)-butane-2,3-diol (250mg) and 4,6-Dichloro-2-[(2,3- difluorobenzyl)thio]pyrimidine (the product of example 1 step (U)) (427mg) in anhydrous tetrahydrofuran (20ml) and 60% sodium hydride (33.4mg) to give the subtitle compound as a colourless gum. Yield: 440mg.
MS: APCI(+ve) 361 [M+H+]
-41-
Example 6
N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[[(2R)-2,3-dihydroxypropyl]oxy]-4- py rinii diny 1] - 1 - azetidinesu 1 fonami de
To a solution of N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(25)-l,4-dioxasρiro[4.5]dec-2- ylmethoxy]-4-pyrimidinyl]-l-azetidinesulfonamide (the product of step ii) (0.34g) in methanol (5mL)/ H2O (O. ImL) was added pyridinium p-toluenesuh°onate (78mg) and the mixture was stirred at reflux for 2h and then ambient temperature for 2Oh. The reaction mixture was evaporated, suspended in H2O and extracted with EtOAc (x2). The combined organic layers were dried (MgSO4), filtered and evaporated. The residue was purified by column chromatography on silica gel using DCM/MeOH (98:2) as eluent to give the title compound as a white solid. Yield: 0.15g MS: APCI(+ve) 463 [M+H+] 1H NMR: δ (DMSO) 2.13 (quintet, 2H), 3.42 (m, 2H), 3.77 (m, IH), 3.82 (t, 4H), 4.16 (dd, IH), 4.35 (dd, IH), 4.46 (s, 2H), 4.67 (t, IH), 4.97 (d,lH), 6.16 (s, IH), 7.17 (m, IH), 7.35 (m, IH), 7.44 (m, IH), 11.13 (br s, IH);
The intermediates for this compound were prepared as follows:
i) 4-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-[(2S)-l,4-dioxaspiro[4.5]dec-2- ylmethoxy ] -pyrimidine
The subtitle compound was prepared according to the procedure outlined in example 1 step iii) using (25)-l,4-dioxaspiro[4.5]decane-2-methanol (0.46g) and 4,6-Dichloro-2-[(2,3- difluorobenzyl)thio]pyrimidine (the product of example 1 step (ii) (0.75g) in THF (8mL) and 60% sodium hydride (39mg) to give the subtitle compound as a pale yellow solid. Yield: 0.7Og. MS: APCI(+ve) 403/405 [M+H+]
-42-
ii) 7V-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(25)-l,4-dioxaspiro[4.5]dec-2-ylmethoxy]- 4-pyrimidinyl]-l-azetidinesulfonainide
The subtitle compound was prepared according to the procedure outlined in example 1 step iv) using a mixture of azetidine- 1- sulfonamide (prepared according to patent WO 2004/011443, 0.25g), tris(dibenzylideneacetone)dipaUadium (0) (83rng), 2- dicyclohexylphosphino-2',4',6'-tri-wopropyl-l,r-biphenyl (XPHOS) (43mg), cesium carbonate (0.44g) and 4-chloro-2-[[(2,3-difluoroρhenyl)methyl]thio]-6-[(25)-l,4- dioxaspiro[4.5]dec-2-ylmethoxy]-pyrimidine (0.4Og) in dioxane (8mL). Purification was by column chromatography on silica gel using EtOAC/irøhexane (1:9 to 1:2 gradient) as eluent to give the subtitle compound as a pale yellow oil. Yield: 0.34g MS: APCI(+ve) 543 [M+H+]
Example 7 iV-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[[(25)-2,3-dihydroxypropyl]oxy]-4- pyrimidinyl] - 1 -azetidinesulfonamide
To a solution of N-[2-[[(2,3-difluoroρhenyl)methyl]thio]-6-[[(4i?)-2,2-dimethyl-l,3-dioxolan- 4-yl]methoxy]-4-pyrirrήdinyl]-l-azetidinesuhcbnamide (the product of step ϋ) (0.48g) in methanol (5 mL)/ H2O (0. ImL) was added pyridinium p-toluenesulfonate (0.12g) and the mixture was stirred at reflux for 2h. The reaction mixture was evaporated, suspended in H2O and extracted with EtOAc (x2). The combined organic layers were dried (MgSO4), filtered and evaporated. The residue was triturated with DCM to give the title compound as a white solid. Yield: 0.3Og MS: APCI(+ve) 463 [M+H+]
1H NMR: δ (DMSO) 2.15 (quintet, 2H), 3.42 (m, 2H), 3.77 (m, IH), 3.90 (t, 4H), 4.17 (dd, IH), 4.35 (dd, IH), 4.46 (s, 2H), 4.67 (t, IH), 4.98 (d,lH), 6.16 (s, IH), 7.16 (m, IH), 7.34 (m, IH), 7.44 (m, IH), 11.13 (br s, IH);
-43-
The intermediates for this compound were prepared as follows:
i) 4-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-[[(4R)-2,2-dimethyl-l,3-dioxolan-4- yl]methoxy]-pyrimidiue
The subtitle compound was prepared according to the procedure outlined in example 1 step iii) using 2,2-dimethyl-(4Λ)-l,3-dioxolane-4-methanol (0.26g) and 4,6-Dichloro-2-[(2,3- difluorobenzyl)thio]pyrimidine (the product of example 1 step ϋ) (0.5Og) in THF (5mL) and 60% sodium hydride (79mg) to give the subtitle compound as a clear, colourless oil. Yield: 0.47g.
MS: APCI(+ve) 403/405 [M+H+]
ii) N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[[(4R)-2,2-dimethyl-l,3-dioxolan-4- yl]methoxy]-4-pyrimidinyl]-l-azetidinesulfonamide The subtitle compound was prepared according to the procedure outlined in example 1 step iv) using a mixture of azetidine-1- sulfonamide (prepared according to patent WO 2004/011443, 0.24g), tris(dibenzylideneacetone)dipalladium (0) (0.1 Ig), 2- dicyclohexylphosphino-2',4',6'-tri-/røpropyl-l,r-biphenyl (XPHOS) (55mg), cesium carbonate (0.57g) and 4-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-[[(4Λ)-2,2-dimethyl- l,3-dioxolan-4-yl]methoxy]-pyrimidine (0.47 g) in dioxane (8mL). Purification was by column chromatography on silica using EtOAcΛrøhexane (3:7) as eluent to give the subtitle compound as a pale yellow solid. Yield: 0.49g MS: APCI(+ve) 503 [M+H+]
Example 8 iV-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[[(2/?)-2,3-dihydroxy-l,l-dimethylpropyl]oxy]- 4-pyriniidinyl]-l-azetidinesulfonamide
-44-
To a suspension of N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[l-[(4/?)-2,2-dimethyl-l,3- dioxolan-4-yl]-l-methylethoxy]-4-pyriinidinyl]-l-azetidinesulfonamide (the product from step ϋi) 0.34g) in DCM (9mL) was added iron (III) chloride hexahydrate (0.6Ig) and the mixture was stirred at ambient temperature for 35min. The reaction mixture was diluted with sat. sodium hydrogencarbonate solution and extracted with DCM (x3). The combined organic layers were dried (MgSO4), filtered and evaporated. The residue was purified by column chromatography on silica gel using MeOH/DCM (99: 1 to 98:2 gradient) as eluent to give the title compound as a white foam. Yield: 0.2Og MS: APCI(+ve) 489 [M+H+]
1H NMR: δ (DMSO) 1.41 (s, 3H), 1.44 (s, 3H), 2.16 (quintet, 2H), 3.32 (m, IH), 3.56 (m, IH), 3.87 (m, IH), 3.91 (t, 4H), 4.46 (m, 3H), 4.98 (d, IH), 6.06 (s, IH), 7.18 (m, IH), 7.37 (m, IH), 7.42 (m, IH), 11.06 (br s, IH)
The intermediates for this compound were prepared as follows:
i) α,α-2,2-tetramethyl-(4/?)-l,3-dioxolane-4-methanol
To anhydrous cerium (III) chloride (8. Ig of heptahydrate dried under high vacuum at 15O0C for 2Oh) was added THF (1OmL) then methyllithium (1.6M, 11.7mL) and the reaction mixture was stirred at ambient temperature for lOmin. A solution of 2,2-dimethyl-(4i?)-l,3-dioxolane- 4-carboxylic acid methyl ester (Ig) in THF (5mL) was added and the mixture was stirred at ambient temperature for 1.5h. The reaction mixture was quenched by a slow addition Of H2O (1OmL) and then extracted with Et2O (x2). The combined organic layers were dried (MgSO4), filtered and evaporated to afford the subtitle compound as a yellow oil. Yield: 0.4Og.
1H NMR: δ (CDCl3) 1.16 (s, 3H), 1.24 (s, 3H), 1.37 (s, 3H), 1.46 (s, 3H), 3.83 (m, IH), 3.96 (m, 2H)
-45-
ii) 4-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-[l-[(4R)-2,2-dimethyl-l,3-dioxolan-4- yl] - 1 -methylethoxy ]-pyrimidine
The subtitle compound was prepared according to the procedure outlined in example 1 step iii) using α,α-2,2-tetramethyl-(4/?)-l,3-dioxolane-4-methanol (0.32g) and 4,6-Dichloro-2- [(2>3-difluorobenzyl)thio]pyrimidine (the product of example 1 step ϋ) (0.56g) in THF (5mL) and 60% sodium hydride (80mg) to give the subtitle compound as a pale yellow oil. Yield: 0.43g.
1H NMR: δ (CDCl3) 1.16 (s, 3H), 1.24 (s, 3H), 1.55 (s, 3H), 1.57 (s, 3H), 3.87 (dd, IH), 4.02 (dd, IH), 4.35 (t, IH), 4.41 (s, 2H), 6.38 (s, IH), 7.04 (m, 2H), 7.26 (m, IH)
iu) N-[2-[[(2,3-diQuorophenyl)methyl]thio]-6-[l-[(4R)-2,2-dimethyl-l,3-dioxolan-4-yl]-l- methylethoxy]-4-pyrimidinyl]-l-azetidinesulfonamide
The subtitle compound was prepared according to the procedure outlined in example 1 step iv) using a mixture of azetidine- 1- sulfonamide (prepared according to patent WO 2004/011443, 0.2Og), tris(dibenzylideneacetone)dipalladium (0) (91mg), 2- dicyclohexylρhosphino-2',4',6'-tri-/røpropyl-l,r-biphenyl (XPHOS) (42mg), cesium carbonate (0.49g) and 4-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-[l-[(4/?)-2,2-dimethyl- l,3-dioxolan-4-yl]-l-methylethoxy]-pyrimidine (0.43g) in dioxane (8mL). Purification was by column chromatography on silica gel using EtOAc/irahexane (2:8) as eluent to give the subtitle compound as a pale yellow foam. Yield: 0.43g MS: APCI(-ve) 529 [M+H']
Example 9 ΛT-[2-[[(2,3-difluoroplienyl)methyl]thio]-6-[[(25)-2,3-dihydroxy-l,l-dimethylpropyl]oxy]- 4-pyrimidinyl] - 1 -azetidinesulfonamide
To a suspension of N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[l-[(45)-2,2-dimethyl-l,3- dioxolan-4-yl]-l-inethylethoxy]-4-pyriinidinyl]-l-azetidinesulfonarnide (the product from step ϋi) (0.37g) in DCM (1OmL) was added iron (III) chloride hexahydrate (0.66g) and the mixture was stirred at ambient temperature for Ih. The reaction mixture was diluted with sat. sodium hydrogencarbonate solution and extracted with DCM (x3). The combined organic layers were dried (MgSO4), filtered and evaporated. The residue was purified by column chromatography on silica gel using a MeOH/DCM (99:1 to 98:2 gradient) as eluent to give the title compound as a white solid. Yield: 0.16g MS: APCI(+ve) 489 [M+H+] 1H NMR: δ (DMSO) 1.42 (s, 3H), 1.44 (s, 3H), 2.15 (quintet, 2H), 3.33 (m, IH), 3.56 (m, IH), 3.87 (m, IH), 3.90 (t, 4H), 4.44 (m, 3H), 4.98 (d, IH), 6.06 (s, IH), 7.17 (m, IH), 7.36 (m, IH), 7.41 (m, IH), 11.06 (br s, IH)
The intermediates for this compound were prepared as follows:
i) α,α-2,2-tetramethyl-(4S)-l,3-dioxolane-4-methanol
To anhydrous cerium (III) chloride (8. Ig of heptahydrate dried under high vacuum at 15O0C for 2Oh) was added THF (1OmL) then methyllithium (1.6M, 11.7mL) and the reaction mixture was stirred at ambient temperature for lOmin. A solution of 2,2-dimethyl- -(45)-l,3- dioxolane-4-carboxylic acid methyl ester (Ig) in THF (5mL) was added and the mixture was stirred at ambient temperature for 1.5h. The reaction mixture was quenched by a slow addition Of H2O (1OmL) and then extracted with Et2O (x2). The combined organic layers were dried (MgSO4), filtered and evaporated to afford the subtitle compound as a yellow oil. Yield: 0.75g. 1K NMR: δ (CDCl3) 1.15 (s, 3H), 1.24 (s, 3H), 1.38 (s, 3H), 1.43 (s, 3H), 3.84 (m, IH), 3.97 (m, 2H)
ii) 4-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-[l-[(4S)-2,2-dimethyl-l,3-dioxolan-4- yl] - 1 -methylethoxy] -py rimidine The subtitle compound was prepared according to the procedure outlined in example 1 step iii) using α,α-2,2-tetramethyl-(4S)-l,3-dioxolane-4-methanol (0.32g) and 4,6-Dichloro-2- [(2,3-difluorobenzyl)thio]pyrimidine (the product of example 1 step ϋ) (0.56g) in THF (5mL)
-47- and 60% sodium hydride (80mg) to give the subtitle compound as a colourless oil. Yield: 0.37g.
1K NMR: δ (CDCl3) 1.15 (s, 3H), 1.24 (s, 3H),.1.55 (s, 3H), 1.57 (s, 3H), 3.88 (dd, IH), 4.02 (dd, IH), 4.35 (t, IH), 4.41 (s, 2H), 6.38 (s, IH), 7.03 (m, 2H), 7.26 (m, IH) 5 iii) N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[l-[(45)-2,2-dimethyl-l,3-dioxolan-4-yl]-l- methylethoxy]-4-pyrimidinyl]-l-azetidinesulfonamide
The subtitle compound was prepared according to the procedure outlined in example 1 step iv) using a mixture of azetidine-1- sulfonamide (prepared according to patent WO
10 2004/011443, 0.17g), tris(dibenzylideneacetone)dipalladium (0) (78 mg), 2- dicyclohexylphosphino-2',4',6'-tri-iiropropyl-l,r-biρhenyl (XPHOS) (40mg), cesium carbonate (0.42g) and 4-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-[l-[(4,S0-2,2-dimethyl- l,3-dioxolan-4-yl]-l-methylethoxy]-pyrimidine (0.37g) in dioxane (8mL). Purification was by column chromatography on silica gel using EtOAc/isohexane (2:8 to 3:7 gradient) as
15 eluent to give the subtitle compound as a pale yellow oil. Yield: 0.37g MS: APCI(-ve) 529 [M+H ]
Example 10 iV-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[2-hydroxy-l-(hydroxymethyl)-l- 20 methylethoxy ] -4-pyrimidinyl] - 1 -azetidinesulfonamide
To a suspension of N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(2,2,5-trimethyl-l,3-dioxan-5- yl)oxy] -4-pyrimidinyl] -1 -azetidinesulfonamide (the product from step ii) (0.46g) in DCM (15mL) was added iron (III) chloride hexahydrate (0.85g) and the mixture was stirred and 25 ambient temperature for 30min. A saturated solution of sodium hydrogencarbonate was added and then extracted with DCM (x4). The combined organic layers were dried (MgSO4),
-48- filtered and evaporated. The residue was purified by column chromatography on silica gel using a MeOH/DCM (99: 1 to 98:2 gradient) as eluent to give the title compound as a white foam. Yield: lOOmg MS: APCI(-ve) 475 [M+IT]
5 1H NMR: δ (DMSO) 1.43 (s, 3H), 2.15 (quintet, 2H), 3.63 (dd, 2H), 3.73 (dd, 2H), 3.92 (t, 4H), 4.44 (s, 2H), 4.78 (t, 2H), 6.09 (s, IH), 7.17 (m, IH), 7.36 (m, IH), 7.43 (m, IH), 11.06 (s, IH)
The intermediates for this compound were prepared as follows: 10 i) 4-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-[(2,2,5-trimethyl-l,3-dioxan-5-yl)oxy]- pyrimidine
The subtitle compound was prepared according to the procedure outlined in example 1 step iii) using 2,2,5-trimethyl-l,3-dioxan-5-ol (as prepared in Synthesis, 1998, p879) (0.29g) and
15 4,6-Dichloro-2-[(2,3-difluorobenzyl)thio]pyrimidine (the product of example 1 step ϋ) (0.5Ig) in THF (5mL) and 60% sodium hydride (80mg) to give the subtitle compound as a yellow oil. Yield: 0.44g.
1H NMR: δ (CDCl3) 1.16 (s, 3H), 1.24 (s, 3H), 1.53 (s, 3H), 3.85 (d, 2H), 4.14 (d, 2H), 4.38 (s, 2H), 6.48 (s, IH), 7.04 (m, 2H), 7.26 (m, IH)
20 u) N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(2,2,5-trimethyl-l,3-dioxan-5-yl)oxy]-4- pyrimidinyl] - 1 -azetidinesulfonamide
The subtitle compound was prepared according to the procedure outlined in example 1 step (iv) using a mixture of azetidine-1- sulfonamide (prepared according to patent WO
25 2004/011443, 0.22g), tris(dibenzylideneacetone)dipalladium (0) (97mg), 2- dicyclohexylphosphino-2',4',6'-tri-irøpropyl-l,r-biphenyl (XPHOS) (50mg), cesium carbonate (0.52g) and 4-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-[(2,2,5-trimethyl-l,3- dioxan-5-yl)oxy]-pyrimidine (0.44g) in dioxane (1OmL). Purification was by column chromatography on silica gel using EtOAc/isohexane (2:8 to 3:7 gradient) as eluent to give
30 the subtitle compound as a pale yellow oil. Yield: 0.46g MS: APCI(+ve) 517 [M+H+]
-49-
Example 11
N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(lR)-2-hydroxy-l-methylethoxy]-4- pyrimidinyl]-2-thiazolesulfonamide
To a solution of 2-[[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(2-thiazolylsulfonyl)amino]-4- pyrirnidinyl]oxy]-(2/?)-propanoic acid ethyl ester (the product from step ii) (0.1 Ig) in THF (3mL) was added lithium borohydride (2M solution in THF, 0.23mL) and the mixture was mixture was stirred at ambient temperature for 2Oh. The reaction mixture was cooled to O
0C, quenched with 0.5M HCl solution and the aqueous was extracted with EtOAc (x2). The combined organic layers were dried (MgSO
4), filtered and evaporated. The residue was purified by column chromatography on silica gel using MeOH/DCM (99:1 to 98:2 gradient) as eluent to give the title compound as a white solid. Yield: 15mg MS: APCI(+ve) 475 [M+H
+]
1H NMR: δ (CDCl
3) 1.44 (d, 3H), 3.72 (m, 2H), 4.34 (q, 2H), 5.25 (m, IH), 5.29 (s, IH), 6.43 (s, IH), 7.03 (m, 2H), 7.17 (t, IH), 7.66 (s, IH), 7.98 (s, IH)
The intermediates for this compound were prepared as follows:
i) 2-[[6-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-4-pyrimidinyl]oxy]-(2/?)-propanoic acid ethyl ester
The subtitle compound was prepared according to the procedure outlined in example 1 step iii) using 2-hydroxy-(2/?)-propanoic acid ethyl ester (1.45mL) and 4,6-Dichloro-2-[(2,3- difluorobenzyl)thio]pyrimidine (the product of example 1 step ii) (3g) in THF (4OmL) and 60% sodium hydride (0.55g) to give the subtitle compound as a clear, colourless oil. Yield: 2.85g.
MS: APCI(+ve) 389/391 [M+H+]
-SO- ii) 2-[[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(2-thiazolylsulfonyl)amino]-4- pyrimidmyl]oxy]-(2R)-propanoic acid ethyl ester
The subtitle compound was prepared according to the procedure outlined in example 1 step (iv) using a mixture of 2-thiazolesulfbnamide (0.17g), tris(dibenzylideneacetone)dipalladium (0) (64mg), 2-dicyclohexylphosphino-2',4',6'-tri-woρropyl-l,r-biphenyl (XPHOS) (33mg), cesium carbonate (0.34g) and 2-[[6-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-4- pyrimidtnyl]oxy]-(2Λ)- propanoic acid ethyl ester (0.27g) in dioxane (5mL). Purification was by column chromatography on silica using EtOAc/jyøhexane (1:9 to 1: 1 gradient) as eluent to give the subtitle compound as a pale yellow oil. Yield: 0.1 Ig MS: APCI(+ve) 517 [M+H+]
Example 12
N- [6-(difluoromethoxy)-2- [ [(2,3-difluorophenyl)methyl] thio] -4-pyrimidinyl] - 1 - azetidinesulfonamide
The title compound was prepared according to the procedure outlined in example 1 step (iv) using a mixture of azetidine-1-sulphonamide (prepared according to patent WO 2004/011443, 0.11 g), tris(dibenzylideneacetone)dipalladium (0) (0.1Og), 2-dicyclohexylphosphino-2',4',6'- tri-isopropyl-l,l'-biphenyl (XPHOS) (60mg), cesium carbonate (0.26g), 4-chloro-6- (difluoromethoxy)-2-[[(2,3-difluorophenyl)methyl]thio]-pyrimidine (product of step ii)
(0.18g) and anhydrous dioxane (5mL). Purification was by column chromatography on silica gel using EtOAc/isøhexane (3:7) as eluent and the relevant fractions were evaporated. The resulting oil was triturated with diethyl ether/irø-hexane to give the title compound as a white solid. Yield: 70mg MS: APCI(+ve) 439 [M+H+]
-51-
1H NMR: δ (DMSO) 2.13 (quintet, 2H), 3.93 (t, 4H), 4.50 (s, 2H), 6.30 (s, IH), 7.19 - 7.12 (m, IH), 7.45 - 7.30 (m, 2H), 7.79 (t, IH), 11.53 (s,lH)
The intermediates for this compound were prepared as follows:
i) 6-(difluoromethoxy)-2-[[(2,3-difluorophenyl)methyl]thio]- 4-pyrimidinol
To a solution of 2-[[(2,3-difluorophenyl)methyl]thio]-4,6-pyriτnidinediol (3g) in DMF (3OmL), cesium carbonate (4.3g) and chlorodifluoro-acetic acid sodium salt (1.9g) was added. The resulting mixture was heated at 100°C for 2h. The reaction mixture was cooled then diluted with H2O and extracted with EtOAc. The organic layer was washed with H2O and dried (MgSO4), filtered and evaporated. Purification was by column chromatography on silica gel using EtOAc/isohexane (2:8) as eluent to give the subtitle compound as a white solid. Yield: 0.4g MS: APCI(+ve) 421 [M+H+] 1K NMR: δ (DMSO) 4.53 (s, 2H), 7.13 - 7.22 (m, IH), 7.30 - 7.42 (m, 2H), 7.75 (t, IH)
ii) 4-chloro-6-(difluoromethoxy)-2-[[(2,3-difluorophenyl)methyl]thio]-pyrimidine
To a solution of 6-(difluoromethoxy)-2-[[(2,3-difluorophenyl)methyl]thio]-4-pyrimidinol (product of step i) (0.4g) in 1,2-dimethoxyethane was added benzyltriethylammonium chloride (3mg) and phosphorous oxychloride (0.23mL). The resulting mixture was heated to reflux for 16 hours. The reaction mixture was cooled then diluted with H2O and extracted with EtOAc. The organic layer was washed with H2O and dried (MgSO4), filtered and evaporated. Purification was by column chromatography on silica gel using EtOAc/wohexane (2:8) as eluent to give the subtitle compound as a clear, colourless oil. Yield: 0.35g 1H NMR: δ (DMSO) 4.54 (s, 2H), 7.12 - 7.22 (m, 2H), 7.25 (s, IH), 7.30 - 7.42 (m, 2H), 7.81 (t, IH)
-52-
Example 13
N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(lR)-2-hydroxy-l-methylethoxy]-4- pyrimidinyl]- 4-pyridinesuIfonamide
To a solution of JV-[2-[[(2,3-difluoroρhenyl)methyl]thio]-6-[(li?)-l-metliyl-2-
(triphenylmethoxy)ethoxy]-4-pyrimidinyl]-4-pyridinesulfonamide (the product from step iv). (lOOmg) in MeOH (1OmL) was added p-toluenesulfonic acid (31mg) and anisole (0.15g). The reaction was then stirred at room temperature for 18h. The reaction was partitioned between EtOAc (10OmL) and H2O (10OmL). The aqueous layer was then further extracted with EtOAc (2xl00mL). The combined organic layers were dried (MgSO4), filtered and evaporated. The residue was purified by reverse phase HPLC using a TFA (0.2%)/MeCN to give the title compound as a white solid. Yield: 50mg. MS: APCI(+ve) 496 [M+H+] 1H NMR: δ (DMSO) 1.13 (d, 3H), 4.30 (s, 2H), 5.06-5.12 (m, IH), 6.0 (s, IH), 7.07-7.38 (m, 3H), 7.84 (d, 2H), 8.86 (d, 2H)
The intermediates for this compound were prepared as follows:
i) 4-pyridinesulfonamide A solution of 4-pyridinethione (3.33g) in c.HCl (22.5mL) and H2O (6mL) was bubbled with chlorine gas at room temperature for 3h. The reaction mixture was then poured onto ice (15g), and the slurry was then transferred to ice-cold 0.88 ammonia (12OmL). This mixture was then stirred at room temperature overnight before being concentrated in vacuo until solid began to precipitate. At this point the reaction mixture was cooled overnight in the refrigerator and the solid collected by filtration as a yellow solid. Yield: 1.5 Ig. 1U NMR: δ (DMSO) 7.73 (s, 2H), 7.75 (d, 2H), 8.84 (d, 2H)
-53- ii) (2/?)-l-(triphenylmethoxy)-2-propanol
To a suspension of (2/?)-l,2-propanediol (1.9mL) in toluene (2OmL) was added triethylamine (8.3mL) and 4-dimethylaminopyridine (32mg). The mixture was ice-cooled and 1,1', 1"- (chloromethylidyne)tris-benzene (6.6g) was added and the mixture stirred at ambient temperature for 2Oh. The reaction mixture was diluted with toluene then extracted with ammonium chloride solution (x2), then brine (xl) and the organic layer was dried (MgSO4), filtered and evaporated. The resulting oil was triturated with iso-hexane to give subtitle compound as a white solid. Yield: 4g
1H NMR: δ (CDCl3) 1.09 (d, 3H), 2.34 (d, IH), 2.97 (dt, IH), 3.15 (dd, IH), 3.97 (m, IH), 7.23 (m, 3H), 7.28 (m, 6H), 7.45 (m, 6H)
iii) 4-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-[(lΛ)-l-methyl-2- (triphenylmethoxy)ethoxy]-pyrimidine
The subtitle compound was prepared according to the procedure outlined in example 1 step (iii) using (2R)- l-(triphenylrnethoxy)-2-propanol ( 1.35g) and 4,6-Dichloro-2-[(2,3- drfluorobenzyl)thio]pyrimidine (the product of example 1 step (ii) (Ig) in THF (15mL) and 60% sodium hydride (0.18g) to give the subtitle compound as a pale yellow oil. Yield: 1.8g. MS: APCI(+ve) 589 [M+H+]
iv) N-[2-[[(2,3-diαuorophenyl)methyl]thio]-6-[(l/?)-l-methyl-2- (triphenylmethoxy)ethoxy ] -4-pyrimidinyl] -4-pyridinesulfonamide
A mixture of 4-pyridinesulfonamide (the product from step i) (0.2Ig), tris(dibenzylideneacetone)dipalladium (0) (50 mg), 2-dicyclohexylphosphino-2',4',6'-tri- «øpropyl-l,r-biρhenyl (XPHOS) (50mg), cesium carbonate (0.66g) and 4- chloro-2-[[(2,3- difluorophenyl)methyl] thio] -6- [( IR)- 1 -methyl-2-(triphenylmethoxy)ethoxy] -pyrimidine (the product from step iii) (0.4Og) in dioxane (2OmL) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for 3h. The reaction mixture was diluted with DCM, filtered through arbocel and the filtrate evaporated. The residue was purified by reverse phase HPLC using a TFA (0.2%)/MeCN system to give the title compound as a yellow solid. Yield: 0.21g.
MS: APCI(+ve) 711 [M+H+]
-54-
1U NMR: δ (DMSO) 8.85 - 8.76 (m, 2H), 7.83 - 7.73 (m, 2H),7.26 - 7.17 (m, 18H), 6.03 (s, IH), 5.44 - 5.35 (m, IH), 4.29 (s, 2H), 3.08 - 3.01 (m, 2H), 1.22 - 1.14 (m,3H)
Example 14 N-[24[(2,3-difluorophenyl)methyl]thio]-6-ethoxy-4-pyrimidinyl]-l-azetidinesulfonamide
The title compound was prepared according to the procedure outlined in example 1 step iv) using a mixture of azetidine-1 -sulfonamide (prepared according to patent WO 2004/011443, 0.17g), tris(dibenzylideneacetone)dipalladium (0) (75mg), 2-dicyclohexylphosphino-2',4',6'- trw,røpropyl-l,l'-biphenyl (XPHOS) (40mg), cesium carbonate (0.4Og) 4-chloro-2-[[(2,3- difluorophenyl)methyl]thio]-6-ethoxy-pyrimidine (the product from step i) (0.26g)) in dioxane (5mL). Purification was by column chromatography on silica gel using EtOAc/irøhexane (1:9) as eluent to give the title compound as a white solid. Yield: 0.17g MS: APCI(+ve) 417 [M+H+] 1H NMR: δ (DMSO) 1.27 (t, 3H), 2.13 (quintet, 2H), 3.90 (t, 4H), 4.34 (q, 2H), 4.47 (s, 2H), 6.12 (s, IH), 7.15 (m, IH), 7.33 (m, IH), 7.42 (m, IH), 11.11 (br s, IH)
The intermediate for this compound was prepared as follows:
i) 4-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-ethoxy-pyrimidme
To a solution of 4,6-Dichloro-2-[(2,3-difluorobenzyl)tnio]pyrimidme (0.5Og) m ethanol (5mL) was added 60% sodium hydride (72mg) and the reaction mixture was stirred at ambient temperature for 6h. The mixture was diluted with H2O and extracted with EtOAc (x2). The combined organic layers were dried (MgSO4), filtered and evaporated to give the subtitle compound as a clear, colourless oil. Yield:0.53g MS: APCI(+ve) 317/319 [M+H+]
-55-
Example 15
N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-ethoxy-4-pyrimidinyl]-l- piperazinesulfonamide
To a solution of 4-[[[2-[[(2,3-difluorophenyl)methyl]thio]-6-ethoxy-4- pyrimidinyl] amino] sulfonyl]-l-piperazinecarboxy lie acid-l,l-dimethylethyl ester (the product from step ϋ) (0.24g) in DCM (2mL) was added trifluro acetic acid (2mL) and the reaction mixture was stirred at ambient temperature for 2.5h. The reaction mixture was evaporated, the residue was azeotroped with DCM (x2) and then purified by reverse phase HPLC eluting with acetonitrile / aq. 0.2% trifluoroacetic acid mixtures to give title compound as a white solid. Yield: 0.18g MS: APCI(+ve) 446 [M+H
+]
1H NMR: δ (DMSO) 1.28 (t, 3H), 3.18 (m, 4H), 3.44 (m, 4H), 4.36 (q, 2H), 4.47 (s, 2H), 6.05 (s, IH), 7.18 (m, IH), 7.37 (m, 2H), 8.73 (br s, IH), 11.33 (br s, IH)
The intermediate for this compound was prepared as follows:
i) 4-(aminosulfonyl)-l,l-dimethylethyl ester-l-piperazinecarboxylic acid
To a solution of 1,1-dimethylethyl ester-l-piperazinecarboxylic acid (2.94g) in dioxane (4OmL) was added sulfamide (4.Og). The reaction mixture was then heated at reflux for 24 h. The reaction mixture was allowed to cool before being reduced in vacuo. The residue was separated between EtOAc (30OmL) and H2O (30OmL) and the aqueous was further extracted (2x300mL) with EtOAc. Combined organic layers were dried (MgSO4), filtered and evaporated. The residue was purified by column chromatography on silica gel using EtOAc/isohexane (1:1) as eluent to give the subtitle compound as a white solid. Yield: 2.03g. 1U NMR: δ (DMSO) 1.41 (s, 9H), 2.89 (t, 4H), 3.40 (t, 4H), 6.81 (s, 2H)
-56- ii) 4-[[[2-[[(2,3-difluorophenyl)methyl]thio]-6-ethoxy-4-pyriniidinyl]aniino]sulfonyl]-l- piperazmecarboxylic add-l,l-dimethylethyl ester
The subtitle compound was prepared according to the procedure outlined in example 1 step (iv) using a mixture of 4-(aminosulfonyl)-l-piperazinecarboxylic acid-l,l-dimethylethyl ester (the product from step i) (0.29g), tris(dibenzylideneacetone)dipalladium (0) (67mg), 2- dicyclohexylphosphino-2',4',6'-tri-i,yopropyl-l,r-biphenyl (XPHOS) (35mg), cesium carbonate (0.36g) and 4-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-ethoxy-pyrimidine (the product from example 14, step i) (0.23g) in dioxane (5mL). Purification was by column chromatography on silica using EtOAc/wohexane (1:9 to 1:3 gradient) as eluent to give the subtitle compound as a yellow oil. Yield: 0.25g MS: APCI(-ve) 544 [M+H ]
Example 16 iV-[2-[[(2,3-difluorophenyl)methyl]thio]-6-(2,2,2-trifluoroethoxy)-4-pyrimidinyl]-l- azetidinesulfonaπiide
The title compound was prepared according to the procedure outlined in example 1 step iv) using a mixture of azetidine-1 -sulfonamide (prepared according to patent WO 2004/011443, 0.14g), tris(dibeπzylideneacetone)dipalladium (0) (60mg), 2-dicyclohexylphosphino-2',4',6'- tri-wopropyl- 1 , 1 '-biphenyl (XPHOS) (30mg), cesium carbonate (0.32g) 4-chloro-2-[[(2,3- dhcluoroρhenyl)methyl]thio]-6-(2,2,2-trifluoroethoxy)-pyrimidine (the product from step i) (0.24g) in dioxane (5mL). Purification was by column chromatography on silica gel using EtOAc/irøhexane (1:9 to 2:8 gradient) as eluent to give the title compound as a white solid. Yield: 0.1 Ig MS: APCI(+ve) 471 [M+H+]
1H NMR: δ (DMSO) 2.1 (quintet, 2H), 3.83 (t, 4H), 4.51 (s, 2H), 5.03 (q, 2H), 6.22 (s, IH), 7.16 (m, IH), 7.36 (m, IH), 7.42 (m, IH), 11.33 (s, IH)
-57- The intermediate for this compound was prepared as follows:
i) 4-chIoro-2-[[(2,3-difluorophenyl)methyl]thio]-6-(2,2,2-trifluoroethoxy)-pyrimidine
The subtitle compound was prepared according to the procedure outlined in example 1 step iii) using 2,2,2-trifluoroethanol (0.16mL) and 4,6-Dichloro-2-[(2,3- difluorobenzyl)thio]pyrimidine (the product of example 1 step ii) (0.6Og) in THF (6mL) and 60% sodium hydride (94mg) to give the subtitle compound as a clear, colourless oil. Yield: 0.6g.
1H NMR: δ (DMSO) 4.54 (s, 2H, 5.14 (m, 2H), 7.13 (s, IH), 7.19 (m, IH), 7.37 (m, 2H)
Example 17
N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-(2,2,2-trifluoroethoxy)-4-pyrimidinyl]-l- piperazinesulfonamide
To a solution of 4-[[[2-[[(2,3-difluorophenyl)methyl]thio]-6-(2,2,2-trifluoroethoxy)-4- pyrimidinyl] amino] sulfonyl]-l-piperazinecarboxy lie acid- 1,1 -dimethylethyl ester (the product from step i) (0.2Ig) in DCM (2mL) was added trifluro acetic acid (2mL) and the reaction mixture was stirred at ambient temperature for 4h. The reaction mixture was evaporated, the residue was azeotroped with Et
2O (x2) and then purified by reverse phase HPLC eluting with acetonitrile / aq. 0.2% trifluoro acetic acid mixtures to give title compound as a white solid. Yield: 0.14g
MS: APCI(+ve) 500 [M+H+]
1H NMR: δ (DMSO) 3.17 (m, 4H), 3.50 (m, 4H), 4.51 (s, 2H), 5.06 (q, 2H), 6.17 (s, IH), 6.96-7.42 (m, 3H), 8.82 (br s, 2H)
The intermediate for this compound was prepared as follows:
-58- i) 4-[[[2-[[(2,3-difluorophenyI)methyl]thio]-6-(2,2,2-trifluoroethoxy)-4- pyrimidinyljaminolsulfonyll-l-piperazinecarboxylic acid-l^-dimethylethyl ester
The subtitle compound was prepared according to the procedure outlined in example 1 step (iv) using a mixture of 4-(aminosulfonyi)-l-ρiperazinecarboxylic acid-l,l-dimethylethyl ester (the product from example 15, step i), 0.4Og), tris(dibenzylideneacetone)dipalladium (0) (91mg), 2-dicyclohexylphosρlύno-2\4\6^ri-iMφroρyl-l,l>-biphenyl (XPHOS) (48mg), cesium carbonate (0.49g) and 4-chIoro-2-[[(2,3-difluorophenyl)methyl]thio]-6-(2,2,2- trifluoroethoxy)-pyrimidine (the product from example 16, step i) (0.37g) in dioxane (6mL). Purification was by column chromatography on silica gel using EtOAc/wohexane (1:9 to 2:8 gradient) as eluent to give the subtitle compound as a yellow solid. Yield: 0.22g MS: APCI(-ve) 598 [M+H"]
Example 18
N-[2-[[(2,3-difluorophenyl)methyl]tMo]-6Kl,l-dimethylethoxy)-4-pyrimidinyl]-l- azetidinesulfonamide
The title compound was prepared according to the procedure outlined in example 1 step iv) using a mixture of azetidine-1- sulfonamide (prepared according to patent WO 2004/011443, 0.16g), tris(dibenzylideneacetone)dipalladiurn (0) (70mg), 2-dicyclohexylphosphino-2',4',6'- tri-wøpropyl- 1 , 1 '-biphenyl (XPHOS) (36mg), cesium carbonate (O.37g) and 4-chloro-2-
[[(2,3-dhcluorophenyl)methyl]thio]-6-(l,l-dimethylethoxy)-pyrnnidine (the product from step i) (0.26g) in dioxane (6mL). Purification was by column chromatography on silica gel using EtOAc/irøhexane (1:9 to 2:8 gradient) as eluent to give the title compound as a white solid. Yield: 0.28g MS: APCI(-ve) 443 [M+H ]
1H NMR: δ (DMSO) 1.48 (s, 9H), 2.16 (quintet, 2H), 3.92 (t, 4H), 4.46 (s, 2H), 6.03 (s, IH), 7.17 (m, IH), 7.35 (m, IH), 7.42 (m, IH), 11.05 (br s, IH)
-59-
The intermediate for this compound was prepared as follows:
i) 4-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-(l,l-dimethylethoxy)-pyrimidine To a solution of 4,6-Dichloro-2-[(2,3-difluorobenzyl)thio]pyrimidine (the product of example 1 step ϋ) (2g) in THF (2OmL) was added potassium tert-butoxide (0.8g) and the reaction mixture was stirred at ambient temperature for 2Oh. Further potassium tert-butoxide (0.8g) was added and the reaction mixture was stirred at ambient temperature for 4h. The mixture was diluted with H2O and extracted with EtOAc (x3). The combined organic layers were washed with H2O and dried (MgSO4), filtered and evaporated. The resulting oil was purified by column chromatography on silica gel using MeOH/DCM (99:1 to 98:2 gradient) as eluent to give the subtitle compound as a clear, colourless oil. Yield: 0.68g 1K NMR: δ (DMSO) 1.50 (s, 9H), 4.47 (s, 2H), 6.70 (s, IH), 7.19 (m, IH), 7.37 (m, 2H)
Example 19 iV-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[[2-hydroxy-l-(hydroxymethyl)ethyl]thio]- pyrirnidin-4-yl]azetidine-l-sulfonamide
A solution of N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(2-phenyl- 1 ,3-dioxan-5-yl)thio] pyrimidin-4-yl]azetidine-l- sulfonamide (the product of step ii) (0.1 Ig) and pyridinium/?αra- toluenesulfonate (99mg) in methanol (5mL) and H2O (2 drops) was heated at 600C for Ih. The solution was cooled and the solvent evaporated under reduced pressure. The residue was dissolved in EtOAc, washed with H2O, dried (MgSO4) and filtered. The solvent was evaporated under reduced pressure and the residue was purified by flash chromatography on silica gel, eluting with EtOAc/iso-hexane (8:2) to give the subtitle product as a yellow gum. The gum was dissolved in DCM and methanol, filtered through charcoal and the filtrate
-60- evaporated under reduced pressure. The residual solid was dried under high vacuum at 40°C to give the title product as a white solid. Yield: 50mg MS: APCI(-ve) 477 [M-H]
1H NMR: δ (DMSO) 2.13 (m, 2H), 3.66 (octet, 4H), 3.92 (t, 5H), 4.49 (s, 2H), 4.99 (t, 2H), 6.65 (s, IH), 7.17 (m, IH), 7.36 (m, 2H), 11.18 (s, IH).
The intermediates for this compound were prepared as follows:
i) 4-Chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-[(2-phenyl-l,3-dioxan-5- yl)thio]pyrimidine
Sodium methoxide (0. ImL of 25 - 30% methanol solution) was added to a solution of ethyl 5'-(2-phenyl-l,3-dioxan-5-yl)ethanethioate (0.12g; prepare according to the procedure in Chem. Pharm. Bull, 2000, 48, (5), p694-707) in THF (2mL). After stirring for 15min, the product of Example 1, step ϋ) (0.12g) was added. The reaction mixture was stirred at room temperature for 18h. The solvent was evaporated under reduced pressure and the residue purified by flash chromatography on silica gel, eluting with Et2O //sø-hexane (1:9) to give the subtitle product as a beige solid. Yield: 0.18g. MS: APCI(+ve) 467/469 [M+H]
ii) N-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-[(2-phenyl-l,3-dioxan-5-yl)thio]pyriraidin- 4-yl]azetidine-l-sulfonamide
The subtitle compound was prepared from azetidine-1 -sulfonamide (prepared according to patent WO 2004/011443, 0.1 Ig) and 4-Chloro-2-[[(2,3-difluoroρhenyl)methyl]thio]-6-[(2- phenyl-l,3-dioxan-5-yl)thio]pyrimidine (the product of step i) (0.17g ) according to the procedure outlined in Example 1, step iv). The residue was purified by flash chromatography on silica gel, eluting with EtOAc/wø-hexane (2:8) to give the subtitle compound as a colourless oil. Yield: 0.1 Ig MS: APCI(+ve) 567 [M+H]
-61-
Example 20 iV-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(lΛ)-2-hydroxy-l-methyIethoxy]-4- pyrimidinyl]- 1-piperazinesulfonamide
To a solution of 4-[[[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(l/?)-2-hydroxy-l- methylethoxy] -4-pyrimidiny 1] amino] sulfonyl] , 1,1 -dimethylethyl esterl -piperazinecarboxylic acid (the product of step ii) (0.23g) in DCM (3mL) was added trifluoroacetic acid (3mL) . The reaction mixture was then stirred at room temperature for Ih . The solvent was removed and the residue purified by reverse phase HPLC using a TFA (0.2%)/MeCN method to give the title compound as a white solid. Yield: 77mg MS: APCI(+ve) 476 [M+H
+]
1H NMR: δ (DMSO) 1.13 (d, 3H), 3.01-3.05 (m, 4H), 3.13-3.17 (m, 4H), 4.34-4.41 (m, 2H), 4.79 (s, IH), 4.97-5.05 (m, IH), 5.84 (s, IH), 7.10-7.16 (m, IH), 7.27-7.34 (m, IH), 7.39-7.45 (m, lH) The intermediates for this compound were prepared as follows:
(i) 4-[[[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(l/?)-2-hydroxy-l-methylethoxy]-4- pyrimidinyl]amino]su]fonyl], 1,1-dimethylethyl esterl -piperazinecarboxylic acid
A mixture of 4-(aminosulfonyl)- 1,1-dimethylethyl ester- 1 -piperazinecarboxylic acid (the product of example 15 step i) (0.4Og), tris(dibenzylideneacetone)dipalladium (0) (50 mg), 2- dicyclohexylphosphino-2',4',6'-tri-irøpropyl-l,r-biphenyl (XPHOS) (50mg), cesium carbonate (Ig) and (2R)- propanoic acid-2-[[6-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-4- pyrimidinyl]oxy]-, ethyl ester (the product from example 11 step i) (0.4Og) in dioxane (2OmL) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for 20min. The reaction was filtered through arbocel and then separated between EtOAc (20OmL) and H2O (20OmL) and the aqueous was then further extracted with EtOAc (2x200mL). The combined
-62- organic layers were dried (MgSO4), filtered and evaporated to give the subtitle compound as a clear oil. Yield: 0.93g.
MS: APCI(+ve) 618 [M+H+]
(ii), 4-[[[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(l/?)-2-hydroxy-l-methylethoxy]-4- pyrimidinyl]amino]sulfonyl]-, 1,1-dimethylethyl esterl-piperazinecarboxylic acid To a solution of 4-[[[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(l/?)-2-hydroxy-l- methylethoxy]-4-pyrimidinyl] amino] sulfonyl], 1,1-dimethylethyl ester- 1- piperazinecarboxylic acid (the product from step i) (0.93g) in THF (2OmL) was added 2M LiBH4 in THF (3.OmL). The reaction mixture was then heated in a microwave at 500C, 300W, open vessel with cooling for lOmin. The reaction mixture was then quenched with 2N HCl and the volatiles evaporated. The residue was then separated between EtOAc (20OmL) and H2O (20OmL), the aqueous was then further extracted with EtOAc (2x200mL). The combined organic layers were dried (MgSO4), filtered and evaporated and the residue purified by reverse phase HPLC using a TFA (0.2%)/MeCN method to give the title compound as a clear on.
Yield: 0.23g.
MS: APCI(+ve) 576 [M+H+]
Example 21
7V-[6-(difluoromethoxy)-2-[[(2,3-difluorophenyl)methyl]thio]-4-pyrimidinyl]-l-methyl- lH-imidazole-4-sulfonamide
The title compound was prepared according to the procedure outlined in example 1 step (iv) using a mixture of l-methyl-lH-imidazole-4-sulfonamide (0.25g), tris(dibenzylideneacetone)dipalladium (0) (O. lOg), 2-dicyclohexylphosphino-2',4',6'-tri- isopropyl-l,l'-biphenyl (XPΗOS) (60mg), cesium carbonate (0.26g), 4-chloro-6- (difluoromethoxy)-2-[[(2,3-difluorophenyl)methyl]thio]-pyrimidine (product of example 12, step ϋ) (0.18g) and anhydrous dioxane (5ml). Purification was by tituration with methanol/DCM. The resulting white solid was diluted with H2O and extracted with EtOAc. The organic layer was washed with H2O (x2) then brine and dried (MgSO4), filtered and evaporated. The resulting oil was triturated with methanol/DCM to give the title compound as a white solid. Yield: 15mg MS: APCI(+ve) 464 [M+H+]
1H NMR: δ (DMSO) 3.67 (s, 3H), 4.45 (s, 2H), 6.38 (s, IH), 7.10-7.18 (m, IH), 7.30-7.40 (m, 2H), 7.70 (t, IH), 7.82 (s, IH), 8.08 (s, IH)
Example 22
N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(lR)-2-hydroxy-l-methylethoxy]-4- pyrimidinyl]-l,6-dihydro-l-methyl-6-oxo-3-pyridinesulfonamide
To a solution of N-[2-[[(2,3-difluoroρhenyl)methyl]thio]-6-[(l/?)-l-methyl-2- (tτiphenyhnethoxy)ethoxy]-4-pyrimidinyl]-l,6-dihydro-l-methyl-6-oxo-3- pyridinesulfonamide (the product from step ϋ) (0.16g) in MeOH (1OmL) was added p- toluenesulfonic acid (50mg) and anisole (0.22g). The reaction was then stirred at room temperature for 18h. The reaction was partitioned between EtOAc (10OmL) and H2O (10OmL). The aqueous layer was then further extracted with EtOAc (2xl00mL). Combined organic layers were dried (MgSO4), filtered and evaporated. The residue was purified by reverse phase HPLC using TFA (0.2%)/MeCN to give a white solid. Yield: 9mg.
-64-
MS: APCI(+ve) 499 [M+H+]
1H NMR: δ (CD3OD) 1.12 (d, 3H), 3.47 (s, 3H), 3.50-3.53 (m, 2H), 4.30-4.33 (m, 2H), 5.08- 5.19 (m, IH), 5.95 (s, IH), 6.45 (d, IH), 6.93-7.11 (m, 2H), 7.14-7.21 (m, IH), 7.69-7.74 (m, IH), 8.39 (d, IH)
The intermediates for this compound were prepared as follows:
i) [(l,6-dihydro-l-methyl-6-oxo-3-pyridinyl)sulfonyl]-, 1,1-dimethylethyl ester carbamic acid Chlorosulfonyl isocyanate (6mL) was added dropwise to a solution of 2-methyl-2-proρanol (6.5mL) in DCM (75mL) at 0°C. After 5min, l-methyl-2(lH)-pyridinone (9mL) was added dropwise followed by N,N-diisopropylethylamine (14.5mL) also added dropwise. The reaction mixture was then allowed to warm to room temperature over 18h. H2O (10OmL) was added to the reaction mixture and the organic layer was separated. The aqueous was then further extracted with DCM (2xl00mL). The combined organic layers were dried (MgSO4), filtered and evaporated to give the subtitle compound as a pale yellow oil. Yield: 7g 1H NMR: δ (CDCl3) 1.45 (s, 9H), 3.62 (s, 3H), 6.60-6.64 (m, IH), 1.69-1.1 A (m, IH), 8.21- 8.24 (m, IH)
ii) 4-pyridinesulfonamide-N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(lR)-l-methyl-2- (triphenylmethoxy)ethoxy]-4-pyrimidinyl]
A mixture of [(l,6-dihydro-l-methyl-6-oxo-3-pyridinyl)sulfonyl]-, 1,1-dimethylethyl ester carbamic acid (the product from step i) (0.6Og), tris(dibenzylideneacetone)dipalladium (0) (50 mg), 2-dicyclohexylphosphino-2',4',6'-tri-irøpropyl-l,r-biphenyl (XPHOS) (50mg), cesium carbonate (1 g) and 4- chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-[(lΛ)-l-methyl-2-
(triphenyhnethoxy)ethoxy]-pyrimidine (the product of example 13 step hi), 0.4Og) in dioxane (2OmL) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for 3h. The reaction mixture was diluted with DCM, filtered through arbocel and the filtrate evaporated. The residue was purified by reverse phase HPLC using a TFA (0.2%)/MeCN system to give the subtitle compound as a yellow solid. Yield: 0.12g. MS: APCI(+ve) 741 [M+H+]
-65-
1H NMR: δ (DMSO) 1.16-1.23 (m, 3H), 3.06 (d, 2H), 3.35 (s, 3H), 4.33-4.41 (m, 2H), 5.39- 5.47 (m, IH), 6.06 (s, IH), 6.47 (d, IH), 7.04-7.11 (m, 2H), 7.17-7.34 (m, 17H), 7.67-7.71 (m, IH), 8.54-8.56 (m, IH)
Example 23
2-[[6-[(l-azetidinylsulfonyl)amino]-2-[[(2,3-dinuorophenyl)methyl]thio]-4- pyrimidinyl]oxy]-(2J?)-propanoic acid ethyl ester
The title compound was prepared according to the procedure outlined in example 1 step (iv) using a mixture of azetidine- 1- sulfonamide (prepared according to patent WO 2004/011443,
0.6Ig), tris(dibenzylideneacetone)dipalladium (0) (0.15g), 2-dicyclohexylphosphino-2',4',6'- trwΛ?propyl-l,l'-biphenyl (XPHOS) (105mg), cesium carbonate (OJIg), 2-[[6-chloro-2-
[[(2,3-difluorophenyl)methyl]thio]-4-pyrimidinyl]oxy]-(2/?)-propanoic acid ethyl ester (the product of example 11, step i) (0.6Ig) and dioxane (15mL). Purification was by column chromatography on silica gel using MeOH/DCM (5:95) as eluent, followed by reverse phase
HPLC (symmetry as the stationary phase and TFA/acetonitrile as the mobile phase) to give the title compound as a white solid. Yield: 46mg
MS: APCI(+ve) 489 [M+H+]
1H NMR: δ (DMSO) 1.12 (t, 3H), 1.49 (d, 3H), 2.14 (quintet, 2H), 3.92 (t, 4H), 4.05 - 4.16 (m, 2H), 4.42 (dd, 2H), 5.26 (q, IH), 6.21 (s, IH), 7.12 - 7.21 (m, IH), 7.31 - 7.41 (m, 2H),
11.24 (s, IH)
-66-
Example 24
N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(lR)-2-hydroxy-l-methylethoxy]-4- pyrimidinyl]- 1-azetidinesulfonamide
To a suspension of 2-[[6-[( l-azetidinylsulfonyl)amino]-2-[[(2,3-difluorophenyl)methyl]thio]- 4-pyrimidinyi]oxy]-(2/?)-propanoic acid ethyl ester, (the product of example 23) (0.4Og) in THF (1OmL) was added 2M lithium borohydride in THF (0.82mL) dropwise and the mixture was stirred at ambient temperature for 2Oh. The reaction mixture was cooled to O
0C and quenched with IM aqueous hydrochloric acid. The resulting mixture was extracted with EtOAc (x2). The combined organic layers were washed with IM aqueous hydrochloric acid then brine and was dried (MgSO
4), filtered and evaporated. The residue was purified by column chromatography on silica gel using EtOAc/i.røhexane (1:1) as eluent. The resulting oil was triturated with DCM to give the title compound as a white solid. Yield: 0.25g MS: APCI(-ve) 445 [M-H ]
1H NMR δ (CD3OD) 1.15 (d, 3H), 2.11 (quintet, 2H), 3.54 (d, 2H), 3.88 (t, 4H), 4.36 (dd, 2H), 5.16 (dt, IH), 6.12 (s, IH), 6.93 - 7.12 (m, 2H), 7.22 - 7.31 (m, IH)
Example 25
N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(15)-2-hydτoxy-l-methylethoxy]-4- py riniidinyl] - 1-azetidinesulfonamide
The title compound was prepared according to the procedure outlined in example 24 using 2- [[6-[(l-azetidinylsulfonyl)arruno]-2-[[(2,3-difluorophenyl)methyl]tMo]-4-pyrimidinyl]oxy]- (2S)-propanoic acid,ethyl ester, (the product of step ii) (0.28g), THF (8mL) and 2M lithium borohydride in THF (0.57mL). Purification was by column chromatography on silica using EtOAc/z\røhexane (2:3) as eluent to give the title compound as a white solid. Yield: 0.15g MS: APCI(-ve) 445 [M-K]
1H NMR: δ (CD3OD) 1.27 (d, 3H), 2.23 (quintet, 2H), 3.66 (d, 2H), 4.00 (t, 4H), 4.48 (dd, 2H), 5.28 (q, IH), 6.24 (s, IH), 7.05 - 7.23 (m, 2H), 7.33 - 7.43 (m, IH)
The intermediates for this compound were prepared as follows:
i) 2-[[6-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-4-pyrimidinyl]oxy]-(2S)-propanoic acid ethyl ester
The subtitle compound was prepared according to the procedure outlined in example 1 step iii) using 4,6-Dichloro-2-[(2, 3-difluorobenzyl)thio]pyrimidine (product of example 1 step ii)
(0.77g), THF (15mL), 2-hydroxy-(2S)-propanoic acid ethyl ester (0.4OmL) and 60% sodium hydride (0.14g) to give the subtitle compound as a clear, colourless oil. Yield: Ig
MS: APCI(+ve) 389/391 [M+H+]
1H NMR: δ (DMSO) 1.13 (t, 3H), 1.51 (d, 3H), 3.99 - 4.17 (m, 2H), 4.37 - 4.50 (m, 2H), 5.28 - 5.38 (m, IH), 7.02 (s, IH), 7.13 - 7.23 (m, IH), 7.28 - 7.42 (m, 2H)
ii) 2-[[6-[(l-azetidinylsulfonyl)amino]-2-[[(2,3-difluorophenyl)methyl]thio]-4- pyrimidinyl]oxy]-(2S)- propanoic acid ethyl ester
The subtitle compound was prepared according to the procedure outlined in example 1 step (iv) using a mixture of azetidine-1 -sulfonamide (prepared according to patent WO 2004/011443, 0.13g), tris(dibenzylideneacetone)dipalladium (0) (58mg), 2- dicyclohexylphosphino-2',4',6'-tri-irøpropyl-l,r-biphenyl (XPHOS) (42mg), cesium carbonate (0.3 Ig), 2-[[6-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-4-pyrimidinyl]oxy]- (25)-propanoic acid ethyl ester (product of step i) (0.25g) and dioxane (1OmL). Purification was by column chromatography on silica gel using EtOAc/isohexane (3:7) as eluent to give the subtitle compound as clear, colourless oil. Yield: 0.28g MS: APCI(+ve) 489 [M+H+]
-68-
Example 26
2-[[6-[(l-azetidinylsulfonyl)amino]-2-[[(2,3-difluorophenyl)methyl]thio]-4- pyrimidinyl]oxy]-(2/?)- propanamide
To a solution of 2-[[6-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-4-pyrimidinyl]oxy]-(27?)- propanoic acid ethyl ester (the product of example 23) (103mg) in methanol (8mL) ammonia gas was bubbled through at 0°C. The resulting mixture was stirred in a sealed tube at ambient temperature for 48h. The solvent was evapourated under reduced pressure and the resulting solid was triturated with ether to give the title compound was a white solid. Yield: 88mg MS: APCI(+ve) 460 [M+H+]
1H NMR: δ (DMSO) 1.43 (d, 3H), 2.13 (quintet, 2H), 3.91 (t, 4H), 4.45 (dd, 2H), 5.21 (q, IH), 6.23 (s, IH), 7.13 - 7.20 (m, 2H), 7.31 - 7.43 (m, 2H), 7.59 (s, IH), 11.17 (s, IH)
Example 27
2-[[6-[(l-azetidinylsulfonyl)amino]-2-[[(2,3-difluorophenyl)methyl]thio]-4- pyrimidinyl]oxy]-N-methyl-(2/?)-propanainide
To a solution of 2-[[6-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-4-pyrimidinyl]oxy]-(2/?)- propanoic acid ethyl ester (the product of example 23) (lOOmg) in ethanol (1.5mL) was added
-69-
8M methylamine in ethanol. The resulting mixture was stirred in a sealed tube at ambient temperature for 16b_ The solvent was evaporated under reduced pressure. Purification was by reverse phase HPLC (symmetry as the stationary phase and TFA/acetonitrile as the mobile phase) to give the title compound as a white solid. Yield: 60mg MS: APCI(+ve) 474 [M+H+]
1H NMR: δ (DMSO) 1.41 (d, 3H), 2.14 (quintet, 2H), 2.57 (d, 3H), 3.92 (t, 4H), 4.43 (dd, 2H), 5.26 (q, IH), 6.23 (s, IH), 7.12 - 7.21 (m, IH), 7.30 - 7.41 (m, 2H), 8.00 - 8.07 (m, IH), 11.18 (s, IH)
Example 28
2-[[6-[(l-azetidinylsulfonyl)amino]-2-[[(2,3-difluorophenyl)methyl]tliio]-4- pyrimidinyl]oxy]-(2/?)- propanoic acid
To a solution of 2-[[6-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-4-pyrimidinyl]oxy]-(2/?)- propanoic acid ethyl ester (the product of example 23) (0.24g) in methanol (ImL) was added IM aqueous sodium hydroxide (ImL). The resulting mixture was stirred at ambient temperature for 16h. The reaction mixture was acidified using 2M aqueous HCl, then extracted with EtOAc (x2). The combined organics were washed with brine then dried (MgSO4), filtered and evaporated. The resulting oil was triturated with DCM//rø-hexane to give the title compound was a white solid. Yield: 0.2Og MS: APCI(-ve) 459 [M-H ]
1H NMR: δ (DMSO) 1.49 (d, 3H), 2.13 (quintet, 2H), 3.91 (t, 4H), 4.43 (dd, 2H), 5.23 (q, IH), 6.19 (s, IH), 7.12 - 7.21 (m, IH), 7.30 - 7.42 (m, 2H)
■70-
Example 29 iV-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(lΛ)-2-hydroxy-l-methylethoxy]-4- pyrimidinyl] -methanesulfonamide
The title compound was prepared according to the procedure outlined in example 24 using a mixture of 2-[[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(methylsuh
conyl)amήio]-4- pyrimidinyl]oxy]-(2/?)-propanoic acid ethyl ester, (the product of step i) (0.28g), THF (8mL) and 2M lithium borohydride in THF (1.3mL). Purification was by reverse phase HPLC (symmetry as the stationary phase and TFA/acetonitrile as the mobile phase). The resulting oil was triturated with toluene, DCM, then ether/iw-hexane to give the title compound as a white solid. Yield: 0.18g MS: APCI(-ve) 440 [M-H ]
1H NMR: δ (DMSO) 1.17 (d, 3H), 3.29 (s, 3H), 3.47 - 3.50 (m, 3H), 4.47 (dd, 2H), 5.09 - 5.18 (m, IH), 5.99 (s, IH), 7.13 - 7.21 (m, IH), 7.29 - 7.43 (m, 2H), 11.14 (s, IH)
The intermediate for this compound was prepared as follows:
i) 2-[[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(methyIsuIfony])amino]-4- pyrimidinyl]oxy]- (2R) -propanoic acid ethyl ester The subtitle compound was prepared according to the procedure outlined in example 1 step iv) using a mixture of methanesulfonamide (93mg), tris(dibenzylideneacetone)dipalladium (0) (71 mg), 2-dicyclohexylphosρhino-2',4',6'-tri-w<?propyl-l,l'-biphenyl (XPHOS) (52mg), cesium carbonate (O.38g), 2-[[6-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-4- pyrimidinyl]oxy]-(2/?)-propanoic acid ethyl ester (the product of Example 23 step i) (0.3Og) and dioxane (1OmL). Purification was by column chromatography on silica using EtOAc/wohexane (1:1) as eluent to give the subtitle compound as an oil. Yield: 0.28g
-71-
MS: APCI(+ve)448 [M+H+]
Example 30
N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(lR)-2-hydroxy-l-methylethoxy]-4- pyrimidinyl]- 4-morpholinesulfonamide
The title compound was prepared according to the procedure outlined in example 24 using a mixture of 2-[[2-[[(2,3-dMuorophenyl)methyl]tMo]-6-[(4-moφhoh^ylsulfonyl)amino]-4- pyrimidinyl]oxy]-(22?)-propanoic acid ethyl ester, (the product of step i) (0.34g), THF (8mL) and 2M lithium borohydride in THF (ImL). Purification was by reverse phase HPLC
(symmetry as the stationary phase and ammonium acetate/acetonitrile as the mobile phase) to give the title compound as a white solid. Yield: 0.25g
MS: APCI(-ve) 475 [M-H ]
1H NMR: δ (DMSO) 1.16 (d, 3H), 3.11 (s, 4H), 3.42 - 3.53 (m, 2H), 3.59 (t, 4H), 4.43 (dd, 2H), 4.84 (t, IH), 5.10 (q, IH), 5.98 (s, IH), 7.12 - 7.19 (m, IH), 7.29 - 7.37 (m, IH), 7.39 -
7.45 (m, IH)
The intermediate for this compound was prepared as follows:
i) 2-[[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(4-morphoIinylsulfonyl)amino]-4- pyrimidinyl]oxy]-(2R)-propanoic acid ethyl ester
The subtitle compound was prepared according to the procedure outlined in example 1 step iv) using a mixture of 4-morρholinesulfonamide (prepared according to patent WO 2004/011443, 0.19g), tris(dibenzylideneacetone)dipalladium (0) (71 mg), 2- dicyclohexylphosphino-2',4',6'-tri-i5opropyl-l,r-biphenyl (XPHOS) (52mg), cesium carbonate (0.38g), 2-[[6-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-4-pyrimidinyl]oxy]-
-72-
(2/?)-propanoic acid ethyl ester (the product of Example 23 step i) (0.3Og) and dioxane (1OmL). Purification was by column chromatography on silica gel using EtOAc/isohexane (1: 1) as eluent to give the subtitle compound as an oil. Yield: 0.34g MS: APCI(+ve) 519 [M+H+]
Example 31
N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(lΛ)-2-hydroxy-l-methylethoxy]-4- py rimidinyl] - 1 - py rrolidinesulf onamide
The title compound was prepared according to the procedure outlined in example 24 using a mixture of 2-[[2-[[(2,3-difluorophenyl)methyl]tMo]-6-[(l-pyrrolidinylsulfonyl)amino]-4- ρyrirnidinyl]oxy]-(2/?)-propanoic acid ethyl ester, (the product of step i) (O.38g), THF (8mL) and 2M lithium borohydride in THF (1.3mL). Purification was by reverse phase HPLC (symmetry as the stationary phase and TFA/acetonitrile as the mobile phase). The resulting oil was titurated with methanol, toluene, DCM, then ether/wo-hexane to give the title compound as a white solid. Yield: 0.15g MS: APCI(-ve) 459 [M-H
']
1H NMR: δ (DMSO) 1.16 (d, 3H), 1.75 - 1.82 (m, 4H), 3.27 - 3.38 (m, 4H), 3.44 - 3.51 (m, 2H), 4.45 (dd, 2H), 5.10 - 5.18 (m, IH), 5.97 (s, IH), 7.13 - 7.20 (m, IH), 7.29 - 7.42 (m, 2H), 10.91 (s, IH)
The intermediate for this compound was prepared as follows:
i) 2-[[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(l-pyrrolidinylsulfonyl)amino]-4- pyrimidinyl]oxy]-(2/?)-propanoic acid ethyl ester
-73-
The subtitle compound was prepared according to the procedure outlined in example 1, step iv) using a mixture of 1-pyrrolidinesulfonamide (prepared according to patent WO 2004/011443, 0.19g), tris(dibenzylideneacetone)dipalladium (0) (71 mg), 2- dicyclohexylphosphino-2',4',6'-tri-J5Opropyl-l,r-biphenyl (XPHOS) (52mg), cesium carbonate (O.38g), 2-[[6-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-4-pyrirnidinyl]oxy]- (2/?)-propanoic acid ethyl ester (the product of Example 23 step i) (0.3Og) and dioxane (1OmL). Purification was by column chromatography on silica gel using EtOAc/isohexane (1: 1) as eluent to give the subtitle compound as an oil. Yield: O.38g MS: APCI(+ve) 475 [M+H+]
Example 32 iV-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(l/?)-2-hydroxy-l-methylethoxy]-4- pyrimidinyl]-cyclopropanesulfonamide
The title compound was prepared according to the procedure outlined in example 24 using 2- [[6- [(cyclopropylsulfony i)arnino] -2- [ [(2,3-difluorophenyl)methyl] thio] -4-pyrimidinyl] oxy] - (2Λ)-propanoic acid ethyl ester, (the product of step i) (0.3Og), THF (8mL) and 2M lithium borohydride in THF (2mL). Purification was by reverse phase HPLC (symmetry as the stationary phase and TFA /acetonitrile as the mobile phase). The resulting oil was triturated with methanol, toluene, DCM, then ether/irø-hexane to give the title compound as a white solid. Yield: 0.2Og MS: APCI(-ve) 430 [M-H ]
1B. NMR: δ (DMSO) 1.00 - 1.10 (m, 4H), 1.17 (d, 3H), 2.93 - 3.04 (m, IH), 3.47 - 3.50 (m, 2H), 4.47 (s, 2H), 5.08 - 5.20 (m, IH), 6.06 (s, IH), 7.11 - 7.21 (m, IH), 7.28 - 7.45 (m, 2H), 11.10 (s, IH)
-74- The intermediate for this compound was prepared as follows:
i) 2-[[6-[(cyclopropylsulfonyl)amino]-2-[[(2,3-difluorophenyl)methyl]thio]-4- pyrimidinyl]oxy]-(2R)-propanoic acid ethyl ester The subtitle compound was prepared according to the procedure outlined in example 1, step iv) using a mixture of cyclopropanesulfonamide (prepared according to patent WO 2003/099274, 0.14g), tris(dibenzylideneacetone)dipalladium (0) (71 mg), 2- dicyclohexylphosphino-2',4',6'-tri-J5Opropyl-l,r-biphenyl (XPHOS) (52mg), cesium carbonate (O.38g), 2-[[6-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-4-pyrimidinyl]oxy]- (2/?)-propanoic acid ethyl ester (the product of Example 23 step i) (0.3Og) and dioxane (1OmL). Purification was by column chromatography on silica gel using EtOAc/isohexane (1: 1) as eluent to give the subtitle compound as an oil. Yield: 0.3Og MS: APCI(+ve) 503 [M+H+]
Example 33
N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(l/?)-2-hydroxy-l-methyletlioxy]-4- pyrimidinyl] - 1 -methyl- lH-imidazole-4-sulfonamide
The title compound was prepared according to the procedure outlined in example 24 using 2- [[2-[[(2,3-difluorophenyl)methyl]tWo]-6-[[(l-methyl-lH-imidazol-4-yl)sulfonyl]amino]-4- pyrimidinyl]oxy]-(2/?)-propanoic acid ethyl ester (the product of step i) (0.28g), TΗF (8mL) and 2M lithium borohydride in TΗF (0.8ImL). Purification was by reverse phase ΗPLC (symmetry as the stationary phase and TFA/acetonitrile as the mobile phase). The resulting oil was triturated with toluene, methanol, then ether/irø-hexane to give the title compound as a white solid. Yield: 0.12g MS: APCI(-ve) 470 [M-H ]
-75-
1H NMR: δ (DMSO) 1.14 (d, 3H), 3.46 (m, 2H), 3.67 (s, 3H), 4.39 (t, 2H), 5.01 - 5.14 (m, IH), 6.17 (s, IH), 7.09 - 7.19 (m, IH), 7.27 - 7.42 (m, 2H), 7.80 (s, IH), 8.01 (s, IH), 11.55 (s, IH)
The intermediate for this compound was prepared as follows:
i) 2-[[2-[[(2,3-difluorophenyl)methyl]thio]-6-[[(l-methyl-lH-imidazol-4- yl)sulfonyl]amino]-4-pyrimidinyl]oxy]- (2R) -propanoic acid ethyl ester
The subtitle compound was prepared according to the procedure outlined in example 1, step iv) using a mixture of l-methyl-lH-imidazole-4-sulfonamide (0.19g), tris(dibenzylideneacetone)dipalladium (0) (71 mg), 2-dicyclohexylphosphino-2',4',6'-tri- i\røproρyl-l,l'-biphenyl (XPΗOS) (52mg), cesium carbonate (0.38g), 2-[[6-chloro-2-[[(2,3- difluorophenyl)methyl]tiiio]-4-pyrirnidinyl]oxy]-(2/?)-propanoic acid ethyl ester (the product of Example 23 step i) (0.30g) and dioxane (1OmL). Purification was by column chromatography on silica gel using EtOAc/isohexane (1: 1) as eluent to give the subtitle compound as an oil. Yield: 0.28g MS: APCI(+ve) 514 [M+Η+]
Example 34 iV-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(15)-2-ethoxy-l-(hydroxymethyl)ethoxy]-4- pyrimidinyl] • 1 -azetidinesulfonamide
To a solution of N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(lR)-2-[[(l,l- dimethylethyl)dimethylsilyl]oxy] - 1 -(ethoxymethyl)ethoxy] -4-pyrimidinyl] - 1 - azetidinesulfonamide, (product from step v) (0.79g) in THF (1OmL) was added a IM solution of tetrabutylammoniumfluoride in THF (2.4mL) with stirring, at ambient temperature, for
-76-
72h. The reaction mixture was diluted with H2O and extracted with EtOAc (x2). The organic layer was washed with H2O then brine and dried (MgSO4), filtered and evaporated. The resulting oil was purified by reverse phase HPLC (symmetry as the stationary phase and TFA/acetortitrile as the mobile phase) then titurated with DCM followed by ether/tJO-hexane to give the title compound as a white solid. Yield: O.28g MS: APCI(-ve) 489 [M-H ]
1H NMR: δ (DMSO) 1.06 (t, 3H), 2.13 (quintet, 2H), 3.36 - 3.46 (m, 2H), 3.54 - 3.59 (m, 4H), 3.91 (t, 4H), 4.46 (dd, 2H), 4.88 (t, IH), 5.25 (quintet, IH), 6.12 (s, IH), 7.12 - 7.19 (m, IH), 7.30 - 7.38 (m, IH), 7.40 - 7.45 (m, IH), 11.14 (s, IH)
The intermediates for this compound were prepared as follows:
i) (4R)- 4-(ethoxymethyl)-2,2-dimethyl-l,3-dioxolane
To a solution of 2,2-dimethyl-l, 3-dioxolane-4-methanol (1.5g), in dimethylfomylamide (3OmL), 60% sodium hydride (0.5Og) was added portion-wise at O0C then warmed to ambient temperature. Iodoethane (3.5mL) was added to the mixture at O0C then stirred for 16h at room temperature. The reaction mixture was filtered then the filtrate was diluted with H2O and extracted with EtOAc. The organic layer was washed with H2O (x2) then brine and dried (MgSO4), filtered and evaporated. Purification was by column chromatography on silica gel using EtOAc/Et2O (1:1) as eluent to give the subtitle compound as clear, colourless oil. Yield:
1H NMR: δ (DMSO) 1.10 (t, 3H), 1.27 (s, 3H), 1.32 (s, 3H), 3.32 - 3.50 (m, 4H), 3.54 - 3.62 (m, IH), 3.93 - 4.01 (m, IH), 4.11 - 4.20 (m, IH)
ϋ) (2S)-3-ethoxy-l,2-propanediol
A solution of (4R)- 4-(ethoxymethyl)-2,2-dimethyl-l,3-dioxolane (product from step i) (Ig) in 80% glacial acetic acid (3OmL) was stirred at ambient temperature for 48h. The solvent was evaporated, azeotroped with methanol, ethanol and toluene then redissolved in DCM, dried (MgSO4), filtered and evaporated to give the subtitle compound as a yellow oil. Yield: 0.55g. 1H NMR: δ (DMSO) 1.10 (t, 3H), 3.22 - 3.44 (m, 6H), 3.54 (quintet, IH), 4.45 (t, IH), 4.58 (d, IH)
-77- iϋ) (2/2)- l-[[(l,l-dimethylethyl)dimethylsilyl]oxy]-3-ethoxy-2-propanol
To a solution of (2S)-3-ethoxy-l,2-propanediol (product from step ϋ) (0.5Og) in DCM (3OmL) was added tert-butyldimethylsilyl chloride (O.88g), triethylamine (0.43mL) and 4- (dimethylamino)pyridine (31mg) at 0°C. The solution was then warmed to ambient temperature and stirred for 16h. The reaction mixture diluted with H2O and extracted with EtOAc. The organic layer was washed with brine and evaporated. Purification was by column chromatography on silica gel using EtOAc/irøhexane (2:8) as eluent to give the subtitle compound as an oil. Yield: 0.69g 1H NMR: δ (DMSO) 0.07 (s, 6H), 0.90 (s, 9H), 1.14 (t, 3H), 3.28 - 3.65 (m, 7H), 4.70 (d, IH)
iv) 4-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-[(lR)-2-[[(l,l- dimethylethyl)dimethylsilyl]oxy]-l-(ethoxymethyl)ethoxy]-pyrimidine
The subtitle compound was prepared according to the procedure outlined in example 1 step iv) using 4,6-Dichloro-2-[(2, 3-difluorobenzyl)thio]pyrimidine (product of example 1 step ii) (0.43g), (2/?)-l-[[(l,l-dimethylethyl)dimethylsilyl]oxy]-3-ethoxy-2-propanol (product of step iii) (0.47 g), THF (2OmL) and 60% sodium hydride (67mg), to give the subtitle compound as a colourless oil. Yield: 0.7g MS: APCI(+ve) 505/507 [M+H+] 1B NMR: δ (DMSO) 0.03 (s, 6H), 0.80 (s, 9H), 1.09 (t, 3H), 3.39 - 3.50 (m, 2H), 3.60 (d, 2H), 3.75 - 3.81 (m, 2H), 4.49 (s, 2H), 5.35 - 5.44 (m, IH), 6.90 (s, IH), 7.14 - 7.23 (m, IH), 7.32 - 7.43 (m, 2H)
v) 7V-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(lR)-2-[[(l,l- dimethylethyl)dimethyIsilyl]oxy]-l-(cthoxymethyl)ethoxy]-4-pyrimidinyl]-l- azetidinesulfonamide
The subtitle compound was prepared according to the procedure outlined in example 1 step (iv) using a mixture of azetidine-1- sulfonamide (prepared according to patent WO 2004/011443, 0.29g), tris(dibenzylideneacetone)dipalladium (0) (O.13g), 2- dicyclohexylphosphino-2',4',6'-tri-/5θpropyl-l,r-biphenyl (XPHOS) (93mg), cesium carbonate (0.68g), 4-chloro-2-[[(2,3-difluoroρhenyl)methyl]thio]-6-[(l/?)-2-[[(l,l- dimethylethyl)dirnethylsilyl]oxy]-l-(ethoxymethyl)ethoxy]-pyrirnidine (the product of step iv) (0.7Og) and dioxane (15mL). Purification was by column chromatography on silica gel
-78- using EtOAc/isohexane (3:7) 70:30 as eluent, to give the title compound as a white solid. Yield: 0.22g
MS: APCI(+ve) 605 [M+H+]
1R NMR: δ (DMSO) 0.02 (s, 6H), 0.84 (s, 9H), 1.04 - 1.11 (m, 3H), 2.08 - 2.18 (m, 2H), 3.53 - 3.59 (m, 2H), 3.72 - 3.77 (m, 2H), 3.86 - 3.94 (m, 4H), 3.99 - 4.07 (m, 2H), 4.49 (s, 2H), 5.34 (s, IH), 6.14 (s, IH), 7.10 - 7.20 (m, IH), 7.29 - 7.45 (m, 2H), 11.17 (s, IH)
Example 35
N-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-methoxypyriinidin-4-yl]azetidine-l- sulfonamide
The title compound was prepared according to the procedure outlined in example 1, step iv). A mixture of azetidine-1-sulfonamide (prepared according to patent WO 2004/011443, 0.15 g), tris(dibenzylideneacetone)dipalladium (0) (44mg), 2-dicyclohexylphosρhino-2'4'6'-tri- /sø-propy 1-1,1 'biphenyl (44mg), cesium carbonate (0.36g) and 4-chloro-2-[[(2,3- difluorophenyl)methyl]thio]-6-methoxypyrimidine (the product of step i) (0.23g) in dioxane (7.2mL). Acetic acid (0.67mL) was added and the reaction mixture was extracted with EtOAc (x3). The combined organic layers were washed with H2O, dried (MgSO4), filtered and the solvent evaporated under reduced pressure. The residue was purified by flash column chromatography on silica gel using EtOAc/isohexane (3:7) as eluent. The resulting solid was further purified by trituration with iso-hexane and dried under high vacuum at 4O0C to give the title compound as a pale yellow solid. Yield: 0.29 g. MS: APCI(+ve) 403 [M+H] 1H NMR: δ (DMSO) 2.12 (m, 2H), 3.9 (m, 7H), 4.49 (s, 2H), 6.15 (s, IH), 7.16 (m, IH), 7.39 (m, 2H), 11.12 (s, IH).
The intermediate for this compound was prepared as follows:
-79-
i) 4-Chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-methoxypyrimidine
To a stirred solution of 4,6-Dichloro-2-[(2,3-difluorobenzyl)thio]pyrimidine (the product of Example 1, step ϋ) (5g) in dry methanol (4OmL) was added 60% sodium hydride (0.68g) batchwise over 5min. The reaction mixture was stirred for 5h, H2O added and the solvents were partially evaporated. The residue was extracted with EtOAc which was washed with H2O, dried (MgSO4) and the solvent evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel, eluting with Et2O /iso-hexane (5:95) to give the subtitle compound as a white solid. Yield: 4.05g. MS: APCI(+ve) 303/305 [M+H]
Example 36
N-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-methoxypyrimidin-4-yl]piperazine-l- sulfonamide, trifluoroacetate salt
1 , 1 -Dimethylethyl 4-[2-[[(2,3-difluorophenyl)methyl]thio]-6-methoxypyrimidin-4-yl sulfarnoyl]piperazine-l-carboxylate (the product of step i) (0.36g) and trifluoroacetic acid
(ImL) in dichloromethane (4mL) were stirred at room temperature for 30min. The solvent was evaporated under reduced pressure and the residue was azeotroped with toluene (3x). The residual pale yellow solid was triturated with EtOAc, filtered and dried at 400C under high vacuum to give the title compound as a cream solid. Yield: 0.24g.
MS: APCI(+ve) 432 [M+H]
1H NMR: δ (DMSO) 3.17 (m, 4H), 3.40 (m, 4H), 3.90 (s, 3H), 4.49 (s, 2H), 6.08 (s, IH), 7.18
(m, IH), 7.38 (m, 2H).
The intermediates for this compound were prepared as follows:
-80- i) 1,1 -Dimethylethyl 4- [2- [ [(2,3-difluorophenyl)methyl] thio] -6-methoxy pyrimidin-4- yl sulfamoyl]piperazine-l-carboxylate.
The subtitle compound was prepared from 1,1 -dimethylethyl 4-sulfamoylpiperazine-l- carboxylate (the product of example 15, step i), 0.22g) and 4-Chloro-2-[[(2,3- difluoroρhenyl)methyl]thio]-6-methoxypyrimidine (the product of Example 35, step i) (0.25g) according to the procedure outlined in Example 1, step iv). The crude material was purified by column chromatography on silica gel using EtOAc/jrøhexane (2:8) as eluent. Yield: 0.36g MS: APCI(-ve) 530 [M-H] 1H NMR: δ (CDCl3) 1.45 (s, 9H), 3.27 (t, 4H), 3.48 (t, 4H), 3.94 (s, 3H), 4.40 (s, 2H), 6.23 (s, IH), 7.04 (m, 2H), 7.22 (m, IH).
Example 37
4-Acetyl-N-[2-[[(2,3-difluorophenyl)methy]thio]-6-methoxypyrimidin-4-yl]piperazine-l- sulfonamide
Acetic anhydride (0.78mL) was added to a mixture of N-[2-[[(2,3-
Difluorophenyl)methyl]tMo]-6-methoxypyrirnidin-4-yl]piρerazine-l-sulfonamide, trifluoro acetate salt (the title product of Example 36, 0.84g) and N,N-diisopropylethylamine (ImL) in DCM (5mL). The reaction mixture was stirred at room temperature for 30min and the solvent evaporated under reduced pressure. The residue was dissolved in EtOAc which was washed with aqueous citric acid, H2O, dried (MgSO4) and the solvent evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel, eluting with EtOAc to give the title compound as a white solid. Yield: 78mg. MS: APCI(+ve) 474 [M+H]
-81-
1H NMR: δ (DMSO) 1.98 (s, 3H), 3.20 (m, 4H), 3.87 (s, 3H), 3.32 (br d, 4H), 4.48 (s, 2H), 6.07 (s, IH), 7.17 (m, IH), 7.33 (m, IH), 7.42 (t, IH), 11.18 (s, IH).
Example 38 N-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-methoxypyrimidin-4-yl]morpholine-4- sulfonamide
The title compound was prepared from morpholine-4-sulfonamide (prepared according to patent WO 2004/011443, 0.2Og) and 4-Chloro-2-[[(2,3-drfluorophenyl)metliyl]thio]-6- methoxypyrimidine (the product of Example 35, step i) (0.25g) according to the procedure outlined in Example 1, step iv). The crude material was purified by column chromatography using EtOAc/isohexane (2:8) as eluent. Yield: 0.26g.
MS: APCI(+ve) 433 [M+H]
1H NMR: δ (DMSO) 3.17 (t, 4H), 3.59 (t, 4H), 3.88 (s, 3H), 4.48 (s, 2H), 6.09 (s, IH), 7.17 (m, IH), 7.34 (m, IH), 7.43 (t, IH), 11.17 (s, IH).
Example 39 N-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-methoxypyrimidin-4-yl]methane-sulfonamide
The title compound was prepared from methane sulfonamide (0.1 Ig) and 4-Chloro-2-[[(2,3- difluorophenyl)methyl]thio]-6-methoxypyrimidine (the product of Example 35, step i) (0.25g) according to the procedure outlined in Example 1, step iv). The crude material was purified by column chromatography using EtOAc/isohexane (2:8) as eluent. Yield: 0.12g.
-82-
MS: APCI(+ve) 362 [M+H]
1H NMR: δ (DMSO) 3.28 (s, 3H), 3.87 (s, 3H), 4.49 (s, 2H), 6.03 (s, IH), 7.17 (m, IH), 7.37
(m, 2H), 11.14 (s, IH).
Example 40
N-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-methoxypyrimidm-4-yl]-l-methyl -lH- imidazole-4-sulfonamide
The title compound was prepared from 1 -methyl- 1 H- imidazole-4-sulfonamide (0.19g) and 4- Chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-methoxyρyrimidine (the product of Example
35, step i) (0.25g) according to the procedure outlined in Example 1, step iv). The crude material was purified by column chromatography using EtOAc/isøhexane (2:8) as eluent.
Yield: 0.1 Ig.
MS: APCI(+ve) 428 [M+H] 1H NMR: δ (DMSO) 3.67 (s, 3H), 3.83 (s, 3H), 4.41 (s, 2H), 6.20 (s, IH), 7.15 (m, IH), 7.36
(m, 2H), 7.78 (s, IH), 8.00 (s, IH), 11.55 (s, IH)
Example 41
N-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-[(S)-isoxazolidin-4-yl)oxy]pyrimidin-4- yl]azetidine-l-sulfonamide
-83- l,l-Dimethylethyl (S)-4-[6-(azetidine-l-sulfonylaπύno)-2-[[(2,3-difluorophenyl) methyl]thio]ρyrimidin-4-yloxy]isoxazolidine-2-carboxylate (the product of step ii) (0.14g) and trifluoroacetic acid (ImL) in dichloromethane (2mL) were stirred at room temperature for 30min. The solvent was evaporated under reduced pressure and the residue was azeotroped with toluene (3x). The residue was purified by reverse phase HPLC eluting with acetonitrile / 0.1% aqueous ammonium acetate mixtures to give the title compound as a white solid. Yield: 80mg.
MS: APCI(+ve) 458 [M+H]
1H NMR: δ (DMSO) 7.37 (m, 2H), 7.18 (m, IH), 6.16 (s, IH), 5.65 (bm, IH), 4.49 (s, 2H), 3.91-3.81 (bs+t, 6H), 3.01 (bs, IH), 2.13 (m, 2H).
The intermediates for this compound were prepared as follows:
i) l,l-Dimethylethyl (S)-4-[6-chloro-2-[[(2,3-difluorophenyl)methyl]thio]pyrimidin-4- yloxy ] -isoxazolidine-2-carboxylate.
To a stirred solution of 4,6-Dichloro-2-[(2,3-difluorobenzyl)thio]pyrimidine (the product of Example 1, step ii) (0.25g) and 1,1-dimethylethyl (S)-4-hydroxyisoxazolidine-2-carboxylate (0.16g) in dry THF (5mL) was added 60% Sodium hydride (0.034g) over 5min. The reaction mixture was stirred and heated at 6O0C for 7 days, H2O was added and the solvents were partially evaporated. The residue was extracted with EtOAc which was washed with H2O, dried (MgSO4) and the solvent evaporated under reduced pressure. The residue was purified by flash chromatography on silica gel, eluting with Et2O /irø-hexane (1:9) to give the subtitle compound as a gum. Yield: 0.15g. MS: APCI(+ve) 460/462 [M+H]
ii) l,l-Dimethylethyl (S)-4-[6-(azetidine-l-sulfonylamino)-2-[[(2,3-difluorophenyl) methyl]thio]pyrimidin-4-yloxy]isoxazolidine-2-carboxylate.
The subtitle compound was prepared from azetidine-1- sulfonamide (prepared according to patent WO 2004/011443, 0.22g) and 1,1-Dimethylethyl (S)-4-[6-chloro-2-[[(2,3- difluorophenyl)methyl]tWo]pyrimidin-4-yloxy]-isoxazolidine-2-carboxylate (the product of step i) (0.13g) according to the procedure outlined in Example 1, step iv). The crude material was purified by column chromatography using EtOAc/isohexane (2:8) as eluent. Yield: 0.14g
-84-
MS: APCI(-ve) 558 [M-H]
Example 42
N-[6-((R)-2-amino-l-methylethoxy)-2-[[(2,3-Difluorophenyl)methyl]thio]pyrimidin-4- yl]azetidine-l-sulfonamide
The title compound was prepared from 1,1-dimethylethyl [(R)-2-[6-azetidine-l-sulfonyl aπiino)-2-[[(2,3-dMuorophenyl)methyl]tWo]pyrirnidin-4-yloxy]propyl}carbamate (0.26g) (the product of step ii) according to the procedure outlined in Example 41. The crude material was purified by column chromatography using EtOAc/isohexane (2:8) as eluent. Yield: 0.1 Ig
MS: APCI(+ve) 446 [M+H]
1H NMR: δ (DMSO) 1.19 (d, 3H), 1.97 (m, 2H), 2.99 (m, 2H), 3.58 (t, 4H), 4.38 (q, 2H), 5.15
(s, IH), 5.97 (s, IH), 7.12 (m, IH), 7.30 (m, IH), 7.43 (t, IH), 7.49 (br s, 3H).
The intermediates for this compound were prepared as follows:
i) l,l-Dimethylethyl [(R)-2-[6-chloro-2-[[(2,3-difluorophenyl)methyl]thio]pyrimidin-4- yloxy]propyl]carbamate The subtitle compound was prepared from 1,1-dimethylethyl ((R)-2-hydroxypropyl) carbamate (0.15g) and 4,6-Dichloro-2-[(2,3-difluorobenzyl)thio]pyrimidine (the product of Example 1, step ii) (0.25g) according to the procedure outlined in Example 41, step i) with heating at 450C for 18h. The crude material was purified by column chromatography using EtOAc/irøhexane (2:8) as eluent. Yield: 0.23g. MS: APCI(-ve) 444/446 [M-H]
-85- u) l,l-Dimethylethyl [(R)-2-[6-azetidine-l-sulfonylamino)-2-[[(2,3-diαuorophenyl) methyl]thio]pyrimidin-4-yloxy]propy]carbamate
The subtitle compound was prepared from azetidine-1 -sulfonamide (prepared according to patent WO 2004/011443, 0.1 Ig) and 1,1-Dimethylethyl [(R)-2-[6-chloro-2-[[(2,3- difluorophenyl)methyl]thio]pyrimidin-4-yloxy]propyl]carbamate (the product of step i) (0.2g) according to the procedure outlined in Example 1, step iv). The crude material was purified by column chromatography using EtOAc/isohexane (2:8) as eluent. Yield: 0.14g MS: APCI(-ve) 544 [M-H]
Example 43
N- [(R)-2- [6- [azetidine- 1 -sulfonylamino] -2- [ [ (2,3-difluorophenyl)methyl] thio] pyrimidin- 4-yloxy]propyl]acetamide
To a suspension of N-[6-((R)-2-amino-l-methylethoxy)-2-[[(2,3- Difluorophenyl)methyl]tMo]pyrirrήdin-4-yl]azetidine-l-suhconamide (the title product of Example 42) (0.05g) in dichloromethane (1OmL) was added pyridine (0.02mL), followed by acetic anhydride (0.02mL). The mixture was stirred overnight at room temperature. Pyridine (0.02mL) and acetic anhydride (0.02mL) were added and the reaction mixture was stirred for a further 2h. Pyridine (1.OmL) and acetic anhydride (0.5OmL) were added and the reaction mixture was stirred for a further 2h. The reaction mixture was diluted with dichloromethane, washed with aqueous citric acid, H2O, dried (MgSO4), filtered and the solvent evaporated under reduced pressure. The residue was purified by flash column chromatography on silica gel using 40% EtOAc in /.røhexane as eluent. The isolated product was further purified by reverse phase HPLC eluting with acetonitrile / aq. 0.1% ammonium acetate mixtures to give the title compound as a white solid. Yield: 55mg. MS: APCI(-ve) 486 [M-H]
-86-
1H NMR: δ (DMSO) 1.11 (d, 3H), 1.80 (s, 3H), 1.96 (m, 2H), 3.20 (m, 2H), 3.55 (t, 4H), 4.34 (q, 2H), 5.02 (m, IH), 5.88 (s, IH), 7.12 (m, IH), 7.29 (m, IH), 7.41 (t, IH), 7.99 (t, IH).
Example 44 N-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-[(R,S)-2-dimethylamino-l-methylethoxy] pyrimidin-4-yl]azetidine-l-sulfonaniide
The title compound was prepared from azetidine-1- sulfonamide (prepared according to patent WO 2004/011443, 0.15g) and 2-[[6-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-4- pyrimidinyl]oxy]-Λf,Λf-dimethyl-l-propanamine (the product of step i) (0.29g) according to the procedure outlined in Example 1, step iv). The reaction product was purified by reverse phase HPLC eluting with acetonitrile / aq. 0.1% ammonium acetate mixtures to give the title compound as a pale yellow solid. Yield: 0.30g MS: APCI(+ve) 474 [M+H]
1H NMR: δ (DMSO) 1.17 (d, 3H), 2.07 (m, 2H), 2.24 (s, 6H), 2.44 (m, IH), 2.64 (m, IH), 3.79 (t, 4H), 4.24 (t, 2H), 5.27 (m, IH), 6.00 (s, IH), 7.15 (m, IH), 7.33 (m, IH), 7.42 (t, IH).
The intermediate for this compound was prepared as follows:
i) 2-[[6-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-4-pyrimidinyl]oxy]-N^V-dimethyH- propanamine
The subtitle compound was prepared according to the procedure outlined in Example 41 step i) using l-dimethylamino-2-propanol (80mg), 4,6-Dichloro-2-[(2,3- difluorobenzyl)thio]pyrimidine (the product of Example 1 step ϋ) (0.25g) and 60% sodium
-87- hydride (30mg) in THF (2mL) at room temperature for 2d to give the subtitle compound as a pale yellow gum. Yield: 0.29g. MS: APCI(+ve) 374 [M+H]
Example 45 iV-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-{[(lR,2R)-2,3-dihydroxy-l- methylpropyl]oxy}-4-pyrimidinyl]-l-azetidinesulfonamide
To a solution of N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(l/?)-l-[(4/?)-2,2-dimethyl-l,3- dioxolan-4-yl]ethoxy]-4-pyrimidinyl]- 1-azetidinesulfonamide (the product of step vi) (0.13g) in DCM (5mL) was added iron (III) chloride hexahydrate (0.26g). The reaction mixture was stirred at ambient temperature for 1.5h, then saturated aqueous sodium bicarbonate (ImL) was added. The layers were separated and the aqueous material extracted with DCM (x3) and ethyl acetate (x3). The combined organic extracts were washed with saturated aqueous sodium chloride, dried (MgSO4), filtered and evaporated. The residual pale yellow solid was slowly precipitated from DCM, filtered and the resulting material washed with minimal cold DCM (2 x ImL) to afford the title compound as a white powder. Yield: 64mg. MS: APCI(+ve) 477 [M+H+]
1H NMR: δ (CDCl3) 1.33 (d, 3H), 2.27 (quintet, 2H), 2.55 (d, IH), 3.61 - 3.70 (m, 2H), 3.74 - 3.82 (m, IH), 4.02 (t, 4H), 4.31 - 4.41 (m, 2H), 5.32 (quintet, IH), 6.34 (s, IH), 6.98 - 7.24 (m, 3H).
The intermediates for this compound were prepared as follows:
i) (2S,3R)-3-(Benzyloxy)-2-hydroxybutanoic acid
To a solution of (2S,3R)-2-amino-3-benzyloxy-butyric acid (l.lg) in 2M sulfuric acid (6.3ImL) was added dropwise over 2h a solution of sodium nitrite (0.65g) in water (6mL),
keeping the internal temperature of the reaction below 00C. The reaction mixture was stirred at -5°C for 6h then allowed to warm to room temperature overnight. The mixture was adjusted to pH 4 with 50% aqueous sodium hydroxide then ethyl acetate was added. The mixture was stirred vigorously and acidified to pH 2 with concentrated sulfuric acid. The layers were separated and the aqueous layer extracted with further ethyl acetate (x2). The combined organic extracts were washed with saturated aqueous sodium chloride, dried (MgSO4), filtered and evaporated to give the subtitle compound as a yellow crystalline solid which was used without further purification. Yield: 0.84g. MS: APCI(+ve) 211, [M+H+] 1H NMR: δ (300 MHz, CDCl3) 1.31 (d, 3H), 3.99 - 4.05 (m, IH), 4.16 (d, IH), 4.51 (d, IH), 4.69 (d, IH), 7.22 - 7.38 (m, 5H).
ii) (2R,3R)-3-(Benzyloxy)butane-l,2-diol
To a solution of (2.?,3/?)-3-(benzyloxy)-2-hydroxybutanoic acid (the product of step i) (0.79g) and trimethyl borate (0.67mL) in anhydrous tetrahydrofuran (4mL) at 00C was added dropwise borane-dimethyl sulfide complex (3mL, 2M in tetrahydrofuran). The reaction mixture was stirred at room temperature overnight, then further borane-dimethyl sulfide complex (3mL, 2M in tetrahydrofuran) was added at 00C and the reaction mixture stirred at room temperature for a further 2d. The mixture was cooled to 00C and methanol (1OmL) slowly added. When effervescence had ceased, the volatiles were evaporated, further methanol added and the mixture concentrated again to give the subtitle compound as a yellow oil which was used without further purification. Yield: 0.68g. MS: APCI(+ve) 197, [M+H+]
1H NMR: δ (CDCl3) 1.25 (d, 3H), 2.17 (t, IH), 2.77 (d, IH), 3.52 - 3.77 (m, 4H), 4.43 (d, IH), 4.69 (d, IH), 7.27 - 7.39 (m, 5H).
iϋ) (4Λ)-4-[(l/?)-l-(Benzyloxy)ethyl]-2,2-dimethyl-l,3-dioxolane
A stirred solution of (2/?,3i?)-3-(benzyloxy)butane-l,2-diol (the product of step ii) (O.68g), p- toluene sulfonic acid monohydrate (34mg) and 2,2-dimethoxypropane (0.43mL) in toluene (1OmL) was heated to reflux for 30min, then anhydrous sodium sulfate was added and reflux continued for 2.5h. The reaction mixture was allowed to cool and diluted with EtOAc and saturated aqueous sodium bicarbonate and the layers separated. The organic extract was
-89- washed with brine, dried (MgSO4), filtered and evaporated. The residue was purified by column chromatography on silica gel using EtOAc/isohexane (1:9 to 1: 1 gradient) as eluent to give the subtitle compound as a colourless liquid. Yield: 0.46g.
1H NMR: δ (CDCl3) 1.13 (d, 3H), 1.37 (s, 3H), 1.42 (s, 3H), 3.60 (quintet, IH), 3.71 (dd, IH), 3.99 (dd, IH), 4.15 (quintet, IH), 4.64 (d, IH), 4.67 (d, IH), 7.24 - 7.38 (m, 5H).
iv) (lR)-l-[(4/?)-2,2-Dimethyl-l,3-dioxolan-4-yl]ethanol
Ammonia (c. 5OmL) was condensed at -78°C into a three-necked flask which had been oven- dried overnight, and to it was added a solution of (4#)-4-[(l/?)-l-(benzyloxy)ethyl]-2,2- dimethyl- 1,3-dio xo lane (the product of step iii) (0.39g) in tetrahydrofuran (7.5mL). Sodium was added in small pieces until the reaction mixture was dark blue, then it was allowed to warm to -400C and kept at this temperature for 1.5h, during which time further sodium was added when the blue colour faded. The reaction mixture was quenched with excess solid ammonium chloride and allowed to warm to room temperature. Ether (2OmL) was added followed by water, cautiously (1OmL). The layers were separated and the aqueous layer extracted with further ether (x3). The combined organic extracts were washed with saturated aqueous sodium chloride, dried (MgSO4), filtered and evaporated to give the subtitle compound as a pale yellow liquid which was used without further purification. Yield: 0.24g. 1H NMR: δ (CDCl3) 1.16 (d, 3H), 1.37 (s, 3H), 1.44 (s, 3H), 3.67 - 3.77 (m, 2H), 3.93 (q, IH), 4.00 - 4.05 (m, IH).
v) 4-Chloro-2-[(2,3-difluorobenzyl)thio]-6-{(lΛ)-l-[(4R)-2,2-dimethyl-l,3-dioxolan-4- yl]ethoxy }pyrimidine
To a solution of (l/?)-l-[(4/?)-2,2-dimethyl-l,3-dioxolan-4-yl]ethanol (the product of step iv) (0.24g) in dry THF (1OmL) at 00C was added in portions sodium hydride (91mg as 60% dispersion in mineral oil) followed in portions by 4,6-dichloro-2-[(2,3- difluorobenzyl)thio]pyrimidine (the product of example 1 step ii) (0.50g). The reaction mixture was stirred at room temperature for 48h then quenched with saturated aqueous ammonium chloride (1OmL) and diluted with ethyl acetate. The layers were separated and the aqueous layer extracted with further ethyl acetate. The combined organic extracts were washed with brine, dried (MgSO4), filtered and evaporated. The residue was purified by
-90- column chromatography on silica gel using EtOAc/irøhexane (1:19 to 1:9 gradient) as eluent to give the subtitle compound as a pale yellow solid. Yield: 0.42g. MS: APCI(+ve) 417/419 [M+H+]
1H NMR: δ (CDCl3) 1.24 (d, 3H), 1.36 (s, 3H), 1.40 (s, 3H), 3.74 (dd, IH), 4.03 (dd, IH), 5 4.21 (q, IH), 4.40 (s, 2H), 5.28 (quintet, IH), 6.44 (s, IH), 6.98 - 7.11 (m, 2H), 7.26 - 7.31 (m, IH).
vi) N-[2-[[(2,3-Diαuorophenyl)methyl]thio]-6-[(l/?)-l-[(4R)-2,2-dimethyl-l,3-dioxolan-4- yl] ethoxy ] -4-pyrimidinyl] - 1 -azetidinesulfonamide
10 A mixture of azetidine- 1-sulphonamide (prepared according to patent WO 2004/011443,
0.2Og), tris(dibenzylideneacetone)-dipalladium (0) (33mg), 2-dicyclohexylphosphino-2',4',6'- tri-wopropyl-l,l'-biphenyl (XPHOS) (17mg), cesium carbonate (0.18g) and 4-chloro-2- [[(2,3-difluorophenyl)-methyl]thio]-6-[(2 -phenyl- 1 ,3-dioxan-5-yl)oxy]-pyrimidine (the product of step v) (0.15g) in dioxane (5mL) was heated at reflux in a microwave at 1000C,
15 300W, open vessel with cooling for 25min. Saturated aqueous ammonium chloride was added and the resulting mixture extracted with ethyl acetate. The combined organic extracts were washed with brine, dried (MgSO4), filtered and evaporated. The residue was purified by column chromatography on silica gel using EtOAc/isøhexane (1: 19 to 3:7 gradient) as eluent to give the subtitle compound as a yellow oil. Yield: 0.14g.
20 MS: APCI(+ve) 517 [M+H+]
1H NMR: δ (CDCl3) 1.23 (d, 3H), 1.38 (s, 3H), 1.43 (s, 3H), 2.25 (quintet, 2H), 3.77 (dd, IH), 3.98 - 4.09 (m, 5H), 4.24 (q, IH), 4.37 (s, 2H), 5.30 (quintet, IH), 6.32 (s, IH), 6.98 - 7.11 (m, 2H), 7.20 - 7.26 (m, IH). Example 46
25 iV-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-[[(lR,2/?)-2,3-dihydroxy-l-methylpropyl]oxy]- 4-py rimidinyl] -methanesulfonamide
To a solution of N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(l/?)-l-[(4/?)-2,2-dimethyl-l,3- dioxolan-4-yl]ethoxy]-4-pyrirπidinyl]-methanesulfonarnide (the product of step i) (0.23g) in DCM (5mL) was added iron (III) chloride hexahydrate (0.25g). The reaction mixture was stirred at ambient temperature for 2h then saturated aqueous sodium bicarbonate (2mL) was added. The layers were separated and the aqueous material extracted with DCM (x3) and ethyl acetate (x3). The combined organic extracts were washed with brine, dried (MgSO4), filtered and evaporated. The residual yellow solid was precipitated from 10% DCM in Et2O, filtered and the resulting material washed with minimal Et2O (2 x ImL) to afford the title compound as a white powder. Yield: 24mg. MS: APCI(+ve) 436, [M+H+]
1H NMR: δ (DMSO) 1.19 (d, 3H), 3.29 (s, 3H), 3.36 - 3.40 (m, 2H), 3.46 - 3.53 (m, IH), 4.43 (d, IH), 4.48 (d, IH), 4.54 - 4.57 (m, IH), 4.88 (d, IH), 5.16 - 5.24 (m, IH), 5.98 (s, IH), 7.12
- 7.19 (m, IH), 7.29 - 7.43 (m, 2H), 11.10 (s, IH).
The intermediate for this compound was prepared as follows:
i) N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(l/?)-l-[(4/?)-2,2-dimethyl-l,3-dioxolan-4- yl]ethoxy]-4-pyrimidinyl]-methanesulfonamide
A mixture of methanesulfonamide (0. Hg), tris(dibenzylideneacetone)dipalladium (0) (26rng), 2-dicyclohexylphosphino-2',4',6'-tri-/5opropyl-l,l'-biphenyl (XPHOS) (14mg), cesium carbonate (0.14g) and 4-chloro-2-[[(2,3-difluorophenyl)-methyl]thio]-6-[(2-phenyl-l,3- dioxan-5-yl)oxy]-pyrimidine (the subtitle product of example 46 step v) (0.12g) in dioxane (6mL) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for 15min. Saturated aqueous ammonium chloride was added and the resulting mixture extracted with ethyl acetate. The combined organic extracts were washed with brine, dried (MgSO4), filtered and evaporated. The residue was purified by column chromatography on silica gel using EtOAc/jΛ?hexane (1:19 to 3:7 gradient) as eluent to give the subtitle compound as a yellow oil. Yield: 0.12g. MS: APCI(+ve) 476 [M+H+] 1H NMR (CDCl3) δ 1.23 (d, 3H), 1.37 (s, 3H), 1.42 (s, 3H), 3.22 (s, 3H), 3.76 (dd, IH), 4.04 (dd, IH), 4.23 (q, IH), 4.38 (s, 2H), 5.30 (quintet, IH), 6.23 (s, IH), 6.97 - 7.11 (m, 2H), 7.22
- 7.28 (m, IH).
-92-
Example 47 yV-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-{[(l/?,25)-2,3-dihydroxy-l-methylpropyl]oxy}-
4-pyrimidinyl]-l-azetidinesulfonainide
To a solution of N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(li?)-l-[(45)-2,2-dimethyl-l,3- dioxolan-4-yl]ethoxy]-4-pyrimidniyl]-l-azetidinesulfonamide (the product of step U) (0.13g) in DCM (5mL) was added iron (III) chloride hexahydrate (0.24g). The reaction mixture was stirred at ambient temperature for Ih, then saturated aqueous sodium bicarbonate (1OmL) was added. The layers were separated and the aqueous material extracted with DCM (3 x 1OmL) and EtOAc (3 x 1OmL). The combined organic extracts were washed with saturated sodium chloride, dried (MgSO4), filtered and evaporated. The residual pale yellow solid was slowly precipitated from DCM, filtered and the resulting material washed with minimal cold DCM (2 x ImL) to afford the title compound as a white powder. Yield: 45mg. MS: APCI(+ve) 477 [M+H+]
1H NMR: δ (CDCl3) 1.36 (d, 3H), 2.27 (quintet, 2H), 2.34 (br s, IH), 2.67 (d, IH), 3.59 - 3.65 (m, IH), 3.67 - 3.78 (m, 2H), 4.02 (t, 4H), 4.36 (s, 2H), 5.23 (quintet, IH), 6.31 (s, IH), 7.00 - 7.10 (m, 2H), 7.19 - 7.23 (m, IH).
The intermediates for this compound were prepared as follows:
i) 4-cMoro-2-[(2,3-difluorobenzyl)thio]-6-{(lΛ)-l-[(45)-2,2-dimethyl-l,3-dioxolan-4- yl] ethoxy }pyrimidine
The subtitle compound was prepared according to the procedure outlined in example 1 step (iii) using (l/?)-l-[(45)-2,2-dimethyl-l,3-dioxolan-4-yl]ethanol (prepared according to Liebigs. Ann. Chem. 1987, 7-14) (0.25g) and 4,6-Dichloro-2-[(2,3-difluorobenzyl)thio]pyrirnidine (the product of example 1 step ϋ) (0.53g) in THF (2OmL) and 60% sodium hydride (80mg). Crude
-93- material was purified by column chromatography on silica gel using EtOAc/isohexane (1:3) as eluent to give the subtitle compound as a clear, colourless oil. Yield: 0.37g. MS: APCI(+ve) 417/419 [M+H+]
ϋ) iV-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-[(l/?)-l-[(45)-2,2-dimethyl-l,3-dioxolan-4- yl] ethoxy ] -4-pyrimidinyl] - 1 -azetidinesulfonamide
A mixture of azetidine-1-sulphonamide (prepared according to patent WO 2004/011443,
0.16g), tris(dibenzylideneacetone)-dipalladium (0) (33mg), 2-dicyclohexylphosphino-2',4',6'- tri-i5Opropyl-l,l'-biphenyl (XPHOS) (17mg), cesium carbonate (0.28g) and 4-chloro-2- [[(2,3-difluorophenyl)-methyl]thio]-6-[(2-phenyl-l,3-dioxan-5-yl)oxy]-pyrimidine (the product of step i) (0.25g) in dioxane (1OmL) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for 20min. Saturated ammonium chloride was added and the resulting mixture extracted with EtOAc. The combined organic extracts were washed with saturated aqueous sodium chloride, dried (MgSO4), filtered and evaporated. The residue was purified by column chromatography on silica gel using EtOAc/irøhexane (3:7) as eluent to give the subtitle compound as a yellow oil. Yield: 0.13g. MS: APCI(+ve) 517 [M+H+]
Example 48 N-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-{[(lR,25)-2,3-dihydroxy-l-methylpropyl]oxy}- 4-py rimidiny I] - 1 -piperazinesulfonamide
A solution of tert-butyl 4-{[(2-[(2,3-difluorobenzyl)thio]-6-{(l/?)-l-[(45)-2,2-dimethyl-l,3- dioxolan-4-yl]ethoxy}pyrirmdin-4-yl)amino]suhconyl}piperazine-l-carboxylate (the product of step i) (0.23g) in 10% trifluoro acetic acid/DCM (5mL) was stirred at room temperature for Ih. The mixture was evaporated to dryness in vacuo. The resulting crude oil was purified by
-94- reverse phase HPLC (75% to 5% gradient of 0.1% aqueous ammonium acetate in acetonitrile as eluent) to give the title compound as a white solid. Yield: 40mg. MS: APCl(+ve) 506 [M+H+]
1H NMR δ (DMSO) 1.14 (d, 3H), 2.99 - 3.05 (m, 4H), 3.11 - 3.17 (m, 4H), 3.25 - 3.40 (m, 2H), 3.54 - 3.61 (m, IH), 4.34 (d, IH), 4.41 (d, IH), 4.54 (br s,lH), 4.81 (d, IH), 5.03 (dq, IH), 5.82 (s, IH), 7.09 - 7.16 (m, IH), 7.26 - 7.35 (m, IH), 7.43 (dd, IH)
The intermediate for this compound was prepared as follows:
i) tert-butyl 4-{[(2-[(2,3-dinuorobenzyl)thio]-6-{(lR)-l-[(4S)-2,2-dimethyl-l,3-dioxolan- 4-yl]ethoxy}pyrimidin-4-yl)amino]sulfonyl}piperazine-l-carboxylate
The subtitle compound was prepared from 1,1-dimethylethyl 4-sulfamoylpiperazine-l- carboxylate (the product of example 15, step i), 0.26g) and 4-chloro-2-[(2,3- difluorobenzyl)tMo]-6-{(l/?)-l-[(45)-2,2-dimethyl-l,3-dioxolan-4-yl]ethoxy}pyrirnidine (the product of Example 47, step i) (0.2Ig) according to the procedure outlined in Example 1, step iv). Yield: 0.28g MS: APCI(-ve) 644 [M-H]
Example 49 5-(azetidin-l-ylcarbonyl)-N-{2-[(2,3-difluorobenzyl)thio]-6-[(lR)-2-hydroxy-l- methylethoxy]pyrimidin-4-yl}fiiran-2-sulfonamide
To a solution of 5-(azetidin-l-ylcarbonyl)-N-{2-[(2,3-difluorobenzyl)thio]-6-[(l/?)-l-methyl- 2-(triphenyLmethyloxy)ethoxy]pyrimidin-4-yl}furan-2-sulfonamide (the product of step iv) (0.24g) in methanol (5mL) was added /?αrα-toluenesulfonic acid hydrate (58mg) and anisole (0.34mL). After stirring at room temperature for 2d, H2O (5mL) was added and the mixture
-95- extracted with EtOAc (3 x 1OmL). The combined organic layers were washed with brine (1OmL), dried (MgSO4), filtered and evaporated to dryness in vacuo. The resulting crude solid was purified by reverse phase HPLC (50% to 5% gradient of 0.1% aqueous ammonium acetate in acetonitrile as eluent) to give the title compound as a white solid. Yield: lOmg. MS: APCI(+ve) 541 [M+H+]
1H NMR δ (CDCl3) 1.28 (d, 3H), 2.39 (quintet, 2H), 3.70 (dd, IH), 3.76 (dd, IH), 4.20 (t, 4H), 4.33 (d, IH), 4.37 (d, IH), 4.51 (t, 2H), 5.31 (dquintet, IH), 6.41 (s, IH), 6.99-7.08 (m, 2H), 7.11 (d, IH), 7.17-7.22 (m, IH), 7.21 (d, IH)
The intermediates for this compound were prepared as follows:
i) methyl 5-[(te/t-butylamino)sulfonyl]-2-fiiroate
To a solution of methyl 5-(chlorosulfonyl)-2-furoate (3.Og) in DCM (10OmL) was added tert- butylamine (3.6mL). After stirring at room temperature for 2 days the mixture was filtered through a pad of celite, washing with DCM (2 x 1OmL). The filtrate was evaporated to dryness in vacuo. The resulting crude residue was purified by column chromatography using EtOAc/jrøhexane (2:8) as eluent to give the subtitle compound as a foam. Yield: 2.75g. MS: APCI(-ve) 260 [M-H]
ii) 5-(azetidin-l-ylcarbonyl)-N-(ϊe/t-butyl)furan-2-sulfonamide
To a solution of methyl 5-[(ter^butylamino)sulfonyl]-2-furoate (the product of step i) (2.15g) in methanol (8OmL) was added azetidine (1.15mL). After stirring at room temperature for 5h the mixture was evaporated to dryness in vacuo. The resulting residue was partitioned between EtOAc (5OmL) and H2O (5OmL). The separated organic layer was dried (MgSO4), filtered and evaporated to dryness in vacuo. The resulting crude material was purified by column chromatography (EtOAC as eluent) to give the subtitle compound as a pale yellow oil. Yield: 3g. MS: APCI(+ve) 287 [M+H+]
iii) 5-(azetidin-l-ylcarbonyl)fiiran-2-sulfonamide
A solution of 5-(azetidin-l-ylcarbonyl)-N-(tert-butyl)furan-2-suhcbnamide (the product of step ii) (3g) in trifluoroacetic acid (9OmL) was stirred at room temperature. After 18h the mixture
-96- was evaporated to dryness in vacuo. The resulting oil was triturated with Et2O and filtered to give the subtitle compound as a white solid. Yield: 1.75g. MS: APCI(+ve) 231 [M+H+]
iv) 5-(azetidin-l-ylcarbonyl)-N-{2-[(2,3-difluorobeiizyl)thio]-6-[(l/?)-l-methyl-2- (triphenylmethyloxy)ethoxy]pyrimidin-4-yl}fiiran-2-sulfonamide
The subtitle compound was prepared from 5-(azetidin-l-ylcarbonyl)furan-2-sulfonamide (the product of step ϋi) (0.4Og) and 4-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-[(li?)-l- methyl-2-(triphenyhnethoxy)ethoxy]-pyrimidine (the product of Example 13, step iii) (0.4Ig) according to the procedure outlined in Example 1, step iv). Yield: 0.25g MS: APCI(-ve) 781 [M-H]
Example 50
N-(^rr-butyl)-5-[({2-[(2,3-difluorobenzyl)thio]-6-[(lR)-2-hydroxy-l- methylethoxy]pyrimidin-4-yl}amino)sulfonyl]-2-fiiramide
To a solution of ethyl (2/?)-2-({6-[({5-[(tert-butylamino)carbonyl]-2-furyl}suhSonyl)amino]-2- [(2,3-difluorobenzyl)thio]pyrimidin-4-yl}oxy)propanoate (the product of step iii) (0.25g) in THF (1OmL) was added a solution of lithium borohydride (0.6mL, 2.0M in hexanes) dropwise at 00C. The mixture was warmed to room temperature and stirred for 18h. After cooling to 00C, IN HCl (2OmL) was added slowly, and the mixture extracted with EtOAc (3 x 2OmL). The combined organic layers were dried (MgSO4), filtered and evaporated to dryness in vacuo. The resulting crude oil was purified by reverse phase HPLC (75% to 5% gradient of 0.2% aqueous trifluoro acetic acid in acetonitrile as eluent) to give the title compound as a white solid. Yield: 90mg. MS: APCI(-ve) 555 [M-H]
-97-
1H NMR δ (CDCl3): 1.27 (d, 3H), 1.44 (s, 9H), 3.69 (dd, IH), 3.75 (dd, IH), 4.33 (d, IH), 4.38 (d, IH), 5.26-5.33 (m, IH), 6.23 (br s, IH), 6.32 (s, IH), 6.98-7.08 (m, 2H), 7.09 (d, IH), 7.17-7.21 (m, IH), 7.23 (d, IH)
The intermediates for this compound were prepared as follows:
i) ΛKterf-butyl)-5-cyanofiiran-2-sulfonainide
A solution of 5-formylfuran-2-sulfonic acid sodium salt (2.97g), and hydro xylamine hydrochloride (1.05g) in H2O (1.35mL) and acetic acid (2ImL) was heated at 6O0C for 4h. After cooling to room temperature the solvent was removed in vacuo. The crude residue was triturated with Et2O (3 x 5OmL) and dried under high vacuum to give a crude pale brown solid. A solution of this material (3.7g) in phosphorusoxychloride (10OmL) was heated at 600C for 18h. After cooling to room temperature the mixture was partitioned between ice- water (10OmL) and EtOAc (10OmL). The aqueous layer was separated and further extracted with EtOAc (2 x 10OmL). The combined organic phases were washed with brine (10OmL), dried (MgSO4), filtered and evaporated to dryness in vacuo to give a crude brown oil. To a solution of this crude oil (1.6g) in DCM (85mL) was added te/t-butylamine (1.8mL). After stirring at room temperature for 2 days the mixture was filtered through celite, washing with DCM (2 x 2OmL). The filtrate was evaporated to dryness in vacuo. The resulting crude material was purified by column chromatography using EtOAc/isohexane (2:8) as eluent to give the subtitle compound as a pale yellow oil. Yield: 1.Og. MS: APCI(-ve) 227 [M-H]
ii) 5-(aminosulfonyl)-N-(tø/t-butyl)-2-furamide A solution of Λr-(tert-butyl)-5-cyanofuran-2-sulfonamide (the product of step i) (1.Og) in trifluoro acetic acid (3OmL) was stirred at room temperature for 24h. The mixture was evaporated to dryness in vacuo to leave a crude yellow oil that was triturated with Et2O and filtered to give the subtitle compound as a white solid. Yield: 0.25g. MS: APCI(-ve) 245 [M-H]
iii) ethyl (2R)-2-({6-[({5-[(^rt-butylamino)carbonyl]-2-furyl}sulfonyl)amino]-2-[(2,3- difluorobenzyl)thio]pyrimidin-4-yl}oxy)propanoate
-98-
The subtitle compound was prepared according to the procedure outlined in example 1 step (iv) using a mixture of 5-(arninosulfonyl)-N-(terf-butyl)-2-furamide (0.25g), tris(dibenzylideneacetone)dipalladium (0) (58mg), 2-dicyclohexylphosphino-2',4',6'-tri- isøpropyl-l,l'-biphenyl (XPHOS) (30mg), cesium carbonate (0.65g) and 2-[[6-chloro-2- [[(2,3-difluorophenyl)methyl]thio]-4-pyrimidinyl]oxy]-(2/?)- propanoic acid ethyl ester (product of Example 11 step i) (0.26g) in dioxane (1OmL). Purification was trituration with Et2O to give the subtitle compound as a white solid. Yield: 0.25g MS: APCI(+ve) 599 [M+H+]
Example 51
5-cyano-iV-{2-[(2,3-difluorobenzyl)thio]-6-[(li?)-2-hydroxy-l-methylethoxy]pyriinidin-4- yl}fiiran-2-sulfonamide
To a solution of 5-cyano-N-{2-[(2,3-difluorobenzyl)tWo]-6-[(l#)-l-methyl-2- (triphenylmethyloxy)ethoxy]pyrirnidin-4-yl}furan-2-sulfonarnide (the product of step ϋ) (0.15g) in methanol (5mL) was added pαrø-toluenesulfonic acid hydrate (39mg) and anisole (0.23mL). After stirring at room temperature for 5h, H2O (5mL) was added and the mixture extracted with EtOAc (3 x 1OmL). The combined organic layers were washed with brine (1OmL), dried (MgSO4), filtered and evaporated to dryness in vacuo. The resulting crude solid was purified by reverse phase HPLC (75% to 5% gradient of 0.1 % aqueous ammonium acetate in acetonitrile as eluent) to give the title compound as a white solid. Yield: 20mg. MS: APCI(-ve) 481 [M+H"]
1H NMR δ (CDCl3): 1.28 (d, 3H), 3.71 (dd, IH), 3.76 (dd, IH), 4.34 (d, IH), 4.38 (d, IH), 5.29-5.33 (m, IH), 6.31 (s, IH), 7.00-7.11 (m, 2H), 7.15 (d, IH), 7.18-7.21 (m, IH), 7.24 (d, IH)
The intermediates for this compound were prepared as follows:
-99-
i) 5-cyanofuran-2-sulfonamide
A solution of N-(tert-butyl)-5-cyanofuran-2-sulfonamide (the product of example 50, step i) (1.Og) in trifluoroacetic acid (3OmL) was stirred at room temperature for 24h. The mixture was evaporated to dryness in vacuo to leave a crude yellow oil that was triturated with Et2O and filtered. The filtrate was evaporated to dryness in vacuo to give the subtitle compound as a pale yellow oil. Yield: 0.29g. MS: APCI(-ve) 171 [M-H ]
ii) 5-cyano-N-{2-[(2,3-difluorobenzyl)thio]-6-[(lR)-l-methyl-2- (triphenyImethyloxy)ethoxy]pyrinιidin-4-yl}fiiraii-2-sulfonaiiiide
The subtitle compound was prepared from 5-cyanofuran-2-sulfonamide (the product of step i) (0.29g) and 4-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-[(l/?)-l-methyl-2- (triphenyhnethoxy)ethoxy]-pyrimidine (the product of Example 13, step ii) (0.15g) according to the procedure outlined in Example 1, step iv). Purification was by column chromatography on silica gel using EtOAc/isohexane (1:4 to 2:3 gradient) to give the subtitle compound as a pale yellow solid. Yield: 0.25g MS: APCI(-ve) 723 [M-H]
Example 52
N-{2-[(2,3-Difluorobenzyl)thio]-6-methoxypyrimidin-4-yl}-4-pyrimidin-2-ylpiperazine-l- sulfonamide
The title compound was prepared according to the procedure outlined in example 1 step (iv) using a mixture of 4-pyrimidin-2-ylpiperazine-l -sulfonamide (0.24g), tris(dibenzylideneacetone)dipalladium (0) (60mg), 2-dicyclohexylphosphino-2',4',6'-tri- jrøρropyl-l,l'-biphenyl (XPHOS) (31mg), cesium carbonate (0.32g) and 4-Chloro-2-[[(2,3-
-100- difluorophenyl)methyl]thio]-6-methoxypyrimidine (the product of Example 35, step i) (0.2Og) in dioxane (6ml). The crude material was purified by column chromatography using EtOAc/isohexane (2:8 to 1:1 gradient) as eluent to give the title compound as a pale yellow foam Yield: 0.19g. MS: APCI(+ve) 510 [M+H]
1H NMR: δ (DMSO) 3.26 (m, 4H), 3.77(m, 4H), 3.86 (s, 3H), 4.47 (s, 2H), 6.08 (s, IH), 6.67 (t, IH), 7.13 (m, IH), 7.33 (m, IH), 7.40 (dt, IH), 8.37 (d, 2H), 11.16 (bs, IH)
The intermediate for this compound was prepared as follows:
i) 4-Py rimidin-2-ylpiperazine- 1 -sulfonamide
The subtitle compound was prepared according to the procedure outlined in example 15 step i) using 2-piperazin-l-ylpyrimidine (3.Og) and sulfamide (1.2g) in dioxane (3OmL) to give the subtitle compound as a white solid. Yield: 2.06g. 1H NMR: δ (DMSO) 3.00 (t, 2H), 3.83 (t, 2H), 6.68 (t, IH), 6.81 (bs, IH), 8.39 (d, 2H)
Example 53
N-{5-[({2-[(2,3-Difluorobenzyl)thio]-6-methoxypyrimidin-4-yl}amino)sulfonyl]-4-methyl- 1 ,3-thiazol-2-yl}acetamide
The title compound was prepared according to the procedure outlined in example 1 step (iv) using a mixture of N-[5-(aminosulfonyl)-4-methyl-l,3-thiazol-2-yl]acetamide (0.25g), tris(dibenzylideneacetone)dipalladium (0) (64mg), 2-dicyclohexylphosphino-2',4',6'-tri- ϊrøρropyl-l,l'-biphenyl (XPHOS) (33mg), cesium carbonate (0.69g) and 4-Chloro-2-[[(2,3- difluorophenyl)methyl]thio]-6-methoxypyrimidine (the product of Example 35, step i) (0.2Ig) in dioxane (6ml). The crude material was purified by column chromatography using EtOAc/isøhexane (2.5:7.5 to 4:6 gradient) as eluent to give the title compound as a pale yellow solid. Yield: 85mg.
-101-
MS: APCI(+ve) 502 [M+H]
1H NMR: δ (CDCl3) 2.26 (s, 3H), 2.57 (s, 3H), 3.90 (s, 3H), 4.38 (s, 2H), 6.32 (s, IH), 7.03 (m, 2H), 7.19 (dt, IH)
The intermediates for this compound were prepared as follows:
i) N-{ 5- [(tert-Butylamino)sulfonyl] -4-methyl- 1 ,3-thiazol-2-yl}acetamide
To a suspension of 2-(acetylamino)-4-methyl-l,3-thiazole-5-sulfonyl chloride (1.Og) in DCM (10ml) was added tert-butylamine (0.92ml) and the mixture was stirred at room temperature for 2d. The mixture was diluted with H2O and extracted with DCM (x3). The combined organic extracts were dried (MgSO4), filtered and evaporated to give the subtitle compound as a beige foam. Yield l.lg. MS: APCI(+ve) 292 [M+H]
ϋ) N-[5-(Aminosulfonyl)-4-methyl-l,3-thiazol-2-yl]acetamide
A solution of N-{5-[(tert-butylamino)sulfonyl]-4-methyl-l,3-thiazol-2-yl}acetamide (l. lg) in TFA (10ml) was stirred at room temperature for 3d. The mixture was evaporated, redissolved in TFA (10ml) and stirred for a further Id. On evaporation the resulting oil was azeotroped with DCM (x2) and triturated with Et2O to give the subtitle compound as a beige solid. Yield 0.7g.
MS: APCI(+ve) 236 [M+H]
Example 54
2-Amino-N-{2-[(2,3-difluorobenzyl)thio]-6-methoxypyrimidin-4-yl}-4-methyl-l,3- thiazole-5-sulfonamide
The title compound was prepared according to the procedure outlined in example 1 step (iv) using a mixture of 2-amino-4-methyl-l,3-thiazole-5-suhconamide (0.23g),
-102- tris(dibenzylideneacetone)dipalladium (0) (73mg), 2-dicyclohexylρhosphino-2',4',6'-tri- woproρyl-l,l'-biρhenyl (XPHOS) (38mg), cesium carbonate (0.39g) and 4-Chloro-2-[[(2,3- difluorophenyl)methyl]thio]-6-methoxypyrimidine (the product of Example 35, step i) (0.24g) in dioxane (6ml). The crude material was purified by column chromatography using EtOAc/ii'øhexane (2:8 to 4.5:5.5 gradient) as eluent and trituration with Et2O to give the title compound as a beige solid. Yield: 0.15g. MS: APCI(+ve) 460 [M+H]
1H NMR: δ (DMSO) 2.42 (s, 3H), 3.91 (s, 3H), 4.56 (s, 2H), 6.07 (s, IH), 7.18 (dq, IH), 7.35 (m, 2H), 7.55 (bs, 2H), 11.91 (bs, IH)
The intermediate for this compound was prepared as follows:
i) 2-Amino-4-methyl-l,3-thiazole-5-sulfonamide
A suspension of iV-[5-(aminosulfonyl)-4- methyl- l,3-thiazol-2-yl]acetamide (the product from Example 53, step ϋ) (0.44g) in hydrazine hydrate (1.5ml) was stirred at room temperature for 4h. The mixture was diluted with H2O and extracted with EtOAc (x4). The combined organic extracts were dried (MgSO4), filtered and evaporated to give the subtitle compound as an off- white solid. Yield 0.23g. MS: APCI(+ve) 194 [M+H]
Example 55 iV-(2-[(2,3-Difluorobenzyl)thio]-6-{[(l/?,25)-2,3-dihydroxy-l- methylpropyl]oxy}pyrimidin-4-yl)methanesulfonamide
To a solution of N-(2-[(2,3-difluorobenzyl)thio]-6-{(l/?)-l-[(45)-2,2-dimethyl-l,3-dioxolan-4- yl]ethoxy}pyrimidin-4-yl)methanesulfonamide (0.23g) in MeOH (2ml) was added TFA (0.4ml) and the reaction was stirred at room temperature for 2Oh. The mixture was
-103- evaporated, suspended in saturated sodium carbonate solution and then re-acidified to pH5 with glacial acetic acid with stirring. The resulting solid was collected, washed with H
2O and dried to give the title compound as a cream solid. Yield 0.17g. MS: APCI(+ve) 436 [M+H]
1U NMR: δ (DMSO) 1.18 (d, 3H), 3.26 (s, 3H), 3.36 (t, 2H), 3.62 (quintet, IH), 4.45 (quintet, 2H), 4.60 (t, IH), 4.93 (d, IH), 5.17 (quintet, IH), 5.97 (s, IH), 7.17 (m, IH), 7.35 (m, IH), 7.40 (m, IH)
The intermediate for this compound was prepared as follows:
i) iV-(2-[(2,3-Diαuorobenzyl)thio]-6-{(l/?)-l-[(4S)-2,2-diπiethyl-l,3-dioxolan-4- yl] ethoxy }py riniidi n-4-yl)methanesulfonamide
The subtitle compound was prepared according to the procedure outlined in example 1 step (iv) using a mixture of methane sulfonamide (0.25g), tris(dibenzylideneacetone)dipalladium (0) (55mg), 2-dicyclohexylρhosphino-2',4',6'-tri-/røpropyl-l,l'-biphenyl (XPHOS) (29mg), cesium carbonate (0.3Og) and 4-chloro-2-[(2,3-difluorobenzyl)thio]-6-{(l/?)-l-[(45)-2,2- dimethyl-l,3-dioxolan-4-yl]ethoxy}pyrimidine (the product of Example 47, step i) (0.25g) in dioxane (5ml). The crude material was purified by column chromatography using EtOAc/isøhexane (2:8) as eluent to give the subtitle compound as a yellow oil. Yield: 0.25g. MS: APCI(+ve) 476 [M+H]
Example 56
N-(2-[(2,3-Difluorobenzyl)thio]-6-{[(l/?,25)-2,3-dihydτoxy-l- methylpropyl]oxy}pyrimidin-4-yl)morpholine-4-sulfonamide
To a solution of N-(2-[(2,3-difluorobenzyl)thio]-6-{(l/?)-l-[(45)-2,2-dimethyl-l,3-dioxolan-4- yl]ethoxy}pyrimidin-4-yl)morphoUne-4-sulfonamide (0.2Og) in MeOH (2ml) was added TFA
-104-
(0.4ml) and the reaction was stirred at room temperature for 2Oh. The mixture was evaporated, suspended in saturated sodium carbonate solution and then re-acidified to pH5 with glacial acetic acid with stirring. The resulting solid was collected, washed with H2O and dried to give the title compound as a white solid. Yield 0.15g. MS: APCI(+ve) 507 [M+H]
1H NMR: δ (DMSO) 1.18 (d, 3H), 3.18 (m, 4H), 3.33 (m, 2H), 3.60 (m, 5H), 4.44 (q, 2H), 4.60 (t, IH), 4.89 (d, IH), 5.20 (quintet, IH), 6.03 (s, IH), 7.16 (m, IH), 7.38 (m, 2H), 11.13 (bs, IH)
The intermediate for this compound was prepared as follows:
i) N-(2-[(2,3-Difluorobenzyl)thio]-6-{(lR)-l-[(4S)-2,2-dimethyl-l,3-dioxolan-4- yl]ethoxy}pyrimidin-4-yl)morphoBne-4-sulfonamidc
The subtitle compound was prepared according to the procedure outlined in example 1 step (iv) using a mixture of 4-morpholine sulfonamide (prepared according to patent WO 2004/011443) (0.15g), tris(dibenzylideneacetone)dipalladium (0) (55mg), 2- dicyclohexylphosphino-2',4',6'-tri-irøpropyl-l,r-biphenyl (XPHOS) (29mg), cesium carbonate (0.30g) and 4-chloro-2-[(2,3-difluorobenzyl)thio]-6-{(l/?)-l-[(45)-2,2-dimethyl- l,3-dioxolan-4-yl]ethoxy}pyrimidine (the product of Example 47, step i) (0.25g) in dioxane (5ml). The crude material was purified by column chromatography using EtOAc/isohexane (1:9 to 2.5:7.5 gradient) as eluent to give the subtitle compound as an off-white foam Yield: 0.2Og. MS: APCI(+ve) 547 [M+H]
Example 57
N-[2-[(2,3-Difluorobenzyl)thio]-6-(methylthio)pyrimidin-4-yl]methanesulfonamide
A mixture of methane sulfonamide (0.22g), tris(dibenzylideneacetone)-dipalladium (0) (33mg), 2-dicyclohexylphosphino-2',4',6'-tri-irøpropyl-l,l'-biphenyl (XPHOS) (17mg), cesium carbonate (0.58g) and 4-chloro-2-[(2,3-difluorobenzyl)thio]-6-(methylthio)pyrimidine (the product of step i) (0.38g) in dioxane (1OmL) was heated at 1000C for 18h. The mixture was cooled and saturated ammonium chloride was added and the resulting mixture extracted with EtOAc. The combined organic extracts were washed with saturated aqueous sodium chloride, dried (MgSO4), filtered and evaporated. The residue was purified by reverse phase HPLC eluting with acetonitrile / aq. 0.1% TFA mixtures to give the title compound as a white solid. Yield: 30mg. MS: APCI(+ve) 378 [M+H+]
1H NMR (CDCl3) δ 2.52 (3H, s), 3.21 (3H, s), 4.44 (2H, s), 6.73 (IH, s), 6.99-7.10 (2H, m), 7.21-7.24 (IH, m)
The intermediate for this compound was prepared as follows:
i) 4-chloro-2-[(2,3-difluorobenzyl)thio]-6-(methylthio)pyrimidine
To a solution of 4,6-dichloro-2-[(2,3-difluorobenzyl)thio]pyrimidine (the product of example 1 step ii) (1.54g) in THF (5OmL) was added sodium methanethio late (0.39g). The mixture was allowed to warm to room temperature and stirring continued for 2h. Saturated ammonium chloride was added and the resulting mixture extracted with EtOAc. The combined organic extracts were washed with saturated aqueous sodium chloride, dried (MgSO4), filtered and evaporated to give the subtitle compound as a pale yellow solid. Yield: 1.5 Ig. MS: APCI(-ve) 317/319 [M-K]
Example 58
N-[2-[(2,3-Difluorobenzyl)thio]-6-(methylthio)pyrimidin-4-yl]azetidine-l-sulfonamide
A mixture of azetidine- 1-sulfonamide (prepared according to patent WO 2004/011443, 0.32g), tris(dibenzylideneacetone)-dipalladium (0) (33mg), 2-dicyclohexylphosphino-2',4',6'- tri-iiOpropyl-l,r-biphenyl (XPHOS) (17mg), cesium carbonate (0.58g) and 4-chloro-2-[(2,3- difluorobenzyl)thio]-6-(methylthio)pyrimidine (the product of example 57, step i) (0.38g) in dioxane (1OmL) was heated at 1000C for 18h. The mixture was cooled and saturated ammonium chloride was added and the resulting mixture extracted with EtOAc. The combined organic extracts were washed with saturated aqueous sodium chloride, dried (MgSO4), filtered and evaporated. The residue was purified by reverse phase HPLC eluting with acetonitrile / aq. 0.1% ammonium acetate mixtures to give the title compound as a white solid. Yield: 50mg.
MS: APCI(+ve) 419 [M+H+]
1H NMR (CDCl3) δ 2.25 (2H, quintet), 2.51 (3H, s), 4.01 (4H, t), 4.43 (2H, s), 6.81 (IH, s),
6.98-7.10 (2H, m), 7.21-7.24 (IH, m)
Example 59
N-[2-[(2,3-Difluorobenzyl)thio]-6-(methylthio)pyrimidin-4-yl]morpholine-4-sulfonamide
A mixture of 4-moφholinesulfonamide (prepared according to patent WO 2004/011443, 0.39g), tris(dibenzylideneacetone)-dipalladium (0) (33mg), 2-dicyclohexylphosphino-2',4',6'- tri-iropropyl-l.l'-biphenyl (XPHOS) (17mg), cesium carbonate (0.58g) and 4-chloro-2-[(2,3- difluorobenzyl)thio]-6-(methylthio)ρyrimidine (the product of example 57, step i) (O.38g) in dioxane (1OmL) was heated at 1000C for 18h. The mixture was cooled and saturated ammonium chloride was added and the resulting mixture extracted with EtOAc. The combined organic extracts were washed with saturated aqueous sodium chloride, dried (MgSO4), filtered and evaporated. The residue was purified by reverse phase HPLC eluting with acetonitrile / aq. 0.1% ammonium acetate mixtures to give the title compound as a white solid. Yield: 30mg. MS: APCI(+ve) 449 [M+H+]
-107-
1R NMR (CDCl3) δ 2.51 (3H, s), 3.30 (4H, t), 3.72 (4H, t), 4.43 (2H, s), 6.73 (IH, s), 7.00- 7.10 (2H, m), 7.21-7.24 (IH, m)
Example 60 N-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-{[(lR,2/?)-2,3-dihydroxy-l- methylpropyl]oxy}-4-pyrimidinyl]-l-morpholinesulfonamide
To a solution of N-[2-[[(2,3-difluorophenyl)methyl]thio]-6-[(l/?)-l-[(4/?)-2,2-dimethyl-l,3- dioxolan-4-yl]ethoxy]-4-pyrimidinyl]-l-morpholinesulfonamide (the product of step i) (0.17g) in DCM (5ml) was added iron (III) chloride hexahydrate (0.25g). The reaction mixture was stirred at room temperature for 3h after which time further iron (III) chloride hexahydrate (0.25g) was added. After a further 3h saturated aqueous sodium bicarbonate (ImI) was added. The layers were separated and the aqueous material extracted with DCM (x3) and EtOAc (x3). The combined organic extracts were washed with saturated aqueous sodium chloride, dried with sodium sulfate, filtered and evaporated. The residual solid was purified by reverse phase HPLC (gradient 25-95% acetonitrile in 0.2% aqueous TFA) to afford the title compound as a white powder. Yield: 23mg MS: APCI(+ve) 507 [M+H+] 1H NMR: δ (400 MHz, CDCl3) 1.31 (d, 3H), 3.29 - 3.32 (m, 4H), 3.60 - 3.80 (m, 7H), 4.36 (>/2Abq, IH), 4.36 C/2Abq, IH), 5.31 (quintet, IH), 6.23 (s, IH), 6.99 - 7.10 (m, 2H), 7.20 - 7.23 (m, IH).
The intermediate for this compound was prepared as follows:
i) N-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-[(l/?)-l-[(4/?)-2,2-dimethyl-l,3-dioxolan-4- yl] ethoxy ] -4-pyrimidinyl] - 1 -morpholinesulfonamide
-108-
A mixture of moφholine-4-sulfonamide (prepared according to patent WO 2004/011443, 0.239g), tris(dibenzylideneacetone)-dipalladium (0) (33mg), 2-dicyclohexylphosphino- 2',4',6'-tri-isopropyl-l,l'-biphenyl (XPHOS) (17mg), cesium carbonate (0.176g) and 4- chloro-2-[(2,3-difluorobenzyl)thio]-6- { (IR)- l-[(4/?)-2,2-dimethyl- 1 ,3-dioxolan-4- yljethoxy Jpyrimidine (the product of example 45 step vϋ) (0.15Og) in anhydrous dioxane (6ml) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for 20 min. Saturated aqueous ammonium chloride was added and the resulting mixture extracted with EtOAc. The combined organic extracts were washed with saturated aqueous sodium chloride, dried with sodium sulfate, filtered and evaporated. The residue was purified by column chromatography on silica using a 1 : 19 to 2:3 mixture of EtOAc and irø-hexane as eluent to give the subtitle compound as a yellow gum. Yield: 0.165 g MS: APCI(+ve) 547 [M+H+]
Example 61 yV-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-{[(15,2R)-2,3-dihydroxy-l-methylpropyl]oxy}- 4-pyrimidinyl] - 1 -azetidinesulfonamide
To a solution of N-(2-[(2,3-difluorobenzyl)thio]-6-{(15)-l-[(2/?)-l,4-dioxaspiro[4.5]dec-2- yl]ethoxy}pyrimidin-4-yl)azetidine-l-sulfonamide (the product of step ϋ) (43mg) in DCM (4ml) was added iron (III) chloride hexahydrate (73mg). The reaction mixture was stirred at room temperature for 2h after which time further iron (HI) chloride hexahydrate (40mg) was added. After 3d at -18 0C, H2O and DCM were added. The layers were separated and the aqueous material extracted with further DCM. The combined organic extracts were washed with saturated aqueous sodium chloride, dried with sodium sulfate, filtered and evaporated. TFA (ImI) and DCM (4ml) were added to the residue and the reaction mixture stirred at room temperature for 2d. The mixture was partitioned between saturated aqueous sodium bicarbonate and DCM, then neutralised with 2M aqueous hydrochloric acid, the layers
-109- separated and the aqueous material extracted with further DCM. The DCM extracts were allowed to slowly evaporate and the resulting solid washed with minimal cold DCM to afford the title compound as a white powder. Yield: 1 lmg MS: APCI(+ve) 477 [M+H+] 1H NMR: δ (300 MHz, CDCl3) 1.36 (d, 3H), 2.27 (quintet, 2H), 3.59 - 3.65 (m, IH), 3.68 - 3.78 (m, 2H), 4.02 (t, 4H), 4.37 (s, 2H), 5.23 (quintet, IH), 6.31 (s, IH), 6.99 - 7.11 (m, 2H), 7.19 - 7.23 (m, IH).
The intermediates for this compound were prepared as follows:
i) 4-Chloro-2-[(2,3-diQuorobenzyl)thio]-6-{(lS)-l-[(2R)-l,4-dioxaspiro[4.5]dec-2- yl]ethoxy}pyrimidine
A solution of (lS)-l-[(2/?)-l,4-dioxaspiro[4.5]dec-2-yi]ethanol (prepared according to /. Org. Chem. 1995, 60, 585-587, 0.183 g of -2: 1 mixture of diastereomers) in dry THF (5ml) was cooled to 00C and to it was added (in portions) sodium hydride (46mg as 60% dispersion in mineral oil) followed in portions by 4,6-dichloro-2-(2,3-difluoro-benzylsulfanyl)-pyrimidine (product of example 1 step ii, 0.252 g). The reaction mixture was stirred at room temperature for 24h then quenched with saturated aqueous ammonium chloride (2ml) and diluted with EtOAc. The layers were separated and the aqueous layer extracted with further EtOAc. The combined organic extracts were washed with saturated aqueous sodium chloride, dried with sodium sulfate, filtered and evaporated to leave a yellow oil which was purified by column chromatography on silica using a 0.5 to 4% mixture of EtOAc in isø-hexane as eluent to afford the subtitle compound as a white solid. Yield: 0.1Og MS: APCI(+ve) 457/459 [M+H+]
u) N-(2-[(2,3-Diαuorobenzyl)thio]-6-{(15)-l-[(2R)-l,4-dioxaspiro[4.5]dec-2- yl]ethoxy}pyrimidin-4-yl)azetidine-l-sulfonaimde
A mixture of azetidine-1 -sulfonamide (prepared according to patent WO 2004/011443,
0.13Ig), tris(dibenzylideneacetone)-dipalladium (0) (22mg), 2-dicyclohexylphosphino- 2',4',6'-tri-wopropyl-l,l'-biphenyl (XPHOS) (llmg), cesium carbonate (0.117g) and 4- chloro-2-[(2,3-difluorobenzyl)thio]-6-{(15)-l-[(2/?)-l,4-dioxasρiro[4.5]dec-2- yl]ethoxy}pyrimidine (the product of step i) (0.10Og) in anhydrous dioxane (5ml) was heated
-110- at reflux in a microwave at 10O0C, 30OW, open vessel with cooling for 15min. Saturated aqueous ammonium chloride was added and the resulting mixture extracted with EtOAc. The combined organic extracts were washed with saturated aqueous sodium chloride, dried with sodium sulfate, filtered and evaporated. The residue was purified by column chromatography on silica using a 1 : 19 to 3:7 mixture of EtOAc and /rø-hexane as eluent and then by reverse phase HPLC (gradient 25-95% acetonitrile in 0.1% ammonium acetate) to give the subtitle compound as a colourless gum. Yield: 43mg MS: APCI(+ve) 557 [M+H+]
Example 62
N-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-methoxypyrimidin-4-yl]-4- ethanesulfonylpiperazine- 1 -sulfonamide
The title compound was prepared from 4-ethanesulfonylpiperazine-l -sulfonamide (the product of step i) (0.3Ig) and 4-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6- methoxypyrimidine (the product of Example 35, step i) (0.25g) according to the procedure outlined in Example 1, step iv). The crude material was purified by column chromatography using EtOAc / /røhexane (1:1) as eluent. Yield: 0.37g MS: APCI (+ve) 524 [M+H
+]
1H NMR: 5 (DMSO) 1.17 (t, 3H), 3.06 (q, 2H), 3.23 (d, 4H), 3.28 (d, 4H), 3.88 (s, 3H), 4.48 (s, 2H), 6.06 (s, IH), 7.17 (m, IH), 7.33 (m, IH), 7.42 (t, IH), 11.22 (bs, IH).
The intermediate for this compound was prepared as follows:
i) 4-Ethanesulfonylpiperazi ne- 1 -sulfonamide
-111-
To a solution of 1-ethanesulfonylpiperazine (1.Og) in dioxane (10ml) was added sulfamide (0.5Ig). The reaction was then heated at 1000C for 24h. The reaction was allowed to cool before being concentrated in vacuo. The residue was stirred in Et2O for 4h and the mixture filtered to give the product as a white solid. Yield: 1.3g. 1H NMR: δ (DMSO) 1.21 (t, 3H), 3.02 (t, 4H), 3.09 (q, 2H), 3.28 (t, 4H), 6.89 (s, 2H).
Example 63
N-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-methoxypyrimidin-4-yl]-3-oxopiperazine-l- sulfonamide
The title compound was prepared from 3-oxopiperazine-l-sulfonamide (the product of step i) (0.22g) and 4-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-methoxypyrimidine (the product of Example 35, step i) (0.25g) according to the procedure outlined in Example 1, step iv). The crude material was purified by column chromatography using EtOAc / isόhtxaae (1 : 1) as eluent to give a white solid. This solid was dissolved in EtOAc and Et2O and extracted with IN sodium hydroxide. The basic solution was washed with Et2O, acidified with dilute hydrochloric acid and extracted with EtOAc. The organic solution was washed with H2O, dried (MgSO4) and the solvent evaporated in vacuo to give the product as a yellow foam. Yield: 40mg
MS: APCI (+ve) 446 [M+H+]
1H NMR: δ (DMSO) 3.19 (s, 2H), 3.43 (t, 2H), 3.80 (s, 2H), 3.88 (s, 3H), 4.48 (s, 2H), 6.03
(s, IH), 7.17 (q, IH), 7.34 (m, IH), 7.41 (m, IH), 8.07 (s, IH), 11.29 (s, IH).
The intermediate for this compound was prepared as follows:
i) 3-Oxopiperazine-l-sulfonamide
-112-
The subtitle compound was prepared according to the procedure outlined in Example 62 step i) using 2-oxopiperazine (0.5g) and sulfamide (0.45g) to give a beige solid. Yield: 0.83g. 1H NMR: δ (DMSO) 3.14 (t, 2H), 3.25 (t, 2H), 3.50 (s, 2H), 7.02 (s, 2H), 8.04 (s, IH).
Example 64
N-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-methoxypyrimidin-4-yl]-l,l- dioxothiomorpholine-4-sulfonamide
The title compound was prepared from l,l-dioxothiomorpholine-4-sulfonamide (0.3 Ig, McManus, J.M. et al, J. Med. Chem (1965) 8 766-776) and 4-chloro-2-[[(2,3-difluoroρhenyl) methyl]thio]-6-methoxypyrimidine (the product of Example 35, step i) (0.25g) according to the procedure outlined in Example 1, step iv). The crude material was purified by column chromatography using EtOAc / /røhexane (1: 1) as eluent. Yield: 0.48g MS: APCI (+ve) 481 [M+H+] 1H NMR: δ (DMSO) 3.24 (bt, 4H), 3.71 (bm, 4H), 3.89 (s, 3H), 4.49 (s, 2H), 6.01 (s, IH), 7.17 (m, IH), 7.34 (m, IH), 7.40 (t, IH).
Example 65
4-O-{6-[(Azetidin-l-ylsulfonyl)amino]-2-[(2,3-difluorobenzyl)thio]pyrimidiii-4-yl}-2,5- dideoxy-D-ϊ7treo-pentitol
To a solution of 4-O-{6-[(azetidin-l-ylsulfonyl)amino]-2-[(2,3-difluorobenzyl)thio] pyrimidin-4-yl}-2,5-dideoxy-l,3-O-(4-methoxybenzylidene)-D-t/ireo-pentitol (the product from step vii) in MeOH (9ml) was added TFA (ImI) dropwise. The reaction mixture was stirred at room temperaturefor 18h. The reaction mixture was reduced in vacuo and the residue redissolved in EtOAc (20ml) before reducing in vacuo directly onto silica and purifying by column chromatography on silica gel 50%EtOAc/50% jio-hexane to give the title compound as a white solid. Yield: 32 mg MS: APCI(-ve) 489 [M+H ]
1H NMR: δ (DMSO) 1.25-1.29 (m, 3H), 1.73-1.80 (m, 2H), 2.25 (q, 2H), 3.88-3.96 (m, 3H), 4.02 (t, 4H), 4.32-4.40 (m, 2H), 5.12-5.20 (m, IH), 6.32 (s, IH), 6.99-7.10 (m, 2H), 7.21-7.25 (πUH)
The intermediates for this compound were prepared as follows:
i) (2/?)-2-{[tøAt-Butyl(diphenyl)silyl]oxy}propanoic acid
To a solution of (2i?)-2-hydroxyρroρanoic acid (5g) in DMF (20ml) was added TBDPSCl (33.Og) and imidazole (16.4g). The reaction was then stirred overnight at RT. The reaction was partitioned between EtOAc (200ml) and H2O (200ml). The organic s were recovered and washed with 10% citric acid (200ml), H2O (200ml) and finally brine (200ml). The organics were then collected, dried (MgSO4) before being reduced in vacuo. The residue was dissolved in MeOH (200ml), cooled in an ice bath and potassium carbonate (6.9g) in H2O was added. After stirring at room temperaturefor 6h the solvent was removed in vacuo and the residue diluted with H2O (100ml). The pH was then adjusted to pH 4 with 10% citric acid, and the aqueous extracted three times with EtOAc (3 x 200ml). The organics were collected dried MgSO4 before reducing in vacuo to give the subtitle compound as a colourless oil. Yield: 7.5g MS: APCI(-ve) 327, [M+H ]
U) Ethyl (4R)-4-{[te/t-butyl(diphenyl)silyl]oxy}-3-oxopentanoate
To a solution of (2i?)-2-{[tert-Butyl(diphenyl)silyl]oxy}propanoic acid (the product from step i, 7.85g) in THF (300ml) was added CDI (4.26g) and the reaction was stirred at room temperaturefor approximately 15 mins. In a separate flask (71.7ml) of a IM heptane solution was added to a solution of ethyl hydrogen malonate (9.47g) in THF (300ml) at O0C. This
-114- solution was then allowed to warm to RT. The acyl imidazole solution was then transferred to the flask which contained the magnesium salt and the reaction was monitored for the next 2d. When the reaction was complete it was quenched by the addition of 250ml of sat. aq NH4Cl solution. The reaction mixture was then extracted with Et2O (3x200ml). The combined organics were dried (MgSO4), filtered and reduced to yield a clear oil which was purified by column chromatography on silica gel 4%EtOAc/96% /.rø-hexane. This gave the subtitle compound as a colourless oil. Yield: 2.Og
1R NMR: δ (DMSO) 1.03 (m, 9H), 1.11-1.19 (m, 6H), 3.31 (s, 2H), 4.07 (q, 2H), 4.23-4.31 (m, IH), 7.37-7.52 (m, 6H), 7.57-7.66 (m, 4H)
iii) 4-0-[te/t-Butyl(diphenyl)sttyl]-2,5-dideoxy-D-g/jcerø-pentitol To a solution of Ethyl (4/?)-4-{[tert-butyl(diphenyl)silyl]oxy}-3-oxopentanoate (the product from step ii) (2.Og) in THF (100ml) was added 2M LiBH4 in THF (12ml) The reaction was then stirred at room temperaturefor 18h. Saturated ammonium chloride (200ml) was added to the reaction mixture to quench any remaining LiBH4. The reaction mixture was then extracted using EtOAc (3x200ml). The organics were recovered and dried (MgSO4) before reducing in vacuo. The residue was purified by column chromatography on silica gel 30% EtOAc/70% /sø-hexaneto yield the subtitle compound as a clear oil. Yield: 540mg 1H NMR: 5 (CDCB) 1.00-1.03 (m, 3H), 1.07 (s, 9H), 1.61-1.68 (m, 2H), 3.63-3.84 (m, 4H), 7.36-7.47 (m, 6H), 7.65-7.70 (m, 4H)
iv) 4-O-[<eft-Butyl(diphenyl)silyl]-2,5-dideoxy-l,3-O-(4-methoxybenzylidene)-D-g(ycero- pentitol
To a solution of 4-O-[tm-butyl(diphenyl)siryl]-2,5-dideoxy-D-g/)>cero-pentitol (the product from step iii) (0.6Og) and l-(dimethoxymethyl)-4-methoxybenzene (0.30g) in DCM (60ml) was added tosic acid (60mg). The reaction was the stirred at room temperaturefor 3h before addition of more l-(dimethoxymethyl)-4-methoxybenzene (0.6Ig) and a further 2h stirring at RT. The reaction was worked up by reducing directly onto silica and purifying by flash column chromatography on silica gel 10% EtOAc/90% /.rø-hexane. This yielded the subtitle compound as a clear colourless oil. Yield: 0.45g MS: APCI(+ve) 477, [M+H+]
-115- v) (lR)-l-[2-(4-Methoxyphenyl)-l,3-dioxan-4-yl]ethanol
To a solution of 4-O-[tert-Butyl(diphenyl)silyl]-2,5-dideoxy-l,3-O-(4-methoxybenzylidene)- D-gZycerø-pentitol (the product from step iv, 0.4Og) in THF was added TBAF (2.76ml). The reaction was then allowed to stir at room temperaturefor 18h. The reaction mixture was then reduced directly onto silica and purified by column chromatography on silica gel 25% EtO Ac/75% /.ro-hexane. This gave the subtitle compound as a clear colourless oil. Yield: 0.19g MS: APCI(+ve) 239 [M+H+]
vi) 2-O-{6-Chloro-2-[(2,3-difluorobenzyl)thio]pyrimidin-4-yl}-l,4-dideoxy-3,5-O-(4- methoxybenzylidene)-D-fΛreø-pentitol
4-O-{6-Chloro-2-[(2,3-difluorobenzyl)tbio]pyrimidin-4-yl}-2,5-dideoxy-l,3-O-(4- methoxybenzylidene)-D-eryi1Λro-pentitol
To a solution of (0.18g) (ltf)-l-[2-(4-Methoxyphenyl)-l,3-dioxan-4-yl]ethanol and 4,6- Dichloro-2-[(2,3-difluorobenzyl)thio]pyrimidine (the product of example 1 step (ii), 0.25g) in anhydrous THF (10ml) at room temperaturewas added 60% sodium hydride (38mg). After stirring for 18h the reaction mixture was partitioned between H2O (50ml) solution and EtOAc (150ml). The organics were separated and the aqueous layer was re-extracted with EtOAc (2xl50ml) The organics collected, dried (MgSO4) and solvents removed in vacuo to give the subtitle compound as a colourless oil. The residue was then purified by column chromatography on silica gel 10%EtOAc/90% /so-hex aneto separate the two diastereoisomers
2-O-{6-Chloro-2-[(2,3-difluorobenzyl)thio]pyrimidin-4-yl}-l,4-dideoxy-3,5-O-(4- methoxybenzylidene)-D-fΛreo-pentitol
1H NMR: 5 (CDCB) 1.36 (d, 3H), 1.52-1.59 (m, IH), 1.83-1.96 (m, IH), 3.80 (s, 3H), 3.90- 3.98 (m, IH), 4.26-4.30 (m, IH), 4.40 (s, 2H), 5.28-5.33 (m, IH), 5.47 (s, IH), 6.43 (s, IH), 6.88 (d, 2H), 6.96-7.10 (m, 2H), 7.24-7.29 (m, IH), 7.37 (d, 2H). Yield: 0.15g.
4-O-{6-Chloro-2-[(2,3-difluorobenzyl)thio]pyrimidin-4-yl}-2,5-dideoxy-l,3-O-(4- methoxybenzylidene)-D-eryf/ιrø-pentitol
-116-
1H NMR: δ (CDC13) 1.32 (d, 3H), 1.52-1.59 (m, IH), 1.86-1.98 (m, IH), 3.79 (s, 3H), 3.91- 4.01 (m, IH), 4.26-4.31 (m, IH), 4.40 (s, 2H), 5.35-5.41 (m, IH), 5.46 (s, IH), 6.43 (s, IH), 6.86 (d, 2H), 6.96-7.09 (m, 2H), 7.25-7.30 (m, IH), 7.34 (d, 2H). Yield: 0.2Og.
vii) 2-O-{6-[(Azetidin-l-ylsulfonyl)amino]-2-[(2,3-difluorobenzyl)thio]pyriniidin-4-yl}- l,4-dideoxy-3,5-0-(4-methoxybenzylidene)-D-fΛrø>-pentitol
A mixture of Azetidine-1- sulfonamide (73mg), tris(dibenzylideneacetone)dipalladium (0) (50 mg), 2-dicyclohexylphosphino-2',4',6'-tri-/5Opropyl-l,r-biphenyl (XPHOS) (50mg), cesium carbonate (0.14g) and 2-O-{6-chloro-2-[(2,3-difluorobenzyl)thio]pyrimidin-4-yl}-l,4- dideoxy-3,5-0-(4-methoxybenzyridene)-D-t/ιreø-pentitol (the product of example 65 step vi the diastereoisomer which eluted first) (0.145g) in dioxane (1OmL) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for 30min. The reaction mixture was partitioned between aq. ammonium chloride solution (50ml) and EtOAc (150ml). The organics were separated and the aqueous layer was re-extracted with EtOAc (2 xl50 ml). The organics collected, dried (MgSO4) and solvents removed in vacuo to give the subtitle compound as a yellow solid. Yield: 0.45g. MS: APCI(+ve) 609 [M+H+]
Example 66 4-O-{6-[(Azetidin-l-ylsulfonyl)amino]-2-[(2,3-difluorobenzyl)thio]pyrimidin-4-yl}-2,5- dideoxy -O-erythro -pentitol
To a solution of 4-O-{6-[(azetidin-l-ylsuhconyl)arnino]-2-[(2,3-difluorobenzyl)thio] ρyrimidin-4-yl}-2,5-dideoxy-l,3-O-(4-methoxybenzylidene)-D-ervt/iro-pentitol (the product from step i) in MeOH (9ml) was added TFA (ImI) dropwise. The reaction mixture was stirred at room temperaturefor 18h. The reaction mixture was reduced in vacuo and the residue
-117- redissolved in EtOAc (20ml) before reducing in vacuo directly onto silica and purifying by column chromatography on silica 50%EtOAc/50% /sø-hexane to give the title compound as a white solid. Yield: 12 mg MS: APCI(+ve) 489 [M+H+] 1R NMR: δ (DMSO) 1.29 (d, 3H), 1.72-1.77 (m, 2H), 2.26 (q, 2H), 3.83-3.94 (m, 2H), 3.99- 4.07 (m, 5H), 4.37 (s, 2H), 5.18-5.24 (m, IH), 6.32 (s, IH), 6.99-7.10 (m, 2H), 7.20-7.24 (m, IH)
The intermediate for this compound was prepared as follows:
i) 4-O-{6-[(Azetidin-l-ylsulfonyl)amino]-2-[(2,3-difluorobenzyl)thio]pyrimidin-4-yl}-2,5- dideoxy-l,3-0-(4-methoxybenzylidene)-D-eryf/ιro-pentitol
A mixture of azetidine-1 -sulfonamide (0.13g), tris(dibenzylideneacetone)dipalladium (0) (50 mg), 2-dicyclohexylphosphino-2',4',6'-tri-/sopropyl-l,l'-biphenyl (XPHOS) (50mg), cesium carbonate (0.23g) and 4-O-{6-CMoro-2-[(2,3-difluoroberizyl)tlήo]pyrirnidin-4-yl}-2,5- dideoxy-l,3-0-(4-methoxybenzylidene)-D-eryt/ι/O-pentitol (the product of example 65 step vi the diastereoisomer which eluted second) (0.2Og) in dioxane (1OmL) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for 30mins. The reaction mixture was partitioned between aq. ammonium chloride solution (50ml) and EtOAc (150ml). The organics were separated and the aqueous layer was re-extracted with EtOAc (2 xl50 ml). The organics collected, dried (MgSO4) and solvents removed in vacuo to give the subtitle compound as a yellow solid. Yield: 0.5Og. MS: APCI(+ve) 609.9 [M+H+]
-118-
Example 67
N-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-[(lR)-2-hydroxy-l-methylethoxy]-4- pyrimidinyl] 4-methyl-piperazine-l-sulfonamide
To a solution of N-{2-[(2,3-difluorobenzyl)thio]-6-[(l/?)-l-methyl-2-(trityloxy)ethoxy] pyriimdin-4-yl}-4-methylpiperazine-l-sulfonamide (the product from step ϋ) (100 mg) in DCM (3 ml) was added TFA (3ml) dropwise. The reaction was then allowed to stir at room temperaturefor the following 3h until complete. The reaction was then reduced in vacuo and the resulting residue was purified by preparative HPLC to yield the title compound as a white solid. Yield: 45 mg
MS: APCI(+ve) 490 [M+H+]
1U NMR: 5 (DMSO) 1.27 (d, 3H), 2.31 (s, 3H), 2.50 (t, 4H), 3.36 (t, 4H), 3.67-3.77 (m, 2H),
4.31-4.41 (m, 2H), 5.26-5.32 (m, IH), 6.24 (s, IH), 7.00-7.10 (m, 2H), 7.19-7.24 (m, IH)
The intermediates for this compound were prepared as follows:
i) 4-Methylpiperazine-l-sulfonamide
To a solution of 1-Methylpiperazine (1.58 g) in dioxane was added sulf amide (4.0 g) and the reaction mixture was then heated at reflux in dioxane for 18h. The reaction mixture was then reduced in vacuo and the residue partitioned between EtOAc (100 ml) and H2O (100 ml). The organics were separated and the aqueous layer was re-extracted with EtOAc (2 xlOO ml). Organics were combined, dried (MgSO4) and reduced in vacuo to give the subtitle compound as a white solid. Yield: 640 mg 1H NMR: δ (DMSO) 2.18 (s, 3H), 2.37 (t, 4H), 2.94 (t, 4H), 6.74 (s, 2H)
ii) N-{2-[(2,3-Difluorobenzyl)thio]-6-[(lR)-l-methyl-2-(trityloxy)ethoxy]pyrimidin-4-yl}-
4-methylpiperazine- 1 -sulfonamide
-119-
A mixture of 4-Methylpiperazine-l -sulfonamide (the product from step i) (0.64g), tris(dibenzylideneacetone)dipalladium (0) (50 mg), 2-dicyclohexylphosphino-2',4',6'-tri- trøpropyl-l.l'-biphenyl (XPHOS) (50mg), cesium carbonate (0.55 g) and 4- chloro-2-[[(2,3- difluorophenyl)methyl]thio]-6-[( IR)- l-methyl-2-(triphenylmethoxy)ethoxy]-pyrimidine ((the product of example 13 step ϋi), 0.5Og) in dioxane (4OmL) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for Ih. The reaction mixture was diluted with DCM, filtered through arbocel and the filtrate evaporated. The residue was purified by reverse phase HPLC using a TFA (0.2%)/MeCN system to give the subtitle compound as a yellow solid. Yield: 0.22g. 1H NMR: 5 (CDCl3) 1.27 (d, 3H), 2.21 (s, 3H), 2.33-2.47 (m, 4H), 3.24-3.35 (m, 4H), 4.29- 4.42 (m, 2H), 5.42-5.54 (m, IH), 6.26 (s, IH), 6.93-7.08 (m, 2H), 7.18-7.31 (m, 10H), 7.37- 7.41 (m, 6H)
Example 68 N-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-[(lR)-2-hydroxy-l-methylethoxy]-4- pyrimidinyl] - 1 ,4-diazepane- 1 -sulfonamide
To a solution of N-[2-[[(2,3-Difluoroρhenyl)methyl]thio]-6-[(l/?)-l-methyl-2-
(trityloxy)ethoxy]-4-pyrimidinyl]- sulfamoyl-l,4-diazepane-l-carboxylic acid tert-butyl ester (the product of step ϋ, 100 mg) in DCM (3 ml) was added TFA (3ml) dropwise. The reaction was then allowed to stir at room temperaturefor the following 3h until complete. The reaction was then reduced in vacuo and the resulting residue was purified by preparative HPLC to yield the title compound as a white solid. Yield: 62 mg MS: APCI(+ve) 490 [M+H+] 1R NMR: δ (DMSO) 1.13 (d, 3H), 1.90-1.97 (m, 2H), 3.34-3.55 (m, 10H), 4.36-4.42 (m, 2H), 5.02-5.08 (m, IH), 5.77 (s, IH) ), 7.11-7.18 (m, IH), 7.29-7.36 (m, IH), 7.37-7.44 (m, IH)
-120-
The intermediates for this compound were prepared as follows:
i) 4-Sulfamoyl-l,4-diazepane-l-carboxylic acid tert-butyl ester
To a solution of 1,4-Diazepane-l-carboxylic acid tert-butyl ester (3.16 g) in dioxane (40ml) was added sulfamide (4.0 g) and the reaction mixture was then heated at reflux for 18h. The reaction mixture was then reduced in vacuo and the residue partitioned between EtOAc (100 ml) and H2O (100 ml). The organics were separated and the aqueous layer was re-extracted with EtOAc (2 xlOO ml). Organics were combined, dried (MgSO4) and reduced in vacuo to give the subtitle compound as a white solid. Yield: 4.27 g 1H NMR: (DMSO) δ 1.40 (s, 9H), 1.70-1.77 (m, 2H), 3.12-3.23 (m, 4H), 3.32-3.44 (m, 2H), 6.72 (s, 2H)
ii) iV-{2-[(2,3-Difluorobenzyl)thio]-6-[(l/?)-l-methyl-2-(trityloxy)ethoxy]pyrimidin-4-yl}- 4-pyrimidinyl] sulfamoyl-l^-diazepane-l-carboxylic acid tert-butyl ester A mixture of 4-Sulfamoyl-l,4-diazeρane-l-carboxylic acid tert-butyl ester (the product from step i) (0.84g), tris(dibenzylideneacetone)dipalladium (0) (50 mg), 2-dicyclohexylphosρhino- 2',4',6'-tri-/.røpropyl-l,l'-biphenyl (XPHOS) (50mg), cesium carbonate (0.55 g) and 4- chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-[(l/?)-l-methyl-2-(triphenyhnethoxy)ethoxy]- pyrimidine (the product of example 13 step iii), 0.5Og) in dioxane (2OmL) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for 3h. The reaction mixture was then reduced in vacuo and the residue partitioned between EtOAc (100 ml) and H2O (100 ml). The organics were separated and the aqueous layer was re-extracted with EtOAc (2 xlOO ml). Organics were combined, dried (MgSO4) and reduced in vacuo and the resulting residue was purified by prep HPLC to give the subtitle compound as a clear colourless oil. Yield: 0.1 Ig
1B NMR: (CDCl3) 5 1.27 (d, 3H), 1.43 (s, 9H), 1.88 (quintet, 2H), 3.09-3.16 (m, IH), 3.24- 3.30 (m, IH), 3.34-3.53 (m, 8H), 4.28-4.44 (m, 2H), 5.47-5.54 (m, IH), 6.03-6.11 (m, 2H), 6.93-7.08 (m, 2H), 7.18-7.30 (m, 10H), 7.35-7.41 (m, 6H)
-121-
Example 69
N-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-[(lR)-2-hydroxy-l-methylethoxy]-4- pyrimidinyl ] -4-ethyl-piperazine- 1 -sulfonamide
To a solution of ethyl(2/?)-2-[(2-[(2,3-Difluorobenzyl)thio]-6-{[(4-ethylpiperazin-l- yl)sulfonyl] amino }pyrimidin-4-yl)oxy]propanoate (the product from step i) (0.72g) in THF
(10 ml) was added 2M LiBH4 in THF (1.3 ml). The reaction was then stirred for 18h at RT.
Saturated NH4Cl (150 ml) was then added to the reaction mixture which was extracted with DCM (3x150 ml). Organics were combined, dried (MgSO4) and reduced in vacuo and the resulting residue was purified by prep HPLC to give the title compound as a white solid.
Yield: 45 mg
MS: APCI(+ve) 504 [M+H+]
1K NMR: (CDCB) δ 1.15 (d, 3H), 1.24 (t, 3H), 3.13 (q, 2H), 2.50 (t, 4H), 3.36 (t, 4H), 3.67- 3.77 (m, 2H), 4.31-4.41 (m, 2H), 5.32-5.26 (m, IH), 6.24 (s, IH), 7.00-7.10 (m, 2H), 7.19-
7.24 (m, IH)
The intermediate for this compound was prepared as follows:
i) Ethyl (2/?)-2-[(2-[(2,3-difluorobenzyl)thio]-6-{[(4-ethylpiperazin-l- yl)sulfonyl]amino}pyrimidin-4-yl)oxy]propanoate
To a solution of 1-Ethylpiperazine (1 g) in dioxane (10 ml) was added sulfamide (0.746 g) and the reaction mixture was then heated at reflux in dioxane for 72h. The reaction mixture was purified by loading onto SCX and eluting with (200 ml) MeOH/NH3. The eluent was then reduced in vacuo to yield 4-Ethylpiρerazine-l- sulfonamide as a white solid. A mixture of 4- ethylpiperazine-1 -sulfonamide (0.289g), tris(dibenzylideneacetone)dipalladium (0) (50 mg),
-122-
2-dicyclohexylphosphino-2',4',6'-tri-irøpropyl-l,r-biphenyl (XPHOS) (50mg), cesium carbonate (0.628 g) and 2-[[6-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-4- pyrimidinyl]oxy]-(2R)-propanoic acid ethyl ester ((the product of example 5 step i), 0.5Og) in dioxane (1OmL) was heated at reflux in a microwave at lOOC, 300W, open vessel with cooling for 30min. The reaction mixture was then reduced in vacuo and the residue was partitioned between EtOAc (150 ml) and H2O (100 ml). The organics were separated and the aqueous layer was re-extracted with EtOAc (2 xl50ml). Organics were combined, dried (MgSO4) and reduced in vacuo to give the subtitle compound as a yellow solid. Yield: 0.720 g MS: APCI(+ve) 546 [M+H+]
Example 70
N-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-[(l/?)-2-hydroxy-l-methylethoxy]-4- pyrimidinyl ] -4-(4-pyridyl)piperazine-l-sulfonamide
To a solution of ylpiperazine)sulfonyl]amino}pyrirnidin-4-yl)oxy]propanoate (the product from step i) (0.72g) in THF (10 ml) was added 2M LiBH
4 in THF (1.3 ml). The reaction was then stirred for 18h at RT. Saturated NH
4Cl (150 ml) was then added to the reaction mixture which was extracted with DCM (3x150 ml). Organics were combined, dried (MgSO
4) and reduced in vacuo and the resulting residue was purified by prep HPLC to give the title compound as a white solid. Yield: 10 mg MS: APCI(+ve) 553 [M+H
+]
1H NMR: (DMSO) δ 1.16 (d, 3H), 3.37-3.41 (m, 4H), 3.46-3.51 (m, 2H), 3.75-3.79 (m, 4H), 4.40-4.48 (m, 2H), 5.11-5.17 (m, IH), 6.01 (s, IH), 7.13-7.21 (m, 3H), 7.31-7.39 (m, 2H), 8.28 (d, 2H)
-123-
The intermediate for this compound was prepared as follows:
^ Ethyl ilRyi-Kl-Kl^-dΑuoTobenzyYfMoπ-iK^pyndm-Λ-ylpipemήn-l- yl)sulfonyl]amino}pyrimidin-4-yl)oxy]propanoate To a solution of l-pyridin-4-ylpiρerazine (1.23 g) in dioxane (10ml) was added sulfamide (0.746 g) and the reaction mixture was then heated at reflux in dioxane for 72h. The reaction mixture was purified by loading onto SCX and eluting with (200 ml) MeOHTNH3. The eluent was then reduced in vacuo to yield 4-pyridin-4-ylpiperazine-l -sulfonamide as a white so Ud. A mixture of 4-pyridin-4-ylpiperazine-l- sulfonamide (0.26Og), tris(dibenzylideneacetone)dipalladium (0) (50 mg), 2-dicyclohexylphosphino-2',4',6'-tri- jrapropyl-l,l'-biphenyl (XPHOS) (50mg), cesium carbonate (0.438 g) and 2-[[6-chloro-2- [[(2,3-difluorophenyl)methyl]thio]-4-pyrimidinyl]oxy]-(2R)-propanoic acid ethyl ester ((the product of example 5 step i), 0.350g) in dioxane (10ml) was heated at reflux in a microwave at lOOC, 300W, open vessel with cooling for 30 mins. The reaction mixture was then reduced in vacuo and the residue was partitioned between EtOAc (150 ml) and H2O (100 ml). The organics were separated and the aqueous layer was re-extracted with EtOAc (2 xl50ml). Organics were combined, dried (MgSO4) and reduced in vacuo to give the subtitle compound as a yellow solid. Yield: 0.720 g MS: APCI(+ve) 595 [M+H+]
Example 71
7V-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-[(l/?)-2-hydroxy-l-methylethoxy]-4- pyrimidinyl ]-(3R)-3-ethylpiperazine-l-sulfonamide
To a solution of ethyl(2Λ)-2-[(2-[(2,3-difluorobenzyl)ihio]-6-{[((3Λ)-3- ethylpiperazine)sulfonyl]amino}pyrirnidin-4-yl)oxy]propanoate (the product from step i)
-124-
(0.7Ig) in THF (10 ml) was added 2M LiBH4 in THF (1.3 ml). The reaction was then stirred for 18h at RT. Saturated NH4Cl (150 ml) was then added to the reaction mixture which was extracted with DCM (3x150 ml). Organics were combined, dried (MgSO4) and reduced in vacuo and the resulting residue was purified by prep HPLC to give the title compound as a white solid. Yield: 70 mg MS: APCI(+ve) 504 [M+H+]
1B. NMR: (CDCl3) 50.87 (t, 3H), 1.28 (d, 3H), 1.53-1.63 (m, IH), 1.63-1.73 (m, IH), 2.79- 2.88 (m, IH), 2.95-3.08 (m, 2H), 3.61-3.80 (m, 4H), 3.97-4.05 (m, IH), 4.08-4.16 (m, IH), 4.33-4.44 (m, 2H), 5.28-5.36 (m, IH), 6.28 (m, IH), 6.99-7.12 (m, 2H), 7.19-7.24 (m, IH)
The intermediate for this compound was prepared as follows:
i) Ethyl (2R)-2-[(2-[(2,3-difluorobenzyl)thio]-6-{[((3R)-3-ethylpiperazine-l- sulfonaimde)sulfonyl]amino}pyrirnidiii-4-yl)oxy]propanoate To a solution of (3/?)-3-ethylρiperazine (0.5g) in dioxane (10ml) was added sulfamide (0.373g) and the reaction mixture was then heated at reflux in dioxane for 3d. The reaction mixture was purified by loading onto SCX and eluting with (200 ml) MeOHZNH3. The eluent was then reduced in vacuo to yield (3i?)-3-ethylpiperazine-l -sulfonamide as a white solid. A mixture of (3/?)-3-ethylpiperazine-l-sulfonamide (0.26Og), tris(dibenzylideneacetone)dipalladium (0) (50 mg), 2-dicyclohexylphosphino-2',4',6'-tri- i.ropropyl-l,l'-biphenyl (XPHOS) (50mg), cesium carbonate (0.438 g) and 2-[[6-chloro-2- [[(2,3-difluorophenyl)methyl]thio]-4-pyrimidinyl]oxy]-(2R)-propanoic acid ethyl ester ((the product of example 5 step i), 0.350g) in dioxane (10ml) was heated at reflux in a microwave at lOOC, 300W, open vessel with cooling for 30min. The reaction mixture was then reduced in vacuo and the residue was partitioned between EtOAc (150 ml) and H2O (100 ml). The organics were separated and the aqueous layer was re-extracted with EtOAc (2 xl50ml). Organics were combined, dried (MgSO4) and reduced in vacuo to give the subtitle compound as a yellow solid. Yield: 0.705 g MS: APCI(+ve) 546 [M+H+]
-125-
Example 72
N-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-[(lR)-2-hydroxy-l-methylethoxy]-4- pyrimidinyl ]- (3R,5S)-3,5-dimethylpiperazine-l -sulfonamide
To a solution of Ethyl(2fl)-2-[(2-[(2,3-dffluorobenzyl)thio]-6- { [((3_R,5S)-3,5- dimethylpiperazine)sulfonyl]amiαo}pyrirnidin-4-yl)oxy]propanoate (the product from step i), 0.5Og) in THF (10 ml) was added 2M LiBH
4 in THF (0.9 ml). The reaction was then stirred for 18h at RT. Saturated NH
4Cl (150 ml) was then added to the reaction mixture which was extracted with DCM (3x150 ml). Organics were combined, dried (MgSO
4) and reduced in vacuo and the resulting residue was purified by prep HPLC to give the title compound as a white solid. Yield: 80 mg MS: APCI(+ve) 504 [M+H
+]
1H NMR: (DMSO) δ 1.13 (d, 6H), 3.01-3.21 (m, 2H), 3.37-3.57 (m, 4H), 4.33-4.43 (m, 2H), 4.76-4.81 (m, 2H), 4.97-5.05 (m, IH), 5.81 (s, IH), 7.09-7.17 (m, IH), 7.26-7.35 (m, IH), 7.39-7.45 (m, IH)
The intermediate for this compound was prepared as follows:
i) Ethyl (2R)-2-[(2-[(2,3-difluorobenzyl)thio]-6-{[((3tf,5S)-3,5- dimethylpiperazine)sulfonyl]amino}pyrimidin-4-yl)oxy]propanoate
To a solution of (2/?,65)-2,6-dimethylpiperazine (Ig) in dioxane (10ml) was added sulfamide (0.746g) and the reaction mixture was then heated at reflux in dioxane for 72h. The reaction mixture was partitioned between EtOAc (150ml) and H2O (150ml) and the aqueous re- extracted with EtOAc (2x150ml). Organics were collected dried and reduced in vacuo to yield (3/?,5S)-3,5-dimethylpiperazine-l-sulfonamide as a white solid (0.29g). A mixture of (3R,5S)- 3,5-dimethylpiperazine-l-sulfonamide (0.29g), tris(dibenzylideneacetone)dipalladium (0) (50
-126- mg), 2-dicyclohexylphosphino-2',4',6'-tri-wopropyl-l,r-biphenyl (XPHOS) (50mg), cesium carbonate (0.628 g) and 2-[[6-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-4- pyrimidinyl]oxy]-(2R)-propanoic acid ethyl ester (the product of example 5 step i), 0.5g) in dioxane (1OmL) was heated at reflux in a microwave at 100°C, 300W, open vessel with cooling for 30min. The reaction mixture was then reduced in vacuo and the residue was partitioned between EtOAc (150 ml) and H2O (100 ml). The organics were separated and the aqueous layer was re-extracted with EtOAc (2 xl50ml). Organics were combined, dried (MgSO4) and reduced in vacuo to give the subtitle compound as a yellow solid. Yield: 0.940 g MS: APCI(+ve) 546 [M+H+]
Example 73 iV-{2-[(2,3-Difluorobenzyl)tMo]-6-[(lR)-2-hydroxy-l-methylethoxy]pyrimidin-4-yl}-9- methyl-3,9-diazabicyclo[4.2.1 ] nonane-3-sulfonamide
To a solution of ethyl (2fl)-2-[(2-[(2,3-difluorobenzyl)thio]-6- { [(9-methyl-3,9- diazabicyclo[4.2. l]non-3-yl)sulfonyl] amino }pyrimidin-4-yl)oxy]propanoate (the product from step i), 0.35g) in THF (10ml) was added 2M LiBH
4 in THF (0.6ml). The reaction was then stirred for 18h at room temperature. Saturated NH
4Cl (150ml) was then added to the reaction mixture which was extracted with DCM (3xl50ml). Organics were combined, dried (MgSO
4) and reduced in vacuo and the resulting residue was purified by prep HPLC to give the title compound as a white solid. Yield 20mg MS: APCI(+ve) 530 [M+H
+]
1H NMR: (CDCL3) δ 1.27 (d, 3H), 1.32-1.40 (m, 2H), 1.65-1.80 (m, 4H), 2.54 (s, 3H), 2.91- 3.05 (m, 2H), 3.39-3.48 (m, 2H), 3.65-3.75 (m, 2H), 4.03-4.11 (m, IH), 4.16-4.24 (m, IH), 4.32-4.46 (m, 2H), 5.22-5.28 (m, IH), 6.19 (s, IH), 6.98-7.08 (m, 2H), 7.20-7.32 (m, IH)
-127-
The intermediate for this compound was prepared as follows:
i) Ethyl (2R)-2-[(2-[(2,3-difluorobenzyl)thio]-6-{[(9-methyl-3,9-diazabicydo[4.2.1]non-3- yl)sulfonyl]amino}pyrimidin-4-yl)oxy]propanoate To a solution of 9-methyl-3,9-diazabicyclo[4.2. l]nonane (0.56g) in 1,4-dioxane (10ml) was added sulfamide (0.37g) and the reaction mixture was then heated at reflux in 1,4-dioxane for 72h. The reaction mixture was purified by loading onto SCX and eluting with 7N NH3ZMeOH (200ml). The eluent was then reduced in vacuo to yield 9-methy 1-3,9- diazabicyclo[4.2.1]nonane-3-sulfonamide (0.13g) as a yellow solid. A mixture of 9-methyl- 3,9-diazabicyclo[4.2. l]nonane-3-sulfonamide (0.13g), tris(dibenzylideneacetone)diρalladium (0) (50mg), 2-dicyclohexylphosphino-2',4',6'-tri-i5Opropyl-l,l'-biphenyl (XPHOS) (50mg), cesium carbonate (0.3Ig) and 2-[[6-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-4- pyrimidinyl]oxy]-(2R)-propanoic acid ethyl ester (the product of example 5 step i), 0.25g) in 1,4-dioxane (1OmL) was heated at reflux in a microwave at 100°C, 300W, open vessel with cooling for 30min. The reaction mixture was then reduced in vacuo and the residue was partitioned between EtOAc (150ml) and H2O (100ml). The organics were separated and the aqueous layer was re-extracted with EtOAc (2 xl50ml). Organics were combined, dried (MgSO4) and reduced in vacuo to give the subtitle compound as a yellow solid. Yield 0.35 g MS: APCI(+ve) 572 [M+H+]
Example 74
N-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-[(lR)-2-hydroxy-l-methylethoxy]-4- py rimidiny 1 ] - (35)-3-methylpiperazine- 1 -sulfonamide
To a solution of Ethyl (2/?)-2-[(2-[(2,3-difluorobenzyl)thio]-6-{[((35)-3-methylpiperazine-l- suh
conamide)sulfonyl]amino}pyrimidin-4-yl)oxy]propanoate (the product from step i) (0.94g) in THF (10 ml) was added 2M LiBH
4 in THF (1.8 ml). The reaction was then stirred
-128- for 18h at RT. Saturated NH
4Cl (150 ml) was then added to the reaction mixture which was extracted with DCM (3x150 ml). Organics were combined, dried (MgSO
4) and reduced in vacuo and the resulting residue was purified by prep HPLC to give the title compound as a white solid. Yield: 35 mg MS: APCI(+ve) 490 [M+H
+]
1H NMR: (CDCl3) δ 1.36 (d, 3H), 1.61 (d, 3H), 2.81-3.03 (m, 3H), 3.20-3.39 (m, 4H), 3.67- 3.84 (m, 2H), 4.22-4.44 (m, 2H), 5.29-5.37 (m, IH), 6.18 (s, IH), 7.02-7.11 (m, 2H), 7.13- 7.19 (m, IH)
The intermediate for this compound was prepared as follows:
i) Ethyl (2R)-2-[(2-[(2,3-difluorobenzyl)thio]-6-{[((3S)-3- methylpipera-dne)sulfonyl]amino}pyrimidiii-4-yl)oxy]propanoate
To a solution of (2S)-2-methylpiρerazine (0.914g) in dioxane (10ml) was added sulfamide (0.746g) and the reaction mixture was then heated at reflux in dioxane for 3d. The reaction mixture was partitioned between EtOAc (150ml) and H2O (150ml) and the aqueous re- extracted with EtOAc (2xl50ml). Organics were collected dried and reduced in vacuo to yield (3S)-3-methylpiperazine-l -sulfonamide as a white solid (0.27g). A mixture of (3S)-3- methylpiperazine-1- sulfonamide (0.27g), tris(dibenzylideneacetone)dipalladium (0) (50 mg), 2-dicyclohexylphosphino-2' ,4' ,6' -tri-irøpropyl- 1,1' -biphenyl (XPHOS) (50mg), cesium carbonate (0.628 g) and 2-[[6-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-4- pyrimidinyl]oxy]-(2R)-propanoic acid ethyl ester ((the product of example 5 step i), 0.5Og) in dioxane (2OmL) was heated at reflux in a microwave at 100°C, 300W, open vessel with cooling for 30 mins. The reaction mixture was then reduced in vacuo and the residue was partitioned between EtOAc (150 ml) and H2O (100 ml). The organics were separated and the aqueous layer was re-extracted with EtOAc (2 xl50ml). Organics were combined, dried (MgSO4) and reduced in vacuo to give the subtitle compound as a yellow solid. Yield: 0.940 g MS: APCI(+ve) 532 [M+H+]
-129-
Example 75
N-(2-[(2,3-Difluorobenzyl)thio]-6-{[(lR,2R)-2-hydroxy-methylpropyl]oxy}pyrimidin-4- yl)- 1 ,4-diazepane- 1 -sulfonamide
N-(2-[(2,3-Difluorobenzyl)thio]-6-{[(lR,2R)-2-hydroxy-methylpropyl]oxy}pyrimidin-4-yl)- tert-bυtyl 4-(aminosulfonyl)-l,4-diazepane-l-carboxylate (1.6g) was dissolved in DCM
(30ml) and stirred until in solution. To this solution was added TFA (30ml). The reaction was then allowed to stir at room temperatureovernight. The reaction was then reduced in vacuo and the resulting yellow residue purified by HPLC to give the title compound as a white solid. Yield: 76mg MS: APCI(+ve) 504 [M+H+]
1H NMR: (DMSO) δ 1.04 (d, 3H), 1.14 (d, 3H), 1.96-2.02 (m, 2H), 3.16-3.24 (m, 2H), 3.41- 3.45 (m, 4H), 3.66-3.74 (m, 2H), 4.41-4.49 (m, 2H), 4.99-5.05 (m, IH), 5.09 (s, IH), 7.14- 7.23 (m, IH) , 7.31-7.40 (m, 2H), 8.65-8.72 (m, 2H)
The intermediate for this compound was prepared as follows:
i) N-(2-[(2,3-Difluorobenzyl)thio]-6-{[(lR,2R)-2-hydroxy-methylpropyl]oxy}pyrimidin- 4-yl)-fert-butyl 4-(aminosulfonyl)-l,4-diazepane-l-carboxylate
A mixture of 4-sulfamoyl-l,4-diazepane-l-carboxylic acid tert-butyl ester (the product from example 68 step i) (0.54Ig), tris(dibenzylideneacetone)dipalladium (0) (50 mg), 2- dicyclohexylρhosphino-2',4',6'-tri-irøpropyl-l,r-biphenyl (XPHOS) (50mg), cesium carbonate (0.55 g) and (2i?,3i?)-3-({6-chloro-2-[(2,3-difluorobenzyl)thio]pyrimidin-4-
-130- yl}oxy)butan-2-ol (the product of example 4 step i), 0.54Ig) in dioxane (4OmL) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for 15 mins. The reaction mixture was then reduced in vacuo and the residue separated between DCM (200 ml) and H2O (150 ml). The organics were separated and the aqueous layer was re-extracted with DCM (2 x 200ml). Organics were combined, dried (MgSO4) and reduced in vacuo and the resulting residue was purified by prep HPLC to give the subtitle compound as a yellow oil. Yield: 1.6g MS: APCI(+ve) 604 [M+H+]
Example 76 N-(2-[(2,3-Difluorobenzyl)thio]-6-{[(lR,2R)-2-hydroxy-methylpropyl]oxy}pyrimidiii-4- yl)- (3R,55)-3,5-dimethylpiperazine-l-sulfonamide
To a solution of (22?,6S)-2,6-Dimethylpiperazine (Ig) in dioxane (10ml) was added sulfamide (0.746g) and the reaction mixture was then heated at reflux in dioxane for 3d. The reaction mixture was partitioned between EtOAc (150ml) and H2O (150ml) and the aqueous re- extracted with EtOAc (2xl50ml). Organics were collected dried and reduced in vacuo to yield (3/?,5S)-3,5-dirnethylpiρerazine-l -sulfonamide as a white solid (1.05g). A mixture of (3R,5S)- 3,5-dimethylpiperazine-l-sulfonamide (0.54Ig), tris(dibenzylideneacetone)dipalladium (0) (50 mg), 2-dicyclohexylρhosρhino-2\4\6'-tri-/søρropyl-l,l'-biprienyl (XPHOS) (50mg), cesium carbonate (0.731 g) and (2/?,3i?)-3-({6-cMoro-2-[(2,3-dmuorobenzyl)tMo]pyrimidin- 4-yl}oxy)butan-2-ol ((the product of example 4 step i), 0.54Ig) in dioxane (4OmL) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for 20min. The reaction mixture was then reduced in vacuo and the residue separated between DCM (200 ml) and H2O (200 ml). The organics were separated and the aqueous layer was re-extracted with DCM (2 x 200ml). Organics were combined, dried (MgSO4) and reduced in vacuo and the resulting residue was purified by prep HPLC to give the title compound as a white solid. Yield: 80mg
-131-
MS: APCI(+ve) 518 [M+H+]
1B. NMR: (CD3OD) 5 1.16 (d, 3H), 1.22 (d, 3H), 1.33 (d, 6H), 2.89-2.96 (m, 2H), 3.37-3.48 (m, 2H), 3.78-3.85 (m, IH), 3.99-4.04 (m, 2H), 4.40-4.50 (m, 2H), 5.09-5.16 (m, IH), 5.99 (s, IH) , 7.07-7.21 (m, 2H), 7.30-7.36 (m, 2H)
Example 77
N-(2-[(2,3-Difluorobenzyl)thio]-6-{[(lR,2R)-2-hydroxymethylpropyl]oxy}pyrimidin-4- yl)- piperazine-1 -sulfonamide
N-(2-[(2,3-difluorobenzyl)thio]-6-{[(lR,2R)-2-hydroxymethylpropyl]oxy}pyrimidin-4-yl) tert-butyl 4-(aminosulfonyl)piperazine-l-carboxylate (1.45g) was dissolved in DCM (10ml) and allowed to stir at room temperatureuntil homogeneous. TFA (10ml) was then slowly added and the reaction mixture stirred overnight. The reaction mixture was reduced in vacuo, dissolved in MeOH and purified by prep HPLC to give the title compound as a white solid Yield: 25mg
MS: APCI(+ve) 490 [M+H+]
1H NMR: (DMSO) 5 1.02 (d, 3H), 1.09 (d, 3H), 3.01-3.05 (m, 4H), 3.14-3.18 (m, 4H), 3.64- 3.71 (m, 2H), 4.32-4.42 (m, 2H), 4.69-4.73 (m, IH), 4.87-4.94 (m, IH), 5.84 (s, IH), 7.10- 7.17 (m, IH) , 7.27-7.34 (m, IH), 7.40-7.46 (m, IH)
The intermediate for this compound was prepared as follows:
i) N-(2-[(2,3-Difluorobenzyl)thio]-6-{[(lR,2R)-2-hydroxymethylpropyl]oxy}pyrimidiii-4- yl) tert-buty\ 4-(aminosulfonyl)piperazine-l-carboxylate A mixture of 4-(Aminosulfonyl)- 1 , 1-dimethylethyl ester- 1-piperazinecarboxy lie acid (0.663g), tris(dibenzylideneacetone)dipalladium (0) (50 mg), 2-dicyclohexylphosphino-
-132-
2',4',6'-tri-i.røpropyl-l,l'-biphenyl (XPHOS) (50mg), cesium carbonate (0.731 g) and (2i?,3/?)-3-({6-chloro-2-[(2,3-difluorobenzyl)thio]pyriinidin-4-yl}oxy)butan-2-ol ((the product of example 4 step i), 0.54Ig) in dioxane (4OmL) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for 1.5h. The reaction mixture was then reduced in vacuo and the residue partitioned between EtOAc (200 ml) and H2O (200 ml). The organics were separated and the aqueous layer was re-extracted with EtOAc (2 x 200ml). Organics were combined, dried (MgSO4) and reduced in vacuo to give the subtitle compound as a yellow solid. Yield: 1.45g MS: APCI(+ve) 590 [M+H+]
Example 78
N-(2-[(2,3-Difluorobenzyl)thio]-6-{[(lR,2R)-2-hydroxymethylpropyl]oxy}pyrimidin-4- yl)-l,2,3,4-tetrahydroisoquinoline-7-sulfonamide
N-(2-[(2,3-Difluorobenzyl)thio]-6- { [( lR,2R)-2-hydroxymethylpropyl]oxy }pyrimidin-4-yl) 2- (trifluoroacetyl)-l
)2,3,4-tetrahydroisoquiαoune-7-suh
conamide (the product from step ϋi), 0.54g) was added to a solution of 7N NH
3 in MeOH (20ml), sealed and stirred at room temperaturefor Ih. The reaction was reduced in vacuo and the resulting residue purified by prep HPLC to give the title compound as a white solid. Yield: 180mg MS: APCI(+ve) 537 [M+H
+]
1H NMR: (DMSO) δ 0.98 (d, 3H), 1.04 (d, 3H), 2.95 (t, 2H), 3.30 (t, 2H), 3.60-3.67 (m, IH),
4.22 (s, 2H), 4.25-4.27 (m, 2H), 4.78-4.85 (m, IH), 5.63 (s, IH), 7.09-7.15 (m, IH) , 7.21-
7.23 (m, IH), 7.26-7.39 (m, 2H), 7.56-7.61 (m, 2H)
The intermediates for this compound were prepared as follows:
i) iV-(^e/t-Butyl)-2-(trifluoroacetyl)-l,2,3,4-tetrahydroisoqiiinoline-7-sulfonamide
-133-
To a solution of 2-(trifluoroacetyl)-l,2,3,4-tetrahydroisoquinoline-7-sulfonyl chloride (3g) in DCM (50ml) was added 2-methylpropan-2-amine (1.73g). The reaction was then allowed to stir at room temperature 18h. The reaction was partitioned between H2O (100ml) and DCM (100ml). The organics were separated and the aqueous layer was re-extracted with DCM (2 x 5 200ml). Organics were combined, dried (MgSO4) and reduced in vacuo to give the subtitle compound as colourless oil. Yield: 3.56g
1H NMR: (DMSO) 8 1.10 (s, 9H), 2.95-3.02 (m, 2H), 3.80-3.86 (m, 2H), 4.79-4.86 (m, 2H), 7.37-7.49 (m, 2H), 7.63-7.72 (m, IH)
0 ii) 1,2,3,4- Tetrahydroisoquinoline-7-sulfonamide
N-(tert-Butyl)-2-(trifluoroacetyl)- 1 ,2,3,4-tetrahydroisoquinoline-7-suhconamide (the product from step i), 1.78g) was dissolved in TFA and stirred at room temperaturefor 96h. The reaction was reduced in vacuo and the residue purified by column chromatography on silica gel 50% EtO Ac/50 % /.rø-hexane to give the subtitle compound as a white solid. Yield: 0.65g 5 1H NMR: (DMSO) δ 2.95-3.02 (m, 2H), 3.80-3.86 (m, 2H), 4.80-4.85 (m, 2H), 7.38-7.43 (m, IH), 7.64-7.77 (m, 2H)
iϋ) N-(2-[(2,3-Difluorobenzyl)thio]-6-{[(lR,2R)-2-hydroxymethylpropyl]oxy}pyrimidin- 4-yl) 2-(trifluoroacetyl)-l,2,3,4-tetrahydroisoquinoline-7-sulfonamide 0 A mixture of l,2,3,4-Tetrahydroisoquinoline-7-sulfonamide (the product from step ii), 0.65g), tris(dibenzylideneacetone)dipalladium (0) (50 mg), 2-dicyclohexylphosphino-2',4',6'-tri- /røρropyl-l,l'-biρhenyl (XPHOS) (50mg), cesium carbonate (0.731 g) and (2R,3R)-3-({6- chloro-2-[(2,3-difluorobenzyl)thio]pyrimidin-4-yl}oxy)butan-2-ol (the product of example 4 step i), 0.432g) in dioxane (4OmL) was heated at reflux in a microwave at 1000C, 300W, open 5 vessel with cooling for 20min. The reaction mixture was then reduced in vacuo and the residue separated between DCM (100 ml) and H2O (100 ml). The organics were separated and the aqueous layer was re-extracted with DCM (2 x 100ml). Organics were combined, dried (MgSO4) and reduced in vacuo and the resulting residue was purified by column chromatography on silica 50% EtOAc/50 % /.rø-hexane to give the subtitle compound as a 0 clear oil. Yield: 0.54g
MS: APCI(+ve) 633 [M+H+]
-134-
Example 79 iV-{2-[(2,3-Diαuorobenzyl)thio]-6-[(lS)-2-hydroxy-l- (isopropoxymethyl)ethoxy]pyrimidin-4-yl}-l-methyl-l/ir-iinidazole-4-sulfonamide
The title compound was prepared according to the procedure outlined in example 34 using N- {6-[(li?)-2-{[tert-butyl(dimethyl)silyl]oxy}-l-(isopropoxymethyl)ethoxy]-2-[(2,3- dMuorobenzyl)tWo]pyriπήdin-4-yl}-l-methyl-lH-iπύdazole-4-sulfonarnide (the product from step (v) (90mg) in TΗF (5mL) and IM solution of tetrabutylammoniumfluoride in TΗF (0.28mL) to give the title compound as a white solid. Yield: 30mg. MS: APCI(+ve) 530 [M+Η
+]
1K NMR:(DMSO) δ 0.98 - 1.04 (m, 6H), 3.47 - 3.56 (m, 4H), 3.67 (s, 3H), 4.40 (s, 2H), 5.14 (q, IH), 6.17 (s, IH), 7.07 - 7.18 (m, IH), 7.28 - 7.41 (m, 2H), 7.79 (d, IH), 8.00 (d, IH), 11.57 (s, IH)
The intermediates for this compound were prepared as follows:
i) (4R)-4-(isøPropoxymethyl)-2,2-dimethyl-l,3-dioxolane
To a solution of 2,2-dimethyl-l, 3-dioxolane-4-methanol (2g), in DMSO (5OmL), powdered potassium hydroxide was added portionwise at 0°C then warmed to room temperature. 2- Iodo-propane (43mL) was added to the mixture at 0°C then stirred for 72h at room temperature. The reaction mixture was diluted with H2O and extracted with EtOAc. The organic layer was washed with H2O then brine (x2) and dried (MgSO4), filtered and evaporated to give the subtitle compound as clear, colourless oil. Yield: 2g 1K NMR:(DMSO) δ 1.08 (d, 6H), 1.26 (d, 3H), 1.31 (s, 3H), 3.30 - 3.43 (m, 2H), 3.51 - 3.61 (m, 2H), 3.94 - 4.00 (m, IH), 4.08 - 4.15 (m, IH)
-135-
ii) (2S)-3-wøPropoxypropane-l,2-diol
Acetyl chloride was added dropwise into a solution of MeOH (3OmL) at O0C with stirring for 5 min. A solution of (42?)-4-(«øpropoxymethyl)-2,2-dimethyl-l,3-dioxolane (1.7g) (the product from step (i), in MeOH (3OmL), was added dropwise to the reaction mixture. The solution was then warmed to room temperature and stirred for 2h. The reaction mixture was evaporated to give the subtitle compound as clear oil. Yield: 0.8g 1H NMR:(DMSO) δ 1.07 (dd, 6H), 3.21 - 3.37 (m, 4H), 3.47 - 3.55 (m, 2H)
iii) (2/?)-l-{[^rt-Butyl(dimethyl)silyl]oxy}-3-isopropoxypropan-2-ol
The subtitle compound was prepared according to the procedure outlined in example 34 step iii) using (2S)-3-irøpropoxyproρane-l,2-diol (0.8Og) (the product from step (ϋ) in DCM (1OmL), tert-butyldimethylsilyl chloride (1.59g), triethylamine (1.43mL) and 4- (dimethylamino)pyridine (50mg) at 0°C to give the subtitle compound as a clear, colourless oH. Yield: 1.86g
1H NMR:(DMSO) δ 0.07 (s, 6H), 0.91 (s, 9H), 1.11 (d, 6H), 3.26 - 3.35 (m, 2H), 3.37 - 3.45 (m, 2H), 3.49 - 3.61 (m, 2H), 4.63 (d, IH)
iv) 4-[(l/?)-2-{[tert-Butyl(dimethyl)silyl]oxy}-l-(isopropoxymethyl)ethoxy]-6-chloro-2- [(2,3-difluorobenzyl)thio]pyrimidine
The subtitle compound was prepared according to the procedure outlined in example 1 step iii) using 4,6-Dichloro-2-[(2, 3-difluorobenzyl)thio]pyrimidine (product of example 1 step ii) (0.46g), (22?)-l-{[tert-butyl(dimethyl)silyl]oxy}-3-/røpropoxypropan-2-ol (product of step iii) (0.66g), THF (5mL) and 60% sodium hydride (80mg), to give the subtitle compound as a colourless oil. Yield: 0.56g
MS: APCI(+ve) 519/521 [M+H+]
v) N-{6-[(l/?)-2-{[^rr-Butyl(dimethyl)silyl]oxy}-l-(isopropoxymethyl)ethoxy]-2-[(2,3- difluorobenzyl)thio]pyrimidin-4-yl}-l-methyl-l/ir-imidazole-4-sulfonaniide
The subtitle compound was prepared according to the procedure outlined in example 1 step (iv) using a mixture of azetidine-1 -sulfonamide (prepared according to patent WO
-136-
2004/011443) (0.19g), tris(dibenzylideneacetone)dipalladium (0) (0.53g), 2- dicyclohexylphosphino-2',4',6'-tri-/røpropyl-l,r-biphenyl (XPHOS) (39mg), cesium carbonate (0.28g), 4-[(l/?)-2-{[tert-butyl(dimethyl)silyl]oxy}-l-(i5opropoxymethyl)ethoxy]- 6-chloro-2-[(2,3-difluorobenzyl)thio]pyrimidine (the product of step iv) (0.3g) and dioxane (15mL). Purification was by column chromatography on silica gel using EtOAc/ira-hexane (2:8) 50:70 as eluent, to give the title compound as a white solid. Yield: 90mg MS: APCI(+ve) 645 [M+H+]
Example 80 iV-{2-[(2,3-Difluorobenzyl)thio]-6-[(l/?)-2-hydroxy-l-methylethoxy]pyrimidin-4-yl}-l,2- dimethyl- lH-iiiiidazole-4-sulfonaniide
The title compound was prepared according to the procedure outlined in example 11 using a mixture of ethyl (2/?)-2-[(2-[(2,3-difluorobenzyl)thio]-6-{[(l,2-dimethyl-lH-imidazol-4- yl)sulfonyl]amino}pyrimidin-4-yl)oxy]propanoate (the product from step (i) (0.25g), lithium borohydride (2M solution in TΗF, 0.48mL) and TΗF (6mL). Purification was by reverse phase ΗPLC (symmetry as the stationary phase and TFA/acetonitrile as the mobile phase) then titurated with Toluene, DCM and then Et2O /isø-hexane to give the title compound as a white solid. Yield: 44mg MS: APCI(+ve) 486 [M+Η+]
1H NMR:(DMS0) 5 1.14 (d, 3H), 2.27 (s, 3H), 3.44 - 3.49 (m, 2H), 3.56 (s, 3H), 4.41 (s, 2H), 5.02 - 5.14 (m, IH), 6.11 (s, IH), 7.08 - 7.20 (m, IH), 7.25 - 7.43 (m, 2H), 7.92 (s, IH), 11.44 (s, IH)
The intermediate for this compound was prepared as follows:
-137- i) Ethyl (2R)-2-[(2-[(2,3-difluorobenzyl)thio]-6-{[(l,2-dimethyl-m-imidazol-4- yl)sulfonyl]amino}pyrimidin-4-yl)oxy]propanoate
The subtitle compound was prepared according to the procedure outlined in example 1 step (iv) using a mixture of l,2-Dimethyl-lH-imidazole-4-sulfonic acid amide (0.19g), tris(dibenzylideneacetone)dipalladium (0) (56mg), 2-dicyclohexylphosphino-2 ' ,4' ,6' -tri- /røpropyl-l,l'-biphenyl (XPHOS) (41mg), cesium carbonate (0.32g), 2-[[6-chloro-2-[[(2,3- difluorophenyl)methyl]thio]-4-pyrimidinyl]oxy]-(2/?)-propanoic acid ethyl ester (the product of example 11 step i) (0.24g) and dioxane (2OmL). Purification was by column chromatography on silica gel using DCM /MeOH (100: 1 to 90: 10 gradient) as eluent, to give the title compound as a pale yellow solid. Yield: 0.25g MS: APCI(+ve) 528 [M+H+]
Example 81
2-{4-[2-(2,3-Difluoro-benzylsulfanyl)-6-methoxy-pyrimidin-4-ylsulfamoyl]-piperazin-l- yl}-N,N-dimethyl-acetamide
To a solution of N,N-Dimethyl-2-piperazin-l-yl-acetamide (0.5Ig), in dioxane (2OmL) was added sulfamide (0.29g). The reaction mixture was then heated at reflux for 24 h. The reaction mixture was allowed to cool before being reduced in vacuo to give the intermediate compound as an off white solid. Yield: 0.65g
The title compound was prepared according to the procedure outlined in example 1 step (iv) using a mixture of the above intermediate compound (0.38g), tris(dibenzylideneacetone)dipalladium (0) (92mg), 2-dicyclohexylphosphino-2',4',6'-tri- irøρropyl-l,l'-biphenyl (XPHOS) (67mg), cesium carbonate (0.49g), 4-Chloro-2-[[(2,3- difluorophenyl)methyl]thio]-6-methoxyρyrimidine (the product of example 35 step i) (0.3Og) and dioxane (1OmL). Purification was by reverse phase HPLC (symmetry as the stationary
-138- phase and TFA/acetonitrile as the mobile phase) then titurated with MeOH followed by DCM to give the title compound as a white solid. Yield: 0.24g MS: APCI(+ve) 517 [M+H+]
1H NMR:(CD30D) δ 3.00 (s, 3H), 3.02 (s, 3H), 3.41 - 3.53 (m, 4H), 3.64 - 3.80 (m, 4H), 3.97 (s, 3H), 4.29 (s, 2H), 4.54 (s, 2H), 6.09 (s, IH), 7.08 - 7.25 (m, 2H), 7.33 - 7.41 (m, IH)
Example 82
4-Pyridin-4-ylmethyl-piperazine-l-sulfonic acid [2-(2,3-difluoro-beiizylsulfanyl)-6- methoxy-pyrimidin-4-yl]-amide
The title compound was prepared according to the procedure outlined in example 81 using a mixture of l-Pyridin-4-ylmethyl-piperazine (0.53g), sulfamide (0.29g) and dioxane (2OmL). Followed by tris(dibenzylideneacetone)dipalladium (0) (92mg), 2-dicyclohexylphosphino- 2',4',6'-tri-wopropyl-l,l'-biphenyl (XPHOS) (67mg), cesium carbonate (0.49g), 4-Chloro-2- [[(2,3-difluorophenyl)methyl]thio]-6-methoxypyrimidine (the product of example 35 step i) (0.30g) and dioxane (1OmL). Purification was by reverse phase HPLC (symmetry as the stationary phase and TFA/acetonitrile as the mobile phase) then titurated with MeOH followed by DCM to give the title compound as a white solid. Yield: 0.23g MS: APCI(+ve) 523 [M+H+] 1H NMR:(CD3OD) δ 2.77 (t, 4H), 3.47 (t, 4H), 3.96 (s, 3H), 4.01 (s, 2H), 4.51 (s, 2H), 6.13 (s, IH), 7.07 - 7.24 (m, 2H), 7.34 - 7.42 (m, IH), 7.99 (d, 2H), 8.76 (d, 2H)
-139-
Example 83 4-(Tetrahydro-fiiran-2-ylniethyl)-piperazine-l-sulfoiiic acid [2-(2,3-difluoro- benzylsulfanyl)-6-methoxy-pyrimidin-4-yl]-aniide
The title compound was prepared according to the procedure outlined in example 81 using a mixture of l-(tetrahydrofuran-2-yl)-l-piperazine (0.5Ig), sulfamide (0.29g) and dioxane (2OmL). Followed by tris(dibenzylideneacetone)dipalladium (0) (92mg), 2- dicyclohexylphosphino-2',4',6'-tri-/røpropyl-l,r-biphenyl (XPHOS) (67mg), cesium carbonate (0.49g), 4-Chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-methoxypyrimidine (the product of example 35 step i) (0.30g) and dioxane (1OmL). Purification was by reverse phase HPLC (symmetry as the stationary phase and TFA/acetonitrile as the mobile phase) then titurated with Toluene, MeOH followed by DCM to give the title compound as a white solid. Yield: 0.15g
MS: APCI(+ve) 516 [M+H+]
1H NMR:(CD3OD) δ 1.53 - 1.67 (m, 2H), 1.92 - 2.03 (m, 2H), 2.10 - 2.22 (m, IH), 3.13 - 3.96 (m, HH), 3.99 (s, 3H), 4.21 - 4.34 (m, IH), 4.55 (s, 2H), 6.08 (s, IH), 7.08 - 7.25 (m, 2H), 7.32 - 7.40 (m, IH)
-140-
Example 84
4-(3-Dimethylamino-propyl)-piperazine-l-sulfonic acid [2-(2,3-difluoro-benzylsulfanyl)- 6-methoxy-pyrimidin-4-yl]-amide
The title compound was prepared according to the procedure outlined in example 81 using a mixture of N, N-dimethyl-3-piρerazi-l-ylpropan-l-amine (0.5Ig), sulfamide (0.29g) and dioxane (2OmL). Followed by tris(dibenzylideneacetone)dipalladium (0) (92mg), 2- dicyclohexylphosphino-2',4',6'-tri-iiOpropyl-l,r-biphenyl (XPHOS) (67mg), cesium carbonate (0.49g), 4-Chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-methoxyρyrimidine (the product of example 35 step i) (0.3Og) and dioxane (1OmL). Purification was by reverse phase BDPLC (symmetry as the stationary phase and TFA/acetonitrile as the mobile phase) then titurated with Toluene, MeOH followed by DCM to give the title compound as a white solid. Yield: 0.14g
MS: APCI(+ve) 517 [MH-H+]
1H NMPv(DMSO) δ 1.85 - 2.00 (m, 2H), 2.48 - 2.53 (m, 10H), 2.78 (s, 6H), 3.02 - 3.11 (m,
2H), 3.90 (s, 3H), 4.49 (s, 2H), 6.12 (s, IH), 7.13 - 7.22 (m, IH), 7.30 - 7.44 (m, 2H)
-141-
Example 85
Piperazine-l,4-disulfoiiic acid [2-(2,3-difluoro-benzylsulfanyl)-6-methoxy-pyrimidin-4- yl] -amide dimethylamide
The title compound was prepared by adding dimethyl sulfamoyl chloride to a solution of N- [2-[[(2,3-Difluorophenyl)methyl]tMo]-6-methoxypyrirnidhi-4-yl]piperazine-l-sulfonamide, trifluoro acetate salt (the product from example 36) (0.25g) in DCM (5mL). Purification was by reverse phase HPLC (symmetry as the stationary phase and TFA/acetonitrile as the mobile phase) then titurated with Toluene, DCM followed by Et2O to give the title compound as a white solid. Yield: 0.1 Ig MS: APCI(+ve) 539 [M+H+]
1H NMR:(DMSO) 52.73 (s, 6H), 3.16 - 3.30 (m, 8H), 3.88 (s, 3H), 4.48 (s, 2H), 6.07 (s, IH),
7.11 - 7.20 (m, IH), 7.29 - 7.45 (m, 2H), 11.28 (s, IH)
Example 86
{4-[2-(2,3-Difluoro-benzylsulfanyl)-6-methoxy-pyrimidin-4-ylsulfamoyl]-piperazin-l-yl}- acetic acid
The title compound was prepared by adding IM NaOH (ImL) to a solution of {4-[2-(2,3- Difluoro-benzylsulfanyl)-6-methoxy-pyrirnidin-4-ylsulfamoyl]-piperazin-l-yl}-acetic acid ethyl ester (the product from step i) (0.3Ig) in MeOH (ImL). Purification was by reverse phase HPLC (symmetry as the stationary phase and TFA/acetonitrile as the mobile phase) then titurated with Toluene, DCM followed by Et2O to give the title compound as a white solid. Yield: 85mg MS: APCI(+ve) 490 [M+H+]
1H NMR:(CD3OD) 5 2.99 - 3.05 (m, 4H), 3.39 (s, 2H), 3.46 - 3.53 (m, 4H), 3.92 (s, 3H), 4.47 (s, 2H), 6.10 (s, IH), 7.05 - 7.24 (m, 3H), 7.35 (t, IH)
The intermediate for this compound was prepared as follows:
i) {4-[2-(2,3-Difluoro-benzylsulfanyl)-6-methoxy-pyrimidin-4-ylsulfamoyl]-piperaziii-l- yl}-acetic add ethyl ester The subtitle compound was prepared by adding 60% sodium hydride (0.18g) portionwise to a solution of N-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-methoxypyrimidin-4-yl]piperazine-l- sulfonamide, trifluoro acetate salt (the product from example 36) (0.53g) and ethyl 2- bromoacetate (0.36mL) in THF (1OmL). The reaction mixture was diluted with H2O and extracted with EtOAc. The organic layer was washed with brine and evaporated to give the subtitle compound as an oil. MS: APCI(+ve) 518 [M+H+] Example 87
4-(2-Hydroxy-ethyl)-piperazine-l-sulfonic acid [2-(2,3-difluoro-benzylsulfanyl)-6- methoxy-pyrimidin-4-yl]-amide
The title compound was prepared according to the procedure outlined in example 24 using a mixture of {4-[2-(2,3-Difluoro-benzylsulfanyl)-6-methoxy-pyrirnidin-4-ylsulfamoyl]-
-143- piperazin-1-yl}- acetic acid ethyl ester (the product from example 86 step i) (0.3Ig) lithium borohydride (IM solution in THF) (1.2mL) in THF (5mL). Purification was by reverse phase HPLC (symmetry as the stationary phase and NH4θAc/acetonitrile as the mobile phase) then titurated with Toluene, MeOH followed by DCM to give the title compound as a white solid. Yield: 13mg
MS: APCI(+ve) 476 [M+H+]
1H NMR:(CD3OD) δ 2.52 - 2.61 (m, 6H), 3.34 (t, 4H), 3.65 (t, 2H), 3.91 (s, 3H), 4.47 (s, 2H),
6.14 (s, IH), 7.04 - 7.19 (m, 2H), 7.35 (t, IH)
Synthesis of examples 88-107
Examples 88-107 were synthesised using the following procedure:- To a solution of the aldehyde (0.2 mmol) in NMP (0.8 mL), N-[2-[[(2,3- Difluorophenyl)methyl]tMo]-6-methoxypyrirnidin-4-yl]piperazine-l-sulfonamide trifluoroacetate salt (the product from example 36) (65mg) was added as an NMP solution (0.4ml) followed by resin bound cyanoborohydride (88mg) and acetic acid (1.8μL). The reaction mixture was agitated for 48 h, then filtered to remove the resin followed by centrifugal evaporation to dryness. The product was purified by LCMS directed purification (XTerra as the stationary phase and ammonia/acetonitrile as the mobile phase) to give the title compound.
-146-
Example 108
N-{2-[(3-Chloro-2-fluorobenzyl)thio]-6-methoxypyrimidin-4-yl}piperazine-l- sulfonamide
The title compound was prepared according to the procedure outlined in example 15 using te^butyl 4-[({2-[(3-chloro-2-fluororjenzyl)trήo]-6-methoxyp}τirnidin-4- yl}amino)sulfonyl]piperazine-l-carboxylate (the product from step ϋ) (0.26g), trifluoroacetic acid (0.5mL) and DCM (1OmL). Purification was by reverse phase HPLC (Symmetry as the
-147- stationary phase and TFA/acetonitrile as the mobile phase) then triturated with MeOH followed by Et2O to give the title compound as a white solid. Yield: 40mg MS: APCI(+ve) 448 [M+H+]
1H NMR:(DMSO) δ 3.15 - 3.24 (m, 4H), 3.36 - 3.48 (m, 4H), 3.92 (s, 3H), 4.50 (s, 2H), 6.10 (s, IH), 7.22 (t, IH), 7.49 - 7.63 (m, 2H), 8.74 (s, IH)
The intermediates for this compound were prepared as follows:
i) 4-Chloro-2-[(3-chloro-2-fluorobenzyl)thio]-6-methoxypyrimidiiie The subtitle compound was prepared according to the procedure outlined in example 35 Step
(i) using 4,6-dichloro-2-[(3-chloro-2-fluorobenzyl)thio]pyrimidine
(prepared according to patent WO 2004/011443) (0.65 g), methanol (8mL) and 60% sodium hydride (88mg). Yield: 0.57g.
1H NMR:(CDC13) δ 3.93 (s, 3H), 4.41 - 4.43 (m, 2H), 6.43 (s, IH), 6.99 - 7.05 (m, IH), 7.25 - 7.32 (m, IH), 7.40 - 7.46 (m, IH)
ii) ter^Butyl 4-[({2-[(3-chloro-2-fluorobenzyl)thio]-6-methoxypyrimidin-4- yl}amino)sulfonyl]piperazine-l-carboxylate
The subtitle compound was prepared according to the procedure outlined in example 1 step (iv) using 4-chloro-2-[(3-chloro-2-fluorobenzyl)thio]-6-methoxypyrimidine (the product from step i) (0.26g), 4-(aminosuhconyl)-l,l-dimethylethyl ester- 1-piperazinecarboxy lie acid (the product of example 15 step i) (0.23g), tris(dibenzylideneacetone)diρalladium (0) (73mg), 2- dicyclohexylphosphmo-2',4',6'-tri-/røpropyl-l,r-biphenyl (XPHOS) (53mg), cesium carbonate (0.33g), and dioxane (8mL). Purification was by column chromatography on silica gel using EtOAc/isøhexane (2:8 to 3:7 gradient) as eluent to give the subtitle compound as a white solid. Yield: 0.26g MS: APCI(-ve) 546 [M-H"]
-148-
Example 109
N-{2-[(2,3-Difluorobenzyl)thio]-6-methoxypyrimidin-4-yl}-3-hydroxyazetidine-l- sulfonamide
The title compound was prepared according to the procedure outlined in example 34 using 3- {[tert-butyl(diphenyl)silyl]oxy}-N-{2-[(2,3-difluorobenzyl)thio]-6-methoxypyrimidin-4- yl}azetidine-l -sulfonamide (the product from step ii) (0.29g) and IM solution of tetrabutylammoniumfluoride in THF (5mL). Purification was by reverse phase HPLC
(symmetry as the stationary phase and TFA/acetonitrile as the mobile phase) then triturated with MeOH, Et2O followed by /rø-hexane to give the title compound as a white solid. Yield:
40mg
MS: APCI(+ve) 419 [M+H+] 1H NMR:(DMSO) δ 3.70 (t, 2H), 3.86 (s, 3H), 3.97 (t, 2H), 4.29 - 4.39 (m, IH), 4.46 (s, 2H), 5.79 (d, IH), 6.16 (s, IH), 7.10 - 7.18 (m, IH), 7.32 (q, IH), 7.41 (t, IH), 11.23 (s, IH)
The intermediates for this compound were prepared as follows:
i) 3-(tø/t-Butyl-diphenyl-silanyloxy)-azetidine-l-sulfonamide
The subtitle compound was prepared according to the procedure outlined in example 15 step (i) using 3-(tert-butyl-diphenyl-silanyloxy)-azetidine (prepared according to patent WO 2003/072557) (0.93g), dioxane (2OmL) and sulfamide (0.34g). Isolation was by filtration to remove excess sulfamide, the filtrate was then reduced in vacuo to give the subtitle compound as a brown oil. Yield: 1.2g MS: APCI(-ve) 389 [M-H]
-149- ii) 3-{[fe/t-Butyl(diphenyl)sUyl]oxy}-N-{2-[(2,3-difluorobenzyl)thio]-6- methoxypyrimidin-4-yl}azetidine-l-sulfonamide
The subtitle compound was prepared according to the procedure outlined in example 1 step (iv) using a mixture of 4-Chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-methoxypyrimidine (the product of example 35 step i) (0.16g), 3-(tert-butyl-diphenyl-silanyloxy)-azetidine-l- sulfonamide (the product from step i) (0.17g), tris(dibenzylideneacetone)dipalladium (0) (33mg), 2-dicyclohexylphosphino-2',4',6'-tri-irøpropyl-l,l'-biphenyl (XPHOS) (24mg), cesium carbonate (0.16g) and dioxane (8mL). Purification was by column chromatography on silica gel using EtOAc/irøhexane (1:9 to 2:8 gradient) as eluent to give the subtitle compound as a yellow oil. Yield: 0.12g MS: APCI(+ve) 657 [M+H+]
Example 110
N'-{2-[(2,3-Difluorobenzyl)thio]-6-methoxypyrimidin-4-yl}-N-[2-(dimethylamino)ethyl]- N-methylsulfamide
The title compound was prepared according to the procedure outlined in example 1 step (iv) using a mixture of 4-Chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-methoxypyrimidine (the product of example 35 step i) (0.35g), N-[2-(dimethylamino)ethyl]-AT-methylsulfamide (prepared according to procedure outlined in Org.Letts 2004, 6 (16), 2705-2708) (0.18g), tris(dibenzylideneacetone)dipalladium (0) (73mg), 2-dicyclohexylphosphino-2',4',6'-tri- i.røpropyl-l,r-biphenyl (XPHOS) (53mg), cesium carbonate (0.39g) and dioxane (2OmL). Purification was by reverse phase HPLC (symmetry as the stationary phase and TFA/acetonitrile as the mobile phase) then titurated with MeOH followed by Et2O to give the title compound as a white solid. Yield: 0.12g MS: APCI(+ve) 448 [M+H+]
1H NMR (DMSO) δ 2.84 (6H, s), 2.86 (3H, s), 3.33 (2H, t), 3.57 (2H, t), 3.93 (3H, s), 4.53 (2H, s), 6.05 (IH, s), 7.16 - 7.24 (IH, m), 7.32 - 7.45 (2H, m)
-150-
Example 111
N-(2-[(2,3-Difluorobenzyl)thio]-6-{[(lR,2R)-2-hydroxymethylpropyl]oxy}pyrimidin-4- yl)-(25)-2-methylpiperaziiie-l-sulfonamide
N-(2-[(2,3-dmuorobenzyl)thio]-6-{[(lR,2R)-2-hydroxymethylpropyl]oxy}pyrimidiB-4-yl) tert-butyl 4-(aminosulfonyl)piperazine-l-carboxylate (the product from step ϋ),0.65g) was dissolved in DCM (15ml) and allowed to stir at room temperatureuntil homogeneous. TFA (15ml) was then slowly added and the reaction mixture stirred overnight. The reaction mixture was reduced in vacuo, dissolved in MeOH and purified by prep HPLC to give the title compound as a white solid Yield 105mg
1H NMR: (DMSO) δ 1.04 (d, 3H), 1.14 (d, 3H), 1.27 (d, 3H), 2.82-2.91 (m, IH), 2.97-3.06 (m, IH), 3.19-3.27 (m, 2H), 3.36-3.44 (m, IH), 3.67-3.77 (m, 2H), 4.14-4.21 (m, IH), 4.41- 4.50 (m, 2H) , 4.98-5.05 (m, IH), 5.91 (s, IH), 7.14-7.21 (m, IH) , 7.31-7.41 (m, 2H), 11.28 (s, IH) MS: APCI(+ve) 504.1 [M+H+]
The intermediates for this compound were prepared as follows:
i) te/t-Butyl (3S)-4-(aminosulfonyl)-3-methylpiperazine-l-carboxylate
To a solution of (2S)-2-methylpiperazine-l -sulfonamide (0.5g) in dioxane (40ml) was added sulfamide (0.288g) and the reaction mixture was then heated at reflux in the microwave at 1000C, 300W, open vessel with cooling for Ah in dioxane. The reaction mixture was partitioned between DCM (100ml) and H2O (100ml) and the aqueous re-extracted with DCM (2x100ml). Organic s were collected dried and reduced in vacuo to give the subtitle compound as a clear colourless oil (745mg)
-151-
1H NMR: (DMSO) δ 1.11 (d, 3H), 1.40 (s, 9H), 2.84-3.13 (m, 3H), 3.32 (s, 2H), 3.64-3.72 (m, IH), 3.78-3.93 (m, IH), 6.80 (s, 2H)
ii) N-(2-[(2,3-Difluorobenzyl)thio]-6-{[(lR,2R)-2-hydroxymethylpropyl]oxy}pyrimidin-4- yl)-<eιt-butyl (3S)-4-(aminosulfonyl)-3-methylpiperazine-l-carboxylate
A mixture of tert-butyl (35)-4-(aminosulfonyl)-3-methylpiperazine-l-carboxylate ((the product from step i), 0.373g), tris(dibenzylideneacetone)dipalladium (0) (50 mg), 2- dicyclohexylphosphino-2',4',6'-tri-/røpropyl-l,r-biphenyl (XPHOS) (50mg), cesium carbonate (0.488 g) and (2/?,32?)-3-({6-chloro-2-[(2,3-difluorobenzyl)thio]ρyrimidin-4- yl}oxy)butan-2-ol ((the product of example 4 step i), 0.36Ig) in dioxane (20ml) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for l.5h. The reaction mixture was then reduced in vacuo and the residue separated between EtOAc (200 ml) and H2O (200 ml). The organics were separated and the aqueous layer was re-extracted with EtOAc (2 x 200ml). Organics were combined, dried (MgSO4) and reduced in vacuo to give the subtitle compound as a yellow solid Yield 0.65g MS: APCI(+ve) 604.5 [M+H+]
Example 112 N-[2-[(2,3-Difluorobenzyl)tMo]-6-(2-hydroxyethoxy)pyrimidin-4-yl]methanesuIfonamide
A mixture of methanesulfonamide (0.228g), tris(dibenzylideneacetone)dipalladium (0) (50 mg), 2-dicyclohexylphosphino-2',4',6'-tri-irøpropyl-l,l'-biphenyl (XPHOS) (50mg), cesium carbonate (0.585 g) and 2-({6-chloro-2-[(2,3-difluorobenzyl)thio]pyrimidin-4-yl}oxy)ethanol ((the product step i), 0.40Og) in dioxane (20ml) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for 30 mins. The reaction mixture was then reduced in vacuo and the residue separated between DCM (150 ml) and H2O (100 ml). The organics were
-152- separated and the aqueous layer was re-extracted with DCM (2 x 150ml). Organics were combined, dried (MgSO4) and reduced in vacuo and the resulting residue was purified by prep HPLC to give the title compound as a white solid. Yield: 102mg
1H NMR: (DMSO) 5 3.28 (s, 3H), 3.63-3.68 (m, 2H), 4.29 (t, 2H), 4.47 (s, 2H), 4.87 (t, IH), 6.03 (s, IH), 7.13-7.19 (m, IH), 7.31-7.43 (m, 2H), 11.12 (s, IH) MS: APCI(+ve) 391.9 [M+H+]
The intermediate for this compound was prepared as follows:
i) 2-({6-Chloro-2-[(2,3-difluorobenzyl)thio]pyrimidin-4-yl}oxy)ethanol
To a solution of 4,6-dichloro-2-[(2,3-difluorobenzyl)thio]pyrimidine ((the product of example 1 step ii), 5g) and ethylene glycol (1.517g) in THF (100ml) was added NaH (1.3g) slowly and the reaction was then allowed to stir overnight at RT. The reaction mixture was then partitioned between EtOAc (200 ml) and H2O (200 ml). The organics were separated and the aqueous layer was re-extracted with EtOAc (2 x 200ml). Organics were combined, dried (MgSO4) and reduced in vacuo and the resulting residue was purified by column chromatography on silica gel 10% EtOAc/90% iso-Hex to give the subtitle compound as a clear oil. Yield: 2.4g MS: APCI(+ve) 332/334 [M+H+] 1H NMR: (DMSO) δ 3.90-3.95 (m, 2H), 4.42 (s, 2H), 4.45-4.48 (m, 2H), 6.48 (s, IH), 6.98- 7.10 (m, 2H), 7.24-7.30 (m, IH)
Example 113
N-[2-[(2,3-Difluorobenzyl)thio]-6-(2-hydroxyethoxy)pyrimidin-4-yl] piperazine-1- sulfonamide
To a solution of N-[2-[(2,3-difluorobenzyl)thio]-6-(2-hydroxyethoxy)pyrimidin-4-yl] tert- butyl 4-(aminosulfonyl)piperazine-l-carboxylate ((the product from step i), 0.7Og) in DCM
-153-
(20ml) was added TFA (20ml). The reaction was then stirred at room temperaturefor 18Λ. The reaction was then reduced in vacuo and the residue dissolved in 7N NH3ZMeOH (20ml) and stirred at room temperature for Ih. The reaction was then reduced in vacuo and the residue purified by prep HPLC to give the title compound as a white solid. Yield: 33mg MS: APCI(+ve) 462 [M+H+]
1H NMR: (DMSO) δ 3.01-3.05 (m, 4H), 3.13-3.17 (m, 4H), 3.60-3.64 (m, 2H), 4.16 (t, 2H), 4.38 (m, 2H), 4.79 (s, IH), 5.87 (s, IH), 7.09-7.17 (m, IH), 7.26-7.35 (m, IH), 7.41-7.46 (m, IH)
The intermediate for this compound was prepared as follows:
i) ΛH2-[(2,3-Dimiorobenzyl)thio]-6-(2-hydroxyethoxy)pyrimidin-4-yl] tert-butyl 4- (aminosulfonyl)piperazine-l-carboxylate
A mixture of 4-(aminosulfonyl)-l,l-dimethylethyl ester- 1-piperazinecarboxy lie acid (the product from example 15 step i) (0.637g), tris(dibenzylideneacetone)dipalladium (0) (50mg),
2-dicyclohexylphosphino-2',4',6'-tri-/5Opropyl-l,r-biphenyl (XPHOS) (50mg), cesium carbonate (0.385 g) and 2-({6-chloro-2-[(2,3-difluorobenzyl)thio]pyrirnidin-4-yl}oxy)ethanol
(the product from example 112 step ϋ), 0.40Og) in dioxane (20ml) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for 30 mins. The reaction mixture was then reduced in vacuo and the residue separated between DCM (150 ml) and H2O (100 ml).
The organics were separated and the aqueous layer was re-extracted with DCM (2 x 150ml).
Organics were combined, dried (MgSO4) and reduced in vacuo to give the subtitle compound as a yellow solid. Yield: 0.7Og
MS: APCI(-ve) 560 [M-H ] 1H NMR: (DMSO) δ 3.28 (s, 3H), 3.63-3.68 (m, 2H), 4.29 (t, 2H), 4.47 (s, 2H), 4.87 (t, IH),
6.03 (s, IH), 7.13-7.19 (m, IH), 7.31-7.43 (m, 2H), 11.12 (s, IH)
Example 114
N-[2-[(2,3-Difluorobenzyl)thio]-6-(2-hydroxyethoxy)pyrimidin-4-yl]morpholine-4- sulfonamide
-154-
A mixture of morpholine-4- sulfonamide (prepared according to patent WO 2004/011443, 0.399g), tris(dibenzylideneacetone)dipalladium (0) (50mg), 2-dicyclohexylphosphino- 2',4',6'-tri-i.røpropyl-l,l'-biphenyl (XPHOS) (50mg), cesium carbonate (0.585 g) and 2-({6- chloro-2-[(2,3-difluorobenzyl)thio]pyriinidin-4-yl}oxy)ethanol (the product from example 112 step ϋ), 0.40Og) in dioxane (20ml) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for 30 mins. The reaction mixture was then reduced in vacuo and the residue separated between DCM (150 ml) and H2O (100 ml). The organics were separated and the aqueous layer was re-extracted with DCM (2 x 150ml). Organics were combined, dried (MgSO4) and reduced in vacuo and the resulting residue purified by prep HPLC to give the title compound as a white solid. Yield: 0.15g MS: APCI(+ve) 463 [M+H+]
1H NMR: (DMSO) 5 3.18 (t, 4H), 3.60 (t, 4H), 3.66 (t, 2H), 4.30 (t, 2H), 4.47 (s, 2H), 4.88 (s, IH), 6.10 (s, IH), 7.13-7.20 (m, IH), 7.31-7.38 (m, IH), 7.39-7.44 (m, IH)
Example 115
N- [2- [(2,3-Difluorobenzyl)thio] -6-(2-hydroxyethoxy)pyrimidin-4-yl] -azetdine- 1 - sulfonamide
A mixture of azetidine-1- sulfonamide (0.33g, prepared according to patent WO2004/011443), tris(dibenzylideneacetone)dipalladium (0) (50mg), 2-dicyclohexylphosphino-2',4',6'-tri- /søpropyl-l,l'-biphenyl (XPHOS) (50mg), cesium carbonate (0.585 g) and 2-({6-chloro-2- [(2,3-difluorobenzyl)thio]pyrimidin-4-yl}oxy)ethanol ((the product from example 112 step ii),
-155-
0.40Og) in dioxane (20ml) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for 30 mins. The reaction mixture was then reduced in vacuo and the residue separated between DCM (150ml) and H2O (150ml). The organics were separated and the aqueous layer was re-extracted with DCM (2 x 150ml). Organics were combined, dried
5 (MgSO4) and reduced in vacuo and the resulting residue purified by prep HPLC to give the title compound as a white solid. Yield: 0.13g MS: APCI(+ve) 433 [M+H+]
1R NMR: (DMSO) 52.13 (quintet, 2H), 3.65-3.68 (m, 2H), 3.91 (t, 4H), 4.30 (t, 2H), 4.47 (s, 2H), 4.91 (s, IH), 6.16 (s, IH), 7.13-7.19 (m, IH), 7.30-7.38 (m, IH), 7.40-7.45 (m, IH),
10 11.13 (s, IH)
Example 116 N-{2-[(2,3-Difluorobenzyl)thio]-6-wopropoxypyrimidin-4-yl}azetidiiie-l-sulfonamide
15 A mixture of azetidine-1- sulfonamide (0.327g), tris(dibenzylideneacetone)dipalladium (0) (50 mg), 2-dicyclohexylphosρhino-2',4',6'-tri-irøpropyl-l,r-biphenyl (XPHOS) (50mg), cesium carbonate (0.585 g) and 4-chloro-2-[(2,3-difluorobenzyl)thio]-6-ii'opropoxypyrirnidine (the product from step i), 0.40Og) in dioxane (20ml) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for 30 mins. The reaction mixture was then reduced in vacuo
20 and the residue separated between DCM (150 ml) and H2O (150 ml). The organics were separated and the aqueous layer was re-extracted with DCM (2 x 150ml). Organics were combined, dried (MgSO4) and reduced in vacuo and the resulting residue purified by prep HPLC to give the title compound as a white solid. Yield: 0.18g MS: APCI(+ve) 432 [M+H+]
25 1H NMR: (DMSO) 51.31 (d, 6H), 2.26 (quintet, 2H), 4.02 (t, 4H), 4.41 (s, 2H), 5.33 (septet, IH), 6.32 (s, IH), 6.98-7.10 (m, 2H), 7.18-7.28 (m, IH)
-156-
The intermediate for this compound was prepared as follows:
i) 4-Chloro-2-[(2,3-difluorobenzyl)thio]-6-wopropoxypyrimidine
To a solution of 4,6-dichloro-2-[(2,3-difluorobenzyl)thio]pyrimidine ((the product of example 1 step ii), 3g) in propan-2-ol (20ml) was added NaH (0.43g) slowly and the reaction was then allowed to stir overnight at RT. The reaction mixture was then partitioned between DCM (100ml) and H2O (100ml). The organics were separated and the aqueous layer was re- extracted with DCM (2 x 100ml). Organics were combined, dried (MgSO4) and reduced in vacuo to give the subtitle compound as a pale yellow solid. Yield: 1.8g 1H NMR: (DMSO) 5 1.26 (d, 6H), 4.45 (s, 2H), 5.23-5.32 (m, IH), 6.77 (s, IH), 7.14-7.22 (m, IH), 7.31-7.39 (m, 2H)
Example 117
(3S)-3-Amino-N-{2-[(2,3-difluorobenzyl)thio]-6-methoxypyrimidin-4-yl}pyrrolidine-l- sulfonamide
To a solution of tert-butyl {(35)-l-[({2-[(2,3-difluorobenzyl)thio]-6-methoxypyrimidin-4- yl}amino)suhconyl]pyrrolidin-3-yl}carbamate (the product from step ii), 0.75g) in DCM (10ml) was added TFA slowly. The reaction then stirred at room temperaturefor 18h. The reaction was reduced in vacuo and the residue purified by prep HPLC to give the title compound as a white solid. Yield: 70mg MS: APCI(+ve) 432 [M+H+]
1H NMR: (DMSO) δl.89-2.02 (m, IH), 2.07-2.20 (m, IH), 3.30-3.56 (m, 4H), 3.74-3.81 (m, IH), 3.82 (s, 3H), 4.43 (s, 2H), 5.89 (s, IH), 7.12-7.20 (m, IH), 7.28-7.43 (m, 2H)
The intermediates for this compound were prepared as follows:
-157- i) tert-Butyl [(35)-l-(aminosulfonyl)pyrrolidin-3-yl]carbamate
To a solution of tert- butyl (3S)-pyrrolidin-3-ylcarbamate (1.3g) in dioxane (50ml) was added sulfamide (1.55g) and the reaction was heated at 110°C for 18h. The reaction mixture was then partitioned between DCM (150ml) and H2O (100ml). The organics were separated and the aqueous layer was re-extracted with DCM (2 x 150ml). Organics were combined, dried (MgSO4) and reduced in vacuo to give the subtitle compound as a pale yellow solid. Yield: 1.44g
1H NMR: (DMSO) δ 1.39 (s, 9H), 1.67-1.77 (m, IH), 1.98-2.07 (m, IH), 2.82-2.87 (m, IH), 3.06-3.13 (m, IH), 3.15-3.22 (m, IH), 3.30-3.35 (m, IH), 3.93-4.00 (m, IH) , 6.72 (s, 2H)
ii) re/t-Butyl {(3S)-l-[({2-[(2,3-difluorobenzyl)thio]-6-methoxypyrimidin-4- yl}amino)sulfonyl]pyrrolidin-3-yl}carbamate
A mixture of tert-butyl [(35)-l-(aminosulfonyl)pyrrolidin-3-yl]carbamate (the product from step i), 0.525g), tris(dibenzylideneacetone)dipalladium (0) (50 mg), 2-dicyclohexylphosphino- 2',4',6'-tri-irøpropyl-l,l'-biphenyl (XPHOS) (50mg), cesium carbonate (0.429 g) and 4- Chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-methoxypyrimidine ((the product from example 35 step i), 0.40Og) in dioxane (20ml) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for 40 mins. The reaction mixture was then reduced in vacuo and the residue separated between DCM (150 ml) and H2O (150 ml). The organics were separated and the aqueous layer was re-extracted with DCM (2 x 150ml). Organics were combined, dried (MgSO4) and reduced in vacuo to give the subtitle compound as a yellow solid. Yield: 0.75g MS: APCI(-ve) 530 [M-H ] Example 118 (3R)-3-Amino-N-{2-[(2,3-difluorobenzyl)thio]-6-methoxypyriniidiii-4-yl}pyrrolidine-l- sulfonamide
To a solution of tert-butyl {(3/?)-l-[({2-[(2,3-difluorobenzyl)thio]-6-methoxypyrimidin-4- yl}amino)sulfonyl]pyrrolidin-3-yl}carbamate (0.75g) in DCM (10ml) was added TFA (10ml) slowly. The reaction then stirred at room temperaturefor 18h. The reaction was reduced in vacuo and the residue purified by prep HPLC to give the subtitle compound as a white solid. Yield: 0.17g
1U NMR: (DMSO) δl.89-2.02 (m, IH), 2.07-2.20 (m, IH), 3.30-3.56 (m, 4H), 3.74-3.81 (m, IH), 3.82 (s, 3H), 4.43 (s, 2H), 5.89 (s, IH), 7.12-7.20 (m, IH), ), 7.28-7.43 (m, 2H) MS: APCI(+ve) 431.9 [M+H+]
The intermediates for this compound were prepared as follows:
i) tert-Butyl [(3R)-l-(aminosulfonyl)pyrrolidin-3-yl]carbamate
To a solution of tert- butyl (3i?)-pyrrolidin-3-ylcarbamate (1.3g) in dioxane (50ml) was added sulfamide (1.55g) and the reaction was heated at 110°C for 18h. The reaction mixture was then partitioned between DCM (100 ml) and H2O (100 ml). The organics were separated and the aqueous layer was re-extracted with DCM (2 x 100ml). Organics were combined, dried (MgSO4) and reduced in vacuo to give the subtitle compound as a pale yellow solid. Yield: 1 69g 1H NMR: (DMSO) 5 1.39 (s, 9H), 1.68-1.76 (m, IH), 1.98-2.07 (m, IH), 2.82-2.87 (m, IH), 3.06-3.13 (m, IH), 3.15-3.22 (m, IH), 3.29-3.35 (m, IH), 3.92-4.00 (m, IH) , 6.72 (s, 2H)
ii) (3S)-3-Amino-N-{2-[(2,3-difluorobenzyl)thio]-6-methoxypyrimidin-4-yl}fø/t-butyl [ (3S)- 1 -(aminosulfonyl)pyrrolidin-3-yl] carbamate
A mixture of tert-butyl [(3-^-l-(arninosulfonyl)pyrrolidin-3-yl]carbamate (0.525g), tris(dibenzylideneacetone)dipalladium (0) (50mg), 2-dicyclohexylphosphiαo-2',4',6'-tri- m>propyl-l,l'-biphenyl (XPHOS) (50mg), cesium carbonate (0.429 g) and 4-Chloro-2-[[(2,3- difluorophenyl)methyl]thio]-6-methoxypyrimidine (the product from example 35 step i), 0.40Og) in dioxane (20ml) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for 40 mins. The reaction mixture was then reduced in vacuo and the residue separated between DCM (150 ml) and H2O (150 ml). The organics were separated and the aqueous layer was re-extracted with DCM (2 x 150ml). Organics were combined, dried (MgSO4) and reduced in vacuo to give the title compound as a yellow solid. Yield: 0.77g
-159-
MS: APCI(-ve) 539 [M-H ]
Example 119
(3R)-3-Amino-N-{2-[(2,3-difluorobenzyl)thio]-6-methoxypyrimidin-4-yl}pyrrolidine-l- sulfonamide
To a solution of tert-butyl (3/?)-4-[({2-[(2,3-difluorobenzyl)thio]-6-methoxypyrimidin-4- yl}amino)sulfonyl]-3-methylpiperazine-l-carboxylate (0.75g) in DCM (10ml) was added TFA (10ml) slowly. The reaction then stirred at room temperaturefor 18h. The reaction was reduced in vacuo and the residue purified by prep HPLC to give the title compound as a white solid. Yield: 0.27g MS: APCI(+ve) 446 [M+H+]
1H NMR: (CDCl3) δ 1.43 (d, 3H), 3.07 (t, IH), 3.15 (d, IH), 3.26 (d, IH), 3.33 (d, IH), 3.60 (t, IH), 3.86 (d, IH), 3.95 (s, 3H), 4.30-4.37 (m, IH), 4.42 (s, 2H), 6.01 (s, IH), 6.99-7.10 (m, 2H), 7.19-7.22 (m, IH)
The intermediates for this compound were prepared as follows:
i) tert-Buty\ (3/?)-3-methylpiperazine-l-carboxylate To a solution of (2i?)-2-methylpiperazine (Ig) in THF (10ml) was added di-tert-butyl dicarbonate (1.45g). The reaction mixture was allowed to stir at room temperaturefor 18h. The reaction mixture was then partitioned between DCM (100 ml) and H2O (100 ml). The organics were separated and the aqueous layer was re-extracted with DCM (2 x 150ml). Organics were combined, dried (MgSO4) and reduced in vacuo to give the subtitle compound as a clear oil. Yield: 1. Ig
1H NMR: (DMSO) 5 0.92 (d, 3H), 1.38 (s, 9H), 2.57-2.70 (m, IH), 2.76-2.81 (m, IH), 2.87- 2.99 (m, IH), 3.66-3.74 (m, 4H)
-160- ii) tert-Bυtyl (3R)-4-(aminosulfonyl)-3-methylpiperazine-l-carboxylate
To a solution of tert-butyl (32?)-3-methylpiperazine-l-carboxylate ((the product from step i), 1. Ig) in dioxane (60ml) was added sulfamide (1.06g) and the reaction was heated at 110°C for 18h. The reaction mixture was then partitioned between DCM (150ml) and H2O (150ml). The organics were separated and the aqueous layer was re-extracted with DCM (2 x 150ml). Organics were combined, dried (MgSO4) and reduced in vacuo to give the subtitle compound as a pale yellow oil. Yield: 1.44g
1H NMR: (DMSO) 5 1.10 (d, 3H), 1.39 (s, 9H), 3.00-3.11 (m, 3H), 3.27-3.31 (m, 2H), 3.63- 3.71 (m, IH), 3.79-3.87 (m, IH), 6.79 (s, 2H)
iii) tert-Butyl (3/2)-4-[({2-[(2,3-difluorobenzyl)thio]-6-methoxypyrimidin-4- yl}amino)sulfonyl]-3-methylpiperazine-l-carboxylate
A mixture of tert-butyl (3/?)-4-(aminosulfonyl)-3-methylpiperazine-l-carboxylate ((the product from step ii), 0.554g), tris(dibenzylideneacetone)dipalladium (0) (50 mg), 2- dicyclohexylphosphino-2',4',6'-tri-irøpropyl-l,r-biphenyl (XPHOS) (50mg), cesium carbonate (0.429 g) and 4-Chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-methoxypyrimidine (the product from example 35 step i), 0.40Og) in dioxane (20ml) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for 60 mins. The reaction mixture was then reduced in vacuo and the residue separated between DCM (150 ml) and H2O (100 ml). The organics were separated and the aqueous layer was re-extracted with DCM (3 x 150ml). Organics were combined, dried (MgSO4) and reduced in vacuo to give the subtitle compound as a yellow oil. Yield: 0.75g MS: APCI(-ve) 543 [M-H-]
Example 120
(3S)-3-Amino-N-{2-[(2,3-difluorobenzyl)thio]-6-methoxypyrimidin-4-yl}-(2S)-2- methylpiperizine- 1 -sulfonamide
To a solution of ten- butyl (35)-4-[({2-[(2,3-difluorobenzyl)thio]-6-methoxypyrimidin-4- yl}amino)sulfonyl]-3-methylpiperazine-l-carboxylate (0.75g) in DCM (10ml) was added TFA (10ml) slowly. The reaction was then stirred at room temperaturefor 18h. The reaction was reduced in vacuo and the residue purified by prep HPLC to give the title compound as a white so Ud. Yield: 0.18g MS: APCI(+ve) 446 [M+H+]
1H NMR: (CDCl3) 5 1.43 (d, 3H), 3.07 (t, IH), 3.15 (d, IH), 3.26 (d, IH), 3.33 (d, IH), 3.60 (t, IH), 3.86 (d, IH), 3.95 (s, 3H), 4.30-4.37 (m, IH), 4.42 (s, 2H), 6.01 (s, IH), 6.99-7.10 (m, 2H), 7.19-7.22 (m, IH)
The intermediates for this compound were prepared as follows:
i) tert-Butyl (3S)-4-(aminosulfonyl)-3-methylpiperazine-l-carboxylate
To a solution of tert-butyl (3S)-3-methylpiperazine-l-carboxylate (0.5g) in dioxane (40ml) was added sulfamide (0.29g) and the reaction was heated at 110°C for 18h. The reaction mixture was then partitioned between DCM (150 ml) and H2O (150 ml).
The organics were separated and the aqueous layer was re-extracted with DCM (2 x 150ml).
Organics were combined, dried (MgSO4) and reduced in vacuo to give the subtitle compound as a pale yellow oil. Yield: 0.66g 1K NMR: (DMSO) 5 1.10 (d, 3H), 1.40 (s, 9H), 3.00-3.11 (m, 3H), 3.26-3.34 (m, 2H), 3.63-
3.71 (m, IH), 3.79-3.87 (m, IH), 6.79 (s, 2H)
ii) ^/t-Butyl (35)-4-[({2-[(2,3-difluorobenzyl)thio]-6-methoxypyriinidin-4- yl}amino)sulfonyl]-3-methylpiperaziiie-l-carboxylate A mixture of tert-butyl (35)-4-(aminosulfonyl)-3-methylpiperazine-l-carboxylate (0.372g), tris(dibenzylideneacetone)dipalladium (0) (50mg), 2-dicyclohexylphosphino-2',4',6'-tri- irøpropyl-U'-biphenyl (XPHOS) (50mg), cesium carbonate (0.286 g) and 4-Chloro-2-[[(2,3- difluorophenyl)methyl]thio]-6-methoxypyrimidine ((the product from example 35 step i), O.373g) in dioxane (20ml) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for 60min. The reaction mixture was then reduced in vacuo and the residue separated between DCM (150ml) and H2O (100ml). The organics were separated and the
-162- aqueous layer was re-extracted with DCM (3 x 150ml). Organics were combined, dried (MgSO4) and reduced in vacuo to give the subtitle compound as a yellow solid. Yield: 0.65g MS: APCI(-ve) 544 [M-H ]
Example 121
N-[6-Methoxy-2-[(2-phenylethyl)thio]pyrimidin-4-yl]azetidine-l-sulfonamide
A solution of N- [(4-methoxyphenyl)methy I]-N- [6-methoxy-2-[(2-phenylethyl)thio] pyrimidin-4-yl]azetidine-l-sulfonamide (the prouct of step iii, 0.17g) in DCM (ImI) and TFA (2ml) was stirred at room temperature for 18h. The solvent was evaporated under reduced pressure. The residue was recrystallised from EtOAc and irø-hexane to give the title product as a white solid. Yield: 50mg.
MS: APCI (+ve) 381 [M+H]
1H NMR: 5 (DMSO) 2.12 (quintet, 2H), 3.00 (m, 2H), 3.35 (m, 2H), 3.91 (t, 7H), 6.13 (s, IH), 7.23 (m, IH), 7.29 (m, 4H), 11.04 (bs, IH).
The intermediates for this compound were prepared as follows:
i) N- [2-[[(2,3-Difluorophenyl)methyl]thio]-6-methoxypyrimidin-4-yl]-N-[(4- methoxyphenyl)methyl]azetidine-l-sulfonamide
60% Sodium hydride (0.42g) was added to a solution of N-[2-[[(2,3-difluorophenyl) methyl] thio]-6-methoxypyrimidin-4-yl]azetidine-l- sulfonamide (the product of Example 35) (3.82g) in anhydrous DMF (38ml) stirred at 0°C under nitrogen. The reaction mixture was stirred for a further 15min when 4-methoxybenzylchloride (2.98g) was added dropwise over one min followed by potassium iodide (1.66g). After stirring at room temperature for 18h. the reaction mixture was partitioned between EtOAc and H2O. The aqueous layer was separated and further extracted with EtOAc (2x). The combined organic extracts were washed with H2O, dried (MgSO4), filtered and the solvent evaporated under reduced pressure. The residue
-163- was purified by flash column chromatography on silica gel using EtOAc/irø-hexane (2:8) as eluent. The product was further purified by flash column chromatography on silica gel using DCM/isohexane (6:4) as eluent to give the subtitle product as a white solid. Yield: 2.4g. MS: APCI (+ve) 523 [M+H]
ϋ) N-[2-[[(2,3-Difluorophenyl)methyl]sulfonyl]-6-methoxypyrimidin-4-yl]-N-[(4- methoxyphenyl)methyl]azetidine-l-sulfonamide
A mixture of the product of step i) (3.3g) and mCPBA (1. Ig) in DCM was stirred at room temperature for 5h. The reaction mixture was washed with aqueous sodium thiosulfate solution (3 x 100ml; 15g/100ml), aqueous NaHCO3, H2O, dried (MgSO4) and filtered. The solvent was evaporated under reduced pressure to give the subtitle product as a yellow foam. Yield: 3.18g. MS: APCI (+ve) 555 [M+H]
iii) N-[(4-Methoxyphenyl)methyl]-N- [6-methoxy-2-[(2-phenylethyl)thio] pyrimidin-4- yl]azetidine-l-sulfonamide
60% Sodium hydride (29mg) was added to a solution of the product of step ii) (0.36g) and 2- phenylethylthiol (0. Ig) in anhydrous DMF (4ml) stirred under nitrogen. The reaction mixture was stirred for 18h., diluted with EtOAc and washed with H2O. The separated organic solution was dried (MgSO4), filtered and the solvent evaporated under reduced pressure. The residue was purified by flash column chromatography on silica gel using Et2O/i.rohexane (3:7) as eluent to give the product as a white solid. Yield: 0.17g. MS: APCI (+ve) 501 [M+H]
Example 122
N-{6-Methoxy-2-[[(pyiidin-4-yl)methyl]thio]pyrimidin-4-yl}azetidine-l-sulfonamide
The title compound was prepared from N- [(4-methoxyphenyl)methy I]-N- [6-methoxy-2- [[(pyridin-4-yl)methyl]thio] pyrimidin-4-yl]azetidine-l-sulfonairiide (the product of step i) (46mg) by the procedure outlined in Example 121. The crude material was purified by preparative plate chromatography using EtOAc with 0.5% of 7N NH3/MeOH as eluent to give the title product as a white solid. Yield: 31mg. MS: APCI (+ve) 368 [M+H]
1H NMR: δ (DMSO) 2.09 (bt, 2H), 3.84 (bm, 7H), 4.39 (bs, 2H), 6.11 (bs, IH), 7.47 (bs, 2H), 8.48 (bs, 2H).
The intermediate for this compound was prepared as follows:
i) N-[(4-Methoxyphenyl)methyl]-N- [6-methoxy-2-[[(pyiidin-4-yl)methyl]thio] py rimidin-4-yl] azetidine- 1 -sulfonamide
60% NaH (27mg) was added batchwise to a solution of 4-pyridylethanethiol hydrochloride (60mg) in anhydrous DMF (2ml) stirred under nitrogen. After 30min. the subtitle product of
Example 121 step ϋ) (0.2g) was added. The reaction mixture was stirred for a further 18h.
KOtBu (40mg) was added and after 30min. a further quantity of KOtBu (40mg) was added.
After lOmin, KOtBu (40mg) followed by 4-pyridylmethyl bromide hydrobromide (96mg) were added. The reaction mixture was stirred for 5min., diluted with EtOAc and washed with H2O and aqueous Na2CO3. The separated organic solution was dried (MgSO4), filtered and the solvent evaporated under reduced pressure. The residue was purified by flash column chromatography on silica gel using EtOAc/iso-hexane (7:3) as eluent to give the product as a yellow gum. Yield: 46mg.
MS: APCI (+ve) 488 [M+H]
Example 123
N-{2-[[(2-Cyanophenyl)methyl]thio]-6-methoxypyrimidiii-4-yl}azetidine-l-sulfonamide
-165-
The title compound was prepared from N- [2-[[(2-cyanophenyl)methyl]thio]-6-methoxy pyrimidin-4-yl]-N-[(4-methoxyphenyl)methyl]-azetidine-l-sulfonamide (60mg) (the product of step i) by the procedure outlined in Example 121. The crude material was purified by preparative plate chromatography using EtOAc/isohexane (3:7) as eluent to give the title product as a yellow gum. Yield: 31mg. MS: APCI (+ve) 392 [M+H]
1H NMR: δ (DMSO) 2.12 (quintet, 2H), 3.90 (m, 7H), 4.59 (s, 2H), 6.15 (s, IH), 7.47 (t, IH), 7.66 (t, IH), 7.84 (m, 2H), 11.13 (bs, IH).
The intermediate for this compound was prepared as follows:
i) N- {2-[[(2-Cyanophenyl)methyl]thio]-6-methoxypyrimidin-4-yl]-N-[(4-methoxy- phenyl)inethyl}azetidine-l-sulfonaniide The subtitle compound was prepared from the product of Example 121 step ii) (0.2Og) and (2- cyanophenyl)methyl bromide (78mg) by the procedure outlined in Example 122 step i). The crude material was purified by flash column chromatography on silica gel using EtOAc/irøhexane (3.5:6.5) as eluent to give the product as a gum. Yield: 60mg MS: APCI (+ve) 512 [M+H]
Example 124 N-{6-Methoxy-2-[(phenylmethyl)thio]pyrimidin-4-yl}azetidine-l-sulfonamide
The title compound was prepared from N-[(4-methoxyphenyl)methyl]-N-[2-[(phenylmethyl) thio]-6-methoxypyrimidin-4-yl]azetidine-l-sulfonamide (the product of step i) (46mg) by the procedure outlined in Example 121. The crude material was purified by preparative plate chromatography using EtOAc/irøhexane (3:7) as eluent to give the title product as a gum. Yield: 18mg.
MS: APCI (+ve) 367 [M+H]
1YL NMR: δ (DMSO) 2.04 (quintet, 2H), 3.74 (t, 4H), 3.81 (s, 3H), 4.36 (s, 2H), 6.02 (s, IH), 7.23 (m, IH), 7.30 (m, 2H), 7.48 (d, 2H).
The intermediate for this compound was prepared as follows:
i) N-[(4-Methoxyphenyl)methyl]-N-[6-methoxy-2-[(phenylmethyl)thio]pyrimidin-4- yl] azetidine- 1 -sulfonamide KOtBu (46mg) was added to a mixture of the product of Example 121 step ϋ) (0.2Og) and phenylmethylthiol (50mg) in DMF (3ml) stirred under nitrogen. After 2.5h, 60% NaH (12mg) was added. The reaction mixture was stirred for a further 18h., diluted with EtOAc and washed with H2O. The separated organic solution was dried (MgSO4), filtered and the solvent evaporated under reduced pressure. The residue was purified by flash column chromatography on silica gel using EtOAc/isohexane (2:8) as eluent to give the product as a gum. Yield: 45mg. MS: APCI (+ve) 487 [M+H]
Example 125 N-{6-Methoxy-2-[[2(-pyrazin-2-yl)etliyl]tliio]pyrimidin-4-yl}azetidine-l-sulfonamide
-167-
The title compound was prepared fromN-[(4-metlioxyphenyl)methyl]-N-[6-methoxy-2-[2- [(pyrazin-2-yl)ethyl]tMo]pyrirnidin-4-yl]azetidine-l-sulfonarnide (the product of step i) (44mg) by the procedure outlined in Example 121. The crude material was purified by flash column chromatography on silica gel using EtOAc/iiohexane (7:3) as eluent to give the product as a white solid. Yield: 15mg. MS: APCI (+ve) 383 [M+H]
1H NMR: δ (DMSO) 2.12 (quintet, 2H), 3.22 (t, 2H), 3.43 (t, 2H), 3.72 (t, 4H), 3.80 (s, 3H), 5.98 (s, IH), 8.49 (s, IH), 8.58 (s, IH), 8.63 (s, IH).
The intermediate for this compound was prepared as follows:
i) N-[(4-Methoxyphenyl)methyl]-N-[6-methoxy-2-[(pyrazin-2-ylethyl)thio]pyrimidin-4- yl] azetidi ne- 1 -sulfonamide The subtitle compound was prepared from the product of Example 121 step ϋ) (0.2Og) and 2- (pyrazin-2-yl)ethanethiol (57mg) by the procedure outlined in Example 124 step i). The crude material was purified by flash column chromatography on silica gel using EtOAc/isohexane (1:1) as eluent to give the product as a gum. Yield: 44mg. MS: APCI (+ve) 503 [M+H]
Example 126
N-{2-[(2,3-Difluorobenzyl)thio]-6-methoxypyrimidin-4-yl}-l,4-diazepane-l-sulfonamide
A solution of tert-Butyl 4-[({2-[(2,3-difluorobenzyl)tMo]-6-methoxypyriirndin-4- yl}amino)sulfonyl]-l,4-diazepane-l-carboxylate (the product of step i, 0.22g) in 1: 1 TFA:methanol (6ml) was stirred at room temperature for 3h then the volatiles evaporated and 7M ammonia in methanol (5ml) added to the residue. The solution was stirred for 30min then the volatiles evaporated and the resulting solid washed with methanol, DCM, dimethyl sulfoxide and H2O to afford the title compound as a white powder. Yield: 51mg MS: APCI(+ve) 446 [M+H+]
1H NMR: δ 4DMSO) 1.90 - 1.98 (2H, m), 3.17 (4H, t, J = 6.0 Hz), 3.36 (2H, t, J = 5.9 Hz), 3.49 (2H, t, J = 5.8 Hz), 3.77 (3H, s), 4.41 (2H, s), 5.78 (IH, s), 7.11 - 7.19 (IH, m), 7.28 - 7.37 (IH, m), 7.42 - 7.46 (IH, m).
The intermediate for this compound was prepared as follows:
i) ϊe/t-Butyl 4-[({2-[(2,3-difluorobenzyl)thio]-6-methoxypyrimidin-4-yl}ainino)sulfonyl]- 1,4-diazepane-l-carboxylate
A mixture of tert-Butyl 4-(aminosulfony I)- 1,4-diazepane-l-carboxylate (the product of example 75, 0.277g), tris(dibenzylideneacetone)-dipalladium (0) (45mg), 2- dicyclohexylphosphino-2',4',6'-tri-i50propyl-l,l'-biphenyl (XPHOS) (24mg), cesium carbonate (0.242g) and 4-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-methoxypyrimidine (the product of example 35 step i, 0.15g) in anhydrous dioxane (6ml) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for 15min. Saturated aqueous ammonium chloride was added and the resulting mixture extracted with EtOAc. The combined organic extracts were washed with saturated aqueous sodium chloride, dried with sodium sulfate, filtered and evaporated. The residue was purified by column chromatography on silica using a 1 : 19 to 3:7 mixture of EtOAc and /sø-hexane as eluent to give the subtitle compound as a yellow oil. Yield: 0.223g MS: APCI(+ve) 546 [M+H+]
Example 127 (3R,5S)-N-{2-[(2,3-Difluorobenzyl)tWo]-6-methoxypyrimidin-4-yl}-3,5-dimethylpiperazine- 1- sulfonamide
-169-
A mixture of (3/?,5S)-3,5-dimethylpiperazine-l-sulfonamide (the product of example 72, 0.26g), tris(dibenzylideneacetone)-dipalladium (0) (61mg), 2-dicyclohexylphosphino-2',4',6'- tri-isOpropyl-lJ'-biphenyl (XPHOS) (32mg), cesium carbonate (0.32g) and 4-chloro-2- [[(2,3-difluorophenyl)methyl]thio]-6-methoxypyrimidine (the product of example 35 step i, 0.2Og) in anhydrous dioxane (8ml) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for 15min. Saturated aqueous ammonium chloride (5ml) and EtOAc (5ml) were added, followed by H2O. The layers were separated and the organic layer extracted with H2O (x3). The organic layer was discarded and the combined aqueous extracts exhaustively extracted with further EtOAc. These extracts were combined, washed with saturated aqueous sodium chloride, dried with sodium sulfate, filtered and evaporated. The resulting solid was washed with H2O to afford the title compound as a white solid. Yield: 0.11 Ig MS: APCI(+ve) 460 [M+H+] 1H NMR: δ (300 MHz, DMSO) 1.15 (d, 6H), 2.44 - 2.51 (m, 2H), 3.08 - 3.23 (m, 2H), 3.57 (dd, 2H), 3.78 (s, 3H), 4.43 (s, 2H), 5.84 (s, IH), 7.12 - 7.19 (m, IH), 7.29 - 7.38 (m, IH), 7.45 - 7.50 (m, IH).
Example 128
3-Amino-N- { 2-[(2,3-difluorobenzyl)thio]-6-methoxypyrimidin-4-yl } azetidine- 1 -sulfonamide
A solution of tert-butyl { l-[({2-[(2,3-difluorobenzyl)thio]-6-methoxypyrimidin-4- yl}amino)sulfonyl]azetidin-3-yl}carbamate (the product of step ϋ, 0.48g) and TFA (2ml) in methanol (6ml) was stirred at room temperature for 1.5h then the volatiles evaporated and 7M ammonia in methanol (6ml) added to the residue. The solution was stirred for 2h then the
-170- volatiles evaporated and the residue purified by column chromatography on silica using a 2- 8% mixture of methanol in DCM and then further purified by reverse phase HPLC (gradient 5-95% acetonitrile in 0.1% aqueous ammonium acetate) to afford the title compound as a white solid. Yield: 73mg MS: APCI(+ve) 418 [M+H+]
1H NMR: δ (300 MHz, DMSO) 3.64 (dd, 2H), 3.75 - 3.83 (m, IH), 3.79 (s, 3H), 3.90 (t, 2H), 4.43 (s, 2H), 5.93 (s, IH), 7.12 - 7.19 (m, IH), 7.28 - 7.38 (m, IH), 7.43 - 7.48 (m, IH).
The intermediates for this compound were prepared as follows:
i) tert-Butyl [l-(aminosulfonyl)azetidin-3-yl]carbamate
A solution of tert-butyl azetidin-3-ylcarbamate hydrochloride (prepared according to J. Antibiot. 1986, 39, 1243-1256, 0.755 g), Proton-Sponge® (0.85g) and sulfamide (0.42 g) in dioxane (23ml) was heated at reflux for 48h. The residue was partitioned between H2O and EtOAc, and the aqueous layer then extracted with further EtOAc (x4). The combined organic extracts were washed quickly with 2M aqueous hydrochloric acid (x3) then with saturated aqueous sodium bicarbonate, H2O and saturated aqueous sodium chloride, dried with sodium sulfate, filtered and evaporated to afford the subtitle compound as a pale brown powder. Yield: 0.44 g 1B. NMR: δ (300 MHz, DMSO) 1.38 (s, 9H), 3.55 (t, 2H), 3.82 (t, 2H), 4.09 - 4.18 (m, IH), 6.87 (s, 2H), 7.53 (d, IH).
ii) tert-Butyl { l-[({2-[(2,3-difluorobenzyl)thio]-6-methoxypyrimidin-4- yl } amino) sulfonyl] azetidin-3-yl } carbamate A mixture of tert-butyl [l-(aminosulfonyl)azetidin-3-yl]carbamate (0.5Og), tris(dibenzylideneacetone)-dipalladium (0) (0.12g), 2-dicyclohexylphosphino-2',4',6'-tri- /5opropyl-l,l'-biphenyl (XPHOS) (63mg), cesium carbonate (0.65g) and 4-chloro-2-[[(2,3- difluorophenyl)methyl]thio]-6-methoxypyrimidine (the product of example 35 step i, 0.40Og) in anhydrous dioxane (17ml) was heated to reflux in a microwave at 1000C, 300W, open vessel with cooling for 15min. Saturated aqueous ammonium chloride was added and the resulting mixture extracted with EtOAc. The combined organic extracts were washed with saturated aqueous sodium chloride, dried with sodium sulfate, filtered and evaporated. The
-171- residue was purified by column chromatography on silica using a 1: 19 to 3:7 mixture of EtOAc and wø-hexane as eluent to give the subtitle compound as a yellow oil. Yield: 0.48g MS: APCI(+ve) 518 [M+H+]
Example 129 iV-il-ICZjS-Difluorobenzy^thiol-ό-methoxypyriinidin^-yll-B-hydroxy-S-methylazetidine- 1 -sulfonamide
A mixture of 3-hydroxy-3-methylazetidine-l -sulfonamide (0.25g) (prepared according to patent WO 2004/011443), tris(dibenzylideneacetone)-dipalladium (0) (13mg), 2- dicyclohexylphosphino-2',4',6'-tri-i51opropyl-l,l'-biphenyl (XPHOS) (lOmg), cesium carbonate (0.68g) was treated with a solution of 4-Chloro-2-[[(2,3-difluorophenyl)methyl] thio]-6-methoxypyrimidine (the product of example 35 step i) (0.4g) in dioxane (10ml) and the whole then heated at reflux for 30min. H2O (10ml) was added followed by IN hydrochloric acid solution (5ml) and the resulting mixture extracted with EtOAc. The combined organic extracts were washed with saturated aqueous sodium chloride, dried (MgSO4), filtered and evaporated. The residue was purified by column chromatography on silica gel using EtOAc/DCM (1:4) as eluent to give the subtitle compound as a white solid. Yield: 0.5g. MS: APCI(+ve) 433 [M+H+], APCI(-ve) 431 [M-H"]
1B NMR δ(DMSO): 1.28 (s, 3H), 3.70 (d, IH), 3.80 (d, IH), 3.85 (s, 3H), 4.30 (s, 2H), 5.70 (s, IH), 6.10 (s, IH), 7.18 (m, IH), 7.35 (dd, IH), 7.43 (t, IH), 11.20 (bs, IH)
Example 130 3-Amino-7V-{2-[(2,3-difluorobenzyl)thio]-6-methoxypyrimidin-4-yl}-3-methylazetidine-l- sulfonamide
-172-
A solution of N-{2-[(2,3-difluorobenzyl)thio]-6-methoxyρyrimidiα-4-yl}-3-hydroxy-3- methylazetidine-1- sulfonamide (the product from example 129, 0.2g) in THF (5ml) was treated with dusøpropylethylamine (0.45ml) and methanesulfonylchloride (0.1 ImI) under nitrogen.
The whole was stirred at room temperature for 4h. The solvents were then evaporated in vacuo to dryness and the residue treated with 7N ammonia in methanol (9ml) and then heated in a sealed vessel at 75°C for 48h. The volatiles were then evaporated in vacuo and the residue purified by silica gel chromatography eluting with 10% methanol in DCM to give the subtitle product as a colourless gum. This was triturated with Et2O and isø-hexane mixtures and filtered to give the title product as a white solid. Yield: 50mg. MS: APCI(+ve) 432 [M+H+], APCI(-ve) 430 [M-H]
1H NMR δ (CDCl3): 1.45 (s, 3H), 2.90 (bs, 2H), 3.80 (q, 4H), 3.94 (s, 3H), 4.40 (s, 2H), 6.30 (s, IH), 7.10 (m, IH), 7.20 (m, 2H)
Example 131 iV-il-tCl^-Difluorobenzy^thiol-o-methoxypyrimidin^-yU-S-inethyl-S-
(methylamino)azetidine-l-sulfonamide
A solution of N-{2-[(2,3-difluorobenzyl)thio]-6-methoxyρyrimidin-4-yl}-3-hydroxy-3- methylazetidine- 1 -sulfonamide (the product from example 129, 0.16g) in THF (8ml) was treated with dusopropylethylamine (0.5ml) and methanesulfonylchloride (0.113ml) under nitrogen.
-173-
The whole was stirred at room temperature for 16h. The mixture was then treated with 33% methylamine in ethanol (10ml) and then heated in a sealed vessel at 7O0C for 24h. The volatiles were then evaporated in vacuo and the residue purified by silica gel chromatography eluting with 10% methanol in DCM to give the subtitle product as a colourless gum. This was triturated with ethanol and filtered to give the title product as a white solid. Yield: 57mg. MS: APCI(+ve) 446 [M+H+], APCI(-ve) 444 [M-H ]
1H NMR δ(DMSO): 1.33 (s, 3H), 2.35 (s, 3H), 3.60 (d, 2H), 3.80 (s, 3H), 3.85 (d, 2H), 4.40 (s, 2H), 5.92 (s, IH), 7.10 (m, IH), 7.30 (m, IH), 7.40 (m, IH)
Example 132 iV-{2-[(2,3-Difluorobenzyl)thio]-6-methoxypyrimidin-4-yl}-4-glycylpiperazine-l- sulfonamide, hydrochloride salt
A solution of tert-butyl (2-{4-[({2-[(2,3-difluorobenzyl)thio]-6-methoxypyrimidin-4- yl}amino)suhcOnyl]piperazin-l-yl}-2-oxoethyl)carbamate (the product of step ϋ, 0.19g) in 10% TFA/DCM (5mL) was stirred at room temperature for 3h. The solution was evaporated, and then redissolved in 4N HCl in dioxane (2mL) and MeOH (8mL). Evaporation gave a crude residue that was triturated in Et2θ, filtered and dried in a vacuum oven at 400C overnight to give the title compound as a white solid. Yield: 140mg. MS: APCI(-ve) 487 [M-H"]
1H NMR (DMSO) δ 3.20-3.27 (4H, m), 3.41-3.46 (2H, m), 3.53-3.58 (2H, m), 3.86 (2H, s), 3.88 (3H, s), 4.48 (2H, s), 6.09 (IH, s), 7.13-7.21 (IH, m), 7.37-7.44 (2H, m), 8.06 (2H, br s), 11.26 (IH, br s)
the intermediates for this compound were prepared as follows
-174- i) N-[2-[[(2,3-Difluorophenyl)methyl]thio]-6-methoxypyrimidin-4-yl]piperazine-l- sulfonamide, hydrochloride salt
A solution of N-[2-[[(2,3-Difluoroρhenyl)methyl]tWo]-6-methoxypyriiriidin-4-yl]ρiρerazήie- 1 -sulfonamide, trifluoro acetate salt (the product of example 36, 0.6g) in 4N HCl/dioxane (2mL) and Et2O (2OmL) was stirred at room temperature for 20min. The resulting suspension was filtered and the residue dried in a vacuum oven at 400C for 2h to give the subtitle compound as a white solid. Yield: 0.55g. MS: APCI(+ve) 432 [M+H+]
ii) tert-Butyl (2-{4-[({2-[(2,3-difluorobenzyl)thio]-6-methoxypyrimidin-4- yl}amino)sulfonyl]piperazin-l-yl}-2-oxoethyl)carbamate
To a solution of N-(tørt-butoxycarbonyl)grycine (0.1 Ig) in DMF (1OmL) was added 1,3- Dicyclohexycarbodiimide (0.14g) and 1 -hydro xybenzotriazo Ie hydrate (94mg). After stirring at room temperature for Ih, a solution of N-[2-[[(2,3-Difluorophenyl)methyl]thio]-6- methoxypyrirnidin-4-yl]piperazine-l-suhconamide, hydrochloride salt (the product of step i, 0.27g) and N-methylmorpholine (78μL) in DMF (5mL) was added dropwise and stirring continued at room temperature for 24h. The mixture was filtered, rinsed with DCM and the filtrate evaporated. The crude material was purified by column chromatography on silica gel using EtOAc/irøhexane (3:2) as eluent to give the subtitle compound as a foam. Yield: 0.24g MS: APCI(-ve) 587 [M-H]
Example 133
4-β-Alanyl-N-{2-[(2,3-difluorobenzyl)thio]-6-methoxypyrimidin-4-yl}piperazine-l- sulfonamide, hydrochloride salt
The title compound was prepared from tert-buty\ (3-{4-[({2-[(2,3-difluorobenzyl)thio]-6- methoxypyrimidin-4-yl}arnino)sulfonyl]piperazin- l-yl}-3-oxopropyl)carbamate (the product
-175- of step i, 0.22g) according to the procedure outlined in example 132 to give a white solid. Yield: 0.15g.
MS: APCI(+ve) 503 [M+H+]
1H NMR (DMSO) δ 2.66 (2H, t), 2.98 (2H, q), 3.19-3.26 (4H, m), 3.45-3.49 (2H, m), 3.51- 3.54 (2H, m), 3.88 (3H, s), 4.48 (2H, s), 6.08 (IH, s), 7.14-7.20 (IH, m), 7.31-7.39 (IH, m), 7.40-7.44 (IH, m), 7.72 (2H, br s), 11.24 (IH, br s)
The intermediate for this compound was prepared as follows:
i) tert-Butyl (3-{4-[({2-[(2,3-difluorobenzyl)thio]-6-methoxypyrimidin-4- yl}amino)sulfonyl]piperazin- 1 -yl }-3-oxopropyl) carbamate
The subtitle compound was prepared from N-(tert-butoxycarbonyl)β-alanine (0.12g) according to the procedure outline in example 132, step ii). The crude material was purified by column chromatography on silica gel using EtOAc/irøhexane (3:2) as eluent to give the subtitle compound as a foam. Yield: 0.22g MS: APCI(-ve) 601 [M-H]
Example 134
N-(2-[(2,3-Difluorobenzyl)thio]-6-methoxypyrimidin-4-yl)-l,2,3,4- tetrahydroisoquinoline-7-sulfonamide
N-(2-[(2,3-difluorobenzyl)thio]-6- methoxypyrimidin-4-yl) 2-(trifluoroacetyl)- 1 ,2,3,4- tetrahydroisoquinoline-7-sulfonamide (0.805g) was added to a solution of 7N NH3 in MeOH (20ml), sealed and stirred at room temperaturefor 2h. The reaction was reduced in vacuo and the resulting residue purified by prep HPLC to give the title compound as a white solid. Yield: 70mg MS: APCI(+ve) 479 [M+H+]
-176-
1H NMR: (DMSO) δ 3.01-3.08 (m, 2H), 3.35-3.42 (m, 2H), 3.83 (s, 3H), 4.33-4.40 (m, 2H), 4.38 (s, 2H), 6.06 (s, IH), 7.09-7.20 (m, IH), 7.31-7.40 (m, 2H), 7.44-7.50 (m, IH), 7.78-7.87 (m, 2H) , 9.00-9.09 (m, 2H)
The intermediate for this compound was prepared as follows:
i) N-(2-[(2,3-Difluorobenzyl)thio]-6- methoxypyrimidin-4-yl) 2-(trifluoroacetyl)-l,2,3,4- tetrahydroisoquinoline-7-sulfonamide
A mixture of l,2,3,4-tetrahydroisoquinoline-7-sulfonamide (the product from example 78 step ii, 0.6Ig), tris(dibenzylideneacetone)dipalladium (0) (50 mg), 2-dicyclohexylphosphino- 2',4',6'-tri-/røpropyl-l,l'-biρhenyl (XPHOS) (50mg), cesium carbonate (0.43g) and, 4- Chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-methoxypyrimidine (the product from example 35 step i, 0.4g) in dioxane (20ml) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for 3h. The reaction mixture was then reduced in vacuo and the residue partitioned between DCM (150 ml) and H2O (150 ml). The organics were separated and the aqueous layer was re-extracted with DCM (2 x 150ml). Organics were combined, dried (MgSO4) and reduced under to give the subtitle compound as a yellow solid. Yield: 0.8 Ig MS: APCI(+ve) 575 [M+H+]
Example 135
N-(2-[(2,3-Difluorobenzyl)thio]-6-methoxypyrimidin-4-yl)-(2S,5R)-2,5- dimethylpiperazi ne- 1 -sulfonamide
To a solution of (2/?,5S)-2,5-dirnethylpiperazine (2g) in dioxane (100ml) was added sulfamide (2.5g) and the reaction mixture was then heated at reflux in dioxane (100ml) for 72h. The reaction mixture was partitioned between EtOAc (150ml) and H2O (150ml) and the aqueous re-extracted with EtOAc (2xl50ml). Organics were collected, dried and reduced in vacuo to
-177- give (2S,5/?)-2,5-dimethylpiperazine-l -sulfonamide as a white solid (1.2g). A mixture of (2S,5/?)-2,5-dimethylpiperazine-l -sulfonamide (0.38g), tris(dibenzylideneacetone)dipalladium (0) (50 mg), 2-dicyclohexylphosphino-2',4',6'-tri- iiOpropyl-l,l'-biphenyl (XPHOS) (50mg), cesium carbonate (0.43g) and, 4-Chloro-2-[[(2,3- difluorophenyl)methyl]thio]-6-methoxypyrimidine (the product from example 35 step i), 0.4g) in dioxane (2OmL) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for 4h. The reaction mixture was then reduced in vacuo and the residue partitioned between DCM (150ml) and H2O (150ml). The organics were separated and the aqueous layer was re-extracted with DCM (2 x 150ml). Organics were combined, dried (MgSO4) and reduced in vacuo to give a yellow solid. This residue was then purified by prep HPLC to give the title compound as a white solid. Yield: 9mg MS: APCI(+ve) 460 [M+H+]
1B. NMR: (DMSO) 5 1.05 (d, 3H), 1.23 (d, 3H), 2.58-2.67 (m, IH), 2.72-2.80 (m, IH), 3.01- 3.54 (m, 4H), 3.77 (s, 3H), 4.40 (s, 2H), 5.83 (s, IH), 7.07-7.21 (m, IH), 7.24-7.47 (m, 2H)
Example 136
N-(2- [(2,3-Difluorobenzyl)thio] -6-methoxypyrimidin-4-yl) -4-
(aminomethyl)benzenesulfonamide
A mixture of 4-(aminomethyl)benzenesuh
cbnamide (0.37g), tris(dibenzylideneacetone)dipalladium (0) (50 mg), 2-dicyclohexylphosphino-2',4',6'-tri- woρropyl-l,l'-biphenyl (XPHOS) (50mg), cesium carbonate (1.Og) and, 4-Chloro-2-[[(2,3- difluorophenyl)methyl]thio]-6-methoxypyrimidine (the product from example 35 step i), (0.25g) in dioxane (20ml) was heated at reflux in a microwave at 100
0C, 300W, open vessel with cooling for 3h. The reaction mixture was then reduced in vacuo and the residue partitioned between DCM (150ml) and H
2O (150ml). The organics were separated and the
-178- aqueous layer was re-extracted with DCM (2 x 150ml). Organics were combined, dried (MgSO
4) and reduced in vacuo to give a yellow solid. This solid was then purified by prep HPLC to give the title compound as a white solid. Yield: 19mg MS: APCI(+ve) 453 [M+H
+]
1H NMR: (DMSO) δ 3.83 (s, 3H), 4.09-4.14 (m, 2H), 4.37 (s, 2H), 6.08 (s, IH), 7.09-7.22 (m, IH), 7.31-7.38 (m, 2H), 7.66 (d, 2H), 7.98 (d, 2H), 8.16-8.24 (m, 2H)
Example 137 N-{2-[[(3-Fluorophenyl)methyl]thio]-6-methoxypyrimidin-4-yl}azetidine-l-sulfonamide
The title compound was prepared from N-{2-[[(3-fluorophenyl)methyl]thio]-6- methoxypyrimidin-4-yl }-N-[(4-methoxyphenyl)methyl]azetidine- 1-sulfonamide (the product of step iv) (42mg) by the procedure outlined in Example 121. The crude material was purified by preparative plate chromatography using EtOAc/irøhexane (4:6) as eluent to give the title product as a gum. Yield: 22mg. MS: APCI (+ve) 385 [M+H]
1H NMR: δ (DMSO) 2.10 (quintet, 2H), 3.87 (m, 7H), 4.41 (s, 2H), 6.12 (s, IH), 7.07 (m, IH), 7.33 (m, 3H), 11.11 (bs, IH).
The intermediates for this compound were prepared as follows:
i) N-[(4-Methoxyphenyl)methyl]-N-[6-methoxy-2-[[(2,3,4-trifluorophenyl)methyl] thio]pyrimidin-4-yl]azetidine-l-suIfonamide
The subtitle compound was prepared from N-[6-Methoxy-2-[[(2,3,4- trifluorophenyl)methyl]thio]pyrimidin-4-yl]azetidiαe- 1-sulfonamide (the product of Example 146, (5. Ig) by the procedure outlined in Example 121 step i). The crude product was purified
-179- by flash column chromatography on silica gel using EtOAc/isohexane (2:8) as eluent to give the product as an oil. Yield: 4.2g. MS: APCI (+ve) 541 [M+H]
ii) N-[(4-Methoxyphenyl)methyl]-N-[6-methoxy-2-[[(2,3,4-trifluorophenyl)methyl] sulfony l]py rimidin-4-yl] azetidine- 1 -sulfonamide
The subtitle compound was prepared from N- [(4-Methoxyphenyl)methy I]-N- [6-methoxy-2- [[(2,3,4-trifluorophenyl)methyl] thio]pyrimidin-4-yl] azetidine- 1-sulfonamide (the subtitle product of step i), (4.2g) by the procedure outlined in Example 121 step ii). The crude product was purified by flash column chromatography on silica gel using EtOAc / /.røhexane (1:1) as eluent to give the product as a white foam. Yield: 3.3g. MS: APCI (+ve) 573 [M+H]
iii) N-[6-Methoxy-2-thiopyrimdin-4-yl]-N-[(4-methoxyphenyl)methyl]azetidine-l- sulfonamide
NaSH (40mg) was added to a solution of N- [(4-Methoxyphenyl)methy I]-N- [6-methoxy-2- [[(2,3,4-trifluorophenyl)methyl] sulfonyl]pyrirnidin-4-yl]azetidine-l-sulfonamide (the subtitle product of step ii), (0.1Og) and stirred in water (ImI) under nitrogen at 95°C for 45min. NaSH (40mg) followed by DMF (ImI) were added. The reaction mixture was stirred for a further 1.5h at 950C, cooled, acidified with dilute HCl and extracted with EtOAc. The separated organic solution was washed with water, dried (MgSO4), filtered and the solvent evaporated under reduced pressure to give the subtitle product as a clear oil. Yield: 90mg MS: APCI (+ve) 397 [M+H]
iv) N-[2-[[(3-Fluorophenyl)methyl]thio]-6-methoxy pyrimidin-4-yl]-N-[(4-methoxy- phenyl) methyl]-azetidine-l-sulfonamide
60% NaH (8mg) was added to a solution of N-[6-Methoxy-2-thiopyrimdin-4-yl]-N-[(4- methoxyphenyl)methyl] azetidine- 1-sulfonamide (the subtitle product of step iii), (90mg) in anhydrous DMF (ImI). After stirring under nitrogen for 5min (3-fluorophenyi)methyl bromide (42mg) was added. The reaction mixture was stirred at room temperature for a further 18h and then diluted with EtOAc. The separated organic solution was washed with
-180- water, dried (MgSO4), filtered and the solvent evaporated under reduced pressure. The crude material was purified by flash column chromatography on silica gel using EtOAc/irøhexane (3:7) as eluent to give the product as a gum. Yield: 47mg MS: APCI (+ve) 505 [M+H]
Example 138
N-{2-[(2,3-Difluorobenzyl)thio]-6-methoxypyrimidin-4-yl}-4-pyrroIidiii-l-ylpiperidine-l- sulfonamide
The title compound was prepared according to the procedure outlined in example 129 using 4-Chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6-methoxypyrimidhie (the product of example 35 step i) (0.3g), tris(dibenzylideneacetone)-dipalladium (0) (20mg), 2- dicyclohexylphosphino-2',4',6'-tri-i5Opropyl-l,r-biphenyl (XPHOS) (15mg), cesium carbonate (0.7Og) and 4-pyrrolidin-l-ylpiperidine-l-surfonamide (the product from step i), (0.4g). The resulting crude material was purified using silica gel chromatography eluting with 5% methanol in DCM and trituration with Et
2O to give the title compound as a white solid. Yield: 0.27g
MS: APCI(+ve) 500 [M+H+], APCI(-ve) 498 [M-H ] 1H NMR δ(DMSO) δ 1.60 (m, 2H), 1.90 (bs, 4H), 2.10 (d, 2H), 3.10 (m, 5H), 3.70 (d, 4H), 3.90 (3, 3H), 4.50 (s, 2H), 6.05 (s, IH), 7.20 (m, IH), 7.40 (m, 2H)
The intermediates for this compound were prepared as follows:
i) 4-Pyrrolidin- 1 -ylpiperidine- 1 -sulfonamide A mixture of 4-pyrrolidin- 1 -ylpiperidine (0.67g) and sulfamide (0.46g) were heated at 115 0C in dry 1,4-dioxane (30ml) for 16h. The solvents were evaporated in vacuo and the residue partitioned between EtOAc (containing a little methanol) and H2O. The organic phase was
-181- collected and the aqueous layer further extracted with EtOAc(x2). The combined organic phases collected, dried (MgSO4) and the solvent evaporated. The residue was triturated with Et2O and filtered to give the subtitle product as a beige solid. Yield: 0.43g 1H NMR δ(DMSO) 5 1.50 (m, 2H), 1.70 (m, 4H), 1.90 (m, 2H), 2.05 (m, IH), 2.50 (m, 2H), 2.60 (m, 2H), 3.40 (m, 4H), 6.70 (s, 2H)
Example 139
N-{2-[(2,3-Difluorobenzyl)thio]-6-methoxypyrimidin-4-yl}-3-morpholin-4-ylazetidine-l- sulfonamide
A solution of N-{2-[(2,3-dMuoroberizyl)tMo]-6-methoxypyrirnidin-4-yl}-3-hydroxyazetidine-
1-sulfonamide (the product from example 109) (0.28g) in DCM (10ml) was treated with triethylamiαe (0.8ml) and methanesulfonylchloride (0.9 ml) under nitrogen. After heating the mixture at 5O0C for 16h the reaction mixture was partitioned between DCM and aqueous NaHCO3. The organic extracts were dried (MgSO4), filtered and the solvent evaporated under reduced pressure. To a solution of the resulting residue in MeOH (1OmL) and morpholine (8ml) K2CO3 (0.19g) was added and heated at 800C for 16h. The reaction mixture was then partitioned between EtOAc and H2O The organic extracts were washed with brine, dried (MgSO4), filtered and the solvent evaporated under reduced pressure. The residue was purified by reverse phase HPLC (symmetry as the stationary phase and NH-tOAc/acetonitrile as the mobile phase) then titurated with Et2O to give the title compound as a white solid. Yield: 15mg
MS: APCI(+ve) 488 [M+H+] 1U NMR δ(DMSO) 2.23 (s, 4H), 3.00 - 3.08 (m, IH), 3.51 (t, 4H), 3.76 - 3.81 (m, 4H), 3.87 (s, 3H), 4.49 (s, 2H), 6.10 (s, IH), 7.12 - 7.19 (m, IH), 7.29 - 7.38 (m, IH), 7.44 (t, IH)
-182- Examnles 140 - 145
Examples 140 -145 were synthesised using the following procedure: - The title compounds, tabulated below, were prepared from the appropriate 2-thio substituted N-[(4-methoxyphenyl)memyl]-N-[6-methoxy-2-tWo]pyrirrύdin-4-yl]azetidine-l-suhconarnides (the products of step i) by the procedure outlined in Example 121. The crude materials were purified by mass directed purification.
The intermediates for compounds 140 -145 were prepared as follows:
i) Thio-substituted N-[(4-Methoxyphenyl)inethyl]-N-[6-methoxy-2-thio]pyriinldin-4- yl]azetidine-l-sulfonamides
Sodium thiolate (30mg) was added to a solution of N-[(4-Methoxyphenyl)methyl]-N-[6- methoxy-2-[[(2,3,4-trifluorophenyl)methyl] sulfonyl]pyrimidin-4-yl]azetidine-l-sulfonamide (the product of Example 137 step ϋ) (0.15g) stirred in anhydrous DMSO under nitrogen. After 30min, the appropriate bromide or chloride (see R" in the table below) (0.8ImM) was added. The reaction mixture was stirred for a further 30min, diluted with water and the product extracted with EtOAc. The separated organic solution was washed with water, dried (MgSO4), filtered and the solvent evaporated under reduced pressure. The crude products were purified by flash column chromatography on silica gel using mixtures of EtOAc/jrøhexane as eluent to give the products, Examples 140 i) - 145 i), tabulated below.
Example 146
N-[6-Methoxy-2-[[(2,3,4-trifluorophenyl)methyl]thio]pyrimidin-4-yl]azetidine-l- sulfonamide
The title compound was prepared from 4-Chloro-6-methoxy-2-[[(2,3,4- trifluorophenyl)methyl]thio]pyrimidine (the subtitle product of step iii) (7.2g) by the
-185- procedure outlined in Example 1 step iv). The crude material was purified by recrystallisation from /søhexane/EtOAc to give the product as a yellow solid. Yield: 5.1g. MS: APCI (+ve) 421 [M+H]
1H NMR: δ (DMSO) 5 2.13 (quintet, 2H), 3.88 (s, 3H), 3.90 (t, 4H), 4.46 (s, 2H), 6.15 (s, IH), 7.32 - 7.24 (m, IH), 7.53 - 7.46 (m, IH), 11.13 (s, IH)
The intermediates for this compound were prepared as follows:
i) 2-[[(2,3,4-Trifluorophenyl)methyl]Mo]pyrimidine-4,6-diol The subtitle compound was prepared from 2-thiopyrimidine-4,6-diol (80.Og) and (2,3,4- trifluorophenyl)methyl bromide (125g) by the procedure outlined in Example 1 step i). Yield: 15Og. 1H NMR: 5 (DMSO) 4.41 (s, 2H), 5.22 (bs, IH), 7.30 (m, IH), 7.49 (m, IH).
ϋ) 4,6-Dichloro-2-[[(2,3,4-trifluorophenyl)methyl]thio]pyriiradine
The subtitle compound was prepared from the subtitle product of step i) (15Og) by the procedure outlined in Example 1 step ϋ). The crude material was purified by flash column chromatography on silica gel using EtOAc/isøhexane (3:7) as eluent to give the product as a white solid. Yield: 7Og. 1H NMR: δ (CDCl3) 4.37 (s, 2H), 6.91 (m, IH), 7.06 (s, IH), 7.26 (m, IH).
iii) 4-Chloro-6-methoxy-2-[[(2,3,4-trifluorophenyl)methyl]thio]pyrimidine
The subtitle compound was prepared from the subtitle product of step ϋ) (25.Og) by the procedure outlined in Example 35 step i). The crude material was purified by recrystallisation from isøhexane to give the product as white crystals. Yield: 16.4g. MS: APCI (+ve) 321/323 [M+H]
Example 147
Nv-2{2-[(2,3-Difluorobenzyl)thio]-6-methoxypyrimidin-4-yl}-N-methyl-N-(l- methylpiperidin-4-yl)sulfamide
■186-
The title compound was prepared from N-methyl-N-(l-methylpiperidin-4-yl)sulfamide (the product of step i) (0.26g) and 4-chloro-2-[[(2,3-difluorophenyl)methyl]thio]-6- methoxypyrimidine (the product of Example 35, step i) (0.25g) according to the procedure outlined in Example 1, step iv). The crude material was purified by column chromatography using EtOAc / MeOH (9:1 to 8.5:1.5) as eluent. Yield: 0.17g MS: APCI (+ve) 474 [M+H]
1H NMR: δ (DMSO) 1.50 (bd, 2H), 1.66 (m, 2H), 2.02 (t, 2H), 2.20 (s, 3H), 2.67 (s, 3H), 2.84 (bd, 2H), 3.63 (m, IH), 3.82 (s, 3H), 4.44 (s, 2H), 5.90 (s, IH), 7.14 (q, IH), 7.33 (q, IH), 7.41 (t, IH).
The intermediate for this compound was prepared as follows:
i) N-Methyl-N-(l-methylpiperidin-4-yl)sulfamide A solution of l-methyl-4-(methylamino)piperidine (2.6g) and sulfamide (4.Og) in 1,4-dioxane (30ml) was heated at 1100C for 18h. The reaction mixture was cooled, the solvent evaporated under reduced pressure and the residue dissolved in water. The aqueous solution was extracted with EtOAc which was washed with a small volume of saturated aqueous brine, dried (MgSO_0 and the solvent evaporated under reduced pressure to give the subtitle product as a pale yellow solid. Yield : 1.5g
1H NMR: δ (CDCl3) 1.80 (m, 4H), 2.04 (dt, 2H), 2.27 (s, 3H), 2.79 (s, 3H), 2.91 (bd, 2H), 3.74 (quintet, IH), 4.44 (bs, 2H).
Example 148 iV-{2-[(2,3-Difluorobenzyl)thio]-6-methoxypyrimidin-4-yl}-4-morpholin-4-ylpiperidine- 1 -sulfonamide
-187-
Sodium triacetoxyborohydride (0.48g) was added to a solution of N-{2-[(2,3- difluorobenzyl)thio]-6-methoxypyrimidhi-4-yl}-4-oxopiperidiiie-l-sulfonamide (the product of step ϋi) (0.249g), morpholine (0.2mL) and 2M aqueous acetic acid (0.5mL) in DCM (12mL). The mixture was stirred at room temperature for 18h then 2M aqueous sodium hydroxide (1OmL) added to the residue. The mixture was shaken vigorously then acidified to pH 8 with 2M aqueous hydrochloric acid and extracted with ethyl acetate. The combined organic extracts were washed with saturated aqueous sodium chloride, dried with sodium sulfate, filtered and evaporated. The residue was purified by column chromatography on silica using a 3:7 to 1:0 mixture of ethyl acetate and tiO-hexane as eluent then precipitated slowly from methanol, filtered and washed with further methanol to afford the title compound as a pale yellow solid. Yield: 53mg MS: APCI(+ve) 516 [M+H+] 1H NMR: δ (300 MHz, DMSO) 1.26 - 1.41 (m, 2H), 1.76 - 1.83 (m, 2H), 2.28 (t, IH), 2.41 - 2.44 (m, 4H), 2.83 (t, 2H), 3.52 - 3.58 (m, 4H), 3.68 (d, 2H), 3.88 (s, 3H), 4.49 (s, 2H), 6.07 (s, IH), 7.14 - 7.21 (m, IH), 7.31 - 7.47 (m, 2H).
The intermediates for this compound were prepared as follows:
i) l,4-Dioxa-8-azaspiro[4.5]decane-8-sulfonamide
A solution of l,4-dioxa-8-aza-spiro[4.5]decane (2 mL) and sulfamide (1.65g) in 1,4-dioxane (28mL) was heated at reflux for 48h, then the volatiles were evaporated to afford the title compound as a pale yellow solid. Yield: 3.4g 1H NMR: δ (300 MHz, DMSO) 1.71 (dd, 4H), 3.08 (dd, 4H), 3.91 (s, 4H), 6.77 (s, 2H).
-188- ii) N-{2-[(2,3-Difluorobenzyl)thio]-6-methoxypyrimidin-4-yl}-l,4-dioxa-8- azaspiro[4.5]decane-8-sulfonamide
A mixture of l,4-dioxa-8-azaspiro[4.5]decane-8-sulfonamide (the product of step i), (0.29g), tris(dibenzylideneacetone)-dipalladium (0) (61mg), 2-dicyclohexylphosphino-2',4',6'-tri- isøpropyl-1 ,1 '-biphenyl (XPHOS) (32 mg), cesium carbonate (0.32g) and 4-chloro-2-[[(2,3- difluoroρhenyl)methyl]thio]-6-methoxypyrimidine (the product of example 35 step i, 0.2Og) in anhydrous dioxane (8mL) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for 15min. Saturated aqueous ammonium chloride was added and the resulting mixture extracted with ethyl acetate. The combined organic extracts were washed with saturated aqueous sodium chloride, dried with sodium sulfate, filtered and evaporated. The residue was purified by column chromatography on silica using a 1:19 to 2:3 mixture of ethyl acetate and /.rø-hexane as eluent to give the subtitle compound as a yellow foam. Yield: 0.27g MS: APCI(+ve) 489 [M+H+]
iii) yV-{2-[(2,3-Difluorobenzyl)thio]-6-methoxypyrimidin-4-yl}-4-oxopiperidine-l- sulfonamide
N- {2-[(2,3-Difluorobenzyl)thio]-6-methoxypyrimidin-4-yl} - 1 ,4-dioxa-8-azaspiro[4.5]decane-
8-sulfonamide (the product of step ϋ) (0.85g) was heated to 500C in a mixture of 2M aqueous hydrochloric acid (17mL) and THF (17mL). After 24h, the reaction was allowed to cool to room temperature then diluted with ethyl acetate, the layers separated and the organic material washed with saturated aqueous sodium bicarbonate, water, saturated aqueous sodium chloride, dried with sodium sulfate, filtered and evaporated to afford the subtitle compound as a yellow oil. Yield: 0.83g MS: APCI(+ve) 445 [M+H+]
Example 149 iV-{2-[(2,3-Difluorobenzyl)thio]-6-methoxypyrimidin-4-yl}-4-(4-methylpiperazin-l- yl)piperidine- 1 -sulfonamide
-189-
A solution of 1-methyl-piperazine (0.13mL) in DCM (2mL) was added to a solution of acetic acid (0.03mL) and N-{2-[(2,3-difluorobenzyl)thio]-6-methoxypyrimidin-4-yl}-4- oxoρiρeridine-1 -sulfonamide (the product of example 148, step iii) (O.lOg) in DCM (2mL). 5 The solution was stirred at room temperature for Ih then sodium triacetoxyborohydride (0.24g) was added in portions. The mixture was stirred at room temperature overnight then the DCM was evaporated and 3M aqueous sodium hydroxide (6mL) added to the residue. The mixture was shaken vigorously then acidified to pH 8 with 2M aqueous hydrochloric acid and extracted with ethyl acetate. The combined organic extracts were washed with water,
10 saturated aqueous sodium chloride, dried with sodium sulfate, filtered and evaporated. The residue was purified by reverse phase HPLC (gradient 25 - 95% acetonitrile in 0.1% aqueous ammonium acetate) to afford the title compound as a white powder. Yield: 22mg MS: APCI(+ve) 529 [M+H+] 1H NMR: δ (300 MHz, DMSO) 1.26 - 1.42 (m, 2H), 1.75 - 1.78 (m, 2H), 2.25 - 2.77 (m,
15 11H), 2.27 (s, 3H), 3.63 (d, 2H), 3.84 (s, 3H), 4.46 (s, 2H), 6.01 (s, IH), 7.13 - 7.20 (m, IH), 7.31 - 7.40 (m, IH), 7.43 - 7.48 (m, IH).
Example 150 iV-{2-[(2,3-Difluorobenzyl)thio]-6-methoxypyrimidin-4-yl}-4-hydroxypiperidine-l- 20 sulfonamide
From the crude material obtained after work up to prepare N-{2-[(2,3-difluorobenzyl)thio]-6- methoxypyrimidin-4-yl}-4-morpholin-4-ylpiperidine-l-sulfonamide (Example 148), a second product was also isolated. This was further purified by reverse phase HPLC (gradient 25 - 95% acetonitrile in 0.1% aqueous ammonium acetate) to afford the title compound as a white powder. Yield: 33mg
MS: APCI(+ve) 447 [M+H+]
1H NMR: δ (300 MHz, DMSO) 1.33 - 1.44 (m, 2H), 1.70 - 1.76 (m, 2H), 3.00 - 3.08 (m, 2H), 3.41 - 3.49 (m, 2H), 3.57 - 3.64 (m, IH), 3.89 (s, 3H), 4.49 (s, 2H), 4.75 (d, IH), 6.08 (s, IH), 7.14 - 7.21 (m, IH), 7.32 - 7.47 (m, 2H), 11.07 (s, IH).
Example 151
4-Azetidin-l-yl-iV-{2-[(2,3-difluorobenzyl)thio]-6-methoxypyrimidin-4-yl}piperidine-l- sulfonamide
Azetidine hydrochloride (0.1 Ig) was added to a solution of acetic acid (0.025mL) and N-{2- [(2,3-difluorobenzyl)thio]-6-methoxypyrimidin-4-yl}-4-oxopiperidine-l-suh
c'onamide (the product of example 148, step iii) (O.lOg) in DCM (4mL). The solution was stirred at room temperature for Ih then sodium triacetoxyborohydride (0.24g) was added in portions. The mixture was stirred at room temperature overnight then 3M aqueous sodium hydroxide (6mL) added to the residue. The mixture was shaken vigorously then acidified to pH 8 with 2M aqueous hydrochloric acid and extracted with ethyl acetate. The combined organic extracts were concentrated then methanol (ImL) was added and the resulting suspension filtered. The solid was washed with water, methanol and ethyl acetate to afford the title compound as a white powder. Yield: 47mg MS: APCI(+ve) 486 [M+H
+]
-191-
1H NMR: δ (300 MHz, DMSO) 1.15 - 1.26 (m, 2H), 1.69 - 1.78 (m, 2H), 2.01 - 2.09 (m, 2H), 2.47 - 3.51 (m, 9H), 3.83 (s, 3H), 4.45 (s, 2H), 5.96 (s, IH), 7.13 - 7.20 (m, IH), 7.30 - 7.39 (m, IH), 7.43 - 7.48 (m, IH).
Example 152 iV-{2-[(2,3-Difluorobenzyl)thio]-6-methoxypyrimidin-4-yl}-4-(ethylamino)piperidine-l- sulfonamide
Ethylamine (0.56mL of 2M solution in methanol) was added to a solution of acetic acid (0.025mL) and iV-{2-[(2,3-difluorobenzyl)tnio]-6-methoxypyrimidin-4-yl}-4-oxopiperidine- 1 -sulfonamide (the product of example 148, step iii) (O.lOg) in DCM (4mL). The solution was stirred at room temperature for Ih then sodium triacetoxyborohydride (0.24g) was added in portions. The mixture was stirred at room temperature overnight then 3M aqueous sodium hydroxide (6mL) added to the residue. The mixture was shaken vigorously then acidified to pH 8 with 2M aqueous hydrochloric acid and extracted with ethyl acetate. The combined organic extracts were concentrated then methanol (ImL) was added and the resulting suspension filtered. The solid was washed with water, methanol and ethyl acetate to afford the title compound as a very pale yellow powder. Yield: 52mg MS: APCI(+ve) 474 [M+H+] 1H NMR: δ (300 MHz, DMSO) 1.17 (t, 3H), 1.41 - 1.52 (m, 2H), 1.96 - 2.01 (m, 2H), 2.57 - 2.61 (m, 2H), 2.94 (q, 2H), 3.00 - 3.08 (m, IH), 3.55 (d, 2H), 3.74 (s, 3H), 4.40 (s, 2H), 5.84 (s, IH), 7.11 - 7.18 (m, IH), 7.28 - 7.36 (m, IH), 7.44 - 7.49 (m, IH).
Example 153 4-(Cyclopropylamino)-N-{2-[(2,3-difluorobenzyl)thio]-6-methoxypyrimidin-4- yl}piperidine-l-sulfonamide
-192-
A solution of cyclopropylamine (0.08mL) in DCM (2 mL) was added to a solution of acetic acid (0.025mL) and N-{2-[(2,3-difluorobenzyl)thio]-6-metlioxypyrimidin-4-yl}-4- oxopiperidine-1 -sulfonamide (the product of example 148, step iii) (O.lOg) in DCM (2mL). The solution was stirred at room temperature for Ih then sodium triacetoxyborohydride (0.24g) was added in portions. The mixture was stirred at room temperature overnight then 3M aqueous sodium hydroxide (6mL) added to the residue. The mixture was shaken vigorously then acidified to pH 8 with 2M aqueous hydrochloric acid and extracted with ethyl acetate. The combined organic extracts were washed with water, saturated aqueous sodium chloride, dried with sodium sulfate, filtered and evaporated. The crude material was purified by reverse phase HPLC (gradient 25 - 95% acetonitrile in 0.1% aqueous ammonium acetate) to afford the title compound as a white powder. Yield: 21mg MS: APCI(+ve) 486 [M+H+] 1B. NMR: δ (300 MHz, DMSO) -0.02 - 0.17 (m, 4H), 0.90 - 1.04 (m, 2H), 1.53 - 1.58 (m, 2H), 1.85 - 1.92 (m, IH), 2.40 - 2.48 (m, 3H), 3.21 (d, 2H), 3.48 (s, 3H), 4.10 (s, 2H), 5.63 (s, IH), 6.77 - 6.84 (m, IH), 6.95 - 7.03 (m, IH), 7.07 - 7.12 (m, IH).
Example 154
N-[2-[(2,3-Difluorobenzyl)thio]-6-(3-hydroxypropoxy)pyrimidin-4-yl]piperazine-l- sulfonamide
To a solution of tert-butyl 4-({[2-[(2,3-difluorobenzyl)thio]-6-(3-hydroxypropoxy)pyrimidin- 4-yl] amino }sulfonyl)piperazine-l-carboxy late (the product from step ϋ), O.83g) in DCM
-193-
(5ml) was added TFA (5ml) slowly. The reaction was then stirred at room temperaturefor 18h. The reaction was reduced in vacuo and the residue purified by prep HPLC to give the title compound as a white solid. Yield 160mg MS: APCI(+ve) 476 [M+H+] 1H NMR: (DMSO) 52.52 (q, 2H), 3.19 (t, 4H), 3.44 (t, 4H), 3.52 (t, 2H), 4.38 (t, 2H), 4.49 (s, 2H), 4.59 (s, IH), 6.07 (s, IH), 6.99 (s, IH), 7.14-7.24 (m, IH), 7.31-7.45 (m, 2H)
The intermediates for this compound was prepared as follows:
(i) 3-({6-Chloro-2-[(2,3-difluorobenzyl)thio]pyrimidin-4-yl}oxy)propan-l-ol
To a solution of 4,6-dichloro-2-[(2,3-difluorobenzyl)thio]pyrimidine (the product of example 1 step ϋ), 3g) and propane- 1,3-diol (l.lg) in THF (50ml) was added NaH (390mg) slowly and the reaction was then allowed to stir at room temperaturefor 18h. The reaction mixture was then partitioned between DCM (150ml) and H2O (100ml). The organics were separated and the aqueous layer was re-extracted with DCM (2 x 150ml). Organics were combined, dried (MgSO4) and reduced in vacuo to give the subtitle compound as a yellow oil. Yield 2.9g MS: APCI(+ve) 347/349 [M+H+]
ii) ϊeft-Butyl 4-({[2-[(2,3-difluorobenzyl)thio]-6-(3-hydroxypropoxy)pyriimdin-4- yl] amino }sulfonyl)piperazine- 1 -carboxylate
A mixture of 4-(aminosulfonyl)-l-piperazinecarboxylic acid-l,l-dimethylethyl ester (the product from example 15, step i), 0.4g), tris(dibenzylideneacetone)dipalladium (0) (50 mg), 2- dicyclohexylρhosρhino-2',4',6'-tri-irøpropyl-l,r-biphenyl (XPHOS) (50mg), cesium carbonate (0.43g) and 3-({6-chloro-2-[(2,3-difluorobenzyl)thio]pyrimidin-4-yl}oxy)propan-l- ol (the product from step i), 0.4g) in 1,4-dioxane (40ml) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for 3h. The reaction mixture was then reduced in vacuo and the residue separated between DCM (150ml) and H2O (150ml). The organics were separated and the aqueous layer was re-extracted with DCM (2 x 150ml). Organics were combined, dried (MgSO4) and reduced in vacuo to give the subtitle compound as a yellow solid. Yield 0.83g
MS: APCI(+ve) 576 [M+H+]
-194-
Example 155 N-{2-[(2,3-Difluorobenzyl)thio]-6-methoxypyrimidin-4-yl}piperidine-4-sulfonamide
A mixture of piperidine-4-sulfonamide (O.33g), tris(dibenzylideneacetone)dipalladium (0) (50 mg), 2-dicyclohexylphosphino-2',4',6'-tri-i.røpropyl-l,r-biphenyl (XPHOS) (50mg), cesium carbonate (0.43g) and, 4-Chloro-2-[[(2,3-difluoroρhenyl)methyl]thio]-6-methoxypyrimidine (the product from example 35 step i), 0.4g) in dioxane (20ml) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for 2h. The reaction mixture was then reduced in vacuo and the residue partitioned between DCM (100ml) and H2O (100ml). The organics were separated and the aqueous layer was re-extracted with DCM (2 x 100ml). Organics were combined, dried (MgSO4) and reduced in vacuo and the resulting yellow residue was purified prep HPLC to give the title compound as a white solid. Yield 13mg MS: APCI(+ve) 431 [M+H+] 1H NMR: (DMSO) δ 1.74-1.86 (m, 2H), 2.00-2.10 (m, 2H), 2.73-2.85 (m, 2H), 3.24-3.60 (m, 3H), 3.73 (s, 3H), 4.40 (s, 2H), 5.71 (s, IH), 7.09-7.18 (m, IH), 7.25-7.36 (m, IH), 7.42-7.50 (m, IH)
The intermediates for this compound was prepared as follows:
i) Benzyl 4-(aminosulfonyl)piperidine-l-carboxylate
To a solution of 0.88 NH3 (50ml) was added benzyl 4-(chlorosulfonyl)piperidine-l- carboxylate (4g) and the reaction stirred for 72h at RT. The reaction was then extracted with DCM (3xl50ml). Organics were recovered, dried (MgSO4) and reduced in vacuo to give the subtitle compound as a clear oil. Yield 3.3g 1H NMR: (DMSO) δl.40-1.52 (m, 2H), 1.97-2.03 (m, 2H), 2.81-2.92 (m, 2H), 3.01-3.09 (m, IH), 4.07-4.12 (m, 2H), 5.07 (s, 2H), 6.77 (s, 2H), 7.28-7.40 (m, 5H)
-195- U) Piperidine-4-sulfonamide
Benzyl 4-(aminosulfonyl)piperidine-l-carboxylate (the product from step i), 3.3g) was dissolved in MeOH (20ml). To this solution was added acetic acid (0.5ml) and a catalytic amount of Pd/C. The reaction mixture was subjected to a pressure of 5 bar under an atmosphere of hydrogen gas for 18h at RT. The reaction was filtered through celite and the filtrate was reduced in vacuo to give the subtitle compound as a white solid. Yield 1.7g 1H NMR: (DMSO) §1.46-1.57 (m, 2H), 1.91-1.98 (m, 2H), 2.48-2.57 (m, 2H), 2.85-2.93 (m, IH), 3.05-3.10 (m, 2H), 5.38 (s, 2H), 6.71 (s, IH)
Example 156
N-(2-[(2,3-Difluorobenzyl)thio]-6-{[(<rαns)-2-hydroxycyclopentyl]oxy}pyrimidin-4- yl)azetidϊne-l-sulfonamide
A mixture of azetidine-1 -sulfonamide (0.27g), tris(dibenzylideneacetone)dipalladium (0) (50 mg), 2-dicyclohexylphosphino-2',4',6'-tri-/røpropyl-l,r-biphenyl (XPHOS) (50mg), cesium carbonate (0.43g) and (tra«5)-2-{6-chloro-2-[(2,3-difluorobenzyl)thio]pyrimidin-4- yl}cyclopentanol (the product from step i), 0.5Og) in dioxane (20ml) was heated at reflux in a microwave at 1000C, 300W, open vessel with cooling for Ih. The reaction mixture was then reduced in vacuo and the residue partitioned between DCM (150ml) and H2O (150ml). The organics were separated and the aqueous layer was re-extracted with DCM (2 x 150ml). Organics were combined, dried (MgSO4) and reduced under vacuo and the resulting residue purified by prep HPLC to give the title compound as a white solid. Yield 74mg MS: APCI(+ve) 473 [M+H+]
1H NMR: (DMSQ)δ 1.61-1.84 (m, 4H), 2.02-2.18 (m, 2H), 2.26 (q, 2H), 4.01 (t, 4H), 4.11- 4.18 (m, IH), 4.38 (s, 2H), 4.98-5.03 (m, IH), 6.34 (s, IH), 6.98-7.11 (m, 2H), 7.17-7.24 (m, IH)
-196-
The intermediate for this compound was prepared as follows:
(i) (rrαns)-2-{6-Chloro-2-[(2,3-difluorobenzyl)thio]pyriniidiii-4-yl}cyclopentanol To a solution of 4,6-dichloro-2-[(2,3-difluorobenzyl)thio]pyrimidine (the product of example 1 step ϋ), 2.3g) and (trøns)-cyclopentane-l,2-diol (Ig) in THF (50ml) was added NaH (0.30g) slowly and the reaction was then allowed to stir for 18h at RT. The reaction mixture was then partitioned between DCM (150ml) and H2O (100ml). The organics were separated and the aqueous layer was re-extracted with DCM (2 x 150ml). Organics were combined, dried (MgSO4) and reduced in vacuo and the resulting clear oil was purified by column chromatography on silica gel EtOAc/iso-Hexane (2:8) to give the subtitle compound as a clear colourless oil. Yield 0.94g
MS: APCI(+ve) 373/375 [M+H+]