WO2006013369A2 - Compositions forming non-lamellar dispersions - Google Patents
Compositions forming non-lamellar dispersions Download PDFInfo
- Publication number
- WO2006013369A2 WO2006013369A2 PCT/GB2005/003056 GB2005003056W WO2006013369A2 WO 2006013369 A2 WO2006013369 A2 WO 2006013369A2 GB 2005003056 W GB2005003056 W GB 2005003056W WO 2006013369 A2 WO2006013369 A2 WO 2006013369A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- composition
- lamellar
- particles
- dispersion
- component
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/127—Liposomes
- A61K9/1274—Non-vesicle bilayer structures, e.g. liquid crystals, tubules, cubic phases, cochleates; Sponge phases
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/35—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom
- A61K31/352—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having six-membered rings with one oxygen as the only ring hetero atom condensed with carbocyclic rings, e.g. methantheline
- A61K31/353—3,4-Dihydrobenzopyrans, e.g. chroman, catechin
- A61K31/355—Tocopherols, e.g. vitamin E
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/10—Antimycotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
- A61K9/107—Emulsions ; Emulsion preconcentrates; Micelles
- A61K9/1075—Microemulsions or submicron emulsions; Preconcentrates or solids thereof; Micelles, e.g. made of phospholipids or block copolymers
Definitions
- the present invention relates to an amphiphile composition suitable for use in preparing formulations for administration to human or animal subjects.
- the present invention relates to such compositions which are capable of self- dispersion to provide dispersions of micrometer and sub-micrometer sized non- lamellar particles.
- Amphiphile-based formulations show considerable potential in the delivery of many substances, especially for in vivo delivery to the human or animal body. Because the amphiphile has both polar and apolar groups which cluster to form polar and apolar regions, it can effectively solubilise both polar and apolar compounds. In addition, many of the structures formed by amphiphiles/structuring agents in polar and/or apolar solvents have a very considerable area of polar/apolar boundary at which other amphiphilic compounds can be adsorbed and stabilised.
- phase diagrams The formation of non-lamellar regions in the amphiphile/water, amphiphile/oil and amphiphile/oil/water phase diagrams is a well known phenomenon.
- Such phases include liquid crystalline phases such as the cubic P, cubic D, cubic G and hexagonal phases, which are fluid at the molecular level but show significant long- range order, and the L3 phase which comprises a multiply interconnected bi- continuous network of bilayer sheets which are non-lamellar but lack the long-range order of the liquid crystalline phases.
- these phases may be described as normal (mean curvature towards the apolar region) or reversed (mean curvature towards the polar region).
- the structures are typically lamellar, such as mono- or multi ⁇ lamellar vesicles and liposomes and where the spontaneous curvature is higher, liquid crystalline phases or micellar phases dominate.
- the non-lamellar liquid crystalline and L3 phases are thermodynamically stable systems. That is to say, they are not simply a meta-stable state that will separate and/or reform into layers, lamellar phases or the like, but are the stable thermodynamic form of the mixture.
- the liquid crystalline order discussed above may be examined by use of small-angle X-ray diffraction (SAX), cryo-Transmission Electron Microscopy (cryo-TEM) or Nuclear Magnetic Resonance (NMR) spectroscopy studies.
- SAX small-angle X-ray diffraction
- cryo-TEM cryo-Transmission Electron Microscopy
- NMR Nuclear Magnetic Resonance
- Cryo-TEM may also be used to examine and identify other amphiphile phase structures.
- the sizes and size distributions of the dispersed particles may be examined by light scattering, particularly by use of laser light scattering instruments.
- a bulk non-lamellar phase is typically a thermodynamically stable system.
- this bulk phase may be dispersed in a polar or non -polar solvent to form particles of a non -lamellar (especially liquid crystalline) phase in a bulk solvent.
- This allows the advantages of bulk non-lamellar phases to be applied in situations where use of a bulk non-miscible phase would cause problems, such as in parenteral applications. Further control of a compound's release profile may also be achieved by such a dispersion.
- the dispersion of non-lamellar phase into particles is essential for this amphiphile phase structure to be of value in certain (particularly in vivo) applications.
- the dispersion of non- lamellar phases requires a relatively high energy input and generally requires specialised apparatus. Typical methods include ultrasonication, homogenisation and high pressure filtration. Examples of such "high energy produced" non-lamellar particles can be found in the literature (Kamo et al., Langmuir, 2003, 19, 9191-95 and Gustafsson et al., Langmuir, 1997, 13, 6964-71).
- a high energy method for the formation of dispersed particles of non -lamellar phase in solvents such as water is described in US 5,531,925.
- Such particles have a non- lamellar liquid crystalline or L3 interior phase and a lamellar or L3 surface phase and may also contain active ingredients.
- compositions and methods allowing dispersions of non-lamellar phase particles to be generated without employing high energy dispersion methods. It would be a significant advantage if the particles formed were colloidal and it would also be a considerable advantage if the resulting particles were well tolerated in vivo.
- compositions comprising monoacyl lipids, diacyl glycerols and/or tocopherols, fragmentation agents and optionally active agents have the surprising property that they form self-dispersing compositions which generate colloidal non-lamellar phase particles upon exposure to aqueous conditions without requiring the use of high energy techniques.
- the present invention thus provides a composition
- a composition comprising a) at least one monoacyl lipid; b) at least one diacyl glycerol, at least one tocopherol, or mixtures thereof and c) at least one fragmentation agent; and optionally an active agent, wherein the composition is capable of self-dispersing to provide (preferably colloidal) non-lamellar particles upon contact with an aqueous fluid.
- the invention provides a composition
- a composition comprising a) at least one monoacyl lipid; b) at least one diacyl glycerol; and c) at least one fragmentation agent; and optionally an active agent, wherein the composition is capable of self-dispersing to provide (preferably colloidal) non-lamellar particles upon contact with an aqueous fluid.
- dispersions of non-lamellar particles can be formed without the need for high energy fragmentation methods or specialised equipment. This allows formation of the dispersion at the time and place required, such as at the point of care.
- the present invention thus provides a method for the formation of a dispersion of non-lamellar particles, said method comprising contacting a composition comprising a) at least one monoacyl lipid; b) at least one diacyl glycerol, at least one tocopherol, or mixtures thereof and c) at least one fragmentation agent; with an aqueous fluid and optionally subjecting the thus-formed mixture to a low energy agitation method.
- suitable methods include manual stirring and/or manual shaking and mechanical shaking at up to 350 rpm.
- component b) is at least one diacyl glycerol.
- non-lamellar particles formed by self-dispersion of the compositions of the present invention are also of a unique composition and thus form a further aspect of the invention.
- the present invention thus provides colloidal non-lamellar particles comprising a) at least one monoacyl lipid; b) at least one diacyl glycerol, at least one tocopherol, or mixtures thereof and ' c) at least one fragmentation agent; optionally and active agent and optionally an aqueous fluid.
- component b) is at least one diacyl glycerol.
- compositions of the present invention are highly suitable for allowing the preparation of colloidal dispersions outside of a dedicated manufacturing facility, such as at the point of care. This offers advantages in case the active agent may not be stable in solution or dispersion for long periods or if there is any concern over the particle-size stability of the dispersion to storage.
- Such "prepared on demand" type dispersions are most easily supplied in the form of a kit containing the essential elements required for dispersion preparation.
- the present invention thus provides a kit for the preparation of a dispersion of non-lamellar particles, said kit comprising a composition comprising; a) at least one monoacyl lipid; b) at least one diacyl glycerol, at least one tocopherol, or mixtures thereof and c) at least one fragmentation agent; and optionally an active agent.
- component b) is at least one diacyl glycerol.
- kits may also, optionally, include items such as at least one vessel suitable for carrying out the preparation of the dispersion (e.g. a re-sealable tube of suitable capacity which can be manually shaken), at least one aqueous fluid (preferably pre- measured) suitable for use in preparing the dispersion (e.g. isotonic saline for injection) and/or instructions regarding preparation of the dispersion.
- a vessel suitable for carrying out the preparation of the dispersion e.g. a re-sealable tube of suitable capacity which can be manually shaken
- at least one aqueous fluid preferably pre- measured
- suitable for use in preparing the dispersion e.g. isotonic saline for injection
- the active agent where present, may be formulated with the amphiphile components or may be present as a separate compartment for inclusion when the dispersion is prepared.
- compositions of the invention will desirably be formulated with an active agent, as indicated herein below.
- an active agent is a drug, diagnostic agent, vaccine, prophylactic agent or similar pharmaceutical active
- the present invention provides a pharmaceutical formulation comprising a composition of the invention, at least one active agent and optionally at least one pharmaceutically acceptable carrier or excipient.
- self dispersing as used herein is indicated a composition which does not require the presence of organic solvents (hydrotropes) or high energy techniques such as homogenization, ultrasonication or vigorous mechanical shaking in order to create a colloidal dispersion.
- a composition can be considered “self dispersing” if the solvent-free composition is capable of forming a dispersion of non -lamellar particles with a monomodal size distribution with an average size no larger than 5 ⁇ m and a distribution width of no larger than 3 ⁇ m at half height by a method comprising forming a 5 wt% solution in an aqueous fluid (such as water or aqueous buffer) and shaking for up to 12 hours at up to 350 rpm.
- aqueous fluid such as water or aqueous buffer
- compositions of the present invention may contain solvents/hydrotropes, the presence of these agents is not necessary for the self-dispersion. Thus, the compositions are all capable of self dispersion in the absence of any solvent or hydrotrope even if such agents are included in the compositions for other reasons (such as to provide a convenient liquid composition).
- self-dispersing indicates self-dispersion from a bulk solid or liquid lipid mixture or solution (e.g. with up to 15%, preferably up to 10% by weight added solvent) but does not encompass self-dispersion where a lipid mixture has previously been fragmented by use of hydrotropes or a high energy technique and subsequently dried in finely divided powder form such that each particle is ready formed for rehydration.
- the compositions of the invention are "self-dispersing" in that they inherently possess the properties required to generate a colloidal dispersion, as indicated above.
- compositions which consist of, for example, coated micron sized particles are not "capable of self-dispersion” unless the particles could be created by self-dispersion, as described herein, followed by drying.
- Previously known compositions use hydrotropes and/or high energy techniques followed by drying and such compositions are thus not capable of self- dispersion.
- non-lamellar compositions are not treated with homogenization, ultra sonication or hydrotropes simply to speed up the process of dispersion but because they are inherently incapable of self-dispersion.
- previously known techniques for producing non-lamellar dispersions employ high energies or hydrotropes, they continue to produce a significant amount of lamellar (vesicular) particles (see, for example Spicer supra) and typically also result in broad and/or ill-defined size distributions (such as bi- or multi-modal distributions and/or quantities of macroscopic particles e.g. particles larger than lOO ⁇ m).
- the storage stability of previously known high-energy or hydrotrope produced non- lamellar dispersions js generally low.
- compositions do not form non-lamellar well-defined particle dispersions of colloidal-type size in the absence of high energy techniques and/or hydrotropes.
- present compositions allow the advantages of non-lamellar phase particles (in particular non-lamellar reversed hexagonal phase particles) to be accessed without the need for high energies, specialised equipment and/or co- solvents/hydrotropes.
- the compositions furthermore generate reproducible and reliable particle size distributions within the colloidal size range, which are highly stable to storage.
- non-lamellar is used to indicate a normal or reversed liquid crystal phase (such as a cubic or hexagonal phase) or the L3 phase or any combination thereof, as opposed to lamellar structures such as vesicles/liposomes.
- a particle is described as having a non-lamellar phase or form, this indicates that at least the internal region of the particle should adopt this form.
- the particles will generally have two distinct regions, an internal region and a surrounding surface region. The surface region, even in a "non-lamellar" particle will often be lamellar or crystalline and may be any phase ranging from a highly ordered crystalline or liquid crystal phase to a virtually orderless fluid layer.
- a "lamellar" particle as described herein is a particle having a solvent, rather than non -lamellar, core-region.
- non-lamellar as used herein may also refer to normal and/or reversed micellar phase structures.
- compositions of the present invention form reversed non -lamellar phase particles and more preferred that the compositions form reversed liquid crystalline phase particles such as bicontinuous cubic or reversed hexagonal liquid crystalline phase. Since the non-lamellar phase structure forms by self-dispersion in aqueous fluid, it is evident that this phase structure should be in or near equilibrium with the bulk solvent phase. This is typically represented by the presence of a multi ⁇ phase region in the phase diagram with a non-lamellar phase co-existing with a bulk solvent phase. This is also clearly reflected in the stability of the dispersions as discussed herein below.
- compositions may be chosen to provide stable reversed hexagonal liquid crystalline particles in colloidal dispersion.
- Reversed hexagonal colloidal particles are less commonly generated with known amphiphile mixtures and few stable dispersions of such particles have been demonstrated.
- the Examples below demonstrate that the present invention provides not only dispersions of hexagonal liquid crystalline particles but such dispersions which have narrow, colloidal, particles size distributions and are stable to prolonged storage.
- compositions of the present invention will self-disperse to form partially non-lamellar particles and partially lamellar and/or micellar particles but more than 50% of the amphiphile should disperse to be comprised in non- lamellar structures. It is preferred that at least 70% of the amphiphile is formed by self-dispersion of the compositions of the present invention into non-lamellar particles, more preferred that at least 75% and most preferred that at least 85% of the amphiphile self-disperses to be comprised in non-lamellar particles.
- compositions of the present invention will be self-dispersing to provide particles with a monomodal size distribution and an average particle size of no more than 5 ⁇ m. It is preferable that this average particle size be no more than 2 ⁇ m and more preferable that this be no more than 1 ⁇ m.
- the width of the particle size distribution should also be narrow, preferably being no more than 3 ⁇ m at half- height, more preferably no more than l ⁇ m and most preferably no wider than 0.5 ⁇ m at half-height.
- Such particles can be considered colloidal and are suitable for administration (as a dispersion in a suitable fluid) directly to a subject, such as by intravenous injection.
- compositions of the present invention can spontaneously disperse to provide particles in the colloidal, micron or sub- micron particle size range with no detectable particles above 8 ⁇ m and in some cases no detectable particles with sizes above l ⁇ m.
- a narrow and predictable size distribution is advantageous in all administration routes (e.g. oral, nasal, buccal etc.) to provide control over active agent transport and release.
- components for use in the compositions of the present invention include a) at least one monoacyl lipid, b) at least one diacyl glycerol, at least one tocopherol, or mixtures thereof and c) at least one fragmentation agent.
- component b) comprises or consists of at least on diacyl glycerol.
- a monoacyl lipid As component a) of the compositions of the present invention is employed a monoacyl lipid.
- Preferred species of such lipids include monoacyl oligoglycerols such as mono- or preferably di-, tri- or tetra-glycerols and pegylated glyceryl fatty acid esters, as well as pegylated fatty acids.
- the acyl/fatty acid chains will typically have 12 to 22 carbons and 0, 1, 2 or 3 unsaturations.
- Preferred acyl/fatty acid groups include, for example, lauroyl (C 12:0), myristoyl (C 14:0), palmitoyl (C 16:0), phytanoyl (C 16:0), palmitoleoyl (C 16: 1), stearoyl (C 18:0), oleoyl (C18:l), elaidoyl (C18:l), linoleoyl (C18:2), linolenoyl (C18:3), arachidonoyl (C20:4), behenoyl (C22:0) groups, where CX:Y indicates a hydrocarbon chain having X carbon atoms and Y unsaturations
- Particularly preferred specific monoacyl lipids include Diglycerolmonooleate (DGMO), Diglycerolmonolinoleate (DGML) and Polyethyleneglycol(5)-glyceryl-monooleate (TMGO-5).
- the component a) typically forms a micellar or preferably lamellar phase on contact with water. This can be tested by adding water to the material, equilibrating the sample and then determining the phase(s) present in the sample by small angle x-ray scattering (SAXS). It is preferred that the monoacyl component forms a lamellar phase upon contact with water. As an example, DGMO forms a lamellar phase that takes up a maximum of about 40 wt% water.
- component b) of the compositions of the invention is employed a diacyl glycerol, a tocopherol or mixtures thereof.
- the diacyl glycerol component are preferred as part or all of component b) and may be symmetrical or non -symmetrical diacyl lipids and each fatty acid group may be saturated or unsaturated.
- Preferred diacyl glycerols include those with acyl groups each having 12 to 22 carbons and 0, 1, 2 or 3 unsaturations.
- Preferred acyl groups include, for example, lauroyl (C 12:0), myristoyl (C 14:0), palmitoyl (C 16:0), phytanoyl (C 16:0), palmitoleoyl (C 16: 1), stearoyl (C18:0), oleoyl (C18:l), elaidoyl (C18: l), linoleoyl (C18:2), linolenoyl (C18:3), arachidonoyl (C20:4), behenoyl (C22:0) where CX:Y indicates a hydrocarbon chain having X carbon atoms and Y unsaturations.
- a particularly preferred diacyl glycerol is glycerol dioleate (GDO).
- a tocopherol is used to indicate the non-ionic lipid tocopherol, often known as vitamin E, and/or any suitable salts and/or analogues thereof.
- the most preferred of the tocopherols is tocopherol itself, having the structure below.
- contaminant there may be a small proportion of non -tocopherol "contaminant” but this will not be sufficient to alter the advantageous self dispersion properties and/or phase-behaviour of the composition.
- a tocopherol will contain no more than 10% of non- tocopherol-analogue compounds, preferably no more than 5% and most preferably no more than 2% by weight.
- the b) component typically forms reversed liquid crystalline phases, such as reversed cubic or hexagonal phases, or a liquid L2 phase on contact with water.
- the b) component may also be an (especially surface active) oil taking up virtually no water. Again this can be tested by SAXS (see above) or determined by visual inspection by someone skilled in the art. It is preferred that the diacyl glycerol and/or the tocopherol forms an oil or L2 phase upon contact with water. As an example, GDO takes up very little water and separates out as an oily material when contacted with water.
- component c) can function any amphiphile capable of serving as a fragmentation agent with the selected components a) and b).
- a fragmentation agent is a (pure or mixed) agent which allows the composition comprising components a) and b) to self-disperse to form non-lamellar particles, as indicated herein.
- fragmentation agents there are a number of different molecular classes that are suitable as fragmentation agents in the present invention. These include;
- Poloxamers preferably Pluronic® F 127, Pluronic® F68, Pluronic® F 108 Pluronic® L44
- 2-Methacryloyloxyethyl phosphorylcholine n- butyl methacrylate co-block polymers such as PUREBRIGHT MB-37-50T and PUREBRIGHT MB-37-100T from NOF Corp.
- pegylated sorbitan fatty acid esters polysorbates, particularly Polysorbate 80
- PEGylated surfactants e.g. Solutol
- HS 15 from BASF pegylated castor oil derivatives (e.g. Cremophor EL, Cremophor RH40), pegylated fatty acids (e.g. PEG-oleate), pegylated phospholipids (including DOPE-PEG(2000), DOPE-PEG(5000) and DSPE- PEG(5000)), polyglycerin(PG)- phospholipids (such as DSPE-PG, for example, SUNBRIGHT DSPE-PG8G from NOF Corp., DOPE-PG), pegylated oligoalkylsorbitols (such as PEG-60 Sorbitoltetraoleate, e.g.
- pegylated castor oil derivatives e.g. Cremophor EL, Cremophor RH40
- pegylated fatty acids e.g. PEG-oleate
- pegylated phospholipids including DOPE-PEG(2000), DOPE-P
- pegylated glyceryl fatty acid esters e.g. TMGO-15 (Nikko Chemicals)
- pegylated tocopherols such as d- alpha tocopheryl polyethylene glycol 1000 succinate (Vitamin E TPGS (Eastman)) and pegylated alkyl ethers;
- sugar derived alkyl esters such as sucrose laurate and sucrose oleate
- sugar derived alkyl ethers e.g. octyl glucoside
- Proteins including casein, sodium caseinate, lysozyme;
- Anionic surfactants Carboxylates of fatty acids (especially sodium oleate, sodium palmitate, sodium stearate, sodium myristate), alkyl sulfates (such as sodium dodecyl sulphate (SDS)); and
- Cationic surfactants alkyl ammonium salts (including dodecyl trimethyl ammonium bromide (DTAB), cetyl trimethyl ammonium bromide (CTAB) and oleyl ammonium chloride).
- DTAB dodecyl trimethyl ammonium bromide
- CTAB cetyl trimethyl ammonium bromide
- oleyl ammonium chloride alkyl ammonium salts (including dodecyl trimethyl ammonium bromide (DTAB), cetyl trimethyl ammonium bromide (CTAB) and oleyl ammonium chloride).
- micellar (Ll) phases on contact with excess water.
- the components need not form micelles to function as fragmentation agents.
- the effective functioning of a fragmentation agent will easily be tested by a skilled worker by preparing appropriate compositions and conducting simple tests as illustrated in the Examples herein.
- component c) may be a monoacyl (especially non-naturally occurring) lipid. This will most commonly be where component c) is a polymeric mono-acyl lipid falling within category 1) above.
- component c) may be fully or partially made up from one of more of the mono-acyl lipid constituents of component a).
- the only essential feature of this embodiment is that there should be sufficient fragmentation effect to provide for effective self-dispersion and/or stabilisation as described herein. Where this fragmentation effect can be provided by one or more mono-acyl lipid constituents of component a), which will typically occur where component a) comprises at least one non-naturally occurring monoacyl lipid, then this component will also serve as fragmentation agent c). Where some but insufficient fragmentation effect is provided by component a) then the content of additional fragmentation agent contributing to component c) will be correspondingly decreased.
- the components a), b) and c) will be present in the following proportions (where a, b and c are the weights of components a), b) and c) respectively); a/(a+b) is between 0.2 (e.g. 0.3) and 0.9 (e.g. 0.8)and c/(a+b+c) is between 0.01 and 0.3 (or corresponding appropriately to all or part of component a) where component a) includes a fragmentation agent). Compositions within this range have a high tendency to self-disperse under relatively mild conditions, without requiring high energy input.
- a/(a+b) is between 0.25 (e.g. 0.35)and 0.80 (e.g. 0.75), more preferably 0.35 (e.g. 0.4) and 0.75 (e.g. 0.65) and c/(a+b+c) is between 0.03 and 0.25 (e.g. 0.2) (where a, b and c are the weights of components a), b) and c) respectively).
- compositions of the present invention may also comprise an active agent and/or other amphiphilic components.
- charged (especially anionic) lipids/fatty acids may be included so that higher loading levels of active agent may be obtained (such as for a cationic peptide e.g. octreotide).
- additional components are charged lipids or surfactants (e.g. Dioleoyl phosphatidylglycerol (DOPG), oleic acid (OA), Dioleoyltrimethyl ammonium propane (DOTAP)) and polymeric surface modifiers.
- DOPG Dioleoyl phosphatidylglycerol
- OA oleic acid
- DOTAP Dioleoyltrimethyl ammonium propane
- Preferred polymeric surface modifiers include polyethylene oxide copolymers and lipids derivatised with polyethylene oxide, polysaccharides (such a chitosan), hydrophobically modified polysaccharides and amphiphilic proteins.
- Poloxamers are particularly suitable as the polymeric components as are PEG-substituted lipids such as PEG-glyceroldioleate, PEG-dioleoyl phosphatidyl ethanolamine (in particular DOPE-PEG2000 and DOPE PEG-5000) or PEG-dioleoyl phosphatidyl serine.
- Suitable polymeric agents also include PEG-sorbitol tetraolete (Nikko), cholesterol pullulan (NOF) and 2-Methacryloyloxyethyl phosphorylcholine n-butyl methacrylate co-block polymers (PUREBRIGHT MB-37-50T and PUREB RI GHT MB-37-100T from NOF).
- compositions of the invention may be solid, for example powder, compositions or may be liquid precursor compositions, of pure amphiphile and active agent (plus optional excipients) without the need for any solvent or hydrotrope.
- present compositions are provided as solvent free formulations either for dispersion prior to administration or for direct administration in solvent- free form. This has advantages in convenience and ease of administration as well as avoiding unnecessary administration of organic solvent
- liquid compositions of the invention may be prepared as solvent mixtures.
- Such liquid precursors will comprise components a, b, c, a cosolvent and optionally an active agent.
- the liquid precursors containing an active agent can, for example, be filled in capsules and because of the self-dispersing ability of the composition, non-lamellar particles form when contacted with GI- fluid.
- a liquid precursor may be provided in an ampoule for dispersion in a fluid (e.g. isotonic saline) prior to injection.
- Co-solvents should generally be miscible, to at least some extent with water and should be tolerable in the application in which the composition will be used (e.g. biotolerable).
- Organic solvents having 1 to 6 carbon atoms and preferably at least one oxygen substituent and water-soluble polymers thereof are preferred.
- Suitable classes of cosolvents are alcohols (including polyols), ketones, esters, ethers, and polymers thereof.
- Typical co-solvents are ethanol, N-methyl-2 -pyrrolidone (NMP), propylene glycol, dimethylacetamide (DMA), glukofurol, transcutol, PEG400 and glycerol added up to about 15%, preferably up to about 10% (by weight) of total lipid.
- the invention provides solid or semi-solid (e.g. gel, waxy solid) compositions which may be prepared by use of a polymeric agent in the compositions of the invention.
- solid or semi-solid precursors will comprise compositions of the invention as described herein and additionally at least one polymeric solidifying agent.
- compsitions comprise components a, b, c, a polymeric agent, optionally a cosolvent and optionally an active agent.
- the solid or semi-solid precursors are typically liquefiable by heat and can, for example, be filled in capsules, moulded etc. Because of the self-dispersing ability of the compositions, non-lamellar particles form when contacted with GI- fluid.
- the polymeric solidifying agent is a preferably biotollerable polymer, preferably having a melting point between 35 and 100 0 C, more preferably 40-95 0 C and most preferably 45-90 0 C.
- a particularly preferred polymeric agent is polyethylene glycol (PEG) with molar mass in the range of 950-35000, most preferably 1000 to 10,000.
- PEG 4000 is a highly preferred example..
- compositions of the present invention are self-dispersing, they do not need to be administered in the form of a dispersion and indeed do not need to have been previously dispersed before administration.
- the compositions may conveniently be administered as a bulk composition in solid or concentrated liquid form (e.g. as a powder, tablet, powder, solid (semi- solid) or liquid filled capsule), rather than as a dispersion, while maintaining the high transport efficiency associated with colloidal dispersions, which are then generated by dispersion in vivo in the body fluid.
- the bulk administration will thus fragment and disperse as a highly uniform distribution of micron and sub-micron particles which are efficiently transported to sites of action.
- the non-lamellar structure of the particles generated by this in vivo dispersion can provide control over active agent release and efficient targeting and delivery across biological membranes.
- the compositions of the present invention are thus formulated to comprise at least 50% by weight of a pharmaceutical formulation with no more than 10% by weight being total organic solvent content (including any solvent present in the composition) and the remainder non-solvent formulating agents.
- Active agents suitable for inclusion in the compositions of the present include human and veterinary drugs and vaccines, diagnostic agents, "alternative" active agents such as plant essential oils, extracts or aromas, cosmetic agents, nutrients, dietary supplements etc.
- suitable drugs include antibacterial agents such ⁇ -lactams or macrocyclic peptide antibiotics, anti fungal agents such as polyene macrolides (e.g. amphotericin B) or azole antifungals, anticancer and/or anti viral drugs such as nucleoside analogues, paclitaxel, and derivatives thereof, anti inflammatorys, such as non-steroidal anti inflammatory drugs, cardiovascular drugs including cholesterol lowering and blood-pressure lowing agents, analgesics, antidepressants including serotonin uptake inhibitors, vaccines and bone modulators.
- antibacterial agents such as ⁇ -lactams or macrocyclic peptide antibiotics
- anti fungal agents such as polyene macrolides (e.g. amphotericin B) or azole antifungals
- anticancer and/or anti viral drugs such as nucleoside analogues, paclitaxel, and derivatives thereof
- anti inflammatorys such as non-steroidal anti inflammatory drugs
- cardiovascular drugs including cholesterol
- anticancer agents such as paclitaxel and docetaxel
- immunosuppressants such as cyclosporine, tacrolimus, or sirolimus and peptide active agents such somatostatin and analogues thereof (e.g. octreotide).
- Diagnostic agents include radionuclide labelled compounds and contrast agents including X-ray, ultrasound and MRI contrast enhancing agents.
- Nutrients include vitamins, coenzymes, dietary supplements etc.
- the active agents for use in the present invention will generally not be any of components a), b) or c) as described herein.
- compositions and particles of the invention thus include at least one temperature sensitive and/or shear sensitive active agent.
- Temperature sensitive active agents may be considered to be those in which exposure to temperatures of 70°C or higher for 20 minutes or more, in aqueous conditions, result in the loss of at least 10% of the original biological activity.
- Peptides and proteins are the most common active agents which are temperature sensitive and thus these form preferred active agents for use in the invention, particularly in this embodiment.
- Shear sensitive active agents will typically be large and/or multi- subunit proteins which become disrupted by high shear conditions.
- Preferred active agents thus include human and veterinary drugs selected from the group consisting of peptides such as adrenocorticotropic hormone (ACTH) and its fragments, angiotensin and its related peptides, antibodies and their fragments, antigens and their fragments, atrial natriuretic peptides, bioadhesive peptides, Bradykinins and their related peptides, peptide T and its related peptides calcitonins and their related peptides, cell surface receptor protein fragments, chemotactic peptides, cyclosporins, cytokines, Dynorphins and their related peptides, endorphins and P-lidotropin fragments, enkephalin and their related proteins, enzyme inhibitors, fibronectin fragments and their related peptides, gastrointestinal peptides, growth hormone releasing peptides, immunostimulating peptides, insulins, insulin analogues and insulin-like growth factors,
- octreotide substance P and its related peptides, transforming growth factors (TGF) and their related peptides, tumour necrosis factor fragments, toxins and toxoids and functional peptides such as anticancer peptides including angiostatins, antihypertension peptides, anti-blood clotting peptides, and antimicrobial peptides; selected from the group consisting of proteins such as immunoglobulins, angiogenins, bone morphogenic proteins, chemokines, colony stimulating factors (CSF), cytokines, growth factors, interferons, interleukins, leptins, leukemia inhibitory factors, stem cell factors, transforming growth factors and tumor necrosis factors; selected from the group consisting of antivirals, steroidal anti -inflammatory drugs (SAID), non-steroidal anti-inflammatory drugs (NSAED), antibiotic, antifungals, antivirals, vitamins, hormones, retinoic acid and retinoic acid derivatives (including tretinoi
- tacrolimus and sirolimus and immunostimulants
- cardiovascular drugs including lipid lowering agents and blood-pressure lowering agents, bone modulators; vaccines, vaccine adjuvants, immunoglobulins and antisera; diagnostic agents; cosmetic agents, sunscreens and self-tanning agents; nutrients; dietary supplements; herbicides, pesticides, and repellents.
- active agents can be found for instance in Martindale, The Extra Pharmacopoeia.
- the aqueous fluid referred to herein for contacting with the compositions of the present invention may be water or may be any other suitable aqueous solution or mixture including, for example, pharmaceutically acceptable carrier solutions.
- suitable solutions include buffers and isotonic solutions for injection e.g. 0.9% saline at around physiological pH. These fluids are highly suitable for preparing a dispersion at the point of care and may be included in the kit of the invention.
- the fluid may be a body fluid such as blood or gastro ⁇ intestinal (GI) fluid.
- GI gastro ⁇ intestinal
- the composition is administered as a lipid/active agent mixture, optionally with a cosolvent or a polymeric agent to render this liquid or solid (semi-solid) or improve the viscosity properties. Suitable co- solvents are discussed herein above.
- the particles of the invention are essentially stable both in terms of phase behaviour and particle size distribution to storage for at least 10 days at 4°C and/or at room temperature.
- This is a considerable advantage over previously known dispersions of non-lamellar particles (requiring high energy fragmentation or hydrotropes) since these known dispersions are typically not stable to storage for more than a short period (e.g. a few days - see for example Kamo, supra).
- the amphiphile particles of the invention are stable to storage for at least 1 or 2 months, preferably at least 3 months and more preferably at least 6 months at both 4°C and room temperature.
- the particles and dispersions of the invention are stable to storage at 4°C because this is the typical refrigerated storage condition practiced and recommended for many biologically active agents and preparations.
- Previously known non-lamellar (especially reversed hexagonal) particles are even less stable at 4°C than they are at room temperature and are thus unsuitable for generating formulations for refrigerated storage.
- previously known glycerol monooleate (GMO) based non-lamellar dispersions are generally unstable at 4°C.
- a particle size distribution can be considered essentially stable to storage if the average (mean) particle size increases no more than two fold during the storage period.
- the average size should increase no more than 50% and more preferably no more than 20% during the storage period.
- the width of the distribution at half-height should preferably increase by no more than 50%, more preferably by no more than 20% and most preferably no more than 10% during the storage period. Where a distribution is monomodal, it should preferably remain monomodal during the storage period.
- particle size distribution of the compositions of the invention alter in average particle size and particle size distribution width at half-height by no more than 10% and remain monomodal on storage for the periods indicated above.
- colloidal dispersions for use in intravenous or intra-arterial administration that the particle size distribution be stable to storage.
- a composition containing even a relatively small component of non-colloidal particles may cause embolism, or at least unpredictable rates of release upon administration directly to the blood stream.
- the controlled release of an active agent may be dependent upon a reliable particle size distribution in a composition for administration by any other route.
- Pharmaceutical, diagnostic and veterinary products are also desirably stable to storage for several months or the cost and availability of the product is significantly adversely affected. The invention thus significantly improves the prospect of an active agent formulated in a dispersion of non-lamellar particles forming a safe and available product.
- the particles of the invention remain non-lamellar upon storage for the periods discussed above.
- “remains non-lamellar” is indicated that no more than 10% of the non -lamellar particles should adopt a lamellar or micellar phase structure upon storage, preferably no more than 5% and more preferably no more than 2%. In some cases the proportion of non-lamellar particles may even increase upon storage.
- the dispersions formed or formable from the compositions of the present invention are further remarkable in that they can both form and stably remain as dispersions in aqueous fluids at surprisingly high lipid concentrations.
- non-lamellar lipid dispersions are formed and remain stable, if at all, at very low total amphiphile concentrations.
- the maximum typical concentration is frequently 1-2% by weight amphiphile in water with 5-6% being an unusually high concentration.
- the dispersions of and formed by the present invention may be stable in aqueous fluids at concentrations of up to 10 wt%, preferably up to 15 wt% and more preferably up to 20% total amphiphile in water.
- stable is meant stable in both particle size and phase behaviour, as discussed herein.
- the monoacyl lipid a) may comprise or consist of components which, in pure form, generate a micellar or preferably lamellar phase upon contact with water.
- the most commonly used monoacyl lipid for the formation of bulk or dispersed non-lamellar phases is glycerol monooleate (GMO).
- GMO glycerol monooleate
- This monoacyl lipid may be used in the compositions of the present invention but is not suitable for this embodiment because it forms a cubic liquid crystalline phase when the pure compound is exposed to water.
- amphiphile particles of the present invention are non-lamellar and are formed or formable by self-dispersion of the compositions of the invention. Following formation of such particles, however, the dispersion may be further treated in a number of ways, depending upon desired application.
- the particles formed or formable from the composition of the invention may be concentrated and/or dried and/or co -melted with suitable polymeric agents to provide a concentrated dispersion, a "dry" powder or a solid (semi-solid e.g. gel or waxy solid) matrix.
- Suitable techniques for concentration, drying and preparation of a solid (semi-solid) include ultrafiltration, solvent evaporation, freeze drying, spray drying and co-melting of the amphiphile components with a polymeric agent (e.g. polyethylene glycol (PEG)) or other suitable agent followed by cooling to form a solid (semi-solid) precursor.
- a polymeric agent e.g. polyethylene glycol (PEG)
- PEG polyethylene glycol
- dry powders are generated, these may be completely or essentially free of aqueous solvent or may continue to contain some solvent as part of the structured core of the particles., Where all or most of the aqueous solvent is removed, the resultant particles may lose their non-lamellar structure but this will be regenerated upon contact with an aqueous fluid.
- Such powders are capable of regenerating amphiphile particles of the invention and thus form a further aspect thereof. Drying may preferably be conducted in the presence of at least one protective agent and/or at least one agent for aiding resuspension of the resulting powder.
- Suitable agents are well known and include sugars and hydrophilic polymers such as polyvinyl pyrollidone or polyethylene glycol.
- compositions of the present invention are in themselves compositions of the invention because they comprise amphiphile mixtures that are "capable of self-dispersion".
- a micronised dried powder may not be required to self-disperse because this dispersion may already be carried out prior to or during the drying process.
- the particles may be present individually, for example within a matrix of a substance such as trehalose.
- the amphiphile mixtures forming the particles are, however, inherently capable of dispersing with out high energy fragmentation and without the need for hydrotropes and so are compositions of the invention.
- powder compositions which must be generated by use of high energies and/or hydrotopes and comprise amphiphile compositions which are incapable of self dispersion.
- the powder precursors generated from the compositions of the present invention are highly suitable for nasal administration by inhalation of the powder.
- Such a powder may also optionally be mixed with carrier or excipient powders as necessary.
- compositions e.g. solid and/or semi-solid or liquid compositions with or without a cosolvent
- dispersions e.g. in sterile containers ready for administration
- concentrated dispersions for dilution before use
- powders for suspension or direct administration e.g. by inhalation
- powder, solid (semi-solid) or liquid filled capsules tablets, coated tablets, suppositories, gels, creams, ointments and other topical compositions such as eye drops, sprays (such as skin, mouth or nasal sprays e.g.
- compositions of the invention are also suitable as carriers for non- pharmaceutical agents such as essential oils, perfumes, aromas etc.
- Suitable formulations and applications for these are also well known and include cosmetic and household applications including skin treatments (either alone or when formulated with at least one cosmetic active agent, perfume etc.), personal cleaners/cleansers such as skin, nail, face, or mouth cleansers, absorbers for internal or external toxins such as balms, and detoxifying suspensions, household or personal washing powders/liquids, bath/shower gels, cleaning liquids, sprays, gels or foams and bath oils.
- a further preferred processing step which may be carried out on the amphiphile particles formed or fprmable by self-dispersion of the compositions of the invention is a heat treatment step.
- a dispersion of amphiphile particles is heated to a temperature in the range of around 75 to 200°C, preferably 90 to 140°C for between 1 minute and 4 hours, generally between 10 minutes and 1 hour and subsequently cooled to room temperature.
- the effects of this heat treatment step are several but they include the conversion of an even greater proportion of particles to non- lamellar phase and/or the narrowing of the particle size distribution.
- the heat treatment may also improve the storage stability of the particles in dispersion, both in terms of their phase behaviour and their particle size distribution.
- the heat treatment step described above may also be used to enhance the loading of active agents into self dispersed particles of the invention.
- the active agent should be heat tolerant and is dissolved in the aqueous medium in which the particles are dispersed.
- the dispersion is then heat treated as described above and the active agent is thereby incorporated into the particles.
- These particles are highly stable and may thereafter be processed by any suitable method, including those described herein, into any appropriate formulation.
- Active agents which are suitable for any embodiment of the invention but are particularly suited for loading by heat treatment include steroids, sparingly soluble weakly basic drugs, fibrins, statins, dipins, and azoles. Specific preferred examples of these include progesterone, testosterone, simvastatin, lovastatin, nifedipin, felodipin, nicardipin, nimodipin, itraconazole, fluconazole, miconazole, econazole, voriconazole, clotrimazole, ketoconazole, fulvestrant, fenofibrate, octreotide, undecanoate, estradiol, cortisone, hydrocortisone, l la-hydroxyprogesterone, clofibrate gemfibrozil, bezafibrate, ciprofibrate.
- amphiphile based particles of the invention may desirably also be modified with surface active agent (especially a polymer) e.g. a starch or starch derivative, a copolymer containing alkylene oxide residues (such as ethylene oxide/propylene oxide block copolymers), cellulose derivatives (e.g. hydroxypropylmethylcellulose, hydroxyethylcellulose, ethylhydroxyethylcellulose, carboxymethylcellulose, etc) or graft hydrophobically modified derivatives thereof, acacia gum, hydrophobically modified polyacrylic acids or polyacrylates, etc.
- surface active agent especially a polymer
- a polymer e.g. a starch or starch derivative, a copolymer containing alkylene oxide residues (such as ethylene oxide/propylene oxide block copolymers), cellulose derivatives (e.g. hydroxypropylmethylcellulose, hydroxyethylcellulose, ethylhydroxyethylcellulose, carboxymethyl
- the surface active polymer may also be used to provide a functional effect on the surface of the particles, for example, in order to selectively bind or target the particles to their desired site of action.
- polymers such as polyacrylic acids, hyaluronic acids or chitosans may be used to provide mucus adhesive particles. Such particles will thus tend to remain localised, thus increasing the spatial control over the active agent release.
- Compositions of the invention comprising such surface modified particles form a further embodiment of the invention.
- the present invention thus also provides a method for increasing the bioavailability of an orally administrable active agent and/or increasing the effectiveness of an active agent having a site of action in the brain.
- This method comprises formulating the appropriate active agent(s) in compositions, dispersions and/or particles of the present invention and thereafter administering these to the subject.
- the present invention can provide compositions with enhanced blood/brain barrier crossing properties (see Example 17) and increased oral bioavailability of at least 5 times, preferably at least 10 times the oral bioavailability of the active agent in saline solution (see Example20).
- it can provide increased bioavilibility of (especially sparingly soluble) active substances even when compared to (commercially available) reference products (see Example 18).
- Such increased bioavailability over existing commercial preparations offers considerable advantages.
- active agents including those discussed herein are administrable orally and/or could be administered orally by means of the present invention.
- active agents having a site of action in the brain include anti-infective agents for treating brain infections (e.g. anti-fungal and/or anti-bacterial antibiotics) and drugs acting directly on the nervous system including analgesics (especially opioid / narcotic analgesics), anaesthetics, mood controlling agents such as antidepressants, and treatments for brain disorders such as Parkinson's disease (e.g. dopamine analogues), Creutzfeldt-Jakob Disease, Alzheimer's disease and cancers to the brain (e.g. anti ⁇ cancer agents such as taxol derivatives).
- analgesics especially opioid / narcotic analgesics
- anaesthetics especially opioid / narcotic analgesics
- mood controlling agents such as antidepressants
- treatments for brain disorders such as Parkinson's disease (e.g. dopamine analogues
- compositions of the present invention are highly effective in the delivery of active agents, especially pharmaceutical agents such as drugs, and diagnostic agents.
- the invention thus provides methods to solubilize, encapsulate, protect and/or stabilize at least one active agent, said method comprising formulating the active agent as a composition as described herein. All of these methods provide improvements in their respective parameter(s) relative to the same active agent prepared as a formulation in the absence of the compositions of the present invention. Typically this comparative formulation will be the standard pharmaceutical formulation for that active.
- compositions of the present invention are also highly effective in delivering active agents to subjects in vivo.
- the compositions may serve to enhance the effect of an active agent by ensuring that a greater proportion of the administered dose takes effect at the site of action relative to other formulations.
- the invention thus provides a method to increase the uptake, permeation, transport, circulation time,duration of action, efficacy, therapeutic index, bioavailability, patient convenience and/or patient complience, for a pharmaceutically active agent, said method comprising administering said active agent as a composition or formulation of the present invention, as described herein.
- Such methods will generally allow a reduced dose of active agent to be used, or allow a particular dose to be administered with a lower frequency or higher efficacy.
- compositions of the invention may continue to have advantages.
- the invention thus also provides methods to provide a more therapeutic pharmacokinetic profile, decreased level of excipients, and/or improved safety profile for a pharmaceutically active agent, said methods comprising formulating and/or administering said active agent as a formulation as herein described.
- Figure 1 shows the phase diagram of the ternary mixture DGMO/GDO/water at
- Figure 2 shows a cryo-transmission electron micrograph of a self-dispersed sample of DGMO/GDO/Pluronic® F 127.
- Figure 3 shows the particle size distributions of a self-dispersed DGMO/GDO/Pluronic® F 127 sample before and after heat treatment.
- Figure 4 shows the particle size distributions of a self-dispersed DGMO/GDO/Polysorbate 80 sample.
- Figure 5 shows the particle size distributions of a self-dispersed DGMO/GDO/Polysorbate 80 sample before and after heat treatment.
- Figure 6 shows cryo-transmission electron micrographs of a self-dispersed sample of DGMO/GDO/Polysorbate 80 after heat treatment.
- Figure 7 shows the particle size distributions of a self-dispersed DGMO/GDO/OA/Pluronic® F 127 sample before and after heat treatment.
- Figure 8 shows cryo-transmission electron micrographs of a self-dispersed sample of DGMO/GDO/OA/Pluronic® F 127 before and after heat treatment.
- Figure 9 shows the particle size distribution of a self-dispersed DGMO/GDO/DOPG/Pluronic® F 127 sample.
- Figure 10 shows the particle size distributions of a self-dispersed DGML/GDO/Pluronic® F 127 sample before and after heat treatment.
- Figure 1 1 shows the particle size distributions of a self-dispersed DGMO/GDO/GO- 460V sample before and after heat treatment.
- Figure 12 shows the particle size distributions of a concentrated (20 wt%) self- dispersed DGMO/GDO/Polysorbate 80 sample before and after filtration.
- Figure 13 shows the particle size distributions of a self-dispersed DGMO/GDO/OA/Pluronic® F 127 sample after storage for 2 months at 4°C and
- Figure 14 shows the particle size distributions of a self-dispersed DGMO/GDO/Polysorbate 80 sample after storage for 6 months at 25 0 C.
- Figure 15 shows the particle size distribution of a non-self-dispersing GMO/Pluronic® F 127 sample.
- Figure 16 shows the particle size distributions of self-dispersed DGMO/GDO/Polysorbate 80 samples with varying Propofol loading.
- Figure 17 shows cryo-transmission electron micrographs of a self-dispersed DGMO/GDO/Polysorbate 80 sample (100 mg amphiphile/mL) containing 20 mg Propofol/mL.
- Figure 18 shows plasma concentration over time for Propofol administered in a non- lamellar nanoparticle dispersion or as the commercially available Propofol Fresenius Kabi product.
- Figure 19 shows the plasma testosterone concentration over time for the reference Undestor Testocaps and the non-lamellar nanoparticle testosterone undecanoate, respectively.
- phase behavior of the ternary system DGMO/GDO/water was determined using small angle x-ray scattering (SAXS) combined with observations between crossed polarizers. Samples were prepared by mixing the lipid components in the correct proportions into small glass vials and thereafter adding water (typical sample weights were 1 g). The vials were immediately sealed and the samples were equilibrated by repeated centrifugation and thereafter stored for at least 2 weeks before the SAXS measurements. The results are collected in the phase diagram shown in Figure 1.
- This figure displays 3 non-lamellar liquid crystalline (Ic) phase regions: The reversed hexagonal phase (Hn) and the two bicontinuous cubic phases Q 224 and Q 230 .
- a further non-lamellar phase was identified as the liquid reversed micellar L 2 phase.
- L ⁇ lamellar phase
- the Hn phase exists at weight ratios of DGM0/GD0 between approximately 65/35 and 40/60 and at water contents equal to or greater than 5 wt%.
- the non-lamellar Hi 1 phase and the non-lamellar cubic Q 224 phase coexist with a dilute water phase in the water corner of the phase diagram. This behavior is commonly necessary for the formation of dispersions of non -lamellar Ic phase particles.
- a dispersion of non-lamellar (> 70% by weight of amphiphile) and lamellar ( ⁇ 30% by weight of amphiphile) particles was formed by mixing 0.60 g of DGMO and 0.40 g of GDO. The components were molecularly mixed by heating for 5 min at 70°C and vortexing. The homogenous lipid melt (0.80 g) was added drop wise to a solution containing 0.08 g of Pluronic® F127 (BASF, U.S. A) in 39.2 g of deionized water. The resulting coarse dispersion was put on a shaking table (350 rpm) and shaken for 12 hours to give a white homogenous dispersion.
- Pluronic® F127 BASF, U.S. A
- the particle size was measured using laser diffraction (Coulter LS230). The size distribution was found to be narrow and monomodal.
- a cryo-TEM image of the dispersion is shown in Figure 2 and particles with dense (dark) inner structures of reversed liquid crystalline phase can be observed together with some lamellar particles (vesicles).
- Example 2.1 A sample of the dispersion generated in Example 2.1 (25 mL) was autoclaved (125°C, 20 min) and cooled to room temperature. The particle size distribution was narrowed and when examined by cryo-TEM, a still greater proportion of the particles showed non-lamellar character (internal reversed hexagonal phase). The particle size distribution before and after heat treatment is shown in Figure 3.
- the particle size was measured using laser diffraction (Coulter LS230). The size distribution was found to be narrow and monomodal as indicated in Figure 4.
- *non-lamellar particles with disordered inner structure consisting of multiply connected bilayers (> 90% by weight of amphiphile).
- a dispersion consisting of DGMO (2.125 g), GDO (2.125 g) and P80 (0.75 g) in 95.0 g of deionized water was prepared according to the methods of Examples 2.1 and 2.2.
- the size distributions obtained before and after heat treatment were both narrow and monomodal as indicated in Figure 5.
- the heat treated sample was investigated using cryo-TEM. Cryo-TEM images are shown in Figures 6a and 6b and clearly evidence the formation of non -lamellar nanoparticles of uniform size containing a disordered inner structure of multiply connected bilayers.
- non-lamellar particles with disordered inner structure consisting of multiply comiected bilayers (> 90% by weight of amphiphile).
- This particular composition is also well suited for preparing a liquid precursor of the non-lamellar phase dispersion.
- the same components were used in the same ratios.
- the components were molecularly mixed by heating to 40 0 C for 15 min and vortexing.
- the liquid formulation was then dispersed into water (5 wt% amphiphile) with gentle shaking resulting in a milky white dispersion of non-lamellar phase particles.
- the liquid precursor formulation was also fortified with 10% by weight of a co-solvent (e.g.
- Example 5 Further composition: Including anionic component (fatty acid)
- a dispersion consisting of DGMO (2.98 g), GDO (2.0 g), Oleic Acid (OA; Apoteket, Sweden) (0.13 g) and Pluronic® F127 (0.553 g) in 100.0 g of deionized water was prepared according to the methods of Examples 2.1 and 2.2.
- the size distributions obtained before and after heat treatment were both monomodal but the heat treated sample displayed a narrower distribution as indicated in Figure 7.
- the heat treatment was also accompanied by an increased proportion of particles with non-lamellar character as was evidenced by cryo-TEM. Cryo-TEM images obtained from the sample before and after heat treatment are shown in Figure 8.
- the reversed hexagonal structure is clearly observed within the particles in the cryo-TEM images and Fast Fourier Transforms (FFTs) of the internal structure indicate a hexagonal spacing of about 58 A ( ⁇ 5A) [5.8nm ( ⁇ 0.5nm)].
- FFTs Fast Fourier Transforms
- rev. hex./lam mixed reversed hexagonal (> 70% by weight of amphiphile) and lamellar ( ⁇ 30% by weight of amphiphile) particles
- Example 6 Further composition: Including anionic phospholipids
- a dispersion consisting of DGMO (0.150 g), GDO (0.100 g), DOPG (Dioleoyl Phosphatidylglycerol; Avanti Polar Lipids, U.S. A) (0.007 g) and Pluronic® F127 (0.0282 g) in 4.75 g of deionized water was prepared according to the method of Example 2.1.
- the size distribution obtained after shaking was narrow and mono- modal as indicated in Figure 9.
- a dispersion consisting of DGML (Diglycerol Monolinoleate; EMULSIFIER TS- PH 038; DANISCO, Denmark) (1.50 g), GDO (1.00 g) and Pluronic® F127 (0.277 g) in 47.5 g of deionized water was prepared according to the methods of Examples 2.1 and 2.2.
- the size distributions obtained before and after heat treatment were both mono-modal but the heat treated sample contained larger particles and displayed a narrower distribution as indicated in Figure 10.
- the heat treatment was also accompanied by an increased proportion of particles with non-lamellar character as was evidenced by cryo-TEM.
- a dispersion consisting of DGMO (1.50 g), GDO (1.0 g) and GO-460V (PEG-60- sorbitol tetraoleate; Nikko Chemicals, Japan) (0.361 g) in 47.5 g of deionized water was prepared according to the methods of Examples 2.1 and 2.2.
- the size distributions obtained before and after heat treatment were both mono-modal but the heat treated sample displayed a narrower distribution as indicated in Figure 1 1.
- This particular composition is also well suited for preparing a liquid precursor of the reversed hexagonal phase dispersion.
- a co-solvent e.g. ethanol, N-m ethyl -2- pyrrolidone (NMP), propylene glycol, PEG400, glycerol.
- the liquid formulation was then dispersed into water (5 wt% amphiphile) with gentle shaking resulting in a milky white dispersion of mainly reversed hexagonal phase particles.
- a dispersion consisting of DGMO (2.55 g), GDO (2.55 g) and P80 (0.9 g) in 24.0 g of deionized water was prepared according to the method of Example 2.1.
- the obtained homogenous milky white dispersion was filtered through a 0.2 ⁇ m filter.
- the size distributions obtained before and after filtration were both narrow and monomodal as indicated in Figure 12.
- the concentrated non-lamellar phase dispersion was found to be stable to storage at room temperature for at least 2 months.
- non-lam. non-lamellar particles with disordered inner structure consisting of multiply connected bilayers (> 90% by weight of amphiphile).
- a dispersion consisting of DGMO (2.98 g), GDO (2.0 g), Oleic Acid (OA) (0.13 g) and Pluronic® F127 (0.553 g) in 100.0 g of deionized water was prepared according to the method of Example 2.1.
- the dispersion was divided into two batches and stored at 25°C and 4 0 C.
- the particle size distribution was measured at regular intervals and found to be consistent with the original size distribution for at least 2 months storage, at 4°C and 25°C, indicating excellent colloidal and storage stability.
- rev. hexVlam mixed reversed hexagonal (> 70% by weight of amphiphile) and lamellar ( ⁇ 30% by weight of amphiphile) particles
- a dispersion consisting of DGMO (0.934 g), GDO (0.764 g) and P80 (0.302 g) in 38.0 g of deionized water was prepared according to the method of Example 2.1.
- the particle size distribution was measured at regular intervals and found to be consistent with the original size distribution for at least 6 months storage at 25 0 C, as shown in Figure 14.
- *non-lamellar particles with disordered inner structure consisting of multiply connected bilayers (> 90% by weight of amphiphile).
- Example 2-10 display non-lamellar reversed phase particles formed by shaking at low speed during 12 h.
- the resulting dispersions display monomodal, narrow size distributions with the average size being in the submicron range and with a majority of the particles being non-lamellar as evidenced by cryo-TEM.
- the dispersions are produced with a minimum of shear/energy input.
- Example 2 - 10 a dispersion of GMO (RyloTM MG Glycerol Monooleate; DANISCO, Denmark) and Pluronic® F 127 (lacking diacyl glycerol component "b") was prepared according to the method of Example 2.1 (12 h shaking at 350 rpm).
- the ratio of GMO to Pluronic® F 127 was 9/1 wt/wt and the total amphiphile concentration was 5 wt%.
- the resulting coarse dispersion (non-lamellar cubic phase dispersion) was milky white and contained some poorly dispersed material in the form of macroscopic particles.
- the size distribution of the bulk dispersion is shown in Figure 15. The size distribution is bimodal with particle sizes in the range from 0.1 - 2.5 ⁇ m. The poorly dispersed material (macroscopic particles > 100 ⁇ m) is not accounted for in the size distribution shown in Figure 15.
- composition examples (in % w/w):
- Example 13 - Surface functionalization The particle surface was functionalized by preparing a composition by the method of Example 2.1 comprising DGMO (1.77 g), GDO (1.17 g) and DOPG (0.077 g). The components were molecularly mixed by heating for 5 min at 70 0 C and vortexing. The homogenous lipid melt (2.5 g) was added drop wise to 22.5 g of deionized water containing Pluronic® F 127 (0.277 g). The resulting coarse dispersion was put on a shaking table and shaken for 12 hours to give a white homogenous dispersion.
- the particles were functionalized with Chitosan (Pronova Biopolymer, Norway) by adding 0.52 g of a 4 wt% Chitosan solution (Chitosan dissolved in 0.5 wt% acetic acid) to the lipid dispersion and equilibrating the solution for 1 h.
- Chitosan Pronova Biopolymer, Norway
- the above non-lamellar Chitosan-funtionalized dispersion was further treated to provide a dried powder precursor using a Spray-Drier (BUCHI Mini Spray Dryer B- 290).
- the dispersion was spray-dried in the presence of trehalose (Sigma-Aldrich, Sweden) to give a fine white powder.
- a gel containing self-dispersed non-lamellar particles was prepared by adding 5 mg Sodium hyaluronate (Sigma-Aldrich, Sweden) to 1 g of a 20 wt% dispersion of non - lamellar particles prepared by the method of Example 2.1 with the following composition: DGMO (42.5 wt%), GDO (42.5 wt%) and P80 (15 wt%). The resulting mixture was stirred at low speed for 24 h forming a turbid and viscous gel.
- a composition comprising comprising DGMO (54% by weight), GDO (36% by weight) and Pluronic® F 127 (10% by weight) was dispersed in 99 times its weight in water by the method of Example 2.1.
- the dispersion was divided into samples and loaded with each of the active agents shown below by each of two techniques: i) A saturated solution of active was equilibrated with the particles in dispersion at 37 0 C for three days by gentle stirring on a rotating table. ii) The samples were dispersed in a solution of excess active agent and heat treated by autoclavation at 125 0 C for 20 minutes and were allowed to temperature equilibrate at 37 0 C for at least one hour.
- Example 16 Further active agent loading
- Non-lamellar particle dispersions containing the anaesthetic active agent Propofol were formed by mixing a composition comprising DGMO (42.5% by weight of amphiphile), GDO (42.5% by weight of amphiphile) and P80 (15% by weight of amphiphile) with Propofol at the proportions indicated in the table below.
- the components were molecularly mixed by heating for 5 min at 70°C and vortexing.
- the homogenous lipid/Propofol melt was added drop wise to an aqueous solution containing 2.5% (by weight of total formulation) of Glycerol (Apoteket, Sweden).
- the resulting coarse dispersions were put on a shaking table (350 rpm) and shaken for 12 hours to give homogenous dispersions.
- the dispersions were filtered through a 0.2 ⁇ m filter and heat-treated by the method of Example 2.2.
- the particle size distributions of the resulting dispersions were narrow and monomodal with mean particle sizes in the range of 100-150 nm as shown in Figure 16.
- the morphology of the Propofol loaded particles was investigated using cryo- TEM. As shown in Figure 17, the cryo-TEM images reveal that the non -lamellar inner particle structure of multiply connected bilayers is retained after Propofol loading (compare with Figure 6).
- the Propofol loaded dispersions were found to be stable to storage at room temperature for at least 1 month.
- a dispersion of non-lamellar particles containing Propofol was prepared with the same composition and by the same method as in Example 16 except that the Propofol concentration in this case was 10 mg/mL and the amphiphile concentration was 100 mg/mL.
- the non-lamellar particle Propofol dispersion was compared for duration of anaesthesia and pharmacokinetics in rats (male SPF Sprague-Dawley rats (MoI: SPRD HAN, M&B Taconic, Lille Skensved, Denmark)) with the reference commercial Propofol Fresenius Kabi emulsion formulation (10 tng Propofol/mL).
- Plasma concentration over time of propofol was similar for the reference formulation and the non-lamellar particle propofol formulation, respectively ( Figure 18). Also, exposure to the drug after bolus i.v.
- the increased initial half-life may suggest an increased circulation time of the drug carrier and/or a decreased release rate for the drug. This may also explain the prolonged average recovering time after induction of anaesthesia (see table above).
- An alternative explanation is a more effective absorption of Propofol across the blood-brain barrier facilitated by the non-lamellar particles.
- Example 18 Further active agent loading
- a homogenous liquid solution containing the anti-inflammatory local anaesthetic agent Benzydamine hydrochloride (Sigma-Aldrich, Sweden) was prepared by molecularly mixing 6.8 mg of Benzydamine hydrochloride with 1.0 g of a mixture of DGMO/GDO/Polysorbat 80 (42.5/42.5/15 wt%) employing gentle stirring over night at room temperature.
- a 30 wt% lipid dispersion of non -lamellar particles was formed by vortexing this solution together with 2.3 g of deionized water.
- Example 19 Loading of testosterone undecanoate (TEU) and oral bioavailability of TEU formulated in liquid non-lamellar phase precursor
- a homogenous liquid formulation of the hormone testosterone undecanoate (TEU) was prepared by dissolving 0.24 g TEU in a liquid non-lamellar phase precursor mixture comprising DGMO (0.75 g), GDO (0.75 g) and P80 (0.26 g). The sample was allowed to mix with gentle stirring for 3 h. The liquid non -lamellar particle precursor containing TEU was compared for bioavailability of TEU in rats with the reference commercial Undestor Testocaps (Apoteket, Sweden) . The animals were given the liquid formulations with the TEU at a dose of 100 mg TEU per kg bodyweight.
- TES Concentration of testosterone
- Plasma samples (0.3 mL) were collected pre-dose, 1 hr, 3 hrs, 5 hrs, 7 hrs, 9 hrs, 12 hrs and 24 hrs. Concentration of testosterone (TES) in plasma was quantified using a commercial assay. Briefly, the principle of the assay is a competitive ELISA where an unknown amount of antigen (TES) in a sample competes for the binding sites of antibodies coated onto the microtiter wells with a fixed amount of added enzyme-labelled antigen. The assay showed no cross- reactivity with TEU. Plasma concentration of TES after administration of TEU in the non-lamellar nanoparticle formulation was significantly greater than for the commercial reference formulation (Figure 19).
- Example 20 Loading of the peptide Octreotide (OCT) and oral bioavailability of OCT formulated in the non-lamellar phase dispersion
- a dispersion of non-lamellar particles containing the peptide active Octreotide (OCT) (Polypeptides, Sweden) was formed by mixing 1.767 g of DGMO, 1.168 g of GDO, 0.077 g of DOPG and 0.00657 g of OCT. The components were molecularly mixed by heating for 5 min at 7O 0 C and vortexing. The homogenous lipid/OCT melt (2.505 g) was added drop wise to a solution containing 0.2771 g of Pluronic® F127 and ,22.5282 g of deionized water.
- OCT peptide active Octreotide
- the resulting coarse dispersion was put on a shaking table (350 rpm) and shaken for 12 hours to give a white homogenous dispersion.
- the dispersion was thereafter heat-treated by the method of Example 2.2. To the heat-treated dispersion was added 0.52 g of a 4 wt% solution of Chitosan (Chitosan dissolved in 0.5% acetic acid) and the dispersion was allowed to equilibrate for 12 h before oral administration to the animals.
- the rats were prepared by inserting a silicon catheter (OD approx. 1 mm) in the jugular vein under isoflurane anaesthesia. The catheter was tunnelled under the skin and exteriorised between the scapulae. After surgery the rats were allowed 48 hours of recovery before dosing. The catheter was rinsed with 0.9% NaCl containing 1 mM EDTA, every morning during the recovery period.
- the rats were dosed intravenously through the venous catheter or by gavage by a plastic ball-tipped gavage tube.
- Intravenously dosed rats were given 0.2 mg OCT per kg bwt in 1.0 mL/kg of sterile saline and gavaged rats were given the dispersion in water of OCT-containing non-lamellar particles or a saline solution of OCT to a dose of 3 mg OCT per kg bwt (dose volume equal to 10 mL per kg bwt).
- Oral dosing was performed under light isoflurane anaesthesia.
- Blood samples (0.5 mL) were collected pre-dose (one day before dosing), 10 minutes, 30 minutes, 1 hr, 3 hrs, 6 hrs and 24 hrs after dosing in EDTA-treated test tubes also containing 500 KIE aprotinin (Trasylol®) per mL sample. All blood samples were gently mixed and held on ice (maximally 10 minutes) before they were centrifuged at 2,000 g for 10 minutes at +4 0 C. Plasma were then immediately transferred to new test tubes and put on dry ice. Samples were stored at -8O 0 C until analysis.
- the content of OCT in all plasma samples was measured by a competitive immunoassay. Briefly, the OCT peptide coated on a microplate competes for the antibody in solution with the OCT present in the plasma sample. The fraction of antibody remaining in solution is removed, and the fraction bound to the immobilized peptide is quantified, the signal obtained being inversely proportional to the concentration of OCT in the sample.
- Plasma OCT concentration data were utilized to calculate area-under-the-curve from 0 to 6 hours (AUC) by the trapezoidal method.
- Availability (F) (AUCorai x Dose lv )/(AUCiv x Dose or ai) x 100 20.6 - Results Rats were dosed with an intravenous OCT solution, and orally with the dispersion of OCT-containing non-lamellar particles and OCT in a saline solution, according to the above described method. Plasma OCT contents were analysed and OCT plasma concentrations were plotted over time. Absolute bioavailability (F) of OCT administered orally in the non-lamellar nanoparticles was around 0.4%, while OCT delivered in the pure saline solution resulted in bioavailability of approx. 0.04%. Hence, the non-lamellar dispersion has an enhancing effect of around a factor 10 for the oral bioavailability of OCT compared to the pure saline solution.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Communicable Diseases (AREA)
- Dispersion Chemistry (AREA)
- Oncology (AREA)
- Pain & Pain Management (AREA)
- Immunology (AREA)
- Neurology (AREA)
- Psychiatry (AREA)
- Biomedical Technology (AREA)
- Virology (AREA)
- Neurosurgery (AREA)
- Rheumatology (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Colloid Chemistry (AREA)
Abstract
Description









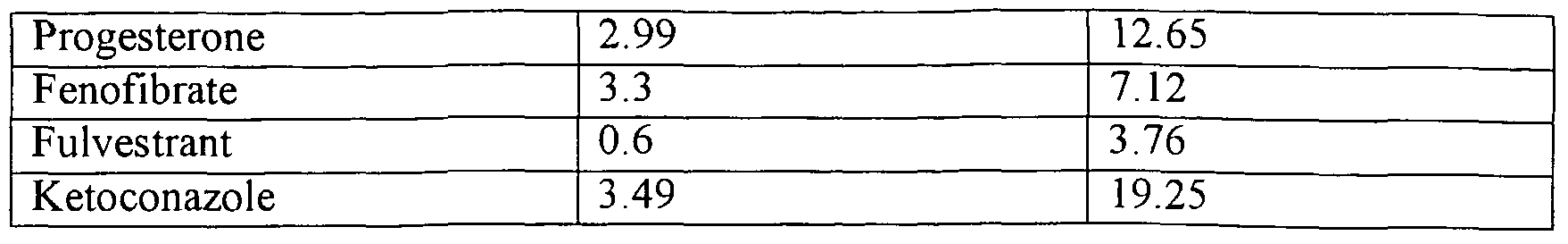


Claims
Priority Applications (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PL05767909T PL1778187T3 (en) | 2004-08-04 | 2005-08-04 | Compositions forming non-lamellar dispersions |
| KR1020077005229A KR101234885B1 (en) | 2004-08-04 | 2005-08-04 | Compositions forming non-lamellar dispersions |
| CN200580029678.1A CN101035511B (en) | 2004-08-04 | 2005-08-04 | Compositions forming non-lamellar dispersions |
| JP2007524396A JP5513713B2 (en) | 2004-08-04 | 2005-08-04 | Compositions that produce non-layered dispersions |
| CA2575906A CA2575906C (en) | 2004-08-04 | 2005-08-04 | Compositions forming non-lamellar dispersions |
| EP05767909A EP1778187B1 (en) | 2004-08-04 | 2005-08-04 | Compositions forming non-lamellar dispersions |
| ES05767909T ES2383530T3 (en) | 2004-08-04 | 2005-08-04 | Compositions that form non-laminar dispersions |
| US11/658,857 US8541400B2 (en) | 2004-08-04 | 2005-08-04 | Compositions forming non-lamellar dispersions |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0417388.6 | 2004-08-04 | ||
| GB0417388A GB0417388D0 (en) | 2004-08-04 | 2004-08-04 | Composition |
| GB0425754.9 | 2004-11-23 | ||
| GB0425754A GB0425754D0 (en) | 2004-11-23 | 2004-11-23 | Composition |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2006013369A2 true WO2006013369A2 (en) | 2006-02-09 |
| WO2006013369A3 WO2006013369A3 (en) | 2006-06-15 |
Family
ID=35520771
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2005/003056 WO2006013369A2 (en) | 2004-08-04 | 2005-08-04 | Compositions forming non-lamellar dispersions |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US8541400B2 (en) |
| EP (1) | EP1778187B1 (en) |
| JP (1) | JP5513713B2 (en) |
| KR (1) | KR101234885B1 (en) |
| CA (1) | CA2575906C (en) |
| ES (1) | ES2383530T3 (en) |
| PL (1) | PL1778187T3 (en) |
| WO (1) | WO2006013369A2 (en) |
Cited By (38)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2052716A1 (en) | 2007-10-24 | 2009-04-29 | Camurus AB | Controlled release formulations |
| WO2011129812A1 (en) * | 2010-04-12 | 2011-10-20 | Clarus Therapeutics, Inc. | Oral testosterone ester formulations and methods of treating testoterone deficiency comprising same |
| EP2428200A2 (en) | 2006-11-29 | 2012-03-14 | Malvern Cosmeceutics Limited | Novel compositions |
| US8241664B2 (en) | 2005-04-15 | 2012-08-14 | Clarus Therapeutics, Inc | Pharmaceutical delivery systems for hydrophobic drugs and compositions comprising same |
| US8492369B2 (en) | 2010-04-12 | 2013-07-23 | Clarus Therapeutics Inc | Oral testosterone ester formulations and methods of treating testosterone deficiency comprising same |
| US8633178B2 (en) | 2011-11-23 | 2014-01-21 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US8778922B2 (en) | 2009-01-08 | 2014-07-15 | Lipocine Inc. | Steroidal compositions |
| US8933059B2 (en) | 2012-06-18 | 2015-01-13 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US9180091B2 (en) | 2012-12-21 | 2015-11-10 | Therapeuticsmd, Inc. | Soluble estradiol capsule for vaginal insertion |
| US9205057B2 (en) | 2010-11-30 | 2015-12-08 | Lipocine Inc. | High-strength testosterone undecanoate compositions |
| US9358241B2 (en) | 2010-11-30 | 2016-06-07 | Lipocine Inc. | High-strength testosterone undecanoate compositions |
| AU2014265072B2 (en) * | 2010-04-12 | 2016-09-15 | Tolmar, Inc. | Oral testosterone ester formulations and methods of treating testoterone deficiency comprising same |
| US9757389B2 (en) | 2014-08-28 | 2017-09-12 | Lipocine Inc. | Bioavailable solid state (17-β)-hydroxy-4-androsten-3-one esters |
| RU2642244C2 (en) * | 2010-04-12 | 2018-01-24 | Кларус Терапеутикс, Инк. | Oral pharmaceutical compositions of testosterone esters and method for testosterone shortage treatment with their use |
| US9931349B2 (en) | 2016-04-01 | 2018-04-03 | Therapeuticsmd, Inc. | Steroid hormone pharmaceutical composition |
| CN108403664A (en) * | 2018-03-16 | 2018-08-17 | 武汉百纳礼康生物制药有限公司 | A kind of gel with liquid crystal structure nanoparticle and preparation method thereof containing opposed polarity drug |
| US10052386B2 (en) | 2012-06-18 | 2018-08-21 | Therapeuticsmd, Inc. | Progesterone formulations |
| US10206932B2 (en) | 2014-05-22 | 2019-02-19 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US10238709B2 (en) | 2015-02-03 | 2019-03-26 | Chiasma, Inc. | Method of treating diseases |
| US10258630B2 (en) | 2014-10-22 | 2019-04-16 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US10286077B2 (en) | 2016-04-01 | 2019-05-14 | Therapeuticsmd, Inc. | Steroid hormone compositions in medium chain oils |
| US10328087B2 (en) | 2015-07-23 | 2019-06-25 | Therapeuticsmd, Inc. | Formulations for solubilizing hormones |
| US10471072B2 (en) | 2012-12-21 | 2019-11-12 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US10471148B2 (en) | 2012-06-18 | 2019-11-12 | Therapeuticsmd, Inc. | Progesterone formulations having a desirable PK profile |
| US10537581B2 (en) | 2012-12-21 | 2020-01-21 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US10561615B2 (en) | 2010-12-10 | 2020-02-18 | Lipocine Inc. | Testosterone undecanoate compositions |
| WO2020082137A1 (en) * | 2018-10-25 | 2020-04-30 | Zeenar Enterprises Pty Ltd | Composition that forms liquid crystalline particles |
| US10682387B2 (en) | 2014-12-10 | 2020-06-16 | Chiasma, Inc. | Oral octreotide administered in combination with other therapeutic agents |
| US10806740B2 (en) | 2012-06-18 | 2020-10-20 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US10806697B2 (en) | 2012-12-21 | 2020-10-20 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US11141457B1 (en) | 2020-12-28 | 2021-10-12 | Amryt Endo, Inc. | Oral octreotide therapy and contraceptive methods |
| US11246875B2 (en) | 2012-12-21 | 2022-02-15 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US11266661B2 (en) | 2012-12-21 | 2022-03-08 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US11400159B2 (en) | 2008-09-17 | 2022-08-02 | Amryt Endo, Inc. | Pharmaceutical compositions and related methods of delivery |
| US11433120B2 (en) | 2011-05-25 | 2022-09-06 | Camurus Ab | Controlled release peptide formulations |
| US11559530B2 (en) | 2016-11-28 | 2023-01-24 | Lipocine Inc. | Oral testosterone undecanoate therapy |
| US11638698B2 (en) | 2017-04-20 | 2023-05-02 | Zeenar Enterprises Pty Ltd | Liquid crystalline dosage form for administering a statin |
| US11707467B2 (en) | 2014-08-28 | 2023-07-25 | Lipocine Inc. | (17-ß)-3-oxoandrost-4-en-17YL tridecanoate compositions and methods of their preparation and use |
Families Citing this family (21)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9649382B2 (en) | 2005-01-14 | 2017-05-16 | Camurus Ab | Topical bioadhesive formulations |
| US8920782B2 (en) * | 2005-01-14 | 2014-12-30 | Camurus Ab | Topical bioadhesive formulations |
| DE602005026998D1 (en) * | 2005-01-14 | 2011-04-28 | Camurus Ab | SOMATOSTATIN-ANALOG FORMULATIONS |
| US9060935B2 (en) * | 2005-01-21 | 2015-06-23 | Camurus Ab | Pharmaceutical lipid compositions |
| JP5198261B2 (en) * | 2005-06-06 | 2013-05-15 | カムルス エービー | GLP-1 analog preparation |
| GB0711656D0 (en) | 2007-06-15 | 2007-07-25 | Camurus Ab | Formulations |
| GB0716385D0 (en) | 2007-08-22 | 2007-10-03 | Camurus Ab | Formulations |
| GB0815435D0 (en) | 2008-08-22 | 2008-10-01 | Camurus Ab | Formulations |
| EP2498754B1 (en) * | 2009-06-25 | 2018-06-06 | Yissum Research Development Company of the Hebrew University of Jerusalem, Ltd. | Reverse hexagonal mesophases (h ii) and uses thereof |
| AU2013265210B2 (en) | 2012-05-25 | 2016-09-15 | Camurus Ab | Somatostatin receptor agonist formulations |
| JP6097803B2 (en) * | 2015-09-17 | 2017-03-15 | クラルス セラピューティクス,インク. | Oral testosterone ester combination and method for treating testosterone deficiency containing the same |
| MX2018013414A (en) * | 2016-05-06 | 2019-06-06 | Eagle Pharmaceuticals Inc | Fulvestrant formulations and methods of their use. |
| US11590077B2 (en) | 2016-05-06 | 2023-02-28 | Eagle Pharmaceuticals, Inc. | Fulvestrant formulations and methods of their use |
| JP7064612B2 (en) | 2018-09-20 | 2022-05-10 | 富士フイルム株式会社 | Biomaterial |
| US11478490B1 (en) | 2021-03-30 | 2022-10-25 | Epalex Corporation | Fospropofol formulations |
| US11628178B2 (en) | 2019-03-26 | 2023-04-18 | Epalex Corporation | Fospropofol methods and compositions |
| US11547714B2 (en) | 2020-02-05 | 2023-01-10 | Epalex Corporation | Fospropofol salts, methods and compositions |
| US11439653B1 (en) | 2021-03-30 | 2022-09-13 | Epalex Corporation | Fospropofol formulations |
| US11633405B2 (en) | 2020-02-07 | 2023-04-25 | Therapeuticsmd, Inc. | Steroid hormone pharmaceutical formulations |
| WO2023189270A1 (en) * | 2022-03-28 | 2023-10-05 | 富士フイルム株式会社 | Biomaterial composition |
| WO2023189273A1 (en) * | 2022-03-28 | 2023-10-05 | 富士フイルム株式会社 | Biological composition |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5531925A (en) * | 1991-10-04 | 1996-07-02 | Gs Biochem Ab | Particles, method of preparing said particles and uses thereof |
| WO1993006921A1 (en) * | 1991-10-04 | 1993-04-15 | Gs Biochem Ab | Particles, method of preparing said particles and uses thereof |
| CZ267796A3 (en) * | 1996-09-12 | 1998-04-15 | Galena A.S. | Medicamentous preparations, particularly for internal application in the form of inherently micro-emulsifying therapeutical systems |
| US6638621B2 (en) | 2000-08-16 | 2003-10-28 | Lyotropic Therapeutics, Inc. | Coated particles, methods of making and using |
| WO2003000236A1 (en) * | 2001-06-23 | 2003-01-03 | Lyotropic Therapeutics, Inc | Particles with improved solubilization capacity |
-
2005
- 2005-08-04 US US11/658,857 patent/US8541400B2/en active Active
- 2005-08-04 ES ES05767909T patent/ES2383530T3/en active Active
- 2005-08-04 PL PL05767909T patent/PL1778187T3/en unknown
- 2005-08-04 JP JP2007524396A patent/JP5513713B2/en not_active Expired - Fee Related
- 2005-08-04 WO PCT/GB2005/003056 patent/WO2006013369A2/en active Application Filing
- 2005-08-04 KR KR1020077005229A patent/KR101234885B1/en not_active IP Right Cessation
- 2005-08-04 CA CA2575906A patent/CA2575906C/en not_active Expired - Fee Related
- 2005-08-04 EP EP05767909A patent/EP1778187B1/en active Active
Non-Patent Citations (1)
| Title |
|---|
| None |
Cited By (104)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8778917B2 (en) | 2005-04-15 | 2014-07-15 | Clarus Therapeutics, Inc. | Pharmaceutical delivery systems for hydrophobic drugs and compositions comprising same |
| US8828428B1 (en) | 2005-04-15 | 2014-09-09 | Clarus Therapeutics, Inc. | Pharmaceutical delivery systems for hydrophobic drugs and compositions comprising same |
| US11331325B2 (en) | 2005-04-15 | 2022-05-17 | Clarus Therapeutics, Inc. | Pharmaceutical delivery systems for hydrophobic drugs and compositions comprising same |
| US8241664B2 (en) | 2005-04-15 | 2012-08-14 | Clarus Therapeutics, Inc | Pharmaceutical delivery systems for hydrophobic drugs and compositions comprising same |
| US11179402B2 (en) | 2005-04-15 | 2021-11-23 | Clarus Therapeutics, Inc. | Pharmaceutical delivery systems for hydrophobic drugs and compositions comprising same |
| US8778916B2 (en) | 2005-04-15 | 2014-07-15 | Clarus Therapeutics, Inc. | Oral testosterone ester formulations and methods of treating testosterone deficiency comprising same |
| EP2428200A2 (en) | 2006-11-29 | 2012-03-14 | Malvern Cosmeceutics Limited | Novel compositions |
| EP2052716A1 (en) | 2007-10-24 | 2009-04-29 | Camurus AB | Controlled release formulations |
| EP2886105A1 (en) | 2007-10-24 | 2015-06-24 | Camurus Ab | Controlled-release formulations |
| US11400159B2 (en) | 2008-09-17 | 2022-08-02 | Amryt Endo, Inc. | Pharmaceutical compositions and related methods of delivery |
| US11969471B2 (en) | 2008-09-17 | 2024-04-30 | Amryt Endo, Inc. | Pharmaceutical compositions and related methods of delivery |
| US11986529B2 (en) | 2008-09-17 | 2024-05-21 | Amryt Endo, Inc. | Pharmaceutical compositions and related methods of delivery |
| US11052096B2 (en) | 2009-01-08 | 2021-07-06 | Lipocine Inc. | Steroidal compositions |
| US11304960B2 (en) | 2009-01-08 | 2022-04-19 | Chandrashekar Giliyar | Steroidal compositions |
| US8778922B2 (en) | 2009-01-08 | 2014-07-15 | Lipocine Inc. | Steroidal compositions |
| US8865695B2 (en) | 2009-01-08 | 2014-10-21 | Lipocine Inc. | Steroidal compositions |
| US11179403B2 (en) | 2010-04-12 | 2021-11-23 | Clarus Therapeutics, Inc. | Oral testosterone ester formulations and methods of treating testosterone deficiency comprising same |
| AU2014265072B2 (en) * | 2010-04-12 | 2016-09-15 | Tolmar, Inc. | Oral testosterone ester formulations and methods of treating testoterone deficiency comprising same |
| WO2011129812A1 (en) * | 2010-04-12 | 2011-10-20 | Clarus Therapeutics, Inc. | Oral testosterone ester formulations and methods of treating testoterone deficiency comprising same |
| US10617696B2 (en) | 2010-04-12 | 2020-04-14 | Clarus Therapeutics, Inc. | Oral testosterone ester formulations and methods of treating testosterone deficiency comprising same |
| US10543219B2 (en) | 2010-04-12 | 2020-01-28 | Clarus Therapeutics, Inc. | Oral testosterone ester formulations and methods of treating testosterone deficiency comprising same |
| US8492369B2 (en) | 2010-04-12 | 2013-07-23 | Clarus Therapeutics Inc | Oral testosterone ester formulations and methods of treating testosterone deficiency comprising same |
| AU2010351080B2 (en) * | 2010-04-12 | 2014-08-28 | Tolmar, Inc. | Oral testosterone ester formulations and methods of treating testoterone deficiency comprising same |
| US11426416B2 (en) | 2010-04-12 | 2022-08-30 | Clarus Therapeutics, Inc. | Oral testosterone ester formulations and methods of treating testosterone deficiency comprising same |
| RU2642244C2 (en) * | 2010-04-12 | 2018-01-24 | Кларус Терапеутикс, Инк. | Oral pharmaceutical compositions of testosterone esters and method for testosterone shortage treatment with their use |
| EP2803350A1 (en) * | 2010-04-12 | 2014-11-19 | Clarus Therapeutics, Inc. | Oral testosterone ester formulations and methods of treating testosterone deficiency comprising same |
| US9480690B2 (en) | 2010-11-30 | 2016-11-01 | Lipocine Inc. | High-strength testosterone undecanoate compositions |
| US9757390B2 (en) | 2010-11-30 | 2017-09-12 | Lipocine Inc. | High-strength testosterone undecanoate compositions |
| US9358241B2 (en) | 2010-11-30 | 2016-06-07 | Lipocine Inc. | High-strength testosterone undecanoate compositions |
| US10799513B2 (en) | 2010-11-30 | 2020-10-13 | Lipocine Inc. | High-strength testosterone undecanoate compositions |
| US10716794B2 (en) | 2010-11-30 | 2020-07-21 | Lipocine Inc. | High-strength testosterone undecanoate compositions |
| US9943527B2 (en) | 2010-11-30 | 2018-04-17 | Lipocine Inc. | High-strength testosterone undecanoate compositions |
| US9949985B2 (en) | 2010-11-30 | 2018-04-24 | Lipocine Inc. | High-strength testosterone undecanoate compositions |
| US10973833B2 (en) | 2010-11-30 | 2021-04-13 | Lipocine Inc. | High-strength testosterone undecanoate compositions |
| US9205057B2 (en) | 2010-11-30 | 2015-12-08 | Lipocine Inc. | High-strength testosterone undecanoate compositions |
| US10226473B2 (en) | 2010-11-30 | 2019-03-12 | Lipocine Inc. | High-strength testosterone undecanoate compositions |
| US10561615B2 (en) | 2010-12-10 | 2020-02-18 | Lipocine Inc. | Testosterone undecanoate compositions |
| US11433120B2 (en) | 2011-05-25 | 2022-09-06 | Camurus Ab | Controlled release peptide formulations |
| US10675288B2 (en) | 2011-11-23 | 2020-06-09 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US9248136B2 (en) | 2011-11-23 | 2016-02-02 | Therapeuticsmd, Inc. | Transdermal hormone replacement therapies |
| US8846648B2 (en) | 2011-11-23 | 2014-09-30 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US8846649B2 (en) | 2011-11-23 | 2014-09-30 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US11103516B2 (en) | 2011-11-23 | 2021-08-31 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US8633178B2 (en) | 2011-11-23 | 2014-01-21 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US11793819B2 (en) | 2011-11-23 | 2023-10-24 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US8987237B2 (en) | 2011-11-23 | 2015-03-24 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US10052386B2 (en) | 2012-06-18 | 2018-08-21 | Therapeuticsmd, Inc. | Progesterone formulations |
| US8933059B2 (en) | 2012-06-18 | 2015-01-13 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US10471148B2 (en) | 2012-06-18 | 2019-11-12 | Therapeuticsmd, Inc. | Progesterone formulations having a desirable PK profile |
| US11166963B2 (en) | 2012-06-18 | 2021-11-09 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US9012434B2 (en) | 2012-06-18 | 2015-04-21 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US11110099B2 (en) | 2012-06-18 | 2021-09-07 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US10639375B2 (en) | 2012-06-18 | 2020-05-05 | Therapeuticsmd, Inc. | Progesterone formulations |
| US11529360B2 (en) | 2012-06-18 | 2022-12-20 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US8987238B2 (en) | 2012-06-18 | 2015-03-24 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US9006222B2 (en) | 2012-06-18 | 2015-04-14 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US11033626B2 (en) | 2012-06-18 | 2021-06-15 | Therapeuticsmd, Inc. | Progesterone formulations having a desirable pk profile |
| US11865179B2 (en) | 2012-06-18 | 2024-01-09 | Therapeuticsmd, Inc. | Progesterone formulations having a desirable PK profile |
| US9301920B2 (en) | 2012-06-18 | 2016-04-05 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US10806740B2 (en) | 2012-06-18 | 2020-10-20 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US11241445B2 (en) | 2012-12-21 | 2022-02-08 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US11246875B2 (en) | 2012-12-21 | 2022-02-15 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US10888516B2 (en) | 2012-12-21 | 2021-01-12 | Therapeuticsmd, Inc. | Soluble estradiol capsule for vaginal insertion |
| US9180091B2 (en) | 2012-12-21 | 2015-11-10 | Therapeuticsmd, Inc. | Soluble estradiol capsule for vaginal insertion |
| US10806697B2 (en) | 2012-12-21 | 2020-10-20 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US11351182B2 (en) | 2012-12-21 | 2022-06-07 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US11497709B2 (en) | 2012-12-21 | 2022-11-15 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US10835487B2 (en) | 2012-12-21 | 2020-11-17 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US11065197B2 (en) | 2012-12-21 | 2021-07-20 | Therapeuticsmd, Inc. | Soluble estradiol capsule for vaginal insertion |
| US11304959B2 (en) | 2012-12-21 | 2022-04-19 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US11266661B2 (en) | 2012-12-21 | 2022-03-08 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US10471072B2 (en) | 2012-12-21 | 2019-11-12 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US11116717B2 (en) | 2012-12-21 | 2021-09-14 | Therapeuticsmd, Inc. | Soluble estradiol capsule for vaginal insertion |
| US11123283B2 (en) | 2012-12-21 | 2021-09-21 | Therapeuticsmd, Inc. | Soluble estradiol capsule for vaginal insertion |
| US11622933B2 (en) | 2012-12-21 | 2023-04-11 | Therapeuticsmd, Inc. | Soluble estradiol capsule for vaginal insertion |
| US10568891B2 (en) | 2012-12-21 | 2020-02-25 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US10537581B2 (en) | 2012-12-21 | 2020-01-21 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US10206932B2 (en) | 2014-05-22 | 2019-02-19 | Therapeuticsmd, Inc. | Natural combination hormone replacement formulations and therapies |
| US11103513B2 (en) | 2014-05-22 | 2021-08-31 | TherapeuticsMD | Natural combination hormone replacement formulations and therapies |
| US11707467B2 (en) | 2014-08-28 | 2023-07-25 | Lipocine Inc. | (17-ß)-3-oxoandrost-4-en-17YL tridecanoate compositions and methods of their preparation and use |
| US11298365B2 (en) | 2014-08-28 | 2022-04-12 | Lipocine Inc. | Bioavailable solid state (17-β)-hydroxy-4-androsten-3-one esters |
| US11872235B1 (en) | 2014-08-28 | 2024-01-16 | Lipocine Inc. | Bioavailable solid state (17-β)-Hydroxy-4-Androsten-3-one esters |
| US9757389B2 (en) | 2014-08-28 | 2017-09-12 | Lipocine Inc. | Bioavailable solid state (17-β)-hydroxy-4-androsten-3-one esters |
| US10258630B2 (en) | 2014-10-22 | 2019-04-16 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US10668082B2 (en) | 2014-10-22 | 2020-06-02 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US10398708B2 (en) | 2014-10-22 | 2019-09-03 | Therapeuticsmd, Inc. | Vaginal inserted estradiol pharmaceutical compositions and methods |
| US10682387B2 (en) | 2014-12-10 | 2020-06-16 | Chiasma, Inc. | Oral octreotide administered in combination with other therapeutic agents |
| US11510963B1 (en) | 2015-02-03 | 2022-11-29 | Amryt Endo, Inc. | Method of treating diseases |
| US11857595B2 (en) | 2015-02-03 | 2024-01-02 | Amryt Endo, Inc. | Method of treating diseases |
| US10238709B2 (en) | 2015-02-03 | 2019-03-26 | Chiasma, Inc. | Method of treating diseases |
| US11338011B2 (en) | 2015-02-03 | 2022-05-24 | Amryt Endo, Inc. | Method of treating diseases |
| US10695397B2 (en) | 2015-02-03 | 2020-06-30 | Chiasma, Inc. | Method of treating diseases |
| US11052126B2 (en) | 2015-02-03 | 2021-07-06 | Chiasma, Inc. | Method of treating diseases |
| US10328087B2 (en) | 2015-07-23 | 2019-06-25 | Therapeuticsmd, Inc. | Formulations for solubilizing hormones |
| US10912783B2 (en) | 2015-07-23 | 2021-02-09 | Therapeuticsmd, Inc. | Formulations for solubilizing hormones |
| US10532059B2 (en) | 2016-04-01 | 2020-01-14 | Therapeuticsmd, Inc. | Steroid hormone pharmaceutical composition |
| US9931349B2 (en) | 2016-04-01 | 2018-04-03 | Therapeuticsmd, Inc. | Steroid hormone pharmaceutical composition |
| US10286077B2 (en) | 2016-04-01 | 2019-05-14 | Therapeuticsmd, Inc. | Steroid hormone compositions in medium chain oils |
| US11559530B2 (en) | 2016-11-28 | 2023-01-24 | Lipocine Inc. | Oral testosterone undecanoate therapy |
| US11638698B2 (en) | 2017-04-20 | 2023-05-02 | Zeenar Enterprises Pty Ltd | Liquid crystalline dosage form for administering a statin |
| CN108403664A (en) * | 2018-03-16 | 2018-08-17 | 武汉百纳礼康生物制药有限公司 | A kind of gel with liquid crystal structure nanoparticle and preparation method thereof containing opposed polarity drug |
| WO2020082137A1 (en) * | 2018-10-25 | 2020-04-30 | Zeenar Enterprises Pty Ltd | Composition that forms liquid crystalline particles |
| US11141457B1 (en) | 2020-12-28 | 2021-10-12 | Amryt Endo, Inc. | Oral octreotide therapy and contraceptive methods |
| US11890316B2 (en) | 2020-12-28 | 2024-02-06 | Amryt Endo, Inc. | Oral octreotide therapy and contraceptive methods |
Also Published As
| Publication number | Publication date |
|---|---|
| US20080161276A1 (en) | 2008-07-03 |
| JP5513713B2 (en) | 2014-06-04 |
| PL1778187T3 (en) | 2012-09-28 |
| CA2575906A1 (en) | 2006-02-09 |
| EP1778187A2 (en) | 2007-05-02 |
| ES2383530T3 (en) | 2012-06-22 |
| US8541400B2 (en) | 2013-09-24 |
| CA2575906C (en) | 2014-04-15 |
| KR101234885B1 (en) | 2013-02-19 |
| WO2006013369A3 (en) | 2006-06-15 |
| EP1778187B1 (en) | 2012-05-23 |
| JP2008509120A (en) | 2008-03-27 |
| KR20070046913A (en) | 2007-05-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA2575906C (en) | Compositions forming non-lamellar dispersions | |
| CA2554052C (en) | Ternary non-lamellar lipid compositions of structure forming amphiphile, structure swelling amphiphile and dispersion stabilising amphiphile | |
| US9526788B2 (en) | Pharmaceutical lipid compositions | |
| Ravichandran | Nanoparticles in drug delivery: potential green nanobiomedicine applications | |
| JP5627039B2 (en) | Nanodispersions of drugs and methods for their preparation | |
| JP4920425B2 (en) | Non-lamellar composition of DOPE and P80 | |
| ITMI980234A1 (en) | PHARMACEUTICAL COMPOSITIONS IN THE FORM OF NANOPARTICLES INCLUDING LIPID SUBSTANCES AND ANTIPHILIC SUBSTANCES AND RELATED PROCESS OF | |
| Ram et al. | A review on solid lipid nanoparticles | |
| MX2007015183A (en) | Pharmaceutical formulations for minimizing drug-drug interactions. | |
| Poovi et al. | Solid lipid nanoparticles and nanostructured lipid carriers: a review of the effect of physicochemical formulation factors in the optimization process, different preparation technique, characterization, and toxicity | |
| CN101035511B (en) | Compositions forming non-lamellar dispersions | |
| Thiruganesh et al. | Solid lipid nanoparticle and nanoparticle lipid carrier for controlled drug delivery–a review of state of art and recent advances | |
| ES2356920T3 (en) | NON-LAMINARY TERNARY LIPID COMPOSITIONS. | |
| Souto et al. | Lipid Matrix Nanoparticles in Diabetes |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KM KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NG NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SM SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU LV MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2575906 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007524396 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005767909 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020077005229 Country of ref document: KR Ref document number: 200580029678.1 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005767909 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 11658857 Country of ref document: US |