WO2005117875A1 - Use of substituted quinoline derivatives for the treatment of drug resistant mycobacterial diseases - Google Patents
Use of substituted quinoline derivatives for the treatment of drug resistant mycobacterial diseases Download PDFInfo
- Publication number
- WO2005117875A1 WO2005117875A1 PCT/EP2005/052371 EP2005052371W WO2005117875A1 WO 2005117875 A1 WO2005117875 A1 WO 2005117875A1 EP 2005052371 W EP2005052371 W EP 2005052371W WO 2005117875 A1 WO2005117875 A1 WO 2005117875A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phenyl
- alkyl
- formula
- bromo
- methoxy
- Prior art date
Links
- 239000003814 drug Substances 0.000 title claims abstract description 81
- 229940079593 drug Drugs 0.000 title claims abstract description 71
- 238000011282 treatment Methods 0.000 title claims abstract description 35
- 125000002943 quinolinyl group Chemical class N1=C(C=CC2=CC=CC=C12)* 0.000 title claims abstract description 8
- 208000027531 mycobacterial infectious disease Diseases 0.000 title claims description 9
- 229940027991 antiseptic and disinfectant quinoline derivative Drugs 0.000 title description 8
- 150000001875 compounds Chemical class 0.000 claims abstract description 96
- 150000003839 salts Chemical class 0.000 claims abstract description 44
- 239000002253 acid Substances 0.000 claims abstract description 39
- 241000186359 Mycobacterium Species 0.000 claims abstract description 23
- 239000003926 antimycobacterial agent Substances 0.000 claims abstract description 19
- 238000002360 preparation method Methods 0.000 claims abstract description 18
- 208000015181 infectious disease Diseases 0.000 claims abstract description 17
- 125000000815 N-oxide group Chemical group 0.000 claims abstract description 16
- 239000004480 active ingredient Substances 0.000 claims abstract description 8
- 239000003937 drug carrier Substances 0.000 claims abstract description 8
- 125000000217 alkyl group Chemical group 0.000 claims description 71
- 150000003254 radicals Chemical class 0.000 claims description 62
- 125000005843 halogen group Chemical group 0.000 claims description 58
- 125000004432 carbon atom Chemical group C* 0.000 claims description 56
- -1 cyano, hydroxy Chemical group 0.000 claims description 52
- 239000001257 hydrogen Substances 0.000 claims description 48
- 229910052739 hydrogen Inorganic materials 0.000 claims description 48
- 125000003545 alkoxy group Chemical group 0.000 claims description 43
- 229930195734 saturated hydrocarbon Natural products 0.000 claims description 36
- 201000008827 tuberculosis Diseases 0.000 claims description 35
- 125000001424 substituent group Chemical group 0.000 claims description 30
- 125000001188 haloalkyl group Chemical group 0.000 claims description 28
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 24
- 125000004122 cyclic group Chemical group 0.000 claims description 23
- 229960005206 pyrazinamide Drugs 0.000 claims description 23
- IPEHBUMCGVEMRF-UHFFFAOYSA-N pyrazinecarboxamide Chemical compound NC(=O)C1=CN=CC=N1 IPEHBUMCGVEMRF-UHFFFAOYSA-N 0.000 claims description 23
- 125000004414 alkyl thio group Chemical group 0.000 claims description 21
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 17
- 239000008194 pharmaceutical composition Substances 0.000 claims description 17
- 150000003248 quinolines Chemical class 0.000 claims description 16
- 229910052799 carbon Inorganic materials 0.000 claims description 15
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 15
- 125000004183 alkoxy alkyl group Chemical group 0.000 claims description 14
- 125000006350 alkyl thio alkyl group Chemical group 0.000 claims description 14
- 125000001624 naphthyl group Chemical group 0.000 claims description 14
- 125000000714 pyrimidinyl group Chemical group 0.000 claims description 13
- 125000002618 bicyclic heterocycle group Chemical group 0.000 claims description 12
- 125000002883 imidazolyl group Chemical group 0.000 claims description 12
- 125000002757 morpholinyl group Chemical group 0.000 claims description 10
- 125000004043 oxo group Chemical group O=* 0.000 claims description 10
- 125000004076 pyridyl group Chemical group 0.000 claims description 10
- 125000003373 pyrazinyl group Chemical group 0.000 claims description 9
- 125000003226 pyrazolyl group Chemical group 0.000 claims description 9
- 125000002098 pyridazinyl group Chemical group 0.000 claims description 9
- 125000000168 pyrrolyl group Chemical group 0.000 claims description 9
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 claims description 8
- 125000001246 bromo group Chemical group Br* 0.000 claims description 8
- 125000004663 dialkyl amino group Chemical group 0.000 claims description 8
- 125000002632 imidazolidinyl group Chemical group 0.000 claims description 8
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 8
- 125000001544 thienyl group Chemical group 0.000 claims description 8
- QUIJNHUBAXPXFS-UHFFFAOYSA-N 1-(6-bromo-2-methoxyquinolin-3-yl)-4-(dimethylamino)-2-naphthalen-1-yl-1-phenylbutan-2-ol Chemical compound COC1=NC2=CC=C(Br)C=C2C=C1C(C(O)(CCN(C)C)C=1C2=CC=CC=C2C=CC=1)C1=CC=CC=C1 QUIJNHUBAXPXFS-UHFFFAOYSA-N 0.000 claims description 7
- 125000001309 chloro group Chemical group Cl* 0.000 claims description 7
- 125000001153 fluoro group Chemical group F* 0.000 claims description 7
- 125000004193 piperazinyl group Chemical group 0.000 claims description 7
- 125000003386 piperidinyl group Chemical group 0.000 claims description 7
- 125000001425 triazolyl group Chemical group 0.000 claims description 7
- 125000005605 benzo group Chemical group 0.000 claims description 6
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 claims description 6
- 125000000597 dioxinyl group Chemical group 0.000 claims description 6
- 125000002541 furyl group Chemical group 0.000 claims description 6
- 125000002950 monocyclic group Chemical group 0.000 claims description 6
- 125000002911 monocyclic heterocycle group Chemical group 0.000 claims description 6
- 125000001712 tetrahydronaphthyl group Chemical group C1(CCCC2=CC=CC=C12)* 0.000 claims description 6
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 5
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims description 5
- 125000004604 benzisothiazolyl group Chemical group S1N=C(C2=C1C=CC=C2)* 0.000 claims description 5
- 125000004603 benzisoxazolyl group Chemical group O1N=C(C2=C1C=CC=C2)* 0.000 claims description 5
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 claims description 5
- 125000001164 benzothiazolyl group Chemical group S1C(=NC2=C1C=CC=C2)* 0.000 claims description 5
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 claims description 5
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 claims description 5
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 5
- 125000004473 dialkylaminocarbonyl group Chemical group 0.000 claims description 5
- 125000004438 haloalkoxy group Chemical group 0.000 claims description 5
- 125000001041 indolyl group Chemical group 0.000 claims description 5
- 125000002346 iodo group Chemical group I* 0.000 claims description 5
- 125000001786 isothiazolyl group Chemical group 0.000 claims description 5
- 125000000842 isoxazolyl group Chemical group 0.000 claims description 5
- 125000002971 oxazolyl group Chemical group 0.000 claims description 5
- 125000000719 pyrrolidinyl group Chemical group 0.000 claims description 5
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 claims description 5
- 125000000335 thiazolyl group Chemical group 0.000 claims description 5
- 125000004364 3-pyrrolinyl group Chemical group [H]C1=C([H])C([H])([H])N(*)C1([H])[H] 0.000 claims description 4
- 241000187479 Mycobacterium tuberculosis Species 0.000 claims description 4
- 125000003072 pyrazolidinyl group Chemical group 0.000 claims description 4
- 229910052717 sulfur Inorganic materials 0.000 claims description 4
- 125000004306 triazinyl group Chemical group 0.000 claims description 4
- QUIJNHUBAXPXFS-XLJNKUFUSA-N bedaquiline Chemical compound C1([C@H](C2=CC3=CC(Br)=CC=C3N=C2OC)[C@@](O)(CCN(C)C)C=2C3=CC=CC=C3C=CC=2)=CC=CC=C1 QUIJNHUBAXPXFS-XLJNKUFUSA-N 0.000 claims description 3
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 claims description 3
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims 6
- VPZIAIQZWHQVKN-UHFFFAOYSA-N 1-(6-bromo-2-methoxyquinolin-3-yl)-4-(dimethylamino)-1,2-diphenylbutan-2-ol Chemical compound COC1=NC2=CC=C(Br)C=C2C=C1C(C(O)(CCN(C)C)C=1C=CC=CC=1)C1=CC=CC=C1 VPZIAIQZWHQVKN-UHFFFAOYSA-N 0.000 claims 1
- BTANRVKWQNVYAZ-UHFFFAOYSA-N Sec-butyl alcohol Natural products CCC(C)O BTANRVKWQNVYAZ-UHFFFAOYSA-N 0.000 claims 1
- 239000000203 mixture Substances 0.000 abstract description 11
- 150000002431 hydrogen Chemical group 0.000 description 26
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 22
- 229940125797 compound 12 Drugs 0.000 description 21
- JQXXHWHPUNPDRT-WLSIYKJHSA-N rifampicin Chemical compound O([C@](C1=O)(C)O/C=C/[C@@H]([C@H]([C@@H](OC(C)=O)[C@H](C)[C@H](O)[C@H](C)[C@@H](O)[C@@H](C)\C=C\C=C(C)/C(=O)NC=2C(O)=C3C([O-])=C4C)C)OC)C4=C1C3=C(O)C=2\C=N\N1CC[NH+](C)CC1 JQXXHWHPUNPDRT-WLSIYKJHSA-N 0.000 description 21
- 229960001225 rifampicin Drugs 0.000 description 21
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 20
- 241000699670 Mus sp. Species 0.000 description 17
- 239000002585 base Substances 0.000 description 15
- QRXWMOHMRWLFEY-UHFFFAOYSA-N isoniazide Chemical compound NNC(=O)C1=CC=NC=C1 QRXWMOHMRWLFEY-UHFFFAOYSA-N 0.000 description 15
- 229960003702 moxifloxacin Drugs 0.000 description 13
- FABPRXSRWADJSP-MEDUHNTESA-N moxifloxacin Chemical compound COC1=C(N2C[C@H]3NCCC[C@H]3C2)C(F)=CC(C(C(C(O)=O)=C2)=O)=C1N2C1CC1 FABPRXSRWADJSP-MEDUHNTESA-N 0.000 description 13
- 238000000034 method Methods 0.000 description 9
- 201000009671 multidrug-resistant tuberculosis Diseases 0.000 description 9
- 229960004821 amikacin Drugs 0.000 description 8
- LKCWBDHBTVXHDL-RMDFUYIESA-N amikacin Chemical compound O([C@@H]1[C@@H](N)C[C@H]([C@@H]([C@H]1O)O[C@@H]1[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O1)O)NC(=O)[C@@H](O)CCN)[C@H]1O[C@H](CN)[C@@H](O)[C@H](O)[C@H]1O LKCWBDHBTVXHDL-RMDFUYIESA-N 0.000 description 8
- 229960003350 isoniazid Drugs 0.000 description 8
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 8
- 210000000952 spleen Anatomy 0.000 description 8
- 239000003795 chemical substances by application Substances 0.000 description 7
- AEOCXXJPGCBFJA-UHFFFAOYSA-N ethionamide Chemical compound CCC1=CC(C(N)=S)=CC=N1 AEOCXXJPGCBFJA-UHFFFAOYSA-N 0.000 description 7
- 229960002001 ethionamide Drugs 0.000 description 7
- 210000004072 lung Anatomy 0.000 description 7
- AEUTYOVWOVBAKS-UWVGGRQHSA-N ethambutol Chemical compound CC[C@@H](CO)NCCN[C@@H](CC)CO AEUTYOVWOVBAKS-UWVGGRQHSA-N 0.000 description 6
- 239000000651 prodrug Substances 0.000 description 6
- 229940002612 prodrug Drugs 0.000 description 6
- 239000007787 solid Substances 0.000 description 6
- 239000000243 solution Substances 0.000 description 6
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 6
- 125000004568 thiomorpholinyl group Chemical group 0.000 description 6
- GSDSWSVVBLHKDQ-UHFFFAOYSA-N 9-fluoro-3-methyl-10-(4-methylpiperazin-1-yl)-7-oxo-2,3-dihydro-7H-[1,4]oxazino[2,3,4-ij]quinoline-6-carboxylic acid Chemical compound FC1=CC(C(C(C(O)=O)=C2)=O)=C3N2C(C)COC3=C1N1CCN(C)CC1 GSDSWSVVBLHKDQ-UHFFFAOYSA-N 0.000 description 5
- 239000002552 dosage form Substances 0.000 description 5
- 238000011081 inoculation Methods 0.000 description 5
- 229960001699 ofloxacin Drugs 0.000 description 5
- 238000000338 in vitro Methods 0.000 description 4
- 230000002401 inhibitory effect Effects 0.000 description 4
- 229910052760 oxygen Inorganic materials 0.000 description 4
- 239000003826 tablet Substances 0.000 description 4
- 238000012360 testing method Methods 0.000 description 4
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 229920001817 Agar Polymers 0.000 description 3
- 0 CC(C)(OC1)OC1C(*)** Chemical compound CC(C)(OC1)OC1C(*)** 0.000 description 3
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 3
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- KGTSLTYUUFWZNW-PPJQWWMSSA-N [(7S,9E,11S,12R,13S,14R,15R,16R,17S,18S,19E,21Z)-2,15,17,27,29-pentahydroxy-11-methoxy-3,7,12,14,16,18,22-heptamethyl-26-[(E)-(4-methylpiperazin-1-yl)iminomethyl]-6,23-dioxo-8,30-dioxa-24-azatetracyclo[23.3.1.14,7.05,28]triaconta-1(29),2,4,9,19,21,25,27-octaen-13-yl] acetate pyridine-4-carbohydrazide Chemical compound NNC(=O)c1ccncc1.CO[C@H]1\C=C\O[C@@]2(C)Oc3c(C2=O)c2c(O)c(\C=N\N4CCN(C)CC4)c(NC(=O)\C(C)=C/C=C/[C@H](C)[C@H](O)[C@@H](C)[C@@H](O)[C@@H](C)[C@H](OC(C)=O)[C@@H]1C)c(O)c2c(O)c3C KGTSLTYUUFWZNW-PPJQWWMSSA-N 0.000 description 3
- 239000008272 agar Substances 0.000 description 3
- 230000001580 bacterial effect Effects 0.000 description 3
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 125000004093 cyano group Chemical group *C#N 0.000 description 3
- 201000010099 disease Diseases 0.000 description 3
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 3
- 230000000694 effects Effects 0.000 description 3
- 229960000285 ethambutol Drugs 0.000 description 3
- 238000001727 in vivo Methods 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 230000035772 mutation Effects 0.000 description 3
- WDZCUPBHRAEYDL-GZAUEHORSA-N rifapentine Chemical compound O([C@](C1=O)(C)O/C=C/[C@@H]([C@H]([C@@H](OC(C)=O)[C@H](C)[C@H](O)[C@H](C)[C@@H](O)[C@@H](C)\C=C\C=C(C)/C(=O)NC=2C(O)=C3C(O)=C4C)C)OC)C4=C1C3=C(O)C=2\C=N\N(CC1)CCN1C1CCCC1 WDZCUPBHRAEYDL-GZAUEHORSA-N 0.000 description 3
- 229960005322 streptomycin Drugs 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 230000004083 survival effect Effects 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- VCOPTHOUUNAYKQ-WBTCAYNUSA-N (3s)-3,6-diamino-n-[[(2s,5s,8e,11s,15s)-15-amino-11-[(6r)-2-amino-1,4,5,6-tetrahydropyrimidin-6-yl]-8-[(carbamoylamino)methylidene]-2-(hydroxymethyl)-3,6,9,12,16-pentaoxo-1,4,7,10,13-pentazacyclohexadec-5-yl]methyl]hexanamide;(3s)-3,6-diamino-n-[[(2s,5s,8 Chemical compound N1C(=O)\C(=C/NC(N)=O)NC(=O)[C@H](CNC(=O)C[C@@H](N)CCCN)NC(=O)[C@H](C)NC(=O)[C@@H](N)CNC(=O)[C@@H]1[C@@H]1NC(N)=NCC1.N1C(=O)\C(=C/NC(N)=O)NC(=O)[C@H](CNC(=O)C[C@@H](N)CCCN)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CNC(=O)[C@@H]1[C@@H]1NC(N)=NCC1 VCOPTHOUUNAYKQ-WBTCAYNUSA-N 0.000 description 2
- WUBBRNOQWQTFEX-UHFFFAOYSA-N 4-aminosalicylic acid Chemical compound NC1=CC=C(C(O)=O)C(O)=C1 WUBBRNOQWQTFEX-UHFFFAOYSA-N 0.000 description 2
- 208000030507 AIDS Diseases 0.000 description 2
- JGLMVXWAHNTPRF-CMDGGOBGSA-N CCN1N=C(C)C=C1C(=O)NC1=NC2=CC(=CC(OC)=C2N1C\C=C\CN1C(NC(=O)C2=CC(C)=NN2CC)=NC2=CC(=CC(OCCCN3CCOCC3)=C12)C(N)=O)C(N)=O Chemical compound CCN1N=C(C)C=C1C(=O)NC1=NC2=CC(=CC(OC)=C2N1C\C=C\CN1C(NC(=O)C2=CC(C)=NN2CC)=NC2=CC(=CC(OCCCN3CCOCC3)=C12)C(N)=O)C(N)=O JGLMVXWAHNTPRF-CMDGGOBGSA-N 0.000 description 2
- 108010065839 Capreomycin Proteins 0.000 description 2
- DYDCUQKUCUHJBH-UWTATZPHSA-N D-Cycloserine Chemical compound N[C@@H]1CONC1=O DYDCUQKUCUHJBH-UWTATZPHSA-N 0.000 description 2
- DYDCUQKUCUHJBH-UHFFFAOYSA-N D-Cycloserine Natural products NC1CONC1=O DYDCUQKUCUHJBH-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- 206010034133 Pathogen resistance Diseases 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 229960004909 aminosalicylic acid Drugs 0.000 description 2
- 229940034014 antimycobacterial agent Drugs 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 230000003115 biocidal effect Effects 0.000 description 2
- 229960004602 capreomycin Drugs 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 238000006243 chemical reaction Methods 0.000 description 2
- MYSWGUAQZAJSOK-UHFFFAOYSA-N ciprofloxacin Chemical compound C12=CC(N3CCNCC3)=C(F)C=C2C(=O)C(C(=O)O)=CN1C1CC1 MYSWGUAQZAJSOK-UHFFFAOYSA-N 0.000 description 2
- 230000001332 colony forming effect Effects 0.000 description 2
- 229960003077 cycloserine Drugs 0.000 description 2
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 2
- 229940072185 drug for treatment of tuberculosis Drugs 0.000 description 2
- 238000011156 evaluation Methods 0.000 description 2
- 229940124307 fluoroquinolone Drugs 0.000 description 2
- 238000003304 gavage Methods 0.000 description 2
- 239000008103 glucose Substances 0.000 description 2
- 230000002458 infectious effect Effects 0.000 description 2
- 239000004615 ingredient Substances 0.000 description 2
- 229930027917 kanamycin Natural products 0.000 description 2
- 229960000318 kanamycin Drugs 0.000 description 2
- SBUJHOSQTJFQJX-NOAMYHISSA-N kanamycin Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CN)O[C@@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](N)[C@H](O)[C@@H](CO)O2)O)[C@H](N)C[C@@H]1N SBUJHOSQTJFQJX-NOAMYHISSA-N 0.000 description 2
- 229930182823 kanamycin A Natural products 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 239000000314 lubricant Substances 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- 239000013641 positive control Substances 0.000 description 2
- 239000000843 powder Substances 0.000 description 2
- 230000002265 prevention Effects 0.000 description 2
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 2
- 102000004169 proteins and genes Human genes 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- 239000012453 solvate Substances 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 239000007858 starting material Substances 0.000 description 2
- 230000001954 sterilising effect Effects 0.000 description 2
- 238000003419 tautomerization reaction Methods 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 2
- WRIDQFICGBMAFQ-UHFFFAOYSA-N (E)-8-Octadecenoic acid Natural products CCCCCCCCCC=CCCCCCCC(O)=O WRIDQFICGBMAFQ-UHFFFAOYSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- CFPPFXNAPDMERR-UHFFFAOYSA-N 1-(6-bromo-2-methoxyquinolin-3-yl)-4-(dimethylamino)-1-(4-methylphenyl)-2-naphthalen-1-ylbutan-2-ol Chemical compound COC1=NC2=CC=C(Br)C=C2C=C1C(C(O)(CCN(C)C)C=1C2=CC=CC=C2C=CC=1)C1=CC=C(C)C=C1 CFPPFXNAPDMERR-UHFFFAOYSA-N 0.000 description 1
- HGUAWYGZOVFIAB-UHFFFAOYSA-N 1-(6-bromo-2-methoxyquinolin-3-yl)-4-(dimethylamino)-2-(2-fluorophenyl)-1-phenylbutan-2-ol Chemical compound COC1=NC2=CC=C(Br)C=C2C=C1C(C(O)(CCN(C)C)C=1C(=CC=CC=1)F)C1=CC=CC=C1 HGUAWYGZOVFIAB-UHFFFAOYSA-N 0.000 description 1
- KMYODCXGVMLGJT-UHFFFAOYSA-N 2-quinolin-3-ylethanol Chemical compound C1=CC=CC2=CC(CCO)=CN=C21 KMYODCXGVMLGJT-UHFFFAOYSA-N 0.000 description 1
- LQJBNNIYVWPHFW-UHFFFAOYSA-N 20:1omega9c fatty acid Natural products CCCCCCCCCCC=CCCCCCCCC(O)=O LQJBNNIYVWPHFW-UHFFFAOYSA-N 0.000 description 1
- BMYNFMYTOJXKLE-UHFFFAOYSA-N 3-azaniumyl-2-hydroxypropanoate Chemical compound NCC(O)C(O)=O BMYNFMYTOJXKLE-UHFFFAOYSA-N 0.000 description 1
- QSBYPNXLFMSGKH-UHFFFAOYSA-N 9-Heptadecensaeure Natural products CCCCCCCC=CCCCCCCCC(O)=O QSBYPNXLFMSGKH-UHFFFAOYSA-N 0.000 description 1
- 102000009027 Albumins Human genes 0.000 description 1
- 108010088751 Albumins Proteins 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 239000004475 Arginine Substances 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 238000009631 Broth culture Methods 0.000 description 1
- 102000016938 Catalase Human genes 0.000 description 1
- 108010053835 Catalase Proteins 0.000 description 1
- HZZVJAQRINQKSD-UHFFFAOYSA-N Clavulanic acid Natural products OC(=O)C1C(=CCO)OC2CC(=O)N21 HZZVJAQRINQKSD-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 101100001678 Emericella variicolor andM gene Proteins 0.000 description 1
- 230000005526 G1 to G0 transition Effects 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- KDXKERNSBIXSRK-UHFFFAOYSA-N Lysine Natural products NCCCCC(N)C(O)=O KDXKERNSBIXSRK-UHFFFAOYSA-N 0.000 description 1
- 239000004472 Lysine Substances 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 241000186367 Mycobacterium avium Species 0.000 description 1
- 241000186366 Mycobacterium bovis Species 0.000 description 1
- 241000186365 Mycobacterium fortuitum Species 0.000 description 1
- 241000186362 Mycobacterium leprae Species 0.000 description 1
- 241001646725 Mycobacterium tuberculosis H37Rv Species 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 150000001204 N-oxides Chemical class 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- 239000005642 Oleic acid Substances 0.000 description 1
- ZQPPMHVWECSIRJ-UHFFFAOYSA-N Oleic acid Natural products CCCCCCCCC=CCCCCCCCC(O)=O ZQPPMHVWECSIRJ-UHFFFAOYSA-N 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical class N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 1
- 229930189077 Rifamycin Natural products 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- MUMGGOZAMZWBJJ-DYKIIFRCSA-N Testostosterone Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 MUMGGOZAMZWBJJ-DYKIIFRCSA-N 0.000 description 1
- KVPNBBUKZBDNBJ-OIXVRUHCSA-N [(7S,9E,11S,12R,13S,14R,15R,16R,17S,18S,19E,21Z)-2,15,17,27,29-pentahydroxy-11-methoxy-3,7,12,14,16,18,22-heptamethyl-26-[(E)-(4-methylpiperazin-1-yl)iminomethyl]-6,23-dioxo-8,30-dioxa-24-azatetracyclo[23.3.1.14,7.05,28]triaconta-1(29),2,4,9,19,21,25,27-octaen-13-yl] acetate pyrazine-2-carboxamide pyridine-4-carbohydrazide Chemical compound NC(=O)c1cnccn1.NNC(=O)c1ccncc1.CO[C@H]1\C=C\O[C@@]2(C)Oc3c(C2=O)c2c(O)c(\C=N\N4CCN(C)CC4)c(NC(=O)\C(C)=C/C=C/[C@H](C)[C@H](O)[C@@H](C)[C@@H](O)[C@@H](C)[C@H](OC(C)=O)[C@@H]1C)c(O)c2c(O)c3C KVPNBBUKZBDNBJ-OIXVRUHCSA-N 0.000 description 1
- ZWBTYMGEBZUQTK-PVLSIAFMSA-N [(7S,9E,11S,12R,13S,14R,15R,16R,17S,18S,19E,21Z)-2,15,17,32-tetrahydroxy-11-methoxy-3,7,12,14,16,18,22-heptamethyl-1'-(2-methylpropyl)-6,23-dioxospiro[8,33-dioxa-24,27,29-triazapentacyclo[23.6.1.14,7.05,31.026,30]tritriaconta-1(32),2,4,9,19,21,24,26,30-nonaene-28,4'-piperidine]-13-yl] acetate Chemical compound CO[C@H]1\C=C\O[C@@]2(C)Oc3c(C2=O)c2c4NC5(CCN(CC(C)C)CC5)N=c4c(=NC(=O)\C(C)=C/C=C/[C@H](C)[C@H](O)[C@@H](C)[C@@H](O)[C@@H](C)[C@H](OC(C)=O)[C@@H]1C)c(O)c2c(O)c3C ZWBTYMGEBZUQTK-PVLSIAFMSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 208000036981 active tuberculosis Diseases 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 150000001413 amino acids Chemical class 0.000 description 1
- 125000003277 amino group Chemical group 0.000 description 1
- 150000003863 ammonium salts Chemical class 0.000 description 1
- 229960003022 amoxicillin Drugs 0.000 description 1
- LSQZJLSUYDQPKJ-NJBDSQKTSA-N amoxicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=C(O)C=C1 LSQZJLSUYDQPKJ-NJBDSQKTSA-N 0.000 description 1
- 230000000844 anti-bacterial effect Effects 0.000 description 1
- 230000002365 anti-tubercular Effects 0.000 description 1
- 238000013459 approach Methods 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- ODKSFYDXXFIFQN-UHFFFAOYSA-N arginine Natural products OC(=O)C(N)CCCNC(N)=N ODKSFYDXXFIFQN-UHFFFAOYSA-N 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 description 1
- 244000052616 bacterial pathogen Species 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-N benzenesulfonic acid Chemical compound OS(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-N 0.000 description 1
- 229940092714 benzenesulfonic acid Drugs 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 239000006172 buffering agent Substances 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 159000000007 calcium salts Chemical class 0.000 description 1
- 238000002512 chemotherapy Methods 0.000 description 1
- 229960003405 ciprofloxacin Drugs 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- AGOYDEPGAOXOCK-KCBOHYOISA-N clarithromycin Chemical compound O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=O)[C@H](C)C[C@](C)([C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)OC)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 AGOYDEPGAOXOCK-KCBOHYOISA-N 0.000 description 1
- 229960002626 clarithromycin Drugs 0.000 description 1
- HZZVJAQRINQKSD-PBFISZAISA-N clavulanic acid Chemical compound OC(=O)[C@H]1C(=C/CO)/O[C@@H]2CC(=O)N21 HZZVJAQRINQKSD-PBFISZAISA-N 0.000 description 1
- 229960003324 clavulanic acid Drugs 0.000 description 1
- WDQPAMHFFCXSNU-BGABXYSRSA-N clofazimine Chemical compound C12=CC=CC=C2N=C2C=C(NC=3C=CC(Cl)=CC=3)C(=N/C(C)C)/C=C2N1C1=CC=C(Cl)C=C1 WDQPAMHFFCXSNU-BGABXYSRSA-N 0.000 description 1
- 229960004287 clofazimine Drugs 0.000 description 1
- 239000003086 colorant Substances 0.000 description 1
- 238000002648 combination therapy Methods 0.000 description 1
- 239000000625 cyclamic acid and its Na and Ca salt Substances 0.000 description 1
- 125000004210 cyclohexylmethyl group Chemical group [H]C([H])(*)C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- HCAJEUSONLESMK-UHFFFAOYSA-N cyclohexylsulfamic acid Chemical compound OS(=O)(=O)NC1CCCCC1 HCAJEUSONLESMK-UHFFFAOYSA-N 0.000 description 1
- 230000002498 deadly effect Effects 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 1
- 231100000676 disease causative agent Toxicity 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 238000012377 drug delivery Methods 0.000 description 1
- 208000015355 drug-resistant tuberculosis Diseases 0.000 description 1
- 239000003995 emulsifying agent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 125000002587 enol group Chemical group 0.000 description 1
- 210000000981 epithelium Anatomy 0.000 description 1
- CCIVGXIOQKPBKL-UHFFFAOYSA-M ethanesulfonate Chemical compound CCS([O-])(=O)=O CCIVGXIOQKPBKL-UHFFFAOYSA-M 0.000 description 1
- 238000013401 experimental design Methods 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 238000001640 fractional crystallisation Methods 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- 229960004275 glycolic acid Drugs 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 230000009036 growth inhibition Effects 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 239000002054 inoculum Substances 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- QXJSBBXBKPUZAA-UHFFFAOYSA-N isooleic acid Natural products CCCCCCCC=CCCCCCCCCC(O)=O QXJSBBXBKPUZAA-UHFFFAOYSA-N 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 125000000468 ketone group Chemical group 0.000 description 1
- 150000002576 ketones Chemical class 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 230000003902 lesion Effects 0.000 description 1
- 231100000518 lethal Toxicity 0.000 description 1
- 230000001665 lethal effect Effects 0.000 description 1
- 238000004811 liquid chromatography Methods 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 239000003120 macrolide antibiotic agent Substances 0.000 description 1
- 229940041033 macrolides Drugs 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- GRVDJDISBSALJP-UHFFFAOYSA-N methyloxidanyl Chemical group [O]C GRVDJDISBSALJP-UHFFFAOYSA-N 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 238000012544 monitoring process Methods 0.000 description 1
- DUWWHGPELOTTOE-UHFFFAOYSA-N n-(5-chloro-2,4-dimethoxyphenyl)-3-oxobutanamide Chemical compound COC1=CC(OC)=C(NC(=O)CC(C)=O)C=C1Cl DUWWHGPELOTTOE-UHFFFAOYSA-N 0.000 description 1
- 239000013642 negative control Substances 0.000 description 1
- 239000002547 new drug Substances 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 description 1
- 125000004433 nitrogen atom Chemical group N* 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- ZQPPMHVWECSIRJ-KTKRTIGZSA-N oleic acid Chemical compound CCCCCCCC\C=C/CCCCCCCC(O)=O ZQPPMHVWECSIRJ-KTKRTIGZSA-N 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 230000008520 organization Effects 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- LSQZJLSUYDQPKJ-UHFFFAOYSA-N p-Hydroxyampicillin Natural products O=C1N2C(C(O)=O)C(C)(C)SC2C1NC(=O)C(N)C1=CC=C(O)C=C1 LSQZJLSUYDQPKJ-UHFFFAOYSA-N 0.000 description 1
- WLJNZVDCPSBLRP-UHFFFAOYSA-N pamoic acid Chemical compound C1=CC=C2C(CC=3C4=CC=CC=C4C=C(C=3O)C(=O)O)=C(O)C(C(O)=O)=CC2=C1 WLJNZVDCPSBLRP-UHFFFAOYSA-N 0.000 description 1
- 230000001717 pathogenic effect Effects 0.000 description 1
- 235000019371 penicillin G benzathine Nutrition 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002335 preservative effect Effects 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 230000005180 public health Effects 0.000 description 1
- 229940107700 pyruvic acid Drugs 0.000 description 1
- 150000007660 quinolones Chemical class 0.000 description 1
- 229960000885 rifabutin Drugs 0.000 description 1
- BTVYFIMKUHNOBZ-QXMMDKDBSA-N rifamycin s Chemical class O=C1C(C(O)=C2C)=C3C(=O)C=C1NC(=O)\C(C)=C/C=C\C(C)C(O)C(C)C(O)C(C)C(OC(C)=O)C(C)C(OC)\C=C/OC1(C)OC2=C3C1=O BTVYFIMKUHNOBZ-QXMMDKDBSA-N 0.000 description 1
- 229940081192 rifamycins Drugs 0.000 description 1
- 229960002599 rifapentine Drugs 0.000 description 1
- 230000000630 rising effect Effects 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 1
- 229920006395 saturated elastomer Polymers 0.000 description 1
- 238000010956 selective crystallization Methods 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 238000009097 single-agent therapy Methods 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- 229960004954 sparfloxacin Drugs 0.000 description 1
- DZZWHBIBMUVIIW-DTORHVGOSA-N sparfloxacin Chemical compound C1[C@@H](C)N[C@@H](C)CN1C1=C(F)C(N)=C2C(=O)C(C(O)=O)=CN(C3CC3)C2=C1F DZZWHBIBMUVIIW-DTORHVGOSA-N 0.000 description 1
- 239000003381 stabilizer Substances 0.000 description 1
- 238000011301 standard therapy Methods 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 230000031068 symbiosis, encompassing mutualism through parasitism Effects 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 229940051104 testim Drugs 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- 229960003231 thioacetazone Drugs 0.000 description 1
- SRVJKTDHMYAMHA-WUXMJOGZSA-N thioacetazone Chemical compound CC(=O)NC1=CC=C(\C=N\NC(N)=S)C=C1 SRVJKTDHMYAMHA-WUXMJOGZSA-N 0.000 description 1
- 231100000331 toxic Toxicity 0.000 description 1
- 230000002588 toxic effect Effects 0.000 description 1
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 1
- 235000012431 wafers Nutrition 0.000 description 1
- 230000003442 weekly effect Effects 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
- A61P31/06—Antibacterial agents for tuberculosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Definitions
- the present invention relates to the use of substituted quinoline derivatives for inhibiting the growth of drug resistant Mycobacterium strains including growth inhibition of multi drug resistant Mycobacterium strains.
- the substituted quinoline derivatives can thus be used for the treatment or the prevention of Mycobacterial diseases caused by drug resistant, particularly multi drug resistant Mycobacteria. More in particular the present quinoline derivatives can be used for the treatment or the prevention of Mycobacterial diseases caused by drug resistant including multi drug resistant Mycobacterium tuberculosis.
- the present invention also relates to a combination of (a) a substituted quinoline derivative according to the present invention and (b) one or more other antimycobacterial agents.
- Mycobacterium tuberculosis is the causative agent of tuberculosis (TB), a serious and potentially fatal infection with a world- wide distribution.
- TB tuberculosis
- Estimates from the World Health Organization indicate that more than 8 million people contract TB each year, and 2 million people die from tuberculosis yearly. In the last decade, TB cases have grown 20% worldwide with the highest burden in the most impoverished communities. If these trends continue, TB incidence will increase by 41% in the next twenty years. Fifty years since the introduction of an effective chemotherapy, TB remains after AIDS, the leading infectious cause of adult mortality in the world. Complicating the TB epidemic is the rising tide of multi-drug- resistant strains, and the deadly symbiosis with HTV. People who are HIV-positive and infected with TB are 30 times more likely to develop active TB than people who are KV-negative and TB is responsible for the death of one out of every three people with HTV/AIDS worldwide
- MDR-TB multi drug- resistant strains
- MDR-TB multi drug- resistant strains
- isomazid and rifampin the most effective drugs of the four-drug standard, isomazid and rifampin.
- MDR-TB is lethal when untreated and can not be adequately treated through the standard therapy, so treatment requires up to 2 years of "second-line" drugs. These drugs are often toxic, expensive and marginally effective.
- infectious MDR-TB patients continue to spread the disease, producing new infections with MDR-TB strains. There is a high medical need for drugs which demonstrate activity against resistant and/or MDR strains.
- a drug resistant Mycobacterium is a Mycobacterium which is no longer susceptible to at least one previously effective drug; which has developed the ability to withstand antibiotic attack by at least one previously effective drug.
- a drug resistant strain may relay that ability to withstand to its progeny. Said resistance may be due to random genetic mutations in the bacterial cell that alters its sensitivity to a single drug or to different drugs.
- MDR tuberculosis is a specific form of drug resistant tuberculosis due to a bacterium resistant to at least isoniazid and rifampicin (with or without resistance to other drugs), which are at present the two most powerful anti-TB drugs.
- drug resistant includes multi drug resistant.
- the substituted quinoline derivatives of the present invention are very useful for inhibiting growth of drug resistant, in particular multi drug resistant, Mycobacteria and therefore useful for the treatment of diseases caused by drug resistant, in particular multi drug resistant, Mycobacteria, particularly those diseases caused by drug resistant, in particular multi drug resistant, pathogenic Mycobacterium ⁇ f.) tuberculosis, M. bovis, M. avium, M.fortuitum, M. leprae andM. marinum, more particularly Mycobacterium tuberculosis.
- substituted quinoline derivatives relating to the present invention were already disclosed in WO 2004/011436. Said document discloses the antirnycobacterial property of the substituted quinoline derivatives against sensitive, susceptible Mycobacterium strains but is silent on their activity against drug resistant, in particular multi drug resistant, Mycobacteria.
- the present invention relates to the use of a substituted quinoline derivative for the preparation of a medicament for the treatment of a warm-blooded mammal infected with a drug resistant Mycobacterium strain wherein the substituted quinoline derivative is a compound according to Formula (la) or Formula (lb)
- R 1 is hydrogen, halo, haloalkyl, cyano, hydroxy, Ar, Het, alkyl, alkyloxy, alkylthio, alkyloxyalkyl, alkylthioalkyl, ⁇ r-alkyl or di(Ar)alkyl ; is an integer equal to 1, 2, 3 or 4 ;
- R is hydrogen, hydroxy, mercapto, alkyloxy, alkyloxyalkyloxy, alkylthio, mono or di(alkyl)amino or a radical of formula wherein Y is CH 2 , O, S, ⁇ H or ⁇ -alkyl ;
- R j is alkyl, Ar, Ar-alkyl, Het or Het-alkyl; q is an integer equal to zero, 1 , 2, 3 or 4 ;
- R 4 and R 5 each independently are hydrogen, alkyl or benzyl; or
- R 4 and R 5 together and including the N to which they are attached may form a radical selected from the group of pyrrolidinyl, 2H-pyrrolyl, 2-pyrrolinyl, 3-pyrrolinyl, pyrrolyl, imidazolidinyl, pyrazolidinyl, 2-imidazolinyl, 2-pyrazolinyl, imidazolyl, pyrazolyl, triazolyl, piperidinyl, pyridinyl, piperazinyl, imidazolidinyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl, morpholinyl and thiomorpholinyl, optionally substituted with alkyl, halo, haloalkyl, hydroxy, alkyloxy, amino, mono- or dialkylamino, alkylthio, alkyloxyalkyl, alkylthioalkyl and pyrimidinyl ;
- R' is hydrogen, alkyl, Ar or Het ;
- R is hydrogen or alkyl ;
- R 9 is oxo ;
- alkyl is a straight or branched saturated hydrocarbon radical having from 1 to 6 carbon atoms ; or is a cyclic saturated hydrocarbon radical having from 3 to 6 carbon atoms ; or is a a cyclic saturated hydrocarbon radical having from 3 to 6 carbon atoms attached to a straight or branched saturated hydrocarbon radical having from 1 to 6 carbon atoms ; wherein each carbon atom can be optionally substituted with halo, hydroxy, alkyloxy or oxo ;
- Ar is a homocycle selected from the group of phenyl, naphthyl, acenaphthyl, tetrahydronaphthyl, each optionally substituted with 1, 2 or 3 substituents, each substituent independently selected from the group of hydroxy, halo, cyano, nitro, amino, mono- or dialkylamino, alkyl, haloalkyl, alkyloxy
- Het is a monocyclic heterocycle selected from the group of N-phenoxypiperidinyl, pyrrolyl, pyrazolyl, imidazolyl, furanyl, thienyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, pyridinyl, pyrimidinyl, pyrazinyl and pyridazinyl; or a bicyclic heterocycle selected from the group of quinolinyl, quinoxalinyl, indolyl, beruimidazolyl, benzoxazolyl, benzisoxazolyl, benzothiazolyl, benzisothiazolyl, benzofuranyl, benzothienyl, 2 J 3-dihydrobenzo[l,4]dioxinyl or benzo[l,3]dioxolyl ; each monocyclic and bicyclic heterocycle may optionally be substituted on a carbon
- the present invention relates to the use of a substituted quinoline derivative for the preparation of a medicament for the treatment of an infection with a drug resistant Mycobacterium strain wherein the substituted quinoline derivative is a compound according to Formula (la) or Formula (lb).
- the present invention also concerns a method of treating a patient suffering from , or at risk of, an infection with a drug resistant mycobacterial strain, which comprises administering to the patient a therapeutically effective amount of a compound or pharmaceutical composition according to the invention.
- the compounds according to Formula (la) and (lb) are interrelated in that e.g. a compound according to Formula (lb), with R 9 equal to oxo is the tautomeric equivalent of a compound according to Formula (la) with R 2 equal to hydroxy (keto-enol tautomerism).
- alkyl is a straight or branched saturated hydrocarbon radical having from 1 to 6 carbon atoms ; or is a cyclic saturated hydrocarbon radical having from 3 to 6 carbon atoms ; or is a cyclic saturated hydrocarbon radical having from 3 to 6 carbon atoms attached to a straight or branched saturated hydrocarbon radical having from 1 to 6 carbon atoms ; wherein each carbon atom can be optionally substituted with halo, hydroxy, alkyloxy or oxo.
- alkyl is methyl, ethyl or cyclohexylmethyl.
- Ar is a homocycle selected from the group of phenyl, naphthyl, acenaphthyl, tetrahydronaphthyl, each optionally substituted with 1, 2 or 3 substituents, each substituent independently selected from the group of hydroxy, halo, cyano, nitro, arnino, mono- or dialkylamino, alkyl, haloalkyl, alkyloxy, haloalkyloxy, carboxyl, alkyloxycarbonyl, aminocarbonyl, morpholinyl and mono- or dialkylaminocarbonyl.
- Ar is naphthyl or phenyl, each optionally substituted with 1 or 2 halo substituents.
- Het is a monocyclic heterocycle selected from the group of N-phenoxypiperidinyl, pyrrolyl, pyrazolyl, imidazolyl, furanyl, thienyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, pyridinyl, pyrimidinyl, pyrazinyl and pyridazinyl; or a bicyclic heterocycle selected from the group of quinolinyl, quinoxalinyl, indolyl, berizimidazolyl, benzoxazolyl, benzisoxazolyl, benzothiazolyl, benzisothiazolyl, benzofuranyl, benzothienyl, 2,3-dihydrobenzo[l,4]dioxinyl or benzo[l,3]dioxolyl ; each monocyclic and bicyclic heterocycle may optionally be substituted
- halo is a substituent selected from the group of fluoro, chloro, bromo and iodo and haloalkyl is a straight or branched saturated hydrocarbon radical having from 1 to 6 carbon atoms or a cyclic saturated hydrocarbon radical having from 3 to 6 carbon atoms, wherein one or more carbon atoms are substituted with one or more halo-atoms.
- halo is bromo, fluoro or chloro and preferably, haloalkyl is trifluoromethyl.
- each halo atom may be the same or different.
- the invention relates to the use as defined hereinabove of compounds of Formula (la)
- R 1 is hydrogen, halo, haloalkyl, cyano, hydroxy, Ar, Het, alkyl, alkyloxy, alkylthio, alkyloxyalkyl, alkylthioalkyl, Ar-alkyl or di(Ar)alkyl ; is an integer equal to 1, 2, 3 or 4 ; is hydrogen, hydroxy, mercapto, alkyloxy, alkyloxyalkyloxy, alkylthio, mono or di(alkyl)amino or a radical of formula wherein Y is CH 2 , O, S, NH orN-alkyl ; is alkyl, Ar, Ar-alkyl, Het or Het-alkyl; q is an integer equal to zero, 1, 2, 3 or 4 ;
- R 4 and R 5 each independently are hydrogen, alkyl or benzyl; or R 4 and R 5 together and including the N to which they are attached may form a radical selected from the group of pyrrolidinyl, 2H-pyrrolyl, 2-pyrrolinyl, 3-pyrrolinyl, pyrrolyl, imidazolidinyl, pyrazolidinyl, 2-imidazolinyl, 2-pyrazolinyl, imidazolyl, pyrazolyl, triazolyl, piperidinyl, pyridinyl, piperazinyl, imidazolidinyl, pyridazinyl, pvrimidinyl, pyrazinyl, triazinyl, morpholinyl and thiomorpholinyl, optionally substituted with alkyl, halo, haloalkyl, hydroxy, alkyloxy, amino, mono- or dialkylamino, alkylthio, alkyloxyalky
- R 7 is hydrogen, alkyl, Ar or ⁇ et ;
- R 8 is hydrogen or alkyl
- R 9 is oxo ;
- alkyl is a straight or branched saturated hydrocarbon radical having from 1 to 6 carbon atoms ; or is a cyclic saturated hydrocarbon radical having from 3 to 6 carbon atoms ; or is a a cyclic saturated hydrocarbon radical having from 3 to 6 carbon atoms attached to a straight or branched saturated hydrocarbon radical having from 1 to 6 carbon atoms ; wherein each carbon atom can be optionally substituted with halo, hydroxy, alkyloxy or oxo ;
- Ar is a homocycle selected from the group of phenyl, naphthyl, acenaphthyl, tetrahydronaphthyl, each optionally substituted with 1, 2 or 3 substituents, each substituent independently selected from the group of hydroxy, halo, cyano, nitro, amino, mono- or dialkylamino, alkyl, haloalkyl, alkyloxy, haloalkyloxy, carboxyl, alkyloxycarbonyl, aminocarbonyl, morpholinyl and mono- or dialkylaminocarbonyl ; Het is a monocyclic heterocycle selected from the group of N-phenoxypiperidinyl, pyrrolyl, pyrazolyl, imidazolyl, furanyl, thienyl, oxazolyl, isoxazolyl, thiazolyl, isothiazolyl, pyridinyl, pyrimidinyl, pyrazinyl and
- the invention also relates to the use as defined hereinabove of compounds of Formula
- R 1 is hydrogen, halo, haloalkyl, cyano, hydroxy, Ar, Het, alkyl, alkyloxy, alkylthio, alkyloxyalkyl, alkylthioalkyl, Ar-alkyl or di(Ar)alkyl ;
- p is an integer equal to 1, 2, 3 or 4 ;
- R 2 is hydrogen, hydroxy, mercapto, alkyloxy, alkyloxyalkyloxy, alkylthio, mono or di(alkyl)amino or a radical of formula wherein Y is CH 2 , O, S, NH or N-alkyl ;
- R 3 is alkyl, Ar, Ar-alkyl, Het or Het-alkyl; q is an integer equal to zero, 1 , 2, 3 or 4 ; R 4 and R 5 each independently are hydrogen, alkyl or benzyl; or
- R 4 and R 5 together and including the ⁇ to which they are attached may form a radical selected from the group of pyrrolidinyl, 2H-pyrrolyl, 2-pyrrolinyl, 3-pyrrolinyl, pyrrolyl, imidazolidinyl, pyrazolidinyl, 2-imidazolinyl, 2-pyrazolinyl, imidazolyl, pyrazolyl, triazolyl, piperidinyl, pyridinyl, piperazinyl, imidazolidinyl, pyridazinyl, pyrimidinyl, pyrazinyl, triazinyl, morpholinyl and thiomorpholinyl, optionally substituted with alkyl, halo, haloalkyl, hydroxy, alkyloxy, amino, mono- or dklkylamino, alkylthio, alkyloxyalkyl, alkylthioalkyl and pyrimidinyl
- R 6 is hydrogen, halo, haloalkyl, hydroxy, Ar, alkyl, alkyloxy, alkylthio, alkyloxyalkyl, alkylthioalkyl, Ar-alkyl or di(Ar)alkyl ; r is an integer equal to 1, 2, 3, 4 or 5 ; and
- R 7 is hydrogen, alkyl, Ar or ⁇ et ;
- R 8 is hydrogen or alkyl
- the invention also relates to the use as defined hereinabove of compounds of Formula (la) or (lb) wherein : R 1 is hydrogen, halo, cyano, Ar, Het, alkyl, and alkyloxy ; p is an integer equal to zero, 1 , 2, 3 or 4 ;
- R 2 is hydrogen, hydroxy, alkyloxy, alkyloxyalkyloxy, alkylthio or a radical of formula wherein Y is O ;
- R 3 is alkyl, Ar, Ar-alkyl or Het ; q is an integer equal to zero, 1 , 2, or 3 ; R 4 and R 5 each independently are hydrogen, alkyl or benzyl; or R 4 and R 5 together and including the N to which they are attached may form a radical selected from the group of pyrrolidinyl, imidazolyl, triazolyl, piperidinyl, piperazinyl, pyrazinyl,morpholinyl and thiomorpholinyl, optionally substituted with alkyl and pyrimidinyl ;
- R 8 is hydrogen or alkyl
- R 9 is oxo ;
- alkyl is a straight or branched saturated hydrocarbon radical having from 1 to 6 carbon atoms ; or is a cyclic saturated hydrocarbon radical having from 3 to 6 carbon atoms ; or is a a cyclic saturated hydrocarbon radical having from 3 to 6 carbon atoms attached to a straight or branched saturated hydrocarbon radical having from 1 to 6 carbon atoms ; wherein each carbon atom can be optionally substituted with halo or hydroxy ;
- Ar is a homocycle selected from the group of phenyl, naphthyl, acenaphthyl, .
- Het is a monocyclic heterocycle selected from the group of N-phenoxypiperidinyl, furanyl, thienyl, pyridinyl, pyrimidinyl ; or a bicyclic heterocycle selected from the group of benzothienyl, 2,3-dihydrobenzo[l,4]dioxinyl or benzo[l,3]- dioxolyl; each monocyclic and bicyclic heterocycle may optionally be substituted on a carbon atom with 1, 2 or 3 alkyl substituents ; and halo is a substituent selected from the group of fluoro, chloro and bromo.
- R 1 is hydrogen, halo, Ar, alkyl or alkyloxy. More preferably, R 1 is halo. Most preferably, R 1 is bromo.
- p is equal to 1.
- R 2 is hydrogen, alkyloxy or alkylthio. More preferably, R 2 is alkyloxy, in particular C alkyloxy. Most preferably, R 2 is methyloxy.
- d ⁇ alkyl is a straight or branched saturated hydrocarbon radical having from 1 to 4 carbon atoms such as for example methyl, ethyl, propyl, 2-methyl-ethyl and the like.
- R 3 is naphthyl, phenyl or thienyl, each optionally substituted with 1 or 2 substituents, that substituent preferably being a halo or haloalkyl, most preferably being a halo. More preferably, R 3 is naphthyl or phenyl, each optionally substituted with halo, preferably 3 -fluoro. Even more preferably, R 3 is naphthyl or phenyl. Most preferably, R 3 is naphthyl.
- q is equal to zero, 1 or 2. More preferably, q is equal to I.
- R 4 and R 5 each independently are hydrogen or alkyl, in particular hydrogen or C M alkyl, more in particular more preferably hydrogen, methyl or ethyl, most preferably methyl.
- d ⁇ alkyl is a straight or branched saturated hydrocarbon radical having from 1 to 4 carbon atoms such as for example methyl, ethyl, propyl, 2-methyl-ethyl and the like.
- R 4 and R 5 together and including the N to which they are attached form a radical selected from the group of imidazolyl, triazolyl, piperidinyl, piperazinyl and thiomorpholinyl, optionally substituted with alkyl, halo, haloalkyl, hydroxy, alkyloxy, alkylthio, alkyloxyalkyl or alkylthioalkyl, preferably substituted with alkyl, most preferably substituted with methyl or ethyl.
- R 6 is hydrogen, alkyl or halo. Most preferably, R 6 is hydrogen. Preferably r is 0, 1 or 2.
- R 7 is hydrogen or methyl, more preferably hydrogen.
- R 8 is alkyl, preferably methyl and R 9 is oxygen.
- An interesting group of compounds are the compounds according to formula (la), the pharmaceutically acceptable acid or base addition salts thereof, the stereochemically isomeric forms thereof, the tautomeric forms thereof or the N-oxide forms thereof.
- the compound is one of the following :
- R 1 is hydrogen, halo, Ar, alkyl or alkyloxy
- p 1
- R 2 is hydrogen, alkyloxy or alkylthio
- R 3 is naphthyl, phenyl or thienyl, each optionally substituted with 1 or 2 substituents selected from the group of halo and haloalkyl
- q 0, 1, 2 or 3
- R 4 and R 5 each independently are hydrogen or alkyl or R 4 and R 5 together and including the ⁇ to which they are attached form a radical selected from the group of imidazolyl, triazolyl, piperidinyl, piperazinyl and thiomorpholinyl
- R 6 is hydrogen, alkyl or halo
- r is equal to 0 or 1
- R 7 is hydrogen.
- the compound is : o l-(6-bromo-2-methoxy-qumolm-3-yl)-2-(3,5-difluoro-phenyl)-4-dimethylamino-l- phenyl-butan-2-ol ; o 1 -(6-bromo-2-methoxy-qumolm-3-yl)-4-dimethylamino-2-naphthalen- 1 -yl- 1 - phenyl-butan-2-ol corresponding to 6-bromo- ⁇ -[2-(dimethylamino)ethyl]-2- methoxy- ⁇ -l-naphfhalenyl- ⁇ -phenyl-3-quinolineethanol; o 1 -(6-bromo-2-methoxy-quinolin-3-yl)-2-(2, 5-difluoro-phenyl)-4-dime ylatnino- 1 - phenyl-butan-2-o
- the compound is o l-(6-bromo-2-methoxy-qumolm-3-yl)-4-dimethylamino-2-(3-fluoro-phenyl)-l- phenyl-butan-2-ol; o 1 -(6-bromo-2-methoxy-qumolm-3-yl)-4-dimethylamino-2-phenyl- 1 -phenyl-butan- 2-ol;
- the compound is ( ⁇ S, ⁇ R)-6-bromo- ⁇ -[2- (djjnethylammo)ethyl]-2-methoxy- ⁇ -l-naphthalenyl- ⁇ -phenyl-3-quinolineethanol which corresponds to (lR,2S)-l-(6-bromo-2-me oxy-qumolm-3-yl)-4- ⁇ ethylamino- 2-na ⁇ hthalen-l-yl-l-phenyl-
- the pharmaceutically acceptable acid addition salts are defined to comprise the therapeutically active non-toxic acid addition salt forms which the compounds according to either Formula (la) and (lb) are able to form.
- Said acid addition salts can be obtained by treating the base form of the compounds according to either Formula (la) and (lb) with appropriate acids, for example inorganic acids, for example hydrohalic acid, in particular hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid and phosphoric acid ; organic acids, for example acetic acid, hydroxyacetic acid, propanoic acid, lactic acid, pyruvic acid, oxalic acid, malonic acid, succinic acid, maleic acid, fumaric acid, malic acid, tartaric acid, citric acid, methanesulfonic acid, ethanesulfonic acid, benzenesulfonic acid, p-toluenesulfonic acid, cyclamic acid, salicyclic acid, p-aminosalicy
- the compounds according to either Formula (la) and (lb) containing acidic protons may also be converted into their therapeutically active non-toxic base addition salt forms by treatment with appropriate orgamc and inorganic bases.
- Appropriate base salts forms comprise, for example, the ammonium salts, the alkaline and earth alkaline metal salts, in particular lithium, sodium, potassium, magnesium and calcium salts, salts with organic bases, e.g. the benzathine, N-methyl-D-glucamine, hybrarnine salts, and salts with amino acids, for example arginine and lysine.
- addition salt as used in the framework of this application also comprises the solvates which the compounds according to either Formula (la) and (lb) as well as the salts thereof, are able to form.
- Such solvates are, for example, hydrates and alcoholates.
- stereochemically isomeric forms as used herein defines all possible isomeric forms which the compounds of either Formula (la) and (lb) may possess. Unless otherwise mentioned or indicated, the chemical designation of compounds denotes the mixture of all possible stereochemically isomeric forms, said mixtures containing all diastereomers and enantiomers of the basic molecular structure . More in particular, stereogenic centers may have the R- or S-configuration; substituents on bivalent cyclic (partially) saturated radicals may have either the cis- or trans- configuration. Stereochemically isomeric forms of the compounds of either Formula (la) and (lb) are obviously intended to be embraced within the scope of this invention.
- an R or S descriptor is assigned (based on Cahn-Ingold-Prelog sequence rule) to the lowest-numbered chiral center, the reference center.
- the configuration of the second stereogenic center is indicated using relative descriptors [R *,R * ] or [R *,S* ⁇ , where R * is always specified as the reference center and [R*R*] indicates centers with the same chirality and [R*,S*] indicates centers of unlike chirality.
- R * is always specified as the reference center
- [R*R*] indicates centers with the same chirality
- [R*,S*] indicates centers of unlike chirality.
- the stereo descriptor would be specified as S-[R*,S* ⁇ .
- the position of the highest priority substituent on the asymmetric carbon atom in the ring system having the lowest ring number is arbitrarily always in the " ⁇ " position of the mean plane determined by the ring system.
- the position of the highest priority substituent on the other asymmetric carbon atom in the ring system relative to the position of the highest priority substituent on the reference atom is denominated “ ", if it is on the same side of the mean plane determined by the ring system, or " ⁇ ", if it is on the other side of the mean plane determined by the ring system.
- Compounds of either Formula (la) and (lb) and some of the intermediate compounds invariably have at least two stereogenic centers in their structure which may lead to at least 4 stereochemically different structures.
- the tautomeric forms of the compounds of either Formula (la) and (lb) are meant to comprise those compounds of either Formula (la) and (lb) wherein e.g. an enol group is converted into a keto group (keto-enol tautomerism).
- N-oxide forms of the compounds according to either Formula (la) and (lb) are meant to comprise those compounds of either Formula (la) and (lb) wherein one or several tertiary nitrogen atoms are oxidized to the so-called N-oxide.
- the compounds of either Formula (la) and (lb) as prepared in the processes described below may be synthesized in the form of racemic mixtures of enantiomers which can be separated from one another following art-known resolution procedures.
- the racemic compounds of either Formula (la) and (Tb) may be converted into the corresponding diastereomeric salt forms by reaction with a suitable chiral acid. Said diastereomeric salt forms are subsequently separated, for example, by selective or fractional crystallization and the enantiomers are liberated therefrom by alkali.
- An alternative manner of separating the enantiomeric forms of the compounds of either Formula (la) and (lb) involves liquid chromatography using a chiral stationary phase.
- Said pure stereochemically isomeric forms may also be derived from the corresponding pure stereochemically isomeric forms of the appropriate starting materials, provided that the reaction occurs stereospecifically.
- said compound will be synthesized by stereospecific methods of preparation. These methods will advantageously employ enantiomerically pure starting materials.
- the invention also comprises derivative compounds (usually called "pro-drugs") of the pharmacologically-active compounds according to the invention, which are degraded in vivo to yield the compounds according to the invention.
- Pro-drugs are usually (but not always) of lower potency at the target receptor than the compounds to which they are degraded.
- Pro-drugs are particularly useful when the desired compound has chemical or physical properties that make its administration difficult or inefficient. For example, the desired compound may be only poorly soluble, it may be poorly transported across the mucosal epithelium, or it may have an undesirably short plasma half-life. Further discussion on pro-drugs may be found in Stella, V. J. et al., "Prodrugs", Drug Delivery Systems, 1985, pp. 112-176, and Drugs, 1985, 29, pp. 455-473.
- Pro-drugs forms of the pharmacologically-active compounds according to the invention will generally be compounds according to either Formula (la) and (lb), the pharmaceutically acceptable acid or base addition salts thereof, the stereochemically isomeric forms thereof, the tautomeric forms thereof and the N-oxide forms thereof, having an acid group which is esterified or amidated. Included in such esterified acid groups are groups of the formula -COOR * , where R x is a Ci- ⁇ alkyl, phenyl, benzyl or one of the following groups :
- Amidated groups include groups of the formula - CO ⁇ R y R z , wherein R y is H, Ci- ⁇ alkyl, phenyl or benzyl and R z is -OH, H, phenyl or benzyl.
- Compounds according to the invention having an amino group may be derivatised with a ketone or an aldehyde such as formaldehyde to form a Mannich base. This base will hydrolyze with first order kinetics in aqueous solution.
- An interesting embodiment of the present invention is the use of a substituted quinoline derivative according to Formula (la) or Formula (lb), in particular ( ⁇ S, ⁇ R)-6-bromo- ⁇ -[2-(dimethylamino)ethyl]-2-methoxy- ⁇ -l-naphthalenyl- ⁇ -phenyl-3- quinolineethanol, for the preparation of a medicament for the treatment of an infection with a drug resistant Mycobacterium strain as defined hereinabove wherein the drug resistant Mycobacterium strain is a drug resistant M. tuberculosis strain.
- a further interesting embodiment of the present invention is the use of a substituted quinoline derivative according to Formula (la) or Formula (lb), in particular ( ⁇ S, ⁇ R)- 6-bromo- ⁇ -[2-(dimethylamino)ethyl]-2-methoxy- ⁇ -l-naphthalenyl- ⁇ -phenyl-3- quinolineethanol, for the preparation of a medicament for the treatment of a human infected with a drug resistant Mycobacterium strain, in particular a drug resistant M. tuberculosis strain.
- Still a further interesting embodiment of the present invention is the use of a substituted quinoline derivative according to Formula (la) or Formula (lb), in particular ( ⁇ S, ⁇ R)- 6-bromo- ⁇ -[2-( ⁇ methylamino)ethyl]-2-methoxy- ⁇ - 1 -naphthalenyl- ⁇ -phenyl-3- quinolineethanol, for the preparation of a medicament for the treatment of an infection with a multi drug resistant Mycobacterium strain, in particular a multi drug resistant M. tuberculosis strain, in particular for the preparation of a medicament for the treatment of a mammal, including a human, infected with a multi drug resistant Mycobacterium strain, in particular a multi drug resistant M. tuberculosis strain.
- the compounds of formula (la) and (lb) can be used to treat drug resistant including multi drug resistant Mycobacterial diseases.
- the exact dosage and frequency of administration depends on the particular compound of formula (la) or (lb) used, the particular condition being treated, the severity of the condition being treated, the age, weight and general physical condition of the particular patient as well as other medication the individual may be taking, as is well known to those skilled in the art
- said effective daily amount may be lowered or increased depending on the response of the treated subject and/or depending on the evaluation of the physician prescribing the compounds of the instant invention.
- the compounds of formula (la) and (lb) are active against drug resistant including multi drug resistant Mycobacterial strains
- the present compounds may be combined with other antimycobacterial agents in order to effectively combat Mycobacterial diseases.
- the present invention also relates.to a combination of (a) a compound of formula (la) or (lb), in particular ( ⁇ S, ⁇ R)-6-bromo- ⁇ -[2-(dimethylamino)ethyl]-2- methoxy- ⁇ -l-naphthalenyl- ⁇ -phenyl-3-quinolineethanol or a pharmaceutically acceptable acid addition salt thereof, and (b) one or more other antimycobacterial agents.
- the present invention also relates to a combination of (a) a compound of formula (la) or (lb), in particular ( ⁇ S, ⁇ R)-6-bromo- ⁇ -[2-(dime1hylamino)ethyl]-2-methoxy- ⁇ -l- naphlhalenyl- ⁇ -phenyl-3-quinolineethanol or a pharmaceutically acceptable acid addition salt thereof, and (b) one or more other antimycobacterial agents for use as a medicine.
- a compound of formula (la) or (lb) in particular ( ⁇ S, ⁇ R)-6-bromo- ⁇ -[2-(dime1hylamino)ethyl]-2-methoxy- ⁇ -l- naphlhalenyl- ⁇ -phenyl-3-quinolineethanol or a pharmaceutically acceptable acid addition salt thereof, and (b) one or more other antimycobacterial agents for use as a medicine.
- a pharmaceutical composition comprising a pharmaceutically acceptable carrier and, as active ingredient, a therapeutically effective amount of (a) a compound of formula (la) or (lb), in particular ( ⁇ S, ⁇ R)-6-bromo- ⁇ -[2-(dimelhylamino)ethyl]-2-methoxy- ⁇ - l-naphthalenyl- ⁇ -phenyl-3-quinolineethanol or a pharmaceutically acceptable acid addition salt thereof, and (b) one or more other antimycobacterial agents, is also comprised by the present invention.
- the present invention also relates to the use of a combination or pharmaceutical composition as defined above for the treatment of an infection with a drug resistant Mycobacterium strain, in particular a drug resistant M tuberculosis strain.
- the above defined combination or pharmaceutical composition may also be used to treat an infection with a susceptible Mycobacterial strain, in particular a susceptible M. tuberculosis strain.
- the compound of formula (la) or (lb) is preferably a compound of formula (la).
- isoniazid pyrazinamide
- amikacin ethionamide
- moxifloxacin et
- the present compounds of formula (la) or (lb), in particular ( ⁇ S, ⁇ R)-6- bromo- ⁇ -[2-(dmethylamino)ethyl]-2-methoxy- ⁇ -l-naphthalenyl- ⁇ -phenyl-3- quin lineethano], are combined with rifapentin and moxifloxacin.
- Another interesting combination according to the present invention is a combination of (a) a compound of formula (la) or (lb), in particular ( ⁇ S, ⁇ R)-6-bromo- ⁇ -[2- (dimethylamino)ethyl]-2-methoxy- ⁇ -l-naphthalenyl- ⁇ -phenyl-3-quinolineethanol or a pharmaceutically acceptable acid addition salt thereof, and (b) one or more other antimycobacterial agents wherein said one or more other antimycobacterial agents comprise pyrazinamide.
- the present invention also relates to a combination of a compound of formula (la) or (lb), in particular ( ⁇ S, ⁇ R)-6-bromo- ⁇ -[2- (dimemylam o)ethyl]-2-methoxy- ⁇ -l-naphthalenyl- ⁇ -phenyl-3-quinolineethanol or a pharmaceutically acceptable acid addition salt thereof, and pyrazinamide and optionally one or more other antimycobacterial agents.
- Examples of such combinations are the combination of ( ⁇ S, ⁇ R)-6-bromo- ⁇ -[2-(dimethylamino)ethyl]-2-methoxy- ⁇ -l- na ⁇ hthalenyl- ⁇ -phenyl-3 -quinolineethanol or a pharmaceutically acceptable acid addition salt thereof, and pyrazinamide; the combination of ( ⁇ S, ⁇ R)-6-bromo- ⁇ -[2- (dimethylan ⁇ no)ethyl]-2-methoxy- ⁇ -l -naphthalenyl- ⁇ -phenyl-3-quinolineethanol or a pharmaceutically acceptable acid addition salt thereof, pyrazinamide and rifapentin; the combination of ( ⁇ S, ⁇ R)-6-bromo- ⁇ -[2-(dimemylamino)ethy]]-2-methoxy- ⁇ -l- naphthalenyl- ⁇ -phenyl-3-quinolineethanol or a pharmaceutically acceptable acid addition salt thereof, pyrazinamide
- a pharmaceutical composition comprising a pharmaceutically acceptable carrier and, as active ingredient, a therapeutically effective amount of the active ingredients listed in the above combinations, is also comprised by the present invention.
- the present pharmaceutical composition may have various pharmaceutical forms for administration purposes. As appropriate compositions there may be cited all compositions usually employed for systemically administering drugs. To prepare the pharmaceutical compositions of this invention, an effective amount of the particular compounds, optionally in addition salt form, as the active ingredient is combined in intimate admixture with a pharmaceutically acceptable carrier, which carrier may take a wide variety of forms depending on the form of preparation desired for administration. These pharmaceutical compositions are desirable in unitary dosage form suitable, in particular, for administration orally or by parenteral injection.
- any of the usual pharmaceutical media may be employed such as, for example, water, glycols, oils, alcohols and the like in the case of oral liquid preparations such as suspensions, syrups, elixirs, emulsions and solutions; or solid carriers such as starches, sugars, kaolin, diluents, lubricants, binders, disintegrating agents and the like in the case of powders, pills, capsules and tablets. Because of their ease in administration, tablets and capsules represent the most advantageous oral unit dosage forms in which case solid pharmaceutical carriers are obviously employed.
- the carrier will usually comprise sterile water, at least in large part, though other ingredients, for example, to aid solubility, may be included.
- injectable solutions for example, may be prepared in which the carrier comprises saline solution, glucose solution or a mixture of saline and glucose solution.
- injectable suspensions may also be prepared in which case appropriate liquid carriers, suspending agents and the like may be employed.
- solid form preparations which are intended to be converted, shortly before use, to liquid form preparations.
- the pharmaceutical composition will preferably comprise from 0.05 to 99 % by weight, more preferably from 0.1 to 70 % by weight of the active ingredients, and, from 1 to 99.95 % by weight, more preferably from 30 to 99.9 weight % of a pharmaceutically acceptable carrier, all percentages being based on the total composition.
- the weight to weight ratio's of the compound of formula (la) or (lb) and (b) the other antimycobacterial agent(s) when given as a combination may be determined by the person skilled in the art. Said ratio and the exact dosage and frequency of administration depends on the particular compound of formula (la) or (lb) and the other antimycobacterial agent(s) used, the particular condition being treated, the severity of the condition being treated, the age, weight and general physical condition of the particular patient as well as other medication the individual may be taking, as is well known to those skilled in the art. Furthermore, it is evident that said effective daily amount may be lowered or increased depending on the response of the treated subject and/or depending on the evaluation of the physician prescribing the compounds of the instant invention.
- the compounds of formula (la) or (lb) and the one or more other antimycobacterial agents may be combined in a single preparation or they may be formulated in separate preparations so that they can be aclministered simultaneously, separately or sequentially.
- the present invention also relates to a product containing (a) a compound of formula (la) or (lb), and (b) one or more other antimycobacterial agents, as a combined preparation for simultaneous, separate or sequential use in the treatment of mycobacterial diseases.
- the pharmaceutical composition may additionally contain various other ingredients known in the art, for example, a lubricant, stabilising agent, buffering agent, emulsifying agent, viscosity-regulating agent, surfactant, preservative, flavouring or colorant.
- a lubricant for example, a lubricant, stabilising agent, buffering agent, emulsifying agent, viscosity-regulating agent, surfactant, preservative, flavouring or colorant.
- Unit dosage form refers to physically discrete units suitable as unitary dosages, each unit containing a predetermined quantity of active ingredient calculated to produce the desired therapeutic effect in association with the required pharmaceutical carrier.
- unit dosage forms are tablets (including scored or coated tablets), capsules, pills, powder packets, wafers, suppositories, injectable solutions or suspensions and the like, and segregated multiples thereof.
- the daily dosage of the compound according to the invention will, of course, vary with the compound employed, the mode of administration, the treatment desired and the mycobacterial disease indicated. However, in general, satisfactory results will be obtained when the compound according to the invention is administered at a daily dosage not exceeding lgram, e.g. in the range from 10 to 50 mg/kg body weight.
- MIC minimal inhibitory concentration
- the following medium 10 % Oleic acid Albumin Dextrose Catalase (OADC)-enriched 7H11 medium.
- OADC Oleic acid Albumin Dextrose Catalase
- the incubations were done at 30°C or 37°C for 3 to 42 days depending on the mycobacterial species.
- Tables 7 and 8 list the MICs (mg/L) against different clinical isolates of resistant Mycobacterium strains.
- Tables 9 and 10 list the MICs (mg L) against different clinical isolates of Mycobacterium strains resistant to fluoroquino tones. In the Tables rifampin and ofloxacin are also included as reference.
- Table 7 Table 7 :
- Compound 12 was also tested against 2 multi-drug resistant M. tuberculosis strains, i.e. a strain resistant to isoniazid 10 mg/L and rifampin and a strain resistant to isoniazid 0.2 mg/L and rifampin.
- the MIC obtained for compound 12 for both strains is 0.03mg/L.
- mice were allocated to the following treatment groups : an untreated control group for survival monitoring, two positive control groups, one with a regimen for susceptible tuberculosis treated with 2 months of isoniazid 25 mgkg, rifampin 10 mg/kg, pyrazinamide 150 mg/kg daily, and the other with a regimen for multi drug resistant tuberculosis treated with 2 months of daily amikacin 150 mg/kg, ethionamide 50 mg/kg, moxifloxacin 100 mg kg and pyrazinamide 150 mg/kg.
- Three negative control groups were treated for 2 months with one of the following drugs, rifampin 10 mgkg daily, moxifloxacin 100 mg/kg daily and compound 1225 mg/kg daily.
- Rifampicin (RMP) 10 mg kg
- Isoniazid (INH) 25 mg/kg
- Table 12 Mean spleen weight and number of CFU per spleen and lung of M.tuberculosis- infected mice and treated with various treatments for 2 months.
- mice sacrificed on day 14 after inoculation Except the pretreatment values were obtained from mice sacrificed on day 14 after inoculation, the remaining results were obtained from mice sacrificed on day 42 after inoculation. Treatment began on day 14, and was administered five time weekly for four weeks. Isoniazid (H), rifampin (R), moxifloxacin (M), pyrazinamide (Z), compound 12 (J), amikacin (A), ethionamide (Et). In vitro testim of susceptibility to compound 12 of fully susceptible and multi drug resistant M.tuberculosis strains in solid medium assay.
- the susceptibility to compound 12 of 73 M. tuberculosis strains was tested in a solid medium assay (agar plates).
- the panel of strains included strains (41) fully susceptible to standard anti-tuberculosis drags as well as multi drug resistant (MDR) strains (32). i.e. strains resistant to at least rifampin and isoniazid.
- MDR multi drug resistant
- Agar plates were welded with solutions containing compound 12 in a concentration ranging from 0.002 mg L to 0.256 mg/L (8 different concentrations tested). M. tuberculosis isolates were then plated on each agar plate and the plates were sealed and incubated at 36°C for 3 weeks.
- Isolate growth was analyzed 3 weeks following plate inoculation and an isolate's MIC was defined as the first concentration at which no growth was observed.
- Table 14 Results for once-a-week group after 2 months lung CFU* % positive mice DO 7.23 J l,99+/-0,75 9/9 M 6,44+/-0,5 7/7 P 3,26+/-0,58 10/10 JP l,63+/-0,92 8/9 JPM l,85+/-0,7 10/10 JPH l,48+/-0,79 10/10 JPZ 0,23+/-0,72 1/10
Landscapes
- Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Epidemiology (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pulmonology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Quinoline Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description

































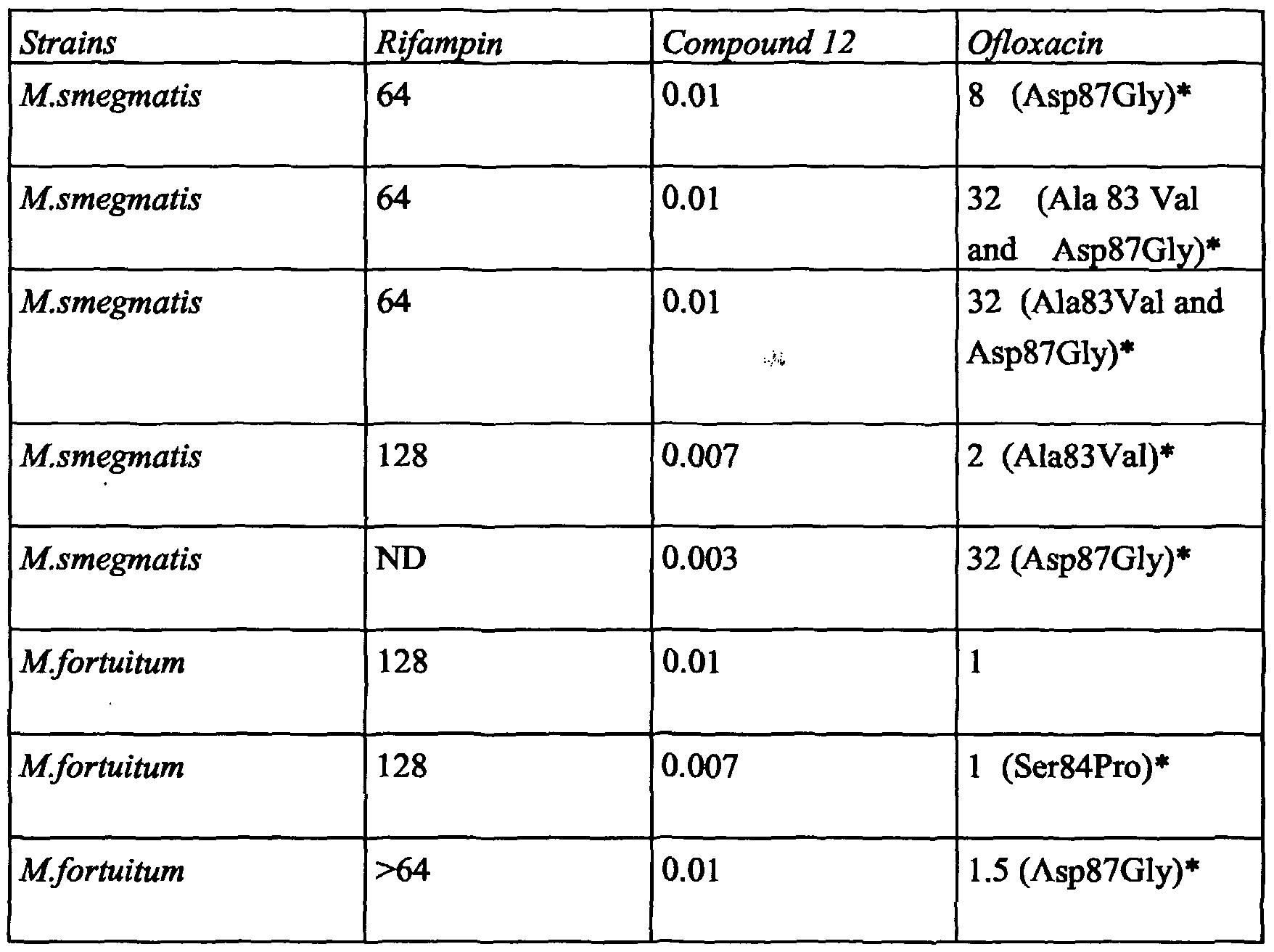
Claims
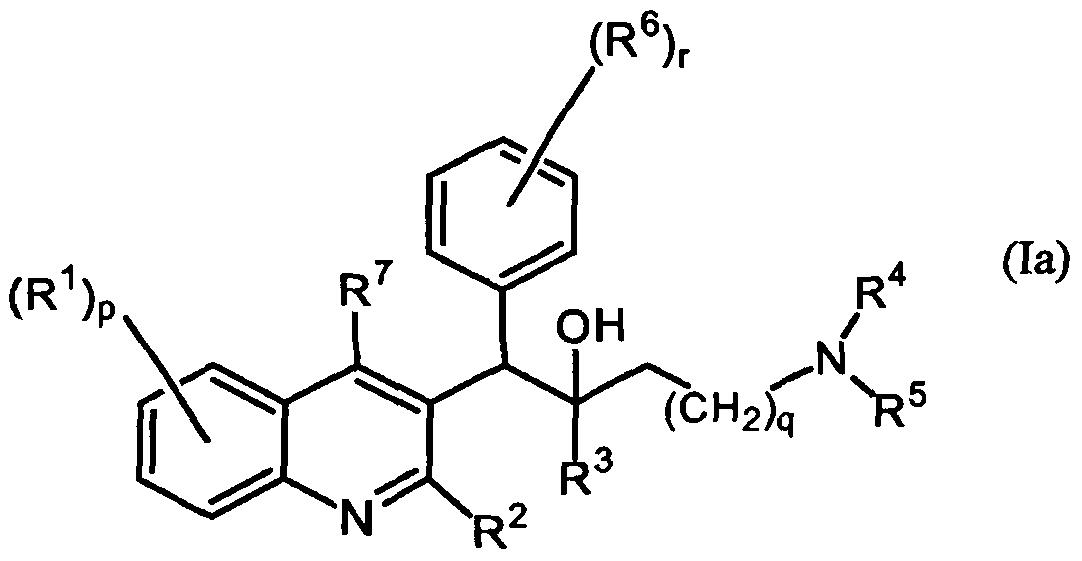


Priority Applications (24)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020137002606A KR20130024969A (en) | 2004-05-28 | 2005-05-24 | Use of substituted quinoline derivatives for the treatment of drug resistant mycobacterial diseases |
| CA2566544A CA2566544C (en) | 2004-05-28 | 2005-05-24 | Use of substituted quinoline derivatives for the treatment of drug resistant mycobacterial diseases |
| DK05743054T DK1753427T3 (en) | 2004-05-28 | 2005-05-24 | Use of Substituted Quinoline Derivatives to Treat Drug-Resistant Mycobacterial Diseases |
| JP2007513922A JP5081617B2 (en) | 2004-05-28 | 2005-05-24 | Use of substituted quinoline derivatives for the treatment of drug-resistant mycobacterial diseases |
| AU2005249231A AU2005249231B2 (en) | 2004-05-28 | 2005-05-24 | Use of substituted quinoline derivatives for the treatment of drug resistant mycobacterial diseases |
| US11/569,681 US20070249667A1 (en) | 2004-05-28 | 2005-05-24 | Use of Substituted Quinoline Derivatives for the Treatment of Drug Resistant Mycobacterial Diseases |
| NZ550840A NZ550840A (en) | 2004-05-28 | 2005-05-24 | Use of substituted quinoline derivatives for the treatment of drug resistant mycobacterial diseases |
| RSP-2008/0291A RS50585B (en) | 2004-05-28 | 2005-05-24 | Use of substituted quinoline derivatives for the treatment of drug resistant mycobacterial diseases |
| EP05743054A EP1753427B1 (en) | 2004-05-28 | 2005-05-24 | Use of substituted quinoline derivatives for the treatment of drug resistant mycobacterial diseases |
| BRPI0510414-9A BRPI0510414B1 (en) | 2004-05-28 | 2005-05-24 | Pharmaceutical combination and composition |
| MEP-2008-89A ME01105B (en) | 2004-05-28 | 2005-05-24 | Use of substituted quinoline derivatives for the treatment of drug resistant mycobacterial diseases |
| EA200602260A EA010651B1 (en) | 2004-05-28 | 2005-05-24 | Use of substituted quinoline derivatives for the treatment of drug resistant mycobacterial diseases |
| ES05743054T ES2306146T3 (en) | 2004-05-28 | 2005-05-24 | USE OF SUBSTITUTED QUINOLINE DERIVATIVES FOR THE TREATMENT OF MICOBACTERIAL DISEASES RESISTANT TO PHARMACOS. |
| PL05743054T PL1753427T3 (en) | 2004-05-28 | 2005-05-24 | Use of substituted quinoline derivatives for the treatment of drug resistant mycobacterial diseases |
| DE602005005810T DE602005005810T2 (en) | 2004-05-28 | 2005-05-24 | USE OF SUBSTITUTED CHINOLIN DERIVATIVES FOR THE TREATMENT OF DRUG-RESISTANT MYCOBACTERIAL DISEASES |
| CN2005800170162A CN1976704B (en) | 2004-05-28 | 2005-05-24 | Use of substituted quinoline derivatives for the treatment of drug resistant mycobacterial diseases |
| MXPA06013888A MXPA06013888A (en) | 2004-05-28 | 2005-05-24 | Use of substituted quinoline derivatives for the treatment of drug resistant mycobacterial diseases. |
| KR1020067024974A KR101371653B1 (en) | 2004-05-28 | 2005-05-24 | Use of substituted quinoline derivatives for the treatment of drug resistant mycobacterial diseases |
| AP2006003828A AP2037A (en) | 2004-05-28 | 2005-05-24 | Use of substituted quinoline derivatives for the treatment of drug resistant mycobacterial diseases |
| IL179630A IL179630A (en) | 2004-05-28 | 2006-11-27 | Use of substituted quinoline derivatives for the preparation of medicaments for the treatment of drug resistant mycobacterial diseases |
| NO20066041A NO338624B1 (en) | 2004-05-28 | 2006-12-28 | Use of substituted quinoline derivatives for the treatment of drug resistant mycobacterial diseases |
| HK07111416.6A HK1106136A1 (en) | 2004-05-28 | 2007-10-23 | Use of substituted quinoline derivatives for the treatment of drug resistant mycobacterial diseases |
| HR20080307T HRP20080307T3 (en) | 2004-05-28 | 2008-06-30 | Use of substituted quinoline derivatives for the treatment of drug resistant mycobacterial diseases |
| US12/719,221 US20100168133A1 (en) | 2004-05-28 | 2010-03-08 | Use of substituted quinoline derivatives for the treatment of drug resistant mycobacterial diseases |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP04102402.7 | 2004-05-28 | ||
| EP04102402 | 2004-05-28 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/719,221 Division US20100168133A1 (en) | 2004-05-28 | 2010-03-08 | Use of substituted quinoline derivatives for the treatment of drug resistant mycobacterial diseases |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2005117875A1 true WO2005117875A1 (en) | 2005-12-15 |
Family
ID=34929150
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2005/052371 WO2005117875A1 (en) | 2004-05-28 | 2005-05-24 | Use of substituted quinoline derivatives for the treatment of drug resistant mycobacterial diseases |
Country Status (35)
| Country | Link |
|---|---|
| US (2) | US20070249667A1 (en) |
| EP (1) | EP1753427B1 (en) |
| JP (2) | JP5081617B2 (en) |
| KR (2) | KR101371653B1 (en) |
| CN (2) | CN102908347A (en) |
| AP (1) | AP2037A (en) |
| AR (1) | AR049127A1 (en) |
| AT (1) | ATE390925T1 (en) |
| AU (1) | AU2005249231B2 (en) |
| BR (1) | BRPI0510414B1 (en) |
| CA (1) | CA2566544C (en) |
| CL (1) | CL2009001980A1 (en) |
| CY (1) | CY1110388T1 (en) |
| DE (1) | DE602005005810T2 (en) |
| DK (1) | DK1753427T3 (en) |
| EA (1) | EA010651B1 (en) |
| ES (1) | ES2306146T3 (en) |
| HK (1) | HK1106136A1 (en) |
| HR (1) | HRP20080307T3 (en) |
| IL (1) | IL179630A (en) |
| JO (1) | JO2524B1 (en) |
| ME (1) | ME01105B (en) |
| MX (1) | MXPA06013888A (en) |
| MY (1) | MY140611A (en) |
| NO (1) | NO338624B1 (en) |
| NZ (1) | NZ550840A (en) |
| PA (1) | PA8635201A1 (en) |
| PL (1) | PL1753427T3 (en) |
| PT (1) | PT1753427E (en) |
| RS (1) | RS50585B (en) |
| SI (1) | SI1753427T1 (en) |
| TW (1) | TWI363625B (en) |
| UA (1) | UA90267C2 (en) |
| WO (1) | WO2005117875A1 (en) |
| ZA (1) | ZA200609899B (en) |
Cited By (27)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006131519A1 (en) * | 2005-06-08 | 2006-12-14 | Janssen Pharmaceutica N.V. | Quinoline derivatives as antibacterial agents |
| JP2006342109A (en) * | 2005-06-09 | 2006-12-21 | Janssen Pharmaceut Nv | Quinoline derivative as antibacterial agent |
| WO2007000434A1 (en) * | 2005-06-28 | 2007-01-04 | Janssen Pharmaceutica N.V. | Quinoline derivatives as antibacterial agents |
| WO2007000436A1 (en) * | 2005-06-28 | 2007-01-04 | Janssen Pharmaceutica N.V. | Quinoline derivatives as antibacterial agents |
| WO2007000435A1 (en) * | 2005-06-28 | 2007-01-04 | Janssen Pharmaceutica N.V. | Quinoline derivatives as antibacterial agents |
| WO2008068267A1 (en) * | 2006-12-06 | 2008-06-12 | Janssen Pharmaceutica N.V. | Antibacterial quinoline derivatives |
| WO2008068268A1 (en) * | 2006-12-06 | 2008-06-12 | Janssen Pharmaceutica N.V. | Antibacterial quinoline derivatives |
| WO2008068270A1 (en) * | 2006-12-06 | 2008-06-12 | Janssen Pharmaceutica N.V. | Antibacterial quinoline derivatives |
| WO2008068272A2 (en) * | 2006-12-06 | 2008-06-12 | Janssen Pharmaceutica N.V. | Antibacterial quinoline derivatives |
| WO2008068231A1 (en) * | 2006-12-05 | 2008-06-12 | Janssen Pharmaceutica N.V. | Fumarate salt of (alpha s, beta r)-6-bromo-alpha-[2-(dimethylamino)ethyl]-2-methoxy-alpha-1-naphthalenyl-beta-phenyl-3-quinolineethanol |
| WO2008068269A1 (en) * | 2006-12-06 | 2008-06-12 | Janssen Pharmaceutica N.V. | Antibacterial quinoline derivatives |
| WO2010064951A1 (en) * | 2008-12-02 | 2010-06-10 | Закрытое Акционерное Общество "Фарм-Синтез" | New quinoline derivatives, a method for the production and the use thereof for treating microbacterial infections and a pharmaceutical composition based on said derivatives |
| WO2011031926A1 (en) * | 2009-09-11 | 2011-03-17 | Vertex Pharmaceuticals Incorporated | Compositions of n-benzyl-3-(4-chlorophenyl)-2-[methyl-[2-oxo-2-(3,4,5-trimethoxyphenyl) acetyl]amino]-n-[3-(4-pyridyl)-1-[2-[4-pyridyl)ethyl]propanamide and uses thereof |
| CN102249935A (en) * | 2010-05-17 | 2011-11-23 | 中国人民解放军军事医学科学院毒物药物研究所 | Aromatic 2-butanols compound and medical application thereof |
| AP2478A (en) * | 2006-12-06 | 2012-09-27 | Janssen Pharmaceutica Nv | Antibacterial quinoline derivatives |
| WO2013160431A1 (en) | 2012-04-27 | 2013-10-31 | Janssen Pharmaceutica Nv | Antibacterial quinoline derivatives |
| WO2013160435A1 (en) | 2012-04-27 | 2013-10-31 | Janssen Pharmaceutica Nv | Antibacterial quinoline derivatives |
| US8686002B2 (en) | 2005-08-21 | 2014-04-01 | AbbVie Deutschland GmbH & Co. KG | Heterocyclic compounds and their use as binding partners for 5-HT5 receptors |
| WO2016120258A1 (en) | 2015-01-27 | 2016-08-04 | Janssen Pharmaceutica Nv | Dispersible compositions |
| TWI572592B (en) * | 2014-07-14 | 2017-03-01 | 辰欣藥業股份有限公司 | Pyridine derivatives and application of anti-macobacterium thereof |
| WO2020047596A1 (en) * | 2018-09-04 | 2020-03-12 | Monash University | Antibacterial compounds and methods of use |
| WO2020144197A1 (en) | 2019-01-09 | 2020-07-16 | Janssen Pharmaceutica Nv | Combination in the treatment of nontuberculous mycobacterial diseases |
| TWI714702B (en) * | 2016-01-13 | 2021-01-01 | 大陸商上海嘉坦醫藥科技有限公司 | Preparation method of pyridine derivates compounds, and intermidiates and structures thereof |
| WO2022101244A1 (en) | 2020-11-12 | 2022-05-19 | Janssen Pharmaceutica Nv | Combination of bedaquiline, ethambutol and a macrolide in the treatment of nontuberculous mycobacterial diseases |
| WO2023217279A1 (en) * | 2022-05-13 | 2023-11-16 | 广州嘉越医药科技有限公司 | Pyridine derivative, intermediate, method for preparing same and use thereof |
| WO2023227123A1 (en) * | 2022-05-27 | 2023-11-30 | 广州嘉越医药科技有限公司 | Use of pyridine derivative |
| WO2023232838A1 (en) | 2022-05-31 | 2023-12-07 | Janssen Pharmaceutica Nv | Bedaquiline for use in the treatment of leprosy |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1753427B1 (en) * | 2004-05-28 | 2008-04-02 | Janssen Pharmaceutica N.V. | Use of substituted quinoline derivatives for the treatment of drug resistant mycobacterial diseases |
| KR101301573B1 (en) * | 2004-12-24 | 2013-09-04 | 얀센 파마슈티카 엔.브이. | Treatment of Latent Tuberculosis |
| MD4009C2 (en) * | 2008-07-15 | 2010-08-31 | Институт Химии Академии Наук Молдовы | Use of 1-methyl-4-(N-methylaminobutyl-4)-b-carboline as antituberculous remedy |
| CN105330595B (en) * | 2014-07-14 | 2019-12-10 | 上海嘉坦医药科技有限公司 | pyridine derivatives and their use as antimycobacterial agents |
| CN106279017A (en) * | 2015-05-21 | 2017-01-04 | 重庆圣华曦药业股份有限公司 | Bedaquinoline crystal form, composition and preparation method thereof |
| EP3366676B1 (en) | 2015-10-20 | 2021-05-26 | Zhejiang Hisun Pharmaceutical Co., Ltd | Crystalline forms of bedaquiline fumarate and preparation methods therefor |
| US10508097B2 (en) * | 2016-03-07 | 2019-12-17 | The Global Alliance For Tb Drug Development, Inc. | Antibacterial compounds and uses thereof |
| CN107243008B (en) * | 2017-04-13 | 2021-03-30 | 中国科学院广州生物医药与健康研究院 | Novel application of pyrazolo [1,5-a ] pyridine compound and composition for treating mycobacterium abscesses infection |
| CN111278985B (en) * | 2017-09-01 | 2023-09-08 | 学校法人北里研究所 | Novel compound having therapeutic activity against mycobacterium avium complex infection disease and method for producing same |
| WO2021107876A1 (en) * | 2019-11-26 | 2021-06-03 | Nanyang Technological University | Compounds for treating tuberculosis |
| CN110804016B (en) * | 2019-12-05 | 2022-11-04 | 福建省微生物研究所 | Diaryl quinoline derivatives against mycobacterium tuberculosis |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004011436A1 (en) * | 2002-07-25 | 2004-02-05 | Janssen Pharmaceutica N.V. | Quinoline derivatives and their use as mycobacterial inhibitors |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5399558A (en) * | 1993-11-24 | 1995-03-21 | Pathogenesis Corporation | Isoflavonoid antibacterial compounds, compositions and use |
| CA2270123A1 (en) * | 1996-10-28 | 1998-05-07 | Department Of The Army, U.S. Government | Compounds, compositions and methods for treating antibiotic-resistant infections |
| US6103905A (en) * | 1997-06-19 | 2000-08-15 | Sepracor, Inc. | Quinoline-indole antimicrobial agents, uses and compositions related thereto |
| EP1753427B1 (en) * | 2004-05-28 | 2008-04-02 | Janssen Pharmaceutica N.V. | Use of substituted quinoline derivatives for the treatment of drug resistant mycobacterial diseases |
| CA2528849C (en) * | 2005-06-08 | 2014-01-14 | Janssen Pharmaceutica N.V. | Quinoline derivatives as antibacterial agents |
-
2005
- 2005-05-24 EP EP05743054A patent/EP1753427B1/en active Active
- 2005-05-24 CN CN2012103943126A patent/CN102908347A/en active Pending
- 2005-05-24 US US11/569,681 patent/US20070249667A1/en not_active Abandoned
- 2005-05-24 AU AU2005249231A patent/AU2005249231B2/en active Active
- 2005-05-24 DE DE602005005810T patent/DE602005005810T2/en active Active
- 2005-05-24 KR KR1020067024974A patent/KR101371653B1/en active Protection Beyond IP Right Term
- 2005-05-24 EA EA200602260A patent/EA010651B1/en active Protection Beyond IP Right Term
- 2005-05-24 DK DK05743054T patent/DK1753427T3/en active
- 2005-05-24 AT AT05743054T patent/ATE390925T1/en active
- 2005-05-24 ES ES05743054T patent/ES2306146T3/en active Active
- 2005-05-24 PT PT05743054T patent/PT1753427E/en unknown
- 2005-05-24 NZ NZ550840A patent/NZ550840A/en unknown
- 2005-05-24 CA CA2566544A patent/CA2566544C/en active Active
- 2005-05-24 BR BRPI0510414-9A patent/BRPI0510414B1/en active IP Right Grant
- 2005-05-24 ME MEP-2008-89A patent/ME01105B/en unknown
- 2005-05-24 JO JO200574A patent/JO2524B1/en active
- 2005-05-24 MX MXPA06013888A patent/MXPA06013888A/en active IP Right Grant
- 2005-05-24 KR KR1020137002606A patent/KR20130024969A/en not_active Application Discontinuation
- 2005-05-24 RS RSP-2008/0291A patent/RS50585B/en unknown
- 2005-05-24 AP AP2006003828A patent/AP2037A/en active
- 2005-05-24 PL PL05743054T patent/PL1753427T3/en unknown
- 2005-05-24 UA UAA200611048A patent/UA90267C2/en unknown
- 2005-05-24 SI SI200530294T patent/SI1753427T1/en unknown
- 2005-05-24 JP JP2007513922A patent/JP5081617B2/en active Active
- 2005-05-24 WO PCT/EP2005/052371 patent/WO2005117875A1/en active Application Filing
- 2005-05-24 CN CN2005800170162A patent/CN1976704B/en active Active
- 2005-05-27 AR ARP050102200A patent/AR049127A1/en unknown
- 2005-05-27 PA PA20058635201A patent/PA8635201A1/en unknown
- 2005-05-27 TW TW094117334A patent/TWI363625B/en active
- 2005-05-27 MY MYPI20052432A patent/MY140611A/en unknown
-
2006
- 2006-11-27 ZA ZA200609899A patent/ZA200609899B/en unknown
- 2006-11-27 IL IL179630A patent/IL179630A/en active IP Right Grant
- 2006-12-28 NO NO20066041A patent/NO338624B1/en unknown
-
2007
- 2007-10-23 HK HK07111416.6A patent/HK1106136A1/en unknown
-
2008
- 2008-06-30 HR HR20080307T patent/HRP20080307T3/en unknown
- 2008-07-02 CY CY20081100696T patent/CY1110388T1/en unknown
-
2009
- 2009-10-20 CL CL2009001980A patent/CL2009001980A1/en unknown
-
2010
- 2010-03-08 US US12/719,221 patent/US20100168133A1/en not_active Abandoned
-
2012
- 2012-07-06 JP JP2012152373A patent/JP5675718B2/en active Active
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004011436A1 (en) * | 2002-07-25 | 2004-02-05 | Janssen Pharmaceutica N.V. | Quinoline derivatives and their use as mycobacterial inhibitors |
Cited By (86)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NO340352B1 (en) * | 2005-06-08 | 2017-04-10 | Janssen Pharmaceutica Nv | Quinoline derivatives as antibacterial agents |
| US7709498B2 (en) | 2005-06-08 | 2010-05-04 | Janssen Pharmaceutica N.V. | Quinoline derivatives as antibacterial agents |
| AP2383A (en) * | 2005-06-08 | 2012-03-16 | Janssen Pharmaceutica Nv | Quinoline derivatives as antibacterial agents. |
| WO2006131519A1 (en) * | 2005-06-08 | 2006-12-14 | Janssen Pharmaceutica N.V. | Quinoline derivatives as antibacterial agents |
| JP2006342109A (en) * | 2005-06-09 | 2006-12-21 | Janssen Pharmaceut Nv | Quinoline derivative as antibacterial agent |
| EA014431B1 (en) * | 2005-06-28 | 2010-12-30 | Янссен Фармацевтика Н.В. | Quinoline derivatives as antibacterial agents |
| US8841321B2 (en) | 2005-06-28 | 2014-09-23 | Janssen Pharmaceutica Nv | Quinoline derivatives as antibacterial agents |
| AU2006263883B2 (en) * | 2005-06-28 | 2012-12-13 | Janssen Pharmaceutica N.V. | Quinoline derivatives as antibacterial agents |
| AP2386A (en) * | 2005-06-28 | 2012-03-23 | Janssen Pharmaceutica Nv | Quinoline derivatives as antibacterial agents. |
| WO2007000434A1 (en) * | 2005-06-28 | 2007-01-04 | Janssen Pharmaceutica N.V. | Quinoline derivatives as antibacterial agents |
| AU2006263882B2 (en) * | 2005-06-28 | 2012-12-13 | Janssen Pharmaceutica N.V | Quinoline derivatives as antibacterial agents |
| NO342701B1 (en) * | 2005-06-28 | 2018-07-09 | Janssen Pharmaceutica Nv | Quinoline derivatives as antibacterial agents |
| NO341285B1 (en) * | 2005-06-28 | 2017-10-02 | Janssen Pharmaceutica Nv | Quinoline derivatives as antibacterial agents |
| NO341283B1 (en) * | 2005-06-28 | 2017-10-02 | Janssen Pharmaceutica Nv | Quinoline derivatives as antibacterial agents |
| WO2007000435A1 (en) * | 2005-06-28 | 2007-01-04 | Janssen Pharmaceutica N.V. | Quinoline derivatives as antibacterial agents |
| AU2006263884B2 (en) * | 2005-06-28 | 2013-01-10 | Janssen Pharmaceutica N.V. | Quinoline derivatives as antibacterial agents |
| WO2007000436A1 (en) * | 2005-06-28 | 2007-01-04 | Janssen Pharmaceutica N.V. | Quinoline derivatives as antibacterial agents |
| EA014423B1 (en) * | 2005-06-28 | 2010-12-30 | Янссен Фармацевтика Н.В. | Quinoline derivatives as antibacterial agents |
| EA014163B1 (en) * | 2005-06-28 | 2010-10-29 | Янссен Фармацевтика Н.В. | Quinoline derivatives as antibacterial agents |
| US8686002B2 (en) | 2005-08-21 | 2014-04-01 | AbbVie Deutschland GmbH & Co. KG | Heterocyclic compounds and their use as binding partners for 5-HT5 receptors |
| AU2007328945B2 (en) * | 2006-12-05 | 2014-04-03 | Janssen Pharmaceutica N.V. | Fumarate salt of (alpha S, beta R)-6-bromo-alpha-[2-(dimethylamino)ethyl]-2-methoxy-alpha-1-naphthalenyl-beta-phenyl-3-quinolineethanol |
| EA017091B9 (en) * | 2006-12-05 | 2014-10-30 | Янссен Фармацевтика Н.В. | Fumarate salt of (alpha s, beta r)-6-bromo-alpha-[2-(dimethylamino)ethyl]-2-methoxy-alpha-1-naphthalenyl-beta-phenyl-3-quinolineethanol |
| US8546428B2 (en) | 2006-12-05 | 2013-10-01 | Janssen Pharmaceutica Nv | Fumarate salt of (alpha S, beta R)-6-bromo-alpha-[2-(dimethylamino)ethyl]-2-methoxy-alpha-1-naphthalenyl-beta-phenyl-3-quinolineethanol |
| JP2015028049A (en) * | 2006-12-05 | 2015-02-12 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプJanssen Pharmaceutica Naamloze Vennootschap | Fumarate salt of (alpha s, beta r)-6-bromo-alpha-[2-(dimethylamino)ethyl]-2-methoxy-alpha-1-naphthalenyl-beta-phenyl-3-quinolineethanol |
| CN105012303A (en) * | 2006-12-05 | 2015-11-04 | 詹森药业有限公司 | Fumarate salt of (alpha S, beta R)-6-bromo-alpha-[2-(dimethylamino)ethyl]-2-methoxy-alpha-1-naphthalenyl-beta-phenyl-3-quinolineethanol |
| NO342773B1 (en) * | 2006-12-05 | 2018-08-06 | Janssen Pharmaceutica Nv | Fumarate salt of (alpha S, beta R) -6-bromo-alpha [2- (dimethylamino) ethyl] -2-methoxy-alpha-1-naphthalenyl-beta-phenyl-3-quinolinethanol |
| JP2010511663A (en) * | 2006-12-05 | 2010-04-15 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ | Fumarate salt of (alpha S, beta R) -6-bromo-alpha- [2- (dimethylamino) ethyl] -2-methoxy-alpha-1-naphthalenyl-beta-phenyl-3-quinoline ethanol |
| WO2008068231A1 (en) * | 2006-12-05 | 2008-06-12 | Janssen Pharmaceutica N.V. | Fumarate salt of (alpha s, beta r)-6-bromo-alpha-[2-(dimethylamino)ethyl]-2-methoxy-alpha-1-naphthalenyl-beta-phenyl-3-quinolineethanol |
| EA017091B1 (en) * | 2006-12-05 | 2012-09-28 | Янссен Фармацевтика Н.В. | Fumarate salt of (alpha s, beta r)-6-bromo-alpha-[2-(dimethylamino)ethyl]-2-methoxy-alpha-1-naphthalenyl-beta-phenyl-3-quinolineethanol |
| AP2498A (en) * | 2006-12-05 | 2012-10-19 | Janssen Pharmaceutica Nv | Fumarate salt of (ALPHA S, BETA R)-6-bromo-alpha-[2-(dimethylamino)ethyl]-2-methoxy-alpha-1-naphthalenyl-beta-phenyl-3-quinolineethanol |
| US8415475B2 (en) | 2006-12-06 | 2013-04-09 | Janssen Pharmaceutica N.V. | Antibacterial quinoline derivatives |
| KR101526175B1 (en) * | 2006-12-06 | 2015-06-05 | 얀센 파마슈티카 엔.브이. | Antibacterial quinoline derivatives |
| AP2478A (en) * | 2006-12-06 | 2012-09-27 | Janssen Pharmaceutica Nv | Antibacterial quinoline derivatives |
| AP2477A (en) * | 2006-12-06 | 2012-09-27 | Janssen Pharmaceutica Nv | Antibacterial quinoline derivatives |
| EA016760B1 (en) * | 2006-12-06 | 2012-07-30 | Янссен Фармацевтика Н.В. | Antibacterial quinoline derivatives |
| US8293732B2 (en) | 2006-12-06 | 2012-10-23 | Janssen Pharmaceutica N.V. | Antibacterial quinoline derivatives |
| AU2007328889B2 (en) * | 2006-12-06 | 2012-12-06 | Janssen Pharmaceutica N.V. | Antibacterial quinoline derivatives |
| EA016733B1 (en) * | 2006-12-06 | 2012-07-30 | Янссен Фармацевтика Н.В. | Antibacterial quinoline derivatives |
| CN104744361B (en) * | 2006-12-06 | 2021-06-08 | 詹森药业有限公司 | Antibacterial quinoline derivatives |
| US7998979B2 (en) | 2006-12-06 | 2011-08-16 | Janssen Pharmaceutical N.V. | Antibacterial quinoline derivatives |
| AU2007328887B2 (en) * | 2006-12-06 | 2013-01-17 | Janssen Pharmaceutica N.V. | Antibacterial quinoline derivatives |
| AU2007328890B2 (en) * | 2006-12-06 | 2013-03-14 | Janssen Pharmaceutica N.V. | Antibacterial quinoline derivatives |
| KR101908350B1 (en) * | 2006-12-06 | 2018-10-16 | 얀센 파마슈티카 엔.브이. | Antibacterial quinoline derivatives |
| AU2007328892B2 (en) * | 2006-12-06 | 2013-05-16 | Janssen Pharmaceutica N.V. | Antibacterial quinoline derivatives |
| CN101541752B (en) * | 2006-12-06 | 2013-07-17 | 詹森药业有限公司 | Antibacterial quinoline derivatives |
| TWI406659B (en) * | 2006-12-06 | 2013-09-01 | Janssen Pharmaceutica Nv | Antibacterial quinoline derivatives |
| NO342774B1 (en) * | 2006-12-06 | 2018-08-06 | Janssen Pharmaceutica Nv | Antibacterial quinoline derivatives |
| WO2008068267A1 (en) * | 2006-12-06 | 2008-06-12 | Janssen Pharmaceutica N.V. | Antibacterial quinoline derivatives |
| WO2008068268A1 (en) * | 2006-12-06 | 2008-06-12 | Janssen Pharmaceutica N.V. | Antibacterial quinoline derivatives |
| JP2010511672A (en) * | 2006-12-06 | 2010-04-15 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ | Antibacterial quinoline derivative |
| JP2010511670A (en) * | 2006-12-06 | 2010-04-15 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ | Antibacterial quinoline derivative |
| JP2010511671A (en) * | 2006-12-06 | 2010-04-15 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ | Antibacterial quinoline derivative |
| JP2010511669A (en) * | 2006-12-06 | 2010-04-15 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ | Antibacterial quinoline derivative |
| KR101490221B1 (en) | 2006-12-06 | 2015-02-05 | 얀센 파마슈티카 엔.브이. | Antibacterial quinoline derivatives |
| KR101490222B1 (en) | 2006-12-06 | 2015-02-05 | 얀센 파마슈티카 엔.브이. | Antibacterial quinoline derivatives |
| JP2010511668A (en) * | 2006-12-06 | 2010-04-15 | ジヤンセン・フアーマシユーチカ・ナームローゼ・フエンノートシヤツプ | Antibacterial quinoline derivative |
| NO341983B1 (en) * | 2006-12-06 | 2018-03-05 | Janssen Pharmaceutica Nv | Antibacterial quinoline derivatives |
| TWI484959B (en) * | 2006-12-06 | 2015-05-21 | Janssen Pharmaceutica Nv | Antibacterial quinoline derivatives |
| AP2479A (en) * | 2006-12-06 | 2012-09-27 | Janssen Pharmaceutica Nv | Antibacterial quinoline derivatives |
| TWI487525B (en) * | 2006-12-06 | 2015-06-11 | Janssen Pharmaceutica Nv | Antibacterial quinoline derivatives |
| CN104744361A (en) * | 2006-12-06 | 2015-07-01 | 詹森药业有限公司 | Antibacterial quinoline derivatives |
| WO2008068270A1 (en) * | 2006-12-06 | 2008-06-12 | Janssen Pharmaceutica N.V. | Antibacterial quinoline derivatives |
| WO2008068272A3 (en) * | 2006-12-06 | 2008-07-24 | Janssen Pharmaceutica Nv | Antibacterial quinoline derivatives |
| KR20150126737A (en) * | 2006-12-06 | 2015-11-12 | 얀센 파마슈티카 엔.브이. | Antibacterial quinoline derivatives |
| KR101574642B1 (en) * | 2006-12-06 | 2015-12-04 | 얀센 파마슈티카 엔.브이. | Antibacterial quinoline derivatives |
| EA022344B1 (en) * | 2006-12-06 | 2015-12-30 | Янссен Фармацевтика Н.В. | Antibacterial quinoline derivatives |
| WO2008068272A2 (en) * | 2006-12-06 | 2008-06-12 | Janssen Pharmaceutica N.V. | Antibacterial quinoline derivatives |
| WO2008068269A1 (en) * | 2006-12-06 | 2008-06-12 | Janssen Pharmaceutica N.V. | Antibacterial quinoline derivatives |
| WO2010064951A1 (en) * | 2008-12-02 | 2010-06-10 | Закрытое Акционерное Общество "Фарм-Синтез" | New quinoline derivatives, a method for the production and the use thereof for treating microbacterial infections and a pharmaceutical composition based on said derivatives |
| WO2011031926A1 (en) * | 2009-09-11 | 2011-03-17 | Vertex Pharmaceuticals Incorporated | Compositions of n-benzyl-3-(4-chlorophenyl)-2-[methyl-[2-oxo-2-(3,4,5-trimethoxyphenyl) acetyl]amino]-n-[3-(4-pyridyl)-1-[2-[4-pyridyl)ethyl]propanamide and uses thereof |
| CN102249935A (en) * | 2010-05-17 | 2011-11-23 | 中国人民解放军军事医学科学院毒物药物研究所 | Aromatic 2-butanols compound and medical application thereof |
| CN102249935B (en) * | 2010-05-17 | 2015-05-20 | 中国人民解放军军事医学科学院毒物药物研究所 | Aromatic 2-butanols compound and medical application thereof |
| US9617244B2 (en) | 2012-04-27 | 2017-04-11 | Janssen Pharmaceutica Nv | Antibacterial quinoline derivatives |
| US9133167B2 (en) | 2012-04-27 | 2015-09-15 | Janssen Pharmaceutica Nv | Antibacterial quinoline derivatives |
| WO2013160435A1 (en) | 2012-04-27 | 2013-10-31 | Janssen Pharmaceutica Nv | Antibacterial quinoline derivatives |
| WO2013160431A1 (en) | 2012-04-27 | 2013-10-31 | Janssen Pharmaceutica Nv | Antibacterial quinoline derivatives |
| RU2664587C1 (en) * | 2014-07-14 | 2018-08-21 | Шанхай Цзя Тань Фарматек Ко. Лтд. | Derivatives of pyridine and application thereof against microbacteria |
| TWI572592B (en) * | 2014-07-14 | 2017-03-01 | 辰欣藥業股份有限公司 | Pyridine derivatives and application of anti-macobacterium thereof |
| WO2016120258A1 (en) | 2015-01-27 | 2016-08-04 | Janssen Pharmaceutica Nv | Dispersible compositions |
| TWI714702B (en) * | 2016-01-13 | 2021-01-01 | 大陸商上海嘉坦醫藥科技有限公司 | Preparation method of pyridine derivates compounds, and intermidiates and structures thereof |
| WO2020047596A1 (en) * | 2018-09-04 | 2020-03-12 | Monash University | Antibacterial compounds and methods of use |
| WO2020144197A1 (en) | 2019-01-09 | 2020-07-16 | Janssen Pharmaceutica Nv | Combination in the treatment of nontuberculous mycobacterial diseases |
| WO2022101244A1 (en) | 2020-11-12 | 2022-05-19 | Janssen Pharmaceutica Nv | Combination of bedaquiline, ethambutol and a macrolide in the treatment of nontuberculous mycobacterial diseases |
| WO2023217279A1 (en) * | 2022-05-13 | 2023-11-16 | 广州嘉越医药科技有限公司 | Pyridine derivative, intermediate, method for preparing same and use thereof |
| WO2023227123A1 (en) * | 2022-05-27 | 2023-11-30 | 广州嘉越医药科技有限公司 | Use of pyridine derivative |
| WO2023232838A1 (en) | 2022-05-31 | 2023-12-07 | Janssen Pharmaceutica Nv | Bedaquiline for use in the treatment of leprosy |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1753427B1 (en) | Use of substituted quinoline derivatives for the treatment of drug resistant mycobacterial diseases | |
| EP1830850B1 (en) | Quinoline derivatives for the treatment of latent tuberculosis | |
| KR100733577B1 (en) | Quinoline derivatives and their use as mycobacterial inhibitors | |
| EP1713776B1 (en) | Quinoline derivatives for use as mycobacterial inhibitors | |
| CA2862193C (en) | Antituberculosis drug combination comprising oxazole compounds | |
| JP6426530B2 (en) | Treatment of latent tuberculosis | |
| MXPA05013413A (en) | Treatment of latent tuberculosis |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KM KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NG NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SM SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 12006502051 Country of ref document: PH Ref document number: 2005249231 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1200601723 Country of ref document: VN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005743054 Country of ref document: EP Ref document number: 550840 Country of ref document: NZ Ref document number: 6315/DELNP/2006 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 2005249231 Country of ref document: AU Date of ref document: 20050524 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005249231 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2566544 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: AP/P/2006/003828 Country of ref document: AP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200580017016.2 Country of ref document: CN Ref document number: 179630 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2006/013888 Country of ref document: MX Ref document number: 11569681 Country of ref document: US Ref document number: 2007513922 Country of ref document: JP Ref document number: 1020067024974 Country of ref document: KR |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200602260 Country of ref document: EA |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020067024974 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005743054 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: PI0510414 Country of ref document: BR |
|
| WWP | Wipo information: published in national office |
Ref document number: 11569681 Country of ref document: US |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2005743054 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P-2008/0291 Country of ref document: RS |