WO2005079761A1 - Compositions for delivering hypnotic agents across the oral mucosa and methods of use thereof - Google Patents
Compositions for delivering hypnotic agents across the oral mucosa and methods of use thereof Download PDFInfo
- Publication number
- WO2005079761A1 WO2005079761A1 PCT/US2005/005021 US2005005021W WO2005079761A1 WO 2005079761 A1 WO2005079761 A1 WO 2005079761A1 US 2005005021 W US2005005021 W US 2005005021W WO 2005079761 A1 WO2005079761 A1 WO 2005079761A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- composition
- group
- salt
- ofthe
- saliva
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/437—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having nitrogen as a ring hetero atom, e.g. indolizine, beta-carboline
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
- A61K9/0056—Mouth soluble or dispersible forms; Suckable, eatable, chewable coherent forms; Forms rapidly disintegrating in the mouth; Lozenges; Lollipops; Bite capsules; Baked products; Baits or other oral forms for animals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
- A61K9/006—Oral mucosa, e.g. mucoadhesive forms, sublingual droplets; Buccal patches or films; Buccal sprays
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/20—Hypnotics; Sedatives
Definitions
- sornnia is a condition that affects a person's ability to fall asleep or to maintain sleep. It is the most common sleep disorder, affecting millions of Americans each year.
- Benzodiazepines which are available as short, intermediate, or long-acting hypnotic agents, have proven useful in treating insomnia. These benzodiazepines are thought to bind non- selectively to benzodiazepin ⁇ i (omega and benzodiazepine 2 (omega ) receptors. This non- selective binding maybe responsible for some of the potential problems associated with the use of benzodiazepine compounds as hypnotics. For example, some benzodiazepines are thought to interfere with memory, cognition, and psychomotor function. In addition, problems with altered sleep architecture, rebound insomnia, hangover effects, and abuse potential have been reported with benzodiazepine use.
- zolpidem Ambien ® ; Searle and Co.
- zaleplon Nonata ® ; Wyeth-Ayerst Co.
- Zolpidem an imidazopyridine
- zolpidem has been demonstrated to reduce sleep latency, increase sleep duration, and reduce nighttime awakenings.
- zolpidem has been found to preserve stage III and stage IV sleep, and to result in less disruption of REM (Rapid Eye Movement) sleep.
- Zaleplon is a pyrazolopyrimidine derivative, which has also proven useful as a hypnotic agent.
- zolpidem and zaleplon are both poorly soluble in aqueous media.
- these hypnotic agents are delivered as oral dosages, which are formulated, for example, as tablets or capsules that are swallowed.
- Oral administration has several disadvantages, such as drug losses during hepatic first pass metabolism, during enzymatic degradation within the GI tract, and during absorption. These drug losses not only increase the variability in drug response, but also often require that the medicament be given in greater initial doses.
- the time to reach a therapeutic effect may be quite long, typically around forty-five minutes or longer.
- the mucous membranes of the oral cavity can be divided into five main regions: the floor of the mouth (sublingual), the cheeks (buccal), the gums (gingival), the roof of the mouth (palatal), and the lining of the lips. These regions differ from each other with respect to their anatomy, drug permeability, and physiological response to drags. For example, in terms of permeability, sublingual is more permeable than buccal, which is more permeable than palatal.
- This permeability is generally based on the relative thickness and degree of keratinization of these membranes, with the sublingual mucosa being relatively thin and non-keratinized, the buccal mucosa being thicker and non-keratinized, and the palatal mucosa being intermediate in thickness, but keratinized.
- the extent of drag delivery is also affected by the properties of the drag to be delivered.
- the ability of a molecule to pass through any mucous membrane is dependent upon its size, its lipid solubility, and the extent to which it is ionized, among other factors.
- transmucosal dosage forms include the use of a single buffering agent in order to change the pH of the saliva and tissues surrounding the buccal mucosa.
- these single buffering agents typically react with an acid or a base to create a final pH that is dependent upon the initial pH of the saliva of the user.
- a buffering agent used to attain a final pH that is dependent upon the initial pH of the user results in great variability.
- the extent of ionization, and hence the extent of absorption across the mucous membranes cannot be predicted with any sort of accuracy. This may pose significant problems when calculating precise doses, minimizing variability in patient response, and proving consistency in drug loading to the regulatory authorities.
- a single buffering agent is typically not capable of sustaining a given pH over a period of time for optimal absorption.
- compositions for delivering hypnotic agents across the oral mucosa having buffer systems that facilitate absorption of the agents in a safe and stable manner there is a need in the art for compositions for delivering hypnotic agents across the oral mucosa having a buffer system that produces a final pH, independent of the initial pH, and sustains that final pH for a given period of time.
- compositions capable of rapidly facilitating substantially complete conversion of the hypnotic agent from its ionized to its un-ionized form The present invention satisfies these and other needs.
- the present invention provides novel compositions for the delivery of a hypnotic agent across the oral mucosa.
- the buffer system in the compositions of the present invention raises the pH of saliva to a pH greater than about 7.8, thereby facilitating the substantially complete conversion of the hypnotic agent from its ionized to its un-ionized form.
- the dose of hypnotic agent is rapidly and efficiently absorbed by the oral mucosa with surprisingly low inter-subject variability (e.g., lower variability than absorption across the gut in the same patients).
- compositions of the present invention for treating sleep disorders such as insomnia are also provided.
- the present invention provides a solid composition for delivery of a hypnotic agent across the oral mucosa, the composition comprising: (a) a hypnotic agent selected from the group consisting of an imidazopyridine, a dihydropyrrolopyrazine, a pyrazolopyrimidine, and a pharmaceutically acceptable salt thereof; (b) a carrier that provides complete buccal or sublingual disintegration in about 5 minutes or less following administration to the mouth; and (c) a binary buffer system comprising a carbonate salt and a bicarbonate salt, wherein the binary buffer system raises the pH of saliva to a pH greater than about 7.8, irrespective of the starting pH of saliva.
- a hypnotic agent selected from the group consisting of an imidazopyridine, a dihydropyrrolopyrazine, a pyrazolopyrimidine, and a pharmaceutically acceptable salt thereof
- a carrier that provides complete buccal or sublingual disintegration in about 5 minutes or less following administration to the mouth
- the present invention provides a composition for delivery of a hypnotic agent across the oral mucosa, the composition comprising: (a) a hypnotic agent selected from the group consisting of an imidazopyridine, a dihydropyrrolopyrazine, a pyrazolopyrimidine, and a pharmaceutically acceptable salt thereof; (b) a carrier; and (c) a binary buffer system comprising a carbonate salt and a bicarbonate salt, wherein the binary buffer system raises the pH of saliva to a pH greater than about 7.8, irrespective of the starting pH of saliva.
- the present invention provides a composition for delivery of a hypnotic agent across the oral mucosa, the composition comprising: (a) a hypnotic agent selected from the group consisting of an imidazopyridine, a dihydropyrrolopyrazine, a pyrazolopyrimidine, and a pharmaceutically acceptable salt thereof; (b) a carrier; and (c) a binary buffer system comprising a carbonate salt or a bicarbonate salt and a second buffering agent, wherein the binary buffer system raises the pH of saliva to a pH greater than about 7.8, irrespective of the starting pH of saliva.
- the present invention provides a composition for delivery of a hypnotic agent across the oral mucosa, the composition comprising: (a) a hypnotic agent selected from the group consisting of an imidazopyridine, a dihydropyrrolopyrazine, a pyrazolopyrimidine, and a pharmaceutically acceptable salt thereof; (b) a carrier; and (c) a binary buffer system comprising a metal oxide and a citrate, phosphate, or borate salt, wherem the binary buffer system raises the pH of saliva to a pH greater than about 7.8, irrespective of the starting pH of saliva.
- the present invention provides a composition for delivery of a hypnotic agent across the oral mucosa, the composition comprising: (a) a hypnotic agent selected from the group consisting of an imidazopyridine, a dihydropyrrolopyrazine, a pyrazolopyrimidine, and a pharmaceutically acceptable salt thereof; (b) a carrier; and (c) a ternary buffer system comprising a carbonate salt, a bicarbonate salt, and a third buffering agent, wherein the ternary buffer system raises the pH of saliva to a pH greater than about 7.8, irrespective of the starting pH of saliva.
- the present invention provides a composition for delivery of a hypnotic agent across the oral mucosa, the composition comprising: (a) a hypnotic agent selected from the group consisting of an imidazopyridine, a dihydropyrrolopyrazine, a pyrazolopyrimidine, and a pharmaceutically acceptable salt thereof; (b) a carrier; and (c) a buffer system comprising a carbonate salt or a bicarbonate salt and two or more buffering agents selected from the group consisting of a metal oxide, a citrate salt, a phosphate salt, and a borate salt, wherein the buffer system raises the pH of saliva to a pH greater than about 7.8, irrespective of the starting pH of saliva.
- a hypnotic agent selected from the group consisting of an imidazopyridine, a dihydropyrrolopyrazine, a pyrazolopyrimidine, and a pharmaceutically acceptable salt thereof
- a carrier comprising a carbonate salt or a bicarbon
- the present invention provides a method for treating a sleep disorder in a subject in need thereof, the method comprising: administering to the subject a composition comprising a therapeutically effective amount of a hypnotic agent selected from the group consisting of an imidazopyridine, a dihydropyrrolopyrazine, a pyrazolopyrimidine, and a pharmaceutically acceptable salt thereof; a carrier; and a binary buffer system comprising a carbonate salt and a bicarbonate salt, wherein the binary buffer system raises the pH of saliva to a pH greater than about 7.8, irrespective of the starting pH of saliva.
- a hypnotic agent selected from the group consisting of an imidazopyridine, a dihydropyrrolopyrazine, a pyrazolopyrimidine, and a pharmaceutically acceptable salt thereof
- a carrier and a binary buffer system comprising a carbonate salt and a bicarbonate salt, wherein the binary buffer system raises the pH of saliva to a pH greater than about 7.8, irrespective
- Figure 1 is a bar chart illustrating the relationship between the pH and membrane permeation for zolpidem tartrate.
- Figure 2 shows the mean dissolution profiles for a zolpidem quick-dissolving tablet and zolpidem lozenge of the present invention.
- Figure 3 shows the plasma concentration over time in each subject for Formulation A (zolpidem sublingual powdered tablet) at a 2 minute swallowing time.
- Figure 4 shows the plasma concentration over time in each subject for Formulation A at a 5 minute swallowing time.
- Figure 5 shows the plasma concentration over time in each subject for Formulation A at a 10 minute swallowing time.
- Figure 6 shows the mean plasma concentration over time for Formulation A at the 3 different swallowing times and for Formulation B (PO Ambien ® ), which was obtained from the literature.
- Figure 7 shows the mean plasma concentration over time for Formulation A at swallowing times of 2 and 5 minutes using the data from all subjects or excluding the data from subjects 3, 6, and 7.
- Figure 8 is an expanded view of the first 90 minutes shown in Figure 4.
- Figure 9 shows a representative plasma concentration over time for Formulation C (SL Tablet) at swallowing times of 2 and 5 minutes and for Formulation B.
- Figure 10 shows a representative plasma concentration over time for Formulation D (FS Tablet) at swallowing times of 2 and 5 minutes and for Formulation B.
- sleep disorder refers to a disruptive pattern of sleep arising from many causes including, without limitation, dysfunctional sleep mechanisms, abnormalities in physiological functions during sleep, abnormalities of the biological clock, and sleep disturbances that are induced by factors extrinsic to the sleep process.
- the term encompasses disorders associated with difficulties in staying asleep and/or falling asleep such as insomnia (e.g., transient, short-term, and chronic), delayed sleep phase syndrome, hypnotic-dependent sleep disorder, and stimulant-dependent sleep disorder; disorders associated with difficulties in staying awake such as sleep apnea, narcolepsy, restless leg syndrome, obstructive sleep apnea, central sleep apnea, idiopathic hypersomnia, respiratory muscle weakness-associated sleep disorder; disorders associated with difficulties in adhering to a regular sleep schedule such as sleep state misperception, shift work sleep disorder, chronic time zone change syndrome, and irregular sleep-wake syndrome; disorders associated with abnormal behaviors such as sleep terror disorder (i.e., parasomnia) and sleepwalking (i.e., somnambulism); and other disorders such as sleep bruxism, fibromyalgia, and nightmares.
- insomnia e.g., transient, short-term, and chronic
- delayed sleep phase syndrome hyp
- insomnia refers to a sleep disorder characterized by symptoms including, without limitation, difficulty in falling asleep, difficulty in staying asleep, intermittent wakefulness, and/or waking up too early. The term also encompasses daytime symptoms such as sleepiness, anxiety, impaired concentration, impaired memory, and irritability. Types of insomnia suitable for treatment with the compositions of the present invention include, without limitation, transient, short-term, and chronic insomnia.
- transient insomnia refers to msomnia lasting for a few nights.
- short-term insomnia refers to insomnia lasting for about two to about four weeks.
- chronic insomnia refers to insomnia lasting for at least one month.
- the terms "therapeutic agent” and “drug” are used interchangeably herein to refer to a substance having a pharmaceutical, pharmacological, psychosomatic, or therapeutic effect.
- the therapeutic agent or drug is a hypnotic agent.
- Suitable hypnotic agents for use in the present invention include, without limitation, an imidazopyridine compound such as zolpidem or alpidem; a dihydropyrrolopyrazine compound such as zopeclon; a pyrazolopyrimidine compound such as zaleplon or indiplon; pharmaceutically acceptable salts thereof; and combinations thereof.
- the hypnotic agent is zolpidem, in all suitable forms.
- a therapeutically effective amount refers to the amount of a hypnotic agent that is capable of achieving a therapeutic effect in a subject in need thereof.
- a therapeutically effective amount of a hypnotic agent can be the amount that is capable of preventing or relieving one or more symptoms associated with a sleep disorder.
- the term "bioavailability” refers to the rate and/or extent to which a drug is absorbed or becomes available to the treatment site in the body.
- the terms “disintegration” and “dissolution” are used interchangeably herein to refer to the reduction of a solid dosage form ofthe present invention to a liquid form. More particularly, a complete disintegration or dissolution of a solid dosage form refers to less than about 25% by weight ofthe solid dosage form remaining in the mouth following an appropriate time period, e.g., 5 minutes or less, after administration.
- Suitable methods known in the art for determining the disintegration profile of a solid dosage form include, e.g., the United States Pharmacopeia (USP) disintegration test.
- Suitable methods known in the art for determining the dissolution profile of a solid dosage form include, e.g., USP dissolution tests such as USP ⁇ 711> Apparatus 1 or USP ⁇ 711> Apparatus 2.
- the phrase "substantially complete conversion ofthe hypnotic agent from its ionized to its un-ionized form” refers to greater than about 50% conversion ofthe hypnotic agent from its ionized form into its un-ionized form.
- the buffer system may favor at least about 50%, 55%, 60%, 65%, 70%, 75%, 80%, 85%, 90%, 95%, or 99% conversion ofthe hypnotic agent from its ionized form into its un-ionized form. In some embodiments, the conversion occurs within about 10 minutes following administration.
- variable refers to inter-subject variability in terms ofthe percent of relative standard deviation (RSD) for the maximum plasma concentration (C max ) and the time to reach the maximum plasma concentration (T max ).
- RSD percent of relative standard deviation
- the compositions ofthe present invention have an RSD for C max of about 27% versus about 45% for commercial oral tablets such as Ambien ® tablets.
- the compositions ofthe present invention have an RSD for ⁇ i ax of about 50% versus about 100% for commercial oral tablets such as Ambien ® tablets.
- administering refers to administration ofthe compositions ofthe present invention to the mucous membranes ofthe oral cavity (i.e., oral mucosa).
- suitable sites of administration within the oral mucosa include, without limitation, the mucous membranes ofthe floor ofthe mouth (sublingual mucosa), the cheeks (buccal mucosa), the gums (gingival mucosa), the roof of the mouth (palatal mucosa), the lining ofthe lips, and combinations thereof.
- the compositions ofthe present invention are administered to the sublingual mucosa, buccal mucosa, or a combination thereof.
- the present invention provides novel compositions for the delivery of a hypnotic agent across the oral mucosa.
- the buffer system in the compositions of he present invention raises the pH of saliva to a pH greater than about 7.8, thereby facilitating the substantially complete conversion ofthe hypnotic agent from its ionized to its un-ionized form.
- the dose of hypnotic agent is rapidly and efficiently absorbed by the oral mucosa with surprisingly low inter-subject variability in terms of maximum plasma concentration (C m a x ) and the time to reach the maximum plasma concentration (T max ).
- delivery of the hypnotic agent across the oral mucosa advantageously bypasses hepatic first pass metabolism ofthe drag and avoids enzymatic degradation ofthe drug within the gastrointestinal tract, resulting in increased bioavailability ofthe hypnotic agent and reduced time to onset of therapeutic activity as compared to traditional dosage forms for oral (e.g., tablet) administration.
- Methods for using the compositions ofthe present invention for treating sleep disorders such as various types of insomnia are also provided.
- the present invention is based upon the surprising discovery that sublingual delivery of zolpidem compositions containing the buffer systems described herein provides both increased bioavailability ofthe therapeutic agent and reduced time to onset of therapeutic activity that far surpass those observed for commercial oral tablets such as Ambien ® tablets and buccal tablets such as zolpidem FlashDose ® tablets (Biovail Technologies Ltd.; Chantilly, VA).
- zolpidem FlashDose ® tablets Biovail Technologies Ltd.; Chantilly, VA
- the zolpidem in the compositions ofthe present invention reaches the systemic circulation in a substantially shorter period of time and at a substantially higher concentration than the zolpidem in either ofthe commercial tablet compositions.
- the zolpidem compositions ofthe present invention are superior to the commercial tablet compositions in reducing the time to onset of therapeutic activity, mamtaining sleep (e.g., total sleep time, number of awakenings), enhancing sleep quality, eliminating the effect of food, and reducing any morning-after residual effects.
- the present invention provides a solid composition for delivery of a hypnotic agent across the oral mucosa, the composition comprising: (a) a hypnotic agent selected from the group consisting of an imidazopyridine, a dihydropyrrolopyrazine, a pyrazolopyrimidine, and a pharmaceutically acceptable salt thereof; (b) a carrier that provides complete buccal or sublingual disintegration in about 5 minutes or less following administration to the mouth; and (c) a binary buffer system comprising a carbonate salt and a bicarbonate salt, wherein the binary buffer system raises the pH of saliva to a pH greater than about 7.8, irrespective of the starting pH of saliva.
- a hypnotic agent selected from the group consisting of an imidazopyridine, a dihydropyrrolopyrazine, a pyrazolopyrimidine, and a pharmaceutically acceptable salt thereof
- a carrier that provides complete buccal or sublingual disintegration in about 5 minutes or less following administration to the mouth
- the binary buffer system raises the pH of saliva to a pH greater than about 8.5, irrespective ofthe starting pH of saliva. In certain other instances, the binary buffer system raises the pH of saliva to a pH greater than about 9 (e.g., about 9-11), irrespective ofthe starting pH of saliva.
- the imidazopyridine hypnotic agent is selected from the group consisting of zolpidem and alpidem. Preferably, the imidazopyridine hypnotic agent is zolpidem.
- zolpidem is suitable for use in the compositions described herein, e.g., a salt form of zolpidem (e.g., zolpidem tartrate), a free base form of zolpidem, or a mixture thereof.
- the dihydropyrrolopyrazine hypnotic agent is zopeclon.
- the pyrazolopyrimidine hypnotic agent is selected from the group consisting of zaleplon and indiplon.
- the carrier provides complete buccal or sublingual disintegration in about 2 minutes or less following administration.
- the carrier comprises at least one binder and at least one disintegrating agent in such relative proportion to provide a buccal or sublingual disintegration time of about 5 minutes or less, preferably about 2 minutes or less, following administration.
- the ratio ofthe binder to the disintegrating agent is from about 0.1 to about 10.0, more preferably from about 0.1 to about 1.0, and most preferably from about 0.26 to about 0.79.
- the compositions ofthe present invention can be made without any binders, e.g., to produce a highly friable dosage form.
- the carbonate salt is selected from the group consisting of sodium carbonate, potassium carbonate, calcium carbonate, ammonium carbonate, and magnesium carbonate.
- the bicarbonate salt is selected from the group consisting of sodium bicarbonate, potassium bicarbonate, calcium bicarbonate, ammonium bicarbonate, and magnesium bicarbonate.
- the binary buffer system comprises sodium carbonate and sodium bicarbonate.
- the sodium bicarbonate is dessicant-coated sodium bicarbonate.
- the compositions ofthe present invention are in a dosage form selected from the group consisting of a lozenge, a chewing gum, a chewable tablet, and a dissolving tablet such as a slow-dissolving tablet or a quick-dissolving tablet.
- the composition is a lozenge or a dissolving tablet.
- a description of lozenge, chewing gum, chewable tablet, slow-dissolving tablet, and quick-dissolving tablet compositions containing a hypnotic agent is provided in the Examples below.
- the hypnotic agent is delivered across an oral mucosa selected from the group consisting ofthe sublingual mucosa, the buccal mucosa, and a combination thereof.
- the composition is administered sublingually so that the hypnotic agent is delivered across the sublingual mucosa.
- the carrier is selected from the group consisting of a binder, a gum base, and combinations thereof.
- Suitable binders for use in the compositions ofthe present invention include, without limitation, sugar alcohols such as mannitol, sorbitol, and xylitol; sugars such as lactose, dextrose, sucrose, glucose, and powdered sugar; natural gums such as acacia gum, xanthan gum, guar gum, tara gum, mesquite gum, fenugreek gum, locust bean gum, ghatti gum, and tragacanth gum; other substances such as inositol, molasses, maltodextrin, starch, cellulose, microcrystalline cellulose, polyvinylpyrrolidone, alginate, extract of Irish moss, panwar gum, mucilage of isapol husks, Veegum ® , larch arabogalactan, gelatin,
- Suitable gum bases for use in the compositions ofthe present invention include, for example, materials selected from among the many water- insoluble and saliva-insoluble gum base materials known in the art.
- the gum base comprises at least one hydrophobic polymer and at least one hydrophilic polymer.
- suitable hydrophobic and hydrophilic polymers for gum bases include both natural and synthetic polymers such as elastomers, rubbers, and combinations thereof.
- suitable natural polymers include, without limitation, substances of plant origin such as chicle, jelutong, gutta percha, crown gum, and combinations thereof.
- Suitable synthetic polymers include elastomers such as butadiene-styrene copolymers, isobutylene and isoprene copolymers (e.g., "butyl rubber"), polyethylene, polyisobutylene, polyvinylester (e.g., polyvinyl acetate and polyvinyl acetate phthalate), and combinations thereof.
- the gum base comprises a mixture of butyl rubber (i.e., isobutylene and isoprene copolymer), polyisobutylene, and optionally, polyvinylacetate (e.g., having a molecular weight of approximately 12,000).
- compositions ofthe present invention can further comprise a sweetening agent, a flavoring agent, a protecting agent, a plasticizer, a wax, an elastomeric solvent, a filler material, a preservative, or combinations thereof.
- the compositions ofthe present invention can further comprise a lubricating agent, a wetting agent, an emulsifying agent, a solubilizing agent, a suspending agent, a coloring agent, a disintegrating agent, or combinations thereof.
- the average particle size ofthe drug in the compositions described herein is about 20 microns, as compared to a typical average drug particle size of from about 75 to about 100 microns.
- the average particle size ofthe drug in the compositions described herein is less than or equal to the average particle size ofthe carrier ingredients (e.g., gum base, binders, etc.).
- the hypnotic agent is zolpidem and the binary buffer system comprises sodium carbonate and sodium bicarbonate.
- Such compositions are preferably formulated in the form of a lozenge, candy, or dissolving tablet (e.g., slow-dissolving tablet or quick-dissolving tablet) for sublingual administration.
- the sodium bicarbonate is dessicant-coated sodium bicarbonate. A combined weight percent of sodium carbonate and sodium bicarbonate that is greater than or equal to the weight percent of zolpidem is also preferred.
- the composition comprises from about 1.0 to about 5.5 weight percent zolpidem; from about 6.0 to about 10.0 weight percent sodium carbonate; and from about 9.0 to about 13.0 weight percent dessicant-coated sodium bicarbonate.
- the composition comprises about 4.5 weight percent zolpidem; about 8.0 weight percent sodium carbonate; and about 11.0 weight percent dessicant-coated sodium bicarbonate.
- compositions are preferably in the form of a lozenge or candy with a mass of from about 100 to about 300 mg, e.g., about 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290, and 300 mg.
- the lozenges or candies dissolve in a subject's mouth at a very rapid rate, e.g., within about 2-3 minutes following administration.
- the composition comprises from about 1.0 to about 5.0 weight percent zolpidem; from about 5.0 to about 9.0 weight percent sodium carbonate; and from about 7.0 to about 11.0 weight percent sodium bicarbonate.
- the composition comprises about 4.0 weight percent zolpidem; about 7.0 weight percent sodium carbonate; and about 9.0 weight percent sodium bicarbonate.
- a dissolving tablet such as a slow-dissolving tablet or a quick- dissolving tablet of from about 100 to about 300 mg, e.g., about 100, 110, 120, 130, 140, 150, 160, 170, 180, 190, 200, 210, 220, 230, 240, 250, 260, 270, 280, 290, and 300 mg.
- the quick-dissolving tablets dissolve in a subject's mouth at a rapid rate, e.g., within about 5 minutes following administration, and the slow-dissolving tablets dissolve in a subject's mouth at a slower rate, e.g., within about 10 minutes following administration.
- the present invention provides a composition for delivery of a hypnotic agent across the oral mucosa, the composition comprising: (a) a hypnotic agent selected from the group consisting of an imidazopyridine, a dihydropyrrolopyrazine, a pyrazolopyrimidine, and a pharmaceutically acceptable salt thereof; (b) a carrier; and (c) a binary buffer system comprising a carbonate salt and a bicarbonate salt, wherein the binary buffer system raises the pH of saliva to a pH greater than about 7.8, irrespective of the starting pH of saliva.
- the binary buffer system raises the pH of saliva to a pH greater than about 8.5, irrespective ofthe starting pH of saliva. In certain other instances, the binary buffer system raises the pH of saliva to a pH greater than about 9 (e.g., about 9-11), irrespective ofthe starting pH of saliva.
- Suitable imidazopyridine, dihydropyrrolopyrazine, and pyrazolopyrimidine hypnotic agents for use in the present invention are described above.
- Suitable carbonate salts and bicarbonate salts for use in the binary buffer systems ofthe present invention are also described above.
- compositions ofthe present invention are in any ofthe dosage forms described above.
- the hypnotic agent is delivered across an oral mucosa as described above.
- the carrier is selected from the group consisting of a binder, a gum base, and combinations thereof. Suitable binders and gum bases for use in the compositions ofthe present invention are described above.
- the carrier provides a buccal or sublingual disintegration time of about 10 minutes or less, preferably about 5 minutes or less, and more preferably about 2 minutes or less, following administration.
- the carrier comprises at least one binder and at least one disintegrating agent in such relative proportion to provide a buccal or sublingual disintegration time of about 10 minutes or less, preferably about 5 minutes or less, and more preferably about 2 minutes or less, following administration.
- compositions ofthe present invention can further comprise one or more ofthe additional agents described above.
- the average particle size ofthe drag in the compositions described herein is about 20 microns and/or is less than or equal to the average particle size ofthe carrier ingredients (e.g., gum base, binders, etc.).
- the hypnotic agent is zolpidem and the binary buffer system comprises sodium carbonate and sodium bicarbonate. Preferred amounts of each of these components is described above, along with preferred dosage forms and their preferred weight.
- the present invention provides a composition for delivery of a hypnotic agent across the oral mucosa, the composition comprising: (a) a hypnotic agent selected from the group consisting of an imidazopyridine, a dihydropyrrolopyrazine, a pyrazolopyrimidine, and a pharmaceutically acceptable salt thereof; (b) a carrier; and (c) a binary buffer system comprising a carbonate salt or a bicarbonate salt and a second buffering agent, wherem the binary buffer system raises the pH of saliva to a pH greater than about 7.8, irrespective of the starting pH of saliva.
- the binary buffer system raises the pH of saliva to a pH greater than about 8.5, irrespective of he starting pH of saliva. In certain other instances, the binary buffer system raises the pH of saliva to a pH greater than about 9 (e.g., about 9-11), irrespective ofthe starting pH of saliva.
- Suitable imidazopyridine, dihydropyrrolopyrazme, and pyrazolopyrimidine hypnotic agents for use in the present invention are described above.
- Suitable carbonate salts and bicarbonate salts for use in the binary buffer systems ofthe present invention are also described above.
- the second buffering agent is selected from the group consisting of a metal oxide, a citrate salt, a phosphate salt, a borate salt, an ascorbate salt, an acetate salt, and alkaline starch.
- Suitable metal oxides include, without limitation, magnesium oxide and aluminum oxide.
- the magnesium oxide is amorphous magnesium oxide.
- Suitable citrate, phosphate, and borate salts include, without limitation, any salt of citric acid, phosphoric acid, or boric acid known in the art.
- the citrate salt is selected from the group consisting of sodium citrate, potassium citrate, calcium citrate, magnesium citrate, and ammonium citrate.
- the phosphate salt is selected from the group consisting of monobasic sodium phosphate, dibasic sodium phosphate, monobasic potassium phosphate, dibasic potassium phosphate, monobasic calcium phosphate, dibasic calcium phosphate, monobasic magnesium phosphate, dibasic magnesium phosphate, monobasic ammonium phosphate, and dibasic ammonium phosphate.
- the borate salt is selected from the group consisting of sodium borate, potassium borate, calcium borate, magnesium borate, and ammonium borate.
- the binary buffer system comprises a carbonate salt and a metal oxide, a citrate salt, a phosphate salt, or a borate salt.
- the binary buffer system comprises a bicarbonate salt and a metal oxide, a citrate salt, a phosphate salt, or a borate salt.
- compositions ofthe present invention are in any ofthe dosage forms described above.
- the hypnotic agent is delivered across an oral mucosa as described above.
- the carrier is selected from the group consisting of a binder, a gum base, and combinations thereof. Suitable binders and gum bases for use in the compositions ofthe present invention are described above.
- the carrier provides a buccal or sublingual disintegration time as described above.
- the carrier comprises at least one binder and at least one disintegrating agent as described above.
- compositions ofthe present invention can further comprise one or more ofthe additional agents described above.
- the average particle size ofthe drug in the compositions described herein is about 20 microns and/or is less than or equal to the average particle size ofthe carrier ingredients (e.g., gum base, binders, etc.).
- the hypnotic agent is zolpidem and the binary buffer system comprises sodium carbonate or sodium bicarbonate and a second buffering agent.
- Such compositions are preferably formulated in the form of a lozenge, candy, or dissolving tablet for sublingual administration.
- the present invention provides a composition for delivery of a hypnotic agent across the oral mucosa, the composition comprising: (a) a hypnotic agent selected from the group consisting of an imidazopyridine, a dihydropyrrolopyrazine, a pyrazolopyrimidine, and a pharmaceutically acceptable salt thereof; (b) a carrier; and (c) a binary buffer system comprising a metal oxide and a citrate, phosphate, or borate salt, wherein the binary buffer system raises the pH of saliva to a pH greater than about 7.8, irrespective ofthe starting pH of saliva.
- the binary buffer system raises the pH of saliva to a pH greater than about 8.5, irrespective ofthe starting pH of saliva. In certain other instances, the binary buffer system raises the pH of saliva to a pH greater than about 9 (e.g., about 9-11), irrespective ofthe starting pH of saliva.
- Suitable imidazopyridine, dihydropynolopyrazine, and pyrazolopyrimidine hypnotic agents for use in the present invention are described above.
- Suitable metal oxides include, without limitation, magnesium oxide and aluminum oxide.
- Suitable citrate, phosphate, and borate salts include, without limitation, any salt of citric acid, phosphoric acid, or boric acid known in the art such as those described above.
- the binary buffer system comprises a metal oxide and a citrate salt.
- the binary buffer system comprises a metal oxide and a phosphate salt.
- the binary buffer system comprises a metal oxide and a borate salt.
- the compositions ofthe present invention are in any ofthe dosage forms described above.
- the hypnotic agent is delivered across an oral mucosa as described above.
- the carrier is selected from the group consisting of a binder, a gum base, and combinations thereof. Suitable binders and gum bases for use in the compositions ofthe present invention are described above.
- the carrier provides a buccal or sublingual disintegration time as described above.
- the carrier comprises at least one binder and at least one disintegrating agent as described above.
- compositions ofthe present invention can further comprise one or more ofthe additional agents described above.
- the average particle size ofthe drag in the compositions described herein is about 20 microns and/or is less than or equal to the average particle size ofthe carrier ingredients (e.g., gum base, binders, etc.).
- the hypnotic agent is zolpidem and the binary buffer system comprises amorphous magnesium oxide and a citrate, phosphate, or borate salt.
- Such compositions are preferably formulated in the form of a lozenge, candy, or dissolving tablet for sublingual aciministration.
- the present invention provides a composition for delivery of a hypnotic agent across the oral mucosa, the composition comprising: (a) a hypnotic agent selected from the group consisting of an imidazopyridine, a dihydropynolopyrazine, a pyrazolopyrimidine, and a pharmaceutically acceptable salt thereof; (b) a carrier; and (c) a ternary buffer system comprising a carbonate salt, a bicarbonate salt, and a third buffering agent, wherein the ternary buffer system raises the pH of saliva to a pH greater than about 7.8, irrespective ofthe starting pH of saliva.
- the binary buffer system raises the pH of saliva to a pH greater than about 8.5, irrespective ofthe starting pH of saliva. In certain other instances, the binary buffer system raises the pH of saliva to a pH greater than about 9 (e.g., about 9-11), rrespective ofthe starting pH of saliva.
- Suitable imidazopyridine, dihydropyrrolopyrazine, and pyrazolopyrimidine hypnotic agents for use in the present invention are described above.
- Suitable carbonate salts and bicarbonate salts for use in the ternary buffer systems ofthe present invention are also described above.
- the third buffering agent is selected from the group consisting of a metal oxide, a citrate salt, a phosphate salt, a borate salt, an ascorbate salt, an acetate salt, and alkaline starch.
- Suitable metal oxides include, without limitation, magnesium oxide and aluminum oxide.
- Suitable citrate, phosphate, and borate salts include, without limitation, any salt of citric acid, phosphoric acid, or boric acid known in the art such as those described above, hi certain instances, the ternary buffer system comprises a carbonate salt, a bicarbonate salt, and a metal oxide. In certain other instances, the ternary buffer system comprises a carbonate salt, a bicarbonate salt, and a citrate, phosphate, or borate salt.
- compositions ofthe present mvention are in any ofthe dosage forms described above.
- the hypnotic agent is delivered across an oral mucosa as described above.
- the carrier is selected from the group consisting of a binder, a gum base, and combinations thereof. Suitable binders and gum bases for use in the compositions ofthe present invention are described above.
- the carrier provides a buccal or sublingual disintegration time as described above.
- the carrier comprises at least one binder and at least one disintegrating agent as described above.
- compositions ofthe present invention can further comprise one or more ofthe additional agents described above.
- the average particle size ofthe drag in the compositions described herein is about 20 microns and/or is less than or equal to the average particle size ofthe carrier ingredients (e.g., gum base, binders, etc.).
- the hypnotic agent is zolpidem and the ternary buffer system comprises sodium carbonate, sodium bicarbonate, and a third buffering agent.
- Such compositions are preferably formulated in the form of a lozenge, candy, or dissolving tablet for sublingual administration.
- the third buffering agent is a metal oxide
- a weight percent ofthe metal oxide that is greater than the combined weight percent of sodium carbonate and sodium bicarbonate is prefened.
- the present invention provides a composition for delivery of a hypnotic agent across the oral mucosa, the composition comprising: (a) a hypnotic agent selected from the group consisting of an imidazopyridine, a dihydropynolopyrazine, a pyrazolopyrimidine, and a pharmaceutically acceptable salt thereof; (b) a carrier; and (c) a buffer system comprising a carbonate salt or a bicarbonate salt and two or more buffering agents selected from the group consisting of a metal oxide, a citrate salt, a phosphate salt, and a borate salt, wherein the buffer system raises the pH of saliva to a pH greater than about 7.8, inespective ofthe starting pH of saliva.
- the binary buffer system raises the pH of saliva to a pH greater than about 8.5, inespective ofthe starting pH of saliva. In certain other instances, the binary buffer system raises the pH of saliva to a pH greater than about 9 (e.g., about 9-11), inespective ofthe starting pH of saliva.
- Suitable imidazopyridine, dihydropynolopyrazine, and pyrazolopyrimidine hypnotic agents for use in the present invention are described above.
- Suitable carbonate salts and bicarbonate salts for use in the buffer systems ofthe present invention are also described above.
- Suitable metal oxides include, without limitation, magnesium oxide and aluminum oxide.
- Suitable citrate, phosphate, and borate salts include, without limitation, any salt of citric acid, phosphoric acid, or boric acid known in the art such as those described above.
- the buffer system comprises a carbonate salt or a bicarbonate salt, a metal oxide, and a citrate, phosphate, or borate salt.
- the buffer system comprises a carbonate salt or a bicarbonate salt, a citrate salt, and a phosphate salt.
- the buffer system comprises a carbonate salt or a bicarbonate salt, a citrate salt, and a borate salt.
- the buffer system comprises a carbonate salt or a bicarbonate salt, a phosphate salt, and a borate salt.
- the compositions ofthe present invention are in any ofthe dosage forms described above.
- the hypnotic agent is delivered across an oral mucosa as described above.
- the carrier is selected from the group consisting of a binder, a gum base, and combinations thereof. Suitable binders and gum bases for use in the compositions ofthe present invention are described above.
- the carrier provides a buccal or sublingual disintegration time as described above.
- the carrier comprises at least one binder and at least one disintegrating agent as described above.
- compositions ofthe present mvention can further comprise one or more ofthe additional agents described above.
- the average particle size ofthe drag in the compositions described herein is about 20 microns and/or is less than or equal to the average particle size ofthe carrier ingredients (e.g., gum base, binders, etc.).
- the hypnotic agent is zolpidem and the buffer system comprises sodium carbonate or sodium bicarbonate and two or more buffering agents selected from the group consisting of a metal oxide, a citrate salt, a phosphate salt, and a borate salt.
- Such compositions are preferably formulated in the form of a lozenge, candy, or dissolving tablet for sublingual administration.
- the present invention provides a method for treating a sleep disorder in a subject in need thereof, the method comprising: administering to the subject a composition comprising a therapeutically effective amount of a hypnotic agent selected from the group consisting of an imidazopyridine, a dihydropynolopyrazine, a pyrazolopyrimidine, and a pharmaceutically acceptable salt thereof; a carrier; and a binary buffer system comprising a carbonate salt and a bicarbonate salt, wherein the binary buffer system raises the pH of saliva to a pH greater than about 7.8, inespective ofthe starting pH of saliva.
- a hypnotic agent selected from the group consisting of an imidazopyridine, a dihydropynolopyrazine, a pyrazolopyrimidine, and a pharmaceutically acceptable salt thereof
- a carrier and a binary buffer system comprising a carbonate salt and a bicarbonate salt, wherein the binary buffer system raises the pH of saliva to a pH greater than about
- the composition delivers the hypnotic agent across the oral mucosa such as, for example, the sublingual mucosa, the buccal mucosa, or a combination thereof.
- the composition is administered sublingually so that the hypnotic agent is delivered across the sublingual mucosa.
- Suitable sleep disorders that can be treated with the compositions ofthe present invention include, without limitation, insomnia such as transient insomnia, short-term insomnia, and chronic insomnia.
- the binary buffer system raises the pH of saliva to a pH greater than about 8.5, inespective ofthe starting pH of saliva. In certain other instances, the binary buffer system raises the pH of saliva to a pH greater than about 9 (e.g., about 9-11), inespective ofthe starting pH of saliva.
- Suitable imidazopyridine, dihydropynolopyrazine, and pyrazolopyrimidine hypnotic agents for use in the present invention are described above.
- Suitable carbonate salts and bicarbonate salts for use in the binary buffer systems ofthe present invention are also described above.
- the binary buffer system comprises a carbonate salt or a bicarbonate salt and a second buffering agent such as a metal oxide, a citrate salt, a phosphate salt, a borate salt, an ascorbate salt, an acetate salt, and alkaline starch.
- the binary buffer system comprises a metal oxide and a citrate, phosphate, or borate salt.
- the buffer system is a ternary buffer system comprising a carbonate salt, a bicarbonate salt, and a third buffering agent such as a metal oxide, a citrate salt, a phosphate salt, a borate salt, an ascorbate salt, an acetate salt, and alkaline starch.
- the buffer system comprises a carbonate salt or a bicarbonate salt and two or more buffering agents selected from the group consisting of a metal oxide, a citrate salt, a phosphate salt, and a borate salt.
- the compositions ofthe present invention are in any ofthe dosage forms described above.
- the hypnotic agent is delivered across an oral mucosa as described above.
- the carrier is selected from the group consisting of a binder, a gum base, and combinations thereof. Suitable binders and gum bases for use in the compositions ofthe present invention are described above.
- the carrier provides a buccal or sublingual disintegration time as described above.
- the carrier comprises at least one binder and at least one disintegrating agent as described above.
- compositions ofthe present invention can further comprise one or more ofthe additional agents described above.
- the average particle size ofthe drug in the compositions described herein is about 20 microns and/or is less than or equal to the average particle size ofthe carrier ingredients (e.g., gum base, binders, etc.).
- the hypnotic agent is zolpidem and the binary buffer system comprises sodium carbonate and sodium bicarbonate. Prefened amounts of each of these components is described above, along with prefened dosage forms and their prefened weight.
- the hypnotic agents ofthe present invention are preferably selected from an imidazopyridine compound such as zolpidem or alpidem; a dihydropynolopyrazine compound such as zopeclon; a pyrazolopyrimidine compound such as zaleplon or indiplon; pharmaceutically acceptable salts thereof; and combinations thereof. More preferably, the hypnotic agent is zolpidem, in all suitable forms. [0092] In general, the hypnotic agents ofthe present invention are basic compounds having an ionized form and an un-ionized form. In certain instances, the hypnotic agent is initially present at least partly in an ionized form.
- the hypnotic agent is initially present in an un-ionized form.
- the buffer system ofthe compositions described herein helps to convert substantially all ofthe hypnotic agent from its ionized form to its un-ionized form.
- the buffer system helps ensure that the hypnotic agent, initially in an un-ionized form, remains in an un-ionized form.
- the term "hypnotic agent” includes all pharmaceutically acceptable forms ofthe hypnotic agent being described.
- the hypnotic agent can be in a racemic or isomeric mixture, a solid complex bound to an ion exchange resin, or the like.
- the hypnotic agent can be in a solvated form.
- the term "hypnotic agent” is also intended to include all pharmaceutically acceptable salts, derivatives, and analogs ofthe hypnotic agent being described, as well as combinations thereof.
- the pharmaceutically acceptable salts ofthe hypnotic agent include, without limitation, the tartrate, succinate, tartarate, bitartarate, dihydrochloride, salicylate, hemisuccinate, citrate, maleate, hydrochloride, carbamate, sulfate, nitrate, and benzoate salt forms thereof, as well as combinations thereof and the like.
- Any form ofthe hypnotic agent is suitable for use in the compositions ofthe present invention, e.g., a pharmaceutically acceptable salt ofthe hypnotic agent (e.g., zolpidem tartrate), a free base ofthe hypnotic agent, or a mixture thereof.
- pH pKa + Log 10 (un-ionized concentration ionized concentration).
- equimolar concentrations ofthe unionized form and ionized form exist.
- the pH is one unit higher than the pKa
- the ratio ofthe un-ionized form to the ionized form is 91:9.
- the pH is two units higher than the pKa
- the ratio of un-ionized form to the ionized form is 100:1.
- the un-ionized form is lipophilic and, therefore, more capable of passing through mucous membranes such as the oral mucosa than the ionized form, which is lipophobic in nature. Accordingly, increasing the pH ofthe saliva favors conversion ofthe ionized form into the un-ionized form for basic compounds such as the hypnotic agents described herein, and the final pH can be determined by making use ofthe above formula.
- the hypnotic agents ofthe present invention are selected from the class of compounds in the imidazopyridine, dihydropynolopyrazine, or pyrazolopyrimidine family and are useful in the treatment of conditions such as sleep disorders.
- imidazopyridine compounds for use in the present invention are zolpidem, alpidem, pharmaceutically acceptable salts thereof, analogs thereof, and derivatives thereof. These imidazopyridine compounds each have an imidazopyridine group, as shown below:
- the nitrogen in the imidazole portion ofthe bicyclic ring ofthe structure controls the extent of ionization and the degree of lipophilicity in any given medium.
- the nitrogen in the imidazole portion imparts a pKa of from about 6.8 to about 7.5 to the molecule. Therefore, using the above formula, it can be demonstrated that about 90% conversion to an un-ionized form can be achieved for these compounds at apH of from about 7.8 to about 8.5.
- dihydropynolopyrazine compounds for use in the present invention are zopeclon, pharmaceutically acceptable salts thereof, analogs thereof, and derivatives thereof. These dihydropynolopyrazines each have a dihydropynolopyrazine group, as shown below: Zopeclon
- pyrazolopyrimidine compounds for use in the present invention are zaleplon, indiplon, pharmaceutically acceptable salts thereof, analogs thereof, and derivatives thereof.
- pyrazolopyrimidmes each have a pyrazolopyrimidine group, as shown below:
- the nitrogen in the pyrimidine group controls the extent of ionization and the degree of lipophilicity in any given medium.
- the nitrogen in the pyrimidine group imparts a pKa of from about 8 to about 9 to the molecule. Therefore, using the above formula, it can be demonstrated that about 90% conversion to an un-ionized form can be achieved for these compounds at a pH of from about 9 to about 10.
- the hypnotic agents ofthe present invention acts as benzodiazepine receptor agonists.
- the hypnotic agents selectively bind to the benzodiazepine ! receptor.
- the therapeutic activity ofthe hypnotic agents ofthe present invention in treating sleep disorders is attributed to an enhancement ofthe inhibitory action of gamma-aminobutyric acid (GAB A) in the central nervous system.
- GAB A gamma-aminobutyric acid
- the buffer systems ofthe compositions described herein are capable of raising the pH of saliva to a pH greater than about 7.8, inespective ofthe starting pH of saliva.
- the buffer system helps convert substantially all ofthe hypnotic agent from its ionized form to its un-ionized form.
- the buffer system helps ensure that the hypnotic agent, initially in an un-ionized form, remains in an un-ionized form.
- basic buffering agents are typically used in the buffer systems ofthe present invention, one skilled in the art will appreciate that acidic agents can also be used to adjust the pH ofthe buffer system as long as the buffer system as a whole raises the pH of saliva to a pH greater than about 7.8.
- the present invention provides binary buffer systems comprising a carbonate salt and a bicarbonate salt.
- concentration of each buffer system component is tailored such that the final salivary pH is achieved and sustained for a period of time, e.g., for at least about 2 minutes, at least about 5 minutes, at least about 10 minutes, at least about 20 minutes, or at least about 60 minutes. This typically involves a sensory and safety trial and enor type of procedure of adding various amounts of each buffer system component and then measuring the final pH over time. In this way, selection of an appropriate weight ratio for each buffer system component can be easily determined in just a few trials.
- the weight ratio of carbonate salt to bicarbonate salt can be from about 1:10 to about 10:1, preferably from about 1:5 to about 5:1, more preferably from about 1 :3 to about 3:1, and still more preferably from about 1 :2 to about 2:1.
- the carbonate salt is generally selected from sodium carbonate, potassium carbonate, calcium carbonate, ammonium carbonate, and magnesium carbonate.
- the carbonate salt is sodium carbonate or potassium carbonate.
- the carbonate salt is sodium carbonate.
- the bicarbonate salt is generally selected from sodium bicarbonate, potassium bicarbonate, calcium bicarbonate, ammonium bicarbonate, and magnesium bicarbonate.
- the bicarbonate salt is sodium bicarbonate or potassium bicarbonate.
- the bicarbonate salt is sodium bicarbonate.
- a dessicant-coated sodium bicarbonate is prefened.
- the amount of carbonate salt and bicarbonate salt used in the binary buffer system is an amount that is sufficient to raise salivary pH to a pH of about 7.8 or more, preferably about 8.5 or more, and more preferably about 9 or more (e.g., about 9-11), inespective ofthe starting pH.
- the amount of bicarbonate salt is greater than or equal to the amount of carbonate salt
- the weight ratio of carbonate salt to bicarbonate salt is from about 1 : 1 to about 1:10, preferably from about 1 : 1 to about 1:5, and more preferably from about 1:1 to about 1:2, e.g., 1:1, 1:1.1, 1:1.2, 1:1.3, 1:1.4, 1:1.5, 1:1.6, 1:1.7, 1:1.8, 1:1.9, or 1 :2.
- the amount of bicarbonate salt is less than or equal to the amount of carbonate salt
- the weight ratio of carbonate salt to bicarbonate salt is from about 1 :1 to about 10:1, preferably from about 1 : 1 to about 5:1, and more preferably from about 1 : 1 to about 2:1, e.g., 1:1, 1.1:1, 1.2:1, 1.3:1, 1.4:1, 1.5:1, 1.6:1, 1.7:1, 1.8:1, 1.9:1, or 2:1.
- the combined amount of carbonate salt and bicarbonate salt is greater than or equal to the amount ofthe hypnotic agent, and the weight ratio of carbonate salt and bicarbonate salt to hypnotic agent is preferably from about 1:1 to about 10:1, e.g., 1:1, 2:1, 3:1, 4:1, 5:1, 6:1, 7:1, 8:1, 9:1, or 10:1.
- the combined amount of carbonate salt and bicarbonate salt is less than or equal to the amount ofthe hypnotic agent, and the weight ratio of carbonate salt and bicarbonate salt to hypnotic agent is preferably from about 1:1 to about 1:10, e.g., 1:1, 1:2, 1:3, 1:4, 1:5, 1:6, 1:7, 1:8, 1:9, or 1:10.
- the buffer systems ofthe present invention are binary buffer systems containing sodium carbonate and sodium bicarbonate.
- the buffer systems ofthe present invention are binary buffer systems comprising a carbonate salt or a bicarbonate salt and a second buffering agent.
- concentration of each buffer system component is tailored such that the final salivary pH is achieved and sustained for a period of time, e.g., for at least about 2 minutes, at least about 5 minutes, at least about 10 minutes, at least about 20 minutes, or at least about 60 minutes.
- Suitable carbonate salts and bicarbonate salts are described above.
- the amount of carbonate salt or bicarbonate salt used in the binary buffer system is an amount that is sufficient, when used with the second buffering agent, to raise salivary pH to a pH of about 7.8 or more, preferably about 8.5 or more, and more preferably about 9 or more (e.g., about 9-11), inespective ofthe starting pH.
- the amount ofthe second buffering agent in the binary buffer system is greater than or equal to the amount ofthe carbonate salt or bicarbonate salt.
- the weight ratio ofthe second buffering agent to the carbonate salt or bicarbonate salt can be from about 1:1 to about 10:1, preferably from about 1 : 1 to about 5:1, and more preferably from about 1 : 1 to about 3 : 1.
- the amount ofthe second buffering agent in the binary buffer system is less than or equal to the amount ofthe carbonate salt or bicarbonate salt.
- the weight ratio ofthe second buffering agent to the carbonate salt or bicarbonate salt can be from about 1 : 1 to about 1:10, preferably from about 1:1 to about 1:5, and more preferably from about 1:1 to about 1:3.
- the second buffering agent is generally selected from a metal oxide such as magnesium oxide or aluminum oxide; a citrate salt such as sodium citrate, potassium citrate, calcium citrate, magnesium citrate, and ammonium citrate; a phosphate salt such as monobasic sodium phosphate, dibasic sodium phosphate, monobasic potassium phosphate, dibasic potassium phosphate, monobasic calcium phosphate, dibasic calcium phosphate, monobasic magnesium phosphate, dibasic magnesium phosphate, monobasic ammonium phosphate, and dibasic ammonium phosphate; a borate salt such as sodium borate, potassium borate, calcium borate, magnesium borate, and ammonium borate; an ascorbate salt such as potassium ascorbate or sodium ascorbate; an acetate salt such as potassium acetate or sodium acetate; and alkaline starch.
- a metal oxide such as magnesium oxide or aluminum oxide
- a citrate salt such as sodium citrate, potassium citrate, calcium citrate, magnesium cit
- any metal oxide or salt of citric acid, phosphoric acid, boric acid, ascorbic acid, or acetic acid is suitable for use in the buffer systems ofthe present invention.
- the amount ofthe second buffering agent used in the binary buffer system is an amount that is sufficient, when used with the carbonate salt or bicarbonate salt, to raise salivary pH to a pH of about 7.8 or more, preferably about 8.5 or more, and more preferably about 9 or more (e.g., about 9-11), inespective ofthe starting pH.
- a metal oxide such as magnesium oxide or aluminum oxide is the prefened second buffering agent.
- the metal oxide is amorphous magnesium oxide.
- the buffer systems ofthe present invention are binary buffer systems comprising a metal oxide and a citrate, phosphate, or borate salt.
- concentration of each buffer system component is tailored such that the final salivary pH is achieved and sustained for a period of time, e.g., for at least about 2 minutes, at least 5 about minutes, at least about 10 minutes, at least about 20 minutes, or at least about 60 minutes.
- Suitable metal oxides include, without limitation, magnesium oxide and aluminum oxide.
- Suitable citrate, phosphate, and borate salts include, without limitation, essentially any salt of citric acid, phosphoric acid, or boric acid known in the art such as those described above.
- the binary buffer system comprises a metal oxide and a citrate salt.
- the binary buffer system comprises a metal oxide and a phosphate salt.
- the binary buffer system comprises a metal oxide and a borate salt.
- the amount ofthe metal oxide used in the binary buffer system is an amount that is sufficient, when used with the citrate, phosphate, or borate salt, to raise salivary pH to a pH of about 7.8 or more, preferably about 8.5 or more, and more preferably about 9 or more (e.g., about 9-11), inespective ofthe starting pH.
- the amount ofthe citrate, phosphate, or borate salt used in the binary buffer system is an amount that is sufficient, when used with the metal oxide, to raise salivary pH to a pH of about 7.8 or more, preferably about 8.5 or more, and more preferably about 9 or more (e.g., about 9-11), inespective ofthe starting pH.
- the amount ofthe metal oxide in the binary buffer system is greater than or equal to the amount ofthe citrate, phosphate, or borate salt.
- the weight ratio ofthe metal oxide to the citrate, phosphate, or borate salt can be from about 1:1 to about 10:1, preferably from about 1:1 to about 5:1, and more preferably from about 1:1 to about 3 : 1.
- the amount of the metal oxide in the binary buffer system is less than or equal to the amount ofthe citrate, phosphate, or borate salt.
- the weight ratio ofthe metal oxide to the citrate, phosphate, or borate salt can be from about 1:1 to about 1:10, preferably from about 1:1 to about 1:5, and more preferably from about 1:1 to about 1:3.
- the buffer systems ofthe present invention are ternary buffer systems comprising a carbonate salt, a bicarbonate salt, and a third buffering agent.
- concentration of each buffer system component is tailored such that the final salivary pH is achieved and sustained for a period of time, e.g., for at least about 2 minutes, at least 5 about minutes, at least about 10 minutes, at least about 20 minutes, or at least about 60 minutes.
- the procedure described above for deterrnining an appropriate weight ratio for each buffer system component can also be applied to ternary buffer systems.
- Suitable carbonate salts and bicarbonate salts are described above.
- the amount of carbonate salt and bicarbonate salt used in the ternary buffer system is an amount that is sufficient, when used with the third buffering agent, to raise salivary pH to a pH of about 7.8 or more, preferably about 8.5 or more, and more preferably about 9 or more (e.g., about 9- 11), inespective ofthe starting pH.
- the third buffering agent is generally selected from a metal oxide, a citrate salt, a phosphate salt, a borate salt, an ascorbate salt such as potassium ascorbate or sodium ascorbate, an acetate salt such as potassium acetate or sodium acetate, and alkaline starch.
- Suitable metal oxides include, without limitation, magnesium oxide and aluminum oxide.
- Suitable citrate, phosphate, and borate salts include, without limitation, any salt of citric acid, phosphoric acid, or boric acid known in the art such as those described above.
- the amount of the third buffering agent used in the ternary buffer system is an amount that is sufficient, when used with the remaining components, to raise salivary pH to a pH of about 7.8 or more, preferably about 8.5 or more, and more preferably about 9 or more (e.g., about 9-11), inespective ofthe starting pH.
- a metal oxide such as magnesium oxide or aluminum oxide is the prefened third buffering agent.
- the metal oxide is amorphous magnesium oxide.
- the amount ofthe carbonate salt or bicarbonate salt in the ternary buffer system is greater than or equal to the amount ofthe third buffering agent.
- the weight ratio ofthe carbonate salt or bicarbonate salt to the third buffering agent can be from about 1 : 1 to about 10:1, preferably from about 1 : 1 to about 5:1, and more preferably from about 1:1 to about 3:1.
- the amount ofthe carbonate salt or bicarbonate salt in the ternary buffer system is less than or equal to the amount ofthe third buffering agent.
- the weight ratio ofthe carbonate salt or bicarbonate salt to the third buffering agent can be from about 1 : 1 to about 1:10, preferably from about 1 : 1 to about 1 :5, and more preferably from about 1 : 1 to about 1:3.
- the ternary buffer systems of the present invention in some of the most prefened embodiments, contain sodium carbonate, sodium bicarbonate, and amorphous magnesium oxide.
- the amount of sodium bicarbonate is greater than or equal to the amount of sodium carbonate.
- the weight ratio of sodium bicarbonate to sodium carbonate can be from about 1 :1 to about 10:1, preferably from about 1 :1 to about 5:1, and more preferably from about 1:1 to about 3:1.
- the amount of amorphous magnesium oxide is greater than or equal to the combined amount of sodium carbonate and sodium bicarbonate.
- the weight ratio of amorphous magnesium oxide to sodium carbonate and sodium bicarbonate can be from about 1:1 to about 10:1, preferably from about 1:1 to about 5:1, and more preferably from about 1:1 to about 3:1.
- the buffer systems ofthe present invention are buffer systems comprising a carbonate salt or a bicarbonate salt and two or more buffering agents selected from the group consisting of a metal oxide, a citrate salt, a phosphate salt, and a borate salt.
- concentration of each buffer system component is tailored such that the final salivary pH is achieved and sustained for a period of time, e.g., for at least about 2 minutes, at least 5 about minutes, at least about 10 minutes, at least about 20 minutes, or at least about 60 minutes.
- Suitable carbonate salts and bicarbonate salts are described above.
- the amount of carbonate salt or bicarbonate salt used in the buffer system is an amount that is sufficient, when used with the remaining components, to raise salivary pH to a pH of about 7.8 or more, preferably about 8.5 or more, and more preferably about 9 or more (e.g., about 9-11), inespective ofthe starting pH.
- the two or more buffering agents are generally selected from a metal oxide, a citrate salt, a phosphate salt, a borate salt, an ascorbate salt, an acetate salt, and alkaline starch.
- Suitable metal oxides include, without limitation, magnesium oxide and aluminum oxide.
- Suitable citrate, phosphate, borate, ascorbate, and acetate salts include, without limitation, essentially any salt of citric acid, phosphoric acid, boric acid, ascorbic acid, or acetic acid known in the art such as those described above.
- the amount ofthe additional buffering agents used in the buffer system is an amount that is sufficient, when used with the carbonate salt or bicarbonate salt, to raise salivary pH to a pH of about 7.8 or more, preferably about 8.5 or more, and more preferably about 9 or more (e.g., about 9-11), inespective ofthe starting pH.
- the buffer system comprises a carbonate salt or a bicarbonate salt, a metal oxide, and a citrate, phosphate, or borate salt.
- the buffer system comprises a carbonate salt or a bicarbonate salt, a citrate salt, and a phosphate salt.
- the buffer system comprises a carbonate salt or a bicarbonate salt, a citrate salt, and a borate salt, h certain other instances, the buffer system comprises a carbonate salt or a bicarbonate salt, a phosphate salt, and a borate salt.
- the metal oxide is amorphous magnesium oxide.
- the amount ofthe carbonate salt or bicarbonate salt in the buffer system is greater than or equal to the amount ofthe metal oxide or the citrate, phosphate, or borate salt.
- the weight ratio ofthe carbonate salt or bicarbonate salt to the metal oxide or the citrate, phosphate, or borate salt can be from about 1 : 1 to about 10:1, preferably from about 1 : 1 to about 5:1, and more preferably from about 1 : 1 to about 3 : 1.
- the amount ofthe carbonate salt or bicarbonate salt in the buffer system is less than or equal to the amount ofthe metal oxide or the citrate, phosphate, or borate salt.
- the weight ratio ofthe carbonate salt or bicarbonate salt to the metal oxide or the citrate, phosphate, or borate salt can be from about 1 : 1 to about 1:10, preferably from about 1 : 1 to about 1:5, and more preferably from about 1 : 1 to about 1:3.
- the buffer system may also have subsidiary beneficial effects on the extent of absorption across the oral mucosa.
- the buffer system may create a final salivary pH that in turn affects the molecular configuration ofthe therapeutic agent in a way in which absorption across the oral mucosa is increased. It is to be understood that these subsidiary beneficial effects ofthe buffer system are within the general scope ofthe buffer system and compositions herein described.
- compositions of the present invention may take the form of solid, semi-solid, lyophilized powder, or liquid dosage forms, such as, for example, tablets (e.g., chewable, slow-dissolving, quick-dissolving), pills, capsules, lozenges, gums, powders, solutions, suspensions, emulsions, aerosols, or the like.
- the dosage form is a chewing gum, dissolving tablet, chewable tablet, candy, or lozenge.
- dosage forms such as chewing gums, chewable tablets, dissolving tablets, or lozenges containing a buffer system described herein offer advantages over the traditional dosage forms for oral administration (i.e., Ambien ® ).
- each of these dosage forms avoids hepatic first pass metabolism, degradation within the gastrointestinal tract, and drug loss during absorption. Consequently, the amount of therapeutic agent required per dose is less than that which would be required if formulated, for example, in a pill or tablet for oral administration.
- the bioavailability ofthe therapeutic agent is increased, thereby reducing the time to onset of therapeutic activity as compared to traditional dosage forms for oral adniinistration (see, Example 5 below).
- the prefened dosage forms ofthe present invention e.g., chewing gums, chewable tablets, dissolving tablets, lozenges
- a buffer system described herein offer advantages over dosage forms for oral mucosal administration that do not contain the buffer system (i.e., zolpidem FlashDose ® tablet)
- the buffer system in the dosage forms ofthe present invention helps convert substantially all ofthe therapeutic agent from its ionized form to its un-ionized form, the bioavailability ofthe therapeutic agent is increased, thereby reducing the time to onset of therapeutic activity as compared to dosage forms for oral mucosal administration that do not contain the buffer system.
- zolpidem FlashDose ® tablet has a pharmacokinetic profile similar to that observed for the orally administered Ambien ® tablet.
- the zolpidem compositions ofthe present invention surpass both commercial tablet compositions by providing an increase in the bioavailability of zolpidem and a reduction in the time to onset of therapeutic activity.
- dosage form refers to physically discrete units suitable as unitary dosages for human subjects and other mammals, each unit containing a predetermined quantity of therapeutic agent calculated to produce the desired onset, tolerability, and therapeutic effects, in association with one or more suitable pharmaceutical excipients such as carriers. Methods for preparing such dosage forms are known or will be apparent to those skilled in the art. For example, in some embodiments, a chewing gum dosage form ofthe present invention can be prepared according to the procedures set forth in U.S. Pat. No. 4,405,647.
- a tablet, lozenge, or candy dosage form ofthe present invention can be prepared according to the procedures set forth, for example, in Remington: The Science and Practice of Pharmacy, 20 th Ed., Lippincott, Williams & Wilkins (2003); Pharmaceutical Dosage Forms, Volume 1: Tablets, 2 nd Ed., Marcel Dekker, Inc., New York, N.Y. (1989); and similar publications.
- the dosage form to be administered will, in any event, contain a quantity ofthe therapeutic agent in a therapeutically effective amount for relief of the condition being treated when administered in accordance with the teachings of this invention.
- carrier refers to a typically inert substance used as a diluent or vehicle for a drug such as a therapeutic agent.
- the term also encompasses a typically inert substance that imparts cohesive qualities to the composition.
- Suitable carriers for use in the compositions ofthe present invention include, without limitation, a binder, a gum base, and combinations thereof.
- Non-limiting examples of binders include mannitol, sorbitol, xylitol, maltodextrin, lactose, dextrose, sucrose, glucose, inositol, powdered sugar, molasses, starch, cellulose, microcrystalline cellulose, polyvinylpynolidone, acacia gum, guar gum, tragacanth gum, alginate, extract of Irish moss, panwar gum, ghatti gum, mucilage of isapol husks, Veegum ® , larch arabogalactan, gelatin, methylcellulose, ethylcellulose, carboxymethylcellulose, hydroxypropylmethylcellulose, polyacrylic acid (e.g., Carbopol), calcium silicate, calcium phosphate, dicalcium phosphate, calcium sulfate, kaolin, sodium chloride, polyethylene glycol, and combinations thereof.
- binders include mannitol,
- binders can be pre-processed to improve their flowability and taste by methods known in the art such as freeze drying (see, e.g., Fundamentals of Freeze-Drying, PA ⁇ . Biotechnol., 14:281-360 (2002); Lyophililization of Unit Dose Pharmaceutical Dosage Forms, Drug. Dev. Ind. Pharm., 29:595-602 (2003)); solid-solution preparation (see, e.g., U.S. Pat. No. 6,264,987); and lubricant dusting and wet-granulation preparation with a suitable lubricating agent (see, e.g., Remington: The Science and Practice of Pharmacy, supra).
- freeze drying see, e.g., Fundamentals of Freeze-Drying, PA ⁇ . Biotechnol., 14:281-360 (2002); Lyophililization of Unit Dose Pharmaceutical Dosage Forms, Drug. Dev. Ind. Pharm., 29:595-602 (2003)
- solid-solution preparation
- compositions ofthe present invention comprise from about 25% to about 90% by weight ofthe binder, and preferably from about 50% to about 80%.
- binders e.g., to produce a highly friable dosage form.
- Non-limiting examples of gum bases include materials selected from among the many water-insoluble and saliva-insoluble gum base materials known in the art.
- the gum base comprises at least one hydrophobic polymer and at least one hydrophilic polymer.
- suitable hydrophobic and hydrophihc polymers for gum bases include both natural and synthetic polymers such as elastomers, rubbers, and combinations thereof.
- suitable natural polymers include, without limitation, substances of plant origin such as chicle, jelutong, gutta percha, crown gum, and combinations thereof.
- Suitable synthetic polymers include elastomers such as butadiene-styrene copolymers, isobutylene and isoprene copolymers (e.g., "butyl rubber"), polyethylene, polyisobutylene, polyvinylester (e.g., polyvinyl acetate and polyvinyl acetate phthalate), and combinations thereof.
- the gum base comprises a mixture of butyl rubber (i.e., isobutylene and isoprene copolymer), polyisobutylene, and optionally, polyvinylacetate (e.g., having a molecular weight of approximately 12,000).
- the gum base comprises from about 25% to about 75% by weight of these polymers, and preferably from about 30% to about 60%.
- compositions ofthe present invention can additionally include lubricating agents; wetting agents; emulsifying agents; solubilizing agents; suspending agents; preserving agents such as methyl-, ethyl-, and propyl-hydroxy-benzoates, butylated hydroxytoluene, and butylated hydroxyanisole; sweetening agents; flavoring agents; coloring agents; and disintegrating agents (i.e., dissolving agents) such as crospovidone as well as croscarmellose sodium and other cross-linked cellulose polymers.
- Lubricating agents can be used to prevent adhesion ofthe dosage form to the surface ofthe dies and punches, and to reduce inter-particle friction. Lubricating agents may also facilitate ejection ofthe dosage form from the die cavity and improve the rate of granulation flow during processing.
- suitable lubricating agents include, without limitation, magnesium stearate, calcium stearate, zinc stearate, stearic acid, simethicone, silicon dioxide, talc, hydrogenated vegetable oil, polyethylene glycol, mineral oil, and combinations thereof.
- the compositions ofthe present invention can comprise from about 0% to about 10% by weight ofthe lubricating agent, and preferably from about 1% to about 5%.
- Sweetening agents can be used to improve the palatability of the composition by masking any unpleasant tastes it may have.
- suitable sweetening agents include, without limitation, compounds selected from the saccharide family such as the mono-, di-, tri-, poly-, and oligosaccharides; sugars such as sucrose, glucose (corn syrup), dextrose, invert sugar, fructose, maltodextrin, and polydextrose; saccharin and salts thereof such as sodium and calcium salts; cyclamic acid and salts thereof; dipeptide sweeteners; chlorinated sugar derivatives such as sucralose and dihydrochalcone; sugar alcohols such as sorbitol, sorbitol syrup, mannitol, xylitol, hexa-resorcinol, and the like, and combinations thereof.
- Hydrogenated starch hydrolysate, and the potassium, calcium, and sodium salts of 3,6- dihydro-6-methyl- 1 - 1 ,2,3 -oxathiazin-4-one-2,2-dioxide may also be used.
- sorbitol, mannitol, and xylitol are prefened sweetening agents.
- the compositions ofthe present invention can comprise from about 0% to about 80% by weight ofthe sweetening agent, preferably from about 5% to about 75%, and more preferably from about 25% to about 50%.
- Flavoring agents can also be used to improve the palatability ofthe composition.
- suitable flavoring agents include, without limitation, natural and or synthetic (i.e., artificial) compounds such as peppermint, spearmint, wintergreen, cinnamon, menthol, cherry, strawberry, watermelon, grape, banana, peach, pineapple, apricot, pear, raspbeny, lemon, grapefruit, orange, plum, apple, fruit punch, passion fruit, chocolate (e.g., white, milk, dark), vanilla, caramel, coffee, hazelnut, combinations thereof, and the like.
- Coloring agents can be used to color code the composition, for example, to indicate the type and dosage ofthe therapeutic agent therein.
- Suitable coloring agents include, without limitation, natural and/or artificial compounds such as FD & C coloring agents, natural juice concentrates, pigments such as titanium oxide, silicon dioxide, and zinc oxide, combinations thereof, and the like.
- the compositions ofthe present invention can comprise from about 0% to about 10% by weight ofthe flavoring and/or coloring agent, preferably from about 0.1% to about 5%, and more preferably from about 2% to about 3%.
- the compositions ofthe present invention comprise a hypnotic agent or a pharmaceutically acceptable salt thereof, a carrier such as a gum base, a binary or ternary buffer system, and optionally a protecting agent.
- the chewing gum composition may further comprise lubricating agents, wetting agents, emulsifying agents, solubilizing agents; suspending agents, preserving agents, sweetening agents, flavoring agents, and coloring agents.
- the chewing gum composition comprises from about 0.001% to about 10.0% by weight ofthe hypnotic agent (in whatever chosen form, measured as per its free base form), more typically from about 0.01% to about 5.0%, and still more typically from about 0.1% to about 3.0%.
- the binary or ternary buffer system ofthe chewing gum composition provides for a final salivary pH in excess of at least about 7.8, preferably at least about 8.5, and more preferably at least about 9 (e.g., about 9-11).
- the chewing gum composition typically comprises from about 20% to about 95% ofthe gum base, more typically from about 30% to about 85%, and most typically from about 50% to about 70% of the gum base.
- the chewing gum composition may further comprise a protecting agent.
- the protecting agent coats at least part ofthe therapeutic agent, typically upon the mixing ofthe two agents.
- the protecting agent may be mixed with the therapeutic agent in a ratio of from about 0.1 to about 100 by weight, preferably in a ratio of from about 1 to about 50, and more preferably in a ratio of about 1 to about 10.
- the protecting agent reduces the adhesion between the therapeutic agent and the gum base so that the therapeutic agent may be more easily released from the gum base.
- the therapeutic agent may be delivered across the mucous membranes ofthe oral cavity within about 5 to about 20 minutes of chewing, preferably within about 10 minutes of chewing.
- a variety of different protecting agents may be used.
- suitable protecting agents include, without limitation, calcium stearate, glycerin monostearate, glyceryl behenate, glyceryl palmitostearate, hydrogenated castor oil, hydrogenated vegetable oil type I, light mineral oil, magnesium lauryl sulfate, magnesium stearate, mineral oil, poloxamer, polyethylene gycol, sodium benzoate, sodium chloride, sodium lauryl sulfate, stearic acid, cab-o-sil, talc, zinc stearate, and combinations thereof.
- the gum base may additionally include plasticizers such as softeners or emulsifiers. Such plasticizers may, for example, help reduce the viscosity ofthe gum base to a desirable consistency and improve its overall texture and bite. Plasticizers may also facilitate the release ofthe therapeutic agent upon mastication. Non-limiting examples of plasticizers include lecithin, mono- and diglycerides, lanolin, stearic acid, sodium stearate, potassium stearate, glycerol triacetate, glycerol monostearate, glycerin, and combinations thereof.
- the gum base typically comprises from about 0% to about 20% by weight ofthe plasticizer, and more typically from about 5% to about 15%.
- the gum base may further comprise waxes such as beeswax and microcrystalline wax, fats or oils such as soybean and cottonseed oil, and combinations thereof.
- waxes such as beeswax and microcrystalline wax
- fats or oils such as soybean and cottonseed oil
- combinations thereof Typically, the gum base comprises from about 0% to about 25% by weight of these waxes and oils, and more typically comprises from about 15% to about 20%.
- the gum base may further comprise one or more elastomeric solvents such as rosins and resins.
- solvents include methyl, glycerol, and pentaerythritol esters of rosins, modified rosins such as hydrogenated, dimerized or polymerized rosins, or combinations thereof (e.g., pentaerythritol ester of partially hydrogenated wood rosin, pentaerythritol ester of wood rosin, glycerol ester of wood rosin, glycerol ester of partially dimerized rosin, glycerol ester of polymerized rosin, glycerol ester of tall oil rosin, glycerol ester of wood rosin and partially hydrogenated wood rosin and partially hydrogenated methyl ester of rosin such as polymers of alpha-pinene or beta-pinene, terpene resins including polyterpene
- the gum base may further comprise a filler material to enhance the chewability of the final chewing gum composition.
- Fillers that are substantially non-reactive with other components ofthe final chewing gum formulation are preferable.
- suitable fillers include, without limitation, calcium carbonate, magnesium silicate (i.e., talc), dicalcium phosphate, metallic mineral salts (e.g., alumina, aluminum hydroxide, and aluminum silicates), and combinations thereof.
- the gum base comprises from about 0% to about 30% by weight ofthe filler, and more typically from about 10% to about 20%.
- the gum base need not be prepared from its individual components.
- the gum base can be purchased with the desired ingredients contained therein, and can be modified to include additional agents.
- Several manufacturers produce gum bases suitable for use with the described chewing gum compositions. Examples of such gum bases include, without limitation, Pharmagum ⁇ M, S, or C (SPI Pharma Group; New Castle, DE).
- Pharmagum ⁇ comprises a mixture of gum base, sweetening agent, plasticizer, and sugar.
- the chewing gum composition includes a therapeutic agent centerf ⁇ ll.
- a centerfill may be particularly suitable when immediate release ofthe therapeutic agent is prefened.
- encapsulating the therapeutic agent in a centerfill may help to mask any undesirable taste that the therapeutic agent may have.
- the gum base surrounds, at least in part, a centerfill.
- the centerfill comprises at least one therapeutic agent, and may be a liquid or semi-liquid material.
- the centerfill material can be a synthetic polymer, a semi-synthetic polymer, low-fat, or fat-free and contain one or more sweetening agents, flavoring agents, coloring agents, and/or scenting agents.
- the centerfill includes a binary or ternary buffer system as described herein. Methods for preparing a centerfill chewing gum are described, for example, in U.S. Pat. No. 3,806,290, which is hereby incorporated by reference in its entirety.
- the chewing gum compositions can have any desired shape, size, and texture.
- the chewing gum can have the shape of a stick, tab, gumball, and the like.
- the chewing gum can be any desirable color.
- the chewing gum can be any shade of red, blue, green, orange, yellow, violet, indigo, and mixtures thereof, and can be color coded to indicate the type and dosage ofthe therapeutic agent therein.
- the chewing gum can be individually wrapped or grouped together in pieces for packaging by methods well known in the art.
- the dosage form is a tablet such as a dissolving tablet (i.e., disintegrating tablet) or chewable tablet
- the compositions ofthe present invention comprise a hypnotic agent or a pharmaceutically acceptable salt thereof, a carrier such as a binder, and a binary or ternary buffer system.
- the tablet composition may further comprise lubricating agents, wetting agents, emulsifying agents, solubilizing agents; suspending agents, preserving agents, sweetening agents, flavoring agents, coloring agents, and disintegrating agents.
- the tablet compositions ofthe present invention comprise from about 0.001% to about 10.0% by weight ofthe hypnotic agent (in whatever chosen form, measured as per its free base form), and more typically from about 1.0% to about 5.0%.
- the tablet composition provides for a final salivary pH in excess of at least about 7.8, preferably at least about 8.5, and more preferably at least about 9 (e.g., about 9-11).
- the tablet is a dissolving tablet such as a slow-dissolving or quick-dissolving tablet that is dissolved by a subject's saliva, without the need for chewing.
- a dissolving tablet placed on the subject's tongue can be used for buccal delivery ofthe therapeutic agent.
- a dissolving tablet placed underneath the subject's tongue can be used for sublingual delivery ofthe therapeutic agent.
- This type of dosage form may be particularly desirable for pediatric and geriatric patients, since small children and aged individuals often have difficulty chewing certain items.
- the dissolving tablet is formulated to dissolve within about 1 to about 15 minutes, preferably within about 2 to about 10 minutes, e.g., within about 2, 3, 4, 5, 6, 7, 8, 9, or 10 minutes, following administration.
- quick-dissolving tablets dissolve faster than slow-dissolving tablets, which are typically dissolved gradually rather than rapidly by a subject's saliva.
- the slow-dissolving or quick- ' dissolving tablet delivers the therapeutic agent across the sublingual mucosa.
- the tablet is a chewable tablet that is chewed by a subject and formulated to dissolve either rapidly or gradually.
- a chewable tablet placed on the subject's tongue can be used for buccal delivery ofthe therapeutic agent.
- the chewable tablet can be moved around within the mouth and can sometimes be parked between the gums and the cheeks or underneath the tongue.
- at least a portion ofthe therapeutic agent contained within a chewable tablet may also be delivered sublingually (i.e., across the sublingual mucosa).
- the chewable tablet is formulated to dissolve within about 1 to about 15 minutes, preferably within about 2 to about 10 minutes, e.g., within about 2, 3, 4, 5, 6, 7, 8, 9, or 10 minutes, following administration.
- the dissolving and chewable tablets ofthe present invention are typically formulated to dissolve within about 1 to 15 minutes following administration.
- these time frames are amenable to maximum exposure ofthe therapeutic agent to the oral mucosa (e.g., to the sublingual and/or buccal mucosa), they are not always amenable to user compliance (e.g., users may swallow too frequently and, therefore, hinder maximal transmucosal absorption). Consequently, in certain instances, it may be desirable to strike a balance between patient compliance and maximum exposure time ofthe therapeutic agent to the oral mucosa.
- subtle changes to the tablet formulation such as, for example, replacing one flavoring agent for another (e.g., chocolate for spearmint) or replacing one binder or sweetening agent for another (e.g., lactose for mannitol or sorbitol) may be used to reduce salivation.
- the carrier present in the tablets ofthe present invention is typically a binder that is useful in keeping the tablet in a semi-solid state, and may be a solid or a liquid, and may for example be a high-melting point fat or waxy material.
- Materials suitable as binders are discussed in detail above and may be used alone or in combination in the tablet compositions ofthe present invention.
- binders such as mannitol, sorbitol, lactose, sucrose, and inositol can impart properties to the tablet that permit or enhance its disintegration in the mouth.
- the tablet composition may further comprise a protecting agent.
- the protecting agent coats at least part ofthe therapeutic agent, typically upon the mixing ofthe two agents.
- the protecting agent may be mixed with the therapeutic agent in a ratio of from about 0.1 to about 100 by weight, preferably in a ratio of from about 1 to about 50, and more preferably in a ratio of about 1 to about 10.
- the protecting agent reduces the adhesion between the therapeutic agent and the binder so that the therapeutic agent may be more easily released from the binder. In this way, the therapeutic agent maybe delivered across the mucous membranes ofthe oral cavity within about 5 to about 20 minutes, preferably within about 10 minutes.
- Materials suitable as protectmg agents are discussed in detail above and may be used alone or in combination in the tablet compositions ofthe present invention.
- the tablet composition may also comprise one or more elastomeric solvents such as rosins and resins. Non-limiting examples of such solvents are discussed in detail above and may be used alone or in combination in the tablet compositions ofthe present invention.
- the tablet composition may further comprise waxes such as beeswax and microcrystalline wax, fats or oils such as soybean and cottonseed oil, and combinations thereof.
- the tablet composition may additionally include plasticizers such as softeners or emulsifiers. Such plasticizers may, for example, help reduce the viscosity ofthe salivary solution ofthe dissolved tablet to a desirable consistency and improve its overall texture and bite and help facilitate the release ofthe therapeutic agent.
- the tablet composition includes a therapeutic agent centerfill.
- a centerfill maybe particularly suitable when immediate release ofthe therapeutic agent is prefened.
- encapsulating the therapeutic agent in a centerfill may help to mask any undesirable taste that the therapeutic agent may have.
- the binder surrounds, at least in part, a centerfill.
- the centerfill comprises at least one therapeutic agent, and may be a liquid or semi-liquid material.
- the centerfill material can be a synthetic polymer, a semi-synthetic polymer, low-fat, or fat-free and contain one or more sweetening agents, flavoring agents, coloring agents, and/or scenting agents.
- the centerfill includes a binary or ternary buffer system as described herein.
- the tablet composition of the present invention is multilayered.
- the dissolving or chewable tablet can be designed to provide more than one therapeutic agent, e.g., two or more hypnotic agents or one or more hypnotic agents in combination with one or more non-hypnotic therapeutic agents.
- the first layer contains a hypnotic agent and the second layer contains the same or different hypnotic agent or a non-hypnotic therapeutic agent.
- the first layer comprises the dissolving or chewable portion ofthe tablet, and the second (i.e., subsequent) layer is coated by the first layer.
- This type of formulation may be particularly suitable when immediate release ofthe hypnotic agent, followed by gastrointestinal absorption of a second therapeutic agent, is desirable. Gastrointestinal absorption ofthe second therapeutic agent may be desirable, for example, in order to mitigate co-morbid symptoms or to sustain the therapeutic benefit ofthe hypnotic agent in the dissolving or the chewable portion ofthe tablet.
- the second layer is present as a layer lateral to the first layer.
- the second layer typically comprises at least one therapeutic agent, and can also comprise one or more sweetening agents, flavoring agents, coloring agents, and scenting agents as described above.
- the second layer further includes a binary or ternary buffer system as described herein.
- the combination of hypnotic agents with or without non- hypnotic therapeutic agents need not take the form of a multilayered tablet, but instead comprises a single homogenous tablet layer.
- This type of formulation may also be used in the case where gastrointestinal absorption of at least one therapeutic agent is desirable.
- the relative extent of ionization ofthe two or more therapeutic agents determines how they are to be absorbed. For example, those therapeutic agents that are un-ionized are absorbed through the oral mucosa, while the ionized agents are swallowed for gastrointestinal absorption.
- the tablet compositions can have any desired shape, size, and texture.
- the tablet can have the shape of a stick, tab, pellet, sphere, and the like.
- the tablet can be any desirable color.
- the tablet can be any shade of red, blue, green, orange, yellow, violet, indigo, and mixtures thereof, and can be color coded to indicate the type and dosage ofthe therapeutic agent therein.
- the tablets can be individually wrapped or grouped together in pieces for packaging by methods well known in the art.
- the compositions ofthe present invention comprise a hypnotic agent or a pharmaceutically acceptable salt thereof, a carrier such as a binder, and a binary or ternary buffer system.
- the lozenge or candy composition may further comprise lubricating agents, wetting agents, emulsifying agents, solubilizing agents; suspending agents, preserving agents, sweetening agents, flavoring agents, coloring agents, and disintegrating agents.
- the lozenge compositions ofthe present invention comprise from about 0.001% to about 10.0% by weight ofthe hypnotic agent (in whatever chosen form, measured as per its free base form), and more typically from about 1.0% to about 5.0%. hi some embodiments, about 4.5% by weight ofthe hypnotic agent is used.
- the binary or ternary buffer system ofthe lozenge composition provides for a final salivary pH in excess of at least about 7.8, preferably at least about 8.5, and more preferably at least about 9 (e.g., about 9-11).
- the lozenge or candy is dissolved by a subject's saliva, without the need for chewing.
- a lozenge placed on the subject's tongue can be used for buccal delivery ofthe therapeutic agent.
- a lozenge placed underneath the subject's tongue can be used for sublingual delivery ofthe therapeutic agent.
- This type of dosage form may be particularly desirable for pediatric and geriatric patients, since small children and aged individuals often have difficulty chewing certain items.
- the lozenge is formulated to dissolve within about 1 to about 15 minutes, preferably within about 2 to about 10 minutes, e.g., within about 2, 3, 4, 5, 6, 7, 8, 9, or 10 minutes, following administration.
- the lozenge or candy delivers the therapeutic agent across the sublingual mucosa.
- the lozenges the present mvention are typically formulated to dissolve within about 1 to 15 minutes following administration.
- these time frames are amenable to maximum exposure ofthe therapeutic agent to the oral mucosa (e.g., to the sublingual and/or buccal mucosa)
- they are not always amenable to user compliance (e.g., users may swallow too frequently and, therefore, hinder maximal transmucosal absorption). Consequently, in certain instances, it may be desirable to strike a balance between patient compliance and maximum exposure time ofthe therapeutic agent to the oral mucosa.
- subtle changes to the lozenge formulation such as, for example, replacing one flavoring agent for another (e.g., chocolate for spearmint) or replacing one binder or sweetening agent for another (e.g., lactose for mannitol or sorbitol) may be used to reduce salivation.
- the carrier present in the lozenges ofthe present invention is typically a binder that is useful in keeping the lozenge in a semi-solid state, and may be a solid or a liquid, and may for example be a high-melting point fat or waxy material.
- Materials suitable as binders are discussed in detail above and may be used alone or in combination in the lozenge compositions ofthe present invention.
- binders such as mannitol, sorbitol, lactose, sucrose, and inositol can impart properties to the lozenge that permit or enhance its disintegration in the mouth.
- the lozenge composition may further comprise a protecting agent.
- the protecting agent coats at least part ofthe therapeutic agent, typically upon the mixing ofthe two agents.
- the protecting agent may be mixed with the therapeutic agent in a ratio of from about 0.1 to about 100 by weight, preferably in a ratio of from about 1 to about 50, and more preferably in a ratio of about 1 to about 10.
- the protecting agent reduces the adhesion between the therapeutic agent and the binder so that the therapeutic agent may be more easily released from the binder. In this way, the therapeutic agent may be delivered across the mucous membranes ofthe oral cavity within about 5 to about 20 minutes, preferably within about 10 minutes.
- Materials suitable as protecting agents are discussed in detail above and may be used alone or in combination in the lozenge compositions ofthe present invention.
- the lozenge composition may also comprise one or more elastomeric solvents such as rosins and resins. Non-limiting examples of such solvents are discussed in detail above and may be used alone or in combination in the tablet compositions ofthe present invention.
- the lozenge composition may further comprise waxes such as beeswax and microcrystalline wax, fats or oils such as soybean and cottonseed oil, and combinations thereof.
- the lozenge composition may additionally include plasticizers such as softeners or emulsifiers. Such plasticizers may, for example, help reduce the viscosity ofthe salivary solution ofthe dissolved lozenge to a desirable consistency and improve its overall texture and bite and help facilitate the release ofthe therapeutic agent. Non-limiting examples of such plasticizers are discussed in detail above and may be used alone or in combination in the lozenge compositions ofthe present invention.
- the lozenge composition includes a therapeutic agent centerfill.
- a centerfill may be particularly suitable when immediate release ofthe therapeutic agent is prefened.
- encapsulating the therapeutic agent in a centerfill may help to mask any undesirable taste that the therapeutic agent may have.
- the binder sunounds, at least in part, a centerfill.
- the centerfill comprises at least one therapeutic agent, and may be a liquid or semi-liquid material.
- the centerfill material can be low-fat or fat free and contain one or more sweetening agents, flavoring agents, coloring agents, and/or scenting agents.
- the centerfill includes a binary or ternary buffer system as described herein.
- the lozenge composition ofthe present invention is multilayered.
- the lozenge can be designed to provide more than one therapeutic agent, e.g., two or more hypnotic agents or one or more hypnotic agents in combination with one or more non-hypnotic therapeutic agents.
- the first layer contains a hypnotic agent and the second layer contains the same or different hypnotic agent or a non-hypnotic therapeutic agent.
- the first layer comprises the dissolving portion ofthe lozenge, and the second (i.e., subsequent) layer is coated by the first layer.
- This type of formulation may be particularly suitable when immediate release ofthe hypnotic agent, followed by gastrointestinal absorption of a second therapeutic agent, is desirable. Gastrointestinal absorption ofthe second therapeutic agent may be desirable, for example, in order to mitigate co-morbid symptoms or to sustain the therapeutic benefit ofthe hypnotic agent in the dissolving portion ofthe lozenge.
- the second layer is present as a layer lateral to the first layer.
- the second layer typically comprises at least one therapeutic agent, and can also comprise one or more sweetening agents, flavoring agents, coloring agents, and scenting agents as described above.
- the second layer further includes a binary or ternary buffer system as described herein.
- the combination of hypnotic agents with or without non- hypnotic therapeutic agents need not take the form of a multilayered lozenge, but instead comprises a single homogenous lozenge layer.
- This type of formulation may also be used in the case where gastrointestinal absorption of at least one therapeutic agent is desirable.
- the relative extent of ionization ofthe two or more therapeutic agents determines how they are to be absorbed. For example, those therapeutic agents that are un-ionized are absorbed through the oral mucosa, while the ionized agents are swallowed for gastrointestinal absorption.
- the lozenge compositions can have any desired shape, size, and texture.
- the lozenge can have the shape of a stick, tab, pellet, sphere, and the like.
- the lozenge can be any desirable color.
- the lozenge can be any shade of red, blue, green, orange, yellow, violet, indigo, and mixtures thereof, and can be color coded to indicate the type and dosage ofthe therapeutic agent therein.
- the lozenges can be individually wrapped or grouped together in pieces for packaging by methods well known in the art.
- compositions ofthe present invention are useful in therapeutic applications, e.g., for treating a sleep disorder.
- the compositions ofthe present invention provide the rapid and predictable delivery of a hypnotic agent across the oral mucosa with surprisingly low inter-subject variability in terms of maximum plasma concentration (C max ) and the time to reach the maximum plasma concentration (T max ) by raising the pH of saliva to a pH greater than about 7.8, inespective ofthe starting pH of saliva.
- the delivery ofthe therapeutic agent across the oral mucosa avoids hepatic first pass metabolism, degradation within the gastrointestinal tract, and drug loss during absorption.
- the therapeutic agent reaches the systemic circulation in a substantially shorter period of time and at a substantially higher concentration than with traditional oral (e.g., tablet) adniinistration.
- compositions ofthe present invention offer advantages over compositions for oral mucosal administration that do not contain the buffer system described herein.
- the buffer system in the compositions ofthe present invention helps convert substantially all ofthe therapeutic agent from its ionized form to its un-ionized form, the therapeutic agent reaches the systemic circulation in a substantially shorter period of time (e.g., reducing the time to onset of therapeutic activity) and at a substantially higher concentration than with compositions for oral mucosal administration that do not contain the buffer system.
- compositions of the present invention have particular utility in the area of human and veterinary therapeutics.
- administered dosages will be effective to deliver picomolar to micromolar concentrations ofthe hypnotic agent to the appropriate site.
- Administration ofthe compositions ofthe present invention is preferably carried out via any ofthe accepted modes of administration to the mucous membranes ofthe oral cavity.
- suitable sites of administration within the oral mucosa include, without limitation, the mucous membranes ofthe floor ofthe mouth (sublingual mucosa), the cheeks (buccal mucosa), the gums (gingival mucosa), the roof of the mouth (palatal mucosa), the lining ofthe lips, and combinations thereof. These regions differ from each other with respect to their anatomy, drug permeability, and physiological response to drugs.
- the compositions ofthe present invention are administered to the sublingual mucosa, buccal mucosa, or a combination thereof.
- the oral mucosa possessing a rich blood supply and suitable drag permeability, is an especially attractive route of administration for systemic drug delivery. Furthermore, delivery of a therapeutic agent across the oral mucosa bypasses hepatic first pass metabolism, avoids enzymatic degradation within the gastrointestinal tract, and provides a more suitable enzymatic flora for drug absorption.
- sublingual delivery refers to the administration of a therapeutic agent across the mucous membranes lining the floor ofthe mouth and/or the ventral tongue.
- uccal delivery refers to the administration of a therapeutic agent across the mucous membranes lining the cheeks.
- the oral mucosa is composed of an outermost layer of stratified squamous epithelium. Beneath this layer lies a basement membrane, i.e., the lamina basement, followed by the submucosa as the innermost layer.
- the epithelium ofthe oral mucosa is similar to the stratified squamous epithelia found in the rest ofthe body in that it contains a mitotically active basal cell layer, advancing through a number of differentiating intermediate layers to the superficial layers, where cells are shed from the surface ofthe epithelium (Gandhi et al, Ind. J. Pharm. Sci., 50:145-152 (1988)).
- the epithelium ofthe buccal mucosa is about 40-50 cell layers thick, while that ofthe sublingual epithelium contains somewhat fewer cell layers.
- the epithelial cells increase in size and become flatter as they travel from the basal layers to the superficial layers.
- the turnover time for buccal mucosal epithelium is representative ofthe turnover time for sublingual mucosal epithelium as well as other epithelia in the oral mucosa (Harris et al, J. Pharm. Sci, 81:1-10 (1992)).
- the thickness of the oral mucosa varies depending on the site in the oral cavity.
- the buccal mucosa measures at about 500-800 ⁇ m in thickness
- the hard and soft palatal mucosa, the sublingual mucosa, the ventral tongue, and the gingival mucosa measure at about 100-200 ⁇ m in thickness.
- the composition ofthe epithelium also varies depending on the site in the oral cavity.
- the mucosae of areas subject to mechanical stress are keratinized similar to the epidermis.
- the mucosae ofthe soft palate, the sublingual region, and the buccal region are not keratinized (Harris et al, supra).
- the keratinized epithelia contain neutral lipids like ceramides and acylceramides, which have been associated with providing a barrier function. As a result, these epithelia are relatively impermeable to water.
- non-keratinized epithelia, such as sublingual and buccal epithelia do not contain acylceramides and have only small amounts of ceramide (Wertz et al, Crit.
- Non-keratinized epithelia also contain small amounts of neutral but polar lipids, e.g., cholesterol sulfate and glucosyl ceramides.
- the oral mucosa is a somewhat leaky epithelia intermediate between that ofthe epidermis and intestinal mucosa.
- the permeability ofthe buccal mucosa is estimated to be about 4-4000 times greater than that of skin (Galey et al, J. Invest. Dermal, 67:713-717 (1976)).
- the permeability of different regions ofthe oral mucosa generally decrease in the order of sublingual mucosa greater than buccal mucosa, and buccal mucosa greater than palatal mucosa (Harris et al, supra). This permeability is generally based upon the relative thickness and degree of keratinization of these membranes, with the sublingual mucosa being relatively thin and non-keratinized, the buccal mucosa being thicker and non-keratinized, and the palatal mucosa being intermediate in thickness, but keratinized.
- the epithelial cells ofthe oral mucosa are surrounded by mucus comprising primarily complexes of proteins and carbohydrates that may or may not be attached to certain regions on the cell surface.
- the mucus may play a role in cell-cell adhesion, as well as acting as a lubricant, allowing cells to move relative to one another (Tabak et al, J. Oral Pathol, 11:1-17 (1982)).
- mucus In stratified squamous epithelia found elsewhere in the body, mucus is synthesized by specialized mucus secreting cells such as goblet cells; however, in the oral mucosa, mucus is secreted by the major and minor salivary glands as part of saliva (Tabak et al, supra; Rathbone et al, Adv. Drug Del. Rev., 13:1-22 (1994)).
- the mucus network carries a negative charge due to the sialic acid and sulfate residues present on the carbohydrates.
- mucus can form a strongly cohesive gel stracture that binds to the epithelial cell surface as a gelatinous layer (Gandhi et al, supra).
- the buffer systems ofthe present invention neutralize the sialic acid residues present on the carbohydrates and prevent them from interacting with the therapeutic agent, thereby further enhancing drag permeation.
- Saliva is the protective fluid for all tissues ofthe oral cavity.
- Saliva is an aqueous fluid with about 1% organic and inorganic materials.
- the major determinant ofthe salivary composition is the flow rate, which in turn depends upon factors such as the time of day, the type of stimulus, and the degree of stimulation.
- the salivary pH typically ranges from about 5.5 to about 7.0, depending on the flow rate. For example, at high flow rates, the sodium and bicarbonate concentrations increase, leading to an increase in the pH. Because the daily salivary volume is between about 0.5 to about 2 liters, the oral cavity provides an aqueous environment for the hydration and/or dissolution ofthe oral mucosal dosage forms ofthe present mvention.
- the sublingual mucosa is the most highly permeable region ofthe oral cavity, and provides rapid absorption and high bioavailability of a drag in a convenient, accessible, and well-accepted route of administration (Harris et al, supra).
- Suitable sublingual dosage forms include, without limitation, tablets (e.g., quick-dissolving, slow-dissolving), lozenges, candy, and soft gelatin capsules filled with liquid drug. Such systems create a very high drug concentration in the sublingual region before they are systemically absorbed across the sublingual mucosa.
- the sublingual mucosa is particularly well-suited for producing a rapid onset of action, and sublingual dosage forms can be used to deliver drugs with shorter delivery period requirements and/or less frequent dosing regimens.
- the buccal mucosa is considerably less permeable than the sublingual area, rapid absorption and high bioavailability of a drug can also be observed with buccal administration.
- Suitable buccal dosage forms include, without limitation, chewing gums, tablets (e.g., quick- dissolving, slow-dissolving), lozenges, candy, and the like. Both the buccal mucosa and the sublingual mucosa are far superior to the gastrointestinal tract for providing increased absorption and bioavailability of a drag.
- penetration enhancers can be included in the dosage forms ofthe present invention.
- the penetration enhancers may be of the type that alters the nature of the oral mucosa to enhance penetration, or ofthe type that alters the nature ofthe therapeutic agent to enhance penetration through the oral mucosa.
- Suitable penetration enhancers include, without limitation, polyoxyethylene 23- lauryl ether, aprotin, azone, benzalkonium chloride, cetylpyridinium chloride, cetyltrimethylammonium bromide, cyclodextrin, dextran sulfate, lauric acid, propylene glycol, lysophosphatidylcholine, menthol, methoxysalicylate, methyloleate, oleic acid, phosphatidylcholine, polyoxyethylene, polysorbate 80, sodium ethylenediaminetetraacetic acid ("EDTA”), sodium deoxycholate, sodium glycocholate, sodium glycodeoxycholate, sodium lauryl suflate, sodium salicylate, sodium taurocholate, sodium taurodeoxycholate, as well as certain sulfoxides and glycosides, and combinations thereof.
- EDTA sodium ethylenediaminetetraacetic acid
- Membrane assays were performed using zolpidem tartrate solutions at a pH of 5.8, 6.8, and 7.8.
- the alkaline pH values of 7.8 were adjusted using freshly prepared 0.01 M sodium bicarbonate/sodium carbonate buffer solution.
- the acidic pH of 5.8 was achieved using a 0.01 M acetate buffer solution (a mixture of sodium acetate and acetic acid).
- the neutral pH of 6.8 was achieved by adding 0.01 M acetate solution to the sodium bicarbonate/sodium carbonate buffer solution.
- Permeation through the membrane was measured by determining the concentration of zolpidem in the acceptor cell and is expressed as P e (effective permeability in centimeters per second).
- Figure 1 shows a bar chart illustrating the relationship between pH and zolpidem membrane permeability.
- Zolpidem can be formulated as a chewing gum composition as described above.
- the unit dose or serving ofthe chewing gum comprises from about 0.1 to about 100 milligrams (mg) zolpidem (as measured in its tartrate salt form), preferably from about 1 to about 50 mg, and more preferably from about 2 to about 25 mg.
- the unit dose comprises from about 2 to about 20 mg zolpidem, preferably from about 5 to about 15 mg.
- Extra zolpidem for example, up to from about 10% to about 25% by weight, can be added as "overage” or as the amount that may be expected to be "washed away” and not otherwise released or absorbed during mastication.
- the unit dose or serving ofthe chewing gum comprises from about 0.81 to about 42 mg zolpidem in its base form, and more preferably from about 1.64 to about 20.5 mg.
- the unit dose comprises from about 1.64 to about 16.4 mg zolpidem in its free base form, preferably from about 1.64 to about 12.3 mg, and more preferably from about 1.64 to about 8.2 mg, e.g., about 1.64, 2.46, 3.28, 4.1, 4.92, 5.78, 6.56, 7.38, or 8.2 mg.
- the unit dose comprises a mixture of zolpidem in free base form and salt form (e.g., zolpidem tartrate).
- the zolpidem chewing gum composition comprises from about 0.001% to about 10.0% zolpidem (in whatever chosen form, measured as per its free base form), preferably from about 0.05% to about 2.0%, and more preferably from about 0.1% to about 1.0%. In some embodiments, about 0.25% zolpidem is used.
- the buffer system ofthe zolpidem chewing gum composition provides for a final salivary pH in excess of at least about 7.8, preferably at least about 8.5, and more preferably at least about 9 (e.g., about 9-11).
- a zolpidem chewing gum was made according to the following procedure. Silicon dioxide USP (0.35 kg) was passed through a #20 mesh screen, and then loaded into a blender containing 0.810 kg mannitol granular USP and 9.569 kg Pharmagum TM C. The material was blended for 10 minutes. Zolpidem tartrate EP (0.034 kg) was ground with the silicon dioxide (0.02 kg) using a mortar and pestle. The remaining silicon dioxide, along with 0.228 kg magnesium stearate, was added into the mortar while continuing to grind. The ground materials were transfe ⁇ ed into a plastic bag, and the mortar was rinsed using 0.01 kg silicone dioxide, and transfe ⁇ ed into the bag.
- the average particle size ofthe drag (i.e., zolpidem) in the chewing gum is about 20 microns, as compared to a typical average drag particle size of from about 75 to about 100 microns.
- the average particle size ofthe drag in the chewing gum is less than or equal to the average particle size ofthe carrier ingredients (e.g., gum base, binders, etc.).
- the zolpidem chewing gum composition ofthe present invention can be used, e.g., for treatment of insomnia; see, Holm et al, Drugs, 59:865-889 (2000).
- the subject chews the chewing gum as is normally done with any non-medicated type of chewing gum for about 5 to about 20 minutes, at approximately an average rate of about 10 to about 45 chews per minute. The gum is then discarded.
- a typical dosage form of the zolpidem chewing gum ofthe present invention is designed to produce an average plasma concentration of at least from about 20 to about 300 nanograms of zolpidem per milliliter of plasma.
- a 5 mg zolpidem chewing gum can be designed to produce a mean peak plasma concentration within the range of from about 20 to about 100 nanograms of zolpidem per milliliter of plasma within about 5 minutes to about 2 hours.
- a 10 mg zolpidem chewing gum can be designed to produce a mean peak plasma concentration within the range of from about 100 to about 300 nanograms of zolpidem per milliliter of plasma within about 5 minutes to about 2 hours.
- the chewing gum compositions ofthe present invention provide a convenient, reliable, practical, and painless system for delivering zolpidem across the oral mucosa.
- the chewing gum compositions are capable of rapidly delivering zolpidem with low inter-subject variability in terms of maximum plasma concentration (C max ) and the time to reach the maximum plasma concentration (T max ) so that a therapeutically effective amount of zolpidem enters the bloodstream within about 30 minutes, 20 minutes, 15 minutes, 10 minutes, 5 minutes, or even within about 1-2 minutes after zolpidem is released from the carrier.
- This example illustrates the slow-dissolving, quick-dissolving, and chewable zolpidem tablet compositions ofthe present invention.
- Zolpidem can be formulated as a tablet composition as described above.
- the unit dose or serving ofthe tablet comprises from about 0.1 to about 100 milligrams (mg) zolpidem (as measured in its tartrate salt form), preferably from about 1 to about 50 mg, and more preferably from about 2 to about 25 mg.
- the unit dose comprises from about 2 to about 20 mg zolpidem, preferably from about 2 to about 15 mg, and more preferably from about 2 to about 10 mg, e.g., about 2, 3, 4, 5, 6, 7, 8, 9, or 10 mg.
- the unit dose comprises a dose of zolpidem that is less than the dose typically used in commercial oral tablets, but possesses comparable or greater bioavailability and onset of therapeutic activity as well as lower variability of drug absorption.
- unit doses of from about 2 to about 5 mg zolpidem are prefened, with unit doses of about 4 mg zolpidem being particularly prefened.
- Extra zolpidem for example, up to from about 10% to about 25% by weight, can be added as "overage” or as the amount that may be expected to be "washed away” and not otherwise released or absorbed during tablet dissolution and/or mastication.
- the unit dose or serving ofthe tablet comprises from about 0.81 to about 42 mg zolpidem in its base form, and more preferably from about 1.64 to about 20.5 mg.
- the unit dose comprises from about 1.64 to about 16.4 mg zolpidem in its free base form, preferably from about 1.64 to about 12.3 mg, and more preferably from about 1.64 to about 8.2 mg, e.g., about 1.64, 2.46, 3.28, 4.1, 4.92, 5.78, 6.56, 7.38, or 8.2 mg.
- the unit dose comprises a mixture of zolpidem in free base form and salt form (e.g., zolpidem tartrate).
- the zolpidem tablet composition comprises from about 0.001% to about 10.0% zolpidem (in whatever chosen form, measured as per its free base form), preferably from about 0.1 % to about 8.0%, more preferably from about 1.0% to about 7.0%, and still more preferably from about 1.0% to about 5.0%. In some embodiments, about 4.0% zolpidem is used.
- the buffer system ofthe zolpidem tablet composition provides for a final salivary pH in excess of at least about 7.8, preferably at least about 8.5, and more preferably at least about 9 (e.g., about 9-11).
- Zolpidem slow-dissolving tablets [0193] A zolpidem slow-dissolving tablet was made according to the following procedure. Magnesium stearate USP (0.35 kg) was passed through a #20 mesh screen, and then loaded into a blender containing 0.810 kg mannitol granular USP and 9.569 kg sorbitol. The material was blended for 10 minutes. Zolpidem tartrate EP (0.034 kg) was ground with the magnesium stearate (0.02 kg) using a mortar and pestle. The remaining silicon dioxide, along with 0.228 kg magnesium stearate was added into the mortar while continuing to grind. The ground materials were transfe ⁇ ed into a plastic bag, and the mortar was rinsed using 0.01 kg silicone dioxide, and transfe ⁇ ed into the bag. The contents ofthe bag were then blended for five minutes.
- Magnesium stearate USP (0.35 kg) was passed through a #20 mesh screen, and then loaded into a blender containing
- the average particle size ofthe drug (i.e., zolpidem) in the slow-dissolving tablet is about 20 microns, as compared to a typical average drug particle size of from about 75 to about 100 microns.
- the average particle size ofthe drug in the slow-dissolving tablet is less than or equal to the average particle size ofthe carrier ingredients (e.g., gum base, binders, etc.).
- a second zolpidem slow-dissolving tablet was made according to the formulation shown in Table 2 and the following procedure. Three separate blends of silicon dioxide with zolpidem, sodium bicarbonate, and sodium carbonate; mannitol and sorbitol; and spearmint flavor, sucralose, stearic acid, and magnesium stearate were prepared. The three blends were screened separately and mixed to form a single blend. The single blend was then compressed into tablets after testing for content uniformity.
- the average particle size ofthe drag (i.e., zolpidem) in the slow-dissolving tablet is about 20 microns, which is less than or equal to the average particle size ofthe carrier ingredients (e.g., gum base, binders, etc.).
- the unit weight for each tablet was 250 mg.
- the pH ofthe tablet was about 9.8 and remained stable. These tablets dissolve within about 10 minutes following subUngual administration.
- a zolpidem quick-dissolving tablet was made according to the following procedure. Mannitol (3.633 kg) and sorbitol (0.469 kg) were blended for ten minutes. Sodium carbonate (0.330 kg), sodium bicarbonate (0.165 kg), natural peppermint flavor (0.125 kg), natural menthol flavor (0.025 kg), and sucralose (0.020 kg) were blended separately for ten minutes. Magnesium stearate (0.075 kg), and zolpidem tartrate (0.034 kg) were blended for ten minutes and then passed through a #12 mesh screen. The blended mixtures were then added together and compressed into tablets.
- the average particle size ofthe drug (i.e., zolpidem) in the quick-dissolving tablet is about 20 microns, as compared to a typical average drug particle size of from about 75 to about 100 microns.
- the average particle size ofthe drug in the quick-dissolving tablet is less than or equal to the average particle size ofthe carrier ingredients (e.g., gum base, binders, etc.).
- a second zolpidem quick-dissolving tablet was made according to the formulation shown in Table 3 and the following procedure. Three separate blends of silicon dioxide with zolpidem, sodium carbonate, and sodium bicarbonate; mannitol and sorbitol; and polyethylene glycol, spearmint flavor, sucralose, magnesium stearate, crospovidone, and croscarmellose sodium were prepared. The three blends were screened separately and mixed to form a single blend. The single blend was then compressed into tablets after testing for content uniformity.
- the average particle size ofthe drag (i.e., zolpidem) in the quick-dissolving tablet is about 20 microns, which is less than or equal to the average particle size ofthe carrier ingredients (e.g., gum base, binders, etc.).
- the unit weight for each tablet was 250 mg.
- the pH ofthe tablet was about 9.8 and remained stable. These tablets dissolve within about 5 minutes following sublingual administration.
- a zolpidem chewable tablet was made according to the following procedure. Magnesium stearate USP (0.35 kg) was passed through a #20 mesh screen, and then loaded into a blender containing 0.810 kg mannitol granular USP, 9.569 kg sorbitol, and 0.020 kg stearic acid. The material was blended for 10 minutes. Zolpidem tartrate EP (0.034 kg) was ground with the magnesium stearate (0.02 kg) using a mortar and pestle. The remaining silicon dioxide, along with 0.228 kg magnesium stearate was added into the mortar while continuing to grind. The ground materials were transfe ⁇ ed into a plastic bag, and the mortar was rinsed using 0.01 kg silicone dioxide, and transfe ⁇ ed into the bag. The contents ofthe bag were then blended for five minutes.
- the blend was passed through a #12 mesh screen and then blended for an additional 15 minutes.
- Magnesium stearate (0.114 kg) was passed through a #20 mesh screen and added to the blend and blended for five minutes.
- the blend was collected and placed in plastic bags.
- Two silica gel desiccant bags were placed around the plastic bags to absorb ambient moisture.
- the blend was then compressed into tablets.
- the average particle size ofthe drug (i.e., zolpidem) in the chewable tablet is about 20 microns, as compared to a typical average drag particle size of from about 75 to about 100 microns.
- the average particle size ofthe drug in the chewable tablet is less than or equal to the average particle size of he carrier ingredients (e.g., gum base, binders, etc.).
- the zolpidem tablet composition ofthe present invention can be used, e.g., for treatment of insomnia.
- the subject chews the chewable tablet as is normally done with any non-medicated type of chewable tablet at approximately an average rate of about 10 to about 45 chews per minute.
- the subject holds the tablet underneath the tongue and either swallows while the tablet is dissolving or swallows after the tablet has dissolved.
- a typical dosage form ofthe zolpidem tablet ofthe present invention is designed to produce an average plasma concentration of at least from about 20 to about 300 nanograms of zolpidem per milliliter of plasma.
- a 5 mg zolpidem tablet can be designed to produce a mean peak plasma concentration within the range of from about 20 to about 100 nanograms of zolpidem per milliliter of plasma within about 5 minutes to about 2 hours.
- a 10 mg zolpidem tablet can be designed to produce a mean peak plasma concentration within the range of from about 100 to about 300 nanograms of zolpidem per milliliter of plasma within about 5 minutes to about 2 hours.
- the tablet compositions ofthe present invention provide a convenient, reliable, practical, and painless system for delivering zolpidem across the oral mucosa.
- the tablet compositions are capable of rapidly delivering zolpidem with low inter-subject variability in terms of maximum plasma concentration (C ma ⁇ ) and the time to reach the maximum plasma concentration (T max ) so that a therapeutically effective amount of zolpidem enters the bloodstream within about 30 minutes, 20 minutes, 15 minutes, 10 minutes, 5 minutes, or even within about 1-2 minutes after zolpidem is released from the carrier.
- This example illustrates the zolpidem lozenge compositions ofthe present invention.
- Zolpidem can be formulated as a lozenge or candy composition as described above.
- the unit dose or serving ofthe lozenge comprises from about 0.1 to about 100 milligrams (mg) zolpidem (as measured in its tartrate salt form), preferably from about 1 to about 50 mg, and more preferably from about 2 to about 25 mg.
- the unit dose comprises from about 2 to about 20 mg zolpidem, preferably from about 2 to about 15 mg, and more preferably from about 2 to about 10 mg, e.g., about 2, 3, 4, 5, 6, 7, 8, 9, or 10 mg.
- the unit dose comprises a dose of zolpidem that is less than the dose typically used in commercial oral tablets, but possesses comparable or greater bioavailability and onset of therapeutic activity as well as lower inter-subject variability of drug absorption.
- unit doses of from about 2 to about 5 mg zolpidem are prefened, with unit doses of about 4 mg zolpidem being particularly prefened.
- Extra zolpidem for example, up to from about 10% to about 25% by weight, can be added as "overage” or as the amount that may be expected to be "washed away” and not otherwise released or absorbed during lozenge dissolution and/or mastication.
- the unit dose or serving ofthe lozenge comprises from about 0.81 to about 42 mg zolpidem in its base form, and more preferably from about 1.64 to about 20.5 mg.
- the unit dose comprises from about 1.64 to about 16.4 mg zolpidem in its free base form, preferably from about 1.64 to about 12.3 mg, and more preferably from about 1.64 to about 8.2 mg, e.g., about 1.64, 2.46, 3.28, 4.1, 4.92, 5.78, 6.56, 7.38, or 8.2 mg.
- the unit dose comprises a mixture of zolpidem in free base form and salt form (e.g., zolpidem tartrate).
- the zolpidem lozenge composition comprises from about 0.001% to about 10.0% zolpidem (in whatever chosen form, measured as per its free base form), preferably from about 0.1% to about 8.0%, more preferably from about 1.0% to about 7.0%, and still more preferably from about 1.0% to about 5.5%. In some embodiments, about 4.5% zolpidem is used.
- the buffer system ofthe zolpidem lozenge composition provides for a final salivary pH in excess of at least about 7.8, preferably at least about 8.5, and more preferably at least about 9 (e.g., about 9-11).
- a zolpidem lozenge was made according to the formulation shown in Table 4 and the following procedure. Three separate blends of silicon dioxide with zolpidem, sodium carbonate, and sodium bicarbonate; Pharmaburst; and spearmint flavor, sucralose, magnesium stearate, and croscarmellose sodium were prepared. The three blends were screened separately and mixed to form a single blend. The single blend was then compressed into lozenges after testing for content uniformity. By using this procedure, the average particle size ofthe drag (i.e., zolpidem) in the lozenge is about 20 microns, as compared to a typical average drug particle size of from about 75 to about 100 microns.
- the average particle size ofthe drag in the lozenge is less than or equal to the average particle size ofthe carrier ingredients (e.g., gum base, binders, etc.).
- the unit weight for each lozenge was 210 mg.
- the pH ofthe lozenge was about 9.8 and remained stable. These lozenges dissolve within about 2-3 minutes following sublingual administration.
- the batch quantity formulation produces 21,000 unit doses.
- the zolpidem lozenge composition ofthe present invention can be used, e.g., for treatment of insomnia.
- the subject holds the lozenge underneath the tongue and either swallows while the lozenge is dissolving or swallows after the lozenge has dissolved.
- the lozenges described herein have a very rapid rate of dissolution, and are capable of dissolving within about 2-3 minutes following sublingual administration.
- a typical dosage form ofthe zolpidem lozenge ofthe present invention is designed to produce an average plasma concentration of at least from about 20 to about 300 nanograms of zolpidem per milliliter of plasma.
- a 5 mg zolpidem lozenge can be designed to produce a mean peak plasma concentration within the range of from about 20 to about 100 nanograms of zolpidem per milliliter of plasma within about 5 minutes to about 2 hours.
- a 10 mg zolpidem lozenge can be designed to produce a mean peak plasma concentration within the range of from about 100 to about 300 nanograms of zolpidem per milliliter of plasma within about 5 minutes to about 2 hours.
- the lozenge compositions ofthe present invention provide a convenient, reliable, practical, and painless system for delivering zolpidem across the oral mucosa.
- the lozenge compositions are capable of very rapidly delivering zolpidem with low inter-subject variability in terms of maximum plasma concentration (C max ) and the time to reach the maximum plasma concentration (T ma ⁇ ) so that a therapeutically effective amount of zolpidem enters the bloodstream within about 30 minutes, 20 minutes, 15 minutes, 10 minutes, 5 minutes, or even within about 1-2 minutes after zolpidem is released from the carrier.
- Example 5 Dissolution Profiles for Zolpidem Tablet and Lozenge Compositions.
- This example illustrates the mean dissolution profiles for a zolpidem quick- dissolving tablet made according to Table 3 and a zolpidem lozenge made according to Table 4.
- compositions tested were as follows: 1. Zolpidem quick-dissolving tablet (typically dissolves sublingually in about 5 minutes). 2. Zolpidem lozenge (typically dissolves sublingually in about 2-3 minutes).
- Table 5 below shows the dissolution data and Figure 2 shows the mean dissolution profiles for a zolpidem quick-dissolving tablet and zolpidem lozenge ofthe present invention at 5, 10, 15, 20, and 30 minutes in phosphate buffered medium (pH 6.8). Table 5. Dissolution data for the zolpidem quick-dissolving tablet and zolpidem lozenge.
- a 10 mg zolpidem powdered tablet buffered at a pH of 9.8 with 23 mg sodium bicarbonate and 17 mg sodium carbonate was determined in eight healthy subjects (5 male, 3 female).
- Formulation A was administered under the subject's tongue and had a very rapid dissolution rate, i.e., within about 1 to about 3 minutes.
- the study performed was a fixed-sequence, open-label pharmacokinetic study in which subjects swallowed saliva at a rate of every 2, 5, or 10 minutes over a 10 minute period of time ("swallowing time").
- a 2 minute swallowing time refers to swallowing saliva every 2 minutes over a 10 minute period (i.e., 5 blocks of 2 minutes each); a 5 minute swallowing time refers to swallowing saliva every 5 minutes over a 10 minute period (i.e., 2 blocks of 5 minutes each); and a 10 minute swallowing time refers to swallowing saliva every 10 minutes over a 10 minute period (i.e., 1 block of 10 minutes).
- Serum blood samples were collected over an 8 hour period and the plasma was assayed for zolpidem levels, e.g., using high pressure liquid chromatography (HPLC)-tandem mass spectrometry (MS).
- HPLC high pressure liquid chromatography
- MS mass spectrometry
- Figures 3-5 show the plasma concentration over time in each subject for Formulation A at swallowing times of 2, 5, and 10 minutes, respectively.
- Tables 6-8 below show the values for the pharmacokinetic parameters determined in each subject for Formulation A at swallowing times of 2, 5, and 10 minutes, respectively.
- Table 6. Pharmacokinetic parameters for Formulation A at a 2 minute swallowing time.
- Values represent the mean.
- the numbers in parentheses for T ⁇ represent the rrnnimum and maximum values, respectively.
- the numbers in parentheses for C max and AUC represent the coefficient of variation percent (CV%).
- Figure 7 shows the mean plasma concentration over time for Formulation A at swallowing times of 2 and 5 minutes using the data from all 8 subjects and the mean plasma concentration over time for Formulation A at swallowing times of 2 and 5 minutes excluding the data from subjects 3, 6, and 7, who apparently swallowed earlier than their scheduled swallowing time.
- Table 10 shows the mean values for the pharmacokinetic parameters determined for Formulation A using the data from all 8 subjects or excluding the data from subjects 3, 6, and 7. When subjects who apparently did not comply with the study protocol were excluded from the analysis, peak plasma zolpidem concentrations for the remaining subjects were achieved within about 30 minutes rather than from about 45 to about 70 minutes following sublingual administration. Table 10. Pharmacokinetic parameters for Formulation A with all subjects or excluding those who swallowed early.
- Figure 8 is an expanded view ofthe first 90 minutes shown in Figure 6.
- Figure 8 illustrates the estimated time for the onset of sleep in subjects taking Formulation A (left dotted line) compared to the time for the onset of sleep in subjects taking Formulation B (right dotted line).
- the mean plasma zolpidem concentration effective for inducing sleep onset is shown by the horizontal line in Figure 8.
- Table 11 shows the reported time for the onset of sleep during the daytime in each subject taking Formulation A at the 3 different swallowing times.
- onset of sleep for subjects taking the zolpidem powdered sublingual tablet is substantially faster than that achieved with the commercial oral tablet.
- the onset of sleep for subj ects taking the sublingual tablets of the present invention can be as early as within about 12.5 minutes following administration, which is more than 3 times faster than the onset of sleep for subjects taking the commercial oral tablet.
- One skilled in the art will appreciate that the onset of sleep observed during the daytime conesponds to the onset of sleep at night.
- the pharmacokinetic profiles for sublingually administered zolpidem provide a softer and longer-lasting peak of zolpidem (see, Figure 6), and thus resemble a pharmacokinetic profile for intravenously administered zolpidem.
- this infusion- like pharmacokinetic profile is equivalent to or even superior to the commercial oral tablet in reducing the time to onset of therapeutic activity, maintaining sleep (e.g., total sleep time, number of awakenings), enhancing sleep quality, eliminating the effect of food, and reducing any morning-after residual effects.
- Formulation B (PO Ambien) was administered perorally with 180 ml of water.
- the study performed was a three-way crossover, fixed- sequence pharmacokinetic study in which subjects swallowed saliva at a rate of every 2 or 5 minutes over a 10 minute period of time ("swallowing time") for Formulations C and D.
- Serum blood samples were collected over a 12 hour period and the plasma was assayed for zolpidem levels, e.g., using high pressure liquid chromatography (HPLC)-tandem mass spectrometry (MS).
- HPLC high pressure liquid chromatography
- MS mass spectrometry
- Figure 9 shows the mean plasma concentration over time for Formulation C (SL Tablet) at swallowing times of 2 and 5 minutes and for Formulation B (PO Ambien).
- Figure 10 shows the mean plasma concentration over time for Formulation D (FS Tablet) at swallowing times of 2 and 5 minutes and for Formulation B (PO Ambien).
- This study demonstrates that delivery of zolpidem across the oral mucosa produced peak plasma zolpidem concentrations at a substantially earlier period in time and at a substantially higher level following sublingual administration than observed for the commercial oral tablet administration.
- the present study shows that zolpidem from both dissolving tablets is rapidly absorbed and has substantially better bioavailability than the commercial oral tablet.
- the onset of sleep for subjects taking either zolpidem dissolving tablet is substantially faster than that achieved with the commercial oral tablet.
- the present study also shows that the improvement in bioavailability is independent of the swallowing time and the formulation ofthe dissolving tablet.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Physiology (AREA)
- Nutrition Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Zoology (AREA)
- Engineering & Computer Science (AREA)
- Anesthesiology (AREA)
- Medicinal Preparation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description
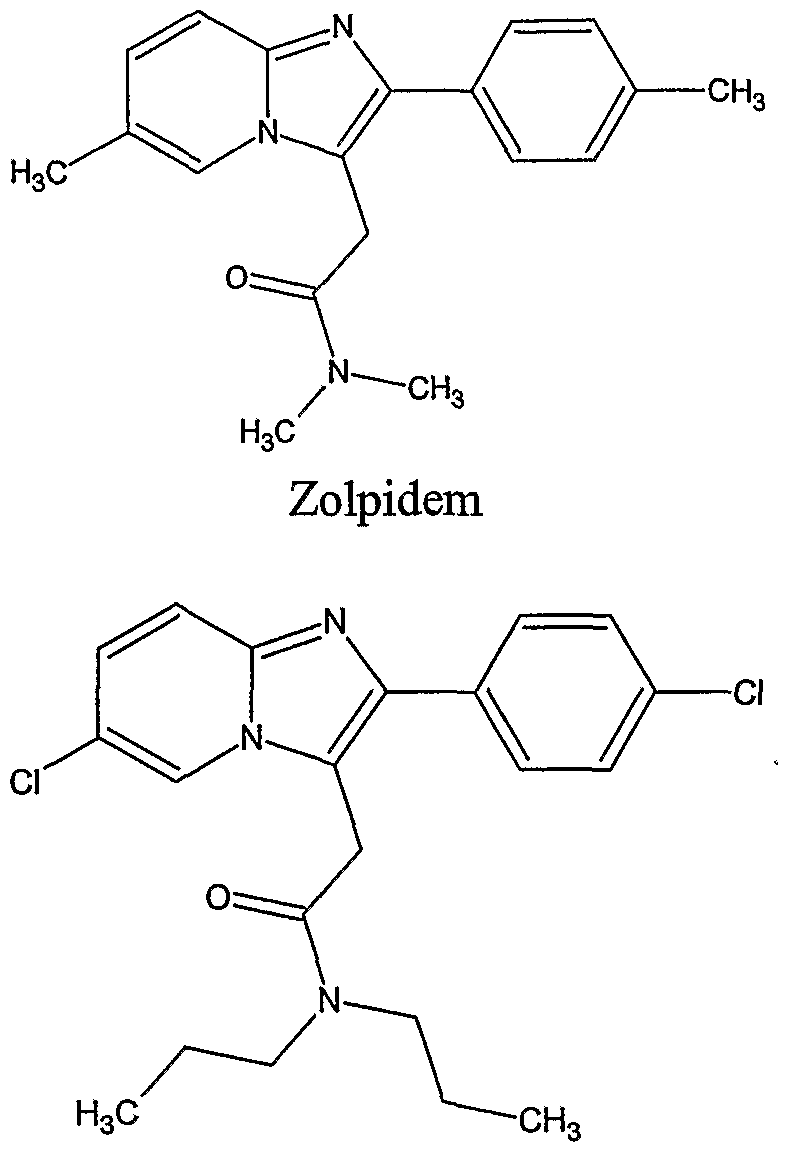







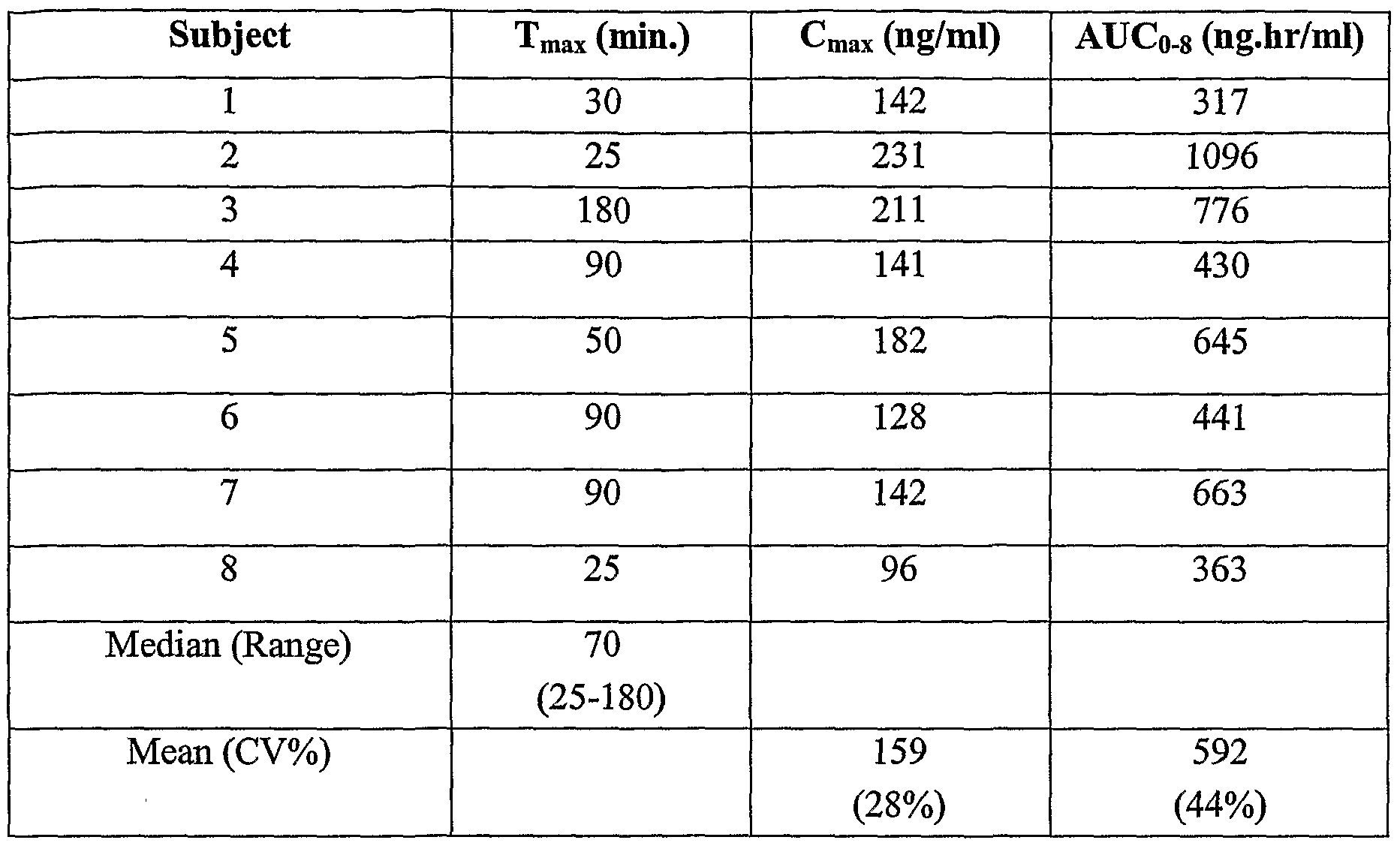
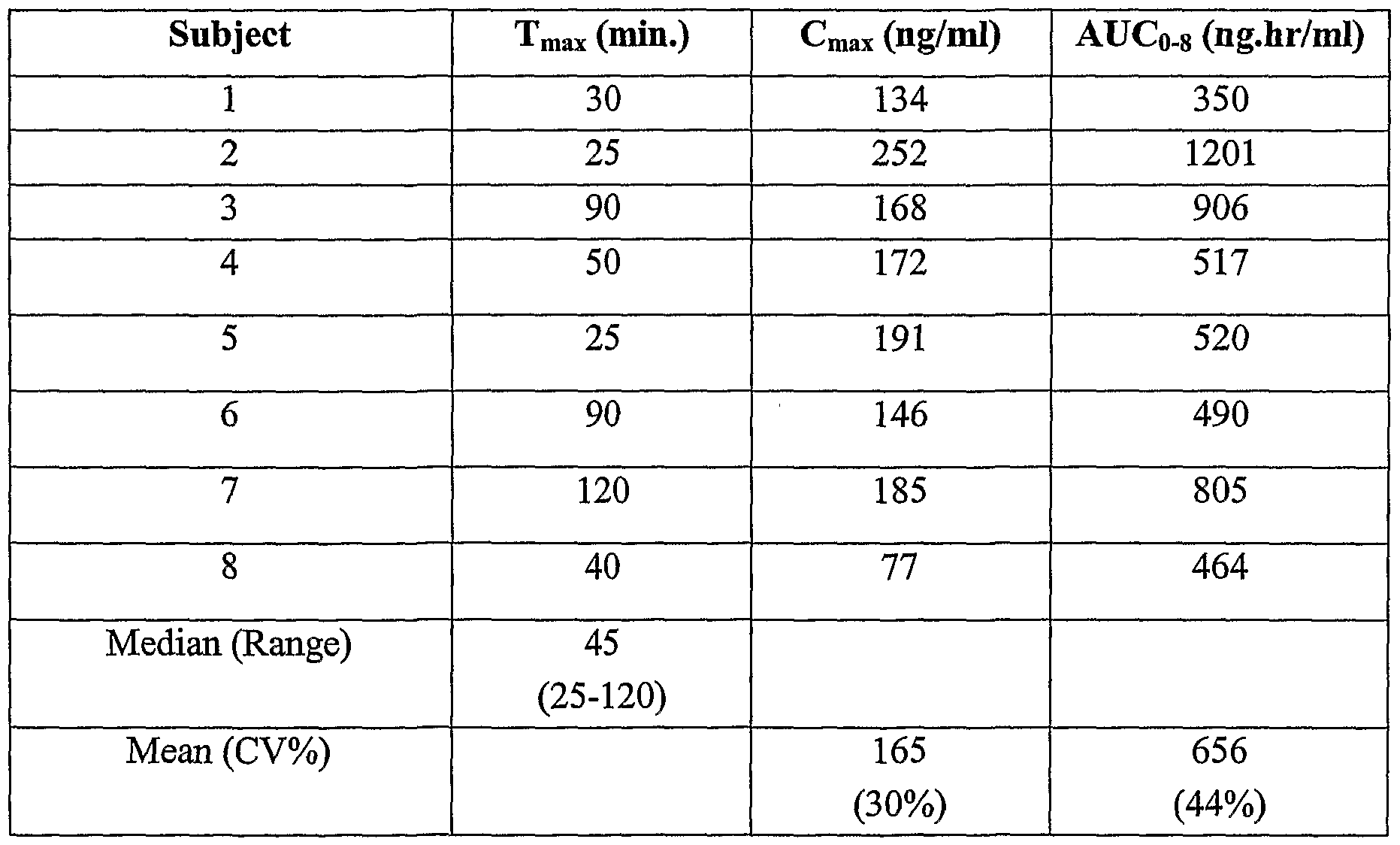



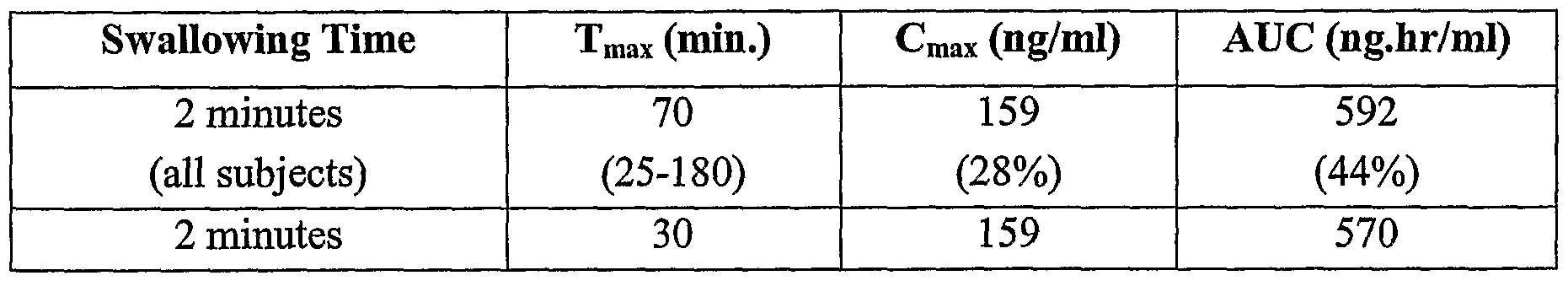


Claims
Priority Applications (13)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020167023779A KR20160105935A (en) | 2004-02-17 | 2005-02-16 | Compositions for delivering hypnotic agents across the oral mucosa and methods of use thereof |
| EP05713712A EP1715853A4 (en) | 2004-02-17 | 2005-02-16 | Compositions for delivering hypnotic agents across the oral mucosa and methods of use thereof |
| BRPI0507733-8A BRPI0507733A (en) | 2004-02-17 | 2005-02-16 | composition for the release of a hypnotic agent through the oral mucosa, and method for treating a sleep disorder in a patient in need thereof |
| KR1020147018143A KR20140104986A (en) | 2004-02-17 | 2005-02-16 | Compositions for delivering hypnotic agents across the oral mucosa and methods of use thereof |
| KR1020127031305A KR20130006523A (en) | 2004-02-17 | 2005-02-16 | Compositions for delivering hypnotic agents across the oral mucosa and methods of use thereof |
| KR1020137025409A KR20130116378A (en) | 2004-02-17 | 2005-02-16 | Compositions for delivering hypnotic agents across the oral mucosa and methods of use thereof |
| JP2006553364A JP5179757B2 (en) | 2004-02-17 | 2005-02-16 | Composition for delivering a hypnotic agent through the oral mucosa and method of use thereof |
| MXPA06009296A MXPA06009296A (en) | 2004-02-17 | 2005-02-16 | Compositions for delivering hypnotic agents across the oral mucosa and methods of use thereof. |
| AU2005215782A AU2005215782B2 (en) | 2004-02-17 | 2005-02-16 | Compositions for delivering hypnotic agents across the oral mucosa and methods of use thereof |
| NZ548856A NZ548856A (en) | 2004-02-17 | 2005-02-16 | Compositions for delivering hypnotic agents across the oral mucosa and methods of use thereof |
| KR1020157007690A KR20150038745A (en) | 2004-02-17 | 2005-02-16 | Compositions for delivering hypnotic agents across the oral mucosa and methods of use thereof |
| CA2556450A CA2556450C (en) | 2004-02-17 | 2005-02-16 | Compositions for delivering hypnotic agents across the oral mucosa and methods of use thereof |
| IL177227A IL177227A (en) | 2004-02-17 | 2006-08-02 | Compositions comprising zolpidem for delivering a hypnotic agent across the oral mucosa for treating insomnia |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US78311804A | 2004-02-17 | 2004-02-17 | |
| US60/608,957 | 2004-02-17 | ||
| US59862904P | 2004-08-03 | 2004-08-03 | |
| US60/598,629 | 2004-08-03 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2005079761A1 true WO2005079761A1 (en) | 2005-09-01 |
Family
ID=34890604
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2005/005021 WO2005079761A1 (en) | 2004-02-17 | 2005-02-16 | Compositions for delivering hypnotic agents across the oral mucosa and methods of use thereof |
Country Status (7)
| Country | Link |
|---|---|
| AU (1) | AU2005215782B2 (en) |
| BR (1) | BRPI0507733A (en) |
| CA (1) | CA2556450C (en) |
| IL (1) | IL177227A (en) |
| MX (1) | MXPA06009296A (en) |
| NZ (1) | NZ548856A (en) |
| WO (1) | WO2005079761A1 (en) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1883408A2 (en) * | 2005-05-25 | 2008-02-06 | Transcept Pharmaceuticals, Inc. | Solid compositions and methods for treating middle-of-the night insomnia |
| US20120238616A1 (en) * | 2007-04-09 | 2012-09-20 | Handley Dean A | Methods and compositions for treating sleep-related breathing disorders |
| US20130280327A1 (en) * | 2010-12-16 | 2013-10-24 | Sanofi | Zolpidem-based orodispersible pharmaceutical tablet |
| CN103919780A (en) * | 2012-12-26 | 2014-07-16 | 上海中西制药有限公司 | Sedative-hypnotic preparation, compound preparation, preparation method and pharmaceutical composition thereof |
| US9265720B2 (en) | 2004-10-27 | 2016-02-23 | Orexo Ab | Pharmaceutical formulations useful in the treatment of insomnia |
| WO2020075183A1 (en) | 2018-10-08 | 2020-04-16 | Troikaa Pharmaceuticals Limited | Oromucosal solutions of zolpidem or pharmaceutically acceptable salts thereof |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6218397B1 (en) * | 1992-12-17 | 2001-04-17 | Pfizer Inc | Pyrazolopyrimidines as CRF antagonists |
| US6441018B2 (en) * | 1992-12-17 | 2002-08-27 | Pfizer Inc. | Pyrazoles and pyrazolopyrimidines having CRF antagonistic activity |
| US6624162B2 (en) * | 2001-10-22 | 2003-09-23 | Pfizer Inc. | Imidazopyridine compounds as 5-HT4 receptor modulators |
-
2005
- 2005-02-16 MX MXPA06009296A patent/MXPA06009296A/en active IP Right Grant
- 2005-02-16 AU AU2005215782A patent/AU2005215782B2/en not_active Ceased
- 2005-02-16 BR BRPI0507733-8A patent/BRPI0507733A/en not_active Application Discontinuation
- 2005-02-16 NZ NZ548856A patent/NZ548856A/en not_active IP Right Cessation
- 2005-02-16 WO PCT/US2005/005021 patent/WO2005079761A1/en active Application Filing
- 2005-02-16 CA CA2556450A patent/CA2556450C/en not_active Expired - Fee Related
-
2006
- 2006-08-02 IL IL177227A patent/IL177227A/en not_active IP Right Cessation
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6218397B1 (en) * | 1992-12-17 | 2001-04-17 | Pfizer Inc | Pyrazolopyrimidines as CRF antagonists |
| US6441018B2 (en) * | 1992-12-17 | 2002-08-27 | Pfizer Inc. | Pyrazoles and pyrazolopyrimidines having CRF antagonistic activity |
| US6624162B2 (en) * | 2001-10-22 | 2003-09-23 | Pfizer Inc. | Imidazopyridine compounds as 5-HT4 receptor modulators |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9265720B2 (en) | 2004-10-27 | 2016-02-23 | Orexo Ab | Pharmaceutical formulations useful in the treatment of insomnia |
| US9597281B2 (en) | 2004-10-27 | 2017-03-21 | Orexo Ab | Pharmaceutical formulations useful in the treatment of insomnia |
| EP1883408A2 (en) * | 2005-05-25 | 2008-02-06 | Transcept Pharmaceuticals, Inc. | Solid compositions and methods for treating middle-of-the night insomnia |
| EP1883408A4 (en) * | 2005-05-25 | 2009-11-11 | Transcept Pharmaceuticals Inc | Solid compositions and methods for treating middle-of-the night insomnia |
| US20120238616A1 (en) * | 2007-04-09 | 2012-09-20 | Handley Dean A | Methods and compositions for treating sleep-related breathing disorders |
| US20130280327A1 (en) * | 2010-12-16 | 2013-10-24 | Sanofi | Zolpidem-based orodispersible pharmaceutical tablet |
| CN103919780A (en) * | 2012-12-26 | 2014-07-16 | 上海中西制药有限公司 | Sedative-hypnotic preparation, compound preparation, preparation method and pharmaceutical composition thereof |
| CN103919780B (en) * | 2012-12-26 | 2016-12-28 | 上海中西制药有限公司 | Calming soporific preparation, its compound preparation, preparation method and pharmaceutical composition |
| WO2020075183A1 (en) | 2018-10-08 | 2020-04-16 | Troikaa Pharmaceuticals Limited | Oromucosal solutions of zolpidem or pharmaceutically acceptable salts thereof |
| CN112996488A (en) * | 2018-10-08 | 2021-06-18 | 特罗伊卡药品有限公司 | Oral mucosal solution of zolpidem or pharmaceutically acceptable salt thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| IL177227A0 (en) | 2006-12-10 |
| MXPA06009296A (en) | 2007-01-26 |
| BRPI0507733A (en) | 2007-07-10 |
| CA2556450A1 (en) | 2005-09-01 |
| IL177227A (en) | 2013-05-30 |
| AU2005215782B2 (en) | 2011-04-21 |
| NZ548856A (en) | 2009-10-30 |
| CA2556450C (en) | 2013-08-06 |
| AU2005215782A1 (en) | 2005-09-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US7658945B2 (en) | Compositions for delivering hypnotic agents across the oral mucosa and methods of use thereof | |
| US8242131B2 (en) | Methods of treating middle-of-the-night insomnia | |
| US20080254101A1 (en) | Pilocarpine compositions and methods of use thereof | |
| WO2005018565A2 (en) | Compositions for delivering 5-ht agonists across the oral mucosa and methods of use thereof | |
| US20120034300A1 (en) | Stabilized zolpidem pharmaceutical compositions | |
| US20110039881A1 (en) | Compositions and methods for treating middle-of-the-night insomnia | |
| AU2005215782B2 (en) | Compositions for delivering hypnotic agents across the oral mucosa and methods of use thereof | |
| CA2816904A1 (en) | Compositions for delivering hypnotic agents across the oral mucosa and methods of use thereof | |
| KR20160105935A (en) | Compositions for delivering hypnotic agents across the oral mucosa and methods of use thereof | |
| AU2016203713A1 (en) | Solid compositions and methods for treating middle-of-the night insomnia | |
| AU2012238306A1 (en) | Solid compositions and methods for treating middle-of-the night insomnia |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2005713712 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 548856 Country of ref document: NZ Ref document number: 2005215782 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006/06411 Country of ref document: ZA Ref document number: 200606411 Country of ref document: ZA Ref document number: 177227 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006553364 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2006/009296 Country of ref document: MX Ref document number: 2556450 Country of ref document: CA |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2005215782 Country of ref document: AU Date of ref document: 20050216 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005215782 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2493/KOLNP/2006 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200580008195.3 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020067018935 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005713712 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020067018935 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: PI0507733 Country of ref document: BR |