WO2005077411A2 - Composition and method for the treatment of carcinoma - Google Patents
Composition and method for the treatment of carcinoma Download PDFInfo
- Publication number
- WO2005077411A2 WO2005077411A2 PCT/IB2005/000509 IB2005000509W WO2005077411A2 WO 2005077411 A2 WO2005077411 A2 WO 2005077411A2 IB 2005000509 W IB2005000509 W IB 2005000509W WO 2005077411 A2 WO2005077411 A2 WO 2005077411A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- cell activator
- compound
- administered
- cell
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/39—Medicinal preparations containing antigens or antibodies characterised by the immunostimulating additives, e.g. chemical adjuvants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/66—Phosphorus compounds
- A61K31/662—Phosphorus acids or esters thereof having P—C bonds, e.g. foscarnet, trichlorfon
- A61K31/663—Compounds having two or more phosphorus acid groups or esters thereof, e.g. clodronic acid, pamidronic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7068—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid
- A61K31/7072—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines having oxo groups directly attached to the pyrimidine ring, e.g. cytidine, cytidylic acid having two oxo groups directly attached to the pyrimidine ring, e.g. uridine, uridylic acid, thymidine, zidovudine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7042—Compounds having saccharide radicals and heterocyclic rings
- A61K31/7052—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides
- A61K31/706—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom
- A61K31/7064—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines
- A61K31/7076—Compounds having saccharide radicals and heterocyclic rings having nitrogen as a ring hetero atom, e.g. nucleosides, nucleotides containing six-membered rings with nitrogen as a ring hetero atom containing condensed or non-condensed pyrimidines containing purines, e.g. adenosine, adenylic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/19—Cytokines; Lymphokines; Interferons
- A61K38/20—Interleukins [IL]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/02—Bacterial antigens
- A61K39/04—Mycobacterium, e.g. Mycobacterium tuberculosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K2039/555—Medicinal preparations containing antigens or antibodies characterised by a specific combination antigen/adjuvant
Definitions
- the present invention relates to compositions and methods useful for treating a carcinoma or viral infection in mammals, including humans.
- the methods and compositions typically comprise use of an immunogenic or immunomodulatory compound, and a ⁇ T cell activator, such that the composition is effective for treating a carcinoma or viral infection.
- the methods comprise use of a a ⁇ T cell activator and a Mycobacterium antigen, for example is an attenuated strain of Mycobacterium bovis (Bacillus Calmette-Guerin (BCG)).
- Carcinomas account for about 85% of all cancers. A significant portion of these carcinomas are carcinoma in situ, or superficial cancers, such as superficial bladder cancer and diseases caused by human papilloma virus (HPV) infection.
- HPV human papilloma virus
- Carcinoma of the bladder accounts for about 2 % of all solid tumors in the United States with more than 50,000 new cases being diagnosed each year.
- the peak prevalence of bladder cancer is in individuals 60-70 years old and several etiologic factors have been implicated including smoking and exposure to industrial chemicals.
- Bladder cancer is the fifth most common neoplasm and the twelfth leading cause of cancer death.
- carcinoma of the bladder is categorized by grade (usually I-IV) and by depth of malignancy (either superficial, invasive, or metastatic bladder cancer).
- Superficial bladder cancer which is confined to the bladder epithelium, usually presents as papillary tumors (stages ta or TI) or carcinoma-in-situ (CIS).
- Diagnosis of bladder cancer is by cytoscopy and biopsy. At the time of diagnosis, about 70 % of patients have only superficial disease, 25 % have locally invasive disease, and 5 % already have distant metastasis.
- Superficial bladder cancer is treated with transurethal resection and/or fulguration. Cytoscopy is usually reserved for those tumors which cannot be resected transurethrally. After transurethal resection, 50 % of patients remain disease free; however the other half will experience multiple recurrences with about 10 % developing invasive or metastatic disease within 3-4 years. Superficial recurrences are treated with transurethal resection, often followed by intravesical chemotherapy to prevent or delay any additional recurrence. Patients who are considered at high risk for recurrence after the initial transurethal resection or those with concurrent CIS are frequently given intravesical adjunct therapy as prophylaxis against recurrence.
- BCG Bacillus Calmette-Guerin
- mitomycin mitomycin
- doxorubicin doxorubicin
- thiotepa thiotepa
- Clinical studies may have various endpoints such as tumor recurrence, tumor progression or patient survival.
- endpoints such as tumor recurrence, tumor progression or patient survival.
- a significant reduction in tumor recurrences was noted in 4 of 5 BCG studies, 2 of 5 mitomycin studies, 2 of 4 doxorubicin studies, and 6 of 10 thiotepa studies; and a significant reduction in tumor progression was documented in 3 of 3 BCG studies, 0 of 2 mitomycin studies, 0 of 2 doxorubicin studies, and 0 of 3 thiotepa studies.
- BCG is the only one shown to result in a survival advantage over transurethral resection alone.
- intravesical BCG is a form of immunotherapy. Intravesical BCG appears to induce tumor regression through a number of specific and non-specific actions. It promotes a local inflammatory reaction with histiocytic and leukocytic infiltration in the urinary bladder that is apparently associated with an elimination or reduction of superficial cancerous lesions.
- HPV infection Human papilloma virus (HPV) infections of the urogenital tract represent the most often sexually transmitted viral disease in humans.
- HPV is a double stranded DNA virus and with the recent developed molecular biological techniques, more than 55 different HPV types have been recognized.
- HPV is associated with a wide spectrum of clinical states including condylomata acuminata, latent and subclinical infection, and Bowen's disease. Subclinical infections gain more importance as they are believed to cause intraepithelial neoplasia, based on the frequent detection of HPV DNA in invasive carcinomas, especially in urogenital region.
- a significant risk for the development of an invasive cancer is ascribed to the infections by HPV types 1 6, 1 8 and 33.
- Condylomata acuminata are visible, multifocal, multicentric and multiform lesions. Predilection sites are penis, scrotum, perineum, urethra, perianal regions, intertriginous zones, and oral mucosa. In uncircumcised men the frenulurn, the coronary sulcus and the inner aspect of the foreskin are most often afflicted, whereas in circumcised patients the shaft of the penis is involved. Genital warts are of great psychological and cosmetic relevance representing a major hindrance to sexual performance.
- Treatment options include surgical methods like excision, electrocautery, cryosurgery or laser vaporization. It has been shown in molecular hybridization studies that HPV DNA sequences exist in adjacent normal tissue after carbon dioxide laser removal of genital warts. These findings and the well known high recurrence rates after initial treatment demonstrate the need for adjuvant therapy to eradicate invisible disease.
- the present invention now discloses particular compositions and methods that can be used to efficiently treat a tumor, particularly a carcinoma or adenocarcinoma, and preferably a bladder cancer, in a subject.
- the invention also provides compositions and methods for the treatment of a viral infection, preferably an HPV infection, and conditions associated therewith such as cell proliferative disorders.
- a ⁇ T cell activating compound can enhance the effects of a locally administered immunomodulatory composition (IMC) or immunogenic composition (IC), regardless of whether the IMC or IC are ⁇ T cell activating or a non- ⁇ T cell activating compounds.
- the invention provides a method for enhancing the effect of a locally administered immunomodulatory composition (IMC) or immunogenic composition (IC) in a mammal, the method comprising administering to the mammal a ⁇ T cell activating compound.
- the invention encompasses a method for killing or inhibiting a proliferating cell, a tumor cell or an infected cell in a mammal, the method comprising administering to the mammal an immunomodulatory composition (IMC) or immunogenic composition (IC) locally to a site of disease, and administering a ⁇ T cell activating compound.
- IMC immunomodulatory composition
- IC immunogenic composition
- the invention further discloses the use of an IMC or IC composition for the manufacture of a pharmaceutical composition or medicament, wherein said pharmaceutical composition or medicament is used or administered in combination with a ⁇ T cell activator.
- the invention encompasses the use of a ⁇ T cell activator for the manufacture of a pharmaceutical composition or medicament, wherein said pharmaceutical composition or medicament is used or administered in combination with an IMC or IC composition.
- the invention further discloses the use of an IMC or IC composition and a ⁇ T cell activator for the manufacture of a pharmaceutical composition or medicament.
- the IMC or IC is for local administration to a site of disease.
- related pharmaceutical compositions and kits comprising such compositions.
- the ⁇ T cell activating compound will be administered by a route or to a site other than the site of disease to which the IMC or IC is administered.
- the ⁇ T cell activating compound is administered by a method other than local administration to a disease site.
- the latter method of treatment comprising local and non-local administration can have beneficial effects, particularly when the locally-administered component is delivered intravesically or to skin, for example for treatment of bladder cancer, HPV infection, cell proliferative conditions such as skin disorders and external genital warts, and actinic keratosis and skin tumors, particularly non-melanoma skin cancers such as superficial basal cell carcinoma (BCC), or intra-tumorally, for the treatment of solid tumors.
- BCC superficial basal cell carcinoma
- the treatments (and pharmaceutical compositions) of the present invention can particularly advantageously be used in the treatment of proliferative disorders, tumors, solid tumors, carcinomas, bladder cancer, HPV infection, cell proliferative conditions such as skin disorders, external genital warts, and actinic keratosis and skin tumors, particularly non-melanoma skin cancers such as superficial basal cell carcinoma (BCC).
- BCC superficial basal cell carcinoma
- the method provides that administration of a ⁇ T cell activating compound locally to a site of disease can have beneficial effects, particularly when delivered intravesically or to skin, for example for treatment of bladder cancer, HPV infection, cell proliferative conditions such as skin disorders and external genital warts, and actinic keratosis and skin tumors, particularly non-melanoma skin cancers such as superficial basal cell carcinoma (BCC).
- BCC superficial basal cell carcinoma
- the invention provides a method comprising administering locally to a site of disease a composition capable of recruiting or preferably regulating ⁇ T cell activity or most preferably activating a ⁇ T cell.
- the composition comprises a ⁇ T cell activating compound, most preferably the compound selected from the group of: a compound capable of selectively activating a ⁇ T cell, a compound capable of activating a ⁇ T cell in a substantially pure culture of ⁇ T cells, and a compound of Formulas I to XVI.
- a ⁇ T cell activating compound most preferably the compound selected from the group of: a compound capable of selectively activating a ⁇ T cell, a compound capable of activating a ⁇ T cell in a substantially pure culture of ⁇ T cells, and a compound of Formulas I to XVI.
- the immunomodulatory composition can generally be any agent that modulates one or more aspects of the immune system, for example by stimulating certain aspects of the immune system, or by suppressing certain other aspects, as further described herein.
- An immunogenic composition (IC) may be any agent capable of eliciting a humoral or cellular immune response, or both, when administered to an animal having an immune system.
- An IMC or IC may be, for example, capable of recruiting or preferably regulating the activity of, including but not limited to regulating cytokine production, activating or inhibiting, directly or indirectly, any type of immune cell, including for example activating ⁇ T cells or modulating an activity of ⁇ T cells, or modulating the activity of, particularly maturation of, dendritic cells.
- an IMC or IC can also be a ⁇ T cell activating compound.
- the IMC or IC can be any other suitable compound; an IMC may be for example a cytokine such as IL-2, IL-12, IL15, DL-21 or an agonist of a toll-like receptor (TLR), such as TLR2, TLR3, TLR4, TLR6, TLR7 or TLR9, or other agents described herein.
- TLR toll-like receptor
- Preferred ICs are polypeptide antigens, particularly microbial or tumor antigens, or a killed or attenuated pathogen, microorganism or parasite such as viruses or bacterial strains. A number of such agents are further described herein. Examples of routes of local administration to a site of disease can include but are not limited to dermal and intradermal, intravesical administration (e.g. bladder cancer), or generally intra-tumoral administration (e.g. solid tumors).
- the ⁇ T cell activating compounds described herein can be any suitable ⁇ T cell activating compound. Such a compound can be prepared for use in local or non-local (to a site of disease) administration, including a range of ⁇ T cell activating compounds described herein.
- mycobacterial antigens of which several compositions are currently approved for human therapy for local administration.
- Administration of mycobacterial antigens may lead to activation of ⁇ T cells and therefore represent an example of the invention where the IC or IMC activates ⁇ T cells.
- a mycobacterial antigen is administered locally to a site of disease (e.g.
- a ⁇ T cell activating compound that stimulates the proliferation and/or biological activity of ⁇ T cells is administered to the patient by a non-local route, preferably systemically, most preferably by intravenous or intramuscular administration.
- the ⁇ T cell activating compound administered systemically can be the same compound as the ⁇ T cell activating compound administered locally, or can be a different compound.
- a mycobacterial antigen for example a mycobacterial strain
- it will be preferably to use a different ⁇ T cell activating compound for systemic administration.
- ⁇ T cell activating compounds for systemic administration, particularly synthetic and selective ⁇ T cell activating compounds are provided herein. Such compounds show little or no toxicity at doses required to activate ⁇ T cells.
- the combination therapy thereby preferably amplifies the ⁇ T cell-mediated effects of the composition that is administered locally.
- the EVIC compound or composition is an imidazoquinoline compound, and/or is an agonist of a toll-like receptor (TLR).
- TLR toll-like receptor
- At least one imidazoquinoline compound is currently approved for human therapy for local administration, and others in clinical development.
- a preferred example is AldaraTM (3M; imiquimod), formulated as a cream for dermal administration, for use in the treatment of superficial basal cell carcinoma, HPV infection, and also in testing for the treatment of cutaneous metastases of malignant melanoma (Bong et al, Dermatology 2002;205:135-138).
- Resiquimod has been shown to induce endogenous production of alpha interferon (IFN- ⁇ ), interleukin 12 (IL-12), tumor necrosis factor alpha, and other cytokines from peripheral blood mononuclear cells, monocytes, and dendritic cells (DCs).
- IFN- ⁇ alpha interferon
- IL-12 interleukin 12
- DCs dendritic cells
- Resiquimod is about 100 times more potent in inducing cytokines in vitro and in vivo than the related imidazoquinoline imiquimod (Aldara R-837) (Sauder et al, (2003) Antimicrobial Agents and Chemotherapy, 47(12): 3846- 3852). Resiquimod and imiquimod also differ in the cytokine induction profile: in peripheral blood mononuclear cell cultures, resiquimod induces larger amounts of IL-12 directly and larger amounts of TFN- ⁇ indirectly. Resiquimod is also more effective in enhancing antigen presentation by DCs.
- TLR Toll-like receptor
- TLR7 Toll-like receptor 7
- TLR8 Toll-like receptor 8
- a TLR agonist such as imiquimod or resiquimod is administered locally to a site of disease (e.g. dermally in the case of skin proliferative disorder or skin cancer, or genital warts or HPV infection), and in conjunction with this local administration a ⁇ T cell activating compound that stimulates the proliferation and/or biological activity of ⁇ T cells is administered to the patient by a non-local route, preferably systemically, most preferably by intravenous or intramuscular administration.
- TLR agonist compounds are known in the art and/or described in references cited herein, including but not limited to nucleic acid-based agonists such as CpG containing nucleic acids (TLR9 agonists) and double-stranded RNA (TLR3 agonists).
- nucleic acid-based agonists such as CpG containing nucleic acids (TLR9 agonists) and double-stranded RNA (TLR3 agonists).
- the locally-administered IMC or IC is administered in an amount effective to treat said disease when used in combination therapy with the second compound which is a ⁇ T cell activator.
- ⁇ T cell activator is administered systemically, preferably by intravenous, subcutaneous or intramuscular injection.
- the IMC comprises a compound capable of activating a ⁇ T cell, a cytokine, or a compound which is an agonist of a toll-like receptor (TLR).
- the agonist of a TLR is an imidazoquinoline compound or analog or derivative thereof, or a mycobacterium antigen.
- Said disease is preferably a proliferative disorder, a carcinoma or a viral infection; preferred examples include respectively a bladder cancer, a skin tumor or cancer, or an HPV infection.
- the invention provides a method for the treatment of a disease comprising: (a) administering to said subject a first ⁇ T cell activator compound in a quantity sufficient to stimulate ⁇ T cell activity; and (b) administering to a subject locally at a site of disease, a second ⁇ T cell activator, said second ⁇ T cell activator being administered in a quantity effective to treat said disease when used in combination therapy with said first ⁇ T cell activator.
- Said first and second ⁇ T cell activators may comprise the same compound or composition or may comprise different compounds or compositions.
- the second ⁇ T cell activator is a mycobacterium antigen, and preferably the first ⁇ T cell activator is a selective ⁇ T cell activator.
- the first ⁇ T cell activator is administered systemically, preferably by intravenous injection.
- Said disease is preferably a carcinoma or a viral infection; preferred examples include respectively a bladder cancer or an HPV infection.
- the invention also provides that a selective ⁇ T cell activator can be used in combination with a mycobacterial antigen, preferably by administration via the same route, e.g. preferably intravesical administration or administration to skin.
- a selective ⁇ T cell activator can be used in combination with a mycobacterial antigen, preferably by administration via the same route, e.g. preferably intravesical administration or administration to skin.
- the invention thus discloses a method for treating bladder cancer or HPV infection in a patient comprising administering to a patient in need thereof an amount of a Mycobacterium antigen and a ⁇ T cell activator effective to treat said disease.
- a mycobacterial antigen and a selective ⁇ T cell activator are both administered at a site of disease (e.g. intravesicularly or to skin).
- the mycobacterial antigen and the selective ⁇ T cell activator can be aclministered at the same time or at different times, and can be provided in separate compositions or as a single composition.
- the invention thus also discloses a pharmaceutical composition comprising an IC or IMC and a ⁇ T cell activator at an effective dose to treat a carcinoma or viral infection, preferaby wherein the IMC or IC is not interleukin-2.
- the invention discloses a pharmaceutical composition comprising a Mycobacterium antigen and a ⁇ T cell activator, preferably at an effective dose to treat a carcinoma such as bladder cancer, urinary cancer or a viral infection such as an HPV infection.
- the invention thus also provides the use of a ⁇ T cell activator for the manufacture of a pharmaceutical composition for the treatment of bladder cancer, urinary cancer or a viral infection such as an HPV infection.
- the invention further discloses the use of a Mycobacterium antigen and a ⁇ T cell activator for the manufacture of a pharmaceutical composition for the treatment of bladder cancer.
- the invention also discloses a kit for the treatment of bladder cancer comprising a Mycobacterium antigen and a ⁇ T cell activator.
- said Mycobacterium antigen is an antigen of Mycobacterium bovis.
- said Mycobacterium antigen is an antigen of Mycobacterium phlei.
- said Mycobacterium antigen is an attenuated strain thereof.
- said Mycobacterium antigen is an attenuated strain of Mycobacterium bovis (BCG).
- said Mycobacterium antigen is mycobacterial cell wall, preferably complexed to Mycobacterium DNA.
- a ⁇ T cell activator is a selective ⁇ T cell activator capable of regulating the activity of a ⁇ T cell in a population of ⁇ T cell in culture, most preferably in a substantially pure population of ⁇ T cells, or in a population of ⁇ T cell clones.
- the ⁇ T cell activator is preferably capable of regulating the activity of a ⁇ T cell population of ⁇ T cell clones at millimolar concentration, preferably when the ⁇ T cell activator is present in culture at a concentration of less than 100 niM.
- a ⁇ T cell activator is capable of regulating the activity of a ⁇ T cell in a population of ⁇ T cell clones at millimolar concentration, preferably when the ⁇ T cell activator is present in culture at a concentration of less than 10 mM, or more preferably less than 1 mM.
- Regulating the activity of a ⁇ T cell can be assessed by any suitable means, preferably by assessing cytokine secretion, most preferably TNF-cc secretion as described herein. Methods for obtaining a population of pure ⁇ T cell clones is described in Davodeau et al, (1993) J. Immunology 151(3): 1214-1223 and Moreau et al, (1986) J. Clin. Invest.
- the activator is capable of causing at least a 20%, 50% or greater increase in the number of ⁇ T cells in culture, or more preferably at least a 2-fold increase in the number of ⁇ T cells in culture.
- said ⁇ T cell activator is a composition comprising a compound of formula (I) :
- m is an integer from 1 to 3;
- B is O, NH, or any group capable to be hydrolyzed;
- Y 0 " Cat+, a -C 3 alkyl group, a group -A-R, or a radical selected from the group consisting of a nucleoside, an oligonucleotide, a nucleic acid, an amino acid, a peptide, a protein, a monosaccharide, an oligosaccharide, a polysaccharide, a fatty acid, a simple lipid, a complex lipid, a folic acid, a tetrahydrofolic acid, a phosphoric acid, an inositol, a vitamin, a co-enzyme, a flavonoid, an aldehyde, an epoxyde and a halohydrin;
- A is O, NH, CHF, CF 2 or CH 2 ;
- R is a linear, branched, or cyclic, aromatic or not, saturated or unsaturated, C 1 -C 50 hydrocarbon group, optionally interrupted by at least one heteroatom, wherein said hydrocarbon group comprises an alkyl, an alkylenyl, or an alkynyl, preferably an alkyl or an alkylene, which can be substituted by one or several substituents selected from the group consisting of : an alkyl, an alkylenyl, an alkynyl, an epoxyalkyl, an aryl, an heterocycle, an alkoxy, an acyl, an alcohol, a carboxylic group (-COOH), an ester, an amine, an amino group (-NH 2 ), an amide (-CONH 2 ), an imine, a nitrile, an hydroxyl (-OH), a aldehyde group (-CHO), an halogen, an halogenoalkyl, a thiol (-SH), a thioalky
- said ⁇ T cell activator is a composition comprising a compound of formula (II):
- X is an halogen (preferably selected from I, Br and CI)
- B is O or NH
- m is an integer from 1 to 3
- RI is a methyl or ethyl group
- Cat+ represents one (or several, identical or different) organic or mineral cation(s) (including the proton)
- n is an integer from 2 to 20
- A is O, NH, CHF, CF 2 or CH 2
- Y is O " Cat+, or a nucleoside.
- said ⁇ T cell activator is selected from the group consisting of BrHPP, CBrHPP and epoxPP.
- said ⁇ T cell activator is BrHPP.
- said ⁇ T cell activator is CBrHPP.
- said ⁇ T cell activator is epoxPP.
- said ⁇ T cell activator is a composition comprising a compound of formula (Xfl): in which R 3 , f , and R 5 , identical or different, are a hydrogen or (C ⁇ -C 3 )alkyl group, W is -CH- or-N-, Re is an (C 2 -C 3 )acyl, an aldehyde, an (C ⁇ -C 3 )alcohol, or an (C 2 -C 3 )ester, Cat+ represents one (or several, identical or different) organic or mineral cation(s) (including the proton), B is O or NH, m is an integer from 1 to 3, A is O, NH, CHF, CF 2 or CH 2 , and Y is O " Cat+, or a nucleoside.
- R 3 , f , and R 5 identical or different, are a hydrogen or (C ⁇ -C 3 )alkyl group
- W is -CH- or-N-
- Re is an (C 2
- said ⁇ T cell activator is selected from the group consisting of HDMAPP and CHDMAPP.
- said ⁇ T cell activator is HDMAPP.
- said ⁇ T cell activator is CHDMAPP.
- the (a) the IMC or IC, or the Mycobacterium antigen and (b) the ⁇ T cell activator are administered within about one week, 3 days, or more preferably 48 hours, or about 24 hours of one another.
- said the IMC or IC, or the Mycobacterium antigen and ⁇ T cell activator are administered simultaneously, or within 6 hours of one another.
- Suitable treatment regimens may specify that the ⁇ T cell activator can be administered before or after said the IMC or IC, or the Mycobacterium antigen.
- the compounds can be administered by the same routes.
- said Mycobacterium antigen and ⁇ T cell activator can be administered by different routes.
- the IMC or IC, or the Mycobacterial antigen is administered locally to a site of disease and a ⁇ T cell activator is administered by a non-local route, preferably by systemic administration.
- a Mycobacterium antigen is administered intravesicularly into the bladder.
- said Mycobacterium antigen is administered after a transurethal resection, still more preferably 1 or 2 weeks following transurethal resection.
- said bladder cancer is a stage 0 bladder cancer. More preferably, said stage 0 bladder cancer is a non-invasive papillomary carcinoma (TaTl) or a carcinoma in situ (CIS).
- FIG. 1 shows the synthesis scheme for the compound referred to herein as CHDMAPP, the complete synthesis of which is described in Example 2.
- a or “an” may mean one or more.
- the words “a” or “an” when used in conjunction with the word “comprising”, the words “a” or “an” may mean one or more than one.
- another may mean at least a second or more.
- “Weekly” stands for “about once a week” (meaning that more than one treatment is made with an interval of about one week between treatments), the about here preferably meaning +/-1 day (that is, translating into “every 6 to 8 days”); most preferably, “weekly” stands for "once every 7 days”.
- the term “about” or “approximately” usually means within 20%, more preferably within 10%, and most preferably still within 5% of a given value or range. Alternatively, especially in biological systems (e.g., when measuring an immune response), the term “about” means within about a log (i.e., an order of magnitude) preferably within a factor of two of a given value.
- references for example to treatment of a particular condition mentioned herein for example cancer, proliferative disorder, carcinoma, bladder cancer, urinary cancer, skin proliferative disorder, basal cell carcinoma, genital warts, actinic keratosis, viral infection or HPV infection, can be substituted in the above definition in the same way as the term tumor, and such will be understood according to the same above definition as exemplified for tumor.
- the expressions "stimulating the activity of ⁇ T cells”, “activating ⁇ T cells” and “regulating the activity of ⁇ T cells” designate causing or favoring an increase in the number and/or biological activity of such cells in a subject.
- Stimulating and regulating thus each include without limitation modulating (e.g., stimulating) expansion of such cells in a subject and/or, for instance, triggering of cytokine secretion (e.g., TNF ⁇ or IFN ⁇ ).
- ⁇ T cells normally represent between about 1-10% of total circulating lymphocytes in a healthy adult human subject.
- the present invention can be used to significantly increase the ⁇ T cells population in a subject, particularly to reach at least 30% of total circulating lymphocytes, typically 40%, more preferably at least 50% or 60%, or from 50%-90%.
- Regulating also includes, in addition or in the alternative, modulating the biological activity of ⁇ T cells in a subject, particularly their cytolytic activity or their cytokine-secretion activity.
- the invention defines novel conditions and strategies for increasing the biological activity of ⁇ T cells towards target cells.
- immunogenic means that an agent is capable of eliciting a humoral or cellular immune response, and preferably both.
- An immunogenic entity is also antigenic.
- An immunogenic composition is a composition that elicits a humoral or cellular immune response, or both, when administered to an animal having an immune system.
- antigen refers to any agent (e.g., protein, peptide, lipid, polysaccharide, glycoprotein, glycolipid, nucleic acid or any combination of any of the foregoing) that, when introduced into a host, animal or human, having an immune system (directly or upon expression as in, e.g., DNA vaccines), is recognized by the immune system of the host and is capable of eliciting an immune response.
- the antigen-induced immune response can be humoral or cell-mediated, or both.
- An agent is termed "antigenic” when it is capable of or comprises a component capable of specifically interacting with an antigen recognition molecule of the immune system, such as an immunoglobulin (antibody) or T cell antigen receptor (TCR).
- antigens examples include "surface antigens", i.e., expressed naturally on the surface of a pathogen, or the surface of an infected cell, or the surface of a tumor cell.
- a molecule that is antigenic need not be itself immunogenic, i.e., capable of eliciting an immune response without an adjuvant or carrier.
- An antigen may be "species-specific", referring to an antigen that is only present in or derived from a particular species.
- vector means the vehicle by which a DNA or RNA sequence (e.g., a foreign gene) can be introduced into a host cell, so as to transform the host and promote expression (e.g., transcription and/or translation) of the introduced sequence.
- Vectors include plasmids, phages, viruses, etc.
- nucleic acid molecule refers to the phosphate ester polymeric form of ribonucleosides (adenosine, guanosine, uridine, or cytidine: "RNA molecules”) or deoxyribonucleosides (deoxyadenosine, deoxyguanosine, deoxythymidine, or deoxycytidine: "DNA molecules”), or any phosphoester analogs thereof, such as phosphorothioates and thioesters, in either single stranded form, or a double-stranded helix.
- Oligonucleotides having fewer than 100 nucleotide constituent units
- polynucleotides are included within the defined term as well as double stranded DNA-DNA, DNA-RNA, and RNA- RNA helices.
- This term includes double-stranded DNA found, inter alia, in linear (e.g., restriction fragments) or circular DNA molecules, plasmids, and chromosomes.
- sequences may be described herein according to the normal convention of giving only the sequence in the 5' to 3' direction along the nontranscribed strand of DNA (i.e., the strand having a sequence homologous to the mRNA).
- a "recombinant DNA molecule” is a DNA molecule that has undergone a molecular biological manipulation.
- polypeptide refers to an amino acid-based polymer, which can be encoded by a nucleic acid or prepared synthetically. Polypeptides can be proteins, protein fragments, chimeric proteins, etc. Generally, the term “protein” refers to a polypeptide expressed endogenously in a cell. Generally, a DNA sequence encoding a particular protein or enzyme is “transcribed” into a corresponding sequence of mRNA. The mRNA sequence is, in turn, “translated” into the sequence of amino acids which form a protein. An “amino acid sequence” is any chain of two or more amino acids.
- polypeptide is usually used for amino acid-based polymers having fewer than 100 amino acid constituent units, whereas the term “polypeptide” is reserved for polymers having at least 100 such units.
- polypeptide will be the generic term.
- the terms "in combination” or “combination therapy”, used interchangeably, refer to the situation where two or more therapeutic agents affect the treatment or prevention of the same disease.
- the use of the term “in combination” does not restrict the order in which therapies (e. g. , prophylactic or therapeutic agents) are administered to a subject with the disease.
- a first therapy can be administered prior to (e. g. , 5 minutes, 15 minutes, 30 minutes, 45 minutes, 1 hour, 2 hours, 4 hours, 6 hours, 12 hours, 24 hours, 48 hours, 72 hours, 96 hours, 1 week, 2 weeks, 3 weeks, 4 weeks, 5 weeks, 6 weeks, 8 weeks, or 12 weeks before), concomitantly with, or subsequent to (e.
- the combined results are additive of the effects observed when each treatment is conducted separately. Although at least additive effects are generally desirable, any increased effect, for example an anti-cancer effect, above one of the single therapies would be of benefit. Also, there is no particular requirement for the combined treatment to exhibit synergistic effects, although this is certainly possible and advantageous.
- the present invention concerns a method for inhibiting the growth of cancer cells in the urinary bladder of a mammal having a bladder cancer comprising administering a Mycobacterium antigen and a ⁇ T cell activator.
- the present invention also concerns a method of preventing, or treating a carcinoma or viral infection, preferably a urinary or bladder cancer or an HPV infection in a mammal comprising administering a Mycobacterium antigen, and a ⁇ T cell activator.
- the present invention further concerns a pharmaceutical composition
- a pharmaceutical composition comprising a Mycobacterium antigen, and a ⁇ T cell activator and the use thereof for treatment or prevention of carcinoma or viral infection, preferably a urinary or bladder cancer or an HPV infection.
- said composition is prepared for separate administration of said Mycobacterium antigen and said ⁇ T cell activator.
- said composition further comprises a cytokine.
- said composition also comprises an additional agent active against carcinoma or viral infection, preferably a urinary or bladder cancer or an HPV infection.
- Such agent includes, but are not limited to, drugs, immunostimulants, antigens, antibodies, vaccines, radiation and chemotherapeutic, genetic, biologically engineered and chemically synthesized agents, and agents that target cell death molecules for activation or inactivation and that inhibit proliferation of and induce apoptosis in responsive cells.
- the present invention concerns a pharmaceutical composition comprising a Mycobacterium antigen, and a ⁇ T cell activator for use as a medicament. More particularly, the invention concerns the use of a Mycobacterium antigen and a ⁇ T cell activator for the manufacture of a medicament for the treatment of carcinoma or viral infection, preferably a urinary or bladder cancer or an HPV infection.
- the invention also contemplates the methods and the compositions comprising several Mycobacterium antigens and/or several ⁇ T cell activators.
- said Mycobacterium antigen and said ⁇ T cell activator are administered simultaneously to said mammal. More particularly, a pharmaceutical composition comprising said Mycobacterium antigen and said ⁇ T cell activator is administered to said mammal.
- said Mycobacterium antigen and said ⁇ T cell activator can be a ⁇ _ministered by separately and are administered by different routes of administration, for example the mycobacterial antigen is administered locally at a disease site and the ⁇ T cell activator is administered systemically, preferably by intravenous (iv) route.
- Said Mycobacterium antigen can be administered to said mammal before or after said ⁇ T cell activator.
- the methods may comprise further administering a cytokine.
- Said cytokine is capable of increasing the expansion of a ⁇ T cell population treated with a ⁇ T cell activator compound.
- a preferred cytokine is an interleukin-2 polypeptide (e.g., Research Diagnostics, NJ, #RDI-202).
- interleukin-2 polypeptide e.g., Research Diagnostics, NJ, #RDI-202
- cytokines for use in accordance with the invention and regimens for their administration are described is PCT patent publication no WO 01/56387, the disclosure of which is incorporated herein by reference.
- the present invention more particularly concerns a freeze-dried (lyophilized) pharmaceutical composition comprising a Mycobacterium antigen, and a ⁇ T cell activator.
- the pharmaceutical composition according to the present invention is administered as an aqueous suspension.
- the pharmaceutical composition according to the present invention is suspended in a pharmaceutically acceptable buffer including, but not limited to, saline and phosphate buffered saline (PBS) and is either asceptically processed or terminally sterilized.
- a pharmaceutically acceptable buffer including, but not limited to, saline and phosphate buffered saline (PBS) and is either asceptically processed or terminally sterilized.
- freeze-dried (lyophilized) pharmaceutical composition according to the present invention may be stored in sealed ampoules or vials requiring only the addition of a carrier, for example sterile water, immediately prior to use.
- the present invention also concerns a kit comprising at least one container and a pharmaceutical composition according to the present invention.
- containers are sealed ampoules or vials.
- the kit can comprise a syringe.
- the kit can comprise a container comprising both Mycobacterium antigen, and ⁇ T cell activator.
- the kit can also comprise a container comprising the Mycobacterium antigen and an other one comprising the ⁇ T cell activator.
- the pharmaceutical composition is freeze-dried (lyophilized).
- the pharmaceutical composition according to the present invention in combination with a pharmaceutically acceptable carrier, is administered to a mammal locally to a site of disease in a dosage effective to treat the carcinoma.
- a pharmaceutically acceptable carrier for example, in bladder cancer, local a ⁇ lministration refers to administration into the bladder
- Routes of administration for the ⁇ T cell activator, the IMC and the IC compounds and mycobacterium antigens include, but are not limited to, oral, dermal, subcutaneous, percutaneous, intramuscular, intraperitoneal, intravenous, intradermal, intrathecal, intralesional, intratumoral, intrabladder, intra-vaginal, intraocular, intrarectal, intrapulmonary, intraspinal, transdermal, subdermal, placement within cavities of the body, nasal inhalation, pulmonary inhalation, impression into skin and electrocorporation.
- any suitable method for administering the mycobacterial antigen can be used, depending on the disease.
- preferred methods are as follows.
- the Mycobacterium antigen is administered by instillation into the urinary bladder by, but not limited to, a urinary tract catheter.
- Other methods for instilling the pharmaceutical composition according to the present invention into the urinary bladder are known to those skilled in the art.
- the ⁇ T cell activator is provided by systemic administration, preferably by intravenous infusion or intramuscular injection.
- the bladder cancer is preferably a stage 0 bladder cancer. More preferably, the bladder cancer is a non-invasive papillomary carcinoma (TaTl) or a carcinoma in situ (CIS).
- the methods of treatment and the pharmaceutical compositions according to the present invention are well adapted for the primary treatment of CIS of the bladder (after transurethial resection) either with or without associated papillary tumors, the secondary treatment of CIS of the bladder in patients treated with other intravesical agents who have relapsed or failed to respond, and the primary or secondary treatment of CIS in patients who have contraindications to radical surgery.
- these methods and compositions are also well adapted for the adjuvant treatment following transurethal resection of stage Ta or TI papillary tumors of the bladder, which are at high risk of recurrence.
- the pharmaceutical compositions according to the present invention are administered to a subject having a bladder cancer after a step of transurethial resection.
- the treatment is administered 7-15 days after the transurethial resection.
- compositions comprising an antigen can be used in the same way as or in place of the Mycobacterium antigen in the methods described herein.
- composition suitable for such use include those comprising a killed, inactivated or attenuated pathogen, microorganism or parasite.
- a composition comprising an antigen preferably comprises an enriched or purified polypeptide, lipid, polysaccharide, glycoprotein, glycolipid or nucleic acid antigen.
- said composition comprises at least 1, 2, 3, 4, 5, 10 or 15 distinct antigens, for example at least 1, 2, 3, 4, 5, 10 or 15 distinct polypeptides, or nucleic acids encoding such polypeptides. Further examples of compositions are provided herein.
- any suitable composition comprising an antigen can be used as the IC component in any of the methods of the invention.
- the composition comprising an antigen is a ⁇ _ministered intra-tumorally for the treatment of a tumor or cancer.
- examples of composition suitable for use include those comprising a killed, inactivated or attenuated pathogen, microorganism or parasite.
- a composition comprising an antigen preferably comprises an enriched or purified polypeptide, lipid, polysaccharide, glycoprotein, glycolipid or nucleic acid antigen.
- composition comprises at least 1, 2, 3, 4, 5, 10 or 15 distinct antigens, for example at least 1, 2, 3, 4, 5, 10 or 15 distinct polypeptides, or nuclei acids encoding such polypeptides.
- the present invention concerns a method for inhibiting the growth of proliferating cells, preferably tumor or cancer cells, in a mammal, comprising (a) administering an antigen to the mammal locally to a site of disease, and (b) administering a ⁇ T cell activator to the mammal.
- step (a) comprises administering a nucleic acid encoding an antigen or administering a polypeptide antigen.
- the ⁇ T cell activator of step (b) is administered by an administration route other than intra-tumoral administration.
- the antigen is administered intra-tumorally.
- Any suitable solid tumor may be treated by this manner, including for example prostate, breast, colorectal, lung, pancreatic, renal or melanoma cancers.
- tumor antigens are particularly preferred.
- PCT patent application no. WO 97/18837 disclose methods to produce gram-negative bacteria having non-pyrogenic Lipid A or LPS.
- Preferred bacteria are capable of eliciting an immune response in an individual.
- a preferred live bacterial vaccine must be immunogenic so that it elicits an immune response; however, the vaccine must not be capable of excessive growth in vivo which might result in adverse reactions.
- some suitable bacterial vaccine vectors are temperature sensitive, having minimal replicative ability at normal physiological ranges of body temperature.
- suitable compositions are further described herein.
- a nucleic acid encoding an immunomodulatory polypeptide, or a vector comprising such nucleic acid is used as the IMC component in any of the methods of the invention.
- a cytokine polypeptide preferably a recombinant, purified or isolated polypeptide, or a fragment, variant or derivative thereof, selected form the group consisting of IFN ⁇ , IL-l ⁇ , IL-l ⁇ , IL-2, E -4, IL-5, IL-6, IL-7, IL-8, IL-9, IL-10, IL-11, IL-12, IL-13, IL-15, IL-18, IL-21, IL-23, IL-24, IL-27, _L-28a, _L-28b and IL-29.
- Other preferred examples include but are not limited to a nucleic acid encoding an antigen polypeptide, and a vector comprising a nucleic acid sequence encoding a cytokine.
- polypeptide or nucleic acid is administered intra-tumorally for the treatment of a tumor or cancer.
- the present invention concerns a method for inhibiting the growth of proliferating cells, preferably tumor or cancer cells, in a mammal, comprising (a) administering an immunogenic compound (IC) to the mammal locally to a site of disease, and (b) administering a ⁇ T cell activator to the mammal.
- the IC is administered to skin or intra-tumorally.
- step (a) comprises administering a nucleic acid encoding an IC or administering a polypeptide IC, preferably wherein the IC is a cytokine or an antigen.
- the ⁇ T cell activator of step (b) is administered by an administration route other than locally to said site of disease, or other than by intra-tumoral administration.
- a composition comprising a nucleic acid encoding an antigen is administered intra- tumorally.
- Any suitable solid tumor may be treated by this manner, including for example prostate, breast, colorectal, lung, pancreatic, renal or melanoma cancers.
- Particularly preferred are nucleic acids encoding viral or tumor antigens or cytokines, or polypeptide cytokines or viral or tumor antigens.
- the nucleic acids can be prepared in any suitable manner.
- the nucleic acids may be formulated for delivery as "naked" DNA, or are preferably inserted into a recombinant vector, for example an adenoviral vector (Ad), adeno- associated viral vector (AAV), vaccinia or poxvirus vectors.
- a recombinant vector for example an adenoviral vector (Ad), adeno- associated viral vector (AAV), vaccinia or poxvirus vectors.
- Ad adenoviral vector
- AAV adeno- associated viral vector
- vaccinia or poxvirus vectors for example, PCT publication no. WO 98/04705 describes recombinant vectors containing inserted DNA fragments coding for a polypeptide from an early region and a polypeptide from a late region of a papillomavirus for treating or preventing a papillomavirus infection or tumour.
- WO 01/18035 describes recombinant vectors encoding polypeptides derived from the MUC-1 polypeptide which are able to activate Cytotoxic T Lymphocyte (CTL) response.
- CTL Cytotoxic T Lymphocyte
- US Patent nos. 6,007,806 and 5,744,133 describe recombinant vaccinia virus vectors comprising a heterologous DNA sequence which codes at least for the essential region of a tumor specific protein.
- PCT publication no. WO 86/07610 describes recombinant poxvirus vectors comprising a nucleic acid sequence encoding a human IL-2 protein.
- WO 95/09241 describes a viral vector comprising a nucleic acid encoding for all or part of an immune and/or inflammatory response modulating polypeptide, for the treatment of cancers in mammals, including a poxvirus-derived viral vector comprising a nucleic acid coding for a cytokine such as IL-2, E - 4, IL-5, IL-6 or IL-7, gamma interferon, colony-stimulating factor or type Tjeta' tumour necrosis factor.
- cytokine such as IL-2, E - 4, IL-5, IL-6 or IL-7
- gamma interferon gamma interferon
- colony-stimulating factor or type Tjeta' tumour necrosis factor.
- suitable cytokines and antigens that can be encoded by the nucleic acids are further described herein.
- the present invention concerns a method for inhibiting the growth of proliferating cells, preferably tumor or cancer cells, in a mammal, comprising (a) administering to the mammal, intra-tumorally, a composition comprising a nucleic acid encoding a cytokine, and (b) administering to the mammal a ⁇ T cell activator by an administration route other than intra-tumoral administration.
- the nucleic acid encodes IL-2, IL-12, IL-15 or IL-21 or a fragment, variant or derivative thereof.
- the present invention concerns a method for inhibiting the growth of proliferating cells, preferably tumor or cancer cells, in a mammal, comprising administering to the mammal a TLR agonist, particularly a imidazoquinoline compound, and a ⁇ T cell activator.
- a TLR agonist particularly a imidazoquinoline compound, and a ⁇ T cell activator.
- the present invention concerns a method for treating or preventing a viral or bacterial infection, an HPV infection, cell proliferative conditions such as skin disorders and external genital warts, and actinic keratosis and skin tumors, particularly non- melanoma skin cancers such as superficial basal cell carcinoma (BCC), in a mammal, comprising administering to the mammal a TLR agonist, particularly a imidazoquinoline compound, and a ⁇ T cell activator.
- the immunomodulatory compound is administered to skin.
- the present invention further concerns a pharmaceutical composition or a kit comprising an immunomodulatory compound (EVIC), preferably a TLR agonist or imidazoquinoline compound, and a ⁇ T cell activator, and the use thereof for treatment or prevention of disease.
- said composition further comprises a cytokine.
- said composition also comprises an additional agent active against the particular disease.
- agent includes, but are not limited to, drugs, immunostimulants, antigens, antibodies, vaccines, radiation and chemotherapeutic, genetic, biologically engineered and chemically synthesized agents, and agents that target cell death molecules for activation or inactivation and that inhibit proliferation of and induce apoptosis in responsive cells.
- the present invention concerns a pharmaceutical composition
- a pharmaceutical composition comprising an immunomodulatory compound (IMC), preferably a TLR agonist or imidazoquinoline compound, and a ⁇ T cell activator for use as a medicament, preferably in separate containers.
- an immunostimulatory compound (IMC) preferably a TLR agonist or imidazoquinoline compound
- a ⁇ T cell activator for the manufacture of a medicament for the treatment or prevention of a viral or bacterial infection, an HPV infection, cell proliferative conditions such as skin disorders and external genital warts, and actinic keratosis and skin tumors, particularly non-melanoma skin cancers such as superficial basal cell carcinoma (BCC).
- BCC superficial basal cell carcinoma
- an imidazoquinoline compound imiquimod (Aldara TM) cream is administered dermally to said mammal at a site of genital warts or basal cell carcinoma.
- the ⁇ T cell activator is administered by intravenous (iv),or intramuscular route.
- the methods may comprise further administering a cytokine. Said cytokine is capable of increasing the expansion of a ⁇ T cell population treated with a ⁇ T cell activator compound.
- a preferred cytokine is an interleukin-2 polypeptide; examples of low dose cytokine regimens for use in accordance with the invention are described is PCT patent publication no WO 01/56387, the disclosure of which is incorporated herein by reference.
- Routes of administration include, but are not limited to, oral, dermal, subcutaneous, percutaneous, intramuscular, intraperitoneal, intravenous, intradermal (PCT patent publication no WO 04/020014), intrathecal, intralesional, intratumoral, intrabladder, intra-vaginal, intraocular, intrarectal, intrapulmonary, intraspinal, transdermal, subdermal, placement within cavities of the body, nasal inhalation, pulmonary inhalation, impression into skin and electroporation.
- the immunomodulatory or immunogenic composition and ⁇ T cell activating compound can therefore be advantageously used in a combination therapy.
- the immunomodulatory or immunogenic composition can be administered in a therapeutically effective amount, simultaneously in one composition, or simultaneously in different compositions, or sequentially.
- the first and second compositions must be administered separated by a time interval that still permits the first composition to be used during a treatment cycle of the second composition, or that permits the first composition to show enhanced activity, particularly therapeutic activity, when compared with the single components alone.
- the immunomodulatory or immunogenic composition is a mycobacterium
- the mycobacterium and the ⁇ T cell activating compound are administered within a week, within 5, 4, or 3 days of one another, or within 48 or 24 hours of one another, preferably within 6 hours of each other, and most preferably simultaneously.
- an immunomodulatory compound for use according to the invention is generally any suitable compound that can be administered locally to a site of diseases.
- the term "immunomodulatory compound", “immunomodulatory composition” or IMC, and variations thereof, including but not limited to immunomodulant or immunomodulatory drug refer to a compound that modulates a subject's immune system.
- an immunomodulatory compound is a compound that alters the ability of a subject's immune system to respond to one or more foreign antigens.
- an immunomodulatory compound is a compound that shifts one aspect of a subject's immune response.
- an immunomodulatory compound is a compound that inhibits or reduces a subject's immune system (i.e., an immunosuppressant (compound).
- an immunomodulatory compound is a compound that enhances or increases a subject's immune system and/or enhances or increases the ability of a subject's immune system to respond to one or more foreign antigens; such compound may also be referred to herein as an immunostimulatory compound.
- immunomodulatory compounds examples include B7 molecules (B7-1, B7-2, variants thereof, and fragments thereof) (see, e.g., Adv Exp Med Biol. 2000;465:381-90), ICOS, and OX40 (see Coyle et al., Springer Semin Immunopathol.
- a negative T cell regulator such as an antibody against CTLA4 or against another negative immune cell regulator, such as BTLA and PD-1, and cytokines and growth factors including but not limited to IFN ⁇ , IL-l ⁇ , IL-l ⁇ , IL-2, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, EL-10, IL-11, IL-12, IL-13, IL- 15, IL-18, IL-21, IL-23, IL-24, IL-27, IL-28a, IL-28b, IL-29, KGF, TGF ⁇ , M-CSF, G-CSF, TNF ⁇ , LAF, TCGF, BCGF, TRF, BAF, BDG, MP, LIF, OSM, TMF, PDGF, IFN-alpha, IFN ⁇ , IFN ⁇ s (e.g., INF ⁇ 2b), GM-CSF, CD40L, Flt
- Suitable chemokines can include Glu-Leu-Arg (ELR)-negative chemokines such as IP-10, MCP-3, MIG, and SDF-1 alpha from the human CXC and C-C chemokine families.
- Suitable cytokines also include cytokine derivatives, cytokine variants, cytokine fragments, and cytokine fusion proteins
- Immunomodulatory compounds may also include compounds that are agonists of a Toll-like receptor (TLR).
- TLR generally refers to any Toll-like receptor of any species of organism.
- TLRs are disclosed in PCT publication no. WO 98/50547. Agonists of human TLRs are also described in Table 1 of Ulevich R, (2004) Nature Reviews: Immunology, 4:512- 520; in Table 1 of Akira and Takeda (2004) Nature Reviews Immunology 4:499-511; in Medzhitov R, (2001) Nature Reviews Immunology 1:345-145; and in PCT publication nos. WO 03/031573 and WO 03/103586. Each of the preceding disclosures are incorporated herein by reference, including particularly the compounds listed in the references and Table 1 of the Ulevich (2004) and Akira and Takeda (2004) references.
- TLR agonist refers to a compound that acts as an agonist of a TLR.
- reference to a TLR agonist compound can include the compound in any pharmaceutically acceptable form, including any isomer (e.g., diastereomer or enantiomer), salt, solvate, polymorph, and the like.
- a compound is optically active
- reference to the compound can include each of the compound's enantiomers as well as racemic mixtures of the enantiomers.
- a compound may be identified as an agonist of one or more particular TLRs (e.g., a TLR7 agonist, a TLR8 agonist, or a TLR7/8 agonist).
- TLRs are known to bind certain pathogen-associated ligands. In some cases the ligands are pathogen-derived, while in other cases the ligands are subjectderived.
- TLR3 recognizes polyinosinic-polycytidylic acid (polylC), a "niimic" of double-stranded viral RNA
- TLR4 recognizes lipopolysaccharide (LPS) of many Gram-negative bacteria
- TLR5 binds certain flagellins
- TLR9 binds certain CpG oligonucleotides.
- Certain small molecule IMC compounds are known to be agonists of one or more TLRs including, for example, TLR6, TLR7, and TLR8.
- the TLR agonist may be an agonist of at least one of TLR6, TLR7, TLR8, and TLR9. In certain embodiment, the TLR agonist can be an agonist of TLR7 and/or TLR8. In alternative embodiments, the TLR agonist may be a TLR8-selective agonist. In other alternative embodiments, the TLR agonist can be a TLR7-selective agonist. As used herein, the term "TLR8-selective agonist" refers to any compound that acts as an agonist of TLR8, but does not act as an agonist of TLR7.
- TLR7-selective agonist refers to a compound that acts as an agonist of TLR7, but does not act as an agonist of TLR8.
- TLR7/8 agonist refers to a compound that acts as an agonist of both TLR7 and TLR8.
- a TLR8- selective agonist or a TLR7-selective agonist may act as an agonist for the indicated TLR and one or more of TLR1, TLR2, TLR3, TLR4, TLR5, TLR6, TLR9, or TLR10.
- TLR8-selective agonist may refer to a compound that acts as an agonist for TLR8 and for no other TLR, it may alternatively refer to a compound that acts as an agonist of TLR8 and, for example, TLR6.
- TLR7-selective agomst may refer to a compound that acts as an agonist for TLR7 and for no other TLR, but it may alternatively refer to a compound that acts as an agonist of TLR7 and, for example, TLR6.
- the TLR agonism for a particular compound may be assessed in any suitable manner. For example, assays for detecting TLR agonism of test compounds are described, for example, in PCT publication nos.
- a compound can be identified as an agonist of a particular TLR if performing the assay with a compound results in at least a threshold increase of some biological activity mediated by the particular TLR.
- a compound may be identified as not acting as an agonist of a specified TLR if, when used to perform an assay designed to detect biological activity mediated by the specified TLR, the compound fails to elicit a threshold increase in the biological activity.
- an increase in biological activity refers to an increase in the same biological activity over that observed in an appropriate control. An assay may or may not be performed in conjunction with the appropriate control.
- the precise threshold increase of TLR-mediated biological activity for determining whether a particular compound is or is not an agonist of a particular TLR in a given assay may vary according to factors known in the art including but not limited to the biological activity observed as the endpoint of the assay, the method used to measure or detect the endpoint of the assay, the signal-to-noise ratio of the assay, the precision of the assay, and whether the same assay is being used to determine the agonism of a compound for multiple TLRs.
- the TLR agonist can be a natural agonist of a TLR or a synthetic IMC compound.
- WO 04/060319 lists a number of compounds suitable for use as IMCs, described as follows.
- IMC that may be useful as TLR agonists in immunostimulatory combinations of the invention are small organic molecules (e.g., molecular weight less than about 1000 Daltons, and less than about 500 Daltons in some cases), as opposed to large biological molecules such as proteins, peptides, and the like. Certain small molecule ICM compounds are disclosed in, for example, U.S. Patent Nos.
- IMCs may also include purine derivatives (such as those described in U.S. Patent Nos. 6,376,501, and 6,028,076), certain imidazoquinoline amide derivatives (such as those described in U.S. Patent No. 6.069,149), certain benzimidazole derivatives (such as those described in U.S. Patent 6,387,938), and certain derivatives of a 4-ammopyrimidine fused to a five membered nitrogen containing heterocyclic ring (such as adenine derivatives described in U. S. Patent Nos. 6,376,501; 6,028,076 and 6,329,381; and in WO 02/085905).
- Other IMCs include large biological molecules such as oligonucleotide sequences.
- Some oligonucleotide sequences contain cytosine-guanine dinucleotides (CpG) and are described, for example, in U.S. Patent Nos. 6,194,388; 6,207,646; 6,239,116; 6,339,068; and 6,406,705.
- CpG-containing oligonucleotides can include synthetic immunomodulatory structural motifs such as those described, for example, in U.S. Pat. Nos. 6,426,334 and 6,476,000.
- Other IMC nucleotide sequences lack CpG and are described, for example, in International Patent Publication No. WO 00/75304.
- CpG nucleic acids are known to be TLR9 agonists.
- a CpG nucleic acid is a nucleic acid molecule, having at least one CpG dinucleotide motif in which at least the C of the dinucleotide is unmethylated.
- CpG nucleic acids include but are not limited to A class, B class and C class CpG nucleic acids. These classes of CpG nucleic acid have differing properties and activation profiles. Any other suitable CpG nucleic acid can be envisioned as well, generally where the nucleic acid molecule has an immunostimulatory property.
- the B class of CpG oligonucleotides are synthesized with nuclease resistant phosphorothioate backbones and are generally characterized by good B-cell and DC activation, but only limited NK cell activation.
- the A class of CpG oligonucleotides are synthesized with a chimeric backbone where the 5' and 3' ends are phosphorothioate and the central CpG motif region is phosphodiester. These oligonucleotides are characterized by good NK cell and DC activation leading to greater production of IFN-gamma but limited B-cell activation.
- the C class of CpG oligonucleotides are synthesized with a phosphorothioate backbone and have stimulatory properties intermediate to the other two classes of CpG oligonucleotides (e.g., good activation of B-cells as well as activation of NK cells and DCs).
- the methods of the invention preferably involve the use of A class, B class and C class CpG immunostimulatory nucleic acids.
- B class CpG nucleic acids see, e.g., U.S. Patent Nos. 6,194,388; 6,207,646; 6,214,806; 6,218,371; 6,239,116; and 6,339,068.
- Small molecule ICM compounds suitable for use as a TLR agonist in immunostimulatory combinations of the invention include compounds having a 2-aminopyridine fused to a five membered nitrogen-containing heterocyclic ring. Preferred example of such compounds are those which are TLR7 and/or TLR8 agonists.
- Such compounds include, for example, imidazoquinoline amines including but not limited to substituted imidazoquinoline amines such as, for example, aminoalkyl-substituted imidazoquinoline amines, amide-substituted imidazoquinoline amines, sulfonamide-substituted imidazoquinoline amines, urea-substituted imidazoquinoline amines, aryl ether-substituted imidazoquinoline amines, heterocyclic ether- substituted imidazoquinoline amines, amido ether-substituted in-Lidazoquinoline amines, sulfonamido ether-substituted irnidazoquinoline amines, urea-substituted iniidazoquinoline ethers, and thioether-substituted imidazoquinoline amines; tetrahydroimidazoquinoline amines including
- the TLR agonist and IMC is imiquimod, whose chemical name is l-(2-amino- 2-methylpropyl)-2-(ethoxymethyl)-lH-imidazo[4,5-c]quinolin-4-amine or 4-Amino- 1-isobutyl- lH-imidazo[4,5-c]quinoline.
- the TLR agonist may be an imidazonaphthyridine arnine, a tetrahydroimidazonaphthyridine amine, an oxazoloquinoline amine, a thiazoloquinoline amine, an oxazolopyridine amine, a thiazolopyridine amine, an oxazolonaphthyridine amine, or a thiazolonaphthyridine amine.
- the TLR agonist can be a sulfonamide-substituted imidazoquinoline amine.
- the TLR agonist can be a urea-substituted imidazoquinoline ether. In another alternative embodiment, the TLR agonist can be an aminoalkyl-substituted imidazoquinoline amine. In one particular embodiment, the TLR agonist is 4-amino- , ⁇ ,2-trimethyl-lH- imidazo[4,5-c]quinolin-l-ethanol. In an alternative particular embodiment, the TLR agonist is N-(2- ⁇ 2-[4-amino-2-(2-methoxyethyl)-lH-imidazo[4,5-c]quinolin-l-yl]ethoxy ⁇ ethyl)-N- methylmo holine-4-carboxamide..
- the TLR agonist is N- [4-(4-amino-2-ethyl-lH-imidazo[4,5-c]quinolin-l-yl)butyl ⁇ methanesulfonamide.
- the TLR agonist is N-[4-(4-amino-2propyl-lH-imidazo[4,5-c]quinolin -1 -yl)butyl]methanesulfonamide.
- the TLR -agonist may be a substituted imidazoquinoline amine, a tetrahydroimidazoquinoline amine, an imidazopyridine amine, a 1,2-bridged imidazoquinoline amine, a 6,7-fused cycloalkylimidazopyridine amine, an imidazonaphthyridine amine, a tetrahydroimidazonaphthyridine amine, an oxazoloquinoline amine, a thiazoloquinoline amine, an oxazolopyridine amine, a thiazolopyridine amine, an oxazolonaphthyridine amine, or a thiazolonaphthyridine amine.
- a substituted imidazoquinoline amine refers to an aminoalkylsubstituted imidazoquinoline amine, an amide-substituted imidazoquinoline amine, a sulfonamide- substituted imidazoquinoline amine, a urea-substituted imidazoquinoline amine, an aryl ether- substituted imidazoquinoline amine, a heterocyclic ether-substituted imidazoquinoline amine, an amido ether-substituted imidazoquinoline amine, a sulfonamido ether-substituted imidazoquinoline amine, a urea-substituted imidazoquinoline ether, or a thioether-substituted imidazoquinoline amines.
- substituted imidalzoquinoline amines specifically and expressly exclude l-(2methylpropyl)-IH-imidazo[4,5-c]quinolin amine and 4-amino-a,a- dirnethyl ethoxymethyl-IH-imidazo[4,5-clquinolin-l-ethanol.
- the TLR agomst can be administered in an amount from about 100 ug/kg to about 100 mg/kg.
- the TLR agonist is administered in an amount from about 10 ug/kg to about 10 mg/kg.
- the TLR agonist is administered in an amount from about 1 mg/kg to about 5 mg/kg.
- Preferred immunogenic compounds (IC) suitable for use according to the invention are antigens, particularly microbial - bacterial, viral and fungal - antigens and tumor or cancer antigens.
- Tumor and cancer antigens are particularly well suited for intra-tumoral administration.
- a “cancer antigen” or “tumor antigen” as used herein is a compound, such as a peptide, associated with a tumor or cancer cell surface and which is capable of provoking an immune response when expressed on the surface of an antigen presenting cell in the context of an MHC molecule.
- Cancer antigens can be prepared from cancer cells either by preparing crude extracts of cancer cells, for example, as described in Cohen, et al., 1994, Cancer Research, 54:1055, by partially purifying the antigens, by recombinant technology, or by de novo synthesis of known antigens.
- Cancer antigens include antigens that are recombinately an immunogenic portion of or a whole tumor or cancer. Such antigens can be isolated or prepared recombinatly or by any other means known in the art.
- Tumor antigens can include tumour rejection antigens such as those for prostate, breast, colorectal, lung, pancreatic, renal or melanoma cancers.
- exemplary antigens include MAGE 1 and MAGE 3 or other MAGE antigens (for the treatment of melanoma), PRAME, BAGE, or
- tumour-specific antigens are suitable for use with the adjuvants of the present invention and include, but are not restricted to tumour-specific gangliosides, Prostate specific antigen (PSA) or Her-2/neu, KSA (GA733), PAP, mammaglobin, MUC-1, carcinoembryonic antigen (CEA). Accordingly in one aspect of the present invention there is provided a vaccine comprising an adjuvant composition according to the invention and a tumour rejection antigen.
- PSA Prostate specific antigen
- KSA Her-2/neu
- PAP mammaglobin
- MUC-1 mammaglobin
- CEA carcinoembryonic antigen
- a "microbial antigen” as used herein is an antigen of a microorganism and includes but is not limited to infectious virus, infectious bacteria, infectious parasites and infectious fungi. Such antigens include the intact microorganism as well as . natural isolates and fragments or derivatives thereof and also synthetic compounds which are identical to or similar to natural microorganism antigens and induce an immune response specific for that microorganism. A compound is similar to a natural microorganism antigen if it induces an immune response (humoral and/or cellular) to a natural microorganism antigen. Most such antigens are used routinely in the art and are well known to those of ordinary skill in the art. Another example is a peptide mimic of a polysaccharide antigen.
- Antigens may be derived from infectious virus of both human and non-human vertebrates, include retroviruses, RNA viruses and DNA viruses.
- This group of retroviruses includes both simple retroviruses and complex retroviruses.
- the simple retroviruses include the subgroups of B-type retroviruses, C-type retroviruses and D-type retroviruses.
- An example of a B-type retrovirus is mouse mammary tumor virus (MMTV).
- the C-type retroviruses include subgroups C-type group A (including Rous sarcoma virus (RSV), avian leukemia virus (ALV), and avian myeloblastosis virus (AMV)) and C-type group B (including murine leukemia virus (MLV), feline leukemia virus (FeLV), murine sarcoma virus (MSV), gibbon ape leukemia virus (GALV), spleen necrosis virus (SNV), reticuloendotheliosis virus (RV) and simian sarcoma virus (SSV)).
- the D-type retroviruses include Mason-Pfizer monkey virus (MPMV) and simian retrovirus type 1 (SRV-1).
- the complex retroviruses include the subgroups of lentiviruses, T- cell leukemia viruses and the foamy viruses.
- Lentiviruses include HJV-l, but also include HTV- 2, SIV, Visna virus, feline immunodeficiency virus (FIV), and equine infectious anemia virus (EIAV).
- the T-cell leukemia viruses include HTLV-1, HTLV-II, simian T-cell leukemia virus (STLV), and bovine leukemia virus (BLV).
- the foamy viruses include human foamy virus (HFV), simian foamy virus (SFV) and bovine foamy virus (BFV).
- Illustrative DNA viruses that are antigens in mammals include, but are not limited to: the family Poxviridae, including the genus Orthopoxvirus (Variola major, Variola minor, Monkey pox Vaccinia, Cowpox, Buffalopox, Rabbitpox, Ectromelia), the genus Leporipoxvirus (Myxoma, Fibroma), the genus Avipoxviras (Fowlpox, other avian poxvirus), the genus Capripoxvirus (sheeppox, goatpox), the genus Suipoxviras (Swinepox), the genus Parapoxvirus (contagious postular dermatitis vims, pseudocowpox, bovine papular stomatitis viras); the family doviridae (African swine fever viras, Frog viruses 2 and 3, Lymphocystis viras of
- HPV antigens from any strain of HPV.
- HPV expresses six or seven non-structural and two structural proteins.
- Viral capsid proteins LI and L2 are the late structural proteins. LI is the major capsid protein, the amino acid sequence of which is highly conserved among different HPV types. There are seven early non-structural proteins. Proteins El, E2, and E4 play an important role in viras replication. Protein E4 also plays a role in viras maturation. The role of E5 is less well known. Proteins E6 and E7 are oncoproteins critical for viral replication, as well as for host cell immortalization and transformation.
- Fusion proteins of the invention can contain either the entire sequence of an HPV protein or a fragment thereof, e.g., a fragment of at least 8 amino acids.
- the HPV antigenic sequence is derived from a "high risk" HPV, such as HPV16 or HPV18 E7 protein.
- the HPV antigenic sequence can include an MHC-binding epitope, e.g., an MHC class I and/or an MHC class JJ binding epitope.
- antigens may be derived from bacteria, parasites or yeast.
- suitable species include Neisseria spp, including N. gonorrhea and N. meningitidis (for example, capsular polysaccharides and conjugates thereof, transferrin-binding proteins, lactoferrin binding proteins, PilC and adhesions can be used as antigens); S. pyogenes (for example M proteins or fragments thereof, C5A protease, lipoteichoic acids), S. agalactiae, S. mutans; H. ducreyi; Moraxella spp, including M.
- catarrhalis also known as Branhamella catarrhalis (for example high and low molecular weight adhesins and invasins); Bordetella spp, including B. pertussis (for example pertactin, pertussis toxin or derivatives thereof, filamenteous hemagglutinin, adenylate cyclase, fimbriae), B. parapertussis and B. bronchiseptica; Mycobacterium spp., including M. tuberculosis (for example ESAT6, Antigen 85A, -B or -Q, M. bovis, M leprae, M avium, M. paratuberculosis, M.
- E. smegmatis Legionella spp, including L. pneumophila
- Escherichia spp including enterotoxic E. coli (for example colonization factors, heat-labile toxin or derivatives thereof, heat-stable toxin or derivatives thereof), enterohemorragic E. coli, enteropathogenic E. coli (for example io shiga toxin-like toxin or derivatives thereof);
- Vibrio spp including V. cholera (for example cholera toxin or derivatives thereop; Shigella spp, including S. sonnei, S. dysenteriae, S.
- Yersinia spp including Y enterocolitica (for example a Yop protein) , Y. pestis, Y. pseudotuberculosis; Campylobacter spp, including C jejuni (for example toxins, adhesins and invasins) and C coli; Salmonella spp, including S. typhip S. paratyphi, S. choleraesuis, S. enteritidis; Listeria spp., including L. monocytogenes; Helicobacter spp, including H. pylori (for example urease, catalase, vacuolating toxin); Pseudomonas spp, including P.
- Y enterocolitica for example a Yop protein
- Campylobacter spp including C jejuni (for example toxins, adhesins and invasins) and C coli
- Salmonella spp including S. typhip S. paratyphi,
- Staphylococcus spp. including S. aureus, S. epidermidis; Enterococcus spp., including E. jaecalis, E. jaecium; Clostridium spp., including C tetani (for example tetanus toxin and derivatives thereof), C bo linum (for example botulinum toxin and derivatives thereof, C difficile (for example clostridium toxins A or B and derivatives thereof); Bacillus spp., including B.
- anthracis for example botulinum toxin and derivatives thereof
- Corynebacterium spp. including C diphtheriae (for example diphtheria toxin and derivatives thereof); Borrelia spp., including B. burgdorferi (for example OspA, OspC, DbpA, DbpB), B. garinii (for example OspA, OspC, DbpA, DbpB), B. afzelii (for example OspA, OspC, DbpA, DbpB), B. andersonii (for example OspA, OspC, DbpA, DbpB), B.
- C trachomatis for example MOMP, heparin-binding proteins
- C. pneumoniae for example MONT, heparin- binding proteins
- C psittaci Leptospira spp., including L. interrogans
- Treponema spp. including T.
- pallidum for example the rare outer membrane proteins
- T denticola for example the rare outer membrane proteins
- T hyodysenteriae or species derived from parasites
- Plasmodium spp. including P. falciparum
- Toxoplasma spp. including T. gondii (for example SAG2, SAG3, Yg34)
- Entamoeba spp. including E. histolytica
- Babesia spp. including B. microti
- Trypanosoma spp. including T cruzi
- Giardia spp. including G. lamblia
- Leshmania spp. including L. major
- Pneumocystis spp. including P.
- Trichomonas spp. including T. vaginalis
- Schisostoma spp. including S. mansoni, or species derived from yeast such as Candida spp., including C albicans
- Cryptococcus spp. including C neoformans.
- the Mycobacterial antigen for use according to the invention can be for example, and without to be limited thereto, live, killed or attenuated mycobacterium compositions, mycobacterial culture supematants, mycobacterial cell extracts, cell wall fractions or cell wall elements, preferably purified cell wall elements, DNA fractions or purified DNA molecules, or mycobacterial peptide or non-peptidic (for example non-peptidic phosphorylated antigens) antigens, in the form of fractions enriched in said antigen or purified antigen.
- attenuated or killed mycobacterial strains it is also possible to use any of a variety of non- pathogenic or non-human infecting strains of Mycobacterium, such as for example M. phlei, M. piscium or M. smegmatis in naturally existing forms.
- Preferred mycobacterial antigens are compositions capable of regulating, preferably stimulating ⁇ T cell activity.
- said Mycobacterium antigen is an antigen of any one of Mycobacterium strains, more particularly a Mycobacterium strain selected from the group consisting of Mycobacterium avium, Mycobacterium bovis, Mycobacterium phlei, Mycobacterium tuberculosis, Mycobacterium paratuberculosis, Mycobacterium heamophilum, Mycobacterium leprae, Mycobacterium chelonae, Mycobacterium fortuitum, Mycobacterium kansasii, Mycobacterium marinum, Mycobacterium scrofulaceum, Mycobacterium smegmatis, Mycobacterium ulcerans, and Mycobacterium xenopi.
- Mycobacterium avium Mycobacterium bovis
- Mycobacterium phlei Mycobacterium tuberculosis
- Mycobacterium paratuberculosis Mycobacterium heamophilum
- Mycobacterium leprae Mycobacterium chelon
- said Mycobacterium antigen is an antigen of a Mycobacterium strain selected from the group consisting of Mycobacterium bovis, Mycobacterium phlei, Mycobacterium tuberculosis, and Mycobacterium paratuberculosis.
- said Mycobacterium antigen is an antigen of Mycobacterium bovis.
- said Mycobacterium antigen is an antigen of Mycobacterium phlei, Mycobacterium vaccae or Mycobacterium piscium.
- said antigen of Mycobacterium is an attenuated strain thereof.
- Methods to prepare an attenuated strain are well-known by the man skilled in the art. For example, one method is disclosed in US 6,403,100.
- the Mycobacterium antigen can be for example any attenuated strain of Mycobacterium bovis (Bacillus Calmette-Guerin - BCG).
- Mycobacterium bovis strain can be a BCG strain of ATCC number selected from the group consisting of: 19015; 19274; 27289; 27290; 27291; 35731; 35732; 35733; 35734; 35735; 35736; 35737; 35738; 35739; 35740; 35741; 35742; 35743; 35744; 3574. More particularly, it can be derived, without to be limited thereto, from the strains of Montreal or Pasteur Institute.
- BCG can be PACIS BCG or TICE BCG.
- PACIS BCG is derived from Montreal strain which originates from a BCG culture given to Dr. Armand Frappier by Dr. C. Guerin in 1937.
- BCG can be prepared by the following process. BCG is grown on glycerinized potato medium followed by further passages on Sauton medium. After harvesting by filtration, BCG are resuspended in a 15% (w/v) lactose solution, filled into container, and preferably lyophilized.
- mycobacterial antigen compositions that can use in accordance with the invention may comprise a major mycobacterial antigen or a recombinant mycobacterial strains and DNA forms.
- major antigens that are targets of the immune response to infection by Mycobacteria have been reported in Kaufman, Immunol. Today 11: 129-136 (1990); Young, Ann. Rev. Immunol. 8: 401-420 (1990); Young et al, Academic Press Ltd., London, pp. 1-35, 1990; Young et al, Mol. Microbiol. 6: 133-145 (1992)].
- Recombinant BCG vaccine vehicles have been proposed (Snapper et al, PNAS. USA.
- the pharmaceutical composition comprising the Mycobacterium antigen, and optionally the ⁇ T cell activator, can be administered locally at the disease site (the bladder) via intravesical treatment as following.
- the subject should not drink fluids for 4 hours before treatment and should empty its bladder prior administration of the pharmaceutical composition.
- the pharmaceutical composition is instilled into the bladder slowly (e.g., by gravity flow) via a catheter.
- the pharmaceutical composition is retained in the bladder for about two hours and then voided.
- the subject should lie for 15 minutes each in the prone and suspine positions and also on each side, to maximize surface exposure to the pharmaceutical composition.
- the treatment cycle consists of one intravesicular instillation per week for six weeks. Thereafter, the treatment can be continued at monthly intervals for 6-12 months.
- the treated subjects can be evaluated, for example at 3, 6, and/or 9 months, after the treatment.
- the evaluation can be performed by cytoscopy, cytology and/or biopsy. Patients may also continue to be treated as maintenance therapy.
- a preferred composition comprising a mycobacterial antigen for use in accordance with the invention is Lnmucyst® (Bacillus Calmette-Gueiin (BCG), substrain Connaught) available from Aventis Pasteur.
- Lnmucyst® Bacillus Calmette-Gueiin (BCG), substrain Connaught
- BCG Bacillus Calmette-Gueiin
- the product is made from a culture of an attenuated strain of living bovine tubercle bacillus Mycobacterium bovis. The bacilli are lyophilized (freeze-dried) and are viable upon reconstimtion.
- each CFU When plated on culture media, the progenitor of each colony is termed a "colony-forming unit" (CFU); each CFU is composed of at least one viable bacillus and may comprise several bacilli, some of which may be viable and some non-viable.
- Each vial contains 81 mg (dry weight) of BCG and 5% w/v monosodium glutamate.
- Each vial of ImmuCyst® is reconstituted with the accompanying diluent (3.0 mL), which consists of approximately 0.85% w/v sodium chloride, 0.025% w/v Tween 80, 0.06% w/v sodium dihydrogen phosphate and 0.25% w/v disodium hydrogen phosphate.
- the product and the diluent contain no preservative.
- One dose consists of one 81 mg vial of reconstituted material further diluted in 50 mL sterile, preservative-free saline.
- the reconstituted dose contains 10.5 ⁇ 8.7 x 10 8 colony forming units (CFU) over the course of its shelf-life.
- BCG When administered intravesically as a cancer therapy, BCG promotes a local acute inflammatory and sub-acute granulomatous reaction with histiocytic and leukocytic infiltration in the urothelium and lamina intestinal of the urinary bladder.
- the local inflammatory effects are associated with an elimination or reduction of superficial cancerous lesions of the urinary bladder.
- ImmuCyst® is commercialised for treatment of superficial transitional cell carcinoma (TCC) of the urinary bladder, including carcinoma in situ (CIS), papillary tumors limited to the mucosa (stage Ta), papillary tumors involving the lamina intestinal but not the muscle layer of the bladder (stage TI), or any combination thereof.
- TCC superficial transitional cell carcinoma
- CIS carcinoma in situ
- stage Ta papillary tumors limited to the mucosa
- stage TI papillary tumors involving the lamina intestinal but not the muscle layer of the bladder
- ImmuCyst® is indicated for the treatment and prophylaxis of primary or recurrent carcinoma in-situ (CIS) of the urinary bladder, and for prophylaxis following TUR of primary or recurrent stage Ta and/or TI papillary tumors.
- ImmuCyst® is preferably dosed and administered according to the manufacturer's instructions as follows. Intravesical treatment of the urinary bladder using ImmuCyst® is recommended to begin between 7 to 14 days after biopsy or transurethral resection.
- the induction treatment comprises 6 weekly intravesical treatments with ImmuCyst®, each treatment dose comprising one 81 mg vial of ImmuCyst®. After a 6-week pause, another dose should be given intravesically once weekly for 1-3 weeks. Three weekly doses should definitely be given to patients who still have evidence of bladder cancer. Clinical studies have demonstrated that the 3 doses given at 3 months significantly increased the complete response rate from 73% to 87% at 6 months. Maintenance therapy following induction is recommended. This consists of 1-3 weekly treatments at 6 months following the initiation of treatment, and then every 6 months thereafter until 36 months.
- Each dose (1 reconstituted vial) is further diluted in an additional 50 mL of sterile, preservative- free saline for a total of 53 mL (see reconstitution instructions below).
- a urethral catheter is inserted into the bladder under aseptic conditions, the bladder is drained, and then 53 mL suspension of ImmuCyst® is instilled slowly by gravity, following which the catheter is withdrawn.
- the patient retains the suspension for as long as possible for a total of up to two hours. During the first 15 minutes following instillation, the patient should lie prone. Thereafter, the patient is then allowed to be up. At the end of 2 hours, all patients should void in a seated position for environmental safety reasons. Patients should be instructed to maintain adequate hydration.
- TICETM BCG (Organon Teknika Co ⁇ .).
- TICETM is commercialised for the treatment of carcinoma in situ of the bladder.
- TICETM can be administered according to the manufacturer's instructions as follows, recommending that the product is administered 7-14 days after bladder biopsy.
- the reconstituted TICETM is installed into the bladder by gravity flow using a catheter, is maintained in the bladder for two hours and then voided.
- the patient While the BCG is in the bladder, the patient should be repositioned from the left side to the right side and the back side to the abdomen every 15 minutes in order to maximize surface exposure to the agent.
- a standard treatment of TICE consists of one intravesicular instillation per week for six weeks. The schedule may be repeated once if tumor remission has not been achieved. Thereafter, TICETM administration can be continued at monthly intervals for 6-12 months.
- PacisTM Another preferred mycobacterial antigen-containing composition made from a culture of an attenuated strain of living BCG is PacisTM (Shire Biologies, Sainte-Foy, Quebec, Canada and Urocor, Inc., Oklahoma USA). PacisTM is supplied as a single dose ampule of 120mg (semi-dry weight) lyophilised BCG (2.4 to 12x10°" C.F.U. per ampule). PacisTM is commercialised for the treatment of bladder cancer and treatment. PacisTM can be administered in accordance with the manufacturer's instructions at a single dose of 120mg according to the same methods as TICETM once weekly for six-weeks, which cycle may be repeated if tumor remission has not been achieved.
- BCG-Medac Another preferred mycobacterial antigen-containing composition is BCG-Medac, made from a culture of an attenuated strain of living BCG, strain RIVM derived from strain 1173-P2 (medac, Hamburg, Germany).
- BCG-medac is commercialised for the treatment of bladder cancer and treatment can be carried out in accordance with the manufacturer's instructions as follows.
- the reconstituted dose of BCG-medac contains 2x10 8 - 3 x 10° colony forming units (CFU) of attenuated BCG and is installed into the bladder 2 to 3 weeks after transurethral resection using a single use catheter under slight pressure.
- a standard treatment of BCG-medac consists of one intravesicular instillation per week for six weeks.
- the schedule may be repeated if tumor remission has not been achieved.
- BCG-medac administration can be continued at monthly intervals for a 12 month period.
- BCG-medac can be given for six weeks followed by weekly injections for three consecutive weeks in months 3, 6, 12, 18, 24, 30 and 36.
- An efficient dose of an attenuated BCG strain according the present invention preferably comprises about 0.1 to 50 x 10 8 colony forming units, more preferably 1 to 15 10 8 colony forming units.
- mycobacterial antigen-containing composition is the product referred to as SRL172 (SR Pharma, London, U.K.), a killed Mycobacterium vaccae suspension also described in PCT application no. WO85/03639, the disclosure of which is inco ⁇ orated herein by reference.
- SRL172 SR Pharma, London, U.K.
- MCWE non-viable mycobacterial cell wall extract
- MCWE is composed primarily of peptidoglycan and glycolipid (Chin et al. Journal of Urology 156:1189-1193, 1996) and contain N-acetylmuramyl-L-alanyl-D-isoglutamine (muramyl dipeptide) and mycolic acid derivatives. Both muramyl dipeptide and mycolic acid derivatives stimulate the immune system by activation of macrophage and monocyte mediated reactions (Mallick et al. Comparative Immunology and Microbiology of Infectious Diseases 8:55-63, 1985; Teware et al. Veterinary Parasitology 62:223-230, 1996).
- Mycobacterium cell wall composition preferably deproteinized and delipidated and optionally complexed to DNA.
- M-DNA Mycobacterium phlei deoxyribonucleic acid
- MCC Mycobacterium phlei cell wall complex
- MY-1 DNA-rich fraction extracted and purified from mycobacterium bovis BCG referred to as MY-1.
- MY-1 is described in Fujeida et al, (1999) Am. J. Respir. Grit. Care med. 160: 2056-2061, and the preparation of MY-1 is described in Tokunaga et al, (1984) J. Natl. Cancer Inst. 72:955-962, both of which disclosures are inco ⁇ orated herein by reference.
- ⁇ T cell activator designates a molecule, preferably artificially produced, which can activate ⁇ T lymphocytes. It is more preferably a ligand of the T receptor of ⁇ T lymphocytes.
- the activator may by of various nature, such as a peptide, lipid, small molecule, etc. It may be a purified or otherwise artificially produced (e.g., by chemical synthesis, or by microbiological process) endogenous ligand, or a fragment or derivative thereof, or an antibody having substantially the same antigenic specificity.
- the ⁇ T cell activator preferably increases the biological activity or causes the proliferation of ⁇ T cells, preferably increasing the activation of ⁇ T cells, particularly increasing cytokine secretion from ⁇ T cells or increasing the cytolytic activity of ⁇ T cells, with or without also stimulating the proliferation or expansion of ⁇ T cells.
- the ⁇ T cell activator is administered in an amount and under conditions sufficient to increase the activity ⁇ T cells in a subject, preferably in an amount and under conditions sufficient to increase cytokine secretion by ⁇ T cells and or to increase the cytolytic activity of ⁇ T cells. Cytokine secretion and cytolytic activity as well as ⁇ T cell proliferation can be assessed using any appropriate in vitro assay.
- the ⁇ T cells referred to in the present specification are V ⁇ 9N ⁇ 2 T cells, and preferably the ⁇ T cell activator is a N ⁇ 9N ⁇ 2 T cell activator.
- ⁇ T cell activation can be assessed by administering a compound to an individual (human or non-human primate) and assessing activation or proliferation of V ⁇ 9 V ⁇ 2 T cell.
- a candidate ⁇ T lymphocyte activator is administered to a non-human primate such as a cynomolgus monkey by intravenous infusion (one administration by slow infusion, 50 ml over 30 minutes) in combination with IL-2 (0.9 million units twice daily by subcutaneous injection for 5 days); peripheral ⁇ lymphocytes are analysed by flow cytometry on total monkey blood, after double staining with anti-CD3-PE antibody and anti-Vgamma9-FrrC antibodies and/or anti Vd2 antibodies, and cells are counted by flow cytometry. Peak expansion of the V ⁇ 9V ⁇ 2 T cell population is observed between days 3 and 8, generally at about days 4-6 after administration of the ⁇ T lymphocyte activator.
- any other suitable tests can be used to assess cell proliferation.
- Assessment of proliferation or peripheral ⁇ lymphocytes can generally be analyzed by flow cytometry on total blood (for example total blood obtained from a monkey), after double staining with anti-CD3-PE antibody and anti-Vgamma9-FITC antibodies and/or anti Vd2 antibodies (CD3-PE : SP34 clone, BD Biosciences Pharmingen, Le Pont de Claix, France).
- Anti Vgamma 9, clone 7B6 is a monoclonal raised to human Vgamma 9 but that cross-reacts with cynomolgus monkey cells. It is purified by affinity chromatography on protein A and coupled to FITC.
- 50 ⁇ l monkey blood is incubated 15 min at RT with 5 ⁇ l anti-CD3-PE and 6 ⁇ l anti-delta2-FITC or lO ⁇ l anti- gamma9-FITC antibodies.
- Antibodies are washed with 3ml IX PBS, centrifuged for 4 min at 1300 ⁇ m at RT and supernatant is discarded. Red cells are lysed with the OptiLyse C reagent (Immunotech-Beckman-Coulter, Marseilles, France) according to the manufacturer's instructions.
- stained white blood cells are recovered by centrifugation and resuspended in 300 ⁇ l PBS + 0.2% PFA.
- 50 ⁇ l calibrated Flow CountTM Fluorospheres (Immunotech-Beckman-Coulter, Marseilles, France) are added to the cells for absolute number counting of the populations of interest.
- a ⁇ T lymphocyte activator is a compound capable of regulating the activity of a ⁇ T cell in a population of ⁇ T cell clones in culture.
- the ⁇ T lymphocyte activator is more preferably capable of regulating the activity of a ⁇ T cell population of ⁇ T cell clones in a at millimolar concentration, preferably when the ⁇ T cell activator is present in culture at a concentration of less than 100 mM.
- cytokine production or release is assessed.
- Vg9Vd2 cells are known producers of TNF ⁇ and IFN ⁇ in vitro upon administration of the ⁇ T cell activator. Shortly after ⁇ . T cell activator treatment, samples of sera are collected from an individual and are assayed by ELISA specific for TNF ⁇ or IFN ⁇ .
- T cell can be assessed by any suitable means, preferably by assessing cytokine secretion, most preferably TNF- ⁇ secretion as described herein. Methods for obtaining a population of pure ⁇ . T cell clones is described in Davodeau et al, ((1993) J. Immunology 151(3): 1214-1223) and Moreau et al, ((1986) J. Clin. Invest. 78:874), the disclosures of which are inco ⁇ orated herein by reference.
- cytokine secretion can be determined according to the methods described in Espinosa et al. (J. Biol. Chem., 2001, Vol. 276, Issue 21, 18337-18344), describing measurement of TNF- ⁇ release in a bioassay using TNF- -sensitive cells. Briefly, 10 4 YfiT cells/well were incubated with stimulus plus 25 units of IL2/well in 100 ⁇ l of culture medium during 24 h at 37 °C.
- a preferred assay for cytolytic activity is a 51 Cr release assay.
- the cytolytic activity of ⁇ T cells is measured against autologous normal and tumor target cell lines, or control sensitive target cell lines such as Daudi and control resistant target cell line such as Raji in 4h 51 Cr release assay.
- target cells were used in amounts of 2xl0 3 cells/well and labeled with lOO ⁇ Ci 51 Cr for 60 minutes. Effector/Target ( E/T) ratio ranged from 30: 1 to 3.75: 1.
- Specific lysis (expressed as percentage) is calculated using the standard formula [(experimental-spontaneous release / total-spontaneous release) xl00].
- the methods of the invention can generally be carried out with any ⁇ T cell activator that is capable of stimulating ⁇ T cell activity.
- This stimulation can be by direct effect on ⁇ T cells as discussed below using compounds that can stimulate ⁇ T cells in a pure ⁇ T cell culture, or the stimulation can be by an indirect mechanism, such as treatment with pharmacological agents such as statins which prevent biosynthesis of the ⁇ T cell-stimulating compound isopentenyl pyrophosphate (IPP) or aminobisphosphonates which lead to IPP accumulation (such as, see below).
- a ⁇ T cell activator is a compound capable of regulating the activity of a ⁇ T cell in a population of ⁇ T cell clones in culture.
- the ⁇ T cell activator is capable of regulating the activity of a ⁇ T cell population of ⁇ T cell clones at millimolar concentration, preferably when the ⁇ T cell activator is present in culture at a concentration of less than 100 mM.
- a ⁇ T cell activator is capable of regulating the activity of a ⁇ T cell in a population of ⁇ T cell clones at millimolar concentration, preferably when the ⁇ T cell activator is present in culture at a concentration of less than 10 mM, or more preferably less than 1 mM.
- Regulating the activity of a ⁇ T cell can be assessed by any suitable means, preferably by assessing cytokine secretion, most preferably TNF- ⁇ secretion as described herein.
- the activator is capable of causing at least a 20%, 50% or greater increase in the number of ⁇ T cells in culture, or more preferably at least a 2-fold increase in the number of ⁇ T cells in culture.
- the activator may be a synthetic chemical compound capable of selectively activating V ⁇ 9V ⁇ 2 T lymphocytes.
- Selective activation of V ⁇ 9V ⁇ 2 T lymphocytes indicates that the compound has a selective action towards specific cell populations, preferably increasing activation of V ⁇ 9V ⁇ 2 T cells at a greater rate or to a greater degree than other T cell types such as V ⁇ l T cells, or not substantially not activation other T cell types.
- selectivity can be assessed in vitro T cell activation assays.
- selectivity suggests that preferred compounds can cause a selective or targeted activation of the proliferation or biological activity of V ⁇ 9V ⁇ 2 T lymphocytes.
- said ⁇ T cell activator is a compound of the formula I, especially a ⁇ T cell activator according to formulas I to XVII, especially ⁇ T cell activator selected from the group consisting of BrHPP, CBrHPP, HDMAPP HDMAPP and epoxPP.
- ⁇ T cell activator selected from the group consisting of BrHPP, CBrHPP, HDMAPP HDMAPP and epoxPP.
- aminobiphosphonate compounds such as pamidronate (Novartis, Nuernberg, Germany) and zoledronate may be used.
- ⁇ T cell activators for use in the present invention are phosphoantigens disclosed in WO95/20673, isopentenyl pyrophosphate (IPP) (US5,639,653), as well as alkylamines (such as ethylamine, iso-propyulamine, n-propylamine, n-butylamine and iso-butylamine, for instance).
- IPP isopentenyl pyrophosphate
- alkylamines such as ethylamine, iso-propyulamine, n-propylamine, n-butylamine and iso-butylamine, for instance.
- Isobutyl amine and 3 -aminopropyl phosphonic acid are obtained from Aldrich (Chicago, IL).
- Examples of preferred ⁇ T cell activators according to the present invention comprise the compounds of formula (I) :
- n is an integer from 1 to 3;
- B is O, NH, or any group capable to be hydrolyzed
- Y O " Cat+, a C]-C 3 alkyl group, a group -A-R, or a radical selected from the group consisting of a nucleoside, an oligonucleotide, a nucleic acid, an amino acid, a peptide, a protein, a monosaccharide, an oligosaccharide, a polysaccharide, a fatty acid, a simple lipid, a complex lipid, a folic acid, a tetrahydrofolic acid, a phosphoric acid, an inositol, a vitamin, a co-enzyme, a flavonoid, an aldehyde, an epoxyde and a halohydrin;
- A is O, NH, CHF, CF 2 or CH 2 ; and, R is a linear, branched, or cyclic, aromatic or not, saturated or unsaturated, C ⁇ -C 50 hydrocarbon group, optionally interrupted by at least one heteroatom, wherein said hydrocarbon group comprises an alkyl, an alkylenyl, or an alkynyl, preferably an alkyl or an alkylene, which can be substituted by one or several substituents selected from the group consisting of : an alkyl, an alkylenyl, an alkynyl, an epoxyalkyl, an aryl, an heterocycle, an alkoxy, an acyl, an alcohol, a carboxylic group (-COOH), an ester, an amine, an amino group (-NH 2 ), an amide (-CONH 2 ), an imine, a nitrile, an hydroxyl (-OH), a aldehyde group (-CHO), an halogen, an halogenoalky
- the substituents as defined above are substituted by at least one of the substituents as specified above.
- the substituents are selected from the group consisting of : an (C ⁇ -C 6 )alkyl, an (C 2 - C 6 )alkylenyl, an (C 2 -C 6 )alkynyl, an (C 2 -C 6 )e ⁇ oxyalkyl, an aryl, an heterocycle, an (C C 6 )alkoxy, an (C 2 -C 6 )acyl, an (C ⁇ -C 6 )alcohol, a carboxylic group (-COOH), an (C 2 -C 6 )ester, an (C ⁇ -C 6 )amine, an amino group (-NH 2 ), an amide (-CONH 2 ), an (C ⁇ -C 6 )imine, a nitrile, an hydroxyl (-OH), a aldehyde group (-CHO), an halogen, an (C ⁇ -C 6 )halogenoalkyl, a thiol (-SH), a (C
- the substituents are selected from the group consisting of : an (C ⁇ -C 6 )alkyl, an (C 2 -C 6 )epoxyalkyl, an (C 2 -C 6 )alkylenyl, an (C ⁇ -C 6 )alkoxy, an (C 2 -Ce)acyl, an (C ⁇ -C ⁇ )alcohol, an (C 2 -Cg)ester, an (C ⁇ -C 6 )amine, an (C ⁇ -C 6 )imine, an hydroxyl, a aldehyde group, an halogen, an (C ⁇ -C 6 )halogenoalkyl, and a combination thereof.
- the substituents are selected from the group consisting of : an (C 3 - C 6 )epoxyalkyl, an (C ⁇ -C 3 )alkoxy, an (C 2 -C 3 )acyl, an (C ⁇ -C 3 )alcohol, an (C 2 -C 3 )ester, an (C C 3 )amine, an (C ⁇ -C 3 )imine, an hydroxyl, an halogen, an ( -C ⁇ halogenoalkyl, and a combination thereof, and a combination thereof.
- R is a (C 3 -C 25 )hydrocarbon group, more preferably a (C 5 -C ⁇ 0 )hydrocarbon group.
- alkyl more specifically means a group such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tert-butyl, pentyl, hexyl, heptyl, octyl, nonyl, decyl, undecyl, dodecyl, tridecyl, tetradecyl, pentadecyl, hexadecyl, heptadecyl, octadecyl, nonadecyl, eicosyl, heneicosyl, docosyl and the other isomeric forms thereof.
- (C ⁇ -C 6 )alkyl more specifically means methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tert-butyl, pentyl, hexyl and the other isomeric forms thereof.
- (C ⁇ -C.)alkyl more specifically means methyl, ethyl, propyl, or isopropyl.
- alkenyl refers to an alkyl group defined hereinabove having at least one unsaturated ethylene bond and the term “alkynyl” refers to an alkyl group defined hereinabove having at least one unsaturated acetylene bond.
- (C 2 -C 6 )alkylene includes a ethenyl, a propenyl (1- propenyl or 2-propenyl), a 1- or 2- methylpropenyl, a butenyl (1 -butenyl, 2-butenyl, or 3- butenyl), a methylbutenyl, a 2-ethylpropenyl, a pentenyl (1-pentenyl, 2-pentenyl, 3-pentenyl, 4- pentenyl), an hexenyl (1-hexenyl, 2-hexenyl, 3-hexenyl, 4-hexenyl, 5-hexenyl), and the other isomeric forms thereof.
- (C 2 -C 6 )alkynyl includes ethynyl, 1-propynyl, 2-propynyl, 1-butynyl, 2- butynyl, 3-butynyl, 1-pentynyl, 2-pentynyl, 3-pentynyl, 4-pentynyl, 1-hexynyl, 2-hexynyl, 3- hexynyl, 4-hexynyl, or 5-hexynyl and the other isomeric forms thereof.
- epoxyalkyl refers to an alkyl group defined hereinabove having an epoxide group. More particularly, (C -C 6 )epoxyalkyl includes epoxyethyl, epoxypropyl, epoxybutyl, epoxypentyl, epoxyhexyl and the other isomeric forms thereof. (C 2 -C 3 )epoxyalkyl includes epoxyethyl and epoxypropyl.
- aryl groups are mono-, bi- or tri-cyclic aromatic hydrocarbons having from 6 to 18 carbon atoms. Examples include a phenyl, ⁇ -naphthyl, ⁇ -naphthyl or anthracenyl group, in particular.
- Heterocycle groups are groups containing 5 to 18 rings comprising one or more heteroatoms, preferably 1 to 5 endocyclic heteroatoms. They may be mono-, bi- or tri-cyclic. They may be aromatic or not. Preferably, and more specifically for R 5 , they are aromatic heterocycles. Examples of aromatic heterocycles include pyridine, pyridazine, pyrimidine, pyrazine, furan, thiophene, pyrrole, oxazole, thiazole, isothiazole, imidazole, pyrazole, oxadiazole, triazole, thiadiazole and triazine groups.
- bicycles include in particular quinoline, isoquinoline and quinazoline groups (for two 6-membered rings) and indole, benzimidazole, benzoxazole, benzothiazole and indazole (for a 6-membered ring and a 5-membered ring).
- Nonaromatic heterocycles comprise in particular piperazine, piperidine, etc.
- Alkoxy groups correspond to the alkyl groups defined hereinabove bonded to the molecule by an -O- (ether) bond.
- (C ⁇ -Ce)alkoxy includes methoxy, ethoxy, propyloxy, butyloxy, pentyloxy, hexyloxy and the other isomeric forms thereof.
- (C ⁇ -C 3 )alkoxy includes methoxy, ethoxy, propyloxy, and isopropyloxy.
- Alcyl groups correspond to the alkyl groups defined hereinabove bonded to the molecule by an -CO- (carbonyl) group.
- (C 2 -C 6 )acyl includes acetyl, propylacyl, butylacyl, pentylacyl, hexylacyl and the other isomeric forms thereof.
- (C 2 -C 3 )acyl includes acetyl, propylacyl and isopropylacyl.
- Alcohol groups correspond to the alkyl groups defined hereinabove containing at least one hydroxyl group.
- Alcohol can be primary, secondary or tertiary.
- C 1 -C ⁇ )alcohol includes methanol, ethanol, propanol, butanol, pentanol, hexanol and the other isomeric forms thereof.
- C ⁇ -C 3 )alcohol includes methanol, ethanol, propanol and isopropanol.
- Ester groups correspond to the alkyl groups defined hereinabove bonded to the molecule by an -COO- (ester) bond.
- C 2 -Ce)ester includes methylester, ethylester, propylester, butylester, pentylester and the other isomeric forms thereof.
- C 2 -C 3 )ester includes methylester and ethylester.
- “Amine” groups correspond to the alkyl groups defined hereinabove bonded to the molecule by an -N- (amine) bond.
- (Ci-C 6 )amine includes methylamine, ethylamine, propylamine, butylamine, pentylamine, hexylamine and the other isomeric forms thereof.
- (C ⁇ -C 3 )amine includes methylamine, ethylamine, and propylamine.
- (Ci- C 6 )imine includes methylimine, ethylimine, propylimine, butylimine, pentylimine, hexylimine and the other isomeric forms thereof.
- (C 1 -C 3 )imine includes methylimine, ethylimine, and propylimine.
- the halogen can be CI, Br, I, or F, more preferably Br or F.
- Halogenoalkyl groups correspond to the alkyl groups defined hereinabove having at least one halogen.
- the groups can be monohalogenated or polyhalogenated containing the same or different halogen atoms.
- the group can be an trifluoroalkyl (CF 3 -R).
- ( - C 6 )halogenoalkyl includes halogenomethyl, halogenoethyl, halogenopropyl, halogenobutyl, halogenopentyl, halogenohexyl and the other isomeric forms thereof.
- (CrC 3 )halogenoalkyl includes halogenomethyl, halogenoethyl, and halogenopropyl.
- Thioalkyl groups correspond to the alkyl groups defined hereinabove bonded to the molecule by an -S- (thioether) bond.
- C ⁇ -C 6 )thioalkyl includes thiomethyl, thioethyl, thiopropyl, thiobutyl, thiopentyl, thiohexyl and the other isomeric forms thereof.
- C ⁇ -C 3 )thioalkyl includes thiomethyl, thioethyl, and thiopropyl.
- Sulfone correspond to the alkyl groups defined hereinabove bonded to the molecule by an -SOO- (sulfone) bond.
- (C ⁇ -C 6 )sulfone includes methylsulfone, ethylsulfone, propylsulfone, butylsulfone, pentylsulfone, hexylsulfone and the other isomeric forms thereof.
- (C ⁇ -C 3 )sulfone includes methylsulfone, ethylsulfone and propylsulfone.
- “Sulfoxyde” groups correspond to the alkyl groups defined hereinabove bonded to the molecule by an -SO- (sulfoxide) group.
- (C ⁇ -C f i)sulfoxide includes methylsulfoxide, ethylsulfoxide, propylsulfoxide, butylsulfoxide, pentylsulfoxide, hexylsulfoxide and the other isomeric forms thereof.
- (CrC 3 )sulfoxide includes methylsulfoxide, ethylsulfoxide, propylsulfoxide and isopropylsulfoxide.
- Heteroatom denotes N, S, or O.
- Nucleoside includes adenosine, thymine, uridine, cytidine and guanosine.
- the hydrocarbon group is a cycloalkylenyl such as a cyclopentadiene or a phenyl, or an heterocycle such as a furan, a pyrrole, a thiophene, a thiazole, an imidazole, a triazole, a pyridine, a pyrimidine, a pyrane, or a pyrazine.
- the cycloalkylenyl or the heterocycle is selected from the group consisting of a cyclopentadiene, a pyrrole or an imidazole.
- the cycloalkylenyl or the heterocycle is sustituted by an alcohol.
- said alcohol is a (C ⁇ -C 3 )alcohol.
- the hydrocarbon group is an alkylenyl with one or several double bonds.
- the alkylenyl group has one double bond.
- the alkylenyl group is a (C 3 -C ⁇ o)alkylenyl group, more preferably a (C -C 7 )alkylenyl group.
- said alkylenyl group is substituted by at least one functional group. More preferably, the functional group is selected from the group consisting of an hydroxy, an (C ⁇ -C 3 )alkoxy, an aldehyde, an (C 2 - C 3 )acyl, or an (C 2 -C 3 )ester.
- the hydrocarbon group is butenyl substituted by a group -CH 2 OH.
- said alkenyl group can be the isoform trans (E) or cis (Z), more preferably a trans isoform (E).
- the alkylenyl group is the (E)-4-hydroxy-3-methyl-2-butenyl.
- the alkylenyl group group is an isopentenyl, an dimethylallyl or an hydroxydimethylallyl.
- the hydrocarbon group is an alkyl group substituted by an acyl. More preferably, the hydrocarbon group is an (C 4 -C 7 )alkyl group substituted by an (C ⁇ -C 3 )acyl.
- R is selected from the group consisting of :
- Ri wherein n is an integer from 2 to 20, Ri is a (C ⁇ -C 3 )alkyl group, and R 2 is an halogenated (d- C 3 )alkyl, a (C ⁇ -C 3 )alkoxy-(C ⁇ -C 3 )alkyl, an halogenated (C 2 -C 3 )acyl or a (C ⁇ -C 3 )alkoxy-(C 2 - C 3 )acyl.
- Ri is a methyl or ethyl group
- R 2 is an halogenated methyl (-CH 2 -X, X being an halogen), an halogenated (C 2 -C 3 )acetyl, or (C ⁇ -C 3 )alkoxy- acetyl.
- the halogenated methyl or acetyl can be mono-, di-, or tri-halogenated.
- n is an integer from 2 to 10, or from 2 to 5. In a more preferred embodiment, n is 2.
- n is 2, Ri is a methyl and R 2 is an halogenated methyl, more preferably a monohalogenated methyl, still more preferably a bromide methyl.
- n is 2, Rj is a methyl, R2 is a methyl bromide.
- R is 3-(bromomethyl)-3- butanol-1-yl.
- n is an integer from 2 to 20, and Ri is a methyl or ethyl group.
- Ri is a methyl or ethyl group.
- n is an integer from 2 to 10, or from 2 to 5.
- RI is a methyl.
- R 3 , R 4 , and R 5 are a hydrogen or (C ⁇ -C 3 )alkyl group
- W is -CH- or -N-
- Re is an (C 2 -C 3 )acyl, an aldehyde, an (C ⁇ -C 3 )alcohol, or an (C 2 -C 3 )ester.
- R 3 and R 5 are a methyl and R t is a hydrogen.
- Re is -CH 2 -OH, - CHO, -CO-CH . or -CO-OCH 3 .
- the double-bond between W and C is in conformation trans (E) or cis (Z). More preferably, the double-bond between W and C is in conformation trans (E).
- Y can allow to design a prodrag. Therefore, Y is enzymolabile group which can be cleaved in particular regions of the subject.
- the group Y can also be targeting group.
- Y is O " Cat+, a group -A-R, or a radical selected from the group consisting of a nucleoside, a monosaccharide, an epoxyde and a halohydrin.
- Y is an enzymolabile group.
- Y is O " Cat+, a group -A-R, or a nucleoside.
- Y is O " Cat+.
- Y is a nucleoside.
- Cat + is H 1" , Na + , NH , K + , Li + , (CH ⁇ H ⁇ NH 1" .
- A is O, CHF, CF 2 or CH 2 . More preferably, A is O or CH .
- B is O or NH. More preferably, B is O.
- n is 1 or 2. More preferably, m is 1.
- synthetic ⁇ T cell activators comprise the compounds of formula (D):
- X is an halogen (preferably selected from I, Br and CI)
- B is O or NH
- m is an integer from 1 to 3
- RI is a methyl or ethyl group
- Cat+ represents one (or several, identical or different) organic or mineral cation(s) (including the proton)
- n is an integer from 2 to 20
- A is O, NH, CHF, CF 2 or CH 2
- Y is O " Cat+, a nucleoside, or a radical -A-R, wherein R is selected from the group of 1), 2) or 3).
- Y is O " Cat+, or a nucleoside. More preferably, Y is O " Cat+.
- RI is a methyl.
- A is O or CH 2 . More preferably, A is O. Preferably, n is 2. Preferably, X is a bromide. Preferably, B is O. Preferably, m is 1 or 2. More preferably, m is 1.
- synthetic ⁇ T cell activators comprise the compounds of formula (HI) or (IV) : wherein X, RI , n, m and Y have the aforementioned meaning.
- synthetic ⁇ T cell activators comprise the compounds of formula (V):
- X is an halogen (preferably selected from I, Br and CI)
- RI is a methyl or ethyl group
- Cat+ represents one (or several, identical or different) organic or mineral cation(s) (including the proton)
- n is an integer from 2 to 20.
- RI is a methyl.
- n is 2.
- X is a bromide.
- synthetic ⁇ T cell activators comprise the compound of formula (VI):
- x Cat+ is 1 or 2 Na +
- synthetic ⁇ T cell activators comprise the compound of formula (VII):
- x Cat+ is 1 or 2 Na +
- synthetic ⁇ T cell activators comprise the compounds of formula
- RI is a methyl or ethyl group
- Cat+ represents one (or several, identical or different) organic or mineral cation(s) (including the proton)
- B is O or ⁇ H
- m is an integer from 1 to 3
- n is an integer from 2 to 20
- A is O, ⁇ H, CHF, CF 2 or CH 2
- Y is O " Cat+, a nucleoside, or a radical -A-R, wherein R is selected from the group of 1), 2) or 3).
- Y is O " Cat+, or a nucleoside. More preferably, Y is O " Cat+.
- RI is a methyl.
- A is O or CH 2 . More preferably, A is O.
- n is 2.
- B is O.
- m is 1 or 2. More preferably, m is 1.
- synthetic ⁇ T cell activators comprise the compounds of formula (IX) or (X) :
- synthetic ⁇ T cell activators comprise the compounds of formula (XI): in which RI is a methyl or ethyl group, Cat+ represents one (or several, identical or different) organic or mineral cation(s) (including the proton), and n is an integer from 2 to 20.
- RI is a methyl or ethyl group
- Cat+ represents one (or several, identical or different) organic or mineral cation(s) (including the proton)
- n is an integer from 2 to 20.
- RI is a methyl.
- n is 2.
- synthetic ⁇ T cell activators comprise the compound of formula (XI):
- x Cat+ is 1 or 2 Na + .
- synthetic ⁇ T cell activators comprise the compounds of formula (X ⁇ ):
- R 3 , R_ t , and R 5 are a hydrogen or (C ⁇ -C 3 )alkyl group
- W is -CH- or-N-
- Re is an (C 2 -C 3 )acyl, an aldehyde, an (C ⁇ -C 3 )alcohol, or an (C 2 -C 3 )ester
- Cat+ represents one (or several, identical or different) organic or mineral cation(s) (including the proton)
- B is O or NH
- m is an integer from 1 to 3
- A is O, NH, CHF, CF 2 or CH 2
- Y is O " Cat+, a nucleoside, or a radical -A-R, wherein R is selected from the group of 1), 2) or 3).
- Y is O " Cat+, or a nucleoside. More preferably, Y is O " Cat+.
- A is O or CH 2 . More preferably, A is O. More preferably, R 3 and R 5 are a methyl and t is a hydrogen. More preferably, Re is -CH 2 -OH, -CHO, -CO-CH, or -CO-OCH 3 .
- B is O.
- m is 1 or 2. More preferably, m is 1.
- the double-bond between W and C is in conformation trans (E) or cis (Z). More preferably, the double-bond between W and C is in conformation trans (E).
- synthetic ⁇ T cell activators comprise the compounds of formula (XHI) or (XIV)
- R3, R4, R5, R6, W, m, and Y have the above mentionned meaning.
- W is - CH-.
- R3 and R4 are hydrogen.
- R5 is a methyl.
- R6 is -CH 2 - OH.
- synthetic ⁇ T cell activators comprise the compound of formula (XV):
- synthetic ⁇ T cell activators comprise the compound of formula (XVI):
- synthetic ⁇ T cell activators can comprise the compound of formula (XVI):
- compounds include : (E)l-pyro ⁇ hosphonobuta-l,3-diene ; (E)l- pyrophosphonopenta-l,3-diene ; (E)l-pyrophosphono-4-methylpenta-l,3-diene ; (E,E)1- pyrophosphono-4,8-dimethylnona-l ,3,7-triene ; (E,E,E) 1 -pyrophosphono-4,8, 12- trimethyltrideca-l,3,7,ll-tetraene ; (E,E)l-triphosphono-4,8-dimethylnona-l,3,7-triene ; 4- triphosphono-2-methylbutene ; ⁇ , ⁇ -di-[3-methylpent-3-enyl]-pyrophosphonate ; 1- pyrophosphono-3-methylbut-2-ene ; ⁇ , ⁇ -di-[3-methylbut-2-ene
- the ⁇ T cell activator can be selected from the group consisting of :
- the ⁇ T cell activator can be selected from the group consisting of : 3- (bromomethyl)-3-butanol-l -yl-diphosphate (BrHPP) ; 5-bromo-4-hydroxy-4-methylpentyl pyrophosphonate (CBrHPP) ; 3-(iodomethyl)-3-butanol-l-yl-diphos ⁇ hate (MPP) ; 3- (chloromethyl)-3-butanol-l -yl-diphosphate (CIHPP) ; 3-(bromomethyl)-3-butanol-l-yl- triphosphate (BrHPPP) ; 3-(iodomethyl)-3-butanol-l-yl-triphosphate (IHPPP) ; ⁇ , ⁇ -di-[3- (bromomethyl)-3-butanol-l-yl]-triphosphate (diBrHTP) ; and ⁇ , ⁇ -di
- the ⁇ T cell activator can be selected from the group consisting of : 3,4-epoxy-3-methyl-l-butyl-diphosphate (Epox-PP) ; 3,4,-epoxy-3-methyl-l- butyl-triphosphate (Epox-PPP) ; ⁇ , ⁇ -di-3,4,-epoxy-3-methyl-l-butyl-triphosphate (di-Epox-TP) ; 3,4-epoxy-3-ethyl-l-butyl-diphosphate ; 4,5-epoxy-4-methyl-l-pentyl-diphosphate ; 4,5- epoxy-4-ethyl-l-pentyl-diphosphate ; 5,6-epoxy-5-methyl-l-hexyl-diphosphate ; 5,6-epoxy-5- ethyl-1-hexyl-diphosphate ; 6,7-epoxy-6-methyl-
- the ⁇ T cell activator can be selected from the group consisting of : 3,4-epoxy-3-methyl-l-butyl-diphosphate (Epox-PP) ; 3,4,-epoxy-3-methyl-l- butyl-triphosphate (Epox-PPP) ; ⁇ , ⁇ -di-3,4,-epoxy-3-methyl-l-butyl-triphosphate (di-Epox-TP) ; and uridine 5'-triphosphate -(3,4-epoxy methyl butyl) (Epox-UTP).
- Epox-PP 3,4-epoxy-3-methyl-l-butyl-diphosphate
- Epox-PPP 3,4,-epoxy-3-methyl-l-butyl-triphosphate
- di-Epox-TP ⁇ , ⁇ -di-3,4,-epoxy-3-methyl-l-butyl-triphosphate
- the ⁇ T cell activator can be selected from the group consisting of : (E)-4-hydroxy-3-methyl-2-butenyl pyrophosphate (HDMAPP) and (E)-5-hydroxy-4- methylpent-3-enyl pyrophosphonate (CHDMAPP).
- the ⁇ T cell activator is selected from the group consisting of HDMAPP, CHDMAPP, Epox-PP, BrHPP and CBrHPP, more preferably HDMAPP, CHDMAPP, BrHPP and CBrHPP, still more preferably HDMAPP.
- activators for use in the present invention are phosphoantigens disclosed in WO 95/20673, isopentenyl pyrophosphate (IPP) (US 5,639,653) and 3-methylbut-3-enyl pyrophosphonate (C-IPP).
- IPP isopentenyl pyrophosphate
- C-IPP 3-methylbut-3-enyl pyrophosphonate
- Compounds comprising a nucleoside as Y group can be prepared, for example, by the following reactions. Depending on the type and reactivity of the functional groups provided by Y, the professional is able to adapt the following examples, if necessary including the phases of protection/non-protection of the sensitive functional groups or those that can interact with the coupling reaction.
- R- A-PP >- R-A-PPO -Nucl acetonitrile Reaction A or Nucl-O-V
- Nucl is a nucleoside.
- Nucl-O-V is selected from the group consisting of : 5'-O-Tosyladenosine, 5'-O- Tosyluridine, 5'-O-Tosylcytidine, 5'-O-Tosylthymidine or 5'-O-Tosyl-2'-deoxyadenosine.
- reaction procedure can be the following: CH 2 CH Nucl-O— V M
- -O-V is a good group beginning with V chosen, for example, from among tosyle, mesyle, triflyle, brosyle or bromium
- PP represents the pyrophosphate group
- Nucl is a nucleoside.
- Nucl-O-V is selected from the group consisting of : 5'-O-
- Tosyladenosine 5'-O-Tosyluridine, 5'-O-Tosylcytidine, 5'-O-Tosylthymidine or 5'-O-Tosyl-
- Neutral pH is a nucleophile substitution reaction that can be carried out in conditions similar to those described by Davisson et al, (1987); and Davisson et al. (1986), the disclosures of which are inco ⁇ orated herein by reference.
- This reaction can also be used to prepare compound comprising a monosaccharide as group Y.
- Nucl-O-V is replaced by MonoSac-O-V, wherein Monosac is monosaccharide.
- MonoSac-O-Y group corresponding to compound Methyl-6-O- tosyl-alpha-D-galactopyranoside as described in publication Nilsson and Mosbach, (1980) , inco ⁇ orated herein by reference, or the commercially available mannose triflate compound.
- This reaction can further be used to prepare compound comprising a ohgosaccharide as group Y.
- Nucl-O-V is replaced by oligoSac-O-V, wherein oligoSac is an ohgosaccharide.
- oligoSac-O-Y group corresponding to compound 6 A -O-p- Toluenesulfonyl- ⁇ -cyclodextrin as described in publication (Organic syntheses, Vol. 77, p 225- 228, the disclosure of which is inco ⁇ orated herein by reference).
- This reaction can be used to prepare compound comprising a polysaccharide as group Y.
- Nucl-O-V is replaced by polySac-O-V, wherein polySac is a polysaccharide.
- polySac-O-Y group corresponding to tosylated polysaccharide as described in publication Nilsson et al., (1981); and Nilsson and Mosbach, (1980), the disclosures of which are inco ⁇ orated herein by reference.
- This coupling technique based on the activation of the hydroxyl groups of a polysaccharide support by tosylation allows for covalent coupling in an aqueous or an organic medium.
- This reaction can also be used for preparing compound comprising an aldehyde derivative as group Y by choosing, instead of Nucl, a derivative including a protected aldehyde function in the form of an acetal or any other group protecting this function.
- compounds comprising a nucleoside as Y group can be prepared by the following reaction: 1) Nucl-O-PPP, carbodiimide DMF/Methanol
- the reaction procedure can be the following: u H 2 1 ) Nucl-O-PPP, carbodiimide , , H 2
- Compounds comprising a nucleic acid as Y group, more particularly a ribonucleic acid can be prepared in conditions similar to those described in publication F. Huang et al (1997).
- the authors describe a universal method from catalytic RNA that is applicable to any molecule comprising a free terminal phosphate group.
- Compounds structurally related to the phosphohalohydrine group such as isopentenyl pyrophosphate or thiamine pyrophosphate are used or mentioned by these authors (see p. 8968 of F. Huang et al (1997)).
- R-A has the above mentionned meaning and R'- SH is an amino acid, a peptide or a protein derivative.
- the first phase can be carried out in conditions similar to those described by Davisson et al. (1987) and Davisson et al, (1986), the disclosures of which are inco ⁇ orated herein by reference, from the tetrabutylammonium salt of the initial compound and commercially available compounds such as glycidyl tosylate or epichlorohydrine. This reaction can also be carried out with thriphosphate compounds. Alternatively, a primary amine R'-NH 2 can be used instead of R'-SH. Without the reaction with R'-SH, the first reaction can be used to prepare compound comprising an epoxyde derivative.
- compounds comprising an amino acid, a peptide or a protein derivative as Y group can be prepared by the following reaction: 1) R-NH 2 , carbodiimide DMF/Methanol ,, ⁇ . folk , etc.
- R- A- PPP *- R- A- PPO -P-NH-R' 2 Triethylamine, DMF Reaction E where PPP represents the triphosphate group, PP represents the pyrophosphate group, P represents the phosphate group, R-A has the above mentionned meaning and R'-NH is an amino acid, a peptide or a protein derivative.
- the reaction can be carried out in conditions similar to those described by Knorre et al. (1976), the disclosure of which is inco ⁇ orated herein by reference, from compound (R-A-PPP) and an amino acid, peptide or a protein with formula R- NH 2 . This reaction involves the protection of the sensitive functions of compound R-NH 2 or can react with the carbodiimide (in particular, the carboxyl function).
- Tri or tetra-n-butylammonium salts of phosphoric, pyrophosphoric, triphosphoric, tetra- phosphoric or polyphosphoric acid can be prepared from commercially available corresponding acids.
- Derivatives with a related structure such as derivatives of methanetrisphosphonic acid described in publication Liu et al (1999), the disclosure of which is inco ⁇ orated herein by reference, can also be prepared according to the reaction procedure.
- the above mentioned reactions can be extrapolated to a very large spectrum of molecules or biomolecules by using the reactivity of the hydroxyl, amine, phosphate or thiol functions.
- inositol derivatives can be prepared according to reactions A or B by activation of the hydroxyl function.
- Derivatives of folic acid (vitamin B9) or tetrahydrofolic acid can be prepared according to reactions D or E by calling on the reactivity of the primary amine function.
- preferred compounds are selected which increase the biological activity of ⁇ T cells, preferably increasing the activation of ⁇ T cells, particularly increasing cytokine secretion from ⁇ T cells or increasing the cytolytic activity of ⁇ T cells, with or without also stimulating the expansion of ⁇ T cells.
- a ⁇ T cell activator allows the cytokine secretion by ⁇ T cells to be increased at least 2, 3, 4, 10, 50, 100-fold, as determined in vitro. Cytokine secretion and cytolytic activity can be assessed using any appropriate in vitro assay, or those described herein.
- the present invention relates to methods for the treatment of a carcinoma or viral infection, preferably a urinary or bladder cancer or an HPV infection, where the ⁇ T cell activator is administered in an amount and under conditions sufficient to stimulate the expansion of the ⁇ T cell population in a subject, particularly to reach 30-90% of total circulating lymphocytes, typically 40-90%, more preferably from 50-90%.
- the invention allows the selective expansion of ⁇ T cells in a subject, to reach at least 20%, 30% or 40% of total circulating lymphocytes. Percentage of total circulating lymphocytes can be determined according to methods known in the art. A preferred method for deteixriining the percentage of ⁇ T cells in total circulating lymphocytes is by flow cytometry.
- the present invention relates to methods for the treatment of a carcinoma or viral infection, preferably a urinary or bladder cancer or an HPV infection, where the ⁇ T cell activator is administered in an amount and under conditions sufficient to stimulate the expansion of the ⁇ T cell population in a subject, particularly to increase by more than 2-fold the number of ⁇ T cells in a subject, typically at least 10-fold, more preferably at least 20-fold.
- the present invention relates to methods for the treatment of a bladder cancer, where the ⁇ T cell activator, especially a ⁇ T cell activator according to formulas I to XVII, is administered in an amount and under conditions sufficient to stimulate the expansion of the ⁇ T cell population in a subject, particularly to reach a circulating ⁇ T cell count of at least 500 ⁇ T cells/mm3 in a subject, typically at least 1000 ⁇ T cells/mm3, more preferably at least 2000 ⁇ T cells/mm3.
- the number of ⁇ T cells and circulating ⁇ T cell count in a subject is preferably assessed by obtaining a blood sample from a patient before and after administration of said ⁇ T cell activator and determining the difference in number of ⁇ T cells present in the sample.
- dosage (single administration) of a ⁇ T cell activator compound of formula I for treatment is between about 1 ⁇ g/kg and about 1.2 g/kg.
- dosages related to a group of compounds, and that each particular compound may vary in optimal doses, as further described herein for exemplary compounds.
- compounds are preferably administered in a dose sufficient to significantly increase the biological activity of ⁇ T cells or to significantly increase the ⁇ T cell population in a subject.
- Said dose is preferably administered to the human by intravenous (i.v.) administration during 2 to 180 min, preferably 2 to 120 min, more preferably during about 5 to about 60 min, or most preferably during about 30 min or during about 60 min.
- a compound of formula II to XI is administered in a dosage (single administration) between about 0.1 mg/kg and about 1.2 g/kg, preferably between about 10 mg/kg and about 1.2 g/kg, more preferably between about 5 mg/kg and about 100 mg/kg, even more preferably between about 5 ⁇ g/kg and 60 mg/kg.
- dosage (single administration) for three-weekly or four-weekly treatment is between about 0.1 mg/kg and about 1.2 g/kg, preferably between about 10 mg/kg and about 1.2 g/kg, more preferably between about 5 mg/kg and about 100 mg/kg, even more preferably between about 5 ⁇ g/kg and 60 mg/kg.
- a compound of formula XII to XVII is administered in a dosage (single administration) between about 1 ⁇ g/kg and about 100 mg/kg, preferably between about 10 ⁇ gkg and about 20 mg/kg, more preferably between about 20 ⁇ g/kg and about 5 mg/kg, even more preferably between about 20 ⁇ g/kg and 2.5 mg/kg.
- dosage (single administration) for three-weekly or four-weekly treatment is between about 1 ⁇ g/kg and about 100 mg/kg, preferably between about 10 ⁇ g/kg and about 20 mg/kg, more preferably between about 20 ⁇ g/kg and about 5 mg/kg, even more preferably between about 20 ⁇ g/kg and 2.5 mg/kg.
- dosages and administration and examples of dose response experiments using ⁇ T cell activator in mice and primate models are provided in co-pending PCT Application no. PCT/FR03/03560 filed 2 December 2003, the disclosure of which is inco ⁇ orated herein by reference.
- active compounds for use in the invention - e.g. IMC and IC compounds of the invention, as well as the ⁇ T cell activator compounds - can be administered by any suitable routes of administration including, but not limited to, oral, dermal, subcutaneous, percutaneous, intramuscular, intraperitoneal, intravenous, intradermal, intrathecal, intralesional, intratumoral, intrabladder, intra-vaginal, intraocular, intrarectal, intrapulmonary, intraspinal, transdermal, subdermal, placement within cavities of the body, nasal inhalation, pulmonary inhalation, impression into skin and electroco ⁇ oration.
- routes of administration including, but not limited to, oral, dermal, subcutaneous, percutaneous, intramuscular, intraperitoneal, intravenous, intradermal, intrathecal, intralesional, intratumoral, intrabladder, intra-vaginal, intraocular, intrarectal, intrapulmonary, intraspinal, transdermal, subdermal, placement within cavities of the body, nasal inhalation
- intra-tumoral administration means that the composition is delivered directly into the tumor, i.e. into actively dividing tumor cells surrounding the necrotic central part of the tumor and not, e.g., only into peritumoral cells or into the center of the tumor.
- tumor in this context does not only refer to the primary tumor but also to metastases.
- Appropriate means for intratumoral administration are, e.g., injection, ballistic tools, electroporation, electroinsertion, wounding, scratching, pressurized insertion tools, dermojets, etc.
- intra-tumoral administration is carried out by injection, preferably by a needle and a syringe.
- local administration and “topical administration” are used interchangeably, and refer to administration of a composition to a definite place or locality on the individual's body.
- active compounds for use in the invention e.g. IMC and IC compounds of the invention, as well as the ⁇ T cell activator compounds - can be inco ⁇ orated into pharmaceutical compositions suitable for administration.
- Such compositions typically comprise a pharmaceutically acceptable carrier.
- pharmaceutically acceptable carrier is intended to include any and all solvents, dispersion media, coatings, antibacterial and antifungal agents, isotonic and abso ⁇ tion delaying agents, and the like, compatible with pharmaceutical administration.
- the use of such media and agents for pharmaceutically active substances is well known in the art. Except insofar as any conventional media or agent is incompatible with the active compound, use thereof in the compositions is contemplated. Supplementary active compounds can also be inco ⁇ orated into the compositions.
- a pharmaceutical composition of the invention is formulated to be compatible with its intended route of administration.
- routes of administration include parenteral, e.g., intravenous, intradermal, subcutaneous, oral (e.g., inhalation), transdermal (topical), transmucosal, and rectal administration.
- Solutions or suspensions used for parenteral, intradernial, or subcutaneous application can include the following components: a sterile diluent such as water for injection, saline solution, fixed oils, polyethylene glycols, glycerine, propylene glycol or other synthetic solvents; antibacterial agents such as benzyl alcohol or methyl parabens; antioxidants such as ascorbic acid or sodium bisulfite; chelating agents such as ethylenediaminetetraacetic acid; buffers such as acetates, citrates or phosphates and agents for the adjustment of tonicity such as sodium chloride or dextrose. pH can be adjusted with acids or bases, such as hydrochloric acid or sodium hydroxide.
- a sterile diluent such as water for injection, saline solution, fixed oils, polyethylene glycols, glycerine, propylene glycol or other synthetic solvents
- antibacterial agents such as benzyl alcohol or methyl parabens
- antioxidants
- compositions suitable for injectable use include sterile aqueous solutions (where water soluble) or dispersions and sterile powders for the extemporaneous preparation of sterile injectable solutions or dispersion.
- suitable carriers include physiological saline, bacteriostatic water, Cremophor EL (BASF, Parsippany, NJ.) or phosphate buffered saline (PBS).
- the composition must be sterile and should be fluid to the extent that easy syringability exists. It must be stable under the conditions of manufacture and storage and must be preserved against the contaminating action of microorganisms such as bacteria and fungi.
- the carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (for example, glycerol, propylene glycol, and liquid polyetheylene glycol, and the like), and suitable mixtures thereof.
- the proper fluidity can be maintained, for example, by the use of a coating such as lecithin, by the maintenance of the required particle size in the case of dispersion and by the use of surfactants.
- Prevention of the action of microorganisms can be achieved by various antibacterial and antifungat agents, for example, parabens, cblorobutanol, phenol, ascorbic acid, thimerosal, and the like.
- isotonic agents for example, sugars, polyalcohols such as manitol, sorbitol, sodium chloride in the composition.
- Prolonged abso ⁇ tion of the injectable compositions can be brought about by including in the composition an agent which delays abso ⁇ tion, for example, aluminum nionostearate and gelatin.
- sterile injectable solutions can be prepared by inco ⁇ orating the active compound in the required amount in an appropriate solvent with one or a combination of ingredients enumerated above, as required, followed by filtered sterilization.
- dispersions are prepared by inco ⁇ orating the active compound into a sterile vehicle which contains a basic dispersion medium and the required other ingredients from those enumerated above.
- the preferred methods of preparation are vacuum drying and freeze drying which yields a powder of the active ingredient plus any additional desired ingredient from a previously sterile-filtered solution thereof.
- Oral compositions generally include an inert diluent or an edible carrier. They can be enclosed in gelatin capsules or compressed into tablets. For the pu ⁇ ose of oral therapeutic ad : ministration, the active compound can be inco ⁇ orated with excipients and used in the form of tablets, troches, or capsules. For administration by inhalation, the compounds are delivered in the form of an aerosol spray from pressured container or dispenser which contains a suitable propellant, e.g., a gas such as carbon dioxide, or a nebulizer. Systemic administration can also be by transmucosal or transdernial means. For transmucosal or transdernial administration, penetrants appropriate to the barrier to be permeated are used in the formulation.
- Such penetrants are generally known in the art, and include, for example, for transmucosal administration, detergents, bile salts, and fusidic acid derivatives.
- Transmucosal aclministration can be accomplished through the use of nasal sprays or suppositories.
- the active compounds are formulated into ointments, salves, gels, or creams as generally known in the art. Most preferably, active compound is delivered to a subject by intravenous injection.
- a composition to be administered will often comprise the physiologically active agent together with a penetration enhancer inco ⁇ orated into a dosage form for topical application to the skin or mucous membranes of animals.
- Suitable dosage forms include creams, lotions, gels, ointments, suppositories, mousses, spray, for example nasal sprays, aerosols, buccal and sublingual tablets, gingival and buccal patches or any one of a variety of transdermal devices for use in the continuous administration of systematically active drugs by abso ⁇ tion through the skin, oral mucosa or other membranes.
- the active compounds are prepared with carriers that will protect the compound against rapid elimination from the body, such as a controlled release formulation, including implants and microencapsulated delivery systems.
- a controlled release formulation including implants and microencapsulated delivery systems.
- Biodegradable, biocompatible polymers can be used, such as ethylene vinyl acetate, polyanhydrides, polyglycolic acid, collagen, polyorthoesters, and polylactic acid. Methods for preparation of such formulations will be apparent to those skilled in the art.
- the materials can also be obtained commercially from Alza Co ⁇ oration and Nova Pharmaceuticals, Inc.
- Liposomal suspensions (including liposomes targeted to infected cells with monoclonal antibodies to viral antigens) can also be used as pharmaceutically acceptable carriers. These can be prepared according to methods known to those skilled in the art, for example, as described in U.S. Pat. No. 4,522,81 1.
- Dosage unit form refers to physically discrete units suited as unitary dosages for the subject to be treated; each unit containing a predetermined quantity of active compound calculated to produce the desired therapeutic,effect in association with the required pharmaceutical carrier.
- the specification for the dosage unit forms of the invention are dictated by and directly dependent on the unique characteristics of the active compound and the particular therapeutic effect to be achieved, and the limitations inherent in the art of compounding such an active compound for the treatment of individuals.
- Bladder Cancer Treatment with a mycobacterial antigen and a ⁇ T cell activator can be carried out according to any suitable administration regimen.
- the ⁇ T cell activator may be administered through any of several different routes, typically by injection or oral adrninistration. Injection may be carried out into various tissues, such as by intravenous, intra-peritoneal, intra-arterial, intra-muscular, intra-dermic, subcutaneous, etc. Particularly preferred is intravenous injection.
- the ⁇ T cell activator can be a ⁇ xninistered before, at the same time or after the mycobacterial antigen is acmiinistered.
- the ⁇ T cell activator will be administered no more than several (4, 5, 6, or 7) days before or after treatment with the mycobacterial antigen. Most preferably, however, the ⁇ T cell activator is administered at substantially the same time as the mycobacterial antigen is administered, preferably within 48 hours, 24 hours or more preferably within 12 or within 6 hours of treatment with the the mycobacterial antigen. For example, in the regimen of the Examples, the ⁇ T cell activator is administered several hours before administration of the mycobacterial antigen.
- the ⁇ T cell activator is administered once during the course of mycobacterial antigen therapy. More preferably, however, the ⁇ T cell activator is administered several times. Most preferably, the ⁇ T cell activator is administered according to a regimen in which ⁇ T cell activity, preferably the ⁇ T cell rate (number of ⁇ T cells), is allowed to return to substantially basal rate prior to a second administration of the compound.
- ⁇ T cell activity preferably the ⁇ T cell rate (number of ⁇ T cells)
- the ⁇ T cell rate number of ⁇ T cells
- the course of a preferred cycle for admiiiistering the ⁇ T cell activator is an at least 1 -weekly cycle, but more preferably at least a 2-weekly cycle (at least about 14 days), or more preferably at least 3-weekly or 4-weekly, though cycles anywhere between 2-weekly and 4-weekly are preferred. Also effective and contemplated are cycles of up to 8-weekly, for example 5-weekly, 6-weekly, 7-weekly or 8-weekly.
- the ⁇ T cell activator is administered only the first day of a 2-weekly to 4-weekly, or preferably 3 weekly, cycle.
- the mycobacterial antigen is administered on a 1 -weekly cycle for 6 weeks, and the ⁇ T cell activator occurs on the first day of a 2-weekly to 4-weekly cycle (that is, an about 14 to 28 day weeks repeating cycle).
- the mycobacterial antigen is administered on a 1 -weekly cycle for 6 weeks and the ⁇ T cell activator is administered only the first day of the 2-weekly to 4-weekly, or preferably 3 weekly, cycle.
- the ⁇ T cell activator is administered for at least substantially the duration of mycobacterial antigen treatment.
- a 3-weekly cycle is used for the ⁇ T cell activator and a 1 -weekly cycle is used for the mycobacterial antigen, both over a course of six weeks according to the following scheme: Day 0: mycobacterial antigen and ⁇ T cell activator Day 7: mycobacterial antigen Day 14: mycobacterial antigen Day 21 : mycobacterial antigen and ⁇ T cell activator Day 28: mycobacterial antigen Day 35 : mycobacterial antigen Day 42: (optional): ⁇ T cell activator
- a 3-weekly cycle is used for both the ⁇ T cell activator and the mycobacterial antigen.
- the ⁇ T cell activator and the mycobacterial antigen are administered on the same day.
- a subject will preferably be treated for at least two cycles of ⁇ T cell activator, or more preferably for at least three cycles, or for at least one cycle of mycobacterial antigen therapy, preferably at least one cycle of 1 -weekly mycobacterial antigen administration for a 6 week treatment cycle.
- treatment may continue for a greater number of cycles, for example at least 4, 5, 6 or more cycles can be envisioned.
- the cycle of dosing may be repeated for as long as clinically tolerated and the tumor is under control or until tumor regression.
- mycobacterial antigen regimens administrations of mycobacterial antigen take place 1 -weekly for 6 weeks, followed by an interval (for example 6 weeks), followed by administrations of mycobacterial antigen 3-weekly for a desired duration, such as at least 6 months or 12 months for maintenance therapy.
- the methods of the invention comprises further administering a cytokine.
- a cytokine can be administered, wherein said cytokine is capable of increasing the expansion of a ⁇ T cell population treated with a ⁇ T cell activator compound, preferably wherein the cytokine is capable of inducing an expansion of a ⁇ T cell population which is greater than the expansion resulting from administration of the ⁇ T cell activator compound in the absence of said cytokine.
- a preferred cytokine is an interleukin-2 polypeptide.
- a cytokine having ⁇ T cell proliferation inducing activity is administered at low doses, typically over a period of time comprised between 1 and 10 days.
- the ⁇ T cell activator is preferably administered in a single dose, and typically at the beginning of a cycle.
- a cytokine is administered daily for up to about 10 days, preferably for a period of between about 3 and 10 days, or most preferably for about 7 days.
- the administration of the cytokine begins on the same day (e.g. within 24 hours of) as administration of the ⁇ T cell activator.
- the cytokine can be administered in any suitable scheme within said regimen of between about 3 and 10 days. For example, in one aspect the cytokine is administered each day, while in other aspects the cytokine need not be administered on each day.
- Infections caused by human papilloma virus (HPV) using the Mycobacterium that can be treated according to the methods of the invention may include cutaneous and genital warts in humans, including verruca vulgaris and condylorna acurninaturn, cervical intraepithelial neoplasia and genital carcinomas.
- the treatment is applicable to any disease condition caused by HPV in humans including penile, intraurethral, perianal, intra-anal or perineal infections in men and cervical, vaginal, perigenital, intra-urethral, intra-anal and perineal infections in women, including condylomata acuminata, penile cancer, Bowen's disease, cervical cancer, head and neck cancer, laryngeal papillomatosis and laryngeal carcinoma.
- the mycobacterial antigen treatment may be effected by application of the Mycobacterium antigen in a suitable carrier to the region of infection, which may involve topical application to cutaneous, penile and perianal areas, or intraurethral application to the urogenital tract.
- the treatment may involve a single or a plurality of doses applied at time intervals.
- the individual dosage level may be about 1 mg to about 500 mg attenuated BCG while the time interval between doses may vary from about 1 to about 30 days.
- the number of treatments applied is from 1 to about 30 treatments.
- the mycobacterial antigen and ⁇ T cell activator combination treatment is can be used alone or may be preceded by laser or other surgical or topical therapy.
- the ⁇ T cell activator may be administered as for the treatment of bladder carcinoma, such as through any of several different routes, typically by injection or oral a ⁇ xninistration. Injection may be carried out into various tissues, such as by intravenous, intra-peritoneal, intra-arterial, intra-muscular, intra-dermic, subcutaneous, etc. Particularly preferred is intravenous injection.
- the ⁇ T cell activator can be administered before, at the same time or after the mycobacterial antigen is administered. Generally, the ⁇ T cell activator will be administered no more than several (4, 5, 6, or 7) days before or after treatment with the mycobacterial antigen.
- the ⁇ T cell activator is administered at substantially the same time as the mycobacterial antigen is adininistered, preferably within 48 hours, 24 hours or more preferably 12 or 6 hours of treatment with the the mycobacterial antigen.
- the ⁇ T cell activator is administered several hours before administration of the mycobacterial antigen.
- the ⁇ T cell activator is administered once during the course of mycobacterial antigen therapy. More preferably, however, the ⁇ T cell activator is administered several times. Most preferably, the ⁇ T cell activator is administered according to a regimen in which ⁇ T cell activity, preferably the ⁇ T cell rate (number of ⁇ T cells), is allowed to return to substantially basal rate prior to a second administration of the compound.
- the course of a preferred cycle for administering the ⁇ T cell activator is an at least 1 -weekly cycle, but more preferably at least a 2-weekly cycle (at least about 14 days), or more preferably at least 3- weekly or 4-weekly, though cycles anywhere between 2-weekly and 4-weekly are preferred.
- ⁇ T cell activator is administered only the first day of a 2-weekly to 4-weekly, or preferably 3 weekly, cycle.
- the mycobacterial antigen is administered on a 1 -weekly cycle for about 6 weeks, or for at least 4, 6, 8, 10 or 12 weeks, and the ⁇ T cell activator occurs on the first day of a 2-weekly to 4-weekly cycle (that is, an about 14 to 28 day weeks repeating cycle).
- the mycobacterial antigen is administered on a 1 -weekly cycle for 6 weeks and the ⁇ T cell activator is administered only the first day of the 2-weekly to 4-weekly, or preferably 3 weekly, cycle.
- the ⁇ T cell activator is administered for at least substantially the duration of mycobacterial antigen treatment.
- a 3-weekly cycle is used for the ⁇ T cell activator and a 1 -weekly cycle is used for the mycobacterial antigen, both over a course of six weeks according to the following scheme: Day 0: mycobacterial antigen and ⁇ T cell activator Day 7: mycobacterial antigen Day 14: mycobacterial antigen Day 21 : mycobacterial antigen and ⁇ T cell activator Day 28: mycobacterial antigen Day 35: mycobacterial antigen Day 42: (optional): ⁇ T cell activator
- a 3-weekly cycle is used for both the ⁇ T cell activator and the mycobacterial antigen.
- the ⁇ T cell activator and the mycobacterial antigen are administered on the same day.
- a cytokine may additionally be administered, according to a regimen as described for the treatment of bladder carcinoma.
- the Mycobacterium may be formulated with a keratolytic agent for topical application to the region of infection, particularly as a cream for adherent application to the region of infection.
- the keratolytic agent may be salicylic acid, which may be powdered.
- the keratolytic agent may be present in an amount of about 0.1 to about 50 wt%, preferably about 1 to about 10 wt%.
- the composition which is applied to the area of infection may take any desired form, for example, a cream, a powder or ointment. Any desired form of application may be employed, including slow-release systems, plasters and transdermal systems.
- (E)-4-Hydroxy-3-methylbut-2-enyl diphosphate is prepared according to the method of Wolff et al, Tetrahedron Letters (2002) 43:2555 or Hecht et al, Tetrahedron Letters (2002) 43: 8929.
- the aqueous solutions of the product are sterilized by filtration through a 0.2 ⁇ m filter and stored at -20 °C.
- the solutions are passed beforehand through a DOWEX 50WX8-200 cationic resin column (sodium form) eluted by two column volumes of deionized water.
- allylic alcohol [1] is converted into a protected form [2] by reaction of [1] with Dihydropyrane (DHP) in the presence of Pyridinium p-Toluenesulfonate (PPTs).
- DHP Dihydropyrane
- PPTs Pyridinium p-Toluenesulfonate
- the phosphonylating agent [3] is prepared by treatment of commercially available methylphosphonic dichloride with mo ⁇ holine and methanol.
- intermediate [4] is prepared by reaction of [2] with methyl lithiomethylphosphonomo ⁇ holidate obtained in situ from the phosphonylating agent [3].
- C-HDMAPP (EV5-hydroxy-4-methylpent-3-enyl pyrophosphonate
- the cmde salt of C-HDMAPP obtained at this stage is converted to the ammonium form by cation-exchange over DOWEX 50WX8-200 resin (ammonium form). Purification of the resulting solution is performed by chromatography over silica gel using 27 % ammonia solution/2-propanol 50/50 (v/v) as eluant.
- the aqueous solutions of the product are sterilized by filtration through a 0.2 ⁇ m filter and stored at -20 °C.
- the solutions are passed beforehand through a DOWEX 50WX8-200 cationic resin column (sodium form) eluted by two column volumes of deionized water.
- Trisodium (R,S)-3-(bromomethyl)-3- butanol-1 -yl-diphosphate (BrHPP) was produced as white amo ⁇ hous powder by the following procedure. Tosyl chloride (4.8 g, 25 mmol) and 4-(N,N-dimethyla ⁇ r_ino-) pyridine (3.4 g, 27.5 mmol; Aldrich) were mixed under magnetic stirring with 90 ml of anhydrous dichloromethane in a 250-ml three-necked flask cooled in an ice bath.
- Disodium dihydrogen pyrophosphate (51.5 mmol, 11.1 g) dissolved in 100 ml of deionized water (adjusted to pH 9 with NH t OH) was passed over a cation exchange DOWEX 50WX8 (42 g, 200 meq of form EL*) column and eluted with 150 ml of deionized water (pH 9).
- the collected solution was neutralized to pH 7.3 using tetra- «-butyl ammonium hydroxide and lyophilized.
- the resulting hygroscopic powder was solubilized with anhydrous acetonitrile and further dried by repeated evaporation under reduced pressure.
- Tris (tetra- «-butyl ammonium) hydrogenopyrophosphate (97.5% purity by HPAEC; see below) was stored (concentration, -0.5 M) at —20 °C in anhydrous conditions under molecular sieves.
- the reaction was analyzed by HPAEC (see below), evaporated, and diluted into 50 ml of a mixture composed of a solution (98 % volume) of ammonium hydrogenocarbonate (25 mM) and 2-propanol (2 volume %).
- the resulting mixture was passed over a cation exchange DOWEX 50WX8 (NH ⁇ , 750 meq) column formerly equilibrated with 200 ml of the solution (98 % volume) of ammonium hydrogenocarbonate (25 mM) and 2-propanol (2 volume %).
- the column was eluted with 250 ml of the same solution at a slow flow and collected in a flask kept in an ice bath.
- the collected liquid was lyophilized, and the resulting white powder was solubilized in 130 ml of ammonium hydrogenocarbonate (0.1 M) and completed by 320 ml of acetonitrile/2-propanol (v/v). After agitation, the white precipitate of inorganic pyro- and mono-phosphates was eliminated by centrifugation (2100 x g, 10 °C, 8 min). This procedure was repeated three times, the supernatant was collected and dried, and the resulting oil was diluted in 120 ml of water.
- the ammonium salt of BrHPP obtained after lyophilization was dissolved in water and separated from bromides by passing through Dionex OnGuard-Ag (2 meq/unit) cartridges and an on-line column of (100 meq, 21 g) DOWEX 50WX8-200 (Na + ) eluted by milli-Q water. Colorless stock solutions of BrHPP (Na + ) were filtered over Acrodisc 25 membranes of 0.2 ⁇ M and kept as aliquots at -20 °C.
- the gradient program was as follows: solvent A, acetonitrile; solvent B, 50 mM ammonium acetate; solvent C, water; 0-7 min, 5% B in C; 7.1-11 min, 100% C; 12-15 min, 100% A; 15-17 min, 100% C.
- ImmuCyst® (Bacillus Calmette-Guerin (BCG), substrain Connaught) is commercialised by Aventis Pasteur SA, France. ImmuCyst® is made from a culture of an attenuated strain of living bovine tubercle bacillus Mycobacterium bovis. Phosphostim (Innate Pharma, Marseille, France) is based on a new chemical entity, the drug substance Bromohydrin Pyrophosphate (BrHPP), which is a specific agonist of immune competent cells namely the V ⁇ 9V ⁇ 2 T cell subpopulation bearing anti-tumor activity. Phosphostim (BrHPP, 200 mg) is the intravenous formulation of BrHPP for cancer immunotherapy.
- BCG Bromohydrin Pyrophosphate
- ImmuCyst® is administered as intravesical treatment of the urinary bladder and Phosphostim is administered intravenously.
- the induction treatment comprises 6 weekly intravesical treatments with ImmuCyst®.
- Each treatment dose of ImmuCyst® comprises one 81 mg vial of ImmuCyst®.
- a patient receives repeated cycles of Phosphostim treatment every 3 weeks. The cycle consists of one administration by infusion of Phosphostim.
- Phosphostim can be administered in a dose of about 200 mg/m 2 or about 600 mg/m 2 , although the Phosphostim dose can be between 200 mg/m 2 (5 mg/kg) (corresponding to 118 mg-equivalent of BrHPP anionic form) and about 1000 mg/ m 2 and will be determined in a dose ranging study.
- each administration of Phosphostim is combined with an administration of 1 million IU/m 2 /day of IL-2 (for a total duration of 7 days).
- ImmuCyst® is preferably dosed and administered according to the manufacturer's inst ctions.
- Each dose (1 reconstituted vial) is further diluted in an additional 50 mL of sterile, preservative- free saline for a total of 53 mL.
- a urethral catheter is inserted into the bladder under aseptic conditions, the bladder is drained, and then 53 mL suspension of ImmuCyst® is instilled slowly by gravity, following which the catheter is withdrawn.
- the patient retains the suspension for as long as possible for a total of up to two hours.
- the patient should lie prone. Thereafter, the patient is then allowed to be up.
- all patients should void in a seated position for environmental safety reasons. Patients should be instmcted to maintain adequate hydration.
- Phosphostim (BrHPP, 200 mg) is a freeze-dried apyrogenic sterile white powder to be reconstituted in solution for infusion.
- Each vial of Phosphostim (BrHPP, 200mg) contains 200 mg of BrHPP anionic form and 50mg the excipient alpha-lactose monohydrate (USP).
- Phosphostim is for immediate and single use following first opening and reconstitution.
- Phosphostim is reconstituted immediately prior to use with 2 ml of water for injections to make a 100 mg/ml solution.
- the needed quantities of reconstituted product are diluted in a total volume 100 ml of ringer lactate buffer infusion vehicle.
- the diluted solution is clear and colorless.
- Phosphostim is administered intravenously over 1 hour.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Immunology (AREA)
- Mycology (AREA)
- Molecular Biology (AREA)
- Microbiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Communicable Diseases (AREA)
- Zoology (AREA)
- Gastroenterology & Hepatology (AREA)
- Engineering & Computer Science (AREA)
- Pulmonology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
Abstract
Description

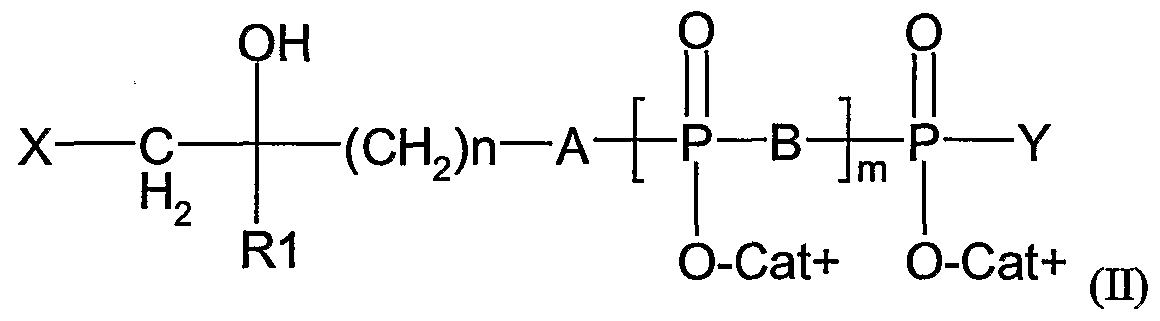





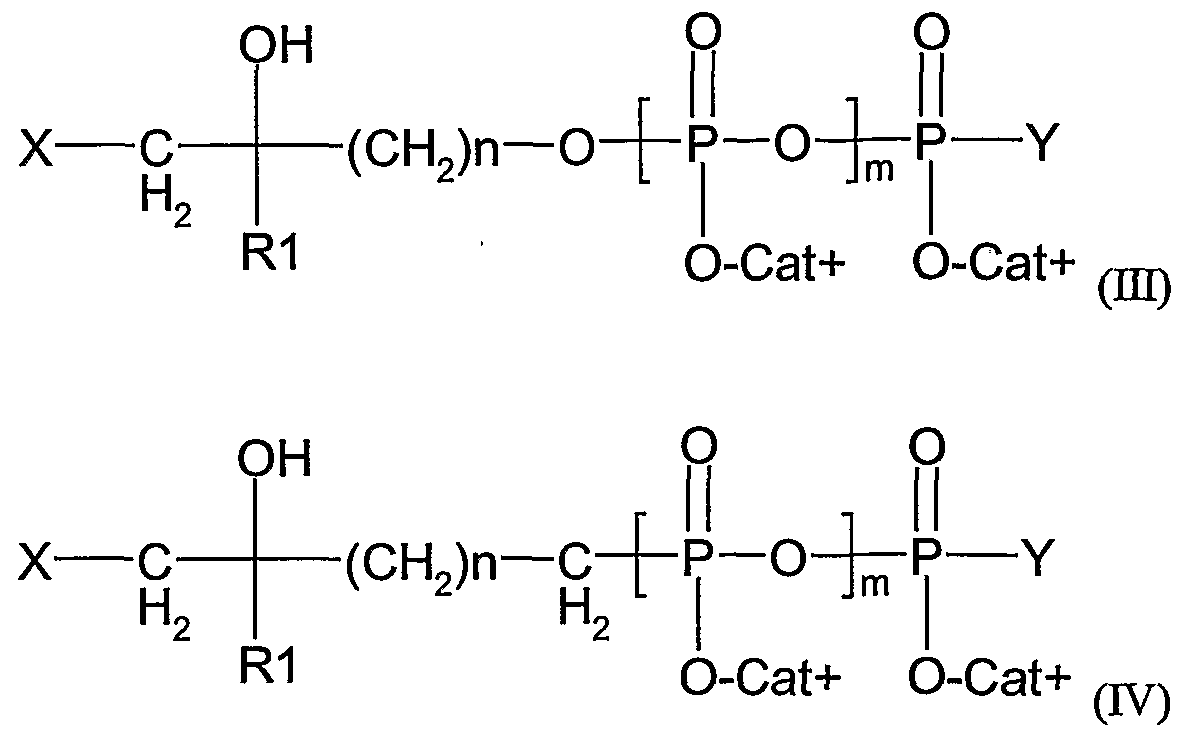




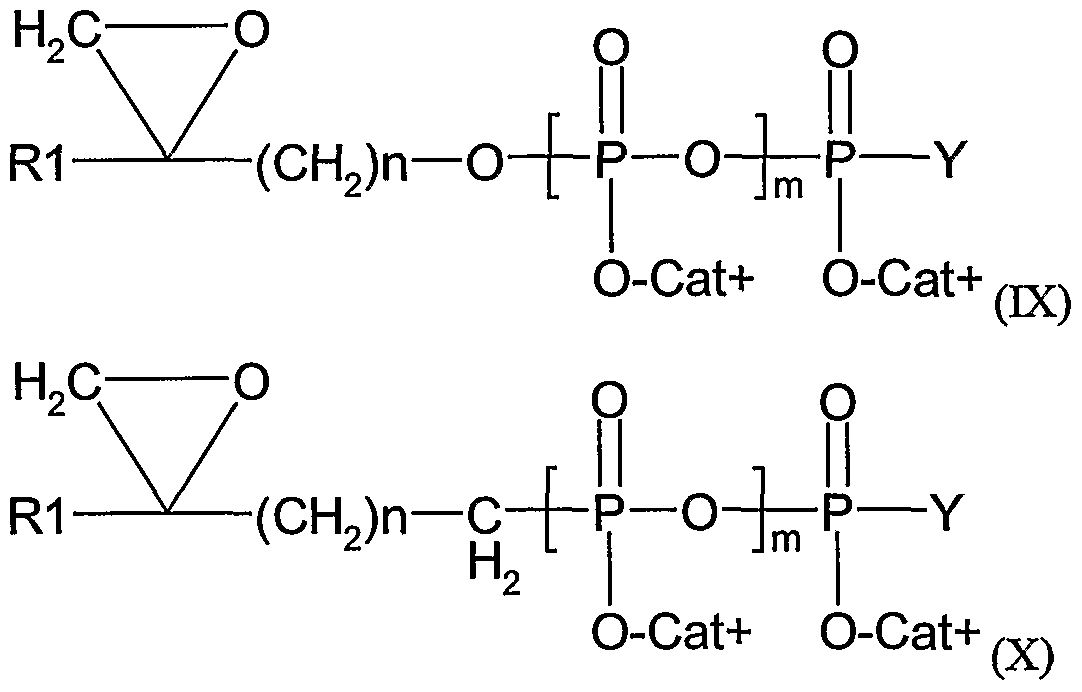







Claims

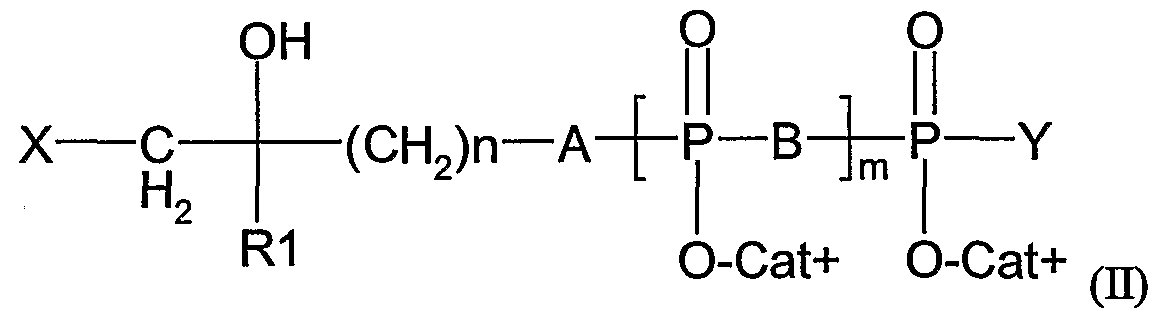




Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10/588,420 US20070134273A1 (en) | 2004-02-10 | 2005-02-08 | Composition and method for the treatment of carcinoma |
| EP05708622A EP1720567A2 (en) | 2004-02-10 | 2005-02-08 | Composition and method for the treatment of carcinoma |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US54285804P | 2004-02-10 | 2004-02-10 | |
| US60/542,858 | 2004-02-10 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2005077411A2 true WO2005077411A2 (en) | 2005-08-25 |
| WO2005077411A3 WO2005077411A3 (en) | 2006-04-13 |
Family
ID=34860346
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2005/000509 WO2005077411A2 (en) | 2004-02-10 | 2005-02-08 | Composition and method for the treatment of carcinoma |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US20070134273A1 (en) |
| EP (1) | EP1720567A2 (en) |
| WO (1) | WO2005077411A2 (en) |
Cited By (9)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006067635A2 (en) * | 2004-12-20 | 2006-06-29 | Innate Pharma S.A. | USE OF Ϝδ T LYMPHOCYTE ACTIVATORS AS VACCINE ADJUVANT |
| WO2007057440A2 (en) * | 2005-11-17 | 2007-05-24 | Innate Pharma | Improved methods of using phosphoantigen for the treatment of cancer |
| WO2008059052A1 (en) * | 2006-11-17 | 2008-05-22 | Innate Pharma | Improved methods of using phosphoantigen for the treatment of cancer |
| EP2059253A2 (en) * | 2006-09-14 | 2009-05-20 | The Trustees Of The University Of Pennsylvania | Modulation of regulatory t cells by human il-18 |
| EP2123285A1 (en) * | 2008-05-21 | 2009-11-25 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Nucleosidic phosphoantigens for use in VGAMMA9DELTA2 T cell-mediated therapy |
| ITMI20081652A1 (en) * | 2008-09-16 | 2010-03-17 | Antica Ritrovati Medicinal I S A R M Srl Soc | TRANSDERMIC COMPOSITIONS FOR HYPOSENSIBILIZING SPECIFIC IMMUNOTHERAPY |
| EP2165710A1 (en) | 2008-09-19 | 2010-03-24 | Institut Curie | Tyrosine kinase receptor Tyro3 as a therapeutic target in the treatment of a bladder tumor |
| WO2013054329A1 (en) | 2011-10-10 | 2013-04-18 | Yeda Research And Development Co. Ltd. | Toll-like receptor 4 (tlr-4) agonist peptides for modulating tlr-4 mediated immune response |
| US9890202B2 (en) | 2010-07-19 | 2018-02-13 | Yeda Research And Development Co. Ltd. | Peptides based on the transmembrane domain of a toll-like receptor (TLR) for treatment of TLR-mediated diseases |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2836483B1 (en) * | 2002-02-22 | 2006-09-15 | Innate Pharma | METHODS FOR PRODUCING GAMMA DELTA T LYMPHOCYTES |
| EP1426052B1 (en) * | 2002-12-02 | 2009-09-02 | Innate Pharma | Compositions comprising interleukin-2 and gamma-delta T cell activator and uses thereof |
| DE602006021408D1 (en) * | 2005-04-27 | 2011-06-01 | Univ Leiden Medical Ct | TREATMENT OF HPV-INDUCED INTRAITITHELIAL ANOGENITAL NEOPLASIA |
| WO2009016486A2 (en) * | 2007-08-02 | 2009-02-05 | Compugen, Ltd | Use of nmda receptor antagonists for treatment of urologic tumors |
| US20100160368A1 (en) * | 2008-08-18 | 2010-06-24 | Gregory Jefferson J | Methods of Treating Dermatological Disorders and Inducing Interferon Biosynthesis With Shorter Durations of Imiquimod Therapy |
| EP2378876B1 (en) | 2008-12-19 | 2018-11-14 | Medicis Pharmaceutical Corporation | Lower dosage strength imiquimod formulations and short dosing regimens for treating actinic keratosis |
| AU2010274097B2 (en) | 2009-07-13 | 2016-06-16 | Medicis Pharmaceutical Corporation | Lower dosage strength imiquimod formulations and short dosing regimens for treating genital and perianal warts |
| CN102639700A (en) | 2009-09-30 | 2012-08-15 | 哈佛大学校长及研究员协会 | Methods for modulation of autophagy through the modulation of autophagy-enhancing gene products |
| US9981018B2 (en) | 2010-06-08 | 2018-05-29 | The Cleveland Clinic Foundation | Compositions and methods for modulating toll-like receptor 2 activation |
| WO2011156451A2 (en) * | 2010-06-08 | 2011-12-15 | The Cleveland Clinic Foundation | Compositions and methods for modulating toll-like receptor 2 activation |
| WO2014072446A1 (en) * | 2012-11-08 | 2014-05-15 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Method for inducing il-2-free proliferation of gamma delta t cells |
| GB201421716D0 (en) | 2014-12-05 | 2015-01-21 | King S College London | Cell expansion procedure |
| WO2021077113A1 (en) * | 2019-10-19 | 2021-04-22 | Texas Biomedical Research Institute | Methods of treatment of bladder cancer by using modified bacillus calmette-guérin |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1995020673A1 (en) * | 1994-01-28 | 1995-08-03 | Centre National De La Recherche Scientifique (C.N.R.S.) | Organo-phosphorus compounds as activators of gamma delta t cells |
| WO2000012519A1 (en) * | 1998-09-01 | 2000-03-09 | Institut National De La Sante Et De La Recherche Medicale | Phosphoepoxides, method for making same and uses |
| WO2000012516A1 (en) * | 1998-09-01 | 2000-03-09 | Institut National De La Sante Et De La Recherche Medicale | Phosphohalohydrins, method for making same and uses |
| WO2001056387A1 (en) * | 2000-02-01 | 2001-08-09 | Donnell Michael A O | Immunotherapeutic treatment methodology for patients afflicted with superficial bladder cancer who previously failed at least one immunostimulatory therapeutic treatment regimen |
| EP1426052A1 (en) * | 2002-12-02 | 2004-06-09 | Innate Pharma | Compositions comprising interleukin-2 and gamma-delta T cell activator and uses thereof |
Family Cites Families (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3591693A (en) * | 1969-05-22 | 1971-07-06 | American Cyanamid Co | Compositions and method for treating mycobacterium tuberculosis with 2,4,6-tris(alkylamino)-s-triazine |
| US5882654A (en) * | 1989-11-03 | 1999-03-16 | Morton; Donald L. | Polyvalent melanoma vaccine |
| IL105503A (en) * | 1992-04-28 | 1999-05-09 | Astra Ab | Peptide-carbohydrate conjugates capable of generating t cell immunity |
| US6809081B1 (en) * | 1998-12-04 | 2004-10-26 | Bioniche Life Sciences, Inc. | Chemotherapeutic composition and method |
| US6949520B1 (en) * | 1999-09-27 | 2005-09-27 | Coley Pharmaceutical Group, Inc. | Methods related to immunostimulatory nucleic acid-induced interferon |
| US6737398B1 (en) * | 1999-09-30 | 2004-05-18 | National Jewish Medical And Research Center | Modulation of γδ T cells to regulate airway hyperresponsiveness |
| US7927612B2 (en) * | 2000-01-19 | 2011-04-19 | Baofa Yu | Combinations and methods for treating neoplasms |
| AU2001251316A1 (en) * | 2000-04-04 | 2001-10-15 | Antigenics Inc. | Isolated and purified nonpeptide antigens from mycobacterium tuberculosis |
| DE10201458A1 (en) * | 2001-04-11 | 2002-10-17 | Adelbert Bacher | New proteins involved in isoprenoid biosynthesis, useful in screening for inhibitors, also new intermediates, potential therapeutic agents, nucleic acids and antibodies |
| EP1408984B1 (en) * | 2001-07-20 | 2008-10-22 | BioAgency AG | Organo-phosphorous compounds for activating gamma/delta t cells |
| FR2833266B1 (en) * | 2001-12-11 | 2004-10-22 | Mayoly Spindler Lab | NOVEL PHOSPHONATE DERIVATIVES, THEIR PREPARATION PROCESS AND THEIR USE AS MODULATORS OF THE ACTIVITY OF TGAMMA9 DELTA2 LYMPHOCYTES |
| FR2836483B1 (en) * | 2002-02-22 | 2006-09-15 | Innate Pharma | METHODS FOR PRODUCING GAMMA DELTA T LYMPHOCYTES |
| US6797722B2 (en) * | 2002-05-03 | 2004-09-28 | Sigma-Tau Industrie Farmaceutiche Riunite S.P.A. | Use of 3-(2-ethylphenyl)-5-(3-methoxyphenyl)-1H-1,2,4-triazole for the treatment of autoimmune diseases |
| RU2348418C2 (en) * | 2002-12-06 | 2009-03-10 | Норсуэст Байотерапьютикс, Инк. | Introduction of dendritic cells exposed to partial aging in vitro, for tumour treatment |
| US20040219629A1 (en) * | 2002-12-19 | 2004-11-04 | Qiong Cheng | Increasing carotenoid production in bacteria via chromosomal integration |
| UY28213A1 (en) * | 2003-02-28 | 2004-09-30 | Bayer Pharmaceuticals Corp | NEW CYANOPIRIDINE DERIVATIVES USEFUL IN THE TREATMENT OF CANCER AND OTHER DISORDERS. |
| EP1469007A1 (en) * | 2003-04-18 | 2004-10-20 | Centre National De La Recherche Scientifique (Cnrs) | Sulfoglycolipid antigens, their extraction from mycobacterieum tuberculosis, and their use against tuberculosis |
| EP1761278B1 (en) * | 2004-04-26 | 2008-12-31 | Innate Pharma | Adjuvant composition and methods for its use |
-
2005
- 2005-02-08 US US10/588,420 patent/US20070134273A1/en not_active Abandoned
- 2005-02-08 EP EP05708622A patent/EP1720567A2/en not_active Withdrawn
- 2005-02-08 WO PCT/IB2005/000509 patent/WO2005077411A2/en active Application Filing
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1995020673A1 (en) * | 1994-01-28 | 1995-08-03 | Centre National De La Recherche Scientifique (C.N.R.S.) | Organo-phosphorus compounds as activators of gamma delta t cells |
| WO2000012519A1 (en) * | 1998-09-01 | 2000-03-09 | Institut National De La Sante Et De La Recherche Medicale | Phosphoepoxides, method for making same and uses |
| WO2000012516A1 (en) * | 1998-09-01 | 2000-03-09 | Institut National De La Sante Et De La Recherche Medicale | Phosphohalohydrins, method for making same and uses |
| WO2001056387A1 (en) * | 2000-02-01 | 2001-08-09 | Donnell Michael A O | Immunotherapeutic treatment methodology for patients afflicted with superficial bladder cancer who previously failed at least one immunostimulatory therapeutic treatment regimen |
| EP1426052A1 (en) * | 2002-12-02 | 2004-06-09 | Innate Pharma | Compositions comprising interleukin-2 and gamma-delta T cell activator and uses thereof |
| WO2004050096A2 (en) * | 2002-12-02 | 2004-06-17 | Innate Pharma | Phosphoantigens for regulating an immune response |
Non-Patent Citations (4)
| Title |
|---|
| KUNZMANN V ET AL: "Stimulation of gammadelta T cells by aminobisphosphonates and induction of antiplasma cell activity in multiple myeloma" BLOOD, W.B.SAUNDERS COMPANY, ORLANDO, FL, US, vol. 96, no. 2, 15 July 2000 (2000-07-15), pages 384-392, XP002223785 ISSN: 0006-4971 * |
| PONTICIELLO ANTONIO ET AL: "Analysis of local T lymphocyte subsets upon stimulation with intravesical BCG: a model to study tuberculosis immunity." RESPIRATORY MEDICINE. JUN 2004, vol. 98, no. 6, June 2004 (2004-06), pages 509-514, XP002350292 ISSN: 0954-6111 * |
| SAINT FABIEN ET AL: "Tolerability of bacille Calmette-Guerin maintenance therapy for superficial bladder cancer" UROLOGY, vol. 57, no. 5, May 2001 (2001-05), pages 883-888, XP002350291 ISSN: 0090-4295 * |
| WILHELM M ET AL: "gammadelta T cells for immune therapy of patients with lymphoid malignancies" BLOOD, W.B.SAUNDERS COMPANY, ORLANDO, FL, US, vol. 102, no. 1, 1 July 2003 (2003-07-01), pages 200-206, XP002276948 ISSN: 0006-4971 * |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006067635A3 (en) * | 2004-12-20 | 2006-08-24 | Innate Pharma Sa | USE OF Ϝδ T LYMPHOCYTE ACTIVATORS AS VACCINE ADJUVANT |
| WO2006067635A2 (en) * | 2004-12-20 | 2006-06-29 | Innate Pharma S.A. | USE OF Ϝδ T LYMPHOCYTE ACTIVATORS AS VACCINE ADJUVANT |
| WO2007057440A2 (en) * | 2005-11-17 | 2007-05-24 | Innate Pharma | Improved methods of using phosphoantigen for the treatment of cancer |
| WO2007057440A3 (en) * | 2005-11-17 | 2008-04-03 | Innate Pharma | Improved methods of using phosphoantigen for the treatment of cancer |
| EP2059253A4 (en) * | 2006-09-14 | 2011-09-14 | Univ Pennsylvania | Modulation of regulatory t cells by human il-18 |
| EP2059253A2 (en) * | 2006-09-14 | 2009-05-20 | The Trustees Of The University Of Pennsylvania | Modulation of regulatory t cells by human il-18 |
| US8679471B2 (en) | 2006-09-14 | 2014-03-25 | The Trustees Of The Univesity Of Pennsylvania | Modulation of regulatory T cells by human IL-18 |
| WO2008059052A1 (en) * | 2006-11-17 | 2008-05-22 | Innate Pharma | Improved methods of using phosphoantigen for the treatment of cancer |
| EP2123285A1 (en) * | 2008-05-21 | 2009-11-25 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Nucleosidic phosphoantigens for use in VGAMMA9DELTA2 T cell-mediated therapy |
| WO2010031519A1 (en) * | 2008-09-16 | 2010-03-25 | Societa' Antica Ritrovati Medicinali S.A.R.M. S.R.L. | Transcutaneous compositions for specific immuno-modulatory treatment |
| ITMI20081652A1 (en) * | 2008-09-16 | 2010-03-17 | Antica Ritrovati Medicinal I S A R M Srl Soc | TRANSDERMIC COMPOSITIONS FOR HYPOSENSIBILIZING SPECIFIC IMMUNOTHERAPY |
| EP2165710A1 (en) | 2008-09-19 | 2010-03-24 | Institut Curie | Tyrosine kinase receptor Tyro3 as a therapeutic target in the treatment of a bladder tumor |
| US9890202B2 (en) | 2010-07-19 | 2018-02-13 | Yeda Research And Development Co. Ltd. | Peptides based on the transmembrane domain of a toll-like receptor (TLR) for treatment of TLR-mediated diseases |
| WO2013054329A1 (en) | 2011-10-10 | 2013-04-18 | Yeda Research And Development Co. Ltd. | Toll-like receptor 4 (tlr-4) agonist peptides for modulating tlr-4 mediated immune response |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2005077411A3 (en) | 2006-04-13 |
| EP1720567A2 (en) | 2006-11-15 |
| US20070134273A1 (en) | 2007-06-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20070134273A1 (en) | Composition and method for the treatment of carcinoma | |
| US6552006B2 (en) | Immunomodulatory polynucleotides in treatment of an infection by an intracellular pathogen | |
| US7108844B2 (en) | Use of stabilized oligonucleotides for preparing a medicament with antitumor activity | |
| US20170340658A1 (en) | Combined use of a chemotherapeutic agent and a cyclic dinucleotide for cancer treatment | |
| US20100254940A1 (en) | Compositions and Methods for Regulating an Immune Response in a Subject | |
| US20050048072A1 (en) | Immunostimulatory combinations and treatments | |
| BR112015013440B1 (en) | compositions comprising cyclic purine dinucleotides with stereochemistry | |
| US20130302278A1 (en) | Use of tlr agonists and/or type 1 interferons to alleviate toxicity of tnf-r agonist therapeutic regimens | |
| AU2005235271A1 (en) | Adjuvant composition and methods for its use | |
| EP1787660A1 (en) | New adjuvants on the basis of bisacyloxypropylcysteine conjugates and their uses in pharmaceutical compositions | |
| WO2006067635A2 (en) | USE OF Ϝδ T LYMPHOCYTE ACTIVATORS AS VACCINE ADJUVANT | |
| EP2793891A1 (en) | Pharmaceutical compositions and methods for treating gastrointestinal infections and disorders | |
| US20090087456A1 (en) | Adjuvanted vaccine | |
| US20210401974A1 (en) | Pharmaceutical compositions, vaccines and their uses in the prevention or treatment of a persistent infection or of cancer | |
| EP3177294B1 (en) | Compositions comprising a pnp inhibitor for use in the treatment of the relapse of malignancy following hematopoietic stem cell transplant | |
| JP2023550544A (en) | Therapy for the treatment of cancer and diseases associated with phagocytosis deficiency |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2007134273 Country of ref document: US Ref document number: 10588420 Country of ref document: US |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Country of ref document: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2005708622 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005708622 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 10588420 Country of ref document: US |