WO2005067900A2 - Azabenzofuran substituted thioureas as inhibitors of viral replication - Google Patents
Azabenzofuran substituted thioureas as inhibitors of viral replication Download PDFInfo
- Publication number
- WO2005067900A2 WO2005067900A2 PCT/US2005/000339 US2005000339W WO2005067900A2 WO 2005067900 A2 WO2005067900 A2 WO 2005067900A2 US 2005000339 W US2005000339 W US 2005000339W WO 2005067900 A2 WO2005067900 A2 WO 2005067900A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- phenyl
- carbonyl
- thiourea
- compound
- Prior art date
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/4355—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a five-membered ring having oxygen as a ring hetero atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
Definitions
- the present invention provides azabenzofuran substituted thiourea compounds, particularly furanylpyridinyl substituted thiourea derivatives and related compounds, useful as antiviral agents.
- Certain azabenzofuran substituted thiourea compounds, including furanylpyridinyl substituted thiourea compounds disclosed herein are potent and/ or selective inhibitors of viral replication, particularly Hepatitis C virus replication.
- the invention also provides pharmaceutical compositions containing one or more azabenzofuran substituted thiourea compounds and one or more pharmaceutically acceptable carriers, excipients, or diluents.
- compositions may contain an azabenzofuran substituted thiourea compound as the only active agent or may contain a combination of an azabenzofuran substituted thiourea compound and one or more other pharmaceutically active agents.
- the invention also provides methods for treating Hepatitis C viral infections in mammals.
- HAV infectious hepatitis
- HBV homologous serum hepatitis
- hepatitis B surface antigen (HBsAg)
- HBV hepatitis B surface antigen
- hepatitis C virus hepatitis C virus, HCV
- HCV hepatitis C virus
- HCV Hepatitis C virus
- HCV is also a common cause of hepatitis in individuals exposed to blood products. There have been an estimated 150,000 new cases of HCV infection each year in the United States alone during the past decade. [0007] The acute phase of HCV infection is usually associated with mild symptoms. However, evidence suggests that only 15%-20% of the infected people will clear HCV. Among the group of chronically infected people, 10-20 % will progress to life-threatening conditions known as cirrhosis and another 1-5% will develop a liver cancer called hepatocellular carcinoma. Unfortunately, the entire infected population is at risk for these life-threatening conditions because no one can predict which individual will eventually progress to any of them.
- HCV is a small, enveloped, single-stranded positive RNA virus in the Flaviviridae family.
- the genome is approximately 10,000 nucleotides and encodes a single polyprotein of about 3,000 amino acids.
- the polyprotein is processed by host cell and viral proteases into three major structural proteins and several non-structural proteins necessary for viral replication.
- Several different genotypes of HCV with slightly different genomic sequences have since been identified that correlate with differences in response to treatment with interferon alpha.
- HCV replicates in infected cells in the cytoplasm, in close association with the endoplasmic reticulum. Incoming positive sense RNA is released and translation is initiated via an internal initiation mechanism.
- RNA element Internal initiation is directed by a cis-acting RNA element at the 5' end of the genome; some reports have suggested that full activity of this internal ribosome entry site, or IRES, is seen with the first 700 nucleotides, which spans the 5' untranslated region (UTR) and the first 123 amino acids of the open reading frame (ORF). All the protein products of HCV are produced by proteolytic cleavage of a large (approximately 3000 amino acid) polyprotein, carried out by one of three proteases: the host signal peptidase, the viral self-cleaving metalloproteinase, NS2, or the viral serine protease NS3/4A .
- IRES internal ribosome entry site
- NS5B is the viral RNA-dependent RNA polymerase (RDRP) that is responsible for the conversion of the input genomic RNA into a negative stranded copy (complimentary RNA, or cRNA; the cRNA then serves as a template for transcription by NS5B of more positive sense genomic/messenger RNA.
- RDRP viral RNA-dependent RNA polymerase
- Alpha interferon is a host protein that is made in response to viral infections and has natural antiviral activity. These standard forms of interferon, however, are now being replaced by pegylated interferons (peginterferons).
- Peginterferon is alpha interferon that has been modified chemically by the addition of a large inert molecule of polyethylene glycol.
- the optimal regimen appears to be a 24- or 48-week course of the combination of pegylated alpha interferon and the nucleoside Ribavarin, an oral antiviral agent that has activity against a broad range of viruses.
- Ribavarin has little effect on HCV, but adding it to interferon increases the sustained response rate by two- to threefold.
- response rates to the combination interferon/ Ribavarin therapy are moderate, in the range 50-60%, although response rates for selected genotypes of HCV (notably genotypes 2 and 3) are typically higher.
- response rates for selected genotypes of HCV notably genotypes 2 and 3 are typically higher.
- Patients receiving interferon often present with flu-like symptoms.
- Pegylated interferon has been associated with bone marrow suppressive effects.
- alpha interferon has multiple neuropsychiatric effects. Prolonged therapy can cause marked irritability, anxiety, personality changes, depression, and even suicide or acute psychosis.
- the invention provides compounds of Formula 1 (shown below) and includes certain azabenzofuran substituted thioureas and related compounds, which possess antiviral activity.
- the invention provides compounds of Formula 1 that are potent and/ or selective inhibitors of Hepatitis C virus replication.
- the invention also provides pharmaceutical compositions containing one or more compound of Formula 1, or a salt, solvate, or acylated prodrug of such compounds, and one or more pharmaceutically acceptable carriers, excipients, or diluents.
- the invention further comprises methods of treating patients suffering from certain infectious diseases by administering to such patients an amount of a compound of Formula 1 effective to reduce signs or symptoms of the disease. These infectious diseases include viral infections, particularly HCV infections.
- the invention is particularly includes methods of treating human patients suffering from an infectious disease, but also encompasses methods of treating other animals, including livestock and domesticated companion animals, suffering from an infectious disease.
- Methods of treatment include administering a compound of Formula 1 as a single active agent or administering a compound of Formula 1 in combination with on or more other therapeutic agent.
- a method of inhibiting HCV replication in vivo by administering a patient infected with HCV a concentration of a compound or salt of Formula 1 that is sufficient to inhibit HCV replicon replication in vitro, is also disclosed herein.
- the invention includes compounds of Formula 1 :
- Formula 1 and the pharmaceutically acceptable salt thereof carry the definitions set forth below.
- X and are independently O, S, NR, or absent, where R is hydrogen, optionally substituted C C ⁇ alkyl, or optionally substituted (aryl)C 0 -C 4 alkyl.
- V is Ci-Ce alkyl, C -C 6 alkenyl, C 3 -C 7 cycloalkyl, or absent.
- Y is Ci-Ce alkyl, C C 6 alkyl substituted with C 3 -C cycloalkyl, C -C 6 alkenyl, or C 3 - C 7 cycloalkyl, or Y is absent; wherein when V is absent, W is absent.
- a 1 is Nitrogen or CRi;
- a 2 is Nitrogen or CR 2 ;
- a 3 is Nitrogen or CR 3 ; and is Nitrogen or CR 4 ; where 1 or 2 of A l5 A 2 , A 3 , or A is Nitrogen.
- R 1 -R 4 when present, are independently chosen from: hydrogen, halogen, hydroxy, cyano, nitro, amino, acetyl, -NHCO 2 , - NHSO 2 , -C haloalkyl, and C ⁇ -C 2 haloalkoxy, and -C ⁇ alkyl, C 2 -C 6 alkenyl, C 2 -C 6 alkynyl, CrCealkoxy, mono- and di-(Cr C 6 alkyl)amino, C 2 -C 6 alkanoyl, C 1 -C 4 alkylthio, C 1 -C 4 alkylsulfinyl, C 1 -C 4 alkylsulfonyl, C 4 alkylcarboxamide, mono- and di(C 1 -C 6 alkyl)carboxamide, (C 3 -C 8 cycloalkyl)Co-C 2 alkyl, C 2 -C monocyclic heterocycloalkyl,
- R 5 is hydrogen, halogen, hydroxy, amino, nitro, cyano, C 1 -C 4 alkyl, Cr alkoxy, Q- C 2 haloalkyl, or d-C ⁇ aloalkoxy.
- R 6 and R are independently hydrogen, or R 6 and R are independently -C ⁇ alkyl, C 2 -C 6 alkenyl, or C 2 -C 6 alkynyl, each of which is substituted with 0 to 3 substituents independently chosen from halogen, hydroxy, amino, C 1 -C 4 alkoxy, -C ⁇ aloalkyl, and - haloalkoxy, or R 6 and R are joined to form a 5- to 7-membered saturated or mono-unsaturated heterocyclic ring optionally containing one additional heteroatom chosen from N, S, and O, which 5- to 7-membered saturated or mono-unsaturated heterocyclic ring is substituted with 0 to 3 substituents independently chosen from halogen, hydroxy
- Certain compounds of Formula 1 disclosed herein exhibit good activity in an HCV replication assay, such as the HCV replicon assay set forth in Example 5, which follows.
- Preferred compounds of Formula 1 exhibit an EC 50 of about 10 micromolar or less, or more preferably an EC 50 of about 1 micromolar or less; or still more preferably an EC 50 of about 500 nanomolar or less in an HCV replicon assay.
- Formula 1 includes all subformulae thereof.
- Formula 1 includes compounds of Formulae 2-12.
- compounds of Formula 1 are referred to herein as azabenzofuran substituted thioureas.
- azobenzofuran is used to indicate a benzo analogue, a six-membered aromatic ring containing one or more nitrogen atom, fused to a furanyl ring.
- the compounds of Formula 1 may contain one or more asymmetric elements such as stereogenic centers, stereogenic axes and the like, e.g. asymmetric carbon atoms, so that the compounds can exist in different stereoisomeric forms. These compounds can be, for example, racemates or optically active forms. For compounds with two or more asymmetric elements, these compounds can additionally be mixtures of diastereomers.
- the single enantiomers i.e., optically active forms
- the single enantiomers can be obtained by asymmetric synthesis, synthesis from optically pure precursors, or by resolution of the racemates. Resolution of the racemates can also be accomplished, for example, by conventional methods such as crystallization in the presence of a resolving agent, or chromatography, using, for example a chiral HPLC column.
- the invention includes compounds of Formula 1 having all possible isotopes of atoms occurring in the compounds. Isotopes include those atoms having the same atomic number but different mass numbers. By way of general example, and without limitation, isotopes of hydrogen include tritium and deuterium and isotopes of carbon include ⁇ C, 13 C, and 14 C. [0026] Certain compounds are described herein using a general formula that includes variables, e.g. Ar, V, W, X, Y, A 1 -A 4 , and R 5 -R 7 .
- each variable within such a Formula 1 is defined independently of other variables.
- a group is said to be substituted, e.g. with 0-2 R*, then said group may be substituted with up to two R* groups and R* at each occurrence is selected independently from the definition of R*.
- combinations of substituents and/or variables are permissible only if such combinations result in stable compounds.
- aromatic moieties are substituted by an oxo group
- the aromatic ring is replaced by the corresponding partially unsaturated ring.
- a pyridyl group substituted by oxo is a pyridone.
- substituents and/or variables are permissible only if such combinations result in stable compounds or useful synthetic intennediates.
- a stable compound or stable structure is meant to imply a compound that is sufficiently robust to survive isolation from a reaction mixture, and subsequent formulation into an effective therapeutic agent.
- the phrase "optionally substituted" indicates that such groups may either be unsubstituted or substituted at one or more of any of the available positions, typically 1, 2, 3, or 4 positions, by one or more suitable groups such as those disclosed herein.
- Suitable groups that may be present on a "substituted" position include, but are not limited to, e.g., halogen; cyano; hydroxyl; nitro; azido; alkanoyl (such as a C 2 -C 6 alkanoyl group such as acyl or the like); carboxamido; alkyl groups (including cycloalkyl groups, having 1 to about 8 carbon atoms, or 1 to about 6 carbon atoms); alkenyl and alkynyl groups (including groups having one or more unsaturated linkages and from 2 to about 8, or 2 to about 6 carbon atoms); alkoxy groups having one or more oxygen linkages and from 1 to about 8, or from 1 to about 6 carbon atoms; aryloxy such as phenoxy, napthyloxy, and 5,6,7,8-tetrahydronapthyloxy; alkylthio groups including those having one or more thioether linkages and from 1 to about 8 carbon atoms
- a dash (“-") that is not between two letters or symbols is used to indicate a point of attachment for a substituent.
- -(CH 2 )C 3 -C 8 cycloalkyl is attached through carbon of the methylene (CH 2 ) group.
- alkyl includes both branched and straight chain saturated aliphatic hydrocarbon groups, having the specified number of carbon atoms, generally from 1 to about 12 carbon atoms.
- the term C ⁇ - C 8 alkyl as used herein indicates an alkyl group having from 1 to about 8 carbon atoms.
- Co-C n alkyl is used herein in conjunction with another group, for example, (aryl)C 0 -C 4 alkyl
- the indicated group in this case aryl
- aryl is either directly bound by a single covalent bond (C 0 ), or attached by an alkyl chain having the specified number of carbon atoms, in this case from 1 to about 4 carbon atoms.
- alkyl include, but are not limited to, methyl, ethyl, n-propyl, isopropyl, n-butyl, 3- methylbutyl, t-butyl, n-pentyl, and sec-pentyl.
- Alkyl groups described herein typically have from 1 to about 12 carbons atoms.
- alkyl groups are lower alkyl groups, those alkyl groups having from 1 to about 8 carbon atoms, from 1 to about 6 carbon atoms, or from 1 to about 4 carbons atoms e.g. Ci-C 8 , CrC 6 , and C ⁇ -C 4 alkyl groups.
- Alkenyl indicates a straight or branched hydrocarbon chain comprising one or more unsaturated carbon-carbon bonds, which may occur in any stable point along the chain. Alkenyl groups described herein typically have from 2 to about 12 carbons atoms.
- Preferred alkenyl groups are lower alkenyl groups, those alkenyl groups having from 2 to about 8 carbon atoms, e.g.
- alkenyl groups include ethenyl, propenyl, and butenyl groups.
- alkenyl groups include ethenyl, propenyl, and butenyl groups.
- Alkynyl indicates a straight or branched hydrocarbon chain comprising one or more triple carbon-carbon bonds that may occur in any stable point along the chain, such as ethynyl and propynyl. Alkynyl groups described herein typically have from 2 to about 12 carbons atoms. Preferred alkynyl groups are lower alkynyl groups, those alkynyl groups having from 2 to about 8 carbon atoms, e.g.
- Alkoxy indicates an alkyl group as defined above with the indicated number of carbon atoms attached through an oxygen bridge (-O-).
- alkoxy include, but are not limited to, methoxy, ethoxy, n- propoxy, i- propoxy, n-butoxy, 2-butoxy, t-butoxy, n- pentoxy, 2-pentoxy, 3- pentoxy, isopentoxy, neopentoxy, n-hexoxy, 2-hexoxy, 3-hexoxy, and 3- methylpentoxy.
- alkenyloxy indicates an alkenyl group as define above with the indicated number of carbon atoms attached through an oxygen bridge (-O-). Examples of alkenyloxy groups include, but are not limited to, prop-1-enyloxy and but-1-enyloxy.
- alkenyloxy groups include, but are not limited to, prop-1-enyloxy and but-1-enyloxy.
- alkanoyloxy indicates an alkanoyl group as defined above, having the indicated number of carbon atoms, attached through an oxygen (-O-) bridge.
- alkylsulfonamide refers to groups of formula -NHSO 2 (alkyl 1 ) and -N(alkyl 2 ) SO (alkyl 1 ) in which the alkyl], and alkyl 2 groups are independently chosen alkyl groups as defined above having the indicated number of carbon atoms.
- alkylsulfinyl means alkyl-(SO)-, where the alkyl group is an alkyl group as defined above having the defined number of carbon atoms.
- An exemplary alkylsulfinyl group is ethylsulfinyl.
- alkylsulfonyl means alkyl-(SO 2 )-, where the alkyl group is an alkyl group as defined above having the defined number of carbon atoms.
- An exemplary alkylsulfonyl group is methylsulfonyl.
- alkylthio means alkyl-S-, where the alkyl group is an alkyl group as defined above having the defined number of carbon atoms.
- An exemplary alkylthio group is methylthio.
- the alkoxy moiety of the alkoxycarbonyl group has the indicated number of carbon atoms; the carbon of the keto bridge is not included in this number.
- aminoalkyl is an alkyl group as defined herein, having the indicated number of carbon atoms, and substituted with at least one amino substituent (-NH ). When indicated, aminoalkyl groups, like other groups described herein, may be additionally substituted.
- the term "mono- and/ or di-alkylamino” indicates secondary or tertiary alkyl amino groups, wherein the alkyl groups are as defined above and have the indicated number of carbon atoms. The point of attacliment of the alkylamino group is on the nitrogen.
- the alkyl groups are independently chosen.
- mono- and di-alkylamino groups examples include ethylamino, dimethylamino, and methyl-propyl-amino.
- "Mono- and/or dialkylaminoalkyl” groups are mono- and/ or di-alkylamino groups attached through an alkyl linker having the specified number of carbon atoms, for example a di-methylaminoethyl group.
- Tertiary amino substituents may by designated by nomenclature of the form N-R-N- R', indicating that the groups R and R' are both attached to a single nitrogen atom.
- mono- and/ or di-alkylaminoalkyl indicates a mono- and/ or di-alkylamino group as described above attached through an alkyl linker having the specified number of carbon atoms.
- aryl indicates aromatic groups containing only carbon in the aromatic ring or rings. Such aromatic groups may be further substituted with carbon or non-carbon atoms or groups. Typical aryl groups contain 1 or 2 separate, fused, or pendant rings and from 6 to about 12 ring atoms, without heteroatoms as ring members. Where indicated aryl groups may be substituted.
- substitution may include fusion to a 5 to 7-membered saturated cyclic group that optionally contains 1 or 2 heteroatoms independently chosen from ⁇ , O, and S, to form, for example, a 3,4-methylenedioxy-phenyl group.
- Aryl groups include, for example, phenyl, naphthyl, including 1- naphthyl and 2- naphthyl, and bi-phenyl. [0049] In the term "(aryl)alkyl", aryl and alkyl are as defined above, and the point of attachment is on the alkyl group.
- (Aryl)Co-C 4 alkyl indicates an aryl group that is directly attached via a single covalent bond (aryl)C 0 alkyl or attached through an alkyl group having from 1 to about 4 carbon atoms.
- the term (aryl)alkyl encompasses, but is not limited to, benzyl, phenylethyl, and piperonyl.
- Cycloalkyl indicates a monocyclic or multicyclic saturated hydrocarbon ring group, having the specified number of carbon atoms, usually from 3 to about 10 ring carbon atoms.
- Monocyclic cycloalkyl groups typically have from 3 to about 8 carbon ring atoms or from 3 to about 7 carbon ring atoms.
- Multicyclic cycloalkyl groups may have 2 or 3 fused cycloalkyl rings or contain bridged or caged cycloalkyl groups.
- Cycloalkyl substituents may be pendant to a substituted nitrogen or carbon atom, or when bound to a substituted carbon atom that may have two substituents a cycloalkyl group may be attached as a spiro group.
- Examples of cycloalkyl groups include cyclopropyl, cyclobutyl, cyclopentyl, or cyclohexyl, as well as bridged or caged saturated ring groups such as norbornane or adamantane.
- cycloalkyl and alkyl are as defined above, and the point of attachment is on the alkyl group. This term encompasses, but is not limited to, cyclopropylmethyl, cyclohexyhnethyl, and cyclohexylmethyl.
- (Cycloalkyl)C 0 -C 2 alkyl indicates a cycloalkyl group that is directly attached via a single covalent bond (i.e. (cycloalkyl)C 0 alkyl) or attached through an alkyl group having from 1 to about 2 carbon atoms.
- 9- to 10-Memberedbicyclic carbocyclic groups refers to saturated, partially unsaturated, and aromatic ring groups have 2 rings, a total of 9 or 10 ring atoms, with all ring members being carbon. Examples include naphthyl, indanyl, and tetrahydronaphthyl.
- the 9- to 10-membered bicyclic carbocyclic group is a group in which no more than one of the two rings is aromatic.
- Haloalkyl indicates both branched and straight-chain alkyl groups having the specified number of carbon atoms, substituted with 1 or more halogen atoms, generally up to the maximum allowable number of halogen atoms. Examples of haloalkyl include, but are not limited to, trifluoromethyl, difluoromethyl, 2-fluoroethyl, and penta-fluoroethyl.
- "Haloalkoxy” indicates a haloalkyl group as defined above attached through an oxygen bridge.
- Heteroaryl indicates a stable 5- to 7-membered monocyclic aromatic ring which contains from 1 to 3, or preferably from 1 to 2, heteroatoms chosen from N, O, and S, with remaining ring atoms being carbon or a stable bicyclic or tricyclic system containing at least one 5- to 7-membered aromatic ring which contains from 1 to 3, or preferably from 1 to 2, heteroatoms chosen from N, O, and S, with remaining ring atoms being carbon.
- the total number of S and O atoms in the heteroaryl group exceeds 1, these heteroatoms are not adjacent to one another. It is preferred that the total number of S and O atoms in the heteroaryl group is not more than 2. It is particularly preferred that the total number of S and O atoms in the aromatic heterocycle is not more than 1.
- heteroaryl groups include, but are not limited to, oxazolyl, pyranyl, pyrazinyl, pyrazolopyrimidinyl, pyrazolyl, pyridizinyl, pyridyl, pyrimidinyl, pynolyl, quinolinyl, tetrazolyl, thiazolyl, thienylpyrazolyl, thiophenyl, triazolyl, benzof ] oxazolyl, benzofuranyl, benzothiazolyl, benzothiophenyl, benzoxadiazolyl, dihydrobenzodioxynyl, furanyl, imidazolyl, indolyl, and isoxazolyl.
- heterocycloalkyl indicates a saturated monocyclic group containing from 1 to about 3 heteroatoms chosen from N, O, and S, with remaining ring atoms being carbon, or a saturated bicyclic ring system having at least one N, O, or S ring atom with remaining atoms being carbon.
- Monocyclic heterocycloalkyl groups have from 4 to about 8 ring atoms, and more typically have from 5 to 7 ring atoms.
- Bicyclic heterocycloalkyl groups typically have from about five to about 12 ring atoms. In certain instances the size of a heterocycloalkyl groups is indicated by the number of ring carbon atoms the group contains.
- a C 2 -C 7 heterocycloalkyl group contains from 2 to about 7 ring carbon atoms with the remaining ring atoms, up to about 3 per ring, being chosen from N, O, and S.
- Prefened heterocycloalkyl groups include C 2 -C 7 monocyclic heterocycloalkyl groups and C 5 - C 10 bicyclic heterocycloalkyl groups.
- heterocycloalkyl groups include morpholinyl, piperazinyl, piperidinyl, and pyrrolidinyl groups.
- 5- to 7- membered saturated or mono-unsaturated heterocyclic ring indicates a cyclic group having from 5 to 7 ring atoms optionally containing one unsaturated bond, such as an alkenyl or alkynyl bond.
- 5- to 7-membered saturated or mono-unsaturated heterocyclic ring contain from 1 to 3 heteroatoms independently selected from N, O, and S. Examples of such rings, which are saturated include piperazine, piperidine, morpholine, and pynolidine. Examples of such groups which are mono-unsaturated include 1,2,3,6- tetrahydropyridine and 2,3-dihydrooxazole.
- 9- to 10-membered bicyclic heterocyclic groups refers to saturated partially unsaturated and aromatic ring groups have 2 rings, a total of 9 or 10 ring atoms, having 1 to about 4 ring atoms independently chosen from N, S, and O, with remaining ring atoms being carbon. Examples include quinolinyl, dihydroquinolinyl, and indolyl.
- the 9- to 10-membered bicyclic heterocyclic group contains one ring Nitrogen atom, with remaining ring atoms being carbon.
- alkylimino contains an alkyl group as defined above covalently bound to the nitrogen atom of an imino group. When specified the alkyl portion of an alkylimino group may be optionally substituted.
- “Pharmaceutically acceptable salts” includes derivatives of the disclosed compounds wherein the parent compound is modified by making non-toxic acid or base salts thereof, and further refers to pharmaceutically acceptable solvates of such compounds and such salts.
- Examples of pharmaceutically acceptable salts include, but are not limited to, mineral or organic acid salts of basic residues such as amines; alkali or organic salts of acidic residues such as carboxylic acids; and the like.
- the pharmaceutically acceptable salts include the conventional non-toxic salts and the quaternary ammonium salts of the parent compound formed, for example, from non-toxic inorganic or organic acids.
- conventional non-toxic acid salts include those derived from inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phosphoric, nitric and the like; and the salts prepared from organic acids such as acetic, propionic, succinic, glycolic, stearic, lactic, malic, tartaric, citric, ascorbic, pamoic, maleic, hydroxymaleic, phenylacetic, glutamic, benzoic, salicylic, mesylic, esylic, besylic, sulfanilic, 2-acetoxybenzoic, fumaric, toluenesulfonic, methanesulfonic, ethane disulfonic, oxalic, isethionic, HOOC-(CH 2 ) n -COOH where n is 0-4, and the like.
- inorganic acids such as hydrochloric, hydrobromic, sulfuric, sulfamic, phospho
- the pharmaceutically acceptable salts of the present invention can be synthesized from a parent compound, a basic or acidic moiety, by conventional chemical methods.
- such salts can be prepared by reacting free acid forms of these compounds with a stoichiometric amount of the appropriate base (such as Na, Ca, Mg, or K hydroxide, carbonate, bicarbonate, or the like), or by reacting free base forms of these compounds with a stoichiometric amount of the appropriate acid.
- a stoichiometric amount of the appropriate base such as Na, Ca, Mg, or K hydroxide, carbonate, bicarbonate, or the like
- Such reactions are typically carried out in water or in an organic solvent, or in a mixture of the two.
- non-aqueous media like ether, ethyl acetate, ethanol, isopropanol, or acetonitrile are prefened, where practicable.
- prodrugs includes any compounds that become compounds of Formula 1 when administered to a mammalian subject, e.g., upon metabolic processing of the prodrug.
- prodrugs include, but are not limited to, acetate, formate and benzoate and like derivatives of functional groups (such as alcohol or amine groups) in the compounds of Formula 1.
- a therapeutically effective amount of a compound of this invention means an amount effective, when administered to a human or non-human patient, to provide a therapeutic benefit such as an amelioration of symptoms, e.g., an amount effective to decrease the symptoms of a viral infection, and preferably an amount sufficient to reduce the symptoms of an HCV infection.
- a therapeutically effective amount of a compound is also an amount sufficient to prevent a significant increase or significantly reduce the detectable level of virus or viral antibodies in the patient's blood, serum, or tissues.
- a significant increase or reduction in the detectable level of virus or viral antibodies is any detectable change that is statistically significant in a standard parametric test of statistical significance such as Student's T-test, where p ⁇ 0.05.
- a "replicon" as used herein includes any genetic element, for example, a plasmid, cosmid, bacmid, phage or virus, that is capable of replication largely under its own control.
- a replicon may be either RNA or DNA and may be single or double stranded.
- Nucleic acid or a “nucleic acid molecule” as used herein refers to any DNA or RNA molecule, either single or double stranded and, if single stranded, the molecule of its complementary sequence in either linear or circular form.
- nucleic acid molecules a sequence or structure of a particular nucleic acid molecule can be described herein according to the normal convention of providing the sequence in the 5' to 3' direction.
- Ar is aryl or heteroaryl substituted with 0 to 5 substituents independently chosen from: (iii) halogen, hydroxy, cyano, nitro, oxo, -C ⁇ aloalkyl, and C haloalkoxy, and (iv) CrC 8 alkyl, C 2 -C 8 alkenyl, -Csalkynyl, Ci-Csalkoxy, C 2 -C 8 alkenyloxy, mono- and di-(C 1 -C 8 alkyl)amino, mono- and di-(C 1 -C 4 alkyl)aminoC 1 -C 4 alkyl, C 2 -C 8 alkanoyl, C 2 - Csalkanoyloxy, C 1 -C 8 alkoxycarbonyl, mono- and di-(C 1 -C 8 alkyl)carboxamide, (C 3 - C cycloalkyl)carboxamide
- the invention includes compounds and salts of Formula 1 in which X is oxygen and Y is -CH 2 -.
- the invention includes compounds and salts of Formula 1 in which X is oxygen and Y is -CH CH 2 -.
- the invention also includes compounds and salts of Formula 1 in which X and Y are absent.
- the invention includes compounds and salts of Formula 1 in which V and W are absent.
- the invention includes compounds and salts of Formula 1 in which V is C ⁇ - C 2 alkyl and W is absent.
- the invention includes compounds and salts of Formula 1, in which is Nitrogen; A 2 is CR 2 ; A 3 is CR 3 ; and 4 is CR 4 , e.g., compounds and salts of Formula 1-A:
- the invention also includes compounds of Formula 1, in which Ai is CR1, A is Nitrogen; A 3 is CR 3 ; and A 4 is CR 4 , e.g., compounds and salts of Formula 1-B:
- the invention includes compounds of Formula 1, in which A 1 is CRi; A 2 is CR 2 ; A 3 is Nitrogen; and A 4 is CR , e.g., compounds and salts of Formula 1-C: Formula 1-C.
- the invention includes compounds of Formula 1, in which Ai is CR 1? A 2 is CR 2 ; A 3 is CR 3 ; and A 4 is Nitrogen, e.g., compounds and salts of Formula 1-D:
- the invention includes compounds of Formula 1, in which A 1 is Nitrogen; A 2 is CR 2 ; A 3 is Nitrogen; and is CR 4 ; e.g., compounds and salts of Formula 1-E.
- the invention includes compounds of Formula 1, in which A 1 is CR ⁇ Ar 2 is
- A is CR 3 ; and A is Nitrogen; e.g., compounds and salts of Formula 1-F.
- the invention includes compounds of Formula 1, in which A ⁇ is Nitrogen; A 2 is CR ; A 3 is CR ; and A 4 is Nitrogen; e.g., compounds and salts of Formula 1-G.
- the invention includes compounds and salts of Fonnula 1, wherein Ri-R ⁇ when present, are independently chosen from (i) hydrogen, halogen, hydroxy, cyano, nitro, amino, acetyl, Cr haloalkyl, and - C 2 haloalkoxy, and (ii) C 1 -C 4 alkyl, C 1 -C 4 alkoxy, mono- and di-(C 1 -C alkyl)amino, C 2 -C 4 alkanoyl, - C 4 alkylthio, C 3 -C cycloalkyl, piperidinyl, piperazinyl, morpholinyl, pynolidinyl, phenyl, pyridyl, and pyrimidinyl; each of which is substituted with 0 to 5 substituents independently chosen from halogen, hydroxy, C 1 -C 4 alkyl, Ci-C 4 alkoxy, mono- and di-(C 1 -
- the invention also includes compounds and salts of Formula 1, wherein R 1 -R 4 when present, are independently chosen from hydrogen, halogen, hydroxy, cyano, nitro, amino, acetyl, trifluoromethyl, trifluoromethoxy, C 1 -C 4 alkyl, C 1 -C 4 alkoxy, mono- and di-(Cr C 4 alkyl) amino, C 2 -C 4 alkanoyl, C 1 -C 4 alkylthio, C 3 -C 7 cycloalkyl, piperidinyl, piperazinyl, morpholinyl, pynolidinyl, phenyl, pyridyl, and pyrimidinyl.
- R 1 -R 4 when present, are independently chosen from hydrogen, halogen, hydroxy, cyano, nitro, amino, acetyl, trifluoromethyl, trifluoromethoxy, C 1 -C 4 alkyl, C 1 -C 4 alkoxy
- the invention also includes compounds and salts of Formula 1, wherein Ri-Rj when present, are independently chosen from hydrogen, halogen, cyano, nitro, amino, acetyl, trifluoromethyl, trifluoromethoxy, C 1 -C 2 alkyl, C 1 -C alkoxy, C 3 -C cycloalkyl, piperidinyl, pynolidinyl, phenyl, and pyridyl.
- Ri-Rj when present, are independently chosen from hydrogen, halogen, cyano, nitro, amino, acetyl, trifluoromethyl, trifluoromethoxy, C 1 -C 2 alkyl, C 1 -C alkoxy, C 3 -C cycloalkyl, piperidinyl, pynolidinyl, phenyl, and pyridyl.
- the invention includes compounds and salts of Formula 1, wherein R 5 is hydrogen or methyl.
- the invention includes compounds and salts of Formula 1, wherein R 6 and R 7 are independently hydrogen, or C 1 -C alkyl, C 2 -C 4 alkenyl, or C 2 -C 4 alkynyl, each of which is substituted with 0 to 3 substituents independently chosen from halogen, hydroxy, amino, - C 4 alkoxy, C ⁇ -C 2 haloalkyl, and Ci-C 2 haloalkoxy.
- the invention also includes compounds and salts of Formula 1, wherein R 6 and R are independently hydrogen, methyl, or ethyl.
- the invention includes compounds and salts of Formula 1, wherein Ar is phenyl, pyridyl, pyrimidinyl, thienyl, pynolyl, furanyl, pyrazolyl, imidazolyl, thiazolyl, triazolyl, thiadiazolyl, oxazolyl, isoxazolyl, benzofuranyl, benzothiazolyl, benzothiophenyl, benzoxadiazolyl, benzo[d]oxazolyl, dihydrobenzodioxynyl, indolyl, pyrazolopyrimidinyl, thienylpyrazolyl, or benzopyranyl, each of which is substituted with 0 to 5 substituents independently chosen from (iii), (iv), and (v).
- the invention also includes compounds and salts of Formula 1, wherein Ar is phenyl or pyridyl; each of which is substituted with 0 to 5 substituents independently chosen from (iii), (iv), and (v).
- Ar is phenyl or pyridyl; each of which is substituted with 0 to 5 substituents independently chosen from (iii), (iv), and (v).
- (iii) represents halogen, hydroxy, cyano, nitro, oxo, C 1 -C 2 haloalkyl, and C 2 haloalkoxy
- (iv) represents CrCsalkyl, C 2 -C 8 alkenyl, C 2 -C 8 alkynyl, -Cgalkoxy, C 2 - Csalkenyloxy, mono- and di-(C 1 -C 8 alkyl)amino, mono- and di-(C 1 -C 4 alkyl)aminoC 1 -C 4 al
- Ai -A carry the definitions set forth above.
- a 8 , A 8 ', A , and A 9 ' are independently Nitrogen or CH, where 0, 1, or 2 of A 8 , A 8 ', A 9 , and A 9 ' are Nitrogen.
- R 5 is hydrogen or methyl.
- R 6 and R are hydrogen or methyl.
- Rio is C ⁇ -C 6 alkyl.
- R 11 represents 0 to 3 substituents independently chosen from halogen, hydroxy, cyano, C ⁇ -C 4 alkyl, C 1 -C 4 alkoxy, mono- and di-(C ⁇ -C 4 alkyl)amino, C ⁇ -C 2 haloalkyl, and Ci- C 2 haloalkoxy.
- R ⁇ may replace the hydrogen atom so that A 8 is CR ⁇ .
- R ⁇ 2 represents 0 to 3 substituents independently chosen from halogen, hydroxy, cyano, C 1 -C 4 alkyl, - alkoxy, mono- and di-(Ci-C 4 alkyl)amino, C 1 -C 2 haloalkyl, and C ⁇ - C 2 haloalkoxy; or in compounds of Formula 4 to 7 and 9 to 10 R 12 may also represent a 5- to 7-membered oxygen containing ring containing 1 or 2 oxygen atoms and fused to the phenyl to which it is attached.
- the 5- to 7 membered oxygen-containing ring is a heterocycloalkyl ring.
- R 13 and R ⁇ 4 are independently hydrogen or methyl, and m is 0, 1, or 2.
- R 15 represents C -C 6 alkoxy or C 3 -C 6 alkyl, each of which is substituted with 0 to 3 substituents independently chosen from halogen, hydroxy, -Csal oxy, and mono- and di- Ri 6 is hydrogen or C ⁇ -C 4 alkyl.
- J is Nitrogen or CH; and Q is O, NR ⁇ 6 , or CH 2 .
- Q is absent, O, -CRi 3 R ⁇ 4 -, or NR ⁇ 6
- R a is a 9- to 10 membered bicyclic carbocyclic group or a 9- to 10- membered bicyclic heterocyclic group containing 1 nitrogen atom, each of which is substituted with 0 to 3 substituents independently chosen from halogen, hydroxy, cyano, C 1 -C 2 alkyl, C 1 -C 2 alkoxy, trifluoromethyl and trifluoromethoxy.
- R a is indanyl or quinolinyl, each each of which is substituted with 0 to 3 substituents independently chosen from halogen, hydroxy, cyano, C 1 -C 2 alkyl, C 1 -C 2 alkoxy, trifluoromethyl and trifluoromethoxy.
- R 12 is a 5- to 7-membered ring containing 1 or 2 oxygen atoms and fused to the phenyl to which it is attached include the following:
- the invention includes compounds and salts of Formulae 2-12, above, wherein when present, are independently chosen from hydrogen, halogen, hydroxy, cyano, nitro, amino, acetyl, trifluoromethyl, trifluoromethoxy, C ⁇ -C 4 alkyl, Ci-C 4 alkoxy, mono- and di-(C 1 -C 4 alkyl)amino, C -C 4 alkanoyl, C ⁇ -C 4 alkylthio, C 3 -C cycloalkyl, piperidinyl, piperazinyl, morpholinyl, pynolidinyl, phenyl, pyridyl, and pyrimidinyl.
- the invention further includes compounds and salts of Formulae 2-12, above, wherein when present, are independently chosen from hydrogen, halogen, cyano, nitro, amino, acetyl, trifluoromethyl, trifluoromethoxy, C ⁇ -C 2 alkyl, C ⁇ -C 2 alkoxy, C 3 -C cycloalkyl, piperidinyl, pynolidinyl, phenyl, and pyridyl.
- the invention includes compounds and salts of Formulae 2-12, above, in which X and Y are both absent. [0098] The invention also includes compounds and salts of Formulae 2-12 in which R 5 , e, and R are all hydrogen. [0099] Compounds of Formula 1 having any combination of definitions for the variables, e.g. A 1 -A 4 , R 5) Re, R 7 , V, W, X, Y, and Ar set forth herein are included within the scope of the invention.
- Ar is phenyl or pyridyl; each of which is substituted with 0 to 5 substituents independently chosen from (iii), (iv), and (v).
- Prefened compounds of Formula 1 exhibit an EC 50 of about 10 micromolar or less, or more preferably an EC 5 0 of about 1 micromolar or less; or an EC50 of about 500 nanomolar or less in an HCV replicon based assay of HCV replication, such as the assay provided in Example 5.
- Prefened compounds of Formula 1 will have certain pharmacological properties. Such properties include, but are not limited to oral bioavailability, low toxicity, low serum protein binding and desirable in vitro and in vivo half-lives.
- the invention includes packaged pharmaceutical formulations. Such packaged formulations include a pharmaceutical composition containing one or more compounds or salts of Formula 1 in a container and instructions for using the composition to treat a patient suffering from Hepatitis C infection (HCV infection).
- HCV infection Hepatitis C infection
- Compounds and salts of Formula 1 can be administered as the neat chemical, but are preferably administered as a pharmaceutical composition or formulation. Accordingly, the invention provides pharmaceutical formulations comprising a compound or pharmaceutically acceptable salt of Formula 1, together with one or more pharmaceutically acceptable canier, excipients, adjuvant, diluent, or other ingredient.
- Compounds of general Formula 1 maybe administered orally, topically, parenterally, by inhalation or spray, sublingually, transdermally, via buccal administration, rectally, as an ophthalmic solution, or by other means, in dosage unit formulations containing conventional non-toxic pharmaceutically acceptable carriers, excipients, adjuvants, and vehicles.
- compositions of the invention may contain a pharmaceutically acceptable carrier, one or more compatible solid or liquid filler diluents or encapsulating substances, which are suitable for administration to an animal.
- Carriers must be of sufficiently high purity and sufficiently low toxicity to render them suitable for administration to the animal being treated.
- the carrier can be inert or it can possess pharmaceutical benefits of its own.
- the amount of carrier employed in conjunction with the compound is sufficient to provide a practical quantity of material for administration per unit dose of the compound.
- Exemplary phannaceutically acceptable carriers or components thereof are sugars, such as lactose, glucose and sucrose; starches, such as corn starch and potato starch; cellulose and its derivatives, such as sodium carboxymethyl cellulose, ethyl cellulose, and methyl cellulose; powdered tragacanth; malt; gelatin; talc; solid lubricants, such as stearic acid and magnesium stearate; calcium sulfate; vegetable oils, such as peanut oil, cottonseed oil, sesame oil, olive oil, and corn oil; polyols such as propylene glycol, glycerine, sorbitol, mannitol, and polyethylene glycol; alginic acid; emulsifiers, such as the TWEENS; wetting agents, such sodium lauryl sulfate; coloring agents; flavoring agents; tableting agents, stabilizers; antioxidants; preservatives; pyrogen-free water; isotonic saline
- pharmaceutically acceptable carriers for systemic administration include sugars, starches, cellulose and its derivatives, malt, gelatin, talc, calcium sulfate, vegetable oils, synthetic oils, polyols, alginic acid, phosphate buffer solutions, emulsifiers, isotonic saline, and pyrogen-free water.
- Prefened carriers for parenteral administration include propylene glycol, ethyl oleate, pynolidone, ethanol, and sesame oil.
- Optional active agents may be included in a pharmaceutical composition, which do not substantially interfere with the activity of the compound of the present invention.
- Effective concentrations of one or more of the compounds of the invention including pharmaceutically acceptable salts, esters or other derivatives thereof are mixed with a suitable pharmaceutical carrier, excipients, adjuvant, or vehicle, h instances in which the compounds exhibit insufficient solubility, methods for solubilizing compounds may be used. Such methods are known to those of skill in this art, and include, but are not limited to, using cosolvents, such as dimethylsulfoxide (DMSO), using surfactants, such as Tween, or dissolution in aqueous sodium bicarbonate. Derivatives of the compounds, such as salts of the compounds or prodrugs of the compounds may also be used in formulating effective pharmaceutical compositions.
- cosolvents such as dimethylsulfoxide (DMSO)
- surfactants such as Tween
- the resulting mixture may be a solution, suspension, emulsion or the like.
- the form of the resulting mixture depends upon a number of factors, including the intended mode of administration and the solubility of the compound in the chosen carrier or vehicle.
- the effective concentration sufficient for ameliorating the symptoms of the disease, disorder or condition treated may be empirically determined.
- the pharmaceutical compositions containing compounds of general Formula 1 may be in a form suitable for oral use, for example, as tablets, troches, lozenges, aqueous or oily suspensions, dispersible powders or granules, emulsions, hard or soft capsules, or syrups or elixirs.
- compositions intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions and such compositions may contain one or more agents, such as sweetening agents, flavoring agents, coloring agents and preserving agents, in order to provide pharmaceutically elegant and palatable preparations.
- agents such as sweetening agents, flavoring agents, coloring agents and preserving agents, in order to provide pharmaceutically elegant and palatable preparations.
- Oral formulations contain between 0.1 and 99% of a compound of the invention and usually at least about 5% (weight %) of a compound of the present invention. Some embodiments contain from about 25% to about 50% or from 5% to 75 % of a compound of invention.
- Liquids formulations [0113] Compounds of the invention can be incorporated into oral liquid preparations such as aqueous or oily suspensions, solutions, emulsions, syrups, or elixirs, for example. Moreover, formulations containing these compounds can be presented as a dry product for constitution with water or other suitable vehicle before use.
- Such liquid preparations can contain conventional additives, such as suspending agents (e.g., sorbitol syrup, methyl cellulose, glucose/sugar, syrup, gelatin, hydroxyethyl cellulose, carboxymethyl cellulose, aluminum stearate gel, and hydrogenated edible fats), emulsifying agents (e.g., lecithin, sorbitan monsoleate, or acacia), non-aqueous vehicles, which can include edible oils (e.g., almond oil, fractionated coconut oil, silyl esters, propylene glycol and ethyl alcohol), and preservatives (e.g., methyl or propyl p-hydroxybenzoate and sorbic acid).
- suspending agents e.g., sorbitol syrup, methyl cellulose, glucose/sugar, syrup, gelatin, hydroxyethyl cellulose, carboxymethyl cellulose, aluminum stearate gel, and hydrogenated edible fats
- emulsifying agents e.g.
- Orally administered compositions also include liquid solutions, emulsions, suspensions, powders, granules, elixirs, tinctures, syrups, and the like.
- the pharmaceutically acceptable carriers suitable for preparation of such compositions are well known in the art.
- Oral formulations may contain preservatives, flavoring agents, sweetening agents, such as sucrose or saccharin, taste-masking agents, and coloring agents.
- Typical components of carriers for syrups, elixirs, emulsions and suspensions include ethanol, glycerol, propylene glycol, polyethylene glycol, liquid sucrose, sorbitol and water.
- Syrups and elixirs may be fonnulated with sweetening agents, for example glycerol, propylene glycol, sorbitol or sucrose.
- Such formulations may also contain a demulcent.
- suspending agents include methylcellulose, sodium carboxymethyl cellulose, AVICEL RC-591, tragacanth and sodium alginate; typical wetting agents include lecithin and polysorbate 80; and typical preservatives include methyl paraben and sodium benzoate.
- Aqueous suspensions contain the active material(s) in admixture with excipients suitable for the manufacture of aqueous suspensions.
- excipients are suspending agents, for example sodium carboxymethylcellulose, methylcellulose, hydropropylmethylcellulose, sodium alginate, polyvinylpynolidone, gum tragacanth and gum acacia; dispersing or wetting agents; may be a naturally-occurring phosphatide, for example, lecithin, or condensation products of an alkylene oxide with fatty acids, for example polyoxyethylene stearate, or condensation products of ethylene oxide with long chain aliphatic alcohols, for example heptadecaethyleneoxycetanol, or condensation products of ethylene oxide with partial esters derived from fatty acids and a hexitol such as polyoxyethylene sorbitol substitute, or condensation products of ethylene oxide with partial esters derived from fatty acids and hexitol anhydrides, for example polyethylene sorbitan substitute.
- phosphatide for example, lecithin
- condensation products of an alkylene oxide with fatty acids for example polyoxyethylene stearate,
- the aqueous suspensions may also contain one or more preservatives, for example ethyl, or n- propyl p-hydroxybenzoate.
- Oily suspensions may be formulated by suspending the active ingredients in a vegetable oil, for example peanut oil, olive oil, sesame oil or coconut oil, or in a mineral oil such as liquid paraffin.
- the oily suspensions may contain a thickening agent, for example beeswax, hard paraffin or cetyl alcohol. Sweetening agents such as those set forth above, and flavoring agents may be added to provide palatable oral preparations. These compositions may be preserved by the addition of an anti-oxidant such as ascorbic acid.
- compositions of the invention may also be in the form of oil- in-water emulsions.
- the oily phase may be a vegetable oil, for example olive oil or peanut oil, or a mineral oil, for example liquid paraffin or mixtures of these.
- Suitable emulsifying agents may be naturally-occurring gums, for example gum acacia or gum tragacanth, naturally-occvtrring phosphatides, for example soy bean, lecithin, and esters or partial esters derived from fatty acids and hexitol, anhydrides, for example sorbitan monoleate, and condensation products of the said partial esters with ethylene oxide, for example polyoxyethylene sorbitan monoleate.
- Dispersible powders [0120] Dispersible powders and granules suitable for preparation of an aqueous suspension by the addition of water provide the active ingredient in admixture with a dispersing or wetting agent, suspending agent and one or more preservatives. Suitable dispersing or wetting agents and suspending agents are exemplified by those already mentioned above.
- Tablets and Capsules typically comprise conventional pharmaceutically compatible adjuvants as inert diluents, such as calcium carbonate, sodium carbonate, mannitol, lactose and cellulose; binders such as starch, gelatin and sucrose; disintegrants such as starch, alginic acid and croscarmelose; lubricants such as magnesium stearate, stearic acid and talc. Glidants such as silicon dioxide can be used to improve flow characteristics of the powder mixture. Coloring agents, such as the FD&C dyes, can be added for appearance.
- inert diluents such as calcium carbonate, sodium carbonate, mannitol, lactose and cellulose
- binders such as starch, gelatin and sucrose
- disintegrants such as starch, alginic acid and croscarmelose
- lubricants such as magnesium stearate, stearic acid and talc.
- Glidants such as silicon dioxide can be
- Sweeteners and flavoring agents such as aspartame, saccharin, menthol, peppermint, and fruit flavors, are useful adjuvants for chewable tablets.
- Capsules typically comprise one or more solid diluents disclosed above. The selection of carrier components often depends on secondary considerations like taste, cost, and shelf stability.
- Such compositions may also be coated by conventional methods, typically with pH or time-dependent coatings, such that the subject compound is released in the gastrointestinal tract in the vicinity of the desired topical application, or at various times to extend the desired action.
- Such dosage forms typically include, but are not limited to, one or more of cellulose acetate phthalate, polyvinylacetate phthalate, hydroxypropyl methylcellulose phthalate, ethyl cellulose, Eudragit coatings, waxes and shellac.
- Formulations for oral use may also be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin, or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, for example peanut oil, liquid paraffin or olive oil.
- compositions maybe in the form of a sterile injectable aqueous or oleaginous suspension.
- This suspension may be formulated according to the known art using those suitable dispersing or wetting agents and suspending agents that have been mentioned above.
- the sterile injectable preparation may also be sterile injectable solution or suspension in a non-toxic parentally acceptable diluent or solvent, for example as a solution in 1,3-butanediol.
- acceptable vehicles and solvents that may be employed are water, Ringer's solution, and isotonic sodium chloride solution.
- sterile, fixed oils are conventionally employed as a solvent or suspending medium.
- compositions for parenteral administration may be administered parenterally in a sterile medium.
- Parenteral administration includes subcutaneous injections, intravenous, intramuscular, intrathecal injection or infusion techniques.
- the drug depending on the vehicle and concentration used, can either be suspended or dissolved in the vehicle.
- adjuvants such as local anesthetics, preservatives and buffering agents can be dissolved in the vehicle.
- the canier comprises at least about 90% by weight of the total composition.
- Compounds of Formula 1 may also be administered in the form of suppositories for rectal administration of the drug.
- These compositions can be prepared by mixing the drug with a suitable non- irritating excipient that is solid at ordinary temperatures but liquid at the rectal temperature and will therefore melt in the rectum to release the drug.
- a suitable non- irritating excipient that is solid at ordinary temperatures but liquid at the rectal temperature and will therefore melt in the rectum to release the drug.
- Such materials are cocoa butter and polyethylene glycols.
- Topical formulations Compounds of the invention may be formulated for local or topical application, such as for topical application to the skin and mucous membranes, such as in the eye, in the form of gels, creams, and lotions and for application to the eye or for intracistemal or intraspinal application.
- Topical compositions of the present invention may be in any form including, for example, solutions, creams, ointments, gels, lotions, milks, cleansers, moisturizers, sprays, skin patches, and the like. [0128] Such solutions maybe formulated as 0.01%) -10% isotonic solutions, pH about 5-7, with appropriate salts. Compounds of the invention may also be formulated for transdermal admimstration as a transdermal patch.
- Topical compositions containing the active compound can be admixed with a variety of carrier materials well known in the art, such as, for example, water, alcohols, aloe vera gel, allantoin, glycerine, vitamin A and E oils, mineral oil, propylene glycol, PPG-2 myristyl propionate, and the like.
- carrier materials such as, for example, water, alcohols, aloe vera gel, allantoin, glycerine, vitamin A and E oils, mineral oil, propylene glycol, PPG-2 myristyl propionate, and the like.
- Other materials suitable for use in topical carriers include, for example, emollients, solvents, humectants, thickeners and powders.
- Emollients such as stearyl alcohol, glyceryl monoricinoleate, glyceryl monostearate, propane- 1,2-diol, butane-l,3-diol, mink oil, cetyl alcohol, iso-propyl isostearate, stearic acid, iso-butyl palmitate, isocetyl stearate, oleyl alcohol, isopropyl laurate, hexyl laurate, decyl oleate, octadecan-2-ol, isocetyl alcohol, cetyl palmitate, dimethylpolysiloxane, di-n-butyl sebacate, iso-propyl myristate, iso-propyl palmitate, isopropyl stearate, butyl stearate, polyethylene glycol
- the compounds of the invention may also be topically administered in the form of liposome delivery systems, such as small unilamellar vesicles, large unilamellar vesicles, and multilamellar vesicles.
- Liposomes can be formed from a variety of phospholipids, such as cholesterol, stearylamine or phosphatidylcholines.
- compositions useful for attaining systemic delivery of the subject compounds include sublingual, buccal and nasal dosage fonns.
- Such compositions typically comprise one or more of soluble filler substances such as sucrose, sorbitol and mannitol, and binders such as acacia, microcrystalline cellulose, carboxymethyl cellulose and hydroxypropyl methylcellulose. Glidants, lubricants, sweeteners, colorants, antioxidants and flavoring agents disclosed above may also be included.
- compositions for inhalation typically can be provided in the form of a solution, suspension or emulsion that can be administered as a dry powder or in the form of an aerosol using a conventional propellant (e.g., dichlorodifluoromethane or trichlorofluoromethane) .
- a conventional propellant e.g., dichlorodifluoromethane or trichlorofluoromethane
- compositions of the present invention may also optionally comprise an activity enhancer.
- the activity enhancer can be chosen from a wide variety of molecules that function in different ways to enhance antimicrobial effects of compounds of the present invention. Particular classes of activity enhancers include skin penetration enhancers and absorbtion enhancers.
- Pharmaceutical compositions of the invention may also contain additional active agents can be chosen from a wide variety of molecules, which can function in different ways to enhance the antimicrobial or therapeutic effects of a compound of the present invention. These optional other active agents, when present, are typically employed in the compositions of the invention at a level ranging from about 0.01% to about 15%. Some embodiments contain from about 0.1% to about 10% by weight of the composition. Other embodiments contain from about 0.5% to about 5% by weight of the composition.
- the invention includes packaged pharmaceutical formulations.
- packaged formulations include a pharmaceutical composition containing one or more compounds or salts of Formula 1 in a container and instructions for using the composition to treat an animal (typically a human patient) suffering from a microorganism infection) or prevent a microorganism infection in an animal.
- an animal typically a human patient
- the compounds of the invention can be administered alone or as mixtures, and the compositions may further include additional drugs or excipients as appropriate for the indication.
- the invention includes methods of treating viral infections, particularly HCV infections, by administering an effective amount of one or more compounds of Formula 1 to patient suffering from a viral infection.
- An effective amount of a compound of Formula 1 may be an amount sufficient to reduce the symptoms of viral infection.
- an effective amount of a compound of Fonnula 1 may be an amoxmt sufficient to significantly reduce the amount of virus or viral antibodies detectable in a patient's tissues or bodily fluids.
- Methods of treatment include administering an amount of a compound of Formula 1 sufficient to reduce or relieve the jaundice, fatigue, dark urine, abdominal pain, loss of appetite, and nausea associated with HCV infection.
- Compounds of Fonnula 1 are thought to ameliorate the HCV disease process by virtue of their inhibition of the replication of the Hepatitis C virus.
- the compounds provided herein may be virucidal, in that they actually kill the active virus, in addition to independently inhibiting viral replication.
- the provided compounds may also function through mechanisms that involve a combination of virucidal activity and inhibition of replication.
- Methods of treatment encompassed by the invention include administering a compound of Formula 1 as the sole active agent and administering a compound of Formula 1 together with one or more other active agents, such another antiviral agent, particularly an anti- viral agent effective against HCV infection.
- the invention includes administering one or more compounds of Formula 1 together with Peg-interferon, Peg-interferon alpha 2b, ribavarin (REBETOL), natural interferon, Albuferon, interferon-alpha-2b recombinant (LNTRON) interferon beta- la, IL-10, interferon gamma-lb, AMANTADINE, or ZADAXIM.
- Methods of Hepatitis C infection treatment particularly include administering a compound of Formula 1 as the sole active agent and combination methods in which a compound of Formula 1 is administered in combination with ribavarin and/ or an interferon, such as peg- interferon, or interferon-alpha-2b recombinant.
- Methods of treatment also include inhibiting HCV replication in vivo, in a patient infected with HCV, by administering a sufficient concentration of a compound of Formula 1 to inhibit HCV replicon replication in vitro.
- sufficient concentration of a compound administered to the patient is meant the concentration of the compound available in the patient's system to combat the infection. Such a concentration by be ascertained experimentally, for example by assaying blood concentration of the compound, or theoretically, by calculating bioavailability.
- Dosage levels of the order of from about 0.1 mg to about 140 mg per kilogram of body weight per day are useful in the treatment of the above-indicated conditions (about 0.5 mg to about 7 g per patient per day).
- Dosage unit forms will generally contain between from about 1 mg to about 500 mg of active agent.
- Frequency of dosage may also vary depending on the compound used and the particular disease treated. However, for treatment of most infectious diseases, a dosage regimen of 4 times daily or less is prefened and a dosage regimen of 1 or 2 times daily or even less frequent is particularly prefened.
- the reaction to form the acid chloride is generally carried out in a solvent. Suitable solvents in this case are inert organic solvents that do not change under the reaction conditions.
- ethers such as diethyl ether or tetrahydrofuranyl, or tertiary butyl methyl ether
- halogenohydrocarbons such as dichloromethane, trichloromethane, tetrachloromethane, 1,2-dichloroethane, trichloro ethane, texrachloroethane, 1,2-dichloroethane or trichloroethylene
- hydrocarbons such as benzene, xylene, toluene, hexane, heptane, cyclohexane or mineral oil fractions, nitromethane, or acetonixrile.
- Reaction of the acid chloride with ammonium or potassium thiocyanate is typically carried out in a solvent in which the inorganic thiocyanate is moderately soluble.
- water may be added to increase solubility. The percentage of water added may vary from one percent to 90 percent, with 50 % (v/v) typically being most prefened.
- Other alkali thiocyanates such as lithium thiocyanates, may be employed. Lithium thiocyanate has increased solubility in tetrahydrofuran and therefore may permit the use of smaller amounts of aqueous components. Cesium, rubidium, strontium, and barium can all be used as counter ions to the thiocyanate, as is well known to one normally skilled in the art.
- Furo[3,2-c]pyridine-2-carbonyl chloride (161 mg, 1 mmol) is added to a solution of ammonium thiocyanate (200 mg, about 3 mmol) in acetone (5 ml) and stined at room temperature for 1 hoxir. Butyl 4-aminobenzoate (180 mg, 0.93 mmol) is added to the reaction mixture. Stirring is continued overnight at room temperature. Solvent is evaporated to dryness and the residue diluted with 10% aqueous NaHCO 3 . The product is filtered, washed with water and methanol, and dried.
- the reaction mixture is diluted with icy water (150 ml) and extracted with benzene (70 ml X 3). The organic layer is dried with anhydrous sodium sulfate, filtered, and concentrated to a small volume ( ⁇ 60 ml).
- the resulting azide compound (5) in benzene is added into a diphenylmethane (40 ml) and tributylamine (7 ml) solution pre-heated to 180 °C. The addition should be controlled so that the temperature doesn't drop below 170 °C. After the addition is complete ( ⁇ 2.5h), the reaction mixture is cooled to room temperature and allowed to stand over night.
- Furopyridine (8, 0.71 g, 5.96 mmol) is treated with n-butyl lithium (2.5M in hexane, 2.86 ml, 7.15 mmol) in anhydrous THF (30 ml) at -78 °C. After 30 minutes, dry carbon dioxide is passed through the reaction mixture and the temperature is allowed to rise gradually to room temperature in 3 hours. The mixture is concentrated to dryness. The residue is dissolved in water ( ⁇ 10 ml) and extracted with ethyl acetate. The aqueous layer is acidified to pH ⁇ 3 with IN HCl and kept in the refrigerator over night.
- azabenzofuran carboxylic acid as a white powder (9)
- Azabenzofuran carboxylic acid (8, 0.054 g, 0.33 mmol) is suspended in DCM (2 ml) and treated with oxalyl chloride (0.058 ml, 0.66 mmol) in the presence DMF (1 drop).
- the reaction is stined at 0 °C for 2 hours xmtil no more bubbles evolve.
- the reaction mixture is concentrated to dryness to yield acid chloride 10, which is treated with ammonium thiocyante (0.05 g, 0.66 mmol) in anhydrous acetone (3 ml) at room temperature for 1 hours.
- EXAMPLE 4 PREPARATION OF ADDITIONAL COMPOUNDS OF FORMULA 1.
- the compounds shown in Table 1 are prepared by methods disclosed in Scheme 1 and in Examples 1-3. Certain compounds shown in Table 1 were analyzed via LC- MS. Retention times and masses of the M + 1 ion are given for those compounds. Retention time (tR) is measured in a gradient that increases from 30-100% B in 3.00 minutes where Buffer A is 0.1% trifluoroacetic acid in water and buffer B is 0.1% trifluoroacetic acid in acetonitrile. An analytical Phenomenex Luna C8 column was used with a flow rate of 2.5 ml /minute. All HPLC/MS analytical data were observed at a wavelength of 220 nm using a Gilson 151 UV/VIS detector followed by a ThermoFinnigan Surveyor MSQ.
- HCV replicon containing cells are treated with different concentrations of the test compound to ascertain the ability of the test compound to suppress replication of the HCV replicon.
- HCV replicon-containing cells are treated with different concentrations of interferon alpha, a known inhibitor of HCV replication.
- the replicon assay system includes Neomycin Phosphotransferase (NPT) as a component of the replicon itself in order to detect the transcription of replicon gene products in the host cell.
- NPT Neomycin Phosphotransferase
- HCV Replicon and Replicon Expression [0165]
- the HCV genome consists of a single ORF that encodes a 3000 amino acid polyprotein.
- the ORF is flanked on the 5' side by an untranslated region that serves as an internal ribosome entry site (IRES) and at the 3' side by a highly conserved sequence necessary for viral replication (3'-NTR).
- IRS internal ribosome entry site
- 3'-NTR highly conserved sequence necessary for viral replication
- the structural proteins, necessary for viral infection are located near the 5' end of the ORF.
- the non-structural proteins, designated NS2 to NS5B comprise the remainder of the ORF.
- the HCV replicon contains, 5'-3', the HCV-IRES, the neomycin phosphotransferase (neo) gene, the IRES of encephalomyocarditis virus, which directs translation of HCV sequences NS3 to NS5B, and the 3'-NTR.
- the sequence of the HCV replicon has been deposited in GenBank (Accession no. AJ242652).
- GenBank accesion no. AJ242652
- the replicon is transfected into Huh-7 cells using standard methods such as electroporation.
- the equipment and materials include, but are not limited to, Huh-7 HCV replicon- containing cells, maintenance media (DMEM (Dulbecco's modified Eagle media) supplemented with 10% FBS, L-glutamine, non-essential amino acids, penicillin (100 units/ml), streptomycin (100 micrograms/ml), and 500 micrograms/ml of Geneticin (G418), screening media (DMEM supplemented with 10% FBS, L-glutamine, non-essential amino acids, penicillin (100 units/ml) and streptomycin (100 micrograms/ml)), 96 well tissue culture plates (flat bottom), 96 well plates (U bottom for drug dilution), Interferon alpha for positive control, fixation reagent (such as methanol: acetone), primary antibody (rabbit anti-NPTII), secondary antibody: Eu-Nl 1, and enhancement solution.
- HCV replicon-containing cells support high levels of viral RNA replicon replication when their density is suitable. Over-confluency causes decreased viral RNA replication. Therefore, cells must be kept growing in log phase in the presence of 500 micrograms/ml of G418. Generally, cells should be passed twice a week at 1: 4-6 dilution. Cell maintenance is conducted as follows: [0170] HCV replicon-containing cells are examined under a microscope to ensure that cells growing well. Cells are rinsed once with PBS and 2 ml trypsin is added. The cell/ trypsin mixture is incubated at 37 °C in a CO 2 incubator for 3-5 minutes.
- HCV-replicon-containing cells are rinsed with once PBS once; 2 mis of trypsin are then added. Cells are incubated at 37°C in a 5% CO 2 incubator for 3-5 minutes. 10 mis of complete medium is added to stop the reaction. Cells are blown gently, put into a 15 ml tube, and spun at 1200 rpm for four minutes.
- the trypsin/medium solution is removed and 5 mis of medium (500 ml DMEM (high glucose)) from BRL catalog #12430-054; 50 mis 10% FBS, 5% Geneticin G418 (50 mg/ml, BRL catalog #10131-035), 5 ml MEM non-essential amino acids (lOOx BRL #11140-050) and 5 ml pen-strep (BRL #15140-148) is added.
- Cells are plated with screening medium (500 ml DMEM (BRL #21063-029), 50 ml FBS (BRL #10082-147) and 5 ml MEM non-essential amino acid (BRL #11140-050) at 6000-7500 cells/100 ⁇ l/well of 96 well plate (6-7.5x105 cells/10 ml/plate). Plates are placed into 37°C 5% CO 2 incubator overnight.
- screening medium 500 ml DMEM (BRL #21063-029), 50 ml FBS (BRL #10082-147) and 5 ml MEM non-essential amino acid (BRL #11140-050) at 6000-7500 cells/100 ⁇ l/well of 96 well plate (6-7.5x105 cells/10 ml/plate). Plates are placed into 37°C 5% CO 2 incubator overnight.
- drugs test compounds or interferon alpha
- media or DMSO/media depending on the final concentration chosen for screening. Generally for 6 concentrations of each test compounds ranging from 10 micromolar to 0.03 micromolar are applied. 100 ⁇ l of the test compound dilution is placed in wells of the 96 well plate containing the HCV replicon cells. Media without drug is added to some wells as a negative controls.
- DMSO is known to affect cell growth. Therefore, if drugs diluted in DMSO are used, all wells, including negative control (media only) and positive control (interferon alpha) wells, must contain the same concentration of DMSO, for single dose screening.
- the plates are incubated at 37°C in a humidified 5% C0 2 environment for three days.
- the NTPII assay is quantitated.
- the medium is poured from the plates and the plates are washed once in 200 ⁇ l of PBS.
- the PBS is then decanted and the plates tapped in a paper towel to remove any remaining PBS.
- Cells are fixed in situ with 100 ⁇ l/well of pre-cooled (-20°C) methanol: acetone (1:1) and the plates are placed at -20°C for 30 minutes.
- the fixing solution is poured from the plates and the plates allowed to air-dry completely (approximately one hour).
- the appearance of the dried cell layer is recorded and the density of the cells in the toxic wells is scored with the naked eye. Alternatively cell viability may be assessed using the MTS assay described below.
- the wells are blocked with 200 ⁇ l of blocking solution (10% FBS; 3% NGS in PBS) for 30 minutes at room temperature.
- the blocking solution is removed and 100 ⁇ l of rabbit anti-NPTII diluted 1:1000 in blocking solution is added to each well.
- the plates are then incubated 45-60 minutes at room temperature. After incubation, wells are washed six times with PBS-0.05% Tween-20 solution.
- Test Results [0179] Compounds shown in Table 1 have been tested in the above assay and found to inhibit replication of the HCV replicon with EC 50 values of less than 10 micromolar.
- EXAMPLE 6 EXAMPLE 6.
- CYTOTOXICITY ASSAYS [0180] To insure that the decrease in replicon replication is due to compound activity against the HCV replicon rather than nonspecific toxicity assays are used to quantitate compound cytotoxicity.
- Example 6A Cellular protein albumin assay for cytotoxicity
- HCV replicon- containing cells are treated for three days with different concentrations of helioxanthin; a compound that is known to be cytotoxic at high concentrations.
- the cells are lysed and the cell lysate used to bind plate-bound goat anti-albumin antibody at room temperature (25 °C to 28 °C) for 3 hours.
- the plate is then washed 6 times with IX PBS. After washing away the unbound proteins, mouse monoclonal anti-human serum albumin is applied to bind the albumin on the plate.
- the complex is then detected using phosphatase-labeled anti-mouse IgG as a second antibody.
- Example 6B MTS Assay for Cytotoxicity
- Cell viability may also be determined by CELLTITER 96 AQUEOUS ONE Solution Cell Proliferation Assay (Promega, Madison WI), a colorimetric assay for determining the number of viable cells, hi this method, before fixing the cells, 10-20 ⁇ l MTS reagent is added to each well according to manufacturer's instructions, plates are incubated at 37°C and read at OD 490 nm. During the incubation period living cells covert the MTS reagent to a formazan product which absorbs at 490 nm. Thus the 490nm absorbance is directly proportional to the number of living cells in culture.
- a direct comparison of the Cellular Album and MTS methods for determining cytotoxicity may be obtained as follows: Cells are treated with different concentrations of test compound or Helioxanthin for a three day-period. Prior to lysis for detection album as described above, the MTS reagent is added according to manufacturer's instruction to each well and incubate at 37 0C and read at OD 490 nm. The cellular album quantitation is then performed as described above.
- EXAMPLE 7 PHARMACEUTICAL FORMULATIONS [0184]
- Examples 7 A through 7G are examples of pharmaceutical compositions containing the compounds of Formula 1.
- the abbreviation "V.I.” stands for the viral inhibitor compounds of Formula 1 of the present invention.
- Example 7 A Oral Drops [0185] 5 grams of V.I. is dissolved in 5 ml of 2-hydroxypropanoic acid and 15 ml polyethylene glycol at about 60 °C to about 80 °C. After cooling to about 30°-40°C, 350 ml polyethylene glycol is added and the mixture was stined well. A solution of 17.5 g sodium saccharin in 25 ml purified water is then added. Flavor and polyethylene glycol q.s. (quantity sufficient) to a volume of 500 ml are added while stirring to provide an oral drop solution comprising 10 mg/ml of V.I.
- Example 7B Example 7A.
- Capsules [0186] 20 grams of the V.I., 6 grams sodium lauryl sulfate, 56 grams starch, 56 grams lactose, 0.8 grams colloidal silicon dioxide, and 1.2 grams magnesium stearate are vigorously stined together. The resulting mixture is subsequently filled into 1000 suitable hardened gelatin capsules, comprising each 20 mg of the active ingredient.
- Example 7C Film-Coated Tablets
- Preparation of tablet core A mixture of 10 grams of the V.I., 57 grams lactose and 20 grams starch is mixed well and thereafter humidified with a solution of 0.5 grams sodium dodecyl sulfate, and 1.0 grams polyvinylpynolidone (KOLLIDON-K 90) in about 20 ml of water. The wet powder mixture is sieved, dried, and sieved again. Then 100 grams microcrystalline cellulose (AVICEL) and 15 grams hydrogenated vegetable oil (STEROTEX) are added. The whole is mixed well and compressed into tablets, giving 1000 tablets, each containing 10 mg of the active ingredient.
- VICEL microcrystalline cellulose
- STEROTEX hydrogenated vegetable oil
- Magnesium Octadecanoate (.25 grams), 0.5 grams polyvinylpynolidone, and 3.0 ml of concentrated color suspension (OPASPRAY K- 1-2109) are added and the whole mixture homogenized.
- the tablet cores are coated with this mixture in a coating apparatus.
- Example 7D Injectible Solutions
- (i) 1.8 grams methyl 4-hydroxybenzoate and 0.2 grams propyl 4-hydroxybenzoate are dissolved in about 0.5 L of boiling water. After cooling to about 50°C, 4 grams lactic acid, 0.05 grams propylene glycol, and 4 grams of viral inhibitor are added while stining. The solution is cooled to room temperature and supplemented with water for injection q.s. giving a solution containing 4 mg/ml of V.I. The solution is sterilized by filtration and filled in sterile containers. [0190] (ii) 100.0 g of an acid salt of an V.I. of the invention is dissolved in boiling water.
- Example 7E Gel
- a compound or salt of the invention may be formed as a gel for topical application.
- a gel is prepared by suspending V.I. (0.2 g - 5.0 g) in benzyl alcohol at room temperature. A mixture of hydroxypropyl cellulose (2.5) grams and demineralized water (q.s. 100 g) is added to the suspension with stirring.
- Phase I contains Sorbitan monostearate (2.0 g), Polyoxyethylene (20) sorbitan monostearate (1.5 g), Synthetic spermaceti (3.0 g) Cetyl stearyl alcohol (10.0 g) and 2- Octyldodecanol (13.5 g). The phase I mixture is heated to 75 °C, stined and mixed.
- Phase II contains V.I. (1.0 g). Phase II is added to phase I, stined and suspended.
- Phase III contains Benzyl alcohol (1.0 g) and demineralized water (q.s. 100 g). Phase III is heated to 75 °C and added to phase II. The cream is mixed intensively and cooled slowly to room temperature, with further stirring. After cooling to room temperature the cream is homogenized.
- Example 7G Sprays
- the active compound solutions or suspensions prepared according to Example 7D can also be processed to sprays.
- a 60 to 90% active compound solution is mixed with 20 to 40% of the usual propellants, for example N 2 , N 2 0, CO 2 , propane, butane, halogenohydrocarbons and the like.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Epidemiology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Virology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Molecular Biology (AREA)
- Gastroenterology & Hepatology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Description



















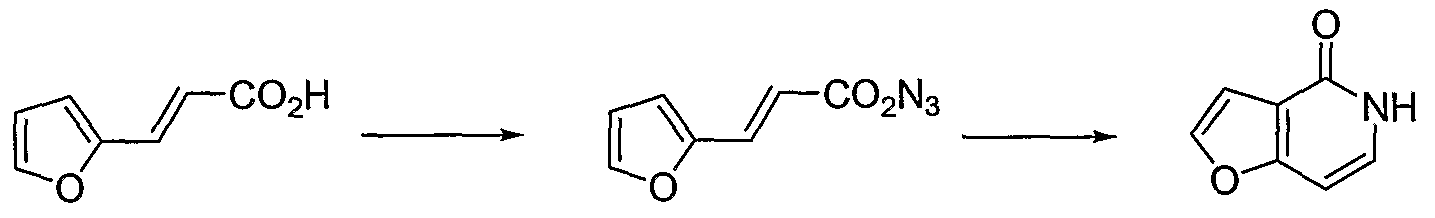
















Claims












Priority Applications (12)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP05705124A EP1709047B1 (en) | 2004-01-06 | 2005-01-05 | Azabenzofuran substituted thioureas as inhibitors of viral replication |
| NZ548375A NZ548375A (en) | 2004-01-06 | 2005-01-05 | Azabenzofuran substituted thioureas as inhibitors of viral replication |
| AP2006003659A AP2006003659A0 (en) | 2004-01-06 | 2005-01-05 | Azabenzofuran substituted thioureas, inhibitors ofviral replication |
| DE602005023266T DE602005023266D1 (en) | 2004-01-06 | 2005-01-05 | AZABENZOFURANS-SUBSTITUTED THIOSCORES AS INHIBITORS OF VIRUS REPLICATION |
| JP2006549390A JP2007517887A (en) | 2004-01-06 | 2005-01-05 | Azabenzofuran substituted thiourea, inhibitor of viral replication |
| BRPI0506705-7A BRPI0506705A (en) | 2004-01-06 | 2005-01-05 | composed of azabenzofuran-substituted thioureas, pharmaceutical compositions and their uses |
| AT05705124T ATE479684T1 (en) | 2004-01-06 | 2005-01-05 | AZABENZOFURAN-SUBSTITUTED THIOUREAS AS INHIBITORS OF VIRUS REPLICATION |
| CA002552002A CA2552002A1 (en) | 2004-01-06 | 2005-01-05 | Azabenzofuran substituted thioureas as inhibitors of viral replication |
| AU2005204369A AU2005204369B2 (en) | 2004-01-06 | 2005-01-05 | Azabenzofuran substituted thioureas as inhibitors of viral replication |
| EA200601281A EA200601281A1 (en) | 2004-01-06 | 2005-01-05 | AZABENZOFURANE-SUBSTITUTED THIOMOPHEIN; VIRUS REPLICATION INHIBITORS |
| IL176486A IL176486A0 (en) | 2004-01-06 | 2006-06-21 | Azabenzofuran substituted thioureas as inhibtitors of viral replication |
| NO20062928A NO20062928L (en) | 2004-01-06 | 2006-06-22 | Azabenzofuran substituted thioureas as inhibitors of viral replication |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US53483904P | 2004-01-06 | 2004-01-06 | |
| US60/534,839 | 2004-01-06 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2005067900A2 true WO2005067900A2 (en) | 2005-07-28 |
| WO2005067900A3 WO2005067900A3 (en) | 2005-09-29 |
Family
ID=34794326
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2005/000339 WO2005067900A2 (en) | 2004-01-06 | 2005-01-05 | Azabenzofuran substituted thioureas as inhibitors of viral replication |
Country Status (19)
| Country | Link |
|---|---|
| US (1) | US7439374B2 (en) |
| EP (1) | EP1709047B1 (en) |
| JP (1) | JP2007517887A (en) |
| KR (1) | KR20060121289A (en) |
| CN (1) | CN1946720A (en) |
| AP (1) | AP2006003659A0 (en) |
| AR (1) | AR047367A1 (en) |
| AT (1) | ATE479684T1 (en) |
| AU (1) | AU2005204369B2 (en) |
| BR (1) | BRPI0506705A (en) |
| CA (1) | CA2552002A1 (en) |
| DE (1) | DE602005023266D1 (en) |
| EA (1) | EA200601281A1 (en) |
| IL (1) | IL176486A0 (en) |
| NO (1) | NO20062928L (en) |
| NZ (1) | NZ548375A (en) |
| OA (1) | OA13357A (en) |
| TW (1) | TW200528459A (en) |
| WO (1) | WO2005067900A2 (en) |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008024763A2 (en) * | 2006-08-25 | 2008-02-28 | Wyeth | Identification and characterization of hcv replicon variants with reduced susceptibility to hcv-796, and methods related thereto |
| WO2008121634A2 (en) | 2007-03-30 | 2008-10-09 | Pharmasset, Inc. | Nucleoside phosphoramidate prodrugs |
| WO2010030538A2 (en) * | 2008-09-11 | 2010-03-18 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis c |
| WO2010075517A2 (en) | 2008-12-23 | 2010-07-01 | Pharmasset, Inc. | Nucleoside analogs |
| WO2010075549A2 (en) | 2008-12-23 | 2010-07-01 | Pharmasset, Inc. | Nucleoside phosphoramidates |
| WO2010075554A1 (en) | 2008-12-23 | 2010-07-01 | Pharmasset, Inc. | Synthesis of purine nucleosides |
| WO2010135569A1 (en) | 2009-05-20 | 2010-11-25 | Pharmasset, Inc. | N- [ (2 ' r) -2 ' -deoxy-2 ' -fluoro-2 ' -methyl-p-phenyl-5 ' -uridylyl] -l-alanine 1-methylethyl ester and process for its production |
| WO2011123645A2 (en) | 2010-03-31 | 2011-10-06 | Pharmasset, Inc. | Nucleoside phosphoramidates |
| WO2011123672A1 (en) | 2010-03-31 | 2011-10-06 | Pharmasset, Inc. | Purine nucleoside phosphoramidate |
| US8324239B2 (en) | 2010-04-21 | 2012-12-04 | Novartis Ag | Furopyridine compounds and uses thereof |
| US8618076B2 (en) | 2009-05-20 | 2013-12-31 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US8889159B2 (en) | 2011-11-29 | 2014-11-18 | Gilead Pharmasset Llc | Compositions and methods for treating hepatitis C virus |
| US11116783B2 (en) | 2013-08-27 | 2021-09-14 | Gilead Pharmasset Llc | Combination formulation of two antiviral compounds |
Families Citing this family (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TW200600492A (en) * | 2004-05-18 | 2006-01-01 | Achillion Pharmaceuticals Inc | Substituted aryl acylthioureas and related compounds; inhibitors of viral replication |
| TWI329102B (en) * | 2006-08-15 | 2010-08-21 | Nat Health Research Institutes | Thiourea compounds and method for inhibiting hepatitis c virus infection |
| US7985763B2 (en) * | 2007-04-10 | 2011-07-26 | National Health Research Institutes | Hepatitis C virus inhibitors |
| WO2008154091A1 (en) * | 2007-06-08 | 2008-12-18 | National Health Research Institutes | Thiourea derivatives |
| TWI361808B (en) * | 2008-01-08 | 2012-04-11 | Nat Health Research Institutes | Imidazolidinone and imidazolidinethione derivatives |
| US8198284B2 (en) * | 2008-04-30 | 2012-06-12 | National Health Research Institutes | Treatment of neurodegenerative disorders with thiourea compounds |
| US8563530B2 (en) | 2010-03-31 | 2013-10-22 | Gilead Pharmassel LLC | Purine nucleoside phosphoramidate |
| JP6069215B2 (en) | 2010-11-30 | 2017-02-01 | ギリアド ファーマセット エルエルシー | Compound |
| UA116087C2 (en) | 2011-09-16 | 2018-02-12 | Гіліад Фармассет Елелсі | Methods for treating hcv |
| JP6077642B2 (en) * | 2012-04-10 | 2017-02-08 | シャンハイ インリ ファーマシューティカル カンパニー リミティド | Fused pyrimidine compounds, methods for their preparation, intermediates, compositions, and uses |
| KR20140119012A (en) | 2013-01-31 | 2014-10-08 | 길리어드 파마셋 엘엘씨 | Combination formulation of two antiviral compounds |
| EP3455218A4 (en) | 2016-05-10 | 2019-12-18 | C4 Therapeutics, Inc. | C3-carbon linked glutarimide degronimers for target protein degradation |
| EP3454856B1 (en) | 2016-05-10 | 2024-09-11 | C4 Therapeutics, Inc. | Heterocyclic degronimers for target protein degradation |
| WO2017197036A1 (en) | 2016-05-10 | 2017-11-16 | C4 Therapeutics, Inc. | Spirocyclic degronimers for target protein degradation |
| KR20210141778A (en) | 2016-06-07 | 2021-11-23 | 자코바이오 파마슈티칼스 컴퍼니 리미티드 | Novel heterocyclic derivatives useful as shp2 inhibitors |
| KR20220113545A (en) | 2017-03-23 | 2022-08-12 | 자코바이오 파마슈티칼스 컴퍼니 리미티드 | Novel heterocyclic derivatives useful as shp2 inhibitors |
Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1408198A (en) * | 1971-08-23 | 1975-10-01 | Medizinska Akad | Antiviral compositions |
| EP0526009A1 (en) * | 1991-07-10 | 1993-02-03 | Eli Lilly And Company | Inhibitors of hiv protease useful for the treatment of aids |
| EP0806205A2 (en) * | 1996-05-06 | 1997-11-12 | Eli Lilly And Company | Urea, thiourea and guanidine compounds and their use as anti-viral agents |
| WO2004041201A2 (en) * | 2002-11-01 | 2004-05-21 | Viropharma Incorporated | Benzofuran compounds, compositions and methods for treatment and prophylaxis of hepatitis c viral infections and associated diseases |
| WO2004046095A1 (en) * | 2002-11-19 | 2004-06-03 | Achillion Pharmaceuticals, Inc. | Substituted aryl thioureas and related compounds; inhibitors of viral replication |
| WO2005007601A2 (en) * | 2003-07-10 | 2005-01-27 | Achillion Pharmaceuticals, Inc. | Substituted arylthiourea derivatives useful as inhibitors of viral replication |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB9504460D0 (en) * | 1995-03-06 | 1995-04-26 | Bayer Ag | N-(3-Benzofuranyl)urea-derivatives |
| US6174905B1 (en) * | 1996-09-30 | 2001-01-16 | Mitsui Chemicals, Inc. | Cell differentiation inducer |
-
2004
- 2004-12-30 TW TW093141409A patent/TW200528459A/en unknown
-
2005
- 2005-01-05 AP AP2006003659A patent/AP2006003659A0/en unknown
- 2005-01-05 US US11/029,910 patent/US7439374B2/en not_active Expired - Fee Related
- 2005-01-05 EP EP05705124A patent/EP1709047B1/en not_active Not-in-force
- 2005-01-05 KR KR1020067013660A patent/KR20060121289A/en not_active Application Discontinuation
- 2005-01-05 AU AU2005204369A patent/AU2005204369B2/en not_active Ceased
- 2005-01-05 DE DE602005023266T patent/DE602005023266D1/en active Active
- 2005-01-05 WO PCT/US2005/000339 patent/WO2005067900A2/en active Application Filing
- 2005-01-05 NZ NZ548375A patent/NZ548375A/en unknown
- 2005-01-05 EA EA200601281A patent/EA200601281A1/en unknown
- 2005-01-05 AT AT05705124T patent/ATE479684T1/en not_active IP Right Cessation
- 2005-01-05 JP JP2006549390A patent/JP2007517887A/en active Pending
- 2005-01-05 CN CNA200580007223XA patent/CN1946720A/en active Pending
- 2005-01-05 AR ARP050100026A patent/AR047367A1/en not_active Application Discontinuation
- 2005-01-05 CA CA002552002A patent/CA2552002A1/en not_active Abandoned
- 2005-01-05 BR BRPI0506705-7A patent/BRPI0506705A/en not_active IP Right Cessation
- 2005-01-05 OA OA1200600222A patent/OA13357A/en unknown
-
2006
- 2006-06-21 IL IL176486A patent/IL176486A0/en unknown
- 2006-06-22 NO NO20062928A patent/NO20062928L/en not_active Application Discontinuation
Patent Citations (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1408198A (en) * | 1971-08-23 | 1975-10-01 | Medizinska Akad | Antiviral compositions |
| EP0526009A1 (en) * | 1991-07-10 | 1993-02-03 | Eli Lilly And Company | Inhibitors of hiv protease useful for the treatment of aids |
| EP0806205A2 (en) * | 1996-05-06 | 1997-11-12 | Eli Lilly And Company | Urea, thiourea and guanidine compounds and their use as anti-viral agents |
| WO2004041201A2 (en) * | 2002-11-01 | 2004-05-21 | Viropharma Incorporated | Benzofuran compounds, compositions and methods for treatment and prophylaxis of hepatitis c viral infections and associated diseases |
| WO2004046095A1 (en) * | 2002-11-19 | 2004-06-03 | Achillion Pharmaceuticals, Inc. | Substituted aryl thioureas and related compounds; inhibitors of viral replication |
| WO2005007601A2 (en) * | 2003-07-10 | 2005-01-27 | Achillion Pharmaceuticals, Inc. | Substituted arylthiourea derivatives useful as inhibitors of viral replication |
Non-Patent Citations (3)
| Title |
|---|
| D. W. LUDOVICI ET AL.: "Evolution of Anti-HIV Drug Candidates. Part 1: From .alpha.-Anilinophenylacetamide (.alpha.-APA) to Imidoyl Thiourea (ITU)" BIOORG. MED. CHEM. LETT., vol. 11, 2001, pages 2225-2228, XP002276467 * |
| DATABASE CAPLUS [Online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; 9 December 1989 (1989-12-09), XP002337572 retrieved from STN Database accession no. 1989:614340 & S. A. ZOTOVA ET AL.: "Synthesis and antiviral activity of 5-methoxybenzofuran derivatives" KHIMIKO-FARMATSEVTICHESKII ZHURNAL, vol. 23, no. 2, 1989, pages 191-195, * |
| P. REYNAUD ET AL.: "Préparation de quelques p-alcoxy benzoyl-1 p-alcoxyphényl-3 thiourées: leur activité "in vitro" sur la souche H37RV du bacille tuberculeux" CHIMIE THERAPEUTIQUE, no. 7, 1966, pages 421-424, XP002276470 * |
Cited By (46)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2008024763A3 (en) * | 2006-08-25 | 2008-12-24 | Wyeth Corp | Identification and characterization of hcv replicon variants with reduced susceptibility to hcv-796, and methods related thereto |
| WO2008024763A2 (en) * | 2006-08-25 | 2008-02-28 | Wyeth | Identification and characterization of hcv replicon variants with reduced susceptibility to hcv-796, and methods related thereto |
| US10183037B2 (en) | 2007-03-30 | 2019-01-22 | Gilead Pharmasset Llc | Nucleoside phosphoramidate prodrugs |
| US8735372B2 (en) | 2007-03-30 | 2014-05-27 | Gilead Pharmasset Llc | Nucleoside phosphoramidate prodrugs |
| US8906880B2 (en) | 2007-03-30 | 2014-12-09 | Gilead Pharmasset Llc | Nucleoside phosphoramidate prodrugs |
| US8580765B2 (en) | 2007-03-30 | 2013-11-12 | Gilead Pharmasset Llc | Nucleoside phosphoramidate prodrugs |
| US11642361B2 (en) | 2007-03-30 | 2023-05-09 | Gilead Sciences, Inc. | Nucleoside phosphoramidate prodrugs |
| WO2008121634A2 (en) | 2007-03-30 | 2008-10-09 | Pharmasset, Inc. | Nucleoside phosphoramidate prodrugs |
| US9085573B2 (en) | 2007-03-30 | 2015-07-21 | Gilead Pharmasset Llc | Nucleoside phosphoramidate prodrugs |
| DE202008018643U1 (en) | 2007-03-30 | 2017-03-16 | Gilead Pharmasset Llc | Nucleosidphosphoramidat prodrugs |
| US9585906B2 (en) | 2007-03-30 | 2017-03-07 | Gilead Pharmasset Llc | Nucleoside phosphoramidate prodrugs |
| WO2010030538A2 (en) * | 2008-09-11 | 2010-03-18 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis c |
| WO2010030538A3 (en) * | 2008-09-11 | 2010-05-06 | Bristol-Myers Squibb Company | Compounds for the treatment of hepatitis c |
| EP3222628A1 (en) | 2008-12-23 | 2017-09-27 | Gilead Pharmasset LLC | Nucleoside phosphoramidates |
| US8716262B2 (en) | 2008-12-23 | 2014-05-06 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US8551973B2 (en) | 2008-12-23 | 2013-10-08 | Gilead Pharmasset Llc | Nucleoside analogs |
| WO2010075554A1 (en) | 2008-12-23 | 2010-07-01 | Pharmasset, Inc. | Synthesis of purine nucleosides |
| EP2671888A1 (en) | 2008-12-23 | 2013-12-11 | Gilead Pharmasset LLC | 3',5'-cyclic nucleoside phosphate analogues |
| US8957045B2 (en) | 2008-12-23 | 2015-02-17 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| WO2010075549A2 (en) | 2008-12-23 | 2010-07-01 | Pharmasset, Inc. | Nucleoside phosphoramidates |
| WO2010075517A2 (en) | 2008-12-23 | 2010-07-01 | Pharmasset, Inc. | Nucleoside analogs |
| US8716263B2 (en) | 2008-12-23 | 2014-05-06 | Gilead Pharmasset Llc | Synthesis of purine nucleosides |
| EP2913337A1 (en) | 2009-05-20 | 2015-09-02 | Gilead Pharmasset LLC | N-[(2'r)-2'-deoxy-2'-fluoro-2'-methyl-p-phenyl-5'-uridylyl]-l-alanine 1-methylethyl ester and process for its production |
| US9284342B2 (en) | 2009-05-20 | 2016-03-15 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US8735569B2 (en) | 2009-05-20 | 2014-05-27 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US8633309B2 (en) | 2009-05-20 | 2014-01-21 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| WO2010135569A1 (en) | 2009-05-20 | 2010-11-25 | Pharmasset, Inc. | N- [ (2 ' r) -2 ' -deoxy-2 ' -fluoro-2 ' -methyl-p-phenyl-5 ' -uridylyl] -l-alanine 1-methylethyl ester and process for its production |
| EP3321275A1 (en) | 2009-05-20 | 2018-05-16 | Gilead Pharmasset LLC | Crystalline form of sofosbuvir |
| US9637512B2 (en) | 2009-05-20 | 2017-05-02 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US8629263B2 (en) | 2009-05-20 | 2014-01-14 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US8618076B2 (en) | 2009-05-20 | 2013-12-31 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| EP2610264A2 (en) | 2009-05-20 | 2013-07-03 | Gilead Pharmasset LLC | N-[(2'r)-2'-deoxy-2'-fluoro-2'-methyl-p-phenyl-5'-uridylyl]-l-alanine 1-methylethyl ester and process for its production |
| EP2910562A1 (en) | 2009-05-20 | 2015-08-26 | Gilead Pharmasset LLC | N-[(2'r)-2'-deoxy-2 '-fluoro-2'-methyl-p-phenyl-5 '-uridylyl]-l-alanine 1-methylethyl ester in crystalline form |
| US8642756B2 (en) | 2009-05-20 | 2014-02-04 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| US9206217B2 (en) | 2009-05-20 | 2015-12-08 | Gilead Pharmasset Llc | Nucleoside phosphoramidates |
| EP2609923A2 (en) | 2010-03-31 | 2013-07-03 | Gilead Pharmasset LLC | Nucleoside Phosphoramidates |
| WO2011123668A2 (en) | 2010-03-31 | 2011-10-06 | Pharmasset, Inc. | Stereoselective synthesis of phosphorus containing actives |
| WO2011123672A1 (en) | 2010-03-31 | 2011-10-06 | Pharmasset, Inc. | Purine nucleoside phosphoramidate |
| EP3290428A1 (en) | 2010-03-31 | 2018-03-07 | Gilead Pharmasset LLC | Tablet comprising crystalline (s)-isopropyl 2-(((s)-(((2r,3r,4r,5r)-5-(2,4-dioxo-3,4-dihydropyrimidin-1 (2h)-yl)-4-fluoro-3-hydroxy-4-methyltetrahydrofuran-2-yl)methoxy)(phenoxy)phosphoryl)amino)propanoate |
| US8859756B2 (en) | 2010-03-31 | 2014-10-14 | Gilead Pharmasset Llc | Stereoselective synthesis of phosphorus containing actives |
| WO2011123645A2 (en) | 2010-03-31 | 2011-10-06 | Pharmasset, Inc. | Nucleoside phosphoramidates |
| EP2752422A1 (en) | 2010-03-31 | 2014-07-09 | Gilead Pharmasset LLC | Stereoselective synthesis of phosphorus containing actives |
| US8324239B2 (en) | 2010-04-21 | 2012-12-04 | Novartis Ag | Furopyridine compounds and uses thereof |
| US8889159B2 (en) | 2011-11-29 | 2014-11-18 | Gilead Pharmasset Llc | Compositions and methods for treating hepatitis C virus |
| US11116783B2 (en) | 2013-08-27 | 2021-09-14 | Gilead Pharmasset Llc | Combination formulation of two antiviral compounds |
| US11707479B2 (en) | 2013-08-27 | 2023-07-25 | Gilead Sciences, Inc. | Combination formulation of two antiviral compounds |
Also Published As
| Publication number | Publication date |
|---|---|
| EA200601281A1 (en) | 2006-12-29 |
| US20050228013A1 (en) | 2005-10-13 |
| KR20060121289A (en) | 2006-11-28 |
| TW200528459A (en) | 2005-09-01 |
| IL176486A0 (en) | 2006-10-05 |
| US7439374B2 (en) | 2008-10-21 |
| AR047367A1 (en) | 2006-01-18 |
| CN1946720A (en) | 2007-04-11 |
| JP2007517887A (en) | 2007-07-05 |
| WO2005067900A3 (en) | 2005-09-29 |
| AU2005204369A1 (en) | 2005-07-28 |
| EP1709047A2 (en) | 2006-10-11 |
| ATE479684T1 (en) | 2010-09-15 |
| NO20062928L (en) | 2006-08-03 |
| OA13357A (en) | 2007-04-13 |
| AP2006003659A0 (en) | 2006-06-30 |
| DE602005023266D1 (en) | 2010-10-14 |
| EP1709047B1 (en) | 2010-09-01 |
| AU2005204369B2 (en) | 2010-06-10 |
| BRPI0506705A (en) | 2007-05-02 |
| NZ548375A (en) | 2010-07-30 |
| CA2552002A1 (en) | 2005-07-28 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1709047B1 (en) | Azabenzofuran substituted thioureas as inhibitors of viral replication | |
| EP1747196B1 (en) | Substituted aryl acylthioureas and related compounds; inhibitors of viral replication | |
| US7094807B2 (en) | Substituted aryl thioureas and related compounds; inhibitors of viral replication | |
| AU2004257277B2 (en) | Substituted arylthiourea derivatives useful as inhibitors of viral replication | |
| US20060100225A1 (en) | Heteroaryl guanidines; inhibitors of viral replication | |
| MXPA06007729A (en) | Azabenzofuran substituted thioureas as inhibitors of viral replication | |
| AU2005326813A1 (en) | Substituted aryl acylthioureas and related compounds; inhibitors of viral replication |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2005705124 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 176486 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2552002 Country of ref document: CA Ref document number: 12006501307 Country of ref document: PH Ref document number: PA/a/2006/007729 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 548375 Country of ref document: NZ Ref document number: 1020067013660 Country of ref document: KR Ref document number: 2005204369 Country of ref document: AU Ref document number: 2006549390 Country of ref document: JP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 4144/DELNP/2006 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 2005204369 Country of ref document: AU Date of ref document: 20050105 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005204369 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200601281 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200580007223.X Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2005705124 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020067013660 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: PI0506705 Country of ref document: BR |