WO2005060963A1 - Benzenesulfonylamino-pyridin-2-yl derivatives and related compounds as inhibitors of 11-beta-hydroxysteroid dehydrogenase type 1 (11-beta-hsd-1) for the treatment of diabetes and obesity - Google Patents
Benzenesulfonylamino-pyridin-2-yl derivatives and related compounds as inhibitors of 11-beta-hydroxysteroid dehydrogenase type 1 (11-beta-hsd-1) for the treatment of diabetes and obesity Download PDFInfo
- Publication number
- WO2005060963A1 WO2005060963A1 PCT/IB2004/004056 IB2004004056W WO2005060963A1 WO 2005060963 A1 WO2005060963 A1 WO 2005060963A1 IB 2004004056 W IB2004004056 W IB 2004004056W WO 2005060963 A1 WO2005060963 A1 WO 2005060963A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- membered heterocyclyl
- alkyl
- aryl
- independently selected
- Prior art date
Links
- WIXBQBJYFZLWLJ-UHFFFAOYSA-N C(C1)CSC1C1C(CC2)OC2C1 Chemical compound C(C1)CSC1C1C(CC2)OC2C1 WIXBQBJYFZLWLJ-UHFFFAOYSA-N 0.000 description 1
- IMNIMPAHZVJRPE-UHFFFAOYSA-N C(C1)N2CCN1CC2 Chemical compound C(C1)N2CCN1CC2 IMNIMPAHZVJRPE-UHFFFAOYSA-N 0.000 description 1
- KIDADRHMWBPVKL-UHFFFAOYSA-N C(C=C1)C=NC=C1C1=CC2OC1C=C2 Chemical compound C(C=C1)C=NC=C1C1=CC2OC1C=C2 KIDADRHMWBPVKL-UHFFFAOYSA-N 0.000 description 1
- CDWOQYIIMKWNEF-UHFFFAOYSA-N C(CO1)N/C1=[O]/C1CN(CCCC2)C2CC1 Chemical compound C(CO1)N/C1=[O]/C1CN(CCCC2)C2CC1 CDWOQYIIMKWNEF-UHFFFAOYSA-N 0.000 description 1
- JAPMJSVZDUYFKL-UHFFFAOYSA-N C1C2C1CCC2 Chemical compound C1C2C1CCC2 JAPMJSVZDUYFKL-UHFFFAOYSA-N 0.000 description 1
- NQRYJNQNLNOLGT-UHFFFAOYSA-N C1CCNCC1 Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 1
- VSWICNJIUPRZIK-UHFFFAOYSA-N C1CNC=CC1 Chemical compound C1CNC=CC1 VSWICNJIUPRZIK-UHFFFAOYSA-N 0.000 description 1
- GLUUGHFHXGJENI-UHFFFAOYSA-N C1NCCNC1 Chemical compound C1NCCNC1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 1
- YNAVUWVOSKDBBP-UHFFFAOYSA-N C1NCCOC1 Chemical compound C1NCCOC1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 1
- UWYZHKAOTLEWKK-UHFFFAOYSA-N C1c2ccccc2CNC1 Chemical compound C1c2ccccc2CNC1 UWYZHKAOTLEWKK-UHFFFAOYSA-N 0.000 description 1
- HBEDSQVIWPRPAY-UHFFFAOYSA-N C1c2ccccc2OC1 Chemical compound C1c2ccccc2OC1 HBEDSQVIWPRPAY-UHFFFAOYSA-N 0.000 description 1
- HREKTUYBUFDTAY-UHFFFAOYSA-N CC(C1)C(S(NC(C(C)C2)=Nc3c2cccc3)(=O)=O)=CC=C1c(cc1)ccc1C#N Chemical compound CC(C1)C(S(NC(C(C)C2)=Nc3c2cccc3)(=O)=O)=CC=C1c(cc1)ccc1C#N HREKTUYBUFDTAY-UHFFFAOYSA-N 0.000 description 1
- CRFGKRNLYWUFHL-UHFFFAOYSA-N CC(CC(c(cc1)ccc1C#N)=C1)C(S(Nc2nc(C)ccc2)(=O)=O)=C1F Chemical compound CC(CC(c(cc1)ccc1C#N)=C1)C(S(Nc2nc(C)ccc2)(=O)=O)=C1F CRFGKRNLYWUFHL-UHFFFAOYSA-N 0.000 description 1
- KRAXJYQQBZVBJS-UHFFFAOYSA-N CC1(C)NC(NS(c2ccc(-c(cc3)ccc3C#N)nc2)(=O)=O)=CC=C1 Chemical compound CC1(C)NC(NS(c2ccc(-c(cc3)ccc3C#N)nc2)(=O)=O)=CC=C1 KRAXJYQQBZVBJS-UHFFFAOYSA-N 0.000 description 1
- JOYWFBBSKVNSAC-UHFFFAOYSA-N CC1(C)OB(c(cc2)ccc2S(Nc2nc(C)ccc2)(=O)=O)OC1(C)C Chemical compound CC1(C)OB(c(cc2)ccc2S(Nc2nc(C)ccc2)(=O)=O)OC1(C)C JOYWFBBSKVNSAC-UHFFFAOYSA-N 0.000 description 1
- UIICNHYOXUCWLU-UHFFFAOYSA-N CCN(CC)C(Cc1cccc(NS(c2c(C)c(cc(C)cc3)c3[s]2)(=O)=O)n1)=O Chemical compound CCN(CC)C(Cc1cccc(NS(c2c(C)c(cc(C)cc3)c3[s]2)(=O)=O)n1)=O UIICNHYOXUCWLU-UHFFFAOYSA-N 0.000 description 1
- KCPIGXCRNTWODY-UHFFFAOYSA-N CCc1cccc(NS(c(cc2)ccc2C(C(C)C2)=CC=C2C#N)(=O)=O)n1 Chemical compound CCc1cccc(NS(c(cc2)ccc2C(C(C)C2)=CC=C2C#N)(=O)=O)n1 KCPIGXCRNTWODY-UHFFFAOYSA-N 0.000 description 1
- PEUZQKRMRMMBLB-UHFFFAOYSA-N Cc(c1cc(C)ccc1[s]1)c1S(Nc1nc(CCO)ccc1)(=O)=O Chemical compound Cc(c1cc(C)ccc1[s]1)c1S(Nc1nc(CCO)ccc1)(=O)=O PEUZQKRMRMMBLB-UHFFFAOYSA-N 0.000 description 1
- NBQWLLOYROUAQA-UHFFFAOYSA-N Cc(cc(cc1)-c(cc2)ccc2C#N)c1S(Nc1nc(C)ccc1)(=O)=O Chemical compound Cc(cc(cc1)-c(cc2)ccc2C#N)c1S(Nc1nc(C)ccc1)(=O)=O NBQWLLOYROUAQA-UHFFFAOYSA-N 0.000 description 1
- DQZHWDQTLLEONO-UHFFFAOYSA-N Cc(cc(cc1)Br)c1S(Nc1nc(C)ccc1)(=O)=O Chemical compound Cc(cc(cc1)Br)c1S(Nc1nc(C)ccc1)(=O)=O DQZHWDQTLLEONO-UHFFFAOYSA-N 0.000 description 1
- JBWDWAQYTUJKQF-UHFFFAOYSA-N Cc1cc(C)nc(NS(c(cc2)ccc2-c(cc2)ccc2C#N)(=O)=O)c1 Chemical compound Cc1cc(C)nc(NS(c(cc2)ccc2-c(cc2)ccc2C#N)(=O)=O)c1 JBWDWAQYTUJKQF-UHFFFAOYSA-N 0.000 description 1
- JGZLAYVUKQROQV-UHFFFAOYSA-N Cc1cccc(N(CCN(C)C)S(c(cc2)ccc2-c(cc2)ccc2C#N)(=O)=O)n1 Chemical compound Cc1cccc(N(CCN(C)C)S(c(cc2)ccc2-c(cc2)ccc2C#N)(=O)=O)n1 JGZLAYVUKQROQV-UHFFFAOYSA-N 0.000 description 1
- OWYWZVRZIUBCOM-UHFFFAOYSA-N Cc1nc(NS(c(cc2)ccc2-c(cc2)ccc2C#N)(=O)=O)ccc1 Chemical compound Cc1nc(NS(c(cc2)ccc2-c(cc2)ccc2C#N)(=O)=O)ccc1 OWYWZVRZIUBCOM-UHFFFAOYSA-N 0.000 description 1
- 0 Cc1nc(NS2(=*c3c2ccc(-c(cc2)ccc2C#N)c3)=O)ccc1 Chemical compound Cc1nc(NS2(=*c3c2ccc(-c(cc2)ccc2C#N)c3)=O)ccc1 0.000 description 1
- UTFIFHIKCKUWFU-UHFFFAOYSA-N N#Cc(cc1)ccc1-c(nc1)ccc1S(Nc1nc(C2CC2)ccc1)(=O)=O Chemical compound N#Cc(cc1)ccc1-c(nc1)ccc1S(Nc1nc(C2CC2)ccc1)(=O)=O UTFIFHIKCKUWFU-UHFFFAOYSA-N 0.000 description 1
- ZKLKHLJWEASUHI-UHFFFAOYSA-N Nc1nc(NS(C(CC2)=CC=C2c(cc2)ccc2C#N)(=O)=O)ccc1 Chemical compound Nc1nc(NS(C(CC2)=CC=C2c(cc2)ccc2C#N)(=O)=O)ccc1 ZKLKHLJWEASUHI-UHFFFAOYSA-N 0.000 description 1
- NTPLEHQIARXKSJ-UHFFFAOYSA-N Nc1nc(NSc(cc2)ccc2-c(cc2)ccc2Cl)ccc1 Chemical compound Nc1nc(NSc(cc2)ccc2-c(cc2)ccc2Cl)ccc1 NTPLEHQIARXKSJ-UHFFFAOYSA-N 0.000 description 1
- XKJCHHZQLQNZHY-UHFFFAOYSA-N O=C(c1ccccc11)NC1=O Chemical compound O=C(c1ccccc11)NC1=O XKJCHHZQLQNZHY-UHFFFAOYSA-N 0.000 description 1
- HNJBEVLQSNELDL-UHFFFAOYSA-N O=C1NCCC1 Chemical compound O=C1NCCC1 HNJBEVLQSNELDL-UHFFFAOYSA-N 0.000 description 1
- REJCLSGCVCNFAX-UHFFFAOYSA-N O=S(c(cc1)cnc1Cl)(Nc1nc(cccc2)c2cc1)=O Chemical compound O=S(c(cc1)cnc1Cl)(Nc1nc(cccc2)c2cc1)=O REJCLSGCVCNFAX-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/08—Indoles; Hydrogenated indoles with only hydrogen atoms or radicals containing only hydrogen and carbon atoms, directly attached to carbon atoms of the hetero ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4402—Non condensed pyridines; Hydrogenated derivatives thereof only substituted in position 2, e.g. pheniramine, bisacodyl
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/02—Drugs for disorders of the urinary system of urine or of the urinary tract, e.g. urine acidifiers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/16—Emollients or protectives, e.g. against radiation
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/06—Antigout agents, e.g. antihyperuricemic or uricosuric agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/04—Centrally acting analgesics, e.g. opioids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/06—Antiglaucoma agents or miotics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/12—Ophthalmic agents for cataracts
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
- A61P31/06—Antibacterial agents for tuberculosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P33/00—Antiparasitic agents
- A61P33/02—Antiprotozoals, e.g. for leishmaniasis, trichomoniasis, toxoplasmosis
- A61P33/06—Antimalarials
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/38—Drugs for disorders of the endocrine system of the suprarenal hormones
- A61P5/46—Drugs for disorders of the endocrine system of the suprarenal hormones for decreasing, blocking or antagonising the activity of glucocorticosteroids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/48—Drugs for disorders of the endocrine system of the pancreatic hormones
- A61P5/50—Drugs for disorders of the endocrine system of the pancreatic hormones for increasing or potentiating the activity of insulin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/60—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D213/72—Nitrogen atoms
- C07D213/76—Nitrogen atoms to which a second hetero atom is attached
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/38—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D235/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings
- C07D235/02—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, condensed with other rings condensed with carbocyclic rings or ring systems
- C07D235/04—Benzimidazoles; Hydrogenated benzimidazoles
- C07D235/06—Benzimidazoles; Hydrogenated benzimidazoles with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached in position 2
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/02—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings
- C07D239/24—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members
- C07D239/28—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings not condensed with other rings having three or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, directly attached to ring carbon atoms
- C07D239/32—One oxygen, sulfur or nitrogen atom
- C07D239/42—One nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D409/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms
- C07D409/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings
- C07D409/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having sulfur atoms as the only ring hetero atoms containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/12—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D491/00—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00
- C07D491/02—Heterocyclic compounds containing in the condensed ring system both one or more rings having oxygen atoms as the only ring hetero atoms and one or more rings having nitrogen atoms as the only ring hetero atoms, not provided for by groups C07D451/00 - C07D459/00, C07D463/00, C07D477/00 or C07D489/00 in which the condensed system contains two hetero rings
- C07D491/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
Definitions
- the present invention relates to novel compounds, to pharmaceutical compositions comprising the compounds, as well as to the use of the compounds in medicine and for the preparation of a medicament which acts on the human 11- ⁇ -hydroxysteroid dehydrogenase type 1 enzyme (11- ⁇ -hsd-1).
- 11- ⁇ -hydroxysteroid dehydrogenase type 1 enzyme 11- ⁇ -hsd-1.
- 10 Background Of The Invention It has been known for more than half a century that glucocorticoids have a central role in diabetes. For example, the removal of the pituitary or the adrenal gland from a diabetic animal alleviates the most severe symptoms of diabetes and lowers the concentration of glucose in the blood (Long, C. D. and F.
- the hepatic insulin sensitivity was improved in healthy human 20 volunteers treated with the non-specific 11- ⁇ -hsd-1 inhibitor carbenoxolone (Walker, B.R., et al. (1995) J. Clin. Endocrinol. Metab. 80: 3155-3159). Furthermore, the expected mechanism has been established by different experiments with mice and rats. These studies showed that the mRNA levels and activities of two key enzymes in hepatic glucose production were reduced, namely the rate-limiting enzyme in gluconeogenesis, phosphoenolpyruvate 25 carboxykinase (PEPCK), and glucose-6-phosphatase (G6Pase) catalyzing the last common step of gluconeogenesis and glycogenolysis.
- PEPCK phosphoenolpyruvate 25 carboxykinase
- G6Pase glucose-6-phosphatase
- the present invention relates to a compound of formula (I):
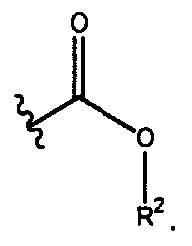
- R 1 N R 2 (") wherein: R 1 is selected from the group consisting of (Ct-Cejalkyl, -(CR 3 R 4 ) t (C 3 -C 12 )cycloalkyl,
- b and k are each independently selected from 1 and 2; j is selected from the group consisting of 0, 1 , and 2; t, u, p, q, and v are each independently selected from the group consisting of 0, 1 , 2, 3, 4, and 5; T is a (6-10)-membered heterocyclyl containing at least one nitrogen atom; R 2 is selected from the group consisting of H, (C -C ⁇ Jalkyl, -(CR 3 R 4 C 3 -C ⁇ 2 )cycloalkyl, -(CR 3 R ),(C 6 -C ⁇ 2 )aryl, and -(CR 3 R ),(4-10)-membered heterocyclyl; each R 3 and R 4 is independently selected from H and (d-C 6 )alkyl; the carbon atoms of T, R 1
- the invention relates to a compound according to formula (I), wherein b is 2.
- the invention relates to a compound according to formula (I), wherein T is a 6-membered heterocyclyl containing at least one nitrogen atom.
- the invention relates to a compound according to formula (I), wherein said T a (6-10)-membered heterocyclyl selected from the group consisting of
- the invention relates to a compound according to formula
- the invention relates to a compound according to formula , N ⁇ * (I), wherein T is r In an embodiment, the invention relates to a compound according to formula (I),
- the invention relates to a compound according to formula (II):
- R 1 is (d-C ⁇ Jalkyl, -(CR 7 R 8 ),(C 3 -C 10 )cycloalkyl, -(CR 7 R 8 ) t (C 6 -C 10 )aryl, or
- each R 2 , R 3 , and R 4 are independently selected from the group consisting of H, (d-C 6 )alkyl, -(CR 7 R 8 ),(C 3 -C 10 )cycloalkyl, -(CR 7 R 8 ),(C 6 -C 10 )aryl, and -(CR 7 R 8 ) t (4-10)-membered heterocyclyl; each R 2 and R 3 may optionally be taken together with the nitrogen to which they are attached to form a (4-10)-membered heterocyclyl; each R 5 and R 6 are independently selected from the group consisting of H, (d-C 6 ) alkyl, -(CR 7 R 8 ),(C 3 -d 0 )cycloalkyl, -(CR 7 R 8 ),(C 6 -C 10 )aryl, and -(CR 7 R 8 ),(4-10)
- the invention relates to a compound according to formula (II),
- the invention relates to a compound according to formula (II), wherein W is a 5-membered heterocyclyl.
- the invention relates to a compound according to formula (II), wherein said 5-membered heterocyclyl is selected from the group consisting of oxazolyl, thiazolyl, pyrazolyl, triazolyl, and oxadiazolyl.
- the invention relates to a compound according to formula (II), wherein b is 2.
- the invention relates to a compound according to formula (II), wherein T is a 6-membered heterocyclyl containing at least one nitrogen atom.
- the invention relates to a compound according to formula (II), wherein said 6-membered heterocyclyl is selected from the group consisting of
- the invention relates to a compound according to formula
- each R 1 is phenyl or napthyl substituted by 1 to 5 R 9 groups; wherein: each R 9 is independently selected from the group consisting of halo, cyano, -CF 3 , hydroxy, (C ⁇ -C ⁇ )alkoxy.
- the invention relates to a compound according to formula (II), wherein R 2 and R 3 are each independently selected from H and (d-C 6 )alkyl; wherein: said (Ci-C ⁇ ) alkyl is optionally substituted by (C 2 -C 6 ) alkenyl or -(CR 7 R 8 ),(C 3 -do)cycloalkyl.
- the invention relates to a compound according to formula (II), wherein R 2 and R 3 are taken together with the nitrogen to which they are attached to form a (4-10)-membered heterocyclyl.
- the invention relates to a compound according to fonnula (II), wherein said (4-10)-membered heterocyclyl is selected from the group consisting of:
- the invention relates to a compound according to formula (II), wherein R 2 is (d-C 6 )alkyl. In an embodiment, the invention relates to a compound according to formula (II), wherein n is 0 and at least one of R 5 and R 6 is H. In another embodiment, the invention relates to a compound selected from the group consisting of:
- An embodiment of the invention relates to a pharmaceutical composition comprising an effective amount of a compound according formula (I) or formula (II), or a pharmaceutically acceptable salt or solvate thereof, and a pharmaceutically acceptable carrier.
- the invention relates to a method of treating a condition that is mediated by the modulation of 11- ⁇ -hsd-1, the method comprising administering to a mammal an effective amount of a compound according formula (I) or formula (II), or a pharmaceutically acceptable salt or solvate thereof.
- the invention relates to a method of treating diabetes, metabolic syndrome, insulin resistance syndrome, obesity, glaucoma, hyperlipidemia, hyperglycemia, hyperinsulinemia, osteoporosis, tuberculosis, atherosclerosis, dementia, depression, virus diseases, inflammatory disorders, or diseases in which the liver is a target organ, the method comprising administering to a mammal an effective amount of a compound according to formula (I) or formula (II), or a pharmaceutically acceptable salt or solvate thereof.
- alkyl as used herein, unless otherwise indicated, includes saturated monovalent hydrocarbon radicals having straight or branched moieties.
- alkenyl as used herein, unless otherwise indicated, includes alkyl moieties having at least one carbon-carbon double bond wherein alkyl is as defined above and including E and Z isomers of said alkenyl moiety.
- alkynyl as used herein, unless otherwise indicated, includes alkyl moieties having at least one carbon-carbon triple bond wherein alkyl is as defined above.
- alkoxy as used herein, unless otherwise indicated, includes O-alkyl groups wherein alkyl is as defined above.
- amino as used herein, is intended to include the -NH 2 radical, and any substitutions of the N atom.
- halogen and halo, as used herein represent chlorine, fluorine, bromine or iodine.
- trifluoromethyl as used herein, is meant to represent a -CF 3 group.
- trifluoromethoxy as used herein, is meant to represent a -OCF 3 group.
- cyano as used herein, is meant to represent a -CN group.
- OMs as used herein, is intended to mean, unless otherwise indicated methanesulfonate.
- Me as used herein, unless otherwise indicated, is intended to mean means methyl.
- MeOH as used herein, unless otherwise indicated, is intended to mean means methanol.
- Et as used herein, unless otherwise indicated, is intended to mean means ethyl.
- Et 2 0 as used herein, unless otherwise indicated, is intended to mean means diethylether.
- EtOH as used herein, unless otherwise indicated, is intended to mean means ethanol.
- Et 3 N as used herein, unless otherwise indicated, is intended to mean means triethylamine.
- EtOAc is ethyl acetate.
- AIMe 2 CI as used herein, unless otherwise indicated, is intended to mean dimethyl aluminum chloride.
- Ac as used herein, unless otherwise indicated, is intended to mean means acetyl.
- TAA trifluoroacetic acid.
- TEA triethanolamine.
- HATU as used herein, unless otherwise indicated, is intended to mean ⁇ /, ⁇ /, ⁇ /' ⁇ /'-tetramethyluronium hexafluorophosphate.
- THF tetrahydrofuran
- TIOH thallium(l) hydroxide
- TIOEt thallium(l) ethoxide
- PCy 3 as used herein, is intended to mean tricyclohexylphosphine.
- Pd 2 (dba) 3 is intended to mean tris(dibenzylideneacetone)dipalladium(0).
- Pd(OAc) 2 is intended to mean palladium(ll) acetate.
- Pd(PPh 3 ) 2 CI 2 is intended to mean dichlorobis(triphenylphosphine)palladium(ll).
- Pd(PPh 3 ) 4 is intended to mean tetrakis(triphenylphophine)palladium(0).
- Pd(dppf)CI 2 is intended to mean (1,1'-bis(diphenylphosphino)ferrocene)dichloropalladium(ll), complex with dichloromethane (1:1).
- G6P is intended to mean glucose-6-phosphate.
- NIDDM is intended to mean non insulin dependent diabetes mellitus
- NADPH is intended to mean nicotinamide adenine dinucleotide phosphate, reduced form.
- CD 3 or CHLORFORM-D is intended to mean deuterochloroform.
- CD 3 OD is intended to mean deuteromethanol.
- CD 3 CN is intended to mean deuteroacetonitrile.
- DEAD is intended to mean diethyl azodicarboxylate.
- TsCH 2 NC is intended to mean tosylmethyl isocyanide.
- CIS0 3 H is intended to mean chlorosulfonic acid.
- DMSO-d 6 or DMSO-D 6 is intended to mean deuterodimethyl sulfoxide.
- DME deuterodimethyl sulfoxide.
- DMF di,2-dimethoxyethane.
- DMF dimethylformamide.
- DMSO dimethylsulfoxide.
- Dl dimethylsulfoxide.
- KOAc potassium acetate.
- nitrogeneat as used herein, is meant to represent an absence of solvent.
- the term “mmol” as used herein, is intended to mean millimole.
- the term “equiv” as used herein, is intended to mean equivalent.
- the term “mL” as used herein, is intended to mean milliliter.
- the term “U” as used herein, is intended to mean units.
- the term “mm” as used herein, is intended to mean millimeter.
- the term “g” as used herein, is intended to mean gram.
- the term “kg” as used herein, is intended to mean kilogram.
- the term “h” as used herein, is intended to mean hour.
- the term “min” as used herein, is intended to mean minute.
- the term “ ⁇ L” as used herein, is intended to mean microliter.
- ⁇ M micromolar.
- ⁇ m as used herein, is intended to mean micrometer.
- M as used herein, is intended to mean molar.
- N as used herein, is intended to mean normal.
- nm as used herein, is intended to mean nanometer.
- nM as used herein, is intended to mean nanoMolar.
- amu as used herein, is intended to mean atomic mass unit.
- °C as used herein, is intended to mean Celsius.
- m z as used herein, is intended to mean, unless otherwise indicated, mass/charge ratio.
- wt/wt is intended to mean weight/weight.
- v/v is intended to mean volume/volume.
- mL/min is intended to mean milliliter/minute.
- UV is intended to mean ultraviolet.
- APCI-MS as used herein, is intended to mean atmospheric pressure chemical ionization mass spectroscopy.
- HPLC as used herein, is intended to mean high performance liquid chromatograph.
- LC as used herein, is intended to mean liquid chromatograph.
- LCMS as used herein, is intended to mean liquid chromatography mass spectroscopy.
- SFC supercritical fluid chromatography
- sat as used herein, is intended to mean saturated.
- aq as used herein, is intended to mean aqueous.
- ELSD as used herein, is intended to mean evaporative light scattering detection.
- MS as used herein, is intended to mean mass spectroscopy.
- HRMS (ESI) as used herein, is intended to mean high resolution mass spectrometry (electrospray ionization).
- Anaal as used herein, is intended to mean analytical.
- Calcd as used herein, is intended to mean calculated.
- NT as used herein, unless otherwise indicated, is intended to mean not tested.
- NA as used herein, unless otherwise indicated, is intended to mean not tested.
- RT as used herein, unless otherwise indicated, is intended to mean room temperature
- Mth as used herein, unless otherwise indicated, is intended to mean Method
- Ce te ® as used herein, unless otherwise indicated, is intended to mean a white solid diatomite filter agent commercially available from World Minerals located in Los Angeles, California USA
- Eg as used herein, unless otherwise indicated, is intended to mean example Terms such as -(CR 3 R 4 ), or -(CR 10 R 11 ) V , for example, are used, R 3 , R 4 , R 10 and R 11 may vary with each iteration of t or v above 1 For instance, where t or v is 2 the terms - (CR 3 R ) V or -(CR 10 R 11 ), may equal -CH
- cycloalkyl refers to a non- aromatic, saturated or partially saturated, monocyclic or fused, spiro or unfused bicyc c or t ⁇ cyc c hydrocarbon referred to herein containing a total of from 3 to 10 carbon atoms, suitably 5-8 ring carbon atoms
- exemplary cycloalkyls include rings having from 3-10 carbon atoms, such as cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, cycloheptyl, and adamantyl
- Illustrative examples of cycloalkyl are derived from, but not limited to, the following
- aryl includes an organic radical derived from an aromatic hydrocarbon by removal of one hydrogen, such as phenyl or naphthyl.
- (3-7)-membered heterocyclyl includes aromatic and non-aromatic heterocyclic groups containing one to four heteroatoms each selected from O, S and N, wherein each heterocyclic group has from 3-7, 6-10, or 4-10 atoms, respectively, in its ring system, and with the proviso that the ring of said group does not contain two adjacent O or S atoms.
- Non-aromatic heterocyclic groups include groups having only 3 atoms in their ring system, but aromatic heterocyclic groups must have at least 5 atoms in their ring system.
- the heterocyclic groups include benzo-fused ring systems.
- An example of a 3 membered heterocyclic group is aziridine, an example of a 4 membered heterocyclic group is azetidinyl (derived from azetidine).
- An example of a 5 membered heterocyclic group is thiazolyl, an example of a 7 membered ring is azepinyl, and an example of a 10 membered heterocyclic group is quinolinyl.
- non-aromatic heterocyclic groups are pyrrolidinyl, tetrahydrofuranyl, dihydrofuranyl, tetrahydrothienyl, tetrahydropyranyl, dihydropyranyl, tetrahydrothiopyranyl, piperidino, morpholino, thiomorpholino, thioxanyl, piperazinyl, azetidinyl, oxetanyl, thietanyl, homopiperidinyl, oxepanyl, thiepanyl, oxazepinyl, diazepinyl, thiazepinyl, 1,2,3,6-tetrahydropyridinyl, 2-pyrrolinyl, 3-pyrrolinyl, indolinyl, 2H-pyranyl, 4H- pyranyl, dioxanyl, 1,3-dioxolanyl, pyrazolinyl, di
- aromatic heterocyclic groups are pyridinyl, imidazolyl, pyrimidinyl, pyrazolyl, triazolyl, pyrazinyl, tetrazolyl, furyl, thienyl, isoxazolyl, thiazolyl, oxazolyl, isothiazolyl, pyrrolyl, quinolinyl, isoquinolinyl, indolyl, benzimidazolyl, benzofuranyl, cinnolinyl, indazolyl, indolizinyl, phthalazinyl, pyridazinyl, triazinyl, isoindolyl, pteridinyl, purinyl, oxadiazolyl, thiadiazolyl, furazanyl, benzofurazanyl, benzothiophenyl, benzothiazolyl, benzoxazolyl, quinazolinyl, quinox
- a group derived from pyrrole may be pyrrol-1-yl (N-attached) or pyrrol-3-yl (C-attached).
- a group derived from imidazole may be imidazol-1-yl (N-attached) or imidazol-3-yl (C-attached).
- the 4-10 membered heterocyclic may be optionally substituted on any ring carbon, sulfur, or nitrogen atom(s) by one to two oxo, per ring.
- heterocyclic group wherein 2 ring carbon atoms are substituted with oxo moieties is 1,1-dioxo-thiomorpholinyl.
- 4-10 membered heterocyclic are derived from, but not limited to, the following:
- solvate is intended to mean a pharmaceutically acceptable solvate form of a specified compound that retains the biological effectiveness of such compound.
- examples of solvates include compounds of the invention in combination with water, isopropanol, ethanol, methanol, DMSO (dimethylsulfoxide), ethyl acetate, acetic acid, or ethanolamine.
- pharmaceutically acceptable salt(s) as used herein, unless otherwise indicated, includes salts of acidic or basic groups which may be present in the compounds of formula (I) or formula (II).
- the compounds of formula (I) or formula (II ) that are basic in nature are capable of forming a wide variety of salts with various inorganic and organic acids.
- the acids that may be used to prepare pharmaceutically acceptable acid addition salts of such basic compounds of formula (I) or formula (II) are those that form non-toxic acid addition salts, Le, salts containing pharmacologically acceptable anions, such as the acetate, benzenesulfonate, benzoate, bicarbonate, bisulfate, bitartrate, borate, bromide, calcium edetate, camsylate, carbonate, chloride, clavulanate, citrate, dihydrochloride, edetate, edislyate, estolate, esylate, ethylsuccinate, fumarate, gluceptate, gluconate, glutamate, glycollylarsanilate, hexylresorcinate, hydrabamine, hydrobromide, hydrochlor
- liver is a target organ
- diabetes hepatitis, liver cancer, liver fibrosis, and malaria.
- Metabolic syndrome as used herein, unless otherwise indicated means psoriasis, diabetes mellitus, wound healing, inflammation, neurodegenerative diseases, galactosemia, maple syrup urine disease, phenylketonuria, hypersarcosinemia, thymine uraciluria, sulfinuria, isovaleric acidemia, saccharopinuria, 4-hydroxybutyric aciduria, glucose-
- treating means reversing, alleviating, inhibiting the progress of, or preventing the disorder or condition to which such term applies, or one or more symptoms of such disorder or condition.
- treatment refers to the act of treating as “treating” is defined immediately above.
- modulate refers to the ability of a modulator for a member of the steroid/thyroid superfamily to either directly (by binding to the receptor as a ligand) or indirectly (as a precursor for a ligand or an inducer which promotes production of ligand from a precursor) induce expression of gene(s) maintained under hormone expression control, or to repress expression of gene(s) maintained under such control.
- modulator refers to the ability of a modulator for a member of the steroid/thyroid superfamily to either directly (by binding to the receptor as a ligand) or indirectly (as a precursor for a ligand or an inducer which promotes production of ligand from a precursor) induce expression of gene(s) maintained under hormone expression control, or to repress expression of gene(s) maintained under such control.
- hormone expression control or to repress expression of gene(s) maintained under such control.
- “obese” is defined, for males, as individuals whose body mass index is greater than 27.8 kg/ m 2 , and for females, as individuals whose body mass index is greater than 27.3 kg/m 2 .
- the invention method is not limited to those who fall within the above criteria. Indeed, the method of the invention can also be advantageously practiced by individuals who fall outside of these traditional criteria, for example, by those who may be prone to obesity.
- inflammatory disorders refers to disorders such as rheumatoid arthritis, ankylosing spondylitis, psoriatic arthritis, psoriasis, chondrocalcinosis, gout, inflammatory bowel disease, ulcerative colitis, Crohn's disease, fibromyalgia, and cachexia.
- therapeutically effective amount refers to that amount of drug or pharmaceutical agent that will elicit the biological or medical response of a tissue, system, animal, or human that is being sought by a researcher, veterinarian, medical doctor or other.
- Tritiated, i.e., 3 H, and carbon-14, i.e., 1 C, isotopes are particularly preferred for their ease of preparation and detectability. Further, substitution with heavier isotopes such as deuterium, i.e., 2 H, can afford certain therapeutic advantages resulting from greater metabolic stability, for example increased in vivo half-life or reduced dosage requirements and, hence, may be preferred in some circumstances.
- Isotopically labeled compounds of fo ⁇ nula (I) or formula (II) of this invention thereof can generally be prepared by carrying out the procedures disclosed in the Schemes and/or in the Examples below, by substituting a readily available isotopically labeled reagent for a non-isotopically labeled reagent.
- the compound of formula la may be prepared by reacting a compound of formula Ic, wherein the group C0 2 R 23 is an ester group such as methyl ester (C0 2 -CH 3 ) or ethyl ester (C0 2 -CH 2 CH 3 ), with aluminum amides (Me 2 AI-NR 2 R 3 ) or (MeAI(CI)-NR 2 R 3 ) in a suitable solvent (e.g. dichloromethane or toluene) advantageously, from room temperature to the boiling point of the solvent, typically from about 20 degrees Celsius to about 100 degrees Celsius.
- a suitable solvent e.g. dichloromethane or toluene
- the compound of formula la may also be prepared by reacting a compound of formula Ic, wherein the group C0 2 R 23 is a carboxylic acid (C0 2 H) with an amine of formula HNR 2 R 3 using standard amide coupling chemistry.
- Compounds of formula Ic may be prepared by reacting a compound of formula lla, wherein the group C0 2 R 23 is an ester group such as methyl ester (C0 2 -CH 3 ) or ethyl ester (C0 2 -CH 2 CH 3 ), with a R 1 - sulfonyl halide or R 1 -sulfinyl halide.
- the compound of formula la may be prepared by reacting a compound of formula Id with a R 1 -sulfonyl halide or R 1 -sulfinyl halide.
- Compounds of formula Id may be prepared by reacting a compound of formula lla, wherein the group C0 2 R 23 is an ester group such as methyl ester (C0 2 -CH 3 ) or ethyl ester (C0 2 - CH 2 CH 3 ), with aluminum amides (Me 2 AI-NR 2 R 3 ) or (MeAI(CI)-NR 2 R 3 ) in a suitable solvent (e.g. dichloromethane or toluene) at a temperature from room temperature to the boiling point of the solvent, typically from about 20 degrees Celsius to about 100 degrees Celsius.
- a suitable solvent e.g. dichloromethane or toluene
- the compound of formula lb may be obtained by cyclodehydration of suitable amide la.
- the compound of formula A may be prepared by reacting B with an R 1 -sulfonyl halide, R 1 -sulfinyl halide, or R 1 -sulfinate in the presence of a base such as an amine.
- a base such as an amine.
- bases include pyridine, triethylamine, and diisopropylethylamine.
- Suitable solvents include pyridine, dichloromethane, or THF.
- the aforementioned reaction can be conducted at room temperature or heated for an appropriate time period, such as 2 to 16 hours, depending on the solvent system used. After the reaction is substantially completed, the base may be removed in vacuo and the resulting residue may be purified using conventional purification techniques.
- R 1 is a non-fused ring system of more than one ring of either an aryl or heterocyclyl.
- the compound of formula A3 may be prepared by a palladium-catalyzed coupling reaction of A2 where X is a halo or trifluoromethylsulfonyl with a reagent Y-N where Y is aryl or heterocyclyl, N is boronic acid, boronate ester, stannane, or zincate.
- Suitable palladium sources for this reaction include Pd(PPh 3 ) 4 , Pd 2 (dba) 3 , Pd(PPh 3 ) 2 CI 2 or Pd(OAc) 2 .
- Ligands such as diphenylphosphinoethane, diphenylphosphinoferrocene, or triphenylphosphine may also be added.
- Suitable solvents for the palladium-catalyzed coupling reaction include dimethylformamide, tetrahydrofuran, or toluene.
- the aforementioned reaction can be conducted at a temperature of about 50 °C to about 150 °C with or without microwave heating for a time period of about 15 min to about 16 hours.
- base additives such as Na 2 C0 3 , Cs 2 C0 3 , TIOH, TIOEt may be added.
- base additives such as Na 2 C0 3 , Cs 2 C0 3 , TIOH, TIOEt may be added.
- Any of the above compounds of formula la, lb, Ic, Id, lla, A, B, A2, and A3 can be converted into another analogous compound by standard chemical manipulations. All starting materials, regents, and solvents are commercially available and are known to those of skill in the art unless otherwise stated. These chemical manipulations are known to those skilled in the art and include (a) removal of a protecting group by methods outlined in T. W. Greene and P.G.M.
- the compounds of the present invention may have asymmetric carbon atoms.
- Diastereomeric mixtures can be separated into their individual diastereomers on the basis of their physical chemical differences by methods known to those skilled in the art, for example, by chromatography or fractional crystallization.
- Enantiomers can be separated by converting the enantiomeric mixtures into a diastereomric mixture by reaction with an appropriate optically active compound (e.g., alcohol), separating the diastereomers and converting (e.g., hydrolyzing) the individual diastereomers to the corresponding pure enantiomers. All such isomers, including diastereomeric mixtures and pure enantiomers are considered as part of the invention.
- the compounds of formula (I) or formula (II) that are basic in nature are capable of forming a wide variety of different salts with various inorganic and organic acids. Although such salts must be pharmaceutically acceptable for administration to animals, it is often desirable in practice to initially isolate the compound of formula (I) or formula (II) from the reaction mixture as a pharmaceutically unacceptable salt and then simply convert the latter back to the free base compound by treatment with an alkaline reagent and subsequently convert the latter free base to a pharmaceutically acceptable acid addition salt.
- the acid addition salts of the base compounds of this invention are readily prepared by treating the base compound with a substantially equivalent amount of the chosen mineral or organic acid in an aqueous solvent medium or in a suitable organic solvent, such as methanol or ethanol.
- the desired solid salt is readily obtained.
- the desired acid salt can also be precipitated from a solution of the free base in an organic solvent by adding to the solution an appropriate mineral or organic acid.
- Those compounds of formula (I) or formula (II) that are acidic in nature are capable of forming base salts with various pharmacologically acceptable cations. Examples of such salts include the alkali metal or alkaline-earth metal salts and particularly, the sodium and potassium salts. These salts are all prepared by conventional techniques.
- the chemical bases which are used as reagents to prepare the pharmaceutically acceptable base salts of this invention are those which form non-toxic base salts with the acidic compounds of formula (I) or formula (II).
- Such non-toxic base salts include those derived from such pharmacologically acceptable cations as sodium, potassium, calcium, and magnesium, etc. These salts can easily be prepared by treating the corresponding acidic compounds with an aqueous solution containing the desired pharmacologically acceptable cations, and then evaporating the resulting solution to dryness, preferably under reduced pressure. Alternatively, they may also be prepared by mixing lower alkanolic solutions of the acidic compounds and the desired alkali metal alkoxide together, and then evaporating the resulting solution to dryness in the same manner as before. In either case, stoichiometric quantities of reagents are preferably employed in order to ensure completeness of reaction and maximum yields of the desired final product.
- the compounds of the present invention may be modulators of 11- ⁇ -hsd-1.
- the compounds of the present invention may modulate processes mediated by 11- ⁇ -hsd-1 , which refer to biological, physiological, endocrinological, and other bodily processes which are mediated by receptor or receptor combinations which are responsive to the 11- ⁇ -hsd-1 inhibitors described herein (e.g., diabetes, hyperlipidemia, obesity, impaired glucose tolerance, hypertension, fatty liver, diabetic complications (e.g. retinopathy, nephropathy, neurosis, cataracts and coronary artery diseases and the like), arteriosclerosis, pregnancy diabetes, polycystic ovary syndrome, cardiovascular diseases (e.g.
- ischemic heart disease and the like cell injury (e.g.) brain injury induced by strokes and the like) induced by atherosclerosis or ischemic heart disease, gout, inflammatory diseases (e.g. arthrosteitis, pain, pyrexia, rheumatoid arthritis, inflammatory enteritis, acne, sunburn, psoriasis, eczema, allergosis, asthma, Gl ulcer, cachexia, autoimmune diseases, pancreatitis and the like), cancer, osteoporosis and cataracts. Modulation of such processes can be accomplished in vitro or in vivo.
- inflammatory diseases e.g. arthrosteitis, pain, pyrexia, rheumatoid arthritis, inflammatory enteritis, acne, sunburn, psoriasis, eczema, allergosis, asthma, Gl ulcer, cachexia, autoimmune diseases, pancreatitis and the like
- cancer osteoporosis and cataracts.
- In vivo modulation can be carried out in a wide range of subjects, such as, for example, humans, rodents, sheep, pigs, cows, and the like.
- the compounds according to the present invention may be used in several indications which involve modulations of 11- ⁇ -hsd-1 enzyme.
- the compounds according to the present invention may be used against dementia (See WO97/07789), osteoporosis (See Canalis E 1996, "Mechanisms of Glucocorticoid Action in Bone: Implications to Glucocorticoid-lnduced Osteoporosis", Journal of Clinical Endocrinology and Metabolism, 81 , 3441-3447) and may also be used disorders in the immune system (see Franchimont, et.
- 11 - ⁇ -hsd-1 is suggested to have a role in aqueous production, rather than drainage, but it is presently unknown if this is by interfering with activation of the glucocorticoid or the mineralocorticoid receptor, or both.
- Bile acids inhibit 11- ⁇ -hydroxysteroid dehydrogenase type 2. This results in a shift in the overall body balance in favor of cortisol over cortisone, as shown by studying the ratio of the urinary metabolites (Quattropani C, Vogt B, Odermatt A, Dick B, Frey B M, Frey F J. 2001. J Clin Invest. Nov; 108(9): 1299-305.
- the compounds of the present invention may also be useful in the treatment of other metabolic disorders associated with impaired glucose utilization and insulin resistance include major late-stage complications of NIDDM, such as diabetic angiopathy, atherosclerosis, diabetic nephropathy, diabetic neuropathy, and diabetic ocular complications such as retinopathy, cataract formation and glaucoma, and many other conditions linked to NIDDM, including dyslipidemia glucocorticoid induced insulin resistance, dyslipidemia, polycysitic ovarian syndrome, obesity, hyperglycemia, hyperlipidemia, hypercholesteremia, hypertriglyceridemia, hyperinsulinemia, and hypertension. Brief definitions of these conditions are available in any medical dictionary, for instance, Stedman's Medical Dictionary (10 th Ed.).
- the 11 ⁇ -hsd-1 assay was performed in a 100mM Triethanolamine buffer pH 8.0, containing 200mM NaCI, 0.02% n-dodecyl ⁇ -D-maltoside, 5% glycerol, 5mM ⁇ - mercaptoethanol.
- a typical reaction for the determination of Kj app values was carried at R.T. in a Corning ® u-bottom 96-well plate and is described as follows: 11 ⁇ -hsd-1 enzyme (5 nM, final concentration) was pre-incubated in the presence of the inhibitor and NADPH (500 ⁇ M, final concentration) for at least 30 minutes in the assay buffer.
- the reaction was initiated by adding the regenerating system (2m M Glucose-6- Phosphate, 1U/mL Glucose-6-Phosphate dehydrogenase, and 6mM MgCI 2, all the concentration reported are final in the assay buffer), and 3H-cortisone (200 nM, final concentration). After 60 minutes, 60 ⁇ L of the assay mixture was transferred to a second 96- well plate and mixed with an equal volume of dimethylsulfoxide to stop the reaction.
- the regenerating system 2m M Glucose-6- Phosphate, 1U/mL Glucose-6-Phosphate dehydrogenase, and 6mM MgCI 2, all the concentration reported are final in the assay buffer
- 3H-cortisone 200 nM, final concentration
- a 15 ⁇ L aliquot from the reaction mixture was loaded into a C-18 column (Polaris C18-A, 50 x 4.6mm, 5 ⁇ , 180 Angstrom from Varian) connected to an automated High-throughput Liquid Chromatography instrument developed by Cohesive Technologies, commercially available from Franklin, Massachusetts USA, with a ⁇ -RAM model 3 Radio-HPLC detector from IN/US, commercially available from Tampa, Florida USA.
- the substrate and product peaks were separated by using an isocratic mixture of 43:57 methanol to water (v/v) at a flow rate of LOmUmin.
- the initial reaction velocities were measured by stopping the reaction at 60 min and by measuring the area of product formation in the absence and the presence of various concentrations of inhibitors.
- the K, a pp values were determined using the equation for tight- binding inhibitor developed by Morrison, JF. (Morrison JF. Biochim Biophys Acta. 1969; 185- 269-86).
- the radiolabeled [1 ,2-3H]-cortisone is commercially available from American
- the a pp values of the compounds of the present invention for the 11 - ⁇ -hsd-1 enzyme may lie typically between about 10 nM and about 10 ⁇ M.
- the compounds of the present invention that were tested all have K a pp's in at least one of the above SPA assays of less than 1 ⁇ M, preferably less than 100 nM. Certain preferred groups of compounds possess differential selectivity toward the various 11- ⁇ -hsd's.
- One group of preferred compounds possesses selective activity towards 11- ⁇ -hsd-1 over 11 ⁇ -hsd-2.
- Another preferred group of compounds possesses selective activity towards 11 ⁇ hsd-2 over 11- ⁇ -hsd- 1. (Morrison JF. Biochim Biophys Acta. 1969; 185: 269-86). Percentage of inhibition was determined in a 100mM Triethanolamine buffer, pH 8.0, 200mM NaCI, 0.02% n-dodecyl ⁇ -D-maltoside and 5mM ⁇ -ME.
- a typical reaction was carried on a Coming ® u-bottom 96-well plate and is described as follows: 11 ⁇ -hsd-1 enzyme (5 nM, final concentration) was pre-incubated in the presence of the inhibitor and NADPH (500 ⁇ M, final concentration) for at least 30 minutes in the assay buffer.
- the reaction was initiated by adding the regenerating system (2mM Glucose-6- Phosphate, 1U/mL Glucose-6-Phosphate dehydrogenase, and 6mM MgCI 2 all the concentration reported are final in the assay buffer), and 3H-cort ⁇ sone (200 nM, final concentration)
- 60 ⁇ L of the assay mixture was transferred to a second 96- well plate and mixed with an equal volume of dimethylsulfoxide to stop the reaction
- a 15 ⁇ L aliquot from the reaction mixture was loaded into a C-18 column (Polaris C18-A, 50 x 4 6mm, 5 ⁇ , 180 Angstrom from Varian) connected to an automated High-throughput Liquid Chromatography instrument developed by Cohesive Technologies commercially available from Franklin, Massachusetts, with a ⁇ -RAM model 3 Radio-HPLC detector from IN/US commercially available from Tampa, Florida
- the substrate and product peaks were separated by using an isocratic mixture
- Percent Inhibition was calculated based on the following equation (100 - (3H-Cort ⁇ sol peak area with ⁇ nh ⁇ b ⁇ tor/3Hcort ⁇ sol peak area without inhibitor) x 100) Certain groups of compounds possess differential selectivity toward the various 11- ⁇ -hsd enzymes One group of compounds possesses selective activity towards 11 - ⁇ -hsd-1 enzyme over 11 ⁇ -hsd-2 enzyme While another group of compounds possesses selective activity towards 11 ⁇ hsd-2 enzymes over 11 - ⁇ -hsd-1 enzymes [1 ,2-3H]-cort ⁇ so ⁇ e is commercially available from American Radiolabeled Chemicals Inc of St Louis, Missouri USA NADPH while Glucose-6-Phosphate and Glucose-6- Phosphate dehydrogenase was purchased from Sigma ® Pharmaceutical Compositions/Formulations.
- Formulations for oral use may be in the form of hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent, for example, calcium carbonate, calcium phosphate or kaolin. They may also be in the form of soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium, such as peanut oil, liquid paraffin or olive oil.
- Aqueous suspensions normally contain active ingredients in admixture with excipients suitable for the manufacture of an aqueous suspension.
- Such excipients may be a suspending agent, such as sodium carboxymethyl cellulose, methyl cellulose, hydroxypropylmethyl cellulose, sodium alginate, polyvinylpyrrolidone, gum tragacanth and gum acacia; a dispersing or wetting agent that may be a naturally occurring phosphatide such as lecithin, a condensation product of ethylene oxide and a long chain fatty acid, for example polyoxyethylene stearate, a condensation product of ethylene oxide and a long chain aliphatic alcohol such as heptadecaethylenoxycetanol, a condensation product of ethylene oxide and a partial ester derived from a fatty acid and hexitol such as polyoxyethylene sorbitol monooleate or a fatty acid hexitol anhydrides such as polyoxyethylene sorbitan monooleate.
- a suspending agent such as sodium carboxymethyl cellulose, methyl cellulose, hydroxypropylmethyl
- the pharmaceutical compositions may be in the form of a sterile injectable aqueous or oleagenous suspension.
- This suspension may be formulated according to know methods using those suitable dispersing or wetting agents and suspending agents that have been mentioned above.
- the sterile injectable preparation may also be formulated as a suspension in a non toxic perenterally-acceptable diluent or solvent, for example as a solution in 1 ,3- butanediol.
- the acceptable vehicles and solvents that may be employed are water, Ringers solution and isotonic sodium chloride solution.
- any bland fixed oil may be employed including synthetic mono- or diglycerides.
- fatty acids such as oleic acid find use in the preparation of injectables.
- the compounds of formula (I) or formula (II) may also be administered in the form of suppositories for rectal administration of the drug.
- These compositions can be prepared by mixing the drug with a suitable non-irritating excipient that is solid at about 25 Celsius but liquid at rectal temperature and will therefore melt in the rectum to release the drug.
- a suitable non-irritating excipient that is solid at about 25 Celsius but liquid at rectal temperature and will therefore melt in the rectum to release the drug.
- Such materials include cocoa butter and other glycerides.
- topical use preparations for example, creams, ointments, jellies solutions, or suspensions, containing the compounds of the present invention are employed.
- the compounds of formula (I) or formula (II) may also be administered in the form of liposome delivery systems such as small unilamellar vesicles, large unilamellar vesicles and multimellar vesicles.
- Liposomes can be formed from a variety of phospholipides, such as cholesterol, stearylamine or phosphatidylcholines.
- Dosage levels of the compounds of the present invention are of the order of about 0.5 mg/kg body weight to about 100 mg/kg body weight.
- a preferred dosage rate is between about 30 mg/kg body weight to about 100 mg/kg body weight.
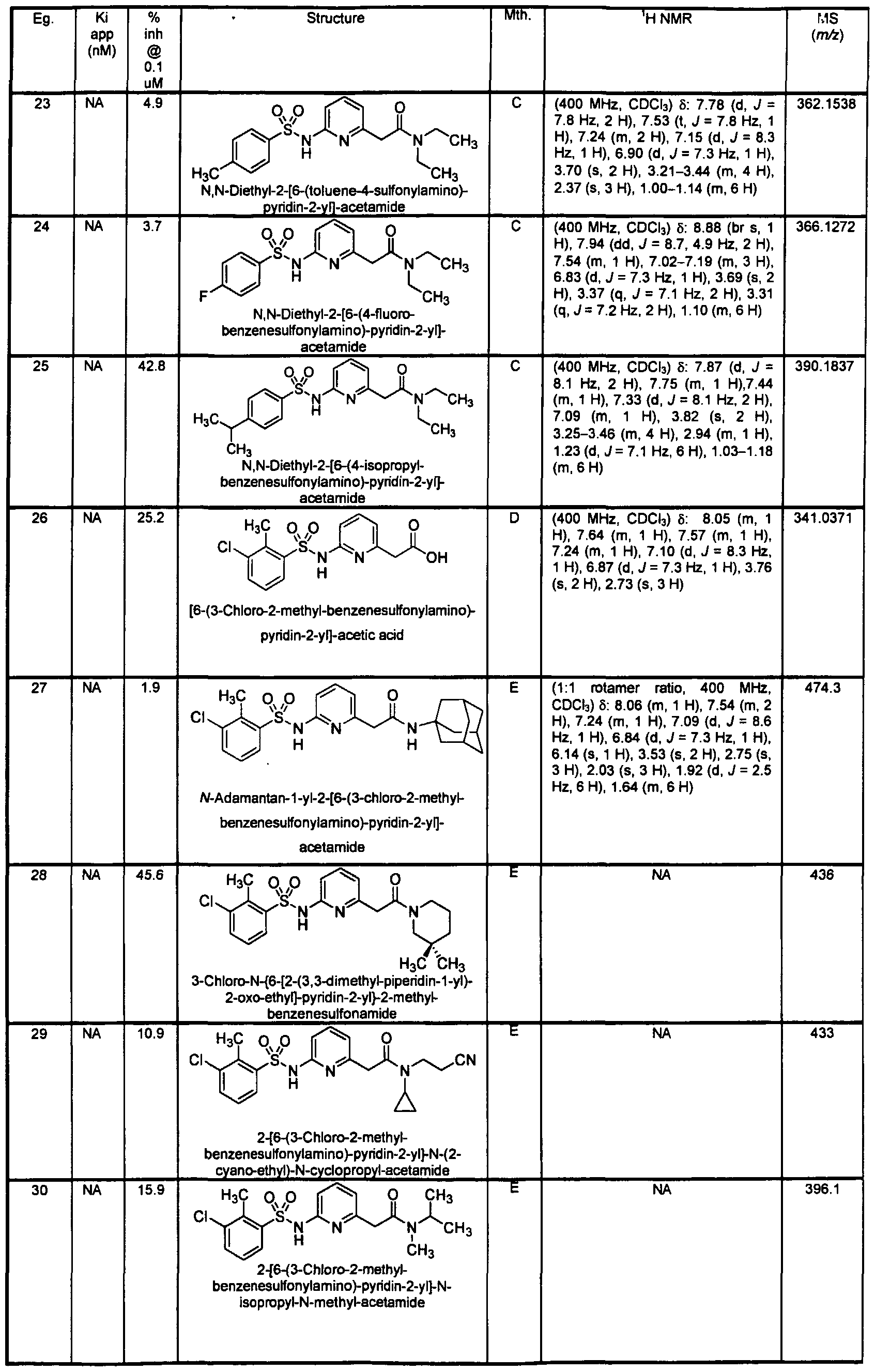
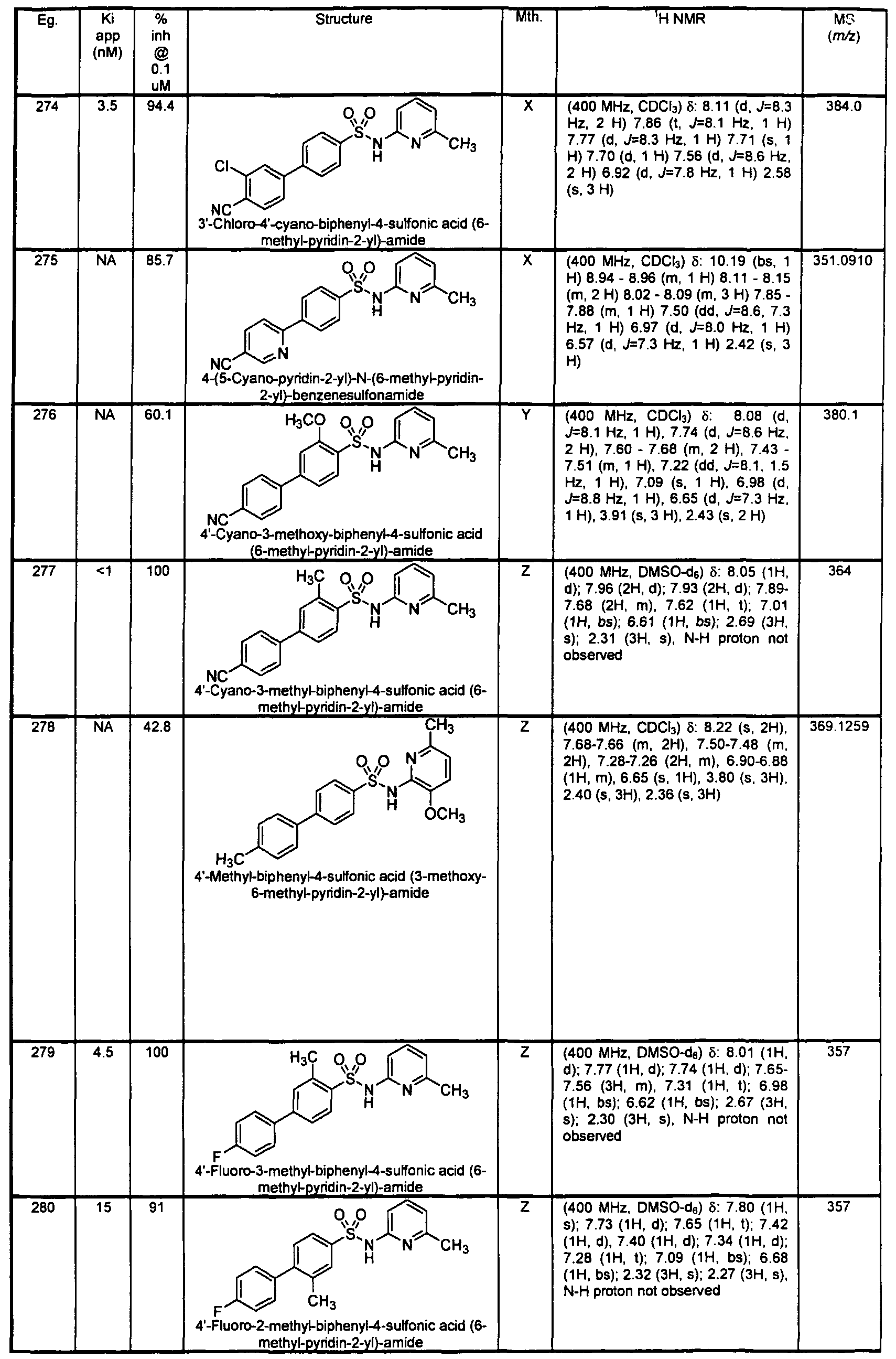
- Example 27 /V-Adama ⁇ tan-1-yl-2-[6-(3-chloro-2-methyl-benzenesulfonylamino)- pyridin-2-yl]-acetamide
- Reactant A Reactant B 1 equiv 1 equiv A stir bar, the amine ⁇ Reactant B, 400 ⁇ L, 80 ⁇ mol, 1.00 equiv 0.2 M in anhydrous DMF), [6-(3-chloro-2-methyl-benzenesulfonylamino)-pyridin-2-yl]-acetic acid (Reactant A
- HATU 160 ⁇ L, 80 ⁇ mol, 1.00 equiv 0.5 M in anhydrous DMF
- 10 x 75 mm test tube The tube was sealed with cellophane and the reaction stirred for 16 h at ambient temperature.
- the solvent was evaporated and the residue dissolved in DMSO containing 0.01 % BHT to yield a 0.05 M solution.
- the solution was injected into an automated HPLC system for purification.
- the solvent of the product containing fraction was evaporated, the residue dissolved in DMSO, analyzed, and submitted for screening.
- the resulting residue was purified with radial chromatography (2 mm silica plate, 2:1 hexanes / ethyl acetate) to yield a clear oil.
- the product was converted to a HCI salt by dissolving in 5 mL diethyl ether and adding 1 HCI in diethyl ether dropwise. The solid was triturated with additional ether and dried on high vacuum to afford the product (0.11 g, 29.5%).
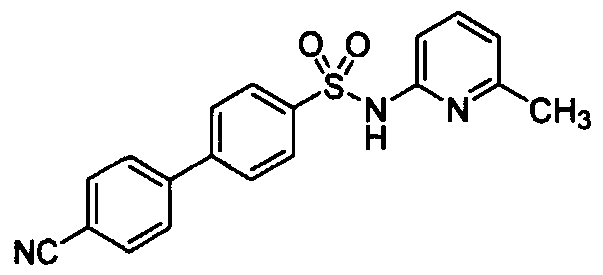
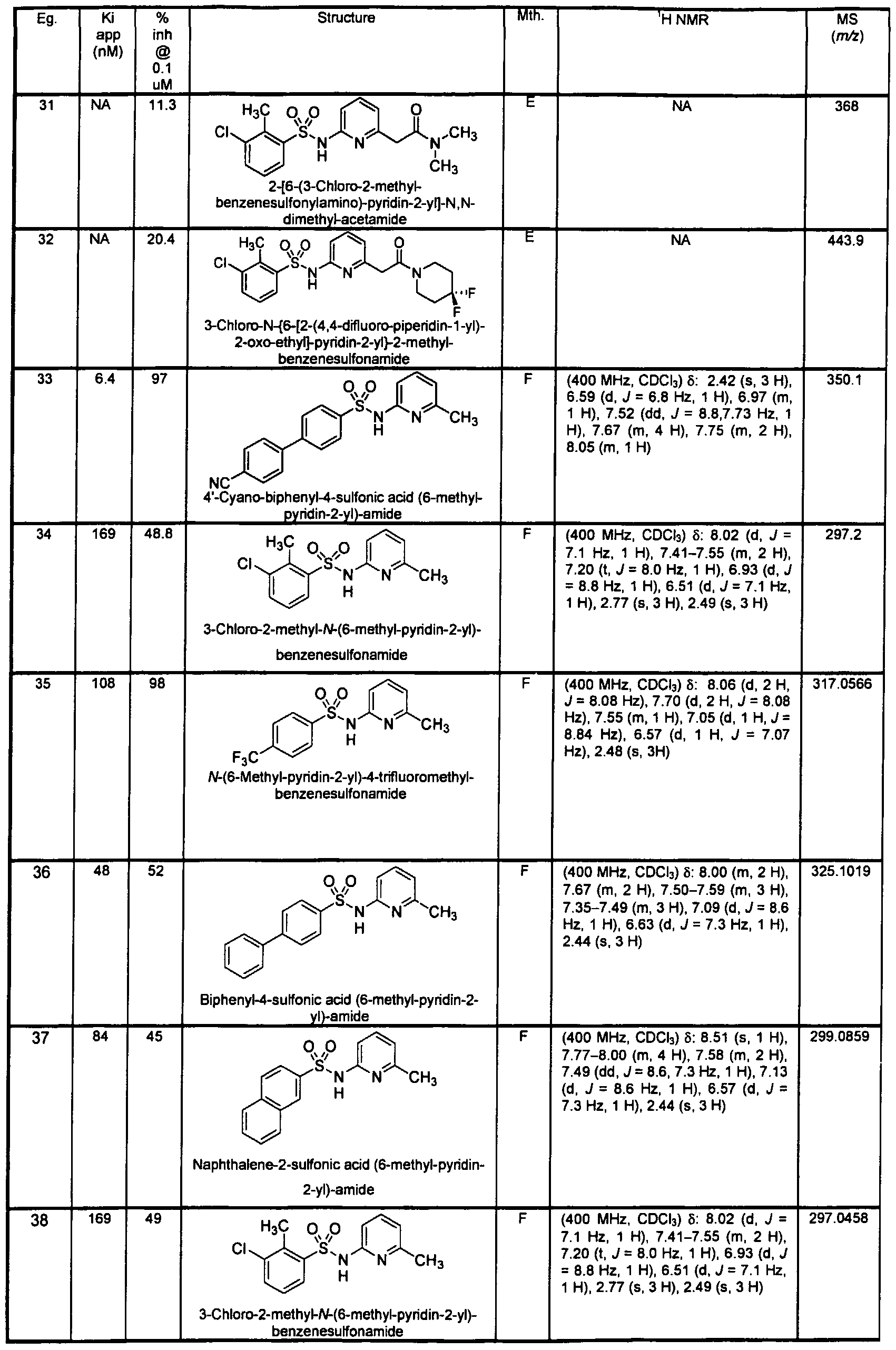
- 4'-Cya ⁇ o-biphenyl-4-sulfonic acid (6-cyclopropyl-pyridin-2-yl)-amide
- 4'-cyano-biphenyl-4- sulfonic acid (6-methyl-pyridin-2-yl)-amide but substituting 6-cyclopropyl-pyridin-2-ylamine and making non-critical variations.
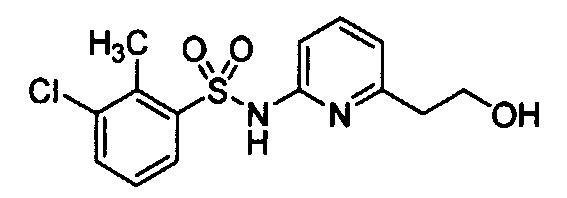
- Example 115 5-Chloro-3-rnethyl-benzo[b]thiophene-2-sulfonic acid [6-(2-hydroxy- ethyl)-pyridin-2-yl]-amide Lithium aluminum hydride (0.015 g, 0.310 mmol, 1.3 equiv) was added in one portion to an ice-cooled solution of [6-(5-Chloro-3-methyl-benzo[b]thiophene-2-sulfonylamino)-pyridin- 2-yl]-acetic acid ethyl ester (0.100 g, 0.235 mmol, 1 equiv) in tetrahydrofuran (4 mL).
- reaction mixture was warmed to 24 °C for 16 h.
- the reaction mixture was cooled to 0 °C, and excess lithium aluminum hydride was quenched with saturated aqueous ammonium chloride solution (10 mL).
- the resulting solution was warmed to 24 °C and stirred for an additional 30 min.
- the reaction mixture was filtered through a plug of Celite ® , and the resulting filtrate was extracted with dichloromethane (60 mL). The organic extract was dried over anhydrous sodium sulfate, filtered, and concentrated. Purification of the residue by high performance flash chromatography (0 ⁇ 1% methanol in dichloromethane) yielded product (0.0421 g, 47%).
- reaction mixtures were heated in a Personal Chemistry Microwave Synthesizer (SmithCreatorTM) for 15 min at 130 °C (energy-control setting for a high absorbing sample).
- the septum caps were removed and the reaction mixture was transferred into a 13x100 mm test tube while leaving any solid material behind.
- the microwave tubes were washed with DMF (1 mL) and the DMF was added to the receiving test tube. Next, the solvent was evaporated (SpeedVac, vaccum, medium heating, 16 h).
- 6-(4-Cyano-phenyl)-pyridi ⁇ e-3-sulfonic acid 6-methyl-pyridin-2-yl)-amide
- 6-chloro-pyridine-3-sulfonic acid 6-methyl-pyridin-2-yl)-amide
- 4-cyanoboronic acid 88 mg, 0.602 mmol
- Pd(PPh 3 ) 33 mg, 0.03 mmol
- aqueous Na 2 C0 3 (0.72 mL, 1.43 mmol
- 6-(4-Cyano-phenyl)-pyridine-3-sulfonic acid quinolin-2-ylamide 148 mg, 0.46 mmol
- 4-cyanophenylboronic acid 136 mg, 0.92 mmol
- DME 1.5 mL
- N,N- dimethylacetamide 2.0 mL
- H 2 0 0.5 mL
- Cs C0 3 451 mg, 1.39 mmol
- 6-(4-Cyano-phenyl)-pyridine-3-sulfonic acid (6-cyclopropyl-pyridin-2-yl)-amide
- 6-chloro-pyridine-3-sulfonic acid (6- cyclopropyl-pyridin-2-yl)-amide and 4-cyanophenyl boronic acid and making non-critical variations.
- Example 317 4-(4-Cyano-phenyl)-piperidine-1 -sulfonic acid (6-amino-pyridin-2-yl)- amide
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Diabetes (AREA)
- Physical Education & Sports Medicine (AREA)
- Rheumatology (AREA)
- Endocrinology (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Ophthalmology & Optometry (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Biomedical Technology (AREA)
- Pulmonology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Pain & Pain Management (AREA)
- Dermatology (AREA)
- Immunology (AREA)
- Urology & Nephrology (AREA)
- Heart & Thoracic Surgery (AREA)
- Oncology (AREA)
- Epidemiology (AREA)
- Communicable Diseases (AREA)
- Cardiology (AREA)
- Tropical Medicine & Parasitology (AREA)
- Psychiatry (AREA)
- Emergency Medicine (AREA)
- Gastroenterology & Hepatology (AREA)
Abstract
Description













































































































Claims




Priority Applications (12)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006544581A JP2007514731A (en) | 2003-12-19 | 2004-12-06 | Benzenesulfonylamino-pyridin-2-yl derivatives and related compounds as inhibitors of 11-beta-hydroxysteroid dehydrogenase type 1 (11-β-hsd-1) for the treatment of diabetes and obesity |
| CA002549651A CA2549651A1 (en) | 2003-12-19 | 2004-12-06 | Benzenesulfonylamino-pyridin-2-yl derivatives and related compounds as inhibitors of 11-beta-hydroxysteroid dehydrogenase type 1 (11-beta-hsd-1) for the treatment of diabetes and obesity |
| BRPI0417687-1A BRPI0417687A (en) | 2003-12-19 | 2004-12-06 | benzenesulfonylamino-pyridin-2-yl derivatives and related compounds as 11-beta-hydroxysteroid dehydrogenase type 1 (11-beta-hsd-1) inhibitors for the treatment of diabetes and obesity |
| EP04801352A EP1696915A1 (en) | 2003-12-19 | 2004-12-06 | Benzenesulfonylamino-pyridin-2-yl derivatives and related compounds as inhibitors of 11-beta-hydroxysteroid dehydrogenase type 1 (11-beta-hsd-1) for the treatment of diabetes and obesity |
| AU2004305321A AU2004305321A1 (en) | 2003-12-19 | 2004-12-06 | Benzenesulfonylamino-pyridin-2-yl derivatives and related compounds as inhibitors of 11-beta-hydroxysteroid dehydrogenase type 1 (11-beta-hsd-1) for the treatment of diabetes and obesity |
| EA200600990A EA200600990A1 (en) | 2003-12-19 | 2004-12-06 | DERIVATIVES OF BENZOSULPHONILYAMINOPYRIDIN-2-EFFLES AND RELATED COMPOUNDS AS 11-BETA-HYDROXYSTEROID-DEGYDROGENASE 1 TYPE (11-BETA-HSD-1) INHIBITORS FOR TREATMENT OF DIABETES FOR TREATMENT OF DIABETES TYPE 1 (11-BETA-HSD-1) FOR DIABETES TREATMENT |
| AP2006003633A AP2006003633A0 (en) | 2003-12-19 | 2004-12-06 | Benzenesulfonylamino-pyridin-2-yl derivatives and related compounds as inhibitors of 11-BETA-hydroxysteroid dehydrogenase type 1 (11-BETA-HSD-1) for the treatment of diabetes and obesity |
| MXPA06007077A MXPA06007077A (en) | 2003-12-19 | 2004-12-06 | Benzenesulfonylamino-pyridin-2-yl derivatives and related compounds as inhibitors of 11-beta-hydroxysteroid dehydrogenase type 1 (11-beta-hsd-1) for the treatment of diabetes and obesity. |
| IS8473A IS8473A (en) | 2003-12-19 | 2006-05-18 | Benzenesulfonylamino-pyridin-2-yl derivatives and related compounds that inhibit type 1 (11-beta-HSD-1) hydrochloric steroids in the treatment of diabetes and obesity |
| IL175949A IL175949A0 (en) | 2003-12-19 | 2006-05-25 | Benzenesulfonylamino-pyridin-2-yl derivatives and related compounds as inhibitors of 11-beta-hydroxy steroid dehydrogenase type1 (11-beta-hsd-1) for the treatment of diabetes and obesity |
| TNP2006000185A TNSN06185A1 (en) | 2003-12-19 | 2006-06-16 | BENZENESULFONYLAMINO-PYRIDINE-2-YLE DERIVATIVES AND RELATED COMPOUNDS AS INHIBITORS OF 11-BETA-HYDROXYSTEROID DEHYDROGENASE TYPE 1 (11-BETA-HSD-1) FOR THE TREATMENT OF DIABETES AND OBESITY |
| NO20063298A NO20063298L (en) | 2003-12-19 | 2006-07-17 | Benzenesulfonylamino-pyridin-2-yl derivatives and related compounds as inhibitors of 11-beta-hydroxy steroid dehydrogenase type 1 (11-beta-hsd-1) for the treatment of diabetes and obesity |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US53118603P | 2003-12-19 | 2003-12-19 | |
| US60/531,186 | 2003-12-19 | ||
| US55692104P | 2004-03-26 | 2004-03-26 | |
| US60/556,921 | 2004-03-26 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2005060963A1 true WO2005060963A1 (en) | 2005-07-07 |
| WO2005060963A8 WO2005060963A8 (en) | 2005-12-01 |
Family
ID=34713786
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2004/004056 WO2005060963A1 (en) | 2003-12-19 | 2004-12-06 | Benzenesulfonylamino-pyridin-2-yl derivatives and related compounds as inhibitors of 11-beta-hydroxysteroid dehydrogenase type 1 (11-beta-hsd-1) for the treatment of diabetes and obesity |
Country Status (24)
| Country | Link |
|---|---|
| US (1) | US20050148631A1 (en) |
| EP (1) | EP1696915A1 (en) |
| JP (1) | JP2007514731A (en) |
| KR (1) | KR20060101772A (en) |
| AP (1) | AP2006003633A0 (en) |
| AR (1) | AR046767A1 (en) |
| AU (1) | AU2004305321A1 (en) |
| BR (1) | BRPI0417687A (en) |
| CA (1) | CA2549651A1 (en) |
| DO (1) | DOP2004001052A (en) |
| EA (1) | EA200600990A1 (en) |
| EC (1) | ECSP066653A (en) |
| IL (1) | IL175949A0 (en) |
| IS (1) | IS8473A (en) |
| MA (1) | MA28271A1 (en) |
| MX (1) | MXPA06007077A (en) |
| NL (1) | NL1027811C2 (en) |
| NO (1) | NO20063298L (en) |
| OA (1) | OA13344A (en) |
| PA (1) | PA8620301A1 (en) |
| PE (1) | PE20050864A1 (en) |
| TW (1) | TW200530185A (en) |
| UY (1) | UY28674A1 (en) |
| WO (1) | WO2005060963A1 (en) |
Cited By (49)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006034804A1 (en) * | 2004-09-29 | 2006-04-06 | F.Hoffmann-La Roche Ag | Indozolone derivatives as 11b-hsd1 inhibitors |
| WO2006037501A1 (en) * | 2004-10-04 | 2006-04-13 | F. Hoffmann-La Roche Ag | Alkil-pyridines as 11-beta inhibitors for diabetes |
| WO2006050908A1 (en) * | 2004-11-08 | 2006-05-18 | Evotec Ag | Inhibitors of 11βετα-hydroxy steroid dehydrogenase type 1 (11beta-hsd1) |
| WO2006095822A1 (en) * | 2005-03-11 | 2006-09-14 | Ono Pharmaceutical Co., Ltd. | Sulfonamide compound and pharmaceutical thereof |
| WO2006106423A2 (en) * | 2005-04-07 | 2006-10-12 | Pfizer Inc. | Amino sulfonyl derivatives as inhibitors of human 11-.beta.-hydrosysteroid dehydrogenase |
| WO2006134467A1 (en) * | 2005-06-16 | 2006-12-21 | Pfizer Inc. | N-(pyridin-2-yl)-sulfonamide derivatives |
| WO2007025892A1 (en) | 2005-08-31 | 2007-03-08 | F. Hoffmann-La Roche Ag | 11-beta-hydroxysteroid dehydrogenase-1-inhibitor-diabetes-type 2-1 |
| WO2007047474A2 (en) | 2005-10-12 | 2007-04-26 | Vertex Pharmaceuticals Incorporated | Biphenyl derivatives as modulators of voltage gated ion channels |
| US7217838B2 (en) | 2005-01-05 | 2007-05-15 | Abbott Laboratories | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| WO2007073935A1 (en) * | 2005-12-29 | 2007-07-05 | Lek Pharmaceuticals D.D. | Heterocyclic compounds |
| WO2007089683A1 (en) * | 2006-01-31 | 2007-08-09 | Incyte Corporation | Amido compounds and their use as pharmaceuticals |
| WO2007114763A1 (en) * | 2006-03-31 | 2007-10-11 | Astrazeneca Ab | Sulphonamide derivates as modulators of the glucocorticoid receptor |
| WO2007128761A2 (en) | 2006-05-04 | 2007-11-15 | Boehringer Ingelheim International Gmbh | Uses of dpp-iv inhibitors |
| JP2009504741A (en) * | 2005-08-16 | 2009-02-05 | アイシーエージェン インコーポレイテッド | Voltage-gated sodium channel inhibitor |
| US7511175B2 (en) | 2005-01-05 | 2009-03-31 | Abbott Laboratories | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| US7645773B2 (en) | 2006-01-18 | 2010-01-12 | Hoffmann-La Roche Inc. | Thiazoles as inhibitors of 11β-hydroxysteroid dehydrogenase |
| US7728030B2 (en) | 2006-12-21 | 2010-06-01 | Astrazeneca Ab | Chemical compounds 572 |
| US7759339B2 (en) | 2005-03-31 | 2010-07-20 | Takeda San Diego, Inc. | Hydroxysteroid dehydrogenase inhibitors |
| US7776874B2 (en) | 2004-05-07 | 2010-08-17 | Incyte Corporation | Amido compounds and their use as pharmaceuticals |
| US7816391B2 (en) | 2007-02-12 | 2010-10-19 | Astrazeneca Ab | Chemical compounds |
| US7838544B2 (en) | 2006-05-17 | 2010-11-23 | Incyte Corporation | Heterocyclic inhibitors of 11-β hydroxyl steroid dehydrogenase type 1 and methods of using the same |
| US7880001B2 (en) | 2004-04-29 | 2011-02-01 | Abbott Laboratories | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase Type 1 enzyme |
| US7964618B2 (en) | 2006-11-03 | 2011-06-21 | Astrazeneca Ab | Chemical compounds |
| US7998959B2 (en) | 2006-01-12 | 2011-08-16 | Incyte Corporation | Modulators of 11-β hydroxyl steroid dehydrogenase type 1, pharmaceutical compositions thereof, and methods of using the same |
| US8030340B2 (en) | 2006-11-23 | 2011-10-04 | Astrazeneca Ab | Indazolyl sulphonamide derivatives useful as glucocorticoid modulators |
| WO2012033070A1 (en) | 2010-09-07 | 2012-03-15 | アステラス製薬株式会社 | Therapeutic agent for pain |
| WO2012035171A2 (en) | 2010-09-17 | 2012-03-22 | Kancera Ab | New compounds |
| US8193207B2 (en) | 2005-12-05 | 2012-06-05 | Incyte Corporation | Lactam compounds and methods of using the same |
| US8198331B2 (en) | 2005-01-05 | 2012-06-12 | Abbott Laboratories | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| US8207167B2 (en) | 2008-09-19 | 2012-06-26 | Pimco 2664 Limited | Aryl-phenyl-sulfonamide-phenylene compounds and their use |
| US8211930B2 (en) | 2008-05-20 | 2012-07-03 | Astrazeneca Ab | Phenyl and benzodioxinyl substituted indazoles derivatives |
| US8288417B2 (en) | 2004-06-24 | 2012-10-16 | Incyte Corporation | N-substituted piperidines and their use as pharmaceuticals |
| US8372841B2 (en) | 2004-04-29 | 2013-02-12 | Abbott Laboratories | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| US8415354B2 (en) | 2004-04-29 | 2013-04-09 | Abbott Laboratories | Methods of use of inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| US8435968B2 (en) | 2008-09-19 | 2013-05-07 | Pimco 2664 Limited | Aryl-phenyl-sulfonamido-cycloalkyl compounds and their use |
| WO2013093095A1 (en) * | 2011-12-22 | 2013-06-27 | Kancera Ab | Bisarylsulfonamides useful in the treatment of inflammation and cancer |
| US8524778B2 (en) | 2007-03-21 | 2013-09-03 | Pimco 2664 Limited | Biphenyl-4-yl-sulfonic acid arylamides and their use as therapeutic agents |
| US8524894B2 (en) | 2009-06-04 | 2013-09-03 | Laboratorios Salvat, S.A. | Inhibitor compounds of 11-beta-hydroxysteroid dehydrogenase type 1 |
| US8716345B2 (en) | 2005-01-05 | 2014-05-06 | Abbvie Inc. | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| US8940902B2 (en) | 2006-04-07 | 2015-01-27 | Abbvie Inc. | Treatment of central nervous system disorders |
| US9067922B2 (en) | 2013-04-19 | 2015-06-30 | Pfizer Limited | Chemical compounds |
| EP2940022A1 (en) * | 2014-04-30 | 2015-11-04 | Masarykova Univerzita | Furopyridines as inhibitors of protein kinases |
| US9371323B2 (en) | 2007-06-21 | 2016-06-21 | Incyte Holdings Corporation | Spirocycles as inhibitors of 11-beta hydroxyl steroid dehydrogenase type 1 |
| US9624167B2 (en) | 2013-06-26 | 2017-04-18 | Pimco 2664 Limited | N-(4-hydroxy-4-methyl-cyclohexyl)-4-phenyl-benzenesulfonamides and N-(4-hydroxy-4-methyl-cyclohexyl)-4-(2-pyridyl)benzenesulfonamides and their therapeutic use |
| EP3235813A1 (en) | 2016-04-19 | 2017-10-25 | Cidqo 2012, S.L. | Aza-tetra-cyclo derivatives |
| US10000449B2 (en) | 2011-12-22 | 2018-06-19 | Kancera Ab | Bisarylsulfonamides useful in the treatment of inflammation and cancer |
| US10005733B2 (en) | 2014-12-17 | 2018-06-26 | Pimco 2664 Limited | N-(4-hydroxy-4-methyl-cyclohexyl)-4-phenyl-benzenesulfonamide and N-(4-hydroxy-4-methyl-cyclohexyl)-4-(2-pyridyl)-benzenesulfonamide compounds and their therapeutic use |
| US10154988B2 (en) | 2012-11-14 | 2018-12-18 | The Johns Hopkins University | Methods and compositions for treating schizophrenia |
| US11434249B1 (en) | 2018-01-02 | 2022-09-06 | Seal Rock Therapeutics, Inc. | ASK1 inhibitor compounds and uses thereof |
Families Citing this family (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2527337A1 (en) * | 2005-04-14 | 2012-11-28 | Bristol-Myers Squibb Company | Inhibitors of 11-beta hydroxysteroid dehydrogenase type I |
| KR101450356B1 (en) * | 2006-11-24 | 2014-10-15 | 에이씨 이뮨 에스.에이. | N-(methyl)-1h-pyrazol-3-amine, n-(methyl)-pyridin-2-amine and n-(methyl)-thiazol-2-amine derivatives for the treatment of diseases associated with amyloid or amyloid-like proteins, like e.g. alzheimer's |
| DE102010033690A1 (en) * | 2010-08-06 | 2012-02-09 | Saltigo Gmbh | Process for the preparation of aminoarylalkyl compounds |
| JPWO2012124744A1 (en) * | 2011-03-14 | 2014-07-24 | 大正製薬株式会社 | Nitrogen-containing fused heterocyclic compounds |
| US20160168132A1 (en) * | 2013-07-31 | 2016-06-16 | Minoryx Therapeutics S.L. | Di(hetero)arylamides and sulfonamides, methods for their preparation and therapeutic uses thereof |
| WO2021092262A1 (en) * | 2019-11-05 | 2021-05-14 | Dermira, Inc. | Mrgprx2 antagonists and uses thereof |
Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0472053A2 (en) * | 1990-08-20 | 1992-02-26 | Eisai Co., Ltd. | Sulfonamide derivatives |
| WO1992011234A1 (en) * | 1990-12-24 | 1992-07-09 | Boehringer Mannheim Gmbh | Novel amines, process for producing them and medicaments containing said compounds |
| WO1993010094A1 (en) * | 1991-11-18 | 1993-05-27 | Kabi Pharmacia Ab | Novel substituted salicyclic acids |
| WO1995026957A1 (en) * | 1994-03-31 | 1995-10-12 | Zeneca Limited | N-heterocyclyl sulphonamide derivatives and their use as endothelin antagonists |
| EP0682016A1 (en) * | 1994-05-13 | 1995-11-15 | Zeneca Limited | Pyridine derivatives |
| WO1996036595A1 (en) * | 1995-05-19 | 1996-11-21 | Chiroscience Limited | 3,4-disubstituted-phenylsulphonamides and their therapeutic use |
| EP0815861A1 (en) * | 1996-06-28 | 1998-01-07 | F. Hoffmann-La Roche Ag | Sulphonamides and their use |
| WO1999047508A1 (en) * | 1998-03-17 | 1999-09-23 | Astrazeneca Ab | Benzenesulfonamide-derivatives and their use as medicaments |
| WO1999050237A1 (en) * | 1998-03-30 | 1999-10-07 | Hiroyoshi Hidaka | Sulfonamide derivatives and drugs containing the same as the active ingredient |
| WO2001017955A2 (en) * | 1999-09-04 | 2001-03-15 | Astrazeneca Ab | Hydroxyacetamidobenzenesulphonamide derivatives |
| WO2001090091A1 (en) * | 2000-05-22 | 2001-11-29 | Biovitrum Ab | Inhibitors of 11-beta-hydroxy steroid dehydrogenase type 1 |
| WO2003043999A1 (en) * | 2001-11-22 | 2003-05-30 | Biovitrum Ab | Inhibitors of 11-beta-hydroxy steroid dehydrogenase type 1 |
| WO2003099773A1 (en) * | 2002-05-24 | 2003-12-04 | Millennium Pharmaceuticals, Inc. | Ccr9 inhibitors and methods of use thereof |
| WO2004056744A1 (en) * | 2002-12-23 | 2004-07-08 | Janssen Pharmaceutica N.V. | Adamantyl acetamides as hydroxysteroid dehydrogenase inhibitors |
| WO2004103980A1 (en) * | 2003-05-21 | 2004-12-02 | Biovitrum Ab | Inhibitors of 11-beta-hydroxy steroid dehydrogenase type i |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6506754B1 (en) * | 2000-04-14 | 2003-01-14 | Corvas International, Inc. | Non-covalent thrombin inhibitors |
| RS44304A (en) * | 2001-11-22 | 2007-06-04 | Biovitrum Ab., | Inhibitors of 11-beta-hydroxy steroid dehydrogenase type 1 |
-
2004
- 2004-12-06 AU AU2004305321A patent/AU2004305321A1/en not_active Abandoned
- 2004-12-06 AP AP2006003633A patent/AP2006003633A0/en unknown
- 2004-12-06 CA CA002549651A patent/CA2549651A1/en not_active Abandoned
- 2004-12-06 EA EA200600990A patent/EA200600990A1/en unknown
- 2004-12-06 OA OA1200600201A patent/OA13344A/en unknown
- 2004-12-06 EP EP04801352A patent/EP1696915A1/en not_active Withdrawn
- 2004-12-06 JP JP2006544581A patent/JP2007514731A/en active Pending
- 2004-12-06 WO PCT/IB2004/004056 patent/WO2005060963A1/en not_active Application Discontinuation
- 2004-12-06 MX MXPA06007077A patent/MXPA06007077A/en not_active Application Discontinuation
- 2004-12-06 KR KR1020067012036A patent/KR20060101772A/en not_active Application Discontinuation
- 2004-12-06 BR BRPI0417687-1A patent/BRPI0417687A/en not_active IP Right Cessation
- 2004-12-10 PE PE2004001240A patent/PE20050864A1/en not_active Application Discontinuation
- 2004-12-15 AR ARP040104669A patent/AR046767A1/en unknown
- 2004-12-16 UY UY28674A patent/UY28674A1/en not_active Application Discontinuation
- 2004-12-16 DO DO2004001052A patent/DOP2004001052A/en unknown
- 2004-12-17 US US11/016,152 patent/US20050148631A1/en not_active Abandoned
- 2004-12-17 PA PA20048620301A patent/PA8620301A1/en unknown
- 2004-12-17 NL NL1027811A patent/NL1027811C2/en not_active IP Right Cessation
- 2004-12-17 TW TW093139461A patent/TW200530185A/en unknown
-
2006
- 2006-05-18 IS IS8473A patent/IS8473A/en unknown
- 2006-05-25 IL IL175949A patent/IL175949A0/en unknown
- 2006-06-16 EC EC2006006653A patent/ECSP066653A/en unknown
- 2006-06-19 MA MA29114A patent/MA28271A1/en unknown
- 2006-07-17 NO NO20063298A patent/NO20063298L/en not_active Application Discontinuation
Patent Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0472053A2 (en) * | 1990-08-20 | 1992-02-26 | Eisai Co., Ltd. | Sulfonamide derivatives |
| WO1992011234A1 (en) * | 1990-12-24 | 1992-07-09 | Boehringer Mannheim Gmbh | Novel amines, process for producing them and medicaments containing said compounds |
| WO1993010094A1 (en) * | 1991-11-18 | 1993-05-27 | Kabi Pharmacia Ab | Novel substituted salicyclic acids |
| WO1995026957A1 (en) * | 1994-03-31 | 1995-10-12 | Zeneca Limited | N-heterocyclyl sulphonamide derivatives and their use as endothelin antagonists |
| EP0682016A1 (en) * | 1994-05-13 | 1995-11-15 | Zeneca Limited | Pyridine derivatives |
| WO1996036595A1 (en) * | 1995-05-19 | 1996-11-21 | Chiroscience Limited | 3,4-disubstituted-phenylsulphonamides and their therapeutic use |
| EP0815861A1 (en) * | 1996-06-28 | 1998-01-07 | F. Hoffmann-La Roche Ag | Sulphonamides and their use |
| WO1999047508A1 (en) * | 1998-03-17 | 1999-09-23 | Astrazeneca Ab | Benzenesulfonamide-derivatives and their use as medicaments |
| WO1999050237A1 (en) * | 1998-03-30 | 1999-10-07 | Hiroyoshi Hidaka | Sulfonamide derivatives and drugs containing the same as the active ingredient |
| WO2001017955A2 (en) * | 1999-09-04 | 2001-03-15 | Astrazeneca Ab | Hydroxyacetamidobenzenesulphonamide derivatives |
| WO2001090091A1 (en) * | 2000-05-22 | 2001-11-29 | Biovitrum Ab | Inhibitors of 11-beta-hydroxy steroid dehydrogenase type 1 |
| WO2003043999A1 (en) * | 2001-11-22 | 2003-05-30 | Biovitrum Ab | Inhibitors of 11-beta-hydroxy steroid dehydrogenase type 1 |
| WO2003099773A1 (en) * | 2002-05-24 | 2003-12-04 | Millennium Pharmaceuticals, Inc. | Ccr9 inhibitors and methods of use thereof |
| WO2004056744A1 (en) * | 2002-12-23 | 2004-07-08 | Janssen Pharmaceutica N.V. | Adamantyl acetamides as hydroxysteroid dehydrogenase inhibitors |
| WO2004103980A1 (en) * | 2003-05-21 | 2004-12-02 | Biovitrum Ab | Inhibitors of 11-beta-hydroxy steroid dehydrogenase type i |
Cited By (95)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8415354B2 (en) | 2004-04-29 | 2013-04-09 | Abbott Laboratories | Methods of use of inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| US7880001B2 (en) | 2004-04-29 | 2011-02-01 | Abbott Laboratories | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase Type 1 enzyme |
| US8372841B2 (en) | 2004-04-29 | 2013-02-12 | Abbott Laboratories | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| US9133145B2 (en) | 2004-04-29 | 2015-09-15 | Abbvie Inc. | Methods of use of inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| US9670154B2 (en) | 2004-05-07 | 2017-06-06 | Incyte Holdings Corporation | Amido compounds and their use as pharmaceuticals |
| US9957229B2 (en) | 2004-05-07 | 2018-05-01 | Incyte Holdings Corporation | Amido compounds and their use as pharmaceuticals |
| US8058288B2 (en) | 2004-05-07 | 2011-11-15 | Incyte Corporation | Amido compounds and their use as pharmaceuticals |
| US9126927B2 (en) | 2004-05-07 | 2015-09-08 | Incyte Holdings Corporation | Amido compounds and their use as pharmaceuticals |
| US7776874B2 (en) | 2004-05-07 | 2010-08-17 | Incyte Corporation | Amido compounds and their use as pharmaceuticals |
| US8288417B2 (en) | 2004-06-24 | 2012-10-16 | Incyte Corporation | N-substituted piperidines and their use as pharmaceuticals |
| WO2006034804A1 (en) * | 2004-09-29 | 2006-04-06 | F.Hoffmann-La Roche Ag | Indozolone derivatives as 11b-hsd1 inhibitors |
| US7528158B2 (en) | 2004-09-29 | 2009-05-05 | Hoffmann-La Roche Inc. | Indazolone derivatives useful as 11b-HSD1 inhibitors |
| US7482341B2 (en) | 2004-10-04 | 2009-01-27 | Hoffmann-La Roche Inc. | Pyridines |
| JP2011157373A (en) * | 2004-10-04 | 2011-08-18 | F Hoffmann La Roche Ag | ALKYL-PYRIDINE AS 11-beta INHIBITOR FOR DIABETES |
| WO2006037501A1 (en) * | 2004-10-04 | 2006-04-13 | F. Hoffmann-La Roche Ag | Alkil-pyridines as 11-beta inhibitors for diabetes |
| JP2008515826A (en) * | 2004-10-04 | 2008-05-15 | エフ.ホフマン−ラ ロシュ アーゲー | Alkylpyridines as 11-beta inhibitors for diabetes |
| WO2006050908A1 (en) * | 2004-11-08 | 2006-05-18 | Evotec Ag | Inhibitors of 11βετα-hydroxy steroid dehydrogenase type 1 (11beta-hsd1) |
| US7855308B2 (en) | 2005-01-05 | 2010-12-21 | Abbott Laboratories | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase Type 1 enzyme |
| US7217838B2 (en) | 2005-01-05 | 2007-05-15 | Abbott Laboratories | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| US7511175B2 (en) | 2005-01-05 | 2009-03-31 | Abbott Laboratories | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| US7528282B2 (en) | 2005-01-05 | 2009-05-05 | Abbott Laboratories | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| US8993632B2 (en) | 2005-01-05 | 2015-03-31 | Abbvie Inc. | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| US9290444B2 (en) | 2005-01-05 | 2016-03-22 | Abbvie Inc. | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| US8716345B2 (en) | 2005-01-05 | 2014-05-06 | Abbvie Inc. | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| USRE41135E1 (en) | 2005-01-05 | 2010-02-16 | Abbott Laboratories | Inhibitors of the 11-β-hydroxysteroid dehydrogenase type 1 enzyme |
| US8314270B2 (en) | 2005-01-05 | 2012-11-20 | Abbott Laboratories | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| US8198331B2 (en) | 2005-01-05 | 2012-06-12 | Abbott Laboratories | Inhibitors of the 11-beta-hydroxysteroid dehydrogenase type 1 enzyme |
| WO2006095822A1 (en) * | 2005-03-11 | 2006-09-14 | Ono Pharmaceutical Co., Ltd. | Sulfonamide compound and pharmaceutical thereof |
| US7759339B2 (en) | 2005-03-31 | 2010-07-20 | Takeda San Diego, Inc. | Hydroxysteroid dehydrogenase inhibitors |
| WO2006106423A3 (en) * | 2005-04-07 | 2007-01-04 | Pfizer | Amino sulfonyl derivatives as inhibitors of human 11-.beta.-hydrosysteroid dehydrogenase |
| WO2006106423A2 (en) * | 2005-04-07 | 2006-10-12 | Pfizer Inc. | Amino sulfonyl derivatives as inhibitors of human 11-.beta.-hydrosysteroid dehydrogenase |
| WO2006134467A1 (en) * | 2005-06-16 | 2006-12-21 | Pfizer Inc. | N-(pyridin-2-yl)-sulfonamide derivatives |
| US7462634B2 (en) | 2005-06-16 | 2008-12-09 | Agouron Pharmaceuticals, Inc. | N-(pyridin-2-yl)-sulfonamide derivatives |
| JP2009504741A (en) * | 2005-08-16 | 2009-02-05 | アイシーエージェン インコーポレイテッド | Voltage-gated sodium channel inhibitor |
| WO2007025892A1 (en) | 2005-08-31 | 2007-03-08 | F. Hoffmann-La Roche Ag | 11-beta-hydroxysteroid dehydrogenase-1-inhibitor-diabetes-type 2-1 |
| US7622492B2 (en) | 2005-08-31 | 2009-11-24 | Hoffmann-La Roche Inc. | Pyrazolones as inhibitors of 11β-hydroxysteroid dehydrogenase |
| JP2009511599A (en) * | 2005-10-12 | 2009-03-19 | バーテックス ファーマシューティカルズ インコーポレイテッド | Biphenyl derivatives as regulators of voltage-gated ion channels |
| WO2007047474A3 (en) * | 2005-10-12 | 2007-07-05 | Vertex Pharma | Biphenyl derivatives as modulators of voltage gated ion channels |
| WO2007047474A2 (en) | 2005-10-12 | 2007-04-26 | Vertex Pharmaceuticals Incorporated | Biphenyl derivatives as modulators of voltage gated ion channels |
| US7683083B2 (en) | 2005-10-12 | 2010-03-23 | Vertex Pharmaceuticals Incorporated | Biphenyl derivatives as modulators of voltage gated ion channels |
| US8193207B2 (en) | 2005-12-05 | 2012-06-05 | Incyte Corporation | Lactam compounds and methods of using the same |
| WO2007073935A1 (en) * | 2005-12-29 | 2007-07-05 | Lek Pharmaceuticals D.D. | Heterocyclic compounds |
| US7998959B2 (en) | 2006-01-12 | 2011-08-16 | Incyte Corporation | Modulators of 11-β hydroxyl steroid dehydrogenase type 1, pharmaceutical compositions thereof, and methods of using the same |
| US7645773B2 (en) | 2006-01-18 | 2010-01-12 | Hoffmann-La Roche Inc. | Thiazoles as inhibitors of 11β-hydroxysteroid dehydrogenase |
| WO2007089683A1 (en) * | 2006-01-31 | 2007-08-09 | Incyte Corporation | Amido compounds and their use as pharmaceuticals |
| WO2007114763A1 (en) * | 2006-03-31 | 2007-10-11 | Astrazeneca Ab | Sulphonamide derivates as modulators of the glucocorticoid receptor |
| US9464072B2 (en) | 2006-04-07 | 2016-10-11 | Abbvie Inc. | Treatment of central nervous system disorders |
| US8940902B2 (en) | 2006-04-07 | 2015-01-27 | Abbvie Inc. | Treatment of central nervous system disorders |
| EP2351568A2 (en) | 2006-05-04 | 2011-08-03 | Boehringer Ingelheim International GmbH | Uses of dpp-iv inhibitors |
| WO2007128761A2 (en) | 2006-05-04 | 2007-11-15 | Boehringer Ingelheim International Gmbh | Uses of dpp-iv inhibitors |
| US7838544B2 (en) | 2006-05-17 | 2010-11-23 | Incyte Corporation | Heterocyclic inhibitors of 11-β hydroxyl steroid dehydrogenase type 1 and methods of using the same |
| US8673938B2 (en) | 2006-11-03 | 2014-03-18 | Astrazeneca Ab | Chemical compounds |
| US7964618B2 (en) | 2006-11-03 | 2011-06-21 | Astrazeneca Ab | Chemical compounds |
| US8030340B2 (en) | 2006-11-23 | 2011-10-04 | Astrazeneca Ab | Indazolyl sulphonamide derivatives useful as glucocorticoid modulators |
| US7728030B2 (en) | 2006-12-21 | 2010-06-01 | Astrazeneca Ab | Chemical compounds 572 |
| US8143290B2 (en) | 2006-12-21 | 2012-03-27 | Astrazeneca Ab | Chemical compounds 572 |
| US8344016B2 (en) | 2007-02-12 | 2013-01-01 | Astrazeneca Ab | Pyrazole derivatives as 11-beta-HSD1 inhibitors |
| US7816391B2 (en) | 2007-02-12 | 2010-10-19 | Astrazeneca Ab | Chemical compounds |
| US8524778B2 (en) | 2007-03-21 | 2013-09-03 | Pimco 2664 Limited | Biphenyl-4-yl-sulfonic acid arylamides and their use as therapeutic agents |
| US9371323B2 (en) | 2007-06-21 | 2016-06-21 | Incyte Holdings Corporation | Spirocycles as inhibitors of 11-beta hydroxyl steroid dehydrogenase type 1 |
| US9873698B2 (en) | 2007-06-21 | 2018-01-23 | Incyte Holdings Corporation | Spirocycles as inhibitors of 11-beta hydroxyl steroid dehydrogenase type 1 |
| US9512110B2 (en) | 2008-05-20 | 2016-12-06 | Astrazeneca Ab | Phenyl and benzodioxinyl substituted indazoles derivatives |
| US8211930B2 (en) | 2008-05-20 | 2012-07-03 | Astrazeneca Ab | Phenyl and benzodioxinyl substituted indazoles derivatives |
| US9738632B2 (en) | 2008-05-20 | 2017-08-22 | Astrazeneca Ab | Phenyl and benzodioxinyl substituted indazoles derivatives |
| US8916600B2 (en) | 2008-05-20 | 2014-12-23 | Astrazeneca Ab | Phenyl and benzodioxinyl substituted indazoles derivatives |
| US8207167B2 (en) | 2008-09-19 | 2012-06-26 | Pimco 2664 Limited | Aryl-phenyl-sulfonamide-phenylene compounds and their use |
| US8435968B2 (en) | 2008-09-19 | 2013-05-07 | Pimco 2664 Limited | Aryl-phenyl-sulfonamido-cycloalkyl compounds and their use |
| US9050329B1 (en) | 2008-09-19 | 2015-06-09 | Pimco 2664 Limited | Aryl-phenyl-sulfonamido-cycloalkyl compounds and their use |
| US9302984B2 (en) | 2008-09-19 | 2016-04-05 | Pimco 2664 Limited | Aryl-phenyl-sulfonamido-cycloalkyl compounds and their use |
| US8822507B2 (en) | 2008-09-19 | 2014-09-02 | Pimco 2664 Limited | Aryl-phenyl-sulfonamido-cycloalkyl compounds and their use |
| US9616037B2 (en) | 2008-09-19 | 2017-04-11 | Pimco 2664 Limited | Aryl-phenyl-sulfonamido-cycloalkyl compounds and their use |
| US8822452B2 (en) | 2009-06-04 | 2014-09-02 | Laboratorios Salvat, S.A. | Inhibitor compounds of 11-beta-hydroxysteroid dehydrogenase type 1 |
| US8524894B2 (en) | 2009-06-04 | 2013-09-03 | Laboratorios Salvat, S.A. | Inhibitor compounds of 11-beta-hydroxysteroid dehydrogenase type 1 |
| WO2012033070A1 (en) | 2010-09-07 | 2012-03-15 | アステラス製薬株式会社 | Therapeutic agent for pain |
| EA022338B1 (en) * | 2010-09-07 | 2015-12-30 | Астеллас Фарма Инк. | Use of triazole derivatives for treating pain |
| US9765040B2 (en) | 2010-09-07 | 2017-09-19 | Astellas Pharma Inc. | Therapeutic agent for pain |
| KR101747486B1 (en) | 2010-09-07 | 2017-06-14 | 아스텔라스세이야쿠 가부시키가이샤 | Therapeutic agent for pain |
| WO2012035171A3 (en) * | 2010-09-17 | 2012-06-21 | Kancera Ab | Sulfonamide compounds |
| US9233946B2 (en) | 2010-09-17 | 2016-01-12 | Kancera Ab | Sulfonamide compounds |
| WO2012035171A2 (en) | 2010-09-17 | 2012-03-22 | Kancera Ab | New compounds |
| US10000449B2 (en) | 2011-12-22 | 2018-06-19 | Kancera Ab | Bisarylsulfonamides useful in the treatment of inflammation and cancer |
| WO2013093095A1 (en) * | 2011-12-22 | 2013-06-27 | Kancera Ab | Bisarylsulfonamides useful in the treatment of inflammation and cancer |
| US10624875B2 (en) | 2012-11-14 | 2020-04-21 | The Johns Hopkins University | Methods and compositions for treating schizophrenia |
| EP3610890A1 (en) | 2012-11-14 | 2020-02-19 | The Johns Hopkins University | Methods and compositions for treating schizophrenia |
| US10154988B2 (en) | 2012-11-14 | 2018-12-18 | The Johns Hopkins University | Methods and compositions for treating schizophrenia |
| US9067922B2 (en) | 2013-04-19 | 2015-06-30 | Pfizer Limited | Chemical compounds |
| US9796670B2 (en) | 2013-06-26 | 2017-10-24 | Pimco 2664 Limited | N-(4-hydroxy-4-methyl-cyclohexyl)-4-phenyl-benzenesulfonamides and N-(4-hydroxy-4-methyl-cyclohexyl)-4-(2-pyridyl)benzenesulfonamides and their therapeutic use |
| US10029979B2 (en) | 2013-06-26 | 2018-07-24 | Pimco 2664 Limited | N-(4-hydroxy-4-methyl-cyclohexyl)-4-phenyl-benzenesulfonamides and N-(4-hydroxy-4-methyl-cyclohexyl)-4-(2-pyridyl)benzenesulfonamides and their therapeutic use |
| US10233147B2 (en) | 2013-06-26 | 2019-03-19 | Pimco 2664 Limited | N-(4-hydroxy-4-methyl-cyclohexyl)-4-phenyl-benzenesulfonamides and N-(4-hydroxy-4-methyl-cyclohexyl)-4-(2-pyridyl)benzenesulfonamides and their therapeutic use |
| US9624167B2 (en) | 2013-06-26 | 2017-04-18 | Pimco 2664 Limited | N-(4-hydroxy-4-methyl-cyclohexyl)-4-phenyl-benzenesulfonamides and N-(4-hydroxy-4-methyl-cyclohexyl)-4-(2-pyridyl)benzenesulfonamides and their therapeutic use |
| EP2940022A1 (en) * | 2014-04-30 | 2015-11-04 | Masarykova Univerzita | Furopyridines as inhibitors of protein kinases |
| US10005733B2 (en) | 2014-12-17 | 2018-06-26 | Pimco 2664 Limited | N-(4-hydroxy-4-methyl-cyclohexyl)-4-phenyl-benzenesulfonamide and N-(4-hydroxy-4-methyl-cyclohexyl)-4-(2-pyridyl)-benzenesulfonamide compounds and their therapeutic use |
| WO2017182464A1 (en) | 2016-04-19 | 2017-10-26 | Cidqo 2012, S.L. | New aza- tetracyclo derivatives |
| EP3235813A1 (en) | 2016-04-19 | 2017-10-25 | Cidqo 2012, S.L. | Aza-tetra-cyclo derivatives |
| US11434249B1 (en) | 2018-01-02 | 2022-09-06 | Seal Rock Therapeutics, Inc. | ASK1 inhibitor compounds and uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| AR046767A1 (en) | 2005-12-21 |
| AU2004305321A1 (en) | 2005-07-07 |
| AP2006003633A0 (en) | 2006-06-30 |
| MXPA06007077A (en) | 2006-08-23 |
| UY28674A1 (en) | 2005-07-29 |
| EA200600990A1 (en) | 2006-10-27 |
| EP1696915A1 (en) | 2006-09-06 |
| DOP2004001052A (en) | 2005-06-30 |
| OA13344A (en) | 2007-04-13 |
| BRPI0417687A (en) | 2007-04-03 |
| ECSP066653A (en) | 2006-10-25 |
| PE20050864A1 (en) | 2005-10-31 |
| WO2005060963A8 (en) | 2005-12-01 |
| IS8473A (en) | 2006-05-18 |
| NL1027811A1 (en) | 2005-06-21 |
| NO20063298L (en) | 2006-09-14 |
| IL175949A0 (en) | 2006-10-05 |
| US20050148631A1 (en) | 2005-07-07 |
| MA28271A1 (en) | 2006-11-01 |
| NL1027811C2 (en) | 2006-03-06 |
| TW200530185A (en) | 2005-09-16 |
| PA8620301A1 (en) | 2005-08-04 |
| CA2549651A1 (en) | 2005-07-07 |
| JP2007514731A (en) | 2007-06-07 |
| KR20060101772A (en) | 2006-09-26 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1696915A1 (en) | Benzenesulfonylamino-pyridin-2-yl derivatives and related compounds as inhibitors of 11-beta-hydroxysteroid dehydrogenase type 1 (11-beta-hsd-1) for the treatment of diabetes and obesity | |
| JP5335426B2 (en) | Diarylamine-containing compounds and compositions and their use as modulators of C-KIT receptors | |
| WO2006106423A2 (en) | Amino sulfonyl derivatives as inhibitors of human 11-.beta.-hydrosysteroid dehydrogenase | |
| JP2921578B2 (en) | Piperidine compound, its production method and its use | |
| EP0544756B1 (en) | 1,2,5,6-tetrahydro-1-methylpyridines, their preparation and use | |
| WO2007057768A2 (en) | Sulfonyl derivatives | |
| WO2006134481A1 (en) | Inhibitors of 11-beta hydroxysteroid dehydrogenase type 1 | |
| US5017573A (en) | Indazole-3-carboxylic acid derivatives | |
| WO2008122115A1 (en) | Inhibitors of histone deacetylase | |
| WO2003037887A1 (en) | Therapeutic isoquinoline compounds | |
| EP2771332B1 (en) | Thiophen and thiazol sulfonamid derivatives as HIV protease inhibitors for the treatment of AIDS | |
| CA2575785A1 (en) | 5-ht7 receptor antagonists | |
| JP2008509961A (en) | 5-HT7 receptor antagonist | |
| ZA200604887B (en) | Benzenesulfonylamino-pyridin-2-yl derivatives and related compounds as inhibitors of 11-beta-hydroxysteriod dehydrogenase type 1(11-beta-HSD-1) for the treatment of diabetes and obesity | |
| US5166341A (en) | 6-amino-1,4-hexahydro-1H-diazepine derivatives | |
| KR20070094949A (en) | 2-(cyclic aminocarbonyl)indoline derivative and medical composition containing the same | |
| Torrens Jover et al. | 5-HT 7 receptor antagonists | |
| AU2006231915A1 (en) | Amino sulfonyl derivatives as inhibitors of human 11-.beta.-hydrosysteroid dehydrogenase | |
| AU2002343313A1 (en) | Therapeutic isoquinoline compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200480038040.X Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| DPEN | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed from 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| CFP | Corrected version of a pamphlet front page | ||
| CR1 | Correction of entry in section i |
Free format text: IN PCT GAZETTE 27/2005 UNDER (71) REPLACE "PFIZER INC.?" BY "PFIZER INC." |
|
| CFP | Corrected version of a pamphlet front page |
Free format text: UNDER (54) PUBLISHED TITLE REPLACED BY CORRECT TITLE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004305321 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2904/DELNP/2006 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 547403 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: AP/P/2006/003633 Country of ref document: AP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 175949 Country of ref document: IL |
|
| ENP | Entry into the national phase |
Ref document number: 2004305321 Country of ref document: AU Date of ref document: 20041206 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004305321 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004801352 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2549651 Country of ref document: CA Ref document number: CR2006-008456 Country of ref document: CR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12006501186 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 06059282 Country of ref document: CO Ref document number: 1020067012036 Country of ref document: KR Ref document number: 2006544581 Country of ref document: JP Ref document number: 200600990 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 9464 Country of ref document: GE Ref document number: 20060222 Country of ref document: UZ Ref document number: PA/a/2006/007077 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: DE |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006/04887 Country of ref document: ZA Ref document number: 200604887 Country of ref document: ZA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1200601196 Country of ref document: VN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004801352 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020067012036 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: PI0417687 Country of ref document: BR |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2004801352 Country of ref document: EP |