PYRAZOLOPYRI Ml DINES AS ANTI - HEPATITIS C AGENTS
The present invention relates to a series of pyrazolo[3,4-d]pyrimidine derivatives which are useful in treating or preventing a hepatitis C infection. WO 94/13677 discloses pyrazolopyrimidines as CRF antagonists. All of the compounds specifically disclosed in that document carry a tnsubstituted phenyl group at the 1 -position. The present invention provides, in a first embodiment, the use of a pyrazolopyrimidine derivative of formula (I), or a pharmaceutically acceptable salt thereof, in the manufacture of a medicament for use in treating or preventing a flaviviridae infection, for example a hepatitis C infection,
wherein: Ri is C6-Cιo aryl, 5- to 10- membered heteroaryl, -(Cι-C4 alkyl)-(C6-C10 aryl) or -(Cι-C4 alkyl)-(5- to 10- membered heteroaryl); R2 is a C6-C10 aryl, C3-C6 carbocyclyl, 5- to 10- membered heteroaryl or 5- to 10- membered heterocyclyl moiety, said moiety being optionally fused to a C6- Cio aryl, C3-C6 carbocyclyl, 5- to 10- membered heteroaryl or 5- to 10- membered heterocyclic ring; and - X is -NR1-, -NR'-CO-NR!'-, -NR'-L- or -NR-CO-L-, wherein R7 and R/; are the same or different and each represent hydrogen or a Cι-C6 alkyl group and L represents a Cι-C6 alkylene group, the aryl, heteroaryl, heterocyclyl and carbocyclyl moieties in the Ri and R substituents being unsubstituted or substituted by 1, 2 or 3 substituents selected from halogen, Cι-C alkyl, Cι-C4 alkoxy, Cι-C haloalkyl, Cι-C4 haloalkoxy, cyano, nitro, C6-Cιo aryl, C3-C6 carbocyclyl, 5- to 10- membered heterocyclyl, 5- to 10- membered heteroaryl, -NR-CO2-R/, -CO2R , -COR;, -NR'-CO-R , -CONRR , -SO2NRR/ ,
- O-ϋRf'' and -0-(CH2)n-R//// substituents, wherein n is from 0 to 4, each R7 is the same or different and is hydrogen or C C6 alkyl, each R7/ is the same or different and is hydrogen, Cι-C6 alkyl, C6-C10 aryl, 5- to 10- membered heterocyclyl or 5- to 10- membered heteroaryl, each R7 is the same or different and is Cι-C6 alkyl, C6-C10 aryl, 5- to 10- membered heterocyclyl or 5- to 10- membered heteroaryl and each R 7 is the same or different and is C6-C10 aryl, 5- to 10- membered heterocyclyl or 5- to 10- membered heteroaryl, the aryl, heteroaryl, heterocyclyl and carbocyclyl moieties in said substituents being unsubstituted or substituted by a further substituent selected from Cι-C4 alkyl, - C hydroxalkyl and Cι-C4 haloalkyl groups. Preferably, the aryl, heteroaryl, heterocyclyl and carbocyclyl moieties in the Ri and R2 substituents are unsubstituted or substituted by 1, 2 or 3 substituents selected from halogen, Cι-C alkyl, Cι-C alkoxy, Cι-C4 haloalkyl, Cι-C4 haloalkoxy, cyano, nitro, C6-C10 aryl, C3-C6 carbocyclyl, 5- to 10- membered heterocyclyl, 5- to 10- membered heteroaryl, -NR-COz-R , -CO2R7, -COR^, -NR -CO-R7 , -CONR R ,
-SO NR
/R
// and -SO
2R
/7/ substituents, wherein each R
; is the same or different and is hydrogen or Cι
.-C
6 alkyl, each R is the same or different and is hydrogen, Ci-C
6 alkyl, Cβ-CiQ aryl, 5- to 10- membered heterocyclyl or 5- to 10- membered heteroaryl and each R
7/is the same or different and is Cι-C
6 alkyl, C
6-C
10 aryl, 5- to 10- membered heterocyclyl or 5- to 10- membered heteroaryl, the aryl, heteroaryl, heterocyclyl and carbocyclyl moieties in said substituents being unsubstituted or substituted by a further substituent selected from Cι-C alkyl, Ci- C
4 hydroxalkyl and Cι-C haloalkyl groups. For the avoidance of doubt, the orientation of the X moiety is such that the left hand side of the depicted groups are attached to the pyrazolopyrimidine ring and the right hand side of the depicted groups are attached to R
2. Thus, for example, when X is -NR -CO-L-, the 4- substituent on the pyrazolopyrimidine moiety is
As used herein, a Cι-C
6 alkyl group or moiety is a linear or branched alkyl group or moiety containing from 1 to 6 carbon atoms, such as a Cι-C
4 alkyl group or moiety. Examples of Cι-C
4 alkyl groups and moieties include methyl, ethyl, n-propyl, i-propyl, n-butyl, i-butyl and t-butyl. For the avoidance of doubt, where two alkyl moieties are present in a group, the alkyl moieties may be the same or different. As used herein, a Cι-C
6 alkylene group or moiety is a linear or branched
alkylene group or moiety, such as a -C
4 alkylene group or moiety. Examples include methylene, ethylene and n-propylene groups and moieties. Typically, as used herein, a C
6-Cι
0 aryl group or moiety is phenyl or naphthyl. Phenyl is preferred. As used herein, a halogen is typically chlorine, fluorine, bromine or iodine and is preferably chlorine, bromine or fluorine. As used herein, an alkoxy group is typically a said alkyl group attached to an oxygen atom. A haloalkyl or haloalkoxy group is typically a said alkyl or alkoxy group substituted by one or more said halogen atoms. Typically, it is substituted by 1, 2 or 3 said halogen atoms. Preferred haloalkyl and haloalkoxy groups include perhaloalkyl and perhaloalkoxy groups such as -CX
3 and -OCX
3 wherein X is a said halogen atom, for example chlorine and fluorine. Particularly preferred haloalkyl groups are -CF
3 and -CC1
3. Particularly preferred haloalkoxy groups are -OCF and -OCCl
3. As used herein, a 5- to 10- membered heteroaryl group or moiety is a monocyclic 5- to 10- membered aromatic ring, such as a 5- or 6- membered ring, containing at least one heteroatom, for example 1, 2 or 3 heteroatoms, selected from O, S and N. Examples include pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, furanyl, thienyl, pyrazolidinyl, pyrrolyl, oxadiazolyl, oxazolyl, isoxazolyl, thiazolyl, thiadiazolyl, imidazolyl and pyrazolyl groups. Pyrrolyl, oxazolyl, thiazolyl and pyrazolyl groups are preferred. As used herein, a 5- to 10- membered heterocyclyl group or moiety is a monocyclic non-aromatic, saturated or unsaturated cyclic group or moiety containing at least one, for example 1, 2 or 3, heteroatoms selected from N, O and S. Typically, it is a 3- to 6- membered ring. Preferably, it is a 5- or 6- membered ring. Suitable heterocyclyl groups and moieties include pyrazolidinyl, piperidyl, piperazinyl, morpholinyl, pyrrolidinyl, pyrrolinyl, imidazolidinyl, imidazolinyl and pyrazolinyl groups and moieties. Piperazinyl and morpholinyl groups and moieties are preferred. As used herein, a C
3-C
6 carbocyclic group or moiety is a monocyclic non- aromatic saturated or unsaturated hydrocarbon ring having from 3 to 6 carbon atoms. Preferably it is a saturated hydrocarbon ring (i.e. a cycloalkyl group) having from 3 to 6 carbon atoms. Examples include cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl. It is preferably cyclohexyl.
Typically, the aryl, heteroaryl, heterocyclyl and carbocyclyl moieties in the Ri and R
2 substituents are unsubstituted or substituted by 1, 2 or 3 substituents selected from halogen, Cι-C
4 alkyl, Cι-C alkoxy, Cι-C haloalkyl, Cι-C haloalkoxy, cyano, 5- to 6- membered heteroaryl, 5- to 6- membered heterocyclyl, -NR-CO
2-R , -CO
2R , -CO-R
;//, -NR-CO-R
7, -CONR'R , -SC^NR'R'', -SO
2R
7 and -0-(CH
2)
n-R
///7 substituents, wherein n is from 0 to 4, each R is the same or different and represents hydrogen or C
1-C
4 alkyl, each R
7 is the same or different and represents hydrogen, Ci- C
4 alkyl, a 5- or 6- membered heteroaryl group or a 5- or 6- membered heterocyclyl group, each R
;/ is the same or different and represents Cι-C
4 alkyl, a 5- or 6- membered heteroaryl group or a 5- or 6- membered heterocyclyl group, and each R ''is the same or different and represents a 5- to 6- membered heteroaryl group or a 5- to 6- membered heterocyclyl group, the heteroaryl and heterocyclyl moieties in said substituents being unsubstituted or substituted by a further substituent selected from Cι-C
4 alkyl and Cι-C
4 hydroxyalkyl groups. More typically, these preferred substituents are selected from selected from halogen, Cι-C
4 alkyl, Cι-C
4 alkoxy, Cι-C
4 haloalkyl, Cι-C haloalkoxy, cyano, 5- to 6- membered heteroaryl, 5- to 6- membered heterocyclyl, -NR
/-CO
2-R
/, -CO^, -CO-R^, -NR'-CO-R
7 , -CONR
/R
//, -SO
2NR
/R
// and -SO
2R
//7 substituents, wherein each R
7 is the same or different and represents hydrogen or Cι-C
4 alkyl, each R
; is the same or different and represents hydrogen, C -C
4 alkyl, a 5- or 6- membered heteroaryl group or a 5- or 6- membered heterocyclyl group, and each R is the same or different and represents Cι-C alkyl, a 5- or 6- membered heteroaryl group or a 5- or 6- membered heterocyclyl group, the heteroaryl and heterocyclyl moieties in said substituents being unsubstituted or substituted by a further substituent selected from -C
4 alkyl and - hydroxyalkyl groups. Typically, only one of the substituents on an aryl, heteroaryl, heterocyclyl or carbocyclyl moiety in the \ and R
2 substituents is a nitro, C
6-C
10 aryl, C
3-C
6 carbocyclyl, 5- to 10- membered heterocyclyl, 5- to 10- membered heteroaryl, -NR!- COz-R , -CO
2R , -COR
;/, -NR^CO-R^, -CONR , -SO
2NRR , -SC^R^ or -O-(CH
2)
n- R
7/ substituent. More typically, only one of the substituents on an aryl, heteroaryl,
heterocyclyl or carbocyclyl moiety in the Ri and R
2 substituents is a cyano, nitro, C
6-C
10 aryl, C
3-C
6 carbocyclyl, 5- to 10- membered heterocyclyl, 5- to 10- membered heteroaryl, -NR-CO
2-R
77, -CO
2R
7, -COR
777, -NR
7-CO-R
777, -CONR
7R
77, -SO
2NRR
//, -SO
2R
777 or -O-(CH
2)n-R
7 substituent. Preferably, the aryl, heteroaryl, heterocyclyl and carbocyclyl moieties in the Ri and R
2 substituents are unsubstituted or substituted by 1 or 2 unsubstituted substituents selected from halogen, Cι-C alkyl, Cι-C
4 alkoxy, Cι-C haloalkyl, Cι-C haloalkoxy, cyano, 5- to 6- membered heteroaryl, for example oxazolyl and thiazolyl, 5- to 6- membered heterocyclyl, for example morpholinyl, -CO
2R
7, -CONFER
77, -NR
7-COR
777, -Sθ
2-NR
7R
7777 and -O-(C
1-C
2 alkyl)-R
/ substituents, wherein each R
7 and R
77 are the same or different and represent hydrogen or Cι-C alkyl, each R is the same or different and represents a Cι-C
4 alkyl group, each R
7777 is the same or different and represents hydrogen, Cι-C
4 alkyl or 5- to 6- membered heteroaryl, for example thiazolyl, and each R
/is the same or different and represents 5- to 6- membered heteroaryl, for example pyridyl. Typically, these preferred unsubstituted substituents are selected from halogen, Cι-C
4 alkyl, Ci-C
4 alkoxy, Cι-C
4 haloalkyl, Cι-C
4 haloalkoxy, cyano, 5- to 6- membered heteroaryl, for example oxazolyl and thiazolyl, 5- to 6- membered heterocyclyl, for example morpholinyl, -CO
2R
7, -CONR
7R
77, -NR
7-COR
777 and -SO
2- NR
7R
7777 substituents, wherein each R
7 and R
77 are the same or different and represent hydrogen or Cι-C
4 alkyl, each R
77 is the same or different and represents a Cι-C
4 alkyl group and each R is the same or different and represents hydrogen, Cι-C alkyl or 5- to 6- membered heteroaryl, for example thiazolyl. Typically, R
1 is a C
6-C
10 aryl or 5- to 10- membered heteroaryl group. More typically, Ri is a C
6-C
10 aryl group, for example a phenyl group. Further, R
1 is typically unsubstituted or substituted by 1, 2 or 3 substituents selected from C
1-C
4 alkyl, C
1-C alkoxy, halogen and Cι-C
4 haloalkyl substituents. Preferably, Ri is unsubstituted or substituted by 1 or 2 substituents selected from Cι-C
4 alkyl, halogen and Cι-C
4 haloalkyl substituents. More preferably, R
\ is a phenyl group which is unsubstituted or substituted by 1 or 2 substituents selected from Cι-C
4 alkyl, halogen and C
1-C
4 haloalkyl substituents.
The R2 substituent is typically monocyclic or bicyclic. Typically, R
2 is a C
3-C
6 carbocyclyl or 5- to 10- membered heterocyclyl group or is a C
6-C
10 aryl or 5- to 10- membered heteroaryl moiety which moiety is optionally fused to a C
3-C
6 carbocyclyl group or to a phenyl ring. Preferably, R
2 is a phenyl or 5- to 6- membered heteroaryl moiety which is optionally fused to a phenyl or C
3-C
6 cycloalkyl group. Most preferably, R
2 is a phenyl, indolyl, indazolyl or 5,6,7,8-tetrahydronapthalenyl group. Typically, when R
2 is a fused ring system it is unsubstituted. When R
2 is a monocyclic moiety, it is typically unsubstitited or carries the substituents set out above as appropriate for the cyclic moieties in the Ri and R
2 substituents. Typically, X is -NR
7-, -NR
7-CO-NR
77-, -NR
7-L- or -NR
7-CO-L-, wherein R
7 and
R77 are the same or different and each represent hydrogen or a Cι-C4 alkyl group, and L represents a Cι-C4 alkylene group. Preferably, R and R77 are the same or different and each represent a C1-C2 alkyl group and L represents a Cι-C2 alkylene group. Preferred compounds of the invention are those in which: - Ri is a C6-C10 aryl or 5- to 10- membered heteroaryl group which is unsubstituted or substituted by 1, 2 or 3 substituents selected from Cι-C4 alkyl, Cι-C4 alkoxy, halogen and Cι-C4 haloalkyl substituents; R2 is a C3-C6 carbocyclyl or 5- to 10- membered heterocyclyl group or is a C6- C10 aryl or 5- to 10- membered heteroaryl moiety which moiety is optionally fused to a C3-C6 carbocyclyl group or to a phenyl ring, the aryl, heteroaryl, heterocyclyl and carbocyclyl moieties in the R2 substituents being unsubstituted or substituted by 1, 2 or 3 substituents selected from halogen, Cι-C4 alkyl, Cι-C alkoxy, CrC4 haloalkyl, Cι-C4 haloalkoxy, cyano, 5- to 6- membered heteroaryl, 5- to 6- membered heterocyclyl, -NR-CO2-R77, -CO2R77, -CO-R777, -NR7-CO-R777, -CONR7R77, -SO2NRR77, -SO2R777 and -O-(CH2)n-R /7 substituents, wherein n is from 0 to 4, each R7 is the same or different and represents hydrogen or Cι-C4 alkyl, each R is the same or different and represents hydrogen, Cι-C alkyl, a 5- or 6- membered heteroaryl group or a 5- or 6- membered heterocyclyl group, each R777 is the same or different and represents Cι-C alkyl, a 5- or 6- membered heteroaryl group or a 5- or 6- membered heterocyclyl group, and each R7777 is the same or different and represents a 5- to 6- membered heteroaryl group or a 5- to 6- membered heterocyclyl group, the heteroaryl and heterocyclyl moieties in said substituents being unsubstituted or
substituted by a further substituent selected from Cj.-C alkyl and Cι-C4 hydroxyalkyl groups; and X is -NR7-, NR7-CO-NR77, -NR-L- or -NR-CO-L- wherein R7 and R77 are the same or different and each represent hydrogen or a Cι-C alkyl group and L represents a Cι-C4 alkylene group. Preferably, in the above preferred compounds of the invention, R2 is a C3-C6 carbocyclyl or 5- to 10- membered heterocyclyl group or is a C6-C10 aryl or 5- to 10- membered heteroaryl moiety which moiety is optionally fused to a C3-C6 carbocyclyl group or to a phenyl ring, the aryl, heteroaryl, heterocyclyl and carbocyclyl moieties in the R2 substituents being unsubstituted or substituted by 1, 2 or 3 substituents selected from halogen, Cι-C4 alkyl, C1-C4 alkoxy, Cι-C haloalkyl, Cι-C haloalkoxy, cyano, 5- to 6- membered heteroaryl, 5- to 6- membered heterocyclyl, -NR-CO2-R77, -CO R77, -CO-R777, -NR7-CO-R777, -CONR7R77, -SO2NR7R77 and -SO2R77 substituents, wherein each R7 is the same or different and represents hydrogen or C1-C4 alkyl, each R77 is the same or different and represents hydrogen, C1-C4 alkyl, a 5- or 6- membered heteroaryl group or a 5- or 6- membered heterocyclyl group, and each R is the same or different and represents Cι-C4 alkyl, a 5- or 6- membered heteroaryl group or a 5- or 6- membered heterocyclyl group, the heteroaryl and heterocyclyl moieties in said substituents being unsubstituted or substituted by a further substituent selected from Cι-C4 alkyl and C1-C4. hydroxyalkyl groups. Most preferred compounds of the invention are those in which: Ri is a phenyl group which is unsubstituted or substituted by 1 or 2 substituents selected from Cι-C4 alkyl, halogen and C1-C4 haloalkyl substituents; R2 is a phenyl, indolyl, indazolyl or 5,6,7,8-tetrahydronaphthalenyl group which is unsubstituted or substituted with 1 or 2 unsubstituted substituents selected from halogen, Cι-C4 alkyl, C1-C4 alkoxy, C1-C4 haloalkyl, Cι-C4 haloalkoxy, cyano, 5- to 6- membered heteroaryl, for example oxazolyl and thiazolyl, 5- to 6- membered heterocyclyl, for example morpholinyl, -CO2R7, -CONRR77, -NR7- COR777, -SO2-NR7R7777 and -O-(C1-C2 alkyl)-R/ substituents, wherein each R7 and R77 are the same or different and represent hydrogen or Cι-C4 alkyl, each R777 is the same or different and represents a d-C4 alkyl group, each R7777 is the same or different and represents hydrogen, Cι-C4 alkyl or 5- to 6- membered heteroaryl,
for example thiazolyl and each R/is the same or different and represents 5- to 6- membered heteroaryl, for example pyridyl; and X is -NR7-, -NR-CO-NR7/, -NR7-L- or -NR-CO-L- wherein R7 and R77 are the same or different and each represent a C1-C2 alkyl group and L represents a Ci- C2 alkylene group. Typically, in these most preferred compounds of the invention, R is a phenyl, indolyl, indazolyl or 5,6,7,8-tetrahydronaphthalenyl group which is unsubstituted or substituted with 1 or 2 substituents selected from halogen, C1-C4 alkyl, C1-C4 alkoxy, C- 1-C4 haloalkyl, Cι-C4 haloalkoxy, cyano, 5- to 6- membered heteroaryl, for example oxazolyl and thiazolyl, 5- to 6- membered heterocyclyl, for example morpholinyl, -
CO2R7, -CONR7R77, -NR7-COR777 and -SO2-NR7R7777 substituents, wherein each R7 and R77 are the same or different and represent hydrogen or C1-C4 alkyl, each R777 is the same or different and represents a C1-C4 alkyl group and each R7777 is the same or different and represents hydrogen, C1-C4 alkyl or 5- to 6- membered heteroaryl, for example thiazolyl. In a further embodiment of the invention, the aryl, heteroaryl, heterocyclyl and carbocyclyl moieties in the R2 substituent do not carry a -CONRR77 substituent when X is -NR- and Ri is an unsubstituted phenyl group. Preferably, in this embodiment, the aryl, heteroaryl, heterocyclyl and carbocyclyl moieties in the Ri and R2 substitutents do not carry a -CONRR77 substituent whatever moieties are present at the X and R\ positions. More preferably, in this embodiment, the aryl, heteroaryl, heterocyclyl and carbocyclyl moieties in the R1 and R2 substituents do not carry an -NR7-CO2-R77, -CO2R77, -COR777, -NR7-CO-R777, -CONR7R77 or -SO2-NR7R77 substituent when X is -NR7-. Most preferably, in this embodiment, the aryl, heteroaryl, heterocyclyl and carbocyclyl moieties in the Ri and R2 substituents do not carry an -NR7-CO2-R77, -CO2R/, -COR777, -NR7-CO-R777, -CONR7R77 or -SO2-NR7R77 substituent whatever moiety is present at the X position. In a further embodiment of the invention, X is other than -NR7-. The medicaments of the present invention are for use in treating or preventing a flaviviridae infection, in particular a hepatitis C infection, in the human or animal body. Preferably, the medicaments are for use in humans.
Compounds of formula (I) containing one or more chiral centre may be used in enantiomerically or diastereoisomerically pure form, or in the form of a mixture of isomers. For the avoidance of doubt, the compounds of formula (I) can, if desired, be used in the form of solvates. Further, for the avoidance of doubt, the compounds of the invention may be used in any tautomeric form. As used herein, a pharmaceutically acceptable salt is a salt with a pharmaceutically acceptable acid or base. Pharmaceutically acceptable acids include both inorganic acids such as hydrochloric, sulphuric, phosphoric, diphosphoric, hydrobromic or nitric acid and organic acids such as citric, fumaric, maleic, malic, ascorbic, succinic, tartaric, benzoic, acetic, methanesulphonic, ethanesulphonic, benzenesulphonic or j9-toluenesulphonic acid. Pharmaceutically acceptable bases include alkali metal (e.g. sodium or potassium) and alkali earth metal (e.g. calcium or magnesium) hydroxides and organic bases such as alkyl amines, aralkyl amines and heterocyclic amines. Particularly preferred compounds of formula (I) include: l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-[2-(lH-indol-3-yl)-ethyl]- amine;
[2-(lH- dol-3-yl)-ethyl]-(l-phenyl-lH-pyrazolo[3,4-d]pyrimidin-4-yl)-amine;
(4-Butyl-phenyl)-(l-phenyl-lH-pyrazolo[3,4-d]pyrimidin-4-yl)-amine; (4-Butyl-phenyl)-[l-(2,4-dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-amine;
(4-Ethoxy-phenyl)-( 1 -phenyl- 1 H-pyrazolo [3 ,4-d]pyrimidin-4-yl)-amine;
[ 1 -(2,4-Dimethyl-phenyl)- 1 H-pyrazolo [3 ,4-d]pyrimidin-4-yl] -(4-ethoxy-phenyl)-amine;
4-[l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-ylamino]-benzoic acid ethyl ester; (5-Chloro-2-methoxy-phenyl)-(l-phenyl-lH-pyrazolo[3,4-d]pyrimidin-4-yl)-amine;
(5-Chloro-2-methoxy-phenyl)-[l-(4-chloro-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]- amine;
[l-(4-Chloro-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-(3-fluoro-phenyl)-amine;
4- {2-[ 1 -(2,4-Dimethyl-phenyl)- lH-pyrazolo[3,4-d]pyrimidin-4-ylamino]-ethyl} - benzenesulfonamide;
[l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-(3-fluoro-phenyl)-amine;
4-{2-[l-(2-Bromo-phenyl)-3H-pyrazolo[3,4-d]pyrimidin-4-ylamino]-ethyl}- benzenesulfonamide;
[ 1 -(2-Bromo-phenyl)- lH-pyrazolo [3 ,4-d]pyrimidin-4-yl] -(4-butyl-phenyl)-amine;
4-[2-(l-o-Tolyl-lH-pyrazolo[3,4-d]pyrimidin-4-ylamino)-ethyl]-benzenesulfonamide;
[2-(lH-lndol-3-yl)-ethyl]-(l-o-tolyl-lH-pyrazolo[3,4-d]pyrimidin-4-yl)-amine;
(4-Butyl-phenyl)-(l-o-tolyl-lH-pyrazolo[3,4-d]pyrimidin-4-yl)-amine; 4-{2-[l-(2,4-Dichloro-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-ylamino]-ethyl}- benzenesulfonamide;
[l-(2,4-Dichloro-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-[2-(lH-indol-3-yl)-ethyl]- amine;
[l-(2,4-Dichloro-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-(4-ethoxy-phenyl)-amine; 4-{2-[l-(3-Chloro-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-ylamino]-ethyl}- benzenesulfonamide;
(4-Butyl-phenyl)- [ 1 -(3 -chloro-phenyl)- 1 H-pyrazolo [3 ,4-d]pyrimidin-4-yl] -amine;
(5-Chloro-2-methoxy-phenyl)-[l-(3-chloro-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]- amine; 4- {2-[ 1 -(2-Chloro-phenyl)- 1 H-pyrazolo[3 ,4-d]pyrimidin-4-ylamino] -ethyl} - benzenesulfonamide;
[l-(2-Chloro-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-[2-(lH-indol-3-yl)-ethyl]- amine;
[l-(2-Chloro-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-(4-ethoxy-phenyl)-amine; (4-Butyl-phenyl)-[l-(2-chloro-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-amine;
(5-Chloro-2-methoxy-phenyl)-[ 1 -(2-chloro-phenyl)- 1 H-pyrazolo [3 ,4-d]pyrimidin-4-yl] - amine;
4- {2-[ 1 -(2-Trifluoromethyl-phenyl)- lH-pyrazolo[3,4-d]pyrimidin-4-ylamino]-ethyl} - benzenesulfonamide; [2-(lH-mdol-3-yl)-ethyl]-[l-(2-trifluoromethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-
4-yl] -amine;
(3-Chloro-phenyl)-[l-(2,4-dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-amine;
(3-Bromo-phenyl)-[l-(2,4-dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-amine;
[l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-(3-trifluoromethyl- phenyl)-amine;
[ 1 -(2,4-Dimethyl-phenyl)- 1 H-pyrazolo [3 ,4-d]pyrimidin-4-yl] -(4-trifluoromethyl- phenyl)-amine;
3-[l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-ylamino]-benzonitrile;
4- [ 1 -(2,4-Dimethyl-phenyl)- 1 H-pyrazolo [3,4-d]pyrimidin-4-ylamino] -benzonitrile;
[l-(4-Bromo-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-(4-butyl-phenyl)-amine;
[ 1 -(4-Bromo-phenyl)- 1 H-pyrazolo [3 ,4-d]pyrimidin-4-yl] -(5-chloro-2-methoxy-phenyι)- amine; [l-(2,4-Difluoro-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-[2-(lH-indol-3-yl)-ethyl]- amine;
(4-Butyl-phenyl)-[l-(2,4-difluoro-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-amine;
(5-Chloro-2-methoxy-phenyl)-[l-(2,4-difluoro-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4- yl] -amine; (3,5-Dichloro-phenyl)-[l-(2,4-dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]- amine;
[l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-(4-oxazol-5-yl-phenyl)- amine;
(3-Chloro-4-fluoro-phenyl)-[l-(2,4-dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4- yl] -amine;
[ 1 -(2,4-Dimethyl-phenyl)- 1 H-pyrazolo [3 ,4-d]pyrimidin-4-yl] -(1 H-indazol- 5 -yl)-amine; l-(4-Chloro-phenyl)-3-[l-(2,4-dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]- urea;
(3 ,4-Dichloro-phenyl)-[ 1 -(2,4-dimethyl-phenyl)- 1 H-pyrazolo [3 ,4-d]pyrimidin-4-yl] - amine;
N-{4-[l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-ylamino]-phenyl}- acetamide;
4-[l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-ylamino]-benzamide;
[ 1 -(2,4-Dimethyl-phenyl)- 1 H-pyrazolo [3 ,4-d]pyrimidin-4-yl] -(3 -oxazol-5 -yl-phenyl)- amine;
2-(2-Bromo-phenyl)-N-[l-(2,4-dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]- acetamide; l-(4-Bromo-phenyl)-3-[l-(2,4-dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]- urea; l-( 2,3-Dichloro-phenyl)-3-[l-(2,4-dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4- yl]-urea; l-(3,4-Dimethyl-phenyl)-3-[l-(2,4-dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4- yl]-urea;
1 -[ 1 -(2,4-Dimethyl-phenyl)- lH-pyrazolo [3 ,4-d]pyrimidin-4-yl] -3 -(4-trifluoromethyl- phenyl)-urea;
4-{3-[l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-ureido}- benzenesulfonamide; (3 ,4-Dichloro-phenyl)- [ 1 -(2,4-dimethyl-phenyl)- 1 H-pyrazolo [3 ,4-d]pyrimidin-4-yl] - methyl-amine;
[l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-(4-trifluoromethoxy- phenyl)-amine;
[ 1 -(2,4-Dimethyl-phenyl)- lH-pyrazolo [3 ,4-d]pyrimidin-4-yl] -(4-morpholin-4-yl- phenyl)-amine;
4-[l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-ylamino]- benzenesulfonamide;
4-[l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-ylamino]-N-thiazol-2-yl- benzenesulfonamide; 2-Chloro-4-[ 1 -(2,4-dimethyl-phenyl)- 1 H-pyrazolo[3 ,4-d]pyrimidin-4-ylamino] - benzonitrile;
[l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-(lH-indol-5-yl)-amine;
1 -(4-Cyano-phenyl)-3-[ 1 -(2,4-dimethyl-phenyl)- 1 H-pyrazolo [3 ,4-d]pyrimidin-4-yl] - urea; l-[l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-3-(3-trifluoromethyl- phenyl)-urea; l-[l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-3-p-tolyl-urea;
(l-Phenyl-lH-pyrazolo[3,4-d]pyrimidin-4-yl)-(5,6,7,8-tetrahydro-naphthalen-l-yl)- amine; (2-Bromo-phenyl)-( 1 -phenyl- 1 H-pyrazolo [3 ,4-d]pyrimidin-4-yl)-amine;
(l-Benzyl-lH-pyrazolo[3,4-d]pyrimidin-4-yl)-(2,4-dimethyl-phenyl)-amine;
[2-(4-Chloro-phenyl)-ethyl]-[l-(2,4-dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4- yl]-amine; l-[l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-3-[4-(pyridin-2- ylmethoxy)-phenyl]-urea;
1 -[ 1 -(2-Chloro-phenyl)- lH-pyrazolo[3,4-d]pyrimidin-4-yl]-3-[4-(pyridin-2-ylmethoxy)- phenyl]-urea;
5-[l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-3-[4-(pyridin-2- ylmethoxy)-phenyl] -urea; l-[l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-(3-fluoro-4-morpholin-
4-yl-phenyl)-amine. and pharmaceutically acceptable salts thereof. The present invention also provides a pharmaceutical composition comprising a pyrazolopyrimidine derivative of formula (I ), or a pharmaceutically acceptable salt thereof, and a pharmaceutically acceptable diluant or carrier,
wherein: - Ri is a phenyl group which is unsubstituted or substituted by 1 or 2 substituents selected from Cι-C
4 alkyl, halogen and C1-C
4 haloalkyl substituents; R
2 is a C
6-C
10 aryl, C
3-C
6 carbocyclyl, 5- to 10- membered heteroaryl or 5- to 10- membered heterocyclyl moiety, said moiety being optionally fused to a C
6- C
10 aryl, C
3-C
6 carbocyclyl, 5- to 10- membered heteroaryl or 5- to 10- membered heterocyclic ring; and X is -NR
7-, -NR
7-CO-NR
77-, -NR
7-L- or -NR-CO-L-, wherein R
7 and R
77 are the same or different and each represent hydrogen or a -C
6 alkyl group and L represents a Cι-C
6 alkylene group, the aryl, heteroaryl, heterocyclyl and carbocyclyl moieties in the R
2 substituents being unsubstituted or substituted by 1, 2 or 3 substituents selected from halogen, Cι-C
4 alkyl, CrC
4 alkoxy, Cι-C
4 haloalkyl, C
1-C
4 haloalkoxy, cyano, nitro, C
6-C
10 aryl, C
3-C
6 carbocyclyl, 5- to 10- membered heterocyclyl, 5- to 10- membered heteroaryl, -NR
7- CO
2-R
77, -CO
2R
77, -COR
777, -NR
7-CO-R
77, -CONRR
77, -SO
2NRR
77, -SO
2R
777 and -O-(CH
2)n-R
777 substituents, wherein n is from 0 to 4, each R
7 is the same or different and is hydrogen or Cι-C
6 alkyl, each R
77 is the same or different and is hydrogen, Ci-Cβ alkyl, C
6-C
10 aryl, 5- to 10- membered heterocyclyl or 5- to 10- membered heteroaryl, each R
77is the same or different and is Cι-C
6 alkyl, C
6-C
10 aryl, 5- to 10- membered
heterocyclyl or 5- to 10- membered heteroaryl, and each R
77 is the same or different and is C
6-Cιo aryl, 5- to 10- membered heterocyclyl or 5- to 10- membered heteroaryl. the aryl, heteroaryl, heterocyclyl and carbocyclyl moieties in said substituents being unsubstituted or substituted by a further substituent selected from C1-C
4 alkyl, - C
4 hydroxalkyl and C1-C
4 haloalkyl groups. Typically, in the compounds of formula (I
7), the aryl, heteroaryl, heterocyclyl and carbocyclyl moieties in the R
2 substituents are unsubstituted or substituted by 1, 2 or 3 substituents selected from halogen, C1-C4 alkyl, C1-C
4 alkoxy, C1
.-C
4 haloalkyl, Ci- C
4 haloalkoxy, cyano, nitro, C
6-C
10 aryl, C
3-C
6 carbocyclyl, 5- to 10- membered heterocyclyl, 5- to 10- membered heteroaiyl, -NR-CO
2-R
77, -CO
2R
77, -COR
777, -NR
7-CO- R
777, -CONRR
77, -SO
2NR
7R
77 and -SO
2R
77/ substituents, wherein each R
7 is the same or different and is hydrogen or Cι-C
6 alkyl, each R is the same or different and is hydrogen, -C
6 alkyl, C
6-C
10 aryl, 5- to 10- membered heterocyclyl or 5- to 10- membered heteroaryl and each R
7is the same or different and is Cι-C
6 alkyl, C
6-Cιo aryl, 5- to 10- membered heterocyclyl or 5- to 10- membered heteroaryl. Preferred Ri, R
2 and X substituents in the formula (I
7) are the same as the preferred Ri, R
2 and X substituents set out above for the formula (I). Preferably, however, X in the formula (I
7) is -NR
7-CO-NR
77- or -NR
7-CO-L- wherein R
7 and R
77 are as defined above. Also provided is a compound of formula (I
7), as defined above, or a pharmaceutically acceptable salt thereof, provided that the compound of formula (I
7) is other than (1 -Phenyl- lH-pyrazo lo[3,4-d]pyrimidin-4-yl)-(5,6,7,8-tetrahydro- naphthalen-l-yl)-amine, (2-Bromo-phenyl)-(l-phenyl-lH-pyrazolo[3,4-d]pyrimidin-4- yl)-amine, l-Benzyl-lH-pyrazolo[3,4-d]pyrimidin-4-yl)-(2,4-dimethyl-phenyl)-amine, [2-(4-Chloro-ρhenyl)-ethyl]-[l-(2,4-dimethyl-ρhenyl)-lH-pyrazolo[3,4-d]pyrimidin-4- yl]-amine and 4-Anilino-l-phenyl-lH-pyrazolo[3,4-d]pyrimidine. The present invention further provides a pharmaceutical composition comprising a compound of formula (I) or a compound of formula (I
7), or a pharmaceutically acceptable salt thereof, interferon and/or ribavirin and a pharmaceutically acceptable carrier or diluent. The compounds of formula (I) and (I
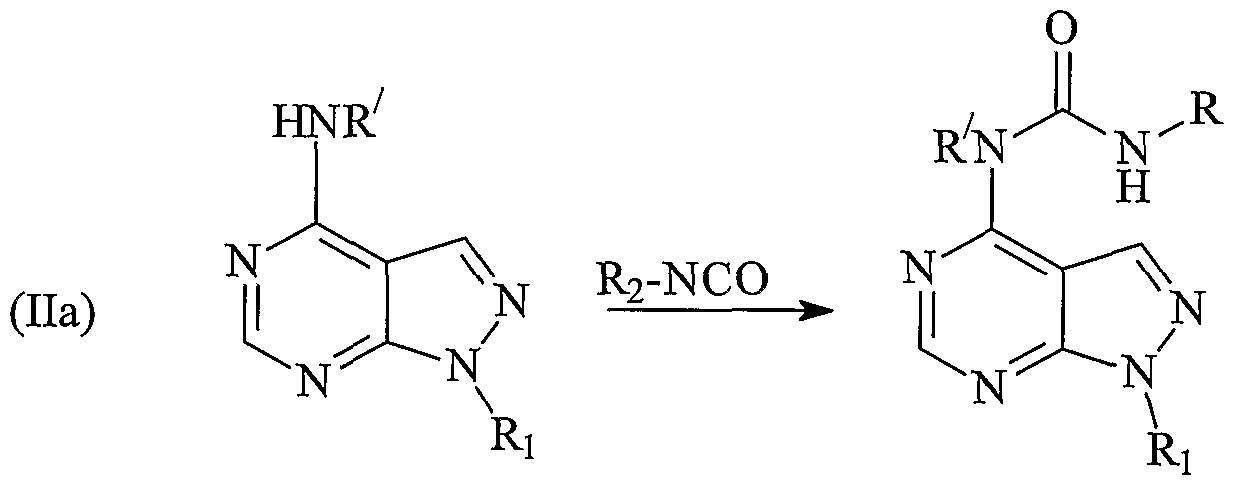
7) can be prepared from intermediates of formula (Ila) or (lib). Compounds of formulae (Ila) and (lib) can be prepared via the
following reaction scheme. Ri in the formulae (Ila) and (lib) is as defined above. R
7 in the formula (Ila) represents hydrogen or a Ci-C
6 alkyl group.
In the above reaction scheme, compounds of formula (Ila) which carry an amine (NH
2) substituent at the 4- position of the pyrazolopyrimidine ring can be prepared from an appropriate 5-amino-4-nitrile-pyrazole intermediate. Compounds of formula (Ila) in which the 4- substituent is an HNR
7 moiety, wherein R
7 is a Ci-C
6 alkyl group, can be prepared either by reacting a corresponding primary amine with an appropriate electrophile (for example R
7-C1) or by reacting a compound of formula (lib) with an appropriate secondary amine. Compounds of formula (I) in which X is -NR
7- or -NR
7-L- can be obtained by reacting a compound of formula (lib) with an amine HNR R, wherein R corresponds to R
2 or -L-R , as follows.
Typically, the reaction is conducting by treating one equivalent of the compound of formula (lib) in dry ethanol or other suitable inert solvent with three equivalents of the appropriate amine and heating to reflux overnight. The resulting solution can then be cooled to room temperature and diluted with water. The resulting precipitate can be isolated by filtration, dried in vacuo and if necessary purified further to give the desired product. Compounds of formula (I) in which X is -NR-CO-NH- can be obtained by reacting a compound of formula (Ila) with a compound R
2-NCO, wherein R
2 is as defined above.
Typically the reaction is conducted either by heating a solution of the appropriate aniline (leq) and triethylamine (2eq) in anhydrous tetrahydrofuran at reflux for five minutes and adding to this triphosgene (leq) in one portion, followed by heating the reaction mixture to generate the isocyanate, or by dissolving a commercially available isocyanate in anhydrous tetrahydrofuran and heating to 85°C. Separately, a solution of amino intermediate (leq) and sodium hydride (1. leq) in anhydrous tetrahydrofuran can be stirred at room temperature for 30 min. After 30 min, the solution of aminopyrazolopyrimidine can be added to the isocyanate solution and the mixture can be stirred under elevated temperature. The crude mixture can then be evaporated and the resulting solid partitioned between ethyl acetate and water. The
combined organic layers can be washed once with brine, and concentrated under reduced pressure, to give a crude product which can be purified by appropriate methods, for example chromatography and HPLC. Compounds of formula (I) in which X is -NR7-CO-NR77- and R77 is other than hydrogen can be prepared by reacting a compound of formula (Ila) with a compound of formula R2(R7)NCO-X, wherein R is as defined above, R7 is a Ci-C6 alkyl group and X is a leaving group, for example chlorine, OPh or OPh-NO2.
Alternatively, compounds of formula (I) in which X is -NR
7-CO-NR
77- may be prepared by reaction of compounds (Ila) with an appropriate difunctional reagent XCOX
7, wherein X and X are leaving groups. Examples of appropriate difunctional reagents include those wherein X=X -CI (phosgene), X=C1 and X -OPh (phenylchloroformate) and X=C1 and X-OPhNO
2 (p-nitrochloroformate). The thus obtained intermediate can subsequently be reacted with an appropriate nucleophile to give the desired compounds.
Alternatively, compounds of formula (I) in which X is -NR-CO-NR77- may be prepared by reaction of a compound of formula (I) in which X is -NR7-CO-NH- with a strong, non-nucleophilic base, for example sodium hydride, and reaction of the resulting anion with an alkylating agent, for example an alkyl iodide. Clearly, if the compound desired has R = H then a temporary protecting group, for example a benzyl group, may be required at the R7 position to prevent the formation of unwanted side products.
Compounds of formula (I) in which X is -NR7-CO-L- can be prepared by reacting a compound of formula (Ila) with R-CO-Cl, wherein R corresponds to -L-R2, as follows.
Typically, the reaction is carried out by treating a suspension of amino intermediate in dry acetonitrile with acid chloride (1.2eq) and heating to reflux for 72h.
The reaction mixtures can be partitioned between ethyl acetate and saturated bicarbonate solution and the organic phase separated and concentrated onto silica gel.
The desired product can then be purified by standard techniques. The starting materials used in the above reaction scheme are known compounds or may be prepared in accordance with known methods. Certain compounds of formulae (Ila) and (lib) are believed to be novel. These novel intermediates therefore also form part of the invention. The compounds in question are:
5-Amino-l-(2,4-dimethyl-phenyl)-lH-pyrazole-4-carbonitrile; 1 -(2,4-Dimethyl-phenyl)- 1 H-pyrazolo [3 ,4-d]pyrimidin-4-ylamine;
1 -(2-Bromo-phenyl)-4-chloro- 1 H-pyrazolo [3 ,4-d]pyrimidine;
4-Chloro- 1 -(2-chloro-phenyl)- 1 H-pyrazolo[3 ,4-d]pyrimidine;
4-Chloro- 1 -(2,4-dichloro-phenyl)- lH-pyrazolo[3 ,4-d]pyrimidine;
4-Chloro- 1 -(2-trifluoromethyl-phenyl)- 1 H-pyrazolo [3 ,4-d]pyrimidine; 4-Chloro- 1 -(3 -chloro-phenyl)- 1 H-pyrazolo [3 ,4-d]pyrimidine;
1 -(4-Bromo-phenyl)-4-chloro- 1 H-pyrazolo[3 ,4-d]pyrimidine;
4-Chloro- 1 -(2,4-difluoro-phenyl)- lH-pyrazolo[3 ,4-d]pyrimidine;
4-Chloro- 1 -p-tolyl- 1 H-pyrazolo [3, 4-d]pyrimidine; and 1 -Benzyl-4-chloro- 1 H-pyrazolo [3 ,4-d]pyrimidine. As explained above, the compounds of the invention are active against flaviviridae, in particular against the hepatitis C virus. The present invention therefore provides a method for treating a patient suffering from or susceptible to a flaviviridae infection, in particular a hepatitis C infection, which method comprises administering to said patient an effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof. Also provided is a method for alleviating or reducing the incidence of a hepatitis C infection in a patient, which method comprises administering to said patient an effective amount of a compound of formula (I) or a pharmaceutically acceptable salt thereof. The compounds of the invention may be administered in a variety of dosage forms. Thus, they can be administered orally, for example as tablets, troches, lozenges, aqueous or oily suspensions, dispersible powders or granules. The compounds of the invention may also be administered parenterally, whether subcutaneously, intravenously, intramuscularly, intrasternally, transdermally or by infusion techniques. The compounds may also be administered as suppositories. The compounds of the invention are typically formulated for administration with a pharmaceutically acceptable carrier or diluent. For example, solid oral forms may contain, together with the active compound, diluents, e.g. lactose, dextrose, saccharose, cellulose, corn starch or potato starch; lubricants, e.g. silica, talc, stearic acid, magnesium or calcium stearate, and/or polyethylene glycols; binding agents; e.g. starches, arable gums, gelatin, methylcellulose, carboxymethylcellulose or polyvinyl pyrrolidone; disaggregating agents, e.g. starch, alginic acid, alginates or sodium starch glycolate; effervescing mixtures; dyestuffs; sweeteners; wetting agents, such as lecithin, polysorbates, laurylsulphates; and, in general, non toxic and pharmacologically inactive substances used in pharmaceutical formulations. Such pharmaceutical preparations may be manufactured in known manner, for example, by means of mixing, granulating, tableting, sugar coating, or film coating processes. Liquid dispersions for oral administration may be syrups, emulsions and suspensions. The syrups may contain as carriers, for example, saccharose or saccharose with glycerine and/or mannitol and/or sorbitol.
Suspensions and emulsions may contain as carrier, for example a natural gum, agar, sodium alginate, pectin, methylcellulose, carboxymethylcellulose, or polyvinyl alcohol. The suspension or solutions for intramuscular injections may contain, together with the active compound, a pharmaceutically acceptable carrier, e.g. sterile water, olive oil, ethyl oleate, glycols, e.g. propylene glycol, and if desired, a suitable amount of lidocaine hydrochloride. Solutions for injection or infusion may contain as carrier, for example, sterile water or preferably they may be in the form of sterile, aqueous, isotonic saline solutions. Compounds of the present invention may be used in conjunction with known anti- viral agents. Preferred known anti- viral agents in this regard are interferon and ribavirin, which are known for the treatment of hepatitis C (Clinical Microbiology Reviews, Jan. 2000, 67-82). The said medicament therefore typically further comprises interferon and/or ribavirin. Further, the present invention provides a pharmaceutical composition comprising: (a) a pyrazolopyrimidine derivative of the formula (I), as defined above, or a pharmaceutically acceptable salt thereof;
(b) interferon and/or ribavirin; and
(c) a pharmaceutically acceptable carrier or diluent. Also provided is a product comprising: (a) a pyrazolopyrimidine derivative of the formula (I), as defined above, or a pharmaceutically acceptable salt thereof; and (b) interferon and/or ribavirin, for separate, simultaneous or sequential use in the treatment of the human or animal body. A therapeutically effective amount of a compound of the invention is administered to a patient. A typical dose is from about 0.01 to 100 mg per kg of body weight, according to the activity of the specific compound, the age, weight and conditions of the subject to be treated, the type and severity of the disease and the frequency and route of administration. Preferably, daily dosage levels are from 0.05 to 16 mg per kg of body weight, more preferably, from 0.05 to 1.25 mg per kg of body weight. The following Examples illustrate the invention. They do not however, limit the invention in any way. In this regard, it is important to understand that the particular
assay used in the Examples section is designed only to provide an indication of anti- hepatitis C activity. There are many assays available to determine such activity, and a negative result in any one particular assay is therefore not determinative.
EXAMPLES
All temperatures are in °C. Thin layer chromatography (TLC) was carried out on Si 60G coated plastic plates with uv254 indicator (Polygram). All NMR spectra were obtained at 250MHz in d6-DMSO unless stated otherwise. LC-MS CONDITIONS
Samples were run on a MicroMass ZMD, using electrospray with simultaneous positive - negative ion detection.
Column : Synergi Hydro-RP, 30 x 4.6mm ID, 4μm. Gradient : 95 : 5 to 5 :95 v/v H2O/CH3CN + 0.05% Formic Acid over 4.0 min, hold 3 min, return to 95:5 v/v H2O/CH3CN + 0.05% Formic Acid over 0.2 min and hold at 95:5 v/v H2O/CH3CN + 0.05% Formic Acid over 3 min. Detection : PDA 250 - 340 nm. Flow rate : 1.5 ml/min
Purification Method A
The crude material was dissolved in ethyl acetate and concentrated onto silica gel and loaded into an empty ISCO cartridge. Using an ISCO Combiflash Sql6x, purification was achieved by chromatography on silica gel with a hexane (solvent A) and ethyl acetate (solvent B) gradient ie Time (min): % solvent B 0 15 3 15 20 80 4 100 1 0 at a flow rate of 20ml/min with peak detection at 254nm.
Purification Method B
The crude material was dissolved in ethyl acetate and concentrated onto silica gel and loaded into an empty ISCO cartridge. Using an ISCO Combiflash Sql6x, purification
was achieved by chromatography on silica gel with a hexane (solvent A) and ethyl acetate (solvent B) gradient ie
Time (min): % solvent B 0 40 3 40 20 100 4 100 at a flow rate of 20ml/min with peak detection at 254nm.
Purification Method C
Preparative HPLC was performed using a system comprising a Synergi Hydro-RP 50 x 21.2mm x 4μM column, Gilson 322 pump, Gilson UV/Vis-155 detection (at 254nM), and a Gilson 215 liquid handler under the control of Gilson Unipoint software in peak collection mode. Elution was performed at 25.6 ml/min with the mobile phase varied over time according to the table below, where solvent A is water + 0.05% formic acid and solvent B is acetonitrile + 0.05% formic acid. Time = (min): % solvent A % solvent B 0 95 5 0.3 95 5 4 3 97 7 3 97 7.2 95 5 9 95 5
Intermediate 1 : 4-chloro-l-(2,4-dimethylphenyl)-pyrazolo[3,4-d]pyrimidine
2,4-Dimethylphenylhydrazine hydrochloride (15.8g, 91.3mmol) was partitioned between 2M sodium hydroxide solution and dichloromethane. The organic layer was separated, dried and reduced under vacuum to give the free hydrazine (12. lg, 90.0mmol). The free hydrazine and ethoxymethylenemalononitrile (10.9g, 89.7mmol) were dissolved in anhydrous ethanol (40ml) and heated at reflux for two hours before
being chilled overnight. The product was filtered, washed with ice-cold ethanol and dried to give 5 -amino- 1 -(2,4-dimethyl-phenyl)- lH-pyrazole-4-carbonitrile (13.8g, 65.1mmol, LC-MS rt 4.32 (MH)+ 213 ). 5-Amino-l-(2,4-dimethyl-phenyl)-lH- pyrazole-4-carbonitrile (10. Og, 47.2mmol) was added portionwise to stirred concentrated sulphuric acid (50ml) at 0°C. On completion of the addition, the mixture was allowed to warm to room temperature and stirred for a further one hour. The reaction solution was poured onto crushed ice and neutralised with concentrated ammonium hydroxide solution, while maintaining a temperature of 10-15°C, before being extracted with ethyl acetate. The organic layer was separated, dried over magnesium sulphate and reduced under vacuum to give 5-amino-l-(2,4- dimethylphenyl)pyrazole-4-carboxamide as a pale yellow solid (12.8g due to persisting organic solvent, LC-MS rt 3.79 (MH)+ 231). A suspension of 5-amino-l-(2,4- dimethylphenyl)-pyrazole-4-carboxamide (12.3g, 53.5mmol) in fonnamide was heated at 150°C for 48 hours. The cooled solution was diluted with water and allowed to stand. The product was filtered, washed with water and dried overnight in a vacuum oven to give 4-hydroxy-l-(2,4-dimethylphenyl)-pyrazolo[3,4-d]pyrimidine (10.8g, LC-MS rt 4.03 (MH)+ 241). A suspension of 4-hydroxy-l-(2,4-dimethylphenyl)pyrazolo[3,4- d]pyrimidine (1.5g, 6.3mmol) and phosphorus pentachloride (1.3g, 6.3mmol) in phosphorus oxychloride (15ml) was heated at reflux overnight. The solvent was removed under vacuum and the residue partitioned between ethyl acetate and water. The organic layer was separated, dried over magnesium sulphate and reduced under vacuum to give 4-chloro-l-(2,4-dimethylphenyl)-pyrazolo[3,4-d]pyrimidine as a yellow solid (1.4g, 5.4mmol) LC-MS rt 5.32 (MH)+ 259/261 1H NMR δ 8.87 (IH, s), 8.73 (IH, s), 7.22-7.36 (3H, m), 2.40 (3H, s), 2.03 (3H, s)
Intermediate 2: l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-ylamine
2,4-Dimethylphenylhydrazine hydrochloride (15.8g, 91.3mmol) was partitioned between 2M sodium hydroxide solution and dichloromethane. The organics were separated, dried and reduced under vacuum to give the free hydrazine (12. lg, 90.0mmol). The free hydrazine and ethoxymethylenemalononitrile (10.9g, 89.7mmol) were dissolved in anhydrous ethanol (40ml) and heated at reflux for two hours before
being chilled overnight. The product was filtered, washed with ice-cold ethanol and dried to give 5-amino-l-(2,4-dimethyl-phenyl)-lH-pyrazole-4-carbonitrile (13.8g, 65.1mmol, LC-MS rt 4.32 (MH)+ 213). A suspension of 5 -amino- 1 -(2,4- dimethyl-phenyl)- lH-pyrazole-4-carbonitrile (4.18g, 19.7mmol) in formamide (15ml) was heated at 150°C overnight. The reaction was cooled to room temperature and diluted with water. The product was filtered and washed with water and diethyl ether to give the title compound as a grey solid (2.86g, 12.0mmol) LC-MS rt 3.55 (MH)+ 240
1H NMR δ 8.31 (IH, s), 8.15 (IH, s), 7.81 (2H, br,d), 7.18-7.25 (3H, m), 2.37 (3H, s), 2.01 (3H, s)
Similarly prepared were the following intermediates:
Intermediate 3: 4-Chloro-l-phenyl-lH-pyrazolo[3,4-d]pyrimidine
Using the method of Intermediate 1 with free phenylhydrazine (4.55ml, 46.3mmol) gave the title compound as a yellow solid (0.81g, 3.5mmol)
LC-MS rt 5.62 (MH)+ 231
1H NMR δ 8.90 (IH, s), 8.64 (IH, s), 8.07 (2H, d), 7.53 (2H, m), 7.35 (IH, t)
Intermediate 4: 4-Chloro-l-o-tolyl-lH-pyrazolo[3,4-d]pyrimidine
Using the method of Intermediate 1 with o-Tolylhydrazine hydrochloride (5.44g, 34.3mmol) gave the title compound (~1.60g - solvent persisted in sample after drying). LC-MS rt 5.39 (MH)+ 245
1H NMR δ 8.80 (IH, s), 8.68 (IH, s), 7.36-7.44 (4H, m), 1.99 (3H, s)
Intermediate 5: l-(2-Bromo-phenyl)-4-chloro-lH-pyrazolo[3,4-d]pyrimidine
Using the method of Intermediate 1 with 2-bromophenylhydrazine hydrochloride (10.92g, 48.9mmol) gave the title compound as an orange solid (1.25g, 4.1mmol). LC-MS rt 5.38 (MH)+ 309/311 1H NMR δ 8.83 (IH, s), 8.73 (IH, s), 7.86 (IH, d,d), 7.51-7.64 (3H, m)
Intermediate 6 : 4-Chloro- 1 -(4-chloro-phenyl)- 1 H-pyrazolo[3 ,4-d]pyrimidine
Using the method of Intermediate 1 with 4-chlorophenylhydrazine hydrochloride (5.62g, 31.4mmol) gave a the title compound as a pale yellow solid (2.85g, 10.7mmol) LC-MS rt 6.05 (MH)+ 265
1H NMR δ 9.06 (IH, s), 8.84 (IH, s), 8.26 (2H, d), 7.74 (2H, d)
Intermediate 7 : 4-Chloro- 1 -(2-chloro-phenyl)- lH-pyrazolo [3 ,4-d]pyrimidine
Using the method of hitermediate 1 with 2-chlorophenylhydrazine hydrochloride (4.25g, 23.7mmol) gave the title compound as a yellow solid (1.15g, 4.3mmol). LC-MS rt 4.97 (MH)+ 265 1H NMR δ 8.96 (IH, s), 8.52 (IH, s), 7.66-7.86 (4H, m)
Intermediate 8: 4-Chloro-l-(2,4-dichloro-phenyl)-lH-pyrazolo[3,4-d]pyrimidine
Using the method of Intermediate 1 with 2,4-dichlorophenylhydrazine hydrochloride (5.40g, 25.3mmol) gave the title compound as a dark orange solid (0.98g, 3.3mmol). LC-MS rt 5.78 (MH)+ 299 1H NMR δ 8.94 (IH, s), 8.84 (IH, s), 8.38 (IH, s), 7.69 (2H, m)
intermediate 9 : 4-Chloro- 1 -(2-trifluoromethyl-phenyl)- 1 H-pyrazolo [3 ,4-d]pyrimidine
Using the method of Intermediate 1 with 2-trifluoromethylphenylhydrazine hydrochloride (4.60g, 21.6mmol) gave the title compound as a crude orange solid (>100%) which was used without further purification. LC-MS rt 5.41 (MH)+ 299 1H NMR δ 8.90 (IH, s), 8.81 (IH, s), 8.60 (IH, d), 8.34 (IH, s), 7.87-7.98 (2H, m)
Intermediate 10: 4-Chloro-l-(3-chloro-phenyl)-lH-pyrazolo[3,4-d]pyrimidine
Using the method of Intermediate 1 with 3-Chlorophenylhydrazine hydrochloride (10.92g, 61.0mmol) gave the title compound as a beige solid (1.02g, 3.8mmol) LC-MS rt 6.15 (MH)+ 265
1H NMR δ 9.05 (IH, s), 8.82 (IH, s), 8.32 (IH, t), 8.18 (IH, d,d), 7.67 (IH, t), 7.53 (IH, d,d)
Intermediate 11: 1 -(4-Bromo-phenyl)-4-chloro- 1 H-pyrazolo [3 ,4-d]pyrimidine
Using the method of Intermediate 1 with 4-bromophenylhydrazine hydrochloride
(5.28g, 23.6mmol) gave the title compound as a beige solid (0.73g, 2.4mmol). LC-MS rt 6.22 (MH)+ 309/311
1H NMR δ 9.07 (IH, s), 8.86 (IH, s), 8.21 (2H, d,d), 7.89 (2H, d,d)
Intermediate 12 : 4-Chloro- 1 -(2,4-difluoro-phenyl)- lH-pyrazolo[3 ,4-d]pyrimidine
Using the method of Intermediate 1 with 2,4-difluorophenylhydrazine hydrochloride (5.46g, 30.2mmol) gave the crude title compound as a beige solid. LC-MS rt 5.33 (MH)+ 267
1H NMR δ 9.00 (IH, s), 8.87 (IH, s), 7.72-7.93 (2H, m), 7.45 (IH, m)
Intermediate 13: 4-ChloiO-l-p-tolyl-lH-pyrazolo[3,4-d]pyrimidine
Using the method of Intermediate 1 with p-tolylhydrazine hydrochloride (5.38g, 33.9mmol) gave the title compound as a brown solid (1.02g, 4.2mmol) LC-MS rt 5.61 (MH)+ 245 1H NMR δ 9.02 (IH, s), 8.79 (IH, s), 8.06 (2H, d), 7.47 (2H, d), 3.42 (3H, s)
Intermediate 14: l-Benzyl-4-chloro-l H-pyrazolo [3 ,4-d]pyrimidine
Using the method of Intermediate 1 with benzylhydrazine dihydrochloride (2.70g, 13.8mmol) gave the title compound as a yellow solid (0.42g, 1.7mmol)
LC-MS rt 5.04 (MH)+ 245
1H NMR δ 8.92 (IH, s), 8.54 (IH, s), 7.22-7.37 (5H, m), 5.71 (2H, s)
Example 1 : [l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-[2-(lH-indol- 3-yl)-ethyl]-amine
A suspension of Intermediate 1 (75mg,leq) in dry ethanol was treated with tryptamine (139mg, 3eq) and heated to reflux overnight. The resulting solution was cooled to room temperature and diluted with water. The resulting precipitate was isolated by filtration, dried in vacuo to give the desired product. LCMS: rt 5.13 (MH)+ 383
1H NMR δ 10.87 (IH, s), 8.56 (IH, t), 8.33 (IH, s), 8.28 (IH, s), 7.61 (IH, d), 7.36 (IH, d), 7.00-7.26 (m, 6H), 3.83 (2H, m), 3.07 (2H, t), 2.38 (3H, s), 2.02 (3H, s)
Prepared similarly were:
Example 2 : [2-( 1 H-Indol-3 -yl)-ethyl] -(1 -phenyl- 1 H-pyrazolo [3 ,4-d]pyrimidin-4-yl)- amine
Reaction of Intermediate 3 (lOOmg, 0.416mmol) with tryptamine (200mg) gave the title compound.
LCMS rt 5.27 (MH)+355
1H NMR δ 10.86 (IH, s), 8.64 (IH, t), 8.42 (2H, d), 8.21 (2H, d), 7.58 (3H, m), 7.35 (2H, m) , 7.22 (IH, d), 6.97-7.12 (2H, m), 3.84 (2H, m), 3.08 (2H, t)
Example 3: (4-Butyl-phenyl)-(l-phenyl-lH-pyrazolo[3,4-d]pyrimidin-4-yl)-amine
Reaction of Intermediate 3 (50mg) with 4-butylaniline (3eq) gave the title compound. LCMS: rt 6.71 (MH) + , 344 1H NMR δ 10.20 (IH, s), 8.52 (2H, m), 8.22 (2H, d), 7.75 (2H, d), 7.58 (2H, t), 7.38 (IH, t), 7.25 (2H, d), 2.62 (2H, t), 1.23-1.50 (4H, m), 0.90 (3H, t)
Example 4: (4-Butyl-phenyl)-[l-(2,4-dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4- yl]-amine
Reaction of Intermediate 1 (75mg) with 4-butylaniline (137uL) gave the title compound after purification by Method A LCMS: rt 6.64 (MH) + , 372
1H NMR δ 10.14 (IH, s), 8.37 (2H, m), 7.73 (2H, d), 7.20-7.28 (5H, m), 2.60 (2H, t), 2.39 (3H, s), 2.03 (3H, s), 1.59 (2H, m), 1.34 (2H, m), 0.92 (3H, t)
Example 5: (4-Ethoxy-phenyl)-(l-phenyl-lH-pyrazolo[3,4-d]pyrimidin-4-yl)-amine
Reaction of Intermediate 3 (lOOmg, 0.416mmol) with p-phenetidine (180uL) gave the title compound. LCMS rt 5.7 (MH)+ 332 1H NMR δ 10.12 (IH, s), 8.48 (IH, s), 8.21 (2H, d), 7.55-7.71 (4H, m), 7.37 (IH, t), 7.00 (IH, d), 4.05 (2H, q), 1.36 (3H, t)
Example 6: [l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-(4-ethoxy- phenyl)-amine
Reaction of Intermediate 1 (75mg) with p-phenetidine (11 luL) gave the title compound after purification by Method A
LCMS rt 5.61 (MH)+ 360
1H NMR δ 10.07 (IH, s), 8.32 (2H, d), 7.68 (2H, d), 7.17-7.27 (3H, m), 6.99 (2H, d), 4.05 (2H, q), 2.39 (3H, s), 2.03 (3H, s), 1.35 (3H, t)
Example 7: 4-[l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-ylamino]- benzoic acid ethyl ester
Reaction of intermediate 1 (75mg) with ethyl 4-aminobenzoate (143mg) gave the title compound after purification by Method A LCMS rt 6.07 (MH)+ 388
1H NMR δ 10.53 (IH, s), 8.63 (IH, s), 8.51 (IH, s), 8.12 (2H, d) 8.02 (2H, d), 7.19-7.31 (3H, m), 4.33 (2H, q), 2.40 (3H, s), 2.04 (3H, s), 1.34 (3H, t)
Example 8: (5-Chloro-2-methoxy-phenyl)-(l-phenyl-lH-pyrazolo[3,4-d]pyrimidin-4- yl)-amine
Reaction of Intermediate 3 (50mg) with 5-chloro-2-methoxyaniline (102mg) gave the title compound after purification by Method A. LCMS rt 6.15 (MH)+ 352/354 1H NMR δ 9.86 (IH, s), 8.49 (2H, s), 8.21 (2H, d), 7.91 (IH, d), 7.58 (2H, m), 7.17-7.41 (3H, m), 3.86 (3H, s)
Example 9 : (5 -Chloro-2-methoxy-phenyl)- [ 1 -(4-chloro-phenyl)- 1 H-pyrazolo [3,4- d]pyrimidin-4-yl] -amine
Reaction of hitermediate 6 (lOOmg) with 5-chloro-2-methoxyaniline (179mg) gave the title compound.
LCMS rt 6.85 (MH)+ 386/388
1H NMR δ 9.91 (IH, s), 8.50 (2H, br s), 8.29 (2H, d), 7.89 (IH, d), 7.65 (2H, d), 7.32 (IH, d,d), 7.19 (IH, d), 3.86 (3H, s)
Example 10: [l-(4-ChloiO-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-(3-fluoro- phenyl)-amine
Reaction of Intermediate 6 (lOOmg) with 3-fluoroaniline (109uL) gave the title compound.
LCMS rt 6.4 (MH)+ 340/342
1H NMR δ 10.46 (IH, s), 8.65 (IH, s), 8.64 (IH, s), 8.30 (2H, d), 8.01 (IH, d,t), 7.59-
7.68 (3H, m), 7.47 (IH, m), 6.99 (IH, t,d)
Example 11: 4- {2-[l -(2,4-Dimethyl-phenyl)- lH-pyrazolo[3,4-d]pyrimidin-4-ylamino]- ethyl} -benzenesulfonamide
Reaction of Intermediate 1 (75mg) with 4-(2-Amino-ethyl)-benzenesulfonamide (173mg) gave the title compound after purification by Method A LCMS rt 4.55 (MH)+ 423
1H NMR δ 8.55 (IH, t), 8.32 (IH, s), 8.27 (IH, s), 7.78 (2H, d), 7.49 (2H, d), 7.18-7.33 (3H, m), 3.80 (2H, q), 3.04 (2H, t), 2.38 (3H, s), 2.01 (3H, s)
Example 12: [l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-(3-fluoro- phenyl)-amine
Reaction of Intermediate 1 (75mg) with 3-fluoroaniline (83.3uL) gave the title compound after purification by Method A LCMS rt 5.86 (MH)+ 334
1H NMR δ 10.38 (IH, s), 8.57 (IH, s), 8.48 (IH, s), 8.02 (IH, d,t), 7.61 (IH, d), 7.45 (IH, m), 7.22-7.30 (3H, m), 6.96 (IH, t,d), 2.39 (3H, s), 2.04 (3H, s)
Example 13 : 4- {2-[ 1 -(2-Bromo-phenyl)-3H-pyrazolo[3,4-d]pyrimidin-4-ylamino]- ethyl} -benzenesulfonamide
Reaction of Intermediate 5 (75mg) with 4-(2-amino-ethyl)-benzenesulfonamide (144mg) gave the title compound after purification by Method A. LCMS rt 4.41 (MH)+ 473/475 1H NMR δ 8.60 (IH, t), 8.36 (IH, s), 8.28 (IH, s), 7.87 (IH, d), 7.78 (2H, d), 7.48-7.61 (5H, m), 3.80 (2H, m), 3.05 (2H, t)
Example 14: [l-(2-Bromo-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-(4-butyl- phenyl)-amine
Reaction of Intermediate 5 (75mg) with 4-butyl aniline (115uL) gave the title compound after purification by Method A LCMS rt 6.26 (MH)+ 422/424
1H NMR δ 10.20 (IH, s), 8.45 (IH, s), 8.38 (IH, s), 7.89 (IH, d), 7.72 (2H, d), 7.53- 7.63 (3H, m), 7.25 (2H, d), 2.60 (2H, t), 1.58 (2H, m), 1.34 (2H, m), 0.93 (3H, t)
Example 15: 4-[2-(l-o-Tolyl-lH-pyrazolo[3,4-d]pyrimidin-4-ylamino)-ethyl]- benzenesulfonamide
Reaction of Intermediate 4 (75mg) with 4-(2-amino-ethyl)-benzenesulfonamide (175mg) gave the title compound after purification by Method A. LCMS rt 4.23 (MH)+ 409 1H NMR δ 8.57 (IH, t), 8.34 (IH, s), 8.28 (IH, s), 7.77 (2H, d), 7.36-7.51 (6H, m), 7.32 (2H, s), 3.80 (2H, m), 3.05 (2H, t), 2.07 (3H, s)
Example 16: [2-(lH-Indol-3-yl)-ethyl]-(l-o-tolyl-lH-pyrazolo[3,4-d]pyrimidin-4-yl)- amine
Reaction of Intermediate 4 (75mg) with tryptamine (141mg) gave the title compound after purification by Method A.
LCMS rt 4.88 (MH)+ 369
1H NMR δ 10.87, (IH, s), 8.58 (IH, t), 8.35 (IH, s), 829 (IH, s), 7.61 (IH, d), 7.35-7.45 (5H, m), 7.23 (IH, d), 7.00-7.09 (2H, m), 3.83 (2H, m), 3.08 (2H, t), 2.08 (3H, t)
Example 17: (4-Butyl-phenyl)-(l-o-tolyl-lH-pyrazolo[3,4-d]pyrimidin-4-yl)-amine
Reaction of Intermediate 4 (75mg) with 4-butylaniline (140uL) gave the title compound after purification by Method A. LCMS rt 6.18 (MH)+ 358
1H NMR δ 10.15 (IH, s), 8.43 (IH, s), 8.38 (IH, s), 7.73 (2H, d), 7.40-7.47 (4H, m), 7.24 (2H, d), 2.56 (2H, t), 2.09 (3H, s), 1.59 (2H, m), 1.34 (2H, m), 0.93 (3H, t)
Example 18: 4- {2- [l-(2,4-Dichloro-phenyl)-l H-pyrazolo [3, 4-d]pyrimidin-4-ylamino]- ethyl} -benzenesulfonamide
Reaction of Intermediate 8 (75mg) with 4-(2-amino-ethyl)-benzenesulfonamide (142mg) gave the title compound (6.5mg) after purification by Method A. LCMS rt 4.83 (MH)+ 463
1H NMR δ 8.63 (IH, t), 8.38 (IH, s), 8.30 (IH, s), 7.94 (IH, s), 7.78 (2H, d), 7.65-7.69 (2H, m), 7.48 (2H, d), 7.32 (2H, s), 3.80 (2H, m), 3.05 (2H, t)
Example 19: [l-(2,4-Dichloro-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-[2-(lH- indol-3 -yl)-ethyl] -amine
Reaction of Intermediate 8 (75mg) with tryptamine (114mg) gave the title compound
(7.1mg) after purification by Method A.
LCMS rt 5.45 (MH)+ 423
1H NMR δ 10.87 (IH, s), 8.65 (IH, t), 8.38 (IH, s), 8.31 (IH, s), 7.94 (IH, s), 7.60-7.69
(3H, m), 7.36 (IH, d), 7.22 (IH, d), 6.97-7.12 (2H, m), 3.83 (2H, m), 3.07 (2H, t)
Example 20: [l-(2,4-Dichloro-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-(4-ethoxy- phenyl)-amine
Reaction of intermediate 8 (75mg) with p-phenetidine (92.4uL) gave the title compound (9.4mg) after purification by Method A.
1H NMR δ 10.25 (IH, s), 8.60 (IH, br,s), 8.47 (IH, s), 8.07 (IH, s), 7.78 (4H, br, m), 7.11 (2H, d), 4.17 (2H, q), 1.47 (3H, t)
Example 21 : 4-{2-[l-(3-Chloro-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-ylamino]- ethyl} -benzenesulfonamide
Reaction of intermediate 10 (75mg) with 4-(2-amino-ethyl)-benzenesulfonamide (170mg) gave the title compound (63.9mg) after purification by Method A. LCMS rt 5.22 (MH)+429/431 1H NMR δ 8.71 (IH, t), 8.50 (IH, s), 8.44 (2H, m), 8.26 (IH, d,d), 7.80 (2H, d), 7.43- 7.66 (4H, m), 7.35 (2H, s), 3.84 (2H, m), 3.08 (2H, t)
Example 22: (4-Butyl-phenyl)-[l-(3-chloro-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]- amine
Reaction of Intermediate 10 (75mg) with 4-butylaniline (135uL) gave the title compound (50.6mg) after purification by Method A. LCMS rt 7.52 (MH)+ 378/380 1H NMR δ 10.09 (IH, s), 8.42 (IH, s), 8.38 (IH, br,s), 8.27 (IH, t), 8.11 (IH, d), 7.62 (2H, d), 7.51 (IH, t), 7.31 (IH, d), 7.16 (2H, d), 2.47 (2H, t), 1.45 (2H, m), 1.22 (2H, m), 0.80 (3H, t)
Example 23: (5-Chloro-2-methoxy-phenyl)-[l-(3-chloro-phenyl)-lH-pyrazolo[3,4- d]pyrimidin-4-yl] -amine
Reaction of hiteraiediate 10 (75mg) with 5-chloro-2-methoxy-aniline (25.3mg) gave the title compound (25.3mg) after purification by Method A.
LCMS rt 6.87 (MH)+ 386/388
1H NMR δ 9.71 (IH, s), 8.31 (IH, s), 8.27 (IH, br,s), 8.20 (IH, t), 8.04 (IH, d), 7.68
(IH, d), 7.40 (IH, t), 7.24 (IH, d,d), 7.12 (IH, d,d), 7.00 (IH, d), 3.66 (3H, s)
Example 24: 4-{2-[l-(2-Chloro-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-ylamino]- ethyl} -b enzenesulfonamide
Reaction of intermediate 7 (75mg) with 4-(2-amino-ethyl)-benzenesulfonamide (170mg) gave the title compound after purification by Method A. LCMS rt 4.45 (MH)+ 429/431
1H NMR δ 8.52 (IH, t), 8.27 (IH, s), 8.19 (IH, s), 7.68 (2H, d), 7.63 (IH, m), 7.4-7.51 (3H, m), 7.39 (2H, d), 7.23 (2H, s), 3.69 (2H, m), 2.95 (2H, t)
Example 25: [l-(2-Chloro-phenyl)-lH-ρyrazolo[3,4-d]pyrimidin-4-yl]-[2-(lH-indol-3- yl)-ethyl] -amine
Reaction of Intermediate 7 (75mg) with tryptamine (137mg) gave the title compound after purification by Method A LCMS rt 5.03 (MH)+ 389/391
1H NMR δ 10.86 (IH, s), 8.61 (IH, t), 8.38 (IH, s), 8.30 (IH, s), 7.58-7.75 (5H, m), 7.36 (IH, d), 7.23 (IH, d), 7.00-7.09 (2H, m), 3.84 (2H, m), 3.08 (2H, t)
Example 26: [l-(2-Chloro-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-(4-ethoxy- phenyl)-amine
Reaction of hitermediate 7 (75mg) with p-phenetidine (1 lOuL) gave the title compound after purification by Method A
LCMS rt 5.37 (MH)+ 366/368
1H NMR δ 10.12 (IH, s), 8.47 (IH, br,s), 8.34 (IH, s), 7.56-7.77 (6H, m), 7.00 (2H, d),
4.06 (2H, q), 1.36 (3H, t)
Example 27 : (4-Butyl-phenyl)-[ 1 -(2-chloro-phenyl)- 1 H-pyrazolo [3 ,4-d]pyrimidin-4-yl] - amine
Reaction of Intermediate 7 (75mg) with 4-butylaniline (135uL) gave the title compound after purification by Method A LCMS rt 6.32 (MH)+ 378/380
1H NMR δ 9.96 (IH, s), 8.24 (IH, br,s), 8.16 (IH, s), 7.49-7.52 (3H, m), 7.34-7.43 (3H,m), 7.03 (2H, d), 2.36 (2H, t), 1.37 (2H, m), 1.12 (2H, m), 0.71 (3H, t)
Example 28: (5-Chloro-2-methoxy-phenyl)-[l-(2-chloro-phenyl)-lH-pyrazolo[3,4- d]pyrimidin-4-yl] -amine
Reaction of Intermediate 7 (75mg) with 5-chloro-2-methoxy-aniline (135mg) gave the title compound after purification by Method A LCMS rt 5.74 (MH)+ 386/388
1H NMR δ 9.91 (IH, s), 8.51 (IH, br,s), 8.40 (IH, s), 7.96 (IH, d), 7.81 (IH, m), 7.65- 7.72 (3H, m), 7.38 (IH, d,d), 7.25 (IH, d), 3.92 (3H, s)
Example 29: 4-{2-[l-(2-Trifluoromethyl-ρhenyl)-lH-ρyrazolo[3,4-d]pyrimidin-4- ylammo]-ethyl}-benzenesulfonamide
Reaction of Intermediate 9 (75mg) with 4-(2-amino-ethyl)-benzenesulfonamide (170mg) gave the title compound (32.4mg) after purification by Method C LCMS rt 4.58 (MH)+ 463
1H NMR δ 8.70 (IH, t), 8.41 (IH, s), 8.33 (IH, s), 7.89-8.07 (3H, m), 7.83 (2H, d), 7.71 (IH, d), 7.54 (2H, d), 7.38 (2H, s), 3.86 (2H, m), 3.11 (2H, t)
Example 30: [2-(lH-Indol-3-yl)-ethyl]-[l-(2-trifluoromethyl-phenyl)-lH-pyrazolo[3,4- d]pyrimidin-4-yl] -amine
Reaction of Intermediate 9 (75mg) with tryptamine (140mg gave the title compound (23.2mg) after purification by Method C LCMS rt 5.14 (MH)+ 423
1H NMR δ 10.91 (IH, s), 8.41 (IH, s), 8.33 (IH, s), 7.64-8.06 (5H, m), 7.41 (IH, d), 7.28 (IH, d), 7.04-7.13 (2H, m), 3.88 (2H, m), 3.13 (2H, t)
Example 31 : (3-Chloro-phenyl)-[l-(2,4-dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin- 4-yl]-amine
Reaction of Intermediate 1 (60mg) with 3-chloroaniline (105uL) gave the title compound after purification by Method A LCMS rt 6.08 (MH)+ 350/352 1H NMR δ 10.33 (IH, s), 8.56 (IH, s), 8.48 (IH, s), 8.19 (IH, t), 7.79 (IH, d,d), 7.45 (IH, t), 7.28 (2H, d), 7.19 (2H, d), 2.40 (3H, s), 2.04 (3H, s)
Example 32 : (3 -Bromo-phenyl)-[ 1 -(2,4-dimethyl-phenyl)- lH-pyrazolo [3 ,4-d]pyrimidin- 4-yl] -amine
Reaction of Intermediate 1 (60mg) with 3-bromoaniline (1 lOuL) gave the title compound after purification by Method A LCMS rt 6.16 (MH)+ 394/396
1H NMR δ 10.32 (IH, s), 8.56 (IH, s), 8.48 (IH, s), 8.31 (IH, t), 7.85 (IH, d,t), 7.22- 7.42 (5H, m), 2.39 (3H, s), 2.04 (3H, s)
Example 33: [l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-(3- trifluoromethyl-phenyl)-amine
Reaction of Intermediate 1 (60mg) with 3-trifluoromethylaniline (125uL) gave the title compound after purification by Method A. LCMS rt 6.17 (MH)+ 384
1H NMR δ 10.42 (IH, s), 8.52 (IH, s), 8.42 (IH, s), 8.32 (IH, s), 8.14 (IH, d), 7.60 (IH, t), 7.43 (IH, d), 7.15-7.24 (3H, m), 2.33 (3H, s), 1.97 (3H, s)
Example 34: [l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-(4- trifluoromethyl-phenyl)-amine
Reaction of intermediate 1 (60mg) with 4-trifluoromethylaniline (125uL) gave the title compound after purification by Method A LCMS rt 6.25 (MH)+ 384 1H NMR δ 10.48 (IH, s), 8.58 (IH, s), 8.46 (IH, s), 8.13 (2H, d), 7.74 (IH, d), 7.18- 7.27 (3H, m), 2.35 (3H, s), 2.00 (3H, s)
Example 35: 3-[l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-ylamino]- benzonitrile
Reaction of Intermediate 1 (60mg) with 3-aminobenzonitrile (120mg) gave the title compound after purification by Method A LCMS rt 5.68 (MH)+ 341
1H NMR δ 10.51 (IH, s), 8.58 (IH, s), 8.52 (IH, s), 8.10 (IH, d,d), 7.62 (2H, m), 7.19- 7.31 (3H, m), 2.40 (3H, s), 2.04 (3H, s)
Example 36: 4-[l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-ylamino]- benzonitrile
Reaction of Intermediate 1 (60mg) with 4-aminobenzonitrile (120mg) gave the title compound after purification by Method A LCMS rt 5.73 (MH)+ 341 1H NMR δ 10.67 (IH, s), 8.69 (IH, s), 8.59 (IH, s), 8.24 (2H, d), 7.94 (2H, d), 7.28- 7.37 (3H, m), 2.45 (3H, s), 2.09 (3H, s)
Example 37 : [ 1 -(4-Bromo-phenyι)- 1 H-pyrazolo [3 ,4-d]pyrimidin-4-yl] -(4-butyl- phenyl)-amine
Reaction of Intermediate 11 (75mg) with 4-butylaniline (115uL) gave the title compound after purification by Method A
LCMS rt 7.62 (MH)+ 422/424
1H NMR δ 10.32 (IH, s), 8.63 (2H, br,s), 8.34 (2H, d,d), 7.81-7.89 (4H, m), 7.33 (2H, d), 2.69 (2H, t), 1.67 (2H, m), 1.41 (2H, m), 1.01 (3H, t)
Example 38 : [ 1 -(4-Bromo-phenyl)- 1 H-pyrazolo[3 ,4-d]pyrimidin-4-yl] -(5 -chloro-2- methoxy-phenyl)-amine
Reaction of Intermediate 11 (75mg) with 5-chloro-2-methoxyaniline (lOOmg) gave the title compound after purification by Method A
LCMS rt 7.07 (MH)+ 430/432
1H NMR δ 9.90 (IH, s), 8.50 (2H, br,s), 8.23 (2H, d,d), 7.89 (IH, d), 7.77 (2H, d,d),
7.31 (lH, d,d), 7.18 (lH, d)
Example 39 : [ 1 -(2,4-Difluoro-phenyl)- 1 H-pyrazolo [3 ,4-d]pyrimidin-4-yl] -[2-( 1 H-indol- 3-yl)-ethyl]-amine
Reaction of Intermediate 12 (lOOmg) with tryptamine (140mg) gave the title compound after purification by Method A LCMS rt 5.11 (MH)+ 391
1H NMR δ 8.57 (IH, t), 8.34 (IH, s), 8.27 (IH, s), 7.50-7.74 (5H, m), 7.43 (2H, d), 7.27 (2H, s), 3.75 (2H, m), 2.99 (2H, t)
Example 40: (4-Butyl-phenyl)-[l-(2,4-difluoro-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4- yl]-amine
Reaction of Intermediate 12 (lOOmg) with 4-butylaniline (130uL) gave the title compound after purification by Method A LCMS rt 6.4 (MH)+ 380 1H NMR δ 10.03 (IH, s), 8.31 (IH, br,s), 8.22 (IH, s), 7.39-7.61 (4H, m), 7.16 (IH, t), 7.07 (2H, d), 2.40 (2H, t), 1.41 (2H, m), 1.17 (2H, m), 0.76 (3H, t)
Example 41 : (5-Chloro-2-methoxy-phenyl)-[l-(2,4-difluoro-phenyl)-lH-pyrazolo[3,4- d]pyrimidin-4-yl] -amine
Reaction of intermediate 12 (lOOmg) with 5-chloro-2-methoxyaniline (135mg) gave the title compound after purification by Method A LCMS rt 5.75 (MH)+ 388/390 1H NMR δ 9.89 (IH, s), 8.48 (IH, br,s), 8.38 (IH, s), 7.88 (IH, d), 7.57-7.83 (2H, m), 7.34 (2H, m), 7.19 (IH, d), 3.86 (3H, s)
Example 42 : (3 ,5 -Dichloro-phenyl)- [ 1 -(2,4-dimethyl-phenyl)- 1 H-pyrazolo [3,4- d]pyrimidin-4-yl] -amine
Reaction of Intermediate 1 (75mg) with 3,5-dichloroaniline (95mg) gave the title compound after purification by Method A LCMS rt 6.22 (M-H)- 382/384
1H NMR δ 10.46 (IH, s), 8.57 (IH, s), 8.55 (IH, s), 7.19-7.35 (4H, m), 2.40 (3H, s), 2.03 (3H, s)
Example 43 : [ 1 -(2,4-Dimethyl-phenyl)- 1 H-pyrazolo [3 ,4-d]pyrimidin-4-yl] -(4-oxazol-5- yl-phenyl)-amine
Reaction of intermediate 1 (75mg) with 4-oxazol-5-yl-phenylamine (95mg) gave the title compound after purification by Method B LCMS rt 5.22 (MH)+ 383 1H NMR δ 10.37 (IH, s), 8.57 (IH, s), 8.45 (2H, d), 8.05 (2H, d), 7.79 (2H, d), 7.65 (IH, s), 7.22-7.30 (3H, m), 2.40 (3H, s), 2.04 (3H, s)
Example 44 : (3 -Chloro-4-fluoro-phenyl)- [ 1 -(2,4-dimethyl-phenyl)- 1 H-pyrazolo [3 ,4- d]pyrimidin-4-yl] -amine
Reaction of Intermediate 1 (75mg) with 3-chloro-4-fluoroaniline (84mg) gave the title compound after purification by Method A
LCMS rt 5.68 (MH)+ 368
1H NMR δ 10.55 (IH, s), 8.72 (IH, s), 8.65 (IH, s), 8.47 (IH, d,d), 7.95 (IH, m), 7.67 (IH, t), 7.40-7.49 (3H, m), 2.58 (3H, s), 2.22 (3H, s)
Example 45: [l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-(lH-indazol- 5-yl)-amine
Reaction of Intermediate 1 (75mg) with 5-aminoindazole (77mg) gave the title compound after purification by Method B
LCMS rt 4.39 (MH)+ 356
1H NMR δ 10.28 (IH, s), 8.43 (IH, s), 8.34 (IH, br,s), 8.18 (IH, s), 7.68 (2H, t), 7.23-
7.34 (3H, m), 2.45 (3H, s), 2.06 (3H, s)
Example 47: (3,4-Dichloro-phenyl)-[l-(2,4-dimethyl-phenyl)-lH-pyrazolo[3,4- d]pyrirnidin-4-yl] -amine
Reaction of Intermediate 1 (75mg) with 3,4-dichloroaniline (95mg) gave the title compound after purification by Method A LCMS rt 6.05 (MH)+ 384/386
1H NMR δ 10.68 (IH, s), 8.81 (IH, s), 8.74 (IH, s), 8.63 (IH, d), 8.09 (IH, d,d), 7.91 (IH, d), 7.46-7.54 (3H, m), 2.63 (3H, s), 2.27 (3H, s)
Example 48: N-{4-[l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-ylamino]- phenyl} -acetamide
Reaction of Intermediate 1 (75mg) withN-(4-amino-phenyl)-acetamide (87mg) gave the title compound after purification by Method A LCMS rt 4.39 (M-H)- 371
1HNMR δ 10.15 (IH, s) ,9.99 (IH, s), 8.40 (IH, br, s), 8.36 (IH, s), 7.74 (2H, d), 7.62 (2H, d), 7.17-7.28 (3H, m), 2.39 (3H, s), 2.06 (3H, s), 2.03 (3H, s)
Example 49 : 4-[ 1 -(2,4-Dimethyl-phenyl)- lH-pyrazolo[3 ,4-d]pyrimidin-4-ylamino ]- benzamide
Reaction of Intermediate 1 (75mg) with 4-amino-benzamide (80mg) gave the title compound after purification by Method A LCMS rt 4.35 (M-H)- 357 1HNMR δ 10.39 (IH, s), 8.59 (IH, s), 8.48 (IH, s), 8.00 (2H, d), 7.93 (2H, d), 7.18- 7.30 (3H, m), 2.39 (3H, s), 2.03 (3H, s)
Example 50 : [ 1 -(2,4-Dimethyl-phenyl)- 1 H-pyrazolo [3 ,4-d]pyrimidin-4-yl] -(3 -oxazol-5 - yl-phenyl)-amine
Reaction of Intermediate 1 (75mg) with 3 -oxazol-5 -yl-phenylamine (95mg) gave the title compound after purification by Method B LCMS rt 5.16 (M-H)- 381
1H NMR δ 10.35 (IH, s), 8.55 (IH, s), 8.51 (IH, s), 8.46 (IH, s), 8.26 (IH, s), 7.95 (IH, m), 7.73 (IH, s), 7.54 (2H, d), 7.18-7.30 (3H, m), 2.39 (3H, s), 2.04 (3H, s)
Example 57: (3,4-Dichloro-phenyl)-[l-(2,4-dimethyl-phenyl)-lH-pyrazolo[3,4- d]pyrimidin-4-yl]-methyl-amine
Reaction of Intermediate 1 (75mg) with (3,4-dichloro-phenyl)-methyl-amine (HOuL) gave the title compound after purification by Method A LCMS rt 5.98 (MH)+398/400 1H NMR δ 8.30 (IH, s), 7.89 (IH, d), 7.79 (IH, d), 7.51 (IH, d,d), 7.10-7.16 (3H, m), 6.78 (IH, s), 3.55 (3H, s), 2.29 (3H, s), 1.89 (3H, s)
Example 58 : [ 1 -(2,4-Dimethyl-phenyl)- 1 H-pyrazolo[3 ,4-d]pyrimidin-4-yl] -(4- trifluoromethoxy-phenyl)-amine
Reaction of intermediate 1 (75mg) with 4-trifluoromethoxyaniline (105uL) gave the title compound after purification by Method A
LCMS rt 5.82 (MH)+ 400
1H NMR δ 10.35 (IH, s), 8.55 (IH, s), 8.43 (IH, s), 8.01 (2H d,d,), 7.43 (2H, d), 7.21- 7.30 (3H, m), 2.39 (3H, s), 2.04 (3H, s)
Example 59: [l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-(4- morpholin-4-yl-phenyl)-amine
Reaction of Intermediate 1 (75mg) with 4-morpholin-4-yl-phenylamine (105uL) gave the title compound after purification by Method A
LCMS rt 4.78 (MH)+ 401
1H NMR δ 10.09 (IH, s), 8.40 (2H, br,s), 7.73 (2H, d), 7.29-7.36 (3H, m), 7.11 (2H, d),
3.87 (4H, t), 3.22 (4H, t), 2.48 (3H, s), 2.12 (3H, s)
Example 60 : 4-[ 1 -(2,4-Dimethyl-phenyl)- 1 H-pyrazolo[3 ,4-d]pyrimidin-4-ylamino] - benzenesulfonamide
Reaction of Intermediate 1 (75mg) with 4-amino-benzenesulfonamide (lOOmg) gave the title compound after purification by Method A LCMS rt 4.57 (MH)+ 395
1H NMR δ 10.63 (IH, s), 8.76 (IH, s), 8.65 (IH, s), 8.26 (2H, d), 8.02 (2H, d), 7.37- 7.46 (3H, m), 2.55 (3H, s), 2.20 (3H, s)
Example 61: 4-[l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-ylamino]-N- thiazol-2-yl-benzenesulfonamide
Reaction of Intermediate 1 (75mg) with 4-amino-N-thiazol-2-yl-benzenesulfonamide (150mg) gave the title compound after purification by Method C LCMS rt 4.63 (MH)+ 478 1H NMR δ 10.66 (IH, s), 8.70 (IH, s), 8.48 (IH, s), 8.11 (2H, d), 7.85 (2H, d), 7.16- 7.30 (5H, m), 6.83 (IH, d), 3.9 (3H, s), 2.03 (3H, s)
Example 62: 2-Chloro-4-[l-(2,4-dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4- ylaminoj-benzonitrile
Reaction of Intermediate 1 (75mg) with 2-chloro-4-aminobenzonitrile (90mg) gave the title compound after purification by Method A LCMS rt 5.76 (MH)+ 375 1H NMR δ 10.73 (IH, s), 8.64 (2H, d), 8.55 (IH, d), 8.00-8.09 (2H, m), 7.20-7.34 (3H, m), 2.43 (3H, s), 2.06 (3H, s)
Example 63: [l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-(lH-indol-5- yl)-amine
Reaction of Intermediate 1 (75mg) with 5-aminoindole (75mg) gave the title compound after purification by Method A LCMS rt 4.62 (MH)+ 355
1H NMR δ 10.91 (IH, s), 9.79 (IH, s), 8.06 (IH, s), 7.68 (IH, br,s), 6.91-7.24 (5H, m), 6.23 (2H, m), 2.14 (3H, s), 1.78 (3H, s)
Example 46: l-(4-Chloro-phenyl)-3-[l-(2,4-dimethyl-ρhenyl)-lH-pyrazolo[3,4- d]pyrimidin-4-yl]-urea
A solution of Intermediate 2 (50mg, 0.208mmol, leq) and sodium hydride (l.leq) in anhydrous tetrahydrofuran (2.5ml) was stirred at room temperature for 30min. This solution was added to a solution of 4-chlorophenyl isocyanate (leq) in anhydrous THF (2.5ml) pre-heated to 85°C and the mixture was stirred for another 30min at 85°C. The crude mixture was evaporated and the resulting solid was partitioned between ethyl acetate and water. The combined organic layers were washed once with brine, and concentrated under reduced pressure, to give a crude white powder which was purified by Method C to give the title compound (12.7mg, 0.03mmol ) LCMS rt 5.78 (MH)+ 393
1H-NMR (CDC13): 11.76 (bs, NH), 10.92 (bs, NH), 8.70 (s, IH), 8.59 (s, IH), 7.63 (d, J=8.75Hz; 2H), 7.36 (d, J=8.75Hz; 2H), 7.24 (d, J-8Hz; 2H), 7.17 (d, J=8Hz; IH), 2.32 (s, 3H), 1.94 (s, 3H)
Similarly prepared were:
Example 52: l-(4-Bromo-phenyl)-3-[l-(2,4-dimethyl-phenyl)-lH-pyrazolo[3,4- d]pyrimidin-4-yl]-urea
Reaction of intermediate 2 (50mg, 0.209mmol) with 4-bromophenyl isocyanate (l.leq) gave the title compound.
LCMS rt 5.90 (MH)+ 437
1H-NMR (CDC13): 11.76 (bs, NH), 10.92 (bs, NH), 8.71 (s, IH), 8.59 (s, IH), 7.58 (d,
J=8.75Hz; 2H), 7.49 (d, J=8.75Hz; 2H), 7.24 (d, J=8Hz; 2H), 7.15 (d, J=8Hz; 1H), 2.32 (s, 3H), 1.93 (s, 3H)
Example 53: l-( 2,3-Dichloro-phenyl)-3-[l-(2,4-dimethyl-phenyl)-lH-pyrazolo[3,4- d]pyrimidin-4-yl] -urea
Reaction of Intermediate 2 (50mg) with 2,3-dichlorophenyl isocyanate (l.leq) gave the title compound.
LCMS rt 6.14 (MH)+ 427
1H NMR δ 11.97 (bs, NH), 11.08 (bs, NH), 8.79 (s, IH), 8.68 (s, IH), 7.56 (d, J=2,5Hz;
2H), 7.19-7.32 (m; 4H), 2.38 (s, 3H), 2.01 (s, 3H)
Example 54: l-(3,4-Dimethyl-phenyl)-3-[l-(2,4-dimethyl-phenyl)-lH-pyrazolo[3,4- d]pyrimidin-4-yl] -urea
Reaction of hitermediate 2 (50mg) with 3,4-dimethylphenyl isocyanate (l.leq) gave the title compound LCMS rt 5.88 (MH)+ 387
1H NMR δ 11.52 (bs, NH), 10.88 (bs, NH), 8.80 (s, IH), 8.67 (s, IH), 7.11-7.41 (m; 6H), 2.40 (s, 3H), 2.25 (s, 3H), 2.19 (s, 3H), 2.02 (s, 3H)
Example 55: l-[l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-3-(4- trifluoromethyl-phenyl)-urea
Reaction of intermediate 2 (50mg) with 4-trifluoromethylphenyl isocyanate (l.leq) gave the title compound LCMS rt 5.86 (MH)+ 427 1H NMR δ 11.74 (bs, NH), 10.86 (bs, NH), 8.60 (s, IH), 8.49 (s, IH), 7.70 (d, J=7.5Hz; 2H), 7.53 (d, J=7.5Hz; 2H), 7.09 (d, J=7.5Hz; 2H), 7.03 (d, J=7.5Hz; IH), 2.20 (s, 3H), 1.81 (s, 3H)
Example 51: 2-(2-Bromo-phenyl)-N-[l-(2,4-dimethyl-phenyl)-lH-pyrazolo[3,4- d]pyrimidin-4-yl]-acetamide
A suspension of Intermediate 2 (50mg) in dry acetonitrile was treated with (2-bromo- phenyl)-acetyl chloride (38uL ,1.2eq) and heated to reflux for 72h. The reaction
mixture was partitioned between ethyl acetate and a saturated sodium bicarbonate solution and the organic phase separated and concentrated. Purification using Method
A gave the title compound
LCMS rt 5.43 (MH)+ 436/438
1H NMR δ 11.71 (IH, s), 8.67 (IH, s), 8.61 (IH, s), 7.66 (IH, d,d) 7.51 (IH, d,d), 7.41
(IH, d,t) 7.19-7.31 (4H, m), 4.16 (2H, s), 2.39 (3H, s), 2.00 (3H, s)
Example 56 : 4- {3 - [ 1 -(2,4-Dimethyl-phenyl)- 1 H-pyrazolo [3 ,4-d]pyrimidin-4-yl] - ureido}-benzenesulfonamide
4-Amino-benzenesulfonamide (36mg, 0.208mmol, leq) and rriethylamine (2eq) in anhydrous THF (2.5ml) was heated at reflux for five minutes followed by the addition of triphosgene (leq) in one portion. The reaction mixture was heated at 85° for 30 minutes to generate the isocyanate. Separately, a solution of Intermediate 2 (50mg, 0.208mmol, leq) and sodium hydride (l.leq) in anhydrous tetrahydrofuran (2.5ml) was stirred at room temperature for 30min then added to the solution of isocyanate at 85°C and the mixture was stirred for another 30min at 85°C. The crude mixture was evaporated and the resulting solid was partitioned between ethyl acetate and water. The combined organic layers were washed once with brine, and concentrated under reduced pressure, to give a crude white powder which was purified by Method C to give the title compound.
LCMS rt 4.67 (MH)+ 438
1H-NMR (CDC13): 11.91 (bs, NH), 11.03 (bs, NH), 8.80 (s, IH), 8.69 (s, IH), 7.84 (s, 4H), 7.23-7.29 (m, 3H), 2.40 (s, 3H), 2.02 (s, 3H)
Prepared similarly were:
Example 64 : 1 -(4-Cyano-phenyl)-3 -[ 1 -(2,4-dimethyl-phenyl)- lH-pyrazolo [3 ,4- d]pyrimidin-4-yl]-urea ~
Reaction of Intermediate 2 (50mg, 0.208mmol) with 4-amino-benzonitrile (leq) gave the title compound LCMS rt 5.34 (MH)+ 384
1H NMR (CDCI3): 11.99 (bs, NH), 11.22 (bs, NH), 8.78 (s, IH), 8.68 (s, IH), 7.86 (s, 5H), 7.31 (d, J=7.5Hz; IH), 7.22 (d, J=7.5Hz; IH), 2.40 (s, 3H), 2.02 (s, 3H)
Example 65: l-[l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-3-(3- trifluoromethyl-phenyl)-urea
Reaction of Intermediate 2 (50mg, 0.208mmol) with 3-trifluoromethyl-phenylamine (leq), as above, gave the title compound LCMS rt 5.87 (MH)+ 427 1H NMR δ ( CDC13): 12.13 (bs, NH), 10.68 (bs, NH), 8.63 (s, 2H), 7.85 (d, J=12.5Hz; 2H), 7.47 (t, J=7.5Hz; IH), 7.37 (d, J=7.5Hz; IH), 7.10-7.25 (m, 3H), 2.35 (s, 3H), 2.04 (s, 3H)
Example 66 : 1 - [ 1 -(2,4-Dimethyl-phenyl)- 1 H-pyrazolo [3 ,4-d]pyrirnidin-4-yl] -3 -p-tolyl- urea
Reaction of Intermediate 2 (50mg, 0.208mmol) with p-tolylamine (leq), as above, gave the title compound LCMS rt 5.70 (MH)+ 373 1H-NMR (CDC13): 12.13 (bs, NH), 10.68 (bs, NH), 8.69 (s, IH), 8.59 (s, IH), 7.49 (d, J=7.5Hz; 2H), 7.13-7.24 (m, 5H), 2.35 (s, 3H), 2.29 (s, 3H), 2.03 (s, 3H)
Example 67: (l-Phenyl-lH-pyrazolo[3,4-d]pyrimidin-4-yl)-(5,6,7,8-tetrahydro- naphthalen- 1 -yl)-amine
Purchased from IFLabs
Example 68 : (2-Bromo-phenyl)-( 1 -phenyl- 1 H-pyrazolo [3 ,4-d]pyrimidin-4-yl)-amine
Purchased from Interbioscreen
Example 69: (l-Benzyl-lH-pyrazolo[3,4-d]pyrimidin-4-yl)-(2,4-dimethyl-phenyl)- amine
Purchased from Interbioscreen
Example 70: [2-(4-Chloro-phenyl)-ethyl]-[l-(2,4-dimethyl-phenyl)-lH-pyrazolo[3,4- d]pyrimidin-4-yl] -amine
Purchased from LFLabs
Example 71 : [l-(2,4-Dimethyl-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-yl]-(3-fluoro-4- morpholin-4-yl-phenyl)-amine
3,4-Difluoronitrobenzene (2.52g) was added to a solution of morpholine ( 6.0ml) in THF (10ml) at 0°. The reaction mixture was allowed to warm to rt and stirred for a further hour. The mixture was diluted with 1M citric acid solution and toluene and the organic phase separated, washed with water, dried and concentrated in vacuo to give an orange solid (2.43 g). This solid was dissolved in 1:1 toluene: ethanol (50ml) and subjected to hydrogenation over 10%/C ( 200mg) at RTP. The catalyst was removed by filtration and the solvent removed to give the desired amine as a beige solid (2.05g) A portion of this amine (75mg) and Intermediate 1 (lOOmg) were combined by the method of Example 1 to give the title compound by purification method 1. LCMS (MH)+ 419 1H NMR δ (DMSO): 10.25 (bs, NH), 8.54 (bs, NH), 8.43 (s, IH), , 7.94 (d, J=15.2Hz; IH), 7.5 (d, J= 7.6, IH), 7.2-7.3 (m, 4H), 3.8 (4H, m), 3.05 (4H, m), 2.41 (3H, s), 2.05 (3H, s)
Example 72 : 5 - [ 1 -(2,4-Dimethyl-phenyl)- 1 H-pyrazolo [3 ,4-d]pyrimidin-4-ylamino ] -2- morpholin-4-yl-benzonitrile
2-Fluoro-5 -nitrobenzene (l.Og) in MeCN (30ml) was treated with morpholine ( 1.0ml) and triethylamine (1.65ml) before being heated to reflux overnight. The mixture was
concentrated and partitioned between 1M citric acid solution and DCM and the organic phase separated, washed with water, dried and concentrated in vacuo to give an orange solid (1.52g). This solid was dissolved in 1:1 toluene: ethanol (50ml) and subjected to hydrogenation over 10%/C ( 150mg) at RTP. The catalyst was removed by filtration and the solvent removed to give the desired amine as a tan solid (1.38g)
A portion of this amine (80mg) and Intermediate 1 (lOOmg) were combined by the method of Example 1 to give the title compound by purification method 1. LCMS (MH)+ 426
1H NMR δ.(DMSO): 10.33 (bs, NH), 8.49 (bs, NH), 8.42 (s, IH), 8.34 (IH, d), 7.96 (dd, J=8.85Hz; IH), 7.15-7.3 (m, 4H), 3.77 (4H, m), 3.13 (4H, m), 2.37 (3H, s), 2.02 (3H, s)
Example 73 : 1 -[ 1 -(2,4-Dimethyl-phenyl)- lH-pyrazolo[3,4-d]pyrimidin-4-yl]-3-[4- (pyridin-2-ylmethoxy)-phenyl]-urea
A solution of di-tert-butyldicarbonate (15g) in THF (50ml) was added to a solution of 4-aminophenol (7.48g) in THF (100ml). Stirred overnight then concentrated and triturated with Et2O / DCM whereupon the product spontaneously crystallised. Petrol was added and the solution concentrated slightly in vacuo and the resulting cold suspension filtered to give a first crop of solid. The filtrate was diluted further with petrol and concentrated to give a second crop that was combined with the first to give 4- tert-butoxycarbonylaminophenol (12.46g). A portion of this material (2.1g) and powdered K2CO3 (6.25g) in MeCN ( 100ml) was treated with 2-bromomethylpyridine (leq, 2.54g) and heated to 80° for 3h. The cooled RM was concentrated and after aqueous workup the residue was purified by column chromatography on silica gel with EtOAc /hexane ( 2:3 to 3:2) as eluant. This gave the desired alkylated product as a pale yellow solid (2.2g) which was dissolved in DCM (10ml), cooled to 0° and treated with TFA (5ml) and stirred for 2h. The mixture was poured into 2N NaOH and extracted in DCM (3x25ml). Concentration of the combined organic phase gave the amine as a brown oil (700mg). This amine (90mg) was used to generate the corresponding isocyanate in situ using triphosgene and triethylamine. This isocyanate was treated with Intermediate 2 by the method of Example 46 to give the desired title compound (12mg)
1H NMR δ 11.4 (IH, s), 10.84 (IH, s), 8.77 (IH, s), 8.63 (IH, s), 8.58 (2H, d), 7.84 (IH, t), 7.53 (2H, m), 7.4-7.0 (5H, m), 5.18 (2H, s), 2.39 (3H, s), 2.0 (3H, s)
Example 74 : 1 - [ 1 -(2-Chloro-phenyl)- 1 H-pyrazolo [3 ,4-d]pyrimidin-4-yl] -3 - [4-(pyridin- 2-ylmethoxy)-phenyl]-urea
In a similar way to Example 73, the amine monomer was used to generate the corresponding isocyanate in situ using triphosgene and triethylamine. This isocyanate was treated with l-(2-Chloro-phenyl)-lH-pyrazolo[3,4-d]pyrimidin-4-ylamine (prepared as per Intermediate 2) by the method of Example 46 to give the desired title compound (lOmg)
1H NMR δ 11.28 (IH, s), 10.86 (IH, s), 8.82 (IH, s), 8.65 (lH,s), 8.57 (2H, m), 8.0-7.5 (8H, m), 7.32 (2H, d), 7.05 (2H, d), 6.94 (IH, d), 5.18 (2H, s)
Activity Example
Cells used: HCV replicon cells Huh 9B (ReBlikon), containing the firefly luciferase - ubiquitin - neomycin phosphotransferase fusion protein and EMCV-IRES driven HCV polyprotein with cell culture adaptive mutations.
Cell culture conditions:
Cells were cultured at 37°C in a 5% CO2 environment and split twice a week on seeding at 2 x 10E6 cells/flask on day 1 and 1 x 10E6 3 days later. Some 0.25mg/ml G418 was added to the culture medium (125ul per 25ml) but not the assay medium. The culture medium consisted of DMEM with 4500g/l glucose and glutamax (Gibco 61965-026) supplemented with 1 x non-essential amino acids, penicillin (100 IU/ml) / streptomycin (100 μg/ml), FCS (10%, 50ml) and 1 mg/ml G418 (Invitrogen cat no 10131-027) & 10 % foetal calf serum.
Assay procedure: A flask of cells was trypsinised and a cell count carried out. Cells were diluted to 100,000 cells/ml and 100 μl of this used to seed one opaque white 96-well plate (for the
replicon assay) and one flat-bottomed clear plate (for the tox assay) for every seven compounds to be tested for IC50. Wells G12 and H12 were left empty in the clear plate as the blank. Plates were then incubated at 37°C in a 5% CO
2 environment for 24 h. On the following day compound dilutions are made up in medium at twice their desired final concentration in a clear round bottomed plate. All dilutions have a final DMSO concentration of 1%. Once the dilution plate had been made up, controls and compounds were transferred to the assay plate (containing the cells) at lOOμl /well in duplicate plates. Exception: in the white (replicon) plate, no compound was added to wells Al and A2 and lOOμl of 1% DMSO was added to these instead, hi the clear (Tox) plate, wells El 2 & F12 only contained the DMSO control. Plates were then incubated at 37°C with 5% CO
2 for 72h. At the end of the incubation time, the cells in the white plate were harvested by washing with 200 μl/ well of warm (37°C) PBS and lysed with 20 μl cell culture lysis buffer (Promega). After 5 min incubation @ RT, luciferin solution was added to the luciferase assay buffer (LARB at 200μl per 10 ml LARB. The M injector of the microplate luminometer (Lmax, Molecular Devices) was primed with 4 x 300μl injections. Plate were inserted into the luminometer and 100 μl luciferase assay reagent was added by the injector on the luminometer. The signal was measured using a 1 second delay followed by a 4 second measurement programme. The IC50, the concentration of the drug required for reducing the replicon level by 50% in relation to the untreated cell control value, can be calculated from the plot of the percentage reduction of the luciferase activity vs. drug concentration. The clear plate was stained with lOOμl 0.5% methylene blue in 50% ethanol at RT for lh, followed by solvation of the absorbed methylene blue in 100 μl per well of 1% lauroylsarcosine. Absorbance of the plate was measured on a microplate spectrophotometer (Molecular Devices) and the absorbance for each concentration of compound expressed as a proportion of the relative DMSO control. The TD50, the concentration of drug required to reduce the total cell area by 50%) relative to the DMSO controls was calculated by plotting the absorbance at 620nm vs drug concentration. Results are shown in the Table below.