WO2005040100A1 - Tetrahydro-naphthalene and urea derivatives - Google Patents
Tetrahydro-naphthalene and urea derivatives Download PDFInfo
- Publication number
- WO2005040100A1 WO2005040100A1 PCT/EP2004/011008 EP2004011008W WO2005040100A1 WO 2005040100 A1 WO2005040100 A1 WO 2005040100A1 EP 2004011008 W EP2004011008 W EP 2004011008W WO 2005040100 A1 WO2005040100 A1 WO 2005040100A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- amino
- halogen
- alkyl
- hydroxy
- alkoxycarbonyl
- Prior art date
Links
- 0 C*(C)N(*)C(N(C)*)=* Chemical compound C*(C)N(*)C(N(C)*)=* 0.000 description 5
- JZGHUWHPHXNQRA-UHFFFAOYSA-N CC(CN=C1CNC(Nc2cccc(CC3)c2CC3=O)=O)(C(F)(F)F)C=C1Cl Chemical compound CC(CN=C1CNC(Nc2cccc(CC3)c2CC3=O)=O)(C(F)(F)F)C=C1Cl JZGHUWHPHXNQRA-UHFFFAOYSA-N 0.000 description 1
- IMEDWEOSHLIWHU-UHFFFAOYSA-N CC(OC1C=CC=CC1)=O Chemical compound CC(OC1C=CC=CC1)=O IMEDWEOSHLIWHU-UHFFFAOYSA-N 0.000 description 1
- DVARSBFAPZPKTR-UHFFFAOYSA-N Nc1c(CC(CO)CC2)c2ccc1 Chemical compound Nc1c(CC(CO)CC2)c2ccc1 DVARSBFAPZPKTR-UHFFFAOYSA-N 0.000 description 1
- LNTXZYMWYQPMOD-CQSZACIVSA-N O[C@H](CC1)Cc2c1cccc2NC(COc(cc1)ccc1Cl)=O Chemical compound O[C@H](CC1)Cc2c1cccc2NC(COc(cc1)ccc1Cl)=O LNTXZYMWYQPMOD-CQSZACIVSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C275/00—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups
- C07C275/28—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of urea groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton
- C07C275/32—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of urea groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton being further substituted by singly-bound oxygen atoms
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/165—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide
- A61K31/167—Amides, e.g. hydroxamic acids having aromatic rings, e.g. colchicine, atenolol, progabide having the nitrogen of a carboxamide group directly attached to the aromatic ring, e.g. lidocaine, paracetamol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/16—Amides, e.g. hydroxamic acids
- A61K31/17—Amides, e.g. hydroxamic acids having the group >N—C(O)—N< or >N—C(S)—N<, e.g. urea, thiourea, carmustine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/335—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin
- A61K31/357—Heterocyclic compounds having oxygen as the only ring hetero atom, e.g. fungichromin having two or more oxygen atoms in the same ring, e.g. crown ethers, guanadrel
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/42—Oxazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/41—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with two or more ring hetero atoms, at least one of which being nitrogen, e.g. tetrazole
- A61K31/433—Thidiazoles
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4406—Non condensed pyridines; Hydrogenated derivatives thereof only substituted in position 3, e.g. zimeldine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4409—Non condensed pyridines; Hydrogenated derivatives thereof only substituted in position 4, e.g. isoniazid, iproniazid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/4412—Non condensed pyridines; Hydrogenated derivatives thereof having oxo groups directly attached to the heterocyclic ring
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/44—Non condensed pyridines; Hydrogenated derivatives thereof
- A61K31/455—Nicotinic acids, e.g. niacin; Derivatives thereof, e.g. esters, amides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/4706—4-Aminoquinolines; 8-Aminoquinolines, e.g. chloroquine, primaquine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/47—Quinolines; Isoquinolines
- A61K31/472—Non-condensed isoquinolines, e.g. papaverine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/517—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with carbocyclic ring systems, e.g. quinazoline, perimidine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/54—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being an organic compound
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/02—Drugs for disorders of the urinary system of urine or of the urinary tract, e.g. urine acidifiers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/10—Drugs for disorders of the urinary system of the bladder
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/01—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms
- C07C233/16—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by singly-bound oxygen atoms
- C07C233/24—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by singly-bound oxygen atoms with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by a carbon atom of a six-membered aromatic ring
- C07C233/29—Carboxylic acid amides having carbon atoms of carboxamide groups bound to hydrogen atoms or to acyclic carbon atoms having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by singly-bound oxygen atoms with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by a carbon atom of a six-membered aromatic ring having the carbon atom of the carboxamide group bound to an acyclic carbon atom of a carbon skeleton containing six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C233/00—Carboxylic acid amides
- C07C233/64—Carboxylic acid amides having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings
- C07C233/67—Carboxylic acid amides having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by singly-bound oxygen atoms
- C07C233/75—Carboxylic acid amides having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a hydrocarbon radical substituted by singly-bound oxygen atoms with the substituted hydrocarbon radical bound to the nitrogen atom of the carboxamide group by a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C235/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms
- C07C235/02—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton
- C07C235/04—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton the carbon skeleton being acyclic and saturated
- C07C235/18—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton the carbon skeleton being acyclic and saturated having at least one of the singly-bound oxygen atoms further bound to a carbon atom of a six-membered aromatic ring, e.g. phenoxyacetamides
- C07C235/24—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton the carbon skeleton being acyclic and saturated having at least one of the singly-bound oxygen atoms further bound to a carbon atom of a six-membered aromatic ring, e.g. phenoxyacetamides having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C235/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms
- C07C235/02—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton
- C07C235/32—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton the carbon skeleton containing six-membered aromatic rings
- C07C235/38—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to acyclic carbon atoms and singly-bound oxygen atoms bound to the same carbon skeleton the carbon skeleton containing six-membered aromatic rings having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C235/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms
- C07C235/42—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings and singly-bound oxygen atoms bound to the same carbon skeleton
- C07C235/44—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings and singly-bound oxygen atoms bound to the same carbon skeleton with carbon atoms of carboxamide groups and singly-bound oxygen atoms bound to carbon atoms of the same non-condensed six-membered aromatic ring
- C07C235/56—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings and singly-bound oxygen atoms bound to the same carbon skeleton with carbon atoms of carboxamide groups and singly-bound oxygen atoms bound to carbon atoms of the same non-condensed six-membered aromatic ring having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C235/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms
- C07C235/42—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings and singly-bound oxygen atoms bound to the same carbon skeleton
- C07C235/44—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings and singly-bound oxygen atoms bound to the same carbon skeleton with carbon atoms of carboxamide groups and singly-bound oxygen atoms bound to carbon atoms of the same non-condensed six-membered aromatic ring
- C07C235/58—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings and singly-bound oxygen atoms bound to the same carbon skeleton with carbon atoms of carboxamide groups and singly-bound oxygen atoms bound to carbon atoms of the same non-condensed six-membered aromatic ring with carbon atoms of carboxamide groups and singly-bound oxygen atoms, bound in ortho-position to carbon atoms of the same non-condensed six-membered aromatic ring
- C07C235/64—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by oxygen atoms having carbon atoms of carboxamide groups bound to carbon atoms of six-membered aromatic rings and singly-bound oxygen atoms bound to the same carbon skeleton with carbon atoms of carboxamide groups and singly-bound oxygen atoms bound to carbon atoms of the same non-condensed six-membered aromatic ring with carbon atoms of carboxamide groups and singly-bound oxygen atoms, bound in ortho-position to carbon atoms of the same non-condensed six-membered aromatic ring having the nitrogen atom of at least one of the carboxamide groups bound to a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C237/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups
- C07C237/02—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton
- C07C237/04—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being acyclic and saturated
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C275/00—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups
- C07C275/28—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of urea groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton
- C07C275/30—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of urea groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton being further substituted by halogen atoms, or by nitro or nitroso groups
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C275/00—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups
- C07C275/28—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of urea groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton
- C07C275/38—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups having nitrogen atoms of urea groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton being further substituted by doubly-bound oxygen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
- C07D209/18—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/36—Radicals substituted by singly-bound nitrogen atoms
- C07D213/40—Acylated substituent nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/04—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D213/24—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen or carbon atoms directly attached to the ring nitrogen atom with substituted hydrocarbon radicals attached to ring carbon atoms
- C07D213/54—Radicals substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D213/56—Amides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D213/00—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members
- C07D213/02—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members
- C07D213/89—Heterocyclic compounds containing six-membered rings, not condensed with other rings, with one nitrogen atom as the only ring hetero atom and three or more double bonds between ring members or between ring members and non-ring members having three double bonds between ring members or between ring members and non-ring members with hetero atoms directly attached to the ring nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D215/00—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems
- C07D215/02—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom
- C07D215/16—Heterocyclic compounds containing quinoline or hydrogenated quinoline ring systems having no bond between the ring nitrogen atom and a non-ring member or having only hydrogen atoms or carbon atoms directly attached to the ring nitrogen atom with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D215/38—Nitrogen atoms
- C07D215/42—Nitrogen atoms attached in position 4
- C07D215/46—Nitrogen atoms attached in position 4 with hydrocarbon radicals, substituted by nitrogen atoms, attached to said nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D217/00—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems
- C07D217/22—Heterocyclic compounds containing isoquinoline or hydrogenated isoquinoline ring systems with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to carbon atoms of the nitrogen-containing ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
- C07D239/72—Quinazolines; Hydrogenated quinazolines
- C07D239/86—Quinazolines; Hydrogenated quinazolines with hetero atoms directly attached in position 4
- C07D239/94—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D261/00—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings
- C07D261/02—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings
- C07D261/06—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members
- C07D261/10—Heterocyclic compounds containing 1,2-oxazole or hydrogenated 1,2-oxazole rings not condensed with other rings having two or more double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D261/14—Nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D285/00—Heterocyclic compounds containing rings having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by groups C07D275/00 - C07D283/00
- C07D285/01—Five-membered rings
- C07D285/02—Thiadiazoles; Hydrogenated thiadiazoles
- C07D285/04—Thiadiazoles; Hydrogenated thiadiazoles not condensed with other rings
- C07D285/06—1,2,3-Thiadiazoles; Hydrogenated 1,2,3-thiadiazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D317/00—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms
- C07D317/08—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3
- C07D317/44—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D317/46—Heterocyclic compounds containing five-membered rings having two oxygen atoms as the only ring hetero atoms having the hetero atoms in positions 1 and 3 ortho- or peri-condensed with carbocyclic rings or ring systems condensed with one six-membered ring
- C07D317/48—Methylenedioxybenzenes or hydrogenated methylenedioxybenzenes, unsubstituted on the hetero ring
- C07D317/62—Methylenedioxybenzenes or hydrogenated methylenedioxybenzenes, unsubstituted on the hetero ring with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to atoms of the carbocyclic ring
- C07D317/66—Nitrogen atoms not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2602/00—Systems containing two condensed rings
- C07C2602/02—Systems containing two condensed rings the rings having only two atoms in common
- C07C2602/04—One of the condensed rings being a six-membered aromatic ring
- C07C2602/08—One of the condensed rings being a six-membered aromatic ring the other ring being five-membered, e.g. indane
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2602/00—Systems containing two condensed rings
- C07C2602/02—Systems containing two condensed rings the rings having only two atoms in common
- C07C2602/04—One of the condensed rings being a six-membered aromatic ring
- C07C2602/10—One of the condensed rings being a six-membered aromatic ring the other ring being six-membered, e.g. tetraline
Definitions
- the present invention relates to a tetrahydro-naphthalene or an urea derivative which is useful as an active ingredient of pharmaceutical preparations.
- the tetrahydro-naphthalene and urea derivatives of the present invention have vanilloid receptor (VR1) antagonistic activity, and can be used for the prophylaxis and treatment of diseases associated with VR1 activity, in particular for the treatment of urological diseases or disorders, such as detrusor overactivity (overactive bladder), urinary incontinence, neurogenic detrusor oeractivity (detrusor hyperflexia), idiopathic detrusor overactivity (detrusor instability), benign prostatic hype ⁇ lasia, and lower urinary tract symptoms; chronic pain, neuropathic pain, postoperative pain, rheumatoid arthritic pain, neuralgia, neuropathies, algesia, nerve injury, ischaemia, neurodegeneration, stroke, and inflammatory disorders such as asthma and chronic obstructive pulmonary (or
- Vanilloid compounds are characterized by the presence of vanillyl group or a functionally equivalent group.
- vanilloid compounds or vanilloid receptor modulators are vanillin (4-hydroxy-3-methoxy-benzaldehyde), guaiacol (2-methoxy-phenol), zingerone (4-/4-hydroxy-3- methoxyphenyl/-2-butanon), eugenol (2-methoxy4-/2-propenyl/phenol), and capsaicin (8-methy-N- vanillyl-6-noneneamide).
- capsaicin the main pungent ingredient in "hot” chili peppers, is a specific neurotoxin that desensitizes C-f ⁇ ber afferent neurons.
- Capsaicin interacts with vanilloid receptors (VR1), which are predominantly expressed in cell bodies of dorsal root ganglia (DRG) or nerve endings of afferent sensory fibers including C-fiber nerve endings [Tominaga M, Caterina MJ, Malmberg AB, Rosen TA, Gilbert H, Skinner K, Raumann BE, Basbaum Al, Julius D: The cloned capsaicin receptor integrates multiple pain-producing stimuli. Neuron. 21: 531-543, 1998].
- VR1 vanilloid receptors
- the VR1 receptor was recently cloned [Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D: Nature 389: 816-824, (1997)] and identified as a nonselective cation channel with six transmembrane domains that is structurally related to the TRP (transient receptor potential) channel family. Binding of capsaicin to VRl allows sodium, calcium and possibly potassium ions to flow down their concentration gradients, causing initial depolarization and release of neurotransmitters from the nerve terminals. VRl can therefore be viewed as a molecular integrator of chemical and physical stimuli that elicit neuronal signals in pathological conditions or diseases.
- antagonists of the VRl receptor can be used for prophylaxis and treatment of the conditions and diseases including chronic pain, neuropathic pain, postoperative pain, rheumatoid arthritic pain, neuralgia, neuropathies, algesia, nerve injury, ischaemia, neuro- degeneration, stroke, inflammatory disorders, urinary incontinence (UI) such as urge urinary incontinence (UUI), and/or overactive bladder.
- UI urinary incontinence
- UUI urge urinary incontinence
- UUI is the involuntary loss of urine.
- UUI is one of the most common types of UI together with stress urinary incontinence (SUI) which is usually caused by a defect in the urethral closure mechanism.
- UUI is often associated with neurological disorders or diseases causing neuronal damages such as dementia, Parkinson's disease, multiple sclerosis, stroke and diabetes, although it also occurs in individuals with no such disorders.
- One of the usual causes of UUI is overactive bladder (OAB) which is a medical condition refening to the symptoms of frequency and urgency derived from abnormal contractions and instability of the detrusor muscle.
- OAB overactive bladder
- Orally active anticholinergic drugs which are commonly prescribed, such as propantheline (ProBanthine), tolterodine tartrate (Detrol) and oxybutynin (Difropan), have serious drawbacks such as unacceptable side effects such as dry mouth, abnormal visions, constipation, and central nervous system disturbances. These side effects lead to poor compliance. Dry mouth symptoms alone are responsible for a 70% non-compliance rate with oxybutynin. The inadequacies of present therapies highlight the need for novel, efficacious, safe, orally available drugs that have fewer side effects.
- WO03/014064 discloses the compounds represented by the general formula:
- X represents C 3-8 cycloalkyl optionally fused by benzene, optionally substituted naphthyl, optionally substituted phenyl, optionally substituted phenyl C I-6 straight alkyl, phenyl fused by cycloalykyl, etc;
- Q aa represents CH or N
- R aa represents hydrogen or methyl
- R b represents hydrogen or methyl
- Y represents substituted naphthyl
- WO03/022809 discloses the compounds having vanilloid receptor antagonist activity represented by the general formula:
- P and P' independently represent aryl or heteroaryl
- R and R independently represent hydrogen, alkoxy, hydroxy, etc;
- WO03/053945 discloses the compounds having vanilloid receptor antagonist activity represented by the general formula:
- R represents hydrogen, alkoxy, hydroxy, etc.
- R represents
- WO03/070247 discloses the compounds having vanilloid receptor antagonist activity represented by the general formula:
- Xc represents N or CR cl ;
- Xc 2 represents N or CR c2 ;
- Xc 3 represents N, NR c3 or CR c3 ;
- Xc 4 represents a bond, N or CR° 4 ;
- Xc 5 represents N or C; provided that at least one of Xci, Xc 2 , Xc 3 and Xc 4 is N;
- Zc_ represents O, NH or S;
- Zc 2 represents a bond, NH or S;
- L c represents alkylene, cycloalkylene, etc;
- R cl , R c2 , R c3 , R c4 , R c5 , R c6 , R c7 , R c8a R cSb are defined in the application; and
- R c9 represents hydrogen, aryl, cycloalkyl, and heterocylcle.
- WO03/080578 discloses the compounds having vanilloid receptor antagonist activity represented by the general formula:
- a d , B d , D and E d are each C or N with the proviso that one or more are N;
- Y ⁇ is an aryl, heteroaryl, carbocyclyl or tased-carbocyclyl;
- n is 0, 1, 2 or 3; and R", R" , R , R a4 ,
- R and R > d6 are defined in the application.
- This invention is to provide a compound of the formula (A), their tautomeric and stereoisomeric form, and salts thereof:
- This invention is to provide an urea derivative of the formula (I), their tautomeric and stereo- isomeric form, and salts thereof:
- n represents an integer of 0 to 6;
- Q] and Q independently represent direct bond or methylene
- Chemical bond between Q 2 __r__rQ 3 is selected from the group consisting of a single bond and a double bond; when Q 2. __z_ .
- Q 3 is a single bond, Q 2 represents CHR 2 , or CO, and Q 3 represents CHR 3 , when Q, — Q, is a double bond, Q 2 represents CR 2 and Q 3 represents CR 3 ;
- R 2 represents hydrogen, hydroxy, C ⁇ . 6 alkoxy or C ⁇ -6 alkanoyloxy
- R 3 represents hydrogen, hydroxy, C 1-6 alkoxy, C ⁇ -6 alkanoyloxy, or C 1-6 alkyl optionally substituted by hydroxy, C_. 6 alkoxy or C__ 6 alkanoyloxy, with the proviso that Qj and Q can not be direct bond at the same time;
- R 2 and R 3 can not be hydrogen at the same time; when Qi and Q 4 are both methylene and R 3 is hydroxy, R 2 is hydroxy, C__ 6 alkoxy or C ⁇ - 6 alkanoyloxy; when Q . is direct bond, R 2 is hydroxy, C ⁇ . 6 alkoxy or C_ -6 alkanoyloxy; and when Q 4 is direct bond, R 2 is hydrogen, C_ -6 alkoxy or Cj._ 6 alkanoyloxy; and R 4 represents aryl optionally having one or two substitaents selected from the group consisting of halogen, hydroxy, .
- the urea derivatives of formula (I) are those wherein; Qi and Q 4 represent methylene;
- Q 2 represents CHR 2 , or CO, wherein
- R 2 represents hydroxy, C ⁇ -6 alkoxy or C 1-6 alkanoyloxy; and Q 3 represents CHR 3 , wherein
- R 3 represents hydrogen, hydroxy, C ⁇ . 6 alkoxy or C ⁇ -6 alkanoyloxy.
- urea derivatives of formula (I) are those wherein;
- Q] represents methylene; Q 4 represents direct bond;
- Q 2 represents CHR 2 or CO, wherein
- R 2 represents hydrogen, C ⁇ . 6 alkoxy or C_.6 alkanoyloxy; and Q 3 represents CHR 3 , wherein
- R 3 represents hydrogen, hydroxy, C 1-6 alkoxy or C ⁇ -6 alkanoyloxy, with the proviso that R 2 and R 3 can not be hydrogen at the same time.
- urea derivatives of formula (I) are those wherein; Qi represents direct bond;
- Q 4 represents methylene; O — 0 ⁇ is a single bond; Q 2 represents CHR 2 or CO, wherein R 2 represents hydroxy, C ⁇ -6 alkoxy or C ⁇ _ 6 alkanoyloxy;
- Q 3 represents CHR 3 , wherein
- R 3 represents hydrogen, hydroxy, C ⁇ _ 6 alkoxy or C_ -6 alkanoyloxy.
- the urea derivatives of formula (T) are those wherein; Qi and Q represent methylene;
- Q- — Q- is a double bond
- Q 2 represents CR 2 , wherein
- R 2 represents C ⁇ _ 6 alkoxy or C ⁇ -6 alkanoyloxy
- Q 3 represents CR 3 , wherein R 3 represents hydrogen, C ⁇ -6 alkoxy or C_. 6 alkanoyloxy.
- the urea derivatives of formula (I) are those wherein;
- Q?— -0? is a single bond or a double bond; when Q 2 r . z_ . Q 3 is a single bond, Q 2 represents CH 2 and Q 3 represents CHR 3 , and when Q 2 _ _rQ 3 is a double bond, Q 2 represents CH and Q 3 represents CR 3 , wherein
- R 3 represents C_ -6 alkyl optionally substituted by hydroxy.
- the urea derivatives of formula (I) are those wherein; n represents an integer of 0 to 1; and R 4 represents phenyl optionally substitated with one or more substitaents selected from the group consisting of chloro, bro o, fluoro, nitro, mthoxy, trifluoromethyl and trifluoromethoxy.
- said urea derivative of the formula (I) is selected from the group consisting of:
- This invention is to provide a hydroxy-tefrahydro-naphthalene derivatives of the formula (I), their tautomeric and stereoisomeric form, and salts thereof:
- n represents an integer of 0 to 6;
- R 1 represents C 3 _ 8 cycloalkyl optionally fused by aryl, wherein said aryl is optionally substitated with one or more substitaents selected from the group consisting of halogen, hydroxy, carboxy, nitro, cyano, amino, C ⁇ _ 6 alkylamino, di(C ⁇ -6 alkyl)amino, C_ -6 alkoxycarbonyl, C__ 6 alkanoyl, C ⁇ -6 alkanoylamino, carbamoyl, C ⁇ -6 alkylcarbamoyl, C ⁇ -6 alkyl optionally substituted by cyano, C_ -6 alkoxycarbonyl, or mono-, di-, or tri- halogen, C .
- .6 alkoxy optionally substitated by mono-, di-, or tri- halogen and C].
- 6 alkylthio optionally substitated by mono-, di-, or tri- halogen; phenyl substitated by heteroaryl, or heteroaryloxy, wherein said heteroaryl and heteroaryloxy are optionally substituted with one or more substitaents selected from the group consisting of halogen, hydroxy, carboxy, nitro, cyano, amino, C_.6 alkylamino, di(C_ -6 alkyl)amino, C_. 6 alkoxycarbonyl, Q.
- hydroxy-tetrahydro-naphthalenylurea derivatives of formula (I) can be those wherein; n represents an integer of 0 or 1 ; and
- R 1 represents C 5- 6cycloalkyl optionally fused by benzene, pyridine, or pyrimidine, wherein said benzene, pyridine, and pyrimidine are optionally substitated by halogen, nitro, or C ⁇ - 6 alkyl optionally substitated by mono-, di-, or tri-halogen.
- hydroxy-tefrahydro-naphthalenylurea derivatives of formula (I) can be those wherein; n represents an integer of 0 or 1; and
- R 1 represents phenyl substituted by thienyl, furyl, pynolyl, thiazolyl, oxazolyl, isoxazolyl, imidazolyl, pyridyl, pyrimidyl, friazolthiadiazolyl, thienyloxy, furyloxy, pynolyl, thiazolyloxy, oxazolyloxy, isoxazolyloxy, imidazolyloxy, pyridyloxy or pyrimidyloxy, wherein said thienyl, furyl, pynolyl, thiazolyl, oxazolyl, isoxazolyl, imidazolyl, pyridyl, pyrimidyl, friazolthiadiazolyl, thienyloxy, furyloxy, pynolyl, thiazolyloxy, oxazolyloxy, isoxazolyloxy, imid
- hydroxy-tetrahydro-naphthalenylurea derivatives of formula (T) can be those wherein; n represents an integer of 0 or 1 ; and
- R 1 represents phenyl fused with thiophene, furan, pyrrole, thiazole, oxazole, isoxazole, imidazole, pyridine, pyrimidine, 1,3-dioxalane, tetrahydrofuran. pynolidine, piperidine, or mo ⁇ holine.
- thiophene, furan, pyrrole, thiazole, oxazole, isoxazole, imidazole, pyridine and pyrimidine are optionally substitated with one or more substitaents selected from the group consisting of halogen, nitro, C ⁇ -6 alkyl optionally substitated by mono-, di-, or tri-halogen, - ⁇ alkoxy optionally substitated by mono-, di-, or tri- halogen, and C ⁇ -6 alkylthio optionally substitated by mono-, di-, or tri- halogen.
- hydroxy-tetrahydro-naphthalenylurea derivatives of formula (I) can be those wherein; n represents an integer of 0 or 1 ; and
- R 1 represents phenyl fused with 1 ,3-dioxalane or tetrahydrofuran.
- the hydroxy-tetrahydro-naphthalenylurea derivatives of formula (I) can be those wherein; n represents an integer of 0 or 1 ;
- R 1 represents thienyl, furyl, pynolyl, thiazolyl, oxazolyl, isoxazolyl, imidazolyl, pyridyl or pyrimidyl, wherein said thienyl, furyl, pyrrolyl, thiazolyl, oxazolyl, isoxazolyl, imidazolyl, pyridyl and pyrimidyl are optionally substitated with one or more substituents selected from the group consisting of halogen, nitro, C ⁇ -6 alkyl optionally substitated by mono-, di-, or tri-halogen, . 6 alkoxy optionally substitated by mono-, di-, or tri- halogen, and C ⁇ - 6 alkylthio optionally substitated by mono-, di-, or tri- halogen.
- hydroxy-tefrahydro-naphthalene derivative of formula (I) are those wherein; n represents an integer of 0 or 1 ; and
- R 1 represents pyridyl or isoxazolyl, wherein said pyridyl and oxazolyl are optionally substitated with one or more substituents selected from the group consisting of halogen, nitro, C_. 6 alkyl optionally substitated by mono-, di-, or tri-halogen, C_ -6 alkoxy optionally substituted by mono-, di-, or tri- halogen, and C 1-6 alkylthio optionally substitated by mono-, di-, or tri- halogen.
- said hydroxy-tefrahydro-naphthalene derivative of the formula (I) is selected from the group consisting of:
- This invention is to provide a hydroxy-tefrahydro-naphthalene derivatives of the formula (I), their tautomeric and stereoisomeric form, and salts thereof:
- R 1 represents aryl or heteroaryl, wherein said aryl and heteroaryl are optionally substitated with one or more substitaents selected from the group consisting of halogen, nitro, hydroxy, carboxy, amino, C_ -6 alkylamino, di(C__ 6 alkyl)amino, C 3 _ 8 cycloalkylamino, C_. 6 alkoxycarbonyl, phenyl (which phenyl is optionally substituted by halogen, trifluoromethyl, trifluoromethoxy, nitro, hydroxy, carboxy, amino, C__ 6 alkylamino, di(Cj . .
- R 11 represents aryl or heteroaryl, wherein said aryl and heteroaryl are optionally substitated with one or more substitaents selected from the group consisting of halogen, nitro, hydroxy, carboxy, amino, C ⁇ -6 alkylamino, di(C ⁇ _ 6 alkyl)amino, C 3-8 cycloalkylamino, C ⁇ -6 alkoxycarbonyl, phenyl (which phenyl is optionally substituted by halogen, nitro, hydroxy, carboxy, amino, C ⁇ -6 alkylamino, di(C ⁇ -6 alkyl)amino, C 3 _ 8 cycloalkylamino, or C_ -6 alkoxycarbonyl), benzyl (in which phenyl moiety is optionally substitated by halogen, nitro, hydroxy, carboxy, amino, C .
- R 12 represents aryl, heteroaryl, or C . .6 alkyl optionally substituted by aryl or heteroaryl, wherein said aryl and heteroaryl are optionally substitated with one or more substitaents selected from the group consisting of halogen, nitro, hydroxy, carboxy, amino, C ⁇ _ 6 alkylamino, di(C ⁇ _ 6 alkyl)amino, C 3-8 cycloalkylamino, C_.
- R 13 represents hydrogen, or C 1-6 alkyl
- said aryl is optionally substitated with one or more substituents selected from the group consisting of halogen, nitro, hydroxy, carboxy, amino, C ⁇ -6 alkylamino, di(C_. 6 alkyl)amino,
- hydroxy-tetrahydro-naphthalenylurea derivatives of formula (I) can be those wherein;
- R 1 represents phenyl, naphthyl, pyridyl, pyrimidyl, indolyl, benzofuranyl, benzo- thiophenyl, quinolinyl or isoquinolinyl, wherein said phenyl, naphthyl, pyridyl, pyrimidyl, indolyl, benzofuranyl, benzothiophenyl, quinolinyl and isoquinolinyl are optionally substitated with one or more substituents selected from the group consisting of halogen, nitro, hydroxy, carboxy, amino, C__ 6 alkylamino, di(C 1-6 alkyl)amino, C 3-8 cycloalkylamino, C ⁇ -6 alkoxycarbonyl, phenyl (which phenyl is optionally substituted by halogen, nitro, hydroxy, carboxy, amino, - ⁇ alkylamino, di(C_.
- the hydroxy-tetrahydro-naphthalenylurea derivatives of formula (I) can be those wherein; R 1 represents phenyl, pyridyl, or pyrimidyl, wherein said phenyl, pyridyl, and pyrimidyl are optionally substitated by one or more of substitaents selected from the group consisting of halogen, nitro, C_ -6 alkyl (which alkyl is optionally substitated by cyano, nitro, or mono-, di-, or tri-halogen), and C ⁇ -6 alkoxy optionally substitated by mono-, di-, or tri- halogen.
- R 1 represents phenyl, pyridyl, or pyrimidyl, wherein said phenyl, pyridyl, and pyrimidyl are optionally substitated by one or more of substitaents selected from the group consisting of hal
- hydroxy-tetrahydro-naphthalenylurea derivatives of formula (I) can be those wherein; represents C 1-6 alkyl optionally substitated by R n , OR 12 , SR 12 or N(R 12 )(R 13 ),
- R 11 represents phenyl, naphthyl, pyridyl or pyrimidyl, wherein said phenyl, naphthyl, pyridyl and pyrimidyl are optionally substitated with one or more substitaents selected from the group consisting of halogen, nitro, hydroxy, carboxy, amino, C_ -6 alkylamino, di(C ⁇ -6 alkylamino, C 3 _s cycloalkylamino, C ⁇ _ 6 alkoxycarbonyl, benzyl (in which phenyl moiety is optionally substitated by halogen, nitro, hydroxy, carboxy, amino, C ⁇ -6 alkylamino, di(C_ -6 alkyl), amino, C 3 .
- .6 alkoxy (which alkoxy is optionally substituted by mono-, di-, or tri- halogen), phenoxy (in which phenyl moiety is optionally substituted by halogen, nitro, hydroxy, carboxy, amino, C__6 alkylamino, di(C ⁇ .6 alkyl)amino, C 3 _ 8 cycloalkylamino, or Cj.6 alkoxycarbonyl or C ⁇ _ 6 alkyl), _6 alkylthio (which alkylthio is optionally substituted by mono-, di-, or tri- halogen), C 3-8 cycloalkyl, and heterocycle;
- R 12 represents pheny, naphthyl, pyridyl, pyrimidyl, or C ⁇ _ 6 alkyl optionally substituted by phenyl, naphthyl, pyridyl or pyrimidyl, wherein said phenyl, naphthyl, pyridyl and pyrimidyl are optionally substitated with one or more substituents selected from the group consisting of halogen, nitro, hydroxy, carboxy, amino, C 1-6 alkylamino, di(C_ -6 alkylamino, C 3-8 cycloalkylamino, C_.6 alkoxycarbonyl, phenyl (which phenyl is optionally substituted by halogen, nitro, hydroxy, carboxy, amino, C_ -6 alkylamino, di(C_ -6 alkyl)amino, C 3-8 cycloalkylamino, or C 1-6 alkoxy- carbonyl), benz
- hydroxy-tetrahydro-naphthalenylurea derivatives of formula (I) can be those wherein;
- R 1 represents C_. alkyl optionally substitated by phenyl (which phenyl is optionally substitated with one or more substitaents selected from the group consisting of halogen, nitro, Cj -6 alkyl optionally substitated by cyano, C 1-6 alkoxycarbonyl or mono-, di-, or tri-halogen, and C ⁇ -6 alkoxy optionally substitated by mono-, di-, or tri- halogen), or N(R 12 )(R 13 ),
- R 12 represents phenyl or C_ -2 alkyl optionally substitated by phenyl, wherein said phenyl is optionally substitated with one or more substitaents selected from the group consisting of halogen, nitro, C ⁇ _ 6 alkyl optionally substituted by mono-, di-, or tri-halogen, and Cj. 6 alkoxy optionally substitated by mono-, di-, or tri- halogen; and
- R 13 represents hydrogen, or C_. 6 alkyl.
- hydroxy-tefrahydro-naphthalene derivative of formula (I) are those wherein;
- R 1 represents C 3 . 8 cycloalkyl optionally fused by phenyl, wherein said phenyl is optionally substitated with one or more substitaents selected from ' the group consisting of halogen, nitro, C ⁇ . 6 alkyl optionally substituted by mono-, di-, or tri-halogen, and C . 6 alkoxy optionally substitated by mono-, di-, or tri- halogen.
- said hydroxy-tefrahydro-naphthalene derivative of the formula (I) is selected from the group consisting of: N-(7-hydroxy-5,6,7,8-tetrahydronaphthalen-l-yl)-2-methoxybenzamide;
- This invention is to provide an urea derivatives of the formula (I), their tautomeric and stereoisomeric form, and salts thereof:
- -X- represents a bond, -O- or -NQR. 1 )- (wherein R 1 is hydrogen or C__ 6 allcyl); with the proviso that when m is 0, -X- represents a bond.
- Qi, Q 2 and Q 3 independently represent ⁇ or CH, with the proviso that at least one of Q_, Q2 and Q 3 is ⁇ ;
- R represents aryl or heteroaryl, wherein said aryl and heteroaryl are optionally substitated with one or more substituents independently selected from the group consisting of halogen, nitro, hydroxy, carboxy, amino, C ⁇ -6 alkylamino, di(C ⁇ -6 alkyl)amino, C 3 .
- urea derivatives of formula (I) can be those wherein;
- n 0, 1, 2, or 3 ;
- p 0 or 1
- -X- represents a bond, -O- or -N ⁇ 1 )- (wherein R 1 is hydrogen or C ⁇ -6 alkyl); with the proviso that when m is 0, -X- represents a bond.
- Qi, Q 2 and Q 3 independently represent N or CH, with the proviso that at least one of Q_, Q 2 and Q 3 is N;
- R represents phenyl, naphthyl, pyridyl, or pyrimidyl, wherein said phenyl, naphthyl, pyridyl and pyrimidyl are optionally substituted with one or more substituents independently selected from the group consisting of halogen, nitro, hydroxy, carboxy, amino, .6 alkylamino, di(C 1-6 alkyl)amino, C 3-8 cyclo- alkylamino, C ⁇ -6 alkoxycarbonyl, phenyl (which phenyl is optionally substituted by halogen, nitro, hydroxy, carboxy, amino, C ⁇ -6 alkylamino, di(C ⁇ -6 alkyl)amino, C 3-8 cycloalkylamino, or C ⁇ -6 alkoxycarbonyl), benzyl (in which phenyl moiety is optionally substitated by halogen, nitro, hydroxy, carboxy, amino, C ⁇
- the urea derivatives of formula (I) can be those wherein; m represents 0, 1, 2, or 3 ; p represents 0 or 1;
- -X- represents a bond, -O- or -N(R ] )- (wherein R 1 is hydrogen or C ⁇ -6 alkyl); with the proviso that when m is 0, -X- represents a bond.
- R represents phenyl, naphthyl, pyridyl, or pyrimidyl, wherein said phenyl, naphthyl, pyridyl and pyrimidyl are optionally substitated with one or more substitaents independently selected from the group consisting of halogen, nitro, hydroxy, carboxy, amino, C ⁇ -6 alkylamino, di(C ⁇ -6 alkyl)amino, C 3 .
- the urea derivatives of formula (I) can be those wherein; m represents 0, 1, 2, or 3; p represents 0 or 1;
- -X- represents a bond, -O- or -N(R')- (wherein R 1 is hydrogen or C ⁇ -6 allcyl); with the proviso that when m is 0, -X- represents a bond.
- R represents phenyl, naphthyl, pyridyl, or pyrimidyl, wherein said phenyl, naphthyl, pyridyl and pyrimidyl are optionally substitated with one or more substituents independently selected from the group consisting of halogen, nitro, hydroxy, carboxy, amino, C ⁇ _ 6 alkylamino, di(C ⁇ . 6 alkyl)amino, C 3-8 cycloalkylamino, Ci.6 alkoxycarbonyl, phenyl (which phenyl is optionally substituted by halogen, nitro, hydroxy, carboxy, amino, C ⁇ -6 alkylamino, di(C ⁇ .
- n 0, 1, 2, or 3 ;
- p 0 or 1
- -X- represents a bond, -O- or -N(R')- (wherein R 1 is hydrogen or C ⁇ -6 alkyl); with the proviso that when m is 0, -X- represents a bond.
- R represents phenyl, naphthyl, pyridyl, or pyrimidyl, wherein said phenyl, naphthyl, pyridyl and pyrimidyl are optionally substitated with one or more substitaents independently selected from the group consisting of halogen, nitro, hydroxy, carboxy, amino, C 1-6 alkylamino, di(C ⁇ -6 alkyl)amino, C 3-8 cycloalkylamino, C ⁇ -6 alkoxycarbonyl, phenyl (which phenyl is optionally substitated by halogen, nitro, hydroxy, carboxy, amino, Cj.
- the urea derivatives of formula (I) can be those wherein; m represents 0, 1, 2, or 3 ; p represents 0 or 1;
- -X- represents a bond, -O- or -N ⁇ 1 )- (wherein R 1 is hydrogen or C__ 6 allcyl); with the proviso that when m is 0, -X- represents a bond.
- Q 2 represents CH;
- Q 3 represents ⁇ ;
- R represents phenyl, naphthyl, pyridyl, or pyrimidyl, wherein said phenyl, naphthyl, pyridyl and pyrimidyl are optionally substituted with one or more substituents independently selected from the group consisting of halogen, nitro, hydroxy, carboxy, amino, Cj.6 alkylamino, di(C_.
- urea derivative of formula (I) are those wherein; m represents 0; p represents 0 or 1;
- Q_, Q 2 and Q 3 independently represent N or CH, with the proviso that at least one of Q b Q 2 and Q 3 is N;
- R represents phenyl, naphthyl, pyridyl, or pyrimidyl, wherein said phenyl, naphthyl, pyridyl and pyrimidyl are optionally substituted with one or more substitaents independently selected from the group consisting of halogen, nitro, hydroxy, carboxy, amino, C 1-6 alkylamino, di(C ⁇ .6 alkyl)amino, C 3-8 cycloalkylamino, .
- the urea derivative of formula (T) are those wherein; m represents 0, 1, 2, or 3 ; p represents 0 or 1;
- -X- represents a bond, -O- or -N(R')- (wherein R 1 is hydrogen or C ⁇ _ 6 alkyl); with the proviso that when m is 0, -X- represents a bond.
- Qi, Q 2 and Q 3 independently represent N or CH, with the proviso that at least one of Qi, Q 2 and Q 3 is N;
- R represents phenyl, naphthyl, pyridyl, or pyrimidyl, wherein said phenyl, naphthyl, pyridyl, or pyrimidyl is optionally substitated by one or more of substituents selected from the group consisting of chloro, bromo, fluoro, nitro, methoxy, trifluoromethyl, trifluoromethoxy and C_. ⁇ alkanoylamino.
- said urea derivative of the formula (I) is selected from the group consisting of:
- the compounds of the present invention are, therefore suitable especially for the prophylaxis and freatment of diseases associated with VRl activity, in particular for the treatment of .
- urological diseases or disorders such as detrusor overactivity (overactive bladder), urinary incontinence, neurogenic detrusor oeractivity (detrusor hyperflexia), idiopathic detrusor overactivity (detrusor instability), benign prostatic hype ⁇ lasia, and lower urinary tract symptoms.
- the compounds of the present invention are also effective for treating or preventing a disease selected from the group consisting of chronic pain, neuropathic pain, postoperative pain, rheumatoid arthritic pain, neuralgia, neuropathies, algesia, nerve injury, ischaemia, neuro- degeneration and/or stroke, as well as inflammatory diseases such as asthma and COPD since the diseases also relate to VRl activity.
- a disease selected from the group consisting of chronic pain, neuropathic pain, postoperative pain, rheumatoid arthritic pain, neuralgia, neuropathies, algesia, nerve injury, ischaemia, neuro- degeneration and/or stroke, as well as inflammatory diseases such as asthma and COPD since the diseases also relate to VRl activity.
- the compounds of the present invention are also useful for the treatment and prophylaxis of neuropathic pain, which is a form of pain often associated with he ⁇ es zoster and post-he ⁇ etic neuralgia, painful diabetic neuropathy, neuropathic low back pain, postfraumatic and postoperative neuralgia, neuralgia due to nerve compression and other neuralgias, phantom pain, complex regional pain syndromes, infectious or parainfectious neuropathies like those associated with HTV infection, pain associated with central nervous system disorders like multiple sclerosis or Parkinson disease or spinal cord injury or traumatic brain injury, and post-stroke pain.
- neuropathic pain which is a form of pain often associated with he ⁇ es zoster and post-he ⁇ etic neuralgia, painful diabetic neuropathy, neuropathic low back pain, postfraumatic and postoperative neuralgia, neuralgia due to nerve compression and other neuralgias, phantom pain, complex regional pain syndromes, infectious or parainfectious neuropath
- the compounds of the present invention are useful for the freatment of musculoskeletal pain, forms of pain often associated with osteoarthritis or rheumatoid arthritis or other forms of arthritis, and back pain.
- the compounds of the present invention are useful for the treatment of pain associated with cancer, including visceral or neuropathic pain associated with cancer or cancer treatment.
- the compounds of the present invention are furthermore useful for the treatment of visceral pain, e.g. pain associated with obstruction of hollow viscus like gallstone colik, pain associated with irritable bowel syndrome, pelvic pain, vulvodynia, orchialgia or prostatodynia, pain associated with inflammatory lesions of joints, skin, muscles or nerves, and orofascial pain and headache, e.g. migraine or tension-type headache.
- visceral pain e.g. pain associated with obstruction of hollow viscus like gallstone colik
- pain associated with irritable bowel syndrome pelvic pain
- vulvodynia orchialgia or prostatodynia
- pain associated with inflammatory lesions of joints, skin, muscles or nerves e.g. migraine or tension-type headache.
- the present invention provides a medicament, which includes one of the compounds, described above and optionally pharmaceutically acceptable excipients.
- Alkyl per se and "alk” and "alkyl” in alkenyl, alkynyl, alkoxy, alkanoyl, alkylamino, alkylamino- carbonyl, alkylaminosulfonyl, alkylsulfonylamino, alkoxycarbonyl, alkoxycarbonylamino and alkanoylamino represent a linear or branched alkyl radical having generally 1 to 6, preferably 1 to 4 and particularly preferably 1 to 3 carbon atoms, representing illustratively and preferably methyl, ethyl, n-propyl, isopropyl, tert-butyl, n-pentyl and n-hexyl.
- Alkoxy illustratively and preferably represents methoxy, ethoxy, n-propoxy, isopropoxy, tert- butoxy, n-pentoxy and n-hexoxy.
- Alkylamino illustratively and preferably represents an alkylamino radical having one or two (independently selected) allcyl substituents, illustratively and preferably representing methylamino, ethylamino, n-propylamino, isopropylamino, tert-butylamino, n-pentylamino, n-hexylamino, N,N- dimethylamino, N ⁇ -diethylamino, ⁇ -ethyl- ⁇ -methylamino, N-methyl- ⁇ -n-propylamino, N- isopropyl- ⁇ -n-propylamino, ⁇ -t-butyl-N-methylamino, N-ethyl-N-n-pentylamino and N-n-hexyl- ⁇ -methylamino.
- Aryl per se and in arylamino and in arylcarbonyl represents a mono- to tricyclic aromatic carbocyclic radical having generally 6 to 14 carbon atoms, illustratively and preferably representing phenyl, naphthyl and phenanthrenyl.
- Cycloalkyl per se and in cycloalkylamino and in cycloalkylcarbonyl represents a cycloalkyl group having generally 3 to 8 and preferably 5 to 7 carbon atoms, illustratively and preferably representing cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl and cycloheptyl.
- Heteroaryl per se and the heteroaryl portion of the heteroaralkyl, heteroaryloxy, heteroaralkyloxy, or heteroarylcarbamoyl represent an aromatic mono- or bicyclic radical having generally 5 to 10 and preferably 5 or 6 ring atoms and up to 5 and preferably up to 4 hetero atoms selected from the group consisting of S, O and N, illustratively and preferably representing thienyl, furyl, pynolyl, thiazolyl, oxazolyl, imidazolyl, pyridyl, pyrimidyl, pyridazinyl, indolyl, isoindolino, indazolyl, benzofuranyl, benzothiophenyl, quinolinyl, isoquinolinyl, tetrazolyl, and triazolyl.
- Heterocyclyl per se and in heterocyclylcarbonyl represents a mono- or polycyclic, preferably mono- or bicyclic, nonaromatic heterocyclic radical having generally 4 to 10 and preferably 5 to 8 ring atoms and up to 3 and preferably up to 2 hetero atoms and/or hetero groups selected from the group consisting of ⁇ , O, S, SO and S0 2 .
- the heterocyclyl radicals can be saturated or partially unsatarated.
- CHAPTER I EMBODIMENT OF THE INVENTION
- the compound of the formula (I) of the present invention can be, but not limited to be, prepared by combining various known methods.
- one or more of the substitaents, such as amino group, carboxyl group, and hydroxyl group of the compounds used as starting materials or intermediates are advantageously protected by a protecting group known to those skilled in the art. Examples of the protecting groups are described in "Protective Groups in Organic Synthesis (3rd Edition)" by Greene and Wuts, John Wiley and Sons, New York 1999.
- the compound of the formula (T) of the present invention can be, but not limited to be, prepared by the Method [A], [B], [C], [D], [E] or [F] below.
- the compound of the formula (I) (wherein n, Q_, Q , Q 3 , Q and R 4 are the same as defined above) can be prepared by the reaction of the compound of the formula (II) (wherein Qj, Q 2 , Q 3 and Q are the same as defined above) and the compound of the formula (HI) (wherein n and R 4 are the same as defined above).
- the reaction may be carried out in a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane; ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane; aromatic hydrocarbons such as benzene, toluene and xylene; nitriles such as acetonitrile; amides such as N, N- dimethylformamide (DMF), ⁇ , ⁇ -dimethylacetamide (DMAC) and ⁇ -methylpynolidone (NMP); urea such as l,3-dimethyl-2-imidazolidinone (DMI); sulfoxides such as dimethylsulfoxide (DMSO); and others.
- a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-d
- the reaction can be carried out in the presence of organic base such as pyridine or triethylamine.
- the reaction temperature can be optionally set depending on the compounds to be reacted.
- the reaction temperatare is usually, but not limited to, about room temperatare to 100°C.
- the reaction may be conducted for, usually, 30 minutes to 48 hours and preferably 1 to 24 hours.
- the compound (II) and (HI) can be prepared by the use of known techniques or are commercially available.
- the compound of the formula (I) (wherein n, Qi, Q 2 , Q 3 , Q 4 and R are the same as defined above) can be prepared by reacting the compound of the formula (IT) (wherein Q_, Q 2 , Q 3 and Q 4 are the same as defined above) with phosgene, diphosgene, triphosgene, 1,1-carbonyldiimidazole (CDI), or l,r-carbonyldi(l,2,4-triazole)(CDT), and then adding the compound of the formula (IV) (wherein n and R 4 are the same as defined above) to the reaction mixture.
- the reaction may be carried out in a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane; ethers such as diethyl ether, iso- propyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane; aromatic hydrocarbons such as benzene, toluene and xylene; nitriles such as acetonitrile; amides such as N, N- dimethylformamide (DMF), N, N-dimefhylacetamide (DMAC) and N-methylpynolidone (NMP); urea such as l,3-dimethyl-2-imidazolidinone (DMI); sulfoxides such as dimethylsulfoxide (DMSO); and others.
- a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-d
- the reaction temperatare can be optionally set depending on the compounds to be reacted.
- the reaction temperatare is usually, but not limited to, about 20°C to 50°C.
- the reaction may be conducted for, usually, 30 minutes to 10 hours and preferably 1 to 24 hours.
- the compound (IV) is commercially available or can be prepared by the use of known techniques and phosgene, diphosgene, triphosgene, CDI, and CDT are commercially available and . [Method C]
- the compound of the formula (I) (wherein n, Q ls Q 2 , Q 3 , Q and R 4 are the same as defined above) can be prepared by reacting the compound of the formula (IT) (wherein Q., Q 2 , Q 3 and Q 4 are the same as defined above) with the compound of the formula (V) (wherein L_ represents halogen atom such as chlorine, bromine, or iodine atom) and then adding the compound of the formula (IV) (wherein n and R 4 are the same as defined above) to the reaction mixture.
- the reaction may be carried out in a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane; ethers such as diethyl ether, iso- propyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane; aromatic hydrocarbons such as benzene, toluene and xylene; nitriles such as acetonifrile; amides such as N, N- dimethylformamide (DMF), N, N-dimethylacetamide (DMAC) and N-methylpynolidone (NMP); urea such as l,3-dimethyl-2-imidazolidinone (DMI); sulfoxides such as dimethylsulfoxide (DMSO); and others.
- a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichlor
- the reaction temperature can be optionally set depending on the compounds to be reacted.
- the reaction temperature is usually, but not limited to, about 20°C to 50°C.
- the reaction may be conducted for, usually, 30 minutes to 10 hours and preferably 1 to 24 hours.
- the reaction can be advantageously carried out in the presence of a base including, for instance, organic amines such as pyridine, friethylamine and N,N-diisopropylethylamine, dimethylaniline, diethylaniline, 4-dimethylaminopyridine, and others.
- a base including, for instance, organic amines such as pyridine, friethylamine and N,N-diisopropylethylamine, dimethylaniline, diethylaniline, 4-dimethylaminopyridine, and others.
- the compound of the formula (I) (wherein n, Q Q 2 , Q 3 , Q and R 4 are the same as defined above) can be prepared by reacting the compound of the formula (IV) (wherein n and R 4 are the same as defined above) with phosgene, diphosgene, triphosgene, 1,1-carbonyldiimidazole (CDI), or 1,1'- carbonyldi(l,2,4-triazole)(CDT) and then adding the compound of the formula (B) (wherein Q_, Q 2 , Q 3 and Q 4 are the same as defined above) to the reaction mixture.
- the reaction may be carried out in a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane; ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane; aromatic hydrocarbons such as benzene, toluene and xylene; nitriles such as acetonitrile; amides such as N, N- dimethylformamide (DMF), N, N-dimethylacetamide (DMAC) and N-methylpynolidone (NMP); urea such as l,3-dimethyl-2-imidazolidinone (DMI); sulfoxides such as dimethylsulfoxide (DMSO); and others.
- a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloro
- the reaction temperature can be optionally set depending on the compounds to be reacted.
- the reaction temperatare is usually, but not limited to, about 20°C to 50°C.
- the reaction may be conducted for, usually, 30 minutes to 10 hours and preferably 1 to 24 hours.
- the compound of the formula (I) (wherein n, Qi, Q 2 , Q 3 , Q 4 and R 4 are the same as defined above) can be prepared by reacting the compound of the formula (TV) (wherein n and R 4 are the same as defined above) with the compound of the formula (V) (wherein Lj is the same as defined above) and then adding the compound of the formula (TT) (wherein Q ls Q 2 , Q 3 and Q are the same as defined above) to the reaction mixture.
- Qi, Q 2 , Q 3 and Q 4 and R 4 can be prepared by reacting the compound of the formula (TV) (wherein n and R 4 are the same as defined above) with the compound of the formula (V) (wherein Lj is the same as defined above) and then adding the compound of the formula (TT) (wherein Q ls Q 2 , Q 3 and Q are the same as defined above) to the reaction mixture.
- the reaction may be carried out in a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane; ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane; aromatic hydrocarbons such as benzene, toluene and xylene; nitriles such as acetonitrile; amides such as N, N- dimethylformamide (DMF), N, N-dimethylacetamide (DMAC) and N-methylpynolidone (NMP); urea such as l,3-dimethyl-2-imidazolidinone (DMT); sulfoxides such as dimethylsulfoxide (DMSO); and others.
- a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloro
- the reaction temperature can be optionally set depending on the compounds to be reacted.
- the reaction temperatare is usually, but not limited to, about 20°C to 50°C.
- the reaction may be conducted for, usually, 30 minutes to 10 hours and preferably 1 to 24 hours.
- the reaction can be advantageously canied out in the presence of a base including, for instance, organic amines such as pyridine, friethylamine and NN-diisopropylethylamine, dimethylaniline, diethylaniline, 4-dimethylaminopyridine, and others.
- a base including, for instance, organic amines such as pyridine, friethylamine and NN-diisopropylethylamine, dimethylaniline, diethylaniline, 4-dimethylaminopyridine, and others.
- the compound of the formula (I-a) (wherein n and R 4 are the same as defined above) can be prepared in the similar manner as described in Method [A], [B], [C], [D] or [E] for the preparation of the compound of the formula (I) by using a compound of the formula (VT) instead of the compound of the formula (IT).
- the compound of the formula (I-b) (wherein n and R 4 are the same as defined above) can be prepared by reacting the compound of the formula (I-a) (wherein n and R 4 are the same as defined above) with an acid such as hydrochloric acid.
- the reaction may be carried out in a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane; ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane; alcohols such as methanol, ethanol; water and others.
- halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane
- ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane
- alcohols such as methanol, ethanol; water and others.
- two or more of the solvents selected from the listed above can be mixed and used.
- the reaction temperature can be optionally set depending on the compounds to be reacted.
- the reaction temperature is usually, but not limited to, about 20°C to 100°C.
- the reaction may be conducted for, usually, 30 minutes to 10 hours and preferably 1 to 24 hours.
- the compound of the formula (I-c) (wherein n and R 4 are the same as defined above) can be prepared by reacting the compound of the formula (I-b) (wherein n and R 4 are the same as defined above) with reducing agent such as sodium borohydride or lithium aluminum hydride.
- the reaction may be carried out in a solvent including, for instance, ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane; alcohols such as methanol, ethanol, isopropanol, and others.
- ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane
- alcohols such as methanol, ethanol, isopropanol, and others.
- two or more of the solvents selected from the listed above can be mixed and used.
- the reaction temperature can be optionally set depending on the compounds to be reacted.
- the reaction temperature is usually, but not limited to, about 20°C to 50°C.
- the reaction may be conducted for, usually, 30 minutes to 10 hours and preferably 1 to 24 hours.
- the compound of the formula (I) of the present invention can be, but not limited to be, prepared by combining various known methods.
- one or more of the substitaents, such as amino group, carboxyl group, and hydroxyl group of the compounds used as starting materials or intermediates are advantageously protected by a protecting group known to those skilled in the art. Examples of the protecting groups are described in "Protective Groups in Organic Synthesis (3rd Edition)" by Greene and Wuts, John Wiley and Sons, New York 1999.
- the compound of the formula (I) of the present invention can be, but not limited to be, prepared by the Method [A] below.
- the compound of the formula (I) of the present invention can be, but not limited to be, prepared by the Method [A], [B], [C], [D], [E], [F], [G] or [H] below.
- the compound of the formula (I) (wherein n and R 1 are the same as defined above) can be prepared by reacting the compound of the formula (II) and the compound of the formula (DI) (wherein Lj represents halogen atom such as chlorine, bromine, or iodine atom) and then adding the compound of the formula (TV) (wherein n, R 1 are the same as defined above) to the reaction mixtare.
- the reaction may be carried out in a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane; ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane; aromatic hydrocarbons such as benzene, toluene and xylene; nitriles such as acetonitrile; amides such as N, N- dimethylformamide (DMF), N, N-dimethylacetamide (DMAC) and N-methylpynolidone (NMP); urea such as l,3-dimethyl-2-imidazolidinone (DMI); sulfoxides such as dimethylsulfoxide (DMSO); and others.
- halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane
- ethers
- reaction temperature can be optionally set depending on the compounds to be reacted.
- the reaction temperatare is usually, but not limited to, about 20°C to 50°C.
- the reaction may be conducted for, usually, 30 minutes to 10 hours and preferably 1 to 24 hours.
- the reaction can be advantageously carried out in the presence of a base including, for instance, organic amines such as pyridine, triethylamine and NN-diisopropylethylamine, dimethylaniline, diethylaniline, 4-dimethylaminopyridine, and others.
- a base including, for instance, organic amines such as pyridine, triethylamine and NN-diisopropylethylamine, dimethylaniline, diethylaniline, 4-dimethylaminopyridine, and others.
- the compound (DI) and (IV) are commercially available or can be prepared by the use of known techniques.
- the compound of the formula (I) (wherein n and R 1 are the same as defined above) can be prepared by the reaction of the compound of the formula (II) and the compound of the formula (V) (wherein n and R 1 are the same as defined above).
- the reaction may be carried out in a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane; ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane; aromatic hydrocarbons such as benzene, toluene and xylene; nitriles such as acetonitrile; amides such as N, N- dimethylformamide (DMF), N, N-dimethylacetamide (DMAC) and N-methylpynolidone (NMP); urea such as l,3-dimethyl-2-imidazolidinone (DMI); sulfoxides such as dimethylsulfoxide (DMSO); and others.
- a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloro
- the reaction can be carried out in the presence of organic base such as pyridine or friethylamine.
- the reaction temperatare can be optionally set depending on the compounds to be reacted.
- the reaction temperatare is usually, but not limited to, about room temperature to 100°C.
- the reaction may be conducted for, usually, 30 minutes to 48 hours and preferably 1 to 24 hours.
- the compound (V) can be prepared by the use of known techniques or are commercially available. [Method C]
- the compound of the formula (I) (wherein n and R 1 are the same as defined above) can be prepared by reacting the compound of the formula (TT) with phosgene, diphosgene, triphosgene, 1,1-carbonyldiimidazole (CDI), or l, -carbonyldi(l,2,4-triazole)(CDT), and then adding the compound of the formula (IV) (wherein n and R 1 are the same as defined above) to the reaction mixture.
- the reaction may be ca ied out in a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane; ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane; aromatic hydrocarbons such as benzene, toluene and xylene; nitriles such as acetonitrile; amides such as N, N- dimethylformamide (DMF), N, N-dimethylacetamide (DMAC) and N-methylpynolidone (NMP); urea such as l,3-dimethyl-2-imidazolidinone (DMI); sulfoxides such as dimethylsulfoxide (DMSO); and others.
- halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane
- the reaction temperature can be optionally set depending on the compounds to be reacted.
- the reaction temperatare is usually, but not limited to, about 20°C to 50°C.
- the reaction may be conducted for, usually, 30 minutes to 10 hours and preferably 1 to 24 hours.
- Phosgene, diphosgene, triphosgene, CDI, and CDT are commercially available.
- the compound of the formula (I) (wherein n and R 1 are the same as defined above) can be prepared by reacting the compound of the formula (IV) (wherein n and R 1 are the same as defined above) with phosgene, diphosgene, triphosgene, 1,1-carbonyldiimidazole (CDI), or 1,1'- carbonyldi(l,2,4-triazole)(CDT) and then adding the compound of the formula (IT) (wherein R 1 is the same as defined above) to the reaction mixture.
- the reaction may be carried out in a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane; ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane; aromatic hydrocarbons such as benzene, toluene and xylene; nitriles such as acetonitrile; amides such as N, N- dimethylformamide (DMF), N, ⁇ -dimethylacetamide (DMAC) and ⁇ -methylpynolidone ( ⁇ MP); urea such as l,3-dimethyl-2-imidazolidinone (DMI); sulfoxides such as dimethylsulfoxide (DMSO); and others.
- a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dich
- the reaction temperature can be optionally set depending on the compounds to be reacted.
- the reaction temperature is usually, but not limited to, about 20°C to 50°C.
- the reaction may be conducted for, usually, 30 minutes to 10 hours and preferably 1 to 24 hours.
- the compound of the formula (I) (wherein n and R 1 are the same as defined above) can be prepared by reacting the compound of the formula (IV) (wherein n and R 1 are the same as defined above) and the compound of the formula (ITT) (wherein , ⁇ is the same as defined above), and then adding the compound of the formula (II) to the reaction mixture.
- the reaction may be carried out in a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane; ethers such as diethyl ether, iso- propyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane; aromatic hydrocarbons such as benzene, toluene and xylene; nitriles such as acetonitrile; amides such as N, N- dimethylformamide (DMF), N, N-dimethylacetamide (DMAC) and N-methylpynolidone (NMP); urea such as l,3-dimethyl-2-imidazolidinone (DMI); sulfoxides such as dimethylsulfoxide (DMSO); and others.
- a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichlor
- the reaction temperature can be optionally set depending on the compounds to be reacted.
- the reaction temperatare is usually, but not limited to, about 20°C to 50 °C.
- the reaction may be conducted for, usually, 30 minutes to 10 hours and preferably 1 to 24 hours.
- the reaction can be advantageously carried out in the presence of a base including, for instance, organic amines such as pyridine, friethylamine and N,N-diisopropylethylamine, dimethylaniline, diethylaniline, 4-dimethylaminopyridine, and others.
- a base including, for instance, organic amines such as pyridine, friethylamine and N,N-diisopropylethylamine, dimethylaniline, diethylaniline, 4-dimethylaminopyridine, and others.
- the compound of the formula (VET) (wherein n and R 1 are the same as defined above) can be prepared by reacting the compound of the formula (VI) with the compound of the formula (V) (wherein n and R 1 are the same as defined above) in a similar manner described in Method B for the preparation of the compound of the formula (I).
- the compound of the formula (VIII) (wherein n and R 1 are the same as defined above) can be prepared by reacting the compound of the formula (VET) (wherein n and R 1 are the same as defined above) with an acid such as hydrochloric acid.
- the reaction may be carried out in a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane; ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane; alcohols such as methanol, ethanol; water and others.
- halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane
- ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane
- alcohols such as methanol, ethanol; water and others.
- two or more of the solvents selected from the listed above can be mixed and used.
- the reaction temperatare can be optionally set depending on the compounds to be reacted.
- the reaction temperatare is usually, but not limited to, about 20°C to 100°C.
- the reaction may be conducted for, usually, 30 minutes to 10 hours and preferably 1 to 24 hours.
- the compound of the formula (I) (wherein n and R 1 are the same as defined above) can be prepared by reacting the compound of the formula (VDI) (wherein n and R 1 are the same as defined above) with reducing agent such as sodium borohydride or lithium aluminum hydride.
- the reaction may be carried out in a solvent including, for instance, ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane; alcohols such as methanol, ethanol, isopropanol, and others.
- ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane
- alcohols such as methanol, ethanol, isopropanol, and others.
- two or more of the solvents selected from the listed above can be mixed and used.
- the reaction temperatare can be optionally set depending on the compounds to be reacted.
- the reaction temperatare is usually, but not limited to, about 20°C to 50°C.
- the reaction may be conducted for, usually, 30 minutes to 10 hours and preferably 1 to 24 hours.
- the compound (VI) is commercially available or can be prepared by the use of known techniques.
- the stereoisomeric form of the compound (I), R form (I-a) (wherein n and R 1 are the same as defined above) can be prepared in the similar manner as described in Method [A], [B], [C], [D], or [E] for the preparation of the compound of the formula (I) by using a compound of the formula (II- a) instead of the compound of the formula (II).
- stereoisomeric form of the compound (I), S form (I-a') (wherein n and R 1 are the same as defined above) can be prepared in the similar manner as described in Method [A], [B], [C], [D], or [E] for the preparation of the compound of the formula (I) by using a compound of the formula (II- a') instead of the compound of the formula (IT).
- the compound (U-a) or (ll-a') can be prepared by the use of known techniques.
- the compound of the formula (T) of the present invention can be, but not limited to be, prepared by combining various known methods.
- one or more of the substituents, such as amino group, carboxyl group, and hydroxyl group of the compounds used as starting materials or intermediates are advantageously protected by a protecting group known to those skilled in the art. Examples of the protecting groups are described in "Protective Groups in Organic Synthesis (3rd Edition)" by Greene and Wuts, John Wiley and Sons, New York 1999.
- the compound of the formula (I) of the present invention can be, but not limited to be, prepared by the Method [A] below. [Method A] o
- the compound of the formula (1) (wherein R 1 is the same as defined above) can be prepared by the reaction of the compound of the formula (IT) with the compound of the formula (HI) (wherein R 1 is the same as defined above and Li represents a leaving group including, for instance, hydroxy, halogen atom such as chlorine, bromine, or iodine atom, or azole such as imidazole or triazole.).
- the reaction may be carried out in a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane; ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane; aromatic hydro- carbons such as benzene, toluene and xylene; nitriles such as acetonitrile; amides such as N,N- dimethylformamide (DMF), N,N-dimethylacetamide (DMAC) and N-methylpynolidone (NMP); ureas such as l,3-dimethyl-2-imidazolidinone (DMI); sulfoxides such as dimethylsulfoxide (DMSO); and others.
- a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dich
- the reaction temperatare can be optionally set depending on the compounds to be reacted.
- the reaction temperatare is usually, but not limited to, about 0°C to 50°C.
- the reaction may be conducted for, usually, 30 minutes to 24 hours and preferably 1 to 10 hours.
- the reaction can be advantageously carried out in the presence of a base including, for instance, organic amines such as pyridine, friethylamine and N,N-diisopropylethylamine, dimethylaniline, diethylaniline, 4-dimethylaminopyridine, and others.
- a base including, for instance, organic amines such as pyridine, friethylamine and N,N-diisopropylethylamine, dimethylaniline, diethylaniline, 4-dimethylaminopyridine, and others.
- the reaction can be advantageously carried out using coupling agent including, for instance, hydroxybenzotriazole, carbodiimides such as N, N-dicyclohexylcarbodi- imide and l-(3-dimethyl-aminopropyl)-3-ethylcarbodiimide; carbonyldiazoles such as 1,1'- carbonyldi(l,3-imiazole)(CDI) and l,l'-carbonyldi(l,2,4-triazole)(CDT), and the like.
- the compound (B) and (IH) are commercially available or can be prepared by the use of known techniques.
- the compound of the formula (I-a) (wherein n is 1 to 6; and Xj is OR 12 , SR 12 or N(R 12 )(R 13 ) (in which R 12 and R 13 are the same as defined above)) can be, but not limited to be, prepared by the following procedures.
- Step B-1 the compound of the formula (V) (wherein n is 1 to 6; L represents a leaving group including, for instance, hydroxy, halogen atom such as chlorine, bromine, or iodine atom, or azole such as imidazole or triazole; and L 2 represents a leaving group including, for instance, halogen atom such as chlorine, bromine, or iodine atom) can be prepared in a similar manner as described in Method [A] by using a compound of the formula (IV) (wherein n, L ⁇ and L 2 are the same as defined above) instead of the compound of the formula (ITT).
- Step B-2 the compound of the formula (I-a) (wherein n and X_ are the same as defined above) can be, but not limited to be, prepared by the reaction of the compound of the formula (V) (wherein n and L 2 are the same as defined above) with the compound of the formula (VI) (wherein Xi is the same as defined above).
- the reaction may be carried out in a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane; ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane; aromatic hydrocarbons such as benzene, toluene and xylene; nitriles such as acetonitrile; amides such as N,N- dimethylformamide (DMF), NN-dimethylacetamide (DMAC) and N-methylpynolidone (NMP); ureas such as l,3-dimethyl-2-imidazolidinone (DMI); sulfoxides such as dimethylsulfoxide (DMSO); and others.
- halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane
- ethers
- reaction temperature can be optionally set depending on the compounds to be reacted.
- the reaction temperatare is usually, but not limited to, about 0°C to 50°C.
- the reaction may be conducted for, usually, 30 minutes to 24 hours and preferably 1 to 10 hours.
- the reaction can be advantageously canied out in the presence of a base including, for instance, organic amines such as pyridine, triethylamine and NN-diisopropylethylamine, dimethylaniline, diethylaniline, 4-dimethylaminopyridine, and others.
- a base including, for instance, organic amines such as pyridine, triethylamine and NN-diisopropylethylamine, dimethylaniline, diethylaniline, 4-dimethylaminopyridine, and others.
- the compound (IV) and (Vf) are commercially available or can be prepared by the use of known techniques.
- the compound of the formula (I) of the present invention can be, but not limited to be, prepared by combining various known methods.
- one or more of the substitaents, such as amino group, carboxyl group, and hydroxyl group of the compounds used as starting materials or intermediates are advantageously protected by a protecting group known to those skilled in the art. Examples of the protecting groups are described in "Protective Groups in Organic Synthesis (3rd Edition)" by Greene and Wuts, John Wiley and Sons, New York 1999.
- the compound of the formula (I) of the present invention can be, but not limited to be, prepared by the Method [A], [B], [C], [D], or [E] below.
- the compound of the formula (I) (wherein m, p, Q b Q 2 , Q 3 , R and X are the same as defined above) can be prepared by reacting the compound of the formula ( T) (wherein Q Q 2 and Q 3 are the same as defined above) and the compound of the formula (DI) (wherein Li represents a leaving group including halogen atom such as chlorine, bromine, or iodine atom) and then adding the compound of the formula (TV) (wherein m, p, R and X are the same as defined above) to the reaction mixture.
- the reaction may be carried out in a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane; ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane; aromatic hydrocarbons such as benzene, toluene and xylene; nitriles such as acetonitrile; amides such as N, N-dimethylformamide (DMF), N, N-dimethylacetamide (DMAC) and ⁇ -methylpyrrolidone ( ⁇ MP); urea such as l,3-dimethyl-2-imidazolidinone (DMI); sulfoxides such as dimethylsulfoxide (DMSO); and others.
- a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichlor
- the reaction temperatare can be optionally set depending on the compounds to be reacted.
- the reaction temperatare is usually, but not limited to, about 20°C to 50 °C.
- the reaction may be conducted for, usually, 30 minutes to 24 hours and preferably 1 to 10 hours.
- the reaction can be advantageously canied out in the presence of a base including, for instance, organic amines such as pyridine, triethylamine and N ⁇ -diisopropylethylamine, dimethylaniline, diethylaniline, 4-dimethylaminopyridine, and others.
- a base including, for instance, organic amines such as pyridine, triethylamine and N ⁇ -diisopropylethylamine, dimethylaniline, diethylaniline, 4-dimethylaminopyridine, and others.
- the compound of the formula (DI) and (IV) are commercially available or can be prepared by the use of known techniques.
- the compound of the formula (I) (wherein m, p, Qi, Q 2 , Q 3 , R and X are the same as defined above) can be prepared by the reaction of the compound of the formula (II) (wherein Q_, Q 2 and Q 3 are the same as defined above) and the compound of the formula (V) (wherein m, p, R and X are the same as defined above).
- the reaction may be carried out in a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane; ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane; aromatic hydrocarbons such as benzene, toluene and xylene; nitriles such as acetonitrile; amides such as N, N- dimethylformamide (DMF), N, N-dimethylacetamide (DMAC) and ⁇ -methylpynolidone ( ⁇ MP); urea such as l,3-dimethyl-2-imidazolidinone (DMT); sulfoxides such as dimethylsulfoxide (DMSO); and others.
- a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichlor
- the reaction can be carried out in the presence of organic base such as pyridine or triethylamine.
- the reaction temperatare can be optionally set depending on the compounds to be reacted.
- the reaction temperatare is usually, but not limited to, about room temperature to 100°C.
- the reaction may be conducted for, usually, 30 minutes to 24 hours and preferably 1 to 10 hours.
- the compound (V) can be prepared by the use of known techniques or are commercially available.
- the compound of the formula (I) (wherein m, p, Qj, Q 2 , Q 3 , R and X are the same as defined above) can be prepared by reacting the compound of the formula (IT) (wherein Qi, Q 2 and Q 3 are the same as defined above) with phosgene, diphosgene, triphosgene, 1,1-carbonyldiimidazole (CDI), or l,r-carbonyldi(l,2,4-triazole)(CDT), and then adding the compound of the formula (IV) (wherein m, p, R and X are the same as defined above) to the reaction mixtare.
- the reaction may be canied out in a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane; ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane; aromatic hydrocarbons such as benzene, toluene and xylene; nitriles such as acetonitrile; amides such as N, N- dimethylformamide (DMF), N, N-dimethylacetamide (DMAC) and N-methylpynolidone (NMP); urea such as l,3-dimethyl-2-imidazolidinone (DMI); sulfoxides such as dimethylsulfoxide (DMSO); and others.
- a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichlor
- the reaction temperatare can be optionally set depending on the compounds to be reacted.
- the reaction temperature is usually, but not limited to, about 20°C to 50°C.
- the reaction may be conducted for, usually, 30 minutes to 24 hours and preferably 1 to 10 hours.
- the compound of the formula (I) (wherein m, p, Qi, Q 2 , Q 3 , R and X are the same as defined above) can be prepared by reacting the compound of the formula (IV) (wherein m, p, R and X are the same as defined above) with phosgene, diphosgene, triphosgene, 1,1-carbonyldiimidazole (CDI), or l, -carbonyldi(l,2,4-triazole)(CDT) and then adding the compound of the formula (TT) (wherein Qj, Q 2 and Q 3 are the same as defined above) to the reaction mixtare.
- the reaction may be carried out in a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane; ethers such as diethyl ether, isopropyl ether, dioxane and tefrahydrofuran (THF) and 1,2-dimethoxyethane; aromatic hydrocarbons such as benzene, toluene and xylene; nitriles such as acetonitrile; amides such as N, N- dimethylformamide (DMF), N, N-dimethylacetamide (DMAC) and N-methylpynolidone (NMP); urea such as l,3-dimethyl-2-imidazolidinone (DMI); sulfoxides such as dimethylsulfoxide (DMSO); and others.
- a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dich
- the reaction temperatare can be optionally set depending on the compounds to be reacted.
- the reaction temperatare is usually, but not limited to, about 20°C to 50°C.
- the reaction may be conducted for, usually, 30 minutes to 24 hours and preferably 1 to 10 hours.
- the compound of the formula (I) (wherein m, p, Qi, Q 2 , Q 3 , R and X are the same as defined above) can be prepared by reacting the compound of the formula (TV) (wherein m, p, R and X are the same as defined above) and the compound of the formula (DT) (wherein Li is the same as defined above), and then adding the compound of the formula (D) (wherein Q Q 2 and Q 3 are the same as defined above) to the reaction mixture.
- the reaction may be carried out in a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane; ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane; aromatic hydrocarbons such as benzene, toluene and xylene; nitriles such as acetonitrile; amides such as N, N- dimethylformamide (DMF), N, N-dimethylacetamide (DMAC) and N-methylpynolidone (NMP); urea such as l,3-dimethyl-2-imidazolidinone (DMI); sulfoxides such as dimethylsulfoxide (DMSO); and others.
- a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloro
- the reaction temperatare can be optionally set depending on the compounds to be reacted.
- the reaction temperature is usually, but not limited to, about 20°C to 50 °C.
- the reaction may be conducted for, usually, 30 minutes to 24 hours and preferably 1 to 10 hours.
- the reaction can be advantageously carried out in the presence of a base including, for instance, organic amines such as pyridine, triethylamine and NN-diisopropylethylamine, dimethylaniline, diethylaniline, 4-dimethylaminopyridine, and others.
- a base including, for instance, organic amines such as pyridine, triethylamine and NN-diisopropylethylamine, dimethylaniline, diethylaniline, 4-dimethylaminopyridine, and others.
- the compound of the formula (VH) (wherein Q_, Q 2 and Q 3 are the same as defined above and P . represents alkyl such as methyl or ethyl) can be prepared by the reduction of the compound of the formula (VI) (wherein Pi, Qi, Q 2 and Q 3 are the same as defined above and P represents amino or nitro).
- the reduction can be carrid out by using the agent including, for instance, metal such as lithium, sodium, and the like.
- the reaction can be carried out in a solvent including, for instance, liquid ammonia; alkylamine such as methylamine, ethylamine, and ethylenediamine (EDA); and alcohols such as methanol, ethanol, isopropanol, tert-butanol and others.
- a solvent including, for instance, liquid ammonia; alkylamine such as methylamine, ethylamine, and ethylenediamine (EDA); and alcohols such as methanol, ethanol, isopropanol, tert-butanol and others.
- a solvent including, for instance, liquid ammonia; alkylamine such as methylamine, ethylamine, and ethylenediamine (EDA); and alcohols such as methanol, ethanol, isopropanol, tert-butanol and others.
- two or more of the solvents selected from the listed above can be mixed and used.
- Solvent including, for instance, ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane can be used as a co-solvent.
- ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane
- THF tetrahydrofuran
- 1,2-dimethoxyethane 1,2-dimethoxyethane
- the reaction temperature can be optionally set depending on the compounds to be reacted.
- the reaction temperature is usually, but not limited to, about -78°C to 50 °C.
- the reaction may be conducted for, usually, 30 minutes to 24 hours and preferably 1 to 10 hours.
- the compound of the formula (VIII) (wherein Qi, Q 2 and Q 3 are the same as defined above) can be prepared by the reaction of the compound of the formula (VH) (wherein Pi, Qi, Q 2 and Q 3 are the same as defined above are the same as defined above) with an acid such as hydrochloric acid.
- the reaction may be canied out in a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane; ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane; alcohols such as methanol, ethanol; water and others.
- halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane
- ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane
- alcohols such as methanol, ethanol; water and others.
- two or more of the solvents selected from the listed above can be mixed and used.
- the reaction temperature can be optionally set depending on the compounds to be reacted.
- the reaction temperature is usually, but not limited to, about 20°C to 100°C.
- the reaction may be conducted for, usually, 30 minutes to 24 hours and preferably 1 to 10 hours.
- the compound of the formula (II) (wherein Qi, Q 2 and Q 3 are the same as defined above) can be prepared by reacting the compound of the formula (VDT) (wherein Q 1 ⁇ Q 2 and Q 3 are the same as defined above) with a reducing agent such as sodium borohydride or lithium aluminum hydride.
- a reducing agent such as sodium borohydride or lithium aluminum hydride.
- the reaction may be carried out in a solvent including, for instance, ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane; alcohols such as methanol, ethanol, isopropanol, and others.
- ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane
- alcohols such as methanol, ethanol, isoprop
- the reaction temperature can be optionally set depending on the compounds to be reacted.
- the reaction temperatare is usually, but not limited to, about 20°C to 50°C.
- the reaction may be conducted for, usually, 30 minutes to 24 hours and preferably 1 to 10 hours.
- the compound (VI) is commercially available or can be prepared by the use of known techniques.
- the compound of the formula (VDT) can also be prepared by the following procedures.
- the compound of the formula (X) (wherein Q Q 2 and Q 3 are the same as defined above) can be prepared by the nitration of the compound of the formula (IX) (wherein Qi, Q 2 and Q 3 are the same as defined above.) using the agent including, for instance, nitroric acid, potassium nitrate, a combination agent of dinifrogen pentoxide and sulphur dioxide, a combination agent of dinifrogen pentoxide, nitromethane and sodium bisulfonate, a combination agent of dimethylsulfoxide, acetic anhydride.
- the agent including, for instance, nitroric acid, potassium nitrate, a combination agent of dinifrogen pentoxide and sulphur dioxide, a combination agent of dinifrogen pentoxide, nitromethane and sodium bisulfonate, a combination agent of dimethylsulfoxide, acetic anhydride.
- the reaction can be carried out without solvent or in a solvent including, for instance, acid such as acetic acid, sulfonic acid, trifluoroacetic acid.
- acid such as acetic acid, sulfonic acid, trifluoroacetic acid.
- two or more of the solvents selected from the listed above can be mixed and used.
- the reaction temperature can be optionally set depending on the compounds to be reacted.
- the reaction temperature is usually, but not limited to, about -15°C to 100°C.
- the reaction may be conducted for, usually, 30 minutes to 24 hours and preferably 1 to 10 hours.
- the compound of the formula (XI) (wherein Q' ⁇ , Q' 2 and Q' 3 independently represent N, N ' -O " or CH, with the proviso that at least one of Q Q 2 and Q 3 is N-O " ) can be prepared by the oxydation of the compound of the formula (DC) (wherein Q_, Q 2 and Q 3 are the same as defined above) using an agent including, for instance, hydrogen peroxide, m-chloro- perbenzoic acid, dimethyldioxirane and the like.
- the reaction can be carried out in a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane; acid such as acetic acid, and water.
- a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane; acid such as acetic acid, and water.
- halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane
- acid such as acetic acid
- the reaction temperature can be optionally set depending on the compounds to be reacted.
- the reaction temperature is usually, but not limited to, about -15°C to 100°C.
- the reaction may be conducted for, usually, 30 minutes to 24 hours and preferably 1 to 10 hours.
- the compound of the formula (XII) (wherein Q' ⁇ , Q' 2 and Q' 3 are the same as defined above) can be prepared by the nitration of the compound of the formula (XT) (wherein Q'j, Q' 2 and Q' 3 are the same as defined above) in a similar manner as described for the preparation of the compound of the formula (X).
- the compound of the formula (X) (wherein Qi, Q 2 and Q 3 are the same as defined above) can be prepared by the reduction of the compound of the formula (XH) (wherein Q'j, Q' 2 and Q' 3 are the same as defined above) using the agent including, for instance, triphenyl phosphine, xriethyl phosphite, trimethyl phosphite, methanesulfonyl chloride, a combination agent of lithium chloride and sodium borohydride, and the like.
- the agent including, for instance, triphenyl phosphine, xriethyl phosphite, trimethyl phosphite, methanesulfonyl chloride, a combination agent of lithium chloride and sodium borohydride, and the like.
- the reaction can be carried out in a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane; ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane; aromatic hydrocarbons such as benzene, toluene and xylene, and the like.
- halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane
- ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane
- aromatic hydrocarbons such as benzene, toluene and xylene, and the like.
- two or more of the solvents selected from the listed above can be mixed and used.
- the reaction temperature can be
- the compound of the formula (VIH) (wherein Qi, Q 2 and Q 3 are the same as defined above) can be prepared by reducing nitro group of the compound of the formula (X) (wherein Q Q 2 and Q 3 are the same as defined above.) using an agent including, for instance, metals such as zinc and iron in the presence of acid including, for instance, hydrochloric acid and acetic acid and stannous chloride, or by hydrogenation using a catalyst including, for instance, palladium on carbon and platinum on carbon.
- an agent including, for instance, metals such as zinc and iron in the presence of acid including, for instance, hydrochloric acid and acetic acid and stannous chloride, or by hydrogenation using a catalyst including, for instance, palladium on carbon and platinum on carbon.
- the reaction can be carried out in a solvent including, for instance, ethers such as diethyl ether, isopropyl ether, dioxane, tetrahydrofuran (THF) and 1,2-dimethoxyethane, aromatic hydrocarbons such as benzene, toluene and xylene, alcohols such as methanol, ethanol, 1-propanol, isopropanol and tert-butanol, water and others.
- ethers such as diethyl ether, isopropyl ether, dioxane, tetrahydrofuran (THF) and 1,2-dimethoxyethane
- aromatic hydrocarbons such as benzene, toluene and xylene
- alcohols such as methanol, ethanol, 1-propanol, isopropanol and tert-butanol, water and others.
- the reaction temperature can be optionally set depending on the compounds to be reacted.
- the reaction temperatare is usually, but not limited to, about 20°C to 120°C.
- the reaction may be conducted for, usually, 30 minutes to 24 hours and preferably 1 to 10 hours.
- the compound of the formula (VHT) (wherein Qi, Q 2 and Q 3 are the same as defined above) can be prepared by reduction of the compound of the formula (XIT) (wherein Q'_, Q' 2 and Q' 3 are the same as defined above) as shown in the Step ii-3.
- the reduction can be carried out using an agent including, for instance, metals such as titanium and iron, and sodium hypophosphite together with a catalyst including, for instance, palladium on carbon and platinum on carbon.
- an agent including, for instance, metals such as titanium and iron, and sodium hypophosphite together with a catalyst including, for instance, palladium on carbon and platinum on carbon.
- the reaction can be canied out in a solvent including, for instance, ethers such as diethyl ether, isopropyl ether, dioxane, tetrahydrofuran (THF) and 1,2-dimethoxyethane, aromatic hydrocarbons such as benzene, toluene and xylene, alcohols such as methanol, ethanol, 1-propanol, isopropanol and tert-butanol, acid such as acetic acid, water and others.
- ethers such as diethyl ether, isopropyl ether, dioxane, tetrahydrofuran (THF) and 1,2-dimethoxyethane
- aromatic hydrocarbons such as benzene, toluene and xylene
- alcohols such as methanol, ethanol, 1-propanol, isopropanol and tert-butanol
- acid such as acetic acid, water and others.
- the reaction temperature can be optionally set depending on the compounds to be reacted.
- the reaction temperature is usually, but not limited to, about 20°C to 120°C.
- the reaction may be conducted for, usually, 30 minutes to 24 hours and preferably 1 to 10 hours.
- the compound (IX) is commercially available or can be prepared by the use of known techniques.
- the compound of the formula (VIH) (wherein Qi, Q 2 and Q 3 are the same as defined above) can be prepared by the reaction of the compound of the formula (XHI) (wherein Q Q 2 and Q 3 are the same as defined above.) using the agent including, for instance, p- toluenesulfonyl isocyanate.
- the reaction can be carried in a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane; ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane; aromatic hydrocarbons such as benzene, toluene and xylene and others.
- halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane
- ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane
- aromatic hydrocarbons such as benzene, toluene and xylene and others.
- two or more of the solvents selected from the listed above can be mixed and used.
- the reaction temperature can be optionally set depending on the compounds to be reacted.
- the reaction temperatare is usually, but not limited to, about 20 °C to 100 °C.
- the reaction may be conducted for, usually, 30 minutes to 24 hours and preferably 1 to 10 hours.
- the compound of the formula (XIV) (wherein Q_, Q 2 and Q 3 are the same as defined above and L 2 represents a leaving group including halogen atom such as chlorine, bromine, or iodine atom; and alkylsulfonyloxy such as frifluoromethylsulfonyloxy) can be prepared by the reaction of the compound of the formula (XHI) (wherein Q 1 ⁇ Q 2 and Q 3 are the same as defined above.) using the agent including, for instance, halogenating reagent such as POCl 3 , POBr 3 , PC1 5 and the like; or sulfonyl chloride such as xrifluoromethylsulfonyl chloride.
- halogenating reagent such as POCl 3 , POBr 3 , PC1 5 and the like
- sulfonyl chloride such as xrifluoromethylsulfonyl chloride.
- the reaction may be carried out in a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and l,2-dichloroethane; such as ethers such as dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane; aromatic hydrocarbons such as benzene, toluene, and xylene, and others.
- halogenated hydrocarbons such as dichloromethane, chloroform and l,2-dichloroethane
- ethers such as dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane
- aromatic hydrocarbons such as benzene, toluene, and xylene, and others.
- two or more of the solvents selected from the listed above can be mixed and used.
- the reaction can be advantageously conducted in the presence of a base, including, for instance, such as pyridine, triethylamine and NN-diisopropylethylamine, dimethylaniline, diethylaniline, and others.
- a base including, for instance, such as pyridine, triethylamine and NN-diisopropylethylamine, dimethylaniline, diethylaniline, and others.
- the reaction temperatare is usually, but not limited to, about 40°C to 200°C and preferably about 20°C to 180°C.
- the reaction may be conducted for, usually, 30 minutes to 24 hours and preferably 2 hours to 10 hours.
- the compound of the formula (XIV) (wherein L 2 , Q_, Q 2 and Q 3 are the same as defined above) can be prepared by the reaction of the compound of the formula (Xlfl) (wherein Qi, Q 2 and Q 3 are the same as defined above) using the agent including, for instance, ammonia.
- the reaction can be advantageously conducted in the presence of a catalyst including, for instance, copper(T) oxide, copper(D) sulfate and the like.
- a catalyst including, for instance, copper(T) oxide, copper(D) sulfate and the like.
- the reaction may be canied out in a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane; such as ethers such as dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane; aromatic hydrocarbons such as benzene, toluene, and xylene, and others.
- halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane
- ethers such as dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane
- aromatic hydrocarbons such as benzene, toluene, and xylene, and others.
- two or more of the solvents selected from the listed above can be mixed and used.
- the reaction temperature is usually, but not limited to, about 40°C to 200°C and preferably about 20°C to 180°C.
- the reaction may be conducted for, usually, 30 minutes to 24 hours and preferably 2 hours to 12 hours.
- the compound of the formula (VIH) can also be prepared by the following procedures.
- the compound of the formula (XVI) (wherein Qi, Q 2 and Q 3 are the same as defined above; and P 3 represents aralkyl such as benzyl, 4-methoxybenzyl, 3,4-dimethoxybenzyl) can be prepared by the reaction of the compound of the formula (XIV) (wherein L 2 , Qi, Q 2 and Q 3 are the same as defined above) with the compound of the formula (XV) (wherein P 3 is the same as defined above).
- the reaction can be carried out in the presence of a palladium catalyst such as tetrakis- (triphenylphosphine)palladium or a combination of a phosphine ligand and a palladium catalyst such as tri-o-tolylphosphine and palladium (H) acetate.
- a palladium catalyst such as tetrakis- (triphenylphosphine)palladium or a combination of a phosphine ligand and a palladium catalyst such as tri-o-tolylphosphine and palladium (H) acetate.
- the reaction can be advantageously carried out in the presence of a base including, for instance, cesium carbonate, sodium carbonate, potassium carbonate, barium hydroxide sodium methoxide, sodium ethoxide, potassium tert-butoxide and the like.
- This reaction can be carried out in a solvent including, for instance, alcohol such as methanol, ethanol, 1-propanol, isopropanol and tert-butanol; ethers, such as dioxane, isopropyl ether, diethyl ether, 1,2-dimethoxyethane and tetrahydrofuran (THF); aromatic hydrocarbons such as benzene, toluene and xylene; nitriles such as acetonitrile; amides such as dimethylformamide (DMF) N, ⁇ - dimethylacetamide and ⁇ -methylpynolidone; sulfoxides such as dimethylsulfoxide (DMSO); water and others.
- a solvent including, for instance, alcohol such as methanol, ethanol, 1-propanol, isopropanol and tert-butanol; ethers, such as dioxane, isopropyl ether, diethyl
- the reaction temperature can be optionally set depending on the compounds to be reacted.
- the reaction temperature is usually, but not limited to, about 10°C to 200°C and preferably about 50°C to 150°C.
- the reaction may be conducted for, usually, 30 minutes to 24 hours and preferably 1 to 12 hours.
- the compound of the formula (VIH) (wherein Qi, Q 2 and Q 3 are the same as defined above) can be prepared by the removal of P 3 of the compound of the formula (XVI) (wherein P 3 , Qi, Q 2 and Q 3 are the same as defined above).
- the removal of P 3 can be done by hydrogenation using a catalyst including, for instance, palladium on carbon and palladium hydroxide. Also, the removal can be done by using a reagent including, for instance, trifluoroacetic acid, eerie ammonium nitrate (CAN) or 2,3-dichloro-5,6-dicyano-l,4- benzoquinone (DDQ), when P 3 is 4-methoxybenzyl or 3,4-dimethoxybenzyl.
- a catalyst including, for instance, palladium on carbon and palladium hydroxide.
- a reagent including, for instance, trifluoroacetic acid, eerie ammonium nitrate (CAN) or 2,3-dichloro-5,6-dicyano-l,4- benzoquinone (DDQ), when P 3 is 4-methoxybenzyl or 3,4-dimethoxybenzyl.
- This reaction can be carried out in a solvent including, for instance, alcohol such as methanol, ethanol, 1-propanol, isopropanol and tert-butanol; ethers, such as dioxane, isopropyl ether, diethyl ether, 1,2-dimethoxyethane and tetrahydrofuran (THF); aromatic hydrocarbons such as benzene, toluene and xylene; ester such as ethyl acetate; water and others.
- a solvent including, for instance, alcohol such as methanol, ethanol, 1-propanol, isopropanol and tert-butanol; ethers, such as dioxane, isopropyl ether, diethyl ether, 1,2-dimethoxyethane and tetrahydrofuran (THF); aromatic hydrocarbons such as benzene, toluene and xylene; ester such as e
- the compound of the formula (I) (wherein m, p, Qi, Q 2 , Q 3 , R and X are the same as defined above) can alternatively be prepared by the following procedures in three steps;
- the compound of the formula (XVH) (wherein m, p, Pi, Q l5 Q 2 , Q 3 , R and X are the same as defined above) can be prepared in a similar manner as described in Method [A], [B], [C], [D] or [E] for the preparation of the compound of the formula (I) by using a compound of the formula (VH) (wherein Pi, Q Q 2 and Q 3 are the same as defined above) instead of the compound of the formula (H).
- the compound of the formula (XVHI) (m, p, Qi, Q 2 , Q 3 , R and X are the same as defined above) can be prepared by reacting the compound of the formula (XVH) (m, p, P b Qi, Q 2 , Q 3 , R and X are the same as defined above) with an acid such as hydrochloric acid.
- the reaction may be canied out in a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane; ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane; alcohols such as methanol, ethanol; water and others.
- halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane
- ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane
- alcohols such as methanol, ethanol; water and others.
- two or more of the solvents selected from the listed above can be mixed and used.
- the reaction temperatare can be optionally set depending on the compounds to be reacted.
- the reaction temperatare is usually, but not limited to, about 20°C to 100°C.
- the reaction may be conducted for, usually, 30 minutes to 24 hours and preferably 1 to 10 hours.
- the compound of the formula (I) (wherein m, p, Q_, Q 2 , Q 3 , R and X are the same as defined above) can be prepared by reacting the compound of the formula (XVIH) (wherein m, p, Qi. Q 2 . Q 3. and X are the same as defined above) with reducing agent such as sodium borohydride or lithium aluminum hydride.
- the reaction may be carried out in a solvent including, for instance, ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane; alcohols such as methanol, ethanol, isopropanol, and others.
- ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane
- alcohols such as methanol, ethanol, isopropanol, and others.
- two or more of the solvents selected from the listed above can be mixed and used.
- the reaction temperatare can be optionally set depending on the compounds to be reacted.
- the reaction temperature is usually, but not limited to, about 20°C to 50°C.
- the reaction may be conducted for, usually, 30 minutes to 24 hours and preferably 1 to 10 hours.
- the compound of the formula (XVHI) (m, p, Qi, Q 2 , Q 3 , R and X are the same as defined above) can alternatively be prepared in a similar manner as described in Method [A], [B], [C], [D] or [E] for the preparation of the compound of the formula (I) by using a compound of the formula (VHT) (wherein Qi, Q 2 and Q 3 are the same as defined above) instead of the compound of the formula (H).
- the compound of the formula (I-a) (wherein m, p, Qi, Q 2 , Q 3 and R are the same as defined above and X' is -O-, or N(R J )-) can be prepared by the following procedures.
- the compound of the formula (XXI) (wherein m, Qi, Q 2 and Q 3 are the same as defined above and L 3 represents leaving group including, for instance, halogen atom such as chlorine, bromine, or iodine atom) can be prepared in a similar manner as described in Method [A], [B], [C], [D] or [E] for the preparation of the compound of the formula (I) by using a compound of the formula (XIX) (wherein m and L 3 are the same as defined above) instead of the compound of the formula (IV), or using a compound of the formula (XX) (wherein m and L 3 are the same as defined above) instead of the compound of the formula (V).
- halogen atom such as chlorine, bromine, or iodine atom
- the compound of the formula (I-a) (wherein m, p, Qi, Q 2 , Q 3 , R and X' are the same as defined above) can be prepared by reacting the compound of the formula (XXI) (wherein m, L 3 , Qi, Q 2 and Q 3 are the same as defined above) and the compound of the formula (XXII) (wherein p, R and X' are the same as defined above).
- the reaction may be carried out in a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloroethane; ethers such as diethyl ether, isopropyl ether, dioxane and tetrahydrofuran (THF) and 1,2-dimethoxyethane; aromatic hydrocarbons such as benzene, toluene and xylene; nitriles such as acetonitrile; amides such as N,N- dimethylformamide (DMF), NN-dimethylacetamide (DMAC) and N-methylpynolidone (NMP); ureas such as l,3-dimethyl-2-imidazolidinone (DMI); sulfoxides such as dimethylsulfoxide (DMSO); and others.
- a solvent including, for instance, halogenated hydrocarbons such as dichloromethane, chloroform and 1,2-dichloro
- the reaction temperature can be optionally set depending on the compounds to be reacted.
- the reaction temperatare is usually, but not limited to, about 0°C to 50°C.
- the reaction may be conducted for, usually, 30 minutes to 24 hours and preferably 1 to 10 hours.
- the reaction can be advantageously carried out in the presence of a base including, for instance, organic amines such as pyridine, triethylamine and NN-diisopropylethylamine, dimethylaniline, diethylaniline, 4-dimethylaminopyridine, and others.
- a base including, for instance, organic amines such as pyridine, triethylamine and NN-diisopropylethylamine, dimethylaniline, diethylaniline, 4-dimethylaminopyridine, and others.
- the stereoisomeric form of the compound (I), R form (I-a) (wherein m, p, Qi, Q 2 , Q 3 , R and X are the same as defined above) can be prepared in a similar manner as described in Method [A], [B], [C], [D], or [E] for the preparation of the compound of the formula (I) by using a compound of the formula (H-a) (wherein Q l9 Q 2 and Q 3 are the same as defined above) instead of the compound of the formula (H).
- the stereoisomeric form of the compound (I), S form (I-a') (wherein m, p, Qi, Q 2 , Q 3 , R and X are the same as defined above) can be prepared in the similar manner as described in Method [A], [B], [C], [D], or [E] for the preparation of the compound of the formula (I) by using a compound of the formula (H-a') (wherein Qi, Q 2 and Q 3 are the same as defined above) instead of the compound of the formula (IT).
- the compound (H-a) or (H-a') can be prepared by the use of known techniques.
- Typical salts of the compound shown by the formula (A) include salts prepared by reaction of the compounds of the present invention with a mineral or organic acid, or an organic or inorganic base. Such salts are known as acid addition and base addition salts, respectively.
- Acids to form acid addition salts include inorganic acids such as, without limitation, sulfuric acid, phosphoric acid, hydrochloric acid, hydrobromic acid, hydriodic acid and the like, and organic acids, such as, without limitation, p-toluenesulfonic acid, methanesulfonic acid, oxalic acid, p- bromophenylsulfonic acid, carbonic acid, succinic acid, citric acid, benzoic acid, acetic acid, and the like.
- inorganic acids such as, without limitation, sulfuric acid, phosphoric acid, hydrochloric acid, hydrobromic acid, hydriodic acid and the like
- organic acids such as, without limitation, p-toluenesulfonic acid, methanesulfonic acid, oxalic acid, p- bromophenylsulfonic acid, carbonic acid, succinic acid, citric acid, benzoic acid, acetic acid, and the like.
- Base addition salts include those derived from inorganic bases, such as, without limitation, ammonium hydroxide, alkaline metal hydroxide, alkaline earth metal hydroxides, carbonates, bicarbonates, and the like, and organic bases, such as, without limitation, ethanolamine, triethylamine, tris(hydroxymethyl)aminomethane, and the like.
- inorganic bases include sodium hydroxide, potassium hydroxide, potassium carbonate, sodium carbonate, sodium bicarbonate, potassium bicarbonate, calcium hydroxide, calcium carbonate, and the like.
- the compound of the present invention or a salt thereof, depending on its substituents, may be modified to form lower alkylesters or known other esters; and/or hydrates or other solvates. Those esters, hydrates, and solvates are included in the scope of the present invention.
- the compound of the present invention may be administered in oral forms, such as, without limitation normal and enteric coated tablets, capsules, pills, powders, granules, elixirs, tinctures, solution, suspensions, syrups, solid and liquid aerosols and emulsions. They may also be administered in parenteral forms, such as, without limitation, intravenous, intraperitoneal, subcutaneous, intramuscular, and the like forms, well-known to those of ordinary skill in the pharmaceutical arts.
- the compounds of the present invention can be administered in infranasal form via topical use of suitable infranasal vehicles, or via transdermal routes, using transdermal delivery systems well-known to those of ordinary skilled in the art.
- the dosage regimen with the use of the compounds of the present invention is selected by one of ordinary skill in the arts, in view of a variety of factors, including, without limitation, age, weight, sex, and medical condition of the recipient, the severity of the condition to be treated, the route of administration, the level of metabolic and excretory function of the recipient, the dosage form employed, the particular compound and salt thereof employed.
- the compounds of the present invention are preferably formulated prior to administration together with one or more pharmaceutically-acceptable excipients.
- Excipients are inert substances such as, without limitation carriers, diluents, flavoring agents, sweeteners, lubricants, solubilizers, suspending agents, binders, tablet disintegrating agents and encapsulating material.
- compositions of the present invention are pharmaceutical formulation comprising a compound of the invention and one or more pharmaceutically-acceptable excipients that are compatible with the other ingredients of the formulation and not deleterious to the recipient thereof.
- Pharmaceutical formulations of the invention are prepared by combining a therapeutically effective amount of the compounds of the invention together with one or more pharmaceutically- acceptable excipients therefore.
- the active ingredient may be mixed with a diluent, or enclosed within a carrier, which may be in the form of a capsule, sachet, paper, or other container.
- the carrier may serve as a diluent, which may be solid, semi-solid, or liquid material which acts as a vehicle, or can be in the form of tablets, pills powders, lozenges, elixirs, suspensions, emulsions, solutions, syrups, aerosols, ointments, containing, for example, up to 10% by weight of the active compound, soft and hard gelatin capsules, suppositories, sterile injectable solutions and sterile packaged powders.
- a diluent which may be solid, semi-solid, or liquid material which acts as a vehicle, or can be in the form of tablets, pills powders, lozenges, elixirs, suspensions, emulsions, solutions, syrups, aerosols, ointments, containing, for example, up to 10% by weight of the active compound, soft and hard gelatin capsules, suppositories, sterile injectable solutions and sterile packaged powders.
- the active ingredient may be combined with an oral, and non-toxic, pharmaceutically-acceptable carrier, such as, without limitation, lactose, starch, sucrose, glucose, sodium carbonate, mannitol, sorbitol, calcium carbonate, calcium phosphate, calcium sulfate, methyl cellulose, and the like; together with, optionally, disintegrating agents, such as, without limitation, maize, starch, methyl cellulose, agar bentonite, xanthan gum, alginic acid, and the like; and optionally, binding agents, for example, without limitation, gelatin, natural sugars, beta- lactose, com sweeteners, natural and synthetic gums, acacia, tragacanth, sodium alginate, carboxymethylcellulose, polyethylene glycol, waxes, and the like; and, optionally, lubricating agents, for example, without limitation, magnesium stearate, sodium stearate, stearic acid, sodium oleate, sodium benzoate
- the carrier may be a finely divided solid which is in admixture with the finely divided active ingredient.
- the active ingredient may be mixed with a carrier having binding properties in suitable proportions and compacted in the shape and size desired to produce tablets.
- the powders and tablets preferably contain from about 1 to about 99 weight percent of the active ingredient which is the novel composition of the present invention.
- Suitable solid carriers are magnesium carboxymethyl cellulose, low melting waxes, and cocoa butter.
- Sterile liquid formulations include suspensions, emulsions, syrups and elixirs.
- the active ingredient can be dissolved or suspended in a pharmaceutically acceptable earners, such as sterile water, sterile organic solvent, or a mixture of both sterile water and sterile organic solvent.
- the active ingredient can also be dissolved in a suitable organic solvent, for example, aqueous propylene glycol.
- a suitable organic solvent for example, aqueous propylene glycol.
- Other compositions can be made by dispersing the finely divided active ingredient in aqueous starch or sodium carboxymethyl cellulose solution or in a suitable oil.
- the formulation may be in unit dosage form, which is a physically discrete unit containing a unit dose, suitable for administration in human or other mammals.
- a unit dosage form can be a capsule or tablets, or a number of capsules or tablets.
- a "unit dose" is a predetermined quantity of the active compound of the present invention, calculated to produce the desired therapeutic effect, in association with one or more excipients.
- the quantity of active ingredient in a unit dose may be varied or adjusted from about 0.1 to about 1000 milligrams or more according to the particular treatment involved.
- Typical oral dosages of the present invention when used for the indicated effects, will range from about O.Olmg /kg/day to about 100 mg/kg/day, preferably from 0.1 mg/kg/day to 30 mg/kg/day, and most preferably from about 0.5 mg/kg/day to about 10 mg/kg/day.
- parenteral administration it has generally proven advantageous to administer quantities of about 0.001 to lOOmg /kg/day, preferably from 0.01 mg/kg/day to 1 mg/kg/day.
- the compounds of the present invention may be administered in a single daily dose, or the total daily dose may be administered in divided doses, two, three, or more times per day. Where delivery is via transdermal forms, of course, administration is continuous.
- Instrument MS Micromass ZQ
- instrument HPLC Waters Alliance 2795
- eluent A water + 500 ⁇ l 50% aqueous formic acid / 1
- eluent B acetonifrile + 500 ⁇ l 50% aqueous formic acid / 1
- oven temp. 35°C
- flow rate 0.0 min 1.0 m_/min- 3.0 min 3.0 ml/min- 4.0 min 3.0 ml/min
- UV-detection 210 nm.
- Instrument MS Micromass ZQ; instrument HPLC: HP 1100 Series; UV DAD; column: Grom-Sil 120 ODS-4 HE 50 mm x 2 mm, 3.0 ⁇ m; eluent A: water + 500 ⁇ l 50% aqueous formic acid / 1, eluent B: acetonifrile + 500 ⁇ l 50% aqueous formic acid / 1; gradient: 0.0 min 0%>B - 2.9 min 70%B -> 3.1 min 90%B -» 4.5 min 90%B; oven temp.: 50 °C; flow rate: 0.8 ml min; UV- detection: 210 nm.
- Preparative HPLC purifications are performed on a GROM-SIL 120 ODS-4 HE 10 ⁇ m, 250 mm x 30 mm column with acetonitrile/water gradients.
- H NMR spectra were recorded using either Bruker DRX-300 (300 MHz for H) spectrometer or Bracker 500 UlfraShieledTM (500 MHz for IH). Chemical shifts are reported in parts per million (ppm) with tetramethylsilane (TMS) as an internal standard at zero ppm. Coupling constant (J) are given in hertz and the abbreviations s, d, t, q, m, and br refer to singlet, doblet, triplet, quartet, multiplex, and broad, respectively.
- TLC was performed on a precoated silica gel plate (Merck silica gel 60 F-254).
- Silica gel WAKO- gel C-200 (75-150 ⁇ m) was used for all column chromatography separations. All chemicals were reagent grade and were purchased from Sigma-Aldrich, Wako pure chemical industries, Ltd., Great Britain, Tokyo kasei kogyo Co., Ltd., Nacalai tesque, Inc., Watanabe Chemical Ind. Ltd., Maybridge pic, Lancaster Synthesis Ltd., Merck KgaA, Germany, or Kanto Chemical Co., Ltd.
- Human vanilloid receptor (hVRl) cDNA was cloned from libraries of axotomized dorsal root ganglia (WO 00/29577). The cloned hVRl cDNA was constructed with pcDNA3 vector and transfected into a CH01uc9aeq cell line. The cell line contains aequorin and CRE-luciferase reporter genes as read-out signals.
- the fransfectants were cloned by limiting dilution in selection medium (DMEM/F12 medium (Gibco BRL) supplemented with 10% FCS, 1.4 mM Sodium pyruvate, 20 mM HEPES, 0.15% Sodium bicarbonate, 100 U/ml penicillin, 100 ⁇ g/ml streptomycin, 2 mM glutamine, non-essential amino acids and 2 mg/ml G418). Ca 2+ influx was examined in the capsaicin-stimulated clones. A high responder clone was selected and used for further experiments in the project.
- the human VRl-CH01uc9aeq cells were maintained in the selection medium and passaged every 3-4 days at l-2.5xl0 5 cells/flask (75 mm 2 ).
- Human VRl-CH01uc9aeq cells were suspended in a culture medium which is the same as the selection medium except for G418 and seeded at a density of 1,000 cells per well into 384-well plates (black walled clear-base / ⁇ alge ⁇ unc International). Following the culture for 48 hrs the medium was changed to 2 ⁇ M Fluo-3 AM (Molecular Probes) and 0.02% Puronic F-127 in assay buffer (Hank's balanced salt solution (HBSS), 17 mM HEPES ( ⁇ H7.4), 1 mM Probenecid, 0.1% BSA) and the cells were incubated for 60 min at 25°C. After washing twice with assay buffer the cells were incubated with a test compound or vehicle for 20 min at 25°C.
- assay buffer Hort's balanced salt solution (HBSS), 17 mM HEPES ( ⁇ H7.4), 1 mM Probenecid, 0.1% BSA
- DRG dorsal root ganglia
- DRG was incubated with 0.1% trypsin (Gibco BRL) in PBS(-) (Gibco BRL) for 30 min at 37°C, then a half volume of fetal calf serum (FCS) was added and the cells were spun down.
- FCS fetal calf serum
- the DRG neuron cells were resuspended in Ham F12/5% FCS/5% horse serum (Gibco BRL) and dispersed by repeated pipetting and passing through 70 ⁇ m mesh (Falcon). The culture plate was incubated for 3 hours at 37°C to remove contaminating Schwann cells.
- ⁇ on-adherent cells were recovered and further cultured in laminin-coated 384 well plates ( ⁇ unc) at lxlO 4 cells/50 ⁇ l well for 2 days in the presence of 50 ng/ml recombinant rat NGF (Sigma) and 50 ⁇ M 5-fluorodeoxyuridine (Sigma).
- Human P2X1 -transfected CH01uc9aeq cell line was established and maintained in Dulbecco's modified Eagle's medium (DMEM/F12) supplemented with 7.5% FCS, 20 mM HEPES-KOH (pH 7.4), 1.4 mM sodium pyruvate, 100 U/ml penicillin, 100 ⁇ g/ml streptomycin, 2 mM glutamine (Gibco BRL) and 0.5 Units/ml apyrase (grade I, Sigma).
- the suspended cells were seeded in each well of 384-well optical bottom black plates (Nalge Nunc International) at 3 x 10 3 / 50 ⁇ l / well. The cells were cultured for following 48 hrs to adhere to the plates.
- P2X1 receptor agonist-mediated increases in cytosolic Ca 2+ levels were measured using a fluorescent Ca 2+ chelating dye, Fluo-3 AM (Molecular Probes).
- the plate-attached cells were washed twice with washing buffer (HBSS, 17 mM HEPES-KOH (pH 7.4), 0.1% BSA and 0.5 units/ml apyrase), and incubated in 40 ⁇ l of loading buffer (1 ⁇ M Fluo-3 AM, 1 mM probenecid, 1 ⁇ M cyclosporin A, 0.01% pluronic (Molecular Probes)in washing buffer) for 1 hour in a dark place.
- Rats were anesthetized by intraperitoneal adminisfration of urethane (Sigma) at 1.2 g/kg.
- the abdomen was opened through a midline incision, and a polyethylene catheter (BECTON DICKINSON, PE50) was implanted into the bladder through the dome.
- a polyethylene catheter (Hibiki, size 5) filled with 2 IU / ml of heparin ( ⁇ ovo Heparin, Aventis Pharma) in saline (Otsuka) was inserted into a common iliac artery.
- the bladder catheter was connected via T-tube to a pressure transducer (Viggo- Spectramed Pte Ltd, DT-XXAD) and a microinjection pump (TERUMO). Saline was infused at room temperature into the bladder at a rate of 2.4 ml/hr. fritravesical pressure was recorded continuously on a chart pen recorder (Yokogawa). At least three reproducible mictarition cycles, conesponding to a 20-minute period, were recorded before a test compound administration and used as baseline values.
- the saline infusion was stopped before administrating compounds.
- a testing compound dissolved in the mixture of ethanol, Tween 80 (IC ⁇ Biomedicals Inc.) and saline (1 : 1 : 8, v/v/v) was administered mtraarterially at 10 mg/kg. 2min after the administration of the compound 10 ⁇ g of capsaicin ( ⁇ acalai Tesque) dissolved in ethanol was administered mtraarterially.
- Cyclo- phosphamide (CYP) dissolved in saline was administered infraperitoneally at 150 mg/kg 48 hours before experiment.
- Rats were anesthetized by intraperitoneal administration of urethane (Sigma) at 1.25 g/kg. The abdomen was opened through a midline incision, and a polyethylene catheter (BECTON DICKINSON PE50) was implanted into the bladder through the dome. In parallel, the inguinal region was incised, and a polyethylene catheter (BECTON DICKINSON, PE50) filled with saline (Otsuka) was inserted into a femoral vein. After the bladder was emptied, the rats were left for 1 hour for recovery from the operation.
- urethane Sigma
- the bladder catheter was connected via T-tabe to a pressure transducer (Viggo- Spectramed Pte Ltd, DT-XXAD) and a microinjection pump (TERUMO). Saline was infused at room temperature into the bladder at a rate of 3.6 ml/hr for 20 min. Intravesical pressure was recorded continuously on a chart pen recorder (Yokogawa). At least three reproducible micturition cycles, conesponding to a 20-minute period, were recorded before a test compound administration.
- a testing compound dissolved in the mixture of ethanol, Tween 80 (ICN Biomedicals Inc.) and saline (1 : 1 : 8, v/v/v) was administered intravenously at 0.05 mg/kg, 0.5 mg/kg or 5 mg/kg.
- saline Nacalai Tesque was infused at room temperatare into the bladder at a rate of 3.6 ml/hr.
- the cystometry parameters were analyzed as described previously [ Lecci A et al: Eur. J. Pharmacol. 259: 129-135, 1994].
- the micturition frequency calculated from micturition interval and the bladder capacity calculated from a volume of infused saline until the first mictarition were analyzed from the cystometry data.
- the testing compounds-mediated inhibition of the frequency and the testing compounds-mediated increase of bladder capacity were evaluated using unpaired Student's t-test. A probability levels less than 5% was accepted as significant difference. Data were analyzed as the mean ⁇ SEM from 4 - 7 rats.
- Acute pain is measured on a hot plate mainly in rats.
- Two variants of hot plate testing are used: In the classical variant animals are put on a hot surface (52 to 56 °C) and the latency time is measured until the animals show nociceptive behavior, such as stepping or foot licking.
- the other variant is an increasing temperature hot plate where the experimental animals are put on a surface of neutral temperature. Subsequently this surface is slowly but constantly heated until the animals begin to lick a hind paw. The temperature which is reached when hind paw licking begins is a measure for pain threshold.
- Compounds are tested against a vehicle treated control group. Substance application is performed at different time points via different application routes (i.v., i.p., p.o., i. , i.c.v., s.c, intradermal, transdermal) prior to pain testing.
- application routes i.v., i.p., p.o., i. , i.c.v., s.c, intradermal, transdermal
- Persistent pain is measured with the formalin or capsaicin test, mainly in rats.
- a solution of 1 to 5% formalin or 10 to 100 ⁇ g capsaicin is injected into one hind paw of the experimental animal.
- the animals show nociceptive reactions like flinching, licking and biting of the affected paw.
- the number of nociceptive reactions within a time frame of up to 90 minutes is a measure for intensity of pain.
- Compounds are tested against a vehicle treated control group. Substance application is performed at different time points via different application routes (i.v., i.p., p.o., i.t., i.c.v., s.c, intradermal, transdermal) prior to formalin or capsaicin administration.
- application routes i.v., i.p., p.o., i.t., i.c.v., s.c, intradermal, transdermal
- Neuropathic pain is induced by different variants of unilateral sciatic nerve injury mainly in rats.
- the operation is performed under anesthesia.
- the first variant of sciatic nerve injury is produced by placing loosely constrictive ligatures around the common sciatic nerve (Bennett and Xie, Pain 33 (1988): 87-107).
- the second variant is the tight ligation of about the half of the diameter of the common sciatic nerve (Seltzer et al., Pain 43 (1990): 205-218).
- a group of models is used in which tight ligations or fransections are made of either the L5 and L6 spinal nerves, or the L5 spinal nerve only (KIM SH; CHUNG JM, AN EXPERIMENTAL-MODEL FOR PERIPHERAL NEUROPATHY PRODUCED BY SEGMENTAL SPINAL NERVE LIGATION IN THE RA, PAIN 50 (3) (1992): 355-363).
- the fourth variant involves an axotomy of two of the three terminal branches of the sciatic nerve (tibial and common peroneal nerves) leaving the remaining sural nerve intact whereas the last variant comprises the axotomy of only the tibial branch leaving the sural and common nerves uninjured. Confrol animals are treated with a sham operation.
- the nerve injured animals develop a chronic mechanical allodynia, cold allodynia, as well as a thermal hyperalgesia.
- Mechanical allodynia is measured by means of a pressure transducer (electronic von Frey Anesthesiometer, HTC fric.-Life Science Instruments, Woodland Hills, SA, USA; Electronic von Frey System, Somedic Sales AB, H ⁇ rby, Sweden).
- Thermal hyperalgesia is measured by means of a radiant heat source (Plantar Test, Ugo Basile, Comerio, Italy), or by means of a cold plate of 5 to 10°C where the nocifensive reactions of the affected hind paw are counted as a measure of pain intensity.
- a further test for cold induced pain is the counting of nocifensive reactions, or duration of nocifensive responses after plantar adminisfration of acetone to the affected hind limb.
- Chronic pain in general is assessed by registering the circadanian rhytms in activity (Surjo and Amdt, Universitat zu K ⁇ ln, Cologne, Germany), and by scoring differences in gait (foot print patterns; FOOTPPTNTS program, Klapdor et al., 1997. A low cost method to analyse footprint patterns. J. Neurosci. Methods 75, 49-54).
- Substance application is performed at different time points via different application routes (i.v., i.p., p.o., i.t., i.c.v., s.c, intradermal, transdermal) prior to pain testing.
- application routes i.v., i.p., p.o., i.t., i.c.v., s.c, intradermal, transdermal
- Inflammatory pain is induced mainly in rats by injection of 0.75 mg carrageenan or complete Freund's adjuvant into one hind paw.
- the animals develop an edema with mechanical allodynia as well as thermal hyperalgesia.
- Mechanical allodynia is measured by means of a pressure transducer (electronic von Frey Anesthesiometer, HTC Lie-Life Science Instruments, Woodland Hills, SA, USA).
- Thermal hyperalgesia is measured by means of a radiant heat source (Plantar Test, Ugo Basile, Comerio, Italy, Paw thermal stimulator, G. Ozaki, University of California, USA).
- Plant Test Ugo Basile, Comerio, Italy
- Paw thermal stimulator G. Ozaki, University of California, USA
- Compoxmds are tested against uninflamed as well as vehicle treated control groups. Substance application is performed at different time points via different application routes (i.v., i.p., p.o., i.t., i.c.v., s.c, intradermal, transdermal) prior to pain testing.
- application routes i.v., i.p., p.o., i.t., i.c.v., s.c, intradermal, transdermal
- Mechanical allodynia is measured by means of a pressure transducer (electronic von Frey Anesthesiometer, HTC Lie-Life Science Instruments, Woodland Hills, SA, USA).
- Compounds are tested against diabetic and non-diabetic vehicle treated control groups. Substance application is performed at different time points via different application routes (i.v., i.p., p.o., i.t., i.c.v., s.c, intradermal, transdermal) prior to pain testing.
- application routes i.v., i.p., p.o., i.t., i.c.v., s.c, intradermal, transdermal
- the compoxmds of the present invention also show excellent selectivity, and strong activity in other assays 2-5 and assays for pain described above.
- Example 1-1 compounds in Example 1-2 to 1-3 as shown in Table 1 were synthesized.
- Example 2-4 to 2-9 as shown in Table 2 were synthesized.
- a mixtare of benzyl [7-(hydroxymethyl)-5,6,7,8-tetrahydronaphthalen-l-yl]carbamate (32.0 mg, 0.10 mmol) and palladium carbon (30 mg) in ethanol (2 ml) was stined under hydrogen at room temperatare for 16 hours.
- the resulting mixtare was filtered through a pad of celite, and the filfrate was concentrated under reduced pressure.
- 2,2-Difluoro-l,3-benzodioxole-5-carbonitrile 1000 mg, 5.46 mol
- ethanol 100 ml
- Pd/C 200 mg
- the catalyst is filtered off.
- the solvent is removed under reduced pressure and the crade mixture is treated with diethyl ether.
- the resulting crystals are separated from the solvent via a glass filter.
- Phenyl-[(7R)-7-hydroxy-5,6,7,8-tefrahydronaphthalen-l-yl]carbamate (100 mg, 0.35 mmol) and 1- [2,2-difluoro-l,3-benzodioxol-5-yl]methanamine (66 mg, 0.35 mmol) are dissolved in dimethylsulfoxide (2.00 ml) and stined at room temperatare for lh. The raw material is purified via HPLC.
- Phenyl-[(7R)-7-hydroxy-5,6,7,8-tefrahydronaphthalen-l-yl]carbamate 300 mg, 1.06 mmol
- l-[3- chloro-5-(trifluoromethyl)pyridin-2-yl]methanamine hydrochloride 261 mg, 1.06 mmol
- N, ⁇ - diisopropylethylamine (191 mg, 1.48 mmol) are dissolved in dimethylsulfoxide (2.00 ml).
- the mixture is reacted at 60 °C for 3h, partitioned between ethyl acetate and water, the organic layer is dried over magnesium sulfate and evaporated to dryness in vacuo.
- the raw material is triturated with diethyl ether, filtered and dried.
- Example 1-1 compounds in Example 1-2 to 1- 4 as shown in Table 1 were synthesized.
- N-Methyl-4-trifluoromethoxyaniline 100 mg, 0.52 mmol
- ethyl bromoacetate 262 mg, 1.57 mmol
- sodium carbonate 166 mg, 1.57 mmol
- the reaction mixtare is partitioned between ethyl acetate and water, the organic layer is dried over magnesium sulfate and evaporated to dryness in vacuo.
- the raw material is purified by preparative HPLC with an acetonitrile/water gradient.
- Ethyl- ⁇ -methyl- ⁇ -[4-(trifluoromethoxy)phenyl]glycinate 200 mg, 0.72 mmol
- potassium hydroxide 81 mg, 1.44 mmol
- the organic extracts are dried over magnesium sulfate and evaporated to dryness in vacuo.
- the raw material is purified by preparative chromatography on silica (eluent: ethyl acetate/methanol, 1:0 - 5:1).
- the compound is obtained accordingly to the procedure for starting compound H from 5-bromo- pyridine-2-carboxylic acid (93 mg, 0.46 mmol) and [4-(frifluoromethyl)phenyl]boronic acid (105 mg, 0.55 mmol).
- (2R)-8-Amino-l,2,3,4-tefrahydro-naphthalen-2-ol (21 mg, 0.13 mmol), N'-(3-dimethylamino- propyl)-N-ethylcarbodiimide hydrochloride (32 mg, 0.17 mmol), 1 -hydroxy- IH-benzotriazole (21 mg, 0.15 mmol) and N-methyl-N-[4-(trifluoromethoxy)phenyl] glycine (35 mg, 0.14 mmol) are dissolved in dimethylacetamide (3 ml).
- the reaction mixture is stined over night at room temperature, partitioned between ethyl acetate and water, dried over magnesium sulfate and evaporated to dryness in vacuo.
- the raw material is purified by chromatography on silica (eluent: cyclohexane/ethyl acetate, 1:1).
- (2R)-8-amino-l,2,3,4-tefrahydro-naphthalen-2-ol 150 mg, 0.92 mmol
- N'-(3-dimethylaminopropyl)-N-ethylcarbodiimide hydrochloride 229 mg, 1.19 mmol
- 1-hydroxy-lH-benzotriazole 149 mg, 1.10 mmol
- (4-chlorophenoxy)acetic acid 189 mg, 1.01 mmol
- Example 3-8 The compound is obtained accordingly to the procedure for Example 3-8 (omitting the first chromatography over silica gel) from (2R)-8-amino-l,2,3,4-tetrahydro-naphthalen-2-ol (100 mg, 0.61 mmol) and [4-(trifluoromethyl)phenoxy]acetic acid (148 mg, 0.67 mmol).
- Example 3-8 The compound is obtained accordingly to the procedure for Example 3-8 (the crude reaction mixture is applied to reversed phase HPLC purification directly) from (2R)-8-amino-l, 2,3,4- tefrahydro-naphthalen-2-ol (25 mg, 0.15 mmol) and 5-[4-(trifluoromethoxy)phenyl]pyridine-2- carboxylic acid (50 mg, 0.18 mmol).
- Example 3-8 The compound is obtained accordingly to the procedure for Example 3-8 (the crude reaction mixtare is applied to reversed phase HPLC purification directly) from (2R)-8-amino-l, 2,3,4- tefrahydro-naphthalen-2-ol (42 mg, 0.26 mmol) and 5-[4-(trifluoromethyl)phenyl]pyridine-2- carboxylic acid (75 mg, 0.28 mmol).
- Example 3-8 The compound is obtained accordingly to the procedure for Example 3-8 (the crade reaction mixture is applied to reversed phase HPLC purification directly) from (2R)-8-amino-l,2,3,4- tefrahydro-naphthalen-2-ol (117 mg, 0.71 mmol) and 6-[4-(trifluoromethyl)phenyl]nicotinic acid (210 mg, 0.79 mmol).
- Example 3-8 The compound is obtained accordingly to the procedure for Example 3-8 (the crade reaction mixture is applied to reversed phase HPLC purification directly) from (2R)-8-amino-l,2,3,4- tefrahydro-naphthalen-2-ol (37 mg, 0.23 mmol) and 6-[4-(trifluoromethoxy)phenyl]nicotinic acid (71 mg, 0.25 mmol).
- a mixture of 4-amino-5,6,7,8-tetrahydroquinolin-6-ol and 4-chloro-3-trifluoromethylphenyl iso- cyanate in tetrahydrofuran is stined at 50°C for 5 hours. After removing the solvent, the resulting residue is purified by silica gel column chromatography to provide N-[4-chloro-3-(trifluoro- methyl)phenyl]-N l -(6-hydroxy-5,6,7,8-tefrahydroquinolin-4-yl)urea.
- Example 1-2 to 1- 8 as shown in Table 1 are synthesized.
- Example 2-1 to 2-8 as shown in Table 2 Example 3-1 to 3-8 as shown in Table 3, and Example 4-1 to 4-8 as shown in Table 4 are synthesized in a similar maimer as as described in Example 1-1.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Urology & Nephrology (AREA)
- Pain & Pain Management (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Pulmonology (AREA)
- Rheumatology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Neurosurgery (AREA)
- Neurology (AREA)
- Immunology (AREA)
- Biomedical Technology (AREA)
- Physical Education & Sports Medicine (AREA)
- Vascular Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen- Or Sulfur-Containing Heterocyclic Ring Compounds With Rings Of Six Or More Members (AREA)
- Other In-Based Heterocyclic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Nitrogen And Oxygen As The Only Ring Hetero Atoms (AREA)
- Heterocyclic Compounds That Contain Two Or More Ring Oxygen Atoms (AREA)
- Quinoline Compounds (AREA)
- Pyridine Compounds (AREA)
- Indole Compounds (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description









































































































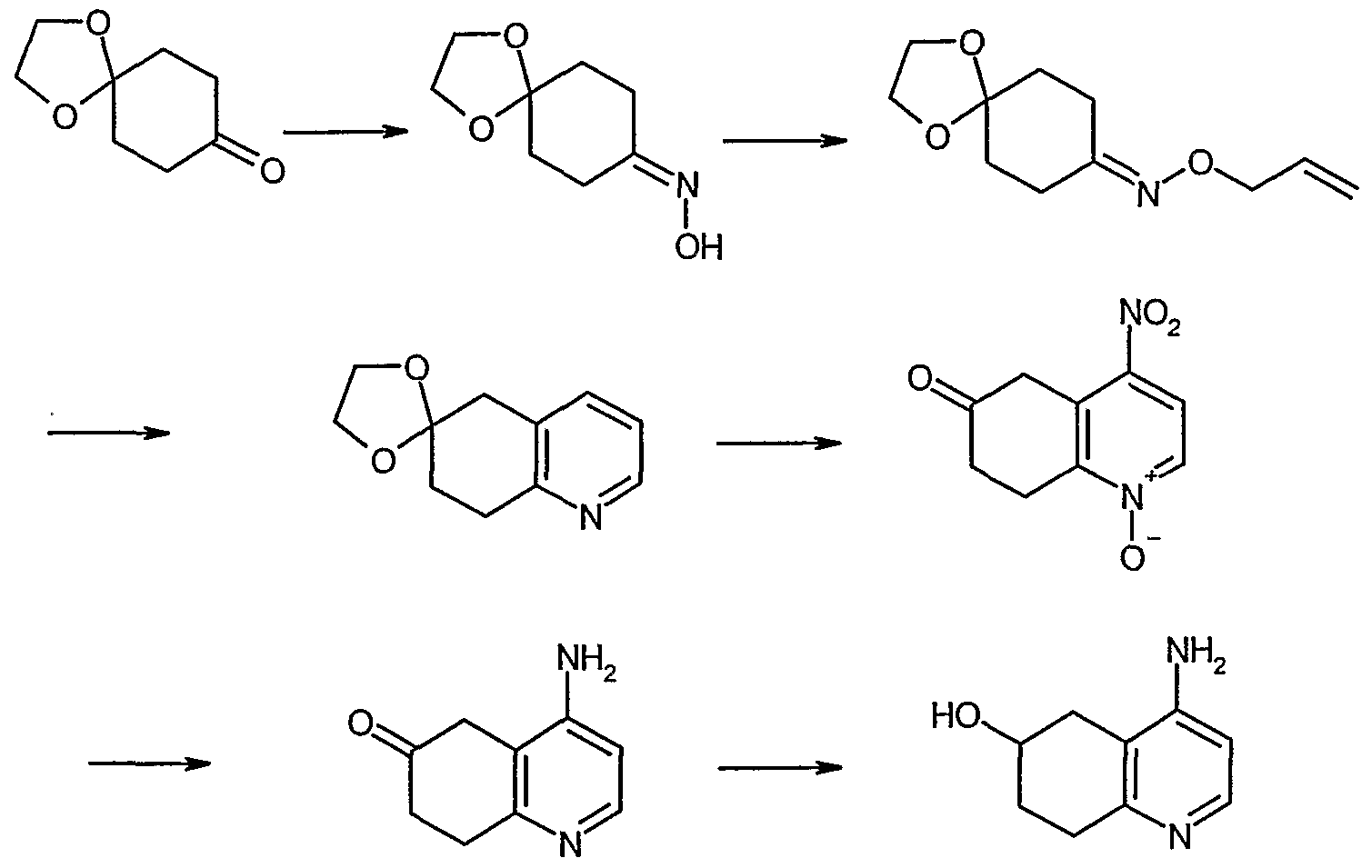









Claims






Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US10/575,027 US20080045546A1 (en) | 2003-10-15 | 2004-10-02 | Tetradydro-Naphthalene And Urea Derivatives |
| CA002542494A CA2542494A1 (en) | 2003-10-15 | 2004-10-02 | Tetrahydro-naphthalene and urea derivatives |
| JP2006534634A JP2007509846A (en) | 2003-10-15 | 2004-10-02 | Tetrahydro-naphthalene and urea derivatives |
| EP04765763A EP1678123A1 (en) | 2003-10-15 | 2004-10-02 | Tetrahydro-naphthalene and urea derivatives |
Applications Claiming Priority (8)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP03023287.0 | 2003-10-15 | ||
| EP03023288.8 | 2003-10-15 | ||
| EP03023288 | 2003-10-15 | ||
| EP03023287 | 2003-10-15 | ||
| EP03025573.1 | 2003-11-08 | ||
| EP03025572 | 2003-11-08 | ||
| EP03025572.3 | 2003-11-08 | ||
| EP03025573 | 2003-11-08 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2005040100A1 true WO2005040100A1 (en) | 2005-05-06 |
Family
ID=34528034
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2004/011008 WO2005040100A1 (en) | 2003-10-15 | 2004-10-02 | Tetrahydro-naphthalene and urea derivatives |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US20080045546A1 (en) |
| EP (1) | EP1678123A1 (en) |
| JP (1) | JP2007509846A (en) |
| CA (1) | CA2542494A1 (en) |
| WO (1) | WO2005040100A1 (en) |
Cited By (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007010383A1 (en) | 2005-07-22 | 2007-01-25 | Mochida Pharmaceutical Co., Ltd. | Novel heterocyclidene acetamide derivative |
| EP1882687A1 (en) * | 2006-07-27 | 2008-01-30 | Amorepacific Corporation | Heterocyclic compounds useful as vanilloid receptor antagonists and pharmaceutical compositions containing the same |
| WO2008079683A2 (en) | 2006-12-20 | 2008-07-03 | Abbott Laboratories | N- (5, 6, 7, 8-tetrahydronaphthalen-1-yl) urea derivatives and related compounds as trpv1 vanilloid receptor antagonists for the treatment of pain |
| WO2009043582A1 (en) * | 2007-10-04 | 2009-04-09 | Pharmeste S.R.L. | Nicotinamide n-oxide derivatives as vanilloid-1 receptor modulators |
| WO2009153721A1 (en) | 2008-06-18 | 2009-12-23 | Pfizer Limited | Nicotinamide derivatives |
| JP2010514742A (en) * | 2006-12-26 | 2010-05-06 | サノフイ−アベンテイス | N- (amino-heteroaryl) -1H-pyrrolopyridine-2-carboxamide derivatives, their preparation, their use in therapy |
| JPWO2008091021A1 (en) * | 2007-01-24 | 2010-05-20 | 持田製薬株式会社 | Heterocyclidene-N- (aryl) acetamide derivatives |
| EP2210878A1 (en) * | 2007-10-16 | 2010-07-28 | Santen Pharmaceutical Co., Ltd | Therapeutic agent for trpv1-mediated disease |
| WO2010137351A1 (en) * | 2009-05-29 | 2010-12-02 | Raqualia Pharma Inc. | Aryl substituted carboxamide derivatives as calcium or sodium channel blockers |
| WO2011120604A1 (en) | 2010-03-30 | 2011-10-06 | Pharmeste S.R.L. | "trpv1 vanilloid receptor antagonists with a bicyclic portion" |
| AU2007277519B2 (en) * | 2006-07-27 | 2011-12-22 | Amorepacific Corporation | Novel compounds, isomer thereof, or pharmaceutically acceptable salts thereof as vanilloid receptor antagonist; and pharmaceutical compositions containing the same |
| WO2013096226A1 (en) | 2011-12-19 | 2013-06-27 | Abbvie Inc. | Trpv1 antagonists |
| WO2013096223A1 (en) | 2011-12-19 | 2013-06-27 | Abbvie Inc. | Trpv1 antagonists |
| WO2013167548A1 (en) * | 2012-05-09 | 2013-11-14 | Bayer Cropscience Ag | Pyrazole tetrahydronaphthyl carboxamides |
| US8796328B2 (en) | 2012-06-20 | 2014-08-05 | Abbvie Inc. | TRPV1 antagonists |
| US8802711B2 (en) | 2011-03-25 | 2014-08-12 | Abbvie Inc. | TRPV1 antagonists |
| CN104870426A (en) * | 2012-12-19 | 2015-08-26 | 拜耳作物科学股份公司 | Tetrahydronaphthyl(Thio) Carboxamides |
| WO2015145371A1 (en) * | 2014-03-27 | 2015-10-01 | Piramal Enterprises Limited | Ror-gamma modulators and uses thereof |
| EP2858501A4 (en) * | 2012-05-22 | 2015-12-09 | Merck Sharp & Dohme | TrkA KINASE INHIBITORS, COMPOSITIONS AND METHODS THEREOF |
| CN109071494A (en) * | 2016-03-31 | 2018-12-21 | 住友化学株式会社 | Heterocyclic compound |
| WO2021039023A1 (en) | 2019-08-23 | 2021-03-04 | 持田製薬株式会社 | Method for producing heterocyclidene acetamide derivatives |
| KR20220051168A (en) | 2019-08-23 | 2022-04-26 | 모찌다 세이야쿠 가부시끼가이샤 | Method for preparing heterocyclideneacetamide derivatives |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8124630B2 (en) * | 1999-01-13 | 2012-02-28 | Bayer Healthcare Llc | ω-carboxyaryl substituted diphenyl ureas as raf kinase inhibitors |
| JP2002534468A (en) * | 1999-01-13 | 2002-10-15 | バイエル コーポレイション | ω-Carboxyaryl-substituted diphenylureas as p38 kinase inhibitors |
| US7371763B2 (en) * | 2001-04-20 | 2008-05-13 | Bayer Pharmaceuticals Corporation | Inhibition of raf kinase using quinolyl, isoquinolyl or pyridyl ureas |
| US20080108672A1 (en) * | 2002-01-11 | 2008-05-08 | Bernd Riedl | Omega-Carboxyaryl Substituted Diphenyl Ureas As Raf Kinase Inhibitors |
| WO2003068228A1 (en) * | 2002-02-11 | 2003-08-21 | Bayer Pharmaceuticals Corporation | Aryl ureas with angiogenesis inhibiting activity |
| US7612113B2 (en) * | 2002-05-08 | 2009-11-03 | Xention Limited | Hydroxy tetrahydro-naphthalenylurea derivatives |
| EP1569896B1 (en) * | 2002-12-06 | 2007-08-15 | Bayer HealthCare AG | Tetrahydro-naphthalene derivatives |
| NZ562414A (en) * | 2003-02-21 | 2009-02-28 | Resmed Ltd | Headgear assembly for nasal pillow mask |
| US7557129B2 (en) * | 2003-02-28 | 2009-07-07 | Bayer Healthcare Llc | Cyanopyridine derivatives useful in the treatment of cancer and other disorders |
| PT1636585E (en) * | 2003-05-20 | 2008-03-27 | Bayer Pharmaceuticals Corp | Diaryl ureas with kinase inhibiting activity |
| US8637553B2 (en) * | 2003-07-23 | 2014-01-28 | Bayer Healthcare Llc | Fluoro substituted omega-carboxyaryl diphenyl urea for the treatment and prevention of diseases and conditions |
| WO2005044802A2 (en) * | 2003-11-08 | 2005-05-19 | Bayer Healthcare Ag | Tetrahydro-quinolinylurea derivatives as vrl antagonists |
| AR062927A1 (en) * | 2006-10-11 | 2008-12-17 | Bayer Healthcare Ag | 4- [4- ([[4- CHLORINE-3- (TRIFLUOROMETILE) PHENYL) CARBAMOIL] AMINO] -3- FLUOROPHENOXY) -N- METHYLPIRIDIN-2-MONOHIDRATED CARBOXAMIDE |
| MX2010012437A (en) | 2008-05-14 | 2011-06-01 | Agriculture Victoria Serv Pty | Use of angiogenin or angiogenin agonists for treating diseases and disorders. |
| AU2013204740C1 (en) | 2012-05-10 | 2015-10-01 | Agriculture Victoria Services Pty Ltd | Methods of treating cancer using angiogenin or an angiogenin agonist |
Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS55161237A (en) * | 1979-06-01 | 1980-12-15 | Konishiroku Photo Ind Co Ltd | Silver halide photographic material |
| WO1997045111A1 (en) * | 1996-05-24 | 1997-12-04 | Neurosearch A/S | Phenyl derivatives useful as blockers of chloride channels |
| WO2003014064A1 (en) * | 2001-07-31 | 2003-02-20 | Bayer Healthcare Ag | Naphthylurea and naphthylacetamide derivatives as vanilloid receptor 1 (vr1) antagonists |
| WO2003022809A2 (en) * | 2001-09-13 | 2003-03-20 | Smithkline Beecham P.L.C. | Urea-compounds active as vanilloid receptor antagonists for the treatment of pain |
| WO2003055848A2 (en) * | 2001-12-26 | 2003-07-10 | Bayer Healthcare Ag | Urea derivatives as vr1- antagonists |
| WO2003070247A1 (en) * | 2002-02-20 | 2003-08-28 | Abbott Laboratories | Fused azabicyclic compounds that inhibit vanilloid receptor subtype 1 (vr1) receptor |
| WO2003095420A1 (en) * | 2002-05-08 | 2003-11-20 | Bayer Healthcare Ag | Hydroxy tetrahydro-naphthalenylurea derivatives |
| WO2004052845A1 (en) * | 2002-12-09 | 2004-06-24 | Bayer Healthcare Ag | Tetrahydro-naphthalene derivatives as vanilloid receptor antagonists |
| WO2004052846A1 (en) * | 2002-12-06 | 2004-06-24 | Bayer Healthcare Ag | Tetrahydro-naphthalene derivatives |
| WO2004072020A1 (en) * | 2003-02-12 | 2004-08-26 | Bayer Healthcare Ag | Hydroxy-tetrahydro-naphthalenylurea derivatives |
| EP1493438A1 (en) * | 2003-07-03 | 2005-01-05 | Bayer HealthCare AG | Vanilloid receptor (VR) inhibitors for treatment of Human Immunodeficiency Virus (HIV)-mediated pain states |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS58140054A (en) * | 1982-02-10 | 1983-08-19 | Sumitomo Chem Co Ltd | Indanylbenzamide derivative, its preparation and agricultural and horticultural fungicide containing said derivative as active component |
| JPS6434962A (en) * | 1986-08-12 | 1989-02-06 | Mitsubishi Chem Ind | Pyridinecarboxylic acid amide derivative and agricultural and horticultural fungicide comprising said derivative as active ingredient |
| JPH089601B2 (en) * | 1987-02-25 | 1996-01-31 | 三菱化学株式会社 | N-Indanylcarboxylic acid amide derivative and agricultural and horticultural fungicide containing the same |
| JP2582863B2 (en) * | 1988-06-13 | 1997-02-19 | 三菱化学株式会社 | Gray mold control agent containing N-indanylcarboxylic acid amide derivative as active ingredient |
| AU3571997A (en) * | 1996-08-01 | 1998-02-25 | Warner-Lambert Company | Novel glutamate receptor antagonists: fused cycloalkyl quinoxalinediones |
| WO1998047879A1 (en) * | 1997-04-22 | 1998-10-29 | Neurosearch A/S | Substituted phenyl derivatives, their preparation and use |
| US6696475B2 (en) * | 1997-04-22 | 2004-02-24 | Neurosearch A/S | Substituted phenyl derivatives, their preparation and use |
| GB9823873D0 (en) * | 1998-10-30 | 1998-12-30 | Pharmacia & Upjohn Spa | 2-ureido-thiazole derivatives,process for their preparation,and their use as antitumour agents |
| EP1240147A1 (en) * | 1999-12-21 | 2002-09-18 | Icagen, Inc. | Potassium channel inhibitors |
| JP2003055209A (en) * | 2001-07-31 | 2003-02-26 | Bayer Ag | Phenylnaphthylurea derivative |
| PL373484A1 (en) * | 2001-12-10 | 2005-09-05 | Amgen Inc. | Vanilloid receptor ligands and their use in treatments |
-
2004
- 2004-10-02 CA CA002542494A patent/CA2542494A1/en not_active Abandoned
- 2004-10-02 EP EP04765763A patent/EP1678123A1/en not_active Withdrawn
- 2004-10-02 JP JP2006534634A patent/JP2007509846A/en active Pending
- 2004-10-02 US US10/575,027 patent/US20080045546A1/en not_active Abandoned
- 2004-10-02 WO PCT/EP2004/011008 patent/WO2005040100A1/en active Application Filing
Patent Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS55161237A (en) * | 1979-06-01 | 1980-12-15 | Konishiroku Photo Ind Co Ltd | Silver halide photographic material |
| WO1997045111A1 (en) * | 1996-05-24 | 1997-12-04 | Neurosearch A/S | Phenyl derivatives useful as blockers of chloride channels |
| WO2003014064A1 (en) * | 2001-07-31 | 2003-02-20 | Bayer Healthcare Ag | Naphthylurea and naphthylacetamide derivatives as vanilloid receptor 1 (vr1) antagonists |
| WO2003022809A2 (en) * | 2001-09-13 | 2003-03-20 | Smithkline Beecham P.L.C. | Urea-compounds active as vanilloid receptor antagonists for the treatment of pain |
| WO2003055848A2 (en) * | 2001-12-26 | 2003-07-10 | Bayer Healthcare Ag | Urea derivatives as vr1- antagonists |
| WO2003070247A1 (en) * | 2002-02-20 | 2003-08-28 | Abbott Laboratories | Fused azabicyclic compounds that inhibit vanilloid receptor subtype 1 (vr1) receptor |
| WO2003095420A1 (en) * | 2002-05-08 | 2003-11-20 | Bayer Healthcare Ag | Hydroxy tetrahydro-naphthalenylurea derivatives |
| WO2004052846A1 (en) * | 2002-12-06 | 2004-06-24 | Bayer Healthcare Ag | Tetrahydro-naphthalene derivatives |
| WO2004052845A1 (en) * | 2002-12-09 | 2004-06-24 | Bayer Healthcare Ag | Tetrahydro-naphthalene derivatives as vanilloid receptor antagonists |
| WO2004072020A1 (en) * | 2003-02-12 | 2004-08-26 | Bayer Healthcare Ag | Hydroxy-tetrahydro-naphthalenylurea derivatives |
| EP1493438A1 (en) * | 2003-07-03 | 2005-01-05 | Bayer HealthCare AG | Vanilloid receptor (VR) inhibitors for treatment of Human Immunodeficiency Virus (HIV)-mediated pain states |
Non-Patent Citations (2)
| Title |
|---|
| DATABASE CAPLUS [online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; "Silver halide photographic materials", XP002320831, retrieved from STN Database accession no. 1981:217541 * |
| DATABASE CAPLUS [online] CHEMICAL ABSTRACTS SERVICE, COLUMBUS, OHIO, US; AMAN, SHUNJI ET AL: "Preparation of urea derivatives as preventive agrochemical pesticides.", XP002320830, retrieved from STN Database accession no. 1992:607160 * |
Cited By (49)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7910751B2 (en) | 2005-07-22 | 2011-03-22 | Mochida Pharmaceutical Co., Ltd. | Heterocyclidene acetamide derivative |
| WO2007010383A1 (en) | 2005-07-22 | 2007-01-25 | Mochida Pharmaceutical Co., Ltd. | Novel heterocyclidene acetamide derivative |
| US8383839B2 (en) | 2005-07-22 | 2013-02-26 | Mochida Pharmaceutical Co., Ltd. | Heterocyclidene acetamide derivative |
| JP4754566B2 (en) * | 2005-07-22 | 2011-08-24 | 持田製薬株式会社 | Novel heterocyclideneacetamide derivatives |
| JP2011153146A (en) * | 2005-07-22 | 2011-08-11 | Mochida Pharmaceut Co Ltd | New heterocyclidene acetamide derivative |
| EP1882687A1 (en) * | 2006-07-27 | 2008-01-30 | Amorepacific Corporation | Heterocyclic compounds useful as vanilloid receptor antagonists and pharmaceutical compositions containing the same |
| AU2007277519B2 (en) * | 2006-07-27 | 2011-12-22 | Amorepacific Corporation | Novel compounds, isomer thereof, or pharmaceutically acceptable salts thereof as vanilloid receptor antagonist; and pharmaceutical compositions containing the same |
| US8350083B2 (en) | 2006-12-20 | 2013-01-08 | Abbvie Inc. | Antagonists of the TRPV1 receptor and uses thereof |
| US8030504B2 (en) | 2006-12-20 | 2011-10-04 | Abbott Laboratories | Antagonists of the TRPV1 receptor and uses thereof |
| EP2450346A1 (en) * | 2006-12-20 | 2012-05-09 | Abbott Laboratories | Antagonists of the TRPV1 receptor and uses thereof |
| WO2008079683A3 (en) * | 2006-12-20 | 2008-08-28 | Abbott Lab | N- (5, 6, 7, 8-tetrahydronaphthalen-1-yl) urea derivatives and related compounds as trpv1 vanilloid receptor antagonists for the treatment of pain |
| WO2008079683A2 (en) | 2006-12-20 | 2008-07-03 | Abbott Laboratories | N- (5, 6, 7, 8-tetrahydronaphthalen-1-yl) urea derivatives and related compounds as trpv1 vanilloid receptor antagonists for the treatment of pain |
| JP2010514742A (en) * | 2006-12-26 | 2010-05-06 | サノフイ−アベンテイス | N- (amino-heteroaryl) -1H-pyrrolopyridine-2-carboxamide derivatives, their preparation, their use in therapy |
| JPWO2008091021A1 (en) * | 2007-01-24 | 2010-05-20 | 持田製薬株式会社 | Heterocyclidene-N- (aryl) acetamide derivatives |
| EP2048133A1 (en) * | 2007-10-04 | 2009-04-15 | Pharmeste S.r.l. | Nicotinamide n-oxide derivatives as vanilloid-1 receptor modulators |
| WO2009043582A1 (en) * | 2007-10-04 | 2009-04-09 | Pharmeste S.R.L. | Nicotinamide n-oxide derivatives as vanilloid-1 receptor modulators |
| US8901155B2 (en) | 2007-10-16 | 2014-12-02 | Santen Pharmaceutical Co., Ltd. | Method for treating a TRPV1-mediated disease |
| EP2666767A1 (en) * | 2007-10-16 | 2013-11-27 | Santen Pharmaceutical Co., Ltd. | Therapeutic agent for TRPV1-mediated disease |
| KR101518525B1 (en) | 2007-10-16 | 2015-05-15 | 산텐 세이야꾸 가부시키가이샤 | Therapeutic agent for trpv1-mediated disease |
| EP2210878A4 (en) * | 2007-10-16 | 2010-12-01 | Santen Pharmaceutical Co Ltd | Therapeutic agent for trpv1-mediated disease |
| EP2210878A1 (en) * | 2007-10-16 | 2010-07-28 | Santen Pharmaceutical Co., Ltd | Therapeutic agent for trpv1-mediated disease |
| WO2009153721A1 (en) | 2008-06-18 | 2009-12-23 | Pfizer Limited | Nicotinamide derivatives |
| CN102448937A (en) * | 2009-05-29 | 2012-05-09 | 拉夸里亚创药株式会社 | Aryl Substituted Carboxamide Derivatives As Calcium Or Sodium Channel Blockers |
| US9522140B2 (en) | 2009-05-29 | 2016-12-20 | Raqualia Pharma Inc. | Aryl substituted carboxamide derivatives as calcium or sodium channel blockers |
| US9101616B2 (en) | 2009-05-29 | 2015-08-11 | Raqualia Pharma Inc. | Aryl substituted carboxamide derivatives as calcium or sodium channel blockers |
| WO2010137351A1 (en) * | 2009-05-29 | 2010-12-02 | Raqualia Pharma Inc. | Aryl substituted carboxamide derivatives as calcium or sodium channel blockers |
| EP2377850A1 (en) * | 2010-03-30 | 2011-10-19 | Pharmeste S.r.l. | TRPV1 vanilloid receptor antagonists with a bicyclic portion |
| WO2011120604A1 (en) | 2010-03-30 | 2011-10-06 | Pharmeste S.R.L. | "trpv1 vanilloid receptor antagonists with a bicyclic portion" |
| US8802711B2 (en) | 2011-03-25 | 2014-08-12 | Abbvie Inc. | TRPV1 antagonists |
| WO2013096223A1 (en) | 2011-12-19 | 2013-06-27 | Abbvie Inc. | Trpv1 antagonists |
| US8859584B2 (en) | 2011-12-19 | 2014-10-14 | Abbvie, Inc. | TRPV1 antagonists |
| US8969325B2 (en) | 2011-12-19 | 2015-03-03 | Abbvie Inc. | TRPV1 antagonists |
| WO2013096226A1 (en) | 2011-12-19 | 2013-06-27 | Abbvie Inc. | Trpv1 antagonists |
| CN104487421A (en) * | 2012-05-09 | 2015-04-01 | 拜耳农作物科学股份公司 | Pyrazole tetrahydronaphthyl carboxamides |
| CN104487421B (en) * | 2012-05-09 | 2017-09-29 | 拜耳农作物科学股份公司 | Pyrazoles tetralyl Carbox amide |
| WO2013167548A1 (en) * | 2012-05-09 | 2013-11-14 | Bayer Cropscience Ag | Pyrazole tetrahydronaphthyl carboxamides |
| US9688608B2 (en) | 2012-05-09 | 2017-06-27 | Bayer Cropscience Ag | Pyrazole tetrahydronaphthyl carboxamides |
| US9204646B2 (en) | 2012-05-09 | 2015-12-08 | Bayer Cropscience Ag | Pyrazole tetrahydronaphthyl carboxamides |
| EP2858501A4 (en) * | 2012-05-22 | 2015-12-09 | Merck Sharp & Dohme | TrkA KINASE INHIBITORS, COMPOSITIONS AND METHODS THEREOF |
| US8796328B2 (en) | 2012-06-20 | 2014-08-05 | Abbvie Inc. | TRPV1 antagonists |
| CN104870426A (en) * | 2012-12-19 | 2015-08-26 | 拜耳作物科学股份公司 | Tetrahydronaphthyl(Thio) Carboxamides |
| WO2015145371A1 (en) * | 2014-03-27 | 2015-10-01 | Piramal Enterprises Limited | Ror-gamma modulators and uses thereof |
| AU2015237788B2 (en) * | 2014-03-27 | 2021-03-11 | Piramal Enterprises Limited | ROR-gamma modulators and uses thereof |
| US11084784B2 (en) | 2014-03-27 | 2021-08-10 | Piramal Enterprises Limited | ROR-gamma modulators and uses thereof |
| CN109071494A (en) * | 2016-03-31 | 2018-12-21 | 住友化学株式会社 | Heterocyclic compound |
| CN109071494B (en) * | 2016-03-31 | 2021-02-12 | 住友化学株式会社 | Heterocyclic compounds |
| US11149027B2 (en) | 2016-03-31 | 2021-10-19 | Sumitomo Chemical Company, Limited | Heterocyclic compound |
| WO2021039023A1 (en) | 2019-08-23 | 2021-03-04 | 持田製薬株式会社 | Method for producing heterocyclidene acetamide derivatives |
| KR20220051168A (en) | 2019-08-23 | 2022-04-26 | 모찌다 세이야쿠 가부시끼가이샤 | Method for preparing heterocyclideneacetamide derivatives |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2007509846A (en) | 2007-04-19 |
| EP1678123A1 (en) | 2006-07-12 |
| US20080045546A1 (en) | 2008-02-21 |
| CA2542494A1 (en) | 2005-05-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1678123A1 (en) | Tetrahydro-naphthalene and urea derivatives | |
| JP4833069B2 (en) | Tetrahydro-naphthalene and urea derivatives | |
| US8088826B2 (en) | Tetrahydro-naphthalene derivatives | |
| US20090209514A1 (en) | Tetrahydro-Naphthalene Derivatives as Vanilloid Receptor Antagonists | |
| US7683076B2 (en) | Tetrahydro-quinolinylurea derivatives | |
| JP4621654B2 (en) | Hydroxy-tetrahydro-naphthalenylurea derivatives |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2004765763 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2542494 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006534634 Country of ref document: JP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004765763 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10575027 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 10575027 Country of ref document: US |