WO2005004802A2 - N-alkyl phenylcarboxamide beta-secretase inhibitors for the treatment of alzheimer's disease - Google Patents
N-alkyl phenylcarboxamide beta-secretase inhibitors for the treatment of alzheimer's disease Download PDFInfo
- Publication number
- WO2005004802A2 WO2005004802A2 PCT/US2004/020234 US2004020234W WO2005004802A2 WO 2005004802 A2 WO2005004802 A2 WO 2005004802A2 US 2004020234 W US2004020234 W US 2004020234W WO 2005004802 A2 WO2005004802 A2 WO 2005004802A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- compound
- group
- mmol
- hydrogen
- compounds
- Prior art date
Links
- 0 *C(Cc1ccccc1)C(O)=O Chemical compound *C(Cc1ccccc1)C(O)=O 0.000 description 1
- KEEOXBOOLAXAIT-XXCBTQFESA-N CCC[C@@H](C(NCC(C)C)=O)NC[C@H](Cc1ccccc1)NC(c1cc(NCC2C(C)C2)cc(N(C)S(C)(=O)=O)c1)=O Chemical compound CCC[C@@H](C(NCC(C)C)=O)NC[C@H](Cc1ccccc1)NC(c1cc(NCC2C(C)C2)cc(N(C)S(C)(=O)=O)c1)=O KEEOXBOOLAXAIT-XXCBTQFESA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C237/00—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups
- C07C237/02—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton
- C07C237/04—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being acyclic and saturated
- C07C237/10—Carboxylic acid amides, the carbon skeleton of the acid part being further substituted by amino groups having the carbon atoms of the carboxamide groups bound to acyclic carbon atoms of the carbon skeleton the carbon skeleton being acyclic and saturated having the nitrogen atom of at least one of the carboxamide groups bound to an acyclic carbon atom of a hydrocarbon radical substituted by nitrogen atoms not being part of nitro or nitroso groups
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C255/00—Carboxylic acid nitriles
- C07C255/49—Carboxylic acid nitriles having cyano groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton
- C07C255/57—Carboxylic acid nitriles having cyano groups bound to carbon atoms of six-membered aromatic rings of a carbon skeleton containing cyano groups and carboxyl groups, other than cyano groups, bound to the carbon skeleton
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C311/00—Amides of sulfonic acids, i.e. compounds having singly-bound oxygen atoms of sulfo groups replaced by nitrogen atoms, not being part of nitro or nitroso groups
- C07C311/01—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms
- C07C311/02—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton
- C07C311/08—Sulfonamides having sulfur atoms of sulfonamide groups bound to acyclic carbon atoms of an acyclic saturated carbon skeleton having the nitrogen atom of at least one of the sulfonamide groups bound to a carbon atom of a six-membered aromatic ring
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/06—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with radicals, containing only hydrogen and carbon atoms, attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/08—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon radicals, substituted by hetero atoms, attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/10—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/02—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D207/04—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D207/10—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D207/14—Nitrogen atoms not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/38—Halogen atoms or nitro radicals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/04—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms
- C07D295/08—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by singly bound oxygen or sulfur atoms
- C07D295/084—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by singly bound oxygen or sulfur atoms with the ring nitrogen atoms and the oxygen or sulfur atoms attached to the same carbon chain, which is not interrupted by carbocyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/16—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms
- C07D295/18—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms acylated on ring nitrogen atoms by radicals derived from carboxylic acids, or sulfur or nitrogen analogues thereof
- C07D295/182—Radicals derived from carboxylic acids
- C07D295/185—Radicals derived from carboxylic acids from aliphatic carboxylic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2601/00—Systems containing only non-condensed rings
- C07C2601/02—Systems containing only non-condensed rings with a three-membered ring
Definitions
- Alzheimer' s disease is characterized by the abnormal deposition of amyloid in the brain in the form of extra-cellular plaques and intra-cellular neurofibrillary tangles.
- the rate of amyloid accumulation is a combination of the rates of formation, aggregation and egress from the brain. It is generally accepted that the main constituent of amyloid plaques is the 4kD amyloid protein ( ⁇ A4, also referred to as A ⁇ , ⁇ -protein and ⁇ AP) which is a proteolytic product of a precursor protein of much larger size.
- the amyloid precursor protein (APP or A ⁇ PP) has a receptor-like structure with a large ectodomain, a membrane spanning region and a short cytoplas ic tail.
- the A ⁇ domain encompasses parts of both extra-cellular and transmembrane domains of APP, thus its release implies the existence of two distinct proteolytic events to generate its NH 2 - and COOH-termini.
- Proteases that release APP and its fragments from the membrane are termed
- secretases Most APP S is released by a putative ⁇ -secretase which cleaves within the A ⁇ protein to release ⁇ -APP s and precludes the release of intact A ⁇ . A minor portion of APPs is released by a ⁇ - secretase, which cleaves near the NH2-terminus of A ⁇ and produces COOH-terminal fragments (CTFs) which contain the whole A ⁇ domain.
- CTFs COOH-terminal fragments
- BACE Alzheimer's disease
- therapeutic agents that can inhibit ⁇ -secretase or BACE may be useful for the treatment of Alzheimer's disease.
- the compounds of the present invention are useful for treating Alzheimer's disease by inhibiting the activity of the ⁇ -secretase or BACE, thus preventing the formation of insoluble A ⁇ and arresting the production of A ⁇ .
- the present invention is directed to compounds that are inhibitors of the ⁇ -secretase enzyme and BACE and which are useful in the treatment of diseases in which the ⁇ -secretase enzyme is involved, such as Alzheimer's disease.
- the invention is also directed to pharmaceutical compositions comprising these compounds and the use of these compounds and compositions in the prevention or treatment of such diseases in which the ⁇ -secretase enzyme is involved.
- Rl is selected from the group consisting of: (1) Ci-6alkyl, unsubstituted or substituted with -OR5 or -S(O)2-C ⁇ _6alkyl, (2) hydrogen, (3) phenyl, and (4) benzyl;
- R2 is selected from the group consisting of: (1) hydrogen, (2) R4-S(O) p -, wherein R4 is independently selected from the group consisting of: (a) C ⁇ _6alkyl, which is unsubstituted or substituted with 1-6 fluoro, (b) phenyl, and (c) benzyl, wherein R5 is independently selected from the group consisting of: (a) hydrogen, (b) -Ci-6alkyl, which is unsubstituted or substituted with 1-6 fluoro, (c) -C3-6cycloalkyl which is unsubstituted or substituted with methyl, (d) phenyl, which is unsubstitued or substituted with halo or methoxy, and (e) benzyl, (4) -CN, (5) -Ci-6alkyl-CN, (6) halogen, (7) wherein R8a and R°>b are independently selected from the group consisting of: (a) hydrogen, (
- n 1, 2, 3 or 4;
- R3 is selected from the group consisting of:
- R6a, R6b ; and R6c are independently selected from the group consisting of: (1) hydrogen, (2) halogen, (3) -OR5, (4) -SR5, and (5) -C ⁇ _6alkyl;
- R9 and RlO are independently selected from the group consisting of: (1) hydrogen, (2) Ci-galkyl, unsubstituted or substituted with -CN or 1-4 halo, (3) -C3_6cycloalkyl, (4) phenyl, which is unsubstitued or substituted with halo or methoxy, and (5) benzyl, or R9 and RlO may be joined together to form a pyrrolidine or piperidine ring which is unsubstituted or substituted with benzyl, -OR5 or 1-4 halo;
- An embodiment of the present invention includes compounds wherein R 1 is C _6alkyl. Another embodiment of the present invention includes compounds wherein R 1 is methyl. Another embodiment of the present invention includes compounds wherein Rl is ethyl. Another embodiment of the present invention includes compounds wherein R 2 is: R4-S(O) 2 -NR5- and wherein R 4 is selected from the group consisting of: (1) C ⁇ _ 6 alkyl, (2) phenyl, and (3) benzyl;
- R 5 is selected from the group consisting of: (1) C ⁇ _6alkyl, (2) phenyl, (3) benzyl, and (4) hydrogen.
- Another embodiment of the present invention includes compounds wherein R 3 is:
- R5 is methyl
- R6a is H or F
- R 6b and R6c are hydrogen.
- Another embodiment of the present invention includes compounds wherein R 3 is:
- Another embodiment of the present invention includes compounds wherein R9 is hydrogen. Another embodiment of the present invention includes compounds wherein RlO is
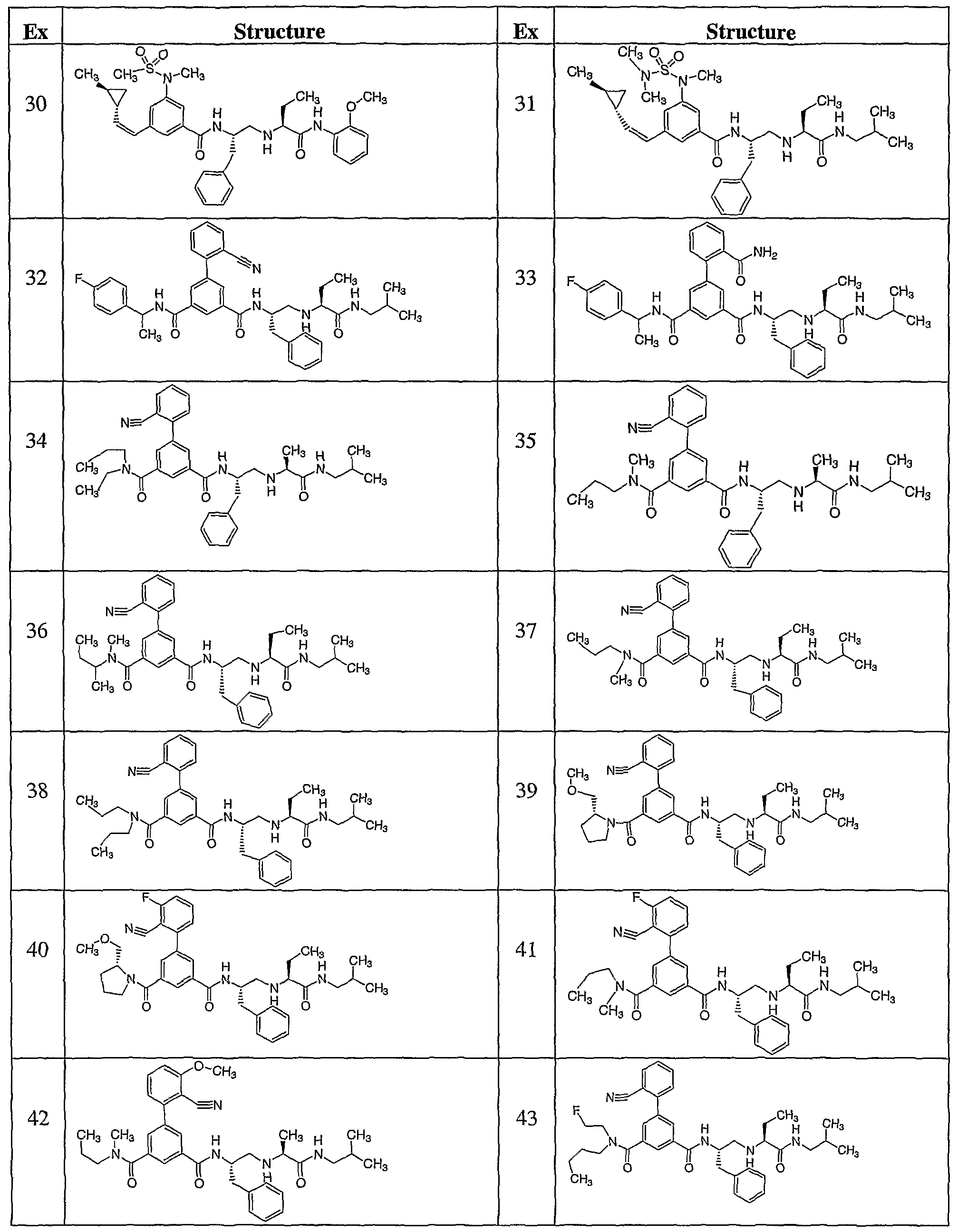
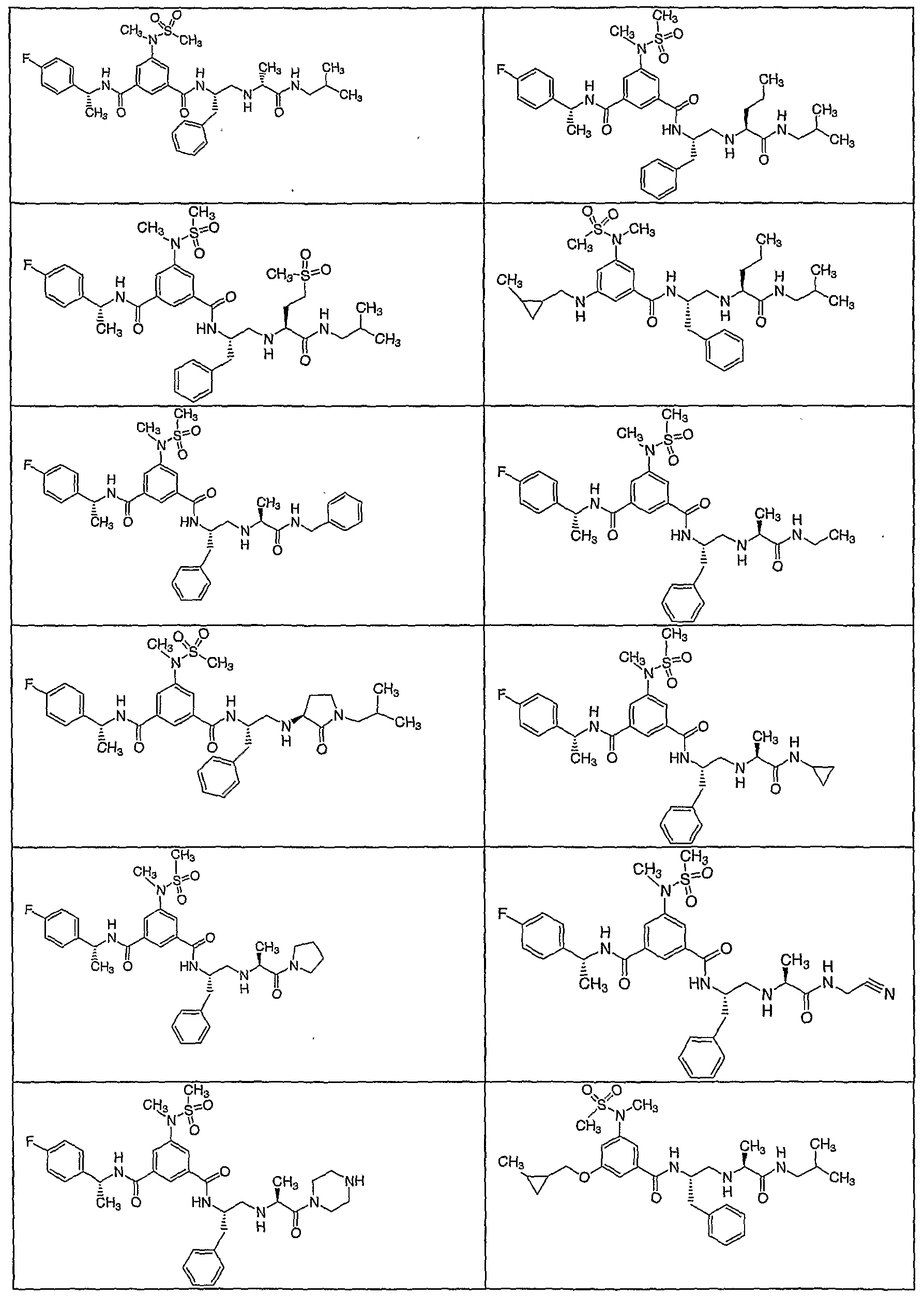
- Ci-galkyl Another embodiment of the present invention includes compounds wherein RlO is iso- butyl. Another embodiment of the present invention includes a compound which is selected from the title compounds of the following Examples and pharmaceutically acceptable salts thereof.
- the compounds of the instant invention have at least one asymmetric center. Additional asymmetric centers may be present depending upon the nature of the various substituents on the molecule. Compounds with asymetric centers give rise to enantiomers (optical isomers), diastereomers (configurational isomers) or both, and it is intended that all of the possible enantiomers and diastereomers in mixtures and as pure or partially purified compounds are included within the scope of this invention. The present invention is meant to encompass all such isomeric forms of these compounds.
- the independent syntheses of the enantiomerically or diastereomerically enriched compounds, or their chromatographic separations, may be achieved as known in the art by appropriate modification of the methodology disclosed herein.
- Their absolute stereochemistry may be determined by the x-ray crystallography of crystalline products or crystalline intermediates that are derivatized, if necessary, with a reagent containing an asymmetric center of known absolute configuration. If desired, racemic mixtures of the compounds may be separated so that the individual enantiomers are isolated.
- the separation can be carried out by methods well known in the art, such as the coupling of a racemic mixture of compounds to an enantiomerically pure compound to form a diastereomeric mixture, followed by separation of the individual diastereomers by standard methods, such as fractional crystallization or chromatography.
- the coupling reaction is often the formation of salts using an enantiomerically pure acid or base.
- the diasteromeric derivatives may then be converted to the pure enantiomers by cleavage of the added chiral residue.
- the racemic mixture of the compounds can also be separated directly by chromatographic methods utilizing chiral stationary phases, which methods are well known in the art.
- any enantiomer of a compound may be obtained by stereoselective synthesis using optically pure starting materials or reagents of known configuration by methods well known in the art.
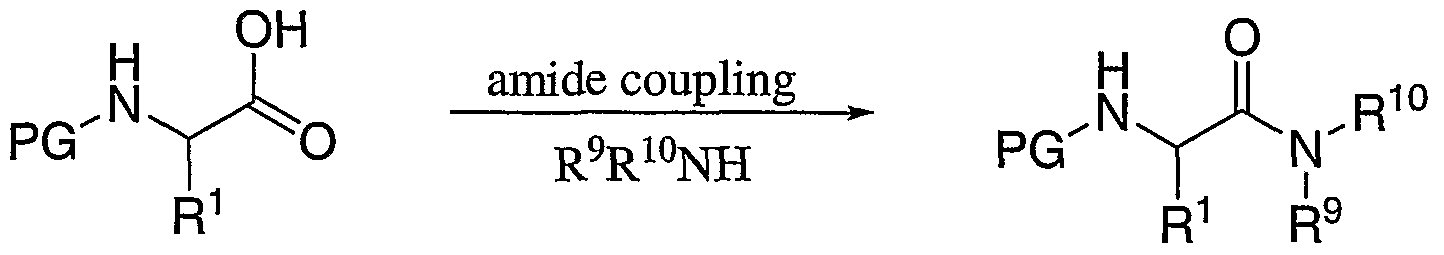
- the compounds of the present invention are prepared by the methods outlined in Scheme
- N-Boc protected amino acids (1-A) are reacted with primary or secondary amines in the presence of a coupling agent such as BOP reagent and an amine base to afford an N-protected amino amide (1-B).
- the Boc group is removed under acidic conditions such as HCl gas in ethyl acetate.
- the resulting amino acid amide salt (1-C) is reductively aminated with boc protected phenylalanine aldehyde using a reducing agent such as sodium cyanoborohydride in methanol.
- the product (1-D) is treated with a strong acid such as HCl gas or trifluoroacetic acid to remove the t- butyloxycarbonyl protecting group to provide the intermediate diamine salt (1-E).
- a strong acid such as HCl gas or trifluoroacetic acid
- Compounds 1-E are coupled to benzoic acid derivatives by standard amide coupling procedures such as BOP reagent and a trialkylamine base to provide final compounds (I).
- Scheme 2 illustrates an alternative process for the synthesis of inhibitors (I).
- Boc-Phe is reduced by standard methods to afford the corresponding alcohol (2-B).
- the resulting alcohol is activated for azide displacement by treatment with methanesulfonyl chloride and an amine base such as triethylamine.
- Azide formation takes place by reacting mesylate 2-C with an excess of sodium azide in a polar aprotic solvent such as DMF at an elevated temperature.
- the product (2-D) is treated with a strong acid such as HCl gas to remove the t-butyloxycarbonyl protecting group to provide the amino azide salt 2-E.
- Standard amide coupling of amine 2-E with a benzoic acid derivative provides 2-F.
- the azide functional group is reduced with a phosphine reagent to provide 2-G which is then alkylated with an appropriately substituted bromoacetate ester and a base such as potassium carbonate.
- the ester 2-H is then saponified with a base such as lithium hydroxide to afford the corresponding carboxylic acid.
- Compounds 2-1 are coupled to a benzoic acid derivative by standard amide coupling procedures such as BOP reagent and a trialkylamine base to provide final compounds I.
- benzoic acids are applicable to schemes 1 and 2 and include examples where R2 is sulfonamide, sulfone, amide, nitrile, alkylnitrile, halogen, phenyl, and cyanocycloalkyl.
- R3 of the benzoic acid in Schemes 1 and 2 is generally selected from a carboxyaminobenzyl group, a substituted olefin, an O or N alkylcyclopropyl, or an alkyl ether, alkylthioether, or secondary amine.
- substantially pure means that the isolated material is at least 90% pure, and preferably 95% pure, and even more preferably 99% pure as assayed by analytical techniques known in the art.
- pharmaceutically acceptable salts refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids including inorganic or organic bases and inorganic or organic acids. Salts derived from inorganic bases include aluminum, ammonium, calcium, copper, ferric, ferrous, lithium, magnesium, manganic salts, manganous, potassium, sodium, zinc, and the like. Particularly preferred are the ammonium, calcium, magnesium, potassium, and sodium salts. Salts in the solid form may exist in more than one crystal structure, and may also be in the form of hydrates.
- Salts derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines, substituted amines including naturally occurring substituted amines, cyclic amines, and basic ion exchange resins, such as arginine, betaine, caffeine, choline, N,N'-dibenzylethylene-diamine, diethylamine, 2-diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N-ethyl- morpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine, tripropylamine, tromethamine, and the like.
- basic ion exchange resins
- salts may be prepared from pharmaceutically acceptable non-toxic acids, including inorganic and organic acids.
- acids include acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethanesulfonic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, nitric, pamoic, pantothenic, phosphoric, succinic, sulfuric, tartaric, p-toluenesulfonic acid, and the like.
- the present invention is directed to the use of the compounds disclosed herein as inhibitors of ⁇ -secretase enzyme activity or ⁇ -site amyloid precursor protein-cleaving enzyme ("BACE") activity, in a patient or subject such as a mammal in need of such inhibition, comprising the administration of an effective amount of the compound.
- BACE ⁇ -site amyloid precursor protein-cleaving enzyme
- ⁇ -secretase enzyme ⁇ -site amyloid precursor protein-cleaving enzyme
- BACE ⁇ -site amyloid precursor protein-cleaving enzyme
- the present invention is further directed to a method for the manufacture of a medicament or a composition for inhibiting ⁇ -secretase enzyme activity in humans and animals comprising combining a compound of the present invention with a pharmaceutical carrier or diluent.
- the compounds of the present invention have utility in treating, preventing, ameliorating, controlling or reducing the risk of Alzheimer' s disease, other diseases mediated by abnormal cleavage of amyloid precursor protein (also referred to as APP), and other conditions that may be treated or prevented by inhibition of ⁇ -secretase.
- Such conditions include mild cognitive impairment, Trisomy 21 (Down Syndrome), cerebral amyloid angiopathy, degenerative dementia, Hereditary Cerebral Hemorrhage with Amyloidosis of the Dutch-Type (HCHWA-D), Creutzfeld-Jakob disease, prion disorders, amyotrophic lateral sclerosis, progressive supranuclear palsy, head trauma, stroke, Down syndrome, pancreatitis, inclusion body myositis, other peripheral amyloidoses, diabetes and atherosclerosis.
- the subject or patient to whom the compounds of the present invention is administered is generally a human being, male or female, in whom inhibition of ⁇ -secretase enzyme activity is desired, but may also encompass other mammals, such as dogs, cats, mice, rats, cattle, horses, sheep, rabbits, monkeys, chimpanzees or other apes or primates, for which inhibition of ⁇ -secretase enzyme activity or treatment of the above noted disorders is desired.
- the compounds of the present invention may be used in combination with one or more other drugs in the treatment, prevention, control, amelioration, or reduction of risk of diseases or conditions for which the compounds of the present invention have utility, where the combination of the drugs together are safer or more effective than either drug alone.
- the compounds of the present invention may be used in combination with one or more other drugs that treat, prevent, control, ameliorate, or reduce the risk of side effects or toxicity of the compounds of the present invention.
- Such other drugs may be administered, by a route and in an amount commonly used therefor, contemporaneously or sequentially with the compounds of the present invention.
- the pharmaceutical compositions of the present invention include those that contain one or more other active ingredients, in addition to the compounds of the present invention.
- the combinations may be administered as part of a unit dosage form combination product, or as a kit or treatment protocol wherein one or more additional drugs are administered in separate dosage forms as part of a treatment regimen.
- Examples of combinations of the compounds of the present invention with other drugs in either unit dose or kit form include combinations with: anti-Alzheimer's agents, for example other beta- secretase inhibitors or gamma-secretase inhibitors; HMG-CoA reductase inhibitors; NSAID's including ibuprofen; vitamin E; anti-amyloid antibodies; CB-1 receptor antagonists or CB-1 receptor inverse agonists; antibiotics such as doxycycline and rifampin; N-methyl-D-aspartate (NMDA) receptor antagonists, such as memantine; cholinesterase inhibitors such as galantamine, rivastigmine, donepezil and tacrine; or other drugs that affect receptors or enzymes that either increase the efficacy, safety, convenience, or reduce unwanted side effects or toxicity of the compounds of the present invention.
- anti-Alzheimer's agents for example other beta- secretase inhibitors or gamma-secretase inhibitors
- composition as used herein is intended to encompass a product comprising specified ingredients in predetermined amounts or proportions, as well as any product which results, directly or indirectly, from combination of the specified ingredients in the specified amounts.
- compositions in relation to pharmaceutical compositions is intended to encompass a product comprising one or more active ingredients, and an optional carrier comprising inert ingredients, as well as any product which results, directly or indirectly, from combination, complexation or aggregation of any two or more of the ingredients.
- compositions are prepared by uniformly and intimately bringing the active ingredient into association with a liquid carrier or a finely divided solid carrier or both, and then, if necessary, shaping the product into the desired formulation.
- the active object compound is included in an amount sufficient to produce the desired effect upon the process or condition of diseases.
- Pharmaceutical compositions intended for oral use may be prepared according to any method known to the art for the manufacture of pharmaceutical compositions and such compositions may contain one or more agents selected from the group consisting of sweetening agents, flavoring agents, coloring agents and preserving agents in order to provide pharmaceutically elegant and palatable preparations. Tablets contain the active ingredient in admixture with non-toxic pharmaceutically acceptable excipients which are suitable for the manufacture of tablets.
- excipients may be for example, inert diluents, granulating and disintegrating agents, binding agents and lubricating agents.
- the tablets may be uncoated or they may be coated by known techniques to delay disintegration and absorption in the gastrointestinal tract and thereby provide a sustained action over a longer period.
- Compositions for oral use may also be presented as hard gelatin capsules wherein the active ingredient is mixed with an inert solid diluent or as soft gelatin capsules wherein the active ingredient is mixed with water or an oil medium.
- Aqueous suspensions contain the active materials in admixture with excipients suitable for the manufacture of aqueous suspensions. Such excipients include suspending agents and dispersing or wetting agents.
- the aqueous suspensions may also contain one or more preservatives, coloring agents, flavoring agents, and sweetening agents.
- Oily suspensions may be formulated by suspending the active ingredient in a vegetable oil or in a mineral oil.
- the oily suspensions may contain a thickening agent.
- Sweetening agents and flavoring agents may be added to provide a palatable oral preparation. These compositions may be preserved by the addition of an anti-oxidant.
- Dispersible powders and granules suitable for preparation of an aqueous suspension by the addition of water provide the active ingredient in admixture with a dispersing or wetting agent, suspending agent and one or more preservatives. Additional excipients, for example sweetening, flavoring and coloring agents, may also be present.
- compositions of the invention may also be in the form of oil-in- water emulsions, which may also contain excipients such as sweetening and flavoring agents.
- the pharmaceutical compositions may also be in the form of a sterile injectable aqueous or oleagenous suspension, which may be formulated according to the known art, or may be administered in the form of suppositories for rectal administration of the drug.
- the compounds of the present invention may also be administered by inhalation, by way of inhalation devices known to those skilled in the art, or transdermally by way of transdermal patch.
- pharmaceutically acceptable it is meant the carrier, diluent or excipient must be compatible with the other ingredients of the formulation and not deleterious to the recipient thereof.
- administering a should be understood to mean providing a compound of the invention to the individual in need of treatment in a form that can be introduced into that individuals body in a therapeutically useful form and therapeutically useful amount, including, but not limited to: oral dosage forms, such as tablets, capsules, syrups, suspensions, and the like; injectable dosage forms, such as IV, IM, or IP, and the like; transdermal dosage forms, including creams, jellies, powders, or patches; buccal dosage forms; inhalation powders, sprays, suspensions, and the like; and rectal suppositories.
- oral dosage forms such as tablets, capsules, syrups, suspensions, and the like
- injectable dosage forms such as IV, IM, or IP, and the like
- transdermal dosage forms including creams, jellies, powders, or patches
- buccal dosage forms inhalation powders, sprays, suspensions, and the like
- rectal suppositories rectal suppositories.
- an effective amount or “therapeutically effective amount” means the amount of the subject compound that will elicit the biological or medical response of a tissue, system, animal or human that is being sought by the researcher, veterinarian, medical doctor or other clinician.
- treatment refers both to the treatment and to the prevention or prophylactic therapy of the mentioned conditions, particularly in a patient who is predisposed to such disease or disorder.
- treatment means any administration of a compound of the present invention and includes (1) inhibiting the disease in an animal that is experiencing or displaying the pathology or symptomatology of the diseased (i.e., arresting further development of the pathology and/or symptomatology), or (2) ameliorating the disease in an animal that is experiencing or displaying the pathology or symptomatology or the diseased (i.e., reversing the pathology and/or symptomatology).
- controlling includes preventing, treating, eradicating, ameliorating or otherwise reducing the severity of the condition being controlled.
- the compositions containing compounds of the present invention may conveniently be presented in unit dosage form and may be prepared by any of the methods well known in the art of pharmacy.
- unit dosage form is taken to mean a single dose wherein all active and inactive ingredients are combined in a suitable system, such that the patient or person adminstering the drug to the patient can open a single container or package with the entire dose contained therein, and does not have to mix any components together from two or more containers or packages.
- Typical examples of unit dosage forms are tablets or capsules for oral administration, single dose vials for injection, or suppositories for rectal administration. This list of unit dosage forms is not intended to be limiting in any way, but merely to represent typical examples in the pharmacy arts of unit dosage forms.
- compositions containing compounds of the present invention may conveniently be presented as a kit, whereby two or more components, which may be active or inactive ingredients, carriers, diluents, and the like, are provided with instructions for preparation of the actual dosage form by the patient or person adminstering the drug to the patient.
- kits may be provided with all necessary materials and ingredients contained therein, or they may contain instructions for using or making materials or components that must be obtained independently by the patient or person administering the drug to the patient.
- the compounds of the present invention are administered at a daily dosage of from about 0.1 milligram to about 100 milligram per kilogram of animal body weight, preferably given as a single daily dose or in divided doses two to six times a day, or in sustained release form.
- the total daily dosage is from about 1.0 milligrams to about 2000 milligrams, preferably from about 0.1 milligrams to about 20 milligrams per kilogram of body weight. In the case of a 70 kg adult human, the total daily dose will generally be from about 7 milligrams to about 1,400 milligrams.
- This dosage regimen may be adjusted to provide the optimal therapeutic response.
- the compounds may be administered on a regimen of 1 to 4 times per day, preferably once or twice per day.
- Specific dosages of the compounds of the present invention, or pharmaceutically acceptable salts thereof, for administration include 1 mg, 5 mg, 10 mg, 30 mg, 80 mg, 100 mg, 150 mg, 300 mg and 500 mg.
- Pharmaceutical compositions of the present invention may be provided in a formulation comprising about 0.5 mg to 1000 mg active ingredient, more preferably comprising about 0.5 mg to 500 mg active ingredient or 0.5 mg to 250 mg active ingredient, or 1 mg to 100 mg active ingredient.
- FRET Assay A homogeneous end point fluorescence resonance energy transfer (FRET) assay is employed with the substrate ([TAMRA-5-CO-EEISEVNLDAEF-NHQSY] QFRET), which is cleaved by BACE 1 to release the fluorescence from TAMRA.
- the Km of the substrate is not determined due to the limit of solubility of the substrate.
- a typical reaction contains approximately 30 nM enzyme, 1.25 ⁇ M of the substrate, and buffer (50 mM NaOAc, pH 4.5, 0.1 mg/ml BSA, 0.2% CHAPS, 15 mM EDTA and 1 mM deferoxamine) in a total reaction volume of 100 ⁇ l.
- the reaction is proceeded for 30 min and the liberation of TAMRA fragment is measured in a 96-well plate LJL Analyst AD using an excitation wavelength of 530 nm and an emission wavelength of 580 nm. Under these conditions, less than 10% of substrate is processed by BACE 1.
- the enzyme used in these studies was soluble (transmembrane domain and cytoplasmic extension excluded) human protein produced in a baculovirus expression system.
- solutions of inhibitor in DMSO four concentrations of the inhibitors were prepared: ImM, 100 ⁇ M, 10 ⁇ M, 1 ⁇ M) were included in the reactions mixture (final DMSO concentration is 0.8%). All experiments were conducted at room temperature using the standard reaction conditions described above.
- HPLC assay A homogeneous end point HPLC assay is employed with the substrate (coumarin-CO-REVNFEVEFR), which is cleaved by BACE 1 to release the N-terminal fragment attached with coumarin.
- the Km of the substrate is greater than 100 ⁇ M and can not be determined due to the limit of solubility of the substrate.
- a typical reaction contains approximately 2 nM enzyme, 1.0 ⁇ M of the substrate, and buffer (50 mM NaOAc, pH 4.5, 0.1 mg/ml BSA, 0.2% CHAPS, 15 mM EDTA and 1 mM deferoxamine) in a total reaction volume of 100 ⁇ l.
- the reaction is proceeded for 30 min and the reaction is stopped by the addition of 25 ⁇ L of 1 M Tris-HCl, pH 8.0.
- the resulting reaction mixture was loaded on the HPLC and the product was separated from substrate with 5 min linear gradient. Under these conditions, less than 10% of substrate is processed by BACE 1.
- the enzyme used in these studies was soluble (transmembrane domain and cytoplasmic extension excluded) human protein produced in a baculovirus expression system.
- Step A To a solution containing 2.0 g (10.0 mmol) of (S)-N-Boc-aminobutyric acid in 50 L of DCM was added 730 mg (10.0 mmol) of isobutyl amine, 4.42 g (10.0 mmol) of BOP Reagent and 4.2 mL (24.0 mmol) of Hunig's base. The reaction mixture was stirred at rt for 30 min before it was extracted with 10% citric acid (10 mL), water (10 mL), saturated NaHCO3 (10 mL) and brine (10 mL). The organic phase was dried over MgSO4, concentrated and chromatographed (1:1 EtOAc / Hexanes) to afford 2.4 g of the desired amide.
- Step B A 0°C solution containing 1.58 g (6.10 mmol) of Boc amide A was dissolved in 40 mL of EtOAc and 4 ml of MeOH and saturated with HCl gas for 10 minutes. The reaction mixture was stirred for 1 h then concentrated to a semi-solid. The residue was triturated with 50 mL of ether to afford 1.10 g of the amine HCl salt as an extremely hygroscopic solid.
- LCMS (M+H) 159. 31
- Step D A 0°C solution containing 201 mg (0.5 mmol) of Boc amine from step C was dissolved in 25 mL of EtOAc and saturated with HCl gas for 5 minutes. The reaction mixture was stirred for 1 h, concentrated and triturated with ether to afford 185 mg of compound I as a white solid.
- Step B To a solution of sodium hydride (0153 g, 3.83 mmol, 60 % oil dispersion) in 10 mL DMF was added sulfonamide (1.0 g, 3.48 mmol) from step A followed by methyl iodide (0.43 mL, 6.97 mmol). After 1 hr the reaction was quenched with H 2 0 (100 mL) and extracted with EtOAc ( 3 x 50 mL). The organic extracts were dried over MgSO4 and evaporated to give 1.03 g of N-methylsulfonamide.
- Step C Diester (1.03 g, 3.38 mmol) from step B was dissolved in 50 mL THF: MeOH (1:1) and cooled to 0° C. IN NaOH (3.38 mL, 3.38 mmol) was added and the reaction was allowed to warm to RT over 8 hours. The solution was acidified with IN HCl (30 mL) and extracted with EtOAc (3 x 50 mL). The combined organic extracts were washed with brine and dried over MgSO , filtered and concentrated in vacuo. Purification on silica gel (5% MeOH/CHCl 3 containing 1% HO Ac) gave 795 mg (82%) of the mono acid.
- Step E To 179 mg (0.438 mmol) of the benzyl amide from step D in 10 mL THF:MeOH (1:1) was added 2 N NaOH (0.66 mL, 1.32 mmol). The solution was heated to 50° C for 1 h. After cooling the solution was acidified by the addition of 1 N HCl (20 mL) and extracted with EtOAc (3 x 30 mL). The combined organic extracts were dried over MgSO4, filtered, and concentrated in vacuo to yield 173 g of the desired carboxylic acid.
- Step F To a solution containing 39.5 mg (0.10 mmol) of the carboxylic acid from step E in 5 mL of DCM was added intermediate amine dihydrochloride IV (35.7 mg, 0.10 mmol), 44.2 mg (0.10 mmol) of BOP reagent and 0.076 mL (0.44 mmol) of diisopropylethyl amine. The reaction mixture was stirred at rt for lh then extracted with 2 x 1 mL IN HCl, 2 x 1 mL water, and 1 mL brine. The organic phase was dried over MgSO4 and subjected to reverse phase chromatography to afford 57.7 mg of the desired product as a white solid.
- Step A To 3-amino-5-nitrobenzoic acid (3.60 g, 19.78 mmol) in 100 mL MeOH was added thionyl chloride (2,59 g, 21.76 mmol). The solution was heated to 65° C for 12 h. Concentration in vacuo afforded the 4.57 g of the methyl ester hydrochloride salt. lH NMR (CD3OD) ⁇ 8.62 (s, IH), 8.28 (s, lH), 8.19 (s, IH), 3.99 (s, 3H).
- Step B To a solution of 3.53 g (18.0 mmol) amino ester from step A in 100 mL CH2Cl2/pyridine (3:1) was added methanesulfonyl chloride (2.07 g, 18.0 mmol). The reaction was stirred at ambient temperature for 1 h followed by evaporation of the solvent. The gummy residue was taken up in EtOAc (100 mL), acidified with IN HCl (100 mL), and extracted with EtOAc (3 x 100 mL). The combined organic extracts were dried over MgSO4, filtered, and concentrated in vacuo to provide 3.97 of the sulfonamide as an off-white solid. lH NMR (CD 3 OD) ⁇ 8.46 (s, IH), 8.30 (s, IH), 8.18 (s, IH), 3.97 (s, 3H), 3.09 (s, 3H).
- Step H To 34 mg (0.10 mmol) of the methyl cyclopropyl methyl aniline from step G in 5 mL THF:MeOH (1:1) was added 2 N NaOH (0.15 mL, 0.30 mmol). The solution was heated to 50°C for 1 h. After cooling the solution was acidified by the addition of 1 N HCl (20 mL) and extracted with EtOAc (3 x 30 mL). The combined organic extracts were dried over MgS04, filtered, and concentrated in vacuo to yield 0.020 g of the desired carboxylic acid.
- LCMS (M + H) 313.2
- Step I A solution containing 31.3 mg (0.10 mmol) of the carboxylic acid from step H, 37.6 mg (0.1 mmol) of intermediate diamine ⁇ , 44.4 mg (0.1 mmol) of BOP reagent and 0.076 mL (0.44 mmol) of
- Step A 3-Nitrobenzoate (35.3 g, 195 mmol) in triflic acid (100 mL) at 0° C was added NIS (43.8 g, 195 mmol) in ten portions. Remove ice bath and stir for 48 hrs. The reaction typically goes to 50% completion. At this time more NIS could be added or cool to 0° C and quench with careful dropwise addition of water. The mixture was extracted three times with EtOAc (250 mL) and the combined extracts were washed with a 10% NaHS0 3 solution, followed by water. The organics were dried over Na2S ⁇ 4, concentrated, and purified on silica gel (10% EtOAc in Hex) affording 24.1 g.
- Step B Tin chloride (88.6 g, 392 mmol) in EtOH (50 mL) was refluxed and the nitrobenzoate from step A (24.1 g, 78.4 mmol) in 1:1 THF:EtOH (100 mL) was added dropwise. The reaction mixture was refluxed for 30 minutes then cooled to 0 C. The resulting solution was basified to pH 8-9 with aq. Na2C03. The aqueous layer was extracted three times with EtOAc (700 mL) and the combined extracts were washed with saturated NaHC03 then brine. The organics were dried over Na2S ⁇ 4 and concentrated • to afford 21 J g of the crude aniline which was used without further purification.
- Step C To a 0° C solution of aniline from step B (21.7 g, 78.3 mmol) in 3:1 CH 2 Cl 2 :pyridine (75 mL) was added methanesulfonyl chloride (6.36 mL, 82.2 mmol). The ice bath was removed after 15 minutes and the solution was stirred overnight at room temperature. The reaction mixture was extracted several times with IN HCl. The organic phase was dried, concentrated, and chromatographed (1:1 EtOAc:Hex) to afford 25.2 g of the desired sulfonamide as a white solid.
- Step D The sulfonamide from step C (23.6 g, 66.5 mmol) in DMF (75 mL) at 0° C was treated with 60% NaH (2.92 g, 73.1 mmol). The solution stirred for 30 minutes before Mel (4.55 mL, 73.1 mmol) was added. The ice bath was removed and the solution was stirred at rt for twelve hours. The reaction was quenched with saturated NH4CI solution and extracted three times with EtOAc (150 mL). The combined organic were washed with water (5 x 50 mL), dried, concentrated to afford 25.3 g of the desired methylated anilide which was used without further purification.
- Step E Jrans-2-methylcyclopropanemethanol (7.0 g, 81 mmol) was added to a solution of PCC (28 g, 130 mmol) in CH 2 C1 2 (225 mL). The solution became black and was stirred for three hours at room temperature. The reaction mixture was diluted with ether (250 mL) and decanted. The liquid solution was filtered through a 4 inch plug of Florisil and the solvent was removed by distillation through a Vigreux column to afford 10 g of the desired aldehyde.
- Step F To a solution of PPI13 (12.4 g, 47.5 mmol) in CH 2 C1 2 (100 mL) at 0° C was added CBr4 (7.88 g, 23.7 mmol). The reaction mixture was stirred for 10 minutes then treated with the carboxaldehyde from step E (1.0 g, 12 mmol). The solution was stirred for 30 minutes at 0° C then 1 hr at room temperature. Hexane was added and the solids were filtered, and the filtrate was concentrated to afford 4.4 g of the dibromide.
- Step H A 100 mL 3-neck round bottom flask was charged with InCl 3 (0.829 g, 10.4 mmol) and dried under vacuum with a heat gun for 2 minutes. THF (16 mL) was added under nitrogen and the flask was immersed in a -78° C ice bath. DEBAL-H (12.4 mL, IM in hexanes) was then added dropwise and the resulting solution was stirred for 30 minutes at -78° C. After this time, the acetylene from step G (10.4 mmol) was added followed by 1.0 M Et 3 B (1.6 mL, IM in hexanes). This reaction mixture was stirred at -78° C for 2.5 hr then warmed to room temperature.
- Step J A solution containing 8.0 mg (0.026 mmol) of the carboxylic acid from step 1, 10.6 mg (0.031 mmol) of intermediate diamine IV, 11.4 mg (0.026 mmol) of BOP reagent and 0.026 mL (0.11 mmol) of Hunig's base was stirred at rt for 1 h in 3 mL of DCM. The solvent was evaporated and the residue was purified by reverse phase chromatography to afford 17.6 mg of the title compound as a white solid.
Abstract
Description























Claims










Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006518678A JP2007516207A (en) | 2003-06-30 | 2004-06-25 | N-alkylphenylcarboxamide β-secretase inhibitors for the treatment of Alzheimer's disease |
| AU2004255183A AU2004255183A1 (en) | 2003-06-30 | 2004-06-25 | N-alkyl phenylcarboxamide beta-secretase inhibitors for the treatment of Alzheimer's disease |
| EP04777003.7A EP1641748B1 (en) | 2003-06-30 | 2004-06-25 | N-alkyl phenylcarboxamide beta-secretase inhibitors for the treatment of alzheimer s disease |
| CA002529994A CA2529994A1 (en) | 2003-06-30 | 2004-06-25 | N-alkyl phenylcarboxamide beta-secretase inhibitors for the treatment of alzheimer's disease |
| US10/563,538 US7291620B2 (en) | 2003-06-30 | 2004-06-25 | N-alkyl phenylcarboxamide beta-secretase inhibitors for the treatment of Alzheimer's disease |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US48399203P | 2003-06-30 | 2003-06-30 | |
| US60/483,992 | 2003-06-30 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2005004802A2 true WO2005004802A2 (en) | 2005-01-20 |
| WO2005004802A3 WO2005004802A3 (en) | 2005-09-22 |
Family
ID=34062007
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2004/020234 WO2005004802A2 (en) | 2003-06-30 | 2004-06-25 | N-alkyl phenylcarboxamide beta-secretase inhibitors for the treatment of alzheimer's disease |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US7291620B2 (en) |
| EP (1) | EP1641748B1 (en) |
| JP (1) | JP2007516207A (en) |
| CN (1) | CN100430377C (en) |
| AU (1) | AU2004255183A1 (en) |
| CA (1) | CA2529994A1 (en) |
| WO (1) | WO2005004802A2 (en) |
Cited By (28)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007017510A2 (en) * | 2005-08-11 | 2007-02-15 | Boehringer Ingelheim International Gmbh | Isophthalic acid diamides for treating alzheimer's disease |
| WO2007017507A1 (en) * | 2005-08-11 | 2007-02-15 | Boehringer Ingelheim International Gmbh | Beta-secretase inhibitors for use in the treatment of alzheimer's disease |
| WO2007017511A2 (en) * | 2005-08-11 | 2007-02-15 | Boehringer Ingelheim International Gmbh | Compounds for treating alzheimer's disease |
| WO2007017509A1 (en) * | 2005-08-11 | 2007-02-15 | Boehringer Ingelheim International Gmbh | Compounds for the treatment of alzheimer's disease |
| WO2007062007A1 (en) | 2005-11-21 | 2007-05-31 | Amgen Inc. | Beta-secretase modulators and methods of use |
| WO2007061930A1 (en) | 2005-11-21 | 2007-05-31 | Amgen Inc. | Beta-secretase modulators and methods of use |
| WO2007061670A1 (en) | 2005-11-21 | 2007-05-31 | Amgen Inc. | Beta-secretase modulators and methods of use |
| US7354942B2 (en) | 2003-11-24 | 2008-04-08 | Merck & Co., Inc. | Benzylether and benzylamino beta-secretase inhibitors for the treatment of Alzheimer's disease |
| WO2008055945A1 (en) | 2006-11-09 | 2008-05-15 | Probiodrug Ag | 3-hydr0xy-1,5-dihydr0-pyrr0l-2-one derivatives as inhibitors of glutaminyl cyclase for the treatment of ulcer, cancer and other diseases |
| WO2008065141A1 (en) | 2006-11-30 | 2008-06-05 | Probiodrug Ag | Novel inhibitors of glutaminyl cyclase |
| JP2008534541A (en) * | 2005-03-30 | 2008-08-28 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Substituted 1,2-ethylenediamine, drugs containing said compounds, their use and methods for their production |
| WO2008104580A1 (en) | 2007-03-01 | 2008-09-04 | Probiodrug Ag | New use of glutaminyl cyclase inhibitors |
| WO2008147544A1 (en) | 2007-05-25 | 2008-12-04 | Amgen Inc. | Substituted hydroxyethyl amine compounds as beta-secretase modulators and methods of use |
| US7550481B2 (en) | 2003-12-19 | 2009-06-23 | Merck & Co., Inc. | Phenylamide and pyridylamide beta-secretase inhibitors for the treatment of Alzheimer's disease |
| US7803809B2 (en) | 2008-11-12 | 2010-09-28 | Amgen Inc. | Substituted pyrano [2,3-b] pyridinamine compounds as beta-secretase modulators and methods of use |
| WO2011029920A1 (en) | 2009-09-11 | 2011-03-17 | Probiodrug Ag | Heterocylcic derivatives as inhibitors of glutaminyl cyclase |
| WO2011063272A1 (en) | 2009-11-23 | 2011-05-26 | Amgen Inc. | Amino heteroaryl compounds as beta-secretase modulators and methods of use |
| WO2011063233A1 (en) | 2009-11-23 | 2011-05-26 | Amgen Inc. | Amino heteroaryl compounds as beta-secretase modulators and methods of use |
| WO2011090911A1 (en) | 2010-01-19 | 2011-07-28 | Amgen Inc. | Amino heteroaryl compounds as beta-secretase modulators and methods of use |
| WO2011107530A2 (en) | 2010-03-03 | 2011-09-09 | Probiodrug Ag | Novel inhibitors |
| WO2011110613A1 (en) | 2010-03-10 | 2011-09-15 | Probiodrug Ag | Heterocyclic inhibitors of glutaminyl cyclase (qc, ec 2.3.2.5) |
| WO2011131748A2 (en) | 2010-04-21 | 2011-10-27 | Probiodrug Ag | Novel inhibitors |
| WO2012123563A1 (en) | 2011-03-16 | 2012-09-20 | Probiodrug Ag | Benz imidazole derivatives as inhibitors of glutaminyl cyclase |
| US8664388B2 (en) | 2007-07-06 | 2014-03-04 | Boehringer Ingelheim International Gmbh | Substituted amino-quinazolinones, medicaments comprising said compound, their use and their method of manufacture |
| US8729071B2 (en) | 2009-10-08 | 2014-05-20 | Merck Sharp & Dohme Corp. | Iminothiadiazine dioxide compounds as BACE inhibitors, compositions and their use |
| EP2865670A1 (en) | 2007-04-18 | 2015-04-29 | Probiodrug AG | Thiourea derivatives as glutaminyl cyclase inhibitors |
| US9833420B2 (en) | 2003-02-27 | 2017-12-05 | JoAnne McLaurin | Methods of preventing, treating, and diagnosing disorders of protein aggregation |
| EP3461819A1 (en) | 2017-09-29 | 2019-04-03 | Probiodrug AG | Inhibitors of glutaminyl cyclase |
Families Citing this family (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7763609B2 (en) | 2003-12-15 | 2010-07-27 | Schering Corporation | Heterocyclic aspartyl protease inhibitors |
| AU2005236020A1 (en) * | 2004-04-20 | 2005-11-03 | Merck & Co., Inc. | 1,3,5-substituted phenyl derivative compounds useful as beta-secretase inhibitors for the treatment of Alzheimer's disease |
| JP4764418B2 (en) | 2004-04-20 | 2011-09-07 | メルク・シャープ・エンド・ドーム・コーポレイション | 2,4,6-substituted pyridyl derivative compounds useful as β-secretase inhibitors for the treatment of Alzheimer's disease |
| WO2006055434A2 (en) * | 2004-11-17 | 2006-05-26 | Merck & Co., Inc. | Macrocyclic tertiary amine beta-secretase inhibitors for the treatment of alzheimer's disease |
| WO2006057945A2 (en) * | 2004-11-23 | 2006-06-01 | Merck & Co., Inc. | 2,3,4,6-substituted pyridyl derivative compounds useful as beta-secretase inhibitors for the treatment of alzheimer's disease |
| EP1817312B1 (en) * | 2004-11-23 | 2011-06-08 | Merck Sharp & Dohme Corp. | Macrocyclic aminopyridyl beta-secretase inhibitors for the treatment of alzheimer's disease |
| ATE528299T1 (en) * | 2005-08-03 | 2011-10-15 | Merck Sharp & Dohme | CYCLIC KETALES AS BETA-SECRETASE INHIBITORS FOR THE TREATMENT OF ALZHEIMER'S DISEASE |
| WO2007019078A2 (en) * | 2005-08-03 | 2007-02-15 | Merck & Co., Inc. | Tricyclic beta-secretase inhibitors for the treatment of alzheimer's disease |
| CA2629317A1 (en) * | 2005-11-16 | 2007-05-24 | Merck & Co., Inc. | Imidazolidinone compounds useful as beta-secretase inhibitors for the treatment of alzheimer's disease |
| US7745484B2 (en) * | 2005-11-21 | 2010-06-29 | Amgen Inc. | Beta-secretase modulators and methods of use |
| AU2007297627A1 (en) * | 2006-09-21 | 2008-03-27 | Merck Sharp & Dohme Corp. | Piperidine and pyrrolidine beta-secretase inhibitors for the treatment of Alzheimer's disease |
| CN101209976B (en) * | 2006-12-29 | 2012-01-11 | 中国人民解放军军事医学科学院毒物药物研究所 | Substituted tartaric acid derivatives and use thereof for preparing beta-secretase inhibitor |
| US20080277393A1 (en) * | 2007-05-07 | 2008-11-13 | True Charles W | Collapsible, stackable, semi-rigid universal cotainer for hazardous and non-hazardous goods |
| WO2008147547A1 (en) | 2007-05-25 | 2008-12-04 | Amgen Inc. | Substituted hydroxyethyl amine compounds as beta-secretase modulators and methods of use |
| CN101348456B (en) * | 2007-07-17 | 2011-05-11 | 中国科学院上海药物研究所 | Benzyl piperidine compound, and preparation and use thereof |
| MX2011002705A (en) * | 2008-09-11 | 2011-09-09 | Amgen Inc | Spiro-tetracyclic ring compounds as betasecretase modulators and methods of use. |
| CA2740107A1 (en) * | 2008-10-10 | 2010-04-15 | Purdue Research Foundation | Compounds for treatment of alzheimer's disease |
| WO2010059953A1 (en) * | 2008-11-20 | 2010-05-27 | Purdue Research Foundation | Quinazoline inhibitors of bace 1 and methods of using |
| US8859590B2 (en) * | 2008-12-05 | 2014-10-14 | Purdue Research Foundation | Inhibitors of BACE1 and methods for treating Alzheimer's disease |
| WO2011006144A1 (en) | 2009-07-10 | 2011-01-13 | Martek Biosciences Corporation | Methods of treating and preventing neurological disorders using docosahexaenoic acid |
| AU2011227511B2 (en) | 2010-03-15 | 2014-02-20 | Amgen Inc. | Spiro-tetracyclic ring compounds as Beta - secretase modulators |
| EP2547686B1 (en) | 2010-03-15 | 2014-01-22 | Amgen Inc. | Amino-dihydrooxazine and amino-dihydrothiazine spiro compounds as beta-secretase modulators and their medical use |
| DE102010024105A1 (en) * | 2010-06-17 | 2011-12-22 | Grünenthal GmbH | Transdermal administration of memantine |
| US9346827B2 (en) | 2011-02-07 | 2016-05-24 | Amgen Inc. | 5-amino-oxazepine and 5-amino-thiazepane compounds as beta secretase antagonists and methods of use |
| US9296759B2 (en) | 2011-09-21 | 2016-03-29 | Amgen Inc. | Amino-oxazine and amino-dihydrothiazine compounds as beta-secretase modulators and methods of use |
| WO2014078314A1 (en) | 2012-11-15 | 2014-05-22 | Amgen Inc. | Amino-oxazine and amino-dihydrothiazine compounds as beta-secretase modulators and methods of use |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003057721A2 (en) | 2002-01-04 | 2003-07-17 | Elan Pharmaceuticals, Inc. | Substituted amino carboxamides for the treatment of alzheimer's disease |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0394311A1 (en) | 1987-11-16 | 1990-10-31 | The Upjohn Company | Renin inhibiting peptides that contain amino and hydroxy dicarboxylic acids |
| ES2174992T3 (en) * | 1992-12-29 | 2002-11-16 | Abbott Lab | INTERMEDIATE PRODUCTS FOR THE OBTAINING OF INHIBITING COMPOUNDS OF RETROVIRAL PROTEASES. |
| US6316440B1 (en) * | 1998-07-30 | 2001-11-13 | Warner-Lambert Company | Reduced dipeptide analogues as calcium channel antagonists |
| EP1194449B1 (en) | 1999-06-28 | 2010-09-22 | Oklahoma Medical Research Foundation | Inhibitors of memapsin 2 and use thereof |
| MXPA02012560A (en) * | 2000-06-30 | 2003-05-14 | Elan Pharm Inc | Compounds to treat alzheimer s disease. |
| WO2003043975A1 (en) * | 2001-11-19 | 2003-05-30 | Elan Pharmaceuticals, Inc. | Amine 1,2- and 1,3-diol compounds and their use for treatment of alzheimer's disease |
| WO2004043916A1 (en) | 2002-11-12 | 2004-05-27 | Merck & Co., Inc. | Phenylcarboxamide beta-secretase inhibitors for the treatment of alzheimer's disease |
-
2004
- 2004-06-25 WO PCT/US2004/020234 patent/WO2005004802A2/en active Application Filing
- 2004-06-25 CN CNB2004800185017A patent/CN100430377C/en not_active Expired - Fee Related
- 2004-06-25 AU AU2004255183A patent/AU2004255183A1/en not_active Abandoned
- 2004-06-25 CA CA002529994A patent/CA2529994A1/en not_active Abandoned
- 2004-06-25 JP JP2006518678A patent/JP2007516207A/en not_active Ceased
- 2004-06-25 US US10/563,538 patent/US7291620B2/en active Active
- 2004-06-25 EP EP04777003.7A patent/EP1641748B1/en active Active
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2003057721A2 (en) | 2002-01-04 | 2003-07-17 | Elan Pharmaceuticals, Inc. | Substituted amino carboxamides for the treatment of alzheimer's disease |
Non-Patent Citations (5)
| Title |
|---|
| H. FUKUMOTO ET AL., ARCH. NEUROL., vol. 59, September 2002 (2002-09-01), pages 1381 - 1389 |
| J.T. HUSE ET AL., J. BIOL. CHEM., vol. 277, no. 18, 3 May 2002 (2002-05-03), pages 16278 - 16284 |
| K.C. CHEN; W.J. HOWE, BIOCHEM. BIOPHYS. RES. COMM, vol. 292, 2002, pages 702 - 708 |
| R. N. ROSENBERG, ARCH. NEUROL., vol. 59, September 2002 (2002-09-01), pages 1367 - 1368 |
| See also references of EP1641748A4 |
Cited By (42)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9833420B2 (en) | 2003-02-27 | 2017-12-05 | JoAnne McLaurin | Methods of preventing, treating, and diagnosing disorders of protein aggregation |
| US7354942B2 (en) | 2003-11-24 | 2008-04-08 | Merck & Co., Inc. | Benzylether and benzylamino beta-secretase inhibitors for the treatment of Alzheimer's disease |
| US7550481B2 (en) | 2003-12-19 | 2009-06-23 | Merck & Co., Inc. | Phenylamide and pyridylamide beta-secretase inhibitors for the treatment of Alzheimer's disease |
| US7713961B2 (en) | 2005-03-30 | 2010-05-11 | Boehringer Ingelheim International Gmbh | Substituted 1,2-ethylenediamines, methods for preparing them and uses thereof |
| JP2008534541A (en) * | 2005-03-30 | 2008-08-28 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Substituted 1,2-ethylenediamine, drugs containing said compounds, their use and methods for their production |
| WO2007017511A3 (en) * | 2005-08-11 | 2007-04-26 | Boehringer Ingelheim Int | Compounds for treating alzheimer's disease |
| WO2007017510A3 (en) * | 2005-08-11 | 2008-02-28 | Boehringer Ingelheim Int | Isophthalic acid diamides for treating alzheimer's disease |
| WO2007017510A2 (en) * | 2005-08-11 | 2007-02-15 | Boehringer Ingelheim International Gmbh | Isophthalic acid diamides for treating alzheimer's disease |
| JP2009504611A (en) * | 2005-08-11 | 2009-02-05 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Β-secretase inhibitor for the treatment of Alzheimer's disease |
| WO2007017509A1 (en) * | 2005-08-11 | 2007-02-15 | Boehringer Ingelheim International Gmbh | Compounds for the treatment of alzheimer's disease |
| WO2007017511A2 (en) * | 2005-08-11 | 2007-02-15 | Boehringer Ingelheim International Gmbh | Compounds for treating alzheimer's disease |
| WO2007017507A1 (en) * | 2005-08-11 | 2007-02-15 | Boehringer Ingelheim International Gmbh | Beta-secretase inhibitors for use in the treatment of alzheimer's disease |
| JP2009504612A (en) * | 2005-08-11 | 2009-02-05 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Compounds for the treatment of Alzheimer's disease |
| JP2009504613A (en) * | 2005-08-11 | 2009-02-05 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Compounds for the treatment of Alzheimer's disease |
| JP2009504614A (en) * | 2005-08-11 | 2009-02-05 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Compounds for the treatment of Alzheimer's disease |
| WO2007061930A1 (en) | 2005-11-21 | 2007-05-31 | Amgen Inc. | Beta-secretase modulators and methods of use |
| WO2007061670A1 (en) | 2005-11-21 | 2007-05-31 | Amgen Inc. | Beta-secretase modulators and methods of use |
| WO2007062007A1 (en) | 2005-11-21 | 2007-05-31 | Amgen Inc. | Beta-secretase modulators and methods of use |
| WO2008055945A1 (en) | 2006-11-09 | 2008-05-15 | Probiodrug Ag | 3-hydr0xy-1,5-dihydr0-pyrr0l-2-one derivatives as inhibitors of glutaminyl cyclase for the treatment of ulcer, cancer and other diseases |
| WO2008065141A1 (en) | 2006-11-30 | 2008-06-05 | Probiodrug Ag | Novel inhibitors of glutaminyl cyclase |
| EP2481408A2 (en) | 2007-03-01 | 2012-08-01 | Probiodrug AG | New use of glutaminyl cyclase inhibitors |
| WO2008104580A1 (en) | 2007-03-01 | 2008-09-04 | Probiodrug Ag | New use of glutaminyl cyclase inhibitors |
| EP2865670A1 (en) | 2007-04-18 | 2015-04-29 | Probiodrug AG | Thiourea derivatives as glutaminyl cyclase inhibitors |
| WO2008147544A1 (en) | 2007-05-25 | 2008-12-04 | Amgen Inc. | Substituted hydroxyethyl amine compounds as beta-secretase modulators and methods of use |
| US8664388B2 (en) | 2007-07-06 | 2014-03-04 | Boehringer Ingelheim International Gmbh | Substituted amino-quinazolinones, medicaments comprising said compound, their use and their method of manufacture |
| US7951838B2 (en) | 2007-11-14 | 2011-05-31 | Amgen Inc. | Substituted spirocyclic chromanamine compounds as Beta-Secretase modulators and methods of use |
| US7803809B2 (en) | 2008-11-12 | 2010-09-28 | Amgen Inc. | Substituted pyrano [2,3-b] pyridinamine compounds as beta-secretase modulators and methods of use |
| WO2011029920A1 (en) | 2009-09-11 | 2011-03-17 | Probiodrug Ag | Heterocylcic derivatives as inhibitors of glutaminyl cyclase |
| US9428475B2 (en) | 2009-10-08 | 2016-08-30 | Merck Sharp & Dohme Corp. | Iminothiadiazine dioxide compounds as BACE inhibitors, compositions, and their use |
| US9029362B2 (en) | 2009-10-08 | 2015-05-12 | Merck Sharp & Dohme Corp. | Iminothiadiazine dioxide compounds as brace inhibitors, compositions, and their use |
| US9475785B2 (en) | 2009-10-08 | 2016-10-25 | Merck Sharp & Dohme Corp. | Iminothiadiazine dioxide compounds as BACE inhibitors, compositions and their use |
| US9687494B2 (en) | 2009-10-08 | 2017-06-27 | Merck Sharp & Dohme Corp. | Iminothiadiazine dioxide compounds as BACE inhibitors, compositions, and their use |
| US8940748B2 (en) | 2009-10-08 | 2015-01-27 | Merck Sharp & Dohme Corp. | Iminothiadiazine dioxide compounds as BACE inhibitors, compositions, and their use |
| US8729071B2 (en) | 2009-10-08 | 2014-05-20 | Merck Sharp & Dohme Corp. | Iminothiadiazine dioxide compounds as BACE inhibitors, compositions and their use |
| WO2011063233A1 (en) | 2009-11-23 | 2011-05-26 | Amgen Inc. | Amino heteroaryl compounds as beta-secretase modulators and methods of use |
| WO2011063272A1 (en) | 2009-11-23 | 2011-05-26 | Amgen Inc. | Amino heteroaryl compounds as beta-secretase modulators and methods of use |
| WO2011090911A1 (en) | 2010-01-19 | 2011-07-28 | Amgen Inc. | Amino heteroaryl compounds as beta-secretase modulators and methods of use |
| WO2011107530A2 (en) | 2010-03-03 | 2011-09-09 | Probiodrug Ag | Novel inhibitors |
| WO2011110613A1 (en) | 2010-03-10 | 2011-09-15 | Probiodrug Ag | Heterocyclic inhibitors of glutaminyl cyclase (qc, ec 2.3.2.5) |
| WO2011131748A2 (en) | 2010-04-21 | 2011-10-27 | Probiodrug Ag | Novel inhibitors |
| WO2012123563A1 (en) | 2011-03-16 | 2012-09-20 | Probiodrug Ag | Benz imidazole derivatives as inhibitors of glutaminyl cyclase |
| EP3461819A1 (en) | 2017-09-29 | 2019-04-03 | Probiodrug AG | Inhibitors of glutaminyl cyclase |
Also Published As
| Publication number | Publication date |
|---|---|
| CN1812963A (en) | 2006-08-02 |
| JP2007516207A (en) | 2007-06-21 |
| US7291620B2 (en) | 2007-11-06 |
| CA2529994A1 (en) | 2005-01-20 |
| AU2004255183A1 (en) | 2005-01-20 |
| EP1641748A2 (en) | 2006-04-05 |
| WO2005004802A3 (en) | 2005-09-22 |
| EP1641748A4 (en) | 2006-11-22 |
| US20060161020A1 (en) | 2006-07-20 |
| EP1641748B1 (en) | 2013-08-28 |
| CN100430377C (en) | 2008-11-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1641748B1 (en) | N-alkyl phenylcarboxamide beta-secretase inhibitors for the treatment of alzheimer s disease | |
| US7348356B2 (en) | Phenylcarboxamide beta-secretase inhibitors for the treatment of Alzheimer's disease | |
| US7348448B2 (en) | Phenylcarboxylate beta-secretase inhibitors for the treatment of alzheimer's disease | |
| JP4673833B2 (en) | Macrocyclic β-secretase inhibitor for the treatment of Alzheimer's disease | |
| US7329746B2 (en) | Macrocyclic beta-secretase inhibitors for the treatment of Alzheimer's disease | |
| EP1673078B1 (en) | Benzylether and benzylamino beta-secretase inhibitors for the treatment of alzheimer's disease | |
| US7449599B2 (en) | Phenyl carboxamide compounds useful as beta-secretase inhibitors for the treatment of alzheimer's disease | |
| EP1689713B1 (en) | Benzylether and benzylamino beta-secretase inhibitors for the treatment of alzheimer s disease |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| ENP | Entry into the national phase |
Ref document number: 2006161020 Country of ref document: US Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10563538 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2529994 Country of ref document: CA Ref document number: 5944/DELNP/2005 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004777003 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006518678 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 20048185017 Country of ref document: CN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004255183 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 2004255183 Country of ref document: AU Date of ref document: 20040625 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004777003 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 10563538 Country of ref document: US |