Reagents and Methods for the formation of disulfide bonds and the glycosylation of proteins
The present application is concerned with reagents and methods for the formation of disulfide bonds and/or for the chemical modification of proteins, in particular reagents and methods for use in the glycosylation of proteins. The co- and post-translational glycosylation of proteins plays a vital role in their biological behaviour and stability (R. Dwek, Chem. Rev., 96:683-720 (1996)) . For example, glycosylation plays a major role in essential biological processes such as cell signalling and regulation, development and immunity. The study of these events is made difficult by the fact that glycoproteins occur naturally as mixtures of so-called glycoforms that possess the same peptide backbone but differ in both the nature and the site of glycosylation. Furthermore, since protein glycosylation is not under direct genetic control, the expression of therapeutic glycoproteins in mammalian cell culture leads to heterogeneous mixtures of glycoforms. The ability to synthesise homogeneous glycoprotein glycoforms is therefore not only a prerequisite for accurate investigation purposes, but is of increasing importance when preparing therapeutic glycoproteins, which are currently marketed as multi- glycoform mixtures (e.g. erythropoietin and interleukins). Other post translational modifications of proteins, such as phosphorylation and methylation, are also of importance. ConuOlling the degree and nature of such modification of a protein therefore allows the possibility of investigating and controlling its behaviour in biological systems (B.G. Davis, Science, Vol 303, p 480-482, 2004). A number of methods for the glycosylation of proteins are known, including chemical synthesis. Chemical synthesis of glycoproteins offers certain advantages, not least the possibility of access to pure glycoprotein glycoforms. One known synthetic method utilises thiol-selective carbohydrate reagents, glycosylmethane thiosulfonate reagents (glyco-MTS). Such glycosylmethane thiosulfonate reagents react with thiol groups in a protein to introduce a glycosyl residue linked to the protein via a disulfide bond (see for example O00/01712). However, glyco-MTS reagents suffer from a number of disadvantages, including occasionally moderate reaction yields, difficulties in their preparation and
problems with stability under the basic conditions in which they are often used. There is therefore a need for further reagents for use in protein glycosylation which are readily prepared, stable and give high yields of the gly cosy lated protein product. There is also a need for alternative methods for protein glycosylation which give high yields of the gly cosy lated protein product, are site-selective, and which allow glycosylation at both single and multiple sites in a wide range of different proteins. We have now surprisingly found that certain sulfur and selenium-containing glycosylation reagents are relatively straightforward to prepare, are generally more stable than the corresponding glyco-MTS reagents and can be used in the glycosylation of a wide range of thiol containing compounds, including proteins, in high yield. In a first aspect, the invention therefore provides a method of forming disulfide bonds (-S-S-), the method comprising reacting an organic compound comprising at least one thiol group (-SH) with a compound of formula I:
R-S— X-R1 wherein: X denotes SO2 or Se, prefereably Se; R denotes an organic moiety, for example an alkyl group, an alkenyl group, an alkynyl group, or a carbohydrate moiety; and R1 denotes an optionally substituted alkyl group, an optionally substituted phenyl group, an optionally substituted pyridyl group or an optionally substituted naphthyl group; with the proviso that when X denotes SO2 then R1 does not denote optionally substituted alkyl. Preferably, the organic compound comprising at least one thiol group is an amino acid, peptide or protein. In a second aspect, the invention further provides a method of chemically modifying a protein, peptide or amino acid comprising at least one thiol group (-SH), the method comprising reacting said protein, peptide or amino acid with a compound of formula I as previously defined.
In a still further aspect, the invention provides compounds of formula I wherein R denotes a carbohydrate moiety. When R denotes an alkenyl or alkenyl group, there is the possibility that the disulphide compound formed by reaction with the compound of formula I may be further elaborated by reaction at the C=C or C≡C bond in the group R. We have also surprisingly found that a thiol containing protein may be converted to the corresponding selenenylsulfide, and that the electrophilic character of the sulfur in the S-Se bond thus created renders it susceptible to nucleophilic substitution by tMol-contairiing compounds including carbohydrates. In a third aspect, the invention therefore provides a method of chemically modifying a protein, peptide or amino acid comprising at least one thiol group (-S-H), the method comprising converting said thiol group into a selenenylsulfide group (-S-Se-R2). The method therefore allows the preparation of a protein, peptide or amino acid comprising at least one selenenylsulfide group. Such proteins, peptides and amino acids comprising at least one selenenylsulfide group form a further feature of the invention. Particularly preferred are proteins or peptides comprising at least one selenenylsulfide group. A selenenylsulfide group in a protein, peptide or amino acid may be further reacted with an organic compound comprising a thiol group to give further chemically modified proteins, peptides or amino acids in which the organic group is attached to the protein, peptide or amino acid via a disulfide bond. Preferably, the organic compound containing the thiol group is a carbohydrate compound, thus providing a method for the glycosylation of an amino acid, peptide or protein. As used herein, "glycosylation" refers to the general process of addition of a glycosyl unit to another moiety via a covalent linkage. In a fourth aspect, the invention therefore provides a method of chemically modifying a protein, peptide or amino acid comprising at least one thiol group (-S-H), the method comprising: (a) converting said thiol group into a selenenylsulfide group (-S-Se-R2); and (b) reacting said selenenylsulfide group with an organic compound containing a thiol group. The method(s) according to the first, second, third and fourth aspects of the invention will hereinafter be referred to as the first method, the second method, the
third method and the fourth method respectively. Unless otherwise stated, all preferred features and definitions herein relate to all these methods. Furthermore, the present invention includes any and all possible combinations of any preferred features referred to herein, whether or not such combinations are specifically disclosed. A generalised reaction scheme for disulfide bond formation according to the first and second methods is shown in Scheme 1: organic moiety -SH + R— S — X -R1
Scheme 1
Preferably, the organic moiety shown in Scheme 1 is a protein, peptide or amino acid. A generalised reaction scheme for the introduction of a selenenylsulfide group into a protein, peptide or amino acid according to the third and fourth methods is shown in Scheme 2:
Scheme 2
The method of Scheme 2 results in covalent linkage of a group R2 to the protein, peptide or amino acid via a selenenylsulfide (-S -Se-) linkage. Such proteins, peptides or amino acids form a further feature of the invention.
Proteins and peptides comprising a selenenylsulfide group may be useful in the determination of protein structure via X-ray diffraction techniques. Currently, MAD (multiple wavelength anomalous dispersion) techniques involve the conversion of any methionine residues in the protein into selenomethionine. Comparison of the X-ray diffraction patterns of the modified and unmodified proteins then allows a determination of the structure of the unmodified protein to be carried out. The method of the invention allows convenient and ready access to alternative selenium-containing proteins or peptides which may be used in such techniques. The methods of the invention provide an easy method for introducing a heavy metal into a protein strucutre, thus making interpretation of the X-ray diffraction data easier. Selenenylsulfide containing proteins, peptides or amino acids may be further reacted with thiol containing organic compounds according to the fourth method as shown in the generalised reaction scheme in Scheme 3:
-S— Se— IT
Q = protein, peptide or amino acid
Scheme 3
The method of Scheme 3 results in covalent linkage of the organic moiety to the protein, peptide or amino acid via a disulfide bond (-S -S-). In this method the protein, peptide or amino acid is acting as an electrophile whilst the thiol-cont-iining organic compound acts as a nucleophile. In contrast, the known reactions utilising glyco-MTS reagents involve reaction of a nucleophilic thiol group in the protein, peptide or amino acid with the electrophilic glyco-MTS reagent. The method of the
invention therefore provides a complementary strategy to the known protein modification strategies utilising glyco-MTS reagents. As used herein, alkyl preferably denotes a straight chain or branched alkyl group containing 1-10 carbon atoms, preferably 1 -6 carbon atoms. Preferred alkyl groups include methyl and ethyl. As used herein, alkenyl preferably denotes a straight chain or branched hydrocarbon group comprising at least one carbon -carbon double bond, and containing 2-20 carbon atoms, preferably 2-10 carbon atoms, and more preferably 2-6 carbon atoms. Preferred alkenyl groups include -(CH
2)CH=CH
2, -CH
2CH
2CH=CH
2, prenyl ((CH
3)
2C=CHCH
2-) and farnesyl ((CH
3)
2C=CH[CH
2CH
2C(CH
3)=CH]
2CH
2-). As used herein, alkynyl preferably denotes a straight chain or branched hydrocarbon group comprising at least one carbon-carbon triple bond, and containing 2-10 carbon atoms, preferably 2 -6 carbon atoms. Preferred alkynyl groups include -CEbC≡CH and -CH
2CH
2C≡CH. When R
1 denotes an optionally substituted moiety, suitable substituents include any substituents which do not interfere with the formation of the compound of formula I or with the disulfide bond forming reaction according to the first or second methods, for example -NO
2, -SO
3H, -CO
2H, -(CH
2CH
2O)
nH and -(CH
2CH
2O)
nMe wherein n denotes 1-100, preferably 1-50, more preferably 1-20, and still more preferably 1-10. The R
1 group may be independently substituted by 1-5, and preferably 1 or 2, substituents. The R
1 group may also optionally be attached to, or form part of, a solid support, for example a resin such as a polystyrene resin. A preferred R
1 group is phenyl. When the group R in the compounds of formula I is phenyl or another aromatic group, then there is the added advantage that the progress of the reaction with the thiol-containing compound according to the first and second methods may be monitored using UN spectroscopy. Thus, for example, the PhSO
2- chromophore displays a maximum in the UN spectrum at approx. 265nm. The PhSO
2- moiety is present in both the compound of formula I and the PhSO
2 " that is the by-product of the disulfide bond forming reaction, but the associated extinction coefficients differ sufficiently for the progress of the reaction to be monitored using UN. Similarly, the third and fourth methods of the invention may be monitored by UV spectroscopy when the group R
2 is phenyl or another aromatic group.
In the compounds of formula I, the group R may be any organic moiety, particularly any organic moiety which is suitable for linkage to a protein, peptide or amino acid. There is no particular limitation on the nature of R. For example, the -S-X- group may be primary, secondary or tertiary. R may be aromatic or aliphatic. The group R may optionally be substituted, for example by phosphoiyl or sulfonyl substituents. When X is Se, R may also be a protein, peptide or amino acid, giving the possibility of linking one protein, peptide or amino acid to another protein, peptide or amino acid via a disulphide linkage. One preferred R group is farnesyl. Farnesylation is a natural post translational modification associated with many proteins, including the oncagenic protein Ras. The methods of the invention therefore allow prepation of farnesy lated proteins, peptides and amino acids. Also preferably, R is a carbohydrate moiety, optionally attached via a linker to the -S-X- group. The linker may contain 1 to 10 atoms between the carbohydrate moiety and the -S-X- group. For example, e lmker may be an alkylene group (for example a -(CH
2)t- group wherein t denotes 1 to 10), or an alkenylene group (for example a -(CH2)CH=CH- or

group). Preferred are compounds in which the -S -X- group is at the anomeric position of a saccharide residue or is attached to the anomeric carbon via a linker. Suitable carbohydrate moieties include monosaccharides, oligos accharides and polysaccharides, and include any carbohydrate moiety which is present in naturally occurring glycoproteins or in biological systems Preferred are optionally protected glycosyl or glycoside derivatives, for example optionally-protected glucosyl, glucoside, galactosyl or galactoside derivatives. Glycosyl and glycoside groups include both α and β groups. Suitable carbohydrate moieties include glucose, galactose, fucose, GlcNAc, GalNAc, sialic acid, and mannose, and oligos accharides or polysaccharides comprising at least one glucose, galactose, fucose, GlcNAc, GalNAc, sialic acid, and/or mannose residue. Any functional groups in the carbohydrate moiety may optionally be protected using protecting groups known in the art (see for example Greene et al, "Protecting groups in organic synthesis", 2nd Edition, Wiley, New York, 1991, the disclosure of which is hereby incorporated by reference). Suitable protecting groups for any -OH groups in the carbohydrate moiety include acetyl (Ac), benzyl (Bn),
pivolyl (piv), silyl (for example tert-butyl dimethylsilyl (TBDMSi) and tert- butyldiphenylsilyl (TMDPSi)), acetals, ketals, and methoxymethyl (MOM). Any protecting groups may be removed before or after attachment of the carbohydrate moiety to the amino acid, peptide or protein. Particularly preferred carbohydrate moieties include Glc(Ac) β -, Glc(Bn)
4β-,
Gal(Ac)4β-, Gal(Bn) β-, Glc(Ac)4α(l,4)Glc(Ac)3α(l,4)Glc(Ac)4β-, β-Glc, β-Gal, α-Man ,α-Man(Ac) , Man(l,6)Manα-, Man(l-6)Man(l-3)Manα-, (Ac)4Man(l-6)(Ac)4Man(l-3)(AC)2Man -, -Et-β-Gal,-Et-β-Glc, Et- -Glc, -Et-α-Man, -Et-Lac, -β-Glc(Ac)2, -β-Glc(Ac)3, -Et-α-Glc(Ac)2, -Et-α-Glc(Ac)3, -Et-α-Glc(Ac)4, -Et-β-Glc(Ac)2, -Et-β-Glc(Ac)3, -Et-β-Glc(Ac)4, -Et-α-Man(Ac)3, -Et-α-Man(Ac)4, -Et-β-Gal(Ac)3, -Et-β-Gal(Ac) , -Et-Lac(Ac)5, -Et-Lac(Ac)6, -Et- Lac(Ac) , and their deprotected equivalents. Preferably, any saccharide units making up the carbohydrate moiety which are derived from naturally occurring sugars will each be in the naturally occurring enantiomeric form, which may be either the D-form (e.g. D -glucose or D-galactose), or the L-form (e.g. L-rhamnose or L -fucose). Any anomeric linkages may be - or β- linkages. The compound comprising a thiol group used in the first or second methods may be any organic compound which comprises at least one thiol group. The thiol group may be primary, secondary or tertiary. The compound may be aromatic or aliphatic. If more than one thiol group is present in the compound, a disulfide bond will potentially be formed at each such thiol group. Preferably, the compound is an amino acid, a peptide or a protein. As used herein, a peptide contains a minimum of two amino acid residues linked together via an amide bond. Any amino acid comprised in the protein, peptide or amino acid is preferably an α-amino acid. Any amino acid may be in the D- or L-form, preferably the L-form. The amino acid, peptide or protein may be any naturally -occurring amino acid, peptide or protein which comprises a thiol group, for example due to the presence of one or more cysteine residues. Alternatively, the amino acid, peptide or protein may be prepared by chemical modification of a precursor non-thiol containing amino acid, peptide or protein. Alternatively, a thiol containing peptide or protein may be prepared via site-directed mutagenesis to introduce a cysteine residue. Site-directed mutagenesis is a known technique in the art (see for example
WO00/01712 and J. Sambrook et al, Molecular Cloning: A Laboratory Manual, 3rd Edition, Cold Springs Harbour Laboratory Press, 2001, the disclosures of which are hereby incorporated by reference). Preferred proteins include enzymes, the selectivity of which may be modified by controlled glycosylation using the methods and reagents according to the invention, and therapeutic proteins. Other preferred proteins include serum albumins and other blood proteins, hormones, interferons, receptors, antibodies, interleukins and erythropoietin. It has been found that the compounds of formula I are normally thiol- selective, and hence that the presence of other functional groups in the thiol - contairring organic compound does not normally interfere with the reaction. However, any other functional groups may optionally be protected using any protecting groups known in the art which are stable under the reaction conditions. The disulfide bond forming reaction in the first or second method is generally carried out in the presence of a buffer at neutral or basic pH (about pH 7 to about 9.5), with slightly basic pHs being preferred (about pH 8 to about 9). Suitable buffers include HEPES, CHES, MES and Tris. If the t ol-contairring compound is a protein, peptide or amino acid, the pH should be such that little or no unwanted denaturation occurs during the reaction. Similarly, the reaction temperature should be selected to avoid any significant damage to any temperature sensitive compounds. For example, a reaction with a protein or peptide is preferably carried out at ambient temperature or below to avoid any denaturation. Aqueous or organic solvent systems may be used, with aqueous solvent systems being preferred for the reaction of proteins, amino acids or peptides to ensure their dissolution. The reaction is generally fairly quick, for example often taking less than 1 hour. In general, an excess of the compound of formula I will be used, for example 10-20 equivalents based on the thiol-contaim'ng compound. In contrast, reactions with glyco-MTS reagents often require the use of approximately 30 equivalents, adding to the cost of the reagents. It has been found that the compounds of formula I wherein R denotes a carbohydrate moiety, X denotes SO2 and R1 denotes phenyl are generally more stable to basic conditions than the corresponding glyco-MTS compounds. Any unreacted or excess compound of formula I may therefore often be recovered from the reaction for reuse, which is particularly advantageous when R denotes a
carbohydrate moiety as such compounds may be relatively expensive and/or time consuming to prepare. Furthermore, the phenyl thiosulfonate compounds of formula I are generally cheaper and easier to prepare than the corresponding MTS compounds. The compounds of formula I may be prepared by a number of different methods. Compounds wherein X denotes SO2 maybe prepared by reacting a compound of formula II:
M(SSO2R1)k II wherein: M denotes a metal, for example Li, Na, K, Cs, Ca, Mg, Zn, or Al, preferably Na or K; and k denotes 1, 2 or 3;
with a compound of formula 111:
R-L IE wherein: R is as defined for the compounds of formula I and L denotes a leaving group. Any leaving group L may be utilised as long as the resultant anion L
" does not unduly interfere with the reaction in any way, for example by reacting with the product. Preferred leaving groups L include halo and sulfonates such as toluenesulfonate (tosylate), methanesulfonate (mesylate) and trifluoromethane sulfonate (triflate), in particular chloro and bromo. Compounds of formula ILT are commercially available or may be prepared using methods known in the art, for example methods for the formation of halo- sugars in general and 1 -halo-sugars in particular. Preferably the compound of formula UI is a glycosyl halide. Examples of suitable compounds of formula HI based on glucose and galactose are shown generically below:
wherein: each R5 independently denotes H, a saccharide moiety, or a suitable protecting group for example Ac or Bn, preferably each R5 denotes H; one of R3 and R4 denotes H and the other denotes OH, O-protecting group or
O-saccharide moiety, preferably H or O -saccharide moiety; and t denotes 1 to 10, preferably 1 to 6, more preferably 2 or 3. The reaction may be carried out in any solvent-system in which the compound of fomiula III is soluble. Preferably, the compound of formula LI is also at least partially soluble in the solvent system. Suitable solvents include alkanols such as ethanol and methanol, NN-dimethylformamide (DMF) and acetonitrile, with acetonitrile being particularly preferred. The compounds of formula II may be prepared by reacting the corresponding sulfinite salt (formula VH) with sulfur, as shown in Scheme 4:
M(SO2R1)k + S → M(SSO2R1)k Nπ π
Scheme 4
Compounds of formula LT which are crystalline are preferred for ease of purification, especially on a large scale. Sulfinite salts of formula VH are available commercially (for example sodium benzenesulfinite) or may be prepared by methods known in the art (see for
example JP 61205249, andM. Uchino et al, Chemical & Pharmaceutical Bulletin, 1978, 26(6), 1837-45, the disclosures of which are hereby incorporated by reference). For example, the corresponding thiolate salt R S- may be p repared by deprotonation of the corresponding thiol compound R^H using a suitable base, for example methyl lithium. The thiolate salt may then be oxidised to the corresponding sulfinite salt using a suitable oxidising agent, for example 2-(phenylsulfonyl)-3-phenyloxaziridine (the "Davis reagent", Sandrinelli et al, Organic Letters (1999), 1(8), 1177-1180, the disclosure of which is hereby incorporated by reference). Alternatively, compounds of formula I in which X denotes SO2 may be prepared by reacting a disulfide of formula Nm with a sulfinite anion R1SO2_ in the presence of silver ions, as shown in Scheme 5:
.— s 1 1 J s P i + ' J τ\?
l 0
clnU2
" R— S — SO,— R
1 J ^
Scheme 5
Disulfide compounds of formula VIU are commercially available or may be prepared using methods known in the art. Compounds of formula I wherein X denotes Se may be formed by reaction of a compound of formula V:
R — SH V wherein R is as defined for the compounds of formula I, with a compound of formula Via or Nib:
R^eL2 R1Se(OH)2 Via VIb
wherein R1 is as defined for the compounds of formula I, and L2 denotes a leaving group, for example OH, Br, CI, CN, or I, preferably Br. The reaction may be carried out in anhydrous dichloiOmethane and then quenched by the addition of triethylamine. A preferred compound of formula lNa is PhSeBr and a preferred compound of formula VIb is PhSe(OH)2. The compounds of formula VI are commercially available (e.g. PhSeBr, PhSeCl, PhSeCΝ, 2-nitrophenyl selenocyanate) or may be prepared by methods known in the art. For example, MeSeBr may be prepared according to the method of Hope, Eric G; Kemmitt, Tim; andLevason, William, in Journal of the Chemical Society, Perkin Transactions 2: Physical Organic Chemistry (1972-1999) (1987), (4), 487-90, the disclosure of which is hereby incorporated by reference. Organic compounds containing at least one thiol group, including compounds of formula V, are commercially available or may be prepared using methods known in the art, for example methods for the preparation of thiol compounds in general, and thio-sugars in particular. For example, thio sugars may be prepared from the corresponding halo sugars by treatment of the halo sugar with thiourea to afford the corresponding isothiouronium salt (W. A. Bonner, J. E. Kahn, J. Am. Chem. Soc. 1951, 73) followed by mild hydrolysis with sodium metabisulfite to give the corresponding thiol. If necessary, suitable protecting groups may be used during the synthesis of any thio-sugars. When R in the compound of formula V denotes a carbohydrate moiety, the thiol group may be at any position in the moiety. Preferably, it is at the anomeric position of a saccharide or is attached to the anomeric carbon via a linker. Examples of suitable compounds of formula V based on glucose and galactose are shown generically below:
wherein: each R
5 independently denotes H, a saccharide moiety, or a suitable protecting group, for example Ac or Bn, preferably each R
5 denotes H; one of R
3 and R
4 denotes H and the other denotes OH, O-protecting group or O-saccharide moiety, preferably H or O -saccharide moiety; and r denotes 2 to 10, preferably 2 to 6, more preferably 2 or 3. Compounds of formula V are also suitable for use as the thiol containing compound in the fourth method of the invention. In the reaction of the compounds of formula V with the compounds of formula VI, any other functional groups in the compound of formula V may be unprotected, or may be protected by protecting groups known in the art. The conversion of the at least one thiol group in the protein, peptide or amino acid to a selenenylsulfide group according to the third or fourth method is highly selective. In addition, the reaction of the thiol containing organic compound with the selenenylsulfide group is highly site-selective. It is not therefore normally necessary for any other functional groups in the protein, peptide or amino acid or in the thiol containing organic compound to be protected whilst practising the methods of the invention. This can be highly advantageous, as it avoids the need for any subsequent deprotection steps to be carried out on the product. If the protein, peptide or amino acid comprises more than one thiol group, then each such thiol group will potentially be converted to the corresponding selenenylsulfide group. Each such selenenylsulfide group may then potentially be reacted with a thiol containing organic compound, leading to attachment of the
organic compound via a disulphide linkage to the protein, peptide or amino acid at multiple sites. The methods of the invention therefore provides a convenient method for the chemical modification of a protein, peptide or amino acid at multiple sites. In particular, the methods of the invention allows glycosylation of a protein, peptide or amino acid at multiple sites. Conversion of the thiol group in the protein, peptide or amino acid to a selenenylsulfide group in the third or fourth methods is conveniently carried out by reacting said protein, peptide or amino acid with a compound of formula Xa or Xb: R
2-Se-L
2 or R
2-Se(OH)
2 Xa Xb wherein: L denotes a leaving group, for example OH, Br, CN, CI or I, preferably Br; and R
2 denotes an optionally substituted alkyl group, an optionally substituted phenyl group, an optionally substituted benzyl group, an optionally substituted pyridyl group or an optionally substituted naphthyl group. A preferred R
2 group is phenyl, a preferred compound of formula Xa is PhSeBr and a preferred compound of formula Xb is PhSe(OH)
2. When R
2 denotes an optionally substituted moiety, suitable substituents include any substituents which do not interfere with the reaction with the thiol containing protein, peptide or amino acid, and preferably also do not interfere with any subsequent reaction of the protein peptide or amino acid , for example reaction with a thiol containing organic compound. Suitable substituents include -NO
2, -SO
3H, -CO
2H, -(CH
2CH
2O)
nH, and -(CH CH2θ)
nMe wherein n denotes 1-100, preferably 1-50, more preferably 1 -20, and still more preferably 1-10. The R
2 group may be independently substituted by 1-5, and preferably 1 or 2, substituents. The R
2 group may also optionally be attached to, or form part of, a solid support. For example, the compound offormulaXa or Xb may be derived from a resin such as a polystyrene resin, as shown below:
The compounds of formula Xa and Xb are commercially available or may be prepared by methods known in the art, as discussed previously for the compounds of formula Via and VIb. At least one mol equivalent of the compound of formula Xa or Xb per thiol group in the protein, peptide or amino acid should be used, to ensure conversion of each such thiol group to the corresponding selenenylsulfide group. The reaction is preferably carried out in an aqueous solvent (such as a mixture of water and acetonitrile) in the presence of a buffer (for example MES, pH 9.5). The pH and temperature of the reaction should be chosen such that undesirable denaturation of the protein or peptide is avoided. Preferably, the reaction is carried out at room temperature or below, at a slightly basic pH (e.g. about pH 8 to about pH 9.5). The organic compound containing a thiol group may be any organic compound which is suitable for linkage to a protein, peptide or amino acid, and in which the sulfur atom of the thiol group can act as a nucleophile to react with a selenenylsulfide group. There is no particular limitation on the nature of the organic compound. For example, the thiol group may be primary, secondary or tertiary. The compound may be aromatic or aliphatic. For example, the compound may be an alkyl, alkenyl (e.g. famesyl) or alkynyl thiol. Preferably, the compound only contains one thiol group. Suitable organic moieties for attachment to a protein, peptide or amino acid include any group which may be useful in modifying the physical or chemical properties of the protein, peptide or amino acid. Suitable moieties include labels (for example fluorescent labels) or groups to aid the stability, processing or solubility of the protein, peptide or amino acid. The organic compound may also be a second protein, peptide or amino acid, giving the possibility of linking one protein,
peptide or amino acid to another protein, peptide or amino acid via a disulphide linkage using the methods of the invention. Preferably, the organic compound containing at least one thiol group is a famesyl derivative, or is a carbohydrate moiety as previously defined, optionally attached via a linker to the thiol (-S -H) group. The linker may contain 1 to 10 atoms between the carbohydrate moiety and the -SH group. For example, the linker may be an alkylene group (for example a -(CH2).- group wherein t denotes 1 to 10), or an alkenylene group (for example a -(CH2)CH=CH- or -CH2CH2CH:=CH- group). Preferred are compounds in which the thiol group is at the anomeric position of a saccharide residue or is attached to the anomeric carbon via a linker. Any functional groups in the carbohydrate moiety may optionally be protected using protecting groups known in the art as discussed oreviously. Any protecting groups may be removed before or after attachment of the carbohydrate moiety to the amino acid, peptide or protein. Preferably, they are removed before reaction with the selenenylsulfide compound, to remove the need for any post- linkage deprotection steps. A further advantage of the glycosylation method of the invention is that it allows for the linkage of unprotected carbohydrate moieties to an amino acid, peptide or protein. The reaction of the selenenylsulfide group with the organic compound containing a thiol group according to the fourth method (i.e. the disulfide bond forming reaction) is generally carried out in the presence of a buffer at neutral or basic pH (e.g. about pH 7 to about pH 9.5), with slightly basic pHs being preferred (e.g. about pH 8 to about pH 9). Suitable buffers include HEPES, CHES, MES and Tris. The pH should be such that little or no unwanted denaturation of the protein or peptide occurs during the reaction. Similarly, the reaction temperature should be selected to avoid any significant damage to any temperature sensitive compounds. For example, a reaction with a protein or peptide is preferably carried out at ambient temperature or below to avoid any denaturation. Aqueous or organic solvent systems may be used, with aqueous solvent systems being preferred to ensure the dissolution of the protein, amino acid or peptide. Aqueous solvent systems are also preferred as they allow the use of unprotected carbohydrate compounds as the organic compound. The reaction is generally fairly quick, for example often taking less than 1 hour.
In general, an excess of the organic compound containing at least one thiol group will be used, for example 10-20 equivalents based on the protein, amino acid or peptide. However, as little as 1 mol equivalent may be used in some cases. Carbohydrate compounds may be expensive and time-consuming to obtain in large quantities. Therefore, when the organic compound containing at least one thiol group is a carbohydrate compound, for reasons of economy it is desirable to use the minimum possible number of equivalents. Prior art methods for protein glycosylation often require use of a very large excess of the carbohydrate compound, for example often of the order of 1000 equivalents (B. G. Davis, Curr. Opin. Biotedinol. 2003, 14, 379). The method of the invention therefore advantageously allows use of fewer equivalents of the glycosyl compound than the prior art methods. The invention will be further illustrated by the following non-limiting Examples.
General Experimental
Melting points were recorded on a Kofler hot block and are uncorrected. Proton nuclear magnetic resonance (5H) spectra 400 MHz spectra were assigned using COSY. Carbon nuclear magnetic resonance (δc) spectra were assigned using
HMQC. Multiplicities were assigned using DEPT sequence. All chemical shifts are quoted on the δ scale in ppm using residual solvent as the internal standard. Infrared spectra adsorption maxima were recorded in wavenumbers (cm" ) and classified as s (strong) and br (broad). Low resolution mass spectra were recorded using electrospray ionisation (ESI), or using chemical ionization (NH3 , CI) techniques as stated. High resolution mass spectra were recorded using chemical ionization (NH3, CI) techniques, or using electrospray ionization (NH3, CI) techniques, or using field ionisation (FI+) as stated. M/z values are reported in Daltons and are followed by their percentage abundance in parentheses. Optical rotations were measured on a polarimeter with a path length of 1 dm.
Concentrations are given in g/100 mL. Thin layer chromatography (tl.c) was carried out on Merck Kieselgel 60F254 pre- coated glassbacked plates. Visulation of the plates was achieved using a UV lamp (λm x = 254 or 365 nm), and/or ammonium molybdate (5% in 2M H2 SO ) or sulfuric
acid (5% in EtOH). Flash column chromatography was carried out using Sorbsil C60 40/60 silica. Dichloromethane (DCM) was distilled from calcium hydride. Acetone was distilled from anhydrous calcium sulfate. Remaining anhydrous solvents were purchased from Fluka. 'Petrol' refers to the fraction of petroleum ether boiling in the range 40-60°C.
Protein Mass spectrometry: Liquid chromatography/mass spectrometry was performed on a Micromass LCT (ESI-TOF-MS) coupled to a Waters Alliance 2790 HPLC using a Phenomenex Jupiter C5 column (150 x 2.1 mm x 5 μm). Water (solvent A) and acetonitrile (solvent B), each containing 0.5% formic acid, were used as the mobile phase at a flow rate of 0.2 ml min"1. The gradient was programmed as follows: 95% A (3 min isocratic) to 100 % B after 16 min then isocratic for 2 min. The electrospray source of the LCT was operated with a capillary voltage of 3 kV and a cone voltage of 30 V. Nitrogen was used as the nebuliser and desolvation gas at a total flow of 400 1 hr"1. Myoglobin (horse heart) was used as a calibration standard and to test the sensitivity of the system.
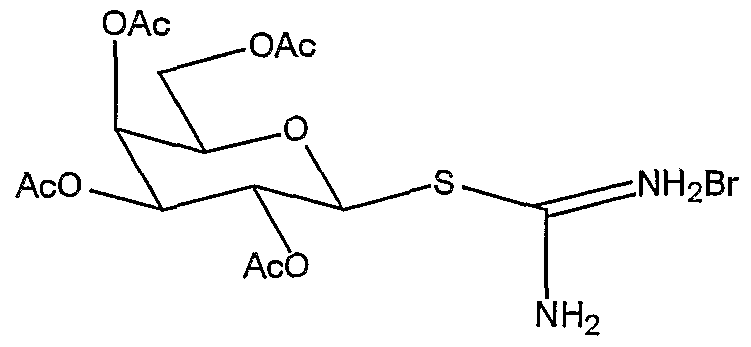
Example 1 : (2,3,4,6-Tetra-O-acetyl-β-D-glucopyranosyl)-l-isothiouronium bromide
2,3,4, 6-Tetra-O-acetyl-α-D-glucopyranosyl bromide (11.0 g, 26.4 mmol) and thiourea (3.10 g, 41.9 mmol) were dissolved in anhydrous acetone (30 mL) under argon and heated to 60°C. After 20 min a white solid precipitated. The precipitate was removed by filtration, the filtrate was returned to reflux, this process was repeated until the solid ceased to precipitate. The off-white crystals were combined and reciystallised from acetone/petrol to afford the title compound (11.4 g, 76%) as a white crystalline solid mp 194-196°C [Lit. 191°C (H. Beyer, U. Schultz, Chem. Ber. 1954, 87, 78)]; [α]D 25 -5.6 (c, 1.0 in H2O) [Lit. [α]D 25 -7.6 (c, 1.4 in H2O) (W. A. Bonner, J. E. Kahn, J Am Chem Soc, 1951 , 73, 2241)]; δH(400 MHz, DMSO-dδ) 1.97, 2.00, 2.02, 2.06 (12H, 4 x s, 4 x CH3 ), 4.06-4.25 (3H, m, H-5, H-6, H-6'),
5.07-5.12 (2H, m, H-2, H-4), 5.31 (IH, at, J9.5 Hz, H-3), 5.77 (IH, d, ,29.9 Hz, H-l), 9.13 (2H, brs, NH2), 9.29 ( 2H, brs, NH2).
Example 2: l-Thio-2.3.4.6-tetra-O-acetyl-β-D-glucopyranose
(2,3,4,6-Tetra-O-acetyl-β-D-glucopyranosyl)-l-isothiouronium bromide (9.0 g, 18.8 mmol) and Na2S2θs (4.93 g, 26.0 mmol) were added to a stirred mixture of DCM (150 mL) and water (70 mL). The mixture was heated to reflux under argon. After 1.5 h the reaction was cooled to room temperature (RT) and the phases were separated. The aqueous layer was re-extracted with DCM (3 x 50 mL). The combined organic layers were washed with water (50 mL), dried over MgSΟ4, filtered and the solvent removed in vacuo to afford the title compound (6.14 g, 90%) as a white solid, mp 112-114°C [Lit. 113-114°C (R. J. Ferrier, R. H. Furneaux, Carbohydr. Res. 1977, 57, 73)]; [α]D 24+6.3 (c, 1.2 in CHC13) [Lit. [α]D 20+5.0 (c, 1.1 in CHC13) (R. J. Ferrier, R. H. Furneaux, Carbohydr. Res. 1977, 57, 73)]; δH(400 MHz, CDC13) 1.99, 2.00, 2.05, 2.06 (12H, 4 x s, 4 CH3), 2.30 (IH, d, Jι,SH 10.2 Hz, SH), 3.71 (IH, ddd, J4,5 10.0 Hz, J5,62.4 Hz, J5β- 4.7 Hz, H-5), 4.10 (IH, dd, J6,& 12.3 Hz, H-6), 4.22 (IH, dd, H-6'), 4.53 (IH, at, J9.9 Hz, H-l), 4.95 (IH, at, J9.5 Hz, H-2), 5.08 (IH, at, J 9.8 Hz, H-4), 5.17 (IH, at, J 9.4 Hz, H-3).
Example 3 : (2 A.6-Tetra-O-acetyl-β-D-galactopyranosyl)-l-isothiouronium bromide
2,3,4,6-Tetra-O-acetyl-D-β-galactopyranosyl bromide (5.4 g, 13.0 mmol) and thiourea (1.25 g, 16.8 mmol) were dissolved in anhydrous acetone (40 mL) under argon and heated to 60°C. After 1 h the reaction was allowed to cool to room temperature and the resulting residue was filtered and recrystallised from acetone/petrol to afford the title compound (4.6 g, 70%, 2 steps) as a white crystalline solid mp 134-137°C [Lit. 170°C from isopropanol (W. A. Bonner, J. E. Kahn, JAm Chem Soc 1951, 73, 2241)];[α]
D 25+40.4 (c, 1.0 in H
2O) [Lit. [α]
D 25 +16.0 (c, 1.6 in EtOH, (W. A. Bonner, J. E. Kahn, JAm Chem Soc 1951 , 73, 2241)); δ
H(500 MHz, DMSO-dg) 1.96, 2.02, 2.09, 2.15 (12H, 4 x s, 4 x CH
3) 4.06-4.13 (2H, , H-6, H-6'), 4.45 (lH, t, J6.2 Hz, H-5), 5.12 (IH, at, J9.9 Hz, H-2), 5.24 (IH, dd, J
2β 10.0 Hz, J
3)4 3.6 Hz, H-3), 5.39 (IH, d, J
3)4 3.1 Hz, H-4), 5.71 (IH, d, J
lj2 10.2 Hz, H-l), 9.12, 9.36 (2 x 2H, 2 x brs, 2 x NH
2).
Example 4: l-Thio-2,3,4,6-tetra-0-acetyl-β-D-galactopyranose
(233,4,6-Tetra-0-acetyl-β-D-galactopvranosyl)-l-isothioxxronium bromide (4.4 g, 8.8 mmol) and Na2S2O5 (2.02 g, 10.6 mmol) were added to a stirred mixture of DCM (60 mL) and water (30 mL). The mixture was heated to reflux under argon. After 2.5 h the reaction was cooled to RT and the phases were separated. The aqueous layer was re-extracted with DCM (3 x 50 mL). The combined organic layers were washed with water (100 mL), brine (100 mL), dried over MgSO4, filtered and the solvent removed in vacuo to afford the title compound (2.65 g, 81%) as a white solid, mp 83-84°C [Lit. 86.5-88°C (J. Frgala, M. Cerny, J. Stanek, Collect. Czech. Chem. Commun. 1975, 40, 1411)]; [α]D 24+30.1 (c, 1.0 in CHCl3) [Lit. [α]D 19 +32.0 (c, 3.5 in CHC13) (J. Frgala, M. Cemy, J. Stanek, Collect. Czech. Chem.
Commun. 1975, 40, 1411)]; δH(400 MHz, CDC13) 1.99, 2.06, 2.10, 2.17 (12H, 4 x s, 4 x CH3), 2.38 (IH, d, J1;SH 10.3 Hz, SH), 3.95 (IH, dt, J4;5 1.2 Hz, J5>6 6.6 Hz, J5>& 6.6 Hz, H-5), 4.09-4.14 (2H, m, H-6, H-6'), 4.53 (IH, at, J9.9 Hz, H-l), 5.02
(IH, dd, J2β 10.1, J3j4 3.4 Hz, H-3), 5.19 (IH, at, J 10.0 Hz, H-2), 5.44 (IH, at, dd, J3)43J Hz, J4)5 1.2 Hz, H-4).
Example 5: (3,4,6-Tri-O-acetyl-2-acetamido-2-deoxy-β-D-glucopyranosyl)-l- isothiouronium chloride
3,4,6-Tri-0-acetyl-2-acetamido-2-deoxy-α-D-glucopyranosoyl chloride (3.0 g, 8.2 mmol) and thiourea (1.21 g, 14.6 mmol) were dissolved in anhydrous acetone (25 mL) under argon and heated to 60° C. After 2 h a white solid precipitated. The precipitate was removed by filtration, the filtrate was returned to reflux, this process was repeated until the solid ceased to precipitate. The off white crystals were combined and reciystallised from acetone/petrol to afford (the title compound (2.19 g, 61%) as a white crystalline solid mp 134-137°C [Lit. 179-181°C from EtOH (D. Horton, M. L. Wolfram, J. Org. Chem. 1962 , 27, 1794)]; [α]D 25 -25.2 (c, 1.0 in H2O) [Lit. [α]D 25 -29.3 (c, 1.1 in MeOH) (D. Horton, M. L. Wolfrom, J. Org. Chem. 1962, 27, 1794)]; δH (400 MHz, DMSO-dg) 1.80 (3H, s, NHCOCH3), 1.94, 1.98, 2.08 (9H, 3 x s, 3 CH3), 4.05 (IH, dd, JSfi 2.4 Hz, J6jS, 12.4 Hz, H-6), 4.17 (IH, dd, J5j6> 5.0 Hz, J6fi , 12.3 Hz, H-6'), 4.26 (IH, ddd, JA>5 10.2 Hz, J5fi 2.3 Hz, JSfi, A.7 Hz, H-5), 4.93 (IH, at, J9.9 Hz, H-4), 5.12 (IH, at, J 9.9 Hz, H-3), 5.73 (IH, d, j1;2 10.4 Hz, H-l), 8.48 (IH, d, J4.7 Hz, NHAc), 9.13 (2H, brs, NH2), 9.29 (2H, brs, NH2).
Example 6: l-T o-3,4,6-fri-0-acetyl-2-acetamido-2-deoxy-β-D-glucopyranose
(3,4,6-Tri-O-acetyl-2-acetarrήdo-2-deoxy-β-D-glucopyranosyl)-l-isothiouronium chloride (1.75 g, 39.8 mmol) andNa^Os (0.91 g, 4.8 mmol) were added to a stirred mixture of DCM (30 mL) and water (15 mL). The mixture was heated to reflux under argon. After 2 h the reaction was cooled to RT and the phases were separated. The aqueous layer was re-extracted with DCM (2 x 50 mL). The combined organic layers were washed with water (50 mL), brine (50 mL), dried over MgSO
4, filtered and the solvent removed in vacuo. Recrystallization from EtOAc/petrol afforded the title compound (1.00 g, 68%) as a white solid, mp 165- 167°C [Lit. 167-168°C (W. M. zu Reckendorf, W. A. Bonner, J. Org. Chem. 1961 , 26, 4596)]; [α]
D 25 -24.8 (c, 1.0 in CHC1
3) [Lit. [α]
D 25 -14.5 (c, 0.9 in CHC1
3) (W. M. zu Reckendorf, W. A. Bonner, J. Org. Chem. 1961 , 26, 4596)]; δ
H(400 MHz, CDC1
3) 1.99, 2.03, 2.05, 2.10 (12H, 4 x s, 4 x CH
3), 2.57 (IH, d, J
ljSH9.2 Hz, SH), 3.67 (IH, ddd, J
>5 9.7 Hz, J
5β 4.8 Hz, J
5 6, 2.3 Hz, H-5), 4.09-4.17 (2H, m, H-2, H-3), 4.24 (IH, dd, J
Sfi 4.8 Hz, J
6>6, 12.4 Hz, H-6), 4.59 (IH, at, J 9.8 Hz, H-l), 5.06-5.15 (2H, m, H-4, H-6'), 5.72 (IH, d, J9.2 Hz, NH).
Example 7: 1-Thio-β-D-galactopyranose
l-Thio-2,3,4,6-tetra-0-acetyl-β-D-galactopyτanose (3.00 g, 7.3 mmol) andNaOMe (40 mg, 0.73 mmol) were added to a stirred solution of MeOH (40 ml). After 2 h, tie. (EtOAc/petrol 1 : 1) indicated the formation of a product (Rf 0.0) with complete consumption of the starting material (Rf 0.5). The reaction was neutralised with the addition of Dowex®-50 ion exchange resin after which point the reaction was filtered and concentrated in vacuo. Recrystallization from MeOH/EtOAc afforded the title compound (1.41 g, 98%) as a white crystalline solid mp. 100-102° C; [α]D 22 +47.6 (c, 1.0 in MeOH; δH (400 MHz, CD3OD), 2.62 (IH, d, J1;SH 8.3 Hz, SH), 3.47 - 3.49 (2H, m, H-2, H-3), 3.57 (IH, at, J 5.9 Hz, H-5), 3.68 (IH, dd, J5fi 5.0 Hz, J6>6, 11.4 Hz, H-6), 3.75 (IH, dd, J5>6, 6.9 Hz, J6>6, 11.5 Hz, H-6'), 3.91 (IH, bs, H-4), 4.37 (IH, bd, J 7.1 Hz, H-l); δc (100 MHz, CD3OD), 61.6 (t, C-6), 69.6 (d, C-4),
74.4, 74.8 (2 x d, C-2, C-3), 80.1 (d, C-5), 81.4 (d, C-l); m/z (ES-) 196 (100%, M-H+); m/z HRMS (ES-) Calcd. for C6H12O5S (M-H+) 195.0327. Found 195.0323.
Example 8: l-Thio-2-acetamido-2-deoxy-β-D-glucopyranose
3,4,6-Tri-O-acetyl-2-acetylamino-2-deoxy-β-D-glucopyranosyl thiol (400 mg, 0.98 mmol) and sodium methoxide (18 mg, 0.3 mmol) were added to a stirred solution of methanol (10ml). After a 30 min period, tl.c. (ethyl acetate) indicated t he formation of a product (Rf 0.0) with complete consumption of the starting material (Rf 0.2). The reaction was neutralised with the addition of Dowex®-50 ion exchange resin after which point the reaction was filtered and concentrated in vacuo. Recrystallisation from methanol/ethyl acetate afforded the title product (230mg, 98%) as awhite crystalline solid; mp. 85-88°C [Lit. 86-88°C]18; [α]D 22 -10.4 (c, 1.0 in MeOH) [Lit. [α]D 25 +177.1 (c, 1.45 in CHC13)]18; H( 00 MHz, MeOH), 2.00 (3H, s, CH3), 3.27-3.37 (2H, m, H-4, H-5), 3.42 (IH, at J9.1 Hz, H-3), 3.64-3.73 (2H, m, H2, H-6), 3.87 (IH, dd, J5fi 2.1 Hz, J6β, 12.0 Hz, H-6'), 4.56 (IH, d, J1>2 10.0 Hz, H-l), 8.11 (lH, bd, JNHj2 9.1 Hz, NH).
Example 9: 1 ,2,3,6-tetra-0-acetyl-4-0-(2,3,6-tri-Ll-acetyl-4-0-(2,3,4,6-tetra-0- acetyl-α-O-glucopyranosyl)-α-D-glucopyranosyl)-D-glucopyranose
Sodium acetate (700 mg, 8.3 mmol) was added to acetic anhydride (50 mL) and heated to reflux, at which point maltotriose (3.00 g, 6.0 mmol) was added and stirred
vigorously. After 90 min, tic. (petro ethyl acetate, 1:2) indicated the formation of a product (Rf 0.3) with complete consumption of the starting material (Rf 0.0). The reaction was allowed to cool to RT and diluted with DCM (50 mL) and partitioned with water (100 mL). The phases were separated and the aqueous layer was re-extracted with DCM (2 x 50 mL). The combined organic layers were washed with sodium hydrogen carbonate (400 mL of a saturated aqueous solution) until pH 8 was obtained, brine (200 mL), dried (MgSO4), filtered and concentrated in vacuo to afford the title product as a mixture of anomers (α/ β, 2/11) as an amorphous white solid; for p compound: δiiC500 MHz, CDC13) 2.05, 2.07, 2.10, 2.14, 2.15, 2.19, 2.21, 2.27 (30H, 8 x s, 10 x OAc), 3.92 (IH, ddd, J4j5 9.5 Hz, J5>6 2.9 Hz, J6β 4.1 Hz,
H-5a), 3.95-4.01 (3H, m, H-4b, H-5b, H-5c), 4.05 (IH, at, J 9.1 Hz, H-4a), 4.09 (IH, dd, J5j6 2.5 Hz, J6J6, 12.7 Hz, H-6c), 4.21 (IH, dd, J5fi 3.4 Hz, J6fi, 12.6 Hz, H-6b), 4.29 (IH, dd, J5,6 3.4 Hz, J6fi, 12.4 Hz, H-6'c), 4.35 (IH, dd, J5>6 4.3 Hz, J6>6, 12.3 Hz, H-6a), 4.48-4.52 (2H, m, H-6'a, H-6'b), 4.78 (IH, dd, J1>2 4.1 Hz, J2j3 10.3 Hz, H-2b), 4.90 (IH, dd, J1)2 4.1 Hz, J2j3 10.6 Hz, H-2c), 5.01 (IH, dd, J 2 8.0 Hz, J2;3 9.0 Hz, H-2a), 5.11 (IH, at, 7 10.1 Hz, H-4c), 5.31 (IH, d, Jlj2 3.9 Hz, H-lb), 5.32-5.44 (3H, m, H-3a, H-3b, H-3c), 5.45 (IH, d, J 2 4.1 Hz, H-lc), 5.79 (IH, d, J1 2 8.2 Hz, H-la); for α compound selected data only: 5H (500 MHz, CDC13) 2.08, 2.09, 2.12, 2.18, 2.21, 2.23, 2.26 (30H, 8 x s, 10 x OAc), 5.07 (IH, at, J 9.9 Hz), 6.28 (IH, d, J1 2 3.8 Hz, H-la). Remaining signals lie in the following multiplet regions, 3.85-3.89, 3.90-3.98, 3.99-4.07, 4.15-4.18, 4.23-4.27, 4.29 -4.32, 4.43-4.49. 4.74-4.76, 4,84-4.87, 4.98-4.94, 5.25-5.54; m/z (ES+) 984 (MNH4 +, 30%), 989 (MNa+, 100%); m/z HRMS (ES+) Calcd. For C40H58O27N (MNH4 +) 984.3196 Found 984.3199.
Example 10: 2,3,6-Tri-0-acetyl-4-0-(2,3,6-tri-0-acetyl-4-O-(2,3,4,6-tetra-0-acetyl-
α-O-glucopyranosyl)-
α-D-glucopyranosyl)-
α-D-glucopyranosyl bromide
l,2,3,6-Tetra-O-acetyl-4-O-(2,3,6-tri-O-acetyl-4-O-(2,3,4,6-tetra-0-acetyl-
α-O- glucopyranosyl)-
α-D-glucopyranosyl)-D-glucopyranose (200 mg, 0.21 mmol) was dissolved in anhydrous DCM (5 mL). To this hydrogen bromide (33% in acetic acid, 2 mL) was added. The mixture was left under argon at RT. After a 30 min period, tic. (petrol: ethyl acetate, 1:2) indicated the formation of a product (R
f 0.6) with complete consumption of the starting material (R
f 0.3). The reaction mixture was partitioned between DCM (10 mL) and water (10 mL), and the aqueous layer re- extracted with DCM (3 x 10 mL). The combined organic layers were washed with sodium hydrogen carbonate (20 mL of a saturated aqueous solution) until pH 8 was obtained, brine (20 mL), dried (MgSΟ
4), filtered and concentrated in vacuo to afford the title product (203 mg, 98%) as a white foam;
+152.2 (c, 1.0 in CHC1
3); 5
H (400 MHz, CDC1
3) 2.03, 2.05, 2.06, 2.08, 2.10, 2.13, 2.18, 2.21 (30H, 10 x COCH
3), 3.93-3.99 (3H, m, H-4b, H-5a, H-5b), 4.05-4.10 (2H, m, H-4c, H-6a), 4.20 (IH, dd, J
5;6 1.8 Hz, J
6fi, 12.2 Hz, H-6b), 4.26-4.34 (2H, m, H-5c, H-6a'), 4.35 (IH, dd, J
5>6 3.5 Hz, J6t6, 12.7 Hz, H-6c), 4.52 (IH, dd, J5fi 0.6 Hz, J6>6, 12.2 Hz, H-6b'), 4.57 (IH, dd, J5;62.1 Hz, J6;6, 12.4 Hz, H-6c55), 4.74 (IH, dd, JU24.1 Hz, J2;39.9 Hz, H-2c), 4.78 (IH, dd, Jl>24.2 Hz, J2β 10.2 Hz, H-2b), 4.88 (IH, dd, JU24.0 Hz, J2;3 10.5 Hz, H-2a), 5.10 (IH, at, 9.7 Hz, H-4a), 5.32 (IH, d, jlj24.0 Hz, H-lb), 5.39 (IH, at, J 9.9 Hz, H-3q), 5.43-5.46 (IH, m, H-3b), 5,45 (IH, d, J1)23.8 Hz, H-la), 5.64 (IH, at, J9.5 Hz, H-3c), 6.53 (IH, d, J1;2 3.9 Hz, H-lc).
Example 11: l-Thio-2,3,6-Tri-0-acetyl-4-O-(2,3,6-tri-O-acetyl-4-0-(2,3,4,6-tetra- O-acetyl-
α-0-glucopyranosyl)-
α-D-glucopyranosyl)-β-D-glucopyranose
2,3,6-Tri-O-acetyl-4-( -(2,3,6-tri-O-acetyl-4-O-(2,3,4,6-tetra-O-acetyl-α-0- glucopyranosyl)-α-D-glucopyranosyl)- α-D-glucopyranosyl bromide (2.10 g, 2.10 mmol) was dissolved in anhydrous acetone (60 mL). To this anhydrous thiourea (315 mg, 4.2 mmol) was added and then heated to reflux under an atmosphere of argon. After a 6.5 h period, tic. (petrol: ethyl acetate, 1 :2) indicated the formation of a product (Rf 0.0) with complete consumption of the starting material (Rf 0.3). The reaction was concentrated in vacuo and titurated with DCM to remove the organics from the excess thiourea. The filtrate was concentrated in vacuo and the residue was purified by column flash chiOmatography (ethyl acetate/methanol, 9: 1) to afford the intermediate 2,3,6-tri-0-acetyl-4-O-(2,3,6-tri-O- acetyl-4-0-(2,3,4,6-tetra-O-acetyl-α-O-glucopyranosyl)-α-D-glucopyranosyl)-β-D- glucopyranosyl-1-isαthiouronium bromide (1.14g, 50%) which was carried on without characterisation. This intermediate (100 mg, 0.09 mmol) and Na2S2O5 (22 mg, 0.11 mmol) were added to a stiπ-ed mixture of DCM (30 mL) and water (15 mL). The mixture was heated to reflux under argon. After 2.5 h, tic. (petrol: ethyl acetate, 1:2) indicated the formation of a product (Rf 0.4) with complete consumption of the starting material (Rf 0.0), at which point the reaction was cooled to RT and the phases separated. The aqueous layer was re-extracted with DCM (2 x 20 mL). The combined organic layers were washed with brine (20 mL), dried (MgSO4), filtered and the solvent removed in vacuo to afford the title product (74 mg, 84%) as a white amorphous solid; [α]D 22 +99.5 (c, 1.0 in CHC13); δH (400 MHz, CDC13) 1.99, 2.00, 2.01, 2.02, 2.03, 2.05, 2.10, 2.15, 2.18 (30H, 9 x s,10 x COCH3), 3.72-3.76 (IH, m, H-5a), 3.90-4.00 (4H, m, H-4a, H-4b, H-5b, H-5c), 4.05 (IH, dd, J5>6 2.2 Hz, J6fi, 12.3 Hz, H-6c), 4.17 (IH, dd, Js>6 3.3 Hz, J6≠, 12.3 Hz, H-6b), 4.25 (IH, dd, J5fi 3.6 Hz, J6fi, 12.5 Hz, H-6c'), 4.30 (IH, JSfi 4.3 Hz, J6;6, 12.2 Hz, H-6c), 4.44 (IH, dd, J5j6 2.2 Hz, J6fi, 12.1 Hz, H-6a'), 4.46 (IH, dd, J5β 2.2 Hz, J6)612.2 Hz, H-6b'), 4.59 (IH, d, J1;2 9.7 Hz, H-la), 4.74 (IH, dd, J1;2
4.1 Hz, J2β 10.6 Hz, H-2b), 4.80 (IH, at, 79.0 Hz, H-2a), 4.85 (IH, dd, 71)2 4.1 Hz, 72j3 10.6 Hz, H-2c), 5.07 (IH, at, 79.9 Hz, H-4c), 5.25 (IH, at, 7 9.0 Hz, H-3a), 5.26 (IH, d, 7lj2 4.1 Hz, H-lb), 5.35 (IH, at, 7 10.0 Hz, H-3b), 5.37-5.41 (2H, m, H-lc, H-3c).
Example 12: l-Thioacetyl-2,3,6-tri-O-acetyl-4-O-(2,3,6-tri-O-acetyl-4-0-(2,3,4,6- tetra-O-acetyl-α-O-glucopyranosyl)-α-D-glucopvranosyl)-β-D-glucopyranose
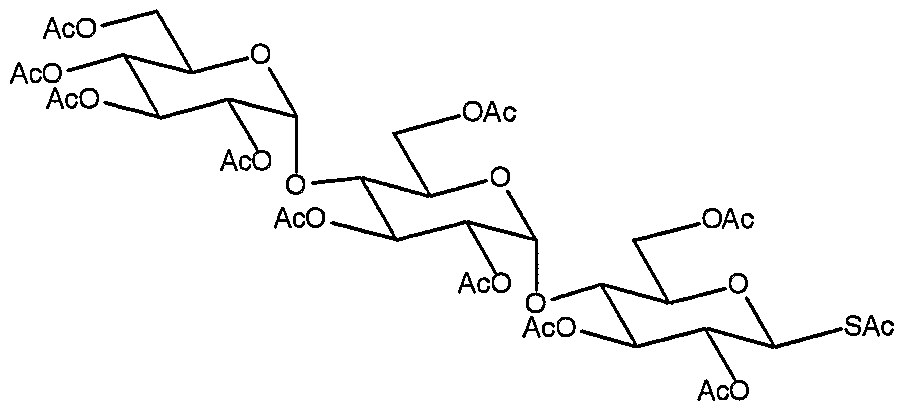
2,3,6-Tri-O-acetyl-4-O-(2,3,6-tri-0-acetyl-4-O-(2,3,4,6-tetra-O-acetyl-α-O- glucopyranosyl)-α-D-glucopyranosyl)- β -D-glucopyranosyl bromide (11.2 g, 11.6 mmol) and potassium thioacetate (3.96 g, 34.8 mmol) were suspended in anhydrous THF (40 ml) and heated to reflux under an inert atmosphere of argon. After 14 h, t.l.c. (petrol EtOAc, 1:2) indicated the formation of a major product (Rf 0.4) along with complete consumption of the starting material (Rf 0.45). The reaction was diluted with water (80 mL) and allowed to cool to RT. The phases were separated and the aqueous phase was re-extracted with DCM (3 x 40 mL). The combined organic layers were washed with sat. NaHCO3 (50 mL) until pH 8 was obtained, brine (50 mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (petrol/EtOAc, 1 :4) to afford the title compound (8.08 g, 71%) as a white foam; [α]D 2S +86.4 (c, 1.0 in CHCl 3); δH (400 MHz, CDC13) 2.01, 2.02, 2.05, 2.08, 2.11, 2.17 (27H, 6 x s, 9 x OAc), 2.40 (3H, s, SAc), 3.88 (IH, ddd, 74;59.8 Hz, J5>6 4.0 Hz, J5>6, 2.1 Hz, H-5a), 3.92-4.01 (4H, m, H-4a, H-4b, H-5b, H-5c), 4.07 (IH, dd, 75>6 2.4 Hz, J6β, 12.3 Hz, H-6c), 4.19 (IH, dd, 75j6 3.5 Hz, J6>6, 12.2 Hz, H-6b), 4.27 (IH, dd, 75)6. 3.8 Hz, 76;6-
12.3 Hz, H-6'c), 4.30 (IH, dd, J5>6 4.2 Hz, 76;6, 12.4 Hz, H-6a), 4.46 (IH, dd, Js>6, 2.6 Hz, J6>6, 12.3 Hz, H-6'b), 4.47 (IH, dd, J5>6, 2.2 Hz, J6β, 12.2 Hz, H-6'a), 4J6 (IH, dd, 7lj2 3.9 Hz, 72>3 10.0 Hz, H-2b), 4.87 (IH, dd, 71>2 3.8 Hz, 72;3 10.6 Hz,
H-2c), 5.99 (IH, dd, 71]2 10.3 Hz, 72,3 9.1 Hz, H-2a), 5.08 (IH, at, 79.9 Hz, H-4c), 5.27 (IH, d, 7lj2 4.0 Hz, H-lb), 5.31 (IH, d, 71)2 10.0 Hz, H-la), 5.33 -5.43 (4H, m, H-lc, H-3a, H-3b, H-3c); δc (125 MHz, CDC13) 20.7, 20.8, 20.9, 21.0, 21.1 (5 x q, 10 x COCH3, SCOCH3), 31.0 (q, SCOCH3) 61.9 (t, C-6c), 62.7 (t, C-6b), 63.3 (t, C-6a), 68.4 (d, C-4c), 69.0 (d, C-5b), 69.5 (d, C-5c), 69.8 (d, C-3c), 70.3 (d, C-2a), 70.5 (d, C-2c), 70.9 (d, C-2a), 72.1 (d, C-3b), 73.0 (d, C-4b), 74.1 (d, C-4a), 76.6 (d, C-3a), 76.9 (d, C-5a), 80.2 (d, C-la), 96.1 (d, C-lc), 96.4 (d, C-lb), 169.4, 169.6, 169.8, 169.9, 170.3, 170.5, 170.6 (7 x s, 10 x COCH3), 196.0 (s, SCOCH3); m/z (ES+) 1000 (MNH4 +, 60%), 1003 (MNa+, 100%).
Example 13: 1-Thio-β-D-maltotriose
l-TlLioacetyl-2,3,6-tri-0-acetyl-4-0-(2,3,6-tri-0-acetyl-4-0-(2,3,4,6-tetra-0-acetyl- α-0-glucopyranosyl)- α-D-glucopyranosyl)-l-thio-β-D-glucopyranose (600 mg, 0.6 mmol) andNaOAc (18 mg, 0.18 mmol) were added to a stirred solution of MeOH (10 ml). After 10 min, tic. (EtOAc/MeOH, 9: 1) indicated the formation of a product (Rf 0.0) with complete consumption of the starting material (Rf 0.9). The reaction was neutralised with the addition of Dowex®-50 ion exchange resin after which point the reaction was filtered and concentrated in vacuo to afford the title compound (305 mg, 98%) as an amorphous solid; [OJD 25 +123 (c, 1.0 in MeOH); H (400 MHz, D2O), 3.15 (IH, at, 79.2 Hz, H-2a), 3.26 (IH, at, 79.3 Hz), 3.41-3.82 (16H, m, H-2b, H-2c, H-3a, H-3b, H-3c, H-4a, H- 4b, H-4c, H-5a, H-5b, H-5c, H-6a, H-6b, H-6c, H-6'a, H-6'b, H-6'c), 4.42 (IH, d, 7 9.6 Hz, H-la), 5.23 (IH, d, 71)2 1.7 Hz, H-l), 5.24 (IH, d, 71)2 1.8 Hz, H-l); δc ( 00 MHz, D2O), 60.8, 70.0 (2 x t, C-6a, C-6b, C-6c), 69.6, 71.5, 71 .8, 72.1, 73.0, 73.2, 73.6, 76.0, 77.1, 77.6, 79.0 (11 x d, C-2a, C-2b, C-2c, C-3a, C-3b, C-3c, C-4a, C-4b, C-4c, C-5a, C-5b, C-5c),
80.2 (d, C-la), 99.8, 100.1 (2 x d, C-lb, C-lc); m/z (ES-) 519 (100%, M-H+); m/z HRMS (ES-) calcd. for C18H31O15S (M-H+) 519.1384. Found 519.1389.
Example 14: 1, 2,3,6-Tetra-Q-acetyl-4-( -(2,3,6-tri-C>-acetyl-4-0-(2,3,6-tri-O-acetyl- 4-O-(2,3,6-tri-0-acetyl-4-O-(2,3,6-tri-0-acetyl-4-O-(2,3,6-tri-0-acetyl-4-O-(2,3,4,6- tetra-O-acetyl-α-O-glucoρyranosyl)-α-D-glucopyranosyl)-α-D-glucopyranosyl)-α- D-glucopyranosyl)-α-D-glucopyranosyl)-α-D-glucopyranosyl)-D-glucopyranose
Sodium acetate (420 mg, 5.2 mmol) was added to acetic anhydride (30 mL) and heated to reflux, at which point maltoheptose (1.00 g, 0.86 mmol) was added and the reaction stirred vigorously. After 90 min t.l.c. (petrol:ethyl acetate, 1:3) indicated the formation of a product (Rf 0.3) with complete consumption of the stalling material (Rf 0.0). The reaction was allowed to cool to RT, diluted with DCM (50 mL) and partitioned with water (100 mL). The phases were separated and the aqueous layer was re-extracted with DCM (2 x 40 mL). The combined organic layers were washed with sodium hydrogen carbonate (200 mL of a saturated aqueous solution) until pH 8 was obtained, brine (100 mL), dried (MgSO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography (petrol: ethyl acetate, 1:3) to afford the title product as an amorphous white solid as a mixture of anomers (α/β, 15/85); δH(500 MHz, CDC13)2.02, 2.03, 2.04, 2.05, 2.06, 2.07, 2.08, 2.10, 2.13, 2.19, 2.22, 2.24 (66H, 12 x s, 22 x OAc), 3.89-4.14 (13H, m, H-4a, H-4b, H-4c, H-4d, H-4e, H-4f, H-5a, H-5b, H-5c, H-5d, H-5e, H-5f, H-5g), 4.25-4.34, 4.39 (IH, dd, 74.0 Hz, 712.3 Hz), 4.52-4.56 (13H, m, H-6a, H-6a\ H-6b, H-6b', H-6c, H -6c',
H-6d, H-6d', H-6e, H-6e', H-6f, H-6f , H-6d, H-6g'), 4.75 -4.79 (5H, m, H-2b, H-2c, H-2d, H-2e, H-2e, H-2f), 4.90 (IH, dd, 71)23.7 Hz, 72j3 10.5 Hz, H-2g), 5.00 (IH, at, 79.4 Hz, H-4g), 5.31-5.45 (13H, m, H-3a, H-3b, H-3c, H-3d, H-3e, H-3f, H-3g, H-lb, H-lc, H-ld, H-le, H-lf, H-lg), 5.19 (0.85H, d, 7lj2 8.1 Hz, H-la β), 6.28 (0.15H, d, 71 24.0 Hz, H-laα).
Example 15: 2,3,6-Tri-O-acetyl-4-O-(2,3,6-tri-0-acetyl-4-O-(2,3,6-tri-( -acetyl-4-O- (2,3,6-tri-O-acetyl-4-O-(2,3,6-tri-O-acetyl-4-O-(2,3,6-tri-0-acetyl-4-O-(2,3,4,6- tetra-O-acetyl-α-O-glucopyranosyl)-α-D-glucopyranosyl)-α-D-glucopyranosyl)-α- D-glucopyranosyl)-α-D-glucopyranosyι)- α-D-glucopyranosyl)-α-D- glucopyranosyl bromide
l,2,3,6-Tetra.-0-acetyl-4-0-(2,3,6-tri-0-acetyl-4-0-(2,3,6-tri-0-acetyl-4-0-(2,3,6- tri- -acetyl-4-0-(2,3,6-tri-0-acetyl-4-O-(2,3,6-tri-0-acetyl-4-0-(2,3,4,6-tetra-0- acetyl-α-O-glucopyranosyl)-α-D-glucopyranosyl)-α-D-glucopyranosyl)-α-D- glucopyranosyl)-α-D-glucopyranosyl)- α-D-glucopyranosyl)-D-glucopyranose (100 mg, 0.05 mmol) was dissolved in anhydrous DCM (5 mL). To this hydrogen bromide (33% in acetic acid, 0.5 mL) was added. The mixture was left stirring under an atmosphere of argon at RT. After a 40 min period, tic. (petrol: ethyl acetate, 1:3) indicated the formation of a product (Rf 0.7) with complete consumption of the starting material (Rf 0.3). The reaction mixture was partitioned between DCM (10 mL) and water (10 mL), and the aqueous layer re-extracted with DCM (3 x 10 mL). The combined organic layers were washed with sodium hydrogen carbonate (150 mL of a saturated aqueous solution) until pH 7 was obtained, brine
(20 mL), dried (MgSO4), filtered and concentrated in vacuo to afford the title product (98 mg, 96%) as a white foam; [α]D 22 +162.0 (c, 1.0 in CHC13); H (400 MHz, CDC13) 2.02, 2.03, 2.04, 2.06, 2.08, 2.10, 2.11, 2.14, 2.19, 2.23, 2.24, 2.25 (66H, 12 x s, 22 x OAc), 3.94-4.04 (12H, m, H-4b, H-4c, H-4d, H-4e, H-4f, H-5b, H-5c, H-5d, H-5e, H-5f, H-5g), 4.08 (IH, dd, 75;6 2.2 Hz, 76 6, 12.6 Hz, H-6), 4.19-4.33, 4.53-4.60 (12H, m, H-5a, H-6b, H-6b', H-6c, H-6c', H -6d, H-6d', H-6e, H-6e', H-6f, H-6f , H-6g, H-6g'), 4.12 (IH, at, 7 9.5 Hz, H-4a), 4.40 (IH, dd, 75;6 3.1 Hz, 76)6. 12.7 Hz, H-6a), 4.64 (IH, dd, J5β 2.3 Hz, J6>6, 12.5 Hz, H-6a'), 4.74 (IH, dd, 71;2 3.9 Hz, 72;3 9.7 Hz, H-2a), 4J5-4.97 (5H, m, H-2b, H-2c, H -2d, H-2e, H-2f), 4.89 (IH, d, 7lj2 4.0 Hz, 72;3 10.6 Hz, H-2g), 5.11 (IH, at, 79.9 Hz, H-4g), 5.32-5.47 (12H, m, H-lb, H-lc, H-ld, H-le, H-lf, H-lg, H -3b, H-3c, H-3d, H-3e, H-3f, H-3g), 5.65 (IH, at, 7 9.4 Hz, H-3a), 6.54 (IH, d, 7lj2 4.3 Hz, H-la).
Example 16: l-Thio-2,3,6-tri-O-acetyl-4-O-(2,3,6-tri-O-acetyl-4-O-(2,3,6-tri-O- acetyl-4-O-(2,3,6-tτi-O-acetyl-4-0-(2,3,6-tri-O-acetyl-4-0-(2,3,6-tri-O-acetyl-4-0- (2,3,4,6-tetra-O-acetyl-α-0-glucopyranosyl)-α-D-glucopyranosyl)-α-D- glucopyranosyl)-α-D-glucopyranosyl)-α-D-glucopyranosyl)- α-D-glucopyranosyl)- β-D-glucopyranose
2,3,6-Tri-O-acetyl-4-O-(2,3,6-rri-0-acetyl-4-O-(2,3,6-tri-O-acetyl-4-O-(2,3,6-tri-O- acetyl-4-O-(2,3,6-tri-O-acetyl-4-O-(2,3,6-tri-0-acetyl-4-O-(2,3,4,6-tetra-O-acetyl-α- 0-glucopyranosyl)-α-D-glucopvranosyl)-α-D-glucopyranosyl)-α-D- glucopyι-anosyl)-α-D-glucopyranosyl)-α-D-glucopyranosyl)-α-D-glucopyranosyl bromide (1.08 g, 0.5 mmol) and tetrabutylammonium iodide (19 mg, 0.05 mmol)
were dissolved in anhydrous acetone (50 mL). To this dried thiourea (52 mg, 0.7 mmol) was added and the reaction was then heated to reflux under an atmosphere of argon. After a 8 h period, t c. (petrol: ethyl acetate, 1:4) indicated the formation of a minor product (Rf 0.0) with complete consumption of the starting material (Rf 0.6) . The reaction was concentrated in vacuo and titurated with DCM to remove the organics from the excess thiourea. The filtrate was concentrated in vacuo and the residue was purified by column flash chromatography (ethyl acetate/methanol, 9: 1) to afford the intermediate 2,3,6-tri-O-acetyl-4-O-(2,3,6-tri-O- acetyl-4-O-(2,3,6-tri-O-acetyl-4-O-(2,3,6-tri-0-acetyl-4-0-(2,3,6-tri-O-acetyl-4-0- (2,3,6-tri-O-acetyl-4-O-(2,3,4,6-tetra-O-acetyl-α-O-glucopyranosyl)-α-D- glucopyranosyl)-α-D-glucopyranosyl)-α-D-glucopyranosyl)-α-D-glucopvranosyl)- α-D-glucopyranosyl)-β-D-glucopyranosyl-l-isothiouronium bromide (212 mg, 19%)) which was taken on further without characterisation. This intermediate (210 mg, 0.09 mmol) andNa2S2Ο5 (22 mg, 0.11 mmol) were added to a stirred mixture of DCM (10 mL) and water (5 mL) . The mixture was heated to reflux under argon. After 4.5 h, tic. (petrol: ethyl acetate, 1:2) indicated the formation of a product (Rf 0.2) with complete consumption of the starting material (Rf 0.0), at which point the reaction was cooled to RT and the phases separated. The aqueous layer was re-extracted with DCM (2 10 mL). The combined organic layers were washed with brine (20 mL), dried (MgSO4), filtered and the solvent removed in vacuo to afford the title product (185 mg, 90%) as a white amorphous solid; [α]D 24 +128.1 (c, 1.0 in CHCLJ; δH (500 MHz, CDC13), 2.00, 2.01, 2.02, 2.03, 2.04, 2.05, 2.07, 2.08, 2.12, 2.17, 2.19, 2.21, 2.22, 2.23 (66H, 14 x s, 22 x COCH3), 2.27 (IH, d, 71;SH 9.8 Hz, SH), 3.76 (IH, dat, 74;5 9.7 Hz, 73.5 Hz, H-5a), 3.92-4.08 (12H, m, H-4a, H-4b, H-4c, H-4d, H-4e, H-4f, H-5b, H-5c, H-5d, H-5e, H-5f, H-5g), 4.17-
4.36, 4.49-4.56 (12H, m, H-6b, H-6b', H-6c, H-6c', H-6d, H -6d', H-6e, H-6e', H-6f, H-6f , H-6g, H-6g'), 4.39 (IH, dd, 75;6 3.6 Hz, 76j6, 12.2 Hz, H-6a), 4.48 (IH, dd, 75j6 3.2 Hz,76j6, 12.3 Hz, H-6a), 4.62 (IH, at, 79.5 Hz, H-la), 4.73-4.78 (5H, m, H-2b, H-2c, H-2d, H-2e, H-2f), 4.82 (IH, at, 79.5 Hz, H-2a), 4.88 (IH, dd, 71>2 4.0 Hz, 72j3 10.4 Hz, H-2g), 5.09 (IH, at, 79.9 Hz, H-4g), 5.27 (IH, at, 79.1 Hz, H-3a), 5.30- 5.44 (12H, m, -lb, H-lc, H-ld, H-le, H-lf, H-lg, H-3b, H-3c, H-3d, H-3e, H-3f, H-3g).
Example 17: Preparation of SBLCysl56-S-SePh
Single site modification was investigated using a model-cysteine-containing protein, serine protease subtilisin Bacillus lentus mutant S156C (SBLCysl56). SBLCysl56 (10 mg) was dissolved in degassed aqueous buffer solution (1 mL, 70 mM CHES, 5 mM MES, 2 mM CaCl2, pH 9.5). PhSeBr (5 mg, 0.02 mmol) was dissolved in acetonitrile (200 μL), of which 150 ML (40 eq) was added to the protein solution and placed on an end-over-end rotator. After 30 min, the absence of free thiol was shown by Ellman's analysis (G. L. Ellman, K. D. Courtney, V. Andres, R. M. Featherstone, Biochem. Pharmacol. 1961, 7, 88). The reaction was placed on an end-over-end rotator for a further 30 min, at which point the reaction mixture was loaded onto a PD10 Sephadex® G25 column and eluted with 70 mM HEPES, 2 mM CaCl2, pH 7.0. The protein fraction was collected and dialysed (MWCO 12-14 KDa) against water, (1 x 4L for 1 h, 2 x 2L for 30 min), to afford SBLS156C-S-SePh; m/z (ES+) found 26864 calcd. 26870.
Example 18: Preparation of SSβGCys344Cys432-(S-SePh)2
Multiple site modifications were investigated using a mutant of the thermophilic β-glycosidase from the archeon Sulfolobus solfataricus containing two cysteine residues (SSβG-Cys344Cys432). SSβG-Cys344Cys432 (1 mg) was dissolved in aqueous buffer solution (1 mL, 70 mM CHES, 5 mM MES, 2 mM CaCl2, pH 9.5).
PhSeBr (2 mg, 0.02 mmol) was dissolved in acetonitrile (200 ML), of which 20 ML
(74 eq) was added to the protein solution and placed on an end-over-end rotator. After 1 h the reaction mixture was loaded onto a PD10 Sephadex® G25 column and eluted with (70 mM HEPES, 2 mM CaCl2, pH 7.0) to afford SSβGCys344Cys432-
(S-SePh)2; m/z (ES+) found 57700 calcd. 57697.
Example 19: Representative protein glycosylation with sugar thiols and reaction with other thiols
SBLCysl56-S-SePh (1 mg) was dissolved in aqueous buffer solution (1 mL, 70 mM CHES, 5 mM MES, 2 mM CaCl2, pH 9.5). The sugar thiol or other thiol was
dissolved in water and added to the protein solution in the stated quantities (see Table below for equivalents) and the mixture placed in an end-over-end rotator. After 1 h the reaction was analysed by mass spectrometry.
Results
1 Activated by reaction with phenyl selenium bromide to give the corresponding protein-S-Se-Ph or protein-(S-Se-Ph)2 compound prior to addition of the thiol. 2 Reacted with PMSF (phenylmethylsulfonyl fluoride) prior to glycosylation to prevent protein degradation due to proteolytic activity.
The results in the above Table demonstrate that the method of the invention provides high percentage conversion to the desired products using as httle as one equivalent of thiol compound. Furthermore, the results demonstrate that the method of the invention can be used for single and multiple site protein glycosylations. The three glycosylation sites in SBL-Cys 156 and SSβGCys344Cys432 are found in very varying protein structures and environments with different levels of exposure, illustrating the broad appUcability of the method of the invention. Example 20 : Representative protein glycosylation of SBLCys 156 using GlcGlcGlcGlcGlcGlcGlc-SH l-Thio-2,3,6-tri-O-acetyl-4-O-(2,3,6-tri-0-acetyl-4-0-(2,3,6-tri-0-acetyl-4-0- (2,3 ,6-tri-0-acetyl-4-0-(2,3 ,6-tri-O-acetyl-4-O-(2,3 ,6-tri-0-acetyl-4-0-(2,3 ,4,6- tetra-0-acetyl-α-0-glucopyranosyl)-α-D-glucopyranosyl)-α-D-glucopyranosyl)-α- D-glucopyranosyl)-α-D-glucopyranosyl)-α-D-glucopyranosyl)-β-D-glucopyranose (15 mg, 0.007 mmol) and sodium methoxide (2 mg, 0.007 mmol) were added to a stirred solution of MeOH (2 ml). After 2 h, tic. (ρetrol:EtOA c, 1:2) indicated the formation of a product (Rf 0.0) with the complete consumption of the starting material (Rf 0.2). The reaction was neutralised with the addition of Dowex®-50 ion exchange resin after which point the reaction was filtered and concentrat ed in vacuo. The crude 1-thio-β-D-maltoheptaose was taken up into water (5 mL) of which
300 μL (11 eq) was added to a solution of SBLCysl 56-S-SePh (1 mg) in 500 μL of aqueous buffer (70 mM CHES, 5 mM MES, 2 mM CaCl2, pH 9.5). The resulting solution was placed on an end-over-end rotator. After 1 h the reaction mixture was loaded onto a PD10 Sephadex® G25 column and eluted with 70 mM HEPES, 2 mM CaCl2, pH 7.0. The protein fraction was collected to afford
GlcGιcGLcGlcGlcGlcGlc-SBLCysl56; m/z (ES+) found 27878 calcd. 27881.
Example 21: Enzymatic extensions of SBLCysl 56 -S-GlcNAc
A. GlcNAc-SBLCysl56 (3 mg) was dissolved in 1 mL of aqueous buffer water. Phenylmethylsulfonyl fluoride (PMSF) was added (50 μL of a 100 mg/mL solution in acetonitrile; 500-fold excess). The reaction mixture was incubated at room temperature for 30 minutes and purified over a Sephadex® G-25 (PD-10) desalting column. The purity of the deactivated protein was assessed by ESI- mass spectrometry (found: 27100, calc. 27104). The protein fraction was lyophilized and re-dissolved in 1.0 mL of 0.1M sodium cacodylate buffer (pH 7.52). MnCl2.4H2O (3.2 mg; 16 μmol) and uridine diphosphate-galactose (UDP-galactose, 2.3 mg; 3.4 μmol, Kyowa Hakko; 30-fold excess) were added. Recombinant bovine β-l,4-galactosyltransferase from Spodoptera Frugiperda (EC 2.4.1.22, 100 mU, Calbiochem) was added and the reaction mixture was incubated at room temperature for 40 min to afford Galβl,4GlcNAc-S-SBL-Cysl 56 (ESI-MS, found 27265, calc. 27266).
B. GDP-fucose (3mg, Kyowa Hakku) and human α-l,3-fucosyltransferase from Spodoptera Frugiperda (EC 2.4.1.65, 10 mU, Calbiochem) were added and the reaction mixture was incubated overnight at room temperature to afford Lewis X-S- SBL-Cysl56 (ESI-MS, found 27410, calc. 27412).
This Example demonstrates that glycosylated proteins prepared according to the method of the invention may be further modified by reaction with suitable carbohydrate modifying enzymes, for example glycosyltransferases such as β-l,4-galactosyltransferase which selectively forms the Galβl,4GlcNAc linkage.
Example 22: Sodium phenylthiosulfonate (NaPTS)
Sodium benzenesulfinate (10 g, 61 mmol) and sulfur (1.95 g, 61 mmol) were dissolved in anhydrous pyridine (60 mL) to give a yellow solution. The reaction was stirred under argon and after 1 h gave a white suspension. The reaction was filtered and washed with anhydrous diethyl ether. Recrystallisation from anhydrous ethanol afforded the title product (10.5 g, 88%) as a white crystalline sohd; mp. 305-306° C [Lit. 287°C, Sato, R.; Goto, T.; Takikawa, Y.; Takizawa, S. Synthesis 1980, 615]; H (200 MHz, DMSO-dg) 7.28-7.76 (5H, m, Ar-H).
Example 23: 2,3,4,6-Tetra-O-acetyl-β-D-glucopyranosyl phenylthiosulfonate
2,3,4,6-Tetra-0-acetyl-α-D-glucopyranosyl bromide (207 mg, 0.5 mmol) was dissolved in anhydrous acetonitrile (5 mL). To this sodium phenylthiosulfonate (201 mg, 1 mmol) and tetrabutylammonium bromide (16 mg, 0.05 mmol) were added. The resulting mixture was stirred under argon at 70° C. After a 4.5 h period, thin layer chromatography (tic.) (petrol: ethyl acetate, 1: 1) indicated the formation of a product (Rf 0.5) with complete consumption of the starting material (Rf 0.3). The solution was concentrated in vacuo. The crude sohd was partitioned between dichloromethane (DCM, 20 mL) and water (20 mL), and the aqueous layer re-extracted with DCM (2 x 20 mL). The combined organics were washed with brine (20 mL), dried over MgSO4, filtered and concentrated in vacuo. The residue was purified by flash column chromatography (petrol: ethyl acetate, 1: 1) to afford the title product (225 mg, 88%) as a white crystalline sohd; mp 129-130° C; [α]D 25 +51.2 (c, 1.0 in CHC13); υ∞ax (KBr) 1754 (s, C=O), 1376 (s, C=C) cm"1. H (400 MHz, C6D6) 1.68, 1.72, 1.73, 1.75 (4 x 3H, 4 x s, 4 x OAc), 3.09 (IH, ddd, 74>5 10.2 Hz, J5 6 2.4 Hz, JSfi, 4.2 Hz, H-5), 3.83 (IH, dd, 75j6 2.4 Hz, J6fi. 12.1 Hz, H-6), 4.08 (IH, dd,
75;6- 4.2 Hz, 76j6, 12.6 Hz, H-6'), 5.17-5.23 (2H, m, H-2, H-4), 5.40 (IH, d, 71;2 10.2 Hz, H-l), 5.44 (IH, at, 79.4 Hz, H-3), 6.98-7.03 (3H, m, Ar-H), 7.90 -7.92 (2H, m, Ar-H). The structure of the product was further confirmed by single crystal X-ray diffraction.
Example 24: 2,3,4,6-Tetra-O-acetyl-β-D-galactopyranosyl phenylthiosulfonate
2,3,4,6-Tetra-O-acetyl-α-D-galactopyranosyl bromide (2.0 g, 5 mmol) was dissolved in anhydrous acetonitrile (80 mL). To this sodium phenylthiosulfonate (2.02 g, 10.3 mmol) and tetrabutylammonium bromide (160 mg, 0.5 mmol) were added. The resulting mixture was stirred under argon at 70° C. After a 5 h period, tic. (petrol: ethyl acetate, 1: 1) indicated the formation of a product (Rf 0.4) with complete consumption of the starting material (Rf 0.6). The solution was concentrated in vacuo. The crude oil was partitioned between DCM (50 mL) and water (50 mL), and the aqueous layer re-extracted with DCM (2 x 50 mL). The combined organics were washed with brine (100 mL), dried (MgSO4), filtered and concentrated in vacuo. The residue was purified by flash column chromatography (petrol: ethyl acetate, 2: 1) to afford the title product (1.7 g, 65%, 2 steps) as a white crystalline sohd; mp 53-54°C;[α]D 27+24.2 (c, 1.0 in CHC13); δH (400 MHz, CDC13) 1.98, 2.03, 2.06, 2.11 (4 x 3H, 4 x s, 4 x OAc), 3.85 (IH, dd, 75)6 8.8 Hz, 76j6> 14.0 Hz, H-6), 3.95-4.00 (2H, m, H-5, H-6), 5.11 (IH, dd, 72j3 9.7 Hz, 73>4 3.3 Hz, H-3), 5.23 (IH, at, 710.3 Hz, H-2), 5.25 (IH, d, 71>2 10.2 Hz, H-l), 5.43 (IH, dd, 73)4 3.6 Hz, 74)5 1.0 Hz, H-4), 7.54-7.68 (3H, m, Ar-H), 7.93-7.97 (2H, m, Ar-H).
Example 25: Ethyl 2,3,4,6-tetra-O-acetyl-l-dithio-β-D-glucopyranosyl disulfide
Method 1: 2,3,4,6-Tetra-O-acetyl-β-D-glucopyranosyl phenylthiosulfonate (100 mg, 0.2 mmol) and triethylamine (0.03 mL, 0.2 mmol) were dissolved in anhydrous DCM (10 mL) and stirred at room temperature (RT) under an atmosphere of argon. A solution of ethane thiol (0.016 mL, 0.2 mmol) in anhydrous DCM (10 mL) was slowly added dropwise via a syringe pump over a 30 min period. After a 40 min period, tic. (petrol: ethyl acetate, 1: 1) indicated the formation of a major product (Rf 0.5) along with complete consumption of the starting material (Rf 0.3). The solution was concentrated in vacuo. The residue was purified by flash column chromatography (petrol: ethyl acetate, 1 : 1) to afford the title product (70 mg, 82%) as a white crystalline sohd; mp 95-96°C [Lit. 100-102°C, (Davis, B. G; Ward, S. I; Rendle, P. M. Chem. Commun. 2001, 189)]; [α]D 22 -164.9 (c, 0.2 in CHC13) [Lit. [α]D 24 -178.0 (c, 1.0 in MeOH) (Davis, B. G; Ward, S. X; Rendle, P. M. Chem. Commun. 2001, 189)]; δH(400 MHz, CDC13) 1.30 (IH, t, 77.4 Hz, CH3), 2.00, 2.02, 2.03, 2.06 (4 x 3H, 4 x s, 4 x CH3), 2.79 (2H, dq, 7CH3.H 7.5 Hz, 7HH 2.7 Hz), 3.73 (IH, ddd, 74)5 10.2 Hz, J5>6 2.5 Hz, JSfi, 4.8 Hz, H-5), 4.14 (IH, dd, J5>6 2.4 Hz, 76;6, 12.4 Hz, H-6), 4.22 (IH, dd, 75;6, 4.7 Hz, J6fi, 12.4 Hz, H-6'), 4.52 (IH, d, 7lj2 9.8 Hz, H-l), 5.10 (IH, at, 79.8 Hz, H-4), 5.21-5.26 (2H, m, H-2, H-3).
Method 2: Phenyl 2,3,4,6-tetra-O-acetyl-l-selenenylsulfide-D-β-glucopyranoside (75 mg, 0.15 mmol) and triethylamine (30 μL, 0.15 mmol) were dissolved in freshly distilled DCM (10 mL). The solution was stirred at RT under an atmosphere of argon. A solution of ethanethiol (11 μL, 0.15 mmol) in anhydrous DCM (10 mL) was added dropwise over 2.5 h. After 3 h, tic. (petrol: EtOAc, 1: 1) indicated the formation of a major product (Rf 0.5) along with complete consumption of the starting material (Rf 0.5). The solution was concentrated in vacuo. The residue was purified by flash column chromatography (petrol: EtOAc, 5:3) to afford the title product (50 mg, 82%) as a white crystalline sohd.
Example 26: Ethyl 2,3,4,6-tetra-O-acetyl-l-dithio-β-D-galactopyranosyl disulfide
Method 1: 2,3,4,6-Tetra-O-acetyl-β-D-galactopyranosyl phenylthiosulfonate (100 mg, 0.2 mmol) and triethylamine (0.03 mL, 0.2 mmol) were dissolved in anhydrous DCM (10 mL) and stirred at RT under an atmosphere of argon. A solution of ethane thiol (0.016 mL, 0.2 mmol) in anhydrous DCM (10 mL) was slowly added dropwise via a syringe pump over a 30 min period. After a 40 min period, tic. (petrol: ethyl acetate, 1 : 1) indicated the formation of a major product (Rf 0.4) along with complete consumption of the starting material (Rf 0.3). The solution was concentrated in vacuo. The residue was purified by flash column chromatography (petrol: ethyl acetate, 1: 1) to afford the title product (78 mg, 91%) as a white crystaUine sohd; mp 65-66°C; [α]D 25 -52.1 (c, 1.4 in CHC13); υmax (KBr) 1746 (s, C=O) cm"1; H( 00 MHz, CDC13) 1.30 (IH, t, 77.4 Hz, CH3), 1.95, 2.01, 2.02, 2.13 (4 x 3H, 4 x s, 4 x CH3), 2.79 (2H, dq, 7CH3-H 7.2 Hz, 7HH 1.7 Hz), 3.94 (IH, td, 74j5 0.9 Hz, 75;6 6.3 Hz, 75;6, 7.0 Hz, H-5), 4.06 (IH, dd, 75>6 6.3 Hz, J6fi, 11.3 Hz, H-6), 4.12 (IH, dd, Jsfi , 7.0 Hz, J6fi. 1 1.2 Hz, H-6'). 4-51 (1 H > d, 7 9.9 Hz, H-l), 5.05 (IH, dd, 72;3 9.9 Hz, 73;4 3.6 Hz, H-3), 5.35-5.40 (2H, m, H-2, H-4).
Method 2: Phenyl 2,3,4,6-tetra-O-acetyl-l-selenenylsulfide-D-β-galactopyranoside (75 mg, 0.15 mmol) and triethylamine (30 μL, 0.15 mmol) were dissolved in freshly distilled DCM (10 mL). The solution was stirred at RT under an atmosphere of argon. A solution of ethanethiol (11 μL, 0.15 mmol) in anhydrous DCM (10 mL) was added dropwise over a 2.5 h. After 3 h, tic. (petrol: EtOAc, 1: 1) indicated the formation of a major product (Rf 0.5) along with complete consumption of the starting material (Rf 0.5). The solution was concentrated in vacuo. The residue was purified by flash column chromatography (petrol: EtOAc, 5:3) to afford the title compound (50 mg, 82%) as a white crystalline sohd.
Example 27: Ethyl 3,4,6-tri-O-acetyl-2-acetamido-2-deoxy-β-D-glucopyranosyl disulfide
Phenyl 3,4,6-tri-O-acetyl-2-acetamido-2-deoxy-l-selenenylsulfide-D-β- glucopyranoside (100 mg, 0.19 mmol) and triethylamine (0.03 mL, 0.19 mmol) were dissolved in freshly distilled DCM (20 mL). The solution was stirred at RT under argon. A solution of ethanethiol (0.014 mL, 0.19 mmol) in anhydrous DCM (10 mL) was added dropwise over 1 h. After 3 h, tic. (EtOAc) indicated the formation of a major product (R
f 0.4) along with complete consumption of the starting material (R
f 0.5). The solution was concentrated in vacuo. The residue was purified by flash column chromatography (EtOAc) to afford the title product.. (75 mg, 93%) as a white amorphous sohd.
(c, 2.5 in CHCl
3); δ
H(400 MHz, CDC1
3), 1.32 (3H, d, 7
CH;CH3 6.6 Hz, CHCH
3), 1.96, 2.04, 2.05, 2.08 (12H, 4 x s, 4 x COCH
3), 2.82 (2H, q, 77.4 Hz, CH
2), 3.75 (IH, ddd, 7
4;5 10.1 Hz, 7
5;6 2.5 Hz, J
5β, 4.7 Hz, H-5), 4.12-4.25 (3H, m, H-2, H-6, H-6'), 4J3 (IH, at, 7
1>2 10.4 Hz, H-l), 5.10 (IH, at, 79.8 Hz, H-4), 5.30 (IH, at, 79.9 Hz, H-3), 5.70 (IH, d, 7
NH)2 9.1 Hz, NH).
Example 28: tø-N-Acetyl-L-cysteinyl-L-serine methylester
bw-L-Cysteinyl-L-serine methylester (100 mg, 0.23 mmol) was dissolved in methanol (5 mL). To this solution acetic anhydride (0.09 mL, 0.92 mmol) and pyridine (0.075 mL, 0.92 mmol) were added. After a 15 min period, tic. (ethyl acetate:methanol 5: 1) indicated the formation of a major product (Rf 0.5) along with complete consumption of the starting material (Rf 0.1). The reaction was concentrated in vacuo. The residue was purified by flash column chromatography (ethyl acetate:methanol 5:1) to afford the title product (60 mg, 50%) as a white
crystalline sohd; mp 145-147°C; [α]D 25 -33.4 (c, 1.0 in CHC13); H(400 MHz, CDC13) 2.04 (3H, s, COCH3), 2.96 (IH, dd, 7CH,H 13 9 Hz, 7CH αH 4.7 Hz, CysCHH), 3.23 (IH, dd, 7CHjH 13.9 Hz, 7CHjαH 4.7 Hz, CysCHH), 3.76 (3H, s, OMe), 3.83 (IH, dd, 7CHjH 11 -4 Hz, 7CH;αH 4.1 Hz, SerCHH), 3.93 (IH, dd, 7CH)H 11.3 Hz, 7CH,αH 4.9 Hz, SerCHH), 4.55 (IH, t, 7 4.3 Hz, αHSer), 4.87 (IH, t, 74.8, αHCys).
Example 29: N-Acetyl-L-cysteinyl-L-serine methylester
bώ-N-Acetyl-L-cysteinyl-L-serine methylester (1.92 g, 3.96 mmol) was dissolved in wet chloroform (100 mL) and methanol (10 mL) and stirred. To this stirred solution tributylphosphine (1.1 mL, 4.36 mmol) was added. After a 2 h period, tic. (ethyl acetate:methanol 10: 1) indicated the formation of a product (Rf 0.6) along with complete consumption of the starting material (Rf 0.3). The reaction was concentrated in vacuo. Recrystallisation from ethyl acetate/methanol afforded the title product (1.77 g, 93%) as awhite crystaUine solid; mp 127-128° C; [α]D 25 -32.0 (c, 1.0 in MeOH); H(400 MHz, CDC13) 1.89 (IH, at, 78.9 Hz, SH), 2.06 (3H, s, COCH3), 2.84-2.93 (IH, m, CysCHH), 2.97-3.04 (IH, m, CysCHH), 3.79 (3H, s, OMe), 3 91 (IH, dd, CHjH 11.4 Hz, 7CH)αH 3.1 Hz, SerCHH), 4.03 (IH, dd, 7CHjH 11.7 Hz, 7CHjαH 4.2 Hz, SerCHH), 4.61 -4.65 (IH, m, αHSer), 4.71-4.76 (IH, m, αHCys), 6.93 (IH, d, 7αHjΝH 7.8 Hz, NHCys), 7.73 (IH, d, 7αH;NH 7.4 Hz, NHSer).
Example 30: N-Acetyl-L-cysteine (2,3,4,6-tetra-O-acetyl-l-dithio-β-D- glucopyranosyl disulfide)-L-serine methylester
2,3,4,6-Tetra-O-acetyl-β-D-glucopyranosyl phenylthiosulfonate (61 mg, 0.12 mmol) was dissolved in anhydrous DCM (5 mL) and stirred at RT under an atmosphere of argon. To this N-acetyl-L-cysteine-L-serine methylester (32 mg, 0.12 mmol) and triethylamine (0.015 mL, 0.11 mmol) in anhydrous DCM (10 mL) and anhydrous methanol (0.5 mL) were slowly added dropwise via a syringe pump over a 4 h period. After a 5 h period, tic. (ethyl acetate:methanol, 10: 1) indicated the formation of a major product (R 0.5) along with complete consumption of the starting material (R
f 0.3, (tic system (petrol: ethyl acetate, 1: 1)). The solution was concentrated in vacuo. The residue was purified by flash column chromatography (ethyl acetate:methanol, 10: 1) to afford the title product (75 mg, 99%) as a white crystaUine sohd; mp 126-128°C [Lit. 125-128°C (Davis, B. G; Ward, S. J.; Rendle, P. M. Chem. Commun. 2001, 189)]; [
α]
D 25 -47.9 (c, 0.7 in CHCl
3) [Lit. [
α]
D 24 -
178-
0 (c, 1.0 in MeOH) (Davis, B. G; Ward, S. X; Rendle, P. M. Chem. Commun. 2001 , 189)];
H(400 MHz, CDC1
3) 2.03, 2.06, 2.07, 2.11 (5 x 3H, 4 x s, 5 x CH
3), 3.05 (IH, dd, 7
CH;H 13.9 Hz, 7
CH)t H 8.8 Hz, CysCHH), 3.28 (IH, dd, 7
CHjH 13.9 Hz, 7
CH;αH 4.8 Hz, CysCHH), 3.80 (3H, s, OMe), 3.89 (IH, ddd, 7
4)5 10.0 Hz, 7
5)62.2 Hz, 7
5;6, 4.1 Hz, H-5), 3.94 (IH, dd, 7
CHjH 11.1 Hz, 7
CHjαH 3.0 Hz, SerCHH), 4.00 (IH, dd, 7
CHjH 13.8 Hz, 7
CH)(χH 3.7 Hz, SerCHH), 4.23 (IH, dd, 7
5;6 4.2 Hz, 7
6>6. 12.4 Hz, H-6), 4.38 (IH, dd, 7
5j6, 2.0 Hz, 7
6;6, 12.5 Hz, H-6'), 4.62-4,65 (IH, m,
αHSer), 4.64 (IH, d, 7
1)2 9.5 Hz, H-l), 4.90-4.94 (IH, m,
αHCys), 5.18 (IH, at, 7 10.1 Hz, H-4), 5.24-5.29 (2H, m, H-2, H-3), 6.94 (IH, d, 7
ΝH,H 7-
9 Hz, NHAc), 7.52 (IH, d, 7
NHjH 7.6 Hz, NHSer).
Example 31: N-Acetyl-L-cysteine (2,3,4,6-tetra-O-acetyl-l-dithio-β-D- galactopyranosyl disulfide)-L-serine methylester
2,3,4,6-Tetra-O-acetyl-β-D-galactopyranosyl phenylthiosulfonate (50 mg, 0.1 mmol) was dissolved in anhydrous DCM (5 mL) and stirred at RT under an atmosphere of argon. A solution of N-acetyl-L-cysteine-L-serine methylester (31 mg, 0.12 mmol) and triethylamine (0.015 mL, O.l lmmol) in anhydrous DCM (10 mL) and anhydrous methanol (0.5 mL) was slowly added dropwise via a syringe pump over a 2 h period. After a 2 h period, tic. (ethyl acetate:methanol, 10: 1) indicated the formation of a major product (R
f 0.5) along with complete consumption of the starting material (R
f 0.5, tic system petrol: ethyl acetate, 1:1). The solution was concentrated in vacuo. The residue was purified by flash column chromatography (ethyl acetate:methanol, 10: 1) to afford the title product (59 mg, 95%) as a white amorphous sohd; [
α]
D 25 -48.8 (c, 0.25 in CHC1
3);
H( 00 MHz, CDC1
3) 1.99, 2.04, 2.05, 2.08, 2.18 (5 x 3H, 4 x s, 5 x CH
3), 2.80 (IH, bs, OH), 2.99 (IH, dd, 7
CH;H 14.1 Hz, 7
CH)(χH 9.2 Hz, CysCHH), 3.32, 3.77 (3H, s, OMe), 3.92 (IH, dd, 7
CH;H l lJ Hz, 7
CH,αH 3.0 Hz, SerCHH), 4.01 (IH, dd, 7
CHjH 11.7 Hz, 7
CH)αH 3.7 Hz, SerCHH), 4.06-4.14 (2H, m, H-5, H-6), 4.20-4.26 (IH, m, H-6'), 4.61 -4.63 (IH, m,
αHSer), 4.65 (IH, d, 7
1)29.8 Hz, H-l), 4.88-4.93 (IH, m,
αHCys), 5.11 (IH, dd, 7
2;3 9.8 Hz, 7
3>4 3.3 Hz, H-3), 5.42-5.47 (2H, m, H-2, H-4), 6.68 (IH, d, 7
ΝHjH 7.8 Hz, NHAc), 7.28 (IH, d, 7
NHjH 8.1 Hz, NHSer).
Example 32: 2,3 ,4,6-Tetra-0-benzyl-α-D-glucopyranosyl bromide
2,3,4,6-Tetra-O-benzyl-D-glucopyranose (1.0 g, 1.9 mmol) was dissolved in anhydrous DCM (6 mL) and anhydrous DMF (0.4 mL) under argon. The resulting solution was stirred at 0°C. Oxalyl bromide (4 mL, 2M in DCM, 24 mmol) was added dropwise over a 5 min period . The reaction was stirred at RT. After a 40 min period, tic. (petrol: ethyl acetate, 2: 1) indicated the formation of a major product (Rf 0.7). The reaction was cooled to 0°C and quenched with ice cold water (30 mL) added over a 5 min period. The reaction was partitioned between DCM (20 mL) and water. The aqueous layer was re-extracted with DCM (3 x 20 mL), the combined
organic layers were washed with brine (40 mL), dried (MgSO4), filtered and concentrated in vacuo to afford the title product (1.10 g, 95%) as a crude yellow oU; δH(400 MHz, CDC13), 3.57 (IH, dd, 7 3.5 Hz, 72;3 9.1 Hz, H-2), 3.68 (IH, dd, J5,6 2.1 Hz, 76,6' 11.0 Hz, H-6), 3.79-3.84 (2H, m, H-4, H-6'), 4.07 (IH, at, 79.1 Hz, H- 3), 4.07-4.11 (IH, m, H-5), 4.47-4.62 (3H, m, PhCHs), 4.74 (s, 2H, PI1CH2), 4.84- 4.89 (2H, m, PhCHb), 5.10 (IH, d, 711.1 Hz, PhCIJa), 6.46 (IH, d, H-l), 7.15-7.41 (20H, m, Ar-H).
Example 33: 2.3.4.6-Tetra-O-benzyl-β-D-glucopyranosyl phenylthiosulfonate
2,3,4,6-Tetra-O-benzyl-D-α-glucopyranosyl bromide (3.55 g, 5.88 mmol) and sodium phenylthiosulfonate (4.76 g, 24.3 mmol) were dissolved in anhydrous 1,4 dioxane (90 mL). The reaction was heated to 70°C under argon. After 20 h, t.l.c. (petrokethyl acetate, 2:1) indicated the formation of a major product (Rf 0.6) with complete consumption of the starting material (Rf 0.7). The reaction was cooled to RT and filtered, the precipitate was washed with petrol/ethyl acetate and the filtrate concentrated in vacuo. The residue was purified by flash column chromatography (petrol:ethyl acetate, 4 :1) to afford 2,3,4, 6-tetra-O-benzyl-D-glucopyranosyl phenylthiosulfonate (3.18 g, 78%) as a white viscous gum as a mixture of α,β compounds in a β :α ratio of 3 : 1. Selective re-crystaUisation from ethyl acetate/petrol afforded pure 2,3,4,6-tetra-O-benzyl-β-D-glucopyranosyl phenylthiosulfonate as a white crystaUine solid; m.p. 106-108°C; [α]D 22+21.4 (c, 0.35 in CHC13); δH(500 MHz, C6D6) 3.21 (IH, ddd, 74,59.7 Hz, 75,6 1.4 Hz, 75j6> 3.8 Hz, H-5), 3.29 (IH, dd, 75,6 1.4 Hz, 76,6' 11.1 Hz, H-6), 3.34 (IH, dd, W 9.9 Hz, 72j3 8.7 Hz, H-2), 3.49 (IH, dd, 75,63.8 Hz, 76j6> 11.1 Hz, H-6'), 3.51 (IH, at, 79.4 Hz, H-3), 3.60 (IH, at, 79.4 Hz, H-4), 4.15, 4.25 (2H, ABq, 712.1 Hz, PhCH2), 4.52, 4.58 (2H, ABq, 711.0 Hz, PhCH2), 4.72, 4.76 (2H, ABq, 711.3 Hz, PhCH2), 4.78, 4.52 (2H, ABq, 711.3 Hz, PhCH2), 5.25 (IH, d, 7 10.2 Hz, H-l), 6.82-6.88 (3H, m, Ar-H), 7.05-7.26 (20H, m, Ar-H), 7.96-7.98 (2H, m, Ar-H).
Example 34: Ethyl 2.3.4.6-tetra-O-benzyl-l-dithio-β-D-glucopyranosyl disulfide
2,3,4,6-Tetra-O-acetyl-β-D-glucopyranosyl phenylthiosulfonate (100 mg, 0.14 mmol) and triethylamine (0.02 mL, 0.14 mmol) were dissolved in anhydrous DCM (10 mL) and stirred at RT under an atmosphere of argon. To this ethane thiol (11 μL, 0.14 mmol) in anhydrous DCM (10 mL) was slowly added dropwise via a syringe pump over a 90 min period. After a 90 min period, tic. (petrol: ethyl acetate, 6:1) indicated the formation of a major product (Rf 0.4) along with complete consumption of the starting material (Rf 0.2). The solution was concentrated in vacuo. The residue was purified by flash column chromatography (petrol: ethyl acetate, 7: 1) to afford the title product (83 mg, 95%) as a clear oU; [ ]o22 -164.9 (c, 0.2 in CHC13) [Lit. [α]D 25 -80.0 (c, 3.0 in MeOH) (Davis, B. G; Ward, S. J.; Rendle, P. M. Chem. Commun. 2001, 189)]; δH(400 MHz, CDC13) 1.22 (IH, t, 77.3 Hz, CH3), 2.68-2.86 (2H, m, CH2), 3.24 (IH, ddd, 74,5 9.7 Hz, 75>6 3.3 Hz, 75,6' 2.1 Hz, H-5), 3.56-3.60 (2H, m, H-6, H-6!), 3.61 (IH, at, 79.1 Hz, H-3), 3.72 (IH, at,
79.4 Hz, H-4), 3.89 (IH, at, 79.1 Hz, H-2), 4.34 (IH, d, 71;2 9.7 Hz, H-l), 4.37, 4.31 (2H, Abq, 712.2 Hz, PhCHs), 4.56, 4.83 (2H, Abq, 711.3 Hz, PhCIfc), 4.77-4.83 (2H, m, PhCϋ), 4.90 (IH, d, 11.1 Hz, PhCHH), 4.97 (IH, d, 710.7 Hz, PhCHH), 7.07-7.21 (14H, m, Ar-H), 7.25-7.27 (2H, m, Ar-H), 7.29-7.31 (2H, m, Ar-H), 7.36-7.38 (2H, m, Ar-H).
Example 35: N-Acetyl-L-cysteine (2,3.4,6-tetra-O-benzyl-l-dithio-β-D- glucopyranosyl disulfideVL-serine methylester
2,3,4,6-Tetra-O-benzyl-β-D-glucopyranosyl phenylthiosulfonate (50 mg, 0.07 mmol) was dissolved in anhydrous DCM (5 mL) and stirred at RT under an atmosphere of Ar. To this N-acetyl-L-cysteine-L-serine methylester (19 mg, 0.07 mmol) and triethylamine (11 μL, 0.08 mmol) in anhydrous DCM (5 mL) and anhydrous methanol (0.5 mL) was slowly added dropwise via a syringe pump over a 5 h period. After a 5 h period, tic. (ethyl acetate) indicated the formation of a major product (R
f 0.6) along with complete consumption of the starting material (R
f 0.9). The solution was concentrated in vacuo. The residue was purified by flash column chromatography (ethyl acetate) to afford the title product (48 mg, 82%) as a white crystalline solid; mp 96-97°C; [α]
D 22 +56.2 (c, 1 in CHC1
3); δ
H (400 MHz, CDC1
3) 2.03 (3H, s, COCH
3), 3.19 (IH, dd, 7
CHjH 14.0 Hz, 7
CH,αH 8.3 Hz, CysCHH), 3.37 (IH, dd, 7
CH,H 14.3 Hz, 7
CH)αH 6.0 Hz, CysCHH), 3.64 (IH, ddd, 7
4j5 9.6 Hz, 7
5,
6 1.8 Hz, 7
5j6. 3.9 Hz, H-5), 3.72 (IH, at, 7 9.2 Hz, H-4), 3.77 (IH, at, 78.8 Hz, H-3), 3.82 (3H, s, OMe), 3.84-3.90 (4H, m, SerCHH. H-2, H-6, H -6'), 3.96 (IH, dd, 7
CH,
H 11.7 Hz, 7
CH,αH 3.3 Hz, SerCHH), 4.50 (IH, d, 7
1) 9.6 Hz, H-l), 4.51, 4.70 (2H, ABq, 711.6 Hz, PhCH
2), 4.55, 4.85 (2H, ABq, 710.4 Hz, PhCH
2), 4.59-4.62 (IH, m, αHSer), 4.81, 4.87 (2H, ABq, 710.6 Hz, PhCH
2), 4.91, 4.97 (2H, ABq, 711.0 Hz, P-1CH2), 4.93-4.98 (IH, m, αHCys), 6.88 (IH, bd, 7
ΝH,
H 7.9 HZ, NHAC), 7.13-7.39 (20H, m, 20 x Ar-C), 7.48 (IH, d, 7
NHJH .6 HZ, NHSer).
Example 36: 2,3,6-Tri-0-acetyl-4-0-(2,3,6-tri-0-acetyl-4-0-(2,3,4,6-tetra-0-acetyl- - 0-glucopyranosyl)-α-D-glucopyranosyl ) - β -D-glucopyranosy 1 pheiryltliiosulf onate
2,3,6-Tri-O-acetyl-4-0-(2,3,6-tri-O-acetyl-4-O-(2,3,4,6-tetra-O-acetyl-α-C- glucopyranosyl)- -D-glucopyranosyl)-α-D-glucopyranosyl bromide (200 mg, 0.21 mmol) was dissolved in anhydrous acetonitrile (10 mL). To this sodium benzenethiosulfonate (80 mg, 0.41 mmol) and tefrabutylammonium iodide (10 mg,
0.02 mmol) were added. The resulting mixture was stirred under argon at 70°C. After a 2 h period, tic. (petrol: ethyl acetate, 1:2) indicated the formation of a UN active product (Rf 0.5) with complete consumption of the starting material (Rf 0.5). At which point the solution was allowed to cool to RT and filtered, the filtrate was concentrated in vacuo. The residue was purified by flash column chromatography (petrol: ethyl acetate, 1:2) to afford the title product (140 mg, 62%) as a white amorphous solid; [α]D 2+69.9 (c, 0.75 in CHC13); δH(500 MHz, CDC13) 2.03, 2.04, 2.06, 2.08, 2.11, 2.15, 2.19, (30H, 10 x COCH3), 3.77-3.79 (IH, m, H-5a), 3.94-4.00 (4H, m, H-4a, H-4c, H-5b, H-5c), 4.10 (IH, dd, 75,62.1 Hz, 76,6' 12.4 Hz, H-6b), 4.17-4.22 (3H, m, H-6a, H-6c, H-6a'), 4.29 (IH, dd, 75)6 3.3 Hz, 76)6> 12.6 Hz,
H-6b'), 4.46 (IH, dd, 75;6 1.9 Hz, 76,6> 12.4 Hz, H-6c'), 4.76 (IH, dd, 71)23.9 Hz, 72,3 10.4 Hz, H-2a), 4.89-4.94 (2H, m, H-2b, H-2c), 5.12 (IH, at, 79.9 Hz, H-4b), 5.28 (IH, d, 71;23.8 Hz, H-la), 5.34 (IH, d, 7lj29.7 Hz, H-lc), 5.37 (IH, at, 79.1 Hz, H-3c), 5.41 (IH, at, 710.1 Hz, H-3b), 5.41-5.45 (2H, m, H-lb, H-3a), 7.62-7.65 (2H, m, Ar-H), 7.71 (IH, m, Ar-H), 8.00-8.02 (2H, m, Ar-H).
Example 37: Ethyl 2,3,6-tri-O-acetyl-4-( -(2,3,6-tri-O-acetyl-4-O-(2,3,4,6-tetra-O- acetyl-α-O-glucopyranosyl)-α-D-glucopyranosyl)-l-dithio-β-D-glucopyranosyl disulfide
2,3,6-Tri-O-acetyl-4-O-(2,3,6-tιi-O-acetyl-4-O-(2,3,4,6-tetra-O-acetyl-α-O- glucopyranosyl)- -D-glucopyranosyl)-β-D-glucopyranosyl phenylthiosulfonate (50 mg, 0.05 mmol) was dissolved in anhydrous DCM (10 mL) and stirred at RT under an atmosphere of argon. A solution of triethylamine (7 μL, 0.05 mmol) and ethane thiol (3 μL, 0.05 mmol) and anhydrous DCM (10 mL) was slowly added dropwise via a syringe pump over a 1 h period. After a lh period, t.l.c. (petrol: ethyl acetate, 1:2) indicated the formation of a major product (Rf 0.6) along with complete consumption of the starting material (Rf 0.4). The solution was concentrated in
vacuo. The residue was purified by flash column chromatography (petrol: ethyl acetate, 1:2) to afford ethyl the title product (43 mg, 93 %) as a clear oil; [c 24 +26.4 (c, 1.5 in CHC13); δH(500 MHz, CDCI3) 1.30 (IH, t, 77.2 Hz, CH3), 2.04, 2.05, 2.06, 2.07, 2.10, 2.14, 2.19, 2.20 (30H, 8 x s, 10 x COCH3), 2.75-2.87 (2H, m, CH2CH3), 3.77-3.81 (IH, m, H-5a), 3.96-4.00 (3H, m, H-4b, H-5c, H-5b), 4.03 (IH, at, 79.3 Hz, H-4a), 4.10 (IH, dd, 75;6 2.3 Hz, 76)6' 12.6 Hz, H-6c), 4.22 (IH, dd, 75,6 2.9 Hz, 76,6' 12.4 Hz, H-6b), 4.29 (IH, dd, 75)6 3.1 Hz, 76)6' 12.4 Hz, H-6'c), 4.33 (IH, dd, 75;6 4.4 Hz, 76,6'12.4 Hz, H-6a), 4.51 (IH, dd, 75,6' 1.8 Hz, 76,6' 12.4 Hz, H- 6b', 4.57 (IH, dd, 75;6 2.3 Hz, 76;6> 12.4 Hz, H-6a'), 4.58 (IH, d, 71;2 9.9 Hz, H-la), 4.79 (IH, dd, 71)2 4.1 Hz, 72)3 10.6 Hz, H-2b), 4.90 (IH, dd, 71)2 4.3 Hz, 72,3 10.4 Hz, H-2c), 5.11 (IH, at, 79.9 Hz, H-4c), 5.16 (IH, at, 79.5 Hz, H-2a), 5.33 (IH, d, 7 4.1 Hz, H-lb), 5.37 (IH, at, 78.9 Hz, H-3a), 5.38- 5.44 (2H, m, H-3b, H-3c), 5.45 (IH, d, 7ιj2 4.1 Hz, H-lc).
Example 38: N-Butoxycarbonyl-L-cysteine (2,3,6-tri-O-acetyl-4-O-(2,3,6-tri-c - acetyl-4-O-(2,3,4,6-tetra-O-acetyl- -c -glucopyranosyl)-α-D-glucopyranosyl)-l- dithio-β-D-glucopyranosyl disulfide)-L-serine methylester
2,3,6-Tri-0-ace1yl-4-O-(2,3,6-tri-0-acetyl-4-O-(2,3,4,6-tetra-O-acetyl-α-0- glucopyranosyl)-α-D-glucopyranosyl)-β-D-glucopyranosyl phenylthiosulfonate (89 mg, 0.08 mmol) was dissolved in anhydrous DCM (5 mL) and stirred at RT under an atmosphere of argon. A solution of triethylamine (0.014 mL, 0.2 mmol) and N-butoxycarbonyl-L-cysteinyl-L-serine methylester (30 mg, 0.09 mmol) in anhydrous DCM (10 mL) and anhydrous methanol (1 mL) was slowly added dropwise via a syringe pump over a 3 h period. After a 3 h period, tic. (ethyl acetate) indicated the formation of a major product (Rf 0.6) along with complete consumption of the starting material (Rf 0.7). The solution was concentrated in
vacuo. The residue was purified by flash column chromatography (ethyl acetate) to afford the title product (66 mg, 74%) as an amorphous white solid; [cφ24 +25.1 (c, 1.25 in CHC13); δH(500 MHz, CDC13) 1.47 (9H, s, C(CH3)3), 2.00, 2.01, 2.02, 2.03, 2.06, 2.09, 2.15, 2.18 (30H, 8 x s, 10 x COCH3), 2.75-2.87 (IH, m, CHHCys), 3.16- 3.19 (lH, m, CHHCys), 3.27 (lH, t, 76.2 Hz, OH), 3.81 (3H, s, OMe), 3.83 -3.85 (IH, m, H-5a), 3.92-4.01 (6H, m, H-4b, H-5b, H-5c, H6a, H-6a', CHHSer), 4.06 (IH, dd, 75)6 2.2 Hz, 76)6' 12.2 Hz, H-6c), 4.09-4.16 (2H, m, H-4a, H-6b), 4.25 (IH, dd, 75,6 3.2 Hz, 76,6' 12.3 Hz, H-6c'), 4.39-4.41 (IH, m, CHHSer), 4.52-4.67 (4H, m, αHSer, αHCys, H-la, H-6'b), 4.74 (IH, dd, 71;24.1 Hz, 72)3 10.3 Hz, H-2b), 4.85 (IH, dd, 71)2 3.7 Hz, 72; 10.5 Hz, H-2c), 5.07 (IH, at, 79.9 HZ, H-4c), 5.11-5.13 (IH, m, H-2a), 5.28 (IH, d, 7ι)2 4.1 Hz, H-lb), 5.32-5.41 (4H, m, H-3a, H-3b, H-3c, NHCys), 5.42 (IH, d, 7ι,2 3.9 Hz, H-lc), 7.25 (IH, bd, 7NH,αH 6.1 Hz, NHSer).
Example 39: Phenyl 2,3,6-tri-O-acetyl-l-selenenylsulfide-4-O-(2,3,6-tri-O-acetyl-4- O-(2,3,4,6-tetra-O-acetyl-α-O-glucopyranosyl)-α-D-glucopyranosyl)-β-D- glucopyranoside
2,3,6-Tri-0-acetyl-4-O-(2,3,6-tri-0-acetyl-4-0-(2,3,4,6-tetra-O-acetyl-α-0- glucopyranosyl) -α-D-glucopyranosyl) -β-D-glucopyranosylthiol (500 mg, 0.53 mmol) and phenyl selenium bromide (200 mg, 0.9 mmol) were dissolved in anhydrous DCM (20 ml). After a 5 min period, tic. (petrol: ethyl acetate 1:2) indicated the formation of a major product (Rf 0.4) along with complete consumption of the starting material (Rf 0.3). The reaction was quenched with the addition of triethylamine (5 ml) and then concentrated in vacuo. The residue was purified by flash column chromatography (petrol: ethyl acetate 1 :2) to afford the title product (527 mg, 91%) as an amorphous off white solid; [O]D 25 -2.6 (c, 1.0 in CHC13); δH (400 MHz, CDC13), 1.99, 2.01, 2.02, 2.04, 2.06, 2.10, 2.14 (30H, 9 x s, 10 x OAc), 3J9 (IH, dat, 74,5 9.7 Hz, 73.4 Hz, H-5a), 3.92 (3H, m, H4b, H-5b,
H-5c), 4.00 (IH, at, 79.3 Hz, H-4a), 4.05 (IH, dd, 75,62.8 Hz, 76,6> 12.8 Hz, H-6c), 4.15 (IH, dd, 75,6 2.8 Hz, 76,6' 12.6 Hz, H-6b), 4.22 (IH, dd, 75)63.7 Hz, 76)6' 12.0 Hz, H-6a), 4.25 (IH, dd, 75,6 3.3 Hz, 76,6' 12.0 Hz, H-6c'), 4.42-4.46 (2H, m, H-6a', H-6b'), 4.66 (IH, d, 7 9.9 Hz, H-la), 4.74 (IH, dd, 7ι)2 4.1 Hz, 72j3 10.4 Hz, H-2b), 4.86 (IH, dd, 7ι,2 4.1 Hz, 72,3 10.5 Hz, H-2c), 5.06 (IH, at, 79.6 Hz, H-4c), 5.07 (IH, at, 79.8 Hz, H-2a), 5.27 (IH, d, 7ι)2 4.4 Hz, H-lb), 5.32-5.39 (3H, m, H-3a, H-3b, H-3c), 5.41 (IH, d, 71;2 4.2 Hz, H-lc), 7.27-7.29 (3H, m, Ar -H), 7.64-7.67 (2H, m, Ar-H).
Example 40: bt-f-N-Butoxycarbonyl-L-cystemyl-L-threonine methylester
bw-N-Butoxycarnoyl-L-Cysteine (4.0 g, 9.1 mmol), L-threonine methylester (2.42 g, 18.2 mmol), DCC (3.75 g, 18.2 mmol) JTOBt (2.46 g, 18.2 mmol) and DIPEA (2.5 ml, 18.2 mmol) was dissolved in freshly distilled DCM (150 mL). After a 18 h period, tic. (ethyl acetate:methanol 9: 1) indicated the formation of a major product (Rf0.5) along with complete consumption of the starting material (Rf 0.0). The reaction was diluted with water (2 x 100 ml) and the phases were partitioned. The organics were washed with brine (100 ml), dried (MgSO ), filtered and the solvent removed in vacuo. The residue was purified by flash column chromatography (ethyl acetate: methanol 9: 1), and recrystallisation from methanol/diethyl ether afforded the title product (3.26 g, 60%) as a white crystalline solid; mp 145-147°C; [α]D 25 +20.8 (c, 1.0 in CHC13); δH(400 MHz, CDC13), 1.23 (3H, d, 7CH)CH3 6.6 Hz, CHCH3), 1.44 (9H, s, C(CH3)3), 3.11-3.12 (2H, m, CH2Cys), 3.26 (IH, bs, OH), 3.75 (3H, s, OMe), 4.32-4.36 (IH, m, CHCH3), 4.61 (dd, 7NH,αThr 8.7 Hz, 7αH,cHCH3 2.15 Hz, CHCH3), 4.63-4.68 (IH, m, αCys), 5.75 (IH, d, 7NH,αHcys 7.4 Hz, NHCys), 7.56 (IH, d, 7NH,αThr 8.6 Hz, NHThr).
Example 41: N-Butoxycarbonyl-L-cystemyl-L-threoiiine methylester
bw-N-Butoxycarbonyl-L-cystemyl-L-threomne methylester (2.0 g, 3.3 mmol) was dissolved in wet chloroform (100 mL) and methanol (10 mL) and stirred. To this stirred solution tributylphosphine (1.0 mL, 4.0 mmol) was added. After a 2 h period, tic. (ethyl acetate:methanol 9: 1) indicated the formation of a product (R
f 0.8) along with complete consumption of the starting material (Rf 0.7). The reaction was concentrated in vacuo. The residue was purified by flash column chromatography (ethyl acetate) to afford the title product (2.0 g, 99%) as a white foam; [αφ
25 -11.4 (c, 1.0 in CHC1
3); δ
H(400 MHz, CDC1
3) 1.09 (3H, d, 7
CH)CH3 6.4 Hz, CH
3), 1.34 (9H, s, C(CH
3)
3), 1.65 (IH, at, 78.7 Hz, SH), 2.72-2.89 (2H, m, CH
2 ), 3.66 (3H, s, OMe), 3.96 (IH, m, OH), 4.24-4.28 (IH, m, CHCH3), 4.34-4.36 (IH, m, αHCys), 4.49 (IH, dd, 7αHThr,ΝH 8.5 Hz, 7
αHThr,CHCH3 2.1 Hz, αHThr), 5.82 (IH, d, 7
αHCys,NH 8.2 Hz, NHCys), 7.38 (IH, d,
8.5 Hz, NHThr).
Example 42: N-Butoxycarbonyl-L-cysteine (2,3,4,6-tetra-O-acetyl-l -dithio-β-D- glucopyranosyl disulfide)-L-threonine methylester
Phenyl 2,3,4,6-tetra-O-acetyl-l -selenenylsulfide-D-β-glucopyranoside (130 mg, 0.25 mmol) and triethylamine (0.02 mL, 0.18 mmol) were dissolved in freshly distilled DCM (10 mL). The resulting solution was stirred at RT. A solution of N-butoxycarbonyl-L-cysteme-L-mreonine methylester (30 mg, 0.089 mmol) in anhydrous methanol (4 mL) was added slowly to the above solution. After a 10 min period, tic. (petrol: ethyl acetate, 1:2) indicated the formation of aproduct (Rf0.2)
along with complete consumption of the starting material (Rf 0.5). The solution was concentrated in vacuo. The residue was purified by flash column chromatography (petrohethyl acetate, 1:2) to afford the title product (32 mg, 51 %) as awhite amorphous solid; [cφ25 -81.2 (c, 0.25 in CHC13); δH(400 MHz, CDC13) 1.28 (3H, d, 7CHCH3 6.1 Hz, CHCH3), 1.51 (9H, s, C(CH3)3), 2.06, 2.08, 2.102.14 (12H, 4 x s, 4 x OAc), 2.86 (IH, bs, OH), 3.06 (IH, dd, 7CHαH 8.8 Hz, 7CHCH 13.4 Hz, CHHCys), 3.31 (IH, dd, 7CΉOH 4.2 Hz, 7CHCH 13.1 Hz, CHHCys), 3.82 (3H, s, OCH3), 3.87- 3.89 (IH, m, H-5), 4.32-4.38 (2H, m, H-6, H-6'), 4.39 (IH, dd, 7CHCH3 6.4 Hz, 7CHαH 2.5 Hz, CHOH), 4.60-4.65 (3H, m, H-l, αHThr, α HCys), 5.20-5.32 (3H, m, H-2, H-3, H-4), 5.42 (IH, d, 7NHαH 8.0 Hz, NHCys), 7.12 (IH, d, 7NHαH 8.9 Hz, NHThr).
Example 43: N-butoxycarbonyl-L-cysteine (2,3,4,6-tetra-O-acetyl-l -dithio-β-D- galactopyranosyl disulfide)-L-threonine methylester
Phenyl 2,3,4,6-tetra-O-acetyl-l-selenenylsulfide-D-β-galactopyranoside (140 mg, 0.27 mmol) and triethylamine (0.01 mL, 0.089 mmol) were dissolved in freshly distilled DCM (5 mL). The resulting solution was stirred at RT. A solution of N-butoxycarbonyl-L-cysteine-L-threonine methylester (26 mg, 0.077 mmol) in anhydrous DCM (5 mL) and anhydrous methanol (4 mL) was added slowly to the above solution. After a 10 min period, tic. (petrobethyl acetate, 1:2) indicated the formation of aproduct (Rf 0.2) along with complete consumption of the starting material (Rf 0.6). The solution was concentrated in vacuo. The residue was purified by flash column chromatography (petrol: ethyl acetate, 1:2) to afford the title product (49 mg, 93%) as a white amorphous solid; [cφ25 -81.2 (c, 0.25 in CHC13); δH (400 MHz, CDC13) 1.24 (3H, d, 7CH,CH3 6.4 Hz, CH3), 1.46 (9H, s, C(CH3)3), 2.01, 2.06, 2.08, 2.20 (12H, 4 x s, 4 x OAc), 2.79 (IH, bd, 7CH,OH 4.1 Hz, OH), 2.99 (IH, dd, 7αH,cH28.8 Hz, 7CH,H 13.9 Hz, CHHCys), 3.32-3.35 (IH, , CHHCys), 3.76 (3H, s,
OCH3), 4.04 (IH, at, 76.2 Hz, H-5), 4.10-4.16 (IH, m, H-6), 4.19 (IH, dd, 75,6> 6.1 Hz, 76,6' 10.8 Hz, H-6'), 4.36-4.46 (IH, m, CHOH), 4.56 (IH, dd, 7αHτ r,CH 2.4 Hz, 7αH)NH 8.9 Hz, αHThr), 4.57-4.64 (IH, m, αHCys), 4.65 (IH, d, 7 9.0 Hz, H-l), 5.13 (IH, dd, 72,3 9.8 Hz, 72;3 9.8 Hz, H-3), 5.31 (IH, d, 7αHcyS)NH 8.3 Hz, NHCys), 5.47 (IH, d, 73,4 3.2 Hz, H-4), 5.52 (IH, at, 79.6 Hz, H-2), 6.91 (IH, d, 7αHT r,NH 9.0 Hz, NHThr).
Example 44: Butoxycarbonyl-L-cysteinyl-(S-3,4,6-tri-0-acetyl-2-acetamido-2- deoxy-β-D-glucopyranosyl disulfide)-L-threonine methylester
The title product was obtained (55mg, 88%) as a white amoφhous solid by a method analogous to that of Example 43 utilising phenyl 3,4,6-tri-0-acetyl-2- acetamido-2-deoxy-l-selenenylsulfide-D-β-as starting material, [cφ25 -47.1 (c, 0.1 in CHC13); δH(400 MHz, CDC13) 1.17 (3H, d, 7CH)CH3 6.4 Hz, CH3), 1.49 (9H, s, C(CH3)3), 1.91, 2.00, 2.02, 2.07 (12H, 4 x s, 4 x, COCH3), 2.99 (IH, dd, 7CHH!CHII 13.5 Hz, 7αH,cH 10.0 Hz, CHH), 3.38 (IH, dd, 7αH,cH4.8 Hz, 7CHH,CHH 13.5 Hz, CHH), 3.88-3.91 (IH, m, H-5), 4.16-4.32 ( H, m, H-2, H-6, H-6', CHCH3), 4.45 (IH, d, 7αH,cH .7 Hz, αHThr), 4.54 (IH, dd, T^CH H 9.1 ΕLz, 7αH,cHH4.7 Hz, αHCys), 4.79 (IH, d, 7ι)2 10.1 Hz, H-l), 5.06 (IH, at, 79.7 Hz, H-4), 5.28 (IH, at, 7 9.7 Hz, H-3).
Example 45: N-Butoxycarbonyl-L-cysteinyl-(S-l-β-D-glucopyranosyl disulfide)-L~ threonine methylester
Phenyl 1-selenenylsulfide-D-β-glucopyranoside (70 mg, 0.2 mmol) and triethylamine (0.01 mL, 0.1 mmol) were dissolved in MeOH (8 mL). The resulting solution was stirred at RT. A solution of N-butoxycarbonyl-L-cysteme-L-mreonine methylester (22 mg, 0.07 mmol) in MeOH (5 mL) was added slowly to the above solution. After 10 min, tic. (EtOAc:MeOH, 9: 1) indicated the formation of a major product (R
f 0.4). The solution was concentrated in vacuo. The residue was purified by flash column chromatography (EtOAc:MeOH, 9: 1) to afford the title compound (32 mg, 91%) as a white amorphous solid; [α]
D 25 - 139.5 (c, 0.6 in MeOH); δ
H(500 MHz, CD
3OD) 1.19 (3H, d, 7
CH,
CH3 6.2 Hz, CHCH3), 1.49 (9H, s, C(CH
3)
3), 2.93 (IH, dd, 7
CHH,
CHH 13.5 Hz, 7
CiLαH9.5 Hz, CH
2Cys), 3.32-3.46 (4H, m, H-3, H-4, H-5, CHH), 3.60-3.63 (IH, m, H-2), 3.13-3.11 (IH, m, H-6), 3.78 (3H, s, OMe), 3.92-3.94 (IH, m, H-6'), 4.31-4.36 (IH, m, CHCH3), 4.39 (IH, d, 7
1)2 9.3 Hz, H-l), 4.48 (IH, d, 7
αH,cH 2.9 Hz, αHThr), 4.69 (IH, dd, 7
«H,
CHH 9.0 HZ, 7
αH,cHH 5.2 Hz, αHCys).
Example 46: N-Butoxycarbonyl-L-cvsteinyl-(S-2-acetamino-2-deoxy-l-β -D- gmcopyranosyl disulfide)-L-threonine methylester
The title compound (32 mg, 91%) was obtained as a white amorphous solid by a method analogous to that of Example 45 utilising phenyl 2-acetamido-2-deoxy-l - selenenylsulfide-β -D-glucopyranoside as starting material. [α]ϋ 25 +6.21 (c, 0.45 in MeOH); δH(500 MHz, CD3OD) 1.19 (3H, d, 7CHCH3 6.7 Hz, CHCH3), 1.49 (9H, s, C(CH3)3), 1.99 (3H, s, COCH3), 2.97 (IH, dd, 7CH,H 13.8 Hz, 7^1x9.6 Hz, CHHCys), 3.31-3.33 (IH, m, CHH), 3.38-3.41 (IH, m, H-5), 3.45 (IH, at, 79.3 Hz, H-4), 3.54 (IH, dd, 72)3 8.6 Hz, 73)4 9.8 Hz, H-3), 3.16-3.11 (IH, m, H-6), 3.78 (3H, s, OMe), 3.79-4.01 (2H, m, H-2, H-6'), 4.33 (IH, dq, 7CHCH3 6.3 Hz, 7CH)αH 3.0 Hz, CHCH3), 4.48 (IH, d, 7αH)cH 3.0 Hz, αHThr), 4.59 (IH, d, 7ι,2 10.3 Hz, H-l), 4.63 - 4.67 (IH, m, αHcys).
Example 47: Phenyl- 1-selenenylsulfide-β-D-glucopyranoside
1-Thio-β-D-glucopyranoside (200 mg, 0.9 mmol) and phenylselenenyl bromide (230 mg, 1.0 mmol) were added to anhydrous 1,4-dioxane (5 mL) stirred under an atmosphere of argon. After a 1 min period, tic. (ethyl acetate) indicated the formation of a major product (Rf 0.2). The reaction was quenched with the addition of friethylamine (2 mL). The solution was concentrated in vacuo. The residue was purified by flash column chromatography (ethyl acetate:methanol, 9: 1) to afford the title product (165 mg, 57%) as an off white amorphous solid; [cφ2 +56.2 (c, 1 in CHC13); δH (400 MHz, MeOD) 3.31-3.33 (2H, m, H-3, H-5), 3.39-3.45 (2H, m, H-2, H-4), 3.62 (IH, dd, 75,6 5.3 Hz, 76j6> 12.1 Hz, H-6), 3.83 (IH, dd, 75j6. 1.9 Hz, 76>β. 12.2 Hz, H-6), 4.47 (IH, d, 71)2 9.4 Hz, H-l), 7.27- 7.34 (3H, m, Ar-H), 7.75-7.78 (2H, m, Ar-H).
Example 48: Phenyl 1-selenenylsulfide-β-D-galactopyranoside
The title compound was obtained (193mg, 20%) as an off white amorphous solid by a method analogous to that of Example 47 utilising 1-thio-β-D-galactopyranoside as starting material. [α]D 25 -111.4 (c, 1 in MeOH); δH(400 MHz, CD3OD) 3.52 (IH, dd, 72;3 9.4 Hz, 73,4 3.3 Hz, H-3), 3.56 (IH, at, 74;5 0.9 Hz, 76.5 Hz, H-5), 3.67-3.69 (2H, d, 76.0 Hz, H-6, H-6'), 3.74 (IH, at, 79.3 Hz, H-2), 3.91 (IH, dd, 73)4 3.2 Hz, 74;5 0.7 Hz, H-4), 4.45 (IH, d, Jl 9.7 Hz, H-l), 7.27-7.30 (3H, m, Ar-H), 7.76-7.79 (2H, m, Ar-H).
Example 49: Phenyl 2,3,4,6-tetra-O-acetyl-l-selenenylsulfide-β-D-glucopyranoside
l-Thio-2,3,4,6-tetra-0-acetyl-β -D-glucopyranose (200 mg, 0.6 mmol) and PhSeBr (150 mg, 0.6 mmol) were added to freshly distilled DCM (5 mL) and stirred under argon at RT. After 5 min, tic. (petrol: EtOAc, 1: 1) indicated the formation of a major product (R
f 0.5) along with complete consumption of the starting material (R
f 0.4). The reaction was quenched with the addition of friethyl-imine (2 mL) and stirred for 5 min. The residue was partitioned between DCM (5 mL) and water (10 mL) and the aqueous phase was re-extracted with DCM (3 x 5 mL). The combined organics were washed with brine (10 mL), dried over MgSO
4, filtered and the solvent removed in vacuo. The resulting residue was purified by flash column chromatography (petrobEtOAc, 2: 1) to afford the title product (260 mg, 93%) as a yellow crystalline solid mp 111-112 °C; [α]
D 25 -250.1 (c, 1.0 in CHC1
3); δ
H (400 MHz, CDCI3) 2.02, 2.01, 2.00 (12H, 4 x s, 4 x CH
3), 3.75 (IH, ddd, 7
4j5 9.9 Hz, 7
5)6 2.4 Hz, 7
5,
6> 4.6 Hz, H-5), 4.08 (IH, dd, 7
5j62.6 Hz, J
6>6> 12.4 Hz, H-6), 4.16 (IH, dd, 7
5!6
' 4.5 Hz, J
6ff 12.4 Hz, H-6'), 4.62 (IH, d, 7
1)2 9.8 Hz, H-l), 5.12 (IH, at, 79.7 Hz, H-4), 5.20-5.30 (2H, m, H-2, H-3), 7.25 -7.28 (3H, m, Ar-H), 7.67-7.70 (2H, m, Ar-H).
Example 50: Phenyl 2,3,4,6-tetra-O-acetyl-l-selenenylsυlfide-β-D- galactopyranoside
The title compound (402 mg, 95%) was obtained as a yellow crystalline solid using a method analogous to that of Example 49 utilising l-xhio-2,3,4,6-tetra-0-acetyl- β- D-galactopyranose as starting material. Mp 123-125 °C; [α]ϋ 25 -172.4 (c, 1.0 in CHCI3); δH(400 MHz, CDCI3) 1.99, 2.02, 2.16 (12H, 4 x s, 4 x CH3), 3.94-4.03 (3H, m, H-5, H-6, H-6'), 4.64 (IH, d, 71)2 10.1 Hz, H-l), 5.04 (IH, dd, 72)3 10.2 Hz,
73,43.3 Hz, H-3), 5.40-5.45 (2H, m, H-2, H-4), 7.27-7.30 (3H, m, Ar-H), 7.69-7.71 (2H, m, Ar-H).
Example 51: Phenyl 3,4,6-tri-0-acetyl-2-acetamido-2-deoxy-l-selenenylsulfide-β- D-glucopyranoside
The title compound (300 mg, 66%) was obtained as a white crystalline solid using a method analogous to that of Example 49 utilising l-thio-3,4,6-tri-0-acetyl-2- acetamido-2-deoxy-β-D-glucopyranose as starting material. Mp 177-179 °C; [α]o
25 -134.0 (c, 1.0 in CHC1
3); δ
H(400 MHz, CDC1
3) 1.90 (3H, s, NHCOCH
3), 1.99, 2.00, 2.03 (9H, 3 x s, 3 x CH
3), 3.76 (IH, ddd, 7
4)5 10.1 Hz, 7
5;62.3 Hz, 7
5,
6' 4.7 Hz, H-5), 4.07 (IH, dd, 7
5,
6 2.3 Hz, 7
6,
6> 12.3 Hz, H-6), 4.15 (IH, dd, 7
5,
6' 4.6 Hz, 7
6)6' 12.2 Hz, H-6'), 4.19-4.24 (IH, m, H-2), 4.78 (IH, d, 7
lj2 10.1 Hz, H-l), 5.09 (IH, at, 79.7 Hz, H-4), 5.28 (IH, at, 79.5 Hz, H-3), 5.19 (IH, d, 79.1 Hz, NHAc), 7.24 -7.28 (3H, m, Ar-H), 7.68-7.70 (2H, m, Ar-H).
Example 52: Phenyl-2-acetylamino-2-deoxy-l-selenenylsulfide-β-D- glucopyranoside
l-Thio-2-acetylamino-2-deoxy-β-D-glucopyranoside (230 mg, 0.98 mmol) and phenylselenenyl bromide (250 mg, 1.08 mmol) were added to anhydrous 1,4-dioxane (5 mL) and anhydrous methanol (3 ml) stirred under an atmosphere of argon. After a 1 min period, tic. (ethyl acetate:methanol, 9: 1) indicated the formation of a major product (Rf 0.4). The reaction was quenched with the addition of friethylamine (5 mL). The solution was concentrated in vacuo. The residue was purified by flash column chromatography (ethyl acetate:methanol, 9: 1) to afford the title product (270 mg, 70%) as a white amorphous solid; [oφ22 -174.0 (c, 1 in
MeOH); δH (400 MHz, MeOD), 1.96 (3H, s, CH3), 3.31-3.39 (2H, m, H-4, H-5), 3.51 (IH, at, 78.1 Hz, H-3), 3.65 (IH, dd, 75)6 5.0 Hz, 76j6. 11.7 Hz, H-6), 3.82-3.90 (2H, m, H-2, H-6'), 4.65 (IH, d, 71)2 10.2 Hz, H-l), 7.27-7.34 (3H, m, ArH), 7.72-7.74 (2H, m, ArH).
Example 53: Ethyl l-τhio-β-D-glucopyranosyl disulfide
Phenyl 1-selenenylsulfide-β-D-glucopyranoside (140 mg, 0.4 mmol) was dissolved in MeOH (10 mL) and stirred at RT. To this solution ethanethiol (10 μL, 0.1 mmol) and triethylamine (60 μL, 0.4 mmol) in MeOH (5 mL) were added dropwise over 1 h. After 1 h, tl.c. (EtOAc:MeOH, 9: 1) indicated the formation of a major product (Rf 0.4) along with complete consumption of the starting material (Rf 0.5). The solution was concentrated in vacuo. The residue was purified by flash column chromatography (EtOAαMeOH, 5: 1) to afford the title product (30 mg, 90%) as a white amoφhous solid; [α]D 22 -65.3 (c, 0.4 in CHCl3); δH (500 MHz, CD3OD) 1.33 (3H, t, 77.4 Hz, CH3), 2.86 (2H, q, 77.4 Hz, CH2), 3.30-3.34 (2H, m, H-4, H-5), 3.41 (IH, at, 79.0 Hz, H-3), 3.49 (IH, at, 7Hz, H-2), 3.67 (IH, dd, 75,6 5.3 Hz, 76.6- 12.0 Hz, H-6), 3.88 (IH, dd, 75,6> 2.1 Hz, 76j6' 12.0 Hz, H-6'), 4.35 (IH, d, ι,2 9.1 Hz, H-l).
Example 54: Ethyl 2-acetamido-2-deoxy-l-disulfide-β-D-glucopyranoside
Phenyl 2-acetamido-2-deoxy-l-selenenylsulfide-β-D-glucopyranoside (140 mg, 0.4 mmol) was dissolved in MeOH (10 mL) and stirred at RT. To this solution ethanethiol (10 μL, 0.13 mmol) and triethylamine (55 μL, 0.4 mmol) in MeOH
(5 mL) were added dropwise over 1 h. After 1 h, tic. (EtOAc:MeOH, 9: 1) indicated the formation of a major product (Rf0.2). The solution was concentrated in vacuo. The resulting residue was purified by flash column chromatography (EtOAc:MeOH, 9: 1) to afford the title product (38 mg, 99%) as a white amorphous solid; [α]ϋ25 -7.9 (c, 1.0 in CHC13); δH (400 MHz, CD3OD) 1.30 (3H, t, 77.3 Hz, CH3), 2.01 (3H, s, OAc), 2.83-2.86 (2H, m, CH2), 3.31-3.39 (2H, m, H-4, H -5), 3.51-3.56 (IH, m, H-3), 3.68-3.72 (IH, m, H-6), 3.84-3.91 (2H, m, H -2, H-6'), 4.57 (IH, d, 71)2 10.3 Hz, H-l).
Example 55: Protein glycosylation procedures using thiosulfonate reagents
A. SBLS156C mutant (24 mg, 0.89 μmol) was dissolved in aqueous buffer solution (2.4 mL, 70 mM HEPES, 2 mM CaCl , pH 6.9). 2,3,4,6-Tetra-O-acetyl-β- D-glucopyranosyl phenylthiosulfonate (50mg, 0.1 mmol) was dissolved in water/acetonitrile (1.6 mL, 9/7 v/v). A portion of the sugar solution (50 μL) was added to the protein solution and placed on an end- over-end rotator. After 25 min, the absence of free thiol was shown by EUman's analysis (Ellman, G. L. Arch. Biochem. Biophys. 1959, 82, 70), at which point another portion of sugar solution (50 μL) was added. The reaction was placed on an end-over-end rotator for a further 5 min, at which point the reaction mixture was loaded onto a PD10 Sephadex® G25 column and eluted with 70 mM HEPES, 2 mM CaCi2, pH 7.0. The protein fraction was collected and dialysed (MWCO 12-14 KDa) against 10 mM MES, 1 mM CaCl2, pH 5.8, (1 x 4L for 1 h, 2 x 2L for 30 min), to afford the glycosylated product m/z (ES) found 27072 calcd. 27078.
B. SBLS156C mutant (24 mg, 0.89 μmol) was dissolved in aqueous buffer solution (2.4 mL, 70 mM HEPES, 2 mM CaCl2, pH 6.9). 2,3,4,6-tetra-O-acetyl-β-D- galactopyranosyl phenylthiosulfonate (50mg, 0.1 mmol) was dissolved in water/acetonitrile (1.0 mL, 1/1 ratio). The sugar solution (50 μL) was added to the protein solution and placed on an end-over-end rotator. After 25 min, the absence of free thiol was shown by EUman's analysis, at which point another portion of sugar solution (50 μl) was added. The reaction was placed on an end-over-end rotator for a further 5 min, at which point the reaction mixture was loaded onto a PD 10
Sephadex® G25 column and eluted with 70 mM HEPES, 2 mM CaCl2ρH 7.0. The protein fraction was collected and dialysed (MWCO 12-14 KDa) against 10 mM MES, 1 mM CaCl2, pH 5.8, (1 x 4L for 1 h, 2 x 2L for 30 min), to afford the gl cosylated product m/z (ES) found 27072 calcd. 27078.
C. SBLS156C mutant (10 mg, 0.37 μmol) was dissolved in degassed aqueous buffer solution (1 mL, 70 mM CHES, 5mM MES, 2 mM CaCl2, pH 9.5). 2,3,6-Tri- 0-acetyl-4-0-(2,3,6-tri-0-acetyl-4-0-(2,3,4,6-tetra-0-acetyl-α-0-glucopyranosyl)- α-D-glucopyranosyl)-β-D-glucopyranosyl phenylthiosulfonate (30mg, 0.03 mmol) was dissolved in acetonitrile (150 μL). The sugar solution (75 μL) was added to the protein solution and placed on an end-over-end rotator. After 30 min, the absence of free thiol was shown by EUman's analysis, at which point the reaction mixture was loaded onto a PD10 Sephadex® G25 column and eluted with 70 mM HEPES, 2 mM CaCl2ρH 7.0. The protein fraction was collected and dialysed (MWCO 12-14 KDa) against 10 mM MES, 1 mM CaCl2, pH 5.8, (1 x 4L for 1 h, 2 x 2L for
30 min), to afford the glycosylated product m/z (ES) found 27654 calcd. 27653.
D. BSA (10 mg, 0.14 μmol) was dissolved in aqueous buffer solution (1 mL, 50 mM Tris, pH 1.1). 2,3,4,6 -Tetra-O-acetyl-β-D-glucopyranosyl phenylthiosulfonate (10mg, 0.02 mmol) was dissolved in water/acetonitrile (1.0 mL, 8/2 ratio). The sugar solution (150 μl) was added to the protein solution and placed on an end-over - end rotator. After 30 min, the absence of free thiol was shown by EUman's analysis, at which point the reaction mixture was loaded onto a PD10 Sephadex- G25 column and eluted with 70 mM HEPES, 2 mM CaCl2 pH 7.0. The protein fraction was collected and dialysed (MWCO 12-14 KDa) against pure water, (1 x 4L for 1 h, 2 x 2L for 30 min), to afford the glycosylated product; m/z (ES) found 66798 calcd. 66794.
E. BSA (10 mg, 0.14 μmol) was dissolved in aqueous buffer solution (1 mL, 50 mM Tris, pH 1.1). 2,3,4,6-Tetra-O-acetyl-β-D-galactopyranosyl phenylthiosulfonate
(25mg, 0.05 mmol) was dissolved in acetonitrile (0.5 mL). The sugar solution (75 μL) was added to the protein solution and placed on an end-over-end rotator. After 30 min, the absence of free thiol was shown by EUman's analysis, at which
point the reaction mixture was loaded onto a PD10 Sephadex® G25 column and eluted with 70 mM HEPES, 2 mM CaCl2ρH 7.0. The protein fraction was collected and dialysed (MWCO 12-14 KDa) against pure water, (1 x 4L for 1 h, 2 x 2L for 30 min), to afford the glycosylated product m z (ES) found 66792 calcd. 66794.
Example 56: Protein glycosylation procedures using selenenylsulfide reagents
A. SBLS 156C mutant (5 mg) was dissolved in degassed aqueous buffer solution (1 mL, 70 mM CHES, 5 mM MES, 2 mM CaCl2, pH 9.5). Phenyl 2,3,4,6 -tetra-O- acetyl-β-D-selenenylsulfide glucopyranoside (10 mg, 0.02 mmol) was dissolved in acetonitrile (500 μl). The sugar solution (500 μl) was added to the protein solution and placed on an end-over-end rotator. After 1 h, the absence of free thiol was shown by EUman's analysis, at which point the reaction mixture was loaded onto a PD10 Sephadex® G25 column and eluted with 70 mM HEPES, 2 mM CaCl2pH 7.0. The protein fraction was collected and dialysed (MWCO 12-14 KDa) against water, (1 x 4L for 1 h, 2 x 2L for 30 min), to afford the glycosylated product, m/z (ES) found 27074 calcd. 27077.
B. BSA (5 mg) was dissolved in degassed aqueous buffer solution (1 mL, 70 mM CHES, 5 mM MES, 2 mM CaCl2, pH 9.5). Phenyl 2,3,4,6-tetra-O-acetyl-β- D-selenenylsulfide glucopyranoside (10 mg, 0.02 mmol) was dissolved in acetonitrile (800 μl). The sugar solution (800 μl) was added to the protein solution and placed on an end-over-end rotator. After 1 h, the absence of free thiol was shown by EUman's analysis, at which point the reaction mixture was loaded onto a PD10 Sephadex® G25 column and eluted with 70 mM HEPES, 2 mM CaCl2ρH 7.0. The protein fraction was collected and dialysed (MWCO 12-14 KDa) against water, (1 x 4L for 1 h, 2 x 2L for 30 min), to afford the glycosylated product m/z (ES) found 66792 calcd. 66794.
C. SBLS 156C mutant (5 mg) was dissolved in degassed aqueous buffer solution (1 mL, 70 mM CHES, 5 mM MES, 2 mM CaCl2, pH 9.5). Phenyl 2,3,4,6 -tetra-O- acetyl-β-D-selenenylsulfide galactopyranoside (10 mg, 0.02 mmol) was dissolved in acetonitrile (500 μl). The sugar solution (500 μl) was added to the protein solution
and placed on an end-over-end rotator. After 1 h, the absence of free thiol was shown by EUman's analysis, at which point the reaction mixture was loaded onto a PD10 Sephadex® G25 column and eluted with 70 mM HEPES, 2 mM CaCl2ρH 7.0. The protein fraction was collected and dialysed (MWCO 12-14 KDa) against water, (1 x 4L for 1 h, 2 x 2L for 30 min), to afford Glc(Ac)4SBLSl56C m/z (ES) found 27074 calcd. 27077.
D. SBLS 156C mutant (10 mg) was dissolved in degassed aqueous buffer solution (1 mL, 70 mM CHES, 5 mM MES, 2 mM CaCl2, pH 9.5). Phenyl- 1- selenenylsulfide-β-D-glucopyranoside (15 mg, 0.02 mmol) was dissolved in water/acetonitrile (0.8 mL, 1/1 ratio). The sugar solution (500 μl) was added to the protein solution and placed on an end-over-end rotator. After 30 min, the absence of free thiol was shown by EUman's analysis, the reaction was placed on an end-over- end rotator for a further 30 min, at which point the reaction mixture was loaded onto a PD 10 Sephadex® G25 column and eluted with 70 mM HEPES, 2 mM CaCl2 pH 7.0. The protein fraction was collected and dialysed (MWCO 12-14 KDa) against water, (1 x 4L for 1 h, 2 x 2L for 30 min), to afford AcGlcSBLS156C m/z (ES) found 27072 calcd. 26911.
E. SBLS 156C mutant (5 mg) was dissolved in degassed aqueous buffer solution (2.4 mL, 70 mM HEPES, 2 mM CaCl2, pH 6.9). Phenyl 2-acetylamino-2-deoxy-l - selenenylsulfide-β-D-glucopyranoside (5 mg, 0.01 mmol) was dissolved in acetonitrile (200 μL, 1/1 ratio). The sugar solution (100 μl) was added to the protein solution and placed on an end-over-end rotator. After 30 min, the absence of free thiol was shown by EUman's analysis, at which point another portion of sugar solution (100 μl) was added. The reaction was placed on an end-over -end rotator for a further 30 min, at which point the reaction mixture was loaded onto a PD10 Sephadex® G25 column and eluted with 70 mM HEPES, 2 mM CaCl2pH 7.0. The protein fraction was collected and dialysed (MWCO 12-14 KDa) against 10 mM MES, 1 mM CaCl2, pH 5.8, (1 x 4L for 1 h, 2 x 2L for 30 min), to afford HOGlcNAcSBLS156C m/z (ES) found 26950 calcd. 26950.
F. SBLS 156C mutant (5 mg) was dissolved in degassed aqueous buffer solution (1 mL, 70 mM CHES, 5 mM MES, 2 mM CaCl2, pH 9.5). Phenyl 3,4,6-tri-O-acetyl- 2-acetylamino-2-deoxy-l-selenenylsulfide-β-D-glucopyranoside (10 mg,
0.02 mmol) was dissolved in acetonitrile (500 μl). The sugar solution (500 μl) was added to the protein solution and placed on an end- over-end rotator. After 1 h, the absence of free thiol was shown by EUman's analysis, at which point the reaction mixture was loaded onto a PD10 Sephadex® G25 column and eluted with 70 mM HEPES, 2 mM CaCkpH 7.0. The protein fraction was collected and dialysed (MWCO 12-14 KDa) against water, (1 x 4L for 1 h, 2 x 2L for 30 min), to afford AcGlcNAcSBLS 156C m/z (ES) found 27074 calcd. 27078.
G. SBLCysl 56 (5 mg) was dissolved in degassed aqueous buffer solution (500 μL, 70 mM CHES, 5 mM MES, 2 mM CaCl2, pH 9.5). Phenyl 2,3,6-tri-O- acetyl-l-selenenylsulfide-4-0-(2,3,6-tri-0-acetyl-4-0-(2,3,4,6-tetra-0-acetyl-α-0- glucopyranosyl)-α-D-glucopyranosyl)-β-D-glucopyranoside (15 mg, 0.015 mmol) was dissolved in acetonitrile (300 μL, 75 eq) and this solution was added to the protein solution and placed on an end-over-end rotator. After 30 min, the absence of free thiol was shown by EUman's analysis. The reaction was placed on an end-over- end rotator for a further 30 min, at which point the reaction mixture was loaded onto a PD10 Sephadex ® G25 column and eluted with 70 mM HEPES, 2 mM CaCl2, pH 7.0. The protein fraction was collected and dialysed (MWCO 12-14 KDa) against water (1 x 4L for 1 h, 2 x 2L for 30 min) to afford Glc(Ac)4Glc(Ac)3Glc(Ac) 3- SBLCysl56 m/z (ES+) found 27644 calcd. 27653.
H. SBLCys 156 (5 mg) was dissolved in degassed aqueous buffer solution (500 μL, 70 mM CHES, 5 mM MES, 2 mM CaCl2, pH 9.5). Phenyl 1- selenenylsulfide-β -D-galactopyranoside (15 mg, 0.04 mmol) was dissolved in water/acetonitrile (600 μL, 1/3 ratio). The sugar solution (600 μL, 230 eq) was added to the protein solution and placed on an end- over-end rotator. After 30 min, the absence of free thiol was shown by EUman's analysis/8-1 the reaction was placed on an end-over-end rotator for a further 30 min, at which point the reaction mixture was loaded onto a PD10 Sephadex ® G25 column and eluted with 70 mM HEPES, 2 mM CaCi2, pH 7.0. The protein fraction was collected and dialysed (MWCO 12-
14 KDa) against water (1 x 4L for 1 h, 2 x 2L for 30 min) to afford Gal-SBLCysl56 m/z (ES+) found 26908 calcd. 26909.
I. 1-Thio-β-D-maltotriose (104 mg, 0.2 mmol) was dissolved in MeOH (5 mL) to which a solution of PhSeBr (70 mg, 0.3 mmol) in EtOAc (2 mL) was added. After 2 min triethylamine (2 mL) was added and the reaction was diluted with water (10 mL) and petrol (5 mL). The phases were separated and the aqueous phase was washed with petrol (3 x 10 mL) and lyophilised. The crude phenyl 1 - selenenylsulfide-maltotriose (m/z 755, 757 (M+B , 100%)) was taken up into water (10 mL) of which 50 μL (25 eq) was added to a solution of SBLCys 156 (1 mg) in 500 μL of buffer (70 mM CHES, 5 mM MES, 2 mM CaCl2, pH 9.5). The resulting solution was placed on an end-over-end rotator. After 2.5 h the reaction mixture was loaded onto a PD10 Sephadex ® G25 column and eluted with 70 mM HEPES, 2 mM CaCi2, pH 7.0. The protein fraction was collected to afford GlcGlcGlc- SBLCysl56 m/z (ES+) found 27226 calcd. 27233.
J. BSA (5 mg) was dissolved in degassed aqueous buffer solution (1 mL,
70 mM CHES, 5 mM MES, 2 mM CaCl2, pH 9.5). Phenyl 1 -selenenylsulfide- β -D- gfαcopyranoside (6 mg, 0.02 mmol) was dissolved in water/acetonitrile (0.7 mL, 2/5 ratio). The sugar solution (700 μL, 225 eq) was added to the protein solution and placed on an end-over-end rotator. After 1 h, the absence of free thiol was shown by EUman's analysis 81 at which point the reaction mixture was loaded onto a PD10 Sephadex ® G25 column and eluted with 70 mM HEPES, 2 mM CaCl2, pH 7.0. The protein fraction was collected and dialysed (MWCO 12-14 KDa) against water (1 x T, for 1 h, 2 x 2L for 30 min) to afford Glc-BSA m/z (ES+) found 66620 calcd. 66625.
Summary of glycosylation reactions utilising selenenyl sulphide reagents
In Tables 1 and 2, MTS denotes CH3-SO2-S-, and PTS denotes Ph-SO2-S-.
Table 1: Preparation
1. from the corresponding parent carbohydrate D -glucose (Glc), D-galactose (Gal) or Glcα(l,4)Glcα(l,4)Glc. 2. Taken from B.G. Davis, R.C. Lloyd and J.B. Jones, 7 Org. Chem., 1998, 63, 9614, and B.G. Davis, M.A.T. Maughan, M.P. Green, A. UUman and J.B. Jones, Tetrahedron Asymmetry, 2000, 11, 245.
3. Taken from B. G. Davis, S. J. Ward and P. M. Randle, Chem. Commun., 2001, 189.
As shown in Table 1, the glyco-PTS reagents according to the invention were synthesis ed in superior yields to the corresponding glyco-MTS reagents. Moreover, the costs of the starting materials for synthesis of the glyco-PTS reagents was approximately ten fold lower than for the corresponding glyco-MTS reagents (at 2003 costs). In Table 2, SBL-Cys 156 is subtilisin Bacillus lentus mutant S 156C, and BSA-Cys58 is bovine serum albumin.
Table 2. Comparison of glycosylation reactions of glyco-MTS and glyco-PTS reagents.
1. Et3N, DCM, RT, 1 equivalent (eq.) of thiosulfonate. 2. Et3N, DCM/MeOH (20: 1), RT, 1 eq. of thiosulfonate; Peptide [P]-Cys-Ser-OMe, [P] = Ac except for reaction with Glc(Ac)4α(l,4)Glc(Ac)3α(l,4)Glc(Ac)3β -PTS where [P] = Boc. 3. 70mM CHES, 5mM MES, 2mM CaCl2 pH 9.5 or 50mM Tris.HCl, pH 7.7, RT, -30 eq. for glyco-MTS, -10 eq. for Glc(Ac)4 β-PTS and Gal(Ac)4β-PTS with SBL- Cysl56, -20 eq. for Glc(Ac)4β-PTS and Gal(Ac)4β-PTS with BSA-Cys58, -40 eq. for Glc(Ac)4α(l,4)Glc(Ac)3α(l,4)Glc(Ac)3 β-PTS with SBL-Cys 156. 4. Taken from B.G. Davis, R.C. Lloyd and J.B. Jones, 7 Org. Chem., 1998, 63, 9614, and B.G. Davis, M.A.T. Maughan, M.P. Green, A. UUman and J.B. Jones, Tetrahedron Asymmetry, 2000, 11, 245. 5. Taken from B. G. Davis, S. J. Ward and P. M. Randle, Chem. Commun., 2001, 189.
As can be seen from Table 2, the glyco-PTS reagents of the invention generally provided a higher yield in the glycosylation reaction than did the corresponding glyco-MTS compound. Example 58: Glycosylation of SBLCys 156 with GlcGlcGlc-S-SePh at varying pH
Reaction conditions: SBLCysl 56 was incubated for 1 h with GlcGlcGlc-S-SePh (20 eq.) in [a] 10 mM Tris pH 7.5; [b] 70 mM CHES, 5mM MES, 2 mM CaCl
2, pH 8.5; [c] 70 mM CHES, 5mM MES, 2 mM CaCl
2, pH 9.5.
Example 59: Representative Protein Farnesylation
SBLCysl 56 (10 mg) was dissolved in aqueous buffer solution (1 mL, 70 mM CHES, 5 mM MES, 2 mM CaCl2, pH 9.5). PMSF (140 μL of a 100 mg/mL solution in acetonitrUe) was added. After 10 minutes the reaction mixture was concentrated on a Vivaspin centrifugal filter (10 kDa MWCO, Sartorius); this step was repeated 3 times with addition of 300 μL of MnTi Q water. A portion of the resulting deactivated SBLCysl 56 (1 mg) was then dissolved in 200 μL of buffer (1 mL, 70 mM CHES, 5 mM MES, 2 mM CaCl2, pH 9.5). Farnesyl phenylthiosulfonates (56 μL of a 5 mg/mL solution in THF, 20 equivalents) were added. The mixture was placed in an end-over-end rotator. After 1 h the reaction was desalted using Nivaspin centrifugal filters (4 filtrations with addition of MUli Q water) and analysed by mass spectrometry.
This Example shows that the methods of the invention can also be used to attach farnesyl groups to proteins. Farnesylation is a natural post translational modification associated with many proteins.
Example 60: D-Mannose pentaacetate
Mannose (50 g, 280 mmol) was suspended in a stirred solution of acetic anhydride (200 mL) and pyridine (200 mL). After 24 h tic. (petrol:ethyl acetate, 1: 1) indicated the formation of a product (R 0.3) with complete consumption of the starting material (Rf 0.0). The reaction was diluted with water (400 mL) and partitioned with ethyl acetate (300 mL). The phases were separated, and the aqueous layer was re-extracted with ethyl acetate (2 x 200 mL). The combined organic layers were washed with dilute hydrochloric acid (2 L, 1M), sodium hydrogen carbonate
(500 mL of a saturated aqueous solution), brine (300 mL), dried over (MgSO4), filtered and concentrated in vacuo to afford the title compound (107.3 g, 98%) as an oU being a mixture of anomers (α/β 2:1); δH(400 MHz, CDC13) 1.95, 1.99, 2.05, 2.16 (15 H, 4 x s, COCH3β), 1.96, 2.00, 2.04, 2.12, 2.13 (15 H, 5 x s, COCH3α), 3.78 (IH, ddd, 74,5 9.9 Hz, 75,6 2.3 Hz, 75,6- 5.4 Hz, H-5β), 3.99-4.03 (m, H-5α),
4.05-4.10 (2H, m, H-6α, H-6β), 4.23 (IH, dd, 75,6> 5.0 Hz, J6fi> 12.1 Hz, H-6α), 4.26 (IH, dd, 75,6' 5.3 Hz, J6fi> 12.4 Hz, H-6'b), 5.10 (IH, dd, 72,3 3.3 Hz, 73,4 10.3 Hz, H-3β), 5.20-5.21 (IH, dd, 71;2 2.1 Hz, 72,3 2.5 Hz, H-2α), 5.24-5.30 (3H, m, H-3α, H-4α, H-4β), 5.43 (IH, dd, 71;2 1.2 Hz, 72,3 3.2 Hz, H-2β), 5.83 (IH, d, 71;2 0.9 Hz, H-lβ), 6.03 (IH, d, 7 2.1 Hz, H-lα).
Example 61: 2.3.4,6-Tetra-O-acetyl-α-D-mannopyranosoyl bromide
D-Mannose pentaacetate (103 g, 264 mmol) was dissolved in anhydrous DCM (200 mL). To this hydrogen bromide (33% in acetic acid, 200 mL) was added. The mixture was left under argon at RT. After a 2 h period, t.l.c. (petrol: ethyl acetate, 2: 1) indicated the formation of a product (Rf 0.3) with complete consumption of the starting material (Rf 0.2). The reaction mixture was partitioned between DCM (100 mL) and ice water (200 mL), and the aqueous layer re-extracted with DCM (3 x 200 mL). The combined organic layers were washed with sodium hydrogen carbonate until pH 8 was obtained, then with brine (300 mL), dried over (MgSO4), filtered and concentrated in vacuo. The resulting title compound, a clear oU, (106.6 g) was used without purification; δH (400 MHz, CDC13) 1.96, 2.03, 2.06, 2.13 (12H, 4 x s, 4 x OAc), 4.09 (IH, dd, 75;6 2.2 Hz, 76,6- 12.5 Hz, H-6), 4.18 (IH, dd, 74j5 10.1 Hz, 75,6 2.2 Hz, 75,6' 4.8 Hz, H-5), 4.28 (IH, dd, 75,6 4.9 Hz, 76,6- 12.5 Hz, H-6'), 5.32 (IH, at, 710.1 Hz, H-4), 5.39 (IH, dd, 71;2 1.6 Hz, 72,3 3.5 Hz, H-2), 5.66 (IH, dd, 72j3 3.5 Hz, 73;4 10.1 Hz, H-3), 6.26 (IH, bs, H-l).
Example 62: (2.3.4, 6-Tetra-O-acetyl-α-D-mannopyranosyl)-l-isothiouronium bromide
The title compound (80.6 g, 60%, 2 steps) was obtained as a white crystaUine sohd using a method analogous to that of Example 3 utilising 2,3,4,6-tetra-O-benzyl-D-α- mannopyranosoyl bromide as starting material. Mp 123-126 °C [Lit. 125-128 °C (H2O)]; [α]D 26+l 19.0 (c, 1.0 in MeOH) [Lit. [α]D 27+103 (c, 1.0 in Acetone)]; δH (400 MHz, DMSO-de) 1.95, 2.02, 2.03, 2.14 (12H, 4 x s, 4 x OAc), 4.08 (IH, dd, 75,6 2.4 Hz, 6,6' 12.3 Hz, H-6), 4.22 (IH, dd, 75,6' 2.4 Hz, 76,6' 12.5 Hz, H-6'), 4.32 (IH, ddd, 74,5 10.0 Hz, 75,6 2.2 Hz, 75,6' 5.2 Hz, H-5), 5.05 (IH, dd, 72,3 3.4 Hz, 73;4 10.0 Hz, H-3), 5.17 (IH, at, 710.0 Hz, H-4), 5.36 (IH, dd, 71;2 1.5 Hz, 72j3 3.4 Hz, H-2), 6.36 (IH, d, 7ι,2 1.2 Hz, H-l), 9.40 (4H, bs, 2 x NH2).
Example 63: 2.3.4.6-Tetra-O-acetyl-α-D-mannopyranosylthiol
The title compound (14.5 g, 98%) was obtained as a colourless oU by a method analogous to that of Example 2 utilising (2,3,4, 6-tetra-O-acetyl-α-D- mannopyranosyl)-l-isothiouronium bromide as starting material. [α]D24+68.7 (c, 1.5 in CHC13) [Lit. [α]D 20+78.6 (c, 0.8 in CHC13)]; δH(400 MHz, CDC13) 1.98, 2.04, 2.08, 2.14 (12H, 4 x s, 4 x OAc), 2.28 (IH, d, 71JSH 6.7 Hz, SH), 4.10 (IH, dd, 75,6 2.4 Hz, 76,6> 12.5 Hz, H-6), 4.28 (IH, dd, 75,6' 5.1 Hz, 76;6- 12.0 Hz, H-6'), 4.32- 4.36 (IH, m, H-5), 5.26-5.34 (3H, m, H-2, H-3, H-4), 5.54 (IH, d, 71;SH 6.9 Hz, H-l).
Example 65: Phenyl 2.3.4.6-tetra-O-acetyl-l-selenenylsulfide-α-D- mannopyrano side
The title compound (590 mg, 83%) was obtained as a yellow oU using a method analogous to that of Example 49 utilising 2,3,4,6-tetra-O-acetyl-α-D- mannopyranosyl thiol as the starting material. [O]D 25 +13.4 (c, 1.0 in CHC13; δπ(400 MHz, CDC13) 1.94, 1.94, 2.02, 2.10 (12H, 4 x s, 4 x OAc), 3.52 (IH, dd, 75,6 2.4 Hz, 76,6' 12.4 Hz, H-6), 3.94 (IH, ddd, 74;5 9.6 Hz, 75,6 2.5 Hz, 75>6< 3.9 Hz, H-5), 4.07 (IH, dd, 75,6- 3.9 Hz, 76,6' 12.4 Hz, H-6'), 5.23 (IH, dd, 72,3 3.2 Hz, 73,49.9 Hz, H-3), 5.28 (IH, at, 79.7 Hz, H-4), 5.38 (IH, d, 71;2 1.6 Hz, H-l), 5.40 (IH, dd, 7 1.5 Hz, 72,3 3.1 Hz, H-2), 7.26-7.28 (3H, m ArH), 7.62-7.65 (2H, m, ArH).
Example 66: 2.3.4.6-Tetra-O-acetyl-α-D-mannopyranoside
D-Mannose pentaacetate (26.4 g, 67.7 mmol) was dissolved in freshly distilled THF (150 mL) and benzylamine (11.1 mL, 101.5 mmol) was added to the stirred solution. After a 24 h period, tl.c. (petrol:ethyl acetate, 1:1) indicated the formation of a product (Rf 0.3) with complete consumption of the starting material (Rf 0.5). The reaction was quenched with the addition of diluted hydrochloric acid (100 mL, 1M) and stirred for 10 min. The reaction was partitioned with DCM (100 mL) and the phases were separated. The aqueous phase was re-extracted with DCM (3 x 100 mL). The combined organics were washed with dilute hydrochloric acid (100 mL, 1M), brine (100 mL) and dried (MgSO ) and concentrated in vacuo. The resulting orange oU was purified by flash column chromatography (petrol: ethyl acetate, 1 : 1). The off white crystals were combined and recrystaUised from petrol/ethyl acetate to afford the title compound (12.4 g, 53%) as a white crystaUine sohd mp 92-94 °C [Lit. 92°C]; [α]D 25 +17.8 (c, 1.0 in CHC13); [Lit. [α]D 25+21.0 (c,
1.0 in CHC13)]; δH(400 MHz, CDC13) 1.98, 2.04, 2.08, 2.14 (12H, 4 x s, 4 OAc), 4.09-4.14 (IH, m, H-6), 4.20-4.26 (2H, m, H-5, H-6'), 4.59-5.00 (IH, m, OH), 5.20-5.23 (2H, m, H-l, H-2), 5.27 (IH, at, 79.9 Hz, H-4), 5.39 (IH, dd, 72;3 2.7 Hz, 73;4 9.6 Hz, H-3).
Example 67: lM'.l'-Trichloro acetimidate 2.3.4.6-tetra-O-acetyl-α-D- mannopyrano side
2,3,4,6-Tetra-O-acetyl-α-D-mannopyranoside (1.01 g, 2.87 mmol), 1,1,1-trichloroacetonitrile (2.9 mL, 28.7 mmol) and activated 4A molecular sieves (ca. 500 mg) were suspended in anhydrous DCM (20 mL) and left stirring at 0 °C for a period of 1 h. At which point DBU (0.085 mL, 0 57 mmol) was added. After a 1.5 h period, t.l.c. (petrol: ethyl acetate, 1:1) indicated the formation of a product (Rf 0.5) with complete consumption of the starting material (Rf 0.2). The reaction was filtered through Celite® and concentrated in vacuo. The resulting residue was purified by flash column chromatography (petrol: ethyl acetate, 1 : 1) to afford the title compound (1.42 g, 99%) as a clear oU; [α]D 25+42.7 (c, 1.0 in CHC13) [Lit. [α]D 21 +50.0 (c, 1.0 in CHCI3)]; δH(400 MHz, CDC13) 2.20, 2.07, 2.09, 2.29 (12H, 4 x s, 4 x OAc), 415-4.22 (2H, m, H-5, H-6), 4.28 (IH, dd, J5j6. 4.3 Hz, 76,6' 11.8 Hz, H-6'), 5.40-5.42 (2H, , H-3, H-4), 5.48 (IH, at, 72.1 Hz, H-2), 6.29 (Hi d, 71>2 1.9 Hz, H-l), 8.80 (IH, s, NH)
Example 68: Benzyl-α-D-mannopyranoside
D-Mannose (30 g, 167 mmol) and acetyl chloride (13 mL, 167 mmol) was dissolved in benzyl alcohol (250 mL) and heated to 50 °C for 1 h. The resulting solution was concentrated by low pressure distillation. The resulting residue was purified by flash column chromatography (ethyl acetate/methanol, 9:1) and recrystaUised from isopropanol/petrol to afford the title compound (29.34 g, 70%) as a white crystaUine sohd m.p. 126-127 °C [Lit 128-129 °C]; [α]
D 26 +102.0 (c, 1.1 in MeOH); [Lit. [αj
D 18 +731 (c, 1.4 in H
2O)]; δ
H(400 MHz, CD
3OD) 3.62 (IH, ddd, 7
4;5 9.5 Hz, 7
5,
6 2.3 Hz, 7
5;6' 5.5 Hz, H-5), 3.68 (IH, at, 79.3 Hz, H-4), 3.733.78 (2H, m, H-3, H-6), 3.85-3.88 (2H, m, H-2, H-6'), 4.75, 4.52 (2H, ABq, 711.6 Hz, CH
2), 4.86 (IH, d, 7
1>2 1.8 Hz, H-l), 7.28-7.38 (5H, m, ArH).
Example 69: Benzyl 4,6-di-O-pivolyl-α-D-mannopyranoside
Benyzl-α-D-mannopryanoside (30.0 g, 111.0 mmol) was suspended in anhydrous pyridine (200 mL) under an atmopshere of inert argon. The resulting suspension was cooled to 0 °C and chlorotriphenyl methane (35 mL, 280 mmol) was added to dropwise. After the addition of the chlorotriphenyl methane, t.l.c. (ethyl acetate) indicated the formation of a major product (Rf 0.7) with complete consumption of the starting material (Rf 0.0). The reaction was partitioned between water (50 mL) and ethyl acetate (100 mL). The phases were separated and the aqueous phase was re-extracted with ethyl acetate (3 x 50 mL). The combined organics were washed with dUute hydrochloric acid (IL, 1M), sodium hydrogen carbonate (800 mL of a saturated aqueous solution) until pH 7 was obtained, brine (200 mL), dried
(MgSO ) and concentrated in vacuo. The resulting residue was recrystaUised from ethyl acetate/petrol to afford the title compound (27.07 g, 56%) as a white crystalline sohd mp 133-135°C; [α]D 25+64.7 (c, 1.0 in CHC13); δH(400 MHz, CDC13) 1.251, 1.254 (18H, 2 x s, 2 x C(CH3)3), 3.85 (IH, at, 79.8 Hz, H-4), 3.92 (IH, ddd, 74,5 9.7 Hz, 75;6 5.6 Hz, J5JS> 2.5 Hz, H-5), 4.05 (IH, dd, 7 1.9 Hz, 72,3
2.1 Hz, H-2), 4.37 (IH, dd, 75,6 5.6 Hz, 76,6' 11.8 Hz, H-6), 4.42 (IH, dd, 75,6- 2.7 Hz,
76,6' 12.0 Hz, H-6'), 4.53, 4.76 (2H, Abq, 711.9 Hz, CH2), 4.90 (IH, d, 7,2 1.8 Hz, H-l), 5.14 (IH, dd, 72,3 3.2 Hz, 73,4 9.7 Hz, H-3), 7.33-7.36 (5H, m, ArH).
Example 70: Benzyl 2.4-di-O-benzyl-3.6-di-O-pivolyl-α-D-mannopyranoside
Benzyl 4,6-di-O-pivolyl-α-D-mannopyranoside (15.0 g, 34.2 mmol) and benzene trichloroacetimidate (17 mL, 91.4 mmol) were dissolved in anhydrous DCM (100 mL) and anhydrous cyclohexane (100 mL) and left stirring for 1 h over 4A molecular sieves (ca 5 g) under an inert atmosphere of argon. After 1 h trimethyl sUyltriflate (0.31 mL, 1.71 mmol) was added. After a 18 h period, t.l.c. (petrol:ethyl acetate, 5:1) indicated the formation of a major product (Rf 0.4) with complete consumption of the starting material (Rf 0.0). The reaction was quenched with triethylamine (ca 30 mL) and the solution was filtered through Celite and concentrated in vacuo.. The resulting residue was purified by flash column chromatography (petrol:ethyl acetate, 5: 1) to afford the title compound (14.4 g, 70%) as a colourless oU; [α]D 25+29.0 (c, 2.0 in CHC13); δH(400 MHz, CDCI3) 1.24, 1.25 (18H, 2 x s, 2 x C(CH3)3), 3.97-4.04 (3H, m, H-2, H-4, H-5), 4.25 (IH, dd, 75,6 4.8 Hz, J5.6- 11.6 Hz, H-6), 4.44 (IH, dd, 75f6' 1.6 Hz, 76.6. 11.7 Hz, H-6'), 4.51, 4.74 (2H, ABq, 12.0 Hz, BnCH2), 4.55, 4.61 (2H, ABq, 7 11.7 Hz, BnCH2), 4.57,4.80 (2H, ABq, 710.7 Hz, BnCH2), 4.92 (IH, d, 71>2 1.8 Hz, H-l), 5.37 (IH, dd, 72,3 3.1 Hz, 73,4 8.8 Hz, H-3), 7.28-7.35 (15H, m, ArH).
Example 71: Benzyl 2.4-di-O-benzyl-α-D-mannopyranoside
Benzyl 2,4-di-0-benzyl-3,6-di-( -pivolyl-α-D-mannopyranoside (8.0 g, 12.9 mmol) and sodium methoxide (1.75 g, 32.4 mmol) were dissolved in methanol (100 mL) and heated to reflux. After a 20 h period, tl.c. (petrol/ethyl acetate, 2: 1) indicated the formation of a major product (R
f 0.2) with complete consumption of the starting material (R
f 0.8). The reaction was neutralised with the addition of Dowex®-50 ion exchange resin after which point the reaction was filtered and concentrated in vacuo. The resulting residue was purified by flash column chromatography (petrol/ethyl acetate, 2: 1) to afford the title compoxmd (4.50 g, 78%) as a clear oil; [α]D
25 +45.2 (c, 1.0 in CHC1
3); δ
H(500 MHz, CDC1
3) 2.83 (2H, bs, 2 x OH), 3.83-3.86 (IH, m, H-5), 3.90-4.00 (4H, m, H-2), H-4), H-6, H-6'), 4.21-4.28 (IH, m, H-3), 4.58 (IH, d, 712.1 Hz, CHH), 472 -4.83 (4H, m, 4 x CIL r), 5.04 (IH, d, 7 11.1 Hz, CHH), 5.09 (IH, bs, H-l), 7.43-7.51 (15H, m, 15 x ArH).
Example 71: Benzyl 2,4-di-O-benzyl-3,6-bt5,-0-(2,3,4,6-tetra-O-acetyl-α-D- mannopyranoside)-α-D-mannopyranoside
Benzyl 2,4-di-
side (255 mg, 0.57 mmol) in
DCM (10 mL) and r,l',r-trichloiOacetimidate-2,3,4,6-tetra-0-acetyl-α-D- mannopyranoside (1.12 g, 2.27 mmol) in DCM (10 mL) were added to a dried flask containing activated 4A molecular sieves (ca 500 mg) via cannular. The resulting solution was stirred for 1 h, after which boron trifluoroetherate (90 μL, 0.85 mmol) was added. After a 16 h period, tl.c. (petrol: ethyl acetate, 2: 1) indicated the formation of amajor product (Rf 0.3) with complete consumption of the starting material (Rf 0.1). The reaction was quenched with triethylamine (ca 5 mL) and the solution was filtered through Celite and concentrated in vacuo. The resulting residue
was purified by flash column chromatography (petro ethyl acetate, 4:3) to afford the title compound (472 mg, 75%) as a white amorphous sohd; [α]D 25 +81.5 (c, 1.0 in CHCI3); δH(500 MHz, CDC13) 1.98, 2.02, 2.05, 2.07, 2.09, 2.10, 2.11, 2.19 (24H, 8 x s, 8 x OAc), 3.74-3.76 (IH, m, H-6a), 3.81-3.87 (3H, m, H-2a, H-5a, H-6'a), 3.92-3.97 (3H, m, H-4a, H-5b, H-6b), 4.03-4.22 (4H, m, H-3a, H-5c, H-6'b, H-6c), 4.27 (IH, dd, 75;6' 5.5 Hz, 76>6' 12.3 Hz, H-6'c), 4.54, 4.75 (2H, Abq, 7119 Hz, CH2), 4.64, 4.81 (2H, Abq, 712.2 Hz, CH2), 4.65, 4.91 (2H, Abq, 7 11.4 Hz, CH2), 4.97 (IH, d, 7i;2 1.7 Hz, H-lc), 5.00 (IH, d, 7 1.6 Hz, H-la), 5.19 (lH,d, 7 1.7 Hz, H-lb), 5.25 (IH, at, 710.0 Hz, H-4b), 5.33 (IH, at, 710.1 Hz, H-4c), 5.36 (IH, dd, 7,2 1.8 Hz, 7,3 3.3 Hz, H-2c), 5.42 (IH, dd, 7 1.5 Hz, 72,3 3.5 Hz, H-2b), 5.44-5.47 (2H, m, H-3b, H3c), 7.32-7.42 (15H, m, ArH).
Example 72: Acetyl 2.4-di-O~acetyl-3.6-tø-O-f2.3.4.6-tetra-O-acetyl-α-D- mannopyranoside)-α/β-D-mannopyranoside
Benzyl 2,4-di-O-benzyl-3,6-bw-O-(2,3,4,6-tetra-O-acetyl-a-D-mannopyranoside)- α-D-mannopyranoside (100 mg, 0.09 mmol) and Pearlman's catalyst (Pd(OH)2, moist, 35 mg) were dissolved in absolute ethanol (5 mL). The resulting solution was degassed and purged with hydrogen gas, then left to stir under an atmosphere of hydrogen. After a 4 day period, t.l.c. (ethyl acetate) indicated the formation of a major product (Rf 0.4) with complete consumption of the starting material (Rf 0.9). The solution was filtered through Cehte and concentrated in vacuo. The resulting residue was purified by flash column chromatography (ethyl acetate) to afford the intermediate 3, 6-bw-(2,3,4,6-tetra-O-acetyl-α-D-mannopyranoside)-α/β-D-
mannopyranoside (74 mg, 98%) as a white amorphous solid; m/z HRMS (ES+) Calcd. for C34H 8O34Na (MNa+) 863.2433. Found 863.2440. This intermediate (74 mg, 0.088 mmol) was resuspended in acetic anhydride (5 mL) and pyridine (5 mL). After 24 h tl.c. (petrol: ethyl acetate, 2:3) indicated the formation of a product (Rf 0.4) with complete consumption of the starting material (Rf 0.0). The reaction was diluted with water (20 mL) and partitioned with ethyl acetate (20 mL) and the phases were separated. The aqueous layer was re-extracted with ethyl acetate (2 x 20 mL). The combined organic layers were washed with dilute hydrochloric acid (500 mL, 1M), sodium hydrogen carbonate (50 mL of a saturated aqueous solution), brine (30 mL), dried over MgSO4, filtered and concentrated in vacuo to give the title compound (83 mg, 98%) as an amoφhous foam being a mixture of anomers (α/β 5: 1); δH (500 MHz, CDC13) α compound, 2.00, 2.02, 2.08, 2.12, 2.17, 2.18, 2.19, 2.26 (33H, 8 x s, 11 x OAc), 3.59 (IH, dd, 75)6 3.0 Hz, 76,6> 11.1 Hz, H-6a), 3.76 (IH, dd, 75,6' 5.2 Hz, 76,6' 11.2 Hz, H-6'a), 3.92 (IH, ddd, 74,5 10.2 Hz, 75,6 3.0 Hz, 75)6' 5.2 Hz, H-5a), 4.04-416 (4H, m, H-5b, H-5c, H-6b, H-6c), 4.21 (IH, dd, 72,3 3.4 Hz, 73,4 9.9 Hz, H-3a), 4.28 (IH, dd, 75)6' 5.5 Hz, 76j6' 12.2 Hz, H-6'b/c), 4.31 (IH, dd, 75]6' 4.7 Hz, 76)6' 12.3 Hz, H-6'b/c), 4.81 (IH, d, 71;2 1.5 Hz, H-lc), 5.06-5.07 (2H, m, H-lb, H-?), 5.20-5.35 (8H, m, H-2a, H-2b, H -2c, H-3b, H- 3c, H-4a, H-4b, H-4c), 6.07 (IH, d, Jι,2 1.8 Hz, H-la). β compound selected data only 3.64 (IH, dd, 75)6 3.7 Hz, 76,6' 10.8 Hz, H-6a), 3.69-3.73 (IH, m, H-5a), 3.76 (IH, dd, Jsjs* 5.2 Hz, 76j6> 11.2 Hz, H-6'a), 4.01 (IH, dd, 72;3 3.2 Hz, 73)4 9.1 Hz, H-3a), 5.50 (IH, dd, 7lj2 0.9 Hz, 72;3 3.2 Hz, H-2a), 5.83 (IH, d, 71>2 0.9 Hz, H-la).
Example 73: 2,4-Di-O-aeetyl-fe-0-(2,3,6-tri-0-aeetyl-- α-O-mannopyranosyl)-α-D- mannopyranosyl bromide
Acetyl 2,4-di-O-acetyl-3,6-bw-O-(2,3,4,6-tetra-O-acetyl-α-D-mannopyranoside)- α/β-D-mannopyranoside (87 mg, 0.09 mmol) was dissolved in anhydrous DCM (5 mL). To this hydrogen bromide (33% in acetic acid, 1 mL) was added. The mixture was stirred under argon at RT. After a 2 h period, tic. (petrol: ethyl acetate, 1:4) indicated the formation of a product (R
f 0.6) with complete consumption of the starting material (R
f 0.4). The reaction mixture was partitioned between DCM (10 mL) and water (10 mL), and the aqueous layer was re-extracted with DCM (3 10 mL). The combined organic layers were washed with sodium hydrogen carbonate (20 mL of a saturated aqueous solution) until pH 8 was obtained, brine (20 mL), dried over MgSO , filtered and concentrated in vacuo to afford the title compound (80 mg, 90%) as a white foam which was taken on without further purification; δ
H(400 MHz, CDC1
3) 1.97, 1.99, 2.05, 2.06, 2.10, 2.12, 2.17, 2.24 (30H, 9 x s, 10 x OAc), 3.60 (IH, dd, 7
5)6 3.0 Hz, 7
6,
6> 11.4 Hz, H-6a), 3.77 (IH, dd, 7
5,6
' 4.5 Hz, 7
6,6
' 11.4 Hz, H-6'a), 4.02-4.09 (5H, m, H-5a, H-5b, H -5c, H-6b, H-6c), 4.24 (IH, dd, 7
5,6
' 6.8 Hz, 7
6j6' 12.2 Hz, H-6'), 4.29 (IH, dd, 7
5,
6' 5.0 Hz, 7
6,
6' 12.6 Hz, H-6'), 4.62 (IH, dd, 7
2)3 3.4 Hz, 7
3)4 10.0 Hz, H-3a), 4.79 (IH, bs, H-lc), 5.02-5.04 (2H, m, H-lb, H-3b), 517-5.30 (5H, m, H-2b, H-2c, H-3c, H-4b, H-4c), 5.39 (IH, at, 7101 Hz, H-4a), 5.43 (IH, dd, 7ι,
2 15 Hz, 7
2)3 3.2 Hz, H-2a), 6.34 (IH, bs, H-la).
Example 74: l-Tlιio-2,4-tetra-Q-acetyl-3,6-0-b/.s,-(2,3,4,6-tetra-Q-acetyl-α-Q- mannopyranosyl)-α-D-mannopyranose
2,4-Tetra-O-acetyl-3,6-O-bM-(2,3,4,6-texra-0-acetyl-α-O-mannopyranosyl)-α-D- mannopyranosyl bromide (850 mg, 0.85 mmol) was dissolved in anhydrous acetone (20 mL). Anhydrous thiourea (115 mg, 1.56 mmol) was added and the mixture was heated to reflux under an atmosphere of argon. After 18 h, tl.c. (petrol: ethyl acetate, 1:3) indicated the formation of a product (R
f 0.0) with complete consumption of the starting material (R
f 0.4). The reaction was concentrated in vacuo and the resutling residue was purified by column flash chromatography (ethyl acetate/methanol, 9: 1) to afford the intermediate 2,4-tetra-O-acetyl-3,6-O-bw-(2,3,4,6-tetra-O-acetyl-α-O- mannopyranosyl)-α-D-mannopyranosyl-l-isothiouronium bromide (550 mg, 60%) which was carried on. This intermediate (550 mg, 0.51 mmol) andNa
2S2θ5
(122 mg, 0.62 mmol) were added to a stiπ-ed mixture of DCM (20 mL) and water (10 mL). The mixture was heated to reflux under argon. After 2.5 h, tl.c. (petrol: ethyl acetate, 1:3) indicated the formation of a product (Rf 0.3) with complete consumption of the starting material (Rf 0.0), at which point the reaction was cooled to RT and the phases separated. The aqueous layer was re-extracted with DCM (2 x 20 mL). The combined organic layers were washed with sodium hydrogen carbonate (20 mL of a saturated aqueous solution), brine (20 mL), dried (MgSO ), filtered and the solvent removed in vacuo. The resulting residue was purified by flash column chromatography (petrol: ethyl acetate, 1:3) to afford the title compound (350 mg, 73%>) as a white amorphous solid; [QJD23 +58.1 (c, 1.2 in CHCI3; δπ (500 MHz, C6D6) 1.74, 1.75, 1.78, 1.82, 1.91, 2.03, 2.06, 2.26 (24H, 8 x s, 10 x Oac), 2.07 (IH, bs, SH), 3.65 (IH, dd, Jsfi 3.2 Hz, J6fi> 11.0 Hz, H-6a), 3.93 (IH, dd, 75;6' 5.3 Hz, 76)6' 11.1 Hz, H-6'a), 4.31-4.38 (4H, m, H-3a, H-5a, H-5b/c, H-6), 4.43-4.45 (IH, m, H-6), 4.51 (IH, dd, 75)6' 5.6 Hz, J6jS> 12.6 Hz, H-6'), 4.56- 4.60 (2H, m, H-5b/c, H-6'), 4.91 (IH, d, 71;2 15 Hz, H-lc), 5.20 (IH, d, 7ι,2 1.8 Hz, H-lb), 5.43 (IH, dd,7 1.8 Hz, 72)3 3.1 Hz, H-2b), 5.45 (IH, bs, H-l), 5.65 (IH, dd, 71)2 1.5 Hz, 72,3 3.1 Hz, H-2a), 5.70-5.82 (5H, m, H-2c, H-3b, H-4a, H-4b, H-4c), 5.85 (IH, dd, 72,3 3.2 Hz, 73,4 10.2 Hz, H-3c).
Example 75: Representative protein glycosylation procedures of SBLCys 156 using Man(l-6)Man(l-3)ManSH
1 -Thio-2,4-tetra-O-acetyl-3 ,6 -0-bis-(2,3 ,4,6-tetra- O-acetyl-α-O-mannopyranosyi)- α-D-mannopyranose (20 mg, 0.02 mmol) and sodium methoxide (2 mg, 0.02mmol)
were added to a stirred solution of methanol (5 mL). After 12 h, (petrol: ethyl acetate, 1:2) indicated the formation of a product (Rf 0.0) with the complete consumption of the starting material (Rf 0.2). The reaction was neutralised with the addition of Dowex®-50 ion exchange resin after which point the reaction was filtered and concentrated in vacuo. The crude sugar thiol was taken up into water (5 mL) of which 38 μL was added to aqueous buffer solution (500 μL, 70 mM CHES, 5 mM MES, 2 mM CaCl2, pH 9.5) containing SBL156CysSePh (1 mg). The resulting solution placed on an end-over-end rotator. After 1 h the reaction mixture was loaded onto a PD10 Sephadex® G25 column and eluted with 70 mM HEPES, 2 mM CaCl2, pH 7..0. The protein fraction was collected to afford Man(Man)Man-S- SBLCysl 56; m/z (ES+) found 27878, calcd. 27881.