WO2004105698A2 - Use of phosphatase inhibitors as adjunct therapy for psychiatric disorders - Google Patents
Use of phosphatase inhibitors as adjunct therapy for psychiatric disorders Download PDFInfo
- Publication number
- WO2004105698A2 WO2004105698A2 PCT/US2004/016492 US2004016492W WO2004105698A2 WO 2004105698 A2 WO2004105698 A2 WO 2004105698A2 US 2004016492 W US2004016492 W US 2004016492W WO 2004105698 A2 WO2004105698 A2 WO 2004105698A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phenyl
- ethyl
- methylsulfonyl
- methyl
- difluoromethoxy
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
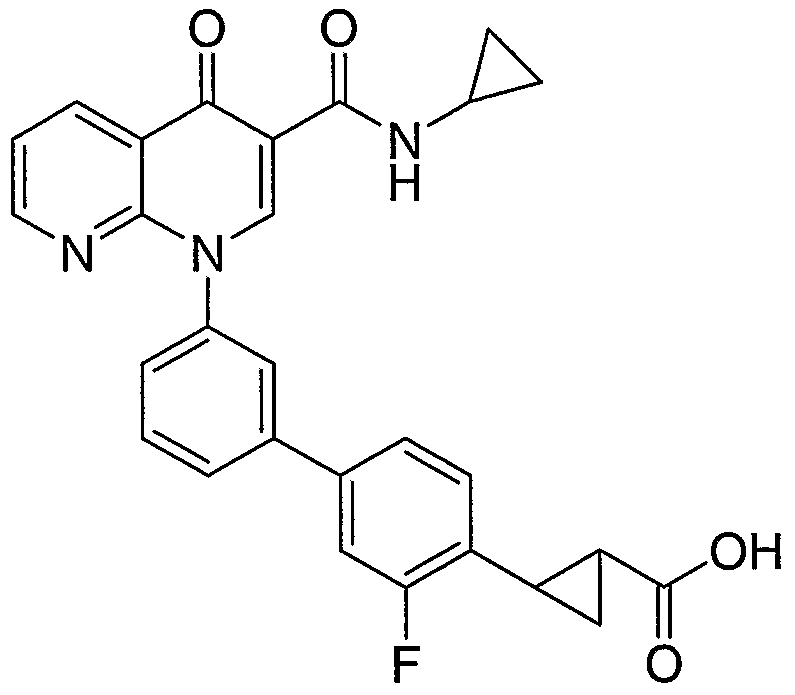
- OAQVFRKCYQNZFP-UHFFFAOYSA-N OC(C(C1)C1C1C=CC(c2cccc(N(C=C3C(NC4CC4)=O)c4ncccc4C3=O)c2)=CC1)=O Chemical compound OC(C(C1)C1C1C=CC(c2cccc(N(C=C3C(NC4CC4)=O)c4ncccc4C3=O)c2)=CC1)=O OAQVFRKCYQNZFP-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/435—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom
- A61K31/4353—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems
- A61K31/436—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with one nitrogen as the only ring hetero atom ortho- or peri-condensed with heterocyclic ring systems the heterocyclic ring system containing a six-membered ring having oxygen as a ring hetero atom, e.g. rapamycin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
Definitions
- This Patent Application relates to the use of phosphatase inhibitors as an adjunct in the treatment of psychiatric disorders.
- this Patent Application relates to the use of Calcineurin (CN) inhibitors and protein phosphatase 1 (PP1) inhibitors as adjunct therapy in behavior psychotherapy, cognitive psychotherapy, psychodynamically oriented psychotherapy and in the treatment of posttraumatic stress disorder.
- CN Calcineurin
- PP1 protein phosphatase 1
- the Classical fear conditioning occurs when an affectively neutral stimulus is paired with a noxious aversive stimulus (unconditioned stimulus (US)) such as footshock.
- US unconditioned stimulus
- the previously neutral stimulus i.e., now the conditioned stimulus (CS)
- CS conditioned stimulus
- a reduced ability to extinguish intense fear memories is a significant clinical problem for a wide range of psychiatric disorders including specific phobias, panic disorder, and posttraumatic stress disorder. Because treatment of these disorders often relies upon the progressive extinction of fear memories, pharmacological enhancement of extinction could be of considerable clinical benefit in these conditions.
- PKA on one hand, appear to be important for the initiation of synaptic plasticity and for learning and memory. PKA also appears to be required for persistent long term potentiation (LPT), important for synaptic strengthening in learning and memory. CN, on the other hand, inhibits synaptic plasticity and memory storage. Similarly, PP1 appears to promote forgetting. Recent studies by Malleret, et. al., Cell, vol. 104, 675-686, 2001, indicate that the balance can be shifted in favor of learning and memory storage by inhibiting CN.
- Lin, et. al appears to be that extinction of fear memory is promoted by enhancing the subjects capacity to either forget the original traumatic event or forget the linkage between original traumatic event and the stimulus that reminds the subject of that event.
- Hormones are compounds that variously affect cellular activity. In many respects, hormones act as messengers to trigger specific cellular responses and activities. Many effects produced by hormones, however, are not caused by the singular effect of just the hormone. Instead, the hormone first binds to a receptor, thereby triggering the release of a second compound that goes on to affect the cellular activity. In this scenario, the hormone is known as the first messenger while the second compound is called the second messenger.
- Cyclic adenosine monophosphate (adenosine 3', 5 '-cyclic monophosphate, "cAMP” or “cyclic AMP”) is known as a second messenger for hormones including epinephrine, glucagon, calcitonin, corticotrophin, lipotropin, luteinizing hormone, norepinephrine, parathyroid hormone, thyroid- stimulating hormone, and vasopressin.
- cAMP mediates cellular responses to hormones.
- Cyclic AMP also mediates cellular responses to various neurotransmitters.
- PDE Phosphodiesterases
- PDE4 Phosphodiesterases
- Tully and Cavallieri disclose in US Patent Application Publication No. WO02/0076398 a method of therapy for cognitive deficits associated with a central nervous system disorder and methods of enhancing cognitive performance by combining cognitive training protocols with administration of CREB pathway-enhancing agents. Tully and Cavallieri does not disclose the use of this combination therapy for psychiatric disorders
- Davis et al disclose in PCT International Application No. WO02/078629 methods for treating an individual with a psychiatric disorder with a pharmacologic agent that enhances learning or conditioning in combination with a session of psychotherapy. Davis et al does not disclose the use of PDE4 inhibitors in combination with psychotherapy. Reines et al disclose in PCT International Application No. WOO 1/64223 the combination of a neurokinin-1 antagonist or an alpha-2 adrenoreceptor agonist with a PDE4 inhibitor for the treatment or prevention of depression and/or anxiety. Reines et al does not disclose the use of PDE4 inhibitors to augment the effect of psychotherapy. In a second aspect, we have concluded that inhibition of phosphatase and PDE4, in conjunction with appropriate therapy is effective in the extinction of fear.
- the present invention provides a method for the treatment of psychiatric disorders using a phosphatase inhibitor in conjunction with psychotherapy.
- a phosphatase inhibitor and optionally a PDE4 inhibitor
- the inclusion of a phosphatase inhibitor (and optionally a PDE4 inhibitor) in the treatment modality enhances the effectiveness of psychotherapy resulting in fewer sessions required to achieve improvement, shorter intervals between sessions, or more pronounced improvement of symptoms, as compared to psychotherapy alone.
- the present invention provides a method for the treatment of psychiatric disorder in a patient, said method comprises administering to said patient a therapeutically effective amount of a phosphatase inhibitor in combination with psychotherapy.
- the method comprises subjecting the individual to one or more sessions of a combination therapy protocol, where the combination therapy protocol comprises administering a therapeutically effective amount of a phosphatase inhibitor in combination with a session of psychotherapy.
- the invention is directed to method for the treatment of psychiatric disorder in a patient, said method comprises administering to said patient a therapeutically effective amount of a phosphatase inhibitor in combination with psychotherapy.
- the psychiatric disorder is posttraumatic stress disorder.
- the psychotherapy is selected from behavior psychotherapy, cognitive psychotherapy, and psychodynamically oriented psychotherapy.
- the psychotherapy is exposure-based psychotherapy.
- the psychotherapy is cognitive psychotherapy.
- the psychotherapy is structured as to enhance acquisition and consolidation, as opposed to enhance the ability forget undesired memories.
- phosphatase is calcineurin or PP 1.
- the method comprises administering to said patient a therapeutically effective amount of a calcineurin inhibitor in combination with psychotherapy.
- the calcineurin inhibitor is selected from Rapamycin and Tacrolimus.
- the PDE4 inhibitor is administered prior to a session of said psychotherapy.
- the PDE4 inhibitor is selected from cilomilast, roflumilast and roflumilast N-oxide.
- a phosphatase inhibitor and optionally a PDE4 inhibitor
- a single dose of the phosphatase inhibitor or the PDE-4 inhibitor or both will be administered in connection with a session of psychotherapy.
- a single dose of a calcineurin inhibitor or a PDE-4 inhibitor or both may be taken 1 to 24 hours (e.g. 1, 2, 4, 6, 8, 12, 16 or 24 hours) before the session.
- psychiatric disorder refers to a disorder that can be treated with the methods of the present invention.
- an individual said to have a psychiatric disorder will have one or more disorders that can be treated with the methods of the present invention.
- an individual may have a single disorder, or may have a constellation of disorders that are to be treated by the methods described herein.
- the psychiatric disorders contemplated in the present invention include, but are not limited to, fear and anxiety disorders, addictive disorders including substance abuse disorders, and mood disorders.
- the invention encompasses the treatment of panic disorder, specific phobia, posttraumatic stress disorder (PTSD), obsessive- compulsive disorder, and movement disorder such as Tourette's syndrome.
- the disorders contemplated herein are defined in, for example, the DSM-IV (Diagnostic and Statistical Manual, 4th edition, American Psychiatric Association).
- the psychiatric disorder to be treated is PTSD.
- Posttraumatic stress disorder is defined by DSM-IV as an anxiety disorder that an individual may develop following exposure to a traumatic event, and is characterized by (1) reexperiencing the traumatic event, such as recurrent nightmares, intrusive recollections of the event, flashbacks, physiological and psychological responses to internal or external cues relating to the event, etc; (2) persistent avoidance of thoughts, people or places associated with the event; (3) numbing of general responsiveness such as emotional detachment, restricted affect or loss of interest in activities; and (4) persistence of increased arousal such as exaggerated startle response, hypervigilence, irritability, difficulty sleeping, etc.
- the lifetime prevalence of PTSD is at least 1%, and in high-risk populations, such as combat veterans or victims of criminal violence, prevalence is reported to be between 3 and 58%; PTSD is therefore of considerable public health concern.
- the methods of the invention encompass the use of any type of psychotherapy that is suitable for the particular psychiatric disorder for which the individual is undergoing treatment, and may be conducted in one or more sessions.
- Suitable methods of psychotherapy include behavior psychotherapy such as exposure-based psychotherapy, cognitive psychotherapy including cognitive training and psychodynamically oriented psychotherapy (see, for example, Foa (2000) J. Clin. Psych. 61(suppl. 5):43-38).
- Exposure based psychotherapy include for example, systematic desensitization, flooding, implosive therapy, and extinction-based therapy.
- Such psychotherapy modalities are well known to one skilled in the art of psychiatry.
- VR exposure therapy has been used to treat a variety of disorders including anxiety disorders such as the fear of heights (Rothbaum and Hodges (1999) Behav. Modif 23(4):507-25), as well as specific phobias, eating disorders, and PTSD (Anderson et al. (2001) Bull. Menninger Clin. 65(1):78-91). Because of the prevalence of PTSD in the general population and the successful use of VR therapy to treat PTSD in, for example, Vietnam veterans (Rothbaum et al. 30 (1999) J.
- Trauma Stress 12(2):263-71) or rape victims (Rothbaum et al. (2001) J. Trauma Stress 14(2):283-93), one embodiment of the present invention specifically contemplates the use of such VR exposure psychotherapy in combination with a PDE4 inhibitor as described elsewhere herein to treat PTSD.
- the phosphatase inhibitors used to practice the present invention may be any that is known, or discovered to inhibit the phosphatase enzyme, and are not limited to any particular structural class of compounds.
- the term "phosphatase inhibitors" includes any pharmaceutically acceptable salts thereof.
- the assay for identifying phosphatase inhibitors is described in the Examples section hereinbelow.
- the utility of phosphatase inhibitors in the present invention may be evaluated using the animal fear conditioning/extinction and clinical experimental protocols disclosed in PCT Application No. WO02/078629, which is hereby incorporated by reference, with the exception that a phosphatase inhibitor is used instead of the pharmacological agent used therein.
- the phosphatase inhibitor may be peptidal or non-peptidal in nature; however, the use of a non-peptidal phosphatase inhibitor is preferred.
- the phosphatase inhibitor is a CNS-penetrant phosphatase inhibitor.
- an orally active phosphatase inhibitor is preferred.
- the phosphatase inhibitor is a long acting phosphatase inhibitor.
- An especially preferred class of phosphatase inhibitors of use in the present invention are those compounds which are orally active and long acting. Representative phosphatase inhibitors of use in the present invention are fully described, for example, in U.S. Patent Nos.
- the PDE4 inhibitors used to practice the present invention may be any that is known, or discovered to inhibit the PDE4 enzyme, and are not limited to any particular structural class of compounds.
- the term "PDE4 inhibitors” includes any pharmaceutically acceptable salts thereof.
- the assay for identifying phosphatase inhibitors is described in the Examples section hereinbelow.
- the utility of PDE4 inhibitors in the present invention may be evaluated using the animal fear conditioning extinction and clinical experimental protocols disclosed in PCT Application No. WO02/078629, which is hereby incorporated by reference, with the exception that a PDE4 inhibitor is used instead of the pharmacological agent used therein.
- the PDE4 inhibitor may be peptidal or non-peptidal in nature; however, the use of a non-peptidal PDE4 inhibitor is preferred.
- the PDE4 inhibitor is a CNS-penetrant PDE4 inhibitor.
- the PDE4 inhibitor is a long acting PDE4 inhibitor.
- An especially preferred class of PDE4 inhibitors of use in the present invention are those compounds which are orally active and long acting. Representative PDE4 inhibitors of use in the present invention are fully described, for example, in U.S. Patent Nos.
- Suitable PDE4 inhibitors include pentoxifylline, isobutylmethylxanthine, cilomilast, roflumilast and its N-oxide, arofylline, lirimilast, GW84247, CP-671305, and terferol.
- Other suitable PDE4 inhibitors for use in the present invention are those presented in the Examples section and their pharmaceutically acceptable salts
- salts refers to salts prepared from pharmaceutically acceptable non-toxic bases or acids.
- the compound of the present invention is acidic, its corresponding salt can be conveniently prepared from pharmaceutically acceptable non-toxic bases, including inorganic bases and organic bases.
- Salts derived from such inorganic bases include aluminum, ammonium, calcium, copper (ic and ous), ferric, ferrous, lithium, magnesium, manganese (ic and ous), potassium, sodium, zinc and the like salts. Particularly preferred are the ammonium, calcium, magnesium, potassium and sodium salts.
- Salts derived from pharmaceutically acceptable organic non-toxic bases include salts of primary, secondary, and tertiary amines, as well as cyclic amines and substituted amines such as naturally occurring and synthesized substituted amines.
- Other pharmaceutically acceptable organic non- toxic bases from which salts can be formed include ion exchange resins such as, for example, arginine, betaine, caffeine, choline, N,N -dibenzylethylenediamine, diethylamine, 2- diethylaminoethanol, 2-dimethylaminoethanol, ethanolamine, ethylenediamine, N- ethylmorpholine, N-ethylpiperidine, glucamine, glucosamine, histidine, hydrabamine, isopropylamine, lysine, methylglucamine, morpholine, piperazine, piperidine, polyamine resins, procaine, purines, theobromine, triethylamine, trimethylamine
- the compound of the present invention When the compound of the present invention is basic, its corresponding salt can be conveniently prepared from pharmaceutically acceptable non-toxic acids, including inorganic and organic acids.
- acids include, for example, acetic, benzenesulfonic, benzoic, camphorsulfonic, citric, ethanesulfonic, fumaric, gluconic, glutamic, hydrobromic, hydrochloric, isethionic, lactic, maleic, malic, mandelic, methanesulfonic, mucic, nitric, pamoic, pantothenic,
- the inhibitor(s) of the invention may be administered to the patient prior to, during or after the psychotherapy session. It is preferably administered within about 24 hours prior to or following the session of psychotherapy, more preferably within about 24 hour prior to initiating psychotherapy, and even more preferably within about 12 hours prior to initiating psychotherapy.
- a full course of treatment of psychiatric disorder entails at least one session of this combination therapy protocol.
- the inhibitor(s) of the invention may be administered in a composition suitable for oral, rectal, topical, and parenteral (including subcutaneous, intramuscular, and intravenous) administration, although the most suitable route in any given case will depend on the particular host, and nature and severity of the conditions for which the active ingredient is being administered.
- the pharmaceutical compositions may be conveniently presented in unit dosage form and prepared by any of the methods well known in the art of pharmacy.
- the inhibitor(s) of the invention are administered in a therapeutically effective amount, which is that amount that provides improved therapeutic benefit relative to that achieved by psychotherapy alone.
- Dosage levels from about O.OOlmg/kg to about 140mg/kg of body weight per day are useful for the purpose of the present invention or about 0.05mg to about 7g per patient per day.
- the amount of active ingredient that may be combined with the carrier materials to produce a single dosage form will vary depending upon the host treated and the particular mode of administration.
- a formulation intended for the oral administration to humans may conveniently contain from about 0.5mg to about 5g of active agent, compounded with an appropriate and convenient amount of carrier material which may vary from about 5 to about 95 percent of the total composition.
- Unit dosage forms will generally contain between from about O.Olmg to about lOOOmg of the active ingredient, typically O.Olmg, 0.05mg, 0.25mg, Img, 5mg, 25mg, 50mg, lOOmg, 200mg, 300mg, 400mg, 500mg, 600mg, 800mg or lOOOmg.
- the specific dose level for any particular patient will depend upon a variety of factors including the age, body weight, general health, sex, diet, time of administration, route of administration, rate of excretion, drug combination and the severity of the particular disease undergoing therapy.
- the The inhibitor(s) of the invention can be combined as the active ingredient in intimate admixture with a pharmaceutical carrier according to conventional pharmaceutical compounding techniques.
- the carrier may take a wide variety of forms depending on the form of preparation desired for administration, e.g., oral or parenteral (including intravenous).
- the pharmaceutical compositions of the present invention can be presented as discrete units suitable for oral administration such as capsules, cachets or tablets each containing a predetermined amount of the active ingredient.
- compositions can be presented as a powder, as granules, as a solution, as a suspension in an aqueous liquid, as a non-aqueous liquid, as an oil-in-water emulsion or as a water-in-oil liquid emulsion.
- the inhibitor(s) of the invention may also be administered by controlled release means and/or delivery devices.
- the compositions may be prepared by any of the methods of pharmacy. In general, such methods include a step of bringing into association the active ingredient with the carrier that constitutes one or more necessary ingredients.
- the compositions are prepared by uniformly and intimately admixing the active ingredient with liquid carriers or finely divided solid carriers or both. The product can then be conveniently shaped into the desired presentation.
- compositions of this invention may include a pharmaceutically acceptable carrier and a compound or a pharmaceutically acceptable salt of a compound of the Examples.
- the compounds or pharmaceutically acceptable salts thereof, can also be included in pharmaceutical compositions in combination with one or more other therapeutically active compounds.
- the pharmaceutical carrier employed can be, for example, a solid, liquid, or gas.
- solid carriers include lactose, terra alba, sucrose, talc, gelatin, agar, pectin, acacia, magnesium stearate, and stearic acid.
- liquid carriers are sugar syrup, peanut oil, olive oil, and water.
- gaseous carriers include carbon dioxide and nitrogen.
- oral liquid preparations such as suspensions, elixirs and solutions
- carriers such as starches, sugars, microcrystalline cellulose, diluents, granulating agents, lubricants, binders, disintegrating agents, and the like may be used to form oral solid preparations such as powders, capsules and tablets.
- tablets and capsules are the preferred oral dosage units whereby solid pharmaceutical carriers are employed.
- tablets may be coated by standard aqueous or nonaqueous techniques
- a tablet containing the composition of this invention may be prepared by compression or molding, optionally with one or more accessory ingredients or adjuvants.
- Compressed tablets may be prepared by compressing, in a suitable machine, the active ingredient in a free-flowing form such as powder or granules, optionally mixed with a binder, lubricant, inert diluent, surface active or dispersing agent. Molded tablets may be made by molding in a suitable machine, a mixture of the powdered compound moistened with an inert liquid diluent.
- compositions of the present invention suitable for parenteral administration may be prepared as solutions or suspensions of the active compounds in water.
- a suitable surfactant can be included such as, for example, hydroxypropylcellulose.
- Dispersions can also be prepared in glycerol, liquid polyethylene glycols, and mixtures thereof in oils. Further, a preservative can be included to prevent the detrimental growth of microorganisms.
- compositions of the present invention suitable for injectable use include sterile aqueous solutions or dispersions.
- the compositions can be in the form of sterile powders for the extemporaneous preparation of such sterile injectable solutions or dispersions.
- the final injectable form must be sterile and must be effectively fluid for easy syringability.
- the pharmaceutical compositions must be stable under the conditions of manufacture and storage; thus, preferably should be preserved against the contaminating action of microorganisms such as bacteria and fungi.
- the carrier can be a solvent or dispersion medium containing, for example, water, ethanol, polyol (e.g. glycerol, propylene glycol and liquid polyethylene glycol), vegetable oils, and suitable mixtures thereof.
- compositions of the present invention can be in a form suitable for topical use such as, for example, an aerosol, cream, ointment, lotion, dusting powder, or the like. Further, the compositions can be in a form suitable for use in transdermal devices prepared via conventional processes As an example, a cream or ointment is prepared by mixing hydrophilic material and water, together with about 5wt% to about 10wt% of the compound, to produce a cream or ointment having a desired consistency.
- compositions of this invention can be in a form suitable for rectal administration wherein the carrier is a solid. It is preferable that the mixture forms unit dose suppositories. Suitable carriers include cocoa butter and other materials commonly used in the art. The suppositories may be conveniently formed by first admixing the composition with the softened or melted carrier(s) followed by chilling and shaping in moulds.
- the pharmaceutical formulations described above may include, as appropriate, one or more additional carrier ingredients such as diluents, buffers, flavoring agents, binders, surface-active agents, thickeners, lubricants, preservatives (including anti-oxidants) and the like.
- additional carrier ingredients such as diluents, buffers, flavoring agents, binders, surface-active agents, thickeners, lubricants, preservatives (including anti-oxidants) and the like.
- additional carrier ingredients such as diluents, buffers, flavoring agents, binders, surface-active agents, thickeners, lubricants, preservatives (including anti-oxidants) and the like.
- additional carrier ingredients such as diluents, buffers, flavoring agents, binders, surface-active agents, thickeners, lubricants, preservatives (including anti-oxidants) and the like.
- other adjuvants can be included to render the formulation isotonic with the blood of the intended recipient
- the compound of this invention can be utilized in combination with other therapeutic compounds.
- the combinations of the inhibitor(s) of the invention can be advantageously used in combination with i) Leukotriene receptor antagonists, ii) Leukotriene biosynthesis inhibitors, iii) COX-2 selective inhibitors, iv) statins, v) NSAIDs, vi) M2/M3 antagonists, vii) corticosteroids, viii) HI (histamine) receptor antagonists and ix) beta 2 adrenoceptor agonist.
- the inhibitor(s) of the invention may be administered with capsules, cachets or tablets each containing Img, 5mg, 25mg, 50mg, lOOmg, 200mg, 300mg, 400mg, or 500mg of the active ingredient of the compound of the present application, or a pharmaceutically acceptable salt thereof, administered prior to, during or after a session of psychotherapy.
- a subject undergoing treatment with the methods of the invention exhibits an improvement in one or more symptoms associated with the psychiatric disorder.
- DSM-IV Diagnostic and Statistical Mamlal of Mental Disorders (4th ea., American Psychiatric Association, Washington D.C.)
- DSM-IV Diagnostic and Statistical Mamlal of Mental Disorders (4th ea., American Psychiatric Association, Washington D.C.)
- the efficacy of the methods of the invention can be assessed using any clinically recognized assessment method for measuring a reduction of one or more symptoms of the particular psychiatric disorder. Examples of such assessment methods are described in, for example, in Experiment 7 of PCT Application WO02/078629.
- PROTOCOL Compounds which inhibit the hydrolysis of cAMP to AMP by the type-IV cAMP- specific phosphodiesterases may be screened in a 96-well plate format as follows:
- test compound dissolved in 2 ⁇ L DMSO
- substrate buffer containing [2,8- H] adenosine 3',5'-cyclic phosphate (cAMP, lOOnM to 50 ⁇ M), lOmM MgCl2, lmM EDTA, 50mM Tris, pH 7.5.
- cAMP adenosine 3',5'-cyclic phosphate
- lOmM MgCl2 lmM EDTA
- 50mM Tris pH 7.5.
- the reaction is initiated by the addition of lOmL of human recombinant PDE4 (the amount was controlled so that -10% product was formed in lOmin.).
- the reaction is stopped after lOmin. by the addition of Img of PDE- SPA beads (Amersham Pharmacia Biotech, Inc., Piscataway, NJ).
- the product AMP generated is quantified on a Wallac Microbeta® 96-well plate counter (EG&G Wallac Co., Gaithersburg, MD).
- the signal in the absence of enzyme is defined as the background.
- 100% activity is defined as the signal detected in the presence of enzyme and DMSO with the background subtracted. Percentage of inhibition is calculated accordingly.
- IC50 value is approximated with a non-linear regression fit using the standard 4-parameter/multiple binding sites equation from a ten point titration.
- the compound examples are comprised of two sub-sets - Example set A and Example set B,
- Examples 1 A through 42 A are characterized and prepared as disclosed in US 6,410,563 Bl, issued June 25, 2002, which is hereby iincorporated by reference. 1A. and 2A. 6-isopropyl-8-(3- ⁇ (Z E)-2-[4-(methylsulfonyl)phenyl]-2- phenylethenyl ⁇ phenyl)quinoline;
- 21A (E)-3- ⁇ 3-[6-(l-cyano-l-methylethyl)-8-quinolinyl]phenyl ⁇ -2-[4- (methylsulfonyl)phenyl]-2-propenoic acid; 22A. 2-methyl-2-[8-(3- ⁇ (E)-2-(3-methyl-l,2,4-oxadiazol-5-yl)-2-[4-
- 26A 6-isopropyl-8-(3- ⁇ (E)-2-(3-methyl-l,2,4-oxadiazol-5-yl)-2-[4- (methylsulfonyl)phenyl]ethenyl ⁇ phenyl)quinoline;
- 27 A (E)-3-(3- ⁇ 6-[l-methyl-l-(methylsulfonyl)ethyl]-8-quinolinyl ⁇ phenyl)-2-[4-
- 33A. and 34A 6-isopro ⁇ yl-8-(3- ⁇ (E/Z)-2-(5-methyl-2-pyridinyl)-2-[4- (methylsulfonyl)phenyl]ethenyl ⁇ phenyl)quinoline; 35A. 8-(3- ⁇ 2,2-bis[4-(methylsulfonyl)phenyl]vinyl ⁇ phenyl)-6- isopropylquinoline;
- 36A. and 37A 2-methyl-2-[8-(3- ⁇ (E/Z)-2-(5-methyl-2-pyridinyl)-2-[4- (methylsulfonyl)phenyl]ethenyl ⁇ phenyl)-6-quinolinyl]propanenitrile; 38A. 2-[8-(3- ⁇ 2,2-bis[4-(methylsulfonyl)phenyl]vinyljphenyl)-6-quinolinyl]-2- methylpropanenitrile;
- Examples IB through 32b are characterized and prepared as disclosed in US 6,399,636 B2, issued June 4, 2002, which is hereby incorporated by reference.
- Step 1 Ethyl 3-(3-bromoanilino)-2-(2-chloronicotinoyl) acrylate.
- Step 2 Ethyl l-(3-bromophenyl)-l,4-dihydro[l,8]naphthyridin-4-one-3-carboxylate.
- the crude compound from Step 1 was dissolved in tetrahydrofuran (0.3 M), the solution was cooled to 0°C, and sodium hydride (as a 60% dispersion in oil , 1.3 eq) was added in portions. After stirring at 0°Cfor 1 hour, the mixture was allowed to warm up to room temperature. After 2 hours, water was added to the suspension and and the insoluble solid was filtered and washed copiously with water. When dry, the solid was stirred in ether at room temperature for 24 hours and filtered to afford the title compound as a cream-colored solid.
- step 1 to 2 can be used:
- Step 3 l-(3-Bromophenyl)-l,4-dihydro[l,8]naphthyridin-4-one-3-carboxylic acid
- Step 2 Ethyl 2-(trans)-[4-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2- yl)phenyl]cyclopropanecarboxylate
- Step 3 2-(trans)-[4-(4,4,5,5-Tetramethyl-l,3,2-dioxaborolan-2- yl)phenyl]cyclopropanecarboxylic acid
- optically active isomers of EXAMPLE IC can be isolated separately by chromatography using chiral column; for example Chiral Pak AD eluting with hexane:EtOH or hexane:iPrOH containing 0.2% TFA.
- Step 1 (trans)-3-(4-Bromo-phenyl)-l-imidazol-l-yl-propenone To a solution of (trans)-3-(4-bromo-phenyl)acrylic acid (1.0 eq) in toluene (0.2M) was added CDI (1.5 eq). The mixture was stirred for 3h at rt. The resulting precipitate was isolated by filtration to afford the title compound as a white solid.
- Step 2 (trans)-3-[3-(4-Bromo-phenyl)-acryloyl]-4-methyl-5-phenyl-oxazolidin-2-one
- Step 3 (trans)-3-[2-(4-Bromo-phenyl)cyclopropanecarbonyl]-4-methyl-5-phenyl-oxazolidin-2- one
- Step 5 2-(trans)- ⁇ 3 , -[3-[(Cyclopropylamino)carbonyl]-4-oxo-l,8-naphthyridin-l(4H)-yl]-l,r- biphenyl-4-yl jcyclopropanecarboxylic acid
- Another alternative is an enantioselective cyclopropanation using for example a bis-oxazoline chiral ligand/copper complex and diazoacetate (Evans et al. J. Am. Chem. Soc. 1991, 113, 726) to prepare optically active ethyl 2-(trans)-(4-bromophenyl)cyclopropanecarboxylate from 4- bromostyrene.
- Step 3 2- ⁇ 3 '-[3-[(Cyclopropylamino)carbonyl]-4-oxo- 1 ,8-naphthyridin- 1 (4H)-yl]- 1 , 1 -biphenyl- 4-ylj-2-methylpropanoic acid
- Step 1 Methyl 2-(cis) -3-(4-bromophenyl)prop-2-enoate
- Step 2 Methyl 2-(cis) -(4-bromophenyl)cyclopropanecarboxylate
- Step 3 Methyl 2- ⁇ 3'-[3-[(cyclopropylamino)carbonyl]-4-oxo-l,8-naphthyridin-l(4H)-yl]-l,r- biphenyl-4-yl ⁇ cyclopropanecarboxylate
- the product was purified by flash chromatography (hexane:EtOAc, 60:40), then vigorous stirring in hexane/ether and isolation by filtration to afford the title compound as a white solid.
- Step 4 2-(cis)- ⁇ 3'-[3-[(Cyclopropylamino)carbonyl]-4-oxo-l,8-naphthyridin-l(4H)-yl]-l,r- biphenyl-4-yl jcyclopropanecarboxylic acid
- optically active diastereoisomers of EXAMPLE 4C can be isolated separately by chromatography using chiral column; for example Chiral Pak AD eluting with hexane:EtOH or hexane PrOH containing 0.2% TFA.
- optically active intermediate can be obtained as follow:
- Step 1 (cis)-l- ⁇ [2-(4-Bromophenyl)cyclopropyl]carbonylj-lH-imidazole
- Step 2 (cis)-3- ⁇ [2-(4-Bromophenyl)cyclopropyl]carbonylj-4-methyl-5-phenyl-l,3-oxazolidin-2- one
- Another alternative is an enantioselective cyclopropanation using for example a bis-oxazoline chiral ligand/copper complex and diazoacetate (Evans et al. J. Am. Chem. Soc. 1991, 113, 726) to prepare optically active ethyl 2-(cis)-(4-bromophenyl)cyclopropanecarboxylate from 4- bromostyrene.
- Step 1 l-(4-Bromophenyl)cyclopropanecarbonitrile
- Step 2 l-(4-Bromophenyl)cyclopropanecarboxylic acid
- Step 3 1 - ⁇ 3 '- [3- [(Cyclopropylamino)carbonyl] -4-oxo- 1 ,8-naphthyridin- 1 (4H)-yl] -1,1 '-biphenyl- 4-yljcyclopropanecarboxylic acid
- Step 2 Methyl (trans)-2- ⁇ 3'-[3-[(cyclopropylamino)carbonyl]-4-oxo-l,8-naphthyridin-l(4H)-yl]- 3-fluoro- 1 , 1 '-biphenyl-4-yl ⁇ cyclopropanecarboxylate
- Step 3 2-(trans)- ⁇ 3'-[3-[(Cyclopropylamino)carbonyl]-4-oxo-l,8-naphthyridin-l(4H)-yl]-3- fluoro- 1 , 1 '-biphenyl-4-yl ⁇ cyclopropanecarboxylic acid
- Optically active (+) or (-)-(trans)-2- ⁇ 3'-[3-[(cyclopropylamino)carbonyl]-4-oxo-l,8-naphthyridin- l(4H)-yl]-3-fluoro-l,r-biphenyl-4-yljcyclopropanecarboxylic acid was obtained using optically active (+) or (-)-(trans)-methyl 2-(4-bromo-2-fluorophenyl)cyclopropanecarboxylate prepared according to procedure described below:
- Step B (-t-)-ethyl 2-(4-bromo-2-fluorophenyl)cyclopropanecarboxylate
- (+)-ethyl 2-(4-bromo-2-fluorophenyl)cyclopropanecarboxylate (mix esters, 1 eq) in tetrahydrofuran-methanol (2:1) was added lithium hydroxide (0.84 eq).
- the reaction was stirred at rt for 2 days.
- the resulting mixture was concentrated, diluted with water, extracted with ether(2x) to obtain the cis ester.
- the aqueous phase was acidified using HCl 10%, extracted with ether (2x) to obtain the (+)-trans acid.
- the organic extract containing the trans acid were combined and washed with brine, dried over MgSO 4 , filtered and concentrated.
- Step D (+)-methyl 2-(4-bromo-2-fluorophenyl)cyclopropanecarboxylate
- Step 1 Ethyl 2-(tr ⁇ ns)-3-(4-bromo-2-chlorophenyl)prop-2-enoate
- Step 2 2-(trans)- ⁇ 3-Chloro-3'-[3-[(cyclopropylamino)carbonyl]-4-oxo-l,8-naphthyridin-l(4H)- yl] - 1 , 1 '-biphenyl-4-yl ⁇ cyclopropanecarboxylic acid
- Step 1 l-(3-bromophenyl)-4-oxo-N-(2,2,2-trifluoroethyl)-l ,4-dihydro-l ,8-naphthyridine-3- carboxamide
- Step 2 2-(trans)- ⁇ 3'-[4-Oxo-3- ⁇ [(2,2,2-trifluoroethyl)amino]carbonylj-l,8-naphthyridin-l(4H)- yl]- 1 , 1 '-biphenyl-4-yl ⁇ cyclopropanecarboxylic acid
- Step 1 Ethyl l-(3-bromophenyl)-l,4-dihydro[l,8]naphthyridin-4-one-3-carboxylate.
- Step 2 l-(3-Bromophenyl)-l,4-dihydro[l,8]naphthyridin-4-one-3-carboxylic acid
- Step 3 1 -(3-bromophenyl)-N-(2,2,2-trifluoroethyl)- 1 ,4-dihydro- 1 ,8-naphthyridin-4-one-3- carboxamide.
- Step 4 N-(2,2,2-trifluoroethyl)-l-[3-(4,4,5,5-tetramethyl-l,3,2-dioxaborolan-2-yl)phenyl]-l,4- dihydro-l,8-naphthyridin-4-one-3-carboxamide
- (+)-ethyl 2-(4-bromo-2-fluorophenyl)cyclopropanecarboxylate (mix esters, 1 eq) in tetrahydrofuran-methanol (2:1) was added lithium hydroxide (0.84 eq).
- the reaction was stirred at rt for 2 days.
- the resulting mixture was concentrated, diluted with water, extracted with ether(2x) to obtain the cis ester.
- the aqueous phase was acidified using HCl 10%, extracted with ether (2x) to obtain the (-t-)-trans acid.
- the organic extract containing the trans acid were combined and washed with brine, dried over MgSO , filtered and concentrated.
- Step 8 (+)-methyl 2-(4-bromo-2-fluorophenyl)cyclopropanecarboxylate
- (+)-2-(4-bromo-2-fluorophenyl)cyclopropanecarboxylic acid from step 7 (1 eq) in methylenechloride was added an ethereal solution of diazomethane until reaction is completed by TLC. The resulting mixture was concentrated. The crude ester was used as such in the next step.
- Step 9 (+)-methyl 2- ⁇ 3-fluoro-3'-[4-oxo-3- ⁇ [(2,2,2-trifluoroethyl)amino]carbonylj-l,8- naphthyridin- 1 (4H)-yl]biphenyl-4-yl ⁇ cyclopropanecarboxylate
- reaction mixture was filtered on Celite and silica gel (1:1) and washed with EtOAc. The filtrate was concentrated in vacuo and remaining solvents were distilled in vacuo. The resulting yellow solid is stirred vigourously in ether. The residue then isolated by filtration and washed with ether. Mother liquors were further purified by flash chromatography (toluene / EtOAc, 100:0 to 70:30).
- Step 10 (+)-2- ⁇ 3-fluoro-3'-[4-oxo-3- ⁇ [(2,2,2-trifluoroethyl)amino]carbonyl ⁇ -l,8-naphthyridin- l(4H)-yl]biphenyl-4-yljcyclopropanecarboxylic acid
- Step 1 ethyl l-(4-bromobenzyl)cyclobutanecarboxylate.
- Step 2 ethyl l-( ⁇ 3'-[3-[(cyclopropylamino)carbonyl]-4-oxo-l,8-naphthyridin-l(4H)-yl]biphenyl- 4-yljmethyl)cyclobutanecarboxylate
- Step 3 l-( ⁇ 3'-[3-[(cyclopropylamino)carbonyl]-4-oxo-l,8-naphthyridin-l(4H)-yl]biphenyl-4- yl ⁇ methyl)cyclobutanecarboxylic acid
Landscapes
- Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
Abstract
Description

















Claims
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP04753335A EP1633306A4 (en) | 2003-05-29 | 2004-05-25 | USE OF PHOSPHATASE INHIBITORS AS ADDITIONAL TREATMENT FOR PSYCHIATRIC DISORDERS |
| US10/557,648 US20060258668A1 (en) | 2003-05-29 | 2004-05-25 | Use of phosphatase inhibitors as adjunct therapy for psychiatric disorders |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US47416803P | 2003-05-29 | 2003-05-29 | |
| US60/474,168 | 2003-05-29 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2004105698A2 true WO2004105698A2 (en) | 2004-12-09 |
| WO2004105698A3 WO2004105698A3 (en) | 2005-06-02 |
Family
ID=33490701
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2004/016492 Ceased WO2004105698A2 (en) | 2003-05-29 | 2004-05-25 | Use of phosphatase inhibitors as adjunct therapy for psychiatric disorders |
Country Status (3)
| Country | Link |
|---|---|
| US (1) | US20060258668A1 (en) |
| EP (1) | EP1633306A4 (en) |
| WO (1) | WO2004105698A2 (en) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2007048225A1 (en) * | 2005-10-27 | 2007-05-03 | Merck Frosst Canada Ltd. | A 4-oxo-1- (3-substituted phenyl) -1, 4-dihydro-1, 8-naphthyridine-3-carboxamide phosphodiesterase-4-inhibitor and a method of preparing same |
| EP1562588A4 (en) * | 2002-11-15 | 2007-10-31 | Merck & Co Inc | USE OF PDE4 INHIBITORS AS ADDITIONAL TREATMENT OF PSYCHIATRIC DISORDERS |
| CN100425591C (en) * | 2005-09-26 | 2008-10-15 | 山东大学 | Sulfonyl diphenylethyllene endocompound and its preparation method and pharmaceutical uses |
| US8273774B2 (en) | 2008-05-27 | 2012-09-25 | Astrazeneca Ab | Phenoxypyridinylamide compounds |
Family Cites Families (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CA2279651A1 (en) * | 1996-09-05 | 1998-03-12 | Massachusetts Institute Of Technology | Compositions and methods for treatment of neurological disorders and neurodegenerative diseases |
| CN1156476C (en) * | 1998-01-29 | 2004-07-07 | 第一三得利制药株式会社 | 1-cycloalkyl-1, 8-naphthyridin-4-one derivatives as phosphodiesterase type IV inhibitors |
| US6376517B1 (en) * | 1998-08-14 | 2002-04-23 | Gpi Nil Holdings, Inc. | Pipecolic acid derivatives for vision and memory disorders |
| US6436971B2 (en) * | 2000-02-09 | 2002-08-20 | Smithkline Beecham Corporation | Use of PDE 4-specific inhibitors to reduce the severity of a bacterial infection after a respiratory viral infection |
-
2004
- 2004-05-25 US US10/557,648 patent/US20060258668A1/en not_active Abandoned
- 2004-05-25 EP EP04753335A patent/EP1633306A4/en not_active Withdrawn
- 2004-05-25 WO PCT/US2004/016492 patent/WO2004105698A2/en not_active Ceased
Non-Patent Citations (1)
| Title |
|---|
| See references of EP1633306A4 * |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1562588A4 (en) * | 2002-11-15 | 2007-10-31 | Merck & Co Inc | USE OF PDE4 INHIBITORS AS ADDITIONAL TREATMENT OF PSYCHIATRIC DISORDERS |
| CN100425591C (en) * | 2005-09-26 | 2008-10-15 | 山东大学 | Sulfonyl diphenylethyllene endocompound and its preparation method and pharmaceutical uses |
| WO2007048225A1 (en) * | 2005-10-27 | 2007-05-03 | Merck Frosst Canada Ltd. | A 4-oxo-1- (3-substituted phenyl) -1, 4-dihydro-1, 8-naphthyridine-3-carboxamide phosphodiesterase-4-inhibitor and a method of preparing same |
| US8273774B2 (en) | 2008-05-27 | 2012-09-25 | Astrazeneca Ab | Phenoxypyridinylamide compounds |
| EP2778156A1 (en) | 2008-05-27 | 2014-09-17 | AstraZeneca AB (Publ) | Phenoxypyridinylamide derivatives and their use in the treatment of PDE4 mediated disease states |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2004105698A3 (en) | 2005-06-02 |
| US20060258668A1 (en) | 2006-11-16 |
| EP1633306A4 (en) | 2007-05-16 |
| EP1633306A2 (en) | 2006-03-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1517895B1 (en) | 8-(biaryl) quinoline pde4 inhibitors | |
| EP1565464B1 (en) | 4-oxo-1-(3-substituted phenyl-1,4-dihydro-1,8-naphthyridine-3-carboxamide phosphodiesterase-4 inhibitors | |
| EP1436290B1 (en) | Alkyne-aryl-naphthyridin-4(1h)-0ne derivatives as type iv phosphodiesterase inhibitor | |
| EP1758883B1 (en) | Substituted 2-quinolyl-oxazoles useful as PDE4 inhibitors | |
| KR102102283B1 (en) | Compounds and methods for antiviral treatment | |
| AU2002322940A1 (en) | Alkyne-aryl phosphodiesterase-4 inhibitors | |
| US20150353546A1 (en) | Metalloenzyme inhibitor compounds | |
| US20130309200A1 (en) | Imidazo [1,2-b] pyridazine and imidazo [4,5-b] pyridine derivatives as jak inhibitors | |
| CN103339115A (en) | Metalloenzyme inhibitor compounds | |
| EP1397359B1 (en) | 1-biaryl-1,8-napthyridin-4-one phosphodiesterase-4 inhibitors | |
| BG65403B1 (en) | Substituted 8-arylquinolines as phosphodiesterase-4 inhibitors | |
| EP1633306A2 (en) | Use of phosphatase inhibitors as adjunct therapy for psychiatric disorders | |
| US20060069115A1 (en) | Use of pde4 inhibitors as adjunct therapy for psychiatric disorders | |
| EP1592419A1 (en) | Use of phosphodiesterase-4 inhibitors as enhancers of cognition | |
| HK40060488B (en) | 3,3-difluoroallylamines or salts thereof and pharmaceutical compositions comprising the same | |
| TW200535136A (en) | 4-oxo-1-(3-substituted) phenyl-1,4-dihydro-1,8-naphthyridine-3-carboxamide phosphodiesterase-4 inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2006258668 Country of ref document: US Ref document number: 10557648 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004753335 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004753335 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 10557648 Country of ref document: US |