WO2004104007A1 - Pyrazolo-quinazoline derivatives,process for their preparation and their use as kinase inhibitors - Google Patents
Pyrazolo-quinazoline derivatives,process for their preparation and their use as kinase inhibitors Download PDFInfo
- Publication number
- WO2004104007A1 WO2004104007A1 PCT/EP2004/050612 EP2004050612W WO2004104007A1 WO 2004104007 A1 WO2004104007 A1 WO 2004104007A1 EP 2004050612 W EP2004050612 W EP 2004050612W WO 2004104007 A1 WO2004104007 A1 WO 2004104007A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- compound
- group
- compounds
- reacting
- Prior art date
Links
- 238000000034 method Methods 0.000 title claims abstract description 100
- 230000008569 process Effects 0.000 title claims abstract description 36
- GNOYRJIXSUNXIH-UHFFFAOYSA-N N1=CNC2=C3C=NN=C3C=CC2=C1 Chemical class N1=CNC2=C3C=NN=C3C=CC2=C1 GNOYRJIXSUNXIH-UHFFFAOYSA-N 0.000 title claims abstract description 17
- 238000002360 preparation method Methods 0.000 title claims abstract description 9
- 229940043355 kinase inhibitor Drugs 0.000 title description 2
- 239000003757 phosphotransferase inhibitor Substances 0.000 title description 2
- 150000001875 compounds Chemical class 0.000 claims abstract description 421
- 230000000694 effects Effects 0.000 claims abstract description 36
- 206010028980 Neoplasm Diseases 0.000 claims abstract description 25
- 238000011282 treatment Methods 0.000 claims abstract description 25
- 150000003839 salts Chemical class 0.000 claims abstract description 20
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 16
- 201000011510 cancer Diseases 0.000 claims abstract description 14
- 102000001253 Protein Kinase Human genes 0.000 claims abstract description 12
- 108060006633 protein kinase Proteins 0.000 claims abstract description 12
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 9
- 201000010099 disease Diseases 0.000 claims abstract description 6
- 239000000203 mixture Substances 0.000 claims description 120
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 92
- 238000006243 chemical reaction Methods 0.000 claims description 79
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 claims description 73
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 claims description 63
- 125000000623 heterocyclic group Chemical group 0.000 claims description 63
- -1 alkylheterocyclyl Chemical group 0.000 claims description 62
- 125000003118 aryl group Chemical group 0.000 claims description 46
- 125000004415 heterocyclylalkyl group Chemical group 0.000 claims description 42
- 239000001257 hydrogen Substances 0.000 claims description 42
- 229910052739 hydrogen Inorganic materials 0.000 claims description 42
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 41
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 39
- 125000000217 alkyl group Chemical group 0.000 claims description 31
- 125000001316 cycloalkyl alkyl group Chemical group 0.000 claims description 30
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 29
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 23
- 108091000080 Phosphotransferase Proteins 0.000 claims description 22
- 102000020233 phosphotransferase Human genes 0.000 claims description 22
- 150000001412 amines Chemical class 0.000 claims description 21
- 125000001301 ethoxy group Chemical group [H]C([H])([H])C([H])([H])O* 0.000 claims description 21
- 230000005764 inhibitory process Effects 0.000 claims description 20
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 18
- ZRALSGWEFCBTJO-UHFFFAOYSA-N Guanidine Chemical compound NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 claims description 18
- 239000003795 chemical substances by application Substances 0.000 claims description 18
- 239000011734 sodium Substances 0.000 claims description 18
- 229910052757 nitrogen Inorganic materials 0.000 claims description 16
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 16
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 claims description 15
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 claims description 14
- 150000002357 guanidines Chemical class 0.000 claims description 14
- 229910052740 iodine Inorganic materials 0.000 claims description 14
- 229910000104 sodium hydride Inorganic materials 0.000 claims description 14
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 claims description 13
- 125000005842 heteroatom Chemical group 0.000 claims description 13
- 229910052760 oxygen Inorganic materials 0.000 claims description 13
- 239000012312 sodium hydride Substances 0.000 claims description 13
- 229910052717 sulfur Inorganic materials 0.000 claims description 13
- 239000000908 ammonium hydroxide Substances 0.000 claims description 12
- 125000006570 (C5-C6) heteroaryl group Chemical group 0.000 claims description 11
- 108090000461 Aurora Kinase A Proteins 0.000 claims description 11
- 102100032311 Aurora kinase A Human genes 0.000 claims description 11
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 11
- 230000002378 acidificating effect Effects 0.000 claims description 11
- WYACBZDAHNBPPB-UHFFFAOYSA-N diethyl oxalate Chemical compound CCOC(=O)C(=O)OCC WYACBZDAHNBPPB-UHFFFAOYSA-N 0.000 claims description 11
- 150000002429 hydrazines Chemical class 0.000 claims description 11
- PJLUWPNLDKJSCF-UHFFFAOYSA-N 2-ethoxycyclohex-2-en-1-one Chemical compound CCOC1=CCCCC1=O PJLUWPNLDKJSCF-UHFFFAOYSA-N 0.000 claims description 10
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 claims description 10
- 229910000024 caesium carbonate Inorganic materials 0.000 claims description 10
- 230000022131 cell cycle Effects 0.000 claims description 10
- 208000035475 disorder Diseases 0.000 claims description 10
- 230000002062 proliferating effect Effects 0.000 claims description 10
- CHJJGSNFBQVOTG-UHFFFAOYSA-N N-methyl-guanidine Natural products CNC(N)=N CHJJGSNFBQVOTG-UHFFFAOYSA-N 0.000 claims description 9
- SWSQBOPZIKWTGO-UHFFFAOYSA-N dimethylaminoamidine Natural products CN(C)C(N)=N SWSQBOPZIKWTGO-UHFFFAOYSA-N 0.000 claims description 9
- 230000002401 inhibitory effect Effects 0.000 claims description 9
- KDLHZDBZIXYQEI-UHFFFAOYSA-N palladium Substances [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 claims description 9
- 125000000547 substituted alkyl group Chemical group 0.000 claims description 9
- 125000002221 trityl group Chemical group [H]C1=C([H])C([H])=C([H])C([H])=C1C([*])(C1=C(C(=C(C(=C1[H])[H])[H])[H])[H])C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 claims description 9
- ZCYVEMRRCGMTRW-UHFFFAOYSA-N 7553-56-2 Chemical group [I] ZCYVEMRRCGMTRW-UHFFFAOYSA-N 0.000 claims description 8
- 125000003545 alkoxy group Chemical group 0.000 claims description 8
- 239000003814 drug Substances 0.000 claims description 8
- 125000000472 sulfonyl group Chemical group *S(*)(=O)=O 0.000 claims description 8
- 239000002246 antineoplastic agent Substances 0.000 claims description 7
- 239000011630 iodine Substances 0.000 claims description 7
- YNESATAKKCNGOF-UHFFFAOYSA-N lithium bis(trimethylsilyl)amide Chemical compound [Li+].C[Si](C)(C)[N-][Si](C)(C)C YNESATAKKCNGOF-UHFFFAOYSA-N 0.000 claims description 7
- 125000003226 pyrazolyl group Chemical group 0.000 claims description 7
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 claims description 6
- 241000124008 Mammalia Species 0.000 claims description 6
- 125000003277 amino group Chemical group 0.000 claims description 6
- 239000003054 catalyst Substances 0.000 claims description 6
- 150000002431 hydrogen Chemical class 0.000 claims description 6
- ITMCEJHCFYSIIV-UHFFFAOYSA-N triflic acid Chemical compound OS(=O)(=O)C(F)(F)F ITMCEJHCFYSIIV-UHFFFAOYSA-N 0.000 claims description 6
- WUSZGSMQYXOBRT-UHFFFAOYSA-N 2-methoxy-4,4-dimethylcyclohex-2-en-1-one Chemical compound COC1=CC(C)(C)CCC1=O WUSZGSMQYXOBRT-UHFFFAOYSA-N 0.000 claims description 5
- JRGZHRJEKVTXCX-UHFFFAOYSA-N 2-methoxy-5,5-dimethylcyclohex-2-en-1-one Chemical compound COC1=CCC(C)(C)CC1=O JRGZHRJEKVTXCX-UHFFFAOYSA-N 0.000 claims description 5
- 125000003282 alkyl amino group Chemical group 0.000 claims description 5
- 125000004414 alkyl thio group Chemical group 0.000 claims description 5
- XQPRBTXUXXVTKB-UHFFFAOYSA-M caesium iodide Chemical compound [I-].[Cs+] XQPRBTXUXXVTKB-UHFFFAOYSA-M 0.000 claims description 5
- 230000004663 cell proliferation Effects 0.000 claims description 5
- 230000001419 dependent effect Effects 0.000 claims description 5
- WBJINCZRORDGAQ-UHFFFAOYSA-N formic acid ethyl ester Natural products CCOC=O WBJINCZRORDGAQ-UHFFFAOYSA-N 0.000 claims description 5
- 125000006517 heterocyclyl carbonyl group Chemical group 0.000 claims description 5
- 229910052763 palladium Inorganic materials 0.000 claims description 5
- 238000001959 radiotherapy Methods 0.000 claims description 5
- 208000024827 Alzheimer disease Diseases 0.000 claims description 4
- 201000001320 Atherosclerosis Diseases 0.000 claims description 4
- XGEGHDBEHXKFPX-UHFFFAOYSA-N N-methyl urea Chemical compound CNC(N)=O XGEGHDBEHXKFPX-UHFFFAOYSA-N 0.000 claims description 4
- 208000031481 Pathologic Constriction Diseases 0.000 claims description 4
- XBDQKXXYIPTUBI-UHFFFAOYSA-M Propionate Chemical compound CCC([O-])=O XBDQKXXYIPTUBI-UHFFFAOYSA-M 0.000 claims description 4
- 201000004681 Psoriasis Diseases 0.000 claims description 4
- 125000004390 alkyl sulfonyl group Chemical group 0.000 claims description 4
- 125000001951 carbamoylamino group Chemical group C(N)(=O)N* 0.000 claims description 4
- 229940127089 cytotoxic agent Drugs 0.000 claims description 4
- 229910052736 halogen Inorganic materials 0.000 claims description 4
- 150000002367 halogens Chemical class 0.000 claims description 4
- 201000005787 hematologic cancer Diseases 0.000 claims description 4
- 208000024200 hematopoietic and lymphoid system neoplasm Diseases 0.000 claims description 4
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical group II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 claims description 4
- OWFXIOWLTKNBAP-UHFFFAOYSA-N isoamyl nitrite Chemical compound CC(C)CCON=O OWFXIOWLTKNBAP-UHFFFAOYSA-N 0.000 claims description 4
- 238000004519 manufacturing process Methods 0.000 claims description 4
- 230000001590 oxidative effect Effects 0.000 claims description 4
- BASFCYQUMIYNBI-UHFFFAOYSA-N platinum Substances [Pt] BASFCYQUMIYNBI-UHFFFAOYSA-N 0.000 claims description 4
- 230000002265 prevention Effects 0.000 claims description 4
- 208000037803 restenosis Diseases 0.000 claims description 4
- BEOOHQFXGBMRKU-UHFFFAOYSA-N sodium cyanoborohydride Chemical compound [Na+].[B-]C#N BEOOHQFXGBMRKU-UHFFFAOYSA-N 0.000 claims description 4
- 230000036262 stenosis Effects 0.000 claims description 4
- 208000037804 stenosis Diseases 0.000 claims description 4
- 210000001685 thyroid gland Anatomy 0.000 claims description 4
- JBWKIWSBJXDJDT-UHFFFAOYSA-N triphenylmethyl chloride Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(Cl)C1=CC=CC=C1 JBWKIWSBJXDJDT-UHFFFAOYSA-N 0.000 claims description 4
- 230000002792 vascular Effects 0.000 claims description 4
- 208000023275 Autoimmune disease Diseases 0.000 claims description 3
- 206010004446 Benign prostatic hyperplasia Diseases 0.000 claims description 3
- 102000003903 Cyclin-dependent kinases Human genes 0.000 claims description 3
- 108090000266 Cyclin-dependent kinases Proteins 0.000 claims description 3
- 206010018364 Glomerulonephritis Diseases 0.000 claims description 3
- 206010027476 Metastases Diseases 0.000 claims description 3
- 208000009905 Neurofibromatoses Diseases 0.000 claims description 3
- 201000010208 Seminoma Diseases 0.000 claims description 3
- 206010052779 Transplant rejections Diseases 0.000 claims description 3
- 208000036142 Viral infection Diseases 0.000 claims description 3
- 201000006083 Xeroderma Pigmentosum Diseases 0.000 claims description 3
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims description 3
- 125000004466 alkoxycarbonylamino group Chemical group 0.000 claims description 3
- 239000002168 alkylating agent Substances 0.000 claims description 3
- 229940100198 alkylating agent Drugs 0.000 claims description 3
- 206010003246 arthritis Diseases 0.000 claims description 3
- 150000001543 aryl boronic acids Chemical class 0.000 claims description 3
- 125000004104 aryloxy group Chemical group 0.000 claims description 3
- 210000003169 central nervous system Anatomy 0.000 claims description 3
- 208000022605 chemotherapy-induced alopecia Diseases 0.000 claims description 3
- 125000005170 cycloalkyloxycarbonyl group Chemical group 0.000 claims description 3
- 239000003085 diluting agent Substances 0.000 claims description 3
- 239000003937 drug carrier Substances 0.000 claims description 3
- 230000003325 follicular Effects 0.000 claims description 3
- 125000005844 heterocyclyloxy group Chemical group 0.000 claims description 3
- 239000012948 isocyanate Substances 0.000 claims description 3
- 150000002513 isocyanates Chemical class 0.000 claims description 3
- 201000001441 melanoma Diseases 0.000 claims description 3
- 230000009401 metastasis Effects 0.000 claims description 3
- 208000015122 neurodegenerative disease Diseases 0.000 claims description 3
- 201000004931 neurofibromatosis Diseases 0.000 claims description 3
- 210000000056 organ Anatomy 0.000 claims description 3
- 201000008968 osteosarcoma Diseases 0.000 claims description 3
- 125000004043 oxo group Chemical group O=* 0.000 claims description 3
- 210000001428 peripheral nervous system Anatomy 0.000 claims description 3
- 229910052697 platinum Inorganic materials 0.000 claims description 3
- 208000015768 polyposis Diseases 0.000 claims description 3
- 208000005069 pulmonary fibrosis Diseases 0.000 claims description 3
- 206010041823 squamous cell carcinoma Diseases 0.000 claims description 3
- 208000001608 teratocarcinoma Diseases 0.000 claims description 3
- 230000005747 tumor angiogenesis Effects 0.000 claims description 3
- 230000009385 viral infection Effects 0.000 claims description 3
- 229910020889 NaBH3 Inorganic materials 0.000 claims description 2
- 102100026379 Neurofibromin Human genes 0.000 claims description 2
- DKGAVHZHDRPRBM-UHFFFAOYSA-N Tert-Butanol Chemical compound CC(C)(C)O DKGAVHZHDRPRBM-UHFFFAOYSA-N 0.000 claims description 2
- 125000005078 alkoxycarbonylalkyl group Chemical group 0.000 claims description 2
- 125000000676 alkoxyimino group Chemical group 0.000 claims description 2
- 125000004457 alkyl amino carbonyl group Chemical group 0.000 claims description 2
- 125000004471 alkyl aminosulfonyl group Chemical group 0.000 claims description 2
- 125000002877 alkyl aryl group Chemical group 0.000 claims description 2
- 125000004448 alkyl carbonyl group Chemical group 0.000 claims description 2
- 125000005197 alkyl carbonyloxy alkyl group Chemical group 0.000 claims description 2
- 125000005196 alkyl carbonyloxy group Chemical group 0.000 claims description 2
- 230000002152 alkylating effect Effects 0.000 claims description 2
- 125000004397 aminosulfonyl group Chemical group NS(=O)(=O)* 0.000 claims description 2
- 150000004982 aromatic amines Chemical class 0.000 claims description 2
- 125000002102 aryl alkyloxo group Chemical group 0.000 claims description 2
- 125000004658 aryl carbonyl amino group Chemical group 0.000 claims description 2
- 125000005129 aryl carbonyl group Chemical group 0.000 claims description 2
- 125000005199 aryl carbonyloxy group Chemical group 0.000 claims description 2
- 125000005002 aryl methyl group Chemical group 0.000 claims description 2
- 125000005161 aryl oxy carbonyl group Chemical group 0.000 claims description 2
- 125000004657 aryl sulfonyl amino group Chemical group 0.000 claims description 2
- 125000004391 aryl sulfonyl group Chemical group 0.000 claims description 2
- 125000005110 aryl thio group Chemical group 0.000 claims description 2
- 125000000852 azido group Chemical group *N=[N+]=[N-] 0.000 claims description 2
- 125000003917 carbamoyl group Chemical group [H]N([H])C(*)=O 0.000 claims description 2
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 claims description 2
- 238000009104 chemotherapy regimen Methods 0.000 claims description 2
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 2
- 125000006254 cycloalkyl carbonyl group Chemical group 0.000 claims description 2
- 239000000824 cytostatic agent Substances 0.000 claims description 2
- 230000001085 cytostatic effect Effects 0.000 claims description 2
- 239000002254 cytotoxic agent Substances 0.000 claims description 2
- 231100000599 cytotoxic agent Toxicity 0.000 claims description 2
- 238000010511 deprotection reaction Methods 0.000 claims description 2
- 125000004472 dialkylaminosulfonyl group Chemical group 0.000 claims description 2
- 125000004986 diarylamino group Chemical group 0.000 claims description 2
- NZZFYRREKKOMAT-UHFFFAOYSA-N diiodomethane Chemical compound ICI NZZFYRREKKOMAT-UHFFFAOYSA-N 0.000 claims description 2
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 claims description 2
- FFUAGWLWBBFQJT-UHFFFAOYSA-N hexamethyldisilazane Chemical compound C[Si](C)(C)N[Si](C)(C)C FFUAGWLWBBFQJT-UHFFFAOYSA-N 0.000 claims description 2
- 230000007062 hydrolysis Effects 0.000 claims description 2
- 238000006460 hydrolysis reaction Methods 0.000 claims description 2
- 125000002768 hydroxyalkyl group Chemical group 0.000 claims description 2
- 229910052744 lithium Inorganic materials 0.000 claims description 2
- 125000004170 methylsulfonyl group Chemical group [H]C([H])([H])S(*)(=O)=O 0.000 claims description 2
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 2
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 2
- 125000003107 substituted aryl group Chemical group 0.000 claims description 2
- 125000006633 tert-butoxycarbonylamino group Chemical group 0.000 claims description 2
- 125000002088 tosyl group Chemical group [H]C1=C([H])C(=C([H])C([H])=C1C([H])([H])[H])S(*)(=O)=O 0.000 claims description 2
- ZDZHCHYQNPQSGG-UHFFFAOYSA-N 1-naphthalen-1-ylnaphthalene Chemical compound C1=CC=C2C(C=3C4=CC=CC=C4C=CC=3)=CC=CC2=C1 ZDZHCHYQNPQSGG-UHFFFAOYSA-N 0.000 claims 1
- 206010039491 Sarcoma Diseases 0.000 claims 1
- 125000000278 alkyl amino alkyl group Chemical group 0.000 claims 1
- 125000003806 alkyl carbonyl amino group Chemical group 0.000 claims 1
- 125000004103 aminoalkyl group Chemical group 0.000 claims 1
- 230000000259 anti-tumor effect Effects 0.000 claims 1
- 238000011319 anticancer therapy Methods 0.000 claims 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 claims 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims 1
- 150000002940 palladium Chemical class 0.000 claims 1
- 238000002560 therapeutic procedure Methods 0.000 abstract description 3
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 344
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 243
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 205
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 description 159
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 149
- 238000005481 NMR spectroscopy Methods 0.000 description 136
- 239000000243 solution Substances 0.000 description 124
- 239000002904 solvent Substances 0.000 description 115
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 111
- 238000005160 1H NMR spectroscopy Methods 0.000 description 106
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 103
- IAZDPXIOMUYVGZ-WFGJKAKNSA-N Dimethyl sulfoxide Chemical compound [2H]C([2H])([2H])S(=O)C([2H])([2H])[2H] IAZDPXIOMUYVGZ-WFGJKAKNSA-N 0.000 description 79
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 75
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 69
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 67
- 229910052938 sodium sulfate Inorganic materials 0.000 description 59
- 229960004132 diethyl ether Drugs 0.000 description 47
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 45
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 42
- 239000007832 Na2SO4 Substances 0.000 description 41
- 239000012044 organic layer Substances 0.000 description 40
- 239000000741 silica gel Substances 0.000 description 40
- 229910002027 silica gel Inorganic materials 0.000 description 40
- BWHMMNNQKKPAPP-UHFFFAOYSA-L potassium carbonate Chemical compound [K+].[K+].[O-]C([O-])=O BWHMMNNQKKPAPP-UHFFFAOYSA-L 0.000 description 39
- 239000007787 solid Substances 0.000 description 38
- 235000019439 ethyl acetate Nutrition 0.000 description 37
- 229940093499 ethyl acetate Drugs 0.000 description 37
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 37
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 36
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 36
- 239000011347 resin Substances 0.000 description 35
- 229920005989 resin Polymers 0.000 description 35
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical class [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 33
- 239000000725 suspension Substances 0.000 description 33
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 31
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 30
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 30
- 239000011541 reaction mixture Substances 0.000 description 30
- 238000010992 reflux Methods 0.000 description 28
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 27
- 239000012267 brine Substances 0.000 description 27
- 239000000047 product Substances 0.000 description 27
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 27
- 238000003756 stirring Methods 0.000 description 27
- 230000002829 reductive effect Effects 0.000 description 26
- 239000003112 inhibitor Substances 0.000 description 24
- 125000001570 methylene group Chemical group [H]C([H])([*:1])[*:2] 0.000 description 24
- 239000000758 substrate Substances 0.000 description 24
- 239000000872 buffer Substances 0.000 description 23
- 238000004587 chromatography analysis Methods 0.000 description 23
- 238000001914 filtration Methods 0.000 description 22
- 229960000583 acetic acid Drugs 0.000 description 21
- 229910000027 potassium carbonate Inorganic materials 0.000 description 21
- 238000011534 incubation Methods 0.000 description 20
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 19
- 238000003818 flash chromatography Methods 0.000 description 19
- 239000003921 oil Substances 0.000 description 19
- 235000019198 oils Nutrition 0.000 description 19
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 18
- 229910052786 argon Inorganic materials 0.000 description 18
- 238000003556 assay Methods 0.000 description 18
- AIYUHDOJVYHVIT-UHFFFAOYSA-M caesium chloride Chemical compound [Cl-].[Cs+] AIYUHDOJVYHVIT-UHFFFAOYSA-M 0.000 description 18
- 235000011152 sodium sulphate Nutrition 0.000 description 18
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 17
- 238000001816 cooling Methods 0.000 description 17
- 239000000284 extract Substances 0.000 description 17
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 16
- 238000004128 high performance liquid chromatography Methods 0.000 description 16
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 15
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 15
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 14
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 14
- 239000011324 bead Substances 0.000 description 14
- 125000002147 dimethylamino group Chemical group [H]C([H])([H])N(*)C([H])([H])[H] 0.000 description 14
- 239000002244 precipitate Substances 0.000 description 13
- XTHFKEDIFFGKHM-UHFFFAOYSA-N Dimethoxyethane Chemical compound COCCOC XTHFKEDIFFGKHM-UHFFFAOYSA-N 0.000 description 12
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 12
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 11
- 102000016736 Cyclin Human genes 0.000 description 11
- 108050006400 Cyclin Proteins 0.000 description 11
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 11
- 0 **c1nc(-c2c(*3)c(C(*)=*)n[n]2*)c3cn1 Chemical compound **c1nc(-c2c(*3)c(C(*)=*)n[n]2*)c3cn1 0.000 description 10
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 10
- 108010090804 Streptavidin Proteins 0.000 description 10
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 10
- 239000002585 base Substances 0.000 description 10
- 210000004027 cell Anatomy 0.000 description 10
- 229910001629 magnesium chloride Inorganic materials 0.000 description 10
- 235000011121 sodium hydroxide Nutrition 0.000 description 10
- 239000007864 aqueous solution Substances 0.000 description 9
- 238000007796 conventional method Methods 0.000 description 9
- 239000000706 filtrate Substances 0.000 description 9
- FVAUCKIRQBBSSJ-UHFFFAOYSA-M sodium iodide Chemical compound [Na+].[I-] FVAUCKIRQBBSSJ-UHFFFAOYSA-M 0.000 description 9
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 9
- VHYFNPMBLIVWCW-UHFFFAOYSA-N 4-Dimethylaminopyridine Chemical compound CN(C)C1=CC=NC=C1 VHYFNPMBLIVWCW-UHFFFAOYSA-N 0.000 description 8
- 108010033040 Histones Proteins 0.000 description 8
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 8
- 239000007983 Tris buffer Substances 0.000 description 8
- 229960004198 guanidine Drugs 0.000 description 8
- WJKHJLXJJJATHN-UHFFFAOYSA-N triflic anhydride Chemical compound FC(F)(F)S(=O)(=O)OS(=O)(=O)C(F)(F)F WJKHJLXJJJATHN-UHFFFAOYSA-N 0.000 description 8
- 102000004190 Enzymes Human genes 0.000 description 7
- 108090000790 Enzymes Proteins 0.000 description 7
- MUALRAIOVNYAIW-UHFFFAOYSA-N binap Chemical compound C1=CC=CC=C1P(C=1C(=C2C=CC=CC2=CC=1)C=1C2=CC=CC=C2C=CC=1P(C=1C=CC=CC=1)C=1C=CC=CC=1)C1=CC=CC=C1 MUALRAIOVNYAIW-UHFFFAOYSA-N 0.000 description 7
- 229910052796 boron Inorganic materials 0.000 description 7
- 239000013058 crude material Substances 0.000 description 7
- 150000002500 ions Chemical class 0.000 description 7
- 235000011118 potassium hydroxide Nutrition 0.000 description 7
- 238000000746 purification Methods 0.000 description 7
- 229910000029 sodium carbonate Inorganic materials 0.000 description 7
- 238000005406 washing Methods 0.000 description 7
- 239000003643 water by type Substances 0.000 description 7
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 6
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 6
- 229920004890 Triton X-100 Polymers 0.000 description 6
- 239000012300 argon atmosphere Substances 0.000 description 6
- 239000012074 organic phase Substances 0.000 description 6
- 239000012071 phase Substances 0.000 description 6
- 239000000126 substance Substances 0.000 description 6
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 6
- 210000004881 tumor cell Anatomy 0.000 description 6
- UCAVIZKQSBARGL-UHFFFAOYSA-N 2h-quinazoline-3-carboxamide Chemical compound C1=CC=CC2=CN(C(=O)N)CN=C21 UCAVIZKQSBARGL-UHFFFAOYSA-N 0.000 description 5
- RMZDXTBIBYDFHR-UHFFFAOYSA-N 2h-quinazoline-3-carboxylic acid Chemical compound C1=CC=CC2=CN(C(=O)O)CN=C21 RMZDXTBIBYDFHR-UHFFFAOYSA-N 0.000 description 5
- XDTMQSROBMDMFD-UHFFFAOYSA-N Cyclohexane Chemical compound C1CCCCC1 XDTMQSROBMDMFD-UHFFFAOYSA-N 0.000 description 5
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 description 5
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 5
- HUMNYLRZRPPJDN-UHFFFAOYSA-N benzaldehyde Chemical compound O=CC1=CC=CC=C1 HUMNYLRZRPPJDN-UHFFFAOYSA-N 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 101150073031 cdk2 gene Proteins 0.000 description 5
- 238000002425 crystallisation Methods 0.000 description 5
- 230000008025 crystallization Effects 0.000 description 5
- 238000001514 detection method Methods 0.000 description 5
- 239000010410 layer Substances 0.000 description 5
- MGJXBDMLVWIYOQ-UHFFFAOYSA-N methylazanide Chemical compound [NH-]C MGJXBDMLVWIYOQ-UHFFFAOYSA-N 0.000 description 5
- XAEFZNCEHLXOMS-UHFFFAOYSA-M potassium benzoate Chemical compound [K+].[O-]C(=O)C1=CC=CC=C1 XAEFZNCEHLXOMS-UHFFFAOYSA-M 0.000 description 5
- 150000003141 primary amines Chemical class 0.000 description 5
- 229920006395 saturated elastomer Polymers 0.000 description 5
- 239000012047 saturated solution Substances 0.000 description 5
- 238000000967 suction filtration Methods 0.000 description 5
- 125000005931 tert-butyloxycarbonyl group Chemical group [H]C([H])([H])C(OC(*)=O)(C([H])([H])[H])C([H])([H])[H] 0.000 description 5
- 238000012360 testing method Methods 0.000 description 5
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 5
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 4
- NLXLAEXVIDQMFP-UHFFFAOYSA-N Ammonia chloride Chemical compound [NH4+].[Cl-] NLXLAEXVIDQMFP-UHFFFAOYSA-N 0.000 description 4
- USFZMSVCRYTOJT-UHFFFAOYSA-N Ammonium acetate Chemical compound N.CC(O)=O USFZMSVCRYTOJT-UHFFFAOYSA-N 0.000 description 4
- 239000005695 Ammonium acetate Substances 0.000 description 4
- 101100005789 Caenorhabditis elegans cdk-4 gene Proteins 0.000 description 4
- 101150053721 Cdk5 gene Proteins 0.000 description 4
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 4
- MHAJPDPJQMAIIY-UHFFFAOYSA-N Hydrogen peroxide Chemical compound OO MHAJPDPJQMAIIY-UHFFFAOYSA-N 0.000 description 4
- 108091054455 MAP kinase family Proteins 0.000 description 4
- 102000043136 MAP kinase family Human genes 0.000 description 4
- 229930006000 Sucrose Natural products 0.000 description 4
- CZMRCDWAGMRECN-UGDNZRGBSA-N Sucrose Chemical compound O[C@H]1[C@H](O)[C@@H](CO)O[C@@]1(CO)O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 CZMRCDWAGMRECN-UGDNZRGBSA-N 0.000 description 4
- 239000002253 acid Substances 0.000 description 4
- 230000004913 activation Effects 0.000 description 4
- 238000005917 acylation reaction Methods 0.000 description 4
- 229940043376 ammonium acetate Drugs 0.000 description 4
- 235000019257 ammonium acetate Nutrition 0.000 description 4
- 229940045988 antineoplastic drug protein kinase inhibitors Drugs 0.000 description 4
- 239000008346 aqueous phase Substances 0.000 description 4
- 239000012298 atmosphere Substances 0.000 description 4
- WARCRYXKINZHGQ-UHFFFAOYSA-N benzohydrazide Chemical compound NNC(=O)C1=CC=CC=C1 WARCRYXKINZHGQ-UHFFFAOYSA-N 0.000 description 4
- 230000003197 catalytic effect Effects 0.000 description 4
- PAFZNILMFXTMIY-UHFFFAOYSA-N cyclohexylamine Chemical compound NC1CCCCC1 PAFZNILMFXTMIY-UHFFFAOYSA-N 0.000 description 4
- 229940079593 drug Drugs 0.000 description 4
- 238000001035 drying Methods 0.000 description 4
- XFLFPYFSAQLHCN-UHFFFAOYSA-N ethyl 7-oxo-2,4,5,6-tetrahydroindazole-3-carboxylate Chemical compound C1CCC(=O)C2=C1C(C(=O)OCC)=NN2 XFLFPYFSAQLHCN-UHFFFAOYSA-N 0.000 description 4
- 238000002474 experimental method Methods 0.000 description 4
- 238000010438 heat treatment Methods 0.000 description 4
- 238000002347 injection Methods 0.000 description 4
- 239000007924 injection Substances 0.000 description 4
- 239000000543 intermediate Substances 0.000 description 4
- 125000002346 iodo group Chemical group I* 0.000 description 4
- 238000011068 loading method Methods 0.000 description 4
- 239000002480 mineral oil Substances 0.000 description 4
- 235000010446 mineral oil Nutrition 0.000 description 4
- HDZGCSFEDULWCS-UHFFFAOYSA-N monomethylhydrazine Chemical compound CNN HDZGCSFEDULWCS-UHFFFAOYSA-N 0.000 description 4
- 230000007935 neutral effect Effects 0.000 description 4
- 229920001223 polyethylene glycol Polymers 0.000 description 4
- 239000011591 potassium Substances 0.000 description 4
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 4
- 239000003909 protein kinase inhibitor Substances 0.000 description 4
- 238000000926 separation method Methods 0.000 description 4
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 4
- 229960004793 sucrose Drugs 0.000 description 4
- 238000003786 synthesis reaction Methods 0.000 description 4
- DYHSDKLCOJIUFX-UHFFFAOYSA-N tert-butoxycarbonyl anhydride Chemical compound CC(C)(C)OC(=O)OC(=O)OC(C)(C)C DYHSDKLCOJIUFX-UHFFFAOYSA-N 0.000 description 4
- 239000012085 test solution Substances 0.000 description 4
- UGNWTBMOAKPKBL-UHFFFAOYSA-N tetrachloro-1,4-benzoquinone Chemical compound ClC1=C(Cl)C(=O)C(Cl)=C(Cl)C1=O UGNWTBMOAKPKBL-UHFFFAOYSA-N 0.000 description 4
- PYOKUURKVVELLB-UHFFFAOYSA-N trimethyl orthoformate Chemical compound COC(OC)OC PYOKUURKVVELLB-UHFFFAOYSA-N 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- 125000003088 (fluoren-9-ylmethoxy)carbonyl group Chemical group 0.000 description 3
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 3
- WADSJYLPJPTMLN-UHFFFAOYSA-N 3-(cycloundecen-1-yl)-1,2-diazacycloundec-2-ene Chemical compound C1CCCCCCCCC=C1C1=NNCCCCCCCC1 WADSJYLPJPTMLN-UHFFFAOYSA-N 0.000 description 3
- XZLRJCSXDLXBSC-UHFFFAOYSA-N 3-[(2-methylpropan-2-yl)oxycarbonylamino]propyl 2-cyanoacetate Chemical group CC(C)(C)OC(=O)NCCCOC(=O)CC#N XZLRJCSXDLXBSC-UHFFFAOYSA-N 0.000 description 3
- XOSBKKVDFISERP-UHFFFAOYSA-N 8-amino-1-methyl-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxamide Chemical compound C1CC2=CN=C(N)N=C2C2=C1C(C(N)=O)=NN2C XOSBKKVDFISERP-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 3
- 102000008130 Cyclic AMP-Dependent Protein Kinases Human genes 0.000 description 3
- 102000002554 Cyclin A Human genes 0.000 description 3
- 108010068192 Cyclin A Proteins 0.000 description 3
- 102000003909 Cyclin E Human genes 0.000 description 3
- 108090000257 Cyclin E Proteins 0.000 description 3
- WSFSSNUMVMOOMR-UHFFFAOYSA-N Formaldehyde Chemical compound O=C WSFSSNUMVMOOMR-UHFFFAOYSA-N 0.000 description 3
- 239000007995 HEPES buffer Substances 0.000 description 3
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 3
- SJRJJKPEHAURKC-UHFFFAOYSA-N N-Methylmorpholine Chemical compound CN1CCOCC1 SJRJJKPEHAURKC-UHFFFAOYSA-N 0.000 description 3
- 229910019501 NaVO3 Inorganic materials 0.000 description 3
- 239000002202 Polyethylene glycol Substances 0.000 description 3
- 102100029008 Putative HTLV-1-related endogenous sequence Human genes 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- 239000004133 Sodium thiosulphate Substances 0.000 description 3
- 239000012317 TBTU Substances 0.000 description 3
- BOGSOFADOWIECK-UHFFFAOYSA-N [N].C=1C=NNC=1 Chemical group [N].C=1C=NNC=1 BOGSOFADOWIECK-UHFFFAOYSA-N 0.000 description 3
- CLZISMQKJZCZDN-UHFFFAOYSA-N [benzotriazol-1-yloxy(dimethylamino)methylidene]-dimethylazanium Chemical compound C1=CC=C2N(OC(N(C)C)=[N+](C)C)N=NC2=C1 CLZISMQKJZCZDN-UHFFFAOYSA-N 0.000 description 3
- 230000010933 acylation Effects 0.000 description 3
- 229910021529 ammonia Inorganic materials 0.000 description 3
- 125000002490 anilino group Chemical group [H]N(*)C1=C([H])C([H])=C([H])C([H])=C1[H] 0.000 description 3
- VHUUQVKOLVNVRT-UHFFFAOYSA-O azanium;hydron;hydroxide Chemical compound [NH4+].O VHUUQVKOLVNVRT-UHFFFAOYSA-O 0.000 description 3
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 3
- IJOOHPMOJXWVHK-UHFFFAOYSA-N chlorotrimethylsilane Chemical compound C[Si](C)(C)Cl IJOOHPMOJXWVHK-UHFFFAOYSA-N 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000003776 cleavage reaction Methods 0.000 description 3
- 101150093523 dbf4 gene Proteins 0.000 description 3
- 235000013681 dietary sucrose Nutrition 0.000 description 3
- VDQVEACBQKUUSU-UHFFFAOYSA-M disodium;sulfanide Chemical compound [Na+].[Na+].[SH-] VDQVEACBQKUUSU-UHFFFAOYSA-M 0.000 description 3
- 238000004821 distillation Methods 0.000 description 3
- CZPGTSIBVXSPJV-UHFFFAOYSA-N ethyl 1-methyl-8-oxo-5,9-dihydro-4h-pyrazolo[4,3-h]quinazoline-3-carboxylate Chemical compound C1CC2=CN=C(O)N=C2C2=C1C(C(=O)OCC)=NN2C CZPGTSIBVXSPJV-UHFFFAOYSA-N 0.000 description 3
- CEAKAIYDERWAJF-UHFFFAOYSA-N ethyl 2-(3-ethoxy-2-oxocyclohex-3-en-1-yl)-2-oxoacetate Chemical compound CCOC(=O)C(=O)C1CCC=C(OCC)C1=O CEAKAIYDERWAJF-UHFFFAOYSA-N 0.000 description 3
- GCEJWVFGMQABRX-UHFFFAOYSA-N ethyl 7-oxo-1-[2-oxo-2-(2-oxobutylamino)ethyl]-5,6-dihydro-4h-indazole-3-carboxylate Chemical compound C1CCC(=O)C2=C1C(C(=O)OCC)=NN2CC(=O)NCC(=O)CC GCEJWVFGMQABRX-UHFFFAOYSA-N 0.000 description 3
- QHRPJYIHTSXHAP-UHFFFAOYSA-N ethyl 8-amino-1-methylpyrazolo[4,3-h]quinazoline-3-carboxylate Chemical compound C1=CC2=CN=C(N)N=C2C2=C1C(C(=O)OCC)=NN2C QHRPJYIHTSXHAP-UHFFFAOYSA-N 0.000 description 3
- CQYIKQCUWSULGR-UHFFFAOYSA-N ethyl 8-iodo-1-methyl-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxylate Chemical compound C1CC2=CN=C(I)N=C2C2=C1C(C(=O)OCC)=NN2C CQYIKQCUWSULGR-UHFFFAOYSA-N 0.000 description 3
- 238000001704 evaporation Methods 0.000 description 3
- 230000008020 evaporation Effects 0.000 description 3
- 238000000605 extraction Methods 0.000 description 3
- 239000007789 gas Substances 0.000 description 3
- 239000012362 glacial acetic acid Substances 0.000 description 3
- 125000000717 hydrazino group Chemical group [H]N([*])N([H])[H] 0.000 description 3
- 239000005457 ice water Substances 0.000 description 3
- 208000032839 leukemia Diseases 0.000 description 3
- GLXDVVHUTZTUQK-UHFFFAOYSA-M lithium;hydroxide;hydrate Chemical compound [Li+].O.[OH-] GLXDVVHUTZTUQK-UHFFFAOYSA-M 0.000 description 3
- 239000012299 nitrogen atmosphere Substances 0.000 description 3
- 229910052700 potassium Inorganic materials 0.000 description 3
- 238000002953 preparative HPLC Methods 0.000 description 3
- 238000012545 processing Methods 0.000 description 3
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 3
- 125000000714 pyrimidinyl group Chemical group 0.000 description 3
- 150000003254 radicals Chemical class 0.000 description 3
- 230000007017 scission Effects 0.000 description 3
- 239000002002 slurry Substances 0.000 description 3
- 235000009518 sodium iodide Nutrition 0.000 description 3
- CMZUMMUJMWNLFH-UHFFFAOYSA-N sodium metavanadate Chemical compound [Na+].[O-][V](=O)=O CMZUMMUJMWNLFH-UHFFFAOYSA-N 0.000 description 3
- 229910052979 sodium sulfide Inorganic materials 0.000 description 3
- AKHNMLFCWUSKQB-UHFFFAOYSA-L sodium thiosulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=S AKHNMLFCWUSKQB-UHFFFAOYSA-L 0.000 description 3
- 235000019345 sodium thiosulphate Nutrition 0.000 description 3
- 239000007858 starting material Substances 0.000 description 3
- 125000001424 substituent group Chemical group 0.000 description 3
- 231100000419 toxicity Toxicity 0.000 description 3
- 230000001988 toxicity Effects 0.000 description 3
- LWIHDJKSTIGBAC-UHFFFAOYSA-K tripotassium phosphate Chemical compound [K+].[K+].[K+].[O-]P([O-])([O-])=O LWIHDJKSTIGBAC-UHFFFAOYSA-K 0.000 description 3
- GPRLSGONYQIRFK-MNYXATJNSA-N triton Chemical compound [3H+] GPRLSGONYQIRFK-MNYXATJNSA-N 0.000 description 3
- 238000001665 trituration Methods 0.000 description 3
- 239000003039 volatile agent Substances 0.000 description 3
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 2
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 2
- NWZSZGALRFJKBT-KNIFDHDWSA-N (2s)-2,6-diaminohexanoic acid;(2s)-2-hydroxybutanedioic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O.NCCCC[C@H](N)C(O)=O NWZSZGALRFJKBT-KNIFDHDWSA-N 0.000 description 2
- DHKHKXVYLBGOIT-UHFFFAOYSA-N 1,1-Diethoxyethane Chemical compound CCOC(C)OCC DHKHKXVYLBGOIT-UHFFFAOYSA-N 0.000 description 2
- WOAHJDHKFWSLKE-UHFFFAOYSA-N 1,2-benzoquinone Chemical compound O=C1C=CC=CC1=O WOAHJDHKFWSLKE-UHFFFAOYSA-N 0.000 description 2
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 description 2
- HZAXFHJVJLSVMW-UHFFFAOYSA-N 2-Aminoethan-1-ol Chemical compound NCCO HZAXFHJVJLSVMW-UHFFFAOYSA-N 0.000 description 2
- ATESHHKXEZPZJE-UHFFFAOYSA-N 2-[3-chloro-4-(4-methylpiperazin-1-yl)phenyl]guanidine Chemical compound C1CN(C)CCN1C1=CC=C(NC(N)=N)C=C1Cl ATESHHKXEZPZJE-UHFFFAOYSA-N 0.000 description 2
- ICSNLGPSRYBMBD-UHFFFAOYSA-N 2-aminopyridine Chemical compound NC1=CC=CC=N1 ICSNLGPSRYBMBD-UHFFFAOYSA-N 0.000 description 2
- AZYZXHAWWJMPHT-UHFFFAOYSA-N 2-benzylsulfanyl-6,7-dihydro-5h-quinazolin-8-one Chemical compound N1=C2C(=O)CCCC2=CN=C1SCC1=CC=CC=C1 AZYZXHAWWJMPHT-UHFFFAOYSA-N 0.000 description 2
- ZQHKVYFTBVJQDQ-UHFFFAOYSA-N 2-benzylsulfanyl-8-ethoxy-5,6-dihydroquinazoline Chemical compound N1=C2C(OCC)=CCCC2=CN=C1SCC1=CC=CC=C1 ZQHKVYFTBVJQDQ-UHFFFAOYSA-N 0.000 description 2
- QAVCQCQQTYJKSO-UHFFFAOYSA-N 2-chloro-11-cyclopropyl-4-methyl-5h-dipyrido[2,3-b:2',3'-f][1,4]diazepin-6-one Chemical compound C12=NC=CC=C2C(=O)NC=2C(C)=CC(Cl)=NC=2N1C1CC1 QAVCQCQQTYJKSO-UHFFFAOYSA-N 0.000 description 2
- XBBJOEVBOOBEBG-UHFFFAOYSA-N 3,3-dimethyl-7-oxabicyclo[4.1.0]heptan-5-one Chemical compound O=C1CC(C)(C)CC2OC21 XBBJOEVBOOBEBG-UHFFFAOYSA-N 0.000 description 2
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 2
- NHQDETIJWKXCTC-UHFFFAOYSA-N 3-chloroperbenzoic acid Chemical compound OOC(=O)C1=CC=CC(Cl)=C1 NHQDETIJWKXCTC-UHFFFAOYSA-N 0.000 description 2
- 125000004575 3-pyrrolidinyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- HAUNPYVLVAIUOO-UHFFFAOYSA-N 4,4-dimethylcyclohex-2-en-1-one Chemical compound CC1(C)CCC(=O)C=C1 HAUNPYVLVAIUOO-UHFFFAOYSA-N 0.000 description 2
- CDDGRARTNILYAB-UHFFFAOYSA-N 5,5-dimethylcyclohex-2-en-1-one Chemical compound CC1(C)CC=CC(=O)C1 CDDGRARTNILYAB-UHFFFAOYSA-N 0.000 description 2
- HWFOUEQKCWVCBH-UHFFFAOYSA-N 8-[(4-methoxyphenyl)methylamino]-1-methyl-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxamide Chemical compound C1=CC(OC)=CC=C1CNC1=NC=C(CCC2=C3N(C)N=C2C(N)=O)C3=N1 HWFOUEQKCWVCBH-UHFFFAOYSA-N 0.000 description 2
- XCWJWGNIBKHDIB-UHFFFAOYSA-N 8-amino-1-methylpyrazolo[4,3-h]quinazoline-3-carboxamide Chemical compound C1=NC(N)=NC2=C3N(C)N=C(C(N)=O)C3=CC=C21 XCWJWGNIBKHDIB-UHFFFAOYSA-N 0.000 description 2
- KRORMGNCPYUHAR-UHFFFAOYSA-N 8-iodo-1-methyl-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxamide Chemical compound C1CC2=CN=C(I)N=C2C2=C1C(C(N)=O)=NN2C KRORMGNCPYUHAR-UHFFFAOYSA-N 0.000 description 2
- PAYRUJLWNCNPSJ-UHFFFAOYSA-N Aniline Chemical compound NC1=CC=CC=C1 PAYRUJLWNCNPSJ-UHFFFAOYSA-N 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical class OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 2
- 101150012716 CDK1 gene Proteins 0.000 description 2
- 108091007914 CDKs Proteins 0.000 description 2
- 101100472050 Caenorhabditis elegans rpl-2 gene Proteins 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 2
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 2
- 201000009030 Carcinoma Diseases 0.000 description 2
- 102000002427 Cyclin B Human genes 0.000 description 2
- 108010068150 Cyclin B Proteins 0.000 description 2
- 108010024986 Cyclin-Dependent Kinase 2 Proteins 0.000 description 2
- 102100036239 Cyclin-dependent kinase 2 Human genes 0.000 description 2
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 2
- 101100059559 Emericella nidulans (strain FGSC A4 / ATCC 38163 / CBS 112.46 / NRRL 194 / M139) nimX gene Proteins 0.000 description 2
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 2
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 2
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 2
- 208000007766 Kaposi sarcoma Diseases 0.000 description 2
- 206010025323 Lymphomas Diseases 0.000 description 2
- 229910021380 Manganese Chloride Inorganic materials 0.000 description 2
- FXHOOIRPVKKKFG-UHFFFAOYSA-N N,N-Dimethylacetamide Chemical compound CN(C)C(C)=O FXHOOIRPVKKKFG-UHFFFAOYSA-N 0.000 description 2
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 2
- 235000019502 Orange oil Nutrition 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- 229910019020 PtO2 Inorganic materials 0.000 description 2
- 230000018199 S phase Effects 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- 101100273808 Xenopus laevis cdk1-b gene Proteins 0.000 description 2
- QMCZMSKKAHWYBJ-GFCCVEGCSA-N [(2s)-2-[(2-methylpropan-2-yl)oxycarbonylamino]-2-phenylethyl] methanesulfonate Chemical compound CC(C)(C)OC(=O)N[C@H](COS(C)(=O)=O)C1=CC=CC=C1 QMCZMSKKAHWYBJ-GFCCVEGCSA-N 0.000 description 2
- LUQGZTAOBMXRGX-UHFFFAOYSA-N [8-(cyclohexylamino)-1-methylpyrazolo[4,3-h]quinazolin-3-yl]-phenylmethanone Chemical compound C12=CC=C3C=NC(NC4CCCCC4)=NC3=C2N(C)N=C1C(=O)C1=CC=CC=C1 LUQGZTAOBMXRGX-UHFFFAOYSA-N 0.000 description 2
- 238000002835 absorbance Methods 0.000 description 2
- 230000001154 acute effect Effects 0.000 description 2
- YKIOKAURTKXMSB-UHFFFAOYSA-N adams's catalyst Chemical compound O=[Pt]=O YKIOKAURTKXMSB-UHFFFAOYSA-N 0.000 description 2
- 235000010443 alginic acid Nutrition 0.000 description 2
- 229920000615 alginic acid Polymers 0.000 description 2
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 2
- 235000019270 ammonium chloride Nutrition 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 239000004037 angiogenesis inhibitor Substances 0.000 description 2
- 230000006907 apoptotic process Effects 0.000 description 2
- 125000001769 aryl amino group Chemical group 0.000 description 2
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 2
- SUMDYPCJJOFFON-UHFFFAOYSA-N beta-hydroxyethanesulfonic acid Natural products OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 2
- 239000011230 binding agent Substances 0.000 description 2
- 210000000481 breast Anatomy 0.000 description 2
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Chemical group BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 description 2
- XJHCXCQVJFPJIK-UHFFFAOYSA-M caesium fluoride Chemical compound [F-].[Cs+] XJHCXCQVJFPJIK-UHFFFAOYSA-M 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 125000005518 carboxamido group Chemical group 0.000 description 2
- 239000001768 carboxy methyl cellulose Substances 0.000 description 2
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 2
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 2
- 238000001311 chemical methods and process Methods 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 239000000460 chlorine Chemical group 0.000 description 2
- 210000001072 colon Anatomy 0.000 description 2
- GBRBMTNGQBKBQE-UHFFFAOYSA-L copper;diiodide Chemical compound I[Cu]I GBRBMTNGQBKBQE-UHFFFAOYSA-L 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- 125000006310 cycloalkyl amino group Chemical group 0.000 description 2
- 125000004663 dialkyl amino group Chemical group 0.000 description 2
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N diphenyl Chemical compound C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 2
- 239000000839 emulsion Substances 0.000 description 2
- 102000052116 epidermal growth factor receptor activity proteins Human genes 0.000 description 2
- 108700015053 epidermal growth factor receptor activity proteins Proteins 0.000 description 2
- QYHWCPQDMFKXIC-UHFFFAOYSA-N ethyl 2-(3-methoxy-5,5-dimethyl-2-oxocyclohex-3-en-1-yl)-2-oxoacetate Chemical compound CCOC(=O)C(=O)C1CC(C)(C)C=C(OC)C1=O QYHWCPQDMFKXIC-UHFFFAOYSA-N 0.000 description 2
- VYUDULDAVWUAHL-UHFFFAOYSA-N ethyl 6-(dimethylaminomethylidene)-1-methyl-7-oxo-4,5-dihydroindazole-3-carboxylate Chemical compound C1CC(=CN(C)C)C(=O)C2=C1C(C(=O)OCC)=NN2C VYUDULDAVWUAHL-UHFFFAOYSA-N 0.000 description 2
- MQLYVMQQHAAGEI-UHFFFAOYSA-N ethyl 8-(carbamoylamino)-1-methylpyrazolo[4,3-h]quinazoline-3-carboxylate Chemical compound C1=CC2=CN=C(NC(N)=O)N=C2C2=C1C(C(=O)OCC)=NN2C MQLYVMQQHAAGEI-UHFFFAOYSA-N 0.000 description 2
- XCLOVJZYWDYYRT-UHFFFAOYSA-N ethyl 8-anilino-1-methyl-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxylate Chemical compound CCOC(=O)C1=NN(C)C(C2=N3)=C1CCC2=CN=C3NC1=CC=CC=C1 XCLOVJZYWDYYRT-UHFFFAOYSA-N 0.000 description 2
- HEQISFCDUZVSDF-UHFFFAOYSA-N ethyl 8-methoxy-1-methyl-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxylate Chemical compound C1CC2=CN=C(OC)N=C2C2=C1C(C(=O)OCC)=NN2C HEQISFCDUZVSDF-UHFFFAOYSA-N 0.000 description 2
- 125000000031 ethylamino group Chemical group [H]C([H])([H])C([H])([H])N([H])[*] 0.000 description 2
- 229910052731 fluorine Inorganic materials 0.000 description 2
- 125000001153 fluoro group Chemical group F* 0.000 description 2
- 229960002949 fluorouracil Drugs 0.000 description 2
- IKDUDTNKRLTJSI-UHFFFAOYSA-N hydrazine monohydrate Substances O.NN IKDUDTNKRLTJSI-UHFFFAOYSA-N 0.000 description 2
- 150000004678 hydrides Chemical class 0.000 description 2
- 229930195733 hydrocarbon Natural products 0.000 description 2
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 229910052742 iron Inorganic materials 0.000 description 2
- 239000000787 lecithin Substances 0.000 description 2
- 235000010445 lecithin Nutrition 0.000 description 2
- 229940067606 lecithin Drugs 0.000 description 2
- LZWQNOHZMQIFBX-UHFFFAOYSA-N lithium;2-methylpropan-2-olate Chemical compound [Li+].CC(C)(C)[O-] LZWQNOHZMQIFBX-UHFFFAOYSA-N 0.000 description 2
- 239000011565 manganese chloride Substances 0.000 description 2
- BDAGIHXWWSANSR-UHFFFAOYSA-N methanoic acid Natural products OC=O BDAGIHXWWSANSR-UHFFFAOYSA-N 0.000 description 2
- WSFSSNUMVMOOMR-BJUDXGSMSA-N methanone Chemical compound O=[11CH2] WSFSSNUMVMOOMR-BJUDXGSMSA-N 0.000 description 2
- JZLONOOYIXEAHM-UHFFFAOYSA-N methyl 2-fluoro-5-nitrobenzoate Chemical compound COC(=O)C1=CC([N+]([O-])=O)=CC=C1F JZLONOOYIXEAHM-UHFFFAOYSA-N 0.000 description 2
- 229920000609 methyl cellulose Polymers 0.000 description 2
- 239000001923 methylcellulose Substances 0.000 description 2
- 235000010981 methylcellulose Nutrition 0.000 description 2
- GIHFHHKDKVDQIT-UHFFFAOYSA-N methylurea;sulfuric acid Chemical compound CNC(O)=N.OS(O)(=O)=O GIHFHHKDKVDQIT-UHFFFAOYSA-N 0.000 description 2
- RXZMYLDMFYNEIM-UHFFFAOYSA-N n,1,4,4-tetramethyl-8-[4-(4-methylpiperazin-1-yl)anilino]-5h-pyrazolo[4,3-h]quinazoline-3-carboxamide Chemical compound CNC(=O)C1=NN(C)C(C2=N3)=C1C(C)(C)CC2=CN=C3NC(C=C1)=CC=C1N1CCN(C)CC1 RXZMYLDMFYNEIM-UHFFFAOYSA-N 0.000 description 2
- YOHYSYJDKVYCJI-UHFFFAOYSA-N n-[3-[[6-[3-(trifluoromethyl)anilino]pyrimidin-4-yl]amino]phenyl]cyclopropanecarboxamide Chemical compound FC(F)(F)C1=CC=CC(NC=2N=CN=C(NC=3C=C(NC(=O)C4CC4)C=CC=3)C=2)=C1 YOHYSYJDKVYCJI-UHFFFAOYSA-N 0.000 description 2
- SYSQUGFVNFXIIT-UHFFFAOYSA-N n-[4-(1,3-benzoxazol-2-yl)phenyl]-4-nitrobenzenesulfonamide Chemical class C1=CC([N+](=O)[O-])=CC=C1S(=O)(=O)NC1=CC=C(C=2OC3=CC=CC=C3N=2)C=C1 SYSQUGFVNFXIIT-UHFFFAOYSA-N 0.000 description 2
- 230000003287 optical effect Effects 0.000 description 2
- 239000010502 orange oil Substances 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- ZRSNZINYAWTAHE-UHFFFAOYSA-N p-methoxybenzaldehyde Chemical compound COC1=CC=C(C=O)C=C1 ZRSNZINYAWTAHE-UHFFFAOYSA-N 0.000 description 2
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 2
- 230000026731 phosphorylation Effects 0.000 description 2
- 238000006366 phosphorylation reaction Methods 0.000 description 2
- 238000010837 poor prognosis Methods 0.000 description 2
- 235000011056 potassium acetate Nutrition 0.000 description 2
- 108090000765 processed proteins & peptides Proteins 0.000 description 2
- 229940002612 prodrug Drugs 0.000 description 2
- 239000000651 prodrug Substances 0.000 description 2
- 230000035755 proliferation Effects 0.000 description 2
- 108090000623 proteins and genes Proteins 0.000 description 2
- 229940083082 pyrimidine derivative acting on arteriolar smooth muscle Drugs 0.000 description 2
- JWVCLYRUEFBMGU-UHFFFAOYSA-N quinazoline Chemical compound N1=CN=CC2=CC=CC=C21 JWVCLYRUEFBMGU-UHFFFAOYSA-N 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 150000003335 secondary amines Chemical class 0.000 description 2
- 235000015424 sodium Nutrition 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- 235000017557 sodium bicarbonate Nutrition 0.000 description 2
- 239000012321 sodium triacetoxyborohydride Substances 0.000 description 2
- 238000010532 solid phase synthesis reaction Methods 0.000 description 2
- 238000004611 spectroscopical analysis Methods 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 239000008223 sterile water Substances 0.000 description 2
- 239000011550 stock solution Substances 0.000 description 2
- 239000000829 suppository Substances 0.000 description 2
- 239000006188 syrup Substances 0.000 description 2
- 235000020357 syrup Nutrition 0.000 description 2
- 238000010998 test method Methods 0.000 description 2
- RMVRSNDYEFQCLF-UHFFFAOYSA-N thiophenol Chemical compound SC1=CC=CC=C1 RMVRSNDYEFQCLF-UHFFFAOYSA-N 0.000 description 2
- 125000001889 triflyl group Chemical group FC(F)(F)S(*)(=O)=O 0.000 description 2
- WOGUASPHMADVMU-AWEZNQCLSA-N (2s)-2-[(2-methylpropan-2-yl)oxycarbonylamino]-2-phenylpropanoic acid Chemical compound CC(C)(C)OC(=O)N[C@](C)(C(O)=O)C1=CC=CC=C1 WOGUASPHMADVMU-AWEZNQCLSA-N 0.000 description 1
- IJXJGQCXFSSHNL-MRVPVSSYSA-N (2s)-2-amino-2-phenylethanol Chemical compound OC[C@@H](N)C1=CC=CC=C1 IJXJGQCXFSSHNL-MRVPVSSYSA-N 0.000 description 1
- 125000004178 (C1-C4) alkyl group Chemical group 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- RXZBMPWDPOLZGW-HEWSMUCTSA-N (Z)-roxithromycin Chemical group O([C@@H]1[C@@H](C)C(=O)O[C@@H]([C@@]([C@H](O)[C@@H](C)C(=N\OCOCCOC)/[C@H](C)C[C@@](C)(O)[C@H](O[C@H]2[C@@H]([C@H](C[C@@H](C)O2)N(C)C)O)[C@H]1C)(C)O)CC)[C@H]1C[C@@](C)(OC)[C@@H](O)[C@H](C)O1 RXZBMPWDPOLZGW-HEWSMUCTSA-N 0.000 description 1
- DIOHEXPTUTVCNX-UHFFFAOYSA-N 1,1,1-trifluoro-n-phenyl-n-(trifluoromethylsulfonyl)methanesulfonamide Chemical compound FC(F)(F)S(=O)(=O)N(S(=O)(=O)C(F)(F)F)C1=CC=CC=C1 DIOHEXPTUTVCNX-UHFFFAOYSA-N 0.000 description 1
- WNXJIVFYUVYPPR-UHFFFAOYSA-N 1,3-dioxolane Chemical compound C1COCO1 WNXJIVFYUVYPPR-UHFFFAOYSA-N 0.000 description 1
- OGYGFUAIIOPWQD-UHFFFAOYSA-N 1,3-thiazolidine Chemical compound C1CSCN1 OGYGFUAIIOPWQD-UHFFFAOYSA-N 0.000 description 1
- XETXLASQWXXIIH-UHFFFAOYSA-N 1-(2-benzylsulfanyl-8-oxo-6,7-dihydro-5h-quinazolin-7-yl)-2-phenylethane-1,2-dione Chemical compound C=1C=CC=CC=1C(=O)C(=O)C(C(C1=N2)=O)CCC1=CN=C2SCC1=CC=CC=C1 XETXLASQWXXIIH-UHFFFAOYSA-N 0.000 description 1
- PVOAHINGSUIXLS-UHFFFAOYSA-N 1-Methylpiperazine Chemical compound CN1CCNCC1 PVOAHINGSUIXLS-UHFFFAOYSA-N 0.000 description 1
- WFQDTOYDVUWQMS-UHFFFAOYSA-N 1-fluoro-4-nitrobenzene Chemical compound [O-][N+](=O)C1=CC=C(F)C=C1 WFQDTOYDVUWQMS-UHFFFAOYSA-N 0.000 description 1
- IXPNQXFRVYWDDI-UHFFFAOYSA-N 1-methyl-2,4-dioxo-1,3-diazinane-5-carboximidamide Chemical compound CN1CC(C(N)=N)C(=O)NC1=O IXPNQXFRVYWDDI-UHFFFAOYSA-N 0.000 description 1
- ZOBRPBVIEUWYJR-UHFFFAOYSA-N 1-methyl-8-(phenylamino)-4,5-dihydro-1h-pyrazolo[4,3-h]quinazoline-3-carboxylic acid Chemical compound N1=C2C=3N(C)N=C(C(O)=O)C=3CCC2=CN=C1NC1=CC=CC=C1 ZOBRPBVIEUWYJR-UHFFFAOYSA-N 0.000 description 1
- VEQZWOXFCXFUCF-UHFFFAOYSA-N 1-methyl-8-[(1-methylpiperidin-4-yl)amino]-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxamide Chemical compound C1CN(C)CCC1NC1=NC=C(CCC2=C3N(C)N=C2C(N)=O)C3=N1 VEQZWOXFCXFUCF-UHFFFAOYSA-N 0.000 description 1
- BAUWRHPMUVYFOD-UHFFFAOYSA-N 1-methylpiperidin-4-ol Chemical compound CN1CCC(O)CC1 BAUWRHPMUVYFOD-UHFFFAOYSA-N 0.000 description 1
- GUKIVPNEPZZJBS-UHFFFAOYSA-N 1-tert-butylpiperazine;dihydrobromide Chemical compound Br.Br.CC(C)(C)N1CCNCC1 GUKIVPNEPZZJBS-UHFFFAOYSA-N 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- GRNOZCCBOFGDCL-UHFFFAOYSA-N 2,2,2-trichloroacetyl isocyanate Chemical compound ClC(Cl)(Cl)C(=O)N=C=O GRNOZCCBOFGDCL-UHFFFAOYSA-N 0.000 description 1
- JKTCBAGSMQIFNL-UHFFFAOYSA-N 2,3-dihydrofuran Chemical compound C1CC=CO1 JKTCBAGSMQIFNL-UHFFFAOYSA-N 0.000 description 1
- OFJBYLCQNJHFMI-UHFFFAOYSA-N 2,5-dihydro-1,2-oxazole Chemical compound C1ONC=C1 OFJBYLCQNJHFMI-UHFFFAOYSA-N 0.000 description 1
- DWLMIHRZURMFAQ-UHFFFAOYSA-N 2-(3-chlorophenyl)guanidine Chemical compound NC(N)=NC1=CC=CC(Cl)=C1 DWLMIHRZURMFAQ-UHFFFAOYSA-N 0.000 description 1
- NGNBDVOYPDDBFK-UHFFFAOYSA-N 2-[2,4-di(pentan-2-yl)phenoxy]acetyl chloride Chemical compound CCCC(C)C1=CC=C(OCC(Cl)=O)C(C(C)CCC)=C1 NGNBDVOYPDDBFK-UHFFFAOYSA-N 0.000 description 1
- LXTOLPBDJRTENC-UHFFFAOYSA-N 2-[3-(4-methylpiperazin-1-yl)phenyl]guanidine Chemical compound C1CN(C)CCN1C1=CC=CC(NC(N)=N)=C1 LXTOLPBDJRTENC-UHFFFAOYSA-N 0.000 description 1
- RWOJNAKNHZUALN-UHFFFAOYSA-N 2-[4-(4-methylpiperazin-1-yl)phenyl]guanidine Chemical compound C1CN(C)CCN1C1=CC=C(NC(N)=N)C=C1 RWOJNAKNHZUALN-UHFFFAOYSA-N 0.000 description 1
- VMZCDNSFRSVYKQ-UHFFFAOYSA-N 2-phenylacetyl chloride Chemical compound ClC(=O)CC1=CC=CC=C1 VMZCDNSFRSVYKQ-UHFFFAOYSA-N 0.000 description 1
- RSEBUVRVKCANEP-UHFFFAOYSA-N 2-pyrroline Chemical compound C1CC=CN1 RSEBUVRVKCANEP-UHFFFAOYSA-N 0.000 description 1
- MGADZUXDNSDTHW-UHFFFAOYSA-N 2H-pyran Chemical compound C1OC=CC=C1 MGADZUXDNSDTHW-UHFFFAOYSA-N 0.000 description 1
- DCTROTZBSDPXSM-UHFFFAOYSA-N 3,3-dimethylbutyl methanesulfonate Chemical compound CC(C)(C)CCOS(C)(=O)=O DCTROTZBSDPXSM-UHFFFAOYSA-N 0.000 description 1
- HLYANMUOYQYPSE-UHFFFAOYSA-N 3,4-dihydro-2h-pyrrole-4-carboxamide Chemical compound NC(=O)C1CCN=C1 HLYANMUOYQYPSE-UHFFFAOYSA-N 0.000 description 1
- ITZYBNPOMFEHOB-UHFFFAOYSA-N 3,4-dihydro-2h-pyrrole-4-carboxylic acid Chemical compound OC(=O)C1CCN=C1 ITZYBNPOMFEHOB-UHFFFAOYSA-N 0.000 description 1
- KEMAETWBRDLVRA-UHFFFAOYSA-N 3-(1-methylpiperidin-4-yl)oxyaniline Chemical compound C1CN(C)CCC1OC1=CC=CC(N)=C1 KEMAETWBRDLVRA-UHFFFAOYSA-N 0.000 description 1
- RJGHJWKQCJAJEP-UHFFFAOYSA-N 3-(4-methylpiperazin-1-yl)aniline Chemical compound C1CN(C)CCN1C1=CC=CC(N)=C1 RJGHJWKQCJAJEP-UHFFFAOYSA-N 0.000 description 1
- ZGPHZHCPWKOKDX-UHFFFAOYSA-N 3-[(4-methylpiperazin-1-yl)methyl]aniline Chemical compound C1CN(C)CCN1CC1=CC=CC(N)=C1 ZGPHZHCPWKOKDX-UHFFFAOYSA-N 0.000 description 1
- SXEZZXWNGLQKSV-UHFFFAOYSA-N 3-methoxy-5,5-dimethylcyclohex-2-en-1-one Chemical compound COC1=CC(=O)CC(C)(C)C1 SXEZZXWNGLQKSV-UHFFFAOYSA-N 0.000 description 1
- VKKPKSPCMVBPTN-UHFFFAOYSA-N 4-(3-chloro-4-methylpiperazin-1-yl)aniline Chemical compound C1C(Cl)N(C)CCN1C1=CC=C(N)C=C1 VKKPKSPCMVBPTN-UHFFFAOYSA-N 0.000 description 1
- OSWFIVFLDKOXQC-UHFFFAOYSA-N 4-(3-methoxyphenyl)aniline Chemical compound COC1=CC=CC(C=2C=CC(N)=CC=2)=C1 OSWFIVFLDKOXQC-UHFFFAOYSA-N 0.000 description 1
- MOZNZNKHRXRLLF-UHFFFAOYSA-N 4-(4-methylpiperazin-1-yl)aniline Chemical compound C1CN(C)CCN1C1=CC=C(N)C=C1 MOZNZNKHRXRLLF-UHFFFAOYSA-N 0.000 description 1
- KHEUKJILBHLJGP-UHFFFAOYSA-N 4-(4-tert-butylpiperazin-1-yl)aniline Chemical compound C1CN(C(C)(C)C)CCN1C1=CC=C(N)C=C1 KHEUKJILBHLJGP-UHFFFAOYSA-N 0.000 description 1
- 125000003143 4-hydroxybenzyl group Chemical group [H]C([*])([H])C1=C([H])C([H])=C(O[H])C([H])=C1[H] 0.000 description 1
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 description 1
- FUQGQRHDOMRUAA-UHFFFAOYSA-N 5,5-dimethyl-7-oxabicyclo[4.1.0]heptan-2-one Chemical compound CC1(C)CCC(=O)C2OC12 FUQGQRHDOMRUAA-UHFFFAOYSA-N 0.000 description 1
- PXRKCOCTEMYUEG-UHFFFAOYSA-N 5-aminoisoindole-1,3-dione Chemical compound NC1=CC=C2C(=O)NC(=O)C2=C1 PXRKCOCTEMYUEG-UHFFFAOYSA-N 0.000 description 1
- SEXGOLJOYUNUBS-UHFFFAOYSA-N 6,8-dimethyl-4,5-dihydro-2H-pyrazolo[4,3-h]quinazoline-3-carboxylic acid Chemical compound CC1=C2CCC3=C(NN=C3C2=NC(=N1)C)C(=O)O SEXGOLJOYUNUBS-UHFFFAOYSA-N 0.000 description 1
- YTQNUVLMVCPPFF-UHFFFAOYSA-N 6-(dimethylaminomethylidene)-2-ethoxycyclohex-2-en-1-one Chemical compound CCOC1=CCCC(=CN(C)C)C1=O YTQNUVLMVCPPFF-UHFFFAOYSA-N 0.000 description 1
- GZYPEUQYTIZWEQ-UHFFFAOYSA-N 8-(4-acetamidoanilino)-1-methyl-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxamide Chemical compound C1=CC(NC(=O)C)=CC=C1NC1=NC=C(CCC2=C3N(C)N=C2C(N)=O)C3=N1 GZYPEUQYTIZWEQ-UHFFFAOYSA-N 0.000 description 1
- QQAAHQLVDAMOQG-UHFFFAOYSA-N 8-(4-hydroxyanilino)-1-methyl-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxamide Chemical compound N1=C2C=3N(C)N=C(C(N)=O)C=3CCC2=CN=C1NC1=CC=C(O)C=C1 QQAAHQLVDAMOQG-UHFFFAOYSA-N 0.000 description 1
- ABEYZUJMIYFBTH-UHFFFAOYSA-N 8-(cyclohexylamino)-1-methyl-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxamide Chemical compound N1=C2C=3N(C)N=C(C(N)=O)C=3CCC2=CN=C1NC1CCCCC1 ABEYZUJMIYFBTH-UHFFFAOYSA-N 0.000 description 1
- YXAVOEZVXUPEKE-UHFFFAOYSA-N 8-(cyclohexylamino)-1-methylpyrazolo[4,3-h]quinazoline-3-carboxamide Chemical compound N=1C2=C3N(C)N=C(C(N)=O)C3=CC=C2C=NC=1NC1CCCCC1 YXAVOEZVXUPEKE-UHFFFAOYSA-N 0.000 description 1
- VIUFPQPXJYQVSE-UHFFFAOYSA-N 8-(cyclopentylamino)-1-methyl-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxamide Chemical compound N1=C2C=3N(C)N=C(C(N)=O)C=3CCC2=CN=C1NC1CCCC1 VIUFPQPXJYQVSE-UHFFFAOYSA-N 0.000 description 1
- VIKKSOKALKRZDM-UHFFFAOYSA-N 8-[(3-cyanophenyl)methylamino]-1-methyl-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxamide Chemical compound N1=C2C=3N(C)N=C(C(N)=O)C=3CCC2=CN=C1NCC1=CC=CC(C#N)=C1 VIKKSOKALKRZDM-UHFFFAOYSA-N 0.000 description 1
- TVSSLJHFDCVIRP-UHFFFAOYSA-N 8-[(4-bromophenyl)methylamino]-1-methyl-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxamide Chemical compound N1=C2C=3N(C)N=C(C(N)=O)C=3CCC2=CN=C1NCC1=CC=C(Br)C=C1 TVSSLJHFDCVIRP-UHFFFAOYSA-N 0.000 description 1
- PCIRJFQKXCEEIU-UHFFFAOYSA-N 8-[(4-hydroxyphenyl)methylamino]-1-methyl-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxamide Chemical compound N1=C2C=3N(C)N=C(C(N)=O)C=3CCC2=CN=C1NCC1=CC=C(O)C=C1 PCIRJFQKXCEEIU-UHFFFAOYSA-N 0.000 description 1
- OQJKPCPEJACKBN-UHFFFAOYSA-N 8-[3-(hydroxymethyl)-4-(4-methylpiperazin-1-yl)anilino]-n,1-dimethyl-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxamide Chemical compound CNC(=O)C1=NN(C)C(C2=N3)=C1CCC2=CN=C3NC(C=C1CO)=CC=C1N1CCN(C)CC1 OQJKPCPEJACKBN-UHFFFAOYSA-N 0.000 description 1
- OAHPPNZKYVGMRY-UHFFFAOYSA-N 8-[3-(hydroxymethyl)anilino]-1-methyl-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxamide Chemical compound N1=C2C=3N(C)N=C(C(N)=O)C=3CCC2=CN=C1NC1=CC=CC(CO)=C1 OAHPPNZKYVGMRY-UHFFFAOYSA-N 0.000 description 1
- WLWQVBSUMWUDAF-UHFFFAOYSA-N 8-[3-chloro-4-(4-methylpiperazin-1-yl)anilino]-1-methyl-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxamide Chemical compound C1CN(C)CCN1C(C(=C1)Cl)=CC=C1NC1=NC=C(CCC2=C3N(C)N=C2C(N)=O)C3=N1 WLWQVBSUMWUDAF-UHFFFAOYSA-N 0.000 description 1
- HWKOTXWDMCHSQT-UHFFFAOYSA-N 8-[4-(diethylamino)anilino]-1-methyl-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxamide Chemical compound C1=CC(N(CC)CC)=CC=C1NC1=NC=C(CCC2=C3N(C)N=C2C(N)=O)C3=N1 HWKOTXWDMCHSQT-UHFFFAOYSA-N 0.000 description 1
- PYLQORVFLYNJJJ-UHFFFAOYSA-N 8-amino-n-ethyl-1-methyl-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxamide Chemical compound C1CC2=CN=C(N)N=C2C2=C1C(C(=O)NCC)=NN2C PYLQORVFLYNJJJ-UHFFFAOYSA-N 0.000 description 1
- QMTMXOGCEWNXQT-UHFFFAOYSA-N 8-amino-n-methyl-4,5-dihydro-2h-pyrazolo[4,3-h]quinazoline-3-carboxamide Chemical compound C1CC2=CN=C(N)N=C2C2=C1C(C(=O)NC)=NN2 QMTMXOGCEWNXQT-UHFFFAOYSA-N 0.000 description 1
- VQKDHTSTSRSCTA-UHFFFAOYSA-N 8-benzamido-1-methyl-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxamide Chemical compound N1=C2C=3N(C)N=C(C(N)=O)C=3CCC2=CN=C1NC(=O)C1=CC=CC=C1 VQKDHTSTSRSCTA-UHFFFAOYSA-N 0.000 description 1
- LTHMFVACGIGYCO-UHFFFAOYSA-N 8-iodo-1-methyl-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carbonyl fluoride Chemical compound C1CC2=CN=C(I)N=C2C2=C1C(C(F)=O)=NN2C LTHMFVACGIGYCO-UHFFFAOYSA-N 0.000 description 1
- XPUCOSSMNRCYIJ-UHFFFAOYSA-N 8-iodo-1-methyl-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxylic acid Chemical compound C1CC2=CN=C(I)N=C2C2=C1C(C(O)=O)=NN2C XPUCOSSMNRCYIJ-UHFFFAOYSA-N 0.000 description 1
- MUBFDKCAYMAVJQ-UHFFFAOYSA-N 8h-quinazolin-7-one Chemical compound C1=NC=C2C=CC(=O)CC2=N1 MUBFDKCAYMAVJQ-UHFFFAOYSA-N 0.000 description 1
- 208000030507 AIDS Diseases 0.000 description 1
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 description 1
- 208000014697 Acute lymphocytic leukaemia Diseases 0.000 description 1
- 208000031261 Acute myeloid leukaemia Diseases 0.000 description 1
- 229920001817 Agar Polymers 0.000 description 1
- GUBGYTABKSRVRQ-XLOQQCSPSA-N Alpha-Lactose Chemical compound O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-XLOQQCSPSA-N 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 206010003571 Astrocytoma Diseases 0.000 description 1
- 102000003989 Aurora kinases Human genes 0.000 description 1
- 108090000433 Aurora kinases Proteins 0.000 description 1
- 208000003950 B-cell lymphoma Diseases 0.000 description 1
- 208000032791 BCR-ABL1 positive chronic myelogenous leukemia Diseases 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- 206010005003 Bladder cancer Diseases 0.000 description 1
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 description 1
- 206010006187 Breast cancer Diseases 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- GNAFJTWHLJCCAN-UHFFFAOYSA-N CC(C)(Cc1cnc(Nc(cc2)ccc2N2CCN(C)CC2)nc1-1)c2c-1[n](C)nc2C(OC)=O Chemical compound CC(C)(Cc1cnc(Nc(cc2)ccc2N2CCN(C)CC2)nc1-1)c2c-1[n](C)nc2C(OC)=O GNAFJTWHLJCCAN-UHFFFAOYSA-N 0.000 description 1
- UZEJUOBTMYJEQW-UHFFFAOYSA-N CC(C)c1cnc(N)nc1C(C)=C Chemical compound CC(C)c1cnc(N)nc1C(C)=C UZEJUOBTMYJEQW-UHFFFAOYSA-N 0.000 description 1
- NAELHJSULMMTGG-UHFFFAOYSA-N CCOC(c1n[n](C)c(C2=N3)c1CCC2=CCCC=C3OS(C(F)(F)F)(=O)=O)=O Chemical compound CCOC(c1n[n](C)c(C2=N3)c1CCC2=CCCC=C3OS(C(F)(F)F)(=O)=O)=O NAELHJSULMMTGG-UHFFFAOYSA-N 0.000 description 1
- LOEPJCJUHSEZBU-UHFFFAOYSA-N CCOC(c1n[n](C)c-2c1CCc1cnc(Nc3ccccn3)nc-21)=O Chemical compound CCOC(c1n[n](C)c-2c1CCc1cnc(Nc3ccccn3)nc-21)=O LOEPJCJUHSEZBU-UHFFFAOYSA-N 0.000 description 1
- 101150006084 CHKB gene Proteins 0.000 description 1
- DEYMRGZIFWARLH-UHFFFAOYSA-N C[n]1nc(C(N)=O)c2c1-c1nc(Nc3ccccn3)ncc1CC2 Chemical compound C[n]1nc(C(N)=O)c2c1-c1nc(Nc3ccccn3)ncc1CC2 DEYMRGZIFWARLH-UHFFFAOYSA-N 0.000 description 1
- 101100326430 Caenorhabditis elegans bub-1 gene Proteins 0.000 description 1
- 101100220616 Caenorhabditis elegans chk-2 gene Proteins 0.000 description 1
- 101100498823 Caenorhabditis elegans ddr-2 gene Proteins 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 1
- 239000004215 Carbon black (E152) Substances 0.000 description 1
- 229940123587 Cell cycle inhibitor Drugs 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- KZBUYRJDOAKODT-UHFFFAOYSA-N Chlorine Chemical group ClCl KZBUYRJDOAKODT-UHFFFAOYSA-N 0.000 description 1
- WBYWAXJHAXSJNI-SREVYHEPSA-N Cinnamic acid Chemical compound OC(=O)\C=C/C1=CC=CC=C1 WBYWAXJHAXSJNI-SREVYHEPSA-N 0.000 description 1
- 229910021595 Copper(I) iodide Inorganic materials 0.000 description 1
- 229920002261 Corn starch Polymers 0.000 description 1
- 102000003910 Cyclin D Human genes 0.000 description 1
- 108090000259 Cyclin D Proteins 0.000 description 1
- 108010025454 Cyclin-Dependent Kinase 5 Proteins 0.000 description 1
- 102000000577 Cyclin-Dependent Kinase Inhibitor p27 Human genes 0.000 description 1
- 108010016777 Cyclin-Dependent Kinase Inhibitor p27 Proteins 0.000 description 1
- 102100032857 Cyclin-dependent kinase 1 Human genes 0.000 description 1
- 101710106279 Cyclin-dependent kinase 1 Proteins 0.000 description 1
- 102100026805 Cyclin-dependent-like kinase 5 Human genes 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 1
- FBPFZTCFMRRESA-JGWLITMVSA-N D-glucitol Chemical compound OC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-JGWLITMVSA-N 0.000 description 1
- 238000005361 D2 NMR spectroscopy Methods 0.000 description 1
- 102100030960 DNA replication licensing factor MCM2 Human genes 0.000 description 1
- 229940123780 DNA topoisomerase I inhibitor Drugs 0.000 description 1
- 229940124087 DNA topoisomerase II inhibitor Drugs 0.000 description 1
- ZAFNJMIOTHYJRJ-UHFFFAOYSA-N Diisopropyl ether Chemical compound CC(C)OC(C)C ZAFNJMIOTHYJRJ-UHFFFAOYSA-N 0.000 description 1
- 102100031480 Dual specificity mitogen-activated protein kinase kinase 1 Human genes 0.000 description 1
- 101710146526 Dual specificity mitogen-activated protein kinase kinase 1 Proteins 0.000 description 1
- LVGKNOAMLMIIKO-UHFFFAOYSA-N Elaidinsaeure-aethylester Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC LVGKNOAMLMIIKO-UHFFFAOYSA-N 0.000 description 1
- 201000008808 Fibrosarcoma Diseases 0.000 description 1
- 239000001828 Gelatine Substances 0.000 description 1
- 208000032612 Glial tumor Diseases 0.000 description 1
- 206010018338 Glioma Diseases 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 208000017604 Hodgkin disease Diseases 0.000 description 1
- 208000021519 Hodgkin lymphoma Diseases 0.000 description 1
- 208000010747 Hodgkins lymphoma Diseases 0.000 description 1
- 101000583807 Homo sapiens DNA replication licensing factor MCM2 Proteins 0.000 description 1
- 101001018431 Homo sapiens DNA replication licensing factor MCM7 Proteins 0.000 description 1
- 101001077604 Homo sapiens Insulin receptor substrate 1 Proteins 0.000 description 1
- 101001012157 Homo sapiens Receptor tyrosine-protein kinase erbB-2 Proteins 0.000 description 1
- WRYCSMQKUKOKBP-UHFFFAOYSA-N Imidazolidine Chemical compound C1CNCN1 WRYCSMQKUKOKBP-UHFFFAOYSA-N 0.000 description 1
- 125000002842 L-seryl group Chemical group O=C([*])[C@](N([H])[H])([H])C([H])([H])O[H] 0.000 description 1
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 229910010084 LiAlH4 Inorganic materials 0.000 description 1
- 229940124647 MEK inhibitor Drugs 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 1
- GLFNIEUTAYBVOC-UHFFFAOYSA-L Manganese chloride Chemical compound Cl[Mn]Cl GLFNIEUTAYBVOC-UHFFFAOYSA-L 0.000 description 1
- 229930195725 Mannitol Natural products 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 239000012359 Methanesulfonyl chloride Substances 0.000 description 1
- 241000699666 Mus <mouse, genus> Species 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- 201000003793 Myelodysplastic syndrome Diseases 0.000 description 1
- 208000033761 Myelogenous Chronic BCR-ABL Positive Leukemia Diseases 0.000 description 1
- 208000033776 Myeloid Acute Leukemia Diseases 0.000 description 1
- SECXISVLQFMRJM-UHFFFAOYSA-N N-Methylpyrrolidone Chemical compound CN1CCCC1=O SECXISVLQFMRJM-UHFFFAOYSA-N 0.000 description 1
- KDJVAIGJQDTBKT-UHFFFAOYSA-N N-cyclopentyl-3-ethyl-1-methyl-4,5-dihydropyrazolo[4,3-h]quinazolin-8-amine Chemical compound C(C)C1=NN(C2=C1CCC=1C=NC(=NC2=1)NC1CCCC1)C KDJVAIGJQDTBKT-UHFFFAOYSA-N 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 229910004879 Na2S2O5 Inorganic materials 0.000 description 1
- 206010029260 Neuroblastoma Diseases 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 208000015914 Non-Hodgkin lymphomas Diseases 0.000 description 1
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 1
- 108091008606 PDGF receptors Proteins 0.000 description 1
- 102000038030 PI3Ks Human genes 0.000 description 1
- 108091007960 PI3Ks Proteins 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- 229920001214 Polysorbate 60 Polymers 0.000 description 1
- 239000004372 Polyvinyl alcohol Substances 0.000 description 1
- 102000001708 Protein Isoforms Human genes 0.000 description 1
- 108010029485 Protein Isoforms Proteins 0.000 description 1
- 102000003923 Protein Kinase C Human genes 0.000 description 1
- 108090000315 Protein Kinase C Proteins 0.000 description 1
- 102100030086 Receptor tyrosine-protein kinase erbB-2 Human genes 0.000 description 1
- 102100027940 Serine/threonine-protein kinase PAK 4 Human genes 0.000 description 1
- 101710148155 Serine/threonine-protein kinase PAK 4 Proteins 0.000 description 1
- 101710148159 Serine/threonine-protein kinase PAK 5 Proteins 0.000 description 1
- 102100026840 Serine/threonine-protein kinase PAK 6 Human genes 0.000 description 1
- 206010041067 Small cell lung cancer Diseases 0.000 description 1
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 1
- 206010042971 T-cell lymphoma Diseases 0.000 description 1
- 208000027585 T-cell non-Hodgkin lymphoma Diseases 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric Acid Chemical compound [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 239000000365 Topoisomerase I Inhibitor Substances 0.000 description 1
- 239000000317 Topoisomerase II Inhibitor Substances 0.000 description 1
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 1
- 239000013504 Triton X-100 Substances 0.000 description 1
- 102000004243 Tubulin Human genes 0.000 description 1
- 108090000704 Tubulin Proteins 0.000 description 1
- 208000007097 Urinary Bladder Neoplasms Diseases 0.000 description 1
- WUDXWASLGUZIMB-UHFFFAOYSA-N [2-(4-methylpiperazino)-5-nitrophenyl]methanol Chemical compound C1CN(C)CCN1C1=CC=C([N+]([O-])=O)C=C1CO WUDXWASLGUZIMB-UHFFFAOYSA-N 0.000 description 1
- MYZFCOGEUAFZRA-UHFFFAOYSA-N [8-(cyclohexylamino)-1-methyl-4,5-dihydropyrazolo[4,3-h]quinazolin-3-yl]-phenylmethanone Chemical compound C1=2CCC3=CN=C(NC4CCCCC4)N=C3C=2N(C)N=C1C(=O)C1=CC=CC=C1 MYZFCOGEUAFZRA-UHFFFAOYSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 235000010489 acacia gum Nutrition 0.000 description 1
- 239000001785 acacia senegal l. willd gum Substances 0.000 description 1
- 238000005903 acid hydrolysis reaction Methods 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 125000002015 acyclic group Chemical group 0.000 description 1
- 239000008272 agar Substances 0.000 description 1
- 235000010419 agar Nutrition 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 150000001299 aldehydes Chemical class 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 239000003513 alkali Substances 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- 125000004656 alkyl sulfonylamino group Chemical group 0.000 description 1
- 230000029936 alkylation Effects 0.000 description 1
- 238000005804 alkylation reaction Methods 0.000 description 1
- AWUCVROLDVIAJX-UHFFFAOYSA-N alpha-glycerophosphate Natural products OCC(O)COP(O)(O)=O AWUCVROLDVIAJX-UHFFFAOYSA-N 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 230000003321 amplification Effects 0.000 description 1
- 229960003116 amyl nitrite Drugs 0.000 description 1
- 229940121369 angiogenesis inhibitor Drugs 0.000 description 1
- 150000001448 anilines Chemical class 0.000 description 1
- 230000002942 anti-growth Effects 0.000 description 1
- 238000011394 anticancer treatment Methods 0.000 description 1
- 239000002256 antimetabolite Substances 0.000 description 1
- 229940041181 antineoplastic drug Drugs 0.000 description 1
- 239000005441 aurora Substances 0.000 description 1
- 230000035578 autophosphorylation Effects 0.000 description 1
- URQAMACHHWVNRD-UHFFFAOYSA-N azane;1-hydroxybenzotriazole Chemical compound N.C1=CC=C2N(O)N=NC2=C1 URQAMACHHWVNRD-UHFFFAOYSA-N 0.000 description 1
- 125000002047 benzodioxolyl group Chemical group O1OC(C2=C1C=CC=C2)* 0.000 description 1
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 description 1
- WXBLLCUINBKULX-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1.OC(=O)C1=CC=CC=C1 WXBLLCUINBKULX-UHFFFAOYSA-N 0.000 description 1
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 description 1
- GTRLQRHWPXEBLF-UHFFFAOYSA-N benzyl carbamimidothioate Chemical compound NC(=N)SCC1=CC=CC=C1 GTRLQRHWPXEBLF-UHFFFAOYSA-N 0.000 description 1
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 1
- 125000002619 bicyclic group Chemical group 0.000 description 1
- 230000008033 biological extinction Effects 0.000 description 1
- 230000033228 biological regulation Effects 0.000 description 1
- 229960002685 biotin Drugs 0.000 description 1
- 235000020958 biotin Nutrition 0.000 description 1
- 239000011616 biotin Substances 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 238000009835 boiling Methods 0.000 description 1
- MCQRPQCQMGVWIQ-UHFFFAOYSA-N boron;methylsulfanylmethane Chemical compound [B].CSC MCQRPQCQMGVWIQ-UHFFFAOYSA-N 0.000 description 1
- 229910052794 bromium Inorganic materials 0.000 description 1
- 125000004063 butyryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 235000011116 calcium hydroxide Nutrition 0.000 description 1
- CJZGTCYPCWQAJB-UHFFFAOYSA-L calcium stearate Chemical compound [Ca+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O CJZGTCYPCWQAJB-UHFFFAOYSA-L 0.000 description 1
- 239000008116 calcium stearate Substances 0.000 description 1
- 235000013539 calcium stearate Nutrition 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 230000005907 cancer growth Effects 0.000 description 1
- 208000035269 cancer or benign tumor Diseases 0.000 description 1
- 239000012830 cancer therapeutic Substances 0.000 description 1
- 239000002775 capsule Substances 0.000 description 1
- 125000002837 carbocyclic group Chemical group 0.000 description 1
- 150000004649 carbonic acid derivatives Chemical class 0.000 description 1
- 125000005392 carboxamide group Chemical group NC(=O)* 0.000 description 1
- 150000003857 carboxamides Chemical class 0.000 description 1
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 1
- 150000001735 carboxylic acids Chemical class 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 230000006369 cell cycle progression Effects 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 239000001913 cellulose Substances 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 210000003793 centrosome Anatomy 0.000 description 1
- 210000003679 cervix uteri Anatomy 0.000 description 1
- 238000004296 chiral HPLC Methods 0.000 description 1
- 229910052801 chlorine Inorganic materials 0.000 description 1
- WORJEOGGNQDSOE-UHFFFAOYSA-N chloroform;methanol Chemical compound OC.ClC(Cl)Cl WORJEOGGNQDSOE-UHFFFAOYSA-N 0.000 description 1
- 230000002759 chromosomal effect Effects 0.000 description 1
- 229940110456 cocoa butter Drugs 0.000 description 1
- 235000019868 cocoa butter Nutrition 0.000 description 1
- 230000002860 competitive effect Effects 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 238000004590 computer program Methods 0.000 description 1
- LSXDOTMGLUJQCM-UHFFFAOYSA-M copper(i) iodide Chemical compound I[Cu] LSXDOTMGLUJQCM-UHFFFAOYSA-M 0.000 description 1
- LBJNMUFDOHXDFG-UHFFFAOYSA-N copper;hydrate Chemical compound O.[Cu].[Cu] LBJNMUFDOHXDFG-UHFFFAOYSA-N 0.000 description 1
- 239000008120 corn starch Substances 0.000 description 1
- 229940111134 coxibs Drugs 0.000 description 1
- WZHCOOQXZCIUNC-UHFFFAOYSA-N cyclandelate Chemical compound C1C(C)(C)CC(C)CC1OC(=O)C(O)C1=CC=CC=C1 WZHCOOQXZCIUNC-UHFFFAOYSA-N 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 239000003255 cyclooxygenase 2 inhibitor Substances 0.000 description 1
- NISGSNTVMOOSJQ-UHFFFAOYSA-N cyclopentanamine Chemical compound NC1CCCC1 NISGSNTVMOOSJQ-UHFFFAOYSA-N 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 238000010908 decantation Methods 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000002950 deficient Effects 0.000 description 1
- 238000006356 dehydrogenation reaction Methods 0.000 description 1
- 238000012217 deletion Methods 0.000 description 1
- 230000037430 deletion Effects 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 125000004473 dialkylaminocarbonyl group Chemical group 0.000 description 1
- KJOZJSGOIJQCGA-UHFFFAOYSA-N dichloromethane;2,2,2-trifluoroacetic acid Chemical compound ClCCl.OC(=O)C(F)(F)F KJOZJSGOIJQCGA-UHFFFAOYSA-N 0.000 description 1
- LZPVNFLWFSSMJC-UHFFFAOYSA-N dichloromethane;n,n-diethylethanamine;methanol Chemical compound OC.ClCCl.CCN(CC)CC LZPVNFLWFSSMJC-UHFFFAOYSA-N 0.000 description 1
- 235000014113 dietary fatty acids Nutrition 0.000 description 1
- HPNMFZURTQLUMO-UHFFFAOYSA-N diethylamine Chemical compound CCNCC HPNMFZURTQLUMO-UHFFFAOYSA-N 0.000 description 1
- SBZXBUIDTXKZTM-UHFFFAOYSA-N diglyme Chemical compound COCCOCCOC SBZXBUIDTXKZTM-UHFFFAOYSA-N 0.000 description 1
- 125000005046 dihydronaphthyl group Chemical group 0.000 description 1
- 125000005044 dihydroquinolinyl group Chemical group N1(CC=CC2=CC=CC=C12)* 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 125000000118 dimethyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 239000006185 dispersion Substances 0.000 description 1
- 238000010494 dissociation reaction Methods 0.000 description 1
- 230000005593 dissociations Effects 0.000 description 1
- ODCCJTMPMUFERV-UHFFFAOYSA-N ditert-butyl carbonate Chemical compound CC(C)(C)OC(=O)OC(C)(C)C ODCCJTMPMUFERV-UHFFFAOYSA-N 0.000 description 1
- MOTZDAYCYVMXPC-UHFFFAOYSA-N dodecyl hydrogen sulfate Chemical class CCCCCCCCCCCCOS(O)(=O)=O MOTZDAYCYVMXPC-UHFFFAOYSA-N 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 229960004679 doxorubicin Drugs 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 238000006735 epoxidation reaction Methods 0.000 description 1
- 150000002118 epoxides Chemical class 0.000 description 1
- 210000003238 esophagus Anatomy 0.000 description 1
- 125000003754 ethoxycarbonyl group Chemical group C(=O)(OCC)* 0.000 description 1
- LNSKFTRABZMJJS-UHFFFAOYSA-N ethyl 2-(3,3-dimethylbutyl)-8-iodo-1,3,4,5-tetrahydropyrazolo[4,3-h]quinazoline-3-carboxylate Chemical compound C1CC2=CN=C(I)N=C2C2=C1C(C(=O)OCC)N(CCC(C)(C)C)N2 LNSKFTRABZMJJS-UHFFFAOYSA-N 0.000 description 1
- OZZSRGIZQGQEKF-UHFFFAOYSA-N ethyl 2-(3-aminopropyl)-4,4-dimethyl-8-[4-(4-methylpiperazin-1-yl)anilino]-3,5-dihydro-1H-pyrazolo[4,3-h]quinazoline-3-carboxylate hydrochloride Chemical compound Cl.CCOC(=O)C1N(CCCN)NC(C2=N3)=C1C(C)(C)CC2=CN=C3NC(C=C1)=CC=C1N1CCN(C)CC1 OZZSRGIZQGQEKF-UHFFFAOYSA-N 0.000 description 1
- LIDHNIWKCABTNH-UHFFFAOYSA-N ethyl 2-(3-aminopropyl)-8-anilino-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxylate;hydrochloride Chemical compound Cl.N1=C2C3=NN(CCCN)C(C(=O)OCC)=C3CCC2=CN=C1NC1=CC=CC=C1 LIDHNIWKCABTNH-UHFFFAOYSA-N 0.000 description 1
- KELJRXJJYZZYTC-UHFFFAOYSA-N ethyl 2-(3-methoxy-6,6-dimethyl-2-oxocyclohex-3-en-1-yl)-2-oxoacetate Chemical compound CCOC(=O)C(=O)C1C(=O)C(OC)=CCC1(C)C KELJRXJJYZZYTC-UHFFFAOYSA-N 0.000 description 1
- AGFSTXULNHROIX-UHFFFAOYSA-N ethyl 4,4-dimethyl-7-oxo-5,6-dihydro-2h-indazole-3-carboxylate Chemical compound CC1(C)CCC(=O)C2=C1C(C(=O)OCC)=NN2 AGFSTXULNHROIX-UHFFFAOYSA-N 0.000 description 1
- VBIKSKQITCMXAH-UHFFFAOYSA-N ethyl 6-(dimethylaminomethylidene)-4,4-dimethyl-7-oxo-2-trityl-3,5-dihydro-1h-indazole-3-carboxylate Chemical compound N1C(C(C(=CN(C)C)CC2(C)C)=O)=C2C(C(=O)OCC)N1C(C=1C=CC=CC=1)(C=1C=CC=CC=1)C1=CC=CC=C1 VBIKSKQITCMXAH-UHFFFAOYSA-N 0.000 description 1
- LMILCBDDCVCYCZ-UHFFFAOYSA-N ethyl 7-ethoxy-4,5-dihydro-2h-indazole-3-carboxylate Chemical compound C1CC=C(OCC)C2=C1C(C(=O)OCC)=NN2 LMILCBDDCVCYCZ-UHFFFAOYSA-N 0.000 description 1
- VIPPTAAWRUMXHX-UHFFFAOYSA-N ethyl 7-oxo-1-phenyl-5,6-dihydro-4h-indazole-3-carboxylate Chemical compound C1CCC(=O)C2=C1C(C(=O)OCC)=NN2C1=CC=CC=C1 VIPPTAAWRUMXHX-UHFFFAOYSA-N 0.000 description 1
- OWJJGDCLFWXAQM-UHFFFAOYSA-N ethyl 7-oxo-2-(2,2,2-trifluoroethyl)-5,6-dihydro-4h-indazole-3-carboxylate Chemical compound C1CCC(=O)C2=NN(CC(F)(F)F)C(C(=O)OCC)=C21 OWJJGDCLFWXAQM-UHFFFAOYSA-N 0.000 description 1
- OSXDWDVYBDDVMH-UHFFFAOYSA-N ethyl 7-oxo-2-[2-oxo-2-(2-oxobutylamino)ethyl]-3,4,5,6-tetrahydro-1h-indazole-3-carboxylate Chemical compound C1CCC(=O)C2=C1C(C(=O)OCC)N(CC(=O)NCC(=O)CC)N2 OSXDWDVYBDDVMH-UHFFFAOYSA-N 0.000 description 1
- RERZBILYXAEMJJ-UHFFFAOYSA-N ethyl 8-(cyclopentylamino)-1-methyl-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxylate Chemical compound CCOC(=O)C1=NN(C)C(C2=N3)=C1CCC2=CN=C3NC1CCCC1 RERZBILYXAEMJJ-UHFFFAOYSA-N 0.000 description 1
- KMCBSWOVKYWTCL-UHFFFAOYSA-N ethyl 8-[3-methoxy-5-(trifluoromethyl)anilino]-1-methyl-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxylate Chemical compound CCOC(=O)C1=NN(C)C(C2=N3)=C1CCC2=CN=C3NC1=CC(OC)=CC(C(F)(F)F)=C1 KMCBSWOVKYWTCL-UHFFFAOYSA-N 0.000 description 1
- LVUMODVSPBTREC-UHFFFAOYSA-N ethyl 8-[4-(4-methylpiperazin-1-yl)-3-(trifluoromethyl)anilino]-4,5-dihydro-2h-pyrazolo[4,3-h]quinazoline-3-carboxylate Chemical compound CCOC(=O)C=1NN=C(C2=N3)C=1CCC2=CN=C3NC(C=C1C(F)(F)F)=CC=C1N1CCN(C)CC1 LVUMODVSPBTREC-UHFFFAOYSA-N 0.000 description 1
- HZBYYUBJXQNHCR-UHFFFAOYSA-N ethyl 8-[4-(4-methylpiperazin-1-yl)anilino]-2-[3-[(2-methylpropan-2-yl)oxycarbonylamino]propyl]-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxylate Chemical compound N1=C2C3=NN(CCCNC(=O)OC(C)(C)C)C(C(=O)OCC)=C3CCC2=CN=C1NC(C=C1)=CC=C1N1CCN(C)CC1 HZBYYUBJXQNHCR-UHFFFAOYSA-N 0.000 description 1
- GUAHQNVJSKHHDA-UHFFFAOYSA-N ethyl 8-amino-1-methyl-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxylate Chemical compound C1CC2=CN=C(N)N=C2C2=C1C(C(=O)OCC)=NN2C GUAHQNVJSKHHDA-UHFFFAOYSA-N 0.000 description 1
- UWHQZMYZCIZYEW-UHFFFAOYSA-N ethyl 8-amino-2-(3-aminopropyl)-1,3,4,5-tetrahydropyrazolo[4,3-h]quinazoline-3-carboxylate hydrochloride Chemical compound Cl.C1CC2=CN=C(N)N=C2C2=C1C(C(=O)OCC)N(CCCN)N2 UWHQZMYZCIZYEW-UHFFFAOYSA-N 0.000 description 1
- JWNUCZJVWXMGSU-UHFFFAOYSA-N ethyl 8-amino-2-(3-aminopropyl)-4,4-dimethyl-3,5-dihydro-1H-pyrazolo[4,3-h]quinazoline-3-carboxylate hydrochloride Chemical compound Cl.CC1(C)CC2=CN=C(N)N=C2C2=C1C(C(=O)OCC)N(CCCN)N2 JWNUCZJVWXMGSU-UHFFFAOYSA-N 0.000 description 1
- CHKSUQVXWUBMBM-UHFFFAOYSA-N ethyl 8-amino-2-[3-[(2-methylpropan-2-yl)oxycarbonylamino]propyl]-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxylate Chemical compound N1=C(N)N=C2C3=NN(CCCNC(=O)OC(C)(C)C)C(C(=O)OCC)=C3CCC2=C1 CHKSUQVXWUBMBM-UHFFFAOYSA-N 0.000 description 1
- RXVFSQHKTCIWTE-UHFFFAOYSA-N ethyl 8-amino-4,4-dimethyl-1-trityl-5h-pyrazolo[4,3-h]quinazoline-3-carboxylate Chemical compound CC1(C)CC2=CN=C(N)N=C2C2=C1C(C(=O)OCC)=NN2C(C=1C=CC=CC=1)(C=1C=CC=CC=1)C1=CC=CC=C1 RXVFSQHKTCIWTE-UHFFFAOYSA-N 0.000 description 1
- ASRLKONJAOKZBY-UHFFFAOYSA-N ethyl 8-amino-4,4-dimethyl-2-trityl-3,5-dihydro-1h-pyrazolo[4,3-h]quinazoline-3-carboxylate Chemical compound N1C(C2=NC(N)=NC=C2CC2(C)C)=C2C(C(=O)OCC)N1C(C=1C=CC=CC=1)(C=1C=CC=CC=1)C1=CC=CC=C1 ASRLKONJAOKZBY-UHFFFAOYSA-N 0.000 description 1
- SKJXAXWOMHRBRX-UHFFFAOYSA-N ethyl 8-amino-4,5-dihydro-2h-pyrazolo[4,3-h]quinazoline-3-carboxylate Chemical compound C1CC2=CN=C(N)N=C2C2=C1C(C(=O)OCC)=NN2 SKJXAXWOMHRBRX-UHFFFAOYSA-N 0.000 description 1
- ZTGSZKZCEONFIE-UHFFFAOYSA-N ethyl 8-anilino-2-[3-[(2-methylpropan-2-yl)oxycarbonylamino]propyl]-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxylate Chemical compound N1=C2C3=NN(CCCNC(=O)OC(C)(C)C)C(C(=O)OCC)=C3CCC2=CN=C1NC1=CC=CC=C1 ZTGSZKZCEONFIE-UHFFFAOYSA-N 0.000 description 1
- KPLUFEYLIYOMJD-UHFFFAOYSA-N ethyl 8-anilino-4,4-dimethyl-2-[3-[(2-methylpropan-2-yl)oxycarbonylamino]propyl]-3,5-dihydro-1h-pyrazolo[4,3-h]quinazoline-3-carboxylate Chemical compound CCOC(=O)C1N(CCCNC(=O)OC(C)(C)C)NC(C2=N3)=C1C(C)(C)CC2=CN=C3NC1=CC=CC=C1 KPLUFEYLIYOMJD-UHFFFAOYSA-N 0.000 description 1
- DRCIMNUQWGTKLV-UHFFFAOYSA-N ethyl 8-anilino-4,5-dihydro-2h-pyrazolo[4,3-h]quinazoline-3-carboxylate Chemical compound CCOC(=O)C1=NNC(C2=N3)=C1CCC2=CN=C3NC1=CC=CC=C1 DRCIMNUQWGTKLV-UHFFFAOYSA-N 0.000 description 1
- USDXBWZWNILIJR-UHFFFAOYSA-N ethyl 8-benzyl-1-methyl-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxylate Chemical compound CCOC(=O)C1=NN(C)C(C2=N3)=C1CCC2=CN=C3CC1=CC=CC=C1 USDXBWZWNILIJR-UHFFFAOYSA-N 0.000 description 1
- UNABKLRSHQIFGV-UHFFFAOYSA-N ethyl 8-ethoxy-1-methyl-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxylate Chemical compound C1CC2=CN=C(OCC)N=C2C2=C1C(C(=O)OCC)=NN2C UNABKLRSHQIFGV-UHFFFAOYSA-N 0.000 description 1
- IIEWJVIFRVWJOD-UHFFFAOYSA-N ethyl cyclohexane Natural products CCC1CCCCC1 IIEWJVIFRVWJOD-UHFFFAOYSA-N 0.000 description 1
- WUDNUHPRLBTKOJ-UHFFFAOYSA-N ethyl isocyanate Chemical compound CCN=C=O WUDNUHPRLBTKOJ-UHFFFAOYSA-N 0.000 description 1
- PQVSTLUFSYVLTO-UHFFFAOYSA-N ethyl n-ethoxycarbonylcarbamate Chemical compound CCOC(=O)NC(=O)OCC PQVSTLUFSYVLTO-UHFFFAOYSA-N 0.000 description 1
- LVGKNOAMLMIIKO-QXMHVHEDSA-N ethyl oleate Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC LVGKNOAMLMIIKO-QXMHVHEDSA-N 0.000 description 1
- 229940093471 ethyl oleate Drugs 0.000 description 1
- QKLCQKPAECHXCQ-UHFFFAOYSA-N ethyl phenylglyoxylate Chemical compound CCOC(=O)C(=O)C1=CC=CC=C1 QKLCQKPAECHXCQ-UHFFFAOYSA-N 0.000 description 1
- 238000013401 experimental design Methods 0.000 description 1
- 239000000194 fatty acid Substances 0.000 description 1
- 229930195729 fatty acid Natural products 0.000 description 1
- 239000007941 film coated tablet Substances 0.000 description 1
- 239000007888 film coating Substances 0.000 description 1
- 238000009501 film coating Methods 0.000 description 1
- FXHZCDUQCKTJIL-UHFFFAOYSA-N fluoren-1-one;piperidine Chemical compound C1CCNCC1.C1=CC=C2C3=CC=CC(=O)C3=CC2=C1 FXHZCDUQCKTJIL-UHFFFAOYSA-N 0.000 description 1
- IRXSLJNXXZKURP-UHFFFAOYSA-N fluorenylmethyloxycarbonyl chloride Chemical compound C1=CC=C2C(COC(=O)Cl)C3=CC=CC=C3C2=C1 IRXSLJNXXZKURP-UHFFFAOYSA-N 0.000 description 1
- 239000011737 fluorine Substances 0.000 description 1
- 235000003599 food sweetener Nutrition 0.000 description 1
- 235000019253 formic acid Nutrition 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 125000002541 furyl group Chemical group 0.000 description 1
- 210000000232 gallbladder Anatomy 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011187 glycerol Nutrition 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 229960000789 guanidine hydrochloride Drugs 0.000 description 1
- PJJJBBJSCAKJQF-UHFFFAOYSA-N guanidinium chloride Chemical compound [Cl-].NC(N)=[NH2+] PJJJBBJSCAKJQF-UHFFFAOYSA-N 0.000 description 1
- 125000002795 guanidino group Chemical group C(N)(=N)N* 0.000 description 1
- 229940083094 guanine derivative acting on arteriolar smooth muscle Drugs 0.000 description 1
- 229940093915 gynecological organic acid Drugs 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 125000005843 halogen group Chemical group 0.000 description 1
- 125000001072 heteroaryl group Chemical group 0.000 description 1
- 125000005241 heteroarylamino group Chemical group 0.000 description 1
- 150000002391 heterocyclic compounds Chemical class 0.000 description 1
- 125000000592 heterocycloalkyl group Chemical group 0.000 description 1
- 229940125697 hormonal agent Drugs 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- WICNYNXYKZNNSN-UHFFFAOYSA-N hydron;4-methylpiperazine-1-carbonyl chloride;chloride Chemical compound Cl.CN1CCN(C(Cl)=O)CC1 WICNYNXYKZNNSN-UHFFFAOYSA-N 0.000 description 1
- RGZRSLKIOCHTSI-UHFFFAOYSA-N hydron;n-methylhydroxylamine;chloride Chemical compound Cl.CNO RGZRSLKIOCHTSI-UHFFFAOYSA-N 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-M hydroxide Chemical compound [OH-] XLYOFNOQVPJJNP-UHFFFAOYSA-M 0.000 description 1
- NPZTUJOABDZTLV-UHFFFAOYSA-N hydroxybenzotriazole Substances O=C1C=CC=C2NNN=C12 NPZTUJOABDZTLV-UHFFFAOYSA-N 0.000 description 1
- 230000003463 hyperproliferative effect Effects 0.000 description 1
- MTNDZQHUAFNZQY-UHFFFAOYSA-N imidazoline Chemical compound C1CN=CN1 MTNDZQHUAFNZQY-UHFFFAOYSA-N 0.000 description 1
- 125000002883 imidazolyl group Chemical group 0.000 description 1
- 239000000677 immunologic agent Substances 0.000 description 1
- 229940124541 immunological agent Drugs 0.000 description 1
- 238000001727 in vivo Methods 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 125000003392 indanyl group Chemical group C1(CCC2=CC=CC=C12)* 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- 125000001041 indolyl group Chemical group 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 239000007927 intramuscular injection Substances 0.000 description 1
- 238000010255 intramuscular injection Methods 0.000 description 1
- 238000007913 intrathecal administration Methods 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- 238000010253 intravenous injection Methods 0.000 description 1
- 238000005040 ion trap Methods 0.000 description 1
- 230000002427 irreversible effect Effects 0.000 description 1
- 229940045996 isethionic acid Drugs 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000005956 isoquinolyl group Chemical group 0.000 description 1
- 125000001786 isothiazolyl group Chemical group 0.000 description 1
- 150000002541 isothioureas Chemical class 0.000 description 1
- 125000000842 isoxazolyl group Chemical group 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 229960004393 lidocaine hydrochloride Drugs 0.000 description 1
- YECIFGHRMFEPJK-UHFFFAOYSA-N lidocaine hydrochloride monohydrate Chemical compound O.[Cl-].CC[NH+](CC)CC(=O)NC1=C(C)C=CC=C1C YECIFGHRMFEPJK-UHFFFAOYSA-N 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 239000006193 liquid solution Substances 0.000 description 1
- 239000006194 liquid suspension Substances 0.000 description 1
- 239000012280 lithium aluminium hydride Substances 0.000 description 1
- AFRJJFRNGGLMDW-UHFFFAOYSA-N lithium amide Chemical compound [Li+].[NH2-] AFRJJFRNGGLMDW-UHFFFAOYSA-N 0.000 description 1
- 229940040692 lithium hydroxide monohydrate Drugs 0.000 description 1
- 210000004185 liver Anatomy 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 210000004072 lung Anatomy 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- VTHJTEIRLNZDEV-UHFFFAOYSA-L magnesium dihydroxide Chemical class [OH-].[OH-].[Mg+2] VTHJTEIRLNZDEV-UHFFFAOYSA-L 0.000 description 1
- 235000012254 magnesium hydroxide Nutrition 0.000 description 1
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Substances [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 1
- 235000019359 magnesium stearate Nutrition 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000000594 mannitol Substances 0.000 description 1
- 235000010355 mannitol Nutrition 0.000 description 1
- 238000001819 mass spectrum Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 239000011159 matrix material Substances 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 230000037353 metabolic pathway Effects 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- QLNWXBAGRTUKKI-UHFFFAOYSA-N metacetamol Chemical compound CC(=O)NC1=CC=CC(O)=C1 QLNWXBAGRTUKKI-UHFFFAOYSA-N 0.000 description 1
- QARBMVPHQWIHKH-UHFFFAOYSA-N methanesulfonyl chloride Chemical compound CS(Cl)(=O)=O QARBMVPHQWIHKH-UHFFFAOYSA-N 0.000 description 1
- 229940087646 methanolamine Drugs 0.000 description 1
- YEWNCBNFPSRCKN-UHFFFAOYSA-N methyl 2-(4-methylpiperazino)-5-nitrobenzenecarboxylate Chemical compound COC(=O)C1=CC([N+]([O-])=O)=CC=C1N1CCN(C)CC1 YEWNCBNFPSRCKN-UHFFFAOYSA-N 0.000 description 1
- NNBBQNFHCVVQHZ-UHFFFAOYSA-N methyl carbamimidothioate;sulfuric acid Chemical compound CSC(N)=N.OS(O)(=O)=O NNBBQNFHCVVQHZ-UHFFFAOYSA-N 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 230000011278 mitosis Effects 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- DBNQIOANXZVWIP-UHFFFAOYSA-N n,n-dimethyl-1,1-bis[(2-methylpropan-2-yl)oxy]methanamine Chemical compound CC(C)(C)OC(N(C)C)OC(C)(C)C DBNQIOANXZVWIP-UHFFFAOYSA-N 0.000 description 1
- ANFLPJIRCJLNJD-UHFFFAOYSA-N n-[(1-acetylpiperidin-4-ylidene)amino]benzamide Chemical compound C1CN(C(=O)C)CCC1=NNC(=O)C1=CC=CC=C1 ANFLPJIRCJLNJD-UHFFFAOYSA-N 0.000 description 1
- XSYJKSNOFBESEE-UHFFFAOYSA-N n-[3-(1-methylpiperidin-4-yl)oxyphenyl]acetamide Chemical compound C1CN(C)CCC1OC1=CC=CC(NC(C)=O)=C1 XSYJKSNOFBESEE-UHFFFAOYSA-N 0.000 description 1
- ABURSMVCWCTGTJ-UHFFFAOYSA-N n-benzyl-1-methyl-8-[4-(4-methylpiperazin-1-yl)anilino]-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxamide Chemical compound C1CN(C)CCN1C(C=C1)=CC=C1NC1=NC=C(CCC2=C3N(C)N=C2C(=O)NCC=2C=CC=CC=2)C3=N1 ABURSMVCWCTGTJ-UHFFFAOYSA-N 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001280 n-hexyl group Chemical group C(CCCCC)* 0.000 description 1
- HJVOAXXWAQCQKG-UHFFFAOYSA-N n-methyl-2h-quinazoline-3-carboxamide Chemical compound C1=CC=CC2=CN(C(=O)NC)CN=C21 HJVOAXXWAQCQKG-UHFFFAOYSA-N 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- ALCSCNYDTSWHDD-UHFFFAOYSA-N n-tritylhydroxylamine Chemical compound C=1C=CC=CC=1C(C=1C=CC=CC=1)(NO)C1=CC=CC=C1 ALCSCNYDTSWHDD-UHFFFAOYSA-N 0.000 description 1
- 229920001206 natural gum Polymers 0.000 description 1
- 208000007538 neurilemmoma Diseases 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 238000003199 nucleic acid amplification method Methods 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 235000005985 organic acids Nutrition 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 210000001672 ovary Anatomy 0.000 description 1
- 125000002971 oxazolyl group Chemical group 0.000 description 1
- 239000007800 oxidant agent Substances 0.000 description 1
- 125000004430 oxygen atom Chemical group O* 0.000 description 1
- 125000003854 p-chlorophenyl group Chemical group [H]C1=C([H])C(*)=C([H])C([H])=C1Cl 0.000 description 1
- UQPUONNXJVWHRM-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 UQPUONNXJVWHRM-UHFFFAOYSA-N 0.000 description 1
- 210000000496 pancreas Anatomy 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 239000001814 pectin Substances 0.000 description 1
- 229920001277 pectin Polymers 0.000 description 1
- 235000010987 pectin Nutrition 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- 150000002989 phenols Chemical class 0.000 description 1
- 125000000951 phenoxy group Chemical group [H]C1=C([H])C([H])=C(O*)C([H])=C1[H] 0.000 description 1
- 125000002467 phosphate group Chemical group [H]OP(=O)(O[H])O[*] 0.000 description 1
- 229940080469 phosphocellulose Drugs 0.000 description 1
- XHXFXVLFKHQFAL-UHFFFAOYSA-N phosphoryl trichloride Chemical compound ClP(Cl)(Cl)=O XHXFXVLFKHQFAL-UHFFFAOYSA-N 0.000 description 1
- XIMLAVRHZBUYBE-UHFFFAOYSA-N piperidin-2-one;hydrate;hydrochloride Chemical compound O.Cl.O=C1CCCCN1 XIMLAVRHZBUYBE-UHFFFAOYSA-N 0.000 description 1
- 125000004482 piperidin-4-yl group Chemical group N1CCC(CC1)* 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 229920000136 polysorbate Polymers 0.000 description 1
- 229940068965 polysorbates Drugs 0.000 description 1
- 229920002451 polyvinyl alcohol Polymers 0.000 description 1
- 235000019422 polyvinyl alcohol Nutrition 0.000 description 1
- 239000001267 polyvinylpyrrolidone Substances 0.000 description 1
- 229920000036 polyvinylpyrrolidone Polymers 0.000 description 1
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 description 1
- CHKVPAROMQMJNQ-UHFFFAOYSA-M potassium bisulfate Chemical compound [K+].OS([O-])(=O)=O CHKVPAROMQMJNQ-UHFFFAOYSA-M 0.000 description 1
- 229910000343 potassium bisulfate Inorganic materials 0.000 description 1
- 229910000160 potassium phosphate Inorganic materials 0.000 description 1
- 235000011009 potassium phosphates Nutrition 0.000 description 1
- LPNYRYFBWFDTMA-UHFFFAOYSA-N potassium tert-butoxide Chemical compound [K+].CC(C)(C)[O-] LPNYRYFBWFDTMA-UHFFFAOYSA-N 0.000 description 1
- UHSONHWLNOHHBJ-UHFFFAOYSA-M potassium;8-(cyclopentylamino)-1-methyl-4,5-dihydropyrazolo[4,3-h]quinazoline-3-carboxylate Chemical compound [K+].N1=C2C=3N(C)N=C(C([O-])=O)C=3CCC2=CN=C1NC1CCCC1 UHSONHWLNOHHBJ-UHFFFAOYSA-M 0.000 description 1
- 229920001592 potato starch Polymers 0.000 description 1
- 239000000843 powder Substances 0.000 description 1
- 239000002243 precursor Substances 0.000 description 1
- 125000002572 propoxy group Chemical group [*]OC([H])([H])C(C([H])([H])[H])([H])[H] 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 239000002599 prostaglandin synthase inhibitor Substances 0.000 description 1
- 210000002307 prostate Anatomy 0.000 description 1
- 239000003528 protein farnesyltransferase inhibitor Substances 0.000 description 1
- 125000000561 purinyl group Chemical group N1=C(N=C2N=CNC2=C1)* 0.000 description 1
- 125000003373 pyrazinyl group Chemical group 0.000 description 1
- LCXHGYDKTPUJPT-UHFFFAOYSA-N pyrazol-1-ylurea Chemical class NC(=O)NN1C=CC=N1 LCXHGYDKTPUJPT-UHFFFAOYSA-N 0.000 description 1
- USPWKWBDZOARPV-UHFFFAOYSA-N pyrazolidine Chemical compound C1CNNC1 USPWKWBDZOARPV-UHFFFAOYSA-N 0.000 description 1
- DNXIASIHZYFFRO-UHFFFAOYSA-N pyrazoline Chemical compound C1CN=NC1 DNXIASIHZYFFRO-UHFFFAOYSA-N 0.000 description 1
- 125000002098 pyridazinyl group Chemical group 0.000 description 1
- 125000004076 pyridyl group Chemical group 0.000 description 1
- 150000003230 pyrimidines Chemical class 0.000 description 1
- DOYOPBSXEIZLRE-UHFFFAOYSA-N pyrrole-3-carboxylic acid Chemical compound OC(=O)C=1C=CNC=1 DOYOPBSXEIZLRE-UHFFFAOYSA-N 0.000 description 1
- ZVJHJDDKYZXRJI-UHFFFAOYSA-N pyrroline Natural products C1CC=NC1 ZVJHJDDKYZXRJI-UHFFFAOYSA-N 0.000 description 1
- LKINCDZXFUXTCZ-UHFFFAOYSA-N pyrrolo[3,4-h]quinazoline-3-carboxylic acid Chemical compound C1=CC2=CN(C(=O)O)C=NC2=C2C=NC=C21 LKINCDZXFUXTCZ-UHFFFAOYSA-N 0.000 description 1
- 125000000168 pyrrolyl group Chemical group 0.000 description 1
- 125000005493 quinolyl group Chemical group 0.000 description 1
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 description 1
- 230000002285 radioactive effect Effects 0.000 description 1
- 239000000376 reactant Substances 0.000 description 1
- 108020003175 receptors Proteins 0.000 description 1
- 102000005962 receptors Human genes 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 201000009410 rhabdomyosarcoma Diseases 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 238000007086 side reaction Methods 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 238000010898 silica gel chromatography Methods 0.000 description 1
- 239000000377 silicon dioxide Substances 0.000 description 1
- 210000003491 skin Anatomy 0.000 description 1
- 208000000587 small cell lung carcinoma Diseases 0.000 description 1
- AWUCVROLDVIAJX-GSVOUGTGSA-N sn-glycerol 3-phosphate Chemical compound OC[C@@H](O)COP(O)(O)=O AWUCVROLDVIAJX-GSVOUGTGSA-N 0.000 description 1
- 235000010413 sodium alginate Nutrition 0.000 description 1
- 239000000661 sodium alginate Substances 0.000 description 1
- 229940005550 sodium alginate Drugs 0.000 description 1
- 239000012279 sodium borohydride Substances 0.000 description 1
- 229910000033 sodium borohydride Inorganic materials 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- AJPJDKMHJJGVTQ-UHFFFAOYSA-M sodium dihydrogen phosphate Chemical compound [Na+].OP(O)([O-])=O AJPJDKMHJJGVTQ-UHFFFAOYSA-M 0.000 description 1
- HRZFUMHJMZEROT-UHFFFAOYSA-L sodium disulfite Chemical compound [Na+].[Na+].[O-]S(=O)S([O-])(=O)=O HRZFUMHJMZEROT-UHFFFAOYSA-L 0.000 description 1
- 229910000162 sodium phosphate Inorganic materials 0.000 description 1
- 229920003109 sodium starch glycolate Polymers 0.000 description 1
- 229940079832 sodium starch glycolate Drugs 0.000 description 1
- 239000008109 sodium starch glycolate Substances 0.000 description 1
- 239000011343 solid material Substances 0.000 description 1
- 238000000527 sonication Methods 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 102000009076 src-Family Kinases Human genes 0.000 description 1
- 108010087686 src-Family Kinases Proteins 0.000 description 1
- 239000008107 starch Substances 0.000 description 1
- 229940032147 starch Drugs 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 238000013517 stratification Methods 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 239000005720 sucrose Substances 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 239000007940 sugar coated tablet Substances 0.000 description 1
- 238000009495 sugar coating Methods 0.000 description 1
- 239000001117 sulphuric acid Substances 0.000 description 1
- 235000011149 sulphuric acid Nutrition 0.000 description 1
- 239000004094 surface-active agent Substances 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 239000003765 sweetening agent Substances 0.000 description 1
- 230000002194 synthesizing effect Effects 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 239000000454 talc Substances 0.000 description 1
- 229910052623 talc Inorganic materials 0.000 description 1
- 235000012222 talc Nutrition 0.000 description 1
- 238000004885 tandem mass spectrometry Methods 0.000 description 1
- 102000013498 tau Proteins Human genes 0.000 description 1
- 108010026424 tau Proteins Proteins 0.000 description 1
- 239000003277 telomerase inhibitor Substances 0.000 description 1
- CMIBWIAICVBURI-UHFFFAOYSA-N tert-butyl 3-aminopyrrolidine-1-carboxylate Chemical compound CC(C)(C)OC(=O)N1CCC(N)C1 CMIBWIAICVBURI-UHFFFAOYSA-N 0.000 description 1
- IOKGWQZQCNXXLD-UHFFFAOYSA-N tert-butyl n-(3-bromopropyl)carbamate Chemical compound CC(C)(C)OC(=O)NCCCBr IOKGWQZQCNXXLD-UHFFFAOYSA-N 0.000 description 1
- BCNZYOJHNLTNEZ-UHFFFAOYSA-N tert-butyldimethylsilyl chloride Chemical compound CC(C)(C)[Si](C)(C)Cl BCNZYOJHNLTNEZ-UHFFFAOYSA-N 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 230000001225 therapeutic effect Effects 0.000 description 1
- CBDKQYKMCICBOF-UHFFFAOYSA-N thiazoline Chemical compound C1CN=CS1 CBDKQYKMCICBOF-UHFFFAOYSA-N 0.000 description 1
- 125000000335 thiazolyl group Chemical group 0.000 description 1
- 125000001544 thienyl group Chemical group 0.000 description 1
- XJDNKRIXUMDJCW-UHFFFAOYSA-J titanium tetrachloride Chemical compound Cl[Ti](Cl)(Cl)Cl XJDNKRIXUMDJCW-UHFFFAOYSA-J 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000007704 transition Effects 0.000 description 1
- 125000001425 triazolyl group Chemical group 0.000 description 1
- GRGCWBWNLSTIEN-UHFFFAOYSA-N trifluoromethanesulfonyl chloride Chemical compound FC(F)(F)S(Cl)(=O)=O GRGCWBWNLSTIEN-UHFFFAOYSA-N 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 229940121358 tyrosine kinase inhibitor Drugs 0.000 description 1
- 239000005483 tyrosine kinase inhibitor Substances 0.000 description 1
- 241000701447 unidentified baculovirus Species 0.000 description 1
- 210000003932 urinary bladder Anatomy 0.000 description 1
- 201000005112 urinary bladder cancer Diseases 0.000 description 1
- LSGOVYNHVSXFFJ-UHFFFAOYSA-N vanadate(3-) Chemical compound [O-][V]([O-])([O-])=O LSGOVYNHVSXFFJ-UHFFFAOYSA-N 0.000 description 1
- 239000000080 wetting agent Substances 0.000 description 1
- WRSWIWOVJBYZAW-UHFFFAOYSA-M zinc;methanidylbenzene;bromide Chemical compound Br[Zn+].[CH2-]C1=CC=CC=C1 WRSWIWOVJBYZAW-UHFFFAOYSA-M 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/12—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains three hetero rings
- C07D487/14—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/535—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one oxygen as the ring hetero atoms, e.g. 1,2-oxazines
- A61K31/5375—1,4-Oxazines, e.g. morpholine
- A61K31/5377—1,4-Oxazines, e.g. morpholine not condensed and containing further heterocyclic rings, e.g. timolol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/55—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole
- A61K31/551—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having seven-membered rings, e.g. azelastine, pentylenetetrazole having two nitrogen atoms, e.g. dilazep
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/08—Drugs for disorders of the urinary system of the prostate
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/14—Drugs for dermatological disorders for baldness or alopecia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/02—Antineoplastic agents specific for leukemia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
- A61P35/04—Antineoplastic agents specific for metastasis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D231/00—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings
- C07D231/54—Heterocyclic compounds containing 1,2-diazole or hydrogenated 1,2-diazole rings condensed with carbocyclic rings or ring systems
- C07D231/56—Benzopyrazoles; Hydrogenated benzopyrazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D239/00—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings
- C07D239/70—Heterocyclic compounds containing 1,3-diazine or hydrogenated 1,3-diazine rings condensed with carbocyclic rings or ring systems
- C07D239/72—Quinazolines; Hydrogenated quinazolines
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/10—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/14—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
Definitions
- the present invention relates to pyrazolo-quinazoline derivatives, to a process for their preparation, to pharmaceutical compositions comprising them, and to their use as therapeutic agents, particularly in the treatment of cancer and cell proliferation disorders.
- cytotoxic drugs such as, e.g., fluorouracil (5-FU), doxorubicin and carnptothecins, damage DNA or affect cellular metabolic pathways and thus cause, in many cases, an indirect block of the cell cycle. Therefore, by producing an irreversible damage to both normal and tumor cells, these agents result in a significant toxicity and side-effects.
- fluorouracil 5-FU
- doxorubicin doxorubicin
- carnptothecins damage DNA or affect cellular metabolic pathways and thus cause, in many cases, an indirect block of the cell cycle. Therefore, by producing an irreversible damage to both normal and tumor cells, these agents result in a significant toxicity and side-effects.
- both the critical Gl-S and G2- M transitions are controlled by the activation of different cyclin/cdk activities.
- both cyclin D/cdk4 and cyclin E/cdk2 are thought to mediate the onset of S-phase. Progression through S-phase requires the activity of cyclin A/cdk2 whereas the activation of cyclin A/cdc2 (cdkl) and cyclin B/cdc2 are required for the onset of mitosis.
- cyclins and cyclin-dependent kinases see, for instance, Kevin R. Webster et al, in Exp. Opin. Invest. Drugs, 1998, Vol. 7(6), 865-887.
- Checkpoint controls are defective in tumor cells due, in part, to disregulation of cdk activity.
- altered expression of cyclin E and cdks has been observed in tumor cells, and deletion of the cdk inhibitor p27 KD? gene in mice has been shown to result in a higher incidence of cancer.
- cdks are rate-limiting enzymes in cell cycle progression and, as such, rapresent molecular targets for therapeutic intervention.
- the direct inhibition of cdk/cyclin kinase activity should be helpful in restricting the unregulated proliferation of a tumor cell.
- Further protein kinases known in the art as being implicated in the growth of cancer cells are the Aurora kinases, in particular Aurora-2.
- Aurora-2 was found to be over-expressed in a number of different tumor types. Its gene locus maps at 20ql3, a chromosomal region frequently amplified in many cancers, including breast [Cancer Res. 1999, 59(9) 2041-4] and colon. 20ql3 amplification correlates with poor prognosis in patients with node-negative breast cancer and increased Aurora-2 expression is indicative of poor prognosis and decreased survival time in bladder cancer patients [J. Natl. Cancer Inst, 2002, 94(17) 1320-9]. For a general reference to Aurora-2 role in the abnormal centrosome function in cancer see also Molecular Cancer Therapeutics, 2003, 2, 589 - 595.
- the pyrazolo-quinazolines of the invention are useful in the treatment of a variety of cancers including, but not limited to: carcinoma such as bladder, breast, colon, kidney, liver, lung, including small cell lung cancer, esophagus, gall-bladder, ovary, pancreas, stomach, cervix, thyroid, prostate, and skin, including squamous cell carcinoma; hematopoietic tumors of lymphoid lineage including leukaemia, acute lymphocitic leukaemia, acute lymphoblastic leukaemia, B-cell lymphoma, T-cell- lymphoma, Hodgkin's lymphoma, non-Hodgkin's lymphoma, hairy cell lymphoma and Burkett's lymphoma; hematopoietic tumors of myeloid lineage, including acute and chronic myelogenous leukemias, myelodysplastic syndrome and promyelocytic leukaemia; tumors of
- these pyrazolo-quinazoline derivatives are also useful in the treatment of a variety of cell proliferative disorders such as, for example, benign prostate hyperplasia, familial adenomatosis, polyposis, neurofibromatosis, psoriasis, vascular smooth cell proliferation associated with atherosclerosis, pulmonary fibrosis, arthritis, glomerulonephritis and post-surgical stenosis and restenosis.
- the compounds of the invention may be useful in treatment of Alzheimer's disease, as suggested by the fact that cdk5 is involved in the phosphorylation of tau protein (J. Biochem. 117, 741-749, 1995).
- the compounds of this invention may also be useful in the treatment of cancer, viral infections, prevention of AIDS development in HIN-infected individuals, autoimmune diseases and neurodegenerative disorders.
- the compounds of this invention may be useful in inhibiting tumor angiogenesis and metastasis, as well as in the treatment of organ transplant rejection and host versus graft disease.
- the compounds of the invention may also act as inhibitor of other protein kinases, e.g., protein kinase C in different isoforms, Met, PAK-4, PAK-5, ZC-1, STLK-2, DDR-2, Bub-1, PLK, Chkl, Chk2, HER2, rafl, MEK1, MAPK, EGF-R, PDGF-R, FGF-R, IGF- R, PI3K, weel kinase, Src, Abl, Akt, MAPK, ILK, MK-2, LKK-2, Cdc7, ⁇ ek, and thus be effective in the treatment of diseases associated with other protein kinases.
- the compounds of the invention are also useful in the treatment and prevention of radiotherapy-induced or chemotherapy-induced alopecia.
- the present invention provides a method for treating cell proliferative disorders caused by and/or associated with an altered protein kinase activity, like for instance Aurora 2 activity and cell cycle dependent kinase activity, by administering to a mammal in need thereof an effective amount of a pyrazolo- quinazoline derivative represented by formula (la) or (lb)
- R is hydrogen or an optionally substituted group selected from amino, straight or branched CrC 6 alkyl, C 3 - 0 cycloalkyl, cycloalkyl-alkyl, aryl, arylalkyl, heterocyclyl or heterocyclylalkyl;
- X is a single bond or a divalent radical selected from -NR'-, -CONR'-, -NH-CO-NH-, -O-, -S- or -SO 2 -, wherein R' is hydrogen or an optionally substituted group selected from straight or branched C t -C ⁇ alkyl, C 3 -C 6 cycloalkyl, cycloalkyl-alkyl, aryl, arylalkyl, heterocyclyl, heterocyclylalkyl or, together with the nitrogen atom to which they are bonded, R and R' may form a 5 to 6 membered heteroaryl or heterocyclyl group optionally containing one additional heteroatom selected among N, O or S; Ri, bonded to any one of the nitrogen atoms of the pyrazole ring as per formulae (la) or (lb), represents a hydrogen atom or an optionally substituted group selected from straight or branched -C ⁇ alkyl, C 3 -C 6
- A is a divalent group selected from -CH 2 -, -(CH 2 ) 2 -, -CH 2 -C(CH 3 ) 2 -, -C(CH 3 ) 2 -CH 2 - or
- the present invention also provides a method for treating cell proliferative disorders caused by and/or associated with an altered protein kinase activity, like cell cycle dependent kinase activity, by administering to a mammal in need thereof an effective amount of a pyrazolo-quinazoline derivative represented by the above formula (la) or (lb).
- the cell proliferative disorder is selected from the group consisting of cancer, Alzheimer's disease, viral infections, auto-immune diseases and neurodegenerative disorders.
- Specific types of cancer that may be treated include carcinoma, squamous cell carcinoma, hematopoietic tumors of myeloid or lymphoid lineage, tumors of mesenchymal origin, tumors of the central and peripheral nervous system, melanoma, seminoma, teratocarcinoma, osteosarcoma, xeroderma pigmentosum, keratoxanthoma, thyroid follicular cancer, and Kaposi's sarcoma.
- the cell proliferative disorder is selected from the group consisting of benign prostate hyperplasia, familial adenomatosis, polyposis, neuro-fibromatosis, psoriasis, vascular smooth cell proliferation associated with atherosclerosis, pulmonary fibrosis, arthritis, glomerulonephritis and post-surgical stenosis and restenosis.
- the inventive method provides tumor angiogenesis and metastasis inhibition as well as treatment of organ transplant rejection and host versus graft disease.
- the inventive methods may also provide cell cycle inhibition or cdk/cyclin dependent inhibition.
- the methods object of the present invention provide treatment and prevention of radiotherapy-induced or chemotherapy-induced alopecia.
- the present invention also provides a pyrazolo-quinazoline derivative represented by formula (la) or (lb)
- R is hydrogen or an optionally substituted group selected from amino, straight or branched C ⁇ -C 6 alkyl, C 3 -C ⁇ o cycloalkyl, cycloalkyl-alkyl, aryl, arylalkyl, heterocyclyl or heterocyclylalkyl;
- X is a single bond or a divalent radical selected from -NR'-, -CONR'-, -NH-CO-NH-, -O-, -S- or -SO 2 -, wherein R' is hydrogen or an optionally substituted group selected from straight or branched -C ⁇ alkyl, C 3 -C 6 cycloalkyl, cycloalkyl-alkyl, aryl, arylalkyl, heterocyclyl, heterocyclylalkyl or, together with the nitrogen atom to which they are bonded, R and R' may form a 5 to 6 membered heteroaryl or heterocyclyl group optionally containing one additional heteroatom
- Ri bonded to any one of the nitrogen atoms of the pyrazole ring as per formulae (la) or (lb), represents a hydrogen atom or an optionally substituted group selected from straight or branched C ⁇ -C 6 alkyl, C 3 -C 6 cycloalkyl, aryl, arylalkyl, heterocyclyl or heterocyclylalkyl or, in formula (lb), Ri is a divalent -(CH ) n -NH- group being linked to R 2 , wherein n is 2 or 3 ;
- R 2 is a group selected from -NR"R'", -N(OH)R", -OR” or -R", wherein R" and R'" are, each independently, hydrogen or an optionally substituted group selected from straight or branched C ⁇ -C 6 alkyl, C 3 -C 6 cycloalkyl or cycloalkyl-alkyl, aryl, arylalkyl, heterocyclyl or heterocyclylalkyl or, together with the nitrogen atom to which they are bonded, R" and R'” may form a 5 to 6 membered heteroaryl or heterocyclyl group, optionally containing one additional heteroatom selected among N, O or S;
- the present invention also includes methods of synthesizing the pyrazolo-quinazoline derivatives represented by formulae (la) or (lb) that, unless otherwise provided, may be conveniently grouped and defined as compounds of formula (I).
- Pharmaceutical compositions comprising the pyrazolo-quinazoline derivatives of formula (I) are also included in the present invention.
- protein kinase inhibitors Several heterocyclic compounds are known in the art as protein kinase inhibitors.
- 2-carboxamido-pyrazoles and 2-ureido-pyrazoles, and derivatives thereof, have been disclosed as protein kinase inhibitors in the international patent applications WO 01/12189, WO 01/12188, WO 02/48114 and WO 02/70515, all in the name of the applicant itself.
- Fused bicyclic compounds comprising pyrazole moieties and possessing kinase inhibitory activity have been also disclosed in WO 00/69846, WO 02/12242 as well as WO 03/028720 and still unpublished US patent application 60/381092 (filed in May 17, 2002), all in the name of the applicant itself.
- Fused tricyclic derivatives possessing kinase inhibitory activity are also disclosed in two co-pending applications PCT/EP03/01594 and PCT/US03/04844 (both claiming February 19, 2002 priority from US applications No. 60/357918 and No. 60/357960, respectively) and herewith incorporated by reference; none of the said applications specifically disclose the derivatives in re.
- fused polycyclic pyrimidine derivatives as protein kinase inhibitors are also disclosed in the international patent applications WO 98/58926 and WO 98/28281, both in the name of Celltech Therapeutics Ltd; though comprised within the general formula of both applications, no specific examples of pyrazolo-quinazolines of the present invention are exemplified therein.
- heterocyclic ring fused pyrimidine derivatives for the treatment of hyperproliferative diseases are disclosed in WO 96/40142 in the name of Pfizer Inc.
- the compounds of formula (I) of the invention may have asymmetric carbon atoms and may therefore exist as individual optical isomers, as racemic admixtures or as any other admixture comprising a majority of one of the two optical isomers, which are all to be intended as within the scope of the present invention.
- the use as an antitumor agent of all the possible isomers and their admixtures and of both the metabolites and the pharmaceutically acceptable bio-precursors (otherwise referred to as pro-drugs) of the compounds of formula (I) are also within the scope of the present invention.
- Prodrugs are any covalently bonded compounds which release the active parent drug, according to formula (I), in vivo.
- Ci-C ⁇ alkyl we intend any of the groups such as, for instance, methyl, ethyl, n-propyl, isopropyl, n-butyl, isobutyl, tert-butyl, sec-butyl, n-pentyl, n-hexyl, and the like.
- C 3 -C 10 cycloalkyl we intend, unless otherwise provided, a cycloaliphatic ring such as cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl, as well as any bridged cycloalkyl group with up to 10 carbon atoms.
- aryl includes carbocyclic or heterocyclic hydrocarbons with from 1 to 2 ring moieties, either fused or linked to each other by single bonds, wherein at least one of the rings is aromatic; if present, any aromatic heterocyclic hydrocarbon also referred to as heteroaryl group, comprises a 5 to 6 membered ring with from 1 to 3 heteroatoms selected among N, O or S.
- aryl groups are, for instance, phenyl, biphenyl, ⁇ - or ⁇ -naphthyl, dihydronaphthyl, thienyl, benzothienyl, furyl, benzofuranyl, pyrrolyl, imidazolyl, pyrazolyl, thiazolyl, isothiazolyl, oxazolyl, isoxazolyl, pyridyl, pyrazinyl, pyrimidinyl, pyridazinyl, indolyl, isoindolyl, purinyl, quinolyl, isoquinolyl, dihydroquinolinyl, quinoxalinyl, benzodioxolyl, indanyl, indenyl, triazolyl, and the like.
- heterocyclyl includes 5 to 6 membered saturated, partly unsaturated or fully unsaturated heterocycles with from 1 to 3 heteroatoms selected among N, O or S.
- saturated or partly unsaturated heterocycles are, for instance, pyran, pyrrolidine, pyrroline, imidazoline, imidazolidine, pyrazolidine, pyrazoline, thiazoline, thiazolidine, dihydrofuran, tetrahydrofuran, 1,3-dioxolane, piperidine, piperazine, morpholine and the like.
- any compound of the invention wherein X represents a single bond has to be intended as having the R group directly linked to the pyrimidine moiety.
- halogen atom we intend a fluorine, chlorine, bromine or iodine atom.
- perfluorinated alkyl we intend any of the above straight or branched Ci-C ⁇ alkyl groups which are substituted by more than one fluorine atom such as, for instance, trifiuoromethyl, triftuoroethyl, 1,1,1,3,3,3-hexafluoropropyl, and the like.
- alkoxy, aryloxy, heterocyclyloxy and derivatives thereof, e.g. perfluorinated alkoxy we intend any of the above alkyl, aryl or heterocyclyl groups linked to the rest of the molecule through an oxygen atom (-O-).
- any group which name is a composite name such as, for instance, arylalkyl or heterocyclylalkyl has to be intended as conventionally construed by the parts from which it derives, e.g. by an alkyl group which is further substituted by aryl or heterocyclyl, wherein alkyl, aryl or heterocyclyl are as above defined.
- any of the terms such as, for instance, alkylthio, alkylamino, dialkylamino, alkoxycarbonyl, alkoxycarbonylamino, heterocyclylcarbonyl, heterocyclylcarbonylamino, cycloalkyloxycarbonyl and the like, include groups wherein the alkyl, alkoxy, aryl, cycloalkyl and heterocyclyl moieties are as above defined.
- Pharmaceutically acceptable salts of the compounds of formula (I) include the acid addition salts with inorganic or organic acids, e.g., nitric, hydrochloric, hydrobromic, sulfuric, perchloric, phosphoric, acetic, trifluoroacetic propionic, glycolic, lactic, oxalic, malonic, malic, maleic, tartaric, citric, benzoic, cinnamic, mandelic, methanesulphonic, isethionic and salicylic acid, as well as the salts with inorganic or organic bases, e.g., alkali or alkaline-earth metals, especially sodium, potassium, calcium or magnesium hydroxides, carbonates or bicarbonates, acyclic or cyclic amines, preferably methylamine, ethylamine, diethylamine, triethylamine, piperidine and the like.
- inorganic or organic acids e.g., nitric, hydrochloric,
- preferred derivatives are those wherein X is a group -NH- and R 2 is a group selected from -NHR", -N(OH)R", -OR” or -R", wherein R" is an optionally substituted group selected from C 3 -C 6 cycloalkyl or cycloalkyl-alkyl, aryl, arylalkyl, heterocyclyl or heterocyclylalkyl; and R, Ri and A are as above defined.
- R 2 is a group selected from -NHR", -N(OH)R", -OR” or -R", wherein R" is an optionally substituted group selected from C 3 -C 6 cycloalkyl or cycloalkyl-alkyl, aryl, arylalkyl, heterocyclyl or heterocyclylalkyl; and R, R t and A are as above defined.
- X is a group -S- and R 2 is a group selected from -NHR", -N(OH)R", -OR” or -R", wherein R" is an optionally substituted group selected from C 3 -C 6 cycloalkyl or cycloalkyl-alkyl, aryl, arylalkyl, heterocyclyl or heterocyclylalkyl; and R, ⁇ and A are as above defined.
- a class of preferred compounds is represented by those derivatives wherein X is a group -NH- and R 2 is a group -NHR" or -N(OH)R" wherein R" is a hydrogen atom or a straight or branched C1-C4 alkyl group; and wherein A, R and Ri are as above defined.
- Another class of preferred compounds of the invention of formula (la) or (lb) is represented by the derivatives wherein X is a group -O- and R 2 is a group -NHR" or
- R is a hydrogen atom or a straight or branched -C4 alkyl group; and wherein A, R and Ri are as above defined.
- Another class of preferred compounds of the invention of formula (la) or (lb) is represented by the derivatives wherein X is a group -S- and R is a group -NHR" or
- R is a hydrogen atom or a straight or branched C ⁇ -C 4 alkyl group; and wherein A, R and Ri are as above defined.
- Another class of preferred compounds of the invention of formula (lb) is represented by the derivatives wherein R, X and A are as above defined and Ri and R 2 are linked together through a divalent -(CH 2 ) n -NH- group so as to give rise to:
- A is a group selected from -CH 2 -C(CH 3 ) 2 - or -C(CH 3 ) 2 -CH 2 -.
- R-X- is amino, R 2 is ethoxy, and Ri is as above defined; and optionally converting them into other derivatives of formula (I); st.4b) with a guanidine derivative of formula (NT)
- Tr stands for trityl (triphenylmethyl); st.14) reacting the compound of formula (XIN) with dimethylformamide-di-tert- butylacetale, as set forth in step (st.3), so as to obtain a compound of formula (XV)
- R is as above defined, A is a -(CH 2 ) 2 - group, -CH 2 -C(CH 3 ) 2 - group, -C(CH 3 ) 2 -
- the compounds of formula (I) which are prepared according to the process object of the invention can be conveniently converted into other compounds of formula (I) by operating according to well-known operative conditions.
- the compounds of formula (I): st.23) wherein R 2 is ethoxy may be converted into the compounds of formula (la) or (lb) wherein R 2 is amino by treatment with ammonium hydroxide
- R 2 is -OH
- R 2 is a group -NR"R'" or -N(OH)R"
- R is hydrogen and X is -NH-
- R is alkyl, cycloalkyl, cycloalkyl-alkyl, arylalkyl, heterocyclyl, heterocyclylalkyl, and X is -NH-, by first converting the amino group to iodine, as described in the previous step (st.29), and by subsequently reacting the iododerivative with an alkyl, cycloalkyl, cycloalkyl-alkyl, arylalkyl, heterocyclyl or heterocyclylalkyl amine of formula RNH 2 (XXVII), wherein R is as therein defined; st.30) wherein R is hydrogen and X is -NH- may be converted into the compounds of formula (I) wherein R is aryl and X is a single bond, by first converting the amino group to iodine, as per the above
- R is as above defined, e.g. methyl
- X is -S-
- R is an optionally substituted alkyl, cycloalkyl, heterocyclyl, cycloalkyl-alkyl, arylalkyl or heterocyclylalkyl group
- R is an optionally substituted alkyl, cycloalkyl, heterocyclyl, cycloalkyl-alkyl, arylalkyl or heterocyclylalkyl group
- R is an optionally substituted alkyl, cycloalkyl, heterocyclyl, cycloalkyl-alkyl, arylalkyl or heterocyclylalkyl group
- R is methyl and X is -O- may be converted into the compounds of formula (I) wherein R is an optionally substituted alkyl, cycloalkyl, heterocyclyl, cycloalkyl-alkyl, heterocyclylalkyl group, and X is -NH-, by first converting the MeO- group into HO-, then by reacting it with a triflating agent so as to obtain the corresponding trifluoromethansulfonate and finally by reacting it with an amine of formula R-NH 2 (XXVII) wherein R is an optionally substituted alkyl, cycloalkyl, heterocyclyl, cycloalkylalkyl, arylalkyl or heterocyclylalkyl group
- step (st.l) of the process 2-ethoxy-2-cyclohexen-l-one is reacted with diethyl oxalate in the presence of LiN(TMS) 2 and of a suitable solvent such as, for instance, dioxane, tetrahydrofuran or diethyl ether.
- step (st.2a) the compound of formula (II) is reacted with a suitable hydrazine derivative of formula (III), in the presence of a lower alcohol such as methanol, ethanol or admixtures thereof.
- the above reaction is carried out in ethanol at refluxing temperature, so as to obtain a mixture of both compounds of formula (INa) and (INb) wherein the former is present in major amounts.
- Their separation into the single compounds (IVa) and (IVb) is carried out under conventional methods, for instance through preparative HPLC.
- step (st.2b) of the process instead, that is by reacting the compound of formula (II) with the hydrazine derivative of formula (III) in the presence of acetic acid, a single compound of formula (INa) is obtained.
- the reaction is preferably carried out at room temperature.
- step (st.2c) of the process the compound of formula (IVa) wherein Ri is hydrogen, is reacted with a suitable compound of formula (INc) in the presence of a base such as sodium hydride in a suitable solvent, for instance tetrahydrofuran, dioxane or dimethylformamide, at a temperature ranging from room temperature to 100°C, so as to obtain a mixture of compounds (INa) and (INb) wherein the former is present in major amounts, and by separating them under conventional methods, for instance through preparative HPLC.
- a base such as sodium hydride
- a suitable solvent for instance tetrahydrofuran, dioxane or dimethylformamide
- step (st.3) of the process the compound of formula (IVa) or (INb) is reacted with dimethylformamide-di-tert-butylacetale, in the presence of a suitable solvent such as, for instance, dimethylfomiamide, so as to get the compounds of formula (Va) or (Vb), respectively.
- a suitable solvent such as, for instance, dimethylfomiamide
- the reaction is carried out at a temperature ranging from room temperature to about 70°C.
- the compound of formula (Va) or (Vb) is reacted with guanidine, guanidine salts or derivatives thereof, alkylisothiourea or methylisourea so as to obtain the corresponding compound of formula (la) or (lb) through pyrimidine ring formation.
- guanidine guanidine salts or derivatives thereof
- alkylisothiourea alkylisothiourea or methylisourea
- the reactions with guanidine or salts thereof such as hydrochloride, carbonate or nitrate, or with the guanidine derivative of formula (VI), as set forth in steps (st.4a) or (st.4b), are carried out in a lower alcoholic solvent under neutral or basic conditions, preferably with ethanol and sodium ethylate or with diazabicycloundecene (DBU) at refluxing temperature or, alternatively, in dimethylformamide at a temperature ranging from 80°C to refluxing temperature in the presence of potassium carbonate.
- a lower alcoholic solvent under neutral or basic conditions, preferably with ethanol and sodium ethylate or with diazabicycloundecene (DBU) at refluxing temperature or, alternatively, in dimethylformamide at a temperature ranging from 80°C to refluxing temperature in the presence of potassium carbonate.
- DBU diazabicycloundecene
- reaction with alkylisothiourea (VH), in (st.4c), is carried out in the presence of potassium acetate and in a suitable solvent such as dimethylformamide at refluxing temperature.
- reaction with methylisourea (st.4d) is carried out in a suitable solvent such as acetonitrile and in the presence of a base such as potassium carbonate at refluxing temperature.
- steps (st.5) and (st.6) are carried out under the operative conditions set forth in steps (st.l), (st.2a) or (st.2b) and lead to the desired compounds of formula (IXa) or (IXb), respectively.
- Step (st.7) of the process is preferably carried out by reacting the derivative of formula (IXa) or (IXb) with ethyl formate under basic conditions, preferably in the presence of sodium ethylate or sodium hydride and of a suitable solvent such as, for instance, diethyl ether, tetrahydrofuran or dioxane, at a temperature ranging from room temperature to refluxing temperature.
- a suitable solvent such as, for instance, diethyl ether, tetrahydrofuran or dioxane
- step (st.8) 2-methoxy-5,5-dimethyl-2-cyclohexen-l-one is reacted with diethyl oxalate in the presence of sodium hydride and in a suitable solvent such as diethyl ether, tetrahydrofuran or dioxane, at refluxing temperature.
- steps (st.10) are essentially those previously reported for steps (st.2a) or (st.2b), and those of steps (st.l 1) and (st.12) correspond to those of (st.3) and (st.4a and st.4b), respectively.
- step (st.13) of the process it is clear to the skilled man that both compounds of formula (IVa) or (IVb) wherein Ri is a hydrogen atom are tautomeric forms of a given compound which can be conveniently identified as having formula (IV).
- this same derivative is reacted with triphenylmethyl chloride so as to obtain a compound of formula (XIV) wherein either one of the two pyrazole nitrogen atoms are alkylated with a trityl (e.g. triphenylmethyl) group.
- step (st.14) and (st.l 5) of the process essentially correspond to those already reported for steps (st.3) and (st.4a and st.4b).
- step (st.16) the trityl group of the compounds of formula (I) is removed under acidic conditions, for instance with trifluoroacetic acid and in the presence of a suitable solvent such as dichloromethane, so as to give rise to the corresponding compound of formula (I) wherein Ri is hydrogen, in both forms:
- step (st.17) of the process allows to selectively alkylate the pyrazole nitrogen atom which is in proximity of the -COOEt group; this reaction may be carried out with lithium tert- butylate and in a suitable solvent, such as dioxane, diethyl ether or tetrahydrofuran.
- a suitable solvent such as dioxane, diethyl ether or tetrahydrofuran.
- step (st.l 8) the above compound is first converted into the free amino derivative by working according to conventional methods, for instance under acidic conditions, preferably with hydrochloric acid, in a suitable solvent such as dioxane at refluxing temperature, and subsequently cyclised to the desired tetracyclic derivative in the presence of a base such as cesium carbonate (CsCO 3 ) and in a suitable solvent such as a lower alcohol, preferably methanol, ranging from room temperature to reflux.
- a base such as cesium carbonate (CsCO 3 )
- a suitable solvent such as a lower alcohol, preferably methanol
- steps (st.19) and (st.20) of the process essentially correspond to those already reported for steps (st.3) and (st.4c); the subsequent acidic treatment of the compound of formula (XVIII) to the compound of formula (XIX) is preferably carried out with an aqueous solution of acetic acid, at a temperature of about 100°C.
- step (st.21) the compound of formula (XIX) is reacted with a suitable derivative of formula (XX) in the presence of sodium hydride and in a suitable solvent, e.g. diethyl ether, tetrahydrofuran or dioxane, at a temperature ranging from about - 50°C to room temperature.
- a suitable solvent e.g. diethyl ether, tetrahydrofuran or dioxane
- step (st.22) essentially correspond to those of step (st.l) of the process.
- the compounds of formula (I) thus prepared may be easily converted into several other compounds of formula (I) of the invention.
- compounds of formula (I) bearing R 2 as an ethoxy group, or even as an alkoxy group can be converted into a variety of derivatives according to methods well- known in the art to convert carboxyester groups (-COOR 2 ) into carboxamides (-CONH 2 ), N-substituted carboxamides (-CONHR”) and carboxylic acids (-COOH), for instance as reported in steps (st.23), (st.24) and (st.25).
- the operative conditions are those widely known in the art and may comprise, for instance in the conversion of a carboxyester group into a carboxamide group, the reaction with ammonia or ammonium hydroxide in the presence of a suitable solvent such as a lower alcohol, dimethylformamide or mixtures thereof; preferably the reaction is carried out with ammonium hydroxide in a methanoVchmethylformarnide mixture, at a temperature ranging from about 50°C to about 100°C.
- step (st.26) of the process compounds of formula (I) wherein R 2 is hydroxy (-COOH) may be converted into carboxamido derivatives (-CONR"R'") or [-CON(OH)R"] wherein R" and R'" are as formerly indicated, also inclusive of compounds wherein R" and R'" form, together with the nitrogen atom to which they are bonded, a 5 or 6 membered heteroaryl or heterocyclyl group optionally containing one additional heteroatom selected among N, O or S.
- the reaction is carried out in the presence of an amine of formula (XXTH) or of a compound of formula (XX1N), as the case may be, under basic conditions, preferably with ⁇ , ⁇ -diisopropyl- ⁇ -ethylamine or triethylamine, in a suitable solvent such as dichloromethane, dimethylformamide, tetrahydrofuran, or dioxane, and in the presence of a suitable condensing agent such as N,N'-dicyclohexylcarbodiimide (DCC), N-(3- dimethylaminopropyl)-N'-ethylcarbodiimide hydrochloride (EDCI) or O-(benzotriazol- l-yl)-N,N,N',N'-tetramethylisouronium tetrafluoroborate (TBTU); catalytic amounts of (benzotriazol-l-yloxy)tripyrrolidinophosphonium he
- reaction with isocyanate is performed with sodium hydride in dimethylformamide whilst the one with the acid chloride may be carried out in a suitable solvent such as pyridine, tetrahydrofuran, ethyl acetate or dioxane, or a mixture of them at room temperature.
- Compounds of formula (I) wherein R-NH- represents an arylamino or heteroarylamino group can be obtained by the corresponding iodo derivatives which, in their turn, may be prepared by the corresponding compounds of formula (I) wherein R-NH- is amino, as per step (st.29) of the process.
- the preparation of the iodo derivatives may be carried out in a- suitable solvent such as tetrahydrofuran, diethyl ether or dimethoxyethane, at a temperature ranging from room temperature to about 70°C, and for a time of about 8 hours to about 48 hours.
- the subsequent conversion of the iododerivative may be carried out in a suitable solvent such as dimethylformamide, dimethoxyethane or acetonitrile and in the presence of catalytic amounts of palladium acetate, (2,2'-bis(diphenylphosphino)-l,r-binaphtalene (BINAP) and a base such as potassium carbonate, potassium phosphate or cesium carbonate, at a temperature ranging from room temperature to 110°C and for a time ranging from about 2 to about 24 hours.
- a suitable solvent such as dimethylformamide, dimethoxyethane or acetonitrile
- a base such as potassium carbonate, potassium phosphate or cesium carbonate
- Compounds of formula (I) wherein R-NH- is amino may be also converted into the corresponding alkylamino or arylmethylamino derivatives of formula (I) as reported in (st.31), by operating in a suitable solvent or in a mixture of solvents, for instance comprising a 1:1:1 mixture of acetic acid, methanol and water.
- Compounds of formula (I) wherein R-NH- is amino may be also converted into the corresponding cycloalkylamino or heterocycloalkylamino derivatives of formula (I) as reported in (st.3 la), by operating in a suitable solvent such as methylene chloride, acetonitrile, dimethylformamide.
- R-X- represents an alkylthio group
- R-S- alkylthio group
- the oxidative step may be carried out with oxone in the presence of a suitable solvent, preferably dimethylformamide or dimethylsulfoxide at room temperature; the subsequent replacement of the alkylsulfonyl group with a suitable amino derivative is preferably carried out in the presence of dimethylsulfoxide, dimethylformamide, dimethoxyethane, dioxane, acetonitrile, N-methyl-pyrrolidone or diglyme, at a temperature ranging from room temperature to about 100°C.
- step (st.33) of the process compounds of formula (I) wherein X is -O- may be easily obtained by reacting the sulfonyl derivative with an alcohol or phenol derivative of formula (XXX) wherein R is as in formula (I).
- the reaction may be carried out in the presence of a base such as potassium or sodium carbonate, butyl lithium, lithium amide, sodium hydride or the like, in a suitable solvent such as dimethylformamide or tetrahydrofuran, and by working at a temperature ranging from room temperature to about 100°C.
- compounds of formula (I) wherein X is -O- may be obtained by reacting the compounds of formula (Va) and (Vb) with methylisourea sulfate by operating in a suitable solvent such as dioxane, dimethylformamide or acetonitrile in the presence of a base such as sodium or potassium carbonate at a temperature ranging from 50°C to 100°C.
- a suitable solvent such as dioxane, dimethylformamide or acetonitrile
- the compounds of formula (I) wherein X is -O- and R is hydrogen may be obtained by reacting the compounds of formula (I) wherein X is -O- and R is methyl with trimethylsilyl chloride in the presence of sodium iodide and in a suitable solvent such as dioxane, tetrahydrofuran or acetonitrile at room temperature.
- the compounds of formula (I) wherein X is -O- ans R is a trifluorosulfonyl group may be obtained by reacting the compounds of formula (I) wherein X is -O- and R is hydrogen with a triflating agent such as trifluoromethanesulfonic anhydride, trifluoromethanesulfonylchloride or N- phenyl-bis(trifluoromethanesulfonimide), optionally in the presence of a base such as triethylamine or N,N-diisopropyl-N-ethylamine (DIPEA), in a suitable solvent such as dichloromethane, tetrahydrofuran or dioxane at a temperature ranging from -78°C to room temperature.
- a triflating agent such as trifluoromethanesulfonic anhydride, trifluoromethanesulfonylchloride or N- phenyl-bis(
- the compounds of formula (I) wherein X is -O- and R is as described above may be obtained by reacting the compounds of formula (I) wherein X is -O- and R is a trifluoromethanesulfonyl group with an alcohol or phenol of formula (XXX) wherein R is as in formula (I), by operating in a suitable solvent such as dioxane, tetrahydrofuran, dimethoxyethane, acetonitrile, dimethylformamide or dimethylsulfoxide, at a temperature ranging from room temperature to abiut 90°C, optionally in the presence of a base such as potassium carbonate, potassium tertbutoxide or sodium hydride.
- a suitable solvent such as dioxane, tetrahydrofuran, dimethoxyethane, acetonitrile, dimethylformamide or dimethylsulfoxide
- reaction may be carried out in a suitable solvent such as toluene, dimethylformamide, dimethoxyethane or acetonitrile, in the presence of palladium acetate, (+)-BINAP and a base such as potassium phosphate (K 3 PO 4 ) or potassium or cesium carbonate (K 2 CO 3 or CsCO 3 ) at a temperature ranging from 0°C to 100°C (st.33c).
- a suitable solvent such as toluene, dimethylformamide, dimethoxyethane or acetonitrile
- a base such as potassium phosphate (K 3 PO 4 ) or potassium or cesium carbonate (K 2 CO 3 or CsCO 3 ) at a temperature ranging from 0°C to 100°C (st.33c).
- the compounds of formula (I) wherein X is -NH- and R is an optionally substituted alkyl, cycloalkyl, heterocyclyl, cycloalkyl-alkyl or a heterocyclylalkyl group may be obtained by reacting the compounds of formula (I) wherein X is -O- and R is a trifluoromethanesulfonyl group with an amine of formula R-NH 2 (XXVII) wherein R is as in formula (I), by operating in a suitable solvent such as dioxane, tetrahydrofuran, dimethoxyethane, acetonitrile, dimethylformamide or dimethylsulfoxide, at a temperature ranging from room temperature to 90°C, optionally in the presence of a base such as potassium carbonate or triemylamine.
- a suitable solvent such as dioxane, tetrahydrofuran, dimethoxyethane, acetonitrile, dimethylform
- Ri is a hydrogen atom or an optionally substituted group selected from straight or branched Ci-C 6 alkyl, C 3 -C 6 cycloalkyl, aryl, arylalkyl, heterocyclyl or heterocyclylalkyl are novel and, hence, represent a further object of the invention.
- the starting material and any other reactant is known or easily prepared according to known methods.
- 2-ethoxy-2-cyclohexen-l-one is a known compound which can be easily obtained by refluxing cyclohexan-l,2-dione with ethanol in toluene, in the presence of catalytic amounts of p-toluenesulfonic acid (TsOH).
- 2-methoxy-4,4-dimethyl-2-cyclohexen-l-one is a known compound which can be prepared through epoxidation of commercially available 4,4-dimethyl-2- cyclohexen-1-one and subsequent treatment of the epoxide with potassium hydroxide in methanol.
- 2-methoxy-5,5-dimethyl-2-cyclohexen-l-one may be prepared according to the following scheme from commercially available 5,5-dimethyl-cyclohexan-l,3-dione:
- the compounds of formula (I) of the invention may be also prepared according to combinatorial chemistry techniques widely known in the art, for instance by accomplishing the aforementioned reactions between the several intermediates in a serial manner and by working under solid-phase-synthesis (SPS) conditions.
- SPS solid-phase-synthesis
- R is hydrogen or an optionally substituted group selected from amino, straight or branched Ci-C 6 alkyl, C 3 -C ⁇ o cycloalkyl, cycloalkyl-alkyl, aryl, arylalkyl, heterocyclyl or heterocyclylalkyl;
- X is a single bond or a divalent radical selected from -NR'-, -CONR'-, -NH-CO-NH-, -O-, -S- or -SO 2 -, wherein R' is hydrogen or an optionally substituted group selected from straight or branched C ⁇ -C 6 alkyl, C 3 -C 6 cycloalkyl, cycloalkyl-alkyl, aryl, arylalkyl, heterocyclyl, heterocyclylalkyl or, together with the nitrogen atom to which they are bonded, R and R' may form a 5 to 6 membered heteroaryl or heterocyclyl group optionally containing one additional heteroatom selected among N, O or S; Ri, bonded to any one of the nitrogen atoms of the pyrazole ring as per formulae (la) or (lb), represents a hydrogen atom or an optionally substituted group selected from straight or branched C ⁇ -C 6 alkyl, C 3 -C 6 cycl
- R 2 is a group selected from -NR"R'", -N(OH)R", -OR” or -R", wherein R" and R'" are, each independently, hydrogen or an optionally substituted group selected from straight or branched - alkyl, C 3 -C 6 cycloalkyl or cycloalkyl-alkyl, aryl, arylalkyl, heterocyclyl or heterocyclylalkyl or, together with the nitrogen atom to which they are bonded, R" and R'” may form a 5 to 6 membered heteroaryl or heterocyclyl group, optionally containing one additional heteroatom selected among N, O or S;
- the compounds of formula (I) are active as protein kinase inhibitors and are therefore useful, for instance, to restrict the unregulated proliferation of tumor cells. In therapy, they may be used in the treatment of various tumors, such as those formerly reported, as well as in the treatment of other cell proliferative disorders such as psoriasis, vascular smooth cell proliferation associated with atherosclerosis and post- surgical stenosis and restenosis and in the treatment of Alzheimer's disease.
- the inhibiting activity of putative cdk/cyclin inhibitors and the potency of selected compounds is determined through a method of assay based on the use of the SPA technology (Amersham Pharmacia Biotech).
- the assay consists of the transfer of radioactivity labelled phosphate moiety by the kinase to a biotinylated substrate.
- the resulting 33P-labelled biotinylated product is allowed to bind to streptavidin-coated SPA beads (biotin capacity 130 pmol/mg), and light emitted was measured in a scintillation counter.
- kinase reaction 4 ⁇ M in house biotinylated histone HI (Sigma # H-5505) substrate, 10 ⁇ M ATP (0.1 microCi P 33 ⁇ -ATP), 1.1 nM Cyclin A/CDK2 complex, inhibitor in a final volume of 30 ⁇ l buffer (TRIS HCI 10 mM pH 7.5, MgCl 2 10 mM, DTT 7.5 mM + 0.2 mg/ml BSA) were added to each well of a 96 U bottom.
- Ki calculation Experimental method: Reaction was carried out in buffer (10 mM Tris, pH 7.5, 10 mM MgCl 2 , 0.2 mg/ml BSA, 7.5 M DTT) containing 3.7 nM enzyme, histone and ATP (constant ratio of coldlabeled ATP 1/3000). Reaction was stopped with EDTA and the substrate captured on phosphomembrane (Multiscreen 96 well plates from Millipore). After extensive washing, the multiscreen plates were read on a top counter. Control (time zero) for each ATP and histone concentrations was measured. Experimental design: Reaction velocities are measured at four ATP, substrate (histone) and inhibitor concentrations.
- An 80-point concentration matrix was designed around the respective ATP and substrate Km values, and the inhibitor IC50 values (0.3, 1, 3, 9 fold the Km or IC50 values).
- a preliminary time course experiment in the absence of inhibitor and at the different ATP and substrate concentrations allows the selection of a single endpoint time (10 min) in the linear range of the reaction for the Ki determination experiment.
- Kinetic parameter estimates were estimated by simultaneous nonlinear least-square regression using [Eq.l] (competitive inhibitor respect to ATP, random mechanism) using the complete data set (80 points):
- the selected compounds are characterized on a panel of ser/thre kinases strictly related to cell cycle (cdk2/cyclin E, cdkl /cyclin Bl, cdk5/p25, cdk4/ cyclin Dl), and also for specificity on MAPK, PKA, EGFR, IGF1-R, Aurora-2 and Cdc 7
- Inhibition assay of cdk2/CvcIin E activity Kinase reaction 10 ⁇ M in house biotinylated histone HI (Sigma # H-5505) substrate, 30 ⁇ M ATP (0.3 microCi P 33 ⁇ -ATP), 4 ng GST-Cyclin E/CDK2 complex, inhibitor in a final volume of 30 ⁇ l buffer (TRIS HCI 10 mM pH 7.5, MgCl 2 10 mM, DTT 7.5 mM + 0.2 mg/ml BSA) were added to each well of a 96 U bottom.
- ATP containing 1 mg SPA beads. Then a volume of 110 ⁇ l is transferred to Optiplate.
- the inhibition assay of cdk5/p25 activity is performed according to the following protocol.
- Inhibition assay of cdk4/Cyclin Dl activity Kinase reaction 0,4 ⁇ M mouse GST-Rb (769-921) (# sc-4112 from Santa Cruz) substrate, 10 ⁇ M ATP (0.5 ⁇ Ci P 33 ⁇ -ATP), 100 ng of baculovirus expressed GST- cdk4/GST-Cyclin Dl, suitable concentrations of inhibitor in a final volume of 50 ⁇ l buffer (TRIS HCI 10 mM pH 7.5, MgCl 2 10 mM, 7.5 mM DTT+ 0.2mg/ml BSA) were added to each well of a 96 U bottom well plate.
- TMS HCI 10 mM pH 7.5, MgCl 2 10 mM, 7.5 mM DTT+ 0.2mg/ml BSA were added to each well of a 96 U bottom well plate.
- reaction was stopped by 20 ⁇ l EDTA 120 mM.
- Capture 60 ⁇ l were transferred from each well to Multiscreen plate, to allow substrate binding to phosphocellulose filter. Plates were then washed 3 times with 150 ⁇ l/well PBS Ca ⁇ Mg " ⁇ free and filtered by Multiscreen filtration system. Detection: filters were allowed to dry at 37°C, then 100 ⁇ l/well scintillant were added and 33 P labeled Rb fragment was detected by radioactivity counting in the Top-Count instrument.
- IC50 determination see above Inhibition assay of PKA activity Kinase reaction: 10 ⁇ M in house biotinylated histone HI (Sigma # H-5505) substrate, 10 ⁇ M ATP (0.2 microM P 33 ⁇ -ATP), 0.45 U PKA (Sigma # 2645), inhibitor in a final volume of 30 ⁇ l buffer (TRIS HCI 10 mM pH 7.5, MgCl 2 10 mM, DTT 7.5 mM + 0.2 mg/ml BSA) were added to each well of a 96 U bottom.
- IC 50 determination see above Inhibition assay of EGFR activity Kinase reaction: 10 ⁇ M in house biotinylated MBP (Sigma # M-1891) substrate, 2 ⁇ M ATP (0.04 microCi P 33 ⁇ -ATP), 36 ng insect cell expressed GST-EGFR, inhibitor in a final volume of 30 ⁇ l buffer (Hepes 50 mM pH 7.5, MgCl 2 3 M, MnCl 2 3 mM, DTT 1 mM, NaVO 3 3 ⁇ M, + 0.2 mg/ml BSA) were added to each well of a 96 U bottom.
- the inhibition assay of IGF1-R activity is performed according to the following protocol.
- IGF1-R must be activated by auto-phosphorylation before starting the experiment. Just prior to the assay, a concentrated enzyme solution (694 nM) is incubated for half a hour at 28°C in the presence of 100 ⁇ M ATP and then brought to the working dilution in the indicated buffer.
- the inhibition assay of Cdc7/dbf4 activity is performed according to the following protocol.
- the Biotin-MCM2 substrate is trans-phosphorylated by the Cdc7/Dbf4 complex in the presence of ATP traced with ⁇ 33 -ATP.
- the phosphorylated Biotin-MCM2 substrate is then captured by Streptavidin-coated SPA beads and the extent of phosphorylation evaluated by ⁇ counting.
- the inhibition assay of Cdc7/dbf4 activity was performed in 96 wells plate according to the following protocol.
- the compounds of the present invention can be administered either as single agents or, alternatively, in combination with known anticancer treatments such as radiation therapy or chemotherapy regimen in combination with cytostatic or cytotoxic agents, antibiotic-type agents, alkylating agents, antimetabolite agents, hormonal agents, immunological agents, interferon-type agents, cyclooxygenase inhibitors (e.g. COX-2 inhibitors), matrixmetalloprotease inhibitors, telomerase inhibitors, tyrosine kinase inhibitors, anti-growth factor receptor agents, anti-HER agents, anti-EGFR agents, anti- angiogenesis agents (e.g.
- cytostatic or cytotoxic agents antibiotic-type agents, alkylating agents, antimetabolite agents, hormonal agents, immunological agents, interferon-type agents, cyclooxygenase inhibitors (e.g. COX-2 inhibitors), matrixmetalloprotease inhibitors, telomerase inhibitors, tyrosine kinase inhibitors, anti-growth factor receptor agents, anti
- angiogenesis inhibitors farnesyl transferase inhibitors, ras-raf signal transduction pathway inhibitors, cell cycle inhibitors, other cdks inhibitors, tubulin binding agents, topoisomerase I inhibitors, topoisomerase II inhibitors, and the like.
- Such combination products employ the compounds of this invention within the dosage range described below and the other pharmaceutically active agent within the approved dosage range.
- Compounds of formula (1) may be used sequentially with known anticancer agents when a combination formulation is inappropriate.
- the compounds of formula (I) of the present invention suitable for administration to a mammal, e.g., to humans, can be administered by the usual routes and the dosage level depends upon the age, weight, conditions of the patient and aclrninistration route.
- a suitable dosage adopted for oral administration of a compound of formula (I) may range from about 10 to about 500 mg per dose, from 1 to 5 times daily.
- the compounds of the invention can be a(lministered in a variety of dosage forms, e.g., orally, in the form tablets, capsules, sugar or film coated tablets, liquid solutions or suspensions; rectally in the form suppositories; parenterally, e.g., intramuscularly, or through intravenous and/or intrathecal and/or intraspinal injection or infusion.
- the present invention also includes pharmaceutical compositions comprising a compound of formula (I) or a pharmaceutically acceptable salt thereof in association with a pharmaceutically acceptable excipient, which may be a carrier or a diluent.
- the pharmaceutical compositions contaming the compounds of the invention are usually prepared following conventional methods and are administered in a suitable pharmaceutical form.
- the solid oral forms may contain, together with the active compound, diluents, e.g., lactose, dextrose saccharose, sucrose, cellulose, corn starch or potato starch; lubricants, e.g., silica, talc, stearic acid, magnesium or calcium stearate, and/or polyethylene glycols; binding agents, e.g., starches, arabic gum, gelatine methylcellulose, carboxymethylcellulose or polyvinyl pyrrolidone; disintegrating agents, e.g., starch, alginic acid, alginates or sodium starch glycolate; effervescing mixtures; dyestuffs; sweeteners; wetting agents such as lecithin, polysorbates, laurylsulphates; and, in general, non-toxic and pharmacologically inactive substances used in pharmaceutical formulations.
- diluents e.g., lactose, dextrose saccharose, suc
- liquid dispersions for oral administration may be, e.g., syrups, emulsions and suspensions.
- the syrups may contain, as carrier, saccharose or saccharose with glycerine and/or mannitol and sorbitol.
- the suspensions and the emulsions may contain, as examples of carriers, natural gum, agar, sodium alginate, pectin, methylcellulose, carboxymethylcellulose, or polyvinyl alcohol.
- the suspension or solutions for intramuscular injections may contain, together with the active compound, a pharmaceutically acceptable carrier, e.g., sterile water, olive oil, ethyl oleate, glycols, e.g., propylene glycol and, if desired, a suitable amount of lidocaine hydrochloride.
- a pharmaceutically acceptable carrier e.g., sterile water, olive oil, ethyl oleate, glycols, e.g., propylene glycol and, if desired, a suitable amount of lidocaine hydrochloride.
- the solutions for intravenous injections or infusions may contain, as a carrier, sterile water or preferably they may be in the form of sterile, aqueous, isotonic, saline solutions or they may contain propylene glycol as a carrier.
- the suppositories may contain, together with the active compound, a pharmaceutically acceptable carrier, e.g., cocoa butter, polyethylene glycol, a polyoxyethylene sorbitan fatty acid ester surfactant or lecithin.
- a pharmaceutically acceptable carrier e.g., cocoa butter, polyethylene glycol, a polyoxyethylene sorbitan fatty acid ester surfactant or lecithin.
- Each code which unambiguosly identifies a single specific compound of formula (I) only, consists of five units B-X-M(C)-D.
- Code B represents any R substituent, as per formula (I), being attached to the rest of the molecule through the X linkage; each B group is represented through the proper chemical formula in the following table I, also indicating its point of attachment to the rest of the molecule X-M.
- Code X just represents the X group in formula (I); its meanings are represented in the following table II, also indicating its point of attachment to the rest of the molecule M.
- Code C represents the Ri group being attached to the rest of the molecule through any one of the pyrazole nitrogen atoms, as per formula (I). Each C group is represented tlirough the proper chemical formula in the following table III, also indicating its point of attachment to the rest of the molecule M.
- Code D represents the R group being attached to the rest of the molecule through the carbonyl group, as per formula (I).
- Each D group is represented through the proper chemical formula in the following table IN, also indicating its point of attachment to the rest of the molecule M.
- code M refers to the central core of the molecule (I) bearing a carbonyl group in position 3. From all of the above it is clear to the skilled person that M is substituted by groups -X- (code X), Ri (code C) and R 2 (code D), as reported in formula (I); each M group is represented through the proper chemical formula, in table N, also indicating the positions of the other substituents.
- the compound B66-X03-M00(C01)-D01 represents the pyrazolo- quinazoline derivative of formula (la) wherein the central core is represented by the moiety M00 of table N, R is the group of formula B66 of table I, X is the divalent group X03 of table II, Rj is the group C01 of table in and R 2 is the group D01 of table IN, having formula
- HPLC HPLC
- the HPLC equipment consisted of a Waters 2790 HPLC system equipped with a 996 Waters PDA detector and Micromass mod. ZQ single quadrupole mass spectrometer, equipped with an electrospray (ESI) ion source. Instrument control, data acquisition and data processing were providen by Empower and MassLynx 4.0 software. HPLC was carried out at 25°C at a flow rate of 1 mL/min using a RPl 8 Waters X Terra (4,6 x 50 mm, 3.5 ⁇ m) column.
- ESI electrospray
- Mobile phase A was ammonium acetate 5 mM buffer (pH 5.5 with acetic acid/acetonitrile 95:5), and Mobile phase B was H 2 O/acetonitrile (5:95); the gradient was from 10 to 90% B in 8 minutes then hold 90% B 2 minutes.
- the injection volume was 10 ⁇ l.
- the mass spectrometer was operated in positive and in negative ion mode, the capillary voltage was set up at 2.5 KV; the source temperature was 120°C; cone was 10 V; full scan, mass range from 100 to 800 amu was set up.
- HPLC/MS Method 2 The HPLC equipment consisted of a Waters 2790 HPLC system equipped with a 996 Waters PDA detector and Micromass mod.
- the mass spectometer was operated in positive and in negative ion mode, the capillary voltage was set up at 2.5 KN; the source temperature was 120°C; cone was 10 N; full scan, mass range from 100 to 800 amu was set up.
- HPLC/MS Method 3 Mass spectra were recorded on a Finnigan LCQ ion trap mass spectrometer using the electrospray (ESI) ionization technique with positive and negative ion detection.
- the mass spectrometer is directly connected to a SSP4000 HPLC system (Thermo Separation), equipped with an LcPal autosampler (CTC Analytics) and a UN 6000LP PDA detector (Thermo Separation).
- Mobile phase A was ammonium acetate 5 mM buffer (pH 5.5 with acetic acid): acetonitrile 90:10
- Mobile phase B was ammonium acetate 5 mM buffer (pH 5.5 with acetic acid): acetonitrile 10:90; the gradient was from 0 to 100% B in 7 minutes then hold 100% B for 2 minutes before requilibration. Total LC time is 12 minutes.
- the injection volume was lO ⁇ l.
- UN Detection was performed between 215 and 400 nm. Ions were generated under the following conditions: ESI sprayer voltage 4.0 kV, heated capillary temperature 255°C, sheath gas nitrogen with a pressure of 5.0 Bar.
- a full scan detection mode (from 50 to 1000 amu) was used with an MS/MS analysis of the most intense ion (normalized collision energy: 35%).
- the aqueous solution was then extracted with diethyl ether (300 mL x 3), dried on Na 2 SO 4 and evaporated under reduced pressure to remove most of the solvent.
- the crude material contained the title compound as a low boiling point oil that was used in the next step without further purification.
- Step 7 Ethyl 6-[(dimethylamino)methylene]-l,4,4-trimethyl-7-oxo-4,5,6,7- tetrahydro-lH-indazoIe-3-carboxylate
- Ethyl 8-iodo-l -methyl-4,5-dihydro-lH-pyrazolo[4,3-h]quinazohne-3-carboxylate (0.5 g, 1.3 mmol) and cyclopentylamine (0.65 mL, 6.5 mmol) were heated at 100°C under nitrogen for 3 hours. The mixture was concentrated under reduced pressure and the residue was purified by chromatography on a silica gel column (eluant: ethyl acetate/cyclohexane 70/30) to give 0.24 g of 8-(cyclopen1ylamino)-l-methyl-lH- pyrazolo[4,3-h]quinazoline-3 -carboxylate (54% yield).
- Example 36 8-(cyclopcntylamino)-l-piperidin-4-yl-4,5-dihydro-lH-pyrazolo[4 -h]quinazolinc- 3-carboxamide_[B73-X00-M00(C18)-D03]
- Step 1 -[(dimcthylamino)methylcnc] -2-cthoxycyclohex-2-cn-l-onc
- the resulting mixture was stirred at room temperature for 1 hour and then heated to 120°C in an oil bath under argon with good stirring for 18 hours. After cooling to room temperature, the reaction mixture was poured into water and extracted with dichloromethane. The organic extracts were washed with brine and dried over Na 2 SO 4 . The solvent was removed under vacuum, the crude solid was taken up with diethyl ether, filtered, washed with diethyl ether and purified by flash chromatography on silica gel (eluant: dichloromethane/methanol 97.5:2.5) to afford 145 mg (63.8% yield) of the title compound.
- reaction mixture was poured into ice- water (200 mL), the pH was adjusted to 10 by addition of saturated sodium carbonate and the solution extracted with ethyl acetate (4 x
- Step 1 8-[(4-Hydroxybenzyl)amino]-l-methyl-4,5-dihydro-lH-pyrazolo[4,3- h]quinazohne-3-carboxamide [B85-X00-M00(C01)-D03]
- the mixture was heated to reflux for 1 hour.
- the reaction mixture was poured into iced water (70 mL) and extracted with dichloromethane; the organic extracts were washed with brine until neutral pH, then with water and dried over Na 2 SO .
- the crude was purified by flash chromatography on silica gel (eluant: dichloromethane/methanol 96:4) to yield a white solid that was crystallized from methanol, affording 47.0 mg of the title compound.
- the reaction mixture was stirred vigorously at 65-70°C for 18 hours. After cooling in an ice- water bath, the solid was filtered off and the filtrate was diluted with dichloromethane (100 mL), washed with 30% ammonium hydroxide (50 mL), sodium thiosulphate (100 mL), brine and dried over anhydrous Na 2 SO 4 .
- the crude was purified by flash chromatography on silica gel (eluant: dichloromethane/methanol 95:59) and 1.48 g of the title compound was isolated (40% yield).
- Step_2 N-[(lS)-2-(dimethylamino)-l-phenylethyl]-l-methyl-8- ⁇ [4-(4- methylpiperazin-l-yl)phenyl] amino ⁇ -4,5-dihydro-lH-pyrazolo [4,3-h] quinazoline- 3-carboxamide [B10-X00-M00(C01)-D72]
- reaction mixture was cooled to 0°C, treated with l-(3-dimethylaminopropyl)-3-ethylcarbodiimide (EDC) (146 mg, 0.760 mmol) and then stirred at room temperature for 18 hours.
- EDC l-(3-dimethylaminopropyl)-3-ethylcarbodiimide
- the reaction mixture was poured into water (10 mL) and the precipitate was filtered, washed with water, dried under vacuum at 40°C for 4 hours. There were obtained 130 mg (yield 76%) of pure title compound.
- Step 1 8-amino-l,4,4-trimethyl-4,5-dihydro-lH-pyrazolo[4,3-h]quinazoIme-3- carboxylic acid potassium salt [B00-X00-M03(C01)-D02]
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Medicinal Chemistry (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Epidemiology (AREA)
- Oncology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Biomedical Technology (AREA)
- Virology (AREA)
- Rheumatology (AREA)
- Urology & Nephrology (AREA)
- Dermatology (AREA)
- Communicable Diseases (AREA)
- Hospice & Palliative Care (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Molecular Biology (AREA)
- Heart & Thoracic Surgery (AREA)
- Cardiology (AREA)
- Tropical Medicine & Parasitology (AREA)
- Psychiatry (AREA)
- Pain & Pain Management (AREA)
- Physical Education & Sports Medicine (AREA)
- AIDS & HIV (AREA)
- Hematology (AREA)
- Pulmonology (AREA)
- Transplantation (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
Description





























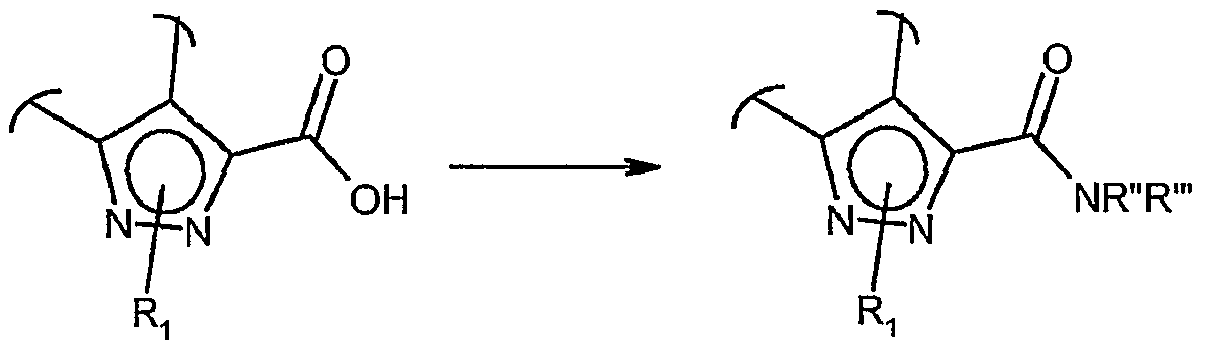






















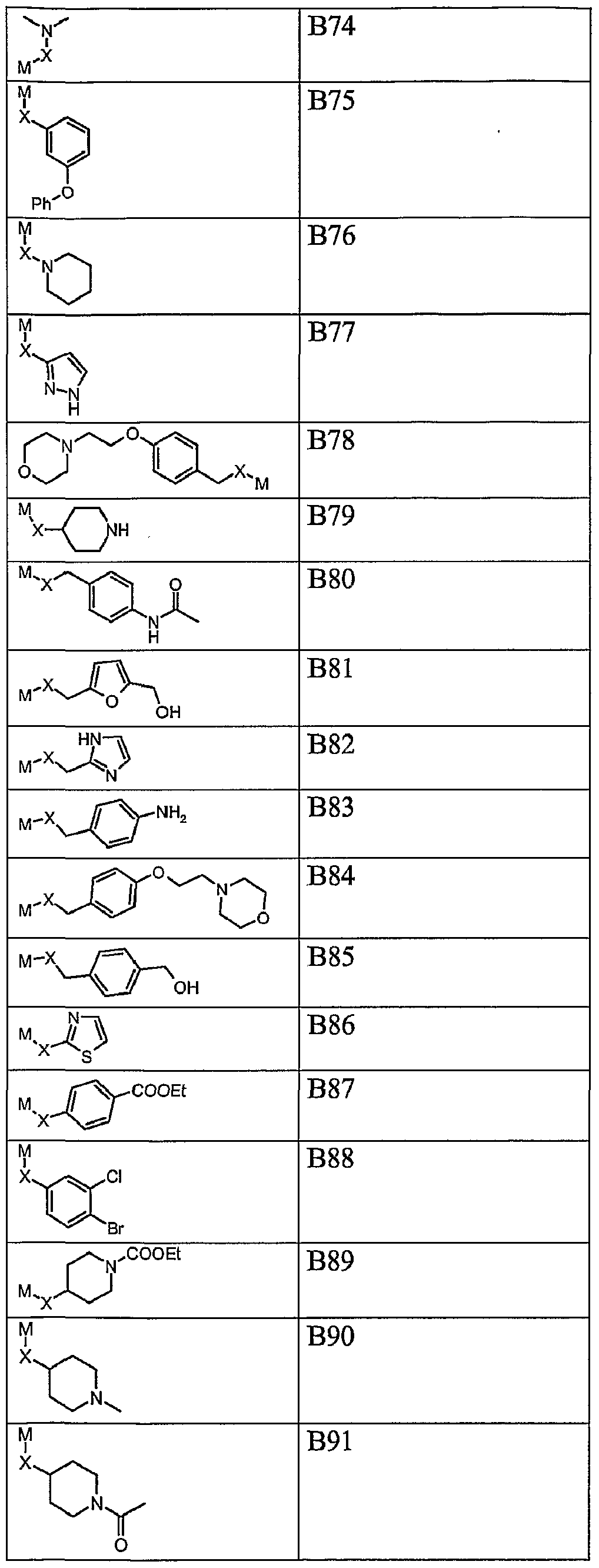


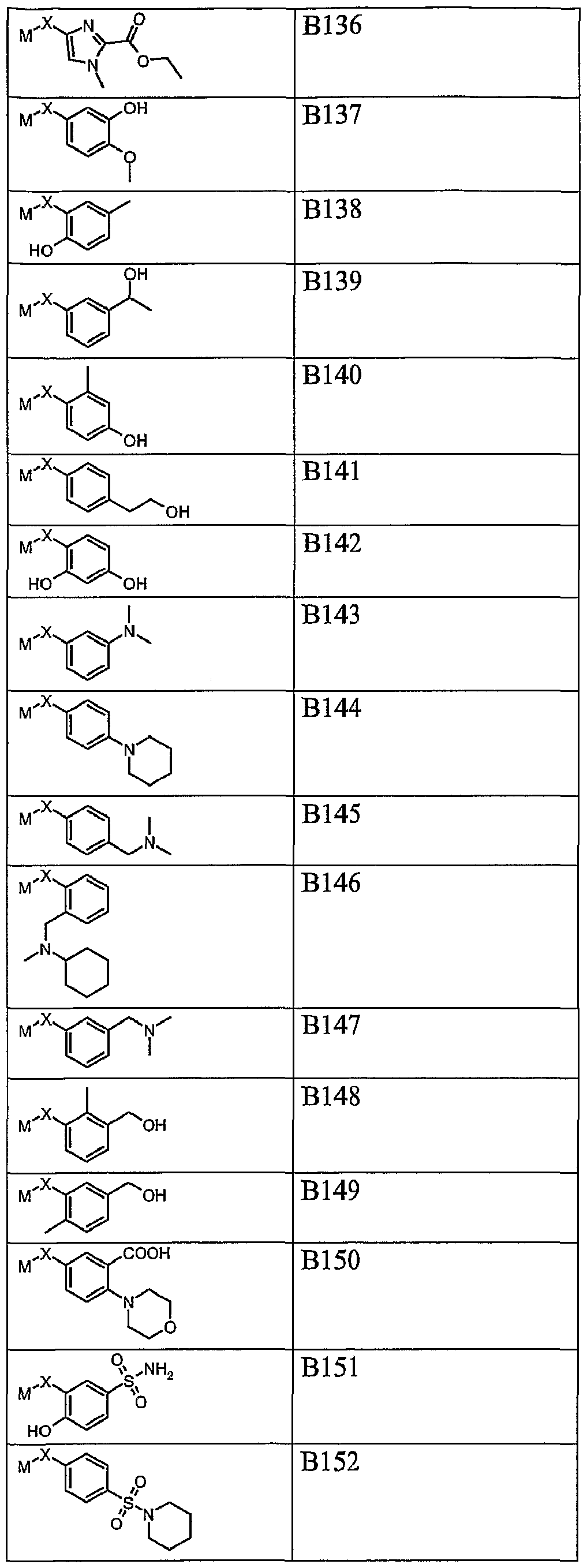









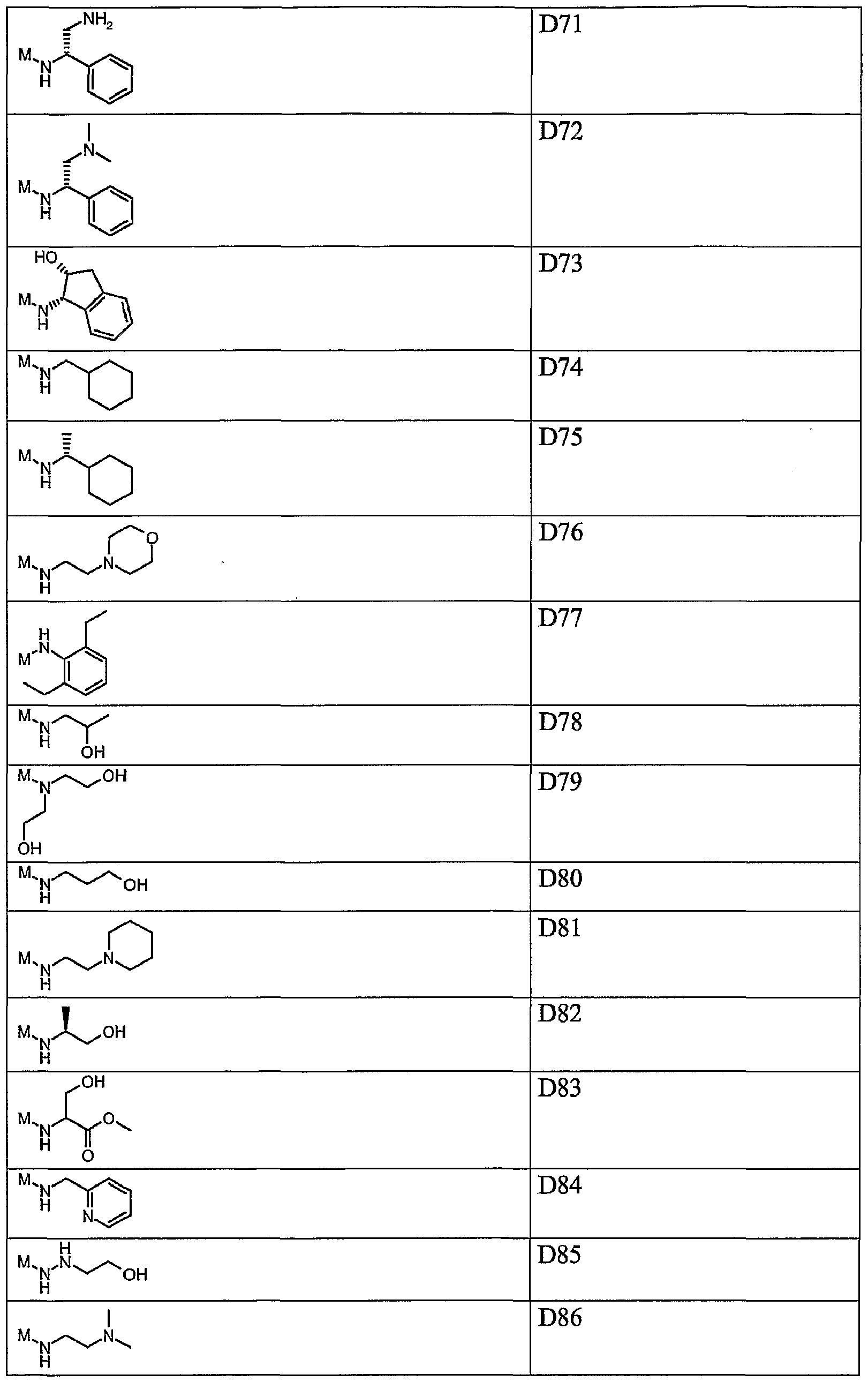

































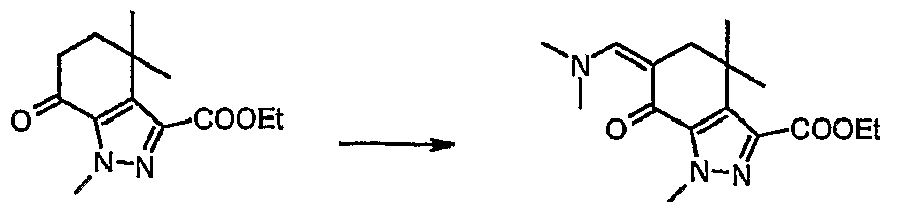












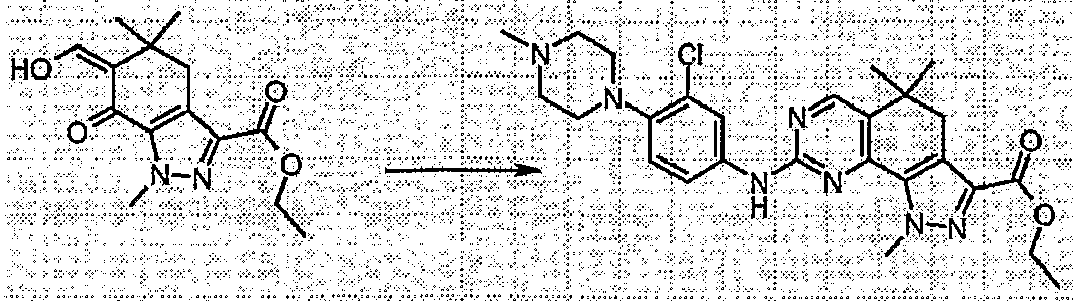
































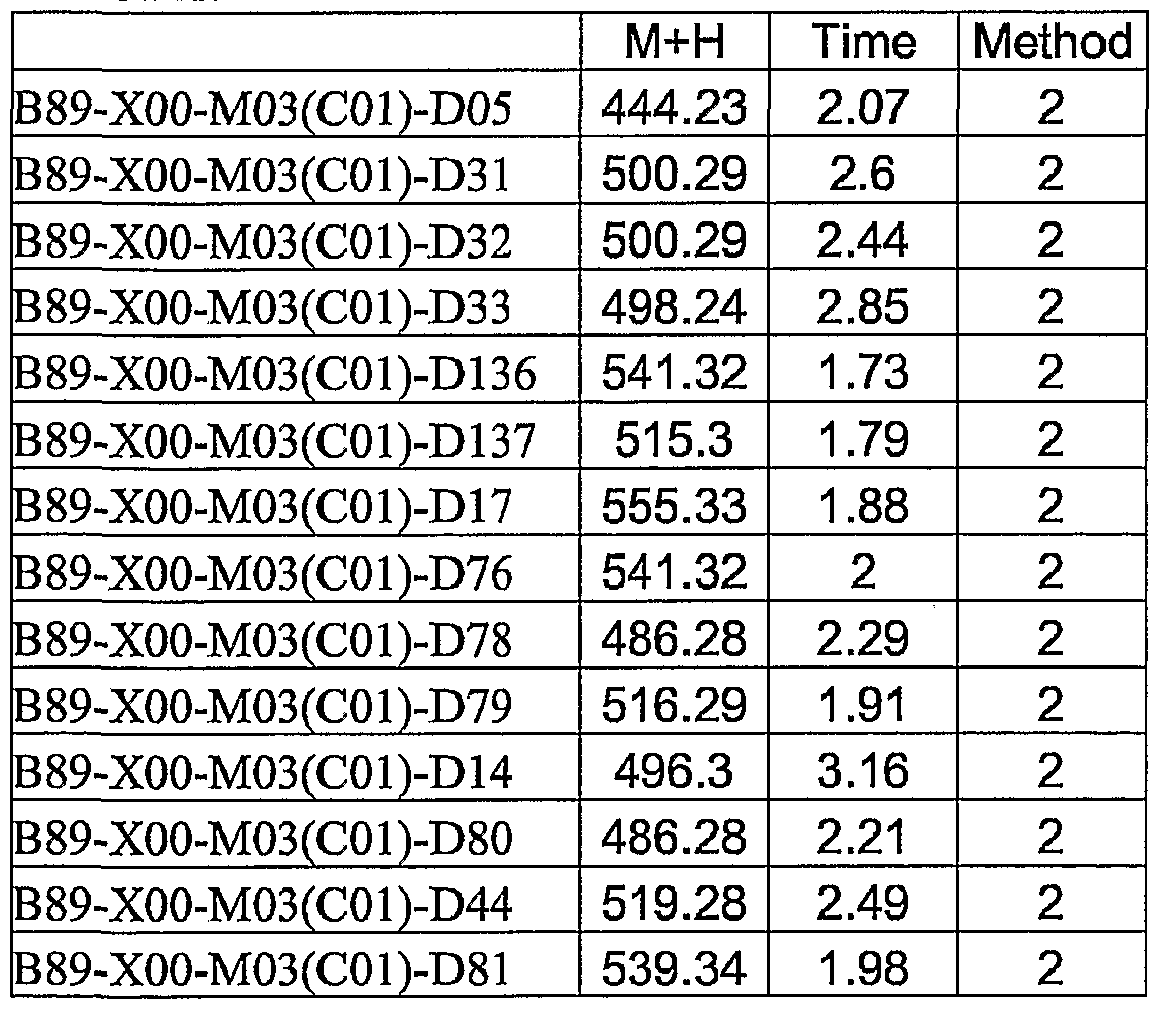







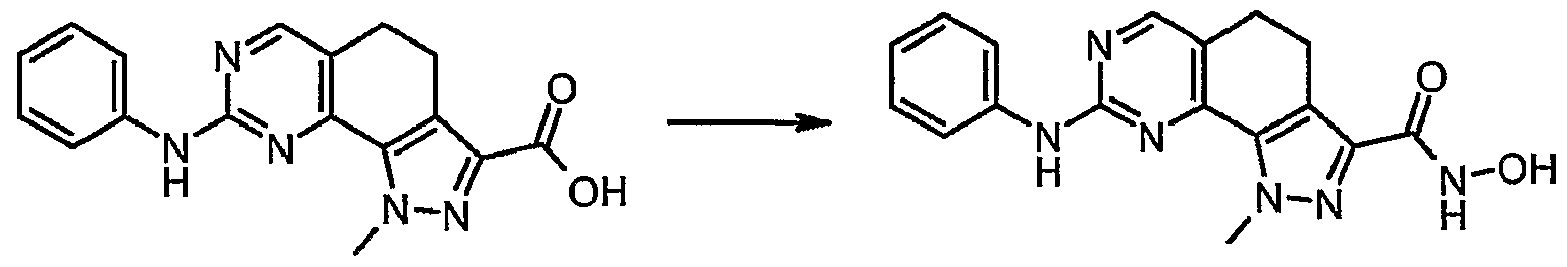








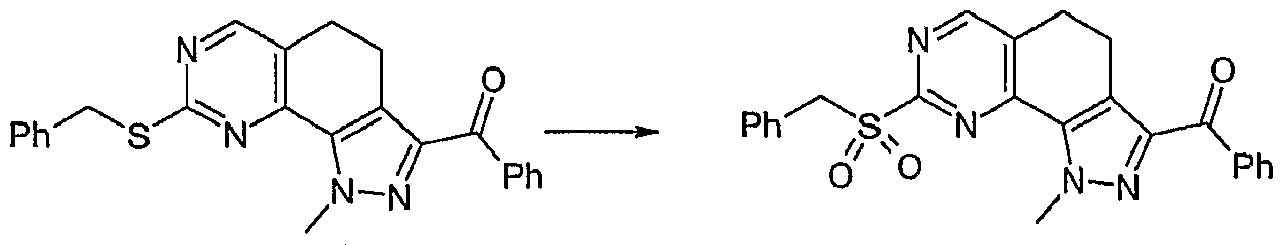













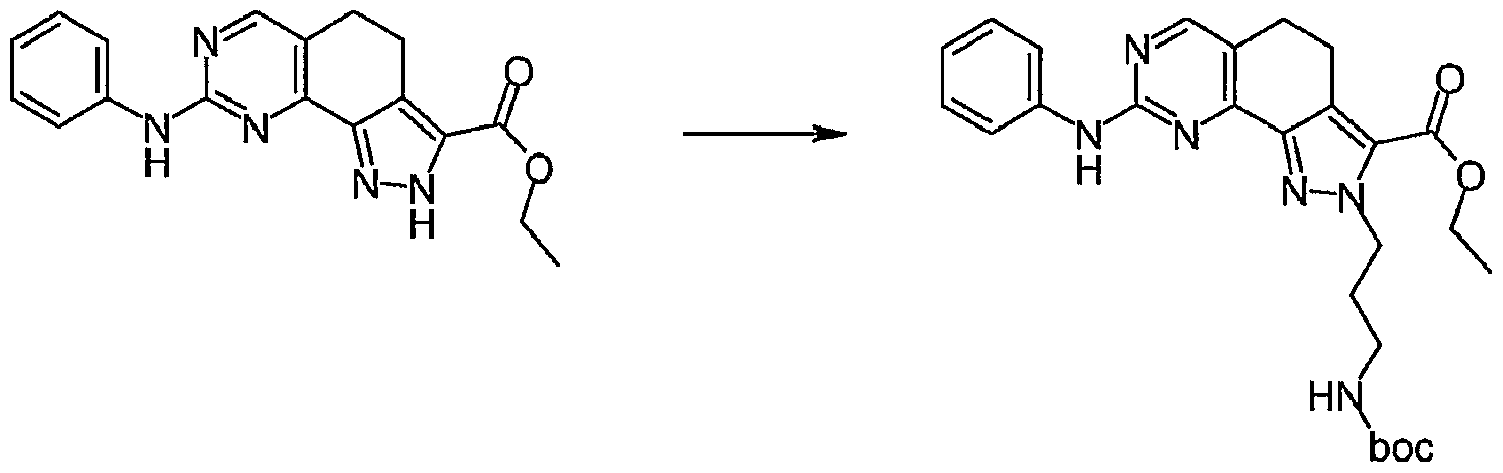
























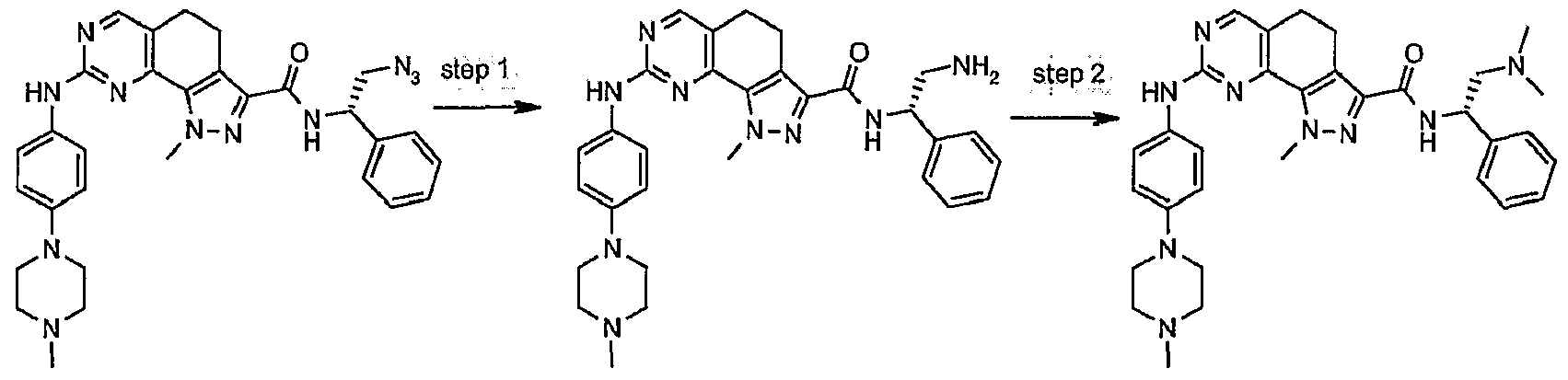
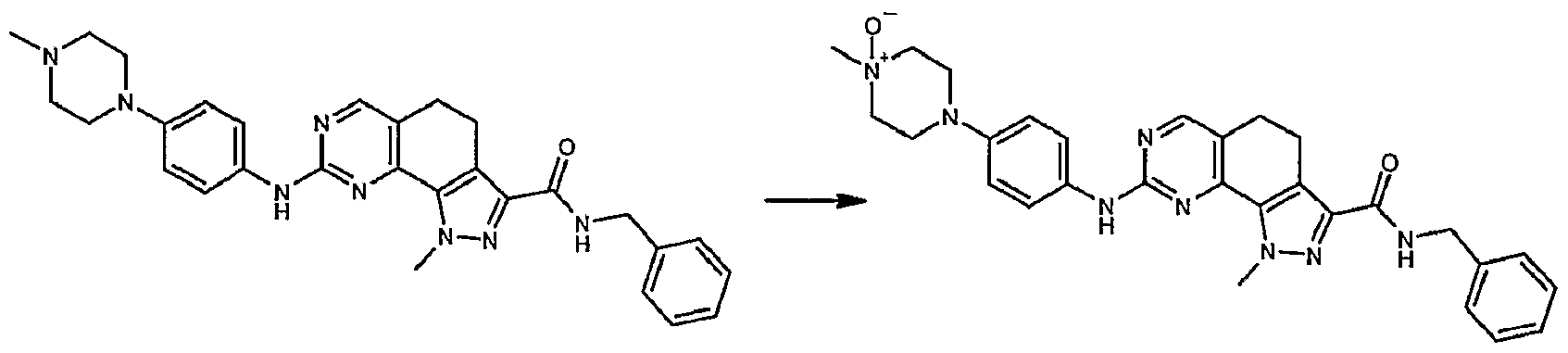





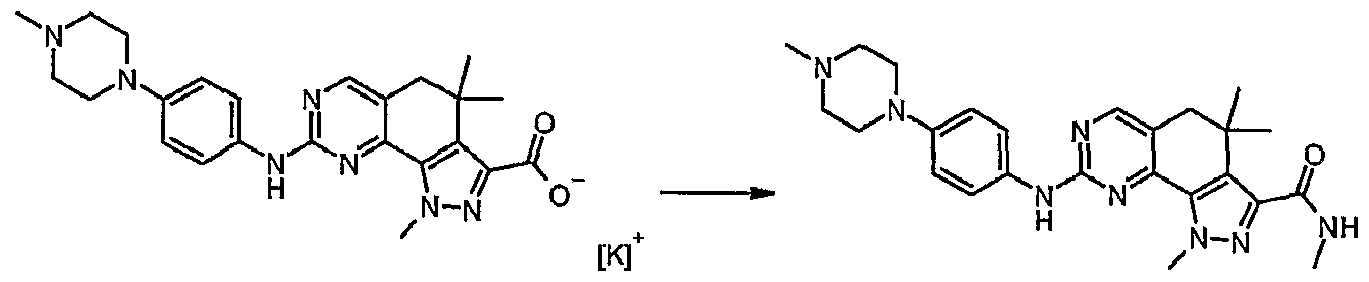








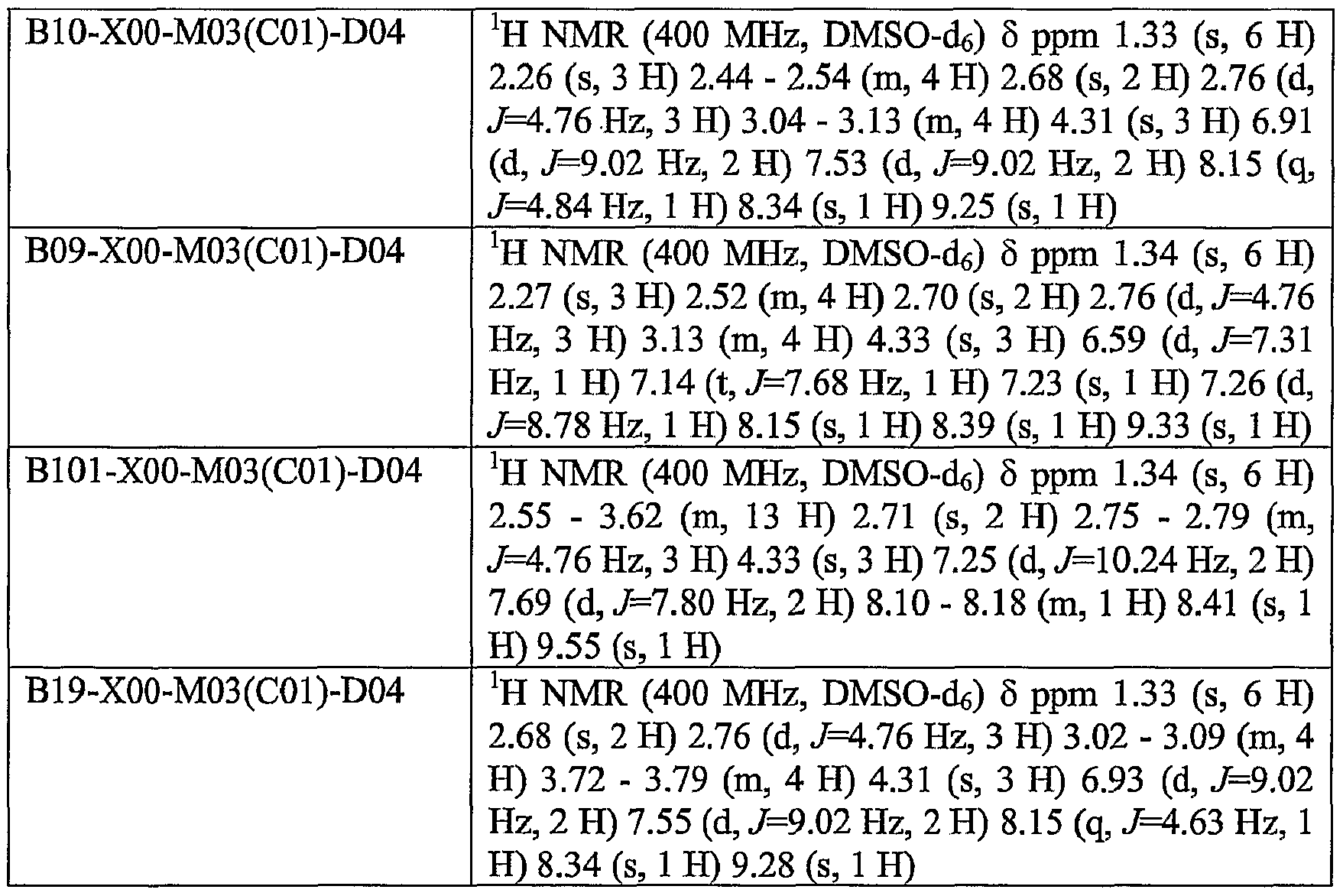























Claims


























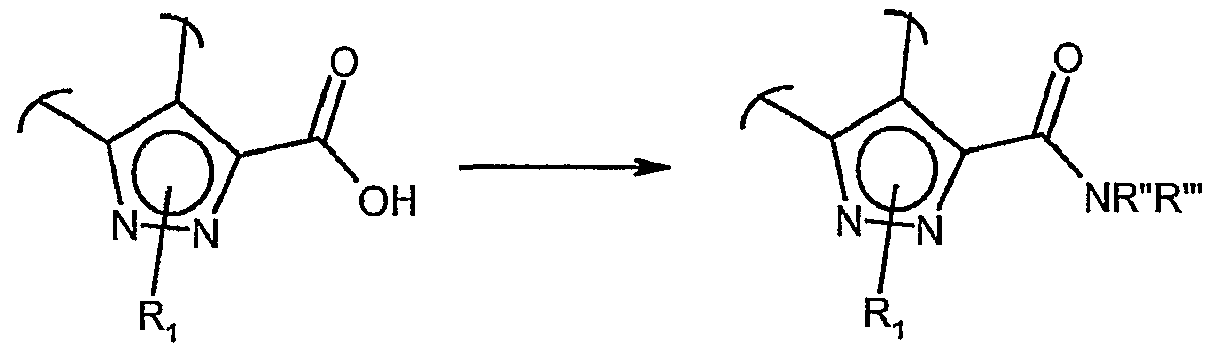


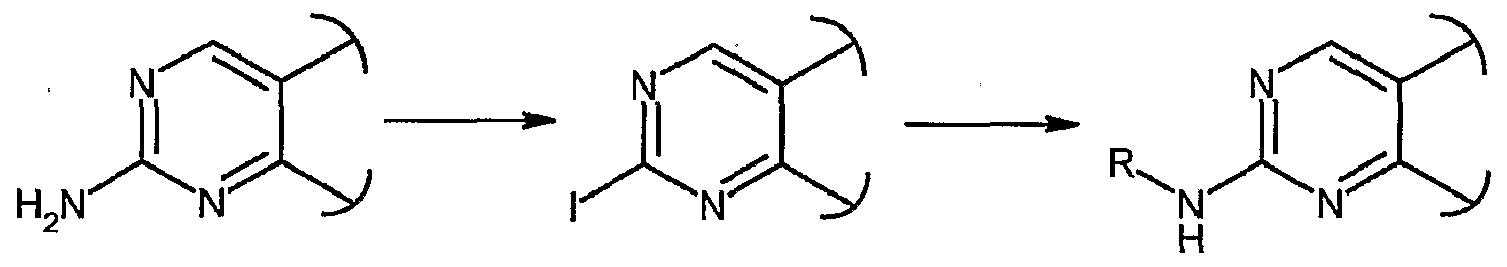


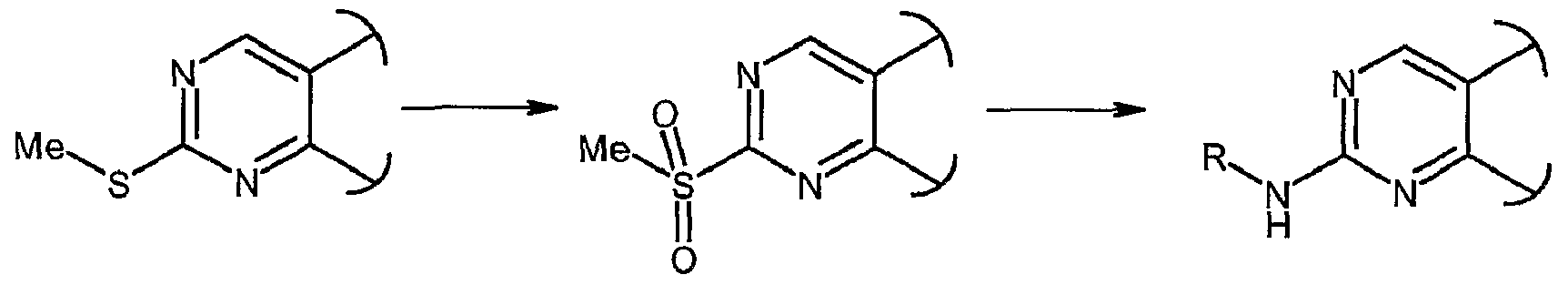





Priority Applications (32)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AP2005003452A AP2064A (en) | 2003-05-22 | 2004-04-27 | Pyrazolo-quinazoline derivatives, process for their preparation and their use as kinase inhibitors |
| PL04741483T PL1636236T3 (en) | 2003-05-22 | 2004-04-27 | Pyrazolo-quinazoline derivatives,process for their preparation and their use as kinase inhibitors |
| YUP-2005/0944A RS20050944A (en) | 2003-05-22 | 2004-04-27 | Pyrazolo-quinazoline derivatives, process for their preparation and their use as kinase inhibitors |
| JP2006530168A JP5043432B2 (en) | 2003-05-22 | 2004-04-27 | Pyrazolo-quinazoline derivatives, process for their preparation and their use as kinase inhibitors |
| UAA200512347A UA80763C2 (en) | 2003-05-22 | 2004-04-27 | Pyrazolo-quinazoline derivatives, process for their preparation and their use as kinase inhibitors |
| NZ543661A NZ543661A (en) | 2003-05-22 | 2004-04-27 | Pyrazolo-quinazoline derivatives having antitumor activity and process for their preparation |
| KR1020057022355A KR101084871B1 (en) | 2003-05-22 | 2004-04-27 | Pyrazolo-quinazoline derivatives, process for their preparation and their use as kinase inhibitors |
| EA200501849A EA010904B1 (en) | 2003-05-22 | 2004-04-27 | Pyrazolo-quinazoline derivatives, process for their preparation and their use as kinase inhibitors |
| BRPI0410563A BRPI0410563B8 (en) | 2003-05-22 | 2004-04-27 | pyrazol-quinazoline compounds, their preparation processes and pharmaceutical compositions |
| DK04741483.4T DK1636236T3 (en) | 2003-05-22 | 2004-04-27 | Pyrazolo-quinazoline derivatives, a process for their preparation, and their use as kinase inhibitors |
| RS20050944A RS52899B (en) | 2003-05-22 | 2004-04-27 | Pyrazolo-quinazoline derivatives, process for their preparation and their use as kinase inhibitors |
| SI200432118T SI1636236T1 (en) | 2003-05-22 | 2004-04-27 | Pyrazolo-quinazoline derivatives,process for their preparation and their use as kinase inhibitors |
| AU2004240772A AU2004240772B2 (en) | 2003-05-22 | 2004-04-27 | Pyrazolo-quinazoline derivatives,process for their preparation and their use as kinase inhibitors |
| CA2526578A CA2526578C (en) | 2003-05-22 | 2004-04-27 | Pyrazolo-quinazoline derivatives, process for their preparation and their use as kinase inhibitors |
| US10/557,565 US7482354B2 (en) | 2003-05-22 | 2004-04-27 | Pyrazolo-quinazoline derivatives, process for their preparation and their use as kinase inhibitors |
| EP04741483.4A EP1636236B1 (en) | 2003-05-22 | 2004-04-27 | Pyrazolo-quinazoline derivatives,process for their preparation and their use as kinase inhibitors |
| ES04741483.4T ES2436524T3 (en) | 2003-05-22 | 2004-04-27 | Pyrazolo-quinazoline derivatives, procedure for its preparation and its use as kinase inhibitors |
| BR122019010200A BR122019010200B8 (en) | 2003-05-22 | 2004-04-27 | pyrazole-quinazoline compounds, their salts, products or kits and pharmaceutical compositions |
| MXPA05012573A MXPA05012573A (en) | 2003-05-22 | 2004-04-27 | Pyrazolo-quinazoline derivatives,process for their preparation and their use as kinase inhibitors. |
| MEP-2008-259A ME00142B (en) | 2003-05-22 | 2004-04-27 | Pyrazolo-quinazoline derivatives,process for their preparation and their use as kinase inhibitors |
| IS8132A IS2939B (en) | 2003-05-22 | 2005-11-18 | Pyrosolo-quinazoline derivatives, their method of production and their use as kinase inhibitors |
| IL172046A IL172046A (en) | 2003-05-22 | 2005-11-20 | Pyrazolo-quinazoline derivatives, process for their preparation and their use in the treatment of cancer |
| NO20055496A NO334992B1 (en) | 2003-05-22 | 2005-11-21 | Pyrazoloquinazoline derivatives, methods of their preparation, intermediates, pharmaceutical compositions and kits, and their use in the treatment of disease |
| HRP20050967AA HRP20050967B8 (en) | 2003-05-22 | 2005-11-21 | Pyrazolo-quinazoline derivatives, process for their preparation and their use as kinase inhibitors |
| TNP2005000298A TNSN05298A1 (en) | 2003-05-22 | 2005-11-21 | PYRAZOLO-QUINAZOLINE DERIVATIVES, PROCESS FOR THEIR PREPARATION AND THEIR USE AS KINASE INHIBITORS |
| US12/262,933 US8541429B2 (en) | 2003-05-22 | 2008-10-31 | Pyrazolo quinazoline derivatives, process for their preparation and their use as kinase inhibitors |
| US13/972,659 US8981089B2 (en) | 2003-05-22 | 2013-08-21 | Pyrazolo-quinazoline derivatives, process for their preparation and their use as kinase inhibitors |
| US14/625,093 US9464090B2 (en) | 2003-05-22 | 2015-02-18 | Pyrazolo-quinazoline derivatives, process for their preparation and their use as kinase inhibitors |
| US15/256,916 US9637497B2 (en) | 2003-05-22 | 2016-09-06 | Pyrazolo-quinazoline derivatives, process for their preparation and their use as kinase inhibitors |
| US15/467,323 US10280176B2 (en) | 2003-05-22 | 2017-03-23 | Pyrazolo-quinazoline derivatives, process for their preparation and their use as kinase inhibitors |
| US16/291,323 US20190194214A1 (en) | 2003-05-22 | 2019-03-04 | Pyrazolo-quinazoline derivatives, process for their preparation and their use as kinase inhibitors |
| US17/084,862 US20210300935A1 (en) | 2003-05-22 | 2020-10-30 | Pyrazolo-quinazoline derivatives, process for their preparation and their use as kinase inhibitors |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US47266103P | 2003-05-22 | 2003-05-22 | |
| US60/472,661 | 2003-05-22 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US10/557,565 A-371-Of-International US7482354B2 (en) | 2003-05-22 | 2004-04-27 | Pyrazolo-quinazoline derivatives, process for their preparation and their use as kinase inhibitors |
| US12/262,933 Division US8541429B2 (en) | 2003-05-22 | 2008-10-31 | Pyrazolo quinazoline derivatives, process for their preparation and their use as kinase inhibitors |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2004104007A1 true WO2004104007A1 (en) | 2004-12-02 |
Family
ID=33476970
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2004/050612 WO2004104007A1 (en) | 2003-05-22 | 2004-04-27 | Pyrazolo-quinazoline derivatives,process for their preparation and their use as kinase inhibitors |
Country Status (36)
| Country | Link |
|---|---|
| US (8) | US7482354B2 (en) |
| EP (1) | EP1636236B1 (en) |
| JP (1) | JP5043432B2 (en) |
| KR (1) | KR101084871B1 (en) |
| CN (2) | CN1826343A (en) |
| AP (1) | AP2064A (en) |
| AR (2) | AR044543A1 (en) |
| AU (1) | AU2004240772B2 (en) |
| BR (2) | BR122019010200B8 (en) |
| CA (1) | CA2526578C (en) |
| CO (1) | CO5721006A2 (en) |
| CR (1) | CR8102A (en) |
| CY (1) | CY1114708T1 (en) |
| DK (1) | DK1636236T3 (en) |
| EA (1) | EA010904B1 (en) |
| EC (1) | ECSP056194A (en) |
| ES (1) | ES2436524T3 (en) |
| GE (1) | GEP20094664B (en) |
| HR (1) | HRP20050967B8 (en) |
| IL (1) | IL172046A (en) |
| IS (1) | IS2939B (en) |
| ME (1) | ME00142B (en) |
| MX (1) | MXPA05012573A (en) |
| MY (1) | MY142019A (en) |
| NO (1) | NO334992B1 (en) |
| NZ (1) | NZ543661A (en) |
| OA (1) | OA13170A (en) |
| PL (1) | PL1636236T3 (en) |
| PT (1) | PT1636236E (en) |
| RS (2) | RS52899B (en) |
| SI (1) | SI1636236T1 (en) |
| TN (1) | TNSN05298A1 (en) |
| TW (1) | TWI349672B (en) |
| UA (1) | UA80763C2 (en) |
| WO (1) | WO2004104007A1 (en) |
| ZA (1) | ZA200509486B (en) |
Cited By (47)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005037843A1 (en) * | 2003-10-14 | 2005-04-28 | Vertex Pharmaceuticals Incorporated | Compositions useful as inhibitors of protein kinases |
| WO2007090794A1 (en) * | 2006-02-10 | 2007-08-16 | Nerviano Medical Sciences S.R.L. | Combinations comprising a cdk inhibitor and a growth factor antibody or anti-mitotic |
| WO2007149395A2 (en) * | 2006-06-20 | 2007-12-27 | Amphora Discovery Corporation | 2,5-substituted oxazole derivatives as protein kinase inhibitors for the treatment of cancer |
| WO2008036277A1 (en) * | 2006-09-18 | 2008-03-27 | Polaris Group | Pyraz0l0(l,5-a) (1, 3, 5) triazine and pyrazolo (1, 5-a) pyrimidine derivatives useful as protein kinase inhibitors |
| WO2008074788A1 (en) | 2006-12-21 | 2008-06-26 | Nerviano Medical Sciences S.R.L. | Substituted pyrazolo-quinazoline derivatives, process for their preparation and their use as kinase inhibitors |
| US7485730B2 (en) * | 2004-05-24 | 2009-02-03 | Neuroscienze Pharmaness S.C. A R.L. | Pharmaceutical compounds |
| WO2009054332A1 (en) | 2007-10-23 | 2009-04-30 | Banyu Pharmaceutical Co., Ltd. | Pyridone-substituted-dihydropyrazolopyrimidinone derivative |
| WO2009156315A1 (en) * | 2008-06-26 | 2009-12-30 | Nerviano Medical Sciences S.R.L. | Pyrazolo-quinazolines |
| WO2010012733A1 (en) | 2008-07-29 | 2010-02-04 | Nerviano Medical Sciences S.R.L. | Use of a cdk inhibitor for the treatment of glioma |
| WO2010012777A1 (en) * | 2008-07-29 | 2010-02-04 | Nerviano Medical Sciences S.R.L. | THERAPEUTIC COMBINATION COMPRISING A CDKs INHIBITOR AND AN ANTINEOPLASTIC AGENT |
| US7713973B2 (en) | 2004-10-15 | 2010-05-11 | Takeda Pharmaceutical Company Limited | Kinase inhibitors |
| WO2010058006A1 (en) | 2008-11-24 | 2010-05-27 | Nerviano Medical Sciences S.R.L. | Cdk inhibitor for the treatment of mesothelioma |
| US7772232B2 (en) | 2004-04-15 | 2010-08-10 | Bristol-Myers Squibb Company | Quinazolinyl compounds as inhibitors of potassium channel function |
| US20100215759A1 (en) * | 2009-02-25 | 2010-08-26 | Neuroscienze Pharmaness S.C.A.R.L. | Pharmaceutical compounds |
| WO2010106028A1 (en) * | 2009-03-20 | 2010-09-23 | Nerviano Medical Sciences S.R.L. | Use of a kinase inhibitor for the treatment of thymoma |
| WO2010125004A1 (en) | 2009-04-29 | 2010-11-04 | Nerviano Medical Sciences S.R.L. | Cdk inhibitor salts |
| WO2010136394A1 (en) * | 2009-05-26 | 2010-12-02 | Nerviano Medical Sciences S.R.L. | Therapeutic combination comprising a plk1 inhibitor and an antineoplastic agent |
| WO2012010704A1 (en) * | 2010-07-23 | 2012-01-26 | Boehringer Ingelheim International Gmbh | New aminopyrazoloquinazolines |
| WO2012013557A1 (en) | 2010-07-30 | 2012-02-02 | Nerviano Medical Sciences S.R.L. | Isoxazolo-quinazolines as modulators of protein kinase activity |
| US8119655B2 (en) | 2005-10-07 | 2012-02-21 | Takeda Pharmaceutical Company Limited | Kinase inhibitors |
| JP2012505857A (en) * | 2008-10-17 | 2012-03-08 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Tetra-aza-heterocycles as phosphatidylinositol-3-kinase (PI-3 kinase) inhibitors |
| WO2012038521A1 (en) | 2010-09-23 | 2012-03-29 | Syngenta Participations Ag | Novel microbiocides |
| WO2012080990A1 (en) | 2010-12-17 | 2012-06-21 | Nerviano Medical Sciences S.R.L. | Substituted pyrazolo-quinazoline derivatives as kinase inhibitors |
| WO2012117021A2 (en) | 2011-03-03 | 2012-09-07 | Syngenta Participations Ag | Novel microbiocidal oxime ethers |
| US8278450B2 (en) | 2007-04-18 | 2012-10-02 | Takeda Pharmaceutical Company Limited | Kinase inhibitors |
| JP2012524756A (en) * | 2009-04-22 | 2012-10-18 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Thia-triaza-as-indacene as a PI3-kinase inhibitor for the treatment of cancer |
| RU2469721C2 (en) * | 2006-05-09 | 2012-12-20 | Новартис Аг | Combination containing iron complex and antineoplastic agent, and use thereof |
| CN102844313A (en) * | 2010-01-28 | 2012-12-26 | 哈佛大学校长及研究员协会 | Compositions and methods for enhancing proteasome activity |
| WO2013092460A1 (en) | 2011-12-20 | 2013-06-27 | Syngenta Participations Ag | Cyclic bisoxime microbicides |
| WO2013110585A1 (en) * | 2012-01-23 | 2013-08-01 | Boehringer Ingelheim International Gmbh | 5, 8 -dihydro- 6h- pyrazolo [3, 4 -h] quinazolines as igf-lr/lr inhibitors |
| EP2641901A1 (en) | 2012-03-22 | 2013-09-25 | Syngenta Participations AG. | Novel microbiocides |
| JP2014503566A (en) * | 2011-01-26 | 2014-02-13 | ネルビアーノ・メデイカル・サイエンシーズ・エツセ・エルレ・エルレ | Tricyclic derivatives, their preparation and their use as kinase inhibitors |
| JP2014503567A (en) * | 2011-01-26 | 2014-02-13 | ネルビアーノ・メデイカル・サイエンシーズ・エツセ・エルレ・エルレ | Tricyclic pyrrolo derivatives, their preparation and their use as kinase inhibitors |
| WO2014072220A1 (en) | 2012-11-07 | 2014-05-15 | Nerviano Medical Sciences S.R.L. | Substituted pyrimidinyl and pyridinyl-pyrrolopyridinones, process for their preparation and their use as kinase inhibitors |
| CN103626777B (en) * | 2009-07-29 | 2015-10-21 | 内尔维阿诺医学科学有限公司 | The salt of PLK inhibitor |
| WO2015181737A1 (en) | 2014-05-28 | 2015-12-03 | Piramal Enterprises Limited | Pharmaceutical combination for the treatment of cancer |
| US9556166B2 (en) | 2011-05-12 | 2017-01-31 | Proteostasis Therapeutics, Inc. | Proteostasis regulators |
| US9850262B2 (en) | 2013-11-12 | 2017-12-26 | Proteostasis Therapeutics, Inc. | Proteasome activity enhancing compounds |
| US9849135B2 (en) | 2013-01-25 | 2017-12-26 | President And Fellows Of Harvard College | USP14 inhibitors for treating or preventing viral infections |
| WO2018011382A1 (en) | 2016-07-15 | 2018-01-18 | Institut Pasteur | 5-hydroxytryptamine 1b receptor-stimulating agent for skin and/or hair repair |
| CN108699067A (en) * | 2016-11-11 | 2018-10-23 | 上海海雁医药科技有限公司 | Miscellaneous tricyclic compound, its preparation method and the purposes pharmaceutically of pyridine amine substitution |
| WO2019011715A1 (en) | 2017-07-11 | 2019-01-17 | Nerviano Medical Sciences S.R.L. | Pyrazolo-quinazoline derivatives as choline kinase inhibitors |
| WO2021043190A1 (en) * | 2019-09-05 | 2021-03-11 | 成都赛诺希亚生物科技有限公司 | Isoxazolo[5,4-h]quinazoline compounds as protein kinase inhibitors |
| EP3666774A4 (en) * | 2017-08-11 | 2021-03-31 | Shengke Pharmaceuticals (Jiangsu) Ltd. | 1h-pyrazolo[4,3-h]quinazoline compound serving as protein kinase inhibitor |
| EP3799873A1 (en) | 2015-07-17 | 2021-04-07 | Institut Pasteur | 5-hydroxytryptamine 1b receptor-stimulating agent for use as a promoter of satellite cells self-renewal and/or differentiation |
| USRE48974E1 (en) | 2014-04-07 | 2022-03-15 | Netherlands Translational Research Center B.V. | (5,6-dihydro)pyrimido[4,5-E]indolizines |
| EP3981770A1 (en) * | 2010-10-25 | 2022-04-13 | G1 Therapeutics, Inc. | Cdk inhibitors |
Families Citing this family (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GEP20094664B (en) | 2003-05-22 | 2009-04-10 | Nerviano Medical Sciences Srl | Pyrazolo-quinazoline derivatives, process for their preparation and their use as kinase inhibitors |
| UA102219C2 (en) * | 2006-12-21 | 2013-06-25 | НЕРВИАНО МЕДИКАЛ САЙЕНСИЗ С.р.л. | Substotuted pyrazolo-quinazoline derivatives, process for the preparation and use thereof as kinase inhibitors |
| WO2009085185A1 (en) * | 2007-12-19 | 2009-07-09 | Amgen Inc. | Fused pyridine, pyrimidine and triazine compounds as cell cycle inhibitors |
| WO2009126584A1 (en) * | 2008-04-07 | 2009-10-15 | Amgen Inc. | Gem-disubstituted and spirocyclic amino pyridines/pyrimidines as cell cycle inhibitors |
| TWI426074B (en) * | 2008-04-30 | 2014-02-11 | Nerviano Medical Sciences Srl | Process for the preparation of 5-(2-amino-pyrimidin-4-yl)-2-aryl-1h-pyrrole-3-carboxamides |
| EP2688887B1 (en) | 2011-03-23 | 2015-05-13 | Amgen Inc. | Fused tricyclic dual inhibitors of cdk 4/6 and flt3 |
| US20140271460A1 (en) | 2013-03-15 | 2014-09-18 | G1 Therapeutics, Inc. | Highly Active Anti-Neoplastic and Anti-Proliferative Agents |
| EP2968291A4 (en) | 2013-03-15 | 2016-09-28 | G1 Therapeutics Inc | Hspc-sparing treatments for rb-positive abnormal cellular proliferation |
| US20150297606A1 (en) | 2014-04-17 | 2015-10-22 | G1 Therapeutics, Inc. | Tricyclic Lactams for Use in the Protection of Hematopoietic Stem and Progenitor Cells Against Ionizing Radiation |
| WO2016040848A1 (en) | 2014-09-12 | 2016-03-17 | G1 Therapeutics, Inc. | Treatment of rb-negative tumors using topoisomerase inhibitors in combination with cyclin dependent kinase 4/6 inhibitors |
| EP3191098A4 (en) | 2014-09-12 | 2018-04-25 | G1 Therapeutics, Inc. | Combinations and dosing regimes to treat rb-positive tumors |
| CN107383019B (en) * | 2017-07-28 | 2019-10-15 | 江苏艾凡生物医药有限公司 | Pyrazolo [4,3-h] quinazoline compounds and application thereof |
| WO2021084541A1 (en) * | 2019-10-31 | 2021-05-06 | Sol-Gel Technologies Ltd. | Treatment of hair loss disorders with a topical egfr inhibitor |
| US10988479B1 (en) | 2020-06-15 | 2021-04-27 | G1 Therapeutics, Inc. | Morphic forms of trilaciclib and methods of manufacture thereof |
| CN114685520B (en) * | 2020-12-25 | 2024-08-30 | 武汉誉祥医药科技有限公司 | Tri-fused ring compound and pharmaceutical composition and application thereof |
| WO2022143576A1 (en) * | 2020-12-31 | 2022-07-07 | 恒元生物医药科技(苏州)有限公司 | Pyrazoloquinazoline compound, and preparation method therefor and use thereof |
| WO2022166725A1 (en) * | 2021-02-08 | 2022-08-11 | 南京明德新药研发有限公司 | 5,6-dihydrothieno[3,4-h]quinazoline compound |
| CN113527310B (en) * | 2021-07-30 | 2022-09-23 | 上海市肺科医院 | Small molecule compound for relieving adverse inflammatory reaction of autoimmune disease patient and application thereof |
| EP4446325A1 (en) * | 2021-12-10 | 2024-10-16 | Shandong Luye Pharmaceutical Co., Ltd. | Protein kinase inhibitor, preparation method therefor, and application thereof |
| WO2024153110A1 (en) * | 2023-01-17 | 2024-07-25 | 北京哲源科技有限责任公司 | Stereoisomer and deuterated derivative of pyrazoloquinazoline compound and use |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20020119975A1 (en) * | 2000-08-11 | 2002-08-29 | Snow Roger John | Heterocyclic compounds useful as inhibitors of tyrosine kinases |
| WO2003070706A1 (en) * | 2002-02-19 | 2003-08-28 | Pharmacia Corporation | Tricyclic pyrazole derivatives for the treatment of inflammation |
| WO2004014352A2 (en) * | 2002-08-07 | 2004-02-19 | Pharmacia Corporation | Methods for treating carbonic anhydrase mediated disorders |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2119975A (en) * | 1936-08-26 | 1938-06-07 | Beatrice S Nazel | Fluid pressure hammer |
| JP3907220B2 (en) * | 1994-06-30 | 2007-04-18 | エーザイ・アール・アンド・ディー・マネジメント株式会社 | Heterocycle-containing compounds |
| MX9709867A (en) | 1995-06-07 | 1998-03-31 | Pfizer | Heterocyclic ring-fused pyrimidine derivatives. |
| WO1998028281A1 (en) | 1996-12-23 | 1998-07-02 | Celltech Therapeutics Limited | Fused polycyclic 2-aminopyrimidine derivatives, their preparation and their use as protein tyrosine kinase inhibitors |
| GB9713087D0 (en) | 1997-06-20 | 1997-08-27 | Celltech Therapeutics Ltd | Chemical compounds |
| GB9911053D0 (en) | 1999-05-12 | 1999-07-14 | Pharmacia & Upjohn Spa | 4,5,6,7-tetrahydroindazole derivatives process for their preparation and their use as antitumour agents |
| US6387900B1 (en) | 1999-08-12 | 2002-05-14 | Pharmacia & Upjohn S.P.A. | 3(5)-ureido-pyrazole derivatives process for their preparation and their use as antitumor agents |
| JP2003507329A (en) | 1999-08-12 | 2003-02-25 | フアルマシア・イタリア・エツセ・ピー・アー | 3 (5) -Amino-pyrazole derivatives, processes for their preparation and their use as antitumor agents |
| PT1320531E (en) | 2000-08-10 | 2010-10-27 | Pfizer Italia Srl | Bicyclo-pyrazoles active as kinase inhibitors, process for their preparation and pharmaceutical compositions comprising them |
| AU2002215053A1 (en) | 2000-11-27 | 2002-06-24 | Pharmacia Italia S.P.A. | Phenylacetamido- pyrazole derivatives and their use as antitumor agents |
| EP1379524A2 (en) | 2001-01-26 | 2004-01-14 | Pharmacia Italia S.p.A. | Chromane derivatives, process for their preparation and their use as antitumor agents |
| WO2003013655A2 (en) * | 2001-08-10 | 2003-02-20 | Pharmacia Corporation | Carbonic anhydrase inhibitors |
| JP4542338B2 (en) | 2001-09-26 | 2010-09-15 | ファイザー イタリア ソシエタ ア レスポンサビリタ リミタータ | Aminoindazole derivatives active as kinase inhibitors, processes for their preparation and pharmaceutical compositions containing them |
| GEP20094664B (en) * | 2003-05-22 | 2009-04-10 | Nerviano Medical Sciences Srl | Pyrazolo-quinazoline derivatives, process for their preparation and their use as kinase inhibitors |
| EP2125822B1 (en) * | 2006-12-21 | 2014-11-19 | Nerviano Medical Sciences S.R.L. | Substituted pyrazolo-quinazoline derivatives, process for their preparation and their use as kinase inhibitors |
-
2004
- 2004-04-27 GE GEAP20049128A patent/GEP20094664B/en unknown
- 2004-04-27 BR BR122019010200A patent/BR122019010200B8/en active IP Right Grant
- 2004-04-27 ME MEP-2008-259A patent/ME00142B/en unknown
- 2004-04-27 WO PCT/EP2004/050612 patent/WO2004104007A1/en active Application Filing
- 2004-04-27 UA UAA200512347A patent/UA80763C2/en unknown
- 2004-04-27 RS RS20050944A patent/RS52899B/en unknown
- 2004-04-27 EP EP04741483.4A patent/EP1636236B1/en not_active Expired - Lifetime
- 2004-04-27 RS YUP-2005/0944A patent/RS20050944A/en unknown
- 2004-04-27 JP JP2006530168A patent/JP5043432B2/en not_active Expired - Lifetime
- 2004-04-27 EA EA200501849A patent/EA010904B1/en unknown
- 2004-04-27 DK DK04741483.4T patent/DK1636236T3/en active
- 2004-04-27 ES ES04741483.4T patent/ES2436524T3/en not_active Expired - Lifetime
- 2004-04-27 PT PT47414834T patent/PT1636236E/en unknown
- 2004-04-27 KR KR1020057022355A patent/KR101084871B1/en active IP Right Grant
- 2004-04-27 BR BRPI0410563A patent/BRPI0410563B8/en active IP Right Grant
- 2004-04-27 OA OA1200500329A patent/OA13170A/en unknown
- 2004-04-27 AU AU2004240772A patent/AU2004240772B2/en not_active Expired
- 2004-04-27 MX MXPA05012573A patent/MXPA05012573A/en active IP Right Grant
- 2004-04-27 US US10/557,565 patent/US7482354B2/en not_active Expired - Lifetime
- 2004-04-27 CN CNA2004800210752A patent/CN1826343A/en active Pending
- 2004-04-27 PL PL04741483T patent/PL1636236T3/en unknown
- 2004-04-27 CA CA2526578A patent/CA2526578C/en not_active Expired - Lifetime
- 2004-04-27 AP AP2005003452A patent/AP2064A/en active
- 2004-04-27 CN CN2010105863199A patent/CN102079746A/en active Pending
- 2004-04-27 NZ NZ543661A patent/NZ543661A/en not_active IP Right Cessation
- 2004-04-27 SI SI200432118T patent/SI1636236T1/en unknown
- 2004-05-04 TW TW093112449A patent/TWI349672B/en not_active IP Right Cessation
- 2004-05-13 MY MYPI20041795A patent/MY142019A/en unknown
- 2004-05-20 AR ARP040101748A patent/AR044543A1/en active IP Right Grant
-
2005
- 2005-11-18 IS IS8132A patent/IS2939B/en unknown
- 2005-11-20 IL IL172046A patent/IL172046A/en active IP Right Grant
- 2005-11-21 TN TNP2005000298A patent/TNSN05298A1/en unknown
- 2005-11-21 HR HRP20050967AA patent/HRP20050967B8/en not_active IP Right Cessation
- 2005-11-21 NO NO20055496A patent/NO334992B1/en unknown
- 2005-11-22 CR CR8102A patent/CR8102A/en unknown
- 2005-11-22 CO CO05118410A patent/CO5721006A2/en active IP Right Grant
- 2005-11-23 ZA ZA200509486A patent/ZA200509486B/en unknown
- 2005-11-30 EC EC2005006194A patent/ECSP056194A/en unknown
-
2008
- 2008-10-31 US US12/262,933 patent/US8541429B2/en active Active
-
2013
- 2013-08-21 US US13/972,659 patent/US8981089B2/en not_active Expired - Lifetime
- 2013-12-09 CY CY20131101108T patent/CY1114708T1/en unknown
-
2015
- 2015-02-18 US US14/625,093 patent/US9464090B2/en not_active Expired - Lifetime
- 2015-11-20 AR ARP150103780A patent/AR102722A2/en unknown
-
2016
- 2016-09-06 US US15/256,916 patent/US9637497B2/en not_active Expired - Lifetime
-
2017
- 2017-03-23 US US15/467,323 patent/US10280176B2/en not_active Expired - Lifetime
-
2019
- 2019-03-04 US US16/291,323 patent/US20190194214A1/en not_active Abandoned
-
2020
- 2020-10-30 US US17/084,862 patent/US20210300935A1/en not_active Abandoned
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20020119975A1 (en) * | 2000-08-11 | 2002-08-29 | Snow Roger John | Heterocyclic compounds useful as inhibitors of tyrosine kinases |
| WO2003070706A1 (en) * | 2002-02-19 | 2003-08-28 | Pharmacia Corporation | Tricyclic pyrazole derivatives for the treatment of inflammation |
| WO2004014352A2 (en) * | 2002-08-07 | 2004-02-19 | Pharmacia Corporation | Methods for treating carbonic anhydrase mediated disorders |
Cited By (114)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8017619B2 (en) | 2003-10-14 | 2011-09-13 | Vertex Pharmaceuticals Incorporated | Compositions useful as inhibitors of protein kinases |
| WO2005037843A1 (en) * | 2003-10-14 | 2005-04-28 | Vertex Pharmaceuticals Incorporated | Compositions useful as inhibitors of protein kinases |
| US7700609B2 (en) | 2003-10-14 | 2010-04-20 | Vertex Pharmaceuticals Incorporated | Compositions useful as inhibitors of protein kinases |
| US8288400B2 (en) | 2003-10-14 | 2012-10-16 | Vertex Pharmaceuticals Incorporated | Compositions useful as inhibitors of protein kinases |
| US7772232B2 (en) | 2004-04-15 | 2010-08-10 | Bristol-Myers Squibb Company | Quinazolinyl compounds as inhibitors of potassium channel function |
| US8357704B2 (en) | 2004-04-15 | 2013-01-22 | Bristol-Myers Squibb Company | Fused heterocyclic compounds as inhibitors of potassium channel function |
| US7485730B2 (en) * | 2004-05-24 | 2009-02-03 | Neuroscienze Pharmaness S.C. A R.L. | Pharmaceutical compounds |
| US8106218B2 (en) | 2004-05-24 | 2012-01-31 | Neuroscienze Pharmaness S.C. A R.L. | Pharmaceutical compounds |
| US8288536B2 (en) | 2004-10-15 | 2012-10-16 | Takeda Pharmaceutical Company Limited | Kinase inhibitors |
| US7713973B2 (en) | 2004-10-15 | 2010-05-11 | Takeda Pharmaceutical Company Limited | Kinase inhibitors |
| US8119655B2 (en) | 2005-10-07 | 2012-02-21 | Takeda Pharmaceutical Company Limited | Kinase inhibitors |
| CN101355936B (en) * | 2006-02-10 | 2013-04-24 | 内尔维阿诺医学科学有限公司 | Combinations comprising a CDK inhibitor and a growth factor antibody or anti-mitotic |
| JP2009526009A (en) * | 2006-02-10 | 2009-07-16 | ネルビアーノ・メデイカル・サイエンシーズ・エツセ・エルレ・エルレ | A combination comprising a CDK inhibitor and a growth factor antibody or anti-mitotic agent |
| US8084027B2 (en) | 2006-02-10 | 2011-12-27 | Nerviano Medical Sciences S.R.L. | Combinations comprising a CDK inhibitor and a growth factor antibody or anti-mitotic |
| EA014231B1 (en) * | 2006-02-10 | 2010-10-29 | НЕРВИАНО МЕДИКАЛ САЙЕНСИЗ С.р.л. | Combinations comprising a cdk inhibitor and a growth factor antibody or anti-mitotic |
| WO2007090794A1 (en) * | 2006-02-10 | 2007-08-16 | Nerviano Medical Sciences S.R.L. | Combinations comprising a cdk inhibitor and a growth factor antibody or anti-mitotic |
| RU2469721C2 (en) * | 2006-05-09 | 2012-12-20 | Новартис Аг | Combination containing iron complex and antineoplastic agent, and use thereof |
| WO2007149395A2 (en) * | 2006-06-20 | 2007-12-27 | Amphora Discovery Corporation | 2,5-substituted oxazole derivatives as protein kinase inhibitors for the treatment of cancer |
| WO2007149395A3 (en) * | 2006-06-20 | 2008-01-31 | Amphora Discovery Corp | 2,5-substituted oxazole derivatives as protein kinase inhibitors for the treatment of cancer |
| US7517882B2 (en) | 2006-09-18 | 2009-04-14 | Polaris Group | Protein kinase inhibitors |
| KR101418619B1 (en) | 2006-09-18 | 2014-07-14 | 폴라리스 그룹 | Pyrazolo(1,5-a)(1,3,5)triazine and pyrazolo(1,5-a)pyrimidine derivatives useful as protein kinase inhibitors |
| AU2007297675B2 (en) * | 2006-09-18 | 2012-09-20 | Polaris Group | Pyrazolo(l,5-a) (1, 3, 5) triazine and pyrazolo (1, 5-a) pyrimidine derivatives useful as protein kinase inhibitors |
| TWI472530B (en) * | 2006-09-18 | 2015-02-11 | Polaris Group | Protein kinase inhibitors |
| JP2010503690A (en) * | 2006-09-18 | 2010-02-04 | ポラリス・グループ | Pyrazolo (1,5-a) (1,3,5) triazine and pyrazolo (1,5-a) pyrimidine derivatives useful as protein kinase inhibitors |
| WO2008036277A1 (en) * | 2006-09-18 | 2008-03-27 | Polaris Group | Pyraz0l0(l,5-a) (1, 3, 5) triazine and pyrazolo (1, 5-a) pyrimidine derivatives useful as protein kinase inhibitors |
| NO342675B1 (en) * | 2006-12-21 | 2018-06-25 | Nerviano Medical Sciences Srl | Substituted pyrazoloquinazoline derivatives with kinase inhibitory effect suitable for the treatment of cancer as well as methods for the preparation thereof |
| JP2010513389A (en) * | 2006-12-21 | 2010-04-30 | ネルビアーノ・メデイカル・サイエンシーズ・エツセ・エルレ・エルレ | Substituted pyrazolo-quinazoline derivatives, methods for their preparation and their use as kinase inhibitors |
| TWI418560B (en) * | 2006-12-21 | 2013-12-11 | Nerviano Medical Sciences Srl | Substituted pyrazolo-quinazoline derivatives, process for their preparation and their use as kinase inhibitors |
| EA017769B1 (en) * | 2006-12-21 | 2013-03-29 | НЕРВИАНО МЕДИКАЛ САЙЕНСИЗ С.р.л. | Substituted pyrazoloquinazoline derivatives, process for their preparation and their use as kinase inhibitors |
| WO2008074788A1 (en) | 2006-12-21 | 2008-06-26 | Nerviano Medical Sciences S.R.L. | Substituted pyrazolo-quinazoline derivatives, process for their preparation and their use as kinase inhibitors |
| AU2007336281B2 (en) * | 2006-12-21 | 2013-05-30 | Nerviano Medical Sciences S.R.L. | Substituted pyrazolo-quinazoline derivatives, process for their preparation and their use as kinase inhibitors |
| US8614220B2 (en) | 2006-12-21 | 2013-12-24 | Nerviano Medical Sciences S.R.L. | Substituted pyrazolo-quinazoline derivatives, process for their preparation and their use as kinase inhibitors |
| US8278450B2 (en) | 2007-04-18 | 2012-10-02 | Takeda Pharmaceutical Company Limited | Kinase inhibitors |
| WO2009054332A1 (en) | 2007-10-23 | 2009-04-30 | Banyu Pharmaceutical Co., Ltd. | Pyridone-substituted-dihydropyrazolopyrimidinone derivative |
| US8846701B2 (en) | 2008-06-26 | 2014-09-30 | Nerviano Medical Sciences S.R.L. | Pyrazolo-quinazolines |
| WO2009156315A1 (en) * | 2008-06-26 | 2009-12-30 | Nerviano Medical Sciences S.R.L. | Pyrazolo-quinazolines |
| US20110105542A1 (en) * | 2008-06-26 | 2011-05-05 | Nerviano Medical Sciences S.R.L. | Pyrazolo--quinazolines |
| AU2009264431B2 (en) * | 2008-06-26 | 2013-11-07 | Nerviano Medical Sciences S.R.L. | Pyrazolo-quinazolines |
| TWI464172B (en) * | 2008-06-26 | 2014-12-11 | Nerviano Medical Sciences Srl | Pyrazolo-quinazolines |
| EA020703B1 (en) * | 2008-06-26 | 2015-01-30 | Ле Лаборатуар Сервье | Pyrazolo-quinazolines |
| EA020703B9 (en) * | 2008-06-26 | 2015-12-30 | Ле Лаборатуар Сервье | Pyrazolo-quinazolines |
| EP3173100A1 (en) | 2008-07-29 | 2017-05-31 | Nerviano Medical Sciences S.r.l. | Therapeutic combination comprising a cdks inhibitor and an antineoplastic agent |
| WO2010012733A1 (en) | 2008-07-29 | 2010-02-04 | Nerviano Medical Sciences S.R.L. | Use of a cdk inhibitor for the treatment of glioma |
| US8946226B2 (en) | 2008-07-29 | 2015-02-03 | Nerviano Medical Sciences S.R.L. | Use of CDK inhibitor for the treatment of glioma |
| WO2010012777A1 (en) * | 2008-07-29 | 2010-02-04 | Nerviano Medical Sciences S.R.L. | THERAPEUTIC COMBINATION COMPRISING A CDKs INHIBITOR AND AN ANTINEOPLASTIC AGENT |
| CN102105151B (en) * | 2008-07-29 | 2013-12-18 | 内尔维阿诺医学科学有限公司 | Use of CDK inhibitor for treatment of glioma |
| US8518930B2 (en) | 2008-07-29 | 2013-08-27 | Nerviano Medical Sciences S.R.L. | Therapeutic combination comprising a CDKS inhibitor and an antineoplastic agent |
| US20110190311A1 (en) * | 2008-07-29 | 2011-08-04 | Nerviano Medical Sciences S.R.L. | Use of cdk inhibitor for the treatment of glioma |
| JP2011529467A (en) * | 2008-07-29 | 2011-12-08 | ネルビアーノ・メデイカル・サイエンシーズ・エツセ・エルレ・エルレ | Use of CDK inhibitors for the treatment of glioma |
| JP2012505857A (en) * | 2008-10-17 | 2012-03-08 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Tetra-aza-heterocycles as phosphatidylinositol-3-kinase (PI-3 kinase) inhibitors |
| WO2010058006A1 (en) | 2008-11-24 | 2010-05-27 | Nerviano Medical Sciences S.R.L. | Cdk inhibitor for the treatment of mesothelioma |
| US20110224222A1 (en) * | 2008-11-24 | 2011-09-15 | Nerviano Medical Sciences S.R.L. | Cdk inhibitor for the treatment of mesothelioma |
| US8912194B2 (en) | 2008-11-24 | 2014-12-16 | Nerviano Medical Sciences S.R.L. | CDK inhibitor for the treatment of mesothelioma |
| US20100215759A1 (en) * | 2009-02-25 | 2010-08-26 | Neuroscienze Pharmaness S.C.A.R.L. | Pharmaceutical compounds |
| US8580793B2 (en) | 2009-03-20 | 2013-11-12 | Nerviano Medical Services S.R.L. | Use of kinase inhibitor for the treatment of thymoma |
| US20120077819A1 (en) * | 2009-03-20 | 2012-03-29 | Nerviano Medical Sciences S.R.L. | Use of kinase inhibitor for the treatment of thymoma |
| CN102356083A (en) * | 2009-03-20 | 2012-02-15 | 内尔维阿诺医学科学有限公司 | Use of kinase inhibitor for treatment of thymoma |
| CN102356083B (en) * | 2009-03-20 | 2014-10-15 | 内尔维阿诺医学科学有限公司 | Use of kinase inhibitor for treatment of thymoma |
| WO2010106028A1 (en) * | 2009-03-20 | 2010-09-23 | Nerviano Medical Sciences S.R.L. | Use of a kinase inhibitor for the treatment of thymoma |
| JP2012524756A (en) * | 2009-04-22 | 2012-10-18 | ベーリンガー インゲルハイム インターナショナル ゲゼルシャフト ミット ベシュレンクテル ハフツング | Thia-triaza-as-indacene as a PI3-kinase inhibitor for the treatment of cancer |
| US9243000B2 (en) | 2009-04-22 | 2016-01-26 | Boehringer Ingelheim International Gmbh | Thia-triaza-indacenes |
| WO2010125004A1 (en) | 2009-04-29 | 2010-11-04 | Nerviano Medical Sciences S.R.L. | Cdk inhibitor salts |
| US8586598B2 (en) | 2009-04-29 | 2013-11-19 | Nerviano Medical Sciences S.R.L. | CDK inhibitor salts |
| US8927530B2 (en) | 2009-05-26 | 2015-01-06 | Nerviano Medical Sciences S.R.L. | Therapeutic combination comprising a PLK1 inhibitor and an antineoplastic agent |
| WO2010136394A1 (en) * | 2009-05-26 | 2010-12-02 | Nerviano Medical Sciences S.R.L. | Therapeutic combination comprising a plk1 inhibitor and an antineoplastic agent |
| CN103626777B (en) * | 2009-07-29 | 2015-10-21 | 内尔维阿诺医学科学有限公司 | The salt of PLK inhibitor |
| US10351568B2 (en) | 2010-01-28 | 2019-07-16 | President And Fellows Of Harvard College | Compositions and methods for enhancing proteasome activity |
| CN102844313A (en) * | 2010-01-28 | 2012-12-26 | 哈佛大学校长及研究员协会 | Compositions and methods for enhancing proteasome activity |
| CN103097388A (en) * | 2010-07-23 | 2013-05-08 | 贝林格尔.英格海姆国际有限公司 | New aminopyrazoloquinazolines |
| WO2012010704A1 (en) * | 2010-07-23 | 2012-01-26 | Boehringer Ingelheim International Gmbh | New aminopyrazoloquinazolines |
| US8735386B2 (en) | 2010-07-23 | 2014-05-27 | Boehringer Ingelheim International Gmbh | Aminopyrazoloquinazolines |
| US9333205B2 (en) | 2010-07-30 | 2016-05-10 | Nerviano Medical Sciences S.R.L. | Isoxazolo-quinazolines as modulators of protein kinase activity |
| WO2012013557A1 (en) | 2010-07-30 | 2012-02-02 | Nerviano Medical Sciences S.R.L. | Isoxazolo-quinazolines as modulators of protein kinase activity |
| WO2012038521A1 (en) | 2010-09-23 | 2012-03-29 | Syngenta Participations Ag | Novel microbiocides |
| EP3981770A1 (en) * | 2010-10-25 | 2022-04-13 | G1 Therapeutics, Inc. | Cdk inhibitors |
| US8541576B2 (en) | 2010-12-17 | 2013-09-24 | Nerviano Medical Sciences Srl | Substituted pyrazolo-quinazoline derivatives as kinase inhibitors |
| WO2012080990A1 (en) | 2010-12-17 | 2012-06-21 | Nerviano Medical Sciences S.R.L. | Substituted pyrazolo-quinazoline derivatives as kinase inhibitors |
| RU2652638C2 (en) * | 2010-12-17 | 2018-04-28 | НЕРВИАНО МЕДИКАЛ САЙЕНСИЗ С.р.л. | Substituted pyrazolo-quinazoline derivatives as kinase inhibitors |
| JP2014503567A (en) * | 2011-01-26 | 2014-02-13 | ネルビアーノ・メデイカル・サイエンシーズ・エツセ・エルレ・エルレ | Tricyclic pyrrolo derivatives, their preparation and their use as kinase inhibitors |
| JP2014503566A (en) * | 2011-01-26 | 2014-02-13 | ネルビアーノ・メデイカル・サイエンシーズ・エツセ・エルレ・エルレ | Tricyclic derivatives, their preparation and their use as kinase inhibitors |
| JP2016053042A (en) * | 2011-01-26 | 2016-04-14 | ネルビアーノ・メデイカル・サイエンシーズ・エツセ・エルレ・エルレ | Tricyclic pyrrolo derivatives, process for their preparation and their use as kinase inhibitors |
| WO2012117021A2 (en) | 2011-03-03 | 2012-09-07 | Syngenta Participations Ag | Novel microbiocidal oxime ethers |
| US9556166B2 (en) | 2011-05-12 | 2017-01-31 | Proteostasis Therapeutics, Inc. | Proteostasis regulators |
| US10532996B2 (en) | 2011-05-12 | 2020-01-14 | Proteostasis Therapeutics, Inc. | Proteostasis regulators |
| WO2013092460A1 (en) | 2011-12-20 | 2013-06-27 | Syngenta Participations Ag | Cyclic bisoxime microbicides |
| WO2013110585A1 (en) * | 2012-01-23 | 2013-08-01 | Boehringer Ingelheim International Gmbh | 5, 8 -dihydro- 6h- pyrazolo [3, 4 -h] quinazolines as igf-lr/lr inhibitors |
| US9150578B2 (en) | 2012-01-23 | 2015-10-06 | Boehringer Ingelheim International Gmbh | 5,8-dihydro-6H-pyrazolo[3,4-h]quinazolines as IGF-1R/IR inhibitors |
| EP2641901A1 (en) | 2012-03-22 | 2013-09-25 | Syngenta Participations AG. | Novel microbiocides |
| WO2013139822A1 (en) | 2012-03-22 | 2013-09-26 | Syngenta Participations Ag | Novel bisoxime microbiocides |
| WO2014072220A1 (en) | 2012-11-07 | 2014-05-15 | Nerviano Medical Sciences S.R.L. | Substituted pyrimidinyl and pyridinyl-pyrrolopyridinones, process for their preparation and their use as kinase inhibitors |
| US9849135B2 (en) | 2013-01-25 | 2017-12-26 | President And Fellows Of Harvard College | USP14 inhibitors for treating or preventing viral infections |
| US9850262B2 (en) | 2013-11-12 | 2017-12-26 | Proteostasis Therapeutics, Inc. | Proteasome activity enhancing compounds |
| US11958873B2 (en) | 2013-11-12 | 2024-04-16 | Kineta, Inc. | Proteasome activity enhancing compounds |
| US11242361B2 (en) | 2013-11-12 | 2022-02-08 | Proteostasis Therapeutics, Inc. | Proteasome activity enhancing compounds |
| USRE50082E1 (en) | 2014-04-07 | 2024-08-20 | Netherlands Translational Research Center B.V. | (5,6-dihydro)pyrimido[4,5-e]indolizines |
| USRE48974E1 (en) | 2014-04-07 | 2022-03-15 | Netherlands Translational Research Center B.V. | (5,6-dihydro)pyrimido[4,5-E]indolizines |
| WO2015181737A1 (en) | 2014-05-28 | 2015-12-03 | Piramal Enterprises Limited | Pharmaceutical combination for the treatment of cancer |
| EP3799873A1 (en) | 2015-07-17 | 2021-04-07 | Institut Pasteur | 5-hydroxytryptamine 1b receptor-stimulating agent for use as a promoter of satellite cells self-renewal and/or differentiation |
| WO2018011382A1 (en) | 2016-07-15 | 2018-01-18 | Institut Pasteur | 5-hydroxytryptamine 1b receptor-stimulating agent for skin and/or hair repair |
| US11168088B2 (en) | 2016-11-11 | 2021-11-09 | Shanghai Haiyan Pharmaceutical Technology Co., Ltd. | Pyridylamino substituted heterotricyclic compounds, and preparation method and pharmaceutical use thereof |
| CN108699067B (en) * | 2016-11-11 | 2021-06-15 | 上海海雁医药科技有限公司 | Pyridinamine-substituted heterotricyclic compounds, their preparation and pharmaceutical use |
| EP3505519A4 (en) * | 2016-11-11 | 2020-01-22 | Shanghai Haiyan Pharmaceutical Technology Co., Ltd. | Pyridinamine-substituted heterotricyclo compounds, preparation thereof, and use in medicines |
| CN108699067A (en) * | 2016-11-11 | 2018-10-23 | 上海海雁医药科技有限公司 | Miscellaneous tricyclic compound, its preparation method and the purposes pharmaceutically of pyridine amine substitution |
| US11117901B2 (en) | 2017-07-11 | 2021-09-14 | Nerviano Medical Sciences S.R.L. | Substituted pyrazolo[4,3-h]quinazolines as choline kinase inhibitors |
| CN110997678A (en) * | 2017-07-11 | 2020-04-10 | 内尔维亚诺医疗科学公司 | Pyrazoloquinazoline derivatives as choline kinase inhibitors |
| CN110997678B (en) * | 2017-07-11 | 2023-02-21 | 内尔维亚诺医疗科学公司 | Pyrazoloquinazoline derivatives as choline kinase inhibitors |
| WO2019011715A1 (en) | 2017-07-11 | 2019-01-17 | Nerviano Medical Sciences S.R.L. | Pyrazolo-quinazoline derivatives as choline kinase inhibitors |
| US11993604B2 (en) | 2017-08-11 | 2024-05-28 | Shengke Pharmaceuticals (Jiangsu) Ltd. | Substituted pyrazolo[4,3-H]quinazolines as protein kinase inhibitors |
| EP3666774A4 (en) * | 2017-08-11 | 2021-03-31 | Shengke Pharmaceuticals (Jiangsu) Ltd. | 1h-pyrazolo[4,3-h]quinazoline compound serving as protein kinase inhibitor |
| US11319323B2 (en) | 2017-08-11 | 2022-05-03 | Shengke Pharmaceuticals (Jiangsu) Ltd. | Substituted pyrazolo[4,3-H]quinazolines as protein kinase inhibitors |
| KR20220088518A (en) * | 2017-08-11 | 2022-06-27 | 셍커 파마슈티컬스 (지앙수) 엘티디. | 1H-pyrazolo[4,3-H]quinazoline compound serving as protein kinase inhibitor |
| KR102464677B1 (en) * | 2017-08-11 | 2022-11-10 | 셍커 파마슈티컬스 (지앙수) 엘티디. | 1H-pyrazolo[4,3-H]quinazoline compound serving as protein kinase inhibitor |
| WO2021043190A1 (en) * | 2019-09-05 | 2021-03-11 | 成都赛诺希亚生物科技有限公司 | Isoxazolo[5,4-h]quinazoline compounds as protein kinase inhibitors |
| CN114423765A (en) * | 2019-09-05 | 2022-04-29 | 成都赛璟生物医药科技有限公司 | Isoxazole [5,4-H ] quinazoline compounds as protein kinase inhibitors |
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2004104007A1 (en) | Pyrazolo-quinazoline derivatives,process for their preparation and their use as kinase inhibitors | |
| KR101223826B1 (en) | Substituted pyrrolo-pyrazole derivatives as kinase inhibitors | |
| EP1660085B1 (en) | Pyridylpyrrole derivatives active as kinase inhibitors | |
| JP5925808B2 (en) | Tricyclic derivatives, their preparation and their use as kinase inhibitors | |
| EP1478357B1 (en) | Tricyclic pyrazole derivatives, process for their preparation and their use as antitumor agents | |
| US20040116497A1 (en) | Chromane derivatives, process for their preparation and their use as antitumor agents |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200480021075.2 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DPEN | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed from 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2004240772 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12005502088 Country of ref document: PH Ref document number: 543661 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 172046 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P20050967A Country of ref document: HR Ref document number: 2526578 Country of ref document: CA Ref document number: PA/a/2005/012573 Country of ref document: MX |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020057022355 Country of ref document: KR Ref document number: 2006530168 Country of ref document: JP Ref document number: 05118410 Country of ref document: CO Ref document number: 5352/DELNP/2005 Country of ref document: IN Ref document number: CR2005-008102 Country of ref document: CR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200509486 Country of ref document: ZA |
|
| ENP | Entry into the national phase |
Ref document number: 2004240772 Country of ref document: AU Date of ref document: 20040427 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004240772 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004741483 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P-2005/0944 Country of ref document: YU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 9128 Country of ref document: GE Ref document number: 200501849 Country of ref document: EA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 20050435 Country of ref document: UZ Ref document number: 1200501901 Country of ref document: VN |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020057022355 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004741483 Country of ref document: EP |
|
| ENP | Entry into the national phase |
Ref document number: PI0410563 Country of ref document: BR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10557565 Country of ref document: US Ref document number: 2007185143 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 10557565 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: CR2011-000081 Country of ref document: CR |