WO2004103279A2 - 3-(2-amino-1-azacyclyl)-5-aryl-1,2,4-oxadiazoles as s1p receptor agonists - Google Patents
3-(2-amino-1-azacyclyl)-5-aryl-1,2,4-oxadiazoles as s1p receptor agonists Download PDFInfo
- Publication number
- WO2004103279A2 WO2004103279A2 PCT/US2004/014837 US2004014837W WO2004103279A2 WO 2004103279 A2 WO2004103279 A2 WO 2004103279A2 US 2004014837 W US2004014837 W US 2004014837W WO 2004103279 A2 WO2004103279 A2 WO 2004103279A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- disease
- group
- halo
- mmol
- compound according
- Prior art date
Links
- 239000000018 receptor agonist Substances 0.000 title description 5
- 229940044601 receptor agonist Drugs 0.000 title description 5
- 150000001875 compounds Chemical class 0.000 claims abstract description 166
- 238000000034 method Methods 0.000 claims abstract description 73
- 210000000056 organ Anatomy 0.000 claims abstract description 23
- 150000003839 salts Chemical class 0.000 claims abstract description 21
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 17
- 201000010099 disease Diseases 0.000 claims abstract description 15
- 210000001519 tissue Anatomy 0.000 claims abstract description 14
- 206010052779 Transplant rejections Diseases 0.000 claims abstract description 10
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 5
- -1 indolazinyl Chemical group 0.000 claims description 45
- 125000005843 halogen group Chemical group 0.000 claims description 31
- 208000006673 asthma Diseases 0.000 claims description 29
- 229910052739 hydrogen Inorganic materials 0.000 claims description 20
- 239000001257 hydrogen Substances 0.000 claims description 20
- 201000004624 Dermatitis Diseases 0.000 claims description 15
- 239000003814 drug Substances 0.000 claims description 14
- 230000004957 immunoregulator effect Effects 0.000 claims description 14
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 14
- 238000002054 transplantation Methods 0.000 claims description 14
- 230000005856 abnormality Effects 0.000 claims description 12
- 230000001363 autoimmune Effects 0.000 claims description 12
- 201000004681 Psoriasis Diseases 0.000 claims description 11
- 125000003118 aryl group Chemical group 0.000 claims description 11
- 210000000988 bone and bone Anatomy 0.000 claims description 11
- 150000002431 hydrogen Chemical class 0.000 claims description 11
- 208000022559 Inflammatory bowel disease Diseases 0.000 claims description 10
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 claims description 10
- 206010046851 Uveitis Diseases 0.000 claims description 10
- 230000001684 chronic effect Effects 0.000 claims description 10
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 10
- 208000015181 infectious disease Diseases 0.000 claims description 10
- 201000006417 multiple sclerosis Diseases 0.000 claims description 10
- 208000023504 respiratory system disease Diseases 0.000 claims description 10
- 206010039073 rheumatoid arthritis Diseases 0.000 claims description 10
- 201000000306 sarcoidosis Diseases 0.000 claims description 10
- 206010019663 Hepatic failure Diseases 0.000 claims description 9
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 9
- 208000007903 liver failure Diseases 0.000 claims description 9
- 231100000835 liver failure Toxicity 0.000 claims description 9
- 201000000596 systemic lupus erythematosus Diseases 0.000 claims description 9
- 208000011231 Crohn disease Diseases 0.000 claims description 8
- 229940079593 drug Drugs 0.000 claims description 8
- 229910052757 nitrogen Inorganic materials 0.000 claims description 8
- 208000023275 Autoimmune disease Diseases 0.000 claims description 7
- 206010009900 Colitis ulcerative Diseases 0.000 claims description 7
- 206010034277 Pemphigoid Diseases 0.000 claims description 7
- 201000006704 Ulcerative Colitis Diseases 0.000 claims description 7
- 208000000594 bullous pemphigoid Diseases 0.000 claims description 7
- 125000001153 fluoro group Chemical group F* 0.000 claims description 7
- 206010021198 ichthyosis Diseases 0.000 claims description 7
- 210000000987 immune system Anatomy 0.000 claims description 7
- 230000001506 immunosuppresive effect Effects 0.000 claims description 7
- 125000001424 substituent group Chemical group 0.000 claims description 7
- 208000011580 syndromic disease Diseases 0.000 claims description 7
- 201000004384 Alopecia Diseases 0.000 claims description 6
- 208000032467 Aplastic anaemia Diseases 0.000 claims description 6
- 208000009137 Behcet syndrome Diseases 0.000 claims description 6
- 206010019799 Hepatitis viral Diseases 0.000 claims description 6
- NTYJJOPFIAHURM-UHFFFAOYSA-N Histamine Chemical compound NCCC1=CN=CN1 NTYJJOPFIAHURM-UHFFFAOYSA-N 0.000 claims description 6
- 206010028980 Neoplasm Diseases 0.000 claims description 6
- 206010029240 Neuritis Diseases 0.000 claims description 6
- 206010006451 bronchitis Diseases 0.000 claims description 6
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 claims description 6
- 208000023589 ischemic disease Diseases 0.000 claims description 6
- 201000005962 mycosis fungoides Diseases 0.000 claims description 6
- 230000036961 partial effect Effects 0.000 claims description 6
- 125000004793 2,2,2-trifluoroethoxy group Chemical group FC(CO*)(F)F 0.000 claims description 5
- 206010012438 Dermatitis atopic Diseases 0.000 claims description 5
- 206010018364 Glomerulonephritis Diseases 0.000 claims description 5
- 208000009329 Graft vs Host Disease Diseases 0.000 claims description 5
- 206010062016 Immunosuppression Diseases 0.000 claims description 5
- 230000001154 acute effect Effects 0.000 claims description 5
- 201000008937 atopic dermatitis Diseases 0.000 claims description 5
- 208000010668 atopic eczema Diseases 0.000 claims description 5
- 201000011510 cancer Diseases 0.000 claims description 5
- 208000037976 chronic inflammation Diseases 0.000 claims description 5
- 208000037893 chronic inflammatory disorder Diseases 0.000 claims description 5
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 5
- 206010023332 keratitis Diseases 0.000 claims description 5
- 229910052760 oxygen Inorganic materials 0.000 claims description 5
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 4
- 208000024827 Alzheimer disease Diseases 0.000 claims description 4
- 206010072579 Granulomatosis with polyangiitis Diseases 0.000 claims description 4
- 208000003084 Graves Ophthalmopathy Diseases 0.000 claims description 4
- 201000002481 Myositis Diseases 0.000 claims description 4
- 208000037979 autoimmune inflammatory disease Diseases 0.000 claims description 4
- 230000000973 chemotherapeutic effect Effects 0.000 claims description 4
- 208000024711 extrinsic asthma Diseases 0.000 claims description 4
- 125000003453 indazolyl group Chemical group N1N=C(C2=C1C=CC=C2)* 0.000 claims description 4
- 210000004185 liver Anatomy 0.000 claims description 4
- 206010025135 lupus erythematosus Diseases 0.000 claims description 4
- 125000002950 monocyclic group Chemical group 0.000 claims description 4
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 4
- 125000001715 oxadiazolyl group Chemical group 0.000 claims description 4
- 125000001544 thienyl group Chemical group 0.000 claims description 4
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 claims description 4
- 208000030507 AIDS Diseases 0.000 claims description 3
- 208000002874 Acne Vulgaris Diseases 0.000 claims description 3
- 208000009304 Acute Kidney Injury Diseases 0.000 claims description 3
- 208000007788 Acute Liver Failure Diseases 0.000 claims description 3
- 206010000804 Acute hepatic failure Diseases 0.000 claims description 3
- 206010001052 Acute respiratory distress syndrome Diseases 0.000 claims description 3
- 208000026872 Addison Disease Diseases 0.000 claims description 3
- 201000010000 Agranulocytosis Diseases 0.000 claims description 3
- 208000035285 Allergic Seasonal Rhinitis Diseases 0.000 claims description 3
- 206010002065 Anaemia megaloblastic Diseases 0.000 claims description 3
- 208000028185 Angioedema Diseases 0.000 claims description 3
- 206010002660 Anoxia Diseases 0.000 claims description 3
- 241000976983 Anoxia Species 0.000 claims description 3
- 206010003210 Arteriosclerosis Diseases 0.000 claims description 3
- 201000001320 Atherosclerosis Diseases 0.000 claims description 3
- 208000032116 Autoimmune Experimental Encephalomyelitis Diseases 0.000 claims description 3
- 206010003827 Autoimmune hepatitis Diseases 0.000 claims description 3
- 208000035143 Bacterial infection Diseases 0.000 claims description 3
- 208000023328 Basedow disease Diseases 0.000 claims description 3
- 206010006458 Bronchitis chronic Diseases 0.000 claims description 3
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 claims description 3
- 208000005623 Carcinogenesis Diseases 0.000 claims description 3
- 201000009030 Carcinoma Diseases 0.000 claims description 3
- 208000031229 Cardiomyopathies Diseases 0.000 claims description 3
- 206010008609 Cholangitis sclerosing Diseases 0.000 claims description 3
- 208000002691 Choroiditis Diseases 0.000 claims description 3
- 208000006545 Chronic Obstructive Pulmonary Disease Diseases 0.000 claims description 3
- 206010009657 Clostridium difficile colitis Diseases 0.000 claims description 3
- 208000015943 Coeliac disease Diseases 0.000 claims description 3
- 206010010744 Conjunctivitis allergic Diseases 0.000 claims description 3
- 206010011224 Cough Diseases 0.000 claims description 3
- 206010011831 Cytomegalovirus infection Diseases 0.000 claims description 3
- 206010012442 Dermatitis contact Diseases 0.000 claims description 3
- 208000007342 Diabetic Nephropathies Diseases 0.000 claims description 3
- 206010014561 Emphysema Diseases 0.000 claims description 3
- 206010014950 Eosinophilia Diseases 0.000 claims description 3
- 206010014989 Epidermolysis bullosa Diseases 0.000 claims description 3
- 206010016228 Fasciitis Diseases 0.000 claims description 3
- 208000024869 Goodpasture syndrome Diseases 0.000 claims description 3
- 206010018691 Granuloma Diseases 0.000 claims description 3
- 208000030836 Hashimoto thyroiditis Diseases 0.000 claims description 3
- 208000035186 Hemolytic Autoimmune Anemia Diseases 0.000 claims description 3
- 208000032759 Hemolytic-Uremic Syndrome Diseases 0.000 claims description 3
- 208000005100 Herpetic Keratitis Diseases 0.000 claims description 3
- 206010020850 Hyperthyroidism Diseases 0.000 claims description 3
- 206010021074 Hypoplastic anaemia Diseases 0.000 claims description 3
- 206010021143 Hypoxia Diseases 0.000 claims description 3
- 201000009794 Idiopathic Pulmonary Fibrosis Diseases 0.000 claims description 3
- 201000003838 Idiopathic interstitial pneumonia Diseases 0.000 claims description 3
- 206010021245 Idiopathic thrombocytopenic purpura Diseases 0.000 claims description 3
- 208000032571 Infant acute respiratory distress syndrome Diseases 0.000 claims description 3
- 102000004877 Insulin Human genes 0.000 claims description 3
- 108090001061 Insulin Proteins 0.000 claims description 3
- 201000002287 Keratoconus Diseases 0.000 claims description 3
- 206010069698 Langerhans' cell histiocytosis Diseases 0.000 claims description 3
- 206010024380 Leukoderma Diseases 0.000 claims description 3
- GWNVDXQDILPJIG-SHSCPDMUSA-N Leukotriene C4 Natural products CCCCCC=C/CC=C/C=C/C=C/C(SCC(NC(=O)CCC(N)C(=O)O)C(=O)NCC(=O)O)C(O)CCCC(=O)O GWNVDXQDILPJIG-SHSCPDMUSA-N 0.000 claims description 3
- 208000012309 Linear IgA disease Diseases 0.000 claims description 3
- 206010058467 Lung neoplasm malignant Diseases 0.000 claims description 3
- 206010025323 Lymphomas Diseases 0.000 claims description 3
- 241000701076 Macacine alphaherpesvirus 1 Species 0.000 claims description 3
- 208000000682 Megaloblastic Anemia Diseases 0.000 claims description 3
- 208000027530 Meniere disease Diseases 0.000 claims description 3
- 206010027476 Metastases Diseases 0.000 claims description 3
- 208000019695 Migraine disease Diseases 0.000 claims description 3
- 206010027910 Mononeuritis Diseases 0.000 claims description 3
- 208000024599 Mooren ulcer Diseases 0.000 claims description 3
- 206010051606 Necrotising colitis Diseases 0.000 claims description 3
- 206010028974 Neonatal respiratory distress syndrome Diseases 0.000 claims description 3
- 208000008589 Obesity Diseases 0.000 claims description 3
- 206010073938 Ophthalmic herpes simplex Diseases 0.000 claims description 3
- 208000001132 Osteoporosis Diseases 0.000 claims description 3
- 206010033645 Pancreatitis Diseases 0.000 claims description 3
- 241000721454 Pemphigus Species 0.000 claims description 3
- 208000031845 Pernicious anaemia Diseases 0.000 claims description 3
- 206010036105 Polyneuropathy Diseases 0.000 claims description 3
- 208000003971 Posterior uveitis Diseases 0.000 claims description 3
- 206010036774 Proctitis Diseases 0.000 claims description 3
- 208000003100 Pseudomembranous Enterocolitis Diseases 0.000 claims description 3
- 206010037128 Pseudomembranous colitis Diseases 0.000 claims description 3
- 208000003670 Pure Red-Cell Aplasia Diseases 0.000 claims description 3
- 208000006311 Pyoderma Diseases 0.000 claims description 3
- 206010037779 Radiculopathy Diseases 0.000 claims description 3
- 208000033626 Renal failure acute Diseases 0.000 claims description 3
- 206010063837 Reperfusion injury Diseases 0.000 claims description 3
- 208000013616 Respiratory Distress Syndrome Diseases 0.000 claims description 3
- 208000002200 Respiratory Hypersensitivity Diseases 0.000 claims description 3
- 208000007014 Retinitis pigmentosa Diseases 0.000 claims description 3
- 206010039705 Scleritis Diseases 0.000 claims description 3
- 206010039710 Scleroderma Diseases 0.000 claims description 3
- 206010039966 Senile dementia Diseases 0.000 claims description 3
- 206010040047 Sepsis Diseases 0.000 claims description 3
- 206010040070 Septic Shock Diseases 0.000 claims description 3
- 208000021386 Sjogren Syndrome Diseases 0.000 claims description 3
- 208000007107 Stomach Ulcer Diseases 0.000 claims description 3
- 208000031673 T-Cell Cutaneous Lymphoma Diseases 0.000 claims description 3
- 208000001106 Takayasu Arteritis Diseases 0.000 claims description 3
- 208000031981 Thrombocytopenic Idiopathic Purpura Diseases 0.000 claims description 3
- 208000007536 Thrombosis Diseases 0.000 claims description 3
- 231100000579 Toxinosis Toxicity 0.000 claims description 3
- 206010064996 Ulcerative keratitis Diseases 0.000 claims description 3
- 208000024780 Urticaria Diseases 0.000 claims description 3
- 206010047115 Vasculitis Diseases 0.000 claims description 3
- 206010000496 acne Diseases 0.000 claims description 3
- 206010069351 acute lung injury Diseases 0.000 claims description 3
- 208000011341 adult acute respiratory distress syndrome Diseases 0.000 claims description 3
- 201000000028 adult respiratory distress syndrome Diseases 0.000 claims description 3
- 206010064930 age-related macular degeneration Diseases 0.000 claims description 3
- 230000032683 aging Effects 0.000 claims description 3
- 230000010085 airway hyperresponsiveness Effects 0.000 claims description 3
- 230000001476 alcoholic effect Effects 0.000 claims description 3
- 239000003513 alkali Substances 0.000 claims description 3
- 201000009961 allergic asthma Diseases 0.000 claims description 3
- 208000002205 allergic conjunctivitis Diseases 0.000 claims description 3
- 231100000360 alopecia Toxicity 0.000 claims description 3
- 208000004631 alopecia areata Diseases 0.000 claims description 3
- 230000007953 anoxia Effects 0.000 claims description 3
- 208000011775 arteriosclerosis disease Diseases 0.000 claims description 3
- 230000003416 augmentation Effects 0.000 claims description 3
- 201000000448 autoimmune hemolytic anemia Diseases 0.000 claims description 3
- 201000003710 autoimmune thrombocytopenic purpura Diseases 0.000 claims description 3
- 208000022362 bacterial infectious disease Diseases 0.000 claims description 3
- 201000009267 bronchiectasis Diseases 0.000 claims description 3
- 230000036952 cancer formation Effects 0.000 claims description 3
- 231100000504 carcinogenesis Toxicity 0.000 claims description 3
- 239000004568 cement Substances 0.000 claims description 3
- 208000007451 chronic bronchitis Diseases 0.000 claims description 3
- 208000020832 chronic kidney disease Diseases 0.000 claims description 3
- 208000025302 chronic primary adrenal insufficiency Diseases 0.000 claims description 3
- 201000010002 cicatricial pemphigoid Diseases 0.000 claims description 3
- 206010009887 colitis Diseases 0.000 claims description 3
- 208000010247 contact dermatitis Diseases 0.000 claims description 3
- 201000009805 cryptogenic organizing pneumonia Diseases 0.000 claims description 3
- 201000007241 cutaneous T cell lymphoma Diseases 0.000 claims description 3
- 201000001981 dermatomyositis Diseases 0.000 claims description 3
- 208000033679 diabetic kidney disease Diseases 0.000 claims description 3
- 239000000428 dust Substances 0.000 claims description 3
- 238000003912 environmental pollution Methods 0.000 claims description 3
- 230000002327 eosinophilic effect Effects 0.000 claims description 3
- 201000001564 eosinophilic gastroenteritis Diseases 0.000 claims description 3
- 208000003401 eosinophilic granuloma Diseases 0.000 claims description 3
- 230000035784 germination Effects 0.000 claims description 3
- 210000004195 gingiva Anatomy 0.000 claims description 3
- 208000007565 gingivitis Diseases 0.000 claims description 3
- 230000003779 hair growth Effects 0.000 claims description 3
- 206010019692 hepatic necrosis Diseases 0.000 claims description 3
- 208000006454 hepatitis Diseases 0.000 claims description 3
- 231100000283 hepatitis Toxicity 0.000 claims description 3
- 201000010884 herpes simplex virus keratitis Diseases 0.000 claims description 3
- 229960001340 histamine Drugs 0.000 claims description 3
- 230000003463 hyperproliferative effect Effects 0.000 claims description 3
- 201000002597 ichthyosis vulgaris Diseases 0.000 claims description 3
- 230000002757 inflammatory effect Effects 0.000 claims description 3
- 208000030603 inherited susceptibility to asthma Diseases 0.000 claims description 3
- 208000014674 injury Diseases 0.000 claims description 3
- 229940125396 insulin Drugs 0.000 claims description 3
- 208000036971 interstitial lung disease 2 Diseases 0.000 claims description 3
- 201000006334 interstitial nephritis Diseases 0.000 claims description 3
- 230000003903 intestinal lesions Effects 0.000 claims description 3
- 201000010659 intrinsic asthma Diseases 0.000 claims description 3
- 208000012947 ischemia reperfusion injury Diseases 0.000 claims description 3
- 208000023569 ischemic bowel disease Diseases 0.000 claims description 3
- 230000000302 ischemic effect Effects 0.000 claims description 3
- 201000010260 leiomyoma Diseases 0.000 claims description 3
- 230000003902 lesion Effects 0.000 claims description 3
- GWNVDXQDILPJIG-NXOLIXFESA-N leukotriene C4 Chemical compound CCCCC\C=C/C\C=C/C=C/C=C/[C@H]([C@@H](O)CCCC(O)=O)SC[C@@H](C(=O)NCC(O)=O)NC(=O)CC[C@H](N)C(O)=O GWNVDXQDILPJIG-NXOLIXFESA-N 0.000 claims description 3
- 201000011486 lichen planus Diseases 0.000 claims description 3
- 231100000149 liver necrosis Toxicity 0.000 claims description 3
- 210000004072 lung Anatomy 0.000 claims description 3
- 201000005202 lung cancer Diseases 0.000 claims description 3
- 208000020816 lung neoplasm Diseases 0.000 claims description 3
- 208000008585 mastocytosis Diseases 0.000 claims description 3
- 231100001016 megaloblastic anemia Toxicity 0.000 claims description 3
- 230000009401 metastasis Effects 0.000 claims description 3
- 206010027599 migraine Diseases 0.000 claims description 3
- 208000013734 mononeuritis simplex Diseases 0.000 claims description 3
- 201000005518 mononeuropathy Diseases 0.000 claims description 3
- 201000006938 muscular dystrophy Diseases 0.000 claims description 3
- 206010028417 myasthenia gravis Diseases 0.000 claims description 3
- 230000017074 necrotic cell death Effects 0.000 claims description 3
- 208000004995 necrotizing enterocolitis Diseases 0.000 claims description 3
- 201000002652 newborn respiratory distress syndrome Diseases 0.000 claims description 3
- 230000000414 obstructive effect Effects 0.000 claims description 3
- 239000001301 oxygen Substances 0.000 claims description 3
- 201000006195 perinatal necrotizing enterocolitis Diseases 0.000 claims description 3
- 201000001245 periodontitis Diseases 0.000 claims description 3
- 210000004261 periodontium Anatomy 0.000 claims description 3
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 3
- 201000006292 polyarteritis nodosa Diseases 0.000 claims description 3
- 208000005987 polymyositis Diseases 0.000 claims description 3
- 208000019629 polyneuritis Diseases 0.000 claims description 3
- 238000004321 preservation Methods 0.000 claims description 3
- 208000025638 primary cutaneous T-cell non-Hodgkin lymphoma Diseases 0.000 claims description 3
- 230000001737 promoting effect Effects 0.000 claims description 3
- 230000005855 radiation Effects 0.000 claims description 3
- 238000002271 resection Methods 0.000 claims description 3
- 206010038718 respiratory syncytial virus bronchiolitis Diseases 0.000 claims description 3
- 230000002441 reversible effect Effects 0.000 claims description 3
- 201000003068 rheumatic fever Diseases 0.000 claims description 3
- 206010039083 rhinitis Diseases 0.000 claims description 3
- 208000010157 sclerosing cholangitis Diseases 0.000 claims description 3
- 230000035945 sensitivity Effects 0.000 claims description 3
- 230000035939 shock Effects 0.000 claims description 3
- 208000004003 siderosis Diseases 0.000 claims description 3
- 208000017520 skin disease Diseases 0.000 claims description 3
- 229910052717 sulfur Inorganic materials 0.000 claims description 3
- 239000003053 toxin Substances 0.000 claims description 3
- 231100000765 toxin Toxicity 0.000 claims description 3
- 230000008733 trauma Effects 0.000 claims description 3
- 208000001072 type 2 diabetes mellitus Diseases 0.000 claims description 3
- 230000003966 vascular damage Effects 0.000 claims description 3
- 201000005539 vernal conjunctivitis Diseases 0.000 claims description 3
- 201000001862 viral hepatitis Diseases 0.000 claims description 3
- 125000005940 1,4-dioxanyl group Chemical group 0.000 claims description 2
- 208000024893 Acute lymphoblastic leukemia Diseases 0.000 claims description 2
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 claims description 2
- 206010015150 Erythema Diseases 0.000 claims description 2
- 206010015218 Erythema multiforme Diseases 0.000 claims description 2
- 208000031422 Lymphocytic Chronic B-Cell Leukemia Diseases 0.000 claims description 2
- 125000004429 atom Chemical group 0.000 claims description 2
- 125000002393 azetidinyl group Chemical group 0.000 claims description 2
- 125000003785 benzimidazolyl group Chemical group N1=C(NC2=C1C=CC=C2)* 0.000 claims description 2
- 125000000499 benzofuranyl group Chemical group O1C(=CC2=C1C=CC=C2)* 0.000 claims description 2
- 125000004196 benzothienyl group Chemical group S1C(=CC2=C1C=CC=C2)* 0.000 claims description 2
- 125000003354 benzotriazolyl group Chemical group N1N=NC2=C1C=CC=C2* 0.000 claims description 2
- 125000004541 benzoxazolyl group Chemical group O1C(=NC2=C1C=CC=C2)* 0.000 claims description 2
- 125000000609 carbazolyl group Chemical group C1(=CC=CC=2C3=CC=CC=C3NC12)* 0.000 claims description 2
- 125000004623 carbolinyl group Chemical group 0.000 claims description 2
- 125000000259 cinnolinyl group Chemical group N1=NC(=CC2=CC=CC=C12)* 0.000 claims description 2
- 125000000723 dihydrobenzofuranyl group Chemical group O1C(CC2=C1C=CC=C2)* 0.000 claims description 2
- 125000005436 dihydrobenzothiophenyl group Chemical group S1C(CC2=C1C=CC=C2)* 0.000 claims description 2
- 125000005435 dihydrobenzoxazolyl group Chemical group O1C(NC2=C1C=CC=C2)* 0.000 claims description 2
- 125000004852 dihydrofuranyl group Chemical group O1C(CC=C1)* 0.000 claims description 2
- 125000005047 dihydroimidazolyl group Chemical group N1(CNC=C1)* 0.000 claims description 2
- 125000001070 dihydroindolyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 claims description 2
- 125000005049 dihydrooxadiazolyl group Chemical group O1N(NC=C1)* 0.000 claims description 2
- 125000005050 dihydrooxazolyl group Chemical group O1C(NC=C1)* 0.000 claims description 2
- 125000005051 dihydropyrazinyl group Chemical group N1(CC=NC=C1)* 0.000 claims description 2
- 125000005052 dihydropyrazolyl group Chemical group N1(NCC=C1)* 0.000 claims description 2
- 125000004655 dihydropyridinyl group Chemical group N1(CC=CC=C1)* 0.000 claims description 2
- 125000005053 dihydropyrimidinyl group Chemical group N1(CN=CC=C1)* 0.000 claims description 2
- 125000005044 dihydroquinolinyl group Chemical group N1(CC=CC2=CC=CC=C12)* 0.000 claims description 2
- 125000005056 dihydrothiazolyl group Chemical group S1C(NC=C1)* 0.000 claims description 2
- 125000005057 dihydrothienyl group Chemical group S1C(CC=C1)* 0.000 claims description 2
- 125000005058 dihydrotriazolyl group Chemical group N1(NNC=C1)* 0.000 claims description 2
- 231100000321 erythema Toxicity 0.000 claims description 2
- 125000002541 furyl group Chemical group 0.000 claims description 2
- 208000024908 graft versus host disease Diseases 0.000 claims description 2
- 125000004634 hexahydroazepinyl group Chemical group N1(CCCCCC1)* 0.000 claims description 2
- 125000004029 hydroxymethyl group Chemical group [H]OC([H])([H])* 0.000 claims description 2
- 125000002883 imidazolyl group Chemical group 0.000 claims description 2
- 125000003387 indolinyl group Chemical group N1(CCC2=CC=CC=C12)* 0.000 claims description 2
- 125000001041 indolyl group Chemical group 0.000 claims description 2
- 125000001977 isobenzofuranyl group Chemical group C=1(OC=C2C=CC=CC12)* 0.000 claims description 2
- 125000000904 isoindolyl group Chemical group C=1(NC=C2C=CC=CC12)* 0.000 claims description 2
- 125000005956 isoquinolyl group Chemical group 0.000 claims description 2
- 125000001786 isothiazolyl group Chemical group 0.000 claims description 2
- 125000000842 isoxazolyl group Chemical group 0.000 claims description 2
- 230000036210 malignancy Effects 0.000 claims description 2
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 2
- 125000004593 naphthyridinyl group Chemical group N1=C(C=CC2=CC=CN=C12)* 0.000 claims description 2
- 125000002971 oxazolyl group Chemical group 0.000 claims description 2
- 125000004193 piperazinyl group Chemical group 0.000 claims description 2
- 125000003386 piperidinyl group Chemical group 0.000 claims description 2
- 125000003373 pyrazinyl group Chemical group 0.000 claims description 2
- 125000003226 pyrazolyl group Chemical group 0.000 claims description 2
- 125000002098 pyridazinyl group Chemical group 0.000 claims description 2
- 125000004076 pyridyl group Chemical group 0.000 claims description 2
- 125000000714 pyrimidinyl group Chemical group 0.000 claims description 2
- 125000002294 quinazolinyl group Chemical group N1=C(N=CC2=CC=CC=C12)* 0.000 claims description 2
- 125000005493 quinolyl group Chemical group 0.000 claims description 2
- 125000001567 quinoxalinyl group Chemical group N1=C(C=NC2=CC=CC=C12)* 0.000 claims description 2
- 125000005958 tetrahydrothienyl group Chemical group 0.000 claims description 2
- 125000001113 thiadiazolyl group Chemical group 0.000 claims description 2
- 125000000335 thiazolyl group Chemical group 0.000 claims description 2
- 125000004568 thiomorpholinyl group Chemical group 0.000 claims description 2
- 125000001425 triazolyl group Chemical group 0.000 claims description 2
- 206010066968 Corneal leukoma Diseases 0.000 claims 1
- 206010053615 Thermal burn Diseases 0.000 claims 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims 1
- 125000002757 morpholinyl group Chemical group 0.000 claims 1
- 125000003718 tetrahydrofuranyl group Chemical group 0.000 claims 1
- 230000036575 thermal burns Effects 0.000 claims 1
- 230000001404 mediated effect Effects 0.000 abstract description 3
- 210000001185 bone marrow Anatomy 0.000 abstract 1
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 81
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical class CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 61
- 239000000203 mixture Substances 0.000 description 55
- 239000000243 solution Substances 0.000 description 52
- 150000001732 carboxylic acid derivatives Chemical class 0.000 description 40
- 235000019439 ethyl acetate Nutrition 0.000 description 39
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 36
- 238000005481 NMR spectroscopy Methods 0.000 description 36
- 210000004027 cell Anatomy 0.000 description 36
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 35
- 101150064129 slp gene Proteins 0.000 description 33
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 30
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 30
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 27
- 102000005962 receptors Human genes 0.000 description 25
- 108020003175 receptors Proteins 0.000 description 25
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 24
- 230000000694 effects Effects 0.000 description 24
- 238000006243 chemical reaction Methods 0.000 description 23
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 21
- 239000000556 agonist Substances 0.000 description 20
- 230000027455 binding Effects 0.000 description 19
- 239000002299 complementary DNA Substances 0.000 description 19
- 238000010898 silica gel chromatography Methods 0.000 description 17
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 16
- 238000002330 electrospray ionisation mass spectrometry Methods 0.000 description 16
- 238000004128 high performance liquid chromatography Methods 0.000 description 16
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 15
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 14
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 14
- 102000036530 EDG receptors Human genes 0.000 description 14
- 108091007263 EDG receptors Proteins 0.000 description 14
- 239000011541 reaction mixture Substances 0.000 description 14
- 239000002904 solvent Substances 0.000 description 14
- DUYSYHSSBDVJSM-KRWOKUGFSA-N sphingosine 1-phosphate Chemical compound CCCCCCCCCCCCC\C=C\[C@@H](O)[C@@H](N)COP(O)(O)=O DUYSYHSSBDVJSM-KRWOKUGFSA-N 0.000 description 14
- 239000004480 active ingredient Substances 0.000 description 13
- VEUMBMHMMCOFAG-UHFFFAOYSA-N 2,3-dihydrooxadiazole Chemical compound N1NC=CO1 VEUMBMHMMCOFAG-UHFFFAOYSA-N 0.000 description 12
- 102100022888 KN motif and ankyrin repeat domain-containing protein 2 Human genes 0.000 description 12
- 239000003446 ligand Substances 0.000 description 12
- 241000699666 Mus <mouse, genus> Species 0.000 description 11
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 11
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 11
- 239000007787 solid Substances 0.000 description 11
- ATHHXGZTWNVVOU-UHFFFAOYSA-N N-methylformamide Chemical compound CNC=O ATHHXGZTWNVVOU-UHFFFAOYSA-N 0.000 description 10
- 239000003795 chemical substances by application Substances 0.000 description 10
- 238000000159 protein binding assay Methods 0.000 description 10
- VYRSLCGXROPJNM-UHFFFAOYSA-N 4-(2,2-difluoropropyl)benzoic acid Chemical compound CC(F)(F)CC1=CC=C(C(O)=O)C=C1 VYRSLCGXROPJNM-UHFFFAOYSA-N 0.000 description 9
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 9
- 239000000543 intermediate Substances 0.000 description 9
- 210000004698 lymphocyte Anatomy 0.000 description 9
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 8
- BAVYZALUXZFZLV-UHFFFAOYSA-N Methylamine Chemical compound NC BAVYZALUXZFZLV-UHFFFAOYSA-N 0.000 description 8
- 238000003556 assay Methods 0.000 description 8
- 239000002585 base Substances 0.000 description 8
- 238000003818 flash chromatography Methods 0.000 description 8
- 239000003018 immunosuppressive agent Substances 0.000 description 8
- 239000012044 organic layer Substances 0.000 description 8
- 238000002360 preparation method Methods 0.000 description 8
- 239000011780 sodium chloride Substances 0.000 description 8
- WEVYAHXRMPXWCK-UHFFFAOYSA-N Acetonitrile Chemical compound CC#N WEVYAHXRMPXWCK-UHFFFAOYSA-N 0.000 description 7
- 239000005711 Benzoic acid Substances 0.000 description 7
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 7
- 241000700159 Rattus Species 0.000 description 7
- 102000011011 Sphingosine 1-phosphate receptors Human genes 0.000 description 7
- 108050001083 Sphingosine 1-phosphate receptors Proteins 0.000 description 7
- 125000004432 carbon atom Chemical group C* 0.000 description 7
- 239000012528 membrane Substances 0.000 description 7
- MXQOYLRVSVOCQT-UHFFFAOYSA-N palladium;tritert-butylphosphane Chemical compound [Pd].CC(C)(C)P(C(C)(C)C)C(C)(C)C.CC(C)(C)P(C(C)(C)C)C(C)(C)C MXQOYLRVSVOCQT-UHFFFAOYSA-N 0.000 description 7
- 101150079914 s1pr2 gene Proteins 0.000 description 7
- 239000000725 suspension Substances 0.000 description 7
- CYPYTURSJDMMMP-WVCUSYJESA-N (1e,4e)-1,5-diphenylpenta-1,4-dien-3-one;palladium Chemical compound [Pd].[Pd].C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1.C=1C=CC=CC=1\C=C\C(=O)\C=C\C1=CC=CC=C1 CYPYTURSJDMMMP-WVCUSYJESA-N 0.000 description 6
- 102000003688 G-Protein-Coupled Receptors Human genes 0.000 description 6
- 108090000045 G-Protein-Coupled Receptors Proteins 0.000 description 6
- AVXURJPOCDRRFD-UHFFFAOYSA-N Hydroxylamine Chemical compound ON AVXURJPOCDRRFD-UHFFFAOYSA-N 0.000 description 6
- WTDHULULXKLSOZ-UHFFFAOYSA-N Hydroxylamine hydrochloride Chemical compound Cl.ON WTDHULULXKLSOZ-UHFFFAOYSA-N 0.000 description 6
- CSNNHWWHGAXBCP-UHFFFAOYSA-L Magnesium sulfate Chemical compound [Mg+2].[O-][S+2]([O-])([O-])[O-] CSNNHWWHGAXBCP-UHFFFAOYSA-L 0.000 description 6
- DNIAPMSPPWPWGF-UHFFFAOYSA-N Propylene glycol Chemical compound CC(O)CO DNIAPMSPPWPWGF-UHFFFAOYSA-N 0.000 description 6
- 102100029802 Sphingosine 1-phosphate receptor 5 Human genes 0.000 description 6
- HEDRZPFGACZZDS-MICDWDOJSA-N Trichloro(2H)methane Chemical compound [2H]C(Cl)(Cl)Cl HEDRZPFGACZZDS-MICDWDOJSA-N 0.000 description 6
- 235000010233 benzoic acid Nutrition 0.000 description 6
- 239000010410 layer Substances 0.000 description 6
- HQKMJHAJHXVSDF-UHFFFAOYSA-L magnesium stearate Chemical compound [Mg+2].CCCCCCCCCCCCCCCCCC([O-])=O.CCCCCCCCCCCCCCCCCC([O-])=O HQKMJHAJHXVSDF-UHFFFAOYSA-L 0.000 description 6
- 239000000843 powder Substances 0.000 description 6
- 238000005160 1H NMR spectroscopy Methods 0.000 description 5
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 5
- PMATZTZNYRCHOR-CGLBZJNRSA-N Cyclosporin A Chemical compound CC[C@@H]1NC(=O)[C@H]([C@H](O)[C@H](C)C\C=C\C)N(C)C(=O)[C@H](C(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](CC(C)C)N(C)C(=O)[C@@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CC(C)C)N(C)C(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)N(C)C(=O)CN(C)C1=O PMATZTZNYRCHOR-CGLBZJNRSA-N 0.000 description 5
- 229930105110 Cyclosporin A Natural products 0.000 description 5
- 108010036949 Cyclosporine Proteins 0.000 description 5
- 239000007995 HEPES buffer Substances 0.000 description 5
- 241001465754 Metazoa Species 0.000 description 5
- 229910019142 PO4 Inorganic materials 0.000 description 5
- 101150043606 S1pr5 gene Proteins 0.000 description 5
- 239000005557 antagonist Substances 0.000 description 5
- 239000012131 assay buffer Substances 0.000 description 5
- JFDZBHWFFUWGJE-UHFFFAOYSA-N benzonitrile Chemical compound N#CC1=CC=CC=C1 JFDZBHWFFUWGJE-UHFFFAOYSA-N 0.000 description 5
- 239000002775 capsule Substances 0.000 description 5
- 229960001265 ciclosporin Drugs 0.000 description 5
- 229940125721 immunosuppressive agent Drugs 0.000 description 5
- 239000010452 phosphate Substances 0.000 description 5
- 230000002265 prevention Effects 0.000 description 5
- 108090000623 proteins and genes Proteins 0.000 description 5
- 230000009919 sequestration Effects 0.000 description 5
- 239000000741 silica gel Substances 0.000 description 5
- 229910002027 silica gel Inorganic materials 0.000 description 5
- 239000003826 tablet Substances 0.000 description 5
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 4
- VUBBCFWWSKOHTH-UHFFFAOYSA-N 4-(2-methylpropyl)benzoic acid Chemical compound CC(C)CC1=CC=C(C(O)=O)C=C1 VUBBCFWWSKOHTH-UHFFFAOYSA-N 0.000 description 4
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 4
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 4
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 4
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 4
- QJJXYPPXXYFBGM-LFZNUXCKSA-N Tacrolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1\C=C(/C)[C@@H]1[C@H](C)[C@@H](O)CC(=O)[C@H](CC=C)/C=C(C)/C[C@H](C)C[C@H](OC)[C@H]([C@H](C[C@H]2C)OC)O[C@@]2(O)C(=O)C(=O)N2CCCC[C@H]2C(=O)O1 QJJXYPPXXYFBGM-LFZNUXCKSA-N 0.000 description 4
- 230000015572 biosynthetic process Effects 0.000 description 4
- 238000004587 chromatography analysis Methods 0.000 description 4
- 229910052681 coesite Inorganic materials 0.000 description 4
- 229910052906 cristobalite Inorganic materials 0.000 description 4
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 4
- 239000003937 drug carrier Substances 0.000 description 4
- 239000013604 expression vector Substances 0.000 description 4
- 239000007903 gelatin capsule Substances 0.000 description 4
- IUYHWZFSGMZEOG-UHFFFAOYSA-M magnesium;propane;chloride Chemical compound [Mg+2].[Cl-].C[CH-]C IUYHWZFSGMZEOG-UHFFFAOYSA-M 0.000 description 4
- 238000005259 measurement Methods 0.000 description 4
- LXCFILQKKLGQFO-UHFFFAOYSA-N p-hydroxybenzoic acid methyl ester Natural products COC(=O)C1=CC=C(O)C=C1 LXCFILQKKLGQFO-UHFFFAOYSA-N 0.000 description 4
- 239000013612 plasmid Substances 0.000 description 4
- 239000000047 product Substances 0.000 description 4
- 239000000377 silicon dioxide Substances 0.000 description 4
- 235000017557 sodium bicarbonate Nutrition 0.000 description 4
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 4
- 229910052682 stishovite Inorganic materials 0.000 description 4
- 229960001967 tacrolimus Drugs 0.000 description 4
- QJJXYPPXXYFBGM-SHYZHZOCSA-N tacrolimus Natural products CO[C@H]1C[C@H](CC[C@@H]1O)C=C(C)[C@H]2OC(=O)[C@H]3CCCCN3C(=O)C(=O)[C@@]4(O)O[C@@H]([C@H](C[C@H]4C)OC)[C@@H](C[C@H](C)CC(=C[C@@H](CC=C)C(=O)C[C@H](O)[C@H]2C)C)OC QJJXYPPXXYFBGM-SHYZHZOCSA-N 0.000 description 4
- 229940124597 therapeutic agent Drugs 0.000 description 4
- 229910052905 tridymite Inorganic materials 0.000 description 4
- 239000003981 vehicle Substances 0.000 description 4
- BTANRVKWQNVYAZ-BYPYZUCNSA-N (2S)-butan-2-ol Chemical compound CC[C@H](C)O BTANRVKWQNVYAZ-BYPYZUCNSA-N 0.000 description 3
- 150000005071 1,2,4-oxadiazoles Chemical class 0.000 description 3
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 3
- ZVYNUGSPFZCYEV-UHFFFAOYSA-N 2,6-dichloro-5-fluoropyridine-3-carboxamide Chemical compound NC(=O)C1=CC(F)=C(Cl)N=C1Cl ZVYNUGSPFZCYEV-UHFFFAOYSA-N 0.000 description 3
- JAUPUQRPBNDMDT-UHFFFAOYSA-N 2-chloropyridine-3-carbonitrile Chemical compound ClC1=NC=CC=C1C#N JAUPUQRPBNDMDT-UHFFFAOYSA-N 0.000 description 3
- XRPVXVRWIDOORM-UHFFFAOYSA-N 2-methylbutanoyl chloride Chemical compound CCC(C)C(Cl)=O XRPVXVRWIDOORM-UHFFFAOYSA-N 0.000 description 3
- QSBHJTCAPWOIIE-UHFFFAOYSA-N 3-fluoro-4-hydroxybenzaldehyde Chemical compound OC1=CC=C(C=O)C=C1F QSBHJTCAPWOIIE-UHFFFAOYSA-N 0.000 description 3
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 3
- CURLTUGMZLYLDI-UHFFFAOYSA-N Carbon dioxide Chemical compound O=C=O CURLTUGMZLYLDI-UHFFFAOYSA-N 0.000 description 3
- 108020004414 DNA Proteins 0.000 description 3
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 3
- OUVXYXNWSVIOSJ-UHFFFAOYSA-N Fluo-4 Chemical compound CC1=CC=C(N(CC(O)=O)CC(O)=O)C(OCCOC=2C(=CC=C(C=2)C2=C3C=C(F)C(=O)C=C3OC3=CC(O)=C(F)C=C32)N(CC(O)=O)CC(O)=O)=C1 OUVXYXNWSVIOSJ-UHFFFAOYSA-N 0.000 description 3
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- 125000002252 acyl group Chemical group 0.000 description 3
- 239000000443 aerosol Substances 0.000 description 3
- 125000003545 alkoxy group Chemical group 0.000 description 3
- 125000000217 alkyl group Chemical group 0.000 description 3
- ZDZHCHYQNPQSGG-UHFFFAOYSA-N binaphthyl group Chemical group C1(=CC=CC2=CC=CC=C12)C1=CC=CC2=CC=CC=C12 ZDZHCHYQNPQSGG-UHFFFAOYSA-N 0.000 description 3
- 210000004369 blood Anatomy 0.000 description 3
- 239000008280 blood Substances 0.000 description 3
- 230000036772 blood pressure Effects 0.000 description 3
- 239000000872 buffer Substances 0.000 description 3
- 239000011575 calcium Substances 0.000 description 3
- 229910052791 calcium Inorganic materials 0.000 description 3
- 230000003185 calcium uptake Effects 0.000 description 3
- 229910052799 carbon Inorganic materials 0.000 description 3
- 235000011089 carbon dioxide Nutrition 0.000 description 3
- 210000000748 cardiovascular system Anatomy 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 238000010367 cloning Methods 0.000 description 3
- 230000008878 coupling Effects 0.000 description 3
- 238000010168 coupling process Methods 0.000 description 3
- 238000005859 coupling reaction Methods 0.000 description 3
- 125000000753 cycloalkyl group Chemical group 0.000 description 3
- 230000001419 dependent effect Effects 0.000 description 3
- 239000002552 dosage form Substances 0.000 description 3
- 230000002526 effect on cardiovascular system Effects 0.000 description 3
- 229960000556 fingolimod Drugs 0.000 description 3
- KKGQTZUTZRNORY-UHFFFAOYSA-N fingolimod Chemical compound CCCCCCCCC1=CC=C(CCC(N)(CO)CO)C=C1 KKGQTZUTZRNORY-UHFFFAOYSA-N 0.000 description 3
- 150000004677 hydrates Chemical class 0.000 description 3
- 229960003444 immunosuppressant agent Drugs 0.000 description 3
- 238000002347 injection Methods 0.000 description 3
- 239000007924 injection Substances 0.000 description 3
- 238000001990 intravenous administration Methods 0.000 description 3
- 239000008101 lactose Substances 0.000 description 3
- 239000008297 liquid dosage form Substances 0.000 description 3
- 235000019359 magnesium stearate Nutrition 0.000 description 3
- 229910052943 magnesium sulfate Inorganic materials 0.000 description 3
- 125000004430 oxygen atom Chemical group O* 0.000 description 3
- 239000012071 phase Substances 0.000 description 3
- 230000003389 potentiating effect Effects 0.000 description 3
- 102000004169 proteins and genes Human genes 0.000 description 3
- 238000000746 purification Methods 0.000 description 3
- 230000002829 reductive effect Effects 0.000 description 3
- 238000010992 reflux Methods 0.000 description 3
- 238000012216 screening Methods 0.000 description 3
- 239000013605 shuttle vector Substances 0.000 description 3
- 101150101769 sip5 gene Proteins 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- 239000007921 spray Substances 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 241001515965 unidentified phage Species 0.000 description 3
- GTLDTDOJJJZVBW-UHFFFAOYSA-N zinc cyanide Chemical compound [Zn+2].N#[C-].N#[C-] GTLDTDOJJJZVBW-UHFFFAOYSA-N 0.000 description 3
- WWUZIQQURGPMPG-UHFFFAOYSA-N (-)-D-erythro-Sphingosine Natural products CCCCCCCCCCCCCC=CC(O)C(N)CO WWUZIQQURGPMPG-UHFFFAOYSA-N 0.000 description 2
- AJKPXKHWCHZLSF-VIFPVBQESA-N (3s)-3-(4-bromophenyl)cyclopentan-1-one Chemical compound C1=CC(Br)=CC=C1[C@@H]1CC(=O)CC1 AJKPXKHWCHZLSF-VIFPVBQESA-N 0.000 description 2
- BBVIDBNAYOIXOE-UHFFFAOYSA-N 1,2,4-oxadiazole Chemical compound C=1N=CON=1 BBVIDBNAYOIXOE-UHFFFAOYSA-N 0.000 description 2
- LJJJURRSDWGEFA-UHFFFAOYSA-N 1-(6-bromopyridin-3-yl)-2-methylpropan-1-one Chemical compound CC(C)C(=O)C1=CC=C(Br)N=C1 LJJJURRSDWGEFA-UHFFFAOYSA-N 0.000 description 2
- ASOKPJOREAFHNY-UHFFFAOYSA-N 1-Hydroxybenzotriazole Chemical compound C1=CC=C2N(O)N=NC2=C1 ASOKPJOREAFHNY-UHFFFAOYSA-N 0.000 description 2
- 125000006018 1-methyl-ethenyl group Chemical group 0.000 description 2
- SUBZTZVHVGYOPM-UHFFFAOYSA-N 2-chloro-5-fluoropyridine-3-carbonitrile Chemical compound FC1=CN=C(Cl)C(C#N)=C1 SUBZTZVHVGYOPM-UHFFFAOYSA-N 0.000 description 2
- KHRZSLCUROLAAR-UHFFFAOYSA-N 2-chloro-5-fluoropyridine-3-carboxamide Chemical compound NC(=O)C1=CC(F)=CN=C1Cl KHRZSLCUROLAAR-UHFFFAOYSA-N 0.000 description 2
- APOYTRAZFJURPB-UHFFFAOYSA-N 2-methoxy-n-(2-methoxyethyl)-n-(trifluoro-$l^{4}-sulfanyl)ethanamine Chemical compound COCCN(S(F)(F)F)CCOC APOYTRAZFJURPB-UHFFFAOYSA-N 0.000 description 2
- DRHKACQBMALHMR-UHFFFAOYSA-N 3-fluoro-4-(2-methylpropanoyl)benzoic acid Chemical compound CC(C)C(=O)C1=CC=C(C(O)=O)C=C1F DRHKACQBMALHMR-UHFFFAOYSA-N 0.000 description 2
- RRWSYSPXNQNVLA-ZETCQYMHSA-N 4-[(2s)-butan-2-yl]oxy-3-(trifluoromethyl)benzoic acid Chemical compound CC[C@H](C)OC1=CC=C(C(O)=O)C=C1C(F)(F)F RRWSYSPXNQNVLA-ZETCQYMHSA-N 0.000 description 2
- BGGNFOLUMKDZHM-UHFFFAOYSA-N 4-[3-(2-chloropyridin-3-yl)-1,2,4-oxadiazol-5-yl]phenol Chemical compound C1=CC(O)=CC=C1C1=NC(C=2C(=NC=CC=2)Cl)=NO1 BGGNFOLUMKDZHM-UHFFFAOYSA-N 0.000 description 2
- VPKDUJJDYNCRTR-UHFFFAOYSA-N 4-[cyclobutyl(difluoro)methyl]benzoic acid Chemical compound C1=CC(C(=O)O)=CC=C1C(F)(F)C1CCC1 VPKDUJJDYNCRTR-UHFFFAOYSA-N 0.000 description 2
- QCIWHVKGVVQHIY-UHFFFAOYSA-N 4-cyclohexylbenzoic acid Chemical group C1=CC(C(=O)O)=CC=C1C1CCCCC1 QCIWHVKGVVQHIY-UHFFFAOYSA-N 0.000 description 2
- 101100496009 Arabidopsis thaliana CIPK11 gene Proteins 0.000 description 2
- CIWBSHSKHKDKBQ-JLAZNSOCSA-N Ascorbic acid Chemical compound OC[C@H](O)[C@H]1OC(=O)C(O)=C1O CIWBSHSKHKDKBQ-JLAZNSOCSA-N 0.000 description 2
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 2
- KZMGYPLQYOPHEL-UHFFFAOYSA-N Boron trifluoride etherate Chemical compound FB(F)F.CCOCC KZMGYPLQYOPHEL-UHFFFAOYSA-N 0.000 description 2
- 108091026890 Coding region Proteins 0.000 description 2
- 108020004635 Complementary DNA Proteins 0.000 description 2
- 208000006069 Corneal Opacity Diseases 0.000 description 2
- 239000003155 DNA primer Substances 0.000 description 2
- QOSSAOTZNIDXMA-UHFFFAOYSA-N Dicylcohexylcarbodiimide Chemical compound C1CCCCC1N=C=NC1CCCCC1 QOSSAOTZNIDXMA-UHFFFAOYSA-N 0.000 description 2
- 108091006027 G proteins Proteins 0.000 description 2
- 102000030782 GTP binding Human genes 0.000 description 2
- 108091000058 GTP-Binding Proteins 0.000 description 2
- 102100039940 Gem-associated protein 7 Human genes 0.000 description 2
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 2
- 101000886583 Homo sapiens Gem-associated protein 7 Proteins 0.000 description 2
- 101000693269 Homo sapiens Sphingosine 1-phosphate receptor 3 Proteins 0.000 description 2
- LELOWRISYMNNSU-UHFFFAOYSA-N Hydrocyanic acid Natural products N#C LELOWRISYMNNSU-UHFFFAOYSA-N 0.000 description 2
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 2
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 2
- AMIMRNSIRUDHCM-UHFFFAOYSA-N Isopropylaldehyde Chemical compound CC(C)C=O AMIMRNSIRUDHCM-UHFFFAOYSA-N 0.000 description 2
- 241001123008 Leukoma Species 0.000 description 2
- 206010025327 Lymphopenia Diseases 0.000 description 2
- 102000018697 Membrane Proteins Human genes 0.000 description 2
- 108010052285 Membrane Proteins Proteins 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 2
- 108091034117 Oligonucleotide Proteins 0.000 description 2
- 101100066912 Oryza sativa subsp. japonica FLO6 gene Proteins 0.000 description 2
- 238000012408 PCR amplification Methods 0.000 description 2
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 2
- 101710097451 Putative G-protein coupled receptor Proteins 0.000 description 2
- 102100039117 Putative vomeronasal receptor-like protein 4 Human genes 0.000 description 2
- 241000283984 Rodentia Species 0.000 description 2
- 101100256977 Saccharomyces cerevisiae (strain ATCC 204508 / S288c) SIP4 gene Proteins 0.000 description 2
- CDBYLPFSWZWCQE-UHFFFAOYSA-L Sodium Carbonate Chemical compound [Na+].[Na+].[O-]C([O-])=O CDBYLPFSWZWCQE-UHFFFAOYSA-L 0.000 description 2
- 102100025747 Sphingosine 1-phosphate receptor 3 Human genes 0.000 description 2
- 229920002472 Starch Polymers 0.000 description 2
- 210000001744 T-lymphocyte Anatomy 0.000 description 2
- 208000001445 Uveomeningoencephalitic Syndrome Diseases 0.000 description 2
- 208000034705 Vogt-Koyanagi-Harada syndrome Diseases 0.000 description 2
- 230000007059 acute toxicity Effects 0.000 description 2
- 231100000403 acute toxicity Toxicity 0.000 description 2
- 230000008484 agonism Effects 0.000 description 2
- 125000004414 alkyl thio group Chemical group 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- 150000001413 amino acids Chemical group 0.000 description 2
- 229910021529 ammonia Inorganic materials 0.000 description 2
- 150000003863 ammonium salts Chemical class 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 125000003435 aroyl group Chemical group 0.000 description 2
- 230000004872 arterial blood pressure Effects 0.000 description 2
- 125000003710 aryl alkyl group Chemical group 0.000 description 2
- 150000001543 aryl boronic acids Chemical class 0.000 description 2
- 239000012298 atmosphere Substances 0.000 description 2
- 230000008901 benefit Effects 0.000 description 2
- 230000000975 bioactive effect Effects 0.000 description 2
- 239000012267 brine Substances 0.000 description 2
- 125000004369 butenyl group Chemical group C(=CCC)* 0.000 description 2
- FJDQFPXHSGXQBY-UHFFFAOYSA-L caesium carbonate Chemical compound [Cs+].[Cs+].[O-]C([O-])=O FJDQFPXHSGXQBY-UHFFFAOYSA-L 0.000 description 2
- 229910000024 caesium carbonate Inorganic materials 0.000 description 2
- PFKFTWBEEFSNDU-UHFFFAOYSA-N carbonyldiimidazole Chemical compound C1=CN=CN1C(=O)N1C=CN=C1 PFKFTWBEEFSNDU-UHFFFAOYSA-N 0.000 description 2
- 150000001735 carboxylic acids Chemical class 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 230000004663 cell proliferation Effects 0.000 description 2
- 230000036755 cellular response Effects 0.000 description 2
- 239000001913 cellulose Substances 0.000 description 2
- 235000010980 cellulose Nutrition 0.000 description 2
- 229920002678 cellulose Polymers 0.000 description 2
- 238000012512 characterization method Methods 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- OSASVXMJTNOKOY-UHFFFAOYSA-N chlorobutanol Chemical compound CC(C)(O)C(Cl)(Cl)Cl OSASVXMJTNOKOY-UHFFFAOYSA-N 0.000 description 2
- 239000007891 compressed tablet Substances 0.000 description 2
- 238000010276 construction Methods 0.000 description 2
- 230000008602 contraction Effects 0.000 description 2
- 238000001816 cooling Methods 0.000 description 2
- 239000006071 cream Substances 0.000 description 2
- 239000013078 crystal Substances 0.000 description 2
- 125000004122 cyclic group Chemical group 0.000 description 2
- 125000000000 cycloalkoxy group Chemical group 0.000 description 2
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 2
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 2
- 238000001514 detection method Methods 0.000 description 2
- 208000035475 disorder Diseases 0.000 description 2
- 239000006185 dispersion Substances 0.000 description 2
- 238000006073 displacement reaction Methods 0.000 description 2
- 238000010494 dissociation reaction Methods 0.000 description 2
- 230000005593 dissociations Effects 0.000 description 2
- 239000006196 drop Substances 0.000 description 2
- 238000001035 drying Methods 0.000 description 2
- JVGGVLGYEVKWJU-UHFFFAOYSA-N ethyl 4-(cyclobutanecarbonyl)benzoate Chemical compound C1=CC(C(=O)OCC)=CC=C1C(=O)C1CCC1 JVGGVLGYEVKWJU-UHFFFAOYSA-N 0.000 description 2
- 239000000706 filtrate Substances 0.000 description 2
- 210000003958 hematopoietic stem cell Anatomy 0.000 description 2
- 230000001861 immunosuppressant effect Effects 0.000 description 2
- 238000001802 infusion Methods 0.000 description 2
- 230000002452 interceptive effect Effects 0.000 description 2
- 230000003834 intracellular effect Effects 0.000 description 2
- 201000010666 keratoconjunctivitis Diseases 0.000 description 2
- 210000000265 leukocyte Anatomy 0.000 description 2
- 238000000670 ligand binding assay Methods 0.000 description 2
- 210000003563 lymphoid tissue Anatomy 0.000 description 2
- 231100001023 lymphopenia Toxicity 0.000 description 2
- 229910001629 magnesium chloride Inorganic materials 0.000 description 2
- 235000019341 magnesium sulphate Nutrition 0.000 description 2
- HSFOKEFAEWVYQS-VIFPVBQESA-N methyl 4-[(2s)-butan-2-yl]oxybenzoate Chemical compound CC[C@H](C)OC1=CC=C(C(=O)OC)C=C1 HSFOKEFAEWVYQS-VIFPVBQESA-N 0.000 description 2
- 235000010270 methyl p-hydroxybenzoate Nutrition 0.000 description 2
- 238000002156 mixing Methods 0.000 description 2
- 238000010369 molecular cloning Methods 0.000 description 2
- 239000002773 nucleotide Substances 0.000 description 2
- 125000003729 nucleotide group Chemical group 0.000 description 2
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 2
- 239000003921 oil Substances 0.000 description 2
- 235000019198 oils Nutrition 0.000 description 2
- 239000002674 ointment Substances 0.000 description 2
- CTSLXHKWHWQRSH-UHFFFAOYSA-N oxalyl chloride Chemical compound ClC(=O)C(Cl)=O CTSLXHKWHWQRSH-UHFFFAOYSA-N 0.000 description 2
- NFHFRUOZVGFOOS-UHFFFAOYSA-N palladium;triphenylphosphane Chemical compound [Pd].C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 NFHFRUOZVGFOOS-UHFFFAOYSA-N 0.000 description 2
- 239000003182 parenteral nutrition solution Substances 0.000 description 2
- 239000008188 pellet Substances 0.000 description 2
- 239000011591 potassium Substances 0.000 description 2
- 229910052700 potassium Inorganic materials 0.000 description 2
- SCVFZCLFOSHCOH-UHFFFAOYSA-M potassium acetate Chemical compound [K+].CC([O-])=O SCVFZCLFOSHCOH-UHFFFAOYSA-M 0.000 description 2
- NROKBHXJSPEDAR-UHFFFAOYSA-M potassium fluoride Chemical compound [F-].[K+] NROKBHXJSPEDAR-UHFFFAOYSA-M 0.000 description 2
- 239000011698 potassium fluoride Substances 0.000 description 2
- 230000008569 process Effects 0.000 description 2
- 125000004368 propenyl group Chemical group C(=CC)* 0.000 description 2
- KRIOVPPHQSLHCZ-UHFFFAOYSA-N propiophenone Chemical compound CCC(=O)C1=CC=CC=C1 KRIOVPPHQSLHCZ-UHFFFAOYSA-N 0.000 description 2
- QELSKZZBTMNZEB-UHFFFAOYSA-N propylparaben Chemical compound CCCOC(=O)C1=CC=C(O)C=C1 QELSKZZBTMNZEB-UHFFFAOYSA-N 0.000 description 2
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 2
- 238000011160 research Methods 0.000 description 2
- 239000000523 sample Substances 0.000 description 2
- 230000037390 scarring Effects 0.000 description 2
- 238000009097 single-agent therapy Methods 0.000 description 2
- 229910052708 sodium Inorganic materials 0.000 description 2
- GEHJYWRUCIMESM-UHFFFAOYSA-L sodium sulfite Chemical compound [Na+].[Na+].[O-]S([O-])=O GEHJYWRUCIMESM-UHFFFAOYSA-L 0.000 description 2
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 2
- WWUZIQQURGPMPG-KRWOKUGFSA-N sphingosine Chemical compound CCCCCCCCCCCCC\C=C\[C@@H](O)[C@@H](N)CO WWUZIQQURGPMPG-KRWOKUGFSA-N 0.000 description 2
- 239000003381 stabilizer Substances 0.000 description 2
- 239000008107 starch Substances 0.000 description 2
- 235000019698 starch Nutrition 0.000 description 2
- 230000000638 stimulation Effects 0.000 description 2
- 230000004083 survival effect Effects 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- FPGGTKZVZWFYPV-UHFFFAOYSA-M tetrabutylammonium fluoride Chemical compound [F-].CCCC[N+](CCCC)(CCCC)CCCC FPGGTKZVZWFYPV-UHFFFAOYSA-M 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- QNMBSXGYAQZCTN-UHFFFAOYSA-N thiophen-3-ylboronic acid Chemical compound OB(O)C=1C=CSC=1 QNMBSXGYAQZCTN-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 230000032258 transport Effects 0.000 description 2
- QAEDZJGFFMLHHQ-UHFFFAOYSA-N trifluoroacetic anhydride Chemical compound FC(F)(F)C(=O)OC(=O)C(F)(F)F QAEDZJGFFMLHHQ-UHFFFAOYSA-N 0.000 description 2
- 239000013598 vector Substances 0.000 description 2
- 239000008096 xylene Substances 0.000 description 2
- 150000003738 xylenes Chemical class 0.000 description 2
- DSDCDMKASWVZHI-UHFFFAOYSA-M zinc;2-methanidylpropane;bromide Chemical compound Br[Zn+].CC(C)[CH2-] DSDCDMKASWVZHI-UHFFFAOYSA-M 0.000 description 2
- BTANRVKWQNVYAZ-SCSAIBSYSA-N (2R)-butan-2-ol Chemical compound CC[C@@H](C)O BTANRVKWQNVYAZ-SCSAIBSYSA-N 0.000 description 1
- POILWHVDKZOXJZ-ARJAWSKDSA-N (3Z)-4-hydroxy-3-penten-2-one Chemical compound C\C(O)=C\C(C)=O POILWHVDKZOXJZ-ARJAWSKDSA-N 0.000 description 1
- QBLFZIBJXUQVRF-UHFFFAOYSA-N (4-bromophenyl)boronic acid Chemical compound OB(O)C1=CC=C(Br)C=C1 QBLFZIBJXUQVRF-UHFFFAOYSA-N 0.000 description 1
- ODIGIKRIUKFKHP-UHFFFAOYSA-N (n-propan-2-yloxycarbonylanilino) acetate Chemical compound CC(C)OC(=O)N(OC(C)=O)C1=CC=CC=C1 ODIGIKRIUKFKHP-UHFFFAOYSA-N 0.000 description 1
- IONHLFTYGWUABZ-UHFFFAOYSA-N 1-(6-bromopyridin-3-yl)-2-methylpropan-1-ol Chemical compound CC(C)C(O)C1=CC=C(Br)N=C1 IONHLFTYGWUABZ-UHFFFAOYSA-N 0.000 description 1
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 1
- MXHBSHPOZYGRMD-UHFFFAOYSA-N 2,2,2-trifluoroethoxy trifluoromethanesulfonate Chemical compound FC(F)(F)COOS(=O)(=O)C(F)(F)F MXHBSHPOZYGRMD-UHFFFAOYSA-N 0.000 description 1
- RTMMSCJWQYWMNK-UHFFFAOYSA-N 2,2,2-trifluoroethyl trifluoromethanesulfonate Chemical compound FC(F)(F)COS(=O)(=O)C(F)(F)F RTMMSCJWQYWMNK-UHFFFAOYSA-N 0.000 description 1
- ZHXUWDPHUQHFOV-UHFFFAOYSA-N 2,5-dibromopyridine Chemical compound BrC1=CC=C(Br)N=C1 ZHXUWDPHUQHFOV-UHFFFAOYSA-N 0.000 description 1
- BXSNGPWULWQNQB-UHFFFAOYSA-N 2-(2-methylpropanoyl)benzoic acid Chemical compound CC(C)C(=O)C1=CC=CC=C1C(O)=O BXSNGPWULWQNQB-UHFFFAOYSA-N 0.000 description 1
- SKMVXFKXYAAJBA-UHFFFAOYSA-N 2-amino-5-fluoro-n'-hydroxypyridine-3-carboximidamide Chemical compound NC1=NC=C(F)C=C1C(=N)NO SKMVXFKXYAAJBA-UHFFFAOYSA-N 0.000 description 1
- KWSANSFLBDJIKU-UHFFFAOYSA-N 2-amino-5-fluoropyridine-3-carbonitrile Chemical compound NC1=NC=C(F)C=C1C#N KWSANSFLBDJIKU-UHFFFAOYSA-N 0.000 description 1
- FLBDUUYQOPBOLR-UHFFFAOYSA-N 2-amino-n'-hydroxypyridine-3-carboximidamide Chemical compound O\N=C(/N)C1=CC=CN=C1N FLBDUUYQOPBOLR-UHFFFAOYSA-N 0.000 description 1
- YYXDQRRDNPRJFL-UHFFFAOYSA-N 2-aminopyridine-3-carbonitrile Chemical compound NC1=NC=CC=C1C#N YYXDQRRDNPRJFL-UHFFFAOYSA-N 0.000 description 1
- REIVHYDACHXPNH-UHFFFAOYSA-N 2-fluoro-4-hydroxybenzonitrile Chemical group OC1=CC=C(C#N)C(F)=C1 REIVHYDACHXPNH-UHFFFAOYSA-N 0.000 description 1
- DGMOBVGABMBZSB-UHFFFAOYSA-N 2-methylpropanoyl chloride Chemical group CC(C)C(Cl)=O DGMOBVGABMBZSB-UHFFFAOYSA-N 0.000 description 1
- MXWUCBHSUUSIDV-UHFFFAOYSA-N 3,5-difluoro-4-(2,2,2-trifluoroethoxy)benzoic acid Chemical compound OC(=O)C1=CC(F)=C(OCC(F)(F)F)C(F)=C1 MXWUCBHSUUSIDV-UHFFFAOYSA-N 0.000 description 1
- DPFXBYKRNMBNBY-UHFFFAOYSA-N 3,5-difluoro-4-(2,2,2-trifluoroethoxy)benzonitrile Chemical compound FC1=CC(C#N)=CC(F)=C1OCC(F)(F)F DPFXBYKRNMBNBY-UHFFFAOYSA-N 0.000 description 1
- IVEWHNWXPMNYRV-UHFFFAOYSA-N 3-(2-chloropyridin-3-yl)-5-[4-(2-methylpropyl)phenyl]-1,2,4-oxadiazole Chemical compound C1=CC(CC(C)C)=CC=C1C1=NC(C=2C(=NC=CC=2)Cl)=NO1 IVEWHNWXPMNYRV-UHFFFAOYSA-N 0.000 description 1
- FPQQSJJWHUJYPU-UHFFFAOYSA-N 3-(dimethylamino)propyliminomethylidene-ethylazanium;chloride Chemical compound Cl.CCN=C=NCCCN(C)C FPQQSJJWHUJYPU-UHFFFAOYSA-N 0.000 description 1
- UZALWJYZYLTEMI-UHFFFAOYSA-N 3-(methylamino)pyrazine-2-carbonitrile Chemical compound CNC1=NC=CN=C1C#N UZALWJYZYLTEMI-UHFFFAOYSA-N 0.000 description 1
- HVMNHAHBZGDUML-UHFFFAOYSA-N 3-[2-(benzotriazol-1-yloxy)pyridin-3-yl]-5-(5-bromopyridin-2-yl)-1,2,4-oxadiazole Chemical compound N1=CC(Br)=CC=C1C1=NC(C=2C(=NC=CC=2)ON2C3=CC=CC=C3N=N2)=NO1 HVMNHAHBZGDUML-UHFFFAOYSA-N 0.000 description 1
- ZVYNMRGBXXJTBZ-UHFFFAOYSA-N 3-[5-(4-bromophenyl)-1,2,4-oxadiazol-3-yl]-n-methylpyridin-2-amine Chemical compound CNC1=NC=CC=C1C1=NOC(C=2C=CC(Br)=CC=2)=N1 ZVYNMRGBXXJTBZ-UHFFFAOYSA-N 0.000 description 1
- HZBXMKNTOPOHDZ-UHFFFAOYSA-N 3-[5-[4-chloro-3-(trifluoromethyl)phenyl]-1,2,4-oxadiazol-3-yl]-n-methylpyridin-2-amine Chemical compound CNC1=NC=CC=C1C1=NOC(C=2C=C(C(Cl)=CC=2)C(F)(F)F)=N1 HZBXMKNTOPOHDZ-UHFFFAOYSA-N 0.000 description 1
- UONXRMTVFCALGF-UHFFFAOYSA-N 3-[5-[4-fluoro-3-(trifluoromethyl)phenyl]-1,2,4-oxadiazol-3-yl]-n-methylpyridin-2-amine Chemical compound CNC1=NC=CC=C1C1=NOC(C=2C=C(C(F)=CC=2)C(F)(F)F)=N1 UONXRMTVFCALGF-UHFFFAOYSA-N 0.000 description 1
- KLDLRDSRCMJKGM-UHFFFAOYSA-N 3-[chloro-(2-oxo-1,3-oxazolidin-3-yl)phosphoryl]-1,3-oxazolidin-2-one Chemical compound C1COC(=O)N1P(=O)(Cl)N1CCOC1=O KLDLRDSRCMJKGM-UHFFFAOYSA-N 0.000 description 1
- USMYBDKCTJNCCZ-UHFFFAOYSA-N 4-(1,1-difluoro-2-methylpropyl)benzoic acid Chemical compound CC(C)C(F)(F)C1=CC=C(C(O)=O)C=C1 USMYBDKCTJNCCZ-UHFFFAOYSA-N 0.000 description 1
- QLRTYSQXRSOYEF-UHFFFAOYSA-N 4-(2-methylbutanoyl)benzoic acid Chemical compound CCC(C)C(=O)C1=CC=C(C(O)=O)C=C1 QLRTYSQXRSOYEF-UHFFFAOYSA-N 0.000 description 1
- FLICZDVBYUMMAS-UHFFFAOYSA-N 4-(2-methylpropanoyl)benzoic acid Chemical compound CC(C)C(=O)C1=CC=C(C(O)=O)C=C1 FLICZDVBYUMMAS-UHFFFAOYSA-N 0.000 description 1
- FOEUJOQGAMKHLJ-UHFFFAOYSA-N 4-(cyclobutanecarbonyl)-2-ethylbenzoic acid Chemical compound C1=C(C(O)=O)C(CC)=CC(C(=O)C2CCC2)=C1 FOEUJOQGAMKHLJ-UHFFFAOYSA-N 0.000 description 1
- PCQXRWWAQBKKOJ-JTQLQIEISA-N 4-[(1s)-3,3-difluorocyclopentyl]benzoic acid Chemical compound C1=CC(C(=O)O)=CC=C1[C@@H]1CC(F)(F)CC1 PCQXRWWAQBKKOJ-JTQLQIEISA-N 0.000 description 1
- OFHGNOFBOJRBAI-LURJTMIESA-N 4-[(2s)-butan-2-yl]oxy-3,5-difluorobenzoic acid Chemical compound CC[C@H](C)OC1=C(F)C=C(C(O)=O)C=C1F OFHGNOFBOJRBAI-LURJTMIESA-N 0.000 description 1
- WBPGDXLBWPYEER-ZETCQYMHSA-N 4-[(2s)-butan-2-yl]oxy-3,5-difluorobenzonitrile Chemical group CC[C@H](C)OC1=C(F)C=C(C#N)C=C1F WBPGDXLBWPYEER-ZETCQYMHSA-N 0.000 description 1
- RMBFBCMMTRYPAT-QMMMGPOBSA-N 4-[(2s)-butan-2-yl]oxy-3-(trifluoromethyl)benzonitrile Chemical compound CC[C@H](C)OC1=CC=C(C#N)C=C1C(F)(F)F RMBFBCMMTRYPAT-QMMMGPOBSA-N 0.000 description 1
- XJYPEAOGEAWBNP-QMMMGPOBSA-N 4-[(2s)-butan-2-yl]oxy-3-fluorobenzaldehyde Chemical compound CC[C@H](C)OC1=CC=C(C=O)C=C1F XJYPEAOGEAWBNP-QMMMGPOBSA-N 0.000 description 1
- CHGBSBAJYKVPJO-ZETCQYMHSA-N 4-[(2s)-butan-2-yl]oxy-3-fluorobenzoic acid Chemical compound CC[C@H](C)OC1=CC=C(C(O)=O)C=C1F CHGBSBAJYKVPJO-ZETCQYMHSA-N 0.000 description 1
- LCMFDJQAHNDLEI-QMMMGPOBSA-N 4-[(2s)-butan-2-yl]oxybenzoic acid Chemical compound CC[C@H](C)OC1=CC=C(C(O)=O)C=C1 LCMFDJQAHNDLEI-QMMMGPOBSA-N 0.000 description 1
- ZDRVLAOYDGQLFI-UHFFFAOYSA-N 4-[[4-(4-chlorophenyl)-1,3-thiazol-2-yl]amino]phenol;hydrochloride Chemical compound Cl.C1=CC(O)=CC=C1NC1=NC(C=2C=CC(Cl)=CC=2)=CS1 ZDRVLAOYDGQLFI-UHFFFAOYSA-N 0.000 description 1
- GPRPSJPFAAGLCA-UHFFFAOYSA-N 4-bromo-2,6-difluorophenol Chemical compound OC1=C(F)C=C(Br)C=C1F GPRPSJPFAAGLCA-UHFFFAOYSA-N 0.000 description 1
- DENKGPBHLYFNGK-UHFFFAOYSA-N 4-bromobenzoyl chloride Chemical compound ClC(=O)C1=CC=C(Br)C=C1 DENKGPBHLYFNGK-UHFFFAOYSA-N 0.000 description 1
- CQZQCORFYSSCFY-UHFFFAOYSA-N 4-fluoro-3-(trifluoromethyl)benzonitrile Chemical compound FC1=CC=C(C#N)C=C1C(F)(F)F CQZQCORFYSSCFY-UHFFFAOYSA-N 0.000 description 1
- BUDISZQHCHGLJW-UHFFFAOYSA-N 4-fluoro-3-(trifluoromethyl)benzoyl chloride Chemical compound FC1=CC=C(C(Cl)=O)C=C1C(F)(F)F BUDISZQHCHGLJW-UHFFFAOYSA-N 0.000 description 1
- 229940090248 4-hydroxybenzoic acid Drugs 0.000 description 1
- 125000005274 4-hydroxybenzoic acid group Chemical group 0.000 description 1
- UPDQVMNODWGPLB-UHFFFAOYSA-N 5-(2-methylpropanoyl)pyridine-2-carbonitrile Chemical compound CC(C)C(=O)C1=CC=C(C#N)N=C1 UPDQVMNODWGPLB-UHFFFAOYSA-N 0.000 description 1
- MNNQIBXLAHVDDL-UHFFFAOYSA-N 5-bromopyridine-2-carboxylic acid Chemical compound OC(=O)C1=CC=C(Br)C=N1 MNNQIBXLAHVDDL-UHFFFAOYSA-N 0.000 description 1
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 1
- 208000036065 Airway Remodeling Diseases 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 206010003591 Ataxia Diseases 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 241000255789 Bombyx mori Species 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- 206010006482 Bronchospasm Diseases 0.000 description 1
- 101100256965 Caenorhabditis elegans sip-1 gene Proteins 0.000 description 1
- UXVMQQNJUSDDNG-UHFFFAOYSA-L Calcium chloride Chemical compound [Cl-].[Cl-].[Ca+2] UXVMQQNJUSDDNG-UHFFFAOYSA-L 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- 241000282693 Cercopithecidae Species 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- BZKFMUIJRXWWQK-UHFFFAOYSA-N Cyclopentenone Chemical compound O=C1CCC=C1 BZKFMUIJRXWWQK-UHFFFAOYSA-N 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- FBPFZTCFMRRESA-FSIIMWSLSA-N D-Glucitol Natural products OC[C@H](O)[C@H](O)[C@@H](O)[C@H](O)CO FBPFZTCFMRRESA-FSIIMWSLSA-N 0.000 description 1
- 241000255581 Drosophila <fruit fly, genus> Species 0.000 description 1
- 206010013975 Dyspnoeas Diseases 0.000 description 1
- ZGTMUACCHSMWAC-UHFFFAOYSA-L EDTA disodium salt (anhydrous) Chemical compound [Na+].[Na+].OC(=O)CN(CC([O-])=O)CCN(CC(O)=O)CC([O-])=O ZGTMUACCHSMWAC-UHFFFAOYSA-L 0.000 description 1
- 241000588724 Escherichia coli Species 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 230000005526 G1 to G0 transition Effects 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 102100039955 Gem-associated protein 6 Human genes 0.000 description 1
- 102000034354 Gi proteins Human genes 0.000 description 1
- 108091006101 Gi proteins Proteins 0.000 description 1
- 241000238631 Hexapoda Species 0.000 description 1
- 101000886614 Homo sapiens Gem-associated protein 6 Proteins 0.000 description 1
- 101000653757 Homo sapiens Sphingosine 1-phosphate receptor 4 Proteins 0.000 description 1
- 101000653759 Homo sapiens Sphingosine 1-phosphate receptor 5 Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- 239000007836 KH2PO4 Substances 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- 102000016994 Lysolipids receptors Human genes 0.000 description 1
- 108010027749 Lysophospholipid Receptors Proteins 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 241001529936 Murinae Species 0.000 description 1
- MZRVEZGGRBJDDB-UHFFFAOYSA-N N-Butyllithium Chemical compound [Li]CCCC MZRVEZGGRBJDDB-UHFFFAOYSA-N 0.000 description 1
- LFTLOKWAGJYHHR-UHFFFAOYSA-N N-methylmorpholine N-oxide Chemical compound CN1(=O)CCOCC1 LFTLOKWAGJYHHR-UHFFFAOYSA-N 0.000 description 1
- 229910017912 NH2OH Inorganic materials 0.000 description 1
- 229910002651 NO3 Inorganic materials 0.000 description 1
- 239000007832 Na2SO4 Substances 0.000 description 1
- 229910020700 Na3VO4 Inorganic materials 0.000 description 1
- 206010029155 Nephropathy toxic Diseases 0.000 description 1
- 206010029350 Neurotoxicity Diseases 0.000 description 1
- NHNBFGGVMKEFGY-UHFFFAOYSA-N Nitrate Chemical compound [O-][N+]([O-])=O NHNBFGGVMKEFGY-UHFFFAOYSA-N 0.000 description 1
- 108091028043 Nucleic acid sequence Proteins 0.000 description 1
- 108020005187 Oligonucleotide Probes Proteins 0.000 description 1
- 206010033799 Paralysis Diseases 0.000 description 1
- 239000004698 Polyethylene Substances 0.000 description 1
- 108010039918 Polylysine Proteins 0.000 description 1
- 229910019020 PtO2 Inorganic materials 0.000 description 1
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 1
- DWAQJAXMDSEUJJ-UHFFFAOYSA-M Sodium bisulfite Chemical compound [Na+].OS([O-])=O DWAQJAXMDSEUJJ-UHFFFAOYSA-M 0.000 description 1
- 102100025749 Sphingosine 1-phosphate receptor 2 Human genes 0.000 description 1
- 101710155462 Sphingosine 1-phosphate receptor 2 Proteins 0.000 description 1
- 102100029803 Sphingosine 1-phosphate receptor 4 Human genes 0.000 description 1
- 235000021355 Stearic acid Nutrition 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 206010044221 Toxic encephalopathy Diseases 0.000 description 1
- 208000003443 Unconsciousness Diseases 0.000 description 1
- 241000700605 Viruses Species 0.000 description 1
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 1
- NCMGULPVECPTGX-UHFFFAOYSA-N [cyclobutyl(difluoro)methyl] benzoate Chemical compound C1(CCC1)C(F)(F)OC(C1=CC=CC=C1)=O NCMGULPVECPTGX-UHFFFAOYSA-N 0.000 description 1
- 206010000059 abdominal discomfort Diseases 0.000 description 1
- 238000010521 absorption reaction Methods 0.000 description 1
- DPXJVFZANSGRMM-UHFFFAOYSA-N acetic acid;2,3,4,5,6-pentahydroxyhexanal;sodium Chemical compound [Na].CC(O)=O.OCC(O)C(O)C(O)C(O)C=O DPXJVFZANSGRMM-UHFFFAOYSA-N 0.000 description 1
- 125000002777 acetyl group Chemical group [H]C([H])([H])C(*)=O 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 230000004913 activation Effects 0.000 description 1
- 230000010933 acylation Effects 0.000 description 1
- 238000005917 acylation reaction Methods 0.000 description 1
- 125000005073 adamantyl group Chemical group C12(CC3CC(CC(C1)C3)C2)* 0.000 description 1
- YKIOKAURTKXMSB-UHFFFAOYSA-N adams's catalyst Chemical compound O=[Pt]=O YKIOKAURTKXMSB-UHFFFAOYSA-N 0.000 description 1
- 210000002867 adherens junction Anatomy 0.000 description 1
- 208000037883 airway inflammation Diseases 0.000 description 1
- 125000003342 alkenyl group Chemical group 0.000 description 1
- 150000003973 alkyl amines Chemical class 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- 125000005278 alkyl sulfonyloxy group Chemical group 0.000 description 1
- 125000000304 alkynyl group Chemical group 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- 150000001409 amidines Chemical class 0.000 description 1
- 230000033115 angiogenesis Effects 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 230000003064 anti-oxidating effect Effects 0.000 description 1
- 230000005875 antibody response Effects 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 239000008346 aqueous phase Substances 0.000 description 1
- 239000007900 aqueous suspension Substances 0.000 description 1
- 125000005333 aroyloxy group Chemical group 0.000 description 1
- 206010003246 arthritis Diseases 0.000 description 1
- 125000005279 aryl sulfonyloxy group Chemical group 0.000 description 1
- 125000005110 aryl thio group Chemical group 0.000 description 1
- 125000004104 aryloxy group Chemical group 0.000 description 1
- 235000010323 ascorbic acid Nutrition 0.000 description 1
- 239000011668 ascorbic acid Substances 0.000 description 1
- 229960005070 ascorbic acid Drugs 0.000 description 1
- 230000006472 autoimmune response Effects 0.000 description 1
- LMEKQMALGUDUQG-UHFFFAOYSA-N azathioprine Chemical compound CN1C=NC([N+]([O-])=O)=C1SC1=NC=NC2=C1NC=N2 LMEKQMALGUDUQG-UHFFFAOYSA-N 0.000 description 1
- 229960002170 azathioprine Drugs 0.000 description 1
- 210000003719 b-lymphocyte Anatomy 0.000 description 1
- 229960000686 benzalkonium chloride Drugs 0.000 description 1
- 125000003236 benzoyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C(*)=O 0.000 description 1
- 125000001231 benzoyloxy group Chemical group C(C1=CC=CC=C1)(=O)O* 0.000 description 1
- KTVOALUCKWXLRT-UHFFFAOYSA-N benzyl 4-bromo-3-fluorobenzoate Chemical compound C1=C(Br)C(F)=CC(C(=O)OCC=2C=CC=CC=2)=C1 KTVOALUCKWXLRT-UHFFFAOYSA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- CADWTSSKOVRVJC-UHFFFAOYSA-N benzyl(dimethyl)azanium;chloride Chemical compound [Cl-].C[NH+](C)CC1=CC=CC=C1 CADWTSSKOVRVJC-UHFFFAOYSA-N 0.000 description 1
- 125000000051 benzyloxy group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])O* 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 230000000903 blocking effect Effects 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- PZOHOALJQOFNTB-UHFFFAOYSA-M brequinar sodium Chemical compound [Na+].N1=C2C=CC(F)=CC2=C(C([O-])=O)C(C)=C1C(C=C1)=CC=C1C1=CC=CC=C1F PZOHOALJQOFNTB-UHFFFAOYSA-M 0.000 description 1
- 239000004044 bronchoconstricting agent Substances 0.000 description 1
- 230000007885 bronchoconstriction Effects 0.000 description 1
- 230000003435 bronchoconstrictive effect Effects 0.000 description 1
- 239000007975 buffered saline Substances 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004063 butyryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000010804 cDNA synthesis Methods 0.000 description 1
- 239000001110 calcium chloride Substances 0.000 description 1
- 235000011148 calcium chloride Nutrition 0.000 description 1
- 229910001628 calcium chloride Inorganic materials 0.000 description 1
- 229940041514 candida albicans extract Drugs 0.000 description 1
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 1
- 150000003857 carboxamides Chemical class 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 150000001244 carboxylic acid anhydrides Chemical class 0.000 description 1
- 238000012754 cardiac puncture Methods 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 230000010261 cell growth Effects 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 238000005119 centrifugation Methods 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- 239000007795 chemical reaction product Substances 0.000 description 1
- 230000035605 chemotaxis Effects 0.000 description 1
- 229960004926 chlorobutanol Drugs 0.000 description 1
- 238000012411 cloning technique Methods 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 229940075614 colloidal silicon dioxide Drugs 0.000 description 1
- 238000004040 coloring Methods 0.000 description 1
- 229940000425 combination drug Drugs 0.000 description 1
- 238000010924 continuous production Methods 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- JFWMYCVMQSLLOO-UHFFFAOYSA-N cyclobutanecarbonyl chloride Chemical group ClC(=O)C1CCC1 JFWMYCVMQSLLOO-UHFFFAOYSA-N 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000004850 cyclobutylmethyl group Chemical group C1(CCC1)C* 0.000 description 1
- 125000001352 cyclobutyloxy group Chemical group C1(CCC1)O* 0.000 description 1
- 125000000582 cycloheptyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 229930182912 cyclosporin Natural products 0.000 description 1
- 230000016396 cytokine production Effects 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 229940127089 cytotoxic agent Drugs 0.000 description 1
- 239000002254 cytotoxic agent Substances 0.000 description 1
- 231100000599 cytotoxic agent Toxicity 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 230000034994 death Effects 0.000 description 1
- 230000007423 decrease Effects 0.000 description 1
- 238000013461 design Methods 0.000 description 1
- 238000011161 development Methods 0.000 description 1
- 230000018109 developmental process Effects 0.000 description 1
- 239000008121 dextrose Substances 0.000 description 1
- 125000005265 dialkylamine group Chemical group 0.000 description 1
- YNHIGQDRGKUECZ-UHFFFAOYSA-N dichloropalladium;triphenylphosphanium Chemical compound Cl[Pd]Cl.C1=CC=CC=C1[PH+](C=1C=CC=CC=1)C1=CC=CC=C1.C1=CC=CC=C1[PH+](C=1C=CC=CC=1)C1=CC=CC=C1 YNHIGQDRGKUECZ-UHFFFAOYSA-N 0.000 description 1
- LCSNDSFWVKMJCT-UHFFFAOYSA-N dicyclohexyl-(2-phenylphenyl)phosphane Chemical group C1CCCCC1P(C=1C(=CC=CC=1)C=1C=CC=CC=1)C1CCCCC1 LCSNDSFWVKMJCT-UHFFFAOYSA-N 0.000 description 1
- 230000004069 differentiation Effects 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 238000010790 dilution Methods 0.000 description 1
- 239000012895 dilution Substances 0.000 description 1
- 238000003113 dilution method Methods 0.000 description 1
- 239000001177 diphosphate Substances 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 239000008298 dragée Substances 0.000 description 1
- 239000003480 eluent Substances 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 210000002889 endothelial cell Anatomy 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- RBEREJVCBYTBMM-UHFFFAOYSA-N ethyl 4-[cyclobutyl(difluoro)methyl]benzoate Chemical compound C1=CC(C(=O)OCC)=CC=C1C(F)(F)C1CCC1 RBEREJVCBYTBMM-UHFFFAOYSA-N 0.000 description 1
- DNJIEGIFACGWOD-UHFFFAOYSA-N ethyl mercaptane Natural products CCS DNJIEGIFACGWOD-UHFFFAOYSA-N 0.000 description 1
- OJCSPXHYDFONPU-UHFFFAOYSA-N etoac etoac Chemical compound CCOC(C)=O.CCOC(C)=O OJCSPXHYDFONPU-UHFFFAOYSA-N 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 239000003889 eye drop Substances 0.000 description 1
- 229940012356 eye drops Drugs 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 238000013100 final test Methods 0.000 description 1
- 238000000799 fluorescence microscopy Methods 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 239000012634 fragment Substances 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 230000002538 fungal effect Effects 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 229960002706 gusperimus Drugs 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 238000003306 harvesting Methods 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- 230000002489 hematologic effect Effects 0.000 description 1
- 125000000623 heterocyclic group Chemical group 0.000 description 1
- 125000003104 hexanoyl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 230000003284 homeostatic effect Effects 0.000 description 1
- 238000000265 homogenisation Methods 0.000 description 1
- 210000005120 human airway smooth muscle cell Anatomy 0.000 description 1
- 230000008348 humoral response Effects 0.000 description 1
- 229930195733 hydrocarbon Natural products 0.000 description 1
- 150000002430 hydrocarbons Chemical class 0.000 description 1
- 125000004435 hydrogen atom Chemical group [H]* 0.000 description 1
- 125000004356 hydroxy functional group Chemical group O* 0.000 description 1
- 230000001900 immune effect Effects 0.000 description 1
- 230000028993 immune response Effects 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 210000004969 inflammatory cell Anatomy 0.000 description 1
- 230000002401 inhibitory effect Effects 0.000 description 1
- 230000000977 initiatory effect Effects 0.000 description 1
- 230000003993 interaction Effects 0.000 description 1
- 238000007918 intramuscular administration Methods 0.000 description 1
- 238000007912 intraperitoneal administration Methods 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 208000032839 leukemia Diseases 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 210000001165 lymph node Anatomy 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 210000004962 mammalian cell Anatomy 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 239000002207 metabolite Substances 0.000 description 1
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Natural products C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 1
- 239000004292 methyl p-hydroxybenzoate Substances 0.000 description 1
- 229960002216 methylparaben Drugs 0.000 description 1
- HPNSFSBZBAHARI-UHFFFAOYSA-N micophenolic acid Natural products OC1=C(CC=C(C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-UHFFFAOYSA-N 0.000 description 1
- 229940016286 microcrystalline cellulose Drugs 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000008108 microcrystalline cellulose Substances 0.000 description 1
- 150000007522 mineralic acids Chemical class 0.000 description 1
- 230000001483 mobilizing effect Effects 0.000 description 1
- 230000004048 modification Effects 0.000 description 1
- 238000012986 modification Methods 0.000 description 1
- 239000003068 molecular probe Substances 0.000 description 1
- 229910000402 monopotassium phosphate Inorganic materials 0.000 description 1
- 235000019796 monopotassium phosphate Nutrition 0.000 description 1
- 230000004899 motility Effects 0.000 description 1
- 229960000951 mycophenolic acid Drugs 0.000 description 1
- HPNSFSBZBAHARI-RUDMXATFSA-N mycophenolic acid Chemical compound OC1=C(C\C=C(/C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-RUDMXATFSA-N 0.000 description 1
- XUSROWNMXDDAOH-UHFFFAOYSA-N n'-hydroxy-2-(methylamino)pyridine-3-carboximidamide Chemical compound CNC1=NC=CC=C1C(=N)NO XUSROWNMXDDAOH-UHFFFAOYSA-N 0.000 description 1
- IDINUJSAMVOPCM-INIZCTEOSA-N n-[(1s)-2-[4-(3-aminopropylamino)butylamino]-1-hydroxy-2-oxoethyl]-7-(diaminomethylideneamino)heptanamide Chemical compound NCCCNCCCCNC(=O)[C@H](O)NC(=O)CCCCCCN=C(N)N IDINUJSAMVOPCM-INIZCTEOSA-N 0.000 description 1
- VRTQLSVABZCBTQ-UHFFFAOYSA-N n-methyl-3-[5-[4-(2,2,2-trifluoroethoxy)phenyl]-1,2,4-oxadiazol-3-yl]pyridin-2-amine Chemical compound CNC1=NC=CC=C1C1=NOC(C=2C=CC(OCC(F)(F)F)=CC=2)=N1 VRTQLSVABZCBTQ-UHFFFAOYSA-N 0.000 description 1
- XVAUTJSDUYZRII-UHFFFAOYSA-N n-methyl-3-[5-[5-(2-methylpropyl)pyridin-2-yl]-1,2,4-oxadiazol-3-yl]pyridin-2-amine Chemical compound CNC1=NC=CC=C1C1=NOC(C=2N=CC(CC(C)C)=CC=2)=N1 XVAUTJSDUYZRII-UHFFFAOYSA-N 0.000 description 1
- 125000001038 naphthoyl group Chemical group C1(=CC=CC2=CC=CC=C12)C(=O)* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 229940105631 nembutal Drugs 0.000 description 1
- 230000007694 nephrotoxicity Effects 0.000 description 1
- 231100000417 nephrotoxicity Toxicity 0.000 description 1
- 210000002241 neurite Anatomy 0.000 description 1
- 230000007135 neurotoxicity Effects 0.000 description 1
- 231100000228 neurotoxicity Toxicity 0.000 description 1
- 150000002825 nitriles Chemical class 0.000 description 1
- 239000012299 nitrogen atmosphere Substances 0.000 description 1
- 239000000041 non-steroidal anti-inflammatory agent Substances 0.000 description 1
- 229940021182 non-steroidal anti-inflammatory drug Drugs 0.000 description 1
- 230000009871 nonspecific binding Effects 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 230000003000 nontoxic effect Effects 0.000 description 1
- 238000000655 nuclear magnetic resonance spectrum Methods 0.000 description 1
- OQCDKBAXFALNLD-UHFFFAOYSA-N octadecanoic acid Natural products CCCCCCCC(C)CCCCCCCCC(O)=O OQCDKBAXFALNLD-UHFFFAOYSA-N 0.000 description 1
- 239000002751 oligonucleotide probe Substances 0.000 description 1
- 239000004006 olive oil Substances 0.000 description 1
- 235000008390 olive oil Nutrition 0.000 description 1
- 229940054534 ophthalmic solution Drugs 0.000 description 1
- 239000002997 ophthalmic solution Substances 0.000 description 1
- 229940100654 ophthalmic suspension Drugs 0.000 description 1
- 150000007524 organic acids Chemical class 0.000 description 1
- 239000012074 organic phase Substances 0.000 description 1
- 238000007427 paired t-test Methods 0.000 description 1
- 235000019629 palatability Nutrition 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- LXNAVEXFUKBNMK-UHFFFAOYSA-N palladium(II) acetate Substances [Pd].CC(O)=O.CC(O)=O LXNAVEXFUKBNMK-UHFFFAOYSA-N 0.000 description 1
- YJVFFLUZDVXJQI-UHFFFAOYSA-L palladium(ii) acetate Chemical compound [Pd+2].CC([O-])=O.CC([O-])=O YJVFFLUZDVXJQI-UHFFFAOYSA-L 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 230000008506 pathogenesis Effects 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- WEXRUCMBJFQVBZ-UHFFFAOYSA-N pentobarbital Chemical compound CCCC(C)C1(CC)C(=O)NC(=O)NC1=O WEXRUCMBJFQVBZ-UHFFFAOYSA-N 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 210000005105 peripheral blood lymphocyte Anatomy 0.000 description 1
- 210000001986 peyer's patch Anatomy 0.000 description 1
- 239000008177 pharmaceutical agent Substances 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 238000007747 plating Methods 0.000 description 1
- 229920000573 polyethylene Polymers 0.000 description 1
- 229920000656 polylysine Polymers 0.000 description 1
- 229920001184 polypeptide Polymers 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 235000011056 potassium acetate Nutrition 0.000 description 1
- 235000015497 potassium bicarbonate Nutrition 0.000 description 1
- 239000011736 potassium bicarbonate Substances 0.000 description 1
- 229910000028 potassium bicarbonate Inorganic materials 0.000 description 1
- GNSKLFRGEWLPPA-UHFFFAOYSA-M potassium dihydrogen phosphate Chemical compound [K+].OP(O)([O-])=O GNSKLFRGEWLPPA-UHFFFAOYSA-M 0.000 description 1
- 235000003270 potassium fluoride Nutrition 0.000 description 1
- TYJJADVDDVDEDZ-UHFFFAOYSA-M potassium hydrogencarbonate Chemical compound [K+].OC([O-])=O TYJJADVDDVDEDZ-UHFFFAOYSA-M 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- DBABZHXKTCFAPX-UHFFFAOYSA-N probenecid Chemical compound CCCN(CCC)S(=O)(=O)C1=CC=C(C(O)=O)C=C1 DBABZHXKTCFAPX-UHFFFAOYSA-N 0.000 description 1
- 102000004196 processed proteins & peptides Human genes 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- VVWRJUBEIPHGQF-UHFFFAOYSA-N propan-2-yl n-propan-2-yloxycarbonyliminocarbamate Chemical compound CC(C)OC(=O)N=NC(=O)OC(C)C VVWRJUBEIPHGQF-UHFFFAOYSA-N 0.000 description 1
- 239000003380 propellant Substances 0.000 description 1
- 125000001501 propionyl group Chemical group O=C([*])C([H])([H])C([H])([H])[H] 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 235000010232 propyl p-hydroxybenzoate Nutrition 0.000 description 1
- 239000004405 propyl p-hydroxybenzoate Substances 0.000 description 1
- 229960003415 propylparaben Drugs 0.000 description 1
- 239000003223 protective agent Substances 0.000 description 1
- OTVZGAXESBAAQQ-UHFFFAOYSA-N pyrazine-2,3-dicarbonitrile Chemical compound N#CC1=NC=CN=C1C#N OTVZGAXESBAAQQ-UHFFFAOYSA-N 0.000 description 1
- ZAHRKKWIAAJSAO-UHFFFAOYSA-N rapamycin Natural products COCC(O)C(=C/C(C)C(=O)CC(OC(=O)C1CCCCN1C(=O)C(=O)C2(O)OC(CC(OC)C(=CC=CC=CC(C)CC(C)C(=O)C)C)CCC2C)C(C)CC3CCC(O)C(C3)OC)C ZAHRKKWIAAJSAO-UHFFFAOYSA-N 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000001105 regulatory effect Effects 0.000 description 1
- 230000027425 release of sequestered calcium ion into cytosol Effects 0.000 description 1
- 238000007634 remodeling Methods 0.000 description 1
- 238000009877 rendering Methods 0.000 description 1
- 230000000241 respiratory effect Effects 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 238000006798 ring closing metathesis reaction Methods 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 150000003335 secondary amines Chemical class 0.000 description 1
- 210000005212 secondary lymphoid organ Anatomy 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- DUIOPKIIICUYRZ-UHFFFAOYSA-N semicarbazide Chemical compound NNC(N)=O DUIOPKIIICUYRZ-UHFFFAOYSA-N 0.000 description 1
- 230000019491 signal transduction Effects 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 230000007781 signaling event Effects 0.000 description 1
- 231100000161 signs of toxicity Toxicity 0.000 description 1
- XGVXKJKTISMIOW-ZDUSSCGKSA-N simurosertib Chemical compound N1N=CC(C=2SC=3C(=O)NC(=NC=3C=2)[C@H]2N3CCC(CC3)C2)=C1C XGVXKJKTISMIOW-ZDUSSCGKSA-N 0.000 description 1
- QFJCIRLUMZQUOT-HPLJOQBZSA-N sirolimus Chemical compound C1C[C@@H](O)[C@H](OC)C[C@@H]1C[C@@H](C)[C@H]1OC(=O)[C@@H]2CCCCN2C(=O)C(=O)[C@](O)(O2)[C@H](C)CC[C@H]2C[C@H](OC)/C(C)=C/C=C/C=C/[C@@H](C)C[C@@H](C)C(=O)[C@H](OC)[C@H](O)/C(C)=C/[C@@H](C)C(=O)C1 QFJCIRLUMZQUOT-HPLJOQBZSA-N 0.000 description 1
- 229960002930 sirolimus Drugs 0.000 description 1
- WXMKPNITSTVMEF-UHFFFAOYSA-M sodium benzoate Chemical compound [Na+].[O-]C(=O)C1=CC=CC=C1 WXMKPNITSTVMEF-UHFFFAOYSA-M 0.000 description 1
- 235000010234 sodium benzoate Nutrition 0.000 description 1
- 239000004299 sodium benzoate Substances 0.000 description 1
- 229910000029 sodium carbonate Inorganic materials 0.000 description 1
- 235000019812 sodium carboxymethyl cellulose Nutrition 0.000 description 1
- 229920001027 sodium carboxymethylcellulose Polymers 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 229910000104 sodium hydride Inorganic materials 0.000 description 1
- 235000010267 sodium hydrogen sulphite Nutrition 0.000 description 1
- 229910052938 sodium sulfate Inorganic materials 0.000 description 1
- 235000011152 sodium sulphate Nutrition 0.000 description 1
- 235000010265 sodium sulphite Nutrition 0.000 description 1
- 239000007909 solid dosage form Substances 0.000 description 1
- 239000012453 solvate Substances 0.000 description 1
- 239000000600 sorbitol Substances 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 150000003408 sphingolipids Chemical class 0.000 description 1
- 108010035597 sphingosine kinase Proteins 0.000 description 1
- 230000003393 splenic effect Effects 0.000 description 1
- 238000010561 standard procedure Methods 0.000 description 1
- 239000008117 stearic acid Substances 0.000 description 1
- 230000000707 stereoselective effect Effects 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 238000005556 structure-activity relationship Methods 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 125000004434 sulfur atom Chemical group 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 238000013268 sustained release Methods 0.000 description 1
- 239000012730 sustained-release form Substances 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 230000009885 systemic effect Effects 0.000 description 1
- UEUXEKPTXMALOB-UHFFFAOYSA-J tetrasodium;2-[2-[bis(carboxylatomethyl)amino]ethyl-(carboxylatomethyl)amino]acetate Chemical compound [Na+].[Na+].[Na+].[Na+].[O-]C(=O)CN(CC([O-])=O)CCN(CC([O-])=O)CC([O-])=O UEUXEKPTXMALOB-UHFFFAOYSA-J 0.000 description 1
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 231100000816 toxic dose Toxicity 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- IHIXIJGXTJIKRB-UHFFFAOYSA-N trisodium vanadate Chemical compound [Na+].[Na+].[Na+].[O-][V]([O-])([O-])=O IHIXIJGXTJIKRB-UHFFFAOYSA-N 0.000 description 1
- 125000003774 valeryl group Chemical group O=C([*])C([H])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- MWOOGOJBHIARFG-UHFFFAOYSA-N vanillin Chemical compound COC1=CC(C=O)=CC=C1O MWOOGOJBHIARFG-UHFFFAOYSA-N 0.000 description 1
- FGQOOHJZONJGDT-UHFFFAOYSA-N vanillin Natural products COC1=CC(O)=CC(C=O)=C1 FGQOOHJZONJGDT-UHFFFAOYSA-N 0.000 description 1
- 235000012141 vanillin Nutrition 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 230000002861 ventricular Effects 0.000 description 1
- 125000000391 vinyl group Chemical group [H]C([*])=C([H])[H] 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
- 239000008215 water for injection Substances 0.000 description 1
- 239000012138 yeast extract Substances 0.000 description 1
- GTJUPSNUGOBNMF-UHFFFAOYSA-M zinc;cyclopentane;bromide Chemical compound Br[Zn+].C1CC[CH-]C1 GTJUPSNUGOBNMF-UHFFFAOYSA-M 0.000 description 1
- DVAYVJGYZRYJEV-UHFFFAOYSA-M zinc;ethyl benzoate;iodide Chemical compound I[Zn+].CCOC(=O)C1=CC=[C-]C=C1 DVAYVJGYZRYJEV-UHFFFAOYSA-M 0.000 description 1
- DGVVWUTYPXICAM-UHFFFAOYSA-N β‐Mercaptoethanol Chemical compound OCCS DGVVWUTYPXICAM-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings
- C07D413/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/14—Drugs for dermatological disorders for baldness or alopecia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
- A61P27/12—Ophthalmic agents for cataracts
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/06—Antianaemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/04—Inotropic agents, i.e. stimulants of cardiac contraction; Drugs for heart failure
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D271/00—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms
- C07D271/02—Heterocyclic compounds containing five-membered rings having two nitrogen atoms and one oxygen atom as the only ring hetero atoms not condensed with other rings
- C07D271/06—1,2,4-Oxadiazoles; Hydrogenated 1,2,4-oxadiazoles
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D413/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms
- C07D413/14—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and oxygen atoms as the only ring hetero atoms containing three or more hetero rings
Definitions
- the present invention is related to compounds that are SlPi/Edgl receptor agonists and thus have immunosuppressive activities by modulating leukocyte trafficking, sequestering lymphocytes in secondary lymphoid tissues, and interfering with celkcell interactions required for an efficient immune response.
- the invention is also directed to pharmaceutical compositions containing such compounds and methods of treatment or prevention.
- Immunosuppressive agents have been shown to be useful in a wide variety of autoimmune and chronic inflammatory diseases, including systemic lupus erythematosis, chronic rheumatoid arthritis, type I diabetes mellitus, inflammatory bowel disease, biliary cinhosis, uveitis, multiple sclerosis and other disorders such as Crohn's disease, ulcerative colitis, bullous pemphigoid, sarcoidosis, psoriasis, autoimmune myositis, Wegener's granulomatosis, ichthyosis, Graves ophthalmopathy, atopic dermatitis and asthma.
- chemotherapeutic regimens for the treatment of cancers, lymphomas and leukemias.
- pathogenesis of each of these conditions may be quite different, they have in common the appearance of a variety of autoantibodies and/or self-reactive lymphocytes. Such self-reactivity may be due, in part, to a loss of the homeostatic controls under which the normal immune system operates.
- the host lymphocytes recognize the foreign tissue antigens and begin to produce both cellular and humoral responses including antibodies, cytokines and cytotoxic lymphocytes which lead to graft rejection.
- autoimmune or a rejection process tissue destruction caused by inflammatory cells and the mediators they release.
- Anti- inflammatory agents such as NSAIDs act principally by blocking the effect or secretion of these mediators but do nothing to modify the immunologic basis of the disease.
- cytotoxic agents such as cyclophosphamide, act in such a nonspecific fashion that both the normal and autoimmune responses are shut off. Indeed, patients treated with such nonspecific immunosuppressive agents are as likely to succumb to infection as they are to their autoimmune disease.
- Cyclosporin A is a drag used to prevent rejection of transplanted organs.
- FK-506 is another drug approved for the prevention of transplant organ rejection, and in particular, liver transplantation. Cyclosporin A and FK-506 act by inhibiting the body's immune system from mobilizing its vast arsenal of natural protecting agents to reject the transplant's foreign protein. Cyclosporin A was approved for the treatment of severe psoriasis and has been approved by European regulatory agencies for the treatment of atopic dermatitis. Though they are effective in delaying or suppressing transplant rejection, Cyclosporin A and FK-506 are known to cause several undesirable side effects including nephrotoxicity, neurotoxicity, and gastrointestinal discomfort. Therefore, an immunosuppressant without these side effects still remains to be developed and would be highly desirable.
- the immunosuppressive compound FTY720 is a lymphocyte sequestration agent cunently in clinical trials.
- FTY720 is metabolized in mammals to a compound that is a potent agonist of sphingosine 1 -phosphate receptors.
- Agonism of sphingosine 1 -phosphate receptors modulates leukocyte trafficking, induces the sequestration of lymphocytes (T-cells and B-cells) in lymph nodes and Peyer's patches without lymphodepletion, and disrapts splenic architecture, thereby interfering with T cell dependent and independent antibody responses.
- Such immunosuppression is desirable to prevent rejection after organ transplantation and in the treatment of autoimmune disorders.
- Sphingosine 1 -phosphate is a bioactive sphingolipid metabolite that is secreted by hematopoietic cells and stored and released from activated platelets.
- SIPi S1P2, SIP3, SIP4, and SIP5, also known as endothelial differentiation genes Edgl, Edg5, Edg3, Edg6, Edg8), that have widespread cellular and tissue distribution and are well conserved in human and rodent species (see Table). Binding to SIP receptors elicits signal transduction through Gq-, Gi/o, G12-, G13-, and Rho-dependent pathways. Ligand-induced activation of SIP 1 and SIP3 has been shown to promote angiogenesis, chemotaxis, and adherens junction assembly through Rac- and Rho-, see Lee, M.-J., S. Thangada, K.P.
- SIP5 is primarily a neuronal receptor with some expression in lymphoid tissue, see hn, D.S., C.E. Heise, N. Ancellin, B.F. O'Dowd, G.J. Shei, R.P. Heavens, M.R. Rigby, T. Hla, S. Mandala, G. McAllister, S. George, and K.R. Lynch. 2000. J. Biol. C em. 275:14281-6.
- Administration of sphingosine 1 -phosphate to animals induces systemic sequestration of peripheral blood lymphocytes into secondary lymphoid organs, thus resulting in therapeutically useful immunosuppression, see Mandala, S., R.
- sphingosine 1-phosphate also has cardiovascular and bronchoconstrictor effects that limit its utility as a therapeutic agent. Intravenous administration of sphingosine 1-phosphate decreases the heart rate, ventricular contraction and blood pressure in rats, see Sugiyama, A., N.N. Aye, Y. Yatomi, Y.
- sphingosine 1-phosphate modulates contraction, cell growth and cytokine production that promote bronchoconstriction, airway inflammation and remodeling in asthma, see Ammit, A.J., A.T. Hastie, L. C. Edsall, R.K. Hoffman, Y. Amrani, V.P. Krymskaya, S.A. Kane, S.P. Peters, R.B. Perm, S. Spiegel, RA. Panettieri. Jr. 2001, FASEB J. 15:1212-1214.
- the undesirable effects of sphingosine 1-phosphate are associated with its non-selective, potent agonist activity on all SIP receptors.
- the present invention encompasses compounds which are agonists of the SlPi/Edgl receptor having selectivity over the SlP3/Edg3 receptor.
- An SlPl/Edgl receptor selective agonist has advantages over cu ⁇ ent therapies and extends the therapeutic window of lymphocyte sequestration agents, allowing better tolerability with higher dosing and thus improving efficacy as monotherapy.
- immunosuppressants While the main use for immunosuppressants is in treating bone ma ⁇ ow, organ and transplant rejection, other uses for such compounds include the treatment of arthritis, in particular, rheumatoid arthritis, insulin and non-insulin dependent diabetes, multiple sclerosis, psoriasis, inflammatory bowel disease, Crohn's disease, lupus erythematosis and the like.
- the present invention is focused on providing immunosuppressant compounds that are safer and more effective than prior compounds.
- the present invention encompasses compounds of Formula I:
- the compounds are useful for treating immune mediated diseases and conditions, such as bone manow, organ and tissue transplant rejection.
- Pharmaceutical compositions and methods of use are included.
- A is C-R3 orN
- D is C-R orN
- E is C-R6 orN and
- G is C-R7 orN
- X, Y and Z are independently selected from the group consisting of: N and C-R8, with the proviso that at least one of X, Y and Z is not N;
- Rl and R2 are each independently selected from the group consisting of:
- Ci-6alkyl optionally substituted with 1 to 3 halo groups, or Rl and R2 may be joined together with the nitrogen atom to which they are attached to form a 3- to 6-membered saturated monocyclic ring;
- R3, R4, R6 and R7 are each independently selected from the group consisting of:
- Ci-4alkyl or Ci-4alkoxy each optionally substituted with 1 to
- R5 is selected from the group consisting of: (1) Ci-6alkyl,
- Ci-4alkyl or Ci-4alkoxy each optionally substituted with oxo, hydroxy or 1 to 3 halo groups,
- R4 and R5 may be joined together with the atoms to which they are attached to form a 5 or 6-membered monocyclic ring, optionally containing 1 to 3 heteratoms selected from O, S and NR8, said ring optionally substituted with 1 to 3 substituents independently selected from the group consisting of: halo, Ci-4alkyl and Ci-4alkoxy, said Ci-4alkyl or Ci-4alkoxy optionally substituted with 1 to 3 halo groups;
- each R8 is independently selected from the group consisting of: hydrogen, halo and Cl-4alkyl, wherein said Cl-4alkyl is optionally substituted with 1 to 3 halo groups;
- HET is selected from the group consisting of: benzimidazolyl, benzofuranyl, benzopyrazolyl, benzotriazolyl, benzothiophenyl, benzoxazolyl, carbazolyl, carbolinyl, cinnolinyl, furanyl, imidazolyl, indolinyl, indolyl, indolazinyl, indazolyl, isobenzofuranyl, isoindolyl, isoquinolyl, isothiazolyl, isoxazolyl, naphthyridinyl, oxadiazolyl, oxazolyl, pyrazinyl, pyrazolyl, pyridopyridinyl, pyridazinyl, pyridyl, pyrimidyl, py ⁇ olyl, quinazolinyl, quinolyl, quinoxalinyl, thiadiazolyl, thiazoly
- A is N
- E is C-R6 and
- G is C-R7.
- A is C-R3
- E is C-R6 and
- G is C-R7.
- Another embodiment of the invention encompasses a compound of Formula I wherein:
- A is C-R3
- E is C-R6 and
- G is C-R7;
- X, Y and Z are C-R8.
- A is C-R3
- E is C-R6 and
- G is C-R7;
- R3, R6 and R7 are hydrogen. Within this embodiment is encompassed a compound of Formula I wherein R is trifluoromethyl or cyano.
- A is C-R3
- D is C-R4
- E is C-R6
- G is C-R7;
- Rl and R2 are each independently selected from the group consisting of hydrogen, methyl and ethyl.
- A is C-R3
- E is C-R6 and
- G is C-R7;
- R5 is selected from the group consisting of:
- A is C-R3
- D is C-R4
- E is C-R6 and
- G is C-R7;
- R5 is selected from the group consisting of:
- phenyl optionally substituted with 1 to 3 substituents independently selected from the group consisting of: halo, methyl, methoxy and hydroxymethyl,
- A is C-R3
- E is C-R6 and
- G is C-R7;
- X is N and Y and Z are both C-R8.
- A is C-R3
- D is C-R4
- E is C-R6 and
- G is C-R7;
- A is C-R3
- E is C-R6 and
- G is C-R7;
- Rl and R2 are each independently selected from the group consisting of: hydrogen and methyl
- R3, R6 and R are hydrogen
- R4 is trifluoromethyl or cyano
- R5 is C2-6alkoxy, optionally substituted with 1 to 5 fluoro groups.
- R5 is selected from 2,2,2- trifluoroethoxy and 2,2,2-trifluoro-l-methylethoxy.
- R5 is selected from 2,2,2-trifluoroethoxy and 2,2,2-trifluoro-l-methylethoxy, X, Y and Z are C-R ⁇ and each R8 is independently selected from hydrogen, methyl and halo.
- R5 is selected from 2,2,2-trifluoroethoxy and 2,2,2-trifluoro-l-methylethoxy
- X is N and Y and Z are both C-R8 and each R8 is independently selected from hydrogen, methyl and halo.
- R5 is selected from 2,2,2-trifluoroethoxy and 2,2,2-trifluoro-l-methylethoxy
- X and Z are both C-R8 and Y is N and each Ro" is independently selected from hydrogen, methyl and halo.
- the invention also encompasses a method of treating an immunoregulatory abnormality in a mammalian patient in need of such treatment comprising administering to said patient a compound of Formula I in an amount that is effective for treating said immunoregulatory abnormality.
- the immunoregulatory abnormality is an autoimmune or chronic inflammatory disease selected from the group consisting of: systemic lupus erythematosis, chronic rheumatoid arthritis, type I diabetes mellitus, inflammatory bowel disease, biliary cinhosis, uveitis, multiple sclerosis, Crohn's disease, ulcerative colitis, bullous pemphigoid, sarcoidosis, psoriasis, autoimmune myositis, Wegener's granulomatosis, ichthyosis, Graves ophthalmopathy and asthma.
- an autoimmune or chronic inflammatory disease selected from the group consisting of: systemic lupus erythematosis, chronic rheumatoid arthritis, type I diabetes mellitus, inflammatory bowel disease, biliary cinhosis, uveitis, multiple sclerosis, Crohn's disease, ulcerative colitis, bullous pemphigoid
- the immunoregulatory abnormality is bone manow or organ transplant rejection or graft-versus-host disease.
- the immunoregulatory abnormality is selected from the group consisting of: transplantation of organs or tissue, graft-versus-host diseases brought about by transplantation, autoimmune syndromes including rheumatoid arthritis, systemic lupus erythematosus, Hashimoto's thyroiditis, multiple sclerosis, myasthenia gravis, type I diabetes, uveitis, posterior uveitis, allergic encephalomyelitis, glomerulonephritis, post-infectious autoimmune diseases including rheumatic fever and post-infectious glomerulonephritis, inflammatory and hyperproliferative skin diseases, psoriasis, atopic dermatitis, contact dermatitis, eczematous de ⁇ natitis, sebonhoeic de ⁇ natitis, lichen planus, pemphigus, bullous pemphigoid, epidermolysis bullosa,
- autoimmune syndromes including rheumato
- the invention also encompasses a method of suppressing the immune system in a mammalian patient in need of immunosuppression comprising administering to said patient an immunosuppressmg effective amount of a compound of Formula I.
- the invention also encompasses a pha ⁇ naceutical composition comprised of a compound of Formula I in combination with a pharmaceutically acceptable carrier.
- the invention also encompasses a method of treating a respiratory disease or condition in a mammalian patient in need of such treatment comprising administering to said patient a compound of Formula I in an amount that is effective for treating said respiratory disease or condition.
- the respiratory disease or condition is selected from the group consisting of: asthma, chronic bronchitis, chronic obstructive pulmonary disease, adult respiratory distress syndrome, infant respiratory distress syndrome, cough, eosinophilic granuloma, respiratory syncytial virus bronchiolitis, bronchiectasis, idiopathic pulmonary fibrosis, acute lung injury and bronchiolitis obliterans organizing pneumonia.
- halogen or halo includes F, CI, Br, and I.
- alkyl means linear or branched structures and combinations thereof, having the indicated number of carbon atoms.
- Ci-6alkyl includes methyl, ethyl, propyl, 2-propyl, s- and t-butyl, butyl, pentyl, hexyl, 1,1- dimethylethyl, cyclopropyl, cyclobutyl, cyclopentyl and cyclohexyl.
- alkoxy means alkoxy groups of a straight, branched or cyclic configuration having the indicated number of carbon atoms. Ci-6alkoxy, for example, includes methoxy, ethoxy, propoxy, isopropoxy, and the like.
- alkylthio means alkylthio groups having the indicated number of carbon atoms of a straight, branched or cyclic configuration.
- Ci- 6 a lkylthio for example, includes methylthio, propylthio, isopropylthio, and the like.
- alkenyl means linear or branched structures and combinations thereof, of the indicated number of carbon atoms, having at least one carbon-to-carbon double bond, wherein hydrogen may be replaced by an additional carbon-to-carbon double bond.
- C2-6alkenyl for example, includes ethenyl, propenyl, 1-methylethenyl, butenyl and the like.
- alkynyl means linear or branched structures and combinations thereof, of the indicated number of carbon atoms, having at least one carbon-to-carbon triple bond.
- C3_6alkynyl for example, includes , propenyl, 1- methylethenyl, butenyl and the like.
- cycloalkyl means mono-, bi- or tri-cyclic structures, optionally combined with linear or branched structures, having the indicated number of carbon atoms.
- cycloalkyl groups include cyclopropyl, cyclopentyl, cycloheptyl, adamantyl, cyclododecylmethyl, 2-ethyl-l- bicyclo[4.4.0]decyl, cyclobutylmethyl and the like.
- cycloalkoxy means cycloalkyl-O- wherein cycloalkyl is as defined above.
- cycloalkoxy includes cyclobutoxy.
- acyl means an alkyl group as defined above substituted at the 1 -position with oxo. Examples include formyl, acetyl, propionyl, butyryl, valeryl and hexanoyl.
- aryl is defined as a mono- or bi-cyclic aromatic ring system and includes, for example, phenyl, naphthyl, and the like.
- aralkyl means an alkyl group as defined above of 1 to 6 carbon atoms with an aryl group as defined above substituted for one of the alkyl hydrogen atoms, for example, benzyl and the like.
- aryloxy means an aryl group as defined above attached to a molecule by an oxygen atom (aryl-O) and includes, for example, phenoxy, naphthoxy and the like.
- aralkoxy means an aralkyl group as defined above attached to a molecule by an oxygen atom (aralkyl-O) and includes, for example, benzyloxy, and the like.
- arylthio is defined as an aryl group as defined above attached to a molecule by an sulfur atom (aryl-S) and includes, for example, thiophenyoxy, thionaphthoxy and the like.
- aroyl means an aryl group as defined above attached to a molecule by an carbonyl group (aryl-C(O)-) and includes, for example, benzoyl, naphthoyl and the like.
- aroyloxy means an aroyl group as defined above attached to a molecule by an oxygen atom (aroyl-O) and includes, for example, benzoyloxy or benzoxy, naphthoyloxy and the like.
- treating encompasses not only treating a patient to relieve the patient of the signs and symptoms of the disease or condition but also prophylactically treating an asymptomatic patient to prevent the onset or progression of the disease or condition.
- amount effective for treating is intended to mean that amount of a drag or pharmaceutical agent that will elicit the biological or medical response of a tissue, a system, animal or human that is being sought by a researcher, veterinarian, medical doctor or other clinician.
- amount of a pharmaceutical dmg that will prevent or reduce the risk of occunence of the biological or medical event that is sought to be prevented in a tissue, a system, animal or human by a researcher, veterinarian, medical doctor or other clinician.
- compositions described herein includes pharmaceutically acceptable salts and hydrates.
- Pharmaceutically acceptable salts include both the metallic (inorganic) salts and organic salts; a list of which is given in Remington's Pharmaceutical Sciences, 17th Edition, pg. 1418 (1985). It is well known to one skilled in the art that an appropriate salt form is chosen based on physical and chemical stability, flowability, hydroscopicity and solubility.
- pharmaceutically acceptable salts include, but are not limited to salts of inorganic acids such as hydrochloride, sulfate, phosphate, diphosphate, hydrobromide, and nitrate or salts of an organic acid such as malate, maleate, fumarate, tartiate, succinate, citrate, acetate, lactate, methanesulfonate, p- toluenesulfonate or pamoate, salicylate and stearate.
- pharmaceutically acceptable cations include, but are not limited to sodium, potassium, calcium, aluminum, lithium and ammonium (especially ammonium salts with secondary amines).
- Prefe ⁇ ed salts of this invention for the reasons cited above include potassium, sodium, calcium and ammonium salts.
- crystal forms, hydrates and solvates of the compounds of Formula I are crystal forms, hydrates and solvates of the compounds of Formula I.
- pharmaceutically acceptable hydrate means the compounds of the instant invention crystallized with one or more molecules of water to form a hydrated form.
- the invention also includes the compounds falling within Formula I in the form of one or more stereoisomers, in substantially pure form or in the form of a mixture of stereoisomers. All such isomers are encompassed within the present invention.
- the compounds of the present invention are immunoregulatory agents useful for treating or preventing automimmune or chronic inflammatory diseases.
- the compounds of the present invention are useful to suppress the immune system in instances where immunosuppression is in order, such as in bone manow, organ or transplant rejection, autoimmune and chronic inflammatory diseases, including systemic lupus erythematosis, chronic rheumatoid arthritis, type I diabetes mellitus, inflammatory bowel disease, biliary cinhosis, uveitis, multiple sclerosis, Crohn's disease, ulcerative colitis, bullous pemphigoid, sarcoidosis, psoriasis, autoimmune myositis, Wegener's granulomatosis, ichthyosis, Graves ophthalmopathy and asthma.
- autoimmune and chronic inflammatory diseases including systemic lupus erythematosis, chronic rheumatoid arthritis, type I diabetes mellitus, inflammatory bowel disease, biliary cinhosis, uveitis, multiple sclerosis, Crohn's disease,
- the compounds of the present invention are useful to treat or prevent a disease or disorder selected from the group consisting of: transplantation of organs or tissue, graft-versus-host diseases brought about by transplantation, autoimmune syndromes including rheumatoid arthritis, systemic lupus erythematosus, Hashimoto's thyroiditis, multiple sclerosis, myasthenia gravis, type I diabetes, uveitis, posterior uveitis, allergic encephalomyelitis, glomeralonephritis, post-infectious autoimmune diseases including rheumatic fever and post-infectious glomeralonephritis, inflammatory and hyperproliferative skin diseases, psoriasis, atopic dermatitis, contact dermatitis, eczematous dermatitis, sebo ⁇ hoeic de ⁇ natitis, lichen planus, pemphigus, bullous pemphigoid, epidermolysis bullosa,
- the compounds of the present invention are also useful for treating or preventing Alzheimer's Disease.
- Also embodied within the present invention is a method of preventing or treating resistance to transplantation or transplantation rejection of organs or tissues in a mammalian patient in need thereof, which comprises administering a therapeutically effective amount of the compound of Formula I.
- a method of suppressing the immune system in a mammalian patient in need thereof, which comprises administering to the patient an immune system suppressing amount of the compound of Formula I is yet another embodiment.
- the method described herein encompasses a method of treating or preventing bone manow or organ transplant rejection which is comprised of admininstering to a mammalian patient in need of such treatment or prevention a compound of Formula I, or a pharmaceutically acceptable salt or hydrate thereof, in an amount that is effective for treating or preventing bone manow or organ transplant rejection.
- the compounds of the present invention are also useful for treating a respiratory dieases or condition, such as asthma, chronic bronchitis, chronic obstructive pulmonary disease, adult respiratory distress syndrome, infant respiratory distress syndrome, cough, eosinophilic granuloma, respiratory syncytial virus bronchiolitis, bronchiectasis, idiopathic pulmonary fibrosis, acute lung injury and bronchiolitis obliterans organizing pneumonia
- a respiratory dieases or condition such as asthma, chronic bronchitis, chronic obstructive pulmonary disease, adult respiratory distress syndrome, infant respiratory distress syndrome, cough, eosinophilic granuloma, respiratory syncytial virus bronchiolitis, bronchiectasis, idiopathic pulmonary fibrosis, acute lung injury and bronchiolitis obliterans organizing pneumonia
- the compounds of the present invention are selective agonists of the SlPi/Edgl receptor having selectivity over SlP3/Edg3 receptor.
- An Edgl selective agonist has advantages over cu ⁇ ent therapies and extends the therapeutic window of lymphocytes sequestration agents, allowing better tolerability with higher dosing and thus improving efficacy as monotherapy.
- the present invention also includes a pharmaceutical formulation comprising a pharmaceutically acceptable carrier and the compound of Formula I or a pharmaceutically acceptable salt or hydrate thereof.
- a prefe ⁇ ed embodiment of the formulation is one where a second immunosuppressive agent is also included.
- second immunosuppressive agents are, but are not limited to azathioprine, brequinar sodium, deoxyspergualin, mizaribine, mycophenolic acid mo ⁇ holino ester, cyclosporin, FK-506, rapamycin, FTY720 and ISAtx247 (Isotechnika).
- the present compounds are useful in the treatment of autoimmune diseases, including the prevention of rejection of bone manow transplant, foreign organ transplants and/or related afflictions, diseases and illnesses.
- the compounds of this invention can be administered by any means that effects contact of the active ingredient compound with the site of action in the body of a warm-blooded animal.
- administration can be oral, topical, including tiansdermal, ocular, buccal, intianasal, inhalation, intravaginal, rectal, intracisternal and parenteral.
- parenteral refers to modes of administration which include subcutaneous, intravenous, intramuscular, intraarticular injection or infusion, intrasternal and intraperitoneal.
- the compounds can be administered by any conventional means available for use in conjunction with pharmaceuticals, either as individual therapeutic agents or in a combination of therapeutic agents. They can be administered alone, but are generally administered with a pharmaceutical carrier selected on the basis of the chosen route of administration and standard pharmaceutical practice.
- the dosage administered will be dependent on the age, health and weight of the recipient, the extent of disease, kind of concu ⁇ ent treatment, if any, frequency of tieatment and the nature of the effect desired.
- a daily dosage of active ingredient compound will be from about 0.1 -2000 milligrams per day.
- the active ingredient can be administered orally in solid dosage forms, such as capsules, tablets, troches, dragees, granules and powders, or in liquid dosage forms, such as elixirs, syrups, emulsions, dispersions, and suspensions.
- the active ingredient can also be administered parenterally, in sterile liquid dosage forms, such as dispersions, suspensions or solutions.
- dosages forms that can also be used to administer the active ingredient as an ointment, cream, drops, tiansdermal patch or powder for topical administration, as an ophthalmic solution or suspension formation, i.e., eye drops, for ocular administration, as an aerosol spray or powder composition for inhalation or intranasal administration, or as a cream, ointment, spray or suppository for rectal or vaginal administration.
- Gelatin capsules contain the active ingredient and powdered carriers, such as lactose, starch, cellulose derivatives, magnesium stearate, stearic acid, and the like. Similar diluents can be used to make compressed tablets. Both tablets and capsules can be manufactured as sustained release products to provide for continuous release of medication over a period of hours. Compressed tablets can be sugar coated or film coated to mask any unpleasant taste and protect the tablet from the atmosphere, or enteric coated for selective disintegration in the gastrointestinal tract. Liquid dosage forms for oral administration can contain coloring and flavoring to increase patient acceptance.
- water a suitable oil, saline, aqueous dextrose (glucose), and related sugar solutions and glycols such as propylene glycol or polyethylene gycols are suitable carriers for parenteral solutions.
- Solutions for parenteral administration preferably contain a water soluble salt of the active ingredient, suitable stabilizing agents, and if necessary, buffer substances.
- Antioxidizing agents such as sodium bisulfite, sodium sulfite, or ascorbic acid, either alone or combined, are suitable stabilizing agents.
- citric acid and its salts and sodium EDTA are also used.
- parenteral solutions can contain preservatives, such as benzalkonium chloride, methyl- or propylparaben, and chlorobutanol.
- Suitable pharmaceutical carriers are described in Remington's Pharmaceutical Sciences, A. Osol, a standard reference text in this field.
- the compounds of the present invention may be conveniently delivered in the form of an aerosol spray presentation from pressurized packs or nebulisers.
- the compounds may also be delivered as powders which may be formulated and the powder composition may be inhaled with the aid of an insufflation powder inhaler device.
- the prefened delivery system for inhalation is a metered dose inhalation (MDI) aerosol, which may be formulated as a suspension or solution of a compound of Formula I in suitable propellants, such as fluorocarbons or hydrocarbons.
- MDI metered dose inhalation
- an ophthalmic preparation may be formulated with an appropriate weight percent solution or suspension of the compounds of Formula I in an appropriate ophthalmic vehicle, such that the compound is maintained in contact with the ocular surface for a sufficient time period to allow the compound to penetrate the comeal and internal regions of the eye.
- a large number of unit capsules are prepared by filling standard two- piece hard gelatin capsules each with 100 milligrams of powdered active ingredient, 150 milligrams of lactose, 50 milligrams of cellulose, and 6 milligrams magnesium stearate.
- SOFT GELATIN CAPSULES 100 milligrams of powdered active ingredient, 150 milligrams of lactose, 50 milligrams of cellulose, and 6 milligrams magnesium stearate.
- a mixture of active ingredient in a digestible oil such as soybean oil, cottonseed oil or olive oil is prepared and injected by means of a positive displacement pump into gelatin to form soft gelatin capsules containing 100 milligrams of the active ingredient.
- the capsules are washed and dried.
- a large number of tablets are prepared by conventional procedures so that the dosage unit is 100 milligrams of active ingredient, 0.2 milligrams of colloidal silicon dioxide, 5 milligrams of magnesium stearate, 275 milligrams of microcrystalline cellulose, 11 milligrams of starch and 98.8 milligrams of lactose.
- Appropriate coatings may be applied to increase palatability or delay absorption.
- a parenteral composition suitable for administration by injection is prepared by stirring 1.5% by weight of active ingredient in 10% by volume propylene glycol. The solution is made to volume with water for injection and sterilized.
- aqueous suspension is prepared for oral administration so that each 5 milliliters contain 100 milligrams of finely divided active ingredient, 100 milligrams of sodium carboxymethyl cellulose, 5 milligrams of sodium benzoate, 1.0 grams of sorbitol solution, U.S.P., and 0.025 milliliters of vanillin.
- the same dosage forms can generally be used when the compounds of this invention are administered stepwise or in conjunction with another therapeutic agent.
- the dosage form and administration route should be selected depending on the compatibility of the combined drugs.
- coadministration is understood to include the administration of the two agents concomitantly or sequentially, or alternatively as a fixed dose combination of the two active components.
- Carboxylic acid ii can be activated for acylation with a reagent such as N,N'-dicyclohexylcarbodiimide, l-(3- dimethylaminopropyl)-3-ethylcarbodiimide, 1,1 '-carbonyldiimidazole, or bis(2-oxo-3- oxazolidinyl)phosphinic chloride in the presence of a suitable base (if necessary) such as triethylamine, N,N-diisopropylethylamine, or sodium bicarbonate in a solvent such as 1,2-dichloroethane, toluene, xylenes, N,N-dimethylformamide or N-methyl pynolidinone.
- a suitable base if necessary
- a 2-(amino)aryl N-hydroxyamidine of general structure iii can then be added which results in the formation of an acyl N-hydroxyamidine iv.
- This intermediate can be isolated using methods known to those skilled in the art (e.g., crystallization, silica gel chromatography, HPLC) and in a subsequent step, cyclized/dehydrated by warming iv in a suitable solvent (e.g., 1,2-dichloroethane, toluene, xylenes, N,N-dimethylformamide or N-methyl pynolidinone) to give a 1,2,4- oxadiazole of structure i.
- a suitable solvent e.g., 1,2-dichloroethane, toluene, xylenes, N,N-dimethylformamide or N-methyl pynolidinone
- Conversion of iii to iv may require added base, in which case reagents such as pyridine, N,N-diisopropylethylarnine or tetrabutylammonium fluoride can be used. It may be more convenient or desirable to not isolate N- hydroxyamidine iv, in which case the teansformation of ii to i can be carried out as a continuous process.
- reagents such as pyridine, N,N-diisopropylethylarnine or tetrabutylammonium fluoride
- acylating agents other than activated carboxylic acid ii it possible to use acylating agents other than activated carboxylic acid ii to give compounds i.
- a carboxylic acid chloride, carboxylic acid anhydride, carboxamide or carbonitrile in the place of carboxylic acid ii and an acyl activating agent to prepare 1,2,4-oxadiazole compounds i as described above.
- Methods to prepare 1,2,4-oxadiazoles using these other acylating agents as well as other methods pertinent to the present invention are known to those skilled in the art and have been reviewed in the literature (see, Clapp, L.B., "1,2,3- and 1,2,4-Oxadiazoles", pp. 366-91 in Comprehensive Heterocyclic Chemistry, Volume 6, Potts, K. T., Editor, Pergamon Press, 1984).
- a second method that can be used to prepare the compounds of the general stracture i in the present invention is shown in Scheme 2.
- Carboxylic acid ii is activated as described in Scheme 1 and used to acylate a 2-(substituted)aryl N- hydroxyamidine v in which the functional group X is a leaving group such as fluoro, chloro, bromo, iodo, cyano, alkylsulfonyloxy or arylsulfonyloxy.
- Conversion to compound iv and ring closure to give i is effected using the methods described above.
- Displacement of the leaving group X is carried out by treating vi with ammonia, an alkylamine or a dialkylamine in a suitable solvent (e.g., methanol, ethanol, N,N- dimethylformamide, dimethylsulfoxide) at or above ambient temperature to give 1,2,4-oxadiazole i.
- a suitable solvent e.g., methanol, ethanol, N,N- dimethylformamide, dimethylsulfoxide
- vi can be treated with N-methylformamide in the presence of diethanolamine at elevated temperature to give compounds i, where -NR1R2 is - NHCH3.
- R3-R8 can be manipulated in compounds i. Examples of this include (but are not limited to): 1) if one or more of R3-R8 is -OH, treatment of i with an alkyl halide or alkyl sulfonate ester in the presence of an appropriate base (e.g., N,N-diisopropylethylamine, triethylamine, pyridine, sodium carbonate) in a suitable solvent (methylene chloride, acetonitrile, toluene, N,N-dimethylformamide) at or above ambient temperature can give compounds i in which one or more of R3-R8 is alkoxy; 2) if one or more of R3- R8 is -CI, -Br, -I, or -OSO2CF3, treatment of i with an aryl boronic acid and a suitable base (sodium hydroxide, potassium bicarbonate) in the presence of
- any of groups R1-R8 may have asymmetric centers, in which case the individual stereoisomers of i can obtained by methods known to those skilled in the art which include (but are not limited to): stereospecific synthesis, resolution of salts of i or any of the intermediates used in its preparation with enantiopure acids or bases, resolution of i or any of the intermediates used in its preparation by HPLC employing enantiopure stationary phases.
- N-HYDROXYAMIDINE 1 2-Chloro-N-hvdroxy-nicotinamidine A mixture of 2-chloro-3-pyridine-carbonitrile (5.00 g, 37 mmol), hydroxylamine hydrochloride (3.73 g, 54 mmol) and sodium bicarbonate (9.10 g, 108 mmol) were sti ⁇ ed together in CH3OH (250 ml) at 50°C for 16 h.
- N-HYDROXYAMIDINEs 2-6 The following N-HYDROXYAMIDINE intermediates were prepared using a procedure analogous to that described for N-HYDROXYAMIDINE 1 substituting the appropriate nitrile for 2-chloro-3-pyridine-carbonitrile.
- N-HYDROXYAMIDINE 7 2-(N-Methylamino)-N-hydroxy-nicotinamidine A mixture of 10 g (72 mmol) 2-chloro-3-pyridine-carbonitrile, 40 mL of 40% methylamine in H2O and 20 mL of iPrOH was sti ⁇ ed at 55 °C for 1.5 h. Aqueous hydroxylamine (6.0 mL, 50 wt. % in H2O) was added and the resulting mixture was sti ⁇ ed at 55 °C for 1 h. The solution was cooled to rt.
- N-HYDROXYAMIDINE 8 2-(Amino)-N-hydroxy-nicotinamidine
- N-HYDROXYAMIDINE 9 3-(N-Methylamino)-pyrazine-2-(N-hydroxyamidine) Step A: 2-(N-Methylamino)-3-cyanopyrazine
- Step B 3-(N-Methylamino)-pyrazine-2-(N-hydroxyamidine)
- N-HYDROXYAMIDINE 10 2-(N-Methylamino)-5-fluoro-N-hvdroxynicotinamidine Step A: 2,6-Dichloro-5-fluoronicotinamide
- Step B 2-Chloro-5-fluoronicotinamide Under a N 2 atmosphere, 2,6-dichloro-5-fluoronicotinamide (500 mg,
- Step C 4-(Cyclobutyldifluoromethyl)benzoic acid A solution of360 mg (1.4 mmol) of ethyl 4-
- CARBOXYLIC ACID 7 3 Trifluoromethyl-4-(2-(S)-butoxy)benzoic acid
- Step A 3-Trifluoromethyl-4-(2-(S)-butoxy)benzonitrile
- a solution of 1.1 g (5.9 mmol) of 4-fluoro-3 - trifluoromethylbenzonitrile and 485 mg (6.5 mmol) of (S)-(+)-2-butanol in 10 mL of THF at-10°C was treated with 235 mg (5.9 mmol) of sodium hydride. The resulting mixture was stined cold for 2 h, then quenched with 10 mL of H2O.
- Step B 3-Trifluoromethyl-4-(l-(S)-methyl-2,2,2-trifluoroethoxy)benzoic acid
- the title compound was prepared using procedures analogous to those described for CARBOXYLIC ACID 7 substituting 1 -(S)-mefhyl-2,2,2-trifluoroethanol (from Step A) for (S)-2-butanol in CARBOXYLIC ACID 7, Step A.
- the enantiomeric purity of the title compound was determined by converting it to the conesponding methyl ester (excess 2.0 M trimethylsilyldiazomethane solution in cyclohexane, THF/MeOH, 5 min) and assaying by HPLC.
- Step B 4-(2-(S)-Butoxy-2-fluoro-benzoic acid
- Step B 5-(2-Methyl-l-oxopropyl)-2-bromopyridine
- Step C 5-(2-Methyl-l-oxopropyl)pyridine-2-carbonitrile
- CARBOXYLIC ACID 22 5-( 1 , 1 -Difluoro-2-methylpropyl pyridine-2-carboxylic acid
- Step B (S)-3-(4-Bromophenyl)-l,l-difluorocyclopentane
- Step B 3-(2-(N-Methylamino)pyridin-3-yl)-5-(4-(2-methylpropyl)phenyl)-l,2,4- oxadiazole
- EXAMPLES 2-9 The following were prepared using procedures analogous to those described in EXAMPLE 1 substituting 4-(cyclohexyl)benzoic acid for 4-(2- methylpropyl)benzoic acid and the appropriate N-HYDROXYAMIDINE for N- HYDROXYAMIDINE 1 in Step A and the appropriate amine for N- methylformamide in Step B.
- EXAMPLE 19 3 -(2-(N-Methylamino)pyridin-3 -yl -5-(4-(2.2-difluoropropyl phenvD- 1.2.4- oxadiazole
- EXAMPLES 20-46 The following were prepared using procedures analogous to those described in EXAMPLE 19 substituting the appropriate CARBOXYLIC ACID for 4-(2,2- difluoropropyl) benzoic acid and the appropriate N-HYDROXYAMIDINE for N- HYDROXYAMIDINE 1.
- Step B 3-(2-(Benzotriazol-l-yloxy) ⁇ yridin-3-yl)-5-(5-(2- methylpropyl)pyridin-2-yl)- 1 ,2,4-oxadiazole
- Step C 3-(2-(N-Methylamino)pyridin-3-yl)-5-(5-(2-methylpropyl)pyridin-2- yl)- 1,2,4-oxadiazole
- Step A 3-(2-(Chloro)pyridin-3-yl)-5-(4-hydroxyphenyl)- 1 ,2,4-oxadiazole
- Step B 3-(2-(Chloro)pyridin-3-yl)-5-(4-(2,2,2-trifluoroethoxy)phenyl)-l,2,4- oxadiazole
- Step C 3-(2-(N-Methylamino)pyridin-3-yl)-5-(4-(2,2,2-trifluoroethoxy)phenyl)- 1,2,4-oxadiazole
- Step B 3-(2-(N-Methylamino)pyridin-3-yl)-5-(4-(2-fluoro-l-fluoromethyl)ethoxy-3- trifluoromethylphenyl) - 1 ,2,4-oxadiazole
- EXAMPLES 73-80 The following were prepared using procedures analogous to those described in EXAMPLE 19 substituting the appropriate N-HYDROXYAMIDINE for N-HYDROXYAMIDINE 1 and the appropriate CARBOXYLIC ACID for 4-(2,2- difluoropropyl)benzoic acid.
- EXAMPLES 88-90 The following were prepared using procedures analogous to those described in EXAMPLE 19 substituting the appropriate N-HYDROXYAMIDINE for N-HYDROXYAMIDINE l and the appropriate CARBOXYLIC ACID for 4-(2,2- difluoropropyl)benzoic acid.
- HYDROXYAMIDINE 1 and the appropriate CARBOXYLIC ACID for 4-(2,2- difluoropropyl)benzoic acid.
- EXAMPLES 103-106 The following were prepared using procedures analogous to those described in EXAMPLE 19 substituting the appropriate N-HYDROXYAMIDINE for N-HYDROXYAMIDINE 1 and the appropriate CARBOXYLIC ACID for 4-(2,2- difluoropropyl)benzoic acid.
- EXAMPLE 107 3-(2-Amino-5-fluoropyridin-3-yl)-5-(3-trifluoromethyl-4-(l,l.l-trifluoro-2-(S)- propyloxy))phenyl)- 1 ,2,4-oxadiazole
- EDC-HC1 34 mg, 0.176 mmol
- the resultant solution was added to a mixture of N-HYDROXYAMIDINE 11 and acetonifrile (1.0 mL) in a sealed tube and heated to 40 °C.
- EXAMPLE 108 3-(2-(TS-Methylamino)-5-fluoropyridin-3-yl)-5-(3-trifluoromethyl-4-(l,l,l-trifluoro- 2-(S)-propyloxy) phenyl)- 1 ,2,4-oxadiazole
- the title compound was prepared using a procedure analogous to that described for EXAMPLE 107 substituting N-HYDROXYAMIDINE 10 for N-
- SlPi/Edgl, SlP3,/Edg3, SlP2/Edg5, SlP4/Edg6 or SIP5 /Edg8 activity of the compounds of the present invention can be evaluated using the following assays:
- Reaction products were extracted with butanol and 33p_ S phingosine-l- phosphate was purified by HPLC.
- Cells expressing EDG/S1P receptors were harvested with enzyme-free dissociation solution (Specialty Media, Lavallette, NJ). They were washed once in cold PBS and suspended in binding assay buffer consisting of 50 mM HEPES-Na, pH 7.5, 5mM MgCl2, lmM CaCl2, and 0.5% fatty acid-free BSA.
- 33p- sp hfngosine-l- phosphate was sonicated with 0.1 nM sphingosine- 1-phosphate in binding assay buffer; 100 ⁇ l of the ligand mixture was added to 100 ⁇ l cells (1 x 106 cells/ml) in a 96 well microtiter dish. Binding was performed for 60 min at room temperature with gentle mixing. Cells were then collected onto GF/B filter plates with a Packard Filtermate Universal Harvester. After drying the filter plates for 30 min, 40 ⁇ l of Microscint 20 was added to each well and binding was measured on a Wallac
- Non-specific binding was defined as the amount of radioactivity remaining in the presence of 0.5 ⁇ M cold sphingosine- 1-phosphate.
- ligand binding assays were performed on membranes prepared from cells expressing Edg/SIP receptors.
- Cells were harvested with enzyme-free dissociation solution and washed once in cold PBS.
- Cells were disrupted by homogenization in ice cold 20 mM HEPES pH 7.4, 10 mM EDTA using a Kinematica polytron (setting 5, for 10 seconds).
- Homogenates were centrifuged at 48,000 x g for 15 min at 40C and the pellet was suspended in 20 mM HEPES pH 7.4, 0.1 mM EDTA.
- the final pellet was suspended in 20 mM HEPES pH 7.4, 100 mM NaCl, 10 mM MgCl2- Ligand binding assays were performed as described above, using 0.5 to 2 ⁇ g of membrane protein.
- Agonists and antagonists of Edg/SIP receptors can be identified in the 33p-sphingosine- 1-phosphate binding assay.
- Compounds diluted in DMSO, methanol, or other solvent, were mixed with probe containing 33p_sphingosine-l- phosphate and binding assay buffer in microtiter dishes.
- Membranes prepared from cells expressing Edg/SIP receptors were added, and binding to 3p_sphingosine-l- phosphate was performed as described. Determination of the amount of binding in the presence of varying concentrations of compound and analysis of the data by nonlinear regression software such as MRLCalc (Merck Research Laboratories) or PRISM (GraphPad Software) was used to measure the affinity of compounds for the receptor.
- Selectivity of compounds for Edg/SIP receptors was determined by measuring the level of 33p_sphingosine- 1-phosphate binding in the presence of the compound using membranes prepared from cells transfected with each respective receptor (SlPi/Edgl, SlP3/Edg3, SlP2/Edg5, SlP4/Edg6, SlPs/Edg8).
- Agonists and antagonists of SlP/Edg receptors can be discriminated in the 35s-GTP ⁇ S binding assay.
- Compounds diluted in DMSO, methanol, or other solvent, were added to microtiter dishes to provide final assay concentrations of 0.01 nM to 10 ⁇ M.
- Membranes prepared from cells expressing SlP/Edg receptors were added, and binding to 5s-GTP ⁇ S was performed as described. When assayed in the absence of the natural ligand or other known agonist, compounds that stimulate 35 s- GTP ⁇ S binding above the endogenous level were considered agonists, while compounds that inhibit the endogenous level of 35s-GTP ⁇ S binding were considered inverse agonists.
- Antagonists were detected in a 35s-GTP ⁇ S binding assay in the presence of a sub-maximal level of natural ligand or known SlP/Edg receptor agonist, where the compounds reduced the level of 35s-GTP ⁇ S binding. Determination of the amount of binding in the presence of varying concentrations of compound was used to measure the potency of compounds as agonists, inverse agonists, or antagonists of SlP/Edg receptors. To evaluate agonists, percent stimulation over basal was calculated as binding in the presence of compound divided by binding in the absence of ligand, multiplied by 100.
- Dose response curves were plotted using a non-linear regression curve fitting program MRLCalc (Merck Research Laboratories), and EC50 values were defined to be the concentration of agonist required to give 50% of its own maximal stimulation.
- Selectivity of compounds for SlP/Edg receptors was determined by measuring the level of 35S-GTP ⁇ S binding in the presence of compound using membranes prepared from cells transfected with each respective receptor.
- a 96-well ligand plate was prepared by diluting sphingosine- 1- phosphate or other agonists into 200 ⁇ l of assay buffer to give a concentration that was 2-fold the final test concentration.
- the ligand plate and the cell plate were loaded into the FLIPR instrument for analysis. Plates were equilibrated to 37°C.
- the assay was initiated by transfening an equal volume of ligand to the cell plate and the calcium flux was recorded over a 3 min interval. Cellular response was quantitated as area (sum) or maximal peak height (max).
- Antagonists were evaluated in the absence of natural ligand by dilution of compounds into the appropriate solvent and transfer to the Fluo-4 labeled cells.
- Antagonists were evaluated by pretreating Fluo-4 labeled cells with varying concentrations of compounds for 15 min prior to the initiation of calcium flux by addition of the natural ligand or other SlP/Edg receptor agonist.
- SlPi/Edgl SlP3/Edg3, SlP2/Edg5, SlP4/Edg6 or SlP5/Edg8.
- RACE PCR cloning technique Frohman, et al., 1988, Proc. Natl. Acad. Sci. USA 85: 8998-9002.
- 5' and/or 3' RACE may be performed to generate a full-length cDNA sequence; (2) direct functional expression of the Edg/SIP cDNA following the construction of an SlP/Edg-containing cDNA library in an appropriate expression vector system; (3) screening an SlP/Edg-containing cDNA library constructed in a bacteriophage or plasmid shuttle vector with a labeled degenerate oligonucleotide probe designed from the amino acid sequence of the SlP/Edg protein; (4) screening an SlP/Edg-containing cDNA library constructed in a bacteriophage or plasmid shuttle vector with a partial cDNA encoding the SlP/Edg protein.
- This partial cDNA is obtained by the specific PCR amplification of SlP/Edg DNA fragments through the design of degenerate oligonucleotide primers from the amino acid sequence known for other proteins which are related to the SlP/Edg protein; (5) screening an SlP/Edg-containing cDNA library constructed in a bacteriophage or plasmid shuttle vector with a partial cDNA or oligonucleotide with homology to a mammalian SlP/Edg protein.
- This strategy may also involve using gene-specific oligonucleotide primers for PCR amplification of SlP/Edg cDNA; or (6) designing 5' and 3' gene specific oligonucleotides using the SlP/Edg nucleotide sequence as a template so that either the full-length cDNA may be generated by known RACE techniques, or a portion of the coding region may be generated by these same known RACE techniques to generate and isolate a portion of the coding region to use as a probe to screen one of numerous types of cDNA and/or genomic libraries in order to isolate a full-length version of the nucleotide sequence encoding SlP/Edg.
- libraries as well as libraries constructed from other cell types-or species types, may be useful for isolating an SlP/Edg-encoding DNA or an SlP/Edg homologue.
- Other types of libraries include, but are not limited to, cDNA libraries derived from other cells.
- suitable cDNA libraries may be prepared from cells or cell lines which have SlP/Edg activity.
- the selection of cells or cell lines for use in preparing a cDNA library to isolate a cDNA encoding SlP/Edg may be done by first measuring cell-associated SlP/Edg activity using any known assay available for such a purpose.
- cDNA libraries can be performed by standard techniques well known in the art. Well known cDNA library construction techniques can be found for example, in Sambrook et al., 1989, Molecular Cloning: A Laboratory Manual; Cold Spring Harbor Laboratory, Cold Spring Harbor, New York. Complementary DNA libraries may also be obtained from numerous commercial sources, including but not limited to Clontech Laboratories, Inc. and Stratagene.
- An expression vector containing DNA encoding an SlP/Edg-like protein may be used for expression of S lP/Edg in a recombinant host cell.
- a recombinant host cell can be cultured under suitable conditions to produce SlP/Edg or a biologically equivalent form.
- Expression vectors may include, but are not limited to, cloning vectors, modified cloning vectors, specifically designed plasmids or viruses. Commercially available mammalian expression vectors may be suitable for recombinant S 1 P/Edg expression.
- Recombinant host cells may be prokaryotic or eukaryotic, including but not limited to, bacteria such as E. coli, fungal cells such as yeast, mammalian cells including, but not limited to, cell lines of bovine, porcine, monkey and rodent origin; and insect cells including but not limited to Drosophila and silkworm derived cell lines.
- bacteria such as E. coli
- fungal cells such as yeast
- mammalian cells including, but not limited to, cell lines of bovine, porcine, monkey and rodent origin
- insect cells including but not limited to Drosophila and silkworm derived cell lines.
- SlP/Edg receptors The nucleotide sequences for the various SlP/Edg receptors are known in the art. See, for example, the following: SlPj/Edgl Human
- EDG6 a novel G-protein- coupled receptor related to receptors for bioactive lysophospholipids, is specifically expressed in lymphoid tissue. Genomics 53: 164-169, hereby incorporated by reference in its entirety. WO 98/48016, published October 29, 1998, hereby incorporated by reference in its entirety.
- Rats were instrumented with femoral arterial and venous catheters for measurement of arterial pressure and intravenous compound administration, respectively. Animals were anesthetized with Nembutal (55 mg/kg, ip). Blood pressure and heart rate were recorded on the Gould Po-Ne-Mah data acquisition system. Heart rate was derived from the arterial pulse wave. Following an acclimation period, a baseline reading was taken (approximately 20 minutes) and the data averaged. Compound was administered intravenously (either bolus injection of approximately 5 seconds or infusion of 15 minutes duration), and data were recorded every 1 minute for 60 minutes post compound administration.
- Nembutal 55 mg/kg, ip
- Heart rate was derived from the arterial pulse wave. Following an acclimation period, a baseline reading was taken (approximately 20 minutes) and the data averaged.
- Compound was administered intravenously (either bolus injection of approximately 5 seconds or infusion of 15 minutes duration), and data were recorded every 1 minute for 60 minutes post
- Data are calculated as either the peak change in heart rate or mean arterial pressure or are calculated as the area under the curve for changes in heart rate or blood pressure versus time. Data are expressed as mean + SEM. A one-tailed Student's paired t-test is used for statistical comparison to baseline values and considered significant at p ⁇ 0.05.
- a single mouse is dosed intravenously (tail vein) with 0.1 ml of test compound dissolved in a non-toxic vehicle and is observed for signs of toxicity. Severe signs may include death, seizure, paralysis or unconciousness. Milder signs are also noted and may include ataxia, labored breathing, ruffling or reduced activity relative to normal.
- the dosing solution is diluted in the same vehicle. The diluted dose is administered in the same fashion to a second mouse and is likewise observed for signs. The process is repeated until a dose is reached that produces no signs. This is considered the estimated no-effect level. An additional mouse is dosed at this level to confirm the absence of signs.
- 0.5 ml of blood is withdrawn via direct cardiac puncture, blood is immediately stabilized with EDTA and hematology is evaluated using a clinical hematology autoanalyzer calibrated for performing murine differential counts (H2000, CARESIDE, Culver City CA).
- H2000, CARESIDE, Culver City CA a clinical hematology autoanalyzer calibrated for performing murine differential counts
- Reduction in lymphocytes by test treatment is established by comparison of hematological parameters of three mice versus three vehicle treated mice.
- the dose used for this evaluation is determined by tolerability using a modification of the dilution method above. For this purpose, no-effect is desirable, mild effects are acceptable and severely toxic doses are serially diluted to levels that produce only mild effects.
- the examples disclosed herein have utility as immunoregulatory agents as demonstrated by their activity as potent and selective agonists of the SlPi/Edgl receptor over the S1PR3/Edg3 receptor as measured in the assays described above.
- the examples disclosed herein possess a selectivity for the SlPi/Edgl receptor over the S1PR3/Edg3 receptor of more than 100 fold as measured by the ratio of EC50 for the SlPi/Edgl receptor to the EC50 for the SlP3/Edg3 receptor as evaluated in the 35s-GTP ⁇ S binding assay described above and possess an EC50 for binding to the SlPi/Edgl receptor of less than 50 nM as evaluated by the 35s-GTP ⁇ S binding assay described above.
Abstract
Description







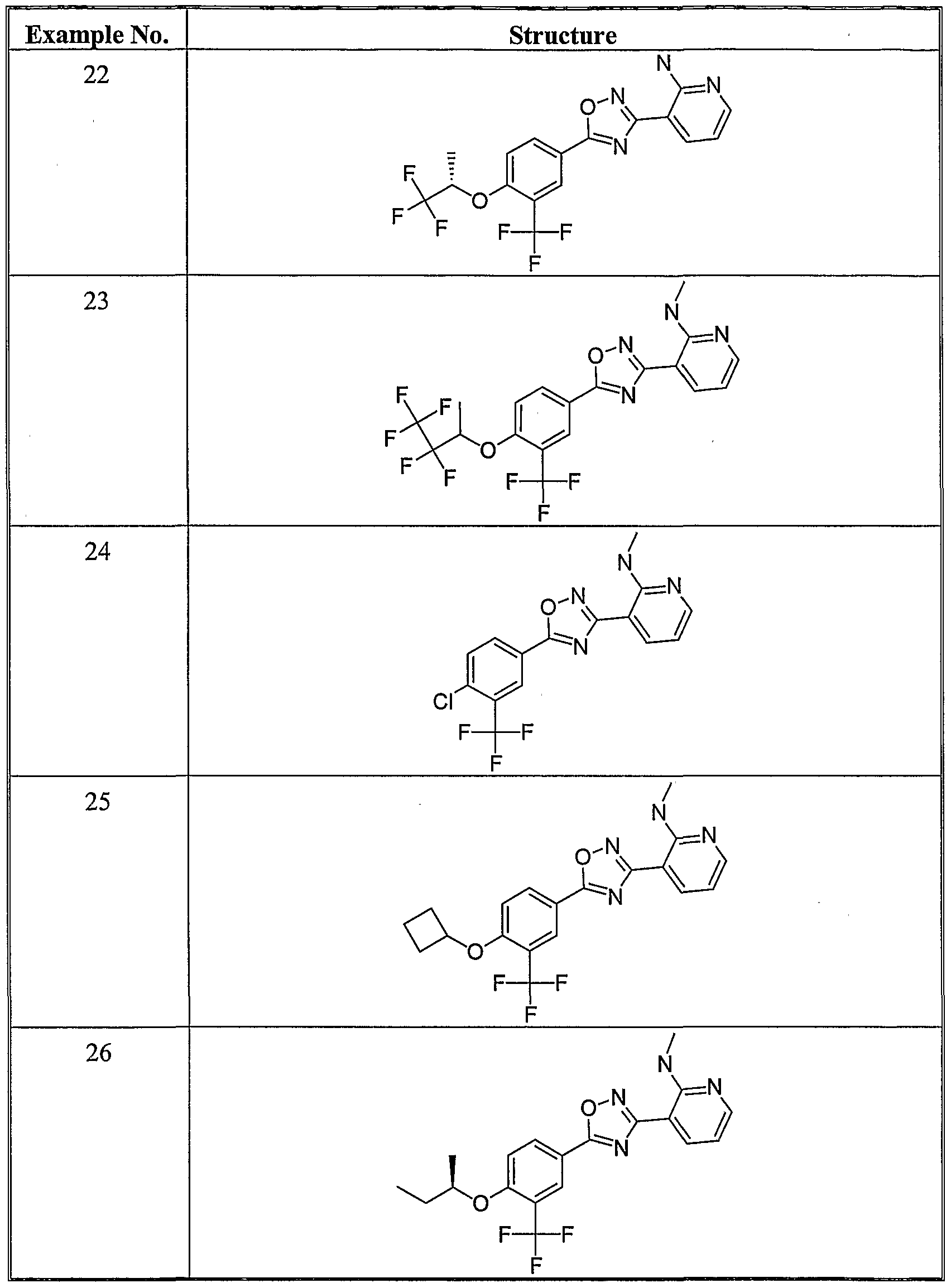























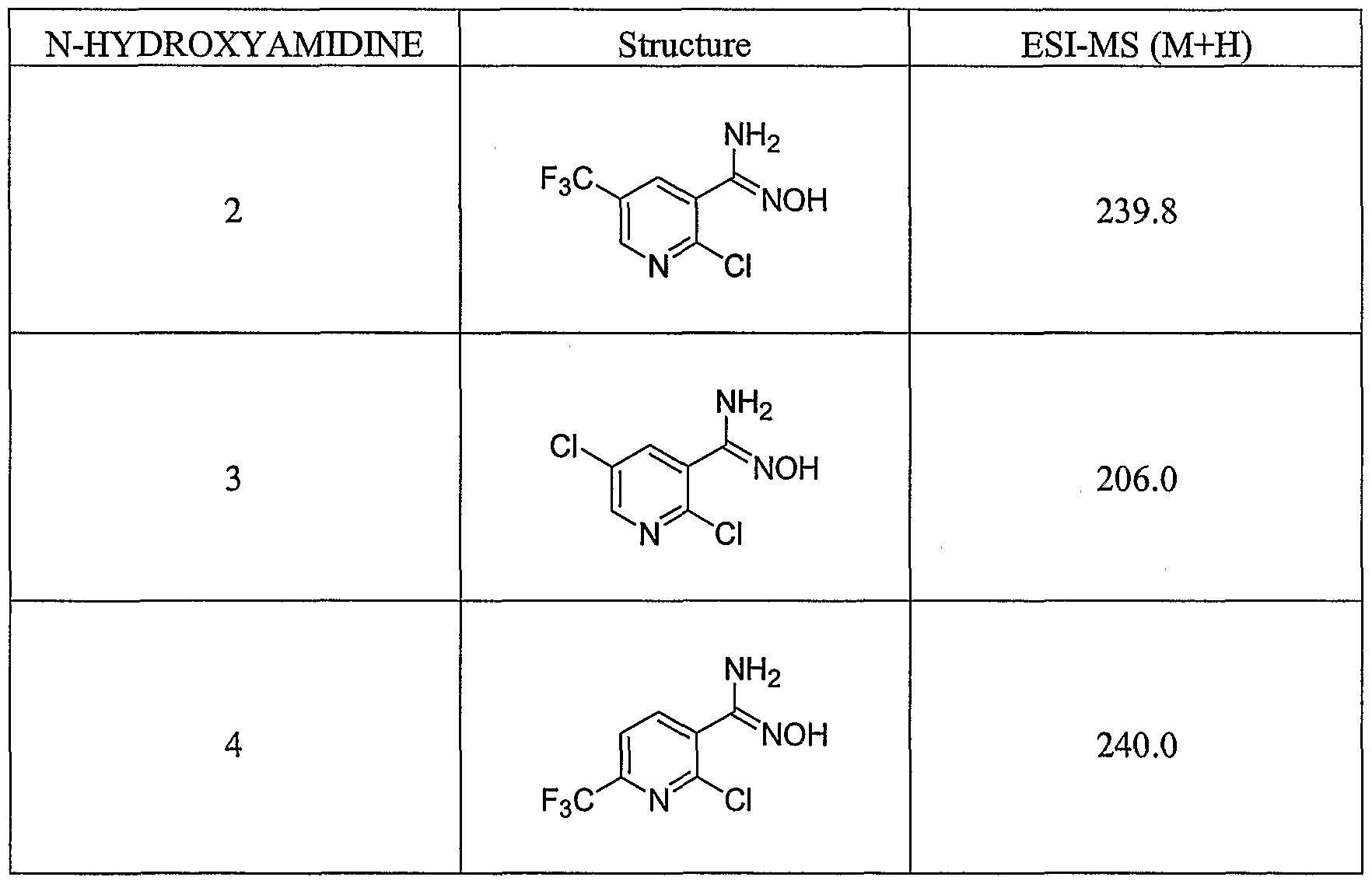











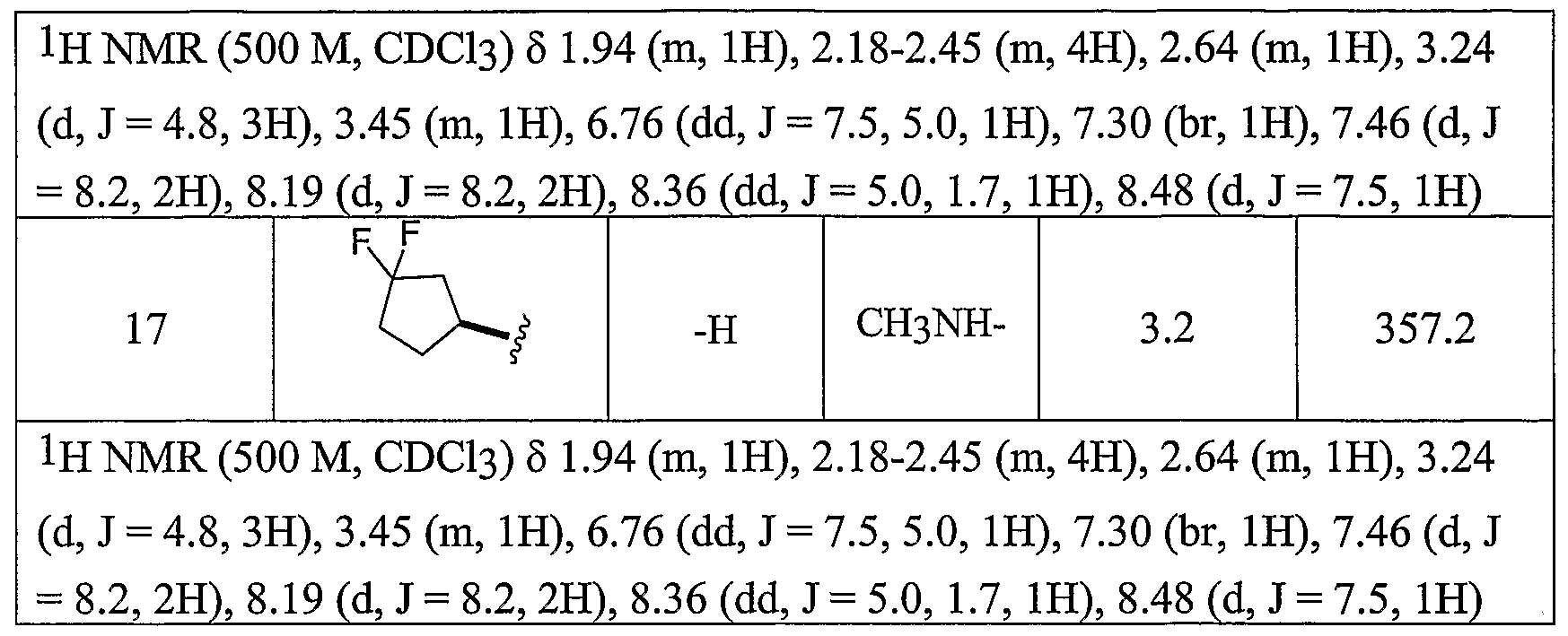





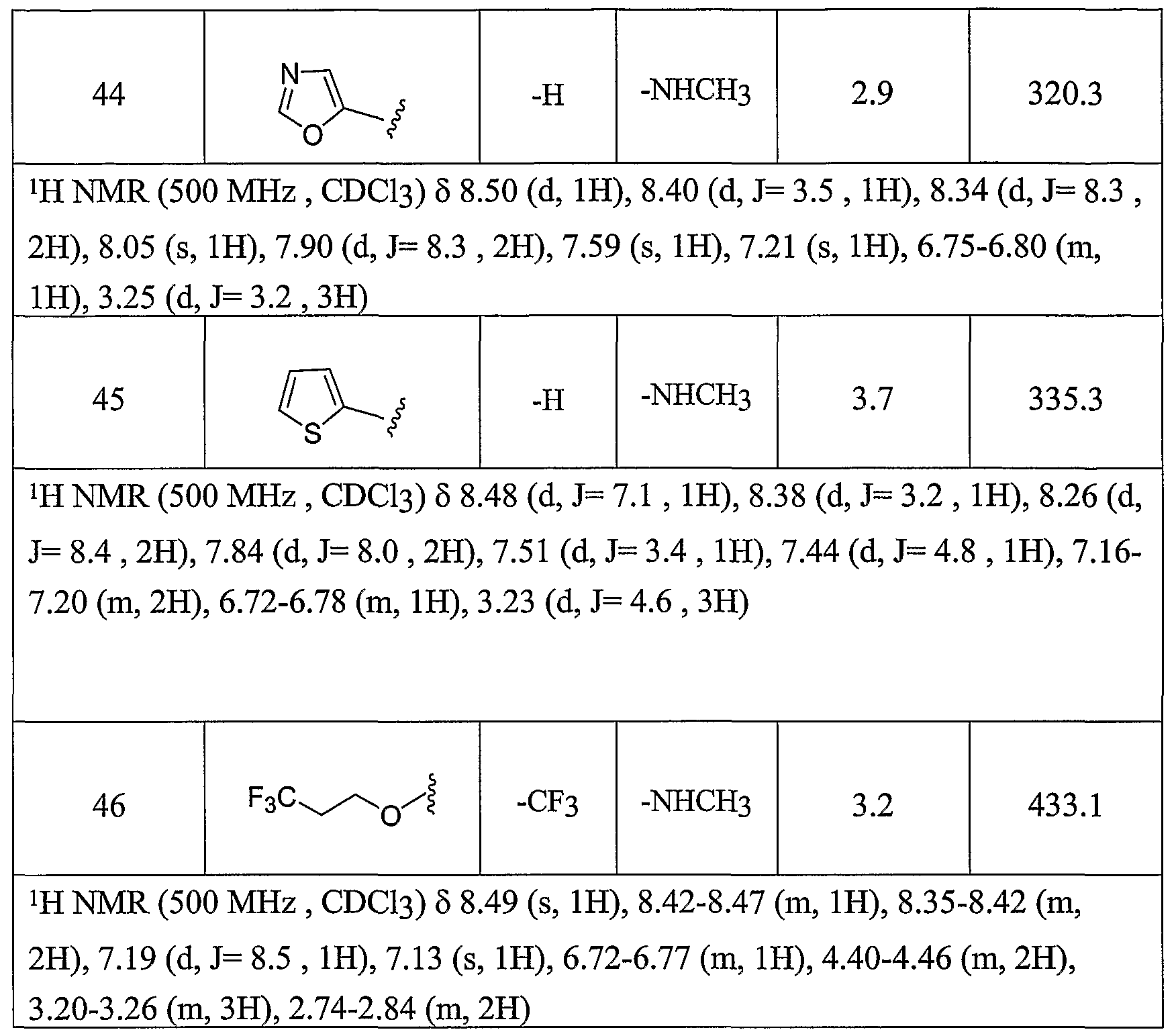





















Claims





















Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006532984A JP2006528980A (en) | 2003-05-15 | 2004-05-12 | 3- (2-Amino-1-azacyclo) -5-aryl-1,2,4-oxadiazoles as S1P receptor agonists |
| EP04751981A EP1625123A4 (en) | 2003-05-15 | 2004-05-12 | 3-(2-amino-1-azacyclyl)-5-aryl-1,2,4-oxadiazoles as s1p receptor agonists |
| CA002524867A CA2524867A1 (en) | 2003-05-15 | 2004-05-12 | 3-(2-amino-1-azacyclyl)-5-aryl-1,2,4-oxadiazoles as s1p receptor agonists |
| AU2004240586A AU2004240586A1 (en) | 2003-05-15 | 2004-05-12 | 3-(2-amino-1-azacyclyl)-5-aryl-1,2,4-oxadiazoles as S1P receptor agonists |
| US10/554,665 US20060252741A1 (en) | 2003-05-15 | 2004-05-12 | 3-(2-amino-1-azacyclyl)-5-aryl-1,2,4-oxadiazoles as s1p receptor agonists |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US47065903P | 2003-05-15 | 2003-05-15 | |
| US60/470,659 | 2003-05-15 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2004103279A2 true WO2004103279A2 (en) | 2004-12-02 |
| WO2004103279A3 WO2004103279A3 (en) | 2005-05-19 |
Family
ID=33476733
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2004/014837 WO2004103279A2 (en) | 2003-05-15 | 2004-05-12 | 3-(2-amino-1-azacyclyl)-5-aryl-1,2,4-oxadiazoles as s1p receptor agonists |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US20060252741A1 (en) |
| EP (1) | EP1625123A4 (en) |
| JP (1) | JP2006528980A (en) |
| CN (1) | CN1788008A (en) |
| AU (1) | AU2004240586A1 (en) |
| CA (1) | CA2524867A1 (en) |
| WO (1) | WO2004103279A2 (en) |
Cited By (95)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005058848A1 (en) * | 2003-12-17 | 2005-06-30 | Merck & Co., Inc. | (3,4-disubstituted)propanoic carboxylates as s1p (edg) receptor agonists |
| WO2006088944A1 (en) * | 2005-02-14 | 2006-08-24 | University Of Virginia Patent Foundation | Sphingosine 1- phos phate agonists comprising cycloalkanes and 5 -membered heterocycles substituted by amino and phenyl groups |
| WO2006100633A1 (en) * | 2005-03-23 | 2006-09-28 | Actelion Pharmaceuticals Ltd | NOVEL THIOPHENE DERIVATIVES AS SPHINGOSINE-l-PHOSPHATE-1 RECEPTOR AGONISTS |
| US7241790B2 (en) | 2002-07-30 | 2007-07-10 | University Of Virginia Patent Foundation | Compounds active in spinigosine 1-phosphate signaling |
| US7241812B2 (en) | 2004-08-13 | 2007-07-10 | Praecis Pharmaceuticals, Inc. | Methods and compositions for modulating sphingosine-1-phosphate (S1P) receptor activity |
| WO2007085451A2 (en) * | 2006-01-27 | 2007-08-02 | Novartis Ag | 3,5-di (aryl or heteroaryl) isoxazoles and 1, 2, 4-oxadiazoles as s1p1 receptor agonists, immunosuppresssive and anti -inflammatory agents |
| WO2007089018A1 (en) | 2006-02-03 | 2007-08-09 | Taisho Pharmaceutical Co., Ltd. | Triazole derivative |
| WO2007092190A2 (en) * | 2006-02-06 | 2007-08-16 | Praecis Pharmaceuticals, Inc. | Methods and compositions for modulating sphingosine-1-phosphate (s1p) receptor activity |
| WO2007091570A1 (en) | 2006-02-06 | 2007-08-16 | Taisho Pharmaceutical Co., Ltd. | Binding inhibitor of sphingosine-1-phosphate |
| WO2007098474A1 (en) * | 2006-02-21 | 2007-08-30 | University Of Virginia Patent Foundation | Phenyl-cycloalkyl and phenyl-heterocyclic derivatives as s1p receptor agonists |
| WO2007116866A1 (en) | 2006-04-03 | 2007-10-18 | Astellas Pharma Inc. | Hetero compound |
| WO2008018447A1 (en) | 2006-08-08 | 2008-02-14 | Kyorin Pharmaceutical Co., Ltd. | Aminoalcohol derivative and immunosuppressant containing the same as active ingredient |
| WO2008018427A1 (en) | 2006-08-08 | 2008-02-14 | Kyorin Pharmaceutical Co., Ltd. | Aminophosphoric acid ester derivative and s1p receptor modulator containing the same as active ingredient |
| WO2008074820A1 (en) * | 2006-12-21 | 2008-06-26 | Glaxo Group Limited | Oxadiazole derivatives as s1p1 receptor agonists |
| WO2008074821A1 (en) | 2006-12-21 | 2008-06-26 | Glaxo Group Limited | Indole derivatives as s1p1 receptor agonists |
| WO2008114157A1 (en) * | 2007-03-16 | 2008-09-25 | Actelion Pharmaceuticals Ltd | Amino- pyridine derivatives as s1p1 /edg1 receptor agonists |
| WO2009017219A1 (en) | 2007-08-01 | 2009-02-05 | Taisho Pharmaceutical Co., Ltd. | Inhibitor of binding of s1p1 |
| WO2009024905A1 (en) * | 2007-08-17 | 2009-02-26 | Actelion Pharmaceuticals Ltd | Pyridine derivatives as s1p1/edg1 receptor modulators |
| WO2009057079A2 (en) * | 2007-11-01 | 2009-05-07 | Actelion Pharmaceuticals Ltd | Novel pyrimidine derivatives |
| WO2009151621A1 (en) * | 2008-06-13 | 2009-12-17 | Arena Pharmaceuticals, Inc. | Substituted (1, 2, 4-0xadiaz0l-3-yl) indolin-1-yl carboxylic acid derivatives useful as s1p1 agonists |
| WO2009151626A1 (en) * | 2008-06-13 | 2009-12-17 | Arena Pharmaceuticals, Inc. | Substituted (1, 2, 4-0xadiaz0l-3-yl) indolin-1-yl carboxylic acid derivatives useful as s1p1 agonists |
| US7638637B2 (en) | 2003-11-03 | 2009-12-29 | University Of Virginia Patent Foundation | Orally available sphingosine 1-phosphate receptor agonists and antagonists |
| JP2010504320A (en) * | 2006-09-21 | 2010-02-12 | アクテリオン ファーマシューティカルズ リミテッド | Phenyl derivatives and their use as immunomodulators |
| US7723378B2 (en) | 2005-03-23 | 2010-05-25 | Actelion Pharmaceuticals Ltd. | Hydrogenated benzo (C) thiophene derivatives as immunomodulators |
| US7750040B2 (en) | 2004-07-29 | 2010-07-06 | Actelion Pharmaceuticals Ltd | Thiophene derivatives |
| WO2010081692A1 (en) * | 2009-01-19 | 2010-07-22 | Almirall, S.A. | Oxadiazole derivatives as slpl receptor agonists |
| US7786173B2 (en) | 2006-11-21 | 2010-08-31 | University Of Virginia Patent Foundation | Tetralin analogs having sphingosine 1-phosphate agonist activity |
| WO2010100142A1 (en) | 2009-03-03 | 2010-09-10 | Merck Serono S.A. | Oxazole pyridine derivatives useful as s1p1 receptor agonists |
| US7915315B2 (en) | 2006-11-21 | 2011-03-29 | University Of Virginia Patent Foundation | Benzocycloheptyl analogs having sphingosine 1-phosphate receptor activity |
| US7964649B2 (en) | 2006-11-21 | 2011-06-21 | University Of Virginia Patent Foundation | Hydrindane analogs having sphingosine 1-phosphate receptor agonist activity |
| US8003800B2 (en) | 2006-01-11 | 2011-08-23 | Actelion Pharmaceuticals Ltd. | Thiophene derivatives as S1P1/EDG1 receptor agonists |
| US8008286B2 (en) | 2006-01-27 | 2011-08-30 | University Of Virginia Patent Foundation | Method for treatment of neuropathic pain |
| US8017631B2 (en) | 2005-04-26 | 2011-09-13 | Neurosearch A/S | Oxadiazole derivatives and their medical use |
| US8022225B2 (en) | 2004-08-04 | 2011-09-20 | Taisho Pharmaceutical Co., Ltd | Triazole derivative |
| US8133910B2 (en) | 2006-09-07 | 2012-03-13 | Actelion Pharmaceuticals Ltd. | Thiophene derivatives as S1P1/EDGE1 receptor agonists |
| US8173710B2 (en) | 2006-02-09 | 2012-05-08 | University Of Virginia Patent Foundation | Bicyclic sphingosine 1-phosphate analogs |
| US8178562B2 (en) | 2006-01-24 | 2012-05-15 | Actelion Pharmaceuticals, Ltd. | Pyridine derivatives |
| US8188279B2 (en) | 2008-05-08 | 2012-05-29 | Allergan, Inc. | Therapeutically useful substituted hydropyrido [3,2,1-ij] quinoline compounds |
| WO2012074719A1 (en) * | 2010-12-03 | 2012-06-07 | Allergan, Inc. | Novel pyridine derivatives as sphingosine 1-phosphate (s1p) receptor modulators |
| WO2012080641A1 (en) * | 2010-12-13 | 2012-06-21 | Centre National De La Recherche Scientifique - Cnrs - | S1p receptor agonists and use thereof in treating hiv infections |
| US8288554B2 (en) | 2006-09-08 | 2012-10-16 | Actelion Pharmaceuticals Ltd. | Pyridin-3-yl derivatives as immunomodulating agents |
| US8329730B2 (en) | 2008-04-30 | 2012-12-11 | Glaxo Group Limited | Compounds |
| US8399451B2 (en) | 2009-08-07 | 2013-03-19 | Bristol-Myers Squibb Company | Heterocyclic compounds |
| US8410112B2 (en) | 2008-11-10 | 2013-04-02 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US8415484B2 (en) | 2008-08-27 | 2013-04-09 | Arena Pharmaceuticals, Inc. | Substituted tricyclic acid derivatives as S1P1 receptor agonists useful in the treatment of autoimmune and inflammatory disorders |
| US8580824B2 (en) | 2006-09-07 | 2013-11-12 | Actelion Pharmaceuticals Ltd. | Pyridin-4-yl derivatives as immunomodulating agents |
| US8580841B2 (en) | 2008-07-23 | 2013-11-12 | Arena Pharmaceuticals, Inc. | Substituted 1,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetic acid derivatives useful in the treatment of autoimmune and inflammatory disorders |
| US8623869B2 (en) | 2010-06-23 | 2014-01-07 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US8658675B2 (en) | 2009-07-16 | 2014-02-25 | Actelion Pharmaceuticals Ltd. | Pyridin-4-yl derivatives |
| US8729062B2 (en) | 2010-12-03 | 2014-05-20 | Allergan, Inc. | Benzyl azetidine derivatives as sphingosine 1-phosphate (S1P) receptor modulators |
| US8735433B1 (en) | 2012-11-14 | 2014-05-27 | Allergan, Inc. | Aryl oxadiazole derivatives as sphingosine 1-phosphate (S1P) receptor modulators |
| US8765751B2 (en) | 2011-09-30 | 2014-07-01 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US8822469B2 (en) | 2011-06-22 | 2014-09-02 | Vertex Pharmaceuticals Incorporated | Pyrrolo[2,3-B]pyrazines useful as inhibitors of ATR kinase |
| US8835470B2 (en) | 2010-04-23 | 2014-09-16 | Bristol-Myers Squibb Company | Mandelamide heterocyclic compounds |
| US8841337B2 (en) | 2011-11-09 | 2014-09-23 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US8841308B2 (en) | 2008-12-19 | 2014-09-23 | Vertex Pharmaceuticals Incorporated | Pyrazin-2-amines useful as inhibitors of ATR kinase |
| US8841450B2 (en) | 2011-11-09 | 2014-09-23 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US8841449B2 (en) | 2011-11-09 | 2014-09-23 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US8846918B2 (en) | 2011-11-09 | 2014-09-30 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US8846686B2 (en) | 2011-09-30 | 2014-09-30 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US8846917B2 (en) | 2011-11-09 | 2014-09-30 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US8846728B2 (en) | 2010-12-03 | 2014-09-30 | Allergan, Inc. | Oxadiazole derivatives as sphingosine 1-phosphate (S1P) receptor modulators |
| US8853419B2 (en) | 2010-01-27 | 2014-10-07 | Arena Pharmaceuticals, Inc. | Processes for the preparation of (R)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-1,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetic acid and salts thereof |
| US8853217B2 (en) | 2011-09-30 | 2014-10-07 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US8871755B2 (en) | 2013-02-12 | 2014-10-28 | Allergan, Inc. | Alkene azetidine derivatives as sphingosine 1-phosphate (S1P) receptor modulators |
| US8877759B2 (en) | 2011-04-05 | 2014-11-04 | Vertex Pharnaceuticals Incorporated | Aminopyrazines as ATR kinase inhibitors |
| US8912198B2 (en) | 2012-10-16 | 2014-12-16 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| JP2015025001A (en) * | 2007-10-04 | 2015-02-05 | メルク セローノ ソシエテ アノニム | Oxadiazole diaryl compounds |
| US8962631B2 (en) | 2010-05-12 | 2015-02-24 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US8969356B2 (en) | 2010-05-12 | 2015-03-03 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9035053B2 (en) | 2011-09-30 | 2015-05-19 | Vertex Pharmaceuticals Incorporated | Processes for making compounds useful as inhibitors of ATR kinase |
| AU2013201157B2 (en) * | 2006-12-21 | 2015-06-11 | Glaxo Group Limited | Indole derivatives as s1p1 receptor agonists |
| US9062008B2 (en) | 2010-05-12 | 2015-06-23 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9085581B2 (en) | 2010-03-03 | 2015-07-21 | Arena Pharmaceuticals, Inc. | Processes for the preparation of S1P1 receptor modulators and crystalline forms thereof |
| US9096584B2 (en) | 2010-05-12 | 2015-08-04 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9096602B2 (en) | 2011-06-22 | 2015-08-04 | Vertex Pharmaceuticals Incorporated | Substituted pyrrolo[2,3-B]pyrazines as ATR kinase inhibitors |
| US9133179B2 (en) | 2011-01-19 | 2015-09-15 | Actelion Pharmaceuticals Ltd. | 2-methoxy-pyridin-4-yl-derivatives |
| US9187437B2 (en) | 2010-09-24 | 2015-11-17 | Bristol-Myers Squibb Company | Substituted oxadiazole compounds |
| US9309250B2 (en) | 2011-06-22 | 2016-04-12 | Vertex Pharmaceuticals Incorporated | Substituted pyrrolo[2,3-b]pyrazines as ATR kinase inhibitors |
| US9334244B2 (en) | 2010-05-12 | 2016-05-10 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9340546B2 (en) | 2012-12-07 | 2016-05-17 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9630956B2 (en) | 2010-05-12 | 2017-04-25 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9663519B2 (en) | 2013-03-15 | 2017-05-30 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9670215B2 (en) | 2014-06-05 | 2017-06-06 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9791456B2 (en) | 2012-10-04 | 2017-10-17 | Vertex Pharmaceuticals Incorporated | Method for measuring ATR inhibition mediated increases in DNA damage |
| US10160760B2 (en) | 2013-12-06 | 2018-12-25 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US10301262B2 (en) | 2015-06-22 | 2019-05-28 | Arena Pharmaceuticals, Inc. | Crystalline L-arginine salt of (R)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-1,2,3,4-tetrahydrocyclo-penta [b]indol-3-yl)acetic acid(Compund1) for use in SIPI receptor-associated disorders |
| US10385043B2 (en) | 2015-05-20 | 2019-08-20 | Idorsia Pharmaceuticals Ltd | Crystalline form of the compound (S)-3-{4-[5-(2-cyclopentyl-6-methoxy-pyridin-4-yl)-[1,2,4]oxadiazol-3-yl]-2-ethyl-6-methyl-phenoxy}-propane-1,2-diol |
| US10478430B2 (en) | 2012-04-05 | 2019-11-19 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase and combination therapies thereof |
| US10813929B2 (en) | 2011-09-30 | 2020-10-27 | Vertex Pharmaceuticals Incorporated | Treating cancer with ATR inhibitors |
| US11007175B2 (en) | 2015-01-06 | 2021-05-18 | Arena Pharmaceuticals, Inc. | Methods of treating conditions related to the S1P1 receptor |
| US11179394B2 (en) | 2014-06-17 | 2021-11-23 | Vertex Pharmaceuticals Incorporated | Method for treating cancer using a combination of Chk1 and ATR inhibitors |
| US11464774B2 (en) | 2015-09-30 | 2022-10-11 | Vertex Pharmaceuticals Incorporated | Method for treating cancer using a combination of DNA damaging agents and ATR inhibitors |
| US11478448B2 (en) | 2017-02-16 | 2022-10-25 | Arena Pharmaceuticals, Inc. | Compounds and methods for treatment of inflammatory bowel disease with extra-intestinal manifestations |
| US11534424B2 (en) | 2017-02-16 | 2022-12-27 | Arena Pharmaceuticals, Inc. | Compounds and methods for treatment of primary biliary cholangitis |
Families Citing this family (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| PL1772145T3 (en) * | 2004-07-16 | 2011-08-31 | Kyorin Seiyaku Kk | Method of effectively using medicine and method concerning prevention of side effect |
| KR101181090B1 (en) * | 2004-10-12 | 2012-09-07 | 교린 세이야꾸 가부시키 가이샤 | Process for producing 2-amino-2-[2-[4-3-benzyloxyphenylthio-2-chlorophenyl]ethyl]-1,3-propanediol hydrochloride or hydrate thereof and intermediate for the same |
| AU2006256968A1 (en) * | 2005-06-08 | 2006-12-14 | Novartis Ag | Polycyclic oxadiazoles or I soxazoles and their use as SIP receptor ligands |
| PT1932522E (en) * | 2005-10-07 | 2012-06-26 | Kyorin Seiyaku Kk | Therapeutic agent for liver disease containing 2-amino-1,3-propanediol derivative as active ingredient |
| TWI389683B (en) * | 2006-02-06 | 2013-03-21 | Kyorin Seiyaku Kk | A therapeutic agent for an inflammatory bowel disease or an inflammatory bowel disease treatment using a 2-amino-1,3-propanediol derivative as an active ingredient |
| GB0625647D0 (en) * | 2006-12-21 | 2007-01-31 | Glaxo Group Ltd | Compounds |
| US8383852B2 (en) * | 2007-02-16 | 2013-02-26 | Emisphere Technologies, Inc. | Compounds having a cyclic moiety and compositions for delivering active agents |
| KR20090130062A (en) * | 2007-04-19 | 2009-12-17 | 글락소 그룹 리미티드 | Oxadiazole substituted indazole derivatives for use as sphingosine 1-phosphate (s1p) agonists |
| AR067762A1 (en) * | 2007-07-31 | 2009-10-21 | Vertex Pharma | PROCESS TO PREPARE 5-FLUORO-1H-PIRAZOLO (3,4-B) PIRIDIN-3-AMINA AND DERIVATIVES OF THE SAME |
| AU2008306885B2 (en) * | 2007-10-04 | 2013-12-05 | Merck Serono S.A. | Oxadiazole derivatives |
| TW200946105A (en) | 2008-02-07 | 2009-11-16 | Kyorin Seiyaku Kk | Therapeutic agent or preventive agent for inflammatory bowel disease containing amino alcohol derivative as active ingredient |
| ES2389042T3 (en) * | 2008-03-06 | 2012-10-22 | Actelion Pharmaceuticals Ltd. | Pyridine compounds |
| JP2012515789A (en) * | 2009-01-23 | 2012-07-12 | ブリストル−マイヤーズ スクイブ カンパニー | Pyrazole-1,2,4-oxadiazole derivatives as sphingosine-1-phosphate agonists |
| US8354398B2 (en) * | 2009-01-23 | 2013-01-15 | Bristol-Myers Squibb Company | Substituted isoxazole compounds |
| US8247436B2 (en) | 2010-03-19 | 2012-08-21 | Novartis Ag | Pyridine and pyrazine derivative for the treatment of CF |
| WO2012061459A1 (en) * | 2010-11-03 | 2012-05-10 | Bristol-Myers Squibb Company | Heterocyclic compounds as s1p1 agonists for the treatment of autoimmune and vascular diseases |
| CN108178759B (en) * | 2018-01-05 | 2020-06-09 | 上海瑞纷医药科技有限责任公司 | Synthesis method of α -adrenoceptor antagonist |
| WO2020051378A1 (en) | 2018-09-06 | 2020-03-12 | Arena Pharmaceuticals, Inc. | Compounds useful in the treatment of autoimmune and inflammatory disorders |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3647809A (en) * | 1968-04-26 | 1972-03-07 | Chinoin Gyogyszer Es Vegyeszet | Certain pyridyl-1 2 4-oxadiazole derivatives |
| DK1210344T3 (en) * | 1999-08-19 | 2006-03-06 | Nps Pharma Inc | Heteropolycyclic Compounds and Their Uses as Metabotropic Glutamate Receptor Antagonists |
| NZ527691A (en) * | 2001-02-21 | 2007-01-26 | Nps Pharma Inc | Heteropolycyclic compounds and their use as metabotropic glutamate receptor antagonists |
| US7479504B2 (en) * | 2002-01-18 | 2009-01-20 | Merck & Co., Inc. | Edg receptor agonists |
| CA2488117A1 (en) * | 2002-06-17 | 2003-12-24 | Merck & Co., Inc. | 1-((5-aryl-1,2,4-oxadiazol-3-yl)benzyl)azetidine-3-carboxylates and 1-((5-aryl-1,2,4-oxadiazol-3-yl)benzyl)pyrrolidine-3-carboxylates as edg receptor agonists |
-
2004
- 2004-05-12 US US10/554,665 patent/US20060252741A1/en not_active Abandoned
- 2004-05-12 CA CA002524867A patent/CA2524867A1/en not_active Abandoned
- 2004-05-12 EP EP04751981A patent/EP1625123A4/en not_active Withdrawn
- 2004-05-12 WO PCT/US2004/014837 patent/WO2004103279A2/en active Application Filing
- 2004-05-12 JP JP2006532984A patent/JP2006528980A/en not_active Withdrawn
- 2004-05-12 CN CNA2004800129905A patent/CN1788008A/en active Pending
- 2004-05-12 AU AU2004240586A patent/AU2004240586A1/en not_active Abandoned
Non-Patent Citations (1)
| Title |
|---|
| See references of EP1625123A4 * |
Cited By (173)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7560477B2 (en) | 2002-07-30 | 2009-07-14 | University Of Virginia Patent Foundation | Compounds active in sphingosine 1-phosphate signaling |
| US7241790B2 (en) | 2002-07-30 | 2007-07-10 | University Of Virginia Patent Foundation | Compounds active in spinigosine 1-phosphate signaling |
| US7638637B2 (en) | 2003-11-03 | 2009-12-29 | University Of Virginia Patent Foundation | Orally available sphingosine 1-phosphate receptor agonists and antagonists |
| US7605171B2 (en) | 2003-12-17 | 2009-10-20 | Merck & Co., Inc. | (3,4-disubstituted)propanoic carboxylates as S1P (Edg) receptor agonists |
| WO2005058848A1 (en) * | 2003-12-17 | 2005-06-30 | Merck & Co., Inc. | (3,4-disubstituted)propanoic carboxylates as s1p (edg) receptor agonists |
| AU2004299456B2 (en) * | 2003-12-17 | 2010-10-07 | Merck Sharp & Dohme Corp. | (3,4-disubstituted)propanoic carboxylates as S1P (Edg) receptor agonists |
| US7750040B2 (en) | 2004-07-29 | 2010-07-06 | Actelion Pharmaceuticals Ltd | Thiophene derivatives |
| US8022225B2 (en) | 2004-08-04 | 2011-09-20 | Taisho Pharmaceutical Co., Ltd | Triazole derivative |
| US7241812B2 (en) | 2004-08-13 | 2007-07-10 | Praecis Pharmaceuticals, Inc. | Methods and compositions for modulating sphingosine-1-phosphate (S1P) receptor activity |
| WO2006088944A1 (en) * | 2005-02-14 | 2006-08-24 | University Of Virginia Patent Foundation | Sphingosine 1- phos phate agonists comprising cycloalkanes and 5 -membered heterocycles substituted by amino and phenyl groups |
| US7754703B2 (en) | 2005-02-14 | 2010-07-13 | University Of Virginia Patent Foundation | Cycloalkane-containing sphingosine 1-phosphate agonists |
| US8329676B2 (en) | 2005-02-14 | 2012-12-11 | University Of Virginia Patent Foundation | Cycloalkane-containing sphingosine 1-phosphate agonists |
| JP2008530135A (en) * | 2005-02-14 | 2008-08-07 | ユニバーシティ オブ バージニア パテント ファンデーション | Sphingosine = 1-phosphate agonist containing a cycloalkane substituted with an amino group and a phenyl group and a 5-membered heterocyclic ring |
| US8039644B2 (en) | 2005-03-23 | 2011-10-18 | Actelion Pharmaceuticals Ltd. | Hydrogenated benzo (C) thiophene derivatives as immunomodulators |
| US7723378B2 (en) | 2005-03-23 | 2010-05-25 | Actelion Pharmaceuticals Ltd. | Hydrogenated benzo (C) thiophene derivatives as immunomodulators |
| US7605269B2 (en) | 2005-03-23 | 2009-10-20 | Actelion Pharmaceuticals Ltd. | Thiophene derivatives as Sphingosine-1-phosphate-1 receptor agonists |
| WO2006100633A1 (en) * | 2005-03-23 | 2006-09-28 | Actelion Pharmaceuticals Ltd | NOVEL THIOPHENE DERIVATIVES AS SPHINGOSINE-l-PHOSPHATE-1 RECEPTOR AGONISTS |
| US8017631B2 (en) | 2005-04-26 | 2011-09-13 | Neurosearch A/S | Oxadiazole derivatives and their medical use |
| US8003800B2 (en) | 2006-01-11 | 2011-08-23 | Actelion Pharmaceuticals Ltd. | Thiophene derivatives as S1P1/EDG1 receptor agonists |
| US8178562B2 (en) | 2006-01-24 | 2012-05-15 | Actelion Pharmaceuticals, Ltd. | Pyridine derivatives |
| US8697732B2 (en) | 2006-01-24 | 2014-04-15 | Actelion Pharmaceuticals Ltd. | Pyridine derivatives |
| WO2007085451A3 (en) * | 2006-01-27 | 2007-12-21 | Novartis Ag | 3,5-di (aryl or heteroaryl) isoxazoles and 1, 2, 4-oxadiazoles as s1p1 receptor agonists, immunosuppresssive and anti -inflammatory agents |
| JP2009524611A (en) * | 2006-01-27 | 2009-07-02 | ノバルティス アクチエンゲゼルシャフト | Reverse isoxazole |
| US7799812B2 (en) | 2006-01-27 | 2010-09-21 | Novartis Ag | Reverse isoxazoles |
| US8008286B2 (en) | 2006-01-27 | 2011-08-30 | University Of Virginia Patent Foundation | Method for treatment of neuropathic pain |
| WO2007085451A2 (en) * | 2006-01-27 | 2007-08-02 | Novartis Ag | 3,5-di (aryl or heteroaryl) isoxazoles and 1, 2, 4-oxadiazoles as s1p1 receptor agonists, immunosuppresssive and anti -inflammatory agents |
| JP5035752B2 (en) * | 2006-02-03 | 2012-09-26 | 大正製薬株式会社 | Triazole derivative |
| WO2007089018A1 (en) | 2006-02-03 | 2007-08-09 | Taisho Pharmaceutical Co., Ltd. | Triazole derivative |
| US8022091B2 (en) | 2006-02-03 | 2011-09-20 | Taisho Pharmaceutical Co., Ltd. | Triazole derivative |
| WO2007091570A1 (en) | 2006-02-06 | 2007-08-16 | Taisho Pharmaceutical Co., Ltd. | Binding inhibitor of sphingosine-1-phosphate |
| WO2007092190A3 (en) * | 2006-02-06 | 2007-11-29 | Praecis Pharm Inc | Methods and compositions for modulating sphingosine-1-phosphate (s1p) receptor activity |
| US7994204B2 (en) | 2006-02-06 | 2011-08-09 | Taisho Pharmaceutical Co., Ltd | Binding inhibitor of sphingosine-1-phosphate |
| WO2007092190A2 (en) * | 2006-02-06 | 2007-08-16 | Praecis Pharmaceuticals, Inc. | Methods and compositions for modulating sphingosine-1-phosphate (s1p) receptor activity |
| US8173710B2 (en) | 2006-02-09 | 2012-05-08 | University Of Virginia Patent Foundation | Bicyclic sphingosine 1-phosphate analogs |
| WO2007098474A1 (en) * | 2006-02-21 | 2007-08-30 | University Of Virginia Patent Foundation | Phenyl-cycloalkyl and phenyl-heterocyclic derivatives as s1p receptor agonists |
| WO2007116866A1 (en) | 2006-04-03 | 2007-10-18 | Astellas Pharma Inc. | Hetero compound |
| US7678820B2 (en) | 2006-04-03 | 2010-03-16 | Astellas Pharma Inc. | Hetero compound |
| US7951825B2 (en) | 2006-04-03 | 2011-05-31 | Astellas Pharma Inc. | Hetero compound |
| WO2008018427A1 (en) | 2006-08-08 | 2008-02-14 | Kyorin Pharmaceutical Co., Ltd. | Aminophosphoric acid ester derivative and s1p receptor modulator containing the same as active ingredient |
| WO2008018447A1 (en) | 2006-08-08 | 2008-02-14 | Kyorin Pharmaceutical Co., Ltd. | Aminoalcohol derivative and immunosuppressant containing the same as active ingredient |
| US8133910B2 (en) | 2006-09-07 | 2012-03-13 | Actelion Pharmaceuticals Ltd. | Thiophene derivatives as S1P1/EDGE1 receptor agonists |
| US8580824B2 (en) | 2006-09-07 | 2013-11-12 | Actelion Pharmaceuticals Ltd. | Pyridin-4-yl derivatives as immunomodulating agents |
| US8288554B2 (en) | 2006-09-08 | 2012-10-16 | Actelion Pharmaceuticals Ltd. | Pyridin-3-yl derivatives as immunomodulating agents |
| US8044076B2 (en) | 2006-09-21 | 2011-10-25 | Actelion Pharmaceuticals Ltd. | Phenyl derivatives and their use as immunomodulators |
| JP2010504320A (en) * | 2006-09-21 | 2010-02-12 | アクテリオン ファーマシューティカルズ リミテッド | Phenyl derivatives and their use as immunomodulators |
| US7915315B2 (en) | 2006-11-21 | 2011-03-29 | University Of Virginia Patent Foundation | Benzocycloheptyl analogs having sphingosine 1-phosphate receptor activity |
| US7964649B2 (en) | 2006-11-21 | 2011-06-21 | University Of Virginia Patent Foundation | Hydrindane analogs having sphingosine 1-phosphate receptor agonist activity |
| US7786173B2 (en) | 2006-11-21 | 2010-08-31 | University Of Virginia Patent Foundation | Tetralin analogs having sphingosine 1-phosphate agonist activity |
| AU2013201157B2 (en) * | 2006-12-21 | 2015-06-11 | Glaxo Group Limited | Indole derivatives as s1p1 receptor agonists |
| WO2008074820A1 (en) * | 2006-12-21 | 2008-06-26 | Glaxo Group Limited | Oxadiazole derivatives as s1p1 receptor agonists |
| US8101775B2 (en) | 2006-12-21 | 2012-01-24 | Glaxo Group Limited | Indole derivatives as S1P1 Receptor |
| JP2010513397A (en) * | 2006-12-21 | 2010-04-30 | グラクソ グループ リミテッド | Indole derivatives as S1P1 receptor agonists |
| EA017406B1 (en) * | 2006-12-21 | 2012-12-28 | Глэксо Груп Лимитед | Indole derivatives as s1p1 receptor agonists |
| WO2008074821A1 (en) | 2006-12-21 | 2008-06-26 | Glaxo Group Limited | Indole derivatives as s1p1 receptor agonists |
| EP2206710A1 (en) * | 2006-12-21 | 2010-07-14 | Glaxo Group Limited | Indole derivatives as S1P1 receptor agonists |
| WO2008114157A1 (en) * | 2007-03-16 | 2008-09-25 | Actelion Pharmaceuticals Ltd | Amino- pyridine derivatives as s1p1 /edg1 receptor agonists |
| AU2008227979B2 (en) * | 2007-03-16 | 2014-02-06 | Actelion Pharmaceuticals Ltd | Amino- pyridine derivatives as S1P1 /EDG1 receptor agonists |
| US8592460B2 (en) | 2007-03-16 | 2013-11-26 | Actelion Pharmaceuticals Ltd. | Amino-pyridine derivatives as S1P1 /EDG1 receptor agonists |
| KR101454944B1 (en) * | 2007-03-16 | 2014-10-27 | 액테리온 파마슈티칼 리미티드 | Amino-pyridine derivatives as s1p1/edg1 receptor agonists |
| US8048898B2 (en) | 2007-08-01 | 2011-11-01 | Taisho Pharmaceutical Co., Ltd | Inhibitor of binding of S1P1 |
| WO2009017219A1 (en) | 2007-08-01 | 2009-02-05 | Taisho Pharmaceutical Co., Ltd. | Inhibitor of binding of s1p1 |
| US8598208B2 (en) | 2007-08-17 | 2013-12-03 | Actelion Pharmaceuticals Ltd. | Pyridine derivatives as S1P1/EDG1 receptor modulators |
| WO2009024905A1 (en) * | 2007-08-17 | 2009-02-26 | Actelion Pharmaceuticals Ltd | Pyridine derivatives as s1p1/edg1 receptor modulators |
| JP2015025001A (en) * | 2007-10-04 | 2015-02-05 | メルク セローノ ソシエテ アノニム | Oxadiazole diaryl compounds |
| WO2009057079A2 (en) * | 2007-11-01 | 2009-05-07 | Actelion Pharmaceuticals Ltd | Novel pyrimidine derivatives |
| US8299086B2 (en) | 2007-11-01 | 2012-10-30 | Actelion Pharmaceuticals Ltd. | Pyrimidine derivatives |
| WO2009057079A3 (en) * | 2007-11-01 | 2009-12-03 | Actelion Pharmaceuticals Ltd | Novel pyrimidine derivatives |
| US8329730B2 (en) | 2008-04-30 | 2012-12-11 | Glaxo Group Limited | Compounds |
| US8431702B2 (en) | 2008-05-08 | 2013-04-30 | Allergan, Inc. | Therapeutically useful substituted hydropyrido [3,2,1-ij] quinoline compounds |
| US8309729B2 (en) | 2008-05-08 | 2012-11-13 | Allergan, Inc. | Therapeutically useful substituted hydropyrido [3,2,1-ij] quinoline compounds |
| US8188279B2 (en) | 2008-05-08 | 2012-05-29 | Allergan, Inc. | Therapeutically useful substituted hydropyrido [3,2,1-ij] quinoline compounds |
| WO2009151626A1 (en) * | 2008-06-13 | 2009-12-17 | Arena Pharmaceuticals, Inc. | Substituted (1, 2, 4-0xadiaz0l-3-yl) indolin-1-yl carboxylic acid derivatives useful as s1p1 agonists |
| WO2009151621A1 (en) * | 2008-06-13 | 2009-12-17 | Arena Pharmaceuticals, Inc. | Substituted (1, 2, 4-0xadiaz0l-3-yl) indolin-1-yl carboxylic acid derivatives useful as s1p1 agonists |
| US9522133B2 (en) | 2008-07-23 | 2016-12-20 | Arena Pharmaceuticals, Inc. | Substituted 1,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetic acid derivatives useful in the treatment of autoimmune and inflammatory disorders |
| US8580841B2 (en) | 2008-07-23 | 2013-11-12 | Arena Pharmaceuticals, Inc. | Substituted 1,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetic acid derivatives useful in the treatment of autoimmune and inflammatory disorders |
| US9126932B2 (en) | 2008-07-23 | 2015-09-08 | Arena Pharmaceuticals, Inc. | Substituted 1,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetic acid derivatives useful in the treatment of autoimmune and inflammatory disorders |
| US8415484B2 (en) | 2008-08-27 | 2013-04-09 | Arena Pharmaceuticals, Inc. | Substituted tricyclic acid derivatives as S1P1 receptor agonists useful in the treatment of autoimmune and inflammatory disorders |
| US9108969B2 (en) | 2008-08-27 | 2015-08-18 | Arena Pharmaceuticals, Inc. | Substituted tricyclic acid derivatives as S1P1 receptor agonists useful in the treatment of autoimmune and inflammatory disorders |
| US8410112B2 (en) | 2008-11-10 | 2013-04-02 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US8841308B2 (en) | 2008-12-19 | 2014-09-23 | Vertex Pharmaceuticals Incorporated | Pyrazin-2-amines useful as inhibitors of ATR kinase |
| US9365557B2 (en) | 2008-12-19 | 2016-06-14 | Vertex Pharmaceuticals Incorporated | Substituted pyrazin-2-amines as inhibitors of ATR kinase |
| US10479784B2 (en) | 2008-12-19 | 2019-11-19 | Vertex Pharmaceuticals Incorporated | Substituted pyrazin-2-amines as inhibitors of ATR kinase |
| US9701674B2 (en) | 2008-12-19 | 2017-07-11 | Vertex Pharmaceuticals Incorporated | Substituted pyrazines as ATR kinase inhibitors |
| US10961232B2 (en) | 2008-12-19 | 2021-03-30 | Vertex Pharmaceuticals Incorporated | Substituted pyrazines as ATR kinase inhibitors |
| EP2210890A1 (en) * | 2009-01-19 | 2010-07-28 | Almirall, S.A. | Oxadiazole derivatives as S1P1 receptor agonists |
| WO2010081692A1 (en) * | 2009-01-19 | 2010-07-22 | Almirall, S.A. | Oxadiazole derivatives as slpl receptor agonists |
| AU2010220338B2 (en) * | 2009-03-03 | 2016-07-21 | Merck Serono S.A. | Oxazole pyridine derivatives useful as S1P1 receptor agonists |
| US8791142B2 (en) | 2009-03-03 | 2014-07-29 | Merck Serono S.A. | Oxazole pyridine derivatives useful as S1P1 receptor agonists |
| WO2010100142A1 (en) | 2009-03-03 | 2010-09-10 | Merck Serono S.A. | Oxazole pyridine derivatives useful as s1p1 receptor agonists |
| US8658675B2 (en) | 2009-07-16 | 2014-02-25 | Actelion Pharmaceuticals Ltd. | Pyridin-4-yl derivatives |
| US8399451B2 (en) | 2009-08-07 | 2013-03-19 | Bristol-Myers Squibb Company | Heterocyclic compounds |
| US9175320B2 (en) | 2010-01-27 | 2015-11-03 | Arena Pharmaceuticals, Inc. | Processes for the preparation of (R)-2-(7-4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-1,2,3,4-tetrahydrocyclopenta[B]indol-3-yl)acetic acid and salts thereof |
| US9447041B2 (en) | 2010-01-27 | 2016-09-20 | Arena Pharmaceuticals, Inc. | Processes for the preparation of (R)-2-(7-4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-1,2,3,4-tetrahydrocyclopenta[B]indol-3-yl)acetic acid and salts thereof |
| US8853419B2 (en) | 2010-01-27 | 2014-10-07 | Arena Pharmaceuticals, Inc. | Processes for the preparation of (R)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-1,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetic acid and salts thereof |
| US11674163B2 (en) | 2010-01-27 | 2023-06-13 | Arena Pharmaceuticals, Inc. | Processes for the preparation of (R)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-1,2,3,4-tetrahydrocyclopenta[b]indol-3-yl)acetic acid and salts thereof |
| US11149292B2 (en) | 2010-01-27 | 2021-10-19 | Arena Pharmaceuticals, Inc. | Processes for the preparation of (R)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-1,2,3,4-tetrahydrocyclopenta[B]indol-3-yl)acetic acid and salts thereof |
| US9085581B2 (en) | 2010-03-03 | 2015-07-21 | Arena Pharmaceuticals, Inc. | Processes for the preparation of S1P1 receptor modulators and crystalline forms thereof |
| US8835470B2 (en) | 2010-04-23 | 2014-09-16 | Bristol-Myers Squibb Company | Mandelamide heterocyclic compounds |
| US9062008B2 (en) | 2010-05-12 | 2015-06-23 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US8969356B2 (en) | 2010-05-12 | 2015-03-03 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9630956B2 (en) | 2010-05-12 | 2017-04-25 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US8962631B2 (en) | 2010-05-12 | 2015-02-24 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9096584B2 (en) | 2010-05-12 | 2015-08-04 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9334244B2 (en) | 2010-05-12 | 2016-05-10 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US8623869B2 (en) | 2010-06-23 | 2014-01-07 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9187437B2 (en) | 2010-09-24 | 2015-11-17 | Bristol-Myers Squibb Company | Substituted oxadiazole compounds |
| WO2012074898A1 (en) * | 2010-12-03 | 2012-06-07 | Allergan, Inc. | Novel alkene derivatives as sphingosine 1-phosphate (s1p) receptor modulators |
| CN103338769A (en) * | 2010-12-03 | 2013-10-02 | 阿勒根公司 | Novel alkene derivatives as sphingosine 1-phosphate (S1P) receptor modulators |
| US8846728B2 (en) | 2010-12-03 | 2014-09-30 | Allergan, Inc. | Oxadiazole derivatives as sphingosine 1-phosphate (S1P) receptor modulators |
| US8697733B2 (en) | 2010-12-03 | 2014-04-15 | Allergan, Inc. | Pyridine derivatives as sphingosine 1-phosphate (S1P) receptor modulators |
| US8288555B2 (en) | 2010-12-03 | 2012-10-16 | Allergan, Inc. | Alkene derivatives as sphingosine 1-phosphate (S1P) receptor modulators |
| US8399492B2 (en) | 2010-12-03 | 2013-03-19 | Allergan, Inc. | Pyridine derivatives as sphingosine 1-phosphate (S1P) receptor modulators |
| CN103338770A (en) * | 2010-12-03 | 2013-10-02 | 阿勒根公司 | Novel pyridine derivatives as sphingosine 1-phosphate (S1P) receptor modulators |
| WO2012074718A1 (en) * | 2010-12-03 | 2012-06-07 | Allergan, Inc. | Novel oxadiazole derivatives as sphingosine 1-phosphate (s1p) receptor modulators |
| US8906899B2 (en) | 2010-12-03 | 2014-12-09 | Allergan, Inc. | Azetidine derivatives as sphingosine 1-phosphate (S1P) receptor modulators |
| CN103370067A (en) * | 2010-12-03 | 2013-10-23 | 阿勒根公司 | Novel oxadiazole derivatives as sphingosine 1-phosphate (S1P) receptor modulators |
| US8492410B2 (en) | 2010-12-03 | 2013-07-23 | Allergan, Inc. | Pyridine derivatives as sphingosine 1-phosphate (S1P) receptor modulators |
| WO2012074719A1 (en) * | 2010-12-03 | 2012-06-07 | Allergan, Inc. | Novel pyridine derivatives as sphingosine 1-phosphate (s1p) receptor modulators |
| US8729062B2 (en) | 2010-12-03 | 2014-05-20 | Allergan, Inc. | Benzyl azetidine derivatives as sphingosine 1-phosphate (S1P) receptor modulators |
| US8618139B2 (en) | 2010-12-03 | 2013-12-31 | Allergan, Inc. | Oxadiazole derivatives as sphingosine 1-phosphate (S1P) receptor modulators |
| WO2012080641A1 (en) * | 2010-12-13 | 2012-06-21 | Centre National De La Recherche Scientifique - Cnrs - | S1p receptor agonists and use thereof in treating hiv infections |
| US9133179B2 (en) | 2011-01-19 | 2015-09-15 | Actelion Pharmaceuticals Ltd. | 2-methoxy-pyridin-4-yl-derivatives |
| US8877759B2 (en) | 2011-04-05 | 2014-11-04 | Vertex Pharnaceuticals Incorporated | Aminopyrazines as ATR kinase inhibitors |
| US9309250B2 (en) | 2011-06-22 | 2016-04-12 | Vertex Pharmaceuticals Incorporated | Substituted pyrrolo[2,3-b]pyrazines as ATR kinase inhibitors |
| US8822469B2 (en) | 2011-06-22 | 2014-09-02 | Vertex Pharmaceuticals Incorporated | Pyrrolo[2,3-B]pyrazines useful as inhibitors of ATR kinase |
| US9096602B2 (en) | 2011-06-22 | 2015-08-04 | Vertex Pharmaceuticals Incorporated | Substituted pyrrolo[2,3-B]pyrazines as ATR kinase inhibitors |
| US10822331B2 (en) | 2011-09-30 | 2020-11-03 | Vertex Pharmaceuticals Incorporated | Processes for preparing ATR inhibitors |
| US9862709B2 (en) | 2011-09-30 | 2018-01-09 | Vertex Pharmaceuticals Incorporated | Processes for making compounds useful as inhibitors of ATR kinase |
| US8765751B2 (en) | 2011-09-30 | 2014-07-01 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US8853217B2 (en) | 2011-09-30 | 2014-10-07 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US8846686B2 (en) | 2011-09-30 | 2014-09-30 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US10208027B2 (en) | 2011-09-30 | 2019-02-19 | Vertex Pharmaceuticals Incorporated | Processes for preparing ATR inhibitors |
| US10813929B2 (en) | 2011-09-30 | 2020-10-27 | Vertex Pharmaceuticals Incorporated | Treating cancer with ATR inhibitors |
| US9035053B2 (en) | 2011-09-30 | 2015-05-19 | Vertex Pharmaceuticals Incorporated | Processes for making compounds useful as inhibitors of ATR kinase |
| US8846917B2 (en) | 2011-11-09 | 2014-09-30 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US8846918B2 (en) | 2011-11-09 | 2014-09-30 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US8841449B2 (en) | 2011-11-09 | 2014-09-23 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US8841450B2 (en) | 2011-11-09 | 2014-09-23 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US8841337B2 (en) | 2011-11-09 | 2014-09-23 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US11110086B2 (en) | 2012-04-05 | 2021-09-07 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase and combination therapies thereof |
| US10478430B2 (en) | 2012-04-05 | 2019-11-19 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase and combination therapies thereof |
| US9791456B2 (en) | 2012-10-04 | 2017-10-17 | Vertex Pharmaceuticals Incorporated | Method for measuring ATR inhibition mediated increases in DNA damage |
| US8912198B2 (en) | 2012-10-16 | 2014-12-16 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US8735433B1 (en) | 2012-11-14 | 2014-05-27 | Allergan, Inc. | Aryl oxadiazole derivatives as sphingosine 1-phosphate (S1P) receptor modulators |
| US11117900B2 (en) | 2012-12-07 | 2021-09-14 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US10392391B2 (en) | 2012-12-07 | 2019-08-27 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US10787452B2 (en) | 2012-12-07 | 2020-09-29 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US11370798B2 (en) | 2012-12-07 | 2022-06-28 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9340546B2 (en) | 2012-12-07 | 2016-05-17 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9718827B2 (en) | 2012-12-07 | 2017-08-01 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9650381B2 (en) | 2012-12-07 | 2017-05-16 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US8871755B2 (en) | 2013-02-12 | 2014-10-28 | Allergan, Inc. | Alkene azetidine derivatives as sphingosine 1-phosphate (S1P) receptor modulators |
| US9663519B2 (en) | 2013-03-15 | 2017-05-30 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US10160760B2 (en) | 2013-12-06 | 2018-12-25 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US11485739B2 (en) | 2013-12-06 | 2022-11-01 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US10815239B2 (en) | 2013-12-06 | 2020-10-27 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US10093676B2 (en) | 2014-06-05 | 2018-10-09 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US9670215B2 (en) | 2014-06-05 | 2017-06-06 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US10800781B2 (en) | 2014-06-05 | 2020-10-13 | Vertex Pharmaceuticals Incorporated | Compounds useful as inhibitors of ATR kinase |
| US11179394B2 (en) | 2014-06-17 | 2021-11-23 | Vertex Pharmaceuticals Incorporated | Method for treating cancer using a combination of Chk1 and ATR inhibitors |
| US11007175B2 (en) | 2015-01-06 | 2021-05-18 | Arena Pharmaceuticals, Inc. | Methods of treating conditions related to the S1P1 receptor |
| US11896578B2 (en) | 2015-01-06 | 2024-02-13 | Arena Pharmaceuticals, Inc. | Methods of treating conditions related to the S1P1 receptor |
| US10836754B2 (en) | 2015-05-20 | 2020-11-17 | Idorsia Pharmaceuticals Ltd | Crystalline form of the compound (S)-3-{4-[5-(2-cyclopentyl-6-methoxy-pyridin-4-yl)-[1,2,4]oxadiazol-3-yl]-2-ethyl-6-methyl-phenoxy}-propane-1,2-diol |
| US10385043B2 (en) | 2015-05-20 | 2019-08-20 | Idorsia Pharmaceuticals Ltd | Crystalline form of the compound (S)-3-{4-[5-(2-cyclopentyl-6-methoxy-pyridin-4-yl)-[1,2,4]oxadiazol-3-yl]-2-ethyl-6-methyl-phenoxy}-propane-1,2-diol |
| US11390615B2 (en) | 2015-05-20 | 2022-07-19 | Idorsia Pharmaceuticals Ltd | Crystalline form of the compound (S)-3-{4-[5-(2-cyclopentyl-6-methoxy-pyridin-4-yl)-[1,2,4]oxadiazol-3-yl]-2-ethyl-6-methyl-phenox |
| US11834443B2 (en) | 2015-05-20 | 2023-12-05 | Idorsia Pharmaceuticals Ltd | Crystalline form of the compound (s)-3-{4-[5-(2-cyclopentyl-6-methoxy-pyridin-4-yl)-[1,2,4]oxadiazol-3-yl]-2-ethyl-6-methyl-phenoxy}-propane-1,2-diol |
| US10301262B2 (en) | 2015-06-22 | 2019-05-28 | Arena Pharmaceuticals, Inc. | Crystalline L-arginine salt of (R)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-1,2,3,4-tetrahydrocyclo-penta [b]indol-3-yl)acetic acid(Compund1) for use in SIPI receptor-associated disorders |
| US10676435B2 (en) | 2015-06-22 | 2020-06-09 | Arena Pharmaceuticals, Inc. | Crystalline L-arginine salt of (R)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-1,2,3,4-tetrahydrocyclo-penta [b]indol-3-yl)acetic acid(Compound 1) for use in SIPI receptor-associated disorders |
| US11884626B2 (en) | 2015-06-22 | 2024-01-30 | Arena Pharmaceuticals, Inc. | Crystalline L-arginine salt of (R)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-1,2,3,4-tetrahydrocyclo-penta [b]indol-3-yl)acetic acid(Compound1) for use in S1P1 receptor-associated disorders |
| US11091435B2 (en) | 2015-06-22 | 2021-08-17 | Arena Pharmaceuticals, Inc. | Crystalline L-arginine salt of (R)-2-(7-(4-cyclopentyl-3-(trifluoromethyl)benzyloxy)-1,2,3, 4-tetrahydrocyclo-penta [b]indol-3-yl)acetic acid(compound1) for use in S1P1 receptor-associated disorders |
| US11464774B2 (en) | 2015-09-30 | 2022-10-11 | Vertex Pharmaceuticals Incorporated | Method for treating cancer using a combination of DNA damaging agents and ATR inhibitors |
| US11534424B2 (en) | 2017-02-16 | 2022-12-27 | Arena Pharmaceuticals, Inc. | Compounds and methods for treatment of primary biliary cholangitis |
| US11478448B2 (en) | 2017-02-16 | 2022-10-25 | Arena Pharmaceuticals, Inc. | Compounds and methods for treatment of inflammatory bowel disease with extra-intestinal manifestations |
Also Published As
| Publication number | Publication date |
|---|---|
| CN1788008A (en) | 2006-06-14 |
| WO2004103279A3 (en) | 2005-05-19 |
| EP1625123A2 (en) | 2006-02-15 |
| JP2006528980A (en) | 2006-12-28 |
| US20060252741A1 (en) | 2006-11-09 |
| EP1625123A4 (en) | 2007-08-29 |
| CA2524867A1 (en) | 2004-12-02 |
| AU2004240586A1 (en) | 2004-12-02 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1625123A2 (en) | 3-(2-amino-1-azacyclyl)-5-aryl-1,2,4-oxadiazoles as s1p receptor agonists | |
| AU2003297232B2 (en) | 1-(amino)indanes and (1,2-dihydro-3-amino)-benzofurans, benzothiophenes and indoles | |
| US7199142B2 (en) | 1-((5-aryl-1,2,4-oxadiazol-3-yl) benzyl)azetidine-3-carboxylates and 1-((5-aryl-1,2,4-oxadiazol-3-yl)benzyl) pyrrolidine-3-carboxylates as edg receptor agonists | |
| EP1670463A2 (en) | 3,5-aryl, heteroaryl or cycloalkyl substituted-1,2,4-oxadiazoles as s1p receptor agonists | |
| JP4709488B2 (en) | N- (benzyl) aminoalkylcarboxylic acid compounds, phosphinic acid compounds, phosphonic acid compounds and tetrazoles as Edg receptor agonists | |
| US7479504B2 (en) | Edg receptor agonists | |
| JP2005531506A (en) | Aminoalkylphosphonates and related compounds as agonists of EDG receptors | |
| EP1804793A2 (en) | 2-(aryl)azacyclylmethyl carboxylates, sulfonates, phosphonates, phosphinates and heterocycles as s1p receptor agonists | |
| AU2003202994A1 (en) | N-(benzyl)aminoalkylcarboxylates, phosphinates, phosphonates and tetrazoles as Edg receptor agonists | |
| WO2003074008A2 (en) | Aminoalkylphosphonates and related compounds as edg receptor agonists |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2004240586 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006252741 Country of ref document: US Ref document number: 10554665 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 4936/DELNP/2005 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2524867 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2004240586 Country of ref document: AU Date of ref document: 20040512 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004240586 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 20048129905 Country of ref document: CN Ref document number: 2006532984 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004751981 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004751981 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 10554665 Country of ref document: US |