WO2004101790A1 - A process for recovering polypeptides that unfold reversibly from a polypeptide repertoire - Google Patents
A process for recovering polypeptides that unfold reversibly from a polypeptide repertoire Download PDFInfo
- Publication number
- WO2004101790A1 WO2004101790A1 PCT/GB2004/002102 GB2004002102W WO2004101790A1 WO 2004101790 A1 WO2004101790 A1 WO 2004101790A1 GB 2004002102 W GB2004002102 W GB 2004002102W WO 2004101790 A1 WO2004101790 A1 WO 2004101790A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- polypeptide
- amino acid
- polypeptides
- variant
- acid sequence
- Prior art date
Links
- 108090000765 processed proteins & peptides Proteins 0.000 title claims abstract description 1002
- 102000004196 processed proteins & peptides Human genes 0.000 title claims abstract description 994
- 229920001184 polypeptide Polymers 0.000 title claims abstract description 993
- 238000000034 method Methods 0.000 title claims abstract description 249
- 230000008569 process Effects 0.000 title claims abstract description 124
- 150000007523 nucleic acids Chemical class 0.000 claims abstract description 114
- 108020004707 nucleic acids Proteins 0.000 claims abstract description 112
- 102000039446 nucleic acids Human genes 0.000 claims abstract description 112
- 125000003275 alpha amino acid group Chemical group 0.000 claims description 285
- 108090000623 proteins and genes Proteins 0.000 claims description 228
- 235000001014 amino acid Nutrition 0.000 claims description 156
- 230000027455 binding Effects 0.000 claims description 125
- 150000001413 amino acids Chemical class 0.000 claims description 120
- 239000003446 ligand Substances 0.000 claims description 119
- 210000004602 germ cell Anatomy 0.000 claims description 107
- 210000004027 cell Anatomy 0.000 claims description 87
- 238000010438 heat treatment Methods 0.000 claims description 72
- 241001515965 unidentified phage Species 0.000 claims description 60
- 125000000539 amino acid group Chemical group 0.000 claims description 59
- 230000002776 aggregation Effects 0.000 claims description 56
- 238000004220 aggregation Methods 0.000 claims description 56
- 108060003951 Immunoglobulin Proteins 0.000 claims description 47
- 239000003795 chemical substances by application Substances 0.000 claims description 47
- 102000018358 immunoglobulin Human genes 0.000 claims description 47
- 239000000243 solution Substances 0.000 claims description 41
- 241000588724 Escherichia coli Species 0.000 claims description 38
- 239000000203 mixture Substances 0.000 claims description 36
- -1 Kabat amino acid Chemical class 0.000 claims description 33
- 230000002829 reductive effect Effects 0.000 claims description 33
- NFGXHKASABOEEW-UHFFFAOYSA-N 1-methylethyl 11-methoxy-3,7,11-trimethyl-2,4-dodecadienoate Chemical compound COC(C)(C)CCCC(C)CC=CC(C)=CC(=O)OC(C)C NFGXHKASABOEEW-UHFFFAOYSA-N 0.000 claims description 31
- 238000002823 phage display Methods 0.000 claims description 30
- 239000000427 antigen Substances 0.000 claims description 28
- 108091007433 antigens Proteins 0.000 claims description 28
- 102000036639 antigens Human genes 0.000 claims description 28
- 238000001816 cooling Methods 0.000 claims description 26
- 238000002844 melting Methods 0.000 claims description 23
- 230000008018 melting Effects 0.000 claims description 23
- 102000004190 Enzymes Human genes 0.000 claims description 22
- 108090000790 Enzymes Proteins 0.000 claims description 22
- 230000000694 effects Effects 0.000 claims description 21
- 230000001580 bacterial effect Effects 0.000 claims description 20
- 235000009508 confectionery Nutrition 0.000 claims description 20
- 230000002209 hydrophobic effect Effects 0.000 claims description 20
- 238000004519 manufacturing process Methods 0.000 claims description 20
- 241000894006 Bacteria Species 0.000 claims description 16
- 239000013612 plasmid Substances 0.000 claims description 15
- 239000000126 substance Substances 0.000 claims description 15
- 238000006467 substitution reaction Methods 0.000 claims description 15
- 235000018417 cysteine Nutrition 0.000 claims description 14
- 102000003675 cytokine receptors Human genes 0.000 claims description 14
- 108010057085 cytokine receptors Proteins 0.000 claims description 14
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 14
- 238000005119 centrifugation Methods 0.000 claims description 13
- XUJNEKJLAYXESH-UHFFFAOYSA-N cysteine Natural products SCC(N)C(O)=O XUJNEKJLAYXESH-UHFFFAOYSA-N 0.000 claims description 13
- 201000010099 disease Diseases 0.000 claims description 13
- 230000001524 infective effect Effects 0.000 claims description 13
- 102000004127 Cytokines Human genes 0.000 claims description 12
- 108090000695 Cytokines Proteins 0.000 claims description 12
- 239000002773 nucleotide Substances 0.000 claims description 12
- 125000003729 nucleotide group Chemical group 0.000 claims description 12
- 230000003197 catalytic effect Effects 0.000 claims description 10
- 230000001965 increasing effect Effects 0.000 claims description 9
- 230000017854 proteolysis Effects 0.000 claims description 9
- RXWNCPJZOCPEPQ-NVWDDTSBSA-N puromycin Chemical compound C1=CC(OC)=CC=C1C[C@H](N)C(=O)N[C@H]1[C@@H](O)[C@H](N2C3=NC=NC(=C3N=C2)N(C)C)O[C@@H]1CO RXWNCPJZOCPEPQ-NVWDDTSBSA-N 0.000 claims description 8
- 238000005406 washing Methods 0.000 claims description 8
- 230000003196 chaotropic effect Effects 0.000 claims description 7
- 230000001404 mediated effect Effects 0.000 claims description 7
- 125000000151 cysteine group Chemical group N[C@@H](CS)C(=O)* 0.000 claims description 6
- 238000012217 deletion Methods 0.000 claims description 6
- 230000037430 deletion Effects 0.000 claims description 6
- 239000003814 drug Substances 0.000 claims description 6
- 239000000839 emulsion Substances 0.000 claims description 6
- 230000002068 genetic effect Effects 0.000 claims description 6
- 239000003960 organic solvent Substances 0.000 claims description 6
- 102000052510 DNA-Binding Proteins Human genes 0.000 claims description 4
- 102000013463 Immunoglobulin Light Chains Human genes 0.000 claims description 4
- 108010065825 Immunoglobulin Light Chains Proteins 0.000 claims description 4
- 206010028980 Neoplasm Diseases 0.000 claims description 4
- 206010067584 Type 1 diabetes mellitus Diseases 0.000 claims description 4
- 238000002819 bacterial display Methods 0.000 claims description 4
- 238000002347 injection Methods 0.000 claims description 4
- 239000007924 injection Substances 0.000 claims description 4
- 238000003780 insertion Methods 0.000 claims description 4
- 230000037431 insertion Effects 0.000 claims description 4
- 238000002818 protein evolution Methods 0.000 claims description 4
- 229950010131 puromycin Drugs 0.000 claims description 4
- 238000002702 ribosome display Methods 0.000 claims description 4
- 201000000596 systemic lupus erythematosus Diseases 0.000 claims description 4
- 101000998953 Homo sapiens Immunoglobulin heavy variable 1-2 Proteins 0.000 claims description 3
- 102100036887 Immunoglobulin heavy variable 1-2 Human genes 0.000 claims description 3
- 239000002253 acid Substances 0.000 claims description 3
- 238000012377 drug delivery Methods 0.000 claims description 3
- 239000008194 pharmaceutical composition Substances 0.000 claims description 3
- 239000000843 powder Substances 0.000 claims description 3
- 230000001629 suppression Effects 0.000 claims description 3
- 238000002560 therapeutic procedure Methods 0.000 claims description 3
- 238000011282 treatment Methods 0.000 claims description 3
- 208000023275 Autoimmune disease Diseases 0.000 claims description 2
- 208000035143 Bacterial infection Diseases 0.000 claims description 2
- 208000011231 Crohn disease Diseases 0.000 claims description 2
- 206010020751 Hypersensitivity Diseases 0.000 claims description 2
- 208000036142 Viral infection Diseases 0.000 claims description 2
- 150000007513 acids Chemical class 0.000 claims description 2
- 208000026935 allergic disease Diseases 0.000 claims description 2
- 230000000172 allergic effect Effects 0.000 claims description 2
- 206010003246 arthritis Diseases 0.000 claims description 2
- 208000010668 atopic eczema Diseases 0.000 claims description 2
- 208000022362 bacterial infectious disease Diseases 0.000 claims description 2
- 201000011510 cancer Diseases 0.000 claims description 2
- 238000010494 dissociation reaction Methods 0.000 claims description 2
- 230000005593 dissociations Effects 0.000 claims description 2
- 230000009610 hypersensitivity Effects 0.000 claims description 2
- 230000002757 inflammatory effect Effects 0.000 claims description 2
- 201000006417 multiple sclerosis Diseases 0.000 claims description 2
- 230000008685 targeting Effects 0.000 claims description 2
- 230000009385 viral infection Effects 0.000 claims description 2
- 101710096438 DNA-binding protein Proteins 0.000 claims 3
- 238000003745 diagnosis Methods 0.000 claims 2
- 241000702374 Enterobacteria phage fd Species 0.000 claims 1
- 101001008255 Homo sapiens Immunoglobulin kappa variable 1D-8 Proteins 0.000 claims 1
- 101001047628 Homo sapiens Immunoglobulin kappa variable 2-29 Proteins 0.000 claims 1
- 101001008321 Homo sapiens Immunoglobulin kappa variable 2D-26 Proteins 0.000 claims 1
- 101001047619 Homo sapiens Immunoglobulin kappa variable 3-20 Proteins 0.000 claims 1
- 101001008263 Homo sapiens Immunoglobulin kappa variable 3D-15 Proteins 0.000 claims 1
- 102000006496 Immunoglobulin Heavy Chains Human genes 0.000 claims 1
- 108010019476 Immunoglobulin Heavy Chains Proteins 0.000 claims 1
- 102100022964 Immunoglobulin kappa variable 3-20 Human genes 0.000 claims 1
- 201000004681 Psoriasis Diseases 0.000 claims 1
- 208000006673 asthma Diseases 0.000 claims 1
- 208000035475 disorder Diseases 0.000 claims 1
- 208000032839 leukemia Diseases 0.000 claims 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims 1
- 238000011321 prophylaxis Methods 0.000 claims 1
- 230000002194 synthesizing effect Effects 0.000 claims 1
- 101150109891 vn gene Proteins 0.000 claims 1
- 102000004169 proteins and genes Human genes 0.000 description 108
- 235000018102 proteins Nutrition 0.000 description 100
- 229940024606 amino acid Drugs 0.000 description 99
- LOKCTEFSRHRXRJ-UHFFFAOYSA-I dipotassium trisodium dihydrogen phosphate hydrogen phosphate dichloride Chemical compound P(=O)(O)(O)[O-].[K+].P(=O)(O)([O-])[O-].[Na+].[Na+].[Cl-].[K+].[Cl-].[Na+] LOKCTEFSRHRXRJ-UHFFFAOYSA-I 0.000 description 57
- 239000002953 phosphate buffered saline Substances 0.000 description 57
- 238000003752 polymerase chain reaction Methods 0.000 description 53
- 239000013598 vector Substances 0.000 description 50
- 238000002965 ELISA Methods 0.000 description 48
- 230000035772 mutation Effects 0.000 description 34
- 230000002441 reversible effect Effects 0.000 description 29
- 108010026228 mRNA guanylyltransferase Proteins 0.000 description 28
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 25
- 239000013615 primer Substances 0.000 description 24
- 239000006228 supernatant Substances 0.000 description 24
- 239000012634 fragment Substances 0.000 description 22
- 108020004705 Codon Proteins 0.000 description 19
- 238000012216 screening Methods 0.000 description 19
- 239000000047 product Substances 0.000 description 17
- 108020004414 DNA Proteins 0.000 description 16
- 229940088598 enzyme Drugs 0.000 description 16
- 239000004098 Tetracycline Substances 0.000 description 15
- 238000002983 circular dichroism Methods 0.000 description 15
- 238000010790 dilution Methods 0.000 description 15
- 239000012895 dilution Substances 0.000 description 15
- 239000013604 expression vector Substances 0.000 description 15
- 229960002180 tetracycline Drugs 0.000 description 15
- 229930101283 tetracycline Natural products 0.000 description 15
- 235000019364 tetracycline Nutrition 0.000 description 15
- 150000003522 tetracyclines Chemical class 0.000 description 15
- 101100112922 Candida albicans CDR3 gene Proteins 0.000 description 13
- 108010047041 Complementarity Determining Regions Proteins 0.000 description 13
- PEDCQBHIVMGVHV-UHFFFAOYSA-N Glycerine Chemical compound OCC(O)CO PEDCQBHIVMGVHV-UHFFFAOYSA-N 0.000 description 13
- 230000003247 decreasing effect Effects 0.000 description 13
- 239000011780 sodium chloride Substances 0.000 description 13
- 108091034117 Oligonucleotide Proteins 0.000 description 12
- XSQUKJJJFZCRTK-UHFFFAOYSA-N Urea Chemical compound NC(N)=O XSQUKJJJFZCRTK-UHFFFAOYSA-N 0.000 description 12
- 239000000872 buffer Substances 0.000 description 12
- 230000010076 replication Effects 0.000 description 12
- 102100035360 Cerebellar degeneration-related antigen 1 Human genes 0.000 description 11
- 102000009465 Growth Factor Receptors Human genes 0.000 description 11
- 108010009202 Growth Factor Receptors Proteins 0.000 description 11
- 108091006905 Human Serum Albumin Proteins 0.000 description 11
- 125000000010 L-asparaginyl group Chemical group O=C([*])[C@](N([H])[H])([H])C([H])([H])C(=O)N([H])[H] 0.000 description 11
- 238000003556 assay Methods 0.000 description 11
- 239000003102 growth factor Substances 0.000 description 11
- 239000011159 matrix material Substances 0.000 description 11
- 238000000746 purification Methods 0.000 description 11
- 241000894007 species Species 0.000 description 11
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 10
- 102000008100 Human Serum Albumin Human genes 0.000 description 10
- 102000018594 Tumour necrosis factor Human genes 0.000 description 10
- 108050007852 Tumour necrosis factor Proteins 0.000 description 10
- 238000010367 cloning Methods 0.000 description 10
- 102000005962 receptors Human genes 0.000 description 10
- 108020003175 receptors Proteins 0.000 description 10
- 239000000758 substrate Substances 0.000 description 10
- 229920001213 Polysorbate 20 Polymers 0.000 description 9
- 238000006243 chemical reaction Methods 0.000 description 9
- 125000002228 disulfide group Chemical group 0.000 description 9
- 238000011534 incubation Methods 0.000 description 9
- 239000000256 polyoxyethylene sorbitan monolaurate Substances 0.000 description 9
- 235000010486 polyoxyethylene sorbitan monolaurate Nutrition 0.000 description 9
- 231100000617 superantigen Toxicity 0.000 description 9
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 8
- 241000724791 Filamentous phage Species 0.000 description 8
- 102000004142 Trypsin Human genes 0.000 description 8
- 108090000631 Trypsin Proteins 0.000 description 8
- 238000013459 approach Methods 0.000 description 8
- 238000004925 denaturation Methods 0.000 description 8
- 238000011084 recovery Methods 0.000 description 8
- 230000007704 transition Effects 0.000 description 8
- 239000012588 trypsin Substances 0.000 description 8
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 7
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 7
- 230000036425 denaturation Effects 0.000 description 7
- 230000001419 dependent effect Effects 0.000 description 7
- 238000001514 detection method Methods 0.000 description 7
- 238000003505 heat denaturation Methods 0.000 description 7
- 230000001976 improved effect Effects 0.000 description 7
- 239000003550 marker Substances 0.000 description 7
- 239000002609 medium Substances 0.000 description 7
- 229910052751 metal Inorganic materials 0.000 description 7
- 239000002184 metal Substances 0.000 description 7
- 239000013642 negative control Substances 0.000 description 7
- 229920001223 polyethylene glycol Polymers 0.000 description 7
- 239000013641 positive control Substances 0.000 description 7
- 238000002360 preparation method Methods 0.000 description 7
- 239000011347 resin Substances 0.000 description 7
- 229920005989 resin Polymers 0.000 description 7
- ZWEHNKRNPOVVGH-UHFFFAOYSA-N 2-Butanone Chemical compound CCC(C)=O ZWEHNKRNPOVVGH-UHFFFAOYSA-N 0.000 description 6
- 108091033380 Coding strand Proteins 0.000 description 6
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 6
- 101000686985 Mouse mammary tumor virus (strain C3H) Protein PR73 Proteins 0.000 description 6
- 102220501005 Nuclear cap-binding protein subunit 2_Y23F_mutation Human genes 0.000 description 6
- 240000004808 Saccharomyces cerevisiae Species 0.000 description 6
- 235000014680 Saccharomyces cerevisiae Nutrition 0.000 description 6
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 6
- 101710120037 Toxin CcdB Proteins 0.000 description 6
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 6
- 238000007792 addition Methods 0.000 description 6
- 230000003321 amplification Effects 0.000 description 6
- 239000004202 carbamide Substances 0.000 description 6
- 238000002523 gelfiltration Methods 0.000 description 6
- 230000003993 interaction Effects 0.000 description 6
- 230000002427 irreversible effect Effects 0.000 description 6
- 238000003199 nucleic acid amplification method Methods 0.000 description 6
- 230000006798 recombination Effects 0.000 description 6
- 238000005215 recombination Methods 0.000 description 6
- 230000000717 retained effect Effects 0.000 description 6
- 239000000725 suspension Substances 0.000 description 6
- 230000001225 therapeutic effect Effects 0.000 description 6
- 238000001712 DNA sequencing Methods 0.000 description 5
- CKLJMWTZIZZHCS-REOHCLBHSA-N L-aspartic acid Chemical group OC(=O)[C@@H](N)CC(O)=O CKLJMWTZIZZHCS-REOHCLBHSA-N 0.000 description 5
- 241001529936 Murinae Species 0.000 description 5
- 241000699666 Mus <mouse, genus> Species 0.000 description 5
- 241000699670 Mus sp. Species 0.000 description 5
- 102100024952 Protein CBFA2T1 Human genes 0.000 description 5
- 108060008683 Tumor Necrosis Factor Receptor Proteins 0.000 description 5
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 description 5
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 description 5
- 238000004458 analytical method Methods 0.000 description 5
- 239000011324 bead Substances 0.000 description 5
- 239000012148 binding buffer Substances 0.000 description 5
- 230000015572 biosynthetic process Effects 0.000 description 5
- 230000008859 change Effects 0.000 description 5
- 238000004520 electroporation Methods 0.000 description 5
- 108020001507 fusion proteins Proteins 0.000 description 5
- 102000037865 fusion proteins Human genes 0.000 description 5
- 230000012010 growth Effects 0.000 description 5
- 239000000833 heterodimer Substances 0.000 description 5
- 150000001455 metallic ions Chemical class 0.000 description 5
- 238000002703 mutagenesis Methods 0.000 description 5
- 231100000350 mutagenesis Toxicity 0.000 description 5
- 108700010839 phage proteins Proteins 0.000 description 5
- 102000003298 tumor necrosis factor receptor Human genes 0.000 description 5
- YBJHBAHKTGYVGT-ZKWXMUAHSA-N (+)-Biotin Chemical compound N1C(=O)N[C@@H]2[C@H](CCCCC(=O)O)SC[C@@H]21 YBJHBAHKTGYVGT-ZKWXMUAHSA-N 0.000 description 4
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 4
- 206010069754 Acquired gene mutation Diseases 0.000 description 4
- 229920000936 Agarose Polymers 0.000 description 4
- 101710132601 Capsid protein Proteins 0.000 description 4
- 102000014914 Carrier Proteins Human genes 0.000 description 4
- 101710094648 Coat protein Proteins 0.000 description 4
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 4
- 102100021181 Golgi phosphoprotein 3 Human genes 0.000 description 4
- 108010001336 Horseradish Peroxidase Proteins 0.000 description 4
- 108010058683 Immobilized Proteins Proteins 0.000 description 4
- 102000004890 Interleukin-8 Human genes 0.000 description 4
- 108090001007 Interleukin-8 Proteins 0.000 description 4
- AYFVYJQAPQTCCC-GBXIJSLDSA-N L-threonine Chemical compound C[C@@H](O)[C@H](N)C(O)=O AYFVYJQAPQTCCC-GBXIJSLDSA-N 0.000 description 4
- 101710125418 Major capsid protein Proteins 0.000 description 4
- 241000124008 Mammalia Species 0.000 description 4
- 241001465754 Metazoa Species 0.000 description 4
- 101710141454 Nucleoprotein Proteins 0.000 description 4
- 101710083689 Probable capsid protein Proteins 0.000 description 4
- 108010090804 Streptavidin Proteins 0.000 description 4
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 4
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 4
- 102100033732 Tumor necrosis factor receptor superfamily member 1A Human genes 0.000 description 4
- 102220541155 Tumor necrosis factor receptor superfamily member 5_S35G_mutation Human genes 0.000 description 4
- 238000002835 absorbance Methods 0.000 description 4
- 229960000723 ampicillin Drugs 0.000 description 4
- AVKUERGKIZMTKX-NJBDSQKTSA-N ampicillin Chemical compound C1([C@@H](N)C(=O)N[C@H]2[C@H]3SC([C@@H](N3C2=O)C(O)=O)(C)C)=CC=CC=C1 AVKUERGKIZMTKX-NJBDSQKTSA-N 0.000 description 4
- 230000008901 benefit Effects 0.000 description 4
- 238000001142 circular dichroism spectrum Methods 0.000 description 4
- 150000001875 compounds Chemical class 0.000 description 4
- 239000001963 growth medium Substances 0.000 description 4
- 239000000710 homodimer Substances 0.000 description 4
- 230000001939 inductive effect Effects 0.000 description 4
- 208000015181 infectious disease Diseases 0.000 description 4
- 210000004962 mammalian cell Anatomy 0.000 description 4
- 230000036961 partial effect Effects 0.000 description 4
- 239000008188 pellet Substances 0.000 description 4
- 238000012163 sequencing technique Methods 0.000 description 4
- 230000037439 somatic mutation Effects 0.000 description 4
- 238000001228 spectrum Methods 0.000 description 4
- 238000003860 storage Methods 0.000 description 4
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- 235000002198 Annona diversifolia Nutrition 0.000 description 3
- 102100021935 C-C motif chemokine 26 Human genes 0.000 description 3
- 108091026890 Coding region Proteins 0.000 description 3
- 102100039498 Cytotoxic T-lymphocyte protein 4 Human genes 0.000 description 3
- 108090001047 Fibroblast growth factor 10 Proteins 0.000 description 3
- 241000238631 Hexapoda Species 0.000 description 3
- 101000897493 Homo sapiens C-C motif chemokine 26 Proteins 0.000 description 3
- 101000889276 Homo sapiens Cytotoxic T-lymphocyte protein 4 Proteins 0.000 description 3
- 108010067060 Immunoglobulin Variable Region Proteins 0.000 description 3
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 3
- 239000008118 PEG 6000 Substances 0.000 description 3
- 229920002584 Polyethylene Glycol 6000 Polymers 0.000 description 3
- 239000004353 Polyethylene glycol 8000 Substances 0.000 description 3
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 3
- 108010076504 Protein Sorting Signals Proteins 0.000 description 3
- 229920002684 Sepharose Polymers 0.000 description 3
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 3
- 108010006785 Taq Polymerase Proteins 0.000 description 3
- 101710187743 Tumor necrosis factor receptor superfamily member 1A Proteins 0.000 description 3
- 241000700605 Viruses Species 0.000 description 3
- 230000002378 acidificating effect Effects 0.000 description 3
- WQZGKKKJIJFFOK-VFUOTHLCSA-N beta-D-glucose Chemical compound OC[C@H]1O[C@@H](O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-VFUOTHLCSA-N 0.000 description 3
- 108091008324 binding proteins Proteins 0.000 description 3
- 238000007413 biotinylation Methods 0.000 description 3
- 230000006287 biotinylation Effects 0.000 description 3
- 230000000903 blocking effect Effects 0.000 description 3
- 239000003153 chemical reaction reagent Substances 0.000 description 3
- 239000011248 coating agent Substances 0.000 description 3
- 238000000576 coating method Methods 0.000 description 3
- 239000012228 culture supernatant Substances 0.000 description 3
- 239000008121 dextrose Substances 0.000 description 3
- 239000010432 diamond Substances 0.000 description 3
- 229940079593 drug Drugs 0.000 description 3
- 238000010828 elution Methods 0.000 description 3
- 239000013613 expression plasmid Substances 0.000 description 3
- 230000004927 fusion Effects 0.000 description 3
- BRZYSWJRSDMWLG-CAXSIQPQSA-N geneticin Natural products O1C[C@@](O)(C)[C@H](NC)[C@@H](O)[C@H]1O[C@@H]1[C@@H](O)[C@H](O[C@@H]2[C@@H]([C@@H](O)[C@H](O)[C@@H](C(C)O)O2)N)[C@@H](N)C[C@H]1N BRZYSWJRSDMWLG-CAXSIQPQSA-N 0.000 description 3
- 239000011521 glass Substances 0.000 description 3
- 230000010354 integration Effects 0.000 description 3
- 230000014759 maintenance of location Effects 0.000 description 3
- 239000012528 membrane Substances 0.000 description 3
- 229960000485 methotrexate Drugs 0.000 description 3
- 238000002156 mixing Methods 0.000 description 3
- 230000000869 mutational effect Effects 0.000 description 3
- 206010028417 myasthenia gravis Diseases 0.000 description 3
- 230000003287 optical effect Effects 0.000 description 3
- 238000004091 panning Methods 0.000 description 3
- 239000002245 particle Substances 0.000 description 3
- 235000019446 polyethylene glycol 8000 Nutrition 0.000 description 3
- 229940085678 polyethylene glycol 8000 Drugs 0.000 description 3
- 210000004896 polypeptide structure Anatomy 0.000 description 3
- 230000000069 prophylactic effect Effects 0.000 description 3
- 230000004845 protein aggregation Effects 0.000 description 3
- 102200090666 rs1556026984 Human genes 0.000 description 3
- 150000003839 salts Chemical class 0.000 description 3
- 239000007787 solid Substances 0.000 description 3
- 238000004611 spectroscopical analysis Methods 0.000 description 3
- 238000004448 titration Methods 0.000 description 3
- 230000002463 transducing effect Effects 0.000 description 3
- 230000014616 translation Effects 0.000 description 3
- 238000013519 translation Methods 0.000 description 3
- 230000003612 virological effect Effects 0.000 description 3
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 2
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 2
- YRNWIFYIFSBPAU-UHFFFAOYSA-N 4-[4-(dimethylamino)phenyl]-n,n-dimethylaniline Chemical compound C1=CC(N(C)C)=CC=C1C1=CC=C(N(C)C)C=C1 YRNWIFYIFSBPAU-UHFFFAOYSA-N 0.000 description 2
- 229920001817 Agar Polymers 0.000 description 2
- 244000153158 Ammi visnaga Species 0.000 description 2
- 235000010585 Ammi visnaga Nutrition 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- 240000003291 Armoracia rusticana Species 0.000 description 2
- 235000011330 Armoracia rusticana Nutrition 0.000 description 2
- 102100023995 Beta-nerve growth factor Human genes 0.000 description 2
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 2
- 102100021943 C-C motif chemokine 2 Human genes 0.000 description 2
- 241000282832 Camelidae Species 0.000 description 2
- 102100035361 Cerebellar degeneration-related protein 2 Human genes 0.000 description 2
- HEDRZPFGACZZDS-UHFFFAOYSA-N Chloroform Chemical compound ClC(Cl)Cl HEDRZPFGACZZDS-UHFFFAOYSA-N 0.000 description 2
- 102000053602 DNA Human genes 0.000 description 2
- 108010014303 DNA-directed DNA polymerase Proteins 0.000 description 2
- 102000016928 DNA-directed DNA polymerase Human genes 0.000 description 2
- 102100031107 Disintegrin and metalloproteinase domain-containing protein 11 Human genes 0.000 description 2
- 101710121366 Disintegrin and metalloproteinase domain-containing protein 11 Proteins 0.000 description 2
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 2
- 241000255581 Drosophila <fruit fly, genus> Species 0.000 description 2
- 241000196324 Embryophyta Species 0.000 description 2
- 241001198387 Escherichia coli BL21(DE3) Species 0.000 description 2
- 241000206602 Eukaryota Species 0.000 description 2
- 102220614871 Extracellular calcium-sensing receptor_S53P_mutation Human genes 0.000 description 2
- 102000004864 Fibroblast growth factor 10 Human genes 0.000 description 2
- 102100037362 Fibronectin Human genes 0.000 description 2
- 108010067306 Fibronectins Proteins 0.000 description 2
- 102100020997 Fractalkine Human genes 0.000 description 2
- 102100034221 Growth-regulated alpha protein Human genes 0.000 description 2
- 101000740205 Homo sapiens Sal-like protein 1 Proteins 0.000 description 2
- 101000801228 Homo sapiens Tumor necrosis factor receptor superfamily member 1A Proteins 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 2
- 108010054477 Immunoglobulin Fab Fragments Proteins 0.000 description 2
- 102000001706 Immunoglobulin Fab Fragments Human genes 0.000 description 2
- 102000017727 Immunoglobulin Variable Region Human genes 0.000 description 2
- 108090000723 Insulin-Like Growth Factor I Proteins 0.000 description 2
- 102100022341 Integrin alpha-E Human genes 0.000 description 2
- 241000235058 Komagataella pastoris Species 0.000 description 2
- QIVBCDIJIAJPQS-VIFPVBQESA-N L-tryptophane Chemical compound C1=CC=C2C(C[C@H](N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-VIFPVBQESA-N 0.000 description 2
- 241000282838 Lama Species 0.000 description 2
- 108090000542 Lymphotoxin-alpha Proteins 0.000 description 2
- 102000004083 Lymphotoxin-alpha Human genes 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- 229930193140 Neomycin Natural products 0.000 description 2
- 241000283973 Oryctolagus cuniculus Species 0.000 description 2
- 238000012408 PCR amplification Methods 0.000 description 2
- 102000035195 Peptidases Human genes 0.000 description 2
- 108091005804 Peptidases Proteins 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- 239000004365 Protease Substances 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- 102100037204 Sal-like protein 1 Human genes 0.000 description 2
- BMKNXTJLHFIAAH-CIUDSAMLSA-N Ser-Ser-Leu Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(O)=O BMKNXTJLHFIAAH-CIUDSAMLSA-N 0.000 description 2
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 2
- 108091008874 T cell receptors Proteins 0.000 description 2
- 102000016266 T-Cell Antigen Receptors Human genes 0.000 description 2
- 241000589500 Thermus aquaticus Species 0.000 description 2
- QIVBCDIJIAJPQS-UHFFFAOYSA-N Tryptophan Natural products C1=CC=C2C(CC(N)C(O)=O)=CNC2=C1 QIVBCDIJIAJPQS-UHFFFAOYSA-N 0.000 description 2
- 108091008605 VEGF receptors Proteins 0.000 description 2
- 102000009484 Vascular Endothelial Growth Factor Receptors Human genes 0.000 description 2
- 230000035508 accumulation Effects 0.000 description 2
- 238000009825 accumulation Methods 0.000 description 2
- 230000009824 affinity maturation Effects 0.000 description 2
- 239000008272 agar Substances 0.000 description 2
- 238000013019 agitation Methods 0.000 description 2
- 235000004279 alanine Nutrition 0.000 description 2
- 125000003295 alanine group Chemical group N[C@@H](C)C(=O)* 0.000 description 2
- 150000001299 aldehydes Chemical class 0.000 description 2
- 108010058061 alpha E integrins Proteins 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 230000000692 anti-sense effect Effects 0.000 description 2
- 229940088710 antibiotic agent Drugs 0.000 description 2
- 229960002685 biotin Drugs 0.000 description 2
- 235000020958 biotin Nutrition 0.000 description 2
- 239000011616 biotin Substances 0.000 description 2
- OWMVSZAMULFTJU-UHFFFAOYSA-N bis-tris Chemical compound OCCN(CCO)C(CO)(CO)CO OWMVSZAMULFTJU-UHFFFAOYSA-N 0.000 description 2
- 210000004369 blood Anatomy 0.000 description 2
- 239000008280 blood Substances 0.000 description 2
- 229940098773 bovine serum albumin Drugs 0.000 description 2
- 210000000234 capsid Anatomy 0.000 description 2
- 239000002738 chelating agent Substances 0.000 description 2
- 210000000349 chromosome Anatomy 0.000 description 2
- 238000003776 cleavage reaction Methods 0.000 description 2
- 238000010276 construction Methods 0.000 description 2
- 229910052802 copper Inorganic materials 0.000 description 2
- 239000010949 copper Substances 0.000 description 2
- 230000009260 cross reactivity Effects 0.000 description 2
- 238000013461 design Methods 0.000 description 2
- 238000012938 design process Methods 0.000 description 2
- 230000003292 diminished effect Effects 0.000 description 2
- 235000013601 eggs Nutrition 0.000 description 2
- 238000001493 electron microscopy Methods 0.000 description 2
- 239000012149 elution buffer Substances 0.000 description 2
- 239000003623 enhancer Substances 0.000 description 2
- 238000009472 formulation Methods 0.000 description 2
- 238000004108 freeze drying Methods 0.000 description 2
- 238000007710 freezing Methods 0.000 description 2
- 230000008014 freezing Effects 0.000 description 2
- 238000002825 functional assay Methods 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 2
- 229960000789 guanidine hydrochloride Drugs 0.000 description 2
- PJJJBBJSCAKJQF-UHFFFAOYSA-N guanidinium chloride Chemical compound [Cl-].NC(N)=[NH2+] PJJJBBJSCAKJQF-UHFFFAOYSA-N 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- 239000001257 hydrogen Substances 0.000 description 2
- 125000004435 hydrogen atom Chemical class [H]* 0.000 description 2
- NBZBKCUXIYYUSX-UHFFFAOYSA-N iminodiacetic acid Chemical compound OC(=O)CNCC(O)=O NBZBKCUXIYYUSX-UHFFFAOYSA-N 0.000 description 2
- 238000000338 in vitro Methods 0.000 description 2
- 210000003000 inclusion body Anatomy 0.000 description 2
- 238000010348 incorporation Methods 0.000 description 2
- 239000000893 inhibin Substances 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- ZPNFWUPYTFPOJU-LPYSRVMUSA-N iniprol Chemical compound C([C@H]1C(=O)NCC(=O)NCC(=O)N[C@H]2CSSC[C@H]3C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@H](C(N[C@H](C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=4C=CC(O)=CC=4)C(=O)N[C@@H](CC=4C=CC=CC=4)C(=O)N[C@@H](CC=4C=CC(O)=CC=4)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CC=4C=CC=CC=4)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC2=O)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CC=2C=CC=CC=2)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H]2N(CCC2)C(=O)[C@@H](N)CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N2[C@@H](CCC2)C(=O)N2[C@@H](CCC2)C(=O)N[C@@H](CC=2C=CC(O)=CC=2)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N2[C@@H](CCC2)C(=O)N3)C(=O)NCC(=O)NCC(=O)N[C@@H](C)C(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@H](C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@H](C(=O)N1)C(C)C)[C@@H](C)O)[C@@H](C)CC)=O)[C@@H](C)CC)C1=CC=C(O)C=C1 ZPNFWUPYTFPOJU-LPYSRVMUSA-N 0.000 description 2
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 2
- 239000000543 intermediate Substances 0.000 description 2
- 150000002576 ketones Chemical class 0.000 description 2
- 230000002147 killing effect Effects 0.000 description 2
- 235000010335 lysozyme Nutrition 0.000 description 2
- WSFSSNUMVMOOMR-NJFSPNSNSA-N methanone Chemical compound O=[14CH2] WSFSSNUMVMOOMR-NJFSPNSNSA-N 0.000 description 2
- 238000012544 monitoring process Methods 0.000 description 2
- 210000001616 monocyte Anatomy 0.000 description 2
- 229960004927 neomycin Drugs 0.000 description 2
- 235000015097 nutrients Nutrition 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- 239000004033 plastic Substances 0.000 description 2
- 229920003023 plastic Polymers 0.000 description 2
- 229920000642 polymer Polymers 0.000 description 2
- 230000001681 protective effect Effects 0.000 description 2
- 230000006432 protein unfolding Effects 0.000 description 2
- 239000011541 reaction mixture Substances 0.000 description 2
- 238000001525 receptor binding assay Methods 0.000 description 2
- 230000022532 regulation of transcription, DNA-dependent Effects 0.000 description 2
- 108091008146 restriction endonucleases Proteins 0.000 description 2
- 102200043501 rs1800450 Human genes 0.000 description 2
- 102200151360 rs878854359 Human genes 0.000 description 2
- 230000007017 scission Effects 0.000 description 2
- 210000002966 serum Anatomy 0.000 description 2
- 239000001632 sodium acetate Substances 0.000 description 2
- 235000017281 sodium acetate Nutrition 0.000 description 2
- 238000002415 sodium dodecyl sulfate polyacrylamide gel electrophoresis Methods 0.000 description 2
- RPENMORRBUTCPR-UHFFFAOYSA-M sodium;1-hydroxy-2,5-dioxopyrrolidine-3-sulfonate Chemical compound [Na+].ON1C(=O)CC(S([O-])(=O)=O)C1=O RPENMORRBUTCPR-UHFFFAOYSA-M 0.000 description 2
- 238000010561 standard procedure Methods 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 2
- 238000013518 transcription Methods 0.000 description 2
- 230000035897 transcription Effects 0.000 description 2
- 230000002103 transcriptional effect Effects 0.000 description 2
- 208000035408 type 1 diabetes mellitus 1 Diseases 0.000 description 2
- 238000005199 ultracentrifugation Methods 0.000 description 2
- 238000002211 ultraviolet spectrum Methods 0.000 description 2
- 238000011144 upstream manufacturing Methods 0.000 description 2
- 238000001262 western blot Methods 0.000 description 2
- 229910052725 zinc Inorganic materials 0.000 description 2
- 239000011701 zinc Substances 0.000 description 2
- YMXHPSHLTSZXKH-RVBZMBCESA-N (2,5-dioxopyrrolidin-1-yl) 5-[(3as,4s,6ar)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoate Chemical compound C([C@H]1[C@H]2NC(=O)N[C@H]2CS1)CCCC(=O)ON1C(=O)CCC1=O YMXHPSHLTSZXKH-RVBZMBCESA-N 0.000 description 1
- HKZAAJSTFUZYTO-LURJTMIESA-N (2s)-2-[[2-[[2-[[2-[(2-aminoacetyl)amino]acetyl]amino]acetyl]amino]acetyl]amino]-3-hydroxypropanoic acid Chemical compound NCC(=O)NCC(=O)NCC(=O)NCC(=O)N[C@@H](CO)C(O)=O HKZAAJSTFUZYTO-LURJTMIESA-N 0.000 description 1
- MZOFCQQQCNRIBI-VMXHOPILSA-N (3s)-4-[[(2s)-1-[[(2s)-1-[[(1s)-1-carboxy-2-hydroxyethyl]amino]-4-methyl-1-oxopentan-2-yl]amino]-5-(diaminomethylideneamino)-1-oxopentan-2-yl]amino]-3-[[2-[[(2s)-2,6-diaminohexanoyl]amino]acetyl]amino]-4-oxobutanoic acid Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@@H](N)CCCCN MZOFCQQQCNRIBI-VMXHOPILSA-N 0.000 description 1
- NVKAWKQGWWIWPM-ABEVXSGRSA-N 17-β-hydroxy-5-α-Androstan-3-one Chemical compound C1C(=O)CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CC[C@H]21 NVKAWKQGWWIWPM-ABEVXSGRSA-N 0.000 description 1
- 125000000980 1H-indol-3-ylmethyl group Chemical group [H]C1=C([H])C([H])=C2N([H])C([H])=C(C([H])([H])[*])C2=C1[H] 0.000 description 1
- DLZKEQQWXODGGZ-KCJUWKMLSA-N 2-[[(2r)-2-[[(2s)-2-amino-3-(4-hydroxyphenyl)propanoyl]amino]propanoyl]amino]acetic acid Chemical compound OC(=O)CNC(=O)[C@@H](C)NC(=O)[C@@H](N)CC1=CC=C(O)C=C1 DLZKEQQWXODGGZ-KCJUWKMLSA-N 0.000 description 1
- KISWVXRQTGLFGD-UHFFFAOYSA-N 2-[[2-[[6-amino-2-[[2-[[2-[[5-amino-2-[[2-[[1-[2-[[6-amino-2-[(2,5-diamino-5-oxopentanoyl)amino]hexanoyl]amino]-5-(diaminomethylideneamino)pentanoyl]pyrrolidine-2-carbonyl]amino]-3-hydroxypropanoyl]amino]-5-oxopentanoyl]amino]-5-(diaminomethylideneamino)p Chemical compound C1CCN(C(=O)C(CCCN=C(N)N)NC(=O)C(CCCCN)NC(=O)C(N)CCC(N)=O)C1C(=O)NC(CO)C(=O)NC(CCC(N)=O)C(=O)NC(CCCN=C(N)N)C(=O)NC(CO)C(=O)NC(CCCCN)C(=O)NC(C(=O)NC(CC(C)C)C(O)=O)CC1=CC=C(O)C=C1 KISWVXRQTGLFGD-UHFFFAOYSA-N 0.000 description 1
- IVLXQGJVBGMLRR-UHFFFAOYSA-N 2-aminoacetic acid;hydron;chloride Chemical compound Cl.NCC(O)=O IVLXQGJVBGMLRR-UHFFFAOYSA-N 0.000 description 1
- UAIUNKRWKOVEES-UHFFFAOYSA-N 3,3',5,5'-tetramethylbenzidine Chemical compound CC1=C(N)C(C)=CC(C=2C=C(C)C(N)=C(C)C=2)=C1 UAIUNKRWKOVEES-UHFFFAOYSA-N 0.000 description 1
- QCVGEOXPDFCNHA-UHFFFAOYSA-N 5,5-dimethyl-2,4-dioxo-1,3-oxazolidine-3-carboxamide Chemical compound CC1(C)OC(=O)N(C(N)=O)C1=O QCVGEOXPDFCNHA-UHFFFAOYSA-N 0.000 description 1
- 101150046224 ABAT gene Proteins 0.000 description 1
- 102100031585 ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Human genes 0.000 description 1
- 102000002260 Alkaline Phosphatase Human genes 0.000 description 1
- 108020004774 Alkaline Phosphatase Proteins 0.000 description 1
- 241000024188 Andala Species 0.000 description 1
- 244000303258 Annona diversifolia Species 0.000 description 1
- 108010005853 Anti-Mullerian Hormone Proteins 0.000 description 1
- 108010025628 Apolipoproteins E Proteins 0.000 description 1
- 102000013918 Apolipoproteins E Human genes 0.000 description 1
- 101000716807 Arabidopsis thaliana Protein SCO1 homolog 1, mitochondrial Proteins 0.000 description 1
- QNFRBNZGVVKBNJ-PEFMBERDSA-N Asp-Ile-Gln Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CCC(=O)N)C(=O)O)NC(=O)[C@H](CC(=O)O)N QNFRBNZGVVKBNJ-PEFMBERDSA-N 0.000 description 1
- YFSLJHLQOALGSY-ZPFDUUQYSA-N Asp-Ile-Lys Chemical compound CC[C@H](C)[C@@H](C(=O)N[C@@H](CCCCN)C(=O)O)NC(=O)[C@H](CC(=O)O)N YFSLJHLQOALGSY-ZPFDUUQYSA-N 0.000 description 1
- 101100136076 Aspergillus oryzae (strain ATCC 42149 / RIB 40) pel1 gene Proteins 0.000 description 1
- 241000228257 Aspergillus sp. Species 0.000 description 1
- 238000012935 Averaging Methods 0.000 description 1
- 108090001008 Avidin Proteins 0.000 description 1
- 235000014469 Bacillus subtilis Nutrition 0.000 description 1
- 108010081589 Becaplermin Proteins 0.000 description 1
- 101710129634 Beta-nerve growth factor Proteins 0.000 description 1
- 102000004506 Blood Proteins Human genes 0.000 description 1
- 108010017384 Blood Proteins Proteins 0.000 description 1
- 101001069913 Bos taurus Growth-regulated protein homolog beta Proteins 0.000 description 1
- 101001069912 Bos taurus Growth-regulated protein homolog gamma Proteins 0.000 description 1
- 102000004219 Brain-derived neurotrophic factor Human genes 0.000 description 1
- 108090000715 Brain-derived neurotrophic factor Proteins 0.000 description 1
- 101710117545 C protein Proteins 0.000 description 1
- 102100023702 C-C motif chemokine 13 Human genes 0.000 description 1
- 101710112613 C-C motif chemokine 13 Proteins 0.000 description 1
- 102100023698 C-C motif chemokine 17 Human genes 0.000 description 1
- 101710155857 C-C motif chemokine 2 Proteins 0.000 description 1
- 102100036846 C-C motif chemokine 21 Human genes 0.000 description 1
- 102100036849 C-C motif chemokine 24 Human genes 0.000 description 1
- 102100032367 C-C motif chemokine 5 Human genes 0.000 description 1
- 102100032366 C-C motif chemokine 7 Human genes 0.000 description 1
- 101710155834 C-C motif chemokine 7 Proteins 0.000 description 1
- 102100034871 C-C motif chemokine 8 Human genes 0.000 description 1
- 101710155833 C-C motif chemokine 8 Proteins 0.000 description 1
- 102100025248 C-X-C motif chemokine 10 Human genes 0.000 description 1
- 102100039398 C-X-C motif chemokine 2 Human genes 0.000 description 1
- 102100036189 C-X-C motif chemokine 3 Human genes 0.000 description 1
- 102100036150 C-X-C motif chemokine 5 Human genes 0.000 description 1
- 102100036153 C-X-C motif chemokine 6 Human genes 0.000 description 1
- 101710085504 C-X-C motif chemokine 6 Proteins 0.000 description 1
- 102100032528 C-type lectin domain family 11 member A Human genes 0.000 description 1
- 108010029697 CD40 Ligand Proteins 0.000 description 1
- 101150013553 CD40 gene Proteins 0.000 description 1
- 102100032937 CD40 ligand Human genes 0.000 description 1
- 101150093802 CXCL1 gene Proteins 0.000 description 1
- 101100008049 Caenorhabditis elegans cut-5 gene Proteins 0.000 description 1
- 101100123850 Caenorhabditis elegans her-1 gene Proteins 0.000 description 1
- 101100314454 Caenorhabditis elegans tra-1 gene Proteins 0.000 description 1
- 241000282836 Camelus dromedarius Species 0.000 description 1
- 241000283707 Capra Species 0.000 description 1
- 235000002568 Capsicum frutescens Nutrition 0.000 description 1
- 108090000565 Capsid Proteins Proteins 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 229920002134 Carboxymethyl cellulose Polymers 0.000 description 1
- 102000005367 Carboxypeptidases Human genes 0.000 description 1
- 108010006303 Carboxypeptidases Proteins 0.000 description 1
- 102100028892 Cardiotrophin-1 Human genes 0.000 description 1
- 102000000844 Cell Surface Receptors Human genes 0.000 description 1
- 108010001857 Cell Surface Receptors Proteins 0.000 description 1
- 101710104316 Cell surface-binding protein Proteins 0.000 description 1
- 108010059892 Cellulase Proteins 0.000 description 1
- 108010083647 Chemokine CCL24 Proteins 0.000 description 1
- 108010055166 Chemokine CCL5 Proteins 0.000 description 1
- 108010078239 Chemokine CX3CL1 Proteins 0.000 description 1
- 101100007328 Cocos nucifera COS-1 gene Proteins 0.000 description 1
- 102000000503 Collagen Type II Human genes 0.000 description 1
- 108010041390 Collagen Type II Proteins 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 241000701022 Cytomegalovirus Species 0.000 description 1
- 239000003155 DNA primer Substances 0.000 description 1
- 108700020911 DNA-Binding Proteins Proteins 0.000 description 1
- 208000016192 Demyelinating disease Diseases 0.000 description 1
- 206010059866 Drug resistance Diseases 0.000 description 1
- 102000001301 EGF receptor Human genes 0.000 description 1
- 108060006698 EGF receptor Proteins 0.000 description 1
- 238000012286 ELISA Assay Methods 0.000 description 1
- 108010000912 Egg Proteins Proteins 0.000 description 1
- 102000002322 Egg Proteins Human genes 0.000 description 1
- 102100023688 Eotaxin Human genes 0.000 description 1
- 101710139422 Eotaxin Proteins 0.000 description 1
- 102000009024 Epidermal Growth Factor Human genes 0.000 description 1
- YQYJSBFKSSDGFO-UHFFFAOYSA-N Epihygromycin Natural products OC1C(O)C(C(=O)C)OC1OC(C(=C1)O)=CC=C1C=C(C)C(=O)NC1C(O)C(O)C2OCOC2C1O YQYJSBFKSSDGFO-UHFFFAOYSA-N 0.000 description 1
- 208000009386 Experimental Arthritis Diseases 0.000 description 1
- 102100028412 Fibroblast growth factor 10 Human genes 0.000 description 1
- 101710115997 Gamma-tubulin complex component 2 Proteins 0.000 description 1
- 108010010803 Gelatin Proteins 0.000 description 1
- 108700028146 Genetic Enhancer Elements Proteins 0.000 description 1
- 102000034615 Glial cell line-derived neurotrophic factor Human genes 0.000 description 1
- 108091010837 Glial cell line-derived neurotrophic factor Proteins 0.000 description 1
- SHERTACNJPYHAR-ACZMJKKPSA-N Gln-Ala-Ser Chemical compound OC[C@@H](C(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CCC(N)=O SHERTACNJPYHAR-ACZMJKKPSA-N 0.000 description 1
- DXJZITDUDUPINW-WHFBIAKZSA-N Gln-Asn Chemical compound NC(=O)CC[C@H](N)C(=O)N[C@@H](CC(N)=O)C(O)=O DXJZITDUDUPINW-WHFBIAKZSA-N 0.000 description 1
- CRRFJBGUGNNOCS-PEFMBERDSA-N Gln-Asp-Ile Chemical compound [H]N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(O)=O CRRFJBGUGNNOCS-PEFMBERDSA-N 0.000 description 1
- NJMYZEJORPYOTO-BQBZGAKWSA-N Gln-Pro Chemical compound NC(=O)CC[C@H](N)C(=O)N1CCC[C@H]1C(O)=O NJMYZEJORPYOTO-BQBZGAKWSA-N 0.000 description 1
- 239000004471 Glycine Substances 0.000 description 1
- 102000003886 Glycoproteins Human genes 0.000 description 1
- 108090000288 Glycoproteins Proteins 0.000 description 1
- 108010017213 Granulocyte-Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 1
- 101150009006 HIS3 gene Proteins 0.000 description 1
- 102220468624 HLA class II histocompatibility antigen, DP beta 1 chain_A84E_mutation Human genes 0.000 description 1
- 102000014702 Haptoglobin Human genes 0.000 description 1
- 108050005077 Haptoglobin Proteins 0.000 description 1
- 102000002812 Heat-Shock Proteins Human genes 0.000 description 1
- 108010004889 Heat-Shock Proteins Proteins 0.000 description 1
- 102000013271 Hemopexin Human genes 0.000 description 1
- 108010026027 Hemopexin Proteins 0.000 description 1
- 102100039869 Histone H2B type F-S Human genes 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101000777636 Homo sapiens ADP-ribosyl cyclase/cyclic ADP-ribose hydrolase 1 Proteins 0.000 description 1
- 101000978362 Homo sapiens C-C motif chemokine 17 Proteins 0.000 description 1
- 101000897480 Homo sapiens C-C motif chemokine 2 Proteins 0.000 description 1
- 101000713085 Homo sapiens C-C motif chemokine 21 Proteins 0.000 description 1
- 101000947193 Homo sapiens C-X-C motif chemokine 3 Proteins 0.000 description 1
- 101000947186 Homo sapiens C-X-C motif chemokine 5 Proteins 0.000 description 1
- 101000942297 Homo sapiens C-type lectin domain family 11 member A Proteins 0.000 description 1
- 101000914324 Homo sapiens Carcinoembryonic antigen-related cell adhesion molecule 5 Proteins 0.000 description 1
- 101000914321 Homo sapiens Carcinoembryonic antigen-related cell adhesion molecule 7 Proteins 0.000 description 1
- 101001057612 Homo sapiens Dual specificity protein phosphatase 5 Proteins 0.000 description 1
- 101001069921 Homo sapiens Growth-regulated alpha protein Proteins 0.000 description 1
- 101001035372 Homo sapiens Histone H2B type F-S Proteins 0.000 description 1
- 101000960954 Homo sapiens Interleukin-18 Proteins 0.000 description 1
- 101000581981 Homo sapiens Neural cell adhesion molecule 1 Proteins 0.000 description 1
- 101000973997 Homo sapiens Nucleosome assembly protein 1-like 4 Proteins 0.000 description 1
- 101000947178 Homo sapiens Platelet basic protein Proteins 0.000 description 1
- 101000617725 Homo sapiens Pregnancy-specific beta-1-glycoprotein 2 Proteins 0.000 description 1
- 101001076715 Homo sapiens RNA-binding protein 39 Proteins 0.000 description 1
- 101000874179 Homo sapiens Syndecan-1 Proteins 0.000 description 1
- 101100425753 Homo sapiens TNFRSF1A gene Proteins 0.000 description 1
- 101000851030 Homo sapiens Vascular endothelial growth factor receptor 3 Proteins 0.000 description 1
- 102000002265 Human Growth Hormone Human genes 0.000 description 1
- 108010000521 Human Growth Hormone Proteins 0.000 description 1
- 239000000854 Human Growth Hormone Substances 0.000 description 1
- 108090001061 Insulin Proteins 0.000 description 1
- 102000004877 Insulin Human genes 0.000 description 1
- 102000004218 Insulin-Like Growth Factor I Human genes 0.000 description 1
- 108090001117 Insulin-Like Growth Factor II Proteins 0.000 description 1
- 102000048143 Insulin-Like Growth Factor II Human genes 0.000 description 1
- 102100037850 Interferon gamma Human genes 0.000 description 1
- 108010047761 Interferon-alpha Proteins 0.000 description 1
- 102000006992 Interferon-alpha Human genes 0.000 description 1
- 108010074328 Interferon-gamma Proteins 0.000 description 1
- 102000003777 Interleukin-1 beta Human genes 0.000 description 1
- 108090000193 Interleukin-1 beta Proteins 0.000 description 1
- 102000019223 Interleukin-1 receptor Human genes 0.000 description 1
- 108050006617 Interleukin-1 receptor Proteins 0.000 description 1
- 108090000174 Interleukin-10 Proteins 0.000 description 1
- 108090000177 Interleukin-11 Proteins 0.000 description 1
- 108010065805 Interleukin-12 Proteins 0.000 description 1
- 108090000176 Interleukin-13 Proteins 0.000 description 1
- 108090000172 Interleukin-15 Proteins 0.000 description 1
- 101800003050 Interleukin-16 Proteins 0.000 description 1
- 108050003558 Interleukin-17 Proteins 0.000 description 1
- 102000013691 Interleukin-17 Human genes 0.000 description 1
- 102000003810 Interleukin-18 Human genes 0.000 description 1
- 108090000171 Interleukin-18 Proteins 0.000 description 1
- 102100039898 Interleukin-18 Human genes 0.000 description 1
- 108010002350 Interleukin-2 Proteins 0.000 description 1
- 102000000588 Interleukin-2 Human genes 0.000 description 1
- 108010002386 Interleukin-3 Proteins 0.000 description 1
- 102000000646 Interleukin-3 Human genes 0.000 description 1
- 108090000978 Interleukin-4 Proteins 0.000 description 1
- 102000004388 Interleukin-4 Human genes 0.000 description 1
- 108090001005 Interleukin-6 Proteins 0.000 description 1
- 108010002586 Interleukin-7 Proteins 0.000 description 1
- 108010002335 Interleukin-9 Proteins 0.000 description 1
- 108010044467 Isoenzymes Proteins 0.000 description 1
- 101150042441 K gene Proteins 0.000 description 1
- QNAYBMKLOCPYGJ-REOHCLBHSA-N L-alanine Chemical compound C[C@H](N)C(O)=O QNAYBMKLOCPYGJ-REOHCLBHSA-N 0.000 description 1
- HNDVDQJCIGZPNO-YFKPBYRVSA-N L-histidine Chemical compound OC(=O)[C@@H](N)CC1=CN=CN1 HNDVDQJCIGZPNO-YFKPBYRVSA-N 0.000 description 1
- 125000000174 L-prolyl group Chemical group [H]N1C([H])([H])C([H])([H])C([H])([H])[C@@]1([H])C(*)=O 0.000 description 1
- 108010063045 Lactoferrin Proteins 0.000 description 1
- 102000010445 Lactoferrin Human genes 0.000 description 1
- GUBGYTABKSRVRQ-QKKXKWKRSA-N Lactose Natural products OC[C@H]1O[C@@H](O[C@H]2[C@H](O)[C@@H](O)C(O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@H]1O GUBGYTABKSRVRQ-QKKXKWKRSA-N 0.000 description 1
- 241000880493 Leptailurus serval Species 0.000 description 1
- UILIPCLTHRPCRB-XUXIUFHCSA-N Leu-Arg-Ile Chemical compound CC[C@H](C)[C@@H](C(=O)O)NC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](CC(C)C)N UILIPCLTHRPCRB-XUXIUFHCSA-N 0.000 description 1
- 108010046938 Macrophage Colony-Stimulating Factor Proteins 0.000 description 1
- 102000007651 Macrophage Colony-Stimulating Factor Human genes 0.000 description 1
- 102000016943 Muramidase Human genes 0.000 description 1
- 108010014251 Muramidase Proteins 0.000 description 1
- 102100038895 Myc proto-oncogene protein Human genes 0.000 description 1
- 101710135898 Myc proto-oncogene protein Proteins 0.000 description 1
- 102000047918 Myelin Basic Human genes 0.000 description 1
- 101710107068 Myelin basic protein Proteins 0.000 description 1
- 108010062010 N-Acetylmuramoyl-L-alanine Amidase Proteins 0.000 description 1
- 102100034296 Natriuretic peptides A Human genes 0.000 description 1
- 108010025020 Nerve Growth Factor Proteins 0.000 description 1
- 102100027347 Neural cell adhesion molecule 1 Human genes 0.000 description 1
- 241000221961 Neurospora crassa Species 0.000 description 1
- 108090000742 Neurotrophin 3 Proteins 0.000 description 1
- 102100029268 Neurotrophin-3 Human genes 0.000 description 1
- 102000003683 Neurotrophin-4 Human genes 0.000 description 1
- 108090000099 Neurotrophin-4 Proteins 0.000 description 1
- 108010015406 Neurturin Proteins 0.000 description 1
- 102100021584 Neurturin Human genes 0.000 description 1
- 244000061176 Nicotiana tabacum Species 0.000 description 1
- 235000002637 Nicotiana tabacum Nutrition 0.000 description 1
- 102220558705 Nuclear protein 1_A33Q_mutation Human genes 0.000 description 1
- 108091028043 Nucleic acid sequence Proteins 0.000 description 1
- 102100036518 Nucleoside diphosphate phosphatase ENTPD5 Human genes 0.000 description 1
- 241001452677 Ogataea methanolica Species 0.000 description 1
- 102000004140 Oncostatin M Human genes 0.000 description 1
- 108090000630 Oncostatin M Proteins 0.000 description 1
- 239000002033 PVDF binder Substances 0.000 description 1
- 102000057297 Pepsin A Human genes 0.000 description 1
- 108090000284 Pepsin A Proteins 0.000 description 1
- 102000045595 Phosphoprotein Phosphatases Human genes 0.000 description 1
- 108700019535 Phosphoprotein Phosphatases Proteins 0.000 description 1
- 108010001014 Plasminogen Activators Proteins 0.000 description 1
- 102000001938 Plasminogen Activators Human genes 0.000 description 1
- 102100036154 Platelet basic protein Human genes 0.000 description 1
- 108090000778 Platelet factor 4 Proteins 0.000 description 1
- 108010038512 Platelet-Derived Growth Factor Proteins 0.000 description 1
- 102000010780 Platelet-Derived Growth Factor Human genes 0.000 description 1
- 239000002202 Polyethylene glycol Substances 0.000 description 1
- 102100022019 Pregnancy-specific beta-1-glycoprotein 2 Human genes 0.000 description 1
- 241000350158 Prioria balsamifera Species 0.000 description 1
- 101710098940 Pro-epidermal growth factor Proteins 0.000 description 1
- ONIBWKKTOPOVIA-UHFFFAOYSA-N Proline Natural products OC(=O)C1CCCN1 ONIBWKKTOPOVIA-UHFFFAOYSA-N 0.000 description 1
- 102100023832 Prolyl endopeptidase FAP Human genes 0.000 description 1
- 101100424383 Rattus norvegicus Taar4 gene Proteins 0.000 description 1
- 108020004511 Recombinant DNA Proteins 0.000 description 1
- 108010091086 Recombinases Proteins 0.000 description 1
- 102000018120 Recombinases Human genes 0.000 description 1
- 101100394989 Rhodopseudomonas palustris (strain ATCC BAA-98 / CGA009) hisI gene Proteins 0.000 description 1
- 102100023361 SAP domain-containing ribonucleoprotein Human genes 0.000 description 1
- 238000011803 SJL/J (JAX™ mice strain) Methods 0.000 description 1
- 206010039491 Sarcoma Diseases 0.000 description 1
- 241000235347 Schizosaccharomyces pombe Species 0.000 description 1
- SSJMZMUVNKEENT-IMJSIDKUSA-N Ser-Ala Chemical compound OC(=O)[C@H](C)NC(=O)[C@@H](N)CO SSJMZMUVNKEENT-IMJSIDKUSA-N 0.000 description 1
- QGMLKFGTGXWAHF-IHRRRGAJSA-N Ser-Arg-Phe Chemical compound [H]N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC1=CC=CC=C1)C(O)=O QGMLKFGTGXWAHF-IHRRRGAJSA-N 0.000 description 1
- UIGMAMGZOJVTDN-WHFBIAKZSA-N Ser-Gly-Ser Chemical compound OC[C@H](N)C(=O)NCC(=O)N[C@@H](CO)C(O)=O UIGMAMGZOJVTDN-WHFBIAKZSA-N 0.000 description 1
- ILVGMCVCQBJPSH-WDSKDSINSA-N Ser-Val Chemical compound CC(C)[C@@H](C(O)=O)NC(=O)[C@@H](N)CO ILVGMCVCQBJPSH-WDSKDSINSA-N 0.000 description 1
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 1
- 108020004682 Single-Stranded DNA Proteins 0.000 description 1
- 102000013275 Somatomedins Human genes 0.000 description 1
- 239000012505 Superdex™ Substances 0.000 description 1
- 102000019197 Superoxide Dismutase Human genes 0.000 description 1
- 108010012715 Superoxide dismutase Proteins 0.000 description 1
- 102100035721 Syndecan-1 Human genes 0.000 description 1
- 101710194518 T4 protein Proteins 0.000 description 1
- 101150084220 TAR2 gene Proteins 0.000 description 1
- 238000003917 TEM image Methods 0.000 description 1
- AYFVYJQAPQTCCC-UHFFFAOYSA-N Threonine Natural products CC(O)C(N)C(O)=O AYFVYJQAPQTCCC-UHFFFAOYSA-N 0.000 description 1
- 239000004473 Threonine Substances 0.000 description 1
- 108010022394 Threonine synthase Proteins 0.000 description 1
- 108010034949 Thyroglobulin Proteins 0.000 description 1
- 102000009843 Thyroglobulin Human genes 0.000 description 1
- 101710150448 Transcriptional regulator Myc Proteins 0.000 description 1
- 102000004338 Transferrin Human genes 0.000 description 1
- 108090000901 Transferrin Proteins 0.000 description 1
- 102000004887 Transforming Growth Factor beta Human genes 0.000 description 1
- 108090001012 Transforming Growth Factor beta Proteins 0.000 description 1
- 102000011117 Transforming Growth Factor beta2 Human genes 0.000 description 1
- 108010009583 Transforming Growth Factors Proteins 0.000 description 1
- 102000009618 Transforming Growth Factors Human genes 0.000 description 1
- 101800004564 Transforming growth factor alpha Proteins 0.000 description 1
- 102400001320 Transforming growth factor alpha Human genes 0.000 description 1
- 101800000304 Transforming growth factor beta-2 Proteins 0.000 description 1
- 102000056172 Transforming growth factor beta-3 Human genes 0.000 description 1
- 108090000097 Transforming growth factor beta-3 Proteins 0.000 description 1
- 108700019146 Transgenes Proteins 0.000 description 1
- 239000007983 Tris buffer Substances 0.000 description 1
- 108060008682 Tumor Necrosis Factor Proteins 0.000 description 1
- 102000000852 Tumor Necrosis Factor-alpha Human genes 0.000 description 1
- 102100040245 Tumor necrosis factor receptor superfamily member 5 Human genes 0.000 description 1
- GAYLGYUVTDMLKC-UWJYBYFXSA-N Tyr-Asp-Ala Chemical compound OC(=O)[C@H](C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@@H](N)CC1=CC=C(O)C=C1 GAYLGYUVTDMLKC-UWJYBYFXSA-N 0.000 description 1
- 229910052770 Uranium Inorganic materials 0.000 description 1
- COQLPRJCUIATTQ-UHFFFAOYSA-N Uranyl acetate Chemical compound O.O.O=[U]=O.CC(O)=O.CC(O)=O COQLPRJCUIATTQ-UHFFFAOYSA-N 0.000 description 1
- 101150117115 V gene Proteins 0.000 description 1
- KZSNJWFQEVHDMF-UHFFFAOYSA-N Valine Chemical group CC(C)C(N)C(O)=O KZSNJWFQEVHDMF-UHFFFAOYSA-N 0.000 description 1
- 102100033179 Vascular endothelial growth factor receptor 3 Human genes 0.000 description 1
- 102000050760 Vitamin D-binding protein Human genes 0.000 description 1
- 101710179590 Vitamin D-binding protein Proteins 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 239000002671 adjuvant Substances 0.000 description 1
- 229940009456 adriamycin Drugs 0.000 description 1
- 239000011543 agarose gel Substances 0.000 description 1
- 230000004931 aggregating effect Effects 0.000 description 1
- 230000001476 alcoholic effect Effects 0.000 description 1
- 235000010443 alginic acid Nutrition 0.000 description 1
- 229920000615 alginic acid Polymers 0.000 description 1
- 238000011166 aliquoting Methods 0.000 description 1
- 230000004075 alteration Effects 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 238000010171 animal model Methods 0.000 description 1
- 235000021120 animal protein Nutrition 0.000 description 1
- 239000000868 anti-mullerian hormone Substances 0.000 description 1
- 239000004599 antimicrobial Substances 0.000 description 1
- 239000003963 antioxidant agent Substances 0.000 description 1
- 108010026054 apolipoprotein SAA Proteins 0.000 description 1
- 239000012062 aqueous buffer Substances 0.000 description 1
- 239000007864 aqueous solution Substances 0.000 description 1
- 210000004507 artificial chromosome Anatomy 0.000 description 1
- 229940009098 aspartate Drugs 0.000 description 1
- 230000001746 atrial effect Effects 0.000 description 1
- 239000005667 attractant Substances 0.000 description 1
- 239000013602 bacteriophage vector Substances 0.000 description 1
- 238000010923 batch production Methods 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- 238000013357 binding ELISA Methods 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 230000005540 biological transmission Effects 0.000 description 1
- 239000002981 blocking agent Substances 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 210000004899 c-terminal region Anatomy 0.000 description 1
- BPKIGYQJPYCAOW-FFJTTWKXSA-I calcium;potassium;disodium;(2s)-2-hydroxypropanoate;dichloride;dihydroxide;hydrate Chemical compound O.[OH-].[OH-].[Na+].[Na+].[Cl-].[Cl-].[K+].[Ca+2].C[C@H](O)C([O-])=O BPKIGYQJPYCAOW-FFJTTWKXSA-I 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 239000001768 carboxy methyl cellulose Substances 0.000 description 1
- 235000010948 carboxy methyl cellulose Nutrition 0.000 description 1
- 239000008112 carboxymethyl-cellulose Substances 0.000 description 1
- 108010031762 cardiodilatin Proteins 0.000 description 1
- 108010041776 cardiotrophin 1 Proteins 0.000 description 1
- 239000000969 carrier Substances 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 238000000423 cell based assay Methods 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- 230000001413 cellular effect Effects 0.000 description 1
- 229940106157 cellulase Drugs 0.000 description 1
- 238000012512 characterization method Methods 0.000 description 1
- 238000007385 chemical modification Methods 0.000 description 1
- 230000031902 chemoattractant activity Effects 0.000 description 1
- 229960005091 chloramphenicol Drugs 0.000 description 1
- WIIZWVCIJKGZOK-RKDXNWHRSA-N chloramphenicol Chemical compound ClC(Cl)C(=O)N[C@H](CO)[C@H](O)C1=CC=C([N+]([O-])=O)C=C1 WIIZWVCIJKGZOK-RKDXNWHRSA-N 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 239000013611 chromosomal DNA Substances 0.000 description 1
- 108010030175 colony inhibiting factor Proteins 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 239000002299 complementary DNA Substances 0.000 description 1
- 238000012258 culturing Methods 0.000 description 1
- 238000005520 cutting process Methods 0.000 description 1
- 150000001945 cysteines Chemical class 0.000 description 1
- 231100000433 cytotoxic Toxicity 0.000 description 1
- 230000001472 cytotoxic effect Effects 0.000 description 1
- 230000007812 deficiency Effects 0.000 description 1
- 239000003398 denaturant Substances 0.000 description 1
- 239000000032 diagnostic agent Substances 0.000 description 1
- 229940039227 diagnostic agent Drugs 0.000 description 1
- 238000000502 dialysis Methods 0.000 description 1
- 125000001142 dicarboxylic acid group Chemical group 0.000 description 1
- 235000015872 dietary supplement Nutrition 0.000 description 1
- 238000000113 differential scanning calorimetry Methods 0.000 description 1
- 230000029087 digestion Effects 0.000 description 1
- 102000004419 dihydrofolate reductase Human genes 0.000 description 1
- 238000007865 diluting Methods 0.000 description 1
- 230000000447 dimerizing effect Effects 0.000 description 1
- 239000002934 diuretic Substances 0.000 description 1
- 108010039433 dolichos biflorus agglutinin Proteins 0.000 description 1
- 238000009510 drug design Methods 0.000 description 1
- 238000001035 drying Methods 0.000 description 1
- 235000014103 egg white Nutrition 0.000 description 1
- 210000000969 egg white Anatomy 0.000 description 1
- 239000003792 electrolyte Substances 0.000 description 1
- 230000008030 elimination Effects 0.000 description 1
- 238000003379 elimination reaction Methods 0.000 description 1
- 238000000295 emission spectrum Methods 0.000 description 1
- 238000012407 engineering method Methods 0.000 description 1
- 238000005516 engineering process Methods 0.000 description 1
- 231100000655 enterotoxin Toxicity 0.000 description 1
- 210000003527 eukaryotic cell Anatomy 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 238000002474 experimental method Methods 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 230000002349 favourable effect Effects 0.000 description 1
- 108010072257 fibroblast activation protein alpha Proteins 0.000 description 1
- 239000012847 fine chemical Substances 0.000 description 1
- 108700014844 flt3 ligand Proteins 0.000 description 1
- 239000012530 fluid Substances 0.000 description 1
- 125000002485 formyl group Chemical group [H]C(*)=O 0.000 description 1
- 230000002538 fungal effect Effects 0.000 description 1
- 239000003626 gastrointestinal polypeptide Substances 0.000 description 1
- 239000008273 gelatin Substances 0.000 description 1
- 229920000159 gelatin Polymers 0.000 description 1
- 235000019322 gelatine Nutrition 0.000 description 1
- 235000011852 gelatine desserts Nutrition 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 239000008187 granular material Substances 0.000 description 1
- 210000003714 granulocyte Anatomy 0.000 description 1
- 230000005484 gravity Effects 0.000 description 1
- 230000036541 health Effects 0.000 description 1
- HNDVDQJCIGZPNO-UHFFFAOYSA-N histidine Natural products OC(=O)C(N)CC1=CN=CN1 HNDVDQJCIGZPNO-UHFFFAOYSA-N 0.000 description 1
- 238000002744 homologous recombination Methods 0.000 description 1
- 230000006801 homologous recombination Effects 0.000 description 1
- 102000043381 human DUSP5 Human genes 0.000 description 1
- 229940106780 human fibrinogen Drugs 0.000 description 1
- 125000001165 hydrophobic group Chemical group 0.000 description 1
- 238000001597 immobilized metal affinity chromatography Methods 0.000 description 1
- 230000003100 immobilizing effect Effects 0.000 description 1
- 230000001900 immune effect Effects 0.000 description 1
- 210000000987 immune system Anatomy 0.000 description 1
- 229940072221 immunoglobulins Drugs 0.000 description 1
- 230000001024 immunotherapeutic effect Effects 0.000 description 1
- 230000002637 immunotoxin Effects 0.000 description 1
- 239000002596 immunotoxin Substances 0.000 description 1
- 231100000608 immunotoxin Toxicity 0.000 description 1
- 229940051026 immunotoxin Drugs 0.000 description 1
- 238000000099 in vitro assay Methods 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 239000000411 inducer Substances 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 239000011261 inert gas Substances 0.000 description 1
- 238000001802 infusion Methods 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 229940125396 insulin Drugs 0.000 description 1
- 230000021995 interleukin-8 production Effects 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- PGLTVOMIXTUURA-UHFFFAOYSA-N iodoacetamide Chemical compound NC(=O)CI PGLTVOMIXTUURA-UHFFFAOYSA-N 0.000 description 1
- JDNTWHVOXJZDSN-UHFFFAOYSA-N iodoacetic acid Chemical compound OC(=O)CI JDNTWHVOXJZDSN-UHFFFAOYSA-N 0.000 description 1
- 238000002955 isolation Methods 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- CSSYQJWUGATIHM-IKGCZBKSSA-N l-phenylalanyl-l-lysyl-l-cysteinyl-l-arginyl-l-arginyl-l-tryptophyl-l-glutaminyl-l-tryptophyl-l-arginyl-l-methionyl-l-lysyl-l-lysyl-l-leucylglycyl-l-alanyl-l-prolyl-l-seryl-l-isoleucyl-l-threonyl-l-cysteinyl-l-valyl-l-arginyl-l-arginyl-l-alanyl-l-phenylal Chemical compound C([C@H](N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](C)C(=O)N1CCC[C@H]1C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CS)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(O)=O)C1=CC=CC=C1 CSSYQJWUGATIHM-IKGCZBKSSA-N 0.000 description 1
- 229940078795 lactoferrin Drugs 0.000 description 1
- 235000021242 lactoferrin Nutrition 0.000 description 1
- 239000008101 lactose Substances 0.000 description 1
- 239000004325 lysozyme Substances 0.000 description 1
- 229960000274 lysozyme Drugs 0.000 description 1
- 230000002101 lytic effect Effects 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- AEUKDPKXTPNBNY-XEYRWQBLSA-N mcp 2 Chemical compound C([C@@H](C(=O)N[C@@H](CS)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CS)C(=O)N[C@@H](CS)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCNC(N)=N)C(O)=O)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CC(C)C)NC(=O)[C@H](CS)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CS)NC(=O)[C@H](C)NC(=O)[C@H](CS)NC(=O)[C@@H](NC(=O)[C@@H](N)C(C)C)C(C)C)C1=CC=CC=C1 AEUKDPKXTPNBNY-XEYRWQBLSA-N 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- 229910021645 metal ion Inorganic materials 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- HPNSFSBZBAHARI-UHFFFAOYSA-N micophenolic acid Natural products OC1=C(CC=C(C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-UHFFFAOYSA-N 0.000 description 1
- 238000002493 microarray Methods 0.000 description 1
- 235000013336 milk Nutrition 0.000 description 1
- 239000008267 milk Substances 0.000 description 1
- 210000004080 milk Anatomy 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 239000000178 monomer Substances 0.000 description 1
- 229940126619 mouse monoclonal antibody Drugs 0.000 description 1
- 239000003471 mutagenic agent Substances 0.000 description 1
- 230000036438 mutation frequency Effects 0.000 description 1
- 229960000951 mycophenolic acid Drugs 0.000 description 1
- HPNSFSBZBAHARI-RUDMXATFSA-N mycophenolic acid Chemical compound OC1=C(C\C=C(/C)CCC(O)=O)C(OC)=C(C)C2=C1C(=O)OC2 HPNSFSBZBAHARI-RUDMXATFSA-N 0.000 description 1
- 230000001452 natriuretic effect Effects 0.000 description 1
- 229940053128 nerve growth factor Drugs 0.000 description 1
- 238000006386 neutralization reaction Methods 0.000 description 1
- 108010028546 nucleoside-diphosphatase Proteins 0.000 description 1
- 230000003606 oligomerizing effect Effects 0.000 description 1
- 239000006174 pH buffer Substances 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 101150040383 pel2 gene Proteins 0.000 description 1
- 101150050446 pelB gene Proteins 0.000 description 1
- 229940111202 pepsin Drugs 0.000 description 1
- 210000001322 periplasm Anatomy 0.000 description 1
- 229940127126 plasminogen activator Drugs 0.000 description 1
- 108010017843 platelet-derived growth factor A Proteins 0.000 description 1
- 108010000685 platelet-derived growth factor AB Proteins 0.000 description 1
- 238000007747 plating Methods 0.000 description 1
- 229920002981 polyvinylidene fluoride Polymers 0.000 description 1
- 230000008092 positive effect Effects 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 238000007639 printing Methods 0.000 description 1
- 210000001236 prokaryotic cell Anatomy 0.000 description 1
- 238000012529 protein A ELISA Methods 0.000 description 1
- 238000000159 protein binding assay Methods 0.000 description 1
- 230000012743 protein tagging Effects 0.000 description 1
- 230000002797 proteolythic effect Effects 0.000 description 1
- 208000007153 proteostasis deficiencies Diseases 0.000 description 1
- 230000002685 pulmonary effect Effects 0.000 description 1
- 238000010791 quenching Methods 0.000 description 1
- 230000000171 quenching effect Effects 0.000 description 1
- 238000002708 random mutagenesis Methods 0.000 description 1
- QGFSVPWZEPKNDV-BRTFOEFASA-N ranp Chemical compound C([C@H]1C(=O)NCC(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@H](C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@H](C(NCC(=O)N[C@@H](C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CSSC[C@@H](C(=O)N1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(O)=O)=O)[C@@H](C)CC)[C@@H](C)CC)C1=CC=CC=C1 QGFSVPWZEPKNDV-BRTFOEFASA-N 0.000 description 1
- 108010043277 recombinant soluble CD4 Proteins 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 238000005057 refrigeration Methods 0.000 description 1
- 238000003303 reheating Methods 0.000 description 1
- 230000003362 replicative effect Effects 0.000 description 1
- 230000001177 retroviral effect Effects 0.000 description 1
- 206010039073 rheumatoid arthritis Diseases 0.000 description 1
- 102220032361 rs104895237 Human genes 0.000 description 1
- 102220229200 rs1064795244 Human genes 0.000 description 1
- 102200145601 rs147394623 Human genes 0.000 description 1
- 102220092683 rs202145133 Human genes 0.000 description 1
- 102200030790 rs546099787 Human genes 0.000 description 1
- 102200132518 rs587777480 Human genes 0.000 description 1
- 102200028844 rs679620 Human genes 0.000 description 1
- 102220147641 rs750280423 Human genes 0.000 description 1
- 102220219512 rs775763888 Human genes 0.000 description 1
- 102220061206 rs786203134 Human genes 0.000 description 1
- 102220072709 rs794729016 Human genes 0.000 description 1
- 102200151359 rs797045153 Human genes 0.000 description 1
- 102200015515 rs797045155 Human genes 0.000 description 1
- 102220009454 rs80356863 Human genes 0.000 description 1
- 102220082690 rs863224215 Human genes 0.000 description 1
- 102220179462 rs886057103 Human genes 0.000 description 1
- 238000001073 sample cooling Methods 0.000 description 1
- 238000003118 sandwich ELISA Methods 0.000 description 1
- 238000007790 scraping Methods 0.000 description 1
- 238000007789 sealing Methods 0.000 description 1
- 230000028327 secretion Effects 0.000 description 1
- 238000010187 selection method Methods 0.000 description 1
- 238000013207 serial dilution Methods 0.000 description 1
- 238000002741 site-directed mutagenesis Methods 0.000 description 1
- 238000001542 size-exclusion chromatography Methods 0.000 description 1
- 235000020183 skimmed milk Nutrition 0.000 description 1
- 239000002002 slurry Substances 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 230000009870 specific binding Effects 0.000 description 1
- 238000001694 spray drying Methods 0.000 description 1
- 230000006641 stabilisation Effects 0.000 description 1
- 238000011105 stabilization Methods 0.000 description 1
- 210000000130 stem cell Anatomy 0.000 description 1
- 210000002784 stomach Anatomy 0.000 description 1
- 238000002198 surface plasmon resonance spectroscopy Methods 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- WWJZWCUNLNYYAU-UHFFFAOYSA-N temephos Chemical compound C1=CC(OP(=S)(OC)OC)=CC=C1SC1=CC=C(OP(=S)(OC)OC)C=C1 WWJZWCUNLNYYAU-UHFFFAOYSA-N 0.000 description 1
- 238000012360 testing method Methods 0.000 description 1
- 101150118377 tet gene Proteins 0.000 description 1
- ZRKFYGHZFMAOKI-QMGMOQQFSA-N tgfbeta Chemical compound C([C@H](NC(=O)[C@H](C(C)C)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CC(C)C)NC(=O)CNC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@@H](NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](N)CCSC)C(C)C)[C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C)C(=O)N[C@@H](CC(C)C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(O)=O)C1=CC=C(O)C=C1 ZRKFYGHZFMAOKI-QMGMOQQFSA-N 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 239000002562 thickening agent Substances 0.000 description 1
- 229960002175 thyroglobulin Drugs 0.000 description 1
- 206010043778 thyroiditis Diseases 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- 231100000765 toxin Toxicity 0.000 description 1
- 108700012359 toxins Proteins 0.000 description 1
- 230000005030 transcription termination Effects 0.000 description 1
- 238000001890 transfection Methods 0.000 description 1
- 239000012581 transferrin Substances 0.000 description 1
- 230000009466 transformation Effects 0.000 description 1
- 230000009261 transgenic effect Effects 0.000 description 1
- 230000001052 transient effect Effects 0.000 description 1
- 238000004627 transmission electron microscopy Methods 0.000 description 1
- LENZDBCJOHFCAS-UHFFFAOYSA-N tris Chemical compound OCC(N)(CO)CO LENZDBCJOHFCAS-UHFFFAOYSA-N 0.000 description 1
- 239000002753 trypsin inhibitor Substances 0.000 description 1
- 108020005087 unfolded proteins Proteins 0.000 description 1
- 241000701161 unidentified adenovirus Species 0.000 description 1
- 241000701447 unidentified baculovirus Species 0.000 description 1
- 229910052720 vanadium Inorganic materials 0.000 description 1
- 238000013022 venting Methods 0.000 description 1
- 239000013603 viral vector Substances 0.000 description 1
- 238000011179 visual inspection Methods 0.000 description 1
- 210000005253 yeast cell Anatomy 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C40—COMBINATORIAL TECHNOLOGY
- C40B—COMBINATORIAL CHEMISTRY; LIBRARIES, e.g. CHEMICAL LIBRARIES
- C40B40/00—Libraries per se, e.g. arrays, mixtures
- C40B40/02—Libraries contained in or displayed by microorganisms, e.g. bacteria or animal cells; Libraries contained in or displayed by vectors, e.g. plasmids; Libraries containing only microorganisms or vectors
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/107—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length by chemical modification of precursor peptides
- C07K1/113—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length by chemical modification of precursor peptides without change of the primary structure
- C07K1/1136—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length by chemical modification of precursor peptides without change of the primary structure by reversible modification of the secondary, tertiary or quarternary structure, e.g. using denaturating or stabilising agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IGs], e.g. monoclonal or polyclonal antibodies
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/10—Processes for the isolation, preparation or purification of DNA or RNA
- C12N15/1034—Isolating an individual clone by screening libraries
- C12N15/1037—Screening libraries presented on the surface of microorganisms, e.g. phage display, E. coli display
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
- C07K2317/567—Framework region [FR]
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
- C07K2317/569—Single domain, e.g. dAb, sdAb, VHH, VNAR or nanobody®
Definitions
- Polypeptides have become increasingly important agents in a variety of applications, including use as medical therapeutic and diagnostic agents and in industrial applications.
- One factor that has hinder further application of polypeptides is their physical and chemical properties.
- polypeptides generally must retain proper folding to be active.
- polypeptides tend to unfold or denature under storage conditions or conditions where they could find utility (e.g., when exposed to heat, organic solvents).
- many polypeptides are produced only with relatively low yield using biological production systems. Accordingly, they can be prohibitively costly to produce.
- a key factor that limits further application of polypeptides is the tendency of unfolded or denatured polypeptides to aggregate irreversibly. Aggregation is influenced by polypeptide concentration and is thought to arise in many cases from partially folded or unfolded intermediates. Factors and conditions that favor partially folded intermediates, such as elevated temperature and high polypeptide concentration, promote irreversible aggregation. (Fink, A.L., Folding & Design 5:R1-R23 (1998).) For example, storing purified polypeptides in concentrated form, such as a lyophilized preparation, f equently results in irreversible aggregation of at least a portion of the polypeptides. Also, production of a polypeptide by expression in biological systems, such as E. coli, often results in the formation of inclusion bodies which contain aggregated polypeptides. Recovering active polypeptides from inclusion bodies can be very difficult and require adding additional steps, such as a refolding step, to a biological production system.
- the invention relates to polypeptides that unfold reversibly, to repertoires containing polypeptides that unfold reversibly and to libraries that contam polypeptides that unfold reversibly or nucleic acids that encode polypeptides that unfold reversibly.
- the invention further relates to processes for producing a library enriched in polypeptides that unfold reversibly or nucleic acids encoding polypeptides that unfold reversibly, processes for selecting and/or isolating polypeptides that unfold reversibly, and to methods for producing a polypeptide that unfolds reversibly.
- the invention is a process for selecting, isolating and/or recovering a polypeptide that unfolds reversibly from a library or a repertoire of polypeptides (e.g., a polypeptide display system).
- the method comprises unfolding a collection of polypeptides (e.g., the polypeptides in a library, a repertoire or a polypeptide display system), refolding at least a portion of the unfolded polypeptides, and selecting, isolating and/or recovering a refolded polypeptide.
- the method comprises providing a collection of unfolded polypeptides (e.g., the polypeptides in a library, a repertoire or a polypeptide display system), refolding at least a portion of the unfolded polypeptides, and selecting, isolating and/or recovering a refolded polypeptide.
- the method comprises providing a polypeptide display system comprising a repertoire, heating the repertoire to a temperature (Ts) at which at least a portion of the displayed polypeptides unfold and cooling the repertoire to a temperature (Tc) that is lower than Ts to produce a cooled repertoire.
- the cooled repertoire comprises at least a portion of polypeptides that have unfolded and refolded and a portion of polypeptides that have aggregated.
- the method further comprises recovering at a temperature (Tr) at least one polypeptide that binds a ligand and unfolds reversibly.
- Tr temperature
- the ligand binds folded polypeptide and does not bind aggregated polypeptides
- the recovered polypeptide has a melting temperature (Tm), and Ts>Tm>Tc, and Ts>Tm>Tr.
- the invention relates to repertoires of polypeptides that unfold reversibly, to libraries of nucleic acids that encode polypeptides that unfold reversibly, and to methods for producing such libraries and repertoires.
- the invention is an isolated polypeptide that unfolds reversibly.
- the polypeptide that unfolds reversibly is a variant of a parental polypeptide that differs from the parental polypeptide in amino acid sequence (e.g., by one or more amino acid replacements, additions and/or deletions), but qualitatively retains function of the parental polypeptide.
- the invention also relates to a process for producing an antibody variable domain library enriched in variable domains that unfold reversibly.
- the process comprises (1) providing a phage display system comprising a plurality of displayed antibody variable region ' s, wherein at least a portion of the displayed variable regions have been unfolded and refolded, (2) selecting phage displaying variable regions that have unfolded, refolded and regained binding function from said phage display system, (3) obtaining nucleic acids encoding CDR1 and /or CDR2 of the variable regions displayed on the recovered phage, and (4) assembling a library of nucleic acids encoding antibody variable domains, wherein said nucleic acids obtained in (3) are operably linked to one or more other nucleic acids to produce a library of constructs that encode antibody variable domains in which CDRl and/or CDR2 are encoded by the nucleic acid obtained in (3).
- substantially all of the displayed variable regions in (1) have been unfolded by heating to about 80°C and refolded by cooling.
- library of nucleic acids assembled in (4) encodes an antibody variable domain in which CDR3 is randomized or is not derived from antibody variable regions that has been selected for the ability to unfold reversibly.
- the polypeptides that unfold reversibly described herein can be produced as soluble proteins in the supernatant of E. coli or yeast cultures with high yield.
- Fig. 1 is an illustration of a nucleic acid encoding human immunoglobulin heavy chain variable region DP47dummy (also referred to herein as DP47d), comprising ge ⁇ nline VH gene segment DP47 and germline VJ gene segment JH4b (SEQ ID NO: 1, coding strand; SEQ ID NO:2 non-coding strand).
- Fig. 1 also presents the amino acid sequence of the encoded V H domain (SEQ ID NO:3). The amino acids are numbered according to the system of abat. (Kabat, E.A. et al, Sequences of Proteins oflmmunological Interest, Fifth Edition, U.S. Department of Health and Human Services, U.S. Government Printing Office (1991).)
- Fig. 2 is an illustration of a nucleic acid encoding human immunoglobulin light chain (K) variable region DPK9 dummy (also referred to herein as DPK9d), comprising germline V K gene segment DP 9 and ge ⁇ nline JK gene segment JK1. (SEQ ID NO:4, coding strand; SEQ ID NO:5 non-coding strand). Fig. 2 also presents the amino acid sequence of the encoded V K domain (SEQ ID NO:6). The amino acids are numbered according to the system of Kabat.
- K human immunoglobulin light chain
- Figs. 3A-3F are histograms that illustrate the relative amount of protein A binding activity retained by phage clones displaying VH domains based on a DP47d scaffold after heat-induced unfolding and refolding.
- a sample of each phage clone was heated to cause the displayed Vu domains to unfold and then cooled to cause refolding, and a sample was left unheated.
- Binding of the heat-treated and the untreated phage samples to protein A was assessed by ELISA.
- the binding activity of the heat-treated clones expressed as a percentage of the binding activity of the untreated clones is illustrated.
- the columns marked "d ⁇ 47" refer to DP47d.
- Figs.4A-4C are panels of a table illustrating the amino acid sequences of the complementarity determining regions (CDR1, CDR2, CDR3) of several of the VH domains displayed on the phage that retained greater than >60% relative binding to protein A in Figs 3A-3F.
- the sequences are identified in Figs 4A-4C by clone number. These clones are also referred to herein using a "pA-" prefix. For example, Clone 13 is also referred to herein as " ⁇ A-C13.”
- the first group of sequences presented in Fig. 4A and 4B are from clones that had a high degree of refolding as assessed by the results ofprotein A binding (group 1).
- the next group of sequences in Fig. 4C are from clones with good refolding. These clones also contain mutations outside the CDRs (group 2).
- the final group of sequences in Fig. 4C are from clones with lower refolding
- Fig. 5 is a graph illustrating the relationship between the capacity of a displayed VH domain to undergo reversible heat unfolding and the hydrophobicity of the amino acid sequence from position 22 to position 36 of the VH domain.
- the Sweet/Eisenberg hydrophobicity score (SE-15 value) for the sequence for position 22 to position 36 of several displayed V H was determined using a window of 15 amino acids.
- the SE-15 value was plotted against the relative protein A binding activity (ELISA) for each clone after undergoing heat-induced unfolding and refolding.
- the graph illustrates that the ability of a displayed V H to undergo heat- induced unfolding and refolding correlates with an SE-15 value of 0 or less for the amino acid sequence from position 22 to position 36.
- Amino acid positions are defined according to Kabat.
- Fig. 6 is a histogram illustrating the Sweet Eiseriberg hydrophobicity score
- VH domains HEL4, pA-C13, pA-C36, pA-C47, pA-C59, pA-C76 and pA-C85 have Sweet/Eisenberg values that are less than 0, and the VH domains of each of these clones undergo reversible heat-induced unfolding.
- Amino acid positions are defined according to Kabat.
- Fig. 7 is an illustration of the characteristic shape of an unfolding curve and a refolding curve.
- the curves are plotted using a measure of the concentration of properly folded polypeptide (e.g., ellipticity or fluorescence) as the abscissa, and the unfolding agent (e.g., heat (temperature)) as the ordinate.
- the unfolding and refolding curves include a region in which the polypeptides are folded, an unfolding/refolding transition in which polypeptides are unfolded to various degrees, and a portion in which the polypeptides are unfolded.
- the y-axis intercept of the refolding curve is the relative amount of refolded protein recovered.
- TM is the melting temperature of the polypeptide
- TM-10 and TM +10 are the melting temperature of the polypeptide minus 10 degrees and plus 10 degrees, respectively.
- the illustrated refolding curve indicates that greater than 75% of the polypeptides refolded.
- Fig. 8 is a graph showing heat-induced unfolding of dAb HEL4.
- dAb FIEL4 was unfolded by heating and ellipticity assessed during heating (filled circles). The unfolded dAb was then refolded by decreasing the temperature. The refolded dAb was then again unfolded by heating and ellipticity assessed during heating (open diamonds).
- the graph shows that the unfolding curves of both heat-induced unfoldings are superimposable, demonstrating that dAb HEL4 undergoes reversible heat-induced unfolding.
- the inset shows the far-UV CD spectra for folded dAb HEL4 at 25°C (Fold) and for unfolded dAb HEL4 at 80°C (Unfold).
- Fig. 9 is a graph showing that a dAb comprising DP47 variant in which Trp47 was replace with Arg (DP47-W47R) does not unfold reversibly upon heating.
- dAb DP47-W47R was unfolded by heating and ellipticity assessed during heating (filled circles). The unfolded dAb was then refolded by decreasing the temperature. The refolded dAb was then again unfolded by heating and ellipticity assessed during heating (open squares). The unfolding curves are not superimposable and have a shape characteristic of denatured polypeptides.
- Fig. 10 is a graph showing that a dAb comprising DP47 variant in which Ser35 was replaced with Gly (DP47-S47G) unfolds reversibly upon heating to a limited extent.
- dAb DP47- S47G was unfolded by heating and ellipticity assessed during heating (filled circles). The unfolded dAb was then refolded by decreasing the temperature. The refolded dAb was then again unfolded by heating and ellipticity assessed during heating (open diamonds). The unfolding curves are not superimposable and reveal that a proportion of the ellipticity (and hence a portion of the original secondary structure) was recovered upon refolding, and that a melting transition is observed upon re-heating the sample.
- Fig. 11 is a graph illustrating the relationship between thermodynamic stability of polypeptides ( ⁇ G folded ⁇ unfolded) and reversible unfolding of polypeptides displayed on phage.
- the graph shows that non-refoldable polypeptides BSA1 and DP47d have high thermodynamic stability, and that refoldable polypeptide HEL4 and several refoldable mutants of DP47 have lower thermodynamic stability.
- Thermodynamic stability of refoldable polypeptides was determined from ellipticity data obtained during heat-induced unfolding.
- Thermodynamic stability of polypeptides that were not refoldable was determined by monitoring fluorescence during urea-induced unfolding.
- Fig. 12 is a graph illustrating the relationship between thermodynamic stability of polypeptides ( ⁇ G folded ⁇ unfolded) and protein expression level in E, coli supernatant.
- the graph shows that non-refoldable polypeptides BSA1 and DP47 have high thermodynamic stability but are expressed at relatively low levels, and that refoldable polypeptide HEL4 and several refoldable mutants of DP47dhave lower thermodynamic stability but are expressed at relatively high levels.
- Thermodynamic stability of refoldable polypeptides was determined from ellipticity data obtained during heat-induced unfolding.
- Protein expression is the amount of polypeptide purified using protein A sepharose from a 1L culture of E. coli, normalized to cell density (OD600) of the culture.
- Fig. 13 is a graph illustrating the relationship between protein expression levels in E, coli supernatant and reversible unfolding of polypeptides displayed on phage.
- the graph shows that non-refoldable polypeptides BSA1 and DP47d are expressed at relatively low levels, and that refoldable polypeptide HEL4 and several refoldable mutants of DP47d have are expressed at relatively high levels.
- the graph reveals a direct correlation between reversible unfolding of polypeptides displayed on phage and the quantity of polypeptide in the supernatant of cultures of £. coli that express the polypeptide.
- Protein expression is the amount of polypeptide purified using protein A sepharose from a 1L culture of E. coli, normalized to cell density (OD600) of the culture.
- Fig. 14 is a graph showing heat-induced unfolding of a single chain Fv (scFv) containing a reversibly unfoldable VK (DPK9-I75N) and a reversibly unfoldable V H (DP47-F27D).
- the scFv was unfolded by heating and ellipticity assessed during heating (filled circles).
- the unfolded scFv was then refolded by decreasing the temperature.
- the refolded scFv was then again unfolded by heating and ellipticity assessed during heating (open diamonds).
- the graph shows that the unfolding curves of both heat-induced unfoldings are superimposable, demonstrating that scFv undergoes reversible heat-induced unfolding, Other scFvs containing germline V H and germline VK, germline V H and unfoldable VK (DPK9-I75N), or reversibly unfoldable V H (DP47-F27D) and germline VK aggregated under the conditions used.
- the inset shows the far-UV CD spectra for folded scFv before heat induced unfolding (dark trace) and for folded scFV following heat induced unfolding and refolding (jighter trace). The spectra are superimposable indicating that the scFv regained all secondary structure following refolding.
- Figs. 15 A and 15B are histograms showing the effect of heat-treatment of phage (80°C for 10 min then cooled to 4°C for 10 min) on binding of displayed dAbs to protein A or antibody
- FIG. 15A phage displaying (5xl0 n TU/ml) either DP47d or HEL4 dAb multivalently were assayed for binding to either anti-c- myc antibody (9E10) or protein A by ELISA.
- 9E10 recognises the c-myc tag peptide tag appended to the dAb as a linear determinant.
- phage displaying DP47d were assayed for binding to protein A by ELISA. Phage concentration was high (5x10 n TU/ml) or low (lxlO 9 TU/ml) and DP47d was displayed in multivalent or monovalent states.
- Fig,15C is a histogram showing the effect of heat-treatment of phage (80°C for 10 min then cooled to 4°C for 10 min) on infectivity. Phage displaying either DP47d or HEL4 multivalently were heated and then cooled, after 10 min a phage sample was treated with 0.9 mg mL trypsin at 22°C for 10 min, and then used to infect £. c ⁇ /j TGI cells.
- FIGS 16A - 16C are copies of transmission electron micrographs of negatively stained phage tips before and after heat treatment (10 min at 80°C).
- FIGS 16A and 16B show that heated DP47d phages form aggregates.
- heated HEL4 phages which display the HEL4 VH domain, which unfolds reversibly, did not form aggregates.
- Fig. 16D is a copy of an image of a Western blot in which 10 10 transducing units (TU) of phage per lane were separated and detected using an anti-pin mouse monoclonal antibody.
- the phage loaded in lanes 1 to 6 were: fd, HEL4
- Fig. 17A is a representative analytical gel-filtration chromatagram of selected human VH3 dAbs.
- the chromatograms for C13 ( ), C36 ( ), C47 ( ), C59 ( -), C76 ( ) and C85 ( ) dAbs (10 ⁇ M in PBS) were obtained using a SUPERDEX-75 column (Amersham Biosciences), apparent Mr for
- C13, C36, C47, C59, C76 and C85 were 22 kDa, 17 J Da, 19 kDa, 10 kDa, 20 kDa, and 15 kDa, respectively.
- Fig. 17B is a graph showing heat induced denaturation curves of the C36 dAb (5 ⁇ M in PBS) recorded by CD at 235 nm: T , mean residual ellipticity upon first heating; 0, mean residual ellipticity upon second heating.
- Fig. 18A is a graph showing that TAR2-10-27 and variants TAR2-10-27 F27D and TAR2-10-27 Y23D bind human Tumor Necrosis Factor Receptor 1
- TNFR1 TNFR1
- TNFR1 TNF-1 and inhibit binding of TNF to the receptor in a receptor binding ELISA.
- TNFR1 was immobilized on the plate and TNF was mixed with TAR2-10-27,
- TAR2-10-27 F27D or TAR2-10-27 Y23D were then added to the wells.
- the amount of TNF that bound the immobilized receptor was quantified using an anti-TNF antibody.
- TAR2-10-27, TAR2-1 -27 F27D and TAR2-10-27 Y23D each bound human TNFR.1 and inhibited the binding of TNF to the receptor.
- Fig. 18B is a graph showing that TAR2-10-27 and variants TAR2-10-27 F27D and TAR2-10-27 Y23D bound human Tumor Necrosis Factor Receptor 1 (TNFR1) expressed on HeLa cells and inhibited TNF-induced production of IL-8 in an in vitro assay.
- TNFR1 Tumor Necrosis Factor Receptor 1
- HeLa cells were plated in microtitre plates and incubated overnight with TAR2-10-27, TAR2-10-27 F27D or TAR2-10-27 Y23D and 300 pg/ml TNF. Post incubation, the supernatant was aspirated off the cells and the amount of IL-8 in the supernatant was measured using a sandwich ELISA.
- TAR2- 10-27, TAR2-10-27 F27D and TAR2-10-27 Y23D each bound human TNFR1 and inhibited TNF-induced IL-8 production.
- the invention relates to polypeptides that unfold reversibly and to methods for selecting and/or designing such polypeptides.
- Polypeptides that unfold reversibly provide several advantages. Notably, such polypeptides are resistant to aggregation or do not aggregate. Due to this resistance to aggregation, polypeptides that unfold reversibly can readily be produced in high yield as soluble proteins by expression using a suitable biological production system, such as E. coli.
- polypeptides that unfold reversibly can be formulated and/or stored at higher concentrations than conventional polypeptides, and with less aggregation and loss of activity.
- polypeptides that unfold reversibly can be selected from a polypeptide display system in which the polypeptides have been unfolded (e.g., by heating) and refolding (e.g., by cooling).
- the selection and design processes described herein yields polypeptides that unfold reversibly and are resistant to aggregation. These selection and design processes are distinct from methods that select polypeptides based on enhanced stability, such as polypeptides that remain folded at elevated temperature. (See, e.g., Jung, S. et al., J. Mol. Biol. -?P4:163-180 (1999).)
- polypeptide display system refers to a system in which a collection of polypeptides are accessible for selection based upon a desired characteristic, such as a physical, chemical or functional characteristic.
- the polypeptide display system can be a suitable repertoire of polypeptides (e.g., in a solution, immobilized on a suitable support).
- the polypeptide display system can also be a biochemical system that employs a cellular expression system (e.g, expression of a library of nucleic acids in, e.g., transformed, infected, transfected or transduced cells and display of the encoded polypeptides on the surface of the cells) or an acellular expression system (e.g., emulsion compartmentalization and display).
- Preferred polypeptide display systems link the coding function of a nucleic acid and physical, chemical and/or functional characteristics of a polypeptide encoded by the nucleic acid.
- polypeptides that have a desired physical, chemical and/or functional characteristic can be selected and a nucleic acid encoding the selected polypeptide can be readily isolated or recovered.
- polypeptide display systems that link the coding function of a nucleic acid and physical, chemical and/or functional characteristics of a polypeptide are known in the art, for example, bacteriophage display (phage display), ribosome display, emulsion compartmentalization and display, yeast display, puromycin display, bacterial display, polypeptide display on plasmid and covalent display and the like.
- bacteriophage display phage display
- ribosome display emulsion compartmentalization and display
- yeast display yeast display
- puromycin display e.g., bacterial display, polypeptide display on plasmid and covalent display and the like.
- the polypeptide display system can comprise a plurality of replicable genetic display packages, as described by McCafferty et al. (e.g., WO 92/01047; U.S. Patent No. 6,172,197).
- a replicable genetic display package (RGDP) is a biological particle which has genetic information providing the particle with the ability to replicate.
- An RGDP can display on its surface at least part of a polypeptide.
- the polypeptide can be encoded by genetic information native to the RGDP and/or artificially placed into the RGDP or an ancestor of it.
- the RGDP can be a vims e.g., a bacteriophage, such as fd or Ml 3.
- the RGDP can be a bacteriophage which displays an antibody variable domain (e.g., VH, VI.) at its surface.
- This type of RGDP can be referred to as a phage antibody (pAb),
- reversibly unfoldable and “unfolds reversibly” refer to polypeptides that can be unfolded (e.g., by heat) and refolded in the method of the invention.
- a reversibly unfoldable polypeptide (a polypeptide that unfolds reversibly) loses function when unfolded but regains function upon refolding.
- Such polypeptides are distinguished from polypeptides that aggregate when unfolded or that improperly refold (misfolded polypeptides), i.e., do not regain function.
- a polypeptide that unfolds reversibly can unfold reversibly when displayed in a polypeptide display system, for example, when display on bacteriophage.
- Particularly preferred polypeptide that unfold reversibly can unfold reversibly when displayed in a polypeptide display system and as a soluble polypeptide (e.g., an autonomous soluble polypeptide).
- oire of polypeptides refers to a collection of polypeptides that are characterized by amino acid sequence diversity.
- the individual members of a repertoire can have common features, such as common structural features (e.g., a common core structure) and/or common functional features (e.g. , capacity to bind a common ligand (e.g. , a generic ligand or a target ligand)).
- common structural features e.g., a common core structure
- common functional features e.g. , capacity to bind a common ligand (e.g. , a generic ligand or a target ligand)).
- library refers to a collection of heterogeneous nucleic acids that can be expressed and preferably are replicable.
- a library can contain a collection of heterogeneous nucleic acids that are incorporated into a suitable vector, such as an expression plasmid, a phagemid and the like. Expression of such a library can produce a repertoire of polypeptides.
- Library also refers to a collection of heterogeneous polypeptides that are displayed in a polypeptide display system that links coding function of a nucleic acid and physical, chemical and/or functional characteristics of a polypeptide encoded by the nucleic acid, and can be selected or screened to provide an individual polypeptide (and nucleic acid encoding same) or a population of polypeptides (and nucleic acids encoding same) that have a desired physical, chemical and/or functional characteristic.
- a collection of phage that displays a collection of heterogeneous polypeptides is one example of such a library.
- a library that is a collection of heterogeneous polypeptides encompasses a repertoire of polypeptides.
- “functional” describes a polypeptide that is properly folded so as to have a specific desired activity, such as ligand-binding activity (e.g., binding generic ligand, binding target ligand), or the biological activity of native or naturally produced protein.
- ligand-binding activity e.g., binding generic ligand, binding target ligand
- the term “functional polypeptide” includes an antibody or antigen-binding fragment thereof that binds a target antigen through its antigen-binding site, and an enzyme that binds its substrate(s).
- “generic ligand” refers to a ligand that binds a substantial portion (e.g., substantially all) of the functional members of a given repertoire.
- a generic ligand (e.g., a common generic ligand) can bind many members of a given repertoire even though the members may not have binding specificity for a common target ligand.
- a functional generic ligand-binding site on a polypeptide indicates that the polypeptide is correctly folded and functional. Accordingly, polypeptides that are correctly folded can be selected or recovered from a repertoire of polypeptides by binding to a generic ligand.
- Suitable examples of generic ligands include superantigens, antibodies that bind an epitope expressed on a substantial portion of functional members of a repertoire, and the like.
- target ligand refers to a ligand which is specifically or selectively bound by a polypeptide.
- a target ligand can be a ligand for which a specific binding polypeptide or polypeptides in a repertoire are identified.
- the target ligand can be any desired antigen or epitope, and when a polypeptide is an enzyme, the target ligand can be any desired substrate. Binding to the target antigen is dependent upon the polypeptide being functional, and upon the specificity of the target antigen-binding site of the polypeptide.
- antibody polypeptide is a polypeptide that is an antibody, a portion of an antibody, or a fusion protein that contains a portion of an antibody (e.g., an antigen-binding portion).
- antibody polypeptides include, for example, an antibody (e.g., an IgG), an antibody heavy chain, an antibody light chain, homodimers and heterodimers of heavy chains and/or light chains, and antigen-binding fragments or portions of an antibody, such as a Fv fragment (e.g.
- single chain Fv scFv
- a disulfide bonded Fv a Fab fragment
- a Fab' fragment a F(ab') 2 fragment
- V H , VL single variable domain
- dAb single variable domain
- antibody format refers to any suitable polypeptide structure in which an antibody variable domain can be incorporated so as to confer binding specificity for antigen on the structure.
- suitable antibody formats are known in the art, such as, chimeric antibodies, humanized antibodies, human antibodies, single chain antibodies, bispecific antibodies, antibody heavy chains, antibody light chains, homodimers and heterodimers of antibody heavy chains and/or light chains, antigen-binding fragments of any of the foregoing (e.g., a Fv fragment (e.g., single chain Fv (scFv), a disulfide bonded Fv), a Fab fragment, a Fab' fragment, a F(ab') 2 fragment), a single variable domain (e.g., VH, V L ), a dAb, and modified versions of any of the foregoing (e.g. , modified by the covalent attachment of polyethylene glycol or other suitable polymer).
- a Fv fragment e.g., single chain Fv (scFv),
- Superantigen is a term of art that refers to generic ligands that interact with members of the immunoglobulin superfamily at a site that is distinct from the conventional ligand-binding sites of these proteins. Staphylococcal enterotoxins are examples of superantigens which interact with T-cell receptors. Superantigens that bind antibodies include Protein G, which binds the IgG constant region (Bjorck and Kronvall, J. Immunol, 133:969 (1984)); Protein A which binds the IgG constant region and VH domains (Forsgren and Sjoquist, J. Immunol, 97:822 (1966)); and Protein L which binds VL domains (Bjorck, J.
- unfolding agent refers to an agent (e.g., compound) or to energy that can cause polypeptide unfolding.
- an unfolding agent is a compound
- the compound can be added to a polypeptide display system in an amount sufficient to cause a desired degree of polypeptide unfolding.
- suitable compounds include, chaotropic agents (e.g., guanidine hydrochloride, urea), acids (e.g. , hydrochloric acid, acetic acid), bases (e.g.
- the unfolding agent can be energy, such as heat and/or pressure.
- the polypeptide system When the unfolding agent is heat, the polypeptide system is exposed to a sufficient amount of energy (e.g., thermal, electromagnetic) to impart enough heat to the polypeptide display system to raise the temperature of the system to a temperature that is sufficient to cause a desired degree of polypeptide unfolding.
- a sufficient amount of energy e.g., thermal, electromagnetic
- Preferred unfolding agents do not substantially inhibit aggregation of unfolded polypeptides that do not unfold reversibly.
- folding gatekeeper refers to an amino acid residue that, by virtue of its biophysical characteristics and by its position in the amino acid sequence of a protein, prevents the irreversible formation of aggregates upon protein unfolding.
- a folding gatekeeper residue blocks off-pathway aggregation, thereby ensuring that the protein can undergo reversible unfolding.
- a folding gate keeper generally reduces the SE score (hydrophobicity score) of the amino acid sequence of the region in which it is found.
- secretable or “secreted” means that when a polypeptide is produced by expression in E. coli., it is produced and exported to the periplastnic space or to the medium.
- polypeptide unfolding and refolding can be assessed, for example, by directly or indirectly detecting polypeptide structure using any suitable method,
- polypeptide structure can be detected by circular dichroism (CD) (e.g., far- UV CD, near-UV CD), fluorescence (e.g., fluorescence of tryptophan side chains), susceptibility to proteolysis, nuclear magnetic resonance (NMR), or by detecting or measuring a polypeptide function that is dependent upon proper folding.
- CD circular dichroism
- fluorescence e.g., fluorescence of tryptophan side chains
- susceptibility to proteolysis e.g., nuclear magnetic resonance (NMR)
- NMR nuclear magnetic resonance
- polypeptide unfolding is assessed using a functional assay in which loss of binding function (e.g., binding a generic and/or target ligand, binding a substrate) indicates that the polypeptide is unfolded.
- the extent of unfolding and refolding of a soluble polypeptide can determined by using an unfolding or denaturation curve.
- An unfolding curve can be produced by plotting the unfolding agent (e.g., temperature, concentration of chaotropic agent, concentration of organic solvent) as the ordinate and the relative concentration of folded polypeptide as the abscissa.
- the relative concentration of folded polypeptide can be determined directly or indirectly using any suitable method (e.g., CD, fluorescence, binding assay).
- a polypeptide solution can be prepared and ellipticity of the solution determined by CD.
- the ellipticity value obtained represents a relative concentration of folded polypeptide of 100%.
- the polypeptide in the solution is then unfolded by incrementally adding unfolding agent (e.g., heat, a chaotropic agent) and ellipticity is determined at suitable increments (e.g., after each increase of one degree in temperature).
- unfolding agent e.g., heat, a chaotropic agent
- ellipticity is determined at suitable increments (e.g., after each increase of one degree in temperature).
- the polypeptide in solution is then refolded by incrementally reducing the unfolding agent and ellipticity is determined at suitable increments.
- the data can be plotted to produce an unfolding curve and a refolding curve. As shown in Fig.
- the unfolding and refolding curves have a characteristic shape that includes a portion in which the polypeptide molecules are folded, an unfolding/refolding transition in which polypeptide molecules are unfolded to various degrees, and a portion in which the polypeptide molecules are unfolded.
- the y-axis intercept of the refolding curve is the relative amount of refolded protein recovered.
- a recovery of at least about 50%, or at least about 60%, or at least about 70%, or at least about 75%, or at least about 80%, or at least about 85%, or at least about 90%, or at least about 95% is indicative that the polypeptide unfolds reversibly.
- the soluble polypeptide unfolds reversibly when heated.
- Reversibility of unfolding of the soluble polypeptide is determined by preparing a polypeptide solution and plotting heat unfolding and refolding curves for the polypeptide.
- the peptide solution can be prepared in any suitable solvent, such as an aqueous buffer that has a pH suitable to allow the peptide to dissolve (e.g., pH that is about 3 units above or below the isoelectric point (pi)).
- the polypeptide solution is concentrated enough to allow unfolding/folding to be detected.
- the polypeptide solution can be about 0.1 ⁇ M to about 100 ⁇ M, or preferably about 1 ⁇ M to about 10 ⁇ M.
- the solution can be heated to about ten degrees below the Tm (Tm-10) and folding assessed by ellipticity or fluorescence (e.g., far-UV CD scan from 200 nm to 250 nm, fixed wavelength CD at 235 nm or 225 nm; tryptophan fluorescent emission spectra at 300 to 450 nm with excitation at 298 nm) to provide 100% relative folded polypeptide.
- Tm melting temperature
- the solution is then heated to at least ten degrees above T (Tm+10) in predetermined increments (e.g., increases of about 0,1 to about 1 degree), and ellipticity or fluorescence is determined at each increment, Then, the polypeptide is refolded by cooling to at least Tm-10 in predetermined increments and ellipticity or fluorescence determined at each increment. If the melting temperature of the polypeptide is not known, the solution can be unfolded by incrementally heating from about 25°C to about 100°C and then refolded by incrementally cooling to at least about 25 Q C, and ellipticity or fluorescence at each heating and cooling increment is determined. The data obtained can be plotted to produce an unfolding curve and a refolding curve, in which the y-axis intercept of the refolding curve is the relative amount of refolded protein recovered.
- polypeptides unfold reversibly as soluble polypeptides, but not as displayed polypeptides (e.g., displayed as phage coat protein fusion proteins on the surface of a bacteriophage).
- polypeptides that undergo reversible unfolding as displayed polypeptides generally also undergo ⁇ eversible unfolding when prepared as soluble polypeptides.
- reversible unfolding in the context of a displayed polypeptide is highly advantageous and affords the ability to select polypeptides that are reversibly unfoldable as soluble polypeptides from a repertoire or library of displayed polypeptides.
- Unfolding and refolding of polypeptides that are contained within a polypeptide display system can be assessed by detecting polypeptide function that is dependent upon proper folding.
- a polypeptide display system comprising displayed polypeptides that have a common function, such as binding a common ligand (e.g., a generic ligand, a target ligand, a substrate), can be unfolded and then refolded, and refolding can be assessed using a functional assay.
- a common ligand e.g., a generic ligand, a target ligand, a substrate
- whether a polypeptide that has a binding activity unfolds reversibly can be determined by displaying the polypeptide on a bacteriophage and measuring or determining binding activity of the displayed polypeptide.
- the displayed polypeptide can be unfolded by heating the phage displaying the polypeptide to about 80°C, and then refolded by cooling the phage to about 20°C or about room temperature, and binding activity of the refolded polypeptide can be measure or determined.
- a recovery of at least about 50%, or at least about 60%, or at least about 70%, or at least about 75%, or at least about 80%, or at least about 85%, or at least about 90%, or at least about 95% of the binding activity is indicative that the polypeptide unfolds reversibly.
- the displayed polypeptide comprises an antibody variable domain, and binding to a generic ligand (e.g., protein A, protein L) or a target ligand is determined.
- polypeptides disclosed herein unfold reversibly in solution and/or when displayed in a suitable polypeptide display system at a polypeptide concentration of at least about 1 ⁇ M to about 1 mM, at least about 1 ⁇ M to about 500 ⁇ M, or at least about ⁇ M to about 100 ⁇ M.
- certain single human antibody variable domains can unfold reversibly when displayed in a multimeric phage display system that produces a local concentration (on the phage tip) of displayed antibody variable domain polypeptide of about 0.5 M.
- the polypeptides unfold reversibly in solution or when displayed on the phage tip at a polypeptide concentration of about 10 ⁇ M, about 20 ⁇ M, about 30 ⁇ M, about 40 ⁇ M, about 50 ⁇ M, about 60 ⁇ M, about 70 ⁇ M, about 80 ⁇ M, about 90 ⁇ M, about 100 ⁇ M, about 200 ⁇ M, about 300 ⁇ M, about 400 ⁇ M or about 500 ⁇ M.
- the invention is a process for selecting, isolating and/or recovering a polypeptide that unfolds reversibly from a library or a repertoire of polypeptides (e.g., a polypeptide display system).
- the method comprises unfolding a collection of polypeptides (e.g., the polypeptides ina library, a repertoire or a polypeptide display system), refolding at least a portion of the unfolded polypeptides, and selecting, isolating and/or recovering a refolded polypeptide.
- the method comprises providing a collection of unfolded polypeptides (e.g., the polypeptides in a library, a repertoire or a polypeptide display system), refolding at least a portion of the unfolded polypeptides, and selecting, isolating and or recovering a refolded polypeptide.
- a collection of unfolded polypeptides e.g., the polypeptides in a library, a repertoire or a polypeptide display system
- the polypeptide that unfolds reversibly is selected, isolated and/or recovered from a repertoire of polypeptides in a suitable polypeptide display system.
- a polypeptide that unfolds reversibly can be selected, isolated and/or recovered from a repertoire of polypeptides that is in solution, or is covalently or noncovalently attached to a suitable surface, such as plastic or glass (e.g., microtiter plate, polypeptide array such as a microarray).
- a suitable surface such as plastic or glass (e.g., microtiter plate, polypeptide array such as a microarray).
- a suitable surface such as plastic or glass (e.g., microtiter plate, polypeptide array such as a microarray).
- an array of peptides on a surface in a manner that places each distinct library member (e.g., unique peptide sequence) at a discrete, predefined location in the array can be used.
- each library member in such an array can be determined by its spatial location in the array.
- the locations in the array where binding interactions between a target ligand, for example, and reactive library members occur can determined, thereby identifying the sequences of the reactive members on the basis of spatial location.
- the method employs a polypeptide display system that links the coding function of a nucleic acid and physical, chemical and/or functional characteristics of the polypeptide encoded by the nucleic acid.
- the polypeptide display system comprises a library, such as a bacteriophage display library.
- Bacteriophage display is a particularly preferred polypeptide display system.
- suitable bacteriophage display systems e.g. , monovalent display and multivalent display systems
- have been described. See, e.g., Griffiths et al., U.S. Patent No. 6,555,313 Bl (incorporated herein by reference); Johnson et al., U.S. Patent No.
- the polypeptides displayed in a bacteriophage display system can be displayed on any suitable bacteriophage, such as a filamentous phage (e.g., fd, M13, FI), a lytic phage (e.g., T4, T7, lambda), or an RNA phage (e.g., MS2), for example.
- a filamentous phage e.g., fd, M13, FI
- a lytic phage e.g., T4, T7, lambda
- RNA phage e.g., MS2
- a library of phage that displays a repertoire of polypeptides, as fusion proteins with a suitable phage coat protein is produced or provided.
- a library can be produced using any suitable methods, such as introducing a library of phage vectors or phagemid vectors encoding the displayed polypetides into suitable host bacteria, and culturing the resulting bacteria to produce phage (e.g., using a suitable helper phage or complementing plasmid if desired).
- the library of phage can be recovered from such a culture using any suitable method, such as precipitation and centrifugation.
- the polypeptide display system can comprise a repertoire of polypeptides that contains any desired amount of diversity.
- the repertoire can contain polypeptides that have amino acid sequences that correspond to naturally occurring polypeptides expressed by an organism, group of organisms, desired tissue or desired cell type, or can contain polypeptides that have random or randomized amino acid sequences.
- the polypeptides can share a common core or scaffold.
- all ploypeptides in the repertoire or library can be based on a scaffold selected from protein A, protein L, protein G, a fibronectin domain, an anticalin, CTLA4, a desired enzyme (e.g., a polymerase, a cellulase), or a polypeptide from the immunoglobulin superfamily, such as an antibody or antibody fragment (e.g., a V H , a V L ).
- polypeptides in such a repertoire or library can comprise defined regions of random or randomized amino acid sequence and regions of common amino acid sequence.
- all or substantially all polypeptides in a repertoire are of a desired type, such as a desired enzyme (e.g., a polymerase) or a desired antigen-binding fragment of an antibody (e.g., human VH or human VL).
- the polypeptide display system comprises a repertoire of polypeptides wherein each polypeptide comprises an antibody variable domain.
- each polypeptide in the repertoire can contain a VH, a V L or an Fv (e.g., a single chain Fv).
- Amino acid sequence diversity can be introduced into any desired region of a desired polypeptide or scaffold using any suitable method.
- amino acid sequence diversity can be introduced into a target region, such as a complementarity determining region of an antibody variable domain or a hydrophobic domain, by preparing a library of nucleic acids that encode the diversified polypeptides using any suitable mutagenesis methods (e.g., low fidelity PCR, oligonucleotide-mediated or site directed mutagenesis, diversification using NNK codons) or any other suitable method.
- a region of a polypeptide to be diversified can be randomized.
- polypeptides that make up the repertoire are largely a matter of choice and uniform polypeptide size is not required.
- the polypeptides in the repertoire contain at least about nine amino acid residues and have secondary structure.
- the polypeptides in the repertoire have at least tertiary structure (form at least one domain).
- the repertoires of polypeptides comprise polypeptides that unfold reversibly and can be unfolded using a suitable unfolding agent as described herein.
- a repertoire of polypeptides can be enriched in polypeptides that unfold reversibly and, for example, are secretable when expressed in E. coli.
- at least about 10% of the polypeptides contained in such an enriched repertoire unfold reversibly. More preferably, at least about 20%, or at least about 30%, or at least about 40%, or at least about 50%, or at least about 60%, or at least about 70%, or at least about 80%, or at least about 90% of the polypeptides in the enriched repertoire unfold reversibly.
- Preferred repertoires contain polypeptides that unfold reversibly when heated.
- substantially all polypeptides in the repertoire share a common selectable characteristic (e.g., physical characteristic, chemical characteristic, functional characteristic).
- the common selectable characteristic is dependent upon proper folding and distinguishes properly folded polypeptides from unfolded and misfolded polypeptides.
- the common selectable characteristic can be a characteristic such as binding affinity which allows properly folded polypeptides to be distinguished from and selected over misfolded and unfolded polypeptides.
- the common selectable characteristic can be used to select properly folded polypeptides but is absent from unfolded and misfolded polypeptides.
- a repertoire of polypeptides in which substantially all polypeptides in the repertoire have a common functional characteristic that distinguishes properly folded polypeptides from unfolded and misfolded polypeptides such as a common binding function (e.g., bind a common generic ligand, bind a common target ligand, bind (or are bound by) a common antibody), a common catalytic activity or resistant to proteolysis (e.g. , proteolysis mediated by a particular protease) can be used in the method.
- a repertoire of polypeptides in which substantially all polypeptides that unfold reversibly in the repertoire have a common selectable characteristic that distinguishes properly folded polypeptides from unfolded and misfolded polypeptides can be used in the method.
- the polypeptide display system comprises a library of polypeptides that comprise immunoglobulin variable domains (e.g. , VH, VL).
- the variable domains can be based on a germline sequence (e.g., DP47dummy (SEQ ID NO:3, DPK9 dummy (SEQ ID NO:6)) and if desired can have one or more diversified regions, such as the complementarity determining regions.
- VH germline sequence
- V ⁇ gene segments DP4, DP7, DPS, DP9, DP10, DP31, DP33, DP45, DP46, DP49, DP50, DP51, DP53, DP54, DP65, DP66, DP67, DP68 and DP69 and the JH segments JHl, JH2, JH3, JH4, JH4b, JH5 and JH6.
- V germline sequences encoded by the VK gene segments DPKl, DPK2, DPK3, DPK4, DPK5, DPK6, DPK7, DPK8, DPK9, DPK10, DPK12, DPK13, DPK15, DPK16, DPK18, DPK19, DPK20, DPK21, DPK22, DPK23, DPK24, DPK25, DPK26 and DPK 28, and the JK sege ents JK 1, JK 2, JK 3, JK 4 and J ⁇ 5.
- VK gene segments DPKl, DPK2, DPK3, DPK4, DPK5, DPK6, DPK7, DPK8, DPK9, DPK10, DPK12, DPK13, DPK15, DPK16, DPK18, DPK19, DPK20, DPK21, DPK22, DPK23, DPK24, DPK25, DPK26 and DPK 28, and the JK sege ents JK 1, JK 2, JK 3, JK 4 and J ⁇ 5.
- One or more of the framework regions (FR) of the variable domain can comprise (a) the amino acid sequence of a human framework region, (b) at least 8 contiguous amino acids of the amino acid sequence of a human framework region, or (c) an amino acid sequence encoded by a human germline antibody gene segment, wherein said framework regions are as defined by Kabat.
- the amino acid sequence of one or more of the framework regions is the same as the amino acid sequence of a corresponding framework region encoded by a human germline antibody gene segment, or the amino acid sequences of one or more of said framework regions collectively comprise up to 5 amino acid differences relative to the amino acid sequence of said corresponding framework region encoded by a human germline antibody gene segment.
- the amino acid sequences of FRl, FR2, FR3 and FR4 are the same as the amino acid sequences of corresponding framework regions encoded by a human ge ⁇ nline antibody gene segment, or the amino acid sequences of FRl , FR2, FR3 and FR4 collectively contain up to 10 amino acid differences relative to the amino acid sequences of corresponding framework regions encoded by said human germline antibody gene segments.
- the amino acid sequence of said FRl, FR2 and FR3 are the same as the amino acid sequences of corresponding framework regions encoded by said human germline antibody gene segment.
- the polypeptides comprising a variable domain preferably comprise a target ligand binding site and/or a generic ligand binding site.
- the generic ligand binding site is a binding site for a superantigen, such as protein A, protein L or protein G.
- variable domain can be based on any desired variable domain, for example a human V H (e.g., V H la, V H lb, V H 2, V H 3, V H 4, V H 5, VH 6), a human V ⁇ (e.g via V ⁇ l, V ⁇ ll, V ⁇ i ⁇ , V ⁇ lV, V ⁇ V or V ⁇ VI) or a human VK (e.g., V ⁇ l, V ⁇ 2, V ⁇ 3, V ⁇ 4, V ⁇ 5, V ⁇ 6, V ⁇ 7, V ⁇ 8, V ⁇ 9 or V ⁇ l0).
- a human V H e.g., V H la, V H lb, V H 2, V H 3, V H 4, V H 5, VH 6
- a human V ⁇ e.g via V ⁇ l, V ⁇ ll, V ⁇ i ⁇ , V ⁇ lV, V ⁇ V or V ⁇ VI
- a human VK e.g., V ⁇ l, V ⁇ 2, V ⁇ 3, V ⁇ 4, V ⁇ 5, V ⁇ 6, V ⁇ 7, V ⁇ 8, V ⁇ 9 or V ⁇
- variable domain is not a Camelid immunoglobulin domain, such as a VH H, or contain one or more amino acids (e.g., frame work amino acids) that are unique to Camelid immunoglobulin variable domains encoded by germline sequences but not, for example, to human immunoglobulin variable domains.
- amino acids e.g., frame work amino acids
- the VH that unfolds reversibly does not comprise one or more amino acids that are unique to murine (e.g., mouse) germline framework regions.
- the variable domain unfolds reversibly when heated.
- the isolated polypeptide comprising a variable domain can be an antibody format.
- the isolated polypeptide comprising a variable domain that unfolds reversibly can be a homodimer of variable domain, a heterodimer comprising a variable domain, an Fv, a scFv, a disulfide bonded Fv, a Fab, a single variable domain or a variable domain fused to an immunoglobulin Fc portion.
- polypeptide display system comprises polypeptides that are nucleic acid polymerases, such as variants of a thermostable DNA polymerase (e.g., Taq polymerase.)
- nucleic acid polymerases such as variants of a thermostable DNA polymerase (e.g., Taq polymerase.)
- the polypeptides can be unfolded using any desired unfolding agent.
- Suitable unfolding agents include, for example, heat and or pressure, low or high pH, chaotropic agents (e.g., guanidine hydrochloride, urea and the like) and organic solvents (e.g, an alcohol (e.g., methanol, ethanol), a ketone (e.g., methyl ethyl ketone), an aldehyde (e.g., formaldehyde, dimethylformaldehyde), tetrahydrofuran, dioxane, toluene and the like).
- chaotropic agents e.g., guanidine hydrochloride, urea and the like
- organic solvents e.g, an alcohol (e.g., methanol, ethanol), a ketone (e.g., methyl ethyl ketone), an aldehyde (e.g., formaldehy
- the displayed polypeptides are unfolded using an unfolding agent, with the proviso that the unfolding agent is not a chaotropic agent.
- unfolding is effectuated by exposing the collection of polypeptides to an amount of unfolding agent (e.g., heat) that is sufficient to cause at least about 10% of the polypeptides in the collection to unfold.
- unfolding is effectuated by exposing the collection of polypeptides to an amount of unfolding agent (e.g., heat) that is sufficient to cause at least about 20%, or at least about 30%, or at least about 40%, or at least about 50%, or at least about 60% or at least about 70%, or at least about 80%, or at least about 90%, or at least about 95%, or at least about 98%, or at least about 99%, or substantially all of the polypeptides in the collection to unfold.
- an amount of unfolding agent e.g., heat
- the polypeptides in the repertoire would have a range of melting temperatures (Tm).
- Tm melting temperatures
- a Tm can be obtained for a repertoire by obtaining a random sample of about 10 to about 100 polypetides from the repertoire and determining the average Tm for the polypeptides in the sample.
- the displayed polypeptides are unfolded by heating the polypeptide display system to a suitable unfolding temperature (Ts), such as a temperature that is sufficient to cause at least about 10% of the displayed polypeptides to unfold.
- Ts unfolding temperature
- a temperature that is sufficient to cause a desired percentage of displayed polypeptides to unfold can readily determined using any suitable methods, for example by reference to an unfolding and/or refolding curve (as described herein).
- the lowest temperature that falls within the unfolded portion of the unfolding and refolding curve for the polypeptide system will generally be sufficient.
- the temperature selected will be dependent upon the thermostability of the displayed polypeptides.
- an unfolding temperature of 100°C or higher can be used if the displayed polypeptides are from, or are variants of polypeptides from, a thermophile or extreme thermophile (e.g., Thermus aquaticus).
- a thermophile or extreme thermophile e.g., Thermus aquaticus.
- the polypeptides are generally unfolded by heating to temperature (Ts) that is at least about the melting temperature (Tm) of the polypeptide to be selected or greater that the Tm of the polypeptide to be selected.
- the displayed polypeptides are unfolded by raising the temperature of the polypeptide display system to an unfolding temperature that is between about 25°C and about 100°C.
- the polypeptide display system is phage display
- Unfolding displayed or soluble polypeptides at high temperatures e.g., at about 100°Q is also prefened and can be advantageous.
- heat sterilizable polypeptides can be selected, isolated and/or sterilized by heating the polypeptide display or system or soluble polypeptide to about lOO'O
- the polypeptide display system can be maintained at that temperature for a period of time (e.g., up to about 10 hours), if desired.
- a period of time e.g., up to about 10 hours
- the polypeptide display system can be maintained at the unfolding temperature for a period of about 100 milliseconds to about 10 hours.
- the polypeptide display system is maintained at the unfolding temperature for about 1 second to about 20 minutes.
- the temperature of the polypeptide display system can be raised at any suitable rate, for example at a rate of about 1°C per millisecond to about 1°C per hour. In a particular embodiment, the temperature is preferably raised at a rate of about 1°C per second.
- the unfolded polypeptides in the polypeptide display system, or a portion of the unfolded polypeptides, can be refolded by decreasing the amount or concentration of unfolding agent in the system.
- the amount or concentration of unfolding agent in the system can be decreased using any suitable method, for example, by dilution, dialysis, buffer exchange, titration or other suitable method.
- Heat can be reduced by cooling at room temperature or under refrigeration (e.g., in a refrigerated cooling block or bath), for example.
- Pressure can be reduced, for example, by venting. It may be desirable to refold only a portion of the unfolded displayed polypeptides before selection.
- highly stable polypeptides that unfold reversibly can be selected, isolated and/recovered when the unfolding agent is decreased minimally, such that only the most stable portion (e.g., about 0.00001% to about 1%) of the unfolded displayed polypeptides refold.
- refolding can be effectuated by decreasing the amount or concentration of unfolding agent in the polypeptide display system to an amount or concentration that results in the desired degree of refolding.
- the amount or concentration of unfolding agent that can remain in the polypeptide display system but permit the desired percentage of unfolded displayed polypeptides to refold can be readily determined using any suitable method, for example by reference to an unfolding and refolding curve (as described herein).
- the amount or concentration of unfolding agent can be reduced to the concentration or amount in the polypeptide display system before unfolding (e.g., the system is cooled to room temperature) or the unfolding agent can be substantially removed.
- refolding is effectuated by decreasing the amount or concentration of unfolding agent (e.g., heat) so that at least about 0.00001% of the unfolded polypeptides refold.
- refolding is effectuated by decreasing the amount or concentration of unfolding agent (e.g., heat) so that at least about 0.0001%, or at least about 0.001%, or at least about 0.01%), or at least about 0.1%, or at least about 1%, or at least about 10%, or about 20%, or at least about 30%), or at least about 40%, or at least about 50%, or at least about 60%) or at least about 70%, or at least about 80%, or at least about 90%, or at least about 95%, or at least about 98%, or at least about 99%, or substantially all of the unfolded polypeptides that can undergo refolding in the polypeptide display system refold.
- unfolding agent e.g., heat
- heat is a preferred unfolding agent.
- the displayed unfolded polypeptides are refolded by lowering the temperature of the polypeptide display system to a refolding temperature (Tc) that is below about 99°C but above the freezing temperature of the polypeptide display system.
- Tc refolding temperature
- the refolding temperature selected will be dependent upon the thermostability of the displayed polypeptides.
- a refolding temperature of 100°C or higher can be used if the displayed polypeptides are from, or are variants of polypeptides from, a thermophile or extreme thermophile (e.g., Thermus aquaticus).
- the unfolding temperature and refolding temperature differ by at least about 10°C to at least about 120°C.
- the polypeptide display system is a phage display system
- the displayed polypeptides are unfolded by heating the system to about 80°C
- the displayed unfolded polypeptides can be refolded by reducing the temperature of the phage display system to a temperature between about 1°C to about 70°C.
- the displayed unfolded polypeptides are refolded by reducing the temperature of the phage display system to a temperature between about 1°C to about 60°C, or about 1°C to about 50°C, or about 1°C to about 40°C, or about 1°C to about 30°C, or about 1°C to about 20°C, or about 1°C to about 10°C.
- the polypeptide display system can be maintained at that temperature for any desired period of time (e.g., up to about 10 hours), if desired.
- the polypeptide display system can be maintained at the refolding temperature for a period of about 100 milliseconds to about 10 hours.
- the polypeptide display system is maintained at the refolding temperature for about 1 minute to about 20 minutes.
- the temperature of the polypeptide display system can be decreased at any suitable rate, for example at a rate of about 1°C per millisecond to about 1°C per hour. In a particular embodiment, the temperature is preferably decreased at a rate of about 1°C per 100 milliseconds or about l°Cper second.
- the polypeptide display system can be maintained at atmospheric pressure ( ⁇ 1 atm) during unfolding and refolding of the displayed polypeptides. However, if desired, the polypeptide display system can be maintained at lower or higher pressure, In such situations, more or less unfolding agent may be required to obtain the desired degree of unfolding. For example, more or less heat may need to be applied to the polypeptide display system (relative to a system at 1 atm) in order to achieve the desired unfolding temperature because lowering or raising the pressure of the system can change the temperature of the system.
- Unfolding and refolding can be carried out under suitable pH or buffer conditions. Generally, unfolding and refolding are carried out at a pH of about 1 to about 13, or about 2 to about 12. Preferred pH conditions for unfolding and refolding are a pH of about 5 to about 9, or about 6 to about 8, or about 7. Folding and unfolding the polypeptides under acidic or alkaline conditions can allow for selection of polypeptides that function under extreme acidic or alkaline conditions, such as polypeptide drugs that can be administered orally and/or have therapeutic action in the stomach.
- a polypeptide that unfolds reversibly can be selected, isolated and/or recovered from a repertoire or library (e.g. , in a polypeptide display system) using any suitable method.
- a polypeptide can be recovered by selecting and/or isolating the polypeptide. Recovery can be carried out at a suitable recovery temperature (Tr).
- a suitable recovery temperature is any temperature that is less than the melting temperature (Tm) of the polypeptide but higher than the freezing temperature of the polypeptide display system.
- a polypeptide that unfold reversibly is selected or isolated based on a selectable characteristic (e.g., physical characteristic, chemical characteristic, functional characteristic) that distinguishes properly folded polypeptides from unfolded and misfolded polypeptides.
- selectable characteristics include fluorescence, susceptibility to fluorescence quenching and susceptibility to chemical modification (e.g., with Iodoacetic acid, Iodoacetamide or other suitable polypeptide modifying agent).
- selectable characteristics include fluorescence, susceptibility to fluorescence quenching and susceptibility to chemical modification (e.g., with Iodoacetic acid, Iodoacetamide or other suitable polypeptide modifying agent).
- a polypeptide that unfolds reversibly is selected, isolated and/or recovered based on a suitable selectable functional characteristic that distinguishes properly folded polypeptides from unfolded and misfolded polypeptides.
- Suitable functional characteristics for selecting or isolating a polypeptide that unfolds reversibly include any function that is dependent on proper folding of the polypeptide. Accordingly, a polypeptide that unfolds reversibly can have the function when properly folded, but the function can be lost or diminished upon unfolding and can be absent or diminished when the polypeptide is unfolded or misfolded.
- Suitable selectable functional characteristics include, for example, binding to a generic ligand (e.g., a superantigen), binding to a target ligand (e.g., an antigen, an epitope, a substrate), binding to an antibody (e.g., through an epitope expressed on the properly folded polypeptide), a catalytic activity and resistance to proteolysis. (See, e.g., Tomlinson ef al., WO 99/20749; WO 01/57065; WO 99/58655.)
- the polypeptide that unfolds reversibly is selected attd/or isolated from a library or repertoire of polypeptides in which substantially all polypeptides that unfold reversibly share a common selectable feature.
- the polypeptide that unfolds reversibly can be selected from a library or repertoire of polypeptides in which substantially all polypeptides that unfold reversibly bind a common generic ligand, bind a common target ligand, bind (or are bound by) a common antibody, possess a common catalytic activity or are each resistant to proteolysis (e.g. , proteolysis mediated by a particular protease). Selection based on binding to a common generic ligand can yield a population of polypeptide that contains all or substantially all polypeptides that unfold reversibly that were components of the original library or repertoire.
- any suitable method can be used to select, isolate and/or recover the polypeptides that unfold reversibly.
- polypeptides that bind a target ligand or a generic ligand, such as protein A, protein L or an antibody can be selected, isolated and/or recovered by panning or using a suitable affinity matrix. Panning can be accomplished by adding a solution of ligand (e.g., generic ligand, target ligand) to a suitable vessel (e.g,, tube, petri dish) and allowing the ligand to become deposited or coated onto the walls of the vessel.
- ligand e.g., generic ligand, target ligand
- Excess ligand can be washed away and polypeptides (e.g., a phage display library) can be added to the vessel and the vessel maintained under conditions suitable for polypeptides to bind the immobilized ligand. Unbound polypeptide can be washed away and bound polypeptides can be recovered using any suitable method, such as scraping or lowering the pH, for example.
- polypeptides e.g., a phage display library
- Suitable ligand affinity matrices generally contain a solid support or bead (e.g. , agarose) to which a ligand is covalently or noncovalently attached,
- the affinity matrix can be combined with polypeptides (e.g., a phage display library) using a batch process, a column process or any other suitable process under conditions suitable for binding of polypeptides to the ligand on the matix.
- Polypeptides that do not bind the affinity matrix can be washed away and bound polypeptides can be eluted and recovered using any suitable method, such as elution with a lower pH buffer, with a mild denaturing agent (e.g., urea), or with a peptide that competes for binding to the ligand.
- a mild denaturing agent e.g., urea
- the generic or target ligand is an antibody or antigen binding fragment thereof.
- Antibodies or antigen binding fragments that bind structural features of polypeptides that are substantially conserved in the polypeptides of a library or repertoire are particularly useful as generic ligands.
- Antibodies and antigen binding fragments suitable for use as ligand for isolating, selecting and or polypeptides that unfold reversibly can be monoclonal or polyclonal and can be prepared using any suitable method,
- Polypeptides that unfold reversibly can also be selected, for example, by binding metal ions.
- immobilized metal affinity chromatography IMAC; Hubert and Porath, J. Chromotography, 98 :247(1 80)
- IMAC immobilized metal affinity chromatography
- a resin typically agarose, comprising a bidentate metal chelator (e.g. iminodiacetic acid, IDA, a dicarboxylic acid group) to which is complexed metallic ions.
- Such resins can be readily prepared, and several such resins are commercially available, such as CHELATING SEPHAROSE 6B (Pharmacia Fine Chemicals; Piscataway, NJ).
- Metallic ion that are of use include, but are not limited to, the divalent cations Ni 2+ , Cu 2+ , Zn 2+ , and Co 2+ .
- a repertoire of polypeptides or library can prepared in binding buffer which consists essentially of salt (typically, NaCl or KCl) at a 0.1 - to 1.OM concentration and a weak ligand (e.g. , Tris, ammonia), the latter of which has affinity for the metallic ions of the resin, but to a lesser degree than does a polypeptide to be selected according to the invention.
- Useful concentrations of the weak ligand range from 0.01- to 0.1M in the binding buffer.
- the repertoire of polypeptides or library can contacted with the resin under conditions which permit polypeptides having metal-binding domains to bind.
- Non- binding polypeptides can be washed away, and the selected polypeptides are eluted with a buffer in which the weak ligand is present in a higher concentration than in the binding buffer, specifically, at a concentration sufficient for the weak ligand to displace the selected polypeptides, despite its lower binding affinity for the metallic ions.
- Useful concentrations of the weak ligand in the elution buffer are 10- to 50- fold higher than in the binding buffer, typically from 0.1 to 0.3 M.
- the concentration of salt in the elution buffer equals that in the binding buffer.
- the metallic ions of the resin typically serve as the generic ligand; however, it is contemplated that they can also be used as the target ligand.
- LMAC can be carried out using a standard chromatography apparatus (columns, through which buffer is drawn by gravity, pulled by a vacuum or driven by pressure), or by batch procedure, in which the metal-bearing resin is mixed, in slurry form, with the repertoire of polypeptides or library.
- Partial purification of a serum T4 protein by IMAC has been described (Staples et al, U.S. Patent No. 5,169,936); however, the broad spectrum of proteins comprising surface-exposed metal-binding domains also encompasses other soluble T4 proteins, human serum proteins (e.g, IgG, haptoglobin, hemopexin, Gc-globulin, Clq, C3, C4), human desmoplasmin, Dolichos biflorus lectin, zinc-inhibited Tyr(P) phosphatases, phenolase, carboxypeptidase isoenzymes, Cu, Zn superoxide dismutases (including those of humans and all other eukaryotes), nucleoside diphosphatase, leukocyte interferon, lactoferrin, human plasma C&-SH glycoprotein, f ⁇ -macroglobulin, ⁇ i-antitrypsin, plasminogen activator, gastrointestinal polypeptides, pep
- extracellular domain sequences of membrane-bound proteins may be purified using IMAC.
- Polypeptides that unfold reversibly and comprise a scaffold from any of the above proteins or metal-binding variants thereof can be selected, isolated or recovered using the methods described herein,
- Polypeptides that unfold reversibly can also be selected from a suitable phage display system (or other suitable polypeptide display system), based on infectivity of phage following unfolding and refolding or based on aggregation of phage displaying polypeptides.
- a high local concentration of displayed polypeptide can be produced by displaying polypeptides on multivalent phage proteins (e.g, pin protein of filamentous bacteriophage), The displayed polypeptides are adjacently located at the phage tip and can interact with each other and form aggregates if they do not unfold reversibly.
- Such aggregates can be intraphage aggregates which form between the polypeptides displayed on a particular phage and or interphage aggregates which form between polypeptides displayed on two or more phage when the phage display system comprises a plurality of phage particles at a sufficient concentration.
- polypeptides that unfold reversibly can be selected from a multivalent phage display system by recovering displayed polypeptides that do not aggregate using any suitable method, such as centrifugation (e.g., ultracentrifugation), or by selecting based on function of the displayed polypeptide or infectivity of the phage. For example, when heated to a suitable temperature (e.g. , about 80°C) the displayed polypeptides unfold.
- polypeptides that unfold reversibly can be selected by unfolding and refolding the polypeptides in a suitable polypeptide display system (e.g., a filamentous bacteriophage display system), and selecting phage with infectivity that is not substantially reduced or that is substantially unchanged.
- a suitable polypeptide display system e.g., a filamentous bacteriophage display system
- the selected polypeptides that unfold reversibly can be further selected for any desired properties, such as binding a desired antigen, catalytic activity, and the like. Selection based on infectivity can also be employed to prepare a library or repertoire that is enriched in polypeptides that unfold reversibly or nucleic acids encoding polypeptides that unfold reversibly.
- Polypeptides that unfold reversibly can also be selected from a suitable phage display system (or other suitable polypeptide display system), based on aggregation of phage displaying polypeptides that do not unfold reversibly.
- a suitable polypeptide display system can be unfolded (e.g., by heating to about 80°C) and refolded by cooling under conditions in which at least a portion of polypeptides that do not unfold reversibly aggregate.
- the presence or degree of aggregation can be determined using any suitable method, such as electron microscopy or an infectivity assay.
- Polypeptides that unfold reversibly can be selected by recovering polypeptides (e.g., displayed on phage) that do not aggregate using any suitable method, such as centrifugation (e.g., ultracentrifugation) or by infeceting suitable host bacteria (when the polypeptides are displayed on phage).
- any suitable method such as centrifugation (e.g., ultracentrifugation) or by infeceting suitable host bacteria (when the polypeptides are displayed on phage).
- polypeptide display systems including systems in which polypeptides are immobilized on a solid support, can be prepared in which displayed polypeptides that do not unfold reversibly can form aggregates.
- the displayed polypeptides in such a system are positioned in close proximity to each other.
- the displayed polypeptides can separated by no more than about twice the length of linear amino acid sequence of the polypeptide.
- the displayed polypeptides are separated by no more that a distance that is determined by multiplying the number of amino acid residues in the polypeptide by the length of a peptide bond (3.8 A) times 2,
- the polypeptides can be separated by no more than about 200 A to about 300 A.
- the polypeptide contains 100 amino acids, and they are separated by no more that 760A.
- the displayed polypeptides are spaced no closer than the distance between the centers of two adjacent identical globular polypetides.
- two immunoglobulin variable domains that are tethered to a substrate by their C-termini should be no closer that 25A.
- polypeptide display systems can be prepared using any suitable method.
- polypeptides can be concatenated (see, e.g., WO 02/30945) or produced as fusion proteins which bring the polypeptides together (e.g., by dimerizing or oligomerizing, by assembly into a viral coat or capsid).
- Such polypeptide display systems can also be prepared by immobilizing polypeptides on to a suitable solid support (e.g., head, plastic, glass) using any suitable method.
- Polypeptides that unfold reversibly can be selected from such a polypeptide display system by recovering displayed polypeptides that do not aggregate using any suitable method. For example, when the polypeptides are displayed on a mobile sold support (e.g., beads) interbead aggregates can form and be removed by as centrifugation or other suitable method.
- a mobile sold support e.g., beads
- the polypeptide display system comprises a plurality of polypeptide species and more than one copy (polypeptide molecule) of each species.
- each polypeptide species is present in a concentration that is sufficient to permit aggregation of species that do not unfold reversibly.
- concentration e.g., local concentration such as on the phage tip
- the process for selecting a polypeptide that binds a ligand and unfolds reversibly from a repertoire of polypeptides comprises, providing a polypeptide display system comprising a repertoire of polypeptides; heating the repertoire to a temperature (Ts) at which at least a portion of the displayed polypeptides are unfolded; cooling the repertoire to a temperature (Tc) wherein Ts>Tc, whereby at least a portion of the polypeptides have refolded and a portion of the polypeptides have aggregated; and recovering at a temperature (Tr) at least one polypeptide that unfolds ⁇ eversibly and binds a ligand,
- the ligand is bound by (binds) folded polypeptide but is not bound by (does not bind) aggregated polypeptides.
- the recovered polypeptide that unfolds reversibly has a melting temperature (Tm), and preferably, the repertoire was heated to Ts, cooled to Tc and the polypeptide that unfolds reversibly was isolated at Tr, such that Ts>Tm>Tc, and Ts>Tm>Tr.
- Tm melting temperature
- the recovered polypeptide is not misfolded. Misfolded polypeptides can be identified based on certain selectable characteristic (e.g. , physical characteristic, chemical characteristic, functional characteristic) that distinguishes properly folded polypeptides from unfolded and misfolded polypeptides as described herein.
- the process further comprises confirming that the recovered polypeptide binds a ligand (e.g., target ligand, generic ligand).
- the process can further comprise confirming that the Tm of the recovered polypeptide is less than the temperature Ts to which the repertoire was heated.
- the Tm of a recovered polypeptide can be determined using any suitable method, e.g., by circular dichroism, change in fluorescence of the polypeptide with increasing temperature, differential scanning calorimetry (DSC).
- the recovered polypeptide has a Tm that is equal to or greater than about 37°C, and more preferably greater that about 37°C, when rounded up to the nearest whole number.
- the polypeptide display system is a phage display system, such as a multivalent phage display system.
- the polypeptide display system, pre-selection repertoire and/or preselection library comprises at least about 10 3 members (species), at least about 10 4 members, at least about 10 5 members, at least about 10 6 members, or at least about 10 7 members.
- a phage display system is provided and at least a portion of the polypeptides that aggregate at Tc form interphage aggregates.
- a phage display system is provided and at least a portion of the polypeptides that aggregate at Tc form interphage aggregates and intraphage aggregates.
- the invention relates to a method for designing and/or preparing a variant polypeptide that unfolds reversibly.
- the method comprises providing the amino acid sequence of a parental polypeptide, identifying one or more regions of said amino acid sequence that are hydrophobic, selecting one or more of the hydrophobic regions, and replacing one or more amino acids in a selected region to produce a variant amino acid sequence in which the hydrophobicity of the selected region is reduced.
- a polypeptide that comprises or consists of the variant amino acid sequence can be produced and, if desired, its ability to unfold reversibly can be assessed or confirmed using any suitable method.
- the variant unfolds reversibly when heated and cooled, as described herein.
- a variant polypeptide that unfolds reversibly can be prepared using any desired parental polypeptide.
- the polypeptide that unfolds reversibly can be prepared using a parental polypeptide that is based on a scaffold selected from an enzyme, protein A, protein L, a fibronectin, an anticalin, a domain of
- CTLA4 or a polypeptide of the immunoglobulin superfamily, such as an antibody or an antibody fragment.
- the parental polypeptide is an antibody variable domain (e.g., a human VH, a human VL), it can comprise V and or D (where the polypeptide comprises a VH domain) and/or J segment sequences encoded by a ge ⁇ nline V, D or J segment respectively, or a sequence that results from naturally occurring or artificial mutations (e.g., somatic mutation or other processes).
- the parental polypeptide can be a parental immunoglobulin variable domain having an amino acid sequence that is encoded by germline gene segments, or that results from insertion and/or deletion of nucleotides during V(D) J recombination (e,g. , N nucleotides, P nucleotides) and/or mutations that arise during affinity maturation.
- the parental polypeptide can be a parental enzyme that has an amino acid sequence encoded by the ge ⁇ nline or that results from naturally occurring or artificial mutation (e.g., somatic mutation).
- Hydrophobic regions of the parental polypeptide can be identified using any suitable scale or algorithm. Preferably, hydrophobic regions are identified using the method of Sweet and Eisenberg. (Sweet, R.M. and Eisenberg, D., J. Mol Biol. 777:479-488 (1983).) The Sweet and Eisenberg method can produce a hydrophobicity score (S/E score) for an amino acid sequence (e.g., an identified hydrophobic region) using a 9 to 18 amino acid window (e.g., a window of 9, 10, 11, 12, 13, 14, 15, 16, 17 or 18 amino acids).
- S/E score hydrophobicity score
- a 15 amino acid window is generally preferred.
- a ⁇ a ⁇ ental amino acid sequence is analyzed using the Sweet and Eisenberg method, and a hydrophobicity plot for a selected hydrophobic region is generated (ordinate is the S/E score using a suitable window, abscissa is the amino acid positions of the selected region).
- a variant amino acid sequence for the selected hydrophobic region is designed in which one or more of the amino acid residues in the parental sequence with a different amino acid residue, and analyzed using the Sweet and Eisenberg method.
- a hydrophobicity plot for the variant amino acid sequence is produces.
- a decrease in the area under the curve of the hydrophobicity plot of a selected hydrophobic region in the variant amino acid sequence relative to the area under the curve of the corresponding region in the parental amino acid sequence is indicative of a reduction in hydrophobicity of the selected region.
- the amino acid sequence of the variant VH or variant V L can comprise one or more of the amino acid replacements for VH or V described herein.
- the amino acid sequence of the variant VH or variant VL can comprise one or more framework regions as described herein.
- a polypeptide comprising a variant antibody variable domain that unfolds reversibly is designed and/or prepared.
- the variant antibody variable domain is a variant VH domain in which the hydrophobicity of the amino acid sequence from position 22 to position 36 is reduced (Kabat amino acid numbering system) relative to the corresponding amino acid sequence of the parental VH.
- the variant antibody variable domain is a variant VH and the hydrophobicity of the HI loop is reduced relative to the HI loop of the parental VH (HI loop is as defined by the AbM amino acid numbering system).
- hydrophobicity is determined using the method of Sweet and Eisenberg with a 15 amino acid window for VH.
- the S/E score of the amino acid sequence from position 22 to position 36 of the variant VH is 0.15 or less, or 0.1 or less for an S/E score rounded to one decimal place, or 0.09 or less, or 0.08 or less, or 0.07 or less, or 0.06 or less, or 0.05 or less, or 0.04 or less, or 0 or less.
- the S/E score of the HI loop of the variant VH is 0 or less.
- the variant antibody variable domain is a variant V and the hydrophobicity of the FR2-CDR2 region and/or FR3 is reduced relative to the FR2-CDR2 region and/or FR3 of the parental V L ( FR2-CDR2 region and FR3 region are as defined by the Kabat amino acid numbering system).
- the variant antibody variable domain is a variant VL and the hydrophobicity of the amino acid sequence from position 44 to position 53 anoVor position 73 to position 76 is reduced relative to the corresponding amino acid sequence of the parental VL (Kabat amino acid numbering system).
- hydrophobicity is determined using the method of Sweet and Eisenberg with an 11 amino acid window for VL- In.
- the S/E score for position 44 to position 53 of the variant V can be than 0,2, less than 0.17, less than 0.15, less fhan0.13, less than 0.10, or less than -0.1.
- the S/E score for FR3 of the variant VL is less than 0.35.
- the S E score for FR3 of the variant V can be less than 0.3, less than 0.25, less than 0.2, less than 0.17, less than 0.15, less than 0.13, less than 0.10, or less than -0.1.
- a variant polypeptide that comprises a variant amino acid sequence and unfolds reversibly can be prepared using any suitable method.
- the amino acid sequence of a parental polypeptide can be altered at particular positions (as described herein with respect to antibody variable domain polypeptides) to produce a variant polypeptide that unfolds reversibly.
- the invention relates to repertoires of polypeptides that unfold reversibly, to libraries that encode polypeptides that unfold reversibly, and to methods for producing such libraries and repertoires
- the libraries and repertoires of polypeptides comprise polypeptides that unfold reversibly (and/or nucleic acids encoding polypeptides that unfold reversibly) using a suitable unfolding agent as described herein.
- At least about 1% of the polypeptides contained in the repertoire or library or encoded by the library unfold reversibly More preferably, at least about 10%, or at least about 20%, or at least about 30%, or at least about 40%, or at least about 50%, or at least about 60%, or at least about 70%, or at least about 80%, or at least about 90%) of the polypeptides in the repertoire or library or encoded by the library unfold reversibly.
- Such libraries are referred to herein as being enriched in polypeptides that unfold reversibly or in nucleic acids encoding polypeptides that unfold reversibly.
- Preferred repertoires and libraries contain polypeptides that unfold reversibly when heated.
- the libraries of the invention comprise heterogeneous nucleic acids that are replicable in a suitable host, such as recombinant vectors that contain nucleic acids encoding polypeptides (e.g., plasmids, phage, phagmids) that are replicable .E. coli, for example.
- a suitable host such as recombinant vectors that contain nucleic acids encoding polypeptides (e.g., plasmids, phage, phagmids) that are replicable .E. coli, for example.
- Libraries that encode and/or contain polypeptides that unfold reversibly can be prepared or obtained using any suitable method.
- the library of the invention can be designed to encode polypeptides that are based on a polypeptide of interest (e.g. , a polypeptide selected from a library) or can be selected from another library using the methods described herein.
- a library enriched in polypeptides that unfold reversibly can be prepared using a suitable polypeptide display system.
- a phage display library comprising a repertoire of displayed polypeptides comprising an antibody variable domain (e.g. , VH, VK, V ⁇ ) is unfolded and refolded as described herein and a collection of refolded polypeptides is recovered thereby yielding a phage display library enriched in polypeptides that unfold reversibly.
- a phage display library comprising a repertoire of displayed polypeptides comprising an antibody variable domain (e.g., VH, VK, V ⁇ ) is first screened to identify members of the repertoire that have binding specificity for a desired target antigen.
- a collection of polypeptides having the desired binding specificity are recovered, the collection is unfolded and refolded, and a collection of polypeptides that unfold reversibly and have the desired binding specificity is recovered, yielding a library enriched in polypeptides that unfold reversibly.
- a polypeptide of interest such as an immunoglobulin variable domain
- the amino acid sequence of the polypeptide is analyzed to identify regions of hydrophobicity.
- Hydrophobicity can be determined using any suitable method, scale or algorithm.
- hydrophobicity is determined using the method of Sweet and Eisenberg. (Sweet, R.M, and Eisenberg, D.,J Mol. Biol. 777:479-488 (1983).)
- a region of hydrophobicity in the polypeptide is selected and a collection of nucleic acids is prepared that contains sequence diversity targeted to the region encoding the hydrophobic region of the polypeptide.
- the collection of nucleic acids can be randomized in the target region or sequence diversity that results in reduced hydrophobicity of the selected region of the polypeptide (through amino acid replacement) can be introduced.
- the collection of nucleic acids can then be inserted into a suitable vector (e.g., a phage, a phagemid) to yield a library.
- a suitable vector e.g., a phage, a phagemid
- the library can be expressed in a suitable polypeptide display system and the polypeptides in the display system can be unfolded, refolded and a collection of polypeptides that unfold reversibly can be selected or recovered as described herein to yield a library enriched in polypeptides that unfold reversibly.
- the hydrophobic region targeted for sequence diversification is a hydrophobic region that is associated with aggregation of unfolded polypeptides.
- Such aggregation prone hydrophobic regions can be identified using any suitable method.
- amino acids in the hydrophobic regions can be replaced with less hydrophobic residues (e.g., Tyr replaced with Asp or Glu).
- the resulting polypeptides can be unfolded and aggregation can be assessed using any suitable method. Decreased aggregation of polypeptide that contains an amino acid replacement indicates that the hydrophobic region that contains the amino acid replacement is an aggregation prone hydrophobic region.
- a particular repertoire of polypeptides or library comprises VH domains (e.g., based on a parental VH domain comprising a germline VH gene segment) containing at least one amino acid replacement that reduces the hydrophobicity of the amino acid sequence from position 22 to position 36, or position 26 to position 35, as defined by the Kabat amino acid numbering scheme.
- Another particular repertoire of polypeptides or library comprises VH domains wherein the hydrophobicity of the amino acid sequence from position 22 to position 36 (Kabat amino acid numbering scheme) has a Sweet/Eisenberg hydrophobicity score of 0 or less.
- At least about 1% of the polypeptides contained in the repertoire or library have a Sweet Eisenberg hydrophobicity score of 0 or less for the amino acid sequence from position 22 to position 36. More preferably, at least about 10%, or at least about 20% > , or at least about 30%, or at least about 40%, or at least about 50%, or at least about 60%, or at least about 70%, or at least about 80%, or at least about 90% of the polypeptides have a Sweet Eisenberg hydrophobicity score of 0 or less for the amino acid sequence from position 22 to position 36.
- Another particular repertoire of polypeptides or library comprises VL domains (e.g., based on a parental VL domain comprising a germline VL gene segment) containing at least one amino acid replacement that reduces the hydrophobicity of the amino acid sequence from position 44 to position 53, as defined by the Kabat amino acid numbering scheme.
- Another repertoire of polypeptides or library comprises V domains (e.g., based on a parental V domain comprising a germline VL gene segment) containing at least one amino acid replacement that reduces the hydrophobicity of framework 3 (FR3), as defined by the Kabat amino acid numbering scheme.
- libraries and repertoires can be produced that contain polypeptides comprising an antibody variable region having amino acid sequence diversity at particular residues or regions as described herein.
- the libraries and repertoires can be based on a polypeptide that unfolds reversibly (e.g., when heated), such as a polypeptide that unfolds reversibly that is selected or designed as described herein.
- a polypeptide that unfolds reversibly e.g., when heated
- one or more amino acid residues are identified in the amino acid sequence or the polypeptide that unfolds reversibly that prevent irreversible aggregation of the polypeptide upon unfolding.
- folding gatekeepers See
- Folding gatekeepers can be identified, for example, by aligning the amino acid sequences of two or more polypeptides that unfold reversibly and that are based on a common amino acid sequence (polypeptide scaffold amino acid sequence) but contain some degree of sequence diversity. Such an alignment can be used to identify amino acid substitutions in the scaffold sequence that are common to polypeptides that unfold reversibly. A polypeptide based on the scaffold sequence but containing one or more the identified amino acid substitutions can be prepared and assessed for reversible unfolding, to confirm that the identified amino acid residues are folding gatekeepers. Folding gatekeeper residues can also be designed into a desired polypeptide, such as a human VH as described herein.
- 1 Tie library can comprise a collection of nucleic acids that encode polypeptides that unfold reversibly and/or a collection of polypeptides that unfold reversibly in which each polypeptide contains a common folding gatekeeper residue (one or more common folding gatekeeper residues).
- a library comprising a collection of heterogeneous nucleic acids that each encode a human antibody variable domain containing a folding gatekeeper residue can be prepared.
- the members of the library can have sequence diversity in a particular region (e.g., in the CDRs).
- the library comprises a collection of heterogeneous nucleic acids encoding antibody variable domains (e.g., VH (human VH), VL, (human VL)) that contain folding gatekeeper residues .
- the library comprises a collection of heterogeneous nucleic acids encoding V H s (human VHS) that contain folding gatekeeper residues in CDR1, CDR1 and CDR2 or CDR2.
- the nucleic acids in such a library encode V H s that contain diverse CDR3s (e.g., randomized CDR3s).
- the gatekeeper residues can be designed into CDR1 and/or CDR2 using the methods described herein or other suitable methods, and nucleic acids produced that encode VHS that contain those gatekeeper residues.
- VH S that unfold reversibly can be isolated or selected, using the methods described herein or other suitable method, and nucleic acid(s) encoding CDR1 and or CDR2 from a VH that unfolds reversibly (the same or different VHS) can be obtained and ligated to one or more nucleic acids encoding suitable framework regions and CDR3.
- the library of nucleic acids encodes a polypeptide that unfolds reversibly, wherein each member of the library encodes a polypeptide comprising the amino acid sequence of a parental polypeptide in which at least one amino acid residue is replaced with a folding gatekeeper residue and at least one other amino acid residue is replaced, added or deleted.
- the parental polypeptide can be an antibody variable domain that comprises framework regions as described herein.
- the parental polypeptide is a human VH and a folding gatekeeper is introduced into CDR1.
- the parental polypeptide is a human V L and a folding gatekeeper is introduced into the FR2-CDR2 region.
- the library of nucleic acids encodes an antibody variable domain that unfolds reversibly, wherein each member of the library comprising a first nucleic acid encoding CDR1 and optionally CDR2 of the variable domain or an antibody variable domain that unfolds reversibly, where said first nucleic acid is operably linked to one or more other nucleic acids to produce a construct that encodes antibody variable domains in which CDR1 and optionally CDR2 are encoded by said first nucleic acid.
- the members of the library encode an antibody variable domain in which CDR3 is diversified (e.g., at targeted positions) or randomized (e.g,, across the entire CDR3 sequence and/or has CDR3s of varying length).
- a nucleic acid sequence that encodes a desired type of polypeptide e.g., a polymerase, an immunoglobulin variable domain
- a collection of nucleic acids that each contain one or more mutations can be prepared, for example by amplifying the nucleic acid using an error-prone polymerase chain reaction (PCR) system, by chemical mutagenesis (Deng et al. J. Biol. Chem. , 269:9533 (1 94)) or using bacterial mutator strains (Low et al. J. Mol. Biol., 260:359 (1996)).
- PCR polymerase chain reaction
- particular regions of the nucleic acid can be targeted for diversification
- Methods for mutating selected positions are also well known in the art and include, for example, the use of mismatched oligonucleotides or degenerate oligonucleotides, with or without the use of PCR.
- synthetic antibody libraries have been created by targeting mutations to the antigen binding loops. Random or semi-random antibody H3 and L3 regions have been appended to ge ⁇ nline immunoglobulin V gene segments to produce large libraries with unmutated framework regions (Hoogenboom and Winter (1992) supra; Nissim et al. (1994) supra; Griffiths et al. (1994) supra; DeKruif et al. (1 95) supra).
- Such diversification has been extended to include some or all of the other antigen binding loops (Crameri et al (1996) Nature Med., 2:100; Riechmann et al. (1995) Bio/Technology, 13:475; Morphosys, WO 97/08320, supra).
- particular regions of the nucleic acid can be targeted for diversification by, for example, a two-step PCR strategy employing the product of the first PCR as a "mega-primer.” (See, e.g., Landt, 0. etal, Gene 95:125-128 (1990).)
- Targeted diversification can also be accomplished, for example, by SOE PCR. (See, e.g,, Horton, R.M. et al, Gene 77:61-68 (1989).)
- sequence diversity at selected positions can be achieved by altering the coding sequence which specifies the sequence of the polypeptide such that a number of possible amino acids (e.g., all 20 or a subset thereof) can be incorporated at that position.
- the most versatile codon is NNK, which encodes all amino acids as well as the TAG stop codon.
- the NNK codon is preferably used in order to introduce the required diversity.
- Other codons which achieve the same ends are also of use, including the NNN codon, which leads to the production of the additional stop codons TGA and TAA.
- Such a targeted approach can allow the full sequence space in a target area to be explored.
- Preferred libraries of polypeptides that unfold reversibly comprise polypeptides that are members of the immunoglobulin superfamily (e.g., antibodies or portions thereof).
- the libraries can comprise antibody polypeptides that unfold reversibly and have an known main-chain conformation. (See, e.g. , Tomlinson et al, WO 99/20749.)
- Libraries can be prepared in a suitable plasmid or vector.
- vector refers to a discrete element that is used to introduce heterologous DNA into cells for the expression and/or replication thereof.
- Any suitable vector can be used, including plasmids (e.g., bacterial plasmids), viral or bacteriophage vectors, artificial chromosomes and episomal vectors.
- plasmids e.g., bacterial plasmids
- viral or bacteriophage vectors e.g., viral or bacteriophage vectors, artificial chromosomes and episomal vectors.
- Such vectors may be used for simple cloning and mutagenesis, or an expression vector can be used to drive expression of the library.
- Vectors and plasmids usually contain one or more cloning sites (e.g., a polylinker), an origin of replication and at least one selectable marker gene.
- Expression vectors can further contain elements to drive transcription and translation of a polypeptide, such as an enhancer element, promoter, transcription termination signal, signal sequences, and the like. These elements can be arranged in such a way as to be operably linked to a cloned insert encoding a polypeptide, such that the polypeptide is expressed and produced when such an expression vector is maintained under conditions suitable for expression (e.g., in a suitable host cell).
- Cloning and expression vectors generally contain nucleic acid sequences that enable the vector to replicate in one or more selected host cells. Typically in cloning vectors, this sequence is one that enables the vector to replicate independently of the host chromosomal DNA and includes origins of replication or autonomously replicating sequences. Such sequences are well known for a variety of bacteria, yeast and viruses.
- the origin of replication from the plasmid pBR322 is suitable for most Gram-negative bacteria, the 2 micron plasmid origin is suitable for yeast, and various viral origins (e.g. SV40, adenovirus) are useful for cloning vectors in mammalian cells.
- the origin of replication is not needed for mammalian expression vectors unless these are used in mammalian cells able to replicate high levels of DNA, such as COS cells.
- Cloning or expression vectors can contain a selection gene also referred to as selectable marker.
- selectable marker genes encodes a protein necessary for the survival or growth of transformed host cells gown in a selective culture medium. Host cells not transformed with the vector containing the selection gene will therefore not survive in the culture medium.
- Typical selection genes encode proteins that confer resistance to antibiotics and other toxins, e.g. ampicillin, neomycin, methotrexate or tetracycline, complement auxotrophic deficiencies, or supply critical nutrients not available in the growth media.
- Suitable expression vectors can contain a number of components, for example, an origin of replication, a selectable marker gene, one or more expression control elements, such as a transcription control element (e.g., promoter, enhancer, terminator) and/or one or more translation signals, a signal sequence or leader sequence, and the like.
- expression control elements and a signal or leader sequence can be provided by the vector or other source.
- the transcriptional and/or translational control sequences of a cloned nucleic acid encoding an antibody chain can be used to direct expression.
- a promoter can be provided for expression in a desired host cell. Promoters can be constitutive or inducible.
- a promoter can be operably linked to a nucleic acid encoding an antibody, antibody chain or portion thereof, such that it directs transcription of the nucleic acid.
- suitable promoters for procaryotic e.g., the /J-lactamase and lactose promoter systems, alkaline phosphatase, the tryptophan (trp) promoter system, lac, tac, T3, T7 promoters for E. coli
- eucaryotic e.g., simian vims 40 early or late promoter, Rous sarcoma vims long terminal repeat promoter, cytomegalovirus promoter, adenovims late promoter, EG- la promoter
- expression vectors typically comprise a selectable marker for selection of host cells carrying the vector, and, in the case of a replicable expression vector, an origin or replication.
- Genes encoding products which confer antibiotic or drug resistance are common selectable markers and may be used in procaryotic (e.g. , 0-lactamase gene (ampicillin resistance), Tet gene for tetracycline resistance) and eucaryotic cells (e.g., neomycin (G418 or geneticin), gpt (mycophenolic acid), ampicillin, or hygromycin resistance genes).
- Dihydrofolate reductase marker genes permit selection with methotrexate in a variety of hosts.
- auxotrophic markers of the host e.g. , LEU2, URA3, HIS3
- vectors which are capable of integrating into the genome of the host cell such as retroviral vectors, are also contemplated.
- Suitable expression vectors for expression in prokaryotic (e.g., bacterial cells such as E. coli) or mammalian cells include, for example, a pET vector (e.g., pET- 12a, ⁇ ET-36, ⁇ ET-37, pET-39, pET-40, Novagen and others), a phage vector (e.g , pCANTAB 5 E, Pharmacia), ⁇ RIT2T (Protein A fusion vector, Pharmacia), pCDM8, pCDNAl.l/amp, pcDNA3.1, pRc RSV, ⁇ EF-1 (Invitrogen, Carlsbad, CA), pCMV-SCRIPT, pFB, pSG5, pXTl (Stratagene, La Jolla, CA), pCDEF3 (Goldman, L.A., et al, Biotechniques, 21: 1013-1015 (1996)), pSVSPORT (GibcoBRL, Rockville,
- Expression vectors which are suitable for use in various expression hosts, such as prokaryotic cells (E. coli), insect cells (Drosophila Schnieder S2 cells, Sf9) and yeast (P. methanolica, P. pastoris, S. cerevisiae) and mammalian cells (eg, COS cells) are available.
- prokaryotic cells E. coli
- insect cells Drosophila Schnieder S2 cells, Sf9
- yeast P. methanolica, P. pastoris, S. cerevisiae
- mammalian cells eg, COS cells
- Preferred vectors are expression vectors that enable the expression of a nucleotide sequence corresponding to a polypeptide library member.
- selection with generic and/or target ligands can be performed by separate propagation and expression of a single clone expressing the polypeptide library member.
- the preferred selection display system is bacteriophage display.
- phage or phagemid vectors may be used.
- the preferred vectors are phagemid vectors which have an E. coli. origin of replication (for double stranded replication) and also a phage origin of replication (for production of single-stranded DNA).
- the vector can contain a /3-lactamase gene to confer selectivity on the phagemid and a lac promoter upstream of an expression cassette that can contain a suitable leader sequence (e.g. , pelB leader sequence), a multiple cloning site, one or more peptide tags, one or more TAG stop codons and the phage protein pill.
- a suitable leader sequence e.g. , pelB leader sequence
- the vector is able to replicate as a plasmid with no expression, produce large quantities of the polypeptide library member only or product phage, some of which contain at least one copy of the polypeptide-pl ⁇ l fusion on their surface.
- the libraries and repertoires of the invention can contain antibody formats.
- the polypeptide contained within the libraries and repertoires can be whole antibodies or fragments therefore, such as Fab, F(ab ' ) 2 , Fv or scFv fragments, separate VH or VL domains, any of which is either modified or unmodified.
- scFv fragments, as well as other antibody polypeptides can be readily produced using any suitable method.
- a number of suitable antibody engineering methods are well known in the art.
- a scFv can be formed by linking nucleic acids encoding two variable domains with an suitable oligonucleotide that encodes an appropriately linker peptide, such as (Gly-Gly-Gly-Gly-Ser) 3 or other suitable linker peptide(s).
- the linker bridges the C-terminal end of the first V region and N- terminal end of the second V region.
- linker bridges the C-terminal end of the first V region and N- terminal end of the second V region.
- Similar techniques for the construction of other antibody formats such as Fv, Fab and F(ab ' ) 2 fragments.
- VH and VL polypeptides can combined with constant region segments, which may be isolated from rearranged genes, germline C genes or synthesized from antibody sequence data.
- a library or repertoire according to the invention can be a V H or VL library or repertoire.
- the libraries and repertoires comprise immunoglobulin variable domains that unfold reversibly (e.g., VH, V L ).
- the variable domains can be based on a ge ⁇ nline sequence (e.g., DP47dummy (SEQ ID NO:3, DPK9 dummy (SEQ ID NO:6)) and if desired can have one or more diversified regions, such as the complementarity determining regions.
- One or more of the framework regions (FR) of the variable domains can comprise (a) the amino acid sequence of a human framework region, (b) at least 8 contiguous amino acids of the amino acid sequence of a human framework region, or (c) an amino acid sequence encoded by a human germline antibody gene segment, wherein said framework regions are as defined by Kabat.
- the amino acid sequence of one or more of the framework regions is the same as the amino acid sequence of a corresponding framework region encoded by a human germline antibody gene segment, or the amino acid sequences of one or more of said framework regions collectively comprise up to 5 amino acid differences relative to the amino acid sequence of said corresponding framework region encoded by a human germline antibody gene segment.
- the arnino acid sequences of FRl, FR2, FR3 and FR4 are the same as the amino acid sequences of corresponding framework regions encoded by a human ge ⁇ nline antibody gene segment, or the amino acid sequences of FRl, FR2, FR3 and FR4 collectively contain up to 10 amino acid differences relative to the amino acid sequences of corresponding framework regions encoded by said human ge ⁇ nline antibody gene segments.
- the amino acid sequence of said FRl, FR2 and FR3 are the same as the amino acid sequences of corresponding framework regions encoded by said human germline antibody gene segment.
- the polypeptides comprising a variable domain that unfolds reversibly preferably comprise a target ligand binding site and/or a generic ligand binding site.
- the generic ligand binding site is a binding site for a superantigen, such as protein A, protein L or protein G.
- variable domains can be based on any desired variable domain, for example a human VH (e.g., V H la, V H lb, V H 2, V H 3, VH 4, V H 5, V H 6), a human V ⁇ (e.g., V ⁇ l, V ⁇ ll, V ⁇ HI, V ⁇ lV, V ⁇ V or V ⁇ VI) or a human VK (e.g., V ⁇ l, V ⁇ 2, V ⁇ 3, V ⁇ 4, V ⁇ 5, V ⁇ 6, V ⁇ 7, V ⁇ 8, V ⁇ 9 or V ⁇ l0).
- a human VH e.g., V H la, V H lb, V H 2, V H 3, VH 4, V H 5, V H 6
- a human V ⁇ e.g., V ⁇ l, V ⁇ ll, V ⁇ HI, V ⁇ lV, V ⁇ V or V ⁇ VI
- a human VK e.g., V ⁇ l, V ⁇ 2, V ⁇ 3, V ⁇ 4, V ⁇ 5, V ⁇ 6, V ⁇ 7, V ⁇ 8, V ⁇ 9 or V
- variable domains are not a Camelid immunoglobulin domain, such as a VH H, or contain one or more amino acids (e.g., framework amino acids) that are unique to Camelid immunoglobulin variable domains encoded by germline sequences but not, for example, to human immunoglobulin variable domains.
- amino acids e.g., framework amino acids
- the VH that unfolds reversibly does not comprise one or more amino acids that are unique to murine (e.g., mouse) germline framework regions.
- the variable domain is unfold reversibly when heated and cooled.
- the isolated polypeptide comprising a variable domain can be an antibody format.
- the isolated polypeptide comprising a variable domain that unfolds reversibly can be a homodimer of variable domain, a heterodimer comprising a variable domain, an Fv, a scFv, a disulfide bonded Fv, a Fab, a single variable domain or a variable domain fused to an immunoglobulin Fc portion.
- the invention is an isolated polypeptide that unfolds reversibly.
- such polypeptides can be expressed in E. coli and recovered in high yield.
- the polypeptides that unfold reversibly e.g. , when heated
- Such polypeptides are also referred to as secretable when expressed in E. coli.
- the polypeptide that unfolds reversibly is secreted in a quantity of at least about 0.5 mg/L when expressed in E. coli.
- the polypeptide that unfolds reversibly is secreted in a quantity of at least about 0.75 mg/L, at least about 1 mg L, at least about 4 mg L, at least about 5 mg/L, at least about 10 mg L, at least about 15 mg L, at least about 20 mg/L, at least about 25 mg L, at least about 30 mg/L, at least about 35 mg L, at least about 40 mg/L, at least about 45 mg/L, or at least about 50 mg/L, or at least about 100 mg/L, or at least about 200 mg/L, or at least about 300 mg/L, or at least about 400 mg/L, or at least about 500 mg/L, or at least about 600 mg/L, or at least about 700 mg/L, or at least about 800 mg/L, at least about 900 mg/L, at least about lg/L when expressed in E.
- the polypeptide that unfolds reversibly is secreted in a quantity of at least about 1 mg/L to at least about lg/L, at least about 1 mg L to at least about 750 mg/L, at least about 100 mg/L to at least about 1 g/L, at least about 200 mg/L to at least about 1 g/L, at least about 300 mg/L to at least about 1 g/L, at least about 400 mg/L to at least about 1 g/L, at least about 500 mg/L to at least about lg/L, at least about 600 mg/L to at least about 1 g/L, at least about 700 mg/L to at least about 1 g/L, at least about 800 mg/L to at least about lg L, or at least about 900 mg L to at least about lg/L when expressed in E.
- the polypeptides described herein can be secretable when expressed in E. coli, they can be produced using any suitable method, such as synthetic chemical methods or biological production methods that do not employ E. coli.
- the polypeptide that unfolds reversibly is a human antibody variable domain (VH, V ) or comprises a human antibody variable domain that unfolds reversibly.
- the polypeptide that unfolds reversibly is a variant of a parental polypeptide that differs from the parental polypeptide in amino acid sequence (e.g., by one or more amino acid replacements, additions and/or deletions), but qualitatively retains function of the parental polypeptide.
- the activity of the variant polypeptide that unfolds reversibly is at least about 25%, at least about 30%, at least about 40%, at least about 50%, at least about 60%, at least about 70%, at least about 80%, at least about 90%, at least about 95%, or essentially the same as of the activity of the parental polypeptide.
- the variant polypeptide can contain an amino acid sequence that differs from the parental enzyme (e.g., by one to about ten amino acid substitutions, deletions and/or insertions) but will retain the catalytic activity of parental enzyme.
- the variant enzyme that unfolds reversibly is characterized by a catalytic rate constant that is at least about 25% of the catalytic rate constant of the parental enzyme.
- a variant polypeptide that unfolds reversibly can be prepared using any desired parental polypeptide.
- the polypeptide that unfolds reversibly can be prepared using a parental polypeptide that is based on a scaffold selected from an enzyme, protein A, protein L, a fibronectm, an anticalin, a domain of CTLA4, or a polypeptide of tlie immunoglobulin superfamily, such as an antibody or an antibody fragment.
- a parental polypeptide that is based on a scaffold selected from an enzyme, protein A, protein L, a fibronectm, an anticalin, a domain of CTLA4, or a polypeptide of tlie immunoglobulin superfamily, such as an antibody or an antibody fragment.
- the parental polypeptide can comprise V and/or D (where the polypeptide comprises a VH domain) and/or J segment sequences encoded by a germline V, D or J segment respectively, or a sequence that results from naturally occurring or artificial mutations (e.g. , somatic mutation or other processes) .
- the parental polypeptide can be a parental immunoglobulin variable domain having an amino acid sequence that is encoded by germline gene segments, or that results from insertion and/or deletion of nucleotides during V(D)J recombination (e.g, N nucleotides, P nucleotides) and/or mutations that arise during affinity maturation.
- the parental polypeptide can be a parental enzyme that has an amino acid sequence encoded by the germline or that results from naturally occurring or artificial mutation (e.g, somatic mutation).
- a variant polypeptide that unfolds reversibly can be prepared using any suitable method.
- the amino acid sequence of a parental polypeptide can be altered at particular positions (as described herein with respect to antibody variable domain polypeptides) to produce a variant polypeptide that unfolds reversibly.
- a variant polypeptide that unfolds reversibly can also be produced, for example, by providing a nucleic acid that encodes a parental polypeptide, preparing a library of nucleic acids that encode variants of the parental polypeptide (e.g., by error prone PCR or other suitable method) and expressing the library in a suitable polypeptide display system.
- a variant polypeptide that unfolds reversibly and retains a desired function of the parental polypeptide can be selected and isolated from such a polypeptide display system using the methods described herein or other suitable methods.
- the isolated polypeptide comprises an immunoglobulin variable domain that unfolds reversibly (e.g., VH, a variant VH, VL and/or variant VL).
- the isolated polypeptide comprises a variant VH and/or a variant V that unfolds reversibly.
- the variable domain that unfolds reversibly e.g. , VH, variant VH, VL, variant VL
- Kj dissociation constant
- K off K off rate
- a suitable K can be about 50 nM to about 20 pM or less, for example about 50 nM, about 1 nM, about 900 pM, about 800 pM, about 700 pM, about 600 pM, about 500 pM, about 400 pM, about 300 pM, about 200 pM, about 100 pM or about 20 pM or less.
- a suitable Koff can be about 1 x 10 "1 s “1 to about 1 x 10 '7 s '1 or less, for example about 1 x 10 '1 s “1 , about 1 x 10 "2 s “1 , about 1 x 10 -3 s “1 , about 1 x 10 "4 about 1 x 10 "5 s "1 , about 1 x 10 "6 s ⁇ or about 1 x 10 "1 s "1 or less.
- d and K off are determined using surface plasmon resonance.
- the isolated polypeptide comprises a VH (e.g. , variant VH) that unfolds reversibly.
- the amino acid sequence of the variant V H that unfolds reversibly differs from the amino acid sequence of the parental VH by at least one amino acid from position 22 to position 36, or differs from the amino acid sequence of the parental VH by at least one amino acid in the HI loop.
- the amino acid positions and CDR (HI , H2 and H3) and framework regions (FRl, FR2, FR3 and FR4) of the VH can be defined using any suitable system, such as the systems of Kabat, Chofhia or AbM.
- positions 22 through 3 are defined according to the amino acid numbering system of Kabat
- the HI loop is defined according to the amino acid numbering system used in the AbM software package (antibody analysis and stractural modeling software; Oxford Molecular).
- the amino acid sequence of the variant VH can contain at least one amino acid replacement from position 22 to position 36 relative to the parental VH.
- the amino acid sequence of the VH that unfolds reversibly contains at least one Pro or Gly residue from position 22 to position 36. In other embodiments, the amino acid sequence of the VH that unfolds reversibly contains at least one Pro or Gly residue in the HI loop. In certain embodiments, the amino acid sequence of the variant VH that unfolds reversibly contains at least one Pro or Gly replacement from position 22 to position 36 relative to the amino acid sequence of the parental VH. In other embodiments, the amino acid sequence of the variant VH that unfolds reversibly contains at least one Pro or Gly replacement in the HI loop relative to the amino acid sequence of the parental VH. In a particular embodiment, the variant VH that unfolds reversibly comprises an amino acid sequence wherein the parental amino acid residue at position 29 is replaced with Pro or Gly.
- the variant V H that unfolds reversibly contains at least one amino acid replacement from position 22 to position 36 relative to the parental VH amino acid sequence, such that the hydrophobicity of the amino acid sequence from position 22 to position 36 of the variant VH is reduced relative to the parental VH.
- Hydrophobicity can be determined using any suitable scale or algorithm.
- hydrophobicity is determined using the method of Sweet and Eisenberg, and the Sweet and Eisenberg hydrophobicity score (S/E score) of the amino acid sequence from position 22 to position 36 of the variant V H is reduced relative to the parental V M .
- the S/E method can use a 9 to 18 amino acid window (e.g., a window of 9, 10, 11, 12, 13, 14, 15, 16, 17 or 18 amino acids).
- a 15 amino acid window is used for V H .
- the S/E score of the amino acid sequence from position 22 to position 36 of the variant VH is 0.15 or less, or 0.1 or less for an S/E score rounded to one decimal place, or 0.09 or less, or 0.08 or less, or 0,07 or less, or 0.06 or less, or 0.05 or less, or 0.04 or less.
- the S/E score or the amino acid sequence from position 22 to position 36 of the variant VH is 0.03 or less, 0.02 or less or 0.01 or less. In more preferred embodiments, the S/E score or the amino acid sequence from position 22 to position 36 of the variant VH is 0 or less,
- Producing VH variants that have a reduced S/E score from position 22 to position 36 relative to a parental VH can yield polypeptides that have several advantages. For example, it has been determined that a lower S E score from position 22 to position 36 correlates with resistance to aggregation and enhanced yields from expression systems (e.g., bacterial expression systems). The capacity of VH domains to resist aggregation at high protein concentrations is associated with a low S/E score.
- a variant V H domain with an S/E score from position 22 to position 36 that is lower than the S/E score of the parental V H domain can display enhanced resistance to aggregation, while lowering the S/E score of this region to 0 or less can produce variant VH domains with superior aggregation resistance at high protein concentrations (e.g., about 200 ⁇ M).
- the VH that unfolds reversibly contains an amino acid sequence from position 22 to position 36 that has an S/E score of 0.15 or less, or 0.1 or less, or 0.09 or less, or 0.08 or less, or 0.07 or less, or 0.06 or less, or 0.05 or less, or 0.04 or less.
- the VH that unfolds reversibly contains an amino acid sequence from position 22 to position 36 that has an S E score of 0.03 or less, 0.02 or less or 0.01 or less.
- the VH that unfolds reversibly contains an amino acid sequence from position 22 to position 36 that has an S/E score of 0 or less.
- the amino acid sequence of the variant VH differs from the amino acid sequence of the parental VH by at least one amino acid in the HI loop.
- the variant VH contains at least one amino acid replacement in the HI loop, such that the hydrophobicity of HI loop is reduced relative to the HI loop of the parental VH.
- the S/E score of the HI loop of the variant VH is 0 or less.
- the VH that unfolds reversibly contains an HI loop that has an S/E score of 0 or less.
- the VH that unfolds reversibly comprises an amino acid sequence in which one or more of the parental amino acid residues at position 27, 29, 30, 31, 32, 33 and 35 (Kabat numbering) is replaced with another amino acid residue as set forth in Table 1.
- the amino acid sequence of the VH that unfolds reversibly can comprise any one or any combination of amino acid replacements set forth in Table 1 , and if desired, a variant VH that unfolds reversibly can further differ from the parental VH by replacement of one or more of the amino acid residues at parental position 22 through parental position 26, parental position 28 and parental position 36 (Kabat numbering).
- the variant VH that unfolds reversibly comprises an amino acid sequence in which the parental amino acid residue at position 27 is replaced with Asp or Glu; the parental amino acid residue at position 29 is replaced with Asp, Glu, Pro or Gly; and/or the parental amino acid residue at position 32 is replaced with Asp or Glu (Kabat numbering).
- the variant VH that unfolds reversibly comprises an amino acid sequence in which the parental amino acid residue at position 27 is replaced with Asp; the parental amino acid residue at position 29 is replaced with Asp, Pro or Gly; and/or the parental amino acid residue at position 32 is replaced with Glu (Kabat numbering).
- the variant VH that unfolds reversibly comprises an amino acid sequence in which the parental amino acid residue at position 27 is replaced with Asp; the parental amino acid residue at position 29 is replaced with Val; and/or the parental amino acid residue at position 32 is replaced with Asp or Glu; and the parental residue at position 35 is replaced with Gly (Kabat numbering).
- the variant VH that unfolds reversibly comprises an amino acid sequence in which the parental amino acid residue at position 27 is replaced with Asp and/or the parental amino acid residue at position 32 is replaced with Asp (Kabat numbering).
- the variant VH that unfolds reversibly can further comprise amino acid replacements relative to the parental sequence at one or more of positions 22-26, 28-31 and 33-36 as described herein (Kabat numbering),e.g. , to bring the S/E score of one or more of these regions to zero or less.
- One or more of the framework regions (FR) of the VH or variant VH that unfold reversibly can comprise (a) the amino acid sequence of a human framework region, (b) at least 8 contiguous amino acids of the amino acid sequence of a human framework region, or (c) an amino acid sequence encoded by a human ge ⁇ nline antibody gene segment, wherein said framework regions are as defined by Kabat.
- the amino acid sequence of one or more of the framework regions is the same as the amino acid sequence of a corresponding framework region encoded by a human germline antibody gene segment, or the amino acid sequences of one or more of said framework regions collectively comprise up to 5 amino acid differences relative to the amino acid sequence of said corresponding framework region encoded by a human germline antibody gene segment.
- the amino acid sequences of FRl , FR2, FR3 and FR4 are the same as the amino acid sequences of co ⁇ esponding framework regions encoded by a human germline antibody gene segment, or the amino acid sequences of FRl, FR2, FR3 and FR4 collectively contain up to 10 amino acid differences relative to the amino acid sequences of co ⁇ esponding framework regions encoded by said human ge ⁇ nline antibody gene segments.
- the amino acid sequence of said FRl, FR2 and FR3 are the same as the amino acid sequences of corresponding framework regions encoded by said human germline antibody gene segment.
- the variant V can be a variant of human DP47 dummy (SEQ ID NO:3).
- the isolated polypeptide comprising a VH that unfolds reversibly comprises a target ligand binding site and/or a generic ligand binding site.
- the generic ligand binding site is a binding site for a superantigen, such as protein A, protein L or protein G.
- the VH that unfolds reversibly can be based on any desired parental VH, for example a human VH (e.g., VH la, VH lb, VH 2, H 3, Vn 4, VH 5, VH 6).
- the VH that unfolds reversibly is not a Camelid immunoglobulin domain, such as a VnH, or does not contain one or more amino acids (e.g., framework amino acids) that are unique to Camelid immunoglobulin variable domains encoded by germline sequences but not, for example, to human immunoglobulin variable domains.
- a human VH e.g., VH la, VH lb, VH 2, H 3, Vn 4, VH 5, VH 6
- the VH that unfolds reversibly is not a Camelid immunoglobulin domain, such as a VnH, or does not contain one or more amino acids (e.g., framework amino acids) that are unique to Camelid immunoglobulin variable domains encoded by
- the VH that unfolds reversibly does not comprise one or more amino acids that are unique to murine (e.g., mouse) germline framework regions.
- the VH unfolds reversibly when heated and cooled.
- the isolated polypeptide comprising a VH that unfolds reversibly can be an antibody format.
- the isolated polypeptide comprising a V H that unfolds reversibly can be a ho odimer of VH, a heterodimer comprising a VH, an Fv, a scFv, a disulfide bonded Fv, a Fab, a VH or a VH fused to an immunoglobulin Fc portion.
- the invention also relates to an isolated polypeptide comprising an immunoglobulin light chain variable domain (VL) (e.g., a variant VL) that unfolds reversibly.
- VL immunoglobulin light chain variable domain
- the isolated polypeptide comprises a VL that unfolds reversibly and comprises an amino acid sequence from position 44 to position 53 that has a Sweet Eisenberg hydrophobicity score (S/E score) of less than 0.23.
- the S/E score for position 44 to position 53 can be less than 0.2, less than 0.17, less than 0.15, less than 0.13, less than 0.10, or less than -0.1 or less.
- the S/E method can use a 9 to 18 amino acid window (e.g.
- V L a window of 9, 10, 11, 12, 13, 14, 15, 16, 17 or 18 amino acids.
- an 11 amino acid window is used for VL-
- the amino acid positions and CDR (LI, L2 and L3) and framework regions (FRl, FR2, FR3 and FR4) of the V L can be defined using any suitable system, such as the systems of Kabat, Chothia or AbM.
- the amino acid positions and CDR and framework regions are according to the amino acid numbering system of Kabat.
- V L that unfolds reversibly further comprises a FR3 that has a Sweet/Eisenberg hydrophobicity score (S/E score) of less than 0,35, and FR3 is as defined by the Kabat amino acid numbering system.
- the S/E score for FR3 can be less than 0.3, less than 0.25, less than 0.2, less than 0.17, less than 0.15, less than 0.13, less than 0.10, or less than -0.1 or less.
- the isolated polypeptide comprises a variant VL that unfolds reversibly.
- the amino acid sequence of the variant VL that unfolds reversibly differs from the amino acid sequence of the parental VL by at least one amino acid from position 44 to position 53 of said variant VL, such that the variant V comprises the amino acid sequence of a parental V wherein at least one amino acid residue from position 44 to position 53 is replaced such that the Sweet Eisenberg hydrophobicity score (S/E score) of the amino acid sequence from position 44 to position 53 of said variant VL is less than 0,23.
- the amino acid sequence of the variant VL that unfolds reversibly can contain at least one amino acid replacement from position 44 to position 53 relative to the parental V 1( .
- the amino acid sequence of the variant VL that unfolds reversibly further differs from the amino acid sequence of the parental V by at least one amino acid in FR3 of said variant VL-
- the variant VL that unfolds reversibly comprises FR3 having the amino acid sequence of a parental VL FR3 wherein at least one amino acid residue is replaced such that the Sweet/Eisenberg hydrophobicity score (S/E score) of FR3 said variant V is less than 0.35.
- the amino acid sequence of the variant V L that unfolds reversibly differs from the amino acid sequence of the parental VL by at least one amino acid in FR3 of said variant VL, such that the variant V comprises the amino acid sequence of a parental V whetein at least one amino acid residue in FR3 is replaced such that the SweetEisenberg hydrophobicity score (S/E score) of the amino acid sequence of FRe of said variant VL is less than 0.35.
- the amino acid sequence of the variant V that unfolds reversibly can differ from the amino acid sequence of the parental VL by at least one amino acid from position 73 to position 76 of said variant VL, such that the variant VL comprises the amino acid sequence of a parental Vi. wherein at least one amino acid residue from position 73 to position 76 is replaced such that the Sweet/Eisenberg hydrophobicity score (S E score) of the amino acid sequence of FR3 of said variant V is less than 0.35.
- the V L that unfolds reversibly comprises an amino acid sequence in which one or more of the amino acid residues at position 10, 13, 20, 23, 26, 27, 29, 31, 32, 35, 36, 39, 40, 42, 45, 46, 47, 8, 49, 50, 57, 59, 60, 68, 75, 79, 80, 83, 89, 90 and 92 (Kabat numbering) is replaced with another amino acid residue as set forth in Table 2.
- the isolated V that unfolds reversibly is a variant V
- it can comprise an amino acid sequence in which one or more of the parental amino acid residues at position 10, 13, 20, 23, 26, 27, 29, 31, 32, 35, 36, 39, 40, 42, 45, 46, 47, 48, 49, 50, 57, 59, 60, 68, 75, 79, 80, 83, 89, 90 and 92 (Kabat numbering) is replaced with another amino acid residue as set forth in Table 2.
- the amino acid sequence of the V or variant V that unfolds reversibly can comprise any one or any combination of amino acid replacements set forth in Table 2, and if desired can further differ from the parental VL by replacement of the amino acid residue at parental position 26 with Asn and or replacement of the amino acid residue at parental position 89 with Arg (Kabat numbering).
- the variant VL that unfolds reversibly comprises an amino acid sequence in which the parental amino acid residue at position 45 is replaced with Glu, Asp, Gta, Pro, Asn, His or Thr; the parental amino acid residue at position 48 is replaced with Asn, Pro, Asp, Thr, Gly or Val; the parental amino acid residue at position 49 is replaced with Asn, Asp, Ser, Cys, Glu, Gly, Lys or Arg; the parental amino acid residue at position 50 is replaced with Pro, Asp, Asn, Glu or Arg; and or the parental amino acid residue at position 75 is replaced with Asn or Met. (Kabat numbering).
- the VL or variant VL that unfolds reversibly comprises an amino acid sequence which contain one of the amino acid replacements set forth in Table 2 and has an amino acid sequences that comprises:
- the amino acid sequence of the variant V that unfolds reversibly contains at least one Pro or Gly replacement from position 44 to position 53 and/or in FR3 (e.g. , from position 73 to position 76) relative to the amino acid sequence of the parental V .
- the amino acid sequence of the variant Vi comprises Pro at position 45, position 48 and/or position 50.
- One or more of the framework regions (FR) of the V or variant VL that unfolds reversibly can comprise (a) the amino acid sequence of a human framework region, (b) at least 8 contiguous amino acids of the amino acid sequence of a human framework region, or (c) an amino acid sequence encoded by a human germline antibody gene segment, wherein said framework regions are as defined by Kabat.
- the amino acid sequence of one or more of the framework regions is the same as the amino acid sequence of a corresponding framework region encoded by a human germline antibody gene segment, or the amino acid sequences of one or more of said framework regions collectively comprise up to 5 amino acid differences relative to the amino acid sequence of said corresponding framework region encoded by a human germline antibody gene segment.
- the amino acid sequences of FRl, FR2, FR3 and FR4 are the same as the amino acid sequences of co ⁇ esponding framework regions encoded by a human germline antibody gene segment, or the amino acid sequences of FRl, FR2, FR3 andFR4 collectively contain up to 10 amino acid differences relative to the amino acid sequences of conesponding framework regions encoded by said human germline antibody gene segments.
- the amino acid sequence of said FRl, FR2 and FR3 are the same as the amino acid sequences of conesponding framework regions encoded by said human germline antibody gene segment.
- the variant V can a variant of human DPK9 dummy (SEQ ID NO:6).
- the isolated polypeptide comprising a V that unfolds reversibly comprises a target ligand binding site and/or a generic ligand binding site.
- the generic ligand binding site is a binding site for a superantigen, such as protein A, protein L or protein G.
- the VL that unfolds reversibly can be based on any desired parental VL, for example a human V ⁇ (e.g., V ⁇ l, V ⁇ ll, V ⁇ ffl, V ⁇ lV, V ⁇ V or V ⁇ VI) or a human VK (e.g, V ⁇ l , V ⁇ 2, V ⁇ 3, V ⁇ 4, V ⁇ 5, V ⁇ 6, V ⁇ 7, V ⁇ 8, V ⁇ 9 or V ⁇ l0).
- the VL that unfolds reversibly is not a Camelid immunoglobulin domain, or contain one or more amino acids (e.g., framework amino acids) that are unique to Camelid immunoglobulin variable domains encoded by germline sequences but not, for example, to human immunoglobulin variable domains.
- the VH that unfolds reversibly does not comprise one or more amino acids that are unique to murine (e.g., mouse) ge ⁇ nline framework regions.
- the VL unfolds reversibly when heated.
- the isolated polypeptide comprising a VL that unfolds reversibly can be an antibody format.
- the isolated polypeptide comprising a V that unfolds reversibly can be a homodimer of V , a heterodimer comprising a VL, an Fv, a scFv, a disulfide bonded Fv, a Fab, a VL or a VL fused to an immunoglobulin Fc portion.
- the invention also relates to an isolated polypeptide comprising an immunoglobulin variable domain (e.g, V H , VL), wherein said variable domain comprises a disulfide bond between a cysteine in CDR2 and a cysteine in CDR3, wherein CDR2 and CDR3 are defined using the Kabat amino acid numbering system.
- the disulfide bond is between cysteine residues at position 52a and position 98; position 51 and position 98; or position 51 and position 100b, wherein said positions are defined using the Kabat amino acid numbering system.
- the disulfide bond is between a cysteine at position 51 and a cysteine in CDR3; or a cysteine in CDR2 and a cysteine at position 98, wherein CDR2, CDR3 and said positions are defined using the Kabat amino acid numbering system.
- the isolated polypeptide comprising a disulfide bonded variable domain can be an antibody format.
- the isolated polypeptide comprising a disulfide bonded variable domain can be a homodimer of variable domains, a heterodimer of variable domains, an Fv, a scFv, a Fab, a single variable domain or a single variable domain fused to an immunoglobulin Fc portion.
- the invention also relates to isolated and/or recombinant nucleic acids encoding polypeptides that unfold reversibly as described herein.
- Nucleic acids refe ⁇ ed to herein as "isolated” are nucleic acids which have been separated away from other material (e.g., other nucleic acids such as genomic DNA, cDNA and/or RNA) in its original environment (e.g., in cells or in a mixture of nucleic acids such as a library).
- An isolated nucleic acid can be isolated as part of a vector (e.g., a plasmid).
- Nucleic acids can be naturally occurring, produced by chemical synthesis, by combinations of biological and chemical methods (e.g., semisynthetic), and be isolated using any suitable methods.
- Nucleic acids refened to herein as "recombinant” are nucleic acids " which have been produced by recombinant DNA methodology, including methods which rely upon artificial recombination, such as cloning into a vector or chromosome using, for example, restriction enzymes, homologous recombination, viruses and the like, and nucleic acids prepared using the polymerase chain reaction (PCR).
- PCR polymerase chain reaction
- Recombinant nucleic acids are also those that result from recombination of endogenous or exogenous nucleic acids through the natural mechanisms of cells or cells modified to allow recombination (e.g., cells modified to express Cre or other suitable recombinase), but are selected for after the introduction to the cells of nucleic acids designed to allow and make recombination probable.
- a functionally rea ⁇ anged human-antibody transgene is a recombinant nucleic acid.
- Nucleic acid molecules of the present invention can be used in the production of antibodies (e.g, human antibodies, humanized antibodies, chimeric antibodies and antigen-binding fragments of the foregoing), e.g., antibodies that bind an ⁇ E integrin or integrin ⁇ E chain (CD103).
- a nucleic acid e.g., DNA
- a suitable construct e.g. , an expression vector
- Expression constructs or expression vectors suitable for the expression of a antibody or antigen-binding fragment are also provided.
- a nucleic acid encoding all or part of a desired antibody can be inserted into a nucleic acid vector, such as a plasmid or virus, for expression.
- the vector can be capable of replication in a suitable biological system (e.g., a replicon).
- suitable vectors are known in the art, including vectors which are maintained in single copy or multiple copy, or which become integrated into the host cell chromosome.
- Suitable expression vectors can contain a number of components, for example, an origin of replication, a selectable marker gene, one or more expression control elements, such as a transcription control element (e.g., promoter, enhancer, terminator) and/or one or more translation signals, a signal sequence or leader sequence, and the like.
- expression control elements and a signal or leader sequence can be provided by the vector or other source.
- the transcriptional and/or translational control sequences of a cloned nucleic acid encoding an antibody chain can be used to direct expression.
- the invention relates to recombinant host cells and a method of preparing an polypeptide of the invention that unfolds reversibly.
- the polypeptide that unfolds reversibly can be obtained, for example, by the expression of one or more recombinant nucleic acids encoding a polypeptide that unfolds reversibly or using other suitable methods.
- the expression constructs described herein can be introduced into a suitable host cell, and the resulting cell can be maintained (e.g., in culture, in an animal, in a plant) under conditions suitable for expression of the constructs.
- Suitable host cells can be prokaryotic, including bacterial cells such as E. coli, B. subtilis and/or other suitable bacteria; eucaryotic cells, such as fungal or yeast cells (e.g. , Pichia pastoris, Aspergillus sp. ,
- the invention also relates to a recombinant host cell which comprises a (one or more) recombinant nucleic acid or expression construct comprising a nucleic acid encoding a polypeptide that unfolds reversibly.
- the invention also includes a method of preparing an polypeptide that unfolds reversibly, comprising maintaining a recombinant host cell of the invention under conditions appropriate for expression of a polypeptide that unfolds reversibly.
- the method can further comprise the step of isolating or recovering the polypeptide that unfolds reversibly, if desired.
- a nucleic acid molecule i.
- nucleic acid molecules encoding a polypeptide that unfolds reversibly, or an expression construct (i.e., one or more constracts) comprising such nucleic acid molecule(s), can be introduced into a suitable host cell to create a recombinant host cell using any method appropriate to the host cell selected (e.g., ttansforrnation, transfection, electroporation, infection), such that the nucleic acid molecule(s) are operably linked to one or more expression control elements (e.g., in a vector, in a construct created by processes in the cell, integrated into the host cell genome).
- expression control elements e.g., in a vector, in a construct created by processes in the cell, integrated into the host cell genome.
- the resulting recombinant host cell can be maintained under conditions suitable for expression (e.g., in the presence of an inducer, in a suitable animal, in suitable culture media supplemented with appropriate salts, growth factors, antibiotics, nutritional supplements, etc.), whereby the encoded polypeptide(s) are produced.
- the encoded protein can be isolated or recovered (e.g., from the animal, the host cell, medium, milk). This process encompasses expression in a host cell of a transgenic animal (see, e.g., WO 92/03918, GenPharm lhtemational).
- polypeptides that unfolds reversibly described herein can also be produced in a suitable in vitro expression system, by chemical synthesis or by any other suitable method.
- the invention does not include a polypeptide having a variable domain comprising a sequence encoded by a ge ⁇ nline VH or germline V gene segment, or consisting of or comprising SEQ LD NOS:7-60.
- polypeptides that unfold reversibly described herein can have binding specificity for a target ligand.
- a polypeptide that comprises an antibody variable region that unfolds reversibly and has binding specificity for a particular target ligand can be selected, isolated and/or recovered using any suitable method, such as the binding methods described herein.
- Exemplary target ligands that polypeptides that unfold reversibly e.g,, polypeptides comprising a reversibly unfoldable VH or VK
- can have binding specificity for include, human or animal proteins, cytokines, cytokine receptors, enzymes, co-factors for enzymes and DNA binding proteins.
- Suitable cytokines and growth factors, cytokine and growth factor receptor and other target ligands include but are not limited to: ApoE, Apo-SAA, BDNF, Cardiotrophin-1, CEA, CD40, CD40 Ligand, CD56, CD38, CD138, EGF, EGF receptor, ENA-78, Eotaxin, Eotaxin-2, Exodus-2, FAP ⁇ , FGF-acidic, FGF- basic, fibroblast growth factor-10, FLT3 ligand, Fractalkine (CX3C), GDNF, G- CSF, GM-CSF, GF- ⁇ l, human serum albumin, insulin, IFN- ⁇ , IGF-I, IGF-II, IL-l ⁇ , IL-1 ⁇ , IL-1 receptor, IL-2, IL-3, IL-4, LL-5, IL-6, IL-7, IL-8 (72 a.a.), LL-8 (77 a.a.), IL-9, IL-10,
- VEGF A VEGF B, VEGF C, VEGF D, VEGF receptor 1, VEGF receptor 2, VEGF receptor 3, GCP-2, GRO/MGSA, GRO- ⁇ , GRO- ⁇ , HCC1, 1-309, HER 1, HER 2, HER 3 and HER 4. It will be appreciated that this list is by no means exhaustive.
- the invention is an isolated polypeptide that comprises an antibody variable domain that unfolds reversibly and binds a target ligand.
- the antibody variable domain that unfolds reversibly binds a target ligand that is a cytokine, growth factor, cytokine receptor or growth factor receptor (e.g., a human cytokine, human growth factor, human cytokine receptor or human growth factor receptor).
- the antibody variable domain that unfolds reversibly neutralizes the activity of the a cytokine, growth factor, cytokine receptor or growth factor receptor with a neutralized dose 50 (ND50) of about 1 ⁇ M or less, or 500 nM or less, in a standard cellular assay, such as the assay that measures TNF-induced IL-8 secretion by HeLa cells described herein.
- ND50 neutralized dose 50
- the antibody variable domain that unfolds reversibly binds a cytokine or growth factor, and inhibits the interaction of the cytokine or growth factor with a cognate cytokine receptor or growth factor receptor with an inhibitory concentration 50 (LC50) of about 1 ⁇ M or less, or about 500 nM or less, in a standard receptor binding assay, such as the assay TNF Receptor 1 (p55) assay described herein.
- LC50 inhibitory concentration 50
- the antibody variable domain that unfolds reversibly binds a cytokine receptor or growth factor receptor, and inhibits the interaction of the cytokine receptor or growth factor receptor with a cognate cytokine or growth factor with an inhibitory concentration 50 (IC50) of about 1 ⁇ M or less, or about 500 nM or less, in a standard receptor binding assay, such as the assay TNF Receptor 1 ( ⁇ 55) assay described herein.
- IC50 inhibitory concentration 50
- compositions comprising a polypeptide that unfolds reversibly, including pharmaceutical or physiological compositions are provided.
- Pharmaceutical or physiological compositions comprise one or more polypeptide that unfolds reversibly and a pharmaceutically or physiologically acceptable carrier.
- these carriers include aqueous or alcoholic/aqueous solutions, emulsions or suspensions, including saline and/or buffered media.
- Parenteral vehicles include sodium chloride solution, Ringer's dextrose, dextrose and sodium chloride and lactated Ringer's.
- Suitable physiologically-acceptable adjuvants if necessary to keep a polypeptide complex in suspension, may be chosen from thickeners such as carboxymethylcellulose, polyvinylpynolidone, gelatin and alginates.
- Intravenous vehicles include fluid and nutrient replenishers and electrolyte replenishers, such as those based on Ringer's dextrose. Preservatives and other additives, such as antimicrobials, antioxidants, chelating agents and inert gases, may also be present (Mack (1982) Remington's Pharmaceutical Sciences, 16th Edition).
- the compositions can comprise a desired amount of polypeptide that unfolds reversibly. For example the compositions can comprise about 5% to about 99% polypeptide that unfolds reversibly by weight.
- the composition can comprise about 10% to about 99%, or about 20% to about 99%, or about 30% to about 99% or about 40% to about 99%, or about 50% to about 99%, or about 60% to about 99%, or about 70% to about 99%, or about 80% to about 99%, or about 90% to about 99%, or about 95% to about 99% polypeptide that unfolds reversibly by weight.
- the composition is a biological washing powder comprising a polypeptide that unfolds reversibly (e.g., a polypeptide comprising an immunoglobulin variable domain that unfolds reversibly).
- the composition is freeze dried (lyophilized).
- the invention also provides a sealed package (e.g., a sealed sterile package) comprising a polypeptide that unfolds reversibly (e.g., when heated (e.g., a polypeptide comprising an immunoglobulin variable domain that unfolds reversibly)).
- a sealed package e.g., a sealed sterile package
- the sealed package further comprises a sterile instrument.
- the sterile instrument is a medical instrument, such as a surgical instrument.
- polypeptides that unfold reversibly described herein will typically find use in preventing, suppressing or treating inflammatory states, allergic hypersensitivity, cancer, bacterial or viral infection, and autoimmune disorders (which include, but are not limited to, Type I diabetes, multiple sclerosis, rheumatoid arthritis, systemic lupus erythematosus, Crohn's disease and myasthenia gravis).
- inflammatory states allergic hypersensitivity, cancer, bacterial or viral infection
- autoimmune disorders which include, but are not limited to, Type I diabetes, multiple sclerosis, rheumatoid arthritis, systemic lupus erythematosus, Crohn's disease and myasthenia gravis.
- prevention involves administration of the protective composition prior to the induction of the disease.
- suppression refers to administration of the composition after an inductive event, but prior to the clinical appearance of the disease.
- Treatment involves administration of the protective composition after disease symptoms become manifest.
- EAE in mouse and rat serves as a model for MS in human.
- the demyelinating disease is induced by administration of myelin basic protein (see Paterson (1986) Textbook oflmmunopathology, Mischer et al., eds., Grune and Stratton, New York, pp. 179-213; McFarlin et al. (1973) Science, 179: 478: and Satoh et al. (1987) 7. Immunol, 138: 179).
- the selected polypeptides of the present invention may be used as separately administered compositions or in conjunction with other agents.
- compositions can include various immunotherapeutic drugs, such as cylcosporine, methotrexate, adriamycin or cisplatinum, and immunotoxins.
- Pharmaceutical compositions can include "cocktails" of various cytotoxic or other agents in conjunction with the selected antibodies, receptors or binding proteins thereof of the present invention, or even combinations of selected polypeptides according to the present invention having different specificities, such as polypeptides selected using different target ligands, whether or not they are pooled prior to administration.
- the route of administration of pharmaceutical compositions according to the invention may be any of those commonly known to those of ordinary skill in the art.
- the selected antibodies, receptors or binding proteins thereof of the invention can be administered to any patient in accordance with standard techniques.
- the administration can be by any appropriate mode, including parenterally, intravenously, intramuscularly, intraperitoneally, transdermally, via the pulmonary route, or also, appropriately, by direct infusion with a catheter.
- the dosage and frequency of administration will depend on the age, sex and condition of the patient, concunent aclministration of other drugs, counterindications and other parameters to be taken into account by the clinician.
- a therapeutically effective amount of a polypeptide that unfolds reversibly is administered.
- a therapeutically effective amount is an amount sufficient to achieve the desired therapeutic effect, under the conditions of administration.
- the invention also provides a kit use in administering a polypeptide that unfold reversibly to a subject (e.g., patient), comprising a polypeptide that unfolds reversibly, a drug delivery device and, optionally, instructions for use.
- a polypeptide that unfolds reversibly can be provided as a formulation, such as a freeze dried formulation.
- the drug delivery device is selected from the group consisting of a syringe, an inhaler, an intranasal or ocular administration device (e.g., a mister, eye or nose dropper) a needleless injection device.
- the selected polypeptides of this invention can be lyophilised for storage and reconstituted in a suitable carrier prior to use. Any suitable lyopbilization method (e.g., spray drying, cake drying) and or reconstitution techniques can be employed. It will be appreciated by those skilled in the art that lyophilisation and reconstitution can lead to varying degrees of antibody activity loss (e.g., with conventional immunoglobulins, IgM antibodies tend to have greater activity loss than IgG antibodies) and that use levels may have to be adjusted upward to compensate.
- the invention provides a composition comprising a lyophilized (freeze dried) polypeptide that unfolds reversibly as described herein.
- the lyophilized (freeze dried) polypeptide loses no more than about 20%, or no more than about 25%, or no more than about 30%, or no more than about 35%, or no more than about 40%, or no more than about 45%, or no more than about 50% of its activity when rehydrated.
- Activity is the amount of polypeptide required to produce the effect of the polypeptide before it was lyophilized. For example, the amount of rehydrated enzyme needed to produce half maximal conversion of a substrate into a product in a give time period, or the amount of a binding polypeptide needed to achieve half saturation of binding sites on a target protein.
- the activity of the polypeptide can be determined using any suitable method before lyophilization, and the activity can be determined using the same method after rehydration to determine amount of lost activity.
- compositions containing the present selected polypeptides or a cocktail thereof can be administered for prophylactic and/or therapeutic treatments, In certain therapeutic applications, an adequate amount to accomplish at least partial inhibition, suppression, modulation, killing, or some other measurable parameter, of a population of selected cells is defined as a "therapeutically-effective dose.” Amounts needed to achieve this dosage will depend upon the severity of the disease and the general state of the patient's own immune system, but generally range from 0.005 to 5.0 mg of selected antibody, receptor (e.g., a T-cell receptor) or binding protein thereof per kilogram of body weight, with doses of 0.05 to 2.0 mg/kg/dose being more commonly used. For prophylactic applications, compositions containing the present selected polypeptides or cocktails thereof may also be administered in similar or slightly lower dosages.
- a composition containing a selected polypeptide according to the present invention may be utilised in prophylactic and therapeutic settings to aid in the alteration, inactivation, killing or removal of a select target cell population in a mammal.
- the selected repertoires of polypeptides described herein may be used extracorporeally or in vitro selectively to kill, deplete or otherwise effectively remove a target cell population from a heterogeneous collection of cells.
- Blood from a mammal may be combined extracorporeally with the selected antibodies, cell-surface receptors or binding proteins thereof whereby the undesired cells are killed or otherwise removed from the blood for return to the mammal in accordance with standard techniques.
- Enor-prone PCR is a random mutagenesis strategy that introduces mutations into a DNA segment. Hence, it is a useful tool for preparing nucleic acids encoding diversified proteins that contain random amino acid substitutions.
- Enor-prone PCR can be carried out in a number of ways, generally by altering buffer conditions (e.g., varying dNTP concentrations) to reduce the fidelity of nucleotide incorporation.
- buffer conditions e.g., varying dNTP concentrations
- an enor-prone PCR library was constructed using the GENEMORPH PCR Mutagenesis Kit (Stratagene). The kit incorporates the MUTAZYME DNA polymerase that has a high intrinsic enor rate of nucleotide incorporation compared to Taq polymerase.
- Mutation frequency using this system can be altered by manipulating the starting concentration of template (VH and VK coding sequence) in the reaction.
- the starting concentration was altered accordingly to give a varying rate of mutational frequency.
- two libraries were constructed for each VH and VK template and cloned into a phagemid vector pR2.
- the VH template was V3- 23/DP47 and J H 4b, and the V L template was 012/02/DPK9 and J ⁇ l .
- the pR2 vector is derived from pHENl . (Hoogenboorn, HR et al., Nucleic Acids Res.
- pR2 contains a lac promoter, a leader sequence upstream of the cloning site which is followed by His6 and VSV tags, an amber stop condon and the gene encoding the pin phage coat protein. These consisted of one having a low mutational frequency ( ⁇ lbp change/template) and the other with a medium mutational frequency ( ⁇ 2b ⁇ changes/template). These libraries were combined to give a diversity of 1 -2 x 10 5 .
- the strategy involved the PCR amplification of the enor-prone PCR libraries from the pR2 phagemid vector followed by subcloning the amplified product into the phage Fd-Myc.
- the enor-prone PCR libraries in the pR2 phagemid in the E. coli host HB2151 , were plated out to give confluent growth on large plates (22x22 cm), The estimated total number of colonies was 10 7 - 10 8 , ensuring that the diversity of the enor-prone libraries was well covered. The colonies were scraped from the plates and the DNA of the phagemid library subsequently isolated.
- the isolated phagemid DNA library was used as the template for PCR amplification of the enor-prone library.
- the library was PCR amplified using synthetic oligonucleotide primers that contained the restriction sites ApaL 1 and Not 1. Thus facilitating subcloning of the amplified products into the corresponding sites in phage Fd-Myc.
- VK Fd-Myc PCR Primers VK Fd-Myc PCR Primers
- the PCR products of the amplified library were purified from an agarose gel and then restriction enzyme digested with ApaL 1 and Not 1.
- the digested product was purified by phenol/chloroform extraction, treated with streptavidin DYNABEADS (superparamagnetic monodisperse polymer beads; Dynal Biotech) to remove cut 5' ends and undigested product, and then subjected to QIAQUICK PCR purification kit (Qiagen).
- the product was ligated into the ApaL 1/Not 1 sites in phage Fd-Myc and transformed into E. coli TGI giving a library size of 10 6 -10 7 .
- the Fd-myc vector was assembled from Fd-tet-Dogl (McCafferty et al. (Nature) 1989) by cutting the vector at ApaLl and Notl, and ligating a synthetic double-stranded DNA cassette composed of 5'-end phosphorylated oligos LJ1012 and LJ1013 (SEQ ID NO:65 and SEQ ID NO:66, respectively) which encode a myc tag and a trypsin cleavage site.
- the resulting vector Fd-myc is very similar to Fd- Tet-Dogl in that ApaLl and Notl sits are present for the cloning of insert in between the leader sequence and gene III.
- the additional feature is the presence of a myc-tag in between the Notl site and gene LU which allows for immunological detection of encoded gene LU fusion protein, and also allows bound phage (e.g., selected using anti-myc antibody) to be eluted by digestion with trypsin since there is a trypsin cleavage site in the myc-tag.
- bound phage e.g., selected using anti-myc antibody
- LJ1012 P-TGCACAGGTCCACTGCAGGAGGCGGCCGCAGAACAAAA ACTCATCTCAGAAGAGGATCTGAATTC (SEQ ID NO:65)
- LJ1013 P- GGCCGAATTCAGATCCTC1TCTGAGATGAGTTTTTGTTCTGCG GCCGCGAGGACGTCACCTGCTG (SEQ LD NO:66) Preparation of the 3.25G Insert and Ligation
- the PCRs were performed on each sub-library using primers LJ1011 and LJ1027, to append an ApaLl site at the 5'-end and a Notl site at the 3'- end.
- the resulting amplified fragments were purified, digested consecutively with ApaLl and with Notl, re-purified and then ligated into the conesponding sites of Fd-myc.
- the 11 ligations were pooled and electro-transformed into E. coli TGI cells. After electroporation, the cells were resuspended in 2XTY and incubated for 1 hour at 37°C for phenotypic expression. The resulting library was then plated on TYE plates supplemented with 1 ⁇ g/ml tetracycline for overnight growth, and an aliquot was taken for titration on TYE plates supplemented with 15 ⁇ g/ml tetracycline. The size of the library was 1.6 x 10 9 clones.
- Aliquots of the library were prepared by resuspending the bacteria at an OD600 of 40, diluting with an equivalent volume of glycerol (final OD600 - 20), and aliquoting as 1 ml samples that were frozen and stored at -80°C until use.
- a 1 ml sample of library 3.25G was thawed and used to inoculate 500 ml of 2XTY supplemented with 15 ⁇ g ml of tetracycline, in a 2.5 L shaker flask.
- the culture was incubated for 20 hours at 30°C for phage production.
- the cells were pelleted by centrifugation at 5,500 g for 15 min to remove bacteria.
- 90 ml PEG/NaCl (20 % Polyethylene glycol 8000 [Sigma; formally sold as PEG 6000], 2.5 M NaCl) were added to 450 ml of culture supernatant, and after mixing, the solution was incubated for 1 hour on ice.
- Immunotubes (Nunc) were coated overnight (about 18 hours) at room temperature with either 4 ml of PBS containing 10 ⁇ g/ml ofprotein A, or 4 ml of PBS containing 10 ⁇ g/ml ofprotein L. In the morning, the solutions were discarded and the tubes were blocked with PBS supplemented with 2% v/v TWEEN 20 (Polyoxyethylensorbitan monolaurate; for protein A-coated immunotubes) or 2% w/v BSA (for protein L-coated immunotubes). The tubes were incubated for 1 hour at 37°C, then washed three times with PBS, before use for phage selection.
- TWEEN 20 Polyoxyethylensorbitan monolaurate
- BSA protein L-coated immunotubes
- the refolded phage solutions were pooled and added to 4 ml of PBS supplemented with either 2% v/v TWEEN 20 (Polyoxyethylensorbitan monolaurate; for selection on protein A) or 2% w/v BSA (for selection on protein L).
- the resulting phage solutions were added to Immunotubes coated with protein A or Immunotubes coated with protein L, after sealing the tubes were rotated end-over- end 30 min at room temperature, and then held on the bench at room temperature for 1.5 hours.
- Unbound phage were removed by washing the tubes ten times with PBS supplemented with 0.1% TWEEN 20 (Polyoxyethylensorbitan monolaurate), and ten times with PBS. Bound phage were eluted by adding 1 ml of PBS supplemented with trypsin (1 mg/ml) and incubating for 10-15 min while gently rotating. The solution containing the eluted phage was then transfened to a fresh microcentrifuge tube and stored on ice.
- TWEEN 20 Polyoxyethylensorbitan monolaurate
- E. coli Infection of Selected fd-Phage From an overnight culture of E. coli TGI cells in 2xTY at 37°C, a 100-fold dilution was prepared in 25 ml of fresh 2XTY medium, and the culture was incubated at 37°C with shaking (250 rpm) until the optical density at 600 nm (OD ⁇ oo) was 0.5-0.7 (mid-log phase). Ten milliliters of this culture was then incubated with 500 ⁇ l of the eluted phage sample (the remaining 500 ⁇ l were kept at 4C) at 37°C for 30 min without shaking to allow for phage infection.
- a 100 ⁇ l aliquot was taken for titration of the phage: lO ⁇ l ofa 1:10 2 dilution and 10 ⁇ l of a 1 : 10 4 dilution in 2xTY were spotted on TYE plates containing 15 ⁇ g/ml tetracycline and grown overnight at 37°C.
- the titre was determined by multiplying the number of colonies by the dilution factor (i.e., 100 or 10,000) then multiplying by 1000 (10 ⁇ l spotted from 10 ml infected culture), to gives the titre for 500 ⁇ l of eluted phage.
- the total number of eluted phage was determined by multiplying by 2 (1 ml total eluate).
- E. coli TGI culture 9.9 ml was transfened to a disposable 14 ml tube, and centrifuged at 3,300 g in for 10 min.
- the cell pellet was resuspended in 2 ml of fresh 2xTY, plated on a large 22 cm 2 dish containing TYE, 15 ⁇ g/ml tetracycline, and incubated overnight at 37°C Amplification of Selected fd-phage Vectors
- the 100 ml overnight culture was centrifuged at 3,300 g for 15 min to remove bacteria.
- the supernatant was filtered through a 0.45 u disposable filter.
- 20 ml PEG/NaCl (20 % Polyethylene glycol 8000; Sigma [formally sold as PEG 6000], 2.5 M NaCl) was added to 80 ml of supernatant, and after mixing, the solution was incubated for 1 hour on ice. Phage were collected from the resulting mixture by centrifugation at 3,300 g for 30 min at 4°C. The supernatant was discarded, the tubes were re-centrifuged briefly, and the remaining PEG/NaCl was carefully removed.
- the pellet was resuspended in 1 ml of PBS, then transfened to a fresh micro-centrifuge tube. Remaining bacterial debris were removed by centrifuging the micro-centrifuge tube at 11,600 g for 10 min. The supernatant was transfe ⁇ ed to a fresh micro-centrifuge tube, and stored at 4°C, until the next selection round, Phage titer was estimated by spectroscopy: a 100-dilution in PBS was prepared and the absorbance at 260 nm was measured. The phage titer (in TU per ml) was calculated using the formula: OD 2 co x 10 ⁇ x 2.214.
- a 96-well culture plate flat bottom, with evaporation lid, Coming
- each well was filled with 175 ⁇ l of 2xTY containing 15 ⁇ g/ml tetracycline, and inoculated with a single colony from the selected bacterial clones obtained using the Phage Selection protocol.
- the plate was incubated overnight (about 18 hours) with shaking at 37°C.
- the next day the cells were pelleted by plate centrifugation for 20 min at 2,000 rpm at room temperature.
- One hundred microliters (100 ⁇ l) of each culture supernatant (containing the phage) were transfened to a fresh 96-well culture plate. .
- phage were chemically derivatized with a biotinylated reagent. This procedure allowed bound phage to be detected using a conjugate of streptavidin and horseradish peroxydase (Str-HRP, Sigma). This strategy was chosen to decrease the possibility of cross reactivity between antibodies used to detect bound phage and immobilized protein A or protein L in the wells. Thus, each 100 ⁇ l sample of culture supernatant was reacted with 100 ⁇ l of PBS containing a 500 ⁇ M concentration of EZ-link Sulfo-NHS biotin (Perbio), for 1 hour at room temperature (with medium agitation on a rotating plate).
- Perbio EZ-link Sulfo-NHS biotin
- Both plates were then treated in parallel: to each well (containing 80 ⁇ l of heated or non-heated phage supernatant), 20 ⁇ l of PBS supplemented with 10% v/v TWEEN 20 (Polyoxyethylensorbitan monolaurate;for protein A-based ELISA) or 10% w/v BSA (for protein L-based ELISA) was added and mixed. The samples were then assayed by ELISA.
- MAXISORB 96-well plates (Nunc) were coated overnight (about 18 hours) at room temperature with either 100 ⁇ l of PBS containing 10 ⁇ g/ml of rotein A, or 100 ⁇ l of PBS containing 10 ⁇ g ml ofprotein L, per individual well. The next day, the plates were emptied, and the wells were blocked with 200 ⁇ l of PBS supplemented with 2% v/v TWEEN 20 (Polyoxyethylensorbitan monolaurate; for protein A-coated wells) or 2% w/v BSA (for protein L-coated wells). The plates are incubated for lh at 37°C, then washed three times with PBS, before use for screening.
- TWEEN 20 Polyoxyethylensorbitan monolaurate
- BSA protein L-coated wells
- VH -DP47 also refe ⁇ ed to as VH -DP47 dummy
- HEL4 a single VH that binds hen egg white lysozyme that was selected from a library based on a VH 3 scaffold (DP47 germline + JH4 segment) with randomised CDR 1, 2 and 3) does unfold reversibly under these conditions.
- Phage clones which yielded significant % refolding were then submitted to DNA sequencing of the VH gene or VK gene insert (see section 6). In addition, selected clones were submitted to a second ELISA (see section 7) to further quantify refolding.
- each tube was filled with 11 ml of 2xTY containing 15 ⁇ g/ml tetracycline, and inoculated with a single colony from the selected bacterial clones obtained using the Phage Selection protocol (Section 3) or after Phage Screening 1 (Section 4).
- the tubes were incubated overnight ( ⁇ 18 hours) with shaking at 37°C. In the morning, the cells were pelleted by centrifugation for 20 min at 3,300 g at 4°C to remove bacteria. The supernatant was filtered through a 0.45 urn disposable filter.
- Phage titer was estimated by spectroscopy: a 100-dilution in PBS was prepared and the absorbance at 260 nm was measured. The phage titer (in TU per ml) was calculated with the formula: OD 2 6o x 10 I x 2.214.
- phage were chemically derivatized with a biotinylated reagent. This procedure allowed bound phage to be detected using a conjugate of streptavidin and horseradish peroxydase (Str-HRP, Sigma). This strategy was chosen to decrease the possibility of cross reactivity between antibodies used to detect bound phage and immobilized protein A or protein L in the wells.
- a 100 ⁇ l sample of phage was transfened to a thin-wall PCR tube (and the remaining biotinylated phage was kept on ice).
- the tube was placed in a PCR apparatus for incubation at 80°C during 10 min (temperature of cover lid: 85°C). After heating, the tube was rapidly cooled down to 4°C in the PCR apparatus.
- Both tubes were then treated in parallel: first eight 4-fold dilutions were prepared for both heat-treated and nonheat-treated samples of the same clone, in the appropriate buffer (PBS supplemented with 2% v/v TWEEN 20 (Polyoxyethylensorbitan monolaurate) for VH -DP47 displaying phage; or PBS supplemented with 2% w/v BSA for VK- DPK9 displaying phage.
- PBS supplemented with 2% v/v TWEEN 20 Polyoxyethylensorbitan monolaurate
- the titer was 10 ⁇ TU per ml.
- the samples were then ready for assay by ELISA.
- ELISA plates were coated and blocked as described in Section 4, The samples were transfened to the empty wells of coated/blocked ELISA plates, and incubated for 2 hours at room temperature. Unbound phage was then removed by washing the wells 6-times with PBS.
- Str-HRP from a 1 mg/ml stock in PBS
- PBS PBS supplemented with 2% v/v TWEEN 20 (Polyoxyethylensorbitan monolaurate; for protein A-based ELISA) or 2% w/v BSA (for protein L-based ELISA)
- 100 ⁇ l of the resulting solution was added to each ELISA well.
- BSA1 is a single VH that binds bovine serum albumin and does not unfold reversibly when heated and cooled, that was selected from a library based on a VH 3 scaffold (DP47 germline + JH4 segment) with randomised CDRs 1 , 2 and 3.
- the scoring was done as follows: for each clone, the phage were tested as un-heated sample and as heat-treated sample in an eight-point dilution series. This approach permitted quantitative deductions about the refoldability of the domain antibody displayed on phage to be made.
- the OD450 were plotted onto a semilog graph (on the X-axis, concentration of phage (in TU) per well according to a semi -log scale; on the Y-axis, OD450 observed at each phage concentration), and linked together by simple linear interpolation between each data points.
- DNAs encoding variable domains from selected clones that display reversible heat unfolding were sequenced as follows: PCR reaction mix: 5 ⁇ l l Ox Buffer
- PCR reaction mix 50 ⁇ l of the PCR reaction mix was aliquoted into each well of a 96 well PCR microplate.
- a colony (Fd-Myc/TGl) was gently touched with a sterile toothpick and transfened into the PCR mix. The toothpick was twisted about 5 times in the mixture.
- the mix was overlayed with mineral oil.
- the PCR parameters were: 94°C for lOmin, followed by 30 cycles of: 94°C 30sec, 50°C 30sec, 72°C 45sec; and a final incubation at 72°C 5min.
- the amplified samples were purified using a QIAQUICK PCR product purification kit (Qiagen). Sequencing was carried out using either of the original PCR primers (LJ212 and/or LJ006).
- Section 7 List of Selected EP Vn
- *m indicates medium ELISA signal and H indicates high ELISA signal.
- Section 8 List of Selected EP VK
- Section 9 List of Selected 3.25G V H
- Clones were analyzed by assessing binding of heat treated and control phage to protein A in an ELISA as described herein.
- FIGs. 3A-3F Clones that had above 60% refolding were further analyzed by sequencing.
- Figs. 4A-4C present the sequence of may of the clones that had above 60% refolding.- The first set of sequences present in Figs. 4A-4C are from clone giving a high OD450 in the ELISA and a high % refolding. These sequences do not contain cysteines. This group of sequences forms the dataset for the analysis of amino acid preferences at all positions of CDR1, CDR2 and CDR3.
- Figs.4A-4C are from clones with excellent ELISA signals and good refolding. These clones contain mutations outside the CDRs.
- Figs. 4A-4C The final group of sequences in Figs. 4A-4C are from with relatively low ELISA signals and/or refolding. From the sequences presented in Figs.4A-4C, six clones were selected and further analyzed in detail on phage (as displayed polypeptide) and as soluble proteins. The six clones selected are refened to as pA-Cl 3, pA-C36, ⁇ A-C47, pA- C59, pA-C76 and pA-C85. (Clones are identified in Figs. 4A-4C using the suffix only, t.e., " P A-C13" is "C13.”)
- mutants were designed into DP47 and analyzed for refolding on phage (as displayed polypeptide) as for some of these mutations, as soluble protein.
- the particular mutations were: F27D, F29V, F27D/F29V, Y32E, S35G, F27D F29V/ ⁇ 32D/S35G, S53P, G54D, S53P/G54D, W47R, FlOOnV, Y102S, F100nV/Y102S, W103R.
- Enor-prone PCR samples a limited sequence space For example, the A50P substitution selected from the enor-prone VK library mat has a frequency of lbp change/ template would sample only 6 additional amino acids in total. Alanine at position 50 is encoded by the codon GCT. Changing this codon by 1 base gives the following codon permutations and amino acids:
- VK Fd-Myc PCR Primers 5' GAG CGC CGT GCA CAG ATC CAG ATG ACC CAG TCT CC 3 ' (SEQ ID NO:7l)
- DPK9 was used as a DNA template.
- the first PCR product or mega-primer was subsequently used in a second PCR reaction together with the second Fd-Myc primer not used in the first reaction. This PCR gave a product containing
- the PCR fragments were ligated into the ApaLl and Notl sites of Fd-myc.
- the ligated DNA was used to transform E. coli TGI cells by electroporation. After one hour of phenotypic expression at 37°C, the cells were plated on TYE plates supplemented with 15 ug/ml of tetracycline.
- cultures were made in 96-well cell culture plates as described in Section 4.
- three wells (usually Al, D6 and HI 1) were inoculated with a positive control phage (eg. Fd- myc-HEL4) and three wells were inoculated with a negative control phage (eg. Fd- myc-DP47d).
- the remaining 90 wells were inoculated with 90 different clones from a mini-library. This plate preparation procedure was repeated with each mini- library.
- Soluble VH or VK dAb were then captured on a protein A or protein L matrix (protein A agarose or protein L agarose). Depending on the culture volume, the supernatant was either loaded directly onto a prepacked protein A or L matrix column, or the matrix was added directly to the supernatant (batch binding). Elution from batch binding can be accomplished directly from collected matrix, or the matrix can be packed into a suitable column. Elution from protein A or protein L matrix was canied out at low pH. The dAbs can be further purified by gel filtration using Superdex75 (Amersham Pharmacia Biotech).
- dAbs were dialyzed overnight in PBS at 4°C, and concentrated (if needed) by centrifugation using Millipore 5K Molecular Weight Cut Off centrifugation concentrator tubes (at 20 °C).
- One and a half ml of dAb at 1-5 ⁇ M in PBS was transfe ⁇ ed to a CD cuvette (1 cm pathlength) and introduced in the Jasco J-720 spectropolarimeter.
- Spectra at room temperature (25°C) or at high temperature (85°C) were recorded in the far-UV from 200 nm to 250 nm (four accumulations followed by averaging) at a scan speed of 12 nm min "1 , with a 2-nm bandwith and a 1 second integration time.
- Heat-induced unfolding curves were recorded at fixed wavelength (usually 23 nm, sometimes at 225 nm) using a 2 nm bandwith.
- the temperature in the cuvette was raised at a rate of 50 °C per hour, from 25 °C to 85 °C.
- Data were acquired with a reading frequency of 1/20 sec '1 , a 1 second integration time and a 2 nm bandwith.
- the sample was rapidly cooled down to 25 °C (15 °C min "1 ), a spectrum was recorded, and a new heat-induced unfolding curves was recorded.
- the first unfolding curve is characterized by a steep transition upon melting and a "noisy" post-transition line (due to the accumulation of aggregates).
- the second unfolding curve is radically different from the first one: a melting transition is barely detectable because no, or very few, unfolded molecules properly refold upon cooling the sample.
- the first and second far-UV spectra differ considerably, the latter being more akin to that observed using a denaturated molecule.
- a typical example of an aggregating dAb is DP47 dummy, or DP47-W47R (Fig. 9).
- Some dAbs do exhibit partial refolding at 1-5 ⁇ M (e.g., DP47-S35G): i.e., upon cooling, a proportion of the ellipticity (and hence a portion of the original secondary structure) is recovered, and a melting transition is observed upon reheating the sample (Fig. 10).
- a proportion of the ellipticity and hence a portion of the original secondary structure
- a melting transition is observed upon reheating the sample (Fig. 10).
- the amount of ellipticity recovered after the first thermal denaturation is divided by the amount of ellipticity of the sample before the first thermal denaturation, and multiplied by 100.
- Phage ELISA and scoring of the clones were done according to protocol of Section 4.
- DNA sequencing of selected clones that showed reversible heat unfolding was done according to protocol of Section 6.
- Section 14 Mini-library in V H results Phage ELISA and scoring of the clones were done according to protocol of
- Section 4 DNA sequencing of selected clones that unfold reversibly when heated and coold was done according to protocol of Section 6.
- a spurious mutation (Ser25 to Pro) was found in clones carrying the following mutations: A84E, or A33 V, or A33E, or A33Q. It is possible that the S25P mutation has a positive effect on refolding, that is most likely surpassing the effect of the mutations at the intended positions.
- Ref. means % refolding on phage as determined by proteinA-binding according to protocol of Section 5.
- SE-15 represents the S/E hydrophobicity score of the segment of sequence from Cys 22 (included) to Trp36 (included): the hydrophobicity scores of each amino acid of the particular clone in that segment are added and then divided by 15.
- Quad means quadruple mutant (i.e., F27D/F29V/S35G/Y32E)
- Section 15 Aggregation resistant domain antibodies selected on phage by heat denaturation Protein aggregation is a problem in biotechnology.
- dAbs antibody heavy chain variable domains
- the dAbs were displayed multivalently at the infective tip of filamentous bacteriophage, and heated transiently to induce unfolding and to promote aggregation of the dAbs.
- the dAbs were selected for binding to protein A (a common generic ligand that binds the folded dAbs). Phage displaying dAbs that unfolded reversibly were thereby enriched with respect to those that did not. From a repertoire of phage dAbs, six dAbs were characterised after selection; all resisted aggregation, were soluble, well expressed from bacteria, and were purified in high yields. These results demonstrate that the methods described herein can be used to produce aggregation resistant polypeptides, and to identify amino acid residues, sequences or features that promote or prevent protein aggregation, including those responsible for protein misfolding diseases (Dobson, CM. Trends Biochem Sci 24, 329-332 (1999); Rochet, J.C & Lansbury, P.T., Jr. Curr Opin Struct Biol 10, 60-68 (2000)).
- HEL4 human VH dAb refe ⁇ ed to as HEL4 has biophysical properties that are similar to those of camels and llamas (see also, Jespers, L., et al JMol Biol 337, 893-903 (2004)).
- the HEL4 dAb unfolded reversibly above 62.1°C (T m ) at concentrations as high as 56 ⁇ M.
- HEL4 and DP47d dAbs were displayed in a multivalent state on the surface of filamentous bacteriophage, thereby providing a link between antibody phenotype and genotype and a powerful means of selection (McCafferty, J., et al. Nature 348, 552-554 (1990)).
- the fusion phage (5xl0 n transducing units per ml (TU/ml) were heated to 80°C for 10 min; the phage capsid withstands this temperature (Holliger, P., etal JMol Biol 288, 649-657 (1999)) but not the dAbs, which unfold above 60°C (Ewert, S., et al Biochemistry 41, 3628- 3636 (2002)), After cooling, the phage-displayed dAbs were assayed for refolding by phage ELISA on protein A (a generic ligand common to these folded dAbs).
- Binding was reduced 3-fold for the HEL4 phage but 560-fold for the DP47d phage (FIG. 15 A). This suggested that the HEL4 dAb had reversibly unfolded on the phage tip to a significant degree, and that the DP47d dAb had not.
- a repertoire of human VH dAbs (1.6xl0 9 clones) was prepared by diversification of the loops comprising the CDRs in the DP47d dAb, and displayed multivalently on phage. After three rounds of heat denaturation followed by selection on protein A, 179 out of 200 colonies secreted dAb phage that retained more than 80% ofprotein A-binding activity after heating. Twenty clones were sequenced and revealed as many unique dAb sequences with a large variability in the CDR sequences and lengths (Table 15). The diversity shows that, as with HEL4, mutations located entirely in the loops comprising the CDRs are sufficient to confer resistance to aggregation.
- dAbs with CDR3s comprising 10 to 20 amino acids were chosen for further characterization (C13, C36, C47, C59, C76, and C85). These proteins were well secreted from bacteria, and the C36, C47 and C59 dAbs were recovered in yields greater than 20 mg/L in bacterial supernatants compared to only 2.9 mg/L for the DP47d dAb (Table 16). After purification on immobilized protein A, the dAbs were subjected to size-exclusion chromatography on a SUPERDEX-75 column (gel filtration column; Amersham Biosciences).
- Protein aggregation is an off-pathway process that competes with the folding pathway, and usually involves association of the unfolded states, or partially unfolded states. Resistance to aggregation can be achieved by introducing mutations that stabilize the native state (increasing -dG t-u, the free energy of folding) and/or that reduce the propensity of the unfolded or partially unfolded states to aggregate (for example by increasing the solubility of these states).
- Several selection strategies have been devised to select for protein variants with improved stability: (i) by linking the infectivity of the phage to the proteolytic resistance of the displayed protein (Kristensen, P. & Winter, G.
- the folding properties of the selected dAbs cannot be attributed to stabilization of the native state, because biophysical analysis of thermodynamic stabilities of the selected dAbs indicates that the selected domains are less stable than typical aggregation-prone human dAbs.
- the free energies of folding at 25°C (from 14 to 23 kJ/mol) (Table 16) are lower than those of the DP47d dAb (35 kJ/rnol) and other aggregation-prone dAbs based on the same human DP47/3-23 germ-line segment (from 39.7 to 52.7 kJ/mol) (Tomlinson, I.M., et al JMol Biol 227, 776-798 (1992); Ewert, S., et al. Biochemistry 41, 3628-3636 (2002)). It appears that the described selection process using heat denaturation selects favorable properties of the unfolded or partially unfolded states.
- polypeptides e.g. , polypeptides expressed in the bacterial periplasm
- filamentous bacteriophage e.g., in a multivalent state
- a ligand that recognizes only the properly folded state of the polypeptide (e.g., an antibody that binds properly folded polypeptide, a receptor that binds properly folded polypeptide)
- Such polypeptides can be diversified (e.g., by engineering random mutations) displayed on phage, denatured, and selected by bio-panning (or other suitable methods) after returning to conditions permissive to the native state (refolding).
- the methods described herein also provide an analytical tool to identify amino acid residues or polypeptide segments involved in off-pathway aggregation of proteins upon folding, including those involved in diseases ofprotein misfolding (Dobson, CM. Trends Biochem Sci 24, 329-332 (1999); Rochet, J.C. & Lansbury, P.T., Jr. Curr Opin Struct Biol 10, 60-68 ⁇ (2000)).
- Phage display of a human VH dAb library The dAb repertoire was created in two- steps by oligonucleotide-mediated diversification of several codons in the sequence of the DP47d dAb as follows (Kabat numbering for the amino acid positions and IUPAC-IUB code for the nucleotides): 27, KWT; 28, ANS; 29, NTT; 30, ANC; 31, NMT; 32, NAS; 33, DHT; 35, RSC; 50, RSC; 52, NNK; 52a, RNS; 53, WW; 54, CGT; 94, RSW; 101, NVS; 102, THT; and NNK codons for all CDR3 positions from 95 to lOOx (where x ranges alphabetically from a to k).
- the DNA inserts were flanked with ApaLl (at 5'-end) and Notl (at 3'-end) sites by PCR, digested and ligated into the conesponding sites of fd-myc, a multivalent phage vector derived from fdCATl (McCafferty, J strictly et al. Nature 348, 552-554 (1990)) that contains a c- myc tag between the Notl site and gene m.
- the ligation products were transformed by electroporation into E. coli TGI cells, and plated on 2xTY plates supplemented with 15 ⁇ g/ml of tetracycline (2xTY-Tet), yielding a library of 1.6xl0 9 clones.
- dAb genes (as Nc ⁇ l-Notl D ⁇ A fragments) were ligated into the conesponding sites of pR2, a phagemid vector derived from pHE ⁇ I (Hoogenboom, H.R. et al Nucleic Acids Res 19, 4133-4137 (1991)) that contains the (His) 8 and VSV tags between the Notl site and gene III. Phage were prepared, purified and stored as described (McCafferty, J., et al Nature 348, 552-554 (1990);
- Phage ELISA assays The ELISA wells were coated overnight at 4°C with one of the following ligands: 10 ⁇ g/ml of protein A in PBS, 10 ⁇ g/ml of mAb 9E10 in PBS, or 3 mg/ml of HEL in 0.1 M ⁇ aHC0 3 buffer, pH 9.6. After blocking the wells with PBS containing 2% Tween-20 (PBST), a dilution series of phage in PBST was incubated for 2h. After washing with PBS, bound phage were detected as follows.
- PBST 2% Tween-20
- phage was detected directly using a conjugate of horseradish peroxidase with an anti-M13 monoclonal antibody (Amersham) using 3 ,3 ' ,5 ,5 ' -tetramethylbenzidine as substrate.
- the phage (4xl0 10 TU/ml in PBS) was first biotinylated at 4°C with biotin-NHS (Perbio) (50 ⁇ M final concentration) and detected by sequential addition of streptavidin- horseradish peroxidase conjugate (1 ⁇ g ml) (Sigma-Aldrich) in PBST, and substrate as above.
- HSA human serum albumin
- PBSM skim- milk powder
- bound phage were detected in two steps using a rabbit anti-fd bacteriophage monoclonal antibody (Sigma, 1/1000 dilution) and goat anti- rabbit IgG serum conjugated with horseradish peroxidase (Sigma, 1/1000 dilution) and substrate as above.
- Phage (lxlO 12 TU/ml in PBS) were heated at 80°C for 10 min and then cooled at 4°C for 10 min or left untreated as control. After dilution to lxlO 10 TU/ml in PBS, phage were adsorbed on glow discharged carbon coated copper grids (SI 60-3, Agar Scientific), washed with PBS and then negatively stained with 2% uranyl acetate (w/v). The samples were studied using a FEl Tecnai 12 transmission electron microscope operating at 120 kV and recorded on film with calibrated magnifications.
- Phage selection Immunotubes (Nunc) were coated overnight with protein A, and blocked with PBST. Purified phage (lxlO 1 ' TU/ml in PBS) were heated as described above, diluted with 3 ml of PBST, and incubated for 2h in the immunotubes. After 10 washes with PBS, protein A-bound phage were eluted in 1 ml of 100 ⁇ g/ml trypsin in PBS during 10 min, then used to infect 10 ml of log-phase E. coli TGI cells at 37°C during 30 min. Serial dilutions (for phage titer) and library plating were performed on 2xTY-Tet agar plates.
- the beads were packed into a column, washed with PBS, and bound dAbs were eluted in 0.1 M glycine-HCl, pH 3.0. After neutralisation to pH 7.4, the protein samples were dialyzed in PBS and concentrated before storage at 4°C Protein purity was estimated by visual inspection after SDS-PAGE on 12% Bis-Tris gel (Invitrogen). To obtain expression yields from normalized cultures, five individual colonies from freshly transformed bacteria were grown overnight at 37°C, and induced for expression as described above (50 ml scale). Following to overnight expression, the cultures were combined and a culture volume co ⁇ esponding to ⁇ OOxODgoonm (120 to 135 ml) was processed for dAb purification as described.
- the amount of purified protein was extrapolated to the protein yield per litre of bacterial culture, conected for a final absorbance of 5.0 at OD ⁇ oo n m-
- 500 ⁇ l of a 7 ⁇ M solution of dAb were loaded on a SUPERDEX-75 (Amersham Biosciences).
- the M t of each of the peaks on the chromatograms were assigned using a scale calibrated with markers, and the yields calculated from the areas under the curve.
- Section 16 Introducing "folding gatekeeper” residues into single variable domain with defined specificity.
- a “folding gatekeeper” is an amino acid that, by the virtue of its biophysical characteristics and by its position in the primary sequence of a protein, prevents the irreversible formation of aggregates upon protein unfolding.
- a folding gatekeeper residue blocks off-pathway aggregation, thereby ensuring that the protein can undergo reversible unfolding.
- the effectiveness of folding gatekeepers influence the maximal concentration at which the unfolded protein can remain in solution without forming aggregates.
- folding gatekeepers have been introduced into single variable domains (DP47d, or VKdummy) lacking antigen-specificity.
- TAR2- 10-27 is a human V ⁇ 3 domain which has binding specificity for human tumor necosis factor receptor 1 (TNFRI, also refe ⁇ ed to as "TAR2").
- the amino acid sequence of TAR2-10-27 is:
- LGGGPNFGYRGQGTLVTVSS (SEQ ID NO:191)
- aggregation is mediated by the hydrophobic segment encompassing the Hl-loop.
- Site-directed mutagenesis and biophysical analysis have shown that even a single point mutation that lower the SE- score over residue 22 to 36 in DP-47 to below zero, can confer proper folding in this variable domain at concentration up to 5 uM.
- the prefened positions for single point mutations are residues Phe27 and Tyr32 and, the prefened substitutions are the hydrophilic Asp, Glu, Gin, Asn residues, or the beta-strand breaking residue Pro.
- four single mutants in the Hl-loop of TAR2-10-27 significantly lower the SE-15 score, and may therefore confer high folding yield. Combinations of mutations would further lower the SE-15 score below zero.
- the F27D, F27Q, Y32D or Y32Q point mutations were introduced into the DP-47- VH3 domain using SOE-PCR, and the nucleic acids encoding the mutated VH3 domains were subcloned, expressed and the encoded proteins were purified
- the PCR-amplified genes encoding these proteins were subcloned into a derivative of the pET-12A vector in which the OmpT lead& ⁇ sequence was replaced with an eukaryotic leader sequence, using the Sail and Notl restriction sites,
- the ligated DNAs were used to transform E. coli BL21(DE3)pLysS, which were then plated on agar plates supplemented with TYE-agar+5%glucose+100 ⁇ g/ml ampicillin and incubated at 37°C overnight.
- a lOmL 2xTY culture was inoculated with a single bacterial colony from the plates and grown overnight at 37°C
- the 2xYT culture contained ampicillin (100 ⁇ g/mL and glucose (5%).
- a 500 mL 2xTY culture in a 2.5L culture flask was inoculated with 5 mL of the over-night culture and grown at 37°C with shaking (250 rpm) until an OD ⁇ oon m of 0.5-0.6.
- the media also contains ampicilin (50 ⁇ g/mL) and glucose (0.1%). When the OD ⁇ oonm reached 0.5-0.6, the culture flasks were further incubated at 30°C with shaking. After about 20 minutes, protein production was induced by the addition of ImM IPTG (final concentration) and the cultures were grown overnight.
- the culture media was transfened to cylindrical 0.5 or 1 L bottles (glass or plastic).
- 0.5-1 mL of a 1:1 slurry of protein A-beads (Amersham Biosciences) was added, and the mixture was rolled on horizontal flask rollers at 4°C overnight. Then the bottles were left upright allowing the resin to settle.
- the resin (beads) was repeatedly washed with lxPBS, 500 mM NaCl (2x), and the washed resin was loaded into a drip column.
- the resin wass washed again with 5 column volumes of lxPBS, 500 M NaCl. After the resin had almost run dry, 0.1 M glycine pH 3.0 was applied to the column and 1ml fractions are collected.
- the collection tubes were filled prior to the elution with 200 ⁇ L IM Tris, pH 7.4 in order to neutralise the solution.
- the OD 28 Q nm was checked for each fraction and the protein elution monitored.
- the fractions that contained protein were combined and dialysed against an appropriate buffer (lx PBS in most cases).
- the protein concentration was again determined (using a calculated exctinction coefficient) and purity was evaluated by SDS-PAGE analysis.
- the protein yields per litre of culture supernatant are shown in Table 18.
- recombinant expression of the DP-47-VH3 domain in the ⁇ ET/BL21(DE3)pLysS system was increased when folding gatekeepers were inserted into the protein sequence.
- Samples of each protein at different concentrations ranging from 1-lOO ⁇ M were prepared. The samples were heated for 10 minutes at 80°C in a PCR block using 0.5 mL thin-walled tubes. The ramping was ⁇ 10°C/min. Duplicate samples were prepared and processed in parallel but omitting the heating step.
- the samples were kept at room temperature or 4"C for a few minutes.
- the samples were transfened to 1.5 mL micro tubes and centrifuged at 4°C for 10 minutes at high speed. The supernatant was recovered without disturbing any pellets that formed.
- 20 ⁇ L of a 1 : 1 slurry of rotein A-beads was added to each tube, the tube was topped up to lmL of PBS and rolled for 1-2 hours on an overhead roller at room temperature. When the protein A beads had settled, the supernatant was removed without disturbing the beads.
- the beads were washed three times with 1 mL PBS and resuspended in 40 ⁇ L of SDSdoading buffer, including DTT.
- the SDS-loading buffer contained a known concentration of BSA which served as a standard to normalise for possible error the loading volume.
- the samples were heated at 90°C for 10 minutes, centrifuged for 10 minutes at 14,000 rpm and then equal amounts of each sample were loaded and separated on a 12% Bis/Tris SDS-NuPage gel. Equal loading, background and intensity of bands were quantified using densitometry.
- the densitometry data was plotted against the concentration of initial protein to produce a sigmoidal curve that was used to determine the protein concentration at which 50%> of the protein refolded after heat- unfolding ([protein] 5 o % ).
- the [proteinjso ⁇ are shown in Table 1 .
- Introducing folding gatekeeper mutations had a beneficial effect on TAR2-10-27 by conferring the ability to resist aggregation at higher protein concentration: 2- to 4- fold higher for TAR210-27 F27D and TAR2-10-27 Y32Q, and 7 to 25-fold higher for TAR210-27 Y32D, relative to the parental TAR2-10-27.
- TNFR1 binding activity of the VH domains was assessed in a receptor binding assay and a cell-based assay.
- TNF receptor 1 Fc fusion protein In the receptor bindng assay, anti-TNFRl VH domains were tested for the ability to inhibit the binding of TNF to recombinant TNF receptor 1 (p55). Briefly, MAXISORP plates were incubated overnight with 30 ⁇ g/ml anti-human Fc mouse monoclonal antibody (Zymed, San Francisco, USA). The wells were washed with phosphate buffered saline (PBS) containing 0.05% TWEEN-20 and then blocked with 1% BSA in PBS before being incubated with lOOng/ml TNF receptor 1 Fc fusion protein (R&D Systems, Minneapolis, USA).
- PBS phosphate buffered saline
- Anti-TNFRl VH domain was mixed with TNF which was added to the washed wells at a final concentration of lOng/ml. TNF binding was detected with 0.2 ⁇ g ml biotinylated anti-TNF antibody (HyCult biotechnology, Uben, Netherlands) followed by 1 in 500 dilution of horse radish peroxidase labelled streptavidin (Amersham Biosciences, UK), and then incubation with TMB substrate (KPL, Gaithersburg, USA). The reaction was stopped by the addition of HCI and the absorbance was read at 450nm. Anti-TNFRl VH domain binding activity lead to a decrease in TNF binding to receptor and therefore a decrease in absorbance compared with the TNF only control. (FIG. 18 A)
- IC50 values can be calculated from the sigmoidal curves with an average precision within a factor of t o.
- This section shows that a folding gatekeeper residue can be introduced, into a VH domain of pre-defined specificity such that the resulting variant combines increased resistance to aggregation with antigen-binding activity. This was achieved without knowing the functional epitope that is bound by the VH domain.
- One variant in particular (TAR2-1027-Y32D) retained full biological activity and had a 10-fold increase in resistance to aggregation fl roteinjso 0 / .).
- Another benefit of introducing folding gatekeepers is the increased expression level of the proteins (e.g., by >10-fold in the pET/BL2l(DE3)pLysS system).
- Section 17 Selection of an antigen-specific VH domain that contains a folding gatekeepers from a synthetic library of VH domains.
- antibody variable domains can be isolated from a synthetic repertoire based on binding to an antigen (e.g., by binding antigen immobilized on in a Petri dish and recovering adherent phage, 'Triopat-ning').
- This approach was used to select a human VH that binds human serum albumin (HSA) from a synthetic human VH repertoire following a heat denaturation step (or without the heat denaturation step for negative control).
- HSA human serum albumin
- the phage library of synthetic VH domains was the libray 4G, which is based on a human VH3 comprising the DP47 germline gene and the J H 4 segment.
- the diversity at the following specific positions was introduced by mutagenesis (using NNK codons; numbering according to Kabat) in CDRl : 30, 31, 33, 5; in CDR2: 50, 52, 52a, 53, 55, 56; and in CDR3: 4-12 diversified residues: e.g.
- the library comprises >lxl0 10 individual clones.
- the 4G library was divided into two sub-libraries of equal number of transducing units (lxlO 1 '). One was used as such for panning against HSA, and the other was heat-unfolded at 80"C for 10 minutes before each panning step.
- the phage sample was dispensed as 100 ⁇ L fractions in a PCR tube. After heating, the fractions were combined. Both samples (heat treated and unheated control) were centrifuges for 10 minutes at 14,000 rpm at 4°C The supernatants were taken further into the selections on immuno-tubes.
- the antigen HSA, Sigma-Aldrich
- was coated in immuno-tubes at 100 ug/mL in PBS), stored at 4°C. The tubes were blocked with 2% skim-milk in PBS (2MPBS).
- a phage ELISA was performed according to the protocol of Section 4, with some modifications.
- the microtiter wells were coated overnight with HSA (1 O ⁇ g/mL in PBS) and blocked with 2MPBS. Each of the samples was washed three times with 0.1% TWEEN-20 in PBS.
- the detection reagents were rabbit-anti- bacteriophage fd full IgG (Sigma) at 1:1000 dilution followed by goat-anti-rabbit conjugated with horseradish peroxidase (full IgG) (Sigma) at 1:1000 dilution. All individual clones were tested against HSA with heat-step or without heat step. They were also tested against BSA (lOug/mL) and plastic. Only 2/37 positive signals were unspecific.
- Clone 10 which has two acidic residues at positions 30 and 31 in the CDRl, exhibits the lowest SE-score (0,06) (15 amino acid window).
- SE-score 0.06
- the mean SE score obtained from all sequences from the unheated phage selection was above 0,15 (was 0.203 ⁇ 0.020; mean ⁇ standar deviation), thereby explaining their greater propensity to aggregate.
- clone #10 shares 92% identity with clones #2, #5, and #6, suggesting a common binding epitope on HSA for these four VH domains. But only clone #10 exhibits the refolding property and has folding gatekeepers at positions 30 and 31.
- phage-displayed VH domains were further analysed with purified phage.
- a phage ELISA was performed according to the protocol of Section 5 with the following modifications. lxlO 10 phage were either heat-treated (10 minutes at 80°C in 100 ⁇ L aliquots) or left untreated. The samples were centrifuged for 10 minutes at 14,000 rpm at 4°C and the supernatants were added to ELISA wells coated with HSA (with a dilution series of 1/3). After developing the ELISA, the OD450nm values (y-axis) for each heat freated/unheated phage pair were plotted against the phage number (x-axis) and the %-refoldability was calculated. The results are shown in Table 20.
- Section 16 demonstrated that a VH domain with good folding properties can be isolated from a repertoire of aggregation-prone VH domains in which the CDRl loop was diversified and hence, a small percentage of the VH domains in the primary repertoire had folding gatekeepers in the CDRl loop.
- This section describers the preparation of a phage repertoire in which the majority of VH domains unfold reversibly.
- VH- 6G Diversity was introduced by randomly combining DNA fragments encoding diversified CDRl, CDR2 and CDR3 using assembly PCR. The resulting fragments were cloned into a phage vector (Fd-myc in which the leader sequence was replaced with an eukaryotic leader sequence; see Section 2), to yield a primary library (VH- 6G).
- the V H library is based on a single human framework (V3-23/DP-47 and Jl ⁇ 4b).
- the canonical structures (Vn: 1-3) encoded by this frameworks are the most common in the human antibody repertoire.
- Side chain diversity was incorporated using NNK diversified codons at positions in the antigen binding site that make contacts with antigen in known structures, and are highly diverse in the mature repertoire. Diversity was incorporated at the following positions (Kabat numbering):
- V H CDRl H30, H31, H32, H33, H35.
- V H CDR2 H50, H52, H52a, H53, H55 and H56.
- V H CDR3 4-12 diversified residues: e.g. H95, H96, H97, and H98 in 4G
- the second stock was first heated at 80'C for 10 minutes, and subsequently panned on protein A.
- the library of phage propagated after panning was enriched for VH domains that refold after heat denaturation.
- the DNA pool obtained from these phages is refened to as DNA pool "H".
- Individual colonies from the primary library and from the selected libraries were separately grown for phage production.
- the phages were tested for their ability to bind to protein A (to assess production as well as co ⁇ ect folding) when untreated or after the heat treatment at 80°C The results are shown in Table 21.
- the na ⁇ ve passage pool was produced by growing phage from the primary library, purifying the phage by precipitation, infecting E. coli with the purified phage and recovering DNA from the bacteria.
- SOE-PCR the pools of CDRl and CDR2s were randomly recombined with the pool of CDR3s, to recreate full VH domain genes. The following combinations were
- the CDRl and CDR2s were systematically taken from the DNA pool H, because the CDRl segment contains the main determinants of aggregation.
- the CDR3s were from the ⁇ ' and 'nP' pools, which are less important for aggregation.
- the fragments were cloned into the phage vector (Fd-myc in which the leader sequence was replaced with an eukaryotic leader sequence; see Section 2), and used to transform E. coli by electroporation in order to obtain large repertoires (>10 10 clones).
- the phages were tested for their ability to bind to protein A (to assess production as well as conect folding) when untreated or after the heat treatment at 80°C. The results are shown in Table 23.
- CAASGFTFQYGPMSW (SEQ ID NO:197) 0.152
- CAASGFTFDDSTMQW (SEQ LD NO:203) -0.075 8 CAASGFTFDNSVMGW (SEQ ID NO:204) 0.046
- CAASGFTFDQTAMHW (SEQ ID NO:205) -0.021
- CAASGFTFPELPMGW (SEQ ID NO:206) 0.105
- CAASGFTFTPGKMNW (SEQ ID NO-.207) 0.013
- CAASGFTFEHRTMGW (SEQ ID NO:208) -0.011
- CAASGFTFPNSJJMV W (SEQ ID NO:209) 0.058
- CAASGFTFGKSTMAW (SEQ ID NO:210) 0.044
- the mean ( ⁇ SD) SE-score for this region of the 14 Clones is 0.007 ( ⁇ 0.018) which is well below the mean SE-score of VH dummy (DP47d) (0.190) and the mean SE score (0.220) calculated for the encoded diversity (NNK codons) for that region.
- the diversity at each position was relatively large: 7, 11, 8, 10 and 9 different amino acids were counted in the dataset, at position 30, 1, 32, 33 and 35, respectively. Strongly hydrophilic and 0-sheet breakers such as Asp, Glu, Pro and Gly accounted for 43% of the amino acids (versus 19% based on the encoded genetic diversity).
- a synthetic library of human VH3 domain was created by recombination of PCR fragments and displayed on the surface of filamentous phage. A majority of the V H 3 dAbs (85%) unfolded reversibly upon heating at 80°C.
- This library was produced by (1) diversification of the CDRl region (at position 30, 31, 32, 33 and 35), (2) heat-treatment of the primary phage library before clean-up on protein A, and (3) PCR-mediated recombination of heat-selected CDRl-CDR2-encoding genes and na ⁇ ve passage- (or simply protein A-cleaned-up) CDR3-encoding genes.
- the CDRl sequences were diverse, but enriched for folding gatekeeper residues such as Asp, Glu, Pro and Gly.
- TARl -5- 19 is a V R I domain which is specific for TNF- ⁇ . Its sequence is:
- folding gatekeepers can be introduced into the region comprising the HI loop of DP47d, or the region comprising FR2- CDR2 of V ⁇ dummy (DPK9).
- DPK9 V ⁇ dummy
- Phage Screening 2 monoclonal phages displaying TARl -5- 19 and variants were heated for 10 min at 80°C or left un-heated, and then incubated in TNF- ⁇ coated wells for ELISA. The % refolding data were calculated as described in Section 5, Phage Screening 2, and the results are shown in Table 30.
- the folding gatekeepers have a beneficial effect on TAR1-5-19 refolding , and the variants that contained folding gatekeepers resisted aggregation when heated to 80°C even when displayed on phage.
- parental TARl -5- 19 also showed good refolding ability due to its low SE-score (0.011).
- the activity assay was a RBA-ELISA (receptor-binding assay where increasing amount of dAb is used to compete with soluble TNF- ⁇ binding to immobilised TNF- -receptor. Bound TNF- ⁇ was then detected with a biotinylated non-competing antibody, and streptavidin-HRP conjugate.
- the IC 50 (in nM) is the dAb dose require to produce a 50% reduction of a non-saturating amount of TNF- ⁇ to the receptor. The results are shown in Table 31. Table 31
- the introduction of the folding gatekeeper into TARl -5-19 affected the in-vitro activity of TARl -5-19. This suggests that residue Y49 in TARl -5-19 may be involved (directly or indirectly) in specific contact with TNF- ⁇ . For TARl -5- 19, Y49 does not appear to be the best choice for introducing a folding gatekeeper (although the mutants are still moderately active). Other positions within the 15 amino acid segment centered around Y49 could be considered to achieve a reduction of the SE-score without reducing the activity of the dAb.
- TARl -5-19 variants in which 148 is replaced with P, D, T, G or N, Y49 is replaced with S, C, E, G, K, N or R and or 175 is replaced with N or M can be produced and characterized as described herein.
- Such a TAR1-5-19 variant can readily be produce using the methods demonstrated herein.
- folding gatekeepers were introduced into a dAb of pre-defined specificity, without prior knowledge of stmcture of the antigen-dAb complex. Binding of variants of TAR1-5-19 was reduced by 6- to 20-fold, but the variants still bound the target with moderate activity, demonstrating that the variants have biological activity. It should be noted that only a two single point mutants were created and studied. Further benefits of introducing folding gatekeepers would be realized by creating and screening additional variants that contain single point mutations or combinations of mutations, for example by positional diversification or by library generation.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Engineering & Computer Science (AREA)
- Biophysics (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Chemical & Material Sciences (AREA)
- Biotechnology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Zoology (AREA)
- Crystallography & Structural Chemistry (AREA)
- Wood Science & Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biomedical Technology (AREA)
- Microbiology (AREA)
- General Engineering & Computer Science (AREA)
- Plant Pathology (AREA)
- Immunology (AREA)
- Physics & Mathematics (AREA)
- Virology (AREA)
- Analytical Chemistry (AREA)
- Peptides Or Proteins (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
Description






































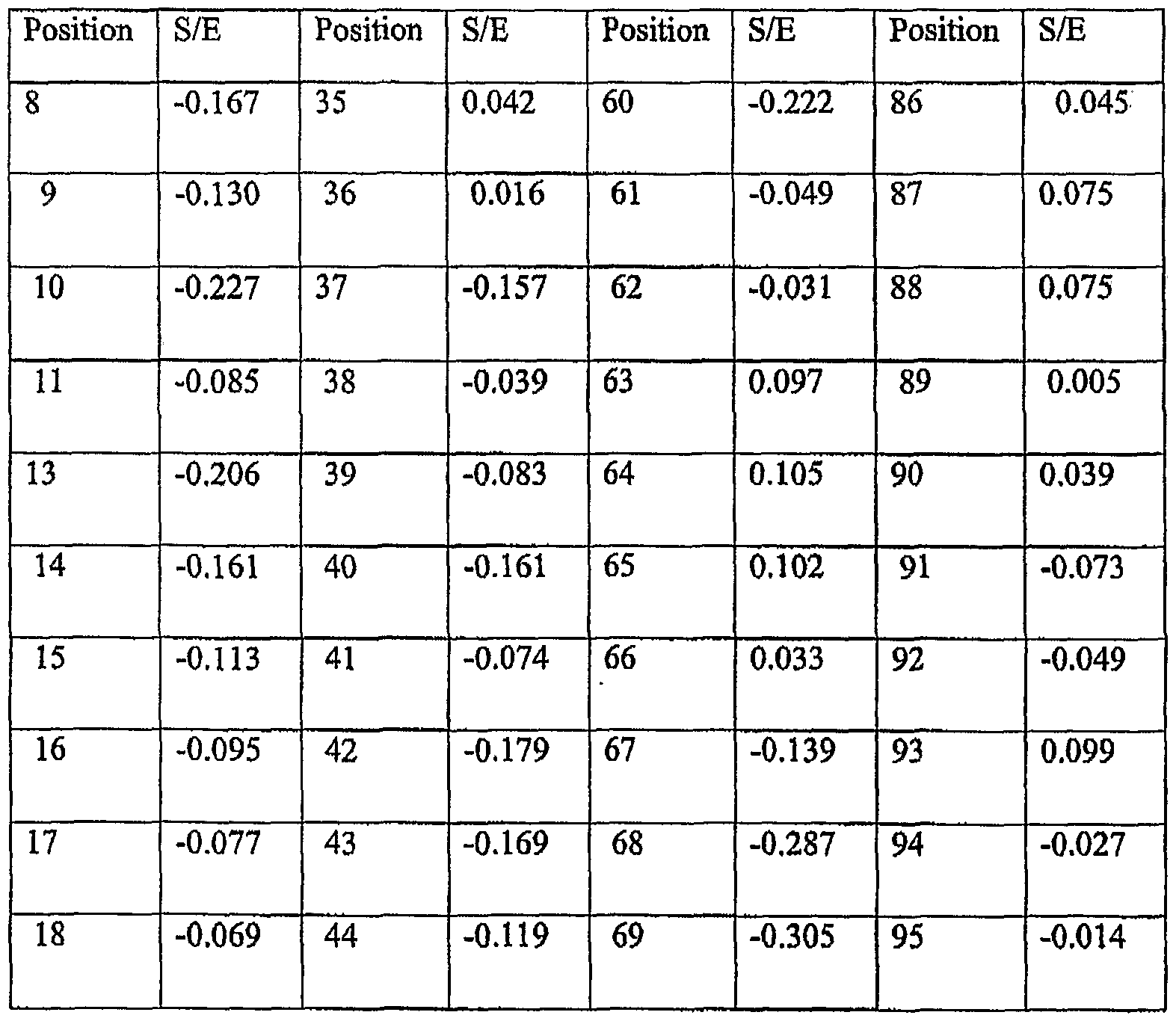
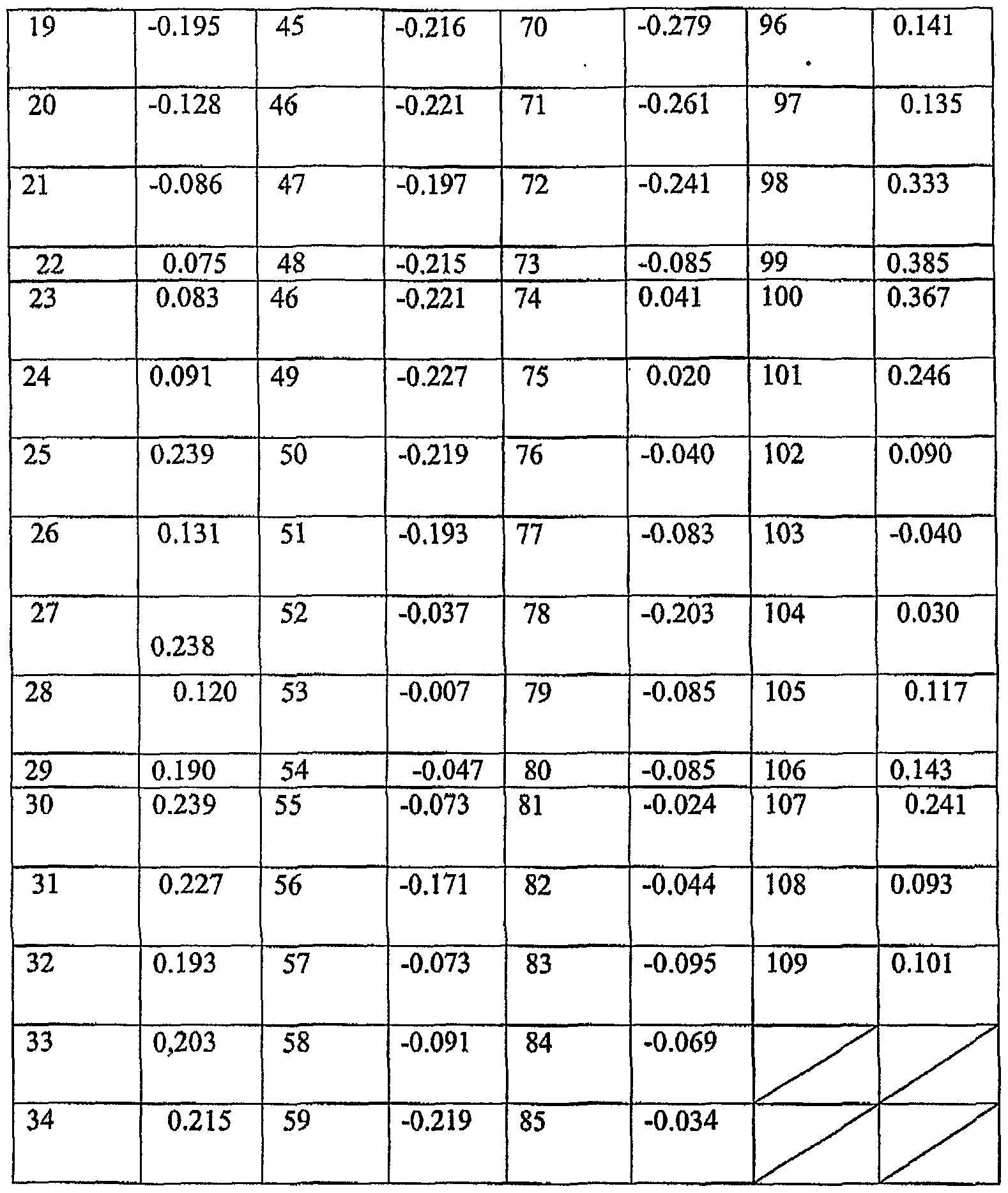


Claims
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CA2525120A CA2525120C (en) | 2003-05-14 | 2004-05-14 | A process for recovering polypeptides that unfold reversibly from a polypeptide repertoire |
| US10/556,360 US20090005257A1 (en) | 2003-05-14 | 2004-05-14 | Process for Recovering Polypeptides that Unfold Reversibly from a Polypeptide Repertoire |
| EP04733030A EP1627062A1 (en) | 2003-05-14 | 2004-05-14 | A process for recovering polypeptides that unfold reversibly from a polypeptide repertoire |
| AU2004239065A AU2004239065B2 (en) | 2003-05-14 | 2004-05-14 | A process for recovering polypeptides that unfold reversibly from a polypeptide repertoire |
| JP2006530506A JP4889493B2 (en) | 2003-05-14 | 2004-05-14 | Method for recovering a polypeptide that unfolds reversibly from a polypeptide repertoire |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US47034003P | 2003-05-14 | 2003-05-14 | |
| US60/470,340 | 2003-05-14 | ||
| US55402104P | 2004-03-17 | 2004-03-17 | |
| US60/554,021 | 2004-03-17 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2004101790A1 true WO2004101790A1 (en) | 2004-11-25 |
Family
ID=33457183
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/GB2004/002102 WO2004101790A1 (en) | 2003-05-14 | 2004-05-14 | A process for recovering polypeptides that unfold reversibly from a polypeptide repertoire |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US20090005257A1 (en) |
| EP (2) | EP2357237A1 (en) |
| JP (3) | JP4889493B2 (en) |
| AU (2) | AU2004239065B2 (en) |
| CA (1) | CA2525120C (en) |
| WO (1) | WO2004101790A1 (en) |
Cited By (86)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006038027A3 (en) * | 2004-10-08 | 2007-01-04 | Domantis Ltd | SINGLE DOMAIN ANTIBODIES AGAINST TNFRl AND METHODS OF USE THEREFOR |
| WO2007141280A2 (en) | 2006-06-06 | 2007-12-13 | Oxford Genome Sciences (Uk) Ltd | Proteins |
| WO2008030611A2 (en) | 2006-09-05 | 2008-03-13 | Medarex, Inc. | Antibodies to bone morphogenic proteins and receptors therefor and methods for their use |
| WO2008052933A2 (en) * | 2006-10-30 | 2008-05-08 | Domantis Limited | Prevention of aggregation of immunoglobulin light or heavy chains |
| WO2008070569A2 (en) | 2006-12-01 | 2008-06-12 | Medarex, Inc. | Human antibodies that bind cd22 and uses thereof |
| WO2008074004A2 (en) | 2006-12-14 | 2008-06-19 | Medarex, Inc. | Human antibodies that bind cd70 and uses thereof |
| WO2009054863A2 (en) | 2006-12-13 | 2009-04-30 | Medarex, Inc. | Human antibodies that bind cd19 and uses thereof |
| JP2009519983A (en) * | 2005-12-20 | 2009-05-21 | アラーナ・テラピューティクス・リミテッド | Chimeric antibodies with partial New World monkey binding regions |
| WO2010011944A2 (en) | 2008-07-25 | 2010-01-28 | Wagner Richard W | Protein screeing methods |
| WO2010046337A2 (en) * | 2008-10-21 | 2010-04-29 | Domantis Limited | Ligands that have binding specificity for dc-sign |
| WO2010085590A1 (en) | 2009-01-23 | 2010-07-29 | Biosynexus Incorporated | Opsonic and protective antibodies specific for lipoteichoic acid gram positive bacteria |
| WO2010084408A2 (en) | 2009-01-21 | 2010-07-29 | Oxford Biotherapeutics Ltd. | Pta089 protein |
| WO2010094720A2 (en) | 2009-02-19 | 2010-08-26 | Glaxo Group Limited | Improved anti-tnfr1 polypeptides, antibody variable domains & antagonists |
| US7785903B2 (en) | 2004-04-09 | 2010-08-31 | Genentech, Inc. | Variable domain library and uses |
| WO2010102175A1 (en) | 2009-03-05 | 2010-09-10 | Medarex, Inc. | Fully human antibodies specific to cadm1 |
| EP2254909A1 (en) * | 2007-12-21 | 2010-12-01 | National Research Council of Canada | NON-AGGREGATING HUMAN Vh DOMAINS |
| WO2011006914A2 (en) | 2009-07-16 | 2011-01-20 | Glaxo Group Limited | Antagonists, uses & methods for partially inhibiting tnfr1 |
| WO2011012609A2 (en) | 2009-07-29 | 2011-02-03 | Glaxo Group Limited | Ligands that bind tgf-beta receptor rii |
| WO2011017835A1 (en) * | 2009-08-11 | 2011-02-17 | Nanjing University | Preparation method of protein or peptide nanoparticles for in vivo drug delivery by unfolding and refolding |
| WO2011023787A1 (en) * | 2009-08-31 | 2011-03-03 | Roche Glycart Ag | Affinity-matured humanized anti cea monoclonal antibodies |
| WO2011047083A1 (en) | 2009-10-13 | 2011-04-21 | Oxford Biotherapeutics Ltd. | Antibodies against epha10 |
| WO2011047442A1 (en) * | 2009-10-23 | 2011-04-28 | Garvan Institute Of Medical Research | Modified variable domain molecules and methods for producing and using same |
| WO2011054007A1 (en) | 2009-11-02 | 2011-05-05 | Oxford Biotherapeutics Ltd. | Ror1 as therapeutic and diagnostic target |
| WO2011051217A1 (en) | 2009-10-27 | 2011-05-05 | Glaxo Group Limited | Stable anti-tnfr1 polypeptides, antibody variable domains & antagonists |
| WO2011054893A2 (en) | 2009-11-05 | 2011-05-12 | Novartis Ag | Biomarkers predictive of progression of fibrosis |
| WO2011086143A2 (en) | 2010-01-14 | 2011-07-21 | Glaxo Group Limited | Liver targeting molecules |
| WO2011098449A1 (en) | 2010-02-10 | 2011-08-18 | Novartis Ag | Methods and compounds for muscle growth |
| WO2011131659A2 (en) | 2010-04-21 | 2011-10-27 | Glaxo Group Limited | Binding domains |
| WO2011138392A1 (en) | 2010-05-06 | 2011-11-10 | Novartis Ag | Compositions and methods of use for therapeutic low density lipoprotein -related protein 6 (lrp6) antibodies |
| EP2420251A2 (en) | 2004-11-10 | 2012-02-22 | Domantis Limited | Ligands that enhance endogenous compounds |
| WO2012022814A1 (en) | 2010-08-20 | 2012-02-23 | Novartis Ag | Antibodies for epidermal growth factor receptor 3 (her3) |
| WO2012032433A1 (en) | 2010-09-09 | 2012-03-15 | Pfizer Inc. | 4-1bb binding molecules |
| EP2441775A1 (en) | 2007-02-26 | 2012-04-18 | Oxford Biotherapeutics Ltd. | Protein |
| EP2447719A1 (en) | 2007-02-26 | 2012-05-02 | Oxford Biotherapeutics Ltd. | Proteins |
| US8188249B2 (en) | 2003-09-10 | 2012-05-29 | Amgen Fremont Inc. | Nucleic acid molecules encoding antibodies to M-CSF |
| WO2012093125A1 (en) | 2011-01-06 | 2012-07-12 | Glaxo Group Limited | Ligands that bind tgf-beta receptor ii |
| WO2012104227A1 (en) | 2011-02-02 | 2012-08-09 | Glaxo Group Limited | Novel antigen binding proteins |
| EP2486941A1 (en) | 2006-10-02 | 2012-08-15 | Medarex, Inc. | Human antibodies that bind CXCR4 and uses thereof |
| WO2012142662A1 (en) | 2011-04-21 | 2012-10-26 | Garvan Institute Of Medical Research | Modified variable domain molecules and methods for producing and using them b |
| GB2490404A (en) * | 2011-04-18 | 2012-10-31 | Evitraproteoma Ab | Methods for determining ligand binding to a target protein using a thermal shift assay |
| WO2013001369A2 (en) | 2011-06-28 | 2013-01-03 | Oxford Biotherapeutics Ltd. | Therapeutic and diagnostic target |
| WO2013003625A2 (en) | 2011-06-28 | 2013-01-03 | Oxford Biotherapeutics Ltd. | Antibodies |
| WO2013041643A1 (en) * | 2011-09-22 | 2013-03-28 | Bioinvent International Ab | Screening methods and uses thereof |
| WO2013067355A1 (en) | 2011-11-04 | 2013-05-10 | Novartis Ag | Low density lipoprotein-related protein 6 (lrp6) - half life extender constructs |
| EP2594590A1 (en) | 2007-12-14 | 2013-05-22 | Bristol-Myers Squibb Company | Binding molecules to the human OX40 receptor |
| WO2013084148A2 (en) | 2011-12-05 | 2013-06-13 | Novartis Ag | Antibodies for epidermal growth factor receptor 3 (her3) directed to domain ii of her3 |
| WO2013084147A2 (en) | 2011-12-05 | 2013-06-13 | Novartis Ag | Antibodies for epidermal growth factor receptor 3 (her3) |
| WO2013171810A1 (en) * | 2012-05-14 | 2013-11-21 | パナソニック株式会社 | Method for obtaining heat-resistant antibody-presenting phage |
| US8642742B2 (en) | 2011-03-02 | 2014-02-04 | Roche Glycart Ag | Anti-CEA antibodies |
| WO2014020331A1 (en) | 2012-08-01 | 2014-02-06 | Oxford Biotherapeutics Ltd. | Therapeutic and diagnostic target |
| EP2740743A2 (en) | 2004-06-01 | 2014-06-11 | Domantis Limited | Bispecific fusion antibodies with enhanced serum half-life |
| EP2769990A2 (en) | 2004-12-02 | 2014-08-27 | Domantis Limited | Bispecific domain antibodies targeting serum albumin and GLP-1 or PYY |
| WO2014133855A1 (en) | 2013-02-28 | 2014-09-04 | Caprion Proteomics Inc. | Tuberculosis biomarkers and uses thereof |
| CN104039809A (en) * | 2011-10-10 | 2014-09-10 | 希望之城公司 | Meditopes and meditope-binding antibodies and uses thereof |
| WO2014159239A2 (en) | 2013-03-14 | 2014-10-02 | Novartis Ag | Antibodies against notch 3 |
| WO2015015401A2 (en) | 2013-08-02 | 2015-02-05 | Pfizer Inc. | Anti-cxcr4 antibodies and antibody-drug conjugates |
| US9028822B2 (en) | 2002-06-28 | 2015-05-12 | Domantis Limited | Antagonists against TNFR1 and methods of use therefor |
| US9179691B2 (en) | 2007-12-14 | 2015-11-10 | Aerodesigns, Inc. | Delivering aerosolizable food products |
| EP3009454A2 (en) | 2009-04-20 | 2016-04-20 | Oxford Bio Therapeutics Limited | Antibodies specific to cadherin-17 |
| US9321832B2 (en) | 2002-06-28 | 2016-04-26 | Domantis Limited | Ligand |
| US9428553B2 (en) | 2012-02-10 | 2016-08-30 | City Of Hope | Meditopes and meditope-binding antibodies and uses thereof |
| US9523693B2 (en) | 2011-04-18 | 2016-12-20 | Biotarget Engagement Interest Group Ab | Methods for determining ligand binding to a target protein using a thermal shift assay |
| US9669108B2 (en) | 2010-10-08 | 2017-06-06 | City Of Hope | Meditopes and meditope-binding antibodies and uses thereof |
| EP3124497A3 (en) * | 2007-09-14 | 2017-06-14 | Adimab, LLC | Rationally designed, synthetic antibody libraries and uses therefor |
| WO2018020000A1 (en) | 2016-07-29 | 2018-02-01 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Antibodies targeting tumor associated macrophages and uses thereof |
| WO2018158398A1 (en) | 2017-03-02 | 2018-09-07 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Antibodies having specificity to nectin-4 and uses thereof |
| WO2019020480A1 (en) | 2017-07-24 | 2019-01-31 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Antibodies and peptides to treat hcmv related diseases |
| US10196635B2 (en) | 2007-09-14 | 2019-02-05 | Adimab, Llc | Rationally designed, synthetic antibody libraries and uses therefor |
| US10316104B2 (en) | 2011-04-29 | 2019-06-11 | Roche Glycart Ag | Immunoconjugates |
| US10329555B2 (en) | 2002-08-12 | 2019-06-25 | Adimab, Llc | High throughput generation and affinity maturation of humanized antibody |
| US10351970B2 (en) | 2002-09-04 | 2019-07-16 | Bioinvent International Ab | Method for screening anti-ligand libraries for identifying anti-ligands specific for differentially and infrequently expressed ligands |
| WO2020010250A2 (en) | 2018-07-03 | 2020-01-09 | Elstar Therapeutics, Inc. | Anti-tcr antibody molecules and uses thereof |
| WO2020053122A1 (en) | 2018-09-10 | 2020-03-19 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Combination of her2/neu antibody with heme for treating cancer |
| WO2020193520A1 (en) | 2019-03-25 | 2020-10-01 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Treatment of taupathy disorders by targeting new tau species |
| EP3736293A1 (en) | 2013-02-12 | 2020-11-11 | Boehringer Ingelheim International Gmbh | Therapeutic and diagnostic target for cancer comprising dll3 binding reagents |
| WO2021058763A1 (en) | 2019-09-27 | 2021-04-01 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Anti-müllerian inhibiting substance antibodies and uses thereof |
| WO2021058729A1 (en) | 2019-09-27 | 2021-04-01 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Anti-müllerian inhibiting substance type i receptor antibodies and uses thereof |
| WO2021116119A1 (en) | 2019-12-09 | 2021-06-17 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Antibodies having specificity to her4 and uses thereof |
| WO2021228956A1 (en) | 2020-05-12 | 2021-11-18 | INSERM (Institut National de la Santé et de la Recherche Médicale) | New method to treat cutaneous t-cell lymphomas and tfh derived lymphomas |
| US11242395B2 (en) | 2017-08-21 | 2022-02-08 | Adagene Inc. | Anti-CD137 molecules and use thereof |
| US11359016B2 (en) | 2018-02-02 | 2022-06-14 | Adagene Inc. | Anti-CTLA4 antibodies and methods of making and using the same |
| WO2022216993A2 (en) | 2021-04-08 | 2022-10-13 | Marengo Therapeutics, Inc. | Multifuntional molecules binding to tcr and uses thereof |
| WO2023175171A1 (en) | 2022-03-18 | 2023-09-21 | Inserm (Institut National De La Sante Et De La Recherche Medicale) | Bk polyomavirus antibodies and uses thereof |
| WO2024052503A1 (en) | 2022-09-08 | 2024-03-14 | Institut National de la Santé et de la Recherche Médicale | Antibodies having specificity to ltbp2 and uses thereof |
| WO2024056668A1 (en) | 2022-09-12 | 2024-03-21 | Institut National de la Santé et de la Recherche Médicale | New anti-itgb8 antibodies and its uses thereof |
| US11952681B2 (en) | 2018-02-02 | 2024-04-09 | Adagene Inc. | Masked activatable CD137 antibodies |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DK1994155T4 (en) | 2006-02-13 | 2022-07-25 | Daiichi Sankyo Co Ltd | POLYNUCLEOTIDE AND POLYPEPTIDE SEQUENCES INVOLVED IN THE BONE MODELING PROCESS |
| US8168181B2 (en) | 2006-02-13 | 2012-05-01 | Alethia Biotherapeutics, Inc. | Methods of impairing osteoclast differentiation using antibodies that bind siglec-15 |
| US8048420B2 (en) | 2007-06-12 | 2011-11-01 | Ac Immune S.A. | Monoclonal antibody |
| US8613923B2 (en) | 2007-06-12 | 2013-12-24 | Ac Immune S.A. | Monoclonal antibody |
| AU2008311367B2 (en) | 2007-10-05 | 2014-11-13 | Ac Immune S.A. | Use of anti-amyloid beta antibody in ocular diseases |
| WO2011020183A1 (en) * | 2009-08-18 | 2011-02-24 | National Research Council Canada | Screening of protein candidates |
| CA2774032C (en) * | 2009-10-23 | 2019-03-26 | Millennium Pharmaceuticals, Inc. | Anti-gcc antibody molecules and related compositions and methods |
| CN103179981B (en) | 2010-07-30 | 2017-02-08 | Ac免疫有限公司 | Safe and functional humanized antibodies |
| CN103501815B (en) | 2010-10-08 | 2016-09-28 | 希望之城公司 | Monoclonal antibody framework combination interface, interposition delivery system and using method thereof for interposition |
| SG11201406855TA (en) | 2012-04-27 | 2014-11-27 | Millennium Pharm Inc | Anti-gcc antibody molecules and use of same to test for susceptibility to gcc-targeted therapy |
| ES2723885T3 (en) | 2012-07-19 | 2019-09-03 | Daiichi Sankyo Co Ltd | Anti-Siglec-15 antibodies |
| US11473081B2 (en) | 2016-12-12 | 2022-10-18 | xCella Biosciences, Inc. | Methods and systems for screening using microcapillary arrays |
| JP2021506976A (en) * | 2017-12-18 | 2021-02-22 | ディストリビューテッド バイオ, インコーポレイテッド | Fundamentally diverse human antibody library |
| US20210230580A1 (en) * | 2018-08-08 | 2021-07-29 | Serimmune Inc. | Quality control reagents and methods for serum antibody profiling |
| KR20220163940A (en) * | 2020-02-04 | 2022-12-12 | 시애틀 칠드런즈 호스피탈 디/비/에이 시애틀 칠드런즈 리서치 인스티튜트 | Anti-dinitrophenol Chimeric Antigen Receptor |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001018058A2 (en) * | 1999-09-07 | 2001-03-15 | Viventia Biotech Inc. | Enhanced phage display library of human vh fragments and methods for producing same |
| WO2001027279A1 (en) * | 1999-10-12 | 2001-04-19 | Cambridge Antibody Technology | Human anti-adipocyte monoclonal antibodies and their use |
| US20020012909A1 (en) * | 1998-11-11 | 2002-01-31 | Daniel Plaksin | Small functional units of antibody heavy chain variable regions |
Family Cites Families (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU4308689A (en) | 1988-09-02 | 1990-04-02 | Protein Engineering Corporation | Generation and selection of recombinant varied binding proteins |
| US5169936A (en) | 1989-04-14 | 1992-12-08 | Biogen, Inc. | Protein purification on immobilized metal affinity resins effected by elution using a weak ligand |
| US5143854A (en) | 1989-06-07 | 1992-09-01 | Affymax Technologies N.V. | Large scale photolithographic solid phase synthesis of polypeptides and receptor binding screening thereof |
| GB9015198D0 (en) | 1990-07-10 | 1990-08-29 | Brien Caroline J O | Binding substance |
| US6172197B1 (en) | 1991-07-10 | 2001-01-09 | Medical Research Council | Methods for producing members of specific binding pairs |
| ATE158021T1 (en) | 1990-08-29 | 1997-09-15 | Genpharm Int | PRODUCTION AND USE OF NON-HUMAN TRANSGENT ANIMALS FOR THE PRODUCTION OF HETEROLOGUE ANTIBODIES |
| EP0562025B1 (en) | 1990-12-06 | 2001-02-07 | Affymetrix, Inc. (a Delaware Corporation) | Compounds and their use in a binary synthesis strategy |
| PT1024191E (en) | 1991-12-02 | 2008-12-22 | Medical Res Council | Production of anti-self antibodies from antibody segment repertoires and displayed on phage |
| US5733743A (en) | 1992-03-24 | 1998-03-31 | Cambridge Antibody Technology Limited | Methods for producing members of specific binding pairs |
| WO1994026087A2 (en) | 1993-05-14 | 1994-11-24 | Connor Kim C O | Recombinant protein production and insect cell culture and process |
| US5702892A (en) | 1995-05-09 | 1997-12-30 | The United States Of America As Represented By The Department Of Health And Human Services | Phage-display of immunoglobulin heavy chain libraries |
| CA2229043C (en) | 1995-08-18 | 2016-06-07 | Morphosys Gesellschaft Fur Proteinoptimierung Mbh | Protein/(poly)peptide libraries |
| EP1496120B1 (en) | 1997-07-07 | 2007-03-28 | Medical Research Council | In vitro sorting method |
| GB9722131D0 (en) | 1997-10-20 | 1997-12-17 | Medical Res Council | Method |
| DK1078051T3 (en) | 1998-05-13 | 2008-04-07 | Domantis Ltd | Phage display selection system for selection of properly folded proteins |
| EP1252319A2 (en) | 2000-02-03 | 2002-10-30 | Domantis Limited | Combinatorial protein domains |
| GB0025144D0 (en) | 2000-10-13 | 2000-11-29 | Medical Res Council | Concatenated nucleic acid sequences |
| DE60237282D1 (en) * | 2001-06-28 | 2010-09-23 | Domantis Ltd | DOUBLE-SPECIFIC LIGAND AND ITS USE |
-
2004
- 2004-05-14 WO PCT/GB2004/002102 patent/WO2004101790A1/en active Application Filing
- 2004-05-14 US US10/556,360 patent/US20090005257A1/en not_active Abandoned
- 2004-05-14 JP JP2006530506A patent/JP4889493B2/en not_active Expired - Fee Related
- 2004-05-14 AU AU2004239065A patent/AU2004239065B2/en not_active Ceased
- 2004-05-14 EP EP10180258A patent/EP2357237A1/en not_active Withdrawn
- 2004-05-14 CA CA2525120A patent/CA2525120C/en not_active Expired - Fee Related
- 2004-05-14 EP EP04733030A patent/EP1627062A1/en not_active Withdrawn
-
2010
- 2010-12-28 JP JP2010292138A patent/JP5049383B2/en not_active Expired - Fee Related
-
2011
- 2011-05-06 AU AU2011202105A patent/AU2011202105A1/en not_active Abandoned
- 2011-11-10 JP JP2011246170A patent/JP2012070743A/en active Pending
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20020012909A1 (en) * | 1998-11-11 | 2002-01-31 | Daniel Plaksin | Small functional units of antibody heavy chain variable regions |
| WO2001018058A2 (en) * | 1999-09-07 | 2001-03-15 | Viventia Biotech Inc. | Enhanced phage display library of human vh fragments and methods for producing same |
| WO2001027279A1 (en) * | 1999-10-12 | 2001-04-19 | Cambridge Antibody Technology | Human anti-adipocyte monoclonal antibodies and their use |
Non-Patent Citations (4)
| Title |
|---|
| DUMOULIN MIREILLE ET AL: "Single-domain antibody fragments with high conformational stability.", PROTEIN SCIENCE : A PUBLICATION OF THE PROTEIN SOCIETY. MAR 2002, vol. 11, no. 3, March 2002 (2002-03-01), pages 500 - 515, XP002296277, ISSN: 0961-8368 * |
| HOOGENBOOM H R ET AL: "Antibody phage display technology and its applications", IMMUNOTECHNOLOGY, ELSEVIER SCIENCE PUBLISHERS BV, NL, vol. 4, no. 1, 1 June 1998 (1998-06-01), pages 1 - 20, XP004127382, ISSN: 1380-2933 * |
| PEDROSO IDOLKA ET AL: "Four-state equilibrium unfolding of an scFv antibody fragment.", BIOCHEMISTRY. 6 AUG 2002, vol. 41, no. 31, 6 August 2002 (2002-08-06), pages 9873 - 9884, XP002296278, ISSN: 0006-2960 * |
| REITER Y ET AL: "An antibody single-domain phage display library of a native heavy chain variable region: isolation of functional single-domain VH molecules with a unique interface", JOURNAL OF MOLECULAR BIOLOGY, LONDON, GB, vol. 290, no. 3, 1 January 1999 (1999-01-01), pages 685 - 698, XP004461990, ISSN: 0022-2836 * |
Cited By (141)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9321832B2 (en) | 2002-06-28 | 2016-04-26 | Domantis Limited | Ligand |
| US9028822B2 (en) | 2002-06-28 | 2015-05-12 | Domantis Limited | Antagonists against TNFR1 and methods of use therefor |
| US10329555B2 (en) | 2002-08-12 | 2019-06-25 | Adimab, Llc | High throughput generation and affinity maturation of humanized antibody |
| US10351970B2 (en) | 2002-09-04 | 2019-07-16 | Bioinvent International Ab | Method for screening anti-ligand libraries for identifying anti-ligands specific for differentially and infrequently expressed ligands |
| US8188249B2 (en) | 2003-09-10 | 2012-05-29 | Amgen Fremont Inc. | Nucleic acid molecules encoding antibodies to M-CSF |
| US10280219B2 (en) | 2003-09-10 | 2019-05-07 | Amgen Fremont Inc. | Antibodies to M-CSF |
| US9718883B2 (en) | 2003-09-10 | 2017-08-01 | Amgen Fremont Inc. | Antibodies to M-CSF |
| US7785903B2 (en) | 2004-04-09 | 2010-08-31 | Genentech, Inc. | Variable domain library and uses |
| EP2740743A2 (en) | 2004-06-01 | 2014-06-11 | Domantis Limited | Bispecific fusion antibodies with enhanced serum half-life |
| EP2371390A3 (en) * | 2004-10-08 | 2013-03-13 | Domantis Limited | Antagonists and methods of use therefor |
| WO2006038027A3 (en) * | 2004-10-08 | 2007-01-04 | Domantis Ltd | SINGLE DOMAIN ANTIBODIES AGAINST TNFRl AND METHODS OF USE THEREFOR |
| EP2420251A2 (en) | 2004-11-10 | 2012-02-22 | Domantis Limited | Ligands that enhance endogenous compounds |
| EP2769990A2 (en) | 2004-12-02 | 2014-08-27 | Domantis Limited | Bispecific domain antibodies targeting serum albumin and GLP-1 or PYY |
| JP2009519720A (en) * | 2005-12-20 | 2009-05-21 | アラーナ・テラピューティクス・リミテッド | Anti-inflammatory domain antibody (dAb) |
| JP2009519983A (en) * | 2005-12-20 | 2009-05-21 | アラーナ・テラピューティクス・リミテッド | Chimeric antibodies with partial New World monkey binding regions |
| WO2007141280A2 (en) | 2006-06-06 | 2007-12-13 | Oxford Genome Sciences (Uk) Ltd | Proteins |
| EP2375255A1 (en) | 2006-06-06 | 2011-10-12 | Oxford Biotherapeutics Ltd. | Proteins |
| WO2008030611A2 (en) | 2006-09-05 | 2008-03-13 | Medarex, Inc. | Antibodies to bone morphogenic proteins and receptors therefor and methods for their use |
| EP2486941A1 (en) | 2006-10-02 | 2012-08-15 | Medarex, Inc. | Human antibodies that bind CXCR4 and uses thereof |
| US8236931B2 (en) | 2006-10-30 | 2012-08-07 | Glaxo Group Limited | Prevention of aggregation of immunoglobulin light or heavy chains |
| JP2010508016A (en) * | 2006-10-30 | 2010-03-18 | ドマンティス リミテッド | Novel polypeptides and their uses |
| WO2008052933A3 (en) * | 2006-10-30 | 2008-11-13 | Domantis Ltd | Prevention of aggregation of immunoglobulin light or heavy chains |
| WO2008052933A2 (en) * | 2006-10-30 | 2008-05-08 | Domantis Limited | Prevention of aggregation of immunoglobulin light or heavy chains |
| WO2008070569A2 (en) | 2006-12-01 | 2008-06-12 | Medarex, Inc. | Human antibodies that bind cd22 and uses thereof |
| WO2009054863A2 (en) | 2006-12-13 | 2009-04-30 | Medarex, Inc. | Human antibodies that bind cd19 and uses thereof |
| WO2008074004A2 (en) | 2006-12-14 | 2008-06-19 | Medarex, Inc. | Human antibodies that bind cd70 and uses thereof |
| EP2441775A1 (en) | 2007-02-26 | 2012-04-18 | Oxford Biotherapeutics Ltd. | Protein |
| EP2447719A1 (en) | 2007-02-26 | 2012-05-02 | Oxford Biotherapeutics Ltd. | Proteins |
| US10196635B2 (en) | 2007-09-14 | 2019-02-05 | Adimab, Llc | Rationally designed, synthetic antibody libraries and uses therefor |
| US11008383B2 (en) | 2007-09-14 | 2021-05-18 | Adimab, Llc | Rationally designed, synthetic antibody libraries and uses therefor |
| US11008568B2 (en) | 2007-09-14 | 2021-05-18 | Adimab, Llc | Rationally designed, synthetic antibody libraries and uses therefor |
| EP3124497A3 (en) * | 2007-09-14 | 2017-06-14 | Adimab, LLC | Rationally designed, synthetic antibody libraries and uses therefor |
| US10189894B2 (en) | 2007-09-14 | 2019-01-29 | Adimab, Llc | Rationally designed, synthetic antibody libraries and uses therefor |
| EP2594590A1 (en) | 2007-12-14 | 2013-05-22 | Bristol-Myers Squibb Company | Binding molecules to the human OX40 receptor |
| EP3239178A1 (en) | 2007-12-14 | 2017-11-01 | Bristol-Myers Squibb Company | Binding molecules to the human ox40 receptor |
| EP2851374A1 (en) | 2007-12-14 | 2015-03-25 | Bristol-Myers Squibb Company | Binding molecules to the human OX40 receptor |
| US9179691B2 (en) | 2007-12-14 | 2015-11-10 | Aerodesigns, Inc. | Delivering aerosolizable food products |
| US20110052565A1 (en) * | 2007-12-21 | 2011-03-03 | National Research Council Of Canada | Non-aggregating human vh domains |
| EP2254909A1 (en) * | 2007-12-21 | 2010-12-01 | National Research Council of Canada | NON-AGGREGATING HUMAN Vh DOMAINS |
| EP2254909A4 (en) * | 2007-12-21 | 2012-05-02 | Ca Nat Research Council | NON-AGGREGATING HUMAN Vh DOMAINS |
| EP3629022A1 (en) | 2008-07-25 | 2020-04-01 | Richard W. Wagner | Protein screening methods |
| WO2010011944A2 (en) | 2008-07-25 | 2010-01-28 | Wagner Richard W | Protein screeing methods |
| CN102257009A (en) * | 2008-10-21 | 2011-11-23 | 杜门蒂斯有限公司 | Ligands that have binding specificity for dc-sign |
| WO2010046337A2 (en) * | 2008-10-21 | 2010-04-29 | Domantis Limited | Ligands that have binding specificity for dc-sign |
| WO2010046337A3 (en) * | 2008-10-21 | 2010-07-01 | Domantis Limited | Ligands that have binding specificity for dc-sign |
| WO2010084408A2 (en) | 2009-01-21 | 2010-07-29 | Oxford Biotherapeutics Ltd. | Pta089 protein |
| WO2010085590A1 (en) | 2009-01-23 | 2010-07-29 | Biosynexus Incorporated | Opsonic and protective antibodies specific for lipoteichoic acid gram positive bacteria |
| WO2010094720A2 (en) | 2009-02-19 | 2010-08-26 | Glaxo Group Limited | Improved anti-tnfr1 polypeptides, antibody variable domains & antagonists |
| WO2010102175A1 (en) | 2009-03-05 | 2010-09-10 | Medarex, Inc. | Fully human antibodies specific to cadm1 |
| EP3009454A2 (en) | 2009-04-20 | 2016-04-20 | Oxford Bio Therapeutics Limited | Antibodies specific to cadherin-17 |
| WO2011006914A2 (en) | 2009-07-16 | 2011-01-20 | Glaxo Group Limited | Antagonists, uses & methods for partially inhibiting tnfr1 |
| WO2011012609A2 (en) | 2009-07-29 | 2011-02-03 | Glaxo Group Limited | Ligands that bind tgf-beta receptor rii |
| WO2011017835A1 (en) * | 2009-08-11 | 2011-02-17 | Nanjing University | Preparation method of protein or peptide nanoparticles for in vivo drug delivery by unfolding and refolding |
| US9068008B2 (en) | 2009-08-31 | 2015-06-30 | Roche Glycart Ag | Antibodies to carcinoembryonic antigen (CEA), methods of making same, and uses thereof |
| CN102741293A (en) * | 2009-08-31 | 2012-10-17 | 罗切格利卡特公司 | Affinity-matured humanized anti cea monoclonal antibodies |
| WO2011023787A1 (en) * | 2009-08-31 | 2011-03-03 | Roche Glycart Ag | Affinity-matured humanized anti cea monoclonal antibodies |
| WO2011047083A1 (en) | 2009-10-13 | 2011-04-21 | Oxford Biotherapeutics Ltd. | Antibodies against epha10 |
| WO2011047442A1 (en) * | 2009-10-23 | 2011-04-28 | Garvan Institute Of Medical Research | Modified variable domain molecules and methods for producing and using same |
| WO2011051217A1 (en) | 2009-10-27 | 2011-05-05 | Glaxo Group Limited | Stable anti-tnfr1 polypeptides, antibody variable domains & antagonists |
| WO2011054007A1 (en) | 2009-11-02 | 2011-05-05 | Oxford Biotherapeutics Ltd. | Ror1 as therapeutic and diagnostic target |
| WO2011054893A2 (en) | 2009-11-05 | 2011-05-12 | Novartis Ag | Biomarkers predictive of progression of fibrosis |
| WO2011086143A2 (en) | 2010-01-14 | 2011-07-21 | Glaxo Group Limited | Liver targeting molecules |
| WO2011098449A1 (en) | 2010-02-10 | 2011-08-18 | Novartis Ag | Methods and compounds for muscle growth |
| WO2011131659A2 (en) | 2010-04-21 | 2011-10-27 | Glaxo Group Limited | Binding domains |
| EP4234698A2 (en) | 2010-05-06 | 2023-08-30 | Novartis AG | Compositions and methods of use for therapeutic low density lipoprotein-related protein 6 (lrp6) antibodies |
| EP3345926A1 (en) | 2010-05-06 | 2018-07-11 | Novartis AG | Compositions and methods of use for therapeutic low density lipoprotein-related protein 6 (lrp6) antibodies |
| WO2011138392A1 (en) | 2010-05-06 | 2011-11-10 | Novartis Ag | Compositions and methods of use for therapeutic low density lipoprotein -related protein 6 (lrp6) antibodies |
| WO2012022814A1 (en) | 2010-08-20 | 2012-02-23 | Novartis Ag | Antibodies for epidermal growth factor receptor 3 (her3) |
| US8821867B2 (en) | 2010-09-09 | 2014-09-02 | Pfizer Inc | 4-1BB binding molecules |
| EP3441404A1 (en) | 2010-09-09 | 2019-02-13 | Pfizer Inc | 4-1bb binding molecules |
| US8337850B2 (en) | 2010-09-09 | 2012-12-25 | Pfizer Inc. | 4-1BB binding molecules |
| WO2012032433A1 (en) | 2010-09-09 | 2012-03-15 | Pfizer Inc. | 4-1bb binding molecules |
| US10640568B2 (en) | 2010-09-09 | 2020-05-05 | Pfizer Inc. | 4-1BB binding molecules |
| US9468678B2 (en) | 2010-09-09 | 2016-10-18 | Pfizer Inc. | Method of producing 4-1BB binding molecules and associated nucleic acids |
| US9669108B2 (en) | 2010-10-08 | 2017-06-06 | City Of Hope | Meditopes and meditope-binding antibodies and uses thereof |
| US11246942B2 (en) | 2010-10-08 | 2022-02-15 | City Of Hope | Meditopes and meditope-binding antibodies and uses thereof |
| WO2012093125A1 (en) | 2011-01-06 | 2012-07-12 | Glaxo Group Limited | Ligands that bind tgf-beta receptor ii |
| WO2012104227A1 (en) | 2011-02-02 | 2012-08-09 | Glaxo Group Limited | Novel antigen binding proteins |
| US8642742B2 (en) | 2011-03-02 | 2014-02-04 | Roche Glycart Ag | Anti-CEA antibodies |
| US9206260B2 (en) | 2011-03-02 | 2015-12-08 | Roche Glycart Ag | Anti-CEA antibodies |
| GB2490404A (en) * | 2011-04-18 | 2012-10-31 | Evitraproteoma Ab | Methods for determining ligand binding to a target protein using a thermal shift assay |
| US9528996B2 (en) | 2011-04-18 | 2016-12-27 | Biotarget Engagement Interest Group Ab | Methods for determining ligand binding to a target protein using a thermal shift assay |
| GB2490404B (en) * | 2011-04-18 | 2013-11-06 | Evitraproteoma Ab | Methods for determining ligand binding to a target protein using a thermal shift assay |
| US8969014B2 (en) | 2011-04-18 | 2015-03-03 | Evitraproteoma Ab | Methods for determining ligand binding to a target protein using a thermal shift assay |
| US9523693B2 (en) | 2011-04-18 | 2016-12-20 | Biotarget Engagement Interest Group Ab | Methods for determining ligand binding to a target protein using a thermal shift assay |
| EP3103810A3 (en) * | 2011-04-21 | 2017-03-08 | Garvan Institute of Medical Research | Modified variable domain molecules and methods for producing and using them |
| US9527908B2 (en) * | 2011-04-21 | 2016-12-27 | Garvan Institute Of Medical Research | Modified variable domain molecules and methods for producing them |
| EP2699597A1 (en) * | 2011-04-21 | 2014-02-26 | Garvan Institute of Medical Research | Modified variable domain molecules and methods for producing and using them b |
| WO2012142662A1 (en) | 2011-04-21 | 2012-10-26 | Garvan Institute Of Medical Research | Modified variable domain molecules and methods for producing and using them b |
| US11332522B2 (en) | 2011-04-21 | 2022-05-17 | Garvan Institute Of Medical Research | Modified variable domain molecules and methods for producing them |
| EP2699597A4 (en) * | 2011-04-21 | 2014-10-01 | Garvan Inst Med Res | Modified variable domain molecules and methods for producing and using them b |
| US20140234218A1 (en) * | 2011-04-21 | 2014-08-21 | Garvan Institute Of Medical Research | Modified variable domain molecules and methods for producing them b |
| US11130822B2 (en) | 2011-04-29 | 2021-09-28 | Roche Glycart Ag | Immunoconjugates |
| US10316104B2 (en) | 2011-04-29 | 2019-06-11 | Roche Glycart Ag | Immunoconjugates |
| WO2013003625A2 (en) | 2011-06-28 | 2013-01-03 | Oxford Biotherapeutics Ltd. | Antibodies |
| WO2013001369A2 (en) | 2011-06-28 | 2013-01-03 | Oxford Biotherapeutics Ltd. | Therapeutic and diagnostic target |
| US10191049B2 (en) | 2011-09-22 | 2019-01-29 | Bioinvent International Ab | Screening methods and uses thereof |
| RU2622089C2 (en) * | 2011-09-22 | 2017-06-09 | Биоинвент Интернэшнл Аб | Screening methods and their application |
| AU2012311542B2 (en) * | 2011-09-22 | 2017-09-21 | Bioinvent International Ab | Screening methods and uses thereof |
| WO2013041643A1 (en) * | 2011-09-22 | 2013-03-28 | Bioinvent International Ab | Screening methods and uses thereof |
| CN104039809A (en) * | 2011-10-10 | 2014-09-10 | 希望之城公司 | Meditopes and meditope-binding antibodies and uses thereof |
| EP3252075A1 (en) | 2011-11-04 | 2017-12-06 | Novartis AG | Low density lipoprotein-related protein 6 (lrp6) - half life extender constructs |
| WO2013067355A1 (en) | 2011-11-04 | 2013-05-10 | Novartis Ag | Low density lipoprotein-related protein 6 (lrp6) - half life extender constructs |
| EP3290442A1 (en) | 2011-11-04 | 2018-03-07 | Novartis AG | Low density lipoprotein-related protein 6 (lrp6) half-life extender constructs |
| WO2013084147A2 (en) | 2011-12-05 | 2013-06-13 | Novartis Ag | Antibodies for epidermal growth factor receptor 3 (her3) |
| WO2013084148A2 (en) | 2011-12-05 | 2013-06-13 | Novartis Ag | Antibodies for epidermal growth factor receptor 3 (her3) directed to domain ii of her3 |
| EP3590538A1 (en) | 2011-12-05 | 2020-01-08 | Novartis AG | Antibodies for epidermal growth factor receptor 3 (her3) |
| US9428553B2 (en) | 2012-02-10 | 2016-08-30 | City Of Hope | Meditopes and meditope-binding antibodies and uses thereof |
| WO2013171810A1 (en) * | 2012-05-14 | 2013-11-21 | パナソニック株式会社 | Method for obtaining heat-resistant antibody-presenting phage |
| US9506054B2 (en) | 2012-05-14 | 2016-11-29 | Panasonic Intellectual Property Management Co., Ltd. | Method for acquiring a heat-stable antibody-displayed phage |
| WO2014020331A1 (en) | 2012-08-01 | 2014-02-06 | Oxford Biotherapeutics Ltd. | Therapeutic and diagnostic target |
| EP3736293A1 (en) | 2013-02-12 | 2020-11-11 | Boehringer Ingelheim International Gmbh | Therapeutic and diagnostic target for cancer comprising dll3 binding reagents |
| WO2014133855A1 (en) | 2013-02-28 | 2014-09-04 | Caprion Proteomics Inc. | Tuberculosis biomarkers and uses thereof |
| WO2014159239A2 (en) | 2013-03-14 | 2014-10-02 | Novartis Ag | Antibodies against notch 3 |
| EP3611189A1 (en) | 2013-03-14 | 2020-02-19 | Novartis AG | Antibodies against notch 3 |
| EP4050033A1 (en) | 2013-08-02 | 2022-08-31 | Pfizer Inc. | Anti-cxcr4 antibodies and antibody-drug conjugates |
| US9708405B2 (en) | 2013-08-02 | 2017-07-18 | Pfizer Inc. | Anti-CXCR4 antibodies and antibody-drug conjugates |
| US10144781B2 (en) | 2013-08-02 | 2018-12-04 | Pfizer Inc. | Anti-CXCR4 antibodies and antibody-drug conjugates |
| WO2015015401A2 (en) | 2013-08-02 | 2015-02-05 | Pfizer Inc. | Anti-cxcr4 antibodies and antibody-drug conjugates |
| WO2018020000A1 (en) | 2016-07-29 | 2018-02-01 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Antibodies targeting tumor associated macrophages and uses thereof |
| WO2018158398A1 (en) | 2017-03-02 | 2018-09-07 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Antibodies having specificity to nectin-4 and uses thereof |
| WO2019020480A1 (en) | 2017-07-24 | 2019-01-31 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Antibodies and peptides to treat hcmv related diseases |
| US11242395B2 (en) | 2017-08-21 | 2022-02-08 | Adagene Inc. | Anti-CD137 molecules and use thereof |
| US11859003B2 (en) | 2017-08-21 | 2024-01-02 | Adagene Inc. | Method for treating cancer using anti-CD137 antibody |
| US11692036B2 (en) | 2018-02-02 | 2023-07-04 | Adagene Inc. | Anti-CTLA4 antibodies and methods of making and using the same |
| US11359016B2 (en) | 2018-02-02 | 2022-06-14 | Adagene Inc. | Anti-CTLA4 antibodies and methods of making and using the same |
| US11952681B2 (en) | 2018-02-02 | 2024-04-09 | Adagene Inc. | Masked activatable CD137 antibodies |
| DE202019005887U1 (en) | 2018-07-03 | 2023-06-14 | Marengo Therapeutics, Inc. | Anti-TCR antibody molecules and uses thereof |
| US11965025B2 (en) | 2018-07-03 | 2024-04-23 | Marengo Therapeutics, Inc. | Method of treating solid cancers with bispecific interleukin-anti-TCRß molecules |
| WO2020010250A2 (en) | 2018-07-03 | 2020-01-09 | Elstar Therapeutics, Inc. | Anti-tcr antibody molecules and uses thereof |
| US11845797B2 (en) | 2018-07-03 | 2023-12-19 | Marengo Therapeutics, Inc. | Anti-TCR antibody molecules and uses thereof |
| WO2020053122A1 (en) | 2018-09-10 | 2020-03-19 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Combination of her2/neu antibody with heme for treating cancer |
| WO2020193520A1 (en) | 2019-03-25 | 2020-10-01 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Treatment of taupathy disorders by targeting new tau species |
| WO2021058763A1 (en) | 2019-09-27 | 2021-04-01 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Anti-müllerian inhibiting substance antibodies and uses thereof |
| WO2021058729A1 (en) | 2019-09-27 | 2021-04-01 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Anti-müllerian inhibiting substance type i receptor antibodies and uses thereof |
| WO2021116119A1 (en) | 2019-12-09 | 2021-06-17 | INSERM (Institut National de la Santé et de la Recherche Médicale) | Antibodies having specificity to her4 and uses thereof |
| WO2021228956A1 (en) | 2020-05-12 | 2021-11-18 | INSERM (Institut National de la Santé et de la Recherche Médicale) | New method to treat cutaneous t-cell lymphomas and tfh derived lymphomas |
| WO2022216993A2 (en) | 2021-04-08 | 2022-10-13 | Marengo Therapeutics, Inc. | Multifuntional molecules binding to tcr and uses thereof |
| WO2023175171A1 (en) | 2022-03-18 | 2023-09-21 | Inserm (Institut National De La Sante Et De La Recherche Medicale) | Bk polyomavirus antibodies and uses thereof |
| WO2024052503A1 (en) | 2022-09-08 | 2024-03-14 | Institut National de la Santé et de la Recherche Médicale | Antibodies having specificity to ltbp2 and uses thereof |
| WO2024056668A1 (en) | 2022-09-12 | 2024-03-21 | Institut National de la Santé et de la Recherche Médicale | New anti-itgb8 antibodies and its uses thereof |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2007535484A (en) | 2007-12-06 |
| JP4889493B2 (en) | 2012-03-07 |
| EP1627062A1 (en) | 2006-02-22 |
| AU2004239065A1 (en) | 2004-11-25 |
| JP2012070743A (en) | 2012-04-12 |
| CA2525120C (en) | 2013-04-30 |
| EP2357237A1 (en) | 2011-08-17 |
| JP2011132233A (en) | 2011-07-07 |
| US20090005257A1 (en) | 2009-01-01 |
| AU2004239065B2 (en) | 2008-05-15 |
| AU2011202105A1 (en) | 2011-05-26 |
| CA2525120A1 (en) | 2004-11-25 |
| JP5049383B2 (en) | 2012-10-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5049383B2 (en) | Method for recovering a polypeptide that unfolds reversibly from a polypeptide repertoire | |
| JP4303105B2 (en) | Dual specific ligands and their use | |
| JP5030782B2 (en) | Single domain antibody against TNFR1 and method of use thereof | |
| RU2401842C2 (en) | Antagonists and method of using said antagonists | |
| EP1345967B1 (en) | Phage display libraries of human v h fragments | |
| EP1025218B9 (en) | Libraries of human antibody polypetides | |
| US20040202995A1 (en) | Nucleic acids, proteins, and screening methods | |
| AU2002319402A1 (en) | Dual-specific ligand and its use | |
| JP2006512895A (en) | Ligand | |
| JP2006523090A (en) | Bispecific single domain antibody specific for ligand and for ligand receptor | |
| JP2009511892A (en) | Screening antibody polypeptide libraries and selected antibody polypeptides | |
| JP2011518565A5 (en) | ||
| JP2014500725A (en) | Collection and its usage | |
| JP2009082141A (en) | Dual-specific ligand and use thereof | |
| CN100575489C (en) | All can recover reversible method of separating folding polypeptide in the library from polypeptide | |
| AU2008201371B2 (en) | A process for recovering polypeptides that unfold reversibly from a polypeptide repertoire | |
| WO2022120024A1 (en) | Variable heavy chain only libraries, methods of preparation thereof, and uses thereof |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200480020375.9 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2525120 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 5170/DELNP/2005 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006530506 Country of ref document: JP Ref document number: 2004239065 Country of ref document: AU |
|
| ENP | Entry into the national phase |
Ref document number: 2004239065 Country of ref document: AU Date of ref document: 20040514 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004239065 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004733030 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004733030 Country of ref document: EP |
|
| DPEN | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed from 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 10556360 Country of ref document: US |