DESCRIPTION MONOSACCHARIDE COMPOUNDS The present invention relates to novel monosaccharides and the pharmaceutically preferable salts , which are as medicaments . This invention relates to inhibitors of sodium-dependent glucose cotransporters (SGLTs) found in the intestine and kidney, especially SGLT II selective inhibitors, which is useful, as hypoglycemic agents, for the therapy of diabetes, both Type I and/or Type II diabetes, as well as hyperglycemia, hyperinsulinemia, obesity, hypertriglyceridemia, diabetic complications, atherosclerosis, Syndrome X and related diseases .
BACKGROUND ART Some glycopyranosides to be useful as an anti-diabetes agent, or the like are known (e.g. EP-0598359, WO-0168660, WO-0236602, WO-03000712, etc. ) .
Diabetes is defined as a disorder that is caused by environmental factors including excessive energy intake and lack of exercise. Therefore diet therapy and/or kinesitherapy are carried out for diabetes patients. But when these therapies do not sufficiently control the patients' condition nor it is not easy to continue these therapies from some situations around the patients, antidiabetic agents are used additionally.
Although biguanide compounds, sulfonylurea compounds, and ameliorants for insulin-resistance etc. are used as such antidiabetics for now, these antidiabetic agents have some side effects, for example biguanides cause lactic acidosis, sulfonylureas cause hypoglycemia, and ameliorants for insulin-resistance cause dropsy and sometimes promote obesity all the more.
Under these circumstances, it has been desired to develop novel antidiabetic agents with an novel action mechanism in order to avoid these side effects.
Desired was a dreamy medicament, if possible, with no side effects or without any side effects, at least, without any grave side effects that affect one's life itself, that is, dead or alive. Recently, it has been reported that a novel type anti-diabetic agent has been in development which lower the blood sugar levels by inhibiting the excessive reabsorption of glucose in kidney, thereby promoting the excretion of glucose in the urine.
Phlorizin is a glycoside, which exists in barks and stems of Rosaceae (e.g. apple, pear, etc.), and was discovered in the 19th century, and has been studied since. Recently, it has been found that Phlorizin is an inhibitor of sodium-dependent glucose cotransporter (SGLT) , which is a monomer with 14 transmembrane segments, and exists only at chorionic membranes of the small
intestines and/or the luminal membranes of the cells in the renal proximal tuble of the kidney, and that phlorizin inhibits the renal tubular glucose reabsorption and promotes the excretion of glucose so that the blood glucose is controlled. However, when phlorizin is administered orally, most of it is hydrolyzed into phloretin, which is the aglycon of phlorizin, and glucose, and hence the amount of phlorizin to be absorbed is so little that the urine glucose excretion effect of phlorizin is very weak. Besides, phloretin has been known to inhibit strongly facilitated diffusion-type glucose transport carrier, for example, when phlorizin is administered to rats, the brain glucose is attenuated. However, when phlorizin is administered for a long time, there may be bad effects on various tissues, and hence phlorizin has not been used as an antidiabetic. It has been .reported that SGLT has two subtypes as being participate in the modulation of blood sugar. The high affinity glucose cotransporter SGLT I, which exists both on the small intestines and the luminal membranes of the cells in the renal proximal tuble, are participate in the modulation of the glucose uptake at small intestine and the glucose reuptake fromthe urine . While the low affinity glucose cotransporter SGLT II, which exists locally on the early SI segment of the renal proximal kidney tuble, appears to be the major transporter responsible for the reuptake of glucose that is filtered at the proximal
tuble glomerulus. Hence, it would be expected that inhibiting human SGLTs could be normalized the blood glucose levels by suppressing the glucose uptake from the small intestine, or by suppressing the excessive reabsorption of glucose in kidney, thereby promoting the excretion the excessive glucose in the urine . It would therefore be expected to prevent or reduce obesity and to promote diuresis by excreting the excessive glucose in the urine, thereby lowering the storage of glucose in various tissues in body. Also, it would be expected to be useful for some related diseases that would be caused following the progress of diabetes or obesity from hyperglycemia .
Additionally, inhibitor of SGLT I may have not only the ameliorative effect for hyperglycemia by inhibiting glucose uptake via SGLT I in the small intestines , but the cause of osmotic diarrhea by flowing of disabsorbed glucose into large intestine . It has been supported by which the patients, who lose SGLT I in the gene, suffer from heavy diarrhea in the most cases and sometimes come to death. While, the patients, who lose SGLT II in the gene, sometimes suffer from renal glycosuria by lower renal threshold of glucose, but this symptom does not cause functional anomaly in the kidney or hypoglycemia, and not made the patients die, so that they do not need to take some treatment specially. So, inhibiting SGLT II selectively for diabetic patients would be expected to normalize plasma glucose by
enhancing the excretion of glucose in the urine, thereby improving insulin sensitivity, and delaying the development of diabetic complications without grave side effects, such as diarrhea and symptom that cannot be rescued by orally glucose administration when hypoglycemia, which may be caused by inhibiting SGLT I.
Consequently, a medicament inhibiting SGLT II selectively is expected to be useful for treatment and/or prevent of not only type II diabetes but type I diabetes by suppressing the glucose reuptake in the kidney, and also expected to be able to overcome the problem on safety that inhibiting SGLT I may cause diarrhea, the symptom that cannot rescue by orally administration of glucose when hypoglycemia, or the like; and if possible, the medicament to be hard to arise hypoglycemia is better.
Being based on these circumstances said above, in the present invention, we provide a monosaccharide compound having SGLTs inhibiting activity (especially, SGLT II selective inhibiting activity) for actively increasing urinary glucose.
DISCLOSURE OF INVENTION The novel monosaccharide compound of the present invention can be shown by the following formula (I) :
wherein R1 is hydrogen or lower alkyl; R2 is hydrogen or lower alkoxy; R3 is hydrogen or hydroxy;
R4 is hydrogen, lower alkoxycarbonyl or hydroxymethyl; and represents single bond or double bond, or a salt thereof.
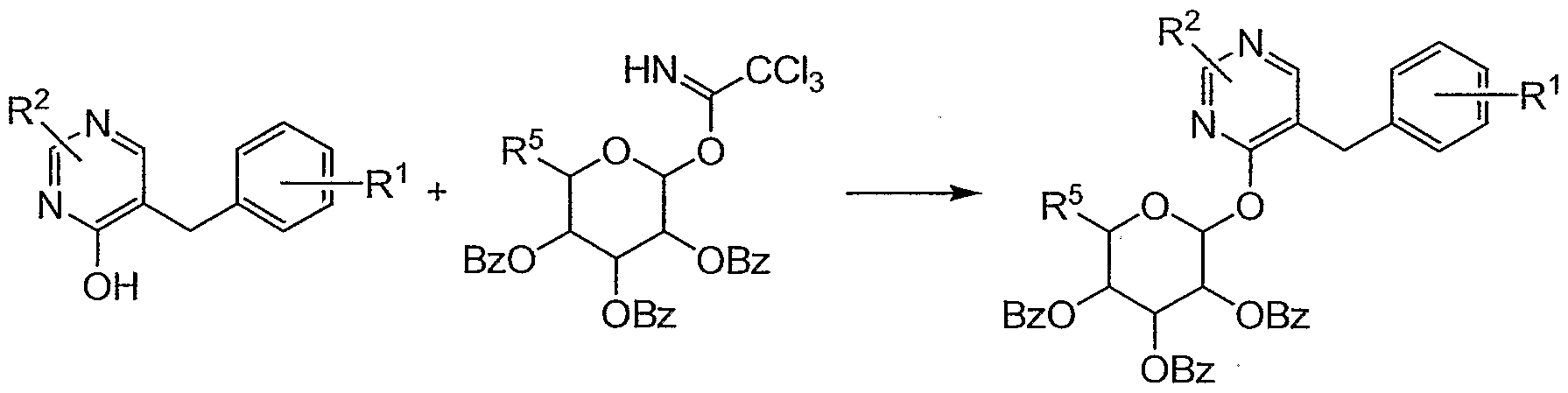
The object compound (.1) and a salt thereof of the present invention can be prepared by the following processes.
Process 1
(ID (I-a) or a salt thereof or a salt thereof
Process 2
(III) (I-b) or a salt thereof or a salt thereof
[wherein R1 and R2 are each as defined above, Bz is benzoyl group; Ac is acetyl group;
R5 is hydrogen or benzoyloxymethyl; R6 is hydrogen or hydroxymethyl; and R7 is lower alkyl.]
The starting compound or a salt thereof is novel and can be prepared, for example, by the following reaction schemes.
Process A
( IV) (VI ) or a salt thereof or a salt thereof
(V) or a salt thereof
Process B
(VI) (VII) or a salt thereof or a salt thereof
(XI) or a salt thereof
(X) or a salt thereof
Process C
(X) (ID or a salt thereof or a salt thereof
(XI) or a salt thereof
Process D
(X) ( III ) or a salt thereof or a salt thereof
(XII ; or ' a salt thereof
[wherein R1, R2, R5, R7, Bz and Ac are each as defined above, and
W is a halogen atom. ]
In addition to the processes as mentioned above, the object compound (I) and a salt thereof can be prepared, for example, according to the procedures as illustrated in Examples in the present specification or in a manner similar thereto.
The starting compounds can be prepared, for example, according to the procedures as illustrated in Preparations in the present specification or in a manner similar thereto.
The object compound (I) and a salt thereof can be prepared according to the methods as shown in a Preparation or Examples, or in a manner similar thereto.
It is to be noted that the solvating form of the compound (I) or its salts (e.g. hydrate, etc.) and any form of the crystal of the compound (I) or its salts are included within the scope of the present invention. 5 Also included in the scope of invention are radiolabelled derivatives of compounds of formula (I) , which are suitable for biological studies .
The compounds of this invention may be converted to salt according to a conventional method. Suitable salts of the object
10. compounds (I) are pharmaceutically acceptable conventional non-toxic salts and include a metal salt such as an alkali metal salt (e.g. sodium salt, potassium salt, etc.) and an alkaline earth metal salt (e.g. calcium salt, magnesium salt, etc.), an ammonium salt, an organic base salt (e.g. trimethylamine salt,
15 triethylamine salt, pyridine salt, picoline salt, dicyclohexylamine salt, etc. ) , anorganic acid salt (e.g. acetate, maleate, tartrate, methanesulfonate, benzenesulfonate, formate, toluenesulfonate, trifluoroacetate, etc. ) , an inorganic acid salt (e.g. hydrochloride, hydrobromide, sulfate, phosphate,
20 etc. ) , a salt with an amino acid (e.g. arginine, aspartic acid, glutamic acid, etc.), and the like.
Suitable examples and illustrations of the various definitions which the present invention includes within the scope thereof
and which appear in the above and following description in the present specification are explained in detail as follows.
The term "lower" used in the description is intended to mean a group having 1 to 6 carbon atom(s) , unless otherwise provided. Suitable examples of "lower alkyl" include straight or branched ones, such as methyl, ethyl, propyl, isopropyl, butyl, isobutyl, tert-butyl, pentyl, hexyl, and the like; in which the preferred one may be (C1-C4) alkyl. Suitable examples of "lower alkoxy" and "lower alkoxy" moiety in the terms "lower alkoxycarbonyl" include straight or branched ones, such as ethoxy, ethoxy, propoxy, isopropoxy, butoxy, isobutoxy, tert-butoxy, pentyloxy, hexyloxy, and the like; in which the preferred one may be (C1-C4) alkoxy. The term "halogen" may be fluoro, chloro, bromo and iodo.
The preferred embodiment of the compound (I) is explained as follows .
1. The compound (I), wherein
R1 is hydrogen, methyl or ethyl;
R2 is hydrogen, methoxy or ethoxy;
R3 is hydrogen, or hydroxy;
R4 is hydrogen, lower alkoxycarbonyl or hydroxymethyl; and
is single bond or double bond.
The more preferred embodiment of the compound (I) is explained as follows.
1. The compound (I), wherein R1 is ethyl; R2 is methoxy; R3 is hydroxy; R4 is hydrogen or hydroxymethyl; and is single bond.
2. The compound (I), wherein R1 is ethyl; R2 is methoxy;
R is hydrogen;
R4 is methoxycarbonyl; and is double bond.
The processes for preparing the object compound (I) are explained in detail as follows.
Process 1
The compound (I-a) or a salt thereof can be prepared by subjecting the compound (II) or a salt thereof to deprotection.
Suitable salt of the compound (II) can be referred to ones as exemplified for the compound (I) .
This reaction is carried out in accordance with a conventional method. The deprotection is preferably carried out in the presence of a base or an acid.
Suitable base employable in this process is not particularly limited so long as it accelerates this process and may include an inorganic base and an organic base such as an alkali metal (e.g. sodium, potassium, cesium, etc.), an alkaline earth metal (e.g. magnesium, calcium, barium, etc.), the hydroxide or methoxide or carbonate or hydrogencarbonate thereof, trialkylamine (e.g. triethylamine, trimethylamine, N, -diisopropylethylamine, etc.), hydrazine, picoline, lutidine, collidine, or the like.
Suitable acid employable in this process is not particularly limited so long as it accelerates this process and may include an organic acid (e.g. formic acid, acetic acid, propionic acid, trifluoroacetic acid, etc.) and inorganic acid (e.g. hydrochloric acid, sulfuric acid, hydrogen bromide, etc.).
The reaction is usually carried out in a solvent such as water, alcohol (e.g. methanol, ethanol, etc.), tetrahydrofuran, dioxane, toluene, methylene chloride, chloroform, N, -dimethylformamide, or any other organic solvents which do
not adversely affect the reaction, or a mixture thereof . A liquid i base and acid 'can be also used as the solvent.
The temperature varies depending on the starting material, the solvent, etc., but it is usually at ambient temperature, under warming or heating.
The reaction- time varies depending on the starting material, solvent, etc., but it is usually from 30 minutes to 24 hours. Process 2
The compound (I-b) or a salt thereof can be prepared by subjecting the compound (III) or a salt thereof to deprotection.
The deprotecting reaction can be carried out in the same manner as in the aforementioned Process 1 or the similarmanners thereto, and therefore the reagents to be used and the reaction conditions (e.g., solvent, reaction temperature, etc.) can be referred to those of Process 1. Process A
The compound (VI) or a salt thereof can be prepared by reacting the compound (IV) or a salt thereof with the compound (V) or a salt thereof. This reaction is usually carried out in the presence of organometallic compound. Suitable ones include lithium diisopropylamide, n-butyllithium, hexamethyldisilazane, alkyl- or aryl-magnesiumbromide which can be a Grignard reagent, etc., more suitably n-butyllithium.
The reaction is usually carried out in a solvent such as water, alcohol (e.g. methanol, ethanol, etc.), tetrahydrofuran, diethyl ether, dioxane, toluene, methylene chloride, chloroform, N, N-dimethylformamide, or any other organic solvents which do not adversely affect the reaction, or a mixture thereof.
The temperature varies depending on the starting material, the solvent, etc., but it is usually under cooling or at ambient temperature.
The reaction time varies depending on the starting material, solvent, etc., but it is usually from 30 minutes to 24 hours. Process B
The compound (X) can be synthesized by functional trans formation, which is obvious to the person skilled in the organic chemistry from the compound (VI) exemplified by Step 1 - 4. Process C
The compound (II) or a salt thereof can be prepared by reacting the compound (X) or a salt thereof with the compound (XI) or a salt thereof.
Suitable salts of the compounds (X) and (XI) can be referred to ones as exemplified for the compound (I) .
This reaction is usually carried out in the presence of Lewis acid (e.g. alminium chloride, boron trifluoride diethyl etherates, trimethylsilyl trifluoromethanesulfonate, silver carbonate, tin tetrachloride, etc.).
The reaction is usually carried out in a solvent such as water, alcohol (e.g. methanol, ethanol, etc. ) , tetrahydrofuran, dioxane, toluene, methylene chloride, chloroform, N, N-dimethylformamide, or any other organic solvents which do not adversely affect the reaction, or a mixture thereof.
The temperature varies depending on the starting material, the solvent, etc., but it is usually at ambient temperature, under warming or heating.
The reaction time varies depending on the starting material, solvent, etc., but it is usually from 30 minutes to 24 hours.' Process D
The compound (III) or a salt thereof can be prepared by reacting the compound (X) or a salt thereof with the compound (XII) or a salt thereof. This reaction can be carried out in the same manner as in the aforementioned Process C or the similar manners thereto, and therefore the reagents to be used and the reaction conditions (e.g., solvent, reaction temperature, etc.) can be referred to those of Process C.
Above processes, all starting materials and product compounds may be salts. The compounds of above processes can be converted to salts according to a conventional method.
For therapeutic purpose, the compound (I) and a
pharmaceutically acceptable salt thereof of the present invention can be used in a form of pharmaceutical preparation containing one of said compounds as an active ingredient, in admixture with a pharmaceutically acceptable carrier such as an organic or inorganic solid, semisolid or liquid excipient suitable for oral, parenteral or external administration. The pharmaceutical preparations may be capsules, tablets, pellets, troches, d agees, granules, inhalant, suppositories, solution, lotion, suspension, emulsion, ointment, gel, cream, ointments, aerosols, or the like suitable for use. If desired, there may be included in these preparations auxiliary substances, stabilizing agents, wetting or emulsifying agents, buffers, thickening agents, coloring agents, perfumes and other commonly used additives .
In order to show the usefulness of the compound (I) of the present invention, the pharmacological test result of the representative compound of-the present invention is shown in the following. Test: Assay for SGLT I and SGLT II activities [I] Test method cDNA sequences of human SGLT I (Genbank #M24847) and SGLT II (Genbank#M95549) were cloned and transfected into CH0-K1 cells. Effect of the test compound on sodium-dependent sugar uptake
of the SGLT I-transfected and SGLT II-transfected cells was
examined by radioactive tracer technique using Cϋ-methyl- D-glucopyranoside as a substrate.
[II] Test compound
(2S, 3R, 4S, 5R, 6R) -2-{ [5- (4-Ethylbenzyl) -2-methoxy- 4-pyrimidinyl] oxy}-6- (hydroxymethyl) tetrahydro-2H-pyran- 3,4,5-triol (Example 2)
[III] Test result
Table 1
human-SGLTs Test compound inhibiting activity
(Example No.) I.C.5.0[.^I) SGLT I / SGLTII
SGLT I SGLT II
2 >100 0.82 >120
The object compound (I) of the present invention is an SGLT II selective inhibiting agent and possesses the various pharmacological actions as stated before.
While the dosage of therapeutically effective amount of the compound (I) will vary depending upon the age and condition of each individual patient to be treated, an average single dose of about 0.01-100 mg of the compound (I) per kg weight of a human being or an animal maybe effective for treating and/or preventing the above-mentioned diseases. In general, amounts between 0.01 mg/body and about 1,000 mg/kg may be administered per day.
The followingpreparation and examples are given for the purpose of illustrating the present invention in more detail.
The abbreviations, symbols and terms used in the preparations and examples have the following meanings . AcOH acetic acid
CHCI3 chloroform
CH2C12 dichloromethane
EtOAc ethyl acetate
MeOH methanol
THF tetrahydrofuran
HC1 hydrochloric acid
MgS04 magnesium sulfate
NaBH4 sodium borohydride
NaHC03 sodium bicarbonate
NaOH sodium hydroxide
Na2S04 sodium sulfate aq. aqueous
Preparation 1 Under nitrogen atmosphere, to a solution of 5-bromo-
2, 4-dimethoxypyrimidine (2.0 g) in THF (35ml) was added dropwise 1.57 n-butyllithium in n-hexane (6.37 ml) over 5 minutes below -70°C. After the mixture was stirred at the same temperature for 30 minutes, a solution of 4-ethylbenzaldehyde (1.23 g) in
THF (4 ml) was added to the mixture at one portion, and the whole was stirred at the same temperature for 30 minutes. The reaction was quenched with saturated aqueous ammonium chloride under ice cooling. The resulting mixture was extracted with EtOAc. The extract was washed withbrine, dried over Na2S0 , and concentrated under reduced pressure. The resulting residue was purified by column chromatography on silica gel (60 g) eluting with a mixed solvent of n-hexane and EtOAc (5:1 to' 5:2). The fractions containing the objective compound were collected and evaporated under reduced pressure to give (2, 4-dimethoxy-5-pyrimidinyl) - (4-ethylphenyl)methanol as oil (2.0 g).
XH NMR (CDC13, 6 ) : 1.23 (3H, t, J=7.8 Hz), 2.61 (2H, q, J=7.8 Hz), 3.97 (3H, s), 3.98 (3H, s) , 5.87 (1H, d, J=4.2 Hz), 7.18 (2H, d, J=8.4 Hz), 7.28 (2H, d, J=8.4 Hz), 8.17 (1H, s) Mass (ESI) : 275(M+H)+ Preparation 2
To a .solution of (2, 4-dimethoxy-5-pyrimidinyl) - (4-ethylphenyl)methanol (1.46 g) in CHC13 (50 ml) was added manganese (IV) oxide chemicals treated (4.63 g) . The mixture was stirred at 50°C for 2.5 hours . Manganese (IV) oxide chemicals treated (1.1 g) was added again, then the whole was stirred at 50°C for 1.5 hours. Manganese (IV) oxide was removedby filtration, the filtrate was concentrated under reduced pressure. The resulting residue was purifiedbycolumn chromatographyon silica
gel (30 g) eluting with a mixed solvent of n-hexane and EtOAc
(5:1 to 5:2) . The fractions containing the objective compound were collected and concentrated under reduced pressure to give
(2, 4-dimethoxy-5-pyrimidinyl) - (4-ethylphenyl) - methanone as colorless solid (865 mg) .
XH NMR (CDC13, δ ) : 1.28 (3H, t, J=7.5 Hz), 2.73 (2H, q, J=7.5 Hz), 3.98 (3H, s) , 4.08 (3H, s) , 7.29 (2H, d, J=8.4 Hz), 7.71 (2H, d, J=8.4 Hz), 8.44 (1H, s) Mass (ESI) : 273(M+H)+ Preparation 3
Under nitrogen atmosphere, to an ice-cooled solution of (2, 4-dimethoxy-5-pyrimidinyl) (4-ethylphenyl) methanone (340 mg) in CH2CI2 (4 ml) was added 1.0 boron tribromide in CH2CI2 (1.56 ml) . After stirring at the same temperature for 30 minutes, 1.0M boron tribromide in CH2C12 (1.2 ml) was added again to the solution, and then the whole was stirred under ice cooling for 30 minutes. The reaction mixture was poured into ice water, and the pH of the aqueous layer was adjusted to 3 with aq. NaHC03. The organic layer was separated, dried over Na2S04, and concentrated under reduced pressure. The resulting residue was purified by column chromatography on silica gel (8 g) eluting with a mixed solvent of CH2C12 and MeOH (30:1) . The fractions containing the objective compound were collected and concentrated under reduced pressure. (4-Ethylphenyl) -
(4-hydroxy-2-methoxy-5-ρyrimidinyl) ethanone (290 mg) was obtained as slight yellowish solid.
XH NMR (CDCI3, 6 ) : 1-27 (3H, t, J=7.5 Hz), 2.72 (2H, q, J=7.5 Hz), 4.08 (3H, s), 7.30 (2H, d, J=8.4 Hz), 7.71 (2H, d, J=8.4 Hz), 8.31 (1H, s)
Mass (ESI) : 259(M+H)+ Preparation 4
To a solution of (4-ethylphenyl) (4-hydroxy-2-methoxy- 5-pyrimidinyl) methanone (127 mg) in a mixed solvent of MeOH (3 ml) and THF (3ml) was added portionwise NaBH4 (23.3mg) at ambient temperature under nitrogen atmosphere . The mixed suspension was stirred at the same temperature for 30 minutes, and then NaBH4 (40 mg) was added again. After stirring at ambient temperature for 30 minutes, NaBH4 (40 mg) was added one more again. After stirring at the same temperature for 30 minutes, the reaction mixture was quenched with ice water and 2N HCl, and then the whole was extractedwith EtOAc. The extract was washedwithbrine, dried over Na2S04, and concentrated under reduced pressure to give 5- [ (4-ethylphenyl) (hydroxy) methyl] -2-methoxy- 4-pyrimidinol as yellowish oil (128 mg) .
XH NMR (CDCI3, δ ) : 1.22 (3H, t, J=7.5 Hz), 2.63 (2H, q, J=7.5 Hz), 3.95 (3H, s) , 5.73 (1H, s) , 7.18 (2H, d, J=8.1 Hz), 7.32 (2H, d, J=8.1 Hz), 7.52 (1H, s) Mass (ESI) : 261(M+H)+
Preparation 5
To a solution of 5- [ (4-ethylphenyl) (hydroxy) methyl] - 2-methoxy-4-pyrimidinol (126 mg) in trifluoroacetic acid (2ml) was added triethylsilane (141 mg) at ambient temperature, then
the mixture was stirred at 53°C for 1 hour. The reaction mixture was poured into ice water, and the pH of the aqueous layer was adjusted to 6 with 15% aq. NaOH. The resulting mixture was extracted with EtOAc. The extract was washed with brine, dried over Na2S04 and evaporated under reduced pressure . The resulting residue was purified by column chromatography on silica gel (8 g) eluting with a mixed solvent of CH2C12 and MeOH (100:1, 20:1). The fractions containing the objective compound were collected and evaporated under reducedpressure to give 5- (4-ethylbenzyl) - 2-methoxy-4-pyrimidinol as yellowish syrup (83 mg) .
XH NMR (CDC13, δ ) : 1.22 (3H, t, J=7.5 Hz), 2.62 (2H, q, J=7.5 Hz), 3.72 (2H, s), 3.95 (3H, s) , 7.12 (2H, d, J=7.8 Hz), 7.18 (2H, d, J=7.8 Hz), 7.47 (1H, s) , 10.78 (1H, br) Mass (ESI) : 245(M+H)+ Preparation 6 To a mixed solution of (2R, 3R, 4S, 5R, 6R) -3, 5-bis (benzoyloxy) - 2- [ (benzoyloxy) methyl] -6- [ (2,2, 2-trichloroethanimidoyl) - oxy] tetrahydro-2iT-pyran-4-yl benzoate (448 mg) and 5- (4-ethylbenzyl) -2-methoxy-4-pyrimidinol (120 mg) in CH2Cl2 (4 ml) was added boron trifluoride diethyl etherate (85.8 mg)
at ambient temperature, and the whole was stirred at the same temperature for 1 hour. Under ice cooling, the reaction was quenched with triethylamine ( 0.12 ml ) . The reaction mixture was poured into ice water, and extracted with CH2C12. The extract was washed with brine, dried over Na2S0 and evaporated under reduced pressure. The resulting residue was purified by column chromatography on silica gel (12 g) eluting with a mixed solvent of n-hexane and EtOAc (4:1, 1:1) . The fractions containing the objective compound were collected and evaporated under reduced pressure to give (2R, 3R, 4S, 5R, 6S) -3, 5-bis (benzoyloxy) - 2- [ (benzoyloxy)methyl] -6-{ [5- (4-ethylbenzyl) -2-methoxy- 4-pyrimidinyl] oxy}tetrahydro-2H-pyran-4-yl benzoate as colorless foam (360 mg) .
XH NMR (CDC13> δ ) : 1.10 (3H, t, J=7.5 Hz), 2.45 (2H, q, J=7.5 Hz) , 3.69, 3.79 (2H, ABq, J=15 Hz) , 3.91 (3H, s) , 4.35-4.50 (2H, m) , 4.60-4.70 (1H, m) , 5.72-5.83 (1H, m) , 5.85-5.94 (1H, m) , 6.03 (1H, t, J=9.5 Hz), 6.54 (1H, d, J=8.1 Hz), 6.86 (2H, d, J=8.4 Hz) , 7.00 (2H, d, J=8.4 Hz) , 7.25-7.60 (12H, m) , 7.80-8.10 (9H, )' Mass (ESI) : 823(M+H)+ Preparation 7
To a solution of 5- (4-ethylbenzyl) -2-methoxy-4-pyrimidinol (60 mg) and (2R, 3R, 4S, 5S, 6R) -4, 5-bis (benzoyloxy) - 6- [ (benzoyloxy) methyl] -2- [ (2,2, 2-trichloroethanimidoyl) -
oxy] tetrahydro-2ff-pyran-3-yl benzoate (224 mg) in CH2Cl2dichloromethane (2.0 ml) was added boron trifluoride etherate (0.038 ml) and the mixture was stirred at ambient temperature for 30 minutes. The reaction was quenched with triethylamine (0.06mL) and the mixture was poured into the mixture of CH2CI2 and water. The organic layer was separated, and dried and concentrated in vacuo. The residue was purified by silica gel column chromatography (eluent; hexane : EtOAc=2 : 1) to give (2R, 3S, 4S, 5R, 6S) -3, 5-bis (benzoyloxy) - 2- [ (benzoyloxy) methyl] -6-{ [5- (4-ethylbenzyl) -2-methoxy-
4-pyrimidinyl] oxy}tetrahydro-2H-pyran-4-yl benzoate (205 mg) as a colorless syrup.
XH NMR (CDCI3, δ ) : 1.10 (3H, t, J=7.7 Hz), 2.45 (2H, q, J=7.7 Hz), 3.74 (IH, d, J=15.0Hz), 3.83 (IH, d, J=15.0Hz), 3.90 (3H, s) , 4.42-4.50 (IH, m) , 4.54-4.62 (2H, m) , 5.73 (IH, dd, J=3.7, 10.3 Hz), 6.07 (IH, d, J=3.7Hz), 6.14 (IH, dd, J=8.4, 10.3 Hz), 6.50 (IH, d, J=8.4 Hz), 6.87 (2H, d, J=8.4 Hz), 7.00 (2H, d, J=8.4Hz), 7.22-7.66 (12H, m) , 7.80 (2H, d, J=8.4Hz), 7.88 (2H, d, J=8.4 Hz), 7.98 (2H, d, J=8.4 Hz), 8.02 (IH, s) , 8.10 (2H, d,. J=8.4 Hz) . Preparation 8
The following compound was obtained in substantially the same manner as that of Preparation 7. (2S, 3R, 4S, 5R) -3, 5-bis (Benzoyloxy) -2-{ [5- (4-ethylbenzyl) -
2-methoxy-4-pyrimidinyl] oxy}tetrahydro-2H-pyran-4-yl benzoate
αH NMR (CDCI3, δ ) : 1.16 (3H, t, J=7.7 Hz), 2.54 (2H, q, J=7.7 Hz), 3.74 (2H, s), 3.85-3.93 (IH, m) , 3.95 (3H, s), 4.42 (IH, dd, J=4.0, 12.5 Hz), 5.37-5.44 (IH, m) , 5.70 (IH, dd, J=5.5, 7.0 Hz), 5.85 (IH, dd, J=7.0, 7.0 Hz), 6.58 (IH, d, J=5.5 Hz), 6.99 (4H, s), 7.26-7.59 (9H, m) , 7.90-8.05 (7H, m) . Mass (ESI) : 689(M+H)+ Preparation 9 The following compound was obtained in substantially the same manner as that of Preparation 7. Methyl (2S, 3S, 4S, 5R, 6S) -3, 4 , 5-tris (acetyloxy) - 6-{ [5- (4-ethylbenzyl) -2-methoxy-4-pyrimidinyl] oxy }- tetrahydro-2H-pyran-2-carboxylate
1H NMR (CDCI3, δ ) : 1.20 (3H, t, J=7.7 Hz), 2.04 (3H, s), 2.05 (3H, s), 2.06 (3H, s) , 2.59 (2H, q, J=7.7 Hz), 3.67-3.84 (2H, m) , 3.71 (3H, s) , 3.95 (3H, s) , 4.23-4.30 (IH, m) , 5.28-5.35 (3H, m) , 6.23-6.28 (IH, m) , 7.05 (2H, d, J=8.3 Hz), 7.09 (2H, d, J=8.3 Hz) , 8.11 (IH, s) . Mass (ESI) : 561(M+H)+ Preparation 10
The following compound was obtained in substantially the same manner as that of Preparation 7. (2S, 3R, 4S, 5S) -3, 5-bis (Benzoyloxy) -2-{ [5- (4-ethylbenzyl) -
2-methoxy-4-pyrimidinyl] oxy}tetrahydro-2H-pyran-4-yl benzoate
XH NMR (CDCI3, δ ) : 1.12 (3H, t, J=7.7 Hz), 2.47 (2H, q, J=7.7 Hz) , 3.72 (IH, d, J=15.4 Hz) , 3.76 (IH, d, J=15.4 Hz) , 3.96 (3H, s), 4.07-4.17 (IH, m) , 4.37 (IH, dd, J=3.7, 12.8 Hz), 5.69 (IH, dd, J=3.7, 8.8 Hz), 5.75-5.80 (IH, m) , 6.05 (IH, dd, J=6.6, 8.8 Hz), 6.48 (IH, d, J=6.6 Hz), 6.90 (2H, d, J=8.4 Hz), 6.97 (2H, d, J=8.4 Hz), 7.25-7.63 (9H, m) , 7.90-8.10 (7H, m) . Mass (ESI) : 689(M+H)+ Example 1
Under nitrogen atmosphere, to an ice-cooled solution of (2R, 3R, 4S, 5R, 6S) -3, 5-bis (benzoyloxy) -2- [ (benzoyloxy) - methyl] -6-{ [5- (4-ethylbenzyl) -2-methoxy-4-pyrimidinyl] oxy}- tetrahydro-2H-pyran-4-yl benzoate (530 mg) in a mixed solvent of MeOH (8 ml) and THF (6 ml) was added 28% sodium methoxide in MeOH solution (436 mg) . After stirring at ambient temperature for 30 minutes, the reaction was quenched with AcOH (135 mg) under ice cooling, and the mixture was evaporated under reduced pressure. To the resulting residue was added a mixed solvent of CH2C12 (100 ml) and MeOH (20 ml) , and insoluble material was removed by filtration. The filtrate was evaporated under reduced pressure. The resulting residue was purified by column chromatography on silica gel (20 g) eluting with a mixed solvent of CH2C12 and MeOH (100:1, 12:1) . The fractions containing the
objective compound were collected and evaporated under reduced pressure to give (2S, 3R, 4S, 5S, 6R) -2-{ [5- (4-ethylbenzyl) - 2-methoxy-4-pyrimidinyl] oxy} -6- (hydroxymethyl) tetrahydro- 27J-pyran-3, 4, 5-triol as colorless powder (181 mg) .
αH NMR (CDC13, δ ) : 1.20 (3H, t, J=7.5 Hz), 2.59 (2H, q, J=7.5 Hz), 3.32-3.70 (5H, m) , 3.75-3.93 (3H, m) , 3.95 (3H, s) , 5.92 (IH, d, J=7.8 Hz), 7.11 (2H, d, J=9.6 Hz), 7.17 (2H, d, J=9.6 Hz), 7.99 (IH, s) Mass (ESI) : 407(M+H)+ Example 2
To a solution of (2R, 3S, 4S, 5R, 6S) -3, 5-bis (benzoyloxy) - 2- [ (benzoyloxy) methyl] -6- { [5- ( 4-ethylbenzyl ) -2-methoxy- 4-pyrimidinyl] oxy} tetrahydro-2H-pyran-4-yl benzoate (195mg) in MeOH (4.8 ml) was added IN NaOH (1.2 ml), and the mixture was stirred at ambient temperature for 1 hour. The mixture was concentrated to ca .3 ml, and then diluted with CHC13 (15 ml) and water (10 ml) . The layers were separated and the aqueous layer was extracted with CHC13 (15 ml) . The combined organic layers were dried over MgS04 and concentrated.- The residue was purified by silica gel column chromatography (eluent; 10% MeOH in CHCI3) to give (2S, 3R, 4S, 5R, 6R) -2-{ [5- (4-ethylbenzyl) - 2-methoxy-4-pyrimidinyl] oxy} -6- (hydroxymethyl) tetrahydro- 2iϊ-pyran-3, 4, 5-triol (40mg) as a colorless crystal.
XH NMR (DMSO-d6, δ ) : 1.14 (3H, t, J=7.7Hz), 2.54 (2H, q, J=7.7
Hz), 3.35-3.78 (8H, m) , 3.84 (3H, s) , 4.60 (IH, d, J=4.4 Hz), 4.64-4.70 (IH, m) , 4.93 (IH, d, J=5.9 Hz), 5.14 (IH, d, J=5.5 Hz), 5.73 (IH, d, J=8.1 Hz), 7.12 (2H, d, J=8.1 Hz), 7.19 (2H, d, J=8.1 Hz) , 8.16 (IH, s) . Mass (ESI) : 407(M+H) + Example 3
To a solution of methyl (2S, 3S, 4S, 5R, 6S) - 3, 4, 5-tris (acetyloxy) -6-{ [5- (4-ethylbenzyl) -2-methoxy- 4-pyrimidinyl] oxy}tetrahydro-2if-pyran-2-carboxylate (155mg) in CH2Cl2-MeOH (1 : 1, 3.0 ml) was added cesium carbonate (270 mg) and the mixture was stirred at ambient temperature for 30 minutes. The resulting mixture was poured into the mixture of CHC13 and water. The organic layer was separated, dried and concentrated in vacuo. The residue was purified by silica gel column chromatography (eluent; n-hexane : EtOAc=l : 1-1 : 2) to give methyl (2S, 3R, 4S) -2-{ [5- (4-ethylbenzyl) -2-methoxy- 4-pyrimidinyl] oxy}-3, 4-dihydroxy-3, 4-dihydro-2H-pyran- 6-carboxylate (35mg) as a colorless crystal. XH NMR (DMS0-d6, <5 ) : 1.13 (3H, t, J=7.5Hz), 2.52 (2H, q, J=7.5 Hz), 3.68 (IH, d, J=14.3Hz), 3.69 (3H, s) , 3.75 (IH, d, J=14.3 Hz), 3.82-3.89 (IH, m) , 3.86 (3H, s) , 4.05-4.13 (IH, m) , 5.42 (1H-, d, J=5.1 Hz), 5.74 (IH, d, J=4.4 Hz), 6.15 (IH, d, J=3.7 Hz), 6.55 (IH, d, J=4.4 Hz), 7.03 (2H, d, J=8.1 Hz), 7.20 (2H, d, J=8.1 Hz) , 8.31 (IH, s) .
Mass (ESI) : 417(M+H)+ Example 4
The following compound was obtained in substantially the same manner as that of Example 3. (2S,3R,4S,5R)-2-{ [5- (4-Ethylbenzyl) -2-methoxy-
4-pyrimidinyl] oxy} tetrahydro-2if-pyran-3, 4, 5-triol E NMR (DMS0-d6, δ ) : 1.14 (3H, t, J=7.3Hz), 2.54 (2H, q, J=7.3 Hz), 3.15-3.43 (4H, m) , 3.69-3.79 (3H, m) , 3.84 (3H, s) , 5.07 (IH, d, J=5.1 Hz), 5.19 (IH, d, J=4.8 Hz), 5.35 (IH, d, J=5.5 Hz), 5.72 (IH, d, J=7.3 Hz), 7.10 (2H, d, J=8.1 Hz), 7.17 (2H, d, J=8.1 Hz) , 8.22 (IH, s) . Mass (ESI) : 377(M+H)+ Example 5
The following compound was obtained in substantially the same manner as that of Example 3.
(2S, 3R, 4S, 5S) -2-{ [5- (4-Ethylbenzyl) -2-methoxy- 4-pyrimidinyl] oxy}tetrahydro-2H-pyran-3, 4, 5-triol
XH NMR (DMSO-de, δ ) : 1.14 (3H, t, J=7.7 Hz), 2.54 (2H, q, J=7.7 Hz), 3.43-3.51 (IH, m) , 3.57-3.64 (IH, br) , 3.67-3.79 (5H, m) , 3.83 (3H, s) , 4.73 (IH, d, J=3.7 Hz), 4.90 (IH, d, J=5.5 Hz), 5.28 (IH, d, J=4.8 Hz), 5.82 (IH, d, J=5.5 Hz), 7.10 (2H, d, J=8.1 Hz), 7.23 (2H, d, J=8.1 Hz), 8.22 (IH, s) . Mass (ESI) : 377(M+H)+