WO2004083172A2 - Process for the preparation of 7-amino (p-hydroxyphenylglyclyamido) cephem compounds - Google Patents
Process for the preparation of 7-amino (p-hydroxyphenylglyclyamido) cephem compounds Download PDFInfo
- Publication number
- WO2004083172A2 WO2004083172A2 PCT/IB2004/000850 IB2004000850W WO2004083172A2 WO 2004083172 A2 WO2004083172 A2 WO 2004083172A2 IB 2004000850 W IB2004000850 W IB 2004000850W WO 2004083172 A2 WO2004083172 A2 WO 2004083172A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- alkyl
- group
- methyl
- solvent
- Prior art date
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D501/00—Heterocyclic compounds containing 5-thia-1-azabicyclo [4.2.0] octane ring systems, i.e. compounds containing a ring system of the formula:, e.g. cephalosporins; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulfur-containing hetero ring
- C07D501/02—Preparation
- C07D501/04—Preparation from compounds already containing the ring or condensed ring systems, e.g. by dehydrogenation of the ring, by introduction, elimination or modification of substituents
- C07D501/06—Acylation of 7-aminocephalosporanic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D501/00—Heterocyclic compounds containing 5-thia-1-azabicyclo [4.2.0] octane ring systems, i.e. compounds containing a ring system of the formula:, e.g. cephalosporins; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulfur-containing hetero ring
- C07D501/14—Compounds having a nitrogen atom directly attached in position 7
- C07D501/16—Compounds having a nitrogen atom directly attached in position 7 with a double bond between positions 2 and 3
- C07D501/20—7-Acylaminocephalosporanic or substituted 7-acylaminocephalosporanic acids in which the acyl radicals are derived from carboxylic acids
- C07D501/22—7-Acylaminocephalosporanic or substituted 7-acylaminocephalosporanic acids in which the acyl radicals are derived from carboxylic acids with radicals containing only hydrogen and carbon atoms, attached in position 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D501/00—Heterocyclic compounds containing 5-thia-1-azabicyclo [4.2.0] octane ring systems, i.e. compounds containing a ring system of the formula:, e.g. cephalosporins; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulfur-containing hetero ring
- C07D501/14—Compounds having a nitrogen atom directly attached in position 7
- C07D501/16—Compounds having a nitrogen atom directly attached in position 7 with a double bond between positions 2 and 3
- C07D501/20—7-Acylaminocephalosporanic or substituted 7-acylaminocephalosporanic acids in which the acyl radicals are derived from carboxylic acids
- C07D501/24—7-Acylaminocephalosporanic or substituted 7-acylaminocephalosporanic acids in which the acyl radicals are derived from carboxylic acids with hydrocarbon radicals, substituted by hetero atoms or hetero rings, attached in position 3
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D501/00—Heterocyclic compounds containing 5-thia-1-azabicyclo [4.2.0] octane ring systems, i.e. compounds containing a ring system of the formula:, e.g. cephalosporins; Such ring systems being further condensed, e.g. 2,3-condensed with an oxygen-, nitrogen- or sulfur-containing hetero ring
- C07D501/14—Compounds having a nitrogen atom directly attached in position 7
- C07D501/16—Compounds having a nitrogen atom directly attached in position 7 with a double bond between positions 2 and 3
- C07D501/20—7-Acylaminocephalosporanic or substituted 7-acylaminocephalosporanic acids in which the acyl radicals are derived from carboxylic acids
- C07D501/24—7-Acylaminocephalosporanic or substituted 7-acylaminocephalosporanic acids in which the acyl radicals are derived from carboxylic acids with hydrocarbon radicals, substituted by hetero atoms or hetero rings, attached in position 3
- C07D501/36—Methylene radicals, substituted by sulfur atoms
Definitions
- the field of the invention relates to pure 7- amino (p-hydroxyphenylglycyl) cephem compounds.
- the invention also relates to processes for the preparation of pure 7- amino (p-hydroxyphenylglycyl) cephem compounds and pharmaceutical compositions that include the pure 7- amino (p-hydroxyphenylglycyl) cephem compounds.
- 7- amino (p-hydroxyphenylglycyl) cephem compounds such as cefprozil, cefadroxil and cefatrizine are generally prepared by reacting a cephem derivative with a reactive derivative, such as a reactive ester; a reactive amide; and a mixed-acidic anhydride of 4-hydroxyphenylglycine.
- a reactive derivative such as a reactive ester; a reactive amide; and a mixed-acidic anhydride of 4-hydroxyphenylglycine.
- British Patent GB 1,240,687 discloses a process involving reacting N-protected 4- hydroxyplienylglycine with ethyl chloroformate to obtain a carbonate derivative which is acylated with a cephem compound.
- this method gives a product of low purity.
- a major impurity formed in such a process has the structure of Formula A,
- R is a group commonly used at the 3-position in cephalosporins, for example, methyl, 1-propenyl and l,2,3-triazol-5-yl tbiornethyl.
- the present invention provides a process which results in pure 7- amino (p- hydroxyphenylglycyl) cephem compounds.
- the process of the present invention avoids purification by tedious and cumbersome processes.
- the process of the present invention reduces the impurity content of the final product, eliminates the costly and time- consuming purification steps.
- R is a group commonly used at the 3-position in cephalosporins and may include C 1 6 alkyl, C 2 6 alkenyl, C 2 _ 6 alkynyl, C 2 6 alkadienyl, cyclic C 3 6 alkyl, aryl, substituted aryl, heteroaryl, or heteroarylthioalkyl . h particular, it may include methyl, 1- propenyl and l,2,3-triazol-5-yl thiomethyl.
- compositions and dosage forms containing a therapeutically effective amount of the cephem compounds of Formula I and which may also contain pharmaceutically acceptable carriers, excipients or diluents which are useful for the treatment of bacterial infections.
- a method of treating bacterial infections comprising administering to a mammal in need thereof, a therapeutically effective amount of cephem compounds of Formula I as described above.
- R is a group commonly used at the 3 -position in cephalosporins and includes C l g alkyl, C 2 6 alkenyl, C 2 6 alkynyl, C 2 6 alkadienyl, cyclic C 3 _ 6 alkyl, aryl, substituted aryl, heteroaryl, or heteroarylthioalkyl . hi particular, it includes methyl, 1- propenyl and l,2,3-triazol-5-yl tl iomethyl.
- the process comprising:
- R' is C 1-4 alkyl and M + is an alkali metal cation
- R is C 1 6 alkyl, C,_ 6 alkenyl, C 2 _ 6 alkynyl, C 2 6 alkadienyl, cyclic C 3 6 alkyl, aryl, substituted aryl, heteroaryl, or heteroarylthioalkyl,
- the preparation of the mixed anhydride is carried out in the presence of a small amount of an acid to minimize the formation of impurity of Formula B, in the step ii) reaction,
- R is a group commonly used at the 3-position in cephalosporins, and includes C. 6 alkyl, C 2 6 alkenyl, C 2 6 alkynyl, C 2 6 alkadienyl, cyclic C 3 6 alkyl, aryl, substituted aryl, heteroaryl, or heteroarylthioalkyl.
- the process is useful for the preparation of a wide variety of 7- amino(p- hydroxyphenylglycyl) -cephalosporins, for example, cefatrizine, cefadroxil, or cefprozil, in good yield and purity.
- Examples of Dane salts of Formula III include sodium or potassium D-N-(l- methoxycarbonylpropen-2-yl)amino-p-hydroxyphenyl- acetate, and sodium or potassium D-N-(l-ethoxycarbonyl- propen-2-yl)-amino-p-hydroxyphenylacetate.
- Examples of alkyl chloro formates include ethyl chloroformate and methyl chloroformate.
- Examples of amines present as a catalyst for mixed carboxylic acid anhydride formation include N-methyl morpholine, N,N-dimethyl benzyl amine, pyridine, picoline, and lutidine.
- the mixed anhydride may be prepared in one or more solvents, including, for example, halogenated hydrocarbon, ketone, ester, ether, nitrile, aromatic hydrocarbon, amide and mixtures thereof.
- chlorinated hydrocarbons include dichoromethane and dichloroethylene;
- ketones include acetone and methyl isobutyl ketone;
- ester include ethyl acetate and isopropylacetate;
- Examples of ether include tetrahydrofuran and dioxane; a nitrile includes acetonitrile;
- aromatic hydrocarbon include toluene.
- a suitable co-solvent may be used with a solvent, for example amide.
- a suitable amide includes one or more of formamide, acetamide, N,N- dimethyl formamide, N-methylacetamide, N,N-dimethylacetamide and N- methylpyrrolidone. Mixtures of all of these solvents are also contemplated.
- the formation of the mixed carboxylic acid anhydride may be carried out at temperatures, from about -80°C to about 50°C, or from about -50°C to about 5°C.
- step i) is generally obtained as a solution or a suspension of the mixed carboxylic acid anhydride, and can be further used as such. If desired, this anhydride may be maintained at from about -60°C to about -20°C.
- the 7-amino-ceph-3-em-4-carboxylic acid of Formula II maybe silylated with silylation agents in a solvent inert under the reaction conditions.
- silylation agents include mono- or bissilylated amides, such as N,0-bis-(trimethylsilyl)acetamide (BSA), N-methyl-N-trimethylsilyl-acetamide (MSA); silylated ureas, such as N,N'-bis- (trimethylsilyl)-urea (BSU); or silazanes, such as 1 1 3,3,3- hexamethyldisilazane (HMDS), in combination with a halosilane such as trimethylchlorsilane, dimethyldichlorosilane, or an amine, such as triethylamine, tert.octylamine.
- a halosilane such as trimethylchlorsilane, dimethyldichlorosilane, or an amine, such
- the solvents used for mixed anhydride preparation above may be used for silylation of the 7-amino-ceph-3-em-4-carboxylic acid, and also for the step ii) reaction.
- the reaction temperatures for the step ii) may be from about -60°C to room temperature or from about -40 ° C to about -10°C.
- the reaction mixture of the step ii) may be worked up in a conventional manner.
- the substituted vinyl group may be split by hydrolysis in aqueous acid.
- the final product may be isolated in a conventional manner, for example by adjusting the pH of the reaction mixture.
- the product of Formula I can be obtained in a very high purity, for example above 98%.
- the 7-amino-ceph-3-em-4-carboxylic acid of Formula II may be prepared in accordance with any of the known methods (see U.S. Patent Nos. 3,867,380; 3 489,752 and 4,520,022).
- the Dane salt of Formula III may be obtained from commercial sources or prepared by methods well known in the art.
- compounds of Formula I having less than 0.5% by weight of the impurity of Formula B can be obtained.
- compounds of Formula I having less than 0.1% by weight of the impurity of Formula B, or less than 0.05% can be obtained.
- the resulting cephem compounds of Formula I may be formulated into ordinary dosage forms such as, for example, tablets, capsules, pills, solutions, etc. hi these cases, the medicaments can be prepared by conventional methods with conventional pharmaceutical excipients.
- compositions include dosage forms suitable for oral and parenteral (including subcutaneous, intramuscular, and ophthalmic) administration.
- the oral dosage forms may include solid dosage forms, like powder, tablets, capsules, as well as liquid suspensions.
- Parenteral dosage forms may include intravenous infusions, sterile solutions for intramuscular, subcutaneous or intravenous administration, dry powders to be reconstituted with sterile water for parenteral administration, and the like.
- the suspension of silylated 7-APCA was added to the above mixed anhydride at - 65 to - 70°C and further stirred for 60 minutes at -40 to -45°C. The temperature was raised to -20 to -25°C and further stirred for 90 minutes.
- a mixture of water (170ml) and 35% hydrochloric acid (35 ml) was added to the reaction mixture and stirred for 15 minutes at 0 to 5°C.
- the aqueous layer was diluted with dimethylformamide(700ml) and acetone (150 ml), and pH of the mixture adjusted to 6.5 with 25% ammonia solution.
- the mixture was stirred at 20-25°C for 2 hours and the separated solid was filtered.
- the solvate was washed dimethylformamide (100 ml) followed by acetone and dried at 40°C to yield 98g of cefprozil as dimethyl formamide (1.5 mole) solvate.
- the suspension of silylated 7-ADCA was added to the above mixed anhydride at - 65 to - 70°C and further stirred for 60 minutes at -40 to -45°C. The temperature was raised to -20 to -25°C and further stirred for 90 minutes.
- a mixture of water (170ml) and 35% hydrochloric acid (35 ml) was added to the reaction mixture and stirred for 15 minutes at 0 to 5°C.
- the aqueous layer was diluted with dimethylformamide (700ml) and acetone (150 ml), and pH of the mixture adjusted to 6.5 with 25% ammonia solution.
- the mixture was stirred at 20-25°C for 2 hours and the separated solid was filtered.
- the solvate was washed dimethylformamide (100 ml) followed by acetone and dried at 40°C to yield 105g of cefadroxil as dimethyl formamide (1.5 mole) solvate.
- cefadroxil dimethyl formamide solvate was converted to white crystalline solid cefadroxil monohydrate by the procedure reported in U.S. Patent No. 4,504,657.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Cephalosporin Compounds (AREA)
Abstract
Description
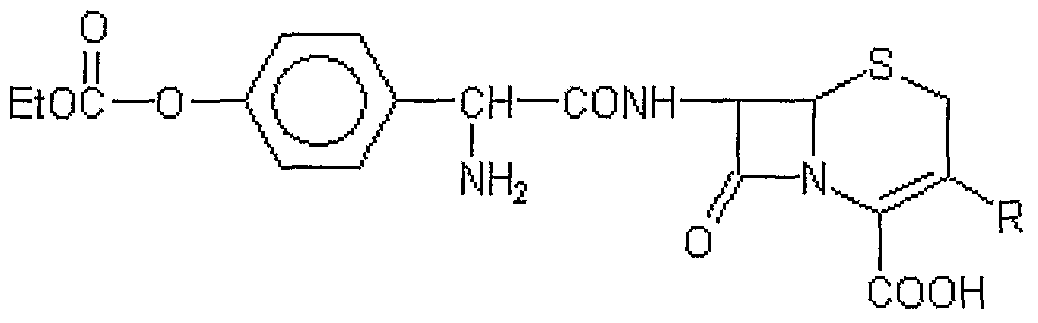





Claims





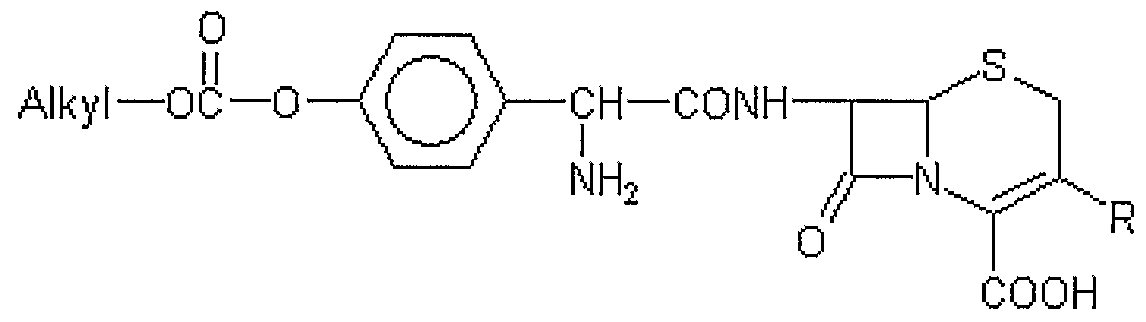
Priority Applications (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP04722335A EP1608661A2 (en) | 2003-03-21 | 2004-03-22 | Process for the preparaton of 7-(p-hydroxyphenylglycylamido)cephem compounds |
| CA002519853A CA2519853A1 (en) | 2003-03-21 | 2004-03-22 | Process for the preparation of 7-amino (p-hydroxyphenylglycylamido) cephem compounds |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| IN353DE2003 | 2003-03-21 | ||
| IN353/DEL/2003 | 2003-03-21 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2004083172A2 true WO2004083172A2 (en) | 2004-09-30 |
| WO2004083172A3 WO2004083172A3 (en) | 2004-12-02 |
Family
ID=33017825
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/IB2004/000850 WO2004083172A2 (en) | 2003-03-21 | 2004-03-22 | Process for the preparation of 7-amino (p-hydroxyphenylglyclyamido) cephem compounds |
Country Status (3)
| Country | Link |
|---|---|
| EP (1) | EP1608661A2 (en) |
| CA (1) | CA2519853A1 (en) |
| WO (1) | WO2004083172A2 (en) |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006048887A1 (en) * | 2004-11-01 | 2006-05-11 | Hetero Drugs Limited | A novel process for preparation of cefprozil intermediate |
| CN102408438A (en) * | 2010-09-26 | 2012-04-11 | 石药集团中奇制药技术(石家庄)有限公司 | Preparation method of cefprozil monohydrate |
| CN102443013A (en) * | 2010-10-10 | 2012-05-09 | 石药集团中奇制药技术(石家庄)有限公司 | Method for preparing cefprozil dimethyl formamide solvate |
| CN102911187A (en) * | 2012-10-11 | 2013-02-06 | 南通康鑫药业有限公司 | Recovery method of cefprozil |
| CN112694487A (en) * | 2020-12-29 | 2021-04-23 | 苏州盛达药业有限公司 | Preparation method of cefprozil |
| CN113533591A (en) * | 2021-06-18 | 2021-10-22 | 山东罗欣药业集团恒欣药业有限公司 | GC analysis method for benzene and paraldehyde in cefprozil |
Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3899394A (en) * | 1972-12-26 | 1975-08-12 | Bristol Myers Co | Production of antibacterial agents |
| US3940354A (en) * | 1973-01-31 | 1976-02-24 | Roussel-Uclaf | Cephalosporan compounds |
| US3988450A (en) * | 1974-04-10 | 1976-10-26 | Beecham Group Limited | Cephalosporins |
| US4139702A (en) * | 1976-12-16 | 1979-02-13 | Dobfar S.P.A. | Process for preparing cephalosporines |
| US4148817A (en) * | 1976-03-25 | 1979-04-10 | Eli Lilly And Company | Process and intermediates for preparing cephalosporin antibiotics |
| WO2004035593A1 (en) * | 2002-10-16 | 2004-04-29 | Orchid Chemicals & Pharmaceuticals Limited | An improved process for the preparation of cefadroxil |
| WO2004039812A1 (en) * | 2002-10-30 | 2004-05-13 | Orchid Chemicals & Pharmaceuticals Limited | Process for the preparation of 3-propenyl cephalosporin dmf solvate |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| KR820001564B1 (en) * | 1981-05-09 | 1982-09-02 | 동신제약 주식회사 | Process for preparing cephalosporin derivatives |
-
2004
- 2004-03-22 CA CA002519853A patent/CA2519853A1/en not_active Abandoned
- 2004-03-22 WO PCT/IB2004/000850 patent/WO2004083172A2/en not_active Application Discontinuation
- 2004-03-22 EP EP04722335A patent/EP1608661A2/en not_active Withdrawn
Patent Citations (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3899394A (en) * | 1972-12-26 | 1975-08-12 | Bristol Myers Co | Production of antibacterial agents |
| US3940354A (en) * | 1973-01-31 | 1976-02-24 | Roussel-Uclaf | Cephalosporan compounds |
| US3988450A (en) * | 1974-04-10 | 1976-10-26 | Beecham Group Limited | Cephalosporins |
| US4148817A (en) * | 1976-03-25 | 1979-04-10 | Eli Lilly And Company | Process and intermediates for preparing cephalosporin antibiotics |
| US4139702A (en) * | 1976-12-16 | 1979-02-13 | Dobfar S.P.A. | Process for preparing cephalosporines |
| WO2004035593A1 (en) * | 2002-10-16 | 2004-04-29 | Orchid Chemicals & Pharmaceuticals Limited | An improved process for the preparation of cefadroxil |
| WO2004039812A1 (en) * | 2002-10-30 | 2004-05-13 | Orchid Chemicals & Pharmaceuticals Limited | Process for the preparation of 3-propenyl cephalosporin dmf solvate |
Non-Patent Citations (4)
| Title |
|---|
| CASTELLVI, J. C. ET AL.: "Highly catalytic species in the formation of symmetrical anhydrides and mixt anhydrides" AFINIDAD, vol. 43, 1986, pages 421-424, XP009036673 * |
| HUANG, R. ET AL: "Synthesis of Cefadroxil" ZHONGGUO YIYAO GONGYE ZAZHI, vol. 21, no. 8, 1990, pages 343-345, XP009036672 * |
| KEMPERMANN, G. J. ET AL.: "Synthesis of Cephalosporin-Type Antibiotics by Coupling of Their beta-Lactam Nucleus and Racemic Amino Acid Side Chains Using a Clathration-Induced Asymmetric Transformation" EUR. J. ORG. CHEM., 2001, pages 1817-1820, XP009036646 * |
| KLEEMANN, A.; ENGEL, J.: "Pharmaceutical Substances; Synthesis, Patents, Application; A-M; 4th ed." 2001, THIEME , STUTTGART, NEW YORK , XP002296968 Pages 368-371 (Cefadroxil, Cefalexin), 376-377 (Cefatrizine), and 396-398 (cis-Cefprozil). * |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006048887A1 (en) * | 2004-11-01 | 2006-05-11 | Hetero Drugs Limited | A novel process for preparation of cefprozil intermediate |
| US7629482B2 (en) | 2004-11-01 | 2009-12-08 | Hetero Drugs Limited | Process for preparation of cefprozil intermediate |
| EP2213676A1 (en) * | 2004-11-01 | 2010-08-04 | Hetero Drugs Limited | A Novel process for preparation of cefprozil |
| CN102408438A (en) * | 2010-09-26 | 2012-04-11 | 石药集团中奇制药技术(石家庄)有限公司 | Preparation method of cefprozil monohydrate |
| CN102408438B (en) * | 2010-09-26 | 2015-01-07 | 石药集团中奇制药技术(石家庄)有限公司 | Preparation method of cefprozil monohydrate |
| CN102443013B (en) * | 2010-10-10 | 2014-09-17 | 石药集团中奇制药技术(石家庄)有限公司 | Method for preparing cefprozil dimethyl formamide solvate |
| CN102443013A (en) * | 2010-10-10 | 2012-05-09 | 石药集团中奇制药技术(石家庄)有限公司 | Method for preparing cefprozil dimethyl formamide solvate |
| CN102911187A (en) * | 2012-10-11 | 2013-02-06 | 南通康鑫药业有限公司 | Recovery method of cefprozil |
| CN102911187B (en) * | 2012-10-11 | 2015-03-11 | 南通康鑫药业有限公司 | Recovery method of cefprozil |
| CN112694487A (en) * | 2020-12-29 | 2021-04-23 | 苏州盛达药业有限公司 | Preparation method of cefprozil |
| CN112694487B (en) * | 2020-12-29 | 2022-04-22 | 苏州盛达药业有限公司 | Preparation method of cefprozil |
| CN113533591A (en) * | 2021-06-18 | 2021-10-22 | 山东罗欣药业集团恒欣药业有限公司 | GC analysis method for benzene and paraldehyde in cefprozil |
| CN113533591B (en) * | 2021-06-18 | 2022-08-23 | 山东罗欣药业集团恒欣药业有限公司 | GC analysis method for benzene and paraldehyde in cefprozil |
Also Published As
| Publication number | Publication date |
|---|---|
| WO2004083172A3 (en) | 2004-12-02 |
| EP1608661A2 (en) | 2005-12-28 |
| CA2519853A1 (en) | 2004-09-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US6552186B2 (en) | β-lactam production | |
| US7427692B2 (en) | Process for preparation of 7-[α-amino (4-hydroxyphenyl) acetamido]-3-substituted-3-cephem-4-carboxylic acid | |
| EP0233271B1 (en) | Process for preparing cephalosporin intermediates | |
| US5461043A (en) | Diastereomers of 1-(isopropoxycarbonyloxy)ethyl 3-cephem-4-carboxylate | |
| EP1068211B1 (en) | Process for purification of a cephalosporin derivative | |
| WO2004083172A2 (en) | Process for the preparation of 7-amino (p-hydroxyphenylglyclyamido) cephem compounds | |
| WO2002083634A2 (en) | Process for the preparation of cefpodoxime acid | |
| CA2123788C (en) | Cephem compound, process for producing the compound, and pharmaceutical composition containing the same | |
| US5998610A (en) | Silylation process | |
| CZ282160B6 (en) | Process for preparing cephepimdihydrochloride hydrate antibiotic and intermediate for preparing thereof | |
| RO109652B1 (en) | Hydrate dihydrochloride cefepim antibiotic preparation process | |
| CA2163080C (en) | Cephem compound, process for producing the compound, and antimicrobial composition containing the same | |
| EP1809638B1 (en) | A novel process for preparation of cefprozil intermediate | |
| US4868294A (en) | Process for preparing cephalosporin intermediates | |
| KR920001769B1 (en) | A process for preparing chlorocefadroxil monohydrate | |
| JP4616844B2 (en) | Production process of intermediates for use in the synthesis of cephalosporin | |
| KR840002046B1 (en) | Process for preparing cepharosporins | |
| JP2758413B2 (en) | Carbapenem compounds | |
| WO2005040175A2 (en) | Process for the preparation of cephem carboxylic acids | |
| US20040077849A1 (en) | Process for the preparation of cefadroxil | |
| EP1299396A1 (en) | Novel cephalosporin compounds and process for preparing the same | |
| AU2002307885A1 (en) | Process for the preparation of cefpodoxime acid | |
| JP2005521692A (en) | Novel cephalosporin compound and method for producing the same | |
| MXPA96005773A (en) | Cephalosporine compounds and processes for the preparation of mis |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): BW GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2519853 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004722335 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 4764/DELNP/2005 Country of ref document: IN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004722335 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2007072945 Country of ref document: US Ref document number: 10549821 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 10549821 Country of ref document: US |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2004722335 Country of ref document: EP |