WO2004082682A1 - Amino cyclobutylamide modulators of chemokine receptor activity - Google Patents
Amino cyclobutylamide modulators of chemokine receptor activity Download PDFInfo
- Publication number
- WO2004082682A1 WO2004082682A1 PCT/US2004/007792 US2004007792W WO2004082682A1 WO 2004082682 A1 WO2004082682 A1 WO 2004082682A1 US 2004007792 W US2004007792 W US 2004007792W WO 2004082682 A1 WO2004082682 A1 WO 2004082682A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- hydroxy
- 6alkyl
- 3alkyl
- substituents
- fluoro
- Prior art date
Links
- 0 C[C@@](C*CC1)[C@@]1(C=C1)c2c1cccc2 Chemical compound C[C@@](C*CC1)[C@@]1(C=C1)c2c1cccc2 0.000 description 4
- PHEYRUKKZRZCPY-UHFFFAOYSA-N C(COCC1)C1NC(c1ccccc1)c1ccccc1 Chemical compound C(COCC1)C1NC(c1ccccc1)c1ccccc1 PHEYRUKKZRZCPY-UHFFFAOYSA-N 0.000 description 1
- XVPVTRVRKBGCPN-UHFFFAOYSA-N CC(C)(C)OC(NCc1cc(C(F)(F)F)cc(F)c1)=O Chemical compound CC(C)(C)OC(NCc1cc(C(F)(F)F)cc(F)c1)=O XVPVTRVRKBGCPN-UHFFFAOYSA-N 0.000 description 1
- ZBEQUADQHVLQLF-UHFFFAOYSA-N CC(C)(CNCC1)C1c(cc1)ccc1F Chemical compound CC(C)(CNCC1)C1c(cc1)ccc1F ZBEQUADQHVLQLF-UHFFFAOYSA-N 0.000 description 1
- PNWOXVZLIZYXQP-UHFFFAOYSA-N CC(C1)(CC1=O)C(OCc1ccccc1)=O Chemical compound CC(C1)(CC1=O)C(OCc1ccccc1)=O PNWOXVZLIZYXQP-UHFFFAOYSA-N 0.000 description 1
- KIJYUDMBYHXDEJ-UHFFFAOYSA-N CN(C(C1)CC1(Cc1ccccc1)C(NCc1cc(F)cc(C(F)(F)F)c1)=O)C1CCOCC1 Chemical compound CN(C(C1)CC1(Cc1ccccc1)C(NCc1cc(F)cc(C(F)(F)F)c1)=O)C1CCOCC1 KIJYUDMBYHXDEJ-UHFFFAOYSA-N 0.000 description 1
- HCIALJVMQBFLCV-UHFFFAOYSA-N COC(C1)(CC1C(OCc1ccccc1)=O)OC Chemical compound COC(C1)(CC1C(OCc1ccccc1)=O)OC HCIALJVMQBFLCV-UHFFFAOYSA-N 0.000 description 1
- ULKJALSRKSAPBD-HFDPZAEDSA-N C[C@@H](CN(CC1)C(C2)CC2(C)C(NCc2cc(C(F)(F)F)cc(C(F)(F)F)c2)=O)[C@@]1(C=C1)c2c1cccc2 Chemical compound C[C@@H](CN(CC1)C(C2)CC2(C)C(NCc2cc(C(F)(F)F)cc(C(F)(F)F)c2)=O)[C@@]1(C=C1)c2c1cccc2 ULKJALSRKSAPBD-HFDPZAEDSA-N 0.000 description 1
- GXBVMAMNZFJTLD-BXKDBHETSA-N C[C@H](CNCC1)[C@@H]1c(cc1)ccc1F Chemical compound C[C@H](CNCC1)[C@@H]1c(cc1)ccc1F GXBVMAMNZFJTLD-BXKDBHETSA-N 0.000 description 1
- BWFBFKPZLKQYPR-UHFFFAOYSA-N N#Cc(cc(C(F)(F)F)cn1)c1Cl Chemical compound N#Cc(cc(C(F)(F)F)cn1)c1Cl BWFBFKPZLKQYPR-UHFFFAOYSA-N 0.000 description 1
- CLWJQHAEFHSMBM-UHFFFAOYSA-N N#Cc(cc(C(F)(F)F)cn1)c1O Chemical compound N#Cc(cc(C(F)(F)F)cn1)c1O CLWJQHAEFHSMBM-UHFFFAOYSA-N 0.000 description 1
- AHVQYHFYQWKUKB-UHFFFAOYSA-N NC1CCOCC1 Chemical compound NC1CCOCC1 AHVQYHFYQWKUKB-UHFFFAOYSA-N 0.000 description 1
- YYZHUSBRLJVFTG-UHFFFAOYSA-N NCc1cncc(C(F)(F)F)c1 Chemical compound NCc1cncc(C(F)(F)F)c1 YYZHUSBRLJVFTG-UHFFFAOYSA-N 0.000 description 1
- HDOWRFHMPULYOA-UHFFFAOYSA-N OC1CCNCC1 Chemical compound OC1CCNCC1 HDOWRFHMPULYOA-UHFFFAOYSA-N 0.000 description 1
- BYRJSCNPUHYZQE-UHFFFAOYSA-N Oc1ccc(C(F)(F)F)cn1 Chemical compound Oc1ccc(C(F)(F)F)cn1 BYRJSCNPUHYZQE-UHFFFAOYSA-N 0.000 description 1
- UWHKLLYQUQPOQK-UHFFFAOYSA-N Oc1ncc(C(F)(F)F)cc1Br Chemical compound Oc1ncc(C(F)(F)F)cc1Br UWHKLLYQUQPOQK-UHFFFAOYSA-N 0.000 description 1
- RFGNOPWHPRCTPY-UHFFFAOYSA-N Oc1ncc(C(F)(F)F)cc1C=O Chemical compound Oc1ncc(C(F)(F)F)cc1C=O RFGNOPWHPRCTPY-UHFFFAOYSA-N 0.000 description 1
- AHVQYHFYQWKUKB-UHFFFAOYSA-O [NH3+]C1CCOCC1 Chemical compound [NH3+]C1CCOCC1 AHVQYHFYQWKUKB-UHFFFAOYSA-O 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D221/00—Heterocyclic compounds containing six-membered rings having one nitrogen atom as the only ring hetero atom, not provided for by groups C07D211/00 - C07D219/00
- C07D221/02—Heterocyclic compounds containing six-membered rings having one nitrogen atom as the only ring hetero atom, not provided for by groups C07D211/00 - C07D219/00 condensed with carbocyclic rings or ring systems
- C07D221/20—Spiro-condensed ring systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/16—Otologicals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/10—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with radicals containing only carbon and hydrogen atoms attached to ring carbon atoms
- C07D211/14—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with radicals containing only carbon and hydrogen atoms attached to ring carbon atoms with hydrocarbon or substituted hydrocarbon radicals attached to the ring nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/08—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms
- C07D211/18—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hydrocarbon or substituted hydrocarbon radicals directly attached to ring carbon atoms with substituted hydrocarbon radicals attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/40—Oxygen atoms
- C07D211/42—Oxygen atoms attached in position 3 or 5
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/40—Oxygen atoms
- C07D211/44—Oxygen atoms attached in position 4
- C07D211/52—Oxygen atoms attached in position 4 having an aryl radical as the second substituent in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/56—Nitrogen atoms
- C07D211/58—Nitrogen atoms attached in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/60—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D211/62—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals attached in position 4
- C07D211/64—Carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals attached in position 4 having an aryl radical as the second substituent in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/68—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member
- C07D211/70—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having one double bond between ring members or between a ring member and a non-ring member with only hydrogen atoms, hydrocarbon or substituted hydrocarbon radicals, directly attached to ring carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D295/00—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms
- C07D295/04—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms
- C07D295/14—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals
- C07D295/155—Heterocyclic compounds containing polymethylene-imine rings with at least five ring members, 3-azabicyclo [3.2.2] nonane, piperazine, morpholine or thiomorpholine rings, having only hydrogen atoms directly attached to the ring carbon atoms with substituted hydrocarbon radicals attached to ring nitrogen atoms substituted by carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals with the ring nitrogen atoms and the carbon atoms with three bonds to hetero atoms separated by carbocyclic rings or by carbon chains interrupted by carbocyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D309/00—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings
- C07D309/02—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members
- C07D309/08—Heterocyclic compounds containing six-membered rings having one oxygen atom as the only ring hetero atom, not condensed with other rings having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D309/14—Nitrogen atoms not forming part of a nitro radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/08—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing alicyclic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D405/00—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom
- C07D405/02—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings
- C07D405/12—Heterocyclic compounds containing both one or more hetero rings having oxygen atoms as the only ring hetero atoms, and one or more rings having nitrogen as the only ring hetero atom containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/10—Spiro-condensed systems
Definitions
- chemokines are a family of small (70-120 amino acids), proinflammatory cytokines, with potent chemotactic activities. Chemokines are chemotactic cytokines that are released by a wide variety of cells to attract various cells, such as monocytes, macrophages, T cells, eosinophils, basophils and neutrophils to sites of inflammation (reviewed in Schall, Cytokine, 3, 165-183 (1991) and
- ⁇ -chemokines such as interleukin-8 (IL-8), neutrophil-activating protein-2 (NAP-2) and melanoma growth stimulatory activity protein (MGSA) are chemotactic primarily for neutrophils
- ⁇ -chemokines such as RANTES, MIP- 1 , MIP- 1 ⁇ , monocyte chemotactic protein- 1 (MCP- 1 ), MCP-2, MCP-3 and eotaxin are chemotactic for macrophages, monocytes, T-cells, eosinophils and basophils (Deng, et al., Nature. 381, 661-666 (1996)).
- the chemokines are secreted by a wide variety of cell types and bind to specific G-protein coupled receptors (GPCRs) (reviewed in Horuk, Trends Pharm. Sci, 15, 159-165 (1994)) present on leukocytes and other cells. These chemokine receptors form a sub-family of GPCRs, which, at present, consists of fifteen characterized members and a number of orphans. Unlike receptors for promiscuous chemoattractants such as C5a, fMLP, PAF, and LTB4, chemokine receptors are more selectively expressed on subsets of leukocytes. Thus, generation of specific chemokines provides a mechanism for recruitment of particular leukocyte subsets.
- GPCRs G-protein coupled receptors
- chemokine receptors On binding their cognate ligands, chemokine receptors transduce an intracellular signal though the associated trimeric G protein, resulting in a rapid increase in intracellular calcium concentration.
- CCR-1 or "CKR-1" or "CC-CKR-1” [M-P-l ⁇ , MEP-l ⁇ , MCP-3, RANTES] (Ben-Barruch, et al., J. Biol.
- the ⁇ - chemokines include eotaxin, MIP ("macrophage inflammatory protein"), MCP ("monocyte chemoattractant protein”) and RANTES ("regulation-upon-activation, normal T expressed and secreted”) among other chemokines.
- Chemokine receptors such as CCR-1, CCR-2, CCR-2A, CCR-2B, CCR-3, CCR-4, CCR-5, CXCR-3, CXCR-4, have been implicated as being important mediators of inflammatory and immunoregulatory disorders and diseases, including asthma, rhinitis and allergic diseases, as well as autoimmune pathologies such as rheumatoid arthritis and atherosclerosis.
- Humans who are homozygous for the 32- basepair deletion in the CCR-5 gene appear to have less susceptibility to rheumatoid arthritis (Gomez, et al. Arthritis & Rheumatism, 42, 989-992 (1999)).
- chemokines are potent chemoattractants for monocytes and macrophages.
- MCP-1 monocyte chemoattractant protein- 1
- CCR2 primary receptor 2
- MCP-1 is produced in a variety of cell types in response to inflammatory stimuli in various species, including rodents and humans, and stimulates chemotaxis in monocytes and a subset of lymphocytes. In particular, MCP-1 production correlates with monocyte and macrophage infiltration at inflammatory sites.
- agents which modulate chemokine receptors such as the CCR-2 receptor would be useful in such disorders and diseases.
- the recruitment of monocytes to inflammatory lesions in the vascular wall is a major component of the pathogenesis of atherogenic plaque formation.
- MCP-1 is produced and secreted by endothelial cells and intimal smooth muscle cells after injury to the vascular wall in hypercholesterolemic conditions.
- Monocytes recruited to the site of injury infiltrate the vascular wall and differentiate to foam cells in response to the released MCP-1.
- CCR2 antagonists may inhibit atherosclerotic lesion formation and pathological progression by impairing monocyte recruitment and differentiation in the arterial wall.
- the present invention is directed to compounds which are modulators of chemokine receptor activity and are useful in the prevention or treatment of certain inflammatory and immunoregulatory disorders and diseases, allergic diseases, atopic conditions including allergic rhinitis, dermatitis, conjunctivitis, and asthma, as well as autoimmune pathologies such as rheumatoid arthritis and atherosclerosis.
- the invention is also directed to pharmaceutical compositions comprising these compounds and the use of these compounds and compositions in the prevention or treatment of such diseases in which chemokine receptors are involved.
- the present invention is directed to compounds of formula I and of formula II:
- X is selected from O, N, S, SO2, or C.
- Y is selected from:
- Rl 1 is independently selected from: hydroxy, hydrogen,
- Rl2 is selected from: hydrogen, C ⁇ _ alkyl, benzyl, phenyl, C3-6 cycloalkyl where the alkyl, phenyl, benzyl, and cycloalkyl groups can be unsubstituted or substituted with 1-3 substituents where the substituents are independently selected from: halo, hydroxy, Ci-3alkyl, Ci-3alkoxy, -CO2H, -CO2-C1-6 alkyl, and triflu
- Z is independently selected from C or N, where at most two of the Z are N.
- R 1 is selected from: hydrogen, -C ⁇ _6alkyl, -Crj-6alkyl-0-Ci-6alkyl, -C()-6alkyl-S-Ci-6alkyl, -(C ⁇ -6alkyl) ⁇ (C3-7cycloalkyl)-(C ⁇ -6alkyl), hydroxy, heterocycle, -CN, -NR 12 Rl2 -NR12C0R 13 , -NRl2S02R 14 , -COR 11 , -CONR12R12, and phenyl; the alkyl and the cycloalkyl are unsubstituted or substituted with 1-7 substituents where the substituents are independently selected from:
- R2 is selected from:
- R3 is selected from:
- R 4 is selected from:
- R5 is selected from: (a) Ci- ⁇ alkyl, where alkyl may be unsubstituted or substituted with
- (k) phenyl which may be unsubstituted or substituted with one or more substituents selected from: halo, trifluoromethyl, Ci-
- R6 is selected from:
- R7 is selected from: hydrogen, (C()-6alkyl)-phenyl, (C ⁇ -6alkyl)-heterocycle, (C ⁇ -6alkyl)-C3_
- R7 and R ⁇ may be joined together to form a ring which is selected from:
- R ⁇ and R ⁇ or R ⁇ and RtO may be joined together to form a ring which is phenyl or heterocycle, wherein the ring is unsubstituted or substituted with 1-7 substituents where the substituents are independently selected from:
- R9 and R 1 ⁇ are independently selected from: (a) hydrogen,
- R 1 ⁇ is selected from:
- Ci- ⁇ alkyl which is unsubstituted or substituted with 1-3 substituents where the substituents are independently selected from: halo, hydroxy, -CO2H, -C ⁇ 2Ci-6alkyl, and -O-C1- 3alkyl;
- R 1 ⁇ is selected from:
- Ci-6alkyl where alkyl may be unsubstituted or substituted with
- substituents where the substituents are selected from: fluoro, Ci-3alkoxy, hydroxy, -COR 11 ,
- R 1 ⁇ is selected from:
- substituents where the substituents are selected from: fluoro, C ⁇ _3alkoxy, hydroxy, -COR 11 ,
- alkyl may be unsubstituted or substituted with 1-6 substituents where the substituents are selected from: fluoro, C ⁇ _3alkoxy, hydroxy, -COR 11 , or R 1 ⁇ and R 1 ⁇ may be joined together by a Ci-4alkyl chain or a C ⁇ -3alkyl-0-C ⁇ -3alkyl chain to form a 3-6 membered ring;
- R 1 ⁇ is selected from:
- Ci-6alkyl where alkyl may be unsubstituted or substituted with
- alkyl may be unsubstituted or substituted with 1-6 fluoro, or R 1 ⁇ and R 1 ⁇ may be joined together by a C2-3alkyl chain to form a 5-6 membered ring, where the alkyl are unsubstituted or substituted with 1-3 substituents where the substiuents are independently selected from: halo, hydroxy, -COR 11 , Ci_3alkyl, and Ci-3alkoxy, or R ⁇ and R 1 ⁇ may be joined together by a Ci-2alkyl-0-Ci-2alkyl chain to form a 6-8 membered ring, where the alkyl are unsubstituted or substituted with 1-3 substituents where the substiuents are independently selected from: halo, hydroxy, -COR 11 , Ci-3alkyl, and Ci-3alkoxy, or R 1 ⁇ and R ⁇ may be joined together by a -0-C ⁇
- R ⁇ is selected from:
- Ci- ⁇ alkyl which may be substituted or unsubstituted with 1-6 of the following substituents: -COR 1 1, hydroxy, fluoro, chloro, -0-Ci-3alkyl; or
- R2 and R!9 can also be joined together to form a heterocycle ring with a linker selected from the following list (with the left side of the linker being bonded to the amide nitrogen at R!9):
- R28 is selected from selected from:
- R28 is connected to the ring via a double bond (in which case the other R28 at the same position is nothing, and where R29 is selected from: (a) hydrogen,
- C ⁇ _6alkyl which may be substituted or unsubstituted with 1-6 of the following substituents: -COR 1 1, hydroxy, fluoro, chloro, -0-Ci-3alkyl;
- n is selected from 1 or 2;
- the dashed line represents a single or a double bond
- a further embodiment of the present invention includes compounds of formula la.
- a further embodiment of the present invention includes compounds of formula Hb.
- Rl, R3, R5, R10 J and Z are described above, and R23 and R24 are independently selected from: (a) hydrogen,
- Rl, R3, R5, R10 ? R23, and R24 are described above, and pharmaceutically acceptable salts and individual diastereomers thereof.
- X is C, O or N.
- X is C.
- Y is -CH2- or -O-
- Z is C.
- R 1 is selected from: hydrogen, phenyl, heterocycle, -Ci-6alkyl, -C ⁇ -6alkyl-0-Ci-6alkyl, and -(Co-6alkyl)-(C3-7cycloalkyl)-(Co-6alkyl), where the alkyl, phenyl, heterocycle, and the cycloalkyl are unsubsti toted or substituted with 1-7 substituents where the substituents are independently selected from:
- R is selected from:
- R 1 is selected from: (a) hydrogen, (b) Ci-6alkyl, which may be unsubstituted or substituted with 1-6 substituents independently selected from: fluoro and hydroxy
- R 1 is selected from:
- R2 is hydrogen
- R3 is nothing.
- R is selected from:
- R3 is selected from:
- R 4 is hydrogen
- R5 is selected from: (a) C ⁇ _6alkyl substituted with 1-6 fluoro, (b) -0-Ci-6alkyl substituted with 1-6 fluoro,
- R ⁇ is selected from:
- R ⁇ is trifluoromethyl
- R ⁇ is hydrogen
- R7 is phenyl, heterocycle, C3-7cycloalkyl
- phenyl, heterocycle, C3_7cycloalkyl, and Ci-6alkyl is unsubstituted or substituted with 1-5 substitoents where the substituents are independently selected from:
- R ⁇ is phenyl, heterocycle, Ci-4alkyl, -COR 1 1, and -CONH-V-COR 1 1;
- V is selected from Ci ⁇ 6alkyl or phenyl; and the phenyl, heterocycle, and C ⁇ _4alkyl is unsubstituted or substituted with 1-3 substituents where the substituents are independently selected from: (a) halo,
- X is C, R ⁇ is hydrogen
- R9 is hydrogem
- R 1 ⁇ is selected from: (a) hydrogen, (b) hydroxy,
- R ⁇ is hydrogen or methyl.
- R 1 ⁇ is selected from:
- Ci-3alkyl which is unsubstituted or substituted with 1-6 fluoro
- R ⁇ is selected from:
- R 1 is hydrogen
- R 1 ⁇ is selected from:
- R 1 ⁇ is hydrogen
- R 1 ⁇ and R 1 ⁇ are joined together by a -CH2CH2- chain or a -CH2CH2CH2- chain to form a cyclopentyl ring or a cyclohexyl ring.
- R 5 is hydrogen.
- R26 is oxygen and connected via a double bond.
- n 1.
- Representative compounds of the present invention include those presented in the Examples and pharmaceutically acceptable salts and individual diastereomers thereof.
- the compounds of the instant invention have at least two asymmetric centers at the 1- and 3-positions of the cyclobutyl ring. Additional asymmetiic centers may be present depending upon the natore of the various substituents on the molecule. Each such asymmetric center will independently produce two optical isomers and it is intended that all of the possible optical isomers and diastereomers in mixtures and as pure or partially purified compounds are included within the ambit of this invention.
- the absolute configurations of selected compounds of this orientation, with substituents on the cyclopentyl ring (amide and amine units), are as depicted below:
- Keto-acid 1-1 (the preparation of which is described in Scheme 2) can first be protected as the corresponding ester, where R27 represents an alkyl such as methyl, ethyl, tert-butyl or benzyl which serves as a protecting group (Greene, T; Wuts, P. G. M. Protective Groups in Organic Synthesis, John Wiley & Sons, Inc., New York, NY 1991).
- Reductive amination of 1-2 with an amine preferably of the from 1-3 (a preparation of which is depicted in Scheme 3) in the presence of a reducing agent such as sodium cyanoborhydride or sodium triacetoxyborohydride gives amino-ester 1-4.
- ester 1-4 Conversion of ester 1-4 to the carboxylic acid 1-5 can be achieved by a number of conditions depending on the nature of the ester.
- methyl or ethyl esters can be readily saponified with sodium hydroxide, or lithium hydroxide; tert- butyl ester can be removed by treatment with TFA.
- Coupling of the acid 1-5 with amine 1-6 (a preparation of which is described in Scheme 4), to give chemokine modulators of the form 1-7, can be accomplished by the standard amide bond formation conditions using a coupling reagent such as DCC, EDC and a catalyst such as DMAP, HOBT or HO AT.
- Alternativly 1-1 can be directly coupled to amine 1-6 to give the keto-amide 1-8.
- Reductive amination with amine 1-3 in the presence of a borohydride such as sodium triacetoxyborohydride or sodium cyanoborohydride then provides the chemokine modulator 1-7.
- Scheme 1A depicts the preparation of chemokine modulators of the form 1-14.
- Intermediate 2-2a (described in Scheme 2) is first reduced to the primary amine 1-9 by catalytic hydrogenation using Raney nickel. Protection of the amine with the appropriate protecting group, such as a tert-butylcarbamate, by treatment with di-tert- butyl dicarbonate followed by reductive amination with amine 1-3 gives intermediate 1-11. Removal of the protecting group, with for example HCl in dioxane or TFA for the boc protected amine gives the free amine 1-12. Acylation of the amine with an acid chloride (1-13) gives the chemokine modulator 1-14. Alternatively the amine can be coupled (as described in Scheme 1) to an appropriate benzoic acid (not shown) to give similar amides 1-14.
- SCHEME 2 Scheme 2
- the keto cyclobutanoic acid (1-1) can be readily synthesized from commercially available materials.
- the initial protected intermediates of the form 2-2 can be made by a double alkylation reaction of an active ester (2- lb) or nitrile (2- la) with 1,3-dibromo 2,2-dimethoxypropane, using a base such as sodium hydride. Removal of the dimethyl acetal and the hydrolysis of the ester or nitrile can be accomplished under acidic conditions in one reaction step to give intermediate 1-1.
- Amines 1-3 were obtained from various sources. Some were commercially available, some were known from the literature and could be prepared according to published procedures, and some were prepared as described herein. Since their structures and the methods for their preparation are diverse, only one Scheme will be outlined in this section; individual syntheses of amines 1-3 can be found in the Experimental Section.
- Scheme 3 shows one method for the synthesis of 4-aryl substitoted piperidines as well as 4-aryl-3-alkyl-piperidines.
- Enol triflate 3-1 (prepared according to Wustrow, D. J., Wise, L. D., Synthesis, (1991), 993-995.) could be coupled to boronic acids 3-2 as described by Wustrow and Wise.
- Hydrogenation of the olefin in 4-3 could be achieved using hydrogen in the presence of a catalyst such as Pd(OH)2/C.
- Oxidation of 3-4 using Ru(IV)oxide hydrate and sodium periodate leads to Boc-lactam 3-5.
- Alkylation with an alkyl halide in the presence of a base such as LDA gives 3-6, with the trans product being predominant.
- Removal of the Boc protecting group could be achieved using standard acidic conditions, such as HCl in dioxane or TFA/DCM.
- Reduction of the lactam 3-7 with, for example borane provides 1-3.2.
- intermediate 3-4 can itself be deprotected under acidic conditions to afford piperidine 1-3.1.
- Amines of the form 1-6 are synthesized in a variety of ways.
- An example of such a synthesis is depicted in Scheme 4.
- the reduction of the nitrile in the presence of an aromatic halide to the corresponding amine can be successfully accomplished e.g. with borane in THF.
- Step B To a cooled (0 °C) solution of ethanolamine (41.8 g, 0.685 mol) in water (90 mL) was added neat (R)-propylene oxide (4.97 g, 85.6 mmol), dropwise. After 1 h at 0 °C the reaction was allowed to rise to room temperature and stirred overnight. " The reaction mixture was concentrated at -80 °C in vacuo to remove the water and most of the ethanolamine, to give 11.79 g of crude product, containing some residual ethanolamine. This material was used without further purification in Step B. Step B
- Step B (860mg, 3.13 mmol) in THF (5mL) was added slowly. The reaction mixture was stirred at -78°C for 30 minutes before Mel (584/ ⁇ L, 9.38 mmol) was added. The reaction was warmed up to room temperatore slowly and stirred overnight. Saturated NH4CI was added and the solution was extracted with EtOAc (3x). Combined organic layer was dried over anhydrous MgS04 and concentrated in vacuo. The crude product was purified by MPLC (20:80, EtOAc :Hexanes) to yield 3-C (383mg, 42.4%).
- reaction mixture was washed with 5% citric acid solution (100 mL), water (2 x 100 mL), saturated NaCl (50 mL), dried over MgS04, filtered and concentrated in vacuo to give 13.5 g crude product, which was used without further purification in step B.
- Step B To a mixture of the amide (Step B) (73 g, 256 mmol) and paraformaldehyde (11.5 g, 385 mmol) was added 200 mL of acetic acid. The reaction mixture was stirred at room temperature for 5 min before concentrated sulfuric acid (200 mL). An exothermic reaction was observed. After 30 min, TLC showed a complete conversion. The mixtore was cooled to RT before poured onto ice water (2000 mL) and extracted with EtOAc (3 x 500 mL). Combined organic layers were washed with water (2x), saturated NaHC03, and brine, dried over MgS04, filtered, evaporated and dried in vacuum.
- EtOAc 3 x 500 mL
- Cis and ttans isomers were also separated (cis 90mg, trans 23mg, 73.4%) Cis was the less polar and more active isomer.
- Cis isomer NMR (500 MHz, CDCI3) ⁇ 7.81 (s, IH), 7.78
- Example 16 A solution of Example 16 (40 mg, 0.08 mmol), 37% formaldehyde (20 ⁇ L, 0.24 mmol), DIEA (25 ⁇ L, 0.12 mmol), TFA (5/iL), NaCNBH (25 mg, 0.40 mmol), and MeOH (1.5 mL) was stirred at room temperature and the reaction was monitored by TLC. The crude reaction was purified by preparative TLC (5:0.5:94.5, MeOH:NH4 ⁇ H:DCM). Cis and ttans isomers were separated with the more polar isomer being the cis and more active isomer. LC-MS for C26H28F6N2O2 MW calculated 514.21, found 515.35.
- Example 42 was synthesized from Example 21 using the procedure detailed in
- Example 41 Cis and trans isomers were synthesized separately from the cis and trans isomers of Example 41, with the ttans being the more polar and active isomer.
- Step D the product from Step D (400 mg, 0.873 mmol) was stirred in a solution of 95% TFA in water (5 mL). The reaction was monitored by HPLC. Upon completion of reaction, the mixture was concenttated in vacuo, redissolved in a minimum amount of DCM, and washed with saturated NaHC03 (4x) to get rid of TFA. The organic layer was dried over anhydrous MgS04 and concentrated in vacuo to yield the desired product (208 mg, 66.5%). The crude product was used on next step.
- Example 57 (15 mg, 0.028 mmol) and Pd/C (5 mg) were stirred in EtOH (7mL) under hydrogen overnight. The reaction was filtered through celite and concenttated in vacuo to yield Example 65 (13.6 mg, 90.7%). LC-MS for C29H32F6N2O MW calculated 538.24, found 5.39.2. EXAMPLE 66
- Example 58 (15 mg, 0.028 mmol) and Pd/C (5 mg) were stirred in EtOH (7mL) under hydrogen overnight. The reaction was filtered through celite and concenttated in vacuo to yield Example 66 (15 mg, 100%). LC-MS for C28H32F4N2O MW calculated 488.25, found 489.25.
- Example 75 A solution of Example 75 (10 mg, 0.025 mmol), formaldehyde (6 ⁇ L, 0.08 mmol), DIEA (7 ⁇ L, 0.04 mmol), TFA (2.5 ⁇ L), NaCNBH (9 mg, 0.1 mmol), and MeOH (l/2mL) was stirred at room temperature. The reaction was monitored by TLC. Upon completion of reaction, the reaction mixture was purified by preparative TLC (3:0.3:96.7, MeOH:NH4 ⁇ H:DCM) to yield Example 77 (6.2mg, 60.2%). LC-MS for C21H28F4N2O2 MW calculated 416.21, found 417.25.
- Example 78 was synthesized from Example 76 according to the procedure detailed in Example 77. The crude product was purified by preparative TLC (5:0.5:94.5, MeOH:NH4 ⁇ H:DCM). LC-MS for C26H30F4N2O2 MW calculated 478.22, found
- Example 79 was synthesized according to the procedure detailed in Example 67, Step F. LC-MS for C30H34F4N2O3 MW calculated 578.22, found 579.25.
- Example 80 (36mg, 46.4%).
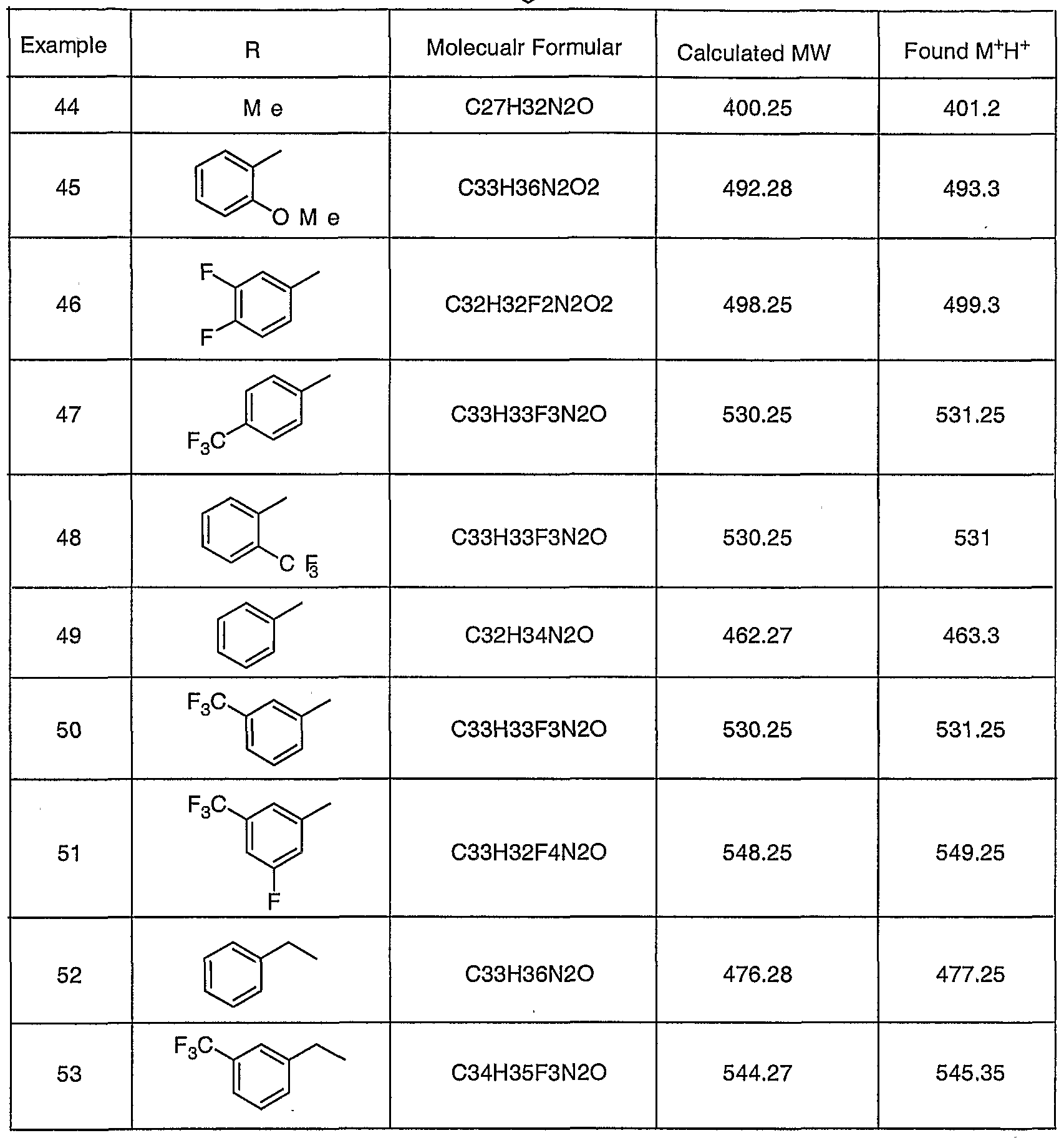
- R2 was derivatized by using either 2-iodoprotane or acetone as the alkylating agent in Step C.
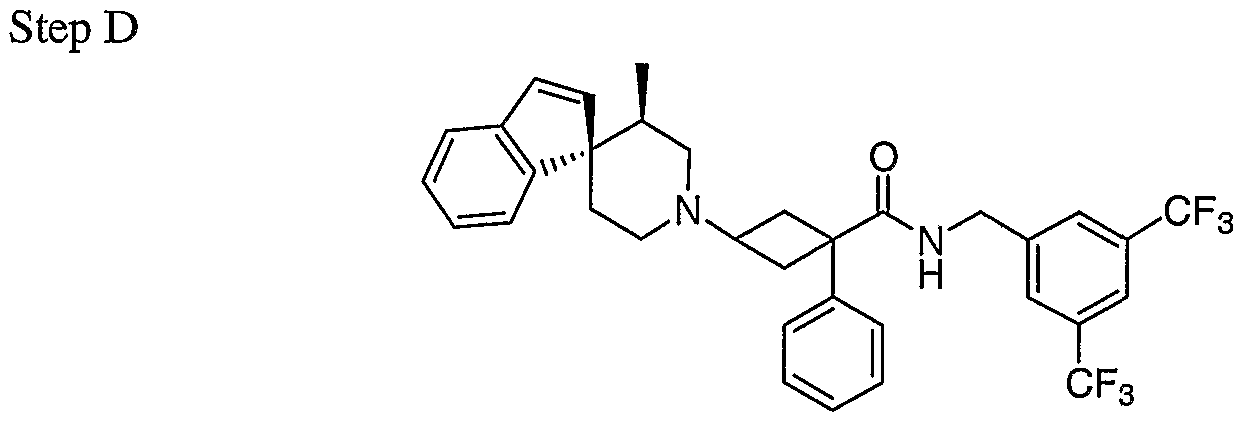
- Rl was derivatized by using different benzylamine in Step E. 1.5 equivalent of DIEA was added for hydrochloride benzylamine.
- R3 was derivatized by incorporating different amines in Step F. All of the components are either commercially available or are described in the Intermediates section. Isomers for some of these compounds were separated by preparative TLC. A few most active ones were resolved using chiral chromatography. A summary of these compounds is listed in the table below.
- Example 83 A solution of Example 83 (40 mg, 0.10 mmol), 37% formaldehyde (25 ⁇ -L, 0.30 mmol), DIEA (23 ⁇ L, 0.15 mmol), TFA (10 ⁇ L), NaCNBH (28 mg, 0.50 mmol), and MeOH (3 mL) was stirred at room temperature and the reaction was monitored by TLC. The crude reaction was purified by preparative TLC (5:0.5:94.5, MeOH:NH4 ⁇ H:DCM). LC-MS for C22H30F4N2O2 MW calculated 430, found
- Example 118 was synthesized from Example 91 using the procedure detailed in Example 117. LC-MS for C23H30F4N2O2 MW calculated 480, found 481.
- Example 85 (15 mg, 0.029 mmol) was stirred in 4 M HCl in dioxane (5 mL). The reaction was monitored by HPLC. Upon completion of reaction, the mixtore was concentrated in vacuo to yield Example 119. LC-MS for C21H29F4N3O2 MW calculated 415, found 416.
- Example 120 was synthesized from Example 92 using the procedure detailed in Example 119. LC-MS for C22H29F6N3O MW calculated 465, found 466.
- the cis isomer was the less polar peak and the more active isomer.
- the cis isomer was further separated by chiral HPLC (AD, 10% EtOH/heptane) to give two enantiomers.
- a variety of compounds were synthesized according to the procedure detailed in Example 122.
- Rl was derivatized by using either 2-iodopropane or acetone as the alkylating agent in Step A.
- R2 was derivatized by using a different amine in Step C.
- R3 was derivatized by incorporating different amines.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Immunology (AREA)
- Rheumatology (AREA)
- Vascular Medicine (AREA)
- Ophthalmology & Optometry (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Pain & Pain Management (AREA)
- Pulmonology (AREA)
- Urology & Nephrology (AREA)
- Dermatology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Physical Education & Sports Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Other In-Based Heterocyclic Compounds (AREA)
- Hydrogenated Pyridines (AREA)
- Pyrane Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Abstract
Description











































































































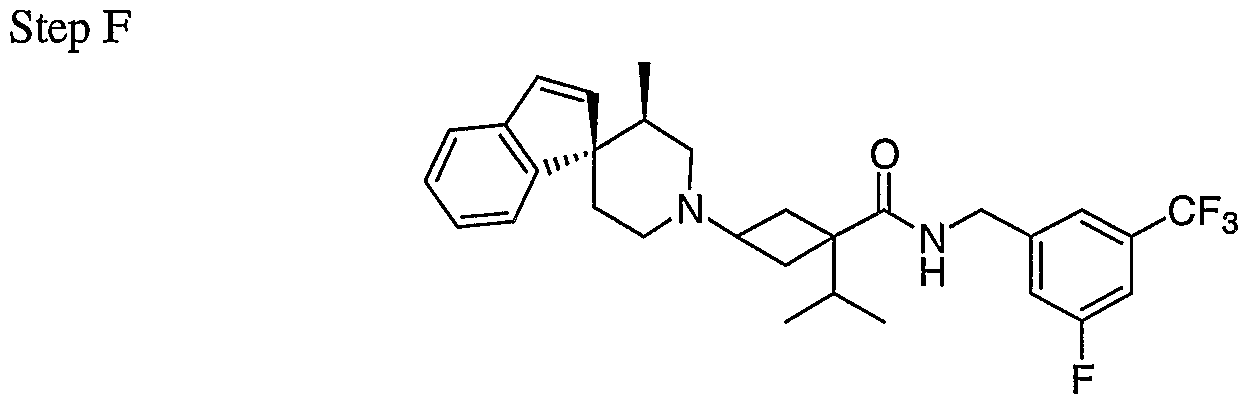

















Claims









Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP04720791A EP1617841A4 (en) | 2003-03-18 | 2004-03-15 | Amino cyclobutylamide modulators of chemokine receptor activity |
| US10/549,739 US7553841B2 (en) | 2003-03-18 | 2004-03-15 | Amino cyclobutylamide modulators of chemokine receptor activity |
| JP2006507176A JP2006520783A (en) | 2003-03-18 | 2004-03-15 | Aminocyclobutyramide modulators of chemokine receptor activity |
| CA002519220A CA2519220A1 (en) | 2003-03-18 | 2004-03-15 | Amino cyclobutylamide modulators of chemokine receptor activity |
| AU2004222336A AU2004222336B2 (en) | 2003-03-18 | 2004-03-15 | Amino cyclobutylamide modulators of chemokine receptor activity |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US45604703P | 2003-03-18 | 2003-03-18 | |
| US60/456,047 | 2003-03-18 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2004082682A1 true WO2004082682A1 (en) | 2004-09-30 |
Family
ID=33030083
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/US2004/007792 WO2004082682A1 (en) | 2003-03-18 | 2004-03-15 | Amino cyclobutylamide modulators of chemokine receptor activity |
Country Status (7)
| Country | Link |
|---|---|
| US (1) | US7553841B2 (en) |
| EP (1) | EP1617841A4 (en) |
| JP (1) | JP2006520783A (en) |
| CN (1) | CN1787818A (en) |
| AU (1) | AU2004222336B2 (en) |
| CA (1) | CA2519220A1 (en) |
| WO (1) | WO2004082682A1 (en) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005070133A3 (en) * | 2004-01-20 | 2005-09-01 | Merck & Co Inc | 2,6-disubstituted piperidines as modulators of chemokine receptor activity |
| JP2010500403A (en) * | 2006-08-16 | 2010-01-07 | エルジー ライフサイエンス リミテッド | Novel process for producing 3-amino-5-fluoro-4-dialkoxypentanoic acid ester |
| US8067415B2 (en) | 2005-11-01 | 2011-11-29 | Millennium Pharmaceuticals, Inc. | Compounds useful as antagonists of CCR2 |
| US8067457B2 (en) | 2005-11-01 | 2011-11-29 | Millennium Pharmaceuticals, Inc. | Compounds useful as antagonists of CCR2 |
| US9783540B2 (en) | 2015-05-21 | 2017-10-10 | Chemocentryx, Inc. | Substituted tetrahydropyrans as CCR2 modulators |
| US11154556B2 (en) | 2018-01-08 | 2021-10-26 | Chemocentryx, Inc. | Methods of treating solid tumors with CCR2 antagonists |
| US11304952B2 (en) | 2017-09-25 | 2022-04-19 | Chemocentryx, Inc. | Combination therapy using a chemokine receptor 2 (CCR2) antagonist and a PD-1/PD-L1 inhibitor |
| US11986466B2 (en) | 2018-01-08 | 2024-05-21 | Chemocentryx, Inc. | Methods of treating solid tumors with CCR2 antagonists |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6374762B1 (en) * | 1997-10-27 | 2002-04-23 | Correct Craft, Inc. | Water sport towing apparatus |
| US7786141B2 (en) * | 2004-08-19 | 2010-08-31 | Vertex Pharmaceuticals Incorporated | Dihydrospiroindene modulators of muscarinic receptors |
| RU2007109794A (en) * | 2004-08-19 | 2008-09-27 | Вертекс Фармасьютикалз, Инкорпорейтед (Us) | MUSCARINE RECEPTOR MODULATORS |
| AU2005309365B2 (en) * | 2004-11-29 | 2011-10-06 | Vertex Pharmaceuticals Incorporated | Modulators of muscarinic receptors |
| AU2006330866A1 (en) * | 2005-12-22 | 2007-07-05 | Vertex Pharmaceuticals Incorporated | Modulators of muscarinic receptors |
| EP1988892A2 (en) | 2006-02-22 | 2008-11-12 | Vertex Pharmaceuticals Incorporated | Modulators of muscarinic receptors |
| ATE517106T1 (en) * | 2006-02-22 | 2011-08-15 | Vertex Pharma | CONDENSED SPIROPIPERIDINE AS MODULATORS OF MUSCARINOUS RECEPTORS |
| US20090105225A1 (en) * | 2006-04-20 | 2009-04-23 | Glaxo Group Limited | 2-Substituted 4-Benzylphthalazinone Derivatives as Histamine H1 and H3 Antagonists |
| CN101500565A (en) * | 2006-06-29 | 2009-08-05 | 弗特克斯药品有限公司 | Modulators of muscarinic receptors |
| CN101553231A (en) * | 2006-08-15 | 2009-10-07 | 弗特克斯药品有限公司 | Modulators of muscarinic receptors |
| JP2010501561A (en) | 2006-08-18 | 2010-01-21 | バーテックス ファーマシューティカルズ インコーポレイテッド | Modulator of muscarinic receptor |
| DE602007005167D1 (en) * | 2006-12-20 | 2010-04-15 | Glaxo Group Ltd | 4-BENZYL-1 (2H) -PHTHALAZINONE AS H1-RECEPTOR ANTAGONISTS |
| CA2700724A1 (en) | 2007-10-03 | 2009-04-09 | Vertex Pharmaceuticals Incorporated | Modulators of muscarinic receptors |
| WO2009047336A1 (en) * | 2007-10-11 | 2009-04-16 | Glaxo Group Limited | Phthalazine and pyrido [3,4-d] pyridaz ine compounds as h1 receptor antagonists |
| US8410139B2 (en) * | 2009-10-07 | 2013-04-02 | Bristol-Myers Squibb Company | Prodrugs of a piperidinyl derivative as modulators of chemokine receptor activity |
| TW201204717A (en) * | 2010-06-17 | 2012-02-01 | Janssen Pharmaceutica Nv | Cyclohexyl-azetidinyl antagonists of CCR2 |
| CN103232340A (en) * | 2013-05-02 | 2013-08-07 | 兰州沃金环保科技有限公司 | Synthesis method of 3-oxocyclobutanecarboxylic acid |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6743794B2 (en) * | 1999-12-22 | 2004-06-01 | Eli Lilly And Company | Methods and compounds for inhibiting MRP1 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001022919A2 (en) * | 1999-09-30 | 2001-04-05 | Merck & Co., Inc. | Spiro[bicyclic -azacycloalkyl and -cycloalkyl] derivatives and uses thereof |
| WO2002013824A1 (en) * | 2000-08-17 | 2002-02-21 | Merck & Co., Inc. | Cyclopentyl modulators of chemokine receptor activity |
| WO2004110376A2 (en) | 2003-06-06 | 2004-12-23 | Merck & Co., Inc. | Ccr-2 antagonists for treatment of neuropathic pain |
-
2004
- 2004-03-15 US US10/549,739 patent/US7553841B2/en not_active Expired - Fee Related
- 2004-03-15 WO PCT/US2004/007792 patent/WO2004082682A1/en active Application Filing
- 2004-03-15 CN CNA2004800131430A patent/CN1787818A/en active Pending
- 2004-03-15 AU AU2004222336A patent/AU2004222336B2/en not_active Expired - Fee Related
- 2004-03-15 EP EP04720791A patent/EP1617841A4/en not_active Withdrawn
- 2004-03-15 JP JP2006507176A patent/JP2006520783A/en active Pending
- 2004-03-15 CA CA002519220A patent/CA2519220A1/en not_active Abandoned
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6743794B2 (en) * | 1999-12-22 | 2004-06-01 | Eli Lilly And Company | Methods and compounds for inhibiting MRP1 |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005070133A3 (en) * | 2004-01-20 | 2005-09-01 | Merck & Co Inc | 2,6-disubstituted piperidines as modulators of chemokine receptor activity |
| US7410961B2 (en) * | 2004-01-20 | 2008-08-12 | Merck & Co., Inc. | 2,6-disubstituted piperiddines as modulators |
| US8067415B2 (en) | 2005-11-01 | 2011-11-29 | Millennium Pharmaceuticals, Inc. | Compounds useful as antagonists of CCR2 |
| US8067457B2 (en) | 2005-11-01 | 2011-11-29 | Millennium Pharmaceuticals, Inc. | Compounds useful as antagonists of CCR2 |
| JP2010500403A (en) * | 2006-08-16 | 2010-01-07 | エルジー ライフサイエンス リミテッド | Novel process for producing 3-amino-5-fluoro-4-dialkoxypentanoic acid ester |
| US9783540B2 (en) | 2015-05-21 | 2017-10-10 | Chemocentryx, Inc. | Substituted tetrahydropyrans as CCR2 modulators |
| US10464934B2 (en) | 2015-05-21 | 2019-11-05 | Chemocentryx, Inc. | Substituted tetrahydropyrans as CCR2 modulators |
| US12054484B2 (en) | 2015-05-21 | 2024-08-06 | Chemocentryx, Inc. | Substituted tetrahydropyrans as CCR2 modulators |
| US11304952B2 (en) | 2017-09-25 | 2022-04-19 | Chemocentryx, Inc. | Combination therapy using a chemokine receptor 2 (CCR2) antagonist and a PD-1/PD-L1 inhibitor |
| US11154556B2 (en) | 2018-01-08 | 2021-10-26 | Chemocentryx, Inc. | Methods of treating solid tumors with CCR2 antagonists |
| US11986466B2 (en) | 2018-01-08 | 2024-05-21 | Chemocentryx, Inc. | Methods of treating solid tumors with CCR2 antagonists |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2519220A1 (en) | 2004-09-30 |
| CN1787818A (en) | 2006-06-14 |
| US7553841B2 (en) | 2009-06-30 |
| US20060211722A1 (en) | 2006-09-21 |
| EP1617841A1 (en) | 2006-01-25 |
| EP1617841A4 (en) | 2008-08-13 |
| JP2006520783A (en) | 2006-09-14 |
| AU2004222336A1 (en) | 2004-09-30 |
| AU2004222336B2 (en) | 2010-06-03 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| AU2004222336B2 (en) | Amino cyclobutylamide modulators of chemokine receptor activity | |
| JP4903333B2 (en) | Chemokine receptor antagonist and method of use thereof | |
| WO2005072361A2 (en) | Aminocyclopentyl pyridopyrazinone modulators of chemokine receptor activity | |
| JP2008534496A (en) | Novel tetrahydro-1H-pyrido [4,3-b] indole derivatives as CB1 'receptor ligands | |
| KR100485147B1 (en) | 1- (1,2-disubstituted piperidinyl) -4- (fused imidazole) -piperidine derivatives | |
| AU2005214319A1 (en) | Amino heterocyclic modulators of chemokine receptor activity | |
| EP1501507A2 (en) | Tetrahydropyranyl cyclopentyl tetrahydropyridopyridine modulators of chemokine receptor activity | |
| EP1501803A2 (en) | Tetrahydropyranyl cyclopentyl tetrahydroisoquinoline modulators of chemokine receptor activity | |
| JP2004506013A (en) | Cyclopentyl chemokine receptor activity modulator | |
| WO2005070133A2 (en) | 2,6-disubstituted piperidines as modulators of chemokine receptor activity | |
| EP1558243A2 (en) | Tetrahydropyranyl cyclopentyl benzylamide modulators of chemokine receptor activity | |
| JP3216889B2 (en) | New 2,9-disubstituted-4H-pyrido [1,2-A] pyrimidin-4-one | |
| AU2003284975A1 (en) | Heteroarylpiperidine modulators of chemokine receptor activity | |
| WO2004094371A2 (en) | Heterocyclic cyclopentyl tetrahydroisoquinoline and tetrahydropyridopyridine modulators of chemokine receptor activity | |
| AU2004215409B2 (en) | Aminocyclopentyl fused heterotricylicamide modulators of chemokine receptor activity | |
| US7598243B2 (en) | Heterocyclic cyclopentyl tetrahydroisoquinoline and tetrahydropyridopyridine modulators of chemokine receptor activity | |
| WO2005014537A2 (en) | Tetrahydropyran heterocyclic cyclopentyl heteroaryl modulators of chemokine receptor activity | |
| JP2013518094A (en) | Aminocyclohexane and aminotetrahydropyran and related compounds as gamma-secretase modulators | |
| KR20000005226A (en) | Hexahydro-pyrido(4,3-b)indole derivatives as antipsychotic drugs |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 3929/DELNP/2005 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004222336 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004720791 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2519220 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2006507176 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10549739 Country of ref document: US |
|
| ENP | Entry into the national phase |
Ref document number: 2004222336 Country of ref document: AU Date of ref document: 20040315 Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004222336 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 20048131430 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004720791 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 10549739 Country of ref document: US |